User login
2015 Update on Parkinson disease
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
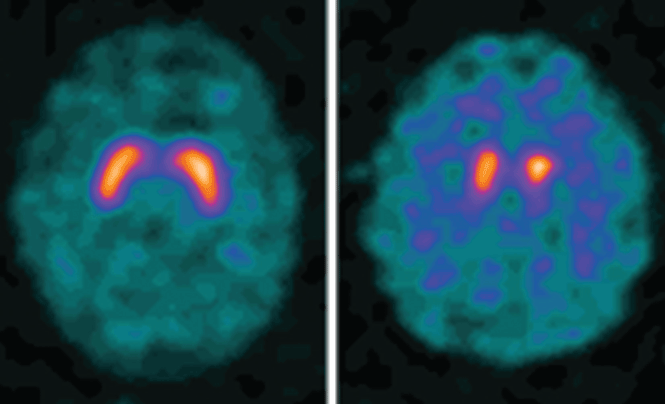
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).

Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).
Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).
Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
KEY POINTS
- Parkinson disease is diagnosed by clinical signs with the mnemonic TRAP: Tremor at rest, Rigidity, Akinesia or bradykinesia, and Postural/gait instability.
- A dopamine transporter functional scan can distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative etiologies such as drug-induced parkinsonism and vascular parkinsonism, and from mimics such as psychogenic parkinsonism and essential tremor.
- Coffee consumption and exercise may benefit patients with Parkinson disease.
- Carbidopa-levodopa combination therapy is still the most effective treatment, but most patients develop dyskinesia after 5 to 10 years of treatment.
- Dyskinesias can be managed by adjusting or changing medications, switching to the new levodopa infusion pump system, or with deep-brain-stimulation surgery.
Nonmotor complications of Parkinson disease
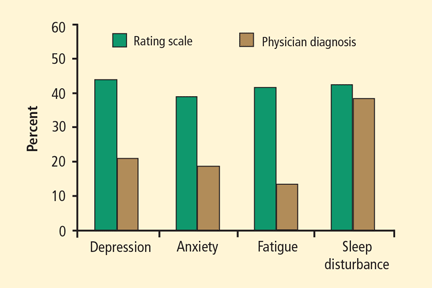
Although the definition of Parkinson disease (PD) is based on the presence of motor features, these are just the “tip of the iceberg.” Nonmotor manifestations are nearly ubiquitous in PD, with behavior problems often being the most malignant. Almost all patients with PD have nonmotor and neuropsychiatric features, including sleep disturbances, compulsive and impulsive behaviors, autonomic dysfunction, and psychosis.
The neuropsychiatric and behavioral features of PD can be classified as intrinsic features, which occur as part of PD, and iatrogenic features, which are complications that arise from treatments used to manage the motor symptoms of PD.
DEMENTIA IN PD
An intrinsic nonmotor feature of PD is dementia, which occurs at a rate four to six times greater in patients with PD than in age-matched controls without PD.1 The prevalence of dementia in PD varies among studies and depends on the demographics of the population being studied. The cross-sectional prevalence of dementia is 40% in patients with PD.2 Seventy-eight percent of a population-based, representative cohort of patients with PD developed dementia during an 8-year study period.3
Dementia is a burden to the caregiver, the patient, and society. Cognitive and behavioral symptoms in patients with PD are the greatest contributors to caregiver distress.4 Dementia and associated behavioral symptoms (ie, hallucinations) hasten nursing home placement, contributing to the financial burden of caring for patients with PD.5 The risk of mortality is increased when dementia develops.6
PSYCHOTIC SYMPTOMS IN PD: AN EFFECT OF EXCESS DOPAMINE STIMULATION
Most of the complications observed in PD can be explained by the dopamine effect of medications and by dopamine deficiencies. An excess of dopamine stimulation caused by administration of prodopaminergic agents manifests as dyskinesias, hallucinations, or delusions. Withdrawal of levodopa will reverse these complications but leads to dopamine deficiency and thus a worsening of PD symptoms. Most patients with PD will tolerate mild dyskinesias or hallucinations if their PD symptoms are well controlled.
The hallucinations in PD tend to be visual as opposed to auditory (as in schizophrenia). They are usually benign and involve figures of people, furry animals, or complex scenes. About 10% to 40% of hallucinations in PD are secondary auditory hallucinations, which tend to be nondistinct, non-paranoid, and often incomprehensible (ie, voices in a crowd).
In the same way, the delusions experienced in patients with PD are distinct from those in schizophrenia. The delusions in PD are usually paranoid in nature and involve stereotyped themes (ie, spousal infidelity, feelings of abandonment) rather than the grandiose delusions that are common in schizophrenia.
The reported prevalence of psychotic symptoms in PD, including hallucinations and delusions, ranges from 20% to 50%.8,9 Auditory hallucinations are a feature in about 10%, and they usually occur with visual hallucinations. Less common are delusions and hallucinations with loss of insight, which are more likely with increasing severity of dementia.
Once a PD patient experiences hallucinations, they are likely to continue. In a 6-year longitudinal study, the prevalence of hallucinations increased from 33% at baseline to 55% at 72 months.10 Persistent psychosis was found in 69% of participants in the Psychosis and Clozapine in PD Study (PSYCLOPS) with 26 months of follow-up.11
High caregiver burden
Psychotic symptoms in PD are associated with high caregiver stress and increased rates of nursing home placement. Goetz et al12 showed that PD patients with psychosis had a much greater risk of nursing home placement than those without psychosis. The prognosis for PD patients in extended-care facilities is worse for those with psychotic symptoms.13
Management of psychotic symptoms
The first step in managing psychosis in PD is to rule out other causes of changes in mental status, such as infection, electrolyte imbalance, or introduction of new medications.
Adjusting anti-PD medications to a tolerable yet effective dose may help to reduce the incidence and severity of psychotic complications. If necessary, selective discontinuation of anti-PD medications may be tried in the following sequence: anticholinergics, amantadine, monoamine oxidase B inhibitors, dopamine agonists, catechol-O-methyltransferase inhibitors, and levodopa/carbidopa.
If motor symptoms prevent dosage minimization or discontinuation of some medications, then the addition of an atypical antipsychotic medication should be considered. Before the advent of atypical antipsychotics, the management of psychosis and hallucinations in PD was unsatisfactory, reflected by a mortality of 100% within 2 years among psychotic PD patients placed in nursing homes compared with 32% among age-matched community dwellers.13 The introduction of atypical antipsychotics has improved survival among PD patients with psychosis. In one study, mortality over 5 years was 44% among PD patients taking long-term clozapine for the treatment of psychosis.14 Recurrence of psychosis is rapid (within 8 weeks) even when PD patients are slowly weaned from atypical antipsychotics.15
Receptor affinities differ among antipsychotics. Because dopamine has been implicated as the principal neurotransmitter in the development of PD psychosis, atypical antipsychotics, with milder dopamine-blocking action, have played a central role in the treatment of PD psychosis. The dopamine D2 receptor is the main target for conventional antipsychotic drugs to exert their clinical effects. Atypical antipsychotics have different affinities for the D2 receptors.16 Occupancy of D2 receptors with atypical antipsychotics is 40% to 70% (risperidone and olanzapine have higher affinity for the D2 receptor than clozapine and quetiapine), and affinity for 5-HT2A receptors can be as high as 70%. This affinity for 5-HT2A receptors relative to D2 receptors may be important for therapeutic efficacy of the atypical antipsychotics. Antagonism of muscarinic, histaminergic, noradrenergic, and other serotonergic receptors also differs among the atypical antipsychotics.
Clozapine remains the gold standard atypical antipsychotic agent, based on results from three relatively small (N = 6 to 60) double-blind, placebo-controlled studies in PD patients with dopaminergic drug-induced psychosis.17–19 Quetiapine improved psychotic symptoms associated with PD in several open-label studies, but has not demonstrated the same success in double-blind clinical trials.20,21
Loss of cholinergic neurons and implications for treatment. In autopsy studies, the loss of cholinergic neurons is more profound in PD than in Alzheimer disease, which suggests that procholinergic drugs may improve symptoms of PD dementia, a major risk factor for hallucinations. In open-label studies, acetylcholinesterase inhibitors have reduced the frequency of hallucinations in patients who have dementia with Lewy bodies (DLB) and in patients with PD dementia. Double-blind trials of patients with DLB and PD dementia concentrated on the effect of cholinesterase inhibitors on dementia and not hallucinations. One concern with the use of a procholinergic drug in patients with PD has been worsening of parkinsonism, but studies of acetylcholinesterase inhibitors have shown no worsening of parkinsonism and only transient worsening of tremor.
Ondansetron, a 5-HT3 receptor antagonist used as an antinausea medication, produced moderate improvements in hallucinations and delusions in an open-label trial for the treatment of psychosis in advanced PD.22 For PD patients with psychosis and comorbid depression, antidepressant therapy and electroconvulsive therapy may be effective options.23,24
MOOD DISTURBANCES IN PD
Depression and apathy occur more frequently in patients with PD than in those who do not have PD.
Depression
Challenges in the management of depression in PD include recognition of depression and distinguishing depressive disorders from mood fluctuations. Whereas a depressive disorder lasts from weeks to years and can occur at any stage of illness, mood fluctuations can change many times daily and appear as nonmotor manifestations during the “off” medication state. Mood fluctuations occur mostly in patients who have developed motor fluctuations. The implication for treatment is that the treatment strategy for a depressive disorder is antidepressant therapy, whereas the strategy for mood fluctuations in PD is to increase the levodopa dose.
Recognition of depression in PD is confounded by the depression criteria of the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition; many of these criteria can be intrinsic features of PD itself—for example, anhedonia, weight/appetite loss or gain, insomnia or hypersomnia, psychomotor retardation, and fatigue. Questions such as “are you feeling sad” or “are you feeling blue” may be superior to questions about associative symptoms when evaluating PD patients for depression.
Most of the medications used for the treatment of depression also work well for depression in patients with PD. Double-blind controlled studies have demonstrated superiority of nortriptyline, citalopram, desipramine, and pramipexole over placebo in improving mood.26–29
Apathy
The overlap between apathy and depressive symptoms can also complicate recognition of apathy, which can be described as a lack of motivation or failure to initiate goal-directed behavior. Apathy involves three domains30:
- Cognitive: expressed as a loss of interest in new experience or a lack of concern about a personal problem
- Diminished affect: flattened affect or a lack of reaction to positive or negative events
- Final: diminished goal-directed cognition, as indicated by a lack of effort or requiring others to structure activities.
Unlike depression, which is similarly representative of PD and other episodic conditions such as dystonia, apathy is more common in PD than in dystonia. In fact, the occurrence of apathy alone distinguishes PD from dystonia. Apathy in PD has no known treatment. If it is associated with depression, apathy may respond to antidepressants.
Repetitive transcranial magnetic stimulation (rTMS) manipulates activity in specific brain neural circuits through the skull to induce changes in behavior. Some studies suggest that modulation of behavior may last beyond the actual stimulation. A randomized, sham-controlled trial of rTMS over the middorsolateral frontal cortex has been conducted with the primary aim of improving apathy in PD. Unfortunately, while patients who were randomized to rTMS experienced some improvement in apathy during the study, the improvement was not significantly different from that observed in patients who received sham treatment.31
IMPULSE CONTROL AND COMPULSIVE DISORDERS IN PD
Impulse control disorders are characterized by the inability to resist an urge to act; the resulting irrational desire to pursue self-gratification may inflict suffering on friends and relatives that compromises relationships and impairs social- and work-related functioning.
Examples of impulse control disorders in PD are pathologic gambling, hypersexuality, compulsive shopping, excessive spending, and binge eating. Patients taking dopamine agonists are two to three times more likely to develop impulse control disorders than those receiving other treatments for PD. Dopamine agonists with relative selectivity for D3 receptors have been implicated in impulse control disorders in PD because D3 receptors are abundant in a region of the brain (ventral striatum) associated with behavioral and substance addictions. Higher levodopa dosages were also associated with impulse control disorders.
Factors associated with impulse control disorders in PD are young age, being single, a family history of impulse control disorders, and levodopa treatment.32 Modifications to dopamine agonist or levodopa therapy are important in the treatment of dopamine agonist–induced impulse disorders.
Compulsive disorders have been described as a class distinct from impulse control disorders and involve repetitive stereotypes and well-ordered acts to decrease inner anxiety and avoid harm. Punding is the engagement of stereotyped behaviors that are repeated compulsively—for example, repetitive manipulation of technical equipment; continual handling, sorting, and examining of objects; grooming; and hoarding. The punder has poor insight into the disruptive and senseless nature of his or her acts. Punding has consistently been related to dopaminergic therapy. Its prevalence in PD patients on dopaminergic therapy ranges from 1.4%33 to 14%.34 An improvement in behavior is observed with a reduction in dosage or discontinuation of levodopa.
Pathologic gambling, or the inability to control gambling, can result in lying to obtain money for gambling, thereby complicating relationships. It can affect up to 8% of patients with PD.35
SUMMARY
Dementia, psychotic symptoms, mood disturbances, and impulse control disorders are important nonmotor manifestations of PD that present management challenges. Some of these manifestations are intrinsic to PD, and some are complications of therapies used to treat the motor manifestations of PD.
Dementia and psychotic symptoms extract a considerable toll on the patient, caregivers, and society. Psychotic symptoms generally manifest as hallucinations (mostly visual) and other sensory disturbances. Initial management involves adjustment of anti-PD medications. The use of atypical antipsychotic drugs has been shown to improve survival among patients with PD. Clozapine is the preferred agent.
Mood disturbances such as depression and apathy may be difficult to diagnose. Depression may be treated similarly to depression unassociated with PD.
Dopamine agonists and levodopa have been associated with impulse control disorders in PD. Compulsive disorders, which are distinct from impulse control disorders, may improve with reduction or discontinuation of levodopa therapy.
- Aarsland D, Andersen K, Larsen JP, Lolk A, Nielsen H, Kragh-Sørensen P. Risk of dementia in Parkinson’s disease: a community-based, prospective study. Neurology 2001; 56:730–736.
- Cummings JL. Intellectual impairment in Parkinson’s disease: clinical, pathologic, and biochemical correlates. J Geriatr Psychiatry Neurol 1988; 1:24–36.
- Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh-Sørensen P. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003; 60:387–392.
- Aarsland D, Larsen JP, Karlsen K, Lim NG, Tandberg E. Mental symptoms in Parkinson’s disease are important contributors to caregiver distress. Int J Geriatr Psychiatry 1999; 14:866–874.
- Aarsland D, Larsen JP, Tandberg E, Laake K. Predictors of nursing home placement in Parkinson’s disease: a population-based, prospective study. J Am Geriatr Soc 2000; 48:938–942.
- Hughes TA, Ross HF, Mindham RH, Spokes EG. Mortality in Parkinson’s disease and its association with dementia and depression. Acta Neurol Scand 2004; 110:118–123.
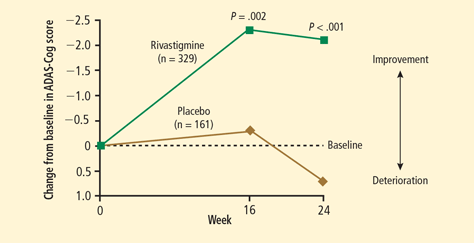
- Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004; 351:2509–2518.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Zahodne LB, Fernandez HH. Pathophysiology and treatment of psychosis in Parkinson’s disease: a review. Drugs Aging 2008; 25:665–682.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Factor SA, Feustel PJ, Friedman JH, et al. Longitudinal outcome of Parkinson’s disease patients with psychosis. Neurology 2003; 60:1756–1761.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Goetz CG, Stebbins GT. Mortality and hallucinations in nursing home patients with advanced Parkinson’s disease. Neurology 1995; 45:669–671.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–115.
- Goldstein JM. Atypical antipsychotic drugs: beyond acute psychosis, new directions. Emerging Drugs 1999; 4:127–151.
- Pollak P, Tison F, Rascol O, et al; on behalf of the French Clozapine Parkinson Study Group. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry 2004; 75:689–695.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Wolters ECh, Hurwitz TA, Mak E, et al. Clozapine in the treatment of parkinsonian patients with dopaminomimetic psychosis. Neurology 1990; 40:832–834.
- Ondo WG, Tintner R, Voung KD, Lai D, Ringholz G. Double-blind, placebo-controlled, unforced titration parallel trial of quetiapine for dopaminergic-induced hallucinations in Parkinson’s disease. Mov Disord 2005; 20:958–963.
- Fernandez HH, Okun MS, Rodriguez RL, Malaty IA, Romrell J. Quetiapine improves visual hallucinations in Parkinson disease but not through normalization of sleep architecture: results from a double-blind clinical-polysomnography study. Int J Neurosci 2009; 119:2196–2205.
- Zoldan J, Friedberg G, Livneh M, Melamed E. Psychosis in advanced Parkinson’s disease: treatment with ondansetron, a 5-HT3 receptor antagonist. Neurology 1995; 45:1305–1308.
- Voon V, Lang AE. Antidepressants in the treatment of psychosis with comorbid depression in Parkinson disease. Clin Neuropharmacol 2004; 27:90–92.
- Ozer F, Meral H, Aydin B, Hanoglu L, Aydemir T, Oral T. Electroconvulsive therapy in drug-induced psychiatric states and neuroleptic malignant syndrome. J ECT 2005; 21:125–127.
- Shulman LM, Taback RL, Rabinstein AA, Weiner WJ. Non-recognition of depression and other non-motor symptoms in Parkinson’s disease. Parkinsonism Relat Disord 2002; 8:193–197.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Barone P, Poewe W, Albrecht S, et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010; 9:573–580.
- Fernandez HH, Merello M. Pramipexole for depression and motor symptoms in Parkinson disease: can we kill two birds with one stone? Lancet Neurol 2010; 9:556–557.
- Marin RS. Apathy: a neuropsychiatric syndrome. J Neuropsychiatry Clin Neurosci 1991; 3:243–254.
- Fernandez HH, Bowers D, Triggs WJ, et al. Repetitive transcranial magnetic stimulation for the treatment of apathy in Parkinson’s disease: results from a double-blind, sham-controlled, randomized, controlled trial. Neurology 2010; 74( suppl 2):352.
- Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 2010; 67:589–595.
- Miyasaki JM, Al Hassan K, Lang AE, Voon V. Punding prevalence in Parkinson’s disease. Mov Disord 2007; 22:1179–1181.
- Evans AH, Katzenschlager R, Paviour D, et al. Punding in Parkinson’s disease: its relation to the dopamine dysregulation syndrome. Mov Disord 2004; 19:397–405.
- Grosset KA, Macphee G, Pal G, et al. Problematic gambling on dopamine agonists: not such a rarity. Mov Disord 2006; 21:2206–2208.
Although the definition of Parkinson disease (PD) is based on the presence of motor features, these are just the “tip of the iceberg.” Nonmotor manifestations are nearly ubiquitous in PD, with behavior problems often being the most malignant. Almost all patients with PD have nonmotor and neuropsychiatric features, including sleep disturbances, compulsive and impulsive behaviors, autonomic dysfunction, and psychosis.
The neuropsychiatric and behavioral features of PD can be classified as intrinsic features, which occur as part of PD, and iatrogenic features, which are complications that arise from treatments used to manage the motor symptoms of PD.
DEMENTIA IN PD
An intrinsic nonmotor feature of PD is dementia, which occurs at a rate four to six times greater in patients with PD than in age-matched controls without PD.1 The prevalence of dementia in PD varies among studies and depends on the demographics of the population being studied. The cross-sectional prevalence of dementia is 40% in patients with PD.2 Seventy-eight percent of a population-based, representative cohort of patients with PD developed dementia during an 8-year study period.3
Dementia is a burden to the caregiver, the patient, and society. Cognitive and behavioral symptoms in patients with PD are the greatest contributors to caregiver distress.4 Dementia and associated behavioral symptoms (ie, hallucinations) hasten nursing home placement, contributing to the financial burden of caring for patients with PD.5 The risk of mortality is increased when dementia develops.6
PSYCHOTIC SYMPTOMS IN PD: AN EFFECT OF EXCESS DOPAMINE STIMULATION
Most of the complications observed in PD can be explained by the dopamine effect of medications and by dopamine deficiencies. An excess of dopamine stimulation caused by administration of prodopaminergic agents manifests as dyskinesias, hallucinations, or delusions. Withdrawal of levodopa will reverse these complications but leads to dopamine deficiency and thus a worsening of PD symptoms. Most patients with PD will tolerate mild dyskinesias or hallucinations if their PD symptoms are well controlled.
The hallucinations in PD tend to be visual as opposed to auditory (as in schizophrenia). They are usually benign and involve figures of people, furry animals, or complex scenes. About 10% to 40% of hallucinations in PD are secondary auditory hallucinations, which tend to be nondistinct, non-paranoid, and often incomprehensible (ie, voices in a crowd).
In the same way, the delusions experienced in patients with PD are distinct from those in schizophrenia. The delusions in PD are usually paranoid in nature and involve stereotyped themes (ie, spousal infidelity, feelings of abandonment) rather than the grandiose delusions that are common in schizophrenia.
The reported prevalence of psychotic symptoms in PD, including hallucinations and delusions, ranges from 20% to 50%.8,9 Auditory hallucinations are a feature in about 10%, and they usually occur with visual hallucinations. Less common are delusions and hallucinations with loss of insight, which are more likely with increasing severity of dementia.
Once a PD patient experiences hallucinations, they are likely to continue. In a 6-year longitudinal study, the prevalence of hallucinations increased from 33% at baseline to 55% at 72 months.10 Persistent psychosis was found in 69% of participants in the Psychosis and Clozapine in PD Study (PSYCLOPS) with 26 months of follow-up.11
High caregiver burden
Psychotic symptoms in PD are associated with high caregiver stress and increased rates of nursing home placement. Goetz et al12 showed that PD patients with psychosis had a much greater risk of nursing home placement than those without psychosis. The prognosis for PD patients in extended-care facilities is worse for those with psychotic symptoms.13
Management of psychotic symptoms
The first step in managing psychosis in PD is to rule out other causes of changes in mental status, such as infection, electrolyte imbalance, or introduction of new medications.
Adjusting anti-PD medications to a tolerable yet effective dose may help to reduce the incidence and severity of psychotic complications. If necessary, selective discontinuation of anti-PD medications may be tried in the following sequence: anticholinergics, amantadine, monoamine oxidase B inhibitors, dopamine agonists, catechol-O-methyltransferase inhibitors, and levodopa/carbidopa.
If motor symptoms prevent dosage minimization or discontinuation of some medications, then the addition of an atypical antipsychotic medication should be considered. Before the advent of atypical antipsychotics, the management of psychosis and hallucinations in PD was unsatisfactory, reflected by a mortality of 100% within 2 years among psychotic PD patients placed in nursing homes compared with 32% among age-matched community dwellers.13 The introduction of atypical antipsychotics has improved survival among PD patients with psychosis. In one study, mortality over 5 years was 44% among PD patients taking long-term clozapine for the treatment of psychosis.14 Recurrence of psychosis is rapid (within 8 weeks) even when PD patients are slowly weaned from atypical antipsychotics.15
Receptor affinities differ among antipsychotics. Because dopamine has been implicated as the principal neurotransmitter in the development of PD psychosis, atypical antipsychotics, with milder dopamine-blocking action, have played a central role in the treatment of PD psychosis. The dopamine D2 receptor is the main target for conventional antipsychotic drugs to exert their clinical effects. Atypical antipsychotics have different affinities for the D2 receptors.16 Occupancy of D2 receptors with atypical antipsychotics is 40% to 70% (risperidone and olanzapine have higher affinity for the D2 receptor than clozapine and quetiapine), and affinity for 5-HT2A receptors can be as high as 70%. This affinity for 5-HT2A receptors relative to D2 receptors may be important for therapeutic efficacy of the atypical antipsychotics. Antagonism of muscarinic, histaminergic, noradrenergic, and other serotonergic receptors also differs among the atypical antipsychotics.
Clozapine remains the gold standard atypical antipsychotic agent, based on results from three relatively small (N = 6 to 60) double-blind, placebo-controlled studies in PD patients with dopaminergic drug-induced psychosis.17–19 Quetiapine improved psychotic symptoms associated with PD in several open-label studies, but has not demonstrated the same success in double-blind clinical trials.20,21
Loss of cholinergic neurons and implications for treatment. In autopsy studies, the loss of cholinergic neurons is more profound in PD than in Alzheimer disease, which suggests that procholinergic drugs may improve symptoms of PD dementia, a major risk factor for hallucinations. In open-label studies, acetylcholinesterase inhibitors have reduced the frequency of hallucinations in patients who have dementia with Lewy bodies (DLB) and in patients with PD dementia. Double-blind trials of patients with DLB and PD dementia concentrated on the effect of cholinesterase inhibitors on dementia and not hallucinations. One concern with the use of a procholinergic drug in patients with PD has been worsening of parkinsonism, but studies of acetylcholinesterase inhibitors have shown no worsening of parkinsonism and only transient worsening of tremor.
Ondansetron, a 5-HT3 receptor antagonist used as an antinausea medication, produced moderate improvements in hallucinations and delusions in an open-label trial for the treatment of psychosis in advanced PD.22 For PD patients with psychosis and comorbid depression, antidepressant therapy and electroconvulsive therapy may be effective options.23,24
MOOD DISTURBANCES IN PD
Depression and apathy occur more frequently in patients with PD than in those who do not have PD.
Depression
Challenges in the management of depression in PD include recognition of depression and distinguishing depressive disorders from mood fluctuations. Whereas a depressive disorder lasts from weeks to years and can occur at any stage of illness, mood fluctuations can change many times daily and appear as nonmotor manifestations during the “off” medication state. Mood fluctuations occur mostly in patients who have developed motor fluctuations. The implication for treatment is that the treatment strategy for a depressive disorder is antidepressant therapy, whereas the strategy for mood fluctuations in PD is to increase the levodopa dose.
Recognition of depression in PD is confounded by the depression criteria of the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition; many of these criteria can be intrinsic features of PD itself—for example, anhedonia, weight/appetite loss or gain, insomnia or hypersomnia, psychomotor retardation, and fatigue. Questions such as “are you feeling sad” or “are you feeling blue” may be superior to questions about associative symptoms when evaluating PD patients for depression.
Most of the medications used for the treatment of depression also work well for depression in patients with PD. Double-blind controlled studies have demonstrated superiority of nortriptyline, citalopram, desipramine, and pramipexole over placebo in improving mood.26–29
Apathy
The overlap between apathy and depressive symptoms can also complicate recognition of apathy, which can be described as a lack of motivation or failure to initiate goal-directed behavior. Apathy involves three domains30:
- Cognitive: expressed as a loss of interest in new experience or a lack of concern about a personal problem
- Diminished affect: flattened affect or a lack of reaction to positive or negative events
- Final: diminished goal-directed cognition, as indicated by a lack of effort or requiring others to structure activities.
Unlike depression, which is similarly representative of PD and other episodic conditions such as dystonia, apathy is more common in PD than in dystonia. In fact, the occurrence of apathy alone distinguishes PD from dystonia. Apathy in PD has no known treatment. If it is associated with depression, apathy may respond to antidepressants.
Repetitive transcranial magnetic stimulation (rTMS) manipulates activity in specific brain neural circuits through the skull to induce changes in behavior. Some studies suggest that modulation of behavior may last beyond the actual stimulation. A randomized, sham-controlled trial of rTMS over the middorsolateral frontal cortex has been conducted with the primary aim of improving apathy in PD. Unfortunately, while patients who were randomized to rTMS experienced some improvement in apathy during the study, the improvement was not significantly different from that observed in patients who received sham treatment.31
IMPULSE CONTROL AND COMPULSIVE DISORDERS IN PD
Impulse control disorders are characterized by the inability to resist an urge to act; the resulting irrational desire to pursue self-gratification may inflict suffering on friends and relatives that compromises relationships and impairs social- and work-related functioning.
Examples of impulse control disorders in PD are pathologic gambling, hypersexuality, compulsive shopping, excessive spending, and binge eating. Patients taking dopamine agonists are two to three times more likely to develop impulse control disorders than those receiving other treatments for PD. Dopamine agonists with relative selectivity for D3 receptors have been implicated in impulse control disorders in PD because D3 receptors are abundant in a region of the brain (ventral striatum) associated with behavioral and substance addictions. Higher levodopa dosages were also associated with impulse control disorders.
Factors associated with impulse control disorders in PD are young age, being single, a family history of impulse control disorders, and levodopa treatment.32 Modifications to dopamine agonist or levodopa therapy are important in the treatment of dopamine agonist–induced impulse disorders.
Compulsive disorders have been described as a class distinct from impulse control disorders and involve repetitive stereotypes and well-ordered acts to decrease inner anxiety and avoid harm. Punding is the engagement of stereotyped behaviors that are repeated compulsively—for example, repetitive manipulation of technical equipment; continual handling, sorting, and examining of objects; grooming; and hoarding. The punder has poor insight into the disruptive and senseless nature of his or her acts. Punding has consistently been related to dopaminergic therapy. Its prevalence in PD patients on dopaminergic therapy ranges from 1.4%33 to 14%.34 An improvement in behavior is observed with a reduction in dosage or discontinuation of levodopa.
Pathologic gambling, or the inability to control gambling, can result in lying to obtain money for gambling, thereby complicating relationships. It can affect up to 8% of patients with PD.35
SUMMARY
Dementia, psychotic symptoms, mood disturbances, and impulse control disorders are important nonmotor manifestations of PD that present management challenges. Some of these manifestations are intrinsic to PD, and some are complications of therapies used to treat the motor manifestations of PD.
Dementia and psychotic symptoms extract a considerable toll on the patient, caregivers, and society. Psychotic symptoms generally manifest as hallucinations (mostly visual) and other sensory disturbances. Initial management involves adjustment of anti-PD medications. The use of atypical antipsychotic drugs has been shown to improve survival among patients with PD. Clozapine is the preferred agent.
Mood disturbances such as depression and apathy may be difficult to diagnose. Depression may be treated similarly to depression unassociated with PD.
Dopamine agonists and levodopa have been associated with impulse control disorders in PD. Compulsive disorders, which are distinct from impulse control disorders, may improve with reduction or discontinuation of levodopa therapy.
Although the definition of Parkinson disease (PD) is based on the presence of motor features, these are just the “tip of the iceberg.” Nonmotor manifestations are nearly ubiquitous in PD, with behavior problems often being the most malignant. Almost all patients with PD have nonmotor and neuropsychiatric features, including sleep disturbances, compulsive and impulsive behaviors, autonomic dysfunction, and psychosis.
The neuropsychiatric and behavioral features of PD can be classified as intrinsic features, which occur as part of PD, and iatrogenic features, which are complications that arise from treatments used to manage the motor symptoms of PD.
DEMENTIA IN PD
An intrinsic nonmotor feature of PD is dementia, which occurs at a rate four to six times greater in patients with PD than in age-matched controls without PD.1 The prevalence of dementia in PD varies among studies and depends on the demographics of the population being studied. The cross-sectional prevalence of dementia is 40% in patients with PD.2 Seventy-eight percent of a population-based, representative cohort of patients with PD developed dementia during an 8-year study period.3
Dementia is a burden to the caregiver, the patient, and society. Cognitive and behavioral symptoms in patients with PD are the greatest contributors to caregiver distress.4 Dementia and associated behavioral symptoms (ie, hallucinations) hasten nursing home placement, contributing to the financial burden of caring for patients with PD.5 The risk of mortality is increased when dementia develops.6
PSYCHOTIC SYMPTOMS IN PD: AN EFFECT OF EXCESS DOPAMINE STIMULATION
Most of the complications observed in PD can be explained by the dopamine effect of medications and by dopamine deficiencies. An excess of dopamine stimulation caused by administration of prodopaminergic agents manifests as dyskinesias, hallucinations, or delusions. Withdrawal of levodopa will reverse these complications but leads to dopamine deficiency and thus a worsening of PD symptoms. Most patients with PD will tolerate mild dyskinesias or hallucinations if their PD symptoms are well controlled.
The hallucinations in PD tend to be visual as opposed to auditory (as in schizophrenia). They are usually benign and involve figures of people, furry animals, or complex scenes. About 10% to 40% of hallucinations in PD are secondary auditory hallucinations, which tend to be nondistinct, non-paranoid, and often incomprehensible (ie, voices in a crowd).
In the same way, the delusions experienced in patients with PD are distinct from those in schizophrenia. The delusions in PD are usually paranoid in nature and involve stereotyped themes (ie, spousal infidelity, feelings of abandonment) rather than the grandiose delusions that are common in schizophrenia.
The reported prevalence of psychotic symptoms in PD, including hallucinations and delusions, ranges from 20% to 50%.8,9 Auditory hallucinations are a feature in about 10%, and they usually occur with visual hallucinations. Less common are delusions and hallucinations with loss of insight, which are more likely with increasing severity of dementia.
Once a PD patient experiences hallucinations, they are likely to continue. In a 6-year longitudinal study, the prevalence of hallucinations increased from 33% at baseline to 55% at 72 months.10 Persistent psychosis was found in 69% of participants in the Psychosis and Clozapine in PD Study (PSYCLOPS) with 26 months of follow-up.11
High caregiver burden
Psychotic symptoms in PD are associated with high caregiver stress and increased rates of nursing home placement. Goetz et al12 showed that PD patients with psychosis had a much greater risk of nursing home placement than those without psychosis. The prognosis for PD patients in extended-care facilities is worse for those with psychotic symptoms.13
Management of psychotic symptoms
The first step in managing psychosis in PD is to rule out other causes of changes in mental status, such as infection, electrolyte imbalance, or introduction of new medications.
Adjusting anti-PD medications to a tolerable yet effective dose may help to reduce the incidence and severity of psychotic complications. If necessary, selective discontinuation of anti-PD medications may be tried in the following sequence: anticholinergics, amantadine, monoamine oxidase B inhibitors, dopamine agonists, catechol-O-methyltransferase inhibitors, and levodopa/carbidopa.
If motor symptoms prevent dosage minimization or discontinuation of some medications, then the addition of an atypical antipsychotic medication should be considered. Before the advent of atypical antipsychotics, the management of psychosis and hallucinations in PD was unsatisfactory, reflected by a mortality of 100% within 2 years among psychotic PD patients placed in nursing homes compared with 32% among age-matched community dwellers.13 The introduction of atypical antipsychotics has improved survival among PD patients with psychosis. In one study, mortality over 5 years was 44% among PD patients taking long-term clozapine for the treatment of psychosis.14 Recurrence of psychosis is rapid (within 8 weeks) even when PD patients are slowly weaned from atypical antipsychotics.15
Receptor affinities differ among antipsychotics. Because dopamine has been implicated as the principal neurotransmitter in the development of PD psychosis, atypical antipsychotics, with milder dopamine-blocking action, have played a central role in the treatment of PD psychosis. The dopamine D2 receptor is the main target for conventional antipsychotic drugs to exert their clinical effects. Atypical antipsychotics have different affinities for the D2 receptors.16 Occupancy of D2 receptors with atypical antipsychotics is 40% to 70% (risperidone and olanzapine have higher affinity for the D2 receptor than clozapine and quetiapine), and affinity for 5-HT2A receptors can be as high as 70%. This affinity for 5-HT2A receptors relative to D2 receptors may be important for therapeutic efficacy of the atypical antipsychotics. Antagonism of muscarinic, histaminergic, noradrenergic, and other serotonergic receptors also differs among the atypical antipsychotics.
Clozapine remains the gold standard atypical antipsychotic agent, based on results from three relatively small (N = 6 to 60) double-blind, placebo-controlled studies in PD patients with dopaminergic drug-induced psychosis.17–19 Quetiapine improved psychotic symptoms associated with PD in several open-label studies, but has not demonstrated the same success in double-blind clinical trials.20,21
Loss of cholinergic neurons and implications for treatment. In autopsy studies, the loss of cholinergic neurons is more profound in PD than in Alzheimer disease, which suggests that procholinergic drugs may improve symptoms of PD dementia, a major risk factor for hallucinations. In open-label studies, acetylcholinesterase inhibitors have reduced the frequency of hallucinations in patients who have dementia with Lewy bodies (DLB) and in patients with PD dementia. Double-blind trials of patients with DLB and PD dementia concentrated on the effect of cholinesterase inhibitors on dementia and not hallucinations. One concern with the use of a procholinergic drug in patients with PD has been worsening of parkinsonism, but studies of acetylcholinesterase inhibitors have shown no worsening of parkinsonism and only transient worsening of tremor.
Ondansetron, a 5-HT3 receptor antagonist used as an antinausea medication, produced moderate improvements in hallucinations and delusions in an open-label trial for the treatment of psychosis in advanced PD.22 For PD patients with psychosis and comorbid depression, antidepressant therapy and electroconvulsive therapy may be effective options.23,24
MOOD DISTURBANCES IN PD
Depression and apathy occur more frequently in patients with PD than in those who do not have PD.
Depression
Challenges in the management of depression in PD include recognition of depression and distinguishing depressive disorders from mood fluctuations. Whereas a depressive disorder lasts from weeks to years and can occur at any stage of illness, mood fluctuations can change many times daily and appear as nonmotor manifestations during the “off” medication state. Mood fluctuations occur mostly in patients who have developed motor fluctuations. The implication for treatment is that the treatment strategy for a depressive disorder is antidepressant therapy, whereas the strategy for mood fluctuations in PD is to increase the levodopa dose.
Recognition of depression in PD is confounded by the depression criteria of the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition; many of these criteria can be intrinsic features of PD itself—for example, anhedonia, weight/appetite loss or gain, insomnia or hypersomnia, psychomotor retardation, and fatigue. Questions such as “are you feeling sad” or “are you feeling blue” may be superior to questions about associative symptoms when evaluating PD patients for depression.
Most of the medications used for the treatment of depression also work well for depression in patients with PD. Double-blind controlled studies have demonstrated superiority of nortriptyline, citalopram, desipramine, and pramipexole over placebo in improving mood.26–29
Apathy
The overlap between apathy and depressive symptoms can also complicate recognition of apathy, which can be described as a lack of motivation or failure to initiate goal-directed behavior. Apathy involves three domains30:
- Cognitive: expressed as a loss of interest in new experience or a lack of concern about a personal problem
- Diminished affect: flattened affect or a lack of reaction to positive or negative events
- Final: diminished goal-directed cognition, as indicated by a lack of effort or requiring others to structure activities.
Unlike depression, which is similarly representative of PD and other episodic conditions such as dystonia, apathy is more common in PD than in dystonia. In fact, the occurrence of apathy alone distinguishes PD from dystonia. Apathy in PD has no known treatment. If it is associated with depression, apathy may respond to antidepressants.
Repetitive transcranial magnetic stimulation (rTMS) manipulates activity in specific brain neural circuits through the skull to induce changes in behavior. Some studies suggest that modulation of behavior may last beyond the actual stimulation. A randomized, sham-controlled trial of rTMS over the middorsolateral frontal cortex has been conducted with the primary aim of improving apathy in PD. Unfortunately, while patients who were randomized to rTMS experienced some improvement in apathy during the study, the improvement was not significantly different from that observed in patients who received sham treatment.31
IMPULSE CONTROL AND COMPULSIVE DISORDERS IN PD
Impulse control disorders are characterized by the inability to resist an urge to act; the resulting irrational desire to pursue self-gratification may inflict suffering on friends and relatives that compromises relationships and impairs social- and work-related functioning.
Examples of impulse control disorders in PD are pathologic gambling, hypersexuality, compulsive shopping, excessive spending, and binge eating. Patients taking dopamine agonists are two to three times more likely to develop impulse control disorders than those receiving other treatments for PD. Dopamine agonists with relative selectivity for D3 receptors have been implicated in impulse control disorders in PD because D3 receptors are abundant in a region of the brain (ventral striatum) associated with behavioral and substance addictions. Higher levodopa dosages were also associated with impulse control disorders.
Factors associated with impulse control disorders in PD are young age, being single, a family history of impulse control disorders, and levodopa treatment.32 Modifications to dopamine agonist or levodopa therapy are important in the treatment of dopamine agonist–induced impulse disorders.
Compulsive disorders have been described as a class distinct from impulse control disorders and involve repetitive stereotypes and well-ordered acts to decrease inner anxiety and avoid harm. Punding is the engagement of stereotyped behaviors that are repeated compulsively—for example, repetitive manipulation of technical equipment; continual handling, sorting, and examining of objects; grooming; and hoarding. The punder has poor insight into the disruptive and senseless nature of his or her acts. Punding has consistently been related to dopaminergic therapy. Its prevalence in PD patients on dopaminergic therapy ranges from 1.4%33 to 14%.34 An improvement in behavior is observed with a reduction in dosage or discontinuation of levodopa.
Pathologic gambling, or the inability to control gambling, can result in lying to obtain money for gambling, thereby complicating relationships. It can affect up to 8% of patients with PD.35
SUMMARY
Dementia, psychotic symptoms, mood disturbances, and impulse control disorders are important nonmotor manifestations of PD that present management challenges. Some of these manifestations are intrinsic to PD, and some are complications of therapies used to treat the motor manifestations of PD.
Dementia and psychotic symptoms extract a considerable toll on the patient, caregivers, and society. Psychotic symptoms generally manifest as hallucinations (mostly visual) and other sensory disturbances. Initial management involves adjustment of anti-PD medications. The use of atypical antipsychotic drugs has been shown to improve survival among patients with PD. Clozapine is the preferred agent.
Mood disturbances such as depression and apathy may be difficult to diagnose. Depression may be treated similarly to depression unassociated with PD.
Dopamine agonists and levodopa have been associated with impulse control disorders in PD. Compulsive disorders, which are distinct from impulse control disorders, may improve with reduction or discontinuation of levodopa therapy.
- Aarsland D, Andersen K, Larsen JP, Lolk A, Nielsen H, Kragh-Sørensen P. Risk of dementia in Parkinson’s disease: a community-based, prospective study. Neurology 2001; 56:730–736.
- Cummings JL. Intellectual impairment in Parkinson’s disease: clinical, pathologic, and biochemical correlates. J Geriatr Psychiatry Neurol 1988; 1:24–36.
- Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh-Sørensen P. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003; 60:387–392.
- Aarsland D, Larsen JP, Karlsen K, Lim NG, Tandberg E. Mental symptoms in Parkinson’s disease are important contributors to caregiver distress. Int J Geriatr Psychiatry 1999; 14:866–874.
- Aarsland D, Larsen JP, Tandberg E, Laake K. Predictors of nursing home placement in Parkinson’s disease: a population-based, prospective study. J Am Geriatr Soc 2000; 48:938–942.
- Hughes TA, Ross HF, Mindham RH, Spokes EG. Mortality in Parkinson’s disease and its association with dementia and depression. Acta Neurol Scand 2004; 110:118–123.
- Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004; 351:2509–2518.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Zahodne LB, Fernandez HH. Pathophysiology and treatment of psychosis in Parkinson’s disease: a review. Drugs Aging 2008; 25:665–682.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Factor SA, Feustel PJ, Friedman JH, et al. Longitudinal outcome of Parkinson’s disease patients with psychosis. Neurology 2003; 60:1756–1761.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Goetz CG, Stebbins GT. Mortality and hallucinations in nursing home patients with advanced Parkinson’s disease. Neurology 1995; 45:669–671.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–115.
- Goldstein JM. Atypical antipsychotic drugs: beyond acute psychosis, new directions. Emerging Drugs 1999; 4:127–151.
- Pollak P, Tison F, Rascol O, et al; on behalf of the French Clozapine Parkinson Study Group. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry 2004; 75:689–695.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Wolters ECh, Hurwitz TA, Mak E, et al. Clozapine in the treatment of parkinsonian patients with dopaminomimetic psychosis. Neurology 1990; 40:832–834.
- Ondo WG, Tintner R, Voung KD, Lai D, Ringholz G. Double-blind, placebo-controlled, unforced titration parallel trial of quetiapine for dopaminergic-induced hallucinations in Parkinson’s disease. Mov Disord 2005; 20:958–963.
- Fernandez HH, Okun MS, Rodriguez RL, Malaty IA, Romrell J. Quetiapine improves visual hallucinations in Parkinson disease but not through normalization of sleep architecture: results from a double-blind clinical-polysomnography study. Int J Neurosci 2009; 119:2196–2205.
- Zoldan J, Friedberg G, Livneh M, Melamed E. Psychosis in advanced Parkinson’s disease: treatment with ondansetron, a 5-HT3 receptor antagonist. Neurology 1995; 45:1305–1308.
- Voon V, Lang AE. Antidepressants in the treatment of psychosis with comorbid depression in Parkinson disease. Clin Neuropharmacol 2004; 27:90–92.
- Ozer F, Meral H, Aydin B, Hanoglu L, Aydemir T, Oral T. Electroconvulsive therapy in drug-induced psychiatric states and neuroleptic malignant syndrome. J ECT 2005; 21:125–127.
- Shulman LM, Taback RL, Rabinstein AA, Weiner WJ. Non-recognition of depression and other non-motor symptoms in Parkinson’s disease. Parkinsonism Relat Disord 2002; 8:193–197.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Barone P, Poewe W, Albrecht S, et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010; 9:573–580.
- Fernandez HH, Merello M. Pramipexole for depression and motor symptoms in Parkinson disease: can we kill two birds with one stone? Lancet Neurol 2010; 9:556–557.
- Marin RS. Apathy: a neuropsychiatric syndrome. J Neuropsychiatry Clin Neurosci 1991; 3:243–254.
- Fernandez HH, Bowers D, Triggs WJ, et al. Repetitive transcranial magnetic stimulation for the treatment of apathy in Parkinson’s disease: results from a double-blind, sham-controlled, randomized, controlled trial. Neurology 2010; 74( suppl 2):352.
- Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 2010; 67:589–595.
- Miyasaki JM, Al Hassan K, Lang AE, Voon V. Punding prevalence in Parkinson’s disease. Mov Disord 2007; 22:1179–1181.
- Evans AH, Katzenschlager R, Paviour D, et al. Punding in Parkinson’s disease: its relation to the dopamine dysregulation syndrome. Mov Disord 2004; 19:397–405.
- Grosset KA, Macphee G, Pal G, et al. Problematic gambling on dopamine agonists: not such a rarity. Mov Disord 2006; 21:2206–2208.
- Aarsland D, Andersen K, Larsen JP, Lolk A, Nielsen H, Kragh-Sørensen P. Risk of dementia in Parkinson’s disease: a community-based, prospective study. Neurology 2001; 56:730–736.
- Cummings JL. Intellectual impairment in Parkinson’s disease: clinical, pathologic, and biochemical correlates. J Geriatr Psychiatry Neurol 1988; 1:24–36.
- Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh-Sørensen P. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003; 60:387–392.
- Aarsland D, Larsen JP, Karlsen K, Lim NG, Tandberg E. Mental symptoms in Parkinson’s disease are important contributors to caregiver distress. Int J Geriatr Psychiatry 1999; 14:866–874.
- Aarsland D, Larsen JP, Tandberg E, Laake K. Predictors of nursing home placement in Parkinson’s disease: a population-based, prospective study. J Am Geriatr Soc 2000; 48:938–942.
- Hughes TA, Ross HF, Mindham RH, Spokes EG. Mortality in Parkinson’s disease and its association with dementia and depression. Acta Neurol Scand 2004; 110:118–123.
- Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004; 351:2509–2518.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Zahodne LB, Fernandez HH. Pathophysiology and treatment of psychosis in Parkinson’s disease: a review. Drugs Aging 2008; 25:665–682.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Factor SA, Feustel PJ, Friedman JH, et al. Longitudinal outcome of Parkinson’s disease patients with psychosis. Neurology 2003; 60:1756–1761.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Goetz CG, Stebbins GT. Mortality and hallucinations in nursing home patients with advanced Parkinson’s disease. Neurology 1995; 45:669–671.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–115.
- Goldstein JM. Atypical antipsychotic drugs: beyond acute psychosis, new directions. Emerging Drugs 1999; 4:127–151.
- Pollak P, Tison F, Rascol O, et al; on behalf of the French Clozapine Parkinson Study Group. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry 2004; 75:689–695.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Wolters ECh, Hurwitz TA, Mak E, et al. Clozapine in the treatment of parkinsonian patients with dopaminomimetic psychosis. Neurology 1990; 40:832–834.
- Ondo WG, Tintner R, Voung KD, Lai D, Ringholz G. Double-blind, placebo-controlled, unforced titration parallel trial of quetiapine for dopaminergic-induced hallucinations in Parkinson’s disease. Mov Disord 2005; 20:958–963.
- Fernandez HH, Okun MS, Rodriguez RL, Malaty IA, Romrell J. Quetiapine improves visual hallucinations in Parkinson disease but not through normalization of sleep architecture: results from a double-blind clinical-polysomnography study. Int J Neurosci 2009; 119:2196–2205.
- Zoldan J, Friedberg G, Livneh M, Melamed E. Psychosis in advanced Parkinson’s disease: treatment with ondansetron, a 5-HT3 receptor antagonist. Neurology 1995; 45:1305–1308.
- Voon V, Lang AE. Antidepressants in the treatment of psychosis with comorbid depression in Parkinson disease. Clin Neuropharmacol 2004; 27:90–92.
- Ozer F, Meral H, Aydin B, Hanoglu L, Aydemir T, Oral T. Electroconvulsive therapy in drug-induced psychiatric states and neuroleptic malignant syndrome. J ECT 2005; 21:125–127.
- Shulman LM, Taback RL, Rabinstein AA, Weiner WJ. Non-recognition of depression and other non-motor symptoms in Parkinson’s disease. Parkinsonism Relat Disord 2002; 8:193–197.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Barone P, Poewe W, Albrecht S, et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010; 9:573–580.
- Fernandez HH, Merello M. Pramipexole for depression and motor symptoms in Parkinson disease: can we kill two birds with one stone? Lancet Neurol 2010; 9:556–557.
- Marin RS. Apathy: a neuropsychiatric syndrome. J Neuropsychiatry Clin Neurosci 1991; 3:243–254.
- Fernandez HH, Bowers D, Triggs WJ, et al. Repetitive transcranial magnetic stimulation for the treatment of apathy in Parkinson’s disease: results from a double-blind, sham-controlled, randomized, controlled trial. Neurology 2010; 74( suppl 2):352.
- Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 2010; 67:589–595.
- Miyasaki JM, Al Hassan K, Lang AE, Voon V. Punding prevalence in Parkinson’s disease. Mov Disord 2007; 22:1179–1181.
- Evans AH, Katzenschlager R, Paviour D, et al. Punding in Parkinson’s disease: its relation to the dopamine dysregulation syndrome. Mov Disord 2004; 19:397–405.
- Grosset KA, Macphee G, Pal G, et al. Problematic gambling on dopamine agonists: not such a rarity. Mov Disord 2006; 21:2206–2208.
In reply: Parkinson disease
In Reply: I thank Dr. Keller for his thoughtful comments. They are most appreciated.
It is true that with availability of generic ropinirole and pramipexole, there are now cheaper alternatives to levodopa. Nonetheless, levodopa remains the cheapest and most efficacious medication for Parkinson disease to date. Whenever levodopa is compared head-to-head with any dopamine agonist, the general results remain consistent: levodopa affords better motor improvement with lesser side effects, but is more likely to lead to motor fluctuations, specifically dyskinesias. Therefore, in general, levodopa is the first choice in elderly patients where tolerability may be an issue, whereas a dopamine agonist may be the initial treatment of choice in younger Parkinson patients, who are able to tolerate the drug better and have a higher likelihood of developing dyskinesias.
It is a tougher task to determine which among the dopamine agonists is superior. The newer dopamine agonists have not been compared head-to-head. Therefore, it is practically a “coin toss” when selecting which dopamine agonist to try. Their mechanism of action (D2 and D3 receptor agonist activity) and frequency of intake (three times per day for generics; once daily for long-acting formulations), cost, and side effect profile are nearly identical, despite minor differences in their half-lives.
Regarding putative neuroprotective agents in Parkinson disease, indeed, isradipine is one of the medications currently undergoing investigation for its potential neuroprotective effect. While I personally have no objection to using it for a Parkinson disease patient who also happens to need an antihypertensive agent, I am more cautious about endorsing it as a neuroprotective agent until results of clinical trials have been released. Similarly, while a large epidemiologic study has shown that people who take ibuprofen are less likely to develop Parkinson disease, there has been no robust human trial that has shown the drug to slow the progression of Parkinson disease among patients who are already suffering from the disorder. Therefore, the current use of ibuprofen in Parkinson disease should be based more on its anti-inflammatory indications rather than its possible neuroprotective effect. Finally, we have shown, in a large, multicenter, global randomized controlled trial with a delayed-start design, that pramipexole is unlikely to possess any meaningful neuroprotective effect. Therefore, I am personally not that optimistic that dexpramipexole would demonstrate such an effect.
While in theory combining the use of catechol-O-methyltransferase (COMT) inhibitors and monoamine oxidase (MAO) type B inhibitors can synergistically work to inhibit the breakdown of other catecholamines and lead to adrenergic crisis when taken concomitantly, this has not been our experience. Perhaps it is because at recommended doses, the MAO inhibition is selective to type B (where receptors are more confined to the brain) and not type A (where receptors are more distributed throughout blood vessels, thereby having a higher likelihood of causing a hypertensive crisis as is seen in the use of nonselective MAO inhibitors). Therefore, at our center, we routinely use the two classes of agents concomitantly with minimal safety concerns.
In Reply: I thank Dr. Keller for his thoughtful comments. They are most appreciated.
It is true that with availability of generic ropinirole and pramipexole, there are now cheaper alternatives to levodopa. Nonetheless, levodopa remains the cheapest and most efficacious medication for Parkinson disease to date. Whenever levodopa is compared head-to-head with any dopamine agonist, the general results remain consistent: levodopa affords better motor improvement with lesser side effects, but is more likely to lead to motor fluctuations, specifically dyskinesias. Therefore, in general, levodopa is the first choice in elderly patients where tolerability may be an issue, whereas a dopamine agonist may be the initial treatment of choice in younger Parkinson patients, who are able to tolerate the drug better and have a higher likelihood of developing dyskinesias.
It is a tougher task to determine which among the dopamine agonists is superior. The newer dopamine agonists have not been compared head-to-head. Therefore, it is practically a “coin toss” when selecting which dopamine agonist to try. Their mechanism of action (D2 and D3 receptor agonist activity) and frequency of intake (three times per day for generics; once daily for long-acting formulations), cost, and side effect profile are nearly identical, despite minor differences in their half-lives.
Regarding putative neuroprotective agents in Parkinson disease, indeed, isradipine is one of the medications currently undergoing investigation for its potential neuroprotective effect. While I personally have no objection to using it for a Parkinson disease patient who also happens to need an antihypertensive agent, I am more cautious about endorsing it as a neuroprotective agent until results of clinical trials have been released. Similarly, while a large epidemiologic study has shown that people who take ibuprofen are less likely to develop Parkinson disease, there has been no robust human trial that has shown the drug to slow the progression of Parkinson disease among patients who are already suffering from the disorder. Therefore, the current use of ibuprofen in Parkinson disease should be based more on its anti-inflammatory indications rather than its possible neuroprotective effect. Finally, we have shown, in a large, multicenter, global randomized controlled trial with a delayed-start design, that pramipexole is unlikely to possess any meaningful neuroprotective effect. Therefore, I am personally not that optimistic that dexpramipexole would demonstrate such an effect.
While in theory combining the use of catechol-O-methyltransferase (COMT) inhibitors and monoamine oxidase (MAO) type B inhibitors can synergistically work to inhibit the breakdown of other catecholamines and lead to adrenergic crisis when taken concomitantly, this has not been our experience. Perhaps it is because at recommended doses, the MAO inhibition is selective to type B (where receptors are more confined to the brain) and not type A (where receptors are more distributed throughout blood vessels, thereby having a higher likelihood of causing a hypertensive crisis as is seen in the use of nonselective MAO inhibitors). Therefore, at our center, we routinely use the two classes of agents concomitantly with minimal safety concerns.
In Reply: I thank Dr. Keller for his thoughtful comments. They are most appreciated.
It is true that with availability of generic ropinirole and pramipexole, there are now cheaper alternatives to levodopa. Nonetheless, levodopa remains the cheapest and most efficacious medication for Parkinson disease to date. Whenever levodopa is compared head-to-head with any dopamine agonist, the general results remain consistent: levodopa affords better motor improvement with lesser side effects, but is more likely to lead to motor fluctuations, specifically dyskinesias. Therefore, in general, levodopa is the first choice in elderly patients where tolerability may be an issue, whereas a dopamine agonist may be the initial treatment of choice in younger Parkinson patients, who are able to tolerate the drug better and have a higher likelihood of developing dyskinesias.
It is a tougher task to determine which among the dopamine agonists is superior. The newer dopamine agonists have not been compared head-to-head. Therefore, it is practically a “coin toss” when selecting which dopamine agonist to try. Their mechanism of action (D2 and D3 receptor agonist activity) and frequency of intake (three times per day for generics; once daily for long-acting formulations), cost, and side effect profile are nearly identical, despite minor differences in their half-lives.
Regarding putative neuroprotective agents in Parkinson disease, indeed, isradipine is one of the medications currently undergoing investigation for its potential neuroprotective effect. While I personally have no objection to using it for a Parkinson disease patient who also happens to need an antihypertensive agent, I am more cautious about endorsing it as a neuroprotective agent until results of clinical trials have been released. Similarly, while a large epidemiologic study has shown that people who take ibuprofen are less likely to develop Parkinson disease, there has been no robust human trial that has shown the drug to slow the progression of Parkinson disease among patients who are already suffering from the disorder. Therefore, the current use of ibuprofen in Parkinson disease should be based more on its anti-inflammatory indications rather than its possible neuroprotective effect. Finally, we have shown, in a large, multicenter, global randomized controlled trial with a delayed-start design, that pramipexole is unlikely to possess any meaningful neuroprotective effect. Therefore, I am personally not that optimistic that dexpramipexole would demonstrate such an effect.
While in theory combining the use of catechol-O-methyltransferase (COMT) inhibitors and monoamine oxidase (MAO) type B inhibitors can synergistically work to inhibit the breakdown of other catecholamines and lead to adrenergic crisis when taken concomitantly, this has not been our experience. Perhaps it is because at recommended doses, the MAO inhibition is selective to type B (where receptors are more confined to the brain) and not type A (where receptors are more distributed throughout blood vessels, thereby having a higher likelihood of causing a hypertensive crisis as is seen in the use of nonselective MAO inhibitors). Therefore, at our center, we routinely use the two classes of agents concomitantly with minimal safety concerns.
In reply: Essential tremor, beta-blockers, calcium channel blockers
In Reply: We agree and thank Dr. Keller for raising this valid point. The two classes of calcium channel blockers are distinct in their actions, and the warning about not combining a calcium channel blocker with a beta-blocker because of the increased risk of developing significant bradycardia applies only to the nondihydropyridine class.
In Reply: We agree and thank Dr. Keller for raising this valid point. The two classes of calcium channel blockers are distinct in their actions, and the warning about not combining a calcium channel blocker with a beta-blocker because of the increased risk of developing significant bradycardia applies only to the nondihydropyridine class.
In Reply: We agree and thank Dr. Keller for raising this valid point. The two classes of calcium channel blockers are distinct in their actions, and the warning about not combining a calcium channel blocker with a beta-blocker because of the increased risk of developing significant bradycardia applies only to the nondihydropyridine class.
Deep brain stimulation: What can patients expect from it?
Deep brain stimulation is an important therapy for Parkinson disease and other movement disorders. It involves implantation of a pulse generator that can be adjusted by telemetry and can be activated and deactivated by clinicians and patients. It is therefore also a good investigational tool, allowing for double-blind, sham-controlled clinical trials by testing the effects of the stimulation with optimal settings compared with no stimulation.
This article will discuss the approved indications for deep brain stimulation (particularly for managing movement disorders), the benefits that can be expected, the risks, the complications, the maintenance required, how candidates for this treatment are evaluated, and the surgical procedure for implantation of the devices.
DEVICE SIMILAR TO HEART PACEMAKERS
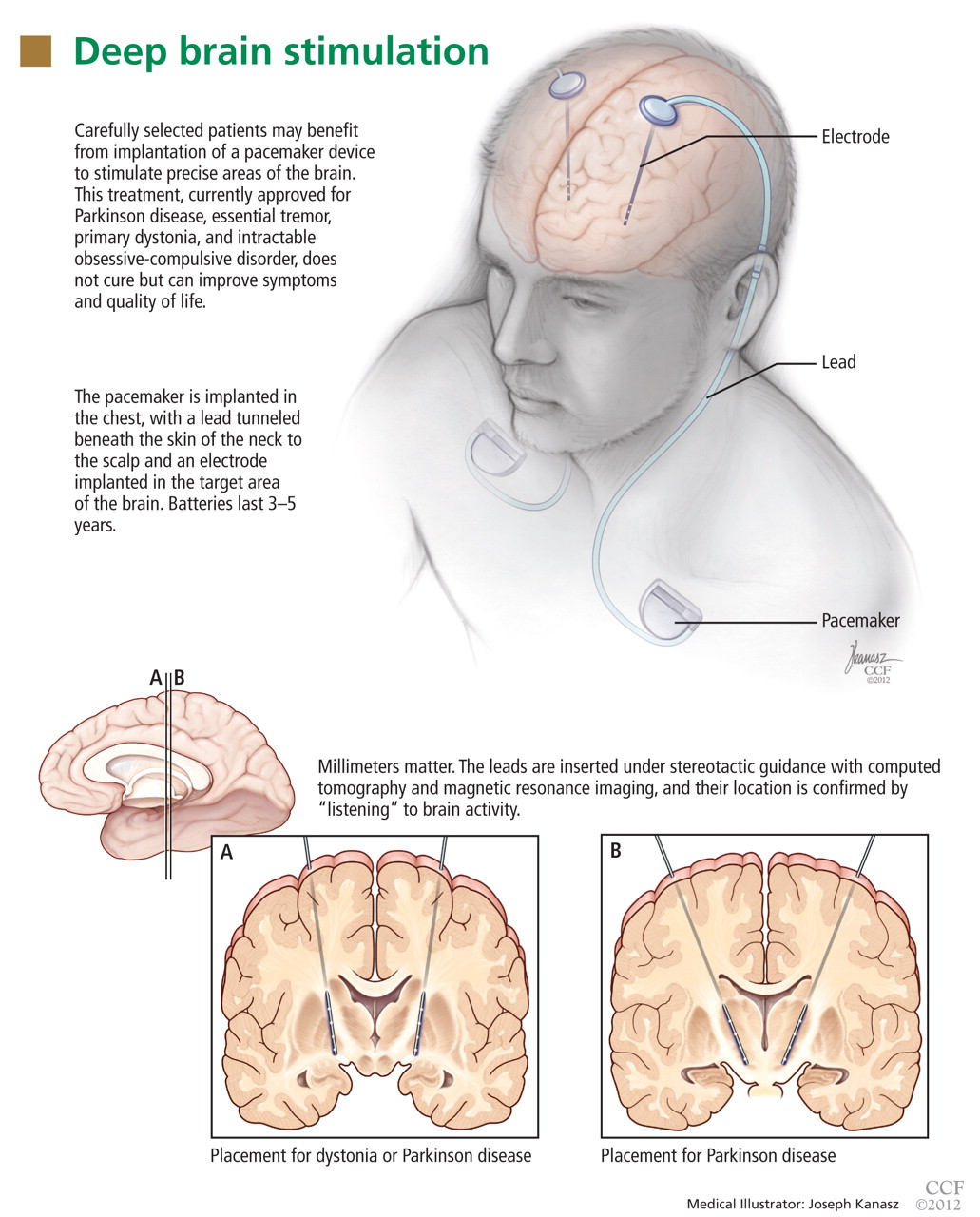
The deep brain stimulation system must be programmed by a physician or midlevel practitioner by observing a symptom and then changing the applied settings to the pulse generator until the symptom improves. This can be a very time-consuming process.
In contrast to heart pacemakers, which run at low frequencies, the brain devices for movement disorders are almost always set to a high frequency, greater than 100 Hz. For this reason, they consume more energy and need larger batteries than those in modern heart pacemakers.
The batteries in these generators typically last 3 to 5 years and are replaced in an outpatient procedure. Newer, smaller, rechargeable devices are expected to last longer but require more maintenance and care by patients, who have to recharge them at home periodically.
INDICATIONS FOR DEEP BRAIN STIMULATION
Deep brain stimulation is approved by the US Food and Drug Administration (FDA) for specific indications:
- Parkinson disease
- Essential tremor
- Primary dystonia (under a humanitarian device exemption)
- Intractable obsessive-compulsive disorder (also under a humanitarian device exemption). We will not discuss this indication further in this paper.
For each of these conditions, deep brain stimulation is considered when nonsurgical management has failed, as is the case for most functional neurosurgical treatments.
Investigations under way in other disorders
Several studies of deep brain stimulation are currently in progress under FDA-approved investigational device exemptions. Some, with funding from industry, are exploring its use in neuropsychiatric conditions other than parkinsonism. Two large clinical trials are evaluating its use for treatment-refractory depression, a common problem and a leading cause of disability in the industrialized world. Multiple investigators are also exploring novel uses of this technology in disorders ranging from obsessive-compulsive disorder to epilepsy.
Investigation is also under way at Cleveland Clinic in a federally funded, prospective, randomized clinical trial of deep brain stimulation for patients with thalamic pain syndrome. The primary hypothesis is that stimulation of the ventral striatal and ventral capsular area will modulate the affective component of this otherwise intractable pain syndrome, reducing pain-related disability and improving quality of life.
DEEP BRAIN STIMULATION VS ABLATION
Before deep brain stimulation became available, the only surgical options for patients with advanced Parkinson disease, tremor, or dystonia were ablative procedures such as pallidotomy (ablation of part of the globus pallidus) and thalamotomy (ablation of part of the thalamus). These procedures had been well known for several decades but fell out of favor when levodopa became available in the 1960s and revolutionized the medical treatment of Parkinson disease.
Surgery for movement disorders, in particular Parkinson disease, had a rebirth in the late 1980s when the limitations and complications associated with the pharmacologic management of Parkinson disease became increasingly evident. Ablative procedures are still used to treat advanced Parkinson disease, but much less commonly in industrialized countries.
Although pallidotomy and thalamotomy can have excellent results, they are not as safe as deep brain stimulation, which has the advantage of being reversible, modulating the function of an area rather than destroying it. Any unwanted effect can be immediately altered or reversed, unlike ablative procedures, in which any change is permanent. In addition, deep brain stimulation is adjustable, and the settings can be optimized as the disease progresses over the years.
Ablative procedures can be risky when performed bilaterally, while deep brain stimulation is routinely done on both hemispheres for patients with bilateral symptoms.
Although deep brain stimulation is today’s surgical treatment of choice, it is not perfect. It has the disadvantage of requiring lifelong maintenance of the hardware, for which the patient remains dependent on a medical center. Patients are usually seen more often at the specialized center in the first few months after surgery for optimization of programming and titration of drugs. (During this time, most patients see a gradual, substantial reduction in medication intake.) They are then followed by their physician and visit the center less often for monitoring of disease status and for further adjustments to the stimulator.
Most patients, to date, receive nonrechargeable pulse generators. As mentioned above, the batteries in these devices typically last 3 to 5 years. Preferably, batteries are replaced before they are completely depleted, to avoid interruption of therapy. Periodic visits to the center allow clinicians to estimate battery expiration ahead of time and plan replacements accordingly.
Rechargeable pulse generators have been recently introduced and are expected to last up to 9 years. They are an option for patients who can comply with the requirements for periodic home recharging of the hardware.
Patients are given a remote control so that they can turn the device on or off and check its status. Most patients keep it turned on all the time, although some turn it off at night to save battery life.
WHAT CAN PARKINSON PATIENTS EXPECT FROM THIS THERAPY?
Typically, some parkinsonian symptoms predominate over others, although some patients with advanced disease present with a severe combination of multiple disabling symptoms. Deep brain stimulation is best suited to address some of the cardinal motor symptoms, particularly tremor, rigidity, and bradykinesia, and motor fluctuations such as “wearing off” and dyskinesia.
Improvement in some motor symptoms
As a general rule, appendicular symptoms such as limb tremor and rigidity are more responsive to this therapy than axial symptoms such as gait and balance problems, but some patients experience improvement in gait as well. Other symptoms, such as swallowing or urinary symptoms, are seldom helped.
Although deep brain stimulation can help manage key motor symptoms and improve quality of life, it does not cure Parkinson disease. Also, there is no evidence to date that it slows disease progression, although this is a topic of ongoing investigation.
Fewer motor fluctuations
A common complaint of patients with advanced Parkinson disease is frequent—and often unpredictable—fluctuations between the “on” state (ie, when the effects of the patient’s levodopa therapy are apparent) and the “off” state (ie, when the levodopa doesn’t seem to be working). Sometimes, in the on state, patients experience involuntary choreic or ballistic movements, called dyskinesias. They also complain that the on time progressively lasts shorter and the day is spent alternating between shorter on states (during which the patient may be dyskinetic) and longer off states, limiting the patient’s independence and quality of life.
Deep brain stimulation can help patients prolong the on time while reducing the amplitude of these fluctuations so that the symptoms are not as severe in the off time and dyskinesias are reduced in the on time.
Some patients undergo deep brain stimulation primarily for managing the adverse effects of levodopa rather than for controlling the symptoms of the disease itself. While these patients need levodopa to address the disabling symptoms of the disease, they also present a greater sensitivity for developing levodopa-induced dyskinesias, quickly fluctuating from a lack of movement (the off state) to a state of uncontrollable movements (during the on state).
Deep brain stimulation typically allows the dosage of levodopa to be significantly reduced and gives patients more on time with fewer side effects and less fluctuation between the on and off states.
Response to levodopa predicts deep brain stimulation’s effects
Whether a patient is likely to be helped by deep brain stimulation can be tested with reasonable predictability by giving a single therapeutic dose of levodopa after the patient has been free of the drug for 12 hours. If there is an obvious difference on objective quantitative testing between the off and on states with a single dose, the patient is likely to benefit from deep brain stimulation. Those who do not respond well or are known to have never been well controlled by levodopa are likely poor candidates.
The test is also used as an indicator of whether the patient’s gait can be improved. Patients whose gait is substantially improved by levodopa, even for only a brief period of time, have a better chance of experiencing improvement in this domain with deep brain stimulation than those who do not show any gait improvement.
An important and notable exception to this rule is tremor control. Even Parkinson patients who do not experience significant improvement in tremor with levodopa (ie, who have medication-resistant tremors) are still likely to benefit from deep brain stimulation. Overall, tremor is the symptom that is most consistently improved with deep brain stimulation.
Results of clinical trials
Several clinical trials have demonstrated that deep brain stimulation plus medication works better than medications alone for advanced Parkinson disease.
Deuschl et al1 conducted a randomized trial in 156 patients with advanced Parkinson disease. Patients receiving subthalamic deep brain stimulation plus medication had significantly greater improvement in motor symptoms as measured by the Unified Parkinson’s Disease Rating Scale as well as in quality-of-life measures than patients receiving medications only.
Krack et al2 reported on the outcomes of 49 patients with advanced Parkinson disease who underwent deep brain stimulation and then were prospectively followed. At 5 years, motor function had improved by approximately 55% from baseline, activities-of-daily-living scores had improved by 49%, and patients continued to need significantly less levodopa and to experience less drug-induced dyskinesia.
Complications related to deep brain stimulation occurred in both studies, including two large intracerebral hemorrhages, one of which was fatal.
Weight gain. During the first 3 months after the device was implanted, patients tended to gain weight (mean 3 kg, maximum 5 kg). Although weight gain is considered an adverse effect, many patients are quite thin by the time they are candidates for deep brain stimulation, and in such cases gaining lean weight can be a benefit.
Patients with poorly controlled Parkinson disease lose weight for several reasons: increased calorie expenditure from shaking and excessive movements; diet modification and protein restriction for some patients who realize that protein competes with levodopa absorption; lack of appetite due to depression or from poor taste sensation (due to anosmia); and decreased overall food consumption due to difficulty swallowing.
DEEP BRAIN STIMULATION FOR ESSENTIAL TREMOR
Essential tremor is more common than Parkinson disease, with a prevalence in the United States estimated at approximately 4,000 per 100,000 people older than 65 years.
The tremor is often bilateral and is characteristically an action tremor, but in many patients it also has a postural, and sometimes a resting, component. It is distinct from parkinsonian tremor, which is usually predominantly a resting tremor. The differential diagnosis includes tremors secondary to central nervous system degenerative disorders as well as psychogenic tremors.
Drinking alcohol tends to relieve essential tremors, a finding that can often be elicited in the patient’s history. Patients whose symptoms improve with an alcoholic beverage are more likely to have essential tremor than another diagnosis.
Response to deep brain stimulation
Most patients with essential tremor respond well to deep brain stimulation of the contralateral ventral intermedius thalamic nucleus.
Treatment is usually started unilaterally, usually aimed at alleviating tremor in the patient’s dominant upper extremity. In selected cases, preference is given to treating the nondominant extremity when it is more severely affected than the dominant extremity.
Implantation of a device on the second side is offered to some patients who continue to be limited in activity and quality of life due to tremor of the untreated extremity. Surgery of the second side can be more complicated than the initial unilateral procedure. In particular, some patients may present with dysarthria, although that seems to be less common in our experience than initially estimated.
In practice, patients with moderate tremors tend to have an excellent response to deep brain stimulation. For this particular indication, if the response is not satisfactory, the treating team tends to consider surgically revising the placement of the lead rather than considering the patient a nonresponder. Patients with very severe tremors may have some residual tremor despite substantial improvement in severity. In our experience, patients with a greater proximal component of tremor tend to have less satisfactory results.
For challenging cases, implantation of additional electrodes in the thalamus or in new targets currently under investigation is sometimes considered, although this is an off-label use.
Treatment of secondary tremors, such as poststroke tremor or tremor due to multiple sclerosis, is sometimes attempted with deep brain stimulation. This is also an off-label option but is considered in selected cases for quality-of-life management.
Patients with axial tremors such as head or voice tremor are less likely to be helped by deep brain stimulation.
DEEP BRAIN STIMULATION FOR PRIMARY DYSTONIA
Generalized dystonia is a less common but severely impairing movement disorder.
Deep brain stimulation is approved for primary dystonia under a humanitarian device exemption, a regulatory mechanism for less common conditions. Deep brain stimulation is an option for patients who have significant impairment related to dystonia and who have not responded to conservative management such as anticholinergic agents, muscle relaxants, benzodiazepines, levodopa, or combinations of these drugs. Surgery has been shown to be effective for patients with primary generalized dystonia, whether or not they tested positive for a dystonia-related gene such as DYT1.
Kupsch et al3 evaluated 40 patients with primary dystonia in a randomized controlled trial of pallidal (globus pallidus pars interna) active deep brain stimulation vs sham stimulation (in which the device was implanted but not activated) for 3 months. Treated patients improved significantly more than controls (39% vs 5%) in the Burke-Fahn- Marsden Dystonia Rating Scale (BFMDRS).4 Similar improvement was noted when those receiving sham stimulation were switched to active stimulation.
During long-term follow-up, the results were generally sustained, with substantial improvement from deep brain stimulation in all movement symptoms evaluated except for speech and swallowing. Unlike improvement in tremor, which is quickly evident during testing in the operating room, the improvement in dystonia occurs gradually, and it may take months for patients to notice a change. Similarly, if stimulation stops because of device malfunction or dead batteries, symptoms sometimes do not recur for weeks or months.
Deep brain stimulation is sometimes offered to patients with dystonia secondary to conditions such as cerebral palsy or trauma (an off-label use). Although benefits are less consistent, deep brain stimulation remains an option for these individuals, aimed at alleviating some of the disabling symptoms. In patients with cerebral palsy or other secondary dystonias, it is sometimes difficult to distinguish how much of the disability is related to spasticity vs dystonia. Deep brain stimulation aims to alleviate the dystonic component; the spasticity may be managed with other options such as intrathecal baclofen (Lioresal).
Patients with tardive dystonia, which is usually secondary to treatment with antipsychotic agents, have been reported to respond well to bilateral deep brain stimulation. Gruber et al5 reported on a series of nine patients with a mean follow-up of 41 months. Patients improved by a mean of approximately 74% on the BFMDRS after 3 to 6 months of deep brain stimulation compared with baseline. None of the patients presented with long-term adverse effects, and quality of life and disability scores also improved significantly.
CANDIDATES ARE EVALUATED BY A MULTIDISCIPLINARY TEAM
Cleveland Clinic conducts a comprehensive 2-day evaluation for patients being considered for deep brain stimulation surgery, including consultations with specialists in neurology, neurosurgery, neuropsychology, and psychiatry.
Patients with significant cognitive deficits—near or meeting the diagnostic criteria for dementia—are usually not recommended to have surgery for Parkinson disease. Deep brain stimulation is not aimed at alleviating cognitive issues related to Parkinson disease or other concomitant dementia. In addition, there is a risk that neurostimulation could further worsen cognitive function in the already compromised brain. Moreover, patients with significant abnormalities detected by neuroimaging may have their diagnosis reconsidered in some cases, and some patients may not be deemed ideal candidates for surgery.
An important part of the process is a discussion with the patient and family about the risks and the potential short-term and long-term benefits. Informed consent requires a good understanding of this equation. Patients are counseled to have realistic expectations about what the procedure can offer. Deep brain stimulation can help some of the symptoms of Parkinson disease but will not cure it, and there is no evidence to date that it reduces its progress. At 5 or 10 years after surgery, patients are expected to be worse overall than they were in the first year after surgery, because of disease progression. However, patients who receive this treatment are expected, in general, to be doing better 5 or 10 years later (or longer) than those who do not receive it.
In addition to the discussion about risks, benefits, and expectations, a careful discussion is also devoted to hardware maintenance, including how to change the batteries. Particularly, younger patients should be informed about the risk of breakage of the leads and the extension wire, as they are likely to outlive their implant. Patients and caregivers should be able to come to the specialized center should hardware malfunction occur.
Patients are also informed that after the system is implanted they cannot undergo magnetic resonance imaging (MRI) except of the head, performed with a specific head coil and under specific parameters. MRI of any other body part and with a body coil is contraindicated.
HOW THE DEVICE IS IMPLANTED
There are several options for implanting a deep brain stimulation device.
Implantation with the patient awake, using a stereotactic headframe
At Cleveland Clinic, we usually prefer implantation with a stereotactic headframe. The base or “halo” of the frame is applied to the head under local anesthesia, followed by imaging via computed tomography (Figure 1). Typically, the tomographic image is fused to a previously acquired MRI image, but the MRI is sometimes either initially performed or repeated on the day of surgery.
Patients are sedated for the beginning of the procedure, while the surgical team is opening the skin and drilling the opening in the skull for placement of the lead. The patient is awakened for placement of the electrodes, which is not painful.
Microelectrode recording is typically performed in order to refine the targeting based on the stereotactic coordinates derived from neuroimaging. Although cadaver atlases exist and provide a guide to the stereotactic localization of subcortical structures, they are not completely accurate in representing the brain anatomy of all patients.
By “listening” to cells and knowing their characteristic signals in specific areas, landmarks can be created, forming an individualized map of the patient’s brain target. Microelectrode recording is invasive and has risks, including the risk of a brain hemorrhage. It is routinely done in most specialized deep brain stimulation centers because it can provide better accuracy and precision in lead placement.
When the target has been located and refined by microelectrode recording, the permanent electrode is inserted. Fluoroscopy is usually used to verify the direction and stability of placement during the procedure.
An intraoperative test of the effects of deep brain stimulation is routinely performed to verify that some benefits can be achieved with the brain lead in its location, to determine the threshold for side effects, or both. For example, the patient may be asked to hold a cup as if trying to drink from it and to write or to draw a spiral on a clipboard to assess for improvements in tremor. Rigidity and bradykinesia can also be tested for improvements.
This intraoperative test is not aimed at assessing the best possible outcome of deep brain stimulation, and not even to see an improvement in all symptoms that burden the patient. Rather, it is to evaluate the likelihood that programming will be feasible with the implanted lead.
Subsequently, implantation of the pulse generator in the chest and connection to the brain lead is completed, usually with the patient under general anesthesia.
Implantation under general anesthesia, with intraoperative MRI
A new alternative to “awake stereotactic surgery” is implantation with the patient under general anesthesia, with intraoperative MRI. We have started to do this procedure in a new operating suite that is attached to an MRI suite. The magnet can be taken in and out of the operating room, allowing the surgeon to verify the location of the implanted leads right at the time of the procedure. In this fashion, intraoperative images are used to guide implantation instead of awake microelectrode recording. This is a new option for patients who cannot tolerate awake surgery and for those who have a contraindication to the regular stereotactic procedure with the patient awake.
Risks of bleeding and infection
The potential complications of implanting a device and leads in the brain can be significant.
Hemorrhage can occur, resulting in a superficial or deep hematoma.
Infection and erosion may require removal of the hardware for antibiotic treatment and possible reimplantation.
Other risks include those related to tunneling the wires from the head to the chest, to implanting the device in the chest, and to serious medical complications after surgery. Hardware failure can occur and requires additional surgery. Finally, environmental risks and risks related to medical devices such as MRI, electrocautery, and cardioversion should also be considered.
Deep brain stimulation is advantageous for its reversibility. If during postoperative programming the brain leads are considered not to be ideally placed, revisions can be done to reposition the leads.
- Deuschl G, Schade-Brittinger C, Krack P, et al; German Parkinson Study Group, Neurostimulation Section. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 2006; 355:896–908.
- Krack P, Batir A, Van Blercom N, et al. Five-year followup of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med 2003; 349:1925–1934.
- Kupsch A, Benecke R, Müller J, et al; Deep-Brain Stimulation for Dystonia Study Group. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med 2006; 355:1978–1990.
- Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scle for the primary torsion dystonias. Neurology 1985; 35:73–77.
- Gruber D, Trottenberg T, Kivi A, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology 2009; 73:53–58.
Deep brain stimulation is an important therapy for Parkinson disease and other movement disorders. It involves implantation of a pulse generator that can be adjusted by telemetry and can be activated and deactivated by clinicians and patients. It is therefore also a good investigational tool, allowing for double-blind, sham-controlled clinical trials by testing the effects of the stimulation with optimal settings compared with no stimulation.
This article will discuss the approved indications for deep brain stimulation (particularly for managing movement disorders), the benefits that can be expected, the risks, the complications, the maintenance required, how candidates for this treatment are evaluated, and the surgical procedure for implantation of the devices.
DEVICE SIMILAR TO HEART PACEMAKERS
The deep brain stimulation system must be programmed by a physician or midlevel practitioner by observing a symptom and then changing the applied settings to the pulse generator until the symptom improves. This can be a very time-consuming process.
In contrast to heart pacemakers, which run at low frequencies, the brain devices for movement disorders are almost always set to a high frequency, greater than 100 Hz. For this reason, they consume more energy and need larger batteries than those in modern heart pacemakers.
The batteries in these generators typically last 3 to 5 years and are replaced in an outpatient procedure. Newer, smaller, rechargeable devices are expected to last longer but require more maintenance and care by patients, who have to recharge them at home periodically.
INDICATIONS FOR DEEP BRAIN STIMULATION
Deep brain stimulation is approved by the US Food and Drug Administration (FDA) for specific indications:
- Parkinson disease
- Essential tremor
- Primary dystonia (under a humanitarian device exemption)
- Intractable obsessive-compulsive disorder (also under a humanitarian device exemption). We will not discuss this indication further in this paper.
For each of these conditions, deep brain stimulation is considered when nonsurgical management has failed, as is the case for most functional neurosurgical treatments.
Investigations under way in other disorders
Several studies of deep brain stimulation are currently in progress under FDA-approved investigational device exemptions. Some, with funding from industry, are exploring its use in neuropsychiatric conditions other than parkinsonism. Two large clinical trials are evaluating its use for treatment-refractory depression, a common problem and a leading cause of disability in the industrialized world. Multiple investigators are also exploring novel uses of this technology in disorders ranging from obsessive-compulsive disorder to epilepsy.
Investigation is also under way at Cleveland Clinic in a federally funded, prospective, randomized clinical trial of deep brain stimulation for patients with thalamic pain syndrome. The primary hypothesis is that stimulation of the ventral striatal and ventral capsular area will modulate the affective component of this otherwise intractable pain syndrome, reducing pain-related disability and improving quality of life.
DEEP BRAIN STIMULATION VS ABLATION
Before deep brain stimulation became available, the only surgical options for patients with advanced Parkinson disease, tremor, or dystonia were ablative procedures such as pallidotomy (ablation of part of the globus pallidus) and thalamotomy (ablation of part of the thalamus). These procedures had been well known for several decades but fell out of favor when levodopa became available in the 1960s and revolutionized the medical treatment of Parkinson disease.
Surgery for movement disorders, in particular Parkinson disease, had a rebirth in the late 1980s when the limitations and complications associated with the pharmacologic management of Parkinson disease became increasingly evident. Ablative procedures are still used to treat advanced Parkinson disease, but much less commonly in industrialized countries.
Although pallidotomy and thalamotomy can have excellent results, they are not as safe as deep brain stimulation, which has the advantage of being reversible, modulating the function of an area rather than destroying it. Any unwanted effect can be immediately altered or reversed, unlike ablative procedures, in which any change is permanent. In addition, deep brain stimulation is adjustable, and the settings can be optimized as the disease progresses over the years.
Ablative procedures can be risky when performed bilaterally, while deep brain stimulation is routinely done on both hemispheres for patients with bilateral symptoms.
Although deep brain stimulation is today’s surgical treatment of choice, it is not perfect. It has the disadvantage of requiring lifelong maintenance of the hardware, for which the patient remains dependent on a medical center. Patients are usually seen more often at the specialized center in the first few months after surgery for optimization of programming and titration of drugs. (During this time, most patients see a gradual, substantial reduction in medication intake.) They are then followed by their physician and visit the center less often for monitoring of disease status and for further adjustments to the stimulator.
Most patients, to date, receive nonrechargeable pulse generators. As mentioned above, the batteries in these devices typically last 3 to 5 years. Preferably, batteries are replaced before they are completely depleted, to avoid interruption of therapy. Periodic visits to the center allow clinicians to estimate battery expiration ahead of time and plan replacements accordingly.
Rechargeable pulse generators have been recently introduced and are expected to last up to 9 years. They are an option for patients who can comply with the requirements for periodic home recharging of the hardware.
Patients are given a remote control so that they can turn the device on or off and check its status. Most patients keep it turned on all the time, although some turn it off at night to save battery life.
WHAT CAN PARKINSON PATIENTS EXPECT FROM THIS THERAPY?
Typically, some parkinsonian symptoms predominate over others, although some patients with advanced disease present with a severe combination of multiple disabling symptoms. Deep brain stimulation is best suited to address some of the cardinal motor symptoms, particularly tremor, rigidity, and bradykinesia, and motor fluctuations such as “wearing off” and dyskinesia.
Improvement in some motor symptoms
As a general rule, appendicular symptoms such as limb tremor and rigidity are more responsive to this therapy than axial symptoms such as gait and balance problems, but some patients experience improvement in gait as well. Other symptoms, such as swallowing or urinary symptoms, are seldom helped.
Although deep brain stimulation can help manage key motor symptoms and improve quality of life, it does not cure Parkinson disease. Also, there is no evidence to date that it slows disease progression, although this is a topic of ongoing investigation.
Fewer motor fluctuations
A common complaint of patients with advanced Parkinson disease is frequent—and often unpredictable—fluctuations between the “on” state (ie, when the effects of the patient’s levodopa therapy are apparent) and the “off” state (ie, when the levodopa doesn’t seem to be working). Sometimes, in the on state, patients experience involuntary choreic or ballistic movements, called dyskinesias. They also complain that the on time progressively lasts shorter and the day is spent alternating between shorter on states (during which the patient may be dyskinetic) and longer off states, limiting the patient’s independence and quality of life.
Deep brain stimulation can help patients prolong the on time while reducing the amplitude of these fluctuations so that the symptoms are not as severe in the off time and dyskinesias are reduced in the on time.
Some patients undergo deep brain stimulation primarily for managing the adverse effects of levodopa rather than for controlling the symptoms of the disease itself. While these patients need levodopa to address the disabling symptoms of the disease, they also present a greater sensitivity for developing levodopa-induced dyskinesias, quickly fluctuating from a lack of movement (the off state) to a state of uncontrollable movements (during the on state).
Deep brain stimulation typically allows the dosage of levodopa to be significantly reduced and gives patients more on time with fewer side effects and less fluctuation between the on and off states.
Response to levodopa predicts deep brain stimulation’s effects
Whether a patient is likely to be helped by deep brain stimulation can be tested with reasonable predictability by giving a single therapeutic dose of levodopa after the patient has been free of the drug for 12 hours. If there is an obvious difference on objective quantitative testing between the off and on states with a single dose, the patient is likely to benefit from deep brain stimulation. Those who do not respond well or are known to have never been well controlled by levodopa are likely poor candidates.
The test is also used as an indicator of whether the patient’s gait can be improved. Patients whose gait is substantially improved by levodopa, even for only a brief period of time, have a better chance of experiencing improvement in this domain with deep brain stimulation than those who do not show any gait improvement.
An important and notable exception to this rule is tremor control. Even Parkinson patients who do not experience significant improvement in tremor with levodopa (ie, who have medication-resistant tremors) are still likely to benefit from deep brain stimulation. Overall, tremor is the symptom that is most consistently improved with deep brain stimulation.
Results of clinical trials
Several clinical trials have demonstrated that deep brain stimulation plus medication works better than medications alone for advanced Parkinson disease.
Deuschl et al1 conducted a randomized trial in 156 patients with advanced Parkinson disease. Patients receiving subthalamic deep brain stimulation plus medication had significantly greater improvement in motor symptoms as measured by the Unified Parkinson’s Disease Rating Scale as well as in quality-of-life measures than patients receiving medications only.
Krack et al2 reported on the outcomes of 49 patients with advanced Parkinson disease who underwent deep brain stimulation and then were prospectively followed. At 5 years, motor function had improved by approximately 55% from baseline, activities-of-daily-living scores had improved by 49%, and patients continued to need significantly less levodopa and to experience less drug-induced dyskinesia.
Complications related to deep brain stimulation occurred in both studies, including two large intracerebral hemorrhages, one of which was fatal.
Weight gain. During the first 3 months after the device was implanted, patients tended to gain weight (mean 3 kg, maximum 5 kg). Although weight gain is considered an adverse effect, many patients are quite thin by the time they are candidates for deep brain stimulation, and in such cases gaining lean weight can be a benefit.
Patients with poorly controlled Parkinson disease lose weight for several reasons: increased calorie expenditure from shaking and excessive movements; diet modification and protein restriction for some patients who realize that protein competes with levodopa absorption; lack of appetite due to depression or from poor taste sensation (due to anosmia); and decreased overall food consumption due to difficulty swallowing.
DEEP BRAIN STIMULATION FOR ESSENTIAL TREMOR
Essential tremor is more common than Parkinson disease, with a prevalence in the United States estimated at approximately 4,000 per 100,000 people older than 65 years.
The tremor is often bilateral and is characteristically an action tremor, but in many patients it also has a postural, and sometimes a resting, component. It is distinct from parkinsonian tremor, which is usually predominantly a resting tremor. The differential diagnosis includes tremors secondary to central nervous system degenerative disorders as well as psychogenic tremors.
Drinking alcohol tends to relieve essential tremors, a finding that can often be elicited in the patient’s history. Patients whose symptoms improve with an alcoholic beverage are more likely to have essential tremor than another diagnosis.
Response to deep brain stimulation
Most patients with essential tremor respond well to deep brain stimulation of the contralateral ventral intermedius thalamic nucleus.
Treatment is usually started unilaterally, usually aimed at alleviating tremor in the patient’s dominant upper extremity. In selected cases, preference is given to treating the nondominant extremity when it is more severely affected than the dominant extremity.
Implantation of a device on the second side is offered to some patients who continue to be limited in activity and quality of life due to tremor of the untreated extremity. Surgery of the second side can be more complicated than the initial unilateral procedure. In particular, some patients may present with dysarthria, although that seems to be less common in our experience than initially estimated.
In practice, patients with moderate tremors tend to have an excellent response to deep brain stimulation. For this particular indication, if the response is not satisfactory, the treating team tends to consider surgically revising the placement of the lead rather than considering the patient a nonresponder. Patients with very severe tremors may have some residual tremor despite substantial improvement in severity. In our experience, patients with a greater proximal component of tremor tend to have less satisfactory results.
For challenging cases, implantation of additional electrodes in the thalamus or in new targets currently under investigation is sometimes considered, although this is an off-label use.
Treatment of secondary tremors, such as poststroke tremor or tremor due to multiple sclerosis, is sometimes attempted with deep brain stimulation. This is also an off-label option but is considered in selected cases for quality-of-life management.
Patients with axial tremors such as head or voice tremor are less likely to be helped by deep brain stimulation.
DEEP BRAIN STIMULATION FOR PRIMARY DYSTONIA
Generalized dystonia is a less common but severely impairing movement disorder.
Deep brain stimulation is approved for primary dystonia under a humanitarian device exemption, a regulatory mechanism for less common conditions. Deep brain stimulation is an option for patients who have significant impairment related to dystonia and who have not responded to conservative management such as anticholinergic agents, muscle relaxants, benzodiazepines, levodopa, or combinations of these drugs. Surgery has been shown to be effective for patients with primary generalized dystonia, whether or not they tested positive for a dystonia-related gene such as DYT1.
Kupsch et al3 evaluated 40 patients with primary dystonia in a randomized controlled trial of pallidal (globus pallidus pars interna) active deep brain stimulation vs sham stimulation (in which the device was implanted but not activated) for 3 months. Treated patients improved significantly more than controls (39% vs 5%) in the Burke-Fahn- Marsden Dystonia Rating Scale (BFMDRS).4 Similar improvement was noted when those receiving sham stimulation were switched to active stimulation.
During long-term follow-up, the results were generally sustained, with substantial improvement from deep brain stimulation in all movement symptoms evaluated except for speech and swallowing. Unlike improvement in tremor, which is quickly evident during testing in the operating room, the improvement in dystonia occurs gradually, and it may take months for patients to notice a change. Similarly, if stimulation stops because of device malfunction or dead batteries, symptoms sometimes do not recur for weeks or months.
Deep brain stimulation is sometimes offered to patients with dystonia secondary to conditions such as cerebral palsy or trauma (an off-label use). Although benefits are less consistent, deep brain stimulation remains an option for these individuals, aimed at alleviating some of the disabling symptoms. In patients with cerebral palsy or other secondary dystonias, it is sometimes difficult to distinguish how much of the disability is related to spasticity vs dystonia. Deep brain stimulation aims to alleviate the dystonic component; the spasticity may be managed with other options such as intrathecal baclofen (Lioresal).
Patients with tardive dystonia, which is usually secondary to treatment with antipsychotic agents, have been reported to respond well to bilateral deep brain stimulation. Gruber et al5 reported on a series of nine patients with a mean follow-up of 41 months. Patients improved by a mean of approximately 74% on the BFMDRS after 3 to 6 months of deep brain stimulation compared with baseline. None of the patients presented with long-term adverse effects, and quality of life and disability scores also improved significantly.
CANDIDATES ARE EVALUATED BY A MULTIDISCIPLINARY TEAM
Cleveland Clinic conducts a comprehensive 2-day evaluation for patients being considered for deep brain stimulation surgery, including consultations with specialists in neurology, neurosurgery, neuropsychology, and psychiatry.
Patients with significant cognitive deficits—near or meeting the diagnostic criteria for dementia—are usually not recommended to have surgery for Parkinson disease. Deep brain stimulation is not aimed at alleviating cognitive issues related to Parkinson disease or other concomitant dementia. In addition, there is a risk that neurostimulation could further worsen cognitive function in the already compromised brain. Moreover, patients with significant abnormalities detected by neuroimaging may have their diagnosis reconsidered in some cases, and some patients may not be deemed ideal candidates for surgery.
An important part of the process is a discussion with the patient and family about the risks and the potential short-term and long-term benefits. Informed consent requires a good understanding of this equation. Patients are counseled to have realistic expectations about what the procedure can offer. Deep brain stimulation can help some of the symptoms of Parkinson disease but will not cure it, and there is no evidence to date that it reduces its progress. At 5 or 10 years after surgery, patients are expected to be worse overall than they were in the first year after surgery, because of disease progression. However, patients who receive this treatment are expected, in general, to be doing better 5 or 10 years later (or longer) than those who do not receive it.
In addition to the discussion about risks, benefits, and expectations, a careful discussion is also devoted to hardware maintenance, including how to change the batteries. Particularly, younger patients should be informed about the risk of breakage of the leads and the extension wire, as they are likely to outlive their implant. Patients and caregivers should be able to come to the specialized center should hardware malfunction occur.
Patients are also informed that after the system is implanted they cannot undergo magnetic resonance imaging (MRI) except of the head, performed with a specific head coil and under specific parameters. MRI of any other body part and with a body coil is contraindicated.
HOW THE DEVICE IS IMPLANTED
There are several options for implanting a deep brain stimulation device.
Implantation with the patient awake, using a stereotactic headframe
At Cleveland Clinic, we usually prefer implantation with a stereotactic headframe. The base or “halo” of the frame is applied to the head under local anesthesia, followed by imaging via computed tomography (Figure 1). Typically, the tomographic image is fused to a previously acquired MRI image, but the MRI is sometimes either initially performed or repeated on the day of surgery.
Patients are sedated for the beginning of the procedure, while the surgical team is opening the skin and drilling the opening in the skull for placement of the lead. The patient is awakened for placement of the electrodes, which is not painful.
Microelectrode recording is typically performed in order to refine the targeting based on the stereotactic coordinates derived from neuroimaging. Although cadaver atlases exist and provide a guide to the stereotactic localization of subcortical structures, they are not completely accurate in representing the brain anatomy of all patients.
By “listening” to cells and knowing their characteristic signals in specific areas, landmarks can be created, forming an individualized map of the patient’s brain target. Microelectrode recording is invasive and has risks, including the risk of a brain hemorrhage. It is routinely done in most specialized deep brain stimulation centers because it can provide better accuracy and precision in lead placement.
When the target has been located and refined by microelectrode recording, the permanent electrode is inserted. Fluoroscopy is usually used to verify the direction and stability of placement during the procedure.
An intraoperative test of the effects of deep brain stimulation is routinely performed to verify that some benefits can be achieved with the brain lead in its location, to determine the threshold for side effects, or both. For example, the patient may be asked to hold a cup as if trying to drink from it and to write or to draw a spiral on a clipboard to assess for improvements in tremor. Rigidity and bradykinesia can also be tested for improvements.
This intraoperative test is not aimed at assessing the best possible outcome of deep brain stimulation, and not even to see an improvement in all symptoms that burden the patient. Rather, it is to evaluate the likelihood that programming will be feasible with the implanted lead.
Subsequently, implantation of the pulse generator in the chest and connection to the brain lead is completed, usually with the patient under general anesthesia.
Implantation under general anesthesia, with intraoperative MRI
A new alternative to “awake stereotactic surgery” is implantation with the patient under general anesthesia, with intraoperative MRI. We have started to do this procedure in a new operating suite that is attached to an MRI suite. The magnet can be taken in and out of the operating room, allowing the surgeon to verify the location of the implanted leads right at the time of the procedure. In this fashion, intraoperative images are used to guide implantation instead of awake microelectrode recording. This is a new option for patients who cannot tolerate awake surgery and for those who have a contraindication to the regular stereotactic procedure with the patient awake.
Risks of bleeding and infection
The potential complications of implanting a device and leads in the brain can be significant.
Hemorrhage can occur, resulting in a superficial or deep hematoma.
Infection and erosion may require removal of the hardware for antibiotic treatment and possible reimplantation.
Other risks include those related to tunneling the wires from the head to the chest, to implanting the device in the chest, and to serious medical complications after surgery. Hardware failure can occur and requires additional surgery. Finally, environmental risks and risks related to medical devices such as MRI, electrocautery, and cardioversion should also be considered.
Deep brain stimulation is advantageous for its reversibility. If during postoperative programming the brain leads are considered not to be ideally placed, revisions can be done to reposition the leads.
Deep brain stimulation is an important therapy for Parkinson disease and other movement disorders. It involves implantation of a pulse generator that can be adjusted by telemetry and can be activated and deactivated by clinicians and patients. It is therefore also a good investigational tool, allowing for double-blind, sham-controlled clinical trials by testing the effects of the stimulation with optimal settings compared with no stimulation.
This article will discuss the approved indications for deep brain stimulation (particularly for managing movement disorders), the benefits that can be expected, the risks, the complications, the maintenance required, how candidates for this treatment are evaluated, and the surgical procedure for implantation of the devices.
DEVICE SIMILAR TO HEART PACEMAKERS
The deep brain stimulation system must be programmed by a physician or midlevel practitioner by observing a symptom and then changing the applied settings to the pulse generator until the symptom improves. This can be a very time-consuming process.
In contrast to heart pacemakers, which run at low frequencies, the brain devices for movement disorders are almost always set to a high frequency, greater than 100 Hz. For this reason, they consume more energy and need larger batteries than those in modern heart pacemakers.
The batteries in these generators typically last 3 to 5 years and are replaced in an outpatient procedure. Newer, smaller, rechargeable devices are expected to last longer but require more maintenance and care by patients, who have to recharge them at home periodically.
INDICATIONS FOR DEEP BRAIN STIMULATION
Deep brain stimulation is approved by the US Food and Drug Administration (FDA) for specific indications:
- Parkinson disease
- Essential tremor
- Primary dystonia (under a humanitarian device exemption)
- Intractable obsessive-compulsive disorder (also under a humanitarian device exemption). We will not discuss this indication further in this paper.
For each of these conditions, deep brain stimulation is considered when nonsurgical management has failed, as is the case for most functional neurosurgical treatments.
Investigations under way in other disorders
Several studies of deep brain stimulation are currently in progress under FDA-approved investigational device exemptions. Some, with funding from industry, are exploring its use in neuropsychiatric conditions other than parkinsonism. Two large clinical trials are evaluating its use for treatment-refractory depression, a common problem and a leading cause of disability in the industrialized world. Multiple investigators are also exploring novel uses of this technology in disorders ranging from obsessive-compulsive disorder to epilepsy.
Investigation is also under way at Cleveland Clinic in a federally funded, prospective, randomized clinical trial of deep brain stimulation for patients with thalamic pain syndrome. The primary hypothesis is that stimulation of the ventral striatal and ventral capsular area will modulate the affective component of this otherwise intractable pain syndrome, reducing pain-related disability and improving quality of life.
DEEP BRAIN STIMULATION VS ABLATION
Before deep brain stimulation became available, the only surgical options for patients with advanced Parkinson disease, tremor, or dystonia were ablative procedures such as pallidotomy (ablation of part of the globus pallidus) and thalamotomy (ablation of part of the thalamus). These procedures had been well known for several decades but fell out of favor when levodopa became available in the 1960s and revolutionized the medical treatment of Parkinson disease.
Surgery for movement disorders, in particular Parkinson disease, had a rebirth in the late 1980s when the limitations and complications associated with the pharmacologic management of Parkinson disease became increasingly evident. Ablative procedures are still used to treat advanced Parkinson disease, but much less commonly in industrialized countries.
Although pallidotomy and thalamotomy can have excellent results, they are not as safe as deep brain stimulation, which has the advantage of being reversible, modulating the function of an area rather than destroying it. Any unwanted effect can be immediately altered or reversed, unlike ablative procedures, in which any change is permanent. In addition, deep brain stimulation is adjustable, and the settings can be optimized as the disease progresses over the years.
Ablative procedures can be risky when performed bilaterally, while deep brain stimulation is routinely done on both hemispheres for patients with bilateral symptoms.
Although deep brain stimulation is today’s surgical treatment of choice, it is not perfect. It has the disadvantage of requiring lifelong maintenance of the hardware, for which the patient remains dependent on a medical center. Patients are usually seen more often at the specialized center in the first few months after surgery for optimization of programming and titration of drugs. (During this time, most patients see a gradual, substantial reduction in medication intake.) They are then followed by their physician and visit the center less often for monitoring of disease status and for further adjustments to the stimulator.
Most patients, to date, receive nonrechargeable pulse generators. As mentioned above, the batteries in these devices typically last 3 to 5 years. Preferably, batteries are replaced before they are completely depleted, to avoid interruption of therapy. Periodic visits to the center allow clinicians to estimate battery expiration ahead of time and plan replacements accordingly.
Rechargeable pulse generators have been recently introduced and are expected to last up to 9 years. They are an option for patients who can comply with the requirements for periodic home recharging of the hardware.
Patients are given a remote control so that they can turn the device on or off and check its status. Most patients keep it turned on all the time, although some turn it off at night to save battery life.
WHAT CAN PARKINSON PATIENTS EXPECT FROM THIS THERAPY?
Typically, some parkinsonian symptoms predominate over others, although some patients with advanced disease present with a severe combination of multiple disabling symptoms. Deep brain stimulation is best suited to address some of the cardinal motor symptoms, particularly tremor, rigidity, and bradykinesia, and motor fluctuations such as “wearing off” and dyskinesia.
Improvement in some motor symptoms
As a general rule, appendicular symptoms such as limb tremor and rigidity are more responsive to this therapy than axial symptoms such as gait and balance problems, but some patients experience improvement in gait as well. Other symptoms, such as swallowing or urinary symptoms, are seldom helped.
Although deep brain stimulation can help manage key motor symptoms and improve quality of life, it does not cure Parkinson disease. Also, there is no evidence to date that it slows disease progression, although this is a topic of ongoing investigation.
Fewer motor fluctuations
A common complaint of patients with advanced Parkinson disease is frequent—and often unpredictable—fluctuations between the “on” state (ie, when the effects of the patient’s levodopa therapy are apparent) and the “off” state (ie, when the levodopa doesn’t seem to be working). Sometimes, in the on state, patients experience involuntary choreic or ballistic movements, called dyskinesias. They also complain that the on time progressively lasts shorter and the day is spent alternating between shorter on states (during which the patient may be dyskinetic) and longer off states, limiting the patient’s independence and quality of life.
Deep brain stimulation can help patients prolong the on time while reducing the amplitude of these fluctuations so that the symptoms are not as severe in the off time and dyskinesias are reduced in the on time.
Some patients undergo deep brain stimulation primarily for managing the adverse effects of levodopa rather than for controlling the symptoms of the disease itself. While these patients need levodopa to address the disabling symptoms of the disease, they also present a greater sensitivity for developing levodopa-induced dyskinesias, quickly fluctuating from a lack of movement (the off state) to a state of uncontrollable movements (during the on state).
Deep brain stimulation typically allows the dosage of levodopa to be significantly reduced and gives patients more on time with fewer side effects and less fluctuation between the on and off states.
Response to levodopa predicts deep brain stimulation’s effects
Whether a patient is likely to be helped by deep brain stimulation can be tested with reasonable predictability by giving a single therapeutic dose of levodopa after the patient has been free of the drug for 12 hours. If there is an obvious difference on objective quantitative testing between the off and on states with a single dose, the patient is likely to benefit from deep brain stimulation. Those who do not respond well or are known to have never been well controlled by levodopa are likely poor candidates.
The test is also used as an indicator of whether the patient’s gait can be improved. Patients whose gait is substantially improved by levodopa, even for only a brief period of time, have a better chance of experiencing improvement in this domain with deep brain stimulation than those who do not show any gait improvement.
An important and notable exception to this rule is tremor control. Even Parkinson patients who do not experience significant improvement in tremor with levodopa (ie, who have medication-resistant tremors) are still likely to benefit from deep brain stimulation. Overall, tremor is the symptom that is most consistently improved with deep brain stimulation.
Results of clinical trials
Several clinical trials have demonstrated that deep brain stimulation plus medication works better than medications alone for advanced Parkinson disease.
Deuschl et al1 conducted a randomized trial in 156 patients with advanced Parkinson disease. Patients receiving subthalamic deep brain stimulation plus medication had significantly greater improvement in motor symptoms as measured by the Unified Parkinson’s Disease Rating Scale as well as in quality-of-life measures than patients receiving medications only.
Krack et al2 reported on the outcomes of 49 patients with advanced Parkinson disease who underwent deep brain stimulation and then were prospectively followed. At 5 years, motor function had improved by approximately 55% from baseline, activities-of-daily-living scores had improved by 49%, and patients continued to need significantly less levodopa and to experience less drug-induced dyskinesia.
Complications related to deep brain stimulation occurred in both studies, including two large intracerebral hemorrhages, one of which was fatal.
Weight gain. During the first 3 months after the device was implanted, patients tended to gain weight (mean 3 kg, maximum 5 kg). Although weight gain is considered an adverse effect, many patients are quite thin by the time they are candidates for deep brain stimulation, and in such cases gaining lean weight can be a benefit.
Patients with poorly controlled Parkinson disease lose weight for several reasons: increased calorie expenditure from shaking and excessive movements; diet modification and protein restriction for some patients who realize that protein competes with levodopa absorption; lack of appetite due to depression or from poor taste sensation (due to anosmia); and decreased overall food consumption due to difficulty swallowing.
DEEP BRAIN STIMULATION FOR ESSENTIAL TREMOR
Essential tremor is more common than Parkinson disease, with a prevalence in the United States estimated at approximately 4,000 per 100,000 people older than 65 years.
The tremor is often bilateral and is characteristically an action tremor, but in many patients it also has a postural, and sometimes a resting, component. It is distinct from parkinsonian tremor, which is usually predominantly a resting tremor. The differential diagnosis includes tremors secondary to central nervous system degenerative disorders as well as psychogenic tremors.
Drinking alcohol tends to relieve essential tremors, a finding that can often be elicited in the patient’s history. Patients whose symptoms improve with an alcoholic beverage are more likely to have essential tremor than another diagnosis.
Response to deep brain stimulation
Most patients with essential tremor respond well to deep brain stimulation of the contralateral ventral intermedius thalamic nucleus.
Treatment is usually started unilaterally, usually aimed at alleviating tremor in the patient’s dominant upper extremity. In selected cases, preference is given to treating the nondominant extremity when it is more severely affected than the dominant extremity.
Implantation of a device on the second side is offered to some patients who continue to be limited in activity and quality of life due to tremor of the untreated extremity. Surgery of the second side can be more complicated than the initial unilateral procedure. In particular, some patients may present with dysarthria, although that seems to be less common in our experience than initially estimated.
In practice, patients with moderate tremors tend to have an excellent response to deep brain stimulation. For this particular indication, if the response is not satisfactory, the treating team tends to consider surgically revising the placement of the lead rather than considering the patient a nonresponder. Patients with very severe tremors may have some residual tremor despite substantial improvement in severity. In our experience, patients with a greater proximal component of tremor tend to have less satisfactory results.
For challenging cases, implantation of additional electrodes in the thalamus or in new targets currently under investigation is sometimes considered, although this is an off-label use.
Treatment of secondary tremors, such as poststroke tremor or tremor due to multiple sclerosis, is sometimes attempted with deep brain stimulation. This is also an off-label option but is considered in selected cases for quality-of-life management.
Patients with axial tremors such as head or voice tremor are less likely to be helped by deep brain stimulation.
DEEP BRAIN STIMULATION FOR PRIMARY DYSTONIA
Generalized dystonia is a less common but severely impairing movement disorder.
Deep brain stimulation is approved for primary dystonia under a humanitarian device exemption, a regulatory mechanism for less common conditions. Deep brain stimulation is an option for patients who have significant impairment related to dystonia and who have not responded to conservative management such as anticholinergic agents, muscle relaxants, benzodiazepines, levodopa, or combinations of these drugs. Surgery has been shown to be effective for patients with primary generalized dystonia, whether or not they tested positive for a dystonia-related gene such as DYT1.
Kupsch et al3 evaluated 40 patients with primary dystonia in a randomized controlled trial of pallidal (globus pallidus pars interna) active deep brain stimulation vs sham stimulation (in which the device was implanted but not activated) for 3 months. Treated patients improved significantly more than controls (39% vs 5%) in the Burke-Fahn- Marsden Dystonia Rating Scale (BFMDRS).4 Similar improvement was noted when those receiving sham stimulation were switched to active stimulation.
During long-term follow-up, the results were generally sustained, with substantial improvement from deep brain stimulation in all movement symptoms evaluated except for speech and swallowing. Unlike improvement in tremor, which is quickly evident during testing in the operating room, the improvement in dystonia occurs gradually, and it may take months for patients to notice a change. Similarly, if stimulation stops because of device malfunction or dead batteries, symptoms sometimes do not recur for weeks or months.
Deep brain stimulation is sometimes offered to patients with dystonia secondary to conditions such as cerebral palsy or trauma (an off-label use). Although benefits are less consistent, deep brain stimulation remains an option for these individuals, aimed at alleviating some of the disabling symptoms. In patients with cerebral palsy or other secondary dystonias, it is sometimes difficult to distinguish how much of the disability is related to spasticity vs dystonia. Deep brain stimulation aims to alleviate the dystonic component; the spasticity may be managed with other options such as intrathecal baclofen (Lioresal).
Patients with tardive dystonia, which is usually secondary to treatment with antipsychotic agents, have been reported to respond well to bilateral deep brain stimulation. Gruber et al5 reported on a series of nine patients with a mean follow-up of 41 months. Patients improved by a mean of approximately 74% on the BFMDRS after 3 to 6 months of deep brain stimulation compared with baseline. None of the patients presented with long-term adverse effects, and quality of life and disability scores also improved significantly.
CANDIDATES ARE EVALUATED BY A MULTIDISCIPLINARY TEAM
Cleveland Clinic conducts a comprehensive 2-day evaluation for patients being considered for deep brain stimulation surgery, including consultations with specialists in neurology, neurosurgery, neuropsychology, and psychiatry.
Patients with significant cognitive deficits—near or meeting the diagnostic criteria for dementia—are usually not recommended to have surgery for Parkinson disease. Deep brain stimulation is not aimed at alleviating cognitive issues related to Parkinson disease or other concomitant dementia. In addition, there is a risk that neurostimulation could further worsen cognitive function in the already compromised brain. Moreover, patients with significant abnormalities detected by neuroimaging may have their diagnosis reconsidered in some cases, and some patients may not be deemed ideal candidates for surgery.
An important part of the process is a discussion with the patient and family about the risks and the potential short-term and long-term benefits. Informed consent requires a good understanding of this equation. Patients are counseled to have realistic expectations about what the procedure can offer. Deep brain stimulation can help some of the symptoms of Parkinson disease but will not cure it, and there is no evidence to date that it reduces its progress. At 5 or 10 years after surgery, patients are expected to be worse overall than they were in the first year after surgery, because of disease progression. However, patients who receive this treatment are expected, in general, to be doing better 5 or 10 years later (or longer) than those who do not receive it.
In addition to the discussion about risks, benefits, and expectations, a careful discussion is also devoted to hardware maintenance, including how to change the batteries. Particularly, younger patients should be informed about the risk of breakage of the leads and the extension wire, as they are likely to outlive their implant. Patients and caregivers should be able to come to the specialized center should hardware malfunction occur.
Patients are also informed that after the system is implanted they cannot undergo magnetic resonance imaging (MRI) except of the head, performed with a specific head coil and under specific parameters. MRI of any other body part and with a body coil is contraindicated.
HOW THE DEVICE IS IMPLANTED
There are several options for implanting a deep brain stimulation device.
Implantation with the patient awake, using a stereotactic headframe
At Cleveland Clinic, we usually prefer implantation with a stereotactic headframe. The base or “halo” of the frame is applied to the head under local anesthesia, followed by imaging via computed tomography (Figure 1). Typically, the tomographic image is fused to a previously acquired MRI image, but the MRI is sometimes either initially performed or repeated on the day of surgery.
Patients are sedated for the beginning of the procedure, while the surgical team is opening the skin and drilling the opening in the skull for placement of the lead. The patient is awakened for placement of the electrodes, which is not painful.
Microelectrode recording is typically performed in order to refine the targeting based on the stereotactic coordinates derived from neuroimaging. Although cadaver atlases exist and provide a guide to the stereotactic localization of subcortical structures, they are not completely accurate in representing the brain anatomy of all patients.
By “listening” to cells and knowing their characteristic signals in specific areas, landmarks can be created, forming an individualized map of the patient’s brain target. Microelectrode recording is invasive and has risks, including the risk of a brain hemorrhage. It is routinely done in most specialized deep brain stimulation centers because it can provide better accuracy and precision in lead placement.
When the target has been located and refined by microelectrode recording, the permanent electrode is inserted. Fluoroscopy is usually used to verify the direction and stability of placement during the procedure.
An intraoperative test of the effects of deep brain stimulation is routinely performed to verify that some benefits can be achieved with the brain lead in its location, to determine the threshold for side effects, or both. For example, the patient may be asked to hold a cup as if trying to drink from it and to write or to draw a spiral on a clipboard to assess for improvements in tremor. Rigidity and bradykinesia can also be tested for improvements.
This intraoperative test is not aimed at assessing the best possible outcome of deep brain stimulation, and not even to see an improvement in all symptoms that burden the patient. Rather, it is to evaluate the likelihood that programming will be feasible with the implanted lead.
Subsequently, implantation of the pulse generator in the chest and connection to the brain lead is completed, usually with the patient under general anesthesia.
Implantation under general anesthesia, with intraoperative MRI
A new alternative to “awake stereotactic surgery” is implantation with the patient under general anesthesia, with intraoperative MRI. We have started to do this procedure in a new operating suite that is attached to an MRI suite. The magnet can be taken in and out of the operating room, allowing the surgeon to verify the location of the implanted leads right at the time of the procedure. In this fashion, intraoperative images are used to guide implantation instead of awake microelectrode recording. This is a new option for patients who cannot tolerate awake surgery and for those who have a contraindication to the regular stereotactic procedure with the patient awake.
Risks of bleeding and infection
The potential complications of implanting a device and leads in the brain can be significant.
Hemorrhage can occur, resulting in a superficial or deep hematoma.
Infection and erosion may require removal of the hardware for antibiotic treatment and possible reimplantation.
Other risks include those related to tunneling the wires from the head to the chest, to implanting the device in the chest, and to serious medical complications after surgery. Hardware failure can occur and requires additional surgery. Finally, environmental risks and risks related to medical devices such as MRI, electrocautery, and cardioversion should also be considered.
Deep brain stimulation is advantageous for its reversibility. If during postoperative programming the brain leads are considered not to be ideally placed, revisions can be done to reposition the leads.
- Deuschl G, Schade-Brittinger C, Krack P, et al; German Parkinson Study Group, Neurostimulation Section. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 2006; 355:896–908.
- Krack P, Batir A, Van Blercom N, et al. Five-year followup of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med 2003; 349:1925–1934.
- Kupsch A, Benecke R, Müller J, et al; Deep-Brain Stimulation for Dystonia Study Group. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med 2006; 355:1978–1990.
- Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scle for the primary torsion dystonias. Neurology 1985; 35:73–77.
- Gruber D, Trottenberg T, Kivi A, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology 2009; 73:53–58.
- Deuschl G, Schade-Brittinger C, Krack P, et al; German Parkinson Study Group, Neurostimulation Section. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 2006; 355:896–908.
- Krack P, Batir A, Van Blercom N, et al. Five-year followup of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med 2003; 349:1925–1934.
- Kupsch A, Benecke R, Müller J, et al; Deep-Brain Stimulation for Dystonia Study Group. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med 2006; 355:1978–1990.
- Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scle for the primary torsion dystonias. Neurology 1985; 35:73–77.
- Gruber D, Trottenberg T, Kivi A, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology 2009; 73:53–58.
KEY POINTS
- Compared with ablative procedures, deep brain stimulation has the advantage of being reversible and adjustable. It is considered safer than ablative surgery, in particular for bilateral procedures, which are often needed for patients with advanced Parkinson disease and other movement disorders.
- For Parkinson disease, deep brain stimulation improves the cardinal motor symptoms, extends medication “on” time, and reduces motor fluctuations during the day.
- In general, patients with Parkinson disease are likely to benefit from this therapy if they show a clear response to levodopa. Patients are therefore asked to stop their Parkinson medications overnight to permit a formal evaluation of their motor response before and after a dose of levodopa.
- Candidates require a thorough evaluation to assess whether they are likely to benefit from deep brain stimulation and if they can comply with the maintenance often required for a successful outcome.
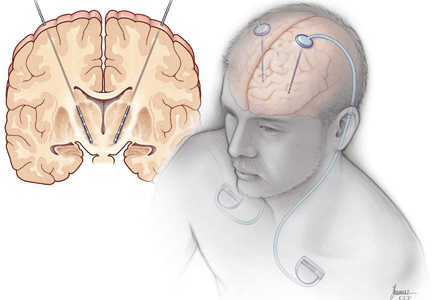
Updates in the medical management of Parkinson disease
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING

Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
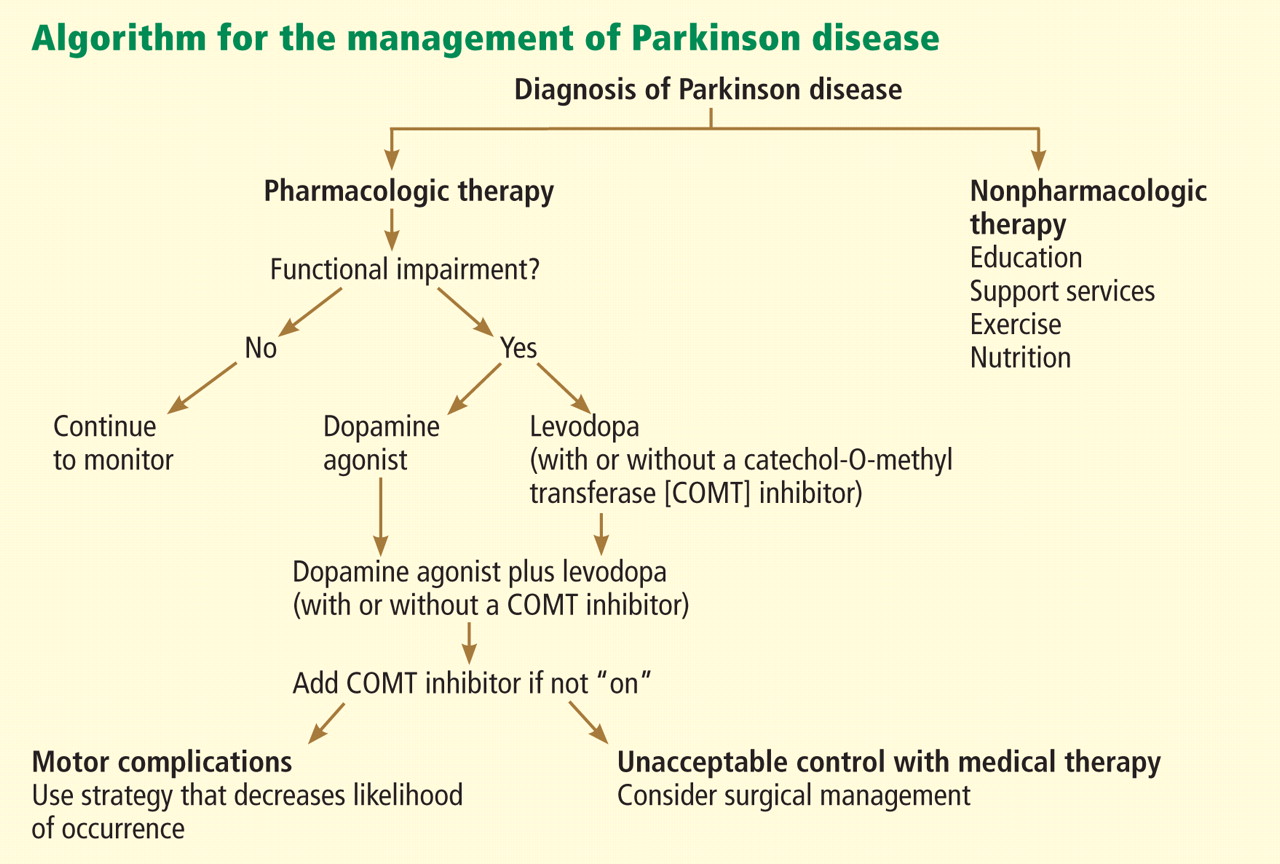
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING
Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING
Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
KEY POINTS
- Parkinson disease can usually be diagnosed on the basis of clinical features: slow movement, resting tremor, rigidity, and asymmetrical presentation, as well as alleviation of symptoms with dopaminergic therapy.
- Early disease can be treated with levodopa, dopamine agonists, anticholinergics, and monoamine oxidase-B inhibitors.
- Advanced Parkinson disease may require a catechol-O-methyltransferase (COMT) inhibitor, apomorphine, and amantadine (Symmetrel). Side effects include motor fluctuations, dyskinesias, and cognitive problems.
Essential tremor: Choosing the right management plan for your patient
Essential tremor, one of the most common movement disorders, affects about 4% of adults 40 years of age and older.1 It is often referred to as familial tremor in patients with a family history of tremor. It has also been called benign tremor to differentiate it from tremor associated with neurodegenerative diseases, particularly Parkinson disease, but this condition is certainly not benign, as it can cause substantial functional impairment and difficulties with routine activities of daily living. The terms “essential” and “idiopathic” refer to the primary nature of the disorder and differentiate it from tremor that is a feature of a distinct neurologic entity or is secondary to a metabolic disease or drug therapy.
Successful management entails exclusion of secondary causes and careful selection of drug therapy. To date, there is no cure for essential tremor; all currently available treatments are purely symptomatic.
In this review, we outline the major diagnostic and therapeutic principles of managing essential tremor, indications for referral to specialists, and alternative and advanced therapeutic options.
CLINICAL PICTURE
Tremor is defined as rhythmic to-and-fro movement in any body part. It can be slow or fast, and its amplitude can be large and coarse, or small or even “fine.” It can appear at rest, with action, or during a sustained posture. In contrast to parkinsonian tremor (which presents mainly at rest), essential tremor is typically but not exclusively postural, kinetic, or both.
Postural tremor refers to tremor seen when the patient holds the affected limb (commonly the arm) unsupported against gravity. Kinetic tremor refers to tremor that appears with active movements. This is often demonstrated clinically by the finger-nose-finger test. Patients with essential tremor commonly have both postural and kinetic tremor.
The tremor commonly involves the arms, hands, and fingers.2 Less commonly, it involves the head, the lips, the tongue, the legs, and the voice. In contrast to parkinsonian tremor, which typically affects one side of the body first, bilateral involvement is the general rule in essential tremor. However, one side of the body may be affected first, or may be more affected than the other. The frequency of the tremor ranges from 4 to 12 Hz (ie, beats per second).
The tremor usually starts in middle age and progresses slowly over time,3 but onset in old age or childhood is also possible.4 Both sexes are equally affected.
The tremor usually gets worse with anxiety, stress, and caffeine intake. It usually gets temporarily better with the consumption of small amounts of alcohol.
The functional impact of essential tremor is judged by its effect on different daily activities, especially writing, eating, drinking, dressing, manual work, and household chores.
In addition to motor dysfunction, the tremor can also have a significant psychological impact on the patient, because it usually gets worse in social situations.
Although it has long been thought that tremor is the sole neurologic sign of essential tremor, recent studies have shown that many patients have additional subtle findings, such as mild gait difficulty,5 slight incoordination,6 mild cognitive impairment,7 and decreased hearing,8 and are more likely to have anxiety and social phobia.9
Although different studies have varied in their findings, it is generally thought that about 50% of patients with essential tremor have a positive family history, often in a first-degree relative, suggesting autosomal dominant inheritance with variable penetrance.10,11 Polygenetic and sporadic variants are also common.
DIFFERENTIAL DIAGNOSIS
Resting tremor
Resting tremor is typically an extrapyramidal sign and, when accompanied by rigidity and bradykinesia, is often part of a parkinsonian syndrome. It is most pronounced at rest when the affected body part is fully supported and stationary. The tremor tends to improve with action or posture. It usually has a “pill-rolling” character and, as mentioned, is associated with other extrapyramidal signs, such as rigidity, slowness, and, later on, postural instability.
About 20% of patients with essential tremor have resting tremor. These patients usually suffer from severe or long-standing disease.12 However, the resting element in these cases is often milder than the postural and kinetic components, and it is typically not associated with other extrapyramidal signs. Also, some patients may have both essential tremor and Parkinson disease.13
Intentional tremor
Pure intentional tremor is usually seen with cerebellar pathology, which includes tumors, stroke, multiple sclerosis, trauma, and spinocerebellar degeneration. The amplitude of this type of tremor increases as the affected limb approaches the final target. It can best be demonstrated clinically by the finger-nose-finger test. The frequency of intentional tremor is slow (2 to 4 Hz) and is usually associated with other cerebellar signs, such as dysmetria, decomposition, rebound, and dysdiadochokinesia (ie, the inability to perform rapid alternating movements in a smooth and coordinated manner).
About 50% of patients with essential tremor have an intentional component to their tremor,6 or it can be mildly present in the form of a slight gait difficulty. However, in essential tremor, other features of cerebellar dysfunction are either absent or only very slight.
Secondary causes of postural-kinetic tremor
Enhanced physiologic tremor. A very mild postural tremor is present in almost all people and is considered “physiologic” since it has almost no clinical significance. This type of tremor is often invisible, but when “enhanced,” it can be visually demonstrated by placing a piece of paper over the stretched hands and watching the ripple from the paper.
Certain conditions can aggravate this physiologic tremor and can make it symptomatic. Common causes include anxiety, sleep deprivation, hypoglycemia, hyperthyroidism, pheochromocytoma, serotonin syndrome, and carcinoid syndrome.
Metabolic tremor. Hyperammonemia can cause tremor in patients with hepatic encephalopathy, and uremia can cause tremor in patients with renal failure. These metabolic conditions classically result in “flappy” tremor (asterixis), a special form of postural tremor characterized by jerking movements with high amplitude. It is best seen when the patient stretches out the arms and extends the wrists as if trying to stop traffic. But even though it may look like tremor, asterixis is thought to be a form of “negative” myoclonus.
Drug-related tremor. Postural-kinetic tremor can be induced by drugs, including lithium (Lithobid), valproate (Depakote), amiodarone (Cordarone), central nervous system stimulants, beta agonists (including inhalers), and some antidepressants. Tremor can also occur with alcohol or sedative withdrawal.
Psychogenic tremor. Tremor can be seen as part of a somatoform disorder commonly referred to as conversion disorder or conversion reaction. Psychogenic tremor is characterized by acute onset, commonly following a psychosocial stressor; it is often atypical, variable in frequency, amplitude, and body-part involvement, and it can readily be interrupted on examination by distracting the patient.
Neurologic disorders. The postural and kinetic elements of essential tremor may also be seen in the following neurologic conditions:
- Holmes (rubral) tremor, a combination of resting, postural, kinetic, and intentional tremor of low frequency and high amplitude. It usually has a proximal component and is often unilateral. It commonly is due to a lesion that involves the brainstem, eg, red nucleus, inferior olive, cerebellum, or thalamus. Common causes include stroke, prolonged hypoxia, and head trauma (including closed-head trauma with negative imaging). This type of tremor is usually associated with ataxia.14
- Dystonic tremor is predominantly postural and is associated with abnormal dystonic posturing of the affected body part, commonly the head, hands, or feet. Unlike the rhythmic oscillations of essential tremor, dystonic tremor is often irregular in rhythm.
- Multiple sclerosis can present with a combination of postural, kinetic, and intentional tremor. Patients usually have a clear history of recurrent neurologic deficits and show a combination of pyramidal, cerebellar, and sensory signs on examination consistent with multiple sclerosis.15
- Neuropathic tremor is seen in a small proportion of patients with peripheral neuropathy, especially demyelinating neuropathy.16 The tremor is usually posturalkinetic and is associated with signs of neuropathy, such as a glove-and-stocking pattern of hypoesthesia, reduced reflexes, and sensory ataxia (including intentional tremor when the eyes are closed).
- Posttraumatic tremor can occur after severe or even mild head trauma, especially in children. It is commonly rubral, but other types have been reported, including a presentation resembling essential tremor.17
- Monosymptomatic or isolated tremor. A number of conditions related to essential tremor with location-specific or task-specific tremor have been described. These rare conditions historically have been classified as “possible essential tremor” or “essential tremor variants” but are now considered separate entities. These include task-specific tremor (eg, writing tremor), isolated head tremor, isolated voice tremor, and orthostatic tremor (tremor in the legs and trunk upon standing in place, but not when sitting or walking).18,19
DIAGNOSIS IS CLINICAL
Essential tremor is a clinical diagnosis. After a thorough review of the medical history and medication exposures, laboratory and imaging tests may be ordered to rule out a secondary cause. A complete metabolic panel, including blood glucose and thyroid-stimulating hormone levels, is usually sufficient. Brain imaging or other imaging is ordered for patients with an atypical presentation.
TREATMENT IS SYMPTOMATIC
Treatment of essential tremor is symptomatic. Several drugs of different pharmacologic classes can reduce the severity of the tremor and improve function.
Choosing the appropriate treatment depends on the type of tremor and the presence of associated conditions. The response to treatment and the development of side effects guide further adjustments. The following is a brief description of the available antitremor agents.
FIRST-LINE AGENTS
Propranolol
Propranolol (Inderal), a nonselective beta blocker, is the most widely used antitremor drug and the only agent approved by the US Food and Drug Administration for essential tremor. It should be started at a low dose and titrated upward gradually. The usual starting dose is 10 mg three times daily. The average effective dose is 120 mg daily. The dose can be increased up to 320 mg if needed and tolerated.
Sustained-release preparations are equally effective and are given as a single daily dose to improve compliance.20
Propranolol improves tremor in 50% to 70% of patients with essential tremor and achieves an average tremor reduction of 50% to 60%.1,21–25 Side effects include bronchoconstriction, bradycardia, hypotension, depression, impotence, fatigue, and gastrointestinal disturbances.
Other beta-blockers, such as nadolol (Corgard) and timolol, are also effective against tremor but are less potent than propranolol.26,27 The selective beta-1-blocker metoprolol (Lopressor) may be effective and has fewer noncardiac side effects than propranolol.28 It can be used in patients who discontinue propranolol because of adverse effects. Atenolol (Tenormin) and pindolol (Visken) have little or no effect on tremor.29
A good candidate for propranolol therapy in essential tremor is:
- A patient with no known contraindication to propranolol
- A patient with hypertension, coronary heart disease, or tachyarrhythmia
- A patient with anxiety or social phobia.
Absolute contraindications to propranolol are:
- Moderate to severe bronchial asthma
- Significant bradycardia or heart block
- Symptomatic hypotension
- End-stage heart failure
- Concurrent use of a calcium channel blocker.
Relative contraindications are:
- Wheezing (eg, chronic obstructive pulmonary disease)
- Depression
- Diabetes mellitus in a patient more prone to hypoglycemia (propranolol masks the warning signs of hypoglycemia)
- Reduced sexual potency in a male patient.
Primidone
Primidone (Mysoline) is an antiepileptic drug structurally similar to barbiturates. Its antitremor effect is equal to that of propranolol, though some studies suggest it is slightly more efficacious.30,31
It should be started at a low dose, ie, 25 mg once daily at bedtime. The dose should then be increased gradually until satisfactory and tolerable tremor control is achieved. Most patients respond to doses of around 250 mg per day.1,22,24–25 The dose can be increased if needed and tolerated.
Primidone reduces tremor by about 50% to 60%.1,22,24–25 Side effects include sedation, dizziness, fatigue, nausea, and depression, as well as ataxia and confusion in severe cases.
A good candidate for primidone in essential tremor is:
- A patient with no known contraindication to primidone
- A patient with contraindications to propranolol
- A younger patient
- A patient with epilepsy.
Absolute contraindications to primidone include:
- Confusion or dementia
- Oral anticoagulant therapy with difficulty controlling the International Normalized Ratio (primidone is a potent enzyme inducer).
Relative contraindications to primidone in essential tremor are:
- Depression
- Alcohol abuse
- Ongoing therapy with sedating drugs
- Ataxia or vertigo.
SECOND-LINE AGENTS
Other antiepileptics
Topiramate (Topamax) is a broad-spectrum antiepileptic shown to be significantly effective against essential tremor.32 It is usually started at a single daily dose of 25 mg and increased gradually to the most effective dose, usually around 300 mg.
Side effects include reduced appetite, weight loss, cognitive dysfunction, and paresthesia.
Favorable candidates include patients who are epileptic or overweight. Contraindications include cognitive impairment and low body weight. It is also not recommended in children so as to avoid any possible negative effect on cognitive development. In rare cases, topiramate has been reported to cause significant visual disturbances.
Gabapentin (Neurontin) is an antiepileptic that is now more often used as a symptomatic treatment for neuropathic pain. Studies have suggested a beneficial effect on essential tremor,33,34 but some investigators have questioned its efficacy.35
Like other antitremor agents, it should be started at a low dose, ie, around 300 mg, and escalated gradually until the tremor is controlled. The usual effective dose is 1,200 mg.
Gabapentin is generally well tolerated, and side effects such as dizziness, drowsiness, sedation, and unsteadiness are rare and usually mild.
The favorable candidate is a patient with associated neuropathy or multiple comorbidities. Gabapentin has also been reported to alleviate neuropathic tremor.
Contraindications are minimal and include intolerability or hypersensitivity to the drug. It also should be avoided in patients at a high risk of falling.
Levetiracetam (Keppra) is a novel antiepileptic effective against partial seizures. Studies have shown contradictory results regarding its antitremor effect. One double-blind, placebo-controlled study demonstrated significant reduction in essential tremor with 1,000 mg of levetiracetam.36 However, its effect on tremor is believed to be short-lived, and some studies argue against its efficacy.37 It has a favorable side-effect profile and is generally very well tolerated. It can be used as an adjunct to other antitremor agents and is preferred for patients with coexisting partial seizures or myoclonus.
Benzodiazepines. Minor tranquilizers are often used to control tremor, especially in coexisting anxiety or insomnia. Alprazolam (Xanax) is the one most widely used for this indication.38 It can be started in a dose of 0.25 mg once at bedtime and increased gradually up to 0.75 to 2 mg. Clonazepam (Klonopin) is particularly useful for orthostatic tremor, a variant of essential tremor characterized by tremor of the legs and trunk upon standing.39
Common side effects of benzodiazepines include sedation, cognitive dysfunction, hypotension, respiratory inhibition, and addiction after prolonged use. In the elderly, they can lead to confusion and disinhibition and can increase the risk of falling. They should be avoided in the elderly and in alcoholic patients and those with a high risk of substance abuse.
Stopping benzodiazepines should be done gradually to avoid withdrawal symptoms, including aggravation of tremor.
THIRD-LINE AGENTS
Clozapine
Clozapine (Clozaril) is a novel antipsychotic drug with no extrapyramidal side effects. It has been reported effective in essential tremor and drug-induced tremor,40,41 but the results of these early studies have not been confirmed.
Clozapine is started as a single daily dose of 12.5 mg and is increased up to 75 mg or 100 mg. It is an attractive option for patients with coexisting psychosis, bipolar disorder, or chorea. Its main side effects are sedation, salivation, weight gain, hypertension, diabetes, and seizures.
One especially serious side effect is agranulocytosis. This potentially fatal effect is rare, occurring in about 1.3% of patients receiving this drug. Weekly monitoring of the white blood cell count is mandated during treatment with clozapine, and this has made clozapine a less attractive option for the routine treatment of essential tremor.
Mirtazapine
Mirtazapine (Remeron) is a novel antidepressant widely used in Parkinson disease as both an antidepressant and a sleeping aid. Case studies have reported efficacy in both essential tremor and parkinsonian tremor,42 but controlled studies have not confirmed this.43 Mirtazapine is a reasonable option in patients with coexisting depression or insomnia. It is usually given as a single bedtime dose of 15 to 30 mg.
Other drugs
Studies of other agents for the treatment of essential tremor—eg, carbonic anhydrase enzyme inhibitors, calcium channel blockers, isoniazid (Tubizid), clonidine (Catapres), phenobarbital, and theophylline—have yielded highly contradictory results. Thus, they are not recommended as first- or second-line agents for essential tremor.
SPECIALTY-LEVEL CARE
When essential tremor does not respond to drug therapy or the patient cannot tolerate drug therapy, the patient should be referred to a center specializing in movement disorders for more advanced treatment options, ie, botulinum toxin injection and deep brain stimulation surgery.
Botulinum toxin
Botulinum toxin type A has been studied for the treatment of essential tremor with variable degrees of success. It has been effective in reducing hand tremor in essential tremor, but without a concomitant improvement in functional disability.44 This limited functional improvement has been attributed to the development of muscle weakness after injection of the neurotoxin. This has also raised questions about unintentional unblinding when interpreting study results. Therefore, most clinicians restrict its use to focal forms of tremor such as voice tremor,45 head tremor, and task-specific tremor.
Side effects are limited and temporary and include muscle weakness, pain at the injection site, dysphagia (when injected for head or voice tremor), and a breathy vocal quality (when injected for voice tremor). Botulinum toxin injection is the treatment of choice for focal dystonia, and therefore would be a good option for dystonic tremor.
Thalamic deep brain stimulation
This technique involves stereotactic implantation of a stimulation lead in the ventral intermediate nucleus of the thalamus. The lead connects via a subcutaneous wire to an intermittent pulse generator, implanted subcutaneously in the infraclavicular region. The stimulation lead produces continuous stimulation of the ventralis intermedius nucleus that is functionally equal to lesional surgery, thus antagonizing the relay of tremor signals at the thalamus.
The battery of the pulse generator must be replaced every 4 to 7 years depending on usage and stimulation parameters. Battery replacement can be performed with minor surgery at the infraclavicular region.
Thalamic deep brain stimulation is indicated for patients with severe, disabling essential tremor who have tremor resistant to drug therapy or who cannot tolerate drug therapy.
The procedure has been shown to provide benefit in 90% of patients, with more than an 80% improvement in tremor severity and functional impact.46–49 Deep brain stimulation is effective against tremor affecting parts of the body other than the limbs, including the head; an exception to this is voice tremor, which usually does not improve dramatically. The procedure can be done unilaterally or bilaterally, depending on symptoms. Patients with asymmetrical tremor and those at risk of side effects can undergo unilateral surgery. Bilateral treatment is recommended for patients with symmetric tremor or significant head tremor, or who are young and healthy.
Surgical risks include brain hemorrhage and infection. Side effects of the stimulation include paresthesias, paresis, imbalance, dysarthria, and, in rare cases, dysphagia.
CHOOSING THE BEST MANAGEMENT PLAN FOR YOUR PATIENT
The choice of treatment may be challenging, given the multiple treatment options and the variability of tremor severity from one patient to another. The following guidelines can be used to help make this decision.
All patients should be advised to reduce caffeine intake, to have sufficient hours of sleep, and to avoid stressful situations.
Patients with minor, nondisabling tremor can be left untreated if the tremors are not bothersome or if the patient prefers not to pursue active treatment.
In patients who have bothersome tremor only when anxious or in certain social situations, give propranolol or alprazolam (or both) to be taken as needed. Relaxation techniques and meditation are also useful for these patients.
Patients with constant bothersome tremor should be started on either propranolol or primidone based on the patient’s profile and propensity to develop side effects from each of these drugs. The dosing should be optimized gradually according to the patient’s response and the drug’s tolerability.
If essential tremor is not sufficiently controlled with one first-line agent (propranolol or primidone), try combining the two first-line agents if the patient finds it tolerable.
A second-line agent can be added to either of the first-line agents or to the combination of both if tremor control is not yet sufficient. A second-line or third-line agent can also be used as the primary treatment if both first-line agents are contraindicated or intolerable. Combining two or more second- and third-line agents is another option. The choice of second- or third-line agent should be guided by the patient’s characteristics and comorbidities in relation to the agent’s side effects and contraindications as detailed in the above section.
Patients should be referred to a movement disorders specialist in cases of resistant tremor, intolerance to oral medications, severe disability, and atypical presentation. Types of tremor known to be poorly responsive to oral medications (eg, head tremor, voice tremor) deserve a specialist evaluation if they contribute significantly to the patient’s morbidity.
The usual specialist treatment of severe voice tremor and head tremor is botulinum toxin injection. Patients with resistant and disabling hand tremor are evaluated for thalamic deep brain stimulation.
Patients with residual disability despite medical and surgical treatment should be referred for occupational therapy. Occupational therapy can improve quality of life through the use of special utensils, pens, computer gadgets, and arm weights, among other devices.
- Zesiewicz TA, Chari A, Jahan I, et al. Overview of essential tremor. Neuropsychiatr Dis Treat 2010; 6:401–408.
- Elble RJ. Essential tremor frequency decreases with time. Neurology 2000; 55:1547–1551.
- Louis ED, Ottman R, Hauser WA. How common is the most common adult movement disorder? Estimates of the prevalence of essential tremor throughout the world. Mov Disord 1998; 13:5–10.
- Louis ED, Dure LS, Pullman S. Essential tremor in childhood: a series of nineteen cases. Mov Disord 2001; 16:921–923.
- Singer C, Sanchez-Ramos J, Weiner WJ. Gait abnormality in essential tremor. Mov Disord 1994; 9:193–196.
- Deuschl G, Wenzelburger R, Löffler K, et al. Essential tremor and cerebellar dysfunction. Clinical and kinematic analysis of intention tremor. Brain 2000; 123:1568–1580.
- Louis ED. Functional correlates of lower cognitive test scores in essential tremor. Mov Disord 2010; 25:481–485.
- Ondo WG, Sutton L, Dat Vuong K, et al. Hearing impairment in essential tremor. Neurology 2003; 61:1093–1097.
- Schneier FR, Barnes LF, Albert SM, et al. Characteristics of social phobia among persons with essential tremor. J Clin Psychiatry 2001; 62:367–372.
- Whaley NR, Putzke JD, Baba Y, et al. Essential tremor: phenotypic expression in a clinical cohort. Parkinsonism Relat Disord 2007; 13:333–339.
- Deng H, Le W, Jankovic J. Genetics of essential tremor. Brain 2007; 130:1456–1464.
- Cohen O, Pullman S, Jurewicz E, et al. Rest tremor in patients with essential tremor: prevalence, clinical correlates, and electrophysiologic characteristics. Arch Neurol 2003; 60:405–410.
- Shahed J, Jankovic J. Exploring the relationship between essential tremor and Parkinson’s disease. Parkinsonism Relat Disord 2007; 13:67–76.
- Yang YW, Chang FC, Tsai CH, et al. Clinical and magnetic resonance imaging manifestations of Holmes tremor. Acta Neurol Taiwan 2005; 14:9–15.
- Alusi SH, Worthington J, Glickman S, et al. A study of tremor in multiple sclerosis. Brain 2001; 124:720–730.
- Breit S, Wächter T, Schöls L, et al. Effective thalamic deep brain stimulation for neuropathic tremor in a patient with severe demyelinating neuropathy. J Neurol Neurosurg Psychiatry 2009; 80:235–236.
- Koller WC, Wong GF, Lang A. Posttraumatic movement disorders: a review. Mov Disord 1989; 4:20–36.
- Jankovic J. Essential tremor: a heterogenous disorder. Mov Disord 2002; 17:638–644.
- Deuschl G, Bain P, Brin M. Consensus statement of the Movement Disorder Society on Tremor. Ad Hoc Scientific Committee. Mov Disord 1998; 13(suppl 3):2–23.
- Calzetti S, Findley LJ, Gresty MA, et al. Effect of a single oral dose of propranolol on essential tremor: a double-blind controlled study. Ann Neurol 1983; 13:165–171.
- Larsen TA, Teräväinen H, Calne DB. Atenolol vs propranolol in essential tremor. A controlled, quantitative study. Acta Neurol Scand 1982; 66:547–554.
- Zesiewicz TA, Elble R, Louis ED, et al. Practice parameter: therapies for essential tremor: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2005; 64:2008–2020.
- Lyons KE, Pahwa R, Comella CL, et al.Benefits and risks of pharmacological treatments for essential tremor. Drug Saf 2003; 26:461–481.
- Pahwa R, Lyons KE. Essential tremor: differential diagnosis and current therapy. Am J Med 2003; 115:134–142.
- Louis ED. Clinical practice. Essential tremor. N Engl J Med 2001; 345:887–891.
- Koller WC. Nadolol in essential tremor. Neurology 1983; 33:1076–1077.
- Dietrichson P, Espen E. Effects of timolol and atenolol on benign essential tremor: placebo-controlled studies based on quantitative tremor recording. J Neurol Neurosurg Psychiatry 1981; 44:677–683.
- Calzetti S, Findley LJ, Gresty MA, et al. Metoprolol and propranolol in essential tremor: a double-blind, controlled study. J Neurol Neurosurg Psychiatry 1981; 44:814–819.
- Teravainen H, Larsen A, Fogelholm R. Comparison between the effects of pindolol and propranolol on essential tremor. Neurology 1977; 27:439–442.
- Gorman WP, Cooper R, Pocock P, et al. A comparison of primidone, propranolol, and placebo in essential tremor, using quantitative analysis. J Neurol Neurosurg Psychiatry 1986; 49:64–68.
- Koller WC, Royse VL. Efficacy of primidone in essential tremor. Neurology 1986; 36:121–124.
- Connor GS. A double-blind placebo-controlled trial of topiramate treatment for essential tremor. Neurology 2002; 59:132–134.
- Gironell A, Kulisevsky J, Barbanoj M, et al. A randomized placebo-controlled comparative trial of gabapentin and propranolol in essential tremor. Arch Neurol 1999; 56:475–480.
- Ondo W, Hunter C, Vuong KD, et al. Gabapentin for essential tremor: a multiple-dose, double-blind, placebo-controlled trial. Mov Disord 2000; 15:678–682.
- Pahwa R, Lyons K, Hubble JP, et al. Double-blind controlled trial of gabapentin in essential tremor. Mov Disord 1998; 13:465–467.
- Bushara KO, Malik T, Exconde RE. The effect of levetiracetam on essential tremor. Neurology 2005; 64:1078–1080.
- Sullivan KL, Hauser RA, Zesiewicz TA. Levetiracetam for the treatment of essential tremor. Mov Disord 2005; 20:640.
- Huber SJ, Paulson GW. Efficacy of alprazolam for essential tremor. Neurology 1988; 38:241–243.
- McManis PG, Sharbrough FW. Orthostatic tremor: clinical and electrophysiologic characteristics. Muscle Nerve 1993; 16:1254–1260.
- Ceravolo R, Salvetti S, Piccini P, et al. Acute and chronic effects of clozapine in essential tremor. Mov Disord 1999; 14:468–472.
- Pakkenberg H, Pakkenberg B. Clozapine in the treatment of tremor. Acta Neurol Scand 1986; 73:295–297.
- Pact V, Giduz T. Mirtazapine treats resting tremor, essential tremor, and levodopa-induced dyskinesias. Neurology 1999; 53:1154.
- Lyons KE, Pahwa R. A double-blind, placebo-controlled, pilot study of mirtazapine in essential tremor. Presented at the 54th Annual Meeting of the American Academy of Neurology, Denver, Colorado. Neurology 2002; 58(suppl 3):A254.
- Brin MF, Lyons KE, Doucette J, et al. A randomized, double masked, controlled trial of botulinum toxin type A in essential hand tremor. Neurology 2001; 56:1523–1528.
- Blitzer A, Brin MF, Stewart C, et al. Abductor laryngeal dystonia: a series treated with botulinum toxin. Laryngoscope 1992; 102:163–167.
- Schuurman PR, Bosch DA, Bossuyt PM, et al. A comparison of continuous thalamic stimulation and thalamotomy for suppression of severe tremor. N Engl J Med 2000; 342:461–468.
- Flora ED, Perera CL, Cameron AL, et al. Deep brain stimulation for essential tremor: a systematic review. Mov Disord 2010; 25:1550–1559.
- Nagaseki Y, Shibazaki T, Hirai T, et al. Long-term follow-up results of selective VIM-thalamotomy. J Neurosurg 1986; 65:296–302.
- Zirh A, Reich SG, Dougherty PM, et al. Stereotactic thalamotomy in the treatment of essential tremor of the upper extremity: reassessment including a blinded measure of outcome. J Neurol Neurosurg Psychiatry 1999; 66:772–775.
Essential tremor, one of the most common movement disorders, affects about 4% of adults 40 years of age and older.1 It is often referred to as familial tremor in patients with a family history of tremor. It has also been called benign tremor to differentiate it from tremor associated with neurodegenerative diseases, particularly Parkinson disease, but this condition is certainly not benign, as it can cause substantial functional impairment and difficulties with routine activities of daily living. The terms “essential” and “idiopathic” refer to the primary nature of the disorder and differentiate it from tremor that is a feature of a distinct neurologic entity or is secondary to a metabolic disease or drug therapy.
Successful management entails exclusion of secondary causes and careful selection of drug therapy. To date, there is no cure for essential tremor; all currently available treatments are purely symptomatic.
In this review, we outline the major diagnostic and therapeutic principles of managing essential tremor, indications for referral to specialists, and alternative and advanced therapeutic options.
CLINICAL PICTURE
Tremor is defined as rhythmic to-and-fro movement in any body part. It can be slow or fast, and its amplitude can be large and coarse, or small or even “fine.” It can appear at rest, with action, or during a sustained posture. In contrast to parkinsonian tremor (which presents mainly at rest), essential tremor is typically but not exclusively postural, kinetic, or both.
Postural tremor refers to tremor seen when the patient holds the affected limb (commonly the arm) unsupported against gravity. Kinetic tremor refers to tremor that appears with active movements. This is often demonstrated clinically by the finger-nose-finger test. Patients with essential tremor commonly have both postural and kinetic tremor.
The tremor commonly involves the arms, hands, and fingers.2 Less commonly, it involves the head, the lips, the tongue, the legs, and the voice. In contrast to parkinsonian tremor, which typically affects one side of the body first, bilateral involvement is the general rule in essential tremor. However, one side of the body may be affected first, or may be more affected than the other. The frequency of the tremor ranges from 4 to 12 Hz (ie, beats per second).
The tremor usually starts in middle age and progresses slowly over time,3 but onset in old age or childhood is also possible.4 Both sexes are equally affected.
The tremor usually gets worse with anxiety, stress, and caffeine intake. It usually gets temporarily better with the consumption of small amounts of alcohol.
The functional impact of essential tremor is judged by its effect on different daily activities, especially writing, eating, drinking, dressing, manual work, and household chores.
In addition to motor dysfunction, the tremor can also have a significant psychological impact on the patient, because it usually gets worse in social situations.
Although it has long been thought that tremor is the sole neurologic sign of essential tremor, recent studies have shown that many patients have additional subtle findings, such as mild gait difficulty,5 slight incoordination,6 mild cognitive impairment,7 and decreased hearing,8 and are more likely to have anxiety and social phobia.9
Although different studies have varied in their findings, it is generally thought that about 50% of patients with essential tremor have a positive family history, often in a first-degree relative, suggesting autosomal dominant inheritance with variable penetrance.10,11 Polygenetic and sporadic variants are also common.
DIFFERENTIAL DIAGNOSIS
Resting tremor
Resting tremor is typically an extrapyramidal sign and, when accompanied by rigidity and bradykinesia, is often part of a parkinsonian syndrome. It is most pronounced at rest when the affected body part is fully supported and stationary. The tremor tends to improve with action or posture. It usually has a “pill-rolling” character and, as mentioned, is associated with other extrapyramidal signs, such as rigidity, slowness, and, later on, postural instability.
About 20% of patients with essential tremor have resting tremor. These patients usually suffer from severe or long-standing disease.12 However, the resting element in these cases is often milder than the postural and kinetic components, and it is typically not associated with other extrapyramidal signs. Also, some patients may have both essential tremor and Parkinson disease.13
Intentional tremor
Pure intentional tremor is usually seen with cerebellar pathology, which includes tumors, stroke, multiple sclerosis, trauma, and spinocerebellar degeneration. The amplitude of this type of tremor increases as the affected limb approaches the final target. It can best be demonstrated clinically by the finger-nose-finger test. The frequency of intentional tremor is slow (2 to 4 Hz) and is usually associated with other cerebellar signs, such as dysmetria, decomposition, rebound, and dysdiadochokinesia (ie, the inability to perform rapid alternating movements in a smooth and coordinated manner).
About 50% of patients with essential tremor have an intentional component to their tremor,6 or it can be mildly present in the form of a slight gait difficulty. However, in essential tremor, other features of cerebellar dysfunction are either absent or only very slight.
Secondary causes of postural-kinetic tremor
Enhanced physiologic tremor. A very mild postural tremor is present in almost all people and is considered “physiologic” since it has almost no clinical significance. This type of tremor is often invisible, but when “enhanced,” it can be visually demonstrated by placing a piece of paper over the stretched hands and watching the ripple from the paper.
Certain conditions can aggravate this physiologic tremor and can make it symptomatic. Common causes include anxiety, sleep deprivation, hypoglycemia, hyperthyroidism, pheochromocytoma, serotonin syndrome, and carcinoid syndrome.
Metabolic tremor. Hyperammonemia can cause tremor in patients with hepatic encephalopathy, and uremia can cause tremor in patients with renal failure. These metabolic conditions classically result in “flappy” tremor (asterixis), a special form of postural tremor characterized by jerking movements with high amplitude. It is best seen when the patient stretches out the arms and extends the wrists as if trying to stop traffic. But even though it may look like tremor, asterixis is thought to be a form of “negative” myoclonus.
Drug-related tremor. Postural-kinetic tremor can be induced by drugs, including lithium (Lithobid), valproate (Depakote), amiodarone (Cordarone), central nervous system stimulants, beta agonists (including inhalers), and some antidepressants. Tremor can also occur with alcohol or sedative withdrawal.
Psychogenic tremor. Tremor can be seen as part of a somatoform disorder commonly referred to as conversion disorder or conversion reaction. Psychogenic tremor is characterized by acute onset, commonly following a psychosocial stressor; it is often atypical, variable in frequency, amplitude, and body-part involvement, and it can readily be interrupted on examination by distracting the patient.
Neurologic disorders. The postural and kinetic elements of essential tremor may also be seen in the following neurologic conditions:
- Holmes (rubral) tremor, a combination of resting, postural, kinetic, and intentional tremor of low frequency and high amplitude. It usually has a proximal component and is often unilateral. It commonly is due to a lesion that involves the brainstem, eg, red nucleus, inferior olive, cerebellum, or thalamus. Common causes include stroke, prolonged hypoxia, and head trauma (including closed-head trauma with negative imaging). This type of tremor is usually associated with ataxia.14
- Dystonic tremor is predominantly postural and is associated with abnormal dystonic posturing of the affected body part, commonly the head, hands, or feet. Unlike the rhythmic oscillations of essential tremor, dystonic tremor is often irregular in rhythm.
- Multiple sclerosis can present with a combination of postural, kinetic, and intentional tremor. Patients usually have a clear history of recurrent neurologic deficits and show a combination of pyramidal, cerebellar, and sensory signs on examination consistent with multiple sclerosis.15
- Neuropathic tremor is seen in a small proportion of patients with peripheral neuropathy, especially demyelinating neuropathy.16 The tremor is usually posturalkinetic and is associated with signs of neuropathy, such as a glove-and-stocking pattern of hypoesthesia, reduced reflexes, and sensory ataxia (including intentional tremor when the eyes are closed).
- Posttraumatic tremor can occur after severe or even mild head trauma, especially in children. It is commonly rubral, but other types have been reported, including a presentation resembling essential tremor.17
- Monosymptomatic or isolated tremor. A number of conditions related to essential tremor with location-specific or task-specific tremor have been described. These rare conditions historically have been classified as “possible essential tremor” or “essential tremor variants” but are now considered separate entities. These include task-specific tremor (eg, writing tremor), isolated head tremor, isolated voice tremor, and orthostatic tremor (tremor in the legs and trunk upon standing in place, but not when sitting or walking).18,19
DIAGNOSIS IS CLINICAL
Essential tremor is a clinical diagnosis. After a thorough review of the medical history and medication exposures, laboratory and imaging tests may be ordered to rule out a secondary cause. A complete metabolic panel, including blood glucose and thyroid-stimulating hormone levels, is usually sufficient. Brain imaging or other imaging is ordered for patients with an atypical presentation.
TREATMENT IS SYMPTOMATIC
Treatment of essential tremor is symptomatic. Several drugs of different pharmacologic classes can reduce the severity of the tremor and improve function.
Choosing the appropriate treatment depends on the type of tremor and the presence of associated conditions. The response to treatment and the development of side effects guide further adjustments. The following is a brief description of the available antitremor agents.
FIRST-LINE AGENTS
Propranolol
Propranolol (Inderal), a nonselective beta blocker, is the most widely used antitremor drug and the only agent approved by the US Food and Drug Administration for essential tremor. It should be started at a low dose and titrated upward gradually. The usual starting dose is 10 mg three times daily. The average effective dose is 120 mg daily. The dose can be increased up to 320 mg if needed and tolerated.
Sustained-release preparations are equally effective and are given as a single daily dose to improve compliance.20
Propranolol improves tremor in 50% to 70% of patients with essential tremor and achieves an average tremor reduction of 50% to 60%.1,21–25 Side effects include bronchoconstriction, bradycardia, hypotension, depression, impotence, fatigue, and gastrointestinal disturbances.
Other beta-blockers, such as nadolol (Corgard) and timolol, are also effective against tremor but are less potent than propranolol.26,27 The selective beta-1-blocker metoprolol (Lopressor) may be effective and has fewer noncardiac side effects than propranolol.28 It can be used in patients who discontinue propranolol because of adverse effects. Atenolol (Tenormin) and pindolol (Visken) have little or no effect on tremor.29
A good candidate for propranolol therapy in essential tremor is:
- A patient with no known contraindication to propranolol
- A patient with hypertension, coronary heart disease, or tachyarrhythmia
- A patient with anxiety or social phobia.
Absolute contraindications to propranolol are:
- Moderate to severe bronchial asthma
- Significant bradycardia or heart block
- Symptomatic hypotension
- End-stage heart failure
- Concurrent use of a calcium channel blocker.
Relative contraindications are:
- Wheezing (eg, chronic obstructive pulmonary disease)
- Depression
- Diabetes mellitus in a patient more prone to hypoglycemia (propranolol masks the warning signs of hypoglycemia)
- Reduced sexual potency in a male patient.
Primidone
Primidone (Mysoline) is an antiepileptic drug structurally similar to barbiturates. Its antitremor effect is equal to that of propranolol, though some studies suggest it is slightly more efficacious.30,31
It should be started at a low dose, ie, 25 mg once daily at bedtime. The dose should then be increased gradually until satisfactory and tolerable tremor control is achieved. Most patients respond to doses of around 250 mg per day.1,22,24–25 The dose can be increased if needed and tolerated.
Primidone reduces tremor by about 50% to 60%.1,22,24–25 Side effects include sedation, dizziness, fatigue, nausea, and depression, as well as ataxia and confusion in severe cases.
A good candidate for primidone in essential tremor is:
- A patient with no known contraindication to primidone
- A patient with contraindications to propranolol
- A younger patient
- A patient with epilepsy.
Absolute contraindications to primidone include:
- Confusion or dementia
- Oral anticoagulant therapy with difficulty controlling the International Normalized Ratio (primidone is a potent enzyme inducer).
Relative contraindications to primidone in essential tremor are:
- Depression
- Alcohol abuse
- Ongoing therapy with sedating drugs
- Ataxia or vertigo.
SECOND-LINE AGENTS
Other antiepileptics
Topiramate (Topamax) is a broad-spectrum antiepileptic shown to be significantly effective against essential tremor.32 It is usually started at a single daily dose of 25 mg and increased gradually to the most effective dose, usually around 300 mg.
Side effects include reduced appetite, weight loss, cognitive dysfunction, and paresthesia.
Favorable candidates include patients who are epileptic or overweight. Contraindications include cognitive impairment and low body weight. It is also not recommended in children so as to avoid any possible negative effect on cognitive development. In rare cases, topiramate has been reported to cause significant visual disturbances.
Gabapentin (Neurontin) is an antiepileptic that is now more often used as a symptomatic treatment for neuropathic pain. Studies have suggested a beneficial effect on essential tremor,33,34 but some investigators have questioned its efficacy.35
Like other antitremor agents, it should be started at a low dose, ie, around 300 mg, and escalated gradually until the tremor is controlled. The usual effective dose is 1,200 mg.
Gabapentin is generally well tolerated, and side effects such as dizziness, drowsiness, sedation, and unsteadiness are rare and usually mild.
The favorable candidate is a patient with associated neuropathy or multiple comorbidities. Gabapentin has also been reported to alleviate neuropathic tremor.
Contraindications are minimal and include intolerability or hypersensitivity to the drug. It also should be avoided in patients at a high risk of falling.
Levetiracetam (Keppra) is a novel antiepileptic effective against partial seizures. Studies have shown contradictory results regarding its antitremor effect. One double-blind, placebo-controlled study demonstrated significant reduction in essential tremor with 1,000 mg of levetiracetam.36 However, its effect on tremor is believed to be short-lived, and some studies argue against its efficacy.37 It has a favorable side-effect profile and is generally very well tolerated. It can be used as an adjunct to other antitremor agents and is preferred for patients with coexisting partial seizures or myoclonus.
Benzodiazepines. Minor tranquilizers are often used to control tremor, especially in coexisting anxiety or insomnia. Alprazolam (Xanax) is the one most widely used for this indication.38 It can be started in a dose of 0.25 mg once at bedtime and increased gradually up to 0.75 to 2 mg. Clonazepam (Klonopin) is particularly useful for orthostatic tremor, a variant of essential tremor characterized by tremor of the legs and trunk upon standing.39
Common side effects of benzodiazepines include sedation, cognitive dysfunction, hypotension, respiratory inhibition, and addiction after prolonged use. In the elderly, they can lead to confusion and disinhibition and can increase the risk of falling. They should be avoided in the elderly and in alcoholic patients and those with a high risk of substance abuse.
Stopping benzodiazepines should be done gradually to avoid withdrawal symptoms, including aggravation of tremor.
THIRD-LINE AGENTS
Clozapine
Clozapine (Clozaril) is a novel antipsychotic drug with no extrapyramidal side effects. It has been reported effective in essential tremor and drug-induced tremor,40,41 but the results of these early studies have not been confirmed.
Clozapine is started as a single daily dose of 12.5 mg and is increased up to 75 mg or 100 mg. It is an attractive option for patients with coexisting psychosis, bipolar disorder, or chorea. Its main side effects are sedation, salivation, weight gain, hypertension, diabetes, and seizures.
One especially serious side effect is agranulocytosis. This potentially fatal effect is rare, occurring in about 1.3% of patients receiving this drug. Weekly monitoring of the white blood cell count is mandated during treatment with clozapine, and this has made clozapine a less attractive option for the routine treatment of essential tremor.
Mirtazapine
Mirtazapine (Remeron) is a novel antidepressant widely used in Parkinson disease as both an antidepressant and a sleeping aid. Case studies have reported efficacy in both essential tremor and parkinsonian tremor,42 but controlled studies have not confirmed this.43 Mirtazapine is a reasonable option in patients with coexisting depression or insomnia. It is usually given as a single bedtime dose of 15 to 30 mg.
Other drugs
Studies of other agents for the treatment of essential tremor—eg, carbonic anhydrase enzyme inhibitors, calcium channel blockers, isoniazid (Tubizid), clonidine (Catapres), phenobarbital, and theophylline—have yielded highly contradictory results. Thus, they are not recommended as first- or second-line agents for essential tremor.
SPECIALTY-LEVEL CARE
When essential tremor does not respond to drug therapy or the patient cannot tolerate drug therapy, the patient should be referred to a center specializing in movement disorders for more advanced treatment options, ie, botulinum toxin injection and deep brain stimulation surgery.
Botulinum toxin
Botulinum toxin type A has been studied for the treatment of essential tremor with variable degrees of success. It has been effective in reducing hand tremor in essential tremor, but without a concomitant improvement in functional disability.44 This limited functional improvement has been attributed to the development of muscle weakness after injection of the neurotoxin. This has also raised questions about unintentional unblinding when interpreting study results. Therefore, most clinicians restrict its use to focal forms of tremor such as voice tremor,45 head tremor, and task-specific tremor.
Side effects are limited and temporary and include muscle weakness, pain at the injection site, dysphagia (when injected for head or voice tremor), and a breathy vocal quality (when injected for voice tremor). Botulinum toxin injection is the treatment of choice for focal dystonia, and therefore would be a good option for dystonic tremor.
Thalamic deep brain stimulation
This technique involves stereotactic implantation of a stimulation lead in the ventral intermediate nucleus of the thalamus. The lead connects via a subcutaneous wire to an intermittent pulse generator, implanted subcutaneously in the infraclavicular region. The stimulation lead produces continuous stimulation of the ventralis intermedius nucleus that is functionally equal to lesional surgery, thus antagonizing the relay of tremor signals at the thalamus.
The battery of the pulse generator must be replaced every 4 to 7 years depending on usage and stimulation parameters. Battery replacement can be performed with minor surgery at the infraclavicular region.
Thalamic deep brain stimulation is indicated for patients with severe, disabling essential tremor who have tremor resistant to drug therapy or who cannot tolerate drug therapy.
The procedure has been shown to provide benefit in 90% of patients, with more than an 80% improvement in tremor severity and functional impact.46–49 Deep brain stimulation is effective against tremor affecting parts of the body other than the limbs, including the head; an exception to this is voice tremor, which usually does not improve dramatically. The procedure can be done unilaterally or bilaterally, depending on symptoms. Patients with asymmetrical tremor and those at risk of side effects can undergo unilateral surgery. Bilateral treatment is recommended for patients with symmetric tremor or significant head tremor, or who are young and healthy.
Surgical risks include brain hemorrhage and infection. Side effects of the stimulation include paresthesias, paresis, imbalance, dysarthria, and, in rare cases, dysphagia.
CHOOSING THE BEST MANAGEMENT PLAN FOR YOUR PATIENT
The choice of treatment may be challenging, given the multiple treatment options and the variability of tremor severity from one patient to another. The following guidelines can be used to help make this decision.
All patients should be advised to reduce caffeine intake, to have sufficient hours of sleep, and to avoid stressful situations.
Patients with minor, nondisabling tremor can be left untreated if the tremors are not bothersome or if the patient prefers not to pursue active treatment.
In patients who have bothersome tremor only when anxious or in certain social situations, give propranolol or alprazolam (or both) to be taken as needed. Relaxation techniques and meditation are also useful for these patients.
Patients with constant bothersome tremor should be started on either propranolol or primidone based on the patient’s profile and propensity to develop side effects from each of these drugs. The dosing should be optimized gradually according to the patient’s response and the drug’s tolerability.
If essential tremor is not sufficiently controlled with one first-line agent (propranolol or primidone), try combining the two first-line agents if the patient finds it tolerable.
A second-line agent can be added to either of the first-line agents or to the combination of both if tremor control is not yet sufficient. A second-line or third-line agent can also be used as the primary treatment if both first-line agents are contraindicated or intolerable. Combining two or more second- and third-line agents is another option. The choice of second- or third-line agent should be guided by the patient’s characteristics and comorbidities in relation to the agent’s side effects and contraindications as detailed in the above section.
Patients should be referred to a movement disorders specialist in cases of resistant tremor, intolerance to oral medications, severe disability, and atypical presentation. Types of tremor known to be poorly responsive to oral medications (eg, head tremor, voice tremor) deserve a specialist evaluation if they contribute significantly to the patient’s morbidity.
The usual specialist treatment of severe voice tremor and head tremor is botulinum toxin injection. Patients with resistant and disabling hand tremor are evaluated for thalamic deep brain stimulation.
Patients with residual disability despite medical and surgical treatment should be referred for occupational therapy. Occupational therapy can improve quality of life through the use of special utensils, pens, computer gadgets, and arm weights, among other devices.
Essential tremor, one of the most common movement disorders, affects about 4% of adults 40 years of age and older.1 It is often referred to as familial tremor in patients with a family history of tremor. It has also been called benign tremor to differentiate it from tremor associated with neurodegenerative diseases, particularly Parkinson disease, but this condition is certainly not benign, as it can cause substantial functional impairment and difficulties with routine activities of daily living. The terms “essential” and “idiopathic” refer to the primary nature of the disorder and differentiate it from tremor that is a feature of a distinct neurologic entity or is secondary to a metabolic disease or drug therapy.
Successful management entails exclusion of secondary causes and careful selection of drug therapy. To date, there is no cure for essential tremor; all currently available treatments are purely symptomatic.
In this review, we outline the major diagnostic and therapeutic principles of managing essential tremor, indications for referral to specialists, and alternative and advanced therapeutic options.
CLINICAL PICTURE
Tremor is defined as rhythmic to-and-fro movement in any body part. It can be slow or fast, and its amplitude can be large and coarse, or small or even “fine.” It can appear at rest, with action, or during a sustained posture. In contrast to parkinsonian tremor (which presents mainly at rest), essential tremor is typically but not exclusively postural, kinetic, or both.
Postural tremor refers to tremor seen when the patient holds the affected limb (commonly the arm) unsupported against gravity. Kinetic tremor refers to tremor that appears with active movements. This is often demonstrated clinically by the finger-nose-finger test. Patients with essential tremor commonly have both postural and kinetic tremor.
The tremor commonly involves the arms, hands, and fingers.2 Less commonly, it involves the head, the lips, the tongue, the legs, and the voice. In contrast to parkinsonian tremor, which typically affects one side of the body first, bilateral involvement is the general rule in essential tremor. However, one side of the body may be affected first, or may be more affected than the other. The frequency of the tremor ranges from 4 to 12 Hz (ie, beats per second).
The tremor usually starts in middle age and progresses slowly over time,3 but onset in old age or childhood is also possible.4 Both sexes are equally affected.
The tremor usually gets worse with anxiety, stress, and caffeine intake. It usually gets temporarily better with the consumption of small amounts of alcohol.
The functional impact of essential tremor is judged by its effect on different daily activities, especially writing, eating, drinking, dressing, manual work, and household chores.
In addition to motor dysfunction, the tremor can also have a significant psychological impact on the patient, because it usually gets worse in social situations.
Although it has long been thought that tremor is the sole neurologic sign of essential tremor, recent studies have shown that many patients have additional subtle findings, such as mild gait difficulty,5 slight incoordination,6 mild cognitive impairment,7 and decreased hearing,8 and are more likely to have anxiety and social phobia.9
Although different studies have varied in their findings, it is generally thought that about 50% of patients with essential tremor have a positive family history, often in a first-degree relative, suggesting autosomal dominant inheritance with variable penetrance.10,11 Polygenetic and sporadic variants are also common.
DIFFERENTIAL DIAGNOSIS
Resting tremor
Resting tremor is typically an extrapyramidal sign and, when accompanied by rigidity and bradykinesia, is often part of a parkinsonian syndrome. It is most pronounced at rest when the affected body part is fully supported and stationary. The tremor tends to improve with action or posture. It usually has a “pill-rolling” character and, as mentioned, is associated with other extrapyramidal signs, such as rigidity, slowness, and, later on, postural instability.
About 20% of patients with essential tremor have resting tremor. These patients usually suffer from severe or long-standing disease.12 However, the resting element in these cases is often milder than the postural and kinetic components, and it is typically not associated with other extrapyramidal signs. Also, some patients may have both essential tremor and Parkinson disease.13
Intentional tremor
Pure intentional tremor is usually seen with cerebellar pathology, which includes tumors, stroke, multiple sclerosis, trauma, and spinocerebellar degeneration. The amplitude of this type of tremor increases as the affected limb approaches the final target. It can best be demonstrated clinically by the finger-nose-finger test. The frequency of intentional tremor is slow (2 to 4 Hz) and is usually associated with other cerebellar signs, such as dysmetria, decomposition, rebound, and dysdiadochokinesia (ie, the inability to perform rapid alternating movements in a smooth and coordinated manner).
About 50% of patients with essential tremor have an intentional component to their tremor,6 or it can be mildly present in the form of a slight gait difficulty. However, in essential tremor, other features of cerebellar dysfunction are either absent or only very slight.
Secondary causes of postural-kinetic tremor
Enhanced physiologic tremor. A very mild postural tremor is present in almost all people and is considered “physiologic” since it has almost no clinical significance. This type of tremor is often invisible, but when “enhanced,” it can be visually demonstrated by placing a piece of paper over the stretched hands and watching the ripple from the paper.
Certain conditions can aggravate this physiologic tremor and can make it symptomatic. Common causes include anxiety, sleep deprivation, hypoglycemia, hyperthyroidism, pheochromocytoma, serotonin syndrome, and carcinoid syndrome.
Metabolic tremor. Hyperammonemia can cause tremor in patients with hepatic encephalopathy, and uremia can cause tremor in patients with renal failure. These metabolic conditions classically result in “flappy” tremor (asterixis), a special form of postural tremor characterized by jerking movements with high amplitude. It is best seen when the patient stretches out the arms and extends the wrists as if trying to stop traffic. But even though it may look like tremor, asterixis is thought to be a form of “negative” myoclonus.
Drug-related tremor. Postural-kinetic tremor can be induced by drugs, including lithium (Lithobid), valproate (Depakote), amiodarone (Cordarone), central nervous system stimulants, beta agonists (including inhalers), and some antidepressants. Tremor can also occur with alcohol or sedative withdrawal.
Psychogenic tremor. Tremor can be seen as part of a somatoform disorder commonly referred to as conversion disorder or conversion reaction. Psychogenic tremor is characterized by acute onset, commonly following a psychosocial stressor; it is often atypical, variable in frequency, amplitude, and body-part involvement, and it can readily be interrupted on examination by distracting the patient.
Neurologic disorders. The postural and kinetic elements of essential tremor may also be seen in the following neurologic conditions:
- Holmes (rubral) tremor, a combination of resting, postural, kinetic, and intentional tremor of low frequency and high amplitude. It usually has a proximal component and is often unilateral. It commonly is due to a lesion that involves the brainstem, eg, red nucleus, inferior olive, cerebellum, or thalamus. Common causes include stroke, prolonged hypoxia, and head trauma (including closed-head trauma with negative imaging). This type of tremor is usually associated with ataxia.14
- Dystonic tremor is predominantly postural and is associated with abnormal dystonic posturing of the affected body part, commonly the head, hands, or feet. Unlike the rhythmic oscillations of essential tremor, dystonic tremor is often irregular in rhythm.
- Multiple sclerosis can present with a combination of postural, kinetic, and intentional tremor. Patients usually have a clear history of recurrent neurologic deficits and show a combination of pyramidal, cerebellar, and sensory signs on examination consistent with multiple sclerosis.15
- Neuropathic tremor is seen in a small proportion of patients with peripheral neuropathy, especially demyelinating neuropathy.16 The tremor is usually posturalkinetic and is associated with signs of neuropathy, such as a glove-and-stocking pattern of hypoesthesia, reduced reflexes, and sensory ataxia (including intentional tremor when the eyes are closed).
- Posttraumatic tremor can occur after severe or even mild head trauma, especially in children. It is commonly rubral, but other types have been reported, including a presentation resembling essential tremor.17
- Monosymptomatic or isolated tremor. A number of conditions related to essential tremor with location-specific or task-specific tremor have been described. These rare conditions historically have been classified as “possible essential tremor” or “essential tremor variants” but are now considered separate entities. These include task-specific tremor (eg, writing tremor), isolated head tremor, isolated voice tremor, and orthostatic tremor (tremor in the legs and trunk upon standing in place, but not when sitting or walking).18,19
DIAGNOSIS IS CLINICAL
Essential tremor is a clinical diagnosis. After a thorough review of the medical history and medication exposures, laboratory and imaging tests may be ordered to rule out a secondary cause. A complete metabolic panel, including blood glucose and thyroid-stimulating hormone levels, is usually sufficient. Brain imaging or other imaging is ordered for patients with an atypical presentation.
TREATMENT IS SYMPTOMATIC
Treatment of essential tremor is symptomatic. Several drugs of different pharmacologic classes can reduce the severity of the tremor and improve function.
Choosing the appropriate treatment depends on the type of tremor and the presence of associated conditions. The response to treatment and the development of side effects guide further adjustments. The following is a brief description of the available antitremor agents.
FIRST-LINE AGENTS
Propranolol
Propranolol (Inderal), a nonselective beta blocker, is the most widely used antitremor drug and the only agent approved by the US Food and Drug Administration for essential tremor. It should be started at a low dose and titrated upward gradually. The usual starting dose is 10 mg three times daily. The average effective dose is 120 mg daily. The dose can be increased up to 320 mg if needed and tolerated.
Sustained-release preparations are equally effective and are given as a single daily dose to improve compliance.20
Propranolol improves tremor in 50% to 70% of patients with essential tremor and achieves an average tremor reduction of 50% to 60%.1,21–25 Side effects include bronchoconstriction, bradycardia, hypotension, depression, impotence, fatigue, and gastrointestinal disturbances.
Other beta-blockers, such as nadolol (Corgard) and timolol, are also effective against tremor but are less potent than propranolol.26,27 The selective beta-1-blocker metoprolol (Lopressor) may be effective and has fewer noncardiac side effects than propranolol.28 It can be used in patients who discontinue propranolol because of adverse effects. Atenolol (Tenormin) and pindolol (Visken) have little or no effect on tremor.29
A good candidate for propranolol therapy in essential tremor is:
- A patient with no known contraindication to propranolol
- A patient with hypertension, coronary heart disease, or tachyarrhythmia
- A patient with anxiety or social phobia.
Absolute contraindications to propranolol are:
- Moderate to severe bronchial asthma
- Significant bradycardia or heart block
- Symptomatic hypotension
- End-stage heart failure
- Concurrent use of a calcium channel blocker.
Relative contraindications are:
- Wheezing (eg, chronic obstructive pulmonary disease)
- Depression
- Diabetes mellitus in a patient more prone to hypoglycemia (propranolol masks the warning signs of hypoglycemia)
- Reduced sexual potency in a male patient.
Primidone
Primidone (Mysoline) is an antiepileptic drug structurally similar to barbiturates. Its antitremor effect is equal to that of propranolol, though some studies suggest it is slightly more efficacious.30,31
It should be started at a low dose, ie, 25 mg once daily at bedtime. The dose should then be increased gradually until satisfactory and tolerable tremor control is achieved. Most patients respond to doses of around 250 mg per day.1,22,24–25 The dose can be increased if needed and tolerated.
Primidone reduces tremor by about 50% to 60%.1,22,24–25 Side effects include sedation, dizziness, fatigue, nausea, and depression, as well as ataxia and confusion in severe cases.
A good candidate for primidone in essential tremor is:
- A patient with no known contraindication to primidone
- A patient with contraindications to propranolol
- A younger patient
- A patient with epilepsy.
Absolute contraindications to primidone include:
- Confusion or dementia
- Oral anticoagulant therapy with difficulty controlling the International Normalized Ratio (primidone is a potent enzyme inducer).
Relative contraindications to primidone in essential tremor are:
- Depression
- Alcohol abuse
- Ongoing therapy with sedating drugs
- Ataxia or vertigo.
SECOND-LINE AGENTS
Other antiepileptics
Topiramate (Topamax) is a broad-spectrum antiepileptic shown to be significantly effective against essential tremor.32 It is usually started at a single daily dose of 25 mg and increased gradually to the most effective dose, usually around 300 mg.
Side effects include reduced appetite, weight loss, cognitive dysfunction, and paresthesia.
Favorable candidates include patients who are epileptic or overweight. Contraindications include cognitive impairment and low body weight. It is also not recommended in children so as to avoid any possible negative effect on cognitive development. In rare cases, topiramate has been reported to cause significant visual disturbances.
Gabapentin (Neurontin) is an antiepileptic that is now more often used as a symptomatic treatment for neuropathic pain. Studies have suggested a beneficial effect on essential tremor,33,34 but some investigators have questioned its efficacy.35
Like other antitremor agents, it should be started at a low dose, ie, around 300 mg, and escalated gradually until the tremor is controlled. The usual effective dose is 1,200 mg.
Gabapentin is generally well tolerated, and side effects such as dizziness, drowsiness, sedation, and unsteadiness are rare and usually mild.
The favorable candidate is a patient with associated neuropathy or multiple comorbidities. Gabapentin has also been reported to alleviate neuropathic tremor.
Contraindications are minimal and include intolerability or hypersensitivity to the drug. It also should be avoided in patients at a high risk of falling.
Levetiracetam (Keppra) is a novel antiepileptic effective against partial seizures. Studies have shown contradictory results regarding its antitremor effect. One double-blind, placebo-controlled study demonstrated significant reduction in essential tremor with 1,000 mg of levetiracetam.36 However, its effect on tremor is believed to be short-lived, and some studies argue against its efficacy.37 It has a favorable side-effect profile and is generally very well tolerated. It can be used as an adjunct to other antitremor agents and is preferred for patients with coexisting partial seizures or myoclonus.
Benzodiazepines. Minor tranquilizers are often used to control tremor, especially in coexisting anxiety or insomnia. Alprazolam (Xanax) is the one most widely used for this indication.38 It can be started in a dose of 0.25 mg once at bedtime and increased gradually up to 0.75 to 2 mg. Clonazepam (Klonopin) is particularly useful for orthostatic tremor, a variant of essential tremor characterized by tremor of the legs and trunk upon standing.39
Common side effects of benzodiazepines include sedation, cognitive dysfunction, hypotension, respiratory inhibition, and addiction after prolonged use. In the elderly, they can lead to confusion and disinhibition and can increase the risk of falling. They should be avoided in the elderly and in alcoholic patients and those with a high risk of substance abuse.
Stopping benzodiazepines should be done gradually to avoid withdrawal symptoms, including aggravation of tremor.
THIRD-LINE AGENTS
Clozapine
Clozapine (Clozaril) is a novel antipsychotic drug with no extrapyramidal side effects. It has been reported effective in essential tremor and drug-induced tremor,40,41 but the results of these early studies have not been confirmed.
Clozapine is started as a single daily dose of 12.5 mg and is increased up to 75 mg or 100 mg. It is an attractive option for patients with coexisting psychosis, bipolar disorder, or chorea. Its main side effects are sedation, salivation, weight gain, hypertension, diabetes, and seizures.
One especially serious side effect is agranulocytosis. This potentially fatal effect is rare, occurring in about 1.3% of patients receiving this drug. Weekly monitoring of the white blood cell count is mandated during treatment with clozapine, and this has made clozapine a less attractive option for the routine treatment of essential tremor.
Mirtazapine
Mirtazapine (Remeron) is a novel antidepressant widely used in Parkinson disease as both an antidepressant and a sleeping aid. Case studies have reported efficacy in both essential tremor and parkinsonian tremor,42 but controlled studies have not confirmed this.43 Mirtazapine is a reasonable option in patients with coexisting depression or insomnia. It is usually given as a single bedtime dose of 15 to 30 mg.
Other drugs
Studies of other agents for the treatment of essential tremor—eg, carbonic anhydrase enzyme inhibitors, calcium channel blockers, isoniazid (Tubizid), clonidine (Catapres), phenobarbital, and theophylline—have yielded highly contradictory results. Thus, they are not recommended as first- or second-line agents for essential tremor.
SPECIALTY-LEVEL CARE
When essential tremor does not respond to drug therapy or the patient cannot tolerate drug therapy, the patient should be referred to a center specializing in movement disorders for more advanced treatment options, ie, botulinum toxin injection and deep brain stimulation surgery.
Botulinum toxin
Botulinum toxin type A has been studied for the treatment of essential tremor with variable degrees of success. It has been effective in reducing hand tremor in essential tremor, but without a concomitant improvement in functional disability.44 This limited functional improvement has been attributed to the development of muscle weakness after injection of the neurotoxin. This has also raised questions about unintentional unblinding when interpreting study results. Therefore, most clinicians restrict its use to focal forms of tremor such as voice tremor,45 head tremor, and task-specific tremor.
Side effects are limited and temporary and include muscle weakness, pain at the injection site, dysphagia (when injected for head or voice tremor), and a breathy vocal quality (when injected for voice tremor). Botulinum toxin injection is the treatment of choice for focal dystonia, and therefore would be a good option for dystonic tremor.
Thalamic deep brain stimulation
This technique involves stereotactic implantation of a stimulation lead in the ventral intermediate nucleus of the thalamus. The lead connects via a subcutaneous wire to an intermittent pulse generator, implanted subcutaneously in the infraclavicular region. The stimulation lead produces continuous stimulation of the ventralis intermedius nucleus that is functionally equal to lesional surgery, thus antagonizing the relay of tremor signals at the thalamus.
The battery of the pulse generator must be replaced every 4 to 7 years depending on usage and stimulation parameters. Battery replacement can be performed with minor surgery at the infraclavicular region.
Thalamic deep brain stimulation is indicated for patients with severe, disabling essential tremor who have tremor resistant to drug therapy or who cannot tolerate drug therapy.
The procedure has been shown to provide benefit in 90% of patients, with more than an 80% improvement in tremor severity and functional impact.46–49 Deep brain stimulation is effective against tremor affecting parts of the body other than the limbs, including the head; an exception to this is voice tremor, which usually does not improve dramatically. The procedure can be done unilaterally or bilaterally, depending on symptoms. Patients with asymmetrical tremor and those at risk of side effects can undergo unilateral surgery. Bilateral treatment is recommended for patients with symmetric tremor or significant head tremor, or who are young and healthy.
Surgical risks include brain hemorrhage and infection. Side effects of the stimulation include paresthesias, paresis, imbalance, dysarthria, and, in rare cases, dysphagia.
CHOOSING THE BEST MANAGEMENT PLAN FOR YOUR PATIENT
The choice of treatment may be challenging, given the multiple treatment options and the variability of tremor severity from one patient to another. The following guidelines can be used to help make this decision.
All patients should be advised to reduce caffeine intake, to have sufficient hours of sleep, and to avoid stressful situations.
Patients with minor, nondisabling tremor can be left untreated if the tremors are not bothersome or if the patient prefers not to pursue active treatment.
In patients who have bothersome tremor only when anxious or in certain social situations, give propranolol or alprazolam (or both) to be taken as needed. Relaxation techniques and meditation are also useful for these patients.
Patients with constant bothersome tremor should be started on either propranolol or primidone based on the patient’s profile and propensity to develop side effects from each of these drugs. The dosing should be optimized gradually according to the patient’s response and the drug’s tolerability.
If essential tremor is not sufficiently controlled with one first-line agent (propranolol or primidone), try combining the two first-line agents if the patient finds it tolerable.
A second-line agent can be added to either of the first-line agents or to the combination of both if tremor control is not yet sufficient. A second-line or third-line agent can also be used as the primary treatment if both first-line agents are contraindicated or intolerable. Combining two or more second- and third-line agents is another option. The choice of second- or third-line agent should be guided by the patient’s characteristics and comorbidities in relation to the agent’s side effects and contraindications as detailed in the above section.
Patients should be referred to a movement disorders specialist in cases of resistant tremor, intolerance to oral medications, severe disability, and atypical presentation. Types of tremor known to be poorly responsive to oral medications (eg, head tremor, voice tremor) deserve a specialist evaluation if they contribute significantly to the patient’s morbidity.
The usual specialist treatment of severe voice tremor and head tremor is botulinum toxin injection. Patients with resistant and disabling hand tremor are evaluated for thalamic deep brain stimulation.
Patients with residual disability despite medical and surgical treatment should be referred for occupational therapy. Occupational therapy can improve quality of life through the use of special utensils, pens, computer gadgets, and arm weights, among other devices.
- Zesiewicz TA, Chari A, Jahan I, et al. Overview of essential tremor. Neuropsychiatr Dis Treat 2010; 6:401–408.
- Elble RJ. Essential tremor frequency decreases with time. Neurology 2000; 55:1547–1551.
- Louis ED, Ottman R, Hauser WA. How common is the most common adult movement disorder? Estimates of the prevalence of essential tremor throughout the world. Mov Disord 1998; 13:5–10.
- Louis ED, Dure LS, Pullman S. Essential tremor in childhood: a series of nineteen cases. Mov Disord 2001; 16:921–923.
- Singer C, Sanchez-Ramos J, Weiner WJ. Gait abnormality in essential tremor. Mov Disord 1994; 9:193–196.
- Deuschl G, Wenzelburger R, Löffler K, et al. Essential tremor and cerebellar dysfunction. Clinical and kinematic analysis of intention tremor. Brain 2000; 123:1568–1580.
- Louis ED. Functional correlates of lower cognitive test scores in essential tremor. Mov Disord 2010; 25:481–485.
- Ondo WG, Sutton L, Dat Vuong K, et al. Hearing impairment in essential tremor. Neurology 2003; 61:1093–1097.
- Schneier FR, Barnes LF, Albert SM, et al. Characteristics of social phobia among persons with essential tremor. J Clin Psychiatry 2001; 62:367–372.
- Whaley NR, Putzke JD, Baba Y, et al. Essential tremor: phenotypic expression in a clinical cohort. Parkinsonism Relat Disord 2007; 13:333–339.
- Deng H, Le W, Jankovic J. Genetics of essential tremor. Brain 2007; 130:1456–1464.
- Cohen O, Pullman S, Jurewicz E, et al. Rest tremor in patients with essential tremor: prevalence, clinical correlates, and electrophysiologic characteristics. Arch Neurol 2003; 60:405–410.
- Shahed J, Jankovic J. Exploring the relationship between essential tremor and Parkinson’s disease. Parkinsonism Relat Disord 2007; 13:67–76.
- Yang YW, Chang FC, Tsai CH, et al. Clinical and magnetic resonance imaging manifestations of Holmes tremor. Acta Neurol Taiwan 2005; 14:9–15.
- Alusi SH, Worthington J, Glickman S, et al. A study of tremor in multiple sclerosis. Brain 2001; 124:720–730.
- Breit S, Wächter T, Schöls L, et al. Effective thalamic deep brain stimulation for neuropathic tremor in a patient with severe demyelinating neuropathy. J Neurol Neurosurg Psychiatry 2009; 80:235–236.
- Koller WC, Wong GF, Lang A. Posttraumatic movement disorders: a review. Mov Disord 1989; 4:20–36.
- Jankovic J. Essential tremor: a heterogenous disorder. Mov Disord 2002; 17:638–644.
- Deuschl G, Bain P, Brin M. Consensus statement of the Movement Disorder Society on Tremor. Ad Hoc Scientific Committee. Mov Disord 1998; 13(suppl 3):2–23.
- Calzetti S, Findley LJ, Gresty MA, et al. Effect of a single oral dose of propranolol on essential tremor: a double-blind controlled study. Ann Neurol 1983; 13:165–171.
- Larsen TA, Teräväinen H, Calne DB. Atenolol vs propranolol in essential tremor. A controlled, quantitative study. Acta Neurol Scand 1982; 66:547–554.
- Zesiewicz TA, Elble R, Louis ED, et al. Practice parameter: therapies for essential tremor: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2005; 64:2008–2020.
- Lyons KE, Pahwa R, Comella CL, et al.Benefits and risks of pharmacological treatments for essential tremor. Drug Saf 2003; 26:461–481.
- Pahwa R, Lyons KE. Essential tremor: differential diagnosis and current therapy. Am J Med 2003; 115:134–142.
- Louis ED. Clinical practice. Essential tremor. N Engl J Med 2001; 345:887–891.
- Koller WC. Nadolol in essential tremor. Neurology 1983; 33:1076–1077.
- Dietrichson P, Espen E. Effects of timolol and atenolol on benign essential tremor: placebo-controlled studies based on quantitative tremor recording. J Neurol Neurosurg Psychiatry 1981; 44:677–683.
- Calzetti S, Findley LJ, Gresty MA, et al. Metoprolol and propranolol in essential tremor: a double-blind, controlled study. J Neurol Neurosurg Psychiatry 1981; 44:814–819.
- Teravainen H, Larsen A, Fogelholm R. Comparison between the effects of pindolol and propranolol on essential tremor. Neurology 1977; 27:439–442.
- Gorman WP, Cooper R, Pocock P, et al. A comparison of primidone, propranolol, and placebo in essential tremor, using quantitative analysis. J Neurol Neurosurg Psychiatry 1986; 49:64–68.
- Koller WC, Royse VL. Efficacy of primidone in essential tremor. Neurology 1986; 36:121–124.
- Connor GS. A double-blind placebo-controlled trial of topiramate treatment for essential tremor. Neurology 2002; 59:132–134.
- Gironell A, Kulisevsky J, Barbanoj M, et al. A randomized placebo-controlled comparative trial of gabapentin and propranolol in essential tremor. Arch Neurol 1999; 56:475–480.
- Ondo W, Hunter C, Vuong KD, et al. Gabapentin for essential tremor: a multiple-dose, double-blind, placebo-controlled trial. Mov Disord 2000; 15:678–682.
- Pahwa R, Lyons K, Hubble JP, et al. Double-blind controlled trial of gabapentin in essential tremor. Mov Disord 1998; 13:465–467.
- Bushara KO, Malik T, Exconde RE. The effect of levetiracetam on essential tremor. Neurology 2005; 64:1078–1080.
- Sullivan KL, Hauser RA, Zesiewicz TA. Levetiracetam for the treatment of essential tremor. Mov Disord 2005; 20:640.
- Huber SJ, Paulson GW. Efficacy of alprazolam for essential tremor. Neurology 1988; 38:241–243.
- McManis PG, Sharbrough FW. Orthostatic tremor: clinical and electrophysiologic characteristics. Muscle Nerve 1993; 16:1254–1260.
- Ceravolo R, Salvetti S, Piccini P, et al. Acute and chronic effects of clozapine in essential tremor. Mov Disord 1999; 14:468–472.
- Pakkenberg H, Pakkenberg B. Clozapine in the treatment of tremor. Acta Neurol Scand 1986; 73:295–297.
- Pact V, Giduz T. Mirtazapine treats resting tremor, essential tremor, and levodopa-induced dyskinesias. Neurology 1999; 53:1154.
- Lyons KE, Pahwa R. A double-blind, placebo-controlled, pilot study of mirtazapine in essential tremor. Presented at the 54th Annual Meeting of the American Academy of Neurology, Denver, Colorado. Neurology 2002; 58(suppl 3):A254.
- Brin MF, Lyons KE, Doucette J, et al. A randomized, double masked, controlled trial of botulinum toxin type A in essential hand tremor. Neurology 2001; 56:1523–1528.
- Blitzer A, Brin MF, Stewart C, et al. Abductor laryngeal dystonia: a series treated with botulinum toxin. Laryngoscope 1992; 102:163–167.
- Schuurman PR, Bosch DA, Bossuyt PM, et al. A comparison of continuous thalamic stimulation and thalamotomy for suppression of severe tremor. N Engl J Med 2000; 342:461–468.
- Flora ED, Perera CL, Cameron AL, et al. Deep brain stimulation for essential tremor: a systematic review. Mov Disord 2010; 25:1550–1559.
- Nagaseki Y, Shibazaki T, Hirai T, et al. Long-term follow-up results of selective VIM-thalamotomy. J Neurosurg 1986; 65:296–302.
- Zirh A, Reich SG, Dougherty PM, et al. Stereotactic thalamotomy in the treatment of essential tremor of the upper extremity: reassessment including a blinded measure of outcome. J Neurol Neurosurg Psychiatry 1999; 66:772–775.
- Zesiewicz TA, Chari A, Jahan I, et al. Overview of essential tremor. Neuropsychiatr Dis Treat 2010; 6:401–408.
- Elble RJ. Essential tremor frequency decreases with time. Neurology 2000; 55:1547–1551.
- Louis ED, Ottman R, Hauser WA. How common is the most common adult movement disorder? Estimates of the prevalence of essential tremor throughout the world. Mov Disord 1998; 13:5–10.
- Louis ED, Dure LS, Pullman S. Essential tremor in childhood: a series of nineteen cases. Mov Disord 2001; 16:921–923.
- Singer C, Sanchez-Ramos J, Weiner WJ. Gait abnormality in essential tremor. Mov Disord 1994; 9:193–196.
- Deuschl G, Wenzelburger R, Löffler K, et al. Essential tremor and cerebellar dysfunction. Clinical and kinematic analysis of intention tremor. Brain 2000; 123:1568–1580.
- Louis ED. Functional correlates of lower cognitive test scores in essential tremor. Mov Disord 2010; 25:481–485.
- Ondo WG, Sutton L, Dat Vuong K, et al. Hearing impairment in essential tremor. Neurology 2003; 61:1093–1097.
- Schneier FR, Barnes LF, Albert SM, et al. Characteristics of social phobia among persons with essential tremor. J Clin Psychiatry 2001; 62:367–372.
- Whaley NR, Putzke JD, Baba Y, et al. Essential tremor: phenotypic expression in a clinical cohort. Parkinsonism Relat Disord 2007; 13:333–339.
- Deng H, Le W, Jankovic J. Genetics of essential tremor. Brain 2007; 130:1456–1464.
- Cohen O, Pullman S, Jurewicz E, et al. Rest tremor in patients with essential tremor: prevalence, clinical correlates, and electrophysiologic characteristics. Arch Neurol 2003; 60:405–410.
- Shahed J, Jankovic J. Exploring the relationship between essential tremor and Parkinson’s disease. Parkinsonism Relat Disord 2007; 13:67–76.
- Yang YW, Chang FC, Tsai CH, et al. Clinical and magnetic resonance imaging manifestations of Holmes tremor. Acta Neurol Taiwan 2005; 14:9–15.
- Alusi SH, Worthington J, Glickman S, et al. A study of tremor in multiple sclerosis. Brain 2001; 124:720–730.
- Breit S, Wächter T, Schöls L, et al. Effective thalamic deep brain stimulation for neuropathic tremor in a patient with severe demyelinating neuropathy. J Neurol Neurosurg Psychiatry 2009; 80:235–236.
- Koller WC, Wong GF, Lang A. Posttraumatic movement disorders: a review. Mov Disord 1989; 4:20–36.
- Jankovic J. Essential tremor: a heterogenous disorder. Mov Disord 2002; 17:638–644.
- Deuschl G, Bain P, Brin M. Consensus statement of the Movement Disorder Society on Tremor. Ad Hoc Scientific Committee. Mov Disord 1998; 13(suppl 3):2–23.
- Calzetti S, Findley LJ, Gresty MA, et al. Effect of a single oral dose of propranolol on essential tremor: a double-blind controlled study. Ann Neurol 1983; 13:165–171.
- Larsen TA, Teräväinen H, Calne DB. Atenolol vs propranolol in essential tremor. A controlled, quantitative study. Acta Neurol Scand 1982; 66:547–554.
- Zesiewicz TA, Elble R, Louis ED, et al. Practice parameter: therapies for essential tremor: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2005; 64:2008–2020.
- Lyons KE, Pahwa R, Comella CL, et al.Benefits and risks of pharmacological treatments for essential tremor. Drug Saf 2003; 26:461–481.
- Pahwa R, Lyons KE. Essential tremor: differential diagnosis and current therapy. Am J Med 2003; 115:134–142.
- Louis ED. Clinical practice. Essential tremor. N Engl J Med 2001; 345:887–891.
- Koller WC. Nadolol in essential tremor. Neurology 1983; 33:1076–1077.
- Dietrichson P, Espen E. Effects of timolol and atenolol on benign essential tremor: placebo-controlled studies based on quantitative tremor recording. J Neurol Neurosurg Psychiatry 1981; 44:677–683.
- Calzetti S, Findley LJ, Gresty MA, et al. Metoprolol and propranolol in essential tremor: a double-blind, controlled study. J Neurol Neurosurg Psychiatry 1981; 44:814–819.
- Teravainen H, Larsen A, Fogelholm R. Comparison between the effects of pindolol and propranolol on essential tremor. Neurology 1977; 27:439–442.
- Gorman WP, Cooper R, Pocock P, et al. A comparison of primidone, propranolol, and placebo in essential tremor, using quantitative analysis. J Neurol Neurosurg Psychiatry 1986; 49:64–68.
- Koller WC, Royse VL. Efficacy of primidone in essential tremor. Neurology 1986; 36:121–124.
- Connor GS. A double-blind placebo-controlled trial of topiramate treatment for essential tremor. Neurology 2002; 59:132–134.
- Gironell A, Kulisevsky J, Barbanoj M, et al. A randomized placebo-controlled comparative trial of gabapentin and propranolol in essential tremor. Arch Neurol 1999; 56:475–480.
- Ondo W, Hunter C, Vuong KD, et al. Gabapentin for essential tremor: a multiple-dose, double-blind, placebo-controlled trial. Mov Disord 2000; 15:678–682.
- Pahwa R, Lyons K, Hubble JP, et al. Double-blind controlled trial of gabapentin in essential tremor. Mov Disord 1998; 13:465–467.
- Bushara KO, Malik T, Exconde RE. The effect of levetiracetam on essential tremor. Neurology 2005; 64:1078–1080.
- Sullivan KL, Hauser RA, Zesiewicz TA. Levetiracetam for the treatment of essential tremor. Mov Disord 2005; 20:640.
- Huber SJ, Paulson GW. Efficacy of alprazolam for essential tremor. Neurology 1988; 38:241–243.
- McManis PG, Sharbrough FW. Orthostatic tremor: clinical and electrophysiologic characteristics. Muscle Nerve 1993; 16:1254–1260.
- Ceravolo R, Salvetti S, Piccini P, et al. Acute and chronic effects of clozapine in essential tremor. Mov Disord 1999; 14:468–472.
- Pakkenberg H, Pakkenberg B. Clozapine in the treatment of tremor. Acta Neurol Scand 1986; 73:295–297.
- Pact V, Giduz T. Mirtazapine treats resting tremor, essential tremor, and levodopa-induced dyskinesias. Neurology 1999; 53:1154.
- Lyons KE, Pahwa R. A double-blind, placebo-controlled, pilot study of mirtazapine in essential tremor. Presented at the 54th Annual Meeting of the American Academy of Neurology, Denver, Colorado. Neurology 2002; 58(suppl 3):A254.
- Brin MF, Lyons KE, Doucette J, et al. A randomized, double masked, controlled trial of botulinum toxin type A in essential hand tremor. Neurology 2001; 56:1523–1528.
- Blitzer A, Brin MF, Stewart C, et al. Abductor laryngeal dystonia: a series treated with botulinum toxin. Laryngoscope 1992; 102:163–167.
- Schuurman PR, Bosch DA, Bossuyt PM, et al. A comparison of continuous thalamic stimulation and thalamotomy for suppression of severe tremor. N Engl J Med 2000; 342:461–468.
- Flora ED, Perera CL, Cameron AL, et al. Deep brain stimulation for essential tremor: a systematic review. Mov Disord 2010; 25:1550–1559.
- Nagaseki Y, Shibazaki T, Hirai T, et al. Long-term follow-up results of selective VIM-thalamotomy. J Neurosurg 1986; 65:296–302.
- Zirh A, Reich SG, Dougherty PM, et al. Stereotactic thalamotomy in the treatment of essential tremor of the upper extremity: reassessment including a blinded measure of outcome. J Neurol Neurosurg Psychiatry 1999; 66:772–775.
KEY POINTS
- In addition to motor dysfunction, the tremor can also have a significant psychological impact on the patient, especially since it usually gets worse in social situations.
- Essential tremor is a clinical diagnosis. After a thorough review of the medical history and medication exposures, laboratory and imaging tests may be ordered to rule out a secondary cause.
- The two first-line agents in drug therapy for essential tremor are the nonselective beta-blocker propranolol (Inderal) and the antiepileptic primidone (Mysoline). They can be used alone or in combination.
- Botulinum toxin injection and deep brain stimulation are reserved for resistant tremor or for patients who do not tolerate drug therapy.
