User login
The efficacy of therapy targeted to a specific oncogene is convincing evidence of “oncogene addiction,” or the concept that some cancers rely on or are “addicted to” a specific gene for their survival and proliferation. In the case of non–small cell lung cancer (NSCLC), drugs that target epidermal growth factor receptor (EGFR) have been proven more effective than conventional chemotherapy in patients with sensitizing EGFR mutations.1
Lung cancer oncogenes can drive oncogenic signaling pathways within tumor cells. Activation of EGFR signaling that drives cell proliferation through pathways such as RAS/RAF/MEK/ERK and the cell survival pathways P13K and AKT has been demonstrated in never-smokers. In heavy smokers, KRAS oncogene mutation is the dominant promoter of activation of oncogenic signaling pathways; it predicts a poor prognosis (especially for lung adenocarcinoma), and it is essentially mutually exclusive with EGFR mutations.
More than 50% of cases of NSCLC have known oncogene mutations for which targeted therapeutics are available.2 For example, gefitinib and erlotinib are the effective inhibitors for the EGFR oncogene mutation, sunitinib for platelet-derived growth factor receptor (PDGFR) amplification, and lapatinib for the less common ERBB2 insertion.
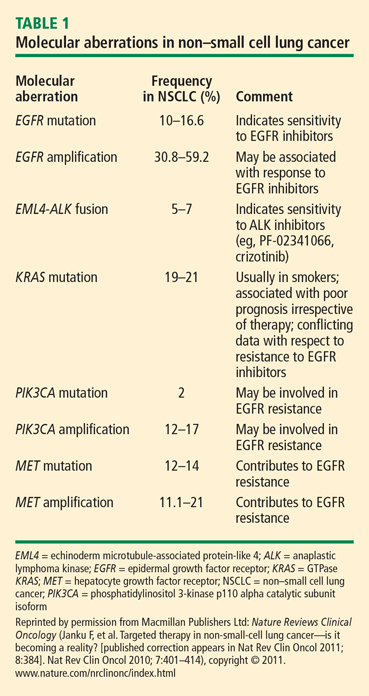
- EGFR mutations and amplifications
- EML4-ALK translocation fusions
- KRAS mutations
- PIK3CA mutations
- MET mutations, alternative splicing, amplification, and overexpression
PLATINUM DOUBLET AS STANDARD
Multiple platinum-based combinations of chemotherapy are in use as first-line therapy for advanced NSCLC. An overall survival (OS) benefit has been established with the use of doublet regimens, but no platinum-based doublet regimen has been proven superior to another on the end point of OS in clinical trials.4
Adding a third agent increases the response rate in advanced NSCLC but does not improve OS; the exception is bevacizumab, a monoclonal antibody targeted to vascular endothelial growth factor (VEGF). Sandler et al5 demonstrated a survival benefit when bevacizumab was added to paclitaxel-carboplatin in a recent study that led to US Food and Drug Administration (FDA) approval of bevacizumab for the treatment of NSCLC.
The next oncogene target explored in advanced NSCLC was EGFR. The tyrosine kinase inhibitors (TKIs) gefitinib, erlotinib, lapatinib, and the monoclonal antibody cetuximab are all clinical inhibitory agents targeting EGFR.
Previously treated NSCLC
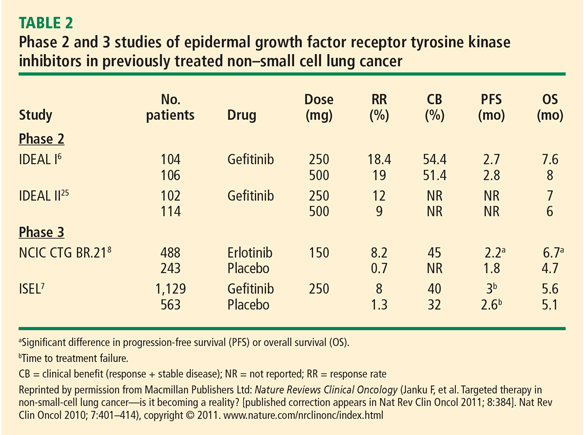
In the phase 3 National Cancer Institute of Canada (NCIC) Clinical Trials Group (CTG) BR.21 trial, erlotinib demonstrated significant superiority over placebo as second- or third-line chemotherapy on PFS and OS in unselected patients with NSCLC. The results led to its approval as treatment for advanced NSCLC in patients who have received at least one prior chemotherapy regimen.8
Patient survival in NCIC CTG BR. 21 was evaluated in a series of patient subsets in exploratory univariate analyses. The effect of erlotinib on survival was similar across most subsets. A greater effect on survival by erlotinib was observed in patients who had never smoked (hazard ratio [HR] = 0.42). In the subgroup of patients who never smoked, EGFR status was predictive of erlotinib survival benefit. Patients who never smoked and were EGFR-positive had a survival benefit with erlotinib (HR = 0.27). There were too few EGFR-negative patients who never smoked to reach a conclusion.
Sensitizing mutations lead to better response
With the approval and clinical use of EGFR-TKIs came knowledge of EGFR kinase mutations that sensitize the mutated RTK to EGFR-TKI; this mechanism translates into dramatic tumor responses.9,10 Certain EGFR mutations contribute to sensitivity to EGFR-TKI treatment. The most common sensitizing mutations are the Exon 21 L858R mutation and the Exon 19 short in-frame deletions. The Exon 20 tends to yield resistant alterations (eg, prototypical T790M mutation and some Exon 20 Dup/Ins) in patients who initially derived benefit from targeted therapeutics. Although advances have been made in targeted therapeutics in lung cancer and other cancers, clinical resistance, particularly acquired or secondary resistance, remains the rule rather than the exception, which inherently limits the long-term clinical success of targeted therapeutics. A higher level of understanding of the mechanisms of resistance could have a substantial impact in maintaining the clinical efficacy of these targeted therapies.
The Iressa Pan-Asia Survival Study (IPASS) was a phase 3 study in which patient selection was based on clinical factors that predict a higher probability of harboring sensitizing EGFR mutations, thus enriching the mutant population, rather than screening for mutation status.11 Patients selected for participation were East Asians with advanced lung adenocarcinoma who were never smokers or former light smokers. They were randomized to treatment with platinum-based doublet chemotherapy (carboplatin-paclitaxel) or the EGFR-TKI gefitinib.
The primary end point—PFS—favored gefitinib over the chemotherapy doublet in the overall patient population. Of the 1,217 patients enrolled, EGFR mutation data for 35.9% could be evaluated. Sixty percent of the patients’ tumors harbored sensitizing EGFR mutations and, in this subset, the benefit of gefitinib was greater than it was in the overall population. In patients without an EGFR mutation, particularly those without Exon 19 deletions or Exon 21:L858R mutations (sensitizing mutations), platinum-based doublet chemotherapy performed better than gefitinib on the PFS end point. Of the mutation-positive patients, 4.2% had T790M mutations, which confers resistance to TKIs; this finding underscored the observation that all mutations cannot be treated the same.
The important message from the IPASS results is that even in a population preselected for EGFR mutation occurrence, the actual presence of the alteration of the target (EGFR mutations) leads to better survival with gefitinib. Molecular profiling of the tumor, therefore, is ultimately superior to profiling by patient phenotype or ethnicity. Thus, molecular selection trumps clinical selection.12
On the basis of IPASS and four similar phase 3 randomized controlled trials, the American Society of Clinical Oncology issued a clinical opinion that “patients with NSCLC who are being considered for first-line therapy with an EGFR-TKI (patients who have not previously received chemotherapy or an EGFR-TKI) should have their tumor tested for EGFR mutations to determine whether an EGFRTKI or chemotherapy is the appropriate first-line therapy.”13
Monoclonal antibody against EGFR
The First-Line Erbitux in Lung Cancer (FLEX) phase 3 worldwide study demonstrated that cetuximab, a monoclonal antibody directed against EGFR, as add-on therapy to a platinum-based doublet (cisplatin and vinorelbine) can extend median OS in patients with advanced EGFR-expressing NSCLC (stage wet IIIB or stage IV).14 As a result, the use of cetuximab combined with cisplatin-vinorelbine has been endorsed by a National Comprehensive Cancer Network (NCCN) guideline as a first-line option for the treatment of advanced NSCLC.
EML4-ALK fusion gene as target for crizotinib
The EML4-ALK fusion translocation oncogene was first identified in 2007 in a small proportion of patients with NSCLC,15,16 and a targeted therapy (crizotinib, an inhibitor of the ALK tyrosine kinase) has been developed. A single-arm phase 1 trial of crizotinib in patients selected for the EML4-ALK fusion gene has been completed.17 Of the approximately 1,500 NSCLC patients screened for the trial, 82 were identified as having advanced ALK-positive disease and were entered. These patients tended to be younger than those with ALK-negative disease, most had little or no exposure to tobacco, and all had adenocarcinoma.
The mean treatment duration was 6.4 months. Most treatment-related side effects were grade 1 or grade 2 gastrointestinal adverse events. The overall response rate was 57%. The disease control rates were 87% at 8 weeks and 66% at 16 weeks. Crizotinib has since moved to phase 2 and phase 3 trials and received FDA approval on August 26, 2011, for treatment of EML4-ALK–positive patients as assayed by a simultaneously approved companion molecular diagnostic test.
Crizotinib-resistant mutations of the ALK-kinase domain have recently been identified; L1196M and C1156Y mutations have been found to confer resistance to crizotinib in initially responsive patients.18
MET: An emerging molecular target
An emerging molecular target being tested in clinical trials is MET, a multifaceted receptor kinase that, when activated, induces tumor cell activities such as cell proliferation and angiogenesis, epithelial-mesenchymal transition, and cell scattering, leading to tumor cell invasion and metastasis.19
MET receptors and EGFR in lung cancer often are coexpressed and coactivated. Dual targeting of MET and EGFR pathways simultaneously is an attractive combined targeted strategy and is being studied in the hope of overcoming secondary resistance to EGFRTKI as well as enhance the primary response to targeted therapy. A number of MET targeting agents, including both small molecular inhibitors and monoclonal antibodies, are currently undergoing various stages of clinical development.20,21
In a phase 2 study, erlotinib plus the MET inhibitor ARQ197 was compared with erlotinib plus placebo in 117 previously treated EGFR-inhibitor–naïve patients with advanced NSCLC.22 In the population of patients with nonsquamous NSCLC, both PFS and OS were extended incrementally by the use of combined inhibition that targeted both the EGFR and MET pathways. Interestingly, the benefit on PFS appears to be more significant in EGFR wild type and KRAS-mutant molecular subgroups. A phase 3 global trial using a similar design, with a goal of enrolling 1,000 patients, has been activated in an attempt to validate the findings from the phase 2 study.
UNDERSTANDING RESISTANCE MECHANISMS WILL OPEN DOORS
Other than novel and more rational combined TKI in lung cancer, a deeper understanding of the resistance mechanisms in the context of oncogene addiction targeting would ultimately have a large impact on the long-term clinical success in lung cancer targeted therapy.
Resistance arises because of cellular heterogeneity within an oncogene-addicted tumor. Tumor shrinkage indicates a response to a molecularly targeted therapy, but residual tumor may be a source of slow-growing drug-tolerant “persistor” cells that promote tumor regrowth, regeneration, and heterogeneity.23,24 Coaddiction, reversible resistance, and addiction-switching models have been proposed to explain resistance, but it is unlikely that a single mechanism can fully explain tumor cell maintenance.
FUTURE OF TARGETED THERAPY IN LUNG CANCER
Technologic advances provide hope for the future of targeted therapy in lung cancer. Some of these advances are cancer genome deep sequencing and tumor molecular profiling. A greater understanding of tumor heterogeneity at the molecular level and tumor-resistant mechanisms, both intrinsic and acquired, should provide further therapeutic opportunity. In the modern era of targeted cancer therapy, identification of novel “druggable” driver oncogene targets can lead to swift development of inhibitors of those targets and adoption of improved and rational combinations of drugs. It is hoped that a better tumor response and more durable responses can be achieved with targeted therapy.
- Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction— a rationale for molecular targeting in cancer therapy. Nat Clin Prac Oncol 2006; 3:448–457.
- Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nature Rev Cancer 2010; 10:241–253.
- Janku F, Stewart DJ, Kurzrock R. Targeted therapy in non-small-cell lung cancer—is it becoming a reality [published correction appears in Nat Rev Clin Oncol 2011; 8:384]? Nat Rev Clin Oncol 2010; 7:401–414.
- Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 2002; 346:92–98.
- Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 2006; 355:2542–2550.
- Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer [published corrections appears in J Clin Oncol 2004; 22:4811[. J Clin Oncol 2003; 21:2237–2246.
- Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005; 366:1527–1537.
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non–small-cell lung cancer. N Engl J Med 2005; 353:123–132.
- Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455:1069–1075.
- Harris TJR, McCormick F. The molecular pathology of cancer. Nat Rev Clin Oncol 2010; 7:251–265.
- Mok TS, Wu Y-L, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361:947–957.
- Shepherd FA. Molecular selection trumps clinical selection. J Clin Oncol 2011; 29:2843–2844.
- Keedy VL, Temin S, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol 2011; 29:2121–2127.
- Von Pawel J, Park K, Pereira JR, et al. Phase III study comparing cisplatin/vinorelbine plus cetuximab versus cisplatin/vinorelbine as first-line treatment for patients with epidermal growth factor (EGFR)-expressing advanced non-small cell lung cancer (NSCLC) (FLEX) [ASCO abstract 7109]. J Clin Oncol 2006; 24( suppl).
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448:561–567.
- Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer 2010; 46:1773–1780.
- Kwak EL, Bang Y-J, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363:1693–1703.
- Choi YL, Soda M, Yamashits Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010; 363:1734–1739.
- Birchmeier C, Birchmeier W, Gherardi E, VandeWoude GF. MET, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4:915–925.
- Feng Y, Ma PC. Anti-MET targeted therapy has come of age: the first durable complete response with MetMAb in metastatic gastric cancer. Cancer Discovery 2011; 1:550–554.
- Feng Y, Thiagarajan PS, Ma PC. MET signaling: novel targeted inhibition and its clinical development in lung cancer. J Thorac Oncol 2012; 7:459–467.
- Sequist LV, Akerley WL, Brugger W, et al. Final results from ARQ 197-209: a global randomized placebo-controlled trial of erlotinib plus ARQ 197 versus erlotinib plus placebo in previously treated EGFR-inhibitor naïve patients with advanced non-small cell lung cancer (NSCLC) [ESMO abstract 3630]. Ann Oncol 2010; 21 (suppl 8):viii122.
- Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev 2007; 21:3214–3231.
- Fan W, Tang Z, Yin L, et al. MET-independent lung cancer cells evading EGRF kinase inhibitors are therapeutically susceptible to BH3 mimetic agents [published online ahead of print May 9, 2011]. Cancer Res 2011; 71:4494–4505. doi: 10.1158/0008-5472.CAN-10-2668
- Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003; 290:2149–2158.
The efficacy of therapy targeted to a specific oncogene is convincing evidence of “oncogene addiction,” or the concept that some cancers rely on or are “addicted to” a specific gene for their survival and proliferation. In the case of non–small cell lung cancer (NSCLC), drugs that target epidermal growth factor receptor (EGFR) have been proven more effective than conventional chemotherapy in patients with sensitizing EGFR mutations.1
Lung cancer oncogenes can drive oncogenic signaling pathways within tumor cells. Activation of EGFR signaling that drives cell proliferation through pathways such as RAS/RAF/MEK/ERK and the cell survival pathways P13K and AKT has been demonstrated in never-smokers. In heavy smokers, KRAS oncogene mutation is the dominant promoter of activation of oncogenic signaling pathways; it predicts a poor prognosis (especially for lung adenocarcinoma), and it is essentially mutually exclusive with EGFR mutations.
More than 50% of cases of NSCLC have known oncogene mutations for which targeted therapeutics are available.2 For example, gefitinib and erlotinib are the effective inhibitors for the EGFR oncogene mutation, sunitinib for platelet-derived growth factor receptor (PDGFR) amplification, and lapatinib for the less common ERBB2 insertion.
- EGFR mutations and amplifications
- EML4-ALK translocation fusions
- KRAS mutations
- PIK3CA mutations
- MET mutations, alternative splicing, amplification, and overexpression
PLATINUM DOUBLET AS STANDARD
Multiple platinum-based combinations of chemotherapy are in use as first-line therapy for advanced NSCLC. An overall survival (OS) benefit has been established with the use of doublet regimens, but no platinum-based doublet regimen has been proven superior to another on the end point of OS in clinical trials.4
Adding a third agent increases the response rate in advanced NSCLC but does not improve OS; the exception is bevacizumab, a monoclonal antibody targeted to vascular endothelial growth factor (VEGF). Sandler et al5 demonstrated a survival benefit when bevacizumab was added to paclitaxel-carboplatin in a recent study that led to US Food and Drug Administration (FDA) approval of bevacizumab for the treatment of NSCLC.
The next oncogene target explored in advanced NSCLC was EGFR. The tyrosine kinase inhibitors (TKIs) gefitinib, erlotinib, lapatinib, and the monoclonal antibody cetuximab are all clinical inhibitory agents targeting EGFR.
Previously treated NSCLC
In the phase 3 National Cancer Institute of Canada (NCIC) Clinical Trials Group (CTG) BR.21 trial, erlotinib demonstrated significant superiority over placebo as second- or third-line chemotherapy on PFS and OS in unselected patients with NSCLC. The results led to its approval as treatment for advanced NSCLC in patients who have received at least one prior chemotherapy regimen.8
Patient survival in NCIC CTG BR. 21 was evaluated in a series of patient subsets in exploratory univariate analyses. The effect of erlotinib on survival was similar across most subsets. A greater effect on survival by erlotinib was observed in patients who had never smoked (hazard ratio [HR] = 0.42). In the subgroup of patients who never smoked, EGFR status was predictive of erlotinib survival benefit. Patients who never smoked and were EGFR-positive had a survival benefit with erlotinib (HR = 0.27). There were too few EGFR-negative patients who never smoked to reach a conclusion.
Sensitizing mutations lead to better response
With the approval and clinical use of EGFR-TKIs came knowledge of EGFR kinase mutations that sensitize the mutated RTK to EGFR-TKI; this mechanism translates into dramatic tumor responses.9,10 Certain EGFR mutations contribute to sensitivity to EGFR-TKI treatment. The most common sensitizing mutations are the Exon 21 L858R mutation and the Exon 19 short in-frame deletions. The Exon 20 tends to yield resistant alterations (eg, prototypical T790M mutation and some Exon 20 Dup/Ins) in patients who initially derived benefit from targeted therapeutics. Although advances have been made in targeted therapeutics in lung cancer and other cancers, clinical resistance, particularly acquired or secondary resistance, remains the rule rather than the exception, which inherently limits the long-term clinical success of targeted therapeutics. A higher level of understanding of the mechanisms of resistance could have a substantial impact in maintaining the clinical efficacy of these targeted therapies.
The Iressa Pan-Asia Survival Study (IPASS) was a phase 3 study in which patient selection was based on clinical factors that predict a higher probability of harboring sensitizing EGFR mutations, thus enriching the mutant population, rather than screening for mutation status.11 Patients selected for participation were East Asians with advanced lung adenocarcinoma who were never smokers or former light smokers. They were randomized to treatment with platinum-based doublet chemotherapy (carboplatin-paclitaxel) or the EGFR-TKI gefitinib.
The primary end point—PFS—favored gefitinib over the chemotherapy doublet in the overall patient population. Of the 1,217 patients enrolled, EGFR mutation data for 35.9% could be evaluated. Sixty percent of the patients’ tumors harbored sensitizing EGFR mutations and, in this subset, the benefit of gefitinib was greater than it was in the overall population. In patients without an EGFR mutation, particularly those without Exon 19 deletions or Exon 21:L858R mutations (sensitizing mutations), platinum-based doublet chemotherapy performed better than gefitinib on the PFS end point. Of the mutation-positive patients, 4.2% had T790M mutations, which confers resistance to TKIs; this finding underscored the observation that all mutations cannot be treated the same.
The important message from the IPASS results is that even in a population preselected for EGFR mutation occurrence, the actual presence of the alteration of the target (EGFR mutations) leads to better survival with gefitinib. Molecular profiling of the tumor, therefore, is ultimately superior to profiling by patient phenotype or ethnicity. Thus, molecular selection trumps clinical selection.12
On the basis of IPASS and four similar phase 3 randomized controlled trials, the American Society of Clinical Oncology issued a clinical opinion that “patients with NSCLC who are being considered for first-line therapy with an EGFR-TKI (patients who have not previously received chemotherapy or an EGFR-TKI) should have their tumor tested for EGFR mutations to determine whether an EGFRTKI or chemotherapy is the appropriate first-line therapy.”13
Monoclonal antibody against EGFR
The First-Line Erbitux in Lung Cancer (FLEX) phase 3 worldwide study demonstrated that cetuximab, a monoclonal antibody directed against EGFR, as add-on therapy to a platinum-based doublet (cisplatin and vinorelbine) can extend median OS in patients with advanced EGFR-expressing NSCLC (stage wet IIIB or stage IV).14 As a result, the use of cetuximab combined with cisplatin-vinorelbine has been endorsed by a National Comprehensive Cancer Network (NCCN) guideline as a first-line option for the treatment of advanced NSCLC.
EML4-ALK fusion gene as target for crizotinib
The EML4-ALK fusion translocation oncogene was first identified in 2007 in a small proportion of patients with NSCLC,15,16 and a targeted therapy (crizotinib, an inhibitor of the ALK tyrosine kinase) has been developed. A single-arm phase 1 trial of crizotinib in patients selected for the EML4-ALK fusion gene has been completed.17 Of the approximately 1,500 NSCLC patients screened for the trial, 82 were identified as having advanced ALK-positive disease and were entered. These patients tended to be younger than those with ALK-negative disease, most had little or no exposure to tobacco, and all had adenocarcinoma.
The mean treatment duration was 6.4 months. Most treatment-related side effects were grade 1 or grade 2 gastrointestinal adverse events. The overall response rate was 57%. The disease control rates were 87% at 8 weeks and 66% at 16 weeks. Crizotinib has since moved to phase 2 and phase 3 trials and received FDA approval on August 26, 2011, for treatment of EML4-ALK–positive patients as assayed by a simultaneously approved companion molecular diagnostic test.
Crizotinib-resistant mutations of the ALK-kinase domain have recently been identified; L1196M and C1156Y mutations have been found to confer resistance to crizotinib in initially responsive patients.18
MET: An emerging molecular target
An emerging molecular target being tested in clinical trials is MET, a multifaceted receptor kinase that, when activated, induces tumor cell activities such as cell proliferation and angiogenesis, epithelial-mesenchymal transition, and cell scattering, leading to tumor cell invasion and metastasis.19
MET receptors and EGFR in lung cancer often are coexpressed and coactivated. Dual targeting of MET and EGFR pathways simultaneously is an attractive combined targeted strategy and is being studied in the hope of overcoming secondary resistance to EGFRTKI as well as enhance the primary response to targeted therapy. A number of MET targeting agents, including both small molecular inhibitors and monoclonal antibodies, are currently undergoing various stages of clinical development.20,21
In a phase 2 study, erlotinib plus the MET inhibitor ARQ197 was compared with erlotinib plus placebo in 117 previously treated EGFR-inhibitor–naïve patients with advanced NSCLC.22 In the population of patients with nonsquamous NSCLC, both PFS and OS were extended incrementally by the use of combined inhibition that targeted both the EGFR and MET pathways. Interestingly, the benefit on PFS appears to be more significant in EGFR wild type and KRAS-mutant molecular subgroups. A phase 3 global trial using a similar design, with a goal of enrolling 1,000 patients, has been activated in an attempt to validate the findings from the phase 2 study.
UNDERSTANDING RESISTANCE MECHANISMS WILL OPEN DOORS
Other than novel and more rational combined TKI in lung cancer, a deeper understanding of the resistance mechanisms in the context of oncogene addiction targeting would ultimately have a large impact on the long-term clinical success in lung cancer targeted therapy.
Resistance arises because of cellular heterogeneity within an oncogene-addicted tumor. Tumor shrinkage indicates a response to a molecularly targeted therapy, but residual tumor may be a source of slow-growing drug-tolerant “persistor” cells that promote tumor regrowth, regeneration, and heterogeneity.23,24 Coaddiction, reversible resistance, and addiction-switching models have been proposed to explain resistance, but it is unlikely that a single mechanism can fully explain tumor cell maintenance.
FUTURE OF TARGETED THERAPY IN LUNG CANCER
Technologic advances provide hope for the future of targeted therapy in lung cancer. Some of these advances are cancer genome deep sequencing and tumor molecular profiling. A greater understanding of tumor heterogeneity at the molecular level and tumor-resistant mechanisms, both intrinsic and acquired, should provide further therapeutic opportunity. In the modern era of targeted cancer therapy, identification of novel “druggable” driver oncogene targets can lead to swift development of inhibitors of those targets and adoption of improved and rational combinations of drugs. It is hoped that a better tumor response and more durable responses can be achieved with targeted therapy.
The efficacy of therapy targeted to a specific oncogene is convincing evidence of “oncogene addiction,” or the concept that some cancers rely on or are “addicted to” a specific gene for their survival and proliferation. In the case of non–small cell lung cancer (NSCLC), drugs that target epidermal growth factor receptor (EGFR) have been proven more effective than conventional chemotherapy in patients with sensitizing EGFR mutations.1
Lung cancer oncogenes can drive oncogenic signaling pathways within tumor cells. Activation of EGFR signaling that drives cell proliferation through pathways such as RAS/RAF/MEK/ERK and the cell survival pathways P13K and AKT has been demonstrated in never-smokers. In heavy smokers, KRAS oncogene mutation is the dominant promoter of activation of oncogenic signaling pathways; it predicts a poor prognosis (especially for lung adenocarcinoma), and it is essentially mutually exclusive with EGFR mutations.
More than 50% of cases of NSCLC have known oncogene mutations for which targeted therapeutics are available.2 For example, gefitinib and erlotinib are the effective inhibitors for the EGFR oncogene mutation, sunitinib for platelet-derived growth factor receptor (PDGFR) amplification, and lapatinib for the less common ERBB2 insertion.
- EGFR mutations and amplifications
- EML4-ALK translocation fusions
- KRAS mutations
- PIK3CA mutations
- MET mutations, alternative splicing, amplification, and overexpression
PLATINUM DOUBLET AS STANDARD
Multiple platinum-based combinations of chemotherapy are in use as first-line therapy for advanced NSCLC. An overall survival (OS) benefit has been established with the use of doublet regimens, but no platinum-based doublet regimen has been proven superior to another on the end point of OS in clinical trials.4
Adding a third agent increases the response rate in advanced NSCLC but does not improve OS; the exception is bevacizumab, a monoclonal antibody targeted to vascular endothelial growth factor (VEGF). Sandler et al5 demonstrated a survival benefit when bevacizumab was added to paclitaxel-carboplatin in a recent study that led to US Food and Drug Administration (FDA) approval of bevacizumab for the treatment of NSCLC.
The next oncogene target explored in advanced NSCLC was EGFR. The tyrosine kinase inhibitors (TKIs) gefitinib, erlotinib, lapatinib, and the monoclonal antibody cetuximab are all clinical inhibitory agents targeting EGFR.
Previously treated NSCLC
In the phase 3 National Cancer Institute of Canada (NCIC) Clinical Trials Group (CTG) BR.21 trial, erlotinib demonstrated significant superiority over placebo as second- or third-line chemotherapy on PFS and OS in unselected patients with NSCLC. The results led to its approval as treatment for advanced NSCLC in patients who have received at least one prior chemotherapy regimen.8
Patient survival in NCIC CTG BR. 21 was evaluated in a series of patient subsets in exploratory univariate analyses. The effect of erlotinib on survival was similar across most subsets. A greater effect on survival by erlotinib was observed in patients who had never smoked (hazard ratio [HR] = 0.42). In the subgroup of patients who never smoked, EGFR status was predictive of erlotinib survival benefit. Patients who never smoked and were EGFR-positive had a survival benefit with erlotinib (HR = 0.27). There were too few EGFR-negative patients who never smoked to reach a conclusion.
Sensitizing mutations lead to better response
With the approval and clinical use of EGFR-TKIs came knowledge of EGFR kinase mutations that sensitize the mutated RTK to EGFR-TKI; this mechanism translates into dramatic tumor responses.9,10 Certain EGFR mutations contribute to sensitivity to EGFR-TKI treatment. The most common sensitizing mutations are the Exon 21 L858R mutation and the Exon 19 short in-frame deletions. The Exon 20 tends to yield resistant alterations (eg, prototypical T790M mutation and some Exon 20 Dup/Ins) in patients who initially derived benefit from targeted therapeutics. Although advances have been made in targeted therapeutics in lung cancer and other cancers, clinical resistance, particularly acquired or secondary resistance, remains the rule rather than the exception, which inherently limits the long-term clinical success of targeted therapeutics. A higher level of understanding of the mechanisms of resistance could have a substantial impact in maintaining the clinical efficacy of these targeted therapies.
The Iressa Pan-Asia Survival Study (IPASS) was a phase 3 study in which patient selection was based on clinical factors that predict a higher probability of harboring sensitizing EGFR mutations, thus enriching the mutant population, rather than screening for mutation status.11 Patients selected for participation were East Asians with advanced lung adenocarcinoma who were never smokers or former light smokers. They were randomized to treatment with platinum-based doublet chemotherapy (carboplatin-paclitaxel) or the EGFR-TKI gefitinib.
The primary end point—PFS—favored gefitinib over the chemotherapy doublet in the overall patient population. Of the 1,217 patients enrolled, EGFR mutation data for 35.9% could be evaluated. Sixty percent of the patients’ tumors harbored sensitizing EGFR mutations and, in this subset, the benefit of gefitinib was greater than it was in the overall population. In patients without an EGFR mutation, particularly those without Exon 19 deletions or Exon 21:L858R mutations (sensitizing mutations), platinum-based doublet chemotherapy performed better than gefitinib on the PFS end point. Of the mutation-positive patients, 4.2% had T790M mutations, which confers resistance to TKIs; this finding underscored the observation that all mutations cannot be treated the same.
The important message from the IPASS results is that even in a population preselected for EGFR mutation occurrence, the actual presence of the alteration of the target (EGFR mutations) leads to better survival with gefitinib. Molecular profiling of the tumor, therefore, is ultimately superior to profiling by patient phenotype or ethnicity. Thus, molecular selection trumps clinical selection.12
On the basis of IPASS and four similar phase 3 randomized controlled trials, the American Society of Clinical Oncology issued a clinical opinion that “patients with NSCLC who are being considered for first-line therapy with an EGFR-TKI (patients who have not previously received chemotherapy or an EGFR-TKI) should have their tumor tested for EGFR mutations to determine whether an EGFRTKI or chemotherapy is the appropriate first-line therapy.”13
Monoclonal antibody against EGFR
The First-Line Erbitux in Lung Cancer (FLEX) phase 3 worldwide study demonstrated that cetuximab, a monoclonal antibody directed against EGFR, as add-on therapy to a platinum-based doublet (cisplatin and vinorelbine) can extend median OS in patients with advanced EGFR-expressing NSCLC (stage wet IIIB or stage IV).14 As a result, the use of cetuximab combined with cisplatin-vinorelbine has been endorsed by a National Comprehensive Cancer Network (NCCN) guideline as a first-line option for the treatment of advanced NSCLC.
EML4-ALK fusion gene as target for crizotinib
The EML4-ALK fusion translocation oncogene was first identified in 2007 in a small proportion of patients with NSCLC,15,16 and a targeted therapy (crizotinib, an inhibitor of the ALK tyrosine kinase) has been developed. A single-arm phase 1 trial of crizotinib in patients selected for the EML4-ALK fusion gene has been completed.17 Of the approximately 1,500 NSCLC patients screened for the trial, 82 were identified as having advanced ALK-positive disease and were entered. These patients tended to be younger than those with ALK-negative disease, most had little or no exposure to tobacco, and all had adenocarcinoma.
The mean treatment duration was 6.4 months. Most treatment-related side effects were grade 1 or grade 2 gastrointestinal adverse events. The overall response rate was 57%. The disease control rates were 87% at 8 weeks and 66% at 16 weeks. Crizotinib has since moved to phase 2 and phase 3 trials and received FDA approval on August 26, 2011, for treatment of EML4-ALK–positive patients as assayed by a simultaneously approved companion molecular diagnostic test.
Crizotinib-resistant mutations of the ALK-kinase domain have recently been identified; L1196M and C1156Y mutations have been found to confer resistance to crizotinib in initially responsive patients.18
MET: An emerging molecular target
An emerging molecular target being tested in clinical trials is MET, a multifaceted receptor kinase that, when activated, induces tumor cell activities such as cell proliferation and angiogenesis, epithelial-mesenchymal transition, and cell scattering, leading to tumor cell invasion and metastasis.19
MET receptors and EGFR in lung cancer often are coexpressed and coactivated. Dual targeting of MET and EGFR pathways simultaneously is an attractive combined targeted strategy and is being studied in the hope of overcoming secondary resistance to EGFRTKI as well as enhance the primary response to targeted therapy. A number of MET targeting agents, including both small molecular inhibitors and monoclonal antibodies, are currently undergoing various stages of clinical development.20,21
In a phase 2 study, erlotinib plus the MET inhibitor ARQ197 was compared with erlotinib plus placebo in 117 previously treated EGFR-inhibitor–naïve patients with advanced NSCLC.22 In the population of patients with nonsquamous NSCLC, both PFS and OS were extended incrementally by the use of combined inhibition that targeted both the EGFR and MET pathways. Interestingly, the benefit on PFS appears to be more significant in EGFR wild type and KRAS-mutant molecular subgroups. A phase 3 global trial using a similar design, with a goal of enrolling 1,000 patients, has been activated in an attempt to validate the findings from the phase 2 study.
UNDERSTANDING RESISTANCE MECHANISMS WILL OPEN DOORS
Other than novel and more rational combined TKI in lung cancer, a deeper understanding of the resistance mechanisms in the context of oncogene addiction targeting would ultimately have a large impact on the long-term clinical success in lung cancer targeted therapy.
Resistance arises because of cellular heterogeneity within an oncogene-addicted tumor. Tumor shrinkage indicates a response to a molecularly targeted therapy, but residual tumor may be a source of slow-growing drug-tolerant “persistor” cells that promote tumor regrowth, regeneration, and heterogeneity.23,24 Coaddiction, reversible resistance, and addiction-switching models have been proposed to explain resistance, but it is unlikely that a single mechanism can fully explain tumor cell maintenance.
FUTURE OF TARGETED THERAPY IN LUNG CANCER
Technologic advances provide hope for the future of targeted therapy in lung cancer. Some of these advances are cancer genome deep sequencing and tumor molecular profiling. A greater understanding of tumor heterogeneity at the molecular level and tumor-resistant mechanisms, both intrinsic and acquired, should provide further therapeutic opportunity. In the modern era of targeted cancer therapy, identification of novel “druggable” driver oncogene targets can lead to swift development of inhibitors of those targets and adoption of improved and rational combinations of drugs. It is hoped that a better tumor response and more durable responses can be achieved with targeted therapy.
- Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction— a rationale for molecular targeting in cancer therapy. Nat Clin Prac Oncol 2006; 3:448–457.
- Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nature Rev Cancer 2010; 10:241–253.
- Janku F, Stewart DJ, Kurzrock R. Targeted therapy in non-small-cell lung cancer—is it becoming a reality [published correction appears in Nat Rev Clin Oncol 2011; 8:384]? Nat Rev Clin Oncol 2010; 7:401–414.
- Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 2002; 346:92–98.
- Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 2006; 355:2542–2550.
- Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer [published corrections appears in J Clin Oncol 2004; 22:4811[. J Clin Oncol 2003; 21:2237–2246.
- Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005; 366:1527–1537.
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non–small-cell lung cancer. N Engl J Med 2005; 353:123–132.
- Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455:1069–1075.
- Harris TJR, McCormick F. The molecular pathology of cancer. Nat Rev Clin Oncol 2010; 7:251–265.
- Mok TS, Wu Y-L, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361:947–957.
- Shepherd FA. Molecular selection trumps clinical selection. J Clin Oncol 2011; 29:2843–2844.
- Keedy VL, Temin S, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol 2011; 29:2121–2127.
- Von Pawel J, Park K, Pereira JR, et al. Phase III study comparing cisplatin/vinorelbine plus cetuximab versus cisplatin/vinorelbine as first-line treatment for patients with epidermal growth factor (EGFR)-expressing advanced non-small cell lung cancer (NSCLC) (FLEX) [ASCO abstract 7109]. J Clin Oncol 2006; 24( suppl).
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448:561–567.
- Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer 2010; 46:1773–1780.
- Kwak EL, Bang Y-J, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363:1693–1703.
- Choi YL, Soda M, Yamashits Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010; 363:1734–1739.
- Birchmeier C, Birchmeier W, Gherardi E, VandeWoude GF. MET, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4:915–925.
- Feng Y, Ma PC. Anti-MET targeted therapy has come of age: the first durable complete response with MetMAb in metastatic gastric cancer. Cancer Discovery 2011; 1:550–554.
- Feng Y, Thiagarajan PS, Ma PC. MET signaling: novel targeted inhibition and its clinical development in lung cancer. J Thorac Oncol 2012; 7:459–467.
- Sequist LV, Akerley WL, Brugger W, et al. Final results from ARQ 197-209: a global randomized placebo-controlled trial of erlotinib plus ARQ 197 versus erlotinib plus placebo in previously treated EGFR-inhibitor naïve patients with advanced non-small cell lung cancer (NSCLC) [ESMO abstract 3630]. Ann Oncol 2010; 21 (suppl 8):viii122.
- Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev 2007; 21:3214–3231.
- Fan W, Tang Z, Yin L, et al. MET-independent lung cancer cells evading EGRF kinase inhibitors are therapeutically susceptible to BH3 mimetic agents [published online ahead of print May 9, 2011]. Cancer Res 2011; 71:4494–4505. doi: 10.1158/0008-5472.CAN-10-2668
- Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003; 290:2149–2158.
- Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction— a rationale for molecular targeting in cancer therapy. Nat Clin Prac Oncol 2006; 3:448–457.
- Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nature Rev Cancer 2010; 10:241–253.
- Janku F, Stewart DJ, Kurzrock R. Targeted therapy in non-small-cell lung cancer—is it becoming a reality [published correction appears in Nat Rev Clin Oncol 2011; 8:384]? Nat Rev Clin Oncol 2010; 7:401–414.
- Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 2002; 346:92–98.
- Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 2006; 355:2542–2550.
- Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer [published corrections appears in J Clin Oncol 2004; 22:4811[. J Clin Oncol 2003; 21:2237–2246.
- Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005; 366:1527–1537.
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non–small-cell lung cancer. N Engl J Med 2005; 353:123–132.
- Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455:1069–1075.
- Harris TJR, McCormick F. The molecular pathology of cancer. Nat Rev Clin Oncol 2010; 7:251–265.
- Mok TS, Wu Y-L, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361:947–957.
- Shepherd FA. Molecular selection trumps clinical selection. J Clin Oncol 2011; 29:2843–2844.
- Keedy VL, Temin S, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol 2011; 29:2121–2127.
- Von Pawel J, Park K, Pereira JR, et al. Phase III study comparing cisplatin/vinorelbine plus cetuximab versus cisplatin/vinorelbine as first-line treatment for patients with epidermal growth factor (EGFR)-expressing advanced non-small cell lung cancer (NSCLC) (FLEX) [ASCO abstract 7109]. J Clin Oncol 2006; 24( suppl).
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448:561–567.
- Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer 2010; 46:1773–1780.
- Kwak EL, Bang Y-J, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363:1693–1703.
- Choi YL, Soda M, Yamashits Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010; 363:1734–1739.
- Birchmeier C, Birchmeier W, Gherardi E, VandeWoude GF. MET, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4:915–925.
- Feng Y, Ma PC. Anti-MET targeted therapy has come of age: the first durable complete response with MetMAb in metastatic gastric cancer. Cancer Discovery 2011; 1:550–554.
- Feng Y, Thiagarajan PS, Ma PC. MET signaling: novel targeted inhibition and its clinical development in lung cancer. J Thorac Oncol 2012; 7:459–467.
- Sequist LV, Akerley WL, Brugger W, et al. Final results from ARQ 197-209: a global randomized placebo-controlled trial of erlotinib plus ARQ 197 versus erlotinib plus placebo in previously treated EGFR-inhibitor naïve patients with advanced non-small cell lung cancer (NSCLC) [ESMO abstract 3630]. Ann Oncol 2010; 21 (suppl 8):viii122.
- Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev 2007; 21:3214–3231.
- Fan W, Tang Z, Yin L, et al. MET-independent lung cancer cells evading EGRF kinase inhibitors are therapeutically susceptible to BH3 mimetic agents [published online ahead of print May 9, 2011]. Cancer Res 2011; 71:4494–4505. doi: 10.1158/0008-5472.CAN-10-2668
- Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003; 290:2149–2158.