User login
Depression can be treated successfully by primary care physicians under “real-world” conditions.
Furthermore, the particular drug or drugs used are not as important as following a rational plan: giving antidepressant medications in adequate doses, monitoring the patient’s symptoms and side effects and adjusting the regimen accordingly, and switching drugs or adding new drugs to the regimen only after an adequate trial.
These are among the lessons learned from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, the largest prospective clinical trial of treatment of major depressive disorder ever conducted. It was funded by the National Institutes of Health and directed by A. John Rush, MD.
WHAT WERE THE AIMS OF STAR*D?
Depression, a common and debilitating condition, affects approximately one in eight people in the United States.1 It is expected2 to be the second-leading cause of disability in the world by the year 2020; today, it is the second-leading cause of disability-adjusted life years in those 15 to 44 years old.3
Nevertheless, the available evidence base for treatment is limited, since most participants in clinical trials are recruited by advertisement rather than from representative practices, and they are often selected to have few comorbid disorders, either medical or psychiatric. In addition, those with chronic depression or current suicidal ideation are excluded.1,4 These uncomplicated and “pristine” participants are unlike typical patients seen by primary care physicians or psychiatrists.
Similarly, the protocols used in these trials do not represent usual clinic practice.Patients in clinical trials undergo more assessment and more frequent follow-up than in real-world practice, they have no say in treatment decisions, the doses are fixed, and the patients and physicians are blinded to the intervention. Consequently, how to translate the results of these efficacy trials into practice is unclear.5
Further, even in relatively uncomplicated cases, only about one-half of outpatients with nonpsychotic major depressive disorder initially treated with a single medication or with psychotherapy will experience a clinically significant improvement in symptoms (ie, a response) during the 8 to 12 weeks of acute-phase treatment,6–10 and only 20% to 35% of patients will reach remission,9 the aim of treatment.8,11 The remission rates are even lower in treatment-resistant depression.12 How to manage most patients—those whose depression does not remit with the first, second, or third step of treatment—is unclear.
Accordingly, the overall objective of STAR*D was to develop and evaluate feasible treatment strategies to improve clinical outcomes for real-world patients with treatment-resistant depression, who were identified prospectively from a pool of patients in a current major depressive episode.13–15 Specifically, STAR*D aimed to determine prospectively which of several treatments is the most effective “next step” for patients who do not reach remission with an initial or subsequent treatment or who cannot tolerate the treatment.
WHY IS STAR*D RELEVANT FOR PRIMARY CARE?
Nearly 10% of all primary care office visits are depression-related.16 Primary care physicians provide nearly half the outpatient care for depressed patients.17 Indeed, primary care physicians log approximately as many outpatient visits for depression as psychiatrists do.18 Medical comorbidity is especially common in primary care settings.19 When to refer to a psychiatrist is not clear.
KEY FEATURES OF THE STUDY DESIGN
STAR*D involved a national consortium of 14 university-based regional centers, which oversaw a total of 23 participating psychiatric and 18 primary care clinics. Enrollment began in 2000, with follow-up completed in 2004.
Entry criteria were broad and inclusive
Patients had to:
- Be between 18 and 75 years of age
- Have a nonpsychotic major depressive disorder, identified by a clinician and confirmed with a symptom checklist based on the Diagnostic and Statistical Manual, fourth edition revised,20 and for which antidepressant treatment is recommended
- Score at least 14 on the 17-item Hamilton Rating Scale for Depression (HAMD17)21
- Not have a primary diagnosis of bipolar disorder, obsessive-compulsive disorder, or an eating disorder, which would require a different treatment strategy, or a seizure disorder (which would preclude bupropion as a second-step treatment).
Dosing recommendations were flexible but vigorous
Medications often were increased to maximally tolerated doses. For example, citalopram (Celexa) was started at 20 mg/day and increased by 20 mg every 2 to 4 weeks if the patient was tolerating it but had not achieved remission, to a maximum dose of 60 mg/day. Treatment could be given for up to 14 weeks, during which side effects22 and clinical ratings23 were assessed by both patients and study coordinators.
Measurement-based care
We used a systematic approach to treatment called “measurement-based care,”24 which involves routinely measuring symptoms23 and side effects22 and using this information to modify the medication doses at critical decision points. This algorithmic approach provided flexible treatment recommendations to ensure that the dosage and duration of antidepressant drug treatment were adequate.25
The severity of depression was assessed by the clinician-rated, 16-item Quick Inventory of Depressive Symptomatology (QIDS-C16). The QIDS-SR16 (the self-report version) can substitute for the QIDS-C1623 to make this approach more feasible. Both tools are available at www.ids-qids.org.
This approach was easily worked into busy primary care and specialty care office workflows (clinic physicians, most with limited research experience, provided the treatment), and could be translated into primary care practice in the community as well.
Four-step protocol
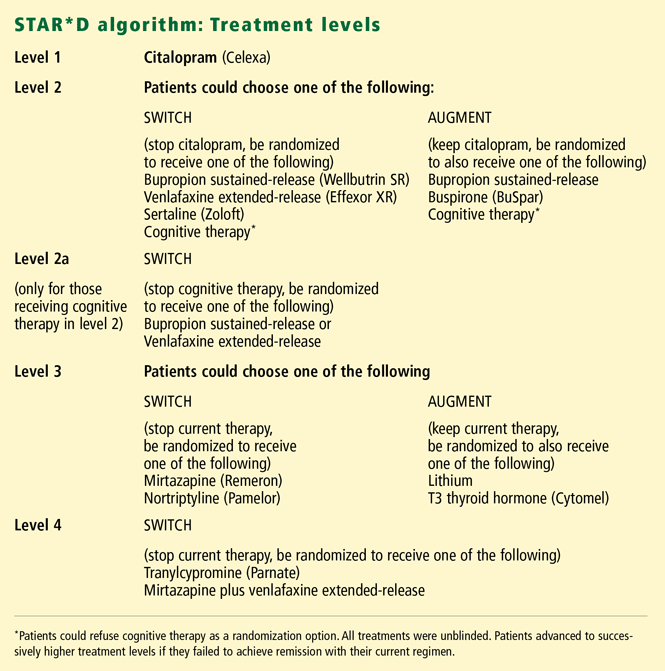
A unique feature of the study design was that the patients, in consultation with their physicians, had some choice in the treatments they received. In this “equipoise-stratified randomized design,”26 at levels 2 and 3 the patient could choose either to switch therapies (stop the current drug and be randomized to receive one of several different treatments) or to augment their current therapy (by adding one of several treatments in a randomized fashion). Patients could decline certain strategies as long as there were at least two possible options to which one might be randomized.
At level 2, one of the options for both switching and augmentation was cognitive therapy, although patients could decline that option. Conversely, if they definitely wanted cognitive therapy, they could choose to be randomized to either cognitive therapy alone or to cognitive therapy added to citalopram. Also, anyone who received cognitive therapy in level 2 and failed to enter remission was additionally randomized to either bupropion or venlafaxine (level 2a) to ensure that all patients had failed trials on two medications before entering level 3.
When switching to medications other than a monoamine oxidase inhibitor (MAOI), the clinician could choose either to stop the current medication and immediately begin the next one, or to decrease the current medication while starting the new one at a low dose and then tapering and titrating over 1 week. (Switching to an MAOI, used only in the final level of treatment, required a 7- to 10-day washout period.)
Outcomes measured
Remission (complete recovery from the depressive episode), the primary study outcome, was defined as a HAM-D17 score of 7 or less, as assessed by treatment-blinded raters.A secondary remission outcome was a QIDS-SR16 score of 5 or less. Of note, the HAM-D17 remission rates were slightly lower than the rates based on the QIDS-SR16, since patients who did not have a HAM-D17 score measured at exit were defined as not being in remission a priori. Thus, the QIDS-SR16 rates might have been a slightly better reflection of actual remission rates.
Response, a secondary outcome, was defined as a reduction of at least 50% in the QIDS-SR16 score from baseline at the last assessment.
FEW DIFFERENCES BETWEEN PSYCHIATRIC, PRIMARY CARE PATIENTS
The patients seen in primary care clinics were surprisingly similar to those seen in psychiatric clinics.27,28 The two groups did not differ in severity of depression, distribution of severity scores, the likelihood of presenting with any of the nine core criteria of a major depressive episode, or the likelihood of having a concomitant axis I psychiatric disorder in addition to depression (about half of participants in each setting had an anxiety disorder).
Recurrent major depressive disorders were common in both groups, though more so in psychiatric patients (78% vs 69%, P < .001), while chronic depression was more common in primary care than in psychiatric patients (30% vs 21%, P < .001). Having either a chronic index episode (ie, lasting > 2 years) or a recurrent major depressive disorder was common in both groups (86% vs 83%, P = .0067).
That said, primary care patients were older (44 years vs 39 years, P < .001), more of them were Hispanic (18% vs 9%, P < .001), and more of them had public insurance (23% vs 9%, P < .001). Fewer of the primary care patients had completed college (20% vs 28%, P < .001), and the primary care patients tended to have greater medical comorbidity. Psychiatric patients were more likely to have attempted suicide in the past and to have had their first depressive illness before age 18.
LEVEL 1: WHAT CAN WE EXPECT FROM INITIAL TREATMENT?
At level 1, all the patients received citalopram. The mean dose was 40.6 ± 16.6 mg/day in the primary care clinics and 42.5 ± 16.8 mg/day in the psychiatric clinics, which are adequate, middle-range doses and higher than the average US dose.29
Approximately 30% of patients achieved remission: 27% as measured on the HAM-D17 and 33% on the QIDS-SR16. The response rate (on the QIDS-SR16) was 47%. There were no differences between primary and psychiatric care settings in remission or response rates.
Patients were more likely to achieve remission if they were white, female, employed, more educated, or wealthier. Longer current episodes, more concurrent psychiatric disorders (especially anxiety disorders or drug abuse), more general medical disorders, and lower baseline function and quality of life were each associated with lower remission rates.
What is an adequate trial?
Longer times than expected were needed to reach response or remission. The average duration required to achieve remission was almost 7 weeks (44 days in primary care; 49 days in psychiatric care). Further, approximately one-third of those who ultimately responded and half of those who entered remission did so after 6 weeks.30 Forty percent of those who entered remission required 8 or more weeks to do so.
These results suggest that longer treatment durations and more vigorous medication dosing than generally used are needed to achieve optimal remission rates. It is imprudent to stop a treatment that the patient is tolerating in a robust dose if the patient reports only partial benefit by 6 weeks; indeed, raising the dose, if tolerated, may help a substantial number of patients respond by 12 or 14 weeks. Instruments to monitor depression severity (eg, self-report measures) can be useful. At least 8 weeks with at least moderately vigorous dosing is recommended.
LEVEL 2: IF THE FIRST TREATMENT FAILS
When switching to a new drug, does it matter which one?
No.
In level 2, if patients had not achieved remission on citalopram alone, they had the choice of switching: stopping citalopram and being randomized to receive either sertraline (Zoloft, another SSRI), venlafaxine extended-release (XR) (Effexor XR, a serotonin and norepinephrine reuptake inhibitor), or bupropion sustained-release (SR) (Wellbutrin SR, a norepinephrine and dopamine reuptake inhibitor). At the last visit the mean daily doses were bupropion SR 282.7 mg/day, sertraline 135.5 mg/day, and venlafaxine-XR 193.6 mg/day.
The remission rate was approximately one-fourth with all three drugs31:
- With bupropion SR—21.3% by HAM-D17, 25.5% by QIDS-SR16
- With sertraline—17.6% by HAM-D17, 26.6% by QIDS-SR16
- With venlafaxine-XR—24.8% by HAM-D17, 25.0% by QIDS-SR16. The remission rates were neither statistically nor clinically different by either measure.
Though the types of side effects related to specific medications may have varied, the overall side-effect burden and the rate of serious adverse events did not differ significantly.
When adding a new drug, does it matter which one?
Again, no.
Instead of switching, patients in level 2 could choose to stay on citalopram and be randomized to add either bupropion SR or buspirone (BuSpar) to the regimen (augmentation). The mean daily doses at the end of level 2 were bupropion SR 267.5 mg and buspirone 40.9 mg.
Rates of remission32:
- With bupropion SR—29.7% on the HAMD-D17, 39.0% on the QIDS-SR16
- With buspirone—30.1% on the HAM-D17, 32.9% on the QIDS-SR16.
However, the QIDS-SR16 scores declined significantly more with bupropion SR than with buspirone (25.3% vs 17.1%, P < .04). The mean total QIDS-SR16 score at the last visit was lower with bupropion SR (8.0) than with buspirone (9.1, P < .02), and augmentation with bupropion SR was better tolerated (the dropout rate due to intolerance was 12.5% with bupropion-SR vs 20.6% with buspirone 20.6%; P < .009).
Can we directly compare the benefits of switching vs augmenting?
No.
Patients could choose whether to switch from citalopram to another drug or to add another drug at the second treatment level.33 Consequently, we could not ensure that the patient groups were equivalent at the point of randomization at the beginning of level 2, and, indeed, they were not.
Those who benefitted more from citalopram treatment and who better tolerated it preferred augmentation, while those who benefitted little or who could not tolerate it preferred to switch. Consequently, those in the augmentation group at level 2 were somewhat less depressed than those who switched. Whether augmentation is better even if the initial treatment is minimally effective could not be evaluated in STAR*D.
What about cognitive therapy?
There was no difference between cognitive therapy (either as a switch or as augmentation) and medication (as a switch or as augmentation).34 Adding another drug was more rapidly effective than adding cognitive therapy. Switching to cognitive therapy was better tolerated than switching to a different antidepressant.
Of note, fewer patients accepted cognitive therapy as a randomization option than we expected, so the sample sizes were small. Possible reasons were that all patients had to receive a medication at study entry (which may have biased selection towards those preferring medication), and cognitive therapy entailed additional copayments and visiting still another provider at another site.
After two levels of treatment, how many patients reach remission?
About 30% of patients in level 1 achieved remission, and of those progressing to level 2, another 30% achieved remission. Together, this adds up to about 50% of patients achieving remission if they remained in treatment (30% in level 1 plus 30% of the roughly 70% remaining in level 2).
IF A SECOND TREATMENT FAILS
If switching again to another drug, does it matter which one?
No.
In level 3, patients could choose to stop the drug they had been taking and be randomized to receive either mirtazapine (Remeron) or nortriptyline (Pamelor).
Switching medications was not as effective as a third step as it was as a second step.35
Remission rates:
- With mirtazapine—12.3% on the HAM-D17, 8.0% on the QIDS-SR16
- With nortriptyline—19.8% on the HAM-D17, 12.4% on the QIDS-SR16.
Response rates were 13.4% with mirtazapine and 16.5% with nortriptyline. Statistically, neither the response nor the remission rates differed by treatment, nor did these two treatments differ in tolerability or side-effect burden.
Does choice of augmentation agent matter: Lithium vs T3?
Similarly, after two failed medication treatments, medication augmentation was less effective than it was at the second step.36 The two augmentation options tested, lithium and T3 thyroid hormone (Cytomel), are commonly considered by psychiatrists but less commonly used by primary care doctors.
Lithium is believed to increase serotonergic function, which may have a synergistic effect on the mechanism of action of antidepressants; a meta-analysis of placebo-controlled studies supports lithium’s effectiveness as adjunctive treatment.37 Its side effects, however, must be closely monitored.38 The primary monitoring concern is the small difference between the therapeutic blood level (0.6–1.2 mEq/L) and potentially toxic blood levels (> 1.5 mEq/L).
Lithium was started at 450 mg/day, and at week 2 it was increased to the recommended dose of 900 mg/day (a dose below the target dose for bipolar disorder). If patients could not tolerate 450 mg/day, the initial dose was 225 mg/day for 1 week before being increased to 450 mg/day, still with the target dose of 900 mg/day. The mean exit dose was 859.9 mg/day, and the median blood level was 0.6 mEq/L.
Thyroid hormone augmentation using T3 is believed to work through both direct and indirect effects on the hypothalamic-pituitary-thyroid axis, which has a strong relationship with depression. The efficacy of T3 augmentation is supported by a meta-analysis of eight studies,39 and T3 is effective whether or not thyroid abnormalities are present.
In STAR*D, T3 was started at 25 μg/day for 1 week, than increased to the recommended dose of 50 μg/day. The mean exit dose was 45.2 μg/day.
Remission rates:
- With lithium augmentation—15.9% by the HAM-D17, 13.2% by the QIDS-SR16
- With T3 augmentation—24.7% by both measures.
Response rates were 16.2% with lithium augmentation and 23.3% with T3 augmentation.
While neither response nor remission rates were statistically significantly different by treatment, lithium was more frequently associated with side effects (P = .045), and more participants in the lithium group left treatment because of side effects (23.2% vs 9.6%; P = .027). These results suggest that in cases in which a clinician is considering an augmentation trial, T3 has slight advantages over lithium in effectiveness and tolerability. T3 also offers the advantages of being easy to use and not necessitating blood level monitoring. These latter benefits are especially relevant to the primary care physician. However, T3’s potential for long-term side effects (eg, osteoporosis, cardiovascular effects) were not examined, and it is not clear when to discontinue it.
LEVEL 4: AFTER THREE FAILURES, HOW SHOULD A CLINICIAN PROCEED?
Switch to mirtazapine plus venlafaxine XR or tranylcypromine?
Patients who reached level 4 were considered to have a highly treatment-resistant depressive illness, so treatments at this level were, by design, more aggressive. Accordingly, at level 4 we investigated treatments that might be considered more demanding than those a primary care physician would use. Approximately 40% of patients in each treatment group were from primary care settings.
Remission rates40:
- With the combination of mirtazapine (mean dose 35.7 mg/day) and venlafaxine XR (mean dose 210.3 mg/day)—13.7% by the HAM-D17 and 15.7% by the QIDS-SR16
- With the MAOI tranylcypromine (Parnate, mean dose 36.9 mg/day)—6.9% by the HAM-D17 and 13.8% by the QIDS-SR16. Response rates were 23.5% with the combination and 12.1% with tranylcypromine. Neither remission nor response rates differed significantly.
However, the percentage reduction in QIDS-SR16 score between baseline and exit was greater with the combination than with tranylcypromine. Further, more patients dropped out of treatment with tranylcypromine because of side effects (P < .03). Tranylcypromine also has the disadvantage of necessitating dietary restrictions.
A significant limitation of this comparison is that patients were less likely to get an adequate trial of tranylcypromine, an MAOI, than of the combination. When the 2-week washout period (required before switching to an MAOI) is subtracted from the total time in treatment, approximately 30% of participants in the tranylcypromine group had less than 2 weeks of treatment, and nearly half had less than 6 weeks of treatment.
Therefore, even though the remission and response rates were similar between groups, the combination of venlafaxine-XR plus mirtazapine therapy might have some advantages over tranylcypromine. These results provided the first evidence of tolerability and at least modest efficacy of this combination for treatment-resistant cases.
Overall, what was the cumulative remission rate?
The theoretical cumulative remission rate after four acute treatment steps was 67%. Remission was more likely to occur during the first two levels of treatment than during the last two. The cumulative remission rates for the first four steps were:
- Level 1—33%
- Level 2—57%
- Level 3—63%
- Level 4—67%.
RESULTS FROM LONG-TERM FOLLOW-UP AFTER REMISSION OR RESPONSE
Patients with a clinically meaningful response or, preferably, remission at any level could enter into a 12-month observational follow-up phase. Those who had required more treatment levels had higher relapse rates during this phase.41 Further, if a patient achieved remission rather than just response to treatment, regardless of the treatment level, the prognosis at follow-up was better, confirming the importance of remission as the goal of treatment.
Results also provided a warning—the greater the number of treatment levels that a patient required, the more likely that patient and physician would settle for response. Whether the greater relapse rates reflect a harder-to-treat depression or the naturalistic design of the follow-up phase (with less control over dosing) is unclear.
WHAT DO THESE RESULTS MEAN FOR PRIMARY CARE PHYSICIANS?
- Measurement-based care is feasible in primary care. Primary care doctors can ensure vigorous but tolerable dosing using a self-report depression scale to monitor response, a side-effects tool to monitor tolerability, and medication adjustments at critical decision points guided by these two measures.
- Remission, ie, complete recovery from a depressive episode, rather than merely substantial improvement, is associated with a better prognosis and is the preferred goal of treatment.
- Pharmacologic differences between psychotropic medications did not translate into substantial clinical differences, although tolerability differed. These findings are consistent with a large-scale systematic evidence review recently completed by the Agency for Healthcare Research and Quality that compared the effectiveness of antidepressants.42 Given the difficulty in predicting what medication will be both efficacious for and tolerated by an individual patient, familiarity with a broad spectrum of antidepressants is prudent.
- Remission of depressive episodes will most likely require repeated trials of sufficiently sustained,vigorously dosed antidepressant medication. From treatment initiation, physicians should ensure maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- If a first treatment doesn’t work, either switching or augmenting it is a reasonable choice. Augmentation may be preferred if the patient is tolerating and receiving partial benefit from the initial medication choice. While bupropion SR and buspirone were not different as augmenters by the primary remission outcome measure, secondary measures (eg, tolerability, depressive symptom change over the course of treatment, clinician-rated Quick Inventory of Depressive Symptomatology) recommended bupropion-SR over buspirone.
- If physicians switch, either a within-class switch (eg, citalopram to sertraline) or an out-of-class switch (eg, citalopram to bupropion SR) is effective, as is a switch to a dual-action agent (eg, venlafaxine XR).
- The likelihood of improvement after two aggressive medication trials is very low and likely requires more complicated medication regimens, and the existing evidence base is quite thin. These primary care patients should likely be referred to psychiatrists for more aggressive and intensive treatment.
- For patients who present with major depressive disorder, STAR*D suggests that with persistence and aggressive yet feasible care, there is hope: after one round, approximately 30% will have a remission; after two rounds, 50%; after three rounds, 60%; and after four rounds, 70%.
- While STAR*D excluded depressed patients with bipolar disorder, a depressive episode in a patient with bipolar disorder can be difficult to distinguish from a depressive episode in a patient with major depressive disorder. Primary care physicians need to consider bipolar disorder both in patients presenting with a depressive episode and in those who fail an adequate trial.43
FUTURE CONSIDERATIONS
Subsequent STAR*D analyses will compare in greater depth outcomes in primary care vs psychiatric settings at each level of treatment. Given the greater risk of depression persistence associated with more successive levels of treatment, subsequent research will focus on ways to more successfully treat depression in the earlier stages, possibly through medication combinations earlier in treatment (somewhat analogous to a “broad-spectrum antibiotic” approach for infections).
- Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003; 289:3095–3105.
- Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997; 349:1498–1504.
- Zimmerman M, Chelminski I, Posternak MA. Generalizability of antidepressant efficacy trials: differences between depressed psychiatric outpatients who would or would not qualify for an efficacy trial. Am J Psychiatry 2005; 162:1370–1372.
- Rothwell PM. External validity of randomised controlled trials: to whom do the results of this trial apply? Lancet 2005; 365:82–93.
- Depression Guideline Panel. Depression in primary care: Volume 1, diagnosis and detection. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Frank E, Karp J, Rush A. Efficacy of treatments for major depression. Psychopharmacol Bull 1993; 29:457–475.
- Depression Guideline Panel. Depression in primary care: Volume 2, Treatment of major depression. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996; 19:179–200.
- Jarrett RB, Rush A. Short-term psychotherapy of depressive disorders: current status and future directions. Psychiatry: Interpers Biol Process 1994; 57:115–132.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depression (revision). Am J Psychiatry 2000; 157(suppl 4):1–45.
- Dunner DL, Rush AJ, Russell JM, et al. Prospective, long-term, multicenter study of the naturalistic outcomes of patients with treatment-resistant depression. J Clin Psychiatry 2006; 67:688–695.
- Fava M, Rush A, Trivedi M, et al. Background and rationale for the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study. Psychiatr Clin North Am 2003; 26:457–494.
- Gaynes B, Davis L, Rush A, Trivedi M, Fava M, Wisniewski S. The aims and design of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study. Prim Psychiatry 2005; 12:36–41.
- Rush A, Fava M, Wisniewski S, et al. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004; 25:119–142.
- Stafford RS, Ausiello JC, Misra B, Saglam D. National Patterns o fDepression Treatment in Primary Care. Prim Care Companion J Clin Psychiatry 2000; 2:211–216.
- Regier D, Narrow W, Rae D, Mandersheid R, Locke B, Goodwin F. The de facto US mental and addictive disorders service system: epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry 1993; 50:85–94.
- Pincus H, Tanielian T, Marcus S, et al. Prescribing trends in psychotropic medications: primary care, psychiatry, and other medical specialities. JAMA 1998; 279:526–531.
- Vuorilehto M, Melartin T, Isometsa E. Depressive disorders in primary care: recurrent, chronic, and co-morbid. Psychol Med 2005; 35:673–682.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–61.
- Wisniewski SR, Rush AJ, Balasubramani GK, Trivedi MH, Nierenberg AA for the STAR*D Investigators. Self-rated global measure of the frequency, intensity, and burden of side effects. J Psychiatric Pract 2006; 12:71–79.
- Rush AJ, Bernstein IH, Trivedi MH, et al. An evaluation of the Quick Inventory of Depressive Symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial report. Biol Psychiatry 2006; 59:493–501.
- Trivedi MH, Rush AJ, Gaynes BN, et al. Maximizing the adequacy of medication treatment in controlled trials and clinical practice: STAR*D measurement-based care. Neuropsychopharmacology 2007/04/04/online 2007.
- Kawamoto K, Houlihan CA, Balas EA, Lobach DF. Improving clinical practice using clinical decision support systems: a systematic review of trials to identify features critical to success. BMJ 2005; 330:765 e-pub March 14 2005.
- Lavori P, Rush A, Wisniewski S, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001; 50:792–801.
- Gaynes BN, Rush AJ, Trivedi MH, et al. Major depression symptoms in primary care and psychiatric care settings: a cross-sectional analysis. Ann Fam Med 2007; 5:126–134.
- Gaynes BN, Rush AJ, Trivedi M, et al. A direct comparison of presenting characteristics of depressed outpatients from primary vs. specialty care settings: preliminary findings from the STAR*D clinical trial. Gen Hosp Psychiatry 2005; 27:87–96.
- Sullivan PW, Valuck R, Saseen J, MacFall HM. A comparison of the direct costs and cost effectiveness of serotonin reuptake inhibitors and associated adverse drug reactions. CNS Drugs 2004; 18:911–932.
- Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163:28–40.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006; 354:1231–1242.
- Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006; 354:1243–1252.
- Wisniewski SR, Fava M, Trivedi MH, et al. Acceptability of second-step treatments to depressed outpatients: a STAR*D report. Am J Psychiatry 2007; 164:753–760.
- Thase ME, Friedman ES, Biggs MM, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry 2007; 164:739–752.
- Fava M, Rush AJ, Wisniewski SR, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry 2006; 163:1161–1172.
- Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T3 augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry 2006; 163:1519–1530.
- Bschor T, Lewitzka U, Sasse J, Adli M, Koberle U, Bauer M. Lithium augmentation in treatment-resistant depression: clinical evidence, serotonergic and endocrine mechanisms. Pharmacopsychiatry 2003; 36(suppl 3):S230–S234.
- Freeman MP, Freeman SA. Lithium: clinical considerations in internal medicine. Am J Med 2006; 119:478–481.
- Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996; 53:842–848.
- McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry 2006; 163:1531–1541.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Gartlehner G, Hansen R, Thieda P, et al. Comparative Effectiveness of Second-generation Antidepressants in the Pharmacologic Treatment of Depression. Agency for Healthcare Research and Quality. http://effectivehealthcare.ahrq.gov/reports/topic.cfm?topic=8&sid=39&rType=3. Accessed December 12, 2007.
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293:956–963.
Depression can be treated successfully by primary care physicians under “real-world” conditions.
Furthermore, the particular drug or drugs used are not as important as following a rational plan: giving antidepressant medications in adequate doses, monitoring the patient’s symptoms and side effects and adjusting the regimen accordingly, and switching drugs or adding new drugs to the regimen only after an adequate trial.
These are among the lessons learned from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, the largest prospective clinical trial of treatment of major depressive disorder ever conducted. It was funded by the National Institutes of Health and directed by A. John Rush, MD.
WHAT WERE THE AIMS OF STAR*D?
Depression, a common and debilitating condition, affects approximately one in eight people in the United States.1 It is expected2 to be the second-leading cause of disability in the world by the year 2020; today, it is the second-leading cause of disability-adjusted life years in those 15 to 44 years old.3
Nevertheless, the available evidence base for treatment is limited, since most participants in clinical trials are recruited by advertisement rather than from representative practices, and they are often selected to have few comorbid disorders, either medical or psychiatric. In addition, those with chronic depression or current suicidal ideation are excluded.1,4 These uncomplicated and “pristine” participants are unlike typical patients seen by primary care physicians or psychiatrists.
Similarly, the protocols used in these trials do not represent usual clinic practice.Patients in clinical trials undergo more assessment and more frequent follow-up than in real-world practice, they have no say in treatment decisions, the doses are fixed, and the patients and physicians are blinded to the intervention. Consequently, how to translate the results of these efficacy trials into practice is unclear.5
Further, even in relatively uncomplicated cases, only about one-half of outpatients with nonpsychotic major depressive disorder initially treated with a single medication or with psychotherapy will experience a clinically significant improvement in symptoms (ie, a response) during the 8 to 12 weeks of acute-phase treatment,6–10 and only 20% to 35% of patients will reach remission,9 the aim of treatment.8,11 The remission rates are even lower in treatment-resistant depression.12 How to manage most patients—those whose depression does not remit with the first, second, or third step of treatment—is unclear.
Accordingly, the overall objective of STAR*D was to develop and evaluate feasible treatment strategies to improve clinical outcomes for real-world patients with treatment-resistant depression, who were identified prospectively from a pool of patients in a current major depressive episode.13–15 Specifically, STAR*D aimed to determine prospectively which of several treatments is the most effective “next step” for patients who do not reach remission with an initial or subsequent treatment or who cannot tolerate the treatment.
WHY IS STAR*D RELEVANT FOR PRIMARY CARE?
Nearly 10% of all primary care office visits are depression-related.16 Primary care physicians provide nearly half the outpatient care for depressed patients.17 Indeed, primary care physicians log approximately as many outpatient visits for depression as psychiatrists do.18 Medical comorbidity is especially common in primary care settings.19 When to refer to a psychiatrist is not clear.
KEY FEATURES OF THE STUDY DESIGN
STAR*D involved a national consortium of 14 university-based regional centers, which oversaw a total of 23 participating psychiatric and 18 primary care clinics. Enrollment began in 2000, with follow-up completed in 2004.
Entry criteria were broad and inclusive
Patients had to:
- Be between 18 and 75 years of age
- Have a nonpsychotic major depressive disorder, identified by a clinician and confirmed with a symptom checklist based on the Diagnostic and Statistical Manual, fourth edition revised,20 and for which antidepressant treatment is recommended
- Score at least 14 on the 17-item Hamilton Rating Scale for Depression (HAMD17)21
- Not have a primary diagnosis of bipolar disorder, obsessive-compulsive disorder, or an eating disorder, which would require a different treatment strategy, or a seizure disorder (which would preclude bupropion as a second-step treatment).
Dosing recommendations were flexible but vigorous
Medications often were increased to maximally tolerated doses. For example, citalopram (Celexa) was started at 20 mg/day and increased by 20 mg every 2 to 4 weeks if the patient was tolerating it but had not achieved remission, to a maximum dose of 60 mg/day. Treatment could be given for up to 14 weeks, during which side effects22 and clinical ratings23 were assessed by both patients and study coordinators.
Measurement-based care
We used a systematic approach to treatment called “measurement-based care,”24 which involves routinely measuring symptoms23 and side effects22 and using this information to modify the medication doses at critical decision points. This algorithmic approach provided flexible treatment recommendations to ensure that the dosage and duration of antidepressant drug treatment were adequate.25
The severity of depression was assessed by the clinician-rated, 16-item Quick Inventory of Depressive Symptomatology (QIDS-C16). The QIDS-SR16 (the self-report version) can substitute for the QIDS-C1623 to make this approach more feasible. Both tools are available at www.ids-qids.org.
This approach was easily worked into busy primary care and specialty care office workflows (clinic physicians, most with limited research experience, provided the treatment), and could be translated into primary care practice in the community as well.
Four-step protocol
A unique feature of the study design was that the patients, in consultation with their physicians, had some choice in the treatments they received. In this “equipoise-stratified randomized design,”26 at levels 2 and 3 the patient could choose either to switch therapies (stop the current drug and be randomized to receive one of several different treatments) or to augment their current therapy (by adding one of several treatments in a randomized fashion). Patients could decline certain strategies as long as there were at least two possible options to which one might be randomized.
At level 2, one of the options for both switching and augmentation was cognitive therapy, although patients could decline that option. Conversely, if they definitely wanted cognitive therapy, they could choose to be randomized to either cognitive therapy alone or to cognitive therapy added to citalopram. Also, anyone who received cognitive therapy in level 2 and failed to enter remission was additionally randomized to either bupropion or venlafaxine (level 2a) to ensure that all patients had failed trials on two medications before entering level 3.
When switching to medications other than a monoamine oxidase inhibitor (MAOI), the clinician could choose either to stop the current medication and immediately begin the next one, or to decrease the current medication while starting the new one at a low dose and then tapering and titrating over 1 week. (Switching to an MAOI, used only in the final level of treatment, required a 7- to 10-day washout period.)
Outcomes measured
Remission (complete recovery from the depressive episode), the primary study outcome, was defined as a HAM-D17 score of 7 or less, as assessed by treatment-blinded raters.A secondary remission outcome was a QIDS-SR16 score of 5 or less. Of note, the HAM-D17 remission rates were slightly lower than the rates based on the QIDS-SR16, since patients who did not have a HAM-D17 score measured at exit were defined as not being in remission a priori. Thus, the QIDS-SR16 rates might have been a slightly better reflection of actual remission rates.
Response, a secondary outcome, was defined as a reduction of at least 50% in the QIDS-SR16 score from baseline at the last assessment.
FEW DIFFERENCES BETWEEN PSYCHIATRIC, PRIMARY CARE PATIENTS
The patients seen in primary care clinics were surprisingly similar to those seen in psychiatric clinics.27,28 The two groups did not differ in severity of depression, distribution of severity scores, the likelihood of presenting with any of the nine core criteria of a major depressive episode, or the likelihood of having a concomitant axis I psychiatric disorder in addition to depression (about half of participants in each setting had an anxiety disorder).
Recurrent major depressive disorders were common in both groups, though more so in psychiatric patients (78% vs 69%, P < .001), while chronic depression was more common in primary care than in psychiatric patients (30% vs 21%, P < .001). Having either a chronic index episode (ie, lasting > 2 years) or a recurrent major depressive disorder was common in both groups (86% vs 83%, P = .0067).
That said, primary care patients were older (44 years vs 39 years, P < .001), more of them were Hispanic (18% vs 9%, P < .001), and more of them had public insurance (23% vs 9%, P < .001). Fewer of the primary care patients had completed college (20% vs 28%, P < .001), and the primary care patients tended to have greater medical comorbidity. Psychiatric patients were more likely to have attempted suicide in the past and to have had their first depressive illness before age 18.
LEVEL 1: WHAT CAN WE EXPECT FROM INITIAL TREATMENT?
At level 1, all the patients received citalopram. The mean dose was 40.6 ± 16.6 mg/day in the primary care clinics and 42.5 ± 16.8 mg/day in the psychiatric clinics, which are adequate, middle-range doses and higher than the average US dose.29
Approximately 30% of patients achieved remission: 27% as measured on the HAM-D17 and 33% on the QIDS-SR16. The response rate (on the QIDS-SR16) was 47%. There were no differences between primary and psychiatric care settings in remission or response rates.
Patients were more likely to achieve remission if they were white, female, employed, more educated, or wealthier. Longer current episodes, more concurrent psychiatric disorders (especially anxiety disorders or drug abuse), more general medical disorders, and lower baseline function and quality of life were each associated with lower remission rates.
What is an adequate trial?
Longer times than expected were needed to reach response or remission. The average duration required to achieve remission was almost 7 weeks (44 days in primary care; 49 days in psychiatric care). Further, approximately one-third of those who ultimately responded and half of those who entered remission did so after 6 weeks.30 Forty percent of those who entered remission required 8 or more weeks to do so.
These results suggest that longer treatment durations and more vigorous medication dosing than generally used are needed to achieve optimal remission rates. It is imprudent to stop a treatment that the patient is tolerating in a robust dose if the patient reports only partial benefit by 6 weeks; indeed, raising the dose, if tolerated, may help a substantial number of patients respond by 12 or 14 weeks. Instruments to monitor depression severity (eg, self-report measures) can be useful. At least 8 weeks with at least moderately vigorous dosing is recommended.
LEVEL 2: IF THE FIRST TREATMENT FAILS
When switching to a new drug, does it matter which one?
No.
In level 2, if patients had not achieved remission on citalopram alone, they had the choice of switching: stopping citalopram and being randomized to receive either sertraline (Zoloft, another SSRI), venlafaxine extended-release (XR) (Effexor XR, a serotonin and norepinephrine reuptake inhibitor), or bupropion sustained-release (SR) (Wellbutrin SR, a norepinephrine and dopamine reuptake inhibitor). At the last visit the mean daily doses were bupropion SR 282.7 mg/day, sertraline 135.5 mg/day, and venlafaxine-XR 193.6 mg/day.
The remission rate was approximately one-fourth with all three drugs31:
- With bupropion SR—21.3% by HAM-D17, 25.5% by QIDS-SR16
- With sertraline—17.6% by HAM-D17, 26.6% by QIDS-SR16
- With venlafaxine-XR—24.8% by HAM-D17, 25.0% by QIDS-SR16. The remission rates were neither statistically nor clinically different by either measure.
Though the types of side effects related to specific medications may have varied, the overall side-effect burden and the rate of serious adverse events did not differ significantly.
When adding a new drug, does it matter which one?
Again, no.
Instead of switching, patients in level 2 could choose to stay on citalopram and be randomized to add either bupropion SR or buspirone (BuSpar) to the regimen (augmentation). The mean daily doses at the end of level 2 were bupropion SR 267.5 mg and buspirone 40.9 mg.
Rates of remission32:
- With bupropion SR—29.7% on the HAMD-D17, 39.0% on the QIDS-SR16
- With buspirone—30.1% on the HAM-D17, 32.9% on the QIDS-SR16.
However, the QIDS-SR16 scores declined significantly more with bupropion SR than with buspirone (25.3% vs 17.1%, P < .04). The mean total QIDS-SR16 score at the last visit was lower with bupropion SR (8.0) than with buspirone (9.1, P < .02), and augmentation with bupropion SR was better tolerated (the dropout rate due to intolerance was 12.5% with bupropion-SR vs 20.6% with buspirone 20.6%; P < .009).
Can we directly compare the benefits of switching vs augmenting?
No.
Patients could choose whether to switch from citalopram to another drug or to add another drug at the second treatment level.33 Consequently, we could not ensure that the patient groups were equivalent at the point of randomization at the beginning of level 2, and, indeed, they were not.
Those who benefitted more from citalopram treatment and who better tolerated it preferred augmentation, while those who benefitted little or who could not tolerate it preferred to switch. Consequently, those in the augmentation group at level 2 were somewhat less depressed than those who switched. Whether augmentation is better even if the initial treatment is minimally effective could not be evaluated in STAR*D.
What about cognitive therapy?
There was no difference between cognitive therapy (either as a switch or as augmentation) and medication (as a switch or as augmentation).34 Adding another drug was more rapidly effective than adding cognitive therapy. Switching to cognitive therapy was better tolerated than switching to a different antidepressant.
Of note, fewer patients accepted cognitive therapy as a randomization option than we expected, so the sample sizes were small. Possible reasons were that all patients had to receive a medication at study entry (which may have biased selection towards those preferring medication), and cognitive therapy entailed additional copayments and visiting still another provider at another site.
After two levels of treatment, how many patients reach remission?
About 30% of patients in level 1 achieved remission, and of those progressing to level 2, another 30% achieved remission. Together, this adds up to about 50% of patients achieving remission if they remained in treatment (30% in level 1 plus 30% of the roughly 70% remaining in level 2).
IF A SECOND TREATMENT FAILS
If switching again to another drug, does it matter which one?
No.
In level 3, patients could choose to stop the drug they had been taking and be randomized to receive either mirtazapine (Remeron) or nortriptyline (Pamelor).
Switching medications was not as effective as a third step as it was as a second step.35
Remission rates:
- With mirtazapine—12.3% on the HAM-D17, 8.0% on the QIDS-SR16
- With nortriptyline—19.8% on the HAM-D17, 12.4% on the QIDS-SR16.
Response rates were 13.4% with mirtazapine and 16.5% with nortriptyline. Statistically, neither the response nor the remission rates differed by treatment, nor did these two treatments differ in tolerability or side-effect burden.
Does choice of augmentation agent matter: Lithium vs T3?
Similarly, after two failed medication treatments, medication augmentation was less effective than it was at the second step.36 The two augmentation options tested, lithium and T3 thyroid hormone (Cytomel), are commonly considered by psychiatrists but less commonly used by primary care doctors.
Lithium is believed to increase serotonergic function, which may have a synergistic effect on the mechanism of action of antidepressants; a meta-analysis of placebo-controlled studies supports lithium’s effectiveness as adjunctive treatment.37 Its side effects, however, must be closely monitored.38 The primary monitoring concern is the small difference between the therapeutic blood level (0.6–1.2 mEq/L) and potentially toxic blood levels (> 1.5 mEq/L).
Lithium was started at 450 mg/day, and at week 2 it was increased to the recommended dose of 900 mg/day (a dose below the target dose for bipolar disorder). If patients could not tolerate 450 mg/day, the initial dose was 225 mg/day for 1 week before being increased to 450 mg/day, still with the target dose of 900 mg/day. The mean exit dose was 859.9 mg/day, and the median blood level was 0.6 mEq/L.
Thyroid hormone augmentation using T3 is believed to work through both direct and indirect effects on the hypothalamic-pituitary-thyroid axis, which has a strong relationship with depression. The efficacy of T3 augmentation is supported by a meta-analysis of eight studies,39 and T3 is effective whether or not thyroid abnormalities are present.
In STAR*D, T3 was started at 25 μg/day for 1 week, than increased to the recommended dose of 50 μg/day. The mean exit dose was 45.2 μg/day.
Remission rates:
- With lithium augmentation—15.9% by the HAM-D17, 13.2% by the QIDS-SR16
- With T3 augmentation—24.7% by both measures.
Response rates were 16.2% with lithium augmentation and 23.3% with T3 augmentation.
While neither response nor remission rates were statistically significantly different by treatment, lithium was more frequently associated with side effects (P = .045), and more participants in the lithium group left treatment because of side effects (23.2% vs 9.6%; P = .027). These results suggest that in cases in which a clinician is considering an augmentation trial, T3 has slight advantages over lithium in effectiveness and tolerability. T3 also offers the advantages of being easy to use and not necessitating blood level monitoring. These latter benefits are especially relevant to the primary care physician. However, T3’s potential for long-term side effects (eg, osteoporosis, cardiovascular effects) were not examined, and it is not clear when to discontinue it.
LEVEL 4: AFTER THREE FAILURES, HOW SHOULD A CLINICIAN PROCEED?
Switch to mirtazapine plus venlafaxine XR or tranylcypromine?
Patients who reached level 4 were considered to have a highly treatment-resistant depressive illness, so treatments at this level were, by design, more aggressive. Accordingly, at level 4 we investigated treatments that might be considered more demanding than those a primary care physician would use. Approximately 40% of patients in each treatment group were from primary care settings.
Remission rates40:
- With the combination of mirtazapine (mean dose 35.7 mg/day) and venlafaxine XR (mean dose 210.3 mg/day)—13.7% by the HAM-D17 and 15.7% by the QIDS-SR16
- With the MAOI tranylcypromine (Parnate, mean dose 36.9 mg/day)—6.9% by the HAM-D17 and 13.8% by the QIDS-SR16. Response rates were 23.5% with the combination and 12.1% with tranylcypromine. Neither remission nor response rates differed significantly.
However, the percentage reduction in QIDS-SR16 score between baseline and exit was greater with the combination than with tranylcypromine. Further, more patients dropped out of treatment with tranylcypromine because of side effects (P < .03). Tranylcypromine also has the disadvantage of necessitating dietary restrictions.
A significant limitation of this comparison is that patients were less likely to get an adequate trial of tranylcypromine, an MAOI, than of the combination. When the 2-week washout period (required before switching to an MAOI) is subtracted from the total time in treatment, approximately 30% of participants in the tranylcypromine group had less than 2 weeks of treatment, and nearly half had less than 6 weeks of treatment.
Therefore, even though the remission and response rates were similar between groups, the combination of venlafaxine-XR plus mirtazapine therapy might have some advantages over tranylcypromine. These results provided the first evidence of tolerability and at least modest efficacy of this combination for treatment-resistant cases.
Overall, what was the cumulative remission rate?
The theoretical cumulative remission rate after four acute treatment steps was 67%. Remission was more likely to occur during the first two levels of treatment than during the last two. The cumulative remission rates for the first four steps were:
- Level 1—33%
- Level 2—57%
- Level 3—63%
- Level 4—67%.
RESULTS FROM LONG-TERM FOLLOW-UP AFTER REMISSION OR RESPONSE
Patients with a clinically meaningful response or, preferably, remission at any level could enter into a 12-month observational follow-up phase. Those who had required more treatment levels had higher relapse rates during this phase.41 Further, if a patient achieved remission rather than just response to treatment, regardless of the treatment level, the prognosis at follow-up was better, confirming the importance of remission as the goal of treatment.
Results also provided a warning—the greater the number of treatment levels that a patient required, the more likely that patient and physician would settle for response. Whether the greater relapse rates reflect a harder-to-treat depression or the naturalistic design of the follow-up phase (with less control over dosing) is unclear.
WHAT DO THESE RESULTS MEAN FOR PRIMARY CARE PHYSICIANS?
- Measurement-based care is feasible in primary care. Primary care doctors can ensure vigorous but tolerable dosing using a self-report depression scale to monitor response, a side-effects tool to monitor tolerability, and medication adjustments at critical decision points guided by these two measures.
- Remission, ie, complete recovery from a depressive episode, rather than merely substantial improvement, is associated with a better prognosis and is the preferred goal of treatment.
- Pharmacologic differences between psychotropic medications did not translate into substantial clinical differences, although tolerability differed. These findings are consistent with a large-scale systematic evidence review recently completed by the Agency for Healthcare Research and Quality that compared the effectiveness of antidepressants.42 Given the difficulty in predicting what medication will be both efficacious for and tolerated by an individual patient, familiarity with a broad spectrum of antidepressants is prudent.
- Remission of depressive episodes will most likely require repeated trials of sufficiently sustained,vigorously dosed antidepressant medication. From treatment initiation, physicians should ensure maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- If a first treatment doesn’t work, either switching or augmenting it is a reasonable choice. Augmentation may be preferred if the patient is tolerating and receiving partial benefit from the initial medication choice. While bupropion SR and buspirone were not different as augmenters by the primary remission outcome measure, secondary measures (eg, tolerability, depressive symptom change over the course of treatment, clinician-rated Quick Inventory of Depressive Symptomatology) recommended bupropion-SR over buspirone.
- If physicians switch, either a within-class switch (eg, citalopram to sertraline) or an out-of-class switch (eg, citalopram to bupropion SR) is effective, as is a switch to a dual-action agent (eg, venlafaxine XR).
- The likelihood of improvement after two aggressive medication trials is very low and likely requires more complicated medication regimens, and the existing evidence base is quite thin. These primary care patients should likely be referred to psychiatrists for more aggressive and intensive treatment.
- For patients who present with major depressive disorder, STAR*D suggests that with persistence and aggressive yet feasible care, there is hope: after one round, approximately 30% will have a remission; after two rounds, 50%; after three rounds, 60%; and after four rounds, 70%.
- While STAR*D excluded depressed patients with bipolar disorder, a depressive episode in a patient with bipolar disorder can be difficult to distinguish from a depressive episode in a patient with major depressive disorder. Primary care physicians need to consider bipolar disorder both in patients presenting with a depressive episode and in those who fail an adequate trial.43
FUTURE CONSIDERATIONS
Subsequent STAR*D analyses will compare in greater depth outcomes in primary care vs psychiatric settings at each level of treatment. Given the greater risk of depression persistence associated with more successive levels of treatment, subsequent research will focus on ways to more successfully treat depression in the earlier stages, possibly through medication combinations earlier in treatment (somewhat analogous to a “broad-spectrum antibiotic” approach for infections).
Depression can be treated successfully by primary care physicians under “real-world” conditions.
Furthermore, the particular drug or drugs used are not as important as following a rational plan: giving antidepressant medications in adequate doses, monitoring the patient’s symptoms and side effects and adjusting the regimen accordingly, and switching drugs or adding new drugs to the regimen only after an adequate trial.
These are among the lessons learned from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, the largest prospective clinical trial of treatment of major depressive disorder ever conducted. It was funded by the National Institutes of Health and directed by A. John Rush, MD.
WHAT WERE THE AIMS OF STAR*D?
Depression, a common and debilitating condition, affects approximately one in eight people in the United States.1 It is expected2 to be the second-leading cause of disability in the world by the year 2020; today, it is the second-leading cause of disability-adjusted life years in those 15 to 44 years old.3
Nevertheless, the available evidence base for treatment is limited, since most participants in clinical trials are recruited by advertisement rather than from representative practices, and they are often selected to have few comorbid disorders, either medical or psychiatric. In addition, those with chronic depression or current suicidal ideation are excluded.1,4 These uncomplicated and “pristine” participants are unlike typical patients seen by primary care physicians or psychiatrists.
Similarly, the protocols used in these trials do not represent usual clinic practice.Patients in clinical trials undergo more assessment and more frequent follow-up than in real-world practice, they have no say in treatment decisions, the doses are fixed, and the patients and physicians are blinded to the intervention. Consequently, how to translate the results of these efficacy trials into practice is unclear.5
Further, even in relatively uncomplicated cases, only about one-half of outpatients with nonpsychotic major depressive disorder initially treated with a single medication or with psychotherapy will experience a clinically significant improvement in symptoms (ie, a response) during the 8 to 12 weeks of acute-phase treatment,6–10 and only 20% to 35% of patients will reach remission,9 the aim of treatment.8,11 The remission rates are even lower in treatment-resistant depression.12 How to manage most patients—those whose depression does not remit with the first, second, or third step of treatment—is unclear.
Accordingly, the overall objective of STAR*D was to develop and evaluate feasible treatment strategies to improve clinical outcomes for real-world patients with treatment-resistant depression, who were identified prospectively from a pool of patients in a current major depressive episode.13–15 Specifically, STAR*D aimed to determine prospectively which of several treatments is the most effective “next step” for patients who do not reach remission with an initial or subsequent treatment or who cannot tolerate the treatment.
WHY IS STAR*D RELEVANT FOR PRIMARY CARE?
Nearly 10% of all primary care office visits are depression-related.16 Primary care physicians provide nearly half the outpatient care for depressed patients.17 Indeed, primary care physicians log approximately as many outpatient visits for depression as psychiatrists do.18 Medical comorbidity is especially common in primary care settings.19 When to refer to a psychiatrist is not clear.
KEY FEATURES OF THE STUDY DESIGN
STAR*D involved a national consortium of 14 university-based regional centers, which oversaw a total of 23 participating psychiatric and 18 primary care clinics. Enrollment began in 2000, with follow-up completed in 2004.
Entry criteria were broad and inclusive
Patients had to:
- Be between 18 and 75 years of age
- Have a nonpsychotic major depressive disorder, identified by a clinician and confirmed with a symptom checklist based on the Diagnostic and Statistical Manual, fourth edition revised,20 and for which antidepressant treatment is recommended
- Score at least 14 on the 17-item Hamilton Rating Scale for Depression (HAMD17)21
- Not have a primary diagnosis of bipolar disorder, obsessive-compulsive disorder, or an eating disorder, which would require a different treatment strategy, or a seizure disorder (which would preclude bupropion as a second-step treatment).
Dosing recommendations were flexible but vigorous
Medications often were increased to maximally tolerated doses. For example, citalopram (Celexa) was started at 20 mg/day and increased by 20 mg every 2 to 4 weeks if the patient was tolerating it but had not achieved remission, to a maximum dose of 60 mg/day. Treatment could be given for up to 14 weeks, during which side effects22 and clinical ratings23 were assessed by both patients and study coordinators.
Measurement-based care
We used a systematic approach to treatment called “measurement-based care,”24 which involves routinely measuring symptoms23 and side effects22 and using this information to modify the medication doses at critical decision points. This algorithmic approach provided flexible treatment recommendations to ensure that the dosage and duration of antidepressant drug treatment were adequate.25
The severity of depression was assessed by the clinician-rated, 16-item Quick Inventory of Depressive Symptomatology (QIDS-C16). The QIDS-SR16 (the self-report version) can substitute for the QIDS-C1623 to make this approach more feasible. Both tools are available at www.ids-qids.org.
This approach was easily worked into busy primary care and specialty care office workflows (clinic physicians, most with limited research experience, provided the treatment), and could be translated into primary care practice in the community as well.
Four-step protocol
A unique feature of the study design was that the patients, in consultation with their physicians, had some choice in the treatments they received. In this “equipoise-stratified randomized design,”26 at levels 2 and 3 the patient could choose either to switch therapies (stop the current drug and be randomized to receive one of several different treatments) or to augment their current therapy (by adding one of several treatments in a randomized fashion). Patients could decline certain strategies as long as there were at least two possible options to which one might be randomized.
At level 2, one of the options for both switching and augmentation was cognitive therapy, although patients could decline that option. Conversely, if they definitely wanted cognitive therapy, they could choose to be randomized to either cognitive therapy alone or to cognitive therapy added to citalopram. Also, anyone who received cognitive therapy in level 2 and failed to enter remission was additionally randomized to either bupropion or venlafaxine (level 2a) to ensure that all patients had failed trials on two medications before entering level 3.
When switching to medications other than a monoamine oxidase inhibitor (MAOI), the clinician could choose either to stop the current medication and immediately begin the next one, or to decrease the current medication while starting the new one at a low dose and then tapering and titrating over 1 week. (Switching to an MAOI, used only in the final level of treatment, required a 7- to 10-day washout period.)
Outcomes measured
Remission (complete recovery from the depressive episode), the primary study outcome, was defined as a HAM-D17 score of 7 or less, as assessed by treatment-blinded raters.A secondary remission outcome was a QIDS-SR16 score of 5 or less. Of note, the HAM-D17 remission rates were slightly lower than the rates based on the QIDS-SR16, since patients who did not have a HAM-D17 score measured at exit were defined as not being in remission a priori. Thus, the QIDS-SR16 rates might have been a slightly better reflection of actual remission rates.
Response, a secondary outcome, was defined as a reduction of at least 50% in the QIDS-SR16 score from baseline at the last assessment.
FEW DIFFERENCES BETWEEN PSYCHIATRIC, PRIMARY CARE PATIENTS
The patients seen in primary care clinics were surprisingly similar to those seen in psychiatric clinics.27,28 The two groups did not differ in severity of depression, distribution of severity scores, the likelihood of presenting with any of the nine core criteria of a major depressive episode, or the likelihood of having a concomitant axis I psychiatric disorder in addition to depression (about half of participants in each setting had an anxiety disorder).
Recurrent major depressive disorders were common in both groups, though more so in psychiatric patients (78% vs 69%, P < .001), while chronic depression was more common in primary care than in psychiatric patients (30% vs 21%, P < .001). Having either a chronic index episode (ie, lasting > 2 years) or a recurrent major depressive disorder was common in both groups (86% vs 83%, P = .0067).
That said, primary care patients were older (44 years vs 39 years, P < .001), more of them were Hispanic (18% vs 9%, P < .001), and more of them had public insurance (23% vs 9%, P < .001). Fewer of the primary care patients had completed college (20% vs 28%, P < .001), and the primary care patients tended to have greater medical comorbidity. Psychiatric patients were more likely to have attempted suicide in the past and to have had their first depressive illness before age 18.
LEVEL 1: WHAT CAN WE EXPECT FROM INITIAL TREATMENT?
At level 1, all the patients received citalopram. The mean dose was 40.6 ± 16.6 mg/day in the primary care clinics and 42.5 ± 16.8 mg/day in the psychiatric clinics, which are adequate, middle-range doses and higher than the average US dose.29
Approximately 30% of patients achieved remission: 27% as measured on the HAM-D17 and 33% on the QIDS-SR16. The response rate (on the QIDS-SR16) was 47%. There were no differences between primary and psychiatric care settings in remission or response rates.
Patients were more likely to achieve remission if they were white, female, employed, more educated, or wealthier. Longer current episodes, more concurrent psychiatric disorders (especially anxiety disorders or drug abuse), more general medical disorders, and lower baseline function and quality of life were each associated with lower remission rates.
What is an adequate trial?
Longer times than expected were needed to reach response or remission. The average duration required to achieve remission was almost 7 weeks (44 days in primary care; 49 days in psychiatric care). Further, approximately one-third of those who ultimately responded and half of those who entered remission did so after 6 weeks.30 Forty percent of those who entered remission required 8 or more weeks to do so.
These results suggest that longer treatment durations and more vigorous medication dosing than generally used are needed to achieve optimal remission rates. It is imprudent to stop a treatment that the patient is tolerating in a robust dose if the patient reports only partial benefit by 6 weeks; indeed, raising the dose, if tolerated, may help a substantial number of patients respond by 12 or 14 weeks. Instruments to monitor depression severity (eg, self-report measures) can be useful. At least 8 weeks with at least moderately vigorous dosing is recommended.
LEVEL 2: IF THE FIRST TREATMENT FAILS
When switching to a new drug, does it matter which one?
No.
In level 2, if patients had not achieved remission on citalopram alone, they had the choice of switching: stopping citalopram and being randomized to receive either sertraline (Zoloft, another SSRI), venlafaxine extended-release (XR) (Effexor XR, a serotonin and norepinephrine reuptake inhibitor), or bupropion sustained-release (SR) (Wellbutrin SR, a norepinephrine and dopamine reuptake inhibitor). At the last visit the mean daily doses were bupropion SR 282.7 mg/day, sertraline 135.5 mg/day, and venlafaxine-XR 193.6 mg/day.
The remission rate was approximately one-fourth with all three drugs31:
- With bupropion SR—21.3% by HAM-D17, 25.5% by QIDS-SR16
- With sertraline—17.6% by HAM-D17, 26.6% by QIDS-SR16
- With venlafaxine-XR—24.8% by HAM-D17, 25.0% by QIDS-SR16. The remission rates were neither statistically nor clinically different by either measure.
Though the types of side effects related to specific medications may have varied, the overall side-effect burden and the rate of serious adverse events did not differ significantly.
When adding a new drug, does it matter which one?
Again, no.
Instead of switching, patients in level 2 could choose to stay on citalopram and be randomized to add either bupropion SR or buspirone (BuSpar) to the regimen (augmentation). The mean daily doses at the end of level 2 were bupropion SR 267.5 mg and buspirone 40.9 mg.
Rates of remission32:
- With bupropion SR—29.7% on the HAMD-D17, 39.0% on the QIDS-SR16
- With buspirone—30.1% on the HAM-D17, 32.9% on the QIDS-SR16.
However, the QIDS-SR16 scores declined significantly more with bupropion SR than with buspirone (25.3% vs 17.1%, P < .04). The mean total QIDS-SR16 score at the last visit was lower with bupropion SR (8.0) than with buspirone (9.1, P < .02), and augmentation with bupropion SR was better tolerated (the dropout rate due to intolerance was 12.5% with bupropion-SR vs 20.6% with buspirone 20.6%; P < .009).
Can we directly compare the benefits of switching vs augmenting?
No.
Patients could choose whether to switch from citalopram to another drug or to add another drug at the second treatment level.33 Consequently, we could not ensure that the patient groups were equivalent at the point of randomization at the beginning of level 2, and, indeed, they were not.
Those who benefitted more from citalopram treatment and who better tolerated it preferred augmentation, while those who benefitted little or who could not tolerate it preferred to switch. Consequently, those in the augmentation group at level 2 were somewhat less depressed than those who switched. Whether augmentation is better even if the initial treatment is minimally effective could not be evaluated in STAR*D.
What about cognitive therapy?
There was no difference between cognitive therapy (either as a switch or as augmentation) and medication (as a switch or as augmentation).34 Adding another drug was more rapidly effective than adding cognitive therapy. Switching to cognitive therapy was better tolerated than switching to a different antidepressant.
Of note, fewer patients accepted cognitive therapy as a randomization option than we expected, so the sample sizes were small. Possible reasons were that all patients had to receive a medication at study entry (which may have biased selection towards those preferring medication), and cognitive therapy entailed additional copayments and visiting still another provider at another site.
After two levels of treatment, how many patients reach remission?
About 30% of patients in level 1 achieved remission, and of those progressing to level 2, another 30% achieved remission. Together, this adds up to about 50% of patients achieving remission if they remained in treatment (30% in level 1 plus 30% of the roughly 70% remaining in level 2).
IF A SECOND TREATMENT FAILS
If switching again to another drug, does it matter which one?
No.
In level 3, patients could choose to stop the drug they had been taking and be randomized to receive either mirtazapine (Remeron) or nortriptyline (Pamelor).
Switching medications was not as effective as a third step as it was as a second step.35
Remission rates:
- With mirtazapine—12.3% on the HAM-D17, 8.0% on the QIDS-SR16
- With nortriptyline—19.8% on the HAM-D17, 12.4% on the QIDS-SR16.
Response rates were 13.4% with mirtazapine and 16.5% with nortriptyline. Statistically, neither the response nor the remission rates differed by treatment, nor did these two treatments differ in tolerability or side-effect burden.
Does choice of augmentation agent matter: Lithium vs T3?
Similarly, after two failed medication treatments, medication augmentation was less effective than it was at the second step.36 The two augmentation options tested, lithium and T3 thyroid hormone (Cytomel), are commonly considered by psychiatrists but less commonly used by primary care doctors.
Lithium is believed to increase serotonergic function, which may have a synergistic effect on the mechanism of action of antidepressants; a meta-analysis of placebo-controlled studies supports lithium’s effectiveness as adjunctive treatment.37 Its side effects, however, must be closely monitored.38 The primary monitoring concern is the small difference between the therapeutic blood level (0.6–1.2 mEq/L) and potentially toxic blood levels (> 1.5 mEq/L).
Lithium was started at 450 mg/day, and at week 2 it was increased to the recommended dose of 900 mg/day (a dose below the target dose for bipolar disorder). If patients could not tolerate 450 mg/day, the initial dose was 225 mg/day for 1 week before being increased to 450 mg/day, still with the target dose of 900 mg/day. The mean exit dose was 859.9 mg/day, and the median blood level was 0.6 mEq/L.
Thyroid hormone augmentation using T3 is believed to work through both direct and indirect effects on the hypothalamic-pituitary-thyroid axis, which has a strong relationship with depression. The efficacy of T3 augmentation is supported by a meta-analysis of eight studies,39 and T3 is effective whether or not thyroid abnormalities are present.
In STAR*D, T3 was started at 25 μg/day for 1 week, than increased to the recommended dose of 50 μg/day. The mean exit dose was 45.2 μg/day.
Remission rates:
- With lithium augmentation—15.9% by the HAM-D17, 13.2% by the QIDS-SR16
- With T3 augmentation—24.7% by both measures.
Response rates were 16.2% with lithium augmentation and 23.3% with T3 augmentation.
While neither response nor remission rates were statistically significantly different by treatment, lithium was more frequently associated with side effects (P = .045), and more participants in the lithium group left treatment because of side effects (23.2% vs 9.6%; P = .027). These results suggest that in cases in which a clinician is considering an augmentation trial, T3 has slight advantages over lithium in effectiveness and tolerability. T3 also offers the advantages of being easy to use and not necessitating blood level monitoring. These latter benefits are especially relevant to the primary care physician. However, T3’s potential for long-term side effects (eg, osteoporosis, cardiovascular effects) were not examined, and it is not clear when to discontinue it.
LEVEL 4: AFTER THREE FAILURES, HOW SHOULD A CLINICIAN PROCEED?
Switch to mirtazapine plus venlafaxine XR or tranylcypromine?
Patients who reached level 4 were considered to have a highly treatment-resistant depressive illness, so treatments at this level were, by design, more aggressive. Accordingly, at level 4 we investigated treatments that might be considered more demanding than those a primary care physician would use. Approximately 40% of patients in each treatment group were from primary care settings.
Remission rates40:
- With the combination of mirtazapine (mean dose 35.7 mg/day) and venlafaxine XR (mean dose 210.3 mg/day)—13.7% by the HAM-D17 and 15.7% by the QIDS-SR16
- With the MAOI tranylcypromine (Parnate, mean dose 36.9 mg/day)—6.9% by the HAM-D17 and 13.8% by the QIDS-SR16. Response rates were 23.5% with the combination and 12.1% with tranylcypromine. Neither remission nor response rates differed significantly.
However, the percentage reduction in QIDS-SR16 score between baseline and exit was greater with the combination than with tranylcypromine. Further, more patients dropped out of treatment with tranylcypromine because of side effects (P < .03). Tranylcypromine also has the disadvantage of necessitating dietary restrictions.
A significant limitation of this comparison is that patients were less likely to get an adequate trial of tranylcypromine, an MAOI, than of the combination. When the 2-week washout period (required before switching to an MAOI) is subtracted from the total time in treatment, approximately 30% of participants in the tranylcypromine group had less than 2 weeks of treatment, and nearly half had less than 6 weeks of treatment.
Therefore, even though the remission and response rates were similar between groups, the combination of venlafaxine-XR plus mirtazapine therapy might have some advantages over tranylcypromine. These results provided the first evidence of tolerability and at least modest efficacy of this combination for treatment-resistant cases.
Overall, what was the cumulative remission rate?
The theoretical cumulative remission rate after four acute treatment steps was 67%. Remission was more likely to occur during the first two levels of treatment than during the last two. The cumulative remission rates for the first four steps were:
- Level 1—33%
- Level 2—57%
- Level 3—63%
- Level 4—67%.
RESULTS FROM LONG-TERM FOLLOW-UP AFTER REMISSION OR RESPONSE
Patients with a clinically meaningful response or, preferably, remission at any level could enter into a 12-month observational follow-up phase. Those who had required more treatment levels had higher relapse rates during this phase.41 Further, if a patient achieved remission rather than just response to treatment, regardless of the treatment level, the prognosis at follow-up was better, confirming the importance of remission as the goal of treatment.
Results also provided a warning—the greater the number of treatment levels that a patient required, the more likely that patient and physician would settle for response. Whether the greater relapse rates reflect a harder-to-treat depression or the naturalistic design of the follow-up phase (with less control over dosing) is unclear.
WHAT DO THESE RESULTS MEAN FOR PRIMARY CARE PHYSICIANS?
- Measurement-based care is feasible in primary care. Primary care doctors can ensure vigorous but tolerable dosing using a self-report depression scale to monitor response, a side-effects tool to monitor tolerability, and medication adjustments at critical decision points guided by these two measures.
- Remission, ie, complete recovery from a depressive episode, rather than merely substantial improvement, is associated with a better prognosis and is the preferred goal of treatment.
- Pharmacologic differences between psychotropic medications did not translate into substantial clinical differences, although tolerability differed. These findings are consistent with a large-scale systematic evidence review recently completed by the Agency for Healthcare Research and Quality that compared the effectiveness of antidepressants.42 Given the difficulty in predicting what medication will be both efficacious for and tolerated by an individual patient, familiarity with a broad spectrum of antidepressants is prudent.
- Remission of depressive episodes will most likely require repeated trials of sufficiently sustained,vigorously dosed antidepressant medication. From treatment initiation, physicians should ensure maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- If a first treatment doesn’t work, either switching or augmenting it is a reasonable choice. Augmentation may be preferred if the patient is tolerating and receiving partial benefit from the initial medication choice. While bupropion SR and buspirone were not different as augmenters by the primary remission outcome measure, secondary measures (eg, tolerability, depressive symptom change over the course of treatment, clinician-rated Quick Inventory of Depressive Symptomatology) recommended bupropion-SR over buspirone.
- If physicians switch, either a within-class switch (eg, citalopram to sertraline) or an out-of-class switch (eg, citalopram to bupropion SR) is effective, as is a switch to a dual-action agent (eg, venlafaxine XR).
- The likelihood of improvement after two aggressive medication trials is very low and likely requires more complicated medication regimens, and the existing evidence base is quite thin. These primary care patients should likely be referred to psychiatrists for more aggressive and intensive treatment.
- For patients who present with major depressive disorder, STAR*D suggests that with persistence and aggressive yet feasible care, there is hope: after one round, approximately 30% will have a remission; after two rounds, 50%; after three rounds, 60%; and after four rounds, 70%.
- While STAR*D excluded depressed patients with bipolar disorder, a depressive episode in a patient with bipolar disorder can be difficult to distinguish from a depressive episode in a patient with major depressive disorder. Primary care physicians need to consider bipolar disorder both in patients presenting with a depressive episode and in those who fail an adequate trial.43
FUTURE CONSIDERATIONS
Subsequent STAR*D analyses will compare in greater depth outcomes in primary care vs psychiatric settings at each level of treatment. Given the greater risk of depression persistence associated with more successive levels of treatment, subsequent research will focus on ways to more successfully treat depression in the earlier stages, possibly through medication combinations earlier in treatment (somewhat analogous to a “broad-spectrum antibiotic” approach for infections).
- Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003; 289:3095–3105.
- Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997; 349:1498–1504.
- Zimmerman M, Chelminski I, Posternak MA. Generalizability of antidepressant efficacy trials: differences between depressed psychiatric outpatients who would or would not qualify for an efficacy trial. Am J Psychiatry 2005; 162:1370–1372.
- Rothwell PM. External validity of randomised controlled trials: to whom do the results of this trial apply? Lancet 2005; 365:82–93.
- Depression Guideline Panel. Depression in primary care: Volume 1, diagnosis and detection. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Frank E, Karp J, Rush A. Efficacy of treatments for major depression. Psychopharmacol Bull 1993; 29:457–475.
- Depression Guideline Panel. Depression in primary care: Volume 2, Treatment of major depression. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996; 19:179–200.
- Jarrett RB, Rush A. Short-term psychotherapy of depressive disorders: current status and future directions. Psychiatry: Interpers Biol Process 1994; 57:115–132.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depression (revision). Am J Psychiatry 2000; 157(suppl 4):1–45.
- Dunner DL, Rush AJ, Russell JM, et al. Prospective, long-term, multicenter study of the naturalistic outcomes of patients with treatment-resistant depression. J Clin Psychiatry 2006; 67:688–695.
- Fava M, Rush A, Trivedi M, et al. Background and rationale for the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study. Psychiatr Clin North Am 2003; 26:457–494.
- Gaynes B, Davis L, Rush A, Trivedi M, Fava M, Wisniewski S. The aims and design of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study. Prim Psychiatry 2005; 12:36–41.
- Rush A, Fava M, Wisniewski S, et al. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004; 25:119–142.
- Stafford RS, Ausiello JC, Misra B, Saglam D. National Patterns o fDepression Treatment in Primary Care. Prim Care Companion J Clin Psychiatry 2000; 2:211–216.
- Regier D, Narrow W, Rae D, Mandersheid R, Locke B, Goodwin F. The de facto US mental and addictive disorders service system: epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry 1993; 50:85–94.
- Pincus H, Tanielian T, Marcus S, et al. Prescribing trends in psychotropic medications: primary care, psychiatry, and other medical specialities. JAMA 1998; 279:526–531.
- Vuorilehto M, Melartin T, Isometsa E. Depressive disorders in primary care: recurrent, chronic, and co-morbid. Psychol Med 2005; 35:673–682.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–61.
- Wisniewski SR, Rush AJ, Balasubramani GK, Trivedi MH, Nierenberg AA for the STAR*D Investigators. Self-rated global measure of the frequency, intensity, and burden of side effects. J Psychiatric Pract 2006; 12:71–79.
- Rush AJ, Bernstein IH, Trivedi MH, et al. An evaluation of the Quick Inventory of Depressive Symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial report. Biol Psychiatry 2006; 59:493–501.
- Trivedi MH, Rush AJ, Gaynes BN, et al. Maximizing the adequacy of medication treatment in controlled trials and clinical practice: STAR*D measurement-based care. Neuropsychopharmacology 2007/04/04/online 2007.
- Kawamoto K, Houlihan CA, Balas EA, Lobach DF. Improving clinical practice using clinical decision support systems: a systematic review of trials to identify features critical to success. BMJ 2005; 330:765 e-pub March 14 2005.
- Lavori P, Rush A, Wisniewski S, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001; 50:792–801.
- Gaynes BN, Rush AJ, Trivedi MH, et al. Major depression symptoms in primary care and psychiatric care settings: a cross-sectional analysis. Ann Fam Med 2007; 5:126–134.
- Gaynes BN, Rush AJ, Trivedi M, et al. A direct comparison of presenting characteristics of depressed outpatients from primary vs. specialty care settings: preliminary findings from the STAR*D clinical trial. Gen Hosp Psychiatry 2005; 27:87–96.
- Sullivan PW, Valuck R, Saseen J, MacFall HM. A comparison of the direct costs and cost effectiveness of serotonin reuptake inhibitors and associated adverse drug reactions. CNS Drugs 2004; 18:911–932.
- Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163:28–40.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006; 354:1231–1242.
- Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006; 354:1243–1252.
- Wisniewski SR, Fava M, Trivedi MH, et al. Acceptability of second-step treatments to depressed outpatients: a STAR*D report. Am J Psychiatry 2007; 164:753–760.
- Thase ME, Friedman ES, Biggs MM, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry 2007; 164:739–752.
- Fava M, Rush AJ, Wisniewski SR, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry 2006; 163:1161–1172.
- Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T3 augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry 2006; 163:1519–1530.
- Bschor T, Lewitzka U, Sasse J, Adli M, Koberle U, Bauer M. Lithium augmentation in treatment-resistant depression: clinical evidence, serotonergic and endocrine mechanisms. Pharmacopsychiatry 2003; 36(suppl 3):S230–S234.
- Freeman MP, Freeman SA. Lithium: clinical considerations in internal medicine. Am J Med 2006; 119:478–481.
- Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996; 53:842–848.
- McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry 2006; 163:1531–1541.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Gartlehner G, Hansen R, Thieda P, et al. Comparative Effectiveness of Second-generation Antidepressants in the Pharmacologic Treatment of Depression. Agency for Healthcare Research and Quality. http://effectivehealthcare.ahrq.gov/reports/topic.cfm?topic=8&sid=39&rType=3. Accessed December 12, 2007.
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293:956–963.
- Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003; 289:3095–3105.
- Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997; 349:1498–1504.
- Zimmerman M, Chelminski I, Posternak MA. Generalizability of antidepressant efficacy trials: differences between depressed psychiatric outpatients who would or would not qualify for an efficacy trial. Am J Psychiatry 2005; 162:1370–1372.
- Rothwell PM. External validity of randomised controlled trials: to whom do the results of this trial apply? Lancet 2005; 365:82–93.
- Depression Guideline Panel. Depression in primary care: Volume 1, diagnosis and detection. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Frank E, Karp J, Rush A. Efficacy of treatments for major depression. Psychopharmacol Bull 1993; 29:457–475.
- Depression Guideline Panel. Depression in primary care: Volume 2, Treatment of major depression. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996; 19:179–200.
- Jarrett RB, Rush A. Short-term psychotherapy of depressive disorders: current status and future directions. Psychiatry: Interpers Biol Process 1994; 57:115–132.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depression (revision). Am J Psychiatry 2000; 157(suppl 4):1–45.
- Dunner DL, Rush AJ, Russell JM, et al. Prospective, long-term, multicenter study of the naturalistic outcomes of patients with treatment-resistant depression. J Clin Psychiatry 2006; 67:688–695.
- Fava M, Rush A, Trivedi M, et al. Background and rationale for the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study. Psychiatr Clin North Am 2003; 26:457–494.
- Gaynes B, Davis L, Rush A, Trivedi M, Fava M, Wisniewski S. The aims and design of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study. Prim Psychiatry 2005; 12:36–41.
- Rush A, Fava M, Wisniewski S, et al. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004; 25:119–142.
- Stafford RS, Ausiello JC, Misra B, Saglam D. National Patterns o fDepression Treatment in Primary Care. Prim Care Companion J Clin Psychiatry 2000; 2:211–216.
- Regier D, Narrow W, Rae D, Mandersheid R, Locke B, Goodwin F. The de facto US mental and addictive disorders service system: epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry 1993; 50:85–94.
- Pincus H, Tanielian T, Marcus S, et al. Prescribing trends in psychotropic medications: primary care, psychiatry, and other medical specialities. JAMA 1998; 279:526–531.
- Vuorilehto M, Melartin T, Isometsa E. Depressive disorders in primary care: recurrent, chronic, and co-morbid. Psychol Med 2005; 35:673–682.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–61.
- Wisniewski SR, Rush AJ, Balasubramani GK, Trivedi MH, Nierenberg AA for the STAR*D Investigators. Self-rated global measure of the frequency, intensity, and burden of side effects. J Psychiatric Pract 2006; 12:71–79.
- Rush AJ, Bernstein IH, Trivedi MH, et al. An evaluation of the Quick Inventory of Depressive Symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial report. Biol Psychiatry 2006; 59:493–501.
- Trivedi MH, Rush AJ, Gaynes BN, et al. Maximizing the adequacy of medication treatment in controlled trials and clinical practice: STAR*D measurement-based care. Neuropsychopharmacology 2007/04/04/online 2007.
- Kawamoto K, Houlihan CA, Balas EA, Lobach DF. Improving clinical practice using clinical decision support systems: a systematic review of trials to identify features critical to success. BMJ 2005; 330:765 e-pub March 14 2005.
- Lavori P, Rush A, Wisniewski S, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001; 50:792–801.
- Gaynes BN, Rush AJ, Trivedi MH, et al. Major depression symptoms in primary care and psychiatric care settings: a cross-sectional analysis. Ann Fam Med 2007; 5:126–134.
- Gaynes BN, Rush AJ, Trivedi M, et al. A direct comparison of presenting characteristics of depressed outpatients from primary vs. specialty care settings: preliminary findings from the STAR*D clinical trial. Gen Hosp Psychiatry 2005; 27:87–96.
- Sullivan PW, Valuck R, Saseen J, MacFall HM. A comparison of the direct costs and cost effectiveness of serotonin reuptake inhibitors and associated adverse drug reactions. CNS Drugs 2004; 18:911–932.
- Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163:28–40.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006; 354:1231–1242.
- Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006; 354:1243–1252.
- Wisniewski SR, Fava M, Trivedi MH, et al. Acceptability of second-step treatments to depressed outpatients: a STAR*D report. Am J Psychiatry 2007; 164:753–760.
- Thase ME, Friedman ES, Biggs MM, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry 2007; 164:739–752.
- Fava M, Rush AJ, Wisniewski SR, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry 2006; 163:1161–1172.
- Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T3 augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry 2006; 163:1519–1530.
- Bschor T, Lewitzka U, Sasse J, Adli M, Koberle U, Bauer M. Lithium augmentation in treatment-resistant depression: clinical evidence, serotonergic and endocrine mechanisms. Pharmacopsychiatry 2003; 36(suppl 3):S230–S234.
- Freeman MP, Freeman SA. Lithium: clinical considerations in internal medicine. Am J Med 2006; 119:478–481.
- Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996; 53:842–848.
- McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry 2006; 163:1531–1541.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Gartlehner G, Hansen R, Thieda P, et al. Comparative Effectiveness of Second-generation Antidepressants in the Pharmacologic Treatment of Depression. Agency for Healthcare Research and Quality. http://effectivehealthcare.ahrq.gov/reports/topic.cfm?topic=8&sid=39&rType=3. Accessed December 12, 2007.
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293:956–963.
KEY POINTS
- Remission (ie, complete relief from a depressive episode) rather than response (merely substantial improvement) should be the goal of treatment, as it is associated with a better prognosis and better function.
- Should the first treatment fail, either switching treat mentor augmenting the current treatment is reasonable.
- For most patients, remission will require repeated trials of sufficiently sustained, vigorously dosed antidepressant medication. Physicians should give maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- After two well-delivered medication trials, the likelihood of remission substantially decreases. Such patients likely require more complicated regimens. Given the thin existing database, these patients are best referred to a psychiatrist for more complex treatments.
- With persistent and vigorous treatment, most patients will enter remission: about 33% after one step, 50% after two steps, 60% after three steps, and 70% after four steps (assuming patients stay in treatment).