User login
Genetics, smoking, and inflammation on MRI predict progression in axial SpA
Progression to structural damage occurs slowly among patients with early axial spondyloarthritis and more often takes place in current smokers, HLAB27 carriers, and those with signs of inflammation on MRI, according to new observations from the French DESIR cohort study.
The findings help to establish risk factors for early progression of nonradiographic axial spondyloarthritis (nr-axSpA) to radiographic disease, said Dr. Maxime Dougados of the department of rheumatology at Cochin Hospital, Paris, and his colleagues (Arthritis Rheumatol. 2016 Mar 18. doi: 10.1002/art.39666).

The study involved 449 patients with recent-onset inflammatory back pain who were participating in the multicenter, longitudinal DESIR study and had x-rays available at baseline and 2-year follow-up.
A total of 16 (4.9%) out of 326 patients who did not fulfill the modified New York criteria (mNY) at baseline progressed from nonradiographic to radiographic axSpA over the 2-year study period.
Of the 123 patients who met the mNY criteria at baseline, 7 (5.7%) no longer met the criteria at follow-up.
Independent factors found to influence the odds of progressing to radiographic axSpA were smoking status (odds ratio, 3.3; 95% confidence interval, 1.0-11.5), HLAB27 positivity (OR, 12.6; 95% CI, 2.3-274), and the presence of inflammation at MRI in the sacroiliac joints (SIJs) at baseline (OR, 48.8; 95% CI, 9.3-904), a multivariate analysis showed.
However, when the authors defined radiographic progression as “a worsening of at least one grade in at least one SIJ,” HLAB27 positivity (OR, 1.68; 95% CI, 0.8-3.71, P = .02), MRI positivity (OR, 5.85; 95% CI, 2.38-15.37, P less than .001), and baseline structural damage at SIJs on pelvic x-rays (OR, 6.35; 95% CI, 2.14-18.88; P less than .001) were predictors of radiographic disease progression. Another analysis confirmed those as risk factors even when the definition of progression also required a final absolute grade of at least 2 in the worsened SIJs.
When the authors considered the change in total score (based on the mean of two readers), only HLAB27 positivity and baseline MRI positivity were highlighted as independent predictors of disease progression. In this analysis, HLAB27 positivity and inflammation on MRI had grading score increases of 0.41 (P = .014) and 1.03 (P less than .001), respectively.
Unlike other studies, the researchers did not see a clear relationship between abnormal C-reactive protein at baseline and radiographic progression.
The findings suggest that, in early SpA, “structural progression does exist but is quite small and observed in a small number of patients,” the study authors wrote.
The authors noted that their findings should be “interpreted with caution” because they were based on a small number of patients, and additional studies with a longer-follow-up period are needed to confirm the findings.
The DESIR cohort was sponsored by the Department de la recherché Clinique et du Development de l’Assistance Publique-Hopitaux de Paris. An unrestricted grant from Pfizer was allocated for the 10 years of follow-up.
Progression to structural damage occurs slowly among patients with early axial spondyloarthritis and more often takes place in current smokers, HLAB27 carriers, and those with signs of inflammation on MRI, according to new observations from the French DESIR cohort study.
The findings help to establish risk factors for early progression of nonradiographic axial spondyloarthritis (nr-axSpA) to radiographic disease, said Dr. Maxime Dougados of the department of rheumatology at Cochin Hospital, Paris, and his colleagues (Arthritis Rheumatol. 2016 Mar 18. doi: 10.1002/art.39666).
The study involved 449 patients with recent-onset inflammatory back pain who were participating in the multicenter, longitudinal DESIR study and had x-rays available at baseline and 2-year follow-up.
A total of 16 (4.9%) out of 326 patients who did not fulfill the modified New York criteria (mNY) at baseline progressed from nonradiographic to radiographic axSpA over the 2-year study period.
Of the 123 patients who met the mNY criteria at baseline, 7 (5.7%) no longer met the criteria at follow-up.
Independent factors found to influence the odds of progressing to radiographic axSpA were smoking status (odds ratio, 3.3; 95% confidence interval, 1.0-11.5), HLAB27 positivity (OR, 12.6; 95% CI, 2.3-274), and the presence of inflammation at MRI in the sacroiliac joints (SIJs) at baseline (OR, 48.8; 95% CI, 9.3-904), a multivariate analysis showed.
However, when the authors defined radiographic progression as “a worsening of at least one grade in at least one SIJ,” HLAB27 positivity (OR, 1.68; 95% CI, 0.8-3.71, P = .02), MRI positivity (OR, 5.85; 95% CI, 2.38-15.37, P less than .001), and baseline structural damage at SIJs on pelvic x-rays (OR, 6.35; 95% CI, 2.14-18.88; P less than .001) were predictors of radiographic disease progression. Another analysis confirmed those as risk factors even when the definition of progression also required a final absolute grade of at least 2 in the worsened SIJs.
When the authors considered the change in total score (based on the mean of two readers), only HLAB27 positivity and baseline MRI positivity were highlighted as independent predictors of disease progression. In this analysis, HLAB27 positivity and inflammation on MRI had grading score increases of 0.41 (P = .014) and 1.03 (P less than .001), respectively.
Unlike other studies, the researchers did not see a clear relationship between abnormal C-reactive protein at baseline and radiographic progression.
The findings suggest that, in early SpA, “structural progression does exist but is quite small and observed in a small number of patients,” the study authors wrote.
The authors noted that their findings should be “interpreted with caution” because they were based on a small number of patients, and additional studies with a longer-follow-up period are needed to confirm the findings.
The DESIR cohort was sponsored by the Department de la recherché Clinique et du Development de l’Assistance Publique-Hopitaux de Paris. An unrestricted grant from Pfizer was allocated for the 10 years of follow-up.
Progression to structural damage occurs slowly among patients with early axial spondyloarthritis and more often takes place in current smokers, HLAB27 carriers, and those with signs of inflammation on MRI, according to new observations from the French DESIR cohort study.
The findings help to establish risk factors for early progression of nonradiographic axial spondyloarthritis (nr-axSpA) to radiographic disease, said Dr. Maxime Dougados of the department of rheumatology at Cochin Hospital, Paris, and his colleagues (Arthritis Rheumatol. 2016 Mar 18. doi: 10.1002/art.39666).
The study involved 449 patients with recent-onset inflammatory back pain who were participating in the multicenter, longitudinal DESIR study and had x-rays available at baseline and 2-year follow-up.
A total of 16 (4.9%) out of 326 patients who did not fulfill the modified New York criteria (mNY) at baseline progressed from nonradiographic to radiographic axSpA over the 2-year study period.
Of the 123 patients who met the mNY criteria at baseline, 7 (5.7%) no longer met the criteria at follow-up.
Independent factors found to influence the odds of progressing to radiographic axSpA were smoking status (odds ratio, 3.3; 95% confidence interval, 1.0-11.5), HLAB27 positivity (OR, 12.6; 95% CI, 2.3-274), and the presence of inflammation at MRI in the sacroiliac joints (SIJs) at baseline (OR, 48.8; 95% CI, 9.3-904), a multivariate analysis showed.
However, when the authors defined radiographic progression as “a worsening of at least one grade in at least one SIJ,” HLAB27 positivity (OR, 1.68; 95% CI, 0.8-3.71, P = .02), MRI positivity (OR, 5.85; 95% CI, 2.38-15.37, P less than .001), and baseline structural damage at SIJs on pelvic x-rays (OR, 6.35; 95% CI, 2.14-18.88; P less than .001) were predictors of radiographic disease progression. Another analysis confirmed those as risk factors even when the definition of progression also required a final absolute grade of at least 2 in the worsened SIJs.
When the authors considered the change in total score (based on the mean of two readers), only HLAB27 positivity and baseline MRI positivity were highlighted as independent predictors of disease progression. In this analysis, HLAB27 positivity and inflammation on MRI had grading score increases of 0.41 (P = .014) and 1.03 (P less than .001), respectively.
Unlike other studies, the researchers did not see a clear relationship between abnormal C-reactive protein at baseline and radiographic progression.
The findings suggest that, in early SpA, “structural progression does exist but is quite small and observed in a small number of patients,” the study authors wrote.
The authors noted that their findings should be “interpreted with caution” because they were based on a small number of patients, and additional studies with a longer-follow-up period are needed to confirm the findings.
The DESIR cohort was sponsored by the Department de la recherché Clinique et du Development de l’Assistance Publique-Hopitaux de Paris. An unrestricted grant from Pfizer was allocated for the 10 years of follow-up.
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point: The switch from nonradiographic to radiographic axSpA, made by about 5% of patients over the 2-year study period, was influenced by HLAB27 positivity, smoking status, and inflammatory sacroiliac joint lesions on MRI.
Major finding: Multivariate analysis showed smoking status (OR, 3.3; 95% CI, 1.0-11.5), HLAB27 positivity (OR, 12.6; 95% CI, 2.3-274), and the presence of inflammation at MRI in the sacroiliac joints (SIJ) at baseline (OR, 48.8; 95% CI, 9.3-904) predicted progression.
Data source: An analysis of 2-year data from 449 patients with recent-onset inflammatory back pain participating in the multicenter, longitudinal DESIR study.
Disclosures: The DESIR cohort was sponsored by the Department de la recherché Clinique et du Development de l’Assistance Publique-Hopitaux de Paris. An unrestricted grant from Pfizer was allocated for the 10 years of follow-up.

Ixekizumab approved for plaque psoriasis
Ixekizumab is approved to treat moderate to severe plaque psoriasis in adults, according to an announcement from the Food and Drug Administration.
“Today’s approval provides patients suffering from plaque psoriasis with another important treatment option to help relieve the skin irritation and discomfort from the condition,” Dr. Julie Beitz, director of the FDA’s Office of Drug Evaluation III, said in a statement .
Ixekizumab is an IgG4 monoclonal antibody that selectively binds with interleukin 17A (IL-17A) cytokines, inhibiting interaction with the IL-17 receptor. It is approved for patients who are candidates for systemic therapy, phototherapy, or a combination of both.

The safety and efficacy of ixekizumab were established in three randomized, placebo-controlled clinical trials with a total of 3,866 participants with plaque psoriasis who were candidates for systemic therapy or phototherapy. Patients treated with ixekizumab achieved greater clinical response than did those who received placebo.
The therapy was approved with a medication guide to inform patients that they may have a greater risk of an infection, or an allergic or autoimmune condition, according to the FDA announcement. The agency advised that physicians should monitor patient for serious allergic reactions and development or worsening of inflammatory bowel disease.
The most common adverse events seen in clinical trials of ixekizumab were upper respiratory infections, injection site reactions, and tinea.
Ixekizumab will be marketed as Taltz by Eli Lilly and Company.
On Twitter @denisefulton
Ixekizumab is approved to treat moderate to severe plaque psoriasis in adults, according to an announcement from the Food and Drug Administration.
“Today’s approval provides patients suffering from plaque psoriasis with another important treatment option to help relieve the skin irritation and discomfort from the condition,” Dr. Julie Beitz, director of the FDA’s Office of Drug Evaluation III, said in a statement .
Ixekizumab is an IgG4 monoclonal antibody that selectively binds with interleukin 17A (IL-17A) cytokines, inhibiting interaction with the IL-17 receptor. It is approved for patients who are candidates for systemic therapy, phototherapy, or a combination of both.
The safety and efficacy of ixekizumab were established in three randomized, placebo-controlled clinical trials with a total of 3,866 participants with plaque psoriasis who were candidates for systemic therapy or phototherapy. Patients treated with ixekizumab achieved greater clinical response than did those who received placebo.
The therapy was approved with a medication guide to inform patients that they may have a greater risk of an infection, or an allergic or autoimmune condition, according to the FDA announcement. The agency advised that physicians should monitor patient for serious allergic reactions and development or worsening of inflammatory bowel disease.
The most common adverse events seen in clinical trials of ixekizumab were upper respiratory infections, injection site reactions, and tinea.
Ixekizumab will be marketed as Taltz by Eli Lilly and Company.
On Twitter @denisefulton
Ixekizumab is approved to treat moderate to severe plaque psoriasis in adults, according to an announcement from the Food and Drug Administration.
“Today’s approval provides patients suffering from plaque psoriasis with another important treatment option to help relieve the skin irritation and discomfort from the condition,” Dr. Julie Beitz, director of the FDA’s Office of Drug Evaluation III, said in a statement .
Ixekizumab is an IgG4 monoclonal antibody that selectively binds with interleukin 17A (IL-17A) cytokines, inhibiting interaction with the IL-17 receptor. It is approved for patients who are candidates for systemic therapy, phototherapy, or a combination of both.
The safety and efficacy of ixekizumab were established in three randomized, placebo-controlled clinical trials with a total of 3,866 participants with plaque psoriasis who were candidates for systemic therapy or phototherapy. Patients treated with ixekizumab achieved greater clinical response than did those who received placebo.
The therapy was approved with a medication guide to inform patients that they may have a greater risk of an infection, or an allergic or autoimmune condition, according to the FDA announcement. The agency advised that physicians should monitor patient for serious allergic reactions and development or worsening of inflammatory bowel disease.
The most common adverse events seen in clinical trials of ixekizumab were upper respiratory infections, injection site reactions, and tinea.
Ixekizumab will be marketed as Taltz by Eli Lilly and Company.
On Twitter @denisefulton

Pushback on Part B drug payment proposal already beginning
Rheumatologists already are voicing concerns regarding a new proposal to test adjustments to how drugs administered in a physician’s office are paid for.
That proposal, published March 11 in the Federal Register, would test a change to the current reimbursement of average sales price plus 6% for Part B drugs with a lower add-on percentage of 2.5% plus $16.50.

In a fact sheet highlighting the proposals, the Centers for Medicare & Medicaid Services said the change to a lower percentage plus a flat fee “will cover the cost of any drug paid under Medicare Part B.”
However, there are already questions about that.
“While this may seem the way in which CMS will control costs, they fail to recognize the cost to facilities in obtaining approval for these treatments, receiving and storing, and ultimately safely administering these therapies in an environment that provides the best outcomes for patients,” Dr. Norman B. Gaylis, a rheumatologist in private practice in Aventura, Fla., said.
In fact, Dr. Gaylis adds that the focus on lowering drug expenses in the Part B space could have the unintended consequence of raising these expenses because it will force a change of venue.
“It has become so prohibitive that ultimately many patients will be referred to more expensive, less efficient outpatient facilities with, in fact, an increase in overall costs,” he said.
More than 300 provider and patient groups covering a range of specialties and including the American College of Rheumatology, the Coalition of State Rheumatology Organizations, and a number of state rheumatology organizations, are calling on Congress to ask CMS to withdraw the proposal.
In a March 17 letter to the majority and minority leaders in both chambers, the group is challenging the CMS assertion in the proposed rule that the current 6% add-on “may encourage the use of more expensive drugs because the 6% add-on generates more revenues for more expensive drugs.”
“This assumption fails to take into account the fact that providers’ prescribing decisions depend on a variety of factors, including clinical characteristics and the complex needs of the Medicare population,” the letter states. “Most importantly, there is no evidence indicating that the payment changes contemplated by the model will improve quality of care, and may adversely impact those patients that lose access to their most appropriate treatments.”
CMS offered two other pricing models that would be tested: indications-based pricing and reference pricing. The former would set payment rates based on the clinical effectiveness of a drug, while the latter would test the impact of setting a benchmark price for a group of drugs in a similar therapeutic class. Related to that is a proposal that CMS enter into voluntary risk-sharing agreements with drug manufacturers to link outcomes with price adjustments.
Dr. Gaylis suggested that CMS is going after the wrong party if cost containment is the ultimate goal here and should be focusing its efforts on the prices of the drugs themselves rather than how much they spend on physician reimbursement.
“Ironically, the major expense, i.e., the cost of drugs themselves, continues to spiral in the absence of any legitimate mechanism between CMS and pharma to contract prices that could save health care billions of dollars,” he said. “Ultimately, in my opinion, the solution rests in creating a fair and equal price for facilities administering these therapies and creating a pass-through where the drugs are not part of the physician’s risk, cost, or benefit and all payers, including CMS, can negotiate drug costs directly with the manufacturer.”
As part of the proposed rule, CMS also is considering creating feedback and decision-support tools to help, such as offering best practices for prescribing certain medications or providing feedback on prescribing patterns relative to local, regional, and national trends.
On the patient side, CMS is proposing to eliminate any patient cost sharing for office-administered drugs.
Comments on the proposals are due May 9.
Rheumatologists already are voicing concerns regarding a new proposal to test adjustments to how drugs administered in a physician’s office are paid for.
That proposal, published March 11 in the Federal Register, would test a change to the current reimbursement of average sales price plus 6% for Part B drugs with a lower add-on percentage of 2.5% plus $16.50.
In a fact sheet highlighting the proposals, the Centers for Medicare & Medicaid Services said the change to a lower percentage plus a flat fee “will cover the cost of any drug paid under Medicare Part B.”
However, there are already questions about that.
“While this may seem the way in which CMS will control costs, they fail to recognize the cost to facilities in obtaining approval for these treatments, receiving and storing, and ultimately safely administering these therapies in an environment that provides the best outcomes for patients,” Dr. Norman B. Gaylis, a rheumatologist in private practice in Aventura, Fla., said.
In fact, Dr. Gaylis adds that the focus on lowering drug expenses in the Part B space could have the unintended consequence of raising these expenses because it will force a change of venue.
“It has become so prohibitive that ultimately many patients will be referred to more expensive, less efficient outpatient facilities with, in fact, an increase in overall costs,” he said.
More than 300 provider and patient groups covering a range of specialties and including the American College of Rheumatology, the Coalition of State Rheumatology Organizations, and a number of state rheumatology organizations, are calling on Congress to ask CMS to withdraw the proposal.
In a March 17 letter to the majority and minority leaders in both chambers, the group is challenging the CMS assertion in the proposed rule that the current 6% add-on “may encourage the use of more expensive drugs because the 6% add-on generates more revenues for more expensive drugs.”
“This assumption fails to take into account the fact that providers’ prescribing decisions depend on a variety of factors, including clinical characteristics and the complex needs of the Medicare population,” the letter states. “Most importantly, there is no evidence indicating that the payment changes contemplated by the model will improve quality of care, and may adversely impact those patients that lose access to their most appropriate treatments.”
CMS offered two other pricing models that would be tested: indications-based pricing and reference pricing. The former would set payment rates based on the clinical effectiveness of a drug, while the latter would test the impact of setting a benchmark price for a group of drugs in a similar therapeutic class. Related to that is a proposal that CMS enter into voluntary risk-sharing agreements with drug manufacturers to link outcomes with price adjustments.
Dr. Gaylis suggested that CMS is going after the wrong party if cost containment is the ultimate goal here and should be focusing its efforts on the prices of the drugs themselves rather than how much they spend on physician reimbursement.
“Ironically, the major expense, i.e., the cost of drugs themselves, continues to spiral in the absence of any legitimate mechanism between CMS and pharma to contract prices that could save health care billions of dollars,” he said. “Ultimately, in my opinion, the solution rests in creating a fair and equal price for facilities administering these therapies and creating a pass-through where the drugs are not part of the physician’s risk, cost, or benefit and all payers, including CMS, can negotiate drug costs directly with the manufacturer.”
As part of the proposed rule, CMS also is considering creating feedback and decision-support tools to help, such as offering best practices for prescribing certain medications or providing feedback on prescribing patterns relative to local, regional, and national trends.
On the patient side, CMS is proposing to eliminate any patient cost sharing for office-administered drugs.
Comments on the proposals are due May 9.
Rheumatologists already are voicing concerns regarding a new proposal to test adjustments to how drugs administered in a physician’s office are paid for.
That proposal, published March 11 in the Federal Register, would test a change to the current reimbursement of average sales price plus 6% for Part B drugs with a lower add-on percentage of 2.5% plus $16.50.
In a fact sheet highlighting the proposals, the Centers for Medicare & Medicaid Services said the change to a lower percentage plus a flat fee “will cover the cost of any drug paid under Medicare Part B.”
However, there are already questions about that.
“While this may seem the way in which CMS will control costs, they fail to recognize the cost to facilities in obtaining approval for these treatments, receiving and storing, and ultimately safely administering these therapies in an environment that provides the best outcomes for patients,” Dr. Norman B. Gaylis, a rheumatologist in private practice in Aventura, Fla., said.
In fact, Dr. Gaylis adds that the focus on lowering drug expenses in the Part B space could have the unintended consequence of raising these expenses because it will force a change of venue.
“It has become so prohibitive that ultimately many patients will be referred to more expensive, less efficient outpatient facilities with, in fact, an increase in overall costs,” he said.
More than 300 provider and patient groups covering a range of specialties and including the American College of Rheumatology, the Coalition of State Rheumatology Organizations, and a number of state rheumatology organizations, are calling on Congress to ask CMS to withdraw the proposal.
In a March 17 letter to the majority and minority leaders in both chambers, the group is challenging the CMS assertion in the proposed rule that the current 6% add-on “may encourage the use of more expensive drugs because the 6% add-on generates more revenues for more expensive drugs.”
“This assumption fails to take into account the fact that providers’ prescribing decisions depend on a variety of factors, including clinical characteristics and the complex needs of the Medicare population,” the letter states. “Most importantly, there is no evidence indicating that the payment changes contemplated by the model will improve quality of care, and may adversely impact those patients that lose access to their most appropriate treatments.”
CMS offered two other pricing models that would be tested: indications-based pricing and reference pricing. The former would set payment rates based on the clinical effectiveness of a drug, while the latter would test the impact of setting a benchmark price for a group of drugs in a similar therapeutic class. Related to that is a proposal that CMS enter into voluntary risk-sharing agreements with drug manufacturers to link outcomes with price adjustments.
Dr. Gaylis suggested that CMS is going after the wrong party if cost containment is the ultimate goal here and should be focusing its efforts on the prices of the drugs themselves rather than how much they spend on physician reimbursement.
“Ironically, the major expense, i.e., the cost of drugs themselves, continues to spiral in the absence of any legitimate mechanism between CMS and pharma to contract prices that could save health care billions of dollars,” he said. “Ultimately, in my opinion, the solution rests in creating a fair and equal price for facilities administering these therapies and creating a pass-through where the drugs are not part of the physician’s risk, cost, or benefit and all payers, including CMS, can negotiate drug costs directly with the manufacturer.”
As part of the proposed rule, CMS also is considering creating feedback and decision-support tools to help, such as offering best practices for prescribing certain medications or providing feedback on prescribing patterns relative to local, regional, and national trends.
On the patient side, CMS is proposing to eliminate any patient cost sharing for office-administered drugs.
Comments on the proposals are due May 9.

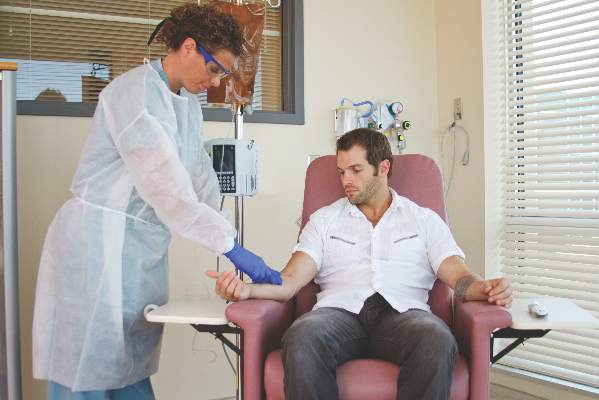
Home infusion policies called out in ACR position statement
Proper administration of intravenous biologics should take place under the close supervision of a physician in a physician’s office, infusion center, or hospital rather than in a patient’s home in order to address potential infusion reactions that can range from mild to life threatening, according to a position statement issued by the American College of Rheumatology’s Committee on Rheumatologic Care.
The “Patient Safety and Site of Service for Infusible Biologics” statement, issued in late February, comes in opposition to “policies that require home infusion” that appear to seek potential cost savings with home infusions rather than meet the standard of care with on-site physician supervision.
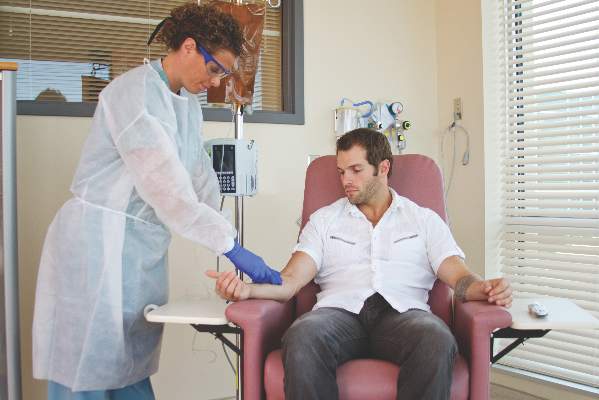
“One observation made by some but not all payers is that infusible biologics are about twice as expensive when infused in a hospital-based infusion center as compared to other locations, such as a clinic-based infusion center or the patient’s home. Thus, some payers are rolling out policies designed to shift patients from hospital-based infusion centers to less expensive sites. The ACR is opposed to policies that would force patients, solely for the purpose of cost containment, to receive infusible biologics in an improperly supervised setting. The purpose of the position statement is to outline that stance,” Dr. Douglas W. White, chair of the ACR’s Committee on Rheumatologic Care, said in an interview.
He noted that he’s “been in on conversations with two payers who are implementing policies to move patients away from hospital-based infusions, but we are aware that others are in various stages of implementing such policies, too. It’s not so much an issue of critical mass for us, rather we’re just trying to keep ahead of the trends, and we think this will be a big trend.”
The potential for adverse reactions is not uncommon during intravenous administration of biologics, the committee wrote, noting, for example, that 10% of patients given infliximab have acute infusion reactions. On-site physicians such as rheumatologists who have experience with the “tremendous heterogeneity of patients with autoimmune disease and the diversity of conditions treated with biologics” can determine the severity of infusion reactions and decide whether or not it is safe to continue a particular biologic agent, in addition to providing reassurance to patients during acute and potentially severe reactions, according to the ACR statement.
Infusion reactions can range in severity from a mild rash to life-threatening anaphylaxis that can involve multiple organ systems leading to respiratory and cardiovascular collapse and requiring immediate treatment with medications such as epinephrine or intravenous glucocorticoids.
The position statement recognizes unusual situations in which home infusion is necessary for a patient to receive treatment because of transportation problems to a medical facility or comorbid conditions in which the risk of no treatment may outweigh the risk of home infusion. In these circumstances, the ACR “encourages providers in such unusual and difficult situations to make the best medical decision based on the individual needs of the patient. Routine home infusion of biologics is considered an unnecessary and dangerous risk to patients and violates our current clinical standards of practice.”
Requirements for using home infusion also threaten “to reduce access to” intravenous biologics, the ACR contends, because “specially trained physicians are less likely to prescribe treatments that are not properly administered in the safest clinical setting [and] patient fear of biologic therapy may lead to noncompliance and inadequate control of disease.”
The ACR noted that home administration of subcutaneous biologics is medically appropriate and the injection site reactions that can occur with their use are often easily managed.
Proper administration of intravenous biologics should take place under the close supervision of a physician in a physician’s office, infusion center, or hospital rather than in a patient’s home in order to address potential infusion reactions that can range from mild to life threatening, according to a position statement issued by the American College of Rheumatology’s Committee on Rheumatologic Care.
The “Patient Safety and Site of Service for Infusible Biologics” statement, issued in late February, comes in opposition to “policies that require home infusion” that appear to seek potential cost savings with home infusions rather than meet the standard of care with on-site physician supervision.
“One observation made by some but not all payers is that infusible biologics are about twice as expensive when infused in a hospital-based infusion center as compared to other locations, such as a clinic-based infusion center or the patient’s home. Thus, some payers are rolling out policies designed to shift patients from hospital-based infusion centers to less expensive sites. The ACR is opposed to policies that would force patients, solely for the purpose of cost containment, to receive infusible biologics in an improperly supervised setting. The purpose of the position statement is to outline that stance,” Dr. Douglas W. White, chair of the ACR’s Committee on Rheumatologic Care, said in an interview.
He noted that he’s “been in on conversations with two payers who are implementing policies to move patients away from hospital-based infusions, but we are aware that others are in various stages of implementing such policies, too. It’s not so much an issue of critical mass for us, rather we’re just trying to keep ahead of the trends, and we think this will be a big trend.”
The potential for adverse reactions is not uncommon during intravenous administration of biologics, the committee wrote, noting, for example, that 10% of patients given infliximab have acute infusion reactions. On-site physicians such as rheumatologists who have experience with the “tremendous heterogeneity of patients with autoimmune disease and the diversity of conditions treated with biologics” can determine the severity of infusion reactions and decide whether or not it is safe to continue a particular biologic agent, in addition to providing reassurance to patients during acute and potentially severe reactions, according to the ACR statement.
Infusion reactions can range in severity from a mild rash to life-threatening anaphylaxis that can involve multiple organ systems leading to respiratory and cardiovascular collapse and requiring immediate treatment with medications such as epinephrine or intravenous glucocorticoids.
The position statement recognizes unusual situations in which home infusion is necessary for a patient to receive treatment because of transportation problems to a medical facility or comorbid conditions in which the risk of no treatment may outweigh the risk of home infusion. In these circumstances, the ACR “encourages providers in such unusual and difficult situations to make the best medical decision based on the individual needs of the patient. Routine home infusion of biologics is considered an unnecessary and dangerous risk to patients and violates our current clinical standards of practice.”
Requirements for using home infusion also threaten “to reduce access to” intravenous biologics, the ACR contends, because “specially trained physicians are less likely to prescribe treatments that are not properly administered in the safest clinical setting [and] patient fear of biologic therapy may lead to noncompliance and inadequate control of disease.”
The ACR noted that home administration of subcutaneous biologics is medically appropriate and the injection site reactions that can occur with their use are often easily managed.
Proper administration of intravenous biologics should take place under the close supervision of a physician in a physician’s office, infusion center, or hospital rather than in a patient’s home in order to address potential infusion reactions that can range from mild to life threatening, according to a position statement issued by the American College of Rheumatology’s Committee on Rheumatologic Care.
The “Patient Safety and Site of Service for Infusible Biologics” statement, issued in late February, comes in opposition to “policies that require home infusion” that appear to seek potential cost savings with home infusions rather than meet the standard of care with on-site physician supervision.
“One observation made by some but not all payers is that infusible biologics are about twice as expensive when infused in a hospital-based infusion center as compared to other locations, such as a clinic-based infusion center or the patient’s home. Thus, some payers are rolling out policies designed to shift patients from hospital-based infusion centers to less expensive sites. The ACR is opposed to policies that would force patients, solely for the purpose of cost containment, to receive infusible biologics in an improperly supervised setting. The purpose of the position statement is to outline that stance,” Dr. Douglas W. White, chair of the ACR’s Committee on Rheumatologic Care, said in an interview.
He noted that he’s “been in on conversations with two payers who are implementing policies to move patients away from hospital-based infusions, but we are aware that others are in various stages of implementing such policies, too. It’s not so much an issue of critical mass for us, rather we’re just trying to keep ahead of the trends, and we think this will be a big trend.”
The potential for adverse reactions is not uncommon during intravenous administration of biologics, the committee wrote, noting, for example, that 10% of patients given infliximab have acute infusion reactions. On-site physicians such as rheumatologists who have experience with the “tremendous heterogeneity of patients with autoimmune disease and the diversity of conditions treated with biologics” can determine the severity of infusion reactions and decide whether or not it is safe to continue a particular biologic agent, in addition to providing reassurance to patients during acute and potentially severe reactions, according to the ACR statement.
Infusion reactions can range in severity from a mild rash to life-threatening anaphylaxis that can involve multiple organ systems leading to respiratory and cardiovascular collapse and requiring immediate treatment with medications such as epinephrine or intravenous glucocorticoids.
The position statement recognizes unusual situations in which home infusion is necessary for a patient to receive treatment because of transportation problems to a medical facility or comorbid conditions in which the risk of no treatment may outweigh the risk of home infusion. In these circumstances, the ACR “encourages providers in such unusual and difficult situations to make the best medical decision based on the individual needs of the patient. Routine home infusion of biologics is considered an unnecessary and dangerous risk to patients and violates our current clinical standards of practice.”
Requirements for using home infusion also threaten “to reduce access to” intravenous biologics, the ACR contends, because “specially trained physicians are less likely to prescribe treatments that are not properly administered in the safest clinical setting [and] patient fear of biologic therapy may lead to noncompliance and inadequate control of disease.”
The ACR noted that home administration of subcutaneous biologics is medically appropriate and the injection site reactions that can occur with their use are often easily managed.
ACR’s 2016-2020 research agenda built through consensus
Therapeutic goals set the tone for the American College of Rheumatology National Research Agenda 2016-2020 by calling for the discovery and development of new therapies for rheumatic disease; finding predictors of response and nonresponse to, and adverse events from therapy; and improving the understanding of how therapies should be used.
Those are the top 3 out of 15 goals facilitated by the ACR’s Committee on Research, which finalized the agenda after seeking input from members of the ACR and Association of Rheumatology Health Professionals (ARHP) living in the United States, and going through several rounds of refining and prioritizing the importance of goals through the input of clinicians, researchers, patients, and stakeholders. The Committee on Research uses the agenda to “set the compass for the organization in terms of research initiatives and facilitate the ACR’s advocacy for the research goals identified.”

Dr. Alexis R. Ogdie-Beatty, who jointly led the development of the agenda for the Committee on Research along with Dr. S. Louis Bridges, said that while the goals for 2016-2020 had a great deal of overlap with those of 2011-2015, “some of the topics that came up were different. Some of the topics were more specific than in the previous agenda. We have some idea how important these issues were to rheumatologists, given that rheumatologists (and patients) rated the importance of the items. Defining new therapeutic targets and developing new therapies for rheumatic diseases was by far the most highly rated goal by rheumatologists. Next most highly rated was to advocate for increased support for rheumatology research and rheumatology investigators – this was included as a supplementary goal that supports the rest of the agenda. Other newer items were those around determining how the changing health care landscape affects rheumatology patients and clinicians. In addition, nonpharmacologic therapy, adult outcomes of pediatric disease, and optimizing patient engagement were topics that were felt to be important. I think these highlight the input of clinicians in identifying research objectives.”
The 2016-2020 agenda is the third set of goals developed by the committee since 2005, and the first to “crowdsource” the important questions to ACR and ARHP members rather than be assembled solely by the committee.

The agenda arose from a multistage process that began with a web-based survey to the ACR/ARHP membership that asked respondents to “list the five most important research questions that need to be addressed over the next 5 years in order to improve the care for patients with rheumatic disease.” A selected group of 100 individuals representing patients, clinicians (academic and community), research (all types with diverse areas/diseases of interest), allied health professionals, pediatric and adult rheumatology, men and women, all career stages, and all regions of the country, used a Delphi exercise to rate 30 statements generated from the survey on a scale from 1 (not important) to 10 (very important). They had the option to provide comments. At a Leadership Summit, stakeholders from various nonprofit foundations associated with rheumatic diseases, the National Institutes of Health, and the president of the Rheumatology Research Foundation gave comments on a draft agenda to the Committee on Research, after which the committee discussed the results and input and then solicited further 1-10 ratings and comments on preliminary agenda goals from the same group of 100 individuals as in the second phase, plus an additional 17 clinicians.
Up next in the rank-ordering after therapeutic goals were three goals about understanding:
• The etiology, pathogenesis, and genetic basis of rheumatic diseases.
• Early disease states to improve early diagnosis, develop biomarkers for early detection, and determine how earlier treatment changes outcomes.
• The immune system and autoimmunity by defining autoimmunity triggers and determining how epigenetics affect disease susceptibility and inflammation.
The 5-year plan proposed developing improved outcome measures that incorporate patient self-reports, imaging, and measures of clinical response and disease activity. The agenda also seeks to gain better understanding of how patients with rheumatic disease, rheumatologists, and rheumatology health professionals are being affected by the changing U.S. health care landscape.
The plan calls for determining the role of nonpharmacologic therapy in the management of rheumatic disease (promoting and improving adherence to physical activity, finding optimal exercise prescriptions, and determining the role of diet on disease activity), as well as evaluating the role of regenerative medicine.
The agenda spells out the need for better engagement of patients in their care as well as for understanding how comorbidities are influenced by rheumatic disease and how pain and fatigue arise in rheumatic disease.
In two separate goals, committee members listed the importance of determining adult outcomes of pediatric rheumatic diseases and the effect of aging on the development, progression, and management of rheumatic diseases.
The Committee on Research identified three supplemental goals that support the others:
• Advocating for increased support for rheumatology research and rheumatology investigators.
• Harmonizing data from existing cohorts and registries to optimize research capabilities.
• Improving patient research partner involvement in research protocols.
Therapeutic goals set the tone for the American College of Rheumatology National Research Agenda 2016-2020 by calling for the discovery and development of new therapies for rheumatic disease; finding predictors of response and nonresponse to, and adverse events from therapy; and improving the understanding of how therapies should be used.
Those are the top 3 out of 15 goals facilitated by the ACR’s Committee on Research, which finalized the agenda after seeking input from members of the ACR and Association of Rheumatology Health Professionals (ARHP) living in the United States, and going through several rounds of refining and prioritizing the importance of goals through the input of clinicians, researchers, patients, and stakeholders. The Committee on Research uses the agenda to “set the compass for the organization in terms of research initiatives and facilitate the ACR’s advocacy for the research goals identified.”
Dr. Alexis R. Ogdie-Beatty, who jointly led the development of the agenda for the Committee on Research along with Dr. S. Louis Bridges, said that while the goals for 2016-2020 had a great deal of overlap with those of 2011-2015, “some of the topics that came up were different. Some of the topics were more specific than in the previous agenda. We have some idea how important these issues were to rheumatologists, given that rheumatologists (and patients) rated the importance of the items. Defining new therapeutic targets and developing new therapies for rheumatic diseases was by far the most highly rated goal by rheumatologists. Next most highly rated was to advocate for increased support for rheumatology research and rheumatology investigators – this was included as a supplementary goal that supports the rest of the agenda. Other newer items were those around determining how the changing health care landscape affects rheumatology patients and clinicians. In addition, nonpharmacologic therapy, adult outcomes of pediatric disease, and optimizing patient engagement were topics that were felt to be important. I think these highlight the input of clinicians in identifying research objectives.”
The 2016-2020 agenda is the third set of goals developed by the committee since 2005, and the first to “crowdsource” the important questions to ACR and ARHP members rather than be assembled solely by the committee.
The agenda arose from a multistage process that began with a web-based survey to the ACR/ARHP membership that asked respondents to “list the five most important research questions that need to be addressed over the next 5 years in order to improve the care for patients with rheumatic disease.” A selected group of 100 individuals representing patients, clinicians (academic and community), research (all types with diverse areas/diseases of interest), allied health professionals, pediatric and adult rheumatology, men and women, all career stages, and all regions of the country, used a Delphi exercise to rate 30 statements generated from the survey on a scale from 1 (not important) to 10 (very important). They had the option to provide comments. At a Leadership Summit, stakeholders from various nonprofit foundations associated with rheumatic diseases, the National Institutes of Health, and the president of the Rheumatology Research Foundation gave comments on a draft agenda to the Committee on Research, after which the committee discussed the results and input and then solicited further 1-10 ratings and comments on preliminary agenda goals from the same group of 100 individuals as in the second phase, plus an additional 17 clinicians.
Up next in the rank-ordering after therapeutic goals were three goals about understanding:
• The etiology, pathogenesis, and genetic basis of rheumatic diseases.
• Early disease states to improve early diagnosis, develop biomarkers for early detection, and determine how earlier treatment changes outcomes.
• The immune system and autoimmunity by defining autoimmunity triggers and determining how epigenetics affect disease susceptibility and inflammation.
The 5-year plan proposed developing improved outcome measures that incorporate patient self-reports, imaging, and measures of clinical response and disease activity. The agenda also seeks to gain better understanding of how patients with rheumatic disease, rheumatologists, and rheumatology health professionals are being affected by the changing U.S. health care landscape.
The plan calls for determining the role of nonpharmacologic therapy in the management of rheumatic disease (promoting and improving adherence to physical activity, finding optimal exercise prescriptions, and determining the role of diet on disease activity), as well as evaluating the role of regenerative medicine.
The agenda spells out the need for better engagement of patients in their care as well as for understanding how comorbidities are influenced by rheumatic disease and how pain and fatigue arise in rheumatic disease.
In two separate goals, committee members listed the importance of determining adult outcomes of pediatric rheumatic diseases and the effect of aging on the development, progression, and management of rheumatic diseases.
The Committee on Research identified three supplemental goals that support the others:
• Advocating for increased support for rheumatology research and rheumatology investigators.
• Harmonizing data from existing cohorts and registries to optimize research capabilities.
• Improving patient research partner involvement in research protocols.
Therapeutic goals set the tone for the American College of Rheumatology National Research Agenda 2016-2020 by calling for the discovery and development of new therapies for rheumatic disease; finding predictors of response and nonresponse to, and adverse events from therapy; and improving the understanding of how therapies should be used.
Those are the top 3 out of 15 goals facilitated by the ACR’s Committee on Research, which finalized the agenda after seeking input from members of the ACR and Association of Rheumatology Health Professionals (ARHP) living in the United States, and going through several rounds of refining and prioritizing the importance of goals through the input of clinicians, researchers, patients, and stakeholders. The Committee on Research uses the agenda to “set the compass for the organization in terms of research initiatives and facilitate the ACR’s advocacy for the research goals identified.”
Dr. Alexis R. Ogdie-Beatty, who jointly led the development of the agenda for the Committee on Research along with Dr. S. Louis Bridges, said that while the goals for 2016-2020 had a great deal of overlap with those of 2011-2015, “some of the topics that came up were different. Some of the topics were more specific than in the previous agenda. We have some idea how important these issues were to rheumatologists, given that rheumatologists (and patients) rated the importance of the items. Defining new therapeutic targets and developing new therapies for rheumatic diseases was by far the most highly rated goal by rheumatologists. Next most highly rated was to advocate for increased support for rheumatology research and rheumatology investigators – this was included as a supplementary goal that supports the rest of the agenda. Other newer items were those around determining how the changing health care landscape affects rheumatology patients and clinicians. In addition, nonpharmacologic therapy, adult outcomes of pediatric disease, and optimizing patient engagement were topics that were felt to be important. I think these highlight the input of clinicians in identifying research objectives.”
The 2016-2020 agenda is the third set of goals developed by the committee since 2005, and the first to “crowdsource” the important questions to ACR and ARHP members rather than be assembled solely by the committee.
The agenda arose from a multistage process that began with a web-based survey to the ACR/ARHP membership that asked respondents to “list the five most important research questions that need to be addressed over the next 5 years in order to improve the care for patients with rheumatic disease.” A selected group of 100 individuals representing patients, clinicians (academic and community), research (all types with diverse areas/diseases of interest), allied health professionals, pediatric and adult rheumatology, men and women, all career stages, and all regions of the country, used a Delphi exercise to rate 30 statements generated from the survey on a scale from 1 (not important) to 10 (very important). They had the option to provide comments. At a Leadership Summit, stakeholders from various nonprofit foundations associated with rheumatic diseases, the National Institutes of Health, and the president of the Rheumatology Research Foundation gave comments on a draft agenda to the Committee on Research, after which the committee discussed the results and input and then solicited further 1-10 ratings and comments on preliminary agenda goals from the same group of 100 individuals as in the second phase, plus an additional 17 clinicians.
Up next in the rank-ordering after therapeutic goals were three goals about understanding:
• The etiology, pathogenesis, and genetic basis of rheumatic diseases.
• Early disease states to improve early diagnosis, develop biomarkers for early detection, and determine how earlier treatment changes outcomes.
• The immune system and autoimmunity by defining autoimmunity triggers and determining how epigenetics affect disease susceptibility and inflammation.
The 5-year plan proposed developing improved outcome measures that incorporate patient self-reports, imaging, and measures of clinical response and disease activity. The agenda also seeks to gain better understanding of how patients with rheumatic disease, rheumatologists, and rheumatology health professionals are being affected by the changing U.S. health care landscape.
The plan calls for determining the role of nonpharmacologic therapy in the management of rheumatic disease (promoting and improving adherence to physical activity, finding optimal exercise prescriptions, and determining the role of diet on disease activity), as well as evaluating the role of regenerative medicine.
The agenda spells out the need for better engagement of patients in their care as well as for understanding how comorbidities are influenced by rheumatic disease and how pain and fatigue arise in rheumatic disease.
In two separate goals, committee members listed the importance of determining adult outcomes of pediatric rheumatic diseases and the effect of aging on the development, progression, and management of rheumatic diseases.
The Committee on Research identified three supplemental goals that support the others:
• Advocating for increased support for rheumatology research and rheumatology investigators.
• Harmonizing data from existing cohorts and registries to optimize research capabilities.
• Improving patient research partner involvement in research protocols.

Sparse, poor evidence supports fumarates for psoriasis
Even though fumaric acid esters are increasingly considered to be a suitable, even a first-line, systemic treatment for moderate to severe psoriasis in some parts of Europe, the evidence supporting their use is sparse and of low quality, according to a report published online in the British Journal of Dermatology.
Fumarates were introduced as anti-psoriasis agents decades ago in Germany. The agents are thought to exert immunomodulating, antiproliferative, and antiangiogenic effects, and they are frequently used off label for psoriasis in the Netherlands and the United Kingdom, said Dr. Deepak M.W. Balak of the department of dermatology, Erasmus University Medical Center, Rotterdam, the Netherlands, and his associates.
To summarize the clinical evidence for this treatment, the investigators performed a systematic review of publications, identifying 68 studies that reported the clinical effects of these agents in comparison with either placebo or other therapies. The researchers were unable to perform a meta-analysis of the data “due to considerable clinical heterogeneity among the studies” in design, patient populations, the drug formulations and dosages examined, the comparator treatments, and the outcomes measured.
Only seven randomized clinical trials were available for review. These had relatively small sample sizes and included only 449 patients in total. They assessed different drug formulations and different, short treatment durations ranging from 2.8 to 4 months. The overall quality of the evidence was rated “moderate.”
All randomized controlled trials reported statistically significant efficacy with fumaric acid ester treatment; mean Psoriasis Area Severity Index (PASI) scores decreased in 42%-65% of patients after 12-16 weeks of treatment. Adverse events were common, affecting 69%-92% of patients, and chiefly involved gastrointestinal complaints, flushing, and laboratory abnormalities such as elevated liver enzymes (up to 62%), eosinophilia (up to 46%), and lymphocytopenia (up to 38%). A total of 8%-39% of patients discontinued treatment because of adverse effects.
There also were 37 observational studies involving a total of 3,457 patients. Almost all were open-label, single-center, uncontrolled cohort studies or retrospective case series with small samples. Treatment duration ranged from 1 month to 14 years. The overall quality of the evidence was rated “very low” (Br J Dermatol. 2016. doi: 10.1111/bjd.14500).
These studies reported similar outcomes to the randomized clinical trials: significant reductions in the extent and severity of psoriasis with fumarate treatment, and frequent adverse effects, predominantly GI problems, flushing, and laboratory abnormalities. Mean reductions in PASI were 13%-86% after 3-4 months of treatment. Several immunosuppressive adverse effects were linked to the treatment, including Kaposi’s sarcoma, organizing pneumonia, tuberculous lymphadenitis, squamous cell carcinoma, melanoma, and seven cases of progressive multifocal leukoencephalopathy. In addition, several renal complications were reported, including six cases of Fanconi syndrome and nine cases of acute renal insufficiency, and there was one case of collagenous colitis.
“Fumaric acid esters have a long history as a systemic psoriasis treatment” dating back to the 1950s, “but their development was not based on high-quality evidence,” Dr. Balak and his associates said.
They added that several randomized clinical trials assessing these agents are currently underway, but their findings haven’t yet been published. And new fumarates for the treatment of psoriasis currently are in development.
No sponsor or funding source was identified for this study. Dr. Balak and his associates reported having no relevant financial disclosures.
Even though fumaric acid esters are increasingly considered to be a suitable, even a first-line, systemic treatment for moderate to severe psoriasis in some parts of Europe, the evidence supporting their use is sparse and of low quality, according to a report published online in the British Journal of Dermatology.
Fumarates were introduced as anti-psoriasis agents decades ago in Germany. The agents are thought to exert immunomodulating, antiproliferative, and antiangiogenic effects, and they are frequently used off label for psoriasis in the Netherlands and the United Kingdom, said Dr. Deepak M.W. Balak of the department of dermatology, Erasmus University Medical Center, Rotterdam, the Netherlands, and his associates.
To summarize the clinical evidence for this treatment, the investigators performed a systematic review of publications, identifying 68 studies that reported the clinical effects of these agents in comparison with either placebo or other therapies. The researchers were unable to perform a meta-analysis of the data “due to considerable clinical heterogeneity among the studies” in design, patient populations, the drug formulations and dosages examined, the comparator treatments, and the outcomes measured.
Only seven randomized clinical trials were available for review. These had relatively small sample sizes and included only 449 patients in total. They assessed different drug formulations and different, short treatment durations ranging from 2.8 to 4 months. The overall quality of the evidence was rated “moderate.”
All randomized controlled trials reported statistically significant efficacy with fumaric acid ester treatment; mean Psoriasis Area Severity Index (PASI) scores decreased in 42%-65% of patients after 12-16 weeks of treatment. Adverse events were common, affecting 69%-92% of patients, and chiefly involved gastrointestinal complaints, flushing, and laboratory abnormalities such as elevated liver enzymes (up to 62%), eosinophilia (up to 46%), and lymphocytopenia (up to 38%). A total of 8%-39% of patients discontinued treatment because of adverse effects.
There also were 37 observational studies involving a total of 3,457 patients. Almost all were open-label, single-center, uncontrolled cohort studies or retrospective case series with small samples. Treatment duration ranged from 1 month to 14 years. The overall quality of the evidence was rated “very low” (Br J Dermatol. 2016. doi: 10.1111/bjd.14500).
These studies reported similar outcomes to the randomized clinical trials: significant reductions in the extent and severity of psoriasis with fumarate treatment, and frequent adverse effects, predominantly GI problems, flushing, and laboratory abnormalities. Mean reductions in PASI were 13%-86% after 3-4 months of treatment. Several immunosuppressive adverse effects were linked to the treatment, including Kaposi’s sarcoma, organizing pneumonia, tuberculous lymphadenitis, squamous cell carcinoma, melanoma, and seven cases of progressive multifocal leukoencephalopathy. In addition, several renal complications were reported, including six cases of Fanconi syndrome and nine cases of acute renal insufficiency, and there was one case of collagenous colitis.
“Fumaric acid esters have a long history as a systemic psoriasis treatment” dating back to the 1950s, “but their development was not based on high-quality evidence,” Dr. Balak and his associates said.
They added that several randomized clinical trials assessing these agents are currently underway, but their findings haven’t yet been published. And new fumarates for the treatment of psoriasis currently are in development.
No sponsor or funding source was identified for this study. Dr. Balak and his associates reported having no relevant financial disclosures.
Even though fumaric acid esters are increasingly considered to be a suitable, even a first-line, systemic treatment for moderate to severe psoriasis in some parts of Europe, the evidence supporting their use is sparse and of low quality, according to a report published online in the British Journal of Dermatology.
Fumarates were introduced as anti-psoriasis agents decades ago in Germany. The agents are thought to exert immunomodulating, antiproliferative, and antiangiogenic effects, and they are frequently used off label for psoriasis in the Netherlands and the United Kingdom, said Dr. Deepak M.W. Balak of the department of dermatology, Erasmus University Medical Center, Rotterdam, the Netherlands, and his associates.
To summarize the clinical evidence for this treatment, the investigators performed a systematic review of publications, identifying 68 studies that reported the clinical effects of these agents in comparison with either placebo or other therapies. The researchers were unable to perform a meta-analysis of the data “due to considerable clinical heterogeneity among the studies” in design, patient populations, the drug formulations and dosages examined, the comparator treatments, and the outcomes measured.
Only seven randomized clinical trials were available for review. These had relatively small sample sizes and included only 449 patients in total. They assessed different drug formulations and different, short treatment durations ranging from 2.8 to 4 months. The overall quality of the evidence was rated “moderate.”
All randomized controlled trials reported statistically significant efficacy with fumaric acid ester treatment; mean Psoriasis Area Severity Index (PASI) scores decreased in 42%-65% of patients after 12-16 weeks of treatment. Adverse events were common, affecting 69%-92% of patients, and chiefly involved gastrointestinal complaints, flushing, and laboratory abnormalities such as elevated liver enzymes (up to 62%), eosinophilia (up to 46%), and lymphocytopenia (up to 38%). A total of 8%-39% of patients discontinued treatment because of adverse effects.
There also were 37 observational studies involving a total of 3,457 patients. Almost all were open-label, single-center, uncontrolled cohort studies or retrospective case series with small samples. Treatment duration ranged from 1 month to 14 years. The overall quality of the evidence was rated “very low” (Br J Dermatol. 2016. doi: 10.1111/bjd.14500).
These studies reported similar outcomes to the randomized clinical trials: significant reductions in the extent and severity of psoriasis with fumarate treatment, and frequent adverse effects, predominantly GI problems, flushing, and laboratory abnormalities. Mean reductions in PASI were 13%-86% after 3-4 months of treatment. Several immunosuppressive adverse effects were linked to the treatment, including Kaposi’s sarcoma, organizing pneumonia, tuberculous lymphadenitis, squamous cell carcinoma, melanoma, and seven cases of progressive multifocal leukoencephalopathy. In addition, several renal complications were reported, including six cases of Fanconi syndrome and nine cases of acute renal insufficiency, and there was one case of collagenous colitis.
“Fumaric acid esters have a long history as a systemic psoriasis treatment” dating back to the 1950s, “but their development was not based on high-quality evidence,” Dr. Balak and his associates said.
They added that several randomized clinical trials assessing these agents are currently underway, but their findings haven’t yet been published. And new fumarates for the treatment of psoriasis currently are in development.
No sponsor or funding source was identified for this study. Dr. Balak and his associates reported having no relevant financial disclosures.
FROM BRITISH JOURNAL OF DERMATOLOGY
Key clinical point: Only sparse, low-quality evidence supports using fumaric acid esters as a treatment for psoriasis.
Major finding: Mean PASI scores decreased 42%-65% in patients treated with fumaric acid esters for 12-16 weeks in randomized controlled trials, but adverse events affected 69%-92% of patients.
Data source: A systematic review of 7 randomized clinical trials and 37 observational studies, involving a total of 3,906 patients.
Disclosures: No sponsor or funding source was identified for this study. Dr. Balak and his associates reported having no relevant financial disclosures.
New CDC opioid guideline targets overprescribing for chronic pain
Nonopioid therapy is the preferred approach for managing chronic pain outside of active cancer, palliative, and end-of-life care, according to a new guideline released today by the Centers for Disease Control and Prevention.
The 12 recommendations included in the guideline center around this principle and two others: using the lowest possible effective dosage when opioids are used, and exercising caution and monitoring patients closely when prescribing opioids.
Specifically, the guideline states that “clinicians should consider opioid therapy only if expected benefits for both pain and function are anticipated to outweigh risks to the patient,” and that “treatment should be combined with nonpharmacologic and nonopioid therapy, as appropriate.”
The guideline also addresses steps to take before starting or continuing opioid therapy, and drug selection, dosage, duration, follow-up, and discontinuation. Recommendations for assessing risk and addressing harms of opioid use are also included.
The CDC developed the guideline as part of the U.S. government’s urgent response to the epidemic of overdose deaths, which has been fueled by a quadrupling of the prescribing and sales of opioids since 1999, according to a CDC press statement. The guideline’s purpose is to help prevent opioid misuse and overdose.
“The CDC Guideline for Prescribing Opioids for Chronic Pain, United States, 2016 will help primary care providers ensure the safest and most effective treatment for their patients,” according to the statement. The CDC’s director, Dr. Tom Frieden, noted that “overprescribing opioids – largely for chronic pain – is a key driver of America’s drug-overdose epidemic.”
In a CDC teleconference marking the release of the guideline, Dr. Frieden said it has become increasingly clear that opioids “carry substantial risks but only uncertain benefits, especially compared with other treatments for chronic pain.
“Beginning treatment with an opioid is a momentous decision, and it should only be done with full understanding by both the clinician and the patient of the substantial risks and uncertain benefits involved,” Dr. Frieden said. He added that he knows of no other medication “that’s routinely used for a nonfatal condition [and] that kills patients so frequently.
“With more than 250 million prescriptions written each year, it’s so important that doctors understand that any one of those prescriptions could potentially end a patient’s life,” he cautioned.
A 2015 study showed that 1 of every 550 patients treated with opioids for noncancer pain – and 1 of 32 who received the highest doses (more than 200 morphine milligram equivalents per day) – died within 2.5 years of the first prescription.
Dr. Frieden noted that opioids do have a place when the potential benefits outweigh the potential harms. “But for most patients – the vast majority of patients – the risks will outweigh the benefits,” he said.
The opioid epidemic is one of the most pressing public health issues in the United States today, said Sylvia M. Burwell, secretary of the Department of Health & Human Services. A year ago, she announced an HHS initiative to reduce prescription opioid and heroin-related drug overdose, death, and dependence.
“Last year, more Americans died from drug overdoses than car crashes,” Ms. Burwell said during the teleconference, noting that families across the nation and from all walks of life have been affected.
Combating the opioid epidemic is a national priority, she said, and the CDC guideline will help in that effort.
“We believe this guideline will help health care professionals provide safer and more effective care for patients dealing with chronic pain, and we also believe it will help these providers drive down the rates of opioid use disorder, overdose, and ... death,” she said.
The American Medical Association greeted the guideline with cautious support.
“While we are largely supportive of the guidelines, we remain concerned about the evidence base informing some of the recommendations,” noted Dr. Patrice A. Harris, chair-elect of the AMA board and chair of the AMA Task Force to Reduce Opioid Abuse, in a statement.
The AMA also cited potential conflicts between the guideline and product labeling and state laws, as well as obstacles such as insurance coverage limits on nonpharmacologic treatments.
“If these guidelines help reduce the deaths resulting from opioids, they will prove to be valuable,” Dr. Harris said in the statement. “If they produce unintended consequences, we will need to mitigate them.”
Of note, the guideline stresses the right of patients with chronic pain to receive safe and effective pain management, and focuses on giving primary care providers – who account for about half of all opioid prescriptions – a road map for providing such pain management by increasing the use of effective nonopioid and nonpharmacologic therapies.
It was developed through a “rigorous scientific process using the best available scientific evidence, consulting with experts, and listening to comments from the public and partner organizations,” according to the CDC statement. The organization “is dedicated to working with partners to improve the evidence base and will refine the recommendations as new research becomes available.
”In conjunction with the release of the guideline, the CDC has provided a checklist for prescribing opioids for chronic pain, and a website with additional tools for implementing the recommendations within the guideline.
The CDC's opioid recommendations
The Centers for Disease Control and Prevention’s new opioid prescription guideline includes 12 recommendations. Here they are, modified slightly for style:
1. Nonpharmacologic therapy and nonopioid pharmacologic therapy are preferred for chronic pain. Providers should only consider adding opioid therapy if expected benefits for both pain and function are anticipated to outweigh risks.
2. Before starting opioid therapy for chronic pain, providers should establish treatment goals with all patients, including realistic goals for pain and function. Providers should not initiate opioid therapy without consideration of how therapy will be discontinued if unsuccessful. Providers should continue opioid therapy only if there is clinically meaningful improvement in pain and function.
3. Before starting and periodically during opioid therapy, providers should discuss with patients known risks and realistic benefits of opioid therapy, and patient and provider responsibilities for managing therapy.
4. When starting opioid therapy for chronic pain, providers should prescribe immediate-release opioids instead of extended-release/long-acting opioids.
5. When opioids are started, providers should prescribe the lowest effective dosage. Providers should use caution when prescribing opioids at any dosage, should implement additional precautions when increasing dosage to 50 or more morphine milligram equivalents (MME) per day, and generally should avoid increasing dosage to 90 or more MME per day.
6. When opioids are used for acute pain, providers should prescribe the lowest effective dose of immediate-release opioids. Three or fewer days often will be sufficient.
7. Providers should evaluate the benefits and harms with patients within 1-4 weeks of starting opioid therapy for chronic pain or of dose escalation. They should reevaluate continued therapy’s benefits and harms every 3 months or more frequently. If continued therapy’s benefits do not outweigh harms, providers should work with patients to reduce dosages or discontinue opioids.
8. During therapy, providers should evaluate risk factors for opioid-related harm. Providers should incorporate into the management plan strategies to mitigate risk, including considering offering naloxone when factors that increase risk for opioid overdose – such as history of overdose, history of substance use disorder, or higher opioid dosage (50 MME or more) – are present.
9. Providers should review the patient’s history of controlled substance prescriptions using state prescription drug monitoring program (PDMP) data to determine whether the patient is receiving high opioid dosages or dangerous combinations that put him or her at high risk for overdose. Providers should review PDMP data when starting opioid therapy for chronic pain and periodically during opioid therapy for chronic pain, ranging from every prescription to every 3 months.
10. When prescribing opioids for chronic pain, providers should use urine drug testing before starting opioid therapy and consider urine drug testing at least annually to assess for prescribed medications, as well as other controlled prescription drugs and illicit drugs.
11. Providers should avoid concurrent prescriptions of opioid pain medication and benzodiazepines whenever possible.
12. Providers should offer or arrange evidence-based treatment (usually medication-assisted treatment with buprenorphine or methadone in combination with behavioral therapies) for patients with opioid use disorder.
M. Alexander Otto contributed to this article.
Nonopioid therapy is the preferred approach for managing chronic pain outside of active cancer, palliative, and end-of-life care, according to a new guideline released today by the Centers for Disease Control and Prevention.
The 12 recommendations included in the guideline center around this principle and two others: using the lowest possible effective dosage when opioids are used, and exercising caution and monitoring patients closely when prescribing opioids.
Specifically, the guideline states that “clinicians should consider opioid therapy only if expected benefits for both pain and function are anticipated to outweigh risks to the patient,” and that “treatment should be combined with nonpharmacologic and nonopioid therapy, as appropriate.”
The guideline also addresses steps to take before starting or continuing opioid therapy, and drug selection, dosage, duration, follow-up, and discontinuation. Recommendations for assessing risk and addressing harms of opioid use are also included.
The CDC developed the guideline as part of the U.S. government’s urgent response to the epidemic of overdose deaths, which has been fueled by a quadrupling of the prescribing and sales of opioids since 1999, according to a CDC press statement. The guideline’s purpose is to help prevent opioid misuse and overdose.
“The CDC Guideline for Prescribing Opioids for Chronic Pain, United States, 2016 will help primary care providers ensure the safest and most effective treatment for their patients,” according to the statement. The CDC’s director, Dr. Tom Frieden, noted that “overprescribing opioids – largely for chronic pain – is a key driver of America’s drug-overdose epidemic.”
In a CDC teleconference marking the release of the guideline, Dr. Frieden said it has become increasingly clear that opioids “carry substantial risks but only uncertain benefits, especially compared with other treatments for chronic pain.
“Beginning treatment with an opioid is a momentous decision, and it should only be done with full understanding by both the clinician and the patient of the substantial risks and uncertain benefits involved,” Dr. Frieden said. He added that he knows of no other medication “that’s routinely used for a nonfatal condition [and] that kills patients so frequently.
“With more than 250 million prescriptions written each year, it’s so important that doctors understand that any one of those prescriptions could potentially end a patient’s life,” he cautioned.
A 2015 study showed that 1 of every 550 patients treated with opioids for noncancer pain – and 1 of 32 who received the highest doses (more than 200 morphine milligram equivalents per day) – died within 2.5 years of the first prescription.
Dr. Frieden noted that opioids do have a place when the potential benefits outweigh the potential harms. “But for most patients – the vast majority of patients – the risks will outweigh the benefits,” he said.
The opioid epidemic is one of the most pressing public health issues in the United States today, said Sylvia M. Burwell, secretary of the Department of Health & Human Services. A year ago, she announced an HHS initiative to reduce prescription opioid and heroin-related drug overdose, death, and dependence.
“Last year, more Americans died from drug overdoses than car crashes,” Ms. Burwell said during the teleconference, noting that families across the nation and from all walks of life have been affected.
Combating the opioid epidemic is a national priority, she said, and the CDC guideline will help in that effort.
“We believe this guideline will help health care professionals provide safer and more effective care for patients dealing with chronic pain, and we also believe it will help these providers drive down the rates of opioid use disorder, overdose, and ... death,” she said.
The American Medical Association greeted the guideline with cautious support.
“While we are largely supportive of the guidelines, we remain concerned about the evidence base informing some of the recommendations,” noted Dr. Patrice A. Harris, chair-elect of the AMA board and chair of the AMA Task Force to Reduce Opioid Abuse, in a statement.
The AMA also cited potential conflicts between the guideline and product labeling and state laws, as well as obstacles such as insurance coverage limits on nonpharmacologic treatments.
“If these guidelines help reduce the deaths resulting from opioids, they will prove to be valuable,” Dr. Harris said in the statement. “If they produce unintended consequences, we will need to mitigate them.”
Of note, the guideline stresses the right of patients with chronic pain to receive safe and effective pain management, and focuses on giving primary care providers – who account for about half of all opioid prescriptions – a road map for providing such pain management by increasing the use of effective nonopioid and nonpharmacologic therapies.
It was developed through a “rigorous scientific process using the best available scientific evidence, consulting with experts, and listening to comments from the public and partner organizations,” according to the CDC statement. The organization “is dedicated to working with partners to improve the evidence base and will refine the recommendations as new research becomes available.
”In conjunction with the release of the guideline, the CDC has provided a checklist for prescribing opioids for chronic pain, and a website with additional tools for implementing the recommendations within the guideline.
The CDC's opioid recommendations
The Centers for Disease Control and Prevention’s new opioid prescription guideline includes 12 recommendations. Here they are, modified slightly for style:
1. Nonpharmacologic therapy and nonopioid pharmacologic therapy are preferred for chronic pain. Providers should only consider adding opioid therapy if expected benefits for both pain and function are anticipated to outweigh risks.
2. Before starting opioid therapy for chronic pain, providers should establish treatment goals with all patients, including realistic goals for pain and function. Providers should not initiate opioid therapy without consideration of how therapy will be discontinued if unsuccessful. Providers should continue opioid therapy only if there is clinically meaningful improvement in pain and function.
3. Before starting and periodically during opioid therapy, providers should discuss with patients known risks and realistic benefits of opioid therapy, and patient and provider responsibilities for managing therapy.
4. When starting opioid therapy for chronic pain, providers should prescribe immediate-release opioids instead of extended-release/long-acting opioids.
5. When opioids are started, providers should prescribe the lowest effective dosage. Providers should use caution when prescribing opioids at any dosage, should implement additional precautions when increasing dosage to 50 or more morphine milligram equivalents (MME) per day, and generally should avoid increasing dosage to 90 or more MME per day.
6. When opioids are used for acute pain, providers should prescribe the lowest effective dose of immediate-release opioids. Three or fewer days often will be sufficient.
7. Providers should evaluate the benefits and harms with patients within 1-4 weeks of starting opioid therapy for chronic pain or of dose escalation. They should reevaluate continued therapy’s benefits and harms every 3 months or more frequently. If continued therapy’s benefits do not outweigh harms, providers should work with patients to reduce dosages or discontinue opioids.
8. During therapy, providers should evaluate risk factors for opioid-related harm. Providers should incorporate into the management plan strategies to mitigate risk, including considering offering naloxone when factors that increase risk for opioid overdose – such as history of overdose, history of substance use disorder, or higher opioid dosage (50 MME or more) – are present.
9. Providers should review the patient’s history of controlled substance prescriptions using state prescription drug monitoring program (PDMP) data to determine whether the patient is receiving high opioid dosages or dangerous combinations that put him or her at high risk for overdose. Providers should review PDMP data when starting opioid therapy for chronic pain and periodically during opioid therapy for chronic pain, ranging from every prescription to every 3 months.
10. When prescribing opioids for chronic pain, providers should use urine drug testing before starting opioid therapy and consider urine drug testing at least annually to assess for prescribed medications, as well as other controlled prescription drugs and illicit drugs.
11. Providers should avoid concurrent prescriptions of opioid pain medication and benzodiazepines whenever possible.
12. Providers should offer or arrange evidence-based treatment (usually medication-assisted treatment with buprenorphine or methadone in combination with behavioral therapies) for patients with opioid use disorder.
M. Alexander Otto contributed to this article.
Nonopioid therapy is the preferred approach for managing chronic pain outside of active cancer, palliative, and end-of-life care, according to a new guideline released today by the Centers for Disease Control and Prevention.
The 12 recommendations included in the guideline center around this principle and two others: using the lowest possible effective dosage when opioids are used, and exercising caution and monitoring patients closely when prescribing opioids.
Specifically, the guideline states that “clinicians should consider opioid therapy only if expected benefits for both pain and function are anticipated to outweigh risks to the patient,” and that “treatment should be combined with nonpharmacologic and nonopioid therapy, as appropriate.”
The guideline also addresses steps to take before starting or continuing opioid therapy, and drug selection, dosage, duration, follow-up, and discontinuation. Recommendations for assessing risk and addressing harms of opioid use are also included.
The CDC developed the guideline as part of the U.S. government’s urgent response to the epidemic of overdose deaths, which has been fueled by a quadrupling of the prescribing and sales of opioids since 1999, according to a CDC press statement. The guideline’s purpose is to help prevent opioid misuse and overdose.
“The CDC Guideline for Prescribing Opioids for Chronic Pain, United States, 2016 will help primary care providers ensure the safest and most effective treatment for their patients,” according to the statement. The CDC’s director, Dr. Tom Frieden, noted that “overprescribing opioids – largely for chronic pain – is a key driver of America’s drug-overdose epidemic.”
In a CDC teleconference marking the release of the guideline, Dr. Frieden said it has become increasingly clear that opioids “carry substantial risks but only uncertain benefits, especially compared with other treatments for chronic pain.
“Beginning treatment with an opioid is a momentous decision, and it should only be done with full understanding by both the clinician and the patient of the substantial risks and uncertain benefits involved,” Dr. Frieden said. He added that he knows of no other medication “that’s routinely used for a nonfatal condition [and] that kills patients so frequently.
“With more than 250 million prescriptions written each year, it’s so important that doctors understand that any one of those prescriptions could potentially end a patient’s life,” he cautioned.
A 2015 study showed that 1 of every 550 patients treated with opioids for noncancer pain – and 1 of 32 who received the highest doses (more than 200 morphine milligram equivalents per day) – died within 2.5 years of the first prescription.
Dr. Frieden noted that opioids do have a place when the potential benefits outweigh the potential harms. “But for most patients – the vast majority of patients – the risks will outweigh the benefits,” he said.
The opioid epidemic is one of the most pressing public health issues in the United States today, said Sylvia M. Burwell, secretary of the Department of Health & Human Services. A year ago, she announced an HHS initiative to reduce prescription opioid and heroin-related drug overdose, death, and dependence.
“Last year, more Americans died from drug overdoses than car crashes,” Ms. Burwell said during the teleconference, noting that families across the nation and from all walks of life have been affected.
Combating the opioid epidemic is a national priority, she said, and the CDC guideline will help in that effort.
“We believe this guideline will help health care professionals provide safer and more effective care for patients dealing with chronic pain, and we also believe it will help these providers drive down the rates of opioid use disorder, overdose, and ... death,” she said.
The American Medical Association greeted the guideline with cautious support.
“While we are largely supportive of the guidelines, we remain concerned about the evidence base informing some of the recommendations,” noted Dr. Patrice A. Harris, chair-elect of the AMA board and chair of the AMA Task Force to Reduce Opioid Abuse, in a statement.
The AMA also cited potential conflicts between the guideline and product labeling and state laws, as well as obstacles such as insurance coverage limits on nonpharmacologic treatments.
“If these guidelines help reduce the deaths resulting from opioids, they will prove to be valuable,” Dr. Harris said in the statement. “If they produce unintended consequences, we will need to mitigate them.”
Of note, the guideline stresses the right of patients with chronic pain to receive safe and effective pain management, and focuses on giving primary care providers – who account for about half of all opioid prescriptions – a road map for providing such pain management by increasing the use of effective nonopioid and nonpharmacologic therapies.
It was developed through a “rigorous scientific process using the best available scientific evidence, consulting with experts, and listening to comments from the public and partner organizations,” according to the CDC statement. The organization “is dedicated to working with partners to improve the evidence base and will refine the recommendations as new research becomes available.
”In conjunction with the release of the guideline, the CDC has provided a checklist for prescribing opioids for chronic pain, and a website with additional tools for implementing the recommendations within the guideline.
The CDC's opioid recommendations
The Centers for Disease Control and Prevention’s new opioid prescription guideline includes 12 recommendations. Here they are, modified slightly for style:
1. Nonpharmacologic therapy and nonopioid pharmacologic therapy are preferred for chronic pain. Providers should only consider adding opioid therapy if expected benefits for both pain and function are anticipated to outweigh risks.
2. Before starting opioid therapy for chronic pain, providers should establish treatment goals with all patients, including realistic goals for pain and function. Providers should not initiate opioid therapy without consideration of how therapy will be discontinued if unsuccessful. Providers should continue opioid therapy only if there is clinically meaningful improvement in pain and function.
3. Before starting and periodically during opioid therapy, providers should discuss with patients known risks and realistic benefits of opioid therapy, and patient and provider responsibilities for managing therapy.
4. When starting opioid therapy for chronic pain, providers should prescribe immediate-release opioids instead of extended-release/long-acting opioids.
5. When opioids are started, providers should prescribe the lowest effective dosage. Providers should use caution when prescribing opioids at any dosage, should implement additional precautions when increasing dosage to 50 or more morphine milligram equivalents (MME) per day, and generally should avoid increasing dosage to 90 or more MME per day.
6. When opioids are used for acute pain, providers should prescribe the lowest effective dose of immediate-release opioids. Three or fewer days often will be sufficient.
7. Providers should evaluate the benefits and harms with patients within 1-4 weeks of starting opioid therapy for chronic pain or of dose escalation. They should reevaluate continued therapy’s benefits and harms every 3 months or more frequently. If continued therapy’s benefits do not outweigh harms, providers should work with patients to reduce dosages or discontinue opioids.
8. During therapy, providers should evaluate risk factors for opioid-related harm. Providers should incorporate into the management plan strategies to mitigate risk, including considering offering naloxone when factors that increase risk for opioid overdose – such as history of overdose, history of substance use disorder, or higher opioid dosage (50 MME or more) – are present.
9. Providers should review the patient’s history of controlled substance prescriptions using state prescription drug monitoring program (PDMP) data to determine whether the patient is receiving high opioid dosages or dangerous combinations that put him or her at high risk for overdose. Providers should review PDMP data when starting opioid therapy for chronic pain and periodically during opioid therapy for chronic pain, ranging from every prescription to every 3 months.
10. When prescribing opioids for chronic pain, providers should use urine drug testing before starting opioid therapy and consider urine drug testing at least annually to assess for prescribed medications, as well as other controlled prescription drugs and illicit drugs.
11. Providers should avoid concurrent prescriptions of opioid pain medication and benzodiazepines whenever possible.
12. Providers should offer or arrange evidence-based treatment (usually medication-assisted treatment with buprenorphine or methadone in combination with behavioral therapies) for patients with opioid use disorder.
M. Alexander Otto contributed to this article.
Anti-Remicade antibodies also cross-react with infliximab biosimilar
Antibodies to the innovator infliximab drug Remicade found in rheumatoid arthritis and spondyloarthritis patients also cross-react with the infliximab biosimilar CT-P13, marketed as Remsima or Inflectra, suggesting that switches from the innovator drug to the biosimilar are not advisable in the presence of anti-infliximab antibodies.
Switching an antibody-positive patient from the innovator drug to the biosimilar could mean that existing infliximab antibodies will “interact with the new drug, enhance clearance, and potentially lead to loss of response and infusion-related reactions,” wrote first author M. Begoña Ruiz-Argüello, Ph.D., an employee of the molecular biology testing company Progenika-Grifols in Derio, Spain, and colleagues (Ann Rheum Dis. 2016 Mar 10. doi: 10.1136/annrheumdis-2015-208684).

In the current study, the investigators set out to discover whether anti-Remicade antibodies cross-reacted with the biosimilar CT-P13, which was approved by the European Medicines Agency in 2013 for the same indications as the originator infliximab biologic Remicade.
They retrospectively selected 250 patients with rheumatoid arthritis (RA) or spondyloarthritis (SpA) who were treated with Remicade and 77 control patients who were infliximab naive.
Antibodies to infliximab were measured at the same time using three bridging ELISA assays: one that used Remicade to detect antibodies (Promonitor-ANTI-IFX kit, Progenika-Grifols, Spain); one that used Remsima (Orion Pharma, Norway); and another that used Inflectra (Hospira, United States).
Overall, 126 (50.4%) patients tested positive for antibodies using the Promonitor-ANTI-IFX kit.
These patients also tested positive for antibodies when the Remsima and Inflectra assays were used. Median antibody concentrations between the assays were not statistically different (P greater than .05). No significant differences were observed between patients with RA and SpA (P greater than .05) or in patients on concomitant immunosuppressive treatment, such as methotrexate.
Contrary to previous research, patients who tested negative for antibodies with the Promonitor-ANTI-IFX kit also tested negative with the Remsima and Inflectra assays. “Although additional epitopes may be present in the biosimilar, results suggest that epitopes influencing the immune response to [infliximab] are also present in the biosimilar,” the researchers said.
The investigators said that their findings also supported the use of therapeutic drug monitoring before considering switching patients between drugs.
Although the researchers recommended not switching between Remicade and Remsima or Inflectra, a small subanalysis in their study suggests it would be okay to switch from adalimumab to the infliximab biosimilar. A control population of 19 patients involved in the study who were anti–adalimumab antibody positive tested negative for antibodies to infliximab across the three assays.
Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.
Antibodies to the innovator infliximab drug Remicade found in rheumatoid arthritis and spondyloarthritis patients also cross-react with the infliximab biosimilar CT-P13, marketed as Remsima or Inflectra, suggesting that switches from the innovator drug to the biosimilar are not advisable in the presence of anti-infliximab antibodies.
Switching an antibody-positive patient from the innovator drug to the biosimilar could mean that existing infliximab antibodies will “interact with the new drug, enhance clearance, and potentially lead to loss of response and infusion-related reactions,” wrote first author M. Begoña Ruiz-Argüello, Ph.D., an employee of the molecular biology testing company Progenika-Grifols in Derio, Spain, and colleagues (Ann Rheum Dis. 2016 Mar 10. doi: 10.1136/annrheumdis-2015-208684).
In the current study, the investigators set out to discover whether anti-Remicade antibodies cross-reacted with the biosimilar CT-P13, which was approved by the European Medicines Agency in 2013 for the same indications as the originator infliximab biologic Remicade.
They retrospectively selected 250 patients with rheumatoid arthritis (RA) or spondyloarthritis (SpA) who were treated with Remicade and 77 control patients who were infliximab naive.
Antibodies to infliximab were measured at the same time using three bridging ELISA assays: one that used Remicade to detect antibodies (Promonitor-ANTI-IFX kit, Progenika-Grifols, Spain); one that used Remsima (Orion Pharma, Norway); and another that used Inflectra (Hospira, United States).
Overall, 126 (50.4%) patients tested positive for antibodies using the Promonitor-ANTI-IFX kit.
These patients also tested positive for antibodies when the Remsima and Inflectra assays were used. Median antibody concentrations between the assays were not statistically different (P greater than .05). No significant differences were observed between patients with RA and SpA (P greater than .05) or in patients on concomitant immunosuppressive treatment, such as methotrexate.
Contrary to previous research, patients who tested negative for antibodies with the Promonitor-ANTI-IFX kit also tested negative with the Remsima and Inflectra assays. “Although additional epitopes may be present in the biosimilar, results suggest that epitopes influencing the immune response to [infliximab] are also present in the biosimilar,” the researchers said.
The investigators said that their findings also supported the use of therapeutic drug monitoring before considering switching patients between drugs.
Although the researchers recommended not switching between Remicade and Remsima or Inflectra, a small subanalysis in their study suggests it would be okay to switch from adalimumab to the infliximab biosimilar. A control population of 19 patients involved in the study who were anti–adalimumab antibody positive tested negative for antibodies to infliximab across the three assays.
Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.
Antibodies to the innovator infliximab drug Remicade found in rheumatoid arthritis and spondyloarthritis patients also cross-react with the infliximab biosimilar CT-P13, marketed as Remsima or Inflectra, suggesting that switches from the innovator drug to the biosimilar are not advisable in the presence of anti-infliximab antibodies.
Switching an antibody-positive patient from the innovator drug to the biosimilar could mean that existing infliximab antibodies will “interact with the new drug, enhance clearance, and potentially lead to loss of response and infusion-related reactions,” wrote first author M. Begoña Ruiz-Argüello, Ph.D., an employee of the molecular biology testing company Progenika-Grifols in Derio, Spain, and colleagues (Ann Rheum Dis. 2016 Mar 10. doi: 10.1136/annrheumdis-2015-208684).
In the current study, the investigators set out to discover whether anti-Remicade antibodies cross-reacted with the biosimilar CT-P13, which was approved by the European Medicines Agency in 2013 for the same indications as the originator infliximab biologic Remicade.
They retrospectively selected 250 patients with rheumatoid arthritis (RA) or spondyloarthritis (SpA) who were treated with Remicade and 77 control patients who were infliximab naive.
Antibodies to infliximab were measured at the same time using three bridging ELISA assays: one that used Remicade to detect antibodies (Promonitor-ANTI-IFX kit, Progenika-Grifols, Spain); one that used Remsima (Orion Pharma, Norway); and another that used Inflectra (Hospira, United States).
Overall, 126 (50.4%) patients tested positive for antibodies using the Promonitor-ANTI-IFX kit.
These patients also tested positive for antibodies when the Remsima and Inflectra assays were used. Median antibody concentrations between the assays were not statistically different (P greater than .05). No significant differences were observed between patients with RA and SpA (P greater than .05) or in patients on concomitant immunosuppressive treatment, such as methotrexate.
Contrary to previous research, patients who tested negative for antibodies with the Promonitor-ANTI-IFX kit also tested negative with the Remsima and Inflectra assays. “Although additional epitopes may be present in the biosimilar, results suggest that epitopes influencing the immune response to [infliximab] are also present in the biosimilar,” the researchers said.
The investigators said that their findings also supported the use of therapeutic drug monitoring before considering switching patients between drugs.
Although the researchers recommended not switching between Remicade and Remsima or Inflectra, a small subanalysis in their study suggests it would be okay to switch from adalimumab to the infliximab biosimilar. A control population of 19 patients involved in the study who were anti–adalimumab antibody positive tested negative for antibodies to infliximab across the three assays.
Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: Rheumatology patients with positive antibodies to Remicade should not be switched to infliximab biosimilar (Remsima, Inflectra).
Major finding: Antibodies to infliximab in Remicade-treated rheumatology patients showed identical reactivity towards the biosimilar CT-P13.
Data source: A retrospective study of 250 consecutive patients with RA and SpA taking Remicade and 77 infliximab-naive controls.
Disclosures: Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.

Fresh evidence of methotrexate efficacy in psoriatic arthritis
MAUI, HAWAII – The effectiveness of methotrexate in psoriatic arthritis is a matter of debate, but Dr. Arthur Kavanaugh is a believer based in part upon a recent subanalysis of the TICOPA study.
Moreover, his new 5-year follow-up analysis from the GO-REVEAL study of golimumab (Simponi) with or without concomitant methotrexate suggests that methotrexate plus the tumor necrosis factor inhibitor provided synergistic efficacy, he said at the 2016 Rheumatology Winter Clinical Symposium.
The 5-year analysis doesn’t provide definitive proof of synergistic benefit because it wasn’t designed or powered with that endpoint in mind (Arthritis Care Res. 2016;68[2]:267–74). No randomized trial completed to date has been. But the first-ever trial set up to test the synergistic efficacy hypothesis is underway. It’s a 52-week, double-blind, multicenter, randomized trial of etanercept (Enbrel) and methotrexate versus either alone in combination with placebo. And while the Amgen-sponsored study won’t be completed until 2018, Dr. Kavanaugh is ready to predict the outcome based in part upon the message contained in his GO-REVEAL findings.

“I’m placing my bet down now that there will be synergy for the X-ray outcome of change in SHS [Sharp/van der Heijde Score] for sure, and maybe for clinical efficacy as well, both joints and skin,” declared Dr. Kavanaugh, the conference director and professor of medicine at the University of California, San Diego.
He pointed to a new subanalysis of the Tight Control of Psoriatic Arthritis (TICOPA) study reported by rheumatologists at the University of Leeds (England) as evidence that methotrexate is effective in psoriatic arthritis. Of the 188 patients in the tight control arm who received methotrexate in the first 12 weeks of the trial, 41% had an ACR 20 response, meaning a 20% improvement in disease signs and symptoms at 12 weeks. A total of 19% had an ACR 50 response. And 27% had at least a 75% improvement in Psoriasis Area and Severity Index, or PASI 75. A 63% reduction in the proportion of patients with dactylitis and a 26% decrease in the proportion of patients with enthesitis was observed in the early methotrexate group. There was a suggestion of a dose-response effect, with better outcomes seen in the 108 participants who received a mean dose greater than 15 mg/week (J Rheumatol. 2016 Feb;43[2]:356-61).
This is a more impressive result than earlier reported from the Methotrexate In Psoriatic Arthritis (MIPA) trial, where the ACR 20 response rate was only 34% (Rheumatology [Oxford]. 2012;51[8]:1368-77). That may well be because methotrexate was given at only 15 mg/week in MIPA, in Dr. Kavanaugh’s view.
“I think methotrexate can work for the peripheral arthritis. This TICOPA analysis gives us a sense of the extent of the improvement, and also the extent of improvement in the skin,” the rheumatologist commented.
Turning to the week 256 results of GO-REVEAL, he said there was no difference in clinical response between psoriatic arthritis patients on golimumab alone or golimumab plus methotrexate at baseline. But among patients who were doing well clinically, with an assessment of minimal disease activity (MDA) on three or more consecutive clinic visits, only those on golimumab plus methotrexate at baseline showed radiologic improvement. The 57 patients on combination therapy who achieved MDA on at least three consecutive visits showed a mean 1.29-point improvement in SHS; the 48 rated as having MDA on four or more consecutive occasions similarly had a mean 1.24-point improvement.
In contrast, the 59 participants who achieved MDA on three or more consecutive visits but were on golimumab without methotrexate at baseline had a 0.25-point increase in SHS, and the 47 who had MDA on at least four consecutive visits had a 0.38-point SHS bump.
Dr. Kavanaugh reported having financial relationships with roughly a dozen pharmaceutical companies
MAUI, HAWAII – The effectiveness of methotrexate in psoriatic arthritis is a matter of debate, but Dr. Arthur Kavanaugh is a believer based in part upon a recent subanalysis of the TICOPA study.
Moreover, his new 5-year follow-up analysis from the GO-REVEAL study of golimumab (Simponi) with or without concomitant methotrexate suggests that methotrexate plus the tumor necrosis factor inhibitor provided synergistic efficacy, he said at the 2016 Rheumatology Winter Clinical Symposium.
The 5-year analysis doesn’t provide definitive proof of synergistic benefit because it wasn’t designed or powered with that endpoint in mind (Arthritis Care Res. 2016;68[2]:267–74). No randomized trial completed to date has been. But the first-ever trial set up to test the synergistic efficacy hypothesis is underway. It’s a 52-week, double-blind, multicenter, randomized trial of etanercept (Enbrel) and methotrexate versus either alone in combination with placebo. And while the Amgen-sponsored study won’t be completed until 2018, Dr. Kavanaugh is ready to predict the outcome based in part upon the message contained in his GO-REVEAL findings.
“I’m placing my bet down now that there will be synergy for the X-ray outcome of change in SHS [Sharp/van der Heijde Score] for sure, and maybe for clinical efficacy as well, both joints and skin,” declared Dr. Kavanaugh, the conference director and professor of medicine at the University of California, San Diego.
He pointed to a new subanalysis of the Tight Control of Psoriatic Arthritis (TICOPA) study reported by rheumatologists at the University of Leeds (England) as evidence that methotrexate is effective in psoriatic arthritis. Of the 188 patients in the tight control arm who received methotrexate in the first 12 weeks of the trial, 41% had an ACR 20 response, meaning a 20% improvement in disease signs and symptoms at 12 weeks. A total of 19% had an ACR 50 response. And 27% had at least a 75% improvement in Psoriasis Area and Severity Index, or PASI 75. A 63% reduction in the proportion of patients with dactylitis and a 26% decrease in the proportion of patients with enthesitis was observed in the early methotrexate group. There was a suggestion of a dose-response effect, with better outcomes seen in the 108 participants who received a mean dose greater than 15 mg/week (J Rheumatol. 2016 Feb;43[2]:356-61).
This is a more impressive result than earlier reported from the Methotrexate In Psoriatic Arthritis (MIPA) trial, where the ACR 20 response rate was only 34% (Rheumatology [Oxford]. 2012;51[8]:1368-77). That may well be because methotrexate was given at only 15 mg/week in MIPA, in Dr. Kavanaugh’s view.
“I think methotrexate can work for the peripheral arthritis. This TICOPA analysis gives us a sense of the extent of the improvement, and also the extent of improvement in the skin,” the rheumatologist commented.
Turning to the week 256 results of GO-REVEAL, he said there was no difference in clinical response between psoriatic arthritis patients on golimumab alone or golimumab plus methotrexate at baseline. But among patients who were doing well clinically, with an assessment of minimal disease activity (MDA) on three or more consecutive clinic visits, only those on golimumab plus methotrexate at baseline showed radiologic improvement. The 57 patients on combination therapy who achieved MDA on at least three consecutive visits showed a mean 1.29-point improvement in SHS; the 48 rated as having MDA on four or more consecutive occasions similarly had a mean 1.24-point improvement.
In contrast, the 59 participants who achieved MDA on three or more consecutive visits but were on golimumab without methotrexate at baseline had a 0.25-point increase in SHS, and the 47 who had MDA on at least four consecutive visits had a 0.38-point SHS bump.
Dr. Kavanaugh reported having financial relationships with roughly a dozen pharmaceutical companies
MAUI, HAWAII – The effectiveness of methotrexate in psoriatic arthritis is a matter of debate, but Dr. Arthur Kavanaugh is a believer based in part upon a recent subanalysis of the TICOPA study.
Moreover, his new 5-year follow-up analysis from the GO-REVEAL study of golimumab (Simponi) with or without concomitant methotrexate suggests that methotrexate plus the tumor necrosis factor inhibitor provided synergistic efficacy, he said at the 2016 Rheumatology Winter Clinical Symposium.
The 5-year analysis doesn’t provide definitive proof of synergistic benefit because it wasn’t designed or powered with that endpoint in mind (Arthritis Care Res. 2016;68[2]:267–74). No randomized trial completed to date has been. But the first-ever trial set up to test the synergistic efficacy hypothesis is underway. It’s a 52-week, double-blind, multicenter, randomized trial of etanercept (Enbrel) and methotrexate versus either alone in combination with placebo. And while the Amgen-sponsored study won’t be completed until 2018, Dr. Kavanaugh is ready to predict the outcome based in part upon the message contained in his GO-REVEAL findings.
“I’m placing my bet down now that there will be synergy for the X-ray outcome of change in SHS [Sharp/van der Heijde Score] for sure, and maybe for clinical efficacy as well, both joints and skin,” declared Dr. Kavanaugh, the conference director and professor of medicine at the University of California, San Diego.
He pointed to a new subanalysis of the Tight Control of Psoriatic Arthritis (TICOPA) study reported by rheumatologists at the University of Leeds (England) as evidence that methotrexate is effective in psoriatic arthritis. Of the 188 patients in the tight control arm who received methotrexate in the first 12 weeks of the trial, 41% had an ACR 20 response, meaning a 20% improvement in disease signs and symptoms at 12 weeks. A total of 19% had an ACR 50 response. And 27% had at least a 75% improvement in Psoriasis Area and Severity Index, or PASI 75. A 63% reduction in the proportion of patients with dactylitis and a 26% decrease in the proportion of patients with enthesitis was observed in the early methotrexate group. There was a suggestion of a dose-response effect, with better outcomes seen in the 108 participants who received a mean dose greater than 15 mg/week (J Rheumatol. 2016 Feb;43[2]:356-61).
This is a more impressive result than earlier reported from the Methotrexate In Psoriatic Arthritis (MIPA) trial, where the ACR 20 response rate was only 34% (Rheumatology [Oxford]. 2012;51[8]:1368-77). That may well be because methotrexate was given at only 15 mg/week in MIPA, in Dr. Kavanaugh’s view.
“I think methotrexate can work for the peripheral arthritis. This TICOPA analysis gives us a sense of the extent of the improvement, and also the extent of improvement in the skin,” the rheumatologist commented.
Turning to the week 256 results of GO-REVEAL, he said there was no difference in clinical response between psoriatic arthritis patients on golimumab alone or golimumab plus methotrexate at baseline. But among patients who were doing well clinically, with an assessment of minimal disease activity (MDA) on three or more consecutive clinic visits, only those on golimumab plus methotrexate at baseline showed radiologic improvement. The 57 patients on combination therapy who achieved MDA on at least three consecutive visits showed a mean 1.29-point improvement in SHS; the 48 rated as having MDA on four or more consecutive occasions similarly had a mean 1.24-point improvement.
In contrast, the 59 participants who achieved MDA on three or more consecutive visits but were on golimumab without methotrexate at baseline had a 0.25-point increase in SHS, and the 47 who had MDA on at least four consecutive visits had a 0.38-point SHS bump.
Dr. Kavanaugh reported having financial relationships with roughly a dozen pharmaceutical companies
EXPERT ANALYSIS FROM RWCS 2016

Expert examines secukinumab’s role in ankylosing spondylitis treatment strategies
MAUI, HAWAII – The most important development within the past year in the treatment of ankylosing spondylitis was the Food and Drug Administration approval of secukinumab (Cosentyx) as the first non-tumor necrosis factor inhibitor biologic for this condition – but the interleukin-17A inhibitor is not going to immediately step into a role as a first-line therapy, Dr. Eric M. Ruderman predicted at the 2016 Rheumatology Winter Clinical Symposium.
“In all likelihood nobody’s going to use this as a first-line drug right out of the gate. It’s a drug you’re going to potentially go to in people who haven’t responded to the things that you’ve been comfortable using for the last 10 or 15 years. So the big practical issue becomes, ‘How does secukinumab perform in TNF inhibitor-naive patients versus prior TNF inhibitor inadequate responders?’ ” according to the rheumatologist, who is professor of medicine at Northwestern University in Chicago.

This question has been addressed in secondary analyses of the pivotal phase III MEASURE 1 and MEASURE 2 trials which have been presented at the annual European League Against Rheumatism and American College of Rheumatology meetings. The bottom line was that the therapeutic response rate in both trials was markedly lower in TNF inhibitor inadequate responders than in TNF inhibitor-naive subjects.
“But there still is a significant response rate in the inadequate responders. It’s clearly better than placebo. So this is a drug that may have a role in your practice at the point where patients have failed on one or two anti-TNF biologics,” according to Dr. Ruderman.
The difference between MEASURE 1 and MEASURE 2 is that MEASURE 1 entailed three intravenous loading doses of the biologic at 2-week intervals before switching to monthly subcutaneous dosing, while MEASURE 2 featured subcutaneous loading doses given weekly for 4 weeks before moving to monthly administration. Interestingly, the FDA approval of secukinumab at 150 mg doesn’t call for a loading dose, even though both pivotal trials relied on them, the rheumatologist observed.
At 16 weeks in MEASURE 1, 66% of TNF inhibitor-naive subjects on secukinumab 150 mg had at least a 20% improvement from baseline in ankylosing spondylitis signs and symptoms, or Assessment of Spondyloarthritis International Society (ASAS) 20, compared with 46% of TNF inhibitor inadequate responders. The week 16 ASAS 20 rate in MEASURE 2 was 68% in TNF inhibitor-naive patients and 50% in those with a prior inadequate response to TNF inhibitor therapy.
How should rheumatologists expect secukinumab to perform in daily clinical practice? In the 181 ankylosing spoindylitis patients who completed 52 weeks in the MEASURE 2 extension study, 74% of those on secukinumab at 150 mg had an ASAS 20 response. In both trials, the secukinumab side effect profile was “reasonably clean,” in Dr. Ruderman’s view, with serious adverse events that were similar to placebo.
Serial MRI scans showed rapid resolution of bone marrow edema and inflammation by 16 weeks, an effect sustained through 52 weeks.
The big unanswered question is whether secukinumab prevents radiographic progression of the disease. Serial cervical and spinal X-rays rated using the modified Stoke Ankylosing Spondylitis Spinal Score showed a mean increase of just 0.30 points at 2 years from a baseline of 10.22, with 80% of patients demonstrating no change over time. But there were no untreated controls for comparison in this analysis, so it’s not possible to say whether the drug actually slowed disease progression or that’s the natural history of disease in those subjects, Dr. Ruderman noted.
Effect of NSAID dosing frequency on progression
On the topic of preventing radiographic progression in ankylosing spondylitis, the rheumatologist highlighted a prospective study presented at last year’s EULAR meeting and published online last summer (Ann Rheum Dis. 2015 Aug 4. doi: 10.1136/annrheumdis-2015-207897) that demonstrated that continuous use of diclofenac didn’t do any better at preventing radiographic spinal disease progression than on-demand use of the nonsteroidal anti-inflammatory drug (NSAID) over the course of 2 years.
“There’s been a lot of noise in the ankylosing spondylitis community about the potential benefit of NSAIDs in preventing structural progression. Previous information suggested that staying on them continuously actually reduced radiographic progression. This diclofenac study has shaken things up a little. It raises the question of whether there is any added benefit for NSAIDs in terms of structural progression,” he commented.
Current ACR/SAA/SPARTAN guidelines, which predate the study, feature a conditional recommendation that patients with active ankylosing spondylitis stay on continuous NSAID therapy.
Secukinumab is also approved for treatment of psoriasis and psoriatic arthritis.
Dr. Ruderman reported serving as a consultant to and/or receiving research grants from numerous pharmaceutical companies, including Novartis, which markets secukinumab.
MAUI, HAWAII – The most important development within the past year in the treatment of ankylosing spondylitis was the Food and Drug Administration approval of secukinumab (Cosentyx) as the first non-tumor necrosis factor inhibitor biologic for this condition – but the interleukin-17A inhibitor is not going to immediately step into a role as a first-line therapy, Dr. Eric M. Ruderman predicted at the 2016 Rheumatology Winter Clinical Symposium.
“In all likelihood nobody’s going to use this as a first-line drug right out of the gate. It’s a drug you’re going to potentially go to in people who haven’t responded to the things that you’ve been comfortable using for the last 10 or 15 years. So the big practical issue becomes, ‘How does secukinumab perform in TNF inhibitor-naive patients versus prior TNF inhibitor inadequate responders?’ ” according to the rheumatologist, who is professor of medicine at Northwestern University in Chicago.
This question has been addressed in secondary analyses of the pivotal phase III MEASURE 1 and MEASURE 2 trials which have been presented at the annual European League Against Rheumatism and American College of Rheumatology meetings. The bottom line was that the therapeutic response rate in both trials was markedly lower in TNF inhibitor inadequate responders than in TNF inhibitor-naive subjects.
“But there still is a significant response rate in the inadequate responders. It’s clearly better than placebo. So this is a drug that may have a role in your practice at the point where patients have failed on one or two anti-TNF biologics,” according to Dr. Ruderman.
The difference between MEASURE 1 and MEASURE 2 is that MEASURE 1 entailed three intravenous loading doses of the biologic at 2-week intervals before switching to monthly subcutaneous dosing, while MEASURE 2 featured subcutaneous loading doses given weekly for 4 weeks before moving to monthly administration. Interestingly, the FDA approval of secukinumab at 150 mg doesn’t call for a loading dose, even though both pivotal trials relied on them, the rheumatologist observed.
At 16 weeks in MEASURE 1, 66% of TNF inhibitor-naive subjects on secukinumab 150 mg had at least a 20% improvement from baseline in ankylosing spondylitis signs and symptoms, or Assessment of Spondyloarthritis International Society (ASAS) 20, compared with 46% of TNF inhibitor inadequate responders. The week 16 ASAS 20 rate in MEASURE 2 was 68% in TNF inhibitor-naive patients and 50% in those with a prior inadequate response to TNF inhibitor therapy.
How should rheumatologists expect secukinumab to perform in daily clinical practice? In the 181 ankylosing spoindylitis patients who completed 52 weeks in the MEASURE 2 extension study, 74% of those on secukinumab at 150 mg had an ASAS 20 response. In both trials, the secukinumab side effect profile was “reasonably clean,” in Dr. Ruderman’s view, with serious adverse events that were similar to placebo.
Serial MRI scans showed rapid resolution of bone marrow edema and inflammation by 16 weeks, an effect sustained through 52 weeks.
The big unanswered question is whether secukinumab prevents radiographic progression of the disease. Serial cervical and spinal X-rays rated using the modified Stoke Ankylosing Spondylitis Spinal Score showed a mean increase of just 0.30 points at 2 years from a baseline of 10.22, with 80% of patients demonstrating no change over time. But there were no untreated controls for comparison in this analysis, so it’s not possible to say whether the drug actually slowed disease progression or that’s the natural history of disease in those subjects, Dr. Ruderman noted.
Effect of NSAID dosing frequency on progression
On the topic of preventing radiographic progression in ankylosing spondylitis, the rheumatologist highlighted a prospective study presented at last year’s EULAR meeting and published online last summer (Ann Rheum Dis. 2015 Aug 4. doi: 10.1136/annrheumdis-2015-207897) that demonstrated that continuous use of diclofenac didn’t do any better at preventing radiographic spinal disease progression than on-demand use of the nonsteroidal anti-inflammatory drug (NSAID) over the course of 2 years.
“There’s been a lot of noise in the ankylosing spondylitis community about the potential benefit of NSAIDs in preventing structural progression. Previous information suggested that staying on them continuously actually reduced radiographic progression. This diclofenac study has shaken things up a little. It raises the question of whether there is any added benefit for NSAIDs in terms of structural progression,” he commented.
Current ACR/SAA/SPARTAN guidelines, which predate the study, feature a conditional recommendation that patients with active ankylosing spondylitis stay on continuous NSAID therapy.
Secukinumab is also approved for treatment of psoriasis and psoriatic arthritis.
Dr. Ruderman reported serving as a consultant to and/or receiving research grants from numerous pharmaceutical companies, including Novartis, which markets secukinumab.
MAUI, HAWAII – The most important development within the past year in the treatment of ankylosing spondylitis was the Food and Drug Administration approval of secukinumab (Cosentyx) as the first non-tumor necrosis factor inhibitor biologic for this condition – but the interleukin-17A inhibitor is not going to immediately step into a role as a first-line therapy, Dr. Eric M. Ruderman predicted at the 2016 Rheumatology Winter Clinical Symposium.
“In all likelihood nobody’s going to use this as a first-line drug right out of the gate. It’s a drug you’re going to potentially go to in people who haven’t responded to the things that you’ve been comfortable using for the last 10 or 15 years. So the big practical issue becomes, ‘How does secukinumab perform in TNF inhibitor-naive patients versus prior TNF inhibitor inadequate responders?’ ” according to the rheumatologist, who is professor of medicine at Northwestern University in Chicago.
This question has been addressed in secondary analyses of the pivotal phase III MEASURE 1 and MEASURE 2 trials which have been presented at the annual European League Against Rheumatism and American College of Rheumatology meetings. The bottom line was that the therapeutic response rate in both trials was markedly lower in TNF inhibitor inadequate responders than in TNF inhibitor-naive subjects.
“But there still is a significant response rate in the inadequate responders. It’s clearly better than placebo. So this is a drug that may have a role in your practice at the point where patients have failed on one or two anti-TNF biologics,” according to Dr. Ruderman.
The difference between MEASURE 1 and MEASURE 2 is that MEASURE 1 entailed three intravenous loading doses of the biologic at 2-week intervals before switching to monthly subcutaneous dosing, while MEASURE 2 featured subcutaneous loading doses given weekly for 4 weeks before moving to monthly administration. Interestingly, the FDA approval of secukinumab at 150 mg doesn’t call for a loading dose, even though both pivotal trials relied on them, the rheumatologist observed.
At 16 weeks in MEASURE 1, 66% of TNF inhibitor-naive subjects on secukinumab 150 mg had at least a 20% improvement from baseline in ankylosing spondylitis signs and symptoms, or Assessment of Spondyloarthritis International Society (ASAS) 20, compared with 46% of TNF inhibitor inadequate responders. The week 16 ASAS 20 rate in MEASURE 2 was 68% in TNF inhibitor-naive patients and 50% in those with a prior inadequate response to TNF inhibitor therapy.
How should rheumatologists expect secukinumab to perform in daily clinical practice? In the 181 ankylosing spoindylitis patients who completed 52 weeks in the MEASURE 2 extension study, 74% of those on secukinumab at 150 mg had an ASAS 20 response. In both trials, the secukinumab side effect profile was “reasonably clean,” in Dr. Ruderman’s view, with serious adverse events that were similar to placebo.
Serial MRI scans showed rapid resolution of bone marrow edema and inflammation by 16 weeks, an effect sustained through 52 weeks.
The big unanswered question is whether secukinumab prevents radiographic progression of the disease. Serial cervical and spinal X-rays rated using the modified Stoke Ankylosing Spondylitis Spinal Score showed a mean increase of just 0.30 points at 2 years from a baseline of 10.22, with 80% of patients demonstrating no change over time. But there were no untreated controls for comparison in this analysis, so it’s not possible to say whether the drug actually slowed disease progression or that’s the natural history of disease in those subjects, Dr. Ruderman noted.
Effect of NSAID dosing frequency on progression
On the topic of preventing radiographic progression in ankylosing spondylitis, the rheumatologist highlighted a prospective study presented at last year’s EULAR meeting and published online last summer (Ann Rheum Dis. 2015 Aug 4. doi: 10.1136/annrheumdis-2015-207897) that demonstrated that continuous use of diclofenac didn’t do any better at preventing radiographic spinal disease progression than on-demand use of the nonsteroidal anti-inflammatory drug (NSAID) over the course of 2 years.
“There’s been a lot of noise in the ankylosing spondylitis community about the potential benefit of NSAIDs in preventing structural progression. Previous information suggested that staying on them continuously actually reduced radiographic progression. This diclofenac study has shaken things up a little. It raises the question of whether there is any added benefit for NSAIDs in terms of structural progression,” he commented.
Current ACR/SAA/SPARTAN guidelines, which predate the study, feature a conditional recommendation that patients with active ankylosing spondylitis stay on continuous NSAID therapy.
Secukinumab is also approved for treatment of psoriasis and psoriatic arthritis.
Dr. Ruderman reported serving as a consultant to and/or receiving research grants from numerous pharmaceutical companies, including Novartis, which markets secukinumab.
EXPERT ANALYSIS FROM RWCS 2016
