User login
Paget disease of bone is a focal disorder of the aging skeleton that can be asymptomatic or can present with pain, bowing deformities, fractures, or nonspecific rheumatic complaints. Physicians often discover it in asymptomatic patients when serum alkaline phosphatase levels are elevated or as an incidental finding on radiography. Despite evidence of germline mutations and polymorphisms that predispose to Paget disease, the environmental determinants that permit disease expression in older people remain unknown.
A STRIKING GEOGRAPHIC DISTRIBUTION
Researchers have been studying the determinants and distribution of Paget disease ever since Sir James Paget first described it in 1877.1
Paget disease has a predilection for the axial skeleton, particularly the lumbosacral spine and pelvis, as well as the skull, femur, and tibia.2 Knowing this, investigators have used screening plain films of the abdomen (kidney-ureter-bladder views) to estimate its prevalence in different populations, as these images capture the lumbosacral spine, pelvis, and proximal femurs. Other means of assessing prevalence have included autopsy series, questionnaires, and screens for biochemical markers of bone turnover, such as elevated serum alkaline phosphatase from bone.3–6
Using these methods, Paget disease has been estimated to occur in 1% to 3% of people over age 55, and in as many as 8% of people over age 80 in certain countries.7
This disease has a striking geographic distribution, being frequent in Europe, Canada, the United States, Australia, New Zealand, and cities of South America, but rare in Scandinavia and Japan. It seems to be equally rare in other countries of the Far East and in India, Russia, and Africa, although its prevalence in these areas has not been thoroughly investigated.8
That it is an ancient disease has been corroborated by excavations in churchyards in Great Britain.9,10 It may be familial or sporadic, but its expression is delayed until late middle age in most persons, and it does not occur in children. For reasons unclear, the prevalence seems to be decreasing in many countries.11–13
GENETICS IS NOT THE WHOLE STORY
The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition, environmental factor, or both.
Mutations in SQSTM1
In 2002, scientists investigating a cohort of French Canadian families found a mutation in the SQSTM1 gene that was present in almost 50% of people with familial Paget disease and in 16% of those with sporadic Paget disease.14 Hocking and his colleagues in the United Kingdom subsequently found the same mutation in 19% of cases of familial Paget disease and in 9% of sporadic cases.15
Further, investigators noted that the mutation was often present on a conserved haplotype, consistent with a stable genetic change occurring in the affected population.16 This observation of a “founder effect” dovetailed with the epidemiology of Paget disease,17 but only with this SQSTM1 mutation.

Throughout Europe, Australia, and the United States, comparable rates of the SQSTM1 mutation were reported in or around the ubiquitin-associated domain. Several specific mutations exist, the most common one being P392L, ie, a prolineto-leucine substitution at amino acid 392. Scientists have tried to correlate severity of disease with genotype, but the findings have been inconsistent.18–21
Investigations into the mechanism of disease have pointed to the role of p62, the product of SQSTM1, in signaling osteoclast activation via nuclear factor kappa B. Since this initial discovery, polymorphisms in the genes affecting osteoclast maturation, activation, and fusion pathways have been shown to predispose to Paget disease. Examples:
- TNFRSF11A, which codes for receptor activator of nuclear factor kappa B, or RANK
- TNFRSF11B, which codes for osteoprotegerin, or OPG
- CSF1, which codes for macrophage colony-stimulating factor 1, and
- OPTN, which codes for optineurin, a member of the nuclear factor kappa B-modulating protein family.
Clinicians interested in these details can read an excellent review of the pathogenesis of Paget disease.22
Other possible factors
Although there is good evidence that measles and canine distemper virus can infect osteoclasts and modify their phenotype, there is no good evidence that these infections by themselves cause Paget disease.23–25 It is, however, tempting to think of these RNA paramyxoviruses as precipitating factors; conceivably, an infectious agent might seed the ends of long bones, accounting for the fixed distribution of Paget disease and its late expression.
Epidemiologic studies from around the world have failed to identify conclusively any environmental exposure that predisposes to Paget disease, although a rural setting, trauma, infection, and milk ingestion have all been proposed.26–28 It is also possible that as bone ages and the marrow becomes less cellular and more fatty, these changes may permit the disease to develop.
The greatest risk factor for Paget disease is perhaps aging, followed by ancestry and a known family history of it. That genetics is not the whole story is evident by reports of people with SQSTM1 mutations who show no clinical evidence of Paget disease in their old age, and patients with Paget disease who have no SQSTM1 mutation.20,29
CLINICAL PRESENTATION
Most patients with Paget disease have no symptoms and come to medical attention because of an elevated serum alkaline phosphatase level or characteristic findings on radiographs ordered for other indications.11 Paget disease is the second most common disorder of aging bone after osteoporosis. Yet unlike osteoporosis, which presents as a systemic fragility of bone, the clinical manifestations of Paget disease depend on which bones are affected and how enlarged or misshapen they have become.
Common complications
As a consequence of this abnormal bone remodeling and overgrowth, many patients present with bone pain. Bone deformity, headache, and hearing loss may also occur (Figure 1), as well as fractures and nerve compression syndromes (eg, spinal stenosis, sciatica, cauda equina syndrome).

It is important to remember that “pagetic” bone may not be the source of pain, and that functional impairment caused by degenerative changes at affected sites is common (Figure 2).30,31
In a study from the New England Registry for Paget’s Disease,32 most patients knew fairly well which bones were affected and what complications resulted from this when deformity, fracture, or total joint replacement had occurred.32 Although Paget disease did affect their quality of life as measured by physical functioning on the Short Form-12 assessment, these impairments did not seem to affect their outlook, which was as good as or better than that in other people their age.
Metabolic complications

Metabolic complications of Paget disease are rare today but can occur in an elderly patient who has active, polyostotic (multibone) disease.33 The accelerated rate of bone remodeling and the increased vascularity of pagetic bone have been reported to lead to high-output heart failure. In theory, treatment should ease this by diminishing blood flow to pagetic bone and restoring bone turnover to more normal levels.34
Hypercalcemia can occur when patients with Paget disease are immobilized for any reason, and there is probably a higher incidence of renal stones in patients with Paget disease.35,36
Malignant complications
Osteosarcoma rarely arises in pagetic bone. Yet Paget disease may account for a significant number of cases of this cancer in the elderly.37 In these cases, osteosarcoma is presumed to be driven by a second genetic mutation, has a genetic signature distinct from that in osteosarcomas occurring in youth, and is quite resistant to treatment.38 In Scandinavia and Japan, where Paget disease is rare, the second peak of osteosarcoma that occurs with aging seems muted as well.39,40 These cancers present with pain, soft-tissue swelling, and variable elevations in serum alkaline phosphatase. Investigations to date suggest that pagetic lesions and osteosarcomas arising in pagetic bone are probably both driven to some extent by stromal cells overexpressing RANK ligand and may not represent defects intrinsic to the osteoclast.41
Giant-cell tumors of bone are also rare but can arise in pagetic bone. A cluster of cases was reported in Avellino and other towns of southern Italy.42 Again, the lesions occur in older individuals and in different sites than those seen in the benign giant-cell tumors recorded in patients without Paget disease.
Metastases from lymphomas, prostate cancer, and breast cancer certainly occur in bone, but rarely in pagetic sites.43 A recent case study noted that patients with prostate cancer who also had Paget disease had a later onset of metastasis to bone than patients without coincident Paget disease.44
A THOUGHTFUL ASSESSMENT
Evaluating a patient with Paget disease requires a thoughtful assessment of its musculoskeletal consequences in an aging skeleton. Pain in Paget disease is often multifactorial. In the elderly, end-stage degenerative disease of the spine, hip, and knees, mechanical instability, compression fractures of the spine, and neuropathies may compound the clinical picture. Therefore, a thorough evaluation is required to plan effective therapy.
Alkaline phosphatase and other markers
A screening serum alkaline phosphatase level is usually sufficient to measure bone turnover. Produced by osteoblasts, alkaline phosphatase is a marker of bone formation, but an imperfect one. Often it is elevated in active Paget disease—but not always.45 Many patients have normal serum alkaline phosphatase levels, particularly if they have monostotic (single-bone) disease. It is unclear why, in a disorder marked by accelerated bone remodeling, the biochemical markers are inconsistent measures of bone turnover.
Research into biochemical markers of Paget disease has had two aims: to identify the single best marker for baseline assessment of pagetic bone activity and to find out whether this measurement responds to therapy.46,47 Measures of bone formation such as bone-specific alkaline phosphatase, osteocalcin, and the procollagen type I peptides, and measures of bone resorption including the pyridinolines, hydroxyproline, and cross-linked collagens, have been analyzed as markers of bone remodeling and show no real advantage over the serum alkaline phosphatase level as reflections of bone turnover. As alkaline phosphatase measurement is inexpensive, available, and reliable, it should be used preferentially, with gamma-glutamyl transpeptidate or 5′ nucleotidase confirming the source as either liver or bone. Readers are directed to a recent review in which the utility of these markers is explored in more detail.48
Imaging studies

Bone scans can give us an idea of the extent, location, and general activity of the disease (Figure 3). Uptake is avid in affected bones, beginning in the subchondral region and spreading throughout the bone. Bone scans can be particularly useful in defining sites of active disease when the serum alkaline phosphatase level is normal.
Plain radiography of the affected bones outlines the anatomy of the problem and gives some insight into the cause of pain (Figure 3).
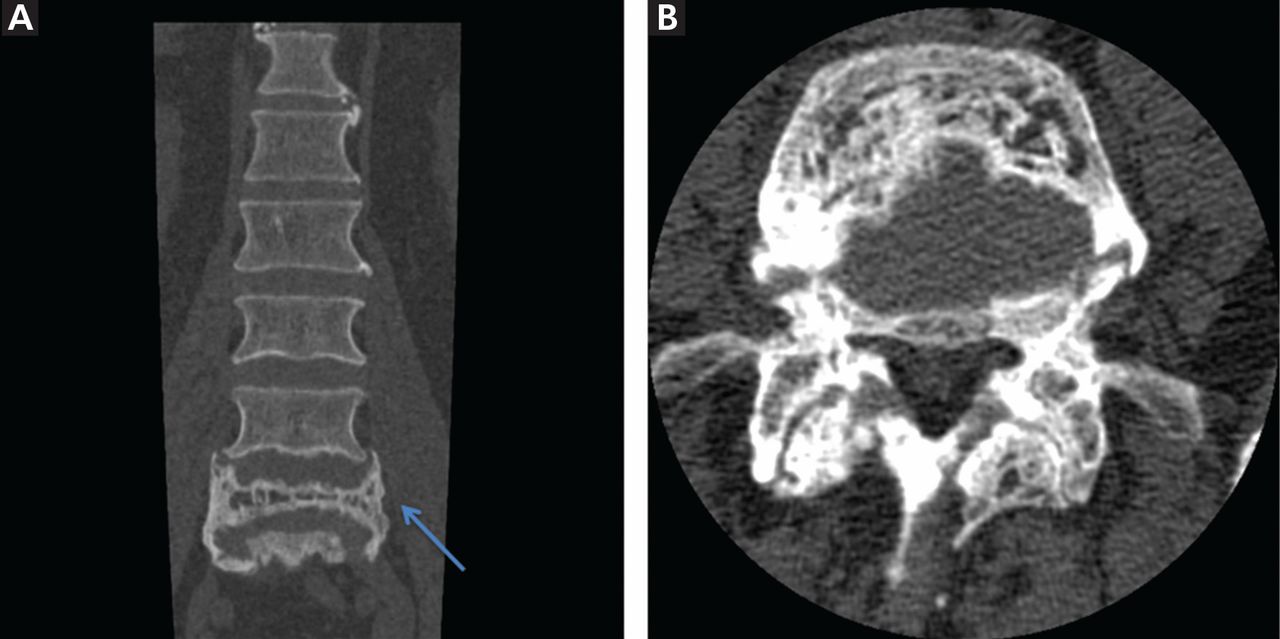
Computed tomography or magnetic resonance imaging may prove useful in cases of spinal stenosis, cauda equina syndrome, compression fractures, or suspected malignancy (Figure 4), but these studies are expensive and generally are not needed.
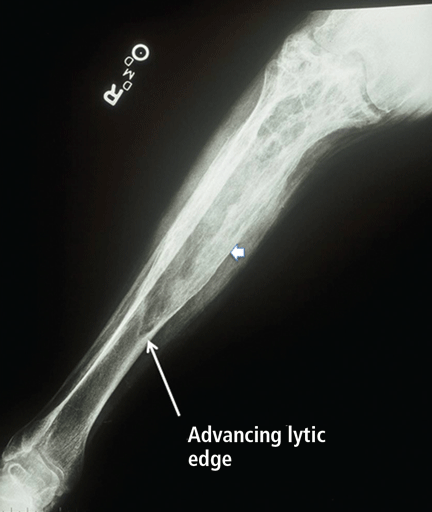
Radiographic features. Paget disease is presumed to be a disease of the osteoclast, and the earliest lesion is described as lytic. In my own experience, it is unusual to see a purely lytic lesion, although sometimes the disease presents in the skull in this way—osteoporosis circumscripta—or in the femur or tibia with an advancing edge of pure osteolysis.
More often, one sees evidence of both resorption by osteoclasts and formation by osteoblasts, reflecting the coupling of these two processes in this disease. Radiographic findings on plain films are usually definitive, showing enlargement of the affected bone, deformity, coarsened trabeculae, and thickened cortices with tunneling (Figure 5).49 In weightbearing bones, pseudofractures may stud the convex surface. These incongruities of bone may persist for years, heralding fracture only when there is focal pain (Figure 6).50
Biopsy is infrequently needed

If these diagnostic findings are not present, then biopsy is indicated. In the United Sates and Canada, where Paget disease is fairly common, biopsy is infrequently needed and is usually reserved for situations in which the differential diagnosis includes cancer, as when the cortex cannot be clearly visualized, the lesions are atypical in pattern or location, or there is a single sclerotic vertebral body on imaging.51
The other indication for biopsy is a “new” pagetic lesion. For reasons unknown, the pattern of skeletal involvement in Paget disease tends to be stable throughout the patient’s lifetime. This is another reason why a baseline bone scan is useful.
TREATMENT WITH BISPHOSPHONATES
Treatment of Paget disease today relies for the most part on the new generation of nitrogen-containing bisphosphonates. As a class, these are antiresorptive agents that inhibit osteoclasts; in this way they slow bone remodeling and enhance the deposition of normal lamellar bone. Their clinical efficacy in Paget disease, coupled with the observation that the earliest lesion in Paget disease is lytic, underscores the principle that Paget disease is a disorder of the osteoclast.
Oral bisphosphonates
Etidronate, approved in 1977, was the first bisphosphonate licensed to treat Paget disease, and it remains available for this indication in the United States. Used in 6-month regimens, it lowers the serum alkaline phosphatase level in some patients, but it has a narrow therapeutic margin. Drug-induced osteomalacia and worsening lytic lesions and fractures in weight-bearing bones are some of the complications.52 When the nitrogen-containing bisphosphonates were developed, they proved to be more potent antiresorptive agents that pose less risk of mineralization defects at prescribed doses.
Alendronate, approved in 1995, is an oral nitrogen-containing bisphosphonate that is effective in treating Paget disease.53 Alendronate is now available in the United States only through special programs (eg, the CVS ProCare Program); the paperwork required to secure this drug is onerous, so the drug is used infrequently. Studies in Paget disease showed that it normalizes the serum alkaline phosphatase level, improves the radiographic appearance, and eases pain in many patients.54 The dosage is 40 mg daily for 6 months.
Risedronate, approved in 1998, is another oral nitrogen-containing bisphosphonate and is comparable to alendronate in efficacy.55 The dosage is 30 mg daily for 2 months.
Tiludronate is another oral bisphosphonate with a different mechanism of action from the nitrogen-containing bisphosphonates.56 It is safe, often effective, but less potent than the newer agents.
The oral bisphosphonates are well tolerated, with few side effects other than gastrointestinal distress. As a class, they are poorly absorbed and so must be taken fasting with a full glass of water on rising, after which the patient should remain upright without food or drink for 30 to 60 minutes. This is a nuisance for elderly patients already on multiple medications and thus makes intravenous agents appealing.
Intravenous bisphosphonates
Pamidronate was approved in 1994. It is quite effective in many patients with Paget disease. There is no consensus around the world on dosing, with regimens ranging from 30 mg to 90 mg or more intravenously in divided doses given over 2 to 4 hours from once a day to once a week. In the United States, 30 mg is given over 4 hours on 3 consecutive days. Resistance to pamidronate has been described; the mechanism is unknown.
Zoledronic acid is a nitrogen-containing bisphosphonate. It is given as a single infusion over 15 minutes, and re-treatment may not be necessary for years. A randomized clinical trial in 2005 demonstrated the efficacy of zoledronic acid 5 mg by infusion compared with oral risedronate in the treatment of Paget disease.57 In observational extension studies lasting as long as 6.5 years, zoledronic acid has been shown to be superior to risedronate in terms of the proportion of patients experiencing a sustained clinical remission.58
While there are many bisphosphonates on the market, an infusion of 5 mg of zoledronic acid seems optimal in most patients who do not have a contraindication or an aversion to intravenous therapy. It tends to normalize the serum alkaline phosphatase level quickly and to leave more patients in sustained biochemical remission than do older bisphosphonates, as noted above. It also tends to be more effective in normalizing the serum alkaline phosphatase level when a patient has used other bisphosphonates in the past or has become resistant to them.
Bisphosphonates reduce bone turnover but do not correct deformities
In randomized clinical trials, bisphosphonates have been shown to restore bone remodeling to more normal levels, to ease pain from pagetic bone, to lower the serum alkaline phosphatase level, and to heal radiographic lesions, but these drugs have not been proven to prevent progression of deformity or to restore the structural integrity of bone (Figure 6).
The Paget’s Disease: Randomized Trial of Intensive Versus Symptomatic Management (PRISM), in 1,324 people with Paget disease in the United Kingdom, showed no difference in the incidence of fracture, orthopedic surgery, quality of life, or hearing thresholds over 2 to 5 years in patients treated with bisphosphonates vs those treated symptomatically, despite a significant difference in serum alkaline phosphatase in the two groups (P < .001).59
In the observational extension study of zoledronic acid described above,58 three of four fractures occurred in the group treated with zoledronic acid, echoing the findings of the PRISM study.
Adverse effects of bisphosphonates
The more potent the bisphosphonate is as an antiresorptive agent, the more it suppresses normal bone remodeling, which can lead to osteonecrosis of the jaw and to atypical femoral fractures.60,61 These complications are unusual in patients with Paget disease because the treatment is intermittent. Sometimes a single dose of zoledronic acid or one course of risedronate or alendronate will last for years.
All the nitrogen-containing bisphosphonates, particularly zoledronic acid, may provoke flulike symptoms of fever, arthralgias, and bone pain. This effect is self-limited, resolves in days, and does not tend to recur. Bone pain may be more sustained, but this also passes, and within weeks the antiresorptive process has abated and pagetic bone pain will ease. Atrial fibrillation is not an anticipated complication of treatment with a bisphosphonate.62 The risk of esophageal cancer is not confirmed at this time.63 Other rare complications of the bisphosphonates include iritis, acute renal failure, and allergy.
Bisphosphonates are not approved for use in patients with creatinine clearance less than 30 mL/min, or in pregnancy.
Other treatments
Calcitonin, an older agent, can still be useful in easing the pain of Paget disease, healing bone lesions, and reducing the metabolic activity of pagetic bone in patients who cannot receive bisphosphonates. It is given by injection in doses of 50 to 100 IU daily or every other day. Although unlikely to effect a sustained clinical remission, calcitonin remains a safe, well-tolerated, and well-studied medication in Paget disease and is approved for this indication.64,65
Denosumab has not been formally studied in Paget disease, but a recent case report indicated it was effective.66
A conservative strategy
Guidelines for treating Paget disease have been written at various times in many countries, including Italy (2007),67 the United Kingdom (2004),68 Japan (2006),69 and Canada (2007).70 Recommendations differ, in part because it is hard to ascertain whether long-term outcomes are improved by treatment, and in part because the prevalence of Paget disease is decreasing and its severity is lessening.11,12 Some guidelines are outdated, since they do not include the newer bisphosphonates.
If the natural history of untreated Paget disease involves the gradual evolution over more than 20 years of bowing deformities in the lower limbs, rigidity and overgrowth of the spine, and softening and enlargement of the skull, as described by Sir James Paget, then treatment should be initiated in hopes that it will modify the outcome. We have no lens to better focus this question on the effect of treatment on the natural history of the disease. We have the PRISM study, designed before zoledronic acid was approved and only 2 to 5 years in duration. And we have the epidemiologic data demonstrating that most patients have no symptoms during their lifetime.
We see the crippling bone disease described by Sir James Paget so infrequently today in the United States that we forget the profound morbidity that may attend the skeletal changes of Paget disease that were common in the early 20th century. Once the bones of the skull are overgrown, the limbs are bowed, and the degenerative joint disease is present, no medication can reverse these changes. Then, the integrity of the bone is lost, and the vulnerability to fracture, early osteoarthritis, nerve compression syndromes, and hearing loss persist. Understanding these consequences prompts the recommendation of early treatment in patients with Paget disease, in hopes of mitigating disease progression.
Patients with active Paget disease, documented either by an elevated serum alkaline phosphatase or by a bone scan, should be treated with a bisphosphonate if the disease is found in sites where remodeling of bone may lead to complications. Such sites include the skull, spine, and long bones of the lower extremity. Paget disease of bone in the pelvis tends to give little trouble (Figure 2) unless it is proximal to a joint, when pain and early arthritis may result. Treatment is safe and, I think, prudent to undertake in any person over age 55 with active disease. To prevent hypocalcemia during treatment, all patients should be repleted with vitamin D and maintained on calcium 1,200 mg daily through diet or supplements with meals.
Throughout the evaluation and treatment, it is important to remember that pain may not emanate from pagetic bone. If medication for Paget disease proves ineffective in the first few months, analgesics, bracing, walking aids, and operative management71 are adjunctive therapies to improve the functional status of these patients.
It is a remarkable clinical observation that treatment of Paget disease may rapidly reverse neurologic syndromes, resolve the erythema or warmth overlying active pagetic bone, and diminish the risk of bleeding with surgery. This response to therapy suggests that there is prompt inhibition and apoptosis of the osteoclasts, accompanied by diminished vascularity of bone. Whatever the mechanism, it is worth treating patients who have spinal stenosis, arthritis, and nerve compression syndromes with calcitonin or bisphosphonates before surgical intervention, whenever possible.34,72
- Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans 1877; 60:37–64.9.
- Guyer PB, Chamberlain AT, Ackery DM, Rolfe EB. The anatomic distribution of osteitis deformans. Clin Orthop Relat Res 1981; 156:141–144.
- Tiegs RD, Lohse CM, Wollan PC, Melton LJ. Long-term trends in the incidence of Paget’s disease of bone. Bone 2000; 27:423–427.
- Altman RD, Bloch DA, Hochberg MC, Murphy WA. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res 2000; 15:461–465.
- Barker DJ. The epidemiology of Paget’s disease of bone. Br Med Bull 1984; 40:396–400.
- Detheridge FM, Guyer PB, Barker DJ. European distribution of Paget’s disease of bone. Br Med J (Clin Res Ed) 1982; 285:1005–1008.
- van Staa TP, Selby P, Leufkens HG, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002; 17:465–471.
- Barker DJ. The epidemiology of Paget’s disease. Metab Bone Dis Relat Res 1981; 3:231–233.
- Rogers J, Jeffrey DR, Watt I. Paget’s disease in an archeological population. J Bone Miner Res 2002; 17:1127–1134.
- Aaron JE, Rogers J, Kanis JA. Paleohistology of Paget’s disease in two medieval skeletons. Am J Phys Anthropol 1992; 89:325–331.
- Poór G, Donáth J, Fornet B, Cooper C. Epidemiology of Paget’s disease in Europe: the prevalence is decreasing. J Bone Miner Res 2006; 21:1545–1549.
- Cundy HR, Gamble G, Wattie D, Rutland M, Cundy T. Paget’s disease of bone in New Zealand: continued decline in disease severity. Calcif Tissue Int 2004; 75:358–364.
- Doyle T, Gunn J, Anderson G, Gill M, Cundy T. Paget’s disease in New Zealand: evidence for declining prevalence. Bone 2002; 31:616–619.
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70:1582–1588.
- Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet 2002; 11:2735–2739.
- Lucas GJ, Hocking LJ, Daroszewska A, et al. Ubiquitin-associated domain mutations of SQSTM1 in Paget’s disease of bone: evidence for a founder effect in patients of British descent. J Bone Miner Res 2005; 20:227–231.
- Mays S. Archaeological skeletons support a northwest European origin for Paget’s disease of bone. J Bone Miner Res 2010; 25:1839–1841.
- Bolland MJ, Tong PC, Naot D, et al. Delayed development of Paget’s disease in offspring inheriting SQSTM1 mutations. J Bone Miner Res 2007; 22:411–415.
- Rea SL, Walsh JP, Ward L, et al. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res 2006; 21:1136–1145.
- Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P38–P44.
- Eekhoff EW, Karperien M, Houtsma D, et al. Familial Paget’s disease in The Netherlands: occurrence, identification of new mutations in the sequestosome 1 gene, and their clinical associations. Arthritis Rheum 2004; 50:1650–1654.
- Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int 2012; 91:97–113.
- Kurihara N, Hiruma Y, Yamana K, et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab 2011; 13:23–34.
- Kurihara N, Zhou H, Reddy SV, et al. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget’s disease-like bone lesions in mice. J Bone Miner Res 2006; 21:446–455.
- Reddy SV, Singer FR, Roodman GD. Bone marrow mononuclear cells from patients with Paget’s disease contain measles virus nucleocapsid messenger ribonucleic acid that has mutations in a specific region of the sequence. J Clin Endocrinol Metab 1995; 80:2108–2111.
- Gennari L, Merlotti D, Martini G, Nuti R. Paget’s disease of bone in Italy. J Bone Miner Res 2006; 21(suppl 2):P14–P21.
- Seton M, Choi HK, Hansen MF, Sebaldt RJ, Cooper C. Analysis of environmental factors in familial versus sporadic Paget’s disease of bone—the New England Registry for Paget’s Disease of Bone. J Bone Miner Res 2003; 18:1519–1524.
- Siris ES. Extensive personal experience: Paget’s disease of bone. J Clin Endocrinol Metab 1995; 80:335–338.
- Lucas GJ, Daroszewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget’s disease of bone and related disorders. J Bone Miner Res 2006; 21(suppl 2):P31–P37.
- Seton M. Diagnosis, complications and treatment of Paget’s disease of bone. Aging Health 2009; 5:497–508.
- Siris E, Roodman GD. Paget’s Disease of Bone. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008.
- Seton M, Moses AM, Bode RK, Schwartz C. Paget’s disease of bone: the skeletal distribution, complications and quality of life as perceived by patients. Bone 2011; 48:281–285.
- Seton M. Paget’s disease of bone. In:Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors. Rheumatology. 5th ed. Philadelphia, PA: Mosby Elsevier; 2010:2021–2028.
- Douglas DL, Duckworth T, Kanis JA, Jefferson AA, Martin TJ, Russell RG. Spinal cord dysfunction in Paget’s disease of bone. Has medical treatment a vascular basis? J Bone Joint Surg Br 1981; 63B:495–503.
- Siris ES. Epidemiological aspects of Paget’s disease: family history and relationship to other medical conditions. Semin Arthritis Rheum 1994; 23:222–225.
- Kanis JA, Evanson JM, Russell RG. Paget’s disease of bone: diagnosis and management. Metab Bone Dis Relat Res 1981; 3:219–230.
- Mangham DC, Davie MW, Grimer RJ. Sarcoma arising in Paget’s disease of bone: declining incidence and increasing age at presentation. Bone 2009; 44:431–436.
- Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P58–P63.
- Price CH. The incidence of osteogenic sarcoma in South-West England and its relationship to Paget’s disease of bone. J Bone Joint Surg Br 1962; 44-B:366–376.
- Ishikawa Y, Tsukuma H, Miller RW. Low rates of Paget’s disease of bone and osteosarcoma in elderly Japanese. Lancet 1996; 347:1559.
- Sun SG, Lau YS, Itonaga I, Sabokbar A, Athanasou NA. Bone stromal cells in pagetic bone and Paget’s sarcoma express RANKL and support human osteoclast formation. J Pathol 2006; 209:114–120.
- Rendina D, Gennari L, De Filippo G, et al. Evidence for increased clinical severity of familial and sporadic Paget’s disease of bone in Campania, southern Italy. J Bone Miner Res 2006; 21:1828–1835.
- Fenton P, Resnick D. Metastases to bone affected by Paget’s disease. A report of three cases. Int Orthop 1991; 15:397–399.
- Tu SM, Som A, Tu B, Logothetis CJ, Lee MH, Yeung SC. Effect of Paget’s disease of bone (osteitis deformans) on the progression of prostate cancer bone metastasis. Br J Cancer 2012; 107:646–651.
- Eekhoff ME, van der Klift M, Kroon HM, et al. Paget’s disease of bone in The Netherlands: a population-based radiological and biochemical survey—the Rotterdam Study. J Bone Miner Res 2004; 19:566–570.
- Reid IR, Davidson JS, Wattie D, et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget’s disease of bone. Bone 2004; 35:224–230.
- Alvarez L, Guañabens N, Peris P, et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget’s disease. Bone 2001; 29:447–452.
- Cundy T, Reid IR. Paget’s disease of bone. Clin Biochem 2012; 45:43–48.
- Cortis K, Micallef K, Mizzi A. Imaging Paget’s disease of bone—from head to toe. Clin Radiol 2011; 66:662–672.
- Redden JF, Dixon J, Vennart W, Hosking DJ. Management of fissure fractures in Paget’s disease. Int Orthop 1981; 5:103–106.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 25-1993. A 67-year-old man with osteolytic lesions of T11 and T12. N Engl J Med 1993; 328:1836–1841.
- Evans RA, Dunstan CR, Hills E, Wong SY. Pathologic fracture due to severe osteomalacia following low-dose diphosphonate treatment of Paget’s disease of bone. Aust N Z J Med 1983; 13:277–279.
- Siris E, Weinstein RS, Altman R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab 1996; 81:961–967.
- Reid IR, Siris E. Alendronate in the treatment of Paget’s disease of bone. Int J Clin Pract Suppl 1999; 101:62–66.
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget’s disease of bone. Paget’s Risedronate/Etidronate Study Group. Am J Med 1999; 106:513–520.
- Peris P, Alvarez L, Vidal S, Martínez MA, Monegal A, Guañabens N. Treatment with tiludronate has a similar effect to risedronate on Paget’s disease activity assessed by bone markers and bone scintigraphy. Clin Exp Rheumatol 2007; 25:206–210.
- Reid IR, Miller P, Lyles K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med 2005; 353:898–908.
- Reid IR, Lyles K, Su G, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011; 26:2261–2270.
- Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH; PRISM Trial Group. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res 2010; 25:20–31.
- Abrahamsen B, Einhorn TA. Beyond a reasonable doubt? Bisphosphonates and atypical femur fractures. Bone 2012; 50:1196–1200.
- Seton M, Krane SM. Use of zoledronic acid in the treatment of Paget’s disease. Ther Clin Risk Manag 2007; 3:913–918.
- Sørensen HT, Christensen S, Mehnert F, et al. Use of bisphosphonates among women and risk of atrial fibrillation and flutter: Population based case-control study. BMJ 2008; 336:813–816.
- Dixon WG, Solomon DH. Bisphosphonates and esophageal cancer—a pathway through the confusion. Nat Rev Rheumatol 2011; 7:369–372.
- Singer FR, Krane SM. Paget’s disease of bone. In:Avioli LV, Krane SM, editors. Metabolic Bone Disease and Clinically Related Disorders. 2nd ed. Philadelphia, PA: W.B. Saunders Company; 1990:546–615.
- Kanis JA, Horn DB, Scott RD, Strong JA. Treatment of Paget’s disease of bone with synthetic salmon calcitonin. Br Med J 1974; 3:727–731.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Adami S, Bartolozzi P, Brandi ML, et al; Societa Italiana di Ortopedia e Traumatologia. [Italian guidelines for the diagnosis and treatment of Paget’s disease of bone.] Reumatismo 2007; 59:153–168. (Article in Italian.)
- Scarsbrok A, Brown M, Wilson D. UK guidelines on management of Paget’s disease of bone. Rheumatology (Oxford) 2004; 43:399–400.
- Takata S, Hashimoto J, Nakatsuka K, et a.l Guidelines for diagnosis and management of Paget’s disease of bone in Japan. J Bone Miner Metab 2006; 24:359–367.
- Josse RG, Hanley DA, Kendler D, Ste Marie L-G, Adachi JD, Brown J. Diagnosis and treatment of Paget’s disease of bone. Clin Invest Med 2007; 30:E210–E223.
- Kaplan FS. Paget’s disease of bone: orthopedic complications. Semin Arthritis Rheum 1994; 23:250–252.
- Kanis JA, Gray RE. Long-term follow-up observations on treatment in Paget’s disease of bone. Clin Orthop Relat Res 1987; 217:99–125.
Paget disease of bone is a focal disorder of the aging skeleton that can be asymptomatic or can present with pain, bowing deformities, fractures, or nonspecific rheumatic complaints. Physicians often discover it in asymptomatic patients when serum alkaline phosphatase levels are elevated or as an incidental finding on radiography. Despite evidence of germline mutations and polymorphisms that predispose to Paget disease, the environmental determinants that permit disease expression in older people remain unknown.
A STRIKING GEOGRAPHIC DISTRIBUTION
Researchers have been studying the determinants and distribution of Paget disease ever since Sir James Paget first described it in 1877.1
Paget disease has a predilection for the axial skeleton, particularly the lumbosacral spine and pelvis, as well as the skull, femur, and tibia.2 Knowing this, investigators have used screening plain films of the abdomen (kidney-ureter-bladder views) to estimate its prevalence in different populations, as these images capture the lumbosacral spine, pelvis, and proximal femurs. Other means of assessing prevalence have included autopsy series, questionnaires, and screens for biochemical markers of bone turnover, such as elevated serum alkaline phosphatase from bone.3–6
Using these methods, Paget disease has been estimated to occur in 1% to 3% of people over age 55, and in as many as 8% of people over age 80 in certain countries.7
This disease has a striking geographic distribution, being frequent in Europe, Canada, the United States, Australia, New Zealand, and cities of South America, but rare in Scandinavia and Japan. It seems to be equally rare in other countries of the Far East and in India, Russia, and Africa, although its prevalence in these areas has not been thoroughly investigated.8
That it is an ancient disease has been corroborated by excavations in churchyards in Great Britain.9,10 It may be familial or sporadic, but its expression is delayed until late middle age in most persons, and it does not occur in children. For reasons unclear, the prevalence seems to be decreasing in many countries.11–13
GENETICS IS NOT THE WHOLE STORY
The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition, environmental factor, or both.
Mutations in SQSTM1
In 2002, scientists investigating a cohort of French Canadian families found a mutation in the SQSTM1 gene that was present in almost 50% of people with familial Paget disease and in 16% of those with sporadic Paget disease.14 Hocking and his colleagues in the United Kingdom subsequently found the same mutation in 19% of cases of familial Paget disease and in 9% of sporadic cases.15
Further, investigators noted that the mutation was often present on a conserved haplotype, consistent with a stable genetic change occurring in the affected population.16 This observation of a “founder effect” dovetailed with the epidemiology of Paget disease,17 but only with this SQSTM1 mutation.
Throughout Europe, Australia, and the United States, comparable rates of the SQSTM1 mutation were reported in or around the ubiquitin-associated domain. Several specific mutations exist, the most common one being P392L, ie, a prolineto-leucine substitution at amino acid 392. Scientists have tried to correlate severity of disease with genotype, but the findings have been inconsistent.18–21
Investigations into the mechanism of disease have pointed to the role of p62, the product of SQSTM1, in signaling osteoclast activation via nuclear factor kappa B. Since this initial discovery, polymorphisms in the genes affecting osteoclast maturation, activation, and fusion pathways have been shown to predispose to Paget disease. Examples:
- TNFRSF11A, which codes for receptor activator of nuclear factor kappa B, or RANK
- TNFRSF11B, which codes for osteoprotegerin, or OPG
- CSF1, which codes for macrophage colony-stimulating factor 1, and
- OPTN, which codes for optineurin, a member of the nuclear factor kappa B-modulating protein family.
Clinicians interested in these details can read an excellent review of the pathogenesis of Paget disease.22
Other possible factors
Although there is good evidence that measles and canine distemper virus can infect osteoclasts and modify their phenotype, there is no good evidence that these infections by themselves cause Paget disease.23–25 It is, however, tempting to think of these RNA paramyxoviruses as precipitating factors; conceivably, an infectious agent might seed the ends of long bones, accounting for the fixed distribution of Paget disease and its late expression.
Epidemiologic studies from around the world have failed to identify conclusively any environmental exposure that predisposes to Paget disease, although a rural setting, trauma, infection, and milk ingestion have all been proposed.26–28 It is also possible that as bone ages and the marrow becomes less cellular and more fatty, these changes may permit the disease to develop.
The greatest risk factor for Paget disease is perhaps aging, followed by ancestry and a known family history of it. That genetics is not the whole story is evident by reports of people with SQSTM1 mutations who show no clinical evidence of Paget disease in their old age, and patients with Paget disease who have no SQSTM1 mutation.20,29
CLINICAL PRESENTATION
Most patients with Paget disease have no symptoms and come to medical attention because of an elevated serum alkaline phosphatase level or characteristic findings on radiographs ordered for other indications.11 Paget disease is the second most common disorder of aging bone after osteoporosis. Yet unlike osteoporosis, which presents as a systemic fragility of bone, the clinical manifestations of Paget disease depend on which bones are affected and how enlarged or misshapen they have become.
Common complications
As a consequence of this abnormal bone remodeling and overgrowth, many patients present with bone pain. Bone deformity, headache, and hearing loss may also occur (Figure 1), as well as fractures and nerve compression syndromes (eg, spinal stenosis, sciatica, cauda equina syndrome).
It is important to remember that “pagetic” bone may not be the source of pain, and that functional impairment caused by degenerative changes at affected sites is common (Figure 2).30,31
In a study from the New England Registry for Paget’s Disease,32 most patients knew fairly well which bones were affected and what complications resulted from this when deformity, fracture, or total joint replacement had occurred.32 Although Paget disease did affect their quality of life as measured by physical functioning on the Short Form-12 assessment, these impairments did not seem to affect their outlook, which was as good as or better than that in other people their age.
Metabolic complications
Metabolic complications of Paget disease are rare today but can occur in an elderly patient who has active, polyostotic (multibone) disease.33 The accelerated rate of bone remodeling and the increased vascularity of pagetic bone have been reported to lead to high-output heart failure. In theory, treatment should ease this by diminishing blood flow to pagetic bone and restoring bone turnover to more normal levels.34
Hypercalcemia can occur when patients with Paget disease are immobilized for any reason, and there is probably a higher incidence of renal stones in patients with Paget disease.35,36
Malignant complications
Osteosarcoma rarely arises in pagetic bone. Yet Paget disease may account for a significant number of cases of this cancer in the elderly.37 In these cases, osteosarcoma is presumed to be driven by a second genetic mutation, has a genetic signature distinct from that in osteosarcomas occurring in youth, and is quite resistant to treatment.38 In Scandinavia and Japan, where Paget disease is rare, the second peak of osteosarcoma that occurs with aging seems muted as well.39,40 These cancers present with pain, soft-tissue swelling, and variable elevations in serum alkaline phosphatase. Investigations to date suggest that pagetic lesions and osteosarcomas arising in pagetic bone are probably both driven to some extent by stromal cells overexpressing RANK ligand and may not represent defects intrinsic to the osteoclast.41
Giant-cell tumors of bone are also rare but can arise in pagetic bone. A cluster of cases was reported in Avellino and other towns of southern Italy.42 Again, the lesions occur in older individuals and in different sites than those seen in the benign giant-cell tumors recorded in patients without Paget disease.
Metastases from lymphomas, prostate cancer, and breast cancer certainly occur in bone, but rarely in pagetic sites.43 A recent case study noted that patients with prostate cancer who also had Paget disease had a later onset of metastasis to bone than patients without coincident Paget disease.44
A THOUGHTFUL ASSESSMENT
Evaluating a patient with Paget disease requires a thoughtful assessment of its musculoskeletal consequences in an aging skeleton. Pain in Paget disease is often multifactorial. In the elderly, end-stage degenerative disease of the spine, hip, and knees, mechanical instability, compression fractures of the spine, and neuropathies may compound the clinical picture. Therefore, a thorough evaluation is required to plan effective therapy.
Alkaline phosphatase and other markers
A screening serum alkaline phosphatase level is usually sufficient to measure bone turnover. Produced by osteoblasts, alkaline phosphatase is a marker of bone formation, but an imperfect one. Often it is elevated in active Paget disease—but not always.45 Many patients have normal serum alkaline phosphatase levels, particularly if they have monostotic (single-bone) disease. It is unclear why, in a disorder marked by accelerated bone remodeling, the biochemical markers are inconsistent measures of bone turnover.
Research into biochemical markers of Paget disease has had two aims: to identify the single best marker for baseline assessment of pagetic bone activity and to find out whether this measurement responds to therapy.46,47 Measures of bone formation such as bone-specific alkaline phosphatase, osteocalcin, and the procollagen type I peptides, and measures of bone resorption including the pyridinolines, hydroxyproline, and cross-linked collagens, have been analyzed as markers of bone remodeling and show no real advantage over the serum alkaline phosphatase level as reflections of bone turnover. As alkaline phosphatase measurement is inexpensive, available, and reliable, it should be used preferentially, with gamma-glutamyl transpeptidate or 5′ nucleotidase confirming the source as either liver or bone. Readers are directed to a recent review in which the utility of these markers is explored in more detail.48
Imaging studies
Bone scans can give us an idea of the extent, location, and general activity of the disease (Figure 3). Uptake is avid in affected bones, beginning in the subchondral region and spreading throughout the bone. Bone scans can be particularly useful in defining sites of active disease when the serum alkaline phosphatase level is normal.
Plain radiography of the affected bones outlines the anatomy of the problem and gives some insight into the cause of pain (Figure 3).
Computed tomography or magnetic resonance imaging may prove useful in cases of spinal stenosis, cauda equina syndrome, compression fractures, or suspected malignancy (Figure 4), but these studies are expensive and generally are not needed.
Radiographic features. Paget disease is presumed to be a disease of the osteoclast, and the earliest lesion is described as lytic. In my own experience, it is unusual to see a purely lytic lesion, although sometimes the disease presents in the skull in this way—osteoporosis circumscripta—or in the femur or tibia with an advancing edge of pure osteolysis.
More often, one sees evidence of both resorption by osteoclasts and formation by osteoblasts, reflecting the coupling of these two processes in this disease. Radiographic findings on plain films are usually definitive, showing enlargement of the affected bone, deformity, coarsened trabeculae, and thickened cortices with tunneling (Figure 5).49 In weightbearing bones, pseudofractures may stud the convex surface. These incongruities of bone may persist for years, heralding fracture only when there is focal pain (Figure 6).50
Biopsy is infrequently needed
If these diagnostic findings are not present, then biopsy is indicated. In the United Sates and Canada, where Paget disease is fairly common, biopsy is infrequently needed and is usually reserved for situations in which the differential diagnosis includes cancer, as when the cortex cannot be clearly visualized, the lesions are atypical in pattern or location, or there is a single sclerotic vertebral body on imaging.51
The other indication for biopsy is a “new” pagetic lesion. For reasons unknown, the pattern of skeletal involvement in Paget disease tends to be stable throughout the patient’s lifetime. This is another reason why a baseline bone scan is useful.
TREATMENT WITH BISPHOSPHONATES
Treatment of Paget disease today relies for the most part on the new generation of nitrogen-containing bisphosphonates. As a class, these are antiresorptive agents that inhibit osteoclasts; in this way they slow bone remodeling and enhance the deposition of normal lamellar bone. Their clinical efficacy in Paget disease, coupled with the observation that the earliest lesion in Paget disease is lytic, underscores the principle that Paget disease is a disorder of the osteoclast.
Oral bisphosphonates
Etidronate, approved in 1977, was the first bisphosphonate licensed to treat Paget disease, and it remains available for this indication in the United States. Used in 6-month regimens, it lowers the serum alkaline phosphatase level in some patients, but it has a narrow therapeutic margin. Drug-induced osteomalacia and worsening lytic lesions and fractures in weight-bearing bones are some of the complications.52 When the nitrogen-containing bisphosphonates were developed, they proved to be more potent antiresorptive agents that pose less risk of mineralization defects at prescribed doses.
Alendronate, approved in 1995, is an oral nitrogen-containing bisphosphonate that is effective in treating Paget disease.53 Alendronate is now available in the United States only through special programs (eg, the CVS ProCare Program); the paperwork required to secure this drug is onerous, so the drug is used infrequently. Studies in Paget disease showed that it normalizes the serum alkaline phosphatase level, improves the radiographic appearance, and eases pain in many patients.54 The dosage is 40 mg daily for 6 months.
Risedronate, approved in 1998, is another oral nitrogen-containing bisphosphonate and is comparable to alendronate in efficacy.55 The dosage is 30 mg daily for 2 months.
Tiludronate is another oral bisphosphonate with a different mechanism of action from the nitrogen-containing bisphosphonates.56 It is safe, often effective, but less potent than the newer agents.
The oral bisphosphonates are well tolerated, with few side effects other than gastrointestinal distress. As a class, they are poorly absorbed and so must be taken fasting with a full glass of water on rising, after which the patient should remain upright without food or drink for 30 to 60 minutes. This is a nuisance for elderly patients already on multiple medications and thus makes intravenous agents appealing.
Intravenous bisphosphonates
Pamidronate was approved in 1994. It is quite effective in many patients with Paget disease. There is no consensus around the world on dosing, with regimens ranging from 30 mg to 90 mg or more intravenously in divided doses given over 2 to 4 hours from once a day to once a week. In the United States, 30 mg is given over 4 hours on 3 consecutive days. Resistance to pamidronate has been described; the mechanism is unknown.
Zoledronic acid is a nitrogen-containing bisphosphonate. It is given as a single infusion over 15 minutes, and re-treatment may not be necessary for years. A randomized clinical trial in 2005 demonstrated the efficacy of zoledronic acid 5 mg by infusion compared with oral risedronate in the treatment of Paget disease.57 In observational extension studies lasting as long as 6.5 years, zoledronic acid has been shown to be superior to risedronate in terms of the proportion of patients experiencing a sustained clinical remission.58
While there are many bisphosphonates on the market, an infusion of 5 mg of zoledronic acid seems optimal in most patients who do not have a contraindication or an aversion to intravenous therapy. It tends to normalize the serum alkaline phosphatase level quickly and to leave more patients in sustained biochemical remission than do older bisphosphonates, as noted above. It also tends to be more effective in normalizing the serum alkaline phosphatase level when a patient has used other bisphosphonates in the past or has become resistant to them.
Bisphosphonates reduce bone turnover but do not correct deformities
In randomized clinical trials, bisphosphonates have been shown to restore bone remodeling to more normal levels, to ease pain from pagetic bone, to lower the serum alkaline phosphatase level, and to heal radiographic lesions, but these drugs have not been proven to prevent progression of deformity or to restore the structural integrity of bone (Figure 6).
The Paget’s Disease: Randomized Trial of Intensive Versus Symptomatic Management (PRISM), in 1,324 people with Paget disease in the United Kingdom, showed no difference in the incidence of fracture, orthopedic surgery, quality of life, or hearing thresholds over 2 to 5 years in patients treated with bisphosphonates vs those treated symptomatically, despite a significant difference in serum alkaline phosphatase in the two groups (P < .001).59
In the observational extension study of zoledronic acid described above,58 three of four fractures occurred in the group treated with zoledronic acid, echoing the findings of the PRISM study.
Adverse effects of bisphosphonates
The more potent the bisphosphonate is as an antiresorptive agent, the more it suppresses normal bone remodeling, which can lead to osteonecrosis of the jaw and to atypical femoral fractures.60,61 These complications are unusual in patients with Paget disease because the treatment is intermittent. Sometimes a single dose of zoledronic acid or one course of risedronate or alendronate will last for years.
All the nitrogen-containing bisphosphonates, particularly zoledronic acid, may provoke flulike symptoms of fever, arthralgias, and bone pain. This effect is self-limited, resolves in days, and does not tend to recur. Bone pain may be more sustained, but this also passes, and within weeks the antiresorptive process has abated and pagetic bone pain will ease. Atrial fibrillation is not an anticipated complication of treatment with a bisphosphonate.62 The risk of esophageal cancer is not confirmed at this time.63 Other rare complications of the bisphosphonates include iritis, acute renal failure, and allergy.
Bisphosphonates are not approved for use in patients with creatinine clearance less than 30 mL/min, or in pregnancy.
Other treatments
Calcitonin, an older agent, can still be useful in easing the pain of Paget disease, healing bone lesions, and reducing the metabolic activity of pagetic bone in patients who cannot receive bisphosphonates. It is given by injection in doses of 50 to 100 IU daily or every other day. Although unlikely to effect a sustained clinical remission, calcitonin remains a safe, well-tolerated, and well-studied medication in Paget disease and is approved for this indication.64,65
Denosumab has not been formally studied in Paget disease, but a recent case report indicated it was effective.66
A conservative strategy
Guidelines for treating Paget disease have been written at various times in many countries, including Italy (2007),67 the United Kingdom (2004),68 Japan (2006),69 and Canada (2007).70 Recommendations differ, in part because it is hard to ascertain whether long-term outcomes are improved by treatment, and in part because the prevalence of Paget disease is decreasing and its severity is lessening.11,12 Some guidelines are outdated, since they do not include the newer bisphosphonates.
If the natural history of untreated Paget disease involves the gradual evolution over more than 20 years of bowing deformities in the lower limbs, rigidity and overgrowth of the spine, and softening and enlargement of the skull, as described by Sir James Paget, then treatment should be initiated in hopes that it will modify the outcome. We have no lens to better focus this question on the effect of treatment on the natural history of the disease. We have the PRISM study, designed before zoledronic acid was approved and only 2 to 5 years in duration. And we have the epidemiologic data demonstrating that most patients have no symptoms during their lifetime.
We see the crippling bone disease described by Sir James Paget so infrequently today in the United States that we forget the profound morbidity that may attend the skeletal changes of Paget disease that were common in the early 20th century. Once the bones of the skull are overgrown, the limbs are bowed, and the degenerative joint disease is present, no medication can reverse these changes. Then, the integrity of the bone is lost, and the vulnerability to fracture, early osteoarthritis, nerve compression syndromes, and hearing loss persist. Understanding these consequences prompts the recommendation of early treatment in patients with Paget disease, in hopes of mitigating disease progression.
Patients with active Paget disease, documented either by an elevated serum alkaline phosphatase or by a bone scan, should be treated with a bisphosphonate if the disease is found in sites where remodeling of bone may lead to complications. Such sites include the skull, spine, and long bones of the lower extremity. Paget disease of bone in the pelvis tends to give little trouble (Figure 2) unless it is proximal to a joint, when pain and early arthritis may result. Treatment is safe and, I think, prudent to undertake in any person over age 55 with active disease. To prevent hypocalcemia during treatment, all patients should be repleted with vitamin D and maintained on calcium 1,200 mg daily through diet or supplements with meals.
Throughout the evaluation and treatment, it is important to remember that pain may not emanate from pagetic bone. If medication for Paget disease proves ineffective in the first few months, analgesics, bracing, walking aids, and operative management71 are adjunctive therapies to improve the functional status of these patients.
It is a remarkable clinical observation that treatment of Paget disease may rapidly reverse neurologic syndromes, resolve the erythema or warmth overlying active pagetic bone, and diminish the risk of bleeding with surgery. This response to therapy suggests that there is prompt inhibition and apoptosis of the osteoclasts, accompanied by diminished vascularity of bone. Whatever the mechanism, it is worth treating patients who have spinal stenosis, arthritis, and nerve compression syndromes with calcitonin or bisphosphonates before surgical intervention, whenever possible.34,72
Paget disease of bone is a focal disorder of the aging skeleton that can be asymptomatic or can present with pain, bowing deformities, fractures, or nonspecific rheumatic complaints. Physicians often discover it in asymptomatic patients when serum alkaline phosphatase levels are elevated or as an incidental finding on radiography. Despite evidence of germline mutations and polymorphisms that predispose to Paget disease, the environmental determinants that permit disease expression in older people remain unknown.
A STRIKING GEOGRAPHIC DISTRIBUTION
Researchers have been studying the determinants and distribution of Paget disease ever since Sir James Paget first described it in 1877.1
Paget disease has a predilection for the axial skeleton, particularly the lumbosacral spine and pelvis, as well as the skull, femur, and tibia.2 Knowing this, investigators have used screening plain films of the abdomen (kidney-ureter-bladder views) to estimate its prevalence in different populations, as these images capture the lumbosacral spine, pelvis, and proximal femurs. Other means of assessing prevalence have included autopsy series, questionnaires, and screens for biochemical markers of bone turnover, such as elevated serum alkaline phosphatase from bone.3–6
Using these methods, Paget disease has been estimated to occur in 1% to 3% of people over age 55, and in as many as 8% of people over age 80 in certain countries.7
This disease has a striking geographic distribution, being frequent in Europe, Canada, the United States, Australia, New Zealand, and cities of South America, but rare in Scandinavia and Japan. It seems to be equally rare in other countries of the Far East and in India, Russia, and Africa, although its prevalence in these areas has not been thoroughly investigated.8
That it is an ancient disease has been corroborated by excavations in churchyards in Great Britain.9,10 It may be familial or sporadic, but its expression is delayed until late middle age in most persons, and it does not occur in children. For reasons unclear, the prevalence seems to be decreasing in many countries.11–13
GENETICS IS NOT THE WHOLE STORY
The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition, environmental factor, or both.
Mutations in SQSTM1
In 2002, scientists investigating a cohort of French Canadian families found a mutation in the SQSTM1 gene that was present in almost 50% of people with familial Paget disease and in 16% of those with sporadic Paget disease.14 Hocking and his colleagues in the United Kingdom subsequently found the same mutation in 19% of cases of familial Paget disease and in 9% of sporadic cases.15
Further, investigators noted that the mutation was often present on a conserved haplotype, consistent with a stable genetic change occurring in the affected population.16 This observation of a “founder effect” dovetailed with the epidemiology of Paget disease,17 but only with this SQSTM1 mutation.
Throughout Europe, Australia, and the United States, comparable rates of the SQSTM1 mutation were reported in or around the ubiquitin-associated domain. Several specific mutations exist, the most common one being P392L, ie, a prolineto-leucine substitution at amino acid 392. Scientists have tried to correlate severity of disease with genotype, but the findings have been inconsistent.18–21
Investigations into the mechanism of disease have pointed to the role of p62, the product of SQSTM1, in signaling osteoclast activation via nuclear factor kappa B. Since this initial discovery, polymorphisms in the genes affecting osteoclast maturation, activation, and fusion pathways have been shown to predispose to Paget disease. Examples:
- TNFRSF11A, which codes for receptor activator of nuclear factor kappa B, or RANK
- TNFRSF11B, which codes for osteoprotegerin, or OPG
- CSF1, which codes for macrophage colony-stimulating factor 1, and
- OPTN, which codes for optineurin, a member of the nuclear factor kappa B-modulating protein family.
Clinicians interested in these details can read an excellent review of the pathogenesis of Paget disease.22
Other possible factors
Although there is good evidence that measles and canine distemper virus can infect osteoclasts and modify their phenotype, there is no good evidence that these infections by themselves cause Paget disease.23–25 It is, however, tempting to think of these RNA paramyxoviruses as precipitating factors; conceivably, an infectious agent might seed the ends of long bones, accounting for the fixed distribution of Paget disease and its late expression.
Epidemiologic studies from around the world have failed to identify conclusively any environmental exposure that predisposes to Paget disease, although a rural setting, trauma, infection, and milk ingestion have all been proposed.26–28 It is also possible that as bone ages and the marrow becomes less cellular and more fatty, these changes may permit the disease to develop.
The greatest risk factor for Paget disease is perhaps aging, followed by ancestry and a known family history of it. That genetics is not the whole story is evident by reports of people with SQSTM1 mutations who show no clinical evidence of Paget disease in their old age, and patients with Paget disease who have no SQSTM1 mutation.20,29
CLINICAL PRESENTATION
Most patients with Paget disease have no symptoms and come to medical attention because of an elevated serum alkaline phosphatase level or characteristic findings on radiographs ordered for other indications.11 Paget disease is the second most common disorder of aging bone after osteoporosis. Yet unlike osteoporosis, which presents as a systemic fragility of bone, the clinical manifestations of Paget disease depend on which bones are affected and how enlarged or misshapen they have become.
Common complications
As a consequence of this abnormal bone remodeling and overgrowth, many patients present with bone pain. Bone deformity, headache, and hearing loss may also occur (Figure 1), as well as fractures and nerve compression syndromes (eg, spinal stenosis, sciatica, cauda equina syndrome).
It is important to remember that “pagetic” bone may not be the source of pain, and that functional impairment caused by degenerative changes at affected sites is common (Figure 2).30,31
In a study from the New England Registry for Paget’s Disease,32 most patients knew fairly well which bones were affected and what complications resulted from this when deformity, fracture, or total joint replacement had occurred.32 Although Paget disease did affect their quality of life as measured by physical functioning on the Short Form-12 assessment, these impairments did not seem to affect their outlook, which was as good as or better than that in other people their age.
Metabolic complications
Metabolic complications of Paget disease are rare today but can occur in an elderly patient who has active, polyostotic (multibone) disease.33 The accelerated rate of bone remodeling and the increased vascularity of pagetic bone have been reported to lead to high-output heart failure. In theory, treatment should ease this by diminishing blood flow to pagetic bone and restoring bone turnover to more normal levels.34
Hypercalcemia can occur when patients with Paget disease are immobilized for any reason, and there is probably a higher incidence of renal stones in patients with Paget disease.35,36
Malignant complications
Osteosarcoma rarely arises in pagetic bone. Yet Paget disease may account for a significant number of cases of this cancer in the elderly.37 In these cases, osteosarcoma is presumed to be driven by a second genetic mutation, has a genetic signature distinct from that in osteosarcomas occurring in youth, and is quite resistant to treatment.38 In Scandinavia and Japan, where Paget disease is rare, the second peak of osteosarcoma that occurs with aging seems muted as well.39,40 These cancers present with pain, soft-tissue swelling, and variable elevations in serum alkaline phosphatase. Investigations to date suggest that pagetic lesions and osteosarcomas arising in pagetic bone are probably both driven to some extent by stromal cells overexpressing RANK ligand and may not represent defects intrinsic to the osteoclast.41
Giant-cell tumors of bone are also rare but can arise in pagetic bone. A cluster of cases was reported in Avellino and other towns of southern Italy.42 Again, the lesions occur in older individuals and in different sites than those seen in the benign giant-cell tumors recorded in patients without Paget disease.
Metastases from lymphomas, prostate cancer, and breast cancer certainly occur in bone, but rarely in pagetic sites.43 A recent case study noted that patients with prostate cancer who also had Paget disease had a later onset of metastasis to bone than patients without coincident Paget disease.44
A THOUGHTFUL ASSESSMENT
Evaluating a patient with Paget disease requires a thoughtful assessment of its musculoskeletal consequences in an aging skeleton. Pain in Paget disease is often multifactorial. In the elderly, end-stage degenerative disease of the spine, hip, and knees, mechanical instability, compression fractures of the spine, and neuropathies may compound the clinical picture. Therefore, a thorough evaluation is required to plan effective therapy.
Alkaline phosphatase and other markers
A screening serum alkaline phosphatase level is usually sufficient to measure bone turnover. Produced by osteoblasts, alkaline phosphatase is a marker of bone formation, but an imperfect one. Often it is elevated in active Paget disease—but not always.45 Many patients have normal serum alkaline phosphatase levels, particularly if they have monostotic (single-bone) disease. It is unclear why, in a disorder marked by accelerated bone remodeling, the biochemical markers are inconsistent measures of bone turnover.
Research into biochemical markers of Paget disease has had two aims: to identify the single best marker for baseline assessment of pagetic bone activity and to find out whether this measurement responds to therapy.46,47 Measures of bone formation such as bone-specific alkaline phosphatase, osteocalcin, and the procollagen type I peptides, and measures of bone resorption including the pyridinolines, hydroxyproline, and cross-linked collagens, have been analyzed as markers of bone remodeling and show no real advantage over the serum alkaline phosphatase level as reflections of bone turnover. As alkaline phosphatase measurement is inexpensive, available, and reliable, it should be used preferentially, with gamma-glutamyl transpeptidate or 5′ nucleotidase confirming the source as either liver or bone. Readers are directed to a recent review in which the utility of these markers is explored in more detail.48
Imaging studies
Bone scans can give us an idea of the extent, location, and general activity of the disease (Figure 3). Uptake is avid in affected bones, beginning in the subchondral region and spreading throughout the bone. Bone scans can be particularly useful in defining sites of active disease when the serum alkaline phosphatase level is normal.
Plain radiography of the affected bones outlines the anatomy of the problem and gives some insight into the cause of pain (Figure 3).
Computed tomography or magnetic resonance imaging may prove useful in cases of spinal stenosis, cauda equina syndrome, compression fractures, or suspected malignancy (Figure 4), but these studies are expensive and generally are not needed.
Radiographic features. Paget disease is presumed to be a disease of the osteoclast, and the earliest lesion is described as lytic. In my own experience, it is unusual to see a purely lytic lesion, although sometimes the disease presents in the skull in this way—osteoporosis circumscripta—or in the femur or tibia with an advancing edge of pure osteolysis.
More often, one sees evidence of both resorption by osteoclasts and formation by osteoblasts, reflecting the coupling of these two processes in this disease. Radiographic findings on plain films are usually definitive, showing enlargement of the affected bone, deformity, coarsened trabeculae, and thickened cortices with tunneling (Figure 5).49 In weightbearing bones, pseudofractures may stud the convex surface. These incongruities of bone may persist for years, heralding fracture only when there is focal pain (Figure 6).50
Biopsy is infrequently needed
If these diagnostic findings are not present, then biopsy is indicated. In the United Sates and Canada, where Paget disease is fairly common, biopsy is infrequently needed and is usually reserved for situations in which the differential diagnosis includes cancer, as when the cortex cannot be clearly visualized, the lesions are atypical in pattern or location, or there is a single sclerotic vertebral body on imaging.51
The other indication for biopsy is a “new” pagetic lesion. For reasons unknown, the pattern of skeletal involvement in Paget disease tends to be stable throughout the patient’s lifetime. This is another reason why a baseline bone scan is useful.
TREATMENT WITH BISPHOSPHONATES
Treatment of Paget disease today relies for the most part on the new generation of nitrogen-containing bisphosphonates. As a class, these are antiresorptive agents that inhibit osteoclasts; in this way they slow bone remodeling and enhance the deposition of normal lamellar bone. Their clinical efficacy in Paget disease, coupled with the observation that the earliest lesion in Paget disease is lytic, underscores the principle that Paget disease is a disorder of the osteoclast.
Oral bisphosphonates
Etidronate, approved in 1977, was the first bisphosphonate licensed to treat Paget disease, and it remains available for this indication in the United States. Used in 6-month regimens, it lowers the serum alkaline phosphatase level in some patients, but it has a narrow therapeutic margin. Drug-induced osteomalacia and worsening lytic lesions and fractures in weight-bearing bones are some of the complications.52 When the nitrogen-containing bisphosphonates were developed, they proved to be more potent antiresorptive agents that pose less risk of mineralization defects at prescribed doses.
Alendronate, approved in 1995, is an oral nitrogen-containing bisphosphonate that is effective in treating Paget disease.53 Alendronate is now available in the United States only through special programs (eg, the CVS ProCare Program); the paperwork required to secure this drug is onerous, so the drug is used infrequently. Studies in Paget disease showed that it normalizes the serum alkaline phosphatase level, improves the radiographic appearance, and eases pain in many patients.54 The dosage is 40 mg daily for 6 months.
Risedronate, approved in 1998, is another oral nitrogen-containing bisphosphonate and is comparable to alendronate in efficacy.55 The dosage is 30 mg daily for 2 months.
Tiludronate is another oral bisphosphonate with a different mechanism of action from the nitrogen-containing bisphosphonates.56 It is safe, often effective, but less potent than the newer agents.
The oral bisphosphonates are well tolerated, with few side effects other than gastrointestinal distress. As a class, they are poorly absorbed and so must be taken fasting with a full glass of water on rising, after which the patient should remain upright without food or drink for 30 to 60 minutes. This is a nuisance for elderly patients already on multiple medications and thus makes intravenous agents appealing.
Intravenous bisphosphonates
Pamidronate was approved in 1994. It is quite effective in many patients with Paget disease. There is no consensus around the world on dosing, with regimens ranging from 30 mg to 90 mg or more intravenously in divided doses given over 2 to 4 hours from once a day to once a week. In the United States, 30 mg is given over 4 hours on 3 consecutive days. Resistance to pamidronate has been described; the mechanism is unknown.
Zoledronic acid is a nitrogen-containing bisphosphonate. It is given as a single infusion over 15 minutes, and re-treatment may not be necessary for years. A randomized clinical trial in 2005 demonstrated the efficacy of zoledronic acid 5 mg by infusion compared with oral risedronate in the treatment of Paget disease.57 In observational extension studies lasting as long as 6.5 years, zoledronic acid has been shown to be superior to risedronate in terms of the proportion of patients experiencing a sustained clinical remission.58
While there are many bisphosphonates on the market, an infusion of 5 mg of zoledronic acid seems optimal in most patients who do not have a contraindication or an aversion to intravenous therapy. It tends to normalize the serum alkaline phosphatase level quickly and to leave more patients in sustained biochemical remission than do older bisphosphonates, as noted above. It also tends to be more effective in normalizing the serum alkaline phosphatase level when a patient has used other bisphosphonates in the past or has become resistant to them.
Bisphosphonates reduce bone turnover but do not correct deformities
In randomized clinical trials, bisphosphonates have been shown to restore bone remodeling to more normal levels, to ease pain from pagetic bone, to lower the serum alkaline phosphatase level, and to heal radiographic lesions, but these drugs have not been proven to prevent progression of deformity or to restore the structural integrity of bone (Figure 6).
The Paget’s Disease: Randomized Trial of Intensive Versus Symptomatic Management (PRISM), in 1,324 people with Paget disease in the United Kingdom, showed no difference in the incidence of fracture, orthopedic surgery, quality of life, or hearing thresholds over 2 to 5 years in patients treated with bisphosphonates vs those treated symptomatically, despite a significant difference in serum alkaline phosphatase in the two groups (P < .001).59
In the observational extension study of zoledronic acid described above,58 three of four fractures occurred in the group treated with zoledronic acid, echoing the findings of the PRISM study.
Adverse effects of bisphosphonates
The more potent the bisphosphonate is as an antiresorptive agent, the more it suppresses normal bone remodeling, which can lead to osteonecrosis of the jaw and to atypical femoral fractures.60,61 These complications are unusual in patients with Paget disease because the treatment is intermittent. Sometimes a single dose of zoledronic acid or one course of risedronate or alendronate will last for years.
All the nitrogen-containing bisphosphonates, particularly zoledronic acid, may provoke flulike symptoms of fever, arthralgias, and bone pain. This effect is self-limited, resolves in days, and does not tend to recur. Bone pain may be more sustained, but this also passes, and within weeks the antiresorptive process has abated and pagetic bone pain will ease. Atrial fibrillation is not an anticipated complication of treatment with a bisphosphonate.62 The risk of esophageal cancer is not confirmed at this time.63 Other rare complications of the bisphosphonates include iritis, acute renal failure, and allergy.
Bisphosphonates are not approved for use in patients with creatinine clearance less than 30 mL/min, or in pregnancy.
Other treatments
Calcitonin, an older agent, can still be useful in easing the pain of Paget disease, healing bone lesions, and reducing the metabolic activity of pagetic bone in patients who cannot receive bisphosphonates. It is given by injection in doses of 50 to 100 IU daily or every other day. Although unlikely to effect a sustained clinical remission, calcitonin remains a safe, well-tolerated, and well-studied medication in Paget disease and is approved for this indication.64,65
Denosumab has not been formally studied in Paget disease, but a recent case report indicated it was effective.66
A conservative strategy
Guidelines for treating Paget disease have been written at various times in many countries, including Italy (2007),67 the United Kingdom (2004),68 Japan (2006),69 and Canada (2007).70 Recommendations differ, in part because it is hard to ascertain whether long-term outcomes are improved by treatment, and in part because the prevalence of Paget disease is decreasing and its severity is lessening.11,12 Some guidelines are outdated, since they do not include the newer bisphosphonates.
If the natural history of untreated Paget disease involves the gradual evolution over more than 20 years of bowing deformities in the lower limbs, rigidity and overgrowth of the spine, and softening and enlargement of the skull, as described by Sir James Paget, then treatment should be initiated in hopes that it will modify the outcome. We have no lens to better focus this question on the effect of treatment on the natural history of the disease. We have the PRISM study, designed before zoledronic acid was approved and only 2 to 5 years in duration. And we have the epidemiologic data demonstrating that most patients have no symptoms during their lifetime.
We see the crippling bone disease described by Sir James Paget so infrequently today in the United States that we forget the profound morbidity that may attend the skeletal changes of Paget disease that were common in the early 20th century. Once the bones of the skull are overgrown, the limbs are bowed, and the degenerative joint disease is present, no medication can reverse these changes. Then, the integrity of the bone is lost, and the vulnerability to fracture, early osteoarthritis, nerve compression syndromes, and hearing loss persist. Understanding these consequences prompts the recommendation of early treatment in patients with Paget disease, in hopes of mitigating disease progression.
Patients with active Paget disease, documented either by an elevated serum alkaline phosphatase or by a bone scan, should be treated with a bisphosphonate if the disease is found in sites where remodeling of bone may lead to complications. Such sites include the skull, spine, and long bones of the lower extremity. Paget disease of bone in the pelvis tends to give little trouble (Figure 2) unless it is proximal to a joint, when pain and early arthritis may result. Treatment is safe and, I think, prudent to undertake in any person over age 55 with active disease. To prevent hypocalcemia during treatment, all patients should be repleted with vitamin D and maintained on calcium 1,200 mg daily through diet or supplements with meals.
Throughout the evaluation and treatment, it is important to remember that pain may not emanate from pagetic bone. If medication for Paget disease proves ineffective in the first few months, analgesics, bracing, walking aids, and operative management71 are adjunctive therapies to improve the functional status of these patients.
It is a remarkable clinical observation that treatment of Paget disease may rapidly reverse neurologic syndromes, resolve the erythema or warmth overlying active pagetic bone, and diminish the risk of bleeding with surgery. This response to therapy suggests that there is prompt inhibition and apoptosis of the osteoclasts, accompanied by diminished vascularity of bone. Whatever the mechanism, it is worth treating patients who have spinal stenosis, arthritis, and nerve compression syndromes with calcitonin or bisphosphonates before surgical intervention, whenever possible.34,72
- Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans 1877; 60:37–64.9.
- Guyer PB, Chamberlain AT, Ackery DM, Rolfe EB. The anatomic distribution of osteitis deformans. Clin Orthop Relat Res 1981; 156:141–144.
- Tiegs RD, Lohse CM, Wollan PC, Melton LJ. Long-term trends in the incidence of Paget’s disease of bone. Bone 2000; 27:423–427.
- Altman RD, Bloch DA, Hochberg MC, Murphy WA. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res 2000; 15:461–465.
- Barker DJ. The epidemiology of Paget’s disease of bone. Br Med Bull 1984; 40:396–400.
- Detheridge FM, Guyer PB, Barker DJ. European distribution of Paget’s disease of bone. Br Med J (Clin Res Ed) 1982; 285:1005–1008.
- van Staa TP, Selby P, Leufkens HG, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002; 17:465–471.
- Barker DJ. The epidemiology of Paget’s disease. Metab Bone Dis Relat Res 1981; 3:231–233.
- Rogers J, Jeffrey DR, Watt I. Paget’s disease in an archeological population. J Bone Miner Res 2002; 17:1127–1134.
- Aaron JE, Rogers J, Kanis JA. Paleohistology of Paget’s disease in two medieval skeletons. Am J Phys Anthropol 1992; 89:325–331.
- Poór G, Donáth J, Fornet B, Cooper C. Epidemiology of Paget’s disease in Europe: the prevalence is decreasing. J Bone Miner Res 2006; 21:1545–1549.
- Cundy HR, Gamble G, Wattie D, Rutland M, Cundy T. Paget’s disease of bone in New Zealand: continued decline in disease severity. Calcif Tissue Int 2004; 75:358–364.
- Doyle T, Gunn J, Anderson G, Gill M, Cundy T. Paget’s disease in New Zealand: evidence for declining prevalence. Bone 2002; 31:616–619.
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70:1582–1588.
- Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet 2002; 11:2735–2739.
- Lucas GJ, Hocking LJ, Daroszewska A, et al. Ubiquitin-associated domain mutations of SQSTM1 in Paget’s disease of bone: evidence for a founder effect in patients of British descent. J Bone Miner Res 2005; 20:227–231.
- Mays S. Archaeological skeletons support a northwest European origin for Paget’s disease of bone. J Bone Miner Res 2010; 25:1839–1841.
- Bolland MJ, Tong PC, Naot D, et al. Delayed development of Paget’s disease in offspring inheriting SQSTM1 mutations. J Bone Miner Res 2007; 22:411–415.
- Rea SL, Walsh JP, Ward L, et al. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res 2006; 21:1136–1145.
- Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P38–P44.
- Eekhoff EW, Karperien M, Houtsma D, et al. Familial Paget’s disease in The Netherlands: occurrence, identification of new mutations in the sequestosome 1 gene, and their clinical associations. Arthritis Rheum 2004; 50:1650–1654.
- Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int 2012; 91:97–113.
- Kurihara N, Hiruma Y, Yamana K, et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab 2011; 13:23–34.
- Kurihara N, Zhou H, Reddy SV, et al. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget’s disease-like bone lesions in mice. J Bone Miner Res 2006; 21:446–455.
- Reddy SV, Singer FR, Roodman GD. Bone marrow mononuclear cells from patients with Paget’s disease contain measles virus nucleocapsid messenger ribonucleic acid that has mutations in a specific region of the sequence. J Clin Endocrinol Metab 1995; 80:2108–2111.
- Gennari L, Merlotti D, Martini G, Nuti R. Paget’s disease of bone in Italy. J Bone Miner Res 2006; 21(suppl 2):P14–P21.
- Seton M, Choi HK, Hansen MF, Sebaldt RJ, Cooper C. Analysis of environmental factors in familial versus sporadic Paget’s disease of bone—the New England Registry for Paget’s Disease of Bone. J Bone Miner Res 2003; 18:1519–1524.
- Siris ES. Extensive personal experience: Paget’s disease of bone. J Clin Endocrinol Metab 1995; 80:335–338.
- Lucas GJ, Daroszewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget’s disease of bone and related disorders. J Bone Miner Res 2006; 21(suppl 2):P31–P37.
- Seton M. Diagnosis, complications and treatment of Paget’s disease of bone. Aging Health 2009; 5:497–508.
- Siris E, Roodman GD. Paget’s Disease of Bone. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008.
- Seton M, Moses AM, Bode RK, Schwartz C. Paget’s disease of bone: the skeletal distribution, complications and quality of life as perceived by patients. Bone 2011; 48:281–285.
- Seton M. Paget’s disease of bone. In:Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors. Rheumatology. 5th ed. Philadelphia, PA: Mosby Elsevier; 2010:2021–2028.
- Douglas DL, Duckworth T, Kanis JA, Jefferson AA, Martin TJ, Russell RG. Spinal cord dysfunction in Paget’s disease of bone. Has medical treatment a vascular basis? J Bone Joint Surg Br 1981; 63B:495–503.
- Siris ES. Epidemiological aspects of Paget’s disease: family history and relationship to other medical conditions. Semin Arthritis Rheum 1994; 23:222–225.
- Kanis JA, Evanson JM, Russell RG. Paget’s disease of bone: diagnosis and management. Metab Bone Dis Relat Res 1981; 3:219–230.
- Mangham DC, Davie MW, Grimer RJ. Sarcoma arising in Paget’s disease of bone: declining incidence and increasing age at presentation. Bone 2009; 44:431–436.
- Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P58–P63.
- Price CH. The incidence of osteogenic sarcoma in South-West England and its relationship to Paget’s disease of bone. J Bone Joint Surg Br 1962; 44-B:366–376.
- Ishikawa Y, Tsukuma H, Miller RW. Low rates of Paget’s disease of bone and osteosarcoma in elderly Japanese. Lancet 1996; 347:1559.
- Sun SG, Lau YS, Itonaga I, Sabokbar A, Athanasou NA. Bone stromal cells in pagetic bone and Paget’s sarcoma express RANKL and support human osteoclast formation. J Pathol 2006; 209:114–120.
- Rendina D, Gennari L, De Filippo G, et al. Evidence for increased clinical severity of familial and sporadic Paget’s disease of bone in Campania, southern Italy. J Bone Miner Res 2006; 21:1828–1835.
- Fenton P, Resnick D. Metastases to bone affected by Paget’s disease. A report of three cases. Int Orthop 1991; 15:397–399.
- Tu SM, Som A, Tu B, Logothetis CJ, Lee MH, Yeung SC. Effect of Paget’s disease of bone (osteitis deformans) on the progression of prostate cancer bone metastasis. Br J Cancer 2012; 107:646–651.
- Eekhoff ME, van der Klift M, Kroon HM, et al. Paget’s disease of bone in The Netherlands: a population-based radiological and biochemical survey—the Rotterdam Study. J Bone Miner Res 2004; 19:566–570.
- Reid IR, Davidson JS, Wattie D, et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget’s disease of bone. Bone 2004; 35:224–230.
- Alvarez L, Guañabens N, Peris P, et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget’s disease. Bone 2001; 29:447–452.
- Cundy T, Reid IR. Paget’s disease of bone. Clin Biochem 2012; 45:43–48.
- Cortis K, Micallef K, Mizzi A. Imaging Paget’s disease of bone—from head to toe. Clin Radiol 2011; 66:662–672.
- Redden JF, Dixon J, Vennart W, Hosking DJ. Management of fissure fractures in Paget’s disease. Int Orthop 1981; 5:103–106.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 25-1993. A 67-year-old man with osteolytic lesions of T11 and T12. N Engl J Med 1993; 328:1836–1841.
- Evans RA, Dunstan CR, Hills E, Wong SY. Pathologic fracture due to severe osteomalacia following low-dose diphosphonate treatment of Paget’s disease of bone. Aust N Z J Med 1983; 13:277–279.
- Siris E, Weinstein RS, Altman R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab 1996; 81:961–967.
- Reid IR, Siris E. Alendronate in the treatment of Paget’s disease of bone. Int J Clin Pract Suppl 1999; 101:62–66.
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget’s disease of bone. Paget’s Risedronate/Etidronate Study Group. Am J Med 1999; 106:513–520.
- Peris P, Alvarez L, Vidal S, Martínez MA, Monegal A, Guañabens N. Treatment with tiludronate has a similar effect to risedronate on Paget’s disease activity assessed by bone markers and bone scintigraphy. Clin Exp Rheumatol 2007; 25:206–210.
- Reid IR, Miller P, Lyles K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med 2005; 353:898–908.
- Reid IR, Lyles K, Su G, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011; 26:2261–2270.
- Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH; PRISM Trial Group. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res 2010; 25:20–31.
- Abrahamsen B, Einhorn TA. Beyond a reasonable doubt? Bisphosphonates and atypical femur fractures. Bone 2012; 50:1196–1200.
- Seton M, Krane SM. Use of zoledronic acid in the treatment of Paget’s disease. Ther Clin Risk Manag 2007; 3:913–918.
- Sørensen HT, Christensen S, Mehnert F, et al. Use of bisphosphonates among women and risk of atrial fibrillation and flutter: Population based case-control study. BMJ 2008; 336:813–816.
- Dixon WG, Solomon DH. Bisphosphonates and esophageal cancer—a pathway through the confusion. Nat Rev Rheumatol 2011; 7:369–372.
- Singer FR, Krane SM. Paget’s disease of bone. In:Avioli LV, Krane SM, editors. Metabolic Bone Disease and Clinically Related Disorders. 2nd ed. Philadelphia, PA: W.B. Saunders Company; 1990:546–615.
- Kanis JA, Horn DB, Scott RD, Strong JA. Treatment of Paget’s disease of bone with synthetic salmon calcitonin. Br Med J 1974; 3:727–731.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Adami S, Bartolozzi P, Brandi ML, et al; Societa Italiana di Ortopedia e Traumatologia. [Italian guidelines for the diagnosis and treatment of Paget’s disease of bone.] Reumatismo 2007; 59:153–168. (Article in Italian.)
- Scarsbrok A, Brown M, Wilson D. UK guidelines on management of Paget’s disease of bone. Rheumatology (Oxford) 2004; 43:399–400.
- Takata S, Hashimoto J, Nakatsuka K, et a.l Guidelines for diagnosis and management of Paget’s disease of bone in Japan. J Bone Miner Metab 2006; 24:359–367.
- Josse RG, Hanley DA, Kendler D, Ste Marie L-G, Adachi JD, Brown J. Diagnosis and treatment of Paget’s disease of bone. Clin Invest Med 2007; 30:E210–E223.
- Kaplan FS. Paget’s disease of bone: orthopedic complications. Semin Arthritis Rheum 1994; 23:250–252.
- Kanis JA, Gray RE. Long-term follow-up observations on treatment in Paget’s disease of bone. Clin Orthop Relat Res 1987; 217:99–125.
- Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans 1877; 60:37–64.9.
- Guyer PB, Chamberlain AT, Ackery DM, Rolfe EB. The anatomic distribution of osteitis deformans. Clin Orthop Relat Res 1981; 156:141–144.
- Tiegs RD, Lohse CM, Wollan PC, Melton LJ. Long-term trends in the incidence of Paget’s disease of bone. Bone 2000; 27:423–427.
- Altman RD, Bloch DA, Hochberg MC, Murphy WA. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res 2000; 15:461–465.
- Barker DJ. The epidemiology of Paget’s disease of bone. Br Med Bull 1984; 40:396–400.
- Detheridge FM, Guyer PB, Barker DJ. European distribution of Paget’s disease of bone. Br Med J (Clin Res Ed) 1982; 285:1005–1008.
- van Staa TP, Selby P, Leufkens HG, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002; 17:465–471.
- Barker DJ. The epidemiology of Paget’s disease. Metab Bone Dis Relat Res 1981; 3:231–233.
- Rogers J, Jeffrey DR, Watt I. Paget’s disease in an archeological population. J Bone Miner Res 2002; 17:1127–1134.
- Aaron JE, Rogers J, Kanis JA. Paleohistology of Paget’s disease in two medieval skeletons. Am J Phys Anthropol 1992; 89:325–331.
- Poór G, Donáth J, Fornet B, Cooper C. Epidemiology of Paget’s disease in Europe: the prevalence is decreasing. J Bone Miner Res 2006; 21:1545–1549.
- Cundy HR, Gamble G, Wattie D, Rutland M, Cundy T. Paget’s disease of bone in New Zealand: continued decline in disease severity. Calcif Tissue Int 2004; 75:358–364.
- Doyle T, Gunn J, Anderson G, Gill M, Cundy T. Paget’s disease in New Zealand: evidence for declining prevalence. Bone 2002; 31:616–619.
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70:1582–1588.
- Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet 2002; 11:2735–2739.
- Lucas GJ, Hocking LJ, Daroszewska A, et al. Ubiquitin-associated domain mutations of SQSTM1 in Paget’s disease of bone: evidence for a founder effect in patients of British descent. J Bone Miner Res 2005; 20:227–231.
- Mays S. Archaeological skeletons support a northwest European origin for Paget’s disease of bone. J Bone Miner Res 2010; 25:1839–1841.
- Bolland MJ, Tong PC, Naot D, et al. Delayed development of Paget’s disease in offspring inheriting SQSTM1 mutations. J Bone Miner Res 2007; 22:411–415.
- Rea SL, Walsh JP, Ward L, et al. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res 2006; 21:1136–1145.
- Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P38–P44.
- Eekhoff EW, Karperien M, Houtsma D, et al. Familial Paget’s disease in The Netherlands: occurrence, identification of new mutations in the sequestosome 1 gene, and their clinical associations. Arthritis Rheum 2004; 50:1650–1654.
- Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int 2012; 91:97–113.
- Kurihara N, Hiruma Y, Yamana K, et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab 2011; 13:23–34.
- Kurihara N, Zhou H, Reddy SV, et al. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget’s disease-like bone lesions in mice. J Bone Miner Res 2006; 21:446–455.
- Reddy SV, Singer FR, Roodman GD. Bone marrow mononuclear cells from patients with Paget’s disease contain measles virus nucleocapsid messenger ribonucleic acid that has mutations in a specific region of the sequence. J Clin Endocrinol Metab 1995; 80:2108–2111.
- Gennari L, Merlotti D, Martini G, Nuti R. Paget’s disease of bone in Italy. J Bone Miner Res 2006; 21(suppl 2):P14–P21.
- Seton M, Choi HK, Hansen MF, Sebaldt RJ, Cooper C. Analysis of environmental factors in familial versus sporadic Paget’s disease of bone—the New England Registry for Paget’s Disease of Bone. J Bone Miner Res 2003; 18:1519–1524.
- Siris ES. Extensive personal experience: Paget’s disease of bone. J Clin Endocrinol Metab 1995; 80:335–338.
- Lucas GJ, Daroszewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget’s disease of bone and related disorders. J Bone Miner Res 2006; 21(suppl 2):P31–P37.
- Seton M. Diagnosis, complications and treatment of Paget’s disease of bone. Aging Health 2009; 5:497–508.
- Siris E, Roodman GD. Paget’s Disease of Bone. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008.
- Seton M, Moses AM, Bode RK, Schwartz C. Paget’s disease of bone: the skeletal distribution, complications and quality of life as perceived by patients. Bone 2011; 48:281–285.
- Seton M. Paget’s disease of bone. In:Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors. Rheumatology. 5th ed. Philadelphia, PA: Mosby Elsevier; 2010:2021–2028.
- Douglas DL, Duckworth T, Kanis JA, Jefferson AA, Martin TJ, Russell RG. Spinal cord dysfunction in Paget’s disease of bone. Has medical treatment a vascular basis? J Bone Joint Surg Br 1981; 63B:495–503.
- Siris ES. Epidemiological aspects of Paget’s disease: family history and relationship to other medical conditions. Semin Arthritis Rheum 1994; 23:222–225.
- Kanis JA, Evanson JM, Russell RG. Paget’s disease of bone: diagnosis and management. Metab Bone Dis Relat Res 1981; 3:219–230.
- Mangham DC, Davie MW, Grimer RJ. Sarcoma arising in Paget’s disease of bone: declining incidence and increasing age at presentation. Bone 2009; 44:431–436.
- Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P58–P63.
- Price CH. The incidence of osteogenic sarcoma in South-West England and its relationship to Paget’s disease of bone. J Bone Joint Surg Br 1962; 44-B:366–376.
- Ishikawa Y, Tsukuma H, Miller RW. Low rates of Paget’s disease of bone and osteosarcoma in elderly Japanese. Lancet 1996; 347:1559.
- Sun SG, Lau YS, Itonaga I, Sabokbar A, Athanasou NA. Bone stromal cells in pagetic bone and Paget’s sarcoma express RANKL and support human osteoclast formation. J Pathol 2006; 209:114–120.
- Rendina D, Gennari L, De Filippo G, et al. Evidence for increased clinical severity of familial and sporadic Paget’s disease of bone in Campania, southern Italy. J Bone Miner Res 2006; 21:1828–1835.
- Fenton P, Resnick D. Metastases to bone affected by Paget’s disease. A report of three cases. Int Orthop 1991; 15:397–399.
- Tu SM, Som A, Tu B, Logothetis CJ, Lee MH, Yeung SC. Effect of Paget’s disease of bone (osteitis deformans) on the progression of prostate cancer bone metastasis. Br J Cancer 2012; 107:646–651.
- Eekhoff ME, van der Klift M, Kroon HM, et al. Paget’s disease of bone in The Netherlands: a population-based radiological and biochemical survey—the Rotterdam Study. J Bone Miner Res 2004; 19:566–570.
- Reid IR, Davidson JS, Wattie D, et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget’s disease of bone. Bone 2004; 35:224–230.
- Alvarez L, Guañabens N, Peris P, et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget’s disease. Bone 2001; 29:447–452.
- Cundy T, Reid IR. Paget’s disease of bone. Clin Biochem 2012; 45:43–48.
- Cortis K, Micallef K, Mizzi A. Imaging Paget’s disease of bone—from head to toe. Clin Radiol 2011; 66:662–672.
- Redden JF, Dixon J, Vennart W, Hosking DJ. Management of fissure fractures in Paget’s disease. Int Orthop 1981; 5:103–106.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 25-1993. A 67-year-old man with osteolytic lesions of T11 and T12. N Engl J Med 1993; 328:1836–1841.
- Evans RA, Dunstan CR, Hills E, Wong SY. Pathologic fracture due to severe osteomalacia following low-dose diphosphonate treatment of Paget’s disease of bone. Aust N Z J Med 1983; 13:277–279.
- Siris E, Weinstein RS, Altman R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab 1996; 81:961–967.
- Reid IR, Siris E. Alendronate in the treatment of Paget’s disease of bone. Int J Clin Pract Suppl 1999; 101:62–66.
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget’s disease of bone. Paget’s Risedronate/Etidronate Study Group. Am J Med 1999; 106:513–520.
- Peris P, Alvarez L, Vidal S, Martínez MA, Monegal A, Guañabens N. Treatment with tiludronate has a similar effect to risedronate on Paget’s disease activity assessed by bone markers and bone scintigraphy. Clin Exp Rheumatol 2007; 25:206–210.
- Reid IR, Miller P, Lyles K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med 2005; 353:898–908.
- Reid IR, Lyles K, Su G, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011; 26:2261–2270.
- Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH; PRISM Trial Group. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res 2010; 25:20–31.
- Abrahamsen B, Einhorn TA. Beyond a reasonable doubt? Bisphosphonates and atypical femur fractures. Bone 2012; 50:1196–1200.
- Seton M, Krane SM. Use of zoledronic acid in the treatment of Paget’s disease. Ther Clin Risk Manag 2007; 3:913–918.
- Sørensen HT, Christensen S, Mehnert F, et al. Use of bisphosphonates among women and risk of atrial fibrillation and flutter: Population based case-control study. BMJ 2008; 336:813–816.
- Dixon WG, Solomon DH. Bisphosphonates and esophageal cancer—a pathway through the confusion. Nat Rev Rheumatol 2011; 7:369–372.
- Singer FR, Krane SM. Paget’s disease of bone. In:Avioli LV, Krane SM, editors. Metabolic Bone Disease and Clinically Related Disorders. 2nd ed. Philadelphia, PA: W.B. Saunders Company; 1990:546–615.
- Kanis JA, Horn DB, Scott RD, Strong JA. Treatment of Paget’s disease of bone with synthetic salmon calcitonin. Br Med J 1974; 3:727–731.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Adami S, Bartolozzi P, Brandi ML, et al; Societa Italiana di Ortopedia e Traumatologia. [Italian guidelines for the diagnosis and treatment of Paget’s disease of bone.] Reumatismo 2007; 59:153–168. (Article in Italian.)
- Scarsbrok A, Brown M, Wilson D. UK guidelines on management of Paget’s disease of bone. Rheumatology (Oxford) 2004; 43:399–400.
- Takata S, Hashimoto J, Nakatsuka K, et a.l Guidelines for diagnosis and management of Paget’s disease of bone in Japan. J Bone Miner Metab 2006; 24:359–367.
- Josse RG, Hanley DA, Kendler D, Ste Marie L-G, Adachi JD, Brown J. Diagnosis and treatment of Paget’s disease of bone. Clin Invest Med 2007; 30:E210–E223.
- Kaplan FS. Paget’s disease of bone: orthopedic complications. Semin Arthritis Rheum 1994; 23:250–252.
- Kanis JA, Gray RE. Long-term follow-up observations on treatment in Paget’s disease of bone. Clin Orthop Relat Res 1987; 217:99–125.
KEY POINTS
- The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition or an environmental factor, or both.
- Because Paget disease tends to occur in an aging skeleton, “pagetic” bone may not always be the source of pain. Rather, the pain may be from secondary degenerative changes of the spine or joints or from compression fractures.
- An elevated serum alkaline phosphatase level may signal Paget disease, but many patients have a normal serum alkaline phosphatase.
- Plain radiography of the affected bones outlines the anatomy of the problem and provides insight into the cause of pain.
- Treatment of Paget disease relies primarily on the new generation of nitrogen-containing bisphosphonates.
