User login
Dust in the Wind
A 52-year-old woman presented with a 4-day history of progressive dyspnea, nonproductive cough, pleuritic chest pain, and subjective fevers. She described dyspnea at rest, which worsened with exertion. She reported no chills, night sweats, weight change, wheezing, hemoptysis, orthopnea, lower extremity edema, or nasal congestion. She also denied myalgia, arthralgia, or joint swelling. She reported no rash, itching, or peripheral lymphadenopathy. She had no seasonal allergies. She was treated for presumed bronchitis with azithromycin by her primary care provider 4 days prior to presentation but experienced progressive dyspnea.
The constellation of dry cough, fever, and dyspnea is often infectious in origin, with the nonproductive, dry cough more suggestive of a viral than bacterial syndrome. Atypical organisms such as Mycoplasma pneumoniae, Legionella pneumophila, and Chlamydia pneumoniae may also present with these symptoms. Noninfectious etiologies should also be considered, including pulmonary embolism, systemic lupus erythematosus, asbestosis, hypersensitivity pneumonitis, sarcoidosis, and lung cancer. The dyspnea at rest stands out as a worrisome feature, as it implies hypoxia; therefore, an oxygen saturation is necessary to quickly determine her peripheral oxygen saturation.
Her past medical history was notable for lung adenocarcinoma, for which she had undergone right upper lobectomy, without chemotherapy or radiation, 13 years ago without recurrence. She had no history of chronic obstructive pulmonary disease, asthma, or pneumonia, nor a family history of chronic obstructive pulmonary disease, asthma, pneumonia, or lung cancer. Her only medication was azithromycin. She drank alcohol on occasion and denied illicit drug use. Three weeks prior to admission, she began smoking 4 to 5 cigarettes per day after 13 years of abstinence. Her smoking history prior to abstinence was 1 pack per day for 20 years. She worked as a department store remodeler; she had no exposure to asbestos, mold, or water-damaged wood. She reported no recent travel, sick contacts, or exposure to animals.
A primary lung neoplasm with a pleural effusion could cause her shortness of breath and pleuritic chest pain. Her history of lung cancer at age 39 raises the possibility of recurrence. For cigarette smokers, a second lung cancer may occur many years after the first diagnosis and treatment, even if they have quit smoking. A review of her original cancer records is essential to confirm the diagnosis of pulmonary adenocarcinoma. What is now being described as pulmonary adenocarcinoma may have been a metastatic lesion arising from outside the lung. Although unlikely, a primary adenocarcinoma may remain active.
Infectious etiologies continue to merit consideration. A parapneumonic effusion from a pneumonia or an empyema are consistent with her symptoms. Systemic lupus erythematosus can cause lung disease with pleural effusions. She does exhibit dyspnea and pleurisy, which are consistent with autoimmune disease, but does not exhibit some of the more typical autoimmune symptoms such as arthralgias, joint swelling, and rash. Pneumothorax could also produce her symptoms; however, pneumothorax usually occurs spontaneously in younger patients or after trauma or a procedure. Remote right upper lobectomy would not be a cause of pneumothorax now. Her reported history makes lung disease or pneumoconiosis due to occupational exposure to mold or aspergillosis a possibility. Legionellosis, histoplasmosis, or coccidioidomycosis should be considered if she lives in or has visited a high-risk area. Pulmonary embolism remains a concern for all patients with new-onset shortness of breath. Decision support tools, such as the Wells criteria, are valuable, but the gestalt of the physician does not lag far behind in accuracy.
Cardiac disease is also in the differential. Bibasilar crackles, third heart sound gallop, and jugular vein distension would suggest heart failure. A pericardial friction rub would be highly suggestive of pericarditis. A paradoxical pulse would raise concern for pericardial tamponade. Pleurisy may be associated with a pericardial effusion, making viral pericarditis and myocarditis possibilities.
She was in moderate distress with tachypnea and increased work of breathing. Her temperature was 36.7°C, heart rate 104 beats per minute, respiratory rate 24 breaths per minute, oxygen saturation was 88% on room air, 94% on 3 liters of oxygen, and blood pressure was 147/61 mmHg. Auscultation of the lungs revealed bibasilar crackles and decreased breath sounds at the bases. She was tachycardic, with a regular rhythm and no appreciable murmurs, rubs, or gallops. There was no jugular venous distention or lower extremity edema. Her thyroid was palpable, without appreciation of nodules. Skin and musculoskeletal examinations were normal.
Unless she is immunocompromised, infection has become lower in the differential, as she is afebrile. Decreased breath sounds at the bases and bibasilar crackles may be due to pleural effusions. Congestive heart failure is a possibility, especially given her dyspnea and bibasilar crackles. Volume overload from renal failure is possible, but she does not have other signs of volume overload such as lower extremity edema or jugular venous distension. It is important to note that crackles may be due to other etiologies, including atelectasis, fibrosis, or pneumonia. Pulmonary embolism may cause hypoxia, tachycardia, and pleural effusions. Additional diseases may present similarly, including human immunodeficiency virus with Pneumocystis jirovecii, causing dyspnea, tachypnea, and tachycardia; hematologic malignancy with anemia, causing dyspnea and tachycardia; and thyrotoxic states with thyromegaly, causing dyspnea and tachycardia. Thyroid storm patients appear in distress, are tachycardic, and may have thyromegaly.
Moderate distress, increased work of breathing, tachycardia, tachypnea, and hypoxia are all worrisome signs. Her temperature is subnormal, although this may not be accurate, as oral temperatures may register lower in patients with increased respiratory rates because of increased air flow across the thermometer. Bibasilar crackles with decreased bibasilar sounds require further investigation. A complete blood count, complete metabolic profile, troponin, arterial blood gas (ABG), electrocardiogram (ECG), and chest radiograph are warranted.
Laboratory studies revealed a white blood cell count of 8600 per mm3 with 11% bands and 7.3% eosinophils, and a hemoglobin count of 15 gm/dL. Basic metabolic panel, liver function tests, coagulation panel, and urinalysis were within normal limits, including serum creatinine 0.7 mg/dL, sodium 143 mmoL/L, chloride 104 mmoL/L, bicarbonate 30 mEq/L, anion gap 9 mmoL/L, and blood urea nitrogen 12 mg/dL. Chest radiograph disclosed diffusely increased interstitial markings and a small left pleural effusion (Figure 1).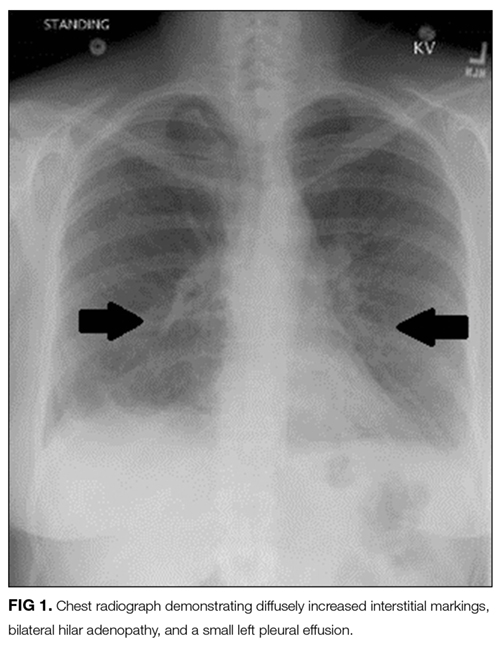
Her bandemia suggests infection. Stress can cause a leukocytosis by demargination of mature white blood cells; however, stress does not often cause immature cells such as bands to appear. Her chest radiograph with diffuse interstitial markings is consistent with a community-acquired pneumonia. Empiric antibiotic therapy should be initiated because of the possibility of community-acquired pneumonia. Recent studies demonstrate that steroids decrease mortality, the need for mechanical ventilation, and the length of stay for patients hospitalized with community-acquired pneumonia; therefore, this patient should also be treated with steroids.
Eosinophilia may be seen in drug reactions, allergies, pulmonary emboli, pleural effusions, and occasionally in malignancy. Eosinophilic pneumonia typically has the “reverse pulmonary edema” picture, with infiltrates in the periphery and not centrally, as in congestive heart failure.
A serum bicarbonate of 30 mEq/L suggests a metabolic compensation for a chronic respiratory acidosis as renal compensation, and rise in bicarbonate generally takes 3 days. She may have been hypoxic longer than her symptoms suggest.
An ABG should be ordered to determine the degree of hypoxia and whether a higher level of care is indicated. The abnormal chest radiograph, along with her hypoxia, merits a closer look at her lung parenchyma with chest computed tomography (CT). A D-dimer would be beneficial to rule out pulmonary embolism. If the D-dimer is positive, chest CT with contrast is indicated to determine if a pulmonary embolism is present. A brain natriuretic peptide would assist in the diagnosis of congestive heart failure. A sputum culture and Gram stain and respiratory viral panel may establish a pathogen for pneumonia. An ECG and troponin to rule out myocardial infarction should be performed as well.
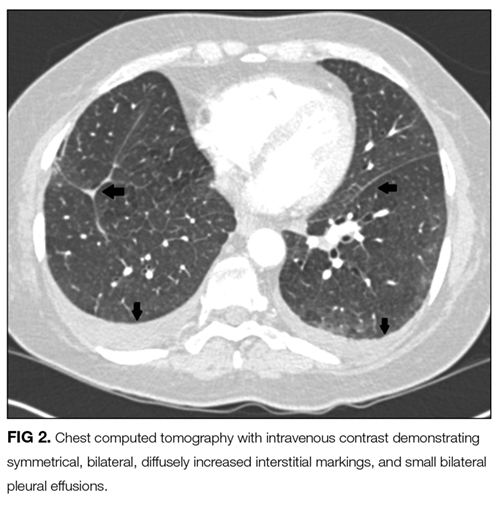
The presence of hilar and subcarinal lymph nodes expands the differential. Stage IV pulmonary sarcoid may present with diffuse infiltrates and nodes, although the acuity in this case makes it less likely. A very aggressive malignancy such as Burkitt lymphoma may have these findings. Acute viral and atypical pneumonias remain possible. Right middle lobe syndrome may cause partial collapse of the right middle lobe. Tuberculosis can be associated with right middle lobe syndrome; however, in this day and age an obstructing mass is more likely the cause. Pulmonary disease, such as cryptogenic organizing pneumonia, idiopathic pulmonary fibrosis, and interstitial lung disease, should be considered in patients with pneumonia unresponsive to antibiotics. Lung biopsy and bronchoalveolar lavage (BAL) would help make the diagnosis and should be the next step, unless her degree of hypoxia is prohibitive. Similarly, thoracentesis with analysis of the pleural fluid for cell count, Gram stain, and culture may help make the diagnosis. Thoracentesis should be done with fluoroscopic guidance, given the risk of pneumothorax, which would further compromise her tenuous respiratory status.
Thoracentesis was attempted, but the pleural effusion was too small to provide a sample. Subsequent serum blood counts with differential showed an increased eosinophilia to 20% and resolved bandemia. Upon further questioning, she recalled several months of extensive, daily, fine-dust exposure from demolition during the remodeling of a new building.
Hypereosinophilia and pulmonary infiltrates narrow the differential considerably to include asthma; parasitic infection, such as the pulmonary phase of ascariasis; exposure, such as to dust, cigarettes, or asbestosis; or hypereosinophilic syndromes characterized by peripheral eosinophilia, along with a tissue eosinophilia, causing organ dysfunction. Idiopathic hypereosinophilic syndrome, a hypereosinophilic syndrome of unknown etiology despite extensive diagnostic testing, is rare, and eosinophilic leukemia even rarer. Her history strongly suggests exposure. Many eosinophilic diseases respond rapidly to steroids, and response to treatment would help narrow the diagnosis. If she does not respond to steroids, a lung and/or bone marrow biopsy would be the next step.
A BAL of the right middle lobe revealed 51% eosinophils, 3% neutrophils, 15% macrophages, and 28% lymphocytes. Gram stain, as well as cultures for bacteria, acid fast bacilli, fungus, herpes simplex virus, and cytomegalovirus cultures, were negative. Transbronchial lung biopsy revealed focal interstitial fibrosis and inflammation, without evidence of infection.
Eosinophils are primarily located in tissues; therefore, peripheral blood eosinophil counts often underestimate the degree of infiltration into end organs such as the lung. With 50% eosinophils, her BAL reflects this. Mold, fungus, chemical, and particle exposure could produce an eosinophilic BAL. She does not appear to be at risk for parasitic exposure. Eosinophilic granulomatosis (previously known as Churg-Strauss) is a consideration, but the lack of signs of vasculitis and wheezing make this less likely. A negative antineutrophil cytoplasmic antibody may provide reassurance. “Fine dust exposure” is consistent with environmental exposure but not a specific antigen. Steroids provide a brisk eosinophil reduction and are appropriate for this patient. There is the possibility of missing infectious or parasitic etiologies; therefore, a culture of BAL fluid should be sent.
Eosinophilic infiltration may lead to fibrosis, as was found on the lung biopsy. She should be counseled to avoid “fine dust exposure” in the future. Follow-up lung imaging and pulmonary function tests (PFTs) should be performed once her acute illness resolves. She should be strongly urged not to smoke tobacco. Interestingly, there are reports that ex-smokers who restart smoking have an increased risk of eosinophilic pneumonia, but in this case dust exposure is the more likely etiology.
She was diagnosed with acute eosinophilic pneumonia (AEP). Antibiotics were discontinued, and oral prednisone was initiated at 40 mg daily, with a brisk response and resolution of her dyspnea. She was discharged with a 6-week prednisone taper. She had no cough, dyspnea, chest pain, or fevers at her follow-up 14 days after discharge. On a 6-week, postdischarge phone call, she continued to report no symptoms, and she maintained abstinence from cigarette smoking.
This case highlights that the very best test in any medical situation is a thorough, detailed history and physical examination. A comprehensive history with physical examination is noninvasive, safe, and cheap. Had the history of fine-dust exposure been known, it is likely that a great deal of testing and money would have been saved. The patient would have been diagnosed and treated earlier, and suffered less.
COMMENTARY
First described in 1989,1,2 AEP is an uncommon cause of acute respiratory failure. Cases have been reported throughout the world, including in the United States, Belgium, Japan, and Iraq.2,3 AEP is an acute febrile illness with cough, chest pain, and dyspnea for fewer than 7 days, diffuse pulmonary infiltrates on chest radiograph, hypoxemia, no history of asthma or atopic disease, no infection, and greater than 25% eosinophils on a BAL.1,3 Physical examination typically reveals fever, tachypnea, and crackles on auscultation.1 Peripheral blood eosinophilia is inconsistently seen at presentation but generally observed as the disease progresses.1 Peripheral eosinophilia at presentation is positively correlated with a milder course of AEP, including higher oxygen saturation and fewer intensive care admissions.4 Acute respiratory failure in AEP progresses rapidly, often within hours.1 Delayed recognition of AEP may lead to respiratory failure, requiring intubation, and even to death.1
Reticular markings with Kerley-B lines, mixed reticular and alveolar infiltrates, and pleural effusions are usually found on chest radiography.1 Bilateral areas of ground-glass attenuation, interlobular septal thickening, bronchovascular bundle thickening, and pleural effusions are seen on chest CT.5 Marked eosinophilic infiltration of the interstitium and alveolar spaces, as well as diffuse alveolar damage with hyaline membrane fibroblast proliferation and inflammatory cells, are present on lung biopsy.1 Restriction with impaired diffusion capacity is found on PFTs. However, PFTs return to normal after recovery.1
AEP is distinguished from other pulmonary diseases by BAL, lung biopsy, symptoms, symptom course, and/or radiographically. AEP is often misdiagnosed as severe community-acquired pneumonia and/or acute respiratory distress syndrome, as AEP tends to occur in previously healthy individuals who have diffuse infiltrates on chest radiograph, fevers, and acute, often severe, respiratory symptoms.1-3 Other eosinophilic lung diseases to rule out include simple pulmonary eosinophilia, chronic eosinophilic pneumonia, eosinophilic granulomatosis with polyangitis (Churg-Strauss), idiopathic hypereosinophilic syndrome, infection, and drug reactions.1,3,5 Simple eosinophilic pneumonia is characterized by no symptoms or very mild pulmonary symptoms and transient patchy infiltrates on radiography.3,5 Patients with simple pulmonary eosinophilia do not have interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions radiographically, as seen with AEP.5 Chronic eosinophilic pneumonia is subacute, with respiratory symptoms of more than 3 months in duration, in contrast with the 7 days of respiratory symptoms for AEP, and is also not associated with interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions on radiography.3,5 Unlike AEP, chronic eosinophilic pneumonia often recurs after the course of steroids has ended.3 In contrast with AEP, eosinophilic granulomatosis with polyangitis is associated with concomitant asthma and the involvement of nonpulmonary organs.3 Idiopathic hypereosinophilic syndrome is characterized by extremely high peripheral eosinophilia and by eosinophilic involvement of multiple organs, and it requires chronic steroid use.3 Patients with allergic bronchopulmonary aspergillosis (ABPA), in contrast with AEP, typically have steroid-dependent asthma and chronic respiratory symptoms.3 ABPA also differs from AEP in that radiographic infiltrates are localized and transient, and the syndrome may relapse after steroid treatment.3 Other infectious etiologies that may present similarly to AEP include invasive pulmonary aspergillosis, pulmonary coccidiodomycosis, Pneumocystis jioveri pneumonia, pulmonary toxocariasis, pulmonary filariasis, paragonimiasis, and Loeffler syndrome (pneumonia due to Strongyloides, Ascaris, or hookworms), highlighting the importance of a thorough travel and exposure history.1,3 Several drugs may cause eosinophilic lung disease, including nitrofurantoin, tetracyclines, phenytoin, L-tryptophan, acetaminophen, ampicillin, heroin, and cocaine, which necessitates a thorough review of medication and illegal drug use.3
Steroids and supportive care are the treatment of choice for AEP, although spontaneous resolution has been seen.1,3 Significant clinical improvement occurs within 24 to 48 hours of steroid initiation.1,3 Optimal dose and duration of therapy have not been determined; however, methylprednisolone 125 mg intravenously every 6 hours until improvement is an often-used option.1 Tapers vary from 2 to 12 weeks with no difference in outcome.1-3 AEP does not recur after appropriate treatment with steroids.1,3
Little is known about the etiology of AEP. It usually occurs in young, healthy individuals and is presumed to be an unusual, acute hypersensitivity reaction to an inhaled allergen.1 A report of 18 US soldiers deployed in or near Iraq proposed dust exposure and cigarette or cigar smoking as a cause of AEP.2 Similar to our patient’s fine-dust exposure and recent onset of cigarette smoking, the soldiers were exposed to the dusty, arid environment for at least 1 day and had been smoking for at least 1 month.2 The authors proposed that small dust particles irritate alveoli, stimulating eosinophils, which are exacerbated by the onset of smoking. Alternatively, cigarette smoke may prime the lung such that dust triggers an inflammatory cascade, resulting in AEP.
TEACHING POINTS
- With the potential for the rapid progression of respiratory failure, it is imperative that the diagnosis of AEP be considered for a patient with diffuse infiltrates on a chest radiograph and acute respiratory failure of unknown cause.
- A thorough history of exposure is key to including AEP in the differential of acute pulmonary disease, with recent-onset cigarette smoking and dust exposure.
- The rapid initiation of steroids leads to a full recovery without recurrence and may be life-saving in AEP.
Disclosure
The authors report no conflicts of interest.
1. Allen J. Acute eosinophilic pneumonia. Semin Respir Crit Care Med. 2006;27:142-147. PubMed
2. Shorr AF, Scoville SL, Cersovsky SB, et al. Acute eosinophilic pneumonia among US military personnel deployed in or near Iraq. JAMA. 2004;292:2997-3005. PubMed
3. Pope-Harman AL, Davis WB, Allen ED, Christoforidis AJ, Allen JN. Acute eosinophilic pneumonia. A summary of 15 cases and review of the literature. Medicine (Baltimore). 1996;75(6):334-342. PubMed
4. Jhun BW, Kim SJ, Kim K, Lee JE. Clinical implications of initial peripheral eosinophilia in acute eosinophilic pneumonia. Respirology. 2014;19:1059-1065. PubMed
5. Daimon T, Johkoh T, Sumikawa H, et al. Acute eosinophilic pneumonia: Thin-section CT findings in 29 patients. Eur J Radiol. 2008;65:462-467. PubMed
A 52-year-old woman presented with a 4-day history of progressive dyspnea, nonproductive cough, pleuritic chest pain, and subjective fevers. She described dyspnea at rest, which worsened with exertion. She reported no chills, night sweats, weight change, wheezing, hemoptysis, orthopnea, lower extremity edema, or nasal congestion. She also denied myalgia, arthralgia, or joint swelling. She reported no rash, itching, or peripheral lymphadenopathy. She had no seasonal allergies. She was treated for presumed bronchitis with azithromycin by her primary care provider 4 days prior to presentation but experienced progressive dyspnea.
The constellation of dry cough, fever, and dyspnea is often infectious in origin, with the nonproductive, dry cough more suggestive of a viral than bacterial syndrome. Atypical organisms such as Mycoplasma pneumoniae, Legionella pneumophila, and Chlamydia pneumoniae may also present with these symptoms. Noninfectious etiologies should also be considered, including pulmonary embolism, systemic lupus erythematosus, asbestosis, hypersensitivity pneumonitis, sarcoidosis, and lung cancer. The dyspnea at rest stands out as a worrisome feature, as it implies hypoxia; therefore, an oxygen saturation is necessary to quickly determine her peripheral oxygen saturation.
Her past medical history was notable for lung adenocarcinoma, for which she had undergone right upper lobectomy, without chemotherapy or radiation, 13 years ago without recurrence. She had no history of chronic obstructive pulmonary disease, asthma, or pneumonia, nor a family history of chronic obstructive pulmonary disease, asthma, pneumonia, or lung cancer. Her only medication was azithromycin. She drank alcohol on occasion and denied illicit drug use. Three weeks prior to admission, she began smoking 4 to 5 cigarettes per day after 13 years of abstinence. Her smoking history prior to abstinence was 1 pack per day for 20 years. She worked as a department store remodeler; she had no exposure to asbestos, mold, or water-damaged wood. She reported no recent travel, sick contacts, or exposure to animals.
A primary lung neoplasm with a pleural effusion could cause her shortness of breath and pleuritic chest pain. Her history of lung cancer at age 39 raises the possibility of recurrence. For cigarette smokers, a second lung cancer may occur many years after the first diagnosis and treatment, even if they have quit smoking. A review of her original cancer records is essential to confirm the diagnosis of pulmonary adenocarcinoma. What is now being described as pulmonary adenocarcinoma may have been a metastatic lesion arising from outside the lung. Although unlikely, a primary adenocarcinoma may remain active.
Infectious etiologies continue to merit consideration. A parapneumonic effusion from a pneumonia or an empyema are consistent with her symptoms. Systemic lupus erythematosus can cause lung disease with pleural effusions. She does exhibit dyspnea and pleurisy, which are consistent with autoimmune disease, but does not exhibit some of the more typical autoimmune symptoms such as arthralgias, joint swelling, and rash. Pneumothorax could also produce her symptoms; however, pneumothorax usually occurs spontaneously in younger patients or after trauma or a procedure. Remote right upper lobectomy would not be a cause of pneumothorax now. Her reported history makes lung disease or pneumoconiosis due to occupational exposure to mold or aspergillosis a possibility. Legionellosis, histoplasmosis, or coccidioidomycosis should be considered if she lives in or has visited a high-risk area. Pulmonary embolism remains a concern for all patients with new-onset shortness of breath. Decision support tools, such as the Wells criteria, are valuable, but the gestalt of the physician does not lag far behind in accuracy.
Cardiac disease is also in the differential. Bibasilar crackles, third heart sound gallop, and jugular vein distension would suggest heart failure. A pericardial friction rub would be highly suggestive of pericarditis. A paradoxical pulse would raise concern for pericardial tamponade. Pleurisy may be associated with a pericardial effusion, making viral pericarditis and myocarditis possibilities.
She was in moderate distress with tachypnea and increased work of breathing. Her temperature was 36.7°C, heart rate 104 beats per minute, respiratory rate 24 breaths per minute, oxygen saturation was 88% on room air, 94% on 3 liters of oxygen, and blood pressure was 147/61 mmHg. Auscultation of the lungs revealed bibasilar crackles and decreased breath sounds at the bases. She was tachycardic, with a regular rhythm and no appreciable murmurs, rubs, or gallops. There was no jugular venous distention or lower extremity edema. Her thyroid was palpable, without appreciation of nodules. Skin and musculoskeletal examinations were normal.
Unless she is immunocompromised, infection has become lower in the differential, as she is afebrile. Decreased breath sounds at the bases and bibasilar crackles may be due to pleural effusions. Congestive heart failure is a possibility, especially given her dyspnea and bibasilar crackles. Volume overload from renal failure is possible, but she does not have other signs of volume overload such as lower extremity edema or jugular venous distension. It is important to note that crackles may be due to other etiologies, including atelectasis, fibrosis, or pneumonia. Pulmonary embolism may cause hypoxia, tachycardia, and pleural effusions. Additional diseases may present similarly, including human immunodeficiency virus with Pneumocystis jirovecii, causing dyspnea, tachypnea, and tachycardia; hematologic malignancy with anemia, causing dyspnea and tachycardia; and thyrotoxic states with thyromegaly, causing dyspnea and tachycardia. Thyroid storm patients appear in distress, are tachycardic, and may have thyromegaly.
Moderate distress, increased work of breathing, tachycardia, tachypnea, and hypoxia are all worrisome signs. Her temperature is subnormal, although this may not be accurate, as oral temperatures may register lower in patients with increased respiratory rates because of increased air flow across the thermometer. Bibasilar crackles with decreased bibasilar sounds require further investigation. A complete blood count, complete metabolic profile, troponin, arterial blood gas (ABG), electrocardiogram (ECG), and chest radiograph are warranted.
Laboratory studies revealed a white blood cell count of 8600 per mm3 with 11% bands and 7.3% eosinophils, and a hemoglobin count of 15 gm/dL. Basic metabolic panel, liver function tests, coagulation panel, and urinalysis were within normal limits, including serum creatinine 0.7 mg/dL, sodium 143 mmoL/L, chloride 104 mmoL/L, bicarbonate 30 mEq/L, anion gap 9 mmoL/L, and blood urea nitrogen 12 mg/dL. Chest radiograph disclosed diffusely increased interstitial markings and a small left pleural effusion (Figure 1).
Her bandemia suggests infection. Stress can cause a leukocytosis by demargination of mature white blood cells; however, stress does not often cause immature cells such as bands to appear. Her chest radiograph with diffuse interstitial markings is consistent with a community-acquired pneumonia. Empiric antibiotic therapy should be initiated because of the possibility of community-acquired pneumonia. Recent studies demonstrate that steroids decrease mortality, the need for mechanical ventilation, and the length of stay for patients hospitalized with community-acquired pneumonia; therefore, this patient should also be treated with steroids.
Eosinophilia may be seen in drug reactions, allergies, pulmonary emboli, pleural effusions, and occasionally in malignancy. Eosinophilic pneumonia typically has the “reverse pulmonary edema” picture, with infiltrates in the periphery and not centrally, as in congestive heart failure.
A serum bicarbonate of 30 mEq/L suggests a metabolic compensation for a chronic respiratory acidosis as renal compensation, and rise in bicarbonate generally takes 3 days. She may have been hypoxic longer than her symptoms suggest.
An ABG should be ordered to determine the degree of hypoxia and whether a higher level of care is indicated. The abnormal chest radiograph, along with her hypoxia, merits a closer look at her lung parenchyma with chest computed tomography (CT). A D-dimer would be beneficial to rule out pulmonary embolism. If the D-dimer is positive, chest CT with contrast is indicated to determine if a pulmonary embolism is present. A brain natriuretic peptide would assist in the diagnosis of congestive heart failure. A sputum culture and Gram stain and respiratory viral panel may establish a pathogen for pneumonia. An ECG and troponin to rule out myocardial infarction should be performed as well.
The presence of hilar and subcarinal lymph nodes expands the differential. Stage IV pulmonary sarcoid may present with diffuse infiltrates and nodes, although the acuity in this case makes it less likely. A very aggressive malignancy such as Burkitt lymphoma may have these findings. Acute viral and atypical pneumonias remain possible. Right middle lobe syndrome may cause partial collapse of the right middle lobe. Tuberculosis can be associated with right middle lobe syndrome; however, in this day and age an obstructing mass is more likely the cause. Pulmonary disease, such as cryptogenic organizing pneumonia, idiopathic pulmonary fibrosis, and interstitial lung disease, should be considered in patients with pneumonia unresponsive to antibiotics. Lung biopsy and bronchoalveolar lavage (BAL) would help make the diagnosis and should be the next step, unless her degree of hypoxia is prohibitive. Similarly, thoracentesis with analysis of the pleural fluid for cell count, Gram stain, and culture may help make the diagnosis. Thoracentesis should be done with fluoroscopic guidance, given the risk of pneumothorax, which would further compromise her tenuous respiratory status.
Thoracentesis was attempted, but the pleural effusion was too small to provide a sample. Subsequent serum blood counts with differential showed an increased eosinophilia to 20% and resolved bandemia. Upon further questioning, she recalled several months of extensive, daily, fine-dust exposure from demolition during the remodeling of a new building.
Hypereosinophilia and pulmonary infiltrates narrow the differential considerably to include asthma; parasitic infection, such as the pulmonary phase of ascariasis; exposure, such as to dust, cigarettes, or asbestosis; or hypereosinophilic syndromes characterized by peripheral eosinophilia, along with a tissue eosinophilia, causing organ dysfunction. Idiopathic hypereosinophilic syndrome, a hypereosinophilic syndrome of unknown etiology despite extensive diagnostic testing, is rare, and eosinophilic leukemia even rarer. Her history strongly suggests exposure. Many eosinophilic diseases respond rapidly to steroids, and response to treatment would help narrow the diagnosis. If she does not respond to steroids, a lung and/or bone marrow biopsy would be the next step.
A BAL of the right middle lobe revealed 51% eosinophils, 3% neutrophils, 15% macrophages, and 28% lymphocytes. Gram stain, as well as cultures for bacteria, acid fast bacilli, fungus, herpes simplex virus, and cytomegalovirus cultures, were negative. Transbronchial lung biopsy revealed focal interstitial fibrosis and inflammation, without evidence of infection.
Eosinophils are primarily located in tissues; therefore, peripheral blood eosinophil counts often underestimate the degree of infiltration into end organs such as the lung. With 50% eosinophils, her BAL reflects this. Mold, fungus, chemical, and particle exposure could produce an eosinophilic BAL. She does not appear to be at risk for parasitic exposure. Eosinophilic granulomatosis (previously known as Churg-Strauss) is a consideration, but the lack of signs of vasculitis and wheezing make this less likely. A negative antineutrophil cytoplasmic antibody may provide reassurance. “Fine dust exposure” is consistent with environmental exposure but not a specific antigen. Steroids provide a brisk eosinophil reduction and are appropriate for this patient. There is the possibility of missing infectious or parasitic etiologies; therefore, a culture of BAL fluid should be sent.
Eosinophilic infiltration may lead to fibrosis, as was found on the lung biopsy. She should be counseled to avoid “fine dust exposure” in the future. Follow-up lung imaging and pulmonary function tests (PFTs) should be performed once her acute illness resolves. She should be strongly urged not to smoke tobacco. Interestingly, there are reports that ex-smokers who restart smoking have an increased risk of eosinophilic pneumonia, but in this case dust exposure is the more likely etiology.
She was diagnosed with acute eosinophilic pneumonia (AEP). Antibiotics were discontinued, and oral prednisone was initiated at 40 mg daily, with a brisk response and resolution of her dyspnea. She was discharged with a 6-week prednisone taper. She had no cough, dyspnea, chest pain, or fevers at her follow-up 14 days after discharge. On a 6-week, postdischarge phone call, she continued to report no symptoms, and she maintained abstinence from cigarette smoking.
This case highlights that the very best test in any medical situation is a thorough, detailed history and physical examination. A comprehensive history with physical examination is noninvasive, safe, and cheap. Had the history of fine-dust exposure been known, it is likely that a great deal of testing and money would have been saved. The patient would have been diagnosed and treated earlier, and suffered less.
COMMENTARY
First described in 1989,1,2 AEP is an uncommon cause of acute respiratory failure. Cases have been reported throughout the world, including in the United States, Belgium, Japan, and Iraq.2,3 AEP is an acute febrile illness with cough, chest pain, and dyspnea for fewer than 7 days, diffuse pulmonary infiltrates on chest radiograph, hypoxemia, no history of asthma or atopic disease, no infection, and greater than 25% eosinophils on a BAL.1,3 Physical examination typically reveals fever, tachypnea, and crackles on auscultation.1 Peripheral blood eosinophilia is inconsistently seen at presentation but generally observed as the disease progresses.1 Peripheral eosinophilia at presentation is positively correlated with a milder course of AEP, including higher oxygen saturation and fewer intensive care admissions.4 Acute respiratory failure in AEP progresses rapidly, often within hours.1 Delayed recognition of AEP may lead to respiratory failure, requiring intubation, and even to death.1
Reticular markings with Kerley-B lines, mixed reticular and alveolar infiltrates, and pleural effusions are usually found on chest radiography.1 Bilateral areas of ground-glass attenuation, interlobular septal thickening, bronchovascular bundle thickening, and pleural effusions are seen on chest CT.5 Marked eosinophilic infiltration of the interstitium and alveolar spaces, as well as diffuse alveolar damage with hyaline membrane fibroblast proliferation and inflammatory cells, are present on lung biopsy.1 Restriction with impaired diffusion capacity is found on PFTs. However, PFTs return to normal after recovery.1
AEP is distinguished from other pulmonary diseases by BAL, lung biopsy, symptoms, symptom course, and/or radiographically. AEP is often misdiagnosed as severe community-acquired pneumonia and/or acute respiratory distress syndrome, as AEP tends to occur in previously healthy individuals who have diffuse infiltrates on chest radiograph, fevers, and acute, often severe, respiratory symptoms.1-3 Other eosinophilic lung diseases to rule out include simple pulmonary eosinophilia, chronic eosinophilic pneumonia, eosinophilic granulomatosis with polyangitis (Churg-Strauss), idiopathic hypereosinophilic syndrome, infection, and drug reactions.1,3,5 Simple eosinophilic pneumonia is characterized by no symptoms or very mild pulmonary symptoms and transient patchy infiltrates on radiography.3,5 Patients with simple pulmonary eosinophilia do not have interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions radiographically, as seen with AEP.5 Chronic eosinophilic pneumonia is subacute, with respiratory symptoms of more than 3 months in duration, in contrast with the 7 days of respiratory symptoms for AEP, and is also not associated with interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions on radiography.3,5 Unlike AEP, chronic eosinophilic pneumonia often recurs after the course of steroids has ended.3 In contrast with AEP, eosinophilic granulomatosis with polyangitis is associated with concomitant asthma and the involvement of nonpulmonary organs.3 Idiopathic hypereosinophilic syndrome is characterized by extremely high peripheral eosinophilia and by eosinophilic involvement of multiple organs, and it requires chronic steroid use.3 Patients with allergic bronchopulmonary aspergillosis (ABPA), in contrast with AEP, typically have steroid-dependent asthma and chronic respiratory symptoms.3 ABPA also differs from AEP in that radiographic infiltrates are localized and transient, and the syndrome may relapse after steroid treatment.3 Other infectious etiologies that may present similarly to AEP include invasive pulmonary aspergillosis, pulmonary coccidiodomycosis, Pneumocystis jioveri pneumonia, pulmonary toxocariasis, pulmonary filariasis, paragonimiasis, and Loeffler syndrome (pneumonia due to Strongyloides, Ascaris, or hookworms), highlighting the importance of a thorough travel and exposure history.1,3 Several drugs may cause eosinophilic lung disease, including nitrofurantoin, tetracyclines, phenytoin, L-tryptophan, acetaminophen, ampicillin, heroin, and cocaine, which necessitates a thorough review of medication and illegal drug use.3
Steroids and supportive care are the treatment of choice for AEP, although spontaneous resolution has been seen.1,3 Significant clinical improvement occurs within 24 to 48 hours of steroid initiation.1,3 Optimal dose and duration of therapy have not been determined; however, methylprednisolone 125 mg intravenously every 6 hours until improvement is an often-used option.1 Tapers vary from 2 to 12 weeks with no difference in outcome.1-3 AEP does not recur after appropriate treatment with steroids.1,3
Little is known about the etiology of AEP. It usually occurs in young, healthy individuals and is presumed to be an unusual, acute hypersensitivity reaction to an inhaled allergen.1 A report of 18 US soldiers deployed in or near Iraq proposed dust exposure and cigarette or cigar smoking as a cause of AEP.2 Similar to our patient’s fine-dust exposure and recent onset of cigarette smoking, the soldiers were exposed to the dusty, arid environment for at least 1 day and had been smoking for at least 1 month.2 The authors proposed that small dust particles irritate alveoli, stimulating eosinophils, which are exacerbated by the onset of smoking. Alternatively, cigarette smoke may prime the lung such that dust triggers an inflammatory cascade, resulting in AEP.
TEACHING POINTS
- With the potential for the rapid progression of respiratory failure, it is imperative that the diagnosis of AEP be considered for a patient with diffuse infiltrates on a chest radiograph and acute respiratory failure of unknown cause.
- A thorough history of exposure is key to including AEP in the differential of acute pulmonary disease, with recent-onset cigarette smoking and dust exposure.
- The rapid initiation of steroids leads to a full recovery without recurrence and may be life-saving in AEP.
Disclosure
The authors report no conflicts of interest.
A 52-year-old woman presented with a 4-day history of progressive dyspnea, nonproductive cough, pleuritic chest pain, and subjective fevers. She described dyspnea at rest, which worsened with exertion. She reported no chills, night sweats, weight change, wheezing, hemoptysis, orthopnea, lower extremity edema, or nasal congestion. She also denied myalgia, arthralgia, or joint swelling. She reported no rash, itching, or peripheral lymphadenopathy. She had no seasonal allergies. She was treated for presumed bronchitis with azithromycin by her primary care provider 4 days prior to presentation but experienced progressive dyspnea.
The constellation of dry cough, fever, and dyspnea is often infectious in origin, with the nonproductive, dry cough more suggestive of a viral than bacterial syndrome. Atypical organisms such as Mycoplasma pneumoniae, Legionella pneumophila, and Chlamydia pneumoniae may also present with these symptoms. Noninfectious etiologies should also be considered, including pulmonary embolism, systemic lupus erythematosus, asbestosis, hypersensitivity pneumonitis, sarcoidosis, and lung cancer. The dyspnea at rest stands out as a worrisome feature, as it implies hypoxia; therefore, an oxygen saturation is necessary to quickly determine her peripheral oxygen saturation.
Her past medical history was notable for lung adenocarcinoma, for which she had undergone right upper lobectomy, without chemotherapy or radiation, 13 years ago without recurrence. She had no history of chronic obstructive pulmonary disease, asthma, or pneumonia, nor a family history of chronic obstructive pulmonary disease, asthma, pneumonia, or lung cancer. Her only medication was azithromycin. She drank alcohol on occasion and denied illicit drug use. Three weeks prior to admission, she began smoking 4 to 5 cigarettes per day after 13 years of abstinence. Her smoking history prior to abstinence was 1 pack per day for 20 years. She worked as a department store remodeler; she had no exposure to asbestos, mold, or water-damaged wood. She reported no recent travel, sick contacts, or exposure to animals.
A primary lung neoplasm with a pleural effusion could cause her shortness of breath and pleuritic chest pain. Her history of lung cancer at age 39 raises the possibility of recurrence. For cigarette smokers, a second lung cancer may occur many years after the first diagnosis and treatment, even if they have quit smoking. A review of her original cancer records is essential to confirm the diagnosis of pulmonary adenocarcinoma. What is now being described as pulmonary adenocarcinoma may have been a metastatic lesion arising from outside the lung. Although unlikely, a primary adenocarcinoma may remain active.
Infectious etiologies continue to merit consideration. A parapneumonic effusion from a pneumonia or an empyema are consistent with her symptoms. Systemic lupus erythematosus can cause lung disease with pleural effusions. She does exhibit dyspnea and pleurisy, which are consistent with autoimmune disease, but does not exhibit some of the more typical autoimmune symptoms such as arthralgias, joint swelling, and rash. Pneumothorax could also produce her symptoms; however, pneumothorax usually occurs spontaneously in younger patients or after trauma or a procedure. Remote right upper lobectomy would not be a cause of pneumothorax now. Her reported history makes lung disease or pneumoconiosis due to occupational exposure to mold or aspergillosis a possibility. Legionellosis, histoplasmosis, or coccidioidomycosis should be considered if she lives in or has visited a high-risk area. Pulmonary embolism remains a concern for all patients with new-onset shortness of breath. Decision support tools, such as the Wells criteria, are valuable, but the gestalt of the physician does not lag far behind in accuracy.
Cardiac disease is also in the differential. Bibasilar crackles, third heart sound gallop, and jugular vein distension would suggest heart failure. A pericardial friction rub would be highly suggestive of pericarditis. A paradoxical pulse would raise concern for pericardial tamponade. Pleurisy may be associated with a pericardial effusion, making viral pericarditis and myocarditis possibilities.
She was in moderate distress with tachypnea and increased work of breathing. Her temperature was 36.7°C, heart rate 104 beats per minute, respiratory rate 24 breaths per minute, oxygen saturation was 88% on room air, 94% on 3 liters of oxygen, and blood pressure was 147/61 mmHg. Auscultation of the lungs revealed bibasilar crackles and decreased breath sounds at the bases. She was tachycardic, with a regular rhythm and no appreciable murmurs, rubs, or gallops. There was no jugular venous distention or lower extremity edema. Her thyroid was palpable, without appreciation of nodules. Skin and musculoskeletal examinations were normal.
Unless she is immunocompromised, infection has become lower in the differential, as she is afebrile. Decreased breath sounds at the bases and bibasilar crackles may be due to pleural effusions. Congestive heart failure is a possibility, especially given her dyspnea and bibasilar crackles. Volume overload from renal failure is possible, but she does not have other signs of volume overload such as lower extremity edema or jugular venous distension. It is important to note that crackles may be due to other etiologies, including atelectasis, fibrosis, or pneumonia. Pulmonary embolism may cause hypoxia, tachycardia, and pleural effusions. Additional diseases may present similarly, including human immunodeficiency virus with Pneumocystis jirovecii, causing dyspnea, tachypnea, and tachycardia; hematologic malignancy with anemia, causing dyspnea and tachycardia; and thyrotoxic states with thyromegaly, causing dyspnea and tachycardia. Thyroid storm patients appear in distress, are tachycardic, and may have thyromegaly.
Moderate distress, increased work of breathing, tachycardia, tachypnea, and hypoxia are all worrisome signs. Her temperature is subnormal, although this may not be accurate, as oral temperatures may register lower in patients with increased respiratory rates because of increased air flow across the thermometer. Bibasilar crackles with decreased bibasilar sounds require further investigation. A complete blood count, complete metabolic profile, troponin, arterial blood gas (ABG), electrocardiogram (ECG), and chest radiograph are warranted.
Laboratory studies revealed a white blood cell count of 8600 per mm3 with 11% bands and 7.3% eosinophils, and a hemoglobin count of 15 gm/dL. Basic metabolic panel, liver function tests, coagulation panel, and urinalysis were within normal limits, including serum creatinine 0.7 mg/dL, sodium 143 mmoL/L, chloride 104 mmoL/L, bicarbonate 30 mEq/L, anion gap 9 mmoL/L, and blood urea nitrogen 12 mg/dL. Chest radiograph disclosed diffusely increased interstitial markings and a small left pleural effusion (Figure 1).
Her bandemia suggests infection. Stress can cause a leukocytosis by demargination of mature white blood cells; however, stress does not often cause immature cells such as bands to appear. Her chest radiograph with diffuse interstitial markings is consistent with a community-acquired pneumonia. Empiric antibiotic therapy should be initiated because of the possibility of community-acquired pneumonia. Recent studies demonstrate that steroids decrease mortality, the need for mechanical ventilation, and the length of stay for patients hospitalized with community-acquired pneumonia; therefore, this patient should also be treated with steroids.
Eosinophilia may be seen in drug reactions, allergies, pulmonary emboli, pleural effusions, and occasionally in malignancy. Eosinophilic pneumonia typically has the “reverse pulmonary edema” picture, with infiltrates in the periphery and not centrally, as in congestive heart failure.
A serum bicarbonate of 30 mEq/L suggests a metabolic compensation for a chronic respiratory acidosis as renal compensation, and rise in bicarbonate generally takes 3 days. She may have been hypoxic longer than her symptoms suggest.
An ABG should be ordered to determine the degree of hypoxia and whether a higher level of care is indicated. The abnormal chest radiograph, along with her hypoxia, merits a closer look at her lung parenchyma with chest computed tomography (CT). A D-dimer would be beneficial to rule out pulmonary embolism. If the D-dimer is positive, chest CT with contrast is indicated to determine if a pulmonary embolism is present. A brain natriuretic peptide would assist in the diagnosis of congestive heart failure. A sputum culture and Gram stain and respiratory viral panel may establish a pathogen for pneumonia. An ECG and troponin to rule out myocardial infarction should be performed as well.
The presence of hilar and subcarinal lymph nodes expands the differential. Stage IV pulmonary sarcoid may present with diffuse infiltrates and nodes, although the acuity in this case makes it less likely. A very aggressive malignancy such as Burkitt lymphoma may have these findings. Acute viral and atypical pneumonias remain possible. Right middle lobe syndrome may cause partial collapse of the right middle lobe. Tuberculosis can be associated with right middle lobe syndrome; however, in this day and age an obstructing mass is more likely the cause. Pulmonary disease, such as cryptogenic organizing pneumonia, idiopathic pulmonary fibrosis, and interstitial lung disease, should be considered in patients with pneumonia unresponsive to antibiotics. Lung biopsy and bronchoalveolar lavage (BAL) would help make the diagnosis and should be the next step, unless her degree of hypoxia is prohibitive. Similarly, thoracentesis with analysis of the pleural fluid for cell count, Gram stain, and culture may help make the diagnosis. Thoracentesis should be done with fluoroscopic guidance, given the risk of pneumothorax, which would further compromise her tenuous respiratory status.
Thoracentesis was attempted, but the pleural effusion was too small to provide a sample. Subsequent serum blood counts with differential showed an increased eosinophilia to 20% and resolved bandemia. Upon further questioning, she recalled several months of extensive, daily, fine-dust exposure from demolition during the remodeling of a new building.
Hypereosinophilia and pulmonary infiltrates narrow the differential considerably to include asthma; parasitic infection, such as the pulmonary phase of ascariasis; exposure, such as to dust, cigarettes, or asbestosis; or hypereosinophilic syndromes characterized by peripheral eosinophilia, along with a tissue eosinophilia, causing organ dysfunction. Idiopathic hypereosinophilic syndrome, a hypereosinophilic syndrome of unknown etiology despite extensive diagnostic testing, is rare, and eosinophilic leukemia even rarer. Her history strongly suggests exposure. Many eosinophilic diseases respond rapidly to steroids, and response to treatment would help narrow the diagnosis. If she does not respond to steroids, a lung and/or bone marrow biopsy would be the next step.
A BAL of the right middle lobe revealed 51% eosinophils, 3% neutrophils, 15% macrophages, and 28% lymphocytes. Gram stain, as well as cultures for bacteria, acid fast bacilli, fungus, herpes simplex virus, and cytomegalovirus cultures, were negative. Transbronchial lung biopsy revealed focal interstitial fibrosis and inflammation, without evidence of infection.
Eosinophils are primarily located in tissues; therefore, peripheral blood eosinophil counts often underestimate the degree of infiltration into end organs such as the lung. With 50% eosinophils, her BAL reflects this. Mold, fungus, chemical, and particle exposure could produce an eosinophilic BAL. She does not appear to be at risk for parasitic exposure. Eosinophilic granulomatosis (previously known as Churg-Strauss) is a consideration, but the lack of signs of vasculitis and wheezing make this less likely. A negative antineutrophil cytoplasmic antibody may provide reassurance. “Fine dust exposure” is consistent with environmental exposure but not a specific antigen. Steroids provide a brisk eosinophil reduction and are appropriate for this patient. There is the possibility of missing infectious or parasitic etiologies; therefore, a culture of BAL fluid should be sent.
Eosinophilic infiltration may lead to fibrosis, as was found on the lung biopsy. She should be counseled to avoid “fine dust exposure” in the future. Follow-up lung imaging and pulmonary function tests (PFTs) should be performed once her acute illness resolves. She should be strongly urged not to smoke tobacco. Interestingly, there are reports that ex-smokers who restart smoking have an increased risk of eosinophilic pneumonia, but in this case dust exposure is the more likely etiology.
She was diagnosed with acute eosinophilic pneumonia (AEP). Antibiotics were discontinued, and oral prednisone was initiated at 40 mg daily, with a brisk response and resolution of her dyspnea. She was discharged with a 6-week prednisone taper. She had no cough, dyspnea, chest pain, or fevers at her follow-up 14 days after discharge. On a 6-week, postdischarge phone call, she continued to report no symptoms, and she maintained abstinence from cigarette smoking.
This case highlights that the very best test in any medical situation is a thorough, detailed history and physical examination. A comprehensive history with physical examination is noninvasive, safe, and cheap. Had the history of fine-dust exposure been known, it is likely that a great deal of testing and money would have been saved. The patient would have been diagnosed and treated earlier, and suffered less.
COMMENTARY
First described in 1989,1,2 AEP is an uncommon cause of acute respiratory failure. Cases have been reported throughout the world, including in the United States, Belgium, Japan, and Iraq.2,3 AEP is an acute febrile illness with cough, chest pain, and dyspnea for fewer than 7 days, diffuse pulmonary infiltrates on chest radiograph, hypoxemia, no history of asthma or atopic disease, no infection, and greater than 25% eosinophils on a BAL.1,3 Physical examination typically reveals fever, tachypnea, and crackles on auscultation.1 Peripheral blood eosinophilia is inconsistently seen at presentation but generally observed as the disease progresses.1 Peripheral eosinophilia at presentation is positively correlated with a milder course of AEP, including higher oxygen saturation and fewer intensive care admissions.4 Acute respiratory failure in AEP progresses rapidly, often within hours.1 Delayed recognition of AEP may lead to respiratory failure, requiring intubation, and even to death.1
Reticular markings with Kerley-B lines, mixed reticular and alveolar infiltrates, and pleural effusions are usually found on chest radiography.1 Bilateral areas of ground-glass attenuation, interlobular septal thickening, bronchovascular bundle thickening, and pleural effusions are seen on chest CT.5 Marked eosinophilic infiltration of the interstitium and alveolar spaces, as well as diffuse alveolar damage with hyaline membrane fibroblast proliferation and inflammatory cells, are present on lung biopsy.1 Restriction with impaired diffusion capacity is found on PFTs. However, PFTs return to normal after recovery.1
AEP is distinguished from other pulmonary diseases by BAL, lung biopsy, symptoms, symptom course, and/or radiographically. AEP is often misdiagnosed as severe community-acquired pneumonia and/or acute respiratory distress syndrome, as AEP tends to occur in previously healthy individuals who have diffuse infiltrates on chest radiograph, fevers, and acute, often severe, respiratory symptoms.1-3 Other eosinophilic lung diseases to rule out include simple pulmonary eosinophilia, chronic eosinophilic pneumonia, eosinophilic granulomatosis with polyangitis (Churg-Strauss), idiopathic hypereosinophilic syndrome, infection, and drug reactions.1,3,5 Simple eosinophilic pneumonia is characterized by no symptoms or very mild pulmonary symptoms and transient patchy infiltrates on radiography.3,5 Patients with simple pulmonary eosinophilia do not have interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions radiographically, as seen with AEP.5 Chronic eosinophilic pneumonia is subacute, with respiratory symptoms of more than 3 months in duration, in contrast with the 7 days of respiratory symptoms for AEP, and is also not associated with interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions on radiography.3,5 Unlike AEP, chronic eosinophilic pneumonia often recurs after the course of steroids has ended.3 In contrast with AEP, eosinophilic granulomatosis with polyangitis is associated with concomitant asthma and the involvement of nonpulmonary organs.3 Idiopathic hypereosinophilic syndrome is characterized by extremely high peripheral eosinophilia and by eosinophilic involvement of multiple organs, and it requires chronic steroid use.3 Patients with allergic bronchopulmonary aspergillosis (ABPA), in contrast with AEP, typically have steroid-dependent asthma and chronic respiratory symptoms.3 ABPA also differs from AEP in that radiographic infiltrates are localized and transient, and the syndrome may relapse after steroid treatment.3 Other infectious etiologies that may present similarly to AEP include invasive pulmonary aspergillosis, pulmonary coccidiodomycosis, Pneumocystis jioveri pneumonia, pulmonary toxocariasis, pulmonary filariasis, paragonimiasis, and Loeffler syndrome (pneumonia due to Strongyloides, Ascaris, or hookworms), highlighting the importance of a thorough travel and exposure history.1,3 Several drugs may cause eosinophilic lung disease, including nitrofurantoin, tetracyclines, phenytoin, L-tryptophan, acetaminophen, ampicillin, heroin, and cocaine, which necessitates a thorough review of medication and illegal drug use.3
Steroids and supportive care are the treatment of choice for AEP, although spontaneous resolution has been seen.1,3 Significant clinical improvement occurs within 24 to 48 hours of steroid initiation.1,3 Optimal dose and duration of therapy have not been determined; however, methylprednisolone 125 mg intravenously every 6 hours until improvement is an often-used option.1 Tapers vary from 2 to 12 weeks with no difference in outcome.1-3 AEP does not recur after appropriate treatment with steroids.1,3
Little is known about the etiology of AEP. It usually occurs in young, healthy individuals and is presumed to be an unusual, acute hypersensitivity reaction to an inhaled allergen.1 A report of 18 US soldiers deployed in or near Iraq proposed dust exposure and cigarette or cigar smoking as a cause of AEP.2 Similar to our patient’s fine-dust exposure and recent onset of cigarette smoking, the soldiers were exposed to the dusty, arid environment for at least 1 day and had been smoking for at least 1 month.2 The authors proposed that small dust particles irritate alveoli, stimulating eosinophils, which are exacerbated by the onset of smoking. Alternatively, cigarette smoke may prime the lung such that dust triggers an inflammatory cascade, resulting in AEP.
TEACHING POINTS
- With the potential for the rapid progression of respiratory failure, it is imperative that the diagnosis of AEP be considered for a patient with diffuse infiltrates on a chest radiograph and acute respiratory failure of unknown cause.
- A thorough history of exposure is key to including AEP in the differential of acute pulmonary disease, with recent-onset cigarette smoking and dust exposure.
- The rapid initiation of steroids leads to a full recovery without recurrence and may be life-saving in AEP.
Disclosure
The authors report no conflicts of interest.
1. Allen J. Acute eosinophilic pneumonia. Semin Respir Crit Care Med. 2006;27:142-147. PubMed
2. Shorr AF, Scoville SL, Cersovsky SB, et al. Acute eosinophilic pneumonia among US military personnel deployed in or near Iraq. JAMA. 2004;292:2997-3005. PubMed
3. Pope-Harman AL, Davis WB, Allen ED, Christoforidis AJ, Allen JN. Acute eosinophilic pneumonia. A summary of 15 cases and review of the literature. Medicine (Baltimore). 1996;75(6):334-342. PubMed
4. Jhun BW, Kim SJ, Kim K, Lee JE. Clinical implications of initial peripheral eosinophilia in acute eosinophilic pneumonia. Respirology. 2014;19:1059-1065. PubMed
5. Daimon T, Johkoh T, Sumikawa H, et al. Acute eosinophilic pneumonia: Thin-section CT findings in 29 patients. Eur J Radiol. 2008;65:462-467. PubMed
1. Allen J. Acute eosinophilic pneumonia. Semin Respir Crit Care Med. 2006;27:142-147. PubMed
2. Shorr AF, Scoville SL, Cersovsky SB, et al. Acute eosinophilic pneumonia among US military personnel deployed in or near Iraq. JAMA. 2004;292:2997-3005. PubMed
3. Pope-Harman AL, Davis WB, Allen ED, Christoforidis AJ, Allen JN. Acute eosinophilic pneumonia. A summary of 15 cases and review of the literature. Medicine (Baltimore). 1996;75(6):334-342. PubMed
4. Jhun BW, Kim SJ, Kim K, Lee JE. Clinical implications of initial peripheral eosinophilia in acute eosinophilic pneumonia. Respirology. 2014;19:1059-1065. PubMed
5. Daimon T, Johkoh T, Sumikawa H, et al. Acute eosinophilic pneumonia: Thin-section CT findings in 29 patients. Eur J Radiol. 2008;65:462-467. PubMed
Morbo Serpentino
A 58-year-old Danish man presented to an urgent care center due to several months of gradually worsening fatigue, weight loss, abdominal pain, and changes in vision . His abdominal pain was diffuse, constant, and moderate in severity. There was no association with meals, and he reported no nausea, vomiting, or change in bowel movements. He also said his vision in both eyes was blurry, but denied diplopia and said the blurring did not improve when either eye w as closed. He denied dysphagia, headache, focal weakness, or sensitivity to bright lights.
Fatigue and weight loss in a middle-aged man are nonspecific complaints that mainly help to alert the clinician that there may be a serious, systemic process lurking. Constant abdominal pain without nausea, vomiting, or change in bowel movements makes intestinal obstruction or a motility disorder less likely. Given that the pain is diffuse, it raises the possibility of an intraperitoneal process or a process within an organ that is irritating the peritoneum.
Worsening of vision can result from disorders anywhere along the visual pathway, including the cornea (keratitis or corneal edema from glaucoma), anterior chamber (uveitis or hyphema), lens (cataracts, dislocations, hyperglycemia), vitreous humor (uveitis), retina (infections, ischemia, detachment, diabetic retinopathy), macula (degenerative disease), optic nerve (optic neuritis), optic chiasm, and the visual projections through the hemispheres to the occipital lobes. To narrow the differential diagnosis, it would be important to inquire about prior eye problems, to measure visual acuity and intraocular pressure, to perform fundoscopic and slit-lamp exams to detect retinal and anterior chamber disorders, respectively, and to assess visual fields. An afferent pupillary defect would suggest optic nerve pathology.
Disorders that could unify the constitutional, abdominal, and visual symptoms include systemic inflammatory diseases, such as sarcoidosis (which has an increased incidence among Northern Europeans), tuberculosis, or cancer. While diabetes mellitus could explain his visual problems, weight loss, and fatigue, the absence of polyuria, polydipsia, or polyphagia argues against this possibility.
The patient had hypercholesterolemia and type 2 diabetes mellitus. Medications were metformin, atorvastatin, and glimepiride. He was a former smoker with 23 pack-years and had quit over 5 years prior. He had not traveled outside of Denmark in 2 years and had no pets at home. He reported being monogamous with his same-sex partner for the past 25 years. He had no significant family history, and he worked at a local hospital as a nurse. He denied any previous ocular history.
On examination, the pulse was 67 beats per minute, temperature was 36.7 degrees Celsius, respiratory rate was 16 breaths per minute, oxygen saturation was 99% while breathing ambient air, and blood pressure was 132/78. Oropharynx demonstrated no thrush or other lesions. The heart rhythm was regular and there were no murmurs. Lungs were clear to auscultation bilaterally. Abdominal exam was normal except for mild tenderness upon palpation in all quadrants, but no masses, organomegaly, rigidity, or rebound tenderness were present. Skin examination revealed several subcutaneous nodules measuring up to 0.5 cm in diameter overlying the right and left posterolateral chest walls. T he nodules were rubbery, pink, nontender, and not warm nor fluctuant. Visual acuity was reduced in both eyes. Extraocular movements were intact, and the pupils reacted to light and accommodated appropriately. The sclerae were injected bilaterally. The remainder of the cranial nerves and neurologic exam were normal. Due to the vision loss , the patient was referred to an ophthalmologist who diagnosed bilateral anterior uveitis.
Though monogamous with his male partner for many years, it is mandatory to consider complications of human immunodeficiency virus infection (HIV ). The absence of oral lesions indicative of a low CD4 count, such as oral hairy leukoplakia or thrush, does not rule out HIV disease. Additional history about his work as a nurse might shed light on his risk of infection, such as airborne exposure to tuberculosis or acquisition of blood-borne pathogens through a needle stick injury. His unremarkable vital signs support the chronicity of his medical condition.
Uveitis can result from numerous causes. When confined to the eye, uncommon hereditary and acquired causes are less likely . In many patients, uveitis arises in the setting of systemic infection or inflammation. The numerous infectious causes of uveitis include syphilis, tuberculosis, toxoplasmosis, cat scratch disease, and viruses such as HIV, West Nile, and Ebola. Among the inflammatory diseases that can cause uveitis are sarcoidosis, inflammatory bowel disease, systemic lupus erythematosus, Behçet disease, and Sjogren syndrome.
Several of these conditions, including tuberculosis and syphilis, may also cause subcutaneous nodules. Both tuberculosis and syphilis can cause skin and gastrointestinal disease. Sarcoidosis could involve the skin, peritoneum, and uvea, and is a possibility in this patient. The dermatologic conditions associated with sarcoidosis are protean and include granulomatous inflammation and nongranulomatous processes such as erythema nodosum. Usually the nodules of erythema nodosum are tender, red or purple, and located on the lower extremities. The lack of tenderness points away from erythema nodosum in this patient. Metastatic cancer can disseminate to the subcutaneous tissue, and the patient’s smoking history and age mandate we consider malignancy. However, skin metastases tend to be hard, not rubbery.
A cost-effective evaluation at this point would include syphilis serologies, HIV testing, testing for tuberculosis with either a purified protein derivative test or interferon gamma release assay, chest radiography, and biopsy of 1 of the lesions on his back.
Laboratory data showed 12,400 white blood cells per cubic milliliter (64% neutrophils, 24% lymphocytes, 9% monocytes, 2% eosinophils, 1% basophils), hemoglobin 7.9 g/dL, mean corpuscular volume 85 fL, platelets 476,000 per cubic milliliter , C-reactive protein 43 mg/ d L (normal < 8 mg/L), gamma-glutamyl-transferase 554 IU/L (normal range 0-45), alkaline phosphatase 865 U/L (normal range 60-200), and erythrocyte sedimentation rate (ESR) 71 mm per hour. International normalized ratio was 1.0, albumin was 3.0 mg/dL, activated partial thromboplastin time was 32 seconds (normal 22 to 35 seconds), and bilirubin was 0.3 mg/dL. Antibodies to HIV , hepatitis C, and hepatitis B surface antigen were not detectable. Electrocardiography ( ECG ) was normal. Plain radiograph of the chest demonstrated multiple nodular lesions bilaterally measuring up to 1 cm with no cavitation. There was a left pleural effusion.
The history and exam findings indicate a serious inflammatory condition affecting his lungs, pleura, eyes, skin, liver, and possibly his peritoneum. In this context, the elevated C-reactive protein and ESR are not helpful in differentiating inflammatory from infectious causes. The constellation of uveitis, pulmonary and cutaneous nodules, and marked abnormalities of liver tests in a middle-aged man of Northern European origin points us toward sarcoidosis. Pleural effusions are not common with sarcoidosis but may occur. However, to avoid premature closure, it is important to consider other possibilities.
Metastatic cancer, including lymphoma, could cause pulmonary and cutaneous nodules and liver involvement, but the chronic time course and uveitis are not consistent with malignancy. Tuberculosis is still a consideration, though one would have expected him to report fevers, night sweats, and, perhaps, exposure to patients with pulmonary tuberculosis in his job as a nurse. Multiple solid pulmonary nodules are also uncommon with pulmonary tuberculosis. Fungal infections such as histoplasmosis can cause skin lesions and pulmonary nodules but do not fit well with uveitis.
At this point, “ tissue is the issue.” A skin nodule would be the easiest site to biopsy. If skin biopsy was not diagnostic, computed tomography (CT) of his chest and abdomen should be performed to identify the next most accessible site for biopsy.
Esophagogastroduodenoscopy (EGD) and colonoscopy showed normal findings, and random biopsies from the stomach and colon were normal. CT of the chest, abdomen, and pelvis performed with the administration of intravenous contrast showed multiple solid opacities in both lung fields up to 1 cm, with enlarged mediastinal and retroperitoneal lymph nodes measuring 1 to 3 cm in diameter, a left pleural effusion, wall thickening in the right colon, and several nonspecific hypodensities in the liver. A punch biopsy taken from the right chest wall lesion demonstrated chronic inflammation without granulomas. The patient underwent CT-guided biopsy of 1 of the right-sided lung nodules, which revealed noncaseating granulomatous inflammation, fibrosis, and necrosis. Neither biopsy contained malignant cells, and additional stains revealed no bacteria, fungi, or acid fast bacilli.
The retroperitoneal and mediastinal adenopathy are indicative of a widely disseminated inflammatory process. Lymphoma continues to be a concern, though uveitis as an initial presenting problem would very unusual. Although biopsy of the chest wall lesion failed to demonstrate granulomatous inflammation, the most parsimonious explanation is that the skin and lung nodules are both related to a single systemic process.
Granulomas form in an attempt to wall off offending agents, whether foreign antigens (talc, certain medications), infectious agents, or self-antigens. Review of histopathology and microbiologic studies are useful first steps. Stains for bacteria, fungi, or acid-fast organisms may diagnose an infectious cause, such as tuberculosis, leprosy, syphilis, fungi, or cat scratch disease. Granulomas in association with vascular inflammation would indicate vasculitis. Other autoimmune considerations include sarcoidosis and Crohn disease. Noncaseating granulomas are typically found in sarcoidosis, cat scratch disease, syphilis, leprosy, or Crohn disease, but do not entirely exclude tuberculosis.
The negative infectious studies and lack of classic features of Crohn disease or other autoimmune diseases further point to sarcoidosis as the etiology of this patient’s illness. A Norwegian dermatologist first described the pathology of sarcoidosis based upon specimens taken from skin nodules. He thought the lesions were sarcoma and described them as, “ multiple benign sarcoid of the skin,” which is where the name “ sarcoidosis” originated.
Diagnosing sarcoidosis requires excluding other mimickers. Additional testing should include syphilis serologies, rheumatoid factor, and antineutrophilic cytoplasmic antibodies. The latter is associated with granulomatosis with polyangiitis and eosinophilic granulomatosis with polyangiitis, either of which may produce granulomatous inflammation of the lungs, skin, and uvea.
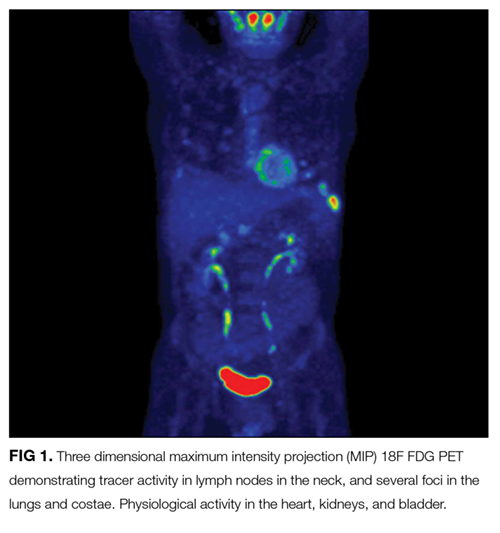
At this juncture, PET-CT represents a costly and unnecessary test that does not narrow our diagnostic possibilities sufficiently to justify its use. Osteolytic lesions would be unusual in sarcoidosis and more likely in lymphoma or infectious processes such as tuberculosis. Tests for syphilis and tuberculosis are required, and are a fraction of the cost of a PET-CT.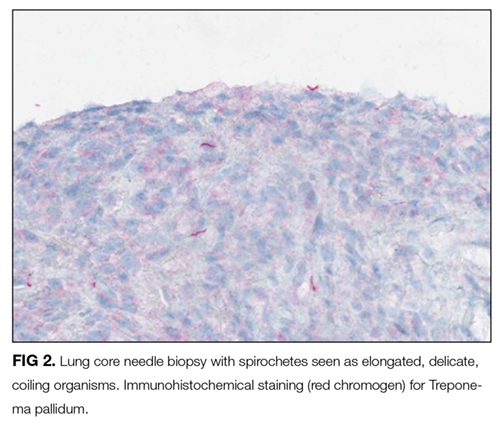
With the biopsy revealing spirochetes, and the positive results of a nontreponemal test (RPR) and confirmatory treponemal results, the diagnosis of syphilis is firmly established. Uveitis indicates neurosyphilis and warrants a longer course of intravenous penicillin. Lumbar puncture should be performed.
A lumbar puncture was performed. Cerebrospinal fluid (CSF) contained 9 white blood cells and 73 red blood cells per cubic milliliter; protein concentration was 73 mg/dL, and glucose was 116 mg/dL. Polymerase chain reaction for T. pallidum was negative. Transthoracic ECG and magnetic resonance imaging of the brain were normal. The patient was treated with intravenous penicillin G at 5 million units 4 times daily for 15 days. A PET-CT scan 3 months later revealed complete resolution of the subcutaneous, pulmonary, liver lesions, lymphadenopathy, and uveitis. Repeat treponemal serologies demonstrated a greater than 4-fold decline in titers.
DISCUSSION
Syphilis is a sexually transmitted disease with increasing incidence worldwide. Untreated infection progresses through 3 stages. The primary stage is characterized by the appearance of a painless chancre after an incubation period of 2 to 3 weeks. Four to 8 weeks later, the secondary stage emerges as a systemic infection, often heralded by a maculopapular rash with desquamation, frequently involving the soles and palms. Hepatitis, iridocyclitis, and early neurosyphilis may also be seen at this stage. Subsequently, syphilis becomes latent. One-third of patients with untreated latent syphilis will develop tertiary syphilis, typified by late neurosyphilis (tabes dorsalis and general paresis), cardiovascular disease (aortitis), or gummatous disease. 1
Gummas are destructive granulomatous lesions that typically present indolently, may occur singly or multiply, and may involve almost any organ. It has been suggested that gummas are the immune system’s defense to slow the bacteria after attempts to kill it have failed. Histologically, gummas are hyalinized nodules with surrounding granulomatous infiltrate of lymphocytes, plasma cells, and multinucleated giant cells with or without necrosis . In the preantibiotic era, gummas were seen in approximately 15% of infected patients, with a latency of 1 to 46 years after primary infection. 2 Penicillin led to a drastic reduction in gummas until the HIV epidemic, which led to the resurgence of gummas at a drastically shortened interval following primary syphilis. 3
Most commonly, gummas affect the skin and bones. In the skin , lesions may be superficial or deep and may progress into ulcerative nodules. In the bones, destructive gummas have a characteristic “moth-eaten” appearance. Less common sequelae of gummas incude gummatous hepatitis, perforated nasal septum (saddle nose deformity), or hard palate erosions. 2,4 R arely, syphilis involves the lungs, appearing as nodules, infiltrates, or pleural effusion. 5
Ocular manifestations occur in approximately 5% of patients with syphilis, more often in secondary and tertiary stages, and are strongly associated with a spread to the central nervous system. Syphilis may affect any structure of the eye, with anterior uveitis as the most frequent manifestation. Partial or complete vision loss is identified in approximately half of the patients with ocular syphilis and may be completely reversed by appropriate treatment. Ophthalmologic findings such as optic neuritis and papilledema imply advanced illness , as do Argyll-Robertson pupils (small pupils that are poorly reactive to light , but with preserved accommodation and convergence). 6,7 The treatment of ocular syphilis is identical to that of neurosyphilis. The Centers for Disease Control and Prevention recommends CSF analysis in any patient with ocular syphilis. Abnormal results should prompt repeat lumbar puncture every 3 to 6 months following treatment until the CSF results normalize. 8
The diagnosis of syphilis relies on indirect serologic tests. T. pallidum cannot be cultured in vitro, and techniques to identify spirochetes directly by using darkfield microscopy or DNA amplification via polymerase chain reaction are limited by availability or by poor sensitivity in advanced syphilis. 1 Imaging modalities including PET cannot reliably differentiate syphilis from other infectious and noninfectious mimickers. 9 F ortunately, syphilis infection can be diagnosed accurately based on reactive treponemal and nontreponemal serum tests. Nontreponemal tests, such as the RPR and Venereal Disease Research Laboratory, have traditionally been utilized as first-line evaluation, followed by a confirmatory treponemal test. However, nontreponemal tests may be nonreactive in a few settings: very early or very late in infection, and in individuals previously treated for syphilis. Thus, newer “reverse testing” algorithms utilize more sensitive and less expensive treponemal tests as the first test, followed by nontreponemal tests if the initial treponemal test is reactive. 8 Regardless of the testing sequence, in patients with no prior history of syphilis, reactive results on both treponemal and nontreponemal assays firmly establish a diagnosis of syphilis, obviating the need for more invasive and costly testing.
In patients with unexplained systemic illness, clinicians should have a low threshold to test for syphilis. Testing should be extended to certain asymptomatic individuals at higher risk of infection, including men who have sex with men, sexual partners of patients infected with syphilis, individuals with HIV or sexually-transmitted diseases, and others with high-risk sexual behavior or a history of sexually-transmitted diseases. 8 As the discussant points out, earlier consideration of and testing for syphilis would have spared the patient from unnecessary and costly EGD, colonoscopy, PET-CT scanning, and 3 biopsies.
Syphilis has been known to be a horribly destructive disease for centuries, earning the moniker “morbo serpentino” (serpentine disease) from the Spanish physician Ruiz Diaz de Isla in the 1500s. 10 In the modern era, physicians must remember to consider the diagnosis of syphilis in order to effectively mitigate the harm from this resurgent disease when it attacks our patients.
TEACHING POINTS
- Syphilis, the great imposter, is rising in incidence and should be on the differential diagnosis in all patients with unexplained multisystem inflammatory disease.
- A cost-effective diagnostic approach to syphilis entails serologic testing with treponemal and nontreponemal assays.
- Unexplained granulomas, especially in the skin, bone, or liver, should prompt consideration of gummatous syphilis.
- Ocular syphilis may involve any part of the visual tract and is treated the same as neurosyphilis.
Disclosure
Dr. Weinreich has received payment for lectures from Boehringer er Ingelheim, Astra Zeneca, TEVA and Novartis in 2016. All other contributors have nothing to report.
1. French P. Syphilis. BMJ. 2007;334:143-147. PubMed
2. Singh AE, Romanowski B. Syphilis: Review with emphasis on clinical, epidemiologic, and some biologic features. Clin Micriobio Rev. 1999;12(2):187-209. PubMed
3. Karp G, Schlaeffer F, Jotkowitz A, Riesenberg K. Syphilis and HIV co-infection. Eur J Int Med. 2009; 20:9-13. PubMed
4. Pilozzi-Edmonds L, Kong LY, Szabo J, Birnbaum LM. Rapid progression to gummatous syphilitic hepatitis and neurosyphilis in a patient with newly diagnosed HIV. Int J STD AIDS. 2014;26(13)985-987. PubMed
5. David G, Perpoint T, Boibieux A, et al. Secondary pulmonary syphilis: report of a likely case and literature review. Clin Infect Dis. 2006;42(3):e11-e15. PubMed
6. Moradi A, Salek S, Daniel E, et al. Clinical features and incidence rates of ocular complications in patients with ocular syphilis. Am J Ophthalmol. 2015;159:334-343. PubMed
7. Aldave AJ, King JA, Cunningham ET Jr. Ocular syphilis. Curr Opin Ophthalmol. 2001;12:433-441. PubMed
8. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137. PubMed
9. Lin M, Darwish B, Chu J. Neurosyphilitic gumma on F18-2-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography: An old disease investigated with new technology. J Clin Neurosc. 2009;16:410-412. PubMed
10. de Ricon‐Ferraz A. Early work on syphilis: Diaz de Ysla’s treatise on the serpentine disease of Hispaniola Island. Int J Dermatol. 1999;38(3):222-227. PubMed
A 58-year-old Danish man presented to an urgent care center due to several months of gradually worsening fatigue, weight loss, abdominal pain, and changes in vision . His abdominal pain was diffuse, constant, and moderate in severity. There was no association with meals, and he reported no nausea, vomiting, or change in bowel movements. He also said his vision in both eyes was blurry, but denied diplopia and said the blurring did not improve when either eye w as closed. He denied dysphagia, headache, focal weakness, or sensitivity to bright lights.
Fatigue and weight loss in a middle-aged man are nonspecific complaints that mainly help to alert the clinician that there may be a serious, systemic process lurking. Constant abdominal pain without nausea, vomiting, or change in bowel movements makes intestinal obstruction or a motility disorder less likely. Given that the pain is diffuse, it raises the possibility of an intraperitoneal process or a process within an organ that is irritating the peritoneum.
Worsening of vision can result from disorders anywhere along the visual pathway, including the cornea (keratitis or corneal edema from glaucoma), anterior chamber (uveitis or hyphema), lens (cataracts, dislocations, hyperglycemia), vitreous humor (uveitis), retina (infections, ischemia, detachment, diabetic retinopathy), macula (degenerative disease), optic nerve (optic neuritis), optic chiasm, and the visual projections through the hemispheres to the occipital lobes. To narrow the differential diagnosis, it would be important to inquire about prior eye problems, to measure visual acuity and intraocular pressure, to perform fundoscopic and slit-lamp exams to detect retinal and anterior chamber disorders, respectively, and to assess visual fields. An afferent pupillary defect would suggest optic nerve pathology.
Disorders that could unify the constitutional, abdominal, and visual symptoms include systemic inflammatory diseases, such as sarcoidosis (which has an increased incidence among Northern Europeans), tuberculosis, or cancer. While diabetes mellitus could explain his visual problems, weight loss, and fatigue, the absence of polyuria, polydipsia, or polyphagia argues against this possibility.
The patient had hypercholesterolemia and type 2 diabetes mellitus. Medications were metformin, atorvastatin, and glimepiride. He was a former smoker with 23 pack-years and had quit over 5 years prior. He had not traveled outside of Denmark in 2 years and had no pets at home. He reported being monogamous with his same-sex partner for the past 25 years. He had no significant family history, and he worked at a local hospital as a nurse. He denied any previous ocular history.
On examination, the pulse was 67 beats per minute, temperature was 36.7 degrees Celsius, respiratory rate was 16 breaths per minute, oxygen saturation was 99% while breathing ambient air, and blood pressure was 132/78. Oropharynx demonstrated no thrush or other lesions. The heart rhythm was regular and there were no murmurs. Lungs were clear to auscultation bilaterally. Abdominal exam was normal except for mild tenderness upon palpation in all quadrants, but no masses, organomegaly, rigidity, or rebound tenderness were present. Skin examination revealed several subcutaneous nodules measuring up to 0.5 cm in diameter overlying the right and left posterolateral chest walls. T he nodules were rubbery, pink, nontender, and not warm nor fluctuant. Visual acuity was reduced in both eyes. Extraocular movements were intact, and the pupils reacted to light and accommodated appropriately. The sclerae were injected bilaterally. The remainder of the cranial nerves and neurologic exam were normal. Due to the vision loss , the patient was referred to an ophthalmologist who diagnosed bilateral anterior uveitis.
Though monogamous with his male partner for many years, it is mandatory to consider complications of human immunodeficiency virus infection (HIV ). The absence of oral lesions indicative of a low CD4 count, such as oral hairy leukoplakia or thrush, does not rule out HIV disease. Additional history about his work as a nurse might shed light on his risk of infection, such as airborne exposure to tuberculosis or acquisition of blood-borne pathogens through a needle stick injury. His unremarkable vital signs support the chronicity of his medical condition.
Uveitis can result from numerous causes. When confined to the eye, uncommon hereditary and acquired causes are less likely . In many patients, uveitis arises in the setting of systemic infection or inflammation. The numerous infectious causes of uveitis include syphilis, tuberculosis, toxoplasmosis, cat scratch disease, and viruses such as HIV, West Nile, and Ebola. Among the inflammatory diseases that can cause uveitis are sarcoidosis, inflammatory bowel disease, systemic lupus erythematosus, Behçet disease, and Sjogren syndrome.
Several of these conditions, including tuberculosis and syphilis, may also cause subcutaneous nodules. Both tuberculosis and syphilis can cause skin and gastrointestinal disease. Sarcoidosis could involve the skin, peritoneum, and uvea, and is a possibility in this patient. The dermatologic conditions associated with sarcoidosis are protean and include granulomatous inflammation and nongranulomatous processes such as erythema nodosum. Usually the nodules of erythema nodosum are tender, red or purple, and located on the lower extremities. The lack of tenderness points away from erythema nodosum in this patient. Metastatic cancer can disseminate to the subcutaneous tissue, and the patient’s smoking history and age mandate we consider malignancy. However, skin metastases tend to be hard, not rubbery.
A cost-effective evaluation at this point would include syphilis serologies, HIV testing, testing for tuberculosis with either a purified protein derivative test or interferon gamma release assay, chest radiography, and biopsy of 1 of the lesions on his back.
Laboratory data showed 12,400 white blood cells per cubic milliliter (64% neutrophils, 24% lymphocytes, 9% monocytes, 2% eosinophils, 1% basophils), hemoglobin 7.9 g/dL, mean corpuscular volume 85 fL, platelets 476,000 per cubic milliliter , C-reactive protein 43 mg/ d L (normal < 8 mg/L), gamma-glutamyl-transferase 554 IU/L (normal range 0-45), alkaline phosphatase 865 U/L (normal range 60-200), and erythrocyte sedimentation rate (ESR) 71 mm per hour. International normalized ratio was 1.0, albumin was 3.0 mg/dL, activated partial thromboplastin time was 32 seconds (normal 22 to 35 seconds), and bilirubin was 0.3 mg/dL. Antibodies to HIV , hepatitis C, and hepatitis B surface antigen were not detectable. Electrocardiography ( ECG ) was normal. Plain radiograph of the chest demonstrated multiple nodular lesions bilaterally measuring up to 1 cm with no cavitation. There was a left pleural effusion.
The history and exam findings indicate a serious inflammatory condition affecting his lungs, pleura, eyes, skin, liver, and possibly his peritoneum. In this context, the elevated C-reactive protein and ESR are not helpful in differentiating inflammatory from infectious causes. The constellation of uveitis, pulmonary and cutaneous nodules, and marked abnormalities of liver tests in a middle-aged man of Northern European origin points us toward sarcoidosis. Pleural effusions are not common with sarcoidosis but may occur. However, to avoid premature closure, it is important to consider other possibilities.
Metastatic cancer, including lymphoma, could cause pulmonary and cutaneous nodules and liver involvement, but the chronic time course and uveitis are not consistent with malignancy. Tuberculosis is still a consideration, though one would have expected him to report fevers, night sweats, and, perhaps, exposure to patients with pulmonary tuberculosis in his job as a nurse. Multiple solid pulmonary nodules are also uncommon with pulmonary tuberculosis. Fungal infections such as histoplasmosis can cause skin lesions and pulmonary nodules but do not fit well with uveitis.
At this point, “ tissue is the issue.” A skin nodule would be the easiest site to biopsy. If skin biopsy was not diagnostic, computed tomography (CT) of his chest and abdomen should be performed to identify the next most accessible site for biopsy.
Esophagogastroduodenoscopy (EGD) and colonoscopy showed normal findings, and random biopsies from the stomach and colon were normal. CT of the chest, abdomen, and pelvis performed with the administration of intravenous contrast showed multiple solid opacities in both lung fields up to 1 cm, with enlarged mediastinal and retroperitoneal lymph nodes measuring 1 to 3 cm in diameter, a left pleural effusion, wall thickening in the right colon, and several nonspecific hypodensities in the liver. A punch biopsy taken from the right chest wall lesion demonstrated chronic inflammation without granulomas. The patient underwent CT-guided biopsy of 1 of the right-sided lung nodules, which revealed noncaseating granulomatous inflammation, fibrosis, and necrosis. Neither biopsy contained malignant cells, and additional stains revealed no bacteria, fungi, or acid fast bacilli.
The retroperitoneal and mediastinal adenopathy are indicative of a widely disseminated inflammatory process. Lymphoma continues to be a concern, though uveitis as an initial presenting problem would very unusual. Although biopsy of the chest wall lesion failed to demonstrate granulomatous inflammation, the most parsimonious explanation is that the skin and lung nodules are both related to a single systemic process.
Granulomas form in an attempt to wall off offending agents, whether foreign antigens (talc, certain medications), infectious agents, or self-antigens. Review of histopathology and microbiologic studies are useful first steps. Stains for bacteria, fungi, or acid-fast organisms may diagnose an infectious cause, such as tuberculosis, leprosy, syphilis, fungi, or cat scratch disease. Granulomas in association with vascular inflammation would indicate vasculitis. Other autoimmune considerations include sarcoidosis and Crohn disease. Noncaseating granulomas are typically found in sarcoidosis, cat scratch disease, syphilis, leprosy, or Crohn disease, but do not entirely exclude tuberculosis.
The negative infectious studies and lack of classic features of Crohn disease or other autoimmune diseases further point to sarcoidosis as the etiology of this patient’s illness. A Norwegian dermatologist first described the pathology of sarcoidosis based upon specimens taken from skin nodules. He thought the lesions were sarcoma and described them as, “ multiple benign sarcoid of the skin,” which is where the name “ sarcoidosis” originated.
Diagnosing sarcoidosis requires excluding other mimickers. Additional testing should include syphilis serologies, rheumatoid factor, and antineutrophilic cytoplasmic antibodies. The latter is associated with granulomatosis with polyangiitis and eosinophilic granulomatosis with polyangiitis, either of which may produce granulomatous inflammation of the lungs, skin, and uvea.
At this juncture, PET-CT represents a costly and unnecessary test that does not narrow our diagnostic possibilities sufficiently to justify its use. Osteolytic lesions would be unusual in sarcoidosis and more likely in lymphoma or infectious processes such as tuberculosis. Tests for syphilis and tuberculosis are required, and are a fraction of the cost of a PET-CT.
With the biopsy revealing spirochetes, and the positive results of a nontreponemal test (RPR) and confirmatory treponemal results, the diagnosis of syphilis is firmly established. Uveitis indicates neurosyphilis and warrants a longer course of intravenous penicillin. Lumbar puncture should be performed.
A lumbar puncture was performed. Cerebrospinal fluid (CSF) contained 9 white blood cells and 73 red blood cells per cubic milliliter; protein concentration was 73 mg/dL, and glucose was 116 mg/dL. Polymerase chain reaction for T. pallidum was negative. Transthoracic ECG and magnetic resonance imaging of the brain were normal. The patient was treated with intravenous penicillin G at 5 million units 4 times daily for 15 days. A PET-CT scan 3 months later revealed complete resolution of the subcutaneous, pulmonary, liver lesions, lymphadenopathy, and uveitis. Repeat treponemal serologies demonstrated a greater than 4-fold decline in titers.
DISCUSSION
Syphilis is a sexually transmitted disease with increasing incidence worldwide. Untreated infection progresses through 3 stages. The primary stage is characterized by the appearance of a painless chancre after an incubation period of 2 to 3 weeks. Four to 8 weeks later, the secondary stage emerges as a systemic infection, often heralded by a maculopapular rash with desquamation, frequently involving the soles and palms. Hepatitis, iridocyclitis, and early neurosyphilis may also be seen at this stage. Subsequently, syphilis becomes latent. One-third of patients with untreated latent syphilis will develop tertiary syphilis, typified by late neurosyphilis (tabes dorsalis and general paresis), cardiovascular disease (aortitis), or gummatous disease. 1
Gummas are destructive granulomatous lesions that typically present indolently, may occur singly or multiply, and may involve almost any organ. It has been suggested that gummas are the immune system’s defense to slow the bacteria after attempts to kill it have failed. Histologically, gummas are hyalinized nodules with surrounding granulomatous infiltrate of lymphocytes, plasma cells, and multinucleated giant cells with or without necrosis . In the preantibiotic era, gummas were seen in approximately 15% of infected patients, with a latency of 1 to 46 years after primary infection. 2 Penicillin led to a drastic reduction in gummas until the HIV epidemic, which led to the resurgence of gummas at a drastically shortened interval following primary syphilis. 3
Most commonly, gummas affect the skin and bones. In the skin , lesions may be superficial or deep and may progress into ulcerative nodules. In the bones, destructive gummas have a characteristic “moth-eaten” appearance. Less common sequelae of gummas incude gummatous hepatitis, perforated nasal septum (saddle nose deformity), or hard palate erosions. 2,4 R arely, syphilis involves the lungs, appearing as nodules, infiltrates, or pleural effusion. 5
Ocular manifestations occur in approximately 5% of patients with syphilis, more often in secondary and tertiary stages, and are strongly associated with a spread to the central nervous system. Syphilis may affect any structure of the eye, with anterior uveitis as the most frequent manifestation. Partial or complete vision loss is identified in approximately half of the patients with ocular syphilis and may be completely reversed by appropriate treatment. Ophthalmologic findings such as optic neuritis and papilledema imply advanced illness , as do Argyll-Robertson pupils (small pupils that are poorly reactive to light , but with preserved accommodation and convergence). 6,7 The treatment of ocular syphilis is identical to that of neurosyphilis. The Centers for Disease Control and Prevention recommends CSF analysis in any patient with ocular syphilis. Abnormal results should prompt repeat lumbar puncture every 3 to 6 months following treatment until the CSF results normalize. 8
The diagnosis of syphilis relies on indirect serologic tests. T. pallidum cannot be cultured in vitro, and techniques to identify spirochetes directly by using darkfield microscopy or DNA amplification via polymerase chain reaction are limited by availability or by poor sensitivity in advanced syphilis. 1 Imaging modalities including PET cannot reliably differentiate syphilis from other infectious and noninfectious mimickers. 9 F ortunately, syphilis infection can be diagnosed accurately based on reactive treponemal and nontreponemal serum tests. Nontreponemal tests, such as the RPR and Venereal Disease Research Laboratory, have traditionally been utilized as first-line evaluation, followed by a confirmatory treponemal test. However, nontreponemal tests may be nonreactive in a few settings: very early or very late in infection, and in individuals previously treated for syphilis. Thus, newer “reverse testing” algorithms utilize more sensitive and less expensive treponemal tests as the first test, followed by nontreponemal tests if the initial treponemal test is reactive. 8 Regardless of the testing sequence, in patients with no prior history of syphilis, reactive results on both treponemal and nontreponemal assays firmly establish a diagnosis of syphilis, obviating the need for more invasive and costly testing.
In patients with unexplained systemic illness, clinicians should have a low threshold to test for syphilis. Testing should be extended to certain asymptomatic individuals at higher risk of infection, including men who have sex with men, sexual partners of patients infected with syphilis, individuals with HIV or sexually-transmitted diseases, and others with high-risk sexual behavior or a history of sexually-transmitted diseases. 8 As the discussant points out, earlier consideration of and testing for syphilis would have spared the patient from unnecessary and costly EGD, colonoscopy, PET-CT scanning, and 3 biopsies.
Syphilis has been known to be a horribly destructive disease for centuries, earning the moniker “morbo serpentino” (serpentine disease) from the Spanish physician Ruiz Diaz de Isla in the 1500s. 10 In the modern era, physicians must remember to consider the diagnosis of syphilis in order to effectively mitigate the harm from this resurgent disease when it attacks our patients.
TEACHING POINTS
- Syphilis, the great imposter, is rising in incidence and should be on the differential diagnosis in all patients with unexplained multisystem inflammatory disease.
- A cost-effective diagnostic approach to syphilis entails serologic testing with treponemal and nontreponemal assays.
- Unexplained granulomas, especially in the skin, bone, or liver, should prompt consideration of gummatous syphilis.
- Ocular syphilis may involve any part of the visual tract and is treated the same as neurosyphilis.
Disclosure
Dr. Weinreich has received payment for lectures from Boehringer er Ingelheim, Astra Zeneca, TEVA and Novartis in 2016. All other contributors have nothing to report.
A 58-year-old Danish man presented to an urgent care center due to several months of gradually worsening fatigue, weight loss, abdominal pain, and changes in vision . His abdominal pain was diffuse, constant, and moderate in severity. There was no association with meals, and he reported no nausea, vomiting, or change in bowel movements. He also said his vision in both eyes was blurry, but denied diplopia and said the blurring did not improve when either eye w as closed. He denied dysphagia, headache, focal weakness, or sensitivity to bright lights.
Fatigue and weight loss in a middle-aged man are nonspecific complaints that mainly help to alert the clinician that there may be a serious, systemic process lurking. Constant abdominal pain without nausea, vomiting, or change in bowel movements makes intestinal obstruction or a motility disorder less likely. Given that the pain is diffuse, it raises the possibility of an intraperitoneal process or a process within an organ that is irritating the peritoneum.
Worsening of vision can result from disorders anywhere along the visual pathway, including the cornea (keratitis or corneal edema from glaucoma), anterior chamber (uveitis or hyphema), lens (cataracts, dislocations, hyperglycemia), vitreous humor (uveitis), retina (infections, ischemia, detachment, diabetic retinopathy), macula (degenerative disease), optic nerve (optic neuritis), optic chiasm, and the visual projections through the hemispheres to the occipital lobes. To narrow the differential diagnosis, it would be important to inquire about prior eye problems, to measure visual acuity and intraocular pressure, to perform fundoscopic and slit-lamp exams to detect retinal and anterior chamber disorders, respectively, and to assess visual fields. An afferent pupillary defect would suggest optic nerve pathology.
Disorders that could unify the constitutional, abdominal, and visual symptoms include systemic inflammatory diseases, such as sarcoidosis (which has an increased incidence among Northern Europeans), tuberculosis, or cancer. While diabetes mellitus could explain his visual problems, weight loss, and fatigue, the absence of polyuria, polydipsia, or polyphagia argues against this possibility.
The patient had hypercholesterolemia and type 2 diabetes mellitus. Medications were metformin, atorvastatin, and glimepiride. He was a former smoker with 23 pack-years and had quit over 5 years prior. He had not traveled outside of Denmark in 2 years and had no pets at home. He reported being monogamous with his same-sex partner for the past 25 years. He had no significant family history, and he worked at a local hospital as a nurse. He denied any previous ocular history.
On examination, the pulse was 67 beats per minute, temperature was 36.7 degrees Celsius, respiratory rate was 16 breaths per minute, oxygen saturation was 99% while breathing ambient air, and blood pressure was 132/78. Oropharynx demonstrated no thrush or other lesions. The heart rhythm was regular and there were no murmurs. Lungs were clear to auscultation bilaterally. Abdominal exam was normal except for mild tenderness upon palpation in all quadrants, but no masses, organomegaly, rigidity, or rebound tenderness were present. Skin examination revealed several subcutaneous nodules measuring up to 0.5 cm in diameter overlying the right and left posterolateral chest walls. T he nodules were rubbery, pink, nontender, and not warm nor fluctuant. Visual acuity was reduced in both eyes. Extraocular movements were intact, and the pupils reacted to light and accommodated appropriately. The sclerae were injected bilaterally. The remainder of the cranial nerves and neurologic exam were normal. Due to the vision loss , the patient was referred to an ophthalmologist who diagnosed bilateral anterior uveitis.
Though monogamous with his male partner for many years, it is mandatory to consider complications of human immunodeficiency virus infection (HIV ). The absence of oral lesions indicative of a low CD4 count, such as oral hairy leukoplakia or thrush, does not rule out HIV disease. Additional history about his work as a nurse might shed light on his risk of infection, such as airborne exposure to tuberculosis or acquisition of blood-borne pathogens through a needle stick injury. His unremarkable vital signs support the chronicity of his medical condition.
Uveitis can result from numerous causes. When confined to the eye, uncommon hereditary and acquired causes are less likely . In many patients, uveitis arises in the setting of systemic infection or inflammation. The numerous infectious causes of uveitis include syphilis, tuberculosis, toxoplasmosis, cat scratch disease, and viruses such as HIV, West Nile, and Ebola. Among the inflammatory diseases that can cause uveitis are sarcoidosis, inflammatory bowel disease, systemic lupus erythematosus, Behçet disease, and Sjogren syndrome.
Several of these conditions, including tuberculosis and syphilis, may also cause subcutaneous nodules. Both tuberculosis and syphilis can cause skin and gastrointestinal disease. Sarcoidosis could involve the skin, peritoneum, and uvea, and is a possibility in this patient. The dermatologic conditions associated with sarcoidosis are protean and include granulomatous inflammation and nongranulomatous processes such as erythema nodosum. Usually the nodules of erythema nodosum are tender, red or purple, and located on the lower extremities. The lack of tenderness points away from erythema nodosum in this patient. Metastatic cancer can disseminate to the subcutaneous tissue, and the patient’s smoking history and age mandate we consider malignancy. However, skin metastases tend to be hard, not rubbery.
A cost-effective evaluation at this point would include syphilis serologies, HIV testing, testing for tuberculosis with either a purified protein derivative test or interferon gamma release assay, chest radiography, and biopsy of 1 of the lesions on his back.
Laboratory data showed 12,400 white blood cells per cubic milliliter (64% neutrophils, 24% lymphocytes, 9% monocytes, 2% eosinophils, 1% basophils), hemoglobin 7.9 g/dL, mean corpuscular volume 85 fL, platelets 476,000 per cubic milliliter , C-reactive protein 43 mg/ d L (normal < 8 mg/L), gamma-glutamyl-transferase 554 IU/L (normal range 0-45), alkaline phosphatase 865 U/L (normal range 60-200), and erythrocyte sedimentation rate (ESR) 71 mm per hour. International normalized ratio was 1.0, albumin was 3.0 mg/dL, activated partial thromboplastin time was 32 seconds (normal 22 to 35 seconds), and bilirubin was 0.3 mg/dL. Antibodies to HIV , hepatitis C, and hepatitis B surface antigen were not detectable. Electrocardiography ( ECG ) was normal. Plain radiograph of the chest demonstrated multiple nodular lesions bilaterally measuring up to 1 cm with no cavitation. There was a left pleural effusion.
The history and exam findings indicate a serious inflammatory condition affecting his lungs, pleura, eyes, skin, liver, and possibly his peritoneum. In this context, the elevated C-reactive protein and ESR are not helpful in differentiating inflammatory from infectious causes. The constellation of uveitis, pulmonary and cutaneous nodules, and marked abnormalities of liver tests in a middle-aged man of Northern European origin points us toward sarcoidosis. Pleural effusions are not common with sarcoidosis but may occur. However, to avoid premature closure, it is important to consider other possibilities.
Metastatic cancer, including lymphoma, could cause pulmonary and cutaneous nodules and liver involvement, but the chronic time course and uveitis are not consistent with malignancy. Tuberculosis is still a consideration, though one would have expected him to report fevers, night sweats, and, perhaps, exposure to patients with pulmonary tuberculosis in his job as a nurse. Multiple solid pulmonary nodules are also uncommon with pulmonary tuberculosis. Fungal infections such as histoplasmosis can cause skin lesions and pulmonary nodules but do not fit well with uveitis.
At this point, “ tissue is the issue.” A skin nodule would be the easiest site to biopsy. If skin biopsy was not diagnostic, computed tomography (CT) of his chest and abdomen should be performed to identify the next most accessible site for biopsy.
Esophagogastroduodenoscopy (EGD) and colonoscopy showed normal findings, and random biopsies from the stomach and colon were normal. CT of the chest, abdomen, and pelvis performed with the administration of intravenous contrast showed multiple solid opacities in both lung fields up to 1 cm, with enlarged mediastinal and retroperitoneal lymph nodes measuring 1 to 3 cm in diameter, a left pleural effusion, wall thickening in the right colon, and several nonspecific hypodensities in the liver. A punch biopsy taken from the right chest wall lesion demonstrated chronic inflammation without granulomas. The patient underwent CT-guided biopsy of 1 of the right-sided lung nodules, which revealed noncaseating granulomatous inflammation, fibrosis, and necrosis. Neither biopsy contained malignant cells, and additional stains revealed no bacteria, fungi, or acid fast bacilli.
The retroperitoneal and mediastinal adenopathy are indicative of a widely disseminated inflammatory process. Lymphoma continues to be a concern, though uveitis as an initial presenting problem would very unusual. Although biopsy of the chest wall lesion failed to demonstrate granulomatous inflammation, the most parsimonious explanation is that the skin and lung nodules are both related to a single systemic process.
Granulomas form in an attempt to wall off offending agents, whether foreign antigens (talc, certain medications), infectious agents, or self-antigens. Review of histopathology and microbiologic studies are useful first steps. Stains for bacteria, fungi, or acid-fast organisms may diagnose an infectious cause, such as tuberculosis, leprosy, syphilis, fungi, or cat scratch disease. Granulomas in association with vascular inflammation would indicate vasculitis. Other autoimmune considerations include sarcoidosis and Crohn disease. Noncaseating granulomas are typically found in sarcoidosis, cat scratch disease, syphilis, leprosy, or Crohn disease, but do not entirely exclude tuberculosis.
The negative infectious studies and lack of classic features of Crohn disease or other autoimmune diseases further point to sarcoidosis as the etiology of this patient’s illness. A Norwegian dermatologist first described the pathology of sarcoidosis based upon specimens taken from skin nodules. He thought the lesions were sarcoma and described them as, “ multiple benign sarcoid of the skin,” which is where the name “ sarcoidosis” originated.
Diagnosing sarcoidosis requires excluding other mimickers. Additional testing should include syphilis serologies, rheumatoid factor, and antineutrophilic cytoplasmic antibodies. The latter is associated with granulomatosis with polyangiitis and eosinophilic granulomatosis with polyangiitis, either of which may produce granulomatous inflammation of the lungs, skin, and uvea.
At this juncture, PET-CT represents a costly and unnecessary test that does not narrow our diagnostic possibilities sufficiently to justify its use. Osteolytic lesions would be unusual in sarcoidosis and more likely in lymphoma or infectious processes such as tuberculosis. Tests for syphilis and tuberculosis are required, and are a fraction of the cost of a PET-CT.
With the biopsy revealing spirochetes, and the positive results of a nontreponemal test (RPR) and confirmatory treponemal results, the diagnosis of syphilis is firmly established. Uveitis indicates neurosyphilis and warrants a longer course of intravenous penicillin. Lumbar puncture should be performed.
A lumbar puncture was performed. Cerebrospinal fluid (CSF) contained 9 white blood cells and 73 red blood cells per cubic milliliter; protein concentration was 73 mg/dL, and glucose was 116 mg/dL. Polymerase chain reaction for T. pallidum was negative. Transthoracic ECG and magnetic resonance imaging of the brain were normal. The patient was treated with intravenous penicillin G at 5 million units 4 times daily for 15 days. A PET-CT scan 3 months later revealed complete resolution of the subcutaneous, pulmonary, liver lesions, lymphadenopathy, and uveitis. Repeat treponemal serologies demonstrated a greater than 4-fold decline in titers.
DISCUSSION
Syphilis is a sexually transmitted disease with increasing incidence worldwide. Untreated infection progresses through 3 stages. The primary stage is characterized by the appearance of a painless chancre after an incubation period of 2 to 3 weeks. Four to 8 weeks later, the secondary stage emerges as a systemic infection, often heralded by a maculopapular rash with desquamation, frequently involving the soles and palms. Hepatitis, iridocyclitis, and early neurosyphilis may also be seen at this stage. Subsequently, syphilis becomes latent. One-third of patients with untreated latent syphilis will develop tertiary syphilis, typified by late neurosyphilis (tabes dorsalis and general paresis), cardiovascular disease (aortitis), or gummatous disease. 1
Gummas are destructive granulomatous lesions that typically present indolently, may occur singly or multiply, and may involve almost any organ. It has been suggested that gummas are the immune system’s defense to slow the bacteria after attempts to kill it have failed. Histologically, gummas are hyalinized nodules with surrounding granulomatous infiltrate of lymphocytes, plasma cells, and multinucleated giant cells with or without necrosis . In the preantibiotic era, gummas were seen in approximately 15% of infected patients, with a latency of 1 to 46 years after primary infection. 2 Penicillin led to a drastic reduction in gummas until the HIV epidemic, which led to the resurgence of gummas at a drastically shortened interval following primary syphilis. 3
Most commonly, gummas affect the skin and bones. In the skin , lesions may be superficial or deep and may progress into ulcerative nodules. In the bones, destructive gummas have a characteristic “moth-eaten” appearance. Less common sequelae of gummas incude gummatous hepatitis, perforated nasal septum (saddle nose deformity), or hard palate erosions. 2,4 R arely, syphilis involves the lungs, appearing as nodules, infiltrates, or pleural effusion. 5
Ocular manifestations occur in approximately 5% of patients with syphilis, more often in secondary and tertiary stages, and are strongly associated with a spread to the central nervous system. Syphilis may affect any structure of the eye, with anterior uveitis as the most frequent manifestation. Partial or complete vision loss is identified in approximately half of the patients with ocular syphilis and may be completely reversed by appropriate treatment. Ophthalmologic findings such as optic neuritis and papilledema imply advanced illness , as do Argyll-Robertson pupils (small pupils that are poorly reactive to light , but with preserved accommodation and convergence). 6,7 The treatment of ocular syphilis is identical to that of neurosyphilis. The Centers for Disease Control and Prevention recommends CSF analysis in any patient with ocular syphilis. Abnormal results should prompt repeat lumbar puncture every 3 to 6 months following treatment until the CSF results normalize. 8
The diagnosis of syphilis relies on indirect serologic tests. T. pallidum cannot be cultured in vitro, and techniques to identify spirochetes directly by using darkfield microscopy or DNA amplification via polymerase chain reaction are limited by availability or by poor sensitivity in advanced syphilis. 1 Imaging modalities including PET cannot reliably differentiate syphilis from other infectious and noninfectious mimickers. 9 F ortunately, syphilis infection can be diagnosed accurately based on reactive treponemal and nontreponemal serum tests. Nontreponemal tests, such as the RPR and Venereal Disease Research Laboratory, have traditionally been utilized as first-line evaluation, followed by a confirmatory treponemal test. However, nontreponemal tests may be nonreactive in a few settings: very early or very late in infection, and in individuals previously treated for syphilis. Thus, newer “reverse testing” algorithms utilize more sensitive and less expensive treponemal tests as the first test, followed by nontreponemal tests if the initial treponemal test is reactive. 8 Regardless of the testing sequence, in patients with no prior history of syphilis, reactive results on both treponemal and nontreponemal assays firmly establish a diagnosis of syphilis, obviating the need for more invasive and costly testing.
In patients with unexplained systemic illness, clinicians should have a low threshold to test for syphilis. Testing should be extended to certain asymptomatic individuals at higher risk of infection, including men who have sex with men, sexual partners of patients infected with syphilis, individuals with HIV or sexually-transmitted diseases, and others with high-risk sexual behavior or a history of sexually-transmitted diseases. 8 As the discussant points out, earlier consideration of and testing for syphilis would have spared the patient from unnecessary and costly EGD, colonoscopy, PET-CT scanning, and 3 biopsies.
Syphilis has been known to be a horribly destructive disease for centuries, earning the moniker “morbo serpentino” (serpentine disease) from the Spanish physician Ruiz Diaz de Isla in the 1500s. 10 In the modern era, physicians must remember to consider the diagnosis of syphilis in order to effectively mitigate the harm from this resurgent disease when it attacks our patients.
TEACHING POINTS
- Syphilis, the great imposter, is rising in incidence and should be on the differential diagnosis in all patients with unexplained multisystem inflammatory disease.
- A cost-effective diagnostic approach to syphilis entails serologic testing with treponemal and nontreponemal assays.
- Unexplained granulomas, especially in the skin, bone, or liver, should prompt consideration of gummatous syphilis.
- Ocular syphilis may involve any part of the visual tract and is treated the same as neurosyphilis.
Disclosure
Dr. Weinreich has received payment for lectures from Boehringer er Ingelheim, Astra Zeneca, TEVA and Novartis in 2016. All other contributors have nothing to report.
1. French P. Syphilis. BMJ. 2007;334:143-147. PubMed
2. Singh AE, Romanowski B. Syphilis: Review with emphasis on clinical, epidemiologic, and some biologic features. Clin Micriobio Rev. 1999;12(2):187-209. PubMed
3. Karp G, Schlaeffer F, Jotkowitz A, Riesenberg K. Syphilis and HIV co-infection. Eur J Int Med. 2009; 20:9-13. PubMed
4. Pilozzi-Edmonds L, Kong LY, Szabo J, Birnbaum LM. Rapid progression to gummatous syphilitic hepatitis and neurosyphilis in a patient with newly diagnosed HIV. Int J STD AIDS. 2014;26(13)985-987. PubMed
5. David G, Perpoint T, Boibieux A, et al. Secondary pulmonary syphilis: report of a likely case and literature review. Clin Infect Dis. 2006;42(3):e11-e15. PubMed
6. Moradi A, Salek S, Daniel E, et al. Clinical features and incidence rates of ocular complications in patients with ocular syphilis. Am J Ophthalmol. 2015;159:334-343. PubMed
7. Aldave AJ, King JA, Cunningham ET Jr. Ocular syphilis. Curr Opin Ophthalmol. 2001;12:433-441. PubMed
8. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137. PubMed
9. Lin M, Darwish B, Chu J. Neurosyphilitic gumma on F18-2-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography: An old disease investigated with new technology. J Clin Neurosc. 2009;16:410-412. PubMed
10. de Ricon‐Ferraz A. Early work on syphilis: Diaz de Ysla’s treatise on the serpentine disease of Hispaniola Island. Int J Dermatol. 1999;38(3):222-227. PubMed
1. French P. Syphilis. BMJ. 2007;334:143-147. PubMed
2. Singh AE, Romanowski B. Syphilis: Review with emphasis on clinical, epidemiologic, and some biologic features. Clin Micriobio Rev. 1999;12(2):187-209. PubMed
3. Karp G, Schlaeffer F, Jotkowitz A, Riesenberg K. Syphilis and HIV co-infection. Eur J Int Med. 2009; 20:9-13. PubMed
4. Pilozzi-Edmonds L, Kong LY, Szabo J, Birnbaum LM. Rapid progression to gummatous syphilitic hepatitis and neurosyphilis in a patient with newly diagnosed HIV. Int J STD AIDS. 2014;26(13)985-987. PubMed
5. David G, Perpoint T, Boibieux A, et al. Secondary pulmonary syphilis: report of a likely case and literature review. Clin Infect Dis. 2006;42(3):e11-e15. PubMed
6. Moradi A, Salek S, Daniel E, et al. Clinical features and incidence rates of ocular complications in patients with ocular syphilis. Am J Ophthalmol. 2015;159:334-343. PubMed
7. Aldave AJ, King JA, Cunningham ET Jr. Ocular syphilis. Curr Opin Ophthalmol. 2001;12:433-441. PubMed
8. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137. PubMed
9. Lin M, Darwish B, Chu J. Neurosyphilitic gumma on F18-2-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography: An old disease investigated with new technology. J Clin Neurosc. 2009;16:410-412. PubMed
10. de Ricon‐Ferraz A. Early work on syphilis: Diaz de Ysla’s treatise on the serpentine disease of Hispaniola Island. Int J Dermatol. 1999;38(3):222-227. PubMed
© 2017 Society of Hospital Medicine
Mass Confusion
A 57-year-old woman presented to the emergency department of a community hospital with a 2-week history of dizziness, blurred vision, and poor coordination following a flu-like illness. Symptoms were initially attributed to complications from a presumed viral illness, but when they persisted for 2 weeks, she underwent magnetic resonance imaging (MRI) of the brain, which was reported as showing a 2.4 x 2.3 x 1.9 cm right frontal lobe mass with mild mass effect and contrast enhancement (Figure 1). She was discharged home at her request with plans for outpatient follow-up.
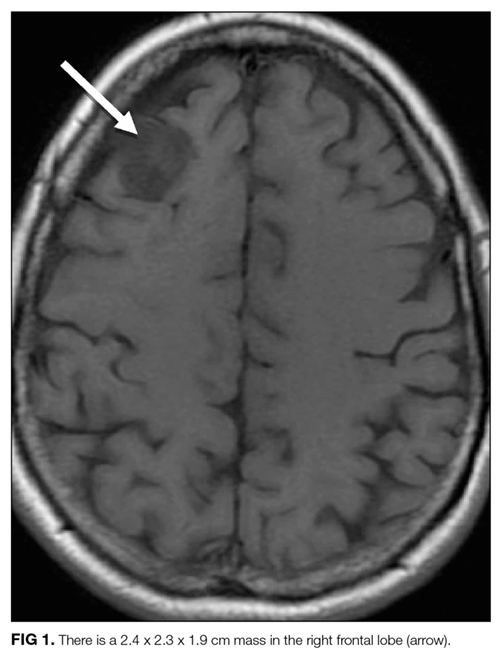
Brain masses are usually neoplastic, infectious, or less commonly, inflammatory. The isolated lesion in the right frontal lobe is unlikely to explain her symptoms, which are more suggestive of multifocal disease or elevated intracranial pressure. Although the frontal eye fields could be affected by the mass, such lesions usually cause tonic eye deviation, not blurry vision; furthermore, coordination, which is impaired here, is not governed by the frontal lobe.
Two weeks later, she returned to the same emergency department with worsening symptoms and new bilateral upper extremity dystonia, confusion, and visual hallucinations. Cerebrospinal fluid (CSF) analysis revealed clear, nonxanthochromic fluid with 4 nucleated cells (a differential was not performed), 113 red blood cells, glucose of 80 mg/dL (normal range, 50-80 mg/dL), and protein of 52 mg/dL (normal range, 15-45 mg/dL).
Confusion is generally caused by a metabolic, infectious, structural, or toxic etiology. Standard CSF test results are usually normal with most toxic or metabolic encephalopathies. The absence of significant CSF inflammation argues against infectious encephalitis; paraneoplastic and autoimmune encephalitis, however, are still possible. The CSF red blood cells were likely due to a mildly traumatic tap, but also may have arisen from the frontal lobe mass or a more diffuse invasive process, although the lack of xanthochromia argues against this. Delirium and red blood cells in the CSF should trigger consideration of herpes simplex virus (HSV) encephalitis, although the time course is a bit too protracted and the reported MRI findings do not suggest typical medial temporal lobe involvement.
The disparate neurologic findings suggest a multifocal process, perhaps embolic (eg, endocarditis), ischemic (eg, intravascular lymphoma), infiltrative (eg, malignancy, neurosarcoidosis), or demyelinating (eg, postinfectious acute disseminated encephalomyelitis, multiple sclerosis). However, most of these would have been detected on the initial MRI. Upper extremity dystonia would likely localize to the basal ganglia, whereas confusion and visual hallucinations are more global. The combination of a movement disorder and visual hallucinations is seen in Lewy body dementia, but this tempo is not typical.
Although the CSF does not have pleocytosis, her original symptoms were flu-like; therefore, CSF testing for viruses (eg, enterovirus) is reasonable. Bacterial, mycobacteria, and fungal studies are apt to be unrevealing, but CSF cytology, IgG index, and oligoclonal bands may be useful. Should the encephalopathy progress further and the general medical evaluation prove to be normal, then tests for autoimmune disorders (eg, antinuclear antibodies, NMDAR, paraneoplastic disorders) and rare causes of rapidly progressive dementias (eg, prion diseases) should be sent.
Additional CSF studies including HSV polymerase chain reaction (PCR), West Nile PCR, Lyme antibody, paraneoplastic antibodies, and cytology were sent. Intravenous acyclovir was administered. The above studies, as well as Gram stain, acid-fast bacillus stain, fungal stain, and cultures, were negative. She was started on levetiracetam for seizure prevention due to the mass lesion. An electroencephalogram (EEG) was reported as showing diffuse background slowing with superimposed semiperiodic sharp waves with a right hemispheric emphasis. Intravenous immunoglobulin (IVIG) 0.4 mg/kg/day over 5 days was administered with no improvement. The patient was transferred to an academic medical center for further evaluation.
The EEG reflects encephalopathy without pointing to a specific diagnosis. Prophylactic antiepileptic medications are not indicated for CNS mass lesions without clinical or electrophysiologic seizure activity. IVIG is often administered when an autoimmune encephalitis is suspected, but the lack of response does not rule out an autoimmune condition.
Her medical history included bilateral cataract extraction, right leg fracture, tonsillectomy, and total abdominal hysterectomy. She had a 25-year smoking history and a family history of lung cancer. She had no history of drug or alcohol use. On examination, her temperature was 37.9°C, blood pressure of 144/98 mm Hg, respiratory rate of 18 breaths per minute, a heart rate of 121 beats per minute, and oxygen saturation of 97% on ambient air. Her eyes were open but she was nonverbal. Her chest was clear to auscultation. Heart sounds were distinct and rhythm was regular. Abdomen was soft and nontender with no organomegaly. Skin examination revealed no rash. Her pupils were equal, round, and reactive to light. She did not follow verbal or gestural commands and intermittently tracked with her eyes, but not consistently enough to characterize extraocular movements. Her face was symmetric. She had a normal gag and blink reflex and an increased jaw jerk reflex. Her arms were flexed with increased tone. She had a positive palmo-mental reflex. She had spontaneous movement of all extremities. She had symmetric, 3+ reflexes of the patella and Achilles tendon with a bilateral Babinski’s sign. Sensation was intact only to withdrawal from noxious stimuli.
The physical exam does not localize to a specific brain region, but suggests a diffuse brain process. There are multiple signs of upper motor neuron involvement, including increased tone, hyperreflexia, and Babinski (plantar flexion) reflexes. A palmo-mental reflex signifies pathology in the cerebrum. Although cranial nerve testing is limited, there are no features of cranial neuropathy; similarly, no pyramidal weakness or sensory deficit has been demonstrated on limited testing. The differential diagnosis of her rapidly progressive encephalopathy includes autoimmune or paraneoplastic encephalitis, diffuse infiltrative malignancy, metabolic diseases (eg, porphyria, heavy metal intoxication), and prion disease.
Her family history of lung cancer and her smoking increases the possibility of paraneoplastic encephalitis, which often has subacute behavioral changes that precede complete neurologic impairment. Inflammatory or hemorrhagic CSF is seen with Balamuthia amoebic infection, which causes a granulomatous encephalitis and is characteristically associated with a mass lesion. Toxoplasmosis causes encephalitis that can be profound, but patients are usually immunocompromised and there are typically multiple lesions.
Laboratory results showed a normal white blood cell count and differential, basic metabolic profile and liver function tests, and C-reactive protein. Human immunodeficiency virus antibody testing was negative. Chest radiography and computed tomography of chest, abdomen, and pelvis were normal. A repeat MRI of the brain with contrast was reported as showing a 2.4 x 2.3 x 1.9 cm heterogeneously enhancing mass in the right frontal lobe with an enhancing dural tail and underlying hyperostosis consistent with a meningioma, and blooming within the mass consistent with prior hemorrhage. No mass effect was present.
The meningioma was resected 3 days after admission but her symptoms did not improve. Routine postoperative MRI was reported to show expected postsurgical changes but no infarct. Brain biopsy at the time of the operation was reported as meningioma and mild gliosis without encephalitis.
The reported MRI findings showing unchanged size and overall appearance of the mass, its connection to the dura and skull, and the pathology results all suggest that the mass is a meningioma. There is no evidence of disease outside of the CNS. Some cancers that provoke a paraneoplastic response can be quite small yet may incite an immune encephalitis; anti-NMDAR-mediated encephalitis can occur with malignancy (often ovarian), although it also arises in the absence of any tumor. Any inclination to definitively exclude conditions not seen on the brain biopsy must be tempered by the limited sensitivity of brain histology examination. Still, what was not seen warrants mention: vascular inflammation suggestive of CNS vasculitis, granulomas that might point to neurosarcoidosis, malignant cells of an infiltrating lymphoma or glioma, or inflammatory cells suggestive of encephalitis. Prion encephalopathy remains possible.
The patient remained unresponsive. A repeat EEG showed bilateral generalized periodic epileptiform discharges with accompanying twitching of the head, face, and left arm, which were suppressed with intravenous propofol and levetiracetam. Three weeks following meningioma resection, a new MRI was read as showing new abnormal signal in the right basal ganglia, abnormality of the cortex on the diffusion weighted images, and progressive generalized volume loss.
Among the aforementioned diagnoses, focal or diffuse periodic epileptiform discharges at 1-2 hertz are most characteristic of prion disease. Striatal and cortical transverse relaxation time (T2)-weighted and diffusion-weighted imaging (DWI) hyperintensities with corresponding restricted diffusion is characteristic of Creutzfeldt-Jakob disease (CJD), although metabolic disorders, seizures, and encephalitis can very rarely show similar MRI findings. The clinical course, the MRI and EEG findings, and nondiagnostic biopsy results, which were initially not assessed for prion disease, collectively point to prion disease. Detection of abnormal prion protein in the brain tissue by immunohistochemistry or molecular methods would confirm the diagnosis.
Review of the original right frontal cortex biopsy specimen at the National Prion Disease Pathology Surveillance Center, including immunostaining with 3F4, a monoclonal antibody to the prion protein, revealed granular deposits typical of prion disease. This finding established a diagnosis of prion disease, likely sporadic CJD. The patient was transitioned to palliative care and died shortly thereafter.
Brain autopsy showed regions with transcortical vacuolation (spongiform change), other cortical regions with varying degrees of vacuolation, abundant reactive astrocytes, paucity of neurons, and dark shrunken neurons. Vacuolation and gliosis were observed in the striatum and were most pronounced in the thalamus. There was no evidence of an inflammatory infiltrate or a neoplastic process. These findings with the positive 3F4 immunohistochemistry and positive Western blot from brain autopsy, as well as the absence of a mutation in the prion protein gene, were diagnostic for CJD.
An investigation was initiated to track the nondisposable surgical instruments used in the meningioma resection that may have been subsequently used in other patients. It was determined that 52 neurosurgical patients may have been exposed to prion-contaminated instruments. The instruments were subsequently processed specifically for prion decontamination. After 7 years, no cases of CJD have been diagnosed in the potentially exposed patients.
DISCUSSION
CJD is a rare neurodegenerative condition1 classified as one of the transmissible spongiform encephalopathies, so called because of the characteristic spongiform pattern (vacuolation) seen on histology, as well as the presence of neuronal loss, reactive gliosis in the gray matter, and the accumulation of the abnormal isoform of the cellular prion protein.2 It affects about one person in every one million people per year worldwide; in the United States there are about 300 cases per year. The most common form of human prion disease, sporadic CJD, is relentlessly progressive and invariably fatal, and in most cases, death occurs less than 5 months from onset.3 There is no cure, although temporizing treatments for symptoms can be helpful.
Sporadic CJD, which accounts for approximately 85% of all cases of prion disease in humans, typically manifests with rapidly progressive dementia and myoclonus after a prolonged incubation period in persons between 55 and 75 years of age. Genetic forms account for approximately 15% and acquired forms less than 1% of human prion diseases.1 Prion diseases have a broad spectrum of clinical manifestations, including dementia, ataxia, parkinsonism, myoclonus, insomnia, paresthesias, and abnormal or changed behavior.4 Given the protean clinical manifestations of prion diseases and rarity, the diagnosis is challenging to make antemortem. One recent study showed that most patients receive about 4 misdiagnoses and are often two-thirds of the way through their disease course before the correct diagnosis of sporadic CJD is made.5
Testing for protein markers of rapid neuronal injury8 in the CSF including 14-3-3, total tau, and neuron-specific enolase can increase suspicion for CJD, although there is a 10%-50% false positive rate with these markers.9 In this case, those tests were not performed; positive results would have been even more nonspecific in the setting of an enhancing brain mass and recent brain surgery.
Although not available at the time this patient was evaluated, the real-time quaking-induced conversion (RT-QuIC) test performed in CSF is diagnostically helpful, and, if positive, supportive of the MRI findings. The sensitivity and specificity of this test have been reported to be between 87%-91% and 98%-100%, respectively, albeit with limited data.10 Applying RT-QuIC to nasal mucosal brushings might lead to even higher sensitivity and specificity.11Seeking a premortem diagnosis for a rare disease with no known cure may seem superfluous, but it has important implications for establishing prognosis, limiting subsequent diagnostic and therapeutic measures, and safeguarding of other patients and operating room personnel. Iatrogenic CJD has occurred following invasive procedures involving neurosurgical instrumentation.12 CJD has been transmitted from grafts of dura mater, transplanted corneas, implantation of inadequately sterilized electrodes in the brain, and in the early 1980s, injections of contaminated pituitary hormones (particularly growth hormone) derived from human pituitary glands taken from cadavers. Since CJD was first described in the 1920s, less than 1% of human prion cases have been acquired iatrogenically.13In patients with rapidly progressive cognitive decline who warrant brain biopsy or surgery, the probability of prion diseases should be assessed based on clinical information and the results of MRI, EEG, and CSF testing. If prion disease is plausible, World Health Organization14 precautions should be employed for neuroinvasive procedures to reduce transmission risk. Disposable equipment should be used when possible, and nondisposable neurosurgical instruments should be quarantined until a nonprion disease diagnosis is identified, or should be regarded as contaminated and reprocessed using the aforementioned protocol.
This case highlights the challenges of seeking the correct diagnosis and its consequences, especially from an infection control perspective. The initial imaging finding of a mass lesion (a meningioma—which is a common incidental finding in older adults15) was a red herring that initially obscured the correct diagnosis. The patient’s progressive cognitive decline, EEG results, and evolving MRI findings, however, prompted further scrutiny of the brain biopsy specimen that eventually steered the clinicians away from mass confusion to diagnostic certainty.
TEACHING POINTS
- Rapidly progressive dementias (RPD) are characterized by cognitive decline over weeks to months. The RPD differential diagnosis includes fulminant forms of common neurodegenerative disorders (eg, Alzheimer’s disease, dementia with Lewy bodies, frontotemporal dementia spectrum), autoimmune encephalidites, CNS cancers, and prion disease.
- Sporadic CJD is the most common human prion disease. It is a rare neurodegenerative condition with onset usually between the ages of 50 and 70 years, and most commonly manifests with rapidly progressive dementia, ataxia, and myoclonus.
- Because of its protean manifestations, the diagnosis of CJD is difficult to make antemortem, and diagnosis is often delayed. Specialist evaluation of brain MRI DWI sequences and new CSF diagnostic tests may allow for earlier diagnosis, which has management and infection control implications.
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. Dr Geschwind’s institution has received R01 grant funding from NIH/NIA; and Alliance Biosecure and the Michael J Homer Family Fund as paid money to his institution, Dr Geschwind has received consulting fees or honoraria from Best Doctors, Kendall Brill & Kelly, CJD Foundation, and Tau Consortium; Dr Geschwind is a consultant for Gerson Lehrman Group, Biohaven Pharmaceuticals, and Advance Medical, outside the submitted work; has grants/grantspending with Quest, Cure PSP, and Tau Consortium, and received payment for lectures from Multiple Grand Rounds Lectures, outside the submitted work. Dr Saint is on a medical advisory board of Doximity, has received honorarium for being a member of the medical advisory board; he is also on the scientifice advisory board of Jvion. Dr Safdar’s institution has received a grant from the VA Patient Safety Center.
1. Brown P, Gibbs CJ, Jr., Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513-529. PubMed
2. Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:913-920. PubMed
3. Johnson RT, Gibbs CJ, Jr. Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998;339:1994-2004. PubMed
4. Will RG, Alpers MP, Dormont D, Schonberger LB. Infectious and sporadic prion diseases. In: Prusiner SB, ed. Prion biology and diseases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999:465-507. \
5. Paterson RW, Torres-Chae CC, Kuo AL, et al. Differential diagnosis of Jakob-Creutzfeldt disease. Arch Neurol. 2012;69:1578-1582. PubMed
6. Tschampa HJ, Kallenberg K, Kretzschmar HA, et al. Pattern of cortical changes in sporadic Creutzfeldt-Jakob disease. AJNR Am J Neuroradiol. 2007;28:1114-1118. PubMed
7. Carswell C, Thompson A, Lukic A, et al. MRI findings are often missed in the diagnosis of Creutzfeldt-Jakob disease. BMC Neurol. 2012;12:153. PubMed
8. Geschwind MD, Martindale J, Miller D, et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2003;60:813-816. PubMed
9. Burkhard PR, Sanchez JC, Landis T, Hochstrasser DF. CSF detection of the 14-3-3 protein in unselected patients with dementia. Neurology. 2001;56:1528-1533. PubMed
10. Orrú CD, Groveman BR, Hughson AG, Zanusso G, Coulthart MB, Caughey B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio. 2015;6:pii: e02451-14 PubMed
11. Orrú CD, Bongianni M, Tonoli G, et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med. 2014;371:519-529. PubMed
12. Brown P, Preece M, Brandel JP, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55:1075-1081. PubMed
13. Brown P, Brandel JP, Sato T, et al. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis. 2012;18:901-907. PubMed
14. WHO infection control guidelines for transmissible spongiform encephalopathies. Report of a WHO consultation, Geneva, Switzerland, 23-26 March 1999. http://www.who.int/csr/resources/publications/bse/whocdscsraph2003.pdf. Accessed on July 10, 2017.
15. Bondy M, Ligon BL. Epidemiology and etiology of intracranial meningiomas: a review. J Neurooncol. 1996;29:197-205. PubMed
A 57-year-old woman presented to the emergency department of a community hospital with a 2-week history of dizziness, blurred vision, and poor coordination following a flu-like illness. Symptoms were initially attributed to complications from a presumed viral illness, but when they persisted for 2 weeks, she underwent magnetic resonance imaging (MRI) of the brain, which was reported as showing a 2.4 x 2.3 x 1.9 cm right frontal lobe mass with mild mass effect and contrast enhancement (Figure 1). She was discharged home at her request with plans for outpatient follow-up.
Brain masses are usually neoplastic, infectious, or less commonly, inflammatory. The isolated lesion in the right frontal lobe is unlikely to explain her symptoms, which are more suggestive of multifocal disease or elevated intracranial pressure. Although the frontal eye fields could be affected by the mass, such lesions usually cause tonic eye deviation, not blurry vision; furthermore, coordination, which is impaired here, is not governed by the frontal lobe.
Two weeks later, she returned to the same emergency department with worsening symptoms and new bilateral upper extremity dystonia, confusion, and visual hallucinations. Cerebrospinal fluid (CSF) analysis revealed clear, nonxanthochromic fluid with 4 nucleated cells (a differential was not performed), 113 red blood cells, glucose of 80 mg/dL (normal range, 50-80 mg/dL), and protein of 52 mg/dL (normal range, 15-45 mg/dL).
Confusion is generally caused by a metabolic, infectious, structural, or toxic etiology. Standard CSF test results are usually normal with most toxic or metabolic encephalopathies. The absence of significant CSF inflammation argues against infectious encephalitis; paraneoplastic and autoimmune encephalitis, however, are still possible. The CSF red blood cells were likely due to a mildly traumatic tap, but also may have arisen from the frontal lobe mass or a more diffuse invasive process, although the lack of xanthochromia argues against this. Delirium and red blood cells in the CSF should trigger consideration of herpes simplex virus (HSV) encephalitis, although the time course is a bit too protracted and the reported MRI findings do not suggest typical medial temporal lobe involvement.
The disparate neurologic findings suggest a multifocal process, perhaps embolic (eg, endocarditis), ischemic (eg, intravascular lymphoma), infiltrative (eg, malignancy, neurosarcoidosis), or demyelinating (eg, postinfectious acute disseminated encephalomyelitis, multiple sclerosis). However, most of these would have been detected on the initial MRI. Upper extremity dystonia would likely localize to the basal ganglia, whereas confusion and visual hallucinations are more global. The combination of a movement disorder and visual hallucinations is seen in Lewy body dementia, but this tempo is not typical.
Although the CSF does not have pleocytosis, her original symptoms were flu-like; therefore, CSF testing for viruses (eg, enterovirus) is reasonable. Bacterial, mycobacteria, and fungal studies are apt to be unrevealing, but CSF cytology, IgG index, and oligoclonal bands may be useful. Should the encephalopathy progress further and the general medical evaluation prove to be normal, then tests for autoimmune disorders (eg, antinuclear antibodies, NMDAR, paraneoplastic disorders) and rare causes of rapidly progressive dementias (eg, prion diseases) should be sent.
Additional CSF studies including HSV polymerase chain reaction (PCR), West Nile PCR, Lyme antibody, paraneoplastic antibodies, and cytology were sent. Intravenous acyclovir was administered. The above studies, as well as Gram stain, acid-fast bacillus stain, fungal stain, and cultures, were negative. She was started on levetiracetam for seizure prevention due to the mass lesion. An electroencephalogram (EEG) was reported as showing diffuse background slowing with superimposed semiperiodic sharp waves with a right hemispheric emphasis. Intravenous immunoglobulin (IVIG) 0.4 mg/kg/day over 5 days was administered with no improvement. The patient was transferred to an academic medical center for further evaluation.
The EEG reflects encephalopathy without pointing to a specific diagnosis. Prophylactic antiepileptic medications are not indicated for CNS mass lesions without clinical or electrophysiologic seizure activity. IVIG is often administered when an autoimmune encephalitis is suspected, but the lack of response does not rule out an autoimmune condition.
Her medical history included bilateral cataract extraction, right leg fracture, tonsillectomy, and total abdominal hysterectomy. She had a 25-year smoking history and a family history of lung cancer. She had no history of drug or alcohol use. On examination, her temperature was 37.9°C, blood pressure of 144/98 mm Hg, respiratory rate of 18 breaths per minute, a heart rate of 121 beats per minute, and oxygen saturation of 97% on ambient air. Her eyes were open but she was nonverbal. Her chest was clear to auscultation. Heart sounds were distinct and rhythm was regular. Abdomen was soft and nontender with no organomegaly. Skin examination revealed no rash. Her pupils were equal, round, and reactive to light. She did not follow verbal or gestural commands and intermittently tracked with her eyes, but not consistently enough to characterize extraocular movements. Her face was symmetric. She had a normal gag and blink reflex and an increased jaw jerk reflex. Her arms were flexed with increased tone. She had a positive palmo-mental reflex. She had spontaneous movement of all extremities. She had symmetric, 3+ reflexes of the patella and Achilles tendon with a bilateral Babinski’s sign. Sensation was intact only to withdrawal from noxious stimuli.
The physical exam does not localize to a specific brain region, but suggests a diffuse brain process. There are multiple signs of upper motor neuron involvement, including increased tone, hyperreflexia, and Babinski (plantar flexion) reflexes. A palmo-mental reflex signifies pathology in the cerebrum. Although cranial nerve testing is limited, there are no features of cranial neuropathy; similarly, no pyramidal weakness or sensory deficit has been demonstrated on limited testing. The differential diagnosis of her rapidly progressive encephalopathy includes autoimmune or paraneoplastic encephalitis, diffuse infiltrative malignancy, metabolic diseases (eg, porphyria, heavy metal intoxication), and prion disease.
Her family history of lung cancer and her smoking increases the possibility of paraneoplastic encephalitis, which often has subacute behavioral changes that precede complete neurologic impairment. Inflammatory or hemorrhagic CSF is seen with Balamuthia amoebic infection, which causes a granulomatous encephalitis and is characteristically associated with a mass lesion. Toxoplasmosis causes encephalitis that can be profound, but patients are usually immunocompromised and there are typically multiple lesions.
Laboratory results showed a normal white blood cell count and differential, basic metabolic profile and liver function tests, and C-reactive protein. Human immunodeficiency virus antibody testing was negative. Chest radiography and computed tomography of chest, abdomen, and pelvis were normal. A repeat MRI of the brain with contrast was reported as showing a 2.4 x 2.3 x 1.9 cm heterogeneously enhancing mass in the right frontal lobe with an enhancing dural tail and underlying hyperostosis consistent with a meningioma, and blooming within the mass consistent with prior hemorrhage. No mass effect was present.
The meningioma was resected 3 days after admission but her symptoms did not improve. Routine postoperative MRI was reported to show expected postsurgical changes but no infarct. Brain biopsy at the time of the operation was reported as meningioma and mild gliosis without encephalitis.
The reported MRI findings showing unchanged size and overall appearance of the mass, its connection to the dura and skull, and the pathology results all suggest that the mass is a meningioma. There is no evidence of disease outside of the CNS. Some cancers that provoke a paraneoplastic response can be quite small yet may incite an immune encephalitis; anti-NMDAR-mediated encephalitis can occur with malignancy (often ovarian), although it also arises in the absence of any tumor. Any inclination to definitively exclude conditions not seen on the brain biopsy must be tempered by the limited sensitivity of brain histology examination. Still, what was not seen warrants mention: vascular inflammation suggestive of CNS vasculitis, granulomas that might point to neurosarcoidosis, malignant cells of an infiltrating lymphoma or glioma, or inflammatory cells suggestive of encephalitis. Prion encephalopathy remains possible.
The patient remained unresponsive. A repeat EEG showed bilateral generalized periodic epileptiform discharges with accompanying twitching of the head, face, and left arm, which were suppressed with intravenous propofol and levetiracetam. Three weeks following meningioma resection, a new MRI was read as showing new abnormal signal in the right basal ganglia, abnormality of the cortex on the diffusion weighted images, and progressive generalized volume loss.
Among the aforementioned diagnoses, focal or diffuse periodic epileptiform discharges at 1-2 hertz are most characteristic of prion disease. Striatal and cortical transverse relaxation time (T2)-weighted and diffusion-weighted imaging (DWI) hyperintensities with corresponding restricted diffusion is characteristic of Creutzfeldt-Jakob disease (CJD), although metabolic disorders, seizures, and encephalitis can very rarely show similar MRI findings. The clinical course, the MRI and EEG findings, and nondiagnostic biopsy results, which were initially not assessed for prion disease, collectively point to prion disease. Detection of abnormal prion protein in the brain tissue by immunohistochemistry or molecular methods would confirm the diagnosis.
Review of the original right frontal cortex biopsy specimen at the National Prion Disease Pathology Surveillance Center, including immunostaining with 3F4, a monoclonal antibody to the prion protein, revealed granular deposits typical of prion disease. This finding established a diagnosis of prion disease, likely sporadic CJD. The patient was transitioned to palliative care and died shortly thereafter.
Brain autopsy showed regions with transcortical vacuolation (spongiform change), other cortical regions with varying degrees of vacuolation, abundant reactive astrocytes, paucity of neurons, and dark shrunken neurons. Vacuolation and gliosis were observed in the striatum and were most pronounced in the thalamus. There was no evidence of an inflammatory infiltrate or a neoplastic process. These findings with the positive 3F4 immunohistochemistry and positive Western blot from brain autopsy, as well as the absence of a mutation in the prion protein gene, were diagnostic for CJD.
An investigation was initiated to track the nondisposable surgical instruments used in the meningioma resection that may have been subsequently used in other patients. It was determined that 52 neurosurgical patients may have been exposed to prion-contaminated instruments. The instruments were subsequently processed specifically for prion decontamination. After 7 years, no cases of CJD have been diagnosed in the potentially exposed patients.
DISCUSSION
CJD is a rare neurodegenerative condition1 classified as one of the transmissible spongiform encephalopathies, so called because of the characteristic spongiform pattern (vacuolation) seen on histology, as well as the presence of neuronal loss, reactive gliosis in the gray matter, and the accumulation of the abnormal isoform of the cellular prion protein.2 It affects about one person in every one million people per year worldwide; in the United States there are about 300 cases per year. The most common form of human prion disease, sporadic CJD, is relentlessly progressive and invariably fatal, and in most cases, death occurs less than 5 months from onset.3 There is no cure, although temporizing treatments for symptoms can be helpful.
Sporadic CJD, which accounts for approximately 85% of all cases of prion disease in humans, typically manifests with rapidly progressive dementia and myoclonus after a prolonged incubation period in persons between 55 and 75 years of age. Genetic forms account for approximately 15% and acquired forms less than 1% of human prion diseases.1 Prion diseases have a broad spectrum of clinical manifestations, including dementia, ataxia, parkinsonism, myoclonus, insomnia, paresthesias, and abnormal or changed behavior.4 Given the protean clinical manifestations of prion diseases and rarity, the diagnosis is challenging to make antemortem. One recent study showed that most patients receive about 4 misdiagnoses and are often two-thirds of the way through their disease course before the correct diagnosis of sporadic CJD is made.5
Testing for protein markers of rapid neuronal injury8 in the CSF including 14-3-3, total tau, and neuron-specific enolase can increase suspicion for CJD, although there is a 10%-50% false positive rate with these markers.9 In this case, those tests were not performed; positive results would have been even more nonspecific in the setting of an enhancing brain mass and recent brain surgery.
Although not available at the time this patient was evaluated, the real-time quaking-induced conversion (RT-QuIC) test performed in CSF is diagnostically helpful, and, if positive, supportive of the MRI findings. The sensitivity and specificity of this test have been reported to be between 87%-91% and 98%-100%, respectively, albeit with limited data.10 Applying RT-QuIC to nasal mucosal brushings might lead to even higher sensitivity and specificity.11Seeking a premortem diagnosis for a rare disease with no known cure may seem superfluous, but it has important implications for establishing prognosis, limiting subsequent diagnostic and therapeutic measures, and safeguarding of other patients and operating room personnel. Iatrogenic CJD has occurred following invasive procedures involving neurosurgical instrumentation.12 CJD has been transmitted from grafts of dura mater, transplanted corneas, implantation of inadequately sterilized electrodes in the brain, and in the early 1980s, injections of contaminated pituitary hormones (particularly growth hormone) derived from human pituitary glands taken from cadavers. Since CJD was first described in the 1920s, less than 1% of human prion cases have been acquired iatrogenically.13In patients with rapidly progressive cognitive decline who warrant brain biopsy or surgery, the probability of prion diseases should be assessed based on clinical information and the results of MRI, EEG, and CSF testing. If prion disease is plausible, World Health Organization14 precautions should be employed for neuroinvasive procedures to reduce transmission risk. Disposable equipment should be used when possible, and nondisposable neurosurgical instruments should be quarantined until a nonprion disease diagnosis is identified, or should be regarded as contaminated and reprocessed using the aforementioned protocol.
This case highlights the challenges of seeking the correct diagnosis and its consequences, especially from an infection control perspective. The initial imaging finding of a mass lesion (a meningioma—which is a common incidental finding in older adults15) was a red herring that initially obscured the correct diagnosis. The patient’s progressive cognitive decline, EEG results, and evolving MRI findings, however, prompted further scrutiny of the brain biopsy specimen that eventually steered the clinicians away from mass confusion to diagnostic certainty.
TEACHING POINTS
- Rapidly progressive dementias (RPD) are characterized by cognitive decline over weeks to months. The RPD differential diagnosis includes fulminant forms of common neurodegenerative disorders (eg, Alzheimer’s disease, dementia with Lewy bodies, frontotemporal dementia spectrum), autoimmune encephalidites, CNS cancers, and prion disease.
- Sporadic CJD is the most common human prion disease. It is a rare neurodegenerative condition with onset usually between the ages of 50 and 70 years, and most commonly manifests with rapidly progressive dementia, ataxia, and myoclonus.
- Because of its protean manifestations, the diagnosis of CJD is difficult to make antemortem, and diagnosis is often delayed. Specialist evaluation of brain MRI DWI sequences and new CSF diagnostic tests may allow for earlier diagnosis, which has management and infection control implications.
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. Dr Geschwind’s institution has received R01 grant funding from NIH/NIA; and Alliance Biosecure and the Michael J Homer Family Fund as paid money to his institution, Dr Geschwind has received consulting fees or honoraria from Best Doctors, Kendall Brill & Kelly, CJD Foundation, and Tau Consortium; Dr Geschwind is a consultant for Gerson Lehrman Group, Biohaven Pharmaceuticals, and Advance Medical, outside the submitted work; has grants/grantspending with Quest, Cure PSP, and Tau Consortium, and received payment for lectures from Multiple Grand Rounds Lectures, outside the submitted work. Dr Saint is on a medical advisory board of Doximity, has received honorarium for being a member of the medical advisory board; he is also on the scientifice advisory board of Jvion. Dr Safdar’s institution has received a grant from the VA Patient Safety Center.
A 57-year-old woman presented to the emergency department of a community hospital with a 2-week history of dizziness, blurred vision, and poor coordination following a flu-like illness. Symptoms were initially attributed to complications from a presumed viral illness, but when they persisted for 2 weeks, she underwent magnetic resonance imaging (MRI) of the brain, which was reported as showing a 2.4 x 2.3 x 1.9 cm right frontal lobe mass with mild mass effect and contrast enhancement (Figure 1). She was discharged home at her request with plans for outpatient follow-up.
Brain masses are usually neoplastic, infectious, or less commonly, inflammatory. The isolated lesion in the right frontal lobe is unlikely to explain her symptoms, which are more suggestive of multifocal disease or elevated intracranial pressure. Although the frontal eye fields could be affected by the mass, such lesions usually cause tonic eye deviation, not blurry vision; furthermore, coordination, which is impaired here, is not governed by the frontal lobe.
Two weeks later, she returned to the same emergency department with worsening symptoms and new bilateral upper extremity dystonia, confusion, and visual hallucinations. Cerebrospinal fluid (CSF) analysis revealed clear, nonxanthochromic fluid with 4 nucleated cells (a differential was not performed), 113 red blood cells, glucose of 80 mg/dL (normal range, 50-80 mg/dL), and protein of 52 mg/dL (normal range, 15-45 mg/dL).
Confusion is generally caused by a metabolic, infectious, structural, or toxic etiology. Standard CSF test results are usually normal with most toxic or metabolic encephalopathies. The absence of significant CSF inflammation argues against infectious encephalitis; paraneoplastic and autoimmune encephalitis, however, are still possible. The CSF red blood cells were likely due to a mildly traumatic tap, but also may have arisen from the frontal lobe mass or a more diffuse invasive process, although the lack of xanthochromia argues against this. Delirium and red blood cells in the CSF should trigger consideration of herpes simplex virus (HSV) encephalitis, although the time course is a bit too protracted and the reported MRI findings do not suggest typical medial temporal lobe involvement.
The disparate neurologic findings suggest a multifocal process, perhaps embolic (eg, endocarditis), ischemic (eg, intravascular lymphoma), infiltrative (eg, malignancy, neurosarcoidosis), or demyelinating (eg, postinfectious acute disseminated encephalomyelitis, multiple sclerosis). However, most of these would have been detected on the initial MRI. Upper extremity dystonia would likely localize to the basal ganglia, whereas confusion and visual hallucinations are more global. The combination of a movement disorder and visual hallucinations is seen in Lewy body dementia, but this tempo is not typical.
Although the CSF does not have pleocytosis, her original symptoms were flu-like; therefore, CSF testing for viruses (eg, enterovirus) is reasonable. Bacterial, mycobacteria, and fungal studies are apt to be unrevealing, but CSF cytology, IgG index, and oligoclonal bands may be useful. Should the encephalopathy progress further and the general medical evaluation prove to be normal, then tests for autoimmune disorders (eg, antinuclear antibodies, NMDAR, paraneoplastic disorders) and rare causes of rapidly progressive dementias (eg, prion diseases) should be sent.
Additional CSF studies including HSV polymerase chain reaction (PCR), West Nile PCR, Lyme antibody, paraneoplastic antibodies, and cytology were sent. Intravenous acyclovir was administered. The above studies, as well as Gram stain, acid-fast bacillus stain, fungal stain, and cultures, were negative. She was started on levetiracetam for seizure prevention due to the mass lesion. An electroencephalogram (EEG) was reported as showing diffuse background slowing with superimposed semiperiodic sharp waves with a right hemispheric emphasis. Intravenous immunoglobulin (IVIG) 0.4 mg/kg/day over 5 days was administered with no improvement. The patient was transferred to an academic medical center for further evaluation.
The EEG reflects encephalopathy without pointing to a specific diagnosis. Prophylactic antiepileptic medications are not indicated for CNS mass lesions without clinical or electrophysiologic seizure activity. IVIG is often administered when an autoimmune encephalitis is suspected, but the lack of response does not rule out an autoimmune condition.
Her medical history included bilateral cataract extraction, right leg fracture, tonsillectomy, and total abdominal hysterectomy. She had a 25-year smoking history and a family history of lung cancer. She had no history of drug or alcohol use. On examination, her temperature was 37.9°C, blood pressure of 144/98 mm Hg, respiratory rate of 18 breaths per minute, a heart rate of 121 beats per minute, and oxygen saturation of 97% on ambient air. Her eyes were open but she was nonverbal. Her chest was clear to auscultation. Heart sounds were distinct and rhythm was regular. Abdomen was soft and nontender with no organomegaly. Skin examination revealed no rash. Her pupils were equal, round, and reactive to light. She did not follow verbal or gestural commands and intermittently tracked with her eyes, but not consistently enough to characterize extraocular movements. Her face was symmetric. She had a normal gag and blink reflex and an increased jaw jerk reflex. Her arms were flexed with increased tone. She had a positive palmo-mental reflex. She had spontaneous movement of all extremities. She had symmetric, 3+ reflexes of the patella and Achilles tendon with a bilateral Babinski’s sign. Sensation was intact only to withdrawal from noxious stimuli.
The physical exam does not localize to a specific brain region, but suggests a diffuse brain process. There are multiple signs of upper motor neuron involvement, including increased tone, hyperreflexia, and Babinski (plantar flexion) reflexes. A palmo-mental reflex signifies pathology in the cerebrum. Although cranial nerve testing is limited, there are no features of cranial neuropathy; similarly, no pyramidal weakness or sensory deficit has been demonstrated on limited testing. The differential diagnosis of her rapidly progressive encephalopathy includes autoimmune or paraneoplastic encephalitis, diffuse infiltrative malignancy, metabolic diseases (eg, porphyria, heavy metal intoxication), and prion disease.
Her family history of lung cancer and her smoking increases the possibility of paraneoplastic encephalitis, which often has subacute behavioral changes that precede complete neurologic impairment. Inflammatory or hemorrhagic CSF is seen with Balamuthia amoebic infection, which causes a granulomatous encephalitis and is characteristically associated with a mass lesion. Toxoplasmosis causes encephalitis that can be profound, but patients are usually immunocompromised and there are typically multiple lesions.
Laboratory results showed a normal white blood cell count and differential, basic metabolic profile and liver function tests, and C-reactive protein. Human immunodeficiency virus antibody testing was negative. Chest radiography and computed tomography of chest, abdomen, and pelvis were normal. A repeat MRI of the brain with contrast was reported as showing a 2.4 x 2.3 x 1.9 cm heterogeneously enhancing mass in the right frontal lobe with an enhancing dural tail and underlying hyperostosis consistent with a meningioma, and blooming within the mass consistent with prior hemorrhage. No mass effect was present.
The meningioma was resected 3 days after admission but her symptoms did not improve. Routine postoperative MRI was reported to show expected postsurgical changes but no infarct. Brain biopsy at the time of the operation was reported as meningioma and mild gliosis without encephalitis.
The reported MRI findings showing unchanged size and overall appearance of the mass, its connection to the dura and skull, and the pathology results all suggest that the mass is a meningioma. There is no evidence of disease outside of the CNS. Some cancers that provoke a paraneoplastic response can be quite small yet may incite an immune encephalitis; anti-NMDAR-mediated encephalitis can occur with malignancy (often ovarian), although it also arises in the absence of any tumor. Any inclination to definitively exclude conditions not seen on the brain biopsy must be tempered by the limited sensitivity of brain histology examination. Still, what was not seen warrants mention: vascular inflammation suggestive of CNS vasculitis, granulomas that might point to neurosarcoidosis, malignant cells of an infiltrating lymphoma or glioma, or inflammatory cells suggestive of encephalitis. Prion encephalopathy remains possible.
The patient remained unresponsive. A repeat EEG showed bilateral generalized periodic epileptiform discharges with accompanying twitching of the head, face, and left arm, which were suppressed with intravenous propofol and levetiracetam. Three weeks following meningioma resection, a new MRI was read as showing new abnormal signal in the right basal ganglia, abnormality of the cortex on the diffusion weighted images, and progressive generalized volume loss.
Among the aforementioned diagnoses, focal or diffuse periodic epileptiform discharges at 1-2 hertz are most characteristic of prion disease. Striatal and cortical transverse relaxation time (T2)-weighted and diffusion-weighted imaging (DWI) hyperintensities with corresponding restricted diffusion is characteristic of Creutzfeldt-Jakob disease (CJD), although metabolic disorders, seizures, and encephalitis can very rarely show similar MRI findings. The clinical course, the MRI and EEG findings, and nondiagnostic biopsy results, which were initially not assessed for prion disease, collectively point to prion disease. Detection of abnormal prion protein in the brain tissue by immunohistochemistry or molecular methods would confirm the diagnosis.
Review of the original right frontal cortex biopsy specimen at the National Prion Disease Pathology Surveillance Center, including immunostaining with 3F4, a monoclonal antibody to the prion protein, revealed granular deposits typical of prion disease. This finding established a diagnosis of prion disease, likely sporadic CJD. The patient was transitioned to palliative care and died shortly thereafter.
Brain autopsy showed regions with transcortical vacuolation (spongiform change), other cortical regions with varying degrees of vacuolation, abundant reactive astrocytes, paucity of neurons, and dark shrunken neurons. Vacuolation and gliosis were observed in the striatum and were most pronounced in the thalamus. There was no evidence of an inflammatory infiltrate or a neoplastic process. These findings with the positive 3F4 immunohistochemistry and positive Western blot from brain autopsy, as well as the absence of a mutation in the prion protein gene, were diagnostic for CJD.
An investigation was initiated to track the nondisposable surgical instruments used in the meningioma resection that may have been subsequently used in other patients. It was determined that 52 neurosurgical patients may have been exposed to prion-contaminated instruments. The instruments were subsequently processed specifically for prion decontamination. After 7 years, no cases of CJD have been diagnosed in the potentially exposed patients.
DISCUSSION
CJD is a rare neurodegenerative condition1 classified as one of the transmissible spongiform encephalopathies, so called because of the characteristic spongiform pattern (vacuolation) seen on histology, as well as the presence of neuronal loss, reactive gliosis in the gray matter, and the accumulation of the abnormal isoform of the cellular prion protein.2 It affects about one person in every one million people per year worldwide; in the United States there are about 300 cases per year. The most common form of human prion disease, sporadic CJD, is relentlessly progressive and invariably fatal, and in most cases, death occurs less than 5 months from onset.3 There is no cure, although temporizing treatments for symptoms can be helpful.
Sporadic CJD, which accounts for approximately 85% of all cases of prion disease in humans, typically manifests with rapidly progressive dementia and myoclonus after a prolonged incubation period in persons between 55 and 75 years of age. Genetic forms account for approximately 15% and acquired forms less than 1% of human prion diseases.1 Prion diseases have a broad spectrum of clinical manifestations, including dementia, ataxia, parkinsonism, myoclonus, insomnia, paresthesias, and abnormal or changed behavior.4 Given the protean clinical manifestations of prion diseases and rarity, the diagnosis is challenging to make antemortem. One recent study showed that most patients receive about 4 misdiagnoses and are often two-thirds of the way through their disease course before the correct diagnosis of sporadic CJD is made.5
Testing for protein markers of rapid neuronal injury8 in the CSF including 14-3-3, total tau, and neuron-specific enolase can increase suspicion for CJD, although there is a 10%-50% false positive rate with these markers.9 In this case, those tests were not performed; positive results would have been even more nonspecific in the setting of an enhancing brain mass and recent brain surgery.
Although not available at the time this patient was evaluated, the real-time quaking-induced conversion (RT-QuIC) test performed in CSF is diagnostically helpful, and, if positive, supportive of the MRI findings. The sensitivity and specificity of this test have been reported to be between 87%-91% and 98%-100%, respectively, albeit with limited data.10 Applying RT-QuIC to nasal mucosal brushings might lead to even higher sensitivity and specificity.11Seeking a premortem diagnosis for a rare disease with no known cure may seem superfluous, but it has important implications for establishing prognosis, limiting subsequent diagnostic and therapeutic measures, and safeguarding of other patients and operating room personnel. Iatrogenic CJD has occurred following invasive procedures involving neurosurgical instrumentation.12 CJD has been transmitted from grafts of dura mater, transplanted corneas, implantation of inadequately sterilized electrodes in the brain, and in the early 1980s, injections of contaminated pituitary hormones (particularly growth hormone) derived from human pituitary glands taken from cadavers. Since CJD was first described in the 1920s, less than 1% of human prion cases have been acquired iatrogenically.13In patients with rapidly progressive cognitive decline who warrant brain biopsy or surgery, the probability of prion diseases should be assessed based on clinical information and the results of MRI, EEG, and CSF testing. If prion disease is plausible, World Health Organization14 precautions should be employed for neuroinvasive procedures to reduce transmission risk. Disposable equipment should be used when possible, and nondisposable neurosurgical instruments should be quarantined until a nonprion disease diagnosis is identified, or should be regarded as contaminated and reprocessed using the aforementioned protocol.
This case highlights the challenges of seeking the correct diagnosis and its consequences, especially from an infection control perspective. The initial imaging finding of a mass lesion (a meningioma—which is a common incidental finding in older adults15) was a red herring that initially obscured the correct diagnosis. The patient’s progressive cognitive decline, EEG results, and evolving MRI findings, however, prompted further scrutiny of the brain biopsy specimen that eventually steered the clinicians away from mass confusion to diagnostic certainty.
TEACHING POINTS
- Rapidly progressive dementias (RPD) are characterized by cognitive decline over weeks to months. The RPD differential diagnosis includes fulminant forms of common neurodegenerative disorders (eg, Alzheimer’s disease, dementia with Lewy bodies, frontotemporal dementia spectrum), autoimmune encephalidites, CNS cancers, and prion disease.
- Sporadic CJD is the most common human prion disease. It is a rare neurodegenerative condition with onset usually between the ages of 50 and 70 years, and most commonly manifests with rapidly progressive dementia, ataxia, and myoclonus.
- Because of its protean manifestations, the diagnosis of CJD is difficult to make antemortem, and diagnosis is often delayed. Specialist evaluation of brain MRI DWI sequences and new CSF diagnostic tests may allow for earlier diagnosis, which has management and infection control implications.
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. Dr Geschwind’s institution has received R01 grant funding from NIH/NIA; and Alliance Biosecure and the Michael J Homer Family Fund as paid money to his institution, Dr Geschwind has received consulting fees or honoraria from Best Doctors, Kendall Brill & Kelly, CJD Foundation, and Tau Consortium; Dr Geschwind is a consultant for Gerson Lehrman Group, Biohaven Pharmaceuticals, and Advance Medical, outside the submitted work; has grants/grantspending with Quest, Cure PSP, and Tau Consortium, and received payment for lectures from Multiple Grand Rounds Lectures, outside the submitted work. Dr Saint is on a medical advisory board of Doximity, has received honorarium for being a member of the medical advisory board; he is also on the scientifice advisory board of Jvion. Dr Safdar’s institution has received a grant from the VA Patient Safety Center.
1. Brown P, Gibbs CJ, Jr., Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513-529. PubMed
2. Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:913-920. PubMed
3. Johnson RT, Gibbs CJ, Jr. Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998;339:1994-2004. PubMed
4. Will RG, Alpers MP, Dormont D, Schonberger LB. Infectious and sporadic prion diseases. In: Prusiner SB, ed. Prion biology and diseases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999:465-507. \
5. Paterson RW, Torres-Chae CC, Kuo AL, et al. Differential diagnosis of Jakob-Creutzfeldt disease. Arch Neurol. 2012;69:1578-1582. PubMed
6. Tschampa HJ, Kallenberg K, Kretzschmar HA, et al. Pattern of cortical changes in sporadic Creutzfeldt-Jakob disease. AJNR Am J Neuroradiol. 2007;28:1114-1118. PubMed
7. Carswell C, Thompson A, Lukic A, et al. MRI findings are often missed in the diagnosis of Creutzfeldt-Jakob disease. BMC Neurol. 2012;12:153. PubMed
8. Geschwind MD, Martindale J, Miller D, et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2003;60:813-816. PubMed
9. Burkhard PR, Sanchez JC, Landis T, Hochstrasser DF. CSF detection of the 14-3-3 protein in unselected patients with dementia. Neurology. 2001;56:1528-1533. PubMed
10. Orrú CD, Groveman BR, Hughson AG, Zanusso G, Coulthart MB, Caughey B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio. 2015;6:pii: e02451-14 PubMed
11. Orrú CD, Bongianni M, Tonoli G, et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med. 2014;371:519-529. PubMed
12. Brown P, Preece M, Brandel JP, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55:1075-1081. PubMed
13. Brown P, Brandel JP, Sato T, et al. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis. 2012;18:901-907. PubMed
14. WHO infection control guidelines for transmissible spongiform encephalopathies. Report of a WHO consultation, Geneva, Switzerland, 23-26 March 1999. http://www.who.int/csr/resources/publications/bse/whocdscsraph2003.pdf. Accessed on July 10, 2017.
15. Bondy M, Ligon BL. Epidemiology and etiology of intracranial meningiomas: a review. J Neurooncol. 1996;29:197-205. PubMed
1. Brown P, Gibbs CJ, Jr., Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513-529. PubMed
2. Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:913-920. PubMed
3. Johnson RT, Gibbs CJ, Jr. Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998;339:1994-2004. PubMed
4. Will RG, Alpers MP, Dormont D, Schonberger LB. Infectious and sporadic prion diseases. In: Prusiner SB, ed. Prion biology and diseases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999:465-507. \
5. Paterson RW, Torres-Chae CC, Kuo AL, et al. Differential diagnosis of Jakob-Creutzfeldt disease. Arch Neurol. 2012;69:1578-1582. PubMed
6. Tschampa HJ, Kallenberg K, Kretzschmar HA, et al. Pattern of cortical changes in sporadic Creutzfeldt-Jakob disease. AJNR Am J Neuroradiol. 2007;28:1114-1118. PubMed
7. Carswell C, Thompson A, Lukic A, et al. MRI findings are often missed in the diagnosis of Creutzfeldt-Jakob disease. BMC Neurol. 2012;12:153. PubMed
8. Geschwind MD, Martindale J, Miller D, et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2003;60:813-816. PubMed
9. Burkhard PR, Sanchez JC, Landis T, Hochstrasser DF. CSF detection of the 14-3-3 protein in unselected patients with dementia. Neurology. 2001;56:1528-1533. PubMed
10. Orrú CD, Groveman BR, Hughson AG, Zanusso G, Coulthart MB, Caughey B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio. 2015;6:pii: e02451-14 PubMed
11. Orrú CD, Bongianni M, Tonoli G, et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med. 2014;371:519-529. PubMed
12. Brown P, Preece M, Brandel JP, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55:1075-1081. PubMed
13. Brown P, Brandel JP, Sato T, et al. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis. 2012;18:901-907. PubMed
14. WHO infection control guidelines for transmissible spongiform encephalopathies. Report of a WHO consultation, Geneva, Switzerland, 23-26 March 1999. http://www.who.int/csr/resources/publications/bse/whocdscsraph2003.pdf. Accessed on July 10, 2017.
15. Bondy M, Ligon BL. Epidemiology and etiology of intracranial meningiomas: a review. J Neurooncol. 1996;29:197-205. PubMed
© 2017 Society of Hospital Medicine
Can’t Shake This Feeling
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
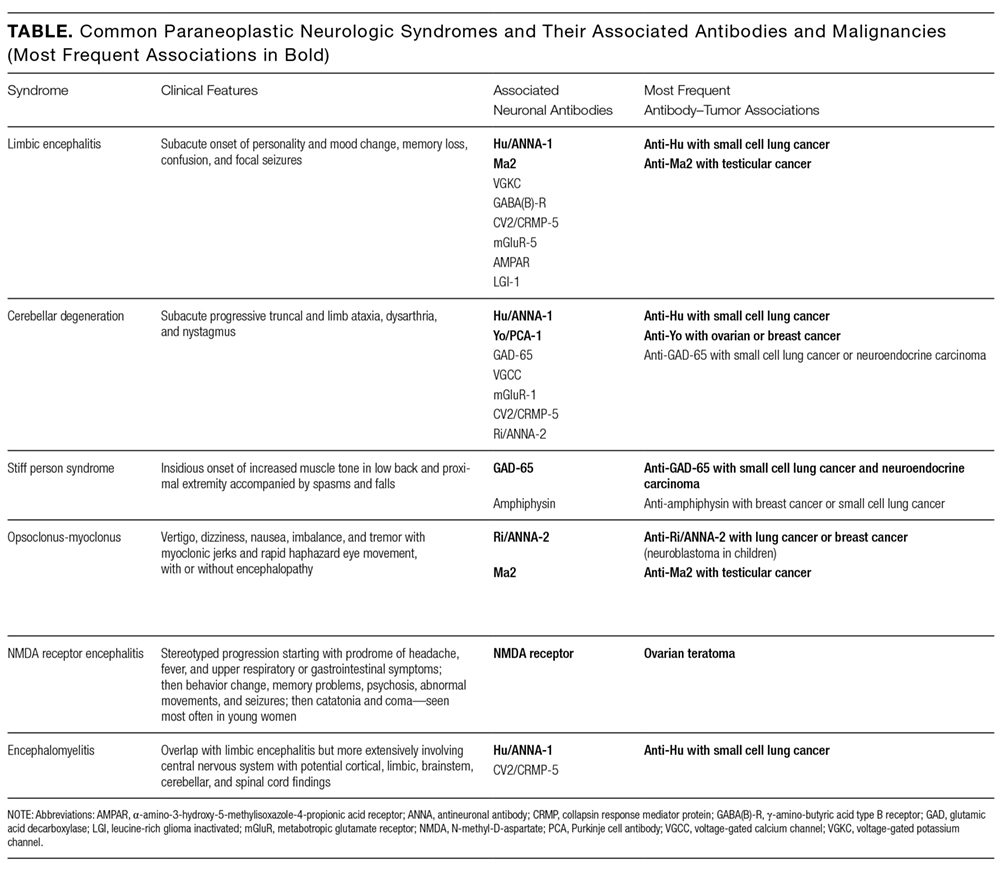
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
© 2017 Society of Hospital Medicine
The plot thickens
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
After losing consciousness at a supermarket, a 70-year-old man was brought to the emergency department by paramedics. He subsequently developed chest pain.
Syncope can be difficult to evaluate, but chest pain may help narrow an otherwise broad differential diagnosis. If this patient has aortic stenosis or hypertrophic cardiomyopathy, effort syncope is the culprit. Cardiac dysrhythmia (eg, ventricular tachycardia), complete heart block, and supraventricular tachycardia each can cause syncope along with chest pain. Myocardial infarction and associated ventricular arrhythmia might also explain both chest pain and syncope. The paramedics might have noted an arrhythmia on the cardiac monitor; if possible, the rhythm strip should be reviewed. A pulmonary embolus can cause chest pain and, if large enough to cause right ventricular compromise, syncope.
According to witnesses at the supermarket, the patient dropped to the ground, lost consciousness, and convulsed for 30 seconds. He had no head trauma, tongue biting, urinary incontinence, or confusion afterward. Electrocardiogram (ECG) performed at the scene showed ST elevations in leads V1 to V3 with ST depressions in the inferior leads. On arrival in the emergency department, the patient described nonradiating substernal chest pressure exacerbated by deep inhalation. The pain did not improve with nitroglycerin. He recalled feeling light-headed before the syncope.
He had not received medical care for 20 years and had no known illnesses other than hypertension. He was not taking any medications. He previously worked as a welder and never smoked tobacco, drank alcohol, or used illicit drugs. The patient’s temperature was 36.4°C. Heart rate was 88 beats per minute, blood pressure 128/72 mm Hg, oxygen saturation 100% on room air, and respiratory rate 22 breaths per minute. The patient had conjunctival pallor. There was a grade 3/6 crescendo- decrescendo systolic murmur loudest at the right upper sternal border without radiation to the carotids. There was no jugular venous distention. Lungs were clear to auscultation bilaterally. There was no peripheral edema, rash, or lymphadenopathy.
Convulsive movements commonly occur during episodes of unconsciousness lasting more than 15 seconds—a phenomenon termed convulsive syncope and often is confused with seizures. These movements are usually clonic jerks of the extremities and trunk and slight twitching of the face, and occasionally tonic extension of the trunk and clenching of the jaw. Absence of tongue biting, urinary incontinence, and confusion in this patient’s case makes seizures less likely.
The distribution of ST segment changes on his ECG are concerning for myocardial infarction in the septal and inferior regions. Right-sided ECG should be performed to assess for right ventricular infarction. Although myocardial ischemia is the primary concern, some features warrant consideration of other etiologies of syncope. First, syncope is an unusual presentation of cardiac ischemia or infarct. The complaint of chest pressure exacerbated by deep inhalation is another atypical feature for myocardial ischemia. Although the patient’s oxygen saturation and heart rate are normal, pulmonary embolism remains a possibility.
The prominent crescendo-decrescendo systolic murmur at the right upper sternal border could indicate aortic stenosis; the carotids should be palpated to assess for pulsus parvus et tardus. A high-flow state associated with anemia could also lead to a midsystolic murmur. Conjunctival pallor typically is seen with hemoglobin levels of 6 g/dL or less. This finding may indicate severe anemia, which has the potential to cause myocardial ischemia and syncope.
Laboratory testing revealed a troponin of 0.04 ng/dL, hemoglobin 4.1 g/dL with MCV of 84.7 fL, white blood cell count 6,500/μL and platelet count 179,000/μL. Serum sodium was 130 mEq/L, urea nitrogen 16 mg/dL, creatinine 1.6 mg/dL, calcium 7.8 mg/dL, total protein 11.4 g/dL (reference range, 6.0-8.2), and albumin 2.2 g/dL. Erythrocyte sedimentation rate (ESR) was 20 mm/h. Serum iron was 48 μg/mL, total iron binding capacity 275 μg/dL, percent iron saturation 17% (reference range, 20-55), and ferritin 10 ng/mL (reference range, 30-400). The international normalized ratio (INR) was 1.5, prothrombin time 15.5 sec (reference range, 9.4-11.6), and partial thromboplastin time 24.7 sec (reference range, 22.9-30.6). Hepatitis C and HIV antibodies were negative as was the urine toxicology screen. Urine protein to creatinine ratio was 0.07. His hemoglobin rose to 7.9 g/dL with transfusion of 4 units packed red blood cells (RBC). His chest pain improved and inferior ST depressions resolved on follow-up ECG. Further history revealed multiple episodes of melena and hematochezia in the preceding weeks without nausea, vomiting, or abdominal pain.
The patient has a strikingly large gamma gap: 9.2. A gap larger than 4 is concerning for the presence of paraproteins. Given the possibility of a paraproteinemia (eg, multiple myeloma, plasmacytoma, Waldenström macroglobulinemia), the first step is to check serum and urine protein electrophoresis. The patient’s anemia is significant and reticulocyte index low. The low ferritin level combined with the inappropriately low reticulocyte count could result from iron deficiency anemia, another bone marrow process, or both. The patient’s syncope likely resulted from severe anemia and hypovolemia associated with hematochezia. The prolonged prothrombin time could be caused by a coagulation factor production problem, from vitamin K deficiency or underlying liver disease, or by a consumptive problem, from low-grade disseminated intravascular coagulation. It is controversial whether inhaling welding fumes causes cancer, but the patient’s age alone makes malignancy a definite possibility.
 and increased background staining secondary to hypergammaglobulinemia.")
Aortic stenosis is intriguing in light of the patient’s painless melena and hematochezia. Heyde syndrome is a phenomenon in which high shear stress causes a reduction in the size of von Willebrand factor predisposing to bleeding from submucosal angiodysplasia. However, Heyde syndrome has been reported to occur in the setting of severe or critical aortic stenosis and is unlikely to be the cause here. The patient needs to be examined with both upper and lower endoscopy to rule out gastrointestinal (GI) malignancy, gastric and esophageal varices, and painless peptic ulcers. High right ventricular systolic blood pressures along with echodense material in the inferior vena cava and right atrium suggest the possibility of malignancy with vascular invasion. Tricuspid regurgitation is consistent with high right-sided pressures. As left ventricular ejection fraction is reduced, some of the high right-sided pressures could also be attributable to left heart failure. CT findings of rectal wall thickening and perirectal lymph nodes could be attributable to cancer with locally metastatic disease. Blood loss caused by this cancer would explain the severe anemia on admission as well as the low ferritin level and the iron deficiency anemia. In this 70-year-old man who has not had routine health care maintenance, the leading diagnosis is colorectal cancer. However, the markedly elevated globulin gap and elevated INR strongly suggest another process (eg, multiple myeloma, other paraproteinemia) is also present.
INR remained elevated (1.5) despite vitamin K supplementation. Peripheral blood smear showed hypochromic and normocytic RBCs with moderate rouleaux formation (Figure 1). Intravenous pantoprazole and octreotide were started. Upper and lower endoscopy revealed multiple esophageal erosions and both a polypoid mass and an ulcer within the rectum with evidence of prior bleeding. The mass was resected and the ulcer biopsied.
 Color fundus photographs show mild dilatation of patient’s retinal veins and retinal hemorrhages. (C,D) Red-free fundus photographs show round blot hemorrhages occurring from vessels within retina (arrow) and linear, fl ame-shaped hemorrhages occurr")
On hospital day 3, the patient was transfused another unit of packed RBCs. Hemoglobin level increased from 7.4 g/dL to 8.2 g/dL, but he began to complain of headache, blurred vision, and worsened chest pain. He did not have weakness, numbness, diplopia, dysphagia, or dysarthria. External examination and extraocular movements of both eyes were normal. Visual acuity was 20/25 bilaterally. Funduscopic examination revealed mild dilation of retinal veins and retinal hemorrhages (Figure 2).
Rouleaux formation occurs as excess cathodal proteins, such as immunoglobulins or fibrinogen, adhere to RBCs and cause the cells to stack together in long chains. Classically this is associated with multiple myeloma, but can occur with Waldenström’s macroglobulinemia and other cancers or infections. It can also occur as an artifact in smear preparation but the large globulin gap in this patient supports pathologic rouleaux formation.
Venous retinopathy with hemorrhages may occur with occlusion of the arterial supply (eg, as with carotid artery obstruction) but also with hyperviscosity syndrome (HVS). Vascular disturbances throughout the body play a major role in HVS, but these changes are most easily visualized in the retina. It is interesting that the patient’s headache and blurred vision began after he received additional blood transfusions. Spuriously low hemoglobin and hematocrit levels may stem from increased plasma volume from high immunoglobulin M (IgM) concentrations in Waldenström macroglobulinemia; thus, RBC transfusions can exacerbate symptoms by elevating total RBC mass. Normocytic, normochromic anemia is characteristic of both multiple myeloma and Waldenström’s macroglobulinemia. That the patient’s chest pain recurred coincidentally with blurred vision and headache suggests the likely cause is cardiac ischemia from hyperviscosity. The serum viscosity level should be checked, and, if it is elevated, urgent serum plasmapheresis should be considered. Determining the source of excess globulin production and treating the underlying disease are crucial at this juncture.
In the general population, rectal adenocarcinoma is the most common cause of a rectal mass. In this patient, presence of a paraproteinemia may point to a different diagnosis. Extramedullary colorectal plasmacytoma can occur in the rectum but is exceedingly rare. Waldenström’s macroglobulinemia, a subtype of lymphoplasmacytic lymphoma, can be associated with a rectal lymphoma. At this point, it is not possible to confidently predict the etiology of the mass.
.")
He was started on cyclophosphamide, bortezomib and dexamethasone for IgG κ myeloma with improvement in his headache, blurred vision, chest pain, and plasma viscosity (4 to 1.8). His hemoglobin remained stable at 10 g/dL. Neoadjuvant Capecitabine and radiation therapy were initiated for his rectal cancer.
DISCUSSION
Multiple myeloma is characterized by monoclonal proliferation of plasma cells, elevated circulating monoclonal immunoglobulin, and end-organ damage.1 It accounts for approximately 0.8% of all new cancer diagnoses; average age at onset is 70 years. The patient described here had an unusual presentation, with GI bleeding and progression to HVS, and known risk factors for multiple myeloma (male sex, low socioeconomic status, welding career).2,3
An early clue in the diagnosis was the patient’s large gamma gap and concurrent anemia. Gamma gap, calculated by subtracting serum albumin from serum total protein, is so named because it often reflects an elevated gamma globulin concentration. However, it actually reflects all nonalbumin serum protein. A gamma gap larger than 3.1 g/dL is an independent risk factor for death4 and may be associated with infection, autoimmunity, and malignancy. Although there are no screening guidelines for multiple myeloma, 73% of cases are brought to attention by anemia discovered on routine laboratory investigation.5 This patient’s lack of prior medical care likely contributed to his atypical presentation. Screening colonoscopy, recommended at age 50, might have identified his rectal cancer at an earlier stage.
The patient’s anemia was likely secondary to GI hemorrhage and bone marrow suppression. His hematochezia might have been partly related to the pathophysiologic interaction of paraproteins with platelets, coagulation factors, and blood vessels.6 Amyloidosis of the GI tract is seen in 8% of AL amyloidosis7 and most frequently manifests as gastrointestinal bleeding, which is thought to be due to ischemia, vascular friability, or mucosal lesions. It less commonly presents as malabsorption or dysmotility.8 Although gastrointestinal amyloid is not typically associated with radiologic abnormalities, occasionally it may cause luminal wall thickening, adenopathy, and inflammatory stranding.9 The gold standard for diagnosis is tissue biopsy. However, presence of amyloidosis does not change the overall treatment strategy for multiple myeloma.
An interesting feature of this case is the development of HVS, which typically manifests with mucosal bleeding, blurred vision, and headache.10 HVS can be diagnosed on retinal examination with findings of venous tortuosity, dilatation, and intraretinal hemorrhage, as occurred in this case,11 and is confirmed with serum viscosity measurement. The first evidence of HVS in this case might have been the spontaneous echo contrast, or “smoke,” detected on echocardiogram. Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates,12 and is associated with conditions that result in left atrial stasis, such as atrial arrhythmias and mitral stenosis. This patient did not have valvular pathology or arrhythmia, and thus the “smoke” likely reflected HVS.
Of the paraproteinemias, Waldenström’s macroglobulinemia is most often associated with HVS, likely because of the pentameric structure of IgM13 and the consequential large size that predisposes to vascular occlusion. Whereas HVS can occur with IgM levels as low as 3 g/dL, it typically does not occur with IgG concentrations under 15 g/dL. This patient presented with an IgG level of 8 g/dL and developed HVS symptoms only after multiple packed RBC transfusions. Elevated IgG level likely made him susceptible to HVS, which ultimately was precipitated by blood transfusion. Therefore, this patient’s initial chest pain most likely was caused by demand cardiac ischemia secondary to anemia, whereas his subsequent, posttransfusion chest pain likely resulted from hyperviscosity angina. Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—has been described in polycythemia and connective tissue disorders.14,15 To our knowledge, however, hyperviscosity angina has not been reported in patients with multiple myeloma. Treatment of hyperviscosity with end-organ damage typically consists of plasmapheresis, but this patient was started on urgent chemotherapy, and his symptoms improved. Untreated HVS can lead to end-organ ischemia and death.
This patient had a multitude of seemingly disparate symptoms and abnormalities that ultimately were united in a diagnosis of IgG κ multiple myeloma. Subsequently diagnosed rectal adenocarcinoma may have led to ongoing blood loss, which worsened the anemia, but had no evident relation to the primary diagnosis of multiple myeloma. This case exemplifies the fact that HVS is a rare but important iatrogenic complication of multiple myeloma treated with blood transfusion. As this patient’s hospital course progressed, the plot, and his blood, thickened.
KEY TEACHING POINTS
- Multiple myeloma is occasionally associated with HVS, which manifests with mucosal bleeding, blurred vision, and headache.
- Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—should be considered in patients with paraproteinemias and chest pain.
- Plasmapheresis reverses the clinical manifestations of HVS but not the underlying disease process (eg, Waldenström’s macroglobulinemia, multiple myeloma, leukemia, polycythemia).
- Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates, and is associated with left atrial stasis, commonly from atrial fibrillation or mitral stenosis, but might be present in HVS.
Acknowledgment
The authors thank Peter Campochiaro, MD, and Whitney Green, MD, for their contributions to the images used in this article.
Disclosure
Dr. Sedighi Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The other authors have nothing to report.
1. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046-1060. PubMed
2. Koessel SL, Theis MK, Vaughan TL, et al. Socioeconomic status and the incidence of multiple myeloma. Epidemiology. 1996;7(1):4-8. PubMed
3. Fritschi L, Siemiatycki J. Lymphoma, myeloma and occupation: results of a case-control study. Int J Cancer. 1996;67(4):498-503. PubMed
4. Juraschek SP, Moliterno AR, Checkley W, Miller ER 3rd. The gamma gap and all-cause mortality. PLoS One. 2015;10(12):e0143494. PubMed
5. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. PubMed
6. Eby CS. Bleeding and thrombosis risks in plasma cell dyscrasias. Hematology Am Soc Hematol Educ Program. 2007:158-164. PubMed
7. Menke DM, Kyle RA, Fleming CR, Wolfe JT 3rd, Kurtin PJ, Oldenburg WA. Symptomatic gastric amyloidosis in patients with primary systemic amyloidosis. Mayo Clin Proc. 1993;68(8):763-767. PubMed
8. Levy DJ, Franklin GO, Rosenthal WS. Gastrointestinal bleeding and amyloidosis. Am J Gastroenterol. 1982;77(6):422-426. PubMed
9. Araoz PA, Batts KP, MacCarty RL. Amyloidosis of the alimentary canal: radiologic-pathologic correlation of CT findings. Abdom Imaging. 2000;25(1):38-44. PubMed
10. Stone MJ, Bogen SA. Evidence-based focused review of management of hyperviscosity syndrome. Blood. 2012;119(10):2205-2208. PubMed
11. Rajagopal R, Apte RS. Seeing through thick and through thin: retinal manifestations of thrombophilic and hyperviscosity syndromes. Surv Ophthalmol. 2016;61(2):236-247. PubMed
12. Black IW. Spontaneous echo contrast: where there’s smoke there’s fire. Echocardiography. 2000;17(4):373-382. PubMed
13. Kwaan HC. Hyperviscosity in plasma cell dyscrasias. Clin Hemorheol Microcirc. 2013;55(1):75-83. PubMed
14. Piccirillo G, Fimognari FL, Valdivia JL, Marigliano V. Effects of phlebotomy on a patient with secondary polycythemia and angina pectoris. Int J Cardiol. 1994;44(2):175-177. PubMed
15. Ovadia S, Lysyy L, Floru S. Emergency plasmapheresis for unstable angina in a patient with hyperviscosity syndrome. Am J Emerg Med. 2005;23(6):811-812. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
After losing consciousness at a supermarket, a 70-year-old man was brought to the emergency department by paramedics. He subsequently developed chest pain.
Syncope can be difficult to evaluate, but chest pain may help narrow an otherwise broad differential diagnosis. If this patient has aortic stenosis or hypertrophic cardiomyopathy, effort syncope is the culprit. Cardiac dysrhythmia (eg, ventricular tachycardia), complete heart block, and supraventricular tachycardia each can cause syncope along with chest pain. Myocardial infarction and associated ventricular arrhythmia might also explain both chest pain and syncope. The paramedics might have noted an arrhythmia on the cardiac monitor; if possible, the rhythm strip should be reviewed. A pulmonary embolus can cause chest pain and, if large enough to cause right ventricular compromise, syncope.
According to witnesses at the supermarket, the patient dropped to the ground, lost consciousness, and convulsed for 30 seconds. He had no head trauma, tongue biting, urinary incontinence, or confusion afterward. Electrocardiogram (ECG) performed at the scene showed ST elevations in leads V1 to V3 with ST depressions in the inferior leads. On arrival in the emergency department, the patient described nonradiating substernal chest pressure exacerbated by deep inhalation. The pain did not improve with nitroglycerin. He recalled feeling light-headed before the syncope.
He had not received medical care for 20 years and had no known illnesses other than hypertension. He was not taking any medications. He previously worked as a welder and never smoked tobacco, drank alcohol, or used illicit drugs. The patient’s temperature was 36.4°C. Heart rate was 88 beats per minute, blood pressure 128/72 mm Hg, oxygen saturation 100% on room air, and respiratory rate 22 breaths per minute. The patient had conjunctival pallor. There was a grade 3/6 crescendo- decrescendo systolic murmur loudest at the right upper sternal border without radiation to the carotids. There was no jugular venous distention. Lungs were clear to auscultation bilaterally. There was no peripheral edema, rash, or lymphadenopathy.
Convulsive movements commonly occur during episodes of unconsciousness lasting more than 15 seconds—a phenomenon termed convulsive syncope and often is confused with seizures. These movements are usually clonic jerks of the extremities and trunk and slight twitching of the face, and occasionally tonic extension of the trunk and clenching of the jaw. Absence of tongue biting, urinary incontinence, and confusion in this patient’s case makes seizures less likely.
The distribution of ST segment changes on his ECG are concerning for myocardial infarction in the septal and inferior regions. Right-sided ECG should be performed to assess for right ventricular infarction. Although myocardial ischemia is the primary concern, some features warrant consideration of other etiologies of syncope. First, syncope is an unusual presentation of cardiac ischemia or infarct. The complaint of chest pressure exacerbated by deep inhalation is another atypical feature for myocardial ischemia. Although the patient’s oxygen saturation and heart rate are normal, pulmonary embolism remains a possibility.
The prominent crescendo-decrescendo systolic murmur at the right upper sternal border could indicate aortic stenosis; the carotids should be palpated to assess for pulsus parvus et tardus. A high-flow state associated with anemia could also lead to a midsystolic murmur. Conjunctival pallor typically is seen with hemoglobin levels of 6 g/dL or less. This finding may indicate severe anemia, which has the potential to cause myocardial ischemia and syncope.
Laboratory testing revealed a troponin of 0.04 ng/dL, hemoglobin 4.1 g/dL with MCV of 84.7 fL, white blood cell count 6,500/μL and platelet count 179,000/μL. Serum sodium was 130 mEq/L, urea nitrogen 16 mg/dL, creatinine 1.6 mg/dL, calcium 7.8 mg/dL, total protein 11.4 g/dL (reference range, 6.0-8.2), and albumin 2.2 g/dL. Erythrocyte sedimentation rate (ESR) was 20 mm/h. Serum iron was 48 μg/mL, total iron binding capacity 275 μg/dL, percent iron saturation 17% (reference range, 20-55), and ferritin 10 ng/mL (reference range, 30-400). The international normalized ratio (INR) was 1.5, prothrombin time 15.5 sec (reference range, 9.4-11.6), and partial thromboplastin time 24.7 sec (reference range, 22.9-30.6). Hepatitis C and HIV antibodies were negative as was the urine toxicology screen. Urine protein to creatinine ratio was 0.07. His hemoglobin rose to 7.9 g/dL with transfusion of 4 units packed red blood cells (RBC). His chest pain improved and inferior ST depressions resolved on follow-up ECG. Further history revealed multiple episodes of melena and hematochezia in the preceding weeks without nausea, vomiting, or abdominal pain.
The patient has a strikingly large gamma gap: 9.2. A gap larger than 4 is concerning for the presence of paraproteins. Given the possibility of a paraproteinemia (eg, multiple myeloma, plasmacytoma, Waldenström macroglobulinemia), the first step is to check serum and urine protein electrophoresis. The patient’s anemia is significant and reticulocyte index low. The low ferritin level combined with the inappropriately low reticulocyte count could result from iron deficiency anemia, another bone marrow process, or both. The patient’s syncope likely resulted from severe anemia and hypovolemia associated with hematochezia. The prolonged prothrombin time could be caused by a coagulation factor production problem, from vitamin K deficiency or underlying liver disease, or by a consumptive problem, from low-grade disseminated intravascular coagulation. It is controversial whether inhaling welding fumes causes cancer, but the patient’s age alone makes malignancy a definite possibility.
Aortic stenosis is intriguing in light of the patient’s painless melena and hematochezia. Heyde syndrome is a phenomenon in which high shear stress causes a reduction in the size of von Willebrand factor predisposing to bleeding from submucosal angiodysplasia. However, Heyde syndrome has been reported to occur in the setting of severe or critical aortic stenosis and is unlikely to be the cause here. The patient needs to be examined with both upper and lower endoscopy to rule out gastrointestinal (GI) malignancy, gastric and esophageal varices, and painless peptic ulcers. High right ventricular systolic blood pressures along with echodense material in the inferior vena cava and right atrium suggest the possibility of malignancy with vascular invasion. Tricuspid regurgitation is consistent with high right-sided pressures. As left ventricular ejection fraction is reduced, some of the high right-sided pressures could also be attributable to left heart failure. CT findings of rectal wall thickening and perirectal lymph nodes could be attributable to cancer with locally metastatic disease. Blood loss caused by this cancer would explain the severe anemia on admission as well as the low ferritin level and the iron deficiency anemia. In this 70-year-old man who has not had routine health care maintenance, the leading diagnosis is colorectal cancer. However, the markedly elevated globulin gap and elevated INR strongly suggest another process (eg, multiple myeloma, other paraproteinemia) is also present.
INR remained elevated (1.5) despite vitamin K supplementation. Peripheral blood smear showed hypochromic and normocytic RBCs with moderate rouleaux formation (Figure 1). Intravenous pantoprazole and octreotide were started. Upper and lower endoscopy revealed multiple esophageal erosions and both a polypoid mass and an ulcer within the rectum with evidence of prior bleeding. The mass was resected and the ulcer biopsied.
On hospital day 3, the patient was transfused another unit of packed RBCs. Hemoglobin level increased from 7.4 g/dL to 8.2 g/dL, but he began to complain of headache, blurred vision, and worsened chest pain. He did not have weakness, numbness, diplopia, dysphagia, or dysarthria. External examination and extraocular movements of both eyes were normal. Visual acuity was 20/25 bilaterally. Funduscopic examination revealed mild dilation of retinal veins and retinal hemorrhages (Figure 2).
Rouleaux formation occurs as excess cathodal proteins, such as immunoglobulins or fibrinogen, adhere to RBCs and cause the cells to stack together in long chains. Classically this is associated with multiple myeloma, but can occur with Waldenström’s macroglobulinemia and other cancers or infections. It can also occur as an artifact in smear preparation but the large globulin gap in this patient supports pathologic rouleaux formation.
Venous retinopathy with hemorrhages may occur with occlusion of the arterial supply (eg, as with carotid artery obstruction) but also with hyperviscosity syndrome (HVS). Vascular disturbances throughout the body play a major role in HVS, but these changes are most easily visualized in the retina. It is interesting that the patient’s headache and blurred vision began after he received additional blood transfusions. Spuriously low hemoglobin and hematocrit levels may stem from increased plasma volume from high immunoglobulin M (IgM) concentrations in Waldenström macroglobulinemia; thus, RBC transfusions can exacerbate symptoms by elevating total RBC mass. Normocytic, normochromic anemia is characteristic of both multiple myeloma and Waldenström’s macroglobulinemia. That the patient’s chest pain recurred coincidentally with blurred vision and headache suggests the likely cause is cardiac ischemia from hyperviscosity. The serum viscosity level should be checked, and, if it is elevated, urgent serum plasmapheresis should be considered. Determining the source of excess globulin production and treating the underlying disease are crucial at this juncture.
In the general population, rectal adenocarcinoma is the most common cause of a rectal mass. In this patient, presence of a paraproteinemia may point to a different diagnosis. Extramedullary colorectal plasmacytoma can occur in the rectum but is exceedingly rare. Waldenström’s macroglobulinemia, a subtype of lymphoplasmacytic lymphoma, can be associated with a rectal lymphoma. At this point, it is not possible to confidently predict the etiology of the mass.
He was started on cyclophosphamide, bortezomib and dexamethasone for IgG κ myeloma with improvement in his headache, blurred vision, chest pain, and plasma viscosity (4 to 1.8). His hemoglobin remained stable at 10 g/dL. Neoadjuvant Capecitabine and radiation therapy were initiated for his rectal cancer.
DISCUSSION
Multiple myeloma is characterized by monoclonal proliferation of plasma cells, elevated circulating monoclonal immunoglobulin, and end-organ damage.1 It accounts for approximately 0.8% of all new cancer diagnoses; average age at onset is 70 years. The patient described here had an unusual presentation, with GI bleeding and progression to HVS, and known risk factors for multiple myeloma (male sex, low socioeconomic status, welding career).2,3
An early clue in the diagnosis was the patient’s large gamma gap and concurrent anemia. Gamma gap, calculated by subtracting serum albumin from serum total protein, is so named because it often reflects an elevated gamma globulin concentration. However, it actually reflects all nonalbumin serum protein. A gamma gap larger than 3.1 g/dL is an independent risk factor for death4 and may be associated with infection, autoimmunity, and malignancy. Although there are no screening guidelines for multiple myeloma, 73% of cases are brought to attention by anemia discovered on routine laboratory investigation.5 This patient’s lack of prior medical care likely contributed to his atypical presentation. Screening colonoscopy, recommended at age 50, might have identified his rectal cancer at an earlier stage.
The patient’s anemia was likely secondary to GI hemorrhage and bone marrow suppression. His hematochezia might have been partly related to the pathophysiologic interaction of paraproteins with platelets, coagulation factors, and blood vessels.6 Amyloidosis of the GI tract is seen in 8% of AL amyloidosis7 and most frequently manifests as gastrointestinal bleeding, which is thought to be due to ischemia, vascular friability, or mucosal lesions. It less commonly presents as malabsorption or dysmotility.8 Although gastrointestinal amyloid is not typically associated with radiologic abnormalities, occasionally it may cause luminal wall thickening, adenopathy, and inflammatory stranding.9 The gold standard for diagnosis is tissue biopsy. However, presence of amyloidosis does not change the overall treatment strategy for multiple myeloma.
An interesting feature of this case is the development of HVS, which typically manifests with mucosal bleeding, blurred vision, and headache.10 HVS can be diagnosed on retinal examination with findings of venous tortuosity, dilatation, and intraretinal hemorrhage, as occurred in this case,11 and is confirmed with serum viscosity measurement. The first evidence of HVS in this case might have been the spontaneous echo contrast, or “smoke,” detected on echocardiogram. Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates,12 and is associated with conditions that result in left atrial stasis, such as atrial arrhythmias and mitral stenosis. This patient did not have valvular pathology or arrhythmia, and thus the “smoke” likely reflected HVS.
Of the paraproteinemias, Waldenström’s macroglobulinemia is most often associated with HVS, likely because of the pentameric structure of IgM13 and the consequential large size that predisposes to vascular occlusion. Whereas HVS can occur with IgM levels as low as 3 g/dL, it typically does not occur with IgG concentrations under 15 g/dL. This patient presented with an IgG level of 8 g/dL and developed HVS symptoms only after multiple packed RBC transfusions. Elevated IgG level likely made him susceptible to HVS, which ultimately was precipitated by blood transfusion. Therefore, this patient’s initial chest pain most likely was caused by demand cardiac ischemia secondary to anemia, whereas his subsequent, posttransfusion chest pain likely resulted from hyperviscosity angina. Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—has been described in polycythemia and connective tissue disorders.14,15 To our knowledge, however, hyperviscosity angina has not been reported in patients with multiple myeloma. Treatment of hyperviscosity with end-organ damage typically consists of plasmapheresis, but this patient was started on urgent chemotherapy, and his symptoms improved. Untreated HVS can lead to end-organ ischemia and death.
This patient had a multitude of seemingly disparate symptoms and abnormalities that ultimately were united in a diagnosis of IgG κ multiple myeloma. Subsequently diagnosed rectal adenocarcinoma may have led to ongoing blood loss, which worsened the anemia, but had no evident relation to the primary diagnosis of multiple myeloma. This case exemplifies the fact that HVS is a rare but important iatrogenic complication of multiple myeloma treated with blood transfusion. As this patient’s hospital course progressed, the plot, and his blood, thickened.
KEY TEACHING POINTS
- Multiple myeloma is occasionally associated with HVS, which manifests with mucosal bleeding, blurred vision, and headache.
- Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—should be considered in patients with paraproteinemias and chest pain.
- Plasmapheresis reverses the clinical manifestations of HVS but not the underlying disease process (eg, Waldenström’s macroglobulinemia, multiple myeloma, leukemia, polycythemia).
- Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates, and is associated with left atrial stasis, commonly from atrial fibrillation or mitral stenosis, but might be present in HVS.
Acknowledgment
The authors thank Peter Campochiaro, MD, and Whitney Green, MD, for their contributions to the images used in this article.
Disclosure
Dr. Sedighi Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The other authors have nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
After losing consciousness at a supermarket, a 70-year-old man was brought to the emergency department by paramedics. He subsequently developed chest pain.
Syncope can be difficult to evaluate, but chest pain may help narrow an otherwise broad differential diagnosis. If this patient has aortic stenosis or hypertrophic cardiomyopathy, effort syncope is the culprit. Cardiac dysrhythmia (eg, ventricular tachycardia), complete heart block, and supraventricular tachycardia each can cause syncope along with chest pain. Myocardial infarction and associated ventricular arrhythmia might also explain both chest pain and syncope. The paramedics might have noted an arrhythmia on the cardiac monitor; if possible, the rhythm strip should be reviewed. A pulmonary embolus can cause chest pain and, if large enough to cause right ventricular compromise, syncope.
According to witnesses at the supermarket, the patient dropped to the ground, lost consciousness, and convulsed for 30 seconds. He had no head trauma, tongue biting, urinary incontinence, or confusion afterward. Electrocardiogram (ECG) performed at the scene showed ST elevations in leads V1 to V3 with ST depressions in the inferior leads. On arrival in the emergency department, the patient described nonradiating substernal chest pressure exacerbated by deep inhalation. The pain did not improve with nitroglycerin. He recalled feeling light-headed before the syncope.
He had not received medical care for 20 years and had no known illnesses other than hypertension. He was not taking any medications. He previously worked as a welder and never smoked tobacco, drank alcohol, or used illicit drugs. The patient’s temperature was 36.4°C. Heart rate was 88 beats per minute, blood pressure 128/72 mm Hg, oxygen saturation 100% on room air, and respiratory rate 22 breaths per minute. The patient had conjunctival pallor. There was a grade 3/6 crescendo- decrescendo systolic murmur loudest at the right upper sternal border without radiation to the carotids. There was no jugular venous distention. Lungs were clear to auscultation bilaterally. There was no peripheral edema, rash, or lymphadenopathy.
Convulsive movements commonly occur during episodes of unconsciousness lasting more than 15 seconds—a phenomenon termed convulsive syncope and often is confused with seizures. These movements are usually clonic jerks of the extremities and trunk and slight twitching of the face, and occasionally tonic extension of the trunk and clenching of the jaw. Absence of tongue biting, urinary incontinence, and confusion in this patient’s case makes seizures less likely.
The distribution of ST segment changes on his ECG are concerning for myocardial infarction in the septal and inferior regions. Right-sided ECG should be performed to assess for right ventricular infarction. Although myocardial ischemia is the primary concern, some features warrant consideration of other etiologies of syncope. First, syncope is an unusual presentation of cardiac ischemia or infarct. The complaint of chest pressure exacerbated by deep inhalation is another atypical feature for myocardial ischemia. Although the patient’s oxygen saturation and heart rate are normal, pulmonary embolism remains a possibility.
The prominent crescendo-decrescendo systolic murmur at the right upper sternal border could indicate aortic stenosis; the carotids should be palpated to assess for pulsus parvus et tardus. A high-flow state associated with anemia could also lead to a midsystolic murmur. Conjunctival pallor typically is seen with hemoglobin levels of 6 g/dL or less. This finding may indicate severe anemia, which has the potential to cause myocardial ischemia and syncope.
Laboratory testing revealed a troponin of 0.04 ng/dL, hemoglobin 4.1 g/dL with MCV of 84.7 fL, white blood cell count 6,500/μL and platelet count 179,000/μL. Serum sodium was 130 mEq/L, urea nitrogen 16 mg/dL, creatinine 1.6 mg/dL, calcium 7.8 mg/dL, total protein 11.4 g/dL (reference range, 6.0-8.2), and albumin 2.2 g/dL. Erythrocyte sedimentation rate (ESR) was 20 mm/h. Serum iron was 48 μg/mL, total iron binding capacity 275 μg/dL, percent iron saturation 17% (reference range, 20-55), and ferritin 10 ng/mL (reference range, 30-400). The international normalized ratio (INR) was 1.5, prothrombin time 15.5 sec (reference range, 9.4-11.6), and partial thromboplastin time 24.7 sec (reference range, 22.9-30.6). Hepatitis C and HIV antibodies were negative as was the urine toxicology screen. Urine protein to creatinine ratio was 0.07. His hemoglobin rose to 7.9 g/dL with transfusion of 4 units packed red blood cells (RBC). His chest pain improved and inferior ST depressions resolved on follow-up ECG. Further history revealed multiple episodes of melena and hematochezia in the preceding weeks without nausea, vomiting, or abdominal pain.
The patient has a strikingly large gamma gap: 9.2. A gap larger than 4 is concerning for the presence of paraproteins. Given the possibility of a paraproteinemia (eg, multiple myeloma, plasmacytoma, Waldenström macroglobulinemia), the first step is to check serum and urine protein electrophoresis. The patient’s anemia is significant and reticulocyte index low. The low ferritin level combined with the inappropriately low reticulocyte count could result from iron deficiency anemia, another bone marrow process, or both. The patient’s syncope likely resulted from severe anemia and hypovolemia associated with hematochezia. The prolonged prothrombin time could be caused by a coagulation factor production problem, from vitamin K deficiency or underlying liver disease, or by a consumptive problem, from low-grade disseminated intravascular coagulation. It is controversial whether inhaling welding fumes causes cancer, but the patient’s age alone makes malignancy a definite possibility.
Aortic stenosis is intriguing in light of the patient’s painless melena and hematochezia. Heyde syndrome is a phenomenon in which high shear stress causes a reduction in the size of von Willebrand factor predisposing to bleeding from submucosal angiodysplasia. However, Heyde syndrome has been reported to occur in the setting of severe or critical aortic stenosis and is unlikely to be the cause here. The patient needs to be examined with both upper and lower endoscopy to rule out gastrointestinal (GI) malignancy, gastric and esophageal varices, and painless peptic ulcers. High right ventricular systolic blood pressures along with echodense material in the inferior vena cava and right atrium suggest the possibility of malignancy with vascular invasion. Tricuspid regurgitation is consistent with high right-sided pressures. As left ventricular ejection fraction is reduced, some of the high right-sided pressures could also be attributable to left heart failure. CT findings of rectal wall thickening and perirectal lymph nodes could be attributable to cancer with locally metastatic disease. Blood loss caused by this cancer would explain the severe anemia on admission as well as the low ferritin level and the iron deficiency anemia. In this 70-year-old man who has not had routine health care maintenance, the leading diagnosis is colorectal cancer. However, the markedly elevated globulin gap and elevated INR strongly suggest another process (eg, multiple myeloma, other paraproteinemia) is also present.
INR remained elevated (1.5) despite vitamin K supplementation. Peripheral blood smear showed hypochromic and normocytic RBCs with moderate rouleaux formation (Figure 1). Intravenous pantoprazole and octreotide were started. Upper and lower endoscopy revealed multiple esophageal erosions and both a polypoid mass and an ulcer within the rectum with evidence of prior bleeding. The mass was resected and the ulcer biopsied.
On hospital day 3, the patient was transfused another unit of packed RBCs. Hemoglobin level increased from 7.4 g/dL to 8.2 g/dL, but he began to complain of headache, blurred vision, and worsened chest pain. He did not have weakness, numbness, diplopia, dysphagia, or dysarthria. External examination and extraocular movements of both eyes were normal. Visual acuity was 20/25 bilaterally. Funduscopic examination revealed mild dilation of retinal veins and retinal hemorrhages (Figure 2).
Rouleaux formation occurs as excess cathodal proteins, such as immunoglobulins or fibrinogen, adhere to RBCs and cause the cells to stack together in long chains. Classically this is associated with multiple myeloma, but can occur with Waldenström’s macroglobulinemia and other cancers or infections. It can also occur as an artifact in smear preparation but the large globulin gap in this patient supports pathologic rouleaux formation.
Venous retinopathy with hemorrhages may occur with occlusion of the arterial supply (eg, as with carotid artery obstruction) but also with hyperviscosity syndrome (HVS). Vascular disturbances throughout the body play a major role in HVS, but these changes are most easily visualized in the retina. It is interesting that the patient’s headache and blurred vision began after he received additional blood transfusions. Spuriously low hemoglobin and hematocrit levels may stem from increased plasma volume from high immunoglobulin M (IgM) concentrations in Waldenström macroglobulinemia; thus, RBC transfusions can exacerbate symptoms by elevating total RBC mass. Normocytic, normochromic anemia is characteristic of both multiple myeloma and Waldenström’s macroglobulinemia. That the patient’s chest pain recurred coincidentally with blurred vision and headache suggests the likely cause is cardiac ischemia from hyperviscosity. The serum viscosity level should be checked, and, if it is elevated, urgent serum plasmapheresis should be considered. Determining the source of excess globulin production and treating the underlying disease are crucial at this juncture.
In the general population, rectal adenocarcinoma is the most common cause of a rectal mass. In this patient, presence of a paraproteinemia may point to a different diagnosis. Extramedullary colorectal plasmacytoma can occur in the rectum but is exceedingly rare. Waldenström’s macroglobulinemia, a subtype of lymphoplasmacytic lymphoma, can be associated with a rectal lymphoma. At this point, it is not possible to confidently predict the etiology of the mass.
He was started on cyclophosphamide, bortezomib and dexamethasone for IgG κ myeloma with improvement in his headache, blurred vision, chest pain, and plasma viscosity (4 to 1.8). His hemoglobin remained stable at 10 g/dL. Neoadjuvant Capecitabine and radiation therapy were initiated for his rectal cancer.
DISCUSSION
Multiple myeloma is characterized by monoclonal proliferation of plasma cells, elevated circulating monoclonal immunoglobulin, and end-organ damage.1 It accounts for approximately 0.8% of all new cancer diagnoses; average age at onset is 70 years. The patient described here had an unusual presentation, with GI bleeding and progression to HVS, and known risk factors for multiple myeloma (male sex, low socioeconomic status, welding career).2,3
An early clue in the diagnosis was the patient’s large gamma gap and concurrent anemia. Gamma gap, calculated by subtracting serum albumin from serum total protein, is so named because it often reflects an elevated gamma globulin concentration. However, it actually reflects all nonalbumin serum protein. A gamma gap larger than 3.1 g/dL is an independent risk factor for death4 and may be associated with infection, autoimmunity, and malignancy. Although there are no screening guidelines for multiple myeloma, 73% of cases are brought to attention by anemia discovered on routine laboratory investigation.5 This patient’s lack of prior medical care likely contributed to his atypical presentation. Screening colonoscopy, recommended at age 50, might have identified his rectal cancer at an earlier stage.
The patient’s anemia was likely secondary to GI hemorrhage and bone marrow suppression. His hematochezia might have been partly related to the pathophysiologic interaction of paraproteins with platelets, coagulation factors, and blood vessels.6 Amyloidosis of the GI tract is seen in 8% of AL amyloidosis7 and most frequently manifests as gastrointestinal bleeding, which is thought to be due to ischemia, vascular friability, or mucosal lesions. It less commonly presents as malabsorption or dysmotility.8 Although gastrointestinal amyloid is not typically associated with radiologic abnormalities, occasionally it may cause luminal wall thickening, adenopathy, and inflammatory stranding.9 The gold standard for diagnosis is tissue biopsy. However, presence of amyloidosis does not change the overall treatment strategy for multiple myeloma.
An interesting feature of this case is the development of HVS, which typically manifests with mucosal bleeding, blurred vision, and headache.10 HVS can be diagnosed on retinal examination with findings of venous tortuosity, dilatation, and intraretinal hemorrhage, as occurred in this case,11 and is confirmed with serum viscosity measurement. The first evidence of HVS in this case might have been the spontaneous echo contrast, or “smoke,” detected on echocardiogram. Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates,12 and is associated with conditions that result in left atrial stasis, such as atrial arrhythmias and mitral stenosis. This patient did not have valvular pathology or arrhythmia, and thus the “smoke” likely reflected HVS.
Of the paraproteinemias, Waldenström’s macroglobulinemia is most often associated with HVS, likely because of the pentameric structure of IgM13 and the consequential large size that predisposes to vascular occlusion. Whereas HVS can occur with IgM levels as low as 3 g/dL, it typically does not occur with IgG concentrations under 15 g/dL. This patient presented with an IgG level of 8 g/dL and developed HVS symptoms only after multiple packed RBC transfusions. Elevated IgG level likely made him susceptible to HVS, which ultimately was precipitated by blood transfusion. Therefore, this patient’s initial chest pain most likely was caused by demand cardiac ischemia secondary to anemia, whereas his subsequent, posttransfusion chest pain likely resulted from hyperviscosity angina. Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—has been described in polycythemia and connective tissue disorders.14,15 To our knowledge, however, hyperviscosity angina has not been reported in patients with multiple myeloma. Treatment of hyperviscosity with end-organ damage typically consists of plasmapheresis, but this patient was started on urgent chemotherapy, and his symptoms improved. Untreated HVS can lead to end-organ ischemia and death.
This patient had a multitude of seemingly disparate symptoms and abnormalities that ultimately were united in a diagnosis of IgG κ multiple myeloma. Subsequently diagnosed rectal adenocarcinoma may have led to ongoing blood loss, which worsened the anemia, but had no evident relation to the primary diagnosis of multiple myeloma. This case exemplifies the fact that HVS is a rare but important iatrogenic complication of multiple myeloma treated with blood transfusion. As this patient’s hospital course progressed, the plot, and his blood, thickened.
KEY TEACHING POINTS
- Multiple myeloma is occasionally associated with HVS, which manifests with mucosal bleeding, blurred vision, and headache.
- Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—should be considered in patients with paraproteinemias and chest pain.
- Plasmapheresis reverses the clinical manifestations of HVS but not the underlying disease process (eg, Waldenström’s macroglobulinemia, multiple myeloma, leukemia, polycythemia).
- Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates, and is associated with left atrial stasis, commonly from atrial fibrillation or mitral stenosis, but might be present in HVS.
Acknowledgment
The authors thank Peter Campochiaro, MD, and Whitney Green, MD, for their contributions to the images used in this article.
Disclosure
Dr. Sedighi Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The other authors have nothing to report.
1. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046-1060. PubMed
2. Koessel SL, Theis MK, Vaughan TL, et al. Socioeconomic status and the incidence of multiple myeloma. Epidemiology. 1996;7(1):4-8. PubMed
3. Fritschi L, Siemiatycki J. Lymphoma, myeloma and occupation: results of a case-control study. Int J Cancer. 1996;67(4):498-503. PubMed
4. Juraschek SP, Moliterno AR, Checkley W, Miller ER 3rd. The gamma gap and all-cause mortality. PLoS One. 2015;10(12):e0143494. PubMed
5. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. PubMed
6. Eby CS. Bleeding and thrombosis risks in plasma cell dyscrasias. Hematology Am Soc Hematol Educ Program. 2007:158-164. PubMed
7. Menke DM, Kyle RA, Fleming CR, Wolfe JT 3rd, Kurtin PJ, Oldenburg WA. Symptomatic gastric amyloidosis in patients with primary systemic amyloidosis. Mayo Clin Proc. 1993;68(8):763-767. PubMed
8. Levy DJ, Franklin GO, Rosenthal WS. Gastrointestinal bleeding and amyloidosis. Am J Gastroenterol. 1982;77(6):422-426. PubMed
9. Araoz PA, Batts KP, MacCarty RL. Amyloidosis of the alimentary canal: radiologic-pathologic correlation of CT findings. Abdom Imaging. 2000;25(1):38-44. PubMed
10. Stone MJ, Bogen SA. Evidence-based focused review of management of hyperviscosity syndrome. Blood. 2012;119(10):2205-2208. PubMed
11. Rajagopal R, Apte RS. Seeing through thick and through thin: retinal manifestations of thrombophilic and hyperviscosity syndromes. Surv Ophthalmol. 2016;61(2):236-247. PubMed
12. Black IW. Spontaneous echo contrast: where there’s smoke there’s fire. Echocardiography. 2000;17(4):373-382. PubMed
13. Kwaan HC. Hyperviscosity in plasma cell dyscrasias. Clin Hemorheol Microcirc. 2013;55(1):75-83. PubMed
14. Piccirillo G, Fimognari FL, Valdivia JL, Marigliano V. Effects of phlebotomy on a patient with secondary polycythemia and angina pectoris. Int J Cardiol. 1994;44(2):175-177. PubMed
15. Ovadia S, Lysyy L, Floru S. Emergency plasmapheresis for unstable angina in a patient with hyperviscosity syndrome. Am J Emerg Med. 2005;23(6):811-812. PubMed
1. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046-1060. PubMed
2. Koessel SL, Theis MK, Vaughan TL, et al. Socioeconomic status and the incidence of multiple myeloma. Epidemiology. 1996;7(1):4-8. PubMed
3. Fritschi L, Siemiatycki J. Lymphoma, myeloma and occupation: results of a case-control study. Int J Cancer. 1996;67(4):498-503. PubMed
4. Juraschek SP, Moliterno AR, Checkley W, Miller ER 3rd. The gamma gap and all-cause mortality. PLoS One. 2015;10(12):e0143494. PubMed
5. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. PubMed
6. Eby CS. Bleeding and thrombosis risks in plasma cell dyscrasias. Hematology Am Soc Hematol Educ Program. 2007:158-164. PubMed
7. Menke DM, Kyle RA, Fleming CR, Wolfe JT 3rd, Kurtin PJ, Oldenburg WA. Symptomatic gastric amyloidosis in patients with primary systemic amyloidosis. Mayo Clin Proc. 1993;68(8):763-767. PubMed
8. Levy DJ, Franklin GO, Rosenthal WS. Gastrointestinal bleeding and amyloidosis. Am J Gastroenterol. 1982;77(6):422-426. PubMed
9. Araoz PA, Batts KP, MacCarty RL. Amyloidosis of the alimentary canal: radiologic-pathologic correlation of CT findings. Abdom Imaging. 2000;25(1):38-44. PubMed
10. Stone MJ, Bogen SA. Evidence-based focused review of management of hyperviscosity syndrome. Blood. 2012;119(10):2205-2208. PubMed
11. Rajagopal R, Apte RS. Seeing through thick and through thin: retinal manifestations of thrombophilic and hyperviscosity syndromes. Surv Ophthalmol. 2016;61(2):236-247. PubMed
12. Black IW. Spontaneous echo contrast: where there’s smoke there’s fire. Echocardiography. 2000;17(4):373-382. PubMed
13. Kwaan HC. Hyperviscosity in plasma cell dyscrasias. Clin Hemorheol Microcirc. 2013;55(1):75-83. PubMed
14. Piccirillo G, Fimognari FL, Valdivia JL, Marigliano V. Effects of phlebotomy on a patient with secondary polycythemia and angina pectoris. Int J Cardiol. 1994;44(2):175-177. PubMed
15. Ovadia S, Lysyy L, Floru S. Emergency plasmapheresis for unstable angina in a patient with hyperviscosity syndrome. Am J Emerg Med. 2005;23(6):811-812. PubMed
© 2017 Society of Hospital Medicine
Hot in the tropics
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 42-year-old Malaysian construction worker with subjective fevers of 4 days’ duration presented to an emergency department in Singapore. He reported nonproductive cough, chills without rigors, sore throat, and body aches. He denied sick contacts. Past medical history included chronic hepatitis B virus (HBV) infection. The patient was not taking any medications.
For this patient presenting acutely with subjective fevers, nonproductive cough, chills, aches, and lethargy, initial considerations include infection with a common virus (influenza virus, adenovirus, Epstein-Barr virus [EBV]), acute human immunodeficiency virus (HIV) infection, emerging infection (severe acute respiratory syndrome [SARS], Middle Eastern respiratory syndrome coronavirus [MERS-CoV] infection, avian influenza), and tropical infection (dengue, chikungunya). Also possible are bacterial infections (eg, with Salmonella typhi or Rickettsia or Mycoplasma species), parasitic infections (eg, malaria), and noninfectious illnesses (eg, autoimmune diseases, thyroiditis, acute leukemia, environmental exposures).
The patient’s temperature was 38.5°C; blood pressure, 133/73 mm Hg; heart rate, 95 beats per minute; respiratory rate, 18 breaths per minute; and oxygen saturation, 100% on ambient air. On physical examination, he appeared comfortable, and heart, lung, abdomen, skin, and extremities were normal. Laboratory test results included white blood cell (WBC) count, 4400/μL (with normal differential); hemoglobin, 16.1 g/dL; and platelet count, 207,000/μL. Serum chemistries were normal. C-reactive protein (CRP) level was 44.6 mg/L (reference range, 0.2-9.1 mg/L), and procalcitonin level was 0.13 ng/mL (reference range, <0.50 ng/mL). Chest radiograph was normal. Dengue antibodies (immunoglobulin M, immunoglobulin G [IgG]) and dengue NS1 antigen were negative. The patient was discharged with a presumptive diagnosis of viral upper respiratory tract infection.
There is no left shift characteristic of bacterial infection or lymphopenia characteristic of rickettsial disease or acute HIV infection. The serologic testing and the patient’s overall appearance make dengue unlikely. The low procalcitonin supports a nonbacterial cause of illness. CRP elevation may indicate an inflammatory process and is relatively nonspecific.
Myalgias, pharyngitis, and cough improved over several days, but fevers persisted, and a rash developed over the lower abdomen. The patient returned to the emergency department and was admitted. He denied weight loss and night sweats. He had multiple female sexual partners, including commercial sex workers, within the previous 6 months. Temperature was 38.5°C. The posterior oropharynx was slightly erythematous. There was no lymphadenopathy. Firm, mildly erythematous macules were present on the anterior abdominal wall (Figure 1). The rest of the physical examination was normal.

Laboratory testing revealed WBC count, 5800/μL (75% neutrophils, 19% lymphocytes, 3% monocytes, 2% atypical mononuclear cells); hemoglobin, 16.3 g/dL; platelet count, 185,000/μL; sodium, 131 mmol/L; potassium, 3.4 mmol/L; creatinine, 0.9 mg/dL; albumin, 3.2 g/dL; alanine aminotransferase (ALT), 99 U/L; aspartate aminotransferase (AST), 137 U/L; alkaline phosphatase (ALP), 63 U/L; and total bilirubin, 1.9 mg/dL. Prothrombin time was 11.1 seconds; partial thromboplastin time, 36.1 seconds; erythrocyte sedimentation rate, 14 mm/h; and CRP, 62.2 mg/L.
EBV, acute HIV, and cytomegalovirus infections often present with adenopathy, which is absent here. Disseminated gonococcal infection can manifest with fever, body aches, and rash, but his rash and the absence of penile discharge, migratory arthritis, and enthesitis are not characteristic. Mycoplasma infection can present with macules, urticaria, or erythema multiforme. Rickettsia illnesses typically cause vasculitis with progression to petechiae or purpura resulting from endothelial damage. Patients with secondary syphilis may have widespread macular lesions, and the accompanying syphilitic hepatitis often manifests with elevations in ALP instead of ALT and AST. The mild elevation in ALT and AST can occur with many systemic viral infections. Sweet syndrome may manifest with febrile illness and rash, but the acuity of this patient’s illness and the rapid evolution favor infection.
The patient’s fevers (35°-40°C) continued without pattern over the next 3 days. Blood and urine cultures were negative. Polymerase chain reaction (PCR) test of the nasal mucosa was negative for respiratory viruses. PCR blood tests for EBV, HIV-1, and cytomegalovirus were also negative. Antistreptolysin O (ASO) titer was 400 IU/mm (reference range, <200 IU/mm). Antinuclear antibodies were negative, and rheumatoid factor was 12.4 U/mL (reference range, <10.3 U/mL). Computed tomography (CT) of the thorax, abdomen, and pelvis was normal. Results of a biopsy of an anterior abdominal wall skin lesion showed perivascular and periadnexal lymphocytic inflammation. Amoxicillin was started for the treatment of possible group A streptococcal infection.
PCR for HIV would be positive at a high level in acute HIV. The skin biopsy is not characteristic of Sweet syndrome, which typically shows neutrophilic infiltrate without leukocytoclastic vasculitis, or of syphilis, which typically shows a plasma cell infiltrate.
The patient’s erythematous oropharynx may indicate recent streptococcal pharyngitis. The fevers, elevated ASO titer, and CRP level are consistent with acute rheumatic fever, but arthritis, carditis, and neurologic manifestations are lacking. Erythema marginatum manifests on the trunk and limbs as macules or papules with central clearing as the lesions spread outward—and differs from the patient’s rash, which is firm and restricted to the abdominal wall.
Fevers persisted through hospital day 7. The WBC count was 1100/μL (75.7% neutrophils, 22.5% lymphocytes), hemoglobin was 10.3 g/dL, and platelet count was 52,000/μL. Additional laboratory test results included ALP, 234 U/L; ALT, 250 U/L; AST, 459 U/L; lactate dehydrogenase, 2303 U/L (reference range, 222-454 U/L); and ferritin, 14,964 ng/mL (reference range, 47-452 ng/mL).
The duration of illness and negative diagnostic tests for infections increases suspicion for a noninfectious illness. Conditions commonly associated with marked hyperferritinemia include adult-onset Still disease (AOSD) and hemophagocytic lymphohistiocytosis (HLH). Of the 9 AOSD diagnostic (Yamaguchi) criteria, 5 are met in this case: fever, rash, sore throat, abnormal liver function tests, and negative rheumatologic tests. However, the patient lacks arthritis, leukocytosis, lymphadenopathy, and hepatosplenomegaly. Except for the elevated ferritin, the AOSD criteria overlap substantially with the criteria for acute rheumatic fever, and still require that infections be adequately excluded. HLH, a state of abnormal immune activation with resultant organ dysfunction, can be a primary disorder, but in adults more often is secondary to underlying infectious, autoimmune, or malignant (often lymphoma) conditions. Elevated ferritin, cytopenias, elevated ALT and AST, elevated CRP and erythrocyte sedimentation rate, and elevated lactate dehydrogenase are consistent with HLH. The HLH diagnosis can be more firmly established with the more specific findings of hypertriglyceridemia, hypofibrinogenemia, and elevated soluble CD25 level. The histopathologic finding of hemophagocytosis in the bone marrow, lymph nodes, or liver may further support the diagnosis of HLH.
Rash and fevers persisted. Hepatitis A, hepatitis C, Rickettsia IgG, Burkholderia pseudomallei (the causative organism of melioidosis), and Leptospira serologies, as well as PCR for herpes simplex virus and parvovirus, were all negative. Hepatitis B viral load was 962 IU/mL (2.98 log), hepatitis B envelope antigen was negative, and hepatitis B envelope antibody was positive. Orientia tsutsugamushi (organism responsible for scrub typhus) IgG titer was elevated at 1:128. Antiliver kidney microsomal antibodies and antineutrophil cytoplasmic antibodies were negative. Fibrinogen level was 0.69 g/L (reference range, 1.8-4.8 g/L), and beta-2 microglobulin level was 5078 ng/mL (reference range, 878-2000 ng/mL). Bone marrow biopsy results showed hypocellular marrow with suppressed myelopoiesis, few atypical lymphoid cells, and few hemophagocytes. Flow cytometry was negative for clonal B lymphocytes and aberrant expression of T lymphocytes. Bone marrow myobacterial PCR and fungal cultures were negative.
The patient’s chronic HBV infection is unlikely to be related to his presentation given his low viral load and absence of signs of hepatic dysfunction. Excluding rickettsial disease requires paired acute and convalescent serologies. O tsutsugamushi, the causative agent of the rickettsial disease scrub typhus, is endemic in Malaysia; thus, his positive O tsutsugamushi IgG may indicate past exposure. His fevers, myalgias, truncal rash, and hepatitis are consistent with scrub typhus, but he lacks the characteristic severe headache and generalized lymphadenopathy. Although eschar formation with evolution of a papular rash is common in scrub typhus, it is often absent in the variant found in Southeast Asia. Although elevated β2 microglobulin level is used as a prognostic marker in multiple myeloma and Waldenström macroglobulinemia, it can be elevated in many immune-active states. The patient likely has HLH, which is supported by the hemophagocytosis seen on bone marrow biopsy, and the hypofibrinogenemia. Potential HLH triggers include O tsutsugamushi infection or recent streptococcal pharyngitis.
A deep-punch skin biopsy of the anterior abdominal wall skin lesion was performed because of the absence of subcutaneous fat in the first biopsy specimen. The latest biopsy results showed irregular interstitial expansion of medium-size lymphocytes in a lobular panniculated pattern. The lymphocytes contained enlarged, irregularly contoured nucleoli and were positive for T-cell markers CD2 and CD3 with reduction in CD5 expression. The lymphomatous cells were of CD8+ with uniform expression of activated cytotoxic granule protein granzyme B and were positive for T-cell hemireceptor β.
Positron emission tomography (PET) CT, obtained for staging purposes, showed multiple hypermetabolic subcutaneous and cutaneous lesions over the torso and upper and lower limbs—compatible with lymphomatous infiltrates (Figure 2). Examination, pathology, and imaging findings suggested a rare neoplasm: subcutaneous panniculitis-like T-cell lymphoma (SPTCL). SPTCL was confirmed by T-cell receptor gene rearrangements studies. with surrounding patchy foci of subcutaneous fat stranding (blue-grey) in anterior abdominal wall and upper left arm, compatible with areas o")
HLH was diagnosed on the basis of the fevers, cytopenias, hypofibrinogenemia, elevated ferritin level, and evidence of hemophagocytosis. SPTCL was suspected as the HLH trigger.
The patient was treated with cyclophosphamide, hydroxydoxorubicin, vincristine, and prednisone. While on this regimen, he developed new skin lesions, and his ferritin level was persistently elevated. He was switched to romidepsin, a histone deacetylase inhibitor that specifically targets cutaneous T-cell lymphoma, but the lesions continued to progress. The patient then was treated with gemcitabine, dexamethasone, and cisplatin, and the rashes resolved. The most recent PET-CT showed nearly complete resolution of the subcutaneous lesions.
DISCUSSION
When residents or visitors to tropical or sub-tropical regions, those located near or between the Tropics of Cancer and Capricorn, present with fever, physicians usually first think of infectious diseases. This patient’s case is a reminder that these important first considerations should not be the last.
Generating a differential diagnosis for tropical illnesses begins with the patient’s history. Factors to be considered include location (regional disease prevalence), exposures (food/water ingestion, outdoor work/recreation, sexual contact, animal contact), and timing (temporal relationship of symptom development to possible exposure). Common tropical infections are malaria, dengue, typhoid, and emerging infections such as chikungunya, avian influenza, and Zika virus infection.1This case underscores the need to analyze diagnostic tests critically. Interpreting tests as simply positive or negative, irrespective of disease features, epidemiology, and test characteristics, can contribute to diagnostic error. For example, the patient’s positive ASO titer requires an understanding of disease features and a nuanced interpretation based on the clinical presentation. The erythematous posterior oropharynx prompted concern for postinfectious sequelae of streptococcal pharyngitis, but his illness was more severe and more prolonged than is typical of that condition. The isolated elevated O tsutsugamushi IgG titer provides an example of the role of epidemiology in test interpretation. Although a single positive value might indicate a new exposure for a visitor to an endemic region, IgG seropositivity in Singapore, where scrub typhus is endemic, likely reflects prior exposure to the organism. Diagnosing an acute scrub typhus infection in a patient in an endemic region requires PCR testing. The skin biopsy results highlight the importance of understanding test characteristics. A skin biopsy specimen must be adequate in order to draw valid and accurate conclusions. In this case, the initial skin biopsy was superficial, and the specimen inadequate, but the test was not “negative.” In the diagnostic skin biopsy, deeper tissue was sampled, and panniculitis (inflammation of subcutaneous fat), which arises in inflammatory, infectious, traumatic, enzymatic, and malignant conditions, was identified. An adequate biopsy specimen that contains subcutaneous fat is essential in making this diagnosis.2This patient eventually manifested several elements of hemophagocytic lymphohistiocytosis (HLH), a syndrome of excessive inflammation and resultant organ injury relating to abnormal immune activation and excessive inflammation. HLH results from deficient down-regulation of activated macrophages and lymphocytes.3 It was initially described in pediatric patients but is now recognized in adults, and associated with mortality as high as 50%.3 A high ferritin level (>2000 ng/mL) has 70% sensitivity and 68% specificity for pediatric HLH and should trigger consideration of HLH in any age group.4 The diagnostic criteria for HLH initially proposed in 2004 by the Histiocyte Society to identify patients for recruitment into a clinical trial included molecular testing consistent with HLH and/or 5 of 8 clinical, laboratory, or histopathologic features (Table 1).5 HScore is a more recent validated scoring system that predicts the probability of HLH (Table 2). A score above 169 signifies diagnostic sensitivity of 93% and specificity of 86%.6
The diagnosis of HLH warrants a search for its underlying cause. Common triggers are viral infections (eg, EBV), autoimmune diseases (eg, systemic lupus erythematosus), and hematologic malignancies. These triggers typically stimulate or suppress the immune system. Initial management involves treatment of the underlying trigger and, potentially, immunosuppression with high
In this case, SPTCL triggered HLH. SPTCL is a rare non-Hodgkin lymphoma characterized by painless subcutaneous nodules or indurated plaques (panniculitis-like) on the trunk or extremities, constitutional symptoms, and, in some cases, HLH.7-10 SPTCL is diagnosed by deep skin biopsy, with immunohistochemistry showing CD8-positive pathologic T cells expressing cytotoxic proteins (eg, granzyme B).9,11 SPTCL can either have an alpha/beta T-cell phenotype (SPTCL-AB) or gamma/delta T-cell phenotype (SPTCL-GD). Seventeen percent of patients with SPTCL-AB and 45% of patients with SPTCL-GD have HLH on diagnosis. Concomitant HLH is associated with decreased 5-year survival.12This patient presented with fevers and was ultimately diagnosed with HLH secondary to SPLTCL. His case is a reminder that not all diseases in the tropics are tropical diseases. In the diagnosis of a febrile illness, a broad evaluative framework and rigorous test results evaluation are essential—no matter where a patient lives or visits.
")
KEY TEACHING POINTS
- A febrile illness acquired in the tropics is not always attributable to a tropical infection.
- To avoid diagnostic error, weigh positive or negative test results against disease features, patient epidemiology, and test characteristics.
- HLH is characterized by fevers, cytopenias, hepatosplenomegaly, hyperferritinemia, hypertriglyceridemia, and hypofibrinogenemia. In tissue specimens, hemophagocytosis may help differentiate HLH from competing conditions.
- After HLH is diagnosed, try to determine its underlying cause, which may be an infection, autoimmunity, or a malignancy (commonly, a lymphoma).
Disclosure
Nothing to report.
1. Centers for Disease Control and Prevention. Destinations [list]. http://wwwnc.cdc.gov/travel/destinations/list/. Accessed April 22, 2016.
2. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22(6):530-549. PubMed
3. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503-1516. PubMed
4. Lehmberg K, McClain KL, Janka GE, Allen CE. Determination of an appropriate cut-off value for ferritin in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2014;61(11):2101-2103. PubMed
5. Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. PubMed
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. PubMed
7. Aronson IK, Worobed CM. Cytophagic histiocytic panniculitis and hemophagocytic lymphohistiocytosis: an overview. Dermatol Ther. 2010;23(4):389-402. PubMed
8. Willemze R, Jansen PM, Cerroni L, et al; EORTC Cutaneous Lymphoma Group. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group study of 83 cases. Blood. 2008;111(2):838-845. PubMed
9. Kumar S, Krenacs L, Medeiros J, et al. Subcutaneous panniculitic T-cell lymphoma is a tumor of cytotoxic T lymphocytes. Hum Pathol. 1998;29(4):397-403. PubMed
10. Salhany KE, Macon WR, Choi JK, et al. Subcutaneous panniculitis-like T-cell lymphoma: clinicopathologic, immunophenotypic, and genotypic analysis of alpha/beta and gamma/delta subtypes. Am J Surg Pathol. 1998;22(7):881-893. PubMed
11. Jaffe ES, Nicolae A, Pittaluga S. Peripheral T-cell and NK-cell lymphomas in the WHO classification: pearls and pitfalls. Mod Pathol. 2013;26(suppl 1):S71-S87. PubMed
12. Willemze R, Hodak E, Zinzani PL, Specht L, Ladetto M; ESMO Guidelines Working Group. Primary cutaneous lymphomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(suppl 6):vi149-vi154. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 42-year-old Malaysian construction worker with subjective fevers of 4 days’ duration presented to an emergency department in Singapore. He reported nonproductive cough, chills without rigors, sore throat, and body aches. He denied sick contacts. Past medical history included chronic hepatitis B virus (HBV) infection. The patient was not taking any medications.
For this patient presenting acutely with subjective fevers, nonproductive cough, chills, aches, and lethargy, initial considerations include infection with a common virus (influenza virus, adenovirus, Epstein-Barr virus [EBV]), acute human immunodeficiency virus (HIV) infection, emerging infection (severe acute respiratory syndrome [SARS], Middle Eastern respiratory syndrome coronavirus [MERS-CoV] infection, avian influenza), and tropical infection (dengue, chikungunya). Also possible are bacterial infections (eg, with Salmonella typhi or Rickettsia or Mycoplasma species), parasitic infections (eg, malaria), and noninfectious illnesses (eg, autoimmune diseases, thyroiditis, acute leukemia, environmental exposures).
The patient’s temperature was 38.5°C; blood pressure, 133/73 mm Hg; heart rate, 95 beats per minute; respiratory rate, 18 breaths per minute; and oxygen saturation, 100% on ambient air. On physical examination, he appeared comfortable, and heart, lung, abdomen, skin, and extremities were normal. Laboratory test results included white blood cell (WBC) count, 4400/μL (with normal differential); hemoglobin, 16.1 g/dL; and platelet count, 207,000/μL. Serum chemistries were normal. C-reactive protein (CRP) level was 44.6 mg/L (reference range, 0.2-9.1 mg/L), and procalcitonin level was 0.13 ng/mL (reference range, <0.50 ng/mL). Chest radiograph was normal. Dengue antibodies (immunoglobulin M, immunoglobulin G [IgG]) and dengue NS1 antigen were negative. The patient was discharged with a presumptive diagnosis of viral upper respiratory tract infection.
There is no left shift characteristic of bacterial infection or lymphopenia characteristic of rickettsial disease or acute HIV infection. The serologic testing and the patient’s overall appearance make dengue unlikely. The low procalcitonin supports a nonbacterial cause of illness. CRP elevation may indicate an inflammatory process and is relatively nonspecific.
Myalgias, pharyngitis, and cough improved over several days, but fevers persisted, and a rash developed over the lower abdomen. The patient returned to the emergency department and was admitted. He denied weight loss and night sweats. He had multiple female sexual partners, including commercial sex workers, within the previous 6 months. Temperature was 38.5°C. The posterior oropharynx was slightly erythematous. There was no lymphadenopathy. Firm, mildly erythematous macules were present on the anterior abdominal wall (Figure 1). The rest of the physical examination was normal.
Laboratory testing revealed WBC count, 5800/μL (75% neutrophils, 19% lymphocytes, 3% monocytes, 2% atypical mononuclear cells); hemoglobin, 16.3 g/dL; platelet count, 185,000/μL; sodium, 131 mmol/L; potassium, 3.4 mmol/L; creatinine, 0.9 mg/dL; albumin, 3.2 g/dL; alanine aminotransferase (ALT), 99 U/L; aspartate aminotransferase (AST), 137 U/L; alkaline phosphatase (ALP), 63 U/L; and total bilirubin, 1.9 mg/dL. Prothrombin time was 11.1 seconds; partial thromboplastin time, 36.1 seconds; erythrocyte sedimentation rate, 14 mm/h; and CRP, 62.2 mg/L.
EBV, acute HIV, and cytomegalovirus infections often present with adenopathy, which is absent here. Disseminated gonococcal infection can manifest with fever, body aches, and rash, but his rash and the absence of penile discharge, migratory arthritis, and enthesitis are not characteristic. Mycoplasma infection can present with macules, urticaria, or erythema multiforme. Rickettsia illnesses typically cause vasculitis with progression to petechiae or purpura resulting from endothelial damage. Patients with secondary syphilis may have widespread macular lesions, and the accompanying syphilitic hepatitis often manifests with elevations in ALP instead of ALT and AST. The mild elevation in ALT and AST can occur with many systemic viral infections. Sweet syndrome may manifest with febrile illness and rash, but the acuity of this patient’s illness and the rapid evolution favor infection.
The patient’s fevers (35°-40°C) continued without pattern over the next 3 days. Blood and urine cultures were negative. Polymerase chain reaction (PCR) test of the nasal mucosa was negative for respiratory viruses. PCR blood tests for EBV, HIV-1, and cytomegalovirus were also negative. Antistreptolysin O (ASO) titer was 400 IU/mm (reference range, <200 IU/mm). Antinuclear antibodies were negative, and rheumatoid factor was 12.4 U/mL (reference range, <10.3 U/mL). Computed tomography (CT) of the thorax, abdomen, and pelvis was normal. Results of a biopsy of an anterior abdominal wall skin lesion showed perivascular and periadnexal lymphocytic inflammation. Amoxicillin was started for the treatment of possible group A streptococcal infection.
PCR for HIV would be positive at a high level in acute HIV. The skin biopsy is not characteristic of Sweet syndrome, which typically shows neutrophilic infiltrate without leukocytoclastic vasculitis, or of syphilis, which typically shows a plasma cell infiltrate.
The patient’s erythematous oropharynx may indicate recent streptococcal pharyngitis. The fevers, elevated ASO titer, and CRP level are consistent with acute rheumatic fever, but arthritis, carditis, and neurologic manifestations are lacking. Erythema marginatum manifests on the trunk and limbs as macules or papules with central clearing as the lesions spread outward—and differs from the patient’s rash, which is firm and restricted to the abdominal wall.
Fevers persisted through hospital day 7. The WBC count was 1100/μL (75.7% neutrophils, 22.5% lymphocytes), hemoglobin was 10.3 g/dL, and platelet count was 52,000/μL. Additional laboratory test results included ALP, 234 U/L; ALT, 250 U/L; AST, 459 U/L; lactate dehydrogenase, 2303 U/L (reference range, 222-454 U/L); and ferritin, 14,964 ng/mL (reference range, 47-452 ng/mL).
The duration of illness and negative diagnostic tests for infections increases suspicion for a noninfectious illness. Conditions commonly associated with marked hyperferritinemia include adult-onset Still disease (AOSD) and hemophagocytic lymphohistiocytosis (HLH). Of the 9 AOSD diagnostic (Yamaguchi) criteria, 5 are met in this case: fever, rash, sore throat, abnormal liver function tests, and negative rheumatologic tests. However, the patient lacks arthritis, leukocytosis, lymphadenopathy, and hepatosplenomegaly. Except for the elevated ferritin, the AOSD criteria overlap substantially with the criteria for acute rheumatic fever, and still require that infections be adequately excluded. HLH, a state of abnormal immune activation with resultant organ dysfunction, can be a primary disorder, but in adults more often is secondary to underlying infectious, autoimmune, or malignant (often lymphoma) conditions. Elevated ferritin, cytopenias, elevated ALT and AST, elevated CRP and erythrocyte sedimentation rate, and elevated lactate dehydrogenase are consistent with HLH. The HLH diagnosis can be more firmly established with the more specific findings of hypertriglyceridemia, hypofibrinogenemia, and elevated soluble CD25 level. The histopathologic finding of hemophagocytosis in the bone marrow, lymph nodes, or liver may further support the diagnosis of HLH.
Rash and fevers persisted. Hepatitis A, hepatitis C, Rickettsia IgG, Burkholderia pseudomallei (the causative organism of melioidosis), and Leptospira serologies, as well as PCR for herpes simplex virus and parvovirus, were all negative. Hepatitis B viral load was 962 IU/mL (2.98 log), hepatitis B envelope antigen was negative, and hepatitis B envelope antibody was positive. Orientia tsutsugamushi (organism responsible for scrub typhus) IgG titer was elevated at 1:128. Antiliver kidney microsomal antibodies and antineutrophil cytoplasmic antibodies were negative. Fibrinogen level was 0.69 g/L (reference range, 1.8-4.8 g/L), and beta-2 microglobulin level was 5078 ng/mL (reference range, 878-2000 ng/mL). Bone marrow biopsy results showed hypocellular marrow with suppressed myelopoiesis, few atypical lymphoid cells, and few hemophagocytes. Flow cytometry was negative for clonal B lymphocytes and aberrant expression of T lymphocytes. Bone marrow myobacterial PCR and fungal cultures were negative.
The patient’s chronic HBV infection is unlikely to be related to his presentation given his low viral load and absence of signs of hepatic dysfunction. Excluding rickettsial disease requires paired acute and convalescent serologies. O tsutsugamushi, the causative agent of the rickettsial disease scrub typhus, is endemic in Malaysia; thus, his positive O tsutsugamushi IgG may indicate past exposure. His fevers, myalgias, truncal rash, and hepatitis are consistent with scrub typhus, but he lacks the characteristic severe headache and generalized lymphadenopathy. Although eschar formation with evolution of a papular rash is common in scrub typhus, it is often absent in the variant found in Southeast Asia. Although elevated β2 microglobulin level is used as a prognostic marker in multiple myeloma and Waldenström macroglobulinemia, it can be elevated in many immune-active states. The patient likely has HLH, which is supported by the hemophagocytosis seen on bone marrow biopsy, and the hypofibrinogenemia. Potential HLH triggers include O tsutsugamushi infection or recent streptococcal pharyngitis.
A deep-punch skin biopsy of the anterior abdominal wall skin lesion was performed because of the absence of subcutaneous fat in the first biopsy specimen. The latest biopsy results showed irregular interstitial expansion of medium-size lymphocytes in a lobular panniculated pattern. The lymphocytes contained enlarged, irregularly contoured nucleoli and were positive for T-cell markers CD2 and CD3 with reduction in CD5 expression. The lymphomatous cells were of CD8+ with uniform expression of activated cytotoxic granule protein granzyme B and were positive for T-cell hemireceptor β.
Positron emission tomography (PET) CT, obtained for staging purposes, showed multiple hypermetabolic subcutaneous and cutaneous lesions over the torso and upper and lower limbs—compatible with lymphomatous infiltrates (Figure 2). Examination, pathology, and imaging findings suggested a rare neoplasm: subcutaneous panniculitis-like T-cell lymphoma (SPTCL). SPTCL was confirmed by T-cell receptor gene rearrangements studies.
HLH was diagnosed on the basis of the fevers, cytopenias, hypofibrinogenemia, elevated ferritin level, and evidence of hemophagocytosis. SPTCL was suspected as the HLH trigger.
The patient was treated with cyclophosphamide, hydroxydoxorubicin, vincristine, and prednisone. While on this regimen, he developed new skin lesions, and his ferritin level was persistently elevated. He was switched to romidepsin, a histone deacetylase inhibitor that specifically targets cutaneous T-cell lymphoma, but the lesions continued to progress. The patient then was treated with gemcitabine, dexamethasone, and cisplatin, and the rashes resolved. The most recent PET-CT showed nearly complete resolution of the subcutaneous lesions.
DISCUSSION
When residents or visitors to tropical or sub-tropical regions, those located near or between the Tropics of Cancer and Capricorn, present with fever, physicians usually first think of infectious diseases. This patient’s case is a reminder that these important first considerations should not be the last.
Generating a differential diagnosis for tropical illnesses begins with the patient’s history. Factors to be considered include location (regional disease prevalence), exposures (food/water ingestion, outdoor work/recreation, sexual contact, animal contact), and timing (temporal relationship of symptom development to possible exposure). Common tropical infections are malaria, dengue, typhoid, and emerging infections such as chikungunya, avian influenza, and Zika virus infection.1This case underscores the need to analyze diagnostic tests critically. Interpreting tests as simply positive or negative, irrespective of disease features, epidemiology, and test characteristics, can contribute to diagnostic error. For example, the patient’s positive ASO titer requires an understanding of disease features and a nuanced interpretation based on the clinical presentation. The erythematous posterior oropharynx prompted concern for postinfectious sequelae of streptococcal pharyngitis, but his illness was more severe and more prolonged than is typical of that condition. The isolated elevated O tsutsugamushi IgG titer provides an example of the role of epidemiology in test interpretation. Although a single positive value might indicate a new exposure for a visitor to an endemic region, IgG seropositivity in Singapore, where scrub typhus is endemic, likely reflects prior exposure to the organism. Diagnosing an acute scrub typhus infection in a patient in an endemic region requires PCR testing. The skin biopsy results highlight the importance of understanding test characteristics. A skin biopsy specimen must be adequate in order to draw valid and accurate conclusions. In this case, the initial skin biopsy was superficial, and the specimen inadequate, but the test was not “negative.” In the diagnostic skin biopsy, deeper tissue was sampled, and panniculitis (inflammation of subcutaneous fat), which arises in inflammatory, infectious, traumatic, enzymatic, and malignant conditions, was identified. An adequate biopsy specimen that contains subcutaneous fat is essential in making this diagnosis.2This patient eventually manifested several elements of hemophagocytic lymphohistiocytosis (HLH), a syndrome of excessive inflammation and resultant organ injury relating to abnormal immune activation and excessive inflammation. HLH results from deficient down-regulation of activated macrophages and lymphocytes.3 It was initially described in pediatric patients but is now recognized in adults, and associated with mortality as high as 50%.3 A high ferritin level (>2000 ng/mL) has 70% sensitivity and 68% specificity for pediatric HLH and should trigger consideration of HLH in any age group.4 The diagnostic criteria for HLH initially proposed in 2004 by the Histiocyte Society to identify patients for recruitment into a clinical trial included molecular testing consistent with HLH and/or 5 of 8 clinical, laboratory, or histopathologic features (Table 1).5 HScore is a more recent validated scoring system that predicts the probability of HLH (Table 2). A score above 169 signifies diagnostic sensitivity of 93% and specificity of 86%.6
The diagnosis of HLH warrants a search for its underlying cause. Common triggers are viral infections (eg, EBV), autoimmune diseases (eg, systemic lupus erythematosus), and hematologic malignancies. These triggers typically stimulate or suppress the immune system. Initial management involves treatment of the underlying trigger and, potentially, immunosuppression with high
In this case, SPTCL triggered HLH. SPTCL is a rare non-Hodgkin lymphoma characterized by painless subcutaneous nodules or indurated plaques (panniculitis-like) on the trunk or extremities, constitutional symptoms, and, in some cases, HLH.7-10 SPTCL is diagnosed by deep skin biopsy, with immunohistochemistry showing CD8-positive pathologic T cells expressing cytotoxic proteins (eg, granzyme B).9,11 SPTCL can either have an alpha/beta T-cell phenotype (SPTCL-AB) or gamma/delta T-cell phenotype (SPTCL-GD). Seventeen percent of patients with SPTCL-AB and 45% of patients with SPTCL-GD have HLH on diagnosis. Concomitant HLH is associated with decreased 5-year survival.12This patient presented with fevers and was ultimately diagnosed with HLH secondary to SPLTCL. His case is a reminder that not all diseases in the tropics are tropical diseases. In the diagnosis of a febrile illness, a broad evaluative framework and rigorous test results evaluation are essential—no matter where a patient lives or visits.
KEY TEACHING POINTS
- A febrile illness acquired in the tropics is not always attributable to a tropical infection.
- To avoid diagnostic error, weigh positive or negative test results against disease features, patient epidemiology, and test characteristics.
- HLH is characterized by fevers, cytopenias, hepatosplenomegaly, hyperferritinemia, hypertriglyceridemia, and hypofibrinogenemia. In tissue specimens, hemophagocytosis may help differentiate HLH from competing conditions.
- After HLH is diagnosed, try to determine its underlying cause, which may be an infection, autoimmunity, or a malignancy (commonly, a lymphoma).
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 42-year-old Malaysian construction worker with subjective fevers of 4 days’ duration presented to an emergency department in Singapore. He reported nonproductive cough, chills without rigors, sore throat, and body aches. He denied sick contacts. Past medical history included chronic hepatitis B virus (HBV) infection. The patient was not taking any medications.
For this patient presenting acutely with subjective fevers, nonproductive cough, chills, aches, and lethargy, initial considerations include infection with a common virus (influenza virus, adenovirus, Epstein-Barr virus [EBV]), acute human immunodeficiency virus (HIV) infection, emerging infection (severe acute respiratory syndrome [SARS], Middle Eastern respiratory syndrome coronavirus [MERS-CoV] infection, avian influenza), and tropical infection (dengue, chikungunya). Also possible are bacterial infections (eg, with Salmonella typhi or Rickettsia or Mycoplasma species), parasitic infections (eg, malaria), and noninfectious illnesses (eg, autoimmune diseases, thyroiditis, acute leukemia, environmental exposures).
The patient’s temperature was 38.5°C; blood pressure, 133/73 mm Hg; heart rate, 95 beats per minute; respiratory rate, 18 breaths per minute; and oxygen saturation, 100% on ambient air. On physical examination, he appeared comfortable, and heart, lung, abdomen, skin, and extremities were normal. Laboratory test results included white blood cell (WBC) count, 4400/μL (with normal differential); hemoglobin, 16.1 g/dL; and platelet count, 207,000/μL. Serum chemistries were normal. C-reactive protein (CRP) level was 44.6 mg/L (reference range, 0.2-9.1 mg/L), and procalcitonin level was 0.13 ng/mL (reference range, <0.50 ng/mL). Chest radiograph was normal. Dengue antibodies (immunoglobulin M, immunoglobulin G [IgG]) and dengue NS1 antigen were negative. The patient was discharged with a presumptive diagnosis of viral upper respiratory tract infection.
There is no left shift characteristic of bacterial infection or lymphopenia characteristic of rickettsial disease or acute HIV infection. The serologic testing and the patient’s overall appearance make dengue unlikely. The low procalcitonin supports a nonbacterial cause of illness. CRP elevation may indicate an inflammatory process and is relatively nonspecific.
Myalgias, pharyngitis, and cough improved over several days, but fevers persisted, and a rash developed over the lower abdomen. The patient returned to the emergency department and was admitted. He denied weight loss and night sweats. He had multiple female sexual partners, including commercial sex workers, within the previous 6 months. Temperature was 38.5°C. The posterior oropharynx was slightly erythematous. There was no lymphadenopathy. Firm, mildly erythematous macules were present on the anterior abdominal wall (Figure 1). The rest of the physical examination was normal.
Laboratory testing revealed WBC count, 5800/μL (75% neutrophils, 19% lymphocytes, 3% monocytes, 2% atypical mononuclear cells); hemoglobin, 16.3 g/dL; platelet count, 185,000/μL; sodium, 131 mmol/L; potassium, 3.4 mmol/L; creatinine, 0.9 mg/dL; albumin, 3.2 g/dL; alanine aminotransferase (ALT), 99 U/L; aspartate aminotransferase (AST), 137 U/L; alkaline phosphatase (ALP), 63 U/L; and total bilirubin, 1.9 mg/dL. Prothrombin time was 11.1 seconds; partial thromboplastin time, 36.1 seconds; erythrocyte sedimentation rate, 14 mm/h; and CRP, 62.2 mg/L.
EBV, acute HIV, and cytomegalovirus infections often present with adenopathy, which is absent here. Disseminated gonococcal infection can manifest with fever, body aches, and rash, but his rash and the absence of penile discharge, migratory arthritis, and enthesitis are not characteristic. Mycoplasma infection can present with macules, urticaria, or erythema multiforme. Rickettsia illnesses typically cause vasculitis with progression to petechiae or purpura resulting from endothelial damage. Patients with secondary syphilis may have widespread macular lesions, and the accompanying syphilitic hepatitis often manifests with elevations in ALP instead of ALT and AST. The mild elevation in ALT and AST can occur with many systemic viral infections. Sweet syndrome may manifest with febrile illness and rash, but the acuity of this patient’s illness and the rapid evolution favor infection.
The patient’s fevers (35°-40°C) continued without pattern over the next 3 days. Blood and urine cultures were negative. Polymerase chain reaction (PCR) test of the nasal mucosa was negative for respiratory viruses. PCR blood tests for EBV, HIV-1, and cytomegalovirus were also negative. Antistreptolysin O (ASO) titer was 400 IU/mm (reference range, <200 IU/mm). Antinuclear antibodies were negative, and rheumatoid factor was 12.4 U/mL (reference range, <10.3 U/mL). Computed tomography (CT) of the thorax, abdomen, and pelvis was normal. Results of a biopsy of an anterior abdominal wall skin lesion showed perivascular and periadnexal lymphocytic inflammation. Amoxicillin was started for the treatment of possible group A streptococcal infection.
PCR for HIV would be positive at a high level in acute HIV. The skin biopsy is not characteristic of Sweet syndrome, which typically shows neutrophilic infiltrate without leukocytoclastic vasculitis, or of syphilis, which typically shows a plasma cell infiltrate.
The patient’s erythematous oropharynx may indicate recent streptococcal pharyngitis. The fevers, elevated ASO titer, and CRP level are consistent with acute rheumatic fever, but arthritis, carditis, and neurologic manifestations are lacking. Erythema marginatum manifests on the trunk and limbs as macules or papules with central clearing as the lesions spread outward—and differs from the patient’s rash, which is firm and restricted to the abdominal wall.
Fevers persisted through hospital day 7. The WBC count was 1100/μL (75.7% neutrophils, 22.5% lymphocytes), hemoglobin was 10.3 g/dL, and platelet count was 52,000/μL. Additional laboratory test results included ALP, 234 U/L; ALT, 250 U/L; AST, 459 U/L; lactate dehydrogenase, 2303 U/L (reference range, 222-454 U/L); and ferritin, 14,964 ng/mL (reference range, 47-452 ng/mL).
The duration of illness and negative diagnostic tests for infections increases suspicion for a noninfectious illness. Conditions commonly associated with marked hyperferritinemia include adult-onset Still disease (AOSD) and hemophagocytic lymphohistiocytosis (HLH). Of the 9 AOSD diagnostic (Yamaguchi) criteria, 5 are met in this case: fever, rash, sore throat, abnormal liver function tests, and negative rheumatologic tests. However, the patient lacks arthritis, leukocytosis, lymphadenopathy, and hepatosplenomegaly. Except for the elevated ferritin, the AOSD criteria overlap substantially with the criteria for acute rheumatic fever, and still require that infections be adequately excluded. HLH, a state of abnormal immune activation with resultant organ dysfunction, can be a primary disorder, but in adults more often is secondary to underlying infectious, autoimmune, or malignant (often lymphoma) conditions. Elevated ferritin, cytopenias, elevated ALT and AST, elevated CRP and erythrocyte sedimentation rate, and elevated lactate dehydrogenase are consistent with HLH. The HLH diagnosis can be more firmly established with the more specific findings of hypertriglyceridemia, hypofibrinogenemia, and elevated soluble CD25 level. The histopathologic finding of hemophagocytosis in the bone marrow, lymph nodes, or liver may further support the diagnosis of HLH.
Rash and fevers persisted. Hepatitis A, hepatitis C, Rickettsia IgG, Burkholderia pseudomallei (the causative organism of melioidosis), and Leptospira serologies, as well as PCR for herpes simplex virus and parvovirus, were all negative. Hepatitis B viral load was 962 IU/mL (2.98 log), hepatitis B envelope antigen was negative, and hepatitis B envelope antibody was positive. Orientia tsutsugamushi (organism responsible for scrub typhus) IgG titer was elevated at 1:128. Antiliver kidney microsomal antibodies and antineutrophil cytoplasmic antibodies were negative. Fibrinogen level was 0.69 g/L (reference range, 1.8-4.8 g/L), and beta-2 microglobulin level was 5078 ng/mL (reference range, 878-2000 ng/mL). Bone marrow biopsy results showed hypocellular marrow with suppressed myelopoiesis, few atypical lymphoid cells, and few hemophagocytes. Flow cytometry was negative for clonal B lymphocytes and aberrant expression of T lymphocytes. Bone marrow myobacterial PCR and fungal cultures were negative.
The patient’s chronic HBV infection is unlikely to be related to his presentation given his low viral load and absence of signs of hepatic dysfunction. Excluding rickettsial disease requires paired acute and convalescent serologies. O tsutsugamushi, the causative agent of the rickettsial disease scrub typhus, is endemic in Malaysia; thus, his positive O tsutsugamushi IgG may indicate past exposure. His fevers, myalgias, truncal rash, and hepatitis are consistent with scrub typhus, but he lacks the characteristic severe headache and generalized lymphadenopathy. Although eschar formation with evolution of a papular rash is common in scrub typhus, it is often absent in the variant found in Southeast Asia. Although elevated β2 microglobulin level is used as a prognostic marker in multiple myeloma and Waldenström macroglobulinemia, it can be elevated in many immune-active states. The patient likely has HLH, which is supported by the hemophagocytosis seen on bone marrow biopsy, and the hypofibrinogenemia. Potential HLH triggers include O tsutsugamushi infection or recent streptococcal pharyngitis.
A deep-punch skin biopsy of the anterior abdominal wall skin lesion was performed because of the absence of subcutaneous fat in the first biopsy specimen. The latest biopsy results showed irregular interstitial expansion of medium-size lymphocytes in a lobular panniculated pattern. The lymphocytes contained enlarged, irregularly contoured nucleoli and were positive for T-cell markers CD2 and CD3 with reduction in CD5 expression. The lymphomatous cells were of CD8+ with uniform expression of activated cytotoxic granule protein granzyme B and were positive for T-cell hemireceptor β.
Positron emission tomography (PET) CT, obtained for staging purposes, showed multiple hypermetabolic subcutaneous and cutaneous lesions over the torso and upper and lower limbs—compatible with lymphomatous infiltrates (Figure 2). Examination, pathology, and imaging findings suggested a rare neoplasm: subcutaneous panniculitis-like T-cell lymphoma (SPTCL). SPTCL was confirmed by T-cell receptor gene rearrangements studies.
HLH was diagnosed on the basis of the fevers, cytopenias, hypofibrinogenemia, elevated ferritin level, and evidence of hemophagocytosis. SPTCL was suspected as the HLH trigger.
The patient was treated with cyclophosphamide, hydroxydoxorubicin, vincristine, and prednisone. While on this regimen, he developed new skin lesions, and his ferritin level was persistently elevated. He was switched to romidepsin, a histone deacetylase inhibitor that specifically targets cutaneous T-cell lymphoma, but the lesions continued to progress. The patient then was treated with gemcitabine, dexamethasone, and cisplatin, and the rashes resolved. The most recent PET-CT showed nearly complete resolution of the subcutaneous lesions.
DISCUSSION
When residents or visitors to tropical or sub-tropical regions, those located near or between the Tropics of Cancer and Capricorn, present with fever, physicians usually first think of infectious diseases. This patient’s case is a reminder that these important first considerations should not be the last.
Generating a differential diagnosis for tropical illnesses begins with the patient’s history. Factors to be considered include location (regional disease prevalence), exposures (food/water ingestion, outdoor work/recreation, sexual contact, animal contact), and timing (temporal relationship of symptom development to possible exposure). Common tropical infections are malaria, dengue, typhoid, and emerging infections such as chikungunya, avian influenza, and Zika virus infection.1This case underscores the need to analyze diagnostic tests critically. Interpreting tests as simply positive or negative, irrespective of disease features, epidemiology, and test characteristics, can contribute to diagnostic error. For example, the patient’s positive ASO titer requires an understanding of disease features and a nuanced interpretation based on the clinical presentation. The erythematous posterior oropharynx prompted concern for postinfectious sequelae of streptococcal pharyngitis, but his illness was more severe and more prolonged than is typical of that condition. The isolated elevated O tsutsugamushi IgG titer provides an example of the role of epidemiology in test interpretation. Although a single positive value might indicate a new exposure for a visitor to an endemic region, IgG seropositivity in Singapore, where scrub typhus is endemic, likely reflects prior exposure to the organism. Diagnosing an acute scrub typhus infection in a patient in an endemic region requires PCR testing. The skin biopsy results highlight the importance of understanding test characteristics. A skin biopsy specimen must be adequate in order to draw valid and accurate conclusions. In this case, the initial skin biopsy was superficial, and the specimen inadequate, but the test was not “negative.” In the diagnostic skin biopsy, deeper tissue was sampled, and panniculitis (inflammation of subcutaneous fat), which arises in inflammatory, infectious, traumatic, enzymatic, and malignant conditions, was identified. An adequate biopsy specimen that contains subcutaneous fat is essential in making this diagnosis.2This patient eventually manifested several elements of hemophagocytic lymphohistiocytosis (HLH), a syndrome of excessive inflammation and resultant organ injury relating to abnormal immune activation and excessive inflammation. HLH results from deficient down-regulation of activated macrophages and lymphocytes.3 It was initially described in pediatric patients but is now recognized in adults, and associated with mortality as high as 50%.3 A high ferritin level (>2000 ng/mL) has 70% sensitivity and 68% specificity for pediatric HLH and should trigger consideration of HLH in any age group.4 The diagnostic criteria for HLH initially proposed in 2004 by the Histiocyte Society to identify patients for recruitment into a clinical trial included molecular testing consistent with HLH and/or 5 of 8 clinical, laboratory, or histopathologic features (Table 1).5 HScore is a more recent validated scoring system that predicts the probability of HLH (Table 2). A score above 169 signifies diagnostic sensitivity of 93% and specificity of 86%.6
The diagnosis of HLH warrants a search for its underlying cause. Common triggers are viral infections (eg, EBV), autoimmune diseases (eg, systemic lupus erythematosus), and hematologic malignancies. These triggers typically stimulate or suppress the immune system. Initial management involves treatment of the underlying trigger and, potentially, immunosuppression with high
In this case, SPTCL triggered HLH. SPTCL is a rare non-Hodgkin lymphoma characterized by painless subcutaneous nodules or indurated plaques (panniculitis-like) on the trunk or extremities, constitutional symptoms, and, in some cases, HLH.7-10 SPTCL is diagnosed by deep skin biopsy, with immunohistochemistry showing CD8-positive pathologic T cells expressing cytotoxic proteins (eg, granzyme B).9,11 SPTCL can either have an alpha/beta T-cell phenotype (SPTCL-AB) or gamma/delta T-cell phenotype (SPTCL-GD). Seventeen percent of patients with SPTCL-AB and 45% of patients with SPTCL-GD have HLH on diagnosis. Concomitant HLH is associated with decreased 5-year survival.12This patient presented with fevers and was ultimately diagnosed with HLH secondary to SPLTCL. His case is a reminder that not all diseases in the tropics are tropical diseases. In the diagnosis of a febrile illness, a broad evaluative framework and rigorous test results evaluation are essential—no matter where a patient lives or visits.
KEY TEACHING POINTS
- A febrile illness acquired in the tropics is not always attributable to a tropical infection.
- To avoid diagnostic error, weigh positive or negative test results against disease features, patient epidemiology, and test characteristics.
- HLH is characterized by fevers, cytopenias, hepatosplenomegaly, hyperferritinemia, hypertriglyceridemia, and hypofibrinogenemia. In tissue specimens, hemophagocytosis may help differentiate HLH from competing conditions.
- After HLH is diagnosed, try to determine its underlying cause, which may be an infection, autoimmunity, or a malignancy (commonly, a lymphoma).
Disclosure
Nothing to report.
1. Centers for Disease Control and Prevention. Destinations [list]. http://wwwnc.cdc.gov/travel/destinations/list/. Accessed April 22, 2016.
2. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22(6):530-549. PubMed
3. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503-1516. PubMed
4. Lehmberg K, McClain KL, Janka GE, Allen CE. Determination of an appropriate cut-off value for ferritin in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2014;61(11):2101-2103. PubMed
5. Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. PubMed
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. PubMed
7. Aronson IK, Worobed CM. Cytophagic histiocytic panniculitis and hemophagocytic lymphohistiocytosis: an overview. Dermatol Ther. 2010;23(4):389-402. PubMed
8. Willemze R, Jansen PM, Cerroni L, et al; EORTC Cutaneous Lymphoma Group. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group study of 83 cases. Blood. 2008;111(2):838-845. PubMed
9. Kumar S, Krenacs L, Medeiros J, et al. Subcutaneous panniculitic T-cell lymphoma is a tumor of cytotoxic T lymphocytes. Hum Pathol. 1998;29(4):397-403. PubMed
10. Salhany KE, Macon WR, Choi JK, et al. Subcutaneous panniculitis-like T-cell lymphoma: clinicopathologic, immunophenotypic, and genotypic analysis of alpha/beta and gamma/delta subtypes. Am J Surg Pathol. 1998;22(7):881-893. PubMed
11. Jaffe ES, Nicolae A, Pittaluga S. Peripheral T-cell and NK-cell lymphomas in the WHO classification: pearls and pitfalls. Mod Pathol. 2013;26(suppl 1):S71-S87. PubMed
12. Willemze R, Hodak E, Zinzani PL, Specht L, Ladetto M; ESMO Guidelines Working Group. Primary cutaneous lymphomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(suppl 6):vi149-vi154. PubMed
1. Centers for Disease Control and Prevention. Destinations [list]. http://wwwnc.cdc.gov/travel/destinations/list/. Accessed April 22, 2016.
2. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22(6):530-549. PubMed
3. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503-1516. PubMed
4. Lehmberg K, McClain KL, Janka GE, Allen CE. Determination of an appropriate cut-off value for ferritin in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2014;61(11):2101-2103. PubMed
5. Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. PubMed
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. PubMed
7. Aronson IK, Worobed CM. Cytophagic histiocytic panniculitis and hemophagocytic lymphohistiocytosis: an overview. Dermatol Ther. 2010;23(4):389-402. PubMed
8. Willemze R, Jansen PM, Cerroni L, et al; EORTC Cutaneous Lymphoma Group. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group study of 83 cases. Blood. 2008;111(2):838-845. PubMed
9. Kumar S, Krenacs L, Medeiros J, et al. Subcutaneous panniculitic T-cell lymphoma is a tumor of cytotoxic T lymphocytes. Hum Pathol. 1998;29(4):397-403. PubMed
10. Salhany KE, Macon WR, Choi JK, et al. Subcutaneous panniculitis-like T-cell lymphoma: clinicopathologic, immunophenotypic, and genotypic analysis of alpha/beta and gamma/delta subtypes. Am J Surg Pathol. 1998;22(7):881-893. PubMed
11. Jaffe ES, Nicolae A, Pittaluga S. Peripheral T-cell and NK-cell lymphomas in the WHO classification: pearls and pitfalls. Mod Pathol. 2013;26(suppl 1):S71-S87. PubMed
12. Willemze R, Hodak E, Zinzani PL, Specht L, Ladetto M; ESMO Guidelines Working Group. Primary cutaneous lymphomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(suppl 6):vi149-vi154. PubMed
© 2017 Society of Hospital Medicine
Rendered speechless
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 63-year-old man at an inpatient rehabilitation center was transferred to an academic tertiary care center for evaluation of slurred speech and episodic confusion. He was accompanied by his wife, who provided the history. Three weeks earlier, the patient had fallen, sustaining a right femur fracture. He underwent surgery and was discharged to rehabilitation on postoperative day 3. During the second week of rehabilitation, he developed a cough and low-grade fevers, which prompted treatment with cefpodoxime for 5 days for presumed pneumonia. The day after completing antimicrobial therapy, he became confused and began to slur his words.
Confusion is a nonspecific symptom that typically has a diffuse or multifocal localization within the cerebral hemispheres and is unlikely to be caused by a single lesion. Slurred speech may accompany global metabolic dysfunction. However, slurred speech typically localizes to the brainstem, the cerebellum in the posterior fossa, the nuclei, or the course of cranial nerves VII, X, or XII, including where these nerves pass through the subarachnoid space.
It seems this patient’s new neurologic symptoms have some relationship to his fall. Long-bone fractures and altered mental status (AMS) lead to consideration of fat emboli, but this syndrome typically presents in the acute period after the fracture. The patient is at risk for a number of complications, related to recent surgery and hospitalization, that could affect the central nervous system (CNS), including systemic infection (possibly with associated meningeal involvement) and venous thromboembolism with concomitant stroke by paradoxical emboli. The episodic nature of the confusion leads to consideration of seizures from structural lesions in the brain. Finally, the circumstances of the fall itself should be explored to determine whether an underlying neurologic dysfunction led to imbalance and gait difficulty.
Over the next 3 days at the inpatient rehabilitation center, the patient’s slurred speech became unintelligible, and he experienced intermittent disorientation to person, place, and time. There was no concomitant fever, dizziness, headache, neck pain, weakness, dyspnea, diarrhea, dysuria, or change in hearing or vision.
Progressive dysarthria argues for an expanding lesion in the posterior fossa, worsening metabolic disturbance, or a problem affecting the cranial nerves (eg, Guillain-Barré syndrome) or neuromuscular junctions (eg, myasthenia gravis). Lack of headache makes a CNS localization less likely, though disorientation must localize to the brain itself. The transient nature of the AMS could signal an ictal phenomenon or a fluctuating toxic or metabolic condition, such as hyperammonemia, drug reaction, or healthcare–acquired delirium.
His past medical history included end-stage liver disease secondary to nonalcoholic steatohepatitis status post transjugular intrahepatic portosystemic shunt (TIPS) procedure three years prior, hepatic encephalopathy, diabetes mellitus type 2, hypertension, previous melanoma excision on his back, and recurrent Clostridium difficile colitis. Two years prior to admission he had been started on an indefinite course of metronidazole 500 mg twice daily without any recurrence. The patient’s other medications were aspirin, furosemide, insulin, lactulose, mirtazapine, pantoprazole, propranolol, spironolactone, and zinc. At the rehabilitation center, he was prescribed oral oxycodone 5 mg as needed every 4 hours for pain. He denied use of tobacco, alcohol, and recreational drugs. He previously worked as a funeral home director and embalmer.
Hyperammonemia and hepatic encephalopathy can present with a fluctuating mental state that often correlates to dietary protein intake or the frequency of bowel movements; the previous TIPS history places the patient at further risk. Use of oxycodone or another narcotic commonly leads to confusion, , especially in patients who are older, have preexisting cognitive decline, or have concomitant medical comorbidities. Mirtazapine and propranolol have been associated more rarely with encephalopathy, and therefore a careful history of adherence, drug interactions, and appropriate dosing should be obtained. Metronidazole is most often associated neurologically with a peripheral neuropathy; however, it is increasingly recognized that some patients can develop a CNS syndrome that features an AMS, which can be severe and accompanied by ataxia, dysarthria, and characteristic brain magnetic resonance imaging (MRI) findings, including hyperintensity surrounding the fourth ventricle on T2-weighted images.
Embalming fluid has a high concentration of formaldehyde, and a recent epidemiologic study suggested a link between formaldehyde exposure and increased risk for amyotrophic lateral sclerosis (ALS). ALS uncommonly presents with isolated dysarthria, but its bulbar form can, usually over a much longer course than is demonstrated here. Finally, the patient’s history of melanoma places him at risk for stroke from hypercoagulability as well as potential brain metastases or carcinomatous meningitis.
Evaluation was initiated at the rehabilitation facility at the onset of the patient’s slurred speech and confusion. Physical examination were negative for focal neurologic deficits, asterixis, and jaundice. Ammonia level was 41 µmol/L (reference range, 11-35 µmol/L). Noncontrast computed tomography (CT) of the head showed no signs of acute infarct or hemorrhage. Symptoms were attributed to hepatic encephalopathy; lactulose was up-titrated to ensure 2 or 3 bowel movements per day, and rifaximin was started.
Hyperammonemia is a cause of non-inflammatory relapsing encephalopathy, but an elevated level is neither a sensitive nor specific indicator of hepatic encephalopathy. Levels of ammonia can fluctuate widely during the day based on the frequency of bowel movements as well as dietary protein intake. In addition, proper handling of samples with prompt delivery to the laboratory is essential to minimize errors.
The ammonia level of 41 µmol/L discovered here is only modestly elevated, but given the patient’s history of TIPS as well as the clinical picture, it is reasonable to aggressively treat hepatic encephalopathy with lactulose to reduce ammonia levels. If he does not improve, an MRI of the brain to exclude a structural lesion and spinal fluid examination looking for inflammatory or infectious conditions would be important next steps. Although CT excludes a large hemorrhage or mass, this screening examination does not visualize many of the findings of the metabolic etiology and the other etiologies under consideration here.
Despite 3 days of therapy for presumed hepatic encephalopathy, the patient’s slurred speech worsened, and he was transferred to an academic tertiary care center for further evaluation. On admission, his temperature was 36.9°C, heart rate was 80 beats per minute, blood pressure was 139/67 mm Hg, respiratory rate was 10 breaths per minute, and oxygen saturation was 99% on room air. He was alert, awake, and oriented to person, place, and time. He was not jaundiced. He exhibited a moderate dysarthria characterized by monotone speech, decreased volume, decreased breath support, and a hoarse vocal quality with intact language function. Motor control of the lips, tongue, and mandible were normal. Motor strength was 5/5 bilaterally in the upper and lower extremities with the exception of right hip flexion, which was 4/5. The patient exhibited mild bilateral dysmetria on finger-to-nose examination, consistent with appendicular ataxia of the upper extremities. Reflexes were depressed throughout, and there was no asterixis. He had 2+ pulses in all extremities and 1+ pitting edema of the right lower extremity to the mid leg. Pulmonary examination revealed inspiratory crackles at the left base. The rest of the examination findings were normal.
The patient’s altered mental state appears to have resolved, and the neurological examination is now mainly characterized by signs that point to the cerebellum. The description of monotone speech typically refers to loss of prosody, the variable stress or intonation of speech, which is characteristic of a cerebellar speech pattern. The hoarseness should be explored to determine if it is a feature of the patient’s speech or is a separate process. Hoarseness may involve the vocal cord and therefore, potentially, cranial nerve X or its nuclei in the brainstem. The appendicular ataxia of the limbs points definitively to the cerebellar hemispheres or their pathways through the brainstem.
Unilateral lower extremity edema, especially in the context of a recent fracture, raises the possibility of deep vein thrombosis. If this patient has a right-to-left intracardiac or intrapulmonary shunt, embolization could lead to an ischemic stroke of the brainstem or cerebellum, potentially causing dysarthria.
Laboratory evaluation revealed hemoglobin level of 10.9 g/dL, white blood cell count of 5.3 × 10 9 /L, platelet count of 169 × 10 9 /L, glucose level of 177 mg/dL, corrected calcium level of 9.0 mg/dL, sodium level of 135 mmol/L, bicarbonate level of 30 mmol/L, creatinine level of 0.9 mg/dL, total bilirubin level of 1.3 mg/dL, direct bilirubin level of 0.4 mg/dL, alkaline phosphatase level of 503 U/L, alanine aminotransferase level of 12 U/L, aspartate aminotransferase level of 33 U/L, ammonia level of 49 µmol/L (range, 0-30 µ mol/L), international normalized ratio of 1.2, and troponin level of <0.01 ng/mL. Electrocardiogram showed normal sinus rhythm.
Some patients with bacterial meningitis do not have a leukocytosis, but patients with meningitis caused by seeding from a systemic infection nearly always do. In this patient’s case, lack of a leukocytosis makes bacterial meningitis very unlikely. The elevated alkaline phosphatase level is expected, as this level peaks about 3 weeks after a long-bone fracture and returns to normal over a few months.
Non-contrast CT scan of the head performed on admission demonstrated no large vessel cortical-based infarct, intracranial hemorrhage, hydrocephalus, mass effect, midline shift, or extra-axial fluid. There was mild cortical atrophy as well as very mild periventricular white matter hypodensity.
The atrophy and mild white-matter hypodensities seen on repeat noncontrast CT are nonspecific for any particular entity in this patient’s age group. MRI is more effective in evaluating toxic encephalopathies, including metronidazole toxicity or Wernicke encephalopathy, and in characterizing small infarcts or inflammatory conditions of the brainstem and cerebellum, which are poorly evaluated by CT due to the bone surrounded space of the posterior fossa. An urgent lumbar puncture is not necessary due to the slow pace of illness, lack of fever, nuchal rigidity, or serum elevated white blood cell count. Rather, performing MRI should be prioritized. If MRI is nondiagnostic, then spinal fluid should be evaluated for evidence of an infectious, autoimmune, paraneoplastic, or neoplastic process.
MRI was subsequently performed. It showed symmetric abnormal T2 hyperintensities involving dentate nuclei (Figure 1), left inferior olivary nuclei (Figure 2), restiform bodies, pontine tegmentum, superior cerebellar peduncles, oculomotor nuclei, and subthalamic nuclei. The most prominent hyperintensity was in the dentate nuclei.

The clinical and radiographic features confirm a diagnosis of metronidazole-associated CNS neurotoxicity. The reason for the predilection for edema in these specific areas of the brainstem and midline cerebellum is unclear but likely is related to selective neuronal vulnerability in these structures. The treatment is to stop metronidazole. In addition, the fluctuating mental status should be evaluated with electroencephalogram to ensure concomitant seizures are not occurring.
These MRI findings were consistent with metronidazole toxicity. Metronidazole was discontinued, and 2 days later the patient’s speech improved. Two weeks after medication discontinuation, his speech was normal. There were no more episodes of confusion.
DISCUSSION
Metronidazole was originally developed in France during the 1950s as an anti-parasitic medication to treat trichomonas infections. In 1962, its antibacterial properties were discovered after a patient with bacterial gingivitis improved while taking metronidazole for treatment of Trichomonas vaginalis.1 Since that time metronidazole has become a first-line treatment for anaerobic bacteria and is now recommended by the Infectious Diseases Society of America2 and the American College of Gastroenterology3 as a first-line therapy for mild and moderate C difficile infections.
Common side effects of metronidazole are nausea, vomiting, decreased appetite, diarrhea, headaches, peripheral neuropathy, and metallic taste; less common is CNS toxicity. Although the incidence of CNS toxicity is unknown, a systematic review of the literature found 64 cases reported between 1965 and 2011.4 CNS toxicity most often occurs between the fifth and sixth decades of life, and about two thirds of the people affected are men.4 CNS adverse effects characteristically fall into 4 categories: cerebellar dysfunction (eg, ataxia, dysarthria, dysmetria, nystagmus; 75%), AMS (33%), seizures (13%), and a combination of the first 3 categories.4
The exact mechanism of metronidazole CNS toxicity is unknown, but vasogenic or cytotoxic edema may be involved.5,6 Other potential etiologies are neural protein inhibition, reversible mitochondrial dysfunction, and modifications of the inhibitory neurotransmitter gamma-aminobutyric acid receptor in the cerebellum.7,8 There is no known genetic predisposition. Although the risk for CNS toxicity traditionally is thought to correlate with therapy duration and cumulative dose,7,9 in 2011 a systemic review found no significant correlation.4 In fact, 26% of patients with CNS toxicity were treated with metronidazole for less than 1 week at time of diagnosis.4
Brain CT is typically normal. On brain MRI, lesions most commonly appear as bilateral symmetric T2 hyperintensities, most often in the cerebellar dentate nuclei (85%) and less often in the midbrain (55%), the splenium of the corpus callosum (50%), the pons (35%), and the medulla (30%).4,10 Radiographic changes have been noted as early as 3 days after symptom onset. Based on damage severity and area affected (white or gray matter), vasogenic edema and cytotoxic edema may in combination be contributing to MRI abnormalities.6,10 Hyperintensities of the bilateral dentate nuclei can help in distinguishing metronidazole-induced encephalopathy from other potential disease processes, such as Wernicke encephalopathy.10
The prognosis for patients with metronidazole-induced neurotoxicity is favorable if metronidazole is discontinued. Approximately two-thirds of patients will have complete resolution of symptoms, which is more commonly observed when patients present with seizures or altered mental status. Approximately one-third will show partial improvement, particularly if the symptoms are due to cerebellar dysfunction. It is rare to experience permanent damage or death.4 Neurologic recovery usually begins within a week after medication discontinuation but may take months for complete recovery to occur.6,8,9,11 Follow-up imaging typically shows reversal of the original lesions, but this does not always correlate with symptom improvement.4,10
Despite its frequent use and long history, metronidazole can have potentially severe toxicity. When patients who are taking this medication present with new signs and symptoms of CNS dysfunction, hospitalists should include metronidazole CNS toxicity in the differential diagnosis and, if they suspect toxicity, have a brain MRI performed. Hospitalists often prescribe metronidazole because of the increasing number of patients being discharged from acute-care hospitals with a diagnosis of C difficile colitis.12 Brain MRI remains the imaging modality of choice for diagnosis. Discontinuation of metronidazole is usually salutary in reversing symptoms. Being keenly aware of this toxicity will help clinicians avoid being rendered speechless by a patient rendered speechless.
TEACHING POINTS
CNS toxicity is a rare but potentially devastating side effect of metronidazole exposure.
Metronidazole CNS adverse effects characteristically fall under 4 categories:
○ Cerebellar dysfunction, such as ataxia, dysarthria, dysmetria, or nystagmus (75%).
○ AMS (33%).
○ Seizures (13%).
○ A combination of the first 3 categories.
- Typically lesions indicating metronidazole toxicity on brain MRI are bilateral symmetric hyperintensities on T2-weighted imaging in the cerebellar dentate nuclei, corpus callosum, midbrain, pons, or medulla.
- Treatment of CNS toxicity is metronidazole discontinuation, which results in a high rate of symptom resolution.
Disclosure
Nothing to report.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541. PubMed
2. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
3. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498. PubMed
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systemic review. Clin Neuropharmacol. 2011;34(6):241-247. PubMed
5. Graves TD, Condon M, Loucaidou M, Perry RJ. Reversible metronidazole-induced cerebellar toxicity in a multiple transplant recipient. J Neurol Sci. 2009;285(1-2):238-240. PubMed
6. Kim DW, Park JM, Yoon BW, Baek MJ, Kim JE, Kim S. Metronidazole-induced encephalopathy. J Neurol Sci. 2004;224(1-2):107-111. PubMed
7. Park KI, Chung JM, Kim JY. Metronidazole neurotoxicity: sequential neuroaxis involvement. Neurol India. 2011;59(1):104-107. PubMed
8. Patel K, Green-Hopkins I, Lu S, Tunkel AR. Cerebellar ataxia following prolonged use of metronidazole: case report and literature review. Int J Infect Dis. 2008;12(6):e111-e114. PubMed
9. Chandak S, Agarwal A, Shukla A, Joon P. A case report of metronidazole induced neurotoxicity in liver abscess patient and the usefulness of MRI for its diagnosis. J Clin Diagn Res. 2016;10(1):TD06-TD07. PubMed
10. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658. PubMed
11. Chacko J, Pramod K, Sinha S, et al. Clinical, neuroimaging and pathological features of 5-nitroimidazole-induced encephalo-neuropathy in two patients: insights into possible pathogenesis. Neurol India. 2011;59(5):743-747. PubMed
12. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 63-year-old man at an inpatient rehabilitation center was transferred to an academic tertiary care center for evaluation of slurred speech and episodic confusion. He was accompanied by his wife, who provided the history. Three weeks earlier, the patient had fallen, sustaining a right femur fracture. He underwent surgery and was discharged to rehabilitation on postoperative day 3. During the second week of rehabilitation, he developed a cough and low-grade fevers, which prompted treatment with cefpodoxime for 5 days for presumed pneumonia. The day after completing antimicrobial therapy, he became confused and began to slur his words.
Confusion is a nonspecific symptom that typically has a diffuse or multifocal localization within the cerebral hemispheres and is unlikely to be caused by a single lesion. Slurred speech may accompany global metabolic dysfunction. However, slurred speech typically localizes to the brainstem, the cerebellum in the posterior fossa, the nuclei, or the course of cranial nerves VII, X, or XII, including where these nerves pass through the subarachnoid space.
It seems this patient’s new neurologic symptoms have some relationship to his fall. Long-bone fractures and altered mental status (AMS) lead to consideration of fat emboli, but this syndrome typically presents in the acute period after the fracture. The patient is at risk for a number of complications, related to recent surgery and hospitalization, that could affect the central nervous system (CNS), including systemic infection (possibly with associated meningeal involvement) and venous thromboembolism with concomitant stroke by paradoxical emboli. The episodic nature of the confusion leads to consideration of seizures from structural lesions in the brain. Finally, the circumstances of the fall itself should be explored to determine whether an underlying neurologic dysfunction led to imbalance and gait difficulty.
Over the next 3 days at the inpatient rehabilitation center, the patient’s slurred speech became unintelligible, and he experienced intermittent disorientation to person, place, and time. There was no concomitant fever, dizziness, headache, neck pain, weakness, dyspnea, diarrhea, dysuria, or change in hearing or vision.
Progressive dysarthria argues for an expanding lesion in the posterior fossa, worsening metabolic disturbance, or a problem affecting the cranial nerves (eg, Guillain-Barré syndrome) or neuromuscular junctions (eg, myasthenia gravis). Lack of headache makes a CNS localization less likely, though disorientation must localize to the brain itself. The transient nature of the AMS could signal an ictal phenomenon or a fluctuating toxic or metabolic condition, such as hyperammonemia, drug reaction, or healthcare–acquired delirium.
His past medical history included end-stage liver disease secondary to nonalcoholic steatohepatitis status post transjugular intrahepatic portosystemic shunt (TIPS) procedure three years prior, hepatic encephalopathy, diabetes mellitus type 2, hypertension, previous melanoma excision on his back, and recurrent Clostridium difficile colitis. Two years prior to admission he had been started on an indefinite course of metronidazole 500 mg twice daily without any recurrence. The patient’s other medications were aspirin, furosemide, insulin, lactulose, mirtazapine, pantoprazole, propranolol, spironolactone, and zinc. At the rehabilitation center, he was prescribed oral oxycodone 5 mg as needed every 4 hours for pain. He denied use of tobacco, alcohol, and recreational drugs. He previously worked as a funeral home director and embalmer.
Hyperammonemia and hepatic encephalopathy can present with a fluctuating mental state that often correlates to dietary protein intake or the frequency of bowel movements; the previous TIPS history places the patient at further risk. Use of oxycodone or another narcotic commonly leads to confusion, , especially in patients who are older, have preexisting cognitive decline, or have concomitant medical comorbidities. Mirtazapine and propranolol have been associated more rarely with encephalopathy, and therefore a careful history of adherence, drug interactions, and appropriate dosing should be obtained. Metronidazole is most often associated neurologically with a peripheral neuropathy; however, it is increasingly recognized that some patients can develop a CNS syndrome that features an AMS, which can be severe and accompanied by ataxia, dysarthria, and characteristic brain magnetic resonance imaging (MRI) findings, including hyperintensity surrounding the fourth ventricle on T2-weighted images.
Embalming fluid has a high concentration of formaldehyde, and a recent epidemiologic study suggested a link between formaldehyde exposure and increased risk for amyotrophic lateral sclerosis (ALS). ALS uncommonly presents with isolated dysarthria, but its bulbar form can, usually over a much longer course than is demonstrated here. Finally, the patient’s history of melanoma places him at risk for stroke from hypercoagulability as well as potential brain metastases or carcinomatous meningitis.
Evaluation was initiated at the rehabilitation facility at the onset of the patient’s slurred speech and confusion. Physical examination were negative for focal neurologic deficits, asterixis, and jaundice. Ammonia level was 41 µmol/L (reference range, 11-35 µmol/L). Noncontrast computed tomography (CT) of the head showed no signs of acute infarct or hemorrhage. Symptoms were attributed to hepatic encephalopathy; lactulose was up-titrated to ensure 2 or 3 bowel movements per day, and rifaximin was started.
Hyperammonemia is a cause of non-inflammatory relapsing encephalopathy, but an elevated level is neither a sensitive nor specific indicator of hepatic encephalopathy. Levels of ammonia can fluctuate widely during the day based on the frequency of bowel movements as well as dietary protein intake. In addition, proper handling of samples with prompt delivery to the laboratory is essential to minimize errors.
The ammonia level of 41 µmol/L discovered here is only modestly elevated, but given the patient’s history of TIPS as well as the clinical picture, it is reasonable to aggressively treat hepatic encephalopathy with lactulose to reduce ammonia levels. If he does not improve, an MRI of the brain to exclude a structural lesion and spinal fluid examination looking for inflammatory or infectious conditions would be important next steps. Although CT excludes a large hemorrhage or mass, this screening examination does not visualize many of the findings of the metabolic etiology and the other etiologies under consideration here.
Despite 3 days of therapy for presumed hepatic encephalopathy, the patient’s slurred speech worsened, and he was transferred to an academic tertiary care center for further evaluation. On admission, his temperature was 36.9°C, heart rate was 80 beats per minute, blood pressure was 139/67 mm Hg, respiratory rate was 10 breaths per minute, and oxygen saturation was 99% on room air. He was alert, awake, and oriented to person, place, and time. He was not jaundiced. He exhibited a moderate dysarthria characterized by monotone speech, decreased volume, decreased breath support, and a hoarse vocal quality with intact language function. Motor control of the lips, tongue, and mandible were normal. Motor strength was 5/5 bilaterally in the upper and lower extremities with the exception of right hip flexion, which was 4/5. The patient exhibited mild bilateral dysmetria on finger-to-nose examination, consistent with appendicular ataxia of the upper extremities. Reflexes were depressed throughout, and there was no asterixis. He had 2+ pulses in all extremities and 1+ pitting edema of the right lower extremity to the mid leg. Pulmonary examination revealed inspiratory crackles at the left base. The rest of the examination findings were normal.
The patient’s altered mental state appears to have resolved, and the neurological examination is now mainly characterized by signs that point to the cerebellum. The description of monotone speech typically refers to loss of prosody, the variable stress or intonation of speech, which is characteristic of a cerebellar speech pattern. The hoarseness should be explored to determine if it is a feature of the patient’s speech or is a separate process. Hoarseness may involve the vocal cord and therefore, potentially, cranial nerve X or its nuclei in the brainstem. The appendicular ataxia of the limbs points definitively to the cerebellar hemispheres or their pathways through the brainstem.
Unilateral lower extremity edema, especially in the context of a recent fracture, raises the possibility of deep vein thrombosis. If this patient has a right-to-left intracardiac or intrapulmonary shunt, embolization could lead to an ischemic stroke of the brainstem or cerebellum, potentially causing dysarthria.
Laboratory evaluation revealed hemoglobin level of 10.9 g/dL, white blood cell count of 5.3 × 10 9 /L, platelet count of 169 × 10 9 /L, glucose level of 177 mg/dL, corrected calcium level of 9.0 mg/dL, sodium level of 135 mmol/L, bicarbonate level of 30 mmol/L, creatinine level of 0.9 mg/dL, total bilirubin level of 1.3 mg/dL, direct bilirubin level of 0.4 mg/dL, alkaline phosphatase level of 503 U/L, alanine aminotransferase level of 12 U/L, aspartate aminotransferase level of 33 U/L, ammonia level of 49 µmol/L (range, 0-30 µ mol/L), international normalized ratio of 1.2, and troponin level of <0.01 ng/mL. Electrocardiogram showed normal sinus rhythm.
Some patients with bacterial meningitis do not have a leukocytosis, but patients with meningitis caused by seeding from a systemic infection nearly always do. In this patient’s case, lack of a leukocytosis makes bacterial meningitis very unlikely. The elevated alkaline phosphatase level is expected, as this level peaks about 3 weeks after a long-bone fracture and returns to normal over a few months.
Non-contrast CT scan of the head performed on admission demonstrated no large vessel cortical-based infarct, intracranial hemorrhage, hydrocephalus, mass effect, midline shift, or extra-axial fluid. There was mild cortical atrophy as well as very mild periventricular white matter hypodensity.
The atrophy and mild white-matter hypodensities seen on repeat noncontrast CT are nonspecific for any particular entity in this patient’s age group. MRI is more effective in evaluating toxic encephalopathies, including metronidazole toxicity or Wernicke encephalopathy, and in characterizing small infarcts or inflammatory conditions of the brainstem and cerebellum, which are poorly evaluated by CT due to the bone surrounded space of the posterior fossa. An urgent lumbar puncture is not necessary due to the slow pace of illness, lack of fever, nuchal rigidity, or serum elevated white blood cell count. Rather, performing MRI should be prioritized. If MRI is nondiagnostic, then spinal fluid should be evaluated for evidence of an infectious, autoimmune, paraneoplastic, or neoplastic process.
MRI was subsequently performed. It showed symmetric abnormal T2 hyperintensities involving dentate nuclei (Figure 1), left inferior olivary nuclei (Figure 2), restiform bodies, pontine tegmentum, superior cerebellar peduncles, oculomotor nuclei, and subthalamic nuclei. The most prominent hyperintensity was in the dentate nuclei.
The clinical and radiographic features confirm a diagnosis of metronidazole-associated CNS neurotoxicity. The reason for the predilection for edema in these specific areas of the brainstem and midline cerebellum is unclear but likely is related to selective neuronal vulnerability in these structures. The treatment is to stop metronidazole. In addition, the fluctuating mental status should be evaluated with electroencephalogram to ensure concomitant seizures are not occurring.
These MRI findings were consistent with metronidazole toxicity. Metronidazole was discontinued, and 2 days later the patient’s speech improved. Two weeks after medication discontinuation, his speech was normal. There were no more episodes of confusion.
DISCUSSION
Metronidazole was originally developed in France during the 1950s as an anti-parasitic medication to treat trichomonas infections. In 1962, its antibacterial properties were discovered after a patient with bacterial gingivitis improved while taking metronidazole for treatment of Trichomonas vaginalis.1 Since that time metronidazole has become a first-line treatment for anaerobic bacteria and is now recommended by the Infectious Diseases Society of America2 and the American College of Gastroenterology3 as a first-line therapy for mild and moderate C difficile infections.
Common side effects of metronidazole are nausea, vomiting, decreased appetite, diarrhea, headaches, peripheral neuropathy, and metallic taste; less common is CNS toxicity. Although the incidence of CNS toxicity is unknown, a systematic review of the literature found 64 cases reported between 1965 and 2011.4 CNS toxicity most often occurs between the fifth and sixth decades of life, and about two thirds of the people affected are men.4 CNS adverse effects characteristically fall into 4 categories: cerebellar dysfunction (eg, ataxia, dysarthria, dysmetria, nystagmus; 75%), AMS (33%), seizures (13%), and a combination of the first 3 categories.4
The exact mechanism of metronidazole CNS toxicity is unknown, but vasogenic or cytotoxic edema may be involved.5,6 Other potential etiologies are neural protein inhibition, reversible mitochondrial dysfunction, and modifications of the inhibitory neurotransmitter gamma-aminobutyric acid receptor in the cerebellum.7,8 There is no known genetic predisposition. Although the risk for CNS toxicity traditionally is thought to correlate with therapy duration and cumulative dose,7,9 in 2011 a systemic review found no significant correlation.4 In fact, 26% of patients with CNS toxicity were treated with metronidazole for less than 1 week at time of diagnosis.4
Brain CT is typically normal. On brain MRI, lesions most commonly appear as bilateral symmetric T2 hyperintensities, most often in the cerebellar dentate nuclei (85%) and less often in the midbrain (55%), the splenium of the corpus callosum (50%), the pons (35%), and the medulla (30%).4,10 Radiographic changes have been noted as early as 3 days after symptom onset. Based on damage severity and area affected (white or gray matter), vasogenic edema and cytotoxic edema may in combination be contributing to MRI abnormalities.6,10 Hyperintensities of the bilateral dentate nuclei can help in distinguishing metronidazole-induced encephalopathy from other potential disease processes, such as Wernicke encephalopathy.10
The prognosis for patients with metronidazole-induced neurotoxicity is favorable if metronidazole is discontinued. Approximately two-thirds of patients will have complete resolution of symptoms, which is more commonly observed when patients present with seizures or altered mental status. Approximately one-third will show partial improvement, particularly if the symptoms are due to cerebellar dysfunction. It is rare to experience permanent damage or death.4 Neurologic recovery usually begins within a week after medication discontinuation but may take months for complete recovery to occur.6,8,9,11 Follow-up imaging typically shows reversal of the original lesions, but this does not always correlate with symptom improvement.4,10
Despite its frequent use and long history, metronidazole can have potentially severe toxicity. When patients who are taking this medication present with new signs and symptoms of CNS dysfunction, hospitalists should include metronidazole CNS toxicity in the differential diagnosis and, if they suspect toxicity, have a brain MRI performed. Hospitalists often prescribe metronidazole because of the increasing number of patients being discharged from acute-care hospitals with a diagnosis of C difficile colitis.12 Brain MRI remains the imaging modality of choice for diagnosis. Discontinuation of metronidazole is usually salutary in reversing symptoms. Being keenly aware of this toxicity will help clinicians avoid being rendered speechless by a patient rendered speechless.
TEACHING POINTS
CNS toxicity is a rare but potentially devastating side effect of metronidazole exposure.
Metronidazole CNS adverse effects characteristically fall under 4 categories:
○ Cerebellar dysfunction, such as ataxia, dysarthria, dysmetria, or nystagmus (75%).
○ AMS (33%).
○ Seizures (13%).
○ A combination of the first 3 categories.
- Typically lesions indicating metronidazole toxicity on brain MRI are bilateral symmetric hyperintensities on T2-weighted imaging in the cerebellar dentate nuclei, corpus callosum, midbrain, pons, or medulla.
- Treatment of CNS toxicity is metronidazole discontinuation, which results in a high rate of symptom resolution.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 63-year-old man at an inpatient rehabilitation center was transferred to an academic tertiary care center for evaluation of slurred speech and episodic confusion. He was accompanied by his wife, who provided the history. Three weeks earlier, the patient had fallen, sustaining a right femur fracture. He underwent surgery and was discharged to rehabilitation on postoperative day 3. During the second week of rehabilitation, he developed a cough and low-grade fevers, which prompted treatment with cefpodoxime for 5 days for presumed pneumonia. The day after completing antimicrobial therapy, he became confused and began to slur his words.
Confusion is a nonspecific symptom that typically has a diffuse or multifocal localization within the cerebral hemispheres and is unlikely to be caused by a single lesion. Slurred speech may accompany global metabolic dysfunction. However, slurred speech typically localizes to the brainstem, the cerebellum in the posterior fossa, the nuclei, or the course of cranial nerves VII, X, or XII, including where these nerves pass through the subarachnoid space.
It seems this patient’s new neurologic symptoms have some relationship to his fall. Long-bone fractures and altered mental status (AMS) lead to consideration of fat emboli, but this syndrome typically presents in the acute period after the fracture. The patient is at risk for a number of complications, related to recent surgery and hospitalization, that could affect the central nervous system (CNS), including systemic infection (possibly with associated meningeal involvement) and venous thromboembolism with concomitant stroke by paradoxical emboli. The episodic nature of the confusion leads to consideration of seizures from structural lesions in the brain. Finally, the circumstances of the fall itself should be explored to determine whether an underlying neurologic dysfunction led to imbalance and gait difficulty.
Over the next 3 days at the inpatient rehabilitation center, the patient’s slurred speech became unintelligible, and he experienced intermittent disorientation to person, place, and time. There was no concomitant fever, dizziness, headache, neck pain, weakness, dyspnea, diarrhea, dysuria, or change in hearing or vision.
Progressive dysarthria argues for an expanding lesion in the posterior fossa, worsening metabolic disturbance, or a problem affecting the cranial nerves (eg, Guillain-Barré syndrome) or neuromuscular junctions (eg, myasthenia gravis). Lack of headache makes a CNS localization less likely, though disorientation must localize to the brain itself. The transient nature of the AMS could signal an ictal phenomenon or a fluctuating toxic or metabolic condition, such as hyperammonemia, drug reaction, or healthcare–acquired delirium.
His past medical history included end-stage liver disease secondary to nonalcoholic steatohepatitis status post transjugular intrahepatic portosystemic shunt (TIPS) procedure three years prior, hepatic encephalopathy, diabetes mellitus type 2, hypertension, previous melanoma excision on his back, and recurrent Clostridium difficile colitis. Two years prior to admission he had been started on an indefinite course of metronidazole 500 mg twice daily without any recurrence. The patient’s other medications were aspirin, furosemide, insulin, lactulose, mirtazapine, pantoprazole, propranolol, spironolactone, and zinc. At the rehabilitation center, he was prescribed oral oxycodone 5 mg as needed every 4 hours for pain. He denied use of tobacco, alcohol, and recreational drugs. He previously worked as a funeral home director and embalmer.
Hyperammonemia and hepatic encephalopathy can present with a fluctuating mental state that often correlates to dietary protein intake or the frequency of bowel movements; the previous TIPS history places the patient at further risk. Use of oxycodone or another narcotic commonly leads to confusion, , especially in patients who are older, have preexisting cognitive decline, or have concomitant medical comorbidities. Mirtazapine and propranolol have been associated more rarely with encephalopathy, and therefore a careful history of adherence, drug interactions, and appropriate dosing should be obtained. Metronidazole is most often associated neurologically with a peripheral neuropathy; however, it is increasingly recognized that some patients can develop a CNS syndrome that features an AMS, which can be severe and accompanied by ataxia, dysarthria, and characteristic brain magnetic resonance imaging (MRI) findings, including hyperintensity surrounding the fourth ventricle on T2-weighted images.
Embalming fluid has a high concentration of formaldehyde, and a recent epidemiologic study suggested a link between formaldehyde exposure and increased risk for amyotrophic lateral sclerosis (ALS). ALS uncommonly presents with isolated dysarthria, but its bulbar form can, usually over a much longer course than is demonstrated here. Finally, the patient’s history of melanoma places him at risk for stroke from hypercoagulability as well as potential brain metastases or carcinomatous meningitis.
Evaluation was initiated at the rehabilitation facility at the onset of the patient’s slurred speech and confusion. Physical examination were negative for focal neurologic deficits, asterixis, and jaundice. Ammonia level was 41 µmol/L (reference range, 11-35 µmol/L). Noncontrast computed tomography (CT) of the head showed no signs of acute infarct or hemorrhage. Symptoms were attributed to hepatic encephalopathy; lactulose was up-titrated to ensure 2 or 3 bowel movements per day, and rifaximin was started.
Hyperammonemia is a cause of non-inflammatory relapsing encephalopathy, but an elevated level is neither a sensitive nor specific indicator of hepatic encephalopathy. Levels of ammonia can fluctuate widely during the day based on the frequency of bowel movements as well as dietary protein intake. In addition, proper handling of samples with prompt delivery to the laboratory is essential to minimize errors.
The ammonia level of 41 µmol/L discovered here is only modestly elevated, but given the patient’s history of TIPS as well as the clinical picture, it is reasonable to aggressively treat hepatic encephalopathy with lactulose to reduce ammonia levels. If he does not improve, an MRI of the brain to exclude a structural lesion and spinal fluid examination looking for inflammatory or infectious conditions would be important next steps. Although CT excludes a large hemorrhage or mass, this screening examination does not visualize many of the findings of the metabolic etiology and the other etiologies under consideration here.
Despite 3 days of therapy for presumed hepatic encephalopathy, the patient’s slurred speech worsened, and he was transferred to an academic tertiary care center for further evaluation. On admission, his temperature was 36.9°C, heart rate was 80 beats per minute, blood pressure was 139/67 mm Hg, respiratory rate was 10 breaths per minute, and oxygen saturation was 99% on room air. He was alert, awake, and oriented to person, place, and time. He was not jaundiced. He exhibited a moderate dysarthria characterized by monotone speech, decreased volume, decreased breath support, and a hoarse vocal quality with intact language function. Motor control of the lips, tongue, and mandible were normal. Motor strength was 5/5 bilaterally in the upper and lower extremities with the exception of right hip flexion, which was 4/5. The patient exhibited mild bilateral dysmetria on finger-to-nose examination, consistent with appendicular ataxia of the upper extremities. Reflexes were depressed throughout, and there was no asterixis. He had 2+ pulses in all extremities and 1+ pitting edema of the right lower extremity to the mid leg. Pulmonary examination revealed inspiratory crackles at the left base. The rest of the examination findings were normal.
The patient’s altered mental state appears to have resolved, and the neurological examination is now mainly characterized by signs that point to the cerebellum. The description of monotone speech typically refers to loss of prosody, the variable stress or intonation of speech, which is characteristic of a cerebellar speech pattern. The hoarseness should be explored to determine if it is a feature of the patient’s speech or is a separate process. Hoarseness may involve the vocal cord and therefore, potentially, cranial nerve X or its nuclei in the brainstem. The appendicular ataxia of the limbs points definitively to the cerebellar hemispheres or their pathways through the brainstem.
Unilateral lower extremity edema, especially in the context of a recent fracture, raises the possibility of deep vein thrombosis. If this patient has a right-to-left intracardiac or intrapulmonary shunt, embolization could lead to an ischemic stroke of the brainstem or cerebellum, potentially causing dysarthria.
Laboratory evaluation revealed hemoglobin level of 10.9 g/dL, white blood cell count of 5.3 × 10 9 /L, platelet count of 169 × 10 9 /L, glucose level of 177 mg/dL, corrected calcium level of 9.0 mg/dL, sodium level of 135 mmol/L, bicarbonate level of 30 mmol/L, creatinine level of 0.9 mg/dL, total bilirubin level of 1.3 mg/dL, direct bilirubin level of 0.4 mg/dL, alkaline phosphatase level of 503 U/L, alanine aminotransferase level of 12 U/L, aspartate aminotransferase level of 33 U/L, ammonia level of 49 µmol/L (range, 0-30 µ mol/L), international normalized ratio of 1.2, and troponin level of <0.01 ng/mL. Electrocardiogram showed normal sinus rhythm.
Some patients with bacterial meningitis do not have a leukocytosis, but patients with meningitis caused by seeding from a systemic infection nearly always do. In this patient’s case, lack of a leukocytosis makes bacterial meningitis very unlikely. The elevated alkaline phosphatase level is expected, as this level peaks about 3 weeks after a long-bone fracture and returns to normal over a few months.
Non-contrast CT scan of the head performed on admission demonstrated no large vessel cortical-based infarct, intracranial hemorrhage, hydrocephalus, mass effect, midline shift, or extra-axial fluid. There was mild cortical atrophy as well as very mild periventricular white matter hypodensity.
The atrophy and mild white-matter hypodensities seen on repeat noncontrast CT are nonspecific for any particular entity in this patient’s age group. MRI is more effective in evaluating toxic encephalopathies, including metronidazole toxicity or Wernicke encephalopathy, and in characterizing small infarcts or inflammatory conditions of the brainstem and cerebellum, which are poorly evaluated by CT due to the bone surrounded space of the posterior fossa. An urgent lumbar puncture is not necessary due to the slow pace of illness, lack of fever, nuchal rigidity, or serum elevated white blood cell count. Rather, performing MRI should be prioritized. If MRI is nondiagnostic, then spinal fluid should be evaluated for evidence of an infectious, autoimmune, paraneoplastic, or neoplastic process.
MRI was subsequently performed. It showed symmetric abnormal T2 hyperintensities involving dentate nuclei (Figure 1), left inferior olivary nuclei (Figure 2), restiform bodies, pontine tegmentum, superior cerebellar peduncles, oculomotor nuclei, and subthalamic nuclei. The most prominent hyperintensity was in the dentate nuclei.
The clinical and radiographic features confirm a diagnosis of metronidazole-associated CNS neurotoxicity. The reason for the predilection for edema in these specific areas of the brainstem and midline cerebellum is unclear but likely is related to selective neuronal vulnerability in these structures. The treatment is to stop metronidazole. In addition, the fluctuating mental status should be evaluated with electroencephalogram to ensure concomitant seizures are not occurring.
These MRI findings were consistent with metronidazole toxicity. Metronidazole was discontinued, and 2 days later the patient’s speech improved. Two weeks after medication discontinuation, his speech was normal. There were no more episodes of confusion.
DISCUSSION
Metronidazole was originally developed in France during the 1950s as an anti-parasitic medication to treat trichomonas infections. In 1962, its antibacterial properties were discovered after a patient with bacterial gingivitis improved while taking metronidazole for treatment of Trichomonas vaginalis.1 Since that time metronidazole has become a first-line treatment for anaerobic bacteria and is now recommended by the Infectious Diseases Society of America2 and the American College of Gastroenterology3 as a first-line therapy for mild and moderate C difficile infections.
Common side effects of metronidazole are nausea, vomiting, decreased appetite, diarrhea, headaches, peripheral neuropathy, and metallic taste; less common is CNS toxicity. Although the incidence of CNS toxicity is unknown, a systematic review of the literature found 64 cases reported between 1965 and 2011.4 CNS toxicity most often occurs between the fifth and sixth decades of life, and about two thirds of the people affected are men.4 CNS adverse effects characteristically fall into 4 categories: cerebellar dysfunction (eg, ataxia, dysarthria, dysmetria, nystagmus; 75%), AMS (33%), seizures (13%), and a combination of the first 3 categories.4
The exact mechanism of metronidazole CNS toxicity is unknown, but vasogenic or cytotoxic edema may be involved.5,6 Other potential etiologies are neural protein inhibition, reversible mitochondrial dysfunction, and modifications of the inhibitory neurotransmitter gamma-aminobutyric acid receptor in the cerebellum.7,8 There is no known genetic predisposition. Although the risk for CNS toxicity traditionally is thought to correlate with therapy duration and cumulative dose,7,9 in 2011 a systemic review found no significant correlation.4 In fact, 26% of patients with CNS toxicity were treated with metronidazole for less than 1 week at time of diagnosis.4
Brain CT is typically normal. On brain MRI, lesions most commonly appear as bilateral symmetric T2 hyperintensities, most often in the cerebellar dentate nuclei (85%) and less often in the midbrain (55%), the splenium of the corpus callosum (50%), the pons (35%), and the medulla (30%).4,10 Radiographic changes have been noted as early as 3 days after symptom onset. Based on damage severity and area affected (white or gray matter), vasogenic edema and cytotoxic edema may in combination be contributing to MRI abnormalities.6,10 Hyperintensities of the bilateral dentate nuclei can help in distinguishing metronidazole-induced encephalopathy from other potential disease processes, such as Wernicke encephalopathy.10
The prognosis for patients with metronidazole-induced neurotoxicity is favorable if metronidazole is discontinued. Approximately two-thirds of patients will have complete resolution of symptoms, which is more commonly observed when patients present with seizures or altered mental status. Approximately one-third will show partial improvement, particularly if the symptoms are due to cerebellar dysfunction. It is rare to experience permanent damage or death.4 Neurologic recovery usually begins within a week after medication discontinuation but may take months for complete recovery to occur.6,8,9,11 Follow-up imaging typically shows reversal of the original lesions, but this does not always correlate with symptom improvement.4,10
Despite its frequent use and long history, metronidazole can have potentially severe toxicity. When patients who are taking this medication present with new signs and symptoms of CNS dysfunction, hospitalists should include metronidazole CNS toxicity in the differential diagnosis and, if they suspect toxicity, have a brain MRI performed. Hospitalists often prescribe metronidazole because of the increasing number of patients being discharged from acute-care hospitals with a diagnosis of C difficile colitis.12 Brain MRI remains the imaging modality of choice for diagnosis. Discontinuation of metronidazole is usually salutary in reversing symptoms. Being keenly aware of this toxicity will help clinicians avoid being rendered speechless by a patient rendered speechless.
TEACHING POINTS
CNS toxicity is a rare but potentially devastating side effect of metronidazole exposure.
Metronidazole CNS adverse effects characteristically fall under 4 categories:
○ Cerebellar dysfunction, such as ataxia, dysarthria, dysmetria, or nystagmus (75%).
○ AMS (33%).
○ Seizures (13%).
○ A combination of the first 3 categories.
- Typically lesions indicating metronidazole toxicity on brain MRI are bilateral symmetric hyperintensities on T2-weighted imaging in the cerebellar dentate nuclei, corpus callosum, midbrain, pons, or medulla.
- Treatment of CNS toxicity is metronidazole discontinuation, which results in a high rate of symptom resolution.
Disclosure
Nothing to report.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541. PubMed
2. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
3. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498. PubMed
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systemic review. Clin Neuropharmacol. 2011;34(6):241-247. PubMed
5. Graves TD, Condon M, Loucaidou M, Perry RJ. Reversible metronidazole-induced cerebellar toxicity in a multiple transplant recipient. J Neurol Sci. 2009;285(1-2):238-240. PubMed
6. Kim DW, Park JM, Yoon BW, Baek MJ, Kim JE, Kim S. Metronidazole-induced encephalopathy. J Neurol Sci. 2004;224(1-2):107-111. PubMed
7. Park KI, Chung JM, Kim JY. Metronidazole neurotoxicity: sequential neuroaxis involvement. Neurol India. 2011;59(1):104-107. PubMed
8. Patel K, Green-Hopkins I, Lu S, Tunkel AR. Cerebellar ataxia following prolonged use of metronidazole: case report and literature review. Int J Infect Dis. 2008;12(6):e111-e114. PubMed
9. Chandak S, Agarwal A, Shukla A, Joon P. A case report of metronidazole induced neurotoxicity in liver abscess patient and the usefulness of MRI for its diagnosis. J Clin Diagn Res. 2016;10(1):TD06-TD07. PubMed
10. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658. PubMed
11. Chacko J, Pramod K, Sinha S, et al. Clinical, neuroimaging and pathological features of 5-nitroimidazole-induced encephalo-neuropathy in two patients: insights into possible pathogenesis. Neurol India. 2011;59(5):743-747. PubMed
12. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3. PubMed
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541. PubMed
2. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
3. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498. PubMed
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systemic review. Clin Neuropharmacol. 2011;34(6):241-247. PubMed
5. Graves TD, Condon M, Loucaidou M, Perry RJ. Reversible metronidazole-induced cerebellar toxicity in a multiple transplant recipient. J Neurol Sci. 2009;285(1-2):238-240. PubMed
6. Kim DW, Park JM, Yoon BW, Baek MJ, Kim JE, Kim S. Metronidazole-induced encephalopathy. J Neurol Sci. 2004;224(1-2):107-111. PubMed
7. Park KI, Chung JM, Kim JY. Metronidazole neurotoxicity: sequential neuroaxis involvement. Neurol India. 2011;59(1):104-107. PubMed
8. Patel K, Green-Hopkins I, Lu S, Tunkel AR. Cerebellar ataxia following prolonged use of metronidazole: case report and literature review. Int J Infect Dis. 2008;12(6):e111-e114. PubMed
9. Chandak S, Agarwal A, Shukla A, Joon P. A case report of metronidazole induced neurotoxicity in liver abscess patient and the usefulness of MRI for its diagnosis. J Clin Diagn Res. 2016;10(1):TD06-TD07. PubMed
10. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658. PubMed
11. Chacko J, Pramod K, Sinha S, et al. Clinical, neuroimaging and pathological features of 5-nitroimidazole-induced encephalo-neuropathy in two patients: insights into possible pathogenesis. Neurol India. 2011;59(5):743-747. PubMed
12. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3. PubMed
© 2017 Society of Hospital Medicine
What are the chances?
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
Two weeks after undergoing a below-knee amputation (BKA) and 10 days after being discharged to a skilled nursing facility (SNF), an 87-year-old man returned to the emergency department (ED) for evaluation of somnolence and altered mental state. In the ED, he was disoriented and unable to provide a detailed history.
The differential diagnosis for acute confusion and altered consciousness is broad. Initial possibilities include toxic-metabolic abnormalities, medication side effects, and infections. Urinary tract infection, pneumonia, and surgical-site infection should be assessed for first, as they are common causes of postoperative altered mentation. Next to be considered are subclinical seizure, ischemic stroke, and infectious encephalitis or meningitis, along with hemorrhagic stroke and subdural hematoma.
During initial assessment, the clinician should ascertain baseline mental state, the timeline of the change in mental status, recent medication changes, history of substance abuse, and concern about any recent trauma, such as a fall. Performing the physical examination, the clinician should assess vital signs and then focus on identifying localizing neurologic deficits.
First steps in the work-up include a complete metabolic panel, complete blood cell count, urinalysis with culture, and a urine toxicology screen. If the patient has a “toxic” appearance, blood cultures should be obtained. An electrocardiogram should be used to screen for drug toxicity or evidence of cardiac ischemia. If laboratory test results do not reveal an obvious infectious or metabolic cause, a noncontrast computed tomography (CT) of the head should be obtained. In terms of early interventions, a low glucose level should be treated with thiamine and then glucose, and naloxone should be given if there is any suspicion of narcotic overdose.
More history was obtained from the patient’s records. The BKA was performed to address a nonhealing transmetatarsal amputation. Two months earlier, the transmetatarsal amputation had been performed as treatment for a diabetic forefoot ulcer with chronic osteomyelitis. The patient’s post-BKA course was uncomplicated. He was started on intravenous (IV) ertapenem on postoperative day 1, and on postoperative day 4 was discharged to the SNF to complete a 6-week course of antibiotics for osteomyelitis. Past medical history included paroxysmal atrial fibrillation, coronary artery disease, congestive heart failure (ejection fraction 40%), and type 2 diabetes mellitus. Medications given at the SNF were oxycodone, acetaminophen, cholecalciferol, melatonin, digoxin, ondansetron, furosemide, gabapentin, correctional insulin, tamsulosin, senna, docusate, warfarin, and metoprolol. While there, the patient’s family expressed concern about his diminishing “mental ability.” They reported he had been fully alert and oriented on arrival at the SNF, and living independently with his wife before the BKA. Then, a week before the ED presentation, he started becoming more somnolent and forgetful. The gabapentin and oxycodone dosages were reduced to minimize their sedative effects, but he showed no improvement. At the SNF, a somnolence work-up was not performed.
Several of the patient’s medications can contribute to altered mental state. Ertapenem can cause seizures as well as profound mental status changes, though these are more likely in the setting of poor renal function. The mental status changes were noticed about a week into the patient’s course of antibiotics, which suggests a possible temporal correlation with the initiation of ertapenem. An electroencephalogram is required to diagnose nonconvulsive seizure activity. Narcotic overdose should still be considered, despite the recent reduction in oxycodone dosage. Digoxin toxicity, though less likely when the dose is stable and there are no changes in renal function, can cause a confused state. Concurrent use of furosemide could potentiate the toxic effects of digoxin.
Non-medication-related concerns include hypoglycemia, hyperglycemia, and, given his history of atrial fibrillation, cardioembolic stroke. Although generalized confusion is not a common manifestation of stroke, a thalamic stroke can alter mental state but be easily missed if not specifically considered. Additional lab work-up should include a digoxin level and, since he is taking warfarin, a prothrombin time/international normalized ratio (PT/INR). If the initial laboratory studies and head CT do not explain the altered mental state, magnetic resonance imaging (MRI) of the brain should be performed to further assess for stroke.
On physical examination in the ED, the patient was resting comfortably with eyes closed, and arousing to voice. He obeyed commands and participated in the examination. His Glasgow Coma Scale score was 13; temperature, 36.8°C, heart rate, 80 beats per minute; respiratory rate, 16 breaths per minute; blood pressure, 90/57 mm Hg; and 100% peripheral capillary oxygen saturation while breathing ambient air. He appeared well developed. His heart rhythm was irregularly irregular, without murmurs, rubs, or gallops. Respiratory and abdominal examination findings were normal. The left BKA incision was well approximated, with no drainage, dehiscence, fluctuance, or erythema. On neurologic examination, the patient was intermittently oriented only to self. Pupils were equal, round, and reactive to light; extraocular movements were intact; face was symmetric; tongue was midline; sensation on face was equal bilaterally; and shoulder shrug was intact. Strength was 5/5 and symmetric in the elbow and hip and 5/5 in the right knee and ankle (not tested on left because of BKA). Deep tendon reflexes were 3+ and symmetrical at the biceps, brachioradialis, and triceps tendons and 3+ in the right patellar and Achilles tendons. Sensation was intact and symmetrical in the upper and lower extremities. The patient’s speech was slow and slurred, and his answers were unrelated to the questions being asked.
The patient’s mental state is best described as lethargic. As he is only intermittently oriented, he meets the criteria for delirium. He is not obtunded or comatose, and his pupils are at least reactive, not pinpoint, so narcotic overdose is less likely. Thalamic stroke remains in the differential diagnosis; despite the seemingly symmetrical sensation examination, hemisensory deficits cannot be definitively ruled out given the patient’s mental state. A rare entity such as carcinomatosis meningitis or another diffuse, infiltrative neoplastic process could be causing his condition. However, because focal deficits other than abnormal speech and diffuse hyperreflexia are absent, toxic, infectious, or metabolic causes are more likely than structural abnormalities. Still possible is a medication toxicity, such as ertapenem toxicity or, less likely, digoxin toxicity. In terms of infectious possibilities, urinary tract infection could certainly present in this fashion, especially if the patient had a somewhat low neurologic reserve at baseline, and hypotension could be secondary to sepsis. Encephalitis or meningitis remains in the differential diagnosis, though the patient appears nontoxic, and therefore a bacterial etiology is very unlikely.
The patient’s hyperreflexia may be an important clue. Although the strength of his reflexes at baseline is unknown, seizures can cause transiently increased reflexes as well as a confused, lethargic mental state. Reflexes can also be increased by a drug overdose that has caused serotonin syndrome. Of the patient’s medications, only ondansetron can cause this reaction. Hyperthyroidism can cause brisk reflexes and confusion, though more typically it causes agitated confusion. A thyroid-stimulating hormone level should be added to the initial laboratory panel.
A complete blood count revealed white blood cell count 11.86 K/uL with neutrophilic predominance and immature granulocytes, hemoglobin 11.5 g/dL, and platelet count 323 K/uL. Serum sodium was 141 mEq/L, potassium 4.2 mEq/L, chloride 103 mEq/L, bicarbonate 30 mEq/L, creatinine 1.14 mg/dL (prior baseline of 0.8-1.0 mg/dL), blood urea nitrogen 26 mg/dL, blood glucose 159 mg/dL, and calcium 9.1 mg/dL. His digoxin level was 1.3 ng/mL (reference range 0.5-1.9 mg/mL) and troponin was undetectable. INR was 2.7 and partial thromboplastin time (PTT) 60 seconds. Vitamin B12 level was 674 pg/mL (reference range >180). A urinalysis had 1+ hyaline casts and was negative for nitrites, leukocyte esterase, blood, and bacteria. An ECG revealed atrial fibrillation with a ventricular rate of 80 beats per minute. A chest radiograph showed clear lung fields. A CT of the head without IV contrast had no evidence of an acute intracranial abnormality. In the ED, 1 liter of IV normal saline was given and blood pressure improved to 127/72 mm Hg.
The head CT does not show intracranial bleeding, and, though it is reassuring that INR is in the therapeutic range, ischemic stroke must remain in the differential diagnosis. Sepsis is less likely given that the criteria for systemic inflammatory response syndrome are not met, and hypotension was rapidly corrected with administration of IV fluids. Urinary tract infection was ruled out with the negative urinalysis. Subclinical seizures remain possible, as does medication-related or other toxicity. A medication overdose, intentional or otherwise, should also be considered.
The patient was admitted to the hospital. On reassessment by the inpatient team, he was oriented only to self, frequently falling asleep, and not recalling earlier conversations when aroused. His speech remained slurred and difficult to understand. Neurologic examination findings were unchanged since the ED examination. On additional cerebellar examination, he had dysmetria with finger-to-nose testing bilaterally and dysdiadochokinesia (impaired rapid alternating movements) of the left hand.
His handedness is not mentioned; the dysdiadochokinesia of the left hand may reflect the patient’s being right-handed, or may signify a focal cerebellar lesion. The cerebellum is also implicated by the bilateral dysmetria. Persistent somnolence in the absence of CT findings suggests a metabolic or infectious process. Metabolic processes that can cause bilateral cerebellar ataxia and somnolence include overdose of a drug or medication. Use of alcohol or a medication such as phenytoin, valproic acid, or a benzodiazepine can cause the symptoms in this case, but was not reported by the family, and there was no documentation of it in the SNF records. Wernicke encephalopathy is rare and is not well supported by the patient’s presentation but should be considered, as it can be easily treated with thiamine. Meningoencephalitis affecting the cerebellum remains possible, but infection is less likely. Both electroencephalogram and brain MRI should be performed, with a specific interest in possible cerebellar lesions. If the MRI is unremarkable, a lumbar puncture should be performed to assess opening pressure and investigate for infectious etiologies.
MRI of the brain showed age-related volume loss and nonspecific white matter disease without acute changes. Lack of a clear explanation for the neurologic findings led to suspicion of a medication side effect. Ertapenem was stopped on admission because it has been reported to rarely cause altered mental status. IV moxifloxacin was started for the osteomyelitis. Over the next 2 days, symptoms began resolving; within 24 hours of ertapenem discontinuation, the patient was awake, alert, and talkative. On examination, he remained dysarthric but was no longer dysmetric. Within 48 hours, the dysarthria was completely resolved, and he was returned to the SNF to complete a course of IV moxifloxacin.
DISCUSSION
Among elderly patients presenting to the ED, altered mental status is a common complaint, accounting for 10% to 30% of visits.1 Medications are a common cause of altered mental status among the elderly and are responsible for 40% of delirium cases.1 The risk of adverse drug events (ADEs) rises with the number of medications prescribed.1-3 Among patients older than 60 years, the incidence of polypharmacy (defined as taking >5 prescription medications) increased from roughly 20% in 1999 to 40% in 2012.4,5 The most common ADEs in the ambulatory setting (25%) are central nervous system (CNS) symptoms, including dizziness, sleep disturbances, and mood changes.6 A medication effect should be suspected in any elderly patient presenting with altered mental state.
The present patient developed a constellation of neurologic symptoms after starting ertapenem, one of the carbapenem antibiotics, which is a class of medications that can cause CNS ADEs. Carbapenems are renally cleared, and adjustments must be made for acute or chronic changes in kidney function. Carbapenems are associated with increased risk of seizure; the incidence of seizure with ertapenem is 0.2%.7,8 Food and Drug Administration postmarketing reports have noted ertapenem can cause somnolence and dyskinesia,9 and several case reports have described ertapenem-associated CNS side effects, including psychosis and encephalopathy.10-13 Symptoms and examination findings can include confusion, disorientation, garbled speech, dysphagia, hallucinations, miosis, myoclonus, tremor, and agitation.10-13 Although reports of dysmetria and dysdiadochokinesia are lacking, suspicion of an ADE in this case was heightened by the timing of the exposure and the absence of alternative infectious, metabolic, and vascular explanations for bilateral cerebellar dysfunction.
The Naranjo Adverse Drug Reaction (ADR) scale may help clinicians differentiate ADEs from other etiologies of symptoms. It uses 10 weighted questions (Table) to estimate the probability that an adverse clinical event is caused by a drug reaction.14 The present case was assigned 1 point for prior reports of neurologic ADEs associated with ertapenem, 2 for the temporal association, 1 for resolution after medication withdrawal, 2 for lack of alternative causes, and 1 for objective evidence of neurologic dysfunction—for a total of 7 points, indicating ertapenem was probably the cause of the patient’s neurologic symptoms. Of 4 prior cases in which carbapenem toxicity was suspected and the Naranjo scale was used, 3 found a probable relationship, and the fourth a highly probable one.10,12 Confusion, disorientation, hallucinations, tangential thoughts, and garbled speech were reported in the 3 probable cases of ADEs. In the highly probable case, tangential thoughts, garbled speech, and miosis were noted on examination, and these findings returned after re-exposure to ertapenem. Of note, these ADEs occurred in patients with normal and abnormal renal function, and in middle-aged and elderly patients.10,11,13
Most medications have a long list of low-frequency and rarely reported adverse effects. The present case reminds clinicians to consider rare adverse effects, or variants of previously reported adverse effects, in a patient with unexplained symptoms. To estimate the probability that a drug is causing harm to a patient, using a validated tool such as the Naranjo scale helps answer the question, What are the chances?
KEY TEACHING POINTS
Clinicians should include rare adverse effects of common medications in the differential diagnosis.
The Naranjo score is a validated tool that can be used to systematically assess the probability of an adverse drug effect at the bedside.
- The presentation of ertapenem-associated neurotoxicity may include features of bilateral cerebellar dysfunction.
Disclosure
Nothing to report.
1. Inouye SK, Fearing MA, Marcantonio ER. Delirium. In: Halter JB, Ouslander JG, Tinetti ME, Studenski S, High KP, Asthana S, eds. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. New York, NY: McGraw-Hill; 2009.
2. Sarkar U, López A, Maselli JH, Gonzales R. Adverse drug events in U.S. adult ambulatory medical care. Health Serv Res. 2011;46(5):1517-1533. PubMed
3. Chrischilles E, Rubenstein L, Van Gilder R, Voelker M, Wright K, Wallace R. Risk factors for adverse drug events in older adults with mobility limitations in the community setting. J Am Geriatr Soc. 2007;55(1):29-34. PubMed
4. Kaufman DW, Kelly JP, Rosenberg L, Anderson TE, Mitchell AA. Recent patterns of medication use in the ambulatory adult population of the United States: the Slone survey. JAMA. 2002;287(3):337-344. PubMed
5. Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314(17):1818-1831. PubMed
6. Thomsen LA, Winterstein AG, Søndergaard B, Haugbølle LS, Melander A. Systematic review of the incidence and characteristics of preventable adverse drug events in ambulatory care. Ann Pharmacother. 2007;41(9):1411-1426. PubMed
7. Zhanel GG, Wiebe R, Dilay L, et al. Comparative review of the carbapenems. Drugs. 2007;67(7):1027-1052. PubMed
8. Cannon JP, Lee TA, Clark NM, Setlak P, Grim SA. The risk of seizures among the carbapenems: a meta-analysis. J Antimicrob Chemother. 2014;69(8):2043-2055. PubMed
9. US Food and Drug Administration. Invanz (ertapenem) injection [safety information]. http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm196605.htm. Published July 2013. Accessed July 6, 2015.
10. Oo Y, Packham D, Yau W, Munckhof WJ. Ertapenem-associated psychosis and encephalopathy. Intern Med J. 2014;44(8):817-819. PubMed
11. Wen MJ, Sung CC, Chau T, Lin SH. Acute prolonged neurotoxicity associated with recommended doses of ertapenem in 2 patients with advanced renal failure. Clin Nephrol. 2013;80(6):474-478. PubMed
12. Duquaine S, Kitchell E, Tate T, Tannen RC, Wickremasinghe IM. Central nervous system toxicity associated with ertapenem use. Ann Pharmacother. 2011;45(1):e6. PubMed
13. Kong V, Beckert L, Awunor-Renner C. A case of beta lactam-induced visual hallucination. N Z Med J. 2009;122(1298):76-77. PubMed
14. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
Two weeks after undergoing a below-knee amputation (BKA) and 10 days after being discharged to a skilled nursing facility (SNF), an 87-year-old man returned to the emergency department (ED) for evaluation of somnolence and altered mental state. In the ED, he was disoriented and unable to provide a detailed history.
The differential diagnosis for acute confusion and altered consciousness is broad. Initial possibilities include toxic-metabolic abnormalities, medication side effects, and infections. Urinary tract infection, pneumonia, and surgical-site infection should be assessed for first, as they are common causes of postoperative altered mentation. Next to be considered are subclinical seizure, ischemic stroke, and infectious encephalitis or meningitis, along with hemorrhagic stroke and subdural hematoma.
During initial assessment, the clinician should ascertain baseline mental state, the timeline of the change in mental status, recent medication changes, history of substance abuse, and concern about any recent trauma, such as a fall. Performing the physical examination, the clinician should assess vital signs and then focus on identifying localizing neurologic deficits.
First steps in the work-up include a complete metabolic panel, complete blood cell count, urinalysis with culture, and a urine toxicology screen. If the patient has a “toxic” appearance, blood cultures should be obtained. An electrocardiogram should be used to screen for drug toxicity or evidence of cardiac ischemia. If laboratory test results do not reveal an obvious infectious or metabolic cause, a noncontrast computed tomography (CT) of the head should be obtained. In terms of early interventions, a low glucose level should be treated with thiamine and then glucose, and naloxone should be given if there is any suspicion of narcotic overdose.
More history was obtained from the patient’s records. The BKA was performed to address a nonhealing transmetatarsal amputation. Two months earlier, the transmetatarsal amputation had been performed as treatment for a diabetic forefoot ulcer with chronic osteomyelitis. The patient’s post-BKA course was uncomplicated. He was started on intravenous (IV) ertapenem on postoperative day 1, and on postoperative day 4 was discharged to the SNF to complete a 6-week course of antibiotics for osteomyelitis. Past medical history included paroxysmal atrial fibrillation, coronary artery disease, congestive heart failure (ejection fraction 40%), and type 2 diabetes mellitus. Medications given at the SNF were oxycodone, acetaminophen, cholecalciferol, melatonin, digoxin, ondansetron, furosemide, gabapentin, correctional insulin, tamsulosin, senna, docusate, warfarin, and metoprolol. While there, the patient’s family expressed concern about his diminishing “mental ability.” They reported he had been fully alert and oriented on arrival at the SNF, and living independently with his wife before the BKA. Then, a week before the ED presentation, he started becoming more somnolent and forgetful. The gabapentin and oxycodone dosages were reduced to minimize their sedative effects, but he showed no improvement. At the SNF, a somnolence work-up was not performed.
Several of the patient’s medications can contribute to altered mental state. Ertapenem can cause seizures as well as profound mental status changes, though these are more likely in the setting of poor renal function. The mental status changes were noticed about a week into the patient’s course of antibiotics, which suggests a possible temporal correlation with the initiation of ertapenem. An electroencephalogram is required to diagnose nonconvulsive seizure activity. Narcotic overdose should still be considered, despite the recent reduction in oxycodone dosage. Digoxin toxicity, though less likely when the dose is stable and there are no changes in renal function, can cause a confused state. Concurrent use of furosemide could potentiate the toxic effects of digoxin.
Non-medication-related concerns include hypoglycemia, hyperglycemia, and, given his history of atrial fibrillation, cardioembolic stroke. Although generalized confusion is not a common manifestation of stroke, a thalamic stroke can alter mental state but be easily missed if not specifically considered. Additional lab work-up should include a digoxin level and, since he is taking warfarin, a prothrombin time/international normalized ratio (PT/INR). If the initial laboratory studies and head CT do not explain the altered mental state, magnetic resonance imaging (MRI) of the brain should be performed to further assess for stroke.
On physical examination in the ED, the patient was resting comfortably with eyes closed, and arousing to voice. He obeyed commands and participated in the examination. His Glasgow Coma Scale score was 13; temperature, 36.8°C, heart rate, 80 beats per minute; respiratory rate, 16 breaths per minute; blood pressure, 90/57 mm Hg; and 100% peripheral capillary oxygen saturation while breathing ambient air. He appeared well developed. His heart rhythm was irregularly irregular, without murmurs, rubs, or gallops. Respiratory and abdominal examination findings were normal. The left BKA incision was well approximated, with no drainage, dehiscence, fluctuance, or erythema. On neurologic examination, the patient was intermittently oriented only to self. Pupils were equal, round, and reactive to light; extraocular movements were intact; face was symmetric; tongue was midline; sensation on face was equal bilaterally; and shoulder shrug was intact. Strength was 5/5 and symmetric in the elbow and hip and 5/5 in the right knee and ankle (not tested on left because of BKA). Deep tendon reflexes were 3+ and symmetrical at the biceps, brachioradialis, and triceps tendons and 3+ in the right patellar and Achilles tendons. Sensation was intact and symmetrical in the upper and lower extremities. The patient’s speech was slow and slurred, and his answers were unrelated to the questions being asked.
The patient’s mental state is best described as lethargic. As he is only intermittently oriented, he meets the criteria for delirium. He is not obtunded or comatose, and his pupils are at least reactive, not pinpoint, so narcotic overdose is less likely. Thalamic stroke remains in the differential diagnosis; despite the seemingly symmetrical sensation examination, hemisensory deficits cannot be definitively ruled out given the patient’s mental state. A rare entity such as carcinomatosis meningitis or another diffuse, infiltrative neoplastic process could be causing his condition. However, because focal deficits other than abnormal speech and diffuse hyperreflexia are absent, toxic, infectious, or metabolic causes are more likely than structural abnormalities. Still possible is a medication toxicity, such as ertapenem toxicity or, less likely, digoxin toxicity. In terms of infectious possibilities, urinary tract infection could certainly present in this fashion, especially if the patient had a somewhat low neurologic reserve at baseline, and hypotension could be secondary to sepsis. Encephalitis or meningitis remains in the differential diagnosis, though the patient appears nontoxic, and therefore a bacterial etiology is very unlikely.
The patient’s hyperreflexia may be an important clue. Although the strength of his reflexes at baseline is unknown, seizures can cause transiently increased reflexes as well as a confused, lethargic mental state. Reflexes can also be increased by a drug overdose that has caused serotonin syndrome. Of the patient’s medications, only ondansetron can cause this reaction. Hyperthyroidism can cause brisk reflexes and confusion, though more typically it causes agitated confusion. A thyroid-stimulating hormone level should be added to the initial laboratory panel.
A complete blood count revealed white blood cell count 11.86 K/uL with neutrophilic predominance and immature granulocytes, hemoglobin 11.5 g/dL, and platelet count 323 K/uL. Serum sodium was 141 mEq/L, potassium 4.2 mEq/L, chloride 103 mEq/L, bicarbonate 30 mEq/L, creatinine 1.14 mg/dL (prior baseline of 0.8-1.0 mg/dL), blood urea nitrogen 26 mg/dL, blood glucose 159 mg/dL, and calcium 9.1 mg/dL. His digoxin level was 1.3 ng/mL (reference range 0.5-1.9 mg/mL) and troponin was undetectable. INR was 2.7 and partial thromboplastin time (PTT) 60 seconds. Vitamin B12 level was 674 pg/mL (reference range >180). A urinalysis had 1+ hyaline casts and was negative for nitrites, leukocyte esterase, blood, and bacteria. An ECG revealed atrial fibrillation with a ventricular rate of 80 beats per minute. A chest radiograph showed clear lung fields. A CT of the head without IV contrast had no evidence of an acute intracranial abnormality. In the ED, 1 liter of IV normal saline was given and blood pressure improved to 127/72 mm Hg.
The head CT does not show intracranial bleeding, and, though it is reassuring that INR is in the therapeutic range, ischemic stroke must remain in the differential diagnosis. Sepsis is less likely given that the criteria for systemic inflammatory response syndrome are not met, and hypotension was rapidly corrected with administration of IV fluids. Urinary tract infection was ruled out with the negative urinalysis. Subclinical seizures remain possible, as does medication-related or other toxicity. A medication overdose, intentional or otherwise, should also be considered.
The patient was admitted to the hospital. On reassessment by the inpatient team, he was oriented only to self, frequently falling asleep, and not recalling earlier conversations when aroused. His speech remained slurred and difficult to understand. Neurologic examination findings were unchanged since the ED examination. On additional cerebellar examination, he had dysmetria with finger-to-nose testing bilaterally and dysdiadochokinesia (impaired rapid alternating movements) of the left hand.
His handedness is not mentioned; the dysdiadochokinesia of the left hand may reflect the patient’s being right-handed, or may signify a focal cerebellar lesion. The cerebellum is also implicated by the bilateral dysmetria. Persistent somnolence in the absence of CT findings suggests a metabolic or infectious process. Metabolic processes that can cause bilateral cerebellar ataxia and somnolence include overdose of a drug or medication. Use of alcohol or a medication such as phenytoin, valproic acid, or a benzodiazepine can cause the symptoms in this case, but was not reported by the family, and there was no documentation of it in the SNF records. Wernicke encephalopathy is rare and is not well supported by the patient’s presentation but should be considered, as it can be easily treated with thiamine. Meningoencephalitis affecting the cerebellum remains possible, but infection is less likely. Both electroencephalogram and brain MRI should be performed, with a specific interest in possible cerebellar lesions. If the MRI is unremarkable, a lumbar puncture should be performed to assess opening pressure and investigate for infectious etiologies.
MRI of the brain showed age-related volume loss and nonspecific white matter disease without acute changes. Lack of a clear explanation for the neurologic findings led to suspicion of a medication side effect. Ertapenem was stopped on admission because it has been reported to rarely cause altered mental status. IV moxifloxacin was started for the osteomyelitis. Over the next 2 days, symptoms began resolving; within 24 hours of ertapenem discontinuation, the patient was awake, alert, and talkative. On examination, he remained dysarthric but was no longer dysmetric. Within 48 hours, the dysarthria was completely resolved, and he was returned to the SNF to complete a course of IV moxifloxacin.
DISCUSSION
Among elderly patients presenting to the ED, altered mental status is a common complaint, accounting for 10% to 30% of visits.1 Medications are a common cause of altered mental status among the elderly and are responsible for 40% of delirium cases.1 The risk of adverse drug events (ADEs) rises with the number of medications prescribed.1-3 Among patients older than 60 years, the incidence of polypharmacy (defined as taking >5 prescription medications) increased from roughly 20% in 1999 to 40% in 2012.4,5 The most common ADEs in the ambulatory setting (25%) are central nervous system (CNS) symptoms, including dizziness, sleep disturbances, and mood changes.6 A medication effect should be suspected in any elderly patient presenting with altered mental state.
The present patient developed a constellation of neurologic symptoms after starting ertapenem, one of the carbapenem antibiotics, which is a class of medications that can cause CNS ADEs. Carbapenems are renally cleared, and adjustments must be made for acute or chronic changes in kidney function. Carbapenems are associated with increased risk of seizure; the incidence of seizure with ertapenem is 0.2%.7,8 Food and Drug Administration postmarketing reports have noted ertapenem can cause somnolence and dyskinesia,9 and several case reports have described ertapenem-associated CNS side effects, including psychosis and encephalopathy.10-13 Symptoms and examination findings can include confusion, disorientation, garbled speech, dysphagia, hallucinations, miosis, myoclonus, tremor, and agitation.10-13 Although reports of dysmetria and dysdiadochokinesia are lacking, suspicion of an ADE in this case was heightened by the timing of the exposure and the absence of alternative infectious, metabolic, and vascular explanations for bilateral cerebellar dysfunction.
The Naranjo Adverse Drug Reaction (ADR) scale may help clinicians differentiate ADEs from other etiologies of symptoms. It uses 10 weighted questions (Table) to estimate the probability that an adverse clinical event is caused by a drug reaction.14 The present case was assigned 1 point for prior reports of neurologic ADEs associated with ertapenem, 2 for the temporal association, 1 for resolution after medication withdrawal, 2 for lack of alternative causes, and 1 for objective evidence of neurologic dysfunction—for a total of 7 points, indicating ertapenem was probably the cause of the patient’s neurologic symptoms. Of 4 prior cases in which carbapenem toxicity was suspected and the Naranjo scale was used, 3 found a probable relationship, and the fourth a highly probable one.10,12 Confusion, disorientation, hallucinations, tangential thoughts, and garbled speech were reported in the 3 probable cases of ADEs. In the highly probable case, tangential thoughts, garbled speech, and miosis were noted on examination, and these findings returned after re-exposure to ertapenem. Of note, these ADEs occurred in patients with normal and abnormal renal function, and in middle-aged and elderly patients.10,11,13
Most medications have a long list of low-frequency and rarely reported adverse effects. The present case reminds clinicians to consider rare adverse effects, or variants of previously reported adverse effects, in a patient with unexplained symptoms. To estimate the probability that a drug is causing harm to a patient, using a validated tool such as the Naranjo scale helps answer the question, What are the chances?
KEY TEACHING POINTS
Clinicians should include rare adverse effects of common medications in the differential diagnosis.
The Naranjo score is a validated tool that can be used to systematically assess the probability of an adverse drug effect at the bedside.
- The presentation of ertapenem-associated neurotoxicity may include features of bilateral cerebellar dysfunction.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
Two weeks after undergoing a below-knee amputation (BKA) and 10 days after being discharged to a skilled nursing facility (SNF), an 87-year-old man returned to the emergency department (ED) for evaluation of somnolence and altered mental state. In the ED, he was disoriented and unable to provide a detailed history.
The differential diagnosis for acute confusion and altered consciousness is broad. Initial possibilities include toxic-metabolic abnormalities, medication side effects, and infections. Urinary tract infection, pneumonia, and surgical-site infection should be assessed for first, as they are common causes of postoperative altered mentation. Next to be considered are subclinical seizure, ischemic stroke, and infectious encephalitis or meningitis, along with hemorrhagic stroke and subdural hematoma.
During initial assessment, the clinician should ascertain baseline mental state, the timeline of the change in mental status, recent medication changes, history of substance abuse, and concern about any recent trauma, such as a fall. Performing the physical examination, the clinician should assess vital signs and then focus on identifying localizing neurologic deficits.
First steps in the work-up include a complete metabolic panel, complete blood cell count, urinalysis with culture, and a urine toxicology screen. If the patient has a “toxic” appearance, blood cultures should be obtained. An electrocardiogram should be used to screen for drug toxicity or evidence of cardiac ischemia. If laboratory test results do not reveal an obvious infectious or metabolic cause, a noncontrast computed tomography (CT) of the head should be obtained. In terms of early interventions, a low glucose level should be treated with thiamine and then glucose, and naloxone should be given if there is any suspicion of narcotic overdose.
More history was obtained from the patient’s records. The BKA was performed to address a nonhealing transmetatarsal amputation. Two months earlier, the transmetatarsal amputation had been performed as treatment for a diabetic forefoot ulcer with chronic osteomyelitis. The patient’s post-BKA course was uncomplicated. He was started on intravenous (IV) ertapenem on postoperative day 1, and on postoperative day 4 was discharged to the SNF to complete a 6-week course of antibiotics for osteomyelitis. Past medical history included paroxysmal atrial fibrillation, coronary artery disease, congestive heart failure (ejection fraction 40%), and type 2 diabetes mellitus. Medications given at the SNF were oxycodone, acetaminophen, cholecalciferol, melatonin, digoxin, ondansetron, furosemide, gabapentin, correctional insulin, tamsulosin, senna, docusate, warfarin, and metoprolol. While there, the patient’s family expressed concern about his diminishing “mental ability.” They reported he had been fully alert and oriented on arrival at the SNF, and living independently with his wife before the BKA. Then, a week before the ED presentation, he started becoming more somnolent and forgetful. The gabapentin and oxycodone dosages were reduced to minimize their sedative effects, but he showed no improvement. At the SNF, a somnolence work-up was not performed.
Several of the patient’s medications can contribute to altered mental state. Ertapenem can cause seizures as well as profound mental status changes, though these are more likely in the setting of poor renal function. The mental status changes were noticed about a week into the patient’s course of antibiotics, which suggests a possible temporal correlation with the initiation of ertapenem. An electroencephalogram is required to diagnose nonconvulsive seizure activity. Narcotic overdose should still be considered, despite the recent reduction in oxycodone dosage. Digoxin toxicity, though less likely when the dose is stable and there are no changes in renal function, can cause a confused state. Concurrent use of furosemide could potentiate the toxic effects of digoxin.
Non-medication-related concerns include hypoglycemia, hyperglycemia, and, given his history of atrial fibrillation, cardioembolic stroke. Although generalized confusion is not a common manifestation of stroke, a thalamic stroke can alter mental state but be easily missed if not specifically considered. Additional lab work-up should include a digoxin level and, since he is taking warfarin, a prothrombin time/international normalized ratio (PT/INR). If the initial laboratory studies and head CT do not explain the altered mental state, magnetic resonance imaging (MRI) of the brain should be performed to further assess for stroke.
On physical examination in the ED, the patient was resting comfortably with eyes closed, and arousing to voice. He obeyed commands and participated in the examination. His Glasgow Coma Scale score was 13; temperature, 36.8°C, heart rate, 80 beats per minute; respiratory rate, 16 breaths per minute; blood pressure, 90/57 mm Hg; and 100% peripheral capillary oxygen saturation while breathing ambient air. He appeared well developed. His heart rhythm was irregularly irregular, without murmurs, rubs, or gallops. Respiratory and abdominal examination findings were normal. The left BKA incision was well approximated, with no drainage, dehiscence, fluctuance, or erythema. On neurologic examination, the patient was intermittently oriented only to self. Pupils were equal, round, and reactive to light; extraocular movements were intact; face was symmetric; tongue was midline; sensation on face was equal bilaterally; and shoulder shrug was intact. Strength was 5/5 and symmetric in the elbow and hip and 5/5 in the right knee and ankle (not tested on left because of BKA). Deep tendon reflexes were 3+ and symmetrical at the biceps, brachioradialis, and triceps tendons and 3+ in the right patellar and Achilles tendons. Sensation was intact and symmetrical in the upper and lower extremities. The patient’s speech was slow and slurred, and his answers were unrelated to the questions being asked.
The patient’s mental state is best described as lethargic. As he is only intermittently oriented, he meets the criteria for delirium. He is not obtunded or comatose, and his pupils are at least reactive, not pinpoint, so narcotic overdose is less likely. Thalamic stroke remains in the differential diagnosis; despite the seemingly symmetrical sensation examination, hemisensory deficits cannot be definitively ruled out given the patient’s mental state. A rare entity such as carcinomatosis meningitis or another diffuse, infiltrative neoplastic process could be causing his condition. However, because focal deficits other than abnormal speech and diffuse hyperreflexia are absent, toxic, infectious, or metabolic causes are more likely than structural abnormalities. Still possible is a medication toxicity, such as ertapenem toxicity or, less likely, digoxin toxicity. In terms of infectious possibilities, urinary tract infection could certainly present in this fashion, especially if the patient had a somewhat low neurologic reserve at baseline, and hypotension could be secondary to sepsis. Encephalitis or meningitis remains in the differential diagnosis, though the patient appears nontoxic, and therefore a bacterial etiology is very unlikely.
The patient’s hyperreflexia may be an important clue. Although the strength of his reflexes at baseline is unknown, seizures can cause transiently increased reflexes as well as a confused, lethargic mental state. Reflexes can also be increased by a drug overdose that has caused serotonin syndrome. Of the patient’s medications, only ondansetron can cause this reaction. Hyperthyroidism can cause brisk reflexes and confusion, though more typically it causes agitated confusion. A thyroid-stimulating hormone level should be added to the initial laboratory panel.
A complete blood count revealed white blood cell count 11.86 K/uL with neutrophilic predominance and immature granulocytes, hemoglobin 11.5 g/dL, and platelet count 323 K/uL. Serum sodium was 141 mEq/L, potassium 4.2 mEq/L, chloride 103 mEq/L, bicarbonate 30 mEq/L, creatinine 1.14 mg/dL (prior baseline of 0.8-1.0 mg/dL), blood urea nitrogen 26 mg/dL, blood glucose 159 mg/dL, and calcium 9.1 mg/dL. His digoxin level was 1.3 ng/mL (reference range 0.5-1.9 mg/mL) and troponin was undetectable. INR was 2.7 and partial thromboplastin time (PTT) 60 seconds. Vitamin B12 level was 674 pg/mL (reference range >180). A urinalysis had 1+ hyaline casts and was negative for nitrites, leukocyte esterase, blood, and bacteria. An ECG revealed atrial fibrillation with a ventricular rate of 80 beats per minute. A chest radiograph showed clear lung fields. A CT of the head without IV contrast had no evidence of an acute intracranial abnormality. In the ED, 1 liter of IV normal saline was given and blood pressure improved to 127/72 mm Hg.
The head CT does not show intracranial bleeding, and, though it is reassuring that INR is in the therapeutic range, ischemic stroke must remain in the differential diagnosis. Sepsis is less likely given that the criteria for systemic inflammatory response syndrome are not met, and hypotension was rapidly corrected with administration of IV fluids. Urinary tract infection was ruled out with the negative urinalysis. Subclinical seizures remain possible, as does medication-related or other toxicity. A medication overdose, intentional or otherwise, should also be considered.
The patient was admitted to the hospital. On reassessment by the inpatient team, he was oriented only to self, frequently falling asleep, and not recalling earlier conversations when aroused. His speech remained slurred and difficult to understand. Neurologic examination findings were unchanged since the ED examination. On additional cerebellar examination, he had dysmetria with finger-to-nose testing bilaterally and dysdiadochokinesia (impaired rapid alternating movements) of the left hand.
His handedness is not mentioned; the dysdiadochokinesia of the left hand may reflect the patient’s being right-handed, or may signify a focal cerebellar lesion. The cerebellum is also implicated by the bilateral dysmetria. Persistent somnolence in the absence of CT findings suggests a metabolic or infectious process. Metabolic processes that can cause bilateral cerebellar ataxia and somnolence include overdose of a drug or medication. Use of alcohol or a medication such as phenytoin, valproic acid, or a benzodiazepine can cause the symptoms in this case, but was not reported by the family, and there was no documentation of it in the SNF records. Wernicke encephalopathy is rare and is not well supported by the patient’s presentation but should be considered, as it can be easily treated with thiamine. Meningoencephalitis affecting the cerebellum remains possible, but infection is less likely. Both electroencephalogram and brain MRI should be performed, with a specific interest in possible cerebellar lesions. If the MRI is unremarkable, a lumbar puncture should be performed to assess opening pressure and investigate for infectious etiologies.
MRI of the brain showed age-related volume loss and nonspecific white matter disease without acute changes. Lack of a clear explanation for the neurologic findings led to suspicion of a medication side effect. Ertapenem was stopped on admission because it has been reported to rarely cause altered mental status. IV moxifloxacin was started for the osteomyelitis. Over the next 2 days, symptoms began resolving; within 24 hours of ertapenem discontinuation, the patient was awake, alert, and talkative. On examination, he remained dysarthric but was no longer dysmetric. Within 48 hours, the dysarthria was completely resolved, and he was returned to the SNF to complete a course of IV moxifloxacin.
DISCUSSION
Among elderly patients presenting to the ED, altered mental status is a common complaint, accounting for 10% to 30% of visits.1 Medications are a common cause of altered mental status among the elderly and are responsible for 40% of delirium cases.1 The risk of adverse drug events (ADEs) rises with the number of medications prescribed.1-3 Among patients older than 60 years, the incidence of polypharmacy (defined as taking >5 prescription medications) increased from roughly 20% in 1999 to 40% in 2012.4,5 The most common ADEs in the ambulatory setting (25%) are central nervous system (CNS) symptoms, including dizziness, sleep disturbances, and mood changes.6 A medication effect should be suspected in any elderly patient presenting with altered mental state.
The present patient developed a constellation of neurologic symptoms after starting ertapenem, one of the carbapenem antibiotics, which is a class of medications that can cause CNS ADEs. Carbapenems are renally cleared, and adjustments must be made for acute or chronic changes in kidney function. Carbapenems are associated with increased risk of seizure; the incidence of seizure with ertapenem is 0.2%.7,8 Food and Drug Administration postmarketing reports have noted ertapenem can cause somnolence and dyskinesia,9 and several case reports have described ertapenem-associated CNS side effects, including psychosis and encephalopathy.10-13 Symptoms and examination findings can include confusion, disorientation, garbled speech, dysphagia, hallucinations, miosis, myoclonus, tremor, and agitation.10-13 Although reports of dysmetria and dysdiadochokinesia are lacking, suspicion of an ADE in this case was heightened by the timing of the exposure and the absence of alternative infectious, metabolic, and vascular explanations for bilateral cerebellar dysfunction.
The Naranjo Adverse Drug Reaction (ADR) scale may help clinicians differentiate ADEs from other etiologies of symptoms. It uses 10 weighted questions (Table) to estimate the probability that an adverse clinical event is caused by a drug reaction.14 The present case was assigned 1 point for prior reports of neurologic ADEs associated with ertapenem, 2 for the temporal association, 1 for resolution after medication withdrawal, 2 for lack of alternative causes, and 1 for objective evidence of neurologic dysfunction—for a total of 7 points, indicating ertapenem was probably the cause of the patient’s neurologic symptoms. Of 4 prior cases in which carbapenem toxicity was suspected and the Naranjo scale was used, 3 found a probable relationship, and the fourth a highly probable one.10,12 Confusion, disorientation, hallucinations, tangential thoughts, and garbled speech were reported in the 3 probable cases of ADEs. In the highly probable case, tangential thoughts, garbled speech, and miosis were noted on examination, and these findings returned after re-exposure to ertapenem. Of note, these ADEs occurred in patients with normal and abnormal renal function, and in middle-aged and elderly patients.10,11,13
Most medications have a long list of low-frequency and rarely reported adverse effects. The present case reminds clinicians to consider rare adverse effects, or variants of previously reported adverse effects, in a patient with unexplained symptoms. To estimate the probability that a drug is causing harm to a patient, using a validated tool such as the Naranjo scale helps answer the question, What are the chances?
KEY TEACHING POINTS
Clinicians should include rare adverse effects of common medications in the differential diagnosis.
The Naranjo score is a validated tool that can be used to systematically assess the probability of an adverse drug effect at the bedside.
- The presentation of ertapenem-associated neurotoxicity may include features of bilateral cerebellar dysfunction.
Disclosure
Nothing to report.
1. Inouye SK, Fearing MA, Marcantonio ER. Delirium. In: Halter JB, Ouslander JG, Tinetti ME, Studenski S, High KP, Asthana S, eds. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. New York, NY: McGraw-Hill; 2009.
2. Sarkar U, López A, Maselli JH, Gonzales R. Adverse drug events in U.S. adult ambulatory medical care. Health Serv Res. 2011;46(5):1517-1533. PubMed
3. Chrischilles E, Rubenstein L, Van Gilder R, Voelker M, Wright K, Wallace R. Risk factors for adverse drug events in older adults with mobility limitations in the community setting. J Am Geriatr Soc. 2007;55(1):29-34. PubMed
4. Kaufman DW, Kelly JP, Rosenberg L, Anderson TE, Mitchell AA. Recent patterns of medication use in the ambulatory adult population of the United States: the Slone survey. JAMA. 2002;287(3):337-344. PubMed
5. Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314(17):1818-1831. PubMed
6. Thomsen LA, Winterstein AG, Søndergaard B, Haugbølle LS, Melander A. Systematic review of the incidence and characteristics of preventable adverse drug events in ambulatory care. Ann Pharmacother. 2007;41(9):1411-1426. PubMed
7. Zhanel GG, Wiebe R, Dilay L, et al. Comparative review of the carbapenems. Drugs. 2007;67(7):1027-1052. PubMed
8. Cannon JP, Lee TA, Clark NM, Setlak P, Grim SA. The risk of seizures among the carbapenems: a meta-analysis. J Antimicrob Chemother. 2014;69(8):2043-2055. PubMed
9. US Food and Drug Administration. Invanz (ertapenem) injection [safety information]. http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm196605.htm. Published July 2013. Accessed July 6, 2015.
10. Oo Y, Packham D, Yau W, Munckhof WJ. Ertapenem-associated psychosis and encephalopathy. Intern Med J. 2014;44(8):817-819. PubMed
11. Wen MJ, Sung CC, Chau T, Lin SH. Acute prolonged neurotoxicity associated with recommended doses of ertapenem in 2 patients with advanced renal failure. Clin Nephrol. 2013;80(6):474-478. PubMed
12. Duquaine S, Kitchell E, Tate T, Tannen RC, Wickremasinghe IM. Central nervous system toxicity associated with ertapenem use. Ann Pharmacother. 2011;45(1):e6. PubMed
13. Kong V, Beckert L, Awunor-Renner C. A case of beta lactam-induced visual hallucination. N Z Med J. 2009;122(1298):76-77. PubMed
14. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245. PubMed
1. Inouye SK, Fearing MA, Marcantonio ER. Delirium. In: Halter JB, Ouslander JG, Tinetti ME, Studenski S, High KP, Asthana S, eds. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. New York, NY: McGraw-Hill; 2009.
2. Sarkar U, López A, Maselli JH, Gonzales R. Adverse drug events in U.S. adult ambulatory medical care. Health Serv Res. 2011;46(5):1517-1533. PubMed
3. Chrischilles E, Rubenstein L, Van Gilder R, Voelker M, Wright K, Wallace R. Risk factors for adverse drug events in older adults with mobility limitations in the community setting. J Am Geriatr Soc. 2007;55(1):29-34. PubMed
4. Kaufman DW, Kelly JP, Rosenberg L, Anderson TE, Mitchell AA. Recent patterns of medication use in the ambulatory adult population of the United States: the Slone survey. JAMA. 2002;287(3):337-344. PubMed
5. Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314(17):1818-1831. PubMed
6. Thomsen LA, Winterstein AG, Søndergaard B, Haugbølle LS, Melander A. Systematic review of the incidence and characteristics of preventable adverse drug events in ambulatory care. Ann Pharmacother. 2007;41(9):1411-1426. PubMed
7. Zhanel GG, Wiebe R, Dilay L, et al. Comparative review of the carbapenems. Drugs. 2007;67(7):1027-1052. PubMed
8. Cannon JP, Lee TA, Clark NM, Setlak P, Grim SA. The risk of seizures among the carbapenems: a meta-analysis. J Antimicrob Chemother. 2014;69(8):2043-2055. PubMed
9. US Food and Drug Administration. Invanz (ertapenem) injection [safety information]. http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm196605.htm. Published July 2013. Accessed July 6, 2015.
10. Oo Y, Packham D, Yau W, Munckhof WJ. Ertapenem-associated psychosis and encephalopathy. Intern Med J. 2014;44(8):817-819. PubMed
11. Wen MJ, Sung CC, Chau T, Lin SH. Acute prolonged neurotoxicity associated with recommended doses of ertapenem in 2 patients with advanced renal failure. Clin Nephrol. 2013;80(6):474-478. PubMed
12. Duquaine S, Kitchell E, Tate T, Tannen RC, Wickremasinghe IM. Central nervous system toxicity associated with ertapenem use. Ann Pharmacother. 2011;45(1):e6. PubMed
13. Kong V, Beckert L, Awunor-Renner C. A case of beta lactam-induced visual hallucination. N Z Med J. 2009;122(1298):76-77. PubMed
14. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245. PubMed
© 2017 Society of Hospital Medicine
Forging ahead
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 45-year-old woman presented to the emergency department with 2 days of generalized, progressive weakness. Her ability to walk and perform daily chores was increasingly limited. On the morning of her presentation, she was unable to stand up without falling.
A complaint of weakness must be classified as either functional weakness related to a systemic process or true neurologic weakness from dysfunction of the central nervous system (eg, brain, spinal cord) or peripheral nervous system (eg, anterior horn cell, nerve, neuromuscular junction, or muscle). More information on her clinical course and a detailed neurologic exam will help clarify this key branch point.
She was 2 weeks status-post laparoscopic Roux-en-Y gastric bypass and gastric band removal performed in Europe. Immediately following surgery, she experienced abdominal discomfort and nausea with occasional nonbloody, nonbilious emesis, attributed to expected postoperative anatomical changes. She developed a postoperative pneumonia treated with amoxicillin-clavulanate. She tolerated her flight back to the United States, but her abdominal discomfort persisted and she had minimal oral intake due to her nausea.
Functional weakness may stem from hypovolemia from insufficient oral intake, anemia related to the recent surgery, electrolyte abnormalities, chronic nutritional issues associated with obesity and weight-reduction surgery, and pneumonia. Prolonged air travel, obesity, and recent surgery place her at risk for venous thromboembolism, which may manifest as reduced exercise tolerance. Nausea, vomiting, and abdominal pain persisting for 2 weeks after a Roux-en-Y gastric bypass surgery raises several concerns, including gastric remnant distension (although hiccups are often prominent); stomal stenosis, which typically presents several weeks after surgery; marginal ulceration; or infection at the surgical site or from an anastomotic leak. She may also have a surgery- or medication-related myopathy.
The patient had a history of obesity, hypertension, hyperlipidemia, migraine headaches, and nonalcoholic steatohepatitis. Four years previously, she had undergone gastric banding complicated by band migration and ulceration at the banding site. Her medications were amlodipine, losartan, ranitidine, acetaminophen, and nadroparin for venous thromboembolism prophylaxis during her flight. She denied alcohol, tobacco, or illicit drug use. On further questioning, she reported diaphoresis, mild dyspnea, loose stools, and a sensation of numbness and “heaviness” in her arms. Her abdominal pain was limited to the surgical incision and was controlled with acetaminophen. She denied fevers, cough, chest pain, diplopia, or dysphagia.
Heaviness in both arms could result from an acutely presenting myopathic or neuropathic process, while the coexistence of numbness suggests a sensorimotor polyneuropathy. Obesity and gastric bypass surgery increase her nutritional risk, and thiamine deficiency may present as an acute axonal polyneuropathy (ie, beriberi). Unlike vitamin B12 deficiency, which may take years to develop, thiamine deficiency can present within 4 weeks of gastric bypass surgery. Her dyspnea may be a manifestation of diaphragmatic weakness, although her ostensibly treated pneumonia or as of yet unproven postoperative anemia may be contributing. Chemoprophylaxis mitigates her risk of venous thromboembolism, which is, nonetheless, unlikely to account for the gastrointestinal symptoms and upper extremity weakness. If she is continuing to take amlodipine and losartan but has become volume-depleted, hypotension may be contributing to the generalized weakness.
Physical examination revealed an obese, pale and diaphoretic woman. Her temperature was 36.9°C, heart rate 77 beats per minute, blood pressure 158/90 mm Hg, respiratory rate 28 breaths per minute, and O2 saturation 99% on ambient air. She had no cervical lymphadenopathy and a normal thyroid exam. There were no murmurs on cardiac examination, and jugular venous pressure was estimated at 10 cm of water. Her lung sounds were clear. Her abdomen was soft, nondistended, with localized tenderness and fluctuance around the midline surgical incision with a small amount of purulent drainage. She was alert and oriented to name, date, place, and situation. Cranial nerves II through XII were grossly intact. Strength was 4/5 in bilateral biceps, triceps and distal hand and finger extensors, 3/5 in bilateral deltoids. Strength in hip flexors was 4/5 and it was 5/5 in distal lower extremities. Sensation was intact to pinprick in upper and lower extremities. Biceps reflexes were absent; patellar and ankle reflexes were 1+ and symmetric. The remainder of the physical exam was unremarkable.
The patient has symmetric proximal muscle weakness with upper extremity predominance and preserved strength in her distal lower extremities. A myopathy could explain this pattern of weakness, further substantiated by absent reflexes and reportedly intact sensation. Subacute causes of myopathy include hypokalemia, hyperkalemia, toxic myopathies from medications, or infection-induced rhabdomyolysis. However, she does not report muscle pain, and the loss of reflexes is faster than would be expected with a myopathy. A more thorough sensory examination would inform the assessment of potential neuropathic processes. Guillain-Barré syndrome (GBS) is possible; it most commonly presents as an ascending, distally predominant acute inflammatory demyelinating polyneuropathy (AIDP), although her upper extremity weakness predominates and there are no clear sensory changes. It remains to be determined how her wound infection might relate to her overall presentation.
Her white blood cell count was 12,600/μL (reference range: 3,400-10,000/μL), hemoglobin was 10.2 g/dL, and platelet count was 698,000/μL. Mean corpuscular volume was 86 fL. Serum chemistries were: sodium 138 mEq/L, potassium 3.8 mEq/L, chloride 106 mmol/L, bicarbonate 15 mmol/L, blood urea nitrogen 5 mg/dL, creatinine 0.65 mg/dL, glucose 125 mg/dL, calcium 8.3 mg/dL, magnesium 1.9 mg/dL, phosphorous 2.4 mg/dL, and lactate 1.8 mmol/L (normal: < 2.0 mmol/L). Creatinine kinase (CK), liver function tests, and coagulation panel were normal. Total protein was 6.4 g/dL, and albumin was 2.7 g/dL. Venous blood gas was: pH 7.39 and PCO2 25 mmHg. Urinalysis revealed ketones. Blood and wound cultures were sent for evaluation. A chest x-ray was unremarkable. An electrocardiogram showed normal sinus rhythm. Computed tomography (CT) of the abdomen and pelvis revealed a multiloculated rim-enhancing fluid collection in the anterior abdominal wall (Figure 1).

She does not have any notable electrolyte derangements that would account for her weakness, and the normal creatinine kinase lowers the probability of a myopathy and excludes rhabdomyolysis. Progression of weakness from proximal to distal muscles in a symmetric fashion is consistent with botulism, and she has an intra-abdominal wound infection that could be harboring Clostridium botulinum. Nonetheless, the normal cranial nerve exam and the rarity of botulism occurring with surgical wounds argue against this diagnosis. She should receive intravenous (IV) thiamine for the possibility of beriberi. A lumbar puncture should be performed to assess for albuminocytologic dissociation, which can be seen in patients with GBS.
The patient received high-dose IV thiamine, IV vancomycin, IV piperacillin-tazobactam, and acetaminophen. Over the subsequent 4 hours, her anion gap acidosis worsened. She declined arterial puncture. Repeat venous blood gas was: pH 7.22, PCO2 28 mmHg, and bicarbonate 11 mmol/L. Lactate and glucose were normal. Serum osmolarity was 292 mmol/kg (reference range: 283-301 mmol/kg). She was started on an IV sodium bicarbonate infusion without improvement in her acidemia.
An acute anion gap metabolic acidosis suggests a limited differential diagnosis that includes lactic acidosis, D-lactic acidosis, severe starvation ketoacidosis, acute renal failure, salicylate, or other drug or poison ingestion. Starvation ketoacidosis may be contributing, but a bicarbonate value this low would be unusual. There is no history of alcohol use or other ingestions, and the normal serum osmolality and low osmolal gap (less than 10 mOsm/kg) argue against a poisoning with ethanol, ethylene glycol, or methanol. The initial combined anion gap metabolic acidosis and respiratory alkalosis is consistent with salicylate toxicity, but she does not report aspirin ingestion. Acetaminophen use in the setting of malnutrition or starvation physiology raises the possibility of 5-oxoproline accumulation.
Routine serum lactate does not detect D-lactate, which is produced by colonic bacteria and has been reported in short bowel syndrome and following intestinal bypass surgery. This may occur weeks to months after intestinal procedures, following ingestion of a heavy carbohydrate load, and almost invariably presents with altered mental status and increased anion gap metabolic acidosis, although generalized weakness has been reported.
A surgical consultant drained her wound infection. Fluid Gram stain was negative. D-lactate, salicylate and acetaminophen levels were undetectable. Thiamine pyrophosphate level was 229 nmol/L (reference range: 78-185 nmol/L). Acetaminophen was discontinued and N-acetylcysteine infusion was started for possible 5-oxoprolinemia. Her anion gap acidosis rapidly improved. Twelve hours after admission, she reported sudden onset of blurry vision. Her vital signs were: temperature 37oC, heart rate 110 beats per minute, respiratory rate 40 breaths per minute, blood pressure 168/90, and oxygen saturation 100% on ambient air. Telemetry showed ventricular bigeminy. On examination, she was unable to abduct her right eye; muscle strength was 1/5 in all extremities; biceps, ankle, and patellar reflexes were absent.
Her neurological deficits have progressed over hours to near complete paralysis, asymmetric cranial nerve paresis, and areflexia. Although botulism can cause blurred vision and absent deep tendon reflexes, patients almost always have symmetrical bulbar findings followed by descending paralysis. Should the “numbness” in her arms reported earlier represent undetected sensory deficits, this, too would be inconsistent with botulism.
A diagnosis of GBS ties together several aspects of her presentation and clinical course. Several variants show different patterns of weakness and may involve cranial nerves. Her tachypnea and dyspnea are concerning signs of potential impending respiratory failure. The ventricular bigeminy and mild hypertension could represent autonomic dysfunction that is seen in many cases of GBS.
She was intubated for airway protection. Computed tomography angiography and magnetic resonance imaging of her brain were normal. Cerebral spinal fluid analysis obtained through lumbar puncture showed the following: white blood cell count 3/μL, red blood cell count 11/μL, protein 63 mg/dL (reference range: 15-60mg/dL), and glucose 128 mg/dL (reference range: 40-80mg/dL).
The lumbar puncture is consistent with GBS given the slightly elevated protein and cell count well below 50/μL. Given the severity of her symptoms, treatment with IV immunoglobulin or plasmapheresis should be initiated. Nerve conduction studies (NCS) and electromyography (EMG) are indicated for diagnostic confirmation.
EMG and NCS revealed a severe sensorimotor polyneuropathy with demyelinating features including a conduction block at a noncompressible site, consistent with AIDP. Left sural nerve biopsy confirmed acute demyelinating and mild axonal neuropathy (Figure 2). On hospital day 2, treatment with IV immunoglobulins (IVIG) was initiated; however, she developed anaphylaxis following her second administration and subsequently received plasmapheresis. A tracheostomy was performed for respiratory muscle weakness, and she was discharged to a nursing facility. C. botulinum cultures from the wound eventually returned negative. Following her hospitalization, a serum 5-oxoproline level sent 10 hours after admission returned as elevated, confirming the additional diagnosis of 5-oxoprolinemia. On follow-up, she can sit up and feed herself without assistance, and her gait continues to improve with physical therapy.

DISCUSSION
This patient presented with rapidly progressive weakness that developed in the 2 weeks following bariatric surgery. In the postsurgical setting, patient complaints of weakness are commonly encountered and can pose a diagnostic challenge. Asthenia (ie, general loss of strength or energy) is frequently reported in the immediate postoperative period, and may result from the stress of surgery, pain, deconditioning, or infection. This must be distinguished from true neurologic weakness, which results from dysfunction of the brain, spinal cord, nerve, neuromuscular junction, or muscle. The initial history can help elucidate the inciting events such as preceding surgery, infections or ingestions, and can also categorize the pattern of weakness. The neurologic examination can localize the pathology within the neuraxis. EMG and NCS can distinguish neuropathy from radiculopathy, and categorize the process as axonal, demyelinating, or mixed. In this case, the oculomotor weakness, sensory abnormalities and areflexia signaled a severe sensorimotor polyneuropathy, and EMG/NCS confirmed a demyelinating process consistent with GBS.
Guillain-Barré syndrome is an acute, immune-mediated polyneuropathy. Patients with GBS often present with a preceding respiratory or diarrheal illness; however, the stress of a recent surgery can serve as an inciting event. The syndrome, acute postgastric reduction surgery (APGARS) neuropathy, was introduced in the literature in 2002, describing 3 patients who presented with progressive vomiting, weakness, and hyporeflexia following bariatric surgery.1 The term has been used to describe bariatric surgery patients who developed postoperative quadriparesis, cranial nerve deficits, and respiratory compromise.2 Given the clinical heterogeneity in the literature with relation to APGARS, it is probable that the cases described could result from multiple etiologies. While GBS is purely immune-mediated and can be precipitated by the stress of surgery itself, postbariatric surgery patients are susceptible to many nutritional deficiencies that can lead to similar presentations.3 For example, thiamine (vitamin B1) and cobalamin (vitamin B12) deficiencies cause distinct postbariatric surgery neuropathies.4 Thiamine deficiency may manifest weeks to months after surgery and can rapidly progress, whereas cobalamin deficiency generally develops over 3 to 5 years. Both of these syndromes demonstrate an axonal pattern of nerve injury on EMG/NCS, in contrast to the demyelinating pattern typically seen in GBS. In addition, bariatric surgery patients are at higher risk for copper deficiency, which usually presents as a myeloneuropathy with subacute gait decline and upper motor neuron signs including spasticity.
Although GBS classically presents with symmetric ascending weakness and sensory abnormalities, it may manifest in myriad ways. Factors influencing the presentation include the types of nerve fibers involved (motor, sensory, cranial or autonomic), the predominant mode of injury (axonal vs demyelinating), and the presence or absence of alteration in consciousness.5 The most common form of GBS is AIDP. The classic presentation involves paresthesias in the fingertips and toes followed by lower extremity weakness that ascends over hours to days to involve the arms and potentially the muscles of respiration. A minority of patients with GBS first experience weakness in the upper extremities or facial muscles, and oculomotor involvement is rare.5 Pain is common and often severe.6 Dysautonomia affects most patients with GBS and may manifest as labile blood pressure or arrhythmias.5 Several variant GBS presentation patterns have been described, including acute motor axonal neuropathy, a pure motor form of GBS; ophthalmoplegia, ataxia, and areflexia in Miller Fisher syndrome; and alteration in consciousness, hyperreflexia, ataxia, and ophthalmoparesis in Bickerstaff’s brain stem encephalitis.5
Patients with GBS can progress rapidly to respiratory failure. Serial neurologic exams may signal the diagnosis and inform triage to the appropriate level of care. Measurement of bedside pulmonary function, including mean inspiratory force and functional vital capacity, help to determine if there is weakness of diaphragmatic muscles. Patients with signs or symptoms of diaphragmatic weakness require monitoring in an intensive care unit and potentially early intubation. Treatment with IVIG or plasmapheresis has been found to hasten recovery from GBS, including earlier improvement in muscle strength and a reduced need for mechanical ventilation.7 Treatment selection is based on available resources as both modalities are felt to be equivalent.The majority of patients with GBS make a full recovery over a period of weeks to months, although many have persistent motor weakness. Despite immunotherapy, up to 20% of patients remain severely disabled and approximately 5% die.8 Advanced age, rapid progression of weakness over a period of less than 72 hours, need for mechanical ventilation, and absent compound muscle action potentials on NCS are all associated with prolonged and incomplete recovery.9
This patient developed respiratory failure within 12 hours of hospitalization, prior to being diagnosed with GBS. Even in that short time, the treating clinicians encountered a series of clinical diversions. The initial proximal pattern of muscle weakness suggested a possible myopathic process; the wound infection introduced the possibility of botulism; obesity and recent bariatric surgery triggered concern for thiamine deficiency; and the anion gap acidosis from 5-oxoprolinemia created yet another clinical detour. While the path from presentation to diagnosis is seldom a straight line, when faced with rapidly progressive weakness, it is paramount to forge ahead with an efficient diagnostic evaluation and timely therapeutic intervention.
KEY TEACHING POINTS
- A complaint of general weakness requires distinction between asthenia (ie, general loss of strength or energy) and true neuromuscular weakness from dysfunction of the brain, spinal cord, nerve, neuromuscular junction, and/or muscle.
- Guillain-Barré syndrome may present in a variety of atypical fashions not limited to ascending, distally predominant weakness.
- Acute postgastric reduction surgery neuropathy should be considered in patients presenting with weakness, vomiting, or hyporeflexia after bariatric surgery.
- Acute inflammatory demyelinating polyneuropathy may rapidly progress to respiratory failure, and warrants serial neurologic examinations, monitoring of pulmonary function, and an expedited diagnostic evaluation.
Disclosure
Nothing to report.
1. Akhtar M, Collins MP, Kissel JT. Acute postgastric reduction surgery (APGARS) Neuropathy: A polynutritional, multisystem disorder. Neurology. 2002;58:A68. PubMed
2. Chang CG, Adams-Huet B, Provost DA. Acute post-gastric reduction surgery (APGARS) neuropathy. Obes Surg. 2004;14(2):182-189. PubMed
3. Chang CG, Helling TS, Black WE, Rymer MM. Weakness after gastric bypass. Obes Surg. 2002;12(4):592-597. PubMed
4. Shankar P, Boylan M, Sriram K. Micronutrient deficiencies after bariatric surgery. Nutrition. 2010;26(11-12):1031-1037. PubMed
5. Dimachkie MM, Barohn RJ. Guillain-Barré syndrome and variants. Neurol Clin. 2013;31(2):491-510. PubMed
6. Ruts L, Drenthen J, Jongen JL, et al. Pain in Guillain-Barré syndrome: a long-term follow-up study. Neurology. 2010;75(16):1439-1447. PubMed
7. Hughes RAC, Wijdicks EFM, Barohn R, et al: Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: immunotherapy for Guillain-Barré syndrome: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003;61:736-740. PubMed
8. Hughes RA, Swan AV, Raphaël JC, Annane D, van Koningsveld R, van Doorn PA. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain. 2007;130(Pt 9):2245-2257. PubMed
9. Rajabally YA, Uncini A. Outcome and predictors in Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 2012;83(7):711-718. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 45-year-old woman presented to the emergency department with 2 days of generalized, progressive weakness. Her ability to walk and perform daily chores was increasingly limited. On the morning of her presentation, she was unable to stand up without falling.
A complaint of weakness must be classified as either functional weakness related to a systemic process or true neurologic weakness from dysfunction of the central nervous system (eg, brain, spinal cord) or peripheral nervous system (eg, anterior horn cell, nerve, neuromuscular junction, or muscle). More information on her clinical course and a detailed neurologic exam will help clarify this key branch point.
She was 2 weeks status-post laparoscopic Roux-en-Y gastric bypass and gastric band removal performed in Europe. Immediately following surgery, she experienced abdominal discomfort and nausea with occasional nonbloody, nonbilious emesis, attributed to expected postoperative anatomical changes. She developed a postoperative pneumonia treated with amoxicillin-clavulanate. She tolerated her flight back to the United States, but her abdominal discomfort persisted and she had minimal oral intake due to her nausea.
Functional weakness may stem from hypovolemia from insufficient oral intake, anemia related to the recent surgery, electrolyte abnormalities, chronic nutritional issues associated with obesity and weight-reduction surgery, and pneumonia. Prolonged air travel, obesity, and recent surgery place her at risk for venous thromboembolism, which may manifest as reduced exercise tolerance. Nausea, vomiting, and abdominal pain persisting for 2 weeks after a Roux-en-Y gastric bypass surgery raises several concerns, including gastric remnant distension (although hiccups are often prominent); stomal stenosis, which typically presents several weeks after surgery; marginal ulceration; or infection at the surgical site or from an anastomotic leak. She may also have a surgery- or medication-related myopathy.
The patient had a history of obesity, hypertension, hyperlipidemia, migraine headaches, and nonalcoholic steatohepatitis. Four years previously, she had undergone gastric banding complicated by band migration and ulceration at the banding site. Her medications were amlodipine, losartan, ranitidine, acetaminophen, and nadroparin for venous thromboembolism prophylaxis during her flight. She denied alcohol, tobacco, or illicit drug use. On further questioning, she reported diaphoresis, mild dyspnea, loose stools, and a sensation of numbness and “heaviness” in her arms. Her abdominal pain was limited to the surgical incision and was controlled with acetaminophen. She denied fevers, cough, chest pain, diplopia, or dysphagia.
Heaviness in both arms could result from an acutely presenting myopathic or neuropathic process, while the coexistence of numbness suggests a sensorimotor polyneuropathy. Obesity and gastric bypass surgery increase her nutritional risk, and thiamine deficiency may present as an acute axonal polyneuropathy (ie, beriberi). Unlike vitamin B12 deficiency, which may take years to develop, thiamine deficiency can present within 4 weeks of gastric bypass surgery. Her dyspnea may be a manifestation of diaphragmatic weakness, although her ostensibly treated pneumonia or as of yet unproven postoperative anemia may be contributing. Chemoprophylaxis mitigates her risk of venous thromboembolism, which is, nonetheless, unlikely to account for the gastrointestinal symptoms and upper extremity weakness. If she is continuing to take amlodipine and losartan but has become volume-depleted, hypotension may be contributing to the generalized weakness.
Physical examination revealed an obese, pale and diaphoretic woman. Her temperature was 36.9°C, heart rate 77 beats per minute, blood pressure 158/90 mm Hg, respiratory rate 28 breaths per minute, and O2 saturation 99% on ambient air. She had no cervical lymphadenopathy and a normal thyroid exam. There were no murmurs on cardiac examination, and jugular venous pressure was estimated at 10 cm of water. Her lung sounds were clear. Her abdomen was soft, nondistended, with localized tenderness and fluctuance around the midline surgical incision with a small amount of purulent drainage. She was alert and oriented to name, date, place, and situation. Cranial nerves II through XII were grossly intact. Strength was 4/5 in bilateral biceps, triceps and distal hand and finger extensors, 3/5 in bilateral deltoids. Strength in hip flexors was 4/5 and it was 5/5 in distal lower extremities. Sensation was intact to pinprick in upper and lower extremities. Biceps reflexes were absent; patellar and ankle reflexes were 1+ and symmetric. The remainder of the physical exam was unremarkable.
The patient has symmetric proximal muscle weakness with upper extremity predominance and preserved strength in her distal lower extremities. A myopathy could explain this pattern of weakness, further substantiated by absent reflexes and reportedly intact sensation. Subacute causes of myopathy include hypokalemia, hyperkalemia, toxic myopathies from medications, or infection-induced rhabdomyolysis. However, she does not report muscle pain, and the loss of reflexes is faster than would be expected with a myopathy. A more thorough sensory examination would inform the assessment of potential neuropathic processes. Guillain-Barré syndrome (GBS) is possible; it most commonly presents as an ascending, distally predominant acute inflammatory demyelinating polyneuropathy (AIDP), although her upper extremity weakness predominates and there are no clear sensory changes. It remains to be determined how her wound infection might relate to her overall presentation.
Her white blood cell count was 12,600/μL (reference range: 3,400-10,000/μL), hemoglobin was 10.2 g/dL, and platelet count was 698,000/μL. Mean corpuscular volume was 86 fL. Serum chemistries were: sodium 138 mEq/L, potassium 3.8 mEq/L, chloride 106 mmol/L, bicarbonate 15 mmol/L, blood urea nitrogen 5 mg/dL, creatinine 0.65 mg/dL, glucose 125 mg/dL, calcium 8.3 mg/dL, magnesium 1.9 mg/dL, phosphorous 2.4 mg/dL, and lactate 1.8 mmol/L (normal: < 2.0 mmol/L). Creatinine kinase (CK), liver function tests, and coagulation panel were normal. Total protein was 6.4 g/dL, and albumin was 2.7 g/dL. Venous blood gas was: pH 7.39 and PCO2 25 mmHg. Urinalysis revealed ketones. Blood and wound cultures were sent for evaluation. A chest x-ray was unremarkable. An electrocardiogram showed normal sinus rhythm. Computed tomography (CT) of the abdomen and pelvis revealed a multiloculated rim-enhancing fluid collection in the anterior abdominal wall (Figure 1).
She does not have any notable electrolyte derangements that would account for her weakness, and the normal creatinine kinase lowers the probability of a myopathy and excludes rhabdomyolysis. Progression of weakness from proximal to distal muscles in a symmetric fashion is consistent with botulism, and she has an intra-abdominal wound infection that could be harboring Clostridium botulinum. Nonetheless, the normal cranial nerve exam and the rarity of botulism occurring with surgical wounds argue against this diagnosis. She should receive intravenous (IV) thiamine for the possibility of beriberi. A lumbar puncture should be performed to assess for albuminocytologic dissociation, which can be seen in patients with GBS.
The patient received high-dose IV thiamine, IV vancomycin, IV piperacillin-tazobactam, and acetaminophen. Over the subsequent 4 hours, her anion gap acidosis worsened. She declined arterial puncture. Repeat venous blood gas was: pH 7.22, PCO2 28 mmHg, and bicarbonate 11 mmol/L. Lactate and glucose were normal. Serum osmolarity was 292 mmol/kg (reference range: 283-301 mmol/kg). She was started on an IV sodium bicarbonate infusion without improvement in her acidemia.
An acute anion gap metabolic acidosis suggests a limited differential diagnosis that includes lactic acidosis, D-lactic acidosis, severe starvation ketoacidosis, acute renal failure, salicylate, or other drug or poison ingestion. Starvation ketoacidosis may be contributing, but a bicarbonate value this low would be unusual. There is no history of alcohol use or other ingestions, and the normal serum osmolality and low osmolal gap (less than 10 mOsm/kg) argue against a poisoning with ethanol, ethylene glycol, or methanol. The initial combined anion gap metabolic acidosis and respiratory alkalosis is consistent with salicylate toxicity, but she does not report aspirin ingestion. Acetaminophen use in the setting of malnutrition or starvation physiology raises the possibility of 5-oxoproline accumulation.
Routine serum lactate does not detect D-lactate, which is produced by colonic bacteria and has been reported in short bowel syndrome and following intestinal bypass surgery. This may occur weeks to months after intestinal procedures, following ingestion of a heavy carbohydrate load, and almost invariably presents with altered mental status and increased anion gap metabolic acidosis, although generalized weakness has been reported.
A surgical consultant drained her wound infection. Fluid Gram stain was negative. D-lactate, salicylate and acetaminophen levels were undetectable. Thiamine pyrophosphate level was 229 nmol/L (reference range: 78-185 nmol/L). Acetaminophen was discontinued and N-acetylcysteine infusion was started for possible 5-oxoprolinemia. Her anion gap acidosis rapidly improved. Twelve hours after admission, she reported sudden onset of blurry vision. Her vital signs were: temperature 37oC, heart rate 110 beats per minute, respiratory rate 40 breaths per minute, blood pressure 168/90, and oxygen saturation 100% on ambient air. Telemetry showed ventricular bigeminy. On examination, she was unable to abduct her right eye; muscle strength was 1/5 in all extremities; biceps, ankle, and patellar reflexes were absent.
Her neurological deficits have progressed over hours to near complete paralysis, asymmetric cranial nerve paresis, and areflexia. Although botulism can cause blurred vision and absent deep tendon reflexes, patients almost always have symmetrical bulbar findings followed by descending paralysis. Should the “numbness” in her arms reported earlier represent undetected sensory deficits, this, too would be inconsistent with botulism.
A diagnosis of GBS ties together several aspects of her presentation and clinical course. Several variants show different patterns of weakness and may involve cranial nerves. Her tachypnea and dyspnea are concerning signs of potential impending respiratory failure. The ventricular bigeminy and mild hypertension could represent autonomic dysfunction that is seen in many cases of GBS.
She was intubated for airway protection. Computed tomography angiography and magnetic resonance imaging of her brain were normal. Cerebral spinal fluid analysis obtained through lumbar puncture showed the following: white blood cell count 3/μL, red blood cell count 11/μL, protein 63 mg/dL (reference range: 15-60mg/dL), and glucose 128 mg/dL (reference range: 40-80mg/dL).
The lumbar puncture is consistent with GBS given the slightly elevated protein and cell count well below 50/μL. Given the severity of her symptoms, treatment with IV immunoglobulin or plasmapheresis should be initiated. Nerve conduction studies (NCS) and electromyography (EMG) are indicated for diagnostic confirmation.
EMG and NCS revealed a severe sensorimotor polyneuropathy with demyelinating features including a conduction block at a noncompressible site, consistent with AIDP. Left sural nerve biopsy confirmed acute demyelinating and mild axonal neuropathy (Figure 2). On hospital day 2, treatment with IV immunoglobulins (IVIG) was initiated; however, she developed anaphylaxis following her second administration and subsequently received plasmapheresis. A tracheostomy was performed for respiratory muscle weakness, and she was discharged to a nursing facility. C. botulinum cultures from the wound eventually returned negative. Following her hospitalization, a serum 5-oxoproline level sent 10 hours after admission returned as elevated, confirming the additional diagnosis of 5-oxoprolinemia. On follow-up, she can sit up and feed herself without assistance, and her gait continues to improve with physical therapy.
DISCUSSION
This patient presented with rapidly progressive weakness that developed in the 2 weeks following bariatric surgery. In the postsurgical setting, patient complaints of weakness are commonly encountered and can pose a diagnostic challenge. Asthenia (ie, general loss of strength or energy) is frequently reported in the immediate postoperative period, and may result from the stress of surgery, pain, deconditioning, or infection. This must be distinguished from true neurologic weakness, which results from dysfunction of the brain, spinal cord, nerve, neuromuscular junction, or muscle. The initial history can help elucidate the inciting events such as preceding surgery, infections or ingestions, and can also categorize the pattern of weakness. The neurologic examination can localize the pathology within the neuraxis. EMG and NCS can distinguish neuropathy from radiculopathy, and categorize the process as axonal, demyelinating, or mixed. In this case, the oculomotor weakness, sensory abnormalities and areflexia signaled a severe sensorimotor polyneuropathy, and EMG/NCS confirmed a demyelinating process consistent with GBS.
Guillain-Barré syndrome is an acute, immune-mediated polyneuropathy. Patients with GBS often present with a preceding respiratory or diarrheal illness; however, the stress of a recent surgery can serve as an inciting event. The syndrome, acute postgastric reduction surgery (APGARS) neuropathy, was introduced in the literature in 2002, describing 3 patients who presented with progressive vomiting, weakness, and hyporeflexia following bariatric surgery.1 The term has been used to describe bariatric surgery patients who developed postoperative quadriparesis, cranial nerve deficits, and respiratory compromise.2 Given the clinical heterogeneity in the literature with relation to APGARS, it is probable that the cases described could result from multiple etiologies. While GBS is purely immune-mediated and can be precipitated by the stress of surgery itself, postbariatric surgery patients are susceptible to many nutritional deficiencies that can lead to similar presentations.3 For example, thiamine (vitamin B1) and cobalamin (vitamin B12) deficiencies cause distinct postbariatric surgery neuropathies.4 Thiamine deficiency may manifest weeks to months after surgery and can rapidly progress, whereas cobalamin deficiency generally develops over 3 to 5 years. Both of these syndromes demonstrate an axonal pattern of nerve injury on EMG/NCS, in contrast to the demyelinating pattern typically seen in GBS. In addition, bariatric surgery patients are at higher risk for copper deficiency, which usually presents as a myeloneuropathy with subacute gait decline and upper motor neuron signs including spasticity.
Although GBS classically presents with symmetric ascending weakness and sensory abnormalities, it may manifest in myriad ways. Factors influencing the presentation include the types of nerve fibers involved (motor, sensory, cranial or autonomic), the predominant mode of injury (axonal vs demyelinating), and the presence or absence of alteration in consciousness.5 The most common form of GBS is AIDP. The classic presentation involves paresthesias in the fingertips and toes followed by lower extremity weakness that ascends over hours to days to involve the arms and potentially the muscles of respiration. A minority of patients with GBS first experience weakness in the upper extremities or facial muscles, and oculomotor involvement is rare.5 Pain is common and often severe.6 Dysautonomia affects most patients with GBS and may manifest as labile blood pressure or arrhythmias.5 Several variant GBS presentation patterns have been described, including acute motor axonal neuropathy, a pure motor form of GBS; ophthalmoplegia, ataxia, and areflexia in Miller Fisher syndrome; and alteration in consciousness, hyperreflexia, ataxia, and ophthalmoparesis in Bickerstaff’s brain stem encephalitis.5
Patients with GBS can progress rapidly to respiratory failure. Serial neurologic exams may signal the diagnosis and inform triage to the appropriate level of care. Measurement of bedside pulmonary function, including mean inspiratory force and functional vital capacity, help to determine if there is weakness of diaphragmatic muscles. Patients with signs or symptoms of diaphragmatic weakness require monitoring in an intensive care unit and potentially early intubation. Treatment with IVIG or plasmapheresis has been found to hasten recovery from GBS, including earlier improvement in muscle strength and a reduced need for mechanical ventilation.7 Treatment selection is based on available resources as both modalities are felt to be equivalent.The majority of patients with GBS make a full recovery over a period of weeks to months, although many have persistent motor weakness. Despite immunotherapy, up to 20% of patients remain severely disabled and approximately 5% die.8 Advanced age, rapid progression of weakness over a period of less than 72 hours, need for mechanical ventilation, and absent compound muscle action potentials on NCS are all associated with prolonged and incomplete recovery.9
This patient developed respiratory failure within 12 hours of hospitalization, prior to being diagnosed with GBS. Even in that short time, the treating clinicians encountered a series of clinical diversions. The initial proximal pattern of muscle weakness suggested a possible myopathic process; the wound infection introduced the possibility of botulism; obesity and recent bariatric surgery triggered concern for thiamine deficiency; and the anion gap acidosis from 5-oxoprolinemia created yet another clinical detour. While the path from presentation to diagnosis is seldom a straight line, when faced with rapidly progressive weakness, it is paramount to forge ahead with an efficient diagnostic evaluation and timely therapeutic intervention.
KEY TEACHING POINTS
- A complaint of general weakness requires distinction between asthenia (ie, general loss of strength or energy) and true neuromuscular weakness from dysfunction of the brain, spinal cord, nerve, neuromuscular junction, and/or muscle.
- Guillain-Barré syndrome may present in a variety of atypical fashions not limited to ascending, distally predominant weakness.
- Acute postgastric reduction surgery neuropathy should be considered in patients presenting with weakness, vomiting, or hyporeflexia after bariatric surgery.
- Acute inflammatory demyelinating polyneuropathy may rapidly progress to respiratory failure, and warrants serial neurologic examinations, monitoring of pulmonary function, and an expedited diagnostic evaluation.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 45-year-old woman presented to the emergency department with 2 days of generalized, progressive weakness. Her ability to walk and perform daily chores was increasingly limited. On the morning of her presentation, she was unable to stand up without falling.
A complaint of weakness must be classified as either functional weakness related to a systemic process or true neurologic weakness from dysfunction of the central nervous system (eg, brain, spinal cord) or peripheral nervous system (eg, anterior horn cell, nerve, neuromuscular junction, or muscle). More information on her clinical course and a detailed neurologic exam will help clarify this key branch point.
She was 2 weeks status-post laparoscopic Roux-en-Y gastric bypass and gastric band removal performed in Europe. Immediately following surgery, she experienced abdominal discomfort and nausea with occasional nonbloody, nonbilious emesis, attributed to expected postoperative anatomical changes. She developed a postoperative pneumonia treated with amoxicillin-clavulanate. She tolerated her flight back to the United States, but her abdominal discomfort persisted and she had minimal oral intake due to her nausea.
Functional weakness may stem from hypovolemia from insufficient oral intake, anemia related to the recent surgery, electrolyte abnormalities, chronic nutritional issues associated with obesity and weight-reduction surgery, and pneumonia. Prolonged air travel, obesity, and recent surgery place her at risk for venous thromboembolism, which may manifest as reduced exercise tolerance. Nausea, vomiting, and abdominal pain persisting for 2 weeks after a Roux-en-Y gastric bypass surgery raises several concerns, including gastric remnant distension (although hiccups are often prominent); stomal stenosis, which typically presents several weeks after surgery; marginal ulceration; or infection at the surgical site or from an anastomotic leak. She may also have a surgery- or medication-related myopathy.
The patient had a history of obesity, hypertension, hyperlipidemia, migraine headaches, and nonalcoholic steatohepatitis. Four years previously, she had undergone gastric banding complicated by band migration and ulceration at the banding site. Her medications were amlodipine, losartan, ranitidine, acetaminophen, and nadroparin for venous thromboembolism prophylaxis during her flight. She denied alcohol, tobacco, or illicit drug use. On further questioning, she reported diaphoresis, mild dyspnea, loose stools, and a sensation of numbness and “heaviness” in her arms. Her abdominal pain was limited to the surgical incision and was controlled with acetaminophen. She denied fevers, cough, chest pain, diplopia, or dysphagia.
Heaviness in both arms could result from an acutely presenting myopathic or neuropathic process, while the coexistence of numbness suggests a sensorimotor polyneuropathy. Obesity and gastric bypass surgery increase her nutritional risk, and thiamine deficiency may present as an acute axonal polyneuropathy (ie, beriberi). Unlike vitamin B12 deficiency, which may take years to develop, thiamine deficiency can present within 4 weeks of gastric bypass surgery. Her dyspnea may be a manifestation of diaphragmatic weakness, although her ostensibly treated pneumonia or as of yet unproven postoperative anemia may be contributing. Chemoprophylaxis mitigates her risk of venous thromboembolism, which is, nonetheless, unlikely to account for the gastrointestinal symptoms and upper extremity weakness. If she is continuing to take amlodipine and losartan but has become volume-depleted, hypotension may be contributing to the generalized weakness.
Physical examination revealed an obese, pale and diaphoretic woman. Her temperature was 36.9°C, heart rate 77 beats per minute, blood pressure 158/90 mm Hg, respiratory rate 28 breaths per minute, and O2 saturation 99% on ambient air. She had no cervical lymphadenopathy and a normal thyroid exam. There were no murmurs on cardiac examination, and jugular venous pressure was estimated at 10 cm of water. Her lung sounds were clear. Her abdomen was soft, nondistended, with localized tenderness and fluctuance around the midline surgical incision with a small amount of purulent drainage. She was alert and oriented to name, date, place, and situation. Cranial nerves II through XII were grossly intact. Strength was 4/5 in bilateral biceps, triceps and distal hand and finger extensors, 3/5 in bilateral deltoids. Strength in hip flexors was 4/5 and it was 5/5 in distal lower extremities. Sensation was intact to pinprick in upper and lower extremities. Biceps reflexes were absent; patellar and ankle reflexes were 1+ and symmetric. The remainder of the physical exam was unremarkable.
The patient has symmetric proximal muscle weakness with upper extremity predominance and preserved strength in her distal lower extremities. A myopathy could explain this pattern of weakness, further substantiated by absent reflexes and reportedly intact sensation. Subacute causes of myopathy include hypokalemia, hyperkalemia, toxic myopathies from medications, or infection-induced rhabdomyolysis. However, she does not report muscle pain, and the loss of reflexes is faster than would be expected with a myopathy. A more thorough sensory examination would inform the assessment of potential neuropathic processes. Guillain-Barré syndrome (GBS) is possible; it most commonly presents as an ascending, distally predominant acute inflammatory demyelinating polyneuropathy (AIDP), although her upper extremity weakness predominates and there are no clear sensory changes. It remains to be determined how her wound infection might relate to her overall presentation.
Her white blood cell count was 12,600/μL (reference range: 3,400-10,000/μL), hemoglobin was 10.2 g/dL, and platelet count was 698,000/μL. Mean corpuscular volume was 86 fL. Serum chemistries were: sodium 138 mEq/L, potassium 3.8 mEq/L, chloride 106 mmol/L, bicarbonate 15 mmol/L, blood urea nitrogen 5 mg/dL, creatinine 0.65 mg/dL, glucose 125 mg/dL, calcium 8.3 mg/dL, magnesium 1.9 mg/dL, phosphorous 2.4 mg/dL, and lactate 1.8 mmol/L (normal: < 2.0 mmol/L). Creatinine kinase (CK), liver function tests, and coagulation panel were normal. Total protein was 6.4 g/dL, and albumin was 2.7 g/dL. Venous blood gas was: pH 7.39 and PCO2 25 mmHg. Urinalysis revealed ketones. Blood and wound cultures were sent for evaluation. A chest x-ray was unremarkable. An electrocardiogram showed normal sinus rhythm. Computed tomography (CT) of the abdomen and pelvis revealed a multiloculated rim-enhancing fluid collection in the anterior abdominal wall (Figure 1).
She does not have any notable electrolyte derangements that would account for her weakness, and the normal creatinine kinase lowers the probability of a myopathy and excludes rhabdomyolysis. Progression of weakness from proximal to distal muscles in a symmetric fashion is consistent with botulism, and she has an intra-abdominal wound infection that could be harboring Clostridium botulinum. Nonetheless, the normal cranial nerve exam and the rarity of botulism occurring with surgical wounds argue against this diagnosis. She should receive intravenous (IV) thiamine for the possibility of beriberi. A lumbar puncture should be performed to assess for albuminocytologic dissociation, which can be seen in patients with GBS.
The patient received high-dose IV thiamine, IV vancomycin, IV piperacillin-tazobactam, and acetaminophen. Over the subsequent 4 hours, her anion gap acidosis worsened. She declined arterial puncture. Repeat venous blood gas was: pH 7.22, PCO2 28 mmHg, and bicarbonate 11 mmol/L. Lactate and glucose were normal. Serum osmolarity was 292 mmol/kg (reference range: 283-301 mmol/kg). She was started on an IV sodium bicarbonate infusion without improvement in her acidemia.
An acute anion gap metabolic acidosis suggests a limited differential diagnosis that includes lactic acidosis, D-lactic acidosis, severe starvation ketoacidosis, acute renal failure, salicylate, or other drug or poison ingestion. Starvation ketoacidosis may be contributing, but a bicarbonate value this low would be unusual. There is no history of alcohol use or other ingestions, and the normal serum osmolality and low osmolal gap (less than 10 mOsm/kg) argue against a poisoning with ethanol, ethylene glycol, or methanol. The initial combined anion gap metabolic acidosis and respiratory alkalosis is consistent with salicylate toxicity, but she does not report aspirin ingestion. Acetaminophen use in the setting of malnutrition or starvation physiology raises the possibility of 5-oxoproline accumulation.
Routine serum lactate does not detect D-lactate, which is produced by colonic bacteria and has been reported in short bowel syndrome and following intestinal bypass surgery. This may occur weeks to months after intestinal procedures, following ingestion of a heavy carbohydrate load, and almost invariably presents with altered mental status and increased anion gap metabolic acidosis, although generalized weakness has been reported.
A surgical consultant drained her wound infection. Fluid Gram stain was negative. D-lactate, salicylate and acetaminophen levels were undetectable. Thiamine pyrophosphate level was 229 nmol/L (reference range: 78-185 nmol/L). Acetaminophen was discontinued and N-acetylcysteine infusion was started for possible 5-oxoprolinemia. Her anion gap acidosis rapidly improved. Twelve hours after admission, she reported sudden onset of blurry vision. Her vital signs were: temperature 37oC, heart rate 110 beats per minute, respiratory rate 40 breaths per minute, blood pressure 168/90, and oxygen saturation 100% on ambient air. Telemetry showed ventricular bigeminy. On examination, she was unable to abduct her right eye; muscle strength was 1/5 in all extremities; biceps, ankle, and patellar reflexes were absent.
Her neurological deficits have progressed over hours to near complete paralysis, asymmetric cranial nerve paresis, and areflexia. Although botulism can cause blurred vision and absent deep tendon reflexes, patients almost always have symmetrical bulbar findings followed by descending paralysis. Should the “numbness” in her arms reported earlier represent undetected sensory deficits, this, too would be inconsistent with botulism.
A diagnosis of GBS ties together several aspects of her presentation and clinical course. Several variants show different patterns of weakness and may involve cranial nerves. Her tachypnea and dyspnea are concerning signs of potential impending respiratory failure. The ventricular bigeminy and mild hypertension could represent autonomic dysfunction that is seen in many cases of GBS.
She was intubated for airway protection. Computed tomography angiography and magnetic resonance imaging of her brain were normal. Cerebral spinal fluid analysis obtained through lumbar puncture showed the following: white blood cell count 3/μL, red blood cell count 11/μL, protein 63 mg/dL (reference range: 15-60mg/dL), and glucose 128 mg/dL (reference range: 40-80mg/dL).
The lumbar puncture is consistent with GBS given the slightly elevated protein and cell count well below 50/μL. Given the severity of her symptoms, treatment with IV immunoglobulin or plasmapheresis should be initiated. Nerve conduction studies (NCS) and electromyography (EMG) are indicated for diagnostic confirmation.
EMG and NCS revealed a severe sensorimotor polyneuropathy with demyelinating features including a conduction block at a noncompressible site, consistent with AIDP. Left sural nerve biopsy confirmed acute demyelinating and mild axonal neuropathy (Figure 2). On hospital day 2, treatment with IV immunoglobulins (IVIG) was initiated; however, she developed anaphylaxis following her second administration and subsequently received plasmapheresis. A tracheostomy was performed for respiratory muscle weakness, and she was discharged to a nursing facility. C. botulinum cultures from the wound eventually returned negative. Following her hospitalization, a serum 5-oxoproline level sent 10 hours after admission returned as elevated, confirming the additional diagnosis of 5-oxoprolinemia. On follow-up, she can sit up and feed herself without assistance, and her gait continues to improve with physical therapy.
DISCUSSION
This patient presented with rapidly progressive weakness that developed in the 2 weeks following bariatric surgery. In the postsurgical setting, patient complaints of weakness are commonly encountered and can pose a diagnostic challenge. Asthenia (ie, general loss of strength or energy) is frequently reported in the immediate postoperative period, and may result from the stress of surgery, pain, deconditioning, or infection. This must be distinguished from true neurologic weakness, which results from dysfunction of the brain, spinal cord, nerve, neuromuscular junction, or muscle. The initial history can help elucidate the inciting events such as preceding surgery, infections or ingestions, and can also categorize the pattern of weakness. The neurologic examination can localize the pathology within the neuraxis. EMG and NCS can distinguish neuropathy from radiculopathy, and categorize the process as axonal, demyelinating, or mixed. In this case, the oculomotor weakness, sensory abnormalities and areflexia signaled a severe sensorimotor polyneuropathy, and EMG/NCS confirmed a demyelinating process consistent with GBS.
Guillain-Barré syndrome is an acute, immune-mediated polyneuropathy. Patients with GBS often present with a preceding respiratory or diarrheal illness; however, the stress of a recent surgery can serve as an inciting event. The syndrome, acute postgastric reduction surgery (APGARS) neuropathy, was introduced in the literature in 2002, describing 3 patients who presented with progressive vomiting, weakness, and hyporeflexia following bariatric surgery.1 The term has been used to describe bariatric surgery patients who developed postoperative quadriparesis, cranial nerve deficits, and respiratory compromise.2 Given the clinical heterogeneity in the literature with relation to APGARS, it is probable that the cases described could result from multiple etiologies. While GBS is purely immune-mediated and can be precipitated by the stress of surgery itself, postbariatric surgery patients are susceptible to many nutritional deficiencies that can lead to similar presentations.3 For example, thiamine (vitamin B1) and cobalamin (vitamin B12) deficiencies cause distinct postbariatric surgery neuropathies.4 Thiamine deficiency may manifest weeks to months after surgery and can rapidly progress, whereas cobalamin deficiency generally develops over 3 to 5 years. Both of these syndromes demonstrate an axonal pattern of nerve injury on EMG/NCS, in contrast to the demyelinating pattern typically seen in GBS. In addition, bariatric surgery patients are at higher risk for copper deficiency, which usually presents as a myeloneuropathy with subacute gait decline and upper motor neuron signs including spasticity.
Although GBS classically presents with symmetric ascending weakness and sensory abnormalities, it may manifest in myriad ways. Factors influencing the presentation include the types of nerve fibers involved (motor, sensory, cranial or autonomic), the predominant mode of injury (axonal vs demyelinating), and the presence or absence of alteration in consciousness.5 The most common form of GBS is AIDP. The classic presentation involves paresthesias in the fingertips and toes followed by lower extremity weakness that ascends over hours to days to involve the arms and potentially the muscles of respiration. A minority of patients with GBS first experience weakness in the upper extremities or facial muscles, and oculomotor involvement is rare.5 Pain is common and often severe.6 Dysautonomia affects most patients with GBS and may manifest as labile blood pressure or arrhythmias.5 Several variant GBS presentation patterns have been described, including acute motor axonal neuropathy, a pure motor form of GBS; ophthalmoplegia, ataxia, and areflexia in Miller Fisher syndrome; and alteration in consciousness, hyperreflexia, ataxia, and ophthalmoparesis in Bickerstaff’s brain stem encephalitis.5
Patients with GBS can progress rapidly to respiratory failure. Serial neurologic exams may signal the diagnosis and inform triage to the appropriate level of care. Measurement of bedside pulmonary function, including mean inspiratory force and functional vital capacity, help to determine if there is weakness of diaphragmatic muscles. Patients with signs or symptoms of diaphragmatic weakness require monitoring in an intensive care unit and potentially early intubation. Treatment with IVIG or plasmapheresis has been found to hasten recovery from GBS, including earlier improvement in muscle strength and a reduced need for mechanical ventilation.7 Treatment selection is based on available resources as both modalities are felt to be equivalent.The majority of patients with GBS make a full recovery over a period of weeks to months, although many have persistent motor weakness. Despite immunotherapy, up to 20% of patients remain severely disabled and approximately 5% die.8 Advanced age, rapid progression of weakness over a period of less than 72 hours, need for mechanical ventilation, and absent compound muscle action potentials on NCS are all associated with prolonged and incomplete recovery.9
This patient developed respiratory failure within 12 hours of hospitalization, prior to being diagnosed with GBS. Even in that short time, the treating clinicians encountered a series of clinical diversions. The initial proximal pattern of muscle weakness suggested a possible myopathic process; the wound infection introduced the possibility of botulism; obesity and recent bariatric surgery triggered concern for thiamine deficiency; and the anion gap acidosis from 5-oxoprolinemia created yet another clinical detour. While the path from presentation to diagnosis is seldom a straight line, when faced with rapidly progressive weakness, it is paramount to forge ahead with an efficient diagnostic evaluation and timely therapeutic intervention.
KEY TEACHING POINTS
- A complaint of general weakness requires distinction between asthenia (ie, general loss of strength or energy) and true neuromuscular weakness from dysfunction of the brain, spinal cord, nerve, neuromuscular junction, and/or muscle.
- Guillain-Barré syndrome may present in a variety of atypical fashions not limited to ascending, distally predominant weakness.
- Acute postgastric reduction surgery neuropathy should be considered in patients presenting with weakness, vomiting, or hyporeflexia after bariatric surgery.
- Acute inflammatory demyelinating polyneuropathy may rapidly progress to respiratory failure, and warrants serial neurologic examinations, monitoring of pulmonary function, and an expedited diagnostic evaluation.
Disclosure
Nothing to report.
1. Akhtar M, Collins MP, Kissel JT. Acute postgastric reduction surgery (APGARS) Neuropathy: A polynutritional, multisystem disorder. Neurology. 2002;58:A68. PubMed
2. Chang CG, Adams-Huet B, Provost DA. Acute post-gastric reduction surgery (APGARS) neuropathy. Obes Surg. 2004;14(2):182-189. PubMed
3. Chang CG, Helling TS, Black WE, Rymer MM. Weakness after gastric bypass. Obes Surg. 2002;12(4):592-597. PubMed
4. Shankar P, Boylan M, Sriram K. Micronutrient deficiencies after bariatric surgery. Nutrition. 2010;26(11-12):1031-1037. PubMed
5. Dimachkie MM, Barohn RJ. Guillain-Barré syndrome and variants. Neurol Clin. 2013;31(2):491-510. PubMed
6. Ruts L, Drenthen J, Jongen JL, et al. Pain in Guillain-Barré syndrome: a long-term follow-up study. Neurology. 2010;75(16):1439-1447. PubMed
7. Hughes RAC, Wijdicks EFM, Barohn R, et al: Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: immunotherapy for Guillain-Barré syndrome: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003;61:736-740. PubMed
8. Hughes RA, Swan AV, Raphaël JC, Annane D, van Koningsveld R, van Doorn PA. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain. 2007;130(Pt 9):2245-2257. PubMed
9. Rajabally YA, Uncini A. Outcome and predictors in Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 2012;83(7):711-718. PubMed
1. Akhtar M, Collins MP, Kissel JT. Acute postgastric reduction surgery (APGARS) Neuropathy: A polynutritional, multisystem disorder. Neurology. 2002;58:A68. PubMed
2. Chang CG, Adams-Huet B, Provost DA. Acute post-gastric reduction surgery (APGARS) neuropathy. Obes Surg. 2004;14(2):182-189. PubMed
3. Chang CG, Helling TS, Black WE, Rymer MM. Weakness after gastric bypass. Obes Surg. 2002;12(4):592-597. PubMed
4. Shankar P, Boylan M, Sriram K. Micronutrient deficiencies after bariatric surgery. Nutrition. 2010;26(11-12):1031-1037. PubMed
5. Dimachkie MM, Barohn RJ. Guillain-Barré syndrome and variants. Neurol Clin. 2013;31(2):491-510. PubMed
6. Ruts L, Drenthen J, Jongen JL, et al. Pain in Guillain-Barré syndrome: a long-term follow-up study. Neurology. 2010;75(16):1439-1447. PubMed
7. Hughes RAC, Wijdicks EFM, Barohn R, et al: Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: immunotherapy for Guillain-Barré syndrome: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003;61:736-740. PubMed
8. Hughes RA, Swan AV, Raphaël JC, Annane D, van Koningsveld R, van Doorn PA. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain. 2007;130(Pt 9):2245-2257. PubMed
9. Rajabally YA, Uncini A. Outcome and predictors in Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 2012;83(7):711-718. PubMed
© 2017 Society of Hospital Medicine