User login
Severe Skin Lesions Complicate Anti-TNF Therapy for IBD
Skin lesions led to discontinuation of anti–tumor necrosis factor-alpha therapy for inflammatory bowel disease in 34% of patients, reported Dr. Jean François Rahier and colleagues in the December issue of Clinical Gastroenterology and Hepatology.
Moreover, the occurrence of psoriasiform and eczematiform lesions associated with anti-TNF-alpha therapy was especially pronounced among patients with a previous history or family history of psoriasis or atopy.
Dr. Rahier of the Cliniques Universitaires de Mont-Godinne, Yvoir, Belgium, studied 85 patients with skin lesions and IBD who were seen at 35 gastroenterology centers in Belgium, France, Switzerland, and Austria.
A total of 69 patients had Crohn’s disease, 15 had ulcerative colitis, and 1 had indeterminate colitis. In all, 23 of the 85 patients were male. Data were collected using a standardized questionnaire. All cases were then reviewed simultaneously by a gastroenterologist and a dermatologist, wrote the authors (Clin. Gastroenterol. Hepatol. 2010 December [doi:10.1016/j.cgh.2010.07.022]).
Psoriasiform lesions (defined as well-delineated, scaly, erythematous plaques, as well as nail pitting, nail discoloration, and palmoplantar pustulosis) were observed in 62 patients. Their median age at the time of lesion development was 32 years, with a female: male ratio of 2:1.
"Fourteen patients (23%) had a previous history of psoriasis before treatment with any anti-TNF agent," wrote the authors. The remainder were diagnosed as having a "new onset of psoriasiform eruption," and among these, eight (17%) reported a history of familial psoriasis, two (4%) reported a history of familial atopy, and six (13%) reported a personal history of atopy.
Overall, 25 of these patients had a good response to topical treatment, whereas the remaining 37 patients had no response. In all, 16 of these nonresponders stopped anti-TNF treatment immediately because of their lesions.
The remaining 21 patients switched to another TNF antagonist; 7 of those patients discontinued this anti-TNF treatment as well. Two patients attempted a third anti-TNF agent, unsuccessfully.
That amounted to an overall discontinuation rate of 40% (25/62) because of psoriasiform skin lesions.
Eczematiform lesions (defined as xerosis and pruriginous, ill-limited plaques with erythematous or squamous macules or vesicles) were noted in 23 patients. The median age at the time of occurrence was 31 years, and the female:male ratio in this group was 6:7.
Of these patients, 10 (44%) had a history of atopy, and 13 were incident cases. Four of these incident cases (31%) had a familial history of atopy, one had a personal history of psoriasis, and one reported familial psoriasis.
A total of 16 patients (70%) had a good response to topical treatment of their lesions, and 7 (30%) had no response.
"Overall a definitive withdrawal of anti-TNF therapies due to uncontrolled [eczematiform] skin lesion was required in 4/23 (17%) patients," wrote the authors.
That equated to a total withdrawal among the entire population of 29 patients, or 34% of the study’s subjects.
The occurrence of lesions was not correlated with disease activity in the bowel. The authors noted that although the majority of lesions occurred in patients taking infliximab, compared with certolizumab or adalimumab, "this most probably reflects the earlier approval of this drug in Europe."
The authors also conceded that the true incidence of withdrawal may have been overstated in this population, since "this paper is a selected cohort of patients presenting with severe psoriasiform and eczematiform skin lesions seen in various centers."
Nevertheless, in light of the high rate of anti-TNF discontinuation shown in this study, they recommended that "all patients should be referred to a dermatologist for clinical evaluation and biopsy of skin lesions if indicated.
"Future research is needed to further explore epidemiological, clinical and fundamental aspects of this vexing paradoxical reaction," they added.
Dr. Rahier disclosed receiving lecture fees from Abbott Laboratories and Schering-Plough. Several other authors also disclosed financial relationships with pharmaceutical companies. They reported that they received no funding for the study.
Skin lesions led to discontinuation of anti–tumor necrosis factor-alpha therapy for inflammatory bowel disease in 34% of patients, reported Dr. Jean François Rahier and colleagues in the December issue of Clinical Gastroenterology and Hepatology.
Moreover, the occurrence of psoriasiform and eczematiform lesions associated with anti-TNF-alpha therapy was especially pronounced among patients with a previous history or family history of psoriasis or atopy.
Dr. Rahier of the Cliniques Universitaires de Mont-Godinne, Yvoir, Belgium, studied 85 patients with skin lesions and IBD who were seen at 35 gastroenterology centers in Belgium, France, Switzerland, and Austria.
A total of 69 patients had Crohn’s disease, 15 had ulcerative colitis, and 1 had indeterminate colitis. In all, 23 of the 85 patients were male. Data were collected using a standardized questionnaire. All cases were then reviewed simultaneously by a gastroenterologist and a dermatologist, wrote the authors (Clin. Gastroenterol. Hepatol. 2010 December [doi:10.1016/j.cgh.2010.07.022]).
Psoriasiform lesions (defined as well-delineated, scaly, erythematous plaques, as well as nail pitting, nail discoloration, and palmoplantar pustulosis) were observed in 62 patients. Their median age at the time of lesion development was 32 years, with a female: male ratio of 2:1.
"Fourteen patients (23%) had a previous history of psoriasis before treatment with any anti-TNF agent," wrote the authors. The remainder were diagnosed as having a "new onset of psoriasiform eruption," and among these, eight (17%) reported a history of familial psoriasis, two (4%) reported a history of familial atopy, and six (13%) reported a personal history of atopy.
Overall, 25 of these patients had a good response to topical treatment, whereas the remaining 37 patients had no response. In all, 16 of these nonresponders stopped anti-TNF treatment immediately because of their lesions.
The remaining 21 patients switched to another TNF antagonist; 7 of those patients discontinued this anti-TNF treatment as well. Two patients attempted a third anti-TNF agent, unsuccessfully.
That amounted to an overall discontinuation rate of 40% (25/62) because of psoriasiform skin lesions.
Eczematiform lesions (defined as xerosis and pruriginous, ill-limited plaques with erythematous or squamous macules or vesicles) were noted in 23 patients. The median age at the time of occurrence was 31 years, and the female:male ratio in this group was 6:7.
Of these patients, 10 (44%) had a history of atopy, and 13 were incident cases. Four of these incident cases (31%) had a familial history of atopy, one had a personal history of psoriasis, and one reported familial psoriasis.
A total of 16 patients (70%) had a good response to topical treatment of their lesions, and 7 (30%) had no response.
"Overall a definitive withdrawal of anti-TNF therapies due to uncontrolled [eczematiform] skin lesion was required in 4/23 (17%) patients," wrote the authors.
That equated to a total withdrawal among the entire population of 29 patients, or 34% of the study’s subjects.
The occurrence of lesions was not correlated with disease activity in the bowel. The authors noted that although the majority of lesions occurred in patients taking infliximab, compared with certolizumab or adalimumab, "this most probably reflects the earlier approval of this drug in Europe."
The authors also conceded that the true incidence of withdrawal may have been overstated in this population, since "this paper is a selected cohort of patients presenting with severe psoriasiform and eczematiform skin lesions seen in various centers."
Nevertheless, in light of the high rate of anti-TNF discontinuation shown in this study, they recommended that "all patients should be referred to a dermatologist for clinical evaluation and biopsy of skin lesions if indicated.
"Future research is needed to further explore epidemiological, clinical and fundamental aspects of this vexing paradoxical reaction," they added.
Dr. Rahier disclosed receiving lecture fees from Abbott Laboratories and Schering-Plough. Several other authors also disclosed financial relationships with pharmaceutical companies. They reported that they received no funding for the study.
Skin lesions led to discontinuation of anti–tumor necrosis factor-alpha therapy for inflammatory bowel disease in 34% of patients, reported Dr. Jean François Rahier and colleagues in the December issue of Clinical Gastroenterology and Hepatology.
Moreover, the occurrence of psoriasiform and eczematiform lesions associated with anti-TNF-alpha therapy was especially pronounced among patients with a previous history or family history of psoriasis or atopy.
Dr. Rahier of the Cliniques Universitaires de Mont-Godinne, Yvoir, Belgium, studied 85 patients with skin lesions and IBD who were seen at 35 gastroenterology centers in Belgium, France, Switzerland, and Austria.
A total of 69 patients had Crohn’s disease, 15 had ulcerative colitis, and 1 had indeterminate colitis. In all, 23 of the 85 patients were male. Data were collected using a standardized questionnaire. All cases were then reviewed simultaneously by a gastroenterologist and a dermatologist, wrote the authors (Clin. Gastroenterol. Hepatol. 2010 December [doi:10.1016/j.cgh.2010.07.022]).
Psoriasiform lesions (defined as well-delineated, scaly, erythematous plaques, as well as nail pitting, nail discoloration, and palmoplantar pustulosis) were observed in 62 patients. Their median age at the time of lesion development was 32 years, with a female: male ratio of 2:1.
"Fourteen patients (23%) had a previous history of psoriasis before treatment with any anti-TNF agent," wrote the authors. The remainder were diagnosed as having a "new onset of psoriasiform eruption," and among these, eight (17%) reported a history of familial psoriasis, two (4%) reported a history of familial atopy, and six (13%) reported a personal history of atopy.
Overall, 25 of these patients had a good response to topical treatment, whereas the remaining 37 patients had no response. In all, 16 of these nonresponders stopped anti-TNF treatment immediately because of their lesions.
The remaining 21 patients switched to another TNF antagonist; 7 of those patients discontinued this anti-TNF treatment as well. Two patients attempted a third anti-TNF agent, unsuccessfully.
That amounted to an overall discontinuation rate of 40% (25/62) because of psoriasiform skin lesions.
Eczematiform lesions (defined as xerosis and pruriginous, ill-limited plaques with erythematous or squamous macules or vesicles) were noted in 23 patients. The median age at the time of occurrence was 31 years, and the female:male ratio in this group was 6:7.
Of these patients, 10 (44%) had a history of atopy, and 13 were incident cases. Four of these incident cases (31%) had a familial history of atopy, one had a personal history of psoriasis, and one reported familial psoriasis.
A total of 16 patients (70%) had a good response to topical treatment of their lesions, and 7 (30%) had no response.
"Overall a definitive withdrawal of anti-TNF therapies due to uncontrolled [eczematiform] skin lesion was required in 4/23 (17%) patients," wrote the authors.
That equated to a total withdrawal among the entire population of 29 patients, or 34% of the study’s subjects.
The occurrence of lesions was not correlated with disease activity in the bowel. The authors noted that although the majority of lesions occurred in patients taking infliximab, compared with certolizumab or adalimumab, "this most probably reflects the earlier approval of this drug in Europe."
The authors also conceded that the true incidence of withdrawal may have been overstated in this population, since "this paper is a selected cohort of patients presenting with severe psoriasiform and eczematiform skin lesions seen in various centers."
Nevertheless, in light of the high rate of anti-TNF discontinuation shown in this study, they recommended that "all patients should be referred to a dermatologist for clinical evaluation and biopsy of skin lesions if indicated.
"Future research is needed to further explore epidemiological, clinical and fundamental aspects of this vexing paradoxical reaction," they added.
Dr. Rahier disclosed receiving lecture fees from Abbott Laboratories and Schering-Plough. Several other authors also disclosed financial relationships with pharmaceutical companies. They reported that they received no funding for the study.
Major Finding: Overall, 34% of patients taking anti-TNF-alpha drugs for inflammatory bowel disease discontinued treatment because of severe psoriasiform and eczematiform skin lesions.
Data Source: A descriptive study of 85 patients with severe skin lesions secondary to anti-TNF treatment who were seen at 35 centers in Europe.
Disclosures: Dr. Rahier disclosed receiving lecture fees from Abbott Laboratories and Schering-Plough. Several other authors also disclosed financial relationships with pharmaceutical companies. They reported that they received no funding for the study.
Briakinumab Boosts Quality of Life for Psoriasis Patients
GOTHENBURG, SWEDEN - Psoriasis patients given the investigational interleukin-12/interleukin-23 inhibitor briakinumab had significantly better scores on health-related quality of life measures than did patients given etanercept in a phase III randomized, double-blind clinical trial.
"These results further enhanced the treatment benefits of briakinumab on patients’ lives beyond the previously described clinical efficacy in significantly reducing psoriasis symptoms versus placebo and etanercept," Yanjun Bao, Ph.D., said at the annual congress of the European Academy of Dermatology and Venereology.
The 12-week study involved 347 psoriasis patients who were randomized double-blind 2:2:1 to briakinumab, etanercept at 50 mg twice weekly, or placebo. Briakinumab was dosed at 200 mg at weeks 0 and 4, then 100 mg at week 8.
Treatment with briakinumab resulted in a mean 10.3-point reduction in Dermatology Life Quality Index scores from a baseline of 12.4, which was significantly greater than the 8.1-point decrease in the etanercept group or the 3.0-point decline in the placebo arm, reported Dr. Bao of Abbott Laboratories in Abbott Park, Ill.
In addition, the briakinumab group’s mean 29.1-point improvement in the visual analog scale for psoriasis-related pain from a baseline score of 34.5 was significantly larger than the 24-point reduction with etanercept and the 6.1-point decrease with placebo. The mean score on the Short Form-36 mental component summary improved by 5.4 points in the briakinumab group from a baseline of 45.9, a significantly greater response than the 3.2-point reduction with etanercept or the 1-point decrease with placebo, she continued.
The phase III study was sponsored by Abbott.
GOTHENBURG, SWEDEN - Psoriasis patients given the investigational interleukin-12/interleukin-23 inhibitor briakinumab had significantly better scores on health-related quality of life measures than did patients given etanercept in a phase III randomized, double-blind clinical trial.
"These results further enhanced the treatment benefits of briakinumab on patients’ lives beyond the previously described clinical efficacy in significantly reducing psoriasis symptoms versus placebo and etanercept," Yanjun Bao, Ph.D., said at the annual congress of the European Academy of Dermatology and Venereology.
The 12-week study involved 347 psoriasis patients who were randomized double-blind 2:2:1 to briakinumab, etanercept at 50 mg twice weekly, or placebo. Briakinumab was dosed at 200 mg at weeks 0 and 4, then 100 mg at week 8.
Treatment with briakinumab resulted in a mean 10.3-point reduction in Dermatology Life Quality Index scores from a baseline of 12.4, which was significantly greater than the 8.1-point decrease in the etanercept group or the 3.0-point decline in the placebo arm, reported Dr. Bao of Abbott Laboratories in Abbott Park, Ill.
In addition, the briakinumab group’s mean 29.1-point improvement in the visual analog scale for psoriasis-related pain from a baseline score of 34.5 was significantly larger than the 24-point reduction with etanercept and the 6.1-point decrease with placebo. The mean score on the Short Form-36 mental component summary improved by 5.4 points in the briakinumab group from a baseline of 45.9, a significantly greater response than the 3.2-point reduction with etanercept or the 1-point decrease with placebo, she continued.
The phase III study was sponsored by Abbott.
GOTHENBURG, SWEDEN - Psoriasis patients given the investigational interleukin-12/interleukin-23 inhibitor briakinumab had significantly better scores on health-related quality of life measures than did patients given etanercept in a phase III randomized, double-blind clinical trial.
"These results further enhanced the treatment benefits of briakinumab on patients’ lives beyond the previously described clinical efficacy in significantly reducing psoriasis symptoms versus placebo and etanercept," Yanjun Bao, Ph.D., said at the annual congress of the European Academy of Dermatology and Venereology.
The 12-week study involved 347 psoriasis patients who were randomized double-blind 2:2:1 to briakinumab, etanercept at 50 mg twice weekly, or placebo. Briakinumab was dosed at 200 mg at weeks 0 and 4, then 100 mg at week 8.
Treatment with briakinumab resulted in a mean 10.3-point reduction in Dermatology Life Quality Index scores from a baseline of 12.4, which was significantly greater than the 8.1-point decrease in the etanercept group or the 3.0-point decline in the placebo arm, reported Dr. Bao of Abbott Laboratories in Abbott Park, Ill.
In addition, the briakinumab group’s mean 29.1-point improvement in the visual analog scale for psoriasis-related pain from a baseline score of 34.5 was significantly larger than the 24-point reduction with etanercept and the 6.1-point decrease with placebo. The mean score on the Short Form-36 mental component summary improved by 5.4 points in the briakinumab group from a baseline of 45.9, a significantly greater response than the 3.2-point reduction with etanercept or the 1-point decrease with placebo, she continued.
The phase III study was sponsored by Abbott.
FROM THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: Scores on the Dermatology Life Quality Index declined by a mean of 10.3points with briakinumab, 8.1 points with etanercept, and 3.0 points with placebo.
Data Source: A 12-week double-blind study of 347 psoriasis patients randomized 2:2:1 to briakinumab, etanercept at 50 mg twice weekly, or placebo. Briakinumab was dosed at 200 mg at weeks 0 and 4, then 100 mg at week 8.
Disclosures: The phase III study was sponsored by Abbott. Dr. Bao is employed by Abbott Laboratories.
Canakinumab Proves Safe, Effective for Long-Term CAPS Treatment
ATLANTA – Canakinumab was safe, well tolerated, and highly effective in the long-term treatment of cryopyrin-associated periodic syndromes in pediatric patients in an open-label phase III extension study.
Treatment of cryopyrin-associated periodic syndromes (CAPS) in 47 patients, aged 3-17 years, who were enrolled in the study was associated with a rapid and sustained clinical and biochemical remission across varying disease severity phenotypes.
Five of the children had familial cold auto-inflammatory syndrome (FCAS), 23 had Muckle-Wells syndrome (MWS), and 18 had chronic infantile neurologic, cutaneous, and articular syndrome/neonatal-onset multisystem inflammatory disease (CINCA/NOMID). One patient was found to not have a CAPS disease phenotype, reported Dr. Jasmin B. Kuemmerle-Deschner at the annual meeting of the American College of Rheumatology.
Of the 47 patients, 38 had not previously been treated with the human interleukin-1 beta monoclonal antibody, and 9 were "rolled over" from earlier phase II or III studies. Of the 38 canakinumab-naive patients, 68% achieved a complete response based on physician global assessment of disease activity and skin assessment, plus normalization of C-reactive protein (CRP) and lowering of serum amyloid A (SAA) to less than 10 mg/L for both measures. More than 80% of the canakinumab-naive patients were relapse-free after a median of 290 days of exposure to treatment, and normal CRP and SAA levels were also maintained throughout the study period in the rollover patients, said Dr. Kuemmerle-Deschner, a pediatric rheumatologist at Universitaetsklinikum Tuebingen (Germany).
The response in the treatment-naive patients was rapid, with levels of CRP and SAA decreasing within 7 days from 14.8 to 2.5 mg/L, and from 40.6 to 7.4 mg/L, respectively, she noted.
Also of note was that 8 of the 11 patients who had an incomplete response within 7 days developed a complete response by 14 days, and the remaining patients – all of whom had the most severe disease phenotype (CINCA/NOMID) – had a partial response, she said.
All patients were treated with 150 mg (or 2 mg/kg for those weighing 40 kg or less) of subcutaneous canakinumab once every 8 weeks. Patients who did not have a complete response were allowed dose adjustments, including an additional dose of 300 mg or 4 mg/kg for those weighing 40 kg or less, or more frequent dosing. Dose adjustments were required in 17 patients, including all of the patients with NOMID; those patients required mean doses of 5.8 mg/kg, compared with 5.5 mg/kg in the MWS patients, and 2.7 mg/kg in the FCAS patients.
Adverse events were generally mild or moderate and transient, and they included abdominal pain, pyrexia, headache, nasopharyngitis, and rhinitis.
"It was remarkable that 92% of patients had no injections-site reaction, and only 8% patients reported mild reaction," Dr. Kuemmerle-Deschner said.
Serious adverse events occurred in six patients, and included nephrotic syndrome, bronchitis, pneumonia, and appendicitis, which occurred in one patient each, and tonsillitis, which occurred in two patients. Only one patient withdrew from the study because of an adverse event.
The findings of this study show that canakinumab, for which earlier phase II and III studies demonstrated high efficacy for treating CAPS, also has long-term safety, tolerability, and efficacy, Dr. Kuemmerle-Deschner said.
"Canakinumab led to rapid and sustained response in the majority of patients," she said, noting that importantly, children with severe CAPS phenotypes required dose adjustment and higher overall doses to sustain remission.
Dr. Kuemmerle-Deschner disclosed that she has received research grants and consulting fees from Novartis Pharmaceuticals, the maker of canakinumab (Ilaris).
ATLANTA – Canakinumab was safe, well tolerated, and highly effective in the long-term treatment of cryopyrin-associated periodic syndromes in pediatric patients in an open-label phase III extension study.
Treatment of cryopyrin-associated periodic syndromes (CAPS) in 47 patients, aged 3-17 years, who were enrolled in the study was associated with a rapid and sustained clinical and biochemical remission across varying disease severity phenotypes.
Five of the children had familial cold auto-inflammatory syndrome (FCAS), 23 had Muckle-Wells syndrome (MWS), and 18 had chronic infantile neurologic, cutaneous, and articular syndrome/neonatal-onset multisystem inflammatory disease (CINCA/NOMID). One patient was found to not have a CAPS disease phenotype, reported Dr. Jasmin B. Kuemmerle-Deschner at the annual meeting of the American College of Rheumatology.
Of the 47 patients, 38 had not previously been treated with the human interleukin-1 beta monoclonal antibody, and 9 were "rolled over" from earlier phase II or III studies. Of the 38 canakinumab-naive patients, 68% achieved a complete response based on physician global assessment of disease activity and skin assessment, plus normalization of C-reactive protein (CRP) and lowering of serum amyloid A (SAA) to less than 10 mg/L for both measures. More than 80% of the canakinumab-naive patients were relapse-free after a median of 290 days of exposure to treatment, and normal CRP and SAA levels were also maintained throughout the study period in the rollover patients, said Dr. Kuemmerle-Deschner, a pediatric rheumatologist at Universitaetsklinikum Tuebingen (Germany).
The response in the treatment-naive patients was rapid, with levels of CRP and SAA decreasing within 7 days from 14.8 to 2.5 mg/L, and from 40.6 to 7.4 mg/L, respectively, she noted.
Also of note was that 8 of the 11 patients who had an incomplete response within 7 days developed a complete response by 14 days, and the remaining patients – all of whom had the most severe disease phenotype (CINCA/NOMID) – had a partial response, she said.
All patients were treated with 150 mg (or 2 mg/kg for those weighing 40 kg or less) of subcutaneous canakinumab once every 8 weeks. Patients who did not have a complete response were allowed dose adjustments, including an additional dose of 300 mg or 4 mg/kg for those weighing 40 kg or less, or more frequent dosing. Dose adjustments were required in 17 patients, including all of the patients with NOMID; those patients required mean doses of 5.8 mg/kg, compared with 5.5 mg/kg in the MWS patients, and 2.7 mg/kg in the FCAS patients.
Adverse events were generally mild or moderate and transient, and they included abdominal pain, pyrexia, headache, nasopharyngitis, and rhinitis.
"It was remarkable that 92% of patients had no injections-site reaction, and only 8% patients reported mild reaction," Dr. Kuemmerle-Deschner said.
Serious adverse events occurred in six patients, and included nephrotic syndrome, bronchitis, pneumonia, and appendicitis, which occurred in one patient each, and tonsillitis, which occurred in two patients. Only one patient withdrew from the study because of an adverse event.
The findings of this study show that canakinumab, for which earlier phase II and III studies demonstrated high efficacy for treating CAPS, also has long-term safety, tolerability, and efficacy, Dr. Kuemmerle-Deschner said.
"Canakinumab led to rapid and sustained response in the majority of patients," she said, noting that importantly, children with severe CAPS phenotypes required dose adjustment and higher overall doses to sustain remission.
Dr. Kuemmerle-Deschner disclosed that she has received research grants and consulting fees from Novartis Pharmaceuticals, the maker of canakinumab (Ilaris).
ATLANTA – Canakinumab was safe, well tolerated, and highly effective in the long-term treatment of cryopyrin-associated periodic syndromes in pediatric patients in an open-label phase III extension study.
Treatment of cryopyrin-associated periodic syndromes (CAPS) in 47 patients, aged 3-17 years, who were enrolled in the study was associated with a rapid and sustained clinical and biochemical remission across varying disease severity phenotypes.
Five of the children had familial cold auto-inflammatory syndrome (FCAS), 23 had Muckle-Wells syndrome (MWS), and 18 had chronic infantile neurologic, cutaneous, and articular syndrome/neonatal-onset multisystem inflammatory disease (CINCA/NOMID). One patient was found to not have a CAPS disease phenotype, reported Dr. Jasmin B. Kuemmerle-Deschner at the annual meeting of the American College of Rheumatology.
Of the 47 patients, 38 had not previously been treated with the human interleukin-1 beta monoclonal antibody, and 9 were "rolled over" from earlier phase II or III studies. Of the 38 canakinumab-naive patients, 68% achieved a complete response based on physician global assessment of disease activity and skin assessment, plus normalization of C-reactive protein (CRP) and lowering of serum amyloid A (SAA) to less than 10 mg/L for both measures. More than 80% of the canakinumab-naive patients were relapse-free after a median of 290 days of exposure to treatment, and normal CRP and SAA levels were also maintained throughout the study period in the rollover patients, said Dr. Kuemmerle-Deschner, a pediatric rheumatologist at Universitaetsklinikum Tuebingen (Germany).
The response in the treatment-naive patients was rapid, with levels of CRP and SAA decreasing within 7 days from 14.8 to 2.5 mg/L, and from 40.6 to 7.4 mg/L, respectively, she noted.
Also of note was that 8 of the 11 patients who had an incomplete response within 7 days developed a complete response by 14 days, and the remaining patients – all of whom had the most severe disease phenotype (CINCA/NOMID) – had a partial response, she said.
All patients were treated with 150 mg (or 2 mg/kg for those weighing 40 kg or less) of subcutaneous canakinumab once every 8 weeks. Patients who did not have a complete response were allowed dose adjustments, including an additional dose of 300 mg or 4 mg/kg for those weighing 40 kg or less, or more frequent dosing. Dose adjustments were required in 17 patients, including all of the patients with NOMID; those patients required mean doses of 5.8 mg/kg, compared with 5.5 mg/kg in the MWS patients, and 2.7 mg/kg in the FCAS patients.
Adverse events were generally mild or moderate and transient, and they included abdominal pain, pyrexia, headache, nasopharyngitis, and rhinitis.
"It was remarkable that 92% of patients had no injections-site reaction, and only 8% patients reported mild reaction," Dr. Kuemmerle-Deschner said.
Serious adverse events occurred in six patients, and included nephrotic syndrome, bronchitis, pneumonia, and appendicitis, which occurred in one patient each, and tonsillitis, which occurred in two patients. Only one patient withdrew from the study because of an adverse event.
The findings of this study show that canakinumab, for which earlier phase II and III studies demonstrated high efficacy for treating CAPS, also has long-term safety, tolerability, and efficacy, Dr. Kuemmerle-Deschner said.
"Canakinumab led to rapid and sustained response in the majority of patients," she said, noting that importantly, children with severe CAPS phenotypes required dose adjustment and higher overall doses to sustain remission.
Dr. Kuemmerle-Deschner disclosed that she has received research grants and consulting fees from Novartis Pharmaceuticals, the maker of canakinumab (Ilaris).
FROM THE ANNUAL MEETING OF THE AMERICAN COLLEGE OF RHEUMATOLOGY
Major Finding: Of 38 canakinumab-naive patients, 68% achieved a complete response based on physician global assessment of disease activity and skin assessment, and normalization of C-reactive protein and serum amyloid A. More than 80% of the canakinumab-naive patients were relapse-free after a median of 290 days of exposure to treatment, and normal levels were also maintained throughout the study period in the rollover patients.
Data Source: From an open-label, single treatment arm, phase III extension study of canakinumab for CAPS.
Disclosures: Dr. Kuemmerle-Deschner disclosed that she has received research grants and consulting fees from Novartis Pharmaceuticals, the maker of canakinumab (Ilaris).
Juvenile Localized Scleroderma: Fewer Flares With Methotrexate
ATLANTA – Methotrexate was an effective and well-tolerated treatment for juvenile localized scleroderma in a randomized, double-blind, placebo-controlled trial involving 70 patients with active disease.
At the end of the 12-month study, 31 of 46 patients randomized to receive methotrexate had responded to treatment and were flare-free, compared with 7 of 24 patients given placebo, Dr. Francesco Zulian reported at the annual meeting of the American College of Rheumatology.
Methotrexate patients received a weekly oral dose of 15 mg/m2 (maximum, 20 mg) for 12 months or until flare of disease. Patients in the placebo group received a placebo administered in the same fashion for 12 months or to flare. Patients in both groups received prednisone at a dose of 1 mg/kg per day, with a maximum dose of 50 mg, for 3 months. They were then tapered to 0 over 1 month. During the initial 3 months of the study, treatment response was comparable for the two groups. Differences in response rates began to emerge after that point, said Dr. Zulian of the University of Padua (Italy).
Patients' lesions were evaluated using a computerized scoring system, and changes were quantified using a skin score rate. Active lesions were monitored based on clinical examination and serial thermography. The mean skin score rates decreased significantly from 1.0 at baseline to 0.79 in the patients on methotrexate, but did not decrease in the patients given placebo. The mean target lesion temperature on thermography decreased by 44% in the methotrexate group and by 12% in the placebo group, a significant difference, he said.
Adverse events occurred in 26 of 46 patients in the methotrexate group, and in 11 of 24 in the placebo group. In the methotrexate group, adverse events included alopecia, nausea, headache, fatigue, hepatotoxicity, weight gain, and striae rubra. The only adverse events in the placebo group were weight gain and striae rubra.
Interestingly, weight gain of more than 5% of body weight occurred in 11% of the methotrexate patients and 42% of the placebo patients, Dr. Zulian said, noting that this may suggest there is some "biological or pathophysiological link" between prednisone and methotrexate that tempers this steroid-related side effect.
None of the side effects was severe enough to stop treatment, but there was a high drop-out rate – due to relapse or lack of response – in both groups; 31 of 46 patients in the methotrexate group, and 7 of 24 in the placebo group, completed the study. At the end of the 12-month study, 17 of 24 patients randomized to placebo had experienced a relapse, compared with 15 of 46 patients randomized to the methotrexate group.
Patients included in the study were aged 6-17 years with active localized scleroderma of linear, generalized, or deep subtypes with onset before age 16. The treatment and placebo groups were similar in regard to clinical and immunological features, as well as mean disease duration of about 2 years, and the absence of immunosuppressive treatment in the 6 months prior to enrollment.
Many treatments have been tried in patients with juvenile localized scleroderma, and methotrexate has emerged as one of the most frequently used drugs for this condition.
"The findings of this study show that a combination of methotrexate and prednisone is of benefit and is well tolerated as treatment for severe course juvenile localized scleroderma," Dr. Zulian said.
Prior studies have shown efficacy ranging from 74% to 93%, but the findings of those studies have been based mostly on clinical judgment, while the current study utilized more objective evaluation criteria, including thermography, and skin score rates calculated based on computerized skin scoring, he said.
"The inclusion of standardized objective parameters represents an innovative way to assess the disease and compare progression in future clinical trials," he added.
Dr. Zulian said he had no relevant financial disclosures.
ATLANTA – Methotrexate was an effective and well-tolerated treatment for juvenile localized scleroderma in a randomized, double-blind, placebo-controlled trial involving 70 patients with active disease.
At the end of the 12-month study, 31 of 46 patients randomized to receive methotrexate had responded to treatment and were flare-free, compared with 7 of 24 patients given placebo, Dr. Francesco Zulian reported at the annual meeting of the American College of Rheumatology.
Methotrexate patients received a weekly oral dose of 15 mg/m2 (maximum, 20 mg) for 12 months or until flare of disease. Patients in the placebo group received a placebo administered in the same fashion for 12 months or to flare. Patients in both groups received prednisone at a dose of 1 mg/kg per day, with a maximum dose of 50 mg, for 3 months. They were then tapered to 0 over 1 month. During the initial 3 months of the study, treatment response was comparable for the two groups. Differences in response rates began to emerge after that point, said Dr. Zulian of the University of Padua (Italy).
Patients' lesions were evaluated using a computerized scoring system, and changes were quantified using a skin score rate. Active lesions were monitored based on clinical examination and serial thermography. The mean skin score rates decreased significantly from 1.0 at baseline to 0.79 in the patients on methotrexate, but did not decrease in the patients given placebo. The mean target lesion temperature on thermography decreased by 44% in the methotrexate group and by 12% in the placebo group, a significant difference, he said.
Adverse events occurred in 26 of 46 patients in the methotrexate group, and in 11 of 24 in the placebo group. In the methotrexate group, adverse events included alopecia, nausea, headache, fatigue, hepatotoxicity, weight gain, and striae rubra. The only adverse events in the placebo group were weight gain and striae rubra.
Interestingly, weight gain of more than 5% of body weight occurred in 11% of the methotrexate patients and 42% of the placebo patients, Dr. Zulian said, noting that this may suggest there is some "biological or pathophysiological link" between prednisone and methotrexate that tempers this steroid-related side effect.
None of the side effects was severe enough to stop treatment, but there was a high drop-out rate – due to relapse or lack of response – in both groups; 31 of 46 patients in the methotrexate group, and 7 of 24 in the placebo group, completed the study. At the end of the 12-month study, 17 of 24 patients randomized to placebo had experienced a relapse, compared with 15 of 46 patients randomized to the methotrexate group.
Patients included in the study were aged 6-17 years with active localized scleroderma of linear, generalized, or deep subtypes with onset before age 16. The treatment and placebo groups were similar in regard to clinical and immunological features, as well as mean disease duration of about 2 years, and the absence of immunosuppressive treatment in the 6 months prior to enrollment.
Many treatments have been tried in patients with juvenile localized scleroderma, and methotrexate has emerged as one of the most frequently used drugs for this condition.
"The findings of this study show that a combination of methotrexate and prednisone is of benefit and is well tolerated as treatment for severe course juvenile localized scleroderma," Dr. Zulian said.
Prior studies have shown efficacy ranging from 74% to 93%, but the findings of those studies have been based mostly on clinical judgment, while the current study utilized more objective evaluation criteria, including thermography, and skin score rates calculated based on computerized skin scoring, he said.
"The inclusion of standardized objective parameters represents an innovative way to assess the disease and compare progression in future clinical trials," he added.
Dr. Zulian said he had no relevant financial disclosures.
ATLANTA – Methotrexate was an effective and well-tolerated treatment for juvenile localized scleroderma in a randomized, double-blind, placebo-controlled trial involving 70 patients with active disease.
At the end of the 12-month study, 31 of 46 patients randomized to receive methotrexate had responded to treatment and were flare-free, compared with 7 of 24 patients given placebo, Dr. Francesco Zulian reported at the annual meeting of the American College of Rheumatology.
Methotrexate patients received a weekly oral dose of 15 mg/m2 (maximum, 20 mg) for 12 months or until flare of disease. Patients in the placebo group received a placebo administered in the same fashion for 12 months or to flare. Patients in both groups received prednisone at a dose of 1 mg/kg per day, with a maximum dose of 50 mg, for 3 months. They were then tapered to 0 over 1 month. During the initial 3 months of the study, treatment response was comparable for the two groups. Differences in response rates began to emerge after that point, said Dr. Zulian of the University of Padua (Italy).
Patients' lesions were evaluated using a computerized scoring system, and changes were quantified using a skin score rate. Active lesions were monitored based on clinical examination and serial thermography. The mean skin score rates decreased significantly from 1.0 at baseline to 0.79 in the patients on methotrexate, but did not decrease in the patients given placebo. The mean target lesion temperature on thermography decreased by 44% in the methotrexate group and by 12% in the placebo group, a significant difference, he said.
Adverse events occurred in 26 of 46 patients in the methotrexate group, and in 11 of 24 in the placebo group. In the methotrexate group, adverse events included alopecia, nausea, headache, fatigue, hepatotoxicity, weight gain, and striae rubra. The only adverse events in the placebo group were weight gain and striae rubra.
Interestingly, weight gain of more than 5% of body weight occurred in 11% of the methotrexate patients and 42% of the placebo patients, Dr. Zulian said, noting that this may suggest there is some "biological or pathophysiological link" between prednisone and methotrexate that tempers this steroid-related side effect.
None of the side effects was severe enough to stop treatment, but there was a high drop-out rate – due to relapse or lack of response – in both groups; 31 of 46 patients in the methotrexate group, and 7 of 24 in the placebo group, completed the study. At the end of the 12-month study, 17 of 24 patients randomized to placebo had experienced a relapse, compared with 15 of 46 patients randomized to the methotrexate group.
Patients included in the study were aged 6-17 years with active localized scleroderma of linear, generalized, or deep subtypes with onset before age 16. The treatment and placebo groups were similar in regard to clinical and immunological features, as well as mean disease duration of about 2 years, and the absence of immunosuppressive treatment in the 6 months prior to enrollment.
Many treatments have been tried in patients with juvenile localized scleroderma, and methotrexate has emerged as one of the most frequently used drugs for this condition.
"The findings of this study show that a combination of methotrexate and prednisone is of benefit and is well tolerated as treatment for severe course juvenile localized scleroderma," Dr. Zulian said.
Prior studies have shown efficacy ranging from 74% to 93%, but the findings of those studies have been based mostly on clinical judgment, while the current study utilized more objective evaluation criteria, including thermography, and skin score rates calculated based on computerized skin scoring, he said.
"The inclusion of standardized objective parameters represents an innovative way to assess the disease and compare progression in future clinical trials," he added.
Dr. Zulian said he had no relevant financial disclosures.
FROM THE ANNUAL MEETING OF THE AMERICAN COLLEGE OF RHEUMATOLOGY
Major Finding: At the end of the 12-month study, 17 of 24 patients randomized to receive placebo and prednisone experienced a relapse, compared with 15 of 46 patients randomized to methotrexate and prednisone.
Data Source: A randomized, double-blind, placebo-controlled trial of 70 children with juvenile localized scleroderma.
Disclosures: Dr. Zulian said he had no relevant financial disclosures.
Coal Tar Making a Psoriasis Comeback
LAS VEGAS – Topical tar therapy for psoriasis may be making a comeback thanks to more user-friendly formulations.
That trend is happening in Europe and may be replicated in the United States, Dr. Linda Stein Gold said at a dermatology seminar sponsored by Skin Disease Education Foundation (SDEF).
"Topical therapy is the bread and butter of psoriasis treatment," and coal tar has been used for centuries to control the symptoms of plaque psoriasis, said Dr. Stein Gold, director of dermatology research at Henry Ford Hospital, Detroit.
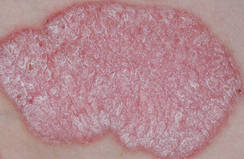
The Goeckerman regimen, a combination of topical tar and ultraviolet light therapy in use since the 1920s, proved "exceptionally" effective and durable, she noted, with an average time of 18 days to 90% clearing of psoriatic lesions and 90% of patients remaining clear for up to 8 months.
But tar therapy was time consuming, a logistical hassle, and aesthetically unpleasing, and its use declined in recent decades with the advent of systemic biologic medications. "People weren't using tar before because it's messy and it smelled. Now we have some better options," Dr. Stein Gold said.
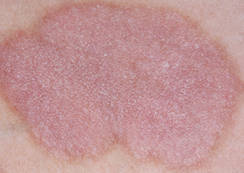
One of the newer topical solutions is a transparent gel of 15% liquor carbonis distillate (LCD), the equivalent of 2.3% coal tar (Psorent, NeoStrata Co.). "It doesn’t discolor bleached hair" when used for scalp psoriasis, it is quick-drying, and it comes in bottles with "dab-on" applicators, she said.
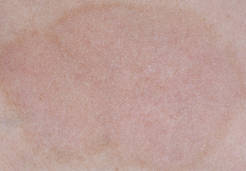
In a controlled comparison with calcipotriol cream in 12 weeks of treatment of 60 adults with moderate plaque psoriasis, the LCD solution was more effective and led to fewer relapses 6 weeks after treatment. Among 55 patients with complete data, mean Psoriasis Area Severity Index (PASI) scores improved by 58% in the LCD solution group and 37% in the calcipotriol group, a significant difference (J. Am. Acad. Dermatol. 2009;60:Ab174 [doi: 10.1016/j.jaad.2008.11.757]).
A 75% improvement in PASI scores was seen in 11 of 27 (41%) of the LCD solution group and none of the 28 patients on calcipotriol. A 50% improvement in PASI scores was seen in 18 of 27 (67%) in the LCD group and 10 of 28 (36%) in the calcipotriol group. These differences between groups were statistically significant.
Among 42 patients with Physician Global Assessment (PGA) scores 6 weeks after treatment, the PGA scores worsened to baseline in 5 of 22 patients (23%) in the LCD solution group and in 14 of 20 patients (70%) in the calcipotriol group, again a significant difference.
A separate study compared the LCD solution in combination with UVB therapy on one side of the body with UVB light therapy alone on the other side of the body in 12 patients in 4 weeks of therapy. "Very quickly, the combo therapy gets more rapid and complete efficacy compared with UVB alone," said Dr. Stein Gold (J. Drugs. Dermatol. 2009;8 [4]:351-7).
Another relatively user-friendly product is an over-the-counter 2% coal tar formulation in a foam base (Scytera, Promius Pharma). The foam is "a much more cosmetically elegant" treatment compared with older tar therapies, she said. It spreads easily, dries quickly, and has an acceptable fragrance, Dr. Stein Gold added.
In a randomized, observer-blind study of 38 patients with chronic plaque-type psoriasis, two lesions on each patient were treated for 8 weeks with a 1% coal tar foam or calcipotriol cream. The treatments appeared to be comparably effective. More patients reported itching, unpleasant odor or staining with the tar foam than with calcipotriol cream, but the foam is considerably less expensive, the investigators noted (Br. J. Dermatol. 2003;149:350-53).
"Tar is something we probably should look at again," Dr. Stein Gold said.
Other useful topical therapies include corticosteroids, she said. While studies with objective measures of atrophy have proved that potent topical corticosteroids do lead to thinning of the skin over time, often this effect is not clinically noticeable, and it reverses over time once treatment is stopped.
Topical vitamin D is complementary to topical steroids for psoriasis therapy and helps counteract some of the side effects of topical steroids on skin.
Dr. Stein Gold said she has had financial associations with Leo Pharma, Medicis Pharmaceutical Corp., Stiefel Laboratories, Galderma, and Novartis. SDEF and this news organization are owned by Elsevier.
LAS VEGAS – Topical tar therapy for psoriasis may be making a comeback thanks to more user-friendly formulations.
That trend is happening in Europe and may be replicated in the United States, Dr. Linda Stein Gold said at a dermatology seminar sponsored by Skin Disease Education Foundation (SDEF).
"Topical therapy is the bread and butter of psoriasis treatment," and coal tar has been used for centuries to control the symptoms of plaque psoriasis, said Dr. Stein Gold, director of dermatology research at Henry Ford Hospital, Detroit.
The Goeckerman regimen, a combination of topical tar and ultraviolet light therapy in use since the 1920s, proved "exceptionally" effective and durable, she noted, with an average time of 18 days to 90% clearing of psoriatic lesions and 90% of patients remaining clear for up to 8 months.
But tar therapy was time consuming, a logistical hassle, and aesthetically unpleasing, and its use declined in recent decades with the advent of systemic biologic medications. "People weren't using tar before because it's messy and it smelled. Now we have some better options," Dr. Stein Gold said.
One of the newer topical solutions is a transparent gel of 15% liquor carbonis distillate (LCD), the equivalent of 2.3% coal tar (Psorent, NeoStrata Co.). "It doesn’t discolor bleached hair" when used for scalp psoriasis, it is quick-drying, and it comes in bottles with "dab-on" applicators, she said.
In a controlled comparison with calcipotriol cream in 12 weeks of treatment of 60 adults with moderate plaque psoriasis, the LCD solution was more effective and led to fewer relapses 6 weeks after treatment. Among 55 patients with complete data, mean Psoriasis Area Severity Index (PASI) scores improved by 58% in the LCD solution group and 37% in the calcipotriol group, a significant difference (J. Am. Acad. Dermatol. 2009;60:Ab174 [doi: 10.1016/j.jaad.2008.11.757]).
A 75% improvement in PASI scores was seen in 11 of 27 (41%) of the LCD solution group and none of the 28 patients on calcipotriol. A 50% improvement in PASI scores was seen in 18 of 27 (67%) in the LCD group and 10 of 28 (36%) in the calcipotriol group. These differences between groups were statistically significant.
Among 42 patients with Physician Global Assessment (PGA) scores 6 weeks after treatment, the PGA scores worsened to baseline in 5 of 22 patients (23%) in the LCD solution group and in 14 of 20 patients (70%) in the calcipotriol group, again a significant difference.
A separate study compared the LCD solution in combination with UVB therapy on one side of the body with UVB light therapy alone on the other side of the body in 12 patients in 4 weeks of therapy. "Very quickly, the combo therapy gets more rapid and complete efficacy compared with UVB alone," said Dr. Stein Gold (J. Drugs. Dermatol. 2009;8 [4]:351-7).
Another relatively user-friendly product is an over-the-counter 2% coal tar formulation in a foam base (Scytera, Promius Pharma). The foam is "a much more cosmetically elegant" treatment compared with older tar therapies, she said. It spreads easily, dries quickly, and has an acceptable fragrance, Dr. Stein Gold added.
In a randomized, observer-blind study of 38 patients with chronic plaque-type psoriasis, two lesions on each patient were treated for 8 weeks with a 1% coal tar foam or calcipotriol cream. The treatments appeared to be comparably effective. More patients reported itching, unpleasant odor or staining with the tar foam than with calcipotriol cream, but the foam is considerably less expensive, the investigators noted (Br. J. Dermatol. 2003;149:350-53).
"Tar is something we probably should look at again," Dr. Stein Gold said.
Other useful topical therapies include corticosteroids, she said. While studies with objective measures of atrophy have proved that potent topical corticosteroids do lead to thinning of the skin over time, often this effect is not clinically noticeable, and it reverses over time once treatment is stopped.
Topical vitamin D is complementary to topical steroids for psoriasis therapy and helps counteract some of the side effects of topical steroids on skin.
Dr. Stein Gold said she has had financial associations with Leo Pharma, Medicis Pharmaceutical Corp., Stiefel Laboratories, Galderma, and Novartis. SDEF and this news organization are owned by Elsevier.
LAS VEGAS – Topical tar therapy for psoriasis may be making a comeback thanks to more user-friendly formulations.
That trend is happening in Europe and may be replicated in the United States, Dr. Linda Stein Gold said at a dermatology seminar sponsored by Skin Disease Education Foundation (SDEF).
"Topical therapy is the bread and butter of psoriasis treatment," and coal tar has been used for centuries to control the symptoms of plaque psoriasis, said Dr. Stein Gold, director of dermatology research at Henry Ford Hospital, Detroit.
The Goeckerman regimen, a combination of topical tar and ultraviolet light therapy in use since the 1920s, proved "exceptionally" effective and durable, she noted, with an average time of 18 days to 90% clearing of psoriatic lesions and 90% of patients remaining clear for up to 8 months.
But tar therapy was time consuming, a logistical hassle, and aesthetically unpleasing, and its use declined in recent decades with the advent of systemic biologic medications. "People weren't using tar before because it's messy and it smelled. Now we have some better options," Dr. Stein Gold said.
One of the newer topical solutions is a transparent gel of 15% liquor carbonis distillate (LCD), the equivalent of 2.3% coal tar (Psorent, NeoStrata Co.). "It doesn’t discolor bleached hair" when used for scalp psoriasis, it is quick-drying, and it comes in bottles with "dab-on" applicators, she said.
In a controlled comparison with calcipotriol cream in 12 weeks of treatment of 60 adults with moderate plaque psoriasis, the LCD solution was more effective and led to fewer relapses 6 weeks after treatment. Among 55 patients with complete data, mean Psoriasis Area Severity Index (PASI) scores improved by 58% in the LCD solution group and 37% in the calcipotriol group, a significant difference (J. Am. Acad. Dermatol. 2009;60:Ab174 [doi: 10.1016/j.jaad.2008.11.757]).
A 75% improvement in PASI scores was seen in 11 of 27 (41%) of the LCD solution group and none of the 28 patients on calcipotriol. A 50% improvement in PASI scores was seen in 18 of 27 (67%) in the LCD group and 10 of 28 (36%) in the calcipotriol group. These differences between groups were statistically significant.
Among 42 patients with Physician Global Assessment (PGA) scores 6 weeks after treatment, the PGA scores worsened to baseline in 5 of 22 patients (23%) in the LCD solution group and in 14 of 20 patients (70%) in the calcipotriol group, again a significant difference.
A separate study compared the LCD solution in combination with UVB therapy on one side of the body with UVB light therapy alone on the other side of the body in 12 patients in 4 weeks of therapy. "Very quickly, the combo therapy gets more rapid and complete efficacy compared with UVB alone," said Dr. Stein Gold (J. Drugs. Dermatol. 2009;8 [4]:351-7).
Another relatively user-friendly product is an over-the-counter 2% coal tar formulation in a foam base (Scytera, Promius Pharma). The foam is "a much more cosmetically elegant" treatment compared with older tar therapies, she said. It spreads easily, dries quickly, and has an acceptable fragrance, Dr. Stein Gold added.
In a randomized, observer-blind study of 38 patients with chronic plaque-type psoriasis, two lesions on each patient were treated for 8 weeks with a 1% coal tar foam or calcipotriol cream. The treatments appeared to be comparably effective. More patients reported itching, unpleasant odor or staining with the tar foam than with calcipotriol cream, but the foam is considerably less expensive, the investigators noted (Br. J. Dermatol. 2003;149:350-53).
"Tar is something we probably should look at again," Dr. Stein Gold said.
Other useful topical therapies include corticosteroids, she said. While studies with objective measures of atrophy have proved that potent topical corticosteroids do lead to thinning of the skin over time, often this effect is not clinically noticeable, and it reverses over time once treatment is stopped.
Topical vitamin D is complementary to topical steroids for psoriasis therapy and helps counteract some of the side effects of topical steroids on skin.
Dr. Stein Gold said she has had financial associations with Leo Pharma, Medicis Pharmaceutical Corp., Stiefel Laboratories, Galderma, and Novartis. SDEF and this news organization are owned by Elsevier.
EXPERT ANALYSIS FROM A DERMATOLOGY SEMINAR SPONSORED BY SKIN DISEASE EDUCATION FOUNDATION
Smoking Cessation Enhances Antimalarial Response in Cutaneous Lupus
SANTA MONICA, Calif. – Quitting smoking is the most important thing patients with cutaneous lupus erythematosus can do to increase their clinical response to antimalarials, according to Dr. Jeffrey P. Callen.
Smokers are already at risk of having more severe disease than their nonsmoking CLE counterparts, Dr. Callen noted at a rheumatology seminar sponsored by Skin Disease Education Foundation (SDEF) and the University of Louisville.

Antimalarial drugs are the basis of therapy for cutaneous lupus erythematosus (CLE). The need for maximizing treatment response has recently become clearer. In the past, certain patients with CLE were considered to be at low risk for progression to systemic LE (SLE). These included patients with fixed lesions with a potential for atrophy. However, data now show that these patients are as likely as others with CLE to progress to SLE.
The data come from a population-based study of inhabitants of Rochester, Minn. The researchers found that the incidence of CLE and SLE were equal. But of the 156 patients with CLE, 12% (19 patients) progressed to SLE within a mean of 8.2 years from the time of their CLE diagnosis, said Dr. Callen, professor of medicine (dermatology) and chief of the division of dermatology at the University of Louisville (Ky).
Of particular interest was the subtype of SLE that the researchers found in the 19 patients who progressed to SLE – there were 9 cases of the localized discoid subtype of CLE, 4 cases of the generalized discoid CLE subtype, 2 with the panniculitis CLE subtype, and 4 with the psoriasiform subtype of CLE (Arch. Dermatol. 2009;145:249-53).

These findings "give us reason to treat these patients more aggressively than we might have done," said Dr. Callen, and part of that more aggressive treatment is to get patients who smoke to stop.
Another tool is to get patients to use sunblock. "This is a photosensitive disease. It’s photodistributed. It’s photoexacerbated. We can reproduce it with phototesting. So sunscreens are important, but even more so is photoprotection. Patients need to change their behavior and wear photoprotective clothing. Whenever we do that, patients are going to have vitamin D deficiency. Data have come out recently that lupus patients in general have vitamin D deficiency. So we need to address that and [ensure that they] get adequate vitamin D. No one has done a study to see if [supplementation with vitamin D] makes a difference," Dr. Callen noted.
Some patients also respond to topical corticosteroids, selected according to the lesion and the site that are being treated. Topical calcineurin inhibitors have been used off label.
But first-line therapy is the antimalarial hydroxychloroquine or chloroquine. "To me, systemic corticosteroid therapy is not a therapy for cutaneous lupus," Dr. Callen said.
Data from one study suggest that giving patients hydroxychloroquine delays the time from onset of CLE to progression to SLE (Lupus 2007;16:401-9). "To me, that means we need to be treating patients with antimalarials earlier than we do," he said.
If those therapies do not work, "we may combine hydroxychloroquine or chloroquine with quinacrine, but we do not combine hydroxychloroquine and chloroquine. I am a little bit fearful of exacerbating potential ocular toxicity."
Or one can use a second-line therapy that ranges from immunosuppressive therapy to thalidomide.
"We ask first if the patients are taking drugs that may exacerbate the disease, especially in the setting of subacute cutaneous LE. And stop those drugs if at all possible. Is the patient smoking? Try to get them to stop," he said.
SDEF and this news organization are owned by Elsevier. Dr. Callen reported that he does not have any relevant financial relationships with any commercial interests.
SANTA MONICA, Calif. – Quitting smoking is the most important thing patients with cutaneous lupus erythematosus can do to increase their clinical response to antimalarials, according to Dr. Jeffrey P. Callen.
Smokers are already at risk of having more severe disease than their nonsmoking CLE counterparts, Dr. Callen noted at a rheumatology seminar sponsored by Skin Disease Education Foundation (SDEF) and the University of Louisville.
Antimalarial drugs are the basis of therapy for cutaneous lupus erythematosus (CLE). The need for maximizing treatment response has recently become clearer. In the past, certain patients with CLE were considered to be at low risk for progression to systemic LE (SLE). These included patients with fixed lesions with a potential for atrophy. However, data now show that these patients are as likely as others with CLE to progress to SLE.
The data come from a population-based study of inhabitants of Rochester, Minn. The researchers found that the incidence of CLE and SLE were equal. But of the 156 patients with CLE, 12% (19 patients) progressed to SLE within a mean of 8.2 years from the time of their CLE diagnosis, said Dr. Callen, professor of medicine (dermatology) and chief of the division of dermatology at the University of Louisville (Ky).
Of particular interest was the subtype of SLE that the researchers found in the 19 patients who progressed to SLE – there were 9 cases of the localized discoid subtype of CLE, 4 cases of the generalized discoid CLE subtype, 2 with the panniculitis CLE subtype, and 4 with the psoriasiform subtype of CLE (Arch. Dermatol. 2009;145:249-53).
These findings "give us reason to treat these patients more aggressively than we might have done," said Dr. Callen, and part of that more aggressive treatment is to get patients who smoke to stop.
Another tool is to get patients to use sunblock. "This is a photosensitive disease. It’s photodistributed. It’s photoexacerbated. We can reproduce it with phototesting. So sunscreens are important, but even more so is photoprotection. Patients need to change their behavior and wear photoprotective clothing. Whenever we do that, patients are going to have vitamin D deficiency. Data have come out recently that lupus patients in general have vitamin D deficiency. So we need to address that and [ensure that they] get adequate vitamin D. No one has done a study to see if [supplementation with vitamin D] makes a difference," Dr. Callen noted.
Some patients also respond to topical corticosteroids, selected according to the lesion and the site that are being treated. Topical calcineurin inhibitors have been used off label.
But first-line therapy is the antimalarial hydroxychloroquine or chloroquine. "To me, systemic corticosteroid therapy is not a therapy for cutaneous lupus," Dr. Callen said.
Data from one study suggest that giving patients hydroxychloroquine delays the time from onset of CLE to progression to SLE (Lupus 2007;16:401-9). "To me, that means we need to be treating patients with antimalarials earlier than we do," he said.
If those therapies do not work, "we may combine hydroxychloroquine or chloroquine with quinacrine, but we do not combine hydroxychloroquine and chloroquine. I am a little bit fearful of exacerbating potential ocular toxicity."
Or one can use a second-line therapy that ranges from immunosuppressive therapy to thalidomide.
"We ask first if the patients are taking drugs that may exacerbate the disease, especially in the setting of subacute cutaneous LE. And stop those drugs if at all possible. Is the patient smoking? Try to get them to stop," he said.
SDEF and this news organization are owned by Elsevier. Dr. Callen reported that he does not have any relevant financial relationships with any commercial interests.
SANTA MONICA, Calif. – Quitting smoking is the most important thing patients with cutaneous lupus erythematosus can do to increase their clinical response to antimalarials, according to Dr. Jeffrey P. Callen.
Smokers are already at risk of having more severe disease than their nonsmoking CLE counterparts, Dr. Callen noted at a rheumatology seminar sponsored by Skin Disease Education Foundation (SDEF) and the University of Louisville.
Antimalarial drugs are the basis of therapy for cutaneous lupus erythematosus (CLE). The need for maximizing treatment response has recently become clearer. In the past, certain patients with CLE were considered to be at low risk for progression to systemic LE (SLE). These included patients with fixed lesions with a potential for atrophy. However, data now show that these patients are as likely as others with CLE to progress to SLE.
The data come from a population-based study of inhabitants of Rochester, Minn. The researchers found that the incidence of CLE and SLE were equal. But of the 156 patients with CLE, 12% (19 patients) progressed to SLE within a mean of 8.2 years from the time of their CLE diagnosis, said Dr. Callen, professor of medicine (dermatology) and chief of the division of dermatology at the University of Louisville (Ky).
Of particular interest was the subtype of SLE that the researchers found in the 19 patients who progressed to SLE – there were 9 cases of the localized discoid subtype of CLE, 4 cases of the generalized discoid CLE subtype, 2 with the panniculitis CLE subtype, and 4 with the psoriasiform subtype of CLE (Arch. Dermatol. 2009;145:249-53).
These findings "give us reason to treat these patients more aggressively than we might have done," said Dr. Callen, and part of that more aggressive treatment is to get patients who smoke to stop.
Another tool is to get patients to use sunblock. "This is a photosensitive disease. It’s photodistributed. It’s photoexacerbated. We can reproduce it with phototesting. So sunscreens are important, but even more so is photoprotection. Patients need to change their behavior and wear photoprotective clothing. Whenever we do that, patients are going to have vitamin D deficiency. Data have come out recently that lupus patients in general have vitamin D deficiency. So we need to address that and [ensure that they] get adequate vitamin D. No one has done a study to see if [supplementation with vitamin D] makes a difference," Dr. Callen noted.
Some patients also respond to topical corticosteroids, selected according to the lesion and the site that are being treated. Topical calcineurin inhibitors have been used off label.
But first-line therapy is the antimalarial hydroxychloroquine or chloroquine. "To me, systemic corticosteroid therapy is not a therapy for cutaneous lupus," Dr. Callen said.
Data from one study suggest that giving patients hydroxychloroquine delays the time from onset of CLE to progression to SLE (Lupus 2007;16:401-9). "To me, that means we need to be treating patients with antimalarials earlier than we do," he said.
If those therapies do not work, "we may combine hydroxychloroquine or chloroquine with quinacrine, but we do not combine hydroxychloroquine and chloroquine. I am a little bit fearful of exacerbating potential ocular toxicity."
Or one can use a second-line therapy that ranges from immunosuppressive therapy to thalidomide.
"We ask first if the patients are taking drugs that may exacerbate the disease, especially in the setting of subacute cutaneous LE. And stop those drugs if at all possible. Is the patient smoking? Try to get them to stop," he said.
SDEF and this news organization are owned by Elsevier. Dr. Callen reported that he does not have any relevant financial relationships with any commercial interests.
EXPERT ANALYSIS FROM A RHEUMATOLOGY SEMINAR
Get an Early Start on Biologics to Treat IBD
SANTA MONICA, Calif. – Gastroenterologists seem to be taking a page from the rheumatologists’ playbook and are starting patients with inflammatory bowel disease on a biologic agent much sooner after diagnosis than has been standard practice.
By doing so, they hope to improve the natural history of the disease, just as rheumatologists have done in rheumatoid arthritis patients, according to Dr. Russell D. Cohen, who spoke at a rheumatology seminar sponsored by Skin Disease Education Foundation (SDEF) and the University of Louisville.
Arthritis and inflammatory bowel disease (IBD) have more in common than the drugs used to treat them. Arthritis is the most common extraintestinal manifestation of IBD. Onset of joint symptoms may precede the onset of IBD, develop in parallel to it, or be unrelated, he said.
Arthritis is most likely to occur in IBD patients who have other extraintestinal manifestations such as dermatologic, ocular, or renal symptoms. The most commonly involved central joints are in the spine, where the arthritis takes the form of ankylosing spondylitis or sacroiliitis. Peripheral joints can develop arthropathies in IBD as well. About 5%-20% of IBD patients get arthritis. The cartilage and bone usually are spared from destruction. Arthritis in IBD usually involves five or more joints and lasts about 3 years.
The incidence of IBD in rheumatoid arthritis is not well defined. One of the few studies to address this question involved a review of the data sets from two large insurance companies involving 17 million people. The researchers found that the prevalence of IBD was about 0.62%-0.65% and the prevalence of RA was about 0.98%-1.08% in patients aged 47-56 years old living in the Midwest or the South. The odds ratio of having both IBD and RA was 2.1-2.7, and of having IBD and ankylosing spondylitis, about 5.8-7.8 (Inflamm. Bowel Dis. 2008;14:738-43).
The advent of biologics has changed the natural history of ulcerative colitis (UC). But before these agents became available, data from a Danish study showed that during the first year after diagnosis, 10% of UC patients lost their colon, about 23% had lost their colon after 10 years, and 31% had lost their colon 18 years out (Gut 1985;26:158-63).
In the prebiologic era, the natural history of Crohn’s disease also was grim. The disease followed an inflammatory path for the first 5 years, then became penetrating with fistula formation between years 5 and 10 in a subset of patients; stricturing could develop after year 10 (Inflamm. Bowel Dis. 2002;8:244-50).
In the past, "virtually all Crohn’s disease patients relapsed and most required one or more surgeries," said Dr. Cohen, who is codirector of the inflammatory bowel disease center at the University of Chicago. He noted that there was a 70% probability that Crohn’s would recur over a 10-year period (Gastroenterology 1985;88:1826-33).
An estimated 10% of Crohn’s patients had their colons removed surgically within 1 year of their diagnosis with IBD, and an estimated 70% of patients who had one surgery for Crohn’s would require another. The disease tended to recur at the site of anastomosis of the previous surgery and to involve as much bowel as had been removed in the previous surgery. About half of the patients needed a second surgery.
Even today, most ulcerative colitis patients are treated with steroids and many of them become steroid dependent. Findings from a study of 63 UC patients placed on steroids showed that at the end of 1 month, 34 achieved complete remission, 19 had a partial remission, and 10 had no response. Follow-up data at 1 year showed that 31 had a prolonged response, 14 were steroid dependent, and 18 needed surgery (Gastroenterology 2001;121:255-60).
These days, there is a move away from making steroids the first drug in the treatment regimen and instead starting with a biologic and adding a steroid only if necessary.
There is some overlap between the biologics used to treat rheumatologic diseases and those used to treat IBD. Recent data show that infliximab in combination with azathioprine induced a steroid-free clinical remission in 44 of 64 patients. In the same study, infliximab plus placebo induced remission in 37 of 65 patients, and azathioprine plus placebo induced remission in 21 of 75 patients. All of the patients had active IBD with a C-reactive protein level of 0.8 mg/dL or higher and lesions that were visible on endoscopy (N. Engl. J. Med. 2010;362:1383-95).
Skin Disease Education Foundation and this news organization are owned by Elsevier. Dr. Cohen disclosed financial relationships with Abbott Labs, Axcan Pharma, Elan, Centocor, Procter & Gamble, Prometheus Laboratories, Salix, Shire, UCB, and Warner Chilcott.
SANTA MONICA, Calif. – Gastroenterologists seem to be taking a page from the rheumatologists’ playbook and are starting patients with inflammatory bowel disease on a biologic agent much sooner after diagnosis than has been standard practice.
By doing so, they hope to improve the natural history of the disease, just as rheumatologists have done in rheumatoid arthritis patients, according to Dr. Russell D. Cohen, who spoke at a rheumatology seminar sponsored by Skin Disease Education Foundation (SDEF) and the University of Louisville.
Arthritis and inflammatory bowel disease (IBD) have more in common than the drugs used to treat them. Arthritis is the most common extraintestinal manifestation of IBD. Onset of joint symptoms may precede the onset of IBD, develop in parallel to it, or be unrelated, he said.
Arthritis is most likely to occur in IBD patients who have other extraintestinal manifestations such as dermatologic, ocular, or renal symptoms. The most commonly involved central joints are in the spine, where the arthritis takes the form of ankylosing spondylitis or sacroiliitis. Peripheral joints can develop arthropathies in IBD as well. About 5%-20% of IBD patients get arthritis. The cartilage and bone usually are spared from destruction. Arthritis in IBD usually involves five or more joints and lasts about 3 years.
The incidence of IBD in rheumatoid arthritis is not well defined. One of the few studies to address this question involved a review of the data sets from two large insurance companies involving 17 million people. The researchers found that the prevalence of IBD was about 0.62%-0.65% and the prevalence of RA was about 0.98%-1.08% in patients aged 47-56 years old living in the Midwest or the South. The odds ratio of having both IBD and RA was 2.1-2.7, and of having IBD and ankylosing spondylitis, about 5.8-7.8 (Inflamm. Bowel Dis. 2008;14:738-43).
The advent of biologics has changed the natural history of ulcerative colitis (UC). But before these agents became available, data from a Danish study showed that during the first year after diagnosis, 10% of UC patients lost their colon, about 23% had lost their colon after 10 years, and 31% had lost their colon 18 years out (Gut 1985;26:158-63).
In the prebiologic era, the natural history of Crohn’s disease also was grim. The disease followed an inflammatory path for the first 5 years, then became penetrating with fistula formation between years 5 and 10 in a subset of patients; stricturing could develop after year 10 (Inflamm. Bowel Dis. 2002;8:244-50).
In the past, "virtually all Crohn’s disease patients relapsed and most required one or more surgeries," said Dr. Cohen, who is codirector of the inflammatory bowel disease center at the University of Chicago. He noted that there was a 70% probability that Crohn’s would recur over a 10-year period (Gastroenterology 1985;88:1826-33).
An estimated 10% of Crohn’s patients had their colons removed surgically within 1 year of their diagnosis with IBD, and an estimated 70% of patients who had one surgery for Crohn’s would require another. The disease tended to recur at the site of anastomosis of the previous surgery and to involve as much bowel as had been removed in the previous surgery. About half of the patients needed a second surgery.
Even today, most ulcerative colitis patients are treated with steroids and many of them become steroid dependent. Findings from a study of 63 UC patients placed on steroids showed that at the end of 1 month, 34 achieved complete remission, 19 had a partial remission, and 10 had no response. Follow-up data at 1 year showed that 31 had a prolonged response, 14 were steroid dependent, and 18 needed surgery (Gastroenterology 2001;121:255-60).
These days, there is a move away from making steroids the first drug in the treatment regimen and instead starting with a biologic and adding a steroid only if necessary.
There is some overlap between the biologics used to treat rheumatologic diseases and those used to treat IBD. Recent data show that infliximab in combination with azathioprine induced a steroid-free clinical remission in 44 of 64 patients. In the same study, infliximab plus placebo induced remission in 37 of 65 patients, and azathioprine plus placebo induced remission in 21 of 75 patients. All of the patients had active IBD with a C-reactive protein level of 0.8 mg/dL or higher and lesions that were visible on endoscopy (N. Engl. J. Med. 2010;362:1383-95).
Skin Disease Education Foundation and this news organization are owned by Elsevier. Dr. Cohen disclosed financial relationships with Abbott Labs, Axcan Pharma, Elan, Centocor, Procter & Gamble, Prometheus Laboratories, Salix, Shire, UCB, and Warner Chilcott.
SANTA MONICA, Calif. – Gastroenterologists seem to be taking a page from the rheumatologists’ playbook and are starting patients with inflammatory bowel disease on a biologic agent much sooner after diagnosis than has been standard practice.
By doing so, they hope to improve the natural history of the disease, just as rheumatologists have done in rheumatoid arthritis patients, according to Dr. Russell D. Cohen, who spoke at a rheumatology seminar sponsored by Skin Disease Education Foundation (SDEF) and the University of Louisville.
Arthritis and inflammatory bowel disease (IBD) have more in common than the drugs used to treat them. Arthritis is the most common extraintestinal manifestation of IBD. Onset of joint symptoms may precede the onset of IBD, develop in parallel to it, or be unrelated, he said.
Arthritis is most likely to occur in IBD patients who have other extraintestinal manifestations such as dermatologic, ocular, or renal symptoms. The most commonly involved central joints are in the spine, where the arthritis takes the form of ankylosing spondylitis or sacroiliitis. Peripheral joints can develop arthropathies in IBD as well. About 5%-20% of IBD patients get arthritis. The cartilage and bone usually are spared from destruction. Arthritis in IBD usually involves five or more joints and lasts about 3 years.
The incidence of IBD in rheumatoid arthritis is not well defined. One of the few studies to address this question involved a review of the data sets from two large insurance companies involving 17 million people. The researchers found that the prevalence of IBD was about 0.62%-0.65% and the prevalence of RA was about 0.98%-1.08% in patients aged 47-56 years old living in the Midwest or the South. The odds ratio of having both IBD and RA was 2.1-2.7, and of having IBD and ankylosing spondylitis, about 5.8-7.8 (Inflamm. Bowel Dis. 2008;14:738-43).
The advent of biologics has changed the natural history of ulcerative colitis (UC). But before these agents became available, data from a Danish study showed that during the first year after diagnosis, 10% of UC patients lost their colon, about 23% had lost their colon after 10 years, and 31% had lost their colon 18 years out (Gut 1985;26:158-63).
In the prebiologic era, the natural history of Crohn’s disease also was grim. The disease followed an inflammatory path for the first 5 years, then became penetrating with fistula formation between years 5 and 10 in a subset of patients; stricturing could develop after year 10 (Inflamm. Bowel Dis. 2002;8:244-50).
In the past, "virtually all Crohn’s disease patients relapsed and most required one or more surgeries," said Dr. Cohen, who is codirector of the inflammatory bowel disease center at the University of Chicago. He noted that there was a 70% probability that Crohn’s would recur over a 10-year period (Gastroenterology 1985;88:1826-33).
An estimated 10% of Crohn’s patients had their colons removed surgically within 1 year of their diagnosis with IBD, and an estimated 70% of patients who had one surgery for Crohn’s would require another. The disease tended to recur at the site of anastomosis of the previous surgery and to involve as much bowel as had been removed in the previous surgery. About half of the patients needed a second surgery.
Even today, most ulcerative colitis patients are treated with steroids and many of them become steroid dependent. Findings from a study of 63 UC patients placed on steroids showed that at the end of 1 month, 34 achieved complete remission, 19 had a partial remission, and 10 had no response. Follow-up data at 1 year showed that 31 had a prolonged response, 14 were steroid dependent, and 18 needed surgery (Gastroenterology 2001;121:255-60).
These days, there is a move away from making steroids the first drug in the treatment regimen and instead starting with a biologic and adding a steroid only if necessary.
There is some overlap between the biologics used to treat rheumatologic diseases and those used to treat IBD. Recent data show that infliximab in combination with azathioprine induced a steroid-free clinical remission in 44 of 64 patients. In the same study, infliximab plus placebo induced remission in 37 of 65 patients, and azathioprine plus placebo induced remission in 21 of 75 patients. All of the patients had active IBD with a C-reactive protein level of 0.8 mg/dL or higher and lesions that were visible on endoscopy (N. Engl. J. Med. 2010;362:1383-95).
Skin Disease Education Foundation and this news organization are owned by Elsevier. Dr. Cohen disclosed financial relationships with Abbott Labs, Axcan Pharma, Elan, Centocor, Procter & Gamble, Prometheus Laboratories, Salix, Shire, UCB, and Warner Chilcott.
EXPERT ANALYSIS FROM A RHEUMATOLOGY SEMINAR
FDA Searches for Clinical Trial Middle Ground at Biosimilar Hearing
SILVER SPRING, MD. – The Food and Drug Administration appeared to be searching for a way to limit the clinical trial requirements for generic biologic therapies – or biosimilars – during a 2-day public hearing on implementing an approval pathway for the products.
In preparing for the hearing, the FDA hinted it might be willing to consider the possibility that a biosimilar application might not require any clinical trials.
Many presenters, including several manufacturers of approved biologic therapies, insisted that biosimilarity could not be established definitively without a clinical trial because analytical methods are not yet sophisticated enough. Others, including some generic pharmaceutical manufacturers, argued the agency should use discretion in deciding whether trials would be necessary.
FDA officials tried to carve out a middle ground between the two camps. Dr. Steven Kozlowski, director of the Office of Biotechnology Products within the agency’s Center for Drug Evaluation and Research (CDER), and Dr. John Jenkins, director of the CDER Office of New Drugs, appeared through their questioning to indicate a strict and extensive clinical data requirement might not get many biosimilars to the market, but an overly lax standard might not uncover some safety concerns before marketing begins.
Bruce Babbitt, Ph.D., principal consultant at Parexel Consulting, said FDA’s questions and statements during the hearing seemed to indicate the agency would be willing to be flexible with requiring trials, unlike in Europe, where trials are required for all applications.
"FDA might be open to establishing comparability using [pharmacokinetic/pharmacodynamic] studies in humans head to head with a reference [product]," he said.
Dr. Babbitt argued during his formal presentation the "totality of the data" of biologic products already establishes a base for proving biosimilarity and should not be ignored.
"We consider it important to balance the value and relevance of these data against the need for clinical safety and efficacy data particularly where minor or modest differences between the biosimilar and reference products are unlikely to translate into any meaningful differences in the clinic," Dr. Babbitt said in written testimony.
Dr. Kozlowski asked if there is a way to use the totality of the data in determining what studies would be required.
"Is there a way of incorporating that totality of the data or prior knowledge to think about what the impact on clinical studies are because usually the impact is some big-step function?" he asked Dr. Babbitt.
The totality of data helps either streamline or expand the data sponsors needed for their application, Dr. Babbitt said.
Dr. Jenkins also pointed out suggestions from innovator companies that could be excessive and likely not allow for many biosimilar approvals.
"For your clinical head-to-head trials you wanted a demonstration of equivalence, and you wanted a small confidence interval, you recommend equivalence for safety and efficacy and the surrogate endpoints are not acceptable unless they’ve been clinically validated," Dr. Jenkins said to Dr. Michael Wenger, global clinical lead at Roche. "Is that really an achievable standard ... or is that essentially closing the door for a biosimilar?"
Dr. Wenger argued the drugs should be assessed on a case-by-case basis.
The issue of extrapolation – allowing a biosimilar to be used in all the indications of its reference product after being thoroughly studied in only one – generated concerns about the extent of the clinical data needed. The clinical trial program for a biosimilar would be extremely large if a sponsor was required to produce data showing similarity for each indication it wanted in an application.
Dr. Kozlowski questioned whether it was possible to extrapolate some indications using data for others.
"Say you had the two most different indications in a product that had five indications and you explore those two. Do you believe you could extrapolate the other three?" he asked.
The idea would depend on the clinical benefit achieved by the innovator drug, according to Dr. Wenger. If it was a benefit that could be achieved easily, it may be possible, he said, adding that a more complicated benefit, like overall survival in oncology, would require more work.
Dr. Kozlowski asked Vijay Tammara, Ph.D., vice president of regulatory affairs at Nuron Biotech Inc., whether requiring pharmacokinetic and pharmacodynamic studies could be sufficient premarket, saving immunogenicity testing for the postmarketing studies.
That concept came up again later, during questioning of Sara Radcliffe, executive vice president of health for the Biotechnology Industry Organization (BIO) when Dr. Kozlowski asked whether it would be useful to use pre-marketing data to establish biosimilarity and postmarketing data to test interchangeability.
Ms. Radcliffe said that idea could be possible if an interchangeability determination was not possible before marketing.
Despite the discussion that appeared to indicate a potential for flexibility at FDA, panel members did not give a definitive answer about which way they were leaning.
Dr, Kozlowski’s questions sometimes presented the case for stronger clinical trial requirements. Rasmus Rojkjaer testified on behalf of the Generic Pharmaceutical Association that data from non-U.S. referenced products should be allowed for applications to the FDA and trials should not be more extensive than those required for an innovator product.
Dr. Kozlowski said there had been cases in Europe where clinical trials were shown to be important because they uncovered information that changed the product’s development. He asked about Mr. Rojkjaer’s view of the idea.
Mr. Rojkjaer reiterated that biosimilar applications should not have over-arching rules governing their reviews.
"I think clinical trials has its role to play, but forcing the industry to fit into a one-[size]-fits-all kind of approach I think would seriously keep the development of these biogeneric compounds back," he said.
When Dr. James Roach, chief medical officer of Momenta Pharmaceuticals Inc., argued the trials should be positioned as supportive to existing data, Dr. Jenkins said the agency still has to decide how it will ensure biosimilars are actually similar enough to the reference product.
"We heard a lot of comments yesterday from patient groups and prescribers that they wanted to be very certain that the biosimilar would be highly similar to the clinical effect of the branded product," Dr. Jenkins said. "How do you reconcile those two viewpoints as far as where we should set the bar on determining the role of the clinical trials in establishing that there’s no clinically meaningful difference as the statute requires between the biosimilar and the reference product?"
Dr. Roach said he thought it was possible to provide a limited clinical data set in addition to the existing data package to gain a product approval.
"The Pink Sheet" and this news organization are both owned by Elsevier.
SILVER SPRING, MD. – The Food and Drug Administration appeared to be searching for a way to limit the clinical trial requirements for generic biologic therapies – or biosimilars – during a 2-day public hearing on implementing an approval pathway for the products.
In preparing for the hearing, the FDA hinted it might be willing to consider the possibility that a biosimilar application might not require any clinical trials.
Many presenters, including several manufacturers of approved biologic therapies, insisted that biosimilarity could not be established definitively without a clinical trial because analytical methods are not yet sophisticated enough. Others, including some generic pharmaceutical manufacturers, argued the agency should use discretion in deciding whether trials would be necessary.
FDA officials tried to carve out a middle ground between the two camps. Dr. Steven Kozlowski, director of the Office of Biotechnology Products within the agency’s Center for Drug Evaluation and Research (CDER), and Dr. John Jenkins, director of the CDER Office of New Drugs, appeared through their questioning to indicate a strict and extensive clinical data requirement might not get many biosimilars to the market, but an overly lax standard might not uncover some safety concerns before marketing begins.
Bruce Babbitt, Ph.D., principal consultant at Parexel Consulting, said FDA’s questions and statements during the hearing seemed to indicate the agency would be willing to be flexible with requiring trials, unlike in Europe, where trials are required for all applications.
"FDA might be open to establishing comparability using [pharmacokinetic/pharmacodynamic] studies in humans head to head with a reference [product]," he said.
Dr. Babbitt argued during his formal presentation the "totality of the data" of biologic products already establishes a base for proving biosimilarity and should not be ignored.
"We consider it important to balance the value and relevance of these data against the need for clinical safety and efficacy data particularly where minor or modest differences between the biosimilar and reference products are unlikely to translate into any meaningful differences in the clinic," Dr. Babbitt said in written testimony.
Dr. Kozlowski asked if there is a way to use the totality of the data in determining what studies would be required.
"Is there a way of incorporating that totality of the data or prior knowledge to think about what the impact on clinical studies are because usually the impact is some big-step function?" he asked Dr. Babbitt.
The totality of data helps either streamline or expand the data sponsors needed for their application, Dr. Babbitt said.
Dr. Jenkins also pointed out suggestions from innovator companies that could be excessive and likely not allow for many biosimilar approvals.
"For your clinical head-to-head trials you wanted a demonstration of equivalence, and you wanted a small confidence interval, you recommend equivalence for safety and efficacy and the surrogate endpoints are not acceptable unless they’ve been clinically validated," Dr. Jenkins said to Dr. Michael Wenger, global clinical lead at Roche. "Is that really an achievable standard ... or is that essentially closing the door for a biosimilar?"
Dr. Wenger argued the drugs should be assessed on a case-by-case basis.
The issue of extrapolation – allowing a biosimilar to be used in all the indications of its reference product after being thoroughly studied in only one – generated concerns about the extent of the clinical data needed. The clinical trial program for a biosimilar would be extremely large if a sponsor was required to produce data showing similarity for each indication it wanted in an application.
Dr. Kozlowski questioned whether it was possible to extrapolate some indications using data for others.
"Say you had the two most different indications in a product that had five indications and you explore those two. Do you believe you could extrapolate the other three?" he asked.
The idea would depend on the clinical benefit achieved by the innovator drug, according to Dr. Wenger. If it was a benefit that could be achieved easily, it may be possible, he said, adding that a more complicated benefit, like overall survival in oncology, would require more work.
Dr. Kozlowski asked Vijay Tammara, Ph.D., vice president of regulatory affairs at Nuron Biotech Inc., whether requiring pharmacokinetic and pharmacodynamic studies could be sufficient premarket, saving immunogenicity testing for the postmarketing studies.
That concept came up again later, during questioning of Sara Radcliffe, executive vice president of health for the Biotechnology Industry Organization (BIO) when Dr. Kozlowski asked whether it would be useful to use pre-marketing data to establish biosimilarity and postmarketing data to test interchangeability.
Ms. Radcliffe said that idea could be possible if an interchangeability determination was not possible before marketing.
Despite the discussion that appeared to indicate a potential for flexibility at FDA, panel members did not give a definitive answer about which way they were leaning.
Dr, Kozlowski’s questions sometimes presented the case for stronger clinical trial requirements. Rasmus Rojkjaer testified on behalf of the Generic Pharmaceutical Association that data from non-U.S. referenced products should be allowed for applications to the FDA and trials should not be more extensive than those required for an innovator product.
Dr. Kozlowski said there had been cases in Europe where clinical trials were shown to be important because they uncovered information that changed the product’s development. He asked about Mr. Rojkjaer’s view of the idea.
Mr. Rojkjaer reiterated that biosimilar applications should not have over-arching rules governing their reviews.
"I think clinical trials has its role to play, but forcing the industry to fit into a one-[size]-fits-all kind of approach I think would seriously keep the development of these biogeneric compounds back," he said.
When Dr. James Roach, chief medical officer of Momenta Pharmaceuticals Inc., argued the trials should be positioned as supportive to existing data, Dr. Jenkins said the agency still has to decide how it will ensure biosimilars are actually similar enough to the reference product.
"We heard a lot of comments yesterday from patient groups and prescribers that they wanted to be very certain that the biosimilar would be highly similar to the clinical effect of the branded product," Dr. Jenkins said. "How do you reconcile those two viewpoints as far as where we should set the bar on determining the role of the clinical trials in establishing that there’s no clinically meaningful difference as the statute requires between the biosimilar and the reference product?"
Dr. Roach said he thought it was possible to provide a limited clinical data set in addition to the existing data package to gain a product approval.
"The Pink Sheet" and this news organization are both owned by Elsevier.
SILVER SPRING, MD. – The Food and Drug Administration appeared to be searching for a way to limit the clinical trial requirements for generic biologic therapies – or biosimilars – during a 2-day public hearing on implementing an approval pathway for the products.
In preparing for the hearing, the FDA hinted it might be willing to consider the possibility that a biosimilar application might not require any clinical trials.
Many presenters, including several manufacturers of approved biologic therapies, insisted that biosimilarity could not be established definitively without a clinical trial because analytical methods are not yet sophisticated enough. Others, including some generic pharmaceutical manufacturers, argued the agency should use discretion in deciding whether trials would be necessary.
FDA officials tried to carve out a middle ground between the two camps. Dr. Steven Kozlowski, director of the Office of Biotechnology Products within the agency’s Center for Drug Evaluation and Research (CDER), and Dr. John Jenkins, director of the CDER Office of New Drugs, appeared through their questioning to indicate a strict and extensive clinical data requirement might not get many biosimilars to the market, but an overly lax standard might not uncover some safety concerns before marketing begins.
Bruce Babbitt, Ph.D., principal consultant at Parexel Consulting, said FDA’s questions and statements during the hearing seemed to indicate the agency would be willing to be flexible with requiring trials, unlike in Europe, where trials are required for all applications.
"FDA might be open to establishing comparability using [pharmacokinetic/pharmacodynamic] studies in humans head to head with a reference [product]," he said.
Dr. Babbitt argued during his formal presentation the "totality of the data" of biologic products already establishes a base for proving biosimilarity and should not be ignored.
"We consider it important to balance the value and relevance of these data against the need for clinical safety and efficacy data particularly where minor or modest differences between the biosimilar and reference products are unlikely to translate into any meaningful differences in the clinic," Dr. Babbitt said in written testimony.
Dr. Kozlowski asked if there is a way to use the totality of the data in determining what studies would be required.
"Is there a way of incorporating that totality of the data or prior knowledge to think about what the impact on clinical studies are because usually the impact is some big-step function?" he asked Dr. Babbitt.
The totality of data helps either streamline or expand the data sponsors needed for their application, Dr. Babbitt said.
Dr. Jenkins also pointed out suggestions from innovator companies that could be excessive and likely not allow for many biosimilar approvals.
"For your clinical head-to-head trials you wanted a demonstration of equivalence, and you wanted a small confidence interval, you recommend equivalence for safety and efficacy and the surrogate endpoints are not acceptable unless they’ve been clinically validated," Dr. Jenkins said to Dr. Michael Wenger, global clinical lead at Roche. "Is that really an achievable standard ... or is that essentially closing the door for a biosimilar?"
Dr. Wenger argued the drugs should be assessed on a case-by-case basis.
The issue of extrapolation – allowing a biosimilar to be used in all the indications of its reference product after being thoroughly studied in only one – generated concerns about the extent of the clinical data needed. The clinical trial program for a biosimilar would be extremely large if a sponsor was required to produce data showing similarity for each indication it wanted in an application.
Dr. Kozlowski questioned whether it was possible to extrapolate some indications using data for others.
"Say you had the two most different indications in a product that had five indications and you explore those two. Do you believe you could extrapolate the other three?" he asked.
The idea would depend on the clinical benefit achieved by the innovator drug, according to Dr. Wenger. If it was a benefit that could be achieved easily, it may be possible, he said, adding that a more complicated benefit, like overall survival in oncology, would require more work.
Dr. Kozlowski asked Vijay Tammara, Ph.D., vice president of regulatory affairs at Nuron Biotech Inc., whether requiring pharmacokinetic and pharmacodynamic studies could be sufficient premarket, saving immunogenicity testing for the postmarketing studies.
That concept came up again later, during questioning of Sara Radcliffe, executive vice president of health for the Biotechnology Industry Organization (BIO) when Dr. Kozlowski asked whether it would be useful to use pre-marketing data to establish biosimilarity and postmarketing data to test interchangeability.
Ms. Radcliffe said that idea could be possible if an interchangeability determination was not possible before marketing.
Despite the discussion that appeared to indicate a potential for flexibility at FDA, panel members did not give a definitive answer about which way they were leaning.
Dr, Kozlowski’s questions sometimes presented the case for stronger clinical trial requirements. Rasmus Rojkjaer testified on behalf of the Generic Pharmaceutical Association that data from non-U.S. referenced products should be allowed for applications to the FDA and trials should not be more extensive than those required for an innovator product.
Dr. Kozlowski said there had been cases in Europe where clinical trials were shown to be important because they uncovered information that changed the product’s development. He asked about Mr. Rojkjaer’s view of the idea.
Mr. Rojkjaer reiterated that biosimilar applications should not have over-arching rules governing their reviews.
"I think clinical trials has its role to play, but forcing the industry to fit into a one-[size]-fits-all kind of approach I think would seriously keep the development of these biogeneric compounds back," he said.
When Dr. James Roach, chief medical officer of Momenta Pharmaceuticals Inc., argued the trials should be positioned as supportive to existing data, Dr. Jenkins said the agency still has to decide how it will ensure biosimilars are actually similar enough to the reference product.
"We heard a lot of comments yesterday from patient groups and prescribers that they wanted to be very certain that the biosimilar would be highly similar to the clinical effect of the branded product," Dr. Jenkins said. "How do you reconcile those two viewpoints as far as where we should set the bar on determining the role of the clinical trials in establishing that there’s no clinically meaningful difference as the statute requires between the biosimilar and the reference product?"
Dr. Roach said he thought it was possible to provide a limited clinical data set in addition to the existing data package to gain a product approval.
"The Pink Sheet" and this news organization are both owned by Elsevier.
FROM A HEARING HELD BY THE FOOD AND DRUG ADMINISTRATION
Favorable 3-Year Safety Update for Ustekinumab in Psoriasis
GOTHENBURG, SWEDEN – Long-term risks of malignancies, serious infections, and cardiovascular events in ustekinumab-treated psoriasis patients was found to be what is expected in the general U.S. population, according to a 3-year safety analysis of the major randomized clinical trials of the biologic agent.
Placebo-treated control groups were utilized during the first 12 weeks of the phase III PHOENIX I and II trials and not at all in the phase III Active Comparator Psoriasis Trial (ACCEPT). For the safety analysis based on 3 years of follow-up – for purposes of comparison – risk estimates for the general U.S. population were derived from the National Institute of Health’s Surveillance, Epidemiology and End Results (SEER) database and MarketScan Research Data, Dr. Kenneth B. Gordon explained at the annual congress of the European Academy of Dermatology and Venereology.
The 3-year safety analysis included 3,117 psoriasis patients treated with ustekinumab (Stelara) at 45 mg or 90 mg per dose based on body weight, in accordance with the product labeling. Among this group were 1,247 patients who received the interleukin-12 and -23 inhibitor for at least 2 years, noted Dr. Gordon, a dermatologist at the University of Chicago.
The cumulative incidence of nonmelanoma skin cancer through year 3 in the 3,117 psoriasis patients was 0.64 cases per 100 patient-years in patients on ustekinumab 45 mg and 0.77 per 100 patient-years in those on ustekinumab 90 mg. During the 12-week controlled period of the studies, the incidence in placebo-treated patients was 1.13 cases per 100 patient-years.
Rates of other malignancies, including melanoma and lymphoma, through 3 years were 0.69 and 0.46 cases per 100 patient-years in patients on ustekinumab 45 and 90 mg, respectively. These rates were consistent with those expected in the U.S. general population based upon SEER data. Rates of malignancies other than nonmelanoma skin cancer were also comparable to those reported in a large population of psoriasis patients treated with systemic agents in the U.K. General Practice Research Database, where the incidence was 0.58 cases per 100 patient-years (J. Invest. Dermatol. 2009;129:2604-12).
The rate of serious infections through 3 years was 0.82 per 100 patient-years in psoriasis patients on ustekinumab 45 mg and 1.5 per 100 patient-years in those on the higher dose, for a combined event rate of 1.19 per 100 patient-years. This compared favorably to the expected rate of 1.51 per 100 patient-years in psoriasis patients with nonbiologic systemic agents derived from the MarketScan database, Dr. Gordon said.
The combined rate of cardiovascular death, MI, or stroke through 3 years in ustekinumab-treated psoriasis patients was 0.41 per 100 patient-years in those on the lower dose and 0.35 per 100 patient-years in those receiving 90 mg. These rates were lower, albeit not significantly so, than rates expected in the general population using data from the Framingham Heart Study and in psoriasis patients using the U.K. General Practice Research Database.
Rates of malignancies, serious infections, and cardiovascular events in ustekinumab-treated patients remained stable over time without any indication of cumulative toxicity. Another safety analysis is planned when participants in the phase III trials reach the 5-year follow-up mark.
The study was funded by Centocor. Dr. Gordon disclosed that he serves as a consultant to the company.
GOTHENBURG, SWEDEN – Long-term risks of malignancies, serious infections, and cardiovascular events in ustekinumab-treated psoriasis patients was found to be what is expected in the general U.S. population, according to a 3-year safety analysis of the major randomized clinical trials of the biologic agent.
Placebo-treated control groups were utilized during the first 12 weeks of the phase III PHOENIX I and II trials and not at all in the phase III Active Comparator Psoriasis Trial (ACCEPT). For the safety analysis based on 3 years of follow-up – for purposes of comparison – risk estimates for the general U.S. population were derived from the National Institute of Health’s Surveillance, Epidemiology and End Results (SEER) database and MarketScan Research Data, Dr. Kenneth B. Gordon explained at the annual congress of the European Academy of Dermatology and Venereology.
The 3-year safety analysis included 3,117 psoriasis patients treated with ustekinumab (Stelara) at 45 mg or 90 mg per dose based on body weight, in accordance with the product labeling. Among this group were 1,247 patients who received the interleukin-12 and -23 inhibitor for at least 2 years, noted Dr. Gordon, a dermatologist at the University of Chicago.
The cumulative incidence of nonmelanoma skin cancer through year 3 in the 3,117 psoriasis patients was 0.64 cases per 100 patient-years in patients on ustekinumab 45 mg and 0.77 per 100 patient-years in those on ustekinumab 90 mg. During the 12-week controlled period of the studies, the incidence in placebo-treated patients was 1.13 cases per 100 patient-years.
Rates of other malignancies, including melanoma and lymphoma, through 3 years were 0.69 and 0.46 cases per 100 patient-years in patients on ustekinumab 45 and 90 mg, respectively. These rates were consistent with those expected in the U.S. general population based upon SEER data. Rates of malignancies other than nonmelanoma skin cancer were also comparable to those reported in a large population of psoriasis patients treated with systemic agents in the U.K. General Practice Research Database, where the incidence was 0.58 cases per 100 patient-years (J. Invest. Dermatol. 2009;129:2604-12).
The rate of serious infections through 3 years was 0.82 per 100 patient-years in psoriasis patients on ustekinumab 45 mg and 1.5 per 100 patient-years in those on the higher dose, for a combined event rate of 1.19 per 100 patient-years. This compared favorably to the expected rate of 1.51 per 100 patient-years in psoriasis patients with nonbiologic systemic agents derived from the MarketScan database, Dr. Gordon said.
The combined rate of cardiovascular death, MI, or stroke through 3 years in ustekinumab-treated psoriasis patients was 0.41 per 100 patient-years in those on the lower dose and 0.35 per 100 patient-years in those receiving 90 mg. These rates were lower, albeit not significantly so, than rates expected in the general population using data from the Framingham Heart Study and in psoriasis patients using the U.K. General Practice Research Database.
Rates of malignancies, serious infections, and cardiovascular events in ustekinumab-treated patients remained stable over time without any indication of cumulative toxicity. Another safety analysis is planned when participants in the phase III trials reach the 5-year follow-up mark.
The study was funded by Centocor. Dr. Gordon disclosed that he serves as a consultant to the company.
GOTHENBURG, SWEDEN – Long-term risks of malignancies, serious infections, and cardiovascular events in ustekinumab-treated psoriasis patients was found to be what is expected in the general U.S. population, according to a 3-year safety analysis of the major randomized clinical trials of the biologic agent.
Placebo-treated control groups were utilized during the first 12 weeks of the phase III PHOENIX I and II trials and not at all in the phase III Active Comparator Psoriasis Trial (ACCEPT). For the safety analysis based on 3 years of follow-up – for purposes of comparison – risk estimates for the general U.S. population were derived from the National Institute of Health’s Surveillance, Epidemiology and End Results (SEER) database and MarketScan Research Data, Dr. Kenneth B. Gordon explained at the annual congress of the European Academy of Dermatology and Venereology.
The 3-year safety analysis included 3,117 psoriasis patients treated with ustekinumab (Stelara) at 45 mg or 90 mg per dose based on body weight, in accordance with the product labeling. Among this group were 1,247 patients who received the interleukin-12 and -23 inhibitor for at least 2 years, noted Dr. Gordon, a dermatologist at the University of Chicago.
The cumulative incidence of nonmelanoma skin cancer through year 3 in the 3,117 psoriasis patients was 0.64 cases per 100 patient-years in patients on ustekinumab 45 mg and 0.77 per 100 patient-years in those on ustekinumab 90 mg. During the 12-week controlled period of the studies, the incidence in placebo-treated patients was 1.13 cases per 100 patient-years.
Rates of other malignancies, including melanoma and lymphoma, through 3 years were 0.69 and 0.46 cases per 100 patient-years in patients on ustekinumab 45 and 90 mg, respectively. These rates were consistent with those expected in the U.S. general population based upon SEER data. Rates of malignancies other than nonmelanoma skin cancer were also comparable to those reported in a large population of psoriasis patients treated with systemic agents in the U.K. General Practice Research Database, where the incidence was 0.58 cases per 100 patient-years (J. Invest. Dermatol. 2009;129:2604-12).
The rate of serious infections through 3 years was 0.82 per 100 patient-years in psoriasis patients on ustekinumab 45 mg and 1.5 per 100 patient-years in those on the higher dose, for a combined event rate of 1.19 per 100 patient-years. This compared favorably to the expected rate of 1.51 per 100 patient-years in psoriasis patients with nonbiologic systemic agents derived from the MarketScan database, Dr. Gordon said.
The combined rate of cardiovascular death, MI, or stroke through 3 years in ustekinumab-treated psoriasis patients was 0.41 per 100 patient-years in those on the lower dose and 0.35 per 100 patient-years in those receiving 90 mg. These rates were lower, albeit not significantly so, than rates expected in the general population using data from the Framingham Heart Study and in psoriasis patients using the U.K. General Practice Research Database.
Rates of malignancies, serious infections, and cardiovascular events in ustekinumab-treated patients remained stable over time without any indication of cumulative toxicity. Another safety analysis is planned when participants in the phase III trials reach the 5-year follow-up mark.
The study was funded by Centocor. Dr. Gordon disclosed that he serves as a consultant to the company.
FROM THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: Rates of malignancies, serious infections, and cardiovascular events in ustekinumab patients remained stable over time without any indication of cumulative toxicity and comparable to expected rates in the general population.
Data Source: The 3-year safety analysis included 3,117 psoriasis patients treated with ustekinumab at 45 mg or 90 mg.
Disclosures: The study was funded by Centocor. Dr. Gordon disclosed that he serves as a consultant to the company.
NIH at Forefront of New Clinical Trials Push, Director Says
WASHINGTON – Breakthroughs in human genetics combined with funding from the Affordable Care Act have poised the National Institutes of Health to make real progress in the areas of orphan human diseases, according to NIH Director Francis S. Collins.
Speaking with enthusiasm to those he addressed as his "peeps" at the annual meeting of the American Society of Human Genetics, Dr. Collins shared his excitement at the state of human genetics in the post-genomic world, in large part driven by technology that has significantly lowered the cost of DNA sequencing, in turn speeding genetic research tremendously.
This, combined with new ACA funding, has enabled NIH to fund and pursue translational research, moving laboratory results toward and into clinical trials, something that is a new way of thinking for the agency, Dr. Collins said.

Rather than relying on pharmaceutical and biotechnology companies to take charge of the translational research, Dr. Collins encouraged academic researchers to consider partnering with NIH, at least for those orphan disease conditions in which the federal government would not be seen as being in competition with private enterprise.
"There is a serious crisis underway in the way in which this pipeline for drug discovery has been floundering. ... Pharma has been investing a larger and larger amount of money – between $40 and $50 billion dollars a year – and yet in spite of that, FDA approvals of new molecular entities, that is genuinely new drug therapeutics, not ‘me-toos,’ have been dropping steadily over the last 15 years," Dr. Collins said.
The reasons for this are complex, he said, but a big part of the problem involves coming up with appropriate targets and targeting compounds. He said this is an area in which NIH is and can be very much involved.
NIH now encourages academic researchers to take their targets to the assay stage and beyond, providing high-throughput screening (HTS) assistance from the NIH Chemical Genomics Center. Subsequent medicinal chemistry assistance is also available to help to modify HTS hits to enable compounds to become more drug-like and to match current ADME (absorption, distribution, metabolism, and excretion) criteria.
With NIH assistance, more than 150 lead compounds have reached this stage over the last 4-5 years, more than half of which are "poised to go to the next step" of preclinical trials in animals, or the "Valley of Death," according to Dr. Collins, "because this is where projects often go to die."
NIH is now able to assist in this high-risk area through the Therapeutics for Rare and Neglected Diseases (TRND) program in its Office of Rare Diseases Research. The TRND was funded at $24 million in fiscal year 2009.
NIH also is positioned to assist researchers in early phase human trials of orphan diseases through its 240-bed Clinical Center, Dr. Collins said.
"And we have 50 and soon we will have 60 Clinical and Translational Science Awards scattered all across the country which will also be set up to conduct these sorts of trials for new molecular entities," he added.
This new direction in research funding has involved unprecedented cooperation with the Food and Drug Administration, Dr. Collins said, with an NIH-FDA leadership council formed to ensure that new drug candidates are most safely and efficiently moved into the clinical trials framework in ways that would best enable FDA analysis and validation, particularly for rare diseases.
Dr. Collins was particularly excited about five instances in which NIH is using this new model of helping "de-risk" the drug development process for orphan or neglected diseases through TRND. These include four rare diseases (Niemann-Pick Disease Type C, hereditary inclusion body myopathy, sickle cell disease, and chronic lymphocytic leukemia) and one neglected disease (schistosomiasis).
If and when these compounds and those for other rare diseases become ready for marketing and production, NIH would then work with private companies to achieve licensing agreement to enable their manufacture and sale, according to Dr. Collins.
Dr. Collins reported having no financial conflicts of interest with regard to his presentation.
WASHINGTON – Breakthroughs in human genetics combined with funding from the Affordable Care Act have poised the National Institutes of Health to make real progress in the areas of orphan human diseases, according to NIH Director Francis S. Collins.
Speaking with enthusiasm to those he addressed as his "peeps" at the annual meeting of the American Society of Human Genetics, Dr. Collins shared his excitement at the state of human genetics in the post-genomic world, in large part driven by technology that has significantly lowered the cost of DNA sequencing, in turn speeding genetic research tremendously.
This, combined with new ACA funding, has enabled NIH to fund and pursue translational research, moving laboratory results toward and into clinical trials, something that is a new way of thinking for the agency, Dr. Collins said.
Rather than relying on pharmaceutical and biotechnology companies to take charge of the translational research, Dr. Collins encouraged academic researchers to consider partnering with NIH, at least for those orphan disease conditions in which the federal government would not be seen as being in competition with private enterprise.
"There is a serious crisis underway in the way in which this pipeline for drug discovery has been floundering. ... Pharma has been investing a larger and larger amount of money – between $40 and $50 billion dollars a year – and yet in spite of that, FDA approvals of new molecular entities, that is genuinely new drug therapeutics, not ‘me-toos,’ have been dropping steadily over the last 15 years," Dr. Collins said.
The reasons for this are complex, he said, but a big part of the problem involves coming up with appropriate targets and targeting compounds. He said this is an area in which NIH is and can be very much involved.
NIH now encourages academic researchers to take their targets to the assay stage and beyond, providing high-throughput screening (HTS) assistance from the NIH Chemical Genomics Center. Subsequent medicinal chemistry assistance is also available to help to modify HTS hits to enable compounds to become more drug-like and to match current ADME (absorption, distribution, metabolism, and excretion) criteria.
With NIH assistance, more than 150 lead compounds have reached this stage over the last 4-5 years, more than half of which are "poised to go to the next step" of preclinical trials in animals, or the "Valley of Death," according to Dr. Collins, "because this is where projects often go to die."
NIH is now able to assist in this high-risk area through the Therapeutics for Rare and Neglected Diseases (TRND) program in its Office of Rare Diseases Research. The TRND was funded at $24 million in fiscal year 2009.
NIH also is positioned to assist researchers in early phase human trials of orphan diseases through its 240-bed Clinical Center, Dr. Collins said.
"And we have 50 and soon we will have 60 Clinical and Translational Science Awards scattered all across the country which will also be set up to conduct these sorts of trials for new molecular entities," he added.
This new direction in research funding has involved unprecedented cooperation with the Food and Drug Administration, Dr. Collins said, with an NIH-FDA leadership council formed to ensure that new drug candidates are most safely and efficiently moved into the clinical trials framework in ways that would best enable FDA analysis and validation, particularly for rare diseases.
Dr. Collins was particularly excited about five instances in which NIH is using this new model of helping "de-risk" the drug development process for orphan or neglected diseases through TRND. These include four rare diseases (Niemann-Pick Disease Type C, hereditary inclusion body myopathy, sickle cell disease, and chronic lymphocytic leukemia) and one neglected disease (schistosomiasis).
If and when these compounds and those for other rare diseases become ready for marketing and production, NIH would then work with private companies to achieve licensing agreement to enable their manufacture and sale, according to Dr. Collins.
Dr. Collins reported having no financial conflicts of interest with regard to his presentation.
WASHINGTON – Breakthroughs in human genetics combined with funding from the Affordable Care Act have poised the National Institutes of Health to make real progress in the areas of orphan human diseases, according to NIH Director Francis S. Collins.
Speaking with enthusiasm to those he addressed as his "peeps" at the annual meeting of the American Society of Human Genetics, Dr. Collins shared his excitement at the state of human genetics in the post-genomic world, in large part driven by technology that has significantly lowered the cost of DNA sequencing, in turn speeding genetic research tremendously.
This, combined with new ACA funding, has enabled NIH to fund and pursue translational research, moving laboratory results toward and into clinical trials, something that is a new way of thinking for the agency, Dr. Collins said.
Rather than relying on pharmaceutical and biotechnology companies to take charge of the translational research, Dr. Collins encouraged academic researchers to consider partnering with NIH, at least for those orphan disease conditions in which the federal government would not be seen as being in competition with private enterprise.
"There is a serious crisis underway in the way in which this pipeline for drug discovery has been floundering. ... Pharma has been investing a larger and larger amount of money – between $40 and $50 billion dollars a year – and yet in spite of that, FDA approvals of new molecular entities, that is genuinely new drug therapeutics, not ‘me-toos,’ have been dropping steadily over the last 15 years," Dr. Collins said.
The reasons for this are complex, he said, but a big part of the problem involves coming up with appropriate targets and targeting compounds. He said this is an area in which NIH is and can be very much involved.
NIH now encourages academic researchers to take their targets to the assay stage and beyond, providing high-throughput screening (HTS) assistance from the NIH Chemical Genomics Center. Subsequent medicinal chemistry assistance is also available to help to modify HTS hits to enable compounds to become more drug-like and to match current ADME (absorption, distribution, metabolism, and excretion) criteria.
With NIH assistance, more than 150 lead compounds have reached this stage over the last 4-5 years, more than half of which are "poised to go to the next step" of preclinical trials in animals, or the "Valley of Death," according to Dr. Collins, "because this is where projects often go to die."
NIH is now able to assist in this high-risk area through the Therapeutics for Rare and Neglected Diseases (TRND) program in its Office of Rare Diseases Research. The TRND was funded at $24 million in fiscal year 2009.
NIH also is positioned to assist researchers in early phase human trials of orphan diseases through its 240-bed Clinical Center, Dr. Collins said.
"And we have 50 and soon we will have 60 Clinical and Translational Science Awards scattered all across the country which will also be set up to conduct these sorts of trials for new molecular entities," he added.
This new direction in research funding has involved unprecedented cooperation with the Food and Drug Administration, Dr. Collins said, with an NIH-FDA leadership council formed to ensure that new drug candidates are most safely and efficiently moved into the clinical trials framework in ways that would best enable FDA analysis and validation, particularly for rare diseases.
Dr. Collins was particularly excited about five instances in which NIH is using this new model of helping "de-risk" the drug development process for orphan or neglected diseases through TRND. These include four rare diseases (Niemann-Pick Disease Type C, hereditary inclusion body myopathy, sickle cell disease, and chronic lymphocytic leukemia) and one neglected disease (schistosomiasis).
If and when these compounds and those for other rare diseases become ready for marketing and production, NIH would then work with private companies to achieve licensing agreement to enable their manufacture and sale, according to Dr. Collins.
Dr. Collins reported having no financial conflicts of interest with regard to his presentation.
FROM THE ANNUAL MEETING OF THE AMERICAN SOCIETY OF HUMAN GENETICS