CCJM delivers practical clinical articles relevant to internists, cardiologists, endocrinologists, and other specialists, all written by known experts.
A 69-year-old diabetic woman with stage 4 non–small-cell lung cancer presented with a 3-day history of abdominal pain and loss of appetite. She was being treated with corticosteroids for a brain metastasis.
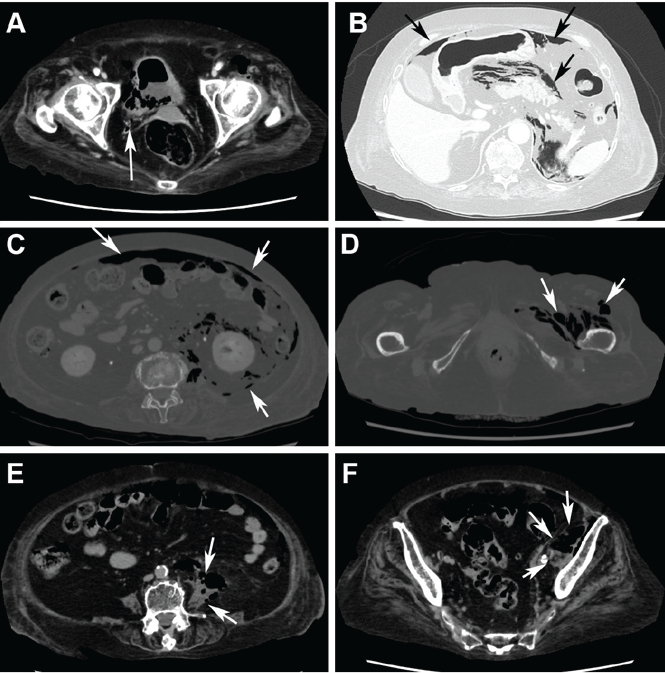
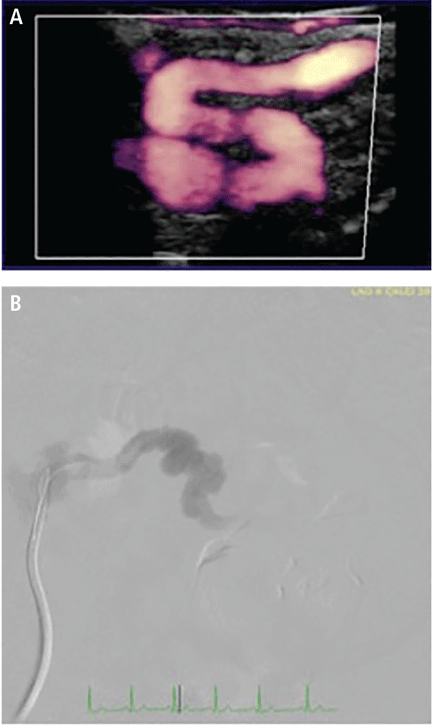
Computed tomography (CT) (Figure 1) revealed air within the bladder wall and lumen; diffuse air in the intraperitoneum and retroperitoneum; air distributed from the left iliopsoas muscle to the left femur that spread around the obturator muscle; air in the left ureter; and an abscess in the psoas major muscle extending to the ala of the ilium. A diagnosis of emphysematous cystitis complicated by extensive abdominal emphysema and abscess was made.
Figure 1. Abdominal computed tomography showed emphysematous lesions in the bladder wall and in several other organs in the abdomen, along with an abscess. (A) An image obtained with mediastinal-window settings showed multiple air bubbles with emphysematous changes in the right posterior wall of the urinary bladder (arrow). (B) An image obtained with lung-window settings showed intraperitoneal air (arrows). (C) An image obtained with bone settings showed intraperitoneal and retroperitoneal air (arrows). (D) An image obtained with bone settings showed air in the left femur that had spread around the obturator muscle (arrows). (E) An image obtained with mediastinal-window settings showed an abscess in the psoas major muscle with air (arrows), and another (F) showed the abscess extending to the ala of the ilium.
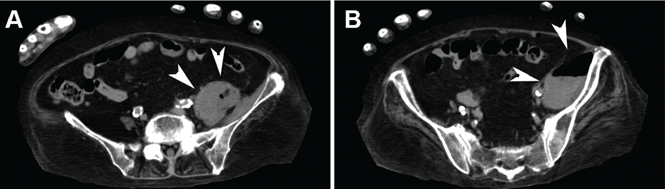
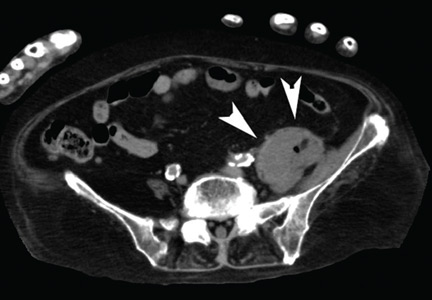
Blood cultures were negative, but urine cultures grew extended-spectrum beta-lactamase-producing Escherichia coli, which was sensitive to meropenem. Meropenem was given intravenously for 24 days and was stopped when levels of inflammatory markers improved and urine cultures were negative. However, on day 29, the patient developed a fever. Follow-up CT showed that the abscess in the psoas muscle had enlarged (Figure 2). We chose not to surgically drain the abscess because the patient had terminal lung cancer. The patient expired 6 days later, 35 days after her hospital admission.
Figure 2. After cessation of meropenem, computed tomography with mediastinal window settings showed enlargement of the abscess in the psoas major muscle (A, arrowheads) and in front of the ilium (B, arrowheads).
EMPHYSEMATOUS CYSTITIS ASSOCIATED WITH A PSOAS MUSCLE ABSCESS
Emphysematous cystitis is an uncommon urinary tract infection characterized by air within the bladder wall and lumen that is caused by gas-producing pathogens.1,2 The disease is often found in elderly diabetic women. Treatment of emphysematous cystitis typically includes intravenous antibiotics, adequate bladder drainage, and, for diabetic patients, appropriate glycemic control.
Psoas muscle abscess is a collection of pus in the retroperitoneal space.3 It can be primary, caused by hematogenous spread from the site of an occult infection, or secondary, caused by contiguous spread from adjacent infected organs, including those of the urinary tract. Psoas muscle abscess associated with emphysematous cystitis, as in our patient, is rare. We have seen only one other report in the medical literature.4
TREATMENT
The treatment of psoas muscle abscess involves the use of broad-spectrum antibiotics and drainage.5 Small abscesses (less than 3.5 cm) can be controlled with antibiotics alone. Image-guided percutaneous drainage is a safe, minimally invasive option. Surgery is indicated for unsuccessful percutaneous drainage, loculated abscesses, and abscesses difficult to approach percutaneously, or when the underlying disease requires definitive surgical management.
As in our patient, the presence of additional comorbid immunosuppressive conditions2 such as lung cancer and treatment with corticosteroids can allow the infection to become widespread and life-threatening.
References
Thomas AA, Lane BR, Thomas AZ, Remer EM, Campbell SC, Shoskes DA. Emphysematous cystitis: a review of 135 cases. BJU Int 2007; 100:17–20.
Grupper M, Kravtsov A, Potasman I. Emphysematous cystitis: illustrative case report and review of the literature. Medicine (Baltimore) 2007; 86:47–53.
Takayo Ota, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Masanobu Nakano, MSc Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Noriko Tanaka, MD Department of Radiology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Tomohiro Suzumura, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Nozomi Miyatake, BPharm Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Yoshikazu Hasegawa, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Tomhiro Ozaki, MD Department of Medical Oncology, Nara Hospital Kinki University Faculty of Medicine, Ikoma, Nara, Japan
Hiroshi Tsukuda, MD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Masahiro Fukuoka, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Address: Takayo Ota, MD, PhD, Department of Medical Oncology, Izumi Municipal Hospital, 4-10-10, Fuchu, Izumi, Osaka, 594-0071 Japan; takayo.ota@gmail.com
Takayo Ota, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Masanobu Nakano, MSc Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Noriko Tanaka, MD Department of Radiology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Tomohiro Suzumura, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Nozomi Miyatake, BPharm Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Yoshikazu Hasegawa, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Tomhiro Ozaki, MD Department of Medical Oncology, Nara Hospital Kinki University Faculty of Medicine, Ikoma, Nara, Japan
Hiroshi Tsukuda, MD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Masahiro Fukuoka, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Address: Takayo Ota, MD, PhD, Department of Medical Oncology, Izumi Municipal Hospital, 4-10-10, Fuchu, Izumi, Osaka, 594-0071 Japan; takayo.ota@gmail.com
Author and Disclosure Information
Takayo Ota, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Masanobu Nakano, MSc Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Noriko Tanaka, MD Department of Radiology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Tomohiro Suzumura, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Nozomi Miyatake, BPharm Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Yoshikazu Hasegawa, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Tomhiro Ozaki, MD Department of Medical Oncology, Nara Hospital Kinki University Faculty of Medicine, Ikoma, Nara, Japan
Hiroshi Tsukuda, MD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Masahiro Fukuoka, MD, PhD Department of Medical Oncology, Izumi Municipal Hospital, Izumi, Osaka, Japan
Address: Takayo Ota, MD, PhD, Department of Medical Oncology, Izumi Municipal Hospital, 4-10-10, Fuchu, Izumi, Osaka, 594-0071 Japan; takayo.ota@gmail.com
A 69-year-old diabetic woman with stage 4 non–small-cell lung cancer presented with a 3-day history of abdominal pain and loss of appetite. She was being treated with corticosteroids for a brain metastasis.
Computed tomography (CT) (Figure 1) revealed air within the bladder wall and lumen; diffuse air in the intraperitoneum and retroperitoneum; air distributed from the left iliopsoas muscle to the left femur that spread around the obturator muscle; air in the left ureter; and an abscess in the psoas major muscle extending to the ala of the ilium. A diagnosis of emphysematous cystitis complicated by extensive abdominal emphysema and abscess was made.
Figure 1. Abdominal computed tomography showed emphysematous lesions in the bladder wall and in several other organs in the abdomen, along with an abscess. (A) An image obtained with mediastinal-window settings showed multiple air bubbles with emphysematous changes in the right posterior wall of the urinary bladder (arrow). (B) An image obtained with lung-window settings showed intraperitoneal air (arrows). (C) An image obtained with bone settings showed intraperitoneal and retroperitoneal air (arrows). (D) An image obtained with bone settings showed air in the left femur that had spread around the obturator muscle (arrows). (E) An image obtained with mediastinal-window settings showed an abscess in the psoas major muscle with air (arrows), and another (F) showed the abscess extending to the ala of the ilium.
Blood cultures were negative, but urine cultures grew extended-spectrum beta-lactamase-producing Escherichia coli, which was sensitive to meropenem. Meropenem was given intravenously for 24 days and was stopped when levels of inflammatory markers improved and urine cultures were negative. However, on day 29, the patient developed a fever. Follow-up CT showed that the abscess in the psoas muscle had enlarged (Figure 2). We chose not to surgically drain the abscess because the patient had terminal lung cancer. The patient expired 6 days later, 35 days after her hospital admission.
Figure 2. After cessation of meropenem, computed tomography with mediastinal window settings showed enlargement of the abscess in the psoas major muscle (A, arrowheads) and in front of the ilium (B, arrowheads).
EMPHYSEMATOUS CYSTITIS ASSOCIATED WITH A PSOAS MUSCLE ABSCESS
Emphysematous cystitis is an uncommon urinary tract infection characterized by air within the bladder wall and lumen that is caused by gas-producing pathogens.1,2 The disease is often found in elderly diabetic women. Treatment of emphysematous cystitis typically includes intravenous antibiotics, adequate bladder drainage, and, for diabetic patients, appropriate glycemic control.
Psoas muscle abscess is a collection of pus in the retroperitoneal space.3 It can be primary, caused by hematogenous spread from the site of an occult infection, or secondary, caused by contiguous spread from adjacent infected organs, including those of the urinary tract. Psoas muscle abscess associated with emphysematous cystitis, as in our patient, is rare. We have seen only one other report in the medical literature.4
TREATMENT
The treatment of psoas muscle abscess involves the use of broad-spectrum antibiotics and drainage.5 Small abscesses (less than 3.5 cm) can be controlled with antibiotics alone. Image-guided percutaneous drainage is a safe, minimally invasive option. Surgery is indicated for unsuccessful percutaneous drainage, loculated abscesses, and abscesses difficult to approach percutaneously, or when the underlying disease requires definitive surgical management.
As in our patient, the presence of additional comorbid immunosuppressive conditions2 such as lung cancer and treatment with corticosteroids can allow the infection to become widespread and life-threatening.
A 69-year-old diabetic woman with stage 4 non–small-cell lung cancer presented with a 3-day history of abdominal pain and loss of appetite. She was being treated with corticosteroids for a brain metastasis.
Computed tomography (CT) (Figure 1) revealed air within the bladder wall and lumen; diffuse air in the intraperitoneum and retroperitoneum; air distributed from the left iliopsoas muscle to the left femur that spread around the obturator muscle; air in the left ureter; and an abscess in the psoas major muscle extending to the ala of the ilium. A diagnosis of emphysematous cystitis complicated by extensive abdominal emphysema and abscess was made.
Figure 1. Abdominal computed tomography showed emphysematous lesions in the bladder wall and in several other organs in the abdomen, along with an abscess. (A) An image obtained with mediastinal-window settings showed multiple air bubbles with emphysematous changes in the right posterior wall of the urinary bladder (arrow). (B) An image obtained with lung-window settings showed intraperitoneal air (arrows). (C) An image obtained with bone settings showed intraperitoneal and retroperitoneal air (arrows). (D) An image obtained with bone settings showed air in the left femur that had spread around the obturator muscle (arrows). (E) An image obtained with mediastinal-window settings showed an abscess in the psoas major muscle with air (arrows), and another (F) showed the abscess extending to the ala of the ilium.
Blood cultures were negative, but urine cultures grew extended-spectrum beta-lactamase-producing Escherichia coli, which was sensitive to meropenem. Meropenem was given intravenously for 24 days and was stopped when levels of inflammatory markers improved and urine cultures were negative. However, on day 29, the patient developed a fever. Follow-up CT showed that the abscess in the psoas muscle had enlarged (Figure 2). We chose not to surgically drain the abscess because the patient had terminal lung cancer. The patient expired 6 days later, 35 days after her hospital admission.
Figure 2. After cessation of meropenem, computed tomography with mediastinal window settings showed enlargement of the abscess in the psoas major muscle (A, arrowheads) and in front of the ilium (B, arrowheads).
EMPHYSEMATOUS CYSTITIS ASSOCIATED WITH A PSOAS MUSCLE ABSCESS
Emphysematous cystitis is an uncommon urinary tract infection characterized by air within the bladder wall and lumen that is caused by gas-producing pathogens.1,2 The disease is often found in elderly diabetic women. Treatment of emphysematous cystitis typically includes intravenous antibiotics, adequate bladder drainage, and, for diabetic patients, appropriate glycemic control.
Psoas muscle abscess is a collection of pus in the retroperitoneal space.3 It can be primary, caused by hematogenous spread from the site of an occult infection, or secondary, caused by contiguous spread from adjacent infected organs, including those of the urinary tract. Psoas muscle abscess associated with emphysematous cystitis, as in our patient, is rare. We have seen only one other report in the medical literature.4
TREATMENT
The treatment of psoas muscle abscess involves the use of broad-spectrum antibiotics and drainage.5 Small abscesses (less than 3.5 cm) can be controlled with antibiotics alone. Image-guided percutaneous drainage is a safe, minimally invasive option. Surgery is indicated for unsuccessful percutaneous drainage, loculated abscesses, and abscesses difficult to approach percutaneously, or when the underlying disease requires definitive surgical management.
As in our patient, the presence of additional comorbid immunosuppressive conditions2 such as lung cancer and treatment with corticosteroids can allow the infection to become widespread and life-threatening.
References
Thomas AA, Lane BR, Thomas AZ, Remer EM, Campbell SC, Shoskes DA. Emphysematous cystitis: a review of 135 cases. BJU Int 2007; 100:17–20.
Grupper M, Kravtsov A, Potasman I. Emphysematous cystitis: illustrative case report and review of the literature. Medicine (Baltimore) 2007; 86:47–53.
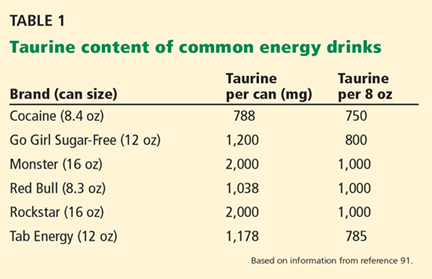
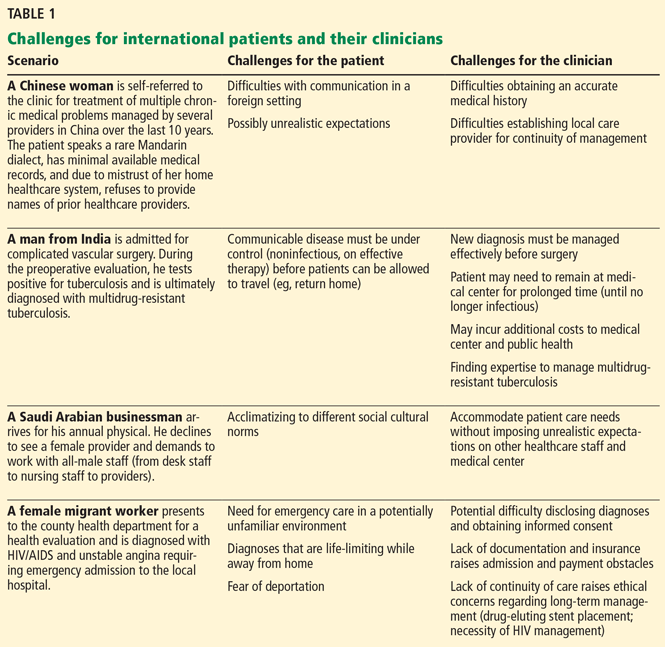
Taurine—an amino acid found in abundance in the human brain, retina, heart, and reproductive organs, as well as in meat and seafood—is also a major ingredient in “energy drinks” (Table 1).1,2 Given the tremendous popularity of these drinks in the United States, it would seem important to know and to recognize taurine’s neuroendocrine effects. Unfortunately, little is known about the effects of taurine supplementation in humans.
This paper reviews the sparse data to provide clinicians some background on the structure, synthesis, distribution, metabolism, mechanisms, effects, safety, and proposed therapeutic targets of taurine.
TAURINE’S THERAPEUTIC POTENTIAL
Taurine has been reported to have widespread anti-inflammatory actions.3,4 Taurine supplementation has been proposed to have beneficial effects in the treatment of epilepsy,5 heart failure,6,7 cystic fibrosis,8 and diabetes9 and has been shown in animal studies to protect against neurotoxic insults from alcohol, ammonia, lead, and other substances.10–16
In addition, taurine analogues such as homotaurine and N-acetyl-homotaurine (acamprosate) have been probed for possible therapeutic applications. Homotaurine has been shown to have antiamyloid activity that could in theory protect against the progression of Alzheimer disease,17 and acamprosate is approved by the US Food and Drug Administration (FDA) for the treatment of alcohol use disorders.18
TAURINE CONSUMPTION
Energy drinks are widely consumed in the United States, with an estimated 354 million gallons sold in 2009, or approximately 5.25 L/year per person over age 10.1 In 2012, US sales of energy drinks exceeded $12 billion,19 with young men, particularly those in the military deployed in war zones, being the biggest consumers.20–22 Analyses have found that of 49 nonalcoholic energy drinks tested, the average concentration of taurine was 3,180 mg/L, or approximately 750 mg per 8-oz serving.23,24 Popular brands include Red Bull, Monster, Rockstar (Table 1), NOS, Amp, and Full Throttle.
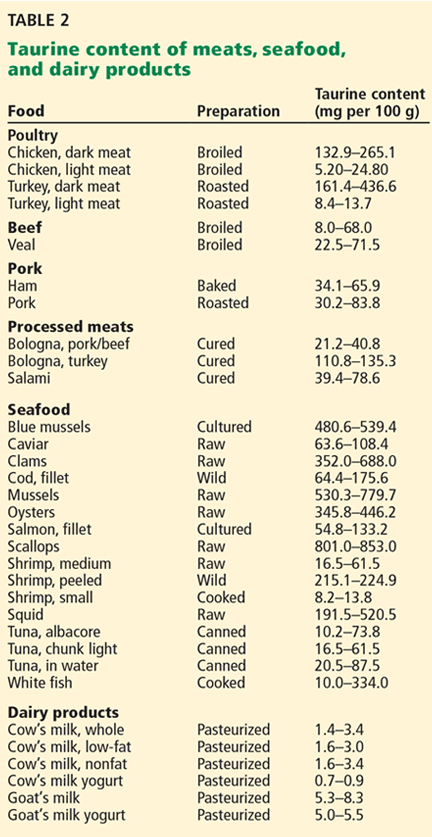
Taurine is plentiful in the human body, which contains up to 1 g of taurine per kg.25 Foods such as poultry, beef, pork, seafood, and processed meats have a high taurine content (Table 2).26–29 People who eat meat and seafood have plentiful taurine intake, whereas vegetarians and vegans consume much less and have significantly lower circulating levels30 because plants do not contain taurine in appreciable amounts.26,29
The typical American diet provides between 123 and 178 mg of taurine daily.26 Consumption of one 8-oz energy drink can increase the average intake 6 to 16 times. A lacto-ovo vegetarian diet provides only about 17 mg of taurine daily, and an 8-oz energy drink can increase the average intake by 44 to 117 mg.26 And since a vegan diet provides essentially no taurine,30 energy drink intake in any amount would constitute a major relative increase in taurine consumption.
ATTEMPTS TO STUDY TAURINE'S EFFECTS
Since most clinical trials to date have looked at the effects of taurine in combination with other ingredients such as caffeine, creatine, and glucose31–35 in drinks such as Red Bull, these studies cannot be used to determine the effects of taurine alone. In the few clinical trials that have tested isolated taurine consumption, data are not sufficient to make a conclusion on direct effects on energy metabolism.
Rutherford et al36 tested the effect of oral taurine supplementation (1,660 mg) on endurance in trained male cyclists 1 hour before exercise, but observed no effect on fluid intake, heart rate, subjective exertion, or time-trial performance. A small increase (16%) in total fat oxidation was observed during the 90-minute exercise period. Since mitochondria are the main location of fatty acid degradation, this effect may be attributed to taurine supplementation, with subsequent improvement in mitochondrial function.
Zhang et al37 found a 30-second increase in cycling energy capacity after 7 days of 6 g oral taurine supplementation, but the study was neither blinded nor placebo-controlled.
Kammerer et al38 tested the effect of 1 g of taurine supplementation on physical and mental performance in young adult soldiers 45 minutes before physical fitness and cognitive testing. This double-blind, placebo-controlled randomized trial found no effect of taurine on cardiorespiratory fitness indices, concentration, or immediate memory, nor did it find any effect of an 80-mg dose of caffeine.
In sum, the available data are far from sufficient to determine the direct effect of taurine consumption on energy metabolism in healthy people.
PHARMACOLOGY OF TAURINE
Chemical structure
Taurine, or 2-aminoethane sulfonic acid, is a conditionally essential amino acid, ie, we can usually make enough in our own bodies. It was first prepared on a large scale for physiologic investigation almost 90 years ago, through the purification of ox bile.39 It can be obtained either exogenously through dietary sources or endogenously through biosynthesis from methionine and cysteine precursors, both essential sulfur-containing alpha-amino acids.40 Both sources are important to maintain physiologic levels of taurine, and either can help compensate for the other in cases of deficiency.41
The structure of taurine has two main differences from the essential amino acids. First, taurine’s amino group is attached to the beta-carbon rather than the alpha-carbon, making it a beta-amino acid instead of an alpha-amino acid.42 Second, the acid group in taurine is sulfonic acid, whereas the essential amino acids have a carboxylic acid.43 Because of its distinctive structure, taurine is not used as a structural unit in proteins,43 existing mostly as a free amino acid within cells, readily positioned to perform several unique functions.
Synthesis
De novo synthesis of taurine involves several enzymes and at least five pathways,44 mostly differing by the order in which sulfur is oxidized and decarboxylated.45
The rate-limiting enzyme of the predominant pathway is thought to be cysteine sulfinate decarboxylase (CSD), and its presence within an organ indicates involvement in taurine production.44 CSD has been found in the liver,46 the primary site of taurine biosynthesis, as well as in the retina,47 brain,48 kidney,49 mammary glands,50,51 and reproductive organs.52
Distribution
Taurine levels are highest in electrically excitable tissues such as the central nervous system, retina, and heart; in secretory structures such as the pineal gland and the pituitary gland (including the posterior lobe or neurohypophysis); and in platelets25 and neutrophils.53
In the fetal brain, the taurine concentration is higher than that of any other amino acid,54 but the concentration in the brain decreases with advancing age, whereas glutamate levels increase over time to make it the predominant amino acid in the adult brain.54 Regardless, taurine is still the second most prevalent amino acid in the adult brain, its levels comparable to those of gamma-aminobutyric acid (GABA).55
Taurine has also been found in variable amounts in the liver, muscle, kidney, pancreas, spleen, small intestine, and lungs,56 as well as in several other locations.45,57
Taurine is also present in the male and female reproductive organs. In male rats, taurine and taurine biosynthesis have been localized to Leydig cells of the testes, the cellular source of testosterone in males, as well as the cremaster muscle, efferent ducts, and peritubular myoid cells surrounding seminiferous tubules.58 More recently, taurine has been detected in the testes of humans59 and is also found in sperm and seminal fluid.60 Levels of taurine in spermatozoa are correlated with sperm quality, presumably by protecting against lipid peroxidation through taurine’s antioxidant effects,61,62 as well as through contribution to the spermatozoa maturation process by facilitating the capacitation, motility, and acrosomal reaction of sperm.63
In female rats, taurine has been found in uterine tissue,64 oviducts,65 uterine fluid (where it is the predominant amino acid),66 and thecal cells of developing follicles of ovaries, cells responsible for the synthesis of androgens such as testosterone and androstenedione.65 Taurine is also a major component of human breast milk67 and is important for proper neonatal nutrition.68
Metabolism and excretion
Ninety-five percent of taurine is excreted in urine, about 70% as taurine itself, and the rest as sulfate. Most of the sulfate derived from taurine is produced by bacterial metabolism in the gut and then absorbed.69 However, taurine can also be conjugated with bile acids to act as a detergent in lipid emulsification.70 In this form, it may be subjected to the enterohepatic circulation, which gives bacteria another chance to convert it into inorganic sulfate for excretion in urine.69
MECHANISMS AND NEUROENDOCRINE EFFECTS
As a free amino acid, taurine has widespread distribution and unique biochemical and physiologic properties and exhibits several organ-specific functions; however, indisputable evidence of a taurine-specific receptor is lacking, and its putative existence71 is controversial.72 Nonetheless, taurine is a neuromodulator with a variety of actions.
Neurotransmission
Taurine is known to be an inhibitory neurotransmitter and neuromodulator.73 It is structurally analogous to GABA, the main inhibitory neurotransmitter in the brain.45 Accordingly, it binds to GABA receptors to serve as an agonist,74,75 causing neuronal hyperpolarization and inhibition. Taurine has an even higher affinity for glycine receptors75 where it has long been known to act as an agonist.76 GABA and glycine receptors both belong to the Cys-loop receptor superfamily,77 with conservation of subunits that allows taurine to bind each receptor, albeit at different affinities. The binding effects of taurine on GABA and glycine receptors have not been well documented quantitatively; however, it is known that taurine has a substantially lower affinity than GABA and glycine for their respective receptors.76
Catecholamines and the sympathetic nervous system
Surprisingly little is known about the effects of taurine on norepinephrine, dopamine, and the human sympathetic nervous system.78 Humans with borderline hypertension given 6 g of taurine orally for 7 days79 experienced decreases in epinephrine secretion and blood pressure, but normotensive study participants did not experience similar results, possibly because of a better ability to regulate sympathetic tone. Mizushima et al80 showed that a longer period of taurine intake (6 g orally for 3 weeks) could elicit a decrease in norepinephrine in healthy men with normal blood pressure. Other similar studies81–83 also suggested interplay between taurine and catecholamines, but the extent is still undetermined.
Growth hormone, prolactin, sex hormones, and cortisol
Taurine appears to have a complex relationship with several hormones, although its direct effects on hormone secretion remain obscure. Clinical studies of the acute and chronic neuroendocrine effects of taurine loading in humans are needed.
In female rats, secretion of prolactin is increased by the intraventricular injection of 5 μL of 2.0 μmol taurine over a 10-minute period.84 Ikuyama et al85 found an increase in prolactin and growth hormone secretion in adult male rats given 10 μL of 0.25 μmol and 1.0 μmol taurine intraventricularly, yet a higher dose of 4.0 μmol had no effect on either hormone. Furthermore, prolactin receptor deficiency is seen in CSD knockout mice, but the receptor is restored with taurine supplementation.86
Mantovani and DeVivo87 reported that 375 to 8,000 mg/day of taurine given orally for 4 to 6 months to epileptic patients stimulated the secretion of growth hormone. However, in another study, a single 75-mg/kg dose of oral taurine did not trigger an acute increase in levels of growth hormone or prolactin in humans.88 Energy drinks may contain up to 1,000 mg of taurine per 8-oz serving, but the effects of larger doses on growth hormone, which is banned as a supplement by major athletic organizations because of its anabolic and possible performance-enhancing effects, remain to be determined.
Taurine may have effects on human sex hormones, based on the limited observations in rodents.89–94
Although human salivary cortisol concentrations were purportedly assessed in response to 2,000 mg of oral taurine,95 the methods and reported data are not adequate to draw any conclusions.
Energy metabolism
Mammals are unable to directly use taurine in energy production because they cannot directly reduce it.25 Instead, bacteria in the gut use it as a source of energy, carbon, nitrogen, and sulfur.96 However, taurine deficiency appears to impair the cellular respiratory chain, resulting in diminished production of adenosine triphosphate and diminished uptake of long-chain fatty acids by mitochondria, at least in the heart.97
Taurine is present in human mitochondria and regulates mitochondrial function. For example, taurine in mitochondria assists in conjugation of transfer RNA for leucine, lysine, glutamate, and glutamine.98 In TauT knockout mice, deficiency of taurine causes mitochondrial dysfunction, triggering a greater than 80% decrease in exercise capacity.99 Several studies in rodents have shown increased exercise capacity after taurine supplementation.100–102 In addition, taurine is critical for the growth of blastocytes, skeletal muscle, and myocardium; it is necessary for mitochondrial development and is also important for muscular endurance.103,104
Antioxidation, anti-inflammation, and other functions
Taurine is a major antioxidant, scavenging reactive oxygen and protecting against oxidative stress to organs including the brain,97,105,106 where it increasingly appears to have neuroprotective effects.107,108
Cellular taurine also has anti-inflammatory actions.3 One of the proposed mechanisms is taurine inhibition of NF-kappa B, an important transcription factor for the synthesis of pro-inflammatory cytokines.4 This function may be important in protecting polyunsaturated fatty acids from oxidative stress—helping to maintain and stabilize the structure and function of plasma membranes within the lungs,109 heart,110 brain,111 liver,112 and spermatozoa.61,62
Taurine is also conjugated to bile acids synthesized in the liver, forming bile salts70 that act as detergents to help emulsify and digest lipids in the body. In addition, taurine facilitates xenobiotic detoxification in the liver by conjugating with several drugs to aid in their excretion.25 Taurine is also implicated in calcium modulation113 and homeostasis.114 Through inhibition of several types of calcium channels, taurine has been shown to decrease calcium influx into cells, effectively serving a cytoprotective role against calcium overload.115,116
TAURINE DEFICIENCY
Fetal and neonatal deficiency
Though taurine is considered nonessential in adults because it can be readily synthesized endogenously, it is thought to be conditionally essential in neonatal nutrition.68 It is the second most abundant free amino acid in human breast milk117 and the most abundant free amino acid in fetal brain.118 In cases of long-term parenteral nutrition, neonates can become drastically taurine deficient119 due to suboptimal CSD activity,118 leading to retinal dysfunction.41 Taurine deficiencies can lead to functional and structural brain damage.118 Moreover, maternal taurine deficiency results in neurologic abnormalities in offspring120 and may lead to oxidative stress throughout life.121
In 1984, the FDA approved the inclusion of taurine in infant formulas based on research showing that taurine-deficient infants had impaired fat absorption, bile acid secretion, retinal function, and hepatic function.122 But still under debate are the amount and duration of taurine supplementation required by preterm and low-birth-weight infants, as several randomized controlled trials failed to show statistically significant effects on growth.123 Nonetheless, given the alleged detrimental ramifications of a lack of taurine supplementation, as well as the ethical dilemma of performing additional research trials on infants, it is presumed that infant formulas and parenteral nutrition for preterm and low-birth-weight infants will continue to contain taurine.
Age- and disease-related deficiency
Although taurine deficiency is rare in neonates, it is perhaps inevitable with advancing age. Healthy elderly patients ages 61 to 81 have up to a 49% decrease in plasma taurine concentration compared with healthy individuals ages 27 to 57.124 While reduced renal retention125 and taurine intake126 can account for depressed taurine levels, Eppler and Dawson127 found that tissue and circulating taurine concentrations decrease over the human life span primarily due to an age-dependent depletion of CSD activity in the liver. This effectively impairs the biosynthesis of endogenous taurine from cysteine or methionine or both, forcing a greater reliance on exogenous sources.
While specific mechanisms have not been fully elucidated, taurine deficiency has also been identified in patients suffering from diseases including but not limited to disorders of bone (osteogenesis imperfecta, osteoporosis),128 blood (acute myelogenous leukemia),129 central nervous system (schizophrenia, Friedreich ataxia-spinocerebellar degeneration),130,131 retina (retinitis pigmentosa),132 circulatory system and heart (essential hypertension, atherosclerosis),133 digestion (Gaucher disease),134 absorption (short-bowel syndrome),135 cellular proliferation (cancer),136 and membrane channels (cystic fibrosis),137 as well as in patients restricted to long-term parenteral nutrition.138 However, the apparent correlation between taurine deficiency and these conditions does not necessarily mean causation; more study is needed to elucidate a direct connection.
SAFETY AND TOXICITY OF TAURINE SUPPLEMENTATION
An upper safe level of intake for taurine has not been established. To date, several studies have involved heavy taurine supplementation without serious adverse effects. While the largest dosage of taurine tested in humans appears to be 10 g/day for 6 months,139 a number of studies have used 1 to 6 g/day for periods of 1 week to 1 year.140 However, the assessment of potential acute, subacute, and chronic adverse effects has not been comprehensive. The Scientific Committee on Food of the European Commission141 reviewed several toxicologic studies on taurine through 2003 and were unable to expose any carcinogenic or teratogenic potential. Nevertheless, based on the available data from trials in humans and lower animals, Shao and Hathcock140 suggested an observed safe level of taurine of 3 g/day, a conservatively smaller dose that carries a higher level of confidence. Because there is no “observed adverse effect level” for daily taurine intake,141 more research must be done to ensure safety of higher amounts of taurine administration and to define a tolerable upper limit of intake.
Interactions with medications
To date, the literature is scarce regarding potential interactions between taurine and commonly used medications.
Although no evidence specifically links taurine with adverse effects when used concurrently with other medications, there may be a link between taurine supplementation and various cytochrome P450 systems responsible for hepatic drug metabolism. Specifically, taurine inhibits cytochrome P450 2E1, a highly conserved xenobiotic-metabolizing P450 responsible for the breakdown of more than 70 substrates, including several commonly used anesthetics, analgesics, antidepressants, antibacterials, and antiepileptics.142 Of note, taurine may contribute to the attenuation of oxidative stress in the liver in the presence of alcohol143 and acetaminophen,144 two substances frequently used and abused. Since the P450 2E1 system catalyzes comparable reactions in rodents and humans,142 rodents should plausibly serve as a model for further testing of the effects of taurine on various substrates.
POTENTIAL THERAPEUTIC APPLICATIONS
More analysis is needed to fully unlock and understand taurine’s potential value in healthcare.
Correction of late-life taurine decline in humans could be beneficial for cognitive performance, energy metabolism, sexual function, and vision, but clinical studies remain to be performed. While a decline in taurine with age may intensify the stress caused by reactive oxygen species, taurine supplementation has been shown to decrease the presence of oxidative markers127 and to serve a neuroprotective role in rodents.145,146 Taurine levels increase in the hippocampus after experimentally induced gliosis147 and are neuroprotective against glutamate excitotoxicity.148,149 Furthermore, data in Alzheimer disease, Huntington disease, and brain ischemia experimental models show that taurine inhibits neuronal death (apoptosis).13,150,151 Taurine has even been proposed as a potential preventive treatment for Alzheimer dementia, as it stabilizes protein conformations to prevent their aggregation and subsequent dysfunction.152 Although improvement in memory and cognitive performance has been linked to taurine supplementation in old mice,145,153 similar results have not been found in adult mice whose taurine levels are within normal limits. Taurine also has transient anticonvulsant effects in some epileptic humans.154
Within the male reproductive organs, the age-related decline in taurine may or may not have implications regarding sexuality, as only very limited rat data are available.89–91
In cats, taurine supplementation has been found to prevent the progressive degeneration of retinal photoreceptors seen in retinitis pigmentosa, a genetic disease that causes the loss of vision.155
While several energy drink companies have advertised that taurine plays a role in improving cognitive and physical performance, there are few human studies that examine this contention in the absence of confounding factors such as caffeine or glucose.36,37,95 Taurine supplementation in patients with heart failure has been shown to increase exercise capacity vs placebo.156 This supports the idea that in cases of taurine deficiency, such as those seen in cardiomyopathy,157 taurine supplementation could have restorative effects. However, we are not aware of any double-blind, placebo-controlled clinical trial of taurine alone in healthy patients that measured energy parameters as clinical outcomes.
Although it remains possible that acute supraphysiologic taurine levels achieved by supplementation could transiently trigger various psychoneuroendocrine responses in healthy people, clinical research is needed in which taurine is the sole intervention. At present, the most compelling clinical reason to prescribe or recommend taurine supplementation is taurine deficiency.
McLellan TM, Lieberman HR. Do energy drinks contain active components other than caffeine? Nutr Rev 2012; 70:730–744.
Park E, Quinn MR, Wright CE, Schuller-Levis G. Taurine chloramine inhibits the synthesis of nitric oxide and the release of tumor necrosis factor in activated RAW 264.7 cells. J Leukoc Biol 1993; 54:119–124.
Kontny E, Szczepanska K, Kowalczewski J, et al. The mechanism of taurine chloramine inhibition of cytokine (interleukin-6, interleukin-8) production by rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheum 2000; 43:2169–2177.
Barbeau A, Inoue N, Tsukada Y, Butterworth RF. The neuropharmacology of taurine. Life Sci 1975; 17:669–677.
Kramer JH, Chovan JP, Schaffer SW. Effect of taurine on calcium paradox and ischemic heart failure. Am J Physiol 1981; 240:H238–H246.
Azuma J, Sawamura A, Awata N, et al. Therapeutic effect of taurine in congestive heart failure: a double-blind crossover trial. Clin Cardiol 1985; 8:276–282.
Darling PB, Lepage G, Leroy C, Masson P, Roy CC. Effect of taurine supplements on fat absorption in cystic fibrosis. Pediatr Res 1985; 19:578–582.
Franconi F, Di Leo MA, Bennardini F, Ghirlanda G. Is taurine beneficial in reducing risk factors for diabetes mellitus? Neurochem Res 2004; 29:143–150.
Malcangio M, Bartolini A, Ghelardini C, et al. Effect of ICV taurine on the impairment of learning, convulsions and death caused by hypoxia. Psychopharmacology (Berl) 1989; 98:316–320.
Rivas-Arancibia S, Dorado-Martínez C, Borgonio-Pérez G, et al. Effects of taurine on ozone-induced memory deficits and lipid peroxidation levels in brains of young, mature, and old rats. Environ Res 2000; 82:7–17.
Vohra BP, Hui X. Improvement of impaired memory in mice by taurine. Neural Plast 2000; 7:245–259.
Tadros MG, Khalifa AE, Abdel-Naim AB, Arafa HM. Neuroprotective effect of taurine in 3-nitropropionic acid-induced experimental animal model of Huntington’s disease phenotype. Pharmacol Biochem Behav 2005; 82:574–582.
Zhu DM, Wang M, She JQ, Yu K, Ruan DY. Protection by a taurine supplemented diet from lead-induced deficits of long-term potentiation/depotentiation in dentate gyrus of rats in vivo. Neuroscience 2005; 134:215–224.
Chepkova AN, Sergeeva OA, Haas HL. Taurine rescues hippocampal long-term potentiation from ammonia-induced impairment. Neurobiol Dis 2006; 23:512–521.
Wang GH, Jiang ZL, Fan XJ, Zhang L, Li X, Ke KF. Neuroprotective effect of taurine against focal cerebral ischemia in rats possibly mediated by activation of both GABAA and glycine receptors. Neuropharmacology 2007; 52:1199–1209.
Caltagirone C, Ferrannini L, Marchionni N, Nappi G, Scapagnini G, Trabucchi M. The potential protective effect of tramiprosate (homotaurine) against Alzheimer’s disease: a review. Aging Clin Exp Res 2012; 24:580–587.
Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA 2014; 311:1889–1900.
Sorkin BC, Camp KM, Haggans CJ, et al. Executive summary of NIH workshop on the use and biology of energy drinks: current knowledge and critical gaps. Nutr Rev 2014; 72(suppl 1):1–8.
Centers for Disease Control and Prevention (CDC). Energy drink consumption and its association with sleep problems among US service members on a combat deployment—Afghanistan, 2010. MMWR Morb Mortal Wkly Rep 2012; 61:895–898.
Bailey RL, Saldanha LG, Dwyer JT. Estimating caffeine intake from energy drinks and dietary supplements in the United States. Nutr Rev 2014; 72(suppl 1):9–13.
Stephens MB, Attipoe S, Jones D, Ledford CJ, Deuster PA. Energy drink and energy shot use in the military. Nutr Rev 2014; 72(suppl 1):72–77.
Triebel S, Sproll C, Reusch H, Godelmann R, Lachenmeier DW. Rapid analysis of taurine in energy drinks using amino acid analyzer and Fourier transform infrared (FTIR) spectroscopy as basis for toxicological evaluation. Amino Acids 2007; 33:451–457.
Huxtable RJ. Physiological actions of taurine. Physiol Rev 1992; 72:101–163.
Laidlaw SA, Grosvenor M, Kopple JD. The taurine content of common foodstuffs. JPEN J Parenter Enteral Nutr 1990; 14:183–188.
Manzi P, Pizzoferrato L. Taurine in milk and yoghurt marketed in Italy. Int J Food Sci Nutr 2013; 64:112–116.
Dragnes BT, Larsen R, Ernstsen MH, Mæhre H, Elvevoll EO. Impact of processing on the taurine content in processed seafood and their corresponding unprocessed raw materials. Int J Food Sci Nutr 2009; 60:143–152.
Laidlaw SA, Shultz TD, Cecchino JT, Kopple JD. Plasma and urine taurine levels in vegans. Am J Clin Nutr 1988; 47:660–663.
Rana SK, Sanders TA. Taurine concentrations in the diet, plasma, urine and breast milk of vegans compared with omnivores. Br J Nutr 1986; 56:17–27.
Alford C, Cox H, Wescott R. The effects of Red Bull energy drink on human performance and mood. Amino Acids 2001; 21:139–150.
Hoffman JR, Ratamess NA, Ross R, Shanklin M, Kang J, Faigenbaum AD. Effect of a pre-exercise energy supplement on the acute hormonal response to resistance exercise. J Strength Cond Res 2008; 22:874–882.
Ivy JL, Kammer L, Ding Z, et al. Improved cycling time-trial performance after ingestion of a caffeine energy drink. Int J Sport Nutr Exerc Metab 2009; 19:61–78.
Gonzalez AM, Walsh AL, Ratamess NA, Kang J, Hoffman JR. Effect of a pre-workout energy supplement on acute multi-joint resistance exercise. J Sports Sci Med 2011; 10:261–266.
Yeh TS, Chan KH, Hsu MC, Liu JF. Supplementation with soybean peptides, taurine, pueraria isoflavone, and ginseng saponin complex improves endurance exercise capacity in humans. J Med Food 2011; 14:219–225.
Rutherford JA, Spriet LL, Stellingwerff T. The effect of acute taurine ingestion on endurance performance and metabolism in well-trained cyclists. Int J Sport Nutr Exerc Metab 2010; 20:322–329.
Zhang M, Izumi I, Kagamimori S, et al. Role of taurine supplementation to prevent exercise-induced oxidative stress in healthy young men. Amino Acids 2004; 26:203–207.
Kammerer M, Jaramillo JA, García A, Calderón JC, Valbuena LH. Effects of energy drink major bioactive compounds on the performance of young adults in fitness and cognitive tests: a randomized controlled trial. J Int Soc Sports Nutr 2014; 11:44.
Kermack WO, Slater RH. The preparation of taurine in considerable quantity. Biochem J 1927; 21:1065–1067.
Huxtable RJ, Lippincott SE. Diet and biosynthesis as sources of taurine in the mouse. J Nutr 1982; 112:1003–1010.
Ohkuma S, Tamura J, Kuriyama K. Roles of endogenous and exogenous taurine and glycine in the formation of conjugated bile acids: analyses using freshly isolated and primary cultured rat hepatocytes. Jpn J Pharmacol 1984; 35:347–358.
Chapman GE, Greenwood CE. Taurine in nutrition and brain development. Nutrition Research 1988; 8:955–968.
Andrews S, Schmidt CLA. Titration curves of taurine and cysteic acid. J Biol Chem 1927; 73:651–654.
Huxtable RJ. Taurine in the central nervous system and the mammalian actions of taurine. Prog Neurobiol 1989; 32:471–533.
Jacobsen JG, Smith LH. Biochemistry and physiology of taurine and taurine derivatives. Physiol Rev 1968; 48:424–511.
Hope DB. Pyridoxal phosphate as the coenzyme of the mammalian decarboxylase for L-cysteine sulphinic and L-cysteic acids. Biochem J 1955; 59:497–500.
Pasantes-Morales H, Lopez-Colome AM, Salceda R, Mandel P. Cysteine sulphinate decarboxylase in chick and rat retina during development. J Neurochem 1976; 27:1103–1106.
Oertel WH, Schmechel DE, Weise VK, et al. Comparison of cysteine sulphinic acid decarboxylase isoenzymes and glutamic acid decarboxylase in rat liver and brain. Neuroscience 1981; 6:2701–2714.
Reymond I, Bitoun M, Levillain O, Tappaz M. Regional expression and histological localization of cysteine sulfinate decarboxylase mRNA in the rat kidney. J Histochem Cytochem 2000; 48:1461–1468.
Hu JM, Ikemura R, Chang KT, Suzuki M, Nishihara M, Takahashi M. Expression of cysteine sulfinate decarboxylase mRNA in rat mammary gland. J Vet Med Sci 2000; 62:829–834.
Ueki I, Stipanuk MH. Enzymes of the taurine biosynthetic pathway are expressed in rat mammary gland. J Nutr 2007; 137:1887–1894.
Li JH, Ling YQ, Fan JJ, Zhang XP, Cui S. Expression of cysteine sulfinate decarboxylase (CSD) in male reproductive organs of mice. Histochem Cell Biol 2006; 125:607–613.
Green TR, Fellman JH, Eicher AL, Pratt KL. Antioxidant role and subcellular location of hypotaurine and taurine in human neutrophils. Biochim Biophys Acta 1991; 1073:91–97.
Lefauconnier JM, Portemer C, Chatagner F. Free amino acids and related substances in human glial tumours and in fetal brain: comparison with normal adult brain. Brain Res 1976; 117:105–113.
Sturman JA, Rassin DK, Gaull GE. Taurine in development. Life Sci 1977; 21:1–22.
Sturman JA. Taurine pool sizes in the rat: effects of vitamin B-6 deficiency and high taurine diet. J Nutr 1973; 103:1566–1580.
Terauchi A, Nakazaw A, Johkura K, Yan L, Usuda N. Immunohistochemical localization of taurine in various tissues of the mouse. Amino Acids 1998; 15:151–160.
Lobo MV, Alonso FJ, del Río RM. Immunohistochemical localization of taurine in the male reproductive organs of the rat. J Histochem Cytochem 2000; 48:313–320.
Aaronson DS, Iman R, Walsh TJ, Kurhanewicz J, Turek PJ. A novel application of 1H magnetic resonance spectroscopy: non-invasive identification of spermatogenesis in men with non-obstructive azoospermia. Hum Reprod 2010; 25:847–852.
Holmes RP, Goodman HO, Shihabi ZK, Jarow JP. The taurine and hypotaurine content of human semen. J Androl 1992; 13:289–292.
Alvarez JG, Storey BT. Taurine, hypotaurine, epinephrine and albumin inhibit lipid peroxidation in rabbit spermatozoa and protect against loss of motility. Biol Reprod 1983; 29:548–555.
Das J, Ghosh J, Manna P, Sinha M, Sil PC. Taurine protects rat testes against NaAsO(2)-induced oxidative stress and apoptosis via mitochondrial dependent and independent pathways. Toxicol Lett 2009; 187:201–210.
Mrsny RJ, Meizel S. Inhibition of hamster sperm Na+, K+-ATPase activity by taurine and hypotaurine. Life Sci 1985; 36:271–275.
Phoenix J, Wray S. Changes in human and rat uterine phosphoethanolamine and taurine with pregnancy and parturition. Exp Physiol 1994; 79:601–604.
Lobo MV, Alonso FJ, Latorre A, del Río RM. Immunohistochemical localization of taurine in the rat ovary, oviduct, and uterus. J Histochem Cytochem 2001; 49:1133–1142.
Casslén BG. Free amino acids in human uterine fluid. Possible role of high taurine concentration. J Reprod Med 1987; 32:181–184.
Pamblanco M, Portolés M, Paredes C, Ten A, Comín J. Free amino acids in preterm and term milk from mothers delivering appropriate- or small-for-gestational-age infants. Am J Clin Nutr 1989; 50:778–781.
Rigo J, Senterre J. Is taurine essential for the neonates? Biol Neonate 1977; 32:73–76.
Sturman JA, Hepner GW, Hofmann AF, Thomas PJ. Metabolism of [35S]taurine in man. J Nutr 1975; 105:1206–1214.
Bremer J. Species differences in the conjugation of free bile acids with taurine and glycine. Biochem J 1956; 63:507–513.
Wu JY, Tang XW, Tsai WH. Taurine receptor: kinetic analysis and pharmacological studies. Adv Exp Med Biol 1992; 315:263–268.
Ripps H, Shen W. Review: taurine: a “very essential” amino acid. Mol Vis 2012; 18:2673–2686.
Haas HL, Hösli L. The depression of brain stem neurones by taurine and its interaction with strychnine and bicuculline. Brain Res 1973; 52:399–402.
Bureau MH, Olsen RW. Taurine acts on a subclass of GABAA receptors in mammalian brain in vitro. Eur J Pharmacol 1991; 207:9–16.
Bhattarai JP, Park SJ, Chun SW, Cho DH, Han SK. Activation of synaptic and extrasynaptic glycine receptors by taurine in preoptic hypothalamic neurons. Neurosci Lett 2015; 608:51–56.
Horikoshi T, Asanuma A, Yanagisawa K, Anzai K, Goto S. Taurine and beta-alanine act on both GABA and glycine receptors in Xenopus oocyte injected with mouse brain messenger RNA. Brain Res 1988; 464:97–105.
Miller PS, Smart TG. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci 2010; 31:161–174.
Fujita T, Sato Y. Hypotensive effect of taurine. Possible involvement of the sympathetic nervous system and endogenous opiates. J Clin Invest 1988; 82:993–997.
Fujita T, Ando K, Noda H, Ito Y, Sato Y. Effects of increased adrenomedullary activity and taurine in young patients with borderline hypertension. Circulation 1987; 75:525–532.
Mizushima S, Nara Y, Sawamura M, Yamori Y. Effects of oral taurine supplementation on lipids and sympathetic nerve tone. Adv Exp Med Biol 1996; 403:615–622.
Fujita T, Sato Y. The antihypertensive effect of taurine in DOCA-salt rats. J Hypertens Suppl 1984; 2:S563–S565.
Fujita T, Sato Y, Ando K. Changes in cardiac and hypothalamic noradrenergic activity with taurine in DOCA-salt rats. Am J Physiol 1986; 251:H926–H933.
Sato Y, Ando K, Fujita T. Role of sympathetic nervous system in hypotensive action of taurine in DOCA-salt rats. Hypertension 1987; 9:81–87.
Scheibel J, Elsasser T, Ondo JG. A neuromodulatory role for taurine in controlling prolactin secretion in female rats. Psychoneuroendocrinology 1981; 6:139–144.
Ikuyama S, Okajima T, Kato K, Ibayashi H. Effect of taurine on growth hormone and prolactin secretion in rats: possible interaction with opioid peptidergic system. Life Sci 1988; 43:807–812.
Park E, Park SY, Dobkin C, Schuller-Levis G. Development of a novel cysteine sulfinic acid decarboxylase knockout mouse: dietary taurine reduces neonatal mortality. J Amino Acids 2014; 2014:346809.
Mantovani J, DeVivo DC. Effects of taurine on seizures and growth hormone release in epileptic patients. Arch Neurol 1979; 36:672–674.
Carlson HE, Miglietta JT, Roginsky MS, Stegink LD. Stimulation of pituitary hormone secretion by neurotransmitter amino acids in humans. Metabolism 1989; 38:1179–1182.
Yang J, Wu G, Feng Y, Lv Q, Lin S, Hu J. Effects of taurine on male reproduction in rats of different ages. J Biomed Sci 2010; 17(suppl 1):S9.
Yang J, Lin S, Feng Y, Wu G, Hu J. Taurine enhances the sexual response and mating ability in aged male rats. Adv Exp Med Biol 2013; 776:347–355.
Yang J, Zong X, Wu G, Lin S, Feng Y, Hu J. Taurine increases testicular function in aged rats by inhibiting oxidative stress and apoptosis. Amino Acids 2015; 47:1549–1558.
Scheibel J, Elsasser T, Ondo JG. Stimulation of LH and FSH secretion following intraventricular injection of cysteic acid but not taurine. Brain Res 1980; 201:99–106.
Price MT, Olney JW, Mitchell MV, Fuller T, Cicero TJ. Luteinizing hormone releasing action of N-methyl aspartate is blocked by GABA or taurine but not by dopamine antagonists. Brain Res 1978; 158:461–465.
Ma Q, Zhao J, Cao W, Liu J, Cui S. Estradiol decreases taurine level by reducing cysteine sulfinic acid decarboxylase via the estrogen receptor-a in female mice liver. Am J Physiol Gastrointest Liver Physiol 2015; 308:G277–G286.
Giles GE, Mahoney CR, Brunyé TT, Gardony AL, Taylor HA, Kanarek RB. Differential cognitive effects of energy drink ingredients: caffeine, taurine, and glucose. Pharmacol Biochem Behav 2012; 102:569–577.
Giehl TJ, Qoronfleh MW, Wilkinson BJ. Transport, nutritional and metabolic studies of taurine in staphylococci. J Gen Microbiol 1987; 133:849–856.
Schaffer SW, Shimada-Takaura K, Jong CJ, Ito T, Takahashi K. Impaired energy metabolism of the taurine-deficient heart. Amino Acids 2015 Oct 16. Epub ahead of print.
Schaffer SW, Jong CJ, Ito T, Azuma J. Role of taurine in the pathologies of MELAS and MERRF. Amino Acids 2014; 46:47–56.
Warskulat U, Heller-Stilb B, Oermann E, et al. Phenotype of the taurine transporter knockout mouse. Methods Enzymol 2007; 428:439–458.
Yatabe Y, Miyakawa S, Miyazaki T, Matsuzaki Y, Ochiai N. Effects of taurine administration in rat skeletal muscles on exercise. J Orthop Sci 2003; 8:415–419.
Miyazaki T, Matsuzaki Y, Ikegami T, et al. Optimal and effective oral dose of taurine to prolong exercise performance in rat. Amino Acids 2004; 27:291–298.
Goodman CA, Horvath D, Stathis C, et al. Taurine supplementation increases skeletal muscle force production and protects muscle function during and after high-frequency in vitro stimulation. J Appl Physiol (1985) 2009; 107:144–154.
Van Winkle LJ, Patel M, Wasserlauf HG, Dickinson HR, Campione AL. Osmotic regulation of taurine transport via system beta and novel processes in mouse preimplantation conceptuses. Biochim Biophys Acta 1994; 1191:244–255.
Ito T, Kimura Y, Uozumi Y, et al. Taurine depletion caused by knocking out the taurine transporter gene leads to cardiomyopathy with cardiac atrophy. J Mol Cell Cardiol 2008; 44:927–937.
Aydın AF, Çoban J, Dogan-Ekici I, Betül-Kalaz E, Dogru-Abbasoglu S, Uysal M. Carnosine and taurine treatments diminished brain oxidative stress and apoptosis in D-galactose aging model. Metab Brain Dis 2016; 31:337–345.
Nagai K, Fukuno S, Oda A, Konishi H. Protective effects of taurine on doxorubicin-induced acute hepatotoxicity through suppression of oxidative stress and apoptotic responses. Anticancer Drugs 2016; 27:17–23.
Caletti G, Almeida FB, Agnes G, Nin MS, Barros HM, Gomez R. Antidepressant dose of taurine increases mRNA expression of GABAA receptor a2 subunit and BDNF in the hippocampus of diabetic rats. Behav Brain Res 2015; 283:11–55.
Gu Y, Zhao Y, Qian K, Sun M. Taurine attenuates hippocampal and corpus callosum damage, and enhances neurological recovery after closed head injury in rats. Neuroscience 2015; 291:331–340.
Banks MA, Porter DW, Pailes WH, Schwegler-Berry D, Martin WG, Castranova V. Taurine content of isolated rat alveolar type I cells. Comp Biochem Physiol B 1991; 100:795–799.
Raschke P, Massoudy P, Becker BF. Taurine protects the heart from neutrophil-induced reperfusion injury. Free Radic Biol Med 1995; 19:461–471.
Pushpakiran G, Mahalakshmi K, Anuradha CV. Taurine restores ethanol-induced depletion of antioxidants and attenuates oxidative stress in rat tissues. Amino Acids 2004; 27:91–96.
Refik Mas M, Comert B, Oncu K, et al. The effect of taurine treatment on oxidative stress in experimental liver fibrosis. Hepatol Res 2004; 28:207–215.
Sebring LA, Huxtable RJ. Taurine modulation of calcium binding to cardiac sarcolemma. J Pharmacol Exp Ther 1985; 232:445–451.
Schaffer SW, Kramer J, Chovan JP. Regulation of calcium homeostasis in the heart by taurine. Fed Proc 1980; 39:2691–2694.
Foos TM, Wu JY. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem Res 2002; 27:21–26.
Ito T, Schaffer S, Azuma J. The effect of taurine on chronic heart failure: actions of taurine against catecholamine and angiotensin II. Amino Acids 2014; 46:111–119.
Gaull GE. Taurine in pediatric nutrition: review and update. Pediatrics 1989; 83:433–442.
Sturman JA, Rassin DK, Gaull GE. Taurine in developing rat brain: transfer of [35S] taurine to pups via the milk. Pediatr Res 1977; 11:28–33.
Geggel HS, Ament ME, Heckenlively JR, Martin DA, Kopple JD. Nutritional requirement for taurine in patients receiving long-term parenteral nutrition. N Engl J Med 1985; 312:142–146.
Sturman JA, Gargano AD, Messing JM, Imaki H. Feline maternal taurine deficiency: effect on mother and offspring. J Nutr 1986; 116:655–667.
Chesney RW, Helms RA, Christensen M, Budreau AM, Han X, Sturman JA. The role of taurine in infant nutrition. Adv Exp Med Biol 1998; 442:463–476.
Verner A, Craig S, McGuire W. Effect of taurine supplementation on growth and development in preterm or low birth weight infants. Cochrane Database Syst Rev 2007; 4:CD006072.
Jeevanandam M, Young DH, Ramias L, Schiller WR. Effect of major trauma on plasma free amino acid concentrations in geriatric patients. Am J Clin Nutr 1990; 51:1040–1045.
Dawson R Jr, Eppler B, Patterson TA, Shih D, Liu S. The effects of taurine in a rodent model of aging. Adv Exp Med Biol 1996; 403:37–50.
Han X, Budreau AM, Chesney RW. Molecular cloning and functional expression of an LLC-PK1 cell taurine transporter that is adaptively regulated by taurine. Adv Exp Med Biol 1998; 442:261–268.
Eppler B, Dawson R Jr. Dietary taurine manipulations in aged male Fischer 344 rat tissue: taurine concentration, taurine biosynthesis, and oxidative markers. Biochem Pharmacol 2001; 62:29–39.
D’Eufemia P, Finocchiaro R, Celli M, et al. Taurine deficiency in thalassemia major-induced osteoporosis treated with neridronate. Biomed Pharmacother 2010; 64:271–274.
Muscaritoli M, Conversano L, Petti MC, et al. Plasma amino acid concentrations in patients with acute myelogenous leukemia. Nutrition 1999; 15:195–199.
Do KQ, Lauer CJ, Schreiber W, et al. Gamma-glutamylglutamine and taurine concentrations are decreased in the cerebrospinal fluid of drug-naive patients with schizophrenic disorders. J Neurochem 1995; 65:2652–2662.
Lemieux B, Giguère R, Shapcott D. Studies on the role of taurine in Friedreich’s ataxia. Can J Neurol Sci 1984; 11(suppl 4):610–615.
Airaksinen EM, Oja SS, Marnela KM, Sihvola P. Taurine and other amino acids of platelets and plasma in retinitis pigmentosa. Ann Clin Res 1980; 12:52–54.
Kohashi N, Katori R. Decrease of urinary taurine in essential hypertension. Jpn Heart J 1983; 24:91–102.
vom Dahl S, Mönnighoff I, Häussinger D. Decrease of plasma taurine in Gaucher disease and its sustained correction during enzyme replacement therapy. Amino Acids 2000; 19:585–592.
Schneider SM, Joly F, Gehrardt MF, et al. Taurine status and response to intravenous taurine supplementation in adults with short-bowel syndrome undergoing long-term parenteral nutrition: a pilot study. Br J Nutr 2006; 96:365–370.
Gray GE, Landel AM, Meguid MM. Taurine-supplemented total parenteral nutrition and taurine status of malnourished cancer patients. Nutrition 1994; 10:11–15.
Thompson GN. Assessment of taurine deficiency in cystic fibrosis. Clin Chim Acta 1988; 171:233–237.
Cho KH, Kim ES, Chen JD. Taurine intake and excretion in patients undergoing long term enteral nutrition. Adv Exp Med Biol 2000; 483:605–612.
Durelli L, Mutani R, Fassio F. The treatment of myotonia: evaluation of chronic oral taurine therapy. Neurology 1983; 33:599–603.
Shao A, Hathcock JN. Risk assessment for the amino acids taurine, L-glutamine and L-arginine. Regul Toxicol Pharmacol 2008; 50:376–399.
Tanaka E, Terada M, Misawa S. Cytochrome P450 2E1: its clinical and toxicological role. J Clin Pharm Ther 2000; 25:165–175.
Kerai MD, Waterfield CJ, Kenyon SH, Asker DS, Timbrell JA. Reversal of ethanol-induced hepatic steatosis and lipid peroxidation by taurine: a study in rats. Alcohol Alcohol 1999; 34:529–541.
Das J, Ghosh J, Manna P, Sil PC. Acetaminophen induced acute liver failure via oxidative stress and JNK activation: protective role of taurine by the suppression of cytochrome P450 2E1. Free Radic Res 2010; 44:340–355.
El Idrissi A, Shen CH, L’amoreaux WJ. Neuroprotective role of taurine during aging. Amino Acids 2013; 45:735–750.
Gharibani P, Modi J, Menzie J, et al. Comparison between single and combined post-treatment with S-methyl-N,N-diethylthiolcarbamate sulfoxide and taurine following transient focal cerebral ischemia in rat brain. Neuroscience 2015; 300:460–473.
Junyent F, De Lemos L, Utrera J, et al. Content and traffic of taurine in hippocampal reactive astrocytes. Hippocampus 2011; 21:185–197.
El Idrissi A, Trenkner E. Growth factors and taurine protect against excitotoxicity by stabilizing calcium homeostasis and energy metabolism. J Neurosci 1999; 19:9459–9468.
Wu H, Jin Y, Wei J, Jin H, Sha D, Wu JY. Mode of action of taurine as a neuroprotector. Brain Res 2005; 1038:123–131.
Paula-Lima AC, De Felice FG, Brito-Moreira J, Ferreira ST. Activation of GABA(A) receptors by taurine and muscimol blocks the neurotoxicity of beta-amyloid in rat hippocampal and cortical neurons. Neuropharmacology 2005; 49:1140–1148.
Takatani T, Takahashi K, Uozumi Y, et al. Taurine inhibits apoptosis by preventing formation of the Apaf-1/caspase-9 apoptosome. Am J Physiol Cell Physiol 2004; 287:C949–C953.
Atamna H, Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J Alzheimers Dis 2010; 20(suppl 2):S439–S452.
El Idrissi A. Taurine improves learning and retention in aged mice. Neurosci Lett 2008; 436:19–22.
Oja SS, Saransaari P. Taurine and epilepsy. Epilepsy Res 2013; 104:187–194.
Berson EL, Hayes KC, Rabin AR, Schmidt SY, Watson G. Retinal degeneration in cats fed casein. II. Supplementation with methionine, cysteine, or taurine. Invest Ophthalmol 1976; 15:52–58.
Beyranvand MR, Khalafi MK, Roshan VD, Choobineh S, Parsa SA, Piranfar MA. Effect of taurine supplementation on exercise capacity of patients with heart failure. J Cardiol 2011; 57:333–337.
Eby G, Halcomb WW. Elimination of cardiac arrhythmias using oral taurine with l-arginine with case histories: hypothesis for nitric oxide stabilization of the sinus node. Med Hypotheses 2006; 67:1200–1204.
Jonathan J. Caine, MD Department of Psychiatry and Behavioral Neurosciences, University of Cincinnati College of Medicine; Department of Veterans Affairs Medical Center, Cincinnati, OH
Thomas D. Geracioti, MD Department of Psychiatry and Behavioral Neurosciences, University of Cincinnati College of Medicine; Department of Veterans Affairs Medical Center, Cincinnati, OH
Address: Jonathan J. Caine, MD, University of Cincinnati Medical Center, Department of Psychiatry and Behavioral Neurosciences, PO Box 670559, Cincinnati, OH 45219-0559; cainejj@mail.uc.edu
Jonathan J. Caine, MD Department of Psychiatry and Behavioral Neurosciences, University of Cincinnati College of Medicine; Department of Veterans Affairs Medical Center, Cincinnati, OH
Thomas D. Geracioti, MD Department of Psychiatry and Behavioral Neurosciences, University of Cincinnati College of Medicine; Department of Veterans Affairs Medical Center, Cincinnati, OH
Address: Jonathan J. Caine, MD, University of Cincinnati Medical Center, Department of Psychiatry and Behavioral Neurosciences, PO Box 670559, Cincinnati, OH 45219-0559; cainejj@mail.uc.edu
Author and Disclosure Information
Jonathan J. Caine, MD Department of Psychiatry and Behavioral Neurosciences, University of Cincinnati College of Medicine; Department of Veterans Affairs Medical Center, Cincinnati, OH
Thomas D. Geracioti, MD Department of Psychiatry and Behavioral Neurosciences, University of Cincinnati College of Medicine; Department of Veterans Affairs Medical Center, Cincinnati, OH
Address: Jonathan J. Caine, MD, University of Cincinnati Medical Center, Department of Psychiatry and Behavioral Neurosciences, PO Box 670559, Cincinnati, OH 45219-0559; cainejj@mail.uc.edu
Taurine—an amino acid found in abundance in the human brain, retina, heart, and reproductive organs, as well as in meat and seafood—is also a major ingredient in “energy drinks” (Table 1).1,2 Given the tremendous popularity of these drinks in the United States, it would seem important to know and to recognize taurine’s neuroendocrine effects. Unfortunately, little is known about the effects of taurine supplementation in humans.
This paper reviews the sparse data to provide clinicians some background on the structure, synthesis, distribution, metabolism, mechanisms, effects, safety, and proposed therapeutic targets of taurine.
TAURINE’S THERAPEUTIC POTENTIAL
Taurine has been reported to have widespread anti-inflammatory actions.3,4 Taurine supplementation has been proposed to have beneficial effects in the treatment of epilepsy,5 heart failure,6,7 cystic fibrosis,8 and diabetes9 and has been shown in animal studies to protect against neurotoxic insults from alcohol, ammonia, lead, and other substances.10–16
In addition, taurine analogues such as homotaurine and N-acetyl-homotaurine (acamprosate) have been probed for possible therapeutic applications. Homotaurine has been shown to have antiamyloid activity that could in theory protect against the progression of Alzheimer disease,17 and acamprosate is approved by the US Food and Drug Administration (FDA) for the treatment of alcohol use disorders.18
TAURINE CONSUMPTION
Energy drinks are widely consumed in the United States, with an estimated 354 million gallons sold in 2009, or approximately 5.25 L/year per person over age 10.1 In 2012, US sales of energy drinks exceeded $12 billion,19 with young men, particularly those in the military deployed in war zones, being the biggest consumers.20–22 Analyses have found that of 49 nonalcoholic energy drinks tested, the average concentration of taurine was 3,180 mg/L, or approximately 750 mg per 8-oz serving.23,24 Popular brands include Red Bull, Monster, Rockstar (Table 1), NOS, Amp, and Full Throttle.
Taurine is plentiful in the human body, which contains up to 1 g of taurine per kg.25 Foods such as poultry, beef, pork, seafood, and processed meats have a high taurine content (Table 2).26–29 People who eat meat and seafood have plentiful taurine intake, whereas vegetarians and vegans consume much less and have significantly lower circulating levels30 because plants do not contain taurine in appreciable amounts.26,29
The typical American diet provides between 123 and 178 mg of taurine daily.26 Consumption of one 8-oz energy drink can increase the average intake 6 to 16 times. A lacto-ovo vegetarian diet provides only about 17 mg of taurine daily, and an 8-oz energy drink can increase the average intake by 44 to 117 mg.26 And since a vegan diet provides essentially no taurine,30 energy drink intake in any amount would constitute a major relative increase in taurine consumption.
ATTEMPTS TO STUDY TAURINE'S EFFECTS
Since most clinical trials to date have looked at the effects of taurine in combination with other ingredients such as caffeine, creatine, and glucose31–35 in drinks such as Red Bull, these studies cannot be used to determine the effects of taurine alone. In the few clinical trials that have tested isolated taurine consumption, data are not sufficient to make a conclusion on direct effects on energy metabolism.
Rutherford et al36 tested the effect of oral taurine supplementation (1,660 mg) on endurance in trained male cyclists 1 hour before exercise, but observed no effect on fluid intake, heart rate, subjective exertion, or time-trial performance. A small increase (16%) in total fat oxidation was observed during the 90-minute exercise period. Since mitochondria are the main location of fatty acid degradation, this effect may be attributed to taurine supplementation, with subsequent improvement in mitochondrial function.
Zhang et al37 found a 30-second increase in cycling energy capacity after 7 days of 6 g oral taurine supplementation, but the study was neither blinded nor placebo-controlled.
Kammerer et al38 tested the effect of 1 g of taurine supplementation on physical and mental performance in young adult soldiers 45 minutes before physical fitness and cognitive testing. This double-blind, placebo-controlled randomized trial found no effect of taurine on cardiorespiratory fitness indices, concentration, or immediate memory, nor did it find any effect of an 80-mg dose of caffeine.
In sum, the available data are far from sufficient to determine the direct effect of taurine consumption on energy metabolism in healthy people.
PHARMACOLOGY OF TAURINE
Chemical structure
Taurine, or 2-aminoethane sulfonic acid, is a conditionally essential amino acid, ie, we can usually make enough in our own bodies. It was first prepared on a large scale for physiologic investigation almost 90 years ago, through the purification of ox bile.39 It can be obtained either exogenously through dietary sources or endogenously through biosynthesis from methionine and cysteine precursors, both essential sulfur-containing alpha-amino acids.40 Both sources are important to maintain physiologic levels of taurine, and either can help compensate for the other in cases of deficiency.41
The structure of taurine has two main differences from the essential amino acids. First, taurine’s amino group is attached to the beta-carbon rather than the alpha-carbon, making it a beta-amino acid instead of an alpha-amino acid.42 Second, the acid group in taurine is sulfonic acid, whereas the essential amino acids have a carboxylic acid.43 Because of its distinctive structure, taurine is not used as a structural unit in proteins,43 existing mostly as a free amino acid within cells, readily positioned to perform several unique functions.
Synthesis
De novo synthesis of taurine involves several enzymes and at least five pathways,44 mostly differing by the order in which sulfur is oxidized and decarboxylated.45
The rate-limiting enzyme of the predominant pathway is thought to be cysteine sulfinate decarboxylase (CSD), and its presence within an organ indicates involvement in taurine production.44 CSD has been found in the liver,46 the primary site of taurine biosynthesis, as well as in the retina,47 brain,48 kidney,49 mammary glands,50,51 and reproductive organs.52
Distribution
Taurine levels are highest in electrically excitable tissues such as the central nervous system, retina, and heart; in secretory structures such as the pineal gland and the pituitary gland (including the posterior lobe or neurohypophysis); and in platelets25 and neutrophils.53
In the fetal brain, the taurine concentration is higher than that of any other amino acid,54 but the concentration in the brain decreases with advancing age, whereas glutamate levels increase over time to make it the predominant amino acid in the adult brain.54 Regardless, taurine is still the second most prevalent amino acid in the adult brain, its levels comparable to those of gamma-aminobutyric acid (GABA).55
Taurine has also been found in variable amounts in the liver, muscle, kidney, pancreas, spleen, small intestine, and lungs,56 as well as in several other locations.45,57
Taurine is also present in the male and female reproductive organs. In male rats, taurine and taurine biosynthesis have been localized to Leydig cells of the testes, the cellular source of testosterone in males, as well as the cremaster muscle, efferent ducts, and peritubular myoid cells surrounding seminiferous tubules.58 More recently, taurine has been detected in the testes of humans59 and is also found in sperm and seminal fluid.60 Levels of taurine in spermatozoa are correlated with sperm quality, presumably by protecting against lipid peroxidation through taurine’s antioxidant effects,61,62 as well as through contribution to the spermatozoa maturation process by facilitating the capacitation, motility, and acrosomal reaction of sperm.63
In female rats, taurine has been found in uterine tissue,64 oviducts,65 uterine fluid (where it is the predominant amino acid),66 and thecal cells of developing follicles of ovaries, cells responsible for the synthesis of androgens such as testosterone and androstenedione.65 Taurine is also a major component of human breast milk67 and is important for proper neonatal nutrition.68
Metabolism and excretion
Ninety-five percent of taurine is excreted in urine, about 70% as taurine itself, and the rest as sulfate. Most of the sulfate derived from taurine is produced by bacterial metabolism in the gut and then absorbed.69 However, taurine can also be conjugated with bile acids to act as a detergent in lipid emulsification.70 In this form, it may be subjected to the enterohepatic circulation, which gives bacteria another chance to convert it into inorganic sulfate for excretion in urine.69
MECHANISMS AND NEUROENDOCRINE EFFECTS
As a free amino acid, taurine has widespread distribution and unique biochemical and physiologic properties and exhibits several organ-specific functions; however, indisputable evidence of a taurine-specific receptor is lacking, and its putative existence71 is controversial.72 Nonetheless, taurine is a neuromodulator with a variety of actions.
Neurotransmission
Taurine is known to be an inhibitory neurotransmitter and neuromodulator.73 It is structurally analogous to GABA, the main inhibitory neurotransmitter in the brain.45 Accordingly, it binds to GABA receptors to serve as an agonist,74,75 causing neuronal hyperpolarization and inhibition. Taurine has an even higher affinity for glycine receptors75 where it has long been known to act as an agonist.76 GABA and glycine receptors both belong to the Cys-loop receptor superfamily,77 with conservation of subunits that allows taurine to bind each receptor, albeit at different affinities. The binding effects of taurine on GABA and glycine receptors have not been well documented quantitatively; however, it is known that taurine has a substantially lower affinity than GABA and glycine for their respective receptors.76
Catecholamines and the sympathetic nervous system
Surprisingly little is known about the effects of taurine on norepinephrine, dopamine, and the human sympathetic nervous system.78 Humans with borderline hypertension given 6 g of taurine orally for 7 days79 experienced decreases in epinephrine secretion and blood pressure, but normotensive study participants did not experience similar results, possibly because of a better ability to regulate sympathetic tone. Mizushima et al80 showed that a longer period of taurine intake (6 g orally for 3 weeks) could elicit a decrease in norepinephrine in healthy men with normal blood pressure. Other similar studies81–83 also suggested interplay between taurine and catecholamines, but the extent is still undetermined.
Growth hormone, prolactin, sex hormones, and cortisol
Taurine appears to have a complex relationship with several hormones, although its direct effects on hormone secretion remain obscure. Clinical studies of the acute and chronic neuroendocrine effects of taurine loading in humans are needed.
In female rats, secretion of prolactin is increased by the intraventricular injection of 5 μL of 2.0 μmol taurine over a 10-minute period.84 Ikuyama et al85 found an increase in prolactin and growth hormone secretion in adult male rats given 10 μL of 0.25 μmol and 1.0 μmol taurine intraventricularly, yet a higher dose of 4.0 μmol had no effect on either hormone. Furthermore, prolactin receptor deficiency is seen in CSD knockout mice, but the receptor is restored with taurine supplementation.86
Mantovani and DeVivo87 reported that 375 to 8,000 mg/day of taurine given orally for 4 to 6 months to epileptic patients stimulated the secretion of growth hormone. However, in another study, a single 75-mg/kg dose of oral taurine did not trigger an acute increase in levels of growth hormone or prolactin in humans.88 Energy drinks may contain up to 1,000 mg of taurine per 8-oz serving, but the effects of larger doses on growth hormone, which is banned as a supplement by major athletic organizations because of its anabolic and possible performance-enhancing effects, remain to be determined.
Taurine may have effects on human sex hormones, based on the limited observations in rodents.89–94
Although human salivary cortisol concentrations were purportedly assessed in response to 2,000 mg of oral taurine,95 the methods and reported data are not adequate to draw any conclusions.
Energy metabolism
Mammals are unable to directly use taurine in energy production because they cannot directly reduce it.25 Instead, bacteria in the gut use it as a source of energy, carbon, nitrogen, and sulfur.96 However, taurine deficiency appears to impair the cellular respiratory chain, resulting in diminished production of adenosine triphosphate and diminished uptake of long-chain fatty acids by mitochondria, at least in the heart.97
Taurine is present in human mitochondria and regulates mitochondrial function. For example, taurine in mitochondria assists in conjugation of transfer RNA for leucine, lysine, glutamate, and glutamine.98 In TauT knockout mice, deficiency of taurine causes mitochondrial dysfunction, triggering a greater than 80% decrease in exercise capacity.99 Several studies in rodents have shown increased exercise capacity after taurine supplementation.100–102 In addition, taurine is critical for the growth of blastocytes, skeletal muscle, and myocardium; it is necessary for mitochondrial development and is also important for muscular endurance.103,104
Antioxidation, anti-inflammation, and other functions
Taurine is a major antioxidant, scavenging reactive oxygen and protecting against oxidative stress to organs including the brain,97,105,106 where it increasingly appears to have neuroprotective effects.107,108
Cellular taurine also has anti-inflammatory actions.3 One of the proposed mechanisms is taurine inhibition of NF-kappa B, an important transcription factor for the synthesis of pro-inflammatory cytokines.4 This function may be important in protecting polyunsaturated fatty acids from oxidative stress—helping to maintain and stabilize the structure and function of plasma membranes within the lungs,109 heart,110 brain,111 liver,112 and spermatozoa.61,62
Taurine is also conjugated to bile acids synthesized in the liver, forming bile salts70 that act as detergents to help emulsify and digest lipids in the body. In addition, taurine facilitates xenobiotic detoxification in the liver by conjugating with several drugs to aid in their excretion.25 Taurine is also implicated in calcium modulation113 and homeostasis.114 Through inhibition of several types of calcium channels, taurine has been shown to decrease calcium influx into cells, effectively serving a cytoprotective role against calcium overload.115,116
TAURINE DEFICIENCY
Fetal and neonatal deficiency
Though taurine is considered nonessential in adults because it can be readily synthesized endogenously, it is thought to be conditionally essential in neonatal nutrition.68 It is the second most abundant free amino acid in human breast milk117 and the most abundant free amino acid in fetal brain.118 In cases of long-term parenteral nutrition, neonates can become drastically taurine deficient119 due to suboptimal CSD activity,118 leading to retinal dysfunction.41 Taurine deficiencies can lead to functional and structural brain damage.118 Moreover, maternal taurine deficiency results in neurologic abnormalities in offspring120 and may lead to oxidative stress throughout life.121
In 1984, the FDA approved the inclusion of taurine in infant formulas based on research showing that taurine-deficient infants had impaired fat absorption, bile acid secretion, retinal function, and hepatic function.122 But still under debate are the amount and duration of taurine supplementation required by preterm and low-birth-weight infants, as several randomized controlled trials failed to show statistically significant effects on growth.123 Nonetheless, given the alleged detrimental ramifications of a lack of taurine supplementation, as well as the ethical dilemma of performing additional research trials on infants, it is presumed that infant formulas and parenteral nutrition for preterm and low-birth-weight infants will continue to contain taurine.
Age- and disease-related deficiency
Although taurine deficiency is rare in neonates, it is perhaps inevitable with advancing age. Healthy elderly patients ages 61 to 81 have up to a 49% decrease in plasma taurine concentration compared with healthy individuals ages 27 to 57.124 While reduced renal retention125 and taurine intake126 can account for depressed taurine levels, Eppler and Dawson127 found that tissue and circulating taurine concentrations decrease over the human life span primarily due to an age-dependent depletion of CSD activity in the liver. This effectively impairs the biosynthesis of endogenous taurine from cysteine or methionine or both, forcing a greater reliance on exogenous sources.
While specific mechanisms have not been fully elucidated, taurine deficiency has also been identified in patients suffering from diseases including but not limited to disorders of bone (osteogenesis imperfecta, osteoporosis),128 blood (acute myelogenous leukemia),129 central nervous system (schizophrenia, Friedreich ataxia-spinocerebellar degeneration),130,131 retina (retinitis pigmentosa),132 circulatory system and heart (essential hypertension, atherosclerosis),133 digestion (Gaucher disease),134 absorption (short-bowel syndrome),135 cellular proliferation (cancer),136 and membrane channels (cystic fibrosis),137 as well as in patients restricted to long-term parenteral nutrition.138 However, the apparent correlation between taurine deficiency and these conditions does not necessarily mean causation; more study is needed to elucidate a direct connection.
SAFETY AND TOXICITY OF TAURINE SUPPLEMENTATION
An upper safe level of intake for taurine has not been established. To date, several studies have involved heavy taurine supplementation without serious adverse effects. While the largest dosage of taurine tested in humans appears to be 10 g/day for 6 months,139 a number of studies have used 1 to 6 g/day for periods of 1 week to 1 year.140 However, the assessment of potential acute, subacute, and chronic adverse effects has not been comprehensive. The Scientific Committee on Food of the European Commission141 reviewed several toxicologic studies on taurine through 2003 and were unable to expose any carcinogenic or teratogenic potential. Nevertheless, based on the available data from trials in humans and lower animals, Shao and Hathcock140 suggested an observed safe level of taurine of 3 g/day, a conservatively smaller dose that carries a higher level of confidence. Because there is no “observed adverse effect level” for daily taurine intake,141 more research must be done to ensure safety of higher amounts of taurine administration and to define a tolerable upper limit of intake.
Interactions with medications
To date, the literature is scarce regarding potential interactions between taurine and commonly used medications.
Although no evidence specifically links taurine with adverse effects when used concurrently with other medications, there may be a link between taurine supplementation and various cytochrome P450 systems responsible for hepatic drug metabolism. Specifically, taurine inhibits cytochrome P450 2E1, a highly conserved xenobiotic-metabolizing P450 responsible for the breakdown of more than 70 substrates, including several commonly used anesthetics, analgesics, antidepressants, antibacterials, and antiepileptics.142 Of note, taurine may contribute to the attenuation of oxidative stress in the liver in the presence of alcohol143 and acetaminophen,144 two substances frequently used and abused. Since the P450 2E1 system catalyzes comparable reactions in rodents and humans,142 rodents should plausibly serve as a model for further testing of the effects of taurine on various substrates.
POTENTIAL THERAPEUTIC APPLICATIONS
More analysis is needed to fully unlock and understand taurine’s potential value in healthcare.
Correction of late-life taurine decline in humans could be beneficial for cognitive performance, energy metabolism, sexual function, and vision, but clinical studies remain to be performed. While a decline in taurine with age may intensify the stress caused by reactive oxygen species, taurine supplementation has been shown to decrease the presence of oxidative markers127 and to serve a neuroprotective role in rodents.145,146 Taurine levels increase in the hippocampus after experimentally induced gliosis147 and are neuroprotective against glutamate excitotoxicity.148,149 Furthermore, data in Alzheimer disease, Huntington disease, and brain ischemia experimental models show that taurine inhibits neuronal death (apoptosis).13,150,151 Taurine has even been proposed as a potential preventive treatment for Alzheimer dementia, as it stabilizes protein conformations to prevent their aggregation and subsequent dysfunction.152 Although improvement in memory and cognitive performance has been linked to taurine supplementation in old mice,145,153 similar results have not been found in adult mice whose taurine levels are within normal limits. Taurine also has transient anticonvulsant effects in some epileptic humans.154
Within the male reproductive organs, the age-related decline in taurine may or may not have implications regarding sexuality, as only very limited rat data are available.89–91
In cats, taurine supplementation has been found to prevent the progressive degeneration of retinal photoreceptors seen in retinitis pigmentosa, a genetic disease that causes the loss of vision.155
While several energy drink companies have advertised that taurine plays a role in improving cognitive and physical performance, there are few human studies that examine this contention in the absence of confounding factors such as caffeine or glucose.36,37,95 Taurine supplementation in patients with heart failure has been shown to increase exercise capacity vs placebo.156 This supports the idea that in cases of taurine deficiency, such as those seen in cardiomyopathy,157 taurine supplementation could have restorative effects. However, we are not aware of any double-blind, placebo-controlled clinical trial of taurine alone in healthy patients that measured energy parameters as clinical outcomes.
Although it remains possible that acute supraphysiologic taurine levels achieved by supplementation could transiently trigger various psychoneuroendocrine responses in healthy people, clinical research is needed in which taurine is the sole intervention. At present, the most compelling clinical reason to prescribe or recommend taurine supplementation is taurine deficiency.
Taurine—an amino acid found in abundance in the human brain, retina, heart, and reproductive organs, as well as in meat and seafood—is also a major ingredient in “energy drinks” (Table 1).1,2 Given the tremendous popularity of these drinks in the United States, it would seem important to know and to recognize taurine’s neuroendocrine effects. Unfortunately, little is known about the effects of taurine supplementation in humans.
This paper reviews the sparse data to provide clinicians some background on the structure, synthesis, distribution, metabolism, mechanisms, effects, safety, and proposed therapeutic targets of taurine.
TAURINE’S THERAPEUTIC POTENTIAL
Taurine has been reported to have widespread anti-inflammatory actions.3,4 Taurine supplementation has been proposed to have beneficial effects in the treatment of epilepsy,5 heart failure,6,7 cystic fibrosis,8 and diabetes9 and has been shown in animal studies to protect against neurotoxic insults from alcohol, ammonia, lead, and other substances.10–16
In addition, taurine analogues such as homotaurine and N-acetyl-homotaurine (acamprosate) have been probed for possible therapeutic applications. Homotaurine has been shown to have antiamyloid activity that could in theory protect against the progression of Alzheimer disease,17 and acamprosate is approved by the US Food and Drug Administration (FDA) for the treatment of alcohol use disorders.18
TAURINE CONSUMPTION
Energy drinks are widely consumed in the United States, with an estimated 354 million gallons sold in 2009, or approximately 5.25 L/year per person over age 10.1 In 2012, US sales of energy drinks exceeded $12 billion,19 with young men, particularly those in the military deployed in war zones, being the biggest consumers.20–22 Analyses have found that of 49 nonalcoholic energy drinks tested, the average concentration of taurine was 3,180 mg/L, or approximately 750 mg per 8-oz serving.23,24 Popular brands include Red Bull, Monster, Rockstar (Table 1), NOS, Amp, and Full Throttle.
Taurine is plentiful in the human body, which contains up to 1 g of taurine per kg.25 Foods such as poultry, beef, pork, seafood, and processed meats have a high taurine content (Table 2).26–29 People who eat meat and seafood have plentiful taurine intake, whereas vegetarians and vegans consume much less and have significantly lower circulating levels30 because plants do not contain taurine in appreciable amounts.26,29
The typical American diet provides between 123 and 178 mg of taurine daily.26 Consumption of one 8-oz energy drink can increase the average intake 6 to 16 times. A lacto-ovo vegetarian diet provides only about 17 mg of taurine daily, and an 8-oz energy drink can increase the average intake by 44 to 117 mg.26 And since a vegan diet provides essentially no taurine,30 energy drink intake in any amount would constitute a major relative increase in taurine consumption.
ATTEMPTS TO STUDY TAURINE'S EFFECTS
Since most clinical trials to date have looked at the effects of taurine in combination with other ingredients such as caffeine, creatine, and glucose31–35 in drinks such as Red Bull, these studies cannot be used to determine the effects of taurine alone. In the few clinical trials that have tested isolated taurine consumption, data are not sufficient to make a conclusion on direct effects on energy metabolism.
Rutherford et al36 tested the effect of oral taurine supplementation (1,660 mg) on endurance in trained male cyclists 1 hour before exercise, but observed no effect on fluid intake, heart rate, subjective exertion, or time-trial performance. A small increase (16%) in total fat oxidation was observed during the 90-minute exercise period. Since mitochondria are the main location of fatty acid degradation, this effect may be attributed to taurine supplementation, with subsequent improvement in mitochondrial function.
Zhang et al37 found a 30-second increase in cycling energy capacity after 7 days of 6 g oral taurine supplementation, but the study was neither blinded nor placebo-controlled.
Kammerer et al38 tested the effect of 1 g of taurine supplementation on physical and mental performance in young adult soldiers 45 minutes before physical fitness and cognitive testing. This double-blind, placebo-controlled randomized trial found no effect of taurine on cardiorespiratory fitness indices, concentration, or immediate memory, nor did it find any effect of an 80-mg dose of caffeine.
In sum, the available data are far from sufficient to determine the direct effect of taurine consumption on energy metabolism in healthy people.
PHARMACOLOGY OF TAURINE
Chemical structure
Taurine, or 2-aminoethane sulfonic acid, is a conditionally essential amino acid, ie, we can usually make enough in our own bodies. It was first prepared on a large scale for physiologic investigation almost 90 years ago, through the purification of ox bile.39 It can be obtained either exogenously through dietary sources or endogenously through biosynthesis from methionine and cysteine precursors, both essential sulfur-containing alpha-amino acids.40 Both sources are important to maintain physiologic levels of taurine, and either can help compensate for the other in cases of deficiency.41
The structure of taurine has two main differences from the essential amino acids. First, taurine’s amino group is attached to the beta-carbon rather than the alpha-carbon, making it a beta-amino acid instead of an alpha-amino acid.42 Second, the acid group in taurine is sulfonic acid, whereas the essential amino acids have a carboxylic acid.43 Because of its distinctive structure, taurine is not used as a structural unit in proteins,43 existing mostly as a free amino acid within cells, readily positioned to perform several unique functions.
Synthesis
De novo synthesis of taurine involves several enzymes and at least five pathways,44 mostly differing by the order in which sulfur is oxidized and decarboxylated.45
The rate-limiting enzyme of the predominant pathway is thought to be cysteine sulfinate decarboxylase (CSD), and its presence within an organ indicates involvement in taurine production.44 CSD has been found in the liver,46 the primary site of taurine biosynthesis, as well as in the retina,47 brain,48 kidney,49 mammary glands,50,51 and reproductive organs.52
Distribution
Taurine levels are highest in electrically excitable tissues such as the central nervous system, retina, and heart; in secretory structures such as the pineal gland and the pituitary gland (including the posterior lobe or neurohypophysis); and in platelets25 and neutrophils.53
In the fetal brain, the taurine concentration is higher than that of any other amino acid,54 but the concentration in the brain decreases with advancing age, whereas glutamate levels increase over time to make it the predominant amino acid in the adult brain.54 Regardless, taurine is still the second most prevalent amino acid in the adult brain, its levels comparable to those of gamma-aminobutyric acid (GABA).55
Taurine has also been found in variable amounts in the liver, muscle, kidney, pancreas, spleen, small intestine, and lungs,56 as well as in several other locations.45,57
Taurine is also present in the male and female reproductive organs. In male rats, taurine and taurine biosynthesis have been localized to Leydig cells of the testes, the cellular source of testosterone in males, as well as the cremaster muscle, efferent ducts, and peritubular myoid cells surrounding seminiferous tubules.58 More recently, taurine has been detected in the testes of humans59 and is also found in sperm and seminal fluid.60 Levels of taurine in spermatozoa are correlated with sperm quality, presumably by protecting against lipid peroxidation through taurine’s antioxidant effects,61,62 as well as through contribution to the spermatozoa maturation process by facilitating the capacitation, motility, and acrosomal reaction of sperm.63
In female rats, taurine has been found in uterine tissue,64 oviducts,65 uterine fluid (where it is the predominant amino acid),66 and thecal cells of developing follicles of ovaries, cells responsible for the synthesis of androgens such as testosterone and androstenedione.65 Taurine is also a major component of human breast milk67 and is important for proper neonatal nutrition.68
Metabolism and excretion
Ninety-five percent of taurine is excreted in urine, about 70% as taurine itself, and the rest as sulfate. Most of the sulfate derived from taurine is produced by bacterial metabolism in the gut and then absorbed.69 However, taurine can also be conjugated with bile acids to act as a detergent in lipid emulsification.70 In this form, it may be subjected to the enterohepatic circulation, which gives bacteria another chance to convert it into inorganic sulfate for excretion in urine.69
MECHANISMS AND NEUROENDOCRINE EFFECTS
As a free amino acid, taurine has widespread distribution and unique biochemical and physiologic properties and exhibits several organ-specific functions; however, indisputable evidence of a taurine-specific receptor is lacking, and its putative existence71 is controversial.72 Nonetheless, taurine is a neuromodulator with a variety of actions.
Neurotransmission
Taurine is known to be an inhibitory neurotransmitter and neuromodulator.73 It is structurally analogous to GABA, the main inhibitory neurotransmitter in the brain.45 Accordingly, it binds to GABA receptors to serve as an agonist,74,75 causing neuronal hyperpolarization and inhibition. Taurine has an even higher affinity for glycine receptors75 where it has long been known to act as an agonist.76 GABA and glycine receptors both belong to the Cys-loop receptor superfamily,77 with conservation of subunits that allows taurine to bind each receptor, albeit at different affinities. The binding effects of taurine on GABA and glycine receptors have not been well documented quantitatively; however, it is known that taurine has a substantially lower affinity than GABA and glycine for their respective receptors.76
Catecholamines and the sympathetic nervous system
Surprisingly little is known about the effects of taurine on norepinephrine, dopamine, and the human sympathetic nervous system.78 Humans with borderline hypertension given 6 g of taurine orally for 7 days79 experienced decreases in epinephrine secretion and blood pressure, but normotensive study participants did not experience similar results, possibly because of a better ability to regulate sympathetic tone. Mizushima et al80 showed that a longer period of taurine intake (6 g orally for 3 weeks) could elicit a decrease in norepinephrine in healthy men with normal blood pressure. Other similar studies81–83 also suggested interplay between taurine and catecholamines, but the extent is still undetermined.
Growth hormone, prolactin, sex hormones, and cortisol
Taurine appears to have a complex relationship with several hormones, although its direct effects on hormone secretion remain obscure. Clinical studies of the acute and chronic neuroendocrine effects of taurine loading in humans are needed.
In female rats, secretion of prolactin is increased by the intraventricular injection of 5 μL of 2.0 μmol taurine over a 10-minute period.84 Ikuyama et al85 found an increase in prolactin and growth hormone secretion in adult male rats given 10 μL of 0.25 μmol and 1.0 μmol taurine intraventricularly, yet a higher dose of 4.0 μmol had no effect on either hormone. Furthermore, prolactin receptor deficiency is seen in CSD knockout mice, but the receptor is restored with taurine supplementation.86
Mantovani and DeVivo87 reported that 375 to 8,000 mg/day of taurine given orally for 4 to 6 months to epileptic patients stimulated the secretion of growth hormone. However, in another study, a single 75-mg/kg dose of oral taurine did not trigger an acute increase in levels of growth hormone or prolactin in humans.88 Energy drinks may contain up to 1,000 mg of taurine per 8-oz serving, but the effects of larger doses on growth hormone, which is banned as a supplement by major athletic organizations because of its anabolic and possible performance-enhancing effects, remain to be determined.
Taurine may have effects on human sex hormones, based on the limited observations in rodents.89–94
Although human salivary cortisol concentrations were purportedly assessed in response to 2,000 mg of oral taurine,95 the methods and reported data are not adequate to draw any conclusions.
Energy metabolism
Mammals are unable to directly use taurine in energy production because they cannot directly reduce it.25 Instead, bacteria in the gut use it as a source of energy, carbon, nitrogen, and sulfur.96 However, taurine deficiency appears to impair the cellular respiratory chain, resulting in diminished production of adenosine triphosphate and diminished uptake of long-chain fatty acids by mitochondria, at least in the heart.97
Taurine is present in human mitochondria and regulates mitochondrial function. For example, taurine in mitochondria assists in conjugation of transfer RNA for leucine, lysine, glutamate, and glutamine.98 In TauT knockout mice, deficiency of taurine causes mitochondrial dysfunction, triggering a greater than 80% decrease in exercise capacity.99 Several studies in rodents have shown increased exercise capacity after taurine supplementation.100–102 In addition, taurine is critical for the growth of blastocytes, skeletal muscle, and myocardium; it is necessary for mitochondrial development and is also important for muscular endurance.103,104
Antioxidation, anti-inflammation, and other functions
Taurine is a major antioxidant, scavenging reactive oxygen and protecting against oxidative stress to organs including the brain,97,105,106 where it increasingly appears to have neuroprotective effects.107,108
Cellular taurine also has anti-inflammatory actions.3 One of the proposed mechanisms is taurine inhibition of NF-kappa B, an important transcription factor for the synthesis of pro-inflammatory cytokines.4 This function may be important in protecting polyunsaturated fatty acids from oxidative stress—helping to maintain and stabilize the structure and function of plasma membranes within the lungs,109 heart,110 brain,111 liver,112 and spermatozoa.61,62
Taurine is also conjugated to bile acids synthesized in the liver, forming bile salts70 that act as detergents to help emulsify and digest lipids in the body. In addition, taurine facilitates xenobiotic detoxification in the liver by conjugating with several drugs to aid in their excretion.25 Taurine is also implicated in calcium modulation113 and homeostasis.114 Through inhibition of several types of calcium channels, taurine has been shown to decrease calcium influx into cells, effectively serving a cytoprotective role against calcium overload.115,116
TAURINE DEFICIENCY
Fetal and neonatal deficiency
Though taurine is considered nonessential in adults because it can be readily synthesized endogenously, it is thought to be conditionally essential in neonatal nutrition.68 It is the second most abundant free amino acid in human breast milk117 and the most abundant free amino acid in fetal brain.118 In cases of long-term parenteral nutrition, neonates can become drastically taurine deficient119 due to suboptimal CSD activity,118 leading to retinal dysfunction.41 Taurine deficiencies can lead to functional and structural brain damage.118 Moreover, maternal taurine deficiency results in neurologic abnormalities in offspring120 and may lead to oxidative stress throughout life.121
In 1984, the FDA approved the inclusion of taurine in infant formulas based on research showing that taurine-deficient infants had impaired fat absorption, bile acid secretion, retinal function, and hepatic function.122 But still under debate are the amount and duration of taurine supplementation required by preterm and low-birth-weight infants, as several randomized controlled trials failed to show statistically significant effects on growth.123 Nonetheless, given the alleged detrimental ramifications of a lack of taurine supplementation, as well as the ethical dilemma of performing additional research trials on infants, it is presumed that infant formulas and parenteral nutrition for preterm and low-birth-weight infants will continue to contain taurine.
Age- and disease-related deficiency
Although taurine deficiency is rare in neonates, it is perhaps inevitable with advancing age. Healthy elderly patients ages 61 to 81 have up to a 49% decrease in plasma taurine concentration compared with healthy individuals ages 27 to 57.124 While reduced renal retention125 and taurine intake126 can account for depressed taurine levels, Eppler and Dawson127 found that tissue and circulating taurine concentrations decrease over the human life span primarily due to an age-dependent depletion of CSD activity in the liver. This effectively impairs the biosynthesis of endogenous taurine from cysteine or methionine or both, forcing a greater reliance on exogenous sources.
While specific mechanisms have not been fully elucidated, taurine deficiency has also been identified in patients suffering from diseases including but not limited to disorders of bone (osteogenesis imperfecta, osteoporosis),128 blood (acute myelogenous leukemia),129 central nervous system (schizophrenia, Friedreich ataxia-spinocerebellar degeneration),130,131 retina (retinitis pigmentosa),132 circulatory system and heart (essential hypertension, atherosclerosis),133 digestion (Gaucher disease),134 absorption (short-bowel syndrome),135 cellular proliferation (cancer),136 and membrane channels (cystic fibrosis),137 as well as in patients restricted to long-term parenteral nutrition.138 However, the apparent correlation between taurine deficiency and these conditions does not necessarily mean causation; more study is needed to elucidate a direct connection.
SAFETY AND TOXICITY OF TAURINE SUPPLEMENTATION
An upper safe level of intake for taurine has not been established. To date, several studies have involved heavy taurine supplementation without serious adverse effects. While the largest dosage of taurine tested in humans appears to be 10 g/day for 6 months,139 a number of studies have used 1 to 6 g/day for periods of 1 week to 1 year.140 However, the assessment of potential acute, subacute, and chronic adverse effects has not been comprehensive. The Scientific Committee on Food of the European Commission141 reviewed several toxicologic studies on taurine through 2003 and were unable to expose any carcinogenic or teratogenic potential. Nevertheless, based on the available data from trials in humans and lower animals, Shao and Hathcock140 suggested an observed safe level of taurine of 3 g/day, a conservatively smaller dose that carries a higher level of confidence. Because there is no “observed adverse effect level” for daily taurine intake,141 more research must be done to ensure safety of higher amounts of taurine administration and to define a tolerable upper limit of intake.
Interactions with medications
To date, the literature is scarce regarding potential interactions between taurine and commonly used medications.
Although no evidence specifically links taurine with adverse effects when used concurrently with other medications, there may be a link between taurine supplementation and various cytochrome P450 systems responsible for hepatic drug metabolism. Specifically, taurine inhibits cytochrome P450 2E1, a highly conserved xenobiotic-metabolizing P450 responsible for the breakdown of more than 70 substrates, including several commonly used anesthetics, analgesics, antidepressants, antibacterials, and antiepileptics.142 Of note, taurine may contribute to the attenuation of oxidative stress in the liver in the presence of alcohol143 and acetaminophen,144 two substances frequently used and abused. Since the P450 2E1 system catalyzes comparable reactions in rodents and humans,142 rodents should plausibly serve as a model for further testing of the effects of taurine on various substrates.
POTENTIAL THERAPEUTIC APPLICATIONS
More analysis is needed to fully unlock and understand taurine’s potential value in healthcare.
Correction of late-life taurine decline in humans could be beneficial for cognitive performance, energy metabolism, sexual function, and vision, but clinical studies remain to be performed. While a decline in taurine with age may intensify the stress caused by reactive oxygen species, taurine supplementation has been shown to decrease the presence of oxidative markers127 and to serve a neuroprotective role in rodents.145,146 Taurine levels increase in the hippocampus after experimentally induced gliosis147 and are neuroprotective against glutamate excitotoxicity.148,149 Furthermore, data in Alzheimer disease, Huntington disease, and brain ischemia experimental models show that taurine inhibits neuronal death (apoptosis).13,150,151 Taurine has even been proposed as a potential preventive treatment for Alzheimer dementia, as it stabilizes protein conformations to prevent their aggregation and subsequent dysfunction.152 Although improvement in memory and cognitive performance has been linked to taurine supplementation in old mice,145,153 similar results have not been found in adult mice whose taurine levels are within normal limits. Taurine also has transient anticonvulsant effects in some epileptic humans.154
Within the male reproductive organs, the age-related decline in taurine may or may not have implications regarding sexuality, as only very limited rat data are available.89–91
In cats, taurine supplementation has been found to prevent the progressive degeneration of retinal photoreceptors seen in retinitis pigmentosa, a genetic disease that causes the loss of vision.155
While several energy drink companies have advertised that taurine plays a role in improving cognitive and physical performance, there are few human studies that examine this contention in the absence of confounding factors such as caffeine or glucose.36,37,95 Taurine supplementation in patients with heart failure has been shown to increase exercise capacity vs placebo.156 This supports the idea that in cases of taurine deficiency, such as those seen in cardiomyopathy,157 taurine supplementation could have restorative effects. However, we are not aware of any double-blind, placebo-controlled clinical trial of taurine alone in healthy patients that measured energy parameters as clinical outcomes.
Although it remains possible that acute supraphysiologic taurine levels achieved by supplementation could transiently trigger various psychoneuroendocrine responses in healthy people, clinical research is needed in which taurine is the sole intervention. At present, the most compelling clinical reason to prescribe or recommend taurine supplementation is taurine deficiency.
McLellan TM, Lieberman HR. Do energy drinks contain active components other than caffeine? Nutr Rev 2012; 70:730–744.
Park E, Quinn MR, Wright CE, Schuller-Levis G. Taurine chloramine inhibits the synthesis of nitric oxide and the release of tumor necrosis factor in activated RAW 264.7 cells. J Leukoc Biol 1993; 54:119–124.
Kontny E, Szczepanska K, Kowalczewski J, et al. The mechanism of taurine chloramine inhibition of cytokine (interleukin-6, interleukin-8) production by rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheum 2000; 43:2169–2177.
Barbeau A, Inoue N, Tsukada Y, Butterworth RF. The neuropharmacology of taurine. Life Sci 1975; 17:669–677.
Kramer JH, Chovan JP, Schaffer SW. Effect of taurine on calcium paradox and ischemic heart failure. Am J Physiol 1981; 240:H238–H246.
Azuma J, Sawamura A, Awata N, et al. Therapeutic effect of taurine in congestive heart failure: a double-blind crossover trial. Clin Cardiol 1985; 8:276–282.
Darling PB, Lepage G, Leroy C, Masson P, Roy CC. Effect of taurine supplements on fat absorption in cystic fibrosis. Pediatr Res 1985; 19:578–582.
Franconi F, Di Leo MA, Bennardini F, Ghirlanda G. Is taurine beneficial in reducing risk factors for diabetes mellitus? Neurochem Res 2004; 29:143–150.
Malcangio M, Bartolini A, Ghelardini C, et al. Effect of ICV taurine on the impairment of learning, convulsions and death caused by hypoxia. Psychopharmacology (Berl) 1989; 98:316–320.
Rivas-Arancibia S, Dorado-Martínez C, Borgonio-Pérez G, et al. Effects of taurine on ozone-induced memory deficits and lipid peroxidation levels in brains of young, mature, and old rats. Environ Res 2000; 82:7–17.
Vohra BP, Hui X. Improvement of impaired memory in mice by taurine. Neural Plast 2000; 7:245–259.
Tadros MG, Khalifa AE, Abdel-Naim AB, Arafa HM. Neuroprotective effect of taurine in 3-nitropropionic acid-induced experimental animal model of Huntington’s disease phenotype. Pharmacol Biochem Behav 2005; 82:574–582.
Zhu DM, Wang M, She JQ, Yu K, Ruan DY. Protection by a taurine supplemented diet from lead-induced deficits of long-term potentiation/depotentiation in dentate gyrus of rats in vivo. Neuroscience 2005; 134:215–224.
Chepkova AN, Sergeeva OA, Haas HL. Taurine rescues hippocampal long-term potentiation from ammonia-induced impairment. Neurobiol Dis 2006; 23:512–521.
Wang GH, Jiang ZL, Fan XJ, Zhang L, Li X, Ke KF. Neuroprotective effect of taurine against focal cerebral ischemia in rats possibly mediated by activation of both GABAA and glycine receptors. Neuropharmacology 2007; 52:1199–1209.
Caltagirone C, Ferrannini L, Marchionni N, Nappi G, Scapagnini G, Trabucchi M. The potential protective effect of tramiprosate (homotaurine) against Alzheimer’s disease: a review. Aging Clin Exp Res 2012; 24:580–587.
Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA 2014; 311:1889–1900.
Sorkin BC, Camp KM, Haggans CJ, et al. Executive summary of NIH workshop on the use and biology of energy drinks: current knowledge and critical gaps. Nutr Rev 2014; 72(suppl 1):1–8.
Centers for Disease Control and Prevention (CDC). Energy drink consumption and its association with sleep problems among US service members on a combat deployment—Afghanistan, 2010. MMWR Morb Mortal Wkly Rep 2012; 61:895–898.
Bailey RL, Saldanha LG, Dwyer JT. Estimating caffeine intake from energy drinks and dietary supplements in the United States. Nutr Rev 2014; 72(suppl 1):9–13.
Stephens MB, Attipoe S, Jones D, Ledford CJ, Deuster PA. Energy drink and energy shot use in the military. Nutr Rev 2014; 72(suppl 1):72–77.
Triebel S, Sproll C, Reusch H, Godelmann R, Lachenmeier DW. Rapid analysis of taurine in energy drinks using amino acid analyzer and Fourier transform infrared (FTIR) spectroscopy as basis for toxicological evaluation. Amino Acids 2007; 33:451–457.
Huxtable RJ. Physiological actions of taurine. Physiol Rev 1992; 72:101–163.
Laidlaw SA, Grosvenor M, Kopple JD. The taurine content of common foodstuffs. JPEN J Parenter Enteral Nutr 1990; 14:183–188.
Manzi P, Pizzoferrato L. Taurine in milk and yoghurt marketed in Italy. Int J Food Sci Nutr 2013; 64:112–116.
Dragnes BT, Larsen R, Ernstsen MH, Mæhre H, Elvevoll EO. Impact of processing on the taurine content in processed seafood and their corresponding unprocessed raw materials. Int J Food Sci Nutr 2009; 60:143–152.
Laidlaw SA, Shultz TD, Cecchino JT, Kopple JD. Plasma and urine taurine levels in vegans. Am J Clin Nutr 1988; 47:660–663.
Rana SK, Sanders TA. Taurine concentrations in the diet, plasma, urine and breast milk of vegans compared with omnivores. Br J Nutr 1986; 56:17–27.
Alford C, Cox H, Wescott R. The effects of Red Bull energy drink on human performance and mood. Amino Acids 2001; 21:139–150.
Hoffman JR, Ratamess NA, Ross R, Shanklin M, Kang J, Faigenbaum AD. Effect of a pre-exercise energy supplement on the acute hormonal response to resistance exercise. J Strength Cond Res 2008; 22:874–882.
Ivy JL, Kammer L, Ding Z, et al. Improved cycling time-trial performance after ingestion of a caffeine energy drink. Int J Sport Nutr Exerc Metab 2009; 19:61–78.
Gonzalez AM, Walsh AL, Ratamess NA, Kang J, Hoffman JR. Effect of a pre-workout energy supplement on acute multi-joint resistance exercise. J Sports Sci Med 2011; 10:261–266.
Yeh TS, Chan KH, Hsu MC, Liu JF. Supplementation with soybean peptides, taurine, pueraria isoflavone, and ginseng saponin complex improves endurance exercise capacity in humans. J Med Food 2011; 14:219–225.
Rutherford JA, Spriet LL, Stellingwerff T. The effect of acute taurine ingestion on endurance performance and metabolism in well-trained cyclists. Int J Sport Nutr Exerc Metab 2010; 20:322–329.
Zhang M, Izumi I, Kagamimori S, et al. Role of taurine supplementation to prevent exercise-induced oxidative stress in healthy young men. Amino Acids 2004; 26:203–207.
Kammerer M, Jaramillo JA, García A, Calderón JC, Valbuena LH. Effects of energy drink major bioactive compounds on the performance of young adults in fitness and cognitive tests: a randomized controlled trial. J Int Soc Sports Nutr 2014; 11:44.
Kermack WO, Slater RH. The preparation of taurine in considerable quantity. Biochem J 1927; 21:1065–1067.
Huxtable RJ, Lippincott SE. Diet and biosynthesis as sources of taurine in the mouse. J Nutr 1982; 112:1003–1010.
Ohkuma S, Tamura J, Kuriyama K. Roles of endogenous and exogenous taurine and glycine in the formation of conjugated bile acids: analyses using freshly isolated and primary cultured rat hepatocytes. Jpn J Pharmacol 1984; 35:347–358.
Chapman GE, Greenwood CE. Taurine in nutrition and brain development. Nutrition Research 1988; 8:955–968.
Andrews S, Schmidt CLA. Titration curves of taurine and cysteic acid. J Biol Chem 1927; 73:651–654.
Huxtable RJ. Taurine in the central nervous system and the mammalian actions of taurine. Prog Neurobiol 1989; 32:471–533.
Jacobsen JG, Smith LH. Biochemistry and physiology of taurine and taurine derivatives. Physiol Rev 1968; 48:424–511.
Hope DB. Pyridoxal phosphate as the coenzyme of the mammalian decarboxylase for L-cysteine sulphinic and L-cysteic acids. Biochem J 1955; 59:497–500.
Pasantes-Morales H, Lopez-Colome AM, Salceda R, Mandel P. Cysteine sulphinate decarboxylase in chick and rat retina during development. J Neurochem 1976; 27:1103–1106.
Oertel WH, Schmechel DE, Weise VK, et al. Comparison of cysteine sulphinic acid decarboxylase isoenzymes and glutamic acid decarboxylase in rat liver and brain. Neuroscience 1981; 6:2701–2714.
Reymond I, Bitoun M, Levillain O, Tappaz M. Regional expression and histological localization of cysteine sulfinate decarboxylase mRNA in the rat kidney. J Histochem Cytochem 2000; 48:1461–1468.
Hu JM, Ikemura R, Chang KT, Suzuki M, Nishihara M, Takahashi M. Expression of cysteine sulfinate decarboxylase mRNA in rat mammary gland. J Vet Med Sci 2000; 62:829–834.
Ueki I, Stipanuk MH. Enzymes of the taurine biosynthetic pathway are expressed in rat mammary gland. J Nutr 2007; 137:1887–1894.
Li JH, Ling YQ, Fan JJ, Zhang XP, Cui S. Expression of cysteine sulfinate decarboxylase (CSD) in male reproductive organs of mice. Histochem Cell Biol 2006; 125:607–613.
Green TR, Fellman JH, Eicher AL, Pratt KL. Antioxidant role and subcellular location of hypotaurine and taurine in human neutrophils. Biochim Biophys Acta 1991; 1073:91–97.
Lefauconnier JM, Portemer C, Chatagner F. Free amino acids and related substances in human glial tumours and in fetal brain: comparison with normal adult brain. Brain Res 1976; 117:105–113.
Sturman JA, Rassin DK, Gaull GE. Taurine in development. Life Sci 1977; 21:1–22.
Sturman JA. Taurine pool sizes in the rat: effects of vitamin B-6 deficiency and high taurine diet. J Nutr 1973; 103:1566–1580.
Terauchi A, Nakazaw A, Johkura K, Yan L, Usuda N. Immunohistochemical localization of taurine in various tissues of the mouse. Amino Acids 1998; 15:151–160.
Lobo MV, Alonso FJ, del Río RM. Immunohistochemical localization of taurine in the male reproductive organs of the rat. J Histochem Cytochem 2000; 48:313–320.
Aaronson DS, Iman R, Walsh TJ, Kurhanewicz J, Turek PJ. A novel application of 1H magnetic resonance spectroscopy: non-invasive identification of spermatogenesis in men with non-obstructive azoospermia. Hum Reprod 2010; 25:847–852.
Holmes RP, Goodman HO, Shihabi ZK, Jarow JP. The taurine and hypotaurine content of human semen. J Androl 1992; 13:289–292.
Alvarez JG, Storey BT. Taurine, hypotaurine, epinephrine and albumin inhibit lipid peroxidation in rabbit spermatozoa and protect against loss of motility. Biol Reprod 1983; 29:548–555.
Das J, Ghosh J, Manna P, Sinha M, Sil PC. Taurine protects rat testes against NaAsO(2)-induced oxidative stress and apoptosis via mitochondrial dependent and independent pathways. Toxicol Lett 2009; 187:201–210.
Mrsny RJ, Meizel S. Inhibition of hamster sperm Na+, K+-ATPase activity by taurine and hypotaurine. Life Sci 1985; 36:271–275.
Phoenix J, Wray S. Changes in human and rat uterine phosphoethanolamine and taurine with pregnancy and parturition. Exp Physiol 1994; 79:601–604.
Lobo MV, Alonso FJ, Latorre A, del Río RM. Immunohistochemical localization of taurine in the rat ovary, oviduct, and uterus. J Histochem Cytochem 2001; 49:1133–1142.
Casslén BG. Free amino acids in human uterine fluid. Possible role of high taurine concentration. J Reprod Med 1987; 32:181–184.
Pamblanco M, Portolés M, Paredes C, Ten A, Comín J. Free amino acids in preterm and term milk from mothers delivering appropriate- or small-for-gestational-age infants. Am J Clin Nutr 1989; 50:778–781.
Rigo J, Senterre J. Is taurine essential for the neonates? Biol Neonate 1977; 32:73–76.
Sturman JA, Hepner GW, Hofmann AF, Thomas PJ. Metabolism of [35S]taurine in man. J Nutr 1975; 105:1206–1214.
Bremer J. Species differences in the conjugation of free bile acids with taurine and glycine. Biochem J 1956; 63:507–513.
Wu JY, Tang XW, Tsai WH. Taurine receptor: kinetic analysis and pharmacological studies. Adv Exp Med Biol 1992; 315:263–268.
Ripps H, Shen W. Review: taurine: a “very essential” amino acid. Mol Vis 2012; 18:2673–2686.
Haas HL, Hösli L. The depression of brain stem neurones by taurine and its interaction with strychnine and bicuculline. Brain Res 1973; 52:399–402.
Bureau MH, Olsen RW. Taurine acts on a subclass of GABAA receptors in mammalian brain in vitro. Eur J Pharmacol 1991; 207:9–16.
Bhattarai JP, Park SJ, Chun SW, Cho DH, Han SK. Activation of synaptic and extrasynaptic glycine receptors by taurine in preoptic hypothalamic neurons. Neurosci Lett 2015; 608:51–56.
Horikoshi T, Asanuma A, Yanagisawa K, Anzai K, Goto S. Taurine and beta-alanine act on both GABA and glycine receptors in Xenopus oocyte injected with mouse brain messenger RNA. Brain Res 1988; 464:97–105.
Miller PS, Smart TG. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci 2010; 31:161–174.
Fujita T, Sato Y. Hypotensive effect of taurine. Possible involvement of the sympathetic nervous system and endogenous opiates. J Clin Invest 1988; 82:993–997.
Fujita T, Ando K, Noda H, Ito Y, Sato Y. Effects of increased adrenomedullary activity and taurine in young patients with borderline hypertension. Circulation 1987; 75:525–532.
Mizushima S, Nara Y, Sawamura M, Yamori Y. Effects of oral taurine supplementation on lipids and sympathetic nerve tone. Adv Exp Med Biol 1996; 403:615–622.
Fujita T, Sato Y. The antihypertensive effect of taurine in DOCA-salt rats. J Hypertens Suppl 1984; 2:S563–S565.
Fujita T, Sato Y, Ando K. Changes in cardiac and hypothalamic noradrenergic activity with taurine in DOCA-salt rats. Am J Physiol 1986; 251:H926–H933.
Sato Y, Ando K, Fujita T. Role of sympathetic nervous system in hypotensive action of taurine in DOCA-salt rats. Hypertension 1987; 9:81–87.
Scheibel J, Elsasser T, Ondo JG. A neuromodulatory role for taurine in controlling prolactin secretion in female rats. Psychoneuroendocrinology 1981; 6:139–144.
Ikuyama S, Okajima T, Kato K, Ibayashi H. Effect of taurine on growth hormone and prolactin secretion in rats: possible interaction with opioid peptidergic system. Life Sci 1988; 43:807–812.
Park E, Park SY, Dobkin C, Schuller-Levis G. Development of a novel cysteine sulfinic acid decarboxylase knockout mouse: dietary taurine reduces neonatal mortality. J Amino Acids 2014; 2014:346809.
Mantovani J, DeVivo DC. Effects of taurine on seizures and growth hormone release in epileptic patients. Arch Neurol 1979; 36:672–674.
Carlson HE, Miglietta JT, Roginsky MS, Stegink LD. Stimulation of pituitary hormone secretion by neurotransmitter amino acids in humans. Metabolism 1989; 38:1179–1182.
Yang J, Wu G, Feng Y, Lv Q, Lin S, Hu J. Effects of taurine on male reproduction in rats of different ages. J Biomed Sci 2010; 17(suppl 1):S9.
Yang J, Lin S, Feng Y, Wu G, Hu J. Taurine enhances the sexual response and mating ability in aged male rats. Adv Exp Med Biol 2013; 776:347–355.
Yang J, Zong X, Wu G, Lin S, Feng Y, Hu J. Taurine increases testicular function in aged rats by inhibiting oxidative stress and apoptosis. Amino Acids 2015; 47:1549–1558.
Scheibel J, Elsasser T, Ondo JG. Stimulation of LH and FSH secretion following intraventricular injection of cysteic acid but not taurine. Brain Res 1980; 201:99–106.
Price MT, Olney JW, Mitchell MV, Fuller T, Cicero TJ. Luteinizing hormone releasing action of N-methyl aspartate is blocked by GABA or taurine but not by dopamine antagonists. Brain Res 1978; 158:461–465.
Ma Q, Zhao J, Cao W, Liu J, Cui S. Estradiol decreases taurine level by reducing cysteine sulfinic acid decarboxylase via the estrogen receptor-a in female mice liver. Am J Physiol Gastrointest Liver Physiol 2015; 308:G277–G286.
Giles GE, Mahoney CR, Brunyé TT, Gardony AL, Taylor HA, Kanarek RB. Differential cognitive effects of energy drink ingredients: caffeine, taurine, and glucose. Pharmacol Biochem Behav 2012; 102:569–577.
Giehl TJ, Qoronfleh MW, Wilkinson BJ. Transport, nutritional and metabolic studies of taurine in staphylococci. J Gen Microbiol 1987; 133:849–856.
Schaffer SW, Shimada-Takaura K, Jong CJ, Ito T, Takahashi K. Impaired energy metabolism of the taurine-deficient heart. Amino Acids 2015 Oct 16. Epub ahead of print.
Schaffer SW, Jong CJ, Ito T, Azuma J. Role of taurine in the pathologies of MELAS and MERRF. Amino Acids 2014; 46:47–56.
Warskulat U, Heller-Stilb B, Oermann E, et al. Phenotype of the taurine transporter knockout mouse. Methods Enzymol 2007; 428:439–458.
Yatabe Y, Miyakawa S, Miyazaki T, Matsuzaki Y, Ochiai N. Effects of taurine administration in rat skeletal muscles on exercise. J Orthop Sci 2003; 8:415–419.
Miyazaki T, Matsuzaki Y, Ikegami T, et al. Optimal and effective oral dose of taurine to prolong exercise performance in rat. Amino Acids 2004; 27:291–298.
Goodman CA, Horvath D, Stathis C, et al. Taurine supplementation increases skeletal muscle force production and protects muscle function during and after high-frequency in vitro stimulation. J Appl Physiol (1985) 2009; 107:144–154.
Van Winkle LJ, Patel M, Wasserlauf HG, Dickinson HR, Campione AL. Osmotic regulation of taurine transport via system beta and novel processes in mouse preimplantation conceptuses. Biochim Biophys Acta 1994; 1191:244–255.
Ito T, Kimura Y, Uozumi Y, et al. Taurine depletion caused by knocking out the taurine transporter gene leads to cardiomyopathy with cardiac atrophy. J Mol Cell Cardiol 2008; 44:927–937.
Aydın AF, Çoban J, Dogan-Ekici I, Betül-Kalaz E, Dogru-Abbasoglu S, Uysal M. Carnosine and taurine treatments diminished brain oxidative stress and apoptosis in D-galactose aging model. Metab Brain Dis 2016; 31:337–345.
Nagai K, Fukuno S, Oda A, Konishi H. Protective effects of taurine on doxorubicin-induced acute hepatotoxicity through suppression of oxidative stress and apoptotic responses. Anticancer Drugs 2016; 27:17–23.
Caletti G, Almeida FB, Agnes G, Nin MS, Barros HM, Gomez R. Antidepressant dose of taurine increases mRNA expression of GABAA receptor a2 subunit and BDNF in the hippocampus of diabetic rats. Behav Brain Res 2015; 283:11–55.
Gu Y, Zhao Y, Qian K, Sun M. Taurine attenuates hippocampal and corpus callosum damage, and enhances neurological recovery after closed head injury in rats. Neuroscience 2015; 291:331–340.
Banks MA, Porter DW, Pailes WH, Schwegler-Berry D, Martin WG, Castranova V. Taurine content of isolated rat alveolar type I cells. Comp Biochem Physiol B 1991; 100:795–799.
Raschke P, Massoudy P, Becker BF. Taurine protects the heart from neutrophil-induced reperfusion injury. Free Radic Biol Med 1995; 19:461–471.
Pushpakiran G, Mahalakshmi K, Anuradha CV. Taurine restores ethanol-induced depletion of antioxidants and attenuates oxidative stress in rat tissues. Amino Acids 2004; 27:91–96.
Refik Mas M, Comert B, Oncu K, et al. The effect of taurine treatment on oxidative stress in experimental liver fibrosis. Hepatol Res 2004; 28:207–215.
Sebring LA, Huxtable RJ. Taurine modulation of calcium binding to cardiac sarcolemma. J Pharmacol Exp Ther 1985; 232:445–451.
Schaffer SW, Kramer J, Chovan JP. Regulation of calcium homeostasis in the heart by taurine. Fed Proc 1980; 39:2691–2694.
Foos TM, Wu JY. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem Res 2002; 27:21–26.
Ito T, Schaffer S, Azuma J. The effect of taurine on chronic heart failure: actions of taurine against catecholamine and angiotensin II. Amino Acids 2014; 46:111–119.
Gaull GE. Taurine in pediatric nutrition: review and update. Pediatrics 1989; 83:433–442.
Sturman JA, Rassin DK, Gaull GE. Taurine in developing rat brain: transfer of [35S] taurine to pups via the milk. Pediatr Res 1977; 11:28–33.
Geggel HS, Ament ME, Heckenlively JR, Martin DA, Kopple JD. Nutritional requirement for taurine in patients receiving long-term parenteral nutrition. N Engl J Med 1985; 312:142–146.
Sturman JA, Gargano AD, Messing JM, Imaki H. Feline maternal taurine deficiency: effect on mother and offspring. J Nutr 1986; 116:655–667.
Chesney RW, Helms RA, Christensen M, Budreau AM, Han X, Sturman JA. The role of taurine in infant nutrition. Adv Exp Med Biol 1998; 442:463–476.
Verner A, Craig S, McGuire W. Effect of taurine supplementation on growth and development in preterm or low birth weight infants. Cochrane Database Syst Rev 2007; 4:CD006072.
Jeevanandam M, Young DH, Ramias L, Schiller WR. Effect of major trauma on plasma free amino acid concentrations in geriatric patients. Am J Clin Nutr 1990; 51:1040–1045.
Dawson R Jr, Eppler B, Patterson TA, Shih D, Liu S. The effects of taurine in a rodent model of aging. Adv Exp Med Biol 1996; 403:37–50.
Han X, Budreau AM, Chesney RW. Molecular cloning and functional expression of an LLC-PK1 cell taurine transporter that is adaptively regulated by taurine. Adv Exp Med Biol 1998; 442:261–268.
Eppler B, Dawson R Jr. Dietary taurine manipulations in aged male Fischer 344 rat tissue: taurine concentration, taurine biosynthesis, and oxidative markers. Biochem Pharmacol 2001; 62:29–39.
D’Eufemia P, Finocchiaro R, Celli M, et al. Taurine deficiency in thalassemia major-induced osteoporosis treated with neridronate. Biomed Pharmacother 2010; 64:271–274.
Muscaritoli M, Conversano L, Petti MC, et al. Plasma amino acid concentrations in patients with acute myelogenous leukemia. Nutrition 1999; 15:195–199.
Do KQ, Lauer CJ, Schreiber W, et al. Gamma-glutamylglutamine and taurine concentrations are decreased in the cerebrospinal fluid of drug-naive patients with schizophrenic disorders. J Neurochem 1995; 65:2652–2662.
Lemieux B, Giguère R, Shapcott D. Studies on the role of taurine in Friedreich’s ataxia. Can J Neurol Sci 1984; 11(suppl 4):610–615.
Airaksinen EM, Oja SS, Marnela KM, Sihvola P. Taurine and other amino acids of platelets and plasma in retinitis pigmentosa. Ann Clin Res 1980; 12:52–54.
Kohashi N, Katori R. Decrease of urinary taurine in essential hypertension. Jpn Heart J 1983; 24:91–102.
vom Dahl S, Mönnighoff I, Häussinger D. Decrease of plasma taurine in Gaucher disease and its sustained correction during enzyme replacement therapy. Amino Acids 2000; 19:585–592.
Schneider SM, Joly F, Gehrardt MF, et al. Taurine status and response to intravenous taurine supplementation in adults with short-bowel syndrome undergoing long-term parenteral nutrition: a pilot study. Br J Nutr 2006; 96:365–370.
Gray GE, Landel AM, Meguid MM. Taurine-supplemented total parenteral nutrition and taurine status of malnourished cancer patients. Nutrition 1994; 10:11–15.
Thompson GN. Assessment of taurine deficiency in cystic fibrosis. Clin Chim Acta 1988; 171:233–237.
Cho KH, Kim ES, Chen JD. Taurine intake and excretion in patients undergoing long term enteral nutrition. Adv Exp Med Biol 2000; 483:605–612.
Durelli L, Mutani R, Fassio F. The treatment of myotonia: evaluation of chronic oral taurine therapy. Neurology 1983; 33:599–603.
Shao A, Hathcock JN. Risk assessment for the amino acids taurine, L-glutamine and L-arginine. Regul Toxicol Pharmacol 2008; 50:376–399.
Tanaka E, Terada M, Misawa S. Cytochrome P450 2E1: its clinical and toxicological role. J Clin Pharm Ther 2000; 25:165–175.
Kerai MD, Waterfield CJ, Kenyon SH, Asker DS, Timbrell JA. Reversal of ethanol-induced hepatic steatosis and lipid peroxidation by taurine: a study in rats. Alcohol Alcohol 1999; 34:529–541.
Das J, Ghosh J, Manna P, Sil PC. Acetaminophen induced acute liver failure via oxidative stress and JNK activation: protective role of taurine by the suppression of cytochrome P450 2E1. Free Radic Res 2010; 44:340–355.
El Idrissi A, Shen CH, L’amoreaux WJ. Neuroprotective role of taurine during aging. Amino Acids 2013; 45:735–750.
Gharibani P, Modi J, Menzie J, et al. Comparison between single and combined post-treatment with S-methyl-N,N-diethylthiolcarbamate sulfoxide and taurine following transient focal cerebral ischemia in rat brain. Neuroscience 2015; 300:460–473.
Junyent F, De Lemos L, Utrera J, et al. Content and traffic of taurine in hippocampal reactive astrocytes. Hippocampus 2011; 21:185–197.
El Idrissi A, Trenkner E. Growth factors and taurine protect against excitotoxicity by stabilizing calcium homeostasis and energy metabolism. J Neurosci 1999; 19:9459–9468.
Wu H, Jin Y, Wei J, Jin H, Sha D, Wu JY. Mode of action of taurine as a neuroprotector. Brain Res 2005; 1038:123–131.
Paula-Lima AC, De Felice FG, Brito-Moreira J, Ferreira ST. Activation of GABA(A) receptors by taurine and muscimol blocks the neurotoxicity of beta-amyloid in rat hippocampal and cortical neurons. Neuropharmacology 2005; 49:1140–1148.
Takatani T, Takahashi K, Uozumi Y, et al. Taurine inhibits apoptosis by preventing formation of the Apaf-1/caspase-9 apoptosome. Am J Physiol Cell Physiol 2004; 287:C949–C953.
Atamna H, Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J Alzheimers Dis 2010; 20(suppl 2):S439–S452.
El Idrissi A. Taurine improves learning and retention in aged mice. Neurosci Lett 2008; 436:19–22.
Oja SS, Saransaari P. Taurine and epilepsy. Epilepsy Res 2013; 104:187–194.
Berson EL, Hayes KC, Rabin AR, Schmidt SY, Watson G. Retinal degeneration in cats fed casein. II. Supplementation with methionine, cysteine, or taurine. Invest Ophthalmol 1976; 15:52–58.
Beyranvand MR, Khalafi MK, Roshan VD, Choobineh S, Parsa SA, Piranfar MA. Effect of taurine supplementation on exercise capacity of patients with heart failure. J Cardiol 2011; 57:333–337.
Eby G, Halcomb WW. Elimination of cardiac arrhythmias using oral taurine with l-arginine with case histories: hypothesis for nitric oxide stabilization of the sinus node. Med Hypotheses 2006; 67:1200–1204.
McLellan TM, Lieberman HR. Do energy drinks contain active components other than caffeine? Nutr Rev 2012; 70:730–744.
Park E, Quinn MR, Wright CE, Schuller-Levis G. Taurine chloramine inhibits the synthesis of nitric oxide and the release of tumor necrosis factor in activated RAW 264.7 cells. J Leukoc Biol 1993; 54:119–124.
Kontny E, Szczepanska K, Kowalczewski J, et al. The mechanism of taurine chloramine inhibition of cytokine (interleukin-6, interleukin-8) production by rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheum 2000; 43:2169–2177.
Barbeau A, Inoue N, Tsukada Y, Butterworth RF. The neuropharmacology of taurine. Life Sci 1975; 17:669–677.
Kramer JH, Chovan JP, Schaffer SW. Effect of taurine on calcium paradox and ischemic heart failure. Am J Physiol 1981; 240:H238–H246.
Azuma J, Sawamura A, Awata N, et al. Therapeutic effect of taurine in congestive heart failure: a double-blind crossover trial. Clin Cardiol 1985; 8:276–282.
Darling PB, Lepage G, Leroy C, Masson P, Roy CC. Effect of taurine supplements on fat absorption in cystic fibrosis. Pediatr Res 1985; 19:578–582.
Franconi F, Di Leo MA, Bennardini F, Ghirlanda G. Is taurine beneficial in reducing risk factors for diabetes mellitus? Neurochem Res 2004; 29:143–150.
Malcangio M, Bartolini A, Ghelardini C, et al. Effect of ICV taurine on the impairment of learning, convulsions and death caused by hypoxia. Psychopharmacology (Berl) 1989; 98:316–320.
Rivas-Arancibia S, Dorado-Martínez C, Borgonio-Pérez G, et al. Effects of taurine on ozone-induced memory deficits and lipid peroxidation levels in brains of young, mature, and old rats. Environ Res 2000; 82:7–17.
Vohra BP, Hui X. Improvement of impaired memory in mice by taurine. Neural Plast 2000; 7:245–259.
Tadros MG, Khalifa AE, Abdel-Naim AB, Arafa HM. Neuroprotective effect of taurine in 3-nitropropionic acid-induced experimental animal model of Huntington’s disease phenotype. Pharmacol Biochem Behav 2005; 82:574–582.
Zhu DM, Wang M, She JQ, Yu K, Ruan DY. Protection by a taurine supplemented diet from lead-induced deficits of long-term potentiation/depotentiation in dentate gyrus of rats in vivo. Neuroscience 2005; 134:215–224.
Chepkova AN, Sergeeva OA, Haas HL. Taurine rescues hippocampal long-term potentiation from ammonia-induced impairment. Neurobiol Dis 2006; 23:512–521.
Wang GH, Jiang ZL, Fan XJ, Zhang L, Li X, Ke KF. Neuroprotective effect of taurine against focal cerebral ischemia in rats possibly mediated by activation of both GABAA and glycine receptors. Neuropharmacology 2007; 52:1199–1209.
Caltagirone C, Ferrannini L, Marchionni N, Nappi G, Scapagnini G, Trabucchi M. The potential protective effect of tramiprosate (homotaurine) against Alzheimer’s disease: a review. Aging Clin Exp Res 2012; 24:580–587.
Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA 2014; 311:1889–1900.
Sorkin BC, Camp KM, Haggans CJ, et al. Executive summary of NIH workshop on the use and biology of energy drinks: current knowledge and critical gaps. Nutr Rev 2014; 72(suppl 1):1–8.
Centers for Disease Control and Prevention (CDC). Energy drink consumption and its association with sleep problems among US service members on a combat deployment—Afghanistan, 2010. MMWR Morb Mortal Wkly Rep 2012; 61:895–898.
Bailey RL, Saldanha LG, Dwyer JT. Estimating caffeine intake from energy drinks and dietary supplements in the United States. Nutr Rev 2014; 72(suppl 1):9–13.
Stephens MB, Attipoe S, Jones D, Ledford CJ, Deuster PA. Energy drink and energy shot use in the military. Nutr Rev 2014; 72(suppl 1):72–77.
Triebel S, Sproll C, Reusch H, Godelmann R, Lachenmeier DW. Rapid analysis of taurine in energy drinks using amino acid analyzer and Fourier transform infrared (FTIR) spectroscopy as basis for toxicological evaluation. Amino Acids 2007; 33:451–457.
Huxtable RJ. Physiological actions of taurine. Physiol Rev 1992; 72:101–163.
Laidlaw SA, Grosvenor M, Kopple JD. The taurine content of common foodstuffs. JPEN J Parenter Enteral Nutr 1990; 14:183–188.
Manzi P, Pizzoferrato L. Taurine in milk and yoghurt marketed in Italy. Int J Food Sci Nutr 2013; 64:112–116.
Dragnes BT, Larsen R, Ernstsen MH, Mæhre H, Elvevoll EO. Impact of processing on the taurine content in processed seafood and their corresponding unprocessed raw materials. Int J Food Sci Nutr 2009; 60:143–152.
Laidlaw SA, Shultz TD, Cecchino JT, Kopple JD. Plasma and urine taurine levels in vegans. Am J Clin Nutr 1988; 47:660–663.
Rana SK, Sanders TA. Taurine concentrations in the diet, plasma, urine and breast milk of vegans compared with omnivores. Br J Nutr 1986; 56:17–27.
Alford C, Cox H, Wescott R. The effects of Red Bull energy drink on human performance and mood. Amino Acids 2001; 21:139–150.
Hoffman JR, Ratamess NA, Ross R, Shanklin M, Kang J, Faigenbaum AD. Effect of a pre-exercise energy supplement on the acute hormonal response to resistance exercise. J Strength Cond Res 2008; 22:874–882.
Ivy JL, Kammer L, Ding Z, et al. Improved cycling time-trial performance after ingestion of a caffeine energy drink. Int J Sport Nutr Exerc Metab 2009; 19:61–78.
Gonzalez AM, Walsh AL, Ratamess NA, Kang J, Hoffman JR. Effect of a pre-workout energy supplement on acute multi-joint resistance exercise. J Sports Sci Med 2011; 10:261–266.
Yeh TS, Chan KH, Hsu MC, Liu JF. Supplementation with soybean peptides, taurine, pueraria isoflavone, and ginseng saponin complex improves endurance exercise capacity in humans. J Med Food 2011; 14:219–225.
Rutherford JA, Spriet LL, Stellingwerff T. The effect of acute taurine ingestion on endurance performance and metabolism in well-trained cyclists. Int J Sport Nutr Exerc Metab 2010; 20:322–329.
Zhang M, Izumi I, Kagamimori S, et al. Role of taurine supplementation to prevent exercise-induced oxidative stress in healthy young men. Amino Acids 2004; 26:203–207.
Kammerer M, Jaramillo JA, García A, Calderón JC, Valbuena LH. Effects of energy drink major bioactive compounds on the performance of young adults in fitness and cognitive tests: a randomized controlled trial. J Int Soc Sports Nutr 2014; 11:44.
Kermack WO, Slater RH. The preparation of taurine in considerable quantity. Biochem J 1927; 21:1065–1067.
Huxtable RJ, Lippincott SE. Diet and biosynthesis as sources of taurine in the mouse. J Nutr 1982; 112:1003–1010.
Ohkuma S, Tamura J, Kuriyama K. Roles of endogenous and exogenous taurine and glycine in the formation of conjugated bile acids: analyses using freshly isolated and primary cultured rat hepatocytes. Jpn J Pharmacol 1984; 35:347–358.
Chapman GE, Greenwood CE. Taurine in nutrition and brain development. Nutrition Research 1988; 8:955–968.
Andrews S, Schmidt CLA. Titration curves of taurine and cysteic acid. J Biol Chem 1927; 73:651–654.
Huxtable RJ. Taurine in the central nervous system and the mammalian actions of taurine. Prog Neurobiol 1989; 32:471–533.
Jacobsen JG, Smith LH. Biochemistry and physiology of taurine and taurine derivatives. Physiol Rev 1968; 48:424–511.
Hope DB. Pyridoxal phosphate as the coenzyme of the mammalian decarboxylase for L-cysteine sulphinic and L-cysteic acids. Biochem J 1955; 59:497–500.
Pasantes-Morales H, Lopez-Colome AM, Salceda R, Mandel P. Cysteine sulphinate decarboxylase in chick and rat retina during development. J Neurochem 1976; 27:1103–1106.
Oertel WH, Schmechel DE, Weise VK, et al. Comparison of cysteine sulphinic acid decarboxylase isoenzymes and glutamic acid decarboxylase in rat liver and brain. Neuroscience 1981; 6:2701–2714.
Reymond I, Bitoun M, Levillain O, Tappaz M. Regional expression and histological localization of cysteine sulfinate decarboxylase mRNA in the rat kidney. J Histochem Cytochem 2000; 48:1461–1468.
Hu JM, Ikemura R, Chang KT, Suzuki M, Nishihara M, Takahashi M. Expression of cysteine sulfinate decarboxylase mRNA in rat mammary gland. J Vet Med Sci 2000; 62:829–834.
Ueki I, Stipanuk MH. Enzymes of the taurine biosynthetic pathway are expressed in rat mammary gland. J Nutr 2007; 137:1887–1894.
Li JH, Ling YQ, Fan JJ, Zhang XP, Cui S. Expression of cysteine sulfinate decarboxylase (CSD) in male reproductive organs of mice. Histochem Cell Biol 2006; 125:607–613.
Green TR, Fellman JH, Eicher AL, Pratt KL. Antioxidant role and subcellular location of hypotaurine and taurine in human neutrophils. Biochim Biophys Acta 1991; 1073:91–97.
Lefauconnier JM, Portemer C, Chatagner F. Free amino acids and related substances in human glial tumours and in fetal brain: comparison with normal adult brain. Brain Res 1976; 117:105–113.
Sturman JA, Rassin DK, Gaull GE. Taurine in development. Life Sci 1977; 21:1–22.
Sturman JA. Taurine pool sizes in the rat: effects of vitamin B-6 deficiency and high taurine diet. J Nutr 1973; 103:1566–1580.
Terauchi A, Nakazaw A, Johkura K, Yan L, Usuda N. Immunohistochemical localization of taurine in various tissues of the mouse. Amino Acids 1998; 15:151–160.
Lobo MV, Alonso FJ, del Río RM. Immunohistochemical localization of taurine in the male reproductive organs of the rat. J Histochem Cytochem 2000; 48:313–320.
Aaronson DS, Iman R, Walsh TJ, Kurhanewicz J, Turek PJ. A novel application of 1H magnetic resonance spectroscopy: non-invasive identification of spermatogenesis in men with non-obstructive azoospermia. Hum Reprod 2010; 25:847–852.
Holmes RP, Goodman HO, Shihabi ZK, Jarow JP. The taurine and hypotaurine content of human semen. J Androl 1992; 13:289–292.
Alvarez JG, Storey BT. Taurine, hypotaurine, epinephrine and albumin inhibit lipid peroxidation in rabbit spermatozoa and protect against loss of motility. Biol Reprod 1983; 29:548–555.
Das J, Ghosh J, Manna P, Sinha M, Sil PC. Taurine protects rat testes against NaAsO(2)-induced oxidative stress and apoptosis via mitochondrial dependent and independent pathways. Toxicol Lett 2009; 187:201–210.
Mrsny RJ, Meizel S. Inhibition of hamster sperm Na+, K+-ATPase activity by taurine and hypotaurine. Life Sci 1985; 36:271–275.
Phoenix J, Wray S. Changes in human and rat uterine phosphoethanolamine and taurine with pregnancy and parturition. Exp Physiol 1994; 79:601–604.
Lobo MV, Alonso FJ, Latorre A, del Río RM. Immunohistochemical localization of taurine in the rat ovary, oviduct, and uterus. J Histochem Cytochem 2001; 49:1133–1142.
Casslén BG. Free amino acids in human uterine fluid. Possible role of high taurine concentration. J Reprod Med 1987; 32:181–184.
Pamblanco M, Portolés M, Paredes C, Ten A, Comín J. Free amino acids in preterm and term milk from mothers delivering appropriate- or small-for-gestational-age infants. Am J Clin Nutr 1989; 50:778–781.
Rigo J, Senterre J. Is taurine essential for the neonates? Biol Neonate 1977; 32:73–76.
Sturman JA, Hepner GW, Hofmann AF, Thomas PJ. Metabolism of [35S]taurine in man. J Nutr 1975; 105:1206–1214.
Bremer J. Species differences in the conjugation of free bile acids with taurine and glycine. Biochem J 1956; 63:507–513.
Wu JY, Tang XW, Tsai WH. Taurine receptor: kinetic analysis and pharmacological studies. Adv Exp Med Biol 1992; 315:263–268.
Ripps H, Shen W. Review: taurine: a “very essential” amino acid. Mol Vis 2012; 18:2673–2686.
Haas HL, Hösli L. The depression of brain stem neurones by taurine and its interaction with strychnine and bicuculline. Brain Res 1973; 52:399–402.
Bureau MH, Olsen RW. Taurine acts on a subclass of GABAA receptors in mammalian brain in vitro. Eur J Pharmacol 1991; 207:9–16.
Bhattarai JP, Park SJ, Chun SW, Cho DH, Han SK. Activation of synaptic and extrasynaptic glycine receptors by taurine in preoptic hypothalamic neurons. Neurosci Lett 2015; 608:51–56.
Horikoshi T, Asanuma A, Yanagisawa K, Anzai K, Goto S. Taurine and beta-alanine act on both GABA and glycine receptors in Xenopus oocyte injected with mouse brain messenger RNA. Brain Res 1988; 464:97–105.
Miller PS, Smart TG. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci 2010; 31:161–174.
Fujita T, Sato Y. Hypotensive effect of taurine. Possible involvement of the sympathetic nervous system and endogenous opiates. J Clin Invest 1988; 82:993–997.
Fujita T, Ando K, Noda H, Ito Y, Sato Y. Effects of increased adrenomedullary activity and taurine in young patients with borderline hypertension. Circulation 1987; 75:525–532.
Mizushima S, Nara Y, Sawamura M, Yamori Y. Effects of oral taurine supplementation on lipids and sympathetic nerve tone. Adv Exp Med Biol 1996; 403:615–622.
Fujita T, Sato Y. The antihypertensive effect of taurine in DOCA-salt rats. J Hypertens Suppl 1984; 2:S563–S565.
Fujita T, Sato Y, Ando K. Changes in cardiac and hypothalamic noradrenergic activity with taurine in DOCA-salt rats. Am J Physiol 1986; 251:H926–H933.
Sato Y, Ando K, Fujita T. Role of sympathetic nervous system in hypotensive action of taurine in DOCA-salt rats. Hypertension 1987; 9:81–87.
Scheibel J, Elsasser T, Ondo JG. A neuromodulatory role for taurine in controlling prolactin secretion in female rats. Psychoneuroendocrinology 1981; 6:139–144.
Ikuyama S, Okajima T, Kato K, Ibayashi H. Effect of taurine on growth hormone and prolactin secretion in rats: possible interaction with opioid peptidergic system. Life Sci 1988; 43:807–812.
Park E, Park SY, Dobkin C, Schuller-Levis G. Development of a novel cysteine sulfinic acid decarboxylase knockout mouse: dietary taurine reduces neonatal mortality. J Amino Acids 2014; 2014:346809.
Mantovani J, DeVivo DC. Effects of taurine on seizures and growth hormone release in epileptic patients. Arch Neurol 1979; 36:672–674.
Carlson HE, Miglietta JT, Roginsky MS, Stegink LD. Stimulation of pituitary hormone secretion by neurotransmitter amino acids in humans. Metabolism 1989; 38:1179–1182.
Yang J, Wu G, Feng Y, Lv Q, Lin S, Hu J. Effects of taurine on male reproduction in rats of different ages. J Biomed Sci 2010; 17(suppl 1):S9.
Yang J, Lin S, Feng Y, Wu G, Hu J. Taurine enhances the sexual response and mating ability in aged male rats. Adv Exp Med Biol 2013; 776:347–355.
Yang J, Zong X, Wu G, Lin S, Feng Y, Hu J. Taurine increases testicular function in aged rats by inhibiting oxidative stress and apoptosis. Amino Acids 2015; 47:1549–1558.
Scheibel J, Elsasser T, Ondo JG. Stimulation of LH and FSH secretion following intraventricular injection of cysteic acid but not taurine. Brain Res 1980; 201:99–106.
Price MT, Olney JW, Mitchell MV, Fuller T, Cicero TJ. Luteinizing hormone releasing action of N-methyl aspartate is blocked by GABA or taurine but not by dopamine antagonists. Brain Res 1978; 158:461–465.
Ma Q, Zhao J, Cao W, Liu J, Cui S. Estradiol decreases taurine level by reducing cysteine sulfinic acid decarboxylase via the estrogen receptor-a in female mice liver. Am J Physiol Gastrointest Liver Physiol 2015; 308:G277–G286.
Giles GE, Mahoney CR, Brunyé TT, Gardony AL, Taylor HA, Kanarek RB. Differential cognitive effects of energy drink ingredients: caffeine, taurine, and glucose. Pharmacol Biochem Behav 2012; 102:569–577.
Giehl TJ, Qoronfleh MW, Wilkinson BJ. Transport, nutritional and metabolic studies of taurine in staphylococci. J Gen Microbiol 1987; 133:849–856.
Schaffer SW, Shimada-Takaura K, Jong CJ, Ito T, Takahashi K. Impaired energy metabolism of the taurine-deficient heart. Amino Acids 2015 Oct 16. Epub ahead of print.
Schaffer SW, Jong CJ, Ito T, Azuma J. Role of taurine in the pathologies of MELAS and MERRF. Amino Acids 2014; 46:47–56.
Warskulat U, Heller-Stilb B, Oermann E, et al. Phenotype of the taurine transporter knockout mouse. Methods Enzymol 2007; 428:439–458.
Yatabe Y, Miyakawa S, Miyazaki T, Matsuzaki Y, Ochiai N. Effects of taurine administration in rat skeletal muscles on exercise. J Orthop Sci 2003; 8:415–419.
Miyazaki T, Matsuzaki Y, Ikegami T, et al. Optimal and effective oral dose of taurine to prolong exercise performance in rat. Amino Acids 2004; 27:291–298.
Goodman CA, Horvath D, Stathis C, et al. Taurine supplementation increases skeletal muscle force production and protects muscle function during and after high-frequency in vitro stimulation. J Appl Physiol (1985) 2009; 107:144–154.
Van Winkle LJ, Patel M, Wasserlauf HG, Dickinson HR, Campione AL. Osmotic regulation of taurine transport via system beta and novel processes in mouse preimplantation conceptuses. Biochim Biophys Acta 1994; 1191:244–255.
Ito T, Kimura Y, Uozumi Y, et al. Taurine depletion caused by knocking out the taurine transporter gene leads to cardiomyopathy with cardiac atrophy. J Mol Cell Cardiol 2008; 44:927–937.
Aydın AF, Çoban J, Dogan-Ekici I, Betül-Kalaz E, Dogru-Abbasoglu S, Uysal M. Carnosine and taurine treatments diminished brain oxidative stress and apoptosis in D-galactose aging model. Metab Brain Dis 2016; 31:337–345.
Nagai K, Fukuno S, Oda A, Konishi H. Protective effects of taurine on doxorubicin-induced acute hepatotoxicity through suppression of oxidative stress and apoptotic responses. Anticancer Drugs 2016; 27:17–23.
Caletti G, Almeida FB, Agnes G, Nin MS, Barros HM, Gomez R. Antidepressant dose of taurine increases mRNA expression of GABAA receptor a2 subunit and BDNF in the hippocampus of diabetic rats. Behav Brain Res 2015; 283:11–55.
Gu Y, Zhao Y, Qian K, Sun M. Taurine attenuates hippocampal and corpus callosum damage, and enhances neurological recovery after closed head injury in rats. Neuroscience 2015; 291:331–340.
Banks MA, Porter DW, Pailes WH, Schwegler-Berry D, Martin WG, Castranova V. Taurine content of isolated rat alveolar type I cells. Comp Biochem Physiol B 1991; 100:795–799.
Raschke P, Massoudy P, Becker BF. Taurine protects the heart from neutrophil-induced reperfusion injury. Free Radic Biol Med 1995; 19:461–471.
Pushpakiran G, Mahalakshmi K, Anuradha CV. Taurine restores ethanol-induced depletion of antioxidants and attenuates oxidative stress in rat tissues. Amino Acids 2004; 27:91–96.
Refik Mas M, Comert B, Oncu K, et al. The effect of taurine treatment on oxidative stress in experimental liver fibrosis. Hepatol Res 2004; 28:207–215.
Sebring LA, Huxtable RJ. Taurine modulation of calcium binding to cardiac sarcolemma. J Pharmacol Exp Ther 1985; 232:445–451.
Schaffer SW, Kramer J, Chovan JP. Regulation of calcium homeostasis in the heart by taurine. Fed Proc 1980; 39:2691–2694.
Foos TM, Wu JY. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem Res 2002; 27:21–26.
Ito T, Schaffer S, Azuma J. The effect of taurine on chronic heart failure: actions of taurine against catecholamine and angiotensin II. Amino Acids 2014; 46:111–119.
Gaull GE. Taurine in pediatric nutrition: review and update. Pediatrics 1989; 83:433–442.
Sturman JA, Rassin DK, Gaull GE. Taurine in developing rat brain: transfer of [35S] taurine to pups via the milk. Pediatr Res 1977; 11:28–33.
Geggel HS, Ament ME, Heckenlively JR, Martin DA, Kopple JD. Nutritional requirement for taurine in patients receiving long-term parenteral nutrition. N Engl J Med 1985; 312:142–146.
Sturman JA, Gargano AD, Messing JM, Imaki H. Feline maternal taurine deficiency: effect on mother and offspring. J Nutr 1986; 116:655–667.
Chesney RW, Helms RA, Christensen M, Budreau AM, Han X, Sturman JA. The role of taurine in infant nutrition. Adv Exp Med Biol 1998; 442:463–476.
Verner A, Craig S, McGuire W. Effect of taurine supplementation on growth and development in preterm or low birth weight infants. Cochrane Database Syst Rev 2007; 4:CD006072.
Jeevanandam M, Young DH, Ramias L, Schiller WR. Effect of major trauma on plasma free amino acid concentrations in geriatric patients. Am J Clin Nutr 1990; 51:1040–1045.
Dawson R Jr, Eppler B, Patterson TA, Shih D, Liu S. The effects of taurine in a rodent model of aging. Adv Exp Med Biol 1996; 403:37–50.
Han X, Budreau AM, Chesney RW. Molecular cloning and functional expression of an LLC-PK1 cell taurine transporter that is adaptively regulated by taurine. Adv Exp Med Biol 1998; 442:261–268.
Eppler B, Dawson R Jr. Dietary taurine manipulations in aged male Fischer 344 rat tissue: taurine concentration, taurine biosynthesis, and oxidative markers. Biochem Pharmacol 2001; 62:29–39.
D’Eufemia P, Finocchiaro R, Celli M, et al. Taurine deficiency in thalassemia major-induced osteoporosis treated with neridronate. Biomed Pharmacother 2010; 64:271–274.
Muscaritoli M, Conversano L, Petti MC, et al. Plasma amino acid concentrations in patients with acute myelogenous leukemia. Nutrition 1999; 15:195–199.
Do KQ, Lauer CJ, Schreiber W, et al. Gamma-glutamylglutamine and taurine concentrations are decreased in the cerebrospinal fluid of drug-naive patients with schizophrenic disorders. J Neurochem 1995; 65:2652–2662.
Lemieux B, Giguère R, Shapcott D. Studies on the role of taurine in Friedreich’s ataxia. Can J Neurol Sci 1984; 11(suppl 4):610–615.
Airaksinen EM, Oja SS, Marnela KM, Sihvola P. Taurine and other amino acids of platelets and plasma in retinitis pigmentosa. Ann Clin Res 1980; 12:52–54.
Kohashi N, Katori R. Decrease of urinary taurine in essential hypertension. Jpn Heart J 1983; 24:91–102.
vom Dahl S, Mönnighoff I, Häussinger D. Decrease of plasma taurine in Gaucher disease and its sustained correction during enzyme replacement therapy. Amino Acids 2000; 19:585–592.
Schneider SM, Joly F, Gehrardt MF, et al. Taurine status and response to intravenous taurine supplementation in adults with short-bowel syndrome undergoing long-term parenteral nutrition: a pilot study. Br J Nutr 2006; 96:365–370.
Gray GE, Landel AM, Meguid MM. Taurine-supplemented total parenteral nutrition and taurine status of malnourished cancer patients. Nutrition 1994; 10:11–15.
Thompson GN. Assessment of taurine deficiency in cystic fibrosis. Clin Chim Acta 1988; 171:233–237.
Cho KH, Kim ES, Chen JD. Taurine intake and excretion in patients undergoing long term enteral nutrition. Adv Exp Med Biol 2000; 483:605–612.
Durelli L, Mutani R, Fassio F. The treatment of myotonia: evaluation of chronic oral taurine therapy. Neurology 1983; 33:599–603.
Shao A, Hathcock JN. Risk assessment for the amino acids taurine, L-glutamine and L-arginine. Regul Toxicol Pharmacol 2008; 50:376–399.
Tanaka E, Terada M, Misawa S. Cytochrome P450 2E1: its clinical and toxicological role. J Clin Pharm Ther 2000; 25:165–175.
Kerai MD, Waterfield CJ, Kenyon SH, Asker DS, Timbrell JA. Reversal of ethanol-induced hepatic steatosis and lipid peroxidation by taurine: a study in rats. Alcohol Alcohol 1999; 34:529–541.
Das J, Ghosh J, Manna P, Sil PC. Acetaminophen induced acute liver failure via oxidative stress and JNK activation: protective role of taurine by the suppression of cytochrome P450 2E1. Free Radic Res 2010; 44:340–355.
El Idrissi A, Shen CH, L’amoreaux WJ. Neuroprotective role of taurine during aging. Amino Acids 2013; 45:735–750.
Gharibani P, Modi J, Menzie J, et al. Comparison between single and combined post-treatment with S-methyl-N,N-diethylthiolcarbamate sulfoxide and taurine following transient focal cerebral ischemia in rat brain. Neuroscience 2015; 300:460–473.
Junyent F, De Lemos L, Utrera J, et al. Content and traffic of taurine in hippocampal reactive astrocytes. Hippocampus 2011; 21:185–197.
El Idrissi A, Trenkner E. Growth factors and taurine protect against excitotoxicity by stabilizing calcium homeostasis and energy metabolism. J Neurosci 1999; 19:9459–9468.
Wu H, Jin Y, Wei J, Jin H, Sha D, Wu JY. Mode of action of taurine as a neuroprotector. Brain Res 2005; 1038:123–131.
Paula-Lima AC, De Felice FG, Brito-Moreira J, Ferreira ST. Activation of GABA(A) receptors by taurine and muscimol blocks the neurotoxicity of beta-amyloid in rat hippocampal and cortical neurons. Neuropharmacology 2005; 49:1140–1148.
Takatani T, Takahashi K, Uozumi Y, et al. Taurine inhibits apoptosis by preventing formation of the Apaf-1/caspase-9 apoptosome. Am J Physiol Cell Physiol 2004; 287:C949–C953.
Atamna H, Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J Alzheimers Dis 2010; 20(suppl 2):S439–S452.
El Idrissi A. Taurine improves learning and retention in aged mice. Neurosci Lett 2008; 436:19–22.
Oja SS, Saransaari P. Taurine and epilepsy. Epilepsy Res 2013; 104:187–194.
Berson EL, Hayes KC, Rabin AR, Schmidt SY, Watson G. Retinal degeneration in cats fed casein. II. Supplementation with methionine, cysteine, or taurine. Invest Ophthalmol 1976; 15:52–58.
Beyranvand MR, Khalafi MK, Roshan VD, Choobineh S, Parsa SA, Piranfar MA. Effect of taurine supplementation on exercise capacity of patients with heart failure. J Cardiol 2011; 57:333–337.
Eby G, Halcomb WW. Elimination of cardiac arrhythmias using oral taurine with l-arginine with case histories: hypothesis for nitric oxide stabilization of the sinus node. Med Hypotheses 2006; 67:1200–1204.
Energy drinks are widely consumed in the United States, with an estimated 354 million gallons sold in 2009, or approximately 5.25 L/year per person over age 10.
Taurine has been reported to have anti-inflammatory action. Supplementation has been proposed to have beneficial effects in epilepsy, heart failure, cystic fibrosis, and diabetes, and has been shown in animal studies to protect against neurotoxic insults from alcohol, ammonia, lead, and other substances.
Taurine is an inhibitory neurotransmitter and neuromodulator. It is structurally analogous to gamma-aminobutyric acid, the main inhibitory neurotransmitter in the brain.
In this issueof the Journal, Ataya et al1 provide a comprehensive review of thrombolysis in submassive pulmonary embolism, a subject of much debate. In massive pulmonary embolism, thrombolytic therapy is usually indicated2; in submassive pulmonary embolism, the decision is not so clear. Which patients with submassive embolism would benefit from thrombolysis, and which patients require only anticoagulant therapy? The answer lies in finding the balance between the potential benefit of thrombolytic therapy—preventing death or hemodynamic collapse—and the numerically low but potentially catastrophic risk of intracranial bleeding.
In general, submassive pulmonary embolism refers to an acute pulmonary embolus serious enough to cause evidence of right ventricular dysfunction or necrosis but not hemodynamic instability (ie, with systolic blood pressure > 90 mm Hg) on presentation.3 It accounts for about 25% of cases of pulmonary embolism,4,5 and perhaps 0.5 to 1% of patients admitted to intensive care units across the country.6 The 30-day mortality rate can be as high as 30%, making it a condition that requires prompt identification and appropriate management.
But clinical trials have failed to demonstrate a substantial improvement in mortality rates with thrombolytic therapy in patients with submassive pulmonary embolism, and have shown improvement only in other clinical end points.7 Part of the problem is that this is a heterogeneous condition, posing a challenge for the optimal design and interpretation of studies.
WHO IS AT RISK OF DEATH OR DETERIORATION?
If clinicians could ascertain in each patient whether the risk-benefit ratio is favorable for thrombolytic therapy, it would be easier to provide optimal care. This is not a straightforward task, and it requires integration of clinical judgment, high index of suspicion for deterioration, and clinical tools.
One of the challenges is that it is difficult to identify normotensive patients at the highest risk of poor outcomes. Several factors are associated with a higher risk of death within 30 days (Table 1). While each of these has a negative predictive value of about 95% or even higher (meaning that it is very good at predicting who will not die), they all have very low positive predictive values (meaning that none of them, by itself, is very good at predicting who will die).
For this reason, a multimodal approach to risk stratification has emerged. For example, Jiménez et al8 showed that normotensive patients with acute pulmonary embolism and a combination of abnormal Simplified Pulmonary Embolism Severity Index, elevated B-type natriuretic peptide level, elevated troponin level, and lower-extremity deep vein thrombosis had a 26% rate of complications (death, hemodynamic collapse, or recurrent pulmonary embolism) within 30 days.
Bova et al9 showed that the combination of borderline low systolic blood pressure (90–100 mm Hg), tachycardia (heart rate ≥ 110 beats per minute), elevated troponin, and right ventricular dysfunction by echocardiography or computed tomography allowed for the separation of three groups with significantly different rates of poor outcomes.
WHO IS AT RISK OF BLEEDING?
Estimation of the risk of bleeding is currently less sophisticated, and we need a bleeding score to use in the setting of acute pulmonary embolism. A few studies have shed some light on this issue beyond the known absolute and relative contraindications to thrombolysis.
Ataya et al1 note a meta-analysis10 showing that systemic thrombolytic therapy was not associated with an increased risk of major bleeding in patients age 65 or younger. Similarly, a large observational study showed a strong association between the risk of intracerebral hemorrhage and increasing age11 and also identified comorbidities such as kidney disease as risk factors. While the frequently cited Pulmonary Embolism Thrombolysis trial12 showed a significantly higher risk of stroke with tenecteplase, careful review of its data reveals that all 10 of the 506 patients in the tenecteplase group who sustained a hemorrhagic stroke were age 65 or older.12
A TEAM APPROACH
Thus, in patients with acute pulmonary embolism, clinicians face the difficult task of assessing the patient’s risk of death and clinical worsening and balancing that risk against the risk of bleeding, to identify those who may benefit from early reperfusion therapies, including systemic thrombolysis, catheter-directed thrombolysis, mechanical treatment, and surgical embolectomy.
Given the absence of high-quality evidence to guide these decisions, several institutions have developed multidisciplinary pulmonary embolism response teams to provide rapid evaluation and risk stratification and to recommend and implement advanced therapies, as appropriate. This is a novel concept that is still evolving but holds promise, as it integrates the experience and expertise of physicians in multiple specialties, such as pulmonary and critical care medicine, vascular medicine, interventional radiology, interventional cardiology, emergency medicine, and cardiothoracic surgery, who can then fill the currently existing knowledge gaps for clinical care and, possibly, research.13
Early published experience has documented the feasibility of this multidisciplinary approach.14 The first 95 patients treated at Cleveland Clinic had a 30-day mortality rate of 3.2%, which was lower than the expected 9% rate predicted by the Pulmonary Embolism Severity Index score (unpublished observation).
Figure 1. Cleveland Clinic pulmonary embolism response team algorithm.
Figure 1 shows the algorithm currently used by Cleveland Clinic’s pulmonary embolism response team, with the caveat that no algorithm can fully capture the extent of the complexities and discussions that each case triggers within the team.
TOWARD BETTER UNDERSTANDING
As Ataya et al point out,1 the current state of the evidence does not allow a clear, simplistic, one-size-fits-all approach. A question that arises from this controversial topic is whether we should look for markers of right ventricular dysfunction in every patient admitted with a diagnosis of pulmonary embolism, or only in those with a significant anatomic burden of clot on imaging. Would testing everyone be an appropriate way to identify patients at risk of further deterioration early and therefore prevent adverse outcomes in a timely manner? Or would it not be cost-effective and translate into ordering more diagnostic testing, as well as an increase in downstream workup with higher healthcare costs?
Once we better understand this condition and the factors that predict a higher risk of deterioration, we should be able to design prospective studies that can help elucidate the most appropriate diagnostic and therapeutic approach for such challenging cases. In the meantime, it is important to appraise the evidence in a critical way, as Ataya et al have done in their review.
References
Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. The role of thrombolytic therapy in patients with submassive pulmonary embolism. Cleve Clin J Med 2016; 83:923–932.
Kucher N, Goldhaber SZ. Management of massive pulmonary embolism. Circulation 2005; 112:e28–e32.
Tapson VF. Acute pulmonary embolism. N Engl J Med 2008; 358:1037–1052.
Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
Bahloul M, Chaari A, Kallel H, et al. Pulmonary embolism in intensive care unit: predictive factors, clinical manifestations and outcome. Ann Thorac Med 2010; 5:97–103.
Piazza G, Goldhaber SZ. Fibrinolysis for acute pulmonary embolism. Vasc Med 2010; 15:419–428.
Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med 2014; 189:718–726.
Bova C, Sanchez O, Prandoni P, et al. Identification of intermediate-risk patients with acute symptomatic pulmonary embolism. Eur Respir J 2014; 44:694–703.
Chatterjee S, Chakraborty A, Weinberg I, et al. Thrombolysis for pulmonary embolism and risk of all-cause mortality, major bleeding, and intracranial hemorrhage: a meta-analysis. JAMA 2014; 311:2414–2421.
Stein PD, Matta F, Steinberger DS, Keyes DC. Intracerebral hemorrhage with thrombolytic therapy for acute pulmonary embolism. Am J Med 2012; 125:50–56.
Meyer G, Vicaut E, Danays T, et al. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
Kabrhel C, Rosovsky R, Channick R, et al. A multidisciplinary pulmonary embolism response team: initial 30-month experience with a novel approach to delivery of care to patients with submassive and massive pulmonary embolism. Chest 2016; 150:384–393.
Carlos L. Alviar, MD Assistant Professor, Division of Cardiovascular Medicine, University of Florida College of Medicine, Gainesville
Gustavo A. Heresi, MD Medical Director, Pulmonary Thromboendarterectomy Program, Departments of Pulmonary Medicine and Critical Care Medicine, Respiratory Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Gustavo A. Heresi, MD, Respiratory Institute, A90, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; heresig@ccf.org
Carlos L. Alviar, MD Assistant Professor, Division of Cardiovascular Medicine, University of Florida College of Medicine, Gainesville
Gustavo A. Heresi, MD Medical Director, Pulmonary Thromboendarterectomy Program, Departments of Pulmonary Medicine and Critical Care Medicine, Respiratory Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Gustavo A. Heresi, MD, Respiratory Institute, A90, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; heresig@ccf.org
Author and Disclosure Information
Carlos L. Alviar, MD Assistant Professor, Division of Cardiovascular Medicine, University of Florida College of Medicine, Gainesville
Gustavo A. Heresi, MD Medical Director, Pulmonary Thromboendarterectomy Program, Departments of Pulmonary Medicine and Critical Care Medicine, Respiratory Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Gustavo A. Heresi, MD, Respiratory Institute, A90, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; heresig@ccf.org
In this issueof the Journal, Ataya et al1 provide a comprehensive review of thrombolysis in submassive pulmonary embolism, a subject of much debate. In massive pulmonary embolism, thrombolytic therapy is usually indicated2; in submassive pulmonary embolism, the decision is not so clear. Which patients with submassive embolism would benefit from thrombolysis, and which patients require only anticoagulant therapy? The answer lies in finding the balance between the potential benefit of thrombolytic therapy—preventing death or hemodynamic collapse—and the numerically low but potentially catastrophic risk of intracranial bleeding.
In general, submassive pulmonary embolism refers to an acute pulmonary embolus serious enough to cause evidence of right ventricular dysfunction or necrosis but not hemodynamic instability (ie, with systolic blood pressure > 90 mm Hg) on presentation.3 It accounts for about 25% of cases of pulmonary embolism,4,5 and perhaps 0.5 to 1% of patients admitted to intensive care units across the country.6 The 30-day mortality rate can be as high as 30%, making it a condition that requires prompt identification and appropriate management.
But clinical trials have failed to demonstrate a substantial improvement in mortality rates with thrombolytic therapy in patients with submassive pulmonary embolism, and have shown improvement only in other clinical end points.7 Part of the problem is that this is a heterogeneous condition, posing a challenge for the optimal design and interpretation of studies.
WHO IS AT RISK OF DEATH OR DETERIORATION?
If clinicians could ascertain in each patient whether the risk-benefit ratio is favorable for thrombolytic therapy, it would be easier to provide optimal care. This is not a straightforward task, and it requires integration of clinical judgment, high index of suspicion for deterioration, and clinical tools.
One of the challenges is that it is difficult to identify normotensive patients at the highest risk of poor outcomes. Several factors are associated with a higher risk of death within 30 days (Table 1). While each of these has a negative predictive value of about 95% or even higher (meaning that it is very good at predicting who will not die), they all have very low positive predictive values (meaning that none of them, by itself, is very good at predicting who will die).
For this reason, a multimodal approach to risk stratification has emerged. For example, Jiménez et al8 showed that normotensive patients with acute pulmonary embolism and a combination of abnormal Simplified Pulmonary Embolism Severity Index, elevated B-type natriuretic peptide level, elevated troponin level, and lower-extremity deep vein thrombosis had a 26% rate of complications (death, hemodynamic collapse, or recurrent pulmonary embolism) within 30 days.
Bova et al9 showed that the combination of borderline low systolic blood pressure (90–100 mm Hg), tachycardia (heart rate ≥ 110 beats per minute), elevated troponin, and right ventricular dysfunction by echocardiography or computed tomography allowed for the separation of three groups with significantly different rates of poor outcomes.
WHO IS AT RISK OF BLEEDING?
Estimation of the risk of bleeding is currently less sophisticated, and we need a bleeding score to use in the setting of acute pulmonary embolism. A few studies have shed some light on this issue beyond the known absolute and relative contraindications to thrombolysis.
Ataya et al1 note a meta-analysis10 showing that systemic thrombolytic therapy was not associated with an increased risk of major bleeding in patients age 65 or younger. Similarly, a large observational study showed a strong association between the risk of intracerebral hemorrhage and increasing age11 and also identified comorbidities such as kidney disease as risk factors. While the frequently cited Pulmonary Embolism Thrombolysis trial12 showed a significantly higher risk of stroke with tenecteplase, careful review of its data reveals that all 10 of the 506 patients in the tenecteplase group who sustained a hemorrhagic stroke were age 65 or older.12
A TEAM APPROACH
Thus, in patients with acute pulmonary embolism, clinicians face the difficult task of assessing the patient’s risk of death and clinical worsening and balancing that risk against the risk of bleeding, to identify those who may benefit from early reperfusion therapies, including systemic thrombolysis, catheter-directed thrombolysis, mechanical treatment, and surgical embolectomy.
Given the absence of high-quality evidence to guide these decisions, several institutions have developed multidisciplinary pulmonary embolism response teams to provide rapid evaluation and risk stratification and to recommend and implement advanced therapies, as appropriate. This is a novel concept that is still evolving but holds promise, as it integrates the experience and expertise of physicians in multiple specialties, such as pulmonary and critical care medicine, vascular medicine, interventional radiology, interventional cardiology, emergency medicine, and cardiothoracic surgery, who can then fill the currently existing knowledge gaps for clinical care and, possibly, research.13
Early published experience has documented the feasibility of this multidisciplinary approach.14 The first 95 patients treated at Cleveland Clinic had a 30-day mortality rate of 3.2%, which was lower than the expected 9% rate predicted by the Pulmonary Embolism Severity Index score (unpublished observation).
Figure 1. Cleveland Clinic pulmonary embolism response team algorithm.
Figure 1 shows the algorithm currently used by Cleveland Clinic’s pulmonary embolism response team, with the caveat that no algorithm can fully capture the extent of the complexities and discussions that each case triggers within the team.
TOWARD BETTER UNDERSTANDING
As Ataya et al point out,1 the current state of the evidence does not allow a clear, simplistic, one-size-fits-all approach. A question that arises from this controversial topic is whether we should look for markers of right ventricular dysfunction in every patient admitted with a diagnosis of pulmonary embolism, or only in those with a significant anatomic burden of clot on imaging. Would testing everyone be an appropriate way to identify patients at risk of further deterioration early and therefore prevent adverse outcomes in a timely manner? Or would it not be cost-effective and translate into ordering more diagnostic testing, as well as an increase in downstream workup with higher healthcare costs?
Once we better understand this condition and the factors that predict a higher risk of deterioration, we should be able to design prospective studies that can help elucidate the most appropriate diagnostic and therapeutic approach for such challenging cases. In the meantime, it is important to appraise the evidence in a critical way, as Ataya et al have done in their review.
In this issueof the Journal, Ataya et al1 provide a comprehensive review of thrombolysis in submassive pulmonary embolism, a subject of much debate. In massive pulmonary embolism, thrombolytic therapy is usually indicated2; in submassive pulmonary embolism, the decision is not so clear. Which patients with submassive embolism would benefit from thrombolysis, and which patients require only anticoagulant therapy? The answer lies in finding the balance between the potential benefit of thrombolytic therapy—preventing death or hemodynamic collapse—and the numerically low but potentially catastrophic risk of intracranial bleeding.
In general, submassive pulmonary embolism refers to an acute pulmonary embolus serious enough to cause evidence of right ventricular dysfunction or necrosis but not hemodynamic instability (ie, with systolic blood pressure > 90 mm Hg) on presentation.3 It accounts for about 25% of cases of pulmonary embolism,4,5 and perhaps 0.5 to 1% of patients admitted to intensive care units across the country.6 The 30-day mortality rate can be as high as 30%, making it a condition that requires prompt identification and appropriate management.
But clinical trials have failed to demonstrate a substantial improvement in mortality rates with thrombolytic therapy in patients with submassive pulmonary embolism, and have shown improvement only in other clinical end points.7 Part of the problem is that this is a heterogeneous condition, posing a challenge for the optimal design and interpretation of studies.
WHO IS AT RISK OF DEATH OR DETERIORATION?
If clinicians could ascertain in each patient whether the risk-benefit ratio is favorable for thrombolytic therapy, it would be easier to provide optimal care. This is not a straightforward task, and it requires integration of clinical judgment, high index of suspicion for deterioration, and clinical tools.
One of the challenges is that it is difficult to identify normotensive patients at the highest risk of poor outcomes. Several factors are associated with a higher risk of death within 30 days (Table 1). While each of these has a negative predictive value of about 95% or even higher (meaning that it is very good at predicting who will not die), they all have very low positive predictive values (meaning that none of them, by itself, is very good at predicting who will die).
For this reason, a multimodal approach to risk stratification has emerged. For example, Jiménez et al8 showed that normotensive patients with acute pulmonary embolism and a combination of abnormal Simplified Pulmonary Embolism Severity Index, elevated B-type natriuretic peptide level, elevated troponin level, and lower-extremity deep vein thrombosis had a 26% rate of complications (death, hemodynamic collapse, or recurrent pulmonary embolism) within 30 days.
Bova et al9 showed that the combination of borderline low systolic blood pressure (90–100 mm Hg), tachycardia (heart rate ≥ 110 beats per minute), elevated troponin, and right ventricular dysfunction by echocardiography or computed tomography allowed for the separation of three groups with significantly different rates of poor outcomes.
WHO IS AT RISK OF BLEEDING?
Estimation of the risk of bleeding is currently less sophisticated, and we need a bleeding score to use in the setting of acute pulmonary embolism. A few studies have shed some light on this issue beyond the known absolute and relative contraindications to thrombolysis.
Ataya et al1 note a meta-analysis10 showing that systemic thrombolytic therapy was not associated with an increased risk of major bleeding in patients age 65 or younger. Similarly, a large observational study showed a strong association between the risk of intracerebral hemorrhage and increasing age11 and also identified comorbidities such as kidney disease as risk factors. While the frequently cited Pulmonary Embolism Thrombolysis trial12 showed a significantly higher risk of stroke with tenecteplase, careful review of its data reveals that all 10 of the 506 patients in the tenecteplase group who sustained a hemorrhagic stroke were age 65 or older.12
A TEAM APPROACH
Thus, in patients with acute pulmonary embolism, clinicians face the difficult task of assessing the patient’s risk of death and clinical worsening and balancing that risk against the risk of bleeding, to identify those who may benefit from early reperfusion therapies, including systemic thrombolysis, catheter-directed thrombolysis, mechanical treatment, and surgical embolectomy.
Given the absence of high-quality evidence to guide these decisions, several institutions have developed multidisciplinary pulmonary embolism response teams to provide rapid evaluation and risk stratification and to recommend and implement advanced therapies, as appropriate. This is a novel concept that is still evolving but holds promise, as it integrates the experience and expertise of physicians in multiple specialties, such as pulmonary and critical care medicine, vascular medicine, interventional radiology, interventional cardiology, emergency medicine, and cardiothoracic surgery, who can then fill the currently existing knowledge gaps for clinical care and, possibly, research.13
Early published experience has documented the feasibility of this multidisciplinary approach.14 The first 95 patients treated at Cleveland Clinic had a 30-day mortality rate of 3.2%, which was lower than the expected 9% rate predicted by the Pulmonary Embolism Severity Index score (unpublished observation).
Figure 1. Cleveland Clinic pulmonary embolism response team algorithm.
Figure 1 shows the algorithm currently used by Cleveland Clinic’s pulmonary embolism response team, with the caveat that no algorithm can fully capture the extent of the complexities and discussions that each case triggers within the team.
TOWARD BETTER UNDERSTANDING
As Ataya et al point out,1 the current state of the evidence does not allow a clear, simplistic, one-size-fits-all approach. A question that arises from this controversial topic is whether we should look for markers of right ventricular dysfunction in every patient admitted with a diagnosis of pulmonary embolism, or only in those with a significant anatomic burden of clot on imaging. Would testing everyone be an appropriate way to identify patients at risk of further deterioration early and therefore prevent adverse outcomes in a timely manner? Or would it not be cost-effective and translate into ordering more diagnostic testing, as well as an increase in downstream workup with higher healthcare costs?
Once we better understand this condition and the factors that predict a higher risk of deterioration, we should be able to design prospective studies that can help elucidate the most appropriate diagnostic and therapeutic approach for such challenging cases. In the meantime, it is important to appraise the evidence in a critical way, as Ataya et al have done in their review.
References
Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. The role of thrombolytic therapy in patients with submassive pulmonary embolism. Cleve Clin J Med 2016; 83:923–932.
Kucher N, Goldhaber SZ. Management of massive pulmonary embolism. Circulation 2005; 112:e28–e32.
Tapson VF. Acute pulmonary embolism. N Engl J Med 2008; 358:1037–1052.
Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
Bahloul M, Chaari A, Kallel H, et al. Pulmonary embolism in intensive care unit: predictive factors, clinical manifestations and outcome. Ann Thorac Med 2010; 5:97–103.
Piazza G, Goldhaber SZ. Fibrinolysis for acute pulmonary embolism. Vasc Med 2010; 15:419–428.
Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med 2014; 189:718–726.
Bova C, Sanchez O, Prandoni P, et al. Identification of intermediate-risk patients with acute symptomatic pulmonary embolism. Eur Respir J 2014; 44:694–703.
Chatterjee S, Chakraborty A, Weinberg I, et al. Thrombolysis for pulmonary embolism and risk of all-cause mortality, major bleeding, and intracranial hemorrhage: a meta-analysis. JAMA 2014; 311:2414–2421.
Stein PD, Matta F, Steinberger DS, Keyes DC. Intracerebral hemorrhage with thrombolytic therapy for acute pulmonary embolism. Am J Med 2012; 125:50–56.
Meyer G, Vicaut E, Danays T, et al. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
Kabrhel C, Rosovsky R, Channick R, et al. A multidisciplinary pulmonary embolism response team: initial 30-month experience with a novel approach to delivery of care to patients with submassive and massive pulmonary embolism. Chest 2016; 150:384–393.
References
Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. The role of thrombolytic therapy in patients with submassive pulmonary embolism. Cleve Clin J Med 2016; 83:923–932.
Kucher N, Goldhaber SZ. Management of massive pulmonary embolism. Circulation 2005; 112:e28–e32.
Tapson VF. Acute pulmonary embolism. N Engl J Med 2008; 358:1037–1052.
Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
Bahloul M, Chaari A, Kallel H, et al. Pulmonary embolism in intensive care unit: predictive factors, clinical manifestations and outcome. Ann Thorac Med 2010; 5:97–103.
Piazza G, Goldhaber SZ. Fibrinolysis for acute pulmonary embolism. Vasc Med 2010; 15:419–428.
Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med 2014; 189:718–726.
Bova C, Sanchez O, Prandoni P, et al. Identification of intermediate-risk patients with acute symptomatic pulmonary embolism. Eur Respir J 2014; 44:694–703.
Chatterjee S, Chakraborty A, Weinberg I, et al. Thrombolysis for pulmonary embolism and risk of all-cause mortality, major bleeding, and intracranial hemorrhage: a meta-analysis. JAMA 2014; 311:2414–2421.
Stein PD, Matta F, Steinberger DS, Keyes DC. Intracerebral hemorrhage with thrombolytic therapy for acute pulmonary embolism. Am J Med 2012; 125:50–56.
Meyer G, Vicaut E, Danays T, et al. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
Kabrhel C, Rosovsky R, Channick R, et al. A multidisciplinary pulmonary embolism response team: initial 30-month experience with a novel approach to delivery of care to patients with submassive and massive pulmonary embolism. Chest 2016; 150:384–393.
Innovations are dominating the early part of the 21st century and the impact on cardiovascular medicine has been especially remarkable. Keeping up and evaluating the relevance of these innovations and the role in patient care is a constant challenge and opportunity for providers and scientists alike.
This Cleveland Clinic Journal of Medicine supplement on cardiovascular disease presents healthcare providers with evidenced-based reviews of important innovations and a glimpse into their potential for an exciting future.
In this supplement, Amar Krishnaswamy, MD, and colleagues look to new frontiers in valve replacement therapies. The success of transcatheter aortic valve replacement has led to extending the technique to the mitral valve. While technical challenges exist with transcatheter mitral valve replacement, methods to overcome these challenges are feasible. The authors review the various valve devices currently under development and examine their potential implications in practice.
The introduction of stents in percutaneous coronary interventions has been one of the most revolutionary innovations in cardiovascular medicine, resulting in impressive outcomes during the past few decades. Despite the dramatic advancement, persistent rates of restenosis and thrombosis continue to cause substantial morbidity and mortality. Stephen Ellis, MD, and Haris Riaz, MD, discuss the evolution of stent design from bare-metal stents through drug-eluting stents and their impact on outcomes. The evolution continues with the development of bioresorbable polymers and stents without polymers. The authors consider the promise of these innovations, especially bioresorbable stents, to further reduce restenosis and stent thrombosis.
Erich Kiehl, MD, and Daniel Cantillon, MD, present information about the latest innovation in cardiac pacing—leadless pacemakers. The first leadless pacemaker was approved earlier this year. In over 50 years of use of transvenous pacemakers, long-term complications have primarily involved the endovascular leads and surgical pocket. The authors discuss the promise of leadless cardiac pacing using catheter-based delivery of a self-contained device in the right ventricle to favorably reduce these complications, as well the current limitation of single-chamber pacing and possible future directions.
Innovations in monoclonal antibody therapy have resulted in a new class of biologic drugs to lower low-density-lipoprotein (LDL) in the blood—PCSK9 inhibitors. These new biologics target the overexpression of the PCSK9 protein in the liver, thereby increasing LDL receptors available to metabolize and remove LDL from the blood. Khendi White, MD, Chaitra Mohan, MD, and Michael Rocco, MD, discuss potential candidates for recently approved PCSK9 inhibitor therapy.
Ellen Brinza, MS, and Heather Gornik, MD, discuss new findings in our understanding of fibromuscular dysplasia (FMD). This uncommon nonatherosclerotic disease leads to narrowing, dissection, or aneurysm of medium-sized arteries. FMD is caused by abnormal development of the arterial cell wall and can cause symptoms if narrowing or a tear decreases blood flow through the artery. The authors discuss evaluation, management, and surveillance strategies as well as important lifestyle modifications and appropriate treatment of symptoms.
We hope this presentation of recent innovations in cardiovascular medicine is useful and informative to you and your clinical practice.
Innovations are dominating the early part of the 21st century and the impact on cardiovascular medicine has been especially remarkable. Keeping up and evaluating the relevance of these innovations and the role in patient care is a constant challenge and opportunity for providers and scientists alike.
This Cleveland Clinic Journal of Medicine supplement on cardiovascular disease presents healthcare providers with evidenced-based reviews of important innovations and a glimpse into their potential for an exciting future.
In this supplement, Amar Krishnaswamy, MD, and colleagues look to new frontiers in valve replacement therapies. The success of transcatheter aortic valve replacement has led to extending the technique to the mitral valve. While technical challenges exist with transcatheter mitral valve replacement, methods to overcome these challenges are feasible. The authors review the various valve devices currently under development and examine their potential implications in practice.
The introduction of stents in percutaneous coronary interventions has been one of the most revolutionary innovations in cardiovascular medicine, resulting in impressive outcomes during the past few decades. Despite the dramatic advancement, persistent rates of restenosis and thrombosis continue to cause substantial morbidity and mortality. Stephen Ellis, MD, and Haris Riaz, MD, discuss the evolution of stent design from bare-metal stents through drug-eluting stents and their impact on outcomes. The evolution continues with the development of bioresorbable polymers and stents without polymers. The authors consider the promise of these innovations, especially bioresorbable stents, to further reduce restenosis and stent thrombosis.
Erich Kiehl, MD, and Daniel Cantillon, MD, present information about the latest innovation in cardiac pacing—leadless pacemakers. The first leadless pacemaker was approved earlier this year. In over 50 years of use of transvenous pacemakers, long-term complications have primarily involved the endovascular leads and surgical pocket. The authors discuss the promise of leadless cardiac pacing using catheter-based delivery of a self-contained device in the right ventricle to favorably reduce these complications, as well the current limitation of single-chamber pacing and possible future directions.
Innovations in monoclonal antibody therapy have resulted in a new class of biologic drugs to lower low-density-lipoprotein (LDL) in the blood—PCSK9 inhibitors. These new biologics target the overexpression of the PCSK9 protein in the liver, thereby increasing LDL receptors available to metabolize and remove LDL from the blood. Khendi White, MD, Chaitra Mohan, MD, and Michael Rocco, MD, discuss potential candidates for recently approved PCSK9 inhibitor therapy.
Ellen Brinza, MS, and Heather Gornik, MD, discuss new findings in our understanding of fibromuscular dysplasia (FMD). This uncommon nonatherosclerotic disease leads to narrowing, dissection, or aneurysm of medium-sized arteries. FMD is caused by abnormal development of the arterial cell wall and can cause symptoms if narrowing or a tear decreases blood flow through the artery. The authors discuss evaluation, management, and surveillance strategies as well as important lifestyle modifications and appropriate treatment of symptoms.
We hope this presentation of recent innovations in cardiovascular medicine is useful and informative to you and your clinical practice.
Innovations are dominating the early part of the 21st century and the impact on cardiovascular medicine has been especially remarkable. Keeping up and evaluating the relevance of these innovations and the role in patient care is a constant challenge and opportunity for providers and scientists alike.
This Cleveland Clinic Journal of Medicine supplement on cardiovascular disease presents healthcare providers with evidenced-based reviews of important innovations and a glimpse into their potential for an exciting future.
In this supplement, Amar Krishnaswamy, MD, and colleagues look to new frontiers in valve replacement therapies. The success of transcatheter aortic valve replacement has led to extending the technique to the mitral valve. While technical challenges exist with transcatheter mitral valve replacement, methods to overcome these challenges are feasible. The authors review the various valve devices currently under development and examine their potential implications in practice.
The introduction of stents in percutaneous coronary interventions has been one of the most revolutionary innovations in cardiovascular medicine, resulting in impressive outcomes during the past few decades. Despite the dramatic advancement, persistent rates of restenosis and thrombosis continue to cause substantial morbidity and mortality. Stephen Ellis, MD, and Haris Riaz, MD, discuss the evolution of stent design from bare-metal stents through drug-eluting stents and their impact on outcomes. The evolution continues with the development of bioresorbable polymers and stents without polymers. The authors consider the promise of these innovations, especially bioresorbable stents, to further reduce restenosis and stent thrombosis.
Erich Kiehl, MD, and Daniel Cantillon, MD, present information about the latest innovation in cardiac pacing—leadless pacemakers. The first leadless pacemaker was approved earlier this year. In over 50 years of use of transvenous pacemakers, long-term complications have primarily involved the endovascular leads and surgical pocket. The authors discuss the promise of leadless cardiac pacing using catheter-based delivery of a self-contained device in the right ventricle to favorably reduce these complications, as well the current limitation of single-chamber pacing and possible future directions.
Innovations in monoclonal antibody therapy have resulted in a new class of biologic drugs to lower low-density-lipoprotein (LDL) in the blood—PCSK9 inhibitors. These new biologics target the overexpression of the PCSK9 protein in the liver, thereby increasing LDL receptors available to metabolize and remove LDL from the blood. Khendi White, MD, Chaitra Mohan, MD, and Michael Rocco, MD, discuss potential candidates for recently approved PCSK9 inhibitor therapy.
Ellen Brinza, MS, and Heather Gornik, MD, discuss new findings in our understanding of fibromuscular dysplasia (FMD). This uncommon nonatherosclerotic disease leads to narrowing, dissection, or aneurysm of medium-sized arteries. FMD is caused by abnormal development of the arterial cell wall and can cause symptoms if narrowing or a tear decreases blood flow through the artery. The authors discuss evaluation, management, and surveillance strategies as well as important lifestyle modifications and appropriate treatment of symptoms.
We hope this presentation of recent innovations in cardiovascular medicine is useful and informative to you and your clinical practice.
In the last 10 years, we have seen a revolution in transcatheter therapies for structural heart disease. The most widely embraced, transcatheter aortic valve replacement (TAVR) was originally intended for patients in whom surgery was considered impossible, but it has now been established as an excellent alternative to surgical aortic valve replacement in patients at high or intermediate risk.1–3 As TAVR has become established, with well-designed devices and acceptable safety and efficacy, it has inspired operators and inventors to push the envelope of innovation to transcatheter mitral valve replacement (TMVR).
This review summarizes the newest data available for the TMVR devices currently being tested in patients with native mitral regurgitation, bioprosthetic degeneration, and degenerative mitral stenosis.
THE MITRAL VALVE: THE NEW FRONTIER
Whereas the pathologic mechanisms of aortic stenosis generally all result in the same anatomic consequence (ie, calcification of the valve leaflets and commissures resulting in reduced mobility), mitral valve regurgitation is much more heterogeneous. Primary (degenerative) mitral regurgitation is caused by intrinsic valve pathology such as myxomatous degeneration, chordal detachment, fibroelastic deficiency, endocarditis, and other conditions that prevent the leaflets from coapting properly. In contrast, in secondary or functional mitral regurgitation, the leaflets are normal but do not coapt properly because of apical tethering to a dilated left ventricle, reduced closing forces with left ventricular dysfunction, or annular dilation as the result of either left ventricular or left atrial dilation.
Surgical mitral valve repair is safe and effective in patients with degenerative mitral regurgitation caused by leaflet prolapse and flail. However, some patients cannot undergo surgery because they have comorbid conditions that place them at extreme risk.4 For example, most patients with functional mitral regurgitation due to ischemic or dilated cardiomyopathy have significant surgical risk and multiple comorbidities, and in this group surgical repair has limited efficacy.5 A sizeable proportion of patients with mitral regurgitation may not be offered surgery because their risk is too high.6 Therefore, alternatives to the current surgical treatments have the potential to benefit a large number of patients.
Similarly, many patients with degenerative mitral stenosis caused by calcification of the mitral annulus also cannot undergo cardiac surgery because of prohibitively high risk. While rheumatic disease is the most common cause of mitral stenosis worldwide, degenerative mitral stenosis may be the cause in up to one-fourth of patients overall and up to 60% of patients older than 80 years.7 In the latter group, not only do old age and comorbidities such as diabetes mellitus and chronic kidney disease pose surgical risks, the technical challenge of surgically implanting a prosthetic mitral valve in the setting of a calcified annulus may be significant.8
The mitral valve is, therefore, the perfect new frontier for percutaneous valve replacement therapies, and TMVR is emerging as a potential option for patients with mitral regurgitation and degenerative mitral stenosis. The currently available percutaneous treatment options for mitral regurgitation include edge-to-edge leaflet repair, direct and indirect annuloplasty, spacers, and left ventricular remodeling devices (Table 1).9,10 As surgical mitral valve repair is strongly preferred over mitral valve replacement, the percutaneous procedures and the devices that are used are engineered to approximate the current standard surgical techniques. However, given the complex pathologies involved, surgical repair often requires the use of multiple repair techniques in the same patient. Therefore, percutaneous repair may also require more than one type of device in the same patient and may not be anatomically feasible in many patients. Replacing the entire valve may obviate some of these challenges.
Reprinted with permission from Wolters Kluwer Health, Inc. (Sud K, et al. Degenerated mitral stenosis: unmet need for percutaneous interventions. Circulation 2016; 133:1594–1604).
Figure 1. Routes of transcatheter mitral valve replacement: (A) transseptal antegrade via the femoral vein; (B) transapical retrograde via direct left ventricular access.
Compared with the aortic valve, the mitral valve poses a greater challenge to percutaneous treatment due to its structure and dynamic relationship with the left ventricle. Some specific challenges facing the development of TMVR are that the mitral valve is large, it is difficult to access, it is asymmetrical, it lacks an anatomically well-defined annulus to which to anchor the replacement valve, its geometry changes throughout the cardiac cycle, and placing a replacement valve in it entails the risk of left ventricular outflow tract obstruction. Despite these challenges, a number of devices are undergoing preclinical testing, a few are in phase 1 clinical trials, and registries are being kept. Depending on the specific device, an antegrade transseptal approach to the mitral valve (via the femoral vein) or a retrograde transapical approach (via direct left ventricular access) may be used (Figure 1).
NATIVE MITRAL VALVE REGURGITATION
For degenerative mitral regurgitation, the standard of care is cardiac surgery at a hospital experienced with mitral valve repair, and with very low rates of mortality and morbidity. For patients in whom the surgical risk is prohibitive, percutaneous edge-to-edge leaflet repair using the MitraClip (Abbott Vascular, Minneapolis, MN) is the best option if the anatomy permits. If the mitral valve pathology is not amenable to MitraClip repair, the patient may be evaluated for TMVR under a clinical trial protocol.
For functional mitral regurgitation, the decisions are more complex. If the patient has chronic atrial fibrillation, electrical cardioversion and antiarrhythmic drug therapy may restore and maintain sinus rhythm, though if the left atrium is large, sinus rhythm may not be possible. If the patient has left ventricular dysfunction, guideline-directed medical therapy should be optimized; this reduces the risk of exacerbations, hospitalizations, and death and may also reduce the degree of regurgitation. If the patient has severe left ventricular dysfunction and a wide QRS duration, cardiac resynchronization therapy (biventricular pacing) may also be beneficial and reduce functional mitral regurgitation. If symptoms and severe functional mitral regurgitation persist despite these measures and the patient’s surgical risk is deemed to be extreme, options include MitraClip placement as part of the randomized Cardiovascular Outcomes Assessment of the MitraClip Percutaneous Therapy (COAPT) trial, which compares guideline-directed medical therapy with guideline-directed therapy plus MitraClip. Another option is enrollment in a clinical trial or registry of TMVR.
At this writing, six TMVR devices have been implanted in humans:
Fortis (Edwards Lifesciences, Irvine, CA)
Tendyne (Tendyne Holding Inc, Roseville, MN)
NaviGate (NaviGate Cardiac Structures, Inc, Lake Forest, CA)
Intrepid (Medtronic, Minneapolis, MN)
CardiAQ (Edwards Lifesciences, Irvine, CA)
Tiara (Neovasc Inc, Richmond, BC).
Most of the early experience with these valves has not yet been published, but some data have been presented at national and international meetings.
The Fortis valve
Courtesy of Edwards Lifesciences.
Fortis valve
The Fortis valve consists of a self-expanding nitinol frame and leaflets made of bovine pericardium and is implanted via a transapical approach.
The device was successfully implanted in three patients in Quebec City, Canada, and at 6 months, all had improved significantly in functional class and none had needed to be hospitalized.11 Echocardiographic assessment demonstrated trace or less mitral regurgitation and a mean transvalvular gradient less than 4 mm Hg in all.
Bapat and colleagues12 attempted to implant the device in 13 patients in Europe and Canada. The average left ventricular ejection fraction was 34%, and 12 of 13 patients (92%) had functional mitral regurgitation. Procedural success was achieved in 10 patients, but five patients died within 30 days. While the deaths were due to nonvalvular issues (multiorgan failure, septic shock, intestinal ischemia after failed valve implantation and conversion to open surgery, malnutrition leading to respiratory failure, and valve thrombosis), the trial is currently on hold as more data are collected and reviewed. Among the eight patients who survived the first month, all were still alive at 6 months, and echocardiography demonstrated no or trivial mitral regurgitation in six patients (80%) and mild regurgitation in two patients (20%); the average mitral gradient was 4 mm Hg, and there was no change in mean left ventricular ejection fraction.
The Tendyne valve is a self-expanding prosthesis with porcine pericardial leaflets. It is delivered transapically and is held in place by a tether from the valve to the left ventricular apex.
In the first 12 patients enrolled in an early feasibility trial,13 the average left ventricular ejection fraction was 40%, and 11 of the 12 patients had functional mitral regurgitation. The device was successfully implanted in 11 patients, while one patient developed left ventricular outflow tract obstruction and the device was uneventfully removed. All patients were still alive at 30 days, and the 11 patients who still had a prosthetic valve did not have any residual mitral regurgitation.
As of this writing, almost 80 patients have received the device, though the data have not yet been presented. Patients are being enrolled in phase 1 trials.
The NaviGate valve
Courtesy of Jose Navia.
NaviGate valve
The NaviGate valve consists of a trileaflet subassembly fabricated from bovine pericardium, mounted on a self-expanding nitinol stent, and is only implanted transatrially.
Figure 2. Transatrial implantation of the NaviGate transcatheter mitral valve replacement prosthesis. (A) Initial unsheathing of the valve (arrow) via the left atrium (LA); (B) no residual mitral regurgitation on left ventriculography (LV). Ao = ascending aorta
NaviGate valves were successfully implanted in two patients via a transatrial approach (Figure 2). Both patients had excellent valve performance without residual mitral regurgitation or left ventricular outflow tract obstruction. The first patient showed significant improvement in functional class and freedom from hospitalization at 6 months, but the second patient died within a week of the implant due to advanced heart failure.14 A US clinical trial is expected soon.
The Intrepid valve
Courtesy of Medtronic.
Intrepid valve
The Intrepid valve consists of an outer stent to provide fixation to the annulus and an inner stent that houses a bovine pericardial valve. The device is a self-expanding system that is delivered transapically.
In a series of 15 patients, 11 had functional mitral regurgitation (with an average left ventricular ejection fraction of 35%) and four had degenerative mitral regurgitation (with an average left ventricular ejection fraction of 57%).15 The device was successfully implanted in 14 patients, after which the average mitral valve gradient was 4 mm Hg. All patients but one were left with no regurgitation (the other patient had 1+ regurgitation).
A trial is currently under way in Europe.
The CardiAQ valve
Courtesy of Edwards Lifesciences.
CardiAQ valve
The CardiAQ is constructed of bovine pericardium and can be delivered by the transseptal or transapical route.
Of 12 patients treated under compassionate use,16 two-thirds (eight patients) had functional mitral regurgitation. Two patients died during the procedure, three died of noncardiac complications within 30 days, and one more died of sepsis shortly after 30 days. This early experience demonstrates the importance of careful patient selection and postprocedural management in the feasibility assessment of these new technologies.
The Tiara valve, a self-expanding prosthesis with bovine pericardial leaflets, is delivered by the transapical route.
Eleven patients underwent Tiara implantation as part of either a Canadian special access registry or an international feasibility trial. Their average Society of Thoracic Surgeons score (ie, their calculated risk of major morbidity or operative mortality) was 15.6%, and their average left ventricular ejection fraction was 29%. Only two patients had degenerative mitral regurgitation. Nine patients had uneventful procedures and demonstrated no residual mitral regurgitation and no left ventricular outflow tract obstruction. The procedure was converted to open surgery in two patients owing to valve malpositioning, and both of them died within 30 days. One patient in whom the procedure was successful suffered erosion of the septum and died on day 4.17
Patients are being enrolled in phase 1 trials.
DEGENERATIVE MITRAL STENOSIS
Reprinted with permission from Wolters Kluwer Health, Inc. (Sud K, et al. Degenerated mitral stenosis: unmet need for percutaneous interventions. Circulation 2016; 133:1594–1604).
Figure 3. Mitral annular calcification (MAC) provides a “frame” for transcatheter mitral valve replacement prosthesis implantation in the mitral position for degenerative mitral stenosis. Ao = aorta; LVOT = left ventricular outflow tract
In patients with degenerative mitral stenosis, extensive mitral annular calcification may provide an adequate “frame” to hold a transcatheter valve prosthesis (Figure 3). Exploiting this feature, numerous investigators have successfully deployed prosthetic valves designed for TAVR in the calcified mitral annulus via the retrograde transapical and antegrade transseptal routes.
Guerrero and colleagues presented results from the first global registry of TMVR in mitral annular calcification at the 2016 EuroPCR Congress.18 Of 104 patients analyzed, almost all received an Edwards’ Sapien balloon-expandable valve (first-generation, Sapien XT, or Sapien 3); the others received Boston Scientific’s Lotus or Direct Flow Medical (Direct Flow Medical, Santa Clara, CA) valves. With an average age of 73 years and a high prevalence of comorbidities such as diabetes, chronic obstructive pulmonary disease, atrial fibrillation, chronic kidney disease, and prior cardiac surgery, the group presented extreme surgical risk, with an average Society of Thoracic Surgeons risk score of 14.4%. Slightly more than 40% of the patients underwent transapical implantation, slightly less than 40% underwent transfemoral or transseptal implantation, and just under 20% had a direct atrial approach.
The implantation was technically successful in 78 of 104 patients (75%); 13 patients (12.5%) required a second mitral valve to be placed, 11 patients (10.5%) had left ventricular outflow tract obstruction, four patients (4%) had valve embolization, and two patients (2%) had left ventricular perforation. At 30 days, 11 of 104 patients (10.6%) had died of cardiac causes and 15 patients (14.4%) had died of noncardiac causes. When divided roughly into three equal groups by chronological order, the last third of patients, compared with the first third of patients, enjoyed greater technical success (80%, n = 32/40 vs 62.5%, n = 20/32), better 30-day survival (85%, n = 34/40 vs 62.5%, n = 20/32), and no conversion to open surgery (0 vs 12.5%, n = 4/32), likely demonstrating both improved patient selection and lessons learned from shared experience. At 1 year, almost 90% of patients had New York Heart Association class I or II symptoms. Prior to the procedure, 91.5% had New York Heart Association class III or IV symptoms.
At present, TMVR in mitral annular calcification is not approved in the United States or elsewhere. However, multiple registries are currently enrolling patients or are in formative stages to push the frontier of the currently available technologies until better, dedicated devices are available for this group of patients.
BIOPROSTHETIC VALVE OR VALVE RING FAILURE
Figure 4. Transfemoral mitral valve-in-valve placement of a balloon-expandable valve. (A) Catheter via femoral vein (white arrow) and crossing the interatrial septum with unexpanded valve in place (black arrow) within the mitral prosthesis (arrowhead); (B) balloon inflation of the TAVR prosthesis (black arrow); (C) fully expanded valve in place; (D) 3D transesophageal echocardiographic view from the left atrium of the stenosed mitral valve (arrow); (E) mitral valve open (arrow) after valve-in-valve placement.
Implantation of a TAVR prosthetic inside a degenerated bioprosthetic mitral valve (valve-in-valve) and mitral valve ring (valve-in-ring) is generally limited to case series with short-term results using the Edwards Sapien series, Boston Scientific Lotus, Medtronic Melody (Medtronic, Minneapolis, MN), and Direct Flow Medical valves (Figure 4).19–23
The largest collective experience was presented in the Valve-in-Valve International Data (VIVID) registry, which included 349 patients who had mitral valve-in-valve placement and 88 patients who had mitral valve-in-ring procedures. Their average age was 74 and the mean Society of Thoracic Surgeons score was 12.9% in both groups.24 Of the 437 patients, 345 patients (78.9%) underwent transapical implantation, and 391 patients (89.5%) received a Sapien XT or Sapien 3 valve. In the valve-in-valve group, 41% of the patients had regurgitation, 25% had stenosis, and 34% had both. In the valve-in-ring group, 60% of the patients had regurgitation, 17% had stenosis, and 23% had both.
Valve placement was successful in most patients. The rate of stroke was low (2.9% with valve-in-valve placement, 1.1% with valve-in-ring placement), though the rate of moderate or greater residual mitral regurgitation was significantly higher in patients undergoing valve-in-ring procedures (14.8% vs 2.6%, P < .001), as was the rate of left ventricular outflow tract obstruction (8% vs 2.6%, P = .03). There was also a trend toward worse 30-day mortality in the valve-in-ring group (11.4% vs 7.7%, P = .15). As with aortic valve-in-valve procedures, small surgical mitral valves (≤ 25 mm) were associated with higher postprocedural gradients.
Eleid and colleagues25 published their experience with antegrade transseptal TMVR in 48 patients with an average Society of Thoracic Surgeons score of 13.2%, 33 of whom underwent valve-in-valve procedures and nine of whom underwent valve-in-ring procedures. (The other six patients underwent mitral valve implantation for severe mitral annular calcification.) In the valve-in-valve group, 31 patients successfully underwent implant procedures, but two patients died during the procedure from left ventricular perforation. Of the nine valve-in-ring patients, two had acute embolization of the valve and were converted to open surgery. Among the seven patients in whom implantation was successful, two developed significant left ventricular outflow tract obstruction; one was treated with surgical resection of the anterior mitral valve leaflet and the other was medically managed.
CONCLUSION
Transcatheter mitral valve replacement in regurgitant mitral valves, failing mitral valve bioprosthetics and rings, and calcified mitral annuli has been effectively conducted in a number of patients who had no surgical options due to prohibitive surgical risk. International registries and our experience have demonstrated that the valve-in-valve procedure using a TAVR prosthesis carries the greatest likelihood of success, given the rigid frame of the surgical bioprosthetic that allows stable valve deployment. While approved in Europe for this indication, use of these devices for this application in the United States is considered “off label” and is performed only in clinically extenuating circumstances. Implantation of TAVR prosthetics in patients with prior mitral ring repair or for native mitral stenosis also has been performed successfully, although left ventricular outflow tract obstruction is a significant risk in this early experience.
Devices designed specifically for TMVR are in their clinical infancy and have been implanted successfully in only small numbers of patients, most of whom had functional mitral regurgitation. Despite reasonable technical success, most of these trials have been plagued by high mortality rates at 30 days in large part due to the extreme risk of the patients in whom these procedures have been conducted. At present, enrollment in TMVR trials for patients with degenerative or functional mitral regurgitation is limited to those without a surgical option and who conform to very specific anatomic criteria.
References
Leon MB, Smith CR, Mack M, et al; PARTNER Trial Investigators. Transcatheter aortic-valve implantation for aortic stenosis in patients who cannot undergo surgery. N Engl J Med 2010; 363:1597–1607.
Smith CR, Leon MB, Mack MJ, et al; PARTNER Trial Investigators. Transcatheter versus surgical aortic-valve replacement in high-risk patients. N Engl J Med 2011; 364:2187–2198.
Thourani VH, Kodali S, Makkar RR, et al. Transcatheter aortic valve replacement versus surgical valve replacement in intermediate-risk patients: a propensity score analysis. Lancet 2016; 387:2218–2225.
Goel SS, Bajaj N, Aggarwal B, et al. Prevalence and outcomes of unoperated patients with severe symptomatic mitral regurgitation and heart failure: comprehensive analysis to determine the potential role of MitraClip for this unmet need. J Am Coll Cardiol 2014; 63:185–186.
DiBardino DJ, ElBardissi AW, McClure RS, Razo-Vasquez OA, Kelly NE, Cohn LH. Four decades of experience with mitral valve repair: analysis of differential indications, technical evolution, and long-term outcome. J Thorac Cardiovasc Surg 2010; 139:76–83; discussion 83–74.
Mirabel M, Iung B, Baron G, et al. What are the characteristics of patients with severe, symptomatic, mitral regurgitation who are denied surgery? Eur Heart J 2007; 28:1358–1365.
Iung B, Baron G, Butchart EG, et al. A prospective survey of patients with valvular heart disease in europe: the Euro Heart Survey on Valvular Heart Disease. Eur Heart J 2003; 24:1231–1243.
Sud K, Agarwal S, Parashar A, et al. Degenerative mitral stenosis: unmet need for percutaneous interventions. Circulation 2016; 133:1594–1604.
Svensson LG, Ye J, Piemonte TC, Kirker-Head C, Leon MB, Webb JG. Mitral valve regurgitation and left ventricular dysfunction treatment with an intravalvular spacer. J Card Surg 2015; 30:53–54.
Raman J, Raghavan J, Chandrashekar P, Sugeng L. Can we repair the mitral valve from outside the heart? A novel extra-cardiac approach to functional mitral regurgitation. Heart Lung Circ 2011; 20:157–162.
Abdul-Jawad Altisent O, Dumont E, Dagenais F, et al. Initial experience of transcatheter mitral valve replacement with a novel transcatheter mitral valve: procedural and 6-month follow-up results. J Am Coll Cardiol 2015; 66:1011–1019.
Bapat V. FORTIS: design, clinical results, and next steps. Presented at CRT (Cardiovascular Research Technologies) 16; Feburary 20–23, 2016; Washington, DC.
Sorajja P. Tendyne: technology and clinical results update. Presented at CRT (Cardiovascular Research Technologies) 16; February 20–23, 2016; Washington, DC.
Navia J. Personal communication.
Bapat V. Medtronic Intrepid transcatheter mitral valve replacement. Presented at EuroPCR 2015; May 19–22, 2015; Paris, France.
Herrmann H. Cardiaq-Edwards TMVR. Presented at CRT (Cardiovascular Research Technologies) 16; February 20–23, 2016; Washington, DC.
Dvir D. Tiara: design, clincal results, and next steps. Presented at CRT (Cardiovascular Research Technologies) 16; February 20–23, 2016; Washington, DC.
Guerrero M, Dvir D, Himbert D, et al. Transcatheter mitral valve replacement in native mitra valve disease with severe mitral annular calcification: results from the first global registry. JACC Cardiovasc Interv 2016; 9:1361–1371.
Seiffert M, Franzen O, Conradi L, et al. Series of transcatheter valve-in-valve implantations in high-risk patients with degenerated bioprostheses in aortic and mitral position. Catheter Cardiovasc Interv 2010; 76:608–615.
Webb JG, Wood DA, Ye J, et al. Transcatheter valve-in-valve implantation for failed bioprosthetic heart valves. Circulation 2010; 121:1848–1857.
Cerillo AG, Chiaramonti F, Murzi M, et al. Transcatheter valve in valve implantation for failed mitral and tricuspid bioprosthesis. Catheter Cardiovasc Interv 2011; 78:987–995.
Seiffert M, Conradi L, Baldus S, et al. Transcatheter mitral valve-in-valve implantation in patients with degenerated bioprostheses. JACC Cardiovasc Interv 2012; 5:341–349.
Wilbring M, Alexiou K, Tugtekin SM, et al. Pushing the limits—further evolutions of transcatheter valve procedures in the mitral position, including valve-in-valve, valve-in-ring, and valve-in-native-ring. J Thorac Cardiovasc Surg 2014; 147:210–219.
Dvir D, on behalf of the VIVID Registry Investigators. Transcatheter mitral valve-in-valve and valve-in-ring implantations. Transcatheter Valve Therapies 2015.
Eleid MF, Cabalka AK, Williams MR, et al. Percutaneous transvenous transseptal transcatheter valve implantation in failed bioprosthetic mitral valves, ring annuloplasty, and severe mitral annular calcification. JACC Cardiovasc Interv 2016; 9:1161–1174.
Amar Krishnaswamy, MD Program Director, Interventional Cardiology Fellowship, Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Stephanie Mick, MD Surgical Director, TAVR, Department of Thoracic and Cardiovascular Surgery, Heart and Vascular Institute, Cleveland Clinic
Jose Navia, MD Departments of Thoracic and Cardiovascular Surgery, Biomedical Engineering, and Transplantation Center, Cleveland Clinic
Marc Gillinov, MD Institute Experience Officer, Department of Thoracic and Cardiovascular Surgery, Heart and Vascular Institute, Cleveland Clinic; Associate Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
E. Murat Tuzcu, MD Chairman, Department of Cardiovascular Medicine, Cleveland Clinic Abu Dhabpeveland, OH
Samir R. Kapadia, MD Director, Sones Catheterization Laboratories, Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic; Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Correspondence: Amar Krishnaswamy, MD, Department of Cardiovascular Medicine, J2-3, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44118; krishna2@ccf.org
Drs. Krishnaswamy, Mick, Tuzcu, and Kapadia reported no financial interests or relationships that pose a potential conflict of interest with this article. Dr. Gillinov reported consulting for Abbott Vascular, Atricure, ClearFlow Inc., Edwards Lifesciences, Medtronic, On-X Life Technologies Inc., and Tendyne Holdings Inc.; ownership interest in ClearFlow Inc.; teaching/speaking for Intuitive Surgical; and research support for St. Jude Medical. Dr. Navia reported receipt of consulting/speaking fees from Edwards Lifesciences and Maquet Cardiovascular and royalty payments from NaviGate Cardiac Structures.
Amar Krishnaswamy, MD Program Director, Interventional Cardiology Fellowship, Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Stephanie Mick, MD Surgical Director, TAVR, Department of Thoracic and Cardiovascular Surgery, Heart and Vascular Institute, Cleveland Clinic
Jose Navia, MD Departments of Thoracic and Cardiovascular Surgery, Biomedical Engineering, and Transplantation Center, Cleveland Clinic
Marc Gillinov, MD Institute Experience Officer, Department of Thoracic and Cardiovascular Surgery, Heart and Vascular Institute, Cleveland Clinic; Associate Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
E. Murat Tuzcu, MD Chairman, Department of Cardiovascular Medicine, Cleveland Clinic Abu Dhabpeveland, OH
Samir R. Kapadia, MD Director, Sones Catheterization Laboratories, Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic; Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Correspondence: Amar Krishnaswamy, MD, Department of Cardiovascular Medicine, J2-3, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44118; krishna2@ccf.org
Drs. Krishnaswamy, Mick, Tuzcu, and Kapadia reported no financial interests or relationships that pose a potential conflict of interest with this article. Dr. Gillinov reported consulting for Abbott Vascular, Atricure, ClearFlow Inc., Edwards Lifesciences, Medtronic, On-X Life Technologies Inc., and Tendyne Holdings Inc.; ownership interest in ClearFlow Inc.; teaching/speaking for Intuitive Surgical; and research support for St. Jude Medical. Dr. Navia reported receipt of consulting/speaking fees from Edwards Lifesciences and Maquet Cardiovascular and royalty payments from NaviGate Cardiac Structures.
Author and Disclosure Information
Amar Krishnaswamy, MD Program Director, Interventional Cardiology Fellowship, Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Stephanie Mick, MD Surgical Director, TAVR, Department of Thoracic and Cardiovascular Surgery, Heart and Vascular Institute, Cleveland Clinic
Jose Navia, MD Departments of Thoracic and Cardiovascular Surgery, Biomedical Engineering, and Transplantation Center, Cleveland Clinic
Marc Gillinov, MD Institute Experience Officer, Department of Thoracic and Cardiovascular Surgery, Heart and Vascular Institute, Cleveland Clinic; Associate Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
E. Murat Tuzcu, MD Chairman, Department of Cardiovascular Medicine, Cleveland Clinic Abu Dhabpeveland, OH
Samir R. Kapadia, MD Director, Sones Catheterization Laboratories, Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic; Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Correspondence: Amar Krishnaswamy, MD, Department of Cardiovascular Medicine, J2-3, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44118; krishna2@ccf.org
Drs. Krishnaswamy, Mick, Tuzcu, and Kapadia reported no financial interests or relationships that pose a potential conflict of interest with this article. Dr. Gillinov reported consulting for Abbott Vascular, Atricure, ClearFlow Inc., Edwards Lifesciences, Medtronic, On-X Life Technologies Inc., and Tendyne Holdings Inc.; ownership interest in ClearFlow Inc.; teaching/speaking for Intuitive Surgical; and research support for St. Jude Medical. Dr. Navia reported receipt of consulting/speaking fees from Edwards Lifesciences and Maquet Cardiovascular and royalty payments from NaviGate Cardiac Structures.
In the last 10 years, we have seen a revolution in transcatheter therapies for structural heart disease. The most widely embraced, transcatheter aortic valve replacement (TAVR) was originally intended for patients in whom surgery was considered impossible, but it has now been established as an excellent alternative to surgical aortic valve replacement in patients at high or intermediate risk.1–3 As TAVR has become established, with well-designed devices and acceptable safety and efficacy, it has inspired operators and inventors to push the envelope of innovation to transcatheter mitral valve replacement (TMVR).
This review summarizes the newest data available for the TMVR devices currently being tested in patients with native mitral regurgitation, bioprosthetic degeneration, and degenerative mitral stenosis.
THE MITRAL VALVE: THE NEW FRONTIER
Whereas the pathologic mechanisms of aortic stenosis generally all result in the same anatomic consequence (ie, calcification of the valve leaflets and commissures resulting in reduced mobility), mitral valve regurgitation is much more heterogeneous. Primary (degenerative) mitral regurgitation is caused by intrinsic valve pathology such as myxomatous degeneration, chordal detachment, fibroelastic deficiency, endocarditis, and other conditions that prevent the leaflets from coapting properly. In contrast, in secondary or functional mitral regurgitation, the leaflets are normal but do not coapt properly because of apical tethering to a dilated left ventricle, reduced closing forces with left ventricular dysfunction, or annular dilation as the result of either left ventricular or left atrial dilation.
Surgical mitral valve repair is safe and effective in patients with degenerative mitral regurgitation caused by leaflet prolapse and flail. However, some patients cannot undergo surgery because they have comorbid conditions that place them at extreme risk.4 For example, most patients with functional mitral regurgitation due to ischemic or dilated cardiomyopathy have significant surgical risk and multiple comorbidities, and in this group surgical repair has limited efficacy.5 A sizeable proportion of patients with mitral regurgitation may not be offered surgery because their risk is too high.6 Therefore, alternatives to the current surgical treatments have the potential to benefit a large number of patients.
Similarly, many patients with degenerative mitral stenosis caused by calcification of the mitral annulus also cannot undergo cardiac surgery because of prohibitively high risk. While rheumatic disease is the most common cause of mitral stenosis worldwide, degenerative mitral stenosis may be the cause in up to one-fourth of patients overall and up to 60% of patients older than 80 years.7 In the latter group, not only do old age and comorbidities such as diabetes mellitus and chronic kidney disease pose surgical risks, the technical challenge of surgically implanting a prosthetic mitral valve in the setting of a calcified annulus may be significant.8
The mitral valve is, therefore, the perfect new frontier for percutaneous valve replacement therapies, and TMVR is emerging as a potential option for patients with mitral regurgitation and degenerative mitral stenosis. The currently available percutaneous treatment options for mitral regurgitation include edge-to-edge leaflet repair, direct and indirect annuloplasty, spacers, and left ventricular remodeling devices (Table 1).9,10 As surgical mitral valve repair is strongly preferred over mitral valve replacement, the percutaneous procedures and the devices that are used are engineered to approximate the current standard surgical techniques. However, given the complex pathologies involved, surgical repair often requires the use of multiple repair techniques in the same patient. Therefore, percutaneous repair may also require more than one type of device in the same patient and may not be anatomically feasible in many patients. Replacing the entire valve may obviate some of these challenges.
Reprinted with permission from Wolters Kluwer Health, Inc. (Sud K, et al. Degenerated mitral stenosis: unmet need for percutaneous interventions. Circulation 2016; 133:1594–1604).
Figure 1. Routes of transcatheter mitral valve replacement: (A) transseptal antegrade via the femoral vein; (B) transapical retrograde via direct left ventricular access.
Compared with the aortic valve, the mitral valve poses a greater challenge to percutaneous treatment due to its structure and dynamic relationship with the left ventricle. Some specific challenges facing the development of TMVR are that the mitral valve is large, it is difficult to access, it is asymmetrical, it lacks an anatomically well-defined annulus to which to anchor the replacement valve, its geometry changes throughout the cardiac cycle, and placing a replacement valve in it entails the risk of left ventricular outflow tract obstruction. Despite these challenges, a number of devices are undergoing preclinical testing, a few are in phase 1 clinical trials, and registries are being kept. Depending on the specific device, an antegrade transseptal approach to the mitral valve (via the femoral vein) or a retrograde transapical approach (via direct left ventricular access) may be used (Figure 1).
NATIVE MITRAL VALVE REGURGITATION
For degenerative mitral regurgitation, the standard of care is cardiac surgery at a hospital experienced with mitral valve repair, and with very low rates of mortality and morbidity. For patients in whom the surgical risk is prohibitive, percutaneous edge-to-edge leaflet repair using the MitraClip (Abbott Vascular, Minneapolis, MN) is the best option if the anatomy permits. If the mitral valve pathology is not amenable to MitraClip repair, the patient may be evaluated for TMVR under a clinical trial protocol.
For functional mitral regurgitation, the decisions are more complex. If the patient has chronic atrial fibrillation, electrical cardioversion and antiarrhythmic drug therapy may restore and maintain sinus rhythm, though if the left atrium is large, sinus rhythm may not be possible. If the patient has left ventricular dysfunction, guideline-directed medical therapy should be optimized; this reduces the risk of exacerbations, hospitalizations, and death and may also reduce the degree of regurgitation. If the patient has severe left ventricular dysfunction and a wide QRS duration, cardiac resynchronization therapy (biventricular pacing) may also be beneficial and reduce functional mitral regurgitation. If symptoms and severe functional mitral regurgitation persist despite these measures and the patient’s surgical risk is deemed to be extreme, options include MitraClip placement as part of the randomized Cardiovascular Outcomes Assessment of the MitraClip Percutaneous Therapy (COAPT) trial, which compares guideline-directed medical therapy with guideline-directed therapy plus MitraClip. Another option is enrollment in a clinical trial or registry of TMVR.
At this writing, six TMVR devices have been implanted in humans:
Fortis (Edwards Lifesciences, Irvine, CA)
Tendyne (Tendyne Holding Inc, Roseville, MN)
NaviGate (NaviGate Cardiac Structures, Inc, Lake Forest, CA)
Intrepid (Medtronic, Minneapolis, MN)
CardiAQ (Edwards Lifesciences, Irvine, CA)
Tiara (Neovasc Inc, Richmond, BC).
Most of the early experience with these valves has not yet been published, but some data have been presented at national and international meetings.
The Fortis valve
Courtesy of Edwards Lifesciences.
Fortis valve
The Fortis valve consists of a self-expanding nitinol frame and leaflets made of bovine pericardium and is implanted via a transapical approach.
The device was successfully implanted in three patients in Quebec City, Canada, and at 6 months, all had improved significantly in functional class and none had needed to be hospitalized.11 Echocardiographic assessment demonstrated trace or less mitral regurgitation and a mean transvalvular gradient less than 4 mm Hg in all.
Bapat and colleagues12 attempted to implant the device in 13 patients in Europe and Canada. The average left ventricular ejection fraction was 34%, and 12 of 13 patients (92%) had functional mitral regurgitation. Procedural success was achieved in 10 patients, but five patients died within 30 days. While the deaths were due to nonvalvular issues (multiorgan failure, septic shock, intestinal ischemia after failed valve implantation and conversion to open surgery, malnutrition leading to respiratory failure, and valve thrombosis), the trial is currently on hold as more data are collected and reviewed. Among the eight patients who survived the first month, all were still alive at 6 months, and echocardiography demonstrated no or trivial mitral regurgitation in six patients (80%) and mild regurgitation in two patients (20%); the average mitral gradient was 4 mm Hg, and there was no change in mean left ventricular ejection fraction.
The Tendyne valve is a self-expanding prosthesis with porcine pericardial leaflets. It is delivered transapically and is held in place by a tether from the valve to the left ventricular apex.
In the first 12 patients enrolled in an early feasibility trial,13 the average left ventricular ejection fraction was 40%, and 11 of the 12 patients had functional mitral regurgitation. The device was successfully implanted in 11 patients, while one patient developed left ventricular outflow tract obstruction and the device was uneventfully removed. All patients were still alive at 30 days, and the 11 patients who still had a prosthetic valve did not have any residual mitral regurgitation.
As of this writing, almost 80 patients have received the device, though the data have not yet been presented. Patients are being enrolled in phase 1 trials.
The NaviGate valve
Courtesy of Jose Navia.
NaviGate valve
The NaviGate valve consists of a trileaflet subassembly fabricated from bovine pericardium, mounted on a self-expanding nitinol stent, and is only implanted transatrially.
Figure 2. Transatrial implantation of the NaviGate transcatheter mitral valve replacement prosthesis. (A) Initial unsheathing of the valve (arrow) via the left atrium (LA); (B) no residual mitral regurgitation on left ventriculography (LV). Ao = ascending aorta
NaviGate valves were successfully implanted in two patients via a transatrial approach (Figure 2). Both patients had excellent valve performance without residual mitral regurgitation or left ventricular outflow tract obstruction. The first patient showed significant improvement in functional class and freedom from hospitalization at 6 months, but the second patient died within a week of the implant due to advanced heart failure.14 A US clinical trial is expected soon.
The Intrepid valve
Courtesy of Medtronic.
Intrepid valve
The Intrepid valve consists of an outer stent to provide fixation to the annulus and an inner stent that houses a bovine pericardial valve. The device is a self-expanding system that is delivered transapically.
In a series of 15 patients, 11 had functional mitral regurgitation (with an average left ventricular ejection fraction of 35%) and four had degenerative mitral regurgitation (with an average left ventricular ejection fraction of 57%).15 The device was successfully implanted in 14 patients, after which the average mitral valve gradient was 4 mm Hg. All patients but one were left with no regurgitation (the other patient had 1+ regurgitation).
A trial is currently under way in Europe.
The CardiAQ valve
Courtesy of Edwards Lifesciences.
CardiAQ valve
The CardiAQ is constructed of bovine pericardium and can be delivered by the transseptal or transapical route.
Of 12 patients treated under compassionate use,16 two-thirds (eight patients) had functional mitral regurgitation. Two patients died during the procedure, three died of noncardiac complications within 30 days, and one more died of sepsis shortly after 30 days. This early experience demonstrates the importance of careful patient selection and postprocedural management in the feasibility assessment of these new technologies.
The Tiara valve, a self-expanding prosthesis with bovine pericardial leaflets, is delivered by the transapical route.
Eleven patients underwent Tiara implantation as part of either a Canadian special access registry or an international feasibility trial. Their average Society of Thoracic Surgeons score (ie, their calculated risk of major morbidity or operative mortality) was 15.6%, and their average left ventricular ejection fraction was 29%. Only two patients had degenerative mitral regurgitation. Nine patients had uneventful procedures and demonstrated no residual mitral regurgitation and no left ventricular outflow tract obstruction. The procedure was converted to open surgery in two patients owing to valve malpositioning, and both of them died within 30 days. One patient in whom the procedure was successful suffered erosion of the septum and died on day 4.17
Patients are being enrolled in phase 1 trials.
DEGENERATIVE MITRAL STENOSIS
Reprinted with permission from Wolters Kluwer Health, Inc. (Sud K, et al. Degenerated mitral stenosis: unmet need for percutaneous interventions. Circulation 2016; 133:1594–1604).
Figure 3. Mitral annular calcification (MAC) provides a “frame” for transcatheter mitral valve replacement prosthesis implantation in the mitral position for degenerative mitral stenosis. Ao = aorta; LVOT = left ventricular outflow tract
In patients with degenerative mitral stenosis, extensive mitral annular calcification may provide an adequate “frame” to hold a transcatheter valve prosthesis (Figure 3). Exploiting this feature, numerous investigators have successfully deployed prosthetic valves designed for TAVR in the calcified mitral annulus via the retrograde transapical and antegrade transseptal routes.
Guerrero and colleagues presented results from the first global registry of TMVR in mitral annular calcification at the 2016 EuroPCR Congress.18 Of 104 patients analyzed, almost all received an Edwards’ Sapien balloon-expandable valve (first-generation, Sapien XT, or Sapien 3); the others received Boston Scientific’s Lotus or Direct Flow Medical (Direct Flow Medical, Santa Clara, CA) valves. With an average age of 73 years and a high prevalence of comorbidities such as diabetes, chronic obstructive pulmonary disease, atrial fibrillation, chronic kidney disease, and prior cardiac surgery, the group presented extreme surgical risk, with an average Society of Thoracic Surgeons risk score of 14.4%. Slightly more than 40% of the patients underwent transapical implantation, slightly less than 40% underwent transfemoral or transseptal implantation, and just under 20% had a direct atrial approach.
The implantation was technically successful in 78 of 104 patients (75%); 13 patients (12.5%) required a second mitral valve to be placed, 11 patients (10.5%) had left ventricular outflow tract obstruction, four patients (4%) had valve embolization, and two patients (2%) had left ventricular perforation. At 30 days, 11 of 104 patients (10.6%) had died of cardiac causes and 15 patients (14.4%) had died of noncardiac causes. When divided roughly into three equal groups by chronological order, the last third of patients, compared with the first third of patients, enjoyed greater technical success (80%, n = 32/40 vs 62.5%, n = 20/32), better 30-day survival (85%, n = 34/40 vs 62.5%, n = 20/32), and no conversion to open surgery (0 vs 12.5%, n = 4/32), likely demonstrating both improved patient selection and lessons learned from shared experience. At 1 year, almost 90% of patients had New York Heart Association class I or II symptoms. Prior to the procedure, 91.5% had New York Heart Association class III or IV symptoms.
At present, TMVR in mitral annular calcification is not approved in the United States or elsewhere. However, multiple registries are currently enrolling patients or are in formative stages to push the frontier of the currently available technologies until better, dedicated devices are available for this group of patients.
BIOPROSTHETIC VALVE OR VALVE RING FAILURE
Figure 4. Transfemoral mitral valve-in-valve placement of a balloon-expandable valve. (A) Catheter via femoral vein (white arrow) and crossing the interatrial septum with unexpanded valve in place (black arrow) within the mitral prosthesis (arrowhead); (B) balloon inflation of the TAVR prosthesis (black arrow); (C) fully expanded valve in place; (D) 3D transesophageal echocardiographic view from the left atrium of the stenosed mitral valve (arrow); (E) mitral valve open (arrow) after valve-in-valve placement.
Implantation of a TAVR prosthetic inside a degenerated bioprosthetic mitral valve (valve-in-valve) and mitral valve ring (valve-in-ring) is generally limited to case series with short-term results using the Edwards Sapien series, Boston Scientific Lotus, Medtronic Melody (Medtronic, Minneapolis, MN), and Direct Flow Medical valves (Figure 4).19–23
The largest collective experience was presented in the Valve-in-Valve International Data (VIVID) registry, which included 349 patients who had mitral valve-in-valve placement and 88 patients who had mitral valve-in-ring procedures. Their average age was 74 and the mean Society of Thoracic Surgeons score was 12.9% in both groups.24 Of the 437 patients, 345 patients (78.9%) underwent transapical implantation, and 391 patients (89.5%) received a Sapien XT or Sapien 3 valve. In the valve-in-valve group, 41% of the patients had regurgitation, 25% had stenosis, and 34% had both. In the valve-in-ring group, 60% of the patients had regurgitation, 17% had stenosis, and 23% had both.
Valve placement was successful in most patients. The rate of stroke was low (2.9% with valve-in-valve placement, 1.1% with valve-in-ring placement), though the rate of moderate or greater residual mitral regurgitation was significantly higher in patients undergoing valve-in-ring procedures (14.8% vs 2.6%, P < .001), as was the rate of left ventricular outflow tract obstruction (8% vs 2.6%, P = .03). There was also a trend toward worse 30-day mortality in the valve-in-ring group (11.4% vs 7.7%, P = .15). As with aortic valve-in-valve procedures, small surgical mitral valves (≤ 25 mm) were associated with higher postprocedural gradients.
Eleid and colleagues25 published their experience with antegrade transseptal TMVR in 48 patients with an average Society of Thoracic Surgeons score of 13.2%, 33 of whom underwent valve-in-valve procedures and nine of whom underwent valve-in-ring procedures. (The other six patients underwent mitral valve implantation for severe mitral annular calcification.) In the valve-in-valve group, 31 patients successfully underwent implant procedures, but two patients died during the procedure from left ventricular perforation. Of the nine valve-in-ring patients, two had acute embolization of the valve and were converted to open surgery. Among the seven patients in whom implantation was successful, two developed significant left ventricular outflow tract obstruction; one was treated with surgical resection of the anterior mitral valve leaflet and the other was medically managed.
CONCLUSION
Transcatheter mitral valve replacement in regurgitant mitral valves, failing mitral valve bioprosthetics and rings, and calcified mitral annuli has been effectively conducted in a number of patients who had no surgical options due to prohibitive surgical risk. International registries and our experience have demonstrated that the valve-in-valve procedure using a TAVR prosthesis carries the greatest likelihood of success, given the rigid frame of the surgical bioprosthetic that allows stable valve deployment. While approved in Europe for this indication, use of these devices for this application in the United States is considered “off label” and is performed only in clinically extenuating circumstances. Implantation of TAVR prosthetics in patients with prior mitral ring repair or for native mitral stenosis also has been performed successfully, although left ventricular outflow tract obstruction is a significant risk in this early experience.
Devices designed specifically for TMVR are in their clinical infancy and have been implanted successfully in only small numbers of patients, most of whom had functional mitral regurgitation. Despite reasonable technical success, most of these trials have been plagued by high mortality rates at 30 days in large part due to the extreme risk of the patients in whom these procedures have been conducted. At present, enrollment in TMVR trials for patients with degenerative or functional mitral regurgitation is limited to those without a surgical option and who conform to very specific anatomic criteria.
In the last 10 years, we have seen a revolution in transcatheter therapies for structural heart disease. The most widely embraced, transcatheter aortic valve replacement (TAVR) was originally intended for patients in whom surgery was considered impossible, but it has now been established as an excellent alternative to surgical aortic valve replacement in patients at high or intermediate risk.1–3 As TAVR has become established, with well-designed devices and acceptable safety and efficacy, it has inspired operators and inventors to push the envelope of innovation to transcatheter mitral valve replacement (TMVR).
This review summarizes the newest data available for the TMVR devices currently being tested in patients with native mitral regurgitation, bioprosthetic degeneration, and degenerative mitral stenosis.
THE MITRAL VALVE: THE NEW FRONTIER
Whereas the pathologic mechanisms of aortic stenosis generally all result in the same anatomic consequence (ie, calcification of the valve leaflets and commissures resulting in reduced mobility), mitral valve regurgitation is much more heterogeneous. Primary (degenerative) mitral regurgitation is caused by intrinsic valve pathology such as myxomatous degeneration, chordal detachment, fibroelastic deficiency, endocarditis, and other conditions that prevent the leaflets from coapting properly. In contrast, in secondary or functional mitral regurgitation, the leaflets are normal but do not coapt properly because of apical tethering to a dilated left ventricle, reduced closing forces with left ventricular dysfunction, or annular dilation as the result of either left ventricular or left atrial dilation.
Surgical mitral valve repair is safe and effective in patients with degenerative mitral regurgitation caused by leaflet prolapse and flail. However, some patients cannot undergo surgery because they have comorbid conditions that place them at extreme risk.4 For example, most patients with functional mitral regurgitation due to ischemic or dilated cardiomyopathy have significant surgical risk and multiple comorbidities, and in this group surgical repair has limited efficacy.5 A sizeable proportion of patients with mitral regurgitation may not be offered surgery because their risk is too high.6 Therefore, alternatives to the current surgical treatments have the potential to benefit a large number of patients.
Similarly, many patients with degenerative mitral stenosis caused by calcification of the mitral annulus also cannot undergo cardiac surgery because of prohibitively high risk. While rheumatic disease is the most common cause of mitral stenosis worldwide, degenerative mitral stenosis may be the cause in up to one-fourth of patients overall and up to 60% of patients older than 80 years.7 In the latter group, not only do old age and comorbidities such as diabetes mellitus and chronic kidney disease pose surgical risks, the technical challenge of surgically implanting a prosthetic mitral valve in the setting of a calcified annulus may be significant.8
The mitral valve is, therefore, the perfect new frontier for percutaneous valve replacement therapies, and TMVR is emerging as a potential option for patients with mitral regurgitation and degenerative mitral stenosis. The currently available percutaneous treatment options for mitral regurgitation include edge-to-edge leaflet repair, direct and indirect annuloplasty, spacers, and left ventricular remodeling devices (Table 1).9,10 As surgical mitral valve repair is strongly preferred over mitral valve replacement, the percutaneous procedures and the devices that are used are engineered to approximate the current standard surgical techniques. However, given the complex pathologies involved, surgical repair often requires the use of multiple repair techniques in the same patient. Therefore, percutaneous repair may also require more than one type of device in the same patient and may not be anatomically feasible in many patients. Replacing the entire valve may obviate some of these challenges.
Reprinted with permission from Wolters Kluwer Health, Inc. (Sud K, et al. Degenerated mitral stenosis: unmet need for percutaneous interventions. Circulation 2016; 133:1594–1604).
Figure 1. Routes of transcatheter mitral valve replacement: (A) transseptal antegrade via the femoral vein; (B) transapical retrograde via direct left ventricular access.
Compared with the aortic valve, the mitral valve poses a greater challenge to percutaneous treatment due to its structure and dynamic relationship with the left ventricle. Some specific challenges facing the development of TMVR are that the mitral valve is large, it is difficult to access, it is asymmetrical, it lacks an anatomically well-defined annulus to which to anchor the replacement valve, its geometry changes throughout the cardiac cycle, and placing a replacement valve in it entails the risk of left ventricular outflow tract obstruction. Despite these challenges, a number of devices are undergoing preclinical testing, a few are in phase 1 clinical trials, and registries are being kept. Depending on the specific device, an antegrade transseptal approach to the mitral valve (via the femoral vein) or a retrograde transapical approach (via direct left ventricular access) may be used (Figure 1).
NATIVE MITRAL VALVE REGURGITATION
For degenerative mitral regurgitation, the standard of care is cardiac surgery at a hospital experienced with mitral valve repair, and with very low rates of mortality and morbidity. For patients in whom the surgical risk is prohibitive, percutaneous edge-to-edge leaflet repair using the MitraClip (Abbott Vascular, Minneapolis, MN) is the best option if the anatomy permits. If the mitral valve pathology is not amenable to MitraClip repair, the patient may be evaluated for TMVR under a clinical trial protocol.
For functional mitral regurgitation, the decisions are more complex. If the patient has chronic atrial fibrillation, electrical cardioversion and antiarrhythmic drug therapy may restore and maintain sinus rhythm, though if the left atrium is large, sinus rhythm may not be possible. If the patient has left ventricular dysfunction, guideline-directed medical therapy should be optimized; this reduces the risk of exacerbations, hospitalizations, and death and may also reduce the degree of regurgitation. If the patient has severe left ventricular dysfunction and a wide QRS duration, cardiac resynchronization therapy (biventricular pacing) may also be beneficial and reduce functional mitral regurgitation. If symptoms and severe functional mitral regurgitation persist despite these measures and the patient’s surgical risk is deemed to be extreme, options include MitraClip placement as part of the randomized Cardiovascular Outcomes Assessment of the MitraClip Percutaneous Therapy (COAPT) trial, which compares guideline-directed medical therapy with guideline-directed therapy plus MitraClip. Another option is enrollment in a clinical trial or registry of TMVR.
At this writing, six TMVR devices have been implanted in humans:
Fortis (Edwards Lifesciences, Irvine, CA)
Tendyne (Tendyne Holding Inc, Roseville, MN)
NaviGate (NaviGate Cardiac Structures, Inc, Lake Forest, CA)
Intrepid (Medtronic, Minneapolis, MN)
CardiAQ (Edwards Lifesciences, Irvine, CA)
Tiara (Neovasc Inc, Richmond, BC).
Most of the early experience with these valves has not yet been published, but some data have been presented at national and international meetings.
The Fortis valve
Courtesy of Edwards Lifesciences.
Fortis valve
The Fortis valve consists of a self-expanding nitinol frame and leaflets made of bovine pericardium and is implanted via a transapical approach.
The device was successfully implanted in three patients in Quebec City, Canada, and at 6 months, all had improved significantly in functional class and none had needed to be hospitalized.11 Echocardiographic assessment demonstrated trace or less mitral regurgitation and a mean transvalvular gradient less than 4 mm Hg in all.
Bapat and colleagues12 attempted to implant the device in 13 patients in Europe and Canada. The average left ventricular ejection fraction was 34%, and 12 of 13 patients (92%) had functional mitral regurgitation. Procedural success was achieved in 10 patients, but five patients died within 30 days. While the deaths were due to nonvalvular issues (multiorgan failure, septic shock, intestinal ischemia after failed valve implantation and conversion to open surgery, malnutrition leading to respiratory failure, and valve thrombosis), the trial is currently on hold as more data are collected and reviewed. Among the eight patients who survived the first month, all were still alive at 6 months, and echocardiography demonstrated no or trivial mitral regurgitation in six patients (80%) and mild regurgitation in two patients (20%); the average mitral gradient was 4 mm Hg, and there was no change in mean left ventricular ejection fraction.
The Tendyne valve is a self-expanding prosthesis with porcine pericardial leaflets. It is delivered transapically and is held in place by a tether from the valve to the left ventricular apex.
In the first 12 patients enrolled in an early feasibility trial,13 the average left ventricular ejection fraction was 40%, and 11 of the 12 patients had functional mitral regurgitation. The device was successfully implanted in 11 patients, while one patient developed left ventricular outflow tract obstruction and the device was uneventfully removed. All patients were still alive at 30 days, and the 11 patients who still had a prosthetic valve did not have any residual mitral regurgitation.
As of this writing, almost 80 patients have received the device, though the data have not yet been presented. Patients are being enrolled in phase 1 trials.
The NaviGate valve
Courtesy of Jose Navia.
NaviGate valve
The NaviGate valve consists of a trileaflet subassembly fabricated from bovine pericardium, mounted on a self-expanding nitinol stent, and is only implanted transatrially.
Figure 2. Transatrial implantation of the NaviGate transcatheter mitral valve replacement prosthesis. (A) Initial unsheathing of the valve (arrow) via the left atrium (LA); (B) no residual mitral regurgitation on left ventriculography (LV). Ao = ascending aorta
NaviGate valves were successfully implanted in two patients via a transatrial approach (Figure 2). Both patients had excellent valve performance without residual mitral regurgitation or left ventricular outflow tract obstruction. The first patient showed significant improvement in functional class and freedom from hospitalization at 6 months, but the second patient died within a week of the implant due to advanced heart failure.14 A US clinical trial is expected soon.
The Intrepid valve
Courtesy of Medtronic.
Intrepid valve
The Intrepid valve consists of an outer stent to provide fixation to the annulus and an inner stent that houses a bovine pericardial valve. The device is a self-expanding system that is delivered transapically.
In a series of 15 patients, 11 had functional mitral regurgitation (with an average left ventricular ejection fraction of 35%) and four had degenerative mitral regurgitation (with an average left ventricular ejection fraction of 57%).15 The device was successfully implanted in 14 patients, after which the average mitral valve gradient was 4 mm Hg. All patients but one were left with no regurgitation (the other patient had 1+ regurgitation).
A trial is currently under way in Europe.
The CardiAQ valve
Courtesy of Edwards Lifesciences.
CardiAQ valve
The CardiAQ is constructed of bovine pericardium and can be delivered by the transseptal or transapical route.
Of 12 patients treated under compassionate use,16 two-thirds (eight patients) had functional mitral regurgitation. Two patients died during the procedure, three died of noncardiac complications within 30 days, and one more died of sepsis shortly after 30 days. This early experience demonstrates the importance of careful patient selection and postprocedural management in the feasibility assessment of these new technologies.
The Tiara valve, a self-expanding prosthesis with bovine pericardial leaflets, is delivered by the transapical route.
Eleven patients underwent Tiara implantation as part of either a Canadian special access registry or an international feasibility trial. Their average Society of Thoracic Surgeons score (ie, their calculated risk of major morbidity or operative mortality) was 15.6%, and their average left ventricular ejection fraction was 29%. Only two patients had degenerative mitral regurgitation. Nine patients had uneventful procedures and demonstrated no residual mitral regurgitation and no left ventricular outflow tract obstruction. The procedure was converted to open surgery in two patients owing to valve malpositioning, and both of them died within 30 days. One patient in whom the procedure was successful suffered erosion of the septum and died on day 4.17
Patients are being enrolled in phase 1 trials.
DEGENERATIVE MITRAL STENOSIS
Reprinted with permission from Wolters Kluwer Health, Inc. (Sud K, et al. Degenerated mitral stenosis: unmet need for percutaneous interventions. Circulation 2016; 133:1594–1604).
Figure 3. Mitral annular calcification (MAC) provides a “frame” for transcatheter mitral valve replacement prosthesis implantation in the mitral position for degenerative mitral stenosis. Ao = aorta; LVOT = left ventricular outflow tract
In patients with degenerative mitral stenosis, extensive mitral annular calcification may provide an adequate “frame” to hold a transcatheter valve prosthesis (Figure 3). Exploiting this feature, numerous investigators have successfully deployed prosthetic valves designed for TAVR in the calcified mitral annulus via the retrograde transapical and antegrade transseptal routes.
Guerrero and colleagues presented results from the first global registry of TMVR in mitral annular calcification at the 2016 EuroPCR Congress.18 Of 104 patients analyzed, almost all received an Edwards’ Sapien balloon-expandable valve (first-generation, Sapien XT, or Sapien 3); the others received Boston Scientific’s Lotus or Direct Flow Medical (Direct Flow Medical, Santa Clara, CA) valves. With an average age of 73 years and a high prevalence of comorbidities such as diabetes, chronic obstructive pulmonary disease, atrial fibrillation, chronic kidney disease, and prior cardiac surgery, the group presented extreme surgical risk, with an average Society of Thoracic Surgeons risk score of 14.4%. Slightly more than 40% of the patients underwent transapical implantation, slightly less than 40% underwent transfemoral or transseptal implantation, and just under 20% had a direct atrial approach.
The implantation was technically successful in 78 of 104 patients (75%); 13 patients (12.5%) required a second mitral valve to be placed, 11 patients (10.5%) had left ventricular outflow tract obstruction, four patients (4%) had valve embolization, and two patients (2%) had left ventricular perforation. At 30 days, 11 of 104 patients (10.6%) had died of cardiac causes and 15 patients (14.4%) had died of noncardiac causes. When divided roughly into three equal groups by chronological order, the last third of patients, compared with the first third of patients, enjoyed greater technical success (80%, n = 32/40 vs 62.5%, n = 20/32), better 30-day survival (85%, n = 34/40 vs 62.5%, n = 20/32), and no conversion to open surgery (0 vs 12.5%, n = 4/32), likely demonstrating both improved patient selection and lessons learned from shared experience. At 1 year, almost 90% of patients had New York Heart Association class I or II symptoms. Prior to the procedure, 91.5% had New York Heart Association class III or IV symptoms.
At present, TMVR in mitral annular calcification is not approved in the United States or elsewhere. However, multiple registries are currently enrolling patients or are in formative stages to push the frontier of the currently available technologies until better, dedicated devices are available for this group of patients.
BIOPROSTHETIC VALVE OR VALVE RING FAILURE
Figure 4. Transfemoral mitral valve-in-valve placement of a balloon-expandable valve. (A) Catheter via femoral vein (white arrow) and crossing the interatrial septum with unexpanded valve in place (black arrow) within the mitral prosthesis (arrowhead); (B) balloon inflation of the TAVR prosthesis (black arrow); (C) fully expanded valve in place; (D) 3D transesophageal echocardiographic view from the left atrium of the stenosed mitral valve (arrow); (E) mitral valve open (arrow) after valve-in-valve placement.
Implantation of a TAVR prosthetic inside a degenerated bioprosthetic mitral valve (valve-in-valve) and mitral valve ring (valve-in-ring) is generally limited to case series with short-term results using the Edwards Sapien series, Boston Scientific Lotus, Medtronic Melody (Medtronic, Minneapolis, MN), and Direct Flow Medical valves (Figure 4).19–23
The largest collective experience was presented in the Valve-in-Valve International Data (VIVID) registry, which included 349 patients who had mitral valve-in-valve placement and 88 patients who had mitral valve-in-ring procedures. Their average age was 74 and the mean Society of Thoracic Surgeons score was 12.9% in both groups.24 Of the 437 patients, 345 patients (78.9%) underwent transapical implantation, and 391 patients (89.5%) received a Sapien XT or Sapien 3 valve. In the valve-in-valve group, 41% of the patients had regurgitation, 25% had stenosis, and 34% had both. In the valve-in-ring group, 60% of the patients had regurgitation, 17% had stenosis, and 23% had both.
Valve placement was successful in most patients. The rate of stroke was low (2.9% with valve-in-valve placement, 1.1% with valve-in-ring placement), though the rate of moderate or greater residual mitral regurgitation was significantly higher in patients undergoing valve-in-ring procedures (14.8% vs 2.6%, P < .001), as was the rate of left ventricular outflow tract obstruction (8% vs 2.6%, P = .03). There was also a trend toward worse 30-day mortality in the valve-in-ring group (11.4% vs 7.7%, P = .15). As with aortic valve-in-valve procedures, small surgical mitral valves (≤ 25 mm) were associated with higher postprocedural gradients.
Eleid and colleagues25 published their experience with antegrade transseptal TMVR in 48 patients with an average Society of Thoracic Surgeons score of 13.2%, 33 of whom underwent valve-in-valve procedures and nine of whom underwent valve-in-ring procedures. (The other six patients underwent mitral valve implantation for severe mitral annular calcification.) In the valve-in-valve group, 31 patients successfully underwent implant procedures, but two patients died during the procedure from left ventricular perforation. Of the nine valve-in-ring patients, two had acute embolization of the valve and were converted to open surgery. Among the seven patients in whom implantation was successful, two developed significant left ventricular outflow tract obstruction; one was treated with surgical resection of the anterior mitral valve leaflet and the other was medically managed.
CONCLUSION
Transcatheter mitral valve replacement in regurgitant mitral valves, failing mitral valve bioprosthetics and rings, and calcified mitral annuli has been effectively conducted in a number of patients who had no surgical options due to prohibitive surgical risk. International registries and our experience have demonstrated that the valve-in-valve procedure using a TAVR prosthesis carries the greatest likelihood of success, given the rigid frame of the surgical bioprosthetic that allows stable valve deployment. While approved in Europe for this indication, use of these devices for this application in the United States is considered “off label” and is performed only in clinically extenuating circumstances. Implantation of TAVR prosthetics in patients with prior mitral ring repair or for native mitral stenosis also has been performed successfully, although left ventricular outflow tract obstruction is a significant risk in this early experience.
Devices designed specifically for TMVR are in their clinical infancy and have been implanted successfully in only small numbers of patients, most of whom had functional mitral regurgitation. Despite reasonable technical success, most of these trials have been plagued by high mortality rates at 30 days in large part due to the extreme risk of the patients in whom these procedures have been conducted. At present, enrollment in TMVR trials for patients with degenerative or functional mitral regurgitation is limited to those without a surgical option and who conform to very specific anatomic criteria.
References
Leon MB, Smith CR, Mack M, et al; PARTNER Trial Investigators. Transcatheter aortic-valve implantation for aortic stenosis in patients who cannot undergo surgery. N Engl J Med 2010; 363:1597–1607.
Smith CR, Leon MB, Mack MJ, et al; PARTNER Trial Investigators. Transcatheter versus surgical aortic-valve replacement in high-risk patients. N Engl J Med 2011; 364:2187–2198.
Thourani VH, Kodali S, Makkar RR, et al. Transcatheter aortic valve replacement versus surgical valve replacement in intermediate-risk patients: a propensity score analysis. Lancet 2016; 387:2218–2225.
Goel SS, Bajaj N, Aggarwal B, et al. Prevalence and outcomes of unoperated patients with severe symptomatic mitral regurgitation and heart failure: comprehensive analysis to determine the potential role of MitraClip for this unmet need. J Am Coll Cardiol 2014; 63:185–186.
DiBardino DJ, ElBardissi AW, McClure RS, Razo-Vasquez OA, Kelly NE, Cohn LH. Four decades of experience with mitral valve repair: analysis of differential indications, technical evolution, and long-term outcome. J Thorac Cardiovasc Surg 2010; 139:76–83; discussion 83–74.
Mirabel M, Iung B, Baron G, et al. What are the characteristics of patients with severe, symptomatic, mitral regurgitation who are denied surgery? Eur Heart J 2007; 28:1358–1365.
Iung B, Baron G, Butchart EG, et al. A prospective survey of patients with valvular heart disease in europe: the Euro Heart Survey on Valvular Heart Disease. Eur Heart J 2003; 24:1231–1243.
Sud K, Agarwal S, Parashar A, et al. Degenerative mitral stenosis: unmet need for percutaneous interventions. Circulation 2016; 133:1594–1604.
Svensson LG, Ye J, Piemonte TC, Kirker-Head C, Leon MB, Webb JG. Mitral valve regurgitation and left ventricular dysfunction treatment with an intravalvular spacer. J Card Surg 2015; 30:53–54.
Raman J, Raghavan J, Chandrashekar P, Sugeng L. Can we repair the mitral valve from outside the heart? A novel extra-cardiac approach to functional mitral regurgitation. Heart Lung Circ 2011; 20:157–162.
Abdul-Jawad Altisent O, Dumont E, Dagenais F, et al. Initial experience of transcatheter mitral valve replacement with a novel transcatheter mitral valve: procedural and 6-month follow-up results. J Am Coll Cardiol 2015; 66:1011–1019.
Bapat V. FORTIS: design, clinical results, and next steps. Presented at CRT (Cardiovascular Research Technologies) 16; Feburary 20–23, 2016; Washington, DC.
Sorajja P. Tendyne: technology and clinical results update. Presented at CRT (Cardiovascular Research Technologies) 16; February 20–23, 2016; Washington, DC.
Navia J. Personal communication.
Bapat V. Medtronic Intrepid transcatheter mitral valve replacement. Presented at EuroPCR 2015; May 19–22, 2015; Paris, France.
Herrmann H. Cardiaq-Edwards TMVR. Presented at CRT (Cardiovascular Research Technologies) 16; February 20–23, 2016; Washington, DC.
Dvir D. Tiara: design, clincal results, and next steps. Presented at CRT (Cardiovascular Research Technologies) 16; February 20–23, 2016; Washington, DC.
Guerrero M, Dvir D, Himbert D, et al. Transcatheter mitral valve replacement in native mitra valve disease with severe mitral annular calcification: results from the first global registry. JACC Cardiovasc Interv 2016; 9:1361–1371.
Seiffert M, Franzen O, Conradi L, et al. Series of transcatheter valve-in-valve implantations in high-risk patients with degenerated bioprostheses in aortic and mitral position. Catheter Cardiovasc Interv 2010; 76:608–615.
Webb JG, Wood DA, Ye J, et al. Transcatheter valve-in-valve implantation for failed bioprosthetic heart valves. Circulation 2010; 121:1848–1857.
Cerillo AG, Chiaramonti F, Murzi M, et al. Transcatheter valve in valve implantation for failed mitral and tricuspid bioprosthesis. Catheter Cardiovasc Interv 2011; 78:987–995.
Seiffert M, Conradi L, Baldus S, et al. Transcatheter mitral valve-in-valve implantation in patients with degenerated bioprostheses. JACC Cardiovasc Interv 2012; 5:341–349.
Wilbring M, Alexiou K, Tugtekin SM, et al. Pushing the limits—further evolutions of transcatheter valve procedures in the mitral position, including valve-in-valve, valve-in-ring, and valve-in-native-ring. J Thorac Cardiovasc Surg 2014; 147:210–219.
Dvir D, on behalf of the VIVID Registry Investigators. Transcatheter mitral valve-in-valve and valve-in-ring implantations. Transcatheter Valve Therapies 2015.
Eleid MF, Cabalka AK, Williams MR, et al. Percutaneous transvenous transseptal transcatheter valve implantation in failed bioprosthetic mitral valves, ring annuloplasty, and severe mitral annular calcification. JACC Cardiovasc Interv 2016; 9:1161–1174.
References
Leon MB, Smith CR, Mack M, et al; PARTNER Trial Investigators. Transcatheter aortic-valve implantation for aortic stenosis in patients who cannot undergo surgery. N Engl J Med 2010; 363:1597–1607.
Smith CR, Leon MB, Mack MJ, et al; PARTNER Trial Investigators. Transcatheter versus surgical aortic-valve replacement in high-risk patients. N Engl J Med 2011; 364:2187–2198.
Thourani VH, Kodali S, Makkar RR, et al. Transcatheter aortic valve replacement versus surgical valve replacement in intermediate-risk patients: a propensity score analysis. Lancet 2016; 387:2218–2225.
Goel SS, Bajaj N, Aggarwal B, et al. Prevalence and outcomes of unoperated patients with severe symptomatic mitral regurgitation and heart failure: comprehensive analysis to determine the potential role of MitraClip for this unmet need. J Am Coll Cardiol 2014; 63:185–186.
DiBardino DJ, ElBardissi AW, McClure RS, Razo-Vasquez OA, Kelly NE, Cohn LH. Four decades of experience with mitral valve repair: analysis of differential indications, technical evolution, and long-term outcome. J Thorac Cardiovasc Surg 2010; 139:76–83; discussion 83–74.
Mirabel M, Iung B, Baron G, et al. What are the characteristics of patients with severe, symptomatic, mitral regurgitation who are denied surgery? Eur Heart J 2007; 28:1358–1365.
Iung B, Baron G, Butchart EG, et al. A prospective survey of patients with valvular heart disease in europe: the Euro Heart Survey on Valvular Heart Disease. Eur Heart J 2003; 24:1231–1243.
Sud K, Agarwal S, Parashar A, et al. Degenerative mitral stenosis: unmet need for percutaneous interventions. Circulation 2016; 133:1594–1604.
Svensson LG, Ye J, Piemonte TC, Kirker-Head C, Leon MB, Webb JG. Mitral valve regurgitation and left ventricular dysfunction treatment with an intravalvular spacer. J Card Surg 2015; 30:53–54.
Raman J, Raghavan J, Chandrashekar P, Sugeng L. Can we repair the mitral valve from outside the heart? A novel extra-cardiac approach to functional mitral regurgitation. Heart Lung Circ 2011; 20:157–162.
Abdul-Jawad Altisent O, Dumont E, Dagenais F, et al. Initial experience of transcatheter mitral valve replacement with a novel transcatheter mitral valve: procedural and 6-month follow-up results. J Am Coll Cardiol 2015; 66:1011–1019.
Bapat V. FORTIS: design, clinical results, and next steps. Presented at CRT (Cardiovascular Research Technologies) 16; Feburary 20–23, 2016; Washington, DC.
Sorajja P. Tendyne: technology and clinical results update. Presented at CRT (Cardiovascular Research Technologies) 16; February 20–23, 2016; Washington, DC.
Navia J. Personal communication.
Bapat V. Medtronic Intrepid transcatheter mitral valve replacement. Presented at EuroPCR 2015; May 19–22, 2015; Paris, France.
Herrmann H. Cardiaq-Edwards TMVR. Presented at CRT (Cardiovascular Research Technologies) 16; February 20–23, 2016; Washington, DC.
Dvir D. Tiara: design, clincal results, and next steps. Presented at CRT (Cardiovascular Research Technologies) 16; February 20–23, 2016; Washington, DC.
Guerrero M, Dvir D, Himbert D, et al. Transcatheter mitral valve replacement in native mitra valve disease with severe mitral annular calcification: results from the first global registry. JACC Cardiovasc Interv 2016; 9:1361–1371.
Seiffert M, Franzen O, Conradi L, et al. Series of transcatheter valve-in-valve implantations in high-risk patients with degenerated bioprostheses in aortic and mitral position. Catheter Cardiovasc Interv 2010; 76:608–615.
Webb JG, Wood DA, Ye J, et al. Transcatheter valve-in-valve implantation for failed bioprosthetic heart valves. Circulation 2010; 121:1848–1857.
Cerillo AG, Chiaramonti F, Murzi M, et al. Transcatheter valve in valve implantation for failed mitral and tricuspid bioprosthesis. Catheter Cardiovasc Interv 2011; 78:987–995.
Seiffert M, Conradi L, Baldus S, et al. Transcatheter mitral valve-in-valve implantation in patients with degenerated bioprostheses. JACC Cardiovasc Interv 2012; 5:341–349.
Wilbring M, Alexiou K, Tugtekin SM, et al. Pushing the limits—further evolutions of transcatheter valve procedures in the mitral position, including valve-in-valve, valve-in-ring, and valve-in-native-ring. J Thorac Cardiovasc Surg 2014; 147:210–219.
Dvir D, on behalf of the VIVID Registry Investigators. Transcatheter mitral valve-in-valve and valve-in-ring implantations. Transcatheter Valve Therapies 2015.
Eleid MF, Cabalka AK, Williams MR, et al. Percutaneous transvenous transseptal transcatheter valve implantation in failed bioprosthetic mitral valves, ring annuloplasty, and severe mitral annular calcification. JACC Cardiovasc Interv 2016; 9:1161–1174.
Cleveland Clinic Journal of Medicine 2017 November; 83(suppl 2):S10-S17
Inside the Article
KEY POINTS
Most TMVR procedures are performed by either a retrograde transapical approach or an antegrade transseptal approach.
In the small number of patients who have undergone TMVR for native mitral valve regurgitation to date, mortality rates at 30 days have been high, reflecting the seriousness of illness in these patients.
At present, none of the new devices for TMVR in patients with native mitral valve regurgitation are approved for general use, although some of them are being tested in phase 1 clinical trials that are enrolling patients.
Valves made for TAVR have been used for TMVR in patients with degenerative mitral stenosis or failure of mitral bioprostheses; however, these are off-label uses of these devices.
Disallow All Ads
Alternative CME
Consolidated Pubs: Do Not Show Source Publication Logo
Interventional cardiology has made great strides in the last few decades. Percutaneous coronary intervention (PCI) is among the most commonly performed medical procedures globally.1 At the time of inception, PCI was plagued by high complication rates—balloon catheters had a 50% target-lesion restenosis rate at 6 months and required emergency bypass surgery in up to 6% patients.2 With passage of time, the complication rate of PCI has markedly decreased.
Figure 1. Reduction of restenosis rates by stent type.
The introduction of stents had a dramatic impact on lowering the complication rates. Initially, the bare-metal stents (BMS) reduced the stent restenosis rate to 10% to 15%. Drug-eluting stents (DES) have further revolutionized the field (Figure 1), significantly lowering rates of stent thrombosis (less than 0.5% in 1 year) and risk of restenosis (less than 5% in 1 year).3–6 The second-generation DES widely used in contemporary practice have made even more reductions owing to their improved designs and metallic and polymer composition; and concurrent advancements in the medical management, including use of antithrombotic and antiproliferative drugs, have further contributed to improved rates.
Figure 2. Second- vs first-generation drug-eluting stents.
What, then, is to be hoped for? Unfortunately, with the advent of stents, complications such as stent thrombosis and stent restenosis also emerged. These complications can be life-threatening in the form of post-procedural or late myocardial infarction and cardiac death. Thus, although the US Food and Drug Administration (FDA) assesses target-lesion failure (defined as a composite of cardiac death, target vessel myocardial infarction, or ischemia-driven target vessel revascularization) at 1 year, patients can have complications for the remainder of their lives. Despite the advancements attained by the second-generation DES over their predecessors, the issue of stent thrombosis and restenosis continues to plague second-generation DES with a 2% to 2.5% increased rate of target-lesion failure each year, seemingly forever (Figure 2).7,8
This article will briefly discuss the stent design and pathophysiology driving stent thrombosis and restenosis along with potential strategies to mitigate the problem. It pays special emphasis to bioresorbable stents, given their increasing interest among interventional cardiologists and patients, and given their potential to transform the practice of PCI.
STENT DESIGN
Contemporary DES essentially consist of three components:
A metallic alloy with a mesh-like design serves as the platform for the stent.
This framework is coated with a multi-layered polymer that holds and releases the active drug in a controlled manner so that its effects can be extended.
Figure 3. Components of drug-eluting and bioresorbable stents.An antiproliferative drug (absent in the bioresorbable stents) that inhibits the smooth muscle proliferation and neointimal hyperplastic response: sirolimus or paclitaxel in first-generation DES; everolimus or zotarolimus in second-generation DES (Figure 3).
WHAT CAUSES STENT THROMBOSIS AND RESTENOSIS?
Several theories and pathophysiological mechanisms have been proposed to explain these late adverse events (Table 1). However, our overall understanding of the cause remains modest at best. The major factor seems to be persistent presence of polymer on the stent and the ensuing inflammation. The second issue appears to be related to neoatherosclerosis that is generally defined as lipid or calcified neointima. Neoatherosclerosis is especially problematic for the second-generation DES. Neoatherosclerosis eventually predisposes to the development of thin cap fibroadenoma, and the rupture of thin cap leads to stent thrombosis and restenosis.
Autopsy studies suggest that approximately 50% of first- and second-generation DES start developing neoatherosclerosis within 1 to 3 years of implantation.9 Turbulence created by thick strutted stents or incomplete impaction of stents to the vessel wall predisposes the stents to platelet aggregation and fibrinogen deposition, thereby increasing the risk of neoatherosclerosis. Despite these pathologic insights, no treatment strategy has been shown to attenuate the problem, with the exception of high-dose statins.
CAN WE SOLVE THE PROBLEM?
Three technological approaches have been proposed to overcome stent thrombosis and restenosis:
Stents coated with bioresorbable polymers that quickly degrade
Stents without polymers
Stents that are completely resorbed.
STENTS WITH BIORESORBABLE POLYMERS
As described above, the presence of a polymer on the stent predisposes it to inflammation. Therefore, it would be logical to hypothesize that a bioresorbable polymer would reduce the inflammation. This approach is typified by the second-generation paclitaxel-eluting stent (Synergy, Boston Scientific). It has a biodegradable coating that resorbs within 4 months and releases everolimus in a dose intensity similar to that seen with the contemporary second-generation DES.
The largest trial of this device to date, the Evolve II study, randomly assigned 1,684 patients to the biostable-polymer, everolimus-eluting chromium stent (Promus, Boston Scientific) or the paclitaxel-eluting stent (Synergy, Boston Scientific).10 Two-year follow-up data suggest that the rate of target-lesion failure was 9.4% in the paclitaxel-eluting stent patients vs 8.5% in the everolimus-eluting stent patients. Notably, no definite stent thrombosis was seen in the Synergy-treated patients 24 hours after the initial device implantation.
STENTS WITHOUT POLYMERS
If polymers predispose to inflammation, stents without polymers should mitigate the risk. Such stent types are exemplified by the BioFreedom (Biosensors International) stainless steel stent, a polymer-free umirolimus (also known as biolimus A9)-eluting stent. These stents have a microstructured surface that holds the drug without a polymer and releases the active drug over a few months.
The LEADERS FREE clinical trial studied this stent in 2,466 patients at high risk of bleeding.11 The patients were randomized to receive either a BMS or the polymer-free stent. All patients were required to receive dual antiplatelet therapy for only 1 month. At 1 year, the composite risk of cardiac death, myocardial infarction, and stent thrombois was 9.4% in patients with BioFreedom stents vs 12.9% in BMS patients. Of note, the primary end point did not include stent restenosis, thereby not disadvantaging the BMS.
Medtronic’s polymer-free, sirolimus-eluting stent is currently under investigation in the RevElution clinical trial.12 It has a cylindrical structure with the core replaced by the active drug sirolimus. Abluminal holes in the stent allow controlled release of the drug. A pharmacokinetic analysis show that 90% of the medication is released within the first 90 days and that tissue concentrations are maintained in the therapeutic range until at least that time.13 This actually exceeds that of the second-generation everolimus-eluting DES.
BIORESORBABLE STENTS
Bioresorbable scaffolds or stents disappear entirely over time and have drawn considerable attention in the interventional cardiology community. The FDA recently approved Abbott’s Poly-L-Lactic Acid (PLLA) everolimus-eluting stent (Absorb). The rate of bioresorption of this device can be controlled by modulating the respective contribution of amorphous and crystalline PLLA backbone. The advantage of bioresorbable stents appears to stem from the fact that with bioresorbable devices, the vessel may actually expand and the purported nidus for inflammation goes away. This has been demonstrated by serial intravascular ultrasound-based studies.14
The return of pulsatility also appears to modulate the transition of smooth muscles from proliferative back to their contractile phenotype. This has been hypothesized to reduce the risk of neoatherosclerosis and, consequently, stent restenosis. The limitation of this device is the large strut size (157 micron for Absorb vs 81 microns for Xience). Dissolving metallic scaffolds also tend to have thicker struts than the current DES (120 vs approximately 80 microns).
The Absorb III trial was a pivotal noninferiority US trial that led to the device approval.15 In this trial, 2,008 patients were randomized to receive the Absorb bioresorbable, everolimus-eluting stent or the DES Xience. The primary study end point was target-lesion failure at 1 year. As is often the case with US landmark studies, patient and lesion complexities were limited. Patients with acute coronary syndrome, elevated cardiac enzymes, high-risk anatomic lesions such as bifurcation lesions, and chronic total occlusion were excluded. Patients with diabetes comprised less than one-third of the patients, and lesions were relatively short at 13 ± 6 mm.
Figure 4. ABSORB learnings: MICAT. Pre-dilation with noncompliant balloon sized 1:1 to normal vessel with complete balloon expansion; post-dilation at 14–16 atmospheres.
Device success per lesion was lower with Absorb than with Xience (94.3% vs 99.3%; P < .0001). This is likely due to the larger strut size. Absorb III did meet the prespecified primary end point for noninferiority (P = .007), although the rate of adverse events was somewhat higher (7.8% vs 6.1%). A subgroup analysis reveals that 19% of all lesions were smaller than what was originally intended, and in these patients, the Absorb device performed poorly with a 4.6% risk of device thrombosis. When limited to patients with the intended reference vessel sizes, the results of target-lesion failure and stent thrombosis were similar (6.6% vs 5.5% and 0.8% vs 0.5%, respectively).15
The implantation technique also seems to have influenced the results, with increased use of post-dilation as the study evolved. Recent observations from the MICAT group have shown that the use of high pressure post-dilation and other procedural advancements may considerably reduce adverse outcomes associated with Absorb (Figure 4).16 Thus, while the pooled analysis in the form of a meta-analysis has suggested an increased risk of device thrombosis,17 the difference is attenuated by selecting lesions of appropriate size, high-pressure post-dilation, and procedural advancements (Table 2).
CONCLUSION AND THE WAY FORWARD
Current first-generation bioresorbable stents can achieve results similar to those of second-generation DES, provided that they are used in patients with noncomplicated coronary lesions and the implant techniques are optimized. We do not know the outcomes of bioresorbable stents in patients with complex lesions. Current experience suggests that other changes in technique would be needed. For example, minimizing scaffold overlap in long and bifurcating lesions. Whether that would translate into diminishing the rate of late adverse events remains to be determined. As of now, we only have data on approximately 100 highly selected patients beyond 3 years (no adverse events 2.5 to 5 years after implantation).
Several investigational second-generation bioresorbable stents, including Elixir’s Dissolve PLLA, Boston Scientific’s FAST, and a newer version of Absorb, are in early clinical trials. Smaller strut thickness holds the promise of attenuating the risk of stent thrombosis. Since the polymer persists, no reduction in dual antiplatelet therapy duration is likely to be achieved.
Results from long-term follow-up of Absorb III and on-going trials are eagerly awaited to ascertain whether the rate of late complications of DES can be mitigated. It would not be surprising if the second-generation bioresorbable stents make DES a thing of the past within the next decade.
References
Palmerini T, Benedetto U, Biondi-Zoccai G, et al. Long-term safety of drug-eluting and bare-metal stents: evidence from a comprehensive network meta-analysis. J Am Coll Cardiol 2015; 65:2496–2507.
Bates ER. Balancing the evidence base on coronary stents. N Engl J Med 2016; 375:1286–1288.
Macaya C, Serruys PW, Ruygrok P, et al. Continued benefit of coronary stenting versus balloon angioplasty: one-year clinical follow-up of BENESTENT trial. BENESTENT Study Group. J Am Coll Cardiol 1996; 27:255–261.
Fajadet J, Wijns W, Laarman GJ; for the ENDEAVOR II Investigators. Randomized, double-blind, multicenter study of the Endeavor zotarolimus-eluting phosphorylcholine-encapsulated stent for treatment of native coronary artery lesions: clinical and angiographic results of the ENDEAVOR II trial. Circulation 2006; 114:798–806.
Kirtane AJ, Leon MB, Ball MW, et al; ENDEAVOR IV Investigators. The “final” 5-year follow-up from the ENDEAVOR IV trial comparing a zotarolimus-eluting stent with a paclitaxel-eluting stent. JACC Cardiovasc Interv 2013; 6:325–333.
Stone GW, Midei M, Newman W, et al; SPIRIT III Investigators. Comparison of an everolimus-eluting stent and a paclitaxel-eluting stent in patients with coronary artery disease: a randomized trial. JAMA 2008; 299:1903–1913.
Gada H, Kirtane AJ, Newman W, et al. 5-year results of a randomized comparison of XIENCE V everolimus-eluting and TAXUS paclitaxel-eluting stents: final results from the SPIRIT III trial (clinical evaluation of the XIENCE V everolimus eluting coronary stent system in the treatment of patients with de novo native coronary artery lesions). JACC Cardiovasc Interv 2013; 6:1263–1266.
Benedetto U, Raja SG, Soliman RFB, et al; on behalf of the Harefield Cardiac Outcomes Research Group. Minimally invasive direct coronary artery bypass improves late survival compared with drug-eluting stents in isolated proximal left anterior descending artery disease: a 10-year follow-up, single-center, propensity score analysis. J Thorac Cardiovasc Surg 2014; 148:1316–1322.
Virmani R, CV Path Institute. Problems encountered with drug-eluting stent. Presented at Transcatheter Cardiovascular Therapeutics (TCT); October 11–15, 2015; San Francisco, CA.
Kereiakes DJ, Meredith IT, Windecker S, et al. Efficacy and safety of a novel bioabsorbable polymer-coated, everolimus-eluting coronary stent: the EVOLVE II Randomized Trial. Circ Cardiovasc Interv 2015; 8:e002372.
Urban P, Meredith IT, Abizaid A, et al; for the LEADERS FREE Investigators. Polymer-free drug-coated coronary stents in patients at high bleeding risk. N Engl J Med 2015; 373:2038–2047.
Worthley SG, Abizaid A, Kirtane AJ, Simon D, Windecker S, Stone GW. Stent strut coverage and stent apposition after implantation of a novel drug-filled coronary stent: optical coherence tomography results from RevElution trial. J Am Coll Cardiol 2015; 66(suppl B):B235 [Abstract TCT-579].
Abizaid A, Costa RA, Schofer J, et al. Serial multimodality imaging and 2-year clinical outcomes of the novel DESolve novolimus-eluting bioresorbable coronary scaffold system for the treatment of single de novo coronary lesions. JACC Cardiovasc Interv 2016; 9:565–574.
Ellis SG, Kereiakes DJ, Metzger DC, et al; for the ABOSRB III Investigators. Everolimus-eluting bioresorbable scaffolds for coronary artery disease. N Engl J Med 2015; 373:1905–1915.
Puricel S, Cuculi F, Weissner M, et al. Bioresorbable coronary scaffold thrombosis: multicenter comprehensive analysis of clinical presentation, mechanisms, and predictors. J Am Coll Cardiol 2016; 67:921−931.
Stone GW, Gao R, Kimura T, et al. 1-year outcomes with the Absorb bioresorbable scaffold in patients with coronary artery disease: a patient-level, pooled meta-analysis. Lancet 2016; 387:1277–1289.
Stephen G. Ellis, MD Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic
Haris Riaz, MD Resident, Department of Internal Medicine, Cleveland Clinic
Correspondence: Stephen G. Ellis, MD, Department of Cardiovascular Medicine, J2-3, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; elliss@ccf.org
Dr. Ellis reported consulting for Abbott Vascular. Dr. Riaz reported no financial interests or relationships that pose a potential conflict of interest with this article.
Stephen G. Ellis, MD Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic
Haris Riaz, MD Resident, Department of Internal Medicine, Cleveland Clinic
Correspondence: Stephen G. Ellis, MD, Department of Cardiovascular Medicine, J2-3, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; elliss@ccf.org
Dr. Ellis reported consulting for Abbott Vascular. Dr. Riaz reported no financial interests or relationships that pose a potential conflict of interest with this article.
Author and Disclosure Information
Stephen G. Ellis, MD Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic
Haris Riaz, MD Resident, Department of Internal Medicine, Cleveland Clinic
Correspondence: Stephen G. Ellis, MD, Department of Cardiovascular Medicine, J2-3, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; elliss@ccf.org
Dr. Ellis reported consulting for Abbott Vascular. Dr. Riaz reported no financial interests or relationships that pose a potential conflict of interest with this article.
Interventional cardiology has made great strides in the last few decades. Percutaneous coronary intervention (PCI) is among the most commonly performed medical procedures globally.1 At the time of inception, PCI was plagued by high complication rates—balloon catheters had a 50% target-lesion restenosis rate at 6 months and required emergency bypass surgery in up to 6% patients.2 With passage of time, the complication rate of PCI has markedly decreased.
Figure 1. Reduction of restenosis rates by stent type.
The introduction of stents had a dramatic impact on lowering the complication rates. Initially, the bare-metal stents (BMS) reduced the stent restenosis rate to 10% to 15%. Drug-eluting stents (DES) have further revolutionized the field (Figure 1), significantly lowering rates of stent thrombosis (less than 0.5% in 1 year) and risk of restenosis (less than 5% in 1 year).3–6 The second-generation DES widely used in contemporary practice have made even more reductions owing to their improved designs and metallic and polymer composition; and concurrent advancements in the medical management, including use of antithrombotic and antiproliferative drugs, have further contributed to improved rates.
Figure 2. Second- vs first-generation drug-eluting stents.
What, then, is to be hoped for? Unfortunately, with the advent of stents, complications such as stent thrombosis and stent restenosis also emerged. These complications can be life-threatening in the form of post-procedural or late myocardial infarction and cardiac death. Thus, although the US Food and Drug Administration (FDA) assesses target-lesion failure (defined as a composite of cardiac death, target vessel myocardial infarction, or ischemia-driven target vessel revascularization) at 1 year, patients can have complications for the remainder of their lives. Despite the advancements attained by the second-generation DES over their predecessors, the issue of stent thrombosis and restenosis continues to plague second-generation DES with a 2% to 2.5% increased rate of target-lesion failure each year, seemingly forever (Figure 2).7,8
This article will briefly discuss the stent design and pathophysiology driving stent thrombosis and restenosis along with potential strategies to mitigate the problem. It pays special emphasis to bioresorbable stents, given their increasing interest among interventional cardiologists and patients, and given their potential to transform the practice of PCI.
STENT DESIGN
Contemporary DES essentially consist of three components:
A metallic alloy with a mesh-like design serves as the platform for the stent.
This framework is coated with a multi-layered polymer that holds and releases the active drug in a controlled manner so that its effects can be extended.
Figure 3. Components of drug-eluting and bioresorbable stents.An antiproliferative drug (absent in the bioresorbable stents) that inhibits the smooth muscle proliferation and neointimal hyperplastic response: sirolimus or paclitaxel in first-generation DES; everolimus or zotarolimus in second-generation DES (Figure 3).
WHAT CAUSES STENT THROMBOSIS AND RESTENOSIS?
Several theories and pathophysiological mechanisms have been proposed to explain these late adverse events (Table 1). However, our overall understanding of the cause remains modest at best. The major factor seems to be persistent presence of polymer on the stent and the ensuing inflammation. The second issue appears to be related to neoatherosclerosis that is generally defined as lipid or calcified neointima. Neoatherosclerosis is especially problematic for the second-generation DES. Neoatherosclerosis eventually predisposes to the development of thin cap fibroadenoma, and the rupture of thin cap leads to stent thrombosis and restenosis.
Autopsy studies suggest that approximately 50% of first- and second-generation DES start developing neoatherosclerosis within 1 to 3 years of implantation.9 Turbulence created by thick strutted stents or incomplete impaction of stents to the vessel wall predisposes the stents to platelet aggregation and fibrinogen deposition, thereby increasing the risk of neoatherosclerosis. Despite these pathologic insights, no treatment strategy has been shown to attenuate the problem, with the exception of high-dose statins.
CAN WE SOLVE THE PROBLEM?
Three technological approaches have been proposed to overcome stent thrombosis and restenosis:
Stents coated with bioresorbable polymers that quickly degrade
Stents without polymers
Stents that are completely resorbed.
STENTS WITH BIORESORBABLE POLYMERS
As described above, the presence of a polymer on the stent predisposes it to inflammation. Therefore, it would be logical to hypothesize that a bioresorbable polymer would reduce the inflammation. This approach is typified by the second-generation paclitaxel-eluting stent (Synergy, Boston Scientific). It has a biodegradable coating that resorbs within 4 months and releases everolimus in a dose intensity similar to that seen with the contemporary second-generation DES.
The largest trial of this device to date, the Evolve II study, randomly assigned 1,684 patients to the biostable-polymer, everolimus-eluting chromium stent (Promus, Boston Scientific) or the paclitaxel-eluting stent (Synergy, Boston Scientific).10 Two-year follow-up data suggest that the rate of target-lesion failure was 9.4% in the paclitaxel-eluting stent patients vs 8.5% in the everolimus-eluting stent patients. Notably, no definite stent thrombosis was seen in the Synergy-treated patients 24 hours after the initial device implantation.
STENTS WITHOUT POLYMERS
If polymers predispose to inflammation, stents without polymers should mitigate the risk. Such stent types are exemplified by the BioFreedom (Biosensors International) stainless steel stent, a polymer-free umirolimus (also known as biolimus A9)-eluting stent. These stents have a microstructured surface that holds the drug without a polymer and releases the active drug over a few months.
The LEADERS FREE clinical trial studied this stent in 2,466 patients at high risk of bleeding.11 The patients were randomized to receive either a BMS or the polymer-free stent. All patients were required to receive dual antiplatelet therapy for only 1 month. At 1 year, the composite risk of cardiac death, myocardial infarction, and stent thrombois was 9.4% in patients with BioFreedom stents vs 12.9% in BMS patients. Of note, the primary end point did not include stent restenosis, thereby not disadvantaging the BMS.
Medtronic’s polymer-free, sirolimus-eluting stent is currently under investigation in the RevElution clinical trial.12 It has a cylindrical structure with the core replaced by the active drug sirolimus. Abluminal holes in the stent allow controlled release of the drug. A pharmacokinetic analysis show that 90% of the medication is released within the first 90 days and that tissue concentrations are maintained in the therapeutic range until at least that time.13 This actually exceeds that of the second-generation everolimus-eluting DES.
BIORESORBABLE STENTS
Bioresorbable scaffolds or stents disappear entirely over time and have drawn considerable attention in the interventional cardiology community. The FDA recently approved Abbott’s Poly-L-Lactic Acid (PLLA) everolimus-eluting stent (Absorb). The rate of bioresorption of this device can be controlled by modulating the respective contribution of amorphous and crystalline PLLA backbone. The advantage of bioresorbable stents appears to stem from the fact that with bioresorbable devices, the vessel may actually expand and the purported nidus for inflammation goes away. This has been demonstrated by serial intravascular ultrasound-based studies.14
The return of pulsatility also appears to modulate the transition of smooth muscles from proliferative back to their contractile phenotype. This has been hypothesized to reduce the risk of neoatherosclerosis and, consequently, stent restenosis. The limitation of this device is the large strut size (157 micron for Absorb vs 81 microns for Xience). Dissolving metallic scaffolds also tend to have thicker struts than the current DES (120 vs approximately 80 microns).
The Absorb III trial was a pivotal noninferiority US trial that led to the device approval.15 In this trial, 2,008 patients were randomized to receive the Absorb bioresorbable, everolimus-eluting stent or the DES Xience. The primary study end point was target-lesion failure at 1 year. As is often the case with US landmark studies, patient and lesion complexities were limited. Patients with acute coronary syndrome, elevated cardiac enzymes, high-risk anatomic lesions such as bifurcation lesions, and chronic total occlusion were excluded. Patients with diabetes comprised less than one-third of the patients, and lesions were relatively short at 13 ± 6 mm.
Figure 4. ABSORB learnings: MICAT. Pre-dilation with noncompliant balloon sized 1:1 to normal vessel with complete balloon expansion; post-dilation at 14–16 atmospheres.
Device success per lesion was lower with Absorb than with Xience (94.3% vs 99.3%; P < .0001). This is likely due to the larger strut size. Absorb III did meet the prespecified primary end point for noninferiority (P = .007), although the rate of adverse events was somewhat higher (7.8% vs 6.1%). A subgroup analysis reveals that 19% of all lesions were smaller than what was originally intended, and in these patients, the Absorb device performed poorly with a 4.6% risk of device thrombosis. When limited to patients with the intended reference vessel sizes, the results of target-lesion failure and stent thrombosis were similar (6.6% vs 5.5% and 0.8% vs 0.5%, respectively).15
The implantation technique also seems to have influenced the results, with increased use of post-dilation as the study evolved. Recent observations from the MICAT group have shown that the use of high pressure post-dilation and other procedural advancements may considerably reduce adverse outcomes associated with Absorb (Figure 4).16 Thus, while the pooled analysis in the form of a meta-analysis has suggested an increased risk of device thrombosis,17 the difference is attenuated by selecting lesions of appropriate size, high-pressure post-dilation, and procedural advancements (Table 2).
CONCLUSION AND THE WAY FORWARD
Current first-generation bioresorbable stents can achieve results similar to those of second-generation DES, provided that they are used in patients with noncomplicated coronary lesions and the implant techniques are optimized. We do not know the outcomes of bioresorbable stents in patients with complex lesions. Current experience suggests that other changes in technique would be needed. For example, minimizing scaffold overlap in long and bifurcating lesions. Whether that would translate into diminishing the rate of late adverse events remains to be determined. As of now, we only have data on approximately 100 highly selected patients beyond 3 years (no adverse events 2.5 to 5 years after implantation).
Several investigational second-generation bioresorbable stents, including Elixir’s Dissolve PLLA, Boston Scientific’s FAST, and a newer version of Absorb, are in early clinical trials. Smaller strut thickness holds the promise of attenuating the risk of stent thrombosis. Since the polymer persists, no reduction in dual antiplatelet therapy duration is likely to be achieved.
Results from long-term follow-up of Absorb III and on-going trials are eagerly awaited to ascertain whether the rate of late complications of DES can be mitigated. It would not be surprising if the second-generation bioresorbable stents make DES a thing of the past within the next decade.
Interventional cardiology has made great strides in the last few decades. Percutaneous coronary intervention (PCI) is among the most commonly performed medical procedures globally.1 At the time of inception, PCI was plagued by high complication rates—balloon catheters had a 50% target-lesion restenosis rate at 6 months and required emergency bypass surgery in up to 6% patients.2 With passage of time, the complication rate of PCI has markedly decreased.
Figure 1. Reduction of restenosis rates by stent type.
The introduction of stents had a dramatic impact on lowering the complication rates. Initially, the bare-metal stents (BMS) reduced the stent restenosis rate to 10% to 15%. Drug-eluting stents (DES) have further revolutionized the field (Figure 1), significantly lowering rates of stent thrombosis (less than 0.5% in 1 year) and risk of restenosis (less than 5% in 1 year).3–6 The second-generation DES widely used in contemporary practice have made even more reductions owing to their improved designs and metallic and polymer composition; and concurrent advancements in the medical management, including use of antithrombotic and antiproliferative drugs, have further contributed to improved rates.
Figure 2. Second- vs first-generation drug-eluting stents.
What, then, is to be hoped for? Unfortunately, with the advent of stents, complications such as stent thrombosis and stent restenosis also emerged. These complications can be life-threatening in the form of post-procedural or late myocardial infarction and cardiac death. Thus, although the US Food and Drug Administration (FDA) assesses target-lesion failure (defined as a composite of cardiac death, target vessel myocardial infarction, or ischemia-driven target vessel revascularization) at 1 year, patients can have complications for the remainder of their lives. Despite the advancements attained by the second-generation DES over their predecessors, the issue of stent thrombosis and restenosis continues to plague second-generation DES with a 2% to 2.5% increased rate of target-lesion failure each year, seemingly forever (Figure 2).7,8
This article will briefly discuss the stent design and pathophysiology driving stent thrombosis and restenosis along with potential strategies to mitigate the problem. It pays special emphasis to bioresorbable stents, given their increasing interest among interventional cardiologists and patients, and given their potential to transform the practice of PCI.
STENT DESIGN
Contemporary DES essentially consist of three components:
A metallic alloy with a mesh-like design serves as the platform for the stent.
This framework is coated with a multi-layered polymer that holds and releases the active drug in a controlled manner so that its effects can be extended.
Figure 3. Components of drug-eluting and bioresorbable stents.An antiproliferative drug (absent in the bioresorbable stents) that inhibits the smooth muscle proliferation and neointimal hyperplastic response: sirolimus or paclitaxel in first-generation DES; everolimus or zotarolimus in second-generation DES (Figure 3).
WHAT CAUSES STENT THROMBOSIS AND RESTENOSIS?
Several theories and pathophysiological mechanisms have been proposed to explain these late adverse events (Table 1). However, our overall understanding of the cause remains modest at best. The major factor seems to be persistent presence of polymer on the stent and the ensuing inflammation. The second issue appears to be related to neoatherosclerosis that is generally defined as lipid or calcified neointima. Neoatherosclerosis is especially problematic for the second-generation DES. Neoatherosclerosis eventually predisposes to the development of thin cap fibroadenoma, and the rupture of thin cap leads to stent thrombosis and restenosis.
Autopsy studies suggest that approximately 50% of first- and second-generation DES start developing neoatherosclerosis within 1 to 3 years of implantation.9 Turbulence created by thick strutted stents or incomplete impaction of stents to the vessel wall predisposes the stents to platelet aggregation and fibrinogen deposition, thereby increasing the risk of neoatherosclerosis. Despite these pathologic insights, no treatment strategy has been shown to attenuate the problem, with the exception of high-dose statins.
CAN WE SOLVE THE PROBLEM?
Three technological approaches have been proposed to overcome stent thrombosis and restenosis:
Stents coated with bioresorbable polymers that quickly degrade
Stents without polymers
Stents that are completely resorbed.
STENTS WITH BIORESORBABLE POLYMERS
As described above, the presence of a polymer on the stent predisposes it to inflammation. Therefore, it would be logical to hypothesize that a bioresorbable polymer would reduce the inflammation. This approach is typified by the second-generation paclitaxel-eluting stent (Synergy, Boston Scientific). It has a biodegradable coating that resorbs within 4 months and releases everolimus in a dose intensity similar to that seen with the contemporary second-generation DES.
The largest trial of this device to date, the Evolve II study, randomly assigned 1,684 patients to the biostable-polymer, everolimus-eluting chromium stent (Promus, Boston Scientific) or the paclitaxel-eluting stent (Synergy, Boston Scientific).10 Two-year follow-up data suggest that the rate of target-lesion failure was 9.4% in the paclitaxel-eluting stent patients vs 8.5% in the everolimus-eluting stent patients. Notably, no definite stent thrombosis was seen in the Synergy-treated patients 24 hours after the initial device implantation.
STENTS WITHOUT POLYMERS
If polymers predispose to inflammation, stents without polymers should mitigate the risk. Such stent types are exemplified by the BioFreedom (Biosensors International) stainless steel stent, a polymer-free umirolimus (also known as biolimus A9)-eluting stent. These stents have a microstructured surface that holds the drug without a polymer and releases the active drug over a few months.
The LEADERS FREE clinical trial studied this stent in 2,466 patients at high risk of bleeding.11 The patients were randomized to receive either a BMS or the polymer-free stent. All patients were required to receive dual antiplatelet therapy for only 1 month. At 1 year, the composite risk of cardiac death, myocardial infarction, and stent thrombois was 9.4% in patients with BioFreedom stents vs 12.9% in BMS patients. Of note, the primary end point did not include stent restenosis, thereby not disadvantaging the BMS.
Medtronic’s polymer-free, sirolimus-eluting stent is currently under investigation in the RevElution clinical trial.12 It has a cylindrical structure with the core replaced by the active drug sirolimus. Abluminal holes in the stent allow controlled release of the drug. A pharmacokinetic analysis show that 90% of the medication is released within the first 90 days and that tissue concentrations are maintained in the therapeutic range until at least that time.13 This actually exceeds that of the second-generation everolimus-eluting DES.
BIORESORBABLE STENTS
Bioresorbable scaffolds or stents disappear entirely over time and have drawn considerable attention in the interventional cardiology community. The FDA recently approved Abbott’s Poly-L-Lactic Acid (PLLA) everolimus-eluting stent (Absorb). The rate of bioresorption of this device can be controlled by modulating the respective contribution of amorphous and crystalline PLLA backbone. The advantage of bioresorbable stents appears to stem from the fact that with bioresorbable devices, the vessel may actually expand and the purported nidus for inflammation goes away. This has been demonstrated by serial intravascular ultrasound-based studies.14
The return of pulsatility also appears to modulate the transition of smooth muscles from proliferative back to their contractile phenotype. This has been hypothesized to reduce the risk of neoatherosclerosis and, consequently, stent restenosis. The limitation of this device is the large strut size (157 micron for Absorb vs 81 microns for Xience). Dissolving metallic scaffolds also tend to have thicker struts than the current DES (120 vs approximately 80 microns).
The Absorb III trial was a pivotal noninferiority US trial that led to the device approval.15 In this trial, 2,008 patients were randomized to receive the Absorb bioresorbable, everolimus-eluting stent or the DES Xience. The primary study end point was target-lesion failure at 1 year. As is often the case with US landmark studies, patient and lesion complexities were limited. Patients with acute coronary syndrome, elevated cardiac enzymes, high-risk anatomic lesions such as bifurcation lesions, and chronic total occlusion were excluded. Patients with diabetes comprised less than one-third of the patients, and lesions were relatively short at 13 ± 6 mm.
Figure 4. ABSORB learnings: MICAT. Pre-dilation with noncompliant balloon sized 1:1 to normal vessel with complete balloon expansion; post-dilation at 14–16 atmospheres.
Device success per lesion was lower with Absorb than with Xience (94.3% vs 99.3%; P < .0001). This is likely due to the larger strut size. Absorb III did meet the prespecified primary end point for noninferiority (P = .007), although the rate of adverse events was somewhat higher (7.8% vs 6.1%). A subgroup analysis reveals that 19% of all lesions were smaller than what was originally intended, and in these patients, the Absorb device performed poorly with a 4.6% risk of device thrombosis. When limited to patients with the intended reference vessel sizes, the results of target-lesion failure and stent thrombosis were similar (6.6% vs 5.5% and 0.8% vs 0.5%, respectively).15
The implantation technique also seems to have influenced the results, with increased use of post-dilation as the study evolved. Recent observations from the MICAT group have shown that the use of high pressure post-dilation and other procedural advancements may considerably reduce adverse outcomes associated with Absorb (Figure 4).16 Thus, while the pooled analysis in the form of a meta-analysis has suggested an increased risk of device thrombosis,17 the difference is attenuated by selecting lesions of appropriate size, high-pressure post-dilation, and procedural advancements (Table 2).
CONCLUSION AND THE WAY FORWARD
Current first-generation bioresorbable stents can achieve results similar to those of second-generation DES, provided that they are used in patients with noncomplicated coronary lesions and the implant techniques are optimized. We do not know the outcomes of bioresorbable stents in patients with complex lesions. Current experience suggests that other changes in technique would be needed. For example, minimizing scaffold overlap in long and bifurcating lesions. Whether that would translate into diminishing the rate of late adverse events remains to be determined. As of now, we only have data on approximately 100 highly selected patients beyond 3 years (no adverse events 2.5 to 5 years after implantation).
Several investigational second-generation bioresorbable stents, including Elixir’s Dissolve PLLA, Boston Scientific’s FAST, and a newer version of Absorb, are in early clinical trials. Smaller strut thickness holds the promise of attenuating the risk of stent thrombosis. Since the polymer persists, no reduction in dual antiplatelet therapy duration is likely to be achieved.
Results from long-term follow-up of Absorb III and on-going trials are eagerly awaited to ascertain whether the rate of late complications of DES can be mitigated. It would not be surprising if the second-generation bioresorbable stents make DES a thing of the past within the next decade.
References
Palmerini T, Benedetto U, Biondi-Zoccai G, et al. Long-term safety of drug-eluting and bare-metal stents: evidence from a comprehensive network meta-analysis. J Am Coll Cardiol 2015; 65:2496–2507.
Bates ER. Balancing the evidence base on coronary stents. N Engl J Med 2016; 375:1286–1288.
Macaya C, Serruys PW, Ruygrok P, et al. Continued benefit of coronary stenting versus balloon angioplasty: one-year clinical follow-up of BENESTENT trial. BENESTENT Study Group. J Am Coll Cardiol 1996; 27:255–261.
Fajadet J, Wijns W, Laarman GJ; for the ENDEAVOR II Investigators. Randomized, double-blind, multicenter study of the Endeavor zotarolimus-eluting phosphorylcholine-encapsulated stent for treatment of native coronary artery lesions: clinical and angiographic results of the ENDEAVOR II trial. Circulation 2006; 114:798–806.
Kirtane AJ, Leon MB, Ball MW, et al; ENDEAVOR IV Investigators. The “final” 5-year follow-up from the ENDEAVOR IV trial comparing a zotarolimus-eluting stent with a paclitaxel-eluting stent. JACC Cardiovasc Interv 2013; 6:325–333.
Stone GW, Midei M, Newman W, et al; SPIRIT III Investigators. Comparison of an everolimus-eluting stent and a paclitaxel-eluting stent in patients with coronary artery disease: a randomized trial. JAMA 2008; 299:1903–1913.
Gada H, Kirtane AJ, Newman W, et al. 5-year results of a randomized comparison of XIENCE V everolimus-eluting and TAXUS paclitaxel-eluting stents: final results from the SPIRIT III trial (clinical evaluation of the XIENCE V everolimus eluting coronary stent system in the treatment of patients with de novo native coronary artery lesions). JACC Cardiovasc Interv 2013; 6:1263–1266.
Benedetto U, Raja SG, Soliman RFB, et al; on behalf of the Harefield Cardiac Outcomes Research Group. Minimally invasive direct coronary artery bypass improves late survival compared with drug-eluting stents in isolated proximal left anterior descending artery disease: a 10-year follow-up, single-center, propensity score analysis. J Thorac Cardiovasc Surg 2014; 148:1316–1322.
Virmani R, CV Path Institute. Problems encountered with drug-eluting stent. Presented at Transcatheter Cardiovascular Therapeutics (TCT); October 11–15, 2015; San Francisco, CA.
Kereiakes DJ, Meredith IT, Windecker S, et al. Efficacy and safety of a novel bioabsorbable polymer-coated, everolimus-eluting coronary stent: the EVOLVE II Randomized Trial. Circ Cardiovasc Interv 2015; 8:e002372.
Urban P, Meredith IT, Abizaid A, et al; for the LEADERS FREE Investigators. Polymer-free drug-coated coronary stents in patients at high bleeding risk. N Engl J Med 2015; 373:2038–2047.
Worthley SG, Abizaid A, Kirtane AJ, Simon D, Windecker S, Stone GW. Stent strut coverage and stent apposition after implantation of a novel drug-filled coronary stent: optical coherence tomography results from RevElution trial. J Am Coll Cardiol 2015; 66(suppl B):B235 [Abstract TCT-579].
Abizaid A, Costa RA, Schofer J, et al. Serial multimodality imaging and 2-year clinical outcomes of the novel DESolve novolimus-eluting bioresorbable coronary scaffold system for the treatment of single de novo coronary lesions. JACC Cardiovasc Interv 2016; 9:565–574.
Ellis SG, Kereiakes DJ, Metzger DC, et al; for the ABOSRB III Investigators. Everolimus-eluting bioresorbable scaffolds for coronary artery disease. N Engl J Med 2015; 373:1905–1915.
Puricel S, Cuculi F, Weissner M, et al. Bioresorbable coronary scaffold thrombosis: multicenter comprehensive analysis of clinical presentation, mechanisms, and predictors. J Am Coll Cardiol 2016; 67:921−931.
Stone GW, Gao R, Kimura T, et al. 1-year outcomes with the Absorb bioresorbable scaffold in patients with coronary artery disease: a patient-level, pooled meta-analysis. Lancet 2016; 387:1277–1289.
References
Palmerini T, Benedetto U, Biondi-Zoccai G, et al. Long-term safety of drug-eluting and bare-metal stents: evidence from a comprehensive network meta-analysis. J Am Coll Cardiol 2015; 65:2496–2507.
Bates ER. Balancing the evidence base on coronary stents. N Engl J Med 2016; 375:1286–1288.
Macaya C, Serruys PW, Ruygrok P, et al. Continued benefit of coronary stenting versus balloon angioplasty: one-year clinical follow-up of BENESTENT trial. BENESTENT Study Group. J Am Coll Cardiol 1996; 27:255–261.
Fajadet J, Wijns W, Laarman GJ; for the ENDEAVOR II Investigators. Randomized, double-blind, multicenter study of the Endeavor zotarolimus-eluting phosphorylcholine-encapsulated stent for treatment of native coronary artery lesions: clinical and angiographic results of the ENDEAVOR II trial. Circulation 2006; 114:798–806.
Kirtane AJ, Leon MB, Ball MW, et al; ENDEAVOR IV Investigators. The “final” 5-year follow-up from the ENDEAVOR IV trial comparing a zotarolimus-eluting stent with a paclitaxel-eluting stent. JACC Cardiovasc Interv 2013; 6:325–333.
Stone GW, Midei M, Newman W, et al; SPIRIT III Investigators. Comparison of an everolimus-eluting stent and a paclitaxel-eluting stent in patients with coronary artery disease: a randomized trial. JAMA 2008; 299:1903–1913.
Gada H, Kirtane AJ, Newman W, et al. 5-year results of a randomized comparison of XIENCE V everolimus-eluting and TAXUS paclitaxel-eluting stents: final results from the SPIRIT III trial (clinical evaluation of the XIENCE V everolimus eluting coronary stent system in the treatment of patients with de novo native coronary artery lesions). JACC Cardiovasc Interv 2013; 6:1263–1266.
Benedetto U, Raja SG, Soliman RFB, et al; on behalf of the Harefield Cardiac Outcomes Research Group. Minimally invasive direct coronary artery bypass improves late survival compared with drug-eluting stents in isolated proximal left anterior descending artery disease: a 10-year follow-up, single-center, propensity score analysis. J Thorac Cardiovasc Surg 2014; 148:1316–1322.
Virmani R, CV Path Institute. Problems encountered with drug-eluting stent. Presented at Transcatheter Cardiovascular Therapeutics (TCT); October 11–15, 2015; San Francisco, CA.
Kereiakes DJ, Meredith IT, Windecker S, et al. Efficacy and safety of a novel bioabsorbable polymer-coated, everolimus-eluting coronary stent: the EVOLVE II Randomized Trial. Circ Cardiovasc Interv 2015; 8:e002372.
Urban P, Meredith IT, Abizaid A, et al; for the LEADERS FREE Investigators. Polymer-free drug-coated coronary stents in patients at high bleeding risk. N Engl J Med 2015; 373:2038–2047.
Worthley SG, Abizaid A, Kirtane AJ, Simon D, Windecker S, Stone GW. Stent strut coverage and stent apposition after implantation of a novel drug-filled coronary stent: optical coherence tomography results from RevElution trial. J Am Coll Cardiol 2015; 66(suppl B):B235 [Abstract TCT-579].
Abizaid A, Costa RA, Schofer J, et al. Serial multimodality imaging and 2-year clinical outcomes of the novel DESolve novolimus-eluting bioresorbable coronary scaffold system for the treatment of single de novo coronary lesions. JACC Cardiovasc Interv 2016; 9:565–574.
Ellis SG, Kereiakes DJ, Metzger DC, et al; for the ABOSRB III Investigators. Everolimus-eluting bioresorbable scaffolds for coronary artery disease. N Engl J Med 2015; 373:1905–1915.
Puricel S, Cuculi F, Weissner M, et al. Bioresorbable coronary scaffold thrombosis: multicenter comprehensive analysis of clinical presentation, mechanisms, and predictors. J Am Coll Cardiol 2016; 67:921−931.
Stone GW, Gao R, Kimura T, et al. 1-year outcomes with the Absorb bioresorbable scaffold in patients with coronary artery disease: a patient-level, pooled meta-analysis. Lancet 2016; 387:1277–1289.
Bioresorbable stents: The future of interventional cardiology?
Display Headline
Bioresorbable stents: The future of interventional cardiology?
Legacy Keywords
stents, percutaneous coronary intervention, PCI, bare metal stent, BMS, drug-eluting stents, DES, thrombosis, everolimus, Promus, paclitaxel, Synergy, Boston Scientific, Medtronic, BioFreedom, Biosensors International, sirolimus, Stephen Ellis, Haris Riaz
Legacy Keywords
stents, percutaneous coronary intervention, PCI, bare metal stent, BMS, drug-eluting stents, DES, thrombosis, everolimus, Promus, paclitaxel, Synergy, Boston Scientific, Medtronic, BioFreedom, Biosensors International, sirolimus, Stephen Ellis, Haris Riaz
Citation Override
Cleveland Clinic Journal of Medicine 2016 November; 83(suppl 2):S18-S23
Inside the Article
KEY POINTS
Stents have dramatically improved outcomes associated with percutaneous coronary angioplasty.
Bare-metal stents were the first stents developed, followed by first- and second-generation drug-eluting stents, which have progressively reduced complication rates.
Despite the improvements with conventional stents, persistent rates of restenosis and stent thrombosis remain, which can lead to increased coronary morbidity and mortality.
New stent technologies include stents coated with bioresorbable polymers, stents without polymers, and completely bioresorbable stents.
Disallow All Ads
Alternative CME
Consolidated Pubs: Do Not Show Source Publication Logo
The first clinical implantation of a cardiac pacemaker was performed surgically in 1958 by Drs. Elmvist and Senning via thoracotomy and direct attachment of electrodes to the myocardium. Transvenous pacing was introduced in 1962 by Drs. Lagergren, Parsonnet, and Welti.1,2 The general configuration of transvenous leads connected to a pulse generator situated in a surgical pocket has remained the standard of care ever since. Despite almost 60 years of technological innovation, contemporary permanent transvenous pacing continues to carry significant short- and long-term morbidity. Long-term composite complication rates are estimated at over 10%,3 further stratified as 12% in the 2 months post-implant (short-term) and 9% thereafter (long-term).4 Transvenous pacing complications are associated with an increase in both hospitalization days (hazard ratio 2.3) and unique hospitalizations (hazard ratio 4.4).5
Short-term complications
Source: Lead fracture and pocket infection images courtesy of Dr. Mohamed Kanj. Hematoma image courtesy of Dr. John Rickard.
Figure 1. Common transvenous pacemaker lead and pocket-related complications.
Short-term complications include lead dislodgment, pocket hematoma, pericardial effusion, and pneumothorax (Figure 1). Pocket hematomas are common with concurrent antiplatelet or anticoagulant administration, with incidence estimates varying from 5% to 33% depending on the definition.6 Morbidity associated with pocket hematoma include prolonged hospitalization, need for re-operation,7 and an almost eightfold increase in the rate of device infection over the long term compared with patients without pocket hematoma.8 New pericardial effusions after implant may affect up to 10% of patients; they are generally small, including 90% attributable to pericarditis or contained microperforation not requiring intervention. Overt lead perforation resulting in cardiac tamponade occurs in about 1% of transvenous pacemaker implants, of which 10% (0.1% overall) require open chest surgery, with the remainder treated with percutaneous drainage.9
Long-term complications
Long-term complications are predominantly lead and pocket-related but also include venous occlusive disease and tricuspid valve pathology.4 The development of primary lead failure due to insulation defects, conductor fracture, or dislodgment has been associated with major adverse events in 16% of patients, and an additional 6% if transvenous lead extraction is needed, which can rarely lead to hemorrhagic death by vascular tears involving the heart or superior vena cava.10 Fibrous tissue growth around the indwelling vascular leads can result in venous obstruction present in up to 14% of patients by 6 months after implant.11 This increases to 26% by the time of device replacement or upgrade, which is typically 5 to 10 years after the original implant, including 17% of patients with a complete venous occlusion.12 In addition, worsened tricuspid regurgitation due to lead impingement on the valve is seen in 7% to 40% of patients depending on definitions,13 with post-implant severe tricuspid regurgitation independently associated with increased mortality risk.14 The rate of device infection is 1% to 2% at 1 year,8,15 and 3% over the lifetime of the initial transvenous system; this increases to more than 10% after generator replacement.16
LEADLESS PACING TECHNOLOGY
Figure 2. Leadless pacemakers (A) Nanostim and (B) Micra.
The principal goal of leadless pacing is to reduce short- and long-term pacemaker complications by eliminating the two most common sources of problems: the transvenous leads and the surgical pocket. Discussion of leadless pacing strategies began as early as 1970.17 Although several preclinical studies demonstrated efficacy with leadless prototypes,18–20 clinical implementation of fully leadless technology did not occur until recently. As shown in Figure 2, there are now two commercially available leadless pacing devices: Nanostim (St. Jude Medical Inc., St. Paul, MN) and Micra (Medtronic Inc., Dublin, Ireland). At the time of this writing, both have commercial approval in Europe. In the United States, Micra received commercial approval from the US Food and Drug Administration on April 6, 2016, with a similar decision expected on Nanostim. The current approved indications for leadless pacing are chronic atrial tachyarrhythmia with advanced atrioventricular (AV) block; advanced AV block with low level of physical activity or short expected lifespan; and infrequent pauses or unexplained syncope with abnormal findings at electrophysiologic study. Although differences exist between Nanostim and Micra, as shown in Table 1,21–27 there are fundamental similarities. Both are single-unit designs encapsulating the electrodes and pulse generator with rate-adaptive functionality. Both are delivered via an endovascular femoral venous approach without the need for incisional access, transvenous leads, or surgical pocket (Figures 3 and 4).21–27
Nanostim: Landmark trials
As the world’s first-in-man leadless pacemaker, Nanostim was evaluated in two prospective, non-randomized, multicenter, single-arm trials abbreviated LEADLESS22 and LEADLESS II.24 The first human feasibility study, LEADLESS, enrolled 33 patients with approved indications for ventricular-only pacing while excluding patients with expected pacemaker dependency. The most common indication was bradycardia in the presence of persistent atrial arrhythmias, thereby obviating the need for atrial pacing. The primary outcome was freedom from serious complications at 90 days. The secondary outcomes were implant success rate and device performance at 3 months. The results demonstrated 94% composite safety (31 of 33 patients) at 3 months. There was one cardiac perforation leading to tamponade and eventually death after prolonged hospitalization, and one inadvertent deployment into the left ventricle via patent foramen ovale that was successfully retrieved and redeployed without complication. The implant success rate was 97%, and the electrical parameters involving sensing, pacing thresholds, and impedance were as expected at 3 months. Results of 1-year follow-up were published for the LEADLESS cohort,25 revealing no additional complications from 3 to 12 months, no adverse changes in electrical performance parameters, and 100% effectiveness of rate-responsive programming.
Figure 3. Fluoroscopic images depicting catheter-based deployment and subsequent release for the (A) Nanostim and (B) Micra.
The subsequent LEADLESS II trial enrolled 526 patients but did not exclude patients with expected pacemaker dependency, and its results were reported in a preplanned interim analysis when 300 patients had reached 6 months of follow-up (mean follow-up 6.9 ± 4.2 months).24 The primary efficacy end point involved electrical performance including capture thresholds and sensing. Initial deployment success was 96% with expected electrical parameters at implant that were stable at 6 months of follow-up. The rate of freedom from serious adverse events was 93%, with complications including device dislodgment (1.7%, mean 8 ± 6 days after implant), perforation (1.3%), performance deficiency requiring device retrieval and replacement (1.3%), and groin complications (1.3%). There were no device-related deaths, and all device dislodgments were successfully treated percutaneously.
Figure 4. Frontal-plane radiographs showing implanted Nanostim (A) and Micra (B) leadless pacing devices and a traditional dual-chamber pacemaker (C). Panel D depicts cardiac deployment.
There was no prospective control arm involving transvenous pacing in either the LEADLESS or LEADLESS II trial. Thus, in an effort to compare Nanostim (n = 718) vs transvenous pacing, complication rates were calculated for a propensity-matched registry cohort of 10,521 transvenous patients, and differences were reported.26 At 1 month, the composite complication rate was 5.8% for Nanostim (1.5% pericardial effusion, 1% dislodgment) and 12.7% for transvenous pacing (7.6% lead-related, 3.9% thoracic trauma, infection 1.9%) (P < .001). Between 1 month and 2 years, complication rates were only 0.6% for Nanostim vs 5.4% for transvenous pacing (P < .001). This lower complication rate at 2 years was driven almost entirely by a 2.6% infection rate and 2.4% lead-complication rate in the transvenous pacemaker group, nonexistent in the leadless group.
Micra: Landmark trials
Micra was evaluated in a prospective, nonrandomized, multicenter, single-arm trial, enrolling 725 patients with indications for ventricular-only pacing; approximately two-thirds of the cohort had bradycardia in the presence of persistent atrial arrhythmias, similar to the Nanostim cohort.27 The efficacy end point was stable capture threshold at 6 months. The safety end point was freedom from major complications resulting in new or prolonged hospitalization at 6 months. The implant success rate was 99%, and 98% of patients met the primary efficacy end point. The safety end point was met in 96% of patients. Complications included perforation or pericardial effusion (1.6%), groin complication (0.7%), elevated threshold (0.3%), venous thromboembolism (0.3%), and others (1.7%). No dislodgments were reported. There was no prospective, randomized control arm to compare Micra and transvenous pacing. A post hoc analysis was performed comparing major complication rates in this study with an unmatched 2,667-patient meta-analysis control cohort.27 The hazard ratio for the leadless pacing strategy was calculated at 0.49 (95% confidence interval 0.33 to 0.75, P = .001) with absolute risk reduction 3.4% at 6 months resulting in a number needed to treat of 29.4 patients. Further broken down, Micra patients compared with the control cohort had reduced rates of both subsequent hospitalizations (3.9% to 2.3%) and device revisions (3.5% to 0.4%).
ADVANTAGES OF LEADLESS PACING
As discussed above, the major observed benefit with both Nanostim and Micra compared with transvenous cohorts is the elimination of lead and pocket-related complications.25,27 Leadless pacing introduces a new 1% to 2% groin complication rate for both devices not present with transvenous pacing, and also a 1% device dislodgment rate in the case of Nanostim (all dislodgments were treated percutaneously). Data from both clinical trials suggest that the complication rates are largely compressed acutely. In contrast, there are considerable mid-term and long-term complications for transvenous systems.3–5 Indeed, the mid- to long-term window is where leadless pacing is expected to have the most favorable impact. As with any new disruptive technology, operator experience may be important, as evidenced by a near halving of the complication rate observed in the LEADLESS II trial after gaining the experience of 10 implants.25
Other benefits of leadless pacing include a generally quick procedure (average implant time was 30 minutes in LEADLESS and LEADLESS II)22,25 and full shoulder mobility afterwards, so that patients can resume driving once groin soreness has subsided, typically within a few days. (Current studies are investigating whether immediate shoulder mobility with leadless pacing is beneficial to older patients suffering from arthritis.) The lack of an incision allows patients to bathe and shower as soon as they desire, whereas after transvenous pacemaker implant, motion in the affected shoulder is usually restricted for several weeks to avoid lead dislodgment, and showering and bathing are restricted to avoid contamination of the incision with nonsterile tap water. (In some cases, a tightly adherent waterproof dressing can be used.) The leadless systems were designed for compatibility with magnetic resonance imaging (MRI), whereas not all transvenous pacemaker generators and leads are MRI compatible.
Leadless devices are not expected to span the tricuspid valve to create incident or worsening tricuspid regurgitation. In a recent small study of 22 patients undergoing Micra implant, there were no new cases of severe tricuspid regurgitation after the procedure, with only a 9% increase in mild and 5% increase in moderate tricuspid regurgitation,28 vs a rate of 40% of worsening tricuspid regurgitation and 10% of new severe tricuspid regurgitation with transvenous pacing.13,14
Transvenous pacemaker implant requires surgery for pulse generator exchange at a mean of 7 years, a procedure carrying significant risk of short- and long-term complications.10
END-OF-SERVICE QUESTIONS: ATTEMPT RETRIEVAL OR NOT?
Both leadless systems have favorable projected in-service battery life: a reported 15.0 years for Nanostim25 and mean 12.5 years for Micra.27 The inevitable question is what to do then. The Nanostim system was designed to be retrievable using a dedicated catheter system. Micra was not designed with an accompanying retrieval system. Pathologic examinations of leadless devices at autopsy or after explant have revealed a range of device endothelialization, from partial at 19 months to full at 4 months.29,30
As of this writing, no extraction complications have been observed with Nanostim explants up to 506 days after implant (n = 12, mean 197 days after implant).31 Needless to say, there is not yet enough experience worldwide with either system to know what the end-of-service will look like in 10 to 15 years. One strategy could involve first attempting percutaneous retrieval and replacement, if retrieval is not possible, abandoning the old device while implanting a new device alongside. Another strategy would be to forgo a retrieval attempt altogether. In the LEADLESS II study,24 the mean patient age was 75. In this cohort, forgoing elective retrieval for those who live to reach the end of pacemaker service between the age of 85 and 90 would seem reasonable assuming the next device provides similar longevity. For younger patients, careful consideration of long-term strategies is needed. It is not known what the replacement technology will look like in another decade with respect to device size or battery longevity. Preclinical studies using swine and human cadaver hearts have demonstrated the feasibility of multiple right-ventricular Micra implants without affecting cardiac function.32,33
OTHER LIMITATIONS AND CAUTIONARY NOTES
At present, leadless pacing is approved for single-chamber right-ventricular pacing. In the developed world, single right-ventricular pacing modes account for only 20% to 30% of new pacemaker implants, which total more than 1 million per year worldwide.34,35 As with any new technology, the up-front cost of leadless pacemaker implant is expected to be significantly higher than transvenous systems, which at this point remains poorly defined, as the field has not caught up in terms of charges, reimbursement, and billing codes. While those concerns fall outside the scope of this review, it is not known if the expected reductions in mid- and long-term complications will make up for an up-front cost difference. However, a cost-efficacy study reported that one complication of a transvenous pacemaker system was more expensive than the initial implant itself.36 The longest-term follow-up data currently available are with Nanostim, showing an absolute complication reduction of 11.7% at 2 years,24 a disparity only expected to widen with prolonged follow-up, particularly after transvenous generator exchange, when complication rates rapidly escalate.
FUTURE DIRECTIONS
The next horizon of leadless technology will be for right-atrial and dual-chamber pacing to treat the far more pervasive pacing indication of sinus node dysfunction with or without AV block. In the latter application, the two devices will communicate. Prototypes and early nonhuman evaluations are ongoing for both. Leadless pacing is also being investigated for use in tachycardia. Tjong et al37 reported on the safety and feasibility of an entirely leadless pacemaker plus an implantable cardioverter-defibrillator (ICD) system in two sheep and one human using both Nanostim and subcutaneous ICD. Currently, two important limitations of subcutaneous ICD are its inability to provide backup bradycardia and antitachycardia pacing (it provides only defibrillation). The EMBLEM PACE study will enroll 250 patients to receive a leadless pacemaker and Emblem subcutaneous ICD (Boston Scientific, Boston, MA), with patients subsequently receiving commanded antitachycardia pacing for ventricular arrhythmias and bradycardia pacing provided by the leadless device as indicated.
CONCLUSIONS
Leadless cardiac pacing is a safe and efficacious alternative to standard transvenous pacing systems. Although long-term data are limited, available short- and mid-term data show that the elimination of transvenous leads and the surgical pocket results in significant reductions in complication rates. Currently, leadless pacing is approved only for right-ventricular pacing, but investigation of right-atrial, dual-chamber, and fully leadless pacemaker-defibrillator hybrid systems is ongoing.
References
Lagergren H. How it happened: my recollection of early pacing. Pacing Clin Electrophysiol 1978; 1:140–143.
Parsonnet V. Permanent transvenous pacing in 1962. Pacing Clin Electrophysiol 1978; 1:265–268.
Kirkfeldt RE, Johansen JB, Nohr EA, Jorgensen OD, Nielsen JC. Complications after cardiac implantable electronic device implantations: an analysis of a complete, nationwide cohort in Denmark. Eur Heart J 2014; 35:1186–1194.
Udo EO, Zuithoff NP, van Hemel NM, et al. Incidence and predictors of short- and long-term complications in pacemaker therapy: the FOLLOWPACE study. Heart Rhythm 2012; 9:728–735.
Palmisano P, Accogli M, Zaccaria M, et al. Rate, causes, and impact on patient outcome of implantable device complications requiring surgical revision: large population survey from two centres in Italy. Europace 2013; 15:531–540.
De Sensi F, Miracapillo G, Cresti A, Severi S, Airaksinen KE. Pocket hematoma: a call for definition. Pacing Clin Electrophysiol Aug 2015; 38:909–913.
Wiegand UK, LeJeune D, Boguschewski F, et al. Pocket hematoma after pacemaker or implantable cardioverter defibrillator surgery: influence of patient morbidity, operation strategy, and perioperative antiplatelet/anticoagulation therapy. Chest 2004; 126:1177–1186.
Essebag V, Verma A, Healey JS, et al. Clinically significant pocket hematoma increases long-term risk of device infection: Bruise Control Infection Study. J Am Coll Cardiol 2016; 67:1300–1308.
Ohlow MA, Lauer B, Brunelli M, Geller JC. Incidence and predictors of pericardial effusion after permanent heart rhythm device implantation: prospective evaluation of 968 consecutive patients. Circ J 2013; 77:975–981.
Hauser RG, Hayes DL, Kallinen LM, et al. Clinical experience with pacemaker pulse generators and transvenous leads: an 8-year prospective multicenter study. Heart Rhythm 2007; 4:154–160.
Korkeila P, Nyman K, Ylitalo A, et al. Venous obstruction after pacemaker implantation. Pacing Clin Electrophysiol 2007; 30:199–206.
Haghjoo M, Nikoo MH, Fazelifar AF, Alizadeh A, Emkanjoo Z, Sadr-Ameli MA. Predictors of venous obstruction following pacemaker or implantable cardioverter-defibrillator implantation: a contrast venographic study on 100 patients admitted for generator change, lead revision, or device upgrade. Europace 2007; 9:328–332.
Al-Mohaissen MA, Chan KL. Prevalence and mechanism of tricuspid regurgitation following implantation of endocardial leads for pacemaker or cardioverter-defibrillator. J Am Soc Echocardiogr 2012; 25:245–252.
Al-Bawardy R, Krishnaswamy A, Rajeswaran J, et al. Tricuspid regurgitation and implantable devices. Pacing Clin Electrophysiol 2015; 38:259–266.
Klug D, Balde M, Pavin D, et al. Risk factors related to infections of implanted pacemakers and cardioverter-defibrillators: results of a large prospective study. Circulation 2007; 116:1349–1355.
Johansen JB, Jorgensen OD, Moller M, Arnsbo P, Mortensen PT, Nielsen JC. Infection after pacemaker implantation: infection rates and risk factors associated with infection in a population-based cohort study of 46,299 consecutive patients. Eur Heart J 2011; 32:991–998.
Lown B, Kosowsky BD. Artificial cardiac pacemakers. I. N Engl J Med 1970; 283:907–916.
Sutton R. The first European journal on cardiac electrophysiology and pacing, the European Journal of Cardiac Pacing and Electrophysiology. Europace 2011; 13:1663–1664.
Vardas PE, Politopoulous C, Manios E, Parthenakis F, Tsagarkis C. A miniature pacemaker introduced intravenously and implanted endocardially. Preliminary findings from an experimental study. Eur J Card Pacing Electrophysiol 1991; 1:27–30.
Eggen MD, Grubac V, Bonner MD. Design and evaluation of a novel fixation mechanism for a transcatheter pacemaker. IEEE Trans Biomed Eng 2015; 62:2316–2323.
Reddy VY, Knops RE, Sperzel J, et al. Permanent leadless cardiac pacing: results of the LEADLESS trial. Circulation 2014; 129:1466–1471.
Ritter P, Duray GZ, Steinwender C, et al. Early performance of a miniaturized leadless cardiac pacemaker: the Micra Transcatheter Pacing Study. Eur Heart J 2015; 36:2510–2519.
Reddy VY, Exner DV, Cantillon DJ, et al. Percutaneous implantation of an entirely intracardiac leadless pacemaker. N Engl J Med 2015; 373:1125–1135.
Knops RE, Tjong FV, Neuzil P, et al. Chronic performance of a leadless cardiac pacemaker: 1-year follow-up of the LEADLESS trial. J Am Coll Cardiol 2015; 65:1497–1504.
Reddy VY, Cantillon DJ, Ip J, et al. A comparative study of acute and mid-term complications of leadless versus transvenous pacemakers. Heart Rhythm 2016 July. [Epub ahead of print].
Reynolds D, Duray GZ, Omar R, et al. A leadless intracardiac transcatheter pacing system. N Engl J Med 2016; 374:533–541.
Garikipati NV, Karve A, Okabe T, et al. Tricuspid regurgitation after leadless pacemaker implantation. Abstract presented at Heart Rhythm Society Scientific Sessions, May 4–7, 2016, San Francisco, CA.
Tjong FV, Stam OC, van der Wal AC, et al. Postmortem histopathological examination of a leadless pacemaker shows partial encapsulation after 19 months. Circ Arrhythm Electrophysiol 2015; 8:1293–1295.
Borgquist R, Ljungstrom E, Koul B, Hoijer CJ. Leadless Medtronic Micra pacemaker almost completely endothelialized already after 4 months: first clinical experience from an explanted heart. Eur Heart J 2016; 37:2503.
Reddy VY, Knops RE, Defaye P, et al. Worldwide clinical experience of the retrieval of leadless cardiac pacemakers. Abstract presented at Heart Rhythm Society Scientific Sessions, May 4–7, 2016, San Francisco, CA.
Chen K, Zheng X, Dai Y, et al. Multiple leadless pacemakers implanted in the right ventricle of swine. Europace 2016 January 31. pii: euv418. [Epub ahead of print].
Omdahl P, Eggen MD, Bonner MD, Iaizzo PA, Wika K. Right ventricular anatomy can accommodate multiple micra transcatheter pacemakers. Pacing Clin Electrophysiol 2016; 39:393–397.
Mond HG, Proclemer A. The 11th world survey of cardiac pacing and implantable cardioverter-defibrillators: calendar year 2009—a World Society of Arrhythmia’s project. Pacing Clin Electrophysiol 2011; 34:1013–1027.
Epstein AE, DiMarco JP, Ellenbogen KA, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Heart Rhythm Society. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2013; 127:e283–352.
Tobin K, Stewart J, Westveer D, Frumin H. Acute complications of permanent pacemaker implantation: their financial implication and relation to volume and operator experience. Am J Cardiol 2000; 85:774–776, A9.
Tjong FV, Brouwer TF, Smeding L, et al. Combined leadless pacemaker and subcutaneous implantable defibrillator therapy: feasibility, safety, and performance. Europace 2016 March 3. [Epub ahead of print].
Correspondence: Daniel J. Cantillon, MD, FACC, FHRS, Department of Cardiovascular Medicine, J2-2, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; cantild@ccf.org
Dr. Kiehl reported no financial interests or relationships that pose a potential conflict of interest with this article. Dr. Cantillon reported consulting for Boston Scientific Corporation and St. Jude Medical; membership on advisory committees for Boston Scientific Corporation and St. Jude Medical; and teaching/speaking for Boston Scientific Corporation.
Correspondence: Daniel J. Cantillon, MD, FACC, FHRS, Department of Cardiovascular Medicine, J2-2, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; cantild@ccf.org
Dr. Kiehl reported no financial interests or relationships that pose a potential conflict of interest with this article. Dr. Cantillon reported consulting for Boston Scientific Corporation and St. Jude Medical; membership on advisory committees for Boston Scientific Corporation and St. Jude Medical; and teaching/speaking for Boston Scientific Corporation.
Author and Disclosure Information
Erich L. Kiehl, MD Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic
Correspondence: Daniel J. Cantillon, MD, FACC, FHRS, Department of Cardiovascular Medicine, J2-2, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; cantild@ccf.org
Dr. Kiehl reported no financial interests or relationships that pose a potential conflict of interest with this article. Dr. Cantillon reported consulting for Boston Scientific Corporation and St. Jude Medical; membership on advisory committees for Boston Scientific Corporation and St. Jude Medical; and teaching/speaking for Boston Scientific Corporation.
The first clinical implantation of a cardiac pacemaker was performed surgically in 1958 by Drs. Elmvist and Senning via thoracotomy and direct attachment of electrodes to the myocardium. Transvenous pacing was introduced in 1962 by Drs. Lagergren, Parsonnet, and Welti.1,2 The general configuration of transvenous leads connected to a pulse generator situated in a surgical pocket has remained the standard of care ever since. Despite almost 60 years of technological innovation, contemporary permanent transvenous pacing continues to carry significant short- and long-term morbidity. Long-term composite complication rates are estimated at over 10%,3 further stratified as 12% in the 2 months post-implant (short-term) and 9% thereafter (long-term).4 Transvenous pacing complications are associated with an increase in both hospitalization days (hazard ratio 2.3) and unique hospitalizations (hazard ratio 4.4).5
Short-term complications
Source: Lead fracture and pocket infection images courtesy of Dr. Mohamed Kanj. Hematoma image courtesy of Dr. John Rickard.
Figure 1. Common transvenous pacemaker lead and pocket-related complications.
Short-term complications include lead dislodgment, pocket hematoma, pericardial effusion, and pneumothorax (Figure 1). Pocket hematomas are common with concurrent antiplatelet or anticoagulant administration, with incidence estimates varying from 5% to 33% depending on the definition.6 Morbidity associated with pocket hematoma include prolonged hospitalization, need for re-operation,7 and an almost eightfold increase in the rate of device infection over the long term compared with patients without pocket hematoma.8 New pericardial effusions after implant may affect up to 10% of patients; they are generally small, including 90% attributable to pericarditis or contained microperforation not requiring intervention. Overt lead perforation resulting in cardiac tamponade occurs in about 1% of transvenous pacemaker implants, of which 10% (0.1% overall) require open chest surgery, with the remainder treated with percutaneous drainage.9
Long-term complications
Long-term complications are predominantly lead and pocket-related but also include venous occlusive disease and tricuspid valve pathology.4 The development of primary lead failure due to insulation defects, conductor fracture, or dislodgment has been associated with major adverse events in 16% of patients, and an additional 6% if transvenous lead extraction is needed, which can rarely lead to hemorrhagic death by vascular tears involving the heart or superior vena cava.10 Fibrous tissue growth around the indwelling vascular leads can result in venous obstruction present in up to 14% of patients by 6 months after implant.11 This increases to 26% by the time of device replacement or upgrade, which is typically 5 to 10 years after the original implant, including 17% of patients with a complete venous occlusion.12 In addition, worsened tricuspid regurgitation due to lead impingement on the valve is seen in 7% to 40% of patients depending on definitions,13 with post-implant severe tricuspid regurgitation independently associated with increased mortality risk.14 The rate of device infection is 1% to 2% at 1 year,8,15 and 3% over the lifetime of the initial transvenous system; this increases to more than 10% after generator replacement.16
LEADLESS PACING TECHNOLOGY
Figure 2. Leadless pacemakers (A) Nanostim and (B) Micra.
The principal goal of leadless pacing is to reduce short- and long-term pacemaker complications by eliminating the two most common sources of problems: the transvenous leads and the surgical pocket. Discussion of leadless pacing strategies began as early as 1970.17 Although several preclinical studies demonstrated efficacy with leadless prototypes,18–20 clinical implementation of fully leadless technology did not occur until recently. As shown in Figure 2, there are now two commercially available leadless pacing devices: Nanostim (St. Jude Medical Inc., St. Paul, MN) and Micra (Medtronic Inc., Dublin, Ireland). At the time of this writing, both have commercial approval in Europe. In the United States, Micra received commercial approval from the US Food and Drug Administration on April 6, 2016, with a similar decision expected on Nanostim. The current approved indications for leadless pacing are chronic atrial tachyarrhythmia with advanced atrioventricular (AV) block; advanced AV block with low level of physical activity or short expected lifespan; and infrequent pauses or unexplained syncope with abnormal findings at electrophysiologic study. Although differences exist between Nanostim and Micra, as shown in Table 1,21–27 there are fundamental similarities. Both are single-unit designs encapsulating the electrodes and pulse generator with rate-adaptive functionality. Both are delivered via an endovascular femoral venous approach without the need for incisional access, transvenous leads, or surgical pocket (Figures 3 and 4).21–27
Nanostim: Landmark trials
As the world’s first-in-man leadless pacemaker, Nanostim was evaluated in two prospective, non-randomized, multicenter, single-arm trials abbreviated LEADLESS22 and LEADLESS II.24 The first human feasibility study, LEADLESS, enrolled 33 patients with approved indications for ventricular-only pacing while excluding patients with expected pacemaker dependency. The most common indication was bradycardia in the presence of persistent atrial arrhythmias, thereby obviating the need for atrial pacing. The primary outcome was freedom from serious complications at 90 days. The secondary outcomes were implant success rate and device performance at 3 months. The results demonstrated 94% composite safety (31 of 33 patients) at 3 months. There was one cardiac perforation leading to tamponade and eventually death after prolonged hospitalization, and one inadvertent deployment into the left ventricle via patent foramen ovale that was successfully retrieved and redeployed without complication. The implant success rate was 97%, and the electrical parameters involving sensing, pacing thresholds, and impedance were as expected at 3 months. Results of 1-year follow-up were published for the LEADLESS cohort,25 revealing no additional complications from 3 to 12 months, no adverse changes in electrical performance parameters, and 100% effectiveness of rate-responsive programming.
Figure 3. Fluoroscopic images depicting catheter-based deployment and subsequent release for the (A) Nanostim and (B) Micra.
The subsequent LEADLESS II trial enrolled 526 patients but did not exclude patients with expected pacemaker dependency, and its results were reported in a preplanned interim analysis when 300 patients had reached 6 months of follow-up (mean follow-up 6.9 ± 4.2 months).24 The primary efficacy end point involved electrical performance including capture thresholds and sensing. Initial deployment success was 96% with expected electrical parameters at implant that were stable at 6 months of follow-up. The rate of freedom from serious adverse events was 93%, with complications including device dislodgment (1.7%, mean 8 ± 6 days after implant), perforation (1.3%), performance deficiency requiring device retrieval and replacement (1.3%), and groin complications (1.3%). There were no device-related deaths, and all device dislodgments were successfully treated percutaneously.
Figure 4. Frontal-plane radiographs showing implanted Nanostim (A) and Micra (B) leadless pacing devices and a traditional dual-chamber pacemaker (C). Panel D depicts cardiac deployment.
There was no prospective control arm involving transvenous pacing in either the LEADLESS or LEADLESS II trial. Thus, in an effort to compare Nanostim (n = 718) vs transvenous pacing, complication rates were calculated for a propensity-matched registry cohort of 10,521 transvenous patients, and differences were reported.26 At 1 month, the composite complication rate was 5.8% for Nanostim (1.5% pericardial effusion, 1% dislodgment) and 12.7% for transvenous pacing (7.6% lead-related, 3.9% thoracic trauma, infection 1.9%) (P < .001). Between 1 month and 2 years, complication rates were only 0.6% for Nanostim vs 5.4% for transvenous pacing (P < .001). This lower complication rate at 2 years was driven almost entirely by a 2.6% infection rate and 2.4% lead-complication rate in the transvenous pacemaker group, nonexistent in the leadless group.
Micra: Landmark trials
Micra was evaluated in a prospective, nonrandomized, multicenter, single-arm trial, enrolling 725 patients with indications for ventricular-only pacing; approximately two-thirds of the cohort had bradycardia in the presence of persistent atrial arrhythmias, similar to the Nanostim cohort.27 The efficacy end point was stable capture threshold at 6 months. The safety end point was freedom from major complications resulting in new or prolonged hospitalization at 6 months. The implant success rate was 99%, and 98% of patients met the primary efficacy end point. The safety end point was met in 96% of patients. Complications included perforation or pericardial effusion (1.6%), groin complication (0.7%), elevated threshold (0.3%), venous thromboembolism (0.3%), and others (1.7%). No dislodgments were reported. There was no prospective, randomized control arm to compare Micra and transvenous pacing. A post hoc analysis was performed comparing major complication rates in this study with an unmatched 2,667-patient meta-analysis control cohort.27 The hazard ratio for the leadless pacing strategy was calculated at 0.49 (95% confidence interval 0.33 to 0.75, P = .001) with absolute risk reduction 3.4% at 6 months resulting in a number needed to treat of 29.4 patients. Further broken down, Micra patients compared with the control cohort had reduced rates of both subsequent hospitalizations (3.9% to 2.3%) and device revisions (3.5% to 0.4%).
ADVANTAGES OF LEADLESS PACING
As discussed above, the major observed benefit with both Nanostim and Micra compared with transvenous cohorts is the elimination of lead and pocket-related complications.25,27 Leadless pacing introduces a new 1% to 2% groin complication rate for both devices not present with transvenous pacing, and also a 1% device dislodgment rate in the case of Nanostim (all dislodgments were treated percutaneously). Data from both clinical trials suggest that the complication rates are largely compressed acutely. In contrast, there are considerable mid-term and long-term complications for transvenous systems.3–5 Indeed, the mid- to long-term window is where leadless pacing is expected to have the most favorable impact. As with any new disruptive technology, operator experience may be important, as evidenced by a near halving of the complication rate observed in the LEADLESS II trial after gaining the experience of 10 implants.25
Other benefits of leadless pacing include a generally quick procedure (average implant time was 30 minutes in LEADLESS and LEADLESS II)22,25 and full shoulder mobility afterwards, so that patients can resume driving once groin soreness has subsided, typically within a few days. (Current studies are investigating whether immediate shoulder mobility with leadless pacing is beneficial to older patients suffering from arthritis.) The lack of an incision allows patients to bathe and shower as soon as they desire, whereas after transvenous pacemaker implant, motion in the affected shoulder is usually restricted for several weeks to avoid lead dislodgment, and showering and bathing are restricted to avoid contamination of the incision with nonsterile tap water. (In some cases, a tightly adherent waterproof dressing can be used.) The leadless systems were designed for compatibility with magnetic resonance imaging (MRI), whereas not all transvenous pacemaker generators and leads are MRI compatible.
Leadless devices are not expected to span the tricuspid valve to create incident or worsening tricuspid regurgitation. In a recent small study of 22 patients undergoing Micra implant, there were no new cases of severe tricuspid regurgitation after the procedure, with only a 9% increase in mild and 5% increase in moderate tricuspid regurgitation,28 vs a rate of 40% of worsening tricuspid regurgitation and 10% of new severe tricuspid regurgitation with transvenous pacing.13,14
Transvenous pacemaker implant requires surgery for pulse generator exchange at a mean of 7 years, a procedure carrying significant risk of short- and long-term complications.10
END-OF-SERVICE QUESTIONS: ATTEMPT RETRIEVAL OR NOT?
Both leadless systems have favorable projected in-service battery life: a reported 15.0 years for Nanostim25 and mean 12.5 years for Micra.27 The inevitable question is what to do then. The Nanostim system was designed to be retrievable using a dedicated catheter system. Micra was not designed with an accompanying retrieval system. Pathologic examinations of leadless devices at autopsy or after explant have revealed a range of device endothelialization, from partial at 19 months to full at 4 months.29,30
As of this writing, no extraction complications have been observed with Nanostim explants up to 506 days after implant (n = 12, mean 197 days after implant).31 Needless to say, there is not yet enough experience worldwide with either system to know what the end-of-service will look like in 10 to 15 years. One strategy could involve first attempting percutaneous retrieval and replacement, if retrieval is not possible, abandoning the old device while implanting a new device alongside. Another strategy would be to forgo a retrieval attempt altogether. In the LEADLESS II study,24 the mean patient age was 75. In this cohort, forgoing elective retrieval for those who live to reach the end of pacemaker service between the age of 85 and 90 would seem reasonable assuming the next device provides similar longevity. For younger patients, careful consideration of long-term strategies is needed. It is not known what the replacement technology will look like in another decade with respect to device size or battery longevity. Preclinical studies using swine and human cadaver hearts have demonstrated the feasibility of multiple right-ventricular Micra implants without affecting cardiac function.32,33
OTHER LIMITATIONS AND CAUTIONARY NOTES
At present, leadless pacing is approved for single-chamber right-ventricular pacing. In the developed world, single right-ventricular pacing modes account for only 20% to 30% of new pacemaker implants, which total more than 1 million per year worldwide.34,35 As with any new technology, the up-front cost of leadless pacemaker implant is expected to be significantly higher than transvenous systems, which at this point remains poorly defined, as the field has not caught up in terms of charges, reimbursement, and billing codes. While those concerns fall outside the scope of this review, it is not known if the expected reductions in mid- and long-term complications will make up for an up-front cost difference. However, a cost-efficacy study reported that one complication of a transvenous pacemaker system was more expensive than the initial implant itself.36 The longest-term follow-up data currently available are with Nanostim, showing an absolute complication reduction of 11.7% at 2 years,24 a disparity only expected to widen with prolonged follow-up, particularly after transvenous generator exchange, when complication rates rapidly escalate.
FUTURE DIRECTIONS
The next horizon of leadless technology will be for right-atrial and dual-chamber pacing to treat the far more pervasive pacing indication of sinus node dysfunction with or without AV block. In the latter application, the two devices will communicate. Prototypes and early nonhuman evaluations are ongoing for both. Leadless pacing is also being investigated for use in tachycardia. Tjong et al37 reported on the safety and feasibility of an entirely leadless pacemaker plus an implantable cardioverter-defibrillator (ICD) system in two sheep and one human using both Nanostim and subcutaneous ICD. Currently, two important limitations of subcutaneous ICD are its inability to provide backup bradycardia and antitachycardia pacing (it provides only defibrillation). The EMBLEM PACE study will enroll 250 patients to receive a leadless pacemaker and Emblem subcutaneous ICD (Boston Scientific, Boston, MA), with patients subsequently receiving commanded antitachycardia pacing for ventricular arrhythmias and bradycardia pacing provided by the leadless device as indicated.
CONCLUSIONS
Leadless cardiac pacing is a safe and efficacious alternative to standard transvenous pacing systems. Although long-term data are limited, available short- and mid-term data show that the elimination of transvenous leads and the surgical pocket results in significant reductions in complication rates. Currently, leadless pacing is approved only for right-ventricular pacing, but investigation of right-atrial, dual-chamber, and fully leadless pacemaker-defibrillator hybrid systems is ongoing.
WHY LEADLESS PACING?
The first clinical implantation of a cardiac pacemaker was performed surgically in 1958 by Drs. Elmvist and Senning via thoracotomy and direct attachment of electrodes to the myocardium. Transvenous pacing was introduced in 1962 by Drs. Lagergren, Parsonnet, and Welti.1,2 The general configuration of transvenous leads connected to a pulse generator situated in a surgical pocket has remained the standard of care ever since. Despite almost 60 years of technological innovation, contemporary permanent transvenous pacing continues to carry significant short- and long-term morbidity. Long-term composite complication rates are estimated at over 10%,3 further stratified as 12% in the 2 months post-implant (short-term) and 9% thereafter (long-term).4 Transvenous pacing complications are associated with an increase in both hospitalization days (hazard ratio 2.3) and unique hospitalizations (hazard ratio 4.4).5
Short-term complications
Source: Lead fracture and pocket infection images courtesy of Dr. Mohamed Kanj. Hematoma image courtesy of Dr. John Rickard.
Figure 1. Common transvenous pacemaker lead and pocket-related complications.
Short-term complications include lead dislodgment, pocket hematoma, pericardial effusion, and pneumothorax (Figure 1). Pocket hematomas are common with concurrent antiplatelet or anticoagulant administration, with incidence estimates varying from 5% to 33% depending on the definition.6 Morbidity associated with pocket hematoma include prolonged hospitalization, need for re-operation,7 and an almost eightfold increase in the rate of device infection over the long term compared with patients without pocket hematoma.8 New pericardial effusions after implant may affect up to 10% of patients; they are generally small, including 90% attributable to pericarditis or contained microperforation not requiring intervention. Overt lead perforation resulting in cardiac tamponade occurs in about 1% of transvenous pacemaker implants, of which 10% (0.1% overall) require open chest surgery, with the remainder treated with percutaneous drainage.9
Long-term complications
Long-term complications are predominantly lead and pocket-related but also include venous occlusive disease and tricuspid valve pathology.4 The development of primary lead failure due to insulation defects, conductor fracture, or dislodgment has been associated with major adverse events in 16% of patients, and an additional 6% if transvenous lead extraction is needed, which can rarely lead to hemorrhagic death by vascular tears involving the heart or superior vena cava.10 Fibrous tissue growth around the indwelling vascular leads can result in venous obstruction present in up to 14% of patients by 6 months after implant.11 This increases to 26% by the time of device replacement or upgrade, which is typically 5 to 10 years after the original implant, including 17% of patients with a complete venous occlusion.12 In addition, worsened tricuspid regurgitation due to lead impingement on the valve is seen in 7% to 40% of patients depending on definitions,13 with post-implant severe tricuspid regurgitation independently associated with increased mortality risk.14 The rate of device infection is 1% to 2% at 1 year,8,15 and 3% over the lifetime of the initial transvenous system; this increases to more than 10% after generator replacement.16
LEADLESS PACING TECHNOLOGY
Figure 2. Leadless pacemakers (A) Nanostim and (B) Micra.
The principal goal of leadless pacing is to reduce short- and long-term pacemaker complications by eliminating the two most common sources of problems: the transvenous leads and the surgical pocket. Discussion of leadless pacing strategies began as early as 1970.17 Although several preclinical studies demonstrated efficacy with leadless prototypes,18–20 clinical implementation of fully leadless technology did not occur until recently. As shown in Figure 2, there are now two commercially available leadless pacing devices: Nanostim (St. Jude Medical Inc., St. Paul, MN) and Micra (Medtronic Inc., Dublin, Ireland). At the time of this writing, both have commercial approval in Europe. In the United States, Micra received commercial approval from the US Food and Drug Administration on April 6, 2016, with a similar decision expected on Nanostim. The current approved indications for leadless pacing are chronic atrial tachyarrhythmia with advanced atrioventricular (AV) block; advanced AV block with low level of physical activity or short expected lifespan; and infrequent pauses or unexplained syncope with abnormal findings at electrophysiologic study. Although differences exist between Nanostim and Micra, as shown in Table 1,21–27 there are fundamental similarities. Both are single-unit designs encapsulating the electrodes and pulse generator with rate-adaptive functionality. Both are delivered via an endovascular femoral venous approach without the need for incisional access, transvenous leads, or surgical pocket (Figures 3 and 4).21–27
Nanostim: Landmark trials
As the world’s first-in-man leadless pacemaker, Nanostim was evaluated in two prospective, non-randomized, multicenter, single-arm trials abbreviated LEADLESS22 and LEADLESS II.24 The first human feasibility study, LEADLESS, enrolled 33 patients with approved indications for ventricular-only pacing while excluding patients with expected pacemaker dependency. The most common indication was bradycardia in the presence of persistent atrial arrhythmias, thereby obviating the need for atrial pacing. The primary outcome was freedom from serious complications at 90 days. The secondary outcomes were implant success rate and device performance at 3 months. The results demonstrated 94% composite safety (31 of 33 patients) at 3 months. There was one cardiac perforation leading to tamponade and eventually death after prolonged hospitalization, and one inadvertent deployment into the left ventricle via patent foramen ovale that was successfully retrieved and redeployed without complication. The implant success rate was 97%, and the electrical parameters involving sensing, pacing thresholds, and impedance were as expected at 3 months. Results of 1-year follow-up were published for the LEADLESS cohort,25 revealing no additional complications from 3 to 12 months, no adverse changes in electrical performance parameters, and 100% effectiveness of rate-responsive programming.
Figure 3. Fluoroscopic images depicting catheter-based deployment and subsequent release for the (A) Nanostim and (B) Micra.
The subsequent LEADLESS II trial enrolled 526 patients but did not exclude patients with expected pacemaker dependency, and its results were reported in a preplanned interim analysis when 300 patients had reached 6 months of follow-up (mean follow-up 6.9 ± 4.2 months).24 The primary efficacy end point involved electrical performance including capture thresholds and sensing. Initial deployment success was 96% with expected electrical parameters at implant that were stable at 6 months of follow-up. The rate of freedom from serious adverse events was 93%, with complications including device dislodgment (1.7%, mean 8 ± 6 days after implant), perforation (1.3%), performance deficiency requiring device retrieval and replacement (1.3%), and groin complications (1.3%). There were no device-related deaths, and all device dislodgments were successfully treated percutaneously.
Figure 4. Frontal-plane radiographs showing implanted Nanostim (A) and Micra (B) leadless pacing devices and a traditional dual-chamber pacemaker (C). Panel D depicts cardiac deployment.
There was no prospective control arm involving transvenous pacing in either the LEADLESS or LEADLESS II trial. Thus, in an effort to compare Nanostim (n = 718) vs transvenous pacing, complication rates were calculated for a propensity-matched registry cohort of 10,521 transvenous patients, and differences were reported.26 At 1 month, the composite complication rate was 5.8% for Nanostim (1.5% pericardial effusion, 1% dislodgment) and 12.7% for transvenous pacing (7.6% lead-related, 3.9% thoracic trauma, infection 1.9%) (P < .001). Between 1 month and 2 years, complication rates were only 0.6% for Nanostim vs 5.4% for transvenous pacing (P < .001). This lower complication rate at 2 years was driven almost entirely by a 2.6% infection rate and 2.4% lead-complication rate in the transvenous pacemaker group, nonexistent in the leadless group.
Micra: Landmark trials
Micra was evaluated in a prospective, nonrandomized, multicenter, single-arm trial, enrolling 725 patients with indications for ventricular-only pacing; approximately two-thirds of the cohort had bradycardia in the presence of persistent atrial arrhythmias, similar to the Nanostim cohort.27 The efficacy end point was stable capture threshold at 6 months. The safety end point was freedom from major complications resulting in new or prolonged hospitalization at 6 months. The implant success rate was 99%, and 98% of patients met the primary efficacy end point. The safety end point was met in 96% of patients. Complications included perforation or pericardial effusion (1.6%), groin complication (0.7%), elevated threshold (0.3%), venous thromboembolism (0.3%), and others (1.7%). No dislodgments were reported. There was no prospective, randomized control arm to compare Micra and transvenous pacing. A post hoc analysis was performed comparing major complication rates in this study with an unmatched 2,667-patient meta-analysis control cohort.27 The hazard ratio for the leadless pacing strategy was calculated at 0.49 (95% confidence interval 0.33 to 0.75, P = .001) with absolute risk reduction 3.4% at 6 months resulting in a number needed to treat of 29.4 patients. Further broken down, Micra patients compared with the control cohort had reduced rates of both subsequent hospitalizations (3.9% to 2.3%) and device revisions (3.5% to 0.4%).
ADVANTAGES OF LEADLESS PACING
As discussed above, the major observed benefit with both Nanostim and Micra compared with transvenous cohorts is the elimination of lead and pocket-related complications.25,27 Leadless pacing introduces a new 1% to 2% groin complication rate for both devices not present with transvenous pacing, and also a 1% device dislodgment rate in the case of Nanostim (all dislodgments were treated percutaneously). Data from both clinical trials suggest that the complication rates are largely compressed acutely. In contrast, there are considerable mid-term and long-term complications for transvenous systems.3–5 Indeed, the mid- to long-term window is where leadless pacing is expected to have the most favorable impact. As with any new disruptive technology, operator experience may be important, as evidenced by a near halving of the complication rate observed in the LEADLESS II trial after gaining the experience of 10 implants.25
Other benefits of leadless pacing include a generally quick procedure (average implant time was 30 minutes in LEADLESS and LEADLESS II)22,25 and full shoulder mobility afterwards, so that patients can resume driving once groin soreness has subsided, typically within a few days. (Current studies are investigating whether immediate shoulder mobility with leadless pacing is beneficial to older patients suffering from arthritis.) The lack of an incision allows patients to bathe and shower as soon as they desire, whereas after transvenous pacemaker implant, motion in the affected shoulder is usually restricted for several weeks to avoid lead dislodgment, and showering and bathing are restricted to avoid contamination of the incision with nonsterile tap water. (In some cases, a tightly adherent waterproof dressing can be used.) The leadless systems were designed for compatibility with magnetic resonance imaging (MRI), whereas not all transvenous pacemaker generators and leads are MRI compatible.
Leadless devices are not expected to span the tricuspid valve to create incident or worsening tricuspid regurgitation. In a recent small study of 22 patients undergoing Micra implant, there were no new cases of severe tricuspid regurgitation after the procedure, with only a 9% increase in mild and 5% increase in moderate tricuspid regurgitation,28 vs a rate of 40% of worsening tricuspid regurgitation and 10% of new severe tricuspid regurgitation with transvenous pacing.13,14
Transvenous pacemaker implant requires surgery for pulse generator exchange at a mean of 7 years, a procedure carrying significant risk of short- and long-term complications.10
END-OF-SERVICE QUESTIONS: ATTEMPT RETRIEVAL OR NOT?
Both leadless systems have favorable projected in-service battery life: a reported 15.0 years for Nanostim25 and mean 12.5 years for Micra.27 The inevitable question is what to do then. The Nanostim system was designed to be retrievable using a dedicated catheter system. Micra was not designed with an accompanying retrieval system. Pathologic examinations of leadless devices at autopsy or after explant have revealed a range of device endothelialization, from partial at 19 months to full at 4 months.29,30
As of this writing, no extraction complications have been observed with Nanostim explants up to 506 days after implant (n = 12, mean 197 days after implant).31 Needless to say, there is not yet enough experience worldwide with either system to know what the end-of-service will look like in 10 to 15 years. One strategy could involve first attempting percutaneous retrieval and replacement, if retrieval is not possible, abandoning the old device while implanting a new device alongside. Another strategy would be to forgo a retrieval attempt altogether. In the LEADLESS II study,24 the mean patient age was 75. In this cohort, forgoing elective retrieval for those who live to reach the end of pacemaker service between the age of 85 and 90 would seem reasonable assuming the next device provides similar longevity. For younger patients, careful consideration of long-term strategies is needed. It is not known what the replacement technology will look like in another decade with respect to device size or battery longevity. Preclinical studies using swine and human cadaver hearts have demonstrated the feasibility of multiple right-ventricular Micra implants without affecting cardiac function.32,33
OTHER LIMITATIONS AND CAUTIONARY NOTES
At present, leadless pacing is approved for single-chamber right-ventricular pacing. In the developed world, single right-ventricular pacing modes account for only 20% to 30% of new pacemaker implants, which total more than 1 million per year worldwide.34,35 As with any new technology, the up-front cost of leadless pacemaker implant is expected to be significantly higher than transvenous systems, which at this point remains poorly defined, as the field has not caught up in terms of charges, reimbursement, and billing codes. While those concerns fall outside the scope of this review, it is not known if the expected reductions in mid- and long-term complications will make up for an up-front cost difference. However, a cost-efficacy study reported that one complication of a transvenous pacemaker system was more expensive than the initial implant itself.36 The longest-term follow-up data currently available are with Nanostim, showing an absolute complication reduction of 11.7% at 2 years,24 a disparity only expected to widen with prolonged follow-up, particularly after transvenous generator exchange, when complication rates rapidly escalate.
FUTURE DIRECTIONS
The next horizon of leadless technology will be for right-atrial and dual-chamber pacing to treat the far more pervasive pacing indication of sinus node dysfunction with or without AV block. In the latter application, the two devices will communicate. Prototypes and early nonhuman evaluations are ongoing for both. Leadless pacing is also being investigated for use in tachycardia. Tjong et al37 reported on the safety and feasibility of an entirely leadless pacemaker plus an implantable cardioverter-defibrillator (ICD) system in two sheep and one human using both Nanostim and subcutaneous ICD. Currently, two important limitations of subcutaneous ICD are its inability to provide backup bradycardia and antitachycardia pacing (it provides only defibrillation). The EMBLEM PACE study will enroll 250 patients to receive a leadless pacemaker and Emblem subcutaneous ICD (Boston Scientific, Boston, MA), with patients subsequently receiving commanded antitachycardia pacing for ventricular arrhythmias and bradycardia pacing provided by the leadless device as indicated.
CONCLUSIONS
Leadless cardiac pacing is a safe and efficacious alternative to standard transvenous pacing systems. Although long-term data are limited, available short- and mid-term data show that the elimination of transvenous leads and the surgical pocket results in significant reductions in complication rates. Currently, leadless pacing is approved only for right-ventricular pacing, but investigation of right-atrial, dual-chamber, and fully leadless pacemaker-defibrillator hybrid systems is ongoing.
References
Lagergren H. How it happened: my recollection of early pacing. Pacing Clin Electrophysiol 1978; 1:140–143.
Parsonnet V. Permanent transvenous pacing in 1962. Pacing Clin Electrophysiol 1978; 1:265–268.
Kirkfeldt RE, Johansen JB, Nohr EA, Jorgensen OD, Nielsen JC. Complications after cardiac implantable electronic device implantations: an analysis of a complete, nationwide cohort in Denmark. Eur Heart J 2014; 35:1186–1194.
Udo EO, Zuithoff NP, van Hemel NM, et al. Incidence and predictors of short- and long-term complications in pacemaker therapy: the FOLLOWPACE study. Heart Rhythm 2012; 9:728–735.
Palmisano P, Accogli M, Zaccaria M, et al. Rate, causes, and impact on patient outcome of implantable device complications requiring surgical revision: large population survey from two centres in Italy. Europace 2013; 15:531–540.
De Sensi F, Miracapillo G, Cresti A, Severi S, Airaksinen KE. Pocket hematoma: a call for definition. Pacing Clin Electrophysiol Aug 2015; 38:909–913.
Wiegand UK, LeJeune D, Boguschewski F, et al. Pocket hematoma after pacemaker or implantable cardioverter defibrillator surgery: influence of patient morbidity, operation strategy, and perioperative antiplatelet/anticoagulation therapy. Chest 2004; 126:1177–1186.
Essebag V, Verma A, Healey JS, et al. Clinically significant pocket hematoma increases long-term risk of device infection: Bruise Control Infection Study. J Am Coll Cardiol 2016; 67:1300–1308.
Ohlow MA, Lauer B, Brunelli M, Geller JC. Incidence and predictors of pericardial effusion after permanent heart rhythm device implantation: prospective evaluation of 968 consecutive patients. Circ J 2013; 77:975–981.
Hauser RG, Hayes DL, Kallinen LM, et al. Clinical experience with pacemaker pulse generators and transvenous leads: an 8-year prospective multicenter study. Heart Rhythm 2007; 4:154–160.
Korkeila P, Nyman K, Ylitalo A, et al. Venous obstruction after pacemaker implantation. Pacing Clin Electrophysiol 2007; 30:199–206.
Haghjoo M, Nikoo MH, Fazelifar AF, Alizadeh A, Emkanjoo Z, Sadr-Ameli MA. Predictors of venous obstruction following pacemaker or implantable cardioverter-defibrillator implantation: a contrast venographic study on 100 patients admitted for generator change, lead revision, or device upgrade. Europace 2007; 9:328–332.
Al-Mohaissen MA, Chan KL. Prevalence and mechanism of tricuspid regurgitation following implantation of endocardial leads for pacemaker or cardioverter-defibrillator. J Am Soc Echocardiogr 2012; 25:245–252.
Al-Bawardy R, Krishnaswamy A, Rajeswaran J, et al. Tricuspid regurgitation and implantable devices. Pacing Clin Electrophysiol 2015; 38:259–266.
Klug D, Balde M, Pavin D, et al. Risk factors related to infections of implanted pacemakers and cardioverter-defibrillators: results of a large prospective study. Circulation 2007; 116:1349–1355.
Johansen JB, Jorgensen OD, Moller M, Arnsbo P, Mortensen PT, Nielsen JC. Infection after pacemaker implantation: infection rates and risk factors associated with infection in a population-based cohort study of 46,299 consecutive patients. Eur Heart J 2011; 32:991–998.
Lown B, Kosowsky BD. Artificial cardiac pacemakers. I. N Engl J Med 1970; 283:907–916.
Sutton R. The first European journal on cardiac electrophysiology and pacing, the European Journal of Cardiac Pacing and Electrophysiology. Europace 2011; 13:1663–1664.
Vardas PE, Politopoulous C, Manios E, Parthenakis F, Tsagarkis C. A miniature pacemaker introduced intravenously and implanted endocardially. Preliminary findings from an experimental study. Eur J Card Pacing Electrophysiol 1991; 1:27–30.
Eggen MD, Grubac V, Bonner MD. Design and evaluation of a novel fixation mechanism for a transcatheter pacemaker. IEEE Trans Biomed Eng 2015; 62:2316–2323.
Reddy VY, Knops RE, Sperzel J, et al. Permanent leadless cardiac pacing: results of the LEADLESS trial. Circulation 2014; 129:1466–1471.
Ritter P, Duray GZ, Steinwender C, et al. Early performance of a miniaturized leadless cardiac pacemaker: the Micra Transcatheter Pacing Study. Eur Heart J 2015; 36:2510–2519.
Reddy VY, Exner DV, Cantillon DJ, et al. Percutaneous implantation of an entirely intracardiac leadless pacemaker. N Engl J Med 2015; 373:1125–1135.
Knops RE, Tjong FV, Neuzil P, et al. Chronic performance of a leadless cardiac pacemaker: 1-year follow-up of the LEADLESS trial. J Am Coll Cardiol 2015; 65:1497–1504.
Reddy VY, Cantillon DJ, Ip J, et al. A comparative study of acute and mid-term complications of leadless versus transvenous pacemakers. Heart Rhythm 2016 July. [Epub ahead of print].
Reynolds D, Duray GZ, Omar R, et al. A leadless intracardiac transcatheter pacing system. N Engl J Med 2016; 374:533–541.
Garikipati NV, Karve A, Okabe T, et al. Tricuspid regurgitation after leadless pacemaker implantation. Abstract presented at Heart Rhythm Society Scientific Sessions, May 4–7, 2016, San Francisco, CA.
Tjong FV, Stam OC, van der Wal AC, et al. Postmortem histopathological examination of a leadless pacemaker shows partial encapsulation after 19 months. Circ Arrhythm Electrophysiol 2015; 8:1293–1295.
Borgquist R, Ljungstrom E, Koul B, Hoijer CJ. Leadless Medtronic Micra pacemaker almost completely endothelialized already after 4 months: first clinical experience from an explanted heart. Eur Heart J 2016; 37:2503.
Reddy VY, Knops RE, Defaye P, et al. Worldwide clinical experience of the retrieval of leadless cardiac pacemakers. Abstract presented at Heart Rhythm Society Scientific Sessions, May 4–7, 2016, San Francisco, CA.
Chen K, Zheng X, Dai Y, et al. Multiple leadless pacemakers implanted in the right ventricle of swine. Europace 2016 January 31. pii: euv418. [Epub ahead of print].
Omdahl P, Eggen MD, Bonner MD, Iaizzo PA, Wika K. Right ventricular anatomy can accommodate multiple micra transcatheter pacemakers. Pacing Clin Electrophysiol 2016; 39:393–397.
Mond HG, Proclemer A. The 11th world survey of cardiac pacing and implantable cardioverter-defibrillators: calendar year 2009—a World Society of Arrhythmia’s project. Pacing Clin Electrophysiol 2011; 34:1013–1027.
Epstein AE, DiMarco JP, Ellenbogen KA, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Heart Rhythm Society. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2013; 127:e283–352.
Tobin K, Stewart J, Westveer D, Frumin H. Acute complications of permanent pacemaker implantation: their financial implication and relation to volume and operator experience. Am J Cardiol 2000; 85:774–776, A9.
Tjong FV, Brouwer TF, Smeding L, et al. Combined leadless pacemaker and subcutaneous implantable defibrillator therapy: feasibility, safety, and performance. Europace 2016 March 3. [Epub ahead of print].
References
Lagergren H. How it happened: my recollection of early pacing. Pacing Clin Electrophysiol 1978; 1:140–143.
Parsonnet V. Permanent transvenous pacing in 1962. Pacing Clin Electrophysiol 1978; 1:265–268.
Kirkfeldt RE, Johansen JB, Nohr EA, Jorgensen OD, Nielsen JC. Complications after cardiac implantable electronic device implantations: an analysis of a complete, nationwide cohort in Denmark. Eur Heart J 2014; 35:1186–1194.
Udo EO, Zuithoff NP, van Hemel NM, et al. Incidence and predictors of short- and long-term complications in pacemaker therapy: the FOLLOWPACE study. Heart Rhythm 2012; 9:728–735.
Palmisano P, Accogli M, Zaccaria M, et al. Rate, causes, and impact on patient outcome of implantable device complications requiring surgical revision: large population survey from two centres in Italy. Europace 2013; 15:531–540.
De Sensi F, Miracapillo G, Cresti A, Severi S, Airaksinen KE. Pocket hematoma: a call for definition. Pacing Clin Electrophysiol Aug 2015; 38:909–913.
Wiegand UK, LeJeune D, Boguschewski F, et al. Pocket hematoma after pacemaker or implantable cardioverter defibrillator surgery: influence of patient morbidity, operation strategy, and perioperative antiplatelet/anticoagulation therapy. Chest 2004; 126:1177–1186.
Essebag V, Verma A, Healey JS, et al. Clinically significant pocket hematoma increases long-term risk of device infection: Bruise Control Infection Study. J Am Coll Cardiol 2016; 67:1300–1308.
Ohlow MA, Lauer B, Brunelli M, Geller JC. Incidence and predictors of pericardial effusion after permanent heart rhythm device implantation: prospective evaluation of 968 consecutive patients. Circ J 2013; 77:975–981.
Hauser RG, Hayes DL, Kallinen LM, et al. Clinical experience with pacemaker pulse generators and transvenous leads: an 8-year prospective multicenter study. Heart Rhythm 2007; 4:154–160.
Korkeila P, Nyman K, Ylitalo A, et al. Venous obstruction after pacemaker implantation. Pacing Clin Electrophysiol 2007; 30:199–206.
Haghjoo M, Nikoo MH, Fazelifar AF, Alizadeh A, Emkanjoo Z, Sadr-Ameli MA. Predictors of venous obstruction following pacemaker or implantable cardioverter-defibrillator implantation: a contrast venographic study on 100 patients admitted for generator change, lead revision, or device upgrade. Europace 2007; 9:328–332.
Al-Mohaissen MA, Chan KL. Prevalence and mechanism of tricuspid regurgitation following implantation of endocardial leads for pacemaker or cardioverter-defibrillator. J Am Soc Echocardiogr 2012; 25:245–252.
Al-Bawardy R, Krishnaswamy A, Rajeswaran J, et al. Tricuspid regurgitation and implantable devices. Pacing Clin Electrophysiol 2015; 38:259–266.
Klug D, Balde M, Pavin D, et al. Risk factors related to infections of implanted pacemakers and cardioverter-defibrillators: results of a large prospective study. Circulation 2007; 116:1349–1355.
Johansen JB, Jorgensen OD, Moller M, Arnsbo P, Mortensen PT, Nielsen JC. Infection after pacemaker implantation: infection rates and risk factors associated with infection in a population-based cohort study of 46,299 consecutive patients. Eur Heart J 2011; 32:991–998.
Lown B, Kosowsky BD. Artificial cardiac pacemakers. I. N Engl J Med 1970; 283:907–916.
Sutton R. The first European journal on cardiac electrophysiology and pacing, the European Journal of Cardiac Pacing and Electrophysiology. Europace 2011; 13:1663–1664.
Vardas PE, Politopoulous C, Manios E, Parthenakis F, Tsagarkis C. A miniature pacemaker introduced intravenously and implanted endocardially. Preliminary findings from an experimental study. Eur J Card Pacing Electrophysiol 1991; 1:27–30.
Eggen MD, Grubac V, Bonner MD. Design and evaluation of a novel fixation mechanism for a transcatheter pacemaker. IEEE Trans Biomed Eng 2015; 62:2316–2323.
Reddy VY, Knops RE, Sperzel J, et al. Permanent leadless cardiac pacing: results of the LEADLESS trial. Circulation 2014; 129:1466–1471.
Ritter P, Duray GZ, Steinwender C, et al. Early performance of a miniaturized leadless cardiac pacemaker: the Micra Transcatheter Pacing Study. Eur Heart J 2015; 36:2510–2519.
Reddy VY, Exner DV, Cantillon DJ, et al. Percutaneous implantation of an entirely intracardiac leadless pacemaker. N Engl J Med 2015; 373:1125–1135.
Knops RE, Tjong FV, Neuzil P, et al. Chronic performance of a leadless cardiac pacemaker: 1-year follow-up of the LEADLESS trial. J Am Coll Cardiol 2015; 65:1497–1504.
Reddy VY, Cantillon DJ, Ip J, et al. A comparative study of acute and mid-term complications of leadless versus transvenous pacemakers. Heart Rhythm 2016 July. [Epub ahead of print].
Reynolds D, Duray GZ, Omar R, et al. A leadless intracardiac transcatheter pacing system. N Engl J Med 2016; 374:533–541.
Garikipati NV, Karve A, Okabe T, et al. Tricuspid regurgitation after leadless pacemaker implantation. Abstract presented at Heart Rhythm Society Scientific Sessions, May 4–7, 2016, San Francisco, CA.
Tjong FV, Stam OC, van der Wal AC, et al. Postmortem histopathological examination of a leadless pacemaker shows partial encapsulation after 19 months. Circ Arrhythm Electrophysiol 2015; 8:1293–1295.
Borgquist R, Ljungstrom E, Koul B, Hoijer CJ. Leadless Medtronic Micra pacemaker almost completely endothelialized already after 4 months: first clinical experience from an explanted heart. Eur Heart J 2016; 37:2503.
Reddy VY, Knops RE, Defaye P, et al. Worldwide clinical experience of the retrieval of leadless cardiac pacemakers. Abstract presented at Heart Rhythm Society Scientific Sessions, May 4–7, 2016, San Francisco, CA.
Chen K, Zheng X, Dai Y, et al. Multiple leadless pacemakers implanted in the right ventricle of swine. Europace 2016 January 31. pii: euv418. [Epub ahead of print].
Omdahl P, Eggen MD, Bonner MD, Iaizzo PA, Wika K. Right ventricular anatomy can accommodate multiple micra transcatheter pacemakers. Pacing Clin Electrophysiol 2016; 39:393–397.
Mond HG, Proclemer A. The 11th world survey of cardiac pacing and implantable cardioverter-defibrillators: calendar year 2009—a World Society of Arrhythmia’s project. Pacing Clin Electrophysiol 2011; 34:1013–1027.
Epstein AE, DiMarco JP, Ellenbogen KA, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Heart Rhythm Society. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2013; 127:e283–352.
Tobin K, Stewart J, Westveer D, Frumin H. Acute complications of permanent pacemaker implantation: their financial implication and relation to volume and operator experience. Am J Cardiol 2000; 85:774–776, A9.
Tjong FV, Brouwer TF, Smeding L, et al. Combined leadless pacemaker and subcutaneous implantable defibrillator therapy: feasibility, safety, and performance. Europace 2016 March 3. [Epub ahead of print].
Leadless cardiac pacing: What primary care providers and non-EP cardiologists should know
Display Headline
Leadless cardiac pacing: What primary care providers and non-EP cardiologists should know
Legacy Keywords
pacemakers, leadless pacemakers, Daniel Cantillon, erich kiehl
Legacy Keywords
pacemakers, leadless pacemakers, Daniel Cantillon, erich kiehl
Citation Override
Cleveland Clinic Journal of Medicine 2016 November; 83(suppl 2):S24-S34
Inside the Article
KEY POINTS
Leadless cardiac pacing has emerged as a safe and effective alternative involving catheter-based delivery of a self-contained device directly into the right ventricle without incisional access, leads, or a surgical pocket. The procedure typically can be performed in 30 minutes or less, with fewer postprocedure restrictions.
Leadless pacing is showing promising results, but it is currently limited to single-chamber pacing.
Future directions include atrial and dual-chamber pacing and combining the procedure with a subcutaneous implantable cardioverter-defibrillator.
Disallow All Ads
Alternative CME
Consolidated Pubs: Do Not Show Source Publication Logo
Statin therapy has been shown to substantially reduce adverse events associated with low-density-lipoprotein cholesterol (LDL-C) and cardiovascular disease (CVD). Statins alone are often not adequate to achieve treatment goals, and residual CVD risk remains high. Combination therapies of statins with ezetimibe and resins to further lower LDL-C, fibrates and omega 3 fatty acids to lower triglycerides, and niacin to lower both and raise high-density-liproprotein cholesterol are available, but additional risk reduction has not been consistently demonstrated in clinical trials.
The link between atherogenic lipoproteins and CVD is strong, and the need to develop therapies in addition to statins to substantially and safely reduce LDL-C is a priority. The association of reduced proprotein convertase subtilisin/kexin type 9 (PCSK9) activity with reduced LDL-C and CVD events has led to the rapid development and approval of monoclonal antibody therapies to inhibit PCSK9.
In this review, we discuss trials of these therapies that have shown durable reductions in LDL-C of more than 50%, with acceptable tolerability. Now that PCSK9 inhibitors are approved by the US Food and Drug Administration (FDA), extended data are needed as to long-term tolerability, safety, and efficacy of these agents and, most importantly, demonstration of additional reduction in CVD events.
A CASE FOR ADDITIONAL THERAPIES
CVD is the leading cause of morbidity and death in the United States, responsible for one in four deaths. Hyperlipidemia and, specifically, elevated LDL-C have been found to be important drivers of atherosclerosis and, in turn, adverse cardiovascular (CV) events. Likewise, numerous observational and clinical trials have shown that reducing LDL-C, particularly with statins, decreases CVD events.1–4 More aggressive lowering with higher doses or more intensive statin therapy further reduces rates of adverse outcomes.3,4 In addition, the pleiotropic effects of statins imply that not all of their benefits are derived from LDL-C lowering alone.5 Consequently, it is now standard practice to use statins at the highest tolerable dose to reach target LDL-C levels and prevent CV events in high-risk patients with CVD or multiple coronary artery disease risk factors, regardless of the LDL-C levels.6,7
The American College of Cardiology (ACC) and the American Heart Association released cholesterol guidelines in 2013 that recommend a risk-based approach for statin therapy rather than targeting specific LDL-C levels.6 Although this evidence-based approach may better conform to clinical trials, the debate that lower LDL-C targets will further prevent CVD continues.
Indeed, it appears that lower is better, as demonstrated by the IMPROVE-IT trial.8 Although the control group receiving simvastatin monotherapy had low LDL-C levels (mean, 69.9 mg/dL; 1.8 mmol/L), the experimental group receiving simvastatin plus ezetimibe achieved even lower levels (mean, 53.2 mg/dL;1.4 mmol/L) after 1 year of therapy and had a significantly lower composite primary end point of CV death, major coronary event, or nonfatal stroke at 7 years (34.7% for simvastatin monotherapy vs 32.7% for combined therapy).9 Furthermore, the event-rate reduction with the addition of ezetimibe was the same as the average predicted by the Cholesterol Treatment Trialists’ meta-analysis: an LDL-C reduction of 1 mmol/L (38.6 mg/dL) yields a 23% risk reduction in major coronary events over 5 years.10 Although only a modest absolute reduction in outcomes, it supports the notion that further reduction of LDL-C levels by more potent therapies may offer greater benefit.
There is strong evidence that statin therapy reduces the risk of developing CVD in patients with or without a previous atherosclerotic event; however, residual CVD risk remains even for those on therapy. A contributing factor to this residual risk is that many statin-treated patients have insufficient response or intolerance and do not achieve adequate LDL-C reductions.
There are three clinically important patient populations who are inadequately managed with current therapies and remain at high risk of subsequent CV events; these are patients who would benefit from additional therapies.
1. Patients with familial hypercholesterolemia (FH). This is the most common genetic disorder in the world, yet it is frequently undiagnosed and untreated. Due to high baseline cholesterol levels, achieving LDL-C treatment goals is challenging.
The prevalence may be closer to 1:200 to 1:250 rather than the often quoted 1:500.11
Fewer than 12% of patients with heterzygous FH achieve the LDL-C goal of < 100 mg/dL with maximal statin treatment alone or with a second agent.12
2. Patients with hyperlipidemia not due to FH who are at elevated CV risk and undertreated. In US and European surveys, between 50% and 60% of patients receiving statins with or without other therapies failed to reach LDL-C reduction goals.13
Variation in response to statin treatment between individuals may be considerable.
Poor adherence to statin therapy is common.
3. Patients with side effects to statins, particularly muscle symptoms that prevent statin use or substantially limit the dose.
Although the incidence of myopathy is low (< 0.1%) and rhabdomyolysis is even less common, observational studies suggest that 10% to 20% of patients may limit statin use due to muscle-associated complaints including muscle aching, cramps, or weakness.14
Side effects may be dose-dependent, limiting the use of the high-intensity statin doses that are frequently necessary to achieve LDL-C goals.
Consequently, there is great interest in developing therapies beyond statins that may further reduce CV events. However, treatments other than ezetimibe for further management of hyperlipidemia and risk reduction have failed to demonstrate consistent benefit when added to statin therapy.15–19 The largest studies were with niacin and fibrates. Unfortunately, most trials demonstrated no overall outcomes benefit or only benefits in subgroup analyses, leaving the door open to other pharmacologic interventions.
Studies with the cholesterol ester transfer protein (CETP) inhibitor torcetrapib, in combination with statin therapy, actually demonstrated an overall increase in all-cause mortality in the treatment group.20 Two large outcome trials of the CETP inhibitors dalcetrapib and evacetrapib were stopped after interim analysis predicted no benefit. Although drugs such as lomitapide (a microsomal triglyceride transfer protein inhibitor) and mipomersen (an antisense oligonucleotide inhibitor of ApoB-100 synthesis) can lower LDL-C by reducing ApoB synthesis,21 they are approved only in the small population of individuals with homozygous FH and liver toxicity and side effects are a concern.
Accordingly, current cholesterol management guidelines continue to offer LDL-C as the main target of lipid-modifying therapy, with statins as the primary treatment choice. The desire to build on statin therapy to prevent further progression of atherosclerosis and clinical CVD has encouraged continued focus on strategies to lower LDL-C to even greater extents.
Fortunately for practitioners, for the first time since lovastatin was approved in 1987, there is a new therapy approved by the FDA that significantly lowers LDL-C and, potentially, improves CV outcomes—the proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors. This review will focus on the PCSK9 inhibitors, a novel therapeutic class that reduces LDL-C through increased hepatic clearance. These drugs are rapidly emerging as an ideal adjunctive therapy to statins for patients at the highest risk and as a highly efficacious alternate therapy in patients intolerant of statins.
PCSK9 INHIBITORS: DISCOVERY, MECHANISM, AND THERAPEUTIC INTERVENTIONS
Two PCSK9 inhibitors have received FDA approval: alirocumab (Praluent) and evolocumab (Repatha). Among new molecular entities for clinical use, PCSK9 inhibitor therapies had one of the shortest durations from discovery to development and approval.
Mutations in the PCSK9 gene associated with autosomal dominant hypercholesterolemia were first identified in 2003 in a French family.22 The PCSK9 protein is now known to be a secreted enzymatic serine protease that is primarily synthesized in the liver and binds to the LDL receptor (LDL-R)/LDL-C complex on the surface of hepatocytes, marking the receptor for lysosomal degradation rather than recycling to the cell surface. Thus, it reduces the quantity of LDL-R that is available to remove LDL-C from circulation.23 As a result, higher levels of PCSK9 are associated with higher levels of plasma LDL-C.
The clinical importance of PCSK9 in regulating LDL-C is supported by observed mutations and polymorphisms. Gain-of-function mutations that increase the activity of PCSK9 have been shown to be associated with elevated LDL-C, premature CVD, and myocardial infarction (MI).24 Conversely, loss-of-function mutations (heterozygotes found in 1% to 3% of the population) result in decreased activity of PCSK9, lower LDL-C, and lower incidence of CVD (Table 1).25–29 These observations, combined with data showing that homozygote loss-of-function individuals with very low LDL-C were generally very healthy, sparked interest in developing inhibition of PCSK9 activity as a therapeutic strategy for hyperlipidemia.
Multiple pharmacologic developments are aimed at inhibiting PCSK9, with many compounds in clinical trials. The approaches include gene silencing with loss-of-function mutations, synthetic peptides, oral small molecules, and monoclonal antibodies. Gene silencing was first observed in 2007 when administration of antisense oligonucleotides targeted to selectively inhibit PCSK9 mRNA was found to up-regulate LDL-R, thereby decreasing serum levels of LDL-C.30
The first study to establish the role of synthetic peptides in PCSK9 inhibition was performed in 2008. In this study, the epidermal growth factor-like A synthetic peptide blocked the interaction between PCSK9 and LDL-R, thereby decreasing the degradation of LDL-R and preserving LDL uptake.31 Although studies are limited, synthetic peptides remain an area of great interest given their promising effects on lipid metabolism. Recently, a synthetic PCSK9-binding adnectin derived from the human fibronectin known as BMS-962476, had favorable results in a phase 1 clinical trial. An RNA interference molecule, subcutaneous ALN-PSC, inhibits PCSK9 gene expression by causing destruction of messenger RNA, thus inhibiting PCSK9 synthesis (Table 2).32
PCSK9 INHIBITORS: CLINICAL TRIALS
Subcutaneously administered monoclonal antibodies targeting PCSK9 currently are the only PCSK9 inhibitors FDA-approved for clinical use. The first study to demonstrate efficacy in enhancing uptake of serum LDL-C was performed in 2009.33 Multiple phase 1 and 2 studies soon followed, demonstrating acceptable safety and 50% to 70% reductions in LDL-C at upper-dose titrations.34 Additionally, there were significant reductions in total cholesterol, ApoB, triglycerides, and lipoprotein(a).
These early developments paved the way for larger phase 3 trials (Table 3).35–48 The PCSK9 inhibitors evolocumab and alirocumab have been shown in multiple phase 3 clinical trials to achieve a consistent dose-dependent 50% to 60% reduction in LDL-C across a broad range of CVD risk, pretreatment LDL-C levels, and background therapy: monotherapy (MENDEL-2, ODYSSEY COMBO I),35,44 added to statin therapy (LAPLACE-2, ODYSSEY CHOICE I),38,46 and in individuals with heterozygous FH (RUTHERFORD-2, ODYSSEY-FH).37,42 Trials with bococizumab are under way.
The GAUSS-2 clinical trial (Goal Achievement after Utilizing an Anti-PCSK9 Antibody in Statin Intolerant Subjects-2) demonstrated similar efficacy in reducing LDL-C in patients with clinically assessed statin intolerance due to muscle-related adverse symptoms.39 In GAUSS-3, patients were first identified as being statin intolerant secondary to muscle-associated symptoms based on a randomized, crossover trial of atorvastatin vs placebo.40 The 43% of participants who experienced intolerable muscle-related symptoms on the statin but not on placebo were then randomized to evolocumab vs ezetimibe. Results showed significant reduction in LDL-C in the evolocumab group (52.8%) compared with the ezetimibe group (16.7%). Additionally, among patients with muscle symptoms on statin therapy, PCSK9 therapy was discontinued for muscle symptoms in only 0.7% of evolocumab recipients and 6.8% of ezetimibe recipients.
Overall, the PCSK9 inhibitors are generally well tolerated with injection site reactions being the most common side effect. A meta-analysis published in 2015 of 25 trials including more than 12,000 patients treated with evolocumab and alirocumab reported no significant difference in adverse events or safety outcomes vs placebo or ezetimibe.49 Antidrug binding or neutralizing antibody production to these agents, thus far, has not been shown to be an issue. Additional analyses have not indicated an adverse effect on gonadal hormone levels or increased incidence of new-onset diabetes.
Two studies published in 2015 offer insight into longer term durability and safety as well as potential CVD outcome benefit (Table 4)50,51:
OSLER-1 and 2: Open-Label Study of Long-Term Evaluation against LDL-Cholesterol (OSLER) trials—evolocumab trial;50
ODYSSEY long term: Long-Term Safety and Tolerability of Alirocumab in High Cardiovascular Risk Patients with Hypercholesterolemia Not Adequately Controlled with Their Lipid Modifying Therapy—alirocumab trial.51
The OSLER trials reported durable LDL-C reductions of 61% and the ODYSSEY trial reported a LDL-C reduction of 62%.50,51 In both studies, the overall occurrence of adverse events was similar to placebo, but both reported a higher rate of neurocognitive effects in the active treatment groups (evolocumab 0.9% vs 0.3% for standard therapy; alirocumab 1.2% vs 0.5% for placebo). It must be noted that although the absolute rate of neurocognitive adverse events is low, it is unclear if these events were related to the drugs themselves or to extreme lowering of LDL-C. Nevertheless, the FDA has raised concerns about neurocognitive events. A sub-study of the ongoing FOURIER trial with evolocumab—EBBINGHAUS—is expected to address this concern.
Figure 1. Effect of PCSK9 inhibitors on cardiovascular events.50,51
In addition, analyses of CV events showed that the PCSK9 inhibitors effectively cut the CV rate in half in both studies (Figure 1).50,51 In the OSLER trials,50 evolocumab recipients had 53% reduction in major CV events (0.95% vs 2.18% in the standard therapy group; P = .003). In ODYSSEY,51 alirocumab recipients had a 48% reduction in major CV events (1.7% vs 3.3% for placebo; P = .02). Furthermore, a 2015 meta-analysis of 24 phase 2 and 3 trials reported a statistically significant 55% reduction in all-cause mortality and 50% reduction in CV mortality with PCSK9 inhibitors.52
For many reasons including short length of follow-up, study design, and small numbers of outcome events, the OSLER and ODYSSEY studies, although enticing, are exploratory and hypothesis-generating only and results need to be interpreted with caution. Nevertheless, they have set the stage for ongoing prospective randomized outcome trials that are studying the CV effects and tolerability of PCSK9 inhibitors over a longer time frame. These include the following trials.
The Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects With Elevated Risk (FOURIER) is an ongoing trial with the primary end point of CV death, MI, hospitalization for unstable angina, stroke, or coronary revascularization in high-risk patients receiving evolocumab or placebo.53
The ODYSSEY trial is examining the effect of alirocumab vs placebo on the composite primary endpoint of coronary heart disease death, non-fatal MI, fatal and nonfatal ischemic stroke, and unstable angina requiring hospitalization in patients who have had an acute coronary syndrome event during the previous 4 to 52 weeks.54
The Evaluation of Bococizumab in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects (SPIRE) trials are investigating the effect of bococizumab, a third PCSK9 “humanized” monoclonal antibody, vs placebo in reducing death, MI, stroke, or unstable angina in patients at high-risk of CVD who are receiving standard lipid-lowering therapy with LDL-C > 70 mg/dL (1.8 mmol/L) (SPIRE-1) or > 100 mg/dL (2.6 mmol/L) (SPIRE-2).55,56
Because these outcome trials are attempting to enroll more than 70,000 patients and are event driven, it is difficult to predict when they will be completed (Table 5).53–56 However, recent estimates indicate completion of at least one trial by the end of 2016 or early 2017, with interim analyses of others expected at that time. It is hoped that they will answer the all-important question of whether PCSK9 inhibitors are associated with further CV event reduction benefit.
CURRENT FDA INDICATIONS AND GUIDELINES
The two PCSK9 inhibitors approved by the FDA—alirocumab (subcutaneous 75 mg every 2 weeks up titrated to 150 mg) and evolocumab (subcutaneous 140 mg every 2 weeks or 420 mg every 4 weeks)—are both indicated for use with statins in patients with heterozygous FH or known atherosclerotic CVD who require further reduction in LDL-C levels despite lifestyle interventions and use of maximally tolerated statins. Evolocumab has also been approved for use in patients with homozygous FH.
Although PCSK9 inhibitors are not specifically approved for patients unable to tolerate statins, the results of GAUSS-3, which documented that statin intolerance is a real, definable entity and very responsive to PCSK9 inhibition, makes these drugs promising agents for patients intolerant of statins and, thus, unable to benefit from high-intensity stain therapy.
In April 2016, the ACC released a clinical consensus update to their 2013 cholesterol guidelines, which is their first recommendation specifically addressing the use of non-statin therapies, including the newer PCSK9 inhibitors.57 For high-risk patients with clinical atherosclerotic CVD or LDL-C > 190 and failure to achieve at least a 50% reduction in LDL-C on maximally tolerated statin, non-statins may be considered. Ezetimibe, given its safety and tolerability, should be the first additional medication added. Bile acid sequestrants may be used as a second-line therapy if ezetimibe is not tolerated and triglycerides are not elevated. If therapy goals are not met on maximally tolerated statin and ezetimibe, either approved PCSK9 inhibitor can be added or used to replace ezetimibe. The document also specifies that given the lack of long-term safety and efficacy data on the PCSK9 inhibitors, they are not recommended for use in primary prevention patients in the absence of FH.
CONCLUSION
Although statin therapy has been shown to substantially reduce LDL-C and CVD adverse events, there remains a high rate of inadequate goal achievement and residual CVD risk in patients receiving statins. Combination therapies with ezetimibe and resins to further lower LDL-C, fibrates and omega 3 fatty acids to lower triglycerides, and niacin to lower both and raise high-density-liproprotein cholesterol are available, even though additional CV risk reduction is minimal or elusive when these drugs are added to statin therapy.
The link between atherogenic lipoproteins and CVD is strong, and the need to develop therapies in addition to statins to substantially and safely reduce LDL-C remains a priority. The association of reduced PCSK9 activity with reduced LDL-C and CV events has led to rapid development and approval of monoclonal antibody therapies to inhibit PCSK9. In trials, these therapies have shown substantial and durable reductions in LDL-C of more than 50%, with acceptable tolerability. Now that PCSK9 inhibitors are approved by the FDA, extended data about long-term tolerability, safety, and efficacy and, most importantly, demonstration of additional reduction in CVD events are needed. It is hoped that the long-term ongoing trials will provide these data.
For the immediate future, statin therapy will continue to be the cornerstone of lipid and CVD risk management based on their low generic cost, proven CVD risk reduction, and clinicians’ comfort with their use. However, the reliable efficacy of PCSK9 inhibitors and the fact that statin therapy itself increases PCSK9 activity makes the addition of PCSK9 inhibitors to statins an attractive approach in high-risk patients failing to reach LDL-C treatment goals.
Although current indications are limited, there are patients at high CVD risk who would be appropriate candidates for these therapies. These include patients with the following:
FH with lifetime burden of elevated LDL-C and associated low likelihood of achieving optimal LDL-C control on current available therapies
Complete or partial statin intolerance with high-intensity statin dosing limited by side effects
High CV risk who are not at LDL-C goal on current therapies.
Now that the first therapies are available, practitioners can expect newer approaches to tackle PCSK9-mediated LDL-C reduction. Bococizumab is lagging in phase 3 trials, but the SPIRE program is moving forward with special population studies expected to conclude in 2016 and simultaneous long-term outcomes trials. Other PCSK9 inhibitors being investigated include agents with more durable effect requiring less frequent injections, RNA-interference therapies, vaccinations, antisense therapies, and oral formulations.
The PCSK9 inhibitors hold promise as an adjunct to statin therapy. Their eventual clinical role will depend on a balance between substantial LDL-C reductions, long-term safety, tolerability, and reduction in CVD events vs the cost (estimated at $14,000 a year), access from payers, acceptance of injectable therapies, and magnitude of incremental benefit when added to current therapies. Nevertheless, initial clinical trial data are encouraging and these drugs may be an important addition to the therapeutic armamentarium against CVD.
References
Scandinavian Simvastatin Survival Study Group. Randomized trial of cholesterol lowering in 4,444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
Sacks FM, Pfeiffer MA, Moye LA, et al; Cholesterol and Recurrent Events Trial Investigators. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial Investigators. N Engl J Med 1996; 335:1001–1009.
Schwartz GG, Olsson AG, Ezekowitz MD, et al; Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) Study Investigators. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
Lipid Research Clinics Program. The Lipid Research Clinics Coronary Primary Prevention Trial results (reduction in incidence of coronary heart disease). JAMA 1984; 251:351–364.
Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation 2004; 109(suppl 1):III39–III43.
Stone N, Robinson J, Lichtenstein A, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl 2):S1–S45.
Jacobson TA, Ito MK, Maki KC, et al. National lipid association recommendations for patient-centered management of dyslipidemia: part 1—full report. J Clin Lipidol 2015; 9:129–169.
Jarcho JA, Keaney JF Jr. Proof that lower is better–LDL cholesterol and IMPROVE-IT. N Engl J Med 2015; 372:2448–2450.
Cannon CP, Blazing MA, Giugliano RP, et al; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
de Ferranti S, Rodday AM, Mendelson M, et al. Prevalence of familial hypercholesterolemia in the 1999 to 2012 United States National Health and Nutrition Examination Surveys (NHANES). Circulation 2016; 133:1067–1072.
Perez de Isla L, Alonso R, Watts GF, et al; SAFEHEART investigators. Attainment of LDL-cholesterol treatment goals in patients with familial hypercholesterolemia: 5-year SAFEHEART registry follow-up. J Am Coll Cardiol 2016; 67:1278–1285.
Unni SK, Quek RGW, Biskupiak J, et al. Assessment of statin therapy, LDL-C levels, and cardiovascular events among high-risk patients in the United States. J Clin Lipidol 2016; 10:63–71.
Mammen AL, Amato AA. Statin myopathy: a review of recent progress. Curr Opin Rheum 2010; 22:644–650.
AIM-HIGH Investigators; Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255–2267.
HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J 2013; 34:1279–1291.
ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
Keech A, Simes RJ, Barter P, et al; FIELD study investigators. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomized controlled trial. Lancet 2005; 366:1849–1861.
Kaur N, Pandey A, Negi H, et al. Effect of HDL-raising drugs on cardiovascular outcomes: a systematic review and meta-regression. PLoS One 2014; 9:e94585.
Barter PJ, Caulfield M, Eriksson M, et al; ILLUMINATE investigators. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
Rader D, Kastelein J. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 2014; 129:1022–1032.
Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34:154–156.
Verbeek R, Stoekenbroek RM, Hovingh GK. PCSK9 inhibitors: novel therapeutic agents for the treatment of hypercholesterolemia. Eur J of Pharm 2015; 763(Pt A):38–47.
Steinberg D, Witztum JL. Inhibition of PCSK9: a powerful weapon for achieving ideal LDL cholesterol levels. Proc Natl Acad Sci USA 2009; 106:9546–9547.
Abifadel M, Rabès J-P, Devillers M, et al. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum Mutat 2009; 30:520–529.
Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
Benn M, Nordestgaard BG, Grande P, Schnohr P, Tybærg-Hansen A. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease. J Am Coll Cardiol 2010; 55:2833–2842.
Mortensen MB, Afzal S, Nordestgaard BG, Falk E. The high-density lipoprotein-adjusted SCORE model worsens SCORE-based risk classification in a contemporary population of 30,824 Europeans: the Copenhagen General Population Study. Eur Heart J 2015; 36:2446–2453.
Victor RG, Haley RW, Willett DL, et al. The Dallas Heart Study: a population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am J Cardiol 2004; 93:1473–1480.
Graham MJ, Lemonidis KM, Whipple CP, et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res 2007; 48:763–767.
Shan L, Pang L, Zhang R, Murgolo NJ, Lan H, Hedrick JA. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Comm 2008; 375:69–73.
Stein EA, Raal F. Reduction of low-density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Annu Rev Med 2014; 65:417–431.
Duff CJ, Scott MJ, Kirby IT, Hutchinson SE, Martin SL, Hooper NM. Antibody-mediated disruption of the interaction between PCSK9 and the low-density lipoprotein receptor. Biochem J 2009; 419:577–584.
Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Eng J Med 2012; 366:1108–1118.
Koren MJ, Lundqvist P, Bolognese M, et al; MENDEL-2 Investigators. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol 2014; 63:2531–2540.
Blom DJ, Hala T, Bolognese M, et al; DESCARTES investigators. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med 2014; 370:1809-1819.
Raal FJ, Stein EA, Dufour R, et al; RUTHERFORD-2 Investigators. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet 2014; 385:331–340.
Robinson JG, Nedergaard BS, Rogers WJ, et al; LAPLA C-2 Investigators. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA 2014; 311:1870–1882.
Stroes E, Colquhoun D, Sullivan D, et al; GAUSS-2 Investigators. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol 2014; 63:2541–2548.
Nissen SE, Stroes E, Dent-Acosta RE, et al; GAUSS-3 Investigators. Efficacy and tolerability of evolocumab vs ezetimibe in patients with muscle-related statin intolerance, the GAUSS-3 randomized clinical trial. JAMA 2016; 315:1580–1590.
Trial assessing long term use of PCSK9 inhibition in subjects with genetic LDL disorders (TAUSSIG). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT-1624142. Updated June 25, 2015. Accessed October 23, 2016.
Kastelein JJ, Ginsberg HN, Langslet G, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J 2015; 36:2996–3003.
Efficacy and safety of alirocumab (SAR236553/REGN727) versus placebo on top of lipid-modifying therapy in patients with heterozygous familial hypercholesterolemia; the ODYSSEY HIGH FH trial. U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01617655. Updated September 27, 2016. Accessed October 23, 2016.
Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: The ODYSSEY COMBO I study. Am Heart J 2015; 169:906–915.
Efficacy and Safety of Alirocumab (SAR236553/REGN727) Versus Ezetimibe on Top of Statin in High Cardiovascular Risk Patients With Hypercholesterolemia (ODYSSEY COMBO II). U.S. National Institutes of Health website. Updated June 23, 2016. https://clinicaltrials.gov/ct2/show/NCT01644188. Accessed October 23, 2016.
Roth EM, Moriarty P, Bergeron J, et al; ODYSSEY CHOICE I investigators. A phase III randomized trial evaluating alirocumab 300 mg every 4 weeks as monotherapy or add-on to statin: ODYSSEY CHOICE I. Atherosclerosis 2016, doi: 10.1016/j.atherosclerosis.2016.08.043.
Phase III Study To Evaluate Alirocumab in Patients With Hypercholesterolemia Not Treated With a Statin (ODYSSEY CHOICE II). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT02023879. Updated November 2, 2015. Accessed October 23, 2016.
Monthly and twice monthly subcutaneous dosing of PF-04950615 (RN316) in hypercholesterolemic subjects on a statin. U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/results?term=NCT01592240. Updated October 14, 2014. Accessed October 23, 2016.
Zhang XL, Zhu QQ, Zhu L, et al. Safety and efficacy of anti-PCSK9 antibodies: a meta-analysis of 25 randomized, controlled trials. BMC Med 2015; 13:123.
Sabatine MS, Giugliano RP, Wiviott SD, et al; OSLER Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
Robinson JG, Farnier M, Krempf M, et al; ODYSSEY LONG TERM Investigators. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1489–1499.
Navarese EP, Kolodziejczak M, Schulze V, et al. Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adults with hypercholesterolemia: a systematic review and meta-analysis. Ann Intern Med 2015; 163:40–51.
Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk (FOURIER). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01764633. Updated July 26, 2016. Accessed October 23, 2016.
ODYSSEY Outcomes: Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab. U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01663402. Updated October 23, 2016. Accessed September 13, 2016.
The Evaluation of Bococizumab (PF-04950615;RN316) in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects (SPIRE-1). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01975376. Updated September 22, 2016. Accessed October 23, 2016.
The Evaluation of Bococizumab (PF-04950615; RN316) in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects (SPIRE-2). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01975389. Updated July 26, 2016. Accessed October 23, 2016.
Lloyd-Jones DM, Morris PB, Ballantyne CM, et al; Writing Committee. 2016 ACC expert consensus decision pathway on the role of non-statin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology task force on clinical expert consensus documents. J Am Coll Cardiol 2016; 68:92–125.
Khendi White, MD Fellow, Department of Cardiovascular Medicine, Cleveland Clinic
Chaitra Mohan, MD Resident, Department of Internal Medicine, Cleveland Clinic
Michael Rocco, MD Medical Director of Cardiac Rehabilitation and Stress Testing, Section of Preventive Cardiology; Staff, Section of Clinical Cardiology and Preventive Cardiology, Department of Cardiovascular Medicine, Cleveland Clinic
Correspondence: Michael Rocco, MD, Department of Cardiovascular Medicine, BD10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; roccom@ccf.org
Drs. White, Mohan, and Rocco reported no financial interests or relationships that pose a potential conflict of interest with this article.
Khendi White, MD Fellow, Department of Cardiovascular Medicine, Cleveland Clinic
Chaitra Mohan, MD Resident, Department of Internal Medicine, Cleveland Clinic
Michael Rocco, MD Medical Director of Cardiac Rehabilitation and Stress Testing, Section of Preventive Cardiology; Staff, Section of Clinical Cardiology and Preventive Cardiology, Department of Cardiovascular Medicine, Cleveland Clinic
Correspondence: Michael Rocco, MD, Department of Cardiovascular Medicine, BD10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; roccom@ccf.org
Drs. White, Mohan, and Rocco reported no financial interests or relationships that pose a potential conflict of interest with this article.
Author and Disclosure Information
Khendi White, MD Fellow, Department of Cardiovascular Medicine, Cleveland Clinic
Chaitra Mohan, MD Resident, Department of Internal Medicine, Cleveland Clinic
Michael Rocco, MD Medical Director of Cardiac Rehabilitation and Stress Testing, Section of Preventive Cardiology; Staff, Section of Clinical Cardiology and Preventive Cardiology, Department of Cardiovascular Medicine, Cleveland Clinic
Correspondence: Michael Rocco, MD, Department of Cardiovascular Medicine, BD10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; roccom@ccf.org
Drs. White, Mohan, and Rocco reported no financial interests or relationships that pose a potential conflict of interest with this article.
Statin therapy has been shown to substantially reduce adverse events associated with low-density-lipoprotein cholesterol (LDL-C) and cardiovascular disease (CVD). Statins alone are often not adequate to achieve treatment goals, and residual CVD risk remains high. Combination therapies of statins with ezetimibe and resins to further lower LDL-C, fibrates and omega 3 fatty acids to lower triglycerides, and niacin to lower both and raise high-density-liproprotein cholesterol are available, but additional risk reduction has not been consistently demonstrated in clinical trials.
The link between atherogenic lipoproteins and CVD is strong, and the need to develop therapies in addition to statins to substantially and safely reduce LDL-C is a priority. The association of reduced proprotein convertase subtilisin/kexin type 9 (PCSK9) activity with reduced LDL-C and CVD events has led to the rapid development and approval of monoclonal antibody therapies to inhibit PCSK9.
In this review, we discuss trials of these therapies that have shown durable reductions in LDL-C of more than 50%, with acceptable tolerability. Now that PCSK9 inhibitors are approved by the US Food and Drug Administration (FDA), extended data are needed as to long-term tolerability, safety, and efficacy of these agents and, most importantly, demonstration of additional reduction in CVD events.
A CASE FOR ADDITIONAL THERAPIES
CVD is the leading cause of morbidity and death in the United States, responsible for one in four deaths. Hyperlipidemia and, specifically, elevated LDL-C have been found to be important drivers of atherosclerosis and, in turn, adverse cardiovascular (CV) events. Likewise, numerous observational and clinical trials have shown that reducing LDL-C, particularly with statins, decreases CVD events.1–4 More aggressive lowering with higher doses or more intensive statin therapy further reduces rates of adverse outcomes.3,4 In addition, the pleiotropic effects of statins imply that not all of their benefits are derived from LDL-C lowering alone.5 Consequently, it is now standard practice to use statins at the highest tolerable dose to reach target LDL-C levels and prevent CV events in high-risk patients with CVD or multiple coronary artery disease risk factors, regardless of the LDL-C levels.6,7
The American College of Cardiology (ACC) and the American Heart Association released cholesterol guidelines in 2013 that recommend a risk-based approach for statin therapy rather than targeting specific LDL-C levels.6 Although this evidence-based approach may better conform to clinical trials, the debate that lower LDL-C targets will further prevent CVD continues.
Indeed, it appears that lower is better, as demonstrated by the IMPROVE-IT trial.8 Although the control group receiving simvastatin monotherapy had low LDL-C levels (mean, 69.9 mg/dL; 1.8 mmol/L), the experimental group receiving simvastatin plus ezetimibe achieved even lower levels (mean, 53.2 mg/dL;1.4 mmol/L) after 1 year of therapy and had a significantly lower composite primary end point of CV death, major coronary event, or nonfatal stroke at 7 years (34.7% for simvastatin monotherapy vs 32.7% for combined therapy).9 Furthermore, the event-rate reduction with the addition of ezetimibe was the same as the average predicted by the Cholesterol Treatment Trialists’ meta-analysis: an LDL-C reduction of 1 mmol/L (38.6 mg/dL) yields a 23% risk reduction in major coronary events over 5 years.10 Although only a modest absolute reduction in outcomes, it supports the notion that further reduction of LDL-C levels by more potent therapies may offer greater benefit.
There is strong evidence that statin therapy reduces the risk of developing CVD in patients with or without a previous atherosclerotic event; however, residual CVD risk remains even for those on therapy. A contributing factor to this residual risk is that many statin-treated patients have insufficient response or intolerance and do not achieve adequate LDL-C reductions.
There are three clinically important patient populations who are inadequately managed with current therapies and remain at high risk of subsequent CV events; these are patients who would benefit from additional therapies.
1. Patients with familial hypercholesterolemia (FH). This is the most common genetic disorder in the world, yet it is frequently undiagnosed and untreated. Due to high baseline cholesterol levels, achieving LDL-C treatment goals is challenging.
The prevalence may be closer to 1:200 to 1:250 rather than the often quoted 1:500.11
Fewer than 12% of patients with heterzygous FH achieve the LDL-C goal of < 100 mg/dL with maximal statin treatment alone or with a second agent.12
2. Patients with hyperlipidemia not due to FH who are at elevated CV risk and undertreated. In US and European surveys, between 50% and 60% of patients receiving statins with or without other therapies failed to reach LDL-C reduction goals.13
Variation in response to statin treatment between individuals may be considerable.
Poor adherence to statin therapy is common.
3. Patients with side effects to statins, particularly muscle symptoms that prevent statin use or substantially limit the dose.
Although the incidence of myopathy is low (< 0.1%) and rhabdomyolysis is even less common, observational studies suggest that 10% to 20% of patients may limit statin use due to muscle-associated complaints including muscle aching, cramps, or weakness.14
Side effects may be dose-dependent, limiting the use of the high-intensity statin doses that are frequently necessary to achieve LDL-C goals.
Consequently, there is great interest in developing therapies beyond statins that may further reduce CV events. However, treatments other than ezetimibe for further management of hyperlipidemia and risk reduction have failed to demonstrate consistent benefit when added to statin therapy.15–19 The largest studies were with niacin and fibrates. Unfortunately, most trials demonstrated no overall outcomes benefit or only benefits in subgroup analyses, leaving the door open to other pharmacologic interventions.
Studies with the cholesterol ester transfer protein (CETP) inhibitor torcetrapib, in combination with statin therapy, actually demonstrated an overall increase in all-cause mortality in the treatment group.20 Two large outcome trials of the CETP inhibitors dalcetrapib and evacetrapib were stopped after interim analysis predicted no benefit. Although drugs such as lomitapide (a microsomal triglyceride transfer protein inhibitor) and mipomersen (an antisense oligonucleotide inhibitor of ApoB-100 synthesis) can lower LDL-C by reducing ApoB synthesis,21 they are approved only in the small population of individuals with homozygous FH and liver toxicity and side effects are a concern.
Accordingly, current cholesterol management guidelines continue to offer LDL-C as the main target of lipid-modifying therapy, with statins as the primary treatment choice. The desire to build on statin therapy to prevent further progression of atherosclerosis and clinical CVD has encouraged continued focus on strategies to lower LDL-C to even greater extents.
Fortunately for practitioners, for the first time since lovastatin was approved in 1987, there is a new therapy approved by the FDA that significantly lowers LDL-C and, potentially, improves CV outcomes—the proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors. This review will focus on the PCSK9 inhibitors, a novel therapeutic class that reduces LDL-C through increased hepatic clearance. These drugs are rapidly emerging as an ideal adjunctive therapy to statins for patients at the highest risk and as a highly efficacious alternate therapy in patients intolerant of statins.
PCSK9 INHIBITORS: DISCOVERY, MECHANISM, AND THERAPEUTIC INTERVENTIONS
Two PCSK9 inhibitors have received FDA approval: alirocumab (Praluent) and evolocumab (Repatha). Among new molecular entities for clinical use, PCSK9 inhibitor therapies had one of the shortest durations from discovery to development and approval.
Mutations in the PCSK9 gene associated with autosomal dominant hypercholesterolemia were first identified in 2003 in a French family.22 The PCSK9 protein is now known to be a secreted enzymatic serine protease that is primarily synthesized in the liver and binds to the LDL receptor (LDL-R)/LDL-C complex on the surface of hepatocytes, marking the receptor for lysosomal degradation rather than recycling to the cell surface. Thus, it reduces the quantity of LDL-R that is available to remove LDL-C from circulation.23 As a result, higher levels of PCSK9 are associated with higher levels of plasma LDL-C.
The clinical importance of PCSK9 in regulating LDL-C is supported by observed mutations and polymorphisms. Gain-of-function mutations that increase the activity of PCSK9 have been shown to be associated with elevated LDL-C, premature CVD, and myocardial infarction (MI).24 Conversely, loss-of-function mutations (heterozygotes found in 1% to 3% of the population) result in decreased activity of PCSK9, lower LDL-C, and lower incidence of CVD (Table 1).25–29 These observations, combined with data showing that homozygote loss-of-function individuals with very low LDL-C were generally very healthy, sparked interest in developing inhibition of PCSK9 activity as a therapeutic strategy for hyperlipidemia.
Multiple pharmacologic developments are aimed at inhibiting PCSK9, with many compounds in clinical trials. The approaches include gene silencing with loss-of-function mutations, synthetic peptides, oral small molecules, and monoclonal antibodies. Gene silencing was first observed in 2007 when administration of antisense oligonucleotides targeted to selectively inhibit PCSK9 mRNA was found to up-regulate LDL-R, thereby decreasing serum levels of LDL-C.30
The first study to establish the role of synthetic peptides in PCSK9 inhibition was performed in 2008. In this study, the epidermal growth factor-like A synthetic peptide blocked the interaction between PCSK9 and LDL-R, thereby decreasing the degradation of LDL-R and preserving LDL uptake.31 Although studies are limited, synthetic peptides remain an area of great interest given their promising effects on lipid metabolism. Recently, a synthetic PCSK9-binding adnectin derived from the human fibronectin known as BMS-962476, had favorable results in a phase 1 clinical trial. An RNA interference molecule, subcutaneous ALN-PSC, inhibits PCSK9 gene expression by causing destruction of messenger RNA, thus inhibiting PCSK9 synthesis (Table 2).32
PCSK9 INHIBITORS: CLINICAL TRIALS
Subcutaneously administered monoclonal antibodies targeting PCSK9 currently are the only PCSK9 inhibitors FDA-approved for clinical use. The first study to demonstrate efficacy in enhancing uptake of serum LDL-C was performed in 2009.33 Multiple phase 1 and 2 studies soon followed, demonstrating acceptable safety and 50% to 70% reductions in LDL-C at upper-dose titrations.34 Additionally, there were significant reductions in total cholesterol, ApoB, triglycerides, and lipoprotein(a).
These early developments paved the way for larger phase 3 trials (Table 3).35–48 The PCSK9 inhibitors evolocumab and alirocumab have been shown in multiple phase 3 clinical trials to achieve a consistent dose-dependent 50% to 60% reduction in LDL-C across a broad range of CVD risk, pretreatment LDL-C levels, and background therapy: monotherapy (MENDEL-2, ODYSSEY COMBO I),35,44 added to statin therapy (LAPLACE-2, ODYSSEY CHOICE I),38,46 and in individuals with heterozygous FH (RUTHERFORD-2, ODYSSEY-FH).37,42 Trials with bococizumab are under way.
The GAUSS-2 clinical trial (Goal Achievement after Utilizing an Anti-PCSK9 Antibody in Statin Intolerant Subjects-2) demonstrated similar efficacy in reducing LDL-C in patients with clinically assessed statin intolerance due to muscle-related adverse symptoms.39 In GAUSS-3, patients were first identified as being statin intolerant secondary to muscle-associated symptoms based on a randomized, crossover trial of atorvastatin vs placebo.40 The 43% of participants who experienced intolerable muscle-related symptoms on the statin but not on placebo were then randomized to evolocumab vs ezetimibe. Results showed significant reduction in LDL-C in the evolocumab group (52.8%) compared with the ezetimibe group (16.7%). Additionally, among patients with muscle symptoms on statin therapy, PCSK9 therapy was discontinued for muscle symptoms in only 0.7% of evolocumab recipients and 6.8% of ezetimibe recipients.
Overall, the PCSK9 inhibitors are generally well tolerated with injection site reactions being the most common side effect. A meta-analysis published in 2015 of 25 trials including more than 12,000 patients treated with evolocumab and alirocumab reported no significant difference in adverse events or safety outcomes vs placebo or ezetimibe.49 Antidrug binding or neutralizing antibody production to these agents, thus far, has not been shown to be an issue. Additional analyses have not indicated an adverse effect on gonadal hormone levels or increased incidence of new-onset diabetes.
Two studies published in 2015 offer insight into longer term durability and safety as well as potential CVD outcome benefit (Table 4)50,51:
OSLER-1 and 2: Open-Label Study of Long-Term Evaluation against LDL-Cholesterol (OSLER) trials—evolocumab trial;50
ODYSSEY long term: Long-Term Safety and Tolerability of Alirocumab in High Cardiovascular Risk Patients with Hypercholesterolemia Not Adequately Controlled with Their Lipid Modifying Therapy—alirocumab trial.51
The OSLER trials reported durable LDL-C reductions of 61% and the ODYSSEY trial reported a LDL-C reduction of 62%.50,51 In both studies, the overall occurrence of adverse events was similar to placebo, but both reported a higher rate of neurocognitive effects in the active treatment groups (evolocumab 0.9% vs 0.3% for standard therapy; alirocumab 1.2% vs 0.5% for placebo). It must be noted that although the absolute rate of neurocognitive adverse events is low, it is unclear if these events were related to the drugs themselves or to extreme lowering of LDL-C. Nevertheless, the FDA has raised concerns about neurocognitive events. A sub-study of the ongoing FOURIER trial with evolocumab—EBBINGHAUS—is expected to address this concern.
Figure 1. Effect of PCSK9 inhibitors on cardiovascular events.50,51
In addition, analyses of CV events showed that the PCSK9 inhibitors effectively cut the CV rate in half in both studies (Figure 1).50,51 In the OSLER trials,50 evolocumab recipients had 53% reduction in major CV events (0.95% vs 2.18% in the standard therapy group; P = .003). In ODYSSEY,51 alirocumab recipients had a 48% reduction in major CV events (1.7% vs 3.3% for placebo; P = .02). Furthermore, a 2015 meta-analysis of 24 phase 2 and 3 trials reported a statistically significant 55% reduction in all-cause mortality and 50% reduction in CV mortality with PCSK9 inhibitors.52
For many reasons including short length of follow-up, study design, and small numbers of outcome events, the OSLER and ODYSSEY studies, although enticing, are exploratory and hypothesis-generating only and results need to be interpreted with caution. Nevertheless, they have set the stage for ongoing prospective randomized outcome trials that are studying the CV effects and tolerability of PCSK9 inhibitors over a longer time frame. These include the following trials.
The Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects With Elevated Risk (FOURIER) is an ongoing trial with the primary end point of CV death, MI, hospitalization for unstable angina, stroke, or coronary revascularization in high-risk patients receiving evolocumab or placebo.53
The ODYSSEY trial is examining the effect of alirocumab vs placebo on the composite primary endpoint of coronary heart disease death, non-fatal MI, fatal and nonfatal ischemic stroke, and unstable angina requiring hospitalization in patients who have had an acute coronary syndrome event during the previous 4 to 52 weeks.54
The Evaluation of Bococizumab in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects (SPIRE) trials are investigating the effect of bococizumab, a third PCSK9 “humanized” monoclonal antibody, vs placebo in reducing death, MI, stroke, or unstable angina in patients at high-risk of CVD who are receiving standard lipid-lowering therapy with LDL-C > 70 mg/dL (1.8 mmol/L) (SPIRE-1) or > 100 mg/dL (2.6 mmol/L) (SPIRE-2).55,56
Because these outcome trials are attempting to enroll more than 70,000 patients and are event driven, it is difficult to predict when they will be completed (Table 5).53–56 However, recent estimates indicate completion of at least one trial by the end of 2016 or early 2017, with interim analyses of others expected at that time. It is hoped that they will answer the all-important question of whether PCSK9 inhibitors are associated with further CV event reduction benefit.
CURRENT FDA INDICATIONS AND GUIDELINES
The two PCSK9 inhibitors approved by the FDA—alirocumab (subcutaneous 75 mg every 2 weeks up titrated to 150 mg) and evolocumab (subcutaneous 140 mg every 2 weeks or 420 mg every 4 weeks)—are both indicated for use with statins in patients with heterozygous FH or known atherosclerotic CVD who require further reduction in LDL-C levels despite lifestyle interventions and use of maximally tolerated statins. Evolocumab has also been approved for use in patients with homozygous FH.
Although PCSK9 inhibitors are not specifically approved for patients unable to tolerate statins, the results of GAUSS-3, which documented that statin intolerance is a real, definable entity and very responsive to PCSK9 inhibition, makes these drugs promising agents for patients intolerant of statins and, thus, unable to benefit from high-intensity stain therapy.
In April 2016, the ACC released a clinical consensus update to their 2013 cholesterol guidelines, which is their first recommendation specifically addressing the use of non-statin therapies, including the newer PCSK9 inhibitors.57 For high-risk patients with clinical atherosclerotic CVD or LDL-C > 190 and failure to achieve at least a 50% reduction in LDL-C on maximally tolerated statin, non-statins may be considered. Ezetimibe, given its safety and tolerability, should be the first additional medication added. Bile acid sequestrants may be used as a second-line therapy if ezetimibe is not tolerated and triglycerides are not elevated. If therapy goals are not met on maximally tolerated statin and ezetimibe, either approved PCSK9 inhibitor can be added or used to replace ezetimibe. The document also specifies that given the lack of long-term safety and efficacy data on the PCSK9 inhibitors, they are not recommended for use in primary prevention patients in the absence of FH.
CONCLUSION
Although statin therapy has been shown to substantially reduce LDL-C and CVD adverse events, there remains a high rate of inadequate goal achievement and residual CVD risk in patients receiving statins. Combination therapies with ezetimibe and resins to further lower LDL-C, fibrates and omega 3 fatty acids to lower triglycerides, and niacin to lower both and raise high-density-liproprotein cholesterol are available, even though additional CV risk reduction is minimal or elusive when these drugs are added to statin therapy.
The link between atherogenic lipoproteins and CVD is strong, and the need to develop therapies in addition to statins to substantially and safely reduce LDL-C remains a priority. The association of reduced PCSK9 activity with reduced LDL-C and CV events has led to rapid development and approval of monoclonal antibody therapies to inhibit PCSK9. In trials, these therapies have shown substantial and durable reductions in LDL-C of more than 50%, with acceptable tolerability. Now that PCSK9 inhibitors are approved by the FDA, extended data about long-term tolerability, safety, and efficacy and, most importantly, demonstration of additional reduction in CVD events are needed. It is hoped that the long-term ongoing trials will provide these data.
For the immediate future, statin therapy will continue to be the cornerstone of lipid and CVD risk management based on their low generic cost, proven CVD risk reduction, and clinicians’ comfort with their use. However, the reliable efficacy of PCSK9 inhibitors and the fact that statin therapy itself increases PCSK9 activity makes the addition of PCSK9 inhibitors to statins an attractive approach in high-risk patients failing to reach LDL-C treatment goals.
Although current indications are limited, there are patients at high CVD risk who would be appropriate candidates for these therapies. These include patients with the following:
FH with lifetime burden of elevated LDL-C and associated low likelihood of achieving optimal LDL-C control on current available therapies
Complete or partial statin intolerance with high-intensity statin dosing limited by side effects
High CV risk who are not at LDL-C goal on current therapies.
Now that the first therapies are available, practitioners can expect newer approaches to tackle PCSK9-mediated LDL-C reduction. Bococizumab is lagging in phase 3 trials, but the SPIRE program is moving forward with special population studies expected to conclude in 2016 and simultaneous long-term outcomes trials. Other PCSK9 inhibitors being investigated include agents with more durable effect requiring less frequent injections, RNA-interference therapies, vaccinations, antisense therapies, and oral formulations.
The PCSK9 inhibitors hold promise as an adjunct to statin therapy. Their eventual clinical role will depend on a balance between substantial LDL-C reductions, long-term safety, tolerability, and reduction in CVD events vs the cost (estimated at $14,000 a year), access from payers, acceptance of injectable therapies, and magnitude of incremental benefit when added to current therapies. Nevertheless, initial clinical trial data are encouraging and these drugs may be an important addition to the therapeutic armamentarium against CVD.
Statin therapy has been shown to substantially reduce adverse events associated with low-density-lipoprotein cholesterol (LDL-C) and cardiovascular disease (CVD). Statins alone are often not adequate to achieve treatment goals, and residual CVD risk remains high. Combination therapies of statins with ezetimibe and resins to further lower LDL-C, fibrates and omega 3 fatty acids to lower triglycerides, and niacin to lower both and raise high-density-liproprotein cholesterol are available, but additional risk reduction has not been consistently demonstrated in clinical trials.
The link between atherogenic lipoproteins and CVD is strong, and the need to develop therapies in addition to statins to substantially and safely reduce LDL-C is a priority. The association of reduced proprotein convertase subtilisin/kexin type 9 (PCSK9) activity with reduced LDL-C and CVD events has led to the rapid development and approval of monoclonal antibody therapies to inhibit PCSK9.
In this review, we discuss trials of these therapies that have shown durable reductions in LDL-C of more than 50%, with acceptable tolerability. Now that PCSK9 inhibitors are approved by the US Food and Drug Administration (FDA), extended data are needed as to long-term tolerability, safety, and efficacy of these agents and, most importantly, demonstration of additional reduction in CVD events.
A CASE FOR ADDITIONAL THERAPIES
CVD is the leading cause of morbidity and death in the United States, responsible for one in four deaths. Hyperlipidemia and, specifically, elevated LDL-C have been found to be important drivers of atherosclerosis and, in turn, adverse cardiovascular (CV) events. Likewise, numerous observational and clinical trials have shown that reducing LDL-C, particularly with statins, decreases CVD events.1–4 More aggressive lowering with higher doses or more intensive statin therapy further reduces rates of adverse outcomes.3,4 In addition, the pleiotropic effects of statins imply that not all of their benefits are derived from LDL-C lowering alone.5 Consequently, it is now standard practice to use statins at the highest tolerable dose to reach target LDL-C levels and prevent CV events in high-risk patients with CVD or multiple coronary artery disease risk factors, regardless of the LDL-C levels.6,7
The American College of Cardiology (ACC) and the American Heart Association released cholesterol guidelines in 2013 that recommend a risk-based approach for statin therapy rather than targeting specific LDL-C levels.6 Although this evidence-based approach may better conform to clinical trials, the debate that lower LDL-C targets will further prevent CVD continues.
Indeed, it appears that lower is better, as demonstrated by the IMPROVE-IT trial.8 Although the control group receiving simvastatin monotherapy had low LDL-C levels (mean, 69.9 mg/dL; 1.8 mmol/L), the experimental group receiving simvastatin plus ezetimibe achieved even lower levels (mean, 53.2 mg/dL;1.4 mmol/L) after 1 year of therapy and had a significantly lower composite primary end point of CV death, major coronary event, or nonfatal stroke at 7 years (34.7% for simvastatin monotherapy vs 32.7% for combined therapy).9 Furthermore, the event-rate reduction with the addition of ezetimibe was the same as the average predicted by the Cholesterol Treatment Trialists’ meta-analysis: an LDL-C reduction of 1 mmol/L (38.6 mg/dL) yields a 23% risk reduction in major coronary events over 5 years.10 Although only a modest absolute reduction in outcomes, it supports the notion that further reduction of LDL-C levels by more potent therapies may offer greater benefit.
There is strong evidence that statin therapy reduces the risk of developing CVD in patients with or without a previous atherosclerotic event; however, residual CVD risk remains even for those on therapy. A contributing factor to this residual risk is that many statin-treated patients have insufficient response or intolerance and do not achieve adequate LDL-C reductions.
There are three clinically important patient populations who are inadequately managed with current therapies and remain at high risk of subsequent CV events; these are patients who would benefit from additional therapies.
1. Patients with familial hypercholesterolemia (FH). This is the most common genetic disorder in the world, yet it is frequently undiagnosed and untreated. Due to high baseline cholesterol levels, achieving LDL-C treatment goals is challenging.
The prevalence may be closer to 1:200 to 1:250 rather than the often quoted 1:500.11
Fewer than 12% of patients with heterzygous FH achieve the LDL-C goal of < 100 mg/dL with maximal statin treatment alone or with a second agent.12
2. Patients with hyperlipidemia not due to FH who are at elevated CV risk and undertreated. In US and European surveys, between 50% and 60% of patients receiving statins with or without other therapies failed to reach LDL-C reduction goals.13
Variation in response to statin treatment between individuals may be considerable.
Poor adherence to statin therapy is common.
3. Patients with side effects to statins, particularly muscle symptoms that prevent statin use or substantially limit the dose.
Although the incidence of myopathy is low (< 0.1%) and rhabdomyolysis is even less common, observational studies suggest that 10% to 20% of patients may limit statin use due to muscle-associated complaints including muscle aching, cramps, or weakness.14
Side effects may be dose-dependent, limiting the use of the high-intensity statin doses that are frequently necessary to achieve LDL-C goals.
Consequently, there is great interest in developing therapies beyond statins that may further reduce CV events. However, treatments other than ezetimibe for further management of hyperlipidemia and risk reduction have failed to demonstrate consistent benefit when added to statin therapy.15–19 The largest studies were with niacin and fibrates. Unfortunately, most trials demonstrated no overall outcomes benefit or only benefits in subgroup analyses, leaving the door open to other pharmacologic interventions.
Studies with the cholesterol ester transfer protein (CETP) inhibitor torcetrapib, in combination with statin therapy, actually demonstrated an overall increase in all-cause mortality in the treatment group.20 Two large outcome trials of the CETP inhibitors dalcetrapib and evacetrapib were stopped after interim analysis predicted no benefit. Although drugs such as lomitapide (a microsomal triglyceride transfer protein inhibitor) and mipomersen (an antisense oligonucleotide inhibitor of ApoB-100 synthesis) can lower LDL-C by reducing ApoB synthesis,21 they are approved only in the small population of individuals with homozygous FH and liver toxicity and side effects are a concern.
Accordingly, current cholesterol management guidelines continue to offer LDL-C as the main target of lipid-modifying therapy, with statins as the primary treatment choice. The desire to build on statin therapy to prevent further progression of atherosclerosis and clinical CVD has encouraged continued focus on strategies to lower LDL-C to even greater extents.
Fortunately for practitioners, for the first time since lovastatin was approved in 1987, there is a new therapy approved by the FDA that significantly lowers LDL-C and, potentially, improves CV outcomes—the proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors. This review will focus on the PCSK9 inhibitors, a novel therapeutic class that reduces LDL-C through increased hepatic clearance. These drugs are rapidly emerging as an ideal adjunctive therapy to statins for patients at the highest risk and as a highly efficacious alternate therapy in patients intolerant of statins.
PCSK9 INHIBITORS: DISCOVERY, MECHANISM, AND THERAPEUTIC INTERVENTIONS
Two PCSK9 inhibitors have received FDA approval: alirocumab (Praluent) and evolocumab (Repatha). Among new molecular entities for clinical use, PCSK9 inhibitor therapies had one of the shortest durations from discovery to development and approval.
Mutations in the PCSK9 gene associated with autosomal dominant hypercholesterolemia were first identified in 2003 in a French family.22 The PCSK9 protein is now known to be a secreted enzymatic serine protease that is primarily synthesized in the liver and binds to the LDL receptor (LDL-R)/LDL-C complex on the surface of hepatocytes, marking the receptor for lysosomal degradation rather than recycling to the cell surface. Thus, it reduces the quantity of LDL-R that is available to remove LDL-C from circulation.23 As a result, higher levels of PCSK9 are associated with higher levels of plasma LDL-C.
The clinical importance of PCSK9 in regulating LDL-C is supported by observed mutations and polymorphisms. Gain-of-function mutations that increase the activity of PCSK9 have been shown to be associated with elevated LDL-C, premature CVD, and myocardial infarction (MI).24 Conversely, loss-of-function mutations (heterozygotes found in 1% to 3% of the population) result in decreased activity of PCSK9, lower LDL-C, and lower incidence of CVD (Table 1).25–29 These observations, combined with data showing that homozygote loss-of-function individuals with very low LDL-C were generally very healthy, sparked interest in developing inhibition of PCSK9 activity as a therapeutic strategy for hyperlipidemia.
Multiple pharmacologic developments are aimed at inhibiting PCSK9, with many compounds in clinical trials. The approaches include gene silencing with loss-of-function mutations, synthetic peptides, oral small molecules, and monoclonal antibodies. Gene silencing was first observed in 2007 when administration of antisense oligonucleotides targeted to selectively inhibit PCSK9 mRNA was found to up-regulate LDL-R, thereby decreasing serum levels of LDL-C.30
The first study to establish the role of synthetic peptides in PCSK9 inhibition was performed in 2008. In this study, the epidermal growth factor-like A synthetic peptide blocked the interaction between PCSK9 and LDL-R, thereby decreasing the degradation of LDL-R and preserving LDL uptake.31 Although studies are limited, synthetic peptides remain an area of great interest given their promising effects on lipid metabolism. Recently, a synthetic PCSK9-binding adnectin derived from the human fibronectin known as BMS-962476, had favorable results in a phase 1 clinical trial. An RNA interference molecule, subcutaneous ALN-PSC, inhibits PCSK9 gene expression by causing destruction of messenger RNA, thus inhibiting PCSK9 synthesis (Table 2).32
PCSK9 INHIBITORS: CLINICAL TRIALS
Subcutaneously administered monoclonal antibodies targeting PCSK9 currently are the only PCSK9 inhibitors FDA-approved for clinical use. The first study to demonstrate efficacy in enhancing uptake of serum LDL-C was performed in 2009.33 Multiple phase 1 and 2 studies soon followed, demonstrating acceptable safety and 50% to 70% reductions in LDL-C at upper-dose titrations.34 Additionally, there were significant reductions in total cholesterol, ApoB, triglycerides, and lipoprotein(a).
These early developments paved the way for larger phase 3 trials (Table 3).35–48 The PCSK9 inhibitors evolocumab and alirocumab have been shown in multiple phase 3 clinical trials to achieve a consistent dose-dependent 50% to 60% reduction in LDL-C across a broad range of CVD risk, pretreatment LDL-C levels, and background therapy: monotherapy (MENDEL-2, ODYSSEY COMBO I),35,44 added to statin therapy (LAPLACE-2, ODYSSEY CHOICE I),38,46 and in individuals with heterozygous FH (RUTHERFORD-2, ODYSSEY-FH).37,42 Trials with bococizumab are under way.
The GAUSS-2 clinical trial (Goal Achievement after Utilizing an Anti-PCSK9 Antibody in Statin Intolerant Subjects-2) demonstrated similar efficacy in reducing LDL-C in patients with clinically assessed statin intolerance due to muscle-related adverse symptoms.39 In GAUSS-3, patients were first identified as being statin intolerant secondary to muscle-associated symptoms based on a randomized, crossover trial of atorvastatin vs placebo.40 The 43% of participants who experienced intolerable muscle-related symptoms on the statin but not on placebo were then randomized to evolocumab vs ezetimibe. Results showed significant reduction in LDL-C in the evolocumab group (52.8%) compared with the ezetimibe group (16.7%). Additionally, among patients with muscle symptoms on statin therapy, PCSK9 therapy was discontinued for muscle symptoms in only 0.7% of evolocumab recipients and 6.8% of ezetimibe recipients.
Overall, the PCSK9 inhibitors are generally well tolerated with injection site reactions being the most common side effect. A meta-analysis published in 2015 of 25 trials including more than 12,000 patients treated with evolocumab and alirocumab reported no significant difference in adverse events or safety outcomes vs placebo or ezetimibe.49 Antidrug binding or neutralizing antibody production to these agents, thus far, has not been shown to be an issue. Additional analyses have not indicated an adverse effect on gonadal hormone levels or increased incidence of new-onset diabetes.
Two studies published in 2015 offer insight into longer term durability and safety as well as potential CVD outcome benefit (Table 4)50,51:
OSLER-1 and 2: Open-Label Study of Long-Term Evaluation against LDL-Cholesterol (OSLER) trials—evolocumab trial;50
ODYSSEY long term: Long-Term Safety and Tolerability of Alirocumab in High Cardiovascular Risk Patients with Hypercholesterolemia Not Adequately Controlled with Their Lipid Modifying Therapy—alirocumab trial.51
The OSLER trials reported durable LDL-C reductions of 61% and the ODYSSEY trial reported a LDL-C reduction of 62%.50,51 In both studies, the overall occurrence of adverse events was similar to placebo, but both reported a higher rate of neurocognitive effects in the active treatment groups (evolocumab 0.9% vs 0.3% for standard therapy; alirocumab 1.2% vs 0.5% for placebo). It must be noted that although the absolute rate of neurocognitive adverse events is low, it is unclear if these events were related to the drugs themselves or to extreme lowering of LDL-C. Nevertheless, the FDA has raised concerns about neurocognitive events. A sub-study of the ongoing FOURIER trial with evolocumab—EBBINGHAUS—is expected to address this concern.
Figure 1. Effect of PCSK9 inhibitors on cardiovascular events.50,51
In addition, analyses of CV events showed that the PCSK9 inhibitors effectively cut the CV rate in half in both studies (Figure 1).50,51 In the OSLER trials,50 evolocumab recipients had 53% reduction in major CV events (0.95% vs 2.18% in the standard therapy group; P = .003). In ODYSSEY,51 alirocumab recipients had a 48% reduction in major CV events (1.7% vs 3.3% for placebo; P = .02). Furthermore, a 2015 meta-analysis of 24 phase 2 and 3 trials reported a statistically significant 55% reduction in all-cause mortality and 50% reduction in CV mortality with PCSK9 inhibitors.52
For many reasons including short length of follow-up, study design, and small numbers of outcome events, the OSLER and ODYSSEY studies, although enticing, are exploratory and hypothesis-generating only and results need to be interpreted with caution. Nevertheless, they have set the stage for ongoing prospective randomized outcome trials that are studying the CV effects and tolerability of PCSK9 inhibitors over a longer time frame. These include the following trials.
The Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects With Elevated Risk (FOURIER) is an ongoing trial with the primary end point of CV death, MI, hospitalization for unstable angina, stroke, or coronary revascularization in high-risk patients receiving evolocumab or placebo.53
The ODYSSEY trial is examining the effect of alirocumab vs placebo on the composite primary endpoint of coronary heart disease death, non-fatal MI, fatal and nonfatal ischemic stroke, and unstable angina requiring hospitalization in patients who have had an acute coronary syndrome event during the previous 4 to 52 weeks.54
The Evaluation of Bococizumab in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects (SPIRE) trials are investigating the effect of bococizumab, a third PCSK9 “humanized” monoclonal antibody, vs placebo in reducing death, MI, stroke, or unstable angina in patients at high-risk of CVD who are receiving standard lipid-lowering therapy with LDL-C > 70 mg/dL (1.8 mmol/L) (SPIRE-1) or > 100 mg/dL (2.6 mmol/L) (SPIRE-2).55,56
Because these outcome trials are attempting to enroll more than 70,000 patients and are event driven, it is difficult to predict when they will be completed (Table 5).53–56 However, recent estimates indicate completion of at least one trial by the end of 2016 or early 2017, with interim analyses of others expected at that time. It is hoped that they will answer the all-important question of whether PCSK9 inhibitors are associated with further CV event reduction benefit.
CURRENT FDA INDICATIONS AND GUIDELINES
The two PCSK9 inhibitors approved by the FDA—alirocumab (subcutaneous 75 mg every 2 weeks up titrated to 150 mg) and evolocumab (subcutaneous 140 mg every 2 weeks or 420 mg every 4 weeks)—are both indicated for use with statins in patients with heterozygous FH or known atherosclerotic CVD who require further reduction in LDL-C levels despite lifestyle interventions and use of maximally tolerated statins. Evolocumab has also been approved for use in patients with homozygous FH.
Although PCSK9 inhibitors are not specifically approved for patients unable to tolerate statins, the results of GAUSS-3, which documented that statin intolerance is a real, definable entity and very responsive to PCSK9 inhibition, makes these drugs promising agents for patients intolerant of statins and, thus, unable to benefit from high-intensity stain therapy.
In April 2016, the ACC released a clinical consensus update to their 2013 cholesterol guidelines, which is their first recommendation specifically addressing the use of non-statin therapies, including the newer PCSK9 inhibitors.57 For high-risk patients with clinical atherosclerotic CVD or LDL-C > 190 and failure to achieve at least a 50% reduction in LDL-C on maximally tolerated statin, non-statins may be considered. Ezetimibe, given its safety and tolerability, should be the first additional medication added. Bile acid sequestrants may be used as a second-line therapy if ezetimibe is not tolerated and triglycerides are not elevated. If therapy goals are not met on maximally tolerated statin and ezetimibe, either approved PCSK9 inhibitor can be added or used to replace ezetimibe. The document also specifies that given the lack of long-term safety and efficacy data on the PCSK9 inhibitors, they are not recommended for use in primary prevention patients in the absence of FH.
CONCLUSION
Although statin therapy has been shown to substantially reduce LDL-C and CVD adverse events, there remains a high rate of inadequate goal achievement and residual CVD risk in patients receiving statins. Combination therapies with ezetimibe and resins to further lower LDL-C, fibrates and omega 3 fatty acids to lower triglycerides, and niacin to lower both and raise high-density-liproprotein cholesterol are available, even though additional CV risk reduction is minimal or elusive when these drugs are added to statin therapy.
The link between atherogenic lipoproteins and CVD is strong, and the need to develop therapies in addition to statins to substantially and safely reduce LDL-C remains a priority. The association of reduced PCSK9 activity with reduced LDL-C and CV events has led to rapid development and approval of monoclonal antibody therapies to inhibit PCSK9. In trials, these therapies have shown substantial and durable reductions in LDL-C of more than 50%, with acceptable tolerability. Now that PCSK9 inhibitors are approved by the FDA, extended data about long-term tolerability, safety, and efficacy and, most importantly, demonstration of additional reduction in CVD events are needed. It is hoped that the long-term ongoing trials will provide these data.
For the immediate future, statin therapy will continue to be the cornerstone of lipid and CVD risk management based on their low generic cost, proven CVD risk reduction, and clinicians’ comfort with their use. However, the reliable efficacy of PCSK9 inhibitors and the fact that statin therapy itself increases PCSK9 activity makes the addition of PCSK9 inhibitors to statins an attractive approach in high-risk patients failing to reach LDL-C treatment goals.
Although current indications are limited, there are patients at high CVD risk who would be appropriate candidates for these therapies. These include patients with the following:
FH with lifetime burden of elevated LDL-C and associated low likelihood of achieving optimal LDL-C control on current available therapies
Complete or partial statin intolerance with high-intensity statin dosing limited by side effects
High CV risk who are not at LDL-C goal on current therapies.
Now that the first therapies are available, practitioners can expect newer approaches to tackle PCSK9-mediated LDL-C reduction. Bococizumab is lagging in phase 3 trials, but the SPIRE program is moving forward with special population studies expected to conclude in 2016 and simultaneous long-term outcomes trials. Other PCSK9 inhibitors being investigated include agents with more durable effect requiring less frequent injections, RNA-interference therapies, vaccinations, antisense therapies, and oral formulations.
The PCSK9 inhibitors hold promise as an adjunct to statin therapy. Their eventual clinical role will depend on a balance between substantial LDL-C reductions, long-term safety, tolerability, and reduction in CVD events vs the cost (estimated at $14,000 a year), access from payers, acceptance of injectable therapies, and magnitude of incremental benefit when added to current therapies. Nevertheless, initial clinical trial data are encouraging and these drugs may be an important addition to the therapeutic armamentarium against CVD.
References
Scandinavian Simvastatin Survival Study Group. Randomized trial of cholesterol lowering in 4,444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
Sacks FM, Pfeiffer MA, Moye LA, et al; Cholesterol and Recurrent Events Trial Investigators. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial Investigators. N Engl J Med 1996; 335:1001–1009.
Schwartz GG, Olsson AG, Ezekowitz MD, et al; Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) Study Investigators. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
Lipid Research Clinics Program. The Lipid Research Clinics Coronary Primary Prevention Trial results (reduction in incidence of coronary heart disease). JAMA 1984; 251:351–364.
Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation 2004; 109(suppl 1):III39–III43.
Stone N, Robinson J, Lichtenstein A, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl 2):S1–S45.
Jacobson TA, Ito MK, Maki KC, et al. National lipid association recommendations for patient-centered management of dyslipidemia: part 1—full report. J Clin Lipidol 2015; 9:129–169.
Jarcho JA, Keaney JF Jr. Proof that lower is better–LDL cholesterol and IMPROVE-IT. N Engl J Med 2015; 372:2448–2450.
Cannon CP, Blazing MA, Giugliano RP, et al; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
de Ferranti S, Rodday AM, Mendelson M, et al. Prevalence of familial hypercholesterolemia in the 1999 to 2012 United States National Health and Nutrition Examination Surveys (NHANES). Circulation 2016; 133:1067–1072.
Perez de Isla L, Alonso R, Watts GF, et al; SAFEHEART investigators. Attainment of LDL-cholesterol treatment goals in patients with familial hypercholesterolemia: 5-year SAFEHEART registry follow-up. J Am Coll Cardiol 2016; 67:1278–1285.
Unni SK, Quek RGW, Biskupiak J, et al. Assessment of statin therapy, LDL-C levels, and cardiovascular events among high-risk patients in the United States. J Clin Lipidol 2016; 10:63–71.
Mammen AL, Amato AA. Statin myopathy: a review of recent progress. Curr Opin Rheum 2010; 22:644–650.
AIM-HIGH Investigators; Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255–2267.
HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J 2013; 34:1279–1291.
ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
Keech A, Simes RJ, Barter P, et al; FIELD study investigators. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomized controlled trial. Lancet 2005; 366:1849–1861.
Kaur N, Pandey A, Negi H, et al. Effect of HDL-raising drugs on cardiovascular outcomes: a systematic review and meta-regression. PLoS One 2014; 9:e94585.
Barter PJ, Caulfield M, Eriksson M, et al; ILLUMINATE investigators. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
Rader D, Kastelein J. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 2014; 129:1022–1032.
Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34:154–156.
Verbeek R, Stoekenbroek RM, Hovingh GK. PCSK9 inhibitors: novel therapeutic agents for the treatment of hypercholesterolemia. Eur J of Pharm 2015; 763(Pt A):38–47.
Steinberg D, Witztum JL. Inhibition of PCSK9: a powerful weapon for achieving ideal LDL cholesterol levels. Proc Natl Acad Sci USA 2009; 106:9546–9547.
Abifadel M, Rabès J-P, Devillers M, et al. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum Mutat 2009; 30:520–529.
Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
Benn M, Nordestgaard BG, Grande P, Schnohr P, Tybærg-Hansen A. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease. J Am Coll Cardiol 2010; 55:2833–2842.
Mortensen MB, Afzal S, Nordestgaard BG, Falk E. The high-density lipoprotein-adjusted SCORE model worsens SCORE-based risk classification in a contemporary population of 30,824 Europeans: the Copenhagen General Population Study. Eur Heart J 2015; 36:2446–2453.
Victor RG, Haley RW, Willett DL, et al. The Dallas Heart Study: a population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am J Cardiol 2004; 93:1473–1480.
Graham MJ, Lemonidis KM, Whipple CP, et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res 2007; 48:763–767.
Shan L, Pang L, Zhang R, Murgolo NJ, Lan H, Hedrick JA. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Comm 2008; 375:69–73.
Stein EA, Raal F. Reduction of low-density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Annu Rev Med 2014; 65:417–431.
Duff CJ, Scott MJ, Kirby IT, Hutchinson SE, Martin SL, Hooper NM. Antibody-mediated disruption of the interaction between PCSK9 and the low-density lipoprotein receptor. Biochem J 2009; 419:577–584.
Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Eng J Med 2012; 366:1108–1118.
Koren MJ, Lundqvist P, Bolognese M, et al; MENDEL-2 Investigators. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol 2014; 63:2531–2540.
Blom DJ, Hala T, Bolognese M, et al; DESCARTES investigators. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med 2014; 370:1809-1819.
Raal FJ, Stein EA, Dufour R, et al; RUTHERFORD-2 Investigators. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet 2014; 385:331–340.
Robinson JG, Nedergaard BS, Rogers WJ, et al; LAPLA C-2 Investigators. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA 2014; 311:1870–1882.
Stroes E, Colquhoun D, Sullivan D, et al; GAUSS-2 Investigators. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol 2014; 63:2541–2548.
Nissen SE, Stroes E, Dent-Acosta RE, et al; GAUSS-3 Investigators. Efficacy and tolerability of evolocumab vs ezetimibe in patients with muscle-related statin intolerance, the GAUSS-3 randomized clinical trial. JAMA 2016; 315:1580–1590.
Trial assessing long term use of PCSK9 inhibition in subjects with genetic LDL disorders (TAUSSIG). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT-1624142. Updated June 25, 2015. Accessed October 23, 2016.
Kastelein JJ, Ginsberg HN, Langslet G, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J 2015; 36:2996–3003.
Efficacy and safety of alirocumab (SAR236553/REGN727) versus placebo on top of lipid-modifying therapy in patients with heterozygous familial hypercholesterolemia; the ODYSSEY HIGH FH trial. U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01617655. Updated September 27, 2016. Accessed October 23, 2016.
Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: The ODYSSEY COMBO I study. Am Heart J 2015; 169:906–915.
Efficacy and Safety of Alirocumab (SAR236553/REGN727) Versus Ezetimibe on Top of Statin in High Cardiovascular Risk Patients With Hypercholesterolemia (ODYSSEY COMBO II). U.S. National Institutes of Health website. Updated June 23, 2016. https://clinicaltrials.gov/ct2/show/NCT01644188. Accessed October 23, 2016.
Roth EM, Moriarty P, Bergeron J, et al; ODYSSEY CHOICE I investigators. A phase III randomized trial evaluating alirocumab 300 mg every 4 weeks as monotherapy or add-on to statin: ODYSSEY CHOICE I. Atherosclerosis 2016, doi: 10.1016/j.atherosclerosis.2016.08.043.
Phase III Study To Evaluate Alirocumab in Patients With Hypercholesterolemia Not Treated With a Statin (ODYSSEY CHOICE II). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT02023879. Updated November 2, 2015. Accessed October 23, 2016.
Monthly and twice monthly subcutaneous dosing of PF-04950615 (RN316) in hypercholesterolemic subjects on a statin. U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/results?term=NCT01592240. Updated October 14, 2014. Accessed October 23, 2016.
Zhang XL, Zhu QQ, Zhu L, et al. Safety and efficacy of anti-PCSK9 antibodies: a meta-analysis of 25 randomized, controlled trials. BMC Med 2015; 13:123.
Sabatine MS, Giugliano RP, Wiviott SD, et al; OSLER Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
Robinson JG, Farnier M, Krempf M, et al; ODYSSEY LONG TERM Investigators. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1489–1499.
Navarese EP, Kolodziejczak M, Schulze V, et al. Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adults with hypercholesterolemia: a systematic review and meta-analysis. Ann Intern Med 2015; 163:40–51.
Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk (FOURIER). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01764633. Updated July 26, 2016. Accessed October 23, 2016.
ODYSSEY Outcomes: Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab. U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01663402. Updated October 23, 2016. Accessed September 13, 2016.
The Evaluation of Bococizumab (PF-04950615;RN316) in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects (SPIRE-1). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01975376. Updated September 22, 2016. Accessed October 23, 2016.
The Evaluation of Bococizumab (PF-04950615; RN316) in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects (SPIRE-2). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01975389. Updated July 26, 2016. Accessed October 23, 2016.
Lloyd-Jones DM, Morris PB, Ballantyne CM, et al; Writing Committee. 2016 ACC expert consensus decision pathway on the role of non-statin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology task force on clinical expert consensus documents. J Am Coll Cardiol 2016; 68:92–125.
References
Scandinavian Simvastatin Survival Study Group. Randomized trial of cholesterol lowering in 4,444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
Sacks FM, Pfeiffer MA, Moye LA, et al; Cholesterol and Recurrent Events Trial Investigators. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial Investigators. N Engl J Med 1996; 335:1001–1009.
Schwartz GG, Olsson AG, Ezekowitz MD, et al; Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) Study Investigators. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
Lipid Research Clinics Program. The Lipid Research Clinics Coronary Primary Prevention Trial results (reduction in incidence of coronary heart disease). JAMA 1984; 251:351–364.
Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation 2004; 109(suppl 1):III39–III43.
Stone N, Robinson J, Lichtenstein A, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl 2):S1–S45.
Jacobson TA, Ito MK, Maki KC, et al. National lipid association recommendations for patient-centered management of dyslipidemia: part 1—full report. J Clin Lipidol 2015; 9:129–169.
Jarcho JA, Keaney JF Jr. Proof that lower is better–LDL cholesterol and IMPROVE-IT. N Engl J Med 2015; 372:2448–2450.
Cannon CP, Blazing MA, Giugliano RP, et al; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
de Ferranti S, Rodday AM, Mendelson M, et al. Prevalence of familial hypercholesterolemia in the 1999 to 2012 United States National Health and Nutrition Examination Surveys (NHANES). Circulation 2016; 133:1067–1072.
Perez de Isla L, Alonso R, Watts GF, et al; SAFEHEART investigators. Attainment of LDL-cholesterol treatment goals in patients with familial hypercholesterolemia: 5-year SAFEHEART registry follow-up. J Am Coll Cardiol 2016; 67:1278–1285.
Unni SK, Quek RGW, Biskupiak J, et al. Assessment of statin therapy, LDL-C levels, and cardiovascular events among high-risk patients in the United States. J Clin Lipidol 2016; 10:63–71.
Mammen AL, Amato AA. Statin myopathy: a review of recent progress. Curr Opin Rheum 2010; 22:644–650.
AIM-HIGH Investigators; Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255–2267.
HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J 2013; 34:1279–1291.
ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
Keech A, Simes RJ, Barter P, et al; FIELD study investigators. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomized controlled trial. Lancet 2005; 366:1849–1861.
Kaur N, Pandey A, Negi H, et al. Effect of HDL-raising drugs on cardiovascular outcomes: a systematic review and meta-regression. PLoS One 2014; 9:e94585.
Barter PJ, Caulfield M, Eriksson M, et al; ILLUMINATE investigators. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
Rader D, Kastelein J. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 2014; 129:1022–1032.
Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34:154–156.
Verbeek R, Stoekenbroek RM, Hovingh GK. PCSK9 inhibitors: novel therapeutic agents for the treatment of hypercholesterolemia. Eur J of Pharm 2015; 763(Pt A):38–47.
Steinberg D, Witztum JL. Inhibition of PCSK9: a powerful weapon for achieving ideal LDL cholesterol levels. Proc Natl Acad Sci USA 2009; 106:9546–9547.
Abifadel M, Rabès J-P, Devillers M, et al. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum Mutat 2009; 30:520–529.
Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
Benn M, Nordestgaard BG, Grande P, Schnohr P, Tybærg-Hansen A. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease. J Am Coll Cardiol 2010; 55:2833–2842.
Mortensen MB, Afzal S, Nordestgaard BG, Falk E. The high-density lipoprotein-adjusted SCORE model worsens SCORE-based risk classification in a contemporary population of 30,824 Europeans: the Copenhagen General Population Study. Eur Heart J 2015; 36:2446–2453.
Victor RG, Haley RW, Willett DL, et al. The Dallas Heart Study: a population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am J Cardiol 2004; 93:1473–1480.
Graham MJ, Lemonidis KM, Whipple CP, et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res 2007; 48:763–767.
Shan L, Pang L, Zhang R, Murgolo NJ, Lan H, Hedrick JA. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Comm 2008; 375:69–73.
Stein EA, Raal F. Reduction of low-density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Annu Rev Med 2014; 65:417–431.
Duff CJ, Scott MJ, Kirby IT, Hutchinson SE, Martin SL, Hooper NM. Antibody-mediated disruption of the interaction between PCSK9 and the low-density lipoprotein receptor. Biochem J 2009; 419:577–584.
Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Eng J Med 2012; 366:1108–1118.
Koren MJ, Lundqvist P, Bolognese M, et al; MENDEL-2 Investigators. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol 2014; 63:2531–2540.
Blom DJ, Hala T, Bolognese M, et al; DESCARTES investigators. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med 2014; 370:1809-1819.
Raal FJ, Stein EA, Dufour R, et al; RUTHERFORD-2 Investigators. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet 2014; 385:331–340.
Robinson JG, Nedergaard BS, Rogers WJ, et al; LAPLA C-2 Investigators. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA 2014; 311:1870–1882.
Stroes E, Colquhoun D, Sullivan D, et al; GAUSS-2 Investigators. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol 2014; 63:2541–2548.
Nissen SE, Stroes E, Dent-Acosta RE, et al; GAUSS-3 Investigators. Efficacy and tolerability of evolocumab vs ezetimibe in patients with muscle-related statin intolerance, the GAUSS-3 randomized clinical trial. JAMA 2016; 315:1580–1590.
Trial assessing long term use of PCSK9 inhibition in subjects with genetic LDL disorders (TAUSSIG). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT-1624142. Updated June 25, 2015. Accessed October 23, 2016.
Kastelein JJ, Ginsberg HN, Langslet G, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J 2015; 36:2996–3003.
Efficacy and safety of alirocumab (SAR236553/REGN727) versus placebo on top of lipid-modifying therapy in patients with heterozygous familial hypercholesterolemia; the ODYSSEY HIGH FH trial. U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01617655. Updated September 27, 2016. Accessed October 23, 2016.
Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: The ODYSSEY COMBO I study. Am Heart J 2015; 169:906–915.
Efficacy and Safety of Alirocumab (SAR236553/REGN727) Versus Ezetimibe on Top of Statin in High Cardiovascular Risk Patients With Hypercholesterolemia (ODYSSEY COMBO II). U.S. National Institutes of Health website. Updated June 23, 2016. https://clinicaltrials.gov/ct2/show/NCT01644188. Accessed October 23, 2016.
Roth EM, Moriarty P, Bergeron J, et al; ODYSSEY CHOICE I investigators. A phase III randomized trial evaluating alirocumab 300 mg every 4 weeks as monotherapy or add-on to statin: ODYSSEY CHOICE I. Atherosclerosis 2016, doi: 10.1016/j.atherosclerosis.2016.08.043.
Phase III Study To Evaluate Alirocumab in Patients With Hypercholesterolemia Not Treated With a Statin (ODYSSEY CHOICE II). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT02023879. Updated November 2, 2015. Accessed October 23, 2016.
Monthly and twice monthly subcutaneous dosing of PF-04950615 (RN316) in hypercholesterolemic subjects on a statin. U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/results?term=NCT01592240. Updated October 14, 2014. Accessed October 23, 2016.
Zhang XL, Zhu QQ, Zhu L, et al. Safety and efficacy of anti-PCSK9 antibodies: a meta-analysis of 25 randomized, controlled trials. BMC Med 2015; 13:123.
Sabatine MS, Giugliano RP, Wiviott SD, et al; OSLER Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
Robinson JG, Farnier M, Krempf M, et al; ODYSSEY LONG TERM Investigators. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1489–1499.
Navarese EP, Kolodziejczak M, Schulze V, et al. Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adults with hypercholesterolemia: a systematic review and meta-analysis. Ann Intern Med 2015; 163:40–51.
Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk (FOURIER). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01764633. Updated July 26, 2016. Accessed October 23, 2016.
ODYSSEY Outcomes: Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab. U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01663402. Updated October 23, 2016. Accessed September 13, 2016.
The Evaluation of Bococizumab (PF-04950615;RN316) in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects (SPIRE-1). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01975376. Updated September 22, 2016. Accessed October 23, 2016.
The Evaluation of Bococizumab (PF-04950615; RN316) in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects (SPIRE-2). U.S. National Institutes of Health website. https://clinicaltrials.gov/ct2/show/NCT01975389. Updated July 26, 2016. Accessed October 23, 2016.
Lloyd-Jones DM, Morris PB, Ballantyne CM, et al; Writing Committee. 2016 ACC expert consensus decision pathway on the role of non-statin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology task force on clinical expert consensus documents. J Am Coll Cardiol 2016; 68:92–125.
Cleveland Clinic Journal of Medicine 2016 November; 83(suppl 2): S36-S44
Inside the Article
KEY POINTS
Potential candidates for PCSK9 inhibitor therapy are patients with familial hypercholesterolemia with a lifetime burden of elevated low-density-lipoprotein cholesterol (LDL-C) and thus a low likelihood of optimal control on current therapies; patients with complete or partial statin intolerance, with high-intensity statin dosing limited by adverse effects; and patients at high CVD risk with LDL-C goals not achieved with current therapies.
Subcutaneously administered monoclonal antibodies targeting PCSK9 are currently the only PCSK9 inhibitors with FDA approval.
PCSK9 inhibitors under study include agents with more durable effect and that require less frequent injections, RNA-interference therapies, vaccinations, antisense therapies, and oral formulations.
Disallow All Ads
Alternative CME
Consolidated Pubs: Do Not Show Source Publication Logo
Fibromuscular dysplasia (FMD) is an uncommon vascular disease that leads to narrowing (with either a beaded appearance or, less commonly, focal stenosis), dissection, or aneurysm of medium-sized arteries. Awareness of FMD within the medical community has rapidly expanded during the past decade owing to heightened interest among clinicians, multicenter coordinated research initiatives, and patient advocacy efforts.
In addition, a better understanding of the clinical manifestations and natural history of the disease along with advances in diagnostic imaging have altered the clinical approach to management. There are many unanswered questions regarding FMD, but this review highlights recent insights and how this information has modified clinical care for those affected.
DISTINCT FROM ATHEROSCLEROSIS
Reprinted with permission from Wolters Kluwer Health, Inc. (Poloskey SL, et al. Fibromuscular dysplasia. Circulation 2012; 125:e636–e639).
Figure 1. Multifocal fibromuscular dysplasia (FMD) involving the internal carotid artery (A) and a renal artery (B) with a “string-of-beads” appearance. The less common type, focal FMD, involving the internal carotid artery (C) and a renal artery (D).
FMD results from abnormal development of the arterial cell wall, most commonly the vessel media and less commonly the vessel intima (Figure 1).1,2 Distinct from atherosclerotic processes, FMD shares few typical cardiovascular risk factors aside from an association with tobacco smoking.3,4
The most common variant of FMD is the multifocal type, with the affected arteries resembling a string of beads due to alternating regions of stenosis and dilation.1,5 FMD can also cause a singular stenosis (focal type FMD) and has more recently been associated with findings of arterial tortuosity, aneurysm, and dissection.6,7
Though the disease typically affects the renal and extracranial carotid arteries, it has been noted in most medium-sized arteries throughout the body, most commonly the mesenteric, external iliac, and brachial arteries.1 The location of diseased segments determines symptoms, which commonly include hypertension, headache, and pulsatile tinnitus.8 The overwhelming majority of people affected (> 90%) are women.8
The diagnosis of FMD should be suspected in the case of young or middle-aged women presenting with migraine headaches, pulsatile tinnitus, or hypertension and for women with cervical bruits without typical risk factors for atherosclerotic disease. The diagnosis should also be suspected among patients who have suffered an arterial dissection or who are found to have a cerebral, carotid, or renal aneurysm.
THE US REGISTRY FOR FMD
Since it began enrolling patients in 2009, the US Registry for Fibromuscular Dysplasia has grown to include 13 active centers. It collects longitudinal data on the clinical characteristics, presentation, vascular bed involvement, vascular procedures, and clinical outcomes of patients with FMD.8,9Table 1 highlights key findings and lessons learned from registry publications, many of which have altered previous concepts of this disease.3,7,8,10–12
EPIDEMIOLOGY AND PATHOPHYSIOLOGY
Prevalence
Although FMD is considered a rare disease (and recognized as such by the National Organization of Rare Diseases), the exact prevalence is unknown. A review of 8 studies conducted from 1963 to 2011 found the prevalence of FMD ranged from 2.0% (3 of 150) to 6.6% (47 of 716) among healthy renal transplant donors for a mean prevalence of 3.3% (268 of 8,029) among all donors.13–21 Findings from the Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial, which studied the effectiveness of medical therapy alone vs medical therapy and stenting for treatment of significant renal artery stenosis and hypertension, found that 5.8% (58 of 997) of participants who underwent angiography had concomitant renal FMD.22 Importantly, patients with FMD were supposed to have been excluded from the trial, suggesting that FMD is often overlooked or underdiagnosed. A review published in 2010 reported the prevalence of cerebrovascular FMD to be 0.3% to 3.2% in patients undergoing cerebral angiography, but it noted significant heterogeneity in patient populations and definitions of FMD across published studies.23
Risk factors for FMD: Female sex and tobacco smoking
The mechanisms underlying the pathogenesis of FMD are still poorly understood, and its development is likely related to a combination of genetic and environmental factors. There seems to be a hormonal component to the pathogenesis of FMD, as most patients with this condition are women: approximately 91.5% of patients enrolled in the US Registry.10 Men with FMD, however, seem to have a more aggressive course with a rate of aneurysm or dissection two times higher than that in women with FMD.7
Studies have reported an increased risk of FMD in patients with a history of tobacco smoking.3,24 A US Registry report notes that FMD patients with a history of smoking had a statistically significant higher rate of aneurysm than those who had never smoked (24.8% vs 18.9%), and there was a trend toward increased prevalence of major vascular events in smokers, including subarachnoid hemorrhage, transient ischemic attack, stroke, mesenteric ischemia, renal infarction, and major coronary event.3 This study also found that patients with FMD who were smokers were more likely to have claudication symptoms (15.1% vs 7.4%) or to have undergone a vascular procedure (45.9% vs 36.7%).3 Further research is needed to fully understand the relationship between smoking and its interaction with other environmental, hormonal, and genetic factors.
FMD and connective tissue features
While studies have suggested a genetic component to the development of FMD, the specific genetic mechanisms are unknown.1 Studies have explored the potential relationship between FMD and genetic connective tissue disorders that can present with vascular manifestations, such as Loeys-Dietz, Marfan, and Ehlers-Danlos syndromes, and isolated case reports have noted concomitant FMD lesions in patients with these classical genetic disorders.25–31 In a series of patients with FMD from Cleveland Clinic who underwent genetic testing for selected connective tissue disorders, including Ehlers-Danlos syndrome and Loeys-Dietz syndrome, the overall yield of these tests was low.31 These studies suggest some overlap of FMD and other vascular connective tissue disorders, as well as the likelihood that the arterial manifestations of FMD may develop through multiple potential genetic pathways.
A series of 47 patients with FMD seen at the National Institutes of Health found a high incidence of connective tissue features on physical examination, with 95.7% of patients exhibiting at least four features of connective tissue disease, including marked hypermobility, scoliosis, craniofacial abnormalities, and pes planus (flat foot deformity).32 A study of a larger cohort of female patients seen at Cleveland Clinic did not find classical connective tissue features (such as pectus deformity, hypermobility, atrophic scaring, and club foot deformity) to a greater extent than what is reported in the general population, but it did find a significant prevalence of severe myopia (near sightedness), high-arched palate, dental crowding, and early-onset arthritis.33 Additional studies are needed to clarify the potential relationship between the spectrum of connective tissue disorders and FMD.
A BROADER SCOPE OF ARTERIAL MANIFESTATIONS
Since FMD was first described in the 1930s,34 most case reports have focused on its renal artery manifestations. In 1964, extrarenal involvement was first reported, which included carotid, iliac, and visceral arteries.35 The medical community has since recognized that the disease can affect medium-sized vessels throughout the body and, more recently, that it is a multifaceted disease with varying arterial manifestations outside of the typical string-of-beads appearance or focal FMD lesions.1 In addition to multifocal or focal narrowings, arterial manifestations of FMD include arterial tortuosity, aneurysm, and dissection.
Arterial tortuosity
Figure 2. (A) Duplex ultrasonography with color power angiography in a patient with fibromuscular dysplasia shows arterial tortuosity (the “S curve”) in the internal carotid artery and areas of beading. This feature can also be seen in renal arteries, as shown on angiography (B).
Tortuosity or redundancy of the arteries, particularly the internal carotid arteries, has recently been reported in association with FMD.6 A study based on vascular ultrasonography findings identified this anatomic variant (described as having the appearance of an S-curvature of the internal carotid artery) in 31.9% (37 of 116) of FMD patients.6 This rate of tortuosity is higher than that in the general population, especially when compared with patients of similar age (under age 70). Arterial tortuosity is a common finding in FMD and may be seen in other arterial segments (Figure 2).
Aneurysm and dissection
Both arterial aneurysm and arterial dissection are recognized as manifestations of FMD. A US Registry report published in 2016 found a high prevalence of aneurysm and dissection in the FMD population.7 Of the 921 patients included in this analysis, 21.6% had an aneurysm, 25.7% had an arterial dissection, and 41.7% had either aneurysm or dissection. The most common locations for aneurysm were the extracranial carotid, renal, and intracranial arteries, whereas dissection commonly occurred in the extracranial carotid, vertebral, renal, and coronary arteries. The authors noted that these data may be an underestimation, because the entire cohort did not undergo comprehensive screening for asymptomatic aneurysm or dissection. Patients with aneurysm were more likely to have a history of smoking and subarachnoid hemorrhage, while those with dissection were younger and more likely to have headache, neck pain, and end-organ ischemia, including stroke, renal infarction, or myocardial infarction.
FMD of the coronary arteries
Figure 3. Spontaneous coronary artery dissection (SCAD) in a 42-year-old woman with fibromuscular dysplasia (FMD) who presented with chest pain and nausea and non-ST- segment elevation myocardial infarction. She was found with coronary angiography to have SCAD of the left circumflex coronary (A, red arrow). Computed tomographic angiography showed a string-of-beads appearance of the left internal carotid artery (B, red arrow). Duplex ultrasonography showed turbulence and tortuosity in the mid to distal left internal carotid artery, consistent with a diagnosis of multifocal carotid FMD (C).
The association between FMD and spontaneous coronary artery dissection (SCAD) has recently been discovered (Figure 3). SCAD typically presents as troponin-positive acute coronary syndrome.36 FMD has been identified as a predisposing condition for SCAD in two case series from Vancouver General Hospital37 and Mayo Clinic.38 The case series from Mayo Clinic found that 45% of SCAD patients had FMD in the extracoronary vessels; the case series from Vancouver General Hospital found that 72% had FMD. A more recent study found that there seems to be other manifestations of FMD in the coronary arteries aside from SCAD.39 In this series, 32 patients with multifocal FMD (in the renal, external iliac, or cerebrovascular arteries) who underwent coronary angiography for suspected symptomatic coronary artery disease (either acute coronary syndrome or stable angina) were found to have coronary artery lesions different from those of atherosclerotic disease. In addition to coronary lesions of dissection (SCAD), the most common findings were marked coronary arterial tortuosity (the “S curve”), followed by areas of atypical-appearing irregular or smooth stenosis. More than half of patients in the series had segments of coronary artery ectasia (enlargement).
APPROACH TO MANAGEMENT
There is no cure for FMD, and thus management strategies focus on thorough evaluation and surveillance, lifestyle modification, and treatment of symptoms. Vascular procedures, such as angioplasty or treatment of aneurysms, are required for some patients. Because patients with FMD present with a diverse set of symptoms, consultation with a specialist who has experience with FMD and who works closely with an interdisciplinary team of experts is recommended.1 The interdisciplinary FMD care team may include a vascular medicine physician, cardiologist, nephrologist, neurologist, neurosurgeon, vascular surgeon, and vascular interventionalist (interventional cardiologist and radiologist).
Imaging and screening the vasculature in FMD patients
Because of the variability in location and manifestations of FMD and the high prevalence of aneurysm and dissection, all patients should undergo comprehensive one-time head-to-pelvis screening during the workup for FMD.1,7 Although the technical standard of diagnostic imaging is catheter angiography, noninvasive imaging—computed tomographic angiography (CTA), magnetic resonance angiography (MRA), duplex ultrasonography—is more commonly used to diagnose and monitor the disease.
A study from our group at Cleveland Clinic assessed the utility of a specialized CTA protocol of the chest, abdomen, and pelvis to image and diagnose manifestations of FMD outside of the cerebrovasculature.40 Incremental findings on imaging included areas of beading or focal narrowing in a new vascular territory and previously undiagnosed arterial aneurysm or dissection. These findings were seen in a variety of vascular beds, including the renal, iliac, and mesenteric arteries, although aortic abnormalities were rare. This study supports the diagnostic value of CTA for FMD to detect asymptomatic aneurysms and areas of arterial dissection, but it also suggests that routine vascular imaging of the thorax may not be necessary.40 In cases of SCAD, on-table renal and iliac angiography (performed after coronary angiography) can assist in diagnosis of FMD as an underlying cause.36 The cerebrovascular arteries (carotid, vertebral, and intracranial vessels) can be imaged later with noninvasive imaging (CTA, MRA).
As a general strategy, once patients with FMD undergo comprehensive imaging, a surveillance program is customized for the patient based on the anatomic location of the disease and the nature of the imaging findings. For example, renal and internal carotid artery FMD may be followed with annual duplex ultrasonography, whereas cerebral and renal or visceral aneurysms require periodic CTA or MRA.
Medical therapies
The medical regimen for patients with FMD varies based on disease location and symptoms, though there are no definitive treatment guidelines because of limited data. A study from the US Registry found that 72.9% of registrants were treated with antiplatelet medications,11 and this is a standard approach in our clinical practice for prevention of thromboembolic events. Antiplatelet drug therapy was more common in elderly patients, patients with a history of coronary artery disease or vascular intervention for FMD, and patients with isolated cerebrovascular FMD.11 Blood pressure management is also important in the medical therapy of patients with FMD who have hypertension. For patients with renal artery involvement, treatment with an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker has been suggested.1
Vascular intervention
The need for vascular intervention (eg, angioplasty or endovascular or surgical aneurysm treatment) is determined primarily by symptoms, with renal artery angioplasty for hypertension the most common FMD-related procedure. It is uncommon for vascular intervention to be performed for cerebrovascular FMD in the absence of recurrent transient ischemic attack or stroke despite antiplatelet therapy, arterial dissection that has failed medical management, or sizable aneurysm that requires treatment to prevent rupture.
When considering intervention for renal artery FMD, it is important to note that the appearance of multifocal FMD (beading) on angiography or noninvasive imaging does not reflect the hemodynamic severity of disease: translesional pressure gradients should be measured across the affected artery to determine if there is actually hemodynamic stenosis caused by an area of beading and to select patients for balloon angioplasty.1 Repeat pressure gradient assessment is done after angioplasty to confirm hemodynamic success.1 Surgical intervention for renal FMD is uncommon. It is generally reserved for complex cases in which endovascular techniques have failed.1
Asymptomatic patients with cerebral, visceral, or arterial aneurysm may require endovascular or surgical treatment. If surgery is indicated, the treatment approach (coiling, stenting, or open surgery) is determined by the size and location of the aneurysm, patient-related factors that may influence the risk of rupture (eg, uncontrolled hypertension, family history of ruptured aneurysm), the anatomic characteristics of the aneurysm, and the feasibility of endovascular vs open surgical repair. A US Registry study of 200 patients with an aneurysm reported that 31.5% underwent intervention to treat the aneurysm.7 Aneurysms requiring intervention were most commonly noted in the extracranial carotid, renal, and intracranial arteries.7
CONCLUSION
Awareness and understanding of FMD have substantially improved in recent years, and this has translated into better care for many patients with FMD. Important advancements have included the recognition of the variability of manifestations of this disease—ranging from an arterial string-of-beads appearance to aneurysm, dissection, and tortuosity—and establishing the need for comprehensive vascular imaging screening in FMD patients. Establishing the association of FMD with SCAD has led to better care for patients with SCAD and presents the opportunity to optimize management of these patients to prevent further vascular events. Research initiatives and heightened awareness have provided valuable insight into this disease, but further work is needed to determine the causal mechanisms of FMD and to continue to develop better management strategies.
References
Olin JW, Gornik HL, Bacharach JM, et al. Fibromuscular dysplasia: state of the science and critical unanswered questions. A scientific statement from the American Heart Association. Circulation 2014; 129:1048–1078
O’Connor S, Gornik HL, Froehlich JB, et al. Smoking and adverse outcomes in fibromuscular dysplasia: US registry report. J Am Coll Cardiol 2016; 67:1750–1751.
Sang CN, Whelton PK, Hamper UM, et al. Etiologic factors in renovascular fibromuscular dysplasia. A case-control study. Hypertension 1989; 14:472–479.
Persu A, Touzé E, Mousseaux E, Barral X, Joffre F, Plouin PF. Diagnosis and management of fibromuscular dysplasia: an expert consensus. Eur J Clin Invest 2012; 42:338–347.
Sethi SS, Lau JF, Godbold J, Gustavson S, Olin JW. The S curve: a novel morphological finding in the internal carotid artery in patients with fibromuscular dysplasia. Vasc Med 2014; 19:356–362.
Kadian-Dodov D, Gornik HL, Gu X, et al. Dissection and aneurysm in patients with fibromuscular dysplasia: findings for the U.S. registry for FMD. J Am Coll Cardiol 2016; 68:176–185.
Olin JW, Froehlich J, Gu X, et al. The United States Registry for Fibromuscular Dysplasia: results in the first 447 patients. Circulation 2012; 125:3182–3190.
Kim ES, Olin JW, Froehlich JB, et al. Clinical manifestations of fibromuscular dysplasia vary by patient sex: a report of the United States Registry for Fibromuscular Dysplasia. J Am Coll Cardiol 2013; 62:2026–2028.
Weinberg I, Gu X, Giri J, et al. Anti-platelet and anti-hypertension medication use in patients with fibromuscular dysplasia: results from the United States Registry for Fibromuscular Dysplasia. Vasc Med 2015; 20:447–453.
Green R, Gu X, Kline-Rogers E, et al. Differences between the pediatric and adult presentation of fibromuscular dysplasia: results from the US Registry. Pediatr Nephrol 2016; 31:641–650.
Shivapour DM, Erwin P, Kim ESH. Epidemiology of fibromuscular dysplasia: a review of the literature. Vasc Med 2016; 21:376–381.
Spring DB, Satvatierra O Jr, Palubinskas AJ, Amend WJ Jr, Vincenti FG, Feduska NJ. Results and significance of angiography in potential kidney donors. Radiology 1979; 133:45–47.
Cragg AH, Smith TP, Thompson BH, et al. Incidental fibromuscular dysplasia in potential renal donors: long-term clinical follow-up. Radiology 1989; 172:145–147.
Neymark E, LaBerge JM, Hirose R, et al. Arteriographic detection of renovascular disease in potential renal donors: incidence and effect on donor surgery. Radiology 2000; 214:755–760.
Andreoni KA, Weeks SM, Gerber DA, et al. Incidence of donor renal fibromuscular dysplasia: does it justify routine angiography? Transplantation 2002; 73:1112–1116.
Blondin D, Lanzman R, Schellhammer F, et al. Fibromuscular dysplasia in living renal donors: still a challenge to computed tomographic angiography. Eur J Radiol 2010; 75:67–71.
Lorenz EC, Vrtiska TJ, Lieske JC, et al. Prevalence of renal artery and kidney abnormalities by computed tomography among healthy adults. Clin J Am Soc Nephrol 2010; 5:431–438.
McKenzie GA, Oderich GS, Kawashima A, Misra S. Renal artery fibromuscular dysplasia in 2,640 renal donor subjects: a CT angiography analysis. J Vasc Interv Radiol 2013; 24:1477–1480.
Frick MP, Goldberg ME. Uro- and angiographic findings in a “normal” population: screening of 151 symptom-free potential transplant donors for renal disease. AJR Am J Roentgenol 1980; 134:503–505.
Hendricks NJ, Matsumoto AH, Angle JF, et al. Is fibromuscular dysplasia underdiagnosed? A comparison of the prevalence of FMD seen in CORAL trial participants versus a single institution population of renal donor candidates. Vasc Med 2014; 19:363–367.
Touzé E, Oppenheim C, Trystram D, et al. Fibromuscular dysplasia of cervical and intracranial arteries. Int J Stroke 2010; 5:296–305.
Savard S, Azarine A, Jeunemaitre X, Azizi M, Plouin PF, Steichen O. Association of smoking with phenotype at diagnosis and vascular interventions in patients with renal artery fibromuscular dysplasia. Hypertension 2013; 61:1227–1232.
Schievink WI, Limburg M. Angiographic abnormalities mimicking fibromuscular dysplasia in a patient with Ehlers-Danlos syndrome, type IV. Neurosurgery 1989; 25:482–483.
Schievink WI, Bjornsson J, Piepgras DG. Coexistence of fibromuscular dysplasia and cystic medial necrosis in a patient with Marfan’s syndrome and bilateral carotid artery dissections. Stroke 1994; 25:2492–2496.
Schievink WI, Bjournsson J, Parisi JE, Prakash UB. Arterial fibromuscular dysplasia associated with severe α1-antitrypsin deficiency. Mayo Clin Proc 1994; 69:1040–1043.
Schievink WI, Puumala MR, Meyer FB, Raffel C, Katzmann JA, Parisi JE. Giant intracranial aneurysm and fibromuscular dysplasia in an adolescent with α1-antitrypsin deficiency. J Neurosurg 1996; 85:503–506.
Schievink WI, Meyer FB, Parisi JE, Wijdicks EFM. Fibromuscular dysplasia of the internal carotid artery associated with alpha1-antitrypsin deficiency. Neurosurgery 1998; 43:229–233; discussion 233–234.
Bofinger A, Hawley C, Fisher P, Daunt N, Stowasser M, Gordon R. Alpha-1-antitrypsin phenotypes in patients with renal arterial fibromuscular dysplasia. J Hum Hypertens 2000; 14:91–94.
Poloskey SL, Kim ES, Sanghani R, et al. Low yield of genetic testing for known vascular connective tissue disorders in patients with fibromuscular dysplasia. Vasc Med 2012; 17:371–378.
Ganesh SK, Morissette R, Xu Z, et al. Clinical and biochemical profiles suggest fibromuscular dysplasia is a systemic disease with altered TGF- expression and connective tissue features. FASEB J 2014; 28:3313–3324.
O’Connor S, Kim ES, Brinza E, et al. Systemic connective tissue features in women with fibromuscular dysplasia. Vasc Med 2015; 20:454–462.
Saw J, Mancini GB, Humphries KH. Contemporary review on spontaneous coronary artery dissection. J Am Coll Cardiol 2016; 68:297–312.
Saw J, Aymong E, Sedlak T, et al. Spontaneous coronary artery dissection: association with predisposing arteriopathies and precipitating stressors and cardiovascular outcomes. Circ Cardiovasc Interv 2014; 7:645–655.
Prasad M, Tweet MS, Hayes SN, et al. Prevalence of extracoronary vascular abnormalities and fibromuscular dysplasia in patients with spontaneous coronary artery dissection. Am J Cardiol 2015; 115:1672–1677.
Bolen MA, Brinza E, Renapurkar RD, Kim ES, Gornik HL. Screening CT angiography of the aorta, visceral branch vessels, and pelvic arteries in fibromuscular dysplasia. JACC Cardiovasc Imaging. 2016; doi: 10.1016/j.jcmg.2016.04.010. [Epub ahead of print].
Ellen K. Brinza, MS Vascular Medicine Section, Department of Cardiovascular Medicine, Cleveland Clinic
Heather L. Gornik, MD Vascular Medicine Section, Department of Cardiovascular Medicine, Cleveland Clinic
Correspondence: Heather L. Gornik, MD, Vascular Medicine Section, Department of Cardiovascular Medicine, J3-5, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; gornikh@ccf.org
Ms. Brinza reported no financial interests or relationships that pose a potential conflict of interest with this article. Dr. Gornik reported membership of the medical advisory board of the Fibromuscular Dysplasia Society of America.
Ellen K. Brinza, MS Vascular Medicine Section, Department of Cardiovascular Medicine, Cleveland Clinic
Heather L. Gornik, MD Vascular Medicine Section, Department of Cardiovascular Medicine, Cleveland Clinic
Correspondence: Heather L. Gornik, MD, Vascular Medicine Section, Department of Cardiovascular Medicine, J3-5, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; gornikh@ccf.org
Ms. Brinza reported no financial interests or relationships that pose a potential conflict of interest with this article. Dr. Gornik reported membership of the medical advisory board of the Fibromuscular Dysplasia Society of America.
Author and Disclosure Information
Ellen K. Brinza, MS Vascular Medicine Section, Department of Cardiovascular Medicine, Cleveland Clinic
Heather L. Gornik, MD Vascular Medicine Section, Department of Cardiovascular Medicine, Cleveland Clinic
Correspondence: Heather L. Gornik, MD, Vascular Medicine Section, Department of Cardiovascular Medicine, J3-5, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; gornikh@ccf.org
Ms. Brinza reported no financial interests or relationships that pose a potential conflict of interest with this article. Dr. Gornik reported membership of the medical advisory board of the Fibromuscular Dysplasia Society of America.
Fibromuscular dysplasia (FMD) is an uncommon vascular disease that leads to narrowing (with either a beaded appearance or, less commonly, focal stenosis), dissection, or aneurysm of medium-sized arteries. Awareness of FMD within the medical community has rapidly expanded during the past decade owing to heightened interest among clinicians, multicenter coordinated research initiatives, and patient advocacy efforts.
In addition, a better understanding of the clinical manifestations and natural history of the disease along with advances in diagnostic imaging have altered the clinical approach to management. There are many unanswered questions regarding FMD, but this review highlights recent insights and how this information has modified clinical care for those affected.
DISTINCT FROM ATHEROSCLEROSIS
Reprinted with permission from Wolters Kluwer Health, Inc. (Poloskey SL, et al. Fibromuscular dysplasia. Circulation 2012; 125:e636–e639).
Figure 1. Multifocal fibromuscular dysplasia (FMD) involving the internal carotid artery (A) and a renal artery (B) with a “string-of-beads” appearance. The less common type, focal FMD, involving the internal carotid artery (C) and a renal artery (D).
FMD results from abnormal development of the arterial cell wall, most commonly the vessel media and less commonly the vessel intima (Figure 1).1,2 Distinct from atherosclerotic processes, FMD shares few typical cardiovascular risk factors aside from an association with tobacco smoking.3,4
The most common variant of FMD is the multifocal type, with the affected arteries resembling a string of beads due to alternating regions of stenosis and dilation.1,5 FMD can also cause a singular stenosis (focal type FMD) and has more recently been associated with findings of arterial tortuosity, aneurysm, and dissection.6,7
Though the disease typically affects the renal and extracranial carotid arteries, it has been noted in most medium-sized arteries throughout the body, most commonly the mesenteric, external iliac, and brachial arteries.1 The location of diseased segments determines symptoms, which commonly include hypertension, headache, and pulsatile tinnitus.8 The overwhelming majority of people affected (> 90%) are women.8
The diagnosis of FMD should be suspected in the case of young or middle-aged women presenting with migraine headaches, pulsatile tinnitus, or hypertension and for women with cervical bruits without typical risk factors for atherosclerotic disease. The diagnosis should also be suspected among patients who have suffered an arterial dissection or who are found to have a cerebral, carotid, or renal aneurysm.
THE US REGISTRY FOR FMD
Since it began enrolling patients in 2009, the US Registry for Fibromuscular Dysplasia has grown to include 13 active centers. It collects longitudinal data on the clinical characteristics, presentation, vascular bed involvement, vascular procedures, and clinical outcomes of patients with FMD.8,9Table 1 highlights key findings and lessons learned from registry publications, many of which have altered previous concepts of this disease.3,7,8,10–12
EPIDEMIOLOGY AND PATHOPHYSIOLOGY
Prevalence
Although FMD is considered a rare disease (and recognized as such by the National Organization of Rare Diseases), the exact prevalence is unknown. A review of 8 studies conducted from 1963 to 2011 found the prevalence of FMD ranged from 2.0% (3 of 150) to 6.6% (47 of 716) among healthy renal transplant donors for a mean prevalence of 3.3% (268 of 8,029) among all donors.13–21 Findings from the Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial, which studied the effectiveness of medical therapy alone vs medical therapy and stenting for treatment of significant renal artery stenosis and hypertension, found that 5.8% (58 of 997) of participants who underwent angiography had concomitant renal FMD.22 Importantly, patients with FMD were supposed to have been excluded from the trial, suggesting that FMD is often overlooked or underdiagnosed. A review published in 2010 reported the prevalence of cerebrovascular FMD to be 0.3% to 3.2% in patients undergoing cerebral angiography, but it noted significant heterogeneity in patient populations and definitions of FMD across published studies.23
Risk factors for FMD: Female sex and tobacco smoking
The mechanisms underlying the pathogenesis of FMD are still poorly understood, and its development is likely related to a combination of genetic and environmental factors. There seems to be a hormonal component to the pathogenesis of FMD, as most patients with this condition are women: approximately 91.5% of patients enrolled in the US Registry.10 Men with FMD, however, seem to have a more aggressive course with a rate of aneurysm or dissection two times higher than that in women with FMD.7
Studies have reported an increased risk of FMD in patients with a history of tobacco smoking.3,24 A US Registry report notes that FMD patients with a history of smoking had a statistically significant higher rate of aneurysm than those who had never smoked (24.8% vs 18.9%), and there was a trend toward increased prevalence of major vascular events in smokers, including subarachnoid hemorrhage, transient ischemic attack, stroke, mesenteric ischemia, renal infarction, and major coronary event.3 This study also found that patients with FMD who were smokers were more likely to have claudication symptoms (15.1% vs 7.4%) or to have undergone a vascular procedure (45.9% vs 36.7%).3 Further research is needed to fully understand the relationship between smoking and its interaction with other environmental, hormonal, and genetic factors.
FMD and connective tissue features
While studies have suggested a genetic component to the development of FMD, the specific genetic mechanisms are unknown.1 Studies have explored the potential relationship between FMD and genetic connective tissue disorders that can present with vascular manifestations, such as Loeys-Dietz, Marfan, and Ehlers-Danlos syndromes, and isolated case reports have noted concomitant FMD lesions in patients with these classical genetic disorders.25–31 In a series of patients with FMD from Cleveland Clinic who underwent genetic testing for selected connective tissue disorders, including Ehlers-Danlos syndrome and Loeys-Dietz syndrome, the overall yield of these tests was low.31 These studies suggest some overlap of FMD and other vascular connective tissue disorders, as well as the likelihood that the arterial manifestations of FMD may develop through multiple potential genetic pathways.
A series of 47 patients with FMD seen at the National Institutes of Health found a high incidence of connective tissue features on physical examination, with 95.7% of patients exhibiting at least four features of connective tissue disease, including marked hypermobility, scoliosis, craniofacial abnormalities, and pes planus (flat foot deformity).32 A study of a larger cohort of female patients seen at Cleveland Clinic did not find classical connective tissue features (such as pectus deformity, hypermobility, atrophic scaring, and club foot deformity) to a greater extent than what is reported in the general population, but it did find a significant prevalence of severe myopia (near sightedness), high-arched palate, dental crowding, and early-onset arthritis.33 Additional studies are needed to clarify the potential relationship between the spectrum of connective tissue disorders and FMD.
A BROADER SCOPE OF ARTERIAL MANIFESTATIONS
Since FMD was first described in the 1930s,34 most case reports have focused on its renal artery manifestations. In 1964, extrarenal involvement was first reported, which included carotid, iliac, and visceral arteries.35 The medical community has since recognized that the disease can affect medium-sized vessels throughout the body and, more recently, that it is a multifaceted disease with varying arterial manifestations outside of the typical string-of-beads appearance or focal FMD lesions.1 In addition to multifocal or focal narrowings, arterial manifestations of FMD include arterial tortuosity, aneurysm, and dissection.
Arterial tortuosity
Figure 2. (A) Duplex ultrasonography with color power angiography in a patient with fibromuscular dysplasia shows arterial tortuosity (the “S curve”) in the internal carotid artery and areas of beading. This feature can also be seen in renal arteries, as shown on angiography (B).
Tortuosity or redundancy of the arteries, particularly the internal carotid arteries, has recently been reported in association with FMD.6 A study based on vascular ultrasonography findings identified this anatomic variant (described as having the appearance of an S-curvature of the internal carotid artery) in 31.9% (37 of 116) of FMD patients.6 This rate of tortuosity is higher than that in the general population, especially when compared with patients of similar age (under age 70). Arterial tortuosity is a common finding in FMD and may be seen in other arterial segments (Figure 2).
Aneurysm and dissection
Both arterial aneurysm and arterial dissection are recognized as manifestations of FMD. A US Registry report published in 2016 found a high prevalence of aneurysm and dissection in the FMD population.7 Of the 921 patients included in this analysis, 21.6% had an aneurysm, 25.7% had an arterial dissection, and 41.7% had either aneurysm or dissection. The most common locations for aneurysm were the extracranial carotid, renal, and intracranial arteries, whereas dissection commonly occurred in the extracranial carotid, vertebral, renal, and coronary arteries. The authors noted that these data may be an underestimation, because the entire cohort did not undergo comprehensive screening for asymptomatic aneurysm or dissection. Patients with aneurysm were more likely to have a history of smoking and subarachnoid hemorrhage, while those with dissection were younger and more likely to have headache, neck pain, and end-organ ischemia, including stroke, renal infarction, or myocardial infarction.
FMD of the coronary arteries
Figure 3. Spontaneous coronary artery dissection (SCAD) in a 42-year-old woman with fibromuscular dysplasia (FMD) who presented with chest pain and nausea and non-ST- segment elevation myocardial infarction. She was found with coronary angiography to have SCAD of the left circumflex coronary (A, red arrow). Computed tomographic angiography showed a string-of-beads appearance of the left internal carotid artery (B, red arrow). Duplex ultrasonography showed turbulence and tortuosity in the mid to distal left internal carotid artery, consistent with a diagnosis of multifocal carotid FMD (C).
The association between FMD and spontaneous coronary artery dissection (SCAD) has recently been discovered (Figure 3). SCAD typically presents as troponin-positive acute coronary syndrome.36 FMD has been identified as a predisposing condition for SCAD in two case series from Vancouver General Hospital37 and Mayo Clinic.38 The case series from Mayo Clinic found that 45% of SCAD patients had FMD in the extracoronary vessels; the case series from Vancouver General Hospital found that 72% had FMD. A more recent study found that there seems to be other manifestations of FMD in the coronary arteries aside from SCAD.39 In this series, 32 patients with multifocal FMD (in the renal, external iliac, or cerebrovascular arteries) who underwent coronary angiography for suspected symptomatic coronary artery disease (either acute coronary syndrome or stable angina) were found to have coronary artery lesions different from those of atherosclerotic disease. In addition to coronary lesions of dissection (SCAD), the most common findings were marked coronary arterial tortuosity (the “S curve”), followed by areas of atypical-appearing irregular or smooth stenosis. More than half of patients in the series had segments of coronary artery ectasia (enlargement).
APPROACH TO MANAGEMENT
There is no cure for FMD, and thus management strategies focus on thorough evaluation and surveillance, lifestyle modification, and treatment of symptoms. Vascular procedures, such as angioplasty or treatment of aneurysms, are required for some patients. Because patients with FMD present with a diverse set of symptoms, consultation with a specialist who has experience with FMD and who works closely with an interdisciplinary team of experts is recommended.1 The interdisciplinary FMD care team may include a vascular medicine physician, cardiologist, nephrologist, neurologist, neurosurgeon, vascular surgeon, and vascular interventionalist (interventional cardiologist and radiologist).
Imaging and screening the vasculature in FMD patients
Because of the variability in location and manifestations of FMD and the high prevalence of aneurysm and dissection, all patients should undergo comprehensive one-time head-to-pelvis screening during the workup for FMD.1,7 Although the technical standard of diagnostic imaging is catheter angiography, noninvasive imaging—computed tomographic angiography (CTA), magnetic resonance angiography (MRA), duplex ultrasonography—is more commonly used to diagnose and monitor the disease.
A study from our group at Cleveland Clinic assessed the utility of a specialized CTA protocol of the chest, abdomen, and pelvis to image and diagnose manifestations of FMD outside of the cerebrovasculature.40 Incremental findings on imaging included areas of beading or focal narrowing in a new vascular territory and previously undiagnosed arterial aneurysm or dissection. These findings were seen in a variety of vascular beds, including the renal, iliac, and mesenteric arteries, although aortic abnormalities were rare. This study supports the diagnostic value of CTA for FMD to detect asymptomatic aneurysms and areas of arterial dissection, but it also suggests that routine vascular imaging of the thorax may not be necessary.40 In cases of SCAD, on-table renal and iliac angiography (performed after coronary angiography) can assist in diagnosis of FMD as an underlying cause.36 The cerebrovascular arteries (carotid, vertebral, and intracranial vessels) can be imaged later with noninvasive imaging (CTA, MRA).
As a general strategy, once patients with FMD undergo comprehensive imaging, a surveillance program is customized for the patient based on the anatomic location of the disease and the nature of the imaging findings. For example, renal and internal carotid artery FMD may be followed with annual duplex ultrasonography, whereas cerebral and renal or visceral aneurysms require periodic CTA or MRA.
Medical therapies
The medical regimen for patients with FMD varies based on disease location and symptoms, though there are no definitive treatment guidelines because of limited data. A study from the US Registry found that 72.9% of registrants were treated with antiplatelet medications,11 and this is a standard approach in our clinical practice for prevention of thromboembolic events. Antiplatelet drug therapy was more common in elderly patients, patients with a history of coronary artery disease or vascular intervention for FMD, and patients with isolated cerebrovascular FMD.11 Blood pressure management is also important in the medical therapy of patients with FMD who have hypertension. For patients with renal artery involvement, treatment with an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker has been suggested.1
Vascular intervention
The need for vascular intervention (eg, angioplasty or endovascular or surgical aneurysm treatment) is determined primarily by symptoms, with renal artery angioplasty for hypertension the most common FMD-related procedure. It is uncommon for vascular intervention to be performed for cerebrovascular FMD in the absence of recurrent transient ischemic attack or stroke despite antiplatelet therapy, arterial dissection that has failed medical management, or sizable aneurysm that requires treatment to prevent rupture.
When considering intervention for renal artery FMD, it is important to note that the appearance of multifocal FMD (beading) on angiography or noninvasive imaging does not reflect the hemodynamic severity of disease: translesional pressure gradients should be measured across the affected artery to determine if there is actually hemodynamic stenosis caused by an area of beading and to select patients for balloon angioplasty.1 Repeat pressure gradient assessment is done after angioplasty to confirm hemodynamic success.1 Surgical intervention for renal FMD is uncommon. It is generally reserved for complex cases in which endovascular techniques have failed.1
Asymptomatic patients with cerebral, visceral, or arterial aneurysm may require endovascular or surgical treatment. If surgery is indicated, the treatment approach (coiling, stenting, or open surgery) is determined by the size and location of the aneurysm, patient-related factors that may influence the risk of rupture (eg, uncontrolled hypertension, family history of ruptured aneurysm), the anatomic characteristics of the aneurysm, and the feasibility of endovascular vs open surgical repair. A US Registry study of 200 patients with an aneurysm reported that 31.5% underwent intervention to treat the aneurysm.7 Aneurysms requiring intervention were most commonly noted in the extracranial carotid, renal, and intracranial arteries.7
CONCLUSION
Awareness and understanding of FMD have substantially improved in recent years, and this has translated into better care for many patients with FMD. Important advancements have included the recognition of the variability of manifestations of this disease—ranging from an arterial string-of-beads appearance to aneurysm, dissection, and tortuosity—and establishing the need for comprehensive vascular imaging screening in FMD patients. Establishing the association of FMD with SCAD has led to better care for patients with SCAD and presents the opportunity to optimize management of these patients to prevent further vascular events. Research initiatives and heightened awareness have provided valuable insight into this disease, but further work is needed to determine the causal mechanisms of FMD and to continue to develop better management strategies.
Fibromuscular dysplasia (FMD) is an uncommon vascular disease that leads to narrowing (with either a beaded appearance or, less commonly, focal stenosis), dissection, or aneurysm of medium-sized arteries. Awareness of FMD within the medical community has rapidly expanded during the past decade owing to heightened interest among clinicians, multicenter coordinated research initiatives, and patient advocacy efforts.
In addition, a better understanding of the clinical manifestations and natural history of the disease along with advances in diagnostic imaging have altered the clinical approach to management. There are many unanswered questions regarding FMD, but this review highlights recent insights and how this information has modified clinical care for those affected.
DISTINCT FROM ATHEROSCLEROSIS
Reprinted with permission from Wolters Kluwer Health, Inc. (Poloskey SL, et al. Fibromuscular dysplasia. Circulation 2012; 125:e636–e639).
Figure 1. Multifocal fibromuscular dysplasia (FMD) involving the internal carotid artery (A) and a renal artery (B) with a “string-of-beads” appearance. The less common type, focal FMD, involving the internal carotid artery (C) and a renal artery (D).
FMD results from abnormal development of the arterial cell wall, most commonly the vessel media and less commonly the vessel intima (Figure 1).1,2 Distinct from atherosclerotic processes, FMD shares few typical cardiovascular risk factors aside from an association with tobacco smoking.3,4
The most common variant of FMD is the multifocal type, with the affected arteries resembling a string of beads due to alternating regions of stenosis and dilation.1,5 FMD can also cause a singular stenosis (focal type FMD) and has more recently been associated with findings of arterial tortuosity, aneurysm, and dissection.6,7
Though the disease typically affects the renal and extracranial carotid arteries, it has been noted in most medium-sized arteries throughout the body, most commonly the mesenteric, external iliac, and brachial arteries.1 The location of diseased segments determines symptoms, which commonly include hypertension, headache, and pulsatile tinnitus.8 The overwhelming majority of people affected (> 90%) are women.8
The diagnosis of FMD should be suspected in the case of young or middle-aged women presenting with migraine headaches, pulsatile tinnitus, or hypertension and for women with cervical bruits without typical risk factors for atherosclerotic disease. The diagnosis should also be suspected among patients who have suffered an arterial dissection or who are found to have a cerebral, carotid, or renal aneurysm.
THE US REGISTRY FOR FMD
Since it began enrolling patients in 2009, the US Registry for Fibromuscular Dysplasia has grown to include 13 active centers. It collects longitudinal data on the clinical characteristics, presentation, vascular bed involvement, vascular procedures, and clinical outcomes of patients with FMD.8,9Table 1 highlights key findings and lessons learned from registry publications, many of which have altered previous concepts of this disease.3,7,8,10–12
EPIDEMIOLOGY AND PATHOPHYSIOLOGY
Prevalence
Although FMD is considered a rare disease (and recognized as such by the National Organization of Rare Diseases), the exact prevalence is unknown. A review of 8 studies conducted from 1963 to 2011 found the prevalence of FMD ranged from 2.0% (3 of 150) to 6.6% (47 of 716) among healthy renal transplant donors for a mean prevalence of 3.3% (268 of 8,029) among all donors.13–21 Findings from the Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial, which studied the effectiveness of medical therapy alone vs medical therapy and stenting for treatment of significant renal artery stenosis and hypertension, found that 5.8% (58 of 997) of participants who underwent angiography had concomitant renal FMD.22 Importantly, patients with FMD were supposed to have been excluded from the trial, suggesting that FMD is often overlooked or underdiagnosed. A review published in 2010 reported the prevalence of cerebrovascular FMD to be 0.3% to 3.2% in patients undergoing cerebral angiography, but it noted significant heterogeneity in patient populations and definitions of FMD across published studies.23
Risk factors for FMD: Female sex and tobacco smoking
The mechanisms underlying the pathogenesis of FMD are still poorly understood, and its development is likely related to a combination of genetic and environmental factors. There seems to be a hormonal component to the pathogenesis of FMD, as most patients with this condition are women: approximately 91.5% of patients enrolled in the US Registry.10 Men with FMD, however, seem to have a more aggressive course with a rate of aneurysm or dissection two times higher than that in women with FMD.7
Studies have reported an increased risk of FMD in patients with a history of tobacco smoking.3,24 A US Registry report notes that FMD patients with a history of smoking had a statistically significant higher rate of aneurysm than those who had never smoked (24.8% vs 18.9%), and there was a trend toward increased prevalence of major vascular events in smokers, including subarachnoid hemorrhage, transient ischemic attack, stroke, mesenteric ischemia, renal infarction, and major coronary event.3 This study also found that patients with FMD who were smokers were more likely to have claudication symptoms (15.1% vs 7.4%) or to have undergone a vascular procedure (45.9% vs 36.7%).3 Further research is needed to fully understand the relationship between smoking and its interaction with other environmental, hormonal, and genetic factors.
FMD and connective tissue features
While studies have suggested a genetic component to the development of FMD, the specific genetic mechanisms are unknown.1 Studies have explored the potential relationship between FMD and genetic connective tissue disorders that can present with vascular manifestations, such as Loeys-Dietz, Marfan, and Ehlers-Danlos syndromes, and isolated case reports have noted concomitant FMD lesions in patients with these classical genetic disorders.25–31 In a series of patients with FMD from Cleveland Clinic who underwent genetic testing for selected connective tissue disorders, including Ehlers-Danlos syndrome and Loeys-Dietz syndrome, the overall yield of these tests was low.31 These studies suggest some overlap of FMD and other vascular connective tissue disorders, as well as the likelihood that the arterial manifestations of FMD may develop through multiple potential genetic pathways.
A series of 47 patients with FMD seen at the National Institutes of Health found a high incidence of connective tissue features on physical examination, with 95.7% of patients exhibiting at least four features of connective tissue disease, including marked hypermobility, scoliosis, craniofacial abnormalities, and pes planus (flat foot deformity).32 A study of a larger cohort of female patients seen at Cleveland Clinic did not find classical connective tissue features (such as pectus deformity, hypermobility, atrophic scaring, and club foot deformity) to a greater extent than what is reported in the general population, but it did find a significant prevalence of severe myopia (near sightedness), high-arched palate, dental crowding, and early-onset arthritis.33 Additional studies are needed to clarify the potential relationship between the spectrum of connective tissue disorders and FMD.
A BROADER SCOPE OF ARTERIAL MANIFESTATIONS
Since FMD was first described in the 1930s,34 most case reports have focused on its renal artery manifestations. In 1964, extrarenal involvement was first reported, which included carotid, iliac, and visceral arteries.35 The medical community has since recognized that the disease can affect medium-sized vessels throughout the body and, more recently, that it is a multifaceted disease with varying arterial manifestations outside of the typical string-of-beads appearance or focal FMD lesions.1 In addition to multifocal or focal narrowings, arterial manifestations of FMD include arterial tortuosity, aneurysm, and dissection.
Arterial tortuosity
Figure 2. (A) Duplex ultrasonography with color power angiography in a patient with fibromuscular dysplasia shows arterial tortuosity (the “S curve”) in the internal carotid artery and areas of beading. This feature can also be seen in renal arteries, as shown on angiography (B).
Tortuosity or redundancy of the arteries, particularly the internal carotid arteries, has recently been reported in association with FMD.6 A study based on vascular ultrasonography findings identified this anatomic variant (described as having the appearance of an S-curvature of the internal carotid artery) in 31.9% (37 of 116) of FMD patients.6 This rate of tortuosity is higher than that in the general population, especially when compared with patients of similar age (under age 70). Arterial tortuosity is a common finding in FMD and may be seen in other arterial segments (Figure 2).
Aneurysm and dissection
Both arterial aneurysm and arterial dissection are recognized as manifestations of FMD. A US Registry report published in 2016 found a high prevalence of aneurysm and dissection in the FMD population.7 Of the 921 patients included in this analysis, 21.6% had an aneurysm, 25.7% had an arterial dissection, and 41.7% had either aneurysm or dissection. The most common locations for aneurysm were the extracranial carotid, renal, and intracranial arteries, whereas dissection commonly occurred in the extracranial carotid, vertebral, renal, and coronary arteries. The authors noted that these data may be an underestimation, because the entire cohort did not undergo comprehensive screening for asymptomatic aneurysm or dissection. Patients with aneurysm were more likely to have a history of smoking and subarachnoid hemorrhage, while those with dissection were younger and more likely to have headache, neck pain, and end-organ ischemia, including stroke, renal infarction, or myocardial infarction.
FMD of the coronary arteries
Figure 3. Spontaneous coronary artery dissection (SCAD) in a 42-year-old woman with fibromuscular dysplasia (FMD) who presented with chest pain and nausea and non-ST- segment elevation myocardial infarction. She was found with coronary angiography to have SCAD of the left circumflex coronary (A, red arrow). Computed tomographic angiography showed a string-of-beads appearance of the left internal carotid artery (B, red arrow). Duplex ultrasonography showed turbulence and tortuosity in the mid to distal left internal carotid artery, consistent with a diagnosis of multifocal carotid FMD (C).
The association between FMD and spontaneous coronary artery dissection (SCAD) has recently been discovered (Figure 3). SCAD typically presents as troponin-positive acute coronary syndrome.36 FMD has been identified as a predisposing condition for SCAD in two case series from Vancouver General Hospital37 and Mayo Clinic.38 The case series from Mayo Clinic found that 45% of SCAD patients had FMD in the extracoronary vessels; the case series from Vancouver General Hospital found that 72% had FMD. A more recent study found that there seems to be other manifestations of FMD in the coronary arteries aside from SCAD.39 In this series, 32 patients with multifocal FMD (in the renal, external iliac, or cerebrovascular arteries) who underwent coronary angiography for suspected symptomatic coronary artery disease (either acute coronary syndrome or stable angina) were found to have coronary artery lesions different from those of atherosclerotic disease. In addition to coronary lesions of dissection (SCAD), the most common findings were marked coronary arterial tortuosity (the “S curve”), followed by areas of atypical-appearing irregular or smooth stenosis. More than half of patients in the series had segments of coronary artery ectasia (enlargement).
APPROACH TO MANAGEMENT
There is no cure for FMD, and thus management strategies focus on thorough evaluation and surveillance, lifestyle modification, and treatment of symptoms. Vascular procedures, such as angioplasty or treatment of aneurysms, are required for some patients. Because patients with FMD present with a diverse set of symptoms, consultation with a specialist who has experience with FMD and who works closely with an interdisciplinary team of experts is recommended.1 The interdisciplinary FMD care team may include a vascular medicine physician, cardiologist, nephrologist, neurologist, neurosurgeon, vascular surgeon, and vascular interventionalist (interventional cardiologist and radiologist).
Imaging and screening the vasculature in FMD patients
Because of the variability in location and manifestations of FMD and the high prevalence of aneurysm and dissection, all patients should undergo comprehensive one-time head-to-pelvis screening during the workup for FMD.1,7 Although the technical standard of diagnostic imaging is catheter angiography, noninvasive imaging—computed tomographic angiography (CTA), magnetic resonance angiography (MRA), duplex ultrasonography—is more commonly used to diagnose and monitor the disease.
A study from our group at Cleveland Clinic assessed the utility of a specialized CTA protocol of the chest, abdomen, and pelvis to image and diagnose manifestations of FMD outside of the cerebrovasculature.40 Incremental findings on imaging included areas of beading or focal narrowing in a new vascular territory and previously undiagnosed arterial aneurysm or dissection. These findings were seen in a variety of vascular beds, including the renal, iliac, and mesenteric arteries, although aortic abnormalities were rare. This study supports the diagnostic value of CTA for FMD to detect asymptomatic aneurysms and areas of arterial dissection, but it also suggests that routine vascular imaging of the thorax may not be necessary.40 In cases of SCAD, on-table renal and iliac angiography (performed after coronary angiography) can assist in diagnosis of FMD as an underlying cause.36 The cerebrovascular arteries (carotid, vertebral, and intracranial vessels) can be imaged later with noninvasive imaging (CTA, MRA).
As a general strategy, once patients with FMD undergo comprehensive imaging, a surveillance program is customized for the patient based on the anatomic location of the disease and the nature of the imaging findings. For example, renal and internal carotid artery FMD may be followed with annual duplex ultrasonography, whereas cerebral and renal or visceral aneurysms require periodic CTA or MRA.
Medical therapies
The medical regimen for patients with FMD varies based on disease location and symptoms, though there are no definitive treatment guidelines because of limited data. A study from the US Registry found that 72.9% of registrants were treated with antiplatelet medications,11 and this is a standard approach in our clinical practice for prevention of thromboembolic events. Antiplatelet drug therapy was more common in elderly patients, patients with a history of coronary artery disease or vascular intervention for FMD, and patients with isolated cerebrovascular FMD.11 Blood pressure management is also important in the medical therapy of patients with FMD who have hypertension. For patients with renal artery involvement, treatment with an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker has been suggested.1
Vascular intervention
The need for vascular intervention (eg, angioplasty or endovascular or surgical aneurysm treatment) is determined primarily by symptoms, with renal artery angioplasty for hypertension the most common FMD-related procedure. It is uncommon for vascular intervention to be performed for cerebrovascular FMD in the absence of recurrent transient ischemic attack or stroke despite antiplatelet therapy, arterial dissection that has failed medical management, or sizable aneurysm that requires treatment to prevent rupture.
When considering intervention for renal artery FMD, it is important to note that the appearance of multifocal FMD (beading) on angiography or noninvasive imaging does not reflect the hemodynamic severity of disease: translesional pressure gradients should be measured across the affected artery to determine if there is actually hemodynamic stenosis caused by an area of beading and to select patients for balloon angioplasty.1 Repeat pressure gradient assessment is done after angioplasty to confirm hemodynamic success.1 Surgical intervention for renal FMD is uncommon. It is generally reserved for complex cases in which endovascular techniques have failed.1
Asymptomatic patients with cerebral, visceral, or arterial aneurysm may require endovascular or surgical treatment. If surgery is indicated, the treatment approach (coiling, stenting, or open surgery) is determined by the size and location of the aneurysm, patient-related factors that may influence the risk of rupture (eg, uncontrolled hypertension, family history of ruptured aneurysm), the anatomic characteristics of the aneurysm, and the feasibility of endovascular vs open surgical repair. A US Registry study of 200 patients with an aneurysm reported that 31.5% underwent intervention to treat the aneurysm.7 Aneurysms requiring intervention were most commonly noted in the extracranial carotid, renal, and intracranial arteries.7
CONCLUSION
Awareness and understanding of FMD have substantially improved in recent years, and this has translated into better care for many patients with FMD. Important advancements have included the recognition of the variability of manifestations of this disease—ranging from an arterial string-of-beads appearance to aneurysm, dissection, and tortuosity—and establishing the need for comprehensive vascular imaging screening in FMD patients. Establishing the association of FMD with SCAD has led to better care for patients with SCAD and presents the opportunity to optimize management of these patients to prevent further vascular events. Research initiatives and heightened awareness have provided valuable insight into this disease, but further work is needed to determine the causal mechanisms of FMD and to continue to develop better management strategies.
References
Olin JW, Gornik HL, Bacharach JM, et al. Fibromuscular dysplasia: state of the science and critical unanswered questions. A scientific statement from the American Heart Association. Circulation 2014; 129:1048–1078
O’Connor S, Gornik HL, Froehlich JB, et al. Smoking and adverse outcomes in fibromuscular dysplasia: US registry report. J Am Coll Cardiol 2016; 67:1750–1751.
Sang CN, Whelton PK, Hamper UM, et al. Etiologic factors in renovascular fibromuscular dysplasia. A case-control study. Hypertension 1989; 14:472–479.
Persu A, Touzé E, Mousseaux E, Barral X, Joffre F, Plouin PF. Diagnosis and management of fibromuscular dysplasia: an expert consensus. Eur J Clin Invest 2012; 42:338–347.
Sethi SS, Lau JF, Godbold J, Gustavson S, Olin JW. The S curve: a novel morphological finding in the internal carotid artery in patients with fibromuscular dysplasia. Vasc Med 2014; 19:356–362.
Kadian-Dodov D, Gornik HL, Gu X, et al. Dissection and aneurysm in patients with fibromuscular dysplasia: findings for the U.S. registry for FMD. J Am Coll Cardiol 2016; 68:176–185.
Olin JW, Froehlich J, Gu X, et al. The United States Registry for Fibromuscular Dysplasia: results in the first 447 patients. Circulation 2012; 125:3182–3190.
Kim ES, Olin JW, Froehlich JB, et al. Clinical manifestations of fibromuscular dysplasia vary by patient sex: a report of the United States Registry for Fibromuscular Dysplasia. J Am Coll Cardiol 2013; 62:2026–2028.
Weinberg I, Gu X, Giri J, et al. Anti-platelet and anti-hypertension medication use in patients with fibromuscular dysplasia: results from the United States Registry for Fibromuscular Dysplasia. Vasc Med 2015; 20:447–453.
Green R, Gu X, Kline-Rogers E, et al. Differences between the pediatric and adult presentation of fibromuscular dysplasia: results from the US Registry. Pediatr Nephrol 2016; 31:641–650.
Shivapour DM, Erwin P, Kim ESH. Epidemiology of fibromuscular dysplasia: a review of the literature. Vasc Med 2016; 21:376–381.
Spring DB, Satvatierra O Jr, Palubinskas AJ, Amend WJ Jr, Vincenti FG, Feduska NJ. Results and significance of angiography in potential kidney donors. Radiology 1979; 133:45–47.
Cragg AH, Smith TP, Thompson BH, et al. Incidental fibromuscular dysplasia in potential renal donors: long-term clinical follow-up. Radiology 1989; 172:145–147.
Neymark E, LaBerge JM, Hirose R, et al. Arteriographic detection of renovascular disease in potential renal donors: incidence and effect on donor surgery. Radiology 2000; 214:755–760.
Andreoni KA, Weeks SM, Gerber DA, et al. Incidence of donor renal fibromuscular dysplasia: does it justify routine angiography? Transplantation 2002; 73:1112–1116.
Blondin D, Lanzman R, Schellhammer F, et al. Fibromuscular dysplasia in living renal donors: still a challenge to computed tomographic angiography. Eur J Radiol 2010; 75:67–71.
Lorenz EC, Vrtiska TJ, Lieske JC, et al. Prevalence of renal artery and kidney abnormalities by computed tomography among healthy adults. Clin J Am Soc Nephrol 2010; 5:431–438.
McKenzie GA, Oderich GS, Kawashima A, Misra S. Renal artery fibromuscular dysplasia in 2,640 renal donor subjects: a CT angiography analysis. J Vasc Interv Radiol 2013; 24:1477–1480.
Frick MP, Goldberg ME. Uro- and angiographic findings in a “normal” population: screening of 151 symptom-free potential transplant donors for renal disease. AJR Am J Roentgenol 1980; 134:503–505.
Hendricks NJ, Matsumoto AH, Angle JF, et al. Is fibromuscular dysplasia underdiagnosed? A comparison of the prevalence of FMD seen in CORAL trial participants versus a single institution population of renal donor candidates. Vasc Med 2014; 19:363–367.
Touzé E, Oppenheim C, Trystram D, et al. Fibromuscular dysplasia of cervical and intracranial arteries. Int J Stroke 2010; 5:296–305.
Savard S, Azarine A, Jeunemaitre X, Azizi M, Plouin PF, Steichen O. Association of smoking with phenotype at diagnosis and vascular interventions in patients with renal artery fibromuscular dysplasia. Hypertension 2013; 61:1227–1232.
Schievink WI, Limburg M. Angiographic abnormalities mimicking fibromuscular dysplasia in a patient with Ehlers-Danlos syndrome, type IV. Neurosurgery 1989; 25:482–483.
Schievink WI, Bjornsson J, Piepgras DG. Coexistence of fibromuscular dysplasia and cystic medial necrosis in a patient with Marfan’s syndrome and bilateral carotid artery dissections. Stroke 1994; 25:2492–2496.
Schievink WI, Bjournsson J, Parisi JE, Prakash UB. Arterial fibromuscular dysplasia associated with severe α1-antitrypsin deficiency. Mayo Clin Proc 1994; 69:1040–1043.
Schievink WI, Puumala MR, Meyer FB, Raffel C, Katzmann JA, Parisi JE. Giant intracranial aneurysm and fibromuscular dysplasia in an adolescent with α1-antitrypsin deficiency. J Neurosurg 1996; 85:503–506.
Schievink WI, Meyer FB, Parisi JE, Wijdicks EFM. Fibromuscular dysplasia of the internal carotid artery associated with alpha1-antitrypsin deficiency. Neurosurgery 1998; 43:229–233; discussion 233–234.
Bofinger A, Hawley C, Fisher P, Daunt N, Stowasser M, Gordon R. Alpha-1-antitrypsin phenotypes in patients with renal arterial fibromuscular dysplasia. J Hum Hypertens 2000; 14:91–94.
Poloskey SL, Kim ES, Sanghani R, et al. Low yield of genetic testing for known vascular connective tissue disorders in patients with fibromuscular dysplasia. Vasc Med 2012; 17:371–378.
Ganesh SK, Morissette R, Xu Z, et al. Clinical and biochemical profiles suggest fibromuscular dysplasia is a systemic disease with altered TGF- expression and connective tissue features. FASEB J 2014; 28:3313–3324.
O’Connor S, Kim ES, Brinza E, et al. Systemic connective tissue features in women with fibromuscular dysplasia. Vasc Med 2015; 20:454–462.
Saw J, Mancini GB, Humphries KH. Contemporary review on spontaneous coronary artery dissection. J Am Coll Cardiol 2016; 68:297–312.
Saw J, Aymong E, Sedlak T, et al. Spontaneous coronary artery dissection: association with predisposing arteriopathies and precipitating stressors and cardiovascular outcomes. Circ Cardiovasc Interv 2014; 7:645–655.
Prasad M, Tweet MS, Hayes SN, et al. Prevalence of extracoronary vascular abnormalities and fibromuscular dysplasia in patients with spontaneous coronary artery dissection. Am J Cardiol 2015; 115:1672–1677.
Bolen MA, Brinza E, Renapurkar RD, Kim ES, Gornik HL. Screening CT angiography of the aorta, visceral branch vessels, and pelvic arteries in fibromuscular dysplasia. JACC Cardiovasc Imaging. 2016; doi: 10.1016/j.jcmg.2016.04.010. [Epub ahead of print].
References
Olin JW, Gornik HL, Bacharach JM, et al. Fibromuscular dysplasia: state of the science and critical unanswered questions. A scientific statement from the American Heart Association. Circulation 2014; 129:1048–1078
O’Connor S, Gornik HL, Froehlich JB, et al. Smoking and adverse outcomes in fibromuscular dysplasia: US registry report. J Am Coll Cardiol 2016; 67:1750–1751.
Sang CN, Whelton PK, Hamper UM, et al. Etiologic factors in renovascular fibromuscular dysplasia. A case-control study. Hypertension 1989; 14:472–479.
Persu A, Touzé E, Mousseaux E, Barral X, Joffre F, Plouin PF. Diagnosis and management of fibromuscular dysplasia: an expert consensus. Eur J Clin Invest 2012; 42:338–347.
Sethi SS, Lau JF, Godbold J, Gustavson S, Olin JW. The S curve: a novel morphological finding in the internal carotid artery in patients with fibromuscular dysplasia. Vasc Med 2014; 19:356–362.
Kadian-Dodov D, Gornik HL, Gu X, et al. Dissection and aneurysm in patients with fibromuscular dysplasia: findings for the U.S. registry for FMD. J Am Coll Cardiol 2016; 68:176–185.
Olin JW, Froehlich J, Gu X, et al. The United States Registry for Fibromuscular Dysplasia: results in the first 447 patients. Circulation 2012; 125:3182–3190.
Kim ES, Olin JW, Froehlich JB, et al. Clinical manifestations of fibromuscular dysplasia vary by patient sex: a report of the United States Registry for Fibromuscular Dysplasia. J Am Coll Cardiol 2013; 62:2026–2028.
Weinberg I, Gu X, Giri J, et al. Anti-platelet and anti-hypertension medication use in patients with fibromuscular dysplasia: results from the United States Registry for Fibromuscular Dysplasia. Vasc Med 2015; 20:447–453.
Green R, Gu X, Kline-Rogers E, et al. Differences between the pediatric and adult presentation of fibromuscular dysplasia: results from the US Registry. Pediatr Nephrol 2016; 31:641–650.
Shivapour DM, Erwin P, Kim ESH. Epidemiology of fibromuscular dysplasia: a review of the literature. Vasc Med 2016; 21:376–381.
Spring DB, Satvatierra O Jr, Palubinskas AJ, Amend WJ Jr, Vincenti FG, Feduska NJ. Results and significance of angiography in potential kidney donors. Radiology 1979; 133:45–47.
Cragg AH, Smith TP, Thompson BH, et al. Incidental fibromuscular dysplasia in potential renal donors: long-term clinical follow-up. Radiology 1989; 172:145–147.
Neymark E, LaBerge JM, Hirose R, et al. Arteriographic detection of renovascular disease in potential renal donors: incidence and effect on donor surgery. Radiology 2000; 214:755–760.
Andreoni KA, Weeks SM, Gerber DA, et al. Incidence of donor renal fibromuscular dysplasia: does it justify routine angiography? Transplantation 2002; 73:1112–1116.
Blondin D, Lanzman R, Schellhammer F, et al. Fibromuscular dysplasia in living renal donors: still a challenge to computed tomographic angiography. Eur J Radiol 2010; 75:67–71.
Lorenz EC, Vrtiska TJ, Lieske JC, et al. Prevalence of renal artery and kidney abnormalities by computed tomography among healthy adults. Clin J Am Soc Nephrol 2010; 5:431–438.
McKenzie GA, Oderich GS, Kawashima A, Misra S. Renal artery fibromuscular dysplasia in 2,640 renal donor subjects: a CT angiography analysis. J Vasc Interv Radiol 2013; 24:1477–1480.
Frick MP, Goldberg ME. Uro- and angiographic findings in a “normal” population: screening of 151 symptom-free potential transplant donors for renal disease. AJR Am J Roentgenol 1980; 134:503–505.
Hendricks NJ, Matsumoto AH, Angle JF, et al. Is fibromuscular dysplasia underdiagnosed? A comparison of the prevalence of FMD seen in CORAL trial participants versus a single institution population of renal donor candidates. Vasc Med 2014; 19:363–367.
Touzé E, Oppenheim C, Trystram D, et al. Fibromuscular dysplasia of cervical and intracranial arteries. Int J Stroke 2010; 5:296–305.
Savard S, Azarine A, Jeunemaitre X, Azizi M, Plouin PF, Steichen O. Association of smoking with phenotype at diagnosis and vascular interventions in patients with renal artery fibromuscular dysplasia. Hypertension 2013; 61:1227–1232.
Schievink WI, Limburg M. Angiographic abnormalities mimicking fibromuscular dysplasia in a patient with Ehlers-Danlos syndrome, type IV. Neurosurgery 1989; 25:482–483.
Schievink WI, Bjornsson J, Piepgras DG. Coexistence of fibromuscular dysplasia and cystic medial necrosis in a patient with Marfan’s syndrome and bilateral carotid artery dissections. Stroke 1994; 25:2492–2496.
Schievink WI, Bjournsson J, Parisi JE, Prakash UB. Arterial fibromuscular dysplasia associated with severe α1-antitrypsin deficiency. Mayo Clin Proc 1994; 69:1040–1043.
Schievink WI, Puumala MR, Meyer FB, Raffel C, Katzmann JA, Parisi JE. Giant intracranial aneurysm and fibromuscular dysplasia in an adolescent with α1-antitrypsin deficiency. J Neurosurg 1996; 85:503–506.
Schievink WI, Meyer FB, Parisi JE, Wijdicks EFM. Fibromuscular dysplasia of the internal carotid artery associated with alpha1-antitrypsin deficiency. Neurosurgery 1998; 43:229–233; discussion 233–234.
Bofinger A, Hawley C, Fisher P, Daunt N, Stowasser M, Gordon R. Alpha-1-antitrypsin phenotypes in patients with renal arterial fibromuscular dysplasia. J Hum Hypertens 2000; 14:91–94.
Poloskey SL, Kim ES, Sanghani R, et al. Low yield of genetic testing for known vascular connective tissue disorders in patients with fibromuscular dysplasia. Vasc Med 2012; 17:371–378.
Ganesh SK, Morissette R, Xu Z, et al. Clinical and biochemical profiles suggest fibromuscular dysplasia is a systemic disease with altered TGF- expression and connective tissue features. FASEB J 2014; 28:3313–3324.
O’Connor S, Kim ES, Brinza E, et al. Systemic connective tissue features in women with fibromuscular dysplasia. Vasc Med 2015; 20:454–462.
Saw J, Mancini GB, Humphries KH. Contemporary review on spontaneous coronary artery dissection. J Am Coll Cardiol 2016; 68:297–312.
Saw J, Aymong E, Sedlak T, et al. Spontaneous coronary artery dissection: association with predisposing arteriopathies and precipitating stressors and cardiovascular outcomes. Circ Cardiovasc Interv 2014; 7:645–655.
Prasad M, Tweet MS, Hayes SN, et al. Prevalence of extracoronary vascular abnormalities and fibromuscular dysplasia in patients with spontaneous coronary artery dissection. Am J Cardiol 2015; 115:1672–1677.
Bolen MA, Brinza E, Renapurkar RD, Kim ES, Gornik HL. Screening CT angiography of the aorta, visceral branch vessels, and pelvic arteries in fibromuscular dysplasia. JACC Cardiovasc Imaging. 2016; doi: 10.1016/j.jcmg.2016.04.010. [Epub ahead of print].
Fibromuscular dysplasia: Advances in understanding and management
Display Headline
Fibromuscular dysplasia: Advances in understanding and management
Legacy Keywords
fibromuscular dysplasia, FMD, stenosis, string of beads, dissection, aneurysm, Ellen Brinza, Heather Gornik
Legacy Keywords
fibromuscular dysplasia, FMD, stenosis, string of beads, dissection, aneurysm, Ellen Brinza, Heather Gornik
Citation Override
Cleveland Clinic Journal of Medicine 2016 November; 83(suppl 2):S45-S51
Inside the Article
KEY POINTS
There is no cure for FMD. Management focuses on thorough evaluation and surveillance, lifestyle modification, and treatment of symptoms. Vascular procedures, such as angioplasty or treatment of aneurysms, are required for some patients.
The overwhelming majority (> 90%) of patients with FMD are women. But men seem to have a more aggressive course, with a rate of aneurysm or dissection two times higher than that in women.
The disease can affect medium-sized vessels throughout the body. In addition to the typical “string-of-beads” appearance or focal lesions, manifestations include arterial tortuosity, aneurysm, and dissection.
Disallow All Ads
Alternative CME
Consolidated Pubs: Do Not Show Source Publication Logo
It is much more important to know what sort of a patient has a disease than what sort of a disease a patient has.
—Attributed to Sir William Osler1
Recent yearshave seen an increase in people traveling away from their home region for healthcare, often for care that is less expensive or unavailable where they live.2–4 Many Americans seek care abroad (engaging in “medical tourism”); conversely, the United States annually receives thousands of foreign travelers for medical evaluations, a trend projected to increase.2,3,5 Additionally, US healthcare providers often see foreign travelers for unexpected ailments that develop during their time here.
Traveling for healthcare can be stressful for patients, and caring for international patients may pose challenges for providers and medical centers. On the other hand, such encounters also provide many mutual benefits. Unfortunately, there is little published guidance addressing these issues.2 In this article, we therefore discuss many of the benefits and challenges, with the hope of improving the quality of care delivered and the clinical experience for both providers and patients.
CHALLENGES FOR INTERNATIONAL PATIENTS AND THEIR PROVIDERS
Some scenarios that illustrate challenges faced by international patients and their healthcare providers are presented in Table 1.
For patients, heightened anxiety
Many international patients feel anxious, isolated, and vulnerable, particularly if they have never been away from home before. These feelings arise from multiple factors, including the stress of traveling, lack of family or social support, an unfamiliar environment, contrasting cultural practices, and high expectations.3,4 Language barriers, especially for patients who speak uncommon dialects, and lack of continuously available interpretive services often augment the unsettled emotions of international patients.
Cultural differences
International patients may quickly notice significant differences from their home country in how healthcare is practiced and culturally applied.4,6 Such differences may include dress codes and the comparatively equal role of women vis-à-vis men in the Western medical profession.
For cultural, personal, or religious reasons, some patients feel uncomfortable with healthcare providers of the opposite sex. This discomfort can be heightened if the patient needs a potentially uncomfortable and humiliating procedure such as a gynecologic or rectal examination.
The multidisciplinary team approach to healthcare, which can include trainees, nurses, and pharmacists, may leave patients confused about who their primary health provider is.
Decision-making also has cultural implications. In Western medicine, we respect individual autonomy and expect patients to participate in decisions about their care. However, in many areas of the world, medical decision-making is deferred to extended family members or cultural leaders.2 Additional and often repeated conversations may be needed with both the patient and family members to ensure appropriate understanding and ethical consent for care.
Some international patients may have expectations that are quite different from those of the healthcare provider and that are sometimes unrealistic.2,6
Institutional challenges
Many medical conditions require prolonged treatment and longitudinal care, a notable challenge when that care is delivered outside of one’s home country. Practice models within a clinic may not allow for prolonged subsequent visits, which may be needed to accommodate language-translation services. Complex multidisciplinary plans of care must somehow effectively utilize available appointment slots and be time-efficient.
Criteria for hospitalization differ widely among different countries, often based on resources, and may necessitate additional dialogue between the patient and healthcare provider.
Obtaining, interpreting the patient’s record
Medical records from foreign institutions are often unavailable, incomplete, or illegible. Further, depending on the country, it may be difficult to contact local providers for supplemental information. Differences in time zones, limited access to technology, language barriers, and handwritten notes all pose problems when trying to obtain additional information.
Many under-resourced foreign medical centers cannot duplicate medical records and radiographic films for the patient to bring to the United States. Medical records from foreign laboratories often raise questions about the quality, accuracy, and methodology of the testing platform used.2 Thus, the provider may need to start over and repeat the entire clinical, radiologic, and laboratory evaluation.
Communicating with the patient
Difficulties in communication between patients and providers can hinder the development of a positive and productive relationship, reducing patient autonomy and complicating informed consent.2 Obtaining a medical history from non–English-speaking patients can be arduous and time-consuming. Colloquial language may further alter interpretation and understanding, even for formally trained interpreters. Language differences may make it more difficult to explain differential diagnoses, diagnostic approaches, and management plans.
Many US medical centers provide interpreters for many languages, but the great number of languages spoken around the world ensures that barriers in communication persist. Telephone language lines and other commercial language services are available but may feel less personal to patients or evoke concerns about medical confidentiality. For less commonly spoken languages and dialects, appropriate translation services may not even be available.6
Filling in information gaps
Medical conditions, medications, and treatments may have different names in different countries. The quality of pharmaceuticals in some regions may be questionable, and herbal supplements may be unique to a particular location. Many medications available abroad are not available in the United States, potentially confusing US providers as to medication appropriateness, efficacy, and potential toxicities.
Lacking adequate medical records and trying to obtain a new medical history from patients and their family members, providers may struggle with continued gaps of information, hindering a timely diagnosis and composition of an appropriate management plan.
A culturally sensitive but complete physical examination
Every effort should be made to complete a thorough and comprehensive physical examination, even if the patient’s culture differs on this point. This may require a “chaperone” to be present or, if available, a clinician of the same sex as the patient to perform the examination. A compromised examination will impede making the correct diagnosis.
Religious, cultural, and other patient-specific attitudes and beliefs that may affect a medical evaluation should ideally be addressed before scheduling the appointment. A preexamination discussion with the patient and family can help avert unintentional actions and behavior misperceived as offensive, while strengthening the level of trust between patient and provider.2
Money matters
Foreign patients typically have limited or no medical insurance coverage and thus may be paying out of pocket or through limited governmental subsidies. Many refugees and asylum-seekers have no insurance or money to pay for care. (A full discussion of refugee care is beyond the scope of this article). Thus, it is necessary to ascertain in advance who will pay for the care.
Clinicians must be sensitive to the exorbitantcosts of medical care and medications in the United States, particularly from the perspective of foreign patients. We strive to provide the best cost-effective care, but what is considered cost-effective and standard care for a patient with US health insurance may be viewed differently by international patients. For some foreign patients, some tests and treatments may be just too expensive, raising personal and institutional ethical concerns regarding how best to evaluate and manage these patients. Ideally, these issues should also be addressed before the patient’s appointment is scheduled.
Clinicians must optimize diagnostic and medical management while minimizing unnecessary testing. This principle further underscores the importance of obtaining a complete medical history and physical examination within a time-sensitive and well-coordinated plan of care.2,4
Continuity of care after the patient leaves
As the medical evaluation and care plan approach completion, ensuring some form of continued medical care can become challenging. Some foreign patients may have the financial or legal means (eg, through an extended medical visa) to remain for further care and follow-up, but most do not.
Finding an available, willing health provider in the patient’s native country for continued management may be difficult and time-consuming. Most US medical centers have no established system to identify available foreign health providers, and usually the patient and family are responsible for arranging continued healthcare back in their home country.
Opportunities for possible improvement of care are noted in Table 2.
ADVANTAGES OF CARING FOR INTERNATIONAL PATIENTS
Despite the possible challenges, there are many benefits of caring for international patients.
Gaining medical knowledge
In US medical centers caring for both regional and referred patients, providers are often exposed to medical conditions that range from common ailments to the rare conditions (or “zebras”) taught during residency training. From the medical education standpoint, international patients provide US health providers heightened opportunities to encounter diseases not commonly seen in the United States (eg, infections such as malaria, schistosomiasis, drug-resistant tuberculosis, and advanced or end-stage forms of noncommunicable diseases). Although not limited to international patients, chronically neglected diseases often give providers first-hand experience in the natural history of select disease progression.
Gaining cultural knowledge
Caring for international patients also enables health providers to learn about different cultures, societal norms, and regional beliefs affecting healthcare. In essence, international patients enable US providers to become more diversified and enlightened with communication skills and assorted managerial strategies on a global scale.
These patients remind us of the stark differences regarding access and quality of medical care globally, particularly in lesser-resourced locations. In a busy domestic medical practice with its own daily challenges, many of us forget these international healthcare disparities, and often take for granted the comparative abundance of healthcare resources available in the United States. Provider frustrations about domestic policies and concerns for a “broken” healthcare system often blind us to the available resources we are fortunate to have at our disposal.
Further, as members of the global community, we have the opportunity to learn from international patients while broadening our view of humanity, thereby enhancing our awareness and empathy toward patients and communities struggling with under-resourced healthcare systems. Healthcare providers are often touched by the gratitude of patients for the opportunity to receive treatments that may otherwise be unavailable. Such experiences may motivate many US health providers to become more engaged in coordinated strategies for global health improvement.
Reimbursement is possible
Caring for international patients should not financially deter US health care centers. Complex, multidisciplinary care evaluations may incur notable expenses; however, alternative and more lucrative payer systems, including government subsidies, can be involved to maintain revenue, reimbursements, and even possibly lead to increased donations.3–5 Given the potential for high costs to be incurred, US providers and institutions need to continually ensure appropriate evidence-based use of resources and cost-effective care without compromising the quality of care provided. The price of certain drugs has been rising astonishingly in the United States, and some patients may therefore prefer to obtain them for long-term use upon return to their home country.
High-quality cost-effective care is satisfying to the patient, provider, and institution, and also may save money that can be reallocated.4 Providers also may find personal fulfillment in striving for and achieving such goals, despite the potential challenges throughout the course of care.
Opportunities for improvement
Regardless of the challenges presented by international patients, participating medical centers often enjoy the prestige and credibility of becoming an “international healthcare center.”4,7 From the standpoint of medical education, these centers have the potential to train providers with increased clinical and cultural competencies along with expanding healthcare services to include clinical, educational and research opportunities abroad.
Research is needed to provide evidence-based guidance on best strategies for patients, clinicians, and healthcare systems to effectively care for international patients.
Suggested opportunities for maximizing advantages are noted in Table 3.
Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006; 81:189–192.
Bower LC, Johnson TJ, Hohmann SF, Garman AN, Allen M, Meurer SJ. An evaluation of international patient length of stay. Int J Healthc Manag 2014; 7:200–205.
Satjapot SP, Johnson TJ, Garman AN. International medical travelers, length of stay, and the continuum of care: inquiry and comparison. Qual Manag Health Care 2011; 20:76–83.
Donohoe M. Luxury primary care, academic medical centers, and the erosion of science and professional ethics. J Gen Intern Med 2004; 19:90–94.
Dogan H, Tschudin V, Hot I, Özkan I. Patients’ transcultural needs and carers’ ethical responses. Nurs Ethics 2009; 16:683–696.
Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010; 61:765–773.
It is much more important to know what sort of a patient has a disease than what sort of a disease a patient has.
—Attributed to Sir William Osler1
Recent yearshave seen an increase in people traveling away from their home region for healthcare, often for care that is less expensive or unavailable where they live.2–4 Many Americans seek care abroad (engaging in “medical tourism”); conversely, the United States annually receives thousands of foreign travelers for medical evaluations, a trend projected to increase.2,3,5 Additionally, US healthcare providers often see foreign travelers for unexpected ailments that develop during their time here.
Traveling for healthcare can be stressful for patients, and caring for international patients may pose challenges for providers and medical centers. On the other hand, such encounters also provide many mutual benefits. Unfortunately, there is little published guidance addressing these issues.2 In this article, we therefore discuss many of the benefits and challenges, with the hope of improving the quality of care delivered and the clinical experience for both providers and patients.
CHALLENGES FOR INTERNATIONAL PATIENTS AND THEIR PROVIDERS
Some scenarios that illustrate challenges faced by international patients and their healthcare providers are presented in Table 1.
For patients, heightened anxiety
Many international patients feel anxious, isolated, and vulnerable, particularly if they have never been away from home before. These feelings arise from multiple factors, including the stress of traveling, lack of family or social support, an unfamiliar environment, contrasting cultural practices, and high expectations.3,4 Language barriers, especially for patients who speak uncommon dialects, and lack of continuously available interpretive services often augment the unsettled emotions of international patients.
Cultural differences
International patients may quickly notice significant differences from their home country in how healthcare is practiced and culturally applied.4,6 Such differences may include dress codes and the comparatively equal role of women vis-à-vis men in the Western medical profession.
For cultural, personal, or religious reasons, some patients feel uncomfortable with healthcare providers of the opposite sex. This discomfort can be heightened if the patient needs a potentially uncomfortable and humiliating procedure such as a gynecologic or rectal examination.
The multidisciplinary team approach to healthcare, which can include trainees, nurses, and pharmacists, may leave patients confused about who their primary health provider is.
Decision-making also has cultural implications. In Western medicine, we respect individual autonomy and expect patients to participate in decisions about their care. However, in many areas of the world, medical decision-making is deferred to extended family members or cultural leaders.2 Additional and often repeated conversations may be needed with both the patient and family members to ensure appropriate understanding and ethical consent for care.
Some international patients may have expectations that are quite different from those of the healthcare provider and that are sometimes unrealistic.2,6
Institutional challenges
Many medical conditions require prolonged treatment and longitudinal care, a notable challenge when that care is delivered outside of one’s home country. Practice models within a clinic may not allow for prolonged subsequent visits, which may be needed to accommodate language-translation services. Complex multidisciplinary plans of care must somehow effectively utilize available appointment slots and be time-efficient.
Criteria for hospitalization differ widely among different countries, often based on resources, and may necessitate additional dialogue between the patient and healthcare provider.
Obtaining, interpreting the patient’s record
Medical records from foreign institutions are often unavailable, incomplete, or illegible. Further, depending on the country, it may be difficult to contact local providers for supplemental information. Differences in time zones, limited access to technology, language barriers, and handwritten notes all pose problems when trying to obtain additional information.
Many under-resourced foreign medical centers cannot duplicate medical records and radiographic films for the patient to bring to the United States. Medical records from foreign laboratories often raise questions about the quality, accuracy, and methodology of the testing platform used.2 Thus, the provider may need to start over and repeat the entire clinical, radiologic, and laboratory evaluation.
Communicating with the patient
Difficulties in communication between patients and providers can hinder the development of a positive and productive relationship, reducing patient autonomy and complicating informed consent.2 Obtaining a medical history from non–English-speaking patients can be arduous and time-consuming. Colloquial language may further alter interpretation and understanding, even for formally trained interpreters. Language differences may make it more difficult to explain differential diagnoses, diagnostic approaches, and management plans.
Many US medical centers provide interpreters for many languages, but the great number of languages spoken around the world ensures that barriers in communication persist. Telephone language lines and other commercial language services are available but may feel less personal to patients or evoke concerns about medical confidentiality. For less commonly spoken languages and dialects, appropriate translation services may not even be available.6
Filling in information gaps
Medical conditions, medications, and treatments may have different names in different countries. The quality of pharmaceuticals in some regions may be questionable, and herbal supplements may be unique to a particular location. Many medications available abroad are not available in the United States, potentially confusing US providers as to medication appropriateness, efficacy, and potential toxicities.
Lacking adequate medical records and trying to obtain a new medical history from patients and their family members, providers may struggle with continued gaps of information, hindering a timely diagnosis and composition of an appropriate management plan.
A culturally sensitive but complete physical examination
Every effort should be made to complete a thorough and comprehensive physical examination, even if the patient’s culture differs on this point. This may require a “chaperone” to be present or, if available, a clinician of the same sex as the patient to perform the examination. A compromised examination will impede making the correct diagnosis.
Religious, cultural, and other patient-specific attitudes and beliefs that may affect a medical evaluation should ideally be addressed before scheduling the appointment. A preexamination discussion with the patient and family can help avert unintentional actions and behavior misperceived as offensive, while strengthening the level of trust between patient and provider.2
Money matters
Foreign patients typically have limited or no medical insurance coverage and thus may be paying out of pocket or through limited governmental subsidies. Many refugees and asylum-seekers have no insurance or money to pay for care. (A full discussion of refugee care is beyond the scope of this article). Thus, it is necessary to ascertain in advance who will pay for the care.
Clinicians must be sensitive to the exorbitantcosts of medical care and medications in the United States, particularly from the perspective of foreign patients. We strive to provide the best cost-effective care, but what is considered cost-effective and standard care for a patient with US health insurance may be viewed differently by international patients. For some foreign patients, some tests and treatments may be just too expensive, raising personal and institutional ethical concerns regarding how best to evaluate and manage these patients. Ideally, these issues should also be addressed before the patient’s appointment is scheduled.
Clinicians must optimize diagnostic and medical management while minimizing unnecessary testing. This principle further underscores the importance of obtaining a complete medical history and physical examination within a time-sensitive and well-coordinated plan of care.2,4
Continuity of care after the patient leaves
As the medical evaluation and care plan approach completion, ensuring some form of continued medical care can become challenging. Some foreign patients may have the financial or legal means (eg, through an extended medical visa) to remain for further care and follow-up, but most do not.
Finding an available, willing health provider in the patient’s native country for continued management may be difficult and time-consuming. Most US medical centers have no established system to identify available foreign health providers, and usually the patient and family are responsible for arranging continued healthcare back in their home country.
Opportunities for possible improvement of care are noted in Table 2.
ADVANTAGES OF CARING FOR INTERNATIONAL PATIENTS
Despite the possible challenges, there are many benefits of caring for international patients.
Gaining medical knowledge
In US medical centers caring for both regional and referred patients, providers are often exposed to medical conditions that range from common ailments to the rare conditions (or “zebras”) taught during residency training. From the medical education standpoint, international patients provide US health providers heightened opportunities to encounter diseases not commonly seen in the United States (eg, infections such as malaria, schistosomiasis, drug-resistant tuberculosis, and advanced or end-stage forms of noncommunicable diseases). Although not limited to international patients, chronically neglected diseases often give providers first-hand experience in the natural history of select disease progression.
Gaining cultural knowledge
Caring for international patients also enables health providers to learn about different cultures, societal norms, and regional beliefs affecting healthcare. In essence, international patients enable US providers to become more diversified and enlightened with communication skills and assorted managerial strategies on a global scale.
These patients remind us of the stark differences regarding access and quality of medical care globally, particularly in lesser-resourced locations. In a busy domestic medical practice with its own daily challenges, many of us forget these international healthcare disparities, and often take for granted the comparative abundance of healthcare resources available in the United States. Provider frustrations about domestic policies and concerns for a “broken” healthcare system often blind us to the available resources we are fortunate to have at our disposal.
Further, as members of the global community, we have the opportunity to learn from international patients while broadening our view of humanity, thereby enhancing our awareness and empathy toward patients and communities struggling with under-resourced healthcare systems. Healthcare providers are often touched by the gratitude of patients for the opportunity to receive treatments that may otherwise be unavailable. Such experiences may motivate many US health providers to become more engaged in coordinated strategies for global health improvement.
Reimbursement is possible
Caring for international patients should not financially deter US health care centers. Complex, multidisciplinary care evaluations may incur notable expenses; however, alternative and more lucrative payer systems, including government subsidies, can be involved to maintain revenue, reimbursements, and even possibly lead to increased donations.3–5 Given the potential for high costs to be incurred, US providers and institutions need to continually ensure appropriate evidence-based use of resources and cost-effective care without compromising the quality of care provided. The price of certain drugs has been rising astonishingly in the United States, and some patients may therefore prefer to obtain them for long-term use upon return to their home country.
High-quality cost-effective care is satisfying to the patient, provider, and institution, and also may save money that can be reallocated.4 Providers also may find personal fulfillment in striving for and achieving such goals, despite the potential challenges throughout the course of care.
Opportunities for improvement
Regardless of the challenges presented by international patients, participating medical centers often enjoy the prestige and credibility of becoming an “international healthcare center.”4,7 From the standpoint of medical education, these centers have the potential to train providers with increased clinical and cultural competencies along with expanding healthcare services to include clinical, educational and research opportunities abroad.
Research is needed to provide evidence-based guidance on best strategies for patients, clinicians, and healthcare systems to effectively care for international patients.
Suggested opportunities for maximizing advantages are noted in Table 3.
It is much more important to know what sort of a patient has a disease than what sort of a disease a patient has.
—Attributed to Sir William Osler1
Recent yearshave seen an increase in people traveling away from their home region for healthcare, often for care that is less expensive or unavailable where they live.2–4 Many Americans seek care abroad (engaging in “medical tourism”); conversely, the United States annually receives thousands of foreign travelers for medical evaluations, a trend projected to increase.2,3,5 Additionally, US healthcare providers often see foreign travelers for unexpected ailments that develop during their time here.
Traveling for healthcare can be stressful for patients, and caring for international patients may pose challenges for providers and medical centers. On the other hand, such encounters also provide many mutual benefits. Unfortunately, there is little published guidance addressing these issues.2 In this article, we therefore discuss many of the benefits and challenges, with the hope of improving the quality of care delivered and the clinical experience for both providers and patients.
CHALLENGES FOR INTERNATIONAL PATIENTS AND THEIR PROVIDERS
Some scenarios that illustrate challenges faced by international patients and their healthcare providers are presented in Table 1.
For patients, heightened anxiety
Many international patients feel anxious, isolated, and vulnerable, particularly if they have never been away from home before. These feelings arise from multiple factors, including the stress of traveling, lack of family or social support, an unfamiliar environment, contrasting cultural practices, and high expectations.3,4 Language barriers, especially for patients who speak uncommon dialects, and lack of continuously available interpretive services often augment the unsettled emotions of international patients.
Cultural differences
International patients may quickly notice significant differences from their home country in how healthcare is practiced and culturally applied.4,6 Such differences may include dress codes and the comparatively equal role of women vis-à-vis men in the Western medical profession.
For cultural, personal, or religious reasons, some patients feel uncomfortable with healthcare providers of the opposite sex. This discomfort can be heightened if the patient needs a potentially uncomfortable and humiliating procedure such as a gynecologic or rectal examination.
The multidisciplinary team approach to healthcare, which can include trainees, nurses, and pharmacists, may leave patients confused about who their primary health provider is.
Decision-making also has cultural implications. In Western medicine, we respect individual autonomy and expect patients to participate in decisions about their care. However, in many areas of the world, medical decision-making is deferred to extended family members or cultural leaders.2 Additional and often repeated conversations may be needed with both the patient and family members to ensure appropriate understanding and ethical consent for care.
Some international patients may have expectations that are quite different from those of the healthcare provider and that are sometimes unrealistic.2,6
Institutional challenges
Many medical conditions require prolonged treatment and longitudinal care, a notable challenge when that care is delivered outside of one’s home country. Practice models within a clinic may not allow for prolonged subsequent visits, which may be needed to accommodate language-translation services. Complex multidisciplinary plans of care must somehow effectively utilize available appointment slots and be time-efficient.
Criteria for hospitalization differ widely among different countries, often based on resources, and may necessitate additional dialogue between the patient and healthcare provider.
Obtaining, interpreting the patient’s record
Medical records from foreign institutions are often unavailable, incomplete, or illegible. Further, depending on the country, it may be difficult to contact local providers for supplemental information. Differences in time zones, limited access to technology, language barriers, and handwritten notes all pose problems when trying to obtain additional information.
Many under-resourced foreign medical centers cannot duplicate medical records and radiographic films for the patient to bring to the United States. Medical records from foreign laboratories often raise questions about the quality, accuracy, and methodology of the testing platform used.2 Thus, the provider may need to start over and repeat the entire clinical, radiologic, and laboratory evaluation.
Communicating with the patient
Difficulties in communication between patients and providers can hinder the development of a positive and productive relationship, reducing patient autonomy and complicating informed consent.2 Obtaining a medical history from non–English-speaking patients can be arduous and time-consuming. Colloquial language may further alter interpretation and understanding, even for formally trained interpreters. Language differences may make it more difficult to explain differential diagnoses, diagnostic approaches, and management plans.
Many US medical centers provide interpreters for many languages, but the great number of languages spoken around the world ensures that barriers in communication persist. Telephone language lines and other commercial language services are available but may feel less personal to patients or evoke concerns about medical confidentiality. For less commonly spoken languages and dialects, appropriate translation services may not even be available.6
Filling in information gaps
Medical conditions, medications, and treatments may have different names in different countries. The quality of pharmaceuticals in some regions may be questionable, and herbal supplements may be unique to a particular location. Many medications available abroad are not available in the United States, potentially confusing US providers as to medication appropriateness, efficacy, and potential toxicities.
Lacking adequate medical records and trying to obtain a new medical history from patients and their family members, providers may struggle with continued gaps of information, hindering a timely diagnosis and composition of an appropriate management plan.
A culturally sensitive but complete physical examination
Every effort should be made to complete a thorough and comprehensive physical examination, even if the patient’s culture differs on this point. This may require a “chaperone” to be present or, if available, a clinician of the same sex as the patient to perform the examination. A compromised examination will impede making the correct diagnosis.
Religious, cultural, and other patient-specific attitudes and beliefs that may affect a medical evaluation should ideally be addressed before scheduling the appointment. A preexamination discussion with the patient and family can help avert unintentional actions and behavior misperceived as offensive, while strengthening the level of trust between patient and provider.2
Money matters
Foreign patients typically have limited or no medical insurance coverage and thus may be paying out of pocket or through limited governmental subsidies. Many refugees and asylum-seekers have no insurance or money to pay for care. (A full discussion of refugee care is beyond the scope of this article). Thus, it is necessary to ascertain in advance who will pay for the care.
Clinicians must be sensitive to the exorbitantcosts of medical care and medications in the United States, particularly from the perspective of foreign patients. We strive to provide the best cost-effective care, but what is considered cost-effective and standard care for a patient with US health insurance may be viewed differently by international patients. For some foreign patients, some tests and treatments may be just too expensive, raising personal and institutional ethical concerns regarding how best to evaluate and manage these patients. Ideally, these issues should also be addressed before the patient’s appointment is scheduled.
Clinicians must optimize diagnostic and medical management while minimizing unnecessary testing. This principle further underscores the importance of obtaining a complete medical history and physical examination within a time-sensitive and well-coordinated plan of care.2,4
Continuity of care after the patient leaves
As the medical evaluation and care plan approach completion, ensuring some form of continued medical care can become challenging. Some foreign patients may have the financial or legal means (eg, through an extended medical visa) to remain for further care and follow-up, but most do not.
Finding an available, willing health provider in the patient’s native country for continued management may be difficult and time-consuming. Most US medical centers have no established system to identify available foreign health providers, and usually the patient and family are responsible for arranging continued healthcare back in their home country.
Opportunities for possible improvement of care are noted in Table 2.
ADVANTAGES OF CARING FOR INTERNATIONAL PATIENTS
Despite the possible challenges, there are many benefits of caring for international patients.
Gaining medical knowledge
In US medical centers caring for both regional and referred patients, providers are often exposed to medical conditions that range from common ailments to the rare conditions (or “zebras”) taught during residency training. From the medical education standpoint, international patients provide US health providers heightened opportunities to encounter diseases not commonly seen in the United States (eg, infections such as malaria, schistosomiasis, drug-resistant tuberculosis, and advanced or end-stage forms of noncommunicable diseases). Although not limited to international patients, chronically neglected diseases often give providers first-hand experience in the natural history of select disease progression.
Gaining cultural knowledge
Caring for international patients also enables health providers to learn about different cultures, societal norms, and regional beliefs affecting healthcare. In essence, international patients enable US providers to become more diversified and enlightened with communication skills and assorted managerial strategies on a global scale.
These patients remind us of the stark differences regarding access and quality of medical care globally, particularly in lesser-resourced locations. In a busy domestic medical practice with its own daily challenges, many of us forget these international healthcare disparities, and often take for granted the comparative abundance of healthcare resources available in the United States. Provider frustrations about domestic policies and concerns for a “broken” healthcare system often blind us to the available resources we are fortunate to have at our disposal.
Further, as members of the global community, we have the opportunity to learn from international patients while broadening our view of humanity, thereby enhancing our awareness and empathy toward patients and communities struggling with under-resourced healthcare systems. Healthcare providers are often touched by the gratitude of patients for the opportunity to receive treatments that may otherwise be unavailable. Such experiences may motivate many US health providers to become more engaged in coordinated strategies for global health improvement.
Reimbursement is possible
Caring for international patients should not financially deter US health care centers. Complex, multidisciplinary care evaluations may incur notable expenses; however, alternative and more lucrative payer systems, including government subsidies, can be involved to maintain revenue, reimbursements, and even possibly lead to increased donations.3–5 Given the potential for high costs to be incurred, US providers and institutions need to continually ensure appropriate evidence-based use of resources and cost-effective care without compromising the quality of care provided. The price of certain drugs has been rising astonishingly in the United States, and some patients may therefore prefer to obtain them for long-term use upon return to their home country.
High-quality cost-effective care is satisfying to the patient, provider, and institution, and also may save money that can be reallocated.4 Providers also may find personal fulfillment in striving for and achieving such goals, despite the potential challenges throughout the course of care.
Opportunities for improvement
Regardless of the challenges presented by international patients, participating medical centers often enjoy the prestige and credibility of becoming an “international healthcare center.”4,7 From the standpoint of medical education, these centers have the potential to train providers with increased clinical and cultural competencies along with expanding healthcare services to include clinical, educational and research opportunities abroad.
Research is needed to provide evidence-based guidance on best strategies for patients, clinicians, and healthcare systems to effectively care for international patients.
Suggested opportunities for maximizing advantages are noted in Table 3.
Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006; 81:189–192.
Bower LC, Johnson TJ, Hohmann SF, Garman AN, Allen M, Meurer SJ. An evaluation of international patient length of stay. Int J Healthc Manag 2014; 7:200–205.
Satjapot SP, Johnson TJ, Garman AN. International medical travelers, length of stay, and the continuum of care: inquiry and comparison. Qual Manag Health Care 2011; 20:76–83.
Donohoe M. Luxury primary care, academic medical centers, and the erosion of science and professional ethics. J Gen Intern Med 2004; 19:90–94.
Dogan H, Tschudin V, Hot I, Özkan I. Patients’ transcultural needs and carers’ ethical responses. Nurs Ethics 2009; 16:683–696.
Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010; 61:765–773.
Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006; 81:189–192.
Bower LC, Johnson TJ, Hohmann SF, Garman AN, Allen M, Meurer SJ. An evaluation of international patient length of stay. Int J Healthc Manag 2014; 7:200–205.
Satjapot SP, Johnson TJ, Garman AN. International medical travelers, length of stay, and the continuum of care: inquiry and comparison. Qual Manag Health Care 2011; 20:76–83.
Donohoe M. Luxury primary care, academic medical centers, and the erosion of science and professional ethics. J Gen Intern Med 2004; 19:90–94.
Dogan H, Tschudin V, Hot I, Özkan I. Patients’ transcultural needs and carers’ ethical responses. Nurs Ethics 2009; 16:683–696.
Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010; 61:765–773.
Challenges in caring for international patients include cultural differences, institutional barriers, communication difficulties, sparse medical records, and financial considerations.
Understanding should be reached beforehand on potentially sensitive issues such as physical examinations, payment, tests, and treatment.
Benefits to the provider and institution include enhanced medical skills, cultural competency, personal satisfaction, and institutional prestige.