User login
SFDA approves product for hemophilia A
The Saudi Food & Drug Authority (SFDA) in the Kingdom of Saudi Arabia has approved efmoroctocog alfa (Elocta®), a recombinant human factor VIII Fc-fusion protein, for the treatment of hemophilia A.
Efmoroctocog alfa is indicated for both on-demand treatment and prophylaxis in hemophilia A patients of all ages.
It is the first extended half-life and recombinant factor VIII Fc fusion protein therapy approved for the treatment of hemophilia A in Saudi Arabia.
The SFDA’s approval of efmoroctocog alfa was based on data from a pair of phase 3 studies: A-LONG and Kids A-LONG.
A-LONG
The A-LONG study included 165 previously treated males 12 years of age and older with severe hemophilia A. Researchers evaluated individualized and weekly prophylaxis to reduce or prevent bleeding episodes and on-demand dosing to treat bleeding episodes.
Prophylaxis with efmoroctocog alfa resulted in low annualized bleeding rates, and a majority of bleeding episodes were controlled with a single injection of efmoroctocog alfa.
None of the patients developed neutralizing antibodies, efmoroctocog alfa was considered well-tolerated, and the product had a prolonged half-life when compared with recombinant factor VIII.
Kids A-LONG
The Kids A-LONG study included 71 boys (younger than 12) with severe hemophilia A who had at least 50 prior exposure days to factor VIII therapies.
The children saw their median annualized bleeding rate decrease with efmoroctocog alfa, and close to half of the children did not have any bleeding episodes while they were receiving efmoroctocog alfa.
None of the patients developed inhibitors, and researchers said adverse events were typical of a pediatric hemophilia population.
Efmoroctocog alfa is being developed and commercialized by Sobi and Bioverativ. ![]()
The Saudi Food & Drug Authority (SFDA) in the Kingdom of Saudi Arabia has approved efmoroctocog alfa (Elocta®), a recombinant human factor VIII Fc-fusion protein, for the treatment of hemophilia A.
Efmoroctocog alfa is indicated for both on-demand treatment and prophylaxis in hemophilia A patients of all ages.
It is the first extended half-life and recombinant factor VIII Fc fusion protein therapy approved for the treatment of hemophilia A in Saudi Arabia.
The SFDA’s approval of efmoroctocog alfa was based on data from a pair of phase 3 studies: A-LONG and Kids A-LONG.
A-LONG
The A-LONG study included 165 previously treated males 12 years of age and older with severe hemophilia A. Researchers evaluated individualized and weekly prophylaxis to reduce or prevent bleeding episodes and on-demand dosing to treat bleeding episodes.
Prophylaxis with efmoroctocog alfa resulted in low annualized bleeding rates, and a majority of bleeding episodes were controlled with a single injection of efmoroctocog alfa.
None of the patients developed neutralizing antibodies, efmoroctocog alfa was considered well-tolerated, and the product had a prolonged half-life when compared with recombinant factor VIII.
Kids A-LONG
The Kids A-LONG study included 71 boys (younger than 12) with severe hemophilia A who had at least 50 prior exposure days to factor VIII therapies.
The children saw their median annualized bleeding rate decrease with efmoroctocog alfa, and close to half of the children did not have any bleeding episodes while they were receiving efmoroctocog alfa.
None of the patients developed inhibitors, and researchers said adverse events were typical of a pediatric hemophilia population.
Efmoroctocog alfa is being developed and commercialized by Sobi and Bioverativ. ![]()
The Saudi Food & Drug Authority (SFDA) in the Kingdom of Saudi Arabia has approved efmoroctocog alfa (Elocta®), a recombinant human factor VIII Fc-fusion protein, for the treatment of hemophilia A.
Efmoroctocog alfa is indicated for both on-demand treatment and prophylaxis in hemophilia A patients of all ages.
It is the first extended half-life and recombinant factor VIII Fc fusion protein therapy approved for the treatment of hemophilia A in Saudi Arabia.
The SFDA’s approval of efmoroctocog alfa was based on data from a pair of phase 3 studies: A-LONG and Kids A-LONG.
A-LONG
The A-LONG study included 165 previously treated males 12 years of age and older with severe hemophilia A. Researchers evaluated individualized and weekly prophylaxis to reduce or prevent bleeding episodes and on-demand dosing to treat bleeding episodes.
Prophylaxis with efmoroctocog alfa resulted in low annualized bleeding rates, and a majority of bleeding episodes were controlled with a single injection of efmoroctocog alfa.
None of the patients developed neutralizing antibodies, efmoroctocog alfa was considered well-tolerated, and the product had a prolonged half-life when compared with recombinant factor VIII.
Kids A-LONG
The Kids A-LONG study included 71 boys (younger than 12) with severe hemophilia A who had at least 50 prior exposure days to factor VIII therapies.
The children saw their median annualized bleeding rate decrease with efmoroctocog alfa, and close to half of the children did not have any bleeding episodes while they were receiving efmoroctocog alfa.
None of the patients developed inhibitors, and researchers said adverse events were typical of a pediatric hemophilia population.
Efmoroctocog alfa is being developed and commercialized by Sobi and Bioverativ. ![]()
Device reduces blood draw contamination
A novel device can significantly reduce contamination of blood cultures, according to research published in Clinical Infectious Diseases.
The SteriPath initial specimen diversion device (ISDD) is a blood collection system that diverts and sequesters the first 1.5 mL to 2 mL of blood drawn, which often carries contaminating skin cells and microbes.
By allowing for the disposal of this blood, the ISDD reduced blood culture contamination by 88%, when compared to standard phlebotomy procedures.
In reducing contamination, the SteriPath ISDD could reduce the unnecessary use of antibiotics, according to researchers.
“A lot of people think this is a minor problem,” said study author Mark Rupp, MD, of the University of Nebraska Medical Center in Omaha.
“However, contaminated blood cultures are a big deal. Physicians can be led astray, and patients may be harmed by additional tests and unnecessary antimicrobial therapy.”
For this study, Dr Rupp and his colleagues compared the ISDD and standard blood draw procedures, collecting a total of 1808 blood samples from 904 patients.
The researchers observed a significantly lower rate of blood culture contamination with the ISDD than with standard procedures—0.22% (2/904) and 1.78% (16/904), respectively (P=0.001).
“The 1.78% baseline rate of contamination may seem small, but we should strive to decrease adverse events to the lowest possible level because of the impact to the patient and the burden to our healthcare system,” Dr Rupp said. “We quite clearly showed the rate of contamination was significantly reduced, and that decrease has a very big impact.”
Dr Rupp and his colleagues also noted that the ISDD was about as sensitive as standard procedures. The ISDD detected true bacteremia in 7.2% (65/904) of patients, and standard procedures detected true bacteremia in 7.6% (69/904) of patients (P=0.41).
“What is important about this device is it can greatly limit the blood culture from being contaminated, so physicians are rarely fooled by false-positive results,” Dr Rupp said. “It gives clinicians confidence that results are accurate.”
This research was supported by Magnolia Medical Technologies, Inc., the company marketing the SteriPath ISDD. ![]()
A novel device can significantly reduce contamination of blood cultures, according to research published in Clinical Infectious Diseases.
The SteriPath initial specimen diversion device (ISDD) is a blood collection system that diverts and sequesters the first 1.5 mL to 2 mL of blood drawn, which often carries contaminating skin cells and microbes.
By allowing for the disposal of this blood, the ISDD reduced blood culture contamination by 88%, when compared to standard phlebotomy procedures.
In reducing contamination, the SteriPath ISDD could reduce the unnecessary use of antibiotics, according to researchers.
“A lot of people think this is a minor problem,” said study author Mark Rupp, MD, of the University of Nebraska Medical Center in Omaha.
“However, contaminated blood cultures are a big deal. Physicians can be led astray, and patients may be harmed by additional tests and unnecessary antimicrobial therapy.”
For this study, Dr Rupp and his colleagues compared the ISDD and standard blood draw procedures, collecting a total of 1808 blood samples from 904 patients.
The researchers observed a significantly lower rate of blood culture contamination with the ISDD than with standard procedures—0.22% (2/904) and 1.78% (16/904), respectively (P=0.001).
“The 1.78% baseline rate of contamination may seem small, but we should strive to decrease adverse events to the lowest possible level because of the impact to the patient and the burden to our healthcare system,” Dr Rupp said. “We quite clearly showed the rate of contamination was significantly reduced, and that decrease has a very big impact.”
Dr Rupp and his colleagues also noted that the ISDD was about as sensitive as standard procedures. The ISDD detected true bacteremia in 7.2% (65/904) of patients, and standard procedures detected true bacteremia in 7.6% (69/904) of patients (P=0.41).
“What is important about this device is it can greatly limit the blood culture from being contaminated, so physicians are rarely fooled by false-positive results,” Dr Rupp said. “It gives clinicians confidence that results are accurate.”
This research was supported by Magnolia Medical Technologies, Inc., the company marketing the SteriPath ISDD. ![]()
A novel device can significantly reduce contamination of blood cultures, according to research published in Clinical Infectious Diseases.
The SteriPath initial specimen diversion device (ISDD) is a blood collection system that diverts and sequesters the first 1.5 mL to 2 mL of blood drawn, which often carries contaminating skin cells and microbes.
By allowing for the disposal of this blood, the ISDD reduced blood culture contamination by 88%, when compared to standard phlebotomy procedures.
In reducing contamination, the SteriPath ISDD could reduce the unnecessary use of antibiotics, according to researchers.
“A lot of people think this is a minor problem,” said study author Mark Rupp, MD, of the University of Nebraska Medical Center in Omaha.
“However, contaminated blood cultures are a big deal. Physicians can be led astray, and patients may be harmed by additional tests and unnecessary antimicrobial therapy.”
For this study, Dr Rupp and his colleagues compared the ISDD and standard blood draw procedures, collecting a total of 1808 blood samples from 904 patients.
The researchers observed a significantly lower rate of blood culture contamination with the ISDD than with standard procedures—0.22% (2/904) and 1.78% (16/904), respectively (P=0.001).
“The 1.78% baseline rate of contamination may seem small, but we should strive to decrease adverse events to the lowest possible level because of the impact to the patient and the burden to our healthcare system,” Dr Rupp said. “We quite clearly showed the rate of contamination was significantly reduced, and that decrease has a very big impact.”
Dr Rupp and his colleagues also noted that the ISDD was about as sensitive as standard procedures. The ISDD detected true bacteremia in 7.2% (65/904) of patients, and standard procedures detected true bacteremia in 7.6% (69/904) of patients (P=0.41).
“What is important about this device is it can greatly limit the blood culture from being contaminated, so physicians are rarely fooled by false-positive results,” Dr Rupp said. “It gives clinicians confidence that results are accurate.”
This research was supported by Magnolia Medical Technologies, Inc., the company marketing the SteriPath ISDD. ![]()
Gene variant reduces risk of severe malaria
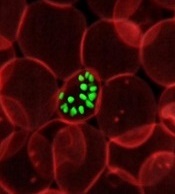
New research indicates that some Africans carry a gene variant that reduces the risk of severe malaria.
The study suggests this variant results from the rearrangement of 2 glycophorin receptors found on the surface of red blood cells.
The malaria parasite Plasmodium falciparum uses these receptors—GYPA and GYPB—to enter the cells.
Researchers identified a gene variant that results in altered GYPA and GYPB receptors and may reduce the risk of severe malaria by 40%.
Ellen Leffler, of the University of Oxford in the UK, and her colleagues reported these findings in Science.
The researchers performed genome sequencing of 765 individuals from 10 ethnic groups in Gambia, Burkina Faso, Cameroon, and Tanzania.
The team also conducted a study across the Gambia, Kenya, and Malawi that included 5310 individuals from the general population and 4579 people who were hospitalized with severe malaria.
These analyses revealed copy number variants affecting GYPA and GYPB.
“[W]e found strong evidence that variation in the glycophorin gene cluster influences malaria susceptibility,” Dr Leffler said.
“We found some people have a complex rearrangement of GYPA and GYPB genes, forming a hybrid glycophorin, and these people are less likely to develop severe complications of the disease.”
The rearrangement involves the loss of GYPB and gain of 2 GYPB-A hybrid genes. The hybrid GYPB-A gene is found in a rare blood group—part of the MNS blood group system—where it is known as Dantu.
DUP4, the most common Dantu gene variant, is a result of the rearrangement. And the researchers found that DUP4 reduced the risk of severe malaria by an estimated 40%.
DUP4 was only present in certain populations, particularly in individuals of East African descent.
The researchers proposed a number of reasons as to why DUP4 may not be more widespread, including the possibility that it emerged recently. Alternatively, it may only protect against certain strains of P falciparum that are specific to east Africa.
Though more research is needed, the team said these findings link the structural variation of glycophorin receptors with resistance to severe malaria.
“We are starting to find that the glycophorin region of the genome has an important role in protecting people against malaria,” said study author Dominic Kwiatkowski, MD, of the University of Oxford.
“Our discovery that a specific variant of glycophorin invasion receptors can give substantial protection against severe malaria will hopefully inspire further research on exactly how Plasmodium falciparum invade red blood cells. This could also help us discover novel parasite weaknesses that could be exploited in future interventions against this deadly disease.” ![]()
New research indicates that some Africans carry a gene variant that reduces the risk of severe malaria.
The study suggests this variant results from the rearrangement of 2 glycophorin receptors found on the surface of red blood cells.
The malaria parasite Plasmodium falciparum uses these receptors—GYPA and GYPB—to enter the cells.
Researchers identified a gene variant that results in altered GYPA and GYPB receptors and may reduce the risk of severe malaria by 40%.
Ellen Leffler, of the University of Oxford in the UK, and her colleagues reported these findings in Science.
The researchers performed genome sequencing of 765 individuals from 10 ethnic groups in Gambia, Burkina Faso, Cameroon, and Tanzania.
The team also conducted a study across the Gambia, Kenya, and Malawi that included 5310 individuals from the general population and 4579 people who were hospitalized with severe malaria.
These analyses revealed copy number variants affecting GYPA and GYPB.
“[W]e found strong evidence that variation in the glycophorin gene cluster influences malaria susceptibility,” Dr Leffler said.
“We found some people have a complex rearrangement of GYPA and GYPB genes, forming a hybrid glycophorin, and these people are less likely to develop severe complications of the disease.”
The rearrangement involves the loss of GYPB and gain of 2 GYPB-A hybrid genes. The hybrid GYPB-A gene is found in a rare blood group—part of the MNS blood group system—where it is known as Dantu.
DUP4, the most common Dantu gene variant, is a result of the rearrangement. And the researchers found that DUP4 reduced the risk of severe malaria by an estimated 40%.
DUP4 was only present in certain populations, particularly in individuals of East African descent.
The researchers proposed a number of reasons as to why DUP4 may not be more widespread, including the possibility that it emerged recently. Alternatively, it may only protect against certain strains of P falciparum that are specific to east Africa.
Though more research is needed, the team said these findings link the structural variation of glycophorin receptors with resistance to severe malaria.
“We are starting to find that the glycophorin region of the genome has an important role in protecting people against malaria,” said study author Dominic Kwiatkowski, MD, of the University of Oxford.
“Our discovery that a specific variant of glycophorin invasion receptors can give substantial protection against severe malaria will hopefully inspire further research on exactly how Plasmodium falciparum invade red blood cells. This could also help us discover novel parasite weaknesses that could be exploited in future interventions against this deadly disease.” ![]()
New research indicates that some Africans carry a gene variant that reduces the risk of severe malaria.
The study suggests this variant results from the rearrangement of 2 glycophorin receptors found on the surface of red blood cells.
The malaria parasite Plasmodium falciparum uses these receptors—GYPA and GYPB—to enter the cells.
Researchers identified a gene variant that results in altered GYPA and GYPB receptors and may reduce the risk of severe malaria by 40%.
Ellen Leffler, of the University of Oxford in the UK, and her colleagues reported these findings in Science.
The researchers performed genome sequencing of 765 individuals from 10 ethnic groups in Gambia, Burkina Faso, Cameroon, and Tanzania.
The team also conducted a study across the Gambia, Kenya, and Malawi that included 5310 individuals from the general population and 4579 people who were hospitalized with severe malaria.
These analyses revealed copy number variants affecting GYPA and GYPB.
“[W]e found strong evidence that variation in the glycophorin gene cluster influences malaria susceptibility,” Dr Leffler said.
“We found some people have a complex rearrangement of GYPA and GYPB genes, forming a hybrid glycophorin, and these people are less likely to develop severe complications of the disease.”
The rearrangement involves the loss of GYPB and gain of 2 GYPB-A hybrid genes. The hybrid GYPB-A gene is found in a rare blood group—part of the MNS blood group system—where it is known as Dantu.
DUP4, the most common Dantu gene variant, is a result of the rearrangement. And the researchers found that DUP4 reduced the risk of severe malaria by an estimated 40%.
DUP4 was only present in certain populations, particularly in individuals of East African descent.
The researchers proposed a number of reasons as to why DUP4 may not be more widespread, including the possibility that it emerged recently. Alternatively, it may only protect against certain strains of P falciparum that are specific to east Africa.
Though more research is needed, the team said these findings link the structural variation of glycophorin receptors with resistance to severe malaria.
“We are starting to find that the glycophorin region of the genome has an important role in protecting people against malaria,” said study author Dominic Kwiatkowski, MD, of the University of Oxford.
“Our discovery that a specific variant of glycophorin invasion receptors can give substantial protection against severe malaria will hopefully inspire further research on exactly how Plasmodium falciparum invade red blood cells. This could also help us discover novel parasite weaknesses that could be exploited in future interventions against this deadly disease.” ![]()
Bivalirudin not superior to heparin in real-world analysis
A new study suggests bivalirudin does not produce better outcomes than heparin in patients undergoing transradial primary percutaneous coronary intervention (PCI) for ST elevation myocardial infarction (STEMI).
Researchers found no significant difference in the rate of a composite of death, myocardial infarction, and stroke in patients who received bivalirudin or heparin, with or without glycoprotein (GP) IIb/IIIa inhibitors.
Likewise, the incidence of bleeding was not significantly different between the heparin and bivalirudin groups.
However, the rate of stent thrombosis was significantly higher in patients who received bivalirudin.
Ion S. Jovin, MD, of Virginia Commonwealth University in Richmond, and colleagues reported these results in JACC: Cardiovascular Interventions.
Using data from the National Cardiovascular Data Registry CathPCI Registry, the researchers examined the records of 67,368 patients with STEMI. The patients underwent primary PCI via radial access at 1584 sites between 2009 to 2015.
Patients received anticoagulation with bivalirudin (n=29,660) or heparin (n=37,708). Twenty-three percent (n=6781) of patients on bivalirudin received GP IIb/IIIa inhibitors, as did 59% of patients on heparin (n=22,416).
Results
In an unadjusted analysis, the researchers found no significant difference in the rate of the composite endpoint, which included death, myocardial infarction, and stroke. The incidence was 4.6% with bivalirudin and 4.7% with heparin (P=0.47).
However, patients who received bivalirudin had a significantly higher rate of acute stent thrombosis—1.03%—than patients who received heparin—0.602% (P<0.001).
And there were significantly fewer bleeding episodes with bivalirudin than with heparin—6.8% and 8.1%, respectively, (P<0.001).
The researchers adjusted their analysis for multiple variables, including a propensity score reflecting the probability of receiving bivalirudin to account for patient differences between groups.
After these adjustments, the odds ratio (OR) of the composite endpoint for bivalirudin versus heparin was 0.95 (P=0.152).
The OR ratio for acute stent thrombosis was 2.11 (P<0.001), and the OR for bleeding was 0.98 (P=0.57).
The researchers did note that, among patients who were not receiving GP IIb/IIIa inhibitors, outcomes were better in patients who received bivalirudin. They had a significantly lower risk of bleeding and the composite endpoint than patients who received heparin.
“Our sensitivity analysis provides some insights into direct comparisons of bivalirudin and heparin when GPIIb/IIIa inhibitors are forced out of the equation and suggests that, in the direct comparison, bivalirudin may have superior outcomes,” Dr Jovin said.
“However, our study showed that, in the real world, over a third of the patients with STEMI undergoing transradial PCI who receive heparin and about a fifth of patients who receive bivalirudin also receive GPIIb/IIIa inhibitors.”
The researchers suggested a need for a randomized trial in patients treated exclusively via transradial primary PCI and anticoagulated with bivalirudin versus heparin as well as a cost-effectiveness analysis comparing heparin and bivalirudin. ![]()
A new study suggests bivalirudin does not produce better outcomes than heparin in patients undergoing transradial primary percutaneous coronary intervention (PCI) for ST elevation myocardial infarction (STEMI).
Researchers found no significant difference in the rate of a composite of death, myocardial infarction, and stroke in patients who received bivalirudin or heparin, with or without glycoprotein (GP) IIb/IIIa inhibitors.
Likewise, the incidence of bleeding was not significantly different between the heparin and bivalirudin groups.
However, the rate of stent thrombosis was significantly higher in patients who received bivalirudin.
Ion S. Jovin, MD, of Virginia Commonwealth University in Richmond, and colleagues reported these results in JACC: Cardiovascular Interventions.
Using data from the National Cardiovascular Data Registry CathPCI Registry, the researchers examined the records of 67,368 patients with STEMI. The patients underwent primary PCI via radial access at 1584 sites between 2009 to 2015.
Patients received anticoagulation with bivalirudin (n=29,660) or heparin (n=37,708). Twenty-three percent (n=6781) of patients on bivalirudin received GP IIb/IIIa inhibitors, as did 59% of patients on heparin (n=22,416).
Results
In an unadjusted analysis, the researchers found no significant difference in the rate of the composite endpoint, which included death, myocardial infarction, and stroke. The incidence was 4.6% with bivalirudin and 4.7% with heparin (P=0.47).
However, patients who received bivalirudin had a significantly higher rate of acute stent thrombosis—1.03%—than patients who received heparin—0.602% (P<0.001).
And there were significantly fewer bleeding episodes with bivalirudin than with heparin—6.8% and 8.1%, respectively, (P<0.001).
The researchers adjusted their analysis for multiple variables, including a propensity score reflecting the probability of receiving bivalirudin to account for patient differences between groups.
After these adjustments, the odds ratio (OR) of the composite endpoint for bivalirudin versus heparin was 0.95 (P=0.152).
The OR ratio for acute stent thrombosis was 2.11 (P<0.001), and the OR for bleeding was 0.98 (P=0.57).
The researchers did note that, among patients who were not receiving GP IIb/IIIa inhibitors, outcomes were better in patients who received bivalirudin. They had a significantly lower risk of bleeding and the composite endpoint than patients who received heparin.
“Our sensitivity analysis provides some insights into direct comparisons of bivalirudin and heparin when GPIIb/IIIa inhibitors are forced out of the equation and suggests that, in the direct comparison, bivalirudin may have superior outcomes,” Dr Jovin said.
“However, our study showed that, in the real world, over a third of the patients with STEMI undergoing transradial PCI who receive heparin and about a fifth of patients who receive bivalirudin also receive GPIIb/IIIa inhibitors.”
The researchers suggested a need for a randomized trial in patients treated exclusively via transradial primary PCI and anticoagulated with bivalirudin versus heparin as well as a cost-effectiveness analysis comparing heparin and bivalirudin. ![]()
A new study suggests bivalirudin does not produce better outcomes than heparin in patients undergoing transradial primary percutaneous coronary intervention (PCI) for ST elevation myocardial infarction (STEMI).
Researchers found no significant difference in the rate of a composite of death, myocardial infarction, and stroke in patients who received bivalirudin or heparin, with or without glycoprotein (GP) IIb/IIIa inhibitors.
Likewise, the incidence of bleeding was not significantly different between the heparin and bivalirudin groups.
However, the rate of stent thrombosis was significantly higher in patients who received bivalirudin.
Ion S. Jovin, MD, of Virginia Commonwealth University in Richmond, and colleagues reported these results in JACC: Cardiovascular Interventions.
Using data from the National Cardiovascular Data Registry CathPCI Registry, the researchers examined the records of 67,368 patients with STEMI. The patients underwent primary PCI via radial access at 1584 sites between 2009 to 2015.
Patients received anticoagulation with bivalirudin (n=29,660) or heparin (n=37,708). Twenty-three percent (n=6781) of patients on bivalirudin received GP IIb/IIIa inhibitors, as did 59% of patients on heparin (n=22,416).
Results
In an unadjusted analysis, the researchers found no significant difference in the rate of the composite endpoint, which included death, myocardial infarction, and stroke. The incidence was 4.6% with bivalirudin and 4.7% with heparin (P=0.47).
However, patients who received bivalirudin had a significantly higher rate of acute stent thrombosis—1.03%—than patients who received heparin—0.602% (P<0.001).
And there were significantly fewer bleeding episodes with bivalirudin than with heparin—6.8% and 8.1%, respectively, (P<0.001).
The researchers adjusted their analysis for multiple variables, including a propensity score reflecting the probability of receiving bivalirudin to account for patient differences between groups.
After these adjustments, the odds ratio (OR) of the composite endpoint for bivalirudin versus heparin was 0.95 (P=0.152).
The OR ratio for acute stent thrombosis was 2.11 (P<0.001), and the OR for bleeding was 0.98 (P=0.57).
The researchers did note that, among patients who were not receiving GP IIb/IIIa inhibitors, outcomes were better in patients who received bivalirudin. They had a significantly lower risk of bleeding and the composite endpoint than patients who received heparin.
“Our sensitivity analysis provides some insights into direct comparisons of bivalirudin and heparin when GPIIb/IIIa inhibitors are forced out of the equation and suggests that, in the direct comparison, bivalirudin may have superior outcomes,” Dr Jovin said.
“However, our study showed that, in the real world, over a third of the patients with STEMI undergoing transradial PCI who receive heparin and about a fifth of patients who receive bivalirudin also receive GPIIb/IIIa inhibitors.”
The researchers suggested a need for a randomized trial in patients treated exclusively via transradial primary PCI and anticoagulated with bivalirudin versus heparin as well as a cost-effectiveness analysis comparing heparin and bivalirudin. ![]()
Drug receives breakthrough designation for relapsed/refractory AML

The US Food and Drug Administration (FDA) has granted GMI-1271 breakthrough therapy designation for the treatment of adults with relapsed/refractory acute myeloid leukemia (AML).
GMI-1271 is an E-selectin antagonist being developed by GlycoMimetics, Inc.
The product already has orphan and fast track designations from the FDA for the treatment of AML.
GMI-1271 is currently being evaluated in a phase 1/2 trial of patients with relapsed or refractory AML and patients age 60 and older with newly diagnosed AML.
The patients are receiving GM-1271 in combination with chemotherapy. The relapsed/refractory group is receiving mitoxantrone, etoposide, and cytarabine. The newly diagnosed patients are receiving cytarabine and idarubicin (7+3).
GlycoMimetics plans to present data from this trial at the 2017 American Society of Clinical Oncology (ASCO) Annual Meeting as abstracts 2520 and 2560.
The company also plans to present the research at the 22nd Congress of the European Hematology Association (EHA) as abstracts P547 and P203.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About fast track designation
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the new drug application or biologic license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted GMI-1271 breakthrough therapy designation for the treatment of adults with relapsed/refractory acute myeloid leukemia (AML).
GMI-1271 is an E-selectin antagonist being developed by GlycoMimetics, Inc.
The product already has orphan and fast track designations from the FDA for the treatment of AML.
GMI-1271 is currently being evaluated in a phase 1/2 trial of patients with relapsed or refractory AML and patients age 60 and older with newly diagnosed AML.
The patients are receiving GM-1271 in combination with chemotherapy. The relapsed/refractory group is receiving mitoxantrone, etoposide, and cytarabine. The newly diagnosed patients are receiving cytarabine and idarubicin (7+3).
GlycoMimetics plans to present data from this trial at the 2017 American Society of Clinical Oncology (ASCO) Annual Meeting as abstracts 2520 and 2560.
The company also plans to present the research at the 22nd Congress of the European Hematology Association (EHA) as abstracts P547 and P203.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About fast track designation
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the new drug application or biologic license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted GMI-1271 breakthrough therapy designation for the treatment of adults with relapsed/refractory acute myeloid leukemia (AML).
GMI-1271 is an E-selectin antagonist being developed by GlycoMimetics, Inc.
The product already has orphan and fast track designations from the FDA for the treatment of AML.
GMI-1271 is currently being evaluated in a phase 1/2 trial of patients with relapsed or refractory AML and patients age 60 and older with newly diagnosed AML.
The patients are receiving GM-1271 in combination with chemotherapy. The relapsed/refractory group is receiving mitoxantrone, etoposide, and cytarabine. The newly diagnosed patients are receiving cytarabine and idarubicin (7+3).
GlycoMimetics plans to present data from this trial at the 2017 American Society of Clinical Oncology (ASCO) Annual Meeting as abstracts 2520 and 2560.
The company also plans to present the research at the 22nd Congress of the European Hematology Association (EHA) as abstracts P547 and P203.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About fast track designation
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the new drug application or biologic license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
Single-cell analysis reveals TKI-resistant CML stem cells

Researchers say they’ve developed a technique for single-cell analysis that has revealed a population of treatment-resistant stem cells in patients with chronic myeloid leukemia (CML).
“It is increasingly recognized that tumors contain a variety of different cell types, including so-called cancer stem cells, that drive the growth and relapse of a patient’s cancer,” said study author Adam Mead, BM BCh, PhD, of University of Oxford in the UK.
“These cells can be very rare and extremely difficult to find after treatment as they become hidden within the normal tissue. We used a new genetic technique to identify and analyze single cancer stem cells in leukemia patients before and after treatment.”
Dr Mead and his colleagues detailed this research in Nature Medicine.
The team’s single-cell analysis technique combines high-sensitivity mutation detection with whole-transcriptome analysis.
The researchers used the method to analyze stem cells from patients with CML and found the cells to be heterogeneous.
In addition, the team was able to identify a subset of CML stem cells that proved resistant to treatment with tyrosine kinase inhibitors (TKIs).
“We found that, even in individual cases of leukemia, there are various types of cancer stem cell that respond differently to the treatment,” Dr Mead said.
“A small number of these cells are highly resistant to the treatment and are likely to be responsible for disease recurrence when the treatment is stopped. Our research allowed us uniquely to analyze these crucial cells that evade treatment so that we might learn how to more effectively eradicate them.”
The researchers said these TKI-resistant CML stem cells were “transcriptionally distinct” from normal hematopoietic stem cells.
The TKI-resistant cells were characterized by dysregulation of specific genes and pathways—TGF-β, TNF-α, JAK–STAT, CTNNB1, and NFKB1A—that could potentially be targeted to improve the treatment of CML.
The researchers also said their single-cell analysis technique can be used beyond CML.
“This technique could be adapted to analyze a range of different cancers to help predict both the likely response to treatment and the risk of the disease returning in the future,” Dr Mead said. “This should eventually enable treatment to be tailored to target each and every type of cancer stem cell that may be present.” ![]()
Researchers say they’ve developed a technique for single-cell analysis that has revealed a population of treatment-resistant stem cells in patients with chronic myeloid leukemia (CML).
“It is increasingly recognized that tumors contain a variety of different cell types, including so-called cancer stem cells, that drive the growth and relapse of a patient’s cancer,” said study author Adam Mead, BM BCh, PhD, of University of Oxford in the UK.
“These cells can be very rare and extremely difficult to find after treatment as they become hidden within the normal tissue. We used a new genetic technique to identify and analyze single cancer stem cells in leukemia patients before and after treatment.”
Dr Mead and his colleagues detailed this research in Nature Medicine.
The team’s single-cell analysis technique combines high-sensitivity mutation detection with whole-transcriptome analysis.
The researchers used the method to analyze stem cells from patients with CML and found the cells to be heterogeneous.
In addition, the team was able to identify a subset of CML stem cells that proved resistant to treatment with tyrosine kinase inhibitors (TKIs).
“We found that, even in individual cases of leukemia, there are various types of cancer stem cell that respond differently to the treatment,” Dr Mead said.
“A small number of these cells are highly resistant to the treatment and are likely to be responsible for disease recurrence when the treatment is stopped. Our research allowed us uniquely to analyze these crucial cells that evade treatment so that we might learn how to more effectively eradicate them.”
The researchers said these TKI-resistant CML stem cells were “transcriptionally distinct” from normal hematopoietic stem cells.
The TKI-resistant cells were characterized by dysregulation of specific genes and pathways—TGF-β, TNF-α, JAK–STAT, CTNNB1, and NFKB1A—that could potentially be targeted to improve the treatment of CML.
The researchers also said their single-cell analysis technique can be used beyond CML.
“This technique could be adapted to analyze a range of different cancers to help predict both the likely response to treatment and the risk of the disease returning in the future,” Dr Mead said. “This should eventually enable treatment to be tailored to target each and every type of cancer stem cell that may be present.” ![]()
Researchers say they’ve developed a technique for single-cell analysis that has revealed a population of treatment-resistant stem cells in patients with chronic myeloid leukemia (CML).
“It is increasingly recognized that tumors contain a variety of different cell types, including so-called cancer stem cells, that drive the growth and relapse of a patient’s cancer,” said study author Adam Mead, BM BCh, PhD, of University of Oxford in the UK.
“These cells can be very rare and extremely difficult to find after treatment as they become hidden within the normal tissue. We used a new genetic technique to identify and analyze single cancer stem cells in leukemia patients before and after treatment.”
Dr Mead and his colleagues detailed this research in Nature Medicine.
The team’s single-cell analysis technique combines high-sensitivity mutation detection with whole-transcriptome analysis.
The researchers used the method to analyze stem cells from patients with CML and found the cells to be heterogeneous.
In addition, the team was able to identify a subset of CML stem cells that proved resistant to treatment with tyrosine kinase inhibitors (TKIs).
“We found that, even in individual cases of leukemia, there are various types of cancer stem cell that respond differently to the treatment,” Dr Mead said.
“A small number of these cells are highly resistant to the treatment and are likely to be responsible for disease recurrence when the treatment is stopped. Our research allowed us uniquely to analyze these crucial cells that evade treatment so that we might learn how to more effectively eradicate them.”
The researchers said these TKI-resistant CML stem cells were “transcriptionally distinct” from normal hematopoietic stem cells.
The TKI-resistant cells were characterized by dysregulation of specific genes and pathways—TGF-β, TNF-α, JAK–STAT, CTNNB1, and NFKB1A—that could potentially be targeted to improve the treatment of CML.
The researchers also said their single-cell analysis technique can be used beyond CML.
“This technique could be adapted to analyze a range of different cancers to help predict both the likely response to treatment and the risk of the disease returning in the future,” Dr Mead said. “This should eventually enable treatment to be tailored to target each and every type of cancer stem cell that may be present.” ![]()
First generic version of clofarabine available in US

Clofarabine Injection, the first-to-market generic version of Sanofi Genzyme’s Clolar, is now available in the US.
The generic, a product of Fresenius Kabi, is available as a single dose vial containing 20 mg per 20 mL clofarabine.
Clofarabine is a purine nucleoside metabolic inhibitor indicated for the treatment of patients ages 1 to 21 with relapsed or refractory acute lymphoblastic leukemia (ALL) who received at least 2 prior treatment regimens.
Clolar was granted accelerated approval for this indication in the US in 2004.
The approval was based on response rates observed in ALL patients. There are no trials verifying that clofarabine confers improvement in survival or disease-related symptoms in ALL patients.
Clofarabine was assessed in a single-arm, phase 2 trial of 61 pediatric patients with relapsed/refractory ALL.
The patients’ median age was 12 (range, 1 to 20 years), and their median number of prior treatment regimens was 3 (range, 2 to 6).
The patients received clofarabine at 52 mg/m2 intravenously over 2 hours daily for 5 days, every 2 to 6 weeks.
The overall response rate was 30%. Seven patient achieved a complete response (CR), 5 had a CR without platelet recovery, and 6 patients had a partial response.
The median duration of CR in patients who did not go on to hematopoietic stem cell transplant was 6 weeks.
The most common grade 3 or higher adverse events were febrile neutropenia, anorexia, hypotension, and nausea.
These results were published in the Journal of Clinical Oncology in 2006. ![]()
Clofarabine Injection, the first-to-market generic version of Sanofi Genzyme’s Clolar, is now available in the US.
The generic, a product of Fresenius Kabi, is available as a single dose vial containing 20 mg per 20 mL clofarabine.
Clofarabine is a purine nucleoside metabolic inhibitor indicated for the treatment of patients ages 1 to 21 with relapsed or refractory acute lymphoblastic leukemia (ALL) who received at least 2 prior treatment regimens.
Clolar was granted accelerated approval for this indication in the US in 2004.
The approval was based on response rates observed in ALL patients. There are no trials verifying that clofarabine confers improvement in survival or disease-related symptoms in ALL patients.
Clofarabine was assessed in a single-arm, phase 2 trial of 61 pediatric patients with relapsed/refractory ALL.
The patients’ median age was 12 (range, 1 to 20 years), and their median number of prior treatment regimens was 3 (range, 2 to 6).
The patients received clofarabine at 52 mg/m2 intravenously over 2 hours daily for 5 days, every 2 to 6 weeks.
The overall response rate was 30%. Seven patient achieved a complete response (CR), 5 had a CR without platelet recovery, and 6 patients had a partial response.
The median duration of CR in patients who did not go on to hematopoietic stem cell transplant was 6 weeks.
The most common grade 3 or higher adverse events were febrile neutropenia, anorexia, hypotension, and nausea.
These results were published in the Journal of Clinical Oncology in 2006. ![]()
Clofarabine Injection, the first-to-market generic version of Sanofi Genzyme’s Clolar, is now available in the US.
The generic, a product of Fresenius Kabi, is available as a single dose vial containing 20 mg per 20 mL clofarabine.
Clofarabine is a purine nucleoside metabolic inhibitor indicated for the treatment of patients ages 1 to 21 with relapsed or refractory acute lymphoblastic leukemia (ALL) who received at least 2 prior treatment regimens.
Clolar was granted accelerated approval for this indication in the US in 2004.
The approval was based on response rates observed in ALL patients. There are no trials verifying that clofarabine confers improvement in survival or disease-related symptoms in ALL patients.
Clofarabine was assessed in a single-arm, phase 2 trial of 61 pediatric patients with relapsed/refractory ALL.
The patients’ median age was 12 (range, 1 to 20 years), and their median number of prior treatment regimens was 3 (range, 2 to 6).
The patients received clofarabine at 52 mg/m2 intravenously over 2 hours daily for 5 days, every 2 to 6 weeks.
The overall response rate was 30%. Seven patient achieved a complete response (CR), 5 had a CR without platelet recovery, and 6 patients had a partial response.
The median duration of CR in patients who did not go on to hematopoietic stem cell transplant was 6 weeks.
The most common grade 3 or higher adverse events were febrile neutropenia, anorexia, hypotension, and nausea.
These results were published in the Journal of Clinical Oncology in 2006.
FDA grants priority review to NDA for copanlisib

The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for copanlisib, an intravenous PI3K inhibitor.
The NDA is for copanlisib as a treatment for patients with relapsed or refractory follicular lymphoma (FL) who have received at least 2 prior therapies.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The application for copanlisib is supported by data from the CHRONOS-1 trial. This phase 2 trial enrolled 141 patients with relapsed/refractory, indolent non-Hodgkin lymphoma. Most of these patients had FL (n=104).
In all patients, copanlisib produced an objective response rate of 59.2%, with a complete response rate of 12%. The median duration of response exceeded 98 weeks.
In the FL subset, copanlisib produced an overall response rate of 58.7%, with a complete response rate of 14.4%. The median duration of response exceeded 52 weeks.
In the entire cohort, there were 3 deaths considered related to copanlisib.
The most common treatment-related adverse events were transient hyperglycemia (all grades: 49%/grade 3-4: 40%) and hypertension (all grades: 29%/grade 3: 23%).
“Patients with relapsed or refractory follicular lymphoma have a poor prognosis, and new treatment options which are well tolerated and effective are needed to prolong progression-free survival and improve quality of life for these patients,” said Martin Dreyling, MD, a professor at the University of Munich Hospital (Grosshadern) in Germany and lead investigator of the CHRONOS-1 study.
“Based on the CHRONOS-1 results, where copanlisib showed durable efficacy with a manageable and distinct safety profile, the compound may have the potential to address this unmet medical need.”
Data from CHRONOS-1 were presented at the AACR Annual Meeting 2017.
Data from the FL subset of the trial are scheduled to be presented at the 2017 ASCO Annual Meeting in June.
Copanlisib is being developed by Bayer. The compound has fast track and orphan drug designations from the FDA.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the NDA or biologic license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for copanlisib, an intravenous PI3K inhibitor.
The NDA is for copanlisib as a treatment for patients with relapsed or refractory follicular lymphoma (FL) who have received at least 2 prior therapies.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The application for copanlisib is supported by data from the CHRONOS-1 trial. This phase 2 trial enrolled 141 patients with relapsed/refractory, indolent non-Hodgkin lymphoma. Most of these patients had FL (n=104).
In all patients, copanlisib produced an objective response rate of 59.2%, with a complete response rate of 12%. The median duration of response exceeded 98 weeks.
In the FL subset, copanlisib produced an overall response rate of 58.7%, with a complete response rate of 14.4%. The median duration of response exceeded 52 weeks.
In the entire cohort, there were 3 deaths considered related to copanlisib.
The most common treatment-related adverse events were transient hyperglycemia (all grades: 49%/grade 3-4: 40%) and hypertension (all grades: 29%/grade 3: 23%).
“Patients with relapsed or refractory follicular lymphoma have a poor prognosis, and new treatment options which are well tolerated and effective are needed to prolong progression-free survival and improve quality of life for these patients,” said Martin Dreyling, MD, a professor at the University of Munich Hospital (Grosshadern) in Germany and lead investigator of the CHRONOS-1 study.
“Based on the CHRONOS-1 results, where copanlisib showed durable efficacy with a manageable and distinct safety profile, the compound may have the potential to address this unmet medical need.”
Data from CHRONOS-1 were presented at the AACR Annual Meeting 2017.
Data from the FL subset of the trial are scheduled to be presented at the 2017 ASCO Annual Meeting in June.
Copanlisib is being developed by Bayer. The compound has fast track and orphan drug designations from the FDA.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the NDA or biologic license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for copanlisib, an intravenous PI3K inhibitor.
The NDA is for copanlisib as a treatment for patients with relapsed or refractory follicular lymphoma (FL) who have received at least 2 prior therapies.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The application for copanlisib is supported by data from the CHRONOS-1 trial. This phase 2 trial enrolled 141 patients with relapsed/refractory, indolent non-Hodgkin lymphoma. Most of these patients had FL (n=104).
In all patients, copanlisib produced an objective response rate of 59.2%, with a complete response rate of 12%. The median duration of response exceeded 98 weeks.
In the FL subset, copanlisib produced an overall response rate of 58.7%, with a complete response rate of 14.4%. The median duration of response exceeded 52 weeks.
In the entire cohort, there were 3 deaths considered related to copanlisib.
The most common treatment-related adverse events were transient hyperglycemia (all grades: 49%/grade 3-4: 40%) and hypertension (all grades: 29%/grade 3: 23%).
“Patients with relapsed or refractory follicular lymphoma have a poor prognosis, and new treatment options which are well tolerated and effective are needed to prolong progression-free survival and improve quality of life for these patients,” said Martin Dreyling, MD, a professor at the University of Munich Hospital (Grosshadern) in Germany and lead investigator of the CHRONOS-1 study.
“Based on the CHRONOS-1 results, where copanlisib showed durable efficacy with a manageable and distinct safety profile, the compound may have the potential to address this unmet medical need.”
Data from CHRONOS-1 were presented at the AACR Annual Meeting 2017.
Data from the FL subset of the trial are scheduled to be presented at the 2017 ASCO Annual Meeting in June.
Copanlisib is being developed by Bayer. The compound has fast track and orphan drug designations from the FDA.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the NDA or biologic license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
Compound could treat lymphoma, myeloma

A nucleoside analog has shown potential for treating primary effusion lymphoma (PEL) and multiple myeloma (MM), according to researchers.
The compound, 6-ethylthioinosine (6-ETI), killed both PEL and MM cells in vitro.
6-ETI also reduced tumor burden and prolonged survival in mouse models of MM and PEL.
Ethel Cesarman, MD, PhD, of Weill Cornell Medicine in New York, New York, and her colleagues conducted this research and disclosed their results in the Journal of Clinical Investigation.
The researchers identified 6-ETI via high-throughput screening, and they initially tested the compound in PEL cell lines. 6-ETI induced necrosis, apoptosis, and autophagy in these cells.
In a xenograft model of PEL, mice treated with 6-ETI experienced “striking and immediate regression of the implanted xenograft within 3 days of treatment,” according to the researchers.
In addition, mice that received 6-ETI had significantly longer progression-free and overall survival than control mice (P<0.0001 for both), and the researchers said there were no obvious toxicities from the treatment.
Looking into the mechanism of 6-ETI, the researchers found that adenosine kinase (ADK) is required to phosphorylate and activate the compound, and PEL cells have high levels of ADK.
Because PEL cells closely resemble plasma cells, the researchers theorized that MM might produce high levels of ADK as well. Experiments in MM cells proved this theory correct.
So the researchers tested 6-ETI in MM cell lines and samples from MM patients. The compound induced apoptosis and autophagy, and it activated a DNA damage response in MM cells.
In a mouse model of MM, 6-ETI treatment significantly reduced tumor burden but did not result in weight loss. And treated mice had significantly longer overall survival than control mice (P<0.005).
Dr Cesarman and her colleagues are now trying to better understand how 6-ETI works and determine what other cancers expressing high levels of ADK might respond to the drug.
“This compound could provide a much-needed approach to treat people with some forms of plasma malignancies as well as other cancers that express ADK,” Dr Cesarman said.
A nucleoside analog has shown potential for treating primary effusion lymphoma (PEL) and multiple myeloma (MM), according to researchers.
The compound, 6-ethylthioinosine (6-ETI), killed both PEL and MM cells in vitro.
6-ETI also reduced tumor burden and prolonged survival in mouse models of MM and PEL.
Ethel Cesarman, MD, PhD, of Weill Cornell Medicine in New York, New York, and her colleagues conducted this research and disclosed their results in the Journal of Clinical Investigation.
The researchers identified 6-ETI via high-throughput screening, and they initially tested the compound in PEL cell lines. 6-ETI induced necrosis, apoptosis, and autophagy in these cells.
In a xenograft model of PEL, mice treated with 6-ETI experienced “striking and immediate regression of the implanted xenograft within 3 days of treatment,” according to the researchers.
In addition, mice that received 6-ETI had significantly longer progression-free and overall survival than control mice (P<0.0001 for both), and the researchers said there were no obvious toxicities from the treatment.
Looking into the mechanism of 6-ETI, the researchers found that adenosine kinase (ADK) is required to phosphorylate and activate the compound, and PEL cells have high levels of ADK.
Because PEL cells closely resemble plasma cells, the researchers theorized that MM might produce high levels of ADK as well. Experiments in MM cells proved this theory correct.
So the researchers tested 6-ETI in MM cell lines and samples from MM patients. The compound induced apoptosis and autophagy, and it activated a DNA damage response in MM cells.
In a mouse model of MM, 6-ETI treatment significantly reduced tumor burden but did not result in weight loss. And treated mice had significantly longer overall survival than control mice (P<0.005).
Dr Cesarman and her colleagues are now trying to better understand how 6-ETI works and determine what other cancers expressing high levels of ADK might respond to the drug.
“This compound could provide a much-needed approach to treat people with some forms of plasma malignancies as well as other cancers that express ADK,” Dr Cesarman said.
A nucleoside analog has shown potential for treating primary effusion lymphoma (PEL) and multiple myeloma (MM), according to researchers.
The compound, 6-ethylthioinosine (6-ETI), killed both PEL and MM cells in vitro.
6-ETI also reduced tumor burden and prolonged survival in mouse models of MM and PEL.
Ethel Cesarman, MD, PhD, of Weill Cornell Medicine in New York, New York, and her colleagues conducted this research and disclosed their results in the Journal of Clinical Investigation.
The researchers identified 6-ETI via high-throughput screening, and they initially tested the compound in PEL cell lines. 6-ETI induced necrosis, apoptosis, and autophagy in these cells.
In a xenograft model of PEL, mice treated with 6-ETI experienced “striking and immediate regression of the implanted xenograft within 3 days of treatment,” according to the researchers.
In addition, mice that received 6-ETI had significantly longer progression-free and overall survival than control mice (P<0.0001 for both), and the researchers said there were no obvious toxicities from the treatment.
Looking into the mechanism of 6-ETI, the researchers found that adenosine kinase (ADK) is required to phosphorylate and activate the compound, and PEL cells have high levels of ADK.
Because PEL cells closely resemble plasma cells, the researchers theorized that MM might produce high levels of ADK as well. Experiments in MM cells proved this theory correct.
So the researchers tested 6-ETI in MM cell lines and samples from MM patients. The compound induced apoptosis and autophagy, and it activated a DNA damage response in MM cells.
In a mouse model of MM, 6-ETI treatment significantly reduced tumor burden but did not result in weight loss. And treated mice had significantly longer overall survival than control mice (P<0.005).
Dr Cesarman and her colleagues are now trying to better understand how 6-ETI works and determine what other cancers expressing high levels of ADK might respond to the drug.
“This compound could provide a much-needed approach to treat people with some forms of plasma malignancies as well as other cancers that express ADK,” Dr Cesarman said.
Drug receives rare pediatric disease designation for SCD

The US Food and Drug Administration (FDA) has granted rare pediatric disease designation to IMR-687, a product intended to treat sickle cell disease (SCD).
IMR-687 is the first SCD candidate to be designated as a drug for a rare pediatric disease, and this designation builds upon the FDA’s earlier granting of orphan designation for IMR-687.
IMR-687 is a selective inhibitor of phosphodiesterase-9 (PDE9i) in blood cells. It was specifically designed to address the underlying pathology of SCD.
In preclinical studies, IMR-687 demonstrated the ability to increase fetal globin. This prevented the polymerization of sickled hemoglobin and reduced red blood cell sickling, leukocytosis, and the occlusion of blood vessels.
Researchers presented these results at the 2016 ASH Annual Meeting.
Imara Inc., the company developing IMR-687, is conducting a phase 1a trial of the product in healthy volunteers.
If the trial has a positive outcome (results are expected this summer), Imara will initiate a phase 2a study in adults with SCD later this year. The company expects to initiate a phase 2 study in pediatric patients in 2018.
About rare pediatric disease designation
Rare pediatric disease designation is granted to drugs that show promise to treat orphan diseases affecting fewer than 200,000 patients in the US, primarily patients age 18 or younger.
The designation provides incentives to advance the development of drugs for rare disease, including access to the FDA’s expedited review and approval programs.
Under the FDA’s Rare Pediatric Disease Priority Review Voucher Program, a sponsor with rare pediatric disease designation who receives an approval of a new drug application is eligible for a voucher that can be redeemed to obtain priority review for any subsequent marketing application.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted rare pediatric disease designation to IMR-687, a product intended to treat sickle cell disease (SCD).
IMR-687 is the first SCD candidate to be designated as a drug for a rare pediatric disease, and this designation builds upon the FDA’s earlier granting of orphan designation for IMR-687.
IMR-687 is a selective inhibitor of phosphodiesterase-9 (PDE9i) in blood cells. It was specifically designed to address the underlying pathology of SCD.
In preclinical studies, IMR-687 demonstrated the ability to increase fetal globin. This prevented the polymerization of sickled hemoglobin and reduced red blood cell sickling, leukocytosis, and the occlusion of blood vessels.
Researchers presented these results at the 2016 ASH Annual Meeting.
Imara Inc., the company developing IMR-687, is conducting a phase 1a trial of the product in healthy volunteers.
If the trial has a positive outcome (results are expected this summer), Imara will initiate a phase 2a study in adults with SCD later this year. The company expects to initiate a phase 2 study in pediatric patients in 2018.
About rare pediatric disease designation
Rare pediatric disease designation is granted to drugs that show promise to treat orphan diseases affecting fewer than 200,000 patients in the US, primarily patients age 18 or younger.
The designation provides incentives to advance the development of drugs for rare disease, including access to the FDA’s expedited review and approval programs.
Under the FDA’s Rare Pediatric Disease Priority Review Voucher Program, a sponsor with rare pediatric disease designation who receives an approval of a new drug application is eligible for a voucher that can be redeemed to obtain priority review for any subsequent marketing application.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted rare pediatric disease designation to IMR-687, a product intended to treat sickle cell disease (SCD).
IMR-687 is the first SCD candidate to be designated as a drug for a rare pediatric disease, and this designation builds upon the FDA’s earlier granting of orphan designation for IMR-687.
IMR-687 is a selective inhibitor of phosphodiesterase-9 (PDE9i) in blood cells. It was specifically designed to address the underlying pathology of SCD.
In preclinical studies, IMR-687 demonstrated the ability to increase fetal globin. This prevented the polymerization of sickled hemoglobin and reduced red blood cell sickling, leukocytosis, and the occlusion of blood vessels.
Researchers presented these results at the 2016 ASH Annual Meeting.
Imara Inc., the company developing IMR-687, is conducting a phase 1a trial of the product in healthy volunteers.
If the trial has a positive outcome (results are expected this summer), Imara will initiate a phase 2a study in adults with SCD later this year. The company expects to initiate a phase 2 study in pediatric patients in 2018.
About rare pediatric disease designation
Rare pediatric disease designation is granted to drugs that show promise to treat orphan diseases affecting fewer than 200,000 patients in the US, primarily patients age 18 or younger.
The designation provides incentives to advance the development of drugs for rare disease, including access to the FDA’s expedited review and approval programs.
Under the FDA’s Rare Pediatric Disease Priority Review Voucher Program, a sponsor with rare pediatric disease designation who receives an approval of a new drug application is eligible for a voucher that can be redeemed to obtain priority review for any subsequent marketing application.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.