User login
NICE recommends bortezomib for untreated MM

Credit: Bill Branson
In a new draft guidance, the UK’s National Institute for Health and Care Excellence (NICE) has recommended bortezomib (Velcade) for certain patients with newly diagnosed multiple myeloma (MM).
NICE is recommending the drug in combination with dexamethasone, or with dexamethasone and thalidomide, as induction treatment for adults with previously untreated MM who are eligible for high-dose chemotherapy with hematopoietic stem cell transplant (HSCT).
An appraisal committee said these regimens are clinically effective for this patient population. Trial data suggest the regimens confer a “clear advantage” over standard therapy with respect to induction response.
And it’s plausible that this may translate to improved survival, the committee said. (Standard treatment in the UK is a combination of cyclophosphamide, thalidomide, and dexamethasone.)
“Clinical specialists told the committee that induction treatment with bortezomib would enable a greater number of patients to proceed to [HSCT] and, consequently, prevent the disease from progressing for longer,” said Sir Andrew Dillon, NICE Chief Executive.
In addition, the committee said the bortezomib regimens are cost-effective for this patient population. The cost of bortezomib is £762.38 per 3.5 mg vial.
On average, a course of treatment with bortezomib given with dexamethasone costs £12,260.91. And a course of bortezomib given with dexamethasone and thalidomide costs £24,840.10.
The cost for bortezomib, thalidomide, and dexamethasone compared to thalidomide and dexamethasone is likely to be below £30,000 per quality-adjusted life-year gained.
The same is true when comparing bortezomib and dexamethasone to cyclophosphamide, thalidomide, and dexamethasone as well as vincristine, doxorubicin, and dexamethasone.
This draft guidance is now with consultees, who have the opportunity to appeal against it.
Other NICE recommendations for MM
NICE already recommends bortezomib monotherapy as a treatment option for MM patients at first relapse who have received one prior therapy and who have undergone, or are unsuitable for, HSCT.
Thalidomide in combination with an alkylating agent and a corticosteroid is recommended as a first-line treatment option in MM patients for whom high-dose chemotherapy with HSCT is considered inappropriate.
Bortezomib is also recommended under these circumstances, if the patient is unable to tolerate or has contraindications to thalidomide.
Lenalidomide in combination with dexamethasone is recommended as a treatment option for people with MM who have received 2 or more prior therapies. ![]()
Credit: Bill Branson
In a new draft guidance, the UK’s National Institute for Health and Care Excellence (NICE) has recommended bortezomib (Velcade) for certain patients with newly diagnosed multiple myeloma (MM).
NICE is recommending the drug in combination with dexamethasone, or with dexamethasone and thalidomide, as induction treatment for adults with previously untreated MM who are eligible for high-dose chemotherapy with hematopoietic stem cell transplant (HSCT).
An appraisal committee said these regimens are clinically effective for this patient population. Trial data suggest the regimens confer a “clear advantage” over standard therapy with respect to induction response.
And it’s plausible that this may translate to improved survival, the committee said. (Standard treatment in the UK is a combination of cyclophosphamide, thalidomide, and dexamethasone.)
“Clinical specialists told the committee that induction treatment with bortezomib would enable a greater number of patients to proceed to [HSCT] and, consequently, prevent the disease from progressing for longer,” said Sir Andrew Dillon, NICE Chief Executive.
In addition, the committee said the bortezomib regimens are cost-effective for this patient population. The cost of bortezomib is £762.38 per 3.5 mg vial.
On average, a course of treatment with bortezomib given with dexamethasone costs £12,260.91. And a course of bortezomib given with dexamethasone and thalidomide costs £24,840.10.
The cost for bortezomib, thalidomide, and dexamethasone compared to thalidomide and dexamethasone is likely to be below £30,000 per quality-adjusted life-year gained.
The same is true when comparing bortezomib and dexamethasone to cyclophosphamide, thalidomide, and dexamethasone as well as vincristine, doxorubicin, and dexamethasone.
This draft guidance is now with consultees, who have the opportunity to appeal against it.
Other NICE recommendations for MM
NICE already recommends bortezomib monotherapy as a treatment option for MM patients at first relapse who have received one prior therapy and who have undergone, or are unsuitable for, HSCT.
Thalidomide in combination with an alkylating agent and a corticosteroid is recommended as a first-line treatment option in MM patients for whom high-dose chemotherapy with HSCT is considered inappropriate.
Bortezomib is also recommended under these circumstances, if the patient is unable to tolerate or has contraindications to thalidomide.
Lenalidomide in combination with dexamethasone is recommended as a treatment option for people with MM who have received 2 or more prior therapies. ![]()
Credit: Bill Branson
In a new draft guidance, the UK’s National Institute for Health and Care Excellence (NICE) has recommended bortezomib (Velcade) for certain patients with newly diagnosed multiple myeloma (MM).
NICE is recommending the drug in combination with dexamethasone, or with dexamethasone and thalidomide, as induction treatment for adults with previously untreated MM who are eligible for high-dose chemotherapy with hematopoietic stem cell transplant (HSCT).
An appraisal committee said these regimens are clinically effective for this patient population. Trial data suggest the regimens confer a “clear advantage” over standard therapy with respect to induction response.
And it’s plausible that this may translate to improved survival, the committee said. (Standard treatment in the UK is a combination of cyclophosphamide, thalidomide, and dexamethasone.)
“Clinical specialists told the committee that induction treatment with bortezomib would enable a greater number of patients to proceed to [HSCT] and, consequently, prevent the disease from progressing for longer,” said Sir Andrew Dillon, NICE Chief Executive.
In addition, the committee said the bortezomib regimens are cost-effective for this patient population. The cost of bortezomib is £762.38 per 3.5 mg vial.
On average, a course of treatment with bortezomib given with dexamethasone costs £12,260.91. And a course of bortezomib given with dexamethasone and thalidomide costs £24,840.10.
The cost for bortezomib, thalidomide, and dexamethasone compared to thalidomide and dexamethasone is likely to be below £30,000 per quality-adjusted life-year gained.
The same is true when comparing bortezomib and dexamethasone to cyclophosphamide, thalidomide, and dexamethasone as well as vincristine, doxorubicin, and dexamethasone.
This draft guidance is now with consultees, who have the opportunity to appeal against it.
Other NICE recommendations for MM
NICE already recommends bortezomib monotherapy as a treatment option for MM patients at first relapse who have received one prior therapy and who have undergone, or are unsuitable for, HSCT.
Thalidomide in combination with an alkylating agent and a corticosteroid is recommended as a first-line treatment option in MM patients for whom high-dose chemotherapy with HSCT is considered inappropriate.
Bortezomib is also recommended under these circumstances, if the patient is unable to tolerate or has contraindications to thalidomide.
Lenalidomide in combination with dexamethasone is recommended as a treatment option for people with MM who have received 2 or more prior therapies. ![]()
NICE wants more info on lenalidomide in MM

Credit: CDC
The UK’s National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending against the use of lenalidomide (Revlimid) in multiple myeloma (MM) patients who have previously received bortezomib.
Based on current information, NICE has said it cannot recommend the drug for patients who have received bortezomib once and are unable to receive thalidomide or undergo hematopoietic stem cell transplant.
NICE’s previous recommendation regarding lenalidomide in MM has not changed. The drug will still be available through the National Health Service (NHS) for MM patients who have received 2 or more prior therapies.
“We are now looking specifically at how well lenalidomide works after someone has received bortezomib, and whether it provides value for money,” said Sir Andrew Dillon, NICE Chief Executive.
“However, from the information provided by the manufacturer, it was unclear if lenalidomide was as effective as re-treatment with bortezomib, and the manufacturer’s own economic model showed that the drug would not be cost effective at this stage.”
“Because of this, we are unable to recommend the drug in preliminary recommendations. We hope that the manufacturer, Celgene, will look again at their submission.”
NICE also pointed out that, since the organization recommended lenalidomide for MM in 2009, there have been no studies comparing lenalidomide to other treatments in these patients. Celgene has only provided data comparing lenalidomide to placebo.
In addition, for the 2009 guidance, Celgene submitted a patient access scheme, where they bear the costs of the drug beyond 26 cycles (normally for a period of 2 years). And this enabled NICE to recommend the drug. But Celgene has not submitted a patient access scheme for the current appraisal.
NICE considered all the cost effectiveness models Celgene submitted to be fundamentally flawed.
NICE concluded that the most plausible costs per quality-adjusted life-year for lenalidomide compared with bortezomib or standard chemotherapies were more than £30,000, whether bortezomib re-treatment was appropriate or not.
Lenalidomide is available as a 21-capsule pack. The cost per pack varies according to capsule size: £3570 (5 mg), £3780 (10 mg), £3969 (15 mg), and £4368 (25 mg). The recommended starting dose is 25 mg orally, once daily on days 1-21 of repeated 28-day cycles.
The draft guidance is open for public comment until April 4. Until a final guidance is issued, NHS bodies should make decisions locally on the funding of specific treatments. Once NICE issues its final guidance, it replaces local recommendations. ![]()
Credit: CDC
The UK’s National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending against the use of lenalidomide (Revlimid) in multiple myeloma (MM) patients who have previously received bortezomib.
Based on current information, NICE has said it cannot recommend the drug for patients who have received bortezomib once and are unable to receive thalidomide or undergo hematopoietic stem cell transplant.
NICE’s previous recommendation regarding lenalidomide in MM has not changed. The drug will still be available through the National Health Service (NHS) for MM patients who have received 2 or more prior therapies.
“We are now looking specifically at how well lenalidomide works after someone has received bortezomib, and whether it provides value for money,” said Sir Andrew Dillon, NICE Chief Executive.
“However, from the information provided by the manufacturer, it was unclear if lenalidomide was as effective as re-treatment with bortezomib, and the manufacturer’s own economic model showed that the drug would not be cost effective at this stage.”
“Because of this, we are unable to recommend the drug in preliminary recommendations. We hope that the manufacturer, Celgene, will look again at their submission.”
NICE also pointed out that, since the organization recommended lenalidomide for MM in 2009, there have been no studies comparing lenalidomide to other treatments in these patients. Celgene has only provided data comparing lenalidomide to placebo.
In addition, for the 2009 guidance, Celgene submitted a patient access scheme, where they bear the costs of the drug beyond 26 cycles (normally for a period of 2 years). And this enabled NICE to recommend the drug. But Celgene has not submitted a patient access scheme for the current appraisal.
NICE considered all the cost effectiveness models Celgene submitted to be fundamentally flawed.
NICE concluded that the most plausible costs per quality-adjusted life-year for lenalidomide compared with bortezomib or standard chemotherapies were more than £30,000, whether bortezomib re-treatment was appropriate or not.
Lenalidomide is available as a 21-capsule pack. The cost per pack varies according to capsule size: £3570 (5 mg), £3780 (10 mg), £3969 (15 mg), and £4368 (25 mg). The recommended starting dose is 25 mg orally, once daily on days 1-21 of repeated 28-day cycles.
The draft guidance is open for public comment until April 4. Until a final guidance is issued, NHS bodies should make decisions locally on the funding of specific treatments. Once NICE issues its final guidance, it replaces local recommendations. ![]()
Credit: CDC
The UK’s National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending against the use of lenalidomide (Revlimid) in multiple myeloma (MM) patients who have previously received bortezomib.
Based on current information, NICE has said it cannot recommend the drug for patients who have received bortezomib once and are unable to receive thalidomide or undergo hematopoietic stem cell transplant.
NICE’s previous recommendation regarding lenalidomide in MM has not changed. The drug will still be available through the National Health Service (NHS) for MM patients who have received 2 or more prior therapies.
“We are now looking specifically at how well lenalidomide works after someone has received bortezomib, and whether it provides value for money,” said Sir Andrew Dillon, NICE Chief Executive.
“However, from the information provided by the manufacturer, it was unclear if lenalidomide was as effective as re-treatment with bortezomib, and the manufacturer’s own economic model showed that the drug would not be cost effective at this stage.”
“Because of this, we are unable to recommend the drug in preliminary recommendations. We hope that the manufacturer, Celgene, will look again at their submission.”
NICE also pointed out that, since the organization recommended lenalidomide for MM in 2009, there have been no studies comparing lenalidomide to other treatments in these patients. Celgene has only provided data comparing lenalidomide to placebo.
In addition, for the 2009 guidance, Celgene submitted a patient access scheme, where they bear the costs of the drug beyond 26 cycles (normally for a period of 2 years). And this enabled NICE to recommend the drug. But Celgene has not submitted a patient access scheme for the current appraisal.
NICE considered all the cost effectiveness models Celgene submitted to be fundamentally flawed.
NICE concluded that the most plausible costs per quality-adjusted life-year for lenalidomide compared with bortezomib or standard chemotherapies were more than £30,000, whether bortezomib re-treatment was appropriate or not.
Lenalidomide is available as a 21-capsule pack. The cost per pack varies according to capsule size: £3570 (5 mg), £3780 (10 mg), £3969 (15 mg), and £4368 (25 mg). The recommended starting dose is 25 mg orally, once daily on days 1-21 of repeated 28-day cycles.
The draft guidance is open for public comment until April 4. Until a final guidance is issued, NHS bodies should make decisions locally on the funding of specific treatments. Once NICE issues its final guidance, it replaces local recommendations. ![]()
FDA approves drug for infantile hemangioma
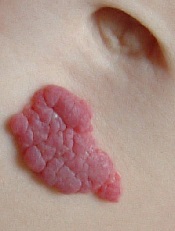
infant’s stomach
The US Food and Drug Administration has approved oral propranolol hydrochloride (Hemangeol) to treat proliferating infantile hemangiomas that require systemic therapy.
The drug, which is also under review in the European Union, will be available in the US in June.
Infantile hemangioma is the most common vascular benign tumor of infancy, affecting 3% to 10% of newborns. The lesions are rarely detectable at birth and start growing noticeably in the first 4 to 6 weeks of life.
While most infantile hemangiomas do not require treatment, approximately 12% do. Depending upon their location, infantile hemangiomas might impair breathing, eating, or vision, or become life-threatening.
Propranolol has long been used in cardiology, but its use in infantile hemangiomas is relatively new. In 2007, Christine Léauté-Labreze, MD, a dermatologist at the Bordeaux University Hospital in France, discovered that propranolol could treat infantile hemangiomas.
Since then, the drug has been used for this indication off-label. And in 2009, Pierre Fabre Dermatologie began developing propranolol hydrochloride for use in infantile hemangiomas.
In a study of 32 children, propranolol slowed the growth of infantile hemangiomas in 100% of patients. Patients had received propranolol at 2 to 3 mg/kg per day for a median of 6.1 months. Side effects were “limited and mild,” according to researchers (V Sans et al. Pediatrics 2009).
Researchers also conducted a randomized, controlled trial of the drug in infants 5 weeks to 5 months old at therapy initiation. The team compared 4 propranolol treatment protocols (1 or 3 mg/kg/day for 3 or 6 months) to placebo.
Propranolol at a daily dose of 3 mg/kg for 6 months had a 60.4% success rate, compared to 3.6% in the placebo group (P<0.0001). Success was defined as complete or nearly complete resolution of the target hemangioma. However, 11.4% of patients needed to be re-treated after stopping propranolol.
The drug is contraindicated in premature infants who have a corrected age of less than 5 weeks, weight less than 2 kg, known hypersensitivity to propranolol or any of its excipients, asthma or a history of bronchospasm, pheochromocytoma, blood pressure less than 50/30 mmHg, a heart rate less than 80 beats per minute, greater than first degree heart block, or decompensated heart failure.
Propranolol hydrochloride can cause serious side effects, including hypoglycemia, bradycardia, hypotension, and bronchospasm. The drug can worsen congestive heart failure and may increase the risk of stroke in children with PHACE syndrome.
The most frequently reported adverse reactions, occurring in more than 10% of infants receiving propranolol hydrochloride, were sleep disorders, diarrhea, vomiting, and aggravated respiratory tract infections, such as bronchitis and bronchiolitis associated with cough and fever. Adverse reactions led to treatment discontinuation in fewer than 2% of treated patients. ![]()
infant’s stomach
The US Food and Drug Administration has approved oral propranolol hydrochloride (Hemangeol) to treat proliferating infantile hemangiomas that require systemic therapy.
The drug, which is also under review in the European Union, will be available in the US in June.
Infantile hemangioma is the most common vascular benign tumor of infancy, affecting 3% to 10% of newborns. The lesions are rarely detectable at birth and start growing noticeably in the first 4 to 6 weeks of life.
While most infantile hemangiomas do not require treatment, approximately 12% do. Depending upon their location, infantile hemangiomas might impair breathing, eating, or vision, or become life-threatening.
Propranolol has long been used in cardiology, but its use in infantile hemangiomas is relatively new. In 2007, Christine Léauté-Labreze, MD, a dermatologist at the Bordeaux University Hospital in France, discovered that propranolol could treat infantile hemangiomas.
Since then, the drug has been used for this indication off-label. And in 2009, Pierre Fabre Dermatologie began developing propranolol hydrochloride for use in infantile hemangiomas.
In a study of 32 children, propranolol slowed the growth of infantile hemangiomas in 100% of patients. Patients had received propranolol at 2 to 3 mg/kg per day for a median of 6.1 months. Side effects were “limited and mild,” according to researchers (V Sans et al. Pediatrics 2009).
Researchers also conducted a randomized, controlled trial of the drug in infants 5 weeks to 5 months old at therapy initiation. The team compared 4 propranolol treatment protocols (1 or 3 mg/kg/day for 3 or 6 months) to placebo.
Propranolol at a daily dose of 3 mg/kg for 6 months had a 60.4% success rate, compared to 3.6% in the placebo group (P<0.0001). Success was defined as complete or nearly complete resolution of the target hemangioma. However, 11.4% of patients needed to be re-treated after stopping propranolol.
The drug is contraindicated in premature infants who have a corrected age of less than 5 weeks, weight less than 2 kg, known hypersensitivity to propranolol or any of its excipients, asthma or a history of bronchospasm, pheochromocytoma, blood pressure less than 50/30 mmHg, a heart rate less than 80 beats per minute, greater than first degree heart block, or decompensated heart failure.
Propranolol hydrochloride can cause serious side effects, including hypoglycemia, bradycardia, hypotension, and bronchospasm. The drug can worsen congestive heart failure and may increase the risk of stroke in children with PHACE syndrome.
The most frequently reported adverse reactions, occurring in more than 10% of infants receiving propranolol hydrochloride, were sleep disorders, diarrhea, vomiting, and aggravated respiratory tract infections, such as bronchitis and bronchiolitis associated with cough and fever. Adverse reactions led to treatment discontinuation in fewer than 2% of treated patients. ![]()
infant’s stomach
The US Food and Drug Administration has approved oral propranolol hydrochloride (Hemangeol) to treat proliferating infantile hemangiomas that require systemic therapy.
The drug, which is also under review in the European Union, will be available in the US in June.
Infantile hemangioma is the most common vascular benign tumor of infancy, affecting 3% to 10% of newborns. The lesions are rarely detectable at birth and start growing noticeably in the first 4 to 6 weeks of life.
While most infantile hemangiomas do not require treatment, approximately 12% do. Depending upon their location, infantile hemangiomas might impair breathing, eating, or vision, or become life-threatening.
Propranolol has long been used in cardiology, but its use in infantile hemangiomas is relatively new. In 2007, Christine Léauté-Labreze, MD, a dermatologist at the Bordeaux University Hospital in France, discovered that propranolol could treat infantile hemangiomas.
Since then, the drug has been used for this indication off-label. And in 2009, Pierre Fabre Dermatologie began developing propranolol hydrochloride for use in infantile hemangiomas.
In a study of 32 children, propranolol slowed the growth of infantile hemangiomas in 100% of patients. Patients had received propranolol at 2 to 3 mg/kg per day for a median of 6.1 months. Side effects were “limited and mild,” according to researchers (V Sans et al. Pediatrics 2009).
Researchers also conducted a randomized, controlled trial of the drug in infants 5 weeks to 5 months old at therapy initiation. The team compared 4 propranolol treatment protocols (1 or 3 mg/kg/day for 3 or 6 months) to placebo.
Propranolol at a daily dose of 3 mg/kg for 6 months had a 60.4% success rate, compared to 3.6% in the placebo group (P<0.0001). Success was defined as complete or nearly complete resolution of the target hemangioma. However, 11.4% of patients needed to be re-treated after stopping propranolol.
The drug is contraindicated in premature infants who have a corrected age of less than 5 weeks, weight less than 2 kg, known hypersensitivity to propranolol or any of its excipients, asthma or a history of bronchospasm, pheochromocytoma, blood pressure less than 50/30 mmHg, a heart rate less than 80 beats per minute, greater than first degree heart block, or decompensated heart failure.
Propranolol hydrochloride can cause serious side effects, including hypoglycemia, bradycardia, hypotension, and bronchospasm. The drug can worsen congestive heart failure and may increase the risk of stroke in children with PHACE syndrome.
The most frequently reported adverse reactions, occurring in more than 10% of infants receiving propranolol hydrochloride, were sleep disorders, diarrhea, vomiting, and aggravated respiratory tract infections, such as bronchitis and bronchiolitis associated with cough and fever. Adverse reactions led to treatment discontinuation in fewer than 2% of treated patients. ![]()
FDA approves IV formulation of antifungal agent
The US Food and Drug Administration has approved an intravenous formulation of posaconazole (Noxafil), which is expected to be available at wholesalers in mid-April.
The antifungal agent is already available as delayed-release tablets and in an oral suspension formulation.
In any formulation, posaconazole is indicated for prophylaxis of invasive Aspergillus and Candida infections in immunocompromised patients who are at high risk of developing these infections.
This includes patients who have developed graft-vs-host disease after hematopoietic stem cell transplant and patients with hematologic malignancies who have prolonged neutropenia resulting from chemotherapy.
Posaconazole injection is indicated for use in patients 18 years of age and older. The delayed-release tablets and oral suspension are indicated for patients 13 years of age and older.
Posaconazole injection is administered with a loading dose of 300 mg (one 300 mg vial) twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by slow intravenous infusion over approximately 90 minutes.
Once combined with a mixture of intravenous solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole injection should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2-8 degrees C (36-46 degrees F).
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
In clinical trials, the adverse reactions reported for posaconazole injection were generally similar to those reported in trials of posaconazole oral suspension. The most frequently reported adverse reactions with an onset during the posaconazole intravenous phase of dosing 300 mg once-daily therapy were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
Patients who are allergic to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given along with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
For more details, see the complete prescribing information. Posaconazole is marketed as Noxafil by Merck. ![]()
The US Food and Drug Administration has approved an intravenous formulation of posaconazole (Noxafil), which is expected to be available at wholesalers in mid-April.
The antifungal agent is already available as delayed-release tablets and in an oral suspension formulation.
In any formulation, posaconazole is indicated for prophylaxis of invasive Aspergillus and Candida infections in immunocompromised patients who are at high risk of developing these infections.
This includes patients who have developed graft-vs-host disease after hematopoietic stem cell transplant and patients with hematologic malignancies who have prolonged neutropenia resulting from chemotherapy.
Posaconazole injection is indicated for use in patients 18 years of age and older. The delayed-release tablets and oral suspension are indicated for patients 13 years of age and older.
Posaconazole injection is administered with a loading dose of 300 mg (one 300 mg vial) twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by slow intravenous infusion over approximately 90 minutes.
Once combined with a mixture of intravenous solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole injection should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2-8 degrees C (36-46 degrees F).
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
In clinical trials, the adverse reactions reported for posaconazole injection were generally similar to those reported in trials of posaconazole oral suspension. The most frequently reported adverse reactions with an onset during the posaconazole intravenous phase of dosing 300 mg once-daily therapy were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
Patients who are allergic to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given along with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
For more details, see the complete prescribing information. Posaconazole is marketed as Noxafil by Merck. ![]()
The US Food and Drug Administration has approved an intravenous formulation of posaconazole (Noxafil), which is expected to be available at wholesalers in mid-April.
The antifungal agent is already available as delayed-release tablets and in an oral suspension formulation.
In any formulation, posaconazole is indicated for prophylaxis of invasive Aspergillus and Candida infections in immunocompromised patients who are at high risk of developing these infections.
This includes patients who have developed graft-vs-host disease after hematopoietic stem cell transplant and patients with hematologic malignancies who have prolonged neutropenia resulting from chemotherapy.
Posaconazole injection is indicated for use in patients 18 years of age and older. The delayed-release tablets and oral suspension are indicated for patients 13 years of age and older.
Posaconazole injection is administered with a loading dose of 300 mg (one 300 mg vial) twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by slow intravenous infusion over approximately 90 minutes.
Once combined with a mixture of intravenous solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole injection should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2-8 degrees C (36-46 degrees F).
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
In clinical trials, the adverse reactions reported for posaconazole injection were generally similar to those reported in trials of posaconazole oral suspension. The most frequently reported adverse reactions with an onset during the posaconazole intravenous phase of dosing 300 mg once-daily therapy were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
Patients who are allergic to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given along with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
For more details, see the complete prescribing information. Posaconazole is marketed as Noxafil by Merck. ![]()
High cost of eculizumab needs explaining, NICE says
Credit: Bill Branson
The UK’s National Institute for Health and Care Excellence (NICE) has asked the manufacturer of eculizumab (Soliris) to explain the high cost of the drug.
Research has suggested that eculizumab can be effective against atypical hemolytic uremic syndrome (aHUS), a rare disease that often proves difficult to treat.
So the National Health Service (NHS) has made eculizumab available for these patients on an interim basis, pending NICE appraisal.
However, an advisory committee for NICE has estimated that routine use of eculizumab would cost the NHS about £58 million in the first year, and costs would exceed £80 million in 5 years.
Therefore, in its draft guidance for eculizumab, the committee has asked the drug’s manufacturer, Alexion Pharma, to explain its costs.
“[The committee has] asked for clarification from the company on aspects of the manufacturing, research, and development costs of a medicinal product for the treatment of a very rare condition,” said Sir Andrew Dillon, Chief Executive at NICE.
“It has also asked NHS England for clarification on treatment costs for a highly specialized technology in the context of a highly specialized service. The information provided will be considered at the next meeting of the evaluation committee in April.”
The committee will also consider comments on its draft guidance at the meeting. The guidance is available for public comment until midday on March 25.
About aHUS
Estimated to affect more than 200 people in England, aHUS is a chronic condition that causes severe inflammation of blood vessels and thrombus formation in small blood vessels throughout the body.
Patients with aHUS can experience significant kidney impairment, thrombosis, heart failure, and brain injury. In about 70% of patients, aHUS is associated with an underlying genetic or acquired abnormality of proteins in the complement immune system.
Before eculizumab became available, plasma therapy (infusion and/or exchange) was the main treatment for aHUS. However, not all patients with aHUS respond to plasma therapy. And up to 40% of patients may die or progress to end-stage renal failure and require dialysis with the first clinical aHUS manifestation, despite the use of plasma therapy.
Some patients may be eligible for a kidney or combined kidney-liver transplantation. However, there is a high risk of organ rejection following recurrent disease.
Eculizumab in aHUS: Treatment and cost
Eculizumab inhibits the disease process by blocking pro-thrombotic and pro-inflammatory processes that can lead to cellular damage in small blood vessels throughout the body, renal failure, and damage to other organs.
Eculizumab is given intravenously in adults as initial treatment at a dose of 900 mg for 4 weeks, then as maintenance treatment at a dose of 1200 mg on week 5 and then every 12 to 16 days. The summary of product characteristics for eculizumab states that treatment should be continued for the patient’s lifetime, unless discontinuation is clinically indicated.
Eculizumab costs £3150 per 30 mL vial, excluding tax, according to the British National Formulary.
“Alexion insisted that its information about the overall cost of eculizumab be kept confidential, and so NICE is unable to share these details of the Alexion submission with stakeholders,” Dillon said.
However, to allow consultees and commentators to properly engage in the consultation process, NICE has prepared an estimate of the possible budget impact eculizumab might have, using information available in the public domain.
This is based on a treatment cost of £340,200 per adult patient in the first year (based on the acquisition cost of the drug and the recommended dosing for an adult), and assumes a patient cohort of 170, as estimated by NHS England in its interim commissioning policy.
Assuming all of these patients receive eculizumab, the budget impact for the first year would be £57.8 million. If an additional 20 new patients are treated the following year (based on a worldwide incidence of 0.4 million), the budget impact will rise to £62.5 million. That is assuming all new patients are treated and all existing patients continue to be treated at the maintenance cost of £327,600 per year.
Using the same assumptions, the budget impact will rise to £69 million in year 3 (190 existing and 20 new patients), £75 million in year 4 (210 existing and 20 new patients) and £82 million in year 5 (230 existing and 20 new patients). ![]()
Credit: Bill Branson
The UK’s National Institute for Health and Care Excellence (NICE) has asked the manufacturer of eculizumab (Soliris) to explain the high cost of the drug.
Research has suggested that eculizumab can be effective against atypical hemolytic uremic syndrome (aHUS), a rare disease that often proves difficult to treat.
So the National Health Service (NHS) has made eculizumab available for these patients on an interim basis, pending NICE appraisal.
However, an advisory committee for NICE has estimated that routine use of eculizumab would cost the NHS about £58 million in the first year, and costs would exceed £80 million in 5 years.
Therefore, in its draft guidance for eculizumab, the committee has asked the drug’s manufacturer, Alexion Pharma, to explain its costs.
“[The committee has] asked for clarification from the company on aspects of the manufacturing, research, and development costs of a medicinal product for the treatment of a very rare condition,” said Sir Andrew Dillon, Chief Executive at NICE.
“It has also asked NHS England for clarification on treatment costs for a highly specialized technology in the context of a highly specialized service. The information provided will be considered at the next meeting of the evaluation committee in April.”
The committee will also consider comments on its draft guidance at the meeting. The guidance is available for public comment until midday on March 25.
About aHUS
Estimated to affect more than 200 people in England, aHUS is a chronic condition that causes severe inflammation of blood vessels and thrombus formation in small blood vessels throughout the body.
Patients with aHUS can experience significant kidney impairment, thrombosis, heart failure, and brain injury. In about 70% of patients, aHUS is associated with an underlying genetic or acquired abnormality of proteins in the complement immune system.
Before eculizumab became available, plasma therapy (infusion and/or exchange) was the main treatment for aHUS. However, not all patients with aHUS respond to plasma therapy. And up to 40% of patients may die or progress to end-stage renal failure and require dialysis with the first clinical aHUS manifestation, despite the use of plasma therapy.
Some patients may be eligible for a kidney or combined kidney-liver transplantation. However, there is a high risk of organ rejection following recurrent disease.
Eculizumab in aHUS: Treatment and cost
Eculizumab inhibits the disease process by blocking pro-thrombotic and pro-inflammatory processes that can lead to cellular damage in small blood vessels throughout the body, renal failure, and damage to other organs.
Eculizumab is given intravenously in adults as initial treatment at a dose of 900 mg for 4 weeks, then as maintenance treatment at a dose of 1200 mg on week 5 and then every 12 to 16 days. The summary of product characteristics for eculizumab states that treatment should be continued for the patient’s lifetime, unless discontinuation is clinically indicated.
Eculizumab costs £3150 per 30 mL vial, excluding tax, according to the British National Formulary.
“Alexion insisted that its information about the overall cost of eculizumab be kept confidential, and so NICE is unable to share these details of the Alexion submission with stakeholders,” Dillon said.
However, to allow consultees and commentators to properly engage in the consultation process, NICE has prepared an estimate of the possible budget impact eculizumab might have, using information available in the public domain.
This is based on a treatment cost of £340,200 per adult patient in the first year (based on the acquisition cost of the drug and the recommended dosing for an adult), and assumes a patient cohort of 170, as estimated by NHS England in its interim commissioning policy.
Assuming all of these patients receive eculizumab, the budget impact for the first year would be £57.8 million. If an additional 20 new patients are treated the following year (based on a worldwide incidence of 0.4 million), the budget impact will rise to £62.5 million. That is assuming all new patients are treated and all existing patients continue to be treated at the maintenance cost of £327,600 per year.
Using the same assumptions, the budget impact will rise to £69 million in year 3 (190 existing and 20 new patients), £75 million in year 4 (210 existing and 20 new patients) and £82 million in year 5 (230 existing and 20 new patients). ![]()
Credit: Bill Branson
The UK’s National Institute for Health and Care Excellence (NICE) has asked the manufacturer of eculizumab (Soliris) to explain the high cost of the drug.
Research has suggested that eculizumab can be effective against atypical hemolytic uremic syndrome (aHUS), a rare disease that often proves difficult to treat.
So the National Health Service (NHS) has made eculizumab available for these patients on an interim basis, pending NICE appraisal.
However, an advisory committee for NICE has estimated that routine use of eculizumab would cost the NHS about £58 million in the first year, and costs would exceed £80 million in 5 years.
Therefore, in its draft guidance for eculizumab, the committee has asked the drug’s manufacturer, Alexion Pharma, to explain its costs.
“[The committee has] asked for clarification from the company on aspects of the manufacturing, research, and development costs of a medicinal product for the treatment of a very rare condition,” said Sir Andrew Dillon, Chief Executive at NICE.
“It has also asked NHS England for clarification on treatment costs for a highly specialized technology in the context of a highly specialized service. The information provided will be considered at the next meeting of the evaluation committee in April.”
The committee will also consider comments on its draft guidance at the meeting. The guidance is available for public comment until midday on March 25.
About aHUS
Estimated to affect more than 200 people in England, aHUS is a chronic condition that causes severe inflammation of blood vessels and thrombus formation in small blood vessels throughout the body.
Patients with aHUS can experience significant kidney impairment, thrombosis, heart failure, and brain injury. In about 70% of patients, aHUS is associated with an underlying genetic or acquired abnormality of proteins in the complement immune system.
Before eculizumab became available, plasma therapy (infusion and/or exchange) was the main treatment for aHUS. However, not all patients with aHUS respond to plasma therapy. And up to 40% of patients may die or progress to end-stage renal failure and require dialysis with the first clinical aHUS manifestation, despite the use of plasma therapy.
Some patients may be eligible for a kidney or combined kidney-liver transplantation. However, there is a high risk of organ rejection following recurrent disease.
Eculizumab in aHUS: Treatment and cost
Eculizumab inhibits the disease process by blocking pro-thrombotic and pro-inflammatory processes that can lead to cellular damage in small blood vessels throughout the body, renal failure, and damage to other organs.
Eculizumab is given intravenously in adults as initial treatment at a dose of 900 mg for 4 weeks, then as maintenance treatment at a dose of 1200 mg on week 5 and then every 12 to 16 days. The summary of product characteristics for eculizumab states that treatment should be continued for the patient’s lifetime, unless discontinuation is clinically indicated.
Eculizumab costs £3150 per 30 mL vial, excluding tax, according to the British National Formulary.
“Alexion insisted that its information about the overall cost of eculizumab be kept confidential, and so NICE is unable to share these details of the Alexion submission with stakeholders,” Dillon said.
However, to allow consultees and commentators to properly engage in the consultation process, NICE has prepared an estimate of the possible budget impact eculizumab might have, using information available in the public domain.
This is based on a treatment cost of £340,200 per adult patient in the first year (based on the acquisition cost of the drug and the recommended dosing for an adult), and assumes a patient cohort of 170, as estimated by NHS England in its interim commissioning policy.
Assuming all of these patients receive eculizumab, the budget impact for the first year would be £57.8 million. If an additional 20 new patients are treated the following year (based on a worldwide incidence of 0.4 million), the budget impact will rise to £62.5 million. That is assuming all new patients are treated and all existing patients continue to be treated at the maintenance cost of £327,600 per year.
Using the same assumptions, the budget impact will rise to £69 million in year 3 (190 existing and 20 new patients), £75 million in year 4 (210 existing and 20 new patients) and £82 million in year 5 (230 existing and 20 new patients). ![]()
FDA approves apixaban to prevent DVT, PE
Credit: CDC
The US Food and Drug Administration (FDA) has approved apixaban (Eliquis) as prophylaxis for deep vein thrombosis (DVT), which may lead to pulmonary embolism (PE), in patients who have undergone hip or knee replacement surgery.
Apixaban is already FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation.
The latest approval is supported by data from 3 trials comprising the ADVANCE clinical trial program.
Results of the first ADVANCE study suggested apixaban was roughly as effective as enoxaparin at preventing DVT and PE in patients who had undergone total knee replacement surgery. But apixaban posed a significantly lower risk of major and nonmajor bleeding.
The ADVANCE-2 study, on the other hand, indicated that apixaban was a more effective means of thromboprophylaxis than enoxaparin in this patient population. And there was no significant difference between the treatment arms in the frequency of major or clinically relevant bleeding.
The ADVANCE-3 study suggested apixaban was more effective than enoxaparin in preventing DVT and PE among patients undergoing hip replacement. And there was no significant difference between the groups with regard to major or clinically relevant bleeding.
The prescribing information for apixaban includes a boxed warning detailing the increased risk of stroke in patients with nonvalvular atrial fibrillation who discontinue the drug without adequate continuous anticoagulation.
The boxed warning also states that, in patients undergoing spinal epidural anesthesia or spinal puncture, apixaban poses an increased risk of epidural or spinal hematoma, which may cause long-term or permanent paralysis.
The risk of these events may be increased by the use of indwelling epidural catheters for the administration of analgesia or by the concomitant use of drugs affecting hemostasis, such as nonsteroidal anti-inflammatory drugs, platelet aggregation inhibitors, or other anticoagulants. The risk also appears to be increased by traumatic or repeated epidural or spinal puncture.
Healthcare professionals should monitor patients for signs and symptoms of neurologic impairment. If neurologic compromise is noted, urgent treatment is necessary.
For more information on adverse events and contraindications, see the full prescribing information for apixaban. The drug is under joint development by Pfizer and Bristol-Myers Squibb. ![]()
Credit: CDC
The US Food and Drug Administration (FDA) has approved apixaban (Eliquis) as prophylaxis for deep vein thrombosis (DVT), which may lead to pulmonary embolism (PE), in patients who have undergone hip or knee replacement surgery.
Apixaban is already FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation.
The latest approval is supported by data from 3 trials comprising the ADVANCE clinical trial program.
Results of the first ADVANCE study suggested apixaban was roughly as effective as enoxaparin at preventing DVT and PE in patients who had undergone total knee replacement surgery. But apixaban posed a significantly lower risk of major and nonmajor bleeding.
The ADVANCE-2 study, on the other hand, indicated that apixaban was a more effective means of thromboprophylaxis than enoxaparin in this patient population. And there was no significant difference between the treatment arms in the frequency of major or clinically relevant bleeding.
The ADVANCE-3 study suggested apixaban was more effective than enoxaparin in preventing DVT and PE among patients undergoing hip replacement. And there was no significant difference between the groups with regard to major or clinically relevant bleeding.
The prescribing information for apixaban includes a boxed warning detailing the increased risk of stroke in patients with nonvalvular atrial fibrillation who discontinue the drug without adequate continuous anticoagulation.
The boxed warning also states that, in patients undergoing spinal epidural anesthesia or spinal puncture, apixaban poses an increased risk of epidural or spinal hematoma, which may cause long-term or permanent paralysis.
The risk of these events may be increased by the use of indwelling epidural catheters for the administration of analgesia or by the concomitant use of drugs affecting hemostasis, such as nonsteroidal anti-inflammatory drugs, platelet aggregation inhibitors, or other anticoagulants. The risk also appears to be increased by traumatic or repeated epidural or spinal puncture.
Healthcare professionals should monitor patients for signs and symptoms of neurologic impairment. If neurologic compromise is noted, urgent treatment is necessary.
For more information on adverse events and contraindications, see the full prescribing information for apixaban. The drug is under joint development by Pfizer and Bristol-Myers Squibb. ![]()
Credit: CDC
The US Food and Drug Administration (FDA) has approved apixaban (Eliquis) as prophylaxis for deep vein thrombosis (DVT), which may lead to pulmonary embolism (PE), in patients who have undergone hip or knee replacement surgery.
Apixaban is already FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation.
The latest approval is supported by data from 3 trials comprising the ADVANCE clinical trial program.
Results of the first ADVANCE study suggested apixaban was roughly as effective as enoxaparin at preventing DVT and PE in patients who had undergone total knee replacement surgery. But apixaban posed a significantly lower risk of major and nonmajor bleeding.
The ADVANCE-2 study, on the other hand, indicated that apixaban was a more effective means of thromboprophylaxis than enoxaparin in this patient population. And there was no significant difference between the treatment arms in the frequency of major or clinically relevant bleeding.
The ADVANCE-3 study suggested apixaban was more effective than enoxaparin in preventing DVT and PE among patients undergoing hip replacement. And there was no significant difference between the groups with regard to major or clinically relevant bleeding.
The prescribing information for apixaban includes a boxed warning detailing the increased risk of stroke in patients with nonvalvular atrial fibrillation who discontinue the drug without adequate continuous anticoagulation.
The boxed warning also states that, in patients undergoing spinal epidural anesthesia or spinal puncture, apixaban poses an increased risk of epidural or spinal hematoma, which may cause long-term or permanent paralysis.
The risk of these events may be increased by the use of indwelling epidural catheters for the administration of analgesia or by the concomitant use of drugs affecting hemostasis, such as nonsteroidal anti-inflammatory drugs, platelet aggregation inhibitors, or other anticoagulants. The risk also appears to be increased by traumatic or repeated epidural or spinal puncture.
Healthcare professionals should monitor patients for signs and symptoms of neurologic impairment. If neurologic compromise is noted, urgent treatment is necessary.
For more information on adverse events and contraindications, see the full prescribing information for apixaban. The drug is under joint development by Pfizer and Bristol-Myers Squibb. ![]()
FDA places imetelstat trials on hold
The US Food and Drug Administration (FDA) has issued a full clinical hold for the telomerase inhibitor imetelstat, citing concerns that long-term exposure to the drug may pose a risk of chronic liver injury.
The hold temporarily suspends all ongoing clinical trials of imetelstat sponsored by the drug’s maker, Geron Corporation.
It’s possible that other studies of imetelstat, such as ongoing investigator-sponsored trials, may be placed on hold as well.
At present, the hold affects the remaining 8 patients enrolled on Geron’s phase 2 study of imetelstat in essential thrombocythemia and polycythemia vera.
It also affects the remaining 2 patients in the company’s phase 2 study of the drug in previously treated multiple myeloma. And Geron believes its planned phase 2 trial of imetelstat in myelofibrosis will likely be delayed as well.
The FDA has not yet issued a written notice of the hold but has given Geron verbal notice.
The agency indicated that the hold is due to persistent low-grade liver function test abnormalities observed in the study of imetelstat in patients with essential thrombocythemia or polycythemia vera and the potential risk of chronic liver injury following long-term exposure to imetelstat.
The FDA expressed concerns about whether these abnormalities are reversible. Geron said it plans to work with the FDA to put an end to the hold. ![]()
The US Food and Drug Administration (FDA) has issued a full clinical hold for the telomerase inhibitor imetelstat, citing concerns that long-term exposure to the drug may pose a risk of chronic liver injury.
The hold temporarily suspends all ongoing clinical trials of imetelstat sponsored by the drug’s maker, Geron Corporation.
It’s possible that other studies of imetelstat, such as ongoing investigator-sponsored trials, may be placed on hold as well.
At present, the hold affects the remaining 8 patients enrolled on Geron’s phase 2 study of imetelstat in essential thrombocythemia and polycythemia vera.
It also affects the remaining 2 patients in the company’s phase 2 study of the drug in previously treated multiple myeloma. And Geron believes its planned phase 2 trial of imetelstat in myelofibrosis will likely be delayed as well.
The FDA has not yet issued a written notice of the hold but has given Geron verbal notice.
The agency indicated that the hold is due to persistent low-grade liver function test abnormalities observed in the study of imetelstat in patients with essential thrombocythemia or polycythemia vera and the potential risk of chronic liver injury following long-term exposure to imetelstat.
The FDA expressed concerns about whether these abnormalities are reversible. Geron said it plans to work with the FDA to put an end to the hold. ![]()
The US Food and Drug Administration (FDA) has issued a full clinical hold for the telomerase inhibitor imetelstat, citing concerns that long-term exposure to the drug may pose a risk of chronic liver injury.
The hold temporarily suspends all ongoing clinical trials of imetelstat sponsored by the drug’s maker, Geron Corporation.
It’s possible that other studies of imetelstat, such as ongoing investigator-sponsored trials, may be placed on hold as well.
At present, the hold affects the remaining 8 patients enrolled on Geron’s phase 2 study of imetelstat in essential thrombocythemia and polycythemia vera.
It also affects the remaining 2 patients in the company’s phase 2 study of the drug in previously treated multiple myeloma. And Geron believes its planned phase 2 trial of imetelstat in myelofibrosis will likely be delayed as well.
The FDA has not yet issued a written notice of the hold but has given Geron verbal notice.
The agency indicated that the hold is due to persistent low-grade liver function test abnormalities observed in the study of imetelstat in patients with essential thrombocythemia or polycythemia vera and the potential risk of chronic liver injury following long-term exposure to imetelstat.
The FDA expressed concerns about whether these abnormalities are reversible. Geron said it plans to work with the FDA to put an end to the hold.
England’s Cancer Drugs Fund raises concerns

Credit: Rhoda Baer
Cancer patients in England are more likely to receive prescriptions for expensive drugs than patients in Wales, according to a study published in the British Journal of Cancer.
The research suggests this disparity is associated with the Cancer Drugs Fund (CDF), money set aside by the English government to pay for drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the National Health Service (NHS).
The governments of Wales, Scotland, and Northern Ireland do not have access to the CDF or have similar programs of their own.
“There’s been much debate surrounding the Cancer Drugs Fund,” said study author Charlotte Chamberlain, MBBS, of the University of Bristol in the UK.
“The vast majority of Cancer Drugs Fund drugs do not cure the cancer but may extend life or improve symptoms in some people. The high cost of these drugs means that the NHS cannot afford other treatments, and, therefore, critics argue that public money is being spent inefficiently.”
To assess the impact of the CDF, Dr Chamberlain and her colleagues analyzed data from hospital pharmacies in England and Wales from August 2007 to December 2012. (The CDF was established in 2010, and the researchers wanted to capture data from before and after its introduction.)
The team evaluated 15 drugs that represent different categories of NICE approval—recommended, not recommended, and not yet appraised.
The results showed that, after the CDF was established, drugs recommended by NICE were not prescribed any more in England than in Wales.
However, drugs that were rejected by NICE because they were not cost-effective were prescribed up to 7 times more often in England than in Wales. For example, in the year before the CDF was introduced, prescription rates of imatinib (which is not recommended by NICE) were substantially higher in England than in Wales.
Immediately before the introduction of the CDF, following the first NICE rejection, imatinib prescribing declined in both countries. But it declined more slowly in England than in Wales, despite 2 additional NICE rejections. Regression analysis showed evidence of an association between the CDF and increased prescribing in England compared to Wales (P<0.001).
The research also revealed surprising information regarding the 3 most recently launched drugs—bendamustine, pazopanib, and abiraterone, which were awaiting NICE appraisal when the CDF was established but have since been approved.
These drugs were prescribed less often in England than in Wales. For instance, prescription rates of bendamustine were 25% lower in England.
This finding suggests that physicians in England have been slower to adopt newer drugs that are cost-effective, the researchers said.
“Our research has highlighted that the CDF has created an inequality between cancer sufferers in England and those in Wales,” Dr Chamberlain noted. “This raises ethical, moral, financial, and policy concerns.”
Credit: Rhoda Baer
Cancer patients in England are more likely to receive prescriptions for expensive drugs than patients in Wales, according to a study published in the British Journal of Cancer.
The research suggests this disparity is associated with the Cancer Drugs Fund (CDF), money set aside by the English government to pay for drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the National Health Service (NHS).
The governments of Wales, Scotland, and Northern Ireland do not have access to the CDF or have similar programs of their own.
“There’s been much debate surrounding the Cancer Drugs Fund,” said study author Charlotte Chamberlain, MBBS, of the University of Bristol in the UK.
“The vast majority of Cancer Drugs Fund drugs do not cure the cancer but may extend life or improve symptoms in some people. The high cost of these drugs means that the NHS cannot afford other treatments, and, therefore, critics argue that public money is being spent inefficiently.”
To assess the impact of the CDF, Dr Chamberlain and her colleagues analyzed data from hospital pharmacies in England and Wales from August 2007 to December 2012. (The CDF was established in 2010, and the researchers wanted to capture data from before and after its introduction.)
The team evaluated 15 drugs that represent different categories of NICE approval—recommended, not recommended, and not yet appraised.
The results showed that, after the CDF was established, drugs recommended by NICE were not prescribed any more in England than in Wales.
However, drugs that were rejected by NICE because they were not cost-effective were prescribed up to 7 times more often in England than in Wales. For example, in the year before the CDF was introduced, prescription rates of imatinib (which is not recommended by NICE) were substantially higher in England than in Wales.
Immediately before the introduction of the CDF, following the first NICE rejection, imatinib prescribing declined in both countries. But it declined more slowly in England than in Wales, despite 2 additional NICE rejections. Regression analysis showed evidence of an association between the CDF and increased prescribing in England compared to Wales (P<0.001).
The research also revealed surprising information regarding the 3 most recently launched drugs—bendamustine, pazopanib, and abiraterone, which were awaiting NICE appraisal when the CDF was established but have since been approved.
These drugs were prescribed less often in England than in Wales. For instance, prescription rates of bendamustine were 25% lower in England.
This finding suggests that physicians in England have been slower to adopt newer drugs that are cost-effective, the researchers said.
“Our research has highlighted that the CDF has created an inequality between cancer sufferers in England and those in Wales,” Dr Chamberlain noted. “This raises ethical, moral, financial, and policy concerns.”
Credit: Rhoda Baer
Cancer patients in England are more likely to receive prescriptions for expensive drugs than patients in Wales, according to a study published in the British Journal of Cancer.
The research suggests this disparity is associated with the Cancer Drugs Fund (CDF), money set aside by the English government to pay for drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the National Health Service (NHS).
The governments of Wales, Scotland, and Northern Ireland do not have access to the CDF or have similar programs of their own.
“There’s been much debate surrounding the Cancer Drugs Fund,” said study author Charlotte Chamberlain, MBBS, of the University of Bristol in the UK.
“The vast majority of Cancer Drugs Fund drugs do not cure the cancer but may extend life or improve symptoms in some people. The high cost of these drugs means that the NHS cannot afford other treatments, and, therefore, critics argue that public money is being spent inefficiently.”
To assess the impact of the CDF, Dr Chamberlain and her colleagues analyzed data from hospital pharmacies in England and Wales from August 2007 to December 2012. (The CDF was established in 2010, and the researchers wanted to capture data from before and after its introduction.)
The team evaluated 15 drugs that represent different categories of NICE approval—recommended, not recommended, and not yet appraised.
The results showed that, after the CDF was established, drugs recommended by NICE were not prescribed any more in England than in Wales.
However, drugs that were rejected by NICE because they were not cost-effective were prescribed up to 7 times more often in England than in Wales. For example, in the year before the CDF was introduced, prescription rates of imatinib (which is not recommended by NICE) were substantially higher in England than in Wales.
Immediately before the introduction of the CDF, following the first NICE rejection, imatinib prescribing declined in both countries. But it declined more slowly in England than in Wales, despite 2 additional NICE rejections. Regression analysis showed evidence of an association between the CDF and increased prescribing in England compared to Wales (P<0.001).
The research also revealed surprising information regarding the 3 most recently launched drugs—bendamustine, pazopanib, and abiraterone, which were awaiting NICE appraisal when the CDF was established but have since been approved.
These drugs were prescribed less often in England than in Wales. For instance, prescription rates of bendamustine were 25% lower in England.
This finding suggests that physicians in England have been slower to adopt newer drugs that are cost-effective, the researchers said.
“Our research has highlighted that the CDF has created an inequality between cancer sufferers in England and those in Wales,” Dr Chamberlain noted. “This raises ethical, moral, financial, and policy concerns.”
Drugs get orphan designation for AML, MM

Credit: FDA
The US Food and Drug Administration (FDA) has granted orphan designation for the histone deacetylase (HDAC) inhibitor pracinostat to treat acute myeloid leukemia (AML) and the proteasome inhibitor marizomib to treat multiple myeloma (MM).
Orphan designation is available for drugs that treat or prevent rare diseases affecting fewer than 200,000 people in the US.
The designation qualifies the sponsor of a drug for development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions, and 7-year marketing exclusivity upon FDA approval.
About pracinostat
The oral HDAC inhibitor pracinostat has been tested in phase 1 and 2 trials of adult and pediatric patients with advanced hematologic disorders and solid tumors.
The drug has been generally well tolerated in more than 200 patients to date, according to the drug’s maker, MEI Pharma.
In a dose-escalation phase 1 trial, pracinostat demonstrated single-agent activity in elderly AML patients. Two of 14 patients (14%) achieved a complete remission, with responses persisting more than 206 days and 362 days.
Researchers are currently conducting a phase 2 trial of pracinostat in combination with azacitidine in elderly patients with newly diagnosed AML. Preliminary data from this trial are expected to be available by December 2014.
About marizomib
The proteasome inhibitor marizomib is under development for the treatment of MM and other malignancies.
Intravenous marizomib has been evaluated in more than 230 patients across 4 phase 1/2 studies, as a single agent or in combination with dexamethasone or an HDAC inhibitor.
Researchers are currently evaluating marizomib in combination with dexamethasone in an ongoing phase 2 trial of highly refractory MM patients, including those who are refractory to carfilzomib.
Marizomib is also being tested in combination with pomalidomide and dexamethasone in a phase 1/2 study of patients with relapsed and refractory MM.
The drug’s maker, Triphase Accelerator Corporation, is currently developing an oral formulation of marizomib.
Credit: FDA
The US Food and Drug Administration (FDA) has granted orphan designation for the histone deacetylase (HDAC) inhibitor pracinostat to treat acute myeloid leukemia (AML) and the proteasome inhibitor marizomib to treat multiple myeloma (MM).
Orphan designation is available for drugs that treat or prevent rare diseases affecting fewer than 200,000 people in the US.
The designation qualifies the sponsor of a drug for development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions, and 7-year marketing exclusivity upon FDA approval.
About pracinostat
The oral HDAC inhibitor pracinostat has been tested in phase 1 and 2 trials of adult and pediatric patients with advanced hematologic disorders and solid tumors.
The drug has been generally well tolerated in more than 200 patients to date, according to the drug’s maker, MEI Pharma.
In a dose-escalation phase 1 trial, pracinostat demonstrated single-agent activity in elderly AML patients. Two of 14 patients (14%) achieved a complete remission, with responses persisting more than 206 days and 362 days.
Researchers are currently conducting a phase 2 trial of pracinostat in combination with azacitidine in elderly patients with newly diagnosed AML. Preliminary data from this trial are expected to be available by December 2014.
About marizomib
The proteasome inhibitor marizomib is under development for the treatment of MM and other malignancies.
Intravenous marizomib has been evaluated in more than 230 patients across 4 phase 1/2 studies, as a single agent or in combination with dexamethasone or an HDAC inhibitor.
Researchers are currently evaluating marizomib in combination with dexamethasone in an ongoing phase 2 trial of highly refractory MM patients, including those who are refractory to carfilzomib.
Marizomib is also being tested in combination with pomalidomide and dexamethasone in a phase 1/2 study of patients with relapsed and refractory MM.
The drug’s maker, Triphase Accelerator Corporation, is currently developing an oral formulation of marizomib.
Credit: FDA
The US Food and Drug Administration (FDA) has granted orphan designation for the histone deacetylase (HDAC) inhibitor pracinostat to treat acute myeloid leukemia (AML) and the proteasome inhibitor marizomib to treat multiple myeloma (MM).
Orphan designation is available for drugs that treat or prevent rare diseases affecting fewer than 200,000 people in the US.
The designation qualifies the sponsor of a drug for development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions, and 7-year marketing exclusivity upon FDA approval.
About pracinostat
The oral HDAC inhibitor pracinostat has been tested in phase 1 and 2 trials of adult and pediatric patients with advanced hematologic disorders and solid tumors.
The drug has been generally well tolerated in more than 200 patients to date, according to the drug’s maker, MEI Pharma.
In a dose-escalation phase 1 trial, pracinostat demonstrated single-agent activity in elderly AML patients. Two of 14 patients (14%) achieved a complete remission, with responses persisting more than 206 days and 362 days.
Researchers are currently conducting a phase 2 trial of pracinostat in combination with azacitidine in elderly patients with newly diagnosed AML. Preliminary data from this trial are expected to be available by December 2014.
About marizomib
The proteasome inhibitor marizomib is under development for the treatment of MM and other malignancies.
Intravenous marizomib has been evaluated in more than 230 patients across 4 phase 1/2 studies, as a single agent or in combination with dexamethasone or an HDAC inhibitor.
Researchers are currently evaluating marizomib in combination with dexamethasone in an ongoing phase 2 trial of highly refractory MM patients, including those who are refractory to carfilzomib.
Marizomib is also being tested in combination with pomalidomide and dexamethasone in a phase 1/2 study of patients with relapsed and refractory MM.
The drug’s maker, Triphase Accelerator Corporation, is currently developing an oral formulation of marizomib.
Researchers create reversible LMWH
Scientists say they’ve created a synthetic form of low-molecular-weight heparin (LMWH) that is both reversible and safe for patients with poor kidney function.
In the event of uncontrolled bleeding, this synthetic heparin can be reversed by an existing drug.
And the LMWH is cleared by the liver rather than the kidneys.
The team described their creation of the drug in Nature Chemical Biology.
“When doctors talk to me about the kind of heparin they want to use during and after surgery, they want it reversible, and they want it to not go through the kidneys,” said study author Jian Liu, PhD, of the University of North Carolina, Chapel Hill.
Dr Liu noted that up to 5% of patients receiving heparin experience some form of uncontrolled bleeding. Patients receiving unfractionated heparin are in less danger because there is an existing FDA-approved antidote available, protamine.
But protamine is not as effective in reversing LMWH. So Dr Liu and his colleagues tweaked the drug’s molecular structure so that protamine is able to deactivate LMWH.
The team used a chemo-enzymatic process to synthesize the LMWH, an approach they developed in research on a simpler anticoagulant published in Science in 2011. Synthesizing the LMWH allowed them to make improvements on the animal-derived form of the drug.
That form of LMWH is cleared from the body by the kidneys, which can make it unsuitable for patients with a weakened renal system. So the researchers made changes that allowed their LMWH to bind to receptors that clear it through the liver.
“If a person’s kidneys aren’t effectively clearing heparin from the blood, the drug stays active in the body for longer than expected,” said study author Nigel Key, MB ChB, also of the University of North Carolina.
“That can represent a potentially dangerous situation for the physician, pharmacist, and patient.”
LMWH did prove dangerous in 2008, when more than 80 people died and hundreds of others suffered adverse reactions to the drug. Authorities linked the problems to a contaminant in raw natural heparin from China.
“Whenever you mix the food chain and the drug chain together, you end up with potential for disaster,” said study author Robert Linhardt, PhD, of the Rensselaer Polytechnic Institute in Troy, New York.
“Whether it comes from contamination, adulteration, impurities like viruses or prions—any of those possibilities are much more likely when you make something in an uncontrolled environment. This is a drug that millions of people rely upon, and it’s important to develop a safe, synthetic alternative to the current supply chain.”
LMWH makes up more than half the US market for heparin. The researchers said the new version they created is a safe, economically viable alternative to the existing animal-derived supply.
“The pig stuff has served us well for 50 years and is very inexpensive, but if we cannot control the supply chain, we cannot ensure the safety of the drug,” Dr Liu said. “I am working for the day when synthetic heparin can be brewed in large laboratories at a low cost.”
Scientists say they’ve created a synthetic form of low-molecular-weight heparin (LMWH) that is both reversible and safe for patients with poor kidney function.
In the event of uncontrolled bleeding, this synthetic heparin can be reversed by an existing drug.
And the LMWH is cleared by the liver rather than the kidneys.
The team described their creation of the drug in Nature Chemical Biology.
“When doctors talk to me about the kind of heparin they want to use during and after surgery, they want it reversible, and they want it to not go through the kidneys,” said study author Jian Liu, PhD, of the University of North Carolina, Chapel Hill.
Dr Liu noted that up to 5% of patients receiving heparin experience some form of uncontrolled bleeding. Patients receiving unfractionated heparin are in less danger because there is an existing FDA-approved antidote available, protamine.
But protamine is not as effective in reversing LMWH. So Dr Liu and his colleagues tweaked the drug’s molecular structure so that protamine is able to deactivate LMWH.
The team used a chemo-enzymatic process to synthesize the LMWH, an approach they developed in research on a simpler anticoagulant published in Science in 2011. Synthesizing the LMWH allowed them to make improvements on the animal-derived form of the drug.
That form of LMWH is cleared from the body by the kidneys, which can make it unsuitable for patients with a weakened renal system. So the researchers made changes that allowed their LMWH to bind to receptors that clear it through the liver.
“If a person’s kidneys aren’t effectively clearing heparin from the blood, the drug stays active in the body for longer than expected,” said study author Nigel Key, MB ChB, also of the University of North Carolina.
“That can represent a potentially dangerous situation for the physician, pharmacist, and patient.”
LMWH did prove dangerous in 2008, when more than 80 people died and hundreds of others suffered adverse reactions to the drug. Authorities linked the problems to a contaminant in raw natural heparin from China.
“Whenever you mix the food chain and the drug chain together, you end up with potential for disaster,” said study author Robert Linhardt, PhD, of the Rensselaer Polytechnic Institute in Troy, New York.
“Whether it comes from contamination, adulteration, impurities like viruses or prions—any of those possibilities are much more likely when you make something in an uncontrolled environment. This is a drug that millions of people rely upon, and it’s important to develop a safe, synthetic alternative to the current supply chain.”
LMWH makes up more than half the US market for heparin. The researchers said the new version they created is a safe, economically viable alternative to the existing animal-derived supply.
“The pig stuff has served us well for 50 years and is very inexpensive, but if we cannot control the supply chain, we cannot ensure the safety of the drug,” Dr Liu said. “I am working for the day when synthetic heparin can be brewed in large laboratories at a low cost.”
Scientists say they’ve created a synthetic form of low-molecular-weight heparin (LMWH) that is both reversible and safe for patients with poor kidney function.
In the event of uncontrolled bleeding, this synthetic heparin can be reversed by an existing drug.
And the LMWH is cleared by the liver rather than the kidneys.
The team described their creation of the drug in Nature Chemical Biology.
“When doctors talk to me about the kind of heparin they want to use during and after surgery, they want it reversible, and they want it to not go through the kidneys,” said study author Jian Liu, PhD, of the University of North Carolina, Chapel Hill.
Dr Liu noted that up to 5% of patients receiving heparin experience some form of uncontrolled bleeding. Patients receiving unfractionated heparin are in less danger because there is an existing FDA-approved antidote available, protamine.
But protamine is not as effective in reversing LMWH. So Dr Liu and his colleagues tweaked the drug’s molecular structure so that protamine is able to deactivate LMWH.
The team used a chemo-enzymatic process to synthesize the LMWH, an approach they developed in research on a simpler anticoagulant published in Science in 2011. Synthesizing the LMWH allowed them to make improvements on the animal-derived form of the drug.
That form of LMWH is cleared from the body by the kidneys, which can make it unsuitable for patients with a weakened renal system. So the researchers made changes that allowed their LMWH to bind to receptors that clear it through the liver.
“If a person’s kidneys aren’t effectively clearing heparin from the blood, the drug stays active in the body for longer than expected,” said study author Nigel Key, MB ChB, also of the University of North Carolina.
“That can represent a potentially dangerous situation for the physician, pharmacist, and patient.”
LMWH did prove dangerous in 2008, when more than 80 people died and hundreds of others suffered adverse reactions to the drug. Authorities linked the problems to a contaminant in raw natural heparin from China.
“Whenever you mix the food chain and the drug chain together, you end up with potential for disaster,” said study author Robert Linhardt, PhD, of the Rensselaer Polytechnic Institute in Troy, New York.
“Whether it comes from contamination, adulteration, impurities like viruses or prions—any of those possibilities are much more likely when you make something in an uncontrolled environment. This is a drug that millions of people rely upon, and it’s important to develop a safe, synthetic alternative to the current supply chain.”
LMWH makes up more than half the US market for heparin. The researchers said the new version they created is a safe, economically viable alternative to the existing animal-derived supply.
“The pig stuff has served us well for 50 years and is very inexpensive, but if we cannot control the supply chain, we cannot ensure the safety of the drug,” Dr Liu said. “I am working for the day when synthetic heparin can be brewed in large laboratories at a low cost.”