User login
Treating thyroid disorders and depression: 3 case studies
Discuss this article at www.facebook.com/CurrentPsychiatry
Many endocrine disorders can manifest as depression, including relatively rare disorders such as Cushing’s syndrome (hypercortisolism) or Conn’s syndrome (primary hyperaldosteronism) as well as common ones such as diabetes mellitus. Most clinicians do not routinely screen for adrenal disorders when evaluating depressed patients because the yield is low, but do screen for thyroid disease because these disorders often mimic depression. The following 3 cases from my practice illustrate some nuances of screening and treating depressed patients with suspected thyroid abnormalities.
CASE 1: Feeling ‘like an 80-year-old’
Ms. A, age 25, has a gastrointestinal stromal tumor (GIST) and states that she feels “like an 80-year-old woman.” She is sore all over with facial swelling, abdominal cramping, and fatigue. This feeling has worsened since she started chemotherapy with sunitinib for the GIST. Her Patient Health Questionnaire-9 (PHQ-9) score is 14 out of 27, indicating moderate depression. As part of a workup for her depression, what general laboratory tests would be most helpful?
Because Ms. A is of menstruating age, check hemoglobin/hematocrit levels to evaluate for anemia. Monitoring electrolytes would allow you to assess for hypernatremia/hyponatremia, hyperkalemia/hypokalemia, and impaired renal function, all of which could cause depressive symptoms. Depending on Ms. A’s habitus or risk of metabolic syndrome, a fasting blood glucose or hemoglobin A1C test to screen for diabetes mellitus might be valuable because depression may be associated with diabetes.1 A1C is a preferred primary screening test for diabetes (≥6.5% constitutes a positive screen) based on revised clinical practice recommendations of the American Diabetes Association. A1C is available as an office-based test that requires just a drop of blood from a finger prick and does not require a fasting blood sample or a full laboratory analysis.
A popular test for a workup of depression is serum 25-hydroxyvitamin D [25(OH)D] (vitamin D), particularly for patients who live in areas with limited exposure to ultraviolet B radiation from sunlight.2 In a study of older adults, vitamin D levels were 14% lower in patients with minor depression and 14% lower in patients with major depressive disorder compared with controls. This study suggests that depression severity is associated with decreased serum vitamin D levels,3 but the association between depression and vitamin D insufficiency and deficiency is unknown. Checking sex hormones also may be helpful depending on the patient’s symptoms, because testosterone deficiency in men and dehydroepiandrosterone deficiency in women can have a direct impact on a patient’s libido and overall sense of well-being. If repleted, improved levels of sex hormones can lead to a dramatic improvement in mood as well.
Because more than one-half of the estimated 27 million Americans with hyperthyroidism or hypothyroidism are undiagnosed, the American Thyroid Association recommends universal screening for thyroid dysfunction after age 35, with a recheck every 5 years.4 However, checking serum thyroid-stimulating hormone (TSH) levels this often may not be cost-effective. Typically, I do not follow this recommendation when assessing or treating asymptomatic individuals, but Ms. A has symptoms of hypothyroidism (Table 1) and is taking a medication—sunitinib—thought to be associated with hypothyroidism.5 Her serum TSH was very high (110 mIU/L; range 0.28 to 5.00) and her serum free T4 (FT4) was low (0.5 ng/dL; range 0.7 to 1.8). These values were consistent with overt hypothyroidism, defined as low FT4 and elevated TSH levels. This is in contrast to subclinical hypothyroidism (SH), which is defined as having an elevated serum TSH with normal thyroid hormone (T3 and T4) levels. SH presents in 5% of young patients (age <45) and increasingly is being diagnoses in older patients (age >55), who are most likely to suffer adverse effects in mood or cognition.6
Table 1
Hypothyroidism symptoms
| Psychiatric overlap |
| Fatigue |
| Hypersomnolence |
| Cognitive impairment (forgetfulness) |
| Difficulty concentrating or learning |
| Weight gain or fluid retention |
| Somatic signs and symptoms |
| Dry, itchy skin |
| Brittle hair and nails |
| Constipation |
| Myalgias |
| Heavy and/or irregular menstrual cycle |
| Increased rate of miscarriage |
| Sensitivity to cold |
CASE 1 CONTINUED: A classic case
Ms. A is started on a full levothyroxine replacement dose of 1.6 μg/kg/d. For hypothyroid patients who do not have cardiac symptoms, weight-based replacement is thought to be safe and more convenient than starting with a low dose and titrating up.7 Ms. A responds quickly. At 6-week follow-up—the recommended time interval for repeat thyroid lab testing after initiating thyroid replacement—her depressive symptoms are markedly improved and her PHQ-9 score is 6, indicating mild depression.
CASE 2: Chronic pain, low mood, and fatigue
Ms. B, age 62, has fibromyalgia and chronic back pain. She takes cyclobenzaprine, 5 mg 2 to 3 times daily, and oxycodone, 40 mg/d, and describes mild depressive symptoms when she presents for routine follow-up. Most of her complaints are related to chronic pain, but she has a history of low mood and fatigue. She says she was prescribed levothyroxine, but is unable to remember if she stopped taking it because of financial constraints or laboratory/clinical improvement. Her neurologist recently checked her serum TSH, which was elevated at 8.1 mIU/L. Is it best to restart thyroid replacement or wait 6 weeks and recheck her thyroid panel?
Mild SH typically is defined as TSH between 4.5 and 10 mIU/L. In contrast, TSH between 10 and 20 mIU/L is considered severe SH. Because Ms. B did not have prominent new symptoms, I felt it was reasonable to wait the recommended 6 weeks before rechecking her thyroid function. At follow-up, Ms. B’s TSH was 4.64 mIU/L and her FT4 was normal: 0.7 ng/dL. Thyroid replacement was not indicated because she did not have obvious symptoms and treating SH does not impact overall mood and cognition until TSH is ≥10 mIU/L.8,9
CASE 2 CONTINUED: Prominent symptoms emerge
Ms. B returns several months later. Another clinician prescribed duloxetine, titrated from 30 mg to 60 mg, for worsening fibromyalgia. Her depressive symptoms are more prominent at this visit, and her PHQ-9 score has risen from 7 to 14, indicating moderate depression. She says previously she failed or poorly tolerated several antidepressants—fluoxetine, sertraline, and citalopram—but was hoping for a pharmacologic adjustment. Most evidence-based augmentation algorithms for treating major depression start with adding a second “traditional” antidepressant such as bupropion, then move to lithium, second-generation antipsychotics, or lamotrigine.10 But what about thyroid hormone augmentation?
Thyroid hormone often is on the lower rungs of depression treatment algorithms despite Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial data. The data suggest triiodothyronine’s (T3) lower side effect burden and ease of use may offer an advantage over lithium augmentation for depressed patients who have failed several medication trials.11 Liothyronine sodium (triiodothyronine) is a relatively benign medication with potential for augmentation when started at 25 to 50 mcg/d concurrently with antidepressants such as sertraline.12 Unfortunately, most augmentation trials with T3 have been short-term—generally 4 to 8 weeks. In my practice, T3 has limited application; I use it mainly for patients with treatment-resistant depression who have failed several other treatments.
Lithium, the comparison medication to thyroid hormone in the third augmentation arm of the STAR*D trial, requires an annual check of thyroid function (TSH testing) to properly monitor for potential lithium-related hypothyroidism or thyroiditis. Hypothyroidism, for which thyroid replacement is required, with lithium therapy is common, affecting 8% to 27% of patients.13 Patients who rapidly gain weight at the beginning of lithium treatment seem to have a higher risk of developing hypothyroidism.13 However, the risk of developing lithium-induced hypothyroidism is tied to the length of treatment; the longer a patient has been treated with lithium, the greater the risk of developing lithium-induced hypothyroidism.
CASE 3: Unable to slow down
Mr. C, age 45, has a 20-year history of major depression controlled reasonably well with paroxetine, 40 mg. He presents with escalating anxiety, depression, and irritability. His wife is concerned about his overwhelming thoughts of death, especially because Mr. C’s father committed suicide 30 years ago under similar circumstances. Mr. C has been tremulous for the past month and has not been sleeping well. He feels like he is “in constant motion” and unable to slow down. He screens in the “highly likely” range for bipolar disorder on the Bipolar Spectrum Diagnostic Scale14 and is started on divalproex ER, 500 mg/d.
His thyroid function tests returns with a suppressed TSH of 0.03 mIU/L and an elevated FT4 of 3.26 ng/dL. Divalproex is discontinued and he is started on the beta blocker atenolol, 25 mg/d, to target his anxiety, tachycardia, and akathisia. TSH receptor antibody testing was positive, which, along with an abnormal radioactive iodine uptake scan, confirmed a diagnosis of Graves’ disease. He receives methimazole, 20 mg/d, as a temporizing measure. An endocrinologist completes a radioactive iodine (I-131) ablation procedure on Mr. C, which resolves his mood and anxiety symptoms.
Although hypothyroidism commonly is associated with depressive symptoms, hyperthyroidism also may present as depression. Most cases of overt hyperthyroidism are directly referred to an endocrinologist because when treating disorders such as Graves’ disease—the most common cause of hyperthyroidism, especially among women age 20 to 40—many nuclear medicine teams require the expert guidance of an endocrinologist before considering radioiodine ablation. Hyperthyroidism often is accompanied by psychiatric and somatic symptoms of an “overactive” nature (Table 2). However, older patients (age >65) with hyperthyroidism may develop apathetic hyperthyroidism, a subset that comprises approximately 10% to 15% of all hyperthyroidism cases in older adults.15 Rather than becoming nervous, jittery, and restless, patients with apathetic hyperthyroidism are depressed, lethargic, and weak, and may develop proximal myopathy or cardiomyopathy. It is essential to differentiate apathetic hyperthyroidism from typical hyperthyroidism because accurately diagnosing and treating apathetic hyperthyroidism will improve outcomes.15
Table 2
Hyperthyroidism symptoms
| Psychiatric overlap |
| Decrease or increase in appetite |
| Insomnia |
| Fatigue |
| Mood instability |
| Irritability |
| Anxiety, nervousness |
| Somatic signs and symptoms |
| Frequent bowel movement, eg, diarrhea |
| Heart palpitations |
| Heat intolerance |
| Increased sweating |
| Light or missed menstrual periods, fertility problems |
| Muscle weakness |
| Shortness of breath |
| Sudden paralysis |
| Tremor, shakiness, dizziness |
| Vision changes |
| Weight loss or gain |
| Thinning of hair |
| Itching and hives |
| Possible increase in blood sugar |
Using beta blockers to treat hyperthyroidism can help control tachycardia or palpitations, tremulousness, and anxiety that often are inherent in hyperthyroidism. But can beta blockers induce depressive symptoms? A 1-year prospective Dutch study of patients who had survived a myocardial infarction did not find evidence that beta blockers induced depressive symptoms.16 However, the long-term and high-dosage effects of beta blockers still are in question.16 In Mr. C’s case, beta blockers had only positive effects on his symptoms and did not exacerbate his depressive symptoms.
Related Resources
- National Women’s Health Resource Center, Inc. Thyroid disorders. www.healthywomen.org/condition/thyroid-disorders.
- American Thyroid Association. www.thyroid.org.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. www.aace.com/files/hypo-hyper.pdf.
Drug Brand Names
- Atenolol • Tenormin
- Bupropion • Wellbutrin, Zyban
- Citalopram • Celexa
- Cyclobenzaprine • Flexeril
- Divalproex ER • Depakote ER
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Lamotrigine • Lamictal
- Levothyroxine • Levoxyl, Synthroid
- Liothyronine sodium • Cytomel, Triostat
- Lithium • Eskalith, Lithobid
- Methimazole • Tapazole
- Oxycodone • OxyContin
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sunitinib • Sutent
Disclosure
Dr. Raj is a speaker for AstraZeneca and Merck.
1. Campayo A, de Jonge P, Roy JF, et al. Depressive disorder and incident diabetes mellitus: the effect of characteristics of depression. Am J Psychiatry. 2010;167(5):580-588.
2. Gallagher JC, Sai AJ. Vitamin D insufficiency deficiency, and bone health. J Clin Endocrinol Metab. 2010;95(6):2630-2633.
3. Hoogendijk WJ, Lips P, Dik MG, et al. Depression is associated with decreased 25-hydroxyvitamin D and increased parathyroid hormone levels in older adults. Arch Gen Psychiatry. 2008;65(5):508-512.
4. Ladenson PW, Singer PA, Ain KB, et al. American Thyroid Association guidelines for detection of thyroid dysfunction. Arch Intern Med. 2000;160(11):1573-1575.
5. Wolter P, Dumez H, Schöffski P. Sunitinib and hypothyroidism. N Engl J Med. 2007;356(15):1580; author reply 1580-1581.
6. Biondi B, Cooper DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev. 2008;29(1):76-131.
7. Roos A, Linn-Rasker SP, van Domburg RT, et al. The starting dose of levothyroxine in primary hypothyroidism treatment: a prospective, randomized, double-blind trial. Arch Intern Med. 2005;165(15):1714-1720.
8. Raj YP. Subclinical hypothyroidism: merely monitor or time to treat? Current Psychiatry. 2009;8(2):47-48.
9. Samuels MH. Cognitive function in subclinical hypothyroidism. J Clin Endocrinol Metab. 2010;95(8):3611-3613.
10. Mann JJ. The medical management of depression. N Engl J Med. 2005;353(17):1819-1834.
11. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530; quiz 1665.
12. Cooper-Kazaz R, Apter JT, Cohen R, et al. Combined treatment with sertraline and liothyronine in major depression: a randomized, double-blind, placebo-controlled trial. Arch Gen Psychiatry. 2007;64(6):679-688.
13. Henry C. Lithium side-effects and predictors of hypothyroidism in patients with bipolar disorder: sex differences. J Psychiatry Neurosci. 2002;27(2):104-107.
14. Ghaemi N, Pies R. The Bipolar Spectrum Diagnostic Scale. http://www.psycheducation.org/depression/BSDS.htm. Published October 2002. Updated June 2003. Accessed October 1 2012.
15. Wu W, Sun Z, Yu J, et al. A clinical retrospective analysis of factors associated with apathetic hyperthyroidism. Pathobiology. 2010;77(1):46-51.
16. van Melle JP, Verbeek DE, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
Discuss this article at www.facebook.com/CurrentPsychiatry
Many endocrine disorders can manifest as depression, including relatively rare disorders such as Cushing’s syndrome (hypercortisolism) or Conn’s syndrome (primary hyperaldosteronism) as well as common ones such as diabetes mellitus. Most clinicians do not routinely screen for adrenal disorders when evaluating depressed patients because the yield is low, but do screen for thyroid disease because these disorders often mimic depression. The following 3 cases from my practice illustrate some nuances of screening and treating depressed patients with suspected thyroid abnormalities.
CASE 1: Feeling ‘like an 80-year-old’
Ms. A, age 25, has a gastrointestinal stromal tumor (GIST) and states that she feels “like an 80-year-old woman.” She is sore all over with facial swelling, abdominal cramping, and fatigue. This feeling has worsened since she started chemotherapy with sunitinib for the GIST. Her Patient Health Questionnaire-9 (PHQ-9) score is 14 out of 27, indicating moderate depression. As part of a workup for her depression, what general laboratory tests would be most helpful?
Because Ms. A is of menstruating age, check hemoglobin/hematocrit levels to evaluate for anemia. Monitoring electrolytes would allow you to assess for hypernatremia/hyponatremia, hyperkalemia/hypokalemia, and impaired renal function, all of which could cause depressive symptoms. Depending on Ms. A’s habitus or risk of metabolic syndrome, a fasting blood glucose or hemoglobin A1C test to screen for diabetes mellitus might be valuable because depression may be associated with diabetes.1 A1C is a preferred primary screening test for diabetes (≥6.5% constitutes a positive screen) based on revised clinical practice recommendations of the American Diabetes Association. A1C is available as an office-based test that requires just a drop of blood from a finger prick and does not require a fasting blood sample or a full laboratory analysis.
A popular test for a workup of depression is serum 25-hydroxyvitamin D [25(OH)D] (vitamin D), particularly for patients who live in areas with limited exposure to ultraviolet B radiation from sunlight.2 In a study of older adults, vitamin D levels were 14% lower in patients with minor depression and 14% lower in patients with major depressive disorder compared with controls. This study suggests that depression severity is associated with decreased serum vitamin D levels,3 but the association between depression and vitamin D insufficiency and deficiency is unknown. Checking sex hormones also may be helpful depending on the patient’s symptoms, because testosterone deficiency in men and dehydroepiandrosterone deficiency in women can have a direct impact on a patient’s libido and overall sense of well-being. If repleted, improved levels of sex hormones can lead to a dramatic improvement in mood as well.
Because more than one-half of the estimated 27 million Americans with hyperthyroidism or hypothyroidism are undiagnosed, the American Thyroid Association recommends universal screening for thyroid dysfunction after age 35, with a recheck every 5 years.4 However, checking serum thyroid-stimulating hormone (TSH) levels this often may not be cost-effective. Typically, I do not follow this recommendation when assessing or treating asymptomatic individuals, but Ms. A has symptoms of hypothyroidism (Table 1) and is taking a medication—sunitinib—thought to be associated with hypothyroidism.5 Her serum TSH was very high (110 mIU/L; range 0.28 to 5.00) and her serum free T4 (FT4) was low (0.5 ng/dL; range 0.7 to 1.8). These values were consistent with overt hypothyroidism, defined as low FT4 and elevated TSH levels. This is in contrast to subclinical hypothyroidism (SH), which is defined as having an elevated serum TSH with normal thyroid hormone (T3 and T4) levels. SH presents in 5% of young patients (age <45) and increasingly is being diagnoses in older patients (age >55), who are most likely to suffer adverse effects in mood or cognition.6
Table 1
Hypothyroidism symptoms
| Psychiatric overlap |
| Fatigue |
| Hypersomnolence |
| Cognitive impairment (forgetfulness) |
| Difficulty concentrating or learning |
| Weight gain or fluid retention |
| Somatic signs and symptoms |
| Dry, itchy skin |
| Brittle hair and nails |
| Constipation |
| Myalgias |
| Heavy and/or irregular menstrual cycle |
| Increased rate of miscarriage |
| Sensitivity to cold |
CASE 1 CONTINUED: A classic case
Ms. A is started on a full levothyroxine replacement dose of 1.6 μg/kg/d. For hypothyroid patients who do not have cardiac symptoms, weight-based replacement is thought to be safe and more convenient than starting with a low dose and titrating up.7 Ms. A responds quickly. At 6-week follow-up—the recommended time interval for repeat thyroid lab testing after initiating thyroid replacement—her depressive symptoms are markedly improved and her PHQ-9 score is 6, indicating mild depression.
CASE 2: Chronic pain, low mood, and fatigue
Ms. B, age 62, has fibromyalgia and chronic back pain. She takes cyclobenzaprine, 5 mg 2 to 3 times daily, and oxycodone, 40 mg/d, and describes mild depressive symptoms when she presents for routine follow-up. Most of her complaints are related to chronic pain, but she has a history of low mood and fatigue. She says she was prescribed levothyroxine, but is unable to remember if she stopped taking it because of financial constraints or laboratory/clinical improvement. Her neurologist recently checked her serum TSH, which was elevated at 8.1 mIU/L. Is it best to restart thyroid replacement or wait 6 weeks and recheck her thyroid panel?
Mild SH typically is defined as TSH between 4.5 and 10 mIU/L. In contrast, TSH between 10 and 20 mIU/L is considered severe SH. Because Ms. B did not have prominent new symptoms, I felt it was reasonable to wait the recommended 6 weeks before rechecking her thyroid function. At follow-up, Ms. B’s TSH was 4.64 mIU/L and her FT4 was normal: 0.7 ng/dL. Thyroid replacement was not indicated because she did not have obvious symptoms and treating SH does not impact overall mood and cognition until TSH is ≥10 mIU/L.8,9
CASE 2 CONTINUED: Prominent symptoms emerge
Ms. B returns several months later. Another clinician prescribed duloxetine, titrated from 30 mg to 60 mg, for worsening fibromyalgia. Her depressive symptoms are more prominent at this visit, and her PHQ-9 score has risen from 7 to 14, indicating moderate depression. She says previously she failed or poorly tolerated several antidepressants—fluoxetine, sertraline, and citalopram—but was hoping for a pharmacologic adjustment. Most evidence-based augmentation algorithms for treating major depression start with adding a second “traditional” antidepressant such as bupropion, then move to lithium, second-generation antipsychotics, or lamotrigine.10 But what about thyroid hormone augmentation?
Thyroid hormone often is on the lower rungs of depression treatment algorithms despite Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial data. The data suggest triiodothyronine’s (T3) lower side effect burden and ease of use may offer an advantage over lithium augmentation for depressed patients who have failed several medication trials.11 Liothyronine sodium (triiodothyronine) is a relatively benign medication with potential for augmentation when started at 25 to 50 mcg/d concurrently with antidepressants such as sertraline.12 Unfortunately, most augmentation trials with T3 have been short-term—generally 4 to 8 weeks. In my practice, T3 has limited application; I use it mainly for patients with treatment-resistant depression who have failed several other treatments.
Lithium, the comparison medication to thyroid hormone in the third augmentation arm of the STAR*D trial, requires an annual check of thyroid function (TSH testing) to properly monitor for potential lithium-related hypothyroidism or thyroiditis. Hypothyroidism, for which thyroid replacement is required, with lithium therapy is common, affecting 8% to 27% of patients.13 Patients who rapidly gain weight at the beginning of lithium treatment seem to have a higher risk of developing hypothyroidism.13 However, the risk of developing lithium-induced hypothyroidism is tied to the length of treatment; the longer a patient has been treated with lithium, the greater the risk of developing lithium-induced hypothyroidism.
CASE 3: Unable to slow down
Mr. C, age 45, has a 20-year history of major depression controlled reasonably well with paroxetine, 40 mg. He presents with escalating anxiety, depression, and irritability. His wife is concerned about his overwhelming thoughts of death, especially because Mr. C’s father committed suicide 30 years ago under similar circumstances. Mr. C has been tremulous for the past month and has not been sleeping well. He feels like he is “in constant motion” and unable to slow down. He screens in the “highly likely” range for bipolar disorder on the Bipolar Spectrum Diagnostic Scale14 and is started on divalproex ER, 500 mg/d.
His thyroid function tests returns with a suppressed TSH of 0.03 mIU/L and an elevated FT4 of 3.26 ng/dL. Divalproex is discontinued and he is started on the beta blocker atenolol, 25 mg/d, to target his anxiety, tachycardia, and akathisia. TSH receptor antibody testing was positive, which, along with an abnormal radioactive iodine uptake scan, confirmed a diagnosis of Graves’ disease. He receives methimazole, 20 mg/d, as a temporizing measure. An endocrinologist completes a radioactive iodine (I-131) ablation procedure on Mr. C, which resolves his mood and anxiety symptoms.
Although hypothyroidism commonly is associated with depressive symptoms, hyperthyroidism also may present as depression. Most cases of overt hyperthyroidism are directly referred to an endocrinologist because when treating disorders such as Graves’ disease—the most common cause of hyperthyroidism, especially among women age 20 to 40—many nuclear medicine teams require the expert guidance of an endocrinologist before considering radioiodine ablation. Hyperthyroidism often is accompanied by psychiatric and somatic symptoms of an “overactive” nature (Table 2). However, older patients (age >65) with hyperthyroidism may develop apathetic hyperthyroidism, a subset that comprises approximately 10% to 15% of all hyperthyroidism cases in older adults.15 Rather than becoming nervous, jittery, and restless, patients with apathetic hyperthyroidism are depressed, lethargic, and weak, and may develop proximal myopathy or cardiomyopathy. It is essential to differentiate apathetic hyperthyroidism from typical hyperthyroidism because accurately diagnosing and treating apathetic hyperthyroidism will improve outcomes.15
Table 2
Hyperthyroidism symptoms
| Psychiatric overlap |
| Decrease or increase in appetite |
| Insomnia |
| Fatigue |
| Mood instability |
| Irritability |
| Anxiety, nervousness |
| Somatic signs and symptoms |
| Frequent bowel movement, eg, diarrhea |
| Heart palpitations |
| Heat intolerance |
| Increased sweating |
| Light or missed menstrual periods, fertility problems |
| Muscle weakness |
| Shortness of breath |
| Sudden paralysis |
| Tremor, shakiness, dizziness |
| Vision changes |
| Weight loss or gain |
| Thinning of hair |
| Itching and hives |
| Possible increase in blood sugar |
Using beta blockers to treat hyperthyroidism can help control tachycardia or palpitations, tremulousness, and anxiety that often are inherent in hyperthyroidism. But can beta blockers induce depressive symptoms? A 1-year prospective Dutch study of patients who had survived a myocardial infarction did not find evidence that beta blockers induced depressive symptoms.16 However, the long-term and high-dosage effects of beta blockers still are in question.16 In Mr. C’s case, beta blockers had only positive effects on his symptoms and did not exacerbate his depressive symptoms.
Related Resources
- National Women’s Health Resource Center, Inc. Thyroid disorders. www.healthywomen.org/condition/thyroid-disorders.
- American Thyroid Association. www.thyroid.org.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. www.aace.com/files/hypo-hyper.pdf.
Drug Brand Names
- Atenolol • Tenormin
- Bupropion • Wellbutrin, Zyban
- Citalopram • Celexa
- Cyclobenzaprine • Flexeril
- Divalproex ER • Depakote ER
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Lamotrigine • Lamictal
- Levothyroxine • Levoxyl, Synthroid
- Liothyronine sodium • Cytomel, Triostat
- Lithium • Eskalith, Lithobid
- Methimazole • Tapazole
- Oxycodone • OxyContin
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sunitinib • Sutent
Disclosure
Dr. Raj is a speaker for AstraZeneca and Merck.
Discuss this article at www.facebook.com/CurrentPsychiatry
Many endocrine disorders can manifest as depression, including relatively rare disorders such as Cushing’s syndrome (hypercortisolism) or Conn’s syndrome (primary hyperaldosteronism) as well as common ones such as diabetes mellitus. Most clinicians do not routinely screen for adrenal disorders when evaluating depressed patients because the yield is low, but do screen for thyroid disease because these disorders often mimic depression. The following 3 cases from my practice illustrate some nuances of screening and treating depressed patients with suspected thyroid abnormalities.
CASE 1: Feeling ‘like an 80-year-old’
Ms. A, age 25, has a gastrointestinal stromal tumor (GIST) and states that she feels “like an 80-year-old woman.” She is sore all over with facial swelling, abdominal cramping, and fatigue. This feeling has worsened since she started chemotherapy with sunitinib for the GIST. Her Patient Health Questionnaire-9 (PHQ-9) score is 14 out of 27, indicating moderate depression. As part of a workup for her depression, what general laboratory tests would be most helpful?
Because Ms. A is of menstruating age, check hemoglobin/hematocrit levels to evaluate for anemia. Monitoring electrolytes would allow you to assess for hypernatremia/hyponatremia, hyperkalemia/hypokalemia, and impaired renal function, all of which could cause depressive symptoms. Depending on Ms. A’s habitus or risk of metabolic syndrome, a fasting blood glucose or hemoglobin A1C test to screen for diabetes mellitus might be valuable because depression may be associated with diabetes.1 A1C is a preferred primary screening test for diabetes (≥6.5% constitutes a positive screen) based on revised clinical practice recommendations of the American Diabetes Association. A1C is available as an office-based test that requires just a drop of blood from a finger prick and does not require a fasting blood sample or a full laboratory analysis.
A popular test for a workup of depression is serum 25-hydroxyvitamin D [25(OH)D] (vitamin D), particularly for patients who live in areas with limited exposure to ultraviolet B radiation from sunlight.2 In a study of older adults, vitamin D levels were 14% lower in patients with minor depression and 14% lower in patients with major depressive disorder compared with controls. This study suggests that depression severity is associated with decreased serum vitamin D levels,3 but the association between depression and vitamin D insufficiency and deficiency is unknown. Checking sex hormones also may be helpful depending on the patient’s symptoms, because testosterone deficiency in men and dehydroepiandrosterone deficiency in women can have a direct impact on a patient’s libido and overall sense of well-being. If repleted, improved levels of sex hormones can lead to a dramatic improvement in mood as well.
Because more than one-half of the estimated 27 million Americans with hyperthyroidism or hypothyroidism are undiagnosed, the American Thyroid Association recommends universal screening for thyroid dysfunction after age 35, with a recheck every 5 years.4 However, checking serum thyroid-stimulating hormone (TSH) levels this often may not be cost-effective. Typically, I do not follow this recommendation when assessing or treating asymptomatic individuals, but Ms. A has symptoms of hypothyroidism (Table 1) and is taking a medication—sunitinib—thought to be associated with hypothyroidism.5 Her serum TSH was very high (110 mIU/L; range 0.28 to 5.00) and her serum free T4 (FT4) was low (0.5 ng/dL; range 0.7 to 1.8). These values were consistent with overt hypothyroidism, defined as low FT4 and elevated TSH levels. This is in contrast to subclinical hypothyroidism (SH), which is defined as having an elevated serum TSH with normal thyroid hormone (T3 and T4) levels. SH presents in 5% of young patients (age <45) and increasingly is being diagnoses in older patients (age >55), who are most likely to suffer adverse effects in mood or cognition.6
Table 1
Hypothyroidism symptoms
| Psychiatric overlap |
| Fatigue |
| Hypersomnolence |
| Cognitive impairment (forgetfulness) |
| Difficulty concentrating or learning |
| Weight gain or fluid retention |
| Somatic signs and symptoms |
| Dry, itchy skin |
| Brittle hair and nails |
| Constipation |
| Myalgias |
| Heavy and/or irregular menstrual cycle |
| Increased rate of miscarriage |
| Sensitivity to cold |
CASE 1 CONTINUED: A classic case
Ms. A is started on a full levothyroxine replacement dose of 1.6 μg/kg/d. For hypothyroid patients who do not have cardiac symptoms, weight-based replacement is thought to be safe and more convenient than starting with a low dose and titrating up.7 Ms. A responds quickly. At 6-week follow-up—the recommended time interval for repeat thyroid lab testing after initiating thyroid replacement—her depressive symptoms are markedly improved and her PHQ-9 score is 6, indicating mild depression.
CASE 2: Chronic pain, low mood, and fatigue
Ms. B, age 62, has fibromyalgia and chronic back pain. She takes cyclobenzaprine, 5 mg 2 to 3 times daily, and oxycodone, 40 mg/d, and describes mild depressive symptoms when she presents for routine follow-up. Most of her complaints are related to chronic pain, but she has a history of low mood and fatigue. She says she was prescribed levothyroxine, but is unable to remember if she stopped taking it because of financial constraints or laboratory/clinical improvement. Her neurologist recently checked her serum TSH, which was elevated at 8.1 mIU/L. Is it best to restart thyroid replacement or wait 6 weeks and recheck her thyroid panel?
Mild SH typically is defined as TSH between 4.5 and 10 mIU/L. In contrast, TSH between 10 and 20 mIU/L is considered severe SH. Because Ms. B did not have prominent new symptoms, I felt it was reasonable to wait the recommended 6 weeks before rechecking her thyroid function. At follow-up, Ms. B’s TSH was 4.64 mIU/L and her FT4 was normal: 0.7 ng/dL. Thyroid replacement was not indicated because she did not have obvious symptoms and treating SH does not impact overall mood and cognition until TSH is ≥10 mIU/L.8,9
CASE 2 CONTINUED: Prominent symptoms emerge
Ms. B returns several months later. Another clinician prescribed duloxetine, titrated from 30 mg to 60 mg, for worsening fibromyalgia. Her depressive symptoms are more prominent at this visit, and her PHQ-9 score has risen from 7 to 14, indicating moderate depression. She says previously she failed or poorly tolerated several antidepressants—fluoxetine, sertraline, and citalopram—but was hoping for a pharmacologic adjustment. Most evidence-based augmentation algorithms for treating major depression start with adding a second “traditional” antidepressant such as bupropion, then move to lithium, second-generation antipsychotics, or lamotrigine.10 But what about thyroid hormone augmentation?
Thyroid hormone often is on the lower rungs of depression treatment algorithms despite Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial data. The data suggest triiodothyronine’s (T3) lower side effect burden and ease of use may offer an advantage over lithium augmentation for depressed patients who have failed several medication trials.11 Liothyronine sodium (triiodothyronine) is a relatively benign medication with potential for augmentation when started at 25 to 50 mcg/d concurrently with antidepressants such as sertraline.12 Unfortunately, most augmentation trials with T3 have been short-term—generally 4 to 8 weeks. In my practice, T3 has limited application; I use it mainly for patients with treatment-resistant depression who have failed several other treatments.
Lithium, the comparison medication to thyroid hormone in the third augmentation arm of the STAR*D trial, requires an annual check of thyroid function (TSH testing) to properly monitor for potential lithium-related hypothyroidism or thyroiditis. Hypothyroidism, for which thyroid replacement is required, with lithium therapy is common, affecting 8% to 27% of patients.13 Patients who rapidly gain weight at the beginning of lithium treatment seem to have a higher risk of developing hypothyroidism.13 However, the risk of developing lithium-induced hypothyroidism is tied to the length of treatment; the longer a patient has been treated with lithium, the greater the risk of developing lithium-induced hypothyroidism.
CASE 3: Unable to slow down
Mr. C, age 45, has a 20-year history of major depression controlled reasonably well with paroxetine, 40 mg. He presents with escalating anxiety, depression, and irritability. His wife is concerned about his overwhelming thoughts of death, especially because Mr. C’s father committed suicide 30 years ago under similar circumstances. Mr. C has been tremulous for the past month and has not been sleeping well. He feels like he is “in constant motion” and unable to slow down. He screens in the “highly likely” range for bipolar disorder on the Bipolar Spectrum Diagnostic Scale14 and is started on divalproex ER, 500 mg/d.
His thyroid function tests returns with a suppressed TSH of 0.03 mIU/L and an elevated FT4 of 3.26 ng/dL. Divalproex is discontinued and he is started on the beta blocker atenolol, 25 mg/d, to target his anxiety, tachycardia, and akathisia. TSH receptor antibody testing was positive, which, along with an abnormal radioactive iodine uptake scan, confirmed a diagnosis of Graves’ disease. He receives methimazole, 20 mg/d, as a temporizing measure. An endocrinologist completes a radioactive iodine (I-131) ablation procedure on Mr. C, which resolves his mood and anxiety symptoms.
Although hypothyroidism commonly is associated with depressive symptoms, hyperthyroidism also may present as depression. Most cases of overt hyperthyroidism are directly referred to an endocrinologist because when treating disorders such as Graves’ disease—the most common cause of hyperthyroidism, especially among women age 20 to 40—many nuclear medicine teams require the expert guidance of an endocrinologist before considering radioiodine ablation. Hyperthyroidism often is accompanied by psychiatric and somatic symptoms of an “overactive” nature (Table 2). However, older patients (age >65) with hyperthyroidism may develop apathetic hyperthyroidism, a subset that comprises approximately 10% to 15% of all hyperthyroidism cases in older adults.15 Rather than becoming nervous, jittery, and restless, patients with apathetic hyperthyroidism are depressed, lethargic, and weak, and may develop proximal myopathy or cardiomyopathy. It is essential to differentiate apathetic hyperthyroidism from typical hyperthyroidism because accurately diagnosing and treating apathetic hyperthyroidism will improve outcomes.15
Table 2
Hyperthyroidism symptoms
| Psychiatric overlap |
| Decrease or increase in appetite |
| Insomnia |
| Fatigue |
| Mood instability |
| Irritability |
| Anxiety, nervousness |
| Somatic signs and symptoms |
| Frequent bowel movement, eg, diarrhea |
| Heart palpitations |
| Heat intolerance |
| Increased sweating |
| Light or missed menstrual periods, fertility problems |
| Muscle weakness |
| Shortness of breath |
| Sudden paralysis |
| Tremor, shakiness, dizziness |
| Vision changes |
| Weight loss or gain |
| Thinning of hair |
| Itching and hives |
| Possible increase in blood sugar |
Using beta blockers to treat hyperthyroidism can help control tachycardia or palpitations, tremulousness, and anxiety that often are inherent in hyperthyroidism. But can beta blockers induce depressive symptoms? A 1-year prospective Dutch study of patients who had survived a myocardial infarction did not find evidence that beta blockers induced depressive symptoms.16 However, the long-term and high-dosage effects of beta blockers still are in question.16 In Mr. C’s case, beta blockers had only positive effects on his symptoms and did not exacerbate his depressive symptoms.
Related Resources
- National Women’s Health Resource Center, Inc. Thyroid disorders. www.healthywomen.org/condition/thyroid-disorders.
- American Thyroid Association. www.thyroid.org.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. www.aace.com/files/hypo-hyper.pdf.
Drug Brand Names
- Atenolol • Tenormin
- Bupropion • Wellbutrin, Zyban
- Citalopram • Celexa
- Cyclobenzaprine • Flexeril
- Divalproex ER • Depakote ER
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Lamotrigine • Lamictal
- Levothyroxine • Levoxyl, Synthroid
- Liothyronine sodium • Cytomel, Triostat
- Lithium • Eskalith, Lithobid
- Methimazole • Tapazole
- Oxycodone • OxyContin
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sunitinib • Sutent
Disclosure
Dr. Raj is a speaker for AstraZeneca and Merck.
1. Campayo A, de Jonge P, Roy JF, et al. Depressive disorder and incident diabetes mellitus: the effect of characteristics of depression. Am J Psychiatry. 2010;167(5):580-588.
2. Gallagher JC, Sai AJ. Vitamin D insufficiency deficiency, and bone health. J Clin Endocrinol Metab. 2010;95(6):2630-2633.
3. Hoogendijk WJ, Lips P, Dik MG, et al. Depression is associated with decreased 25-hydroxyvitamin D and increased parathyroid hormone levels in older adults. Arch Gen Psychiatry. 2008;65(5):508-512.
4. Ladenson PW, Singer PA, Ain KB, et al. American Thyroid Association guidelines for detection of thyroid dysfunction. Arch Intern Med. 2000;160(11):1573-1575.
5. Wolter P, Dumez H, Schöffski P. Sunitinib and hypothyroidism. N Engl J Med. 2007;356(15):1580; author reply 1580-1581.
6. Biondi B, Cooper DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev. 2008;29(1):76-131.
7. Roos A, Linn-Rasker SP, van Domburg RT, et al. The starting dose of levothyroxine in primary hypothyroidism treatment: a prospective, randomized, double-blind trial. Arch Intern Med. 2005;165(15):1714-1720.
8. Raj YP. Subclinical hypothyroidism: merely monitor or time to treat? Current Psychiatry. 2009;8(2):47-48.
9. Samuels MH. Cognitive function in subclinical hypothyroidism. J Clin Endocrinol Metab. 2010;95(8):3611-3613.
10. Mann JJ. The medical management of depression. N Engl J Med. 2005;353(17):1819-1834.
11. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530; quiz 1665.
12. Cooper-Kazaz R, Apter JT, Cohen R, et al. Combined treatment with sertraline and liothyronine in major depression: a randomized, double-blind, placebo-controlled trial. Arch Gen Psychiatry. 2007;64(6):679-688.
13. Henry C. Lithium side-effects and predictors of hypothyroidism in patients with bipolar disorder: sex differences. J Psychiatry Neurosci. 2002;27(2):104-107.
14. Ghaemi N, Pies R. The Bipolar Spectrum Diagnostic Scale. http://www.psycheducation.org/depression/BSDS.htm. Published October 2002. Updated June 2003. Accessed October 1 2012.
15. Wu W, Sun Z, Yu J, et al. A clinical retrospective analysis of factors associated with apathetic hyperthyroidism. Pathobiology. 2010;77(1):46-51.
16. van Melle JP, Verbeek DE, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
1. Campayo A, de Jonge P, Roy JF, et al. Depressive disorder and incident diabetes mellitus: the effect of characteristics of depression. Am J Psychiatry. 2010;167(5):580-588.
2. Gallagher JC, Sai AJ. Vitamin D insufficiency deficiency, and bone health. J Clin Endocrinol Metab. 2010;95(6):2630-2633.
3. Hoogendijk WJ, Lips P, Dik MG, et al. Depression is associated with decreased 25-hydroxyvitamin D and increased parathyroid hormone levels in older adults. Arch Gen Psychiatry. 2008;65(5):508-512.
4. Ladenson PW, Singer PA, Ain KB, et al. American Thyroid Association guidelines for detection of thyroid dysfunction. Arch Intern Med. 2000;160(11):1573-1575.
5. Wolter P, Dumez H, Schöffski P. Sunitinib and hypothyroidism. N Engl J Med. 2007;356(15):1580; author reply 1580-1581.
6. Biondi B, Cooper DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev. 2008;29(1):76-131.
7. Roos A, Linn-Rasker SP, van Domburg RT, et al. The starting dose of levothyroxine in primary hypothyroidism treatment: a prospective, randomized, double-blind trial. Arch Intern Med. 2005;165(15):1714-1720.
8. Raj YP. Subclinical hypothyroidism: merely monitor or time to treat? Current Psychiatry. 2009;8(2):47-48.
9. Samuels MH. Cognitive function in subclinical hypothyroidism. J Clin Endocrinol Metab. 2010;95(8):3611-3613.
10. Mann JJ. The medical management of depression. N Engl J Med. 2005;353(17):1819-1834.
11. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530; quiz 1665.
12. Cooper-Kazaz R, Apter JT, Cohen R, et al. Combined treatment with sertraline and liothyronine in major depression: a randomized, double-blind, placebo-controlled trial. Arch Gen Psychiatry. 2007;64(6):679-688.
13. Henry C. Lithium side-effects and predictors of hypothyroidism in patients with bipolar disorder: sex differences. J Psychiatry Neurosci. 2002;27(2):104-107.
14. Ghaemi N, Pies R. The Bipolar Spectrum Diagnostic Scale. http://www.psycheducation.org/depression/BSDS.htm. Published October 2002. Updated June 2003. Accessed October 1 2012.
15. Wu W, Sun Z, Yu J, et al. A clinical retrospective analysis of factors associated with apathetic hyperthyroidism. Pathobiology. 2010;77(1):46-51.
16. van Melle JP, Verbeek DE, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
How to target psychiatric symptoms of Huntington’s disease
Discuss this article at www.facebook.com/CurrentPsychiatry
Psychiatric symptoms are a common and debilitating manifestation of Huntington’s disease (HD), a progressive, inherited neurodegenerative disorder also characterized by chorea (involuntary, nonrepetitive movements) and cognitive decline. The prevalence of HD is 4 to 8 patients per 100,000 persons in most populations of European descent, with lower prevalence among non-Europeans.1 HD is caused by an abnormal expansion of a trinucleotide (CAG) repeat sequence on chromosome 4, and is inherited in an autosomal dominant fashion, meaning a HD patient’s child has a 50% chance of inheriting the mutation. The expansion is located in the gene that encodes the “huntingtin” protein, the normal function of which is not well understood.
There’s no cure for HD, and treatments primarily are directed at symptom control. Psychiatric symptoms include depression, apathy, anxiety, and psychosis (Table).2-4 Treating patients with HD can be challenging because most psychiatrists will see only a handful of patients with this multifaceted illness during their careers. See Box 1 for a case study of a patient with HD.
Table
Psychiatric symptoms of HD
| Anxiety |
| Apathy |
| Delusions |
| Disinhibitions, impulsivity, aggressive behavior |
| Dysphoria |
| Euphoria |
| Hallucinations |
| Irritability |
| Obsessions and compulsions |
| HD: Huntington’s disease Source: References 2-4 |
Mr. M, age 50, was diagnosed with Huntington’s disease (HD) 1 year ago. He returns to our psychiatric clinic for treatment of depressive symptoms and temper. Previously, he was prescribed citalopram, 40 mg/d; eventually low-dose olanzapine, 2.5 mg at night, was added. Mr. M reported better temper control, but his low mood, irritability, hopelessness, and amotivation were not significantly improved.
Mr. M left his job at a software company because he had difficulty completing tasks as the result of mood and cognitive changes. He wants to return to work, but feels that he would be unable to complete his job duties.
He begins a trial of bupropion, 150 mg/d, to improve the vegetative component of his mood symptoms to help him return to work. Mr. M now complains of worsening chorea, irritability, and insomnia, with continued difficulty completing tasks. He is intermittently tearful throughout the interview.
Mr. M continues to struggle with mood symptoms that likely are related to the stressful experience of declining function and the intrinsic evolution of HD. His chorea worsens on bupropion; this agent is discontinued and replaced with mirtazapine, 15 mg at night, for his depressive symptoms and insomnia. Citalopram and olanzapine are unchanged. Mr. M is advised to follow up with our HD psychiatry team in 1 month, and is referred for brief psychotherapy. We remind him—as we do for all of our HD patients—to call the HD clinic or 911 if he becomes suicidal. Ongoing treatment efforts likely will be complex, given the multifaceted and progressive nature of his disease.
Psychiatric sequelae
In general, psychiatric symptoms of HD become increasingly prevalent over time (Box 2).3,5 In a 2001 study of 52 HD patients by Paulsen et al,2 51 patients had ≥1 psychiatric symptom, such as dysphoria (69.2%), agitation (67.3%), irritability (65.4%), apathy (55.8%), and anxiety (51.9%); delusions (11.5%) and hallucinations (1.9%) were less prevalent.2 Similarly, Thompson et al3 followed 111 HD patients for ≥3 years and all experienced psychiatric symptoms.
According to Thompson et al,3 the presence and severity of apathy, irritability, and depression trend differently across the course of Huntington’s disease (HD). Apathy worsens with disease progression, closely following cognitive and motor symptoms. Irritability increases significantly, but this effect seems confined to early stages of HD. Depressive symptoms appear to decline slightly as HD advances, although it is unclear if this is because of antidepressants’ effects, increasing emotional blunting, and waning insight in later stages of HD, or another unknown factor.3 This study did not examine psychotic symptoms over time because few patients were experiencing delusions or hallucinations.
Similar to Thompson et al, Naarding et al5 found that apathy and depression in HD follow distinct time courses. Depression is a feature of early HD and apathy worsens with overall disease progression.
Depressed mood and functional ability—not cognitive or motor symptoms6—are the 2 most critical factors linked to health-related quality of life in HD. Hamilton et al7 found that apathy or executive dysfunction in HD patients is strongly related to decline in ability to complete activities of daily living, and may be severely debilitating.
Apathy. Often mistaken for a symptom of depression, apathy’s presentation may resemble anhedonia or fatigue; however, research suggests that depression and apathy are distinct conditions. Naarding et al
5 noted that apathy is more common than depressive symptoms in HD patients and may be a hallmark symptom of HD.
Depression affects most HD patients, and often is most severe early in the disease course. Hubers et al8 found that 20% of 100 HD patients had suicidal ideation. The strongest predictor was depressed mood.
Sleep disturbances and daytime somnolence are common among HD patients, and patients with comorbid depression report more disturbed sleep. Managing disturbed sleep and daytime somnolence in HD, with emphasis on comorbid depression, may improve the quality of life of patients and their caregivers.9
Anxiety was present in >50% of HD patients in a study by Paulsen et al2 and 37% evaluated by Craufurd et al.10 Craufurd et al10 also reported that 61% of patients were “physically tense and unable to relax.”
Among HD patients, 5% report obsessions and 10% report compulsive behaviors; these symptoms appear to become increasingly common as HD progresses.4,10
Impulsivity and disinhibition. Craufurd et al10 found that 71% of HD patients experienced poor judgment and self-monitoring, 40% had poor temper control and verbal outbursts, 22% exhibited threatening behavior or violence, and 6% had disinhibited or inappropriate sexual behavior.10
Recent studies have shown higher rates of disinhibition in “presymptomatic” gene-positive subjects vs gene-negative controls, suggesting that these symptoms may arise early in HD.11 Further, researchers demonstrated that patients lack symptom awareness and rate themselves as less impaired than their caregivers do.11
In our clinical experience, impulsivity frequently is encountered and creates significant conflict between patients and their caregivers. We speculate that when coupled with depressive symptoms of HD, impulsivity and disinhibition may play an important role in the high rates of suicidality seen in these patients.
Psychosis. Delusions and hallucinations are less common in HD than other psychiatric symptoms. Craufurd et al10 reported 3% of HD patients had delusions, 3% had auditory hallucinations, 2% had tactile hallucinations, and no patients had visual hallucinations.
A few case reports and a small study by Tsuang et al12 suggested that psychotic features in HD may be similar to those seen in paranoid schizophrenia. Tsuang et al12 also noted that more severe HD-related psychosis tends to cluster in families, which suggests that susceptibility to HD psychosis may be heritable.
Treating psychiatric symptoms
High-quality randomized controlled trials of pharmacotherapies for psychiatric symptoms in HD patients are lacking. Decisions regarding which agents to use often are based on case reports or clinical experience. The suggestions below are based on available evidence and our clinical experience.
Depression. Depressive symptoms in HD seem to respond to conventional pharmacologic treatments for major depressive disorder (MDD). A small trial of venlafaxine extended-release (XR) in 26 HD patients with MDD showed statistically significant improvements in depressive symptoms; however, this trial was not blinded and did not have a placebo group.13 In addition, 1 in 5 patients developed significant side effects—nausea, irritability, or worsening chorea.13
Evidence for selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors, and tricyclic antidepressants (TCAs) is lacking. Antidepressant choice should be based on patient response, side effect profile, and the need for secondary therapeutic effects.14
We often prescribe sertraline, citalopram, or escitalopram for our HD patients because of the relative absence of drug-drug interactions and favorable safety profile in medically and surgically ill patients. However, it’s important to tailor the treatment approach to your patient’s needs—eg, patients prone to forgetting their medicine may benefit from a drug with a longer half-life, such as fluoxetine. We avoid TCAs because of their anticholinergic effects, which may worsen dementia symptoms. Because HD patients have high rates of suicidality, agents that are highly toxic when taken in overdose should be used with caution.
One small study of HD patients with MDD or bipolar disorder showed clinical improvement in depressive symptoms after electroconvulsive therapy (ECT).15 Patients who suffered from comorbid delusions had the best improvements in mood.15 ECT likely is a good choice for HD patients who have failed several antidepressants, are suicidal, or who have depression with psychotic features.16
Apathy. A 2011 review concluded that no evidence-based recommendations regarding pharmacologic treatment for apathy in HD can be made because of lack of research.7 The Huntington’s Disease Society of America’s (HDSA) A Physician’s Guide to Managing Huntington’s Disease includes recommendations for treating apathy based on clinical experience.16 It suggests a nonsedating SSRI, followed by a trial of methylphenidate, pemoline, or dextroamphetamine if SSRIs were unsuccessful.
16 The HDSA guide notes psychostimulants may worsen irritability in HD and have a high potential for abuse. ECT appears to have little effect on apathy.15
Anxiety. A small, open-label study of 11 patients found that olanzapine, 5 mg/d, significantly improved depression, anxiety, irritability, and obsessive behavior in HD patients.17
The HDSA guide suggests treating anxiety and obsessive-compulsive symptoms as you would in patients without HD. For anxiety, SSRIs and possibly a short-term trial of a low-dose benzodiazepine (ie, lorazepam, clonazepam) are suggested.16 Benzodiazepines may increase the risk of falls and delirium in this population. Anecdotally, buspirone is helpful in some patients, with a starting dose of 5 mg 2 to 3 times per day and increased to 20 to 30 mg/d in divided doses.16 For obsessive-compulsive symptoms, SSRIs are recommended; atypical antipsychotics are reserved for severe or refractory symptoms.16
Disinhibition and impulsivity. There’s no research on treating disinhibition and impulsivity in HD. In our clinical experience, atypical antipsychotics are the most helpful. Factors regarding choosing an agent and dosing levels are similar to those for psychotic symptoms.
Psychotic symptoms. Most studies of typical and atypical antipsychotics for HD psychosis have shown beneficial effects.14,16-21 Neurologists frequently use these agents for managing chorea. Both neurologic and psychiatric features of the patient’s presentation must be considered when selecting a drug because treatment directed at 1 component of the disease may inadvertently exacerbate another. Specifically, higher potency antipsychotics (eg, haloperidol) are effective for chorea but can dramatically worsen bradykinesia; lower potency agents (eg, quetiapine) are less helpful for chorea but do not significantly worsen rigidity symptoms.
Olanzapine has been shown to improve chorea, anxiety, irritability, depression, sleep dysfunction, and weight loss in addition to psychotic symptoms.14,17 We find that olanzapine treats a constellation of symptoms common among HD patients, and we prescribe it frequently. Because olanzapine is considered a mid-potency agent, we find it’s best suited for concurrent control of psychotic symptoms and mild to moderate chorea in patients with minimal bradykinesia. Start olanzapine at 2.5 mg/d and gradually increase to 5 to 10 mg/d as tolerated.14
Risperidone is effective for treating psychosis and chorea. It can be started at 0.5 to 1 mg/d, and gradually increased to 6 to 8 mg/d.14 The depot formulation of risperidone has been shown to be effective in HD, which may help patients adhere to their medication.18 Risperidone is a mid-high potency antipsychotic, and in our experience is best used to control psychotic symptoms in patients with moderate chorea and few or no symptoms of bradykinesia or rigidity.
Quetiapine reduces psychotic symptoms, agitation, irritability, and insomnia without worsening bradykinesia or rigidity,19 but it is not beneficial for chorea. It can be started at 12.5 mg/d and gradually increased for effect as tolerated, up to 600 mg/d (depending on indication), in 2 or 3 divided doses.14
Haloperidol is a high-potency typical antipsychotic and may help psychotic patients with severe chorea; it should not be used in patients with bradykinesia. Start haloperidol at 0.5 to 1 mg/d and gradually increase to 6 to 8 mg/d as tolerated.14 Because of higher likelihood of side effects with typical antipsychotics, we often reserve its use for patients whose psychosis does not respond to atypical agents.
Other antipsychotics. Aripiprazole in HD has been examined in only 2 single- patient case reports20,21; the drug appeared to reduce psychosis and possibly chorea. Clozapine’s effectiveness for HD psychosis is not well known. It does not appear to be helpful for chorea and can cause agranulocytosis.22
Because one of the hallmarks of HD is dementia, it is worth noting that the FDA has issued a “black-box” warning on the use of antipsychotic drugs in patients with dementia because of concerns regarding increased mortality. However, drawing specific conclusions is difficult because the FDA warning is based on studies that looked primarily at Alzheimer’s disease and vascular dementia, not HD.
Other pharmacotherapies
Tetrabenazine is the only FDA-approved drug for treating HD. However, it carries a “black-box” warning for increased risk of depression and suicidal ideation and is contraindicated in suicidal patients and those with untreated or inadequately treated depression.
Although several small trials have had conflicting results regarding its benefit, amantadine sometimes is used to treat chorea.23-25 For more information about tetrabenazine and amantadine, see Box 3.
Tetrabenazine, the only FDA-approved drug for treating Huntington’s disease (HD), is a dopamine-depleting agent given to control chorea. In a 12-week, randomized, double-blind, placebo-controlled clinical trial, tetrabenazine was shown to be effective in HD patients.a Treatment with tetrabenazine results in symptomatic improvement of chorea, but does not slow or alter the course of the disease. Tetrabenazine can provide relief from choreiform movements, but these benefits should be balanced with the risks of depression and suicidality.a Tetrabenazine is known to prolong QTc interval, and should be used with caution in combination with other drugs that have the potential to do the same (eg, antipsychotics).a
Several case reports have found an association between tetrabenazine and development of neuroleptic malignant syndrome (NMS).b-d Be aware of the clinical characteristics of NMS—mental status change, rigidity, fever, and dysautonomia—and use caution when starting patients taking tetrabenazine on antipsychotics or other agents known to cause NMS.
Amantadine also has been used to treat chorea in HD patients who are unable to tolerate tetrabenazine or antipsychotics. Our neurologists sometimes have found it to be beneficial in patients with juvenile-onset HD because these patients often have debilitating dystonia. Be aware that amantadine is known to precipitate or worsen psychosis.e
References
- Food and Drug Administration. NDA 21-894 Xenazine® (tetrabenazine). Risk evaluation and mitigation strategy (REMS). Click here. Published August 15, 2008. Updated April 2011. Accessed June 20, 2012.
- Stevens E, Roman A, Houa M, et al. Severe hyperthermia during tetrabenazine therapy for tardive dyskinesia. Intensive Care Med. 1998;24(4):369-371.
- Petzinger GM, Bressman SB. A case of tetrabenazine-induced neuroleptic malignant syndrome after prolonged treatment. Mov Disord. 1997;12(2):246-248.
- Ossemann M, Sindic CJ, Laterre C. Tetrabenazine as a cause of neuroleptic malignant syndrome. Mov Disord. 1996;11(1):95.
- Wolters EC. Dopaminomimetic psychosis in Parkinson’s disease patients: diagnosis and treatment. Neurology. 1999;52 (7 suppl 3):S10-S13.
Related Resources
- Huntington’s Disease Society of America. www.hdsa.org.
- Family Caregiver Alliance. Huntington’s disease. www.caregiver.org/caregiver/jsp/content_node.jsp?nodeid=574.
- Huntington Study Group. www.huntington-study-group.org.
- Huntington’s Disease Advocacy Center. www.hdac.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Wellbutrin XL, others
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
- Methylphenidate • Concerta, Ritalin, others
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Pemoline • Cylert
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tetrabenazine • Xenazine
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Scher is a consultant to the advisory board for Lundbeck.
Ms. Kocsis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Harper PS. The epidemiology of Huntington’s disease. Hum Genet. 1992;89(4):365-376.
2. Paulsen JS, Ready RE, Hamilton JM, et al. Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2001;71(3):310-314.
3. Thompson JC, Harris J, Sollom AC, et al. Longitudinal evaluation of neuropsychiatric symptoms in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2012;24(1):53-60.
4. Beglinger LJ, Langbehn DR, Duff K, et al. Probability of obsessive and compulsive symptoms in Huntington’s disease. Biol Psychiatry. 2007;61(3):415-418.
5. Naarding P, Janzing JG, Eling P, et al. Apathy is not depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2009;21(3):266-270.
6. Ho AK, Gilbert AS, Mason SL, et al. Health-related quality of life in Huntington’s disease: which factors matter most? Mov Disord. 2009;24(4):574-578.
7. Hamilton JM, Salmon DP, Corey-Bloom J, et al. Behavioural abnormalities contribute to functional decline in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2003;74(1):120-122.
8. Hubers AA, Reedeker N, Giltay EJ, et al. Suicidality in Huntington’s disease. J Affect Disord. 2012;136(3):550-557.
9. Videnovic A, Leurgans S, Fan W, et al. Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat Disord. 2009;15(6):471-474.
10. Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(4):219-226.
11. Duff K, Paulsen JS, Beglinger LJ, et al. “Frontal” behaviors before the diagnosis of Huntington’s disease and their relationship to markers of disease progression: evidence of early lack of awareness. J Neuropsychiatry Clin Neurosci. 2010;22(2):196-207.
12. Tsuang D, Almqvist EW, Lipe H, et al. Familial aggregation of psychotic symptoms in Huntington’s disease. Am J Psychiatry. 2000;157(12):1955-1959.
13. Holl AK, Wilkinson L, Painold A, et al. Combating depression in Huntington’s disease: effective antidepressive treatment with venlafaxine XR. Int Clin Psychopharmacol. 2010;25(1):46-50.
14. Killoran A, Biglan KM. Therapeutics in Huntington’s disease. Curr Treat Options Neurol. 2012;14(2):137-149.
15. Ranen NG, Peyser CE, Folstein SE. ECT as a treatment for depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 1994;6(2):154-159.
16. Rosenblatt A, Ranen NG, Nance MA, et al. A physician’s guide to the management of Huntington’s disease. 2nd edition. http://www.hdsa.org/images/content/1/1/11289.pdf. Published 1999. Accessed July 27, 2012.
17. Squitieri F, Cannella M, Piorcellini A, et al. Short-term effects of olanzapine in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(1):69-72.
18. Johnston TG. Risperidone long-acting injection and Huntington’s disease: case series with significant psychiatric and behavioural symptoms. Int Clin Psychopharmacol. 2011;26(2):114-119.
19. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
20. Lin WC, Chou YH. Aripiprazole effects on psychosis and chorea in a patient with Huntington’s disease. Am J Psychiatry. 2008;165(9):1207-1208.
21. Oulis P, Mourikis I, Konstantakopoulos G, et al. Aripiprazole in the treatment of olanzapine-resistant psychotic and motor symptoms of Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2010;22(3):352c.e4-352c.e5.
22. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomised comparative study. J Neurol Neurosurg Psychiatry. 1997;63(1):35-39.
23. Verhagen Metman L, Morris MJ, Farmer C, et al. Huntington’s disease: a randomized, controlled trial using the NMDA-antagonist amantadine. Neurology. 2002;59(5):694-699.
24. Lucetti C, Del Dotto P, Gambaccini G, et al. IV amantadine improves chorea in Huntington’s disease: an acute randomized, controlled study. Neurology. 2003;60(12):1995-1997.
25. O’Suilleabhain P, Dewey RB, Jr. A randomized trial of amantadine in Huntington disease. Arch Neurol. 2003;60(7):996-998.
Discuss this article at www.facebook.com/CurrentPsychiatry
Psychiatric symptoms are a common and debilitating manifestation of Huntington’s disease (HD), a progressive, inherited neurodegenerative disorder also characterized by chorea (involuntary, nonrepetitive movements) and cognitive decline. The prevalence of HD is 4 to 8 patients per 100,000 persons in most populations of European descent, with lower prevalence among non-Europeans.1 HD is caused by an abnormal expansion of a trinucleotide (CAG) repeat sequence on chromosome 4, and is inherited in an autosomal dominant fashion, meaning a HD patient’s child has a 50% chance of inheriting the mutation. The expansion is located in the gene that encodes the “huntingtin” protein, the normal function of which is not well understood.
There’s no cure for HD, and treatments primarily are directed at symptom control. Psychiatric symptoms include depression, apathy, anxiety, and psychosis (Table).2-4 Treating patients with HD can be challenging because most psychiatrists will see only a handful of patients with this multifaceted illness during their careers. See Box 1 for a case study of a patient with HD.
Table
Psychiatric symptoms of HD
| Anxiety |
| Apathy |
| Delusions |
| Disinhibitions, impulsivity, aggressive behavior |
| Dysphoria |
| Euphoria |
| Hallucinations |
| Irritability |
| Obsessions and compulsions |
| HD: Huntington’s disease Source: References 2-4 |
Mr. M, age 50, was diagnosed with Huntington’s disease (HD) 1 year ago. He returns to our psychiatric clinic for treatment of depressive symptoms and temper. Previously, he was prescribed citalopram, 40 mg/d; eventually low-dose olanzapine, 2.5 mg at night, was added. Mr. M reported better temper control, but his low mood, irritability, hopelessness, and amotivation were not significantly improved.
Mr. M left his job at a software company because he had difficulty completing tasks as the result of mood and cognitive changes. He wants to return to work, but feels that he would be unable to complete his job duties.
He begins a trial of bupropion, 150 mg/d, to improve the vegetative component of his mood symptoms to help him return to work. Mr. M now complains of worsening chorea, irritability, and insomnia, with continued difficulty completing tasks. He is intermittently tearful throughout the interview.
Mr. M continues to struggle with mood symptoms that likely are related to the stressful experience of declining function and the intrinsic evolution of HD. His chorea worsens on bupropion; this agent is discontinued and replaced with mirtazapine, 15 mg at night, for his depressive symptoms and insomnia. Citalopram and olanzapine are unchanged. Mr. M is advised to follow up with our HD psychiatry team in 1 month, and is referred for brief psychotherapy. We remind him—as we do for all of our HD patients—to call the HD clinic or 911 if he becomes suicidal. Ongoing treatment efforts likely will be complex, given the multifaceted and progressive nature of his disease.
Psychiatric sequelae
In general, psychiatric symptoms of HD become increasingly prevalent over time (Box 2).3,5 In a 2001 study of 52 HD patients by Paulsen et al,2 51 patients had ≥1 psychiatric symptom, such as dysphoria (69.2%), agitation (67.3%), irritability (65.4%), apathy (55.8%), and anxiety (51.9%); delusions (11.5%) and hallucinations (1.9%) were less prevalent.2 Similarly, Thompson et al3 followed 111 HD patients for ≥3 years and all experienced psychiatric symptoms.
According to Thompson et al,3 the presence and severity of apathy, irritability, and depression trend differently across the course of Huntington’s disease (HD). Apathy worsens with disease progression, closely following cognitive and motor symptoms. Irritability increases significantly, but this effect seems confined to early stages of HD. Depressive symptoms appear to decline slightly as HD advances, although it is unclear if this is because of antidepressants’ effects, increasing emotional blunting, and waning insight in later stages of HD, or another unknown factor.3 This study did not examine psychotic symptoms over time because few patients were experiencing delusions or hallucinations.
Similar to Thompson et al, Naarding et al5 found that apathy and depression in HD follow distinct time courses. Depression is a feature of early HD and apathy worsens with overall disease progression.
Depressed mood and functional ability—not cognitive or motor symptoms6—are the 2 most critical factors linked to health-related quality of life in HD. Hamilton et al7 found that apathy or executive dysfunction in HD patients is strongly related to decline in ability to complete activities of daily living, and may be severely debilitating.
Apathy. Often mistaken for a symptom of depression, apathy’s presentation may resemble anhedonia or fatigue; however, research suggests that depression and apathy are distinct conditions. Naarding et al
5 noted that apathy is more common than depressive symptoms in HD patients and may be a hallmark symptom of HD.
Depression affects most HD patients, and often is most severe early in the disease course. Hubers et al8 found that 20% of 100 HD patients had suicidal ideation. The strongest predictor was depressed mood.
Sleep disturbances and daytime somnolence are common among HD patients, and patients with comorbid depression report more disturbed sleep. Managing disturbed sleep and daytime somnolence in HD, with emphasis on comorbid depression, may improve the quality of life of patients and their caregivers.9
Anxiety was present in >50% of HD patients in a study by Paulsen et al2 and 37% evaluated by Craufurd et al.10 Craufurd et al10 also reported that 61% of patients were “physically tense and unable to relax.”
Among HD patients, 5% report obsessions and 10% report compulsive behaviors; these symptoms appear to become increasingly common as HD progresses.4,10
Impulsivity and disinhibition. Craufurd et al10 found that 71% of HD patients experienced poor judgment and self-monitoring, 40% had poor temper control and verbal outbursts, 22% exhibited threatening behavior or violence, and 6% had disinhibited or inappropriate sexual behavior.10
Recent studies have shown higher rates of disinhibition in “presymptomatic” gene-positive subjects vs gene-negative controls, suggesting that these symptoms may arise early in HD.11 Further, researchers demonstrated that patients lack symptom awareness and rate themselves as less impaired than their caregivers do.11
In our clinical experience, impulsivity frequently is encountered and creates significant conflict between patients and their caregivers. We speculate that when coupled with depressive symptoms of HD, impulsivity and disinhibition may play an important role in the high rates of suicidality seen in these patients.
Psychosis. Delusions and hallucinations are less common in HD than other psychiatric symptoms. Craufurd et al10 reported 3% of HD patients had delusions, 3% had auditory hallucinations, 2% had tactile hallucinations, and no patients had visual hallucinations.
A few case reports and a small study by Tsuang et al12 suggested that psychotic features in HD may be similar to those seen in paranoid schizophrenia. Tsuang et al12 also noted that more severe HD-related psychosis tends to cluster in families, which suggests that susceptibility to HD psychosis may be heritable.
Treating psychiatric symptoms
High-quality randomized controlled trials of pharmacotherapies for psychiatric symptoms in HD patients are lacking. Decisions regarding which agents to use often are based on case reports or clinical experience. The suggestions below are based on available evidence and our clinical experience.
Depression. Depressive symptoms in HD seem to respond to conventional pharmacologic treatments for major depressive disorder (MDD). A small trial of venlafaxine extended-release (XR) in 26 HD patients with MDD showed statistically significant improvements in depressive symptoms; however, this trial was not blinded and did not have a placebo group.13 In addition, 1 in 5 patients developed significant side effects—nausea, irritability, or worsening chorea.13
Evidence for selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors, and tricyclic antidepressants (TCAs) is lacking. Antidepressant choice should be based on patient response, side effect profile, and the need for secondary therapeutic effects.14
We often prescribe sertraline, citalopram, or escitalopram for our HD patients because of the relative absence of drug-drug interactions and favorable safety profile in medically and surgically ill patients. However, it’s important to tailor the treatment approach to your patient’s needs—eg, patients prone to forgetting their medicine may benefit from a drug with a longer half-life, such as fluoxetine. We avoid TCAs because of their anticholinergic effects, which may worsen dementia symptoms. Because HD patients have high rates of suicidality, agents that are highly toxic when taken in overdose should be used with caution.
One small study of HD patients with MDD or bipolar disorder showed clinical improvement in depressive symptoms after electroconvulsive therapy (ECT).15 Patients who suffered from comorbid delusions had the best improvements in mood.15 ECT likely is a good choice for HD patients who have failed several antidepressants, are suicidal, or who have depression with psychotic features.16
Apathy. A 2011 review concluded that no evidence-based recommendations regarding pharmacologic treatment for apathy in HD can be made because of lack of research.7 The Huntington’s Disease Society of America’s (HDSA) A Physician’s Guide to Managing Huntington’s Disease includes recommendations for treating apathy based on clinical experience.16 It suggests a nonsedating SSRI, followed by a trial of methylphenidate, pemoline, or dextroamphetamine if SSRIs were unsuccessful.
16 The HDSA guide notes psychostimulants may worsen irritability in HD and have a high potential for abuse. ECT appears to have little effect on apathy.15
Anxiety. A small, open-label study of 11 patients found that olanzapine, 5 mg/d, significantly improved depression, anxiety, irritability, and obsessive behavior in HD patients.17
The HDSA guide suggests treating anxiety and obsessive-compulsive symptoms as you would in patients without HD. For anxiety, SSRIs and possibly a short-term trial of a low-dose benzodiazepine (ie, lorazepam, clonazepam) are suggested.16 Benzodiazepines may increase the risk of falls and delirium in this population. Anecdotally, buspirone is helpful in some patients, with a starting dose of 5 mg 2 to 3 times per day and increased to 20 to 30 mg/d in divided doses.16 For obsessive-compulsive symptoms, SSRIs are recommended; atypical antipsychotics are reserved for severe or refractory symptoms.16
Disinhibition and impulsivity. There’s no research on treating disinhibition and impulsivity in HD. In our clinical experience, atypical antipsychotics are the most helpful. Factors regarding choosing an agent and dosing levels are similar to those for psychotic symptoms.
Psychotic symptoms. Most studies of typical and atypical antipsychotics for HD psychosis have shown beneficial effects.14,16-21 Neurologists frequently use these agents for managing chorea. Both neurologic and psychiatric features of the patient’s presentation must be considered when selecting a drug because treatment directed at 1 component of the disease may inadvertently exacerbate another. Specifically, higher potency antipsychotics (eg, haloperidol) are effective for chorea but can dramatically worsen bradykinesia; lower potency agents (eg, quetiapine) are less helpful for chorea but do not significantly worsen rigidity symptoms.
Olanzapine has been shown to improve chorea, anxiety, irritability, depression, sleep dysfunction, and weight loss in addition to psychotic symptoms.14,17 We find that olanzapine treats a constellation of symptoms common among HD patients, and we prescribe it frequently. Because olanzapine is considered a mid-potency agent, we find it’s best suited for concurrent control of psychotic symptoms and mild to moderate chorea in patients with minimal bradykinesia. Start olanzapine at 2.5 mg/d and gradually increase to 5 to 10 mg/d as tolerated.14
Risperidone is effective for treating psychosis and chorea. It can be started at 0.5 to 1 mg/d, and gradually increased to 6 to 8 mg/d.14 The depot formulation of risperidone has been shown to be effective in HD, which may help patients adhere to their medication.18 Risperidone is a mid-high potency antipsychotic, and in our experience is best used to control psychotic symptoms in patients with moderate chorea and few or no symptoms of bradykinesia or rigidity.
Quetiapine reduces psychotic symptoms, agitation, irritability, and insomnia without worsening bradykinesia or rigidity,19 but it is not beneficial for chorea. It can be started at 12.5 mg/d and gradually increased for effect as tolerated, up to 600 mg/d (depending on indication), in 2 or 3 divided doses.14
Haloperidol is a high-potency typical antipsychotic and may help psychotic patients with severe chorea; it should not be used in patients with bradykinesia. Start haloperidol at 0.5 to 1 mg/d and gradually increase to 6 to 8 mg/d as tolerated.14 Because of higher likelihood of side effects with typical antipsychotics, we often reserve its use for patients whose psychosis does not respond to atypical agents.
Other antipsychotics. Aripiprazole in HD has been examined in only 2 single- patient case reports20,21; the drug appeared to reduce psychosis and possibly chorea. Clozapine’s effectiveness for HD psychosis is not well known. It does not appear to be helpful for chorea and can cause agranulocytosis.22
Because one of the hallmarks of HD is dementia, it is worth noting that the FDA has issued a “black-box” warning on the use of antipsychotic drugs in patients with dementia because of concerns regarding increased mortality. However, drawing specific conclusions is difficult because the FDA warning is based on studies that looked primarily at Alzheimer’s disease and vascular dementia, not HD.
Other pharmacotherapies
Tetrabenazine is the only FDA-approved drug for treating HD. However, it carries a “black-box” warning for increased risk of depression and suicidal ideation and is contraindicated in suicidal patients and those with untreated or inadequately treated depression.
Although several small trials have had conflicting results regarding its benefit, amantadine sometimes is used to treat chorea.23-25 For more information about tetrabenazine and amantadine, see Box 3.
Tetrabenazine, the only FDA-approved drug for treating Huntington’s disease (HD), is a dopamine-depleting agent given to control chorea. In a 12-week, randomized, double-blind, placebo-controlled clinical trial, tetrabenazine was shown to be effective in HD patients.a Treatment with tetrabenazine results in symptomatic improvement of chorea, but does not slow or alter the course of the disease. Tetrabenazine can provide relief from choreiform movements, but these benefits should be balanced with the risks of depression and suicidality.a Tetrabenazine is known to prolong QTc interval, and should be used with caution in combination with other drugs that have the potential to do the same (eg, antipsychotics).a
Several case reports have found an association between tetrabenazine and development of neuroleptic malignant syndrome (NMS).b-d Be aware of the clinical characteristics of NMS—mental status change, rigidity, fever, and dysautonomia—and use caution when starting patients taking tetrabenazine on antipsychotics or other agents known to cause NMS.
Amantadine also has been used to treat chorea in HD patients who are unable to tolerate tetrabenazine or antipsychotics. Our neurologists sometimes have found it to be beneficial in patients with juvenile-onset HD because these patients often have debilitating dystonia. Be aware that amantadine is known to precipitate or worsen psychosis.e
References
- Food and Drug Administration. NDA 21-894 Xenazine® (tetrabenazine). Risk evaluation and mitigation strategy (REMS). Click here. Published August 15, 2008. Updated April 2011. Accessed June 20, 2012.
- Stevens E, Roman A, Houa M, et al. Severe hyperthermia during tetrabenazine therapy for tardive dyskinesia. Intensive Care Med. 1998;24(4):369-371.
- Petzinger GM, Bressman SB. A case of tetrabenazine-induced neuroleptic malignant syndrome after prolonged treatment. Mov Disord. 1997;12(2):246-248.
- Ossemann M, Sindic CJ, Laterre C. Tetrabenazine as a cause of neuroleptic malignant syndrome. Mov Disord. 1996;11(1):95.
- Wolters EC. Dopaminomimetic psychosis in Parkinson’s disease patients: diagnosis and treatment. Neurology. 1999;52 (7 suppl 3):S10-S13.
Related Resources
- Huntington’s Disease Society of America. www.hdsa.org.
- Family Caregiver Alliance. Huntington’s disease. www.caregiver.org/caregiver/jsp/content_node.jsp?nodeid=574.
- Huntington Study Group. www.huntington-study-group.org.
- Huntington’s Disease Advocacy Center. www.hdac.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Wellbutrin XL, others
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
- Methylphenidate • Concerta, Ritalin, others
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Pemoline • Cylert
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tetrabenazine • Xenazine
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Scher is a consultant to the advisory board for Lundbeck.
Ms. Kocsis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Psychiatric symptoms are a common and debilitating manifestation of Huntington’s disease (HD), a progressive, inherited neurodegenerative disorder also characterized by chorea (involuntary, nonrepetitive movements) and cognitive decline. The prevalence of HD is 4 to 8 patients per 100,000 persons in most populations of European descent, with lower prevalence among non-Europeans.1 HD is caused by an abnormal expansion of a trinucleotide (CAG) repeat sequence on chromosome 4, and is inherited in an autosomal dominant fashion, meaning a HD patient’s child has a 50% chance of inheriting the mutation. The expansion is located in the gene that encodes the “huntingtin” protein, the normal function of which is not well understood.
There’s no cure for HD, and treatments primarily are directed at symptom control. Psychiatric symptoms include depression, apathy, anxiety, and psychosis (Table).2-4 Treating patients with HD can be challenging because most psychiatrists will see only a handful of patients with this multifaceted illness during their careers. See Box 1 for a case study of a patient with HD.
Table
Psychiatric symptoms of HD
| Anxiety |
| Apathy |
| Delusions |
| Disinhibitions, impulsivity, aggressive behavior |
| Dysphoria |
| Euphoria |
| Hallucinations |
| Irritability |
| Obsessions and compulsions |
| HD: Huntington’s disease Source: References 2-4 |
Mr. M, age 50, was diagnosed with Huntington’s disease (HD) 1 year ago. He returns to our psychiatric clinic for treatment of depressive symptoms and temper. Previously, he was prescribed citalopram, 40 mg/d; eventually low-dose olanzapine, 2.5 mg at night, was added. Mr. M reported better temper control, but his low mood, irritability, hopelessness, and amotivation were not significantly improved.
Mr. M left his job at a software company because he had difficulty completing tasks as the result of mood and cognitive changes. He wants to return to work, but feels that he would be unable to complete his job duties.
He begins a trial of bupropion, 150 mg/d, to improve the vegetative component of his mood symptoms to help him return to work. Mr. M now complains of worsening chorea, irritability, and insomnia, with continued difficulty completing tasks. He is intermittently tearful throughout the interview.
Mr. M continues to struggle with mood symptoms that likely are related to the stressful experience of declining function and the intrinsic evolution of HD. His chorea worsens on bupropion; this agent is discontinued and replaced with mirtazapine, 15 mg at night, for his depressive symptoms and insomnia. Citalopram and olanzapine are unchanged. Mr. M is advised to follow up with our HD psychiatry team in 1 month, and is referred for brief psychotherapy. We remind him—as we do for all of our HD patients—to call the HD clinic or 911 if he becomes suicidal. Ongoing treatment efforts likely will be complex, given the multifaceted and progressive nature of his disease.
Psychiatric sequelae
In general, psychiatric symptoms of HD become increasingly prevalent over time (Box 2).3,5 In a 2001 study of 52 HD patients by Paulsen et al,2 51 patients had ≥1 psychiatric symptom, such as dysphoria (69.2%), agitation (67.3%), irritability (65.4%), apathy (55.8%), and anxiety (51.9%); delusions (11.5%) and hallucinations (1.9%) were less prevalent.2 Similarly, Thompson et al3 followed 111 HD patients for ≥3 years and all experienced psychiatric symptoms.
According to Thompson et al,3 the presence and severity of apathy, irritability, and depression trend differently across the course of Huntington’s disease (HD). Apathy worsens with disease progression, closely following cognitive and motor symptoms. Irritability increases significantly, but this effect seems confined to early stages of HD. Depressive symptoms appear to decline slightly as HD advances, although it is unclear if this is because of antidepressants’ effects, increasing emotional blunting, and waning insight in later stages of HD, or another unknown factor.3 This study did not examine psychotic symptoms over time because few patients were experiencing delusions or hallucinations.
Similar to Thompson et al, Naarding et al5 found that apathy and depression in HD follow distinct time courses. Depression is a feature of early HD and apathy worsens with overall disease progression.
Depressed mood and functional ability—not cognitive or motor symptoms6—are the 2 most critical factors linked to health-related quality of life in HD. Hamilton et al7 found that apathy or executive dysfunction in HD patients is strongly related to decline in ability to complete activities of daily living, and may be severely debilitating.
Apathy. Often mistaken for a symptom of depression, apathy’s presentation may resemble anhedonia or fatigue; however, research suggests that depression and apathy are distinct conditions. Naarding et al
5 noted that apathy is more common than depressive symptoms in HD patients and may be a hallmark symptom of HD.
Depression affects most HD patients, and often is most severe early in the disease course. Hubers et al8 found that 20% of 100 HD patients had suicidal ideation. The strongest predictor was depressed mood.
Sleep disturbances and daytime somnolence are common among HD patients, and patients with comorbid depression report more disturbed sleep. Managing disturbed sleep and daytime somnolence in HD, with emphasis on comorbid depression, may improve the quality of life of patients and their caregivers.9
Anxiety was present in >50% of HD patients in a study by Paulsen et al2 and 37% evaluated by Craufurd et al.10 Craufurd et al10 also reported that 61% of patients were “physically tense and unable to relax.”
Among HD patients, 5% report obsessions and 10% report compulsive behaviors; these symptoms appear to become increasingly common as HD progresses.4,10
Impulsivity and disinhibition. Craufurd et al10 found that 71% of HD patients experienced poor judgment and self-monitoring, 40% had poor temper control and verbal outbursts, 22% exhibited threatening behavior or violence, and 6% had disinhibited or inappropriate sexual behavior.10
Recent studies have shown higher rates of disinhibition in “presymptomatic” gene-positive subjects vs gene-negative controls, suggesting that these symptoms may arise early in HD.11 Further, researchers demonstrated that patients lack symptom awareness and rate themselves as less impaired than their caregivers do.11
In our clinical experience, impulsivity frequently is encountered and creates significant conflict between patients and their caregivers. We speculate that when coupled with depressive symptoms of HD, impulsivity and disinhibition may play an important role in the high rates of suicidality seen in these patients.
Psychosis. Delusions and hallucinations are less common in HD than other psychiatric symptoms. Craufurd et al10 reported 3% of HD patients had delusions, 3% had auditory hallucinations, 2% had tactile hallucinations, and no patients had visual hallucinations.
A few case reports and a small study by Tsuang et al12 suggested that psychotic features in HD may be similar to those seen in paranoid schizophrenia. Tsuang et al12 also noted that more severe HD-related psychosis tends to cluster in families, which suggests that susceptibility to HD psychosis may be heritable.
Treating psychiatric symptoms
High-quality randomized controlled trials of pharmacotherapies for psychiatric symptoms in HD patients are lacking. Decisions regarding which agents to use often are based on case reports or clinical experience. The suggestions below are based on available evidence and our clinical experience.
Depression. Depressive symptoms in HD seem to respond to conventional pharmacologic treatments for major depressive disorder (MDD). A small trial of venlafaxine extended-release (XR) in 26 HD patients with MDD showed statistically significant improvements in depressive symptoms; however, this trial was not blinded and did not have a placebo group.13 In addition, 1 in 5 patients developed significant side effects—nausea, irritability, or worsening chorea.13
Evidence for selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors, and tricyclic antidepressants (TCAs) is lacking. Antidepressant choice should be based on patient response, side effect profile, and the need for secondary therapeutic effects.14
We often prescribe sertraline, citalopram, or escitalopram for our HD patients because of the relative absence of drug-drug interactions and favorable safety profile in medically and surgically ill patients. However, it’s important to tailor the treatment approach to your patient’s needs—eg, patients prone to forgetting their medicine may benefit from a drug with a longer half-life, such as fluoxetine. We avoid TCAs because of their anticholinergic effects, which may worsen dementia symptoms. Because HD patients have high rates of suicidality, agents that are highly toxic when taken in overdose should be used with caution.
One small study of HD patients with MDD or bipolar disorder showed clinical improvement in depressive symptoms after electroconvulsive therapy (ECT).15 Patients who suffered from comorbid delusions had the best improvements in mood.15 ECT likely is a good choice for HD patients who have failed several antidepressants, are suicidal, or who have depression with psychotic features.16
Apathy. A 2011 review concluded that no evidence-based recommendations regarding pharmacologic treatment for apathy in HD can be made because of lack of research.7 The Huntington’s Disease Society of America’s (HDSA) A Physician’s Guide to Managing Huntington’s Disease includes recommendations for treating apathy based on clinical experience.16 It suggests a nonsedating SSRI, followed by a trial of methylphenidate, pemoline, or dextroamphetamine if SSRIs were unsuccessful.
16 The HDSA guide notes psychostimulants may worsen irritability in HD and have a high potential for abuse. ECT appears to have little effect on apathy.15
Anxiety. A small, open-label study of 11 patients found that olanzapine, 5 mg/d, significantly improved depression, anxiety, irritability, and obsessive behavior in HD patients.17
The HDSA guide suggests treating anxiety and obsessive-compulsive symptoms as you would in patients without HD. For anxiety, SSRIs and possibly a short-term trial of a low-dose benzodiazepine (ie, lorazepam, clonazepam) are suggested.16 Benzodiazepines may increase the risk of falls and delirium in this population. Anecdotally, buspirone is helpful in some patients, with a starting dose of 5 mg 2 to 3 times per day and increased to 20 to 30 mg/d in divided doses.16 For obsessive-compulsive symptoms, SSRIs are recommended; atypical antipsychotics are reserved for severe or refractory symptoms.16
Disinhibition and impulsivity. There’s no research on treating disinhibition and impulsivity in HD. In our clinical experience, atypical antipsychotics are the most helpful. Factors regarding choosing an agent and dosing levels are similar to those for psychotic symptoms.
Psychotic symptoms. Most studies of typical and atypical antipsychotics for HD psychosis have shown beneficial effects.14,16-21 Neurologists frequently use these agents for managing chorea. Both neurologic and psychiatric features of the patient’s presentation must be considered when selecting a drug because treatment directed at 1 component of the disease may inadvertently exacerbate another. Specifically, higher potency antipsychotics (eg, haloperidol) are effective for chorea but can dramatically worsen bradykinesia; lower potency agents (eg, quetiapine) are less helpful for chorea but do not significantly worsen rigidity symptoms.
Olanzapine has been shown to improve chorea, anxiety, irritability, depression, sleep dysfunction, and weight loss in addition to psychotic symptoms.14,17 We find that olanzapine treats a constellation of symptoms common among HD patients, and we prescribe it frequently. Because olanzapine is considered a mid-potency agent, we find it’s best suited for concurrent control of psychotic symptoms and mild to moderate chorea in patients with minimal bradykinesia. Start olanzapine at 2.5 mg/d and gradually increase to 5 to 10 mg/d as tolerated.14
Risperidone is effective for treating psychosis and chorea. It can be started at 0.5 to 1 mg/d, and gradually increased to 6 to 8 mg/d.14 The depot formulation of risperidone has been shown to be effective in HD, which may help patients adhere to their medication.18 Risperidone is a mid-high potency antipsychotic, and in our experience is best used to control psychotic symptoms in patients with moderate chorea and few or no symptoms of bradykinesia or rigidity.
Quetiapine reduces psychotic symptoms, agitation, irritability, and insomnia without worsening bradykinesia or rigidity,19 but it is not beneficial for chorea. It can be started at 12.5 mg/d and gradually increased for effect as tolerated, up to 600 mg/d (depending on indication), in 2 or 3 divided doses.14
Haloperidol is a high-potency typical antipsychotic and may help psychotic patients with severe chorea; it should not be used in patients with bradykinesia. Start haloperidol at 0.5 to 1 mg/d and gradually increase to 6 to 8 mg/d as tolerated.14 Because of higher likelihood of side effects with typical antipsychotics, we often reserve its use for patients whose psychosis does not respond to atypical agents.
Other antipsychotics. Aripiprazole in HD has been examined in only 2 single- patient case reports20,21; the drug appeared to reduce psychosis and possibly chorea. Clozapine’s effectiveness for HD psychosis is not well known. It does not appear to be helpful for chorea and can cause agranulocytosis.22
Because one of the hallmarks of HD is dementia, it is worth noting that the FDA has issued a “black-box” warning on the use of antipsychotic drugs in patients with dementia because of concerns regarding increased mortality. However, drawing specific conclusions is difficult because the FDA warning is based on studies that looked primarily at Alzheimer’s disease and vascular dementia, not HD.
Other pharmacotherapies
Tetrabenazine is the only FDA-approved drug for treating HD. However, it carries a “black-box” warning for increased risk of depression and suicidal ideation and is contraindicated in suicidal patients and those with untreated or inadequately treated depression.
Although several small trials have had conflicting results regarding its benefit, amantadine sometimes is used to treat chorea.23-25 For more information about tetrabenazine and amantadine, see Box 3.
Tetrabenazine, the only FDA-approved drug for treating Huntington’s disease (HD), is a dopamine-depleting agent given to control chorea. In a 12-week, randomized, double-blind, placebo-controlled clinical trial, tetrabenazine was shown to be effective in HD patients.a Treatment with tetrabenazine results in symptomatic improvement of chorea, but does not slow or alter the course of the disease. Tetrabenazine can provide relief from choreiform movements, but these benefits should be balanced with the risks of depression and suicidality.a Tetrabenazine is known to prolong QTc interval, and should be used with caution in combination with other drugs that have the potential to do the same (eg, antipsychotics).a
Several case reports have found an association between tetrabenazine and development of neuroleptic malignant syndrome (NMS).b-d Be aware of the clinical characteristics of NMS—mental status change, rigidity, fever, and dysautonomia—and use caution when starting patients taking tetrabenazine on antipsychotics or other agents known to cause NMS.
Amantadine also has been used to treat chorea in HD patients who are unable to tolerate tetrabenazine or antipsychotics. Our neurologists sometimes have found it to be beneficial in patients with juvenile-onset HD because these patients often have debilitating dystonia. Be aware that amantadine is known to precipitate or worsen psychosis.e
References
- Food and Drug Administration. NDA 21-894 Xenazine® (tetrabenazine). Risk evaluation and mitigation strategy (REMS). Click here. Published August 15, 2008. Updated April 2011. Accessed June 20, 2012.
- Stevens E, Roman A, Houa M, et al. Severe hyperthermia during tetrabenazine therapy for tardive dyskinesia. Intensive Care Med. 1998;24(4):369-371.
- Petzinger GM, Bressman SB. A case of tetrabenazine-induced neuroleptic malignant syndrome after prolonged treatment. Mov Disord. 1997;12(2):246-248.
- Ossemann M, Sindic CJ, Laterre C. Tetrabenazine as a cause of neuroleptic malignant syndrome. Mov Disord. 1996;11(1):95.
- Wolters EC. Dopaminomimetic psychosis in Parkinson’s disease patients: diagnosis and treatment. Neurology. 1999;52 (7 suppl 3):S10-S13.
Related Resources
- Huntington’s Disease Society of America. www.hdsa.org.
- Family Caregiver Alliance. Huntington’s disease. www.caregiver.org/caregiver/jsp/content_node.jsp?nodeid=574.
- Huntington Study Group. www.huntington-study-group.org.
- Huntington’s Disease Advocacy Center. www.hdac.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Wellbutrin XL, others
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
- Methylphenidate • Concerta, Ritalin, others
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Pemoline • Cylert
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tetrabenazine • Xenazine
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Scher is a consultant to the advisory board for Lundbeck.
Ms. Kocsis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Harper PS. The epidemiology of Huntington’s disease. Hum Genet. 1992;89(4):365-376.
2. Paulsen JS, Ready RE, Hamilton JM, et al. Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2001;71(3):310-314.
3. Thompson JC, Harris J, Sollom AC, et al. Longitudinal evaluation of neuropsychiatric symptoms in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2012;24(1):53-60.
4. Beglinger LJ, Langbehn DR, Duff K, et al. Probability of obsessive and compulsive symptoms in Huntington’s disease. Biol Psychiatry. 2007;61(3):415-418.
5. Naarding P, Janzing JG, Eling P, et al. Apathy is not depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2009;21(3):266-270.
6. Ho AK, Gilbert AS, Mason SL, et al. Health-related quality of life in Huntington’s disease: which factors matter most? Mov Disord. 2009;24(4):574-578.
7. Hamilton JM, Salmon DP, Corey-Bloom J, et al. Behavioural abnormalities contribute to functional decline in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2003;74(1):120-122.
8. Hubers AA, Reedeker N, Giltay EJ, et al. Suicidality in Huntington’s disease. J Affect Disord. 2012;136(3):550-557.
9. Videnovic A, Leurgans S, Fan W, et al. Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat Disord. 2009;15(6):471-474.
10. Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(4):219-226.
11. Duff K, Paulsen JS, Beglinger LJ, et al. “Frontal” behaviors before the diagnosis of Huntington’s disease and their relationship to markers of disease progression: evidence of early lack of awareness. J Neuropsychiatry Clin Neurosci. 2010;22(2):196-207.
12. Tsuang D, Almqvist EW, Lipe H, et al. Familial aggregation of psychotic symptoms in Huntington’s disease. Am J Psychiatry. 2000;157(12):1955-1959.
13. Holl AK, Wilkinson L, Painold A, et al. Combating depression in Huntington’s disease: effective antidepressive treatment with venlafaxine XR. Int Clin Psychopharmacol. 2010;25(1):46-50.
14. Killoran A, Biglan KM. Therapeutics in Huntington’s disease. Curr Treat Options Neurol. 2012;14(2):137-149.
15. Ranen NG, Peyser CE, Folstein SE. ECT as a treatment for depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 1994;6(2):154-159.
16. Rosenblatt A, Ranen NG, Nance MA, et al. A physician’s guide to the management of Huntington’s disease. 2nd edition. http://www.hdsa.org/images/content/1/1/11289.pdf. Published 1999. Accessed July 27, 2012.
17. Squitieri F, Cannella M, Piorcellini A, et al. Short-term effects of olanzapine in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(1):69-72.
18. Johnston TG. Risperidone long-acting injection and Huntington’s disease: case series with significant psychiatric and behavioural symptoms. Int Clin Psychopharmacol. 2011;26(2):114-119.
19. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
20. Lin WC, Chou YH. Aripiprazole effects on psychosis and chorea in a patient with Huntington’s disease. Am J Psychiatry. 2008;165(9):1207-1208.
21. Oulis P, Mourikis I, Konstantakopoulos G, et al. Aripiprazole in the treatment of olanzapine-resistant psychotic and motor symptoms of Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2010;22(3):352c.e4-352c.e5.
22. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomised comparative study. J Neurol Neurosurg Psychiatry. 1997;63(1):35-39.
23. Verhagen Metman L, Morris MJ, Farmer C, et al. Huntington’s disease: a randomized, controlled trial using the NMDA-antagonist amantadine. Neurology. 2002;59(5):694-699.
24. Lucetti C, Del Dotto P, Gambaccini G, et al. IV amantadine improves chorea in Huntington’s disease: an acute randomized, controlled study. Neurology. 2003;60(12):1995-1997.
25. O’Suilleabhain P, Dewey RB, Jr. A randomized trial of amantadine in Huntington disease. Arch Neurol. 2003;60(7):996-998.
1. Harper PS. The epidemiology of Huntington’s disease. Hum Genet. 1992;89(4):365-376.
2. Paulsen JS, Ready RE, Hamilton JM, et al. Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2001;71(3):310-314.
3. Thompson JC, Harris J, Sollom AC, et al. Longitudinal evaluation of neuropsychiatric symptoms in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2012;24(1):53-60.
4. Beglinger LJ, Langbehn DR, Duff K, et al. Probability of obsessive and compulsive symptoms in Huntington’s disease. Biol Psychiatry. 2007;61(3):415-418.
5. Naarding P, Janzing JG, Eling P, et al. Apathy is not depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2009;21(3):266-270.
6. Ho AK, Gilbert AS, Mason SL, et al. Health-related quality of life in Huntington’s disease: which factors matter most? Mov Disord. 2009;24(4):574-578.
7. Hamilton JM, Salmon DP, Corey-Bloom J, et al. Behavioural abnormalities contribute to functional decline in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2003;74(1):120-122.
8. Hubers AA, Reedeker N, Giltay EJ, et al. Suicidality in Huntington’s disease. J Affect Disord. 2012;136(3):550-557.
9. Videnovic A, Leurgans S, Fan W, et al. Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat Disord. 2009;15(6):471-474.
10. Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(4):219-226.
11. Duff K, Paulsen JS, Beglinger LJ, et al. “Frontal” behaviors before the diagnosis of Huntington’s disease and their relationship to markers of disease progression: evidence of early lack of awareness. J Neuropsychiatry Clin Neurosci. 2010;22(2):196-207.
12. Tsuang D, Almqvist EW, Lipe H, et al. Familial aggregation of psychotic symptoms in Huntington’s disease. Am J Psychiatry. 2000;157(12):1955-1959.
13. Holl AK, Wilkinson L, Painold A, et al. Combating depression in Huntington’s disease: effective antidepressive treatment with venlafaxine XR. Int Clin Psychopharmacol. 2010;25(1):46-50.
14. Killoran A, Biglan KM. Therapeutics in Huntington’s disease. Curr Treat Options Neurol. 2012;14(2):137-149.
15. Ranen NG, Peyser CE, Folstein SE. ECT as a treatment for depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 1994;6(2):154-159.
16. Rosenblatt A, Ranen NG, Nance MA, et al. A physician’s guide to the management of Huntington’s disease. 2nd edition. http://www.hdsa.org/images/content/1/1/11289.pdf. Published 1999. Accessed July 27, 2012.
17. Squitieri F, Cannella M, Piorcellini A, et al. Short-term effects of olanzapine in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(1):69-72.
18. Johnston TG. Risperidone long-acting injection and Huntington’s disease: case series with significant psychiatric and behavioural symptoms. Int Clin Psychopharmacol. 2011;26(2):114-119.
19. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
20. Lin WC, Chou YH. Aripiprazole effects on psychosis and chorea in a patient with Huntington’s disease. Am J Psychiatry. 2008;165(9):1207-1208.
21. Oulis P, Mourikis I, Konstantakopoulos G, et al. Aripiprazole in the treatment of olanzapine-resistant psychotic and motor symptoms of Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2010;22(3):352c.e4-352c.e5.
22. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomised comparative study. J Neurol Neurosurg Psychiatry. 1997;63(1):35-39.
23. Verhagen Metman L, Morris MJ, Farmer C, et al. Huntington’s disease: a randomized, controlled trial using the NMDA-antagonist amantadine. Neurology. 2002;59(5):694-699.
24. Lucetti C, Del Dotto P, Gambaccini G, et al. IV amantadine improves chorea in Huntington’s disease: an acute randomized, controlled study. Neurology. 2003;60(12):1995-1997.
25. O’Suilleabhain P, Dewey RB, Jr. A randomized trial of amantadine in Huntington disease. Arch Neurol. 2003;60(7):996-998.
Managing chronic pain: Consider psychotropics and other non-opioids
Discuss this article at www.facebook.com/CurrentPsychiatry
Of the 56 million American adults who report living with chronic pain almost 60% also exhibit psychiatric disorders such as depression or anxiety.1,2 Because patients with chronic pain suffer from a mixture of physical and psychological components, managing such conditions is complicated, and using opioids is tempting. However, treatment needs to address the underlying pathology along with social and psychological factors.
Because substance abuse treatment admissions increased by 400% from 1998 to 2008,3 many physicians look to non-opioids and other treatment modalities to control chronic non-cancer pain. Common pharmacologic therapies used to treat chronic pain include tricyclic antidepressants (TCAs), serotonin-norepinephrine reuptake inhibitors (SNRIs), antiepileptic drugs (AEDs), nonsteroidal anti-inflammatory drugs (NSAIDs), and, to a lesser extent, atypical antipsychotics. TCAs, SNRIs, AEDs, NSAIDs, and atypical antipsychotics influence a variety of presumed underlying pathophysiological processes, including inflammatory mediators, activity of N-methyl-d-aspartate (NMDA) receptors, and voltage-gated calcium channels. In addition, they increase activity of descending inhibitory pain pathways. Animal studies suggest dysfunction of these inhibitory mechanisms contributes to the central sensitization and hyperexcitability of pain transmitting pathways.4
In this article, we discuss psychotropics and other non-opioid agents for treating pain. However, no single solution is best for all patients with chronic pain and this article is not a “how to” guide to avoid administering opioid medication. Also incorporate a multimodal, non-pharmacologic approach whenever possible.
Tricyclic antidepressants
Although this class acts primarily by increasing serotonin levels, norepinephrine and dopamine also are affected depending on the particular medication. Studies have shown that amitriptyline, nortriptyline, and desipramine function well as analgesics independent of their antidepressant effects.5 TCAs may improve pain symptoms at lower therapeutic dosages than those used for treating depression.5
Although researchers have not elucidated TCAs’ mechanism of action with regards to analgesia, they are thought to act within the concept of the gating theory of pain control,6 which functions by activation and inhibition of pain signal transmission. It is believed TCAs act on nociceptive pathways by blocking serotonin and norepinephrine reuptake. Although researchers previously thought that TCAs’ analgesic mechanism was correlated to serotonin reuptake inhibition, this theory has changed. Selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine have not demonstrated substantial effectiveness in neuropathic pain when compared with TCAs and SNRIs. Recent studies have shown that TCAs may work by blocking sodium channels, similar to local anesthetics and antiarrhythmic agents.7
Psychiatrists prescribe TCAs infrequently because of these drugs’ unfavorable side effect profile compared with SSRIs and SNRIs. However, TCAs often are prescribed for pain management as an adjunct to other medications for neuropathic conditions and at lower dosages than those used for treating depression (Table 1).8
Table 1
Tricyclic antidepressants used to treat pain
| Drug | Dosage range for pain (off-label) | Comments |
|---|---|---|
| Amitriptyline | 10 to 100 mg/d | High sedation, high anticholinergic side effects |
| Amoxapine | 50 to 100 mg/d | Low sedation, moderate anticholinergic side effects |
| Clomipramine | 25 to 100 mg/d | Low sedation, low anticholinergic side effects |
| Desipramine | 25 to 100 mg/d | Low sedation, low anticholinergic side effects |
| Imipramine | 25 to 100 mg/d | Moderate sedation, moderate anticholinergic side effects |
| Nortriptyline | 10 to 75 mg/d | Moderate sedation, low anticholinergic side effects |
| Source: Reference 8 | ||
SNRIs
Evidence supports using duloxetine, a potent SNRI that mediates pain inhibition in the descending pathways, for 4 chronic pain conditions:
- diabetic peripheral neuropathic pain
- fibromyalgia
- mechanical low back pain
- pain associated with osteoarthritis.9
Titrate the dosage to 60 mg/d and maintain the patient at this dose for at least 4 weeks. Thereafter, according to patient response, the dosage may be titrated to 120 mg/d (off-label) with appropriate vital sign monitoring and routine lab analysis.
Venlafaxine also can mediate pain response in a similar manner to duloxetine, but is not FDA-approved for treating pain. Use caution when prescribing venlafaxine for patients with a history of hypertension. Milnacipran is a relatively new SNRI that has been shown to be effective in treating fibromyalgia in divided doses of 100 to 200 mg/d (Table 2).9-11
Table 2
Treating pain with serotonin-norepinephrine reuptake inhibitors
| Drug | Dosage range for pain | Comments |
|---|---|---|
| Duloxetine | 60 to 120 mg/d9 | FDA maximum recommended dose is 60 mg/d |
| Milnacipran | 25 to 200 mg/d10 | Approved for treating depression outside the United States |
| Venlafaxine | 75 to 225 mg/d11 | Monitor blood pressure, LFTs, and kidney function |
| LFTs: liver function tests | ||
Antiepileptic drugs
Several AEDs are used for pain management (Table 3).12-16 Gabapentin and pregabalin work by binding to voltage-gated calcium channels and decreasing excitatory neurotransmitter release. Along with TCAs, they are considered a first-line treatment for managing neuropathic pain.17 Gabapentin is FDA-approved for seizures and postherpetic neuralgia, but evidence supports its use in most types of neuropathic pain. Pregabalin is FDA-approved for treating seizures, diabetic peripheral neuropathy, central neuropathic pain, postherpetic neuralgia, and fibromyalgia.
Topiramate inhibits excitatory neurotransmission by enhancing the effects of gamma-aminobutyric acid, and also by blocking NMDA receptors. Topiramate is FDA-approved for seizures and migraine prophylaxis, and is used off-label for treating neuropathic pain. A 12-week trial of topiramate for diabetic neuropathy found significant analgesia in 50% of patients taking the drug, compared with 34% receiving placebo.18
Lamotrigine is approved for several types of seizures and maintenance of bipolar I disorder, and is used off-label for neuropathic pain. A recent Cochrane database review concluded that lamotrigine is ineffective for neuropathic pain14; however, some guidelines recommend using lamotrigine to treat neuropathies that do not respond to treatment with carbamazepine.19
Carbamazepine is a complex AED that is structurally similar to TCAs. It blocks sodium channels and has various pharmacologic properties, including anticholinergic, muscle relaxant, antidepressant, and sedative effects. Carbamazepine has analgesic effects through blockade of synaptic transmission in the trigeminal nucleus and is FDA-approved for seizures, bipolar disorder, neuropathic pain, and trigeminal neuralgia. In a systematic review of 12 trials of carbamazepine that included 4 placebo-controlled trials for trigeminal neuralgia, 2 studies showed a number needed to treat (NNT) of 1.8.20 For diabetic neuropathy, there was insufficient data to calculate NNT.
Oxcarbazepine, an analog of carbamazepine, also is FDA-approved for seizures and is used off-label for neuropathic pain. In the only double-blind trial with positive results, oxcarbazepine titrated to 1,800 mg/d reduced diabetic neuropathy pain scores on a visual analog scale by 24 points—roughly 25%.15
Table 3
Antiepileptic drugs for pain treatment
| Drug | Dosage range for pain | Comments |
|---|---|---|
| Carbamazepine | Starting dose: 100 mg twice a day, doses titrated to 400 to 800 mg/d usually are adequate. Maximum of 1,200 mg/d12 | Anticholinergic effects, blood dyscrasias, hyponatremia, increase in LFTs, ECG changes. CYP450 inducer, many DDIs |
| Gabapentin | Starting dose: 100 to 300 mg at bedtime or 100 to 300 mg 3 times a day, slow titration, maximum of 3,600 mg/d13 | Dizziness, sedation, weight gain, peripheral edema. Adjust dose in renal insufficiency |
| Lamotrigine | 200 to 400 mg/d14 | Sedation, headache, dizziness, ataxia, GI upset, blurred vision. Risk of life-threatening rash |
| Oxcarbazepine | Starting dose: 300 mg/d, then titrated as tolerated to a maximum of 1,800 mg/d15 | Adverse drug reactions similar to carbamazepine, less anticholinergic effects, more hyponatremia. Fewer DDIs than carbamazepine |
| Pregabalin | Starting dose: 50 mg 3 times a day or 75 mg twice a day, may increase every 3 to 7 days as tolerated, maximum of 600 mg/d13 | Same adverse drug reactions as gabapentin, less sedation. Adjust dose in renal insufficiency. More costly than gabapentin |
| Topiramate | Starting dose: 12.5 to 25 mg once or twice a day for 4 weeks; then double the dose every 4 weeks to reach a maximum dose of 100 to 200 mg/d in divided doses16 | Weight loss, anorexia, nephrolithiasis, cognitive impairment |
| CYP450: cytochrome P450; DDIs: drug-drug interactions; GI: gastrointestinal; LFTs: liver function tests | ||
Non-opioid analgesics
NSAIDs have antipyretic, analgesic, and anti-inflammatory effects and are used for fever, headache, mild-to-moderate pain, musculoskeletal pain, menstrual pain, and dental pain. They are particularly useful in treating acute pain, often in combination with opioid analgesics. NSAIDs exert their analgesic action through blockade of prostaglandin production via reversible inhibition of cyclooxygenase-1 and cyclooxygenase-2.
The most common side effects of NSAIDs are the result of gastrointestinal (GI) toxicity and include dyspepsia, heartburn, nausea, anorexia, and epigastric pain.21 GI ulceration and bleeding are rare but serious complications. To decrease these risks, tell patients to take NSAIDs with food. Add a GI protective agent, such as an H2 blocker or proton pump inhibitor, for patients at higher risk for GI complications.22
In addition, inhibition of renal prostaglandins by NSAIDs can cause renal toxicity, fluid retention, and edema, potentially exacerbating existing cardiovascular conditions such as hypertension and heart failure. NSAIDs may increase the risk of serious thrombotic events such as myocardial infarction and stroke. Use NSAIDs at the lowest effective dose for the shortest duration possible and generally avoid prescribing in patients at high risk for cardiovascular disease and pregnant women, especially those in their third trimester.23,24
NSAIDs may cause pharmacodynamic and pharmacokinetic drug-drug interactions. The risk of GI toxicity and bleeding increases when NSAIDs are administered with drugs that also irritate the gastric mucosa or have antiplatelet/anticoagulant effects.21 Plasma concentrations of drugs with a narrow therapeutic index that are renally eliminated, such as methotrexate and lithium, can increase to potentially toxic levels with concurrent NSAID use because NSAIDs decrease renal perfusion.21 Also, the therapeutic effects of antihypertensives may be attenuated because NSAIDs cause fluid retention.25
Acetaminophen (APAP) is available in several dosage forms as a single ingredient and in combination with opioids in prescription products. For more information about APAP, see the Box below.
Atypical antipsychotics
Although atypical antipsychotics are not often used to treat pain, studies indicate that fibromyalgia patients may benefit from ziprasidone26 and olanzapine,27 most often as an adjunctive treatment rather than monotherapy. Randomized controlled studies indicate poor tolerability with several atypical antipsychotics. Weight gain, akathisia, and somnolence are side effects of some atypical antipsychotics. Additionally, ziprasidone has been associated with QTc prolongation. For chronic pain patients, atypical antipsychotics are most useful for treating psychiatric comorbidities.
Although its mechanism of action is not well understood, acetaminophen (APAP) works by blocking prostaglandin syntheses via inhibition of cyclooxygenase-1 and cyclooxygenase-2 in the CNS.a Therefore, in contrast to NSAIDs, APAP does not possess peripheral anti-inflammatory effects or affect platelet function and is effective for treating fever, headache, and acute and chronic mild pain. The American Geriatrics Society recommends APAP for minor and persistent pain in older patientsb and the American College of Rheumatology recommends it as first-line therapy for osteoarthritis of the hip or knee.c
APAP has few clinically significant drug interactions, an excellent safety profile, and a long history of safe and effective use. When used within the recommended dosage range, APAP has few side effects. However, overuse of APAP is the leading cause of acute liver failure in the United States.d APAP hepatotoxicity can be accompanied by nephrotoxicity, is dose-dependent, and can be caused by acute overdose or chronic ingestion at doses over the recommended maximum of 4 g/d. Patients have experienced elevated liver transaminases with coadministration of APAP with phenytoin and phenobarbital.e,f Alcohol and other potentially hepatotoxic drugs also can increase the risk of liver toxicity when combined with APAP.d APAP is pregnancy category B and is considered the drug of choice for treating pain or fever during pregnancy and breast-feeding.g
References
- Amadio P Jr. Peripherally acting analgesics. Am J Med. 1984;77(3A):17-26.
- American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc. 2009;57(8):1331-1346.
- Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. Arthritis Rheum. 2000;43(9):1905-1915.
- Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42(6):1364-1372.
- Pirotte JH. Apparent potentiation of hepatotoxicity from small doses of acetaminophen by phenobarbital. Ann Intern Med. 1984;101(3):403.
- Brackett CC, Bloch JD. Phenytoin as a possible cause of acetaminophen hepatotoxicity: case report and review of the literature. Pharmacotherapy. 2000;20(2):229-233.
- Hersh EV, Moore PA, Ross GL. Over-the-counter analgesics and antipyretics: a critical assessment. Clin Ther. 2000; 22(5):500-548.
Related Resources
- Leo RJ. Chronic nonmalignant pain: How to ‘turn down’ its physiologic triggers. Current Psychiatry. 2008;7(8):19-36.
- Nikolaus T, Zeyfang A. Pharmacological treatments for persistent non-malignant pain in older persons. Drugs Aging. 2004;21(1):19-41.
- World Health Organization. WHO’s pain ladder. www.who.int/cancer/palliative/painladder/en.
Drug Brand Names
- Acetaminophen • Tylenol
- Amitriptyline • Elavil, others
- Amoxapine • Asendin
- Carbamazepine • Tegretol, Carbatrol, others
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Gabapentin • Neurontin, Gralise
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Methotrexate • Rheumatrex, Trexall
- Milnacipran • Savella
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Pregabalin • Lyrica
- Topiramate • Topamax, Topiragen
- Venlafaxine • Effexor
- iprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Brennan F, Carr DB, Cousins M. Pain management: a fundamental human right. Anesth Analg. 2007;105(1):205-221.
2. Thieme K, Turk DC, Flor H. Comorbid depression and anxiety in fibromyalgia syndrome: relationship to somatic and psychosocial variables. Psychosom Med. 2004;66(6):837-844.
3. Substance Abuse and Mental Health Services Administration, Office of Applied Studies Treatment episode data set (TEDS). 1998-2008. National admissions to substance abuse treatment services. Rockville MD: Substance Abuse and Mental Health Services Administration, Office of Applied Studies; 2010.
4. Iyengar S, Webster AA, Hemrick-Luecke SK, et al. Efficacy of duloxetine, a potent and balanced serotonin-norepinephrine reuptake inhibitor in persistent pain models in rats. J Pharmacol Exp Ther. 2004;311(2):576-584.
5. Guay DR. Adjunctive agents in the management of chronic pain. Pharmacotherapy. 2001;21(9):1070-1081.
6. Campbell LC, Clauw DJ, Keefe FJ. Persistent pain and depression: a biopsychosocial perspective. Biol Psychiatry. 2003;54(3):399-409.
7. Dick IE, Brochu RM, Purohit Y, et al. Sodium channel blockade may contribute to the analgesic efficacy of antidepressants. J Pain. 2007;8(4):315-324.
8. Stahl SM. Essential psychopharmacology: the prescriber’s guide. New York NY: Cambridge University Press; 2006.
9. Skljarevski V, Desaiah D, Liu-Seifert H, et al. Efficacy and safety of duloxetine in patients with chronic low back pain. Spine (Phila Pa 1976). 2010;35(13):E578-E585.
10. Hsu ES. Acute and chronic pain management in fibromyalgia: updates on pharmacotherapy. Am J Ther. 2011;18(6):487-509.
11. Bomholt SF, Mikkelsen JD, Blackburn-Munro G. Antinociceptive effects of the antidepressants amitriptyline duloxetine, mirtazapine and citalopram in animal models of acute, persistent and neuropathic pain. Neuropharmacology. 2005;48(2):252-263.
12. Campbell FG, Graham JG, Zilkha KJ. Clinical trial of carbazepine (tegretol) in trigeminal neuralgia. J Neurol Neurosurg Psychiatry. 1966;29(3):265-267.
13. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(10 suppl):S22-S32.
14. Dogra S, Beydoun S, Mazzola J, et al. Oxcarbazepine in painful diabetic neuropathy: a randomized, placebo-controlled study. Eur J Pain. 2005;9(5):543-554.
15. Kline KM, Carroll DG, Malnar KF. Painful diabetic peripheral neuropathy relieved with use of oral topiramate. South Med J. 2003;96(6):602-605.
16. Wiffen PJ, Derry S, Moore RA. Lamotrigine for acute and chronic pain. Cochrane Database Syst Rev. 2011;(2):CD006044.-
17. Dworkin RH, O’Connor AB, Audette J, et al. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc. 2010;85(3 suppl):S3-S14.
18. Raskin P, Donofrio PD, Rosenthal NR, et al. Topiramate vs placebo in painful diabetic neuropathy: analgesic and metabolic effects. Neurology. 2004;63(5):865-873.
19. Moulin DE, Clark AJ, Gilron I, et al. Pharmacological management of chronic neuropathic pain - consensus statement and guidelines from the Canadian Pain Society. Pain Res Manag. 2007;12(1):13-21.
20. Wiffen PJ, Derry S, Moore RA, et al. Carbamazepine for acute and chronic pain in adults. Cochrane Database Syst Rev. 2011;(1):CD005451.-
21. Hersh EV, Moore PA, Ross GL. Over-the-counter analgesics and antipyretics: a critical assessment. Clin Ther. 2000;22(5):500-548.
22. Lanas AI. Current approaches to reducing gastrointestinal toxicity of low-dose aspirin. Am J Med. 2001;110(1A):70S-73S.
23. Antman EM, Bennett JS, Daugherty A, et al. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115(12):1634-1642.
24. Briggs G, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation. 8th ed. Baltimore MD: Lippincott Williams and Wilkins; 2008.
25. Frishman WH. Effects of nonsteroidal anti-inflammatory drug therapy on blood pressure and peripheral edema. Am J Cardiol. 2002;89(6A):18D-25D.
26. Calandre EP, Hidalgo J, Rico-Villademoros F. Use of ziprasidone in patients with fibromyalgia: a case series. Rheumatol Int. 2007;27(5):473-476.
27. Rico-Villademoros F, Hidalgo J, Dominguez I, et al. Atypical antipsychotics in the treatment of fibromyalgia: a case series with olanzapine. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(1):161-164.
Discuss this article at www.facebook.com/CurrentPsychiatry
Of the 56 million American adults who report living with chronic pain almost 60% also exhibit psychiatric disorders such as depression or anxiety.1,2 Because patients with chronic pain suffer from a mixture of physical and psychological components, managing such conditions is complicated, and using opioids is tempting. However, treatment needs to address the underlying pathology along with social and psychological factors.
Because substance abuse treatment admissions increased by 400% from 1998 to 2008,3 many physicians look to non-opioids and other treatment modalities to control chronic non-cancer pain. Common pharmacologic therapies used to treat chronic pain include tricyclic antidepressants (TCAs), serotonin-norepinephrine reuptake inhibitors (SNRIs), antiepileptic drugs (AEDs), nonsteroidal anti-inflammatory drugs (NSAIDs), and, to a lesser extent, atypical antipsychotics. TCAs, SNRIs, AEDs, NSAIDs, and atypical antipsychotics influence a variety of presumed underlying pathophysiological processes, including inflammatory mediators, activity of N-methyl-d-aspartate (NMDA) receptors, and voltage-gated calcium channels. In addition, they increase activity of descending inhibitory pain pathways. Animal studies suggest dysfunction of these inhibitory mechanisms contributes to the central sensitization and hyperexcitability of pain transmitting pathways.4
In this article, we discuss psychotropics and other non-opioid agents for treating pain. However, no single solution is best for all patients with chronic pain and this article is not a “how to” guide to avoid administering opioid medication. Also incorporate a multimodal, non-pharmacologic approach whenever possible.
Tricyclic antidepressants
Although this class acts primarily by increasing serotonin levels, norepinephrine and dopamine also are affected depending on the particular medication. Studies have shown that amitriptyline, nortriptyline, and desipramine function well as analgesics independent of their antidepressant effects.5 TCAs may improve pain symptoms at lower therapeutic dosages than those used for treating depression.5
Although researchers have not elucidated TCAs’ mechanism of action with regards to analgesia, they are thought to act within the concept of the gating theory of pain control,6 which functions by activation and inhibition of pain signal transmission. It is believed TCAs act on nociceptive pathways by blocking serotonin and norepinephrine reuptake. Although researchers previously thought that TCAs’ analgesic mechanism was correlated to serotonin reuptake inhibition, this theory has changed. Selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine have not demonstrated substantial effectiveness in neuropathic pain when compared with TCAs and SNRIs. Recent studies have shown that TCAs may work by blocking sodium channels, similar to local anesthetics and antiarrhythmic agents.7
Psychiatrists prescribe TCAs infrequently because of these drugs’ unfavorable side effect profile compared with SSRIs and SNRIs. However, TCAs often are prescribed for pain management as an adjunct to other medications for neuropathic conditions and at lower dosages than those used for treating depression (Table 1).8
Table 1
Tricyclic antidepressants used to treat pain
| Drug | Dosage range for pain (off-label) | Comments |
|---|---|---|
| Amitriptyline | 10 to 100 mg/d | High sedation, high anticholinergic side effects |
| Amoxapine | 50 to 100 mg/d | Low sedation, moderate anticholinergic side effects |
| Clomipramine | 25 to 100 mg/d | Low sedation, low anticholinergic side effects |
| Desipramine | 25 to 100 mg/d | Low sedation, low anticholinergic side effects |
| Imipramine | 25 to 100 mg/d | Moderate sedation, moderate anticholinergic side effects |
| Nortriptyline | 10 to 75 mg/d | Moderate sedation, low anticholinergic side effects |
| Source: Reference 8 | ||
SNRIs
Evidence supports using duloxetine, a potent SNRI that mediates pain inhibition in the descending pathways, for 4 chronic pain conditions:
- diabetic peripheral neuropathic pain
- fibromyalgia
- mechanical low back pain
- pain associated with osteoarthritis.9
Titrate the dosage to 60 mg/d and maintain the patient at this dose for at least 4 weeks. Thereafter, according to patient response, the dosage may be titrated to 120 mg/d (off-label) with appropriate vital sign monitoring and routine lab analysis.
Venlafaxine also can mediate pain response in a similar manner to duloxetine, but is not FDA-approved for treating pain. Use caution when prescribing venlafaxine for patients with a history of hypertension. Milnacipran is a relatively new SNRI that has been shown to be effective in treating fibromyalgia in divided doses of 100 to 200 mg/d (Table 2).9-11
Table 2
Treating pain with serotonin-norepinephrine reuptake inhibitors
| Drug | Dosage range for pain | Comments |
|---|---|---|
| Duloxetine | 60 to 120 mg/d9 | FDA maximum recommended dose is 60 mg/d |
| Milnacipran | 25 to 200 mg/d10 | Approved for treating depression outside the United States |
| Venlafaxine | 75 to 225 mg/d11 | Monitor blood pressure, LFTs, and kidney function |
| LFTs: liver function tests | ||
Antiepileptic drugs
Several AEDs are used for pain management (Table 3).12-16 Gabapentin and pregabalin work by binding to voltage-gated calcium channels and decreasing excitatory neurotransmitter release. Along with TCAs, they are considered a first-line treatment for managing neuropathic pain.17 Gabapentin is FDA-approved for seizures and postherpetic neuralgia, but evidence supports its use in most types of neuropathic pain. Pregabalin is FDA-approved for treating seizures, diabetic peripheral neuropathy, central neuropathic pain, postherpetic neuralgia, and fibromyalgia.
Topiramate inhibits excitatory neurotransmission by enhancing the effects of gamma-aminobutyric acid, and also by blocking NMDA receptors. Topiramate is FDA-approved for seizures and migraine prophylaxis, and is used off-label for treating neuropathic pain. A 12-week trial of topiramate for diabetic neuropathy found significant analgesia in 50% of patients taking the drug, compared with 34% receiving placebo.18
Lamotrigine is approved for several types of seizures and maintenance of bipolar I disorder, and is used off-label for neuropathic pain. A recent Cochrane database review concluded that lamotrigine is ineffective for neuropathic pain14; however, some guidelines recommend using lamotrigine to treat neuropathies that do not respond to treatment with carbamazepine.19
Carbamazepine is a complex AED that is structurally similar to TCAs. It blocks sodium channels and has various pharmacologic properties, including anticholinergic, muscle relaxant, antidepressant, and sedative effects. Carbamazepine has analgesic effects through blockade of synaptic transmission in the trigeminal nucleus and is FDA-approved for seizures, bipolar disorder, neuropathic pain, and trigeminal neuralgia. In a systematic review of 12 trials of carbamazepine that included 4 placebo-controlled trials for trigeminal neuralgia, 2 studies showed a number needed to treat (NNT) of 1.8.20 For diabetic neuropathy, there was insufficient data to calculate NNT.
Oxcarbazepine, an analog of carbamazepine, also is FDA-approved for seizures and is used off-label for neuropathic pain. In the only double-blind trial with positive results, oxcarbazepine titrated to 1,800 mg/d reduced diabetic neuropathy pain scores on a visual analog scale by 24 points—roughly 25%.15
Table 3
Antiepileptic drugs for pain treatment
| Drug | Dosage range for pain | Comments |
|---|---|---|
| Carbamazepine | Starting dose: 100 mg twice a day, doses titrated to 400 to 800 mg/d usually are adequate. Maximum of 1,200 mg/d12 | Anticholinergic effects, blood dyscrasias, hyponatremia, increase in LFTs, ECG changes. CYP450 inducer, many DDIs |
| Gabapentin | Starting dose: 100 to 300 mg at bedtime or 100 to 300 mg 3 times a day, slow titration, maximum of 3,600 mg/d13 | Dizziness, sedation, weight gain, peripheral edema. Adjust dose in renal insufficiency |
| Lamotrigine | 200 to 400 mg/d14 | Sedation, headache, dizziness, ataxia, GI upset, blurred vision. Risk of life-threatening rash |
| Oxcarbazepine | Starting dose: 300 mg/d, then titrated as tolerated to a maximum of 1,800 mg/d15 | Adverse drug reactions similar to carbamazepine, less anticholinergic effects, more hyponatremia. Fewer DDIs than carbamazepine |
| Pregabalin | Starting dose: 50 mg 3 times a day or 75 mg twice a day, may increase every 3 to 7 days as tolerated, maximum of 600 mg/d13 | Same adverse drug reactions as gabapentin, less sedation. Adjust dose in renal insufficiency. More costly than gabapentin |
| Topiramate | Starting dose: 12.5 to 25 mg once or twice a day for 4 weeks; then double the dose every 4 weeks to reach a maximum dose of 100 to 200 mg/d in divided doses16 | Weight loss, anorexia, nephrolithiasis, cognitive impairment |
| CYP450: cytochrome P450; DDIs: drug-drug interactions; GI: gastrointestinal; LFTs: liver function tests | ||
Non-opioid analgesics
NSAIDs have antipyretic, analgesic, and anti-inflammatory effects and are used for fever, headache, mild-to-moderate pain, musculoskeletal pain, menstrual pain, and dental pain. They are particularly useful in treating acute pain, often in combination with opioid analgesics. NSAIDs exert their analgesic action through blockade of prostaglandin production via reversible inhibition of cyclooxygenase-1 and cyclooxygenase-2.
The most common side effects of NSAIDs are the result of gastrointestinal (GI) toxicity and include dyspepsia, heartburn, nausea, anorexia, and epigastric pain.21 GI ulceration and bleeding are rare but serious complications. To decrease these risks, tell patients to take NSAIDs with food. Add a GI protective agent, such as an H2 blocker or proton pump inhibitor, for patients at higher risk for GI complications.22
In addition, inhibition of renal prostaglandins by NSAIDs can cause renal toxicity, fluid retention, and edema, potentially exacerbating existing cardiovascular conditions such as hypertension and heart failure. NSAIDs may increase the risk of serious thrombotic events such as myocardial infarction and stroke. Use NSAIDs at the lowest effective dose for the shortest duration possible and generally avoid prescribing in patients at high risk for cardiovascular disease and pregnant women, especially those in their third trimester.23,24
NSAIDs may cause pharmacodynamic and pharmacokinetic drug-drug interactions. The risk of GI toxicity and bleeding increases when NSAIDs are administered with drugs that also irritate the gastric mucosa or have antiplatelet/anticoagulant effects.21 Plasma concentrations of drugs with a narrow therapeutic index that are renally eliminated, such as methotrexate and lithium, can increase to potentially toxic levels with concurrent NSAID use because NSAIDs decrease renal perfusion.21 Also, the therapeutic effects of antihypertensives may be attenuated because NSAIDs cause fluid retention.25
Acetaminophen (APAP) is available in several dosage forms as a single ingredient and in combination with opioids in prescription products. For more information about APAP, see the Box below.
Atypical antipsychotics
Although atypical antipsychotics are not often used to treat pain, studies indicate that fibromyalgia patients may benefit from ziprasidone26 and olanzapine,27 most often as an adjunctive treatment rather than monotherapy. Randomized controlled studies indicate poor tolerability with several atypical antipsychotics. Weight gain, akathisia, and somnolence are side effects of some atypical antipsychotics. Additionally, ziprasidone has been associated with QTc prolongation. For chronic pain patients, atypical antipsychotics are most useful for treating psychiatric comorbidities.
Although its mechanism of action is not well understood, acetaminophen (APAP) works by blocking prostaglandin syntheses via inhibition of cyclooxygenase-1 and cyclooxygenase-2 in the CNS.a Therefore, in contrast to NSAIDs, APAP does not possess peripheral anti-inflammatory effects or affect platelet function and is effective for treating fever, headache, and acute and chronic mild pain. The American Geriatrics Society recommends APAP for minor and persistent pain in older patientsb and the American College of Rheumatology recommends it as first-line therapy for osteoarthritis of the hip or knee.c
APAP has few clinically significant drug interactions, an excellent safety profile, and a long history of safe and effective use. When used within the recommended dosage range, APAP has few side effects. However, overuse of APAP is the leading cause of acute liver failure in the United States.d APAP hepatotoxicity can be accompanied by nephrotoxicity, is dose-dependent, and can be caused by acute overdose or chronic ingestion at doses over the recommended maximum of 4 g/d. Patients have experienced elevated liver transaminases with coadministration of APAP with phenytoin and phenobarbital.e,f Alcohol and other potentially hepatotoxic drugs also can increase the risk of liver toxicity when combined with APAP.d APAP is pregnancy category B and is considered the drug of choice for treating pain or fever during pregnancy and breast-feeding.g
References
- Amadio P Jr. Peripherally acting analgesics. Am J Med. 1984;77(3A):17-26.
- American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc. 2009;57(8):1331-1346.
- Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. Arthritis Rheum. 2000;43(9):1905-1915.
- Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42(6):1364-1372.
- Pirotte JH. Apparent potentiation of hepatotoxicity from small doses of acetaminophen by phenobarbital. Ann Intern Med. 1984;101(3):403.
- Brackett CC, Bloch JD. Phenytoin as a possible cause of acetaminophen hepatotoxicity: case report and review of the literature. Pharmacotherapy. 2000;20(2):229-233.
- Hersh EV, Moore PA, Ross GL. Over-the-counter analgesics and antipyretics: a critical assessment. Clin Ther. 2000; 22(5):500-548.
Related Resources
- Leo RJ. Chronic nonmalignant pain: How to ‘turn down’ its physiologic triggers. Current Psychiatry. 2008;7(8):19-36.
- Nikolaus T, Zeyfang A. Pharmacological treatments for persistent non-malignant pain in older persons. Drugs Aging. 2004;21(1):19-41.
- World Health Organization. WHO’s pain ladder. www.who.int/cancer/palliative/painladder/en.
Drug Brand Names
- Acetaminophen • Tylenol
- Amitriptyline • Elavil, others
- Amoxapine • Asendin
- Carbamazepine • Tegretol, Carbatrol, others
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Gabapentin • Neurontin, Gralise
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Methotrexate • Rheumatrex, Trexall
- Milnacipran • Savella
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Pregabalin • Lyrica
- Topiramate • Topamax, Topiragen
- Venlafaxine • Effexor
- iprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Of the 56 million American adults who report living with chronic pain almost 60% also exhibit psychiatric disorders such as depression or anxiety.1,2 Because patients with chronic pain suffer from a mixture of physical and psychological components, managing such conditions is complicated, and using opioids is tempting. However, treatment needs to address the underlying pathology along with social and psychological factors.
Because substance abuse treatment admissions increased by 400% from 1998 to 2008,3 many physicians look to non-opioids and other treatment modalities to control chronic non-cancer pain. Common pharmacologic therapies used to treat chronic pain include tricyclic antidepressants (TCAs), serotonin-norepinephrine reuptake inhibitors (SNRIs), antiepileptic drugs (AEDs), nonsteroidal anti-inflammatory drugs (NSAIDs), and, to a lesser extent, atypical antipsychotics. TCAs, SNRIs, AEDs, NSAIDs, and atypical antipsychotics influence a variety of presumed underlying pathophysiological processes, including inflammatory mediators, activity of N-methyl-d-aspartate (NMDA) receptors, and voltage-gated calcium channels. In addition, they increase activity of descending inhibitory pain pathways. Animal studies suggest dysfunction of these inhibitory mechanisms contributes to the central sensitization and hyperexcitability of pain transmitting pathways.4
In this article, we discuss psychotropics and other non-opioid agents for treating pain. However, no single solution is best for all patients with chronic pain and this article is not a “how to” guide to avoid administering opioid medication. Also incorporate a multimodal, non-pharmacologic approach whenever possible.
Tricyclic antidepressants
Although this class acts primarily by increasing serotonin levels, norepinephrine and dopamine also are affected depending on the particular medication. Studies have shown that amitriptyline, nortriptyline, and desipramine function well as analgesics independent of their antidepressant effects.5 TCAs may improve pain symptoms at lower therapeutic dosages than those used for treating depression.5
Although researchers have not elucidated TCAs’ mechanism of action with regards to analgesia, they are thought to act within the concept of the gating theory of pain control,6 which functions by activation and inhibition of pain signal transmission. It is believed TCAs act on nociceptive pathways by blocking serotonin and norepinephrine reuptake. Although researchers previously thought that TCAs’ analgesic mechanism was correlated to serotonin reuptake inhibition, this theory has changed. Selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine have not demonstrated substantial effectiveness in neuropathic pain when compared with TCAs and SNRIs. Recent studies have shown that TCAs may work by blocking sodium channels, similar to local anesthetics and antiarrhythmic agents.7
Psychiatrists prescribe TCAs infrequently because of these drugs’ unfavorable side effect profile compared with SSRIs and SNRIs. However, TCAs often are prescribed for pain management as an adjunct to other medications for neuropathic conditions and at lower dosages than those used for treating depression (Table 1).8
Table 1
Tricyclic antidepressants used to treat pain
| Drug | Dosage range for pain (off-label) | Comments |
|---|---|---|
| Amitriptyline | 10 to 100 mg/d | High sedation, high anticholinergic side effects |
| Amoxapine | 50 to 100 mg/d | Low sedation, moderate anticholinergic side effects |
| Clomipramine | 25 to 100 mg/d | Low sedation, low anticholinergic side effects |
| Desipramine | 25 to 100 mg/d | Low sedation, low anticholinergic side effects |
| Imipramine | 25 to 100 mg/d | Moderate sedation, moderate anticholinergic side effects |
| Nortriptyline | 10 to 75 mg/d | Moderate sedation, low anticholinergic side effects |
| Source: Reference 8 | ||
SNRIs
Evidence supports using duloxetine, a potent SNRI that mediates pain inhibition in the descending pathways, for 4 chronic pain conditions:
- diabetic peripheral neuropathic pain
- fibromyalgia
- mechanical low back pain
- pain associated with osteoarthritis.9
Titrate the dosage to 60 mg/d and maintain the patient at this dose for at least 4 weeks. Thereafter, according to patient response, the dosage may be titrated to 120 mg/d (off-label) with appropriate vital sign monitoring and routine lab analysis.
Venlafaxine also can mediate pain response in a similar manner to duloxetine, but is not FDA-approved for treating pain. Use caution when prescribing venlafaxine for patients with a history of hypertension. Milnacipran is a relatively new SNRI that has been shown to be effective in treating fibromyalgia in divided doses of 100 to 200 mg/d (Table 2).9-11
Table 2
Treating pain with serotonin-norepinephrine reuptake inhibitors
| Drug | Dosage range for pain | Comments |
|---|---|---|
| Duloxetine | 60 to 120 mg/d9 | FDA maximum recommended dose is 60 mg/d |
| Milnacipran | 25 to 200 mg/d10 | Approved for treating depression outside the United States |
| Venlafaxine | 75 to 225 mg/d11 | Monitor blood pressure, LFTs, and kidney function |
| LFTs: liver function tests | ||
Antiepileptic drugs
Several AEDs are used for pain management (Table 3).12-16 Gabapentin and pregabalin work by binding to voltage-gated calcium channels and decreasing excitatory neurotransmitter release. Along with TCAs, they are considered a first-line treatment for managing neuropathic pain.17 Gabapentin is FDA-approved for seizures and postherpetic neuralgia, but evidence supports its use in most types of neuropathic pain. Pregabalin is FDA-approved for treating seizures, diabetic peripheral neuropathy, central neuropathic pain, postherpetic neuralgia, and fibromyalgia.
Topiramate inhibits excitatory neurotransmission by enhancing the effects of gamma-aminobutyric acid, and also by blocking NMDA receptors. Topiramate is FDA-approved for seizures and migraine prophylaxis, and is used off-label for treating neuropathic pain. A 12-week trial of topiramate for diabetic neuropathy found significant analgesia in 50% of patients taking the drug, compared with 34% receiving placebo.18
Lamotrigine is approved for several types of seizures and maintenance of bipolar I disorder, and is used off-label for neuropathic pain. A recent Cochrane database review concluded that lamotrigine is ineffective for neuropathic pain14; however, some guidelines recommend using lamotrigine to treat neuropathies that do not respond to treatment with carbamazepine.19
Carbamazepine is a complex AED that is structurally similar to TCAs. It blocks sodium channels and has various pharmacologic properties, including anticholinergic, muscle relaxant, antidepressant, and sedative effects. Carbamazepine has analgesic effects through blockade of synaptic transmission in the trigeminal nucleus and is FDA-approved for seizures, bipolar disorder, neuropathic pain, and trigeminal neuralgia. In a systematic review of 12 trials of carbamazepine that included 4 placebo-controlled trials for trigeminal neuralgia, 2 studies showed a number needed to treat (NNT) of 1.8.20 For diabetic neuropathy, there was insufficient data to calculate NNT.
Oxcarbazepine, an analog of carbamazepine, also is FDA-approved for seizures and is used off-label for neuropathic pain. In the only double-blind trial with positive results, oxcarbazepine titrated to 1,800 mg/d reduced diabetic neuropathy pain scores on a visual analog scale by 24 points—roughly 25%.15
Table 3
Antiepileptic drugs for pain treatment
| Drug | Dosage range for pain | Comments |
|---|---|---|
| Carbamazepine | Starting dose: 100 mg twice a day, doses titrated to 400 to 800 mg/d usually are adequate. Maximum of 1,200 mg/d12 | Anticholinergic effects, blood dyscrasias, hyponatremia, increase in LFTs, ECG changes. CYP450 inducer, many DDIs |
| Gabapentin | Starting dose: 100 to 300 mg at bedtime or 100 to 300 mg 3 times a day, slow titration, maximum of 3,600 mg/d13 | Dizziness, sedation, weight gain, peripheral edema. Adjust dose in renal insufficiency |
| Lamotrigine | 200 to 400 mg/d14 | Sedation, headache, dizziness, ataxia, GI upset, blurred vision. Risk of life-threatening rash |
| Oxcarbazepine | Starting dose: 300 mg/d, then titrated as tolerated to a maximum of 1,800 mg/d15 | Adverse drug reactions similar to carbamazepine, less anticholinergic effects, more hyponatremia. Fewer DDIs than carbamazepine |
| Pregabalin | Starting dose: 50 mg 3 times a day or 75 mg twice a day, may increase every 3 to 7 days as tolerated, maximum of 600 mg/d13 | Same adverse drug reactions as gabapentin, less sedation. Adjust dose in renal insufficiency. More costly than gabapentin |
| Topiramate | Starting dose: 12.5 to 25 mg once or twice a day for 4 weeks; then double the dose every 4 weeks to reach a maximum dose of 100 to 200 mg/d in divided doses16 | Weight loss, anorexia, nephrolithiasis, cognitive impairment |
| CYP450: cytochrome P450; DDIs: drug-drug interactions; GI: gastrointestinal; LFTs: liver function tests | ||
Non-opioid analgesics
NSAIDs have antipyretic, analgesic, and anti-inflammatory effects and are used for fever, headache, mild-to-moderate pain, musculoskeletal pain, menstrual pain, and dental pain. They are particularly useful in treating acute pain, often in combination with opioid analgesics. NSAIDs exert their analgesic action through blockade of prostaglandin production via reversible inhibition of cyclooxygenase-1 and cyclooxygenase-2.
The most common side effects of NSAIDs are the result of gastrointestinal (GI) toxicity and include dyspepsia, heartburn, nausea, anorexia, and epigastric pain.21 GI ulceration and bleeding are rare but serious complications. To decrease these risks, tell patients to take NSAIDs with food. Add a GI protective agent, such as an H2 blocker or proton pump inhibitor, for patients at higher risk for GI complications.22
In addition, inhibition of renal prostaglandins by NSAIDs can cause renal toxicity, fluid retention, and edema, potentially exacerbating existing cardiovascular conditions such as hypertension and heart failure. NSAIDs may increase the risk of serious thrombotic events such as myocardial infarction and stroke. Use NSAIDs at the lowest effective dose for the shortest duration possible and generally avoid prescribing in patients at high risk for cardiovascular disease and pregnant women, especially those in their third trimester.23,24
NSAIDs may cause pharmacodynamic and pharmacokinetic drug-drug interactions. The risk of GI toxicity and bleeding increases when NSAIDs are administered with drugs that also irritate the gastric mucosa or have antiplatelet/anticoagulant effects.21 Plasma concentrations of drugs with a narrow therapeutic index that are renally eliminated, such as methotrexate and lithium, can increase to potentially toxic levels with concurrent NSAID use because NSAIDs decrease renal perfusion.21 Also, the therapeutic effects of antihypertensives may be attenuated because NSAIDs cause fluid retention.25
Acetaminophen (APAP) is available in several dosage forms as a single ingredient and in combination with opioids in prescription products. For more information about APAP, see the Box below.
Atypical antipsychotics
Although atypical antipsychotics are not often used to treat pain, studies indicate that fibromyalgia patients may benefit from ziprasidone26 and olanzapine,27 most often as an adjunctive treatment rather than monotherapy. Randomized controlled studies indicate poor tolerability with several atypical antipsychotics. Weight gain, akathisia, and somnolence are side effects of some atypical antipsychotics. Additionally, ziprasidone has been associated with QTc prolongation. For chronic pain patients, atypical antipsychotics are most useful for treating psychiatric comorbidities.
Although its mechanism of action is not well understood, acetaminophen (APAP) works by blocking prostaglandin syntheses via inhibition of cyclooxygenase-1 and cyclooxygenase-2 in the CNS.a Therefore, in contrast to NSAIDs, APAP does not possess peripheral anti-inflammatory effects or affect platelet function and is effective for treating fever, headache, and acute and chronic mild pain. The American Geriatrics Society recommends APAP for minor and persistent pain in older patientsb and the American College of Rheumatology recommends it as first-line therapy for osteoarthritis of the hip or knee.c
APAP has few clinically significant drug interactions, an excellent safety profile, and a long history of safe and effective use. When used within the recommended dosage range, APAP has few side effects. However, overuse of APAP is the leading cause of acute liver failure in the United States.d APAP hepatotoxicity can be accompanied by nephrotoxicity, is dose-dependent, and can be caused by acute overdose or chronic ingestion at doses over the recommended maximum of 4 g/d. Patients have experienced elevated liver transaminases with coadministration of APAP with phenytoin and phenobarbital.e,f Alcohol and other potentially hepatotoxic drugs also can increase the risk of liver toxicity when combined with APAP.d APAP is pregnancy category B and is considered the drug of choice for treating pain or fever during pregnancy and breast-feeding.g
References
- Amadio P Jr. Peripherally acting analgesics. Am J Med. 1984;77(3A):17-26.
- American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc. 2009;57(8):1331-1346.
- Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. Arthritis Rheum. 2000;43(9):1905-1915.
- Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42(6):1364-1372.
- Pirotte JH. Apparent potentiation of hepatotoxicity from small doses of acetaminophen by phenobarbital. Ann Intern Med. 1984;101(3):403.
- Brackett CC, Bloch JD. Phenytoin as a possible cause of acetaminophen hepatotoxicity: case report and review of the literature. Pharmacotherapy. 2000;20(2):229-233.
- Hersh EV, Moore PA, Ross GL. Over-the-counter analgesics and antipyretics: a critical assessment. Clin Ther. 2000; 22(5):500-548.
Related Resources
- Leo RJ. Chronic nonmalignant pain: How to ‘turn down’ its physiologic triggers. Current Psychiatry. 2008;7(8):19-36.
- Nikolaus T, Zeyfang A. Pharmacological treatments for persistent non-malignant pain in older persons. Drugs Aging. 2004;21(1):19-41.
- World Health Organization. WHO’s pain ladder. www.who.int/cancer/palliative/painladder/en.
Drug Brand Names
- Acetaminophen • Tylenol
- Amitriptyline • Elavil, others
- Amoxapine • Asendin
- Carbamazepine • Tegretol, Carbatrol, others
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Gabapentin • Neurontin, Gralise
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Methotrexate • Rheumatrex, Trexall
- Milnacipran • Savella
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Pregabalin • Lyrica
- Topiramate • Topamax, Topiragen
- Venlafaxine • Effexor
- iprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Brennan F, Carr DB, Cousins M. Pain management: a fundamental human right. Anesth Analg. 2007;105(1):205-221.
2. Thieme K, Turk DC, Flor H. Comorbid depression and anxiety in fibromyalgia syndrome: relationship to somatic and psychosocial variables. Psychosom Med. 2004;66(6):837-844.
3. Substance Abuse and Mental Health Services Administration, Office of Applied Studies Treatment episode data set (TEDS). 1998-2008. National admissions to substance abuse treatment services. Rockville MD: Substance Abuse and Mental Health Services Administration, Office of Applied Studies; 2010.
4. Iyengar S, Webster AA, Hemrick-Luecke SK, et al. Efficacy of duloxetine, a potent and balanced serotonin-norepinephrine reuptake inhibitor in persistent pain models in rats. J Pharmacol Exp Ther. 2004;311(2):576-584.
5. Guay DR. Adjunctive agents in the management of chronic pain. Pharmacotherapy. 2001;21(9):1070-1081.
6. Campbell LC, Clauw DJ, Keefe FJ. Persistent pain and depression: a biopsychosocial perspective. Biol Psychiatry. 2003;54(3):399-409.
7. Dick IE, Brochu RM, Purohit Y, et al. Sodium channel blockade may contribute to the analgesic efficacy of antidepressants. J Pain. 2007;8(4):315-324.
8. Stahl SM. Essential psychopharmacology: the prescriber’s guide. New York NY: Cambridge University Press; 2006.
9. Skljarevski V, Desaiah D, Liu-Seifert H, et al. Efficacy and safety of duloxetine in patients with chronic low back pain. Spine (Phila Pa 1976). 2010;35(13):E578-E585.
10. Hsu ES. Acute and chronic pain management in fibromyalgia: updates on pharmacotherapy. Am J Ther. 2011;18(6):487-509.
11. Bomholt SF, Mikkelsen JD, Blackburn-Munro G. Antinociceptive effects of the antidepressants amitriptyline duloxetine, mirtazapine and citalopram in animal models of acute, persistent and neuropathic pain. Neuropharmacology. 2005;48(2):252-263.
12. Campbell FG, Graham JG, Zilkha KJ. Clinical trial of carbazepine (tegretol) in trigeminal neuralgia. J Neurol Neurosurg Psychiatry. 1966;29(3):265-267.
13. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(10 suppl):S22-S32.
14. Dogra S, Beydoun S, Mazzola J, et al. Oxcarbazepine in painful diabetic neuropathy: a randomized, placebo-controlled study. Eur J Pain. 2005;9(5):543-554.
15. Kline KM, Carroll DG, Malnar KF. Painful diabetic peripheral neuropathy relieved with use of oral topiramate. South Med J. 2003;96(6):602-605.
16. Wiffen PJ, Derry S, Moore RA. Lamotrigine for acute and chronic pain. Cochrane Database Syst Rev. 2011;(2):CD006044.-
17. Dworkin RH, O’Connor AB, Audette J, et al. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc. 2010;85(3 suppl):S3-S14.
18. Raskin P, Donofrio PD, Rosenthal NR, et al. Topiramate vs placebo in painful diabetic neuropathy: analgesic and metabolic effects. Neurology. 2004;63(5):865-873.
19. Moulin DE, Clark AJ, Gilron I, et al. Pharmacological management of chronic neuropathic pain - consensus statement and guidelines from the Canadian Pain Society. Pain Res Manag. 2007;12(1):13-21.
20. Wiffen PJ, Derry S, Moore RA, et al. Carbamazepine for acute and chronic pain in adults. Cochrane Database Syst Rev. 2011;(1):CD005451.-
21. Hersh EV, Moore PA, Ross GL. Over-the-counter analgesics and antipyretics: a critical assessment. Clin Ther. 2000;22(5):500-548.
22. Lanas AI. Current approaches to reducing gastrointestinal toxicity of low-dose aspirin. Am J Med. 2001;110(1A):70S-73S.
23. Antman EM, Bennett JS, Daugherty A, et al. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115(12):1634-1642.
24. Briggs G, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation. 8th ed. Baltimore MD: Lippincott Williams and Wilkins; 2008.
25. Frishman WH. Effects of nonsteroidal anti-inflammatory drug therapy on blood pressure and peripheral edema. Am J Cardiol. 2002;89(6A):18D-25D.
26. Calandre EP, Hidalgo J, Rico-Villademoros F. Use of ziprasidone in patients with fibromyalgia: a case series. Rheumatol Int. 2007;27(5):473-476.
27. Rico-Villademoros F, Hidalgo J, Dominguez I, et al. Atypical antipsychotics in the treatment of fibromyalgia: a case series with olanzapine. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(1):161-164.
1. Brennan F, Carr DB, Cousins M. Pain management: a fundamental human right. Anesth Analg. 2007;105(1):205-221.
2. Thieme K, Turk DC, Flor H. Comorbid depression and anxiety in fibromyalgia syndrome: relationship to somatic and psychosocial variables. Psychosom Med. 2004;66(6):837-844.
3. Substance Abuse and Mental Health Services Administration, Office of Applied Studies Treatment episode data set (TEDS). 1998-2008. National admissions to substance abuse treatment services. Rockville MD: Substance Abuse and Mental Health Services Administration, Office of Applied Studies; 2010.
4. Iyengar S, Webster AA, Hemrick-Luecke SK, et al. Efficacy of duloxetine, a potent and balanced serotonin-norepinephrine reuptake inhibitor in persistent pain models in rats. J Pharmacol Exp Ther. 2004;311(2):576-584.
5. Guay DR. Adjunctive agents in the management of chronic pain. Pharmacotherapy. 2001;21(9):1070-1081.
6. Campbell LC, Clauw DJ, Keefe FJ. Persistent pain and depression: a biopsychosocial perspective. Biol Psychiatry. 2003;54(3):399-409.
7. Dick IE, Brochu RM, Purohit Y, et al. Sodium channel blockade may contribute to the analgesic efficacy of antidepressants. J Pain. 2007;8(4):315-324.
8. Stahl SM. Essential psychopharmacology: the prescriber’s guide. New York NY: Cambridge University Press; 2006.
9. Skljarevski V, Desaiah D, Liu-Seifert H, et al. Efficacy and safety of duloxetine in patients with chronic low back pain. Spine (Phila Pa 1976). 2010;35(13):E578-E585.
10. Hsu ES. Acute and chronic pain management in fibromyalgia: updates on pharmacotherapy. Am J Ther. 2011;18(6):487-509.
11. Bomholt SF, Mikkelsen JD, Blackburn-Munro G. Antinociceptive effects of the antidepressants amitriptyline duloxetine, mirtazapine and citalopram in animal models of acute, persistent and neuropathic pain. Neuropharmacology. 2005;48(2):252-263.
12. Campbell FG, Graham JG, Zilkha KJ. Clinical trial of carbazepine (tegretol) in trigeminal neuralgia. J Neurol Neurosurg Psychiatry. 1966;29(3):265-267.
13. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(10 suppl):S22-S32.
14. Dogra S, Beydoun S, Mazzola J, et al. Oxcarbazepine in painful diabetic neuropathy: a randomized, placebo-controlled study. Eur J Pain. 2005;9(5):543-554.
15. Kline KM, Carroll DG, Malnar KF. Painful diabetic peripheral neuropathy relieved with use of oral topiramate. South Med J. 2003;96(6):602-605.
16. Wiffen PJ, Derry S, Moore RA. Lamotrigine for acute and chronic pain. Cochrane Database Syst Rev. 2011;(2):CD006044.-
17. Dworkin RH, O’Connor AB, Audette J, et al. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc. 2010;85(3 suppl):S3-S14.
18. Raskin P, Donofrio PD, Rosenthal NR, et al. Topiramate vs placebo in painful diabetic neuropathy: analgesic and metabolic effects. Neurology. 2004;63(5):865-873.
19. Moulin DE, Clark AJ, Gilron I, et al. Pharmacological management of chronic neuropathic pain - consensus statement and guidelines from the Canadian Pain Society. Pain Res Manag. 2007;12(1):13-21.
20. Wiffen PJ, Derry S, Moore RA, et al. Carbamazepine for acute and chronic pain in adults. Cochrane Database Syst Rev. 2011;(1):CD005451.-
21. Hersh EV, Moore PA, Ross GL. Over-the-counter analgesics and antipyretics: a critical assessment. Clin Ther. 2000;22(5):500-548.
22. Lanas AI. Current approaches to reducing gastrointestinal toxicity of low-dose aspirin. Am J Med. 2001;110(1A):70S-73S.
23. Antman EM, Bennett JS, Daugherty A, et al. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115(12):1634-1642.
24. Briggs G, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation. 8th ed. Baltimore MD: Lippincott Williams and Wilkins; 2008.
25. Frishman WH. Effects of nonsteroidal anti-inflammatory drug therapy on blood pressure and peripheral edema. Am J Cardiol. 2002;89(6A):18D-25D.
26. Calandre EP, Hidalgo J, Rico-Villademoros F. Use of ziprasidone in patients with fibromyalgia: a case series. Rheumatol Int. 2007;27(5):473-476.
27. Rico-Villademoros F, Hidalgo J, Dominguez I, et al. Atypical antipsychotics in the treatment of fibromyalgia: a case series with olanzapine. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(1):161-164.
Psychotropic-induced dry mouth: Don’t overlook this potentially serious side effect
Discuss this article at www.facebook.com/CurrentPsychiatry
Xerostomia, commonly known as “dry mouth,” is a reported side effect of >1,800 drugs from >80 classes.1 This condition often goes unrecognized and untreated, but it can significantly affect patients’ quality of life and cause oral and medical health problems.2,3 Although psychotropic medications are not the only offenders, they comprise a large portion of the agents that can cause dry mouth. Antidepressants, anticonvulsants, anxiolytics, antipsychotics, anticholinergics, and alpha agonists can cause xerostomia.4 The risk of salivary hypofunction increases with polypharmacy and may be especially likely when ≥3 drugs are taken per day.5
Among all reported side effects of antidepressants and antipsychotics, dry mouth often is the most prevalent complaint. For example, in a study of 5 antidepressants 35% to 46% of patients reported dry mouth.6 Rates are similar in users of various antipsychotics. Patients with severe, persistent mental illness often cite side effects as the primary reason for psychotropic noncompliance.7-9
Few psychiatrists routinely screen patients for xerostomia, and if a patient reports this side effect, they may be unlikely to address it or understand its implications because of more pressing concerns such as psychosis or risk of suicide. Historically, education in general medical training about the effects of oral health on a patient’s overall health has been limited. It is crucial for psychiatrists to be aware of potential problems related to dry mouth and the impact it can have on their patients. In this article, we:
- describe how dry mouth can impact a patient’s oral, medical, and psychiatric health
- provide psychiatrists with an understanding of pathology related to xerostomia
- explain how psychiatrists can screen for xerostomia
- discuss the benefits patients may receive when psychiatrists collaborate with dental clinicians to manage this condition.
Implications of xerostomia
Saliva provides a protective function. It is an antimicrobial, buffering, and lubricating agent that aids cleansing and removal of food debris within the mouth. It also helps maintain oral mucosa and remineralizing of tooth structure.10
Psychotropics can affect the amount of saliva secreted and may alter the composition of saliva via their receptor affects on the dual sympathetic and parasympathetic innervations of the salivary glands.11 When the protective environment produced by saliva is altered, patients may start to develop oral problems before experiencing dryness. A 50% reduction in saliva flow may occur before they become aware of the problem.12,13
Patients may not taste food properly, experience cracked lips, or have trouble eating, oral pain, or dentures that no longer fit well.14 Additionally, oral diseases such as dental decay and periodontal disease (Photos 1 and 2), inflamed soft tissue, and candidiasis (Photo 3) also may occur.10,15 Patients may begin to notice dry mouth when they wake at night, which could disrupt sleep. Patients with xerostomia can accumulate excessive amounts of plaque on their teeth and the dorsum of the tongue. The increased bacterial count and release of volatile sulfide gases that occur with dry mouth may explain some cases of halitosis.16,17 Patients also may have difficulty swallowing or speaking and be unaware of the oral health destruction occurring as a result of reduced saliva. Some experts report oral bacteria levels can skyrocket as much as 10-fold in people who take medications that cause dry mouth.18
Infections of the mouth can create havoc elsewhere in the body. The evidence base that establishes an association between periodontal disease and other chronic inflammatory conditions such as diabetes, cardiovascular disease, cancer, and rheumatoid arthritis is steadily growing.19-22 Periodontal disease also is a risk factor for preeclampsia and other illnesses that can negatively affect neonatal health.23,24
Failure to recognize xerostomia caused by psychotropic medications may lead to an increase in cavities, periodontal disease, and chronic systemic inflammatory conditions that can shorten a patient’s life span. Recognizing and treating causes of xerostomia is vital because doing so may halt this chain of events.

Photo 1
This patient complained of dry mouth and exhibits decay (a) and evidence of periodontal disease. Plaque and calculus is present (b), along with gingival recession from the loss of attachment and bone (c). This patient was taking venlafaxine, zolpidem, and alprazolam
Photo 2
Dental cavities were restored with tooth-colored restorations (arrows) on this patient, who has xerostomia. Every effort must be made to manage this patient’s dry mouth or the restorations may fail due to recurrent decay
Photo 3
This partial denture wearer, who complained of dry mouth, has evidence of palatal irritation and sores as a result of xerostomia and use of a partial denture. This patient was taking bupropion, esomeprazole, and tolterodine
Psychiatric patients’ oral health
Psychiatric patients’ oral health status often is poor. Several studies found that compared with the general population, patients who have severe, persistent mental illness are at higher risk to be missing teeth, schedule fewer visits to the dentist, and neglect oral hygiene.25-28 Periodontal disease also could be a problem in these patients.29 Although some evidence suggests mental illness may make patients less likely to go to the dentist, psychotropic medications also may contribute to their dental difficulties.
Screening for xerostomia
Simply advising patients of the problems related to xerostomia and asking several questions may help prevent pain and deterioration in function within the oral cavity (Table 1).14,30
You can perform a simple in-office assessment of the oral cavity by visual inspection and by placing a dry tongue blade against the inside of the cheek mucosa. If the blade sticks to the mucosa and a gentle tug is needed to lift it away, xerostomia may be present.30 Conversely, a healthy mouth will have a collection of saliva on the floor of the oral cavity, and pulling a tongue blade away from the inside of the cheek will not require any effort (Photos 4 and 5).
Table 1
Screening questions for xerostomia
| Does the amount of saliva in your mouth seem to have decreased? |
| Do you have any trouble swallowing, speaking, or eating dry foods? |
| Do you sip liquids more often to help you swallow? |
| Do you notice any dryness or cracking of your lips? |
| Do you have mouth sores or a burning feeling in the mouth? |
| When was the last time you saw your dentist? (Patients with xerostomia may need to see their dentist more frequently) |
| Are you aware of any halitosis (ie, mouth odor)? |
| Source: Reference 14 |
Photo 4
The arrow shows the normal appearance of saliva collecting on the floor of the mouth
Photo 5
This patient complained of dry mouth. Note the floor of the mouth is free of saliva (a). Decay is present (b), and the patient is missing posterior teeth (c). This patient was taking clonidine, metoprolol, hydrochlorothiazide, amlodipine, and irbesartan
Treatment options
Patients who have reduced salivary flow as a result of a medication may become so affected by dryness that their drug regimen may need to be changed. However, the greatest concern is for deteriorating oral health among patients who may be unaware xerostomia is occurring.31
Counsel patients who take medications that can affect their salivary function about the importance of seeing a dentist regularly, and provide referrals when appropriate. Depending upon the patient’s oral health, dentists recommend patients with xerostomia have their teeth cleaned/examined 3 or 4 times per year, rather than the 2 times per year allowed by third-party payers (ie, insurance companies). Also advise patients to be diligent in their oral hygiene practices, including flossing and brushing the teeth and tongue, and to avoid foods that are sticky and/or have high sucrose content (Table 2). Recommend using a toothpaste containing fluoride—preferably one free of sodium lauryl sulfate, which could contribute to mouth sores14—and drinking fluoridated water. Explain to patients that their dentist may recommend in-office high-fluoride applications, high-fluoride prescription toothpaste, and/or “mouth trays” that contain high fluoride gel. Tell patients to avoid cigarettes and caffeinated beverages, which can increase dryness. Alcohol use should be minimized and mouth rinses containing alcohol should not be used.
Many over-the-counter products are available to address xerostomia, including toothpastes, mouth rinses, and gels. Salivary substitutes—which are available as sprays, liquids, tablets, and swab sticks—imitate saliva and may provide a temporary reprieve from dryness. Although none of these products will cure dry mouth, they may help manage the condition. Advise patients to eat foods that stimulate saliva production, such as carrots, apples, and celery, and to chew sugarless gum and candies, which also will stimulate salivary flow.
The FDA has approved 2 prescription drugs for treating xerostomia: cevimeline and pilocarpine. Cevimeline is approved for treating dry mouth associated with Sjögren’s syndrome and pilocarpine is approved for treating dry mouth caused by head and neck radiation therapy; however, these medications’ role in treating dry mouth in psychiatric patients has not been investigated. Both agents are contraindicated in patients with narrow-angle glaucoma, uncontrolled asthma, or liver disease, and should be prescribed with caution for patients with cardiovascular disease, chronic respiratory conditions, or kidney disease.32
Acupuncture and electrostimulation are being studied as a treatment for xerostomia. Trials have found acupuncture improves symptoms of xerostomia,33,34 and 1 study found electrostimulation improved xerostomia in patients with Sjögren’s syndrome.35 Both approaches require more study to confirm their effectiveness.33-35
Table 2
Managing dry mouth: What to tell patients
| Oral hygiene. Tell patients to be diligent in their oral hygiene practices, including brushing and flossing. They should use a toothpaste containing fluoride—preferably one free of sodium lauryl sulfate—and schedule regular dental visits, where they can receive high-fluoride applications or be prescribed high-fluoride prescription toothpastes |
| Diet. Advise patients to avoid foods high in sucrose content, rinse their mouth with water soon after eating, and drink fluoridated water regularly. Tell them that they may be able to stimulate saliva flow with sugarless gum, candies, and foods such as celery and carrots |
| Drying agents. Instruct patients to avoid cigarettes, caffeinated beverages, and mouth rinses that contain alcohol. Explain that some patients may benefit from sleeping in a room with a cool air humidifier |
| Over-the-counter products. Suggest patients try salivary substitutes, which are dispensed in spray bottles, rinses, swish bottles, or oral swab sticks. In addition, products such as dry-mouth toothpaste and moisturizing gels also may help relieve their symptoms |
- Persson K, Axtelius B, Söderfeldt B, et al. Monitoring oral health and dental attendance in an outpatient psychiatric population. J Psychiatr Ment Health Nurs. 2009;16(3):263-271.
- Keene JJ Jr, Galasko GT, Land MF. Antidepressant use in psychiatry and medicine: importance for dental practice. J Am Dent Assoc. 2003;134(1):71-79.
Drug Brand Names
- Alprazolam • Xanax
- Amlodipine • Norvasc
- Bupropion • Wellbutrin, Zyban
- Cevimeline • Evoxac
- Clonidine • Catapres, Kapvay, others
- Esomeprazole • Nexium
- Irbesartan • Avapro
- Metoprolol • Lopressor, Toprol
- Pilocarpine • Salagen
- Tolterodine • Detrol
- Venlafaxine • Effexor
- Zolpidem • Ambien
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Drymouth.info. Overview of drugs and dry mouth. http://drymouth.info/practitioner/overview.asp. Accessed September 2, 2011.
2. Stewart CM, Berg KM, Cha S, et al. Salivary dysfunction and quality of life in Sjögren syndrome: a critical oral-systemic connection. J Am Dent Assoc. 2008;139(3):291-299.
3. Friedman PK. Xerostomia: The invisible oral health condition. http://www.dentistryiq.com/index/display/article-display/295922/articles/woman-dentist-journal/health/xerostomia-the-invisible-oral-health-condition.html. Accessed September 6, 2011.
4. Physician Desk Reference. Montvale NJ: PDR Network LLC.; 2011.
5. Bardow A, Lagerlof F, Nauntofte B, et al. The role of saliva. In: Fejerskov O, Kidd E, eds. Dental caries: the disease and its clinical management. Oxford, United Kingdom: Blackwell Munksgaard; 2008:195.
6. Vanderkooy JD, Kennedy SH, Bagby RM. Antidepressant side effects in depression patients treated in a naturalistic setting: a study of bupropion moclobemide, paroxetine, sertraline, and venlafaxine. Can J Psychiatry. 2002;47(2):174-180.
7. Löffler W, Kilian R, Toumi M, et al. Schizophrenic patients’ subjective reasons for compliance and noncompliance with neuroleptic treatment. Pharmacopsychiatry. 2003;36(3):105-112.
8. Lambert M, Conus P, Eide P, et al. Impact of present and past antipsychotic side effects on attitude toward typical antipsychotic treatment and adherence. Eur Psychiatry. 2004;19(7):415-422.
9. Rettenbacher MA, Hofer A, Eder U, et al. Compliance in schizophrenia: psychopathology, side effects, and patients’ attitudes toward the illness and medication. J Clin Psychiatry. 2004;65(9):1211-1218.
10. Bulkacz J, Carranza FA. Defense mechanisms of the gingiva. In: Newman MG, Takei HH, Klokkevold PR, et al, eds. Carranza’s clinical periodontology. St. Louis, MO: Elsevier Saunders; 2011:69–70.
11. Szabadi E, Tavernor S. Hypo-and hyper-salivation induced by psychoactive drugs. CNS Drugs. 1999;11(6):449-466.
12. Guggenheimer J, Moore PA. Xerostomia: etiology recognition and treatment. J Am Dent Assoc. 2003;134(1):61-69.
13. Dawes C. Physiological factors affecting salivary flow rate oral sugar clearance, and the sensation of dry mouth in man. J Dent Res. 1987;66:648-653.
14. Bartels CL. Xerostomia information for dentists. http://www.homesteadschools.com/dental/courses/Xerostomia/Course.htm. Accessed August 15, 2011.
15. Sitheeque MA, Samaranayake LP. Chronic hyperplastic candidosis/candidiasis (candidal leukoplakia). Crit Rev Oral Biol Med. 2003;14(4):253-267.
16. Porter SR, Scully C. Oral malodour (halitosis). BMJ. 2006;333(7569):632-635.
17. Quirynen M, Van den Veide S, Vanderkerckhove B, et al. Oral malodor. In: Newman MG, Takei HH, Klokkevold PR, et al, eds. Carranza’s clinical periodontology. St. Louis, MO: Elsevier Saunders; 2011:333.
18. Papas A. Dry mouth from drugs: more than just an annoying side effect. Tufts University Heath and Nutrition Letter. 2000;3.-
19. American Academy of Periodontology. Gum disease information from the American Academy of Periodontology http://perio.org. Accessed August 12, 2011.
20. Geismar K, Stoltze K, Sigurd B, et al. Periodontal disease and coronary heart disease. J Periodontol. 2006;77(9):1547-1554.
21. Lee HJ, Garcia RI, Janket SJ, et al. The association between cumulative periodontal disease and stroke history in older adults. J Periodontol. 2006;77(10):1744-1754.
22. Friedewald VE, Kornman KS, Beck JD, et al. The American Journal of Cardiology and Journal of Periodontology editors’ consensus: periodontitis and atherosclerotic cardiovascular disease. J Periodontol. 2009;80(7):1021-1032.
23. Contreras A, Herrera JA, Soto JE, et al. Periodontitis is associated with preeclampsia in pregnant women. J Periodontol. 2006;77(2):182-188.
24. Dasanayake AP, Li Y, Wiener H, et al. Salivary Actinomyces naeslundii genospecies 2 and Lactobacillus casei levels predict pregnancy outcomes. J Periodontol. 2005;76(2):171-177.
25. McCreadie RG, Stevens H, Henderson J, et al. The dental health of people with schizophrenia. Acta Psychiatr Scand. 2004;110(4):306-310.
26. Anttila S, Knuuttila M, Ylöstalo P, et al. Symptoms of depression and anxiety in relation to dental health behavior and self-perceived dental treatment need. Eur J Oral Sci. 2006;114(2):109-114.
27. Sjögren R, Nordström G. Oral health status of psychiatric patients. J Clin Nurs. 2000;9(4):632-638.
28. Ramon T, Grinshpoon A, Zusman SP, et al. Oral health and treatment needs of institutionalized chronic psychiatric patients in Israel. Eur Psychiatry. 2003;18(3):101-105.
29. Portilla MI, Mafla AC, Arteaga JJ. Periodontal status in female psychiatric patients. Colomb Med. 2009;40(2):167-176.
30. Navazesh M. ADA Council on Scientific Affairs and Division of Science. How can oral health care providers determine if patients have dry mouth? J Am Dent Assoc. 2003;134(5):613-620.
31. Mignogna MD, Fedele S, Lo Russo L, et al. Sjögren’s syndrome: the diagnostic potential of early oral manifestations preceding hyposalivation/xerostomia. J Oral Pathol Med. 2005;34(1):1-6.
32. Spolarich AE. Managing the side effects of medications. J Dent Hyg. 2000;74(1):57-69.
33. Johnstone PA, Niemtzow RC, Riffenburgh RH. Acupuncture for xerostomia: clinical update. Cancer. 2002;94(4):1151-1156.
34. Garcia MK, Chiang JS, Cohen L, et al. Acupuncture for radiation-induced xerostomia in patients with cancer: a pilot study. Head Neck. 2009;31(10):1360-1368.
35. Strietzel FP, Lafaurie GI, Mendoza GR, et al. Efficacy and safety of an intraoral electrostimulation device for xerostomia relief: a multicenter, randomized trial. Arthritis Rheum. 2011;63(1):180-190.
Discuss this article at www.facebook.com/CurrentPsychiatry
Xerostomia, commonly known as “dry mouth,” is a reported side effect of >1,800 drugs from >80 classes.1 This condition often goes unrecognized and untreated, but it can significantly affect patients’ quality of life and cause oral and medical health problems.2,3 Although psychotropic medications are not the only offenders, they comprise a large portion of the agents that can cause dry mouth. Antidepressants, anticonvulsants, anxiolytics, antipsychotics, anticholinergics, and alpha agonists can cause xerostomia.4 The risk of salivary hypofunction increases with polypharmacy and may be especially likely when ≥3 drugs are taken per day.5
Among all reported side effects of antidepressants and antipsychotics, dry mouth often is the most prevalent complaint. For example, in a study of 5 antidepressants 35% to 46% of patients reported dry mouth.6 Rates are similar in users of various antipsychotics. Patients with severe, persistent mental illness often cite side effects as the primary reason for psychotropic noncompliance.7-9
Few psychiatrists routinely screen patients for xerostomia, and if a patient reports this side effect, they may be unlikely to address it or understand its implications because of more pressing concerns such as psychosis or risk of suicide. Historically, education in general medical training about the effects of oral health on a patient’s overall health has been limited. It is crucial for psychiatrists to be aware of potential problems related to dry mouth and the impact it can have on their patients. In this article, we:
- describe how dry mouth can impact a patient’s oral, medical, and psychiatric health
- provide psychiatrists with an understanding of pathology related to xerostomia
- explain how psychiatrists can screen for xerostomia
- discuss the benefits patients may receive when psychiatrists collaborate with dental clinicians to manage this condition.
Implications of xerostomia
Saliva provides a protective function. It is an antimicrobial, buffering, and lubricating agent that aids cleansing and removal of food debris within the mouth. It also helps maintain oral mucosa and remineralizing of tooth structure.10
Psychotropics can affect the amount of saliva secreted and may alter the composition of saliva via their receptor affects on the dual sympathetic and parasympathetic innervations of the salivary glands.11 When the protective environment produced by saliva is altered, patients may start to develop oral problems before experiencing dryness. A 50% reduction in saliva flow may occur before they become aware of the problem.12,13
Patients may not taste food properly, experience cracked lips, or have trouble eating, oral pain, or dentures that no longer fit well.14 Additionally, oral diseases such as dental decay and periodontal disease (Photos 1 and 2), inflamed soft tissue, and candidiasis (Photo 3) also may occur.10,15 Patients may begin to notice dry mouth when they wake at night, which could disrupt sleep. Patients with xerostomia can accumulate excessive amounts of plaque on their teeth and the dorsum of the tongue. The increased bacterial count and release of volatile sulfide gases that occur with dry mouth may explain some cases of halitosis.16,17 Patients also may have difficulty swallowing or speaking and be unaware of the oral health destruction occurring as a result of reduced saliva. Some experts report oral bacteria levels can skyrocket as much as 10-fold in people who take medications that cause dry mouth.18
Infections of the mouth can create havoc elsewhere in the body. The evidence base that establishes an association between periodontal disease and other chronic inflammatory conditions such as diabetes, cardiovascular disease, cancer, and rheumatoid arthritis is steadily growing.19-22 Periodontal disease also is a risk factor for preeclampsia and other illnesses that can negatively affect neonatal health.23,24
Failure to recognize xerostomia caused by psychotropic medications may lead to an increase in cavities, periodontal disease, and chronic systemic inflammatory conditions that can shorten a patient’s life span. Recognizing and treating causes of xerostomia is vital because doing so may halt this chain of events.
Photo 1
This patient complained of dry mouth and exhibits decay (a) and evidence of periodontal disease. Plaque and calculus is present (b), along with gingival recession from the loss of attachment and bone (c). This patient was taking venlafaxine, zolpidem, and alprazolam
Photo 2
Dental cavities were restored with tooth-colored restorations (arrows) on this patient, who has xerostomia. Every effort must be made to manage this patient’s dry mouth or the restorations may fail due to recurrent decay
Photo 3
This partial denture wearer, who complained of dry mouth, has evidence of palatal irritation and sores as a result of xerostomia and use of a partial denture. This patient was taking bupropion, esomeprazole, and tolterodine
Psychiatric patients’ oral health
Psychiatric patients’ oral health status often is poor. Several studies found that compared with the general population, patients who have severe, persistent mental illness are at higher risk to be missing teeth, schedule fewer visits to the dentist, and neglect oral hygiene.25-28 Periodontal disease also could be a problem in these patients.29 Although some evidence suggests mental illness may make patients less likely to go to the dentist, psychotropic medications also may contribute to their dental difficulties.
Screening for xerostomia
Simply advising patients of the problems related to xerostomia and asking several questions may help prevent pain and deterioration in function within the oral cavity (Table 1).14,30
You can perform a simple in-office assessment of the oral cavity by visual inspection and by placing a dry tongue blade against the inside of the cheek mucosa. If the blade sticks to the mucosa and a gentle tug is needed to lift it away, xerostomia may be present.30 Conversely, a healthy mouth will have a collection of saliva on the floor of the oral cavity, and pulling a tongue blade away from the inside of the cheek will not require any effort (Photos 4 and 5).
Table 1
Screening questions for xerostomia
| Does the amount of saliva in your mouth seem to have decreased? |
| Do you have any trouble swallowing, speaking, or eating dry foods? |
| Do you sip liquids more often to help you swallow? |
| Do you notice any dryness or cracking of your lips? |
| Do you have mouth sores or a burning feeling in the mouth? |
| When was the last time you saw your dentist? (Patients with xerostomia may need to see their dentist more frequently) |
| Are you aware of any halitosis (ie, mouth odor)? |
| Source: Reference 14 |
Photo 4
The arrow shows the normal appearance of saliva collecting on the floor of the mouth
Photo 5
This patient complained of dry mouth. Note the floor of the mouth is free of saliva (a). Decay is present (b), and the patient is missing posterior teeth (c). This patient was taking clonidine, metoprolol, hydrochlorothiazide, amlodipine, and irbesartan
Treatment options
Patients who have reduced salivary flow as a result of a medication may become so affected by dryness that their drug regimen may need to be changed. However, the greatest concern is for deteriorating oral health among patients who may be unaware xerostomia is occurring.31
Counsel patients who take medications that can affect their salivary function about the importance of seeing a dentist regularly, and provide referrals when appropriate. Depending upon the patient’s oral health, dentists recommend patients with xerostomia have their teeth cleaned/examined 3 or 4 times per year, rather than the 2 times per year allowed by third-party payers (ie, insurance companies). Also advise patients to be diligent in their oral hygiene practices, including flossing and brushing the teeth and tongue, and to avoid foods that are sticky and/or have high sucrose content (Table 2). Recommend using a toothpaste containing fluoride—preferably one free of sodium lauryl sulfate, which could contribute to mouth sores14—and drinking fluoridated water. Explain to patients that their dentist may recommend in-office high-fluoride applications, high-fluoride prescription toothpaste, and/or “mouth trays” that contain high fluoride gel. Tell patients to avoid cigarettes and caffeinated beverages, which can increase dryness. Alcohol use should be minimized and mouth rinses containing alcohol should not be used.
Many over-the-counter products are available to address xerostomia, including toothpastes, mouth rinses, and gels. Salivary substitutes—which are available as sprays, liquids, tablets, and swab sticks—imitate saliva and may provide a temporary reprieve from dryness. Although none of these products will cure dry mouth, they may help manage the condition. Advise patients to eat foods that stimulate saliva production, such as carrots, apples, and celery, and to chew sugarless gum and candies, which also will stimulate salivary flow.
The FDA has approved 2 prescription drugs for treating xerostomia: cevimeline and pilocarpine. Cevimeline is approved for treating dry mouth associated with Sjögren’s syndrome and pilocarpine is approved for treating dry mouth caused by head and neck radiation therapy; however, these medications’ role in treating dry mouth in psychiatric patients has not been investigated. Both agents are contraindicated in patients with narrow-angle glaucoma, uncontrolled asthma, or liver disease, and should be prescribed with caution for patients with cardiovascular disease, chronic respiratory conditions, or kidney disease.32
Acupuncture and electrostimulation are being studied as a treatment for xerostomia. Trials have found acupuncture improves symptoms of xerostomia,33,34 and 1 study found electrostimulation improved xerostomia in patients with Sjögren’s syndrome.35 Both approaches require more study to confirm their effectiveness.33-35
Table 2
Managing dry mouth: What to tell patients
| Oral hygiene. Tell patients to be diligent in their oral hygiene practices, including brushing and flossing. They should use a toothpaste containing fluoride—preferably one free of sodium lauryl sulfate—and schedule regular dental visits, where they can receive high-fluoride applications or be prescribed high-fluoride prescription toothpastes |
| Diet. Advise patients to avoid foods high in sucrose content, rinse their mouth with water soon after eating, and drink fluoridated water regularly. Tell them that they may be able to stimulate saliva flow with sugarless gum, candies, and foods such as celery and carrots |
| Drying agents. Instruct patients to avoid cigarettes, caffeinated beverages, and mouth rinses that contain alcohol. Explain that some patients may benefit from sleeping in a room with a cool air humidifier |
| Over-the-counter products. Suggest patients try salivary substitutes, which are dispensed in spray bottles, rinses, swish bottles, or oral swab sticks. In addition, products such as dry-mouth toothpaste and moisturizing gels also may help relieve their symptoms |
- Persson K, Axtelius B, Söderfeldt B, et al. Monitoring oral health and dental attendance in an outpatient psychiatric population. J Psychiatr Ment Health Nurs. 2009;16(3):263-271.
- Keene JJ Jr, Galasko GT, Land MF. Antidepressant use in psychiatry and medicine: importance for dental practice. J Am Dent Assoc. 2003;134(1):71-79.
Drug Brand Names
- Alprazolam • Xanax
- Amlodipine • Norvasc
- Bupropion • Wellbutrin, Zyban
- Cevimeline • Evoxac
- Clonidine • Catapres, Kapvay, others
- Esomeprazole • Nexium
- Irbesartan • Avapro
- Metoprolol • Lopressor, Toprol
- Pilocarpine • Salagen
- Tolterodine • Detrol
- Venlafaxine • Effexor
- Zolpidem • Ambien
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Xerostomia, commonly known as “dry mouth,” is a reported side effect of >1,800 drugs from >80 classes.1 This condition often goes unrecognized and untreated, but it can significantly affect patients’ quality of life and cause oral and medical health problems.2,3 Although psychotropic medications are not the only offenders, they comprise a large portion of the agents that can cause dry mouth. Antidepressants, anticonvulsants, anxiolytics, antipsychotics, anticholinergics, and alpha agonists can cause xerostomia.4 The risk of salivary hypofunction increases with polypharmacy and may be especially likely when ≥3 drugs are taken per day.5
Among all reported side effects of antidepressants and antipsychotics, dry mouth often is the most prevalent complaint. For example, in a study of 5 antidepressants 35% to 46% of patients reported dry mouth.6 Rates are similar in users of various antipsychotics. Patients with severe, persistent mental illness often cite side effects as the primary reason for psychotropic noncompliance.7-9
Few psychiatrists routinely screen patients for xerostomia, and if a patient reports this side effect, they may be unlikely to address it or understand its implications because of more pressing concerns such as psychosis or risk of suicide. Historically, education in general medical training about the effects of oral health on a patient’s overall health has been limited. It is crucial for psychiatrists to be aware of potential problems related to dry mouth and the impact it can have on their patients. In this article, we:
- describe how dry mouth can impact a patient’s oral, medical, and psychiatric health
- provide psychiatrists with an understanding of pathology related to xerostomia
- explain how psychiatrists can screen for xerostomia
- discuss the benefits patients may receive when psychiatrists collaborate with dental clinicians to manage this condition.
Implications of xerostomia
Saliva provides a protective function. It is an antimicrobial, buffering, and lubricating agent that aids cleansing and removal of food debris within the mouth. It also helps maintain oral mucosa and remineralizing of tooth structure.10
Psychotropics can affect the amount of saliva secreted and may alter the composition of saliva via their receptor affects on the dual sympathetic and parasympathetic innervations of the salivary glands.11 When the protective environment produced by saliva is altered, patients may start to develop oral problems before experiencing dryness. A 50% reduction in saliva flow may occur before they become aware of the problem.12,13
Patients may not taste food properly, experience cracked lips, or have trouble eating, oral pain, or dentures that no longer fit well.14 Additionally, oral diseases such as dental decay and periodontal disease (Photos 1 and 2), inflamed soft tissue, and candidiasis (Photo 3) also may occur.10,15 Patients may begin to notice dry mouth when they wake at night, which could disrupt sleep. Patients with xerostomia can accumulate excessive amounts of plaque on their teeth and the dorsum of the tongue. The increased bacterial count and release of volatile sulfide gases that occur with dry mouth may explain some cases of halitosis.16,17 Patients also may have difficulty swallowing or speaking and be unaware of the oral health destruction occurring as a result of reduced saliva. Some experts report oral bacteria levels can skyrocket as much as 10-fold in people who take medications that cause dry mouth.18
Infections of the mouth can create havoc elsewhere in the body. The evidence base that establishes an association between periodontal disease and other chronic inflammatory conditions such as diabetes, cardiovascular disease, cancer, and rheumatoid arthritis is steadily growing.19-22 Periodontal disease also is a risk factor for preeclampsia and other illnesses that can negatively affect neonatal health.23,24
Failure to recognize xerostomia caused by psychotropic medications may lead to an increase in cavities, periodontal disease, and chronic systemic inflammatory conditions that can shorten a patient’s life span. Recognizing and treating causes of xerostomia is vital because doing so may halt this chain of events.
Photo 1
This patient complained of dry mouth and exhibits decay (a) and evidence of periodontal disease. Plaque and calculus is present (b), along with gingival recession from the loss of attachment and bone (c). This patient was taking venlafaxine, zolpidem, and alprazolam
Photo 2
Dental cavities were restored with tooth-colored restorations (arrows) on this patient, who has xerostomia. Every effort must be made to manage this patient’s dry mouth or the restorations may fail due to recurrent decay
Photo 3
This partial denture wearer, who complained of dry mouth, has evidence of palatal irritation and sores as a result of xerostomia and use of a partial denture. This patient was taking bupropion, esomeprazole, and tolterodine
Psychiatric patients’ oral health
Psychiatric patients’ oral health status often is poor. Several studies found that compared with the general population, patients who have severe, persistent mental illness are at higher risk to be missing teeth, schedule fewer visits to the dentist, and neglect oral hygiene.25-28 Periodontal disease also could be a problem in these patients.29 Although some evidence suggests mental illness may make patients less likely to go to the dentist, psychotropic medications also may contribute to their dental difficulties.
Screening for xerostomia
Simply advising patients of the problems related to xerostomia and asking several questions may help prevent pain and deterioration in function within the oral cavity (Table 1).14,30
You can perform a simple in-office assessment of the oral cavity by visual inspection and by placing a dry tongue blade against the inside of the cheek mucosa. If the blade sticks to the mucosa and a gentle tug is needed to lift it away, xerostomia may be present.30 Conversely, a healthy mouth will have a collection of saliva on the floor of the oral cavity, and pulling a tongue blade away from the inside of the cheek will not require any effort (Photos 4 and 5).
Table 1
Screening questions for xerostomia
| Does the amount of saliva in your mouth seem to have decreased? |
| Do you have any trouble swallowing, speaking, or eating dry foods? |
| Do you sip liquids more often to help you swallow? |
| Do you notice any dryness or cracking of your lips? |
| Do you have mouth sores or a burning feeling in the mouth? |
| When was the last time you saw your dentist? (Patients with xerostomia may need to see their dentist more frequently) |
| Are you aware of any halitosis (ie, mouth odor)? |
| Source: Reference 14 |
Photo 4
The arrow shows the normal appearance of saliva collecting on the floor of the mouth
Photo 5
This patient complained of dry mouth. Note the floor of the mouth is free of saliva (a). Decay is present (b), and the patient is missing posterior teeth (c). This patient was taking clonidine, metoprolol, hydrochlorothiazide, amlodipine, and irbesartan
Treatment options
Patients who have reduced salivary flow as a result of a medication may become so affected by dryness that their drug regimen may need to be changed. However, the greatest concern is for deteriorating oral health among patients who may be unaware xerostomia is occurring.31
Counsel patients who take medications that can affect their salivary function about the importance of seeing a dentist regularly, and provide referrals when appropriate. Depending upon the patient’s oral health, dentists recommend patients with xerostomia have their teeth cleaned/examined 3 or 4 times per year, rather than the 2 times per year allowed by third-party payers (ie, insurance companies). Also advise patients to be diligent in their oral hygiene practices, including flossing and brushing the teeth and tongue, and to avoid foods that are sticky and/or have high sucrose content (Table 2). Recommend using a toothpaste containing fluoride—preferably one free of sodium lauryl sulfate, which could contribute to mouth sores14—and drinking fluoridated water. Explain to patients that their dentist may recommend in-office high-fluoride applications, high-fluoride prescription toothpaste, and/or “mouth trays” that contain high fluoride gel. Tell patients to avoid cigarettes and caffeinated beverages, which can increase dryness. Alcohol use should be minimized and mouth rinses containing alcohol should not be used.
Many over-the-counter products are available to address xerostomia, including toothpastes, mouth rinses, and gels. Salivary substitutes—which are available as sprays, liquids, tablets, and swab sticks—imitate saliva and may provide a temporary reprieve from dryness. Although none of these products will cure dry mouth, they may help manage the condition. Advise patients to eat foods that stimulate saliva production, such as carrots, apples, and celery, and to chew sugarless gum and candies, which also will stimulate salivary flow.
The FDA has approved 2 prescription drugs for treating xerostomia: cevimeline and pilocarpine. Cevimeline is approved for treating dry mouth associated with Sjögren’s syndrome and pilocarpine is approved for treating dry mouth caused by head and neck radiation therapy; however, these medications’ role in treating dry mouth in psychiatric patients has not been investigated. Both agents are contraindicated in patients with narrow-angle glaucoma, uncontrolled asthma, or liver disease, and should be prescribed with caution for patients with cardiovascular disease, chronic respiratory conditions, or kidney disease.32
Acupuncture and electrostimulation are being studied as a treatment for xerostomia. Trials have found acupuncture improves symptoms of xerostomia,33,34 and 1 study found electrostimulation improved xerostomia in patients with Sjögren’s syndrome.35 Both approaches require more study to confirm their effectiveness.33-35
Table 2
Managing dry mouth: What to tell patients
| Oral hygiene. Tell patients to be diligent in their oral hygiene practices, including brushing and flossing. They should use a toothpaste containing fluoride—preferably one free of sodium lauryl sulfate—and schedule regular dental visits, where they can receive high-fluoride applications or be prescribed high-fluoride prescription toothpastes |
| Diet. Advise patients to avoid foods high in sucrose content, rinse their mouth with water soon after eating, and drink fluoridated water regularly. Tell them that they may be able to stimulate saliva flow with sugarless gum, candies, and foods such as celery and carrots |
| Drying agents. Instruct patients to avoid cigarettes, caffeinated beverages, and mouth rinses that contain alcohol. Explain that some patients may benefit from sleeping in a room with a cool air humidifier |
| Over-the-counter products. Suggest patients try salivary substitutes, which are dispensed in spray bottles, rinses, swish bottles, or oral swab sticks. In addition, products such as dry-mouth toothpaste and moisturizing gels also may help relieve their symptoms |
- Persson K, Axtelius B, Söderfeldt B, et al. Monitoring oral health and dental attendance in an outpatient psychiatric population. J Psychiatr Ment Health Nurs. 2009;16(3):263-271.
- Keene JJ Jr, Galasko GT, Land MF. Antidepressant use in psychiatry and medicine: importance for dental practice. J Am Dent Assoc. 2003;134(1):71-79.
Drug Brand Names
- Alprazolam • Xanax
- Amlodipine • Norvasc
- Bupropion • Wellbutrin, Zyban
- Cevimeline • Evoxac
- Clonidine • Catapres, Kapvay, others
- Esomeprazole • Nexium
- Irbesartan • Avapro
- Metoprolol • Lopressor, Toprol
- Pilocarpine • Salagen
- Tolterodine • Detrol
- Venlafaxine • Effexor
- Zolpidem • Ambien
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Drymouth.info. Overview of drugs and dry mouth. http://drymouth.info/practitioner/overview.asp. Accessed September 2, 2011.
2. Stewart CM, Berg KM, Cha S, et al. Salivary dysfunction and quality of life in Sjögren syndrome: a critical oral-systemic connection. J Am Dent Assoc. 2008;139(3):291-299.
3. Friedman PK. Xerostomia: The invisible oral health condition. http://www.dentistryiq.com/index/display/article-display/295922/articles/woman-dentist-journal/health/xerostomia-the-invisible-oral-health-condition.html. Accessed September 6, 2011.
4. Physician Desk Reference. Montvale NJ: PDR Network LLC.; 2011.
5. Bardow A, Lagerlof F, Nauntofte B, et al. The role of saliva. In: Fejerskov O, Kidd E, eds. Dental caries: the disease and its clinical management. Oxford, United Kingdom: Blackwell Munksgaard; 2008:195.
6. Vanderkooy JD, Kennedy SH, Bagby RM. Antidepressant side effects in depression patients treated in a naturalistic setting: a study of bupropion moclobemide, paroxetine, sertraline, and venlafaxine. Can J Psychiatry. 2002;47(2):174-180.
7. Löffler W, Kilian R, Toumi M, et al. Schizophrenic patients’ subjective reasons for compliance and noncompliance with neuroleptic treatment. Pharmacopsychiatry. 2003;36(3):105-112.
8. Lambert M, Conus P, Eide P, et al. Impact of present and past antipsychotic side effects on attitude toward typical antipsychotic treatment and adherence. Eur Psychiatry. 2004;19(7):415-422.
9. Rettenbacher MA, Hofer A, Eder U, et al. Compliance in schizophrenia: psychopathology, side effects, and patients’ attitudes toward the illness and medication. J Clin Psychiatry. 2004;65(9):1211-1218.
10. Bulkacz J, Carranza FA. Defense mechanisms of the gingiva. In: Newman MG, Takei HH, Klokkevold PR, et al, eds. Carranza’s clinical periodontology. St. Louis, MO: Elsevier Saunders; 2011:69–70.
11. Szabadi E, Tavernor S. Hypo-and hyper-salivation induced by psychoactive drugs. CNS Drugs. 1999;11(6):449-466.
12. Guggenheimer J, Moore PA. Xerostomia: etiology recognition and treatment. J Am Dent Assoc. 2003;134(1):61-69.
13. Dawes C. Physiological factors affecting salivary flow rate oral sugar clearance, and the sensation of dry mouth in man. J Dent Res. 1987;66:648-653.
14. Bartels CL. Xerostomia information for dentists. http://www.homesteadschools.com/dental/courses/Xerostomia/Course.htm. Accessed August 15, 2011.
15. Sitheeque MA, Samaranayake LP. Chronic hyperplastic candidosis/candidiasis (candidal leukoplakia). Crit Rev Oral Biol Med. 2003;14(4):253-267.
16. Porter SR, Scully C. Oral malodour (halitosis). BMJ. 2006;333(7569):632-635.
17. Quirynen M, Van den Veide S, Vanderkerckhove B, et al. Oral malodor. In: Newman MG, Takei HH, Klokkevold PR, et al, eds. Carranza’s clinical periodontology. St. Louis, MO: Elsevier Saunders; 2011:333.
18. Papas A. Dry mouth from drugs: more than just an annoying side effect. Tufts University Heath and Nutrition Letter. 2000;3.-
19. American Academy of Periodontology. Gum disease information from the American Academy of Periodontology http://perio.org. Accessed August 12, 2011.
20. Geismar K, Stoltze K, Sigurd B, et al. Periodontal disease and coronary heart disease. J Periodontol. 2006;77(9):1547-1554.
21. Lee HJ, Garcia RI, Janket SJ, et al. The association between cumulative periodontal disease and stroke history in older adults. J Periodontol. 2006;77(10):1744-1754.
22. Friedewald VE, Kornman KS, Beck JD, et al. The American Journal of Cardiology and Journal of Periodontology editors’ consensus: periodontitis and atherosclerotic cardiovascular disease. J Periodontol. 2009;80(7):1021-1032.
23. Contreras A, Herrera JA, Soto JE, et al. Periodontitis is associated with preeclampsia in pregnant women. J Periodontol. 2006;77(2):182-188.
24. Dasanayake AP, Li Y, Wiener H, et al. Salivary Actinomyces naeslundii genospecies 2 and Lactobacillus casei levels predict pregnancy outcomes. J Periodontol. 2005;76(2):171-177.
25. McCreadie RG, Stevens H, Henderson J, et al. The dental health of people with schizophrenia. Acta Psychiatr Scand. 2004;110(4):306-310.
26. Anttila S, Knuuttila M, Ylöstalo P, et al. Symptoms of depression and anxiety in relation to dental health behavior and self-perceived dental treatment need. Eur J Oral Sci. 2006;114(2):109-114.
27. Sjögren R, Nordström G. Oral health status of psychiatric patients. J Clin Nurs. 2000;9(4):632-638.
28. Ramon T, Grinshpoon A, Zusman SP, et al. Oral health and treatment needs of institutionalized chronic psychiatric patients in Israel. Eur Psychiatry. 2003;18(3):101-105.
29. Portilla MI, Mafla AC, Arteaga JJ. Periodontal status in female psychiatric patients. Colomb Med. 2009;40(2):167-176.
30. Navazesh M. ADA Council on Scientific Affairs and Division of Science. How can oral health care providers determine if patients have dry mouth? J Am Dent Assoc. 2003;134(5):613-620.
31. Mignogna MD, Fedele S, Lo Russo L, et al. Sjögren’s syndrome: the diagnostic potential of early oral manifestations preceding hyposalivation/xerostomia. J Oral Pathol Med. 2005;34(1):1-6.
32. Spolarich AE. Managing the side effects of medications. J Dent Hyg. 2000;74(1):57-69.
33. Johnstone PA, Niemtzow RC, Riffenburgh RH. Acupuncture for xerostomia: clinical update. Cancer. 2002;94(4):1151-1156.
34. Garcia MK, Chiang JS, Cohen L, et al. Acupuncture for radiation-induced xerostomia in patients with cancer: a pilot study. Head Neck. 2009;31(10):1360-1368.
35. Strietzel FP, Lafaurie GI, Mendoza GR, et al. Efficacy and safety of an intraoral electrostimulation device for xerostomia relief: a multicenter, randomized trial. Arthritis Rheum. 2011;63(1):180-190.
1. Drymouth.info. Overview of drugs and dry mouth. http://drymouth.info/practitioner/overview.asp. Accessed September 2, 2011.
2. Stewart CM, Berg KM, Cha S, et al. Salivary dysfunction and quality of life in Sjögren syndrome: a critical oral-systemic connection. J Am Dent Assoc. 2008;139(3):291-299.
3. Friedman PK. Xerostomia: The invisible oral health condition. http://www.dentistryiq.com/index/display/article-display/295922/articles/woman-dentist-journal/health/xerostomia-the-invisible-oral-health-condition.html. Accessed September 6, 2011.
4. Physician Desk Reference. Montvale NJ: PDR Network LLC.; 2011.
5. Bardow A, Lagerlof F, Nauntofte B, et al. The role of saliva. In: Fejerskov O, Kidd E, eds. Dental caries: the disease and its clinical management. Oxford, United Kingdom: Blackwell Munksgaard; 2008:195.
6. Vanderkooy JD, Kennedy SH, Bagby RM. Antidepressant side effects in depression patients treated in a naturalistic setting: a study of bupropion moclobemide, paroxetine, sertraline, and venlafaxine. Can J Psychiatry. 2002;47(2):174-180.
7. Löffler W, Kilian R, Toumi M, et al. Schizophrenic patients’ subjective reasons for compliance and noncompliance with neuroleptic treatment. Pharmacopsychiatry. 2003;36(3):105-112.
8. Lambert M, Conus P, Eide P, et al. Impact of present and past antipsychotic side effects on attitude toward typical antipsychotic treatment and adherence. Eur Psychiatry. 2004;19(7):415-422.
9. Rettenbacher MA, Hofer A, Eder U, et al. Compliance in schizophrenia: psychopathology, side effects, and patients’ attitudes toward the illness and medication. J Clin Psychiatry. 2004;65(9):1211-1218.
10. Bulkacz J, Carranza FA. Defense mechanisms of the gingiva. In: Newman MG, Takei HH, Klokkevold PR, et al, eds. Carranza’s clinical periodontology. St. Louis, MO: Elsevier Saunders; 2011:69–70.
11. Szabadi E, Tavernor S. Hypo-and hyper-salivation induced by psychoactive drugs. CNS Drugs. 1999;11(6):449-466.
12. Guggenheimer J, Moore PA. Xerostomia: etiology recognition and treatment. J Am Dent Assoc. 2003;134(1):61-69.
13. Dawes C. Physiological factors affecting salivary flow rate oral sugar clearance, and the sensation of dry mouth in man. J Dent Res. 1987;66:648-653.
14. Bartels CL. Xerostomia information for dentists. http://www.homesteadschools.com/dental/courses/Xerostomia/Course.htm. Accessed August 15, 2011.
15. Sitheeque MA, Samaranayake LP. Chronic hyperplastic candidosis/candidiasis (candidal leukoplakia). Crit Rev Oral Biol Med. 2003;14(4):253-267.
16. Porter SR, Scully C. Oral malodour (halitosis). BMJ. 2006;333(7569):632-635.
17. Quirynen M, Van den Veide S, Vanderkerckhove B, et al. Oral malodor. In: Newman MG, Takei HH, Klokkevold PR, et al, eds. Carranza’s clinical periodontology. St. Louis, MO: Elsevier Saunders; 2011:333.
18. Papas A. Dry mouth from drugs: more than just an annoying side effect. Tufts University Heath and Nutrition Letter. 2000;3.-
19. American Academy of Periodontology. Gum disease information from the American Academy of Periodontology http://perio.org. Accessed August 12, 2011.
20. Geismar K, Stoltze K, Sigurd B, et al. Periodontal disease and coronary heart disease. J Periodontol. 2006;77(9):1547-1554.
21. Lee HJ, Garcia RI, Janket SJ, et al. The association between cumulative periodontal disease and stroke history in older adults. J Periodontol. 2006;77(10):1744-1754.
22. Friedewald VE, Kornman KS, Beck JD, et al. The American Journal of Cardiology and Journal of Periodontology editors’ consensus: periodontitis and atherosclerotic cardiovascular disease. J Periodontol. 2009;80(7):1021-1032.
23. Contreras A, Herrera JA, Soto JE, et al. Periodontitis is associated with preeclampsia in pregnant women. J Periodontol. 2006;77(2):182-188.
24. Dasanayake AP, Li Y, Wiener H, et al. Salivary Actinomyces naeslundii genospecies 2 and Lactobacillus casei levels predict pregnancy outcomes. J Periodontol. 2005;76(2):171-177.
25. McCreadie RG, Stevens H, Henderson J, et al. The dental health of people with schizophrenia. Acta Psychiatr Scand. 2004;110(4):306-310.
26. Anttila S, Knuuttila M, Ylöstalo P, et al. Symptoms of depression and anxiety in relation to dental health behavior and self-perceived dental treatment need. Eur J Oral Sci. 2006;114(2):109-114.
27. Sjögren R, Nordström G. Oral health status of psychiatric patients. J Clin Nurs. 2000;9(4):632-638.
28. Ramon T, Grinshpoon A, Zusman SP, et al. Oral health and treatment needs of institutionalized chronic psychiatric patients in Israel. Eur Psychiatry. 2003;18(3):101-105.
29. Portilla MI, Mafla AC, Arteaga JJ. Periodontal status in female psychiatric patients. Colomb Med. 2009;40(2):167-176.
30. Navazesh M. ADA Council on Scientific Affairs and Division of Science. How can oral health care providers determine if patients have dry mouth? J Am Dent Assoc. 2003;134(5):613-620.
31. Mignogna MD, Fedele S, Lo Russo L, et al. Sjögren’s syndrome: the diagnostic potential of early oral manifestations preceding hyposalivation/xerostomia. J Oral Pathol Med. 2005;34(1):1-6.
32. Spolarich AE. Managing the side effects of medications. J Dent Hyg. 2000;74(1):57-69.
33. Johnstone PA, Niemtzow RC, Riffenburgh RH. Acupuncture for xerostomia: clinical update. Cancer. 2002;94(4):1151-1156.
34. Garcia MK, Chiang JS, Cohen L, et al. Acupuncture for radiation-induced xerostomia in patients with cancer: a pilot study. Head Neck. 2009;31(10):1360-1368.
35. Strietzel FP, Lafaurie GI, Mendoza GR, et al. Efficacy and safety of an intraoral electrostimulation device for xerostomia relief: a multicenter, randomized trial. Arthritis Rheum. 2011;63(1):180-190.
Medically unexplained physical symptoms: Evidence-based interventions
Discuss this article at www.facebook.com/CurrentPsychiatry
Mrs. B, age 45, is referred by her primary care physician (PCP) for treatment of depressive symptoms that have worsened over the last 6 months. Her depressed mood is associated with worsening of multiple chronic, physical symptoms that began 4 years ago with several musculoskeletal complaints. Three years ago she developed recurrent abdominal discomfort and bloating, followed by recurrent chest pain. These symptoms have resulted in multiple trips to the hospital, several invasive procedures, and extensive medical consultation. After repeated workups, her symptoms are medically unexplained. These symptoms interfere with her ability to engage in and enjoy life.
Even after thorough investigation, up to one-third of patients’ physical symptoms remain unexplained.1-3 Most patients with unexplained symptoms improve; however, a small proportion do not. Such patients often are referred for psychiatric consultation.
Workup may reveal psychiatric etiology of a patient’s medically unexplained physical symptoms (MUPS). Clinicians need to take a unique approach to caring for patients whose symptoms remain unexplained after workup because diagnostic features may emerge over time and a collaborative, unbiased, integrated approach eventually may reveal a treatable diagnosis. This type of approach also is important when no medical or psychiatric diagnosis can be reached.
This article reviews the prevalence, comorbidity, and treatment challenges of patients whose physical symptoms are medically unexplained, and recommends evidence-based treatment strategies.
Inconsistent terminology
The terms MUPS, medically unexplained symptoms, somatoform disorder, somatization, and the functional syndromes (eg, irritable bowel syndrome [IBS], fibromyalgia, interstitial cystitis, chronic fatigue, etc.) often are used interchangeably. This inconsistent nomenclature creates classification difficulties because several of these terms assume a different etiology for the patient’s physical symptoms (ie, medical vs psychiatric).
Physical symptoms typically are explained by:
- medical pathophysiology
- psychopathology, or
- unknown etiology (Figure).
MUPS typically are defined as physical or somatic symptoms without a known etiology after appropriate testing, workup, and referrals. Workup may be limited or extensive, may evolve over time (eg, diagnosis may be made 2 years after primary symptom onset), and often involves collaboration among several specialists.
The above definition does not specify severity, medical/psychiatric comorbidity, or number or duration of symptoms. A proposed classification, medically unexplained symptoms spectrum disorder, attempts to categorize patients with MUPS based on severity and duration of symptoms, as well as medical and/or psychiatric comorbidities (Table).4 MUPS may account for up to two-thirds of physical symptoms in specialty clinics; to read about the prevalence of MUPS, see the Box below.
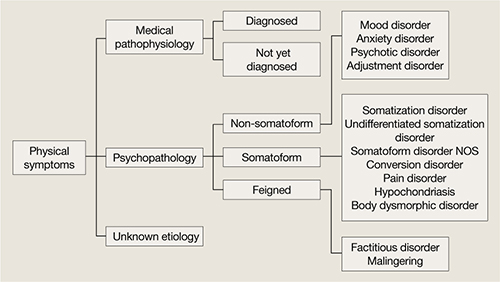
Figure: Typical etiology of physical symptoms
NOS: Not otherwise specifiedTable
Medically unexplained symptoms spectrum disorder
| Severity | Mild, moderate, severe |
| Duration | Acute (days to weeks), subacute (<6 months), chronic (>6 months) |
| Comorbidity | Psychiatric, medical, none |
| Source: Reference 4 | |
CASE CONTINUED: Abuse and assault
Mrs. B has been married for 10 years and has 2 children. She denies using tobacco, alcohol, or illicit drugs. Both of her parents were in good physical health. As a child Mrs. B was physically and verbally abused by her father, and her parents divorced when she was 15. She was sexually assaulted while in college.
Mrs. B has had 1 previous depressive episode, which occurred shortly after the sexual assault. During that time she was hospitalized twice for attempting suicide by overdosing on prescription medications. She was stabilized on fluoxetine, 40 mg/d, which was tapered and discontinued after 2 years.
Factors linked to MUPS
Young women (age 16 to 25) are more likely to receive an MUPS diagnosis than men or older individuals. Employment, socioeconomic background, and educational level are not consistently associated with MUPS.5 Patients with MUPS have higher rates of physical and sexual abuse.6
Several studies have shown an association with childhood parental ill health and development of MUPS, but the exact nature of “ill health” was not clearly defined. Parental death was not associated with MUPS, which suggests that the association to parental ill health is related to non-threatening physical disease.6
Approximately 60% of patients with MUPS have a comorbid non-somatoform DSM-IV-TR diagnosis.7-9 Symptoms and rates of depressive, anxiety, and panic disorders are higher in patients with MUPS than either healthy controls or patients with similar diseases of known organic pathology.7,8
The estimated prevalence of somatoform disorder in patients with MUPS is approximately 4%, which is higher than in the general population (.2% to 2%).7,8 On measures of mental and physical function, patients with MUPS who have somatoform disorders have been found to be more distressed than normal controls and patients with MUPS without somatoform disorders.8
Although few studies have directly examined the relationship between personality disorders and MUPS, there is evidence of an association between certain personality traits (eg, neuroticism, alexithymia, negative affect) and MUPS.10,11
CASE CONTINUED: Rejected advice
Mrs. B has been worked up multiple times for acute coronary syndrome; been unsuccessfully treated for gastroesophageal reflux disease, lactose intolerance, and IBS; had a negative rheumatologic workup; and tried several medication regimens with no improvement in symptoms. Three years ago Mrs. B’s gastroenterologist implied her abdominal symptoms were caused by her history of sexual assault and suggested she seek psychiatric consultation. Offended, Mrs. B sought a second opinion and no longer sees her first gastroenterologist.
Barriers to treatment
Despite having high levels of psychosocial distress, health care utilization, and medical disability, patients with MUPS often are suboptimally treated. Factors that might contribute to this include:
- inadequate identification
- bias in diagnosis and treatment
- poor follow-up on referrals
- an absence of treatment guidelines.7,12,13
Many clinicians are unaware of the high prevalence of MUPS, which often leads to repeated referral to specialty clinics, even when patients already have received an MUPS diagnosis.12,14 Additionally, clinicians often are unaware of how individual biases influence their diagnostic thought process. A “difficult patient” may receive a MUPS diagnosis more readily than a “pleasant patient,” which could contribute to an incomplete workup. An epidemiologic study revealed that the strongest predictor of misdiagnosing MUPS is doctor dissatisfaction with the clinical encounter.15 Younger, unmarried, anxious patients receiving disability benefits are more likely to be incorrectly labeled as having MUPS, only to later receive a non-MUPS diagnosis.15
Bias in treatment and intervention also exists. Qualitative analysis of consultations suggests that physicians’ decisions to offer patients somatic treatments (eg, investigation, add/change medications, referral to specialists) are responses to patients’ extended and complex accounts of their symptoms.17 The likelihood of intervention was unrelated to patients’ request for treatment, and intervention became less likely when patients described psychosocial problems.16
Patients with MUPS and comorbid psychiatric disorders often are referred for psychosocial treatment, but 1 study found that as few as 10% of such patients follow up on a referral.17 In that study, 81% of MUPS patients were willing to receive psychosocial treatment in a primary care setting by their physician. Although there are many reasons patients with MUPS resist referral to mental health professionals, be aware that many of these individuals do not attribute their symptoms to psychosocial problems or experience their symptoms psychologically. To these patients, psychiatric referral may seem inappropriate or be perceived as belittling and minimizing their symptoms.
CASE CONTINUED: Frustration and guilt
Mrs. B’s depressive symptoms began 18 months ago with fatigue, poor sleep, and withdrawal from her children and husband. She struggles with hopelessness that her physical symptoms will not resolve and guilt because of the financial strain her medical care has placed on the family. She is extremely frustrated that her doctors are unable to find a medical diagnosis for her symptoms and fears that without a diagnosis she will be perceived as “crazy.” She is not certain if there is a medical explanation for her symptoms but vehemently believes they are not associated with her mood or psychosocial stress.
Treatment strategies
A collaborative, unbiased, integrated approach to treatment can address some of the challenges that arise when patients with MUPS confront the limitations of modern medicine. Integrated care involves ongoing communication among medical and psychiatric specialists, as well as collaboration with social workers, physical therapists, nutritionists, or pain management specialists when indicated.
Although the primary care provider often coordinates a MUPS patient’s medical treatment, a consulting psychiatrist plays an important educational, diagnostic, and therapeutic role. The therapeutic role is especially important because patients with MUPS frequently view their general practitioner as having a limited role in managing psychosocial problems.18
Because physical illness and psychosocial stress frequently coexist and compound each other, diagnostic efforts should focus on medical and psychiatric illness. Review the patient’s medical workup of the unexplained symptoms and, when indicated, request further testing. Evaluate the risks and benefits of additional testing and discuss them with the patient; additional testing carries a risk of iatrogenic harm, higher false-positive rates, and increased costs. Avoiding iatrogenic harm and unnecessary, overly aggressive testing is essential.
Identifying primary or comorbid psychiatric disease and psychosocial issues also is integral to managing patients with MUPS. This may be difficult because some patients might be hesitant to discuss psychosocial issues, whereas others may be unaware of psychiatric symptomatology or the connection between mental and physical illness. When possible, it may be useful to clarify symptomatology as:
- primarily somatic (expression of psychological illness through physical means)
- primarily psychiatric (psychiatric illness presenting with physical symptoms) or
- bordering between somatic and psychiatric.
CASE CONTINUED: Collaboration and improvement
You diagnose Mrs. B with major depressive disorder and prescribe fluoxetine, titrating her up to 40 mg/d. Mrs. B also begins weekly psychodynamic psychotherapy. In collaboration with her PCP, you decide to refer Mrs. B to physical therapy and direct psychotherapy toward coping strategies, with the hope of improving functionality. Although she continues to have musculoskeletal symptoms after completing physical therapy, Mrs. B notices moderate improvement and feels less distressed by these symptoms.
After 1 year of fluoxetine treatment, Mrs. B’s depressive symptoms improve. In psychotherapy, her fixation on physical symptoms and desire to establish a diagnosis gradually lessen. As her emotional trauma from childhood abuse unravels, psychotherapy shifts toward improving affect regulation. During this time Mrs. B experiences an increase in unexplained chest pain and shortness of breath, which later abate.
Continued follow-up with a gastroenterologist leads to a diagnosis of celiac disease. With treatment, her GI symptoms resolve.
What do patients want?
Begin MUPS treatment by developing a supportive, empathic relationship with the patient. Carefully listen to the patient’s description of his or her symptoms. Elucidating patients’ experience often is challenging because their narratives frequently are complex, nonlinear, and limited by time.18 Patients’ models for understanding their symptoms also may be complex.18 They may be reluctant to share their explanations, fearing they will be unable to communicate the complexity of their beliefs or their symptoms will be oversimplified.18
Focus on understanding what the patient seeks from the physician—emotional support vs diagnosis vs treatment. In a prospective naturalistic study, the content of MUPS patients’ narratives was correlated with what they sought from their physician.17 Patients who sought emotional support frequently discussed psychosocial problems, issues, and management. Patients who wanted an explanation for their symptoms often mentioned physical symptoms, explanations, and diseases. Those who were looking for additional testing or intervention often directly addressed this with the physician.17
Although many patients desire a diagnosis and somatic treatment, this is not always their primary agenda. Many MUPS patients seek emotional support or confirmation of their explanatory model.17,18 Patients’ desires for emotional support, medical explanation, diagnosis, or somatic intervention often are neither clearly nor explicitly stated. Despite this, patients hope their physician understands the extent of their problems and value those who help them make sense of their narratives.18 Misunderstanding patients’ agendas can result in a mismatch of treatment expectations and fracture the patient-physician relationship. Developing mutual expectations is crucial to building rapport, improving collaborative care, and avoiding unnecessary, potentially harmful interventions.
Psychotherapic interventions
Psychopharmacologic treatment is indicated for MUPS patients who have comorbid psychiatric conditions.
Research of psychotherapy in MUPS has been plagued by methodologic problems and inconsistent results.3 Group therapy, short-term dynamic therapy, hypnotherapy, and cognitive-behavioral therapy (CBT) have been studied. In a trial of 140 MUPS patients who received 1 session of CBT, 71% experienced improvement in physical symptoms, 47% in functional status, and 38% in measures of psychological distress.19 A review of 34 randomized controlled trials involving 3,922 patients with somatoform disorders who received CBT found that some patients with MUPS responded after 5 to 6 sessions.3
Cognitive techniques focus on identifying and restructuring automatic, dysfunctional thoughts that may compound, perpetuate, or worsen somatic symptoms. Behavioral techniques include relaxation and efforts to increase motivation. A CBT treatment plan may involve establishing goals, addressing patients’ understanding of their symptoms, obtaining a commitment for treatment, and negotiating the details of the treatment plan.8,12
Supportive techniques also are valuable in treating MUPS patients. Educate patients and treating physicians that there is a neurophysiologic basis for the patient’s physical symptoms and that symptoms may wax and wane. Reinforcement of functional improvement through concrete, practical solutions can help patients develop healthy, adaptive coping skills. Encouraging patients to move beyond somatic complaints to discuss social and personal difficulties can lead to more effective management of these problems.
Clearly communicate your initial impressions, diagnoses, and treatment plan to other members of the treatment team. A consultation letter from the psychiatrist to the PCP has been shown to decrease costs and slightly improve the patient’s functional status, symptoms, and quality of life.20 When possible, educate the PCP and specialists about the dynamics, challenges, biases, and frustrations physicians commonly face when caring for MUPS patients.
Related Resources
- Burton C. Beyond somatization: a review of the understanding and treatment of medically unexplained physical symptoms. Brit J Gen Pract. 2003;53:233-239.
- Creed F. The outcome of medically unexplained symptoms—will DSM-V improve on DSM-IV somatoform disorders? J Psychosom Res. 2009;66:379-381.
- Katon W, Sullivan M, Walker E. Medical symptoms without identified pathology: relationship to psychiatric disorders, childhood and adult trauma, and personality traits. Ann Intern Med. 2001;134(9 Pt 2):917-925.
- Sharpe M, Mayou R, Walker J. Bodily symptoms: new approaches to classification. J Psychosom Res. 2006;60: 353-356.
Drug Brand Names
- Fluoxetine • Prozac
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Medically unexplained physical symptoms (MUPS) has been found to make up 10% to 30% of the physical symptoms in primary care clinics and 37% to 66% in specialty clinics.a-c The latter statistic is based on a cross-sectional survey of 899 consecutive new patients from 7 outpatient clinics in London, United Kingdom. Sixty-five percent responded and 52% of respondents had at least 1 medically unexplained symptom, diagnosed 3 months after initial clinic presentation.c
Patients with MUPS carry significant clinical importance. They are more likely to have a relatively poor quality of life and higher rates of disability.d,e They tend to be higher utilizers of health care.c,f High utilization of services and potentially unnecessary lab testing and consultation result in increased costs and high rates of iatrogenic complications.d-f
References
a. McCarron RM, Xiong GL, Bourgeois JA. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott Williams and Wilkins; 2009.
b. Richardson RD, Engel CC. Evaluation and management of medically unexplained physical symptoms. Neurologist. 2004;10:18-30.
c. Nimnuan C, Hotopf M, Wessely S. Medically unexplained symptoms. An epidemiological study in seven specialties. J Psychosom Res. 2001;51:361-367.
d. Reid S, Wessely S, Crayford T, et al. Medically unexplained symptoms in frequent attenders of secondary health care: retrospective cohort study. BMJ. 2001;322:1-4.
e. Smith BJ, McGorn KJ, Weller D, et al. The identification in primary care of patients who have been repeatedly referred to hospital for medically unexplained symptoms: a pilot study. J Psychosom Res. 2009;67:207-211.
f. Smith RC, Lein C, Collins C, et al. Treating patients with medically unexplained symptoms in primary care. J Gen Intern Med. 2003;18:478-489.
1. Jackson JL, Kroenke K. Prevalence impact, and prognosis of multisomatoform disorder in primary care: a 5-year follow-up study. Psychosom Med. 2008;70:430-434.
2. Jackson JL, George S, Hinchey S. Medically unexplained physical symptoms. J Gen Intern Med. 2009;24:540-542.
3. Kroenke K. Efficacy of treatment for somatoform disorders: a review of randomized controlled trials. Psychosom Med. 2007;69:881-888.
4. Smith RC, Dwamena FC. Classification and diagnosis of patients with medically unexplained symptoms. J Gen Intern Med. 2007;22:685-691.
5. Nimnuan C, Hotopf M, Wessely S. Medically unexplained symptoms. An epidemiological study in seven specialties. J Psychosom Res. 2001;51:361-367.
6. Hotopf M, Mayou R, Wadsworth M, et al. Childhood risk factors for adults with medically unexplained symptoms: results from a national birth cohort study. Am J Psychiatry. 1999;156(11):1796-1800.
7. Smith RC, Gardiner JC, Lyles JS, et al. Exploration of DSM-IV criteria in primary care patients with medically unexplained symptoms. Psychosom Med. 2005;67:123-129.
8. Smith RC, Lyles JS, Gardiner JC, et al. Primary care clinicians treat patients with medically unexplained symptoms: a randomized controlled trial. J Gen Intern Med. 2006;21:671-677.
9. Henningsen P, Zimmermann T, Sattel H. Medically unexplained physical symptoms anxiety, and depression. A meta-analytic review. Psychosom Med. 2003;65:528-533.
10. Costa PT, McCrae RR. Neuroticism somatic complaints, and disease: is the bark worse than the bite? J Pers. 1987;55(2):299-315.
11. Gucht VD, Fischler B, Heiser W. Personality and affect as determinants of medically unexplained symptoms in primary care. A follow-up study. J Psychosom Res. 2004;56:279-285.
12. Smith RC, Lein C, Collins C, et al. Treating patients with medically unexplained symptoms in primary care. J Gen Intern Med. 2003;18:478-489.
13. McFarlane AC, Ellis N, Fasphm F, et al. The conundrum of medically unexplained symptoms: questions to consider. Psychosomatics. 2008;49(5):369-377.
14. Reid S, Wessely S, Crayford T, et al. Medically unexplained symptoms in frequent attenders of secondary health care: retrospective cohort study. BMJ. 2001;322:1-4.
15. Nimnuan C, Hotopf M, Wessely S. Medically unexplained symptoms: how often and why are they missed? QJM. 2000;93:21-28.
16. Salmon P, Humphris GM, Ring A, et al. Primary care consultations about medically unexplained symptoms: patient presentations and doctor responses that influence the probability of somatic intervention. Psychosom Med. 2007;69:571-577.
17. Salmon P, Ring A, Humphris GM, et al. Primary care consultations about medically unexplained symptoms: how do patients indicate what they want? J Gen Intern Med. 2009;24(4):450-456.
18. Peters S, Rogers A, Salmon P, et al. What do patients choose to tell their doctors? Qualitative analysis of potential barriers to reattributing medically unexplained symptoms. J Gen Intern Med. 2008;24(4):443-449.
19. Martin A, Rauh E, Fichter M, et al. A one-session treatment for patients suffering from medically unexplained symptoms in primary care: a randomized clinical trial. Psychosomatics. 2007;48:294-303.
20. Smith BJ, McGorn KJ, Weller D, et al. The identification in primary care of patients who have been repeatedly referred to hospital for medically unexplained symptoms: a pilot study. J Psychosom Res. 2009;67:207-211.
Discuss this article at www.facebook.com/CurrentPsychiatry
Mrs. B, age 45, is referred by her primary care physician (PCP) for treatment of depressive symptoms that have worsened over the last 6 months. Her depressed mood is associated with worsening of multiple chronic, physical symptoms that began 4 years ago with several musculoskeletal complaints. Three years ago she developed recurrent abdominal discomfort and bloating, followed by recurrent chest pain. These symptoms have resulted in multiple trips to the hospital, several invasive procedures, and extensive medical consultation. After repeated workups, her symptoms are medically unexplained. These symptoms interfere with her ability to engage in and enjoy life.
Even after thorough investigation, up to one-third of patients’ physical symptoms remain unexplained.1-3 Most patients with unexplained symptoms improve; however, a small proportion do not. Such patients often are referred for psychiatric consultation.
Workup may reveal psychiatric etiology of a patient’s medically unexplained physical symptoms (MUPS). Clinicians need to take a unique approach to caring for patients whose symptoms remain unexplained after workup because diagnostic features may emerge over time and a collaborative, unbiased, integrated approach eventually may reveal a treatable diagnosis. This type of approach also is important when no medical or psychiatric diagnosis can be reached.
This article reviews the prevalence, comorbidity, and treatment challenges of patients whose physical symptoms are medically unexplained, and recommends evidence-based treatment strategies.
Inconsistent terminology
The terms MUPS, medically unexplained symptoms, somatoform disorder, somatization, and the functional syndromes (eg, irritable bowel syndrome [IBS], fibromyalgia, interstitial cystitis, chronic fatigue, etc.) often are used interchangeably. This inconsistent nomenclature creates classification difficulties because several of these terms assume a different etiology for the patient’s physical symptoms (ie, medical vs psychiatric).
Physical symptoms typically are explained by:
- medical pathophysiology
- psychopathology, or
- unknown etiology (Figure).
MUPS typically are defined as physical or somatic symptoms without a known etiology after appropriate testing, workup, and referrals. Workup may be limited or extensive, may evolve over time (eg, diagnosis may be made 2 years after primary symptom onset), and often involves collaboration among several specialists.
The above definition does not specify severity, medical/psychiatric comorbidity, or number or duration of symptoms. A proposed classification, medically unexplained symptoms spectrum disorder, attempts to categorize patients with MUPS based on severity and duration of symptoms, as well as medical and/or psychiatric comorbidities (Table).4 MUPS may account for up to two-thirds of physical symptoms in specialty clinics; to read about the prevalence of MUPS, see the Box below.
Figure: Typical etiology of physical symptoms
NOS: Not otherwise specifiedTable
Medically unexplained symptoms spectrum disorder
| Severity | Mild, moderate, severe |
| Duration | Acute (days to weeks), subacute (<6 months), chronic (>6 months) |
| Comorbidity | Psychiatric, medical, none |
| Source: Reference 4 | |
CASE CONTINUED: Abuse and assault
Mrs. B has been married for 10 years and has 2 children. She denies using tobacco, alcohol, or illicit drugs. Both of her parents were in good physical health. As a child Mrs. B was physically and verbally abused by her father, and her parents divorced when she was 15. She was sexually assaulted while in college.
Mrs. B has had 1 previous depressive episode, which occurred shortly after the sexual assault. During that time she was hospitalized twice for attempting suicide by overdosing on prescription medications. She was stabilized on fluoxetine, 40 mg/d, which was tapered and discontinued after 2 years.
Factors linked to MUPS
Young women (age 16 to 25) are more likely to receive an MUPS diagnosis than men or older individuals. Employment, socioeconomic background, and educational level are not consistently associated with MUPS.5 Patients with MUPS have higher rates of physical and sexual abuse.6
Several studies have shown an association with childhood parental ill health and development of MUPS, but the exact nature of “ill health” was not clearly defined. Parental death was not associated with MUPS, which suggests that the association to parental ill health is related to non-threatening physical disease.6
Approximately 60% of patients with MUPS have a comorbid non-somatoform DSM-IV-TR diagnosis.7-9 Symptoms and rates of depressive, anxiety, and panic disorders are higher in patients with MUPS than either healthy controls or patients with similar diseases of known organic pathology.7,8
The estimated prevalence of somatoform disorder in patients with MUPS is approximately 4%, which is higher than in the general population (.2% to 2%).7,8 On measures of mental and physical function, patients with MUPS who have somatoform disorders have been found to be more distressed than normal controls and patients with MUPS without somatoform disorders.8
Although few studies have directly examined the relationship between personality disorders and MUPS, there is evidence of an association between certain personality traits (eg, neuroticism, alexithymia, negative affect) and MUPS.10,11
CASE CONTINUED: Rejected advice
Mrs. B has been worked up multiple times for acute coronary syndrome; been unsuccessfully treated for gastroesophageal reflux disease, lactose intolerance, and IBS; had a negative rheumatologic workup; and tried several medication regimens with no improvement in symptoms. Three years ago Mrs. B’s gastroenterologist implied her abdominal symptoms were caused by her history of sexual assault and suggested she seek psychiatric consultation. Offended, Mrs. B sought a second opinion and no longer sees her first gastroenterologist.
Barriers to treatment
Despite having high levels of psychosocial distress, health care utilization, and medical disability, patients with MUPS often are suboptimally treated. Factors that might contribute to this include:
- inadequate identification
- bias in diagnosis and treatment
- poor follow-up on referrals
- an absence of treatment guidelines.7,12,13
Many clinicians are unaware of the high prevalence of MUPS, which often leads to repeated referral to specialty clinics, even when patients already have received an MUPS diagnosis.12,14 Additionally, clinicians often are unaware of how individual biases influence their diagnostic thought process. A “difficult patient” may receive a MUPS diagnosis more readily than a “pleasant patient,” which could contribute to an incomplete workup. An epidemiologic study revealed that the strongest predictor of misdiagnosing MUPS is doctor dissatisfaction with the clinical encounter.15 Younger, unmarried, anxious patients receiving disability benefits are more likely to be incorrectly labeled as having MUPS, only to later receive a non-MUPS diagnosis.15
Bias in treatment and intervention also exists. Qualitative analysis of consultations suggests that physicians’ decisions to offer patients somatic treatments (eg, investigation, add/change medications, referral to specialists) are responses to patients’ extended and complex accounts of their symptoms.17 The likelihood of intervention was unrelated to patients’ request for treatment, and intervention became less likely when patients described psychosocial problems.16
Patients with MUPS and comorbid psychiatric disorders often are referred for psychosocial treatment, but 1 study found that as few as 10% of such patients follow up on a referral.17 In that study, 81% of MUPS patients were willing to receive psychosocial treatment in a primary care setting by their physician. Although there are many reasons patients with MUPS resist referral to mental health professionals, be aware that many of these individuals do not attribute their symptoms to psychosocial problems or experience their symptoms psychologically. To these patients, psychiatric referral may seem inappropriate or be perceived as belittling and minimizing their symptoms.
CASE CONTINUED: Frustration and guilt
Mrs. B’s depressive symptoms began 18 months ago with fatigue, poor sleep, and withdrawal from her children and husband. She struggles with hopelessness that her physical symptoms will not resolve and guilt because of the financial strain her medical care has placed on the family. She is extremely frustrated that her doctors are unable to find a medical diagnosis for her symptoms and fears that without a diagnosis she will be perceived as “crazy.” She is not certain if there is a medical explanation for her symptoms but vehemently believes they are not associated with her mood or psychosocial stress.
Treatment strategies
A collaborative, unbiased, integrated approach to treatment can address some of the challenges that arise when patients with MUPS confront the limitations of modern medicine. Integrated care involves ongoing communication among medical and psychiatric specialists, as well as collaboration with social workers, physical therapists, nutritionists, or pain management specialists when indicated.
Although the primary care provider often coordinates a MUPS patient’s medical treatment, a consulting psychiatrist plays an important educational, diagnostic, and therapeutic role. The therapeutic role is especially important because patients with MUPS frequently view their general practitioner as having a limited role in managing psychosocial problems.18
Because physical illness and psychosocial stress frequently coexist and compound each other, diagnostic efforts should focus on medical and psychiatric illness. Review the patient’s medical workup of the unexplained symptoms and, when indicated, request further testing. Evaluate the risks and benefits of additional testing and discuss them with the patient; additional testing carries a risk of iatrogenic harm, higher false-positive rates, and increased costs. Avoiding iatrogenic harm and unnecessary, overly aggressive testing is essential.
Identifying primary or comorbid psychiatric disease and psychosocial issues also is integral to managing patients with MUPS. This may be difficult because some patients might be hesitant to discuss psychosocial issues, whereas others may be unaware of psychiatric symptomatology or the connection between mental and physical illness. When possible, it may be useful to clarify symptomatology as:
- primarily somatic (expression of psychological illness through physical means)
- primarily psychiatric (psychiatric illness presenting with physical symptoms) or
- bordering between somatic and psychiatric.
CASE CONTINUED: Collaboration and improvement
You diagnose Mrs. B with major depressive disorder and prescribe fluoxetine, titrating her up to 40 mg/d. Mrs. B also begins weekly psychodynamic psychotherapy. In collaboration with her PCP, you decide to refer Mrs. B to physical therapy and direct psychotherapy toward coping strategies, with the hope of improving functionality. Although she continues to have musculoskeletal symptoms after completing physical therapy, Mrs. B notices moderate improvement and feels less distressed by these symptoms.
After 1 year of fluoxetine treatment, Mrs. B’s depressive symptoms improve. In psychotherapy, her fixation on physical symptoms and desire to establish a diagnosis gradually lessen. As her emotional trauma from childhood abuse unravels, psychotherapy shifts toward improving affect regulation. During this time Mrs. B experiences an increase in unexplained chest pain and shortness of breath, which later abate.
Continued follow-up with a gastroenterologist leads to a diagnosis of celiac disease. With treatment, her GI symptoms resolve.
What do patients want?
Begin MUPS treatment by developing a supportive, empathic relationship with the patient. Carefully listen to the patient’s description of his or her symptoms. Elucidating patients’ experience often is challenging because their narratives frequently are complex, nonlinear, and limited by time.18 Patients’ models for understanding their symptoms also may be complex.18 They may be reluctant to share their explanations, fearing they will be unable to communicate the complexity of their beliefs or their symptoms will be oversimplified.18
Focus on understanding what the patient seeks from the physician—emotional support vs diagnosis vs treatment. In a prospective naturalistic study, the content of MUPS patients’ narratives was correlated with what they sought from their physician.17 Patients who sought emotional support frequently discussed psychosocial problems, issues, and management. Patients who wanted an explanation for their symptoms often mentioned physical symptoms, explanations, and diseases. Those who were looking for additional testing or intervention often directly addressed this with the physician.17
Although many patients desire a diagnosis and somatic treatment, this is not always their primary agenda. Many MUPS patients seek emotional support or confirmation of their explanatory model.17,18 Patients’ desires for emotional support, medical explanation, diagnosis, or somatic intervention often are neither clearly nor explicitly stated. Despite this, patients hope their physician understands the extent of their problems and value those who help them make sense of their narratives.18 Misunderstanding patients’ agendas can result in a mismatch of treatment expectations and fracture the patient-physician relationship. Developing mutual expectations is crucial to building rapport, improving collaborative care, and avoiding unnecessary, potentially harmful interventions.
Psychotherapic interventions
Psychopharmacologic treatment is indicated for MUPS patients who have comorbid psychiatric conditions.
Research of psychotherapy in MUPS has been plagued by methodologic problems and inconsistent results.3 Group therapy, short-term dynamic therapy, hypnotherapy, and cognitive-behavioral therapy (CBT) have been studied. In a trial of 140 MUPS patients who received 1 session of CBT, 71% experienced improvement in physical symptoms, 47% in functional status, and 38% in measures of psychological distress.19 A review of 34 randomized controlled trials involving 3,922 patients with somatoform disorders who received CBT found that some patients with MUPS responded after 5 to 6 sessions.3
Cognitive techniques focus on identifying and restructuring automatic, dysfunctional thoughts that may compound, perpetuate, or worsen somatic symptoms. Behavioral techniques include relaxation and efforts to increase motivation. A CBT treatment plan may involve establishing goals, addressing patients’ understanding of their symptoms, obtaining a commitment for treatment, and negotiating the details of the treatment plan.8,12
Supportive techniques also are valuable in treating MUPS patients. Educate patients and treating physicians that there is a neurophysiologic basis for the patient’s physical symptoms and that symptoms may wax and wane. Reinforcement of functional improvement through concrete, practical solutions can help patients develop healthy, adaptive coping skills. Encouraging patients to move beyond somatic complaints to discuss social and personal difficulties can lead to more effective management of these problems.
Clearly communicate your initial impressions, diagnoses, and treatment plan to other members of the treatment team. A consultation letter from the psychiatrist to the PCP has been shown to decrease costs and slightly improve the patient’s functional status, symptoms, and quality of life.20 When possible, educate the PCP and specialists about the dynamics, challenges, biases, and frustrations physicians commonly face when caring for MUPS patients.
Related Resources
- Burton C. Beyond somatization: a review of the understanding and treatment of medically unexplained physical symptoms. Brit J Gen Pract. 2003;53:233-239.
- Creed F. The outcome of medically unexplained symptoms—will DSM-V improve on DSM-IV somatoform disorders? J Psychosom Res. 2009;66:379-381.
- Katon W, Sullivan M, Walker E. Medical symptoms without identified pathology: relationship to psychiatric disorders, childhood and adult trauma, and personality traits. Ann Intern Med. 2001;134(9 Pt 2):917-925.
- Sharpe M, Mayou R, Walker J. Bodily symptoms: new approaches to classification. J Psychosom Res. 2006;60: 353-356.
Drug Brand Names
- Fluoxetine • Prozac
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Medically unexplained physical symptoms (MUPS) has been found to make up 10% to 30% of the physical symptoms in primary care clinics and 37% to 66% in specialty clinics.a-c The latter statistic is based on a cross-sectional survey of 899 consecutive new patients from 7 outpatient clinics in London, United Kingdom. Sixty-five percent responded and 52% of respondents had at least 1 medically unexplained symptom, diagnosed 3 months after initial clinic presentation.c
Patients with MUPS carry significant clinical importance. They are more likely to have a relatively poor quality of life and higher rates of disability.d,e They tend to be higher utilizers of health care.c,f High utilization of services and potentially unnecessary lab testing and consultation result in increased costs and high rates of iatrogenic complications.d-f
References
a. McCarron RM, Xiong GL, Bourgeois JA. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott Williams and Wilkins; 2009.
b. Richardson RD, Engel CC. Evaluation and management of medically unexplained physical symptoms. Neurologist. 2004;10:18-30.
c. Nimnuan C, Hotopf M, Wessely S. Medically unexplained symptoms. An epidemiological study in seven specialties. J Psychosom Res. 2001;51:361-367.
d. Reid S, Wessely S, Crayford T, et al. Medically unexplained symptoms in frequent attenders of secondary health care: retrospective cohort study. BMJ. 2001;322:1-4.
e. Smith BJ, McGorn KJ, Weller D, et al. The identification in primary care of patients who have been repeatedly referred to hospital for medically unexplained symptoms: a pilot study. J Psychosom Res. 2009;67:207-211.
f. Smith RC, Lein C, Collins C, et al. Treating patients with medically unexplained symptoms in primary care. J Gen Intern Med. 2003;18:478-489.
Discuss this article at www.facebook.com/CurrentPsychiatry
Mrs. B, age 45, is referred by her primary care physician (PCP) for treatment of depressive symptoms that have worsened over the last 6 months. Her depressed mood is associated with worsening of multiple chronic, physical symptoms that began 4 years ago with several musculoskeletal complaints. Three years ago she developed recurrent abdominal discomfort and bloating, followed by recurrent chest pain. These symptoms have resulted in multiple trips to the hospital, several invasive procedures, and extensive medical consultation. After repeated workups, her symptoms are medically unexplained. These symptoms interfere with her ability to engage in and enjoy life.
Even after thorough investigation, up to one-third of patients’ physical symptoms remain unexplained.1-3 Most patients with unexplained symptoms improve; however, a small proportion do not. Such patients often are referred for psychiatric consultation.
Workup may reveal psychiatric etiology of a patient’s medically unexplained physical symptoms (MUPS). Clinicians need to take a unique approach to caring for patients whose symptoms remain unexplained after workup because diagnostic features may emerge over time and a collaborative, unbiased, integrated approach eventually may reveal a treatable diagnosis. This type of approach also is important when no medical or psychiatric diagnosis can be reached.
This article reviews the prevalence, comorbidity, and treatment challenges of patients whose physical symptoms are medically unexplained, and recommends evidence-based treatment strategies.
Inconsistent terminology
The terms MUPS, medically unexplained symptoms, somatoform disorder, somatization, and the functional syndromes (eg, irritable bowel syndrome [IBS], fibromyalgia, interstitial cystitis, chronic fatigue, etc.) often are used interchangeably. This inconsistent nomenclature creates classification difficulties because several of these terms assume a different etiology for the patient’s physical symptoms (ie, medical vs psychiatric).
Physical symptoms typically are explained by:
- medical pathophysiology
- psychopathology, or
- unknown etiology (Figure).
MUPS typically are defined as physical or somatic symptoms without a known etiology after appropriate testing, workup, and referrals. Workup may be limited or extensive, may evolve over time (eg, diagnosis may be made 2 years after primary symptom onset), and often involves collaboration among several specialists.
The above definition does not specify severity, medical/psychiatric comorbidity, or number or duration of symptoms. A proposed classification, medically unexplained symptoms spectrum disorder, attempts to categorize patients with MUPS based on severity and duration of symptoms, as well as medical and/or psychiatric comorbidities (Table).4 MUPS may account for up to two-thirds of physical symptoms in specialty clinics; to read about the prevalence of MUPS, see the Box below.
Figure: Typical etiology of physical symptoms
NOS: Not otherwise specifiedTable
Medically unexplained symptoms spectrum disorder
| Severity | Mild, moderate, severe |
| Duration | Acute (days to weeks), subacute (<6 months), chronic (>6 months) |
| Comorbidity | Psychiatric, medical, none |
| Source: Reference 4 | |
CASE CONTINUED: Abuse and assault
Mrs. B has been married for 10 years and has 2 children. She denies using tobacco, alcohol, or illicit drugs. Both of her parents were in good physical health. As a child Mrs. B was physically and verbally abused by her father, and her parents divorced when she was 15. She was sexually assaulted while in college.
Mrs. B has had 1 previous depressive episode, which occurred shortly after the sexual assault. During that time she was hospitalized twice for attempting suicide by overdosing on prescription medications. She was stabilized on fluoxetine, 40 mg/d, which was tapered and discontinued after 2 years.
Factors linked to MUPS
Young women (age 16 to 25) are more likely to receive an MUPS diagnosis than men or older individuals. Employment, socioeconomic background, and educational level are not consistently associated with MUPS.5 Patients with MUPS have higher rates of physical and sexual abuse.6
Several studies have shown an association with childhood parental ill health and development of MUPS, but the exact nature of “ill health” was not clearly defined. Parental death was not associated with MUPS, which suggests that the association to parental ill health is related to non-threatening physical disease.6
Approximately 60% of patients with MUPS have a comorbid non-somatoform DSM-IV-TR diagnosis.7-9 Symptoms and rates of depressive, anxiety, and panic disorders are higher in patients with MUPS than either healthy controls or patients with similar diseases of known organic pathology.7,8
The estimated prevalence of somatoform disorder in patients with MUPS is approximately 4%, which is higher than in the general population (.2% to 2%).7,8 On measures of mental and physical function, patients with MUPS who have somatoform disorders have been found to be more distressed than normal controls and patients with MUPS without somatoform disorders.8
Although few studies have directly examined the relationship between personality disorders and MUPS, there is evidence of an association between certain personality traits (eg, neuroticism, alexithymia, negative affect) and MUPS.10,11
CASE CONTINUED: Rejected advice
Mrs. B has been worked up multiple times for acute coronary syndrome; been unsuccessfully treated for gastroesophageal reflux disease, lactose intolerance, and IBS; had a negative rheumatologic workup; and tried several medication regimens with no improvement in symptoms. Three years ago Mrs. B’s gastroenterologist implied her abdominal symptoms were caused by her history of sexual assault and suggested she seek psychiatric consultation. Offended, Mrs. B sought a second opinion and no longer sees her first gastroenterologist.
Barriers to treatment
Despite having high levels of psychosocial distress, health care utilization, and medical disability, patients with MUPS often are suboptimally treated. Factors that might contribute to this include:
- inadequate identification
- bias in diagnosis and treatment
- poor follow-up on referrals
- an absence of treatment guidelines.7,12,13
Many clinicians are unaware of the high prevalence of MUPS, which often leads to repeated referral to specialty clinics, even when patients already have received an MUPS diagnosis.12,14 Additionally, clinicians often are unaware of how individual biases influence their diagnostic thought process. A “difficult patient” may receive a MUPS diagnosis more readily than a “pleasant patient,” which could contribute to an incomplete workup. An epidemiologic study revealed that the strongest predictor of misdiagnosing MUPS is doctor dissatisfaction with the clinical encounter.15 Younger, unmarried, anxious patients receiving disability benefits are more likely to be incorrectly labeled as having MUPS, only to later receive a non-MUPS diagnosis.15
Bias in treatment and intervention also exists. Qualitative analysis of consultations suggests that physicians’ decisions to offer patients somatic treatments (eg, investigation, add/change medications, referral to specialists) are responses to patients’ extended and complex accounts of their symptoms.17 The likelihood of intervention was unrelated to patients’ request for treatment, and intervention became less likely when patients described psychosocial problems.16
Patients with MUPS and comorbid psychiatric disorders often are referred for psychosocial treatment, but 1 study found that as few as 10% of such patients follow up on a referral.17 In that study, 81% of MUPS patients were willing to receive psychosocial treatment in a primary care setting by their physician. Although there are many reasons patients with MUPS resist referral to mental health professionals, be aware that many of these individuals do not attribute their symptoms to psychosocial problems or experience their symptoms psychologically. To these patients, psychiatric referral may seem inappropriate or be perceived as belittling and minimizing their symptoms.
CASE CONTINUED: Frustration and guilt
Mrs. B’s depressive symptoms began 18 months ago with fatigue, poor sleep, and withdrawal from her children and husband. She struggles with hopelessness that her physical symptoms will not resolve and guilt because of the financial strain her medical care has placed on the family. She is extremely frustrated that her doctors are unable to find a medical diagnosis for her symptoms and fears that without a diagnosis she will be perceived as “crazy.” She is not certain if there is a medical explanation for her symptoms but vehemently believes they are not associated with her mood or psychosocial stress.
Treatment strategies
A collaborative, unbiased, integrated approach to treatment can address some of the challenges that arise when patients with MUPS confront the limitations of modern medicine. Integrated care involves ongoing communication among medical and psychiatric specialists, as well as collaboration with social workers, physical therapists, nutritionists, or pain management specialists when indicated.
Although the primary care provider often coordinates a MUPS patient’s medical treatment, a consulting psychiatrist plays an important educational, diagnostic, and therapeutic role. The therapeutic role is especially important because patients with MUPS frequently view their general practitioner as having a limited role in managing psychosocial problems.18
Because physical illness and psychosocial stress frequently coexist and compound each other, diagnostic efforts should focus on medical and psychiatric illness. Review the patient’s medical workup of the unexplained symptoms and, when indicated, request further testing. Evaluate the risks and benefits of additional testing and discuss them with the patient; additional testing carries a risk of iatrogenic harm, higher false-positive rates, and increased costs. Avoiding iatrogenic harm and unnecessary, overly aggressive testing is essential.
Identifying primary or comorbid psychiatric disease and psychosocial issues also is integral to managing patients with MUPS. This may be difficult because some patients might be hesitant to discuss psychosocial issues, whereas others may be unaware of psychiatric symptomatology or the connection between mental and physical illness. When possible, it may be useful to clarify symptomatology as:
- primarily somatic (expression of psychological illness through physical means)
- primarily psychiatric (psychiatric illness presenting with physical symptoms) or
- bordering between somatic and psychiatric.
CASE CONTINUED: Collaboration and improvement
You diagnose Mrs. B with major depressive disorder and prescribe fluoxetine, titrating her up to 40 mg/d. Mrs. B also begins weekly psychodynamic psychotherapy. In collaboration with her PCP, you decide to refer Mrs. B to physical therapy and direct psychotherapy toward coping strategies, with the hope of improving functionality. Although she continues to have musculoskeletal symptoms after completing physical therapy, Mrs. B notices moderate improvement and feels less distressed by these symptoms.
After 1 year of fluoxetine treatment, Mrs. B’s depressive symptoms improve. In psychotherapy, her fixation on physical symptoms and desire to establish a diagnosis gradually lessen. As her emotional trauma from childhood abuse unravels, psychotherapy shifts toward improving affect regulation. During this time Mrs. B experiences an increase in unexplained chest pain and shortness of breath, which later abate.
Continued follow-up with a gastroenterologist leads to a diagnosis of celiac disease. With treatment, her GI symptoms resolve.
What do patients want?
Begin MUPS treatment by developing a supportive, empathic relationship with the patient. Carefully listen to the patient’s description of his or her symptoms. Elucidating patients’ experience often is challenging because their narratives frequently are complex, nonlinear, and limited by time.18 Patients’ models for understanding their symptoms also may be complex.18 They may be reluctant to share their explanations, fearing they will be unable to communicate the complexity of their beliefs or their symptoms will be oversimplified.18
Focus on understanding what the patient seeks from the physician—emotional support vs diagnosis vs treatment. In a prospective naturalistic study, the content of MUPS patients’ narratives was correlated with what they sought from their physician.17 Patients who sought emotional support frequently discussed psychosocial problems, issues, and management. Patients who wanted an explanation for their symptoms often mentioned physical symptoms, explanations, and diseases. Those who were looking for additional testing or intervention often directly addressed this with the physician.17
Although many patients desire a diagnosis and somatic treatment, this is not always their primary agenda. Many MUPS patients seek emotional support or confirmation of their explanatory model.17,18 Patients’ desires for emotional support, medical explanation, diagnosis, or somatic intervention often are neither clearly nor explicitly stated. Despite this, patients hope their physician understands the extent of their problems and value those who help them make sense of their narratives.18 Misunderstanding patients’ agendas can result in a mismatch of treatment expectations and fracture the patient-physician relationship. Developing mutual expectations is crucial to building rapport, improving collaborative care, and avoiding unnecessary, potentially harmful interventions.
Psychotherapic interventions
Psychopharmacologic treatment is indicated for MUPS patients who have comorbid psychiatric conditions.
Research of psychotherapy in MUPS has been plagued by methodologic problems and inconsistent results.3 Group therapy, short-term dynamic therapy, hypnotherapy, and cognitive-behavioral therapy (CBT) have been studied. In a trial of 140 MUPS patients who received 1 session of CBT, 71% experienced improvement in physical symptoms, 47% in functional status, and 38% in measures of psychological distress.19 A review of 34 randomized controlled trials involving 3,922 patients with somatoform disorders who received CBT found that some patients with MUPS responded after 5 to 6 sessions.3
Cognitive techniques focus on identifying and restructuring automatic, dysfunctional thoughts that may compound, perpetuate, or worsen somatic symptoms. Behavioral techniques include relaxation and efforts to increase motivation. A CBT treatment plan may involve establishing goals, addressing patients’ understanding of their symptoms, obtaining a commitment for treatment, and negotiating the details of the treatment plan.8,12
Supportive techniques also are valuable in treating MUPS patients. Educate patients and treating physicians that there is a neurophysiologic basis for the patient’s physical symptoms and that symptoms may wax and wane. Reinforcement of functional improvement through concrete, practical solutions can help patients develop healthy, adaptive coping skills. Encouraging patients to move beyond somatic complaints to discuss social and personal difficulties can lead to more effective management of these problems.
Clearly communicate your initial impressions, diagnoses, and treatment plan to other members of the treatment team. A consultation letter from the psychiatrist to the PCP has been shown to decrease costs and slightly improve the patient’s functional status, symptoms, and quality of life.20 When possible, educate the PCP and specialists about the dynamics, challenges, biases, and frustrations physicians commonly face when caring for MUPS patients.
Related Resources
- Burton C. Beyond somatization: a review of the understanding and treatment of medically unexplained physical symptoms. Brit J Gen Pract. 2003;53:233-239.
- Creed F. The outcome of medically unexplained symptoms—will DSM-V improve on DSM-IV somatoform disorders? J Psychosom Res. 2009;66:379-381.
- Katon W, Sullivan M, Walker E. Medical symptoms without identified pathology: relationship to psychiatric disorders, childhood and adult trauma, and personality traits. Ann Intern Med. 2001;134(9 Pt 2):917-925.
- Sharpe M, Mayou R, Walker J. Bodily symptoms: new approaches to classification. J Psychosom Res. 2006;60: 353-356.
Drug Brand Names
- Fluoxetine • Prozac
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Medically unexplained physical symptoms (MUPS) has been found to make up 10% to 30% of the physical symptoms in primary care clinics and 37% to 66% in specialty clinics.a-c The latter statistic is based on a cross-sectional survey of 899 consecutive new patients from 7 outpatient clinics in London, United Kingdom. Sixty-five percent responded and 52% of respondents had at least 1 medically unexplained symptom, diagnosed 3 months after initial clinic presentation.c
Patients with MUPS carry significant clinical importance. They are more likely to have a relatively poor quality of life and higher rates of disability.d,e They tend to be higher utilizers of health care.c,f High utilization of services and potentially unnecessary lab testing and consultation result in increased costs and high rates of iatrogenic complications.d-f
References
a. McCarron RM, Xiong GL, Bourgeois JA. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott Williams and Wilkins; 2009.
b. Richardson RD, Engel CC. Evaluation and management of medically unexplained physical symptoms. Neurologist. 2004;10:18-30.
c. Nimnuan C, Hotopf M, Wessely S. Medically unexplained symptoms. An epidemiological study in seven specialties. J Psychosom Res. 2001;51:361-367.
d. Reid S, Wessely S, Crayford T, et al. Medically unexplained symptoms in frequent attenders of secondary health care: retrospective cohort study. BMJ. 2001;322:1-4.
e. Smith BJ, McGorn KJ, Weller D, et al. The identification in primary care of patients who have been repeatedly referred to hospital for medically unexplained symptoms: a pilot study. J Psychosom Res. 2009;67:207-211.
f. Smith RC, Lein C, Collins C, et al. Treating patients with medically unexplained symptoms in primary care. J Gen Intern Med. 2003;18:478-489.
1. Jackson JL, Kroenke K. Prevalence impact, and prognosis of multisomatoform disorder in primary care: a 5-year follow-up study. Psychosom Med. 2008;70:430-434.
2. Jackson JL, George S, Hinchey S. Medically unexplained physical symptoms. J Gen Intern Med. 2009;24:540-542.
3. Kroenke K. Efficacy of treatment for somatoform disorders: a review of randomized controlled trials. Psychosom Med. 2007;69:881-888.
4. Smith RC, Dwamena FC. Classification and diagnosis of patients with medically unexplained symptoms. J Gen Intern Med. 2007;22:685-691.
5. Nimnuan C, Hotopf M, Wessely S. Medically unexplained symptoms. An epidemiological study in seven specialties. J Psychosom Res. 2001;51:361-367.
6. Hotopf M, Mayou R, Wadsworth M, et al. Childhood risk factors for adults with medically unexplained symptoms: results from a national birth cohort study. Am J Psychiatry. 1999;156(11):1796-1800.
7. Smith RC, Gardiner JC, Lyles JS, et al. Exploration of DSM-IV criteria in primary care patients with medically unexplained symptoms. Psychosom Med. 2005;67:123-129.
8. Smith RC, Lyles JS, Gardiner JC, et al. Primary care clinicians treat patients with medically unexplained symptoms: a randomized controlled trial. J Gen Intern Med. 2006;21:671-677.
9. Henningsen P, Zimmermann T, Sattel H. Medically unexplained physical symptoms anxiety, and depression. A meta-analytic review. Psychosom Med. 2003;65:528-533.
10. Costa PT, McCrae RR. Neuroticism somatic complaints, and disease: is the bark worse than the bite? J Pers. 1987;55(2):299-315.
11. Gucht VD, Fischler B, Heiser W. Personality and affect as determinants of medically unexplained symptoms in primary care. A follow-up study. J Psychosom Res. 2004;56:279-285.
12. Smith RC, Lein C, Collins C, et al. Treating patients with medically unexplained symptoms in primary care. J Gen Intern Med. 2003;18:478-489.
13. McFarlane AC, Ellis N, Fasphm F, et al. The conundrum of medically unexplained symptoms: questions to consider. Psychosomatics. 2008;49(5):369-377.
14. Reid S, Wessely S, Crayford T, et al. Medically unexplained symptoms in frequent attenders of secondary health care: retrospective cohort study. BMJ. 2001;322:1-4.
15. Nimnuan C, Hotopf M, Wessely S. Medically unexplained symptoms: how often and why are they missed? QJM. 2000;93:21-28.
16. Salmon P, Humphris GM, Ring A, et al. Primary care consultations about medically unexplained symptoms: patient presentations and doctor responses that influence the probability of somatic intervention. Psychosom Med. 2007;69:571-577.
17. Salmon P, Ring A, Humphris GM, et al. Primary care consultations about medically unexplained symptoms: how do patients indicate what they want? J Gen Intern Med. 2009;24(4):450-456.
18. Peters S, Rogers A, Salmon P, et al. What do patients choose to tell their doctors? Qualitative analysis of potential barriers to reattributing medically unexplained symptoms. J Gen Intern Med. 2008;24(4):443-449.
19. Martin A, Rauh E, Fichter M, et al. A one-session treatment for patients suffering from medically unexplained symptoms in primary care: a randomized clinical trial. Psychosomatics. 2007;48:294-303.
20. Smith BJ, McGorn KJ, Weller D, et al. The identification in primary care of patients who have been repeatedly referred to hospital for medically unexplained symptoms: a pilot study. J Psychosom Res. 2009;67:207-211.
1. Jackson JL, Kroenke K. Prevalence impact, and prognosis of multisomatoform disorder in primary care: a 5-year follow-up study. Psychosom Med. 2008;70:430-434.
2. Jackson JL, George S, Hinchey S. Medically unexplained physical symptoms. J Gen Intern Med. 2009;24:540-542.
3. Kroenke K. Efficacy of treatment for somatoform disorders: a review of randomized controlled trials. Psychosom Med. 2007;69:881-888.
4. Smith RC, Dwamena FC. Classification and diagnosis of patients with medically unexplained symptoms. J Gen Intern Med. 2007;22:685-691.
5. Nimnuan C, Hotopf M, Wessely S. Medically unexplained symptoms. An epidemiological study in seven specialties. J Psychosom Res. 2001;51:361-367.
6. Hotopf M, Mayou R, Wadsworth M, et al. Childhood risk factors for adults with medically unexplained symptoms: results from a national birth cohort study. Am J Psychiatry. 1999;156(11):1796-1800.
7. Smith RC, Gardiner JC, Lyles JS, et al. Exploration of DSM-IV criteria in primary care patients with medically unexplained symptoms. Psychosom Med. 2005;67:123-129.
8. Smith RC, Lyles JS, Gardiner JC, et al. Primary care clinicians treat patients with medically unexplained symptoms: a randomized controlled trial. J Gen Intern Med. 2006;21:671-677.
9. Henningsen P, Zimmermann T, Sattel H. Medically unexplained physical symptoms anxiety, and depression. A meta-analytic review. Psychosom Med. 2003;65:528-533.
10. Costa PT, McCrae RR. Neuroticism somatic complaints, and disease: is the bark worse than the bite? J Pers. 1987;55(2):299-315.
11. Gucht VD, Fischler B, Heiser W. Personality and affect as determinants of medically unexplained symptoms in primary care. A follow-up study. J Psychosom Res. 2004;56:279-285.
12. Smith RC, Lein C, Collins C, et al. Treating patients with medically unexplained symptoms in primary care. J Gen Intern Med. 2003;18:478-489.
13. McFarlane AC, Ellis N, Fasphm F, et al. The conundrum of medically unexplained symptoms: questions to consider. Psychosomatics. 2008;49(5):369-377.
14. Reid S, Wessely S, Crayford T, et al. Medically unexplained symptoms in frequent attenders of secondary health care: retrospective cohort study. BMJ. 2001;322:1-4.
15. Nimnuan C, Hotopf M, Wessely S. Medically unexplained symptoms: how often and why are they missed? QJM. 2000;93:21-28.
16. Salmon P, Humphris GM, Ring A, et al. Primary care consultations about medically unexplained symptoms: patient presentations and doctor responses that influence the probability of somatic intervention. Psychosom Med. 2007;69:571-577.
17. Salmon P, Ring A, Humphris GM, et al. Primary care consultations about medically unexplained symptoms: how do patients indicate what they want? J Gen Intern Med. 2009;24(4):450-456.
18. Peters S, Rogers A, Salmon P, et al. What do patients choose to tell their doctors? Qualitative analysis of potential barriers to reattributing medically unexplained symptoms. J Gen Intern Med. 2008;24(4):443-449.
19. Martin A, Rauh E, Fichter M, et al. A one-session treatment for patients suffering from medically unexplained symptoms in primary care: a randomized clinical trial. Psychosomatics. 2007;48:294-303.
20. Smith BJ, McGorn KJ, Weller D, et al. The identification in primary care of patients who have been repeatedly referred to hospital for medically unexplained symptoms: a pilot study. J Psychosom Res. 2009;67:207-211.
HIV screening: Latest testing guidelines
• Patients with mental illness are at elevated risk for HIV infection.
• Fourth-generation (p24) HIV testing allows accurate, office-based screening during the period of highest infectivity.
• Psychiatrists may be uniquely able to discuss and alter patients’ high-risk behaviors related to HIV transmission and treatment.
Substance use and high-risk sexual behaviors are common among persons with severe mental illness and make them vulnerable to human immunodeficiency virus (HIV) infection.1-3 The prevalence of HIV is 3 times higher among these patients compared with individuals who are not mentally ill. This article reviews risk factors for HIV transmission, HIV screening recommendations, and current testing options, including the fourth-generation HIV test.
According to the World Health Organization, in 2009:
- 33 million people were infected with HIV
- there were 2.6 million new infections
- 1.8 million people died from causes related to acquired immune deficiency syndrome (AIDS).4
In the United States, approximately 1 million people are living with HIV; 55, 000 new cases were diagnosed in North America in 2009.5 The demographics of those infected with HIV in the United States have changed substantially. Most newly infected individuals are African American, and younger adults have higher rates of new infections.6 With the introduction of standardized antiviral medication combinations and federal programs to provide universal access to medications in the mid 1990s, the number of AIDS cases and mortality dropped, but there have not been similar gains in the past decade.6
In countries With access to antiviral medications, medical advances in HIV treatment have transformed an infection with a high mortality rate into a chronic illness. However, the rate of new cases has not changed significantly in the past 10 years. Early detection and treatment initiation reduces high-risk behavior and subsequent transmission in patients with HIV. Evidence suggests that detecting HIV in the acute phase of the illness, when the viral load is high, reduces HIV transmission.1,5,6
HIV screening
HIV screening should be part of routine psychiatric practice, especially in community and forensic settings. We recommend that all psychiatrists understand HIV screening guidelines from the Centers for Disease Control and Prevention (CDC)7 and the American College of Physicians (ACP).8 The CDC (2006)7 and ACP (2009)8 guidelines recommend HIV screening for:
- all patients age 13 to 64, unless the prevalence of undiagnosed HIV infection in your patients has been documented to be <0. 1%7
- all patients seeking treatment for sexually transmitted diseases, including each visit for a new complaint7
- all pregnant women7
- any patient who received a blood transfusion between 1978 and 1985.8
Rescreening frequency is determined on an individual basis, taking into consideration the patient’s risk of contracting HIV. The ACP guidelines recommend using rapid tests when available.8 Although the false positive rates are higher with rapid tests, the benefits of early detection and reducing transmission outweigh the cost of confirming positive tests.8
The fourth-generation test
The gold standard for HIV screening is the enzyme-linked immunosorbent assay antibody test followed by a western blot.8 This has a high sensitivity and specificity but there is a window of time between infection and detectable antibodies in serum during which patients have high virus levels (Figure).9 The combined antibody and antigen p24 test is a fourth-generation screening tool that detects HIV in the acute phase of the illness without requiring expensive ribonucleic acid viral tests.10 The fourth-generation test’s capacity to detect infection during the antibody-negative window when infectivity is highest may help decrease HIV transmission.11
Figure When can HIV tests detect the virus?
EIA: enzyme immunoassay; HIV: human immunodeficiency virus; RNA: ribonucleic acid
Source: Adapted from reference 9
Psychiatrists’ role
Because psychiatrists routinely interact with patients who have an increased risk of HIV, they may be uniquely qualified to help these individuals. Psychiatrists often are comfortable discussing patients’ sexual and substance use history, which allows them to uncover and effectively address high-risk behaviors with appropriate testing and counseling. If testing confirms HIV infection, psychiatrists also can discuss altering high-risk behaviors (Table),1,2 which has significant implications not only for patients, but also for public health.
Table
HIV transmission risk factors
| Intravenous drug use |
| Nonintravenous cocaine use |
| Sex without barrier protection, especially in patients with multiple partners |
| Sex trade or sex work |
| HIV: human immunodeficiency virus |
| Source: References 1,2 |
Related Resource
- Centers for Disease Control and Prevention. HIV/AIDS. www.cdc.gov/hiv.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Health. 2001;91:31-37.
2. Quinn TC, Glasser D, Cannon RO, et al. Human immunodeficiency virus infection among patients attending clinics for sexually transmitted diseases. N Eng J Med. 1988;318:197-203.
3. Erickson B, Wasserheit JN, Rompalo AM, et al. Routine voluntary HIV screening in STD clinic clients: characterization of infected clients. Sex Transm Dis. 1990;17:194-199.
4. World Health Organization. Global summary of the AIDS epidemic. Available at: http://www.who.int/hiv/data/2009_global_summary.png. Accessed February 10 2011.
5. Hall HI, Song R, Rhodes P, et al. Estimation of HIV incidence in the United States. JAMA. 2008;300:520-529.
6. Glynn MK, Lee LM, McKenna MT. The status of national HIV case surveillance United States 2006. Public Health Rep. 2007;122(suppl 1):63-71.
7. Branson BM, Handsfield HH, Lampe MA, et al. and the Centers for Disease Control and Prevention. Revised recommendations for HIV testing of adults adolescents, and pregnant women in health-care settings. MMWR Recomm Rep. 2006;55(RR-14):1-17; quiz CE1-CE4.
8. Qaseem A, Snow V, Shekelle P, et al. and the Clinical Efficacy Assessment Subcommittee American College of Physicians. Screening for HIV in health care settings: a guidance statement from the American College of Physicians and HIV Medicine Association. Ann Intern Med. 2009;150:125-131.
9. Branson BM. The future of HIV testing. J Acquir Immune Defic Syndr. 2010;55:S102-S105.
10. Eshleman S, Khaki L, Laeyendecker O, et al. Detection of individuals with acute HIV-1 infection using the ARCHITECT HIV Ag/Ab combo assay. J Acquir Immune Defic Syndr. 2009;52:121-124.
11. Pandori MW, Hackett J, Jr, Louie B, et al. Assessment of the ability of a fourth-generation immunoassay for human immunodeficiency virus (HIV) antibody and p24 antigen to detect both acute and recent HIV infections in a high-risk setting. J Clin Microbiol. 2009;47:2639-2642.
John Onate, MD
Dr. Onate is Health Sciences Assistant Clinical Professor, Department of Psychiatry and Behavioral Sciences
Travis Fisher, MD
Dr. Fisher is Fellow, Department of Psychosomatic Medicine, University of California Davis Medical Center, Sacramento, CA.
Robert M. McCarron, DO
Series Editor
John Onate, MD
Dr. Onate is Health Sciences Assistant Clinical Professor, Department of Psychiatry and Behavioral Sciences
Travis Fisher, MD
Dr. Fisher is Fellow, Department of Psychosomatic Medicine, University of California Davis Medical Center, Sacramento, CA.
Robert M. McCarron, DO
Series Editor
John Onate, MD
Dr. Onate is Health Sciences Assistant Clinical Professor, Department of Psychiatry and Behavioral Sciences
Travis Fisher, MD
Dr. Fisher is Fellow, Department of Psychosomatic Medicine, University of California Davis Medical Center, Sacramento, CA.
Robert M. McCarron, DO
Series Editor
• Patients with mental illness are at elevated risk for HIV infection.
• Fourth-generation (p24) HIV testing allows accurate, office-based screening during the period of highest infectivity.
• Psychiatrists may be uniquely able to discuss and alter patients’ high-risk behaviors related to HIV transmission and treatment.
Substance use and high-risk sexual behaviors are common among persons with severe mental illness and make them vulnerable to human immunodeficiency virus (HIV) infection.1-3 The prevalence of HIV is 3 times higher among these patients compared with individuals who are not mentally ill. This article reviews risk factors for HIV transmission, HIV screening recommendations, and current testing options, including the fourth-generation HIV test.
According to the World Health Organization, in 2009:
- 33 million people were infected with HIV
- there were 2.6 million new infections
- 1.8 million people died from causes related to acquired immune deficiency syndrome (AIDS).4
In the United States, approximately 1 million people are living with HIV; 55, 000 new cases were diagnosed in North America in 2009.5 The demographics of those infected with HIV in the United States have changed substantially. Most newly infected individuals are African American, and younger adults have higher rates of new infections.6 With the introduction of standardized antiviral medication combinations and federal programs to provide universal access to medications in the mid 1990s, the number of AIDS cases and mortality dropped, but there have not been similar gains in the past decade.6
In countries With access to antiviral medications, medical advances in HIV treatment have transformed an infection with a high mortality rate into a chronic illness. However, the rate of new cases has not changed significantly in the past 10 years. Early detection and treatment initiation reduces high-risk behavior and subsequent transmission in patients with HIV. Evidence suggests that detecting HIV in the acute phase of the illness, when the viral load is high, reduces HIV transmission.1,5,6
HIV screening
HIV screening should be part of routine psychiatric practice, especially in community and forensic settings. We recommend that all psychiatrists understand HIV screening guidelines from the Centers for Disease Control and Prevention (CDC)7 and the American College of Physicians (ACP).8 The CDC (2006)7 and ACP (2009)8 guidelines recommend HIV screening for:
- all patients age 13 to 64, unless the prevalence of undiagnosed HIV infection in your patients has been documented to be <0. 1%7
- all patients seeking treatment for sexually transmitted diseases, including each visit for a new complaint7
- all pregnant women7
- any patient who received a blood transfusion between 1978 and 1985.8
Rescreening frequency is determined on an individual basis, taking into consideration the patient’s risk of contracting HIV. The ACP guidelines recommend using rapid tests when available.8 Although the false positive rates are higher with rapid tests, the benefits of early detection and reducing transmission outweigh the cost of confirming positive tests.8
The fourth-generation test
The gold standard for HIV screening is the enzyme-linked immunosorbent assay antibody test followed by a western blot.8 This has a high sensitivity and specificity but there is a window of time between infection and detectable antibodies in serum during which patients have high virus levels (Figure).9 The combined antibody and antigen p24 test is a fourth-generation screening tool that detects HIV in the acute phase of the illness without requiring expensive ribonucleic acid viral tests.10 The fourth-generation test’s capacity to detect infection during the antibody-negative window when infectivity is highest may help decrease HIV transmission.11
Figure When can HIV tests detect the virus?
EIA: enzyme immunoassay; HIV: human immunodeficiency virus; RNA: ribonucleic acid
Source: Adapted from reference 9
Psychiatrists’ role
Because psychiatrists routinely interact with patients who have an increased risk of HIV, they may be uniquely qualified to help these individuals. Psychiatrists often are comfortable discussing patients’ sexual and substance use history, which allows them to uncover and effectively address high-risk behaviors with appropriate testing and counseling. If testing confirms HIV infection, psychiatrists also can discuss altering high-risk behaviors (Table),1,2 which has significant implications not only for patients, but also for public health.
Table
HIV transmission risk factors
| Intravenous drug use |
| Nonintravenous cocaine use |
| Sex without barrier protection, especially in patients with multiple partners |
| Sex trade or sex work |
| HIV: human immunodeficiency virus |
| Source: References 1,2 |
Related Resource
- Centers for Disease Control and Prevention. HIV/AIDS. www.cdc.gov/hiv.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
• Patients with mental illness are at elevated risk for HIV infection.
• Fourth-generation (p24) HIV testing allows accurate, office-based screening during the period of highest infectivity.
• Psychiatrists may be uniquely able to discuss and alter patients’ high-risk behaviors related to HIV transmission and treatment.
Substance use and high-risk sexual behaviors are common among persons with severe mental illness and make them vulnerable to human immunodeficiency virus (HIV) infection.1-3 The prevalence of HIV is 3 times higher among these patients compared with individuals who are not mentally ill. This article reviews risk factors for HIV transmission, HIV screening recommendations, and current testing options, including the fourth-generation HIV test.
According to the World Health Organization, in 2009:
- 33 million people were infected with HIV
- there were 2.6 million new infections
- 1.8 million people died from causes related to acquired immune deficiency syndrome (AIDS).4
In the United States, approximately 1 million people are living with HIV; 55, 000 new cases were diagnosed in North America in 2009.5 The demographics of those infected with HIV in the United States have changed substantially. Most newly infected individuals are African American, and younger adults have higher rates of new infections.6 With the introduction of standardized antiviral medication combinations and federal programs to provide universal access to medications in the mid 1990s, the number of AIDS cases and mortality dropped, but there have not been similar gains in the past decade.6
In countries With access to antiviral medications, medical advances in HIV treatment have transformed an infection with a high mortality rate into a chronic illness. However, the rate of new cases has not changed significantly in the past 10 years. Early detection and treatment initiation reduces high-risk behavior and subsequent transmission in patients with HIV. Evidence suggests that detecting HIV in the acute phase of the illness, when the viral load is high, reduces HIV transmission.1,5,6
HIV screening
HIV screening should be part of routine psychiatric practice, especially in community and forensic settings. We recommend that all psychiatrists understand HIV screening guidelines from the Centers for Disease Control and Prevention (CDC)7 and the American College of Physicians (ACP).8 The CDC (2006)7 and ACP (2009)8 guidelines recommend HIV screening for:
- all patients age 13 to 64, unless the prevalence of undiagnosed HIV infection in your patients has been documented to be <0. 1%7
- all patients seeking treatment for sexually transmitted diseases, including each visit for a new complaint7
- all pregnant women7
- any patient who received a blood transfusion between 1978 and 1985.8
Rescreening frequency is determined on an individual basis, taking into consideration the patient’s risk of contracting HIV. The ACP guidelines recommend using rapid tests when available.8 Although the false positive rates are higher with rapid tests, the benefits of early detection and reducing transmission outweigh the cost of confirming positive tests.8
The fourth-generation test
The gold standard for HIV screening is the enzyme-linked immunosorbent assay antibody test followed by a western blot.8 This has a high sensitivity and specificity but there is a window of time between infection and detectable antibodies in serum during which patients have high virus levels (Figure).9 The combined antibody and antigen p24 test is a fourth-generation screening tool that detects HIV in the acute phase of the illness without requiring expensive ribonucleic acid viral tests.10 The fourth-generation test’s capacity to detect infection during the antibody-negative window when infectivity is highest may help decrease HIV transmission.11
Figure When can HIV tests detect the virus?
EIA: enzyme immunoassay; HIV: human immunodeficiency virus; RNA: ribonucleic acid
Source: Adapted from reference 9
Psychiatrists’ role
Because psychiatrists routinely interact with patients who have an increased risk of HIV, they may be uniquely qualified to help these individuals. Psychiatrists often are comfortable discussing patients’ sexual and substance use history, which allows them to uncover and effectively address high-risk behaviors with appropriate testing and counseling. If testing confirms HIV infection, psychiatrists also can discuss altering high-risk behaviors (Table),1,2 which has significant implications not only for patients, but also for public health.
Table
HIV transmission risk factors
| Intravenous drug use |
| Nonintravenous cocaine use |
| Sex without barrier protection, especially in patients with multiple partners |
| Sex trade or sex work |
| HIV: human immunodeficiency virus |
| Source: References 1,2 |
Related Resource
- Centers for Disease Control and Prevention. HIV/AIDS. www.cdc.gov/hiv.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Health. 2001;91:31-37.
2. Quinn TC, Glasser D, Cannon RO, et al. Human immunodeficiency virus infection among patients attending clinics for sexually transmitted diseases. N Eng J Med. 1988;318:197-203.
3. Erickson B, Wasserheit JN, Rompalo AM, et al. Routine voluntary HIV screening in STD clinic clients: characterization of infected clients. Sex Transm Dis. 1990;17:194-199.
4. World Health Organization. Global summary of the AIDS epidemic. Available at: http://www.who.int/hiv/data/2009_global_summary.png. Accessed February 10 2011.
5. Hall HI, Song R, Rhodes P, et al. Estimation of HIV incidence in the United States. JAMA. 2008;300:520-529.
6. Glynn MK, Lee LM, McKenna MT. The status of national HIV case surveillance United States 2006. Public Health Rep. 2007;122(suppl 1):63-71.
7. Branson BM, Handsfield HH, Lampe MA, et al. and the Centers for Disease Control and Prevention. Revised recommendations for HIV testing of adults adolescents, and pregnant women in health-care settings. MMWR Recomm Rep. 2006;55(RR-14):1-17; quiz CE1-CE4.
8. Qaseem A, Snow V, Shekelle P, et al. and the Clinical Efficacy Assessment Subcommittee American College of Physicians. Screening for HIV in health care settings: a guidance statement from the American College of Physicians and HIV Medicine Association. Ann Intern Med. 2009;150:125-131.
9. Branson BM. The future of HIV testing. J Acquir Immune Defic Syndr. 2010;55:S102-S105.
10. Eshleman S, Khaki L, Laeyendecker O, et al. Detection of individuals with acute HIV-1 infection using the ARCHITECT HIV Ag/Ab combo assay. J Acquir Immune Defic Syndr. 2009;52:121-124.
11. Pandori MW, Hackett J, Jr, Louie B, et al. Assessment of the ability of a fourth-generation immunoassay for human immunodeficiency virus (HIV) antibody and p24 antigen to detect both acute and recent HIV infections in a high-risk setting. J Clin Microbiol. 2009;47:2639-2642.
1. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Health. 2001;91:31-37.
2. Quinn TC, Glasser D, Cannon RO, et al. Human immunodeficiency virus infection among patients attending clinics for sexually transmitted diseases. N Eng J Med. 1988;318:197-203.
3. Erickson B, Wasserheit JN, Rompalo AM, et al. Routine voluntary HIV screening in STD clinic clients: characterization of infected clients. Sex Transm Dis. 1990;17:194-199.
4. World Health Organization. Global summary of the AIDS epidemic. Available at: http://www.who.int/hiv/data/2009_global_summary.png. Accessed February 10 2011.
5. Hall HI, Song R, Rhodes P, et al. Estimation of HIV incidence in the United States. JAMA. 2008;300:520-529.
6. Glynn MK, Lee LM, McKenna MT. The status of national HIV case surveillance United States 2006. Public Health Rep. 2007;122(suppl 1):63-71.
7. Branson BM, Handsfield HH, Lampe MA, et al. and the Centers for Disease Control and Prevention. Revised recommendations for HIV testing of adults adolescents, and pregnant women in health-care settings. MMWR Recomm Rep. 2006;55(RR-14):1-17; quiz CE1-CE4.
8. Qaseem A, Snow V, Shekelle P, et al. and the Clinical Efficacy Assessment Subcommittee American College of Physicians. Screening for HIV in health care settings: a guidance statement from the American College of Physicians and HIV Medicine Association. Ann Intern Med. 2009;150:125-131.
9. Branson BM. The future of HIV testing. J Acquir Immune Defic Syndr. 2010;55:S102-S105.
10. Eshleman S, Khaki L, Laeyendecker O, et al. Detection of individuals with acute HIV-1 infection using the ARCHITECT HIV Ag/Ab combo assay. J Acquir Immune Defic Syndr. 2009;52:121-124.
11. Pandori MW, Hackett J, Jr, Louie B, et al. Assessment of the ability of a fourth-generation immunoassay for human immunodeficiency virus (HIV) antibody and p24 antigen to detect both acute and recent HIV infections in a high-risk setting. J Clin Microbiol. 2009;47:2639-2642.
Evaluating for conversion disorder: When to suspect Creutzfeldt-Jakob disease
Consider this rare disorder in patients with rapid-onset neurologic symptoms
Ms. J, age 63, is admitted to Neurology with progressive dizziness and cognitive impairment. She had developed word-finding difficulties, weakness, memory problems, and an episode of arm shaking, which prompted referral for inpatient workup. Ms. J has a history of hypertension, palpitations, and diabetes mellitus.
Her neurologic examination is variable; some examiners find pronounced aphasia and right-sided weakness, whereas others document a nearly normal examination. Lumbar puncture (LP) shows normal cell count, glucose, protein, and negative Gram’s stain; MRI of the brain is normal. Enterovirus polymerase chain reaction, cryptococcal antigen, and Lyme antibody are negative. Electroencephalography (EEG) demonstrates diffuse slowing. The primary team requests psychiatric consultation to assess for conversion disorder.
Ms. J is cooperative with psychiatric evaluation. She denies current or past psychiatric symptomatology and does not meet criteria for major depression, dysthymia, adjustment disorder, anxiety disorder, psychosis, or mania. She denies personal or family history of suicidal or homicidal ideation, intent, or plan. Her youngest son died 5 years earlier; she is financially secure and her 40-year marriage is stable. Ms. J denies having a history of substance use, physical or sexual abuse, or trauma.
In the Cardiology clinic 2 months ago, Ms. J denied having neurologic symptoms and was noted to be doing well. Her neurologic symptoms began shortly after that visit and steadily progressed. She is unable to identify an inciting event or stressor. Ms. J worked until 2 weeks before this admission. Neurologic examination at the time of psychiatric consultation is notable for waxing and waning expressive aphasia, right homonymous hemianopsia, and mildly decreased strength in the left biceps and forearm.
Ms. J presented to her cardiologist reporting dizziness and blurred vision 6 weeks ago, and she was observed in the hospital 3 weeks earlier for further evaluation. Laboratory testing during that hospitalization included blood counts, basic metabolic panel, thyroid function studies, erythrocyte sedimentation rate, thiamine, folic acid and vitamin B12, rapid plasma reagin and human immunodeficiency virus antibody, and LP, all reported as within normal limits.
Thorough review of Ms. J’s medical records reveals abnormalities that would be difficult to ascribe to conversion disorder. Specifically, 6 weeks ago, MRI of the brain demonstrated restricted diffusion in the left occipital lobe, and cerebrospinal fluid (CSF) neuron-specific enolase was moderately elevated at 34 ng/mL. The psychiatric consultant discusses these findings and concern for possible rapidly progressive dementia or Creutzfeldt-Jakob disease (CJD) with the primary team, Ms. J, and her family.
Ms. J is discharged with testing for CSF protein 14-3-3 pending and medical follow-up in 10 days. At follow-up 1 week later, her symptoms are worse; she is completely aphasic and wheelchair-bound. Antithyroglobulin and antimicrosomal thyroid antibodies and paraneoplastic antibody panel return normal. CSF protein 14-3-3 ultimately returns positive, supporting a clinical diagnosis of CJD. Ms. J dies shortly after hospital follow-up, less than 4 months after her first complaint of neurologic symptoms. No autopsy is performed.
Patients with conversion disorder may present with neurologic symptoms such as blindness, seizures, paralysis, or sensory loss with no identifiable anatomical or medical explanation.1 Conversion seizures—also known as pseudoseizures or nonepileptic seizures—may be clinically indistinguishable from generalized tonic-clonic seizures, but no EEG correlate can be identified.1,2 Conversion disorder is conceptualized as an unconscious manifestation of psychological conflict or stress—patients are not aware they are producing symptoms—and has been associated with emotional, sexual, and physical trauma.3,4
Conversion disorder is a diagnosis of exclusion and requires thorough evaluation to rule out neurologic or medical etiologies. The differential diagnosis for conversion disorder includes the broad medical differential diagnosis for the symptom, whether it be paralysis, seizures, sensory loss, or other presenting symptoms. Therefore, when evaluating patients for conversion disorder, be vigilant to the possibility of not only underlying psychological stress or trauma but also undiscovered medical or neurologic illness.
In Ms. J’s case, the primary team began to suspect there was no organic cause of her neurologic symptoms. However, psychiatric evaluation revealed that Ms. J had no history of stress or trauma that typically would be associated with conversion disorder, nor did she manifest other psychiatric symptoms, except waxing and waning mental status, which raised concerns for possible delirium or encephalopathy. Additionally, slowing on EEG was a nonspecific but abnormal finding that made conversion disorder unlikely. The consulting psychiatrist discussed this slowing, in conjunction with the abnormal MRI and elevated CSF neuron-specific enolase, with members of the referring Neurology service, who ordered additional testing of CSF for protein 14-3-3.
Creutzfeldt-Jakob disease
CJD is a rapidly progressive neurodegenerative disorder characterized by cognitive changes, behavioral changes, gait disturbances, akinetic mutism, and myoclonus.5 CJD results from the transition of prion proteins, which are present in the normal human brain, to disease-associated forms that aggregate and propagate and result in neurotoxicity with spongiform changes in neurons.6 The transition of normal prions to disease-associated prions may be hereditary, iatrogenic, infectious, or sporadic. Because the pathologic prion protein can be transmitted and normal sterilization procedures do not prevent the spread of CJD, special precautions should be taken to avoid contact with blood or CSF from patients suspected of having CJD.5
CJD most commonly occurs in the sporadic form, for which there are no identifiable risk factors, with an average age of onset between age 50 and 70. The disease affects women and men equally at a rate of 1 to 2 persons per million per year worldwide.6,7 Most patients with CJD die within 12 months of diagnosis8; median survival is 4 to 5 months.7,9 Although there is no approved or standard effective treatment for this uniformly fatal disease, research into the possibility of genetic or post-translational treatments is ongoing. One group reported inhibition of prion propagation by quinacrine and chlorpromazine in vitro.10 Clinical studies of quinacrine have demonstrated tolerability but no impact on the course of CJD.6
Clues to diagnosis. Although there is no treatment for CJD, early diagnosis can help patients and families understand the relentless progression of symptoms and also permits end-of-life planning and palliative care.11 Diagnosing CJD requires a high level of suspicion and traditionally has required brain biopsy or autopsy for conclusive diagnosis, although in some cases rare EEG findings of periodic sharp wave complexes or generalized periodic epileptiform discharges (GPEDs) have suggested the diagnosis.7,8,12 Recently, specific MRI findings have been described with fluid attenuated inversion recovery (FLAIR) and diffusion sequences.9,13,14
Routine LP for CSF examination (including cell count, protein, and glucose) frequently is normal.8 Specific testing to assess for CJD is required. Elevated levels of CSF neuron-specific enolase (normal <30 ng/mL) and protein 14-3-3 (normal <8 ng/mL) are fairly sensitive and specific for CJD when assessed in patients with the proper clinical history, although normal levels of these proteins have been detected in patients later confirmed to have CJD.7,15 A large multinational collaborative study of confirmed CJD cases that evaluated diagnostic test characteristics recommended that because each test has limitations and can be falsely negative—even in a case of later-confirmed CJD—a rational approach to diagnosis includes brain MRI with diffusion-weighted imaging, CSF analysis for protein 14-3-3, and EEG to assess for periodic sharp wave complexes or GPEDs.16
Because CJD presentation varies widely, most clinicians will not consider the diagnosis until the disease has progressed or the patient has died. Patients who present with psychological symptoms or predominant language disturbances and dysphagia may be referred to a psychiatrist or an ear, nose, and throat specialist before seeing a neurologist.9 Patients may be extensively evaluated and treated for conversion disorder when the correct diagnosis is CJD.17
Sporadic CJD traditionally is associated with neurologic presentations, whereas variant CJD is believed to present with psychiatric symptomatology. However, in a 25-year retrospective review of 126 patients with sporadic CJD, 80% of cases demonstrated psychiatric symptoms within the first 100 days of the disease course.18 Of these, nearly 25% showed psychiatric symptoms at presentation, including sleep disturbances, psychotic symptoms, agitation, and anxiety.
Psychiatrists should be aware of distinguishing features of rapidly progressive dementias and CJD, especially in the setting of psychiatric consultation, to rule out somatic etiologies of unexplained neurologic symptoms. It is important to obtain a history of baseline functioning, duration of decline, and psychiatric symptomatology to differentiate between organic and somatic causes. Differential diagnosis for rapidly progressive cognitive impairment is broad and includes delirium from diverse medical causes; rapidly progressive dementia such as accelerated Alzheimer’s disease, Lewy body disease, or frontotemporal dementia; and psychogenic causes, including conversion disorder (Table 1).7,8,12Table 2 provides distinguishing features of CJD, Alzheimer’s disease, Lewy body disease, and frontotemporal dementia with motor neuron disease.7,8,19
Table 1
Differential diagnosis of rapidly progressive dementia
| Celiac disease |
| Central nervous system vasculitis |
| Creutzfeldt-Jakob disease |
| Delirium (numerous possible etiologies) |
| Focal status epilepticus |
| Hashimoto’s encephalopathy |
Infection
|
Intoxication
|
| Limbic encephalopathy from paraneoplastic antibody syndrome |
| Lymphomatoid granulomatosis |
Malignancy
|
| Porphyria |
| Progressive supranuclear palsy |
Psychiatric disorder
|
| Sarcoidosis |
| Stroke |
| Vitamin deficiency (vitamin E, thiamine) |
| EBV: Epstein-Barr virus; HIV: human immunodeficiency virus; HSV: herpes simplex virus Source: References 7,8,12 |
Table 2
Distinguishing features of Creutzfeldt-Jakob disease
| Sporadic CJD | AD | DLBD | FTD-MND | |
|---|---|---|---|---|
| Time course | Rapid progression (median survival 4 to 5 months) | Insidious onset; progressive decline | Insidious onset; progressive decline | May experience rapid course to death |
| Age at onset | Age 50 to 70 | Incidence increases with age (usual onset age 65 to 85) | Older (age ~80) | Young age at onset |
| EEG findings | Periodic atypical triphasic waves; GPEDs | Normal or diffuse abnormalities | Rarely atypical triphasic waves | Increased slow activity, decreased fast activity |
| MRI findings | Restricted diffusion | Generalized atrophy | Generalized atrophy | Frontal atrophy |
| AD: Alzheimer’s disease; CJD: Creutzfeldt-Jakob disease; DLBD: diffuse Lewy body dementia; EEG: electroencephalography; FTD-MND: frontotemporal dementia with motor neuron disease; GPEDs: generalized periodic epileptiform discharges Source: References 7,8,19 | ||||
Related Resources
National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
Centers for Disease Control and Prevention. About CJD. www.cdc.gov/ncidod/dvrd/cjd.
Drug Brand Names
Chlorpromazine • Thorazine, Largactil
Quinacine • Atabrine
Disclosure
Dr. Gagliardi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry. 2006;163(9):1510-1517.
2. Teo WY, Choong CT. Neurological presentations of conversion disorders in a group of Singapore children. Pediatr Int. 2008;50(4):533-536.
3. Brown RJ, Cardena E, Nuenhuis E, et al. Should conversion disorder be reclassified as a dissociative disorder in DSM-V? Psychosomatics. 2007;48:369-378.
4. Stone J, Carson A, Aditya H, et al. The role of physical injury in motor and sensory conversion symptoms: a systematic and narrative review. J Psychosom Res. 2009;66:383-390.
5. National Institute of Neurological Disorders and Stroke Creutzfeldt-Jakob disease fact sheet. Available at: http://www.ninds.nih.gov/disorders/cjd/detail_cjd.htm. Accessed August 7, 2010.
6. Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8:334-344.
7. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64:98-108.
8. Josephs KA, Ahlskog E, Parisi JE, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol. 2009;66(2):201-207.
9. Martindale JL, Geschwind MD, Miller BL. Psychiatric and neuroimaging findings in Creutzfeldt-Jakob disease.Curr Psychiatry Rep. 2003;5:43-46.
10. Korth C, May BCH, Cohen FE, et al. Acridine and phenothiazine derivatives as pharmacotherapies for prion disease. PNAS. 2001;98:9836-9841.
11. Cumbler E, Furfari K, Guerrasio J. Creutzfeldt-Jacob disease presenting as severe depression: a case report. Cases J. 2009;2:122-124.
12. Tan KM, Worrell GA, Parisi JE, et al. Creutzfeldt-Jakob disease with focal electroencephalographic and magnetic resonance imaging findings. Arch Neurol. 2007;64:600-601.
13. Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004;63:443-449.
14. Manners DN, Parchi P, Tonon C, et al. Pathologic correlates of diffusion MRI changes in Creutzfeldt-Jakob disease. Neurology. 2009;72:1425-1431.
15. Aksamit AJ, Preissner CM, Homburger HA. Quantitation of 14-3-3 and neuron-specific enolase proteins in CSF in Creutzfeldt-Jakob disease. Neurology. 2001;57:728-730.
16. Collins SJ, Sanchez-Juan P, Masters CL, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129:2278-2287.
17. Solvason HB, Harris B, Zeifert P, et al. Psychological versus biological clinical interpretation: a patient with prion disease. Am J Psychiatry. 2002;159(4):528-537.
18. Wall CA, Rummans TA, Aksamit AJ, et al. Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci. 2005;17:489-495.
19. Liedorp M, van der Flier WM, Hoogervorst EL, et al. Associations between patterns of EEG abnormalities and diagnosis in a large memory clinic cohort. Dement Geriatr Cogn Disord. 2009;27:18-23.
Consider this rare disorder in patients with rapid-onset neurologic symptoms
Ms. J, age 63, is admitted to Neurology with progressive dizziness and cognitive impairment. She had developed word-finding difficulties, weakness, memory problems, and an episode of arm shaking, which prompted referral for inpatient workup. Ms. J has a history of hypertension, palpitations, and diabetes mellitus.
Her neurologic examination is variable; some examiners find pronounced aphasia and right-sided weakness, whereas others document a nearly normal examination. Lumbar puncture (LP) shows normal cell count, glucose, protein, and negative Gram’s stain; MRI of the brain is normal. Enterovirus polymerase chain reaction, cryptococcal antigen, and Lyme antibody are negative. Electroencephalography (EEG) demonstrates diffuse slowing. The primary team requests psychiatric consultation to assess for conversion disorder.
Ms. J is cooperative with psychiatric evaluation. She denies current or past psychiatric symptomatology and does not meet criteria for major depression, dysthymia, adjustment disorder, anxiety disorder, psychosis, or mania. She denies personal or family history of suicidal or homicidal ideation, intent, or plan. Her youngest son died 5 years earlier; she is financially secure and her 40-year marriage is stable. Ms. J denies having a history of substance use, physical or sexual abuse, or trauma.
In the Cardiology clinic 2 months ago, Ms. J denied having neurologic symptoms and was noted to be doing well. Her neurologic symptoms began shortly after that visit and steadily progressed. She is unable to identify an inciting event or stressor. Ms. J worked until 2 weeks before this admission. Neurologic examination at the time of psychiatric consultation is notable for waxing and waning expressive aphasia, right homonymous hemianopsia, and mildly decreased strength in the left biceps and forearm.
Ms. J presented to her cardiologist reporting dizziness and blurred vision 6 weeks ago, and she was observed in the hospital 3 weeks earlier for further evaluation. Laboratory testing during that hospitalization included blood counts, basic metabolic panel, thyroid function studies, erythrocyte sedimentation rate, thiamine, folic acid and vitamin B12, rapid plasma reagin and human immunodeficiency virus antibody, and LP, all reported as within normal limits.
Thorough review of Ms. J’s medical records reveals abnormalities that would be difficult to ascribe to conversion disorder. Specifically, 6 weeks ago, MRI of the brain demonstrated restricted diffusion in the left occipital lobe, and cerebrospinal fluid (CSF) neuron-specific enolase was moderately elevated at 34 ng/mL. The psychiatric consultant discusses these findings and concern for possible rapidly progressive dementia or Creutzfeldt-Jakob disease (CJD) with the primary team, Ms. J, and her family.
Ms. J is discharged with testing for CSF protein 14-3-3 pending and medical follow-up in 10 days. At follow-up 1 week later, her symptoms are worse; she is completely aphasic and wheelchair-bound. Antithyroglobulin and antimicrosomal thyroid antibodies and paraneoplastic antibody panel return normal. CSF protein 14-3-3 ultimately returns positive, supporting a clinical diagnosis of CJD. Ms. J dies shortly after hospital follow-up, less than 4 months after her first complaint of neurologic symptoms. No autopsy is performed.
Patients with conversion disorder may present with neurologic symptoms such as blindness, seizures, paralysis, or sensory loss with no identifiable anatomical or medical explanation.1 Conversion seizures—also known as pseudoseizures or nonepileptic seizures—may be clinically indistinguishable from generalized tonic-clonic seizures, but no EEG correlate can be identified.1,2 Conversion disorder is conceptualized as an unconscious manifestation of psychological conflict or stress—patients are not aware they are producing symptoms—and has been associated with emotional, sexual, and physical trauma.3,4
Conversion disorder is a diagnosis of exclusion and requires thorough evaluation to rule out neurologic or medical etiologies. The differential diagnosis for conversion disorder includes the broad medical differential diagnosis for the symptom, whether it be paralysis, seizures, sensory loss, or other presenting symptoms. Therefore, when evaluating patients for conversion disorder, be vigilant to the possibility of not only underlying psychological stress or trauma but also undiscovered medical or neurologic illness.
In Ms. J’s case, the primary team began to suspect there was no organic cause of her neurologic symptoms. However, psychiatric evaluation revealed that Ms. J had no history of stress or trauma that typically would be associated with conversion disorder, nor did she manifest other psychiatric symptoms, except waxing and waning mental status, which raised concerns for possible delirium or encephalopathy. Additionally, slowing on EEG was a nonspecific but abnormal finding that made conversion disorder unlikely. The consulting psychiatrist discussed this slowing, in conjunction with the abnormal MRI and elevated CSF neuron-specific enolase, with members of the referring Neurology service, who ordered additional testing of CSF for protein 14-3-3.
Creutzfeldt-Jakob disease
CJD is a rapidly progressive neurodegenerative disorder characterized by cognitive changes, behavioral changes, gait disturbances, akinetic mutism, and myoclonus.5 CJD results from the transition of prion proteins, which are present in the normal human brain, to disease-associated forms that aggregate and propagate and result in neurotoxicity with spongiform changes in neurons.6 The transition of normal prions to disease-associated prions may be hereditary, iatrogenic, infectious, or sporadic. Because the pathologic prion protein can be transmitted and normal sterilization procedures do not prevent the spread of CJD, special precautions should be taken to avoid contact with blood or CSF from patients suspected of having CJD.5
CJD most commonly occurs in the sporadic form, for which there are no identifiable risk factors, with an average age of onset between age 50 and 70. The disease affects women and men equally at a rate of 1 to 2 persons per million per year worldwide.6,7 Most patients with CJD die within 12 months of diagnosis8; median survival is 4 to 5 months.7,9 Although there is no approved or standard effective treatment for this uniformly fatal disease, research into the possibility of genetic or post-translational treatments is ongoing. One group reported inhibition of prion propagation by quinacrine and chlorpromazine in vitro.10 Clinical studies of quinacrine have demonstrated tolerability but no impact on the course of CJD.6
Clues to diagnosis. Although there is no treatment for CJD, early diagnosis can help patients and families understand the relentless progression of symptoms and also permits end-of-life planning and palliative care.11 Diagnosing CJD requires a high level of suspicion and traditionally has required brain biopsy or autopsy for conclusive diagnosis, although in some cases rare EEG findings of periodic sharp wave complexes or generalized periodic epileptiform discharges (GPEDs) have suggested the diagnosis.7,8,12 Recently, specific MRI findings have been described with fluid attenuated inversion recovery (FLAIR) and diffusion sequences.9,13,14
Routine LP for CSF examination (including cell count, protein, and glucose) frequently is normal.8 Specific testing to assess for CJD is required. Elevated levels of CSF neuron-specific enolase (normal <30 ng/mL) and protein 14-3-3 (normal <8 ng/mL) are fairly sensitive and specific for CJD when assessed in patients with the proper clinical history, although normal levels of these proteins have been detected in patients later confirmed to have CJD.7,15 A large multinational collaborative study of confirmed CJD cases that evaluated diagnostic test characteristics recommended that because each test has limitations and can be falsely negative—even in a case of later-confirmed CJD—a rational approach to diagnosis includes brain MRI with diffusion-weighted imaging, CSF analysis for protein 14-3-3, and EEG to assess for periodic sharp wave complexes or GPEDs.16
Because CJD presentation varies widely, most clinicians will not consider the diagnosis until the disease has progressed or the patient has died. Patients who present with psychological symptoms or predominant language disturbances and dysphagia may be referred to a psychiatrist or an ear, nose, and throat specialist before seeing a neurologist.9 Patients may be extensively evaluated and treated for conversion disorder when the correct diagnosis is CJD.17
Sporadic CJD traditionally is associated with neurologic presentations, whereas variant CJD is believed to present with psychiatric symptomatology. However, in a 25-year retrospective review of 126 patients with sporadic CJD, 80% of cases demonstrated psychiatric symptoms within the first 100 days of the disease course.18 Of these, nearly 25% showed psychiatric symptoms at presentation, including sleep disturbances, psychotic symptoms, agitation, and anxiety.
Psychiatrists should be aware of distinguishing features of rapidly progressive dementias and CJD, especially in the setting of psychiatric consultation, to rule out somatic etiologies of unexplained neurologic symptoms. It is important to obtain a history of baseline functioning, duration of decline, and psychiatric symptomatology to differentiate between organic and somatic causes. Differential diagnosis for rapidly progressive cognitive impairment is broad and includes delirium from diverse medical causes; rapidly progressive dementia such as accelerated Alzheimer’s disease, Lewy body disease, or frontotemporal dementia; and psychogenic causes, including conversion disorder (Table 1).7,8,12Table 2 provides distinguishing features of CJD, Alzheimer’s disease, Lewy body disease, and frontotemporal dementia with motor neuron disease.7,8,19
Table 1
Differential diagnosis of rapidly progressive dementia
| Celiac disease |
| Central nervous system vasculitis |
| Creutzfeldt-Jakob disease |
| Delirium (numerous possible etiologies) |
| Focal status epilepticus |
| Hashimoto’s encephalopathy |
Infection
|
Intoxication
|
| Limbic encephalopathy from paraneoplastic antibody syndrome |
| Lymphomatoid granulomatosis |
Malignancy
|
| Porphyria |
| Progressive supranuclear palsy |
Psychiatric disorder
|
| Sarcoidosis |
| Stroke |
| Vitamin deficiency (vitamin E, thiamine) |
| EBV: Epstein-Barr virus; HIV: human immunodeficiency virus; HSV: herpes simplex virus Source: References 7,8,12 |
Table 2
Distinguishing features of Creutzfeldt-Jakob disease
| Sporadic CJD | AD | DLBD | FTD-MND | |
|---|---|---|---|---|
| Time course | Rapid progression (median survival 4 to 5 months) | Insidious onset; progressive decline | Insidious onset; progressive decline | May experience rapid course to death |
| Age at onset | Age 50 to 70 | Incidence increases with age (usual onset age 65 to 85) | Older (age ~80) | Young age at onset |
| EEG findings | Periodic atypical triphasic waves; GPEDs | Normal or diffuse abnormalities | Rarely atypical triphasic waves | Increased slow activity, decreased fast activity |
| MRI findings | Restricted diffusion | Generalized atrophy | Generalized atrophy | Frontal atrophy |
| AD: Alzheimer’s disease; CJD: Creutzfeldt-Jakob disease; DLBD: diffuse Lewy body dementia; EEG: electroencephalography; FTD-MND: frontotemporal dementia with motor neuron disease; GPEDs: generalized periodic epileptiform discharges Source: References 7,8,19 | ||||
Related Resources
National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
Centers for Disease Control and Prevention. About CJD. www.cdc.gov/ncidod/dvrd/cjd.
Drug Brand Names
Chlorpromazine • Thorazine, Largactil
Quinacine • Atabrine
Disclosure
Dr. Gagliardi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Consider this rare disorder in patients with rapid-onset neurologic symptoms
Ms. J, age 63, is admitted to Neurology with progressive dizziness and cognitive impairment. She had developed word-finding difficulties, weakness, memory problems, and an episode of arm shaking, which prompted referral for inpatient workup. Ms. J has a history of hypertension, palpitations, and diabetes mellitus.
Her neurologic examination is variable; some examiners find pronounced aphasia and right-sided weakness, whereas others document a nearly normal examination. Lumbar puncture (LP) shows normal cell count, glucose, protein, and negative Gram’s stain; MRI of the brain is normal. Enterovirus polymerase chain reaction, cryptococcal antigen, and Lyme antibody are negative. Electroencephalography (EEG) demonstrates diffuse slowing. The primary team requests psychiatric consultation to assess for conversion disorder.
Ms. J is cooperative with psychiatric evaluation. She denies current or past psychiatric symptomatology and does not meet criteria for major depression, dysthymia, adjustment disorder, anxiety disorder, psychosis, or mania. She denies personal or family history of suicidal or homicidal ideation, intent, or plan. Her youngest son died 5 years earlier; she is financially secure and her 40-year marriage is stable. Ms. J denies having a history of substance use, physical or sexual abuse, or trauma.
In the Cardiology clinic 2 months ago, Ms. J denied having neurologic symptoms and was noted to be doing well. Her neurologic symptoms began shortly after that visit and steadily progressed. She is unable to identify an inciting event or stressor. Ms. J worked until 2 weeks before this admission. Neurologic examination at the time of psychiatric consultation is notable for waxing and waning expressive aphasia, right homonymous hemianopsia, and mildly decreased strength in the left biceps and forearm.
Ms. J presented to her cardiologist reporting dizziness and blurred vision 6 weeks ago, and she was observed in the hospital 3 weeks earlier for further evaluation. Laboratory testing during that hospitalization included blood counts, basic metabolic panel, thyroid function studies, erythrocyte sedimentation rate, thiamine, folic acid and vitamin B12, rapid plasma reagin and human immunodeficiency virus antibody, and LP, all reported as within normal limits.
Thorough review of Ms. J’s medical records reveals abnormalities that would be difficult to ascribe to conversion disorder. Specifically, 6 weeks ago, MRI of the brain demonstrated restricted diffusion in the left occipital lobe, and cerebrospinal fluid (CSF) neuron-specific enolase was moderately elevated at 34 ng/mL. The psychiatric consultant discusses these findings and concern for possible rapidly progressive dementia or Creutzfeldt-Jakob disease (CJD) with the primary team, Ms. J, and her family.
Ms. J is discharged with testing for CSF protein 14-3-3 pending and medical follow-up in 10 days. At follow-up 1 week later, her symptoms are worse; she is completely aphasic and wheelchair-bound. Antithyroglobulin and antimicrosomal thyroid antibodies and paraneoplastic antibody panel return normal. CSF protein 14-3-3 ultimately returns positive, supporting a clinical diagnosis of CJD. Ms. J dies shortly after hospital follow-up, less than 4 months after her first complaint of neurologic symptoms. No autopsy is performed.
Patients with conversion disorder may present with neurologic symptoms such as blindness, seizures, paralysis, or sensory loss with no identifiable anatomical or medical explanation.1 Conversion seizures—also known as pseudoseizures or nonepileptic seizures—may be clinically indistinguishable from generalized tonic-clonic seizures, but no EEG correlate can be identified.1,2 Conversion disorder is conceptualized as an unconscious manifestation of psychological conflict or stress—patients are not aware they are producing symptoms—and has been associated with emotional, sexual, and physical trauma.3,4
Conversion disorder is a diagnosis of exclusion and requires thorough evaluation to rule out neurologic or medical etiologies. The differential diagnosis for conversion disorder includes the broad medical differential diagnosis for the symptom, whether it be paralysis, seizures, sensory loss, or other presenting symptoms. Therefore, when evaluating patients for conversion disorder, be vigilant to the possibility of not only underlying psychological stress or trauma but also undiscovered medical or neurologic illness.
In Ms. J’s case, the primary team began to suspect there was no organic cause of her neurologic symptoms. However, psychiatric evaluation revealed that Ms. J had no history of stress or trauma that typically would be associated with conversion disorder, nor did she manifest other psychiatric symptoms, except waxing and waning mental status, which raised concerns for possible delirium or encephalopathy. Additionally, slowing on EEG was a nonspecific but abnormal finding that made conversion disorder unlikely. The consulting psychiatrist discussed this slowing, in conjunction with the abnormal MRI and elevated CSF neuron-specific enolase, with members of the referring Neurology service, who ordered additional testing of CSF for protein 14-3-3.
Creutzfeldt-Jakob disease
CJD is a rapidly progressive neurodegenerative disorder characterized by cognitive changes, behavioral changes, gait disturbances, akinetic mutism, and myoclonus.5 CJD results from the transition of prion proteins, which are present in the normal human brain, to disease-associated forms that aggregate and propagate and result in neurotoxicity with spongiform changes in neurons.6 The transition of normal prions to disease-associated prions may be hereditary, iatrogenic, infectious, or sporadic. Because the pathologic prion protein can be transmitted and normal sterilization procedures do not prevent the spread of CJD, special precautions should be taken to avoid contact with blood or CSF from patients suspected of having CJD.5
CJD most commonly occurs in the sporadic form, for which there are no identifiable risk factors, with an average age of onset between age 50 and 70. The disease affects women and men equally at a rate of 1 to 2 persons per million per year worldwide.6,7 Most patients with CJD die within 12 months of diagnosis8; median survival is 4 to 5 months.7,9 Although there is no approved or standard effective treatment for this uniformly fatal disease, research into the possibility of genetic or post-translational treatments is ongoing. One group reported inhibition of prion propagation by quinacrine and chlorpromazine in vitro.10 Clinical studies of quinacrine have demonstrated tolerability but no impact on the course of CJD.6
Clues to diagnosis. Although there is no treatment for CJD, early diagnosis can help patients and families understand the relentless progression of symptoms and also permits end-of-life planning and palliative care.11 Diagnosing CJD requires a high level of suspicion and traditionally has required brain biopsy or autopsy for conclusive diagnosis, although in some cases rare EEG findings of periodic sharp wave complexes or generalized periodic epileptiform discharges (GPEDs) have suggested the diagnosis.7,8,12 Recently, specific MRI findings have been described with fluid attenuated inversion recovery (FLAIR) and diffusion sequences.9,13,14
Routine LP for CSF examination (including cell count, protein, and glucose) frequently is normal.8 Specific testing to assess for CJD is required. Elevated levels of CSF neuron-specific enolase (normal <30 ng/mL) and protein 14-3-3 (normal <8 ng/mL) are fairly sensitive and specific for CJD when assessed in patients with the proper clinical history, although normal levels of these proteins have been detected in patients later confirmed to have CJD.7,15 A large multinational collaborative study of confirmed CJD cases that evaluated diagnostic test characteristics recommended that because each test has limitations and can be falsely negative—even in a case of later-confirmed CJD—a rational approach to diagnosis includes brain MRI with diffusion-weighted imaging, CSF analysis for protein 14-3-3, and EEG to assess for periodic sharp wave complexes or GPEDs.16
Because CJD presentation varies widely, most clinicians will not consider the diagnosis until the disease has progressed or the patient has died. Patients who present with psychological symptoms or predominant language disturbances and dysphagia may be referred to a psychiatrist or an ear, nose, and throat specialist before seeing a neurologist.9 Patients may be extensively evaluated and treated for conversion disorder when the correct diagnosis is CJD.17
Sporadic CJD traditionally is associated with neurologic presentations, whereas variant CJD is believed to present with psychiatric symptomatology. However, in a 25-year retrospective review of 126 patients with sporadic CJD, 80% of cases demonstrated psychiatric symptoms within the first 100 days of the disease course.18 Of these, nearly 25% showed psychiatric symptoms at presentation, including sleep disturbances, psychotic symptoms, agitation, and anxiety.
Psychiatrists should be aware of distinguishing features of rapidly progressive dementias and CJD, especially in the setting of psychiatric consultation, to rule out somatic etiologies of unexplained neurologic symptoms. It is important to obtain a history of baseline functioning, duration of decline, and psychiatric symptomatology to differentiate between organic and somatic causes. Differential diagnosis for rapidly progressive cognitive impairment is broad and includes delirium from diverse medical causes; rapidly progressive dementia such as accelerated Alzheimer’s disease, Lewy body disease, or frontotemporal dementia; and psychogenic causes, including conversion disorder (Table 1).7,8,12Table 2 provides distinguishing features of CJD, Alzheimer’s disease, Lewy body disease, and frontotemporal dementia with motor neuron disease.7,8,19
Table 1
Differential diagnosis of rapidly progressive dementia
| Celiac disease |
| Central nervous system vasculitis |
| Creutzfeldt-Jakob disease |
| Delirium (numerous possible etiologies) |
| Focal status epilepticus |
| Hashimoto’s encephalopathy |
Infection
|
Intoxication
|
| Limbic encephalopathy from paraneoplastic antibody syndrome |
| Lymphomatoid granulomatosis |
Malignancy
|
| Porphyria |
| Progressive supranuclear palsy |
Psychiatric disorder
|
| Sarcoidosis |
| Stroke |
| Vitamin deficiency (vitamin E, thiamine) |
| EBV: Epstein-Barr virus; HIV: human immunodeficiency virus; HSV: herpes simplex virus Source: References 7,8,12 |
Table 2
Distinguishing features of Creutzfeldt-Jakob disease
| Sporadic CJD | AD | DLBD | FTD-MND | |
|---|---|---|---|---|
| Time course | Rapid progression (median survival 4 to 5 months) | Insidious onset; progressive decline | Insidious onset; progressive decline | May experience rapid course to death |
| Age at onset | Age 50 to 70 | Incidence increases with age (usual onset age 65 to 85) | Older (age ~80) | Young age at onset |
| EEG findings | Periodic atypical triphasic waves; GPEDs | Normal or diffuse abnormalities | Rarely atypical triphasic waves | Increased slow activity, decreased fast activity |
| MRI findings | Restricted diffusion | Generalized atrophy | Generalized atrophy | Frontal atrophy |
| AD: Alzheimer’s disease; CJD: Creutzfeldt-Jakob disease; DLBD: diffuse Lewy body dementia; EEG: electroencephalography; FTD-MND: frontotemporal dementia with motor neuron disease; GPEDs: generalized periodic epileptiform discharges Source: References 7,8,19 | ||||
Related Resources
National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
Centers for Disease Control and Prevention. About CJD. www.cdc.gov/ncidod/dvrd/cjd.
Drug Brand Names
Chlorpromazine • Thorazine, Largactil
Quinacine • Atabrine
Disclosure
Dr. Gagliardi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry. 2006;163(9):1510-1517.
2. Teo WY, Choong CT. Neurological presentations of conversion disorders in a group of Singapore children. Pediatr Int. 2008;50(4):533-536.
3. Brown RJ, Cardena E, Nuenhuis E, et al. Should conversion disorder be reclassified as a dissociative disorder in DSM-V? Psychosomatics. 2007;48:369-378.
4. Stone J, Carson A, Aditya H, et al. The role of physical injury in motor and sensory conversion symptoms: a systematic and narrative review. J Psychosom Res. 2009;66:383-390.
5. National Institute of Neurological Disorders and Stroke Creutzfeldt-Jakob disease fact sheet. Available at: http://www.ninds.nih.gov/disorders/cjd/detail_cjd.htm. Accessed August 7, 2010.
6. Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8:334-344.
7. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64:98-108.
8. Josephs KA, Ahlskog E, Parisi JE, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol. 2009;66(2):201-207.
9. Martindale JL, Geschwind MD, Miller BL. Psychiatric and neuroimaging findings in Creutzfeldt-Jakob disease.Curr Psychiatry Rep. 2003;5:43-46.
10. Korth C, May BCH, Cohen FE, et al. Acridine and phenothiazine derivatives as pharmacotherapies for prion disease. PNAS. 2001;98:9836-9841.
11. Cumbler E, Furfari K, Guerrasio J. Creutzfeldt-Jacob disease presenting as severe depression: a case report. Cases J. 2009;2:122-124.
12. Tan KM, Worrell GA, Parisi JE, et al. Creutzfeldt-Jakob disease with focal electroencephalographic and magnetic resonance imaging findings. Arch Neurol. 2007;64:600-601.
13. Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004;63:443-449.
14. Manners DN, Parchi P, Tonon C, et al. Pathologic correlates of diffusion MRI changes in Creutzfeldt-Jakob disease. Neurology. 2009;72:1425-1431.
15. Aksamit AJ, Preissner CM, Homburger HA. Quantitation of 14-3-3 and neuron-specific enolase proteins in CSF in Creutzfeldt-Jakob disease. Neurology. 2001;57:728-730.
16. Collins SJ, Sanchez-Juan P, Masters CL, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129:2278-2287.
17. Solvason HB, Harris B, Zeifert P, et al. Psychological versus biological clinical interpretation: a patient with prion disease. Am J Psychiatry. 2002;159(4):528-537.
18. Wall CA, Rummans TA, Aksamit AJ, et al. Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci. 2005;17:489-495.
19. Liedorp M, van der Flier WM, Hoogervorst EL, et al. Associations between patterns of EEG abnormalities and diagnosis in a large memory clinic cohort. Dement Geriatr Cogn Disord. 2009;27:18-23.
1. Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry. 2006;163(9):1510-1517.
2. Teo WY, Choong CT. Neurological presentations of conversion disorders in a group of Singapore children. Pediatr Int. 2008;50(4):533-536.
3. Brown RJ, Cardena E, Nuenhuis E, et al. Should conversion disorder be reclassified as a dissociative disorder in DSM-V? Psychosomatics. 2007;48:369-378.
4. Stone J, Carson A, Aditya H, et al. The role of physical injury in motor and sensory conversion symptoms: a systematic and narrative review. J Psychosom Res. 2009;66:383-390.
5. National Institute of Neurological Disorders and Stroke Creutzfeldt-Jakob disease fact sheet. Available at: http://www.ninds.nih.gov/disorders/cjd/detail_cjd.htm. Accessed August 7, 2010.
6. Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8:334-344.
7. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64:98-108.
8. Josephs KA, Ahlskog E, Parisi JE, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol. 2009;66(2):201-207.
9. Martindale JL, Geschwind MD, Miller BL. Psychiatric and neuroimaging findings in Creutzfeldt-Jakob disease.Curr Psychiatry Rep. 2003;5:43-46.
10. Korth C, May BCH, Cohen FE, et al. Acridine and phenothiazine derivatives as pharmacotherapies for prion disease. PNAS. 2001;98:9836-9841.
11. Cumbler E, Furfari K, Guerrasio J. Creutzfeldt-Jacob disease presenting as severe depression: a case report. Cases J. 2009;2:122-124.
12. Tan KM, Worrell GA, Parisi JE, et al. Creutzfeldt-Jakob disease with focal electroencephalographic and magnetic resonance imaging findings. Arch Neurol. 2007;64:600-601.
13. Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004;63:443-449.
14. Manners DN, Parchi P, Tonon C, et al. Pathologic correlates of diffusion MRI changes in Creutzfeldt-Jakob disease. Neurology. 2009;72:1425-1431.
15. Aksamit AJ, Preissner CM, Homburger HA. Quantitation of 14-3-3 and neuron-specific enolase proteins in CSF in Creutzfeldt-Jakob disease. Neurology. 2001;57:728-730.
16. Collins SJ, Sanchez-Juan P, Masters CL, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129:2278-2287.
17. Solvason HB, Harris B, Zeifert P, et al. Psychological versus biological clinical interpretation: a patient with prion disease. Am J Psychiatry. 2002;159(4):528-537.
18. Wall CA, Rummans TA, Aksamit AJ, et al. Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci. 2005;17:489-495.
19. Liedorp M, van der Flier WM, Hoogervorst EL, et al. Associations between patterns of EEG abnormalities and diagnosis in a large memory clinic cohort. Dement Geriatr Cogn Disord. 2009;27:18-23.
How often should women be screened for breast cancer?
- Starting at age 20, women should undergo clinical breast exam every 3 years and be counseled about awareness of breast changes.
- Average risk women should undergo clinical breast examination and screening mammography annually starting at age 40.
- Health care providers should inform women about the benefits and limitations of mammography and the potential for false positives.
- Women at high risk include those with inherited susceptibility to breast cancer or chest radiation at a young age. They should be screened with mammography and breast MRI annually starting at age 30.
Breast cancer is the most widespread cancer effecting women in the United States.1 The high prevalence and inherent “cost” of breast cancer mandates physicians to be aware of effective screening tools, existing guidelines, and potential adverse effects.
Mammography screening and improvements in breast cancer treatments have contributed to improved survival rates, but2,3 mammography screening has declined since 2000. Potential reasons for this decrease include:
- poor access to medical care
- fear of radiation exposure
- concern of undesirable test results
- anticipated pain
- misconceptions of cancer risk
- changes in recommendations regarding mammography screening.
Patients with psychiatric illnesses are less likely to receive mammography screening.4,5 Cancer patients with schizophrenia, particularly women with breast cancer, have an increased risk of mortality.6
Risk assessment
Age, genetic predisposition, and factors that affect endogenous estrogen exposure such as early menarche, late menopause, and nulliparity are among the most important breast cancer risk factors (Table 1). Explore these and other risk factors with your patient before making screening recommendations.
Tools such as the Breast Cancer Risk Assessment Tool (BCRAT) can assist in stratifying your patient’s risk. The BCRAT, available at www.cancer.gov/bcrisktool, takes into account, age, race, family history, and previous breast abnormalities. Women at average risk for breast cancer include those with an estimated lifetime risk of <15%. Women with an estimated lifetime risk of 15% to 20% are at moderate risk. Women >20% are at high risk and should consider more intensive screening (Table 2).7,8
Other examples of high-risk features include chest radiation therapy (eg, for Hodgkin’s lymphoma) between age 10 to 30 or a breast cancer 1, early onset (BRCA1) or breast cancer 2, early onset (BRCA2) mutation carried by the patient or a first-degree family member, which can leave patients more susceptible to breast cancer.
Table 1
Breast cancer risk factors
| Female sex |
| Older age |
| Genetic risk factors (eg, BRCA1 and BRCA2 gene mutation)–5% to 10% of breast cancers |
| Family history of breast cancer |
| Personal history of breast cancer |
| Race (eg, Whites have highest incidence, African Americans have highest mortality) |
| Certain benign breast diseases (eg, atypical hyperplasia) |
| Early menarche, late menopause |
| Prior chest radiation (eg, for Hodgkin’s lymphoma; especially age 10 to 30) |
| Nulliparity, late child-bearing |
| Oral contraceptive use |
| Hormone replacement therapy (combined estrogen/progesterone) |
| Not breastfeeding |
| Alcohol (2 to 5 drinks daily increases risk 1.5 times) |
| Obesity |
| BRCA1: breast cancer 1, early onset; BRCA2: breast cancer 2, early onset Source: Adapted from the American Cancer Society; available at www.cancer.org |
Breast cancer screening
Choice of screening is guided by an individualized risk assessment. For women with average risk for breast cancer, the major components of breast cancer screening are clinical breast examination (CBE) and screening mammography.
Breast self-examination is not routinely recommended by expert groups. The American Cancer Society (ACS) recommends that clinicians discuss the benefits and limitations of breast self-exam with patients. The National Comprehensive Cancer Network (NCCN) recommends that women maintain breast health awareness but no longer advocates instruction in self-examination.
CBE by a trained provider, when coupled to routine screening mammography, may add modest benefit in terms of detecting cancer. The ACS and the NCCN suggest CBE along with annual mammography for all women starting at age 40.
Mammography has been to shown to reduce breast cancer mortality.8 A United States Preventive Services Task Force (USPSTF) review found statistically significant reductions in breast cancer mortality for women age 39 to 69.9
Because the USPSTF found a small net benefit of screening mammography in women age 40 to 49, their recent guidelines recommend against routine mammograms for this age group. Instead, the USPSTF suggests that screening be based on individualized risk assessment and discussion of the benefits and risks (false positive tests, overdiagnosis, and psychological harms) of screening.10 Other groups continue to recommend annual mammography starting at age 40 for women at average risk (Table 2).
MRI is more sensitive screening than mammography and the combination of MRI and routine mammograms is more sensitive than either test alone. In 2007, the ACS recommended annual breast MRI screening in addition to mammogram for women at high risk for breast cancer (Table 2). For women with moderately increased risk (15% to 20% lifetime) there is insufficient evidence to recommend for or against MRI for screening, but one may consider it on a case-by-case basis; for example, for women with personal history of breast cancer, atypical hyperplasia, or with mammographically dense breasts.
Table 2
American Cancer Society breast cancer screening recommendations
| Women at average risk* | Women at high risk* | |
|---|---|---|
| Breast self-exam | Not routinely recommended. Discuss the benefits and limitations starting with patients in their 20s. Emphasize the importance of reporting new breast symptoms to a health care provider | |
| Clinical breast exam | At least every 3 years for women in their 20s and 30s. Annually starting at age 40 | Annually, starting at age 30 |
| Mammography | Annually, starting at age 40 | Annually, starting at age 30 |
| Breast MRI | Not recommended | Annually, starting at age 30, along with mammogram |
| *Women at average risk for breast cancer include those with an estimated lifetime risk of <15%. Women with an estimated lifetime risk of 15% to 20% are at moderate risk. Women >20% are at high risk and should consider more intensive screening | ||
| Source: References 7,8, American Cancer Society (www.cancer.org) | ||
Potential harms
Potential mammography harms include the possibility of a false positive result, anxiety as one awaits the test result, and anticipation of discomfort associated with the procedure. There also is the potential for “overdiagnosis” or detection of a cancer that would not have adversely impacted the patient if it had not been discovered. There is also a small risk of radiation exposure from repeated mammograms, but this has not been firmly established in the literature.
False-positive results—an abnormal finding on mammogram that does not result in a breast cancer diagnosis—is a significant issue. One study estimated that 11% of screening mammograms return abnormal findings that lead to additional workup, the majority (90%) of which ultimately result in benign diagnoses.11 Workup often leads to additional mammograms, ultrasound, breast MRI, and invasive procedures such as needle biopsies. False-positive mammograms have been associated with increased symptoms of depression and anxiety.12 Patients may be more apprehensive about breast cancer following a false-positive result, but this does not appear to lead to chronic anxiety.13
The vulnerability of patients experiencing psychiatric illness coupled with the potential psychological consequences of breast cancer make it imperative that psychiatrists remain up-to-date on breast cancer screening guidelines. Reported poor adherence to screening recommendations for mammography may increase the burden of illness and mortality from breast cancer in individuals with mental illness.
Conversations about health maintenance measures always should include careful discussion of the benefits and potential harms associated with the recommended screening tools. Because psychiatrists work closely with patients who may be less likely to undergo mammography, it is important to provide support and advocate for access to health care screening.
Related Resource
- National Cancer Institute. www.cancer.gov.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Table 3
Number needed to screen (NNS) with mammography to prevent 1 breast cancer death
| Age | NNS |
|---|---|
| 39 to 49 | 1,904 |
| 50 to 59 | 1,339 |
| 60 to 69 | 337 |
| Source: Reference 9 | |
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225-249.
2. Chu KC, Tarone RE, Kessler LG, et al. Recent trends in U. S. breast cancer incidence, survival, and mortality rates. J Natl Cancer Inst. 1996;88:1571-1579.
3. Berry DA, Cronin KA, Plevritis SK, et al. Effect of screening and adjuvant therapy on mortality from breast cancer. N Eng J Med. 2005;353:1784-1792.
4. Ludman EJ, Ichikawa LE, Simon GE, et al. Breast and cervical cancer screening specific effects of depression and obesity. Am J Prev Med. 2010;38:303-310.
5. Lindamer LA, Wear E, Robins-Sadler G. Mammography stages of change in middle-aged women with schizophrenia: an exploratory analysis. BMC Psychiatry. 2006;6:49.-
6. Tran E, Rouillon F, Loze JY, et al. Cancer mortality in patients with schizophrenia: an 11-year prospective cohort study. Cancer. 2009;15:3555-3562.
7. Smith RA, Cokkinides V, Brooks D, et al. Cancer screening in the United States, 2010: a review of current American Cancer Society guidelines and issues in cancer screening. CA Cancer J Clin. 2010;60(2):99-119.
8. Saslow D, Boetes C, Burke W, et al. American Cancer Society guidelines for screening with MRI as an adjunct to mammography. CA Cancer J Clin. 2007;57:75-89.
9. Nelson HD, Tyne K, Naik A, et al. Screening for breast cancer: an update from the U.S. Preventive Services Task Force. Ann Intern Med. 2009;151:727-737W237-W242.
10. US Preventive Services Task Force. Screening for breast cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151:716-726W236.-
11. Brown ML, Houn F, Sickles EA, et al. Screening mammography in community practice: positive predictive value of abnormal findings and yield of follow-up diagnostic procedures. AJR Am J Roentgenol. 1995;165:1373-1377.
12. Jatoi I, Zhu K, Shah M, et al. Psychological distress in U.S. women who have experienced false-positive mammograms. Breast Cancer Res Treat. 2006;100:191-200.
13. Brewer NT, Salz T, Lillie SE, et al. Systematic review: the long-term effects of false-positive mammograms. Ann Intern Med. 2007;146:502-510.
John Charlson, MD
Dr. Charlson is assistant professor of medicine, division of neoplastic diseases and related disorders
Thomas W. Heinrich, MD
Dr. Heinrich is assistant professor of psychiatry, chief of consultation psychiatry and director, psychosomatic medicine, Medical College of Wisconsin, Milwaukee, WI.
Robert M. McCarron, DO
Series Editor
John Charlson, MD
Dr. Charlson is assistant professor of medicine, division of neoplastic diseases and related disorders
Thomas W. Heinrich, MD
Dr. Heinrich is assistant professor of psychiatry, chief of consultation psychiatry and director, psychosomatic medicine, Medical College of Wisconsin, Milwaukee, WI.
Robert M. McCarron, DO
Series Editor
John Charlson, MD
Dr. Charlson is assistant professor of medicine, division of neoplastic diseases and related disorders
Thomas W. Heinrich, MD
Dr. Heinrich is assistant professor of psychiatry, chief of consultation psychiatry and director, psychosomatic medicine, Medical College of Wisconsin, Milwaukee, WI.
Robert M. McCarron, DO
Series Editor
- Starting at age 20, women should undergo clinical breast exam every 3 years and be counseled about awareness of breast changes.
- Average risk women should undergo clinical breast examination and screening mammography annually starting at age 40.
- Health care providers should inform women about the benefits and limitations of mammography and the potential for false positives.
- Women at high risk include those with inherited susceptibility to breast cancer or chest radiation at a young age. They should be screened with mammography and breast MRI annually starting at age 30.
Breast cancer is the most widespread cancer effecting women in the United States.1 The high prevalence and inherent “cost” of breast cancer mandates physicians to be aware of effective screening tools, existing guidelines, and potential adverse effects.
Mammography screening and improvements in breast cancer treatments have contributed to improved survival rates, but2,3 mammography screening has declined since 2000. Potential reasons for this decrease include:
- poor access to medical care
- fear of radiation exposure
- concern of undesirable test results
- anticipated pain
- misconceptions of cancer risk
- changes in recommendations regarding mammography screening.
Patients with psychiatric illnesses are less likely to receive mammography screening.4,5 Cancer patients with schizophrenia, particularly women with breast cancer, have an increased risk of mortality.6
Risk assessment
Age, genetic predisposition, and factors that affect endogenous estrogen exposure such as early menarche, late menopause, and nulliparity are among the most important breast cancer risk factors (Table 1). Explore these and other risk factors with your patient before making screening recommendations.
Tools such as the Breast Cancer Risk Assessment Tool (BCRAT) can assist in stratifying your patient’s risk. The BCRAT, available at www.cancer.gov/bcrisktool, takes into account, age, race, family history, and previous breast abnormalities. Women at average risk for breast cancer include those with an estimated lifetime risk of <15%. Women with an estimated lifetime risk of 15% to 20% are at moderate risk. Women >20% are at high risk and should consider more intensive screening (Table 2).7,8
Other examples of high-risk features include chest radiation therapy (eg, for Hodgkin’s lymphoma) between age 10 to 30 or a breast cancer 1, early onset (BRCA1) or breast cancer 2, early onset (BRCA2) mutation carried by the patient or a first-degree family member, which can leave patients more susceptible to breast cancer.
Table 1
Breast cancer risk factors
| Female sex |
| Older age |
| Genetic risk factors (eg, BRCA1 and BRCA2 gene mutation)–5% to 10% of breast cancers |
| Family history of breast cancer |
| Personal history of breast cancer |
| Race (eg, Whites have highest incidence, African Americans have highest mortality) |
| Certain benign breast diseases (eg, atypical hyperplasia) |
| Early menarche, late menopause |
| Prior chest radiation (eg, for Hodgkin’s lymphoma; especially age 10 to 30) |
| Nulliparity, late child-bearing |
| Oral contraceptive use |
| Hormone replacement therapy (combined estrogen/progesterone) |
| Not breastfeeding |
| Alcohol (2 to 5 drinks daily increases risk 1.5 times) |
| Obesity |
| BRCA1: breast cancer 1, early onset; BRCA2: breast cancer 2, early onset Source: Adapted from the American Cancer Society; available at www.cancer.org |
Breast cancer screening
Choice of screening is guided by an individualized risk assessment. For women with average risk for breast cancer, the major components of breast cancer screening are clinical breast examination (CBE) and screening mammography.
Breast self-examination is not routinely recommended by expert groups. The American Cancer Society (ACS) recommends that clinicians discuss the benefits and limitations of breast self-exam with patients. The National Comprehensive Cancer Network (NCCN) recommends that women maintain breast health awareness but no longer advocates instruction in self-examination.
CBE by a trained provider, when coupled to routine screening mammography, may add modest benefit in terms of detecting cancer. The ACS and the NCCN suggest CBE along with annual mammography for all women starting at age 40.
Mammography has been to shown to reduce breast cancer mortality.8 A United States Preventive Services Task Force (USPSTF) review found statistically significant reductions in breast cancer mortality for women age 39 to 69.9
Because the USPSTF found a small net benefit of screening mammography in women age 40 to 49, their recent guidelines recommend against routine mammograms for this age group. Instead, the USPSTF suggests that screening be based on individualized risk assessment and discussion of the benefits and risks (false positive tests, overdiagnosis, and psychological harms) of screening.10 Other groups continue to recommend annual mammography starting at age 40 for women at average risk (Table 2).
MRI is more sensitive screening than mammography and the combination of MRI and routine mammograms is more sensitive than either test alone. In 2007, the ACS recommended annual breast MRI screening in addition to mammogram for women at high risk for breast cancer (Table 2). For women with moderately increased risk (15% to 20% lifetime) there is insufficient evidence to recommend for or against MRI for screening, but one may consider it on a case-by-case basis; for example, for women with personal history of breast cancer, atypical hyperplasia, or with mammographically dense breasts.
Table 2
American Cancer Society breast cancer screening recommendations
| Women at average risk* | Women at high risk* | |
|---|---|---|
| Breast self-exam | Not routinely recommended. Discuss the benefits and limitations starting with patients in their 20s. Emphasize the importance of reporting new breast symptoms to a health care provider | |
| Clinical breast exam | At least every 3 years for women in their 20s and 30s. Annually starting at age 40 | Annually, starting at age 30 |
| Mammography | Annually, starting at age 40 | Annually, starting at age 30 |
| Breast MRI | Not recommended | Annually, starting at age 30, along with mammogram |
| *Women at average risk for breast cancer include those with an estimated lifetime risk of <15%. Women with an estimated lifetime risk of 15% to 20% are at moderate risk. Women >20% are at high risk and should consider more intensive screening | ||
| Source: References 7,8, American Cancer Society (www.cancer.org) | ||
Potential harms
Potential mammography harms include the possibility of a false positive result, anxiety as one awaits the test result, and anticipation of discomfort associated with the procedure. There also is the potential for “overdiagnosis” or detection of a cancer that would not have adversely impacted the patient if it had not been discovered. There is also a small risk of radiation exposure from repeated mammograms, but this has not been firmly established in the literature.
False-positive results—an abnormal finding on mammogram that does not result in a breast cancer diagnosis—is a significant issue. One study estimated that 11% of screening mammograms return abnormal findings that lead to additional workup, the majority (90%) of which ultimately result in benign diagnoses.11 Workup often leads to additional mammograms, ultrasound, breast MRI, and invasive procedures such as needle biopsies. False-positive mammograms have been associated with increased symptoms of depression and anxiety.12 Patients may be more apprehensive about breast cancer following a false-positive result, but this does not appear to lead to chronic anxiety.13
The vulnerability of patients experiencing psychiatric illness coupled with the potential psychological consequences of breast cancer make it imperative that psychiatrists remain up-to-date on breast cancer screening guidelines. Reported poor adherence to screening recommendations for mammography may increase the burden of illness and mortality from breast cancer in individuals with mental illness.
Conversations about health maintenance measures always should include careful discussion of the benefits and potential harms associated with the recommended screening tools. Because psychiatrists work closely with patients who may be less likely to undergo mammography, it is important to provide support and advocate for access to health care screening.
Related Resource
- National Cancer Institute. www.cancer.gov.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Table 3
Number needed to screen (NNS) with mammography to prevent 1 breast cancer death
| Age | NNS |
|---|---|
| 39 to 49 | 1,904 |
| 50 to 59 | 1,339 |
| 60 to 69 | 337 |
| Source: Reference 9 | |
- Starting at age 20, women should undergo clinical breast exam every 3 years and be counseled about awareness of breast changes.
- Average risk women should undergo clinical breast examination and screening mammography annually starting at age 40.
- Health care providers should inform women about the benefits and limitations of mammography and the potential for false positives.
- Women at high risk include those with inherited susceptibility to breast cancer or chest radiation at a young age. They should be screened with mammography and breast MRI annually starting at age 30.
Breast cancer is the most widespread cancer effecting women in the United States.1 The high prevalence and inherent “cost” of breast cancer mandates physicians to be aware of effective screening tools, existing guidelines, and potential adverse effects.
Mammography screening and improvements in breast cancer treatments have contributed to improved survival rates, but2,3 mammography screening has declined since 2000. Potential reasons for this decrease include:
- poor access to medical care
- fear of radiation exposure
- concern of undesirable test results
- anticipated pain
- misconceptions of cancer risk
- changes in recommendations regarding mammography screening.
Patients with psychiatric illnesses are less likely to receive mammography screening.4,5 Cancer patients with schizophrenia, particularly women with breast cancer, have an increased risk of mortality.6
Risk assessment
Age, genetic predisposition, and factors that affect endogenous estrogen exposure such as early menarche, late menopause, and nulliparity are among the most important breast cancer risk factors (Table 1). Explore these and other risk factors with your patient before making screening recommendations.
Tools such as the Breast Cancer Risk Assessment Tool (BCRAT) can assist in stratifying your patient’s risk. The BCRAT, available at www.cancer.gov/bcrisktool, takes into account, age, race, family history, and previous breast abnormalities. Women at average risk for breast cancer include those with an estimated lifetime risk of <15%. Women with an estimated lifetime risk of 15% to 20% are at moderate risk. Women >20% are at high risk and should consider more intensive screening (Table 2).7,8
Other examples of high-risk features include chest radiation therapy (eg, for Hodgkin’s lymphoma) between age 10 to 30 or a breast cancer 1, early onset (BRCA1) or breast cancer 2, early onset (BRCA2) mutation carried by the patient or a first-degree family member, which can leave patients more susceptible to breast cancer.
Table 1
Breast cancer risk factors
| Female sex |
| Older age |
| Genetic risk factors (eg, BRCA1 and BRCA2 gene mutation)–5% to 10% of breast cancers |
| Family history of breast cancer |
| Personal history of breast cancer |
| Race (eg, Whites have highest incidence, African Americans have highest mortality) |
| Certain benign breast diseases (eg, atypical hyperplasia) |
| Early menarche, late menopause |
| Prior chest radiation (eg, for Hodgkin’s lymphoma; especially age 10 to 30) |
| Nulliparity, late child-bearing |
| Oral contraceptive use |
| Hormone replacement therapy (combined estrogen/progesterone) |
| Not breastfeeding |
| Alcohol (2 to 5 drinks daily increases risk 1.5 times) |
| Obesity |
| BRCA1: breast cancer 1, early onset; BRCA2: breast cancer 2, early onset Source: Adapted from the American Cancer Society; available at www.cancer.org |
Breast cancer screening
Choice of screening is guided by an individualized risk assessment. For women with average risk for breast cancer, the major components of breast cancer screening are clinical breast examination (CBE) and screening mammography.
Breast self-examination is not routinely recommended by expert groups. The American Cancer Society (ACS) recommends that clinicians discuss the benefits and limitations of breast self-exam with patients. The National Comprehensive Cancer Network (NCCN) recommends that women maintain breast health awareness but no longer advocates instruction in self-examination.
CBE by a trained provider, when coupled to routine screening mammography, may add modest benefit in terms of detecting cancer. The ACS and the NCCN suggest CBE along with annual mammography for all women starting at age 40.
Mammography has been to shown to reduce breast cancer mortality.8 A United States Preventive Services Task Force (USPSTF) review found statistically significant reductions in breast cancer mortality for women age 39 to 69.9
Because the USPSTF found a small net benefit of screening mammography in women age 40 to 49, their recent guidelines recommend against routine mammograms for this age group. Instead, the USPSTF suggests that screening be based on individualized risk assessment and discussion of the benefits and risks (false positive tests, overdiagnosis, and psychological harms) of screening.10 Other groups continue to recommend annual mammography starting at age 40 for women at average risk (Table 2).
MRI is more sensitive screening than mammography and the combination of MRI and routine mammograms is more sensitive than either test alone. In 2007, the ACS recommended annual breast MRI screening in addition to mammogram for women at high risk for breast cancer (Table 2). For women with moderately increased risk (15% to 20% lifetime) there is insufficient evidence to recommend for or against MRI for screening, but one may consider it on a case-by-case basis; for example, for women with personal history of breast cancer, atypical hyperplasia, or with mammographically dense breasts.
Table 2
American Cancer Society breast cancer screening recommendations
| Women at average risk* | Women at high risk* | |
|---|---|---|
| Breast self-exam | Not routinely recommended. Discuss the benefits and limitations starting with patients in their 20s. Emphasize the importance of reporting new breast symptoms to a health care provider | |
| Clinical breast exam | At least every 3 years for women in their 20s and 30s. Annually starting at age 40 | Annually, starting at age 30 |
| Mammography | Annually, starting at age 40 | Annually, starting at age 30 |
| Breast MRI | Not recommended | Annually, starting at age 30, along with mammogram |
| *Women at average risk for breast cancer include those with an estimated lifetime risk of <15%. Women with an estimated lifetime risk of 15% to 20% are at moderate risk. Women >20% are at high risk and should consider more intensive screening | ||
| Source: References 7,8, American Cancer Society (www.cancer.org) | ||
Potential harms
Potential mammography harms include the possibility of a false positive result, anxiety as one awaits the test result, and anticipation of discomfort associated with the procedure. There also is the potential for “overdiagnosis” or detection of a cancer that would not have adversely impacted the patient if it had not been discovered. There is also a small risk of radiation exposure from repeated mammograms, but this has not been firmly established in the literature.
False-positive results—an abnormal finding on mammogram that does not result in a breast cancer diagnosis—is a significant issue. One study estimated that 11% of screening mammograms return abnormal findings that lead to additional workup, the majority (90%) of which ultimately result in benign diagnoses.11 Workup often leads to additional mammograms, ultrasound, breast MRI, and invasive procedures such as needle biopsies. False-positive mammograms have been associated with increased symptoms of depression and anxiety.12 Patients may be more apprehensive about breast cancer following a false-positive result, but this does not appear to lead to chronic anxiety.13
The vulnerability of patients experiencing psychiatric illness coupled with the potential psychological consequences of breast cancer make it imperative that psychiatrists remain up-to-date on breast cancer screening guidelines. Reported poor adherence to screening recommendations for mammography may increase the burden of illness and mortality from breast cancer in individuals with mental illness.
Conversations about health maintenance measures always should include careful discussion of the benefits and potential harms associated with the recommended screening tools. Because psychiatrists work closely with patients who may be less likely to undergo mammography, it is important to provide support and advocate for access to health care screening.
Related Resource
- National Cancer Institute. www.cancer.gov.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Table 3
Number needed to screen (NNS) with mammography to prevent 1 breast cancer death
| Age | NNS |
|---|---|
| 39 to 49 | 1,904 |
| 50 to 59 | 1,339 |
| 60 to 69 | 337 |
| Source: Reference 9 | |
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225-249.
2. Chu KC, Tarone RE, Kessler LG, et al. Recent trends in U. S. breast cancer incidence, survival, and mortality rates. J Natl Cancer Inst. 1996;88:1571-1579.
3. Berry DA, Cronin KA, Plevritis SK, et al. Effect of screening and adjuvant therapy on mortality from breast cancer. N Eng J Med. 2005;353:1784-1792.
4. Ludman EJ, Ichikawa LE, Simon GE, et al. Breast and cervical cancer screening specific effects of depression and obesity. Am J Prev Med. 2010;38:303-310.
5. Lindamer LA, Wear E, Robins-Sadler G. Mammography stages of change in middle-aged women with schizophrenia: an exploratory analysis. BMC Psychiatry. 2006;6:49.-
6. Tran E, Rouillon F, Loze JY, et al. Cancer mortality in patients with schizophrenia: an 11-year prospective cohort study. Cancer. 2009;15:3555-3562.
7. Smith RA, Cokkinides V, Brooks D, et al. Cancer screening in the United States, 2010: a review of current American Cancer Society guidelines and issues in cancer screening. CA Cancer J Clin. 2010;60(2):99-119.
8. Saslow D, Boetes C, Burke W, et al. American Cancer Society guidelines for screening with MRI as an adjunct to mammography. CA Cancer J Clin. 2007;57:75-89.
9. Nelson HD, Tyne K, Naik A, et al. Screening for breast cancer: an update from the U.S. Preventive Services Task Force. Ann Intern Med. 2009;151:727-737W237-W242.
10. US Preventive Services Task Force. Screening for breast cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151:716-726W236.-
11. Brown ML, Houn F, Sickles EA, et al. Screening mammography in community practice: positive predictive value of abnormal findings and yield of follow-up diagnostic procedures. AJR Am J Roentgenol. 1995;165:1373-1377.
12. Jatoi I, Zhu K, Shah M, et al. Psychological distress in U.S. women who have experienced false-positive mammograms. Breast Cancer Res Treat. 2006;100:191-200.
13. Brewer NT, Salz T, Lillie SE, et al. Systematic review: the long-term effects of false-positive mammograms. Ann Intern Med. 2007;146:502-510.
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225-249.
2. Chu KC, Tarone RE, Kessler LG, et al. Recent trends in U. S. breast cancer incidence, survival, and mortality rates. J Natl Cancer Inst. 1996;88:1571-1579.
3. Berry DA, Cronin KA, Plevritis SK, et al. Effect of screening and adjuvant therapy on mortality from breast cancer. N Eng J Med. 2005;353:1784-1792.
4. Ludman EJ, Ichikawa LE, Simon GE, et al. Breast and cervical cancer screening specific effects of depression and obesity. Am J Prev Med. 2010;38:303-310.
5. Lindamer LA, Wear E, Robins-Sadler G. Mammography stages of change in middle-aged women with schizophrenia: an exploratory analysis. BMC Psychiatry. 2006;6:49.-
6. Tran E, Rouillon F, Loze JY, et al. Cancer mortality in patients with schizophrenia: an 11-year prospective cohort study. Cancer. 2009;15:3555-3562.
7. Smith RA, Cokkinides V, Brooks D, et al. Cancer screening in the United States, 2010: a review of current American Cancer Society guidelines and issues in cancer screening. CA Cancer J Clin. 2010;60(2):99-119.
8. Saslow D, Boetes C, Burke W, et al. American Cancer Society guidelines for screening with MRI as an adjunct to mammography. CA Cancer J Clin. 2007;57:75-89.
9. Nelson HD, Tyne K, Naik A, et al. Screening for breast cancer: an update from the U.S. Preventive Services Task Force. Ann Intern Med. 2009;151:727-737W237-W242.
10. US Preventive Services Task Force. Screening for breast cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151:716-726W236.-
11. Brown ML, Houn F, Sickles EA, et al. Screening mammography in community practice: positive predictive value of abnormal findings and yield of follow-up diagnostic procedures. AJR Am J Roentgenol. 1995;165:1373-1377.
12. Jatoi I, Zhu K, Shah M, et al. Psychological distress in U.S. women who have experienced false-positive mammograms. Breast Cancer Res Treat. 2006;100:191-200.
13. Brewer NT, Salz T, Lillie SE, et al. Systematic review: the long-term effects of false-positive mammograms. Ann Intern Med. 2007;146:502-510.
The truth about treating low back pain
Dr. Lau is chief resident and Dr. Han is residency training director, departments of family and community medicine and psychiatry and behavioral sciences, University of California, Davis, Sacramento, CA.
Principal Source: Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147(7):478-491.
- Acute low back pain generally has a favorable prognosis.
- Accurate categorizations of symptoms as well as self-education and self-care options are pillars of back pain treatment.
- Reserve imaging for patients with ’red flag’ symptoms.
- First-line pharmacotherapy involves acetaminophen and nonsteroidal anti-inflammatory drugs.
- Nonpharmacologic interventions may be appropriate depending on the duration of symptoms.
Low back pain is a common, unrelenting concern for many patients and accounts for a high percentage of health care visits. Although low back pain has a generally favorable prognosis, some patients develop long-term debilitating symptoms that exacerbate or initiate psychiatric conditions. Chronic low back pain has been associated with depression1 and rising rates of depression may contribute to the increasing prevalence of low back pain.2,3
The recent Stepped Care for Affective Disorders and Musculoskeletal Pain (SCAMP) trial showed that optimization of antidepressants in conjunction with a self-management behavioral program reduced depressive and pain symptoms.4 Understanding current diagnostic and treatment recommendations for physical aspects of low back pain will allow psychiatrists to intervene more effectively in somatic and behavioral aspects of the disease and improve functional outcomes.
This article reviews American College of Physicians guidelines on diagnosing and treating low back pain. Most episodes of acute low back pain are self-limited and do not require medical care, with symptom resolution and functional return occurring within the first month. However, 7.6% adult patients report at least 1 episode of severe acute low back pain over 1 year, and one-third of patients who have suffered an acute back pain episode report persistent, moderately intense symptoms and many suffer functional limitations.
Categorizing pain
Back pain can be grouped into 3 categories:
- non-specific low back pain
- back pain associated with radiculopathy or spinal stenosis
- back pain associated with another specific cause.
Low back pain frequently cannot be attributed to a specific disease or spinal abnormality, and conditions such as cancer, compression fracture, spinal stenosis, herniated disks, spinal infection, and ankylosing spondylitis comprise <10% of diagnosed causes of back pain.5 In the absence of “red flag” symptoms that may indicate more serious conditions (Table 1), there is no need to attribute low back pain symptoms to an anatomical source because often there is no associated improvement in outcomes.
Table 1
Back pain symptoms that may indicate a more serious condition
| Progressive loss of motor or sensory function |
| Bilateral sciatica or leg weakness |
| Saddle anesthesia |
| Urinary or fecal incontinence |
| History of substantial trauma |
| Unrelenting pain at night or during rest |
| Unexplained weight loss |
| No improvement after 6 to 8 weeks of conservative therapy |
When imaging is warranted
Although patients often request imaging as part of their workup, routine imaging or other diagnostic tests do not improve outcomes in patients with nonspecific back pain. When patients present with “red flag” symptoms or you suspect another underlying condition, imaging is warranted. MRI generally is preferred over CT. In patients with possible malignancy but no signs of spinal cord compression, multiple strategies have been proposed but not validated. First check plain radiography or erythrocyte sedimentation rate, followed by MRI if abnormalities are found. For patients with low back pain and signs of radiculopathy or spinal stenosis, MRI or CT is appropriate only if patients are candidates for surgery or epidural steroid injection, because symptoms tend to improve within 4 weeks with conservative, noninvasive management.
Selecting treatment
Education and counseling are essential when treating low back pain. Provide your patient with evidence-based information about low back pain, including self-care options such as support measures for pain relief (applying ice packs and heating or pads/blankets) and back-focused stretching and exercise programs (see Related Resources). Remaining as active as possible is more effective than prolonged (>1 to 2 days) bed rest in promoting return to function.6 Consider recommending self-care educational books such as The back book.7 The prognosis of acute low back pain with or without sciatica generally is favorable, and improvement is likely within the first month.8
Pharmacotherapy for low back pain is used in conjunction with—not in lieu of—back care education. However, there is a relative lack of long-term efficacy and safety. Acetaminophen or nonsteroidal anti-inflammatory drugs are typical first-line options.9 Other medications have moderate, mostly short-term benefits. Opioid analgesics or tramadol should be used occasionally and intermittently. When a patient does not respond to a time-limited opioid trial, reassess the symptoms and consider alternate therapies. Muscle relaxants such as cyclobenzaprine offer short-term relief but are associated with CNS side effects, most commonly drowsiness and dizziness but also fatigue, somnolence, confusion, and irritability. Tricyclic antidepressants are options to relieve chronic low back pain.10
Multiple nonpharmacologic therapies have small-to-moderate benefits for low back pain (Table 2). In acute low back pain (<4 weeks), spinal manipulation often is useful. Subacute low back pain (4 to 8 weeks) may improve with intensive interdisciplinary rehabilitation, including cognitive-behavioral therapy (CBT), and can increase functional status and reduce work absenteeism. For chronic low back pain, CBT or progressive relaxation, spinal manipulation, acupuncture, and other modalities have mild to moderate effectiveness.
Table 2
Nonpharmacologic modalities for low back pain
| Duration of back pain | Treatment modality |
|---|---|
| Acute (<4 weeks) | Spinal manipulation |
| Subacute (4 to 8 weeks) | Intensive interdisciplinary rehabilitation (physician consultation, psychological and physical therapy, social and vocational intervention, cognitive-behavioral therapy [CBT]) |
| Chronic (>8 weeks) | Acupuncture, exercise, massage therapy, yoga, CBT, progressive relaxation, spinal manipulation, intensive interdisciplinary rehabilitation |
| Source: Reference 5 | |
- Last AR, Hulbert K. Chronic low back pain: evaluation and management. Am Fam Physician. 2009;79(12):1067-1074.
- Exercise for a better back. www.backcare.org.uk/CMS/files/702-exercise-for-a-better-back.pdf. Accessed April 12, 2010.
Drug brand names
- Cyclobenzaprine • Flexeril
- Tramadol • Ultram, Ultram ER
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bair MJ, Robinson RL, Katon W, et al. Depression and pain comorbidity: a literature review. Arch Intern Med. 2003;163(20):2433-2445.
2. Freburger JK, Holmes GM, Agans RP, et al. The rising prevalence of chronic low back pain. Arch Intern Med. 2009;169(3):251-258.
3. Rush AJ, Polatin P, Gatchel RJ. Depression and chronic low back pain: establishing priorities in treatment. Spine (Phila Pa 1976). 2000;25(20):2566-2571.
4. Kroenke K, Bair MJ, Damush TM, et al. Optimized antidepressant therapy and pain self-management in primary care patients with depression and musculoskeletal pain: a randomized controlled trial. JAMA. 2009;301(20):2099-2110.
5. Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147(7):478-491.
6. Hagen KB, Hilde G, Jamtvedt G, et al. Bed rest for acute low-back pain and sciatica. Cochrane Database Syst Rev. 2004;(4):CD001254.-
7. Burton AK, Waddell G, Tillotson KM, et al. Information and advice to patients with back pain can have a positive effect. A randomized controlled trial of a novel educational booklet in primary care. Spine (Phila Pa 1976). 1999;24(23):2484-2491.
8. Vroomen PC, de Krom MC, Knottnerus JA. Predicting the outcome of sciatica at short-term follow-up. Br J Gen Pract. 2002;52:119-123.
9. van Tulder MW, Scholten RJ, Koes BW, et al. Nonsteroidal anti-inflammatory drugs for low back pain: a systematic review within the framework of the Cochrane Collaboration Back Review Group. Spine (Phila Pa 1976). 2000;25:2501-2513.
10. Staiger TO, Gaster B, Sullivan MD, et al. Systematic review of antidepressants in the treatment of chronic low back pain. Spine (Phila Pa 1976). 2003;28:2540-2545.
Alvin Lau, MD
Jaesu Han, MD
Robert M. McCarron, DO
Series Editor
Alvin Lau, MD
Jaesu Han, MD
Robert M. McCarron, DO
Series Editor
Alvin Lau, MD
Jaesu Han, MD
Robert M. McCarron, DO
Series Editor
Dr. Lau is chief resident and Dr. Han is residency training director, departments of family and community medicine and psychiatry and behavioral sciences, University of California, Davis, Sacramento, CA.
Principal Source: Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147(7):478-491.
- Acute low back pain generally has a favorable prognosis.
- Accurate categorizations of symptoms as well as self-education and self-care options are pillars of back pain treatment.
- Reserve imaging for patients with ’red flag’ symptoms.
- First-line pharmacotherapy involves acetaminophen and nonsteroidal anti-inflammatory drugs.
- Nonpharmacologic interventions may be appropriate depending on the duration of symptoms.
Low back pain is a common, unrelenting concern for many patients and accounts for a high percentage of health care visits. Although low back pain has a generally favorable prognosis, some patients develop long-term debilitating symptoms that exacerbate or initiate psychiatric conditions. Chronic low back pain has been associated with depression1 and rising rates of depression may contribute to the increasing prevalence of low back pain.2,3
The recent Stepped Care for Affective Disorders and Musculoskeletal Pain (SCAMP) trial showed that optimization of antidepressants in conjunction with a self-management behavioral program reduced depressive and pain symptoms.4 Understanding current diagnostic and treatment recommendations for physical aspects of low back pain will allow psychiatrists to intervene more effectively in somatic and behavioral aspects of the disease and improve functional outcomes.
This article reviews American College of Physicians guidelines on diagnosing and treating low back pain. Most episodes of acute low back pain are self-limited and do not require medical care, with symptom resolution and functional return occurring within the first month. However, 7.6% adult patients report at least 1 episode of severe acute low back pain over 1 year, and one-third of patients who have suffered an acute back pain episode report persistent, moderately intense symptoms and many suffer functional limitations.
Categorizing pain
Back pain can be grouped into 3 categories:
- non-specific low back pain
- back pain associated with radiculopathy or spinal stenosis
- back pain associated with another specific cause.
Low back pain frequently cannot be attributed to a specific disease or spinal abnormality, and conditions such as cancer, compression fracture, spinal stenosis, herniated disks, spinal infection, and ankylosing spondylitis comprise <10% of diagnosed causes of back pain.5 In the absence of “red flag” symptoms that may indicate more serious conditions (Table 1), there is no need to attribute low back pain symptoms to an anatomical source because often there is no associated improvement in outcomes.
Table 1
Back pain symptoms that may indicate a more serious condition
| Progressive loss of motor or sensory function |
| Bilateral sciatica or leg weakness |
| Saddle anesthesia |
| Urinary or fecal incontinence |
| History of substantial trauma |
| Unrelenting pain at night or during rest |
| Unexplained weight loss |
| No improvement after 6 to 8 weeks of conservative therapy |
When imaging is warranted
Although patients often request imaging as part of their workup, routine imaging or other diagnostic tests do not improve outcomes in patients with nonspecific back pain. When patients present with “red flag” symptoms or you suspect another underlying condition, imaging is warranted. MRI generally is preferred over CT. In patients with possible malignancy but no signs of spinal cord compression, multiple strategies have been proposed but not validated. First check plain radiography or erythrocyte sedimentation rate, followed by MRI if abnormalities are found. For patients with low back pain and signs of radiculopathy or spinal stenosis, MRI or CT is appropriate only if patients are candidates for surgery or epidural steroid injection, because symptoms tend to improve within 4 weeks with conservative, noninvasive management.
Selecting treatment
Education and counseling are essential when treating low back pain. Provide your patient with evidence-based information about low back pain, including self-care options such as support measures for pain relief (applying ice packs and heating or pads/blankets) and back-focused stretching and exercise programs (see Related Resources). Remaining as active as possible is more effective than prolonged (>1 to 2 days) bed rest in promoting return to function.6 Consider recommending self-care educational books such as The back book.7 The prognosis of acute low back pain with or without sciatica generally is favorable, and improvement is likely within the first month.8
Pharmacotherapy for low back pain is used in conjunction with—not in lieu of—back care education. However, there is a relative lack of long-term efficacy and safety. Acetaminophen or nonsteroidal anti-inflammatory drugs are typical first-line options.9 Other medications have moderate, mostly short-term benefits. Opioid analgesics or tramadol should be used occasionally and intermittently. When a patient does not respond to a time-limited opioid trial, reassess the symptoms and consider alternate therapies. Muscle relaxants such as cyclobenzaprine offer short-term relief but are associated with CNS side effects, most commonly drowsiness and dizziness but also fatigue, somnolence, confusion, and irritability. Tricyclic antidepressants are options to relieve chronic low back pain.10
Multiple nonpharmacologic therapies have small-to-moderate benefits for low back pain (Table 2). In acute low back pain (<4 weeks), spinal manipulation often is useful. Subacute low back pain (4 to 8 weeks) may improve with intensive interdisciplinary rehabilitation, including cognitive-behavioral therapy (CBT), and can increase functional status and reduce work absenteeism. For chronic low back pain, CBT or progressive relaxation, spinal manipulation, acupuncture, and other modalities have mild to moderate effectiveness.
Table 2
Nonpharmacologic modalities for low back pain
| Duration of back pain | Treatment modality |
|---|---|
| Acute (<4 weeks) | Spinal manipulation |
| Subacute (4 to 8 weeks) | Intensive interdisciplinary rehabilitation (physician consultation, psychological and physical therapy, social and vocational intervention, cognitive-behavioral therapy [CBT]) |
| Chronic (>8 weeks) | Acupuncture, exercise, massage therapy, yoga, CBT, progressive relaxation, spinal manipulation, intensive interdisciplinary rehabilitation |
| Source: Reference 5 | |
- Last AR, Hulbert K. Chronic low back pain: evaluation and management. Am Fam Physician. 2009;79(12):1067-1074.
- Exercise for a better back. www.backcare.org.uk/CMS/files/702-exercise-for-a-better-back.pdf. Accessed April 12, 2010.
Drug brand names
- Cyclobenzaprine • Flexeril
- Tramadol • Ultram, Ultram ER
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Lau is chief resident and Dr. Han is residency training director, departments of family and community medicine and psychiatry and behavioral sciences, University of California, Davis, Sacramento, CA.
Principal Source: Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147(7):478-491.
- Acute low back pain generally has a favorable prognosis.
- Accurate categorizations of symptoms as well as self-education and self-care options are pillars of back pain treatment.
- Reserve imaging for patients with ’red flag’ symptoms.
- First-line pharmacotherapy involves acetaminophen and nonsteroidal anti-inflammatory drugs.
- Nonpharmacologic interventions may be appropriate depending on the duration of symptoms.
Low back pain is a common, unrelenting concern for many patients and accounts for a high percentage of health care visits. Although low back pain has a generally favorable prognosis, some patients develop long-term debilitating symptoms that exacerbate or initiate psychiatric conditions. Chronic low back pain has been associated with depression1 and rising rates of depression may contribute to the increasing prevalence of low back pain.2,3
The recent Stepped Care for Affective Disorders and Musculoskeletal Pain (SCAMP) trial showed that optimization of antidepressants in conjunction with a self-management behavioral program reduced depressive and pain symptoms.4 Understanding current diagnostic and treatment recommendations for physical aspects of low back pain will allow psychiatrists to intervene more effectively in somatic and behavioral aspects of the disease and improve functional outcomes.
This article reviews American College of Physicians guidelines on diagnosing and treating low back pain. Most episodes of acute low back pain are self-limited and do not require medical care, with symptom resolution and functional return occurring within the first month. However, 7.6% adult patients report at least 1 episode of severe acute low back pain over 1 year, and one-third of patients who have suffered an acute back pain episode report persistent, moderately intense symptoms and many suffer functional limitations.
Categorizing pain
Back pain can be grouped into 3 categories:
- non-specific low back pain
- back pain associated with radiculopathy or spinal stenosis
- back pain associated with another specific cause.
Low back pain frequently cannot be attributed to a specific disease or spinal abnormality, and conditions such as cancer, compression fracture, spinal stenosis, herniated disks, spinal infection, and ankylosing spondylitis comprise <10% of diagnosed causes of back pain.5 In the absence of “red flag” symptoms that may indicate more serious conditions (Table 1), there is no need to attribute low back pain symptoms to an anatomical source because often there is no associated improvement in outcomes.
Table 1
Back pain symptoms that may indicate a more serious condition
| Progressive loss of motor or sensory function |
| Bilateral sciatica or leg weakness |
| Saddle anesthesia |
| Urinary or fecal incontinence |
| History of substantial trauma |
| Unrelenting pain at night or during rest |
| Unexplained weight loss |
| No improvement after 6 to 8 weeks of conservative therapy |
When imaging is warranted
Although patients often request imaging as part of their workup, routine imaging or other diagnostic tests do not improve outcomes in patients with nonspecific back pain. When patients present with “red flag” symptoms or you suspect another underlying condition, imaging is warranted. MRI generally is preferred over CT. In patients with possible malignancy but no signs of spinal cord compression, multiple strategies have been proposed but not validated. First check plain radiography or erythrocyte sedimentation rate, followed by MRI if abnormalities are found. For patients with low back pain and signs of radiculopathy or spinal stenosis, MRI or CT is appropriate only if patients are candidates for surgery or epidural steroid injection, because symptoms tend to improve within 4 weeks with conservative, noninvasive management.
Selecting treatment
Education and counseling are essential when treating low back pain. Provide your patient with evidence-based information about low back pain, including self-care options such as support measures for pain relief (applying ice packs and heating or pads/blankets) and back-focused stretching and exercise programs (see Related Resources). Remaining as active as possible is more effective than prolonged (>1 to 2 days) bed rest in promoting return to function.6 Consider recommending self-care educational books such as The back book.7 The prognosis of acute low back pain with or without sciatica generally is favorable, and improvement is likely within the first month.8
Pharmacotherapy for low back pain is used in conjunction with—not in lieu of—back care education. However, there is a relative lack of long-term efficacy and safety. Acetaminophen or nonsteroidal anti-inflammatory drugs are typical first-line options.9 Other medications have moderate, mostly short-term benefits. Opioid analgesics or tramadol should be used occasionally and intermittently. When a patient does not respond to a time-limited opioid trial, reassess the symptoms and consider alternate therapies. Muscle relaxants such as cyclobenzaprine offer short-term relief but are associated with CNS side effects, most commonly drowsiness and dizziness but also fatigue, somnolence, confusion, and irritability. Tricyclic antidepressants are options to relieve chronic low back pain.10
Multiple nonpharmacologic therapies have small-to-moderate benefits for low back pain (Table 2). In acute low back pain (<4 weeks), spinal manipulation often is useful. Subacute low back pain (4 to 8 weeks) may improve with intensive interdisciplinary rehabilitation, including cognitive-behavioral therapy (CBT), and can increase functional status and reduce work absenteeism. For chronic low back pain, CBT or progressive relaxation, spinal manipulation, acupuncture, and other modalities have mild to moderate effectiveness.
Table 2
Nonpharmacologic modalities for low back pain
| Duration of back pain | Treatment modality |
|---|---|
| Acute (<4 weeks) | Spinal manipulation |
| Subacute (4 to 8 weeks) | Intensive interdisciplinary rehabilitation (physician consultation, psychological and physical therapy, social and vocational intervention, cognitive-behavioral therapy [CBT]) |
| Chronic (>8 weeks) | Acupuncture, exercise, massage therapy, yoga, CBT, progressive relaxation, spinal manipulation, intensive interdisciplinary rehabilitation |
| Source: Reference 5 | |
- Last AR, Hulbert K. Chronic low back pain: evaluation and management. Am Fam Physician. 2009;79(12):1067-1074.
- Exercise for a better back. www.backcare.org.uk/CMS/files/702-exercise-for-a-better-back.pdf. Accessed April 12, 2010.
Drug brand names
- Cyclobenzaprine • Flexeril
- Tramadol • Ultram, Ultram ER
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bair MJ, Robinson RL, Katon W, et al. Depression and pain comorbidity: a literature review. Arch Intern Med. 2003;163(20):2433-2445.
2. Freburger JK, Holmes GM, Agans RP, et al. The rising prevalence of chronic low back pain. Arch Intern Med. 2009;169(3):251-258.
3. Rush AJ, Polatin P, Gatchel RJ. Depression and chronic low back pain: establishing priorities in treatment. Spine (Phila Pa 1976). 2000;25(20):2566-2571.
4. Kroenke K, Bair MJ, Damush TM, et al. Optimized antidepressant therapy and pain self-management in primary care patients with depression and musculoskeletal pain: a randomized controlled trial. JAMA. 2009;301(20):2099-2110.
5. Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147(7):478-491.
6. Hagen KB, Hilde G, Jamtvedt G, et al. Bed rest for acute low-back pain and sciatica. Cochrane Database Syst Rev. 2004;(4):CD001254.-
7. Burton AK, Waddell G, Tillotson KM, et al. Information and advice to patients with back pain can have a positive effect. A randomized controlled trial of a novel educational booklet in primary care. Spine (Phila Pa 1976). 1999;24(23):2484-2491.
8. Vroomen PC, de Krom MC, Knottnerus JA. Predicting the outcome of sciatica at short-term follow-up. Br J Gen Pract. 2002;52:119-123.
9. van Tulder MW, Scholten RJ, Koes BW, et al. Nonsteroidal anti-inflammatory drugs for low back pain: a systematic review within the framework of the Cochrane Collaboration Back Review Group. Spine (Phila Pa 1976). 2000;25:2501-2513.
10. Staiger TO, Gaster B, Sullivan MD, et al. Systematic review of antidepressants in the treatment of chronic low back pain. Spine (Phila Pa 1976). 2003;28:2540-2545.
1. Bair MJ, Robinson RL, Katon W, et al. Depression and pain comorbidity: a literature review. Arch Intern Med. 2003;163(20):2433-2445.
2. Freburger JK, Holmes GM, Agans RP, et al. The rising prevalence of chronic low back pain. Arch Intern Med. 2009;169(3):251-258.
3. Rush AJ, Polatin P, Gatchel RJ. Depression and chronic low back pain: establishing priorities in treatment. Spine (Phila Pa 1976). 2000;25(20):2566-2571.
4. Kroenke K, Bair MJ, Damush TM, et al. Optimized antidepressant therapy and pain self-management in primary care patients with depression and musculoskeletal pain: a randomized controlled trial. JAMA. 2009;301(20):2099-2110.
5. Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147(7):478-491.
6. Hagen KB, Hilde G, Jamtvedt G, et al. Bed rest for acute low-back pain and sciatica. Cochrane Database Syst Rev. 2004;(4):CD001254.-
7. Burton AK, Waddell G, Tillotson KM, et al. Information and advice to patients with back pain can have a positive effect. A randomized controlled trial of a novel educational booklet in primary care. Spine (Phila Pa 1976). 1999;24(23):2484-2491.
8. Vroomen PC, de Krom MC, Knottnerus JA. Predicting the outcome of sciatica at short-term follow-up. Br J Gen Pract. 2002;52:119-123.
9. van Tulder MW, Scholten RJ, Koes BW, et al. Nonsteroidal anti-inflammatory drugs for low back pain: a systematic review within the framework of the Cochrane Collaboration Back Review Group. Spine (Phila Pa 1976). 2000;25:2501-2513.
10. Staiger TO, Gaster B, Sullivan MD, et al. Systematic review of antidepressants in the treatment of chronic low back pain. Spine (Phila Pa 1976). 2003;28:2540-2545.
Osteoporosis in depression: Which patients are at risk?
Ms. P, age 44, is concerned about her risk of osteoporosis after her 70-year-old mother is hospitalized for a hip fracture. Ms. P has been taking fluoxetine, 40 mg/d, for 10 years to treat recurrent major depressive episodes that began at age 25. She was diagnosed with anorexia nervosa as a teenager, but recovered after 2 years of psychotherapy. She is lactose intolerant, has mild asthma that does not require steroids, and has no history of thyroid disease or bone fracture. Ms. P smokes 10 cigarettes a day but denies using alcohol or illicit drugs. She does not exercise, and her menses occur every 28 to 30 days.
Osteoporosis is a skeletal disease characterized by low bone mineralization and deteriorating bone architecture that results in increased susceptibility to fracture. Approximately 1 in 2 women and 1 in 5 men in the United States will have an osteoporosis-related fracture.1 Proximal femur and vertebral fractures are most common—1.5 million per year—but other bones may be involved.2
Osteoporosis-related fractures are associated with substantial morbidity and mortality. After a hip fracture, osteoporosis patients have a 10% to 20% risk of death within a year.3 Those who recover from hip fracture have a 2.5-fold increased risk of recurrent fracture and often struggle with chronic pain, disability, and loss of self-esteem and independence.1,3–5
Evidence links osteoporosis and depression
Research has shown that patients with major depression are at higher risk of osteoporosis.6 In one study, bone mineral density among 70 depressed outpatients was 15% lower than among age-matched controls.7 In a cross-sectional study, Michelson et al8 found that compared with nondepressed controls, women with current or past major depression had a lower mean bone mineral density—6.5% lower at the spine and 13.6% lower at the femoral neck.
Fewer prospective studies exist; however, most found depression has some impact on bone health. Whooley et al9 prospectively evaluated changes in bone mineral density among 7,414 Caucasian women age ≥65 for 6 years. Depressed women—those who scored ≥6 on the Geriatric Depression Scale—had a 40% higher risk of nonvertebral fracture after adjusting for history of fracture, weight, physical activity level, smoking, alcohol use, nutritional status, and cognitive function. The depressed cohort also had an increased risk of vertebral fracture. In a prospective study of 21,441 Norwegian female and male subjects, women who reported being depressed at 2 of 3 time points—from 1980 until 1995—had 2.5 times the risk of sustaining a nonvertebral fracture compared with those who did not report depression.10
Depressed women also have greater bone loss over time. Mean hip bone mineral density decreased by 0.69% per year in nondepressed women vs 0.96% in depressed women in a study of 4,177 women age ≥69.11 These findings were significant after adjusting for age, functional status, cognitive function, smoking, calcium intake, vitamin D supplement use, weight, antidepressant use, and bisphosphonate use. These findings have been replicated.12
Behavioral factors such as tobacco use and physical inactivity play a role in the risk of osteoporosis; however, emerging findings suggest a pathophysiologic link between depression and poor bone health. Depression is associated with lower estrogen and testosterone levels, which have been linked to decreased bone formation.6 Similarly, compared with matched controls, depressed women with low bone mineral density have higher urinary cortisol levels, suggesting that hypercortisolemia accelerates bone turnover.6,9,13 Finally, evidence suggests that depression is a pro-inflammatory state associated with production of numerous cytokines. Interleukin-6 and tumor necrosis factor-alpha, for example, inhibit osteoclast apoptosis and accelerate bone turnover.6
Fracture risk and psychotropics
Many psychotropic medications—including anticonvulsants, barbiturates, narcotics, and neuroleptics14–16—are associated with increased risk of falls, fractures, and osteoporosis. In this article we focus on selective serotonin reuptake inhibitors (SSRIs) and tricyclic antidepressants (TCAs) because little data is available on other antidepressants (Table 1).17–22
SSRIs are associated with increased fracture risk. In a cohort of 5,995 men age ≥65, Haney et al23 showed that men taking SSRIs have lower bone mineral density at the hip (3.9% lower) and spine (5.6% lower) compared with non-users after adjusting for age, weight, and race. Current SSRI use carries a greater risk than past use. In a prospective study of 7,983 men and women age ≥55, Ziere et al24 reported that risk of nonvertebral fracture among current SSRI users was 28% higher than among past users over a mean follow-up of 8.4 years. In the same study, the risk ratio of nonvertebral fracture was 2.10 for patients using SSRIs within the previous 6 months and 2.98 for use >6 months.
Table 1
Psychotropic medications associated with osteoporosis risk
| Medication/class | Odds ratio (95% confidence interval) |
|---|---|
| Selective serotonin reuptake inhibitors (SSRIs) | 1.45 (1.32 to 1.59) |
| Carbamazepine | 1.18 (1.10 to 1.26) |
| Non-SSRIs (eg, tricyclics, atypicals) | 1.15 (1.07 to 1.24) |
| Valproate | 1.15 (1.05 to 1.26) |
| Oxcarbazepine | 1.14 (1.03 to 1.26) |
| Benzodiazepines | 1.10 (1.04 to 1.16) |
| Lamotrigine | 1.04 (0.91 to 1.19) |
| Typical antipsychotics | 1.01 (0.86 to 1.19) |
| Atypical antipsychotics | 0.96 (0.79 to 1.17) |
| Lithium | 0.63 (0.43 to 0.93) |
| Source: References 17-22 | |
Increased fracture risk with SSRIs may be partially explained by the greater risk of osteoporosis in major depression.25 SSRI use has been linked to higher risk of fracture in the absence of depressive symptoms, however.26 Bolton et al17 revealed a trend of increasing fracture risk with higher SSRI dose. In this study, SSRI users had 45% greater likelihood of fracture than controls after adjusting for a diagnosis of depression.
Researchers are studying the mechanism by which SSRIs affect bone mineralization. Serotonin receptors—including 5-HT2A, 5-HT2B, and 5-HT2C—are present in bone.27 Preliminary investigations suggest SSRIs are concentrated in bone and impact fibroblast formation and osteoblast activity. High bone marrow concentrations of fluoxetine inhibit human osteoblast proliferation. Osteoblasts contribute to bone production.28 Fluoxetine concentrations in bone marrow can be up to 100-fold higher than serum levels, and the drug can be detected in bone up to 3 months after discontinuation.
TCAs. U.S. veterans with prior hip fracture are twice as likely to have received TCAs than age- and sex-matched controls.14In prospective studies, the risk of hip fracture among men and women age ≥65 is 50% higher in patients exposed to TCAs.30 Other investigations have revealed a dose-response relationship between TCA use and risk of fracture.31 A direct comparison of TCAs and SSRIs has found an equivalent increase in fracture risk in these 2 classes.30
A direct effect of TCAs on bone metabolism has not been elucidated. However, side effects of TCAs include orthostatic hypotension, impaired cognition, dizziness, and altered balance, all of which increase the risk of falls and fractures, particularly in elderly patients.31 Most studies of TCAs, however, do not account for depression’s role in fracture risk. Some patients in these studies may have received TCAs for disorders other than major depression, such as peripheral neuropathy or prophylaxis of migraine headaches.
Benzodiazepine use is associated with confusion, ataxia, and vertigo, which may increase the incidence of falls. Even low doses pose a risk. In one case-control study of 1,222 hip fracture patients age ≥65, use of >3 mg/d diazepam equivalents increased risk of hip fracture by 50% after adjusting for confounding factors.30 Although the data are mixed, benzodiazepines with shorter half-lives (eg, lorazepam) might not be safer than those with longer half-lives (eg, clonazepam).31,32
Other psychotropics. Some anticonvulsants may lead to bone demineralization via induction of the cytochrome P450 hepatic enzyme system, which accelerates conversion of vitamin D to an inactive metabolite that cannot adequately facilitate absorption of ingested calcium. The subsequent release of parathyroid hormone causes bone resorption.33 Patients taking anticonvulsants have nearly double the serum parathyroid hormone level of matched controls.34 Carbamazepine, oxcarbazepine, and valproate have been associated with increased risk of fracture.32 Although lamotrigine has not been widely studied, evidence suggests that its impact on bone metabolism is negligible.35
Many antipsychotics, including risperidone and haloperidol, have been associated with osteoporosis. The mechanism by which antipsychotics accelerate bone turnover has not been described; hyperprolactinemia likely plays a role.36
Screening and treatment
Effective pharmacotherapy for osteoporosis includes bisphosphonates (eg, alendronate), selective estrogen receptor modulators (eg, raloxifene), recombinant parathyroid hormone (eg, teriparatide), as well as calcium and vitamin D supplementation. Consider recommending bone density evaluation for depressed patients who have predisposing risk factors (Table 2)1 and those with long-term exposure to psychotropic agents. Dual energy X-ray absorptiometry is the preferred screening method. Refer patients whose results indicate osteopenia or osteoporosis to primary care. Although pharmacotherapy for osteoporosis should be managed by primary care practitioners, psychiatrists can serve an important role by promoting healthy lifestyle behaviors—such as regular exercise and adequate dietary vitamin D and calcium intake (Table 3).1
Table 2
Risk factors for osteoporosis-related fracture*
| Clinical factors |
| Age >50 Female sex Amenorrhea Cognitive impairment Family history of osteoporosis-related fracture Malnutrition Poor visual acuity Previous falls Low body mass index Glucocorticoid use (prednisone >5 mg/d for ≥3 months) |
| Secondary medical conditions |
| Hyperprolactinemia Anorexia nervosa Postmenopausal status Adrenal insufficiency Diabetes mellitus Hyperparathyroidism Celiac disease Inflammatory bowel disease Malabsorption syndromes Multiple myeloma End-stage renal disease |
| Behavioral factors |
| Low calcium intake Tobacco abuse Physical inactivity Excessive alcohol intake (>3 drinks per day) Vitamin D deficiency Immobilization |
| *Italics indicate conditions commonly encountered in psychiatric patients Source: Reference 1 |
Table 3
Reducing osteoporosis risk: Recommendations for patients age >50
| Assess dietary calcium (at least 1,200 mg/d) and dietary vitamin D intake (800 to 1,000 IU/d) |
| Exercise regularly, especially weight-bearing and muscle-strengthening activities (eg, walking, jogging, stair climbing, weight-lifting) |
| Stop using tobacco |
| Avoid heavy alcohol use |
| Implement fall precautions such as rubber-soled shoes when walking, handrails for staircases, and removing tripping hazards, including loose rugs |
| Source: Reference 1 |
CASE CONTINUED: High risk can be lowered
Ms. P’s family history, antidepressant use, smoking, and low dietary calcium intake associated with lactose intolerance increase her risk for osteoporosis. Her history of anorexia nervosa also increases her risk if she experiences amenorrhea. You advise her that she can ameliorate some of these factors by quitting smoking, exercising regularly, and taking calcium and vitamin D supplements. You refer her to her primary care physician because she wishes to undergo bone mineral density screening.
Related resources
- National Osteoporosis Foundation. Clinician‘s guide to prevention and treatment of osteoporosis. www.nof.org/professionals/Clinicians_Guide.htm.
- World Health Organization Fracture Risk Assessment Tool. Calculates a 10-year probability of hip fracture using demographic data, family history, comorbid medication and predisposing medical conditions. www.shef.ac.uk/FRAX.
Drug brand names
- Alendronate • Fosamax
- Carbamazepine • Tegretol
- Clonazepam • Klonopin
- Diazepam • Valium
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Oxcarbazepine • Trileptal
- Prednisone • Deltasone, Meticorten
- Raloxifene • Evista
- Risperidone • Risperdal
- Teriparatide • Forteo
- Valproate • Depakote
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC: National Osteoporosis Foundation; 2008.
2. Lane NE. Epidemiology, etiology, and diagnosis of osteoporosis. Amer J Obstet Gynecol. 2006;194(2 suppl):S3-11.
3. Sambrook P, Cooper C. Osteoporosis. Lancet. 2006;367:2010-2018.
4. Salaffi F, Cimmino MA, Malavolta N, et al. The burden of prevalent fractures on health-related quality of life in postmenopausal women with osteoporosis: the IMOF study. J Rheumatol. 2007;34(7):1551-1560.
5. Gold DT, Stegmaier K, Bales CW, et al. Psychosocial functioning and osteoporosis in late life: results of a multidisciplinary intervention. Journal of Women’s Health. 1993;2:149-155.
6. Mezuk B, Eaton WW, Golden SH. Depression and osteoporosis: epidemiology and potential mediating pathways. Osteoporos Int. 2008;19:1-12.
7. Schweiger U, Deuschle M, Korner A, et al. Low lumbar bone mineral density in patients with major depression. Am J Psychiatry. 1994;151:1691-1693.
8. Michelson D, Stratakis C, Hill L, et al. Bone mineral density in women with depression. N Engl J Med. 1996;335:1176-1181.
9. Whooley MA, Kip KE, Cauley JA, et al. Depression, falls, and risk of fracture in older women. Arch Intern Med. 1999;159:484-490.
10. Søgaard AJ, Joakimsen RM, Tverdal A, et al. Long-term mental distress, bone mineral density, and non-vertebral fractures. The Tromsø Study. Osteoporos Int. 2005;16(8):887-897.
11. Diem SJ, Blackwell TL, Stone KL, et al. Depressive symptoms and rates of bone loss at the hip in older women. J Am Geriatr Soc. 2007;55:824-831.
12. Eskandari F, Martinez PE, Torvik S, et al. Low bone mass in premenopausal women with depression. Arch Intern Med. 2007;167:2329-2336.
13. Yirmiya R, Goshen I, Bajayo A, et al. Depression induces bone loss through stimulation of the sympathetic nervous system. Proc Natl Acad Sci U S A. 2006;103(45):16876-16881.
14. French DD, Campbell R, Spehar A, et al. Outpatient medications and hip fractures in the US: a national veterans study. Drugs Aging. 2005;22(10):877-885.
15. Ensrud KE, Blackwell T, Mangione CM, et al. Central nervous system active medications and risk for fractures in older women. Arch Intern Med. 2003;163:949-957.
16. O’Keane V. Antipsychotic-induced hyperprolactinemia, hypogonadism, and osteoporosis in the treatment of schizophrenia. J Psychopharmacol. 2008;22(2 suppl):70-75.
17. Bolton JM, Metge C, Lix L, et al. Fracture risk from psychotropic medications: a population-based analysis. J Clin Psychopharmacol. 2008;28:384-391.
18. Heidrich FE, Stergachis A, Gross KM. Diuretic drug use and the risk for hip fracture. Ann Intern Med. 1991;115(1):1-6.
19. Yang YX, Lewis JD, Epstein S, et al. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA. 2006;296(24):2947-2953.
20. Schoofs MW, van der Klift M, Hofman A, et al. Thiazide diuretics and the risk for hip fracture. Ann Intern Med. 2003;139(6):476-482.
21. Schlienger RG, Kraenzlin ME, Jick SS, et al. Use of beta blockers and risk of fractures. JAMA. 2004;292(11):1326-1332.
22. Vestergaard P, Rejnmark L, Mosekilde L. Fracture risk associated with different types of oral corticosteroids and effect of termination of corticosteroids on risk of fracture. Calcif Tissue Int. 2008;82:249-257.
23. Haney EM, Chan BK, Diem SJ, et al. Association of low bone mineral density with selective serotonin reuptake inhibitor use by older men. Arch Intern Med. 2007;167:1246-1251.
24. Ziere G, Dieleman JP, van der Cammen TJ, et al. Selective serotonin reuptake inhibiting antidepressants are associated with an increased risk of nonvertebral fractures. J Clin Psychopharmacol. 2008;28:411-417.
25. Richards JB, Papaioannou A, Adachi JD, et al. Effect of selective serotonin reuptake inhibitors on the risk of fracture. Arch Intern Med. 2007;167:188-194.
26. Kerse N, Flicker L, Pfaff JJ, et al. Falls, depression, and antidepressants in later life: a large primary care appraisal. PLoS One. 2008;3(6):e2423.-
27. Westbroek I, van der Plas A, de Rooij KE, et al. Expression of serotonin receptors in bone. J Biol Chem. 2001;276(31):28961-28968.
28. Gustafsson BI, Thommesen L, Stunes AK, et al. Serotonin and fluoxetine modulate bone cell function in vitro. J Cell Biochem. 2006;98:139-151.
29. Bolo NR, Hodé Y, Macher JP. Long-term sequestration of fluorinated compounds in tissues after fluvoxamine or fluoxetine treatment: a fluorine magnetic resonance spectroscopy study in vivo. MAGMA. 2004;16:268-276.
30. Liu B, Anderson G, Mittmann N, et al. Use of selective serotonin-reuptake inhibitors or tricyclic antidepressants and risk of hip fractures in elderly people. Lancet. 1998;351:1303-1307.
31. Vestergaard P, Rejnmark L, Mosekilde L. Anxiolytics, sedatives, antidepressants, neuroleptics, and the risk of fracture. Osteoporos Int. 2006;17:807-816.
32. Wang PS, Bohn RL, Glynn RJ, et al. Hazardous benzodiazepine regimens in the elderly: effects of half-life, dosage, and duration on risk of hip fracture. Am J Psychiatry. 2001;158:892-898.
33. Pack AM. The association between antiepileptic drugs and bone disease. Epilepsy Curr. 2003;3(3):91-95.
34. Kim SH, Lee JW, Choi KG, et al. A 6-month longitudinal study of bone mineral density with antiepileptic drug monotherapy. Epilepsy Behav. 2007;10:291-295.
35. Vestergaard P, Rejnmark L, Mosekilde L. Fracture risk associated with use of antiepileptic drugs. Epilepsia. 2004;45(11):1330-1337.
36. Meaney AM, Smith S, Howes OD, et al. Effects of long-term prolactin-raising antipsychotic medication on bone mineral density in patients with schizophrenia. Br J Psychiatry. 2004;184:503-508.
Ms. P, age 44, is concerned about her risk of osteoporosis after her 70-year-old mother is hospitalized for a hip fracture. Ms. P has been taking fluoxetine, 40 mg/d, for 10 years to treat recurrent major depressive episodes that began at age 25. She was diagnosed with anorexia nervosa as a teenager, but recovered after 2 years of psychotherapy. She is lactose intolerant, has mild asthma that does not require steroids, and has no history of thyroid disease or bone fracture. Ms. P smokes 10 cigarettes a day but denies using alcohol or illicit drugs. She does not exercise, and her menses occur every 28 to 30 days.
Osteoporosis is a skeletal disease characterized by low bone mineralization and deteriorating bone architecture that results in increased susceptibility to fracture. Approximately 1 in 2 women and 1 in 5 men in the United States will have an osteoporosis-related fracture.1 Proximal femur and vertebral fractures are most common—1.5 million per year—but other bones may be involved.2
Osteoporosis-related fractures are associated with substantial morbidity and mortality. After a hip fracture, osteoporosis patients have a 10% to 20% risk of death within a year.3 Those who recover from hip fracture have a 2.5-fold increased risk of recurrent fracture and often struggle with chronic pain, disability, and loss of self-esteem and independence.1,3–5
Evidence links osteoporosis and depression
Research has shown that patients with major depression are at higher risk of osteoporosis.6 In one study, bone mineral density among 70 depressed outpatients was 15% lower than among age-matched controls.7 In a cross-sectional study, Michelson et al8 found that compared with nondepressed controls, women with current or past major depression had a lower mean bone mineral density—6.5% lower at the spine and 13.6% lower at the femoral neck.
Fewer prospective studies exist; however, most found depression has some impact on bone health. Whooley et al9 prospectively evaluated changes in bone mineral density among 7,414 Caucasian women age ≥65 for 6 years. Depressed women—those who scored ≥6 on the Geriatric Depression Scale—had a 40% higher risk of nonvertebral fracture after adjusting for history of fracture, weight, physical activity level, smoking, alcohol use, nutritional status, and cognitive function. The depressed cohort also had an increased risk of vertebral fracture. In a prospective study of 21,441 Norwegian female and male subjects, women who reported being depressed at 2 of 3 time points—from 1980 until 1995—had 2.5 times the risk of sustaining a nonvertebral fracture compared with those who did not report depression.10
Depressed women also have greater bone loss over time. Mean hip bone mineral density decreased by 0.69% per year in nondepressed women vs 0.96% in depressed women in a study of 4,177 women age ≥69.11 These findings were significant after adjusting for age, functional status, cognitive function, smoking, calcium intake, vitamin D supplement use, weight, antidepressant use, and bisphosphonate use. These findings have been replicated.12
Behavioral factors such as tobacco use and physical inactivity play a role in the risk of osteoporosis; however, emerging findings suggest a pathophysiologic link between depression and poor bone health. Depression is associated with lower estrogen and testosterone levels, which have been linked to decreased bone formation.6 Similarly, compared with matched controls, depressed women with low bone mineral density have higher urinary cortisol levels, suggesting that hypercortisolemia accelerates bone turnover.6,9,13 Finally, evidence suggests that depression is a pro-inflammatory state associated with production of numerous cytokines. Interleukin-6 and tumor necrosis factor-alpha, for example, inhibit osteoclast apoptosis and accelerate bone turnover.6
Fracture risk and psychotropics
Many psychotropic medications—including anticonvulsants, barbiturates, narcotics, and neuroleptics14–16—are associated with increased risk of falls, fractures, and osteoporosis. In this article we focus on selective serotonin reuptake inhibitors (SSRIs) and tricyclic antidepressants (TCAs) because little data is available on other antidepressants (Table 1).17–22
SSRIs are associated with increased fracture risk. In a cohort of 5,995 men age ≥65, Haney et al23 showed that men taking SSRIs have lower bone mineral density at the hip (3.9% lower) and spine (5.6% lower) compared with non-users after adjusting for age, weight, and race. Current SSRI use carries a greater risk than past use. In a prospective study of 7,983 men and women age ≥55, Ziere et al24 reported that risk of nonvertebral fracture among current SSRI users was 28% higher than among past users over a mean follow-up of 8.4 years. In the same study, the risk ratio of nonvertebral fracture was 2.10 for patients using SSRIs within the previous 6 months and 2.98 for use >6 months.
Table 1
Psychotropic medications associated with osteoporosis risk
| Medication/class | Odds ratio (95% confidence interval) |
|---|---|
| Selective serotonin reuptake inhibitors (SSRIs) | 1.45 (1.32 to 1.59) |
| Carbamazepine | 1.18 (1.10 to 1.26) |
| Non-SSRIs (eg, tricyclics, atypicals) | 1.15 (1.07 to 1.24) |
| Valproate | 1.15 (1.05 to 1.26) |
| Oxcarbazepine | 1.14 (1.03 to 1.26) |
| Benzodiazepines | 1.10 (1.04 to 1.16) |
| Lamotrigine | 1.04 (0.91 to 1.19) |
| Typical antipsychotics | 1.01 (0.86 to 1.19) |
| Atypical antipsychotics | 0.96 (0.79 to 1.17) |
| Lithium | 0.63 (0.43 to 0.93) |
| Source: References 17-22 | |
Increased fracture risk with SSRIs may be partially explained by the greater risk of osteoporosis in major depression.25 SSRI use has been linked to higher risk of fracture in the absence of depressive symptoms, however.26 Bolton et al17 revealed a trend of increasing fracture risk with higher SSRI dose. In this study, SSRI users had 45% greater likelihood of fracture than controls after adjusting for a diagnosis of depression.
Researchers are studying the mechanism by which SSRIs affect bone mineralization. Serotonin receptors—including 5-HT2A, 5-HT2B, and 5-HT2C—are present in bone.27 Preliminary investigations suggest SSRIs are concentrated in bone and impact fibroblast formation and osteoblast activity. High bone marrow concentrations of fluoxetine inhibit human osteoblast proliferation. Osteoblasts contribute to bone production.28 Fluoxetine concentrations in bone marrow can be up to 100-fold higher than serum levels, and the drug can be detected in bone up to 3 months after discontinuation.
TCAs. U.S. veterans with prior hip fracture are twice as likely to have received TCAs than age- and sex-matched controls.14In prospective studies, the risk of hip fracture among men and women age ≥65 is 50% higher in patients exposed to TCAs.30 Other investigations have revealed a dose-response relationship between TCA use and risk of fracture.31 A direct comparison of TCAs and SSRIs has found an equivalent increase in fracture risk in these 2 classes.30
A direct effect of TCAs on bone metabolism has not been elucidated. However, side effects of TCAs include orthostatic hypotension, impaired cognition, dizziness, and altered balance, all of which increase the risk of falls and fractures, particularly in elderly patients.31 Most studies of TCAs, however, do not account for depression’s role in fracture risk. Some patients in these studies may have received TCAs for disorders other than major depression, such as peripheral neuropathy or prophylaxis of migraine headaches.
Benzodiazepine use is associated with confusion, ataxia, and vertigo, which may increase the incidence of falls. Even low doses pose a risk. In one case-control study of 1,222 hip fracture patients age ≥65, use of >3 mg/d diazepam equivalents increased risk of hip fracture by 50% after adjusting for confounding factors.30 Although the data are mixed, benzodiazepines with shorter half-lives (eg, lorazepam) might not be safer than those with longer half-lives (eg, clonazepam).31,32
Other psychotropics. Some anticonvulsants may lead to bone demineralization via induction of the cytochrome P450 hepatic enzyme system, which accelerates conversion of vitamin D to an inactive metabolite that cannot adequately facilitate absorption of ingested calcium. The subsequent release of parathyroid hormone causes bone resorption.33 Patients taking anticonvulsants have nearly double the serum parathyroid hormone level of matched controls.34 Carbamazepine, oxcarbazepine, and valproate have been associated with increased risk of fracture.32 Although lamotrigine has not been widely studied, evidence suggests that its impact on bone metabolism is negligible.35
Many antipsychotics, including risperidone and haloperidol, have been associated with osteoporosis. The mechanism by which antipsychotics accelerate bone turnover has not been described; hyperprolactinemia likely plays a role.36
Screening and treatment
Effective pharmacotherapy for osteoporosis includes bisphosphonates (eg, alendronate), selective estrogen receptor modulators (eg, raloxifene), recombinant parathyroid hormone (eg, teriparatide), as well as calcium and vitamin D supplementation. Consider recommending bone density evaluation for depressed patients who have predisposing risk factors (Table 2)1 and those with long-term exposure to psychotropic agents. Dual energy X-ray absorptiometry is the preferred screening method. Refer patients whose results indicate osteopenia or osteoporosis to primary care. Although pharmacotherapy for osteoporosis should be managed by primary care practitioners, psychiatrists can serve an important role by promoting healthy lifestyle behaviors—such as regular exercise and adequate dietary vitamin D and calcium intake (Table 3).1
Table 2
Risk factors for osteoporosis-related fracture*
| Clinical factors |
| Age >50 Female sex Amenorrhea Cognitive impairment Family history of osteoporosis-related fracture Malnutrition Poor visual acuity Previous falls Low body mass index Glucocorticoid use (prednisone >5 mg/d for ≥3 months) |
| Secondary medical conditions |
| Hyperprolactinemia Anorexia nervosa Postmenopausal status Adrenal insufficiency Diabetes mellitus Hyperparathyroidism Celiac disease Inflammatory bowel disease Malabsorption syndromes Multiple myeloma End-stage renal disease |
| Behavioral factors |
| Low calcium intake Tobacco abuse Physical inactivity Excessive alcohol intake (>3 drinks per day) Vitamin D deficiency Immobilization |
| *Italics indicate conditions commonly encountered in psychiatric patients Source: Reference 1 |
Table 3
Reducing osteoporosis risk: Recommendations for patients age >50
| Assess dietary calcium (at least 1,200 mg/d) and dietary vitamin D intake (800 to 1,000 IU/d) |
| Exercise regularly, especially weight-bearing and muscle-strengthening activities (eg, walking, jogging, stair climbing, weight-lifting) |
| Stop using tobacco |
| Avoid heavy alcohol use |
| Implement fall precautions such as rubber-soled shoes when walking, handrails for staircases, and removing tripping hazards, including loose rugs |
| Source: Reference 1 |
CASE CONTINUED: High risk can be lowered
Ms. P’s family history, antidepressant use, smoking, and low dietary calcium intake associated with lactose intolerance increase her risk for osteoporosis. Her history of anorexia nervosa also increases her risk if she experiences amenorrhea. You advise her that she can ameliorate some of these factors by quitting smoking, exercising regularly, and taking calcium and vitamin D supplements. You refer her to her primary care physician because she wishes to undergo bone mineral density screening.
Related resources
- National Osteoporosis Foundation. Clinician‘s guide to prevention and treatment of osteoporosis. www.nof.org/professionals/Clinicians_Guide.htm.
- World Health Organization Fracture Risk Assessment Tool. Calculates a 10-year probability of hip fracture using demographic data, family history, comorbid medication and predisposing medical conditions. www.shef.ac.uk/FRAX.
Drug brand names
- Alendronate • Fosamax
- Carbamazepine • Tegretol
- Clonazepam • Klonopin
- Diazepam • Valium
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Oxcarbazepine • Trileptal
- Prednisone • Deltasone, Meticorten
- Raloxifene • Evista
- Risperidone • Risperdal
- Teriparatide • Forteo
- Valproate • Depakote
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Ms. P, age 44, is concerned about her risk of osteoporosis after her 70-year-old mother is hospitalized for a hip fracture. Ms. P has been taking fluoxetine, 40 mg/d, for 10 years to treat recurrent major depressive episodes that began at age 25. She was diagnosed with anorexia nervosa as a teenager, but recovered after 2 years of psychotherapy. She is lactose intolerant, has mild asthma that does not require steroids, and has no history of thyroid disease or bone fracture. Ms. P smokes 10 cigarettes a day but denies using alcohol or illicit drugs. She does not exercise, and her menses occur every 28 to 30 days.
Osteoporosis is a skeletal disease characterized by low bone mineralization and deteriorating bone architecture that results in increased susceptibility to fracture. Approximately 1 in 2 women and 1 in 5 men in the United States will have an osteoporosis-related fracture.1 Proximal femur and vertebral fractures are most common—1.5 million per year—but other bones may be involved.2
Osteoporosis-related fractures are associated with substantial morbidity and mortality. After a hip fracture, osteoporosis patients have a 10% to 20% risk of death within a year.3 Those who recover from hip fracture have a 2.5-fold increased risk of recurrent fracture and often struggle with chronic pain, disability, and loss of self-esteem and independence.1,3–5
Evidence links osteoporosis and depression
Research has shown that patients with major depression are at higher risk of osteoporosis.6 In one study, bone mineral density among 70 depressed outpatients was 15% lower than among age-matched controls.7 In a cross-sectional study, Michelson et al8 found that compared with nondepressed controls, women with current or past major depression had a lower mean bone mineral density—6.5% lower at the spine and 13.6% lower at the femoral neck.
Fewer prospective studies exist; however, most found depression has some impact on bone health. Whooley et al9 prospectively evaluated changes in bone mineral density among 7,414 Caucasian women age ≥65 for 6 years. Depressed women—those who scored ≥6 on the Geriatric Depression Scale—had a 40% higher risk of nonvertebral fracture after adjusting for history of fracture, weight, physical activity level, smoking, alcohol use, nutritional status, and cognitive function. The depressed cohort also had an increased risk of vertebral fracture. In a prospective study of 21,441 Norwegian female and male subjects, women who reported being depressed at 2 of 3 time points—from 1980 until 1995—had 2.5 times the risk of sustaining a nonvertebral fracture compared with those who did not report depression.10
Depressed women also have greater bone loss over time. Mean hip bone mineral density decreased by 0.69% per year in nondepressed women vs 0.96% in depressed women in a study of 4,177 women age ≥69.11 These findings were significant after adjusting for age, functional status, cognitive function, smoking, calcium intake, vitamin D supplement use, weight, antidepressant use, and bisphosphonate use. These findings have been replicated.12
Behavioral factors such as tobacco use and physical inactivity play a role in the risk of osteoporosis; however, emerging findings suggest a pathophysiologic link between depression and poor bone health. Depression is associated with lower estrogen and testosterone levels, which have been linked to decreased bone formation.6 Similarly, compared with matched controls, depressed women with low bone mineral density have higher urinary cortisol levels, suggesting that hypercortisolemia accelerates bone turnover.6,9,13 Finally, evidence suggests that depression is a pro-inflammatory state associated with production of numerous cytokines. Interleukin-6 and tumor necrosis factor-alpha, for example, inhibit osteoclast apoptosis and accelerate bone turnover.6
Fracture risk and psychotropics
Many psychotropic medications—including anticonvulsants, barbiturates, narcotics, and neuroleptics14–16—are associated with increased risk of falls, fractures, and osteoporosis. In this article we focus on selective serotonin reuptake inhibitors (SSRIs) and tricyclic antidepressants (TCAs) because little data is available on other antidepressants (Table 1).17–22
SSRIs are associated with increased fracture risk. In a cohort of 5,995 men age ≥65, Haney et al23 showed that men taking SSRIs have lower bone mineral density at the hip (3.9% lower) and spine (5.6% lower) compared with non-users after adjusting for age, weight, and race. Current SSRI use carries a greater risk than past use. In a prospective study of 7,983 men and women age ≥55, Ziere et al24 reported that risk of nonvertebral fracture among current SSRI users was 28% higher than among past users over a mean follow-up of 8.4 years. In the same study, the risk ratio of nonvertebral fracture was 2.10 for patients using SSRIs within the previous 6 months and 2.98 for use >6 months.
Table 1
Psychotropic medications associated with osteoporosis risk
| Medication/class | Odds ratio (95% confidence interval) |
|---|---|
| Selective serotonin reuptake inhibitors (SSRIs) | 1.45 (1.32 to 1.59) |
| Carbamazepine | 1.18 (1.10 to 1.26) |
| Non-SSRIs (eg, tricyclics, atypicals) | 1.15 (1.07 to 1.24) |
| Valproate | 1.15 (1.05 to 1.26) |
| Oxcarbazepine | 1.14 (1.03 to 1.26) |
| Benzodiazepines | 1.10 (1.04 to 1.16) |
| Lamotrigine | 1.04 (0.91 to 1.19) |
| Typical antipsychotics | 1.01 (0.86 to 1.19) |
| Atypical antipsychotics | 0.96 (0.79 to 1.17) |
| Lithium | 0.63 (0.43 to 0.93) |
| Source: References 17-22 | |
Increased fracture risk with SSRIs may be partially explained by the greater risk of osteoporosis in major depression.25 SSRI use has been linked to higher risk of fracture in the absence of depressive symptoms, however.26 Bolton et al17 revealed a trend of increasing fracture risk with higher SSRI dose. In this study, SSRI users had 45% greater likelihood of fracture than controls after adjusting for a diagnosis of depression.
Researchers are studying the mechanism by which SSRIs affect bone mineralization. Serotonin receptors—including 5-HT2A, 5-HT2B, and 5-HT2C—are present in bone.27 Preliminary investigations suggest SSRIs are concentrated in bone and impact fibroblast formation and osteoblast activity. High bone marrow concentrations of fluoxetine inhibit human osteoblast proliferation. Osteoblasts contribute to bone production.28 Fluoxetine concentrations in bone marrow can be up to 100-fold higher than serum levels, and the drug can be detected in bone up to 3 months after discontinuation.
TCAs. U.S. veterans with prior hip fracture are twice as likely to have received TCAs than age- and sex-matched controls.14In prospective studies, the risk of hip fracture among men and women age ≥65 is 50% higher in patients exposed to TCAs.30 Other investigations have revealed a dose-response relationship between TCA use and risk of fracture.31 A direct comparison of TCAs and SSRIs has found an equivalent increase in fracture risk in these 2 classes.30
A direct effect of TCAs on bone metabolism has not been elucidated. However, side effects of TCAs include orthostatic hypotension, impaired cognition, dizziness, and altered balance, all of which increase the risk of falls and fractures, particularly in elderly patients.31 Most studies of TCAs, however, do not account for depression’s role in fracture risk. Some patients in these studies may have received TCAs for disorders other than major depression, such as peripheral neuropathy or prophylaxis of migraine headaches.
Benzodiazepine use is associated with confusion, ataxia, and vertigo, which may increase the incidence of falls. Even low doses pose a risk. In one case-control study of 1,222 hip fracture patients age ≥65, use of >3 mg/d diazepam equivalents increased risk of hip fracture by 50% after adjusting for confounding factors.30 Although the data are mixed, benzodiazepines with shorter half-lives (eg, lorazepam) might not be safer than those with longer half-lives (eg, clonazepam).31,32
Other psychotropics. Some anticonvulsants may lead to bone demineralization via induction of the cytochrome P450 hepatic enzyme system, which accelerates conversion of vitamin D to an inactive metabolite that cannot adequately facilitate absorption of ingested calcium. The subsequent release of parathyroid hormone causes bone resorption.33 Patients taking anticonvulsants have nearly double the serum parathyroid hormone level of matched controls.34 Carbamazepine, oxcarbazepine, and valproate have been associated with increased risk of fracture.32 Although lamotrigine has not been widely studied, evidence suggests that its impact on bone metabolism is negligible.35
Many antipsychotics, including risperidone and haloperidol, have been associated with osteoporosis. The mechanism by which antipsychotics accelerate bone turnover has not been described; hyperprolactinemia likely plays a role.36
Screening and treatment
Effective pharmacotherapy for osteoporosis includes bisphosphonates (eg, alendronate), selective estrogen receptor modulators (eg, raloxifene), recombinant parathyroid hormone (eg, teriparatide), as well as calcium and vitamin D supplementation. Consider recommending bone density evaluation for depressed patients who have predisposing risk factors (Table 2)1 and those with long-term exposure to psychotropic agents. Dual energy X-ray absorptiometry is the preferred screening method. Refer patients whose results indicate osteopenia or osteoporosis to primary care. Although pharmacotherapy for osteoporosis should be managed by primary care practitioners, psychiatrists can serve an important role by promoting healthy lifestyle behaviors—such as regular exercise and adequate dietary vitamin D and calcium intake (Table 3).1
Table 2
Risk factors for osteoporosis-related fracture*
| Clinical factors |
| Age >50 Female sex Amenorrhea Cognitive impairment Family history of osteoporosis-related fracture Malnutrition Poor visual acuity Previous falls Low body mass index Glucocorticoid use (prednisone >5 mg/d for ≥3 months) |
| Secondary medical conditions |
| Hyperprolactinemia Anorexia nervosa Postmenopausal status Adrenal insufficiency Diabetes mellitus Hyperparathyroidism Celiac disease Inflammatory bowel disease Malabsorption syndromes Multiple myeloma End-stage renal disease |
| Behavioral factors |
| Low calcium intake Tobacco abuse Physical inactivity Excessive alcohol intake (>3 drinks per day) Vitamin D deficiency Immobilization |
| *Italics indicate conditions commonly encountered in psychiatric patients Source: Reference 1 |
Table 3
Reducing osteoporosis risk: Recommendations for patients age >50
| Assess dietary calcium (at least 1,200 mg/d) and dietary vitamin D intake (800 to 1,000 IU/d) |
| Exercise regularly, especially weight-bearing and muscle-strengthening activities (eg, walking, jogging, stair climbing, weight-lifting) |
| Stop using tobacco |
| Avoid heavy alcohol use |
| Implement fall precautions such as rubber-soled shoes when walking, handrails for staircases, and removing tripping hazards, including loose rugs |
| Source: Reference 1 |
CASE CONTINUED: High risk can be lowered
Ms. P’s family history, antidepressant use, smoking, and low dietary calcium intake associated with lactose intolerance increase her risk for osteoporosis. Her history of anorexia nervosa also increases her risk if she experiences amenorrhea. You advise her that she can ameliorate some of these factors by quitting smoking, exercising regularly, and taking calcium and vitamin D supplements. You refer her to her primary care physician because she wishes to undergo bone mineral density screening.
Related resources
- National Osteoporosis Foundation. Clinician‘s guide to prevention and treatment of osteoporosis. www.nof.org/professionals/Clinicians_Guide.htm.
- World Health Organization Fracture Risk Assessment Tool. Calculates a 10-year probability of hip fracture using demographic data, family history, comorbid medication and predisposing medical conditions. www.shef.ac.uk/FRAX.
Drug brand names
- Alendronate • Fosamax
- Carbamazepine • Tegretol
- Clonazepam • Klonopin
- Diazepam • Valium
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Oxcarbazepine • Trileptal
- Prednisone • Deltasone, Meticorten
- Raloxifene • Evista
- Risperidone • Risperdal
- Teriparatide • Forteo
- Valproate • Depakote
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC: National Osteoporosis Foundation; 2008.
2. Lane NE. Epidemiology, etiology, and diagnosis of osteoporosis. Amer J Obstet Gynecol. 2006;194(2 suppl):S3-11.
3. Sambrook P, Cooper C. Osteoporosis. Lancet. 2006;367:2010-2018.
4. Salaffi F, Cimmino MA, Malavolta N, et al. The burden of prevalent fractures on health-related quality of life in postmenopausal women with osteoporosis: the IMOF study. J Rheumatol. 2007;34(7):1551-1560.
5. Gold DT, Stegmaier K, Bales CW, et al. Psychosocial functioning and osteoporosis in late life: results of a multidisciplinary intervention. Journal of Women’s Health. 1993;2:149-155.
6. Mezuk B, Eaton WW, Golden SH. Depression and osteoporosis: epidemiology and potential mediating pathways. Osteoporos Int. 2008;19:1-12.
7. Schweiger U, Deuschle M, Korner A, et al. Low lumbar bone mineral density in patients with major depression. Am J Psychiatry. 1994;151:1691-1693.
8. Michelson D, Stratakis C, Hill L, et al. Bone mineral density in women with depression. N Engl J Med. 1996;335:1176-1181.
9. Whooley MA, Kip KE, Cauley JA, et al. Depression, falls, and risk of fracture in older women. Arch Intern Med. 1999;159:484-490.
10. Søgaard AJ, Joakimsen RM, Tverdal A, et al. Long-term mental distress, bone mineral density, and non-vertebral fractures. The Tromsø Study. Osteoporos Int. 2005;16(8):887-897.
11. Diem SJ, Blackwell TL, Stone KL, et al. Depressive symptoms and rates of bone loss at the hip in older women. J Am Geriatr Soc. 2007;55:824-831.
12. Eskandari F, Martinez PE, Torvik S, et al. Low bone mass in premenopausal women with depression. Arch Intern Med. 2007;167:2329-2336.
13. Yirmiya R, Goshen I, Bajayo A, et al. Depression induces bone loss through stimulation of the sympathetic nervous system. Proc Natl Acad Sci U S A. 2006;103(45):16876-16881.
14. French DD, Campbell R, Spehar A, et al. Outpatient medications and hip fractures in the US: a national veterans study. Drugs Aging. 2005;22(10):877-885.
15. Ensrud KE, Blackwell T, Mangione CM, et al. Central nervous system active medications and risk for fractures in older women. Arch Intern Med. 2003;163:949-957.
16. O’Keane V. Antipsychotic-induced hyperprolactinemia, hypogonadism, and osteoporosis in the treatment of schizophrenia. J Psychopharmacol. 2008;22(2 suppl):70-75.
17. Bolton JM, Metge C, Lix L, et al. Fracture risk from psychotropic medications: a population-based analysis. J Clin Psychopharmacol. 2008;28:384-391.
18. Heidrich FE, Stergachis A, Gross KM. Diuretic drug use and the risk for hip fracture. Ann Intern Med. 1991;115(1):1-6.
19. Yang YX, Lewis JD, Epstein S, et al. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA. 2006;296(24):2947-2953.
20. Schoofs MW, van der Klift M, Hofman A, et al. Thiazide diuretics and the risk for hip fracture. Ann Intern Med. 2003;139(6):476-482.
21. Schlienger RG, Kraenzlin ME, Jick SS, et al. Use of beta blockers and risk of fractures. JAMA. 2004;292(11):1326-1332.
22. Vestergaard P, Rejnmark L, Mosekilde L. Fracture risk associated with different types of oral corticosteroids and effect of termination of corticosteroids on risk of fracture. Calcif Tissue Int. 2008;82:249-257.
23. Haney EM, Chan BK, Diem SJ, et al. Association of low bone mineral density with selective serotonin reuptake inhibitor use by older men. Arch Intern Med. 2007;167:1246-1251.
24. Ziere G, Dieleman JP, van der Cammen TJ, et al. Selective serotonin reuptake inhibiting antidepressants are associated with an increased risk of nonvertebral fractures. J Clin Psychopharmacol. 2008;28:411-417.
25. Richards JB, Papaioannou A, Adachi JD, et al. Effect of selective serotonin reuptake inhibitors on the risk of fracture. Arch Intern Med. 2007;167:188-194.
26. Kerse N, Flicker L, Pfaff JJ, et al. Falls, depression, and antidepressants in later life: a large primary care appraisal. PLoS One. 2008;3(6):e2423.-
27. Westbroek I, van der Plas A, de Rooij KE, et al. Expression of serotonin receptors in bone. J Biol Chem. 2001;276(31):28961-28968.
28. Gustafsson BI, Thommesen L, Stunes AK, et al. Serotonin and fluoxetine modulate bone cell function in vitro. J Cell Biochem. 2006;98:139-151.
29. Bolo NR, Hodé Y, Macher JP. Long-term sequestration of fluorinated compounds in tissues after fluvoxamine or fluoxetine treatment: a fluorine magnetic resonance spectroscopy study in vivo. MAGMA. 2004;16:268-276.
30. Liu B, Anderson G, Mittmann N, et al. Use of selective serotonin-reuptake inhibitors or tricyclic antidepressants and risk of hip fractures in elderly people. Lancet. 1998;351:1303-1307.
31. Vestergaard P, Rejnmark L, Mosekilde L. Anxiolytics, sedatives, antidepressants, neuroleptics, and the risk of fracture. Osteoporos Int. 2006;17:807-816.
32. Wang PS, Bohn RL, Glynn RJ, et al. Hazardous benzodiazepine regimens in the elderly: effects of half-life, dosage, and duration on risk of hip fracture. Am J Psychiatry. 2001;158:892-898.
33. Pack AM. The association between antiepileptic drugs and bone disease. Epilepsy Curr. 2003;3(3):91-95.
34. Kim SH, Lee JW, Choi KG, et al. A 6-month longitudinal study of bone mineral density with antiepileptic drug monotherapy. Epilepsy Behav. 2007;10:291-295.
35. Vestergaard P, Rejnmark L, Mosekilde L. Fracture risk associated with use of antiepileptic drugs. Epilepsia. 2004;45(11):1330-1337.
36. Meaney AM, Smith S, Howes OD, et al. Effects of long-term prolactin-raising antipsychotic medication on bone mineral density in patients with schizophrenia. Br J Psychiatry. 2004;184:503-508.
1. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC: National Osteoporosis Foundation; 2008.
2. Lane NE. Epidemiology, etiology, and diagnosis of osteoporosis. Amer J Obstet Gynecol. 2006;194(2 suppl):S3-11.
3. Sambrook P, Cooper C. Osteoporosis. Lancet. 2006;367:2010-2018.
4. Salaffi F, Cimmino MA, Malavolta N, et al. The burden of prevalent fractures on health-related quality of life in postmenopausal women with osteoporosis: the IMOF study. J Rheumatol. 2007;34(7):1551-1560.
5. Gold DT, Stegmaier K, Bales CW, et al. Psychosocial functioning and osteoporosis in late life: results of a multidisciplinary intervention. Journal of Women’s Health. 1993;2:149-155.
6. Mezuk B, Eaton WW, Golden SH. Depression and osteoporosis: epidemiology and potential mediating pathways. Osteoporos Int. 2008;19:1-12.
7. Schweiger U, Deuschle M, Korner A, et al. Low lumbar bone mineral density in patients with major depression. Am J Psychiatry. 1994;151:1691-1693.
8. Michelson D, Stratakis C, Hill L, et al. Bone mineral density in women with depression. N Engl J Med. 1996;335:1176-1181.
9. Whooley MA, Kip KE, Cauley JA, et al. Depression, falls, and risk of fracture in older women. Arch Intern Med. 1999;159:484-490.
10. Søgaard AJ, Joakimsen RM, Tverdal A, et al. Long-term mental distress, bone mineral density, and non-vertebral fractures. The Tromsø Study. Osteoporos Int. 2005;16(8):887-897.
11. Diem SJ, Blackwell TL, Stone KL, et al. Depressive symptoms and rates of bone loss at the hip in older women. J Am Geriatr Soc. 2007;55:824-831.
12. Eskandari F, Martinez PE, Torvik S, et al. Low bone mass in premenopausal women with depression. Arch Intern Med. 2007;167:2329-2336.
13. Yirmiya R, Goshen I, Bajayo A, et al. Depression induces bone loss through stimulation of the sympathetic nervous system. Proc Natl Acad Sci U S A. 2006;103(45):16876-16881.
14. French DD, Campbell R, Spehar A, et al. Outpatient medications and hip fractures in the US: a national veterans study. Drugs Aging. 2005;22(10):877-885.
15. Ensrud KE, Blackwell T, Mangione CM, et al. Central nervous system active medications and risk for fractures in older women. Arch Intern Med. 2003;163:949-957.
16. O’Keane V. Antipsychotic-induced hyperprolactinemia, hypogonadism, and osteoporosis in the treatment of schizophrenia. J Psychopharmacol. 2008;22(2 suppl):70-75.
17. Bolton JM, Metge C, Lix L, et al. Fracture risk from psychotropic medications: a population-based analysis. J Clin Psychopharmacol. 2008;28:384-391.
18. Heidrich FE, Stergachis A, Gross KM. Diuretic drug use and the risk for hip fracture. Ann Intern Med. 1991;115(1):1-6.
19. Yang YX, Lewis JD, Epstein S, et al. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA. 2006;296(24):2947-2953.
20. Schoofs MW, van der Klift M, Hofman A, et al. Thiazide diuretics and the risk for hip fracture. Ann Intern Med. 2003;139(6):476-482.
21. Schlienger RG, Kraenzlin ME, Jick SS, et al. Use of beta blockers and risk of fractures. JAMA. 2004;292(11):1326-1332.
22. Vestergaard P, Rejnmark L, Mosekilde L. Fracture risk associated with different types of oral corticosteroids and effect of termination of corticosteroids on risk of fracture. Calcif Tissue Int. 2008;82:249-257.
23. Haney EM, Chan BK, Diem SJ, et al. Association of low bone mineral density with selective serotonin reuptake inhibitor use by older men. Arch Intern Med. 2007;167:1246-1251.
24. Ziere G, Dieleman JP, van der Cammen TJ, et al. Selective serotonin reuptake inhibiting antidepressants are associated with an increased risk of nonvertebral fractures. J Clin Psychopharmacol. 2008;28:411-417.
25. Richards JB, Papaioannou A, Adachi JD, et al. Effect of selective serotonin reuptake inhibitors on the risk of fracture. Arch Intern Med. 2007;167:188-194.
26. Kerse N, Flicker L, Pfaff JJ, et al. Falls, depression, and antidepressants in later life: a large primary care appraisal. PLoS One. 2008;3(6):e2423.-
27. Westbroek I, van der Plas A, de Rooij KE, et al. Expression of serotonin receptors in bone. J Biol Chem. 2001;276(31):28961-28968.
28. Gustafsson BI, Thommesen L, Stunes AK, et al. Serotonin and fluoxetine modulate bone cell function in vitro. J Cell Biochem. 2006;98:139-151.
29. Bolo NR, Hodé Y, Macher JP. Long-term sequestration of fluorinated compounds in tissues after fluvoxamine or fluoxetine treatment: a fluorine magnetic resonance spectroscopy study in vivo. MAGMA. 2004;16:268-276.
30. Liu B, Anderson G, Mittmann N, et al. Use of selective serotonin-reuptake inhibitors or tricyclic antidepressants and risk of hip fractures in elderly people. Lancet. 1998;351:1303-1307.
31. Vestergaard P, Rejnmark L, Mosekilde L. Anxiolytics, sedatives, antidepressants, neuroleptics, and the risk of fracture. Osteoporos Int. 2006;17:807-816.
32. Wang PS, Bohn RL, Glynn RJ, et al. Hazardous benzodiazepine regimens in the elderly: effects of half-life, dosage, and duration on risk of hip fracture. Am J Psychiatry. 2001;158:892-898.
33. Pack AM. The association between antiepileptic drugs and bone disease. Epilepsy Curr. 2003;3(3):91-95.
34. Kim SH, Lee JW, Choi KG, et al. A 6-month longitudinal study of bone mineral density with antiepileptic drug monotherapy. Epilepsy Behav. 2007;10:291-295.
35. Vestergaard P, Rejnmark L, Mosekilde L. Fracture risk associated with use of antiepileptic drugs. Epilepsia. 2004;45(11):1330-1337.
36. Meaney AM, Smith S, Howes OD, et al. Effects of long-term prolactin-raising antipsychotic medication on bone mineral density in patients with schizophrenia. Br J Psychiatry. 2004;184:503-508.