User login
Study: Reference pricing does reduce prescription costs

Reference pricing effectively encourages patients to spend significantly less on prescription drugs, according to research published in NEJM.
Under the reference pricing strategy, the insurer or employer establishes its maximum contribution toward the price of therapeutically similar drugs, and the patient must pay the remainder out of pocket.
The insurer/employer contribution is based on the price of the lowest-priced drug in the therapeutic class, called the reference drug.
“If the patient chooses a cheap or moderately priced option, the employer’s contribution will cover most of the cost,” said study author James C. Robinson, PhD, of the University of California at Berkeley.
“However, if the patient insists on a particularly high-priced option, he or she will need to make a meaningful payment from personal resources.”
It has been theorized that this policy would encourage patients to save money by selecting cheaper drugs. However, little is known about how the policy has actually influenced patient spending.
The new study showed that reference pricing was associated with significant changes in drug selection and spending for patients covered by employment-based insurance in the US.
Researchers analyzed changes in prescriptions and pricing for 1302 drugs in 78 therapeutic classes in the US, before and after an alliance of private employers began using reference pricing.
The trends were compared to a cohort without reference pricing. The study’s dataset included 1.1 million prescriptions reimbursed from 2010 to 2014.
Implementation of reference pricing was associated with a 7% increase in prescriptions filled for the low-price reference drug within its therapeutic class, a 14% decrease in average price paid, and a 5% increase in consumer cost-sharing.
In the first 18 months after implementation, employers’ spending dropped $1.34 million, and employees’ cost-sharing increased $120,000.
Based on these findings, Dr Robinson and his colleagues concluded that reference pricing may be one instrument for influencing patients’ drug choices and drug prices paid by employers and insurers. The team believes that, in the future, pharmaceutical companies charging “premium prices” may need to demonstrate that their drugs provide “premium performance.”
“There is huge and unjustified variation within and across geographic areas in the prices charged for almost every test and treatment, drug and device, office visit and hospitalization,” Dr Robinson said.
“It’s not a surprise when one considers that most patients are covered by health insurance and, hence, do not shop among competing providers on the basis of price. Some providers look at price-unconscious consumer demand and ask themselves, ‘Why don’t we raise our prices?’”
This research was funded by the US Agency for Healthcare Research and Quality and the Genentech Foundation. ![]()
Reference pricing effectively encourages patients to spend significantly less on prescription drugs, according to research published in NEJM.
Under the reference pricing strategy, the insurer or employer establishes its maximum contribution toward the price of therapeutically similar drugs, and the patient must pay the remainder out of pocket.
The insurer/employer contribution is based on the price of the lowest-priced drug in the therapeutic class, called the reference drug.
“If the patient chooses a cheap or moderately priced option, the employer’s contribution will cover most of the cost,” said study author James C. Robinson, PhD, of the University of California at Berkeley.
“However, if the patient insists on a particularly high-priced option, he or she will need to make a meaningful payment from personal resources.”
It has been theorized that this policy would encourage patients to save money by selecting cheaper drugs. However, little is known about how the policy has actually influenced patient spending.
The new study showed that reference pricing was associated with significant changes in drug selection and spending for patients covered by employment-based insurance in the US.
Researchers analyzed changes in prescriptions and pricing for 1302 drugs in 78 therapeutic classes in the US, before and after an alliance of private employers began using reference pricing.
The trends were compared to a cohort without reference pricing. The study’s dataset included 1.1 million prescriptions reimbursed from 2010 to 2014.
Implementation of reference pricing was associated with a 7% increase in prescriptions filled for the low-price reference drug within its therapeutic class, a 14% decrease in average price paid, and a 5% increase in consumer cost-sharing.
In the first 18 months after implementation, employers’ spending dropped $1.34 million, and employees’ cost-sharing increased $120,000.
Based on these findings, Dr Robinson and his colleagues concluded that reference pricing may be one instrument for influencing patients’ drug choices and drug prices paid by employers and insurers. The team believes that, in the future, pharmaceutical companies charging “premium prices” may need to demonstrate that their drugs provide “premium performance.”
“There is huge and unjustified variation within and across geographic areas in the prices charged for almost every test and treatment, drug and device, office visit and hospitalization,” Dr Robinson said.
“It’s not a surprise when one considers that most patients are covered by health insurance and, hence, do not shop among competing providers on the basis of price. Some providers look at price-unconscious consumer demand and ask themselves, ‘Why don’t we raise our prices?’”
This research was funded by the US Agency for Healthcare Research and Quality and the Genentech Foundation. ![]()
Reference pricing effectively encourages patients to spend significantly less on prescription drugs, according to research published in NEJM.
Under the reference pricing strategy, the insurer or employer establishes its maximum contribution toward the price of therapeutically similar drugs, and the patient must pay the remainder out of pocket.
The insurer/employer contribution is based on the price of the lowest-priced drug in the therapeutic class, called the reference drug.
“If the patient chooses a cheap or moderately priced option, the employer’s contribution will cover most of the cost,” said study author James C. Robinson, PhD, of the University of California at Berkeley.
“However, if the patient insists on a particularly high-priced option, he or she will need to make a meaningful payment from personal resources.”
It has been theorized that this policy would encourage patients to save money by selecting cheaper drugs. However, little is known about how the policy has actually influenced patient spending.
The new study showed that reference pricing was associated with significant changes in drug selection and spending for patients covered by employment-based insurance in the US.
Researchers analyzed changes in prescriptions and pricing for 1302 drugs in 78 therapeutic classes in the US, before and after an alliance of private employers began using reference pricing.
The trends were compared to a cohort without reference pricing. The study’s dataset included 1.1 million prescriptions reimbursed from 2010 to 2014.
Implementation of reference pricing was associated with a 7% increase in prescriptions filled for the low-price reference drug within its therapeutic class, a 14% decrease in average price paid, and a 5% increase in consumer cost-sharing.
In the first 18 months after implementation, employers’ spending dropped $1.34 million, and employees’ cost-sharing increased $120,000.
Based on these findings, Dr Robinson and his colleagues concluded that reference pricing may be one instrument for influencing patients’ drug choices and drug prices paid by employers and insurers. The team believes that, in the future, pharmaceutical companies charging “premium prices” may need to demonstrate that their drugs provide “premium performance.”
“There is huge and unjustified variation within and across geographic areas in the prices charged for almost every test and treatment, drug and device, office visit and hospitalization,” Dr Robinson said.
“It’s not a surprise when one considers that most patients are covered by health insurance and, hence, do not shop among competing providers on the basis of price. Some providers look at price-unconscious consumer demand and ask themselves, ‘Why don’t we raise our prices?’”
This research was funded by the US Agency for Healthcare Research and Quality and the Genentech Foundation. ![]()
Triplet eradicates B-ALL, prolongs survival in mice

A 3-drug combination has demonstrated long-term efficacy against acute lymphoblastic leukemia (ALL) in mice, according to research published in Nature Communications.
Researchers found that when the production of nucleotides is stopped, a DNA replication stress response is activated, and this allows ALL cells to survive.
The team used these findings to devise a 3-drug regimen that blocks both of the nucleotide biosynthetic pathways and inhibits the replication stress response.
One of the drugs is triapine (3-AP), which inhibits ribonucleotide reductase (RNR) and enhances salvage nucleotide biosynthesis.
Another drug is DI-82, which inhibits the nucleoside salvage kinase deoxycytidine kinase (dCK).
And the third drug is VE-822, which inhibits the replication stress-sensing kinase ataxia telangiectasia and Rad3-related protein (ATR).
The researchers tested these 3 drugs in a mouse model of pre-B-ALL. Starting on day 7 after inoculation of pre-B-ALL, the mice received 3-AP and DI-82 twice a day and VE-822 once a day.
The team said mice in the control group succumbed to their disease within 17 days. However, all of the mice that received the 3-drug combination were still disease-free 442 days after they had stopped treatment (on day 42).
The researchers said the combination was well-tolerated, as mice maintained their body weight and enjoyed long-term survival without any detectable pathology.
The team also tested a dosing regimen in which all 3 drugs were given once a day.
Although this was less effective than the first regimen, 4 of 5 mice had no detectable disease 313 days after stopping treatment.
Two of the researchers involved in this study are co-founders and equity holders of Trethera Corp, a company developing a dCK inhibitor known as TRE-515.
The University of California (where most of the study authors work) has patented intellectual property for small-molecule dCK inhibitors, and it was licensed by Trethera. ![]()
A 3-drug combination has demonstrated long-term efficacy against acute lymphoblastic leukemia (ALL) in mice, according to research published in Nature Communications.
Researchers found that when the production of nucleotides is stopped, a DNA replication stress response is activated, and this allows ALL cells to survive.
The team used these findings to devise a 3-drug regimen that blocks both of the nucleotide biosynthetic pathways and inhibits the replication stress response.
One of the drugs is triapine (3-AP), which inhibits ribonucleotide reductase (RNR) and enhances salvage nucleotide biosynthesis.
Another drug is DI-82, which inhibits the nucleoside salvage kinase deoxycytidine kinase (dCK).
And the third drug is VE-822, which inhibits the replication stress-sensing kinase ataxia telangiectasia and Rad3-related protein (ATR).
The researchers tested these 3 drugs in a mouse model of pre-B-ALL. Starting on day 7 after inoculation of pre-B-ALL, the mice received 3-AP and DI-82 twice a day and VE-822 once a day.
The team said mice in the control group succumbed to their disease within 17 days. However, all of the mice that received the 3-drug combination were still disease-free 442 days after they had stopped treatment (on day 42).
The researchers said the combination was well-tolerated, as mice maintained their body weight and enjoyed long-term survival without any detectable pathology.
The team also tested a dosing regimen in which all 3 drugs were given once a day.
Although this was less effective than the first regimen, 4 of 5 mice had no detectable disease 313 days after stopping treatment.
Two of the researchers involved in this study are co-founders and equity holders of Trethera Corp, a company developing a dCK inhibitor known as TRE-515.
The University of California (where most of the study authors work) has patented intellectual property for small-molecule dCK inhibitors, and it was licensed by Trethera. ![]()
A 3-drug combination has demonstrated long-term efficacy against acute lymphoblastic leukemia (ALL) in mice, according to research published in Nature Communications.
Researchers found that when the production of nucleotides is stopped, a DNA replication stress response is activated, and this allows ALL cells to survive.
The team used these findings to devise a 3-drug regimen that blocks both of the nucleotide biosynthetic pathways and inhibits the replication stress response.
One of the drugs is triapine (3-AP), which inhibits ribonucleotide reductase (RNR) and enhances salvage nucleotide biosynthesis.
Another drug is DI-82, which inhibits the nucleoside salvage kinase deoxycytidine kinase (dCK).
And the third drug is VE-822, which inhibits the replication stress-sensing kinase ataxia telangiectasia and Rad3-related protein (ATR).
The researchers tested these 3 drugs in a mouse model of pre-B-ALL. Starting on day 7 after inoculation of pre-B-ALL, the mice received 3-AP and DI-82 twice a day and VE-822 once a day.
The team said mice in the control group succumbed to their disease within 17 days. However, all of the mice that received the 3-drug combination were still disease-free 442 days after they had stopped treatment (on day 42).
The researchers said the combination was well-tolerated, as mice maintained their body weight and enjoyed long-term survival without any detectable pathology.
The team also tested a dosing regimen in which all 3 drugs were given once a day.
Although this was less effective than the first regimen, 4 of 5 mice had no detectable disease 313 days after stopping treatment.
Two of the researchers involved in this study are co-founders and equity holders of Trethera Corp, a company developing a dCK inhibitor known as TRE-515.
The University of California (where most of the study authors work) has patented intellectual property for small-molecule dCK inhibitors, and it was licensed by Trethera. ![]()
Drug granted orphan designation for chemo-induced ototoxicity

The US Food and Drug Administration (FDA) has granted orphan drug designation to SENS-401 to be used for the prevention of platinum-induced ototoxicity in pediatric patients.
Platinum-based chemotherapies, particularly cisplatin, can induce severe hearing loss in cancer patients, but there is no pharmaceutical agent approved to treat this side effect.
“Hearing loss in pediatric oncology patients is one of the most frequent side effects of cisplatin treatment and may disable them for the rest of their lives,” said Nawal Ouzren, CEO of Sensorion, the company developing SENS-401.
“Based on its unique profile and the data generated to date, we believe SENS-401 has the potential to be a safe and effective treatment for this serious medical condition where a significant unmet need exists. As such, we look forward to working with the FDA and EMA [European Medicines Agency] to set up an IND [investigational new drug application] and design a phase 2 clinical trial in order to evaluate SENS-401 in this indication.”
About SENS-401
SENS-401 (R-azasetron besylate) is a small molecule intended to protect and preserve inner ear tissue when lesions cause progressive or sequelar hearing impairments. The drug can be taken orally or via an injection.
SENS-401 is one of the two enantiomer forms of SENS-218 (azasetron), a racemic molecule belonging to the family of setrons marketed in Asia under the name Serotone. Pharmacological and pharmacokinetic tests have shown a superior profile for SENS-401 compared with the other enantiomer or the racemic form.
Healthy subjects demonstrated a “very good clinical tolerance” to SENS-401 in a phase 1 study, according to Sensorion. The company is planning to launch a phase 2 trial of the drug for platinum-induced ototoxicity in 2018.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to SENS-401 to be used for the prevention of platinum-induced ototoxicity in pediatric patients.
Platinum-based chemotherapies, particularly cisplatin, can induce severe hearing loss in cancer patients, but there is no pharmaceutical agent approved to treat this side effect.
“Hearing loss in pediatric oncology patients is one of the most frequent side effects of cisplatin treatment and may disable them for the rest of their lives,” said Nawal Ouzren, CEO of Sensorion, the company developing SENS-401.
“Based on its unique profile and the data generated to date, we believe SENS-401 has the potential to be a safe and effective treatment for this serious medical condition where a significant unmet need exists. As such, we look forward to working with the FDA and EMA [European Medicines Agency] to set up an IND [investigational new drug application] and design a phase 2 clinical trial in order to evaluate SENS-401 in this indication.”
About SENS-401
SENS-401 (R-azasetron besylate) is a small molecule intended to protect and preserve inner ear tissue when lesions cause progressive or sequelar hearing impairments. The drug can be taken orally or via an injection.
SENS-401 is one of the two enantiomer forms of SENS-218 (azasetron), a racemic molecule belonging to the family of setrons marketed in Asia under the name Serotone. Pharmacological and pharmacokinetic tests have shown a superior profile for SENS-401 compared with the other enantiomer or the racemic form.
Healthy subjects demonstrated a “very good clinical tolerance” to SENS-401 in a phase 1 study, according to Sensorion. The company is planning to launch a phase 2 trial of the drug for platinum-induced ototoxicity in 2018.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to SENS-401 to be used for the prevention of platinum-induced ototoxicity in pediatric patients.
Platinum-based chemotherapies, particularly cisplatin, can induce severe hearing loss in cancer patients, but there is no pharmaceutical agent approved to treat this side effect.
“Hearing loss in pediatric oncology patients is one of the most frequent side effects of cisplatin treatment and may disable them for the rest of their lives,” said Nawal Ouzren, CEO of Sensorion, the company developing SENS-401.
“Based on its unique profile and the data generated to date, we believe SENS-401 has the potential to be a safe and effective treatment for this serious medical condition where a significant unmet need exists. As such, we look forward to working with the FDA and EMA [European Medicines Agency] to set up an IND [investigational new drug application] and design a phase 2 clinical trial in order to evaluate SENS-401 in this indication.”
About SENS-401
SENS-401 (R-azasetron besylate) is a small molecule intended to protect and preserve inner ear tissue when lesions cause progressive or sequelar hearing impairments. The drug can be taken orally or via an injection.
SENS-401 is one of the two enantiomer forms of SENS-218 (azasetron), a racemic molecule belonging to the family of setrons marketed in Asia under the name Serotone. Pharmacological and pharmacokinetic tests have shown a superior profile for SENS-401 compared with the other enantiomer or the racemic form.
Healthy subjects demonstrated a “very good clinical tolerance” to SENS-401 in a phase 1 study, according to Sensorion. The company is planning to launch a phase 2 trial of the drug for platinum-induced ototoxicity in 2018.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
Team uses genetic barcodes to track hematopoiesis

New research suggests the classical yet contested “tree” model of hematopoiesis is accurate.
In this model, the tree trunk is composed of hematopoietic stem cells (HSCs), and the branches consist of various progenitor cells that give rise to a number of distinct cell types.
Because previous research raised doubts about this model, investigators set out to track hematopoiesis more effectively than ever before.
The team used a “random generator” to label HSCs with genetic barcodes, and this allowed them to trace which cell types arise from an HSC.
Hans-Reimer Rodewald, PhD, of the German Cancer Research Center (Deutsches Krebsforschungszentrum, DKFZ) in Heidelberg, and his colleagues described this research in Nature.
“Genetic barcodes have been developed and applied before, but they were based on methods that can also change cellular properties,” Dr Rodewald said. “Our barcodes are different. They can be induced tissue-specifically and directly in the genome of mice–without influencing the animals’ physiological development.”
The basis of the new technology is the Cre/loxP system that is used to rearrange or remove specially labeled DNA segments.
The investigators bred mice whose genomes exhibit the basic elements of the barcode. At a selected site, where no genes are encoded, it contains 9 DNA fragments from a plant called Arabidopsis thaliana. These elements are flanked by 10 genetic cutting sites called IoxP sites.
By administering a pharmacological agent, the matching molecular scissors, called “Cre,” can be activated in the animals’ HSCs. Then, code elements are randomly rearranged or cut out.
“This genetic, random DNA barcode generator can generate up to 1.8 million genetic barcodes, and we can identify the codes that arise only once in an experiment,” said study author Thomas Höfer, PhD, also of DKFZ.
When these specially labeled HSCs divide and mature, the barcodes are preserved.
The investigators performed comprehensive barcode analyses in order to trace an individual blood cell back to the HSC from which it originated.
These analyses revealed 2 large developmental branches “growing” from the HSC “tree trunk.” In one branch, T cells and B cells develop. In the other, red blood cells and other white blood cells, such as granulocytes and monocytes, form.
“Our findings show that the classical model of a hierarchical developmental tree that starts from multipotent stem cells holds true for hematopoiesis,” Dr Rodewald said.
He and his colleagues believe their genetic barcode system can be used for purposes other than studying blood cell development. In the future, it might be used for experimentally tracing the origin of leukemias and other cancers. ![]()
New research suggests the classical yet contested “tree” model of hematopoiesis is accurate.
In this model, the tree trunk is composed of hematopoietic stem cells (HSCs), and the branches consist of various progenitor cells that give rise to a number of distinct cell types.
Because previous research raised doubts about this model, investigators set out to track hematopoiesis more effectively than ever before.
The team used a “random generator” to label HSCs with genetic barcodes, and this allowed them to trace which cell types arise from an HSC.
Hans-Reimer Rodewald, PhD, of the German Cancer Research Center (Deutsches Krebsforschungszentrum, DKFZ) in Heidelberg, and his colleagues described this research in Nature.
“Genetic barcodes have been developed and applied before, but they were based on methods that can also change cellular properties,” Dr Rodewald said. “Our barcodes are different. They can be induced tissue-specifically and directly in the genome of mice–without influencing the animals’ physiological development.”
The basis of the new technology is the Cre/loxP system that is used to rearrange or remove specially labeled DNA segments.
The investigators bred mice whose genomes exhibit the basic elements of the barcode. At a selected site, where no genes are encoded, it contains 9 DNA fragments from a plant called Arabidopsis thaliana. These elements are flanked by 10 genetic cutting sites called IoxP sites.
By administering a pharmacological agent, the matching molecular scissors, called “Cre,” can be activated in the animals’ HSCs. Then, code elements are randomly rearranged or cut out.
“This genetic, random DNA barcode generator can generate up to 1.8 million genetic barcodes, and we can identify the codes that arise only once in an experiment,” said study author Thomas Höfer, PhD, also of DKFZ.
When these specially labeled HSCs divide and mature, the barcodes are preserved.
The investigators performed comprehensive barcode analyses in order to trace an individual blood cell back to the HSC from which it originated.
These analyses revealed 2 large developmental branches “growing” from the HSC “tree trunk.” In one branch, T cells and B cells develop. In the other, red blood cells and other white blood cells, such as granulocytes and monocytes, form.
“Our findings show that the classical model of a hierarchical developmental tree that starts from multipotent stem cells holds true for hematopoiesis,” Dr Rodewald said.
He and his colleagues believe their genetic barcode system can be used for purposes other than studying blood cell development. In the future, it might be used for experimentally tracing the origin of leukemias and other cancers. ![]()
New research suggests the classical yet contested “tree” model of hematopoiesis is accurate.
In this model, the tree trunk is composed of hematopoietic stem cells (HSCs), and the branches consist of various progenitor cells that give rise to a number of distinct cell types.
Because previous research raised doubts about this model, investigators set out to track hematopoiesis more effectively than ever before.
The team used a “random generator” to label HSCs with genetic barcodes, and this allowed them to trace which cell types arise from an HSC.
Hans-Reimer Rodewald, PhD, of the German Cancer Research Center (Deutsches Krebsforschungszentrum, DKFZ) in Heidelberg, and his colleagues described this research in Nature.
“Genetic barcodes have been developed and applied before, but they were based on methods that can also change cellular properties,” Dr Rodewald said. “Our barcodes are different. They can be induced tissue-specifically and directly in the genome of mice–without influencing the animals’ physiological development.”
The basis of the new technology is the Cre/loxP system that is used to rearrange or remove specially labeled DNA segments.
The investigators bred mice whose genomes exhibit the basic elements of the barcode. At a selected site, where no genes are encoded, it contains 9 DNA fragments from a plant called Arabidopsis thaliana. These elements are flanked by 10 genetic cutting sites called IoxP sites.
By administering a pharmacological agent, the matching molecular scissors, called “Cre,” can be activated in the animals’ HSCs. Then, code elements are randomly rearranged or cut out.
“This genetic, random DNA barcode generator can generate up to 1.8 million genetic barcodes, and we can identify the codes that arise only once in an experiment,” said study author Thomas Höfer, PhD, also of DKFZ.
When these specially labeled HSCs divide and mature, the barcodes are preserved.
The investigators performed comprehensive barcode analyses in order to trace an individual blood cell back to the HSC from which it originated.
These analyses revealed 2 large developmental branches “growing” from the HSC “tree trunk.” In one branch, T cells and B cells develop. In the other, red blood cells and other white blood cells, such as granulocytes and monocytes, form.
“Our findings show that the classical model of a hierarchical developmental tree that starts from multipotent stem cells holds true for hematopoiesis,” Dr Rodewald said.
He and his colleagues believe their genetic barcode system can be used for purposes other than studying blood cell development. In the future, it might be used for experimentally tracing the origin of leukemias and other cancers. ![]()
Antibody could treat AML, MM, and NHL

An investigational antibody has demonstrated activity against acute myeloid leukemia (AML), multiple myeloma (MM), and non-Hodgkin lymphoma (NHL), according to researchers.
PF-06747143 is a humanized CXCR4 immunoglobulin G1 (IgG1) antibody that binds to CXCR4 and inhibits both CXCL12-mediated signaling pathways and cell migration.
Whether given alone or in combination with chemotherapy, PF-06747143 demonstrated efficacy in mouse models of NHL, MM, and AML.
Treatment involving PF-06747143—alone or in combination—eradicated more cancer cells than did standard treatment options.
These results were published in Blood Advances. The research was sponsored by Pfizer, Inc., the company developing PF-06747143.
“One of the major limitations we see in treating blood cancers is the failure to clear cancer cells from the bone marrow,” said study author Flavia Pernasetti, PhD, of Pfizer Oncology Research and Development.
“Because the bone marrow allows the cancer cells to flourish, removing these cells is an essential step in treating these malignancies effectively.”
With this goal in mind, Dr Pernasetti and her colleagues looked to the mechanisms that control the movement of cells into the bone marrow (BM) in the first place—the chemokine receptor CXCR4 and its ligand CXCL12.
The researchers created PF-06747143, which attacks and kills cancer cells directly but also removes cancer cells from the BM so they can be killed by other treatments.
Results in NHL
The researchers first tested PF-06747143 in an NHL Ramos xenograft model. Mice received PF-06747143 or a control IgG1 antibody at 10 mg/kg on days 1 and 8.
PF-06747143 significantly inhibited tumor growth compared to the control antibody (P<0.0001). Seventy percent of PF-06747143-treated mice had tumor volumes below their initial size at the end of the study.
PF-06747143 produced a dose-dependent response that was sustained until the end of the study, even after treatment was stopped.
Results in MM
The researchers tested PF-06747143 in a disseminated MM model, in which the OPM2-Luc tumor cells were implanted intravenously and migrated spontaneously to the BM.
Mice received PF-06747143 or IgG1 control at 10 mg/kg weekly for 5 doses. Other mice received melphalan at 1 mg/kg twice a week for a total of 4 cycles.
On day 30, PF-06747143 had significantly inhibited BM tumor growth compared to the control antibody or melphalan (P<0.0001).
PF-06747143-treated mice also had a significant survival benefit. The median survival was 33.5 days for mice that received the control antibody and 36 days for mice treated with melphalan. However, there were no deaths in the PF-06747143-treated mice by day 50, which marked the end of the study (P<0.0001).
The researchers also tested PF-06747143 at a lower dose (1 mg/kg weekly for a total of 7 doses), both alone and in combination with bortezomib (0.5 mg/kg twice a week for a total of 4 cycles).
The median survival was 34 days in the control mice, 44 days in mice that received bortezomib alone, and 47 days in mice that received PF-06747143 alone. However, there were no deaths in the combination arm at day 51, which was the end of the study (P<0.0003).
Results in AML
The researchers tested PF-06747143 in an AML disseminated tumor model using MV4-11 cells.
They compared PF-06747143 (given at 0.1, 1, or 10 mg/kg weekly for 4 doses) to the chemotherapeutic agent daunorubicin (2 mg/kg on days 1, 3, and 5), the FLT3 inhibitor crenolanib (7.5 mg/kg twice a day, on days 11-15 and 25-29), and a control IgG1 antibody (10 mg/kg weekly for 4 doses).
PF-06747143 (at 10 mg/kg), daunorubicin, and crenolanib all significantly reduced the number of tumor cells in the peripheral blood and BM when compared with the control antibody (P<0.05).
PF-06746143 treatment (at 10 mg/kg) reduced the number of AML cells in the BM by 95.9%, while daunorubicin reduced them by 84.5% and crenolanib by 80.5%.
The median survival was 36 days for mice that received PF-06747143 at 0.1 mg/kg, 41 days for mice that received the control antibody, 47 days for mice that received PF-06747143 at 1 mg/kg, and 63 days for mice that received PF-06747143 at 10 mg/kg.
The researchers also found that PF-06747143 had a “strong combinatorial effect” with daunorubicin and cytarabine in a chemotherapy-resistant model of AML. The team noted that only 36% of BM cells are positive for CXCR4 in this model.
Treatment with PF-06747143 alone reduced the percentage of AML cells in the BM to 80%, combination daunorubicin and cytarabine reduced it to 27%, and combination PF-06747143, daunorubicin, and cytarabine reduced the percentage of AML cells in the BM to 0.3%.
“Our preliminary preclinical results are encouraging,” Dr Pernasetti said, “and we are very excited to see how our antibody fares in clinical testing.”
PF-06747143 is currently being evaluated in a phase 1 trial of AML patients. ![]()
An investigational antibody has demonstrated activity against acute myeloid leukemia (AML), multiple myeloma (MM), and non-Hodgkin lymphoma (NHL), according to researchers.
PF-06747143 is a humanized CXCR4 immunoglobulin G1 (IgG1) antibody that binds to CXCR4 and inhibits both CXCL12-mediated signaling pathways and cell migration.
Whether given alone or in combination with chemotherapy, PF-06747143 demonstrated efficacy in mouse models of NHL, MM, and AML.
Treatment involving PF-06747143—alone or in combination—eradicated more cancer cells than did standard treatment options.
These results were published in Blood Advances. The research was sponsored by Pfizer, Inc., the company developing PF-06747143.
“One of the major limitations we see in treating blood cancers is the failure to clear cancer cells from the bone marrow,” said study author Flavia Pernasetti, PhD, of Pfizer Oncology Research and Development.
“Because the bone marrow allows the cancer cells to flourish, removing these cells is an essential step in treating these malignancies effectively.”
With this goal in mind, Dr Pernasetti and her colleagues looked to the mechanisms that control the movement of cells into the bone marrow (BM) in the first place—the chemokine receptor CXCR4 and its ligand CXCL12.
The researchers created PF-06747143, which attacks and kills cancer cells directly but also removes cancer cells from the BM so they can be killed by other treatments.
Results in NHL
The researchers first tested PF-06747143 in an NHL Ramos xenograft model. Mice received PF-06747143 or a control IgG1 antibody at 10 mg/kg on days 1 and 8.
PF-06747143 significantly inhibited tumor growth compared to the control antibody (P<0.0001). Seventy percent of PF-06747143-treated mice had tumor volumes below their initial size at the end of the study.
PF-06747143 produced a dose-dependent response that was sustained until the end of the study, even after treatment was stopped.
Results in MM
The researchers tested PF-06747143 in a disseminated MM model, in which the OPM2-Luc tumor cells were implanted intravenously and migrated spontaneously to the BM.
Mice received PF-06747143 or IgG1 control at 10 mg/kg weekly for 5 doses. Other mice received melphalan at 1 mg/kg twice a week for a total of 4 cycles.
On day 30, PF-06747143 had significantly inhibited BM tumor growth compared to the control antibody or melphalan (P<0.0001).
PF-06747143-treated mice also had a significant survival benefit. The median survival was 33.5 days for mice that received the control antibody and 36 days for mice treated with melphalan. However, there were no deaths in the PF-06747143-treated mice by day 50, which marked the end of the study (P<0.0001).
The researchers also tested PF-06747143 at a lower dose (1 mg/kg weekly for a total of 7 doses), both alone and in combination with bortezomib (0.5 mg/kg twice a week for a total of 4 cycles).
The median survival was 34 days in the control mice, 44 days in mice that received bortezomib alone, and 47 days in mice that received PF-06747143 alone. However, there were no deaths in the combination arm at day 51, which was the end of the study (P<0.0003).
Results in AML
The researchers tested PF-06747143 in an AML disseminated tumor model using MV4-11 cells.
They compared PF-06747143 (given at 0.1, 1, or 10 mg/kg weekly for 4 doses) to the chemotherapeutic agent daunorubicin (2 mg/kg on days 1, 3, and 5), the FLT3 inhibitor crenolanib (7.5 mg/kg twice a day, on days 11-15 and 25-29), and a control IgG1 antibody (10 mg/kg weekly for 4 doses).
PF-06747143 (at 10 mg/kg), daunorubicin, and crenolanib all significantly reduced the number of tumor cells in the peripheral blood and BM when compared with the control antibody (P<0.05).
PF-06746143 treatment (at 10 mg/kg) reduced the number of AML cells in the BM by 95.9%, while daunorubicin reduced them by 84.5% and crenolanib by 80.5%.
The median survival was 36 days for mice that received PF-06747143 at 0.1 mg/kg, 41 days for mice that received the control antibody, 47 days for mice that received PF-06747143 at 1 mg/kg, and 63 days for mice that received PF-06747143 at 10 mg/kg.
The researchers also found that PF-06747143 had a “strong combinatorial effect” with daunorubicin and cytarabine in a chemotherapy-resistant model of AML. The team noted that only 36% of BM cells are positive for CXCR4 in this model.
Treatment with PF-06747143 alone reduced the percentage of AML cells in the BM to 80%, combination daunorubicin and cytarabine reduced it to 27%, and combination PF-06747143, daunorubicin, and cytarabine reduced the percentage of AML cells in the BM to 0.3%.
“Our preliminary preclinical results are encouraging,” Dr Pernasetti said, “and we are very excited to see how our antibody fares in clinical testing.”
PF-06747143 is currently being evaluated in a phase 1 trial of AML patients. ![]()
An investigational antibody has demonstrated activity against acute myeloid leukemia (AML), multiple myeloma (MM), and non-Hodgkin lymphoma (NHL), according to researchers.
PF-06747143 is a humanized CXCR4 immunoglobulin G1 (IgG1) antibody that binds to CXCR4 and inhibits both CXCL12-mediated signaling pathways and cell migration.
Whether given alone or in combination with chemotherapy, PF-06747143 demonstrated efficacy in mouse models of NHL, MM, and AML.
Treatment involving PF-06747143—alone or in combination—eradicated more cancer cells than did standard treatment options.
These results were published in Blood Advances. The research was sponsored by Pfizer, Inc., the company developing PF-06747143.
“One of the major limitations we see in treating blood cancers is the failure to clear cancer cells from the bone marrow,” said study author Flavia Pernasetti, PhD, of Pfizer Oncology Research and Development.
“Because the bone marrow allows the cancer cells to flourish, removing these cells is an essential step in treating these malignancies effectively.”
With this goal in mind, Dr Pernasetti and her colleagues looked to the mechanisms that control the movement of cells into the bone marrow (BM) in the first place—the chemokine receptor CXCR4 and its ligand CXCL12.
The researchers created PF-06747143, which attacks and kills cancer cells directly but also removes cancer cells from the BM so they can be killed by other treatments.
Results in NHL
The researchers first tested PF-06747143 in an NHL Ramos xenograft model. Mice received PF-06747143 or a control IgG1 antibody at 10 mg/kg on days 1 and 8.
PF-06747143 significantly inhibited tumor growth compared to the control antibody (P<0.0001). Seventy percent of PF-06747143-treated mice had tumor volumes below their initial size at the end of the study.
PF-06747143 produced a dose-dependent response that was sustained until the end of the study, even after treatment was stopped.
Results in MM
The researchers tested PF-06747143 in a disseminated MM model, in which the OPM2-Luc tumor cells were implanted intravenously and migrated spontaneously to the BM.
Mice received PF-06747143 or IgG1 control at 10 mg/kg weekly for 5 doses. Other mice received melphalan at 1 mg/kg twice a week for a total of 4 cycles.
On day 30, PF-06747143 had significantly inhibited BM tumor growth compared to the control antibody or melphalan (P<0.0001).
PF-06747143-treated mice also had a significant survival benefit. The median survival was 33.5 days for mice that received the control antibody and 36 days for mice treated with melphalan. However, there were no deaths in the PF-06747143-treated mice by day 50, which marked the end of the study (P<0.0001).
The researchers also tested PF-06747143 at a lower dose (1 mg/kg weekly for a total of 7 doses), both alone and in combination with bortezomib (0.5 mg/kg twice a week for a total of 4 cycles).
The median survival was 34 days in the control mice, 44 days in mice that received bortezomib alone, and 47 days in mice that received PF-06747143 alone. However, there were no deaths in the combination arm at day 51, which was the end of the study (P<0.0003).
Results in AML
The researchers tested PF-06747143 in an AML disseminated tumor model using MV4-11 cells.
They compared PF-06747143 (given at 0.1, 1, or 10 mg/kg weekly for 4 doses) to the chemotherapeutic agent daunorubicin (2 mg/kg on days 1, 3, and 5), the FLT3 inhibitor crenolanib (7.5 mg/kg twice a day, on days 11-15 and 25-29), and a control IgG1 antibody (10 mg/kg weekly for 4 doses).
PF-06747143 (at 10 mg/kg), daunorubicin, and crenolanib all significantly reduced the number of tumor cells in the peripheral blood and BM when compared with the control antibody (P<0.05).
PF-06746143 treatment (at 10 mg/kg) reduced the number of AML cells in the BM by 95.9%, while daunorubicin reduced them by 84.5% and crenolanib by 80.5%.
The median survival was 36 days for mice that received PF-06747143 at 0.1 mg/kg, 41 days for mice that received the control antibody, 47 days for mice that received PF-06747143 at 1 mg/kg, and 63 days for mice that received PF-06747143 at 10 mg/kg.
The researchers also found that PF-06747143 had a “strong combinatorial effect” with daunorubicin and cytarabine in a chemotherapy-resistant model of AML. The team noted that only 36% of BM cells are positive for CXCR4 in this model.
Treatment with PF-06747143 alone reduced the percentage of AML cells in the BM to 80%, combination daunorubicin and cytarabine reduced it to 27%, and combination PF-06747143, daunorubicin, and cytarabine reduced the percentage of AML cells in the BM to 0.3%.
“Our preliminary preclinical results are encouraging,” Dr Pernasetti said, “and we are very excited to see how our antibody fares in clinical testing.”
PF-06747143 is currently being evaluated in a phase 1 trial of AML patients. ![]()
FDA grants drug orphan designation for AML

The US Food and Drug Administration (FDA) has granted orphan drug designation to SY-1425 for the treatment of acute myeloid leukemia (AML).
SY-1425 is an oral, first-in-class, selective retinoic acid receptor alpha (RARα) agonist. It is currently under investigation in a phase 2 trial of patients with AML and myelodysplastic syndromes (MDS).
“We believe that SY-1425 may provide a meaningful benefit for subsets of AML patients whose disease is driven by abnormally high expression of the RARA or IRF8 genes,” said David A. Roth, MD, chief medical officer at Syros Pharmaceuticals, the company developing SY-1425.
“Receiving orphan drug designation is an important regulatory milestone in the development of SY-1425. We’re pleased with the continued progress of the ongoing phase 2 clinical trial, and we look forward to presenting initial clinical data in the fourth quarter of this year.”
Using its gene control platform, Syros discovered subsets of AML and MDS patients with super-enhancers associated with RARA or IRF8. These super-enhancers are believed to drive overexpression of the RARA or IRF8 genes, locking cells in an immature, undifferentiated, and proliferative state and leading to disease.
In preclinical studies, SY-1425 promoted differentiation of AML cells with high RARA or IRF8 expression and inhibited tumor growth and prolonged survival in patient-derived xenograft models of AML with high RARA expression.
In the ongoing phase 2 trial, researchers are assessing the safety and efficacy of SY-1425 as a single agent in 4 AML and MDS patient populations, as well as SY-1425 in combination with azacitidine in newly diagnosed AML patients who are not suitable candidates for standard chemotherapy.
All patients are prospectively selected using biomarkers for high expression of RARA or IRF8. Additional details about the trial can be found at https://clinicaltrials.gov/ct2/show/NCT02807558.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to SY-1425 for the treatment of acute myeloid leukemia (AML).
SY-1425 is an oral, first-in-class, selective retinoic acid receptor alpha (RARα) agonist. It is currently under investigation in a phase 2 trial of patients with AML and myelodysplastic syndromes (MDS).
“We believe that SY-1425 may provide a meaningful benefit for subsets of AML patients whose disease is driven by abnormally high expression of the RARA or IRF8 genes,” said David A. Roth, MD, chief medical officer at Syros Pharmaceuticals, the company developing SY-1425.
“Receiving orphan drug designation is an important regulatory milestone in the development of SY-1425. We’re pleased with the continued progress of the ongoing phase 2 clinical trial, and we look forward to presenting initial clinical data in the fourth quarter of this year.”
Using its gene control platform, Syros discovered subsets of AML and MDS patients with super-enhancers associated with RARA or IRF8. These super-enhancers are believed to drive overexpression of the RARA or IRF8 genes, locking cells in an immature, undifferentiated, and proliferative state and leading to disease.
In preclinical studies, SY-1425 promoted differentiation of AML cells with high RARA or IRF8 expression and inhibited tumor growth and prolonged survival in patient-derived xenograft models of AML with high RARA expression.
In the ongoing phase 2 trial, researchers are assessing the safety and efficacy of SY-1425 as a single agent in 4 AML and MDS patient populations, as well as SY-1425 in combination with azacitidine in newly diagnosed AML patients who are not suitable candidates for standard chemotherapy.
All patients are prospectively selected using biomarkers for high expression of RARA or IRF8. Additional details about the trial can be found at https://clinicaltrials.gov/ct2/show/NCT02807558.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to SY-1425 for the treatment of acute myeloid leukemia (AML).
SY-1425 is an oral, first-in-class, selective retinoic acid receptor alpha (RARα) agonist. It is currently under investigation in a phase 2 trial of patients with AML and myelodysplastic syndromes (MDS).
“We believe that SY-1425 may provide a meaningful benefit for subsets of AML patients whose disease is driven by abnormally high expression of the RARA or IRF8 genes,” said David A. Roth, MD, chief medical officer at Syros Pharmaceuticals, the company developing SY-1425.
“Receiving orphan drug designation is an important regulatory milestone in the development of SY-1425. We’re pleased with the continued progress of the ongoing phase 2 clinical trial, and we look forward to presenting initial clinical data in the fourth quarter of this year.”
Using its gene control platform, Syros discovered subsets of AML and MDS patients with super-enhancers associated with RARA or IRF8. These super-enhancers are believed to drive overexpression of the RARA or IRF8 genes, locking cells in an immature, undifferentiated, and proliferative state and leading to disease.
In preclinical studies, SY-1425 promoted differentiation of AML cells with high RARA or IRF8 expression and inhibited tumor growth and prolonged survival in patient-derived xenograft models of AML with high RARA expression.
In the ongoing phase 2 trial, researchers are assessing the safety and efficacy of SY-1425 as a single agent in 4 AML and MDS patient populations, as well as SY-1425 in combination with azacitidine in newly diagnosed AML patients who are not suitable candidates for standard chemotherapy.
All patients are prospectively selected using biomarkers for high expression of RARA or IRF8. Additional details about the trial can be found at https://clinicaltrials.gov/ct2/show/NCT02807558.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
Vitamin C could help treat TET2-mutant leukemias

Preclinical research suggests high-dose vitamin C may be effective against TET2-mutant leukemias.
Investigators found that vitamin C mimics TET2 restoration, thereby suppressing leukemic colony formation, inhibiting leukemia progression in mice, and enhancing leukemia cells’ sensitivity to treatment with a PARP inhibitor.
“We’re excited by the prospect that high-dose vitamin C might become a safe treatment for blood diseases caused by TET2-deficient leukemia stem cells, most likely in combination with other targeted therapies,” said study author Benjamin G. Neel, MD, PhD, of NYU School of Medicine in New York, New York.
Dr Neel and his colleagues reported their findings in Cell.
Previous research had shown that vitamin C could stimulate the activity of TET2 as well as TET1 and TET3.
Because only 1 copy of the TET2 gene in each stem cell is usually affected in TET2-mutant blood diseases, the investigators hypothesized that high doses of vitamin C might reverse the effects of TET2 deficiency by turning up the action of the remaining functional gene.
Indeed, the team found that vitamin C had the same effect as restoring TET2 function genetically. By promoting DNA demethylation, high-dose vitamin C induced stem cells to mature and suppressed the growth of leukemic stem cells (LSCs) implanted in mice.
“Interestingly, we also found that vitamin C treatment had an effect on leukemic stem cells that resembled damage to their DNA,” said study author Luisa Cimmino, PhD, of NYU School of Medicine.
“For this reason, we decided to combine vitamin C with a PARP inhibitor, a drug type known to cause cancer cell death by blocking the repair of DNA damage, and already approved for treating certain patients with ovarian cancer.”
The combination had an enhanced effect on LSCs, further shifting them from self-renewal back toward maturity and cell death.
Dr Cimmino said these results suggest vitamin C might also be effective against leukemias without TET2 mutations. As vitamin C turns up any TET2 activity normally in place, it might drive LSCs without TET2 mutations toward death as well. ![]()
Preclinical research suggests high-dose vitamin C may be effective against TET2-mutant leukemias.
Investigators found that vitamin C mimics TET2 restoration, thereby suppressing leukemic colony formation, inhibiting leukemia progression in mice, and enhancing leukemia cells’ sensitivity to treatment with a PARP inhibitor.
“We’re excited by the prospect that high-dose vitamin C might become a safe treatment for blood diseases caused by TET2-deficient leukemia stem cells, most likely in combination with other targeted therapies,” said study author Benjamin G. Neel, MD, PhD, of NYU School of Medicine in New York, New York.
Dr Neel and his colleagues reported their findings in Cell.
Previous research had shown that vitamin C could stimulate the activity of TET2 as well as TET1 and TET3.
Because only 1 copy of the TET2 gene in each stem cell is usually affected in TET2-mutant blood diseases, the investigators hypothesized that high doses of vitamin C might reverse the effects of TET2 deficiency by turning up the action of the remaining functional gene.
Indeed, the team found that vitamin C had the same effect as restoring TET2 function genetically. By promoting DNA demethylation, high-dose vitamin C induced stem cells to mature and suppressed the growth of leukemic stem cells (LSCs) implanted in mice.
“Interestingly, we also found that vitamin C treatment had an effect on leukemic stem cells that resembled damage to their DNA,” said study author Luisa Cimmino, PhD, of NYU School of Medicine.
“For this reason, we decided to combine vitamin C with a PARP inhibitor, a drug type known to cause cancer cell death by blocking the repair of DNA damage, and already approved for treating certain patients with ovarian cancer.”
The combination had an enhanced effect on LSCs, further shifting them from self-renewal back toward maturity and cell death.
Dr Cimmino said these results suggest vitamin C might also be effective against leukemias without TET2 mutations. As vitamin C turns up any TET2 activity normally in place, it might drive LSCs without TET2 mutations toward death as well. ![]()
Preclinical research suggests high-dose vitamin C may be effective against TET2-mutant leukemias.
Investigators found that vitamin C mimics TET2 restoration, thereby suppressing leukemic colony formation, inhibiting leukemia progression in mice, and enhancing leukemia cells’ sensitivity to treatment with a PARP inhibitor.
“We’re excited by the prospect that high-dose vitamin C might become a safe treatment for blood diseases caused by TET2-deficient leukemia stem cells, most likely in combination with other targeted therapies,” said study author Benjamin G. Neel, MD, PhD, of NYU School of Medicine in New York, New York.
Dr Neel and his colleagues reported their findings in Cell.
Previous research had shown that vitamin C could stimulate the activity of TET2 as well as TET1 and TET3.
Because only 1 copy of the TET2 gene in each stem cell is usually affected in TET2-mutant blood diseases, the investigators hypothesized that high doses of vitamin C might reverse the effects of TET2 deficiency by turning up the action of the remaining functional gene.
Indeed, the team found that vitamin C had the same effect as restoring TET2 function genetically. By promoting DNA demethylation, high-dose vitamin C induced stem cells to mature and suppressed the growth of leukemic stem cells (LSCs) implanted in mice.
“Interestingly, we also found that vitamin C treatment had an effect on leukemic stem cells that resembled damage to their DNA,” said study author Luisa Cimmino, PhD, of NYU School of Medicine.
“For this reason, we decided to combine vitamin C with a PARP inhibitor, a drug type known to cause cancer cell death by blocking the repair of DNA damage, and already approved for treating certain patients with ovarian cancer.”
The combination had an enhanced effect on LSCs, further shifting them from self-renewal back toward maturity and cell death.
Dr Cimmino said these results suggest vitamin C might also be effective against leukemias without TET2 mutations. As vitamin C turns up any TET2 activity normally in place, it might drive LSCs without TET2 mutations toward death as well.
Researchers compare world health authorities

A new study has revealed substantial differences between health authorities in different regions of the world.
A pair of researchers compared 12 different regulatory authorities responsible for approving drugs and medical products.
The researchers collected data* on annual budgets, new drug approvals per year, numbers of reviewers, standard and median review times, fees for new drug applications (NDAs), and other measurements.
The results were published in Nature Reviews Drug Discovery.
For the 2015 fiscal year, the US Food and Drug Administration (FDA) had the highest budget—$1.19 billion—and India’s Central Drugs Standard Control Organization (CDSCO) had the lowest—$26 million.
In 2016, the FDA again had the highest budget—$1.23 billion—while Health Canada and Switzerland’s SwissMedic had the lowest—$108 million.
In 2016, the European Medicines Agency (EMA) had the highest number of reviewers—4500—and SwissMedic had the lowest—60. (Data from 2015 were not included.)
In 2015, Japan’s Pharmaceuticals and Medical Devices Agency had the highest number of NDA submissions—127—and Health Canada had the lowest—27. Meanwhile, the Chinese FDA had the highest number of new drug approvals—72—and India’s CDSCO had the lowest—17.
The UK’s Medicines and Healthcare products Regulatory Agency (MHRA) technically had the most new drug approvals in 2015, at 146, but not all of these were unique, as the number included all decentralized applications, both with the UK as the reference member state and approvals from concerned member states.
In 2016, the EMA had the highest number of NDA submissions—68—and Health Canada had the lowest—25. Singapore’s Health Sciences Authority had the highest number of new drug approvals—72—while the US FDA and India’s CDSCO had the lowest—22.
The shortest standard review period was 210 days. This is the standard for the EMA, the UK’s MHRA, and Russia’s Roszdravnadzor. The regulatory agency with the longest standard review time—900 days—is the Chinese FDA.
The shortest median time to new drug approval in 2015 was 230 days, for the UK’s MHRA. The longest was 834 days, for the Brazilian Health Surveillance Agency.
The highest NDA review fees were those charged by the US FDA—$2.3 million. The lowest were those charged by India’s CDSCO—50,000 Indian rupees or about USD$1000.
The researchers noted that these data suggest products are being evaluated via different processes and according to different standards, which makes it challenging for pharmaceutical companies to develop drugs for simultaneous submission to all regulatory authorities.
Therefore, a harmonization of approval requirements and processes could significantly improve efficiency.
“Patients would profit especially since new drugs would be available faster and at lower prices,” said study author Thomas D. Szucs, MD, PhD, of the University of Basel in Switzerland.
“This suggests that companies and authorities should strengthen their international collaboration and communicate better with each other.”
*Some data were missing for most of the 12 agencies studied.
A new study has revealed substantial differences between health authorities in different regions of the world.
A pair of researchers compared 12 different regulatory authorities responsible for approving drugs and medical products.
The researchers collected data* on annual budgets, new drug approvals per year, numbers of reviewers, standard and median review times, fees for new drug applications (NDAs), and other measurements.
The results were published in Nature Reviews Drug Discovery.
For the 2015 fiscal year, the US Food and Drug Administration (FDA) had the highest budget—$1.19 billion—and India’s Central Drugs Standard Control Organization (CDSCO) had the lowest—$26 million.
In 2016, the FDA again had the highest budget—$1.23 billion—while Health Canada and Switzerland’s SwissMedic had the lowest—$108 million.
In 2016, the European Medicines Agency (EMA) had the highest number of reviewers—4500—and SwissMedic had the lowest—60. (Data from 2015 were not included.)
In 2015, Japan’s Pharmaceuticals and Medical Devices Agency had the highest number of NDA submissions—127—and Health Canada had the lowest—27. Meanwhile, the Chinese FDA had the highest number of new drug approvals—72—and India’s CDSCO had the lowest—17.
The UK’s Medicines and Healthcare products Regulatory Agency (MHRA) technically had the most new drug approvals in 2015, at 146, but not all of these were unique, as the number included all decentralized applications, both with the UK as the reference member state and approvals from concerned member states.
In 2016, the EMA had the highest number of NDA submissions—68—and Health Canada had the lowest—25. Singapore’s Health Sciences Authority had the highest number of new drug approvals—72—while the US FDA and India’s CDSCO had the lowest—22.
The shortest standard review period was 210 days. This is the standard for the EMA, the UK’s MHRA, and Russia’s Roszdravnadzor. The regulatory agency with the longest standard review time—900 days—is the Chinese FDA.
The shortest median time to new drug approval in 2015 was 230 days, for the UK’s MHRA. The longest was 834 days, for the Brazilian Health Surveillance Agency.
The highest NDA review fees were those charged by the US FDA—$2.3 million. The lowest were those charged by India’s CDSCO—50,000 Indian rupees or about USD$1000.
The researchers noted that these data suggest products are being evaluated via different processes and according to different standards, which makes it challenging for pharmaceutical companies to develop drugs for simultaneous submission to all regulatory authorities.
Therefore, a harmonization of approval requirements and processes could significantly improve efficiency.
“Patients would profit especially since new drugs would be available faster and at lower prices,” said study author Thomas D. Szucs, MD, PhD, of the University of Basel in Switzerland.
“This suggests that companies and authorities should strengthen their international collaboration and communicate better with each other.”
*Some data were missing for most of the 12 agencies studied.
A new study has revealed substantial differences between health authorities in different regions of the world.
A pair of researchers compared 12 different regulatory authorities responsible for approving drugs and medical products.
The researchers collected data* on annual budgets, new drug approvals per year, numbers of reviewers, standard and median review times, fees for new drug applications (NDAs), and other measurements.
The results were published in Nature Reviews Drug Discovery.
For the 2015 fiscal year, the US Food and Drug Administration (FDA) had the highest budget—$1.19 billion—and India’s Central Drugs Standard Control Organization (CDSCO) had the lowest—$26 million.
In 2016, the FDA again had the highest budget—$1.23 billion—while Health Canada and Switzerland’s SwissMedic had the lowest—$108 million.
In 2016, the European Medicines Agency (EMA) had the highest number of reviewers—4500—and SwissMedic had the lowest—60. (Data from 2015 were not included.)
In 2015, Japan’s Pharmaceuticals and Medical Devices Agency had the highest number of NDA submissions—127—and Health Canada had the lowest—27. Meanwhile, the Chinese FDA had the highest number of new drug approvals—72—and India’s CDSCO had the lowest—17.
The UK’s Medicines and Healthcare products Regulatory Agency (MHRA) technically had the most new drug approvals in 2015, at 146, but not all of these were unique, as the number included all decentralized applications, both with the UK as the reference member state and approvals from concerned member states.
In 2016, the EMA had the highest number of NDA submissions—68—and Health Canada had the lowest—25. Singapore’s Health Sciences Authority had the highest number of new drug approvals—72—while the US FDA and India’s CDSCO had the lowest—22.
The shortest standard review period was 210 days. This is the standard for the EMA, the UK’s MHRA, and Russia’s Roszdravnadzor. The regulatory agency with the longest standard review time—900 days—is the Chinese FDA.
The shortest median time to new drug approval in 2015 was 230 days, for the UK’s MHRA. The longest was 834 days, for the Brazilian Health Surveillance Agency.
The highest NDA review fees were those charged by the US FDA—$2.3 million. The lowest were those charged by India’s CDSCO—50,000 Indian rupees or about USD$1000.
The researchers noted that these data suggest products are being evaluated via different processes and according to different standards, which makes it challenging for pharmaceutical companies to develop drugs for simultaneous submission to all regulatory authorities.
Therefore, a harmonization of approval requirements and processes could significantly improve efficiency.
“Patients would profit especially since new drugs would be available faster and at lower prices,” said study author Thomas D. Szucs, MD, PhD, of the University of Basel in Switzerland.
“This suggests that companies and authorities should strengthen their international collaboration and communicate better with each other.”
*Some data were missing for most of the 12 agencies studied.
Test uses nanotechnology to diagnose Zika virus
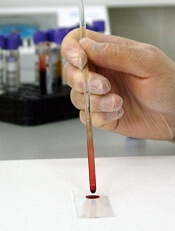
Researchers say they have developed a point-of-care, paper-based test that quickly detects the presence of Zika virus in blood.
Currently, testing for Zika requires that a blood sample be refrigerated and shipped to a medical center or laboratory, delaying diagnosis and possible treatment.
The new test, on the other hand, does not require refrigeration and can produce results in minutes.
“If an assay requires electricity and refrigeration, it defeats the purpose of developing something to use in a resource-limited setting, especially in tropical areas of the world,” said Srikanth Singamaneni, PhD, of Washington University in St. Louis, Missouri.
“We wanted to make the test immune from variations in temperature and humidity.”
Dr Singamaneni and his colleagues described this test in Advanced Biosystems.
The researchers used the test on blood samples from 4 subjects known to be infected with Zika virus and samples from 5 subjects who did not have the virus.
The test showed positive results for the Zika-infected patients and negative results for controls. There were no false-positives.
How the test works
The test uses gold nanorods mounted on paper to detect Zika infection in the blood.
The test relies on a protein made by Zika virus that causes an immune response in infected individuals— ZIKV-nonstructural protein 1 (NS1). The ZIKV-NS1 protein is attached to gold nanorods mounted on a piece of paper.
The paper is then covered with protective nanocrystals. The nanocrystals allow the diagnostic nanorods to be shipped and stored without refrigeration prior to use.
To use the test, a technician rinses the paper with slightly acidic water, removing the protective crystals and exposing the protein mounted on the nanorods.
Then, a drop of the patient’s blood is applied. If the patient has come into contact with the virus, the blood will contain immunoglobulins that react with the ZIKV-NS1 protein.
“We’re taking advantage of the fact that patients mount an immune attack against this viral protein,” said study author Jeremiah J. Morrissey, PhD, of Washington University.
“The immunoglobulins persist in the blood for a few months, and when they come into contact with the gold nanorods, the nanorods undergo a slight color change that can be detected with a hand-held spectrophotometer. With this test, results will be clear before the patient leaves the clinic, allowing immediate counseling and access to treatment.”
The color change cannot be seen with the naked eye, but the researchers are working to change that. They’re also working on developing ways to use saliva rather than blood.
Although the test uses gold, the nanorods are very small. The researchers estimate the cost of the gold used in one of the assays would be 10 to 15 cents.
Researchers say they have developed a point-of-care, paper-based test that quickly detects the presence of Zika virus in blood.
Currently, testing for Zika requires that a blood sample be refrigerated and shipped to a medical center or laboratory, delaying diagnosis and possible treatment.
The new test, on the other hand, does not require refrigeration and can produce results in minutes.
“If an assay requires electricity and refrigeration, it defeats the purpose of developing something to use in a resource-limited setting, especially in tropical areas of the world,” said Srikanth Singamaneni, PhD, of Washington University in St. Louis, Missouri.
“We wanted to make the test immune from variations in temperature and humidity.”
Dr Singamaneni and his colleagues described this test in Advanced Biosystems.
The researchers used the test on blood samples from 4 subjects known to be infected with Zika virus and samples from 5 subjects who did not have the virus.
The test showed positive results for the Zika-infected patients and negative results for controls. There were no false-positives.
How the test works
The test uses gold nanorods mounted on paper to detect Zika infection in the blood.
The test relies on a protein made by Zika virus that causes an immune response in infected individuals— ZIKV-nonstructural protein 1 (NS1). The ZIKV-NS1 protein is attached to gold nanorods mounted on a piece of paper.
The paper is then covered with protective nanocrystals. The nanocrystals allow the diagnostic nanorods to be shipped and stored without refrigeration prior to use.
To use the test, a technician rinses the paper with slightly acidic water, removing the protective crystals and exposing the protein mounted on the nanorods.
Then, a drop of the patient’s blood is applied. If the patient has come into contact with the virus, the blood will contain immunoglobulins that react with the ZIKV-NS1 protein.
“We’re taking advantage of the fact that patients mount an immune attack against this viral protein,” said study author Jeremiah J. Morrissey, PhD, of Washington University.
“The immunoglobulins persist in the blood for a few months, and when they come into contact with the gold nanorods, the nanorods undergo a slight color change that can be detected with a hand-held spectrophotometer. With this test, results will be clear before the patient leaves the clinic, allowing immediate counseling and access to treatment.”
The color change cannot be seen with the naked eye, but the researchers are working to change that. They’re also working on developing ways to use saliva rather than blood.
Although the test uses gold, the nanorods are very small. The researchers estimate the cost of the gold used in one of the assays would be 10 to 15 cents.
Researchers say they have developed a point-of-care, paper-based test that quickly detects the presence of Zika virus in blood.
Currently, testing for Zika requires that a blood sample be refrigerated and shipped to a medical center or laboratory, delaying diagnosis and possible treatment.
The new test, on the other hand, does not require refrigeration and can produce results in minutes.
“If an assay requires electricity and refrigeration, it defeats the purpose of developing something to use in a resource-limited setting, especially in tropical areas of the world,” said Srikanth Singamaneni, PhD, of Washington University in St. Louis, Missouri.
“We wanted to make the test immune from variations in temperature and humidity.”
Dr Singamaneni and his colleagues described this test in Advanced Biosystems.
The researchers used the test on blood samples from 4 subjects known to be infected with Zika virus and samples from 5 subjects who did not have the virus.
The test showed positive results for the Zika-infected patients and negative results for controls. There were no false-positives.
How the test works
The test uses gold nanorods mounted on paper to detect Zika infection in the blood.
The test relies on a protein made by Zika virus that causes an immune response in infected individuals— ZIKV-nonstructural protein 1 (NS1). The ZIKV-NS1 protein is attached to gold nanorods mounted on a piece of paper.
The paper is then covered with protective nanocrystals. The nanocrystals allow the diagnostic nanorods to be shipped and stored without refrigeration prior to use.
To use the test, a technician rinses the paper with slightly acidic water, removing the protective crystals and exposing the protein mounted on the nanorods.
Then, a drop of the patient’s blood is applied. If the patient has come into contact with the virus, the blood will contain immunoglobulins that react with the ZIKV-NS1 protein.
“We’re taking advantage of the fact that patients mount an immune attack against this viral protein,” said study author Jeremiah J. Morrissey, PhD, of Washington University.
“The immunoglobulins persist in the blood for a few months, and when they come into contact with the gold nanorods, the nanorods undergo a slight color change that can be detected with a hand-held spectrophotometer. With this test, results will be clear before the patient leaves the clinic, allowing immediate counseling and access to treatment.”
The color change cannot be seen with the naked eye, but the researchers are working to change that. They’re also working on developing ways to use saliva rather than blood.
Although the test uses gold, the nanorods are very small. The researchers estimate the cost of the gold used in one of the assays would be 10 to 15 cents.
FDA approves inotuzumab ozogamicin for rel/ref ALL

The US Food and Drug Administration (FDA) has approved inotuzumab ozogamicin (Besponsa®) for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The labeling for inotuzumab ozogamicin includes a boxed warning stating that the drug poses a risk of hepatotoxicity, including hepatic veno-occlusive disease (or sinusoidal obstruction syndrome), as well as an increased risk of post-transplant non-relapse mortality.
The full prescribing information for inotuzumab ozogamicin is available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf.
Inotuzumab ozogamicin is an antibody-drug conjugate that consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
The product originates from a collaboration between Pfizer and Celltech (now UCB), but Pfizer has sole responsibility for all manufacturing and clinical development activities.
Inotuzumab ozogamicin was reviewed and approved under the FDA’s breakthrough therapy designation and priority review programs.
The application for inotuzumab ozogamicin was supported by results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell ALL. Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens (high-dose cytarabine, cytarabine plus mitoxantrone, or fludarabine, cytarabine, and granulocyte colony-stimulating factor).
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were thought to be related to chemotherapy.
The US Food and Drug Administration (FDA) has approved inotuzumab ozogamicin (Besponsa®) for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The labeling for inotuzumab ozogamicin includes a boxed warning stating that the drug poses a risk of hepatotoxicity, including hepatic veno-occlusive disease (or sinusoidal obstruction syndrome), as well as an increased risk of post-transplant non-relapse mortality.
The full prescribing information for inotuzumab ozogamicin is available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf.
Inotuzumab ozogamicin is an antibody-drug conjugate that consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
The product originates from a collaboration between Pfizer and Celltech (now UCB), but Pfizer has sole responsibility for all manufacturing and clinical development activities.
Inotuzumab ozogamicin was reviewed and approved under the FDA’s breakthrough therapy designation and priority review programs.
The application for inotuzumab ozogamicin was supported by results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell ALL. Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens (high-dose cytarabine, cytarabine plus mitoxantrone, or fludarabine, cytarabine, and granulocyte colony-stimulating factor).
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were thought to be related to chemotherapy.
The US Food and Drug Administration (FDA) has approved inotuzumab ozogamicin (Besponsa®) for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The labeling for inotuzumab ozogamicin includes a boxed warning stating that the drug poses a risk of hepatotoxicity, including hepatic veno-occlusive disease (or sinusoidal obstruction syndrome), as well as an increased risk of post-transplant non-relapse mortality.
The full prescribing information for inotuzumab ozogamicin is available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf.
Inotuzumab ozogamicin is an antibody-drug conjugate that consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
The product originates from a collaboration between Pfizer and Celltech (now UCB), but Pfizer has sole responsibility for all manufacturing and clinical development activities.
Inotuzumab ozogamicin was reviewed and approved under the FDA’s breakthrough therapy designation and priority review programs.
The application for inotuzumab ozogamicin was supported by results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell ALL. Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens (high-dose cytarabine, cytarabine plus mitoxantrone, or fludarabine, cytarabine, and granulocyte colony-stimulating factor).
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were thought to be related to chemotherapy.