User login
Overcoming glucocorticoid resistance in lymphoma

Image by Ed Uthman
Targeting RUNX1 could combat glucocorticoid resistance in patients with lymphoma, according to research published in the Journal of Cellular Biochemistry.
Researchers found an over activity of RUNX1 in lymphoma cells interfered with sphingolipids and caused cells to become resistant to dexamethasone.
Dexamethasone works, in part, through the control of sphingolipid enzymes, which play a role in instructing cells to live or die.
Specifically, the researchers said they found that ectopic expression of RUNX1 in lymphoma cells consistently perturbs the sphingolipid rheostat and confers increased resistance to glucocorticoid-mediated apoptosis.
The team also described the mechanism of cross-talk between glucocorticoid and sphingolipid metabolism through the enzyme Sgpp1.
The researchers said dexamethasone induces expression of Sgpp1 in T-lymphoma cells and drives cell death, which is reduced by partial knockdown of Sgpp1 with short hairpin RNA or direct transcriptional repression of Sgpp1 by ectopic RUNX1.
These findings suggest that drugs targeting RUNX1 may be able to reverse glucocorticoid resistance in lymphoma patients.
“The possibility of making existing therapies more active and specific by combining [them] with drugs that inhibit RUNX is a new and exciting prospect,” said study author James Neil, of The University of Glasgow in Scotland.
“Our collaborators in the US have recently developed drugs that inhibit RUNX, and we plan to test these with existing therapies in blood cancers where MYC and RUNX are both implicated, including multiple myeloma and Burkitt lymphoma.”
An earlier study by Dr Neil and his colleagues suggested that RUNX1 was a potential therapeutic target in MYC-driven lymphomas. ![]()
Image by Ed Uthman
Targeting RUNX1 could combat glucocorticoid resistance in patients with lymphoma, according to research published in the Journal of Cellular Biochemistry.
Researchers found an over activity of RUNX1 in lymphoma cells interfered with sphingolipids and caused cells to become resistant to dexamethasone.
Dexamethasone works, in part, through the control of sphingolipid enzymes, which play a role in instructing cells to live or die.
Specifically, the researchers said they found that ectopic expression of RUNX1 in lymphoma cells consistently perturbs the sphingolipid rheostat and confers increased resistance to glucocorticoid-mediated apoptosis.
The team also described the mechanism of cross-talk between glucocorticoid and sphingolipid metabolism through the enzyme Sgpp1.
The researchers said dexamethasone induces expression of Sgpp1 in T-lymphoma cells and drives cell death, which is reduced by partial knockdown of Sgpp1 with short hairpin RNA or direct transcriptional repression of Sgpp1 by ectopic RUNX1.
These findings suggest that drugs targeting RUNX1 may be able to reverse glucocorticoid resistance in lymphoma patients.
“The possibility of making existing therapies more active and specific by combining [them] with drugs that inhibit RUNX is a new and exciting prospect,” said study author James Neil, of The University of Glasgow in Scotland.
“Our collaborators in the US have recently developed drugs that inhibit RUNX, and we plan to test these with existing therapies in blood cancers where MYC and RUNX are both implicated, including multiple myeloma and Burkitt lymphoma.”
An earlier study by Dr Neil and his colleagues suggested that RUNX1 was a potential therapeutic target in MYC-driven lymphomas. ![]()
Image by Ed Uthman
Targeting RUNX1 could combat glucocorticoid resistance in patients with lymphoma, according to research published in the Journal of Cellular Biochemistry.
Researchers found an over activity of RUNX1 in lymphoma cells interfered with sphingolipids and caused cells to become resistant to dexamethasone.
Dexamethasone works, in part, through the control of sphingolipid enzymes, which play a role in instructing cells to live or die.
Specifically, the researchers said they found that ectopic expression of RUNX1 in lymphoma cells consistently perturbs the sphingolipid rheostat and confers increased resistance to glucocorticoid-mediated apoptosis.
The team also described the mechanism of cross-talk between glucocorticoid and sphingolipid metabolism through the enzyme Sgpp1.
The researchers said dexamethasone induces expression of Sgpp1 in T-lymphoma cells and drives cell death, which is reduced by partial knockdown of Sgpp1 with short hairpin RNA or direct transcriptional repression of Sgpp1 by ectopic RUNX1.
These findings suggest that drugs targeting RUNX1 may be able to reverse glucocorticoid resistance in lymphoma patients.
“The possibility of making existing therapies more active and specific by combining [them] with drugs that inhibit RUNX is a new and exciting prospect,” said study author James Neil, of The University of Glasgow in Scotland.
“Our collaborators in the US have recently developed drugs that inhibit RUNX, and we plan to test these with existing therapies in blood cancers where MYC and RUNX are both implicated, including multiple myeloma and Burkitt lymphoma.”
An earlier study by Dr Neil and his colleagues suggested that RUNX1 was a potential therapeutic target in MYC-driven lymphomas. ![]()
Reducing the risk of device-related thrombosis

“superomniphobic” surface.
Photo from the Kota lab
at Colorado State University
Researchers say they have engineered “superhemophobic” titanium surfaces that could be used to create implantable medical devices that don’t pose a risk of thrombosis.
The team described this work in Advanced Healthcare Materials.
The undesirable interaction of blood with foreign materials is an ongoing problem in medical research, said study author Ketul Popat, PhD, of Colorado State University in Fort Collins, Colorado.
He and his colleagues noted that, when implanted medical devices come in contact with blood, platelet adhesion and activation occur, which may lead to thrombosis and device failure.
“If we can design materials where blood barely contacts the surface, there is virtually no chance of clotting, which is a coordinated set of events,” Dr Popat said. “Here, we’re targeting the prevention of the first set of events.”
Dr Popat and his colleagues started with sheets of titanium, which are commonly used in medical devices. The team then grew chemically altered surfaces that act as barriers between the titanium and blood.
They analyzed variations of titanium surfaces, including different textures and chemistries, and compared the extent of platelet adhesion and activation.
These experiments revealed that fluorinated nanotubes offered the best protection against clotting.
Having implantable medical devices that repel blood might seem counterintuitive, the researchers noted, as biomedical scientists often use materials with an affinity to blood to make them biologically compatible.
“What we are doing is the exact opposite,” said study author Arun Kota, PhD, of Colorado State University.
“We are taking a material that blood hates to come in contact with, in order to make it compatible with blood.”
In essence, the titanium surface is so repellent that blood is “tricked” into “believing” there’s virtually no foreign material there at all.
Growing a surface and testing it in the lab is only the beginning, the researchers said. They want to continue examining other clotting factors, and eventually, to test real medical devices. ![]()
“superomniphobic” surface.
Photo from the Kota lab
at Colorado State University
Researchers say they have engineered “superhemophobic” titanium surfaces that could be used to create implantable medical devices that don’t pose a risk of thrombosis.
The team described this work in Advanced Healthcare Materials.
The undesirable interaction of blood with foreign materials is an ongoing problem in medical research, said study author Ketul Popat, PhD, of Colorado State University in Fort Collins, Colorado.
He and his colleagues noted that, when implanted medical devices come in contact with blood, platelet adhesion and activation occur, which may lead to thrombosis and device failure.
“If we can design materials where blood barely contacts the surface, there is virtually no chance of clotting, which is a coordinated set of events,” Dr Popat said. “Here, we’re targeting the prevention of the first set of events.”
Dr Popat and his colleagues started with sheets of titanium, which are commonly used in medical devices. The team then grew chemically altered surfaces that act as barriers between the titanium and blood.
They analyzed variations of titanium surfaces, including different textures and chemistries, and compared the extent of platelet adhesion and activation.
These experiments revealed that fluorinated nanotubes offered the best protection against clotting.
Having implantable medical devices that repel blood might seem counterintuitive, the researchers noted, as biomedical scientists often use materials with an affinity to blood to make them biologically compatible.
“What we are doing is the exact opposite,” said study author Arun Kota, PhD, of Colorado State University.
“We are taking a material that blood hates to come in contact with, in order to make it compatible with blood.”
In essence, the titanium surface is so repellent that blood is “tricked” into “believing” there’s virtually no foreign material there at all.
Growing a surface and testing it in the lab is only the beginning, the researchers said. They want to continue examining other clotting factors, and eventually, to test real medical devices. ![]()
“superomniphobic” surface.
Photo from the Kota lab
at Colorado State University
Researchers say they have engineered “superhemophobic” titanium surfaces that could be used to create implantable medical devices that don’t pose a risk of thrombosis.
The team described this work in Advanced Healthcare Materials.
The undesirable interaction of blood with foreign materials is an ongoing problem in medical research, said study author Ketul Popat, PhD, of Colorado State University in Fort Collins, Colorado.
He and his colleagues noted that, when implanted medical devices come in contact with blood, platelet adhesion and activation occur, which may lead to thrombosis and device failure.
“If we can design materials where blood barely contacts the surface, there is virtually no chance of clotting, which is a coordinated set of events,” Dr Popat said. “Here, we’re targeting the prevention of the first set of events.”
Dr Popat and his colleagues started with sheets of titanium, which are commonly used in medical devices. The team then grew chemically altered surfaces that act as barriers between the titanium and blood.
They analyzed variations of titanium surfaces, including different textures and chemistries, and compared the extent of platelet adhesion and activation.
These experiments revealed that fluorinated nanotubes offered the best protection against clotting.
Having implantable medical devices that repel blood might seem counterintuitive, the researchers noted, as biomedical scientists often use materials with an affinity to blood to make them biologically compatible.
“What we are doing is the exact opposite,” said study author Arun Kota, PhD, of Colorado State University.
“We are taking a material that blood hates to come in contact with, in order to make it compatible with blood.”
In essence, the titanium surface is so repellent that blood is “tricked” into “believing” there’s virtually no foreign material there at all.
Growing a surface and testing it in the lab is only the beginning, the researchers said. They want to continue examining other clotting factors, and eventually, to test real medical devices. ![]()
Vaccine candidate can protect humans from malaria
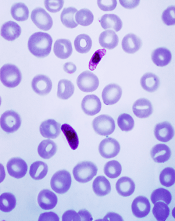
Plasmodium falciparum
Photo by Mae Melvin, CDC
In a phase 2 trial, a malaria vaccine candidate was able to prevent volunteers from contracting Plasmodium falciparum malaria.
The vaccine, known as PfSPZ vaccine, is composed of live but weakened P falciparum sporozoites.
Three to 5 doses of PfSPZ vaccine protected subjects against malaria parasites similar to those in the vaccine as well as parasites different from those in the vaccine.
A majority of subjects were protected at 3 weeks after vaccination. For some subjects, this protection was sustained at 24 weeks.
PfSPZ vaccine was considered well-tolerated, as all adverse events (AEs) in this trial were grade 1 or 2.
These results were published in JCI Insight.
The study was funded by the US Department of Defense through the Joint Warfighter Program, the Military Infectious Disease Research Program, and US Navy Advanced Medical Development, with additional support from Sanaria, Inc., the company developing PfSPZ vaccine.
“Our military continues to be at risk from malaria as it deploys worldwide,” said Kenneth A. Bertram, MD, of the US Army Medical Research and Materiel Command in Ft Detrick, Maryland.
“We are excited about the results of this clinical trial and are now investing in the ongoing clinical trial to finalize the vaccination regimen for PfSPZ vaccine.”
The current trial included 67 volunteers with a median age of 29.6 (range, 19 to 45).
They were randomized to 3 treatment groups. Forty-five subjects were set to receive the PfSPZ vaccine, and 22 subjects served as controls.
The volunteers underwent controlled human malaria infection (CHMI) 3 weeks after vaccinated subjects received their final vaccine dose and then again at 24 weeks.
Efficacy at 3 weeks
Group 1
In this group, 13 subjects received 5 doses of the vaccine at 2.7 × 105. They and 6 control subjects underwent CHMI with a homologous strain of P falciparum, Pf3D7.
Twelve of the 13 fully immunized subjects, or 92.3%, did not develop parasitemia. However, all 6 control subjects did, with a median prepatent period of 11.6 days. The prepatent period in the immunized subject who developed parasitemia was 13.9 days.
Group 2
In this group, 5 subjects received 5 doses of the vaccine at 2.7 × 105. They and 4 control subjects underwent CHMI with a heterologous strain of P falciparum, Pf7G8.
Four of the 5 fully immunized subjects, or 80%, did not develop parasitemia. However, all 4 control subjects did, with a median prepatent period of 11.9 days. The prepatent period in the fully immunized subject who developed parasitemia was 11.9 days.
Group 3
In this group, 15 subjects received 3 doses of the vaccine at 4.5 × 105 and underwent homologous Pf3D7 CHMI. The control subjects for this group were the same as those in group 1.
Thirteen of the 15 immunized subjects, or 86.7%, did not develop parasitemia. The prepatent periods in the 2 immunized subjects who did develop parasitemia were 13.9 days and 16.9 days.
Efficacy at 24 weeks
Study subjects underwent a second CHMI at 24 weeks after immunized participants received their final dose of vaccine.
Group 1
Seven of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (70%). However, all 6 control subjects did, with a median prepatent period of 11.6 days. The median prepatent period in the 3 fully immunized subjects who developed parasitemia was 15.4 days.
Group 2
One of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (10%). However, all control subjects did, with a median prepatent period of 10.9 days. The median prepatent period in the 9 fully immunized subjects who developed parasitemia was 11.9 days.
Group 3
Eight of the 14 fully immunized subjects who underwent a second CHMI did not develop parasitemia (57.1%). The median prepatent period in the 6 fully immunized subjects who did develop parasitemia was 14.0 days.
Safety
There were 66 solicited AEs reported within 7 days of immunization that were considered possibly, probably, or definitely related to vaccination. Ninety-two percent of these AEs were grade 1, and 8% were grade 2. All unsolicited AEs reported within 7 days of immunization were grade 1.
The incidence of AEs was not higher in group 3 than in groups 1 or 2, and the incidence of AEs did not increase as subjects received additional doses of the vaccine.
The most common AEs (with an incidence of 10% or higher in at least 1 group) were injection site pain, headache, fatigue, malaise, myalgia, injection site hemorrhage, and cough.
“The results of this clinical trial, along with recent results from other trials of this vaccine in the US and Africa, were critical to our decision to move forward with a trial involving 400 infants in Kenya,” said Tina Oneko, MD, of the Kenya Medical Research Institute, who is the principal investigator of the Kenya trial but was not involved in the current trial.
“This represents significant progress toward the development of a regimen for PfSPZ vaccine that we anticipate will provide a high level [of] efficacy for malaria prevention in all age groups in Africa.” ![]()
Plasmodium falciparum
Photo by Mae Melvin, CDC
In a phase 2 trial, a malaria vaccine candidate was able to prevent volunteers from contracting Plasmodium falciparum malaria.
The vaccine, known as PfSPZ vaccine, is composed of live but weakened P falciparum sporozoites.
Three to 5 doses of PfSPZ vaccine protected subjects against malaria parasites similar to those in the vaccine as well as parasites different from those in the vaccine.
A majority of subjects were protected at 3 weeks after vaccination. For some subjects, this protection was sustained at 24 weeks.
PfSPZ vaccine was considered well-tolerated, as all adverse events (AEs) in this trial were grade 1 or 2.
These results were published in JCI Insight.
The study was funded by the US Department of Defense through the Joint Warfighter Program, the Military Infectious Disease Research Program, and US Navy Advanced Medical Development, with additional support from Sanaria, Inc., the company developing PfSPZ vaccine.
“Our military continues to be at risk from malaria as it deploys worldwide,” said Kenneth A. Bertram, MD, of the US Army Medical Research and Materiel Command in Ft Detrick, Maryland.
“We are excited about the results of this clinical trial and are now investing in the ongoing clinical trial to finalize the vaccination regimen for PfSPZ vaccine.”
The current trial included 67 volunteers with a median age of 29.6 (range, 19 to 45).
They were randomized to 3 treatment groups. Forty-five subjects were set to receive the PfSPZ vaccine, and 22 subjects served as controls.
The volunteers underwent controlled human malaria infection (CHMI) 3 weeks after vaccinated subjects received their final vaccine dose and then again at 24 weeks.
Efficacy at 3 weeks
Group 1
In this group, 13 subjects received 5 doses of the vaccine at 2.7 × 105. They and 6 control subjects underwent CHMI with a homologous strain of P falciparum, Pf3D7.
Twelve of the 13 fully immunized subjects, or 92.3%, did not develop parasitemia. However, all 6 control subjects did, with a median prepatent period of 11.6 days. The prepatent period in the immunized subject who developed parasitemia was 13.9 days.
Group 2
In this group, 5 subjects received 5 doses of the vaccine at 2.7 × 105. They and 4 control subjects underwent CHMI with a heterologous strain of P falciparum, Pf7G8.
Four of the 5 fully immunized subjects, or 80%, did not develop parasitemia. However, all 4 control subjects did, with a median prepatent period of 11.9 days. The prepatent period in the fully immunized subject who developed parasitemia was 11.9 days.
Group 3
In this group, 15 subjects received 3 doses of the vaccine at 4.5 × 105 and underwent homologous Pf3D7 CHMI. The control subjects for this group were the same as those in group 1.
Thirteen of the 15 immunized subjects, or 86.7%, did not develop parasitemia. The prepatent periods in the 2 immunized subjects who did develop parasitemia were 13.9 days and 16.9 days.
Efficacy at 24 weeks
Study subjects underwent a second CHMI at 24 weeks after immunized participants received their final dose of vaccine.
Group 1
Seven of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (70%). However, all 6 control subjects did, with a median prepatent period of 11.6 days. The median prepatent period in the 3 fully immunized subjects who developed parasitemia was 15.4 days.
Group 2
One of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (10%). However, all control subjects did, with a median prepatent period of 10.9 days. The median prepatent period in the 9 fully immunized subjects who developed parasitemia was 11.9 days.
Group 3
Eight of the 14 fully immunized subjects who underwent a second CHMI did not develop parasitemia (57.1%). The median prepatent period in the 6 fully immunized subjects who did develop parasitemia was 14.0 days.
Safety
There were 66 solicited AEs reported within 7 days of immunization that were considered possibly, probably, or definitely related to vaccination. Ninety-two percent of these AEs were grade 1, and 8% were grade 2. All unsolicited AEs reported within 7 days of immunization were grade 1.
The incidence of AEs was not higher in group 3 than in groups 1 or 2, and the incidence of AEs did not increase as subjects received additional doses of the vaccine.
The most common AEs (with an incidence of 10% or higher in at least 1 group) were injection site pain, headache, fatigue, malaise, myalgia, injection site hemorrhage, and cough.
“The results of this clinical trial, along with recent results from other trials of this vaccine in the US and Africa, were critical to our decision to move forward with a trial involving 400 infants in Kenya,” said Tina Oneko, MD, of the Kenya Medical Research Institute, who is the principal investigator of the Kenya trial but was not involved in the current trial.
“This represents significant progress toward the development of a regimen for PfSPZ vaccine that we anticipate will provide a high level [of] efficacy for malaria prevention in all age groups in Africa.” ![]()
Plasmodium falciparum
Photo by Mae Melvin, CDC
In a phase 2 trial, a malaria vaccine candidate was able to prevent volunteers from contracting Plasmodium falciparum malaria.
The vaccine, known as PfSPZ vaccine, is composed of live but weakened P falciparum sporozoites.
Three to 5 doses of PfSPZ vaccine protected subjects against malaria parasites similar to those in the vaccine as well as parasites different from those in the vaccine.
A majority of subjects were protected at 3 weeks after vaccination. For some subjects, this protection was sustained at 24 weeks.
PfSPZ vaccine was considered well-tolerated, as all adverse events (AEs) in this trial were grade 1 or 2.
These results were published in JCI Insight.
The study was funded by the US Department of Defense through the Joint Warfighter Program, the Military Infectious Disease Research Program, and US Navy Advanced Medical Development, with additional support from Sanaria, Inc., the company developing PfSPZ vaccine.
“Our military continues to be at risk from malaria as it deploys worldwide,” said Kenneth A. Bertram, MD, of the US Army Medical Research and Materiel Command in Ft Detrick, Maryland.
“We are excited about the results of this clinical trial and are now investing in the ongoing clinical trial to finalize the vaccination regimen for PfSPZ vaccine.”
The current trial included 67 volunteers with a median age of 29.6 (range, 19 to 45).
They were randomized to 3 treatment groups. Forty-five subjects were set to receive the PfSPZ vaccine, and 22 subjects served as controls.
The volunteers underwent controlled human malaria infection (CHMI) 3 weeks after vaccinated subjects received their final vaccine dose and then again at 24 weeks.
Efficacy at 3 weeks
Group 1
In this group, 13 subjects received 5 doses of the vaccine at 2.7 × 105. They and 6 control subjects underwent CHMI with a homologous strain of P falciparum, Pf3D7.
Twelve of the 13 fully immunized subjects, or 92.3%, did not develop parasitemia. However, all 6 control subjects did, with a median prepatent period of 11.6 days. The prepatent period in the immunized subject who developed parasitemia was 13.9 days.
Group 2
In this group, 5 subjects received 5 doses of the vaccine at 2.7 × 105. They and 4 control subjects underwent CHMI with a heterologous strain of P falciparum, Pf7G8.
Four of the 5 fully immunized subjects, or 80%, did not develop parasitemia. However, all 4 control subjects did, with a median prepatent period of 11.9 days. The prepatent period in the fully immunized subject who developed parasitemia was 11.9 days.
Group 3
In this group, 15 subjects received 3 doses of the vaccine at 4.5 × 105 and underwent homologous Pf3D7 CHMI. The control subjects for this group were the same as those in group 1.
Thirteen of the 15 immunized subjects, or 86.7%, did not develop parasitemia. The prepatent periods in the 2 immunized subjects who did develop parasitemia were 13.9 days and 16.9 days.
Efficacy at 24 weeks
Study subjects underwent a second CHMI at 24 weeks after immunized participants received their final dose of vaccine.
Group 1
Seven of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (70%). However, all 6 control subjects did, with a median prepatent period of 11.6 days. The median prepatent period in the 3 fully immunized subjects who developed parasitemia was 15.4 days.
Group 2
One of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (10%). However, all control subjects did, with a median prepatent period of 10.9 days. The median prepatent period in the 9 fully immunized subjects who developed parasitemia was 11.9 days.
Group 3
Eight of the 14 fully immunized subjects who underwent a second CHMI did not develop parasitemia (57.1%). The median prepatent period in the 6 fully immunized subjects who did develop parasitemia was 14.0 days.
Safety
There were 66 solicited AEs reported within 7 days of immunization that were considered possibly, probably, or definitely related to vaccination. Ninety-two percent of these AEs were grade 1, and 8% were grade 2. All unsolicited AEs reported within 7 days of immunization were grade 1.
The incidence of AEs was not higher in group 3 than in groups 1 or 2, and the incidence of AEs did not increase as subjects received additional doses of the vaccine.
The most common AEs (with an incidence of 10% or higher in at least 1 group) were injection site pain, headache, fatigue, malaise, myalgia, injection site hemorrhage, and cough.
“The results of this clinical trial, along with recent results from other trials of this vaccine in the US and Africa, were critical to our decision to move forward with a trial involving 400 infants in Kenya,” said Tina Oneko, MD, of the Kenya Medical Research Institute, who is the principal investigator of the Kenya trial but was not involved in the current trial.
“This represents significant progress toward the development of a regimen for PfSPZ vaccine that we anticipate will provide a high level [of] efficacy for malaria prevention in all age groups in Africa.” ![]()
Meta-analysis supports statins as VTE prophylaxis

Photo courtesy of the CDC
Results of a meta-analysis support the idea that statins can reduce the risk of venous thromboembolism (VTE).
Researchers analyzed data from 36 studies involving more than 3.2 million people and found an “extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE.”
The team believes these findings could potentially lead to new guidelines and an expansion of the use of statins.
“These findings underscore a potential beneficial role of statin therapy on VTE in addition to its established role in cardiovascular disease prevention,” said Kamlesh Khunti, MD, PhD, of the University of Leicester in the UK.
Dr Khunti and his colleagues reported these findings in The Lancet Haematology.
The researchers analyzed data from 36 studies. This included 13 cohort studies with a total of 3,148,259 subjects and 23 randomized, controlled trials (RCTs) of 118,464 subjects in which statins were compared to placebo or no treatment.
In the observational studies, statin use was associated with a significant reduction in the risk of VTE, when compared to no statin use. The pooled relative risk (RR) was 0.75 (P<0.0001).
Statin use was associated with a significant reduction in the risk of deep vein thrombosis (DVT), with an RR of 0.77 (P<0.0001). (There were only 2 observational studies looking specifically at DVT outcomes.)
However, there was no significant reduction in the risk of pulmonary embolism (PE) with statin use. The RR was 1.02 (P=0.90). (There was only 1 observational study reporting specifically on PE outcomes.)
In the RCTs, statin use was associated with a significant reduction in the risk of VTE, with an RR of 0.85 (P=0.038).
Statin use was also associated with a significant reduction in DVT risk, with an RR of 0.45, but not PE risk, with an RR of 0.77 (no P values available). (There was only 1 RCT that reported specifically on DVT and PE outcomes.)
The RCTs also showed significant differences in VTE risk according to type of statin used. Patients receiving rosuvastatin had the lowest risk of VTE when compared to those receiving other statins, with an RR of 0.57 (P=0.015).
“Currently, statins are only approved for lipid lowering in the primary and secondary prevention of cardiovascular disease,” said study author Setor Kunutsor, MBChB, PhD, of the University of Bristol in the UK.
“But they have shown great promise beyond their established lipid-lowering effects, and these include potential beneficial impact on multiple disease conditions. These results provide an extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE and may support a true protective effect.” ![]()
Photo courtesy of the CDC
Results of a meta-analysis support the idea that statins can reduce the risk of venous thromboembolism (VTE).
Researchers analyzed data from 36 studies involving more than 3.2 million people and found an “extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE.”
The team believes these findings could potentially lead to new guidelines and an expansion of the use of statins.
“These findings underscore a potential beneficial role of statin therapy on VTE in addition to its established role in cardiovascular disease prevention,” said Kamlesh Khunti, MD, PhD, of the University of Leicester in the UK.
Dr Khunti and his colleagues reported these findings in The Lancet Haematology.
The researchers analyzed data from 36 studies. This included 13 cohort studies with a total of 3,148,259 subjects and 23 randomized, controlled trials (RCTs) of 118,464 subjects in which statins were compared to placebo or no treatment.
In the observational studies, statin use was associated with a significant reduction in the risk of VTE, when compared to no statin use. The pooled relative risk (RR) was 0.75 (P<0.0001).
Statin use was associated with a significant reduction in the risk of deep vein thrombosis (DVT), with an RR of 0.77 (P<0.0001). (There were only 2 observational studies looking specifically at DVT outcomes.)
However, there was no significant reduction in the risk of pulmonary embolism (PE) with statin use. The RR was 1.02 (P=0.90). (There was only 1 observational study reporting specifically on PE outcomes.)
In the RCTs, statin use was associated with a significant reduction in the risk of VTE, with an RR of 0.85 (P=0.038).
Statin use was also associated with a significant reduction in DVT risk, with an RR of 0.45, but not PE risk, with an RR of 0.77 (no P values available). (There was only 1 RCT that reported specifically on DVT and PE outcomes.)
The RCTs also showed significant differences in VTE risk according to type of statin used. Patients receiving rosuvastatin had the lowest risk of VTE when compared to those receiving other statins, with an RR of 0.57 (P=0.015).
“Currently, statins are only approved for lipid lowering in the primary and secondary prevention of cardiovascular disease,” said study author Setor Kunutsor, MBChB, PhD, of the University of Bristol in the UK.
“But they have shown great promise beyond their established lipid-lowering effects, and these include potential beneficial impact on multiple disease conditions. These results provide an extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE and may support a true protective effect.” ![]()
Photo courtesy of the CDC
Results of a meta-analysis support the idea that statins can reduce the risk of venous thromboembolism (VTE).
Researchers analyzed data from 36 studies involving more than 3.2 million people and found an “extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE.”
The team believes these findings could potentially lead to new guidelines and an expansion of the use of statins.
“These findings underscore a potential beneficial role of statin therapy on VTE in addition to its established role in cardiovascular disease prevention,” said Kamlesh Khunti, MD, PhD, of the University of Leicester in the UK.
Dr Khunti and his colleagues reported these findings in The Lancet Haematology.
The researchers analyzed data from 36 studies. This included 13 cohort studies with a total of 3,148,259 subjects and 23 randomized, controlled trials (RCTs) of 118,464 subjects in which statins were compared to placebo or no treatment.
In the observational studies, statin use was associated with a significant reduction in the risk of VTE, when compared to no statin use. The pooled relative risk (RR) was 0.75 (P<0.0001).
Statin use was associated with a significant reduction in the risk of deep vein thrombosis (DVT), with an RR of 0.77 (P<0.0001). (There were only 2 observational studies looking specifically at DVT outcomes.)
However, there was no significant reduction in the risk of pulmonary embolism (PE) with statin use. The RR was 1.02 (P=0.90). (There was only 1 observational study reporting specifically on PE outcomes.)
In the RCTs, statin use was associated with a significant reduction in the risk of VTE, with an RR of 0.85 (P=0.038).
Statin use was also associated with a significant reduction in DVT risk, with an RR of 0.45, but not PE risk, with an RR of 0.77 (no P values available). (There was only 1 RCT that reported specifically on DVT and PE outcomes.)
The RCTs also showed significant differences in VTE risk according to type of statin used. Patients receiving rosuvastatin had the lowest risk of VTE when compared to those receiving other statins, with an RR of 0.57 (P=0.015).
“Currently, statins are only approved for lipid lowering in the primary and secondary prevention of cardiovascular disease,” said study author Setor Kunutsor, MBChB, PhD, of the University of Bristol in the UK.
“But they have shown great promise beyond their established lipid-lowering effects, and these include potential beneficial impact on multiple disease conditions. These results provide an extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE and may support a true protective effect.” ![]()
FDA approves ibrutinib to treat rel/ref MZL

Photo courtesy of
Janssen Biotech, Inc.
The US Food and Drug Administration (FDA) has approved the Bruton’s tyrosine kinase inhibitor ibrutinib (Imbruvica®) for the treatment of marginal zone lymphoma (MZL).
The drug is now approved to treat patients with relapsed/refractory MZL who require systemic therapy and have received at least 1 prior anti-CD20-based therapy.
Ibrutinib has accelerated approval for this indication, based on the overall response rate the drug produced in a phase 2 trial.
Continued approval of ibrutinib as a treatment for MZL may be contingent upon verification and description of clinical benefit in a confirmatory trial.
The FDA’s approval of ibrutinib for MZL makes it the first treatment approved specifically for patients with this disease. It also marks the seventh FDA approval and fifth disease indication for ibrutinib since the drug was first approved in 2013.
Ibrutinib is also FDA-approved to treat chronic lymphocytic leukemia/small lymphocytic lymphoma, patients with mantle cell lymphoma who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia. The approval for mantle cell lymphoma is an accelerated approval.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
Phase 2 trial
The FDA’s approval of ibrutinib for MZL is based on data from the phase 2, single-arm PCYC-1121 study, in which researchers evaluated the drug in MZL patients who required systemic therapy and had received at least 1 prior anti-CD20-based therapy.
Results from this study were presented at the 2016 ASH Annual Meeting (abstract 1213).
The efficacy analysis included 63 patients with 3 subtypes of MZL: mucosa-associated lymphoid tissue (n=32), nodal (n=17), and splenic (n=14).
The overall response rate was 46%, with a partial response rate of 42.9% and a complete response rate of 3.2%. Responses were observed across all 3 MZL subtypes.
The median time to response was 4.5 months (range, 2.3-16.4 months). And the median duration of response was not reached (range, 16.7 months to not reached).
Overall, the safety data from this study was consistent with the known safety profile of ibrutinib in B-cell malignancies.
The most common adverse events of all grades (occurring in >20% of patients) were thrombocytopenia (49%), fatigue (44%), anemia (43%), diarrhea (43%), bruising (41%), musculoskeletal pain (40%), hemorrhage (30%), rash (29%), nausea (25%), peripheral edema (24%), arthralgia (24%), neutropenia (22%), cough (22%), dyspnea (21%), and upper respiratory tract infection (21%).
The most common (>10%) grade 3 or 4 events were decreases in hemoglobin and neutrophils (13% each) and pneumonia (10%).
The risks associated with ibrutinib as listed in the Warnings and Precautions section of the prescribing information are hemorrhage, infections, cytopenias, atrial fibrillation, hypertension, secondary primary malignancies, tumor lysis syndrome, and embryo fetal toxicities. ![]()
Photo courtesy of
Janssen Biotech, Inc.
The US Food and Drug Administration (FDA) has approved the Bruton’s tyrosine kinase inhibitor ibrutinib (Imbruvica®) for the treatment of marginal zone lymphoma (MZL).
The drug is now approved to treat patients with relapsed/refractory MZL who require systemic therapy and have received at least 1 prior anti-CD20-based therapy.
Ibrutinib has accelerated approval for this indication, based on the overall response rate the drug produced in a phase 2 trial.
Continued approval of ibrutinib as a treatment for MZL may be contingent upon verification and description of clinical benefit in a confirmatory trial.
The FDA’s approval of ibrutinib for MZL makes it the first treatment approved specifically for patients with this disease. It also marks the seventh FDA approval and fifth disease indication for ibrutinib since the drug was first approved in 2013.
Ibrutinib is also FDA-approved to treat chronic lymphocytic leukemia/small lymphocytic lymphoma, patients with mantle cell lymphoma who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia. The approval for mantle cell lymphoma is an accelerated approval.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
Phase 2 trial
The FDA’s approval of ibrutinib for MZL is based on data from the phase 2, single-arm PCYC-1121 study, in which researchers evaluated the drug in MZL patients who required systemic therapy and had received at least 1 prior anti-CD20-based therapy.
Results from this study were presented at the 2016 ASH Annual Meeting (abstract 1213).
The efficacy analysis included 63 patients with 3 subtypes of MZL: mucosa-associated lymphoid tissue (n=32), nodal (n=17), and splenic (n=14).
The overall response rate was 46%, with a partial response rate of 42.9% and a complete response rate of 3.2%. Responses were observed across all 3 MZL subtypes.
The median time to response was 4.5 months (range, 2.3-16.4 months). And the median duration of response was not reached (range, 16.7 months to not reached).
Overall, the safety data from this study was consistent with the known safety profile of ibrutinib in B-cell malignancies.
The most common adverse events of all grades (occurring in >20% of patients) were thrombocytopenia (49%), fatigue (44%), anemia (43%), diarrhea (43%), bruising (41%), musculoskeletal pain (40%), hemorrhage (30%), rash (29%), nausea (25%), peripheral edema (24%), arthralgia (24%), neutropenia (22%), cough (22%), dyspnea (21%), and upper respiratory tract infection (21%).
The most common (>10%) grade 3 or 4 events were decreases in hemoglobin and neutrophils (13% each) and pneumonia (10%).
The risks associated with ibrutinib as listed in the Warnings and Precautions section of the prescribing information are hemorrhage, infections, cytopenias, atrial fibrillation, hypertension, secondary primary malignancies, tumor lysis syndrome, and embryo fetal toxicities. ![]()
Photo courtesy of
Janssen Biotech, Inc.
The US Food and Drug Administration (FDA) has approved the Bruton’s tyrosine kinase inhibitor ibrutinib (Imbruvica®) for the treatment of marginal zone lymphoma (MZL).
The drug is now approved to treat patients with relapsed/refractory MZL who require systemic therapy and have received at least 1 prior anti-CD20-based therapy.
Ibrutinib has accelerated approval for this indication, based on the overall response rate the drug produced in a phase 2 trial.
Continued approval of ibrutinib as a treatment for MZL may be contingent upon verification and description of clinical benefit in a confirmatory trial.
The FDA’s approval of ibrutinib for MZL makes it the first treatment approved specifically for patients with this disease. It also marks the seventh FDA approval and fifth disease indication for ibrutinib since the drug was first approved in 2013.
Ibrutinib is also FDA-approved to treat chronic lymphocytic leukemia/small lymphocytic lymphoma, patients with mantle cell lymphoma who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia. The approval for mantle cell lymphoma is an accelerated approval.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
Phase 2 trial
The FDA’s approval of ibrutinib for MZL is based on data from the phase 2, single-arm PCYC-1121 study, in which researchers evaluated the drug in MZL patients who required systemic therapy and had received at least 1 prior anti-CD20-based therapy.
Results from this study were presented at the 2016 ASH Annual Meeting (abstract 1213).
The efficacy analysis included 63 patients with 3 subtypes of MZL: mucosa-associated lymphoid tissue (n=32), nodal (n=17), and splenic (n=14).
The overall response rate was 46%, with a partial response rate of 42.9% and a complete response rate of 3.2%. Responses were observed across all 3 MZL subtypes.
The median time to response was 4.5 months (range, 2.3-16.4 months). And the median duration of response was not reached (range, 16.7 months to not reached).
Overall, the safety data from this study was consistent with the known safety profile of ibrutinib in B-cell malignancies.
The most common adverse events of all grades (occurring in >20% of patients) were thrombocytopenia (49%), fatigue (44%), anemia (43%), diarrhea (43%), bruising (41%), musculoskeletal pain (40%), hemorrhage (30%), rash (29%), nausea (25%), peripheral edema (24%), arthralgia (24%), neutropenia (22%), cough (22%), dyspnea (21%), and upper respiratory tract infection (21%).
The most common (>10%) grade 3 or 4 events were decreases in hemoglobin and neutrophils (13% each) and pneumonia (10%).
The risks associated with ibrutinib as listed in the Warnings and Precautions section of the prescribing information are hemorrhage, infections, cytopenias, atrial fibrillation, hypertension, secondary primary malignancies, tumor lysis syndrome, and embryo fetal toxicities. ![]()
PI ties to industry linked to positive trial results

capsules for a clinical trial
Photo by Esther Dyson
Financial ties between principal investigators (PIs) and drug companies are independently associated with positive clinical trial results, according to new research.
The study showed a significant association between positive trial outcomes and PIs having financial ties to the manufacturer of the study drug, even after accounting for the source of research funding.
Researchers reported these findings in The BMJ.
Salomeh Keyhani, MD, of the University of California, San Francisco, and her colleagues conducted this research, analyzing a random sample of 195 drug trials published in 2013.
The team found financial ties between PIs and manufacturers of the study drug for 67.7% of the studies (n=132). In all, 58% of the PIs had financial ties to the manufacturers (231/397).
Types of financial ties included:
- Advisor/consultancy payments (39%)
- Speakers’ fees (20%)
- Unspecified financial ties (20%)
- Honoraria (13%)
- Employee relationships (13%)
- Travel fees (13%)
- Stock ownership (10%)
- Having a patent related to the study drug (5%).
PIs reported financial ties to the drug manufacturer in 76% (103/136) of studies with positive results and 49% (29/59) of studies with negative results (P<0.001).
In a multivariate analysis adjusted for the study’s funding source, a financial tie was significantly associated with a positive trial outcome. The odds ratio was 3.57 (P=0.001).
In a multivariate analysis adjusted for a range of other study-related factors as well, a financial tie remained significantly associated with a positive trial outcome. The odds ratio was 3.37 (P=0.006).
Dr Keyhani and her colleagues stressed that this analysis was observational and cannot be used to draw conclusions about causation.
However, they said, given the importance of industry and academic collaboration in advancing the development of new treatments, “more thought needs to be given to the roles that investigators, policy makers, and journal editors can play in ensuring the credibility of the evidence base.”
Authors of a related editorial said more research is needed to determine how industry funding and financial ties could influence trial results.
The authors—Andreas Lundh, PhD, of the University of Southern Denmark, and Lisa Bero, PhD, of the University of Sydney in Australia—urged trial investigators to share their data and participate in industry-funded trials only if data are made publicly available.
The authors also suggested journals could help by rejecting research by investigators who are unwilling to share their data and by penalizing investigators who fail to disclose financial ties. The role of sponsors, or companies with which investigators have ties, in the research must also be transparent.
In the meantime, trials with industry funding or investigators with financial ties “should be interpreted with caution until all relevant information is fully disclosed and easily accessible,” the authors concluded. ![]()
capsules for a clinical trial
Photo by Esther Dyson
Financial ties between principal investigators (PIs) and drug companies are independently associated with positive clinical trial results, according to new research.
The study showed a significant association between positive trial outcomes and PIs having financial ties to the manufacturer of the study drug, even after accounting for the source of research funding.
Researchers reported these findings in The BMJ.
Salomeh Keyhani, MD, of the University of California, San Francisco, and her colleagues conducted this research, analyzing a random sample of 195 drug trials published in 2013.
The team found financial ties between PIs and manufacturers of the study drug for 67.7% of the studies (n=132). In all, 58% of the PIs had financial ties to the manufacturers (231/397).
Types of financial ties included:
- Advisor/consultancy payments (39%)
- Speakers’ fees (20%)
- Unspecified financial ties (20%)
- Honoraria (13%)
- Employee relationships (13%)
- Travel fees (13%)
- Stock ownership (10%)
- Having a patent related to the study drug (5%).
PIs reported financial ties to the drug manufacturer in 76% (103/136) of studies with positive results and 49% (29/59) of studies with negative results (P<0.001).
In a multivariate analysis adjusted for the study’s funding source, a financial tie was significantly associated with a positive trial outcome. The odds ratio was 3.57 (P=0.001).
In a multivariate analysis adjusted for a range of other study-related factors as well, a financial tie remained significantly associated with a positive trial outcome. The odds ratio was 3.37 (P=0.006).
Dr Keyhani and her colleagues stressed that this analysis was observational and cannot be used to draw conclusions about causation.
However, they said, given the importance of industry and academic collaboration in advancing the development of new treatments, “more thought needs to be given to the roles that investigators, policy makers, and journal editors can play in ensuring the credibility of the evidence base.”
Authors of a related editorial said more research is needed to determine how industry funding and financial ties could influence trial results.
The authors—Andreas Lundh, PhD, of the University of Southern Denmark, and Lisa Bero, PhD, of the University of Sydney in Australia—urged trial investigators to share their data and participate in industry-funded trials only if data are made publicly available.
The authors also suggested journals could help by rejecting research by investigators who are unwilling to share their data and by penalizing investigators who fail to disclose financial ties. The role of sponsors, or companies with which investigators have ties, in the research must also be transparent.
In the meantime, trials with industry funding or investigators with financial ties “should be interpreted with caution until all relevant information is fully disclosed and easily accessible,” the authors concluded. ![]()
capsules for a clinical trial
Photo by Esther Dyson
Financial ties between principal investigators (PIs) and drug companies are independently associated with positive clinical trial results, according to new research.
The study showed a significant association between positive trial outcomes and PIs having financial ties to the manufacturer of the study drug, even after accounting for the source of research funding.
Researchers reported these findings in The BMJ.
Salomeh Keyhani, MD, of the University of California, San Francisco, and her colleagues conducted this research, analyzing a random sample of 195 drug trials published in 2013.
The team found financial ties between PIs and manufacturers of the study drug for 67.7% of the studies (n=132). In all, 58% of the PIs had financial ties to the manufacturers (231/397).
Types of financial ties included:
- Advisor/consultancy payments (39%)
- Speakers’ fees (20%)
- Unspecified financial ties (20%)
- Honoraria (13%)
- Employee relationships (13%)
- Travel fees (13%)
- Stock ownership (10%)
- Having a patent related to the study drug (5%).
PIs reported financial ties to the drug manufacturer in 76% (103/136) of studies with positive results and 49% (29/59) of studies with negative results (P<0.001).
In a multivariate analysis adjusted for the study’s funding source, a financial tie was significantly associated with a positive trial outcome. The odds ratio was 3.57 (P=0.001).
In a multivariate analysis adjusted for a range of other study-related factors as well, a financial tie remained significantly associated with a positive trial outcome. The odds ratio was 3.37 (P=0.006).
Dr Keyhani and her colleagues stressed that this analysis was observational and cannot be used to draw conclusions about causation.
However, they said, given the importance of industry and academic collaboration in advancing the development of new treatments, “more thought needs to be given to the roles that investigators, policy makers, and journal editors can play in ensuring the credibility of the evidence base.”
Authors of a related editorial said more research is needed to determine how industry funding and financial ties could influence trial results.
The authors—Andreas Lundh, PhD, of the University of Southern Denmark, and Lisa Bero, PhD, of the University of Sydney in Australia—urged trial investigators to share their data and participate in industry-funded trials only if data are made publicly available.
The authors also suggested journals could help by rejecting research by investigators who are unwilling to share their data and by penalizing investigators who fail to disclose financial ties. The role of sponsors, or companies with which investigators have ties, in the research must also be transparent.
In the meantime, trials with industry funding or investigators with financial ties “should be interpreted with caution until all relevant information is fully disclosed and easily accessible,” the authors concluded. ![]()
US agencies update regulations on research subjects

tumor in a test tube
Photo by Rhoda Baer
The US Department of Health and Human Services (HHS) and 15 other federal agencies have issued a final rule to update regulations intended to safeguard individuals who participate in research.
Most provisions in the new rule will go into effect in 2018.
The HHS says the new rule strengthens protections for people who volunteer to participate in research, while ensuring that the oversight system does not add inappropriate administrative burdens.
The current regulations, which have been in place since 1991, are often referred to as the “Common Rule.”
In September 2015, HHS and the other Common Rule agencies published a proposed new rule regarding research subjects—a notice of proposed rulemaking (NPRM)—which drew more than 2100 comments.
In response to concerns raised during the review process, the final rule contains a number of significant changes from the NPRM.
Research covered
The final rule does not cover clinical trials that are not federally funded.
The Common Rule has historically applied only to research conducted or supported by a Common Rule department or agency. And, although the NPRM proposed changing this policy, the final rule remains in line with the Common Rule.
Consent
The final rule requires consent forms to provide potential research subjects with a better understanding of a project’s scope so they can make a more fully informed decision about whether to participate.
Consent forms should include a concise explanation—at the beginning of the document—of the information that would be most important to individuals contemplating participation in a particular study, including the purpose of the research, the risks and benefits, and appropriate alternative treatments that might be beneficial to the prospective subject.
The rule also requires that consent forms for certain federally funded clinical trials be posted on a public website.
Institutional review boards
The final rule requires, in many cases, use of a single institutional review board (IRB) for multi-institutional research studies.
However, the final rule has been modified from the NPRM to add substantial increased flexibility in now allowing broad groups of studies (instead of just specific studies) to be removed from this requirement.
Privacy
The final rule does not include the standardized privacy safeguards for identifiable private information and identifiable biospecimens that were proposed in the NPRM.
In most respects, the final rule retains the current approach to privacy standards.
For studies on stored identifiable data or identifiable biospecimens, researchers will have the option of relying on broad consent obtained for future research as an alternative to seeking IRB approval to waive the consent requirement.
As under the current rule, researchers will not have to obtain consent for studies on non-identified stored data or biospecimens.
Exemptions
The final rule establishes new exempt categories of research based on the level of risk they pose to participants.
For example, to reduce unnecessary regulatory burden and allow IRBs to focus their attention on higher-risk studies, there is a new exemption for secondary research involving identifiable private information if the research is regulated by and participants are protected under the HIPAA rules.
Review
The final rule removes the requirement to conduct continuing review of ongoing research studies in certain instances where such review does little to protect subjects.
For more details, see the final rule. ![]()
tumor in a test tube
Photo by Rhoda Baer
The US Department of Health and Human Services (HHS) and 15 other federal agencies have issued a final rule to update regulations intended to safeguard individuals who participate in research.
Most provisions in the new rule will go into effect in 2018.
The HHS says the new rule strengthens protections for people who volunteer to participate in research, while ensuring that the oversight system does not add inappropriate administrative burdens.
The current regulations, which have been in place since 1991, are often referred to as the “Common Rule.”
In September 2015, HHS and the other Common Rule agencies published a proposed new rule regarding research subjects—a notice of proposed rulemaking (NPRM)—which drew more than 2100 comments.
In response to concerns raised during the review process, the final rule contains a number of significant changes from the NPRM.
Research covered
The final rule does not cover clinical trials that are not federally funded.
The Common Rule has historically applied only to research conducted or supported by a Common Rule department or agency. And, although the NPRM proposed changing this policy, the final rule remains in line with the Common Rule.
Consent
The final rule requires consent forms to provide potential research subjects with a better understanding of a project’s scope so they can make a more fully informed decision about whether to participate.
Consent forms should include a concise explanation—at the beginning of the document—of the information that would be most important to individuals contemplating participation in a particular study, including the purpose of the research, the risks and benefits, and appropriate alternative treatments that might be beneficial to the prospective subject.
The rule also requires that consent forms for certain federally funded clinical trials be posted on a public website.
Institutional review boards
The final rule requires, in many cases, use of a single institutional review board (IRB) for multi-institutional research studies.
However, the final rule has been modified from the NPRM to add substantial increased flexibility in now allowing broad groups of studies (instead of just specific studies) to be removed from this requirement.
Privacy
The final rule does not include the standardized privacy safeguards for identifiable private information and identifiable biospecimens that were proposed in the NPRM.
In most respects, the final rule retains the current approach to privacy standards.
For studies on stored identifiable data or identifiable biospecimens, researchers will have the option of relying on broad consent obtained for future research as an alternative to seeking IRB approval to waive the consent requirement.
As under the current rule, researchers will not have to obtain consent for studies on non-identified stored data or biospecimens.
Exemptions
The final rule establishes new exempt categories of research based on the level of risk they pose to participants.
For example, to reduce unnecessary regulatory burden and allow IRBs to focus their attention on higher-risk studies, there is a new exemption for secondary research involving identifiable private information if the research is regulated by and participants are protected under the HIPAA rules.
Review
The final rule removes the requirement to conduct continuing review of ongoing research studies in certain instances where such review does little to protect subjects.
For more details, see the final rule. ![]()
tumor in a test tube
Photo by Rhoda Baer
The US Department of Health and Human Services (HHS) and 15 other federal agencies have issued a final rule to update regulations intended to safeguard individuals who participate in research.
Most provisions in the new rule will go into effect in 2018.
The HHS says the new rule strengthens protections for people who volunteer to participate in research, while ensuring that the oversight system does not add inappropriate administrative burdens.
The current regulations, which have been in place since 1991, are often referred to as the “Common Rule.”
In September 2015, HHS and the other Common Rule agencies published a proposed new rule regarding research subjects—a notice of proposed rulemaking (NPRM)—which drew more than 2100 comments.
In response to concerns raised during the review process, the final rule contains a number of significant changes from the NPRM.
Research covered
The final rule does not cover clinical trials that are not federally funded.
The Common Rule has historically applied only to research conducted or supported by a Common Rule department or agency. And, although the NPRM proposed changing this policy, the final rule remains in line with the Common Rule.
Consent
The final rule requires consent forms to provide potential research subjects with a better understanding of a project’s scope so they can make a more fully informed decision about whether to participate.
Consent forms should include a concise explanation—at the beginning of the document—of the information that would be most important to individuals contemplating participation in a particular study, including the purpose of the research, the risks and benefits, and appropriate alternative treatments that might be beneficial to the prospective subject.
The rule also requires that consent forms for certain federally funded clinical trials be posted on a public website.
Institutional review boards
The final rule requires, in many cases, use of a single institutional review board (IRB) for multi-institutional research studies.
However, the final rule has been modified from the NPRM to add substantial increased flexibility in now allowing broad groups of studies (instead of just specific studies) to be removed from this requirement.
Privacy
The final rule does not include the standardized privacy safeguards for identifiable private information and identifiable biospecimens that were proposed in the NPRM.
In most respects, the final rule retains the current approach to privacy standards.
For studies on stored identifiable data or identifiable biospecimens, researchers will have the option of relying on broad consent obtained for future research as an alternative to seeking IRB approval to waive the consent requirement.
As under the current rule, researchers will not have to obtain consent for studies on non-identified stored data or biospecimens.
Exemptions
The final rule establishes new exempt categories of research based on the level of risk they pose to participants.
For example, to reduce unnecessary regulatory burden and allow IRBs to focus their attention on higher-risk studies, there is a new exemption for secondary research involving identifiable private information if the research is regulated by and participants are protected under the HIPAA rules.
Review
The final rule removes the requirement to conduct continuing review of ongoing research studies in certain instances where such review does little to protect subjects.
For more details, see the final rule.
Group creates library of SCD-specific iPSCs

Photo from Salk Institute
Researchers have created an induced pluripotent stem cell (iPSC) library intended to aid the study of sickle cell disease (SCD).
The library consists of iPSCs generated from blood samples taken from ethnically diverse SCD patients from around the world.
The researchers say these iPSCs can be used to create disease models, which may allow scientists to better understand how SCD occurs and develop and test new treatments for the disease.
As a complement to the library, the researchers also designed CRISPR/Cas gene editing tools to correct the sickle hemoglobin mutation.
The team described this work in Stem Cell Reports.
“Sickle cell disease affects millions of people worldwide and is an emerging global health burden,” said study author George Murphy, PhD, of the Center for Regenerative Medicine at Boston University School of Medicine in Massachusetts.
“iPSCs have the potential to revolutionize the way we study human development, model life-threatening diseases, and, eventually, treat patients.”
The researchers’ library includes SCD-specific iPSCs from patients of different ethnicities with different β-globin gene haplotypes and fetal hemoglobin levels.
The researchers generated 54 iPSC lines from blood samples collected from individuals of African American, Brazilian, and Saudi Arabian descent. Both genders were represented, as well as a range of ages (3 to 53 years of age).
Most of the cell lines in the library, along with accompanying genetic and hematologic data, are freely available via the WiCell website.
“In addition to the library, we’ve designed and are using gene editing tools to correct the sickle hemoglobin mutation using the stem cell lines,” said Gustavo Mostoslavsky, MD, PhD, also of the Center for Regenerative Medicine at Boston University School of Medicine.
“When coupled with corrected sickle cell disease-specific iPSCs, these tools could one day provide a functional cure for the disorder.”
Photo from Salk Institute
Researchers have created an induced pluripotent stem cell (iPSC) library intended to aid the study of sickle cell disease (SCD).
The library consists of iPSCs generated from blood samples taken from ethnically diverse SCD patients from around the world.
The researchers say these iPSCs can be used to create disease models, which may allow scientists to better understand how SCD occurs and develop and test new treatments for the disease.
As a complement to the library, the researchers also designed CRISPR/Cas gene editing tools to correct the sickle hemoglobin mutation.
The team described this work in Stem Cell Reports.
“Sickle cell disease affects millions of people worldwide and is an emerging global health burden,” said study author George Murphy, PhD, of the Center for Regenerative Medicine at Boston University School of Medicine in Massachusetts.
“iPSCs have the potential to revolutionize the way we study human development, model life-threatening diseases, and, eventually, treat patients.”
The researchers’ library includes SCD-specific iPSCs from patients of different ethnicities with different β-globin gene haplotypes and fetal hemoglobin levels.
The researchers generated 54 iPSC lines from blood samples collected from individuals of African American, Brazilian, and Saudi Arabian descent. Both genders were represented, as well as a range of ages (3 to 53 years of age).
Most of the cell lines in the library, along with accompanying genetic and hematologic data, are freely available via the WiCell website.
“In addition to the library, we’ve designed and are using gene editing tools to correct the sickle hemoglobin mutation using the stem cell lines,” said Gustavo Mostoslavsky, MD, PhD, also of the Center for Regenerative Medicine at Boston University School of Medicine.
“When coupled with corrected sickle cell disease-specific iPSCs, these tools could one day provide a functional cure for the disorder.”
Photo from Salk Institute
Researchers have created an induced pluripotent stem cell (iPSC) library intended to aid the study of sickle cell disease (SCD).
The library consists of iPSCs generated from blood samples taken from ethnically diverse SCD patients from around the world.
The researchers say these iPSCs can be used to create disease models, which may allow scientists to better understand how SCD occurs and develop and test new treatments for the disease.
As a complement to the library, the researchers also designed CRISPR/Cas gene editing tools to correct the sickle hemoglobin mutation.
The team described this work in Stem Cell Reports.
“Sickle cell disease affects millions of people worldwide and is an emerging global health burden,” said study author George Murphy, PhD, of the Center for Regenerative Medicine at Boston University School of Medicine in Massachusetts.
“iPSCs have the potential to revolutionize the way we study human development, model life-threatening diseases, and, eventually, treat patients.”
The researchers’ library includes SCD-specific iPSCs from patients of different ethnicities with different β-globin gene haplotypes and fetal hemoglobin levels.
The researchers generated 54 iPSC lines from blood samples collected from individuals of African American, Brazilian, and Saudi Arabian descent. Both genders were represented, as well as a range of ages (3 to 53 years of age).
Most of the cell lines in the library, along with accompanying genetic and hematologic data, are freely available via the WiCell website.
“In addition to the library, we’ve designed and are using gene editing tools to correct the sickle hemoglobin mutation using the stem cell lines,” said Gustavo Mostoslavsky, MD, PhD, also of the Center for Regenerative Medicine at Boston University School of Medicine.
“When coupled with corrected sickle cell disease-specific iPSCs, these tools could one day provide a functional cure for the disorder.”
Prolonged work-related stress linked to NHL, other cancers in men

burning building in Quebec
Photo by Sylvain Pedneault
New research suggests that prolonged exposure to work-related stress may increase a man’s risk of several cancers.
The study showed a significant association between work-related stress lasting 15 years or more and non-Hodgkin lymphoma (NHL) as well as lung, colon, rectal, and stomach cancers.
Men who had worked as firefighters, engineers, mechanics, and repair workers were most likely to report work-related stress.
Marie-Élise Parent, PhD, of Institut national de la recherche scientifique (INRS) in Laval, Quebec, Canada, and her colleagues conducted this research and published
the results in Preventive Medicine.
The researchers studied 3103 men with 11 different types of cancer who were diagnosed from 1979 to 1985. The team compared these men to 512 control subjects from the general population.
Both cases and controls were interviewed and asked to describe each job they had during their lifetime, including the occurrence of stress related to a job and the reason for that stress.
The researchers then calculated odds ratios (OR) for the association between perceived workplace stress and its duration, and each cancer site. The analyses were adjusted for lifestyle and occupational factors.
The team found that having at least one stressful job in a lifetime was associated with increased odds of 5 cancers:
- Lung—OR=1.33
- Colon—OR=1.51
- Bladder—OR=1.37
- Rectal—OR=1.52
- Stomach—OR=1.53.
When the researchers looked at the duration of stress, they found no significant association between any of the cancers and work-related stress lasting less than 15 years.
However, there were significant associations for several cancers and work-related stress lasting 15 to 30 years or more than 30 years. These included:
- NHL—15-30 years, OR=1.47; >30 years, OR=1.69 (P=0.02)
- Lung cancer—15-30 years, OR=1.47; >30 years, OR=1.51 (P=0.01)
- Colon cancer—15-30 years, OR=1.32; >30 years, OR=1.64 (P<0.01)
- Rectal cancer—15-30 years, OR=1.84; >30 years, OR=1.48 (P=0.01)
- Stomach cancer—15-30 years, OR=2.15; >30 years, OR=1.48 (P=0.01).
The occupations with the highest prevalence of work-related stress were firefighter (40% of firefighting jobs reported as stressful), industrial and aerospace engineer (31%), and motor vehicle and rail transport mechanic/repair worker (28%).
For the same individual, stress varied depending on the job held.
The study also showed that perceived stress was not limited to high work load and time constraints. Customer service, sales commissions, responsibilities, having an anxious temperament, job insecurity, financial problems, challenging or dangerous work conditions, employee supervision, interpersonal conflict, and a difficult commute were all sources of stress listed by study participants.
The researchers said one of the biggest flaws in previous studies of this kind is that none of them assessed work-related stress over a full working lifetime. The team said this made it impossible to determine how the duration of exposure to work-related stress affects cancer development.
This study, on the other hand, shows the importance of measuring stress at different points in an individual’s working life, the researchers said. They added that their results raise the question of whether chronic psychological stress should be viewed as a public health issue.
However, the team also pointed out that these results are unsubstantiated because they are based on a summary assessment of work-related stress for a given job. There is a need for epidemiological studies based on reliable stress measurements, repeated over time, and that take all sources of stress into account.
burning building in Quebec
Photo by Sylvain Pedneault
New research suggests that prolonged exposure to work-related stress may increase a man’s risk of several cancers.
The study showed a significant association between work-related stress lasting 15 years or more and non-Hodgkin lymphoma (NHL) as well as lung, colon, rectal, and stomach cancers.
Men who had worked as firefighters, engineers, mechanics, and repair workers were most likely to report work-related stress.
Marie-Élise Parent, PhD, of Institut national de la recherche scientifique (INRS) in Laval, Quebec, Canada, and her colleagues conducted this research and published
the results in Preventive Medicine.
The researchers studied 3103 men with 11 different types of cancer who were diagnosed from 1979 to 1985. The team compared these men to 512 control subjects from the general population.
Both cases and controls were interviewed and asked to describe each job they had during their lifetime, including the occurrence of stress related to a job and the reason for that stress.
The researchers then calculated odds ratios (OR) for the association between perceived workplace stress and its duration, and each cancer site. The analyses were adjusted for lifestyle and occupational factors.
The team found that having at least one stressful job in a lifetime was associated with increased odds of 5 cancers:
- Lung—OR=1.33
- Colon—OR=1.51
- Bladder—OR=1.37
- Rectal—OR=1.52
- Stomach—OR=1.53.
When the researchers looked at the duration of stress, they found no significant association between any of the cancers and work-related stress lasting less than 15 years.
However, there were significant associations for several cancers and work-related stress lasting 15 to 30 years or more than 30 years. These included:
- NHL—15-30 years, OR=1.47; >30 years, OR=1.69 (P=0.02)
- Lung cancer—15-30 years, OR=1.47; >30 years, OR=1.51 (P=0.01)
- Colon cancer—15-30 years, OR=1.32; >30 years, OR=1.64 (P<0.01)
- Rectal cancer—15-30 years, OR=1.84; >30 years, OR=1.48 (P=0.01)
- Stomach cancer—15-30 years, OR=2.15; >30 years, OR=1.48 (P=0.01).
The occupations with the highest prevalence of work-related stress were firefighter (40% of firefighting jobs reported as stressful), industrial and aerospace engineer (31%), and motor vehicle and rail transport mechanic/repair worker (28%).
For the same individual, stress varied depending on the job held.
The study also showed that perceived stress was not limited to high work load and time constraints. Customer service, sales commissions, responsibilities, having an anxious temperament, job insecurity, financial problems, challenging or dangerous work conditions, employee supervision, interpersonal conflict, and a difficult commute were all sources of stress listed by study participants.
The researchers said one of the biggest flaws in previous studies of this kind is that none of them assessed work-related stress over a full working lifetime. The team said this made it impossible to determine how the duration of exposure to work-related stress affects cancer development.
This study, on the other hand, shows the importance of measuring stress at different points in an individual’s working life, the researchers said. They added that their results raise the question of whether chronic psychological stress should be viewed as a public health issue.
However, the team also pointed out that these results are unsubstantiated because they are based on a summary assessment of work-related stress for a given job. There is a need for epidemiological studies based on reliable stress measurements, repeated over time, and that take all sources of stress into account.
burning building in Quebec
Photo by Sylvain Pedneault
New research suggests that prolonged exposure to work-related stress may increase a man’s risk of several cancers.
The study showed a significant association between work-related stress lasting 15 years or more and non-Hodgkin lymphoma (NHL) as well as lung, colon, rectal, and stomach cancers.
Men who had worked as firefighters, engineers, mechanics, and repair workers were most likely to report work-related stress.
Marie-Élise Parent, PhD, of Institut national de la recherche scientifique (INRS) in Laval, Quebec, Canada, and her colleagues conducted this research and published
the results in Preventive Medicine.
The researchers studied 3103 men with 11 different types of cancer who were diagnosed from 1979 to 1985. The team compared these men to 512 control subjects from the general population.
Both cases and controls were interviewed and asked to describe each job they had during their lifetime, including the occurrence of stress related to a job and the reason for that stress.
The researchers then calculated odds ratios (OR) for the association between perceived workplace stress and its duration, and each cancer site. The analyses were adjusted for lifestyle and occupational factors.
The team found that having at least one stressful job in a lifetime was associated with increased odds of 5 cancers:
- Lung—OR=1.33
- Colon—OR=1.51
- Bladder—OR=1.37
- Rectal—OR=1.52
- Stomach—OR=1.53.
When the researchers looked at the duration of stress, they found no significant association between any of the cancers and work-related stress lasting less than 15 years.
However, there were significant associations for several cancers and work-related stress lasting 15 to 30 years or more than 30 years. These included:
- NHL—15-30 years, OR=1.47; >30 years, OR=1.69 (P=0.02)
- Lung cancer—15-30 years, OR=1.47; >30 years, OR=1.51 (P=0.01)
- Colon cancer—15-30 years, OR=1.32; >30 years, OR=1.64 (P<0.01)
- Rectal cancer—15-30 years, OR=1.84; >30 years, OR=1.48 (P=0.01)
- Stomach cancer—15-30 years, OR=2.15; >30 years, OR=1.48 (P=0.01).
The occupations with the highest prevalence of work-related stress were firefighter (40% of firefighting jobs reported as stressful), industrial and aerospace engineer (31%), and motor vehicle and rail transport mechanic/repair worker (28%).
For the same individual, stress varied depending on the job held.
The study also showed that perceived stress was not limited to high work load and time constraints. Customer service, sales commissions, responsibilities, having an anxious temperament, job insecurity, financial problems, challenging or dangerous work conditions, employee supervision, interpersonal conflict, and a difficult commute were all sources of stress listed by study participants.
The researchers said one of the biggest flaws in previous studies of this kind is that none of them assessed work-related stress over a full working lifetime. The team said this made it impossible to determine how the duration of exposure to work-related stress affects cancer development.
This study, on the other hand, shows the importance of measuring stress at different points in an individual’s working life, the researchers said. They added that their results raise the question of whether chronic psychological stress should be viewed as a public health issue.
However, the team also pointed out that these results are unsubstantiated because they are based on a summary assessment of work-related stress for a given job. There is a need for epidemiological studies based on reliable stress measurements, repeated over time, and that take all sources of stress into account.
Pharma is gaming the system for orphan drugs, investigation suggests

Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs.
Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs.
Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs.