User login
Granulomatous Pigmented Purpuric Dermatosis
Pigmented purpuric dermatoses (PPDs) are a spectrum of chronic disorders that present as speckled brown to purpuric lesions and orange-brown discoloration of the skin.1 Eruptions generally occur in middle-aged to elderly patients and commonly follow a chronic waxing and waning course.2 Lesions usually are found in a localized distribution on the legs. Histologically, PPD presents with perivascular infiltrates of lymphocytes and macrophages centered around the superficial small blood vessels with narrowing of the lumina. Extravasation of red blood cells and hemosiderin deposition are commonly seen in the absence of vasculitis.
The etiology of PPD is unknown; however, important cofactors include venous hypertension, exercise and gravitational dependency, capillary fragility, focal infections, and chemical ingestions.1 Drugs are the most important provoking factors, including acetaminophen, aspirin, adalin, carbromal, chlordiazepoxide, glipizide, glybuzole, hydralazine, meprobamate, dipyridamole, reserpine, thiamine, and interferon-alfa, as well as medroxyprogesterone acetate injection. Other phenomena include contact allergy and alcohol ingestion.1
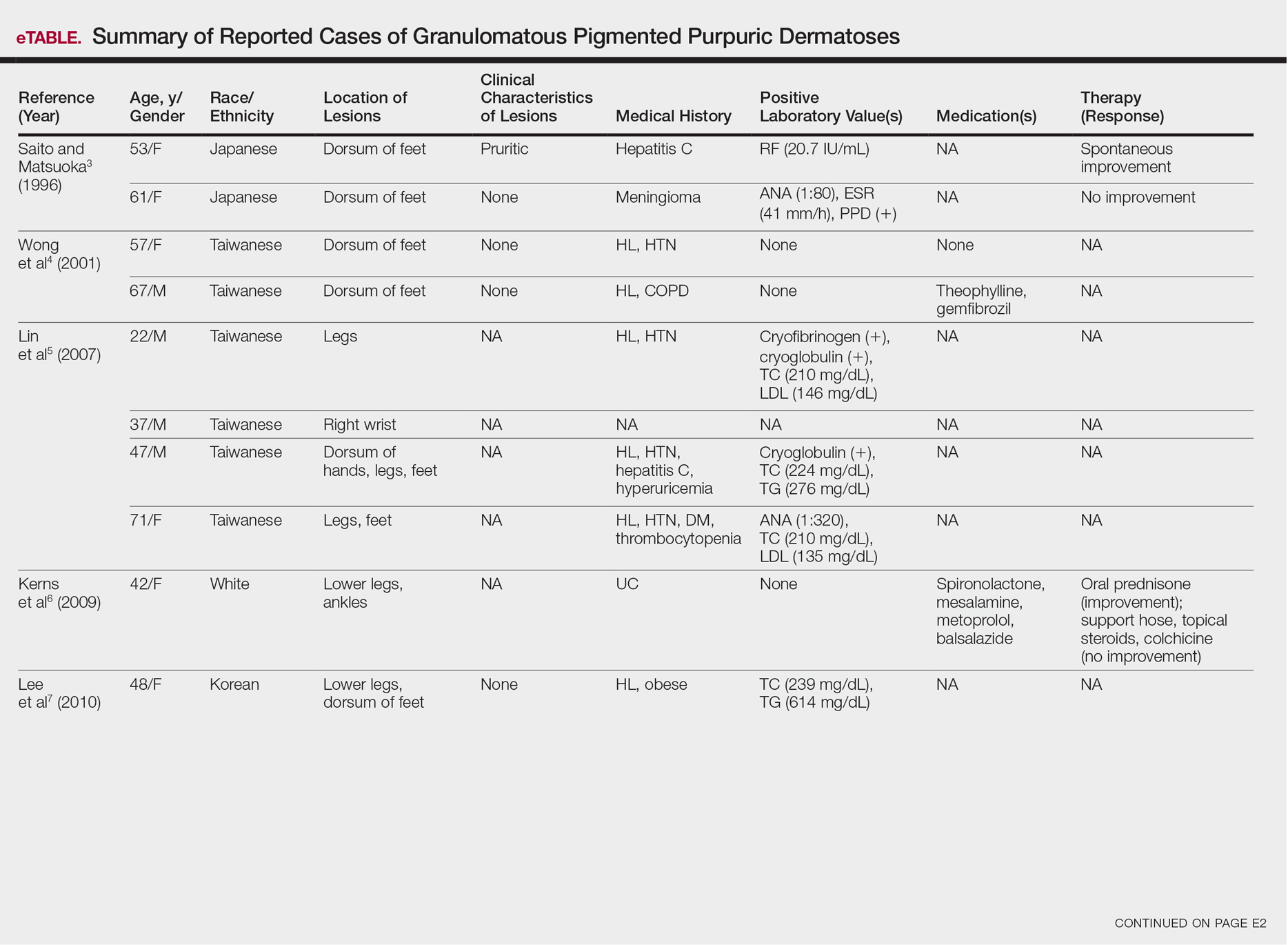
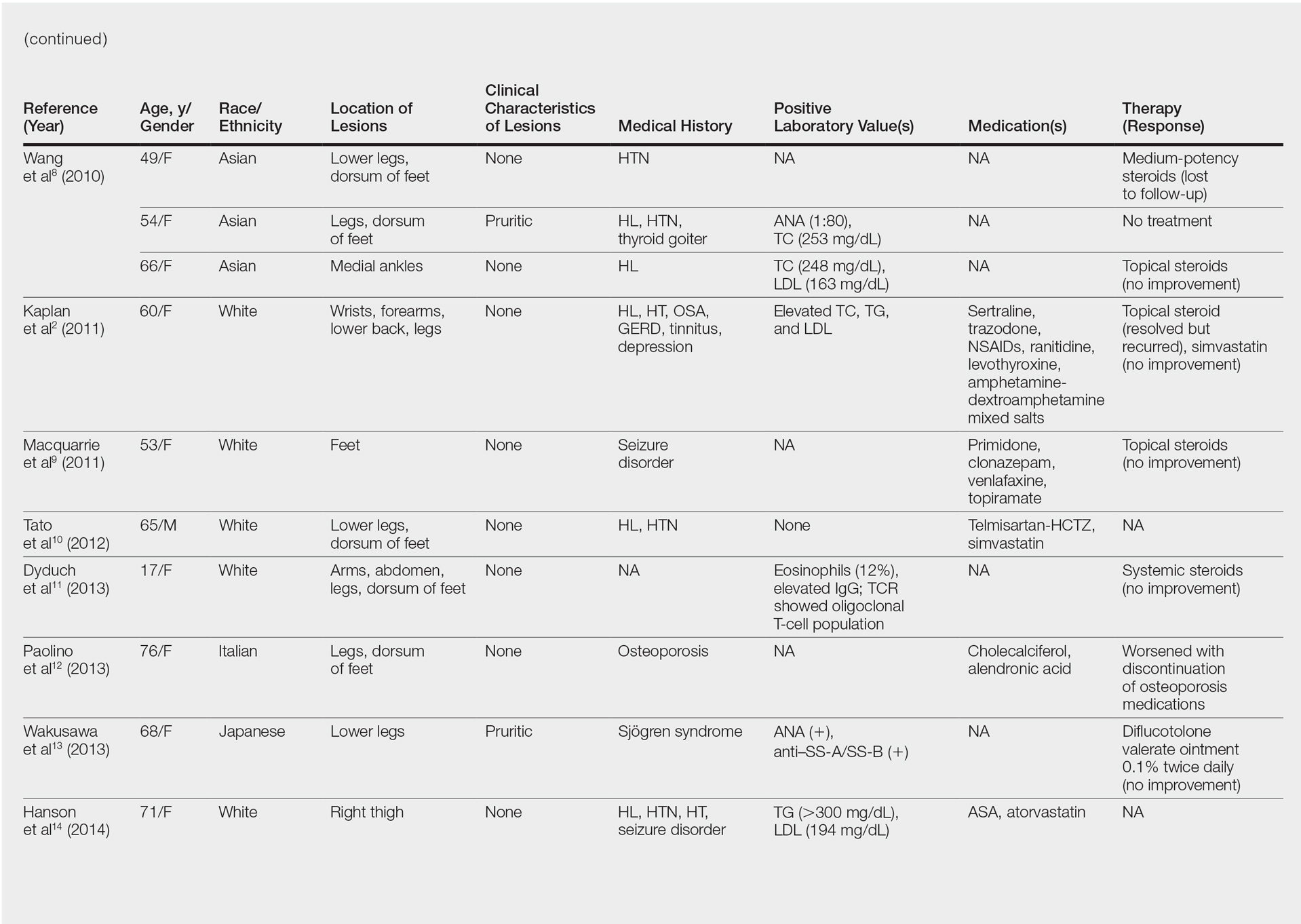
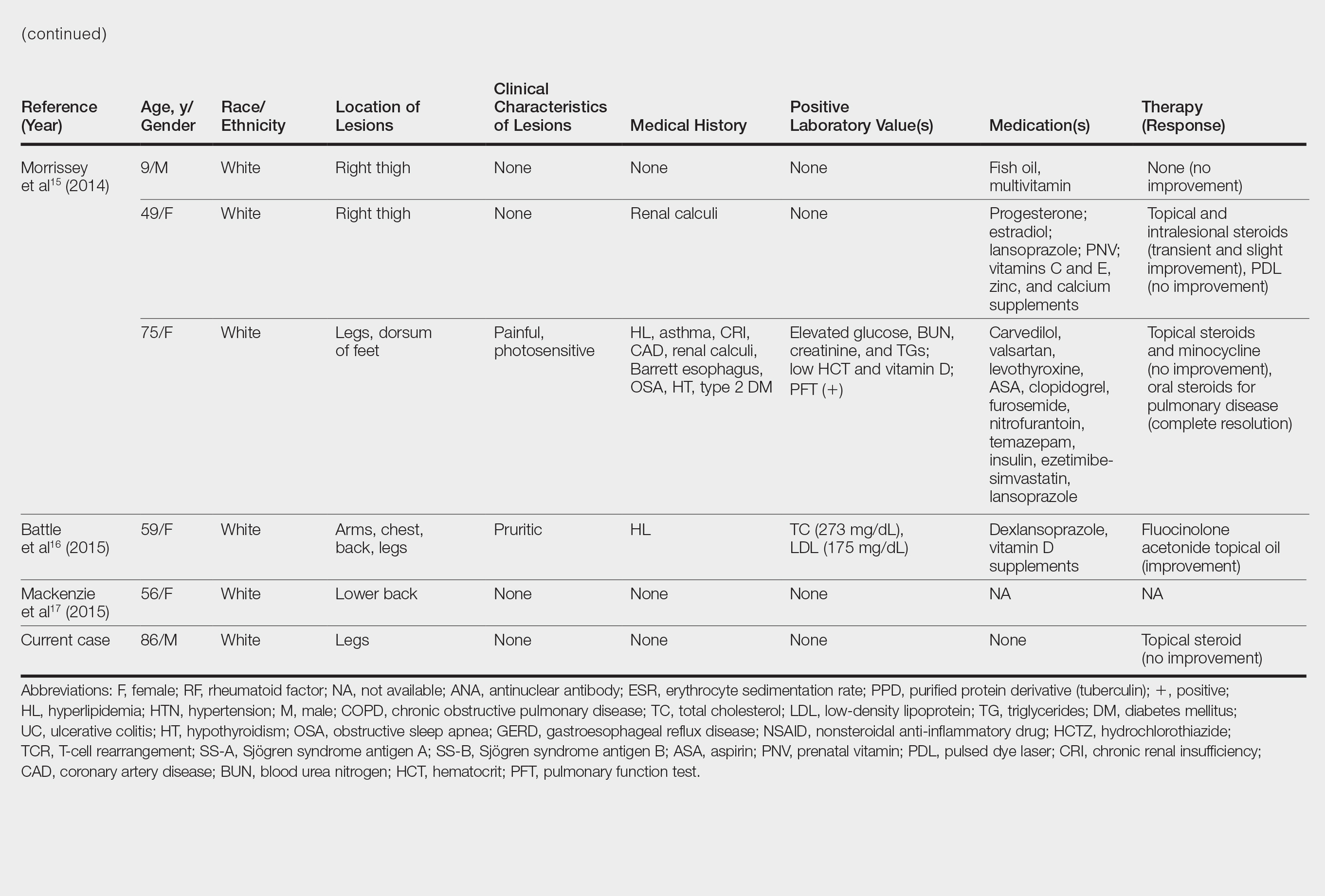
Although the diagnosis often is made clinically, many forms of PPD exist. The 4 main forms include Schaumberg disease, purpura annularis telangiectaticum of Majocchi, pigmented purpuric lichenoid dermatitis of Gougerot and Blum, and eczematoidlike purpura of Doucas and Kapetanakis. Less common variants include itching purpura of Lowenthal, lichen purpuricus, lichen aureus, granulomatous pigmented purpura, transitory pigmented purpuric dermatosis, and linear pigmented purpura.1Granulomatous PPD (GPPD) is a rare histologic variant of PPD. Clinically, it is indistinguishable from other forms of PPD but reveals itself histologically with granulomatous infiltrates superimposed on classic PPD. We report a case of GPPD and provide a thorough literature review focusing on epidemiology, clinical symptoms, and treatment.2-17 The eTable summarizes all reported cases of GPPD.
Case Report
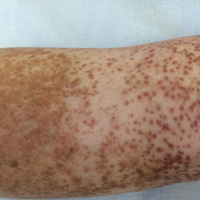
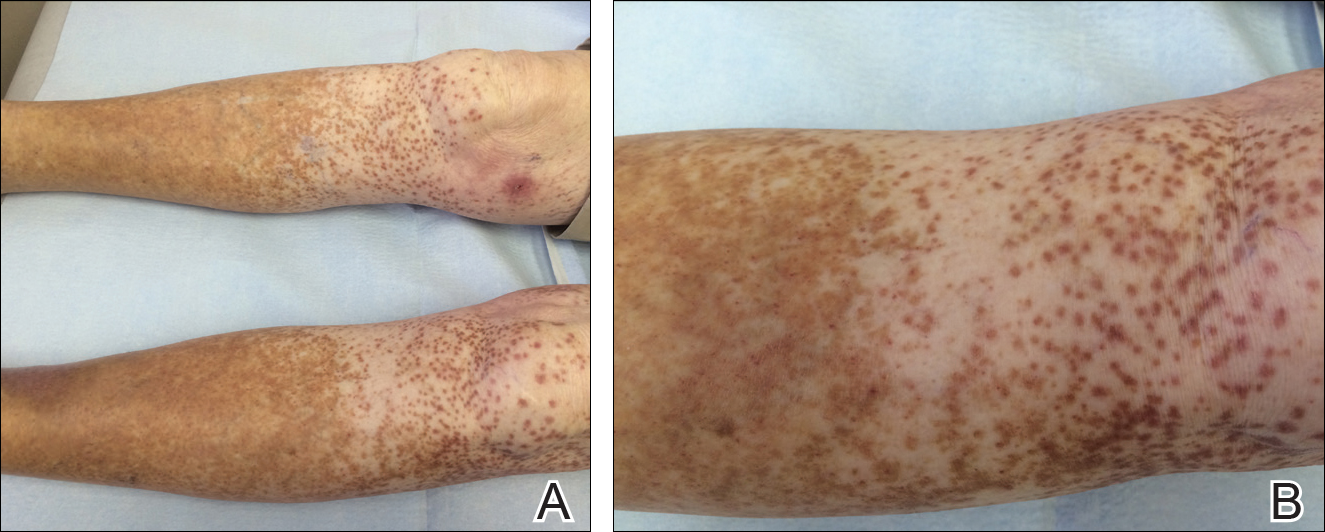
An 86-year-old white man with no remarkable medical history presented with an asymptomatic eruption over the bilateral shins extending up both thighs of 6 years’ duration (Figure 1). It began as a 15-cm patch on the right medial thigh that rapidly spread over 1 year to involve the majority of the legs. Physical examination revealed scattered 1- to 2-mm brown macules coalescing into patches on both legs. The patches increased in density distally and extended from the bilateral thighs to the ankles. Edema of the legs was absent, and lesions were nonblanchable and without scale or induration. The differential diagnoses included stasis dermatitis, vasculitis, and PPD. All laboratory values were within reference range, including complete blood cell count, comprehensive metabolic panel, urine analysis, and lipid profile.
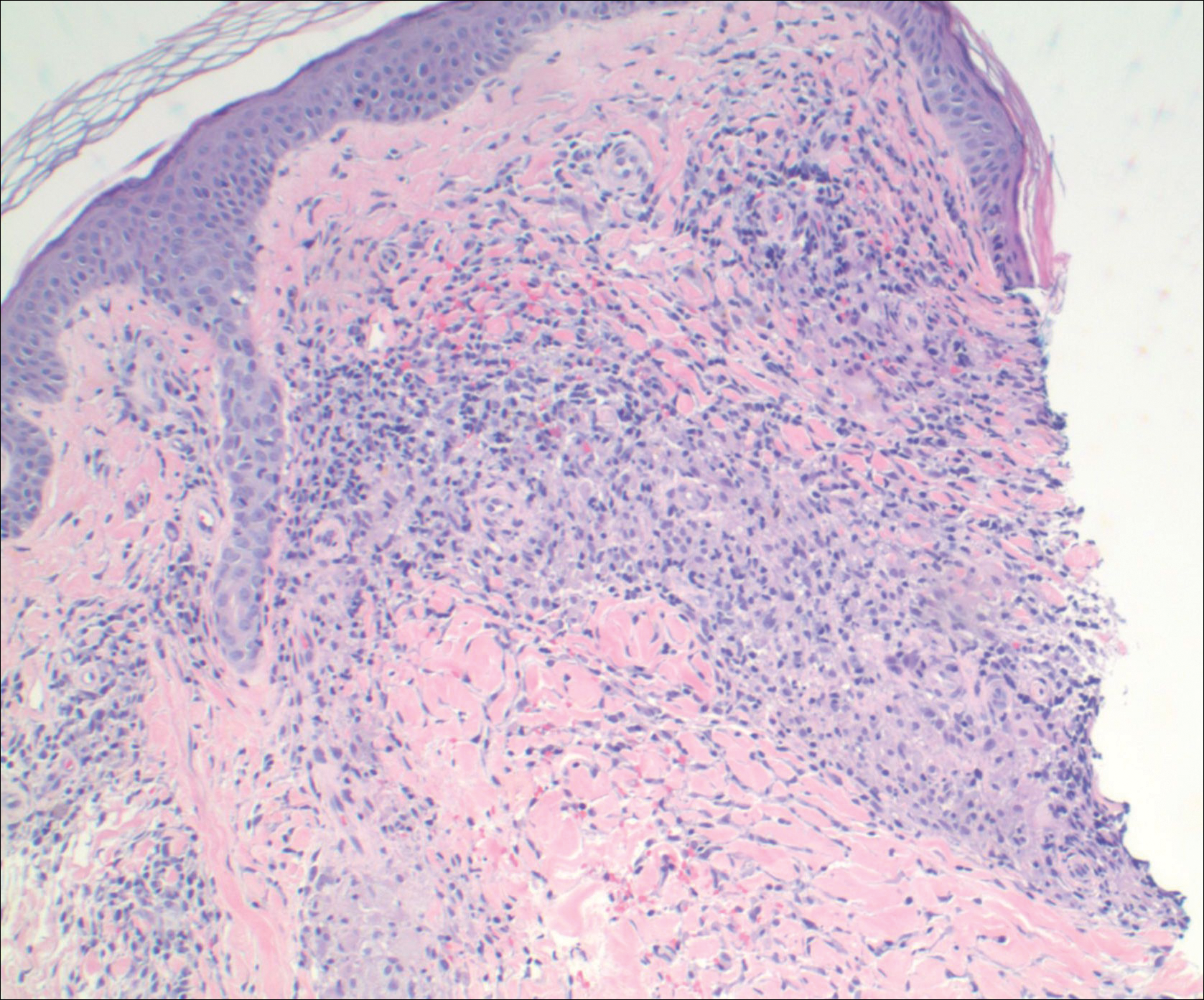
A punch biopsy from the distal right thigh revealed a superficial to mid dermal perivascular lymphocyte-predominant infiltrate with associated siderophages and a focal granulomatous infiltrate comprised of histiocytes (Figure 2). Periodic acid–Schiff, acid-fast bacilli, and Fite stains were negative for microorganisms. No eosinophils or leukocytoclasia were seen. The patient applied betamethasone dipropionate cream 0.05% twice daily for several weeks without improvement. Because the lesions were asymptomatic, he discontinued the topical medication.
Comment
Pathogenesis/Etiology of GPPD
Granulomatous PPD is a rare histological variant of PPD, which was first reported in 1996 by Saito and Matsuoka.3 Originally, GPPD was mainly thought to affect individuals in the Far East and be associated with the hepatitis C virus, antinuclear antibodies, or rheumatoid factor.3 Since its initial description, GPPD continues to predominantly be seen on the distal legs. According to a PubMed search of articles indexed for MEDLINE and the Michigan State University library database using the terms granulomatous pigmented purpuric dermatosis and pigmented purpuric dermatosis, 26 known cases including the current case (Asian, n=13; white, n=13) have been reported. The mean age of onset was 54.5 years and the female to male ratio was 2.5 to 1.
Currently, the etiology of GPPD is unknown; however, 13 reported cases have been associated with hyperlipidemia,2,4,5,7,8,10,14-16 which has led to the speculation that they may be related. Previous investigators have postulated that the granulomatous infiltrate is a response to lipid deposition in the endothelial cells or that the elevated lipid levels launch an incompetent helper T cell (TH1) response, leading to granuloma formation.5,7,8 Currently, hyperlipidemia is present in 50% of patients and appears to be trending downward as more cases present in the literature.
Medications have been implicated in the pathogenesis of PPD and may have a possible role in the development of the granulomatous variant.9 One case report noted preceding medication changes, alluding to the possibility of aminosalicylates being the culprit.6
Another case described GPPD appearing after an upper respiratory tract infection.11 Comorbidities are not uncommon in patients presenting with GPPD. Although the majority of cases are single reports, they include systemic derangements such as hepatitis C,3,5 Sjögren syndrome,13 hypertension,2,4,5,8,10,14,15 seizure disorder,9,14 ulcerative colitis,6 diabetes mellitus,5,15 meningioma,3 renal calculi,15 thrombocytopenia,5 chronic obstructive pulmonary disease,4 thyroid goiter,8 obstructive sleep apnea,2,15 osteoporosis,12 asthma,15 gastroesophageal reflux disease/Barrett esophagus,2,15 hypothyroidism,2,14,15 and hyperuricemia.5
Clinical Presentation
Clinically, GPPD commonly presents as asymptomatic petechiae and bronze discoloration of the lower legs. The clinical presentation can vary from a solitary lesion to a localized eruption typically on the lower legs or rarely a widespread eruption. A review of the literature revealed 5 cases presenting on the upper arms2,5,11,16 and 4 on the trunk.2,11,16,17 Four patients presented with pruritus3,8,13,16 and 1 described pain and photosensitive lesions.15 No other clinical signs of hyperlipidemia were described (eg, xanthomas). The duration of the disease has a wide spectrum, ranging from 3 weeks to 20 years.4,16
Histopathology
With the increasing trend toward dermatoscopic evaluation, 2 reviews evaluated dermatoscopic features of GPPD. These reports described scattered, round to oval, red dots, globules, and patches with a diffuse red-brown or coppery background of pigmentation.14,17
The granulomatous variant of PPD is characterized histopathologically by ill-defined, nonnecrotizing granulomas admixed with a lymphocytic infiltrate. Commonly, erythrocyte extravasation and hemosiderin are seen with granulomas superimposed on classic changes of PPD.15 Vasculitis features including endothelial swelling, fibrinoid necrosis, and leukocytoclasia are absent. Rarely, eosinophils are seen.6 Mild epidermal spongiosis and exocytosis of lymphocytes may be seen in all variants of PPD, except lichen aureus.1 This exocytosis was observed focally in one case of GPPD.4 Although loosely formed granulomas in the papillary dermis are characteristic, 7 cases have had a concomitant lichenoid infiltrate.2,9-11,15,16
Kaplan et al2 reported granulomatous and nongranulomatous PPD occurring together in different areas of the body. A new granulomatous variant was proposed in a 2015 report that revealed 2 patients with granulomatous infiltrates in the mid to deep dermis rather than the classic superficial dermis.15 One case of GPPD was suspicious for progression into mycosis fungoides (MF) and described a lichenoid infiltrate with mild atypical and small lymphocytes migrating into the epidermis.11 Follow-up biopsy lacked epidermotropism and quantitative representation of T-cell subsets. The diagnosis of early-phase MF was based on the progressive clinical course rather than immunohistologic and molecular findings.11 One other case exhibited minimal epidermotropism.15
Management of GPPD should require a lipid profile with other tests to assess cardiovascular risk.10 A thorough medication review and a punch rather than a shave biopsy should be performed, especially because granulomatous infiltrates have been found in the mid to deep dermis.15 With the lack of rebiopsies documented, follow-up and rebiopsy has been suggested if there is suspicion of MF; however, we favor rebiopsy at a later time to help reveal the course of this disease and rule out progression into MF.
Therapy
Thus far, therapy has mostly been with oral and topical steroids. Five case reports noted improvement,2,3,6,15,16 2 with oral and 3 with topical steroids. However, therapy has been discouraging, with clinical improvement being transient in most treatment-responsive patients. One case spontaneously resolved.3 Ten cases did not document therapy or follow-up.4,5,7,10,14,17 Only 1 case reported follow-up after treatment with simvastatin; unfortunately, the patient had no improvement.2 Our case revealed no improvement with topical steroids.
Conclusion
The exact pathogenesis of GPPD is unknown. The initial impression that GPPD was a disease in Far East Asians and patients with hyperlipidemia is becoming less clear. Based on the current literature including the addition of our case, the prevalence appears to be equal among white individuals and Asians, possibly due to increased awareness of this condition and documentation in the literature. Correlation with systemic disorders such as hyperlipidemia and hypertensive medications needs further review. Eight cases reported a medical history of hypertension.4,5,8,10,14 With antihypertensive medications being a potential culprit of PPD, this etiology should not be overlooked. A punch biopsy should be performed, especially because granulomatous infiltrates may be lurking in the mid to deep dermis.15 Granulomatous PPD has a chronic course with a disappointing response to therapy but appears to be benign in nature.12 A rebiopsy is recommended if MF is suspected. Evaluation of GPPD following therapy for hyperlipidemia is not well documented and should be pursued. Clinicians and pathologists should be aware of the suspected associations and consider this variant when dermal granulomatous infiltrates are present with a background of PPD.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Kaplan J, Burgin S, Sepehr A. Granulomatous pigmented purpura: report of a case and review of the literature. J Cutan Pathol. 2011;38:984-989.
- Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
- Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
- Lin WL, Kuo TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia. Clin Exp Dermatol. 2007;32:513-515.
- Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
- Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidaemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
- Wang J, Wu Y, Hsiao P, et al. Granulomatous pigmented purpuric dermatoses: report of three cases and review of the literature. Dermatologica Sinica. 2010;28:77-81.
- Macquarrie EK, Pasternak S, Torok M, et al. Persistent pigmented purpuric dermatitis: granulomatous variant. J Cutan Pathol. 2011;38:979-983.
- Tato BP, Marinero Escobedo S, Pérez González YC, et al. Granulomatous variant of pigmented purpuric dermatosis. Am J Dermatopathol. 2012;34:746-748.
- Dyduch G, Zuber Z, Turowska-Heydel D, et al. Granulomatous pigmented purpura in an adolescent girl: a precursor of mycosis fungoides? Pol J Pathol. 2013;64:157-159; answer 160.
- Paolino S, Cinotti E, Merlo V, et al. Progressive petechial and pigmented macules and papules on the lower extremities. Am J Dermatopathol. 2013;35:370, 388.
- Wakusawa C, Fujimura T, Haga T, et al. Granulomatous pigmented purpuric dermatitis associated with primary Sjögren’s syndrome. Acta Derm Venereol. 2013;93:95-96.
- Hanson C, Fischer R, Fraga G, et al. Granulomatous pigmented purpuric dermatosis: an unusual variant associated with hyperlipidemia. Dermatol Online J. 2014;21. pii:13030/qt0tp272d1.
- Morrissey K, Rosenbach M, DeHoratius D, et al. Granulomatous changes associated with pigmented purpuric dermatosis. Cutis. 2014;94:197-202.
- Battle LR, Shalin SC, Gao L. Granulomatous pigmented purpuric dermatosis [published online December 18, 2014]. Clin Exp Dermatol. 2015;40:387-390.
- Mackenzie AI, Biswas A. Granulomatous pigmented purpuric dermatosis: report of a case with atypical clinical presentation including dermoscopic findings. Am J Dermatopathol. 2015;37:311-314.
Pigmented purpuric dermatoses (PPDs) are a spectrum of chronic disorders that present as speckled brown to purpuric lesions and orange-brown discoloration of the skin.1 Eruptions generally occur in middle-aged to elderly patients and commonly follow a chronic waxing and waning course.2 Lesions usually are found in a localized distribution on the legs. Histologically, PPD presents with perivascular infiltrates of lymphocytes and macrophages centered around the superficial small blood vessels with narrowing of the lumina. Extravasation of red blood cells and hemosiderin deposition are commonly seen in the absence of vasculitis.
The etiology of PPD is unknown; however, important cofactors include venous hypertension, exercise and gravitational dependency, capillary fragility, focal infections, and chemical ingestions.1 Drugs are the most important provoking factors, including acetaminophen, aspirin, adalin, carbromal, chlordiazepoxide, glipizide, glybuzole, hydralazine, meprobamate, dipyridamole, reserpine, thiamine, and interferon-alfa, as well as medroxyprogesterone acetate injection. Other phenomena include contact allergy and alcohol ingestion.1
Although the diagnosis often is made clinically, many forms of PPD exist. The 4 main forms include Schaumberg disease, purpura annularis telangiectaticum of Majocchi, pigmented purpuric lichenoid dermatitis of Gougerot and Blum, and eczematoidlike purpura of Doucas and Kapetanakis. Less common variants include itching purpura of Lowenthal, lichen purpuricus, lichen aureus, granulomatous pigmented purpura, transitory pigmented purpuric dermatosis, and linear pigmented purpura.1Granulomatous PPD (GPPD) is a rare histologic variant of PPD. Clinically, it is indistinguishable from other forms of PPD but reveals itself histologically with granulomatous infiltrates superimposed on classic PPD. We report a case of GPPD and provide a thorough literature review focusing on epidemiology, clinical symptoms, and treatment.2-17 The eTable summarizes all reported cases of GPPD.
Case Report
An 86-year-old white man with no remarkable medical history presented with an asymptomatic eruption over the bilateral shins extending up both thighs of 6 years’ duration (Figure 1). It began as a 15-cm patch on the right medial thigh that rapidly spread over 1 year to involve the majority of the legs. Physical examination revealed scattered 1- to 2-mm brown macules coalescing into patches on both legs. The patches increased in density distally and extended from the bilateral thighs to the ankles. Edema of the legs was absent, and lesions were nonblanchable and without scale or induration. The differential diagnoses included stasis dermatitis, vasculitis, and PPD. All laboratory values were within reference range, including complete blood cell count, comprehensive metabolic panel, urine analysis, and lipid profile.
A punch biopsy from the distal right thigh revealed a superficial to mid dermal perivascular lymphocyte-predominant infiltrate with associated siderophages and a focal granulomatous infiltrate comprised of histiocytes (Figure 2). Periodic acid–Schiff, acid-fast bacilli, and Fite stains were negative for microorganisms. No eosinophils or leukocytoclasia were seen. The patient applied betamethasone dipropionate cream 0.05% twice daily for several weeks without improvement. Because the lesions were asymptomatic, he discontinued the topical medication.
Comment
Pathogenesis/Etiology of GPPD
Granulomatous PPD is a rare histological variant of PPD, which was first reported in 1996 by Saito and Matsuoka.3 Originally, GPPD was mainly thought to affect individuals in the Far East and be associated with the hepatitis C virus, antinuclear antibodies, or rheumatoid factor.3 Since its initial description, GPPD continues to predominantly be seen on the distal legs. According to a PubMed search of articles indexed for MEDLINE and the Michigan State University library database using the terms granulomatous pigmented purpuric dermatosis and pigmented purpuric dermatosis, 26 known cases including the current case (Asian, n=13; white, n=13) have been reported. The mean age of onset was 54.5 years and the female to male ratio was 2.5 to 1.
Currently, the etiology of GPPD is unknown; however, 13 reported cases have been associated with hyperlipidemia,2,4,5,7,8,10,14-16 which has led to the speculation that they may be related. Previous investigators have postulated that the granulomatous infiltrate is a response to lipid deposition in the endothelial cells or that the elevated lipid levels launch an incompetent helper T cell (TH1) response, leading to granuloma formation.5,7,8 Currently, hyperlipidemia is present in 50% of patients and appears to be trending downward as more cases present in the literature.
Medications have been implicated in the pathogenesis of PPD and may have a possible role in the development of the granulomatous variant.9 One case report noted preceding medication changes, alluding to the possibility of aminosalicylates being the culprit.6
Another case described GPPD appearing after an upper respiratory tract infection.11 Comorbidities are not uncommon in patients presenting with GPPD. Although the majority of cases are single reports, they include systemic derangements such as hepatitis C,3,5 Sjögren syndrome,13 hypertension,2,4,5,8,10,14,15 seizure disorder,9,14 ulcerative colitis,6 diabetes mellitus,5,15 meningioma,3 renal calculi,15 thrombocytopenia,5 chronic obstructive pulmonary disease,4 thyroid goiter,8 obstructive sleep apnea,2,15 osteoporosis,12 asthma,15 gastroesophageal reflux disease/Barrett esophagus,2,15 hypothyroidism,2,14,15 and hyperuricemia.5
Clinical Presentation
Clinically, GPPD commonly presents as asymptomatic petechiae and bronze discoloration of the lower legs. The clinical presentation can vary from a solitary lesion to a localized eruption typically on the lower legs or rarely a widespread eruption. A review of the literature revealed 5 cases presenting on the upper arms2,5,11,16 and 4 on the trunk.2,11,16,17 Four patients presented with pruritus3,8,13,16 and 1 described pain and photosensitive lesions.15 No other clinical signs of hyperlipidemia were described (eg, xanthomas). The duration of the disease has a wide spectrum, ranging from 3 weeks to 20 years.4,16
Histopathology
With the increasing trend toward dermatoscopic evaluation, 2 reviews evaluated dermatoscopic features of GPPD. These reports described scattered, round to oval, red dots, globules, and patches with a diffuse red-brown or coppery background of pigmentation.14,17
The granulomatous variant of PPD is characterized histopathologically by ill-defined, nonnecrotizing granulomas admixed with a lymphocytic infiltrate. Commonly, erythrocyte extravasation and hemosiderin are seen with granulomas superimposed on classic changes of PPD.15 Vasculitis features including endothelial swelling, fibrinoid necrosis, and leukocytoclasia are absent. Rarely, eosinophils are seen.6 Mild epidermal spongiosis and exocytosis of lymphocytes may be seen in all variants of PPD, except lichen aureus.1 This exocytosis was observed focally in one case of GPPD.4 Although loosely formed granulomas in the papillary dermis are characteristic, 7 cases have had a concomitant lichenoid infiltrate.2,9-11,15,16
Kaplan et al2 reported granulomatous and nongranulomatous PPD occurring together in different areas of the body. A new granulomatous variant was proposed in a 2015 report that revealed 2 patients with granulomatous infiltrates in the mid to deep dermis rather than the classic superficial dermis.15 One case of GPPD was suspicious for progression into mycosis fungoides (MF) and described a lichenoid infiltrate with mild atypical and small lymphocytes migrating into the epidermis.11 Follow-up biopsy lacked epidermotropism and quantitative representation of T-cell subsets. The diagnosis of early-phase MF was based on the progressive clinical course rather than immunohistologic and molecular findings.11 One other case exhibited minimal epidermotropism.15
Management of GPPD should require a lipid profile with other tests to assess cardiovascular risk.10 A thorough medication review and a punch rather than a shave biopsy should be performed, especially because granulomatous infiltrates have been found in the mid to deep dermis.15 With the lack of rebiopsies documented, follow-up and rebiopsy has been suggested if there is suspicion of MF; however, we favor rebiopsy at a later time to help reveal the course of this disease and rule out progression into MF.
Therapy
Thus far, therapy has mostly been with oral and topical steroids. Five case reports noted improvement,2,3,6,15,16 2 with oral and 3 with topical steroids. However, therapy has been discouraging, with clinical improvement being transient in most treatment-responsive patients. One case spontaneously resolved.3 Ten cases did not document therapy or follow-up.4,5,7,10,14,17 Only 1 case reported follow-up after treatment with simvastatin; unfortunately, the patient had no improvement.2 Our case revealed no improvement with topical steroids.
Conclusion
The exact pathogenesis of GPPD is unknown. The initial impression that GPPD was a disease in Far East Asians and patients with hyperlipidemia is becoming less clear. Based on the current literature including the addition of our case, the prevalence appears to be equal among white individuals and Asians, possibly due to increased awareness of this condition and documentation in the literature. Correlation with systemic disorders such as hyperlipidemia and hypertensive medications needs further review. Eight cases reported a medical history of hypertension.4,5,8,10,14 With antihypertensive medications being a potential culprit of PPD, this etiology should not be overlooked. A punch biopsy should be performed, especially because granulomatous infiltrates may be lurking in the mid to deep dermis.15 Granulomatous PPD has a chronic course with a disappointing response to therapy but appears to be benign in nature.12 A rebiopsy is recommended if MF is suspected. Evaluation of GPPD following therapy for hyperlipidemia is not well documented and should be pursued. Clinicians and pathologists should be aware of the suspected associations and consider this variant when dermal granulomatous infiltrates are present with a background of PPD.
Pigmented purpuric dermatoses (PPDs) are a spectrum of chronic disorders that present as speckled brown to purpuric lesions and orange-brown discoloration of the skin.1 Eruptions generally occur in middle-aged to elderly patients and commonly follow a chronic waxing and waning course.2 Lesions usually are found in a localized distribution on the legs. Histologically, PPD presents with perivascular infiltrates of lymphocytes and macrophages centered around the superficial small blood vessels with narrowing of the lumina. Extravasation of red blood cells and hemosiderin deposition are commonly seen in the absence of vasculitis.
The etiology of PPD is unknown; however, important cofactors include venous hypertension, exercise and gravitational dependency, capillary fragility, focal infections, and chemical ingestions.1 Drugs are the most important provoking factors, including acetaminophen, aspirin, adalin, carbromal, chlordiazepoxide, glipizide, glybuzole, hydralazine, meprobamate, dipyridamole, reserpine, thiamine, and interferon-alfa, as well as medroxyprogesterone acetate injection. Other phenomena include contact allergy and alcohol ingestion.1
Although the diagnosis often is made clinically, many forms of PPD exist. The 4 main forms include Schaumberg disease, purpura annularis telangiectaticum of Majocchi, pigmented purpuric lichenoid dermatitis of Gougerot and Blum, and eczematoidlike purpura of Doucas and Kapetanakis. Less common variants include itching purpura of Lowenthal, lichen purpuricus, lichen aureus, granulomatous pigmented purpura, transitory pigmented purpuric dermatosis, and linear pigmented purpura.1Granulomatous PPD (GPPD) is a rare histologic variant of PPD. Clinically, it is indistinguishable from other forms of PPD but reveals itself histologically with granulomatous infiltrates superimposed on classic PPD. We report a case of GPPD and provide a thorough literature review focusing on epidemiology, clinical symptoms, and treatment.2-17 The eTable summarizes all reported cases of GPPD.
Case Report
An 86-year-old white man with no remarkable medical history presented with an asymptomatic eruption over the bilateral shins extending up both thighs of 6 years’ duration (Figure 1). It began as a 15-cm patch on the right medial thigh that rapidly spread over 1 year to involve the majority of the legs. Physical examination revealed scattered 1- to 2-mm brown macules coalescing into patches on both legs. The patches increased in density distally and extended from the bilateral thighs to the ankles. Edema of the legs was absent, and lesions were nonblanchable and without scale or induration. The differential diagnoses included stasis dermatitis, vasculitis, and PPD. All laboratory values were within reference range, including complete blood cell count, comprehensive metabolic panel, urine analysis, and lipid profile.
A punch biopsy from the distal right thigh revealed a superficial to mid dermal perivascular lymphocyte-predominant infiltrate with associated siderophages and a focal granulomatous infiltrate comprised of histiocytes (Figure 2). Periodic acid–Schiff, acid-fast bacilli, and Fite stains were negative for microorganisms. No eosinophils or leukocytoclasia were seen. The patient applied betamethasone dipropionate cream 0.05% twice daily for several weeks without improvement. Because the lesions were asymptomatic, he discontinued the topical medication.
Comment
Pathogenesis/Etiology of GPPD
Granulomatous PPD is a rare histological variant of PPD, which was first reported in 1996 by Saito and Matsuoka.3 Originally, GPPD was mainly thought to affect individuals in the Far East and be associated with the hepatitis C virus, antinuclear antibodies, or rheumatoid factor.3 Since its initial description, GPPD continues to predominantly be seen on the distal legs. According to a PubMed search of articles indexed for MEDLINE and the Michigan State University library database using the terms granulomatous pigmented purpuric dermatosis and pigmented purpuric dermatosis, 26 known cases including the current case (Asian, n=13; white, n=13) have been reported. The mean age of onset was 54.5 years and the female to male ratio was 2.5 to 1.
Currently, the etiology of GPPD is unknown; however, 13 reported cases have been associated with hyperlipidemia,2,4,5,7,8,10,14-16 which has led to the speculation that they may be related. Previous investigators have postulated that the granulomatous infiltrate is a response to lipid deposition in the endothelial cells or that the elevated lipid levels launch an incompetent helper T cell (TH1) response, leading to granuloma formation.5,7,8 Currently, hyperlipidemia is present in 50% of patients and appears to be trending downward as more cases present in the literature.
Medications have been implicated in the pathogenesis of PPD and may have a possible role in the development of the granulomatous variant.9 One case report noted preceding medication changes, alluding to the possibility of aminosalicylates being the culprit.6
Another case described GPPD appearing after an upper respiratory tract infection.11 Comorbidities are not uncommon in patients presenting with GPPD. Although the majority of cases are single reports, they include systemic derangements such as hepatitis C,3,5 Sjögren syndrome,13 hypertension,2,4,5,8,10,14,15 seizure disorder,9,14 ulcerative colitis,6 diabetes mellitus,5,15 meningioma,3 renal calculi,15 thrombocytopenia,5 chronic obstructive pulmonary disease,4 thyroid goiter,8 obstructive sleep apnea,2,15 osteoporosis,12 asthma,15 gastroesophageal reflux disease/Barrett esophagus,2,15 hypothyroidism,2,14,15 and hyperuricemia.5
Clinical Presentation
Clinically, GPPD commonly presents as asymptomatic petechiae and bronze discoloration of the lower legs. The clinical presentation can vary from a solitary lesion to a localized eruption typically on the lower legs or rarely a widespread eruption. A review of the literature revealed 5 cases presenting on the upper arms2,5,11,16 and 4 on the trunk.2,11,16,17 Four patients presented with pruritus3,8,13,16 and 1 described pain and photosensitive lesions.15 No other clinical signs of hyperlipidemia were described (eg, xanthomas). The duration of the disease has a wide spectrum, ranging from 3 weeks to 20 years.4,16
Histopathology
With the increasing trend toward dermatoscopic evaluation, 2 reviews evaluated dermatoscopic features of GPPD. These reports described scattered, round to oval, red dots, globules, and patches with a diffuse red-brown or coppery background of pigmentation.14,17
The granulomatous variant of PPD is characterized histopathologically by ill-defined, nonnecrotizing granulomas admixed with a lymphocytic infiltrate. Commonly, erythrocyte extravasation and hemosiderin are seen with granulomas superimposed on classic changes of PPD.15 Vasculitis features including endothelial swelling, fibrinoid necrosis, and leukocytoclasia are absent. Rarely, eosinophils are seen.6 Mild epidermal spongiosis and exocytosis of lymphocytes may be seen in all variants of PPD, except lichen aureus.1 This exocytosis was observed focally in one case of GPPD.4 Although loosely formed granulomas in the papillary dermis are characteristic, 7 cases have had a concomitant lichenoid infiltrate.2,9-11,15,16
Kaplan et al2 reported granulomatous and nongranulomatous PPD occurring together in different areas of the body. A new granulomatous variant was proposed in a 2015 report that revealed 2 patients with granulomatous infiltrates in the mid to deep dermis rather than the classic superficial dermis.15 One case of GPPD was suspicious for progression into mycosis fungoides (MF) and described a lichenoid infiltrate with mild atypical and small lymphocytes migrating into the epidermis.11 Follow-up biopsy lacked epidermotropism and quantitative representation of T-cell subsets. The diagnosis of early-phase MF was based on the progressive clinical course rather than immunohistologic and molecular findings.11 One other case exhibited minimal epidermotropism.15
Management of GPPD should require a lipid profile with other tests to assess cardiovascular risk.10 A thorough medication review and a punch rather than a shave biopsy should be performed, especially because granulomatous infiltrates have been found in the mid to deep dermis.15 With the lack of rebiopsies documented, follow-up and rebiopsy has been suggested if there is suspicion of MF; however, we favor rebiopsy at a later time to help reveal the course of this disease and rule out progression into MF.
Therapy
Thus far, therapy has mostly been with oral and topical steroids. Five case reports noted improvement,2,3,6,15,16 2 with oral and 3 with topical steroids. However, therapy has been discouraging, with clinical improvement being transient in most treatment-responsive patients. One case spontaneously resolved.3 Ten cases did not document therapy or follow-up.4,5,7,10,14,17 Only 1 case reported follow-up after treatment with simvastatin; unfortunately, the patient had no improvement.2 Our case revealed no improvement with topical steroids.
Conclusion
The exact pathogenesis of GPPD is unknown. The initial impression that GPPD was a disease in Far East Asians and patients with hyperlipidemia is becoming less clear. Based on the current literature including the addition of our case, the prevalence appears to be equal among white individuals and Asians, possibly due to increased awareness of this condition and documentation in the literature. Correlation with systemic disorders such as hyperlipidemia and hypertensive medications needs further review. Eight cases reported a medical history of hypertension.4,5,8,10,14 With antihypertensive medications being a potential culprit of PPD, this etiology should not be overlooked. A punch biopsy should be performed, especially because granulomatous infiltrates may be lurking in the mid to deep dermis.15 Granulomatous PPD has a chronic course with a disappointing response to therapy but appears to be benign in nature.12 A rebiopsy is recommended if MF is suspected. Evaluation of GPPD following therapy for hyperlipidemia is not well documented and should be pursued. Clinicians and pathologists should be aware of the suspected associations and consider this variant when dermal granulomatous infiltrates are present with a background of PPD.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Kaplan J, Burgin S, Sepehr A. Granulomatous pigmented purpura: report of a case and review of the literature. J Cutan Pathol. 2011;38:984-989.
- Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
- Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
- Lin WL, Kuo TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia. Clin Exp Dermatol. 2007;32:513-515.
- Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
- Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidaemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
- Wang J, Wu Y, Hsiao P, et al. Granulomatous pigmented purpuric dermatoses: report of three cases and review of the literature. Dermatologica Sinica. 2010;28:77-81.
- Macquarrie EK, Pasternak S, Torok M, et al. Persistent pigmented purpuric dermatitis: granulomatous variant. J Cutan Pathol. 2011;38:979-983.
- Tato BP, Marinero Escobedo S, Pérez González YC, et al. Granulomatous variant of pigmented purpuric dermatosis. Am J Dermatopathol. 2012;34:746-748.
- Dyduch G, Zuber Z, Turowska-Heydel D, et al. Granulomatous pigmented purpura in an adolescent girl: a precursor of mycosis fungoides? Pol J Pathol. 2013;64:157-159; answer 160.
- Paolino S, Cinotti E, Merlo V, et al. Progressive petechial and pigmented macules and papules on the lower extremities. Am J Dermatopathol. 2013;35:370, 388.
- Wakusawa C, Fujimura T, Haga T, et al. Granulomatous pigmented purpuric dermatitis associated with primary Sjögren’s syndrome. Acta Derm Venereol. 2013;93:95-96.
- Hanson C, Fischer R, Fraga G, et al. Granulomatous pigmented purpuric dermatosis: an unusual variant associated with hyperlipidemia. Dermatol Online J. 2014;21. pii:13030/qt0tp272d1.
- Morrissey K, Rosenbach M, DeHoratius D, et al. Granulomatous changes associated with pigmented purpuric dermatosis. Cutis. 2014;94:197-202.
- Battle LR, Shalin SC, Gao L. Granulomatous pigmented purpuric dermatosis [published online December 18, 2014]. Clin Exp Dermatol. 2015;40:387-390.
- Mackenzie AI, Biswas A. Granulomatous pigmented purpuric dermatosis: report of a case with atypical clinical presentation including dermoscopic findings. Am J Dermatopathol. 2015;37:311-314.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Kaplan J, Burgin S, Sepehr A. Granulomatous pigmented purpura: report of a case and review of the literature. J Cutan Pathol. 2011;38:984-989.
- Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
- Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
- Lin WL, Kuo TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia. Clin Exp Dermatol. 2007;32:513-515.
- Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
- Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidaemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
- Wang J, Wu Y, Hsiao P, et al. Granulomatous pigmented purpuric dermatoses: report of three cases and review of the literature. Dermatologica Sinica. 2010;28:77-81.
- Macquarrie EK, Pasternak S, Torok M, et al. Persistent pigmented purpuric dermatitis: granulomatous variant. J Cutan Pathol. 2011;38:979-983.
- Tato BP, Marinero Escobedo S, Pérez González YC, et al. Granulomatous variant of pigmented purpuric dermatosis. Am J Dermatopathol. 2012;34:746-748.
- Dyduch G, Zuber Z, Turowska-Heydel D, et al. Granulomatous pigmented purpura in an adolescent girl: a precursor of mycosis fungoides? Pol J Pathol. 2013;64:157-159; answer 160.
- Paolino S, Cinotti E, Merlo V, et al. Progressive petechial and pigmented macules and papules on the lower extremities. Am J Dermatopathol. 2013;35:370, 388.
- Wakusawa C, Fujimura T, Haga T, et al. Granulomatous pigmented purpuric dermatitis associated with primary Sjögren’s syndrome. Acta Derm Venereol. 2013;93:95-96.
- Hanson C, Fischer R, Fraga G, et al. Granulomatous pigmented purpuric dermatosis: an unusual variant associated with hyperlipidemia. Dermatol Online J. 2014;21. pii:13030/qt0tp272d1.
- Morrissey K, Rosenbach M, DeHoratius D, et al. Granulomatous changes associated with pigmented purpuric dermatosis. Cutis. 2014;94:197-202.
- Battle LR, Shalin SC, Gao L. Granulomatous pigmented purpuric dermatosis [published online December 18, 2014]. Clin Exp Dermatol. 2015;40:387-390.
- Mackenzie AI, Biswas A. Granulomatous pigmented purpuric dermatosis: report of a case with atypical clinical presentation including dermoscopic findings. Am J Dermatopathol. 2015;37:311-314.
Practice Points
- Granulomatous pigmented purpuric dermatosis is not only seen in Far East Asians and patients with hyperlipidemia.
- Suspected pigmented purpuric dermatoses should be managed with a punch biopsy to exclude the granulomatous variant.