User login
Brentuximab vedotin bests standard of care in CTCL

Photo by Larry Young
SAN FRANCISCO—The phase 3 ALCANZA trial is the first to convincingly demonstrate that a new systemic agent can be more effective than standard of care (SOC) options for cutaneous T-cell lymphoma (CTCL), according to a speaker at the 9th Annual T-cell Lymphoma Forum.
The trial showed significant improvements in response, symptom burden, and progression-free survival (PFS) in patients with CD30-expressing CTCL who received brentuximab vedotin (BV), as compared to patients who received either bexarotene or methotrexate.
“[These are] compelling results that potentially may have practice-changing implications for the use of brentuximab in managing CD30-expressing CTCL patients who require systemic therapy,” said Youn H. Kim, MD, of Stanford University School of Medicine in California.
Dr Kim presented these results at this year’s T-cell Lymphoma Forum. The data were also presented at the recent ASH Annual Meeting (abstract 182).
The ALCANZA trial was sponsored by Millennium Pharmaceuticals, Inc. (now a part of Takeda Pharmaceutical Company Limited) and Seattle Genetics, Inc.
The study was designed to compare BV to the SOC options of methotrexate or bexarotene in patients with CD30-positive CTCL, including mycosis fungoides (MF) and primary cutaneous anaplastic large-cell lymphoma (pcALCL).
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg IV every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive methotrexate at 5 mg to 50 mg PO weekly or bexarotene at a target dose of 300 mg/m² PO daily for up to 48 weeks.
Patients received BV for a median of 36 weeks (12 cycles), bexarotene for a median of 17 weeks, and methotrexate for a median of 9 weeks. Three patients in the BV arm were still on treatment at the time of analysis.
Patient characteristics
The median age was 62 (range, 22-83) in the BV am and 59 (range, 22-83) in the SOC arm. More than half of patients in each arm were male—52% and 58%, respectively. And most patients in both arms had an ECOG performance status of 0-1—95% and 97%, respectively.
The median number of prior therapies was 4 (range, 0-13) in the BV arm and 3.5 (range, 1-15) in the SOC arm. The median number of systemic therapies was 2 in the BV arm (range, 0-11) and the SOC arm (range, 1-8).
“It was pretty well balanced in terms of baseline characteristics between the 2 arms,” Dr Kim said. “The brentuximab arm had more stage IV patients—in fact, 7 stage IVB in brentuximab and none in the standard of care. And more patients with ALCL [treated with BV] had extracutaneous disease.”
Among pcALCL patients, 44% in the BV arm had extracutaneous disease, compared to 27% in the SOC arm. Among MF patients, 67% in the BV arm had stage IIB-IVB disease, compared to 61% in the SOC arm.
Response
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4).
“[ORR4] was felt to be more meaningful than ORR because it includes not only the response rate but also a duration element in a single endpoint,” Dr Kim said.
ORR4 was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Symptoms
“In CTCL, there’s significant quality of life issues that are not captured adequately by objective response measures, and this patient outcome is very important,” Dr Kim said. “[Quality of life in this study] was captured by Skindex-29, which is an established quality of life measure in skin diseases.”
Patients in the BV arm had a significantly higher reduction in symptom burden according to Skindex-29 than patients receiving SOC. The mean maximum reduction in Skindex-29 symptom domain was -27.96 points in the BV arm and -8.62 points in the SOC arm (P<0.0001).
PFS
PFS was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
Safety
The overall rate of adverse events (AEs) was 95% in the BV arm and 90% in the SOC arm. The rate of grade 3 or higher AEs was 41% and 47%, respectively. And the rate of serious AEs was 29% in both arms.
AEs resulting in discontinuation occurred in 24% of patients in the BV arm and 8% in the SOC arm. In the BV arm, this included peripheral neuropathy (n=9), skin-related hypersensitivity (n=3), E coli infection (n=1), impetigo (n=1), pulmonary embolism (n=1), urticaria (n=1), and vertigo (n=1).
In the SOC arm, AEs leading to discontinuation included maculo-papular rash (n=1), asthenia (n=1), hematuria (n=1), hypernatremia (n=1), neutropenia (n=1), periorbital infection (n=1), and somnolence (n=1). One patient in each arm experienced more than 1 AE resulting in discontinuation.
The most common AEs of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
The majority of the peripheral neuropathy events in the BV arm were grade 1 or 2—26% and 32%, respectively. The rate of grade 3 peripheral neuropathy events was 9%, and there were no grade 4 events.
Eighty-two percent of patients reported resolution or improvement in peripheral neuropathy events in the BV arm at a median of 22.9 months of follow-up.
There were no on-study deaths (occurring within 30 days of the last dose) in the SOC arm, but there were 4 in the BV arm. Three of the BV deaths were considered unrelated to the drug.
The 1 BV-related death was a result of multiple organ dysfunction syndrome attributed to tumor necrosis at visceral disease sites in a patient with T3bN0M1 pcALCL. The other 3 deaths were due to lymphoma progression, pulmonary embolism, and sepsis. ![]()
Photo by Larry Young
SAN FRANCISCO—The phase 3 ALCANZA trial is the first to convincingly demonstrate that a new systemic agent can be more effective than standard of care (SOC) options for cutaneous T-cell lymphoma (CTCL), according to a speaker at the 9th Annual T-cell Lymphoma Forum.
The trial showed significant improvements in response, symptom burden, and progression-free survival (PFS) in patients with CD30-expressing CTCL who received brentuximab vedotin (BV), as compared to patients who received either bexarotene or methotrexate.
“[These are] compelling results that potentially may have practice-changing implications for the use of brentuximab in managing CD30-expressing CTCL patients who require systemic therapy,” said Youn H. Kim, MD, of Stanford University School of Medicine in California.
Dr Kim presented these results at this year’s T-cell Lymphoma Forum. The data were also presented at the recent ASH Annual Meeting (abstract 182).
The ALCANZA trial was sponsored by Millennium Pharmaceuticals, Inc. (now a part of Takeda Pharmaceutical Company Limited) and Seattle Genetics, Inc.
The study was designed to compare BV to the SOC options of methotrexate or bexarotene in patients with CD30-positive CTCL, including mycosis fungoides (MF) and primary cutaneous anaplastic large-cell lymphoma (pcALCL).
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg IV every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive methotrexate at 5 mg to 50 mg PO weekly or bexarotene at a target dose of 300 mg/m² PO daily for up to 48 weeks.
Patients received BV for a median of 36 weeks (12 cycles), bexarotene for a median of 17 weeks, and methotrexate for a median of 9 weeks. Three patients in the BV arm were still on treatment at the time of analysis.
Patient characteristics
The median age was 62 (range, 22-83) in the BV am and 59 (range, 22-83) in the SOC arm. More than half of patients in each arm were male—52% and 58%, respectively. And most patients in both arms had an ECOG performance status of 0-1—95% and 97%, respectively.
The median number of prior therapies was 4 (range, 0-13) in the BV arm and 3.5 (range, 1-15) in the SOC arm. The median number of systemic therapies was 2 in the BV arm (range, 0-11) and the SOC arm (range, 1-8).
“It was pretty well balanced in terms of baseline characteristics between the 2 arms,” Dr Kim said. “The brentuximab arm had more stage IV patients—in fact, 7 stage IVB in brentuximab and none in the standard of care. And more patients with ALCL [treated with BV] had extracutaneous disease.”
Among pcALCL patients, 44% in the BV arm had extracutaneous disease, compared to 27% in the SOC arm. Among MF patients, 67% in the BV arm had stage IIB-IVB disease, compared to 61% in the SOC arm.
Response
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4).
“[ORR4] was felt to be more meaningful than ORR because it includes not only the response rate but also a duration element in a single endpoint,” Dr Kim said.
ORR4 was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Symptoms
“In CTCL, there’s significant quality of life issues that are not captured adequately by objective response measures, and this patient outcome is very important,” Dr Kim said. “[Quality of life in this study] was captured by Skindex-29, which is an established quality of life measure in skin diseases.”
Patients in the BV arm had a significantly higher reduction in symptom burden according to Skindex-29 than patients receiving SOC. The mean maximum reduction in Skindex-29 symptom domain was -27.96 points in the BV arm and -8.62 points in the SOC arm (P<0.0001).
PFS
PFS was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
Safety
The overall rate of adverse events (AEs) was 95% in the BV arm and 90% in the SOC arm. The rate of grade 3 or higher AEs was 41% and 47%, respectively. And the rate of serious AEs was 29% in both arms.
AEs resulting in discontinuation occurred in 24% of patients in the BV arm and 8% in the SOC arm. In the BV arm, this included peripheral neuropathy (n=9), skin-related hypersensitivity (n=3), E coli infection (n=1), impetigo (n=1), pulmonary embolism (n=1), urticaria (n=1), and vertigo (n=1).
In the SOC arm, AEs leading to discontinuation included maculo-papular rash (n=1), asthenia (n=1), hematuria (n=1), hypernatremia (n=1), neutropenia (n=1), periorbital infection (n=1), and somnolence (n=1). One patient in each arm experienced more than 1 AE resulting in discontinuation.
The most common AEs of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
The majority of the peripheral neuropathy events in the BV arm were grade 1 or 2—26% and 32%, respectively. The rate of grade 3 peripheral neuropathy events was 9%, and there were no grade 4 events.
Eighty-two percent of patients reported resolution or improvement in peripheral neuropathy events in the BV arm at a median of 22.9 months of follow-up.
There were no on-study deaths (occurring within 30 days of the last dose) in the SOC arm, but there were 4 in the BV arm. Three of the BV deaths were considered unrelated to the drug.
The 1 BV-related death was a result of multiple organ dysfunction syndrome attributed to tumor necrosis at visceral disease sites in a patient with T3bN0M1 pcALCL. The other 3 deaths were due to lymphoma progression, pulmonary embolism, and sepsis. ![]()
Photo by Larry Young
SAN FRANCISCO—The phase 3 ALCANZA trial is the first to convincingly demonstrate that a new systemic agent can be more effective than standard of care (SOC) options for cutaneous T-cell lymphoma (CTCL), according to a speaker at the 9th Annual T-cell Lymphoma Forum.
The trial showed significant improvements in response, symptom burden, and progression-free survival (PFS) in patients with CD30-expressing CTCL who received brentuximab vedotin (BV), as compared to patients who received either bexarotene or methotrexate.
“[These are] compelling results that potentially may have practice-changing implications for the use of brentuximab in managing CD30-expressing CTCL patients who require systemic therapy,” said Youn H. Kim, MD, of Stanford University School of Medicine in California.
Dr Kim presented these results at this year’s T-cell Lymphoma Forum. The data were also presented at the recent ASH Annual Meeting (abstract 182).
The ALCANZA trial was sponsored by Millennium Pharmaceuticals, Inc. (now a part of Takeda Pharmaceutical Company Limited) and Seattle Genetics, Inc.
The study was designed to compare BV to the SOC options of methotrexate or bexarotene in patients with CD30-positive CTCL, including mycosis fungoides (MF) and primary cutaneous anaplastic large-cell lymphoma (pcALCL).
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg IV every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive methotrexate at 5 mg to 50 mg PO weekly or bexarotene at a target dose of 300 mg/m² PO daily for up to 48 weeks.
Patients received BV for a median of 36 weeks (12 cycles), bexarotene for a median of 17 weeks, and methotrexate for a median of 9 weeks. Three patients in the BV arm were still on treatment at the time of analysis.
Patient characteristics
The median age was 62 (range, 22-83) in the BV am and 59 (range, 22-83) in the SOC arm. More than half of patients in each arm were male—52% and 58%, respectively. And most patients in both arms had an ECOG performance status of 0-1—95% and 97%, respectively.
The median number of prior therapies was 4 (range, 0-13) in the BV arm and 3.5 (range, 1-15) in the SOC arm. The median number of systemic therapies was 2 in the BV arm (range, 0-11) and the SOC arm (range, 1-8).
“It was pretty well balanced in terms of baseline characteristics between the 2 arms,” Dr Kim said. “The brentuximab arm had more stage IV patients—in fact, 7 stage IVB in brentuximab and none in the standard of care. And more patients with ALCL [treated with BV] had extracutaneous disease.”
Among pcALCL patients, 44% in the BV arm had extracutaneous disease, compared to 27% in the SOC arm. Among MF patients, 67% in the BV arm had stage IIB-IVB disease, compared to 61% in the SOC arm.
Response
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4).
“[ORR4] was felt to be more meaningful than ORR because it includes not only the response rate but also a duration element in a single endpoint,” Dr Kim said.
ORR4 was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Symptoms
“In CTCL, there’s significant quality of life issues that are not captured adequately by objective response measures, and this patient outcome is very important,” Dr Kim said. “[Quality of life in this study] was captured by Skindex-29, which is an established quality of life measure in skin diseases.”
Patients in the BV arm had a significantly higher reduction in symptom burden according to Skindex-29 than patients receiving SOC. The mean maximum reduction in Skindex-29 symptom domain was -27.96 points in the BV arm and -8.62 points in the SOC arm (P<0.0001).
PFS
PFS was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
Safety
The overall rate of adverse events (AEs) was 95% in the BV arm and 90% in the SOC arm. The rate of grade 3 or higher AEs was 41% and 47%, respectively. And the rate of serious AEs was 29% in both arms.
AEs resulting in discontinuation occurred in 24% of patients in the BV arm and 8% in the SOC arm. In the BV arm, this included peripheral neuropathy (n=9), skin-related hypersensitivity (n=3), E coli infection (n=1), impetigo (n=1), pulmonary embolism (n=1), urticaria (n=1), and vertigo (n=1).
In the SOC arm, AEs leading to discontinuation included maculo-papular rash (n=1), asthenia (n=1), hematuria (n=1), hypernatremia (n=1), neutropenia (n=1), periorbital infection (n=1), and somnolence (n=1). One patient in each arm experienced more than 1 AE resulting in discontinuation.
The most common AEs of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
The majority of the peripheral neuropathy events in the BV arm were grade 1 or 2—26% and 32%, respectively. The rate of grade 3 peripheral neuropathy events was 9%, and there were no grade 4 events.
Eighty-two percent of patients reported resolution or improvement in peripheral neuropathy events in the BV arm at a median of 22.9 months of follow-up.
There were no on-study deaths (occurring within 30 days of the last dose) in the SOC arm, but there were 4 in the BV arm. Three of the BV deaths were considered unrelated to the drug.
The 1 BV-related death was a result of multiple organ dysfunction syndrome attributed to tumor necrosis at visceral disease sites in a patient with T3bN0M1 pcALCL. The other 3 deaths were due to lymphoma progression, pulmonary embolism, and sepsis. ![]()
CHMP recommends hybrid drug for ALL, other disorders

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for an oral formulation of methotrexate (Jylamvo) as a treatment for acute lymphoblastic leukemia (ALL) and other disorders.
Jylamvo is a hybrid medicine of Methotrexat “Lederle” 25 mg-Stechampulle and Methotrexate “Lederle” 2.5 mg tablets, which have been authorized in the European Union since 1984 and 1959, respectively.
Jylamvo contains the same active substance as these reference medicines—the antineoplastic and immunomodulating agent methotrexate—but is given by mouth as a solution (2 mg/mL).
Jylamvo is intended for use as maintenance treatment in ALL patients age 3 and older.
The drug is also intended to treat:
- Active rheumatoid arthritis in adults
- Polyarthritic forms of active, severe juvenile idiopathic arthritis in adolescents and children age 3 and older when the response to non-steroidal anti-inflammatory drugs has been inadequate
- Severe, treatment-refractory, disabling psoriasis that does not respond sufficiently to other forms of treatment (such as phototherapy, retinoids, and psoralen and ultraviolet A radiation therapy) and severe psoriatic arthritis in adults.
The applicant for Jylamvo is Therakind Limited. Applications for hybrid medicines rely, in part, on the results of preclinical tests and clinical trials for a reference product and, in part, on new data.
The CHMP said studies have demonstrated the satisfactory quality of Jylamvo and its bioequivalence to Methotrexate “Lederle” 2.5 mg tablets and a third product, Ebetrexat 10 mg tablets, which is authorized for similar indications.
The CHMP has proposed that Jylamvo be prescribed by physicians with experience of the various properties of the medicinal product and its mode of action.
Detailed recommendations for the use of Jylamvo will be described in the summary of product characteristics, which will be published in the European public assessment report and made available in all official European Union languages if the European Commission grants marketing authorization for Jylamvo.
The European Commission typically makes a decision on a product within 67 days of the time the CHMP adopts its opinion. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for an oral formulation of methotrexate (Jylamvo) as a treatment for acute lymphoblastic leukemia (ALL) and other disorders.
Jylamvo is a hybrid medicine of Methotrexat “Lederle” 25 mg-Stechampulle and Methotrexate “Lederle” 2.5 mg tablets, which have been authorized in the European Union since 1984 and 1959, respectively.
Jylamvo contains the same active substance as these reference medicines—the antineoplastic and immunomodulating agent methotrexate—but is given by mouth as a solution (2 mg/mL).
Jylamvo is intended for use as maintenance treatment in ALL patients age 3 and older.
The drug is also intended to treat:
- Active rheumatoid arthritis in adults
- Polyarthritic forms of active, severe juvenile idiopathic arthritis in adolescents and children age 3 and older when the response to non-steroidal anti-inflammatory drugs has been inadequate
- Severe, treatment-refractory, disabling psoriasis that does not respond sufficiently to other forms of treatment (such as phototherapy, retinoids, and psoralen and ultraviolet A radiation therapy) and severe psoriatic arthritis in adults.
The applicant for Jylamvo is Therakind Limited. Applications for hybrid medicines rely, in part, on the results of preclinical tests and clinical trials for a reference product and, in part, on new data.
The CHMP said studies have demonstrated the satisfactory quality of Jylamvo and its bioequivalence to Methotrexate “Lederle” 2.5 mg tablets and a third product, Ebetrexat 10 mg tablets, which is authorized for similar indications.
The CHMP has proposed that Jylamvo be prescribed by physicians with experience of the various properties of the medicinal product and its mode of action.
Detailed recommendations for the use of Jylamvo will be described in the summary of product characteristics, which will be published in the European public assessment report and made available in all official European Union languages if the European Commission grants marketing authorization for Jylamvo.
The European Commission typically makes a decision on a product within 67 days of the time the CHMP adopts its opinion. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for an oral formulation of methotrexate (Jylamvo) as a treatment for acute lymphoblastic leukemia (ALL) and other disorders.
Jylamvo is a hybrid medicine of Methotrexat “Lederle” 25 mg-Stechampulle and Methotrexate “Lederle” 2.5 mg tablets, which have been authorized in the European Union since 1984 and 1959, respectively.
Jylamvo contains the same active substance as these reference medicines—the antineoplastic and immunomodulating agent methotrexate—but is given by mouth as a solution (2 mg/mL).
Jylamvo is intended for use as maintenance treatment in ALL patients age 3 and older.
The drug is also intended to treat:
- Active rheumatoid arthritis in adults
- Polyarthritic forms of active, severe juvenile idiopathic arthritis in adolescents and children age 3 and older when the response to non-steroidal anti-inflammatory drugs has been inadequate
- Severe, treatment-refractory, disabling psoriasis that does not respond sufficiently to other forms of treatment (such as phototherapy, retinoids, and psoralen and ultraviolet A radiation therapy) and severe psoriatic arthritis in adults.
The applicant for Jylamvo is Therakind Limited. Applications for hybrid medicines rely, in part, on the results of preclinical tests and clinical trials for a reference product and, in part, on new data.
The CHMP said studies have demonstrated the satisfactory quality of Jylamvo and its bioequivalence to Methotrexate “Lederle” 2.5 mg tablets and a third product, Ebetrexat 10 mg tablets, which is authorized for similar indications.
The CHMP has proposed that Jylamvo be prescribed by physicians with experience of the various properties of the medicinal product and its mode of action.
Detailed recommendations for the use of Jylamvo will be described in the summary of product characteristics, which will be published in the European public assessment report and made available in all official European Union languages if the European Commission grants marketing authorization for Jylamvo.
The European Commission typically makes a decision on a product within 67 days of the time the CHMP adopts its opinion. ![]()
Targeting disease stem cells in AML, MDS
Image by Robert Paulson
The cell surface molecule CD99 occurs more frequently than normal on stem cells responsible for acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS), according to research published in Science Translational Medicine.
Building on this discovery, researchers designed anti-CD99 monoclonal antibodies (mAbs).
In vitro and in vivo experiments
showed that these mAbs can recognize
and destroy AML and MDS stem/progenitor cells.
“Our findings not only identify a new molecule expressed on stem cells that drive these human malignancies, but we show that antibodies against this target can directly kill human AML stem cells,” said study author Christopher Y. Park, MD, PhD, of NYU Langone Medical Center in New York, New York.
“While we still have important details to work out, CD99 is likely to be an exploitable therapeutic target for most AML and MDS patients, and we are working urgently to finalize a therapy for human testing.”
Dr Park and his colleagues first examined stem cell populations from 79 patients with AML and 24 with MDS. More than 80% of stem cells in both groups expressed high levels of CD99.
The levels were so high that leukemia stem cells could be cleanly separated from normal hematopoietic stem cells in AML samples.
Upon confirming that CD99 was abundant on AML and MDS stem cells, the researchers made several anti-CD99 mAbs and tested them in vitro and in mouse models.
The mAbs destroyed AML and MDS stem cells by causing a sudden spike in the activity of SRC family kinases—a group of proteins that are implicated in invasion, tumor progression, and metastasis in a variety of cancers.
However, the mAbs had minimal effects on normal hematopoietic stem cells.
“With the appropriate support, we believe we can rapidly determine the best antibodies for use in patients, produce them at the quality needed to verify our results, and apply for permission to begin clinical trials,” Dr Park said. ![]()
Image by Robert Paulson
The cell surface molecule CD99 occurs more frequently than normal on stem cells responsible for acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS), according to research published in Science Translational Medicine.
Building on this discovery, researchers designed anti-CD99 monoclonal antibodies (mAbs).
In vitro and in vivo experiments
showed that these mAbs can recognize
and destroy AML and MDS stem/progenitor cells.
“Our findings not only identify a new molecule expressed on stem cells that drive these human malignancies, but we show that antibodies against this target can directly kill human AML stem cells,” said study author Christopher Y. Park, MD, PhD, of NYU Langone Medical Center in New York, New York.
“While we still have important details to work out, CD99 is likely to be an exploitable therapeutic target for most AML and MDS patients, and we are working urgently to finalize a therapy for human testing.”
Dr Park and his colleagues first examined stem cell populations from 79 patients with AML and 24 with MDS. More than 80% of stem cells in both groups expressed high levels of CD99.
The levels were so high that leukemia stem cells could be cleanly separated from normal hematopoietic stem cells in AML samples.
Upon confirming that CD99 was abundant on AML and MDS stem cells, the researchers made several anti-CD99 mAbs and tested them in vitro and in mouse models.
The mAbs destroyed AML and MDS stem cells by causing a sudden spike in the activity of SRC family kinases—a group of proteins that are implicated in invasion, tumor progression, and metastasis in a variety of cancers.
However, the mAbs had minimal effects on normal hematopoietic stem cells.
“With the appropriate support, we believe we can rapidly determine the best antibodies for use in patients, produce them at the quality needed to verify our results, and apply for permission to begin clinical trials,” Dr Park said. ![]()
Image by Robert Paulson
The cell surface molecule CD99 occurs more frequently than normal on stem cells responsible for acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS), according to research published in Science Translational Medicine.
Building on this discovery, researchers designed anti-CD99 monoclonal antibodies (mAbs).
In vitro and in vivo experiments
showed that these mAbs can recognize
and destroy AML and MDS stem/progenitor cells.
“Our findings not only identify a new molecule expressed on stem cells that drive these human malignancies, but we show that antibodies against this target can directly kill human AML stem cells,” said study author Christopher Y. Park, MD, PhD, of NYU Langone Medical Center in New York, New York.
“While we still have important details to work out, CD99 is likely to be an exploitable therapeutic target for most AML and MDS patients, and we are working urgently to finalize a therapy for human testing.”
Dr Park and his colleagues first examined stem cell populations from 79 patients with AML and 24 with MDS. More than 80% of stem cells in both groups expressed high levels of CD99.
The levels were so high that leukemia stem cells could be cleanly separated from normal hematopoietic stem cells in AML samples.
Upon confirming that CD99 was abundant on AML and MDS stem cells, the researchers made several anti-CD99 mAbs and tested them in vitro and in mouse models.
The mAbs destroyed AML and MDS stem cells by causing a sudden spike in the activity of SRC family kinases—a group of proteins that are implicated in invasion, tumor progression, and metastasis in a variety of cancers.
However, the mAbs had minimal effects on normal hematopoietic stem cells.
“With the appropriate support, we believe we can rapidly determine the best antibodies for use in patients, produce them at the quality needed to verify our results, and apply for permission to begin clinical trials,” Dr Park said. ![]()
Gene therapy granted breakthrough designation to treat hemophilia B

Image by Spencer Phillips
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the investigational gene therapy AMT-060 as a treatment for patients with severe hemophilia B.
AMT-060 consists of a codon-optimized wild-type factor IX (FIX) gene cassette, the LP1 liver promoter, and an AAV5 viral vector manufactured by uniQure using its proprietary insect cell-based technology platform. uniQure is the company developing AMT-060.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Phase 1/2 trial
The breakthrough designation for AMT-060 is based on results from an ongoing phase 1/2 study. Updated data from this study were recently presented at the 2016 ASH Annual Meeting (abstract 2314).
In this trial, researchers are testing AMT-060 in 10 patients. All patients had severe or moderately severe hemophilia at baseline, including documented FIX levels less than 1% to 2% of normal, and required chronic infusions of prophylactic or on-demand FIX therapy at the time of enrollment.
Each patient received a 1-time, 30-minute, intravenous dose of AMT-060, without the use of corticosteroids. Five patients received AMT-060 at 5 x 1012 gc/kg, and 5 received AMT-060 at 2 x 1013 gc/kg.
The data presented at ASH included up to 52 weeks of follow-up from the low-dose cohort and up to 31 weeks of follow-up from the higher-dose cohort.
Data from the higher-dose cohort show a dose response with improvement in disease state in all 5 patients. Four patients who previously required prophylactic FIX replacement therapy were able to stop this therapy.
As of the data cutoff date for the ASH presentation, 1 unconfirmed spontaneous bleed had been reported during an aggregate of 94 weeks of follow-up after the discontinuation of prophylaxis.
Researchers previously reported that 4 patients in the low-dose cohort were able to discontinue prophylactic therapy. The 1 patient who
remained on prophylaxis sustained an improved disease
phenotype and also required materially less FIX concentrate after
treatment with AMT-060.
According to uniQure, all 5 patients in the low-dose cohort continue to maintain “constant and clinically meaningful” levels of FIX activity for up to 52 weeks post-treatment. In fact, there were no spontaneous bleeds in these patients in the last 14 weeks of observation.
uniQure also said AMT-060 continues to be well-tolerated, and there have been no severe adverse events.
Three patients (2 in the higher-dose cohort and 1 previously reported from the low-dose cohort) experienced mild, asymptomatic elevations of alanine aminotransferase and received a tapering course of corticosteroids per protocol.
These temporary alanine aminotransferase elevations were not associated with any loss of endogenous FIX activity or T-cell response to the AAV5 capsid.
None of the patients in either cohort have developed inhibitory antibodies against FIX, and none of the patients screened tested positive for anti-AAV5 antibodies. ![]()
Image by Spencer Phillips
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the investigational gene therapy AMT-060 as a treatment for patients with severe hemophilia B.
AMT-060 consists of a codon-optimized wild-type factor IX (FIX) gene cassette, the LP1 liver promoter, and an AAV5 viral vector manufactured by uniQure using its proprietary insect cell-based technology platform. uniQure is the company developing AMT-060.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Phase 1/2 trial
The breakthrough designation for AMT-060 is based on results from an ongoing phase 1/2 study. Updated data from this study were recently presented at the 2016 ASH Annual Meeting (abstract 2314).
In this trial, researchers are testing AMT-060 in 10 patients. All patients had severe or moderately severe hemophilia at baseline, including documented FIX levels less than 1% to 2% of normal, and required chronic infusions of prophylactic or on-demand FIX therapy at the time of enrollment.
Each patient received a 1-time, 30-minute, intravenous dose of AMT-060, without the use of corticosteroids. Five patients received AMT-060 at 5 x 1012 gc/kg, and 5 received AMT-060 at 2 x 1013 gc/kg.
The data presented at ASH included up to 52 weeks of follow-up from the low-dose cohort and up to 31 weeks of follow-up from the higher-dose cohort.
Data from the higher-dose cohort show a dose response with improvement in disease state in all 5 patients. Four patients who previously required prophylactic FIX replacement therapy were able to stop this therapy.
As of the data cutoff date for the ASH presentation, 1 unconfirmed spontaneous bleed had been reported during an aggregate of 94 weeks of follow-up after the discontinuation of prophylaxis.
Researchers previously reported that 4 patients in the low-dose cohort were able to discontinue prophylactic therapy. The 1 patient who
remained on prophylaxis sustained an improved disease
phenotype and also required materially less FIX concentrate after
treatment with AMT-060.
According to uniQure, all 5 patients in the low-dose cohort continue to maintain “constant and clinically meaningful” levels of FIX activity for up to 52 weeks post-treatment. In fact, there were no spontaneous bleeds in these patients in the last 14 weeks of observation.
uniQure also said AMT-060 continues to be well-tolerated, and there have been no severe adverse events.
Three patients (2 in the higher-dose cohort and 1 previously reported from the low-dose cohort) experienced mild, asymptomatic elevations of alanine aminotransferase and received a tapering course of corticosteroids per protocol.
These temporary alanine aminotransferase elevations were not associated with any loss of endogenous FIX activity or T-cell response to the AAV5 capsid.
None of the patients in either cohort have developed inhibitory antibodies against FIX, and none of the patients screened tested positive for anti-AAV5 antibodies. ![]()
Image by Spencer Phillips
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the investigational gene therapy AMT-060 as a treatment for patients with severe hemophilia B.
AMT-060 consists of a codon-optimized wild-type factor IX (FIX) gene cassette, the LP1 liver promoter, and an AAV5 viral vector manufactured by uniQure using its proprietary insect cell-based technology platform. uniQure is the company developing AMT-060.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Phase 1/2 trial
The breakthrough designation for AMT-060 is based on results from an ongoing phase 1/2 study. Updated data from this study were recently presented at the 2016 ASH Annual Meeting (abstract 2314).
In this trial, researchers are testing AMT-060 in 10 patients. All patients had severe or moderately severe hemophilia at baseline, including documented FIX levels less than 1% to 2% of normal, and required chronic infusions of prophylactic or on-demand FIX therapy at the time of enrollment.
Each patient received a 1-time, 30-minute, intravenous dose of AMT-060, without the use of corticosteroids. Five patients received AMT-060 at 5 x 1012 gc/kg, and 5 received AMT-060 at 2 x 1013 gc/kg.
The data presented at ASH included up to 52 weeks of follow-up from the low-dose cohort and up to 31 weeks of follow-up from the higher-dose cohort.
Data from the higher-dose cohort show a dose response with improvement in disease state in all 5 patients. Four patients who previously required prophylactic FIX replacement therapy were able to stop this therapy.
As of the data cutoff date for the ASH presentation, 1 unconfirmed spontaneous bleed had been reported during an aggregate of 94 weeks of follow-up after the discontinuation of prophylaxis.
Researchers previously reported that 4 patients in the low-dose cohort were able to discontinue prophylactic therapy. The 1 patient who
remained on prophylaxis sustained an improved disease
phenotype and also required materially less FIX concentrate after
treatment with AMT-060.
According to uniQure, all 5 patients in the low-dose cohort continue to maintain “constant and clinically meaningful” levels of FIX activity for up to 52 weeks post-treatment. In fact, there were no spontaneous bleeds in these patients in the last 14 weeks of observation.
uniQure also said AMT-060 continues to be well-tolerated, and there have been no severe adverse events.
Three patients (2 in the higher-dose cohort and 1 previously reported from the low-dose cohort) experienced mild, asymptomatic elevations of alanine aminotransferase and received a tapering course of corticosteroids per protocol.
These temporary alanine aminotransferase elevations were not associated with any loss of endogenous FIX activity or T-cell response to the AAV5 capsid.
None of the patients in either cohort have developed inhibitory antibodies against FIX, and none of the patients screened tested positive for anti-AAV5 antibodies. ![]()
New recommendations for managing adult AML

Photo courtesy of CDC
The European LeukemiaNet (ELN) has released updated recommendations for the diagnosis and treatment of acute myeloid leukemia (AML) in adults.
The recommendations include revised ELN genetic categories, a proposed response category based on minimal residual disease status, and a proposed category for progressive disease for clinical trials.
They also include the updated World Health Organization classification of myeloid neoplasms and acute leukemia.
The recommendations are published in Blood.
“These guidelines are an important update of the current and widely used recommendations for managing AML, for constructing clinical trials, and for predicting outcomes of AML patients,” said Clara D. Bloomfield, MD, of The Ohio State University Comprehensive Cancer Center in Columbus.
“They will be the new standard of care and will replace the 2010 ELN recommendations for managing AML patients and designing clinical trials.”
Dr Bloomfield said updating the ELN recommendations was prompted by new insights into the molecular and genomic causes of AML, by the development of new genetic tests and tests for detecting minimal residual disease, and by the development of novel anti-leukemic agents.
Changes of note, according to Dr Bloomfield, are that there are now 3 genetic risk categories rather than 4, and the FLT3-ITD mutation has been added as a marker of risk.
In addition, “complete remission with no evidence of minimal residual disease” is a new proposed response category. This criterion requires that genetic markers present at diagnosis are no longer detectable.
“It is no longer good enough to examine bone marrow samples and say the leukemia is gone,” Dr Bloomfield said. “We must also see the loss of genetic markers.”
Another change is that “progressive disease” is a new provisional response category to be used in clinical trials only. The purpose of the category is to harmonize the various definitions of progressive disease that are used in different clinical trials. ![]()
Photo courtesy of CDC
The European LeukemiaNet (ELN) has released updated recommendations for the diagnosis and treatment of acute myeloid leukemia (AML) in adults.
The recommendations include revised ELN genetic categories, a proposed response category based on minimal residual disease status, and a proposed category for progressive disease for clinical trials.
They also include the updated World Health Organization classification of myeloid neoplasms and acute leukemia.
The recommendations are published in Blood.
“These guidelines are an important update of the current and widely used recommendations for managing AML, for constructing clinical trials, and for predicting outcomes of AML patients,” said Clara D. Bloomfield, MD, of The Ohio State University Comprehensive Cancer Center in Columbus.
“They will be the new standard of care and will replace the 2010 ELN recommendations for managing AML patients and designing clinical trials.”
Dr Bloomfield said updating the ELN recommendations was prompted by new insights into the molecular and genomic causes of AML, by the development of new genetic tests and tests for detecting minimal residual disease, and by the development of novel anti-leukemic agents.
Changes of note, according to Dr Bloomfield, are that there are now 3 genetic risk categories rather than 4, and the FLT3-ITD mutation has been added as a marker of risk.
In addition, “complete remission with no evidence of minimal residual disease” is a new proposed response category. This criterion requires that genetic markers present at diagnosis are no longer detectable.
“It is no longer good enough to examine bone marrow samples and say the leukemia is gone,” Dr Bloomfield said. “We must also see the loss of genetic markers.”
Another change is that “progressive disease” is a new provisional response category to be used in clinical trials only. The purpose of the category is to harmonize the various definitions of progressive disease that are used in different clinical trials. ![]()
Photo courtesy of CDC
The European LeukemiaNet (ELN) has released updated recommendations for the diagnosis and treatment of acute myeloid leukemia (AML) in adults.
The recommendations include revised ELN genetic categories, a proposed response category based on minimal residual disease status, and a proposed category for progressive disease for clinical trials.
They also include the updated World Health Organization classification of myeloid neoplasms and acute leukemia.
The recommendations are published in Blood.
“These guidelines are an important update of the current and widely used recommendations for managing AML, for constructing clinical trials, and for predicting outcomes of AML patients,” said Clara D. Bloomfield, MD, of The Ohio State University Comprehensive Cancer Center in Columbus.
“They will be the new standard of care and will replace the 2010 ELN recommendations for managing AML patients and designing clinical trials.”
Dr Bloomfield said updating the ELN recommendations was prompted by new insights into the molecular and genomic causes of AML, by the development of new genetic tests and tests for detecting minimal residual disease, and by the development of novel anti-leukemic agents.
Changes of note, according to Dr Bloomfield, are that there are now 3 genetic risk categories rather than 4, and the FLT3-ITD mutation has been added as a marker of risk.
In addition, “complete remission with no evidence of minimal residual disease” is a new proposed response category. This criterion requires that genetic markers present at diagnosis are no longer detectable.
“It is no longer good enough to examine bone marrow samples and say the leukemia is gone,” Dr Bloomfield said. “We must also see the loss of genetic markers.”
Another change is that “progressive disease” is a new provisional response category to be used in clinical trials only. The purpose of the category is to harmonize the various definitions of progressive disease that are used in different clinical trials. ![]()
Study provides new insight into B-cell metabolism
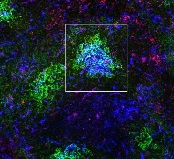
the spleen of a mouse, showing
inactivated GSK3 (magenta)
in B cells (blue) near follicular
dendritic cells (green).
Image from the lab of
Robert Rickert, PhD
Research published in Nature Immunology helps explain how B-cell metabolism adapts to different environments.
The study suggests the protein GSK3 acts as a metabolic checkpoint regulator in B cells, promoting the survival of circulating B cells while limiting the growth and proliferation of B cells in germinal centers.
“Our research shows that the protein GSK3 plays a crucial role in helping B cells meet the energy needs of their distinct states,” said study author Robert Rickert, PhD, of Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California.
“The findings are particularly relevant for certain B-cell pathologies, including lymphoma subtypes, where there is an increased demand for energy to support the hyperproliferation of cells in a microenvironment that may be limited in nutrients.”
Dr Rickert and his colleagues noted that B cells predominate in a quiescent state until they encounter an antigen, which prompts the cells to grow, proliferate, and differentiate.
The team’s new study showed that GSK3 adjusts B-cell metabolism to match the needs of these different cell states.
In circulating B cells, GSK3 limits overall metabolic activity. In proliferating B cells in germinal centers, GSK3 slows glycolysis and the production of mitochondria.
In fact, GSK3 function is essential for B-cell survival in germinal centers. To understand why, the researchers looked at how B cells in these regions generate energy.
The team found that because these B cells are so metabolically active, they consume nearly all available glucose. That switches on glycolysis.
High glycolytic activity leads to an accumulation of toxic reactive oxygen species, as does rapid manufacture of mitochondria, which tend to leak the same chemicals.
Thus, by restraining the metabolism in specific ways, GSK3 prevents cell death induced by reactive oxygen species.
“Our results were really surprising,” Dr Rickert said. “Until now, we would have thought that slowing metabolism would only be important for preventing B cells from becoming cancerous, which it indeed may be. These studies provide insight into the dynamic nature of B-cell metabolism that literally ‘fuels’ differentiation in the germinal center to produce an effective antibody response.”
“It’s not yet clear whether or how GSK3 might be a target for future therapies for B cell-related diseases, but this research opens a lot of doors for further studies. To start with, we plan to investigate how GSK3 is regulated in lymphoma and how that relates to changes in metabolism. That research could lead to new approaches to treating lymphoma.”
This research was performed in collaboration with scientists at Eli Lilly and the Lunenfeld-Tanenbaum Research Institute at the University of Toronto. Funding was provided by the National Institutes of Health, the Lilly Research Award Program, the Arthritis National Research Foundation, and the Canadian Institutes of Health Research. ![]()
the spleen of a mouse, showing
inactivated GSK3 (magenta)
in B cells (blue) near follicular
dendritic cells (green).
Image from the lab of
Robert Rickert, PhD
Research published in Nature Immunology helps explain how B-cell metabolism adapts to different environments.
The study suggests the protein GSK3 acts as a metabolic checkpoint regulator in B cells, promoting the survival of circulating B cells while limiting the growth and proliferation of B cells in germinal centers.
“Our research shows that the protein GSK3 plays a crucial role in helping B cells meet the energy needs of their distinct states,” said study author Robert Rickert, PhD, of Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California.
“The findings are particularly relevant for certain B-cell pathologies, including lymphoma subtypes, where there is an increased demand for energy to support the hyperproliferation of cells in a microenvironment that may be limited in nutrients.”
Dr Rickert and his colleagues noted that B cells predominate in a quiescent state until they encounter an antigen, which prompts the cells to grow, proliferate, and differentiate.
The team’s new study showed that GSK3 adjusts B-cell metabolism to match the needs of these different cell states.
In circulating B cells, GSK3 limits overall metabolic activity. In proliferating B cells in germinal centers, GSK3 slows glycolysis and the production of mitochondria.
In fact, GSK3 function is essential for B-cell survival in germinal centers. To understand why, the researchers looked at how B cells in these regions generate energy.
The team found that because these B cells are so metabolically active, they consume nearly all available glucose. That switches on glycolysis.
High glycolytic activity leads to an accumulation of toxic reactive oxygen species, as does rapid manufacture of mitochondria, which tend to leak the same chemicals.
Thus, by restraining the metabolism in specific ways, GSK3 prevents cell death induced by reactive oxygen species.
“Our results were really surprising,” Dr Rickert said. “Until now, we would have thought that slowing metabolism would only be important for preventing B cells from becoming cancerous, which it indeed may be. These studies provide insight into the dynamic nature of B-cell metabolism that literally ‘fuels’ differentiation in the germinal center to produce an effective antibody response.”
“It’s not yet clear whether or how GSK3 might be a target for future therapies for B cell-related diseases, but this research opens a lot of doors for further studies. To start with, we plan to investigate how GSK3 is regulated in lymphoma and how that relates to changes in metabolism. That research could lead to new approaches to treating lymphoma.”
This research was performed in collaboration with scientists at Eli Lilly and the Lunenfeld-Tanenbaum Research Institute at the University of Toronto. Funding was provided by the National Institutes of Health, the Lilly Research Award Program, the Arthritis National Research Foundation, and the Canadian Institutes of Health Research. ![]()
the spleen of a mouse, showing
inactivated GSK3 (magenta)
in B cells (blue) near follicular
dendritic cells (green).
Image from the lab of
Robert Rickert, PhD
Research published in Nature Immunology helps explain how B-cell metabolism adapts to different environments.
The study suggests the protein GSK3 acts as a metabolic checkpoint regulator in B cells, promoting the survival of circulating B cells while limiting the growth and proliferation of B cells in germinal centers.
“Our research shows that the protein GSK3 plays a crucial role in helping B cells meet the energy needs of their distinct states,” said study author Robert Rickert, PhD, of Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California.
“The findings are particularly relevant for certain B-cell pathologies, including lymphoma subtypes, where there is an increased demand for energy to support the hyperproliferation of cells in a microenvironment that may be limited in nutrients.”
Dr Rickert and his colleagues noted that B cells predominate in a quiescent state until they encounter an antigen, which prompts the cells to grow, proliferate, and differentiate.
The team’s new study showed that GSK3 adjusts B-cell metabolism to match the needs of these different cell states.
In circulating B cells, GSK3 limits overall metabolic activity. In proliferating B cells in germinal centers, GSK3 slows glycolysis and the production of mitochondria.
In fact, GSK3 function is essential for B-cell survival in germinal centers. To understand why, the researchers looked at how B cells in these regions generate energy.
The team found that because these B cells are so metabolically active, they consume nearly all available glucose. That switches on glycolysis.
High glycolytic activity leads to an accumulation of toxic reactive oxygen species, as does rapid manufacture of mitochondria, which tend to leak the same chemicals.
Thus, by restraining the metabolism in specific ways, GSK3 prevents cell death induced by reactive oxygen species.
“Our results were really surprising,” Dr Rickert said. “Until now, we would have thought that slowing metabolism would only be important for preventing B cells from becoming cancerous, which it indeed may be. These studies provide insight into the dynamic nature of B-cell metabolism that literally ‘fuels’ differentiation in the germinal center to produce an effective antibody response.”
“It’s not yet clear whether or how GSK3 might be a target for future therapies for B cell-related diseases, but this research opens a lot of doors for further studies. To start with, we plan to investigate how GSK3 is regulated in lymphoma and how that relates to changes in metabolism. That research could lead to new approaches to treating lymphoma.”
This research was performed in collaboration with scientists at Eli Lilly and the Lunenfeld-Tanenbaum Research Institute at the University of Toronto. Funding was provided by the National Institutes of Health, the Lilly Research Award Program, the Arthritis National Research Foundation, and the Canadian Institutes of Health Research. ![]()
CHMP recommends lenalidomide maintenance

Photo courtesy of Celgene
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has recommended a new indication for lenalidomide (Revlimid®).
The CHMP advised the European Commission (EC) to approve the use of lenalidomide as maintenance therapy in adults who had newly diagnosed multiple myeloma (MM) prior to receiving an autologous stem cell transplant (ASCT).
If approved by the EC, lenalidomide will be the first licensed maintenance treatment available to this patient population in the European Union.
The EC, which generally follows the CHMP’s recommendations, is expected to make its final decision on this use of lenalidomide in approximately 2 months.
If approval is granted, detailed conditions for the use of lenalidomide will be described in the Summary of Product Characteristics, which will be published in the revised European Public Assessment Report.
Lenalidomide is a product of Celgene.
The CHMP’s recommendation to approve lenalidomide as maintenance in MM was based on the results of 2 cooperative group-led studies, CALGB 10010410 and IFM 2005-0211. Results from both studies were published in NEJM in May 2012.
CALGB 100104 was a phase 3, double-blind study of 460 patients with newly diagnosed MM undergoing ASCT. The patients received continuous daily treatment with lenalidomide or placebo until relapse.
IFM 2005-02 was a phase 3, double-blind study of 614 patients newly diagnosed with MM. The patients were randomized to receive a 2-month consolidation regimen post-ASCT of lenalidomide monotherapy, followed by continuous daily treatment with lenalidomide or placebo until relapse.
“Studies show that maintenance treatment after ASCT with Revlimid may help control residual malignant cells and delay tumor growth by enhancing immune function,” said Michel Attal, MD, of the Institut Universitaire du Cancer Toulouse Oncopole and Institut Claudius Regaud in France.
“Our primary goal is to delay disease progression for as long as possible, and we have seen in several independent studies that Revlimid maintenance after ASCT can halve the risk of disease progression by sustaining the response.” ![]()
Photo courtesy of Celgene
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has recommended a new indication for lenalidomide (Revlimid®).
The CHMP advised the European Commission (EC) to approve the use of lenalidomide as maintenance therapy in adults who had newly diagnosed multiple myeloma (MM) prior to receiving an autologous stem cell transplant (ASCT).
If approved by the EC, lenalidomide will be the first licensed maintenance treatment available to this patient population in the European Union.
The EC, which generally follows the CHMP’s recommendations, is expected to make its final decision on this use of lenalidomide in approximately 2 months.
If approval is granted, detailed conditions for the use of lenalidomide will be described in the Summary of Product Characteristics, which will be published in the revised European Public Assessment Report.
Lenalidomide is a product of Celgene.
The CHMP’s recommendation to approve lenalidomide as maintenance in MM was based on the results of 2 cooperative group-led studies, CALGB 10010410 and IFM 2005-0211. Results from both studies were published in NEJM in May 2012.
CALGB 100104 was a phase 3, double-blind study of 460 patients with newly diagnosed MM undergoing ASCT. The patients received continuous daily treatment with lenalidomide or placebo until relapse.
IFM 2005-02 was a phase 3, double-blind study of 614 patients newly diagnosed with MM. The patients were randomized to receive a 2-month consolidation regimen post-ASCT of lenalidomide monotherapy, followed by continuous daily treatment with lenalidomide or placebo until relapse.
“Studies show that maintenance treatment after ASCT with Revlimid may help control residual malignant cells and delay tumor growth by enhancing immune function,” said Michel Attal, MD, of the Institut Universitaire du Cancer Toulouse Oncopole and Institut Claudius Regaud in France.
“Our primary goal is to delay disease progression for as long as possible, and we have seen in several independent studies that Revlimid maintenance after ASCT can halve the risk of disease progression by sustaining the response.” ![]()
Photo courtesy of Celgene
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has recommended a new indication for lenalidomide (Revlimid®).
The CHMP advised the European Commission (EC) to approve the use of lenalidomide as maintenance therapy in adults who had newly diagnosed multiple myeloma (MM) prior to receiving an autologous stem cell transplant (ASCT).
If approved by the EC, lenalidomide will be the first licensed maintenance treatment available to this patient population in the European Union.
The EC, which generally follows the CHMP’s recommendations, is expected to make its final decision on this use of lenalidomide in approximately 2 months.
If approval is granted, detailed conditions for the use of lenalidomide will be described in the Summary of Product Characteristics, which will be published in the revised European Public Assessment Report.
Lenalidomide is a product of Celgene.
The CHMP’s recommendation to approve lenalidomide as maintenance in MM was based on the results of 2 cooperative group-led studies, CALGB 10010410 and IFM 2005-0211. Results from both studies were published in NEJM in May 2012.
CALGB 100104 was a phase 3, double-blind study of 460 patients with newly diagnosed MM undergoing ASCT. The patients received continuous daily treatment with lenalidomide or placebo until relapse.
IFM 2005-02 was a phase 3, double-blind study of 614 patients newly diagnosed with MM. The patients were randomized to receive a 2-month consolidation regimen post-ASCT of lenalidomide monotherapy, followed by continuous daily treatment with lenalidomide or placebo until relapse.
“Studies show that maintenance treatment after ASCT with Revlimid may help control residual malignant cells and delay tumor growth by enhancing immune function,” said Michel Attal, MD, of the Institut Universitaire du Cancer Toulouse Oncopole and Institut Claudius Regaud in France.
“Our primary goal is to delay disease progression for as long as possible, and we have seen in several independent studies that Revlimid maintenance after ASCT can halve the risk of disease progression by sustaining the response.”
Software could improve image analysis, team says
Researchers say they have developed new software that will analyze medical and scientific images faster and more accurately than ever before.
The team says this software, Tracking Equilibrium and Nonequilibrium shifts in Data (TREND), can analyze any series of images, including nuclear magnetic resonance images, computerized tomography scans, ultrasound images, video images, and imaging from scientific equipment of all kinds.
The researchers described the TREND software in Biophysical Journal.
The team said TREND can study sets of images to resolve and track the changes among the images.
And the software can analyze videos to plot and resolve changes as well as reconstruct videos to focus only on the individual processes and changes of interest.
“TREND allows accurate, rapid analysis of incredibly complex and nuanced images, which can potentially save doctors, patients, and scientists countless hours and money,” said Steve Van Doren, PhD, of the University of Missouri in Columbia, Missouri.
“TREND has allowed us to advance our own research into enzyme interactions considerably. Previously, it would take us weeks to analyze a single group of images. With TREND, that analysis now takes only a few minutes and is more accurate and consistent than if a human performed the work.”
Researchers say they have developed new software that will analyze medical and scientific images faster and more accurately than ever before.
The team says this software, Tracking Equilibrium and Nonequilibrium shifts in Data (TREND), can analyze any series of images, including nuclear magnetic resonance images, computerized tomography scans, ultrasound images, video images, and imaging from scientific equipment of all kinds.
The researchers described the TREND software in Biophysical Journal.
The team said TREND can study sets of images to resolve and track the changes among the images.
And the software can analyze videos to plot and resolve changes as well as reconstruct videos to focus only on the individual processes and changes of interest.
“TREND allows accurate, rapid analysis of incredibly complex and nuanced images, which can potentially save doctors, patients, and scientists countless hours and money,” said Steve Van Doren, PhD, of the University of Missouri in Columbia, Missouri.
“TREND has allowed us to advance our own research into enzyme interactions considerably. Previously, it would take us weeks to analyze a single group of images. With TREND, that analysis now takes only a few minutes and is more accurate and consistent than if a human performed the work.”
Researchers say they have developed new software that will analyze medical and scientific images faster and more accurately than ever before.
The team says this software, Tracking Equilibrium and Nonequilibrium shifts in Data (TREND), can analyze any series of images, including nuclear magnetic resonance images, computerized tomography scans, ultrasound images, video images, and imaging from scientific equipment of all kinds.
The researchers described the TREND software in Biophysical Journal.
The team said TREND can study sets of images to resolve and track the changes among the images.
And the software can analyze videos to plot and resolve changes as well as reconstruct videos to focus only on the individual processes and changes of interest.
“TREND allows accurate, rapid analysis of incredibly complex and nuanced images, which can potentially save doctors, patients, and scientists countless hours and money,” said Steve Van Doren, PhD, of the University of Missouri in Columbia, Missouri.
“TREND has allowed us to advance our own research into enzyme interactions considerably. Previously, it would take us weeks to analyze a single group of images. With TREND, that analysis now takes only a few minutes and is more accurate and consistent than if a human performed the work.”
Combo granted orphan designation for DLBCL

The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab), an anti-CD20 monoclonal antibody, and TGR-1202, a PI3K delta inhibitor, in the treatment of diffuse large B-cell lymphoma (DLBCL).
The combination is currently being evaluated in patients with relapsed or refractory DLBCL in the phase 2b UNITY-DLBCL trial.
Ublituximab and TGR-1202 are both products of TG Therapeutics, Inc.
Updated results from a phase 1 study of ublituximab and TGR-1202 in patients with DLBCL and other malignancies were presented at the 21st Congress of the European Hematology Association.
The data included 165 patients treated with varying doses of TGR-1202 alone (n=90) or in combination with ublituximab (n=75). The patients were heavily pretreated, with the majority having 3 or more prior lines of therapy.
There were 7 evaluable patients with DLBCL who received the combination at the phase 3 doses— ublituximab at 900 mg and TGR-1202 at 800 mg micronized.
The overall response rate for this group was 57%. Of the 4 responders, 1 patient had a complete response, and 3 had a partial response. Two patients had stable disease, and 1 progressed.
In the overall study population, the most common adverse events were diarrhea (47%), nausea (45%), fatigue (37%), vomiting (27%), and neutropenia (21%). The most common grade 3/4 adverse events were neutropenia (18%) and anemia (5%).
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to
treat, diagnose, or prevent rare diseases/disorders affecting fewer than
200,000 people in the US.
Orphan designation provides companies
with certain incentives to develop products for rare diseases. This
includes a 50% tax break on research and development, a fee waiver,
access to federal grants, and 7 years of market exclusivity if the
product is approved.
The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab), an anti-CD20 monoclonal antibody, and TGR-1202, a PI3K delta inhibitor, in the treatment of diffuse large B-cell lymphoma (DLBCL).
The combination is currently being evaluated in patients with relapsed or refractory DLBCL in the phase 2b UNITY-DLBCL trial.
Ublituximab and TGR-1202 are both products of TG Therapeutics, Inc.
Updated results from a phase 1 study of ublituximab and TGR-1202 in patients with DLBCL and other malignancies were presented at the 21st Congress of the European Hematology Association.
The data included 165 patients treated with varying doses of TGR-1202 alone (n=90) or in combination with ublituximab (n=75). The patients were heavily pretreated, with the majority having 3 or more prior lines of therapy.
There were 7 evaluable patients with DLBCL who received the combination at the phase 3 doses— ublituximab at 900 mg and TGR-1202 at 800 mg micronized.
The overall response rate for this group was 57%. Of the 4 responders, 1 patient had a complete response, and 3 had a partial response. Two patients had stable disease, and 1 progressed.
In the overall study population, the most common adverse events were diarrhea (47%), nausea (45%), fatigue (37%), vomiting (27%), and neutropenia (21%). The most common grade 3/4 adverse events were neutropenia (18%) and anemia (5%).
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to
treat, diagnose, or prevent rare diseases/disorders affecting fewer than
200,000 people in the US.
Orphan designation provides companies
with certain incentives to develop products for rare diseases. This
includes a 50% tax break on research and development, a fee waiver,
access to federal grants, and 7 years of market exclusivity if the
product is approved.
The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab), an anti-CD20 monoclonal antibody, and TGR-1202, a PI3K delta inhibitor, in the treatment of diffuse large B-cell lymphoma (DLBCL).
The combination is currently being evaluated in patients with relapsed or refractory DLBCL in the phase 2b UNITY-DLBCL trial.
Ublituximab and TGR-1202 are both products of TG Therapeutics, Inc.
Updated results from a phase 1 study of ublituximab and TGR-1202 in patients with DLBCL and other malignancies were presented at the 21st Congress of the European Hematology Association.
The data included 165 patients treated with varying doses of TGR-1202 alone (n=90) or in combination with ublituximab (n=75). The patients were heavily pretreated, with the majority having 3 or more prior lines of therapy.
There were 7 evaluable patients with DLBCL who received the combination at the phase 3 doses— ublituximab at 900 mg and TGR-1202 at 800 mg micronized.
The overall response rate for this group was 57%. Of the 4 responders, 1 patient had a complete response, and 3 had a partial response. Two patients had stable disease, and 1 progressed.
In the overall study population, the most common adverse events were diarrhea (47%), nausea (45%), fatigue (37%), vomiting (27%), and neutropenia (21%). The most common grade 3/4 adverse events were neutropenia (18%) and anemia (5%).
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to
treat, diagnose, or prevent rare diseases/disorders affecting fewer than
200,000 people in the US.
Orphan designation provides companies
with certain incentives to develop products for rare diseases. This
includes a 50% tax break on research and development, a fee waiver,
access to federal grants, and 7 years of market exclusivity if the
product is approved.
Distress linked to higher risk of death from leukemia, other cancers

patient and her father
Photo by Rhoda Baer
A study published in The BMJ suggests that higher levels of psychological distress (anxiety and depression) may be associated with an increased risk of death from leukemia and other cancers.
The findings are observational, so no firm conclusions about cause and effect can be drawn.
However, the researchers said the findings add to the growing evidence that psychological distress could have some predictive capacity for certain physical conditions.
There is some evidence that psychological distress (anxiety and depression) is related to increased rates of cardiovascular disease, but links with different types of cancer are either unclear or untested.
So David Batty, PhD, of University College London in the UK, and his colleagues set out to examine if psychological distress is a potential predictor of site-specific cancer mortality.
The researchers analyzed data from 16 studies (13 from England and 3 from Scotland), which started between 1994 and 2008. The data included 163,363 men and women age 16 or over who were free from cancer at the start of the study.
Psychological distress scores were measured using the general health questionnaire (GHQ-12), and participants were monitored for an average of 9.5 years. During this time, there were 4353 deaths from cancer.
Several factors that could have influenced the results were taken into account, including age, sex, education, socioeconomic status, body mass index, smoking, and alcohol intake.
“After statistical control for these factors, the results show that, compared with people in the least distressed group, death rates in the most distressed group were consistently higher for cancer of the bowel, prostate, pancreas, and esophagus and for leukemia,” Dr Batty said.
He and his colleagues pointed out that this association may also be affected by reverse causality, where undiagnosed (early) cancer might have had an underlying impact on mood.
In a bid to correct for this, the team conducted a further analysis excluding study participants who died in the first 5 years of follow-up, but this made no difference to the findings. The links between distress and cancer remained.
Specifically, compared to people in the least distressed group (GHQ-12 score 0-6), death rates in the most distressed group (score 7-12) were consistently increased for cancer of all sites combined, with a multivariable adjusted hazard ratio (HR) of 1.32.
Death rates were also increased for colorectal (HR=1.84), prostate (HR=2.42), pancreatic (HR=2.76), and esophageal cancers (HR=2.59), as well as for leukemia (HR=3.86).
“Our findings contribute to the evidence that poor mental health might have some predictive capacity for certain physical diseases,” Dr Batty said, “but we are a long way off from knowing if these relationships are truly causal.”
patient and her father
Photo by Rhoda Baer
A study published in The BMJ suggests that higher levels of psychological distress (anxiety and depression) may be associated with an increased risk of death from leukemia and other cancers.
The findings are observational, so no firm conclusions about cause and effect can be drawn.
However, the researchers said the findings add to the growing evidence that psychological distress could have some predictive capacity for certain physical conditions.
There is some evidence that psychological distress (anxiety and depression) is related to increased rates of cardiovascular disease, but links with different types of cancer are either unclear or untested.
So David Batty, PhD, of University College London in the UK, and his colleagues set out to examine if psychological distress is a potential predictor of site-specific cancer mortality.
The researchers analyzed data from 16 studies (13 from England and 3 from Scotland), which started between 1994 and 2008. The data included 163,363 men and women age 16 or over who were free from cancer at the start of the study.
Psychological distress scores were measured using the general health questionnaire (GHQ-12), and participants were monitored for an average of 9.5 years. During this time, there were 4353 deaths from cancer.
Several factors that could have influenced the results were taken into account, including age, sex, education, socioeconomic status, body mass index, smoking, and alcohol intake.
“After statistical control for these factors, the results show that, compared with people in the least distressed group, death rates in the most distressed group were consistently higher for cancer of the bowel, prostate, pancreas, and esophagus and for leukemia,” Dr Batty said.
He and his colleagues pointed out that this association may also be affected by reverse causality, where undiagnosed (early) cancer might have had an underlying impact on mood.
In a bid to correct for this, the team conducted a further analysis excluding study participants who died in the first 5 years of follow-up, but this made no difference to the findings. The links between distress and cancer remained.
Specifically, compared to people in the least distressed group (GHQ-12 score 0-6), death rates in the most distressed group (score 7-12) were consistently increased for cancer of all sites combined, with a multivariable adjusted hazard ratio (HR) of 1.32.
Death rates were also increased for colorectal (HR=1.84), prostate (HR=2.42), pancreatic (HR=2.76), and esophageal cancers (HR=2.59), as well as for leukemia (HR=3.86).
“Our findings contribute to the evidence that poor mental health might have some predictive capacity for certain physical diseases,” Dr Batty said, “but we are a long way off from knowing if these relationships are truly causal.”
patient and her father
Photo by Rhoda Baer
A study published in The BMJ suggests that higher levels of psychological distress (anxiety and depression) may be associated with an increased risk of death from leukemia and other cancers.
The findings are observational, so no firm conclusions about cause and effect can be drawn.
However, the researchers said the findings add to the growing evidence that psychological distress could have some predictive capacity for certain physical conditions.
There is some evidence that psychological distress (anxiety and depression) is related to increased rates of cardiovascular disease, but links with different types of cancer are either unclear or untested.
So David Batty, PhD, of University College London in the UK, and his colleagues set out to examine if psychological distress is a potential predictor of site-specific cancer mortality.
The researchers analyzed data from 16 studies (13 from England and 3 from Scotland), which started between 1994 and 2008. The data included 163,363 men and women age 16 or over who were free from cancer at the start of the study.
Psychological distress scores were measured using the general health questionnaire (GHQ-12), and participants were monitored for an average of 9.5 years. During this time, there were 4353 deaths from cancer.
Several factors that could have influenced the results were taken into account, including age, sex, education, socioeconomic status, body mass index, smoking, and alcohol intake.
“After statistical control for these factors, the results show that, compared with people in the least distressed group, death rates in the most distressed group were consistently higher for cancer of the bowel, prostate, pancreas, and esophagus and for leukemia,” Dr Batty said.
He and his colleagues pointed out that this association may also be affected by reverse causality, where undiagnosed (early) cancer might have had an underlying impact on mood.
In a bid to correct for this, the team conducted a further analysis excluding study participants who died in the first 5 years of follow-up, but this made no difference to the findings. The links between distress and cancer remained.
Specifically, compared to people in the least distressed group (GHQ-12 score 0-6), death rates in the most distressed group (score 7-12) were consistently increased for cancer of all sites combined, with a multivariable adjusted hazard ratio (HR) of 1.32.
Death rates were also increased for colorectal (HR=1.84), prostate (HR=2.42), pancreatic (HR=2.76), and esophageal cancers (HR=2.59), as well as for leukemia (HR=3.86).
“Our findings contribute to the evidence that poor mental health might have some predictive capacity for certain physical diseases,” Dr Batty said, “but we are a long way off from knowing if these relationships are truly causal.”