User login
Analysis identifies gaps in CV risk screening of patients with psoriasis
Just , according to an analysis of 10 years of national survey data.
From 2007 to 2016, national screening rates for four CV risk factors at 14.8 million psoriasis-related visits to dermatology providers were 11% (body-mass index), 7.4% (blood pressure), 2.9% (cholesterol), and 1.7% (glucose). Data from the National Ambulatory Medical Care Survey showed that at least one of the four factors was screened at 16% of dermatology visits, said William B. Song, BS, of the department of dermatology, University of Pennsylvania, Philadelphia, and associates.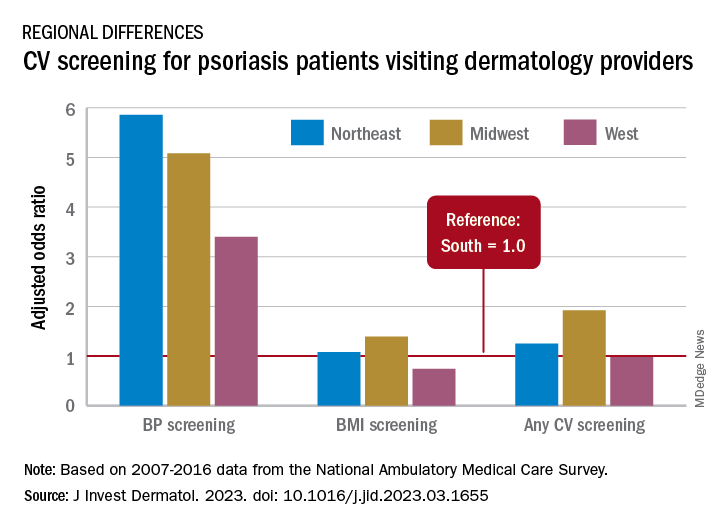
The main focus of their study, however, was regional differences. “CV risk factor screening by dermatology providers for patients with psoriasis is low across all regions of the United States and lowest in the South, the region that experiences the highest CVD burden in the United States,” they wrote in a letter to the editor.
Compared with the South, the adjusted odds of any CV screening were 0.98 in the West, 1.25 in the Northeast, and 1.92 in the Midwest. Blood pressure screening was significantly higher in all three regions, compared with the South, while BMI screening was actually lower in the West (0.74), the investigators reported. Odds ratios were not available for cholesterol and glucose screening because of sample size limitations.
The regional variation in screening rates “is not explained by patient demographics or disease severity,” they noted, adding that 2.8 million visits with BP screening would have been added over the 10-year study period “if providers in the South screened patients with psoriasis for high blood pressure at the same rate as providers in the Northeast.”
Guidelines published in 2019 by the American Academy of Dermatology and the National Psoriasis Foundation – which were cowritten by Joel M. Gelfand, MD, senior author of the current study – noted that dermatologists “play an important role in evidence-based screening of CV risk factors in patients with psoriasis,” the investigators wrote. But the regional variations suggest “that some regions experience barriers to appropriate screening or challenges in adhering to guidelines for managing psoriasis and CV risk.”
While the lack of data from after 2016 is one of the study limitations, they added, “continued efforts to develop effective interventions to improve CV screening and care for people with psoriasis in all regions of the U.S. are needed to more effectively address the burden of CV disease experienced by people with psoriasis.”
The study was partly funded by the National Psoriasis Foundation. Three of the seven investigators disclosed earnings from private companies in the form of consultant fees, research support, and honoraria. Dr. Gelfand is a deputy editor for the Journal of Investigative Dermatology.
Just , according to an analysis of 10 years of national survey data.
From 2007 to 2016, national screening rates for four CV risk factors at 14.8 million psoriasis-related visits to dermatology providers were 11% (body-mass index), 7.4% (blood pressure), 2.9% (cholesterol), and 1.7% (glucose). Data from the National Ambulatory Medical Care Survey showed that at least one of the four factors was screened at 16% of dermatology visits, said William B. Song, BS, of the department of dermatology, University of Pennsylvania, Philadelphia, and associates.
The main focus of their study, however, was regional differences. “CV risk factor screening by dermatology providers for patients with psoriasis is low across all regions of the United States and lowest in the South, the region that experiences the highest CVD burden in the United States,” they wrote in a letter to the editor.
Compared with the South, the adjusted odds of any CV screening were 0.98 in the West, 1.25 in the Northeast, and 1.92 in the Midwest. Blood pressure screening was significantly higher in all three regions, compared with the South, while BMI screening was actually lower in the West (0.74), the investigators reported. Odds ratios were not available for cholesterol and glucose screening because of sample size limitations.
The regional variation in screening rates “is not explained by patient demographics or disease severity,” they noted, adding that 2.8 million visits with BP screening would have been added over the 10-year study period “if providers in the South screened patients with psoriasis for high blood pressure at the same rate as providers in the Northeast.”
Guidelines published in 2019 by the American Academy of Dermatology and the National Psoriasis Foundation – which were cowritten by Joel M. Gelfand, MD, senior author of the current study – noted that dermatologists “play an important role in evidence-based screening of CV risk factors in patients with psoriasis,” the investigators wrote. But the regional variations suggest “that some regions experience barriers to appropriate screening or challenges in adhering to guidelines for managing psoriasis and CV risk.”
While the lack of data from after 2016 is one of the study limitations, they added, “continued efforts to develop effective interventions to improve CV screening and care for people with psoriasis in all regions of the U.S. are needed to more effectively address the burden of CV disease experienced by people with psoriasis.”
The study was partly funded by the National Psoriasis Foundation. Three of the seven investigators disclosed earnings from private companies in the form of consultant fees, research support, and honoraria. Dr. Gelfand is a deputy editor for the Journal of Investigative Dermatology.
Just , according to an analysis of 10 years of national survey data.
From 2007 to 2016, national screening rates for four CV risk factors at 14.8 million psoriasis-related visits to dermatology providers were 11% (body-mass index), 7.4% (blood pressure), 2.9% (cholesterol), and 1.7% (glucose). Data from the National Ambulatory Medical Care Survey showed that at least one of the four factors was screened at 16% of dermatology visits, said William B. Song, BS, of the department of dermatology, University of Pennsylvania, Philadelphia, and associates.
The main focus of their study, however, was regional differences. “CV risk factor screening by dermatology providers for patients with psoriasis is low across all regions of the United States and lowest in the South, the region that experiences the highest CVD burden in the United States,” they wrote in a letter to the editor.
Compared with the South, the adjusted odds of any CV screening were 0.98 in the West, 1.25 in the Northeast, and 1.92 in the Midwest. Blood pressure screening was significantly higher in all three regions, compared with the South, while BMI screening was actually lower in the West (0.74), the investigators reported. Odds ratios were not available for cholesterol and glucose screening because of sample size limitations.
The regional variation in screening rates “is not explained by patient demographics or disease severity,” they noted, adding that 2.8 million visits with BP screening would have been added over the 10-year study period “if providers in the South screened patients with psoriasis for high blood pressure at the same rate as providers in the Northeast.”
Guidelines published in 2019 by the American Academy of Dermatology and the National Psoriasis Foundation – which were cowritten by Joel M. Gelfand, MD, senior author of the current study – noted that dermatologists “play an important role in evidence-based screening of CV risk factors in patients with psoriasis,” the investigators wrote. But the regional variations suggest “that some regions experience barriers to appropriate screening or challenges in adhering to guidelines for managing psoriasis and CV risk.”
While the lack of data from after 2016 is one of the study limitations, they added, “continued efforts to develop effective interventions to improve CV screening and care for people with psoriasis in all regions of the U.S. are needed to more effectively address the burden of CV disease experienced by people with psoriasis.”
The study was partly funded by the National Psoriasis Foundation. Three of the seven investigators disclosed earnings from private companies in the form of consultant fees, research support, and honoraria. Dr. Gelfand is a deputy editor for the Journal of Investigative Dermatology.
FROM THE JOURNAL OF INVESTIGATIVE DERMATOLOGY
Frustration over iPLEDGE evident at FDA meeting
During 2 days of after the chaotic rollout of the new REMS platform at the end of 2021.
On March 29, at the end of the FDA’s joint meeting of two advisory committees that addressed ways to improve the iPLEDGE program, most panelists voted to change the 19-day lockout period for patients who can become pregnant, and the requirement that every month, providers must document counseling of those who cannot get pregnant and are taking the drug for acne.
However, there was no consensus on whether there should be a lockout at all or for how long, and what an appropriate interval for counseling those who cannot get pregnant would be, if not monthly. Those voting on the questions repeatedly cited a lack of data to make well-informed decisions.
The meeting of the two panels, the FDA’s Drug Safety and Risk Management Advisory Committee and the Dermatologic and Ophthalmic Drugs Advisory Committee, was held March 28-29, to discuss proposed changes to iPLEDGE requirements, to minimize the program’s burden on patients, prescribers, and pharmacies – while maintaining safe use of the highly teratogenic drug.
Lockout based on outdated reasoning
John S. Barbieri, MD, a dermatologist and epidemiologist, and director of the Advanced Acne Therapeutics Clinic at Brigham and Women’s Hospital in Boston, speaking as deputy chair of the American Academy of Dermatology Association (AADA) iPLEDGE work group, described the burden of getting the drug to patients. He was not on the panel, but spoke during the open public hearing.
“Compared to other acne medications, the time it takes to successfully go from prescribed (isotretinoin) to when the patient actually has it in their hands is 5- to 10-fold higher,” he said.

Among the barriers is the 19-day lockout period for people who can get pregnant and miss the 7-day window for picking up their prescriptions. They must then wait 19 days to get a pregnancy test to clear them for receiving the medication.
Gregory Wedin, PharmD, pharmacovigilance and risk management director of Upsher-Smith Laboratories, who spoke on behalf of the Isotretinoin Products Manufacturer Group (IPMG), which manages iPLEDGE, said, “The rationale for the 19-day wait is to ensure the next confirmatory pregnancy test is completed after the most fertile period of the menstrual cycle is passed.”
Many don’t have a monthly cycle
But Dr. Barbieri said that reasoning is outdated.
“The current program’s focus on the menstrual cycle is really an antiquated approach,” he said. “Many patients do not have a monthly cycle due to medical conditions like polycystic ovarian syndrome, or due to [certain kinds of] contraception.”
He added, “By removing this 19-day lockout and, really, the archaic timing around the menstrual cycle in general in this program, we can simplify the program, improve it, and better align it with the real-world biology of our patients.” He added that patients are often missing the 7-day window for picking up their prescriptions through no fault of their own. Speakers at the hearing also mentioned insurance hassles and ordering delays.
Communication with IPMG
Ilona Frieden, MD, professor of dermatology and pediatrics at the University of California, San Francisco, and outgoing chair of the AADA iPLEDGE work group, cited difficulty in working with IPMG on modifications as another barrier. She also spoke during the open public hearing.

“Despite many, many attempts to work with the IPMG, we are not aware of any organizational structure or key leaders to communicate with. Instead we have been given repeatedly a generic email address for trying to establish a working relationship and we believe this may explain the inaction of the IPMG since our proposals 4 years ago in 2019.”
Among those proposals, she said, were allowing telemedicine visits as part of the iPLEDGE REMS program and reducing counseling attestation to every 6 months instead of monthly for those who cannot become pregnant.
She pointed to the chaotic rollout of modifications to the iPLEDGE program on a new website at the end of 2021.
In 2021, she said, “despite 6 months of notification, no prescriber input was solicited before revamping the website. This lack of transparency and accountability has been a major hurdle in improving iPLEDGE.”
Dr. Barbieri called the rollout “a debacle” that could have been mitigated with communication with IPMG. “We warned about every issue that happened and talked about ways to mitigate it and were largely ignored,” he said.
“By including dermatologists and key stakeholders in these discussions, as we move forward with changes to improve this program, we can make sure that it’s patient-centered.”
IPMG did not address the specific complaints about the working relationship with the AADA workgroup at the meeting.
Monthly attestation for counseling patients who cannot get pregnant
Dr. Barbieri said the monthly requirement to counsel patients who cannot get pregnant and document that counseling unfairly burdens clinicians and patients. “We’re essentially asking patients to come in monthly just to tell them not to share their drugs [or] donate blood,” he said.
Ken Katz, MD, MSc, a dermatologist at Kaiser Permanente in San Francisco, was among the panel members voting not to continue the 19-day lockout.
“I think this places an unduly high burden physically and psychologically on our patients. It seems arbitrary,” he said. “Likely we will miss some pregnancies; we are missing some already. But the burden is not matched by the benefit.”
IPMG representative Dr. Wedin, said, “while we cannot support eliminating or extending the confirmation interval to a year, the [iPLEDGE] sponsors are agreeable [to] a 120-day confirmation interval.”
He said that while an extension to 120 days would reduce burden on prescribers, it comes with the risk in reducing oversight by a certified iPLEDGE prescriber and potentially increasing the risk for drug sharing.
“A patient may be more likely to share their drug with another person the further along with therapy they get as their condition improves,” Dr. Wedin said.
Home pregnancy testing
The advisory groups were also tasked with discussing whether home pregnancy tests, allowed during the COVID-19 public health emergency, should continue to be allowed. Most committee members and those in the public hearing who spoke on the issue agreed that home tests should continue in an effort to increase access and decrease burden.
During the pandemic, iPLEDGE rules have been relaxed from having a pregnancy test done only at a Clinical Laboratory Improvement Amendments–certified laboratory.
Lindsey Crist, PharmD, a risk management analyst at the FDA, who presented the FDA review committee’s analysis, said that the FDA’s review committee recommends ending the allowance of home tests, citing insufficient data on use and the discovery of instances of falsification of pregnancy tests.
“One study at an academic medical center reviewed the medical records of 89 patients who used home pregnancy tests while taking isotretinoin during the public health emergency. It found that 15.7% submitted falsified pregnancy test results,” Dr. Crist said.
Dr. Crist added, however, that the review committee recommends allowing the tests to be done in a provider’s office as an alternative.
Workaround to avoid falsification
Advisory committee member Brian P. Green, DO, associate professor of dermatology at Penn State University, Hershey, Pa., spoke in support of home pregnancy tests.
“What we have people do for telemedicine is take the stick, write their name, write the date on it, and send a picture of that the same day as their visit,” he said. “That way we have the pregnancy test the same day. Allowing this to continue to happen at home is important. Bringing people in is burdensome and costly.”
Emmy Graber, MD, a dermatologist who practices in Boston, and a director of the American Acne and Rosacea Society (AARS), relayed an example of the burden for a patient using isotretinoin who lives 1.5 hours away from the dermatology office. She is able to meet the requirements of iPLEDGE only through telehealth.

“Home pregnancy tests are highly sensitive, equal to the ones done in CLIA-certified labs, and highly accurate when interpreted by a dermatology provider,” said Dr. Graber, who spoke on behalf of the AARS during the open public hearing.
“Notably, CLIA [Clinical Laboratory Improvement Amendments] certification is not required by other REMS programs” for teratogenic drugs, she added.
Dr. Graber said it’s important to note that in the time the pandemic exceptions have been made for isotretinoin patients, “there has been no reported spike in pregnancy in the past three years.
“We do have some data to show that it is not imposing additional harms,” she said.
Suggestions for improvement
At the end of the hearing, advisory committee members were asked to propose improvements to the iPLEDGE REMS program.
Dr. Green advocated for the addition of an iPLEDGE mobile app.
“Most people go to their phones rather than their computers, particularly teenagers and younger people,” he noted.
Advisory committee member Megha M. Tollefson, MD, professor of dermatology and pediatric and adolescent medicine at Mayo Clinic in Rochester, Minn., echoed the need for an iPLEDGE app.
The young patients getting isotretinoin “don’t respond to email, they don’t necessarily go onto web pages. If we’re going to be as effective as possible, it’s going to have to be through an app-based system.”
Dr. Tollefson said she would like to see patient counseling standardized through the app. “I think there’s a lot of variability in what counseling is given when it’s left to the individual prescriber or practice,” she said.
Exceptions for long-acting contraceptives?
Advisory committee member Abbey B. Berenson, MD, PhD, professor of obstetrics and gynecology at University of Texas Medical Branch in Galveston, said that patients taking long-acting reversible contraceptives (LARCs) may need to be considered differently when deciding the intervals for attestation or whether to have a lockout period.
“LARC methods’ rate of failure is extremely low,” she said. “While it is true, as it has been pointed out, that all methods can fail, when they’re over 99% effective, I think that we can treat those methods differently than we treat methods such as birth control pills or abstinence that fail far more often. That is one way we could minimize burden on the providers and the patients.”
She also suggested using members of the health care team other than physicians to complete counseling, such as a nurse or pharmacist.
Prescriptions for emergency contraception
Advisory committee member Sascha Dublin, MD, PhD, senior scientific investigator for Kaiser Permanente Washington Health Research Institute in Seattle, said most patients taking the drug who can get pregnant should get a prescription for emergency contraception at the time of the first isotretinoin prescription.
“They don’t have to buy it, but to make it available at the very beginning sets the expectation that it would be good to have in your medicine cabinet, particularly if the [contraception] choice is abstinence or birth control pills.”
Dr. Dublin also called for better transparency surrounding the role of IPMG.
She said IPMG should be expected to collect data in a way that allows examination of health disparities, including by race and ethnicity and insurance status. Dr. Dublin added that she was concerned about the poor communication between dermatological societies and IPMG.
“The FDA should really require that IPMG hold periodic, regularly scheduled stakeholder forums,” she said. “There has to be a mechanism in place for IPMG to listen to those concerns in real time and respond.”
The advisory committees’ recommendations to the FDA are nonbinding, but the FDA generally follows the recommendations of advisory panels.
During 2 days of after the chaotic rollout of the new REMS platform at the end of 2021.
On March 29, at the end of the FDA’s joint meeting of two advisory committees that addressed ways to improve the iPLEDGE program, most panelists voted to change the 19-day lockout period for patients who can become pregnant, and the requirement that every month, providers must document counseling of those who cannot get pregnant and are taking the drug for acne.
However, there was no consensus on whether there should be a lockout at all or for how long, and what an appropriate interval for counseling those who cannot get pregnant would be, if not monthly. Those voting on the questions repeatedly cited a lack of data to make well-informed decisions.
The meeting of the two panels, the FDA’s Drug Safety and Risk Management Advisory Committee and the Dermatologic and Ophthalmic Drugs Advisory Committee, was held March 28-29, to discuss proposed changes to iPLEDGE requirements, to minimize the program’s burden on patients, prescribers, and pharmacies – while maintaining safe use of the highly teratogenic drug.
Lockout based on outdated reasoning
John S. Barbieri, MD, a dermatologist and epidemiologist, and director of the Advanced Acne Therapeutics Clinic at Brigham and Women’s Hospital in Boston, speaking as deputy chair of the American Academy of Dermatology Association (AADA) iPLEDGE work group, described the burden of getting the drug to patients. He was not on the panel, but spoke during the open public hearing.
“Compared to other acne medications, the time it takes to successfully go from prescribed (isotretinoin) to when the patient actually has it in their hands is 5- to 10-fold higher,” he said.
Among the barriers is the 19-day lockout period for people who can get pregnant and miss the 7-day window for picking up their prescriptions. They must then wait 19 days to get a pregnancy test to clear them for receiving the medication.
Gregory Wedin, PharmD, pharmacovigilance and risk management director of Upsher-Smith Laboratories, who spoke on behalf of the Isotretinoin Products Manufacturer Group (IPMG), which manages iPLEDGE, said, “The rationale for the 19-day wait is to ensure the next confirmatory pregnancy test is completed after the most fertile period of the menstrual cycle is passed.”
Many don’t have a monthly cycle
But Dr. Barbieri said that reasoning is outdated.
“The current program’s focus on the menstrual cycle is really an antiquated approach,” he said. “Many patients do not have a monthly cycle due to medical conditions like polycystic ovarian syndrome, or due to [certain kinds of] contraception.”
He added, “By removing this 19-day lockout and, really, the archaic timing around the menstrual cycle in general in this program, we can simplify the program, improve it, and better align it with the real-world biology of our patients.” He added that patients are often missing the 7-day window for picking up their prescriptions through no fault of their own. Speakers at the hearing also mentioned insurance hassles and ordering delays.
Communication with IPMG
Ilona Frieden, MD, professor of dermatology and pediatrics at the University of California, San Francisco, and outgoing chair of the AADA iPLEDGE work group, cited difficulty in working with IPMG on modifications as another barrier. She also spoke during the open public hearing.
“Despite many, many attempts to work with the IPMG, we are not aware of any organizational structure or key leaders to communicate with. Instead we have been given repeatedly a generic email address for trying to establish a working relationship and we believe this may explain the inaction of the IPMG since our proposals 4 years ago in 2019.”
Among those proposals, she said, were allowing telemedicine visits as part of the iPLEDGE REMS program and reducing counseling attestation to every 6 months instead of monthly for those who cannot become pregnant.
She pointed to the chaotic rollout of modifications to the iPLEDGE program on a new website at the end of 2021.
In 2021, she said, “despite 6 months of notification, no prescriber input was solicited before revamping the website. This lack of transparency and accountability has been a major hurdle in improving iPLEDGE.”
Dr. Barbieri called the rollout “a debacle” that could have been mitigated with communication with IPMG. “We warned about every issue that happened and talked about ways to mitigate it and were largely ignored,” he said.
“By including dermatologists and key stakeholders in these discussions, as we move forward with changes to improve this program, we can make sure that it’s patient-centered.”
IPMG did not address the specific complaints about the working relationship with the AADA workgroup at the meeting.
Monthly attestation for counseling patients who cannot get pregnant
Dr. Barbieri said the monthly requirement to counsel patients who cannot get pregnant and document that counseling unfairly burdens clinicians and patients. “We’re essentially asking patients to come in monthly just to tell them not to share their drugs [or] donate blood,” he said.
Ken Katz, MD, MSc, a dermatologist at Kaiser Permanente in San Francisco, was among the panel members voting not to continue the 19-day lockout.
“I think this places an unduly high burden physically and psychologically on our patients. It seems arbitrary,” he said. “Likely we will miss some pregnancies; we are missing some already. But the burden is not matched by the benefit.”
IPMG representative Dr. Wedin, said, “while we cannot support eliminating or extending the confirmation interval to a year, the [iPLEDGE] sponsors are agreeable [to] a 120-day confirmation interval.”
He said that while an extension to 120 days would reduce burden on prescribers, it comes with the risk in reducing oversight by a certified iPLEDGE prescriber and potentially increasing the risk for drug sharing.
“A patient may be more likely to share their drug with another person the further along with therapy they get as their condition improves,” Dr. Wedin said.
Home pregnancy testing
The advisory groups were also tasked with discussing whether home pregnancy tests, allowed during the COVID-19 public health emergency, should continue to be allowed. Most committee members and those in the public hearing who spoke on the issue agreed that home tests should continue in an effort to increase access and decrease burden.
During the pandemic, iPLEDGE rules have been relaxed from having a pregnancy test done only at a Clinical Laboratory Improvement Amendments–certified laboratory.
Lindsey Crist, PharmD, a risk management analyst at the FDA, who presented the FDA review committee’s analysis, said that the FDA’s review committee recommends ending the allowance of home tests, citing insufficient data on use and the discovery of instances of falsification of pregnancy tests.
“One study at an academic medical center reviewed the medical records of 89 patients who used home pregnancy tests while taking isotretinoin during the public health emergency. It found that 15.7% submitted falsified pregnancy test results,” Dr. Crist said.
Dr. Crist added, however, that the review committee recommends allowing the tests to be done in a provider’s office as an alternative.
Workaround to avoid falsification
Advisory committee member Brian P. Green, DO, associate professor of dermatology at Penn State University, Hershey, Pa., spoke in support of home pregnancy tests.
“What we have people do for telemedicine is take the stick, write their name, write the date on it, and send a picture of that the same day as their visit,” he said. “That way we have the pregnancy test the same day. Allowing this to continue to happen at home is important. Bringing people in is burdensome and costly.”
Emmy Graber, MD, a dermatologist who practices in Boston, and a director of the American Acne and Rosacea Society (AARS), relayed an example of the burden for a patient using isotretinoin who lives 1.5 hours away from the dermatology office. She is able to meet the requirements of iPLEDGE only through telehealth.
“Home pregnancy tests are highly sensitive, equal to the ones done in CLIA-certified labs, and highly accurate when interpreted by a dermatology provider,” said Dr. Graber, who spoke on behalf of the AARS during the open public hearing.
“Notably, CLIA [Clinical Laboratory Improvement Amendments] certification is not required by other REMS programs” for teratogenic drugs, she added.
Dr. Graber said it’s important to note that in the time the pandemic exceptions have been made for isotretinoin patients, “there has been no reported spike in pregnancy in the past three years.
“We do have some data to show that it is not imposing additional harms,” she said.
Suggestions for improvement
At the end of the hearing, advisory committee members were asked to propose improvements to the iPLEDGE REMS program.
Dr. Green advocated for the addition of an iPLEDGE mobile app.
“Most people go to their phones rather than their computers, particularly teenagers and younger people,” he noted.
Advisory committee member Megha M. Tollefson, MD, professor of dermatology and pediatric and adolescent medicine at Mayo Clinic in Rochester, Minn., echoed the need for an iPLEDGE app.
The young patients getting isotretinoin “don’t respond to email, they don’t necessarily go onto web pages. If we’re going to be as effective as possible, it’s going to have to be through an app-based system.”
Dr. Tollefson said she would like to see patient counseling standardized through the app. “I think there’s a lot of variability in what counseling is given when it’s left to the individual prescriber or practice,” she said.
Exceptions for long-acting contraceptives?
Advisory committee member Abbey B. Berenson, MD, PhD, professor of obstetrics and gynecology at University of Texas Medical Branch in Galveston, said that patients taking long-acting reversible contraceptives (LARCs) may need to be considered differently when deciding the intervals for attestation or whether to have a lockout period.
“LARC methods’ rate of failure is extremely low,” she said. “While it is true, as it has been pointed out, that all methods can fail, when they’re over 99% effective, I think that we can treat those methods differently than we treat methods such as birth control pills or abstinence that fail far more often. That is one way we could minimize burden on the providers and the patients.”
She also suggested using members of the health care team other than physicians to complete counseling, such as a nurse or pharmacist.
Prescriptions for emergency contraception
Advisory committee member Sascha Dublin, MD, PhD, senior scientific investigator for Kaiser Permanente Washington Health Research Institute in Seattle, said most patients taking the drug who can get pregnant should get a prescription for emergency contraception at the time of the first isotretinoin prescription.
“They don’t have to buy it, but to make it available at the very beginning sets the expectation that it would be good to have in your medicine cabinet, particularly if the [contraception] choice is abstinence or birth control pills.”
Dr. Dublin also called for better transparency surrounding the role of IPMG.
She said IPMG should be expected to collect data in a way that allows examination of health disparities, including by race and ethnicity and insurance status. Dr. Dublin added that she was concerned about the poor communication between dermatological societies and IPMG.
“The FDA should really require that IPMG hold periodic, regularly scheduled stakeholder forums,” she said. “There has to be a mechanism in place for IPMG to listen to those concerns in real time and respond.”
The advisory committees’ recommendations to the FDA are nonbinding, but the FDA generally follows the recommendations of advisory panels.
During 2 days of after the chaotic rollout of the new REMS platform at the end of 2021.
On March 29, at the end of the FDA’s joint meeting of two advisory committees that addressed ways to improve the iPLEDGE program, most panelists voted to change the 19-day lockout period for patients who can become pregnant, and the requirement that every month, providers must document counseling of those who cannot get pregnant and are taking the drug for acne.
However, there was no consensus on whether there should be a lockout at all or for how long, and what an appropriate interval for counseling those who cannot get pregnant would be, if not monthly. Those voting on the questions repeatedly cited a lack of data to make well-informed decisions.
The meeting of the two panels, the FDA’s Drug Safety and Risk Management Advisory Committee and the Dermatologic and Ophthalmic Drugs Advisory Committee, was held March 28-29, to discuss proposed changes to iPLEDGE requirements, to minimize the program’s burden on patients, prescribers, and pharmacies – while maintaining safe use of the highly teratogenic drug.
Lockout based on outdated reasoning
John S. Barbieri, MD, a dermatologist and epidemiologist, and director of the Advanced Acne Therapeutics Clinic at Brigham and Women’s Hospital in Boston, speaking as deputy chair of the American Academy of Dermatology Association (AADA) iPLEDGE work group, described the burden of getting the drug to patients. He was not on the panel, but spoke during the open public hearing.
“Compared to other acne medications, the time it takes to successfully go from prescribed (isotretinoin) to when the patient actually has it in their hands is 5- to 10-fold higher,” he said.
Among the barriers is the 19-day lockout period for people who can get pregnant and miss the 7-day window for picking up their prescriptions. They must then wait 19 days to get a pregnancy test to clear them for receiving the medication.
Gregory Wedin, PharmD, pharmacovigilance and risk management director of Upsher-Smith Laboratories, who spoke on behalf of the Isotretinoin Products Manufacturer Group (IPMG), which manages iPLEDGE, said, “The rationale for the 19-day wait is to ensure the next confirmatory pregnancy test is completed after the most fertile period of the menstrual cycle is passed.”
Many don’t have a monthly cycle
But Dr. Barbieri said that reasoning is outdated.
“The current program’s focus on the menstrual cycle is really an antiquated approach,” he said. “Many patients do not have a monthly cycle due to medical conditions like polycystic ovarian syndrome, or due to [certain kinds of] contraception.”
He added, “By removing this 19-day lockout and, really, the archaic timing around the menstrual cycle in general in this program, we can simplify the program, improve it, and better align it with the real-world biology of our patients.” He added that patients are often missing the 7-day window for picking up their prescriptions through no fault of their own. Speakers at the hearing also mentioned insurance hassles and ordering delays.
Communication with IPMG
Ilona Frieden, MD, professor of dermatology and pediatrics at the University of California, San Francisco, and outgoing chair of the AADA iPLEDGE work group, cited difficulty in working with IPMG on modifications as another barrier. She also spoke during the open public hearing.
“Despite many, many attempts to work with the IPMG, we are not aware of any organizational structure or key leaders to communicate with. Instead we have been given repeatedly a generic email address for trying to establish a working relationship and we believe this may explain the inaction of the IPMG since our proposals 4 years ago in 2019.”
Among those proposals, she said, were allowing telemedicine visits as part of the iPLEDGE REMS program and reducing counseling attestation to every 6 months instead of monthly for those who cannot become pregnant.
She pointed to the chaotic rollout of modifications to the iPLEDGE program on a new website at the end of 2021.
In 2021, she said, “despite 6 months of notification, no prescriber input was solicited before revamping the website. This lack of transparency and accountability has been a major hurdle in improving iPLEDGE.”
Dr. Barbieri called the rollout “a debacle” that could have been mitigated with communication with IPMG. “We warned about every issue that happened and talked about ways to mitigate it and were largely ignored,” he said.
“By including dermatologists and key stakeholders in these discussions, as we move forward with changes to improve this program, we can make sure that it’s patient-centered.”
IPMG did not address the specific complaints about the working relationship with the AADA workgroup at the meeting.
Monthly attestation for counseling patients who cannot get pregnant
Dr. Barbieri said the monthly requirement to counsel patients who cannot get pregnant and document that counseling unfairly burdens clinicians and patients. “We’re essentially asking patients to come in monthly just to tell them not to share their drugs [or] donate blood,” he said.
Ken Katz, MD, MSc, a dermatologist at Kaiser Permanente in San Francisco, was among the panel members voting not to continue the 19-day lockout.
“I think this places an unduly high burden physically and psychologically on our patients. It seems arbitrary,” he said. “Likely we will miss some pregnancies; we are missing some already. But the burden is not matched by the benefit.”
IPMG representative Dr. Wedin, said, “while we cannot support eliminating or extending the confirmation interval to a year, the [iPLEDGE] sponsors are agreeable [to] a 120-day confirmation interval.”
He said that while an extension to 120 days would reduce burden on prescribers, it comes with the risk in reducing oversight by a certified iPLEDGE prescriber and potentially increasing the risk for drug sharing.
“A patient may be more likely to share their drug with another person the further along with therapy they get as their condition improves,” Dr. Wedin said.
Home pregnancy testing
The advisory groups were also tasked with discussing whether home pregnancy tests, allowed during the COVID-19 public health emergency, should continue to be allowed. Most committee members and those in the public hearing who spoke on the issue agreed that home tests should continue in an effort to increase access and decrease burden.
During the pandemic, iPLEDGE rules have been relaxed from having a pregnancy test done only at a Clinical Laboratory Improvement Amendments–certified laboratory.
Lindsey Crist, PharmD, a risk management analyst at the FDA, who presented the FDA review committee’s analysis, said that the FDA’s review committee recommends ending the allowance of home tests, citing insufficient data on use and the discovery of instances of falsification of pregnancy tests.
“One study at an academic medical center reviewed the medical records of 89 patients who used home pregnancy tests while taking isotretinoin during the public health emergency. It found that 15.7% submitted falsified pregnancy test results,” Dr. Crist said.
Dr. Crist added, however, that the review committee recommends allowing the tests to be done in a provider’s office as an alternative.
Workaround to avoid falsification
Advisory committee member Brian P. Green, DO, associate professor of dermatology at Penn State University, Hershey, Pa., spoke in support of home pregnancy tests.
“What we have people do for telemedicine is take the stick, write their name, write the date on it, and send a picture of that the same day as their visit,” he said. “That way we have the pregnancy test the same day. Allowing this to continue to happen at home is important. Bringing people in is burdensome and costly.”
Emmy Graber, MD, a dermatologist who practices in Boston, and a director of the American Acne and Rosacea Society (AARS), relayed an example of the burden for a patient using isotretinoin who lives 1.5 hours away from the dermatology office. She is able to meet the requirements of iPLEDGE only through telehealth.
“Home pregnancy tests are highly sensitive, equal to the ones done in CLIA-certified labs, and highly accurate when interpreted by a dermatology provider,” said Dr. Graber, who spoke on behalf of the AARS during the open public hearing.
“Notably, CLIA [Clinical Laboratory Improvement Amendments] certification is not required by other REMS programs” for teratogenic drugs, she added.
Dr. Graber said it’s important to note that in the time the pandemic exceptions have been made for isotretinoin patients, “there has been no reported spike in pregnancy in the past three years.
“We do have some data to show that it is not imposing additional harms,” she said.
Suggestions for improvement
At the end of the hearing, advisory committee members were asked to propose improvements to the iPLEDGE REMS program.
Dr. Green advocated for the addition of an iPLEDGE mobile app.
“Most people go to their phones rather than their computers, particularly teenagers and younger people,” he noted.
Advisory committee member Megha M. Tollefson, MD, professor of dermatology and pediatric and adolescent medicine at Mayo Clinic in Rochester, Minn., echoed the need for an iPLEDGE app.
The young patients getting isotretinoin “don’t respond to email, they don’t necessarily go onto web pages. If we’re going to be as effective as possible, it’s going to have to be through an app-based system.”
Dr. Tollefson said she would like to see patient counseling standardized through the app. “I think there’s a lot of variability in what counseling is given when it’s left to the individual prescriber or practice,” she said.
Exceptions for long-acting contraceptives?
Advisory committee member Abbey B. Berenson, MD, PhD, professor of obstetrics and gynecology at University of Texas Medical Branch in Galveston, said that patients taking long-acting reversible contraceptives (LARCs) may need to be considered differently when deciding the intervals for attestation or whether to have a lockout period.
“LARC methods’ rate of failure is extremely low,” she said. “While it is true, as it has been pointed out, that all methods can fail, when they’re over 99% effective, I think that we can treat those methods differently than we treat methods such as birth control pills or abstinence that fail far more often. That is one way we could minimize burden on the providers and the patients.”
She also suggested using members of the health care team other than physicians to complete counseling, such as a nurse or pharmacist.
Prescriptions for emergency contraception
Advisory committee member Sascha Dublin, MD, PhD, senior scientific investigator for Kaiser Permanente Washington Health Research Institute in Seattle, said most patients taking the drug who can get pregnant should get a prescription for emergency contraception at the time of the first isotretinoin prescription.
“They don’t have to buy it, but to make it available at the very beginning sets the expectation that it would be good to have in your medicine cabinet, particularly if the [contraception] choice is abstinence or birth control pills.”
Dr. Dublin also called for better transparency surrounding the role of IPMG.
She said IPMG should be expected to collect data in a way that allows examination of health disparities, including by race and ethnicity and insurance status. Dr. Dublin added that she was concerned about the poor communication between dermatological societies and IPMG.
“The FDA should really require that IPMG hold periodic, regularly scheduled stakeholder forums,” she said. “There has to be a mechanism in place for IPMG to listen to those concerns in real time and respond.”
The advisory committees’ recommendations to the FDA are nonbinding, but the FDA generally follows the recommendations of advisory panels.
Biosimilars and patients: Discussions should address safety, cost, and anxiety about change
Rheumatologist Marcus Snow, MD, is comfortable with prescribing biosimilars as a first-line, first-time biologic, and discussing them with patients.
“If a biosimilar is on the market, it has gone through rigorous study proving its effectiveness and equivalence to a bio-originator,” said Dr. Snow, a rheumatologist with the University of Nebraska Medical Center, Omaha, and chair of the American College of Rheumatology’s Committee on Rheumatologic Care.

The formulary makes a big difference in the conversation about options, he said. “The formularies dictate what we can prescribe. It may not be appropriate, but it is reality. The cost of biologics for a patient without insurance coverage makes it impossible to afford.”
He will often tell patients that he’ll fight any changes or formulary restrictions he does not agree with. “However, when I see patients in follow-up, even if there is no known change on the horizon, I may bring up biosimilars when we have a moment to chat about them to familiarize them with what may happen in the future.”
The need for patient education on biosimilars presents a barrier to realizing their potential to save money and expand choice, noted Cardinal Health in its 2023 biosimilars report. Of 103 rheumatologists who responded to a Cardinal Health survey, 85% agreed that patient education was important. But those conversations can take an uncomfortable turn if the patient pushes back against taking a biosimilar owing to cost or safety concerns.
It’s not uncommon for a patient to express some anxiety about biosimilars, especially if they’re doing well on a current treatment plan. Most patients do not want any changes that may lead to worsening disease control, Dr. Snow said.

Patients and physicians alike often don’t understand the mechanics of biosimilars. “There’s a lot of misinformation about this,” said Sameer Awsare, MD, an associate executive director for The Permanente Medical Group in Campbell, Calif. Patients should know that a biosimilar will be as clinically efficacious as the medicine they’ve been on, with the same safety profiles, said Dr. Awsare, who works with Kaiser Permanente’s pharmacy partners on biosimilars.
Insurance often drives the conversation
The global anti-inflammatory biologics market is anticipated to reach $150 billion by 2027, according to a recent CVS report. As of March 2023, the Food and Drug Administration had approved 40 biosimilars to 11 different reference products. There are 28 on the U.S. market and 100 more in development. Projected to save more than $180 billion over the next 5 years, they are anticipated to expand choice and drive competition.
Rheumatologists, dermatologists, and gastroenterologists are frequent prescribers, although their choices for immune-mediated inflammatory diseases are limited to tumor necrosis factor inhibitors (infliximab [Remicade] originator and adalimumab [Humira] originator) and anti-CD20 agents, such as rituximab (Rituxan) originator.

Benefit design or formulary usually dictates what medicine a patient receives. “Because of significantly higher out-of-pocket cost or formulary positioning, patients may end up with a generic or a biosimilar instead of a brand-name medicine or branded biologic,” said Robert Popovian, PharmD, MS, chief science policy officer of the Global Healthy Living Foundation.
Insurers rarely offer both Remicade and biosimilar infliximab, allowing the doctor to choose, said Miguel Regueiro, MD, chair of the Cleveland Clinic’s Digestive Disease & Surgery Institute, who prescribes infliximab biosimilars. Most often, the payer will choose the lower-cost biosimilar. “I am fine with the biosimilar, either as a new start or a switch from the reference product.”

However, the patient might feel differently. They can form an attachment to the reference medication if it has prevented severe illness. “They do not want to change, as they feel they are going on a ‘new’ medication that will not work as well,” Dr. Regueiro said.
This is where the education comes in: to reassure patients that a biosimilar will work just as well as the reference product. “For patients who have done well for years on a biologic, more time needs to be spent reassuring them and answering questions,” compared with a patient just starting on a biosimilar, he advised.
But not all physicians are quick to prescribe biosimilars.

Especially with psoriasis, which has so many strong options for reference drugs, a switch may be hard to justify, said dermatologist Stephanie K. Fabbro, MD, assistant professor at Northeast Ohio Medical University, Rootstown. “If I have a preference, I would rather switch a patient to a drug from a different class without a biosimilar option to reduce the possibility of pushback.”
Dr. Fabbro, part of the core faculty in the Riverside Methodist Hospital Dermatology Residency Program in Columbus, will share data from clinical trials and postmarket surveillance with patients to support her decision.
Conversations about cost
Patients may also push back if they don’t save money when switching to a biosimilar. “This dilemma raises the question of who is profiting when a biosimilar is dispensed,” Dr. Popovian said. Insurers and pharmacy benefit managers (PBMs) that take additional concessions from biopharmaceutical manufacturers in the form of rebates and fees will often pocket this money as profit instead of passing savings back to the patient to help reduce their out-of-pocket requirement, he added.
If an originator biologic and a biosimilar are available, “as a pharmacist, I will choose the medicine that will incur the lowest out-of-pocket cost for the patient,” Dr. Popovian said.

Discussing cost – and who dictates which biosimilar is on the formulary – is an important conversation to have with patients, said Vivek Kaul, MD, Segal-Watson Professor of Medicine at the University of Rochester (N.Y.) Medical Center.
Providing equivalent clinical efficacy while saving costs is the economic reality of biosimilars, Dr. Kaul said. Third-party payers regularly evaluate how to provide the same quality of care while saving money. Physicians and patients alike “must be mindful that as time goes on, if the science on biosimilars stays robust, if the adoption is more widespread and the cost-saving proposition turns out to be true, more formularies will be attracted to replacing the reference product with the biosimilar counterpart.”
Providers and patients can weigh the options if a formulary suddenly switches to a biosimilar, Dr. Kaul continued. “You can accept the novel product on the formulary or may have to face out-of-pocket expenses as a patient.” If providers and patients have concerns about the biosimilar, they can always appeal if there’s solid scientific evidence that supports reverting back to the reference product.
“If you think the biosimilar is equally efficacious, comes at a lower cost, and is right for the patient, then the providers should tell the patient that,” he added.
Some studies have questioned whether the biosimilars will save money, compared with the reference drug, Dr. Fabbro noted. Medicare, for example, may pay only for a certain percentage of an approved biosimilar, saddling the patient with a monthly copay costing thousands of dollars. “It is unclear whether biosimilar manufacturers will have the same level of patient support programs as the reference drug companies.”
For that reason, physicians should also inform patients about the robust patient assistance and copay assistance programs many reference drug manufacturers offer, she said.
Biosimilars 101: Familiarizing patients
Safety and ease of use are other common concerns about biosimilars. Patients may ask if the application is different, or why it’s advantageous to switch to a biosimilar, Dr. Awsare said.
Sometimes the syringe or injector for a biosimilar might look different from that of the originator drug, he said.
Anecdotally, Dr. Fabbro has heard stories of patients having injection reactions that they did not experience with the reference drug or having a disease flare-up after starting a biosimilar. 
As is the case with reference products, in their conversations with patients, clinicians should address the adverse event profile of biosimilars, offering data points from published studies and clinical guidelines that support the use of these products. “There should be an emphasis on patient education around efficacy and any side effects, and how the profile of the reference product compares with a proposed biosimilar,” Dr. Kaul suggested.
When Dr. Snow discusses biosimilars and generics, “I make sure to share this in an understandable way based on the patient’s scientific background, or lack thereof,” he said. If there is enough time, he also discusses how European- and U.S.-sourced biologics are slightly different.
Pharmacists should tell patients to expect the same clinical outcomes from a biosimilar, Dr. Popovian said. However, if they have any reduction in efficacy or potential safety concerns, they should communicate with their physician or pharmacist immediately.
In Dr. Regueiro’s practice, a pharmacist specializing in inflammatory bowel disease often has a one-on-one meeting with patients to educate and answer questions. “Additionally, we provide them the Crohn’s and Colitis Foundation web link on biosimilars,” said Dr. Regueiro.
A village approach to education
When biosimilars first came out, there were no formal education materials, Dr. Awsare said. Kaiser Permanente decided to create its own educational materials, not just for patients but also to help educate its primary care doctors; the rheumatologists, dermatologists, and gastroenterologists using the biosimilars; the nurses infusing patients; and the pharmacists preparing the biosimilars.
The health system also has a different approach to choosing medication. Instead of having an insurance company or PBM decide what’s in the formulary, clinicians work with the pharmacists at Kaiser to look at clinical evidence and decide which biosimilar to use. Most of its plans also provide lower copays to patients when they use the biosimilar.
This was the approach for Humira biosimilars, Dr. Awsare said. Eight will be on the market in 2023. “Our rheumatologists, dermatologists, and gastroenterologists looked at the data from Europe, looked at some real-world evidence, and then said: ‘We think this one’s going to be the best one for our patients.’ ”
Having clinicians choose the biosimilar instead of a health plan makes it a lot easier to have conversations with patients, he said. “Once we’ve moved that market share to that particular biosimilar, we give our physicians the time to have those discussions.”
Clinical pharmacists also provide educational support, offering guidance on issues such as side effects, as patients transition to the biosimilar. “We like to use the word ‘transition’ because it’s essentially the same biologic. So, you’re not actually switching,” Dr. Awsare said.
No consensus on interchangeability
Whether the conversation on interchangeability will affect patient conversations with physicians depends on who you ask.
If a biosimilar has an interchangeability designation, it means that the pharmacist can substitute it without the intervention of the clinician who prescribed the reference product. It does not relate to the quality, safety, or effectiveness of biosimilars or interchangeable biosimilar products, Dr. Popovian said.
The United States is the only country that has this designation. Even though it’s not identical to the originator drug, a biosimilar has the same clinical efficacy and safety profile. “So clinically, interchangeability is meaningless,” Dr. Awsare said.
In its report on biosimilars in the autoimmune category, CVS acknowledged that interchangeability was important but would not be a significant factor in driving adoption of biosimilars. However, in a Cardinal Health survey of 72 gastroenterologists, 38% cited the interchangeability of biosimilars as a top concern for adalimumab biosimilars, along with transitioning patients from Humira to a biosimilar (44%).
“Patient education regarding biosimilar safety, efficacy, and interchangeability appears paramount to the acceptance of these products, particularly for patients who are switched from a reference product,” Dr. Kaul noted in the Cardinal Health report.
Wherever supported by data, Dr. Kaul recommends incorporating biosimilar use and interchangeability into best practice guidelines going forward. “That will go a long way in disseminating the latest information on this topic and position this paradigm for increased adoption among providers.”
Some physicians like Dr. Snow aren’t that concerned with interchangeability. This hasn’t affected conversations with patients, he said. Multiple studies demonstrating the lack of antibody formation with multiple switches from different biosimilar drugs has eased his concern about multiple switches causing problems.
“Initially, there was a gap in demonstrating the long-term effect of multiple switches on antibody production and drug effectiveness. That gap has started to close as more data from Europe’s experience with biosimilars becomes available,” Dr. Snow said.
Resources for physicians, patients
The federal government has taken steps to advance biosimilars education and adoption. In 2021, President Biden signed the Advancing Education on Biosimilars Act into law, which directs the FDA to develop or improve continuing education programs that address prescribing of biosimilars and biological products.
The FDA provides educational materials on its website, including a comprehensive curriculum toolkit. The Accreditation Council for Medical Affairs has also created an online 40-hour curriculum for health care professionals called the Board-Certified Biologics and Biosimilars Specialist Program.
Dr. Fabbro recommended patients use the FDA page Biosimilar Basics for Patients to educate themselves on biosimilars. The Global Healthy Living Foundation’s podcast, Breaking Down Biosimilars, is another free resource for patients.
“While much has changed, the continued need for multistakeholder education, awareness, and dedicated research remains even more important as we expand into newer therapeutic areas and classes,” wrote the authors of the Cardinal Health report.
Help patients understand biologics and biosimilars by using AGA resources for providers and patients available at gastro.org/biosimilars.
Dr. Regueiro is on advisory boards and consults for AbbVie, Janssen, UCB, Takeda, Pfizer, Bristol-Myers Squibb, Organon, Amgen, Genentech, Gilead, Salix, Prometheus, Lilly, Celgene, TARGET PharmaSolutions, Trellis, and Boehringer Ingelheim. Dr. Fabbro is a principal investigator for Castle Biosciences, on the speakers bureau for Valchlor, and on the advisory boards of Janssen and Bristol-Myers Squibb. Dr. Popovian, Dr. Snow, Dr. Awsare, and Dr. Kaul had no disclosures.
A version of this article originally appeared on Medscape.com.
Rheumatologist Marcus Snow, MD, is comfortable with prescribing biosimilars as a first-line, first-time biologic, and discussing them with patients.
“If a biosimilar is on the market, it has gone through rigorous study proving its effectiveness and equivalence to a bio-originator,” said Dr. Snow, a rheumatologist with the University of Nebraska Medical Center, Omaha, and chair of the American College of Rheumatology’s Committee on Rheumatologic Care.
The formulary makes a big difference in the conversation about options, he said. “The formularies dictate what we can prescribe. It may not be appropriate, but it is reality. The cost of biologics for a patient without insurance coverage makes it impossible to afford.”
He will often tell patients that he’ll fight any changes or formulary restrictions he does not agree with. “However, when I see patients in follow-up, even if there is no known change on the horizon, I may bring up biosimilars when we have a moment to chat about them to familiarize them with what may happen in the future.”
The need for patient education on biosimilars presents a barrier to realizing their potential to save money and expand choice, noted Cardinal Health in its 2023 biosimilars report. Of 103 rheumatologists who responded to a Cardinal Health survey, 85% agreed that patient education was important. But those conversations can take an uncomfortable turn if the patient pushes back against taking a biosimilar owing to cost or safety concerns.
It’s not uncommon for a patient to express some anxiety about biosimilars, especially if they’re doing well on a current treatment plan. Most patients do not want any changes that may lead to worsening disease control, Dr. Snow said.
Patients and physicians alike often don’t understand the mechanics of biosimilars. “There’s a lot of misinformation about this,” said Sameer Awsare, MD, an associate executive director for The Permanente Medical Group in Campbell, Calif. Patients should know that a biosimilar will be as clinically efficacious as the medicine they’ve been on, with the same safety profiles, said Dr. Awsare, who works with Kaiser Permanente’s pharmacy partners on biosimilars.
Insurance often drives the conversation
The global anti-inflammatory biologics market is anticipated to reach $150 billion by 2027, according to a recent CVS report. As of March 2023, the Food and Drug Administration had approved 40 biosimilars to 11 different reference products. There are 28 on the U.S. market and 100 more in development. Projected to save more than $180 billion over the next 5 years, they are anticipated to expand choice and drive competition.
Rheumatologists, dermatologists, and gastroenterologists are frequent prescribers, although their choices for immune-mediated inflammatory diseases are limited to tumor necrosis factor inhibitors (infliximab [Remicade] originator and adalimumab [Humira] originator) and anti-CD20 agents, such as rituximab (Rituxan) originator.
Benefit design or formulary usually dictates what medicine a patient receives. “Because of significantly higher out-of-pocket cost or formulary positioning, patients may end up with a generic or a biosimilar instead of a brand-name medicine or branded biologic,” said Robert Popovian, PharmD, MS, chief science policy officer of the Global Healthy Living Foundation.
Insurers rarely offer both Remicade and biosimilar infliximab, allowing the doctor to choose, said Miguel Regueiro, MD, chair of the Cleveland Clinic’s Digestive Disease & Surgery Institute, who prescribes infliximab biosimilars. Most often, the payer will choose the lower-cost biosimilar. “I am fine with the biosimilar, either as a new start or a switch from the reference product.”
However, the patient might feel differently. They can form an attachment to the reference medication if it has prevented severe illness. “They do not want to change, as they feel they are going on a ‘new’ medication that will not work as well,” Dr. Regueiro said.
This is where the education comes in: to reassure patients that a biosimilar will work just as well as the reference product. “For patients who have done well for years on a biologic, more time needs to be spent reassuring them and answering questions,” compared with a patient just starting on a biosimilar, he advised.
But not all physicians are quick to prescribe biosimilars.
Especially with psoriasis, which has so many strong options for reference drugs, a switch may be hard to justify, said dermatologist Stephanie K. Fabbro, MD, assistant professor at Northeast Ohio Medical University, Rootstown. “If I have a preference, I would rather switch a patient to a drug from a different class without a biosimilar option to reduce the possibility of pushback.”
Dr. Fabbro, part of the core faculty in the Riverside Methodist Hospital Dermatology Residency Program in Columbus, will share data from clinical trials and postmarket surveillance with patients to support her decision.
Conversations about cost
Patients may also push back if they don’t save money when switching to a biosimilar. “This dilemma raises the question of who is profiting when a biosimilar is dispensed,” Dr. Popovian said. Insurers and pharmacy benefit managers (PBMs) that take additional concessions from biopharmaceutical manufacturers in the form of rebates and fees will often pocket this money as profit instead of passing savings back to the patient to help reduce their out-of-pocket requirement, he added.
If an originator biologic and a biosimilar are available, “as a pharmacist, I will choose the medicine that will incur the lowest out-of-pocket cost for the patient,” Dr. Popovian said.
Discussing cost – and who dictates which biosimilar is on the formulary – is an important conversation to have with patients, said Vivek Kaul, MD, Segal-Watson Professor of Medicine at the University of Rochester (N.Y.) Medical Center.
Providing equivalent clinical efficacy while saving costs is the economic reality of biosimilars, Dr. Kaul said. Third-party payers regularly evaluate how to provide the same quality of care while saving money. Physicians and patients alike “must be mindful that as time goes on, if the science on biosimilars stays robust, if the adoption is more widespread and the cost-saving proposition turns out to be true, more formularies will be attracted to replacing the reference product with the biosimilar counterpart.”
Providers and patients can weigh the options if a formulary suddenly switches to a biosimilar, Dr. Kaul continued. “You can accept the novel product on the formulary or may have to face out-of-pocket expenses as a patient.” If providers and patients have concerns about the biosimilar, they can always appeal if there’s solid scientific evidence that supports reverting back to the reference product.
“If you think the biosimilar is equally efficacious, comes at a lower cost, and is right for the patient, then the providers should tell the patient that,” he added.
Some studies have questioned whether the biosimilars will save money, compared with the reference drug, Dr. Fabbro noted. Medicare, for example, may pay only for a certain percentage of an approved biosimilar, saddling the patient with a monthly copay costing thousands of dollars. “It is unclear whether biosimilar manufacturers will have the same level of patient support programs as the reference drug companies.”
For that reason, physicians should also inform patients about the robust patient assistance and copay assistance programs many reference drug manufacturers offer, she said.
Biosimilars 101: Familiarizing patients
Safety and ease of use are other common concerns about biosimilars. Patients may ask if the application is different, or why it’s advantageous to switch to a biosimilar, Dr. Awsare said.
Sometimes the syringe or injector for a biosimilar might look different from that of the originator drug, he said.
Anecdotally, Dr. Fabbro has heard stories of patients having injection reactions that they did not experience with the reference drug or having a disease flare-up after starting a biosimilar.
As is the case with reference products, in their conversations with patients, clinicians should address the adverse event profile of biosimilars, offering data points from published studies and clinical guidelines that support the use of these products. “There should be an emphasis on patient education around efficacy and any side effects, and how the profile of the reference product compares with a proposed biosimilar,” Dr. Kaul suggested.
When Dr. Snow discusses biosimilars and generics, “I make sure to share this in an understandable way based on the patient’s scientific background, or lack thereof,” he said. If there is enough time, he also discusses how European- and U.S.-sourced biologics are slightly different.
Pharmacists should tell patients to expect the same clinical outcomes from a biosimilar, Dr. Popovian said. However, if they have any reduction in efficacy or potential safety concerns, they should communicate with their physician or pharmacist immediately.
In Dr. Regueiro’s practice, a pharmacist specializing in inflammatory bowel disease often has a one-on-one meeting with patients to educate and answer questions. “Additionally, we provide them the Crohn’s and Colitis Foundation web link on biosimilars,” said Dr. Regueiro.
A village approach to education
When biosimilars first came out, there were no formal education materials, Dr. Awsare said. Kaiser Permanente decided to create its own educational materials, not just for patients but also to help educate its primary care doctors; the rheumatologists, dermatologists, and gastroenterologists using the biosimilars; the nurses infusing patients; and the pharmacists preparing the biosimilars.
The health system also has a different approach to choosing medication. Instead of having an insurance company or PBM decide what’s in the formulary, clinicians work with the pharmacists at Kaiser to look at clinical evidence and decide which biosimilar to use. Most of its plans also provide lower copays to patients when they use the biosimilar.
This was the approach for Humira biosimilars, Dr. Awsare said. Eight will be on the market in 2023. “Our rheumatologists, dermatologists, and gastroenterologists looked at the data from Europe, looked at some real-world evidence, and then said: ‘We think this one’s going to be the best one for our patients.’ ”
Having clinicians choose the biosimilar instead of a health plan makes it a lot easier to have conversations with patients, he said. “Once we’ve moved that market share to that particular biosimilar, we give our physicians the time to have those discussions.”
Clinical pharmacists also provide educational support, offering guidance on issues such as side effects, as patients transition to the biosimilar. “We like to use the word ‘transition’ because it’s essentially the same biologic. So, you’re not actually switching,” Dr. Awsare said.
No consensus on interchangeability
Whether the conversation on interchangeability will affect patient conversations with physicians depends on who you ask.
If a biosimilar has an interchangeability designation, it means that the pharmacist can substitute it without the intervention of the clinician who prescribed the reference product. It does not relate to the quality, safety, or effectiveness of biosimilars or interchangeable biosimilar products, Dr. Popovian said.
The United States is the only country that has this designation. Even though it’s not identical to the originator drug, a biosimilar has the same clinical efficacy and safety profile. “So clinically, interchangeability is meaningless,” Dr. Awsare said.
In its report on biosimilars in the autoimmune category, CVS acknowledged that interchangeability was important but would not be a significant factor in driving adoption of biosimilars. However, in a Cardinal Health survey of 72 gastroenterologists, 38% cited the interchangeability of biosimilars as a top concern for adalimumab biosimilars, along with transitioning patients from Humira to a biosimilar (44%).
“Patient education regarding biosimilar safety, efficacy, and interchangeability appears paramount to the acceptance of these products, particularly for patients who are switched from a reference product,” Dr. Kaul noted in the Cardinal Health report.
Wherever supported by data, Dr. Kaul recommends incorporating biosimilar use and interchangeability into best practice guidelines going forward. “That will go a long way in disseminating the latest information on this topic and position this paradigm for increased adoption among providers.”
Some physicians like Dr. Snow aren’t that concerned with interchangeability. This hasn’t affected conversations with patients, he said. Multiple studies demonstrating the lack of antibody formation with multiple switches from different biosimilar drugs has eased his concern about multiple switches causing problems.
“Initially, there was a gap in demonstrating the long-term effect of multiple switches on antibody production and drug effectiveness. That gap has started to close as more data from Europe’s experience with biosimilars becomes available,” Dr. Snow said.
Resources for physicians, patients
The federal government has taken steps to advance biosimilars education and adoption. In 2021, President Biden signed the Advancing Education on Biosimilars Act into law, which directs the FDA to develop or improve continuing education programs that address prescribing of biosimilars and biological products.
The FDA provides educational materials on its website, including a comprehensive curriculum toolkit. The Accreditation Council for Medical Affairs has also created an online 40-hour curriculum for health care professionals called the Board-Certified Biologics and Biosimilars Specialist Program.
Dr. Fabbro recommended patients use the FDA page Biosimilar Basics for Patients to educate themselves on biosimilars. The Global Healthy Living Foundation’s podcast, Breaking Down Biosimilars, is another free resource for patients.
“While much has changed, the continued need for multistakeholder education, awareness, and dedicated research remains even more important as we expand into newer therapeutic areas and classes,” wrote the authors of the Cardinal Health report.
Help patients understand biologics and biosimilars by using AGA resources for providers and patients available at gastro.org/biosimilars.
Dr. Regueiro is on advisory boards and consults for AbbVie, Janssen, UCB, Takeda, Pfizer, Bristol-Myers Squibb, Organon, Amgen, Genentech, Gilead, Salix, Prometheus, Lilly, Celgene, TARGET PharmaSolutions, Trellis, and Boehringer Ingelheim. Dr. Fabbro is a principal investigator for Castle Biosciences, on the speakers bureau for Valchlor, and on the advisory boards of Janssen and Bristol-Myers Squibb. Dr. Popovian, Dr. Snow, Dr. Awsare, and Dr. Kaul had no disclosures.
A version of this article originally appeared on Medscape.com.
Rheumatologist Marcus Snow, MD, is comfortable with prescribing biosimilars as a first-line, first-time biologic, and discussing them with patients.
“If a biosimilar is on the market, it has gone through rigorous study proving its effectiveness and equivalence to a bio-originator,” said Dr. Snow, a rheumatologist with the University of Nebraska Medical Center, Omaha, and chair of the American College of Rheumatology’s Committee on Rheumatologic Care.
The formulary makes a big difference in the conversation about options, he said. “The formularies dictate what we can prescribe. It may not be appropriate, but it is reality. The cost of biologics for a patient without insurance coverage makes it impossible to afford.”
He will often tell patients that he’ll fight any changes or formulary restrictions he does not agree with. “However, when I see patients in follow-up, even if there is no known change on the horizon, I may bring up biosimilars when we have a moment to chat about them to familiarize them with what may happen in the future.”
The need for patient education on biosimilars presents a barrier to realizing their potential to save money and expand choice, noted Cardinal Health in its 2023 biosimilars report. Of 103 rheumatologists who responded to a Cardinal Health survey, 85% agreed that patient education was important. But those conversations can take an uncomfortable turn if the patient pushes back against taking a biosimilar owing to cost or safety concerns.
It’s not uncommon for a patient to express some anxiety about biosimilars, especially if they’re doing well on a current treatment plan. Most patients do not want any changes that may lead to worsening disease control, Dr. Snow said.
Patients and physicians alike often don’t understand the mechanics of biosimilars. “There’s a lot of misinformation about this,” said Sameer Awsare, MD, an associate executive director for The Permanente Medical Group in Campbell, Calif. Patients should know that a biosimilar will be as clinically efficacious as the medicine they’ve been on, with the same safety profiles, said Dr. Awsare, who works with Kaiser Permanente’s pharmacy partners on biosimilars.
Insurance often drives the conversation
The global anti-inflammatory biologics market is anticipated to reach $150 billion by 2027, according to a recent CVS report. As of March 2023, the Food and Drug Administration had approved 40 biosimilars to 11 different reference products. There are 28 on the U.S. market and 100 more in development. Projected to save more than $180 billion over the next 5 years, they are anticipated to expand choice and drive competition.
Rheumatologists, dermatologists, and gastroenterologists are frequent prescribers, although their choices for immune-mediated inflammatory diseases are limited to tumor necrosis factor inhibitors (infliximab [Remicade] originator and adalimumab [Humira] originator) and anti-CD20 agents, such as rituximab (Rituxan) originator.
Benefit design or formulary usually dictates what medicine a patient receives. “Because of significantly higher out-of-pocket cost or formulary positioning, patients may end up with a generic or a biosimilar instead of a brand-name medicine or branded biologic,” said Robert Popovian, PharmD, MS, chief science policy officer of the Global Healthy Living Foundation.
Insurers rarely offer both Remicade and biosimilar infliximab, allowing the doctor to choose, said Miguel Regueiro, MD, chair of the Cleveland Clinic’s Digestive Disease & Surgery Institute, who prescribes infliximab biosimilars. Most often, the payer will choose the lower-cost biosimilar. “I am fine with the biosimilar, either as a new start or a switch from the reference product.”
However, the patient might feel differently. They can form an attachment to the reference medication if it has prevented severe illness. “They do not want to change, as they feel they are going on a ‘new’ medication that will not work as well,” Dr. Regueiro said.
This is where the education comes in: to reassure patients that a biosimilar will work just as well as the reference product. “For patients who have done well for years on a biologic, more time needs to be spent reassuring them and answering questions,” compared with a patient just starting on a biosimilar, he advised.
But not all physicians are quick to prescribe biosimilars.
Especially with psoriasis, which has so many strong options for reference drugs, a switch may be hard to justify, said dermatologist Stephanie K. Fabbro, MD, assistant professor at Northeast Ohio Medical University, Rootstown. “If I have a preference, I would rather switch a patient to a drug from a different class without a biosimilar option to reduce the possibility of pushback.”
Dr. Fabbro, part of the core faculty in the Riverside Methodist Hospital Dermatology Residency Program in Columbus, will share data from clinical trials and postmarket surveillance with patients to support her decision.
Conversations about cost
Patients may also push back if they don’t save money when switching to a biosimilar. “This dilemma raises the question of who is profiting when a biosimilar is dispensed,” Dr. Popovian said. Insurers and pharmacy benefit managers (PBMs) that take additional concessions from biopharmaceutical manufacturers in the form of rebates and fees will often pocket this money as profit instead of passing savings back to the patient to help reduce their out-of-pocket requirement, he added.
If an originator biologic and a biosimilar are available, “as a pharmacist, I will choose the medicine that will incur the lowest out-of-pocket cost for the patient,” Dr. Popovian said.
Discussing cost – and who dictates which biosimilar is on the formulary – is an important conversation to have with patients, said Vivek Kaul, MD, Segal-Watson Professor of Medicine at the University of Rochester (N.Y.) Medical Center.
Providing equivalent clinical efficacy while saving costs is the economic reality of biosimilars, Dr. Kaul said. Third-party payers regularly evaluate how to provide the same quality of care while saving money. Physicians and patients alike “must be mindful that as time goes on, if the science on biosimilars stays robust, if the adoption is more widespread and the cost-saving proposition turns out to be true, more formularies will be attracted to replacing the reference product with the biosimilar counterpart.”
Providers and patients can weigh the options if a formulary suddenly switches to a biosimilar, Dr. Kaul continued. “You can accept the novel product on the formulary or may have to face out-of-pocket expenses as a patient.” If providers and patients have concerns about the biosimilar, they can always appeal if there’s solid scientific evidence that supports reverting back to the reference product.
“If you think the biosimilar is equally efficacious, comes at a lower cost, and is right for the patient, then the providers should tell the patient that,” he added.
Some studies have questioned whether the biosimilars will save money, compared with the reference drug, Dr. Fabbro noted. Medicare, for example, may pay only for a certain percentage of an approved biosimilar, saddling the patient with a monthly copay costing thousands of dollars. “It is unclear whether biosimilar manufacturers will have the same level of patient support programs as the reference drug companies.”
For that reason, physicians should also inform patients about the robust patient assistance and copay assistance programs many reference drug manufacturers offer, she said.
Biosimilars 101: Familiarizing patients
Safety and ease of use are other common concerns about biosimilars. Patients may ask if the application is different, or why it’s advantageous to switch to a biosimilar, Dr. Awsare said.
Sometimes the syringe or injector for a biosimilar might look different from that of the originator drug, he said.
Anecdotally, Dr. Fabbro has heard stories of patients having injection reactions that they did not experience with the reference drug or having a disease flare-up after starting a biosimilar.
As is the case with reference products, in their conversations with patients, clinicians should address the adverse event profile of biosimilars, offering data points from published studies and clinical guidelines that support the use of these products. “There should be an emphasis on patient education around efficacy and any side effects, and how the profile of the reference product compares with a proposed biosimilar,” Dr. Kaul suggested.
When Dr. Snow discusses biosimilars and generics, “I make sure to share this in an understandable way based on the patient’s scientific background, or lack thereof,” he said. If there is enough time, he also discusses how European- and U.S.-sourced biologics are slightly different.
Pharmacists should tell patients to expect the same clinical outcomes from a biosimilar, Dr. Popovian said. However, if they have any reduction in efficacy or potential safety concerns, they should communicate with their physician or pharmacist immediately.
In Dr. Regueiro’s practice, a pharmacist specializing in inflammatory bowel disease often has a one-on-one meeting with patients to educate and answer questions. “Additionally, we provide them the Crohn’s and Colitis Foundation web link on biosimilars,” said Dr. Regueiro.
A village approach to education
When biosimilars first came out, there were no formal education materials, Dr. Awsare said. Kaiser Permanente decided to create its own educational materials, not just for patients but also to help educate its primary care doctors; the rheumatologists, dermatologists, and gastroenterologists using the biosimilars; the nurses infusing patients; and the pharmacists preparing the biosimilars.
The health system also has a different approach to choosing medication. Instead of having an insurance company or PBM decide what’s in the formulary, clinicians work with the pharmacists at Kaiser to look at clinical evidence and decide which biosimilar to use. Most of its plans also provide lower copays to patients when they use the biosimilar.
This was the approach for Humira biosimilars, Dr. Awsare said. Eight will be on the market in 2023. “Our rheumatologists, dermatologists, and gastroenterologists looked at the data from Europe, looked at some real-world evidence, and then said: ‘We think this one’s going to be the best one for our patients.’ ”
Having clinicians choose the biosimilar instead of a health plan makes it a lot easier to have conversations with patients, he said. “Once we’ve moved that market share to that particular biosimilar, we give our physicians the time to have those discussions.”
Clinical pharmacists also provide educational support, offering guidance on issues such as side effects, as patients transition to the biosimilar. “We like to use the word ‘transition’ because it’s essentially the same biologic. So, you’re not actually switching,” Dr. Awsare said.
No consensus on interchangeability
Whether the conversation on interchangeability will affect patient conversations with physicians depends on who you ask.
If a biosimilar has an interchangeability designation, it means that the pharmacist can substitute it without the intervention of the clinician who prescribed the reference product. It does not relate to the quality, safety, or effectiveness of biosimilars or interchangeable biosimilar products, Dr. Popovian said.
The United States is the only country that has this designation. Even though it’s not identical to the originator drug, a biosimilar has the same clinical efficacy and safety profile. “So clinically, interchangeability is meaningless,” Dr. Awsare said.
In its report on biosimilars in the autoimmune category, CVS acknowledged that interchangeability was important but would not be a significant factor in driving adoption of biosimilars. However, in a Cardinal Health survey of 72 gastroenterologists, 38% cited the interchangeability of biosimilars as a top concern for adalimumab biosimilars, along with transitioning patients from Humira to a biosimilar (44%).
“Patient education regarding biosimilar safety, efficacy, and interchangeability appears paramount to the acceptance of these products, particularly for patients who are switched from a reference product,” Dr. Kaul noted in the Cardinal Health report.
Wherever supported by data, Dr. Kaul recommends incorporating biosimilar use and interchangeability into best practice guidelines going forward. “That will go a long way in disseminating the latest information on this topic and position this paradigm for increased adoption among providers.”
Some physicians like Dr. Snow aren’t that concerned with interchangeability. This hasn’t affected conversations with patients, he said. Multiple studies demonstrating the lack of antibody formation with multiple switches from different biosimilar drugs has eased his concern about multiple switches causing problems.
“Initially, there was a gap in demonstrating the long-term effect of multiple switches on antibody production and drug effectiveness. That gap has started to close as more data from Europe’s experience with biosimilars becomes available,” Dr. Snow said.
Resources for physicians, patients
The federal government has taken steps to advance biosimilars education and adoption. In 2021, President Biden signed the Advancing Education on Biosimilars Act into law, which directs the FDA to develop or improve continuing education programs that address prescribing of biosimilars and biological products.
The FDA provides educational materials on its website, including a comprehensive curriculum toolkit. The Accreditation Council for Medical Affairs has also created an online 40-hour curriculum for health care professionals called the Board-Certified Biologics and Biosimilars Specialist Program.
Dr. Fabbro recommended patients use the FDA page Biosimilar Basics for Patients to educate themselves on biosimilars. The Global Healthy Living Foundation’s podcast, Breaking Down Biosimilars, is another free resource for patients.
“While much has changed, the continued need for multistakeholder education, awareness, and dedicated research remains even more important as we expand into newer therapeutic areas and classes,” wrote the authors of the Cardinal Health report.
Help patients understand biologics and biosimilars by using AGA resources for providers and patients available at gastro.org/biosimilars.
Dr. Regueiro is on advisory boards and consults for AbbVie, Janssen, UCB, Takeda, Pfizer, Bristol-Myers Squibb, Organon, Amgen, Genentech, Gilead, Salix, Prometheus, Lilly, Celgene, TARGET PharmaSolutions, Trellis, and Boehringer Ingelheim. Dr. Fabbro is a principal investigator for Castle Biosciences, on the speakers bureau for Valchlor, and on the advisory boards of Janssen and Bristol-Myers Squibb. Dr. Popovian, Dr. Snow, Dr. Awsare, and Dr. Kaul had no disclosures.
A version of this article originally appeared on Medscape.com.
FDA panels vote to modify isotretinoin iPLEDGE REMS
At a joint meeting of a drug for severe, nodular acne that is highly teratogenic.
The first vote was on whether to continue the 19-day lockout period for patients who can become pregnant and do not pick up their first prescription of isotretinoin within the 7-day prescription window. Those patients currently have to wait 19 days to get their second pregnancy test and receive the medication.
Most (17) of the 22 voting members voted not to continue the 19-day period; 4 voted to keep it; and 1 abstained. But there was no consensus on when the second pregnancy test should occur if the 19-day lockout is changed.
Ken Katz, MD, MSc, a dermatologist at Kaiser Permanente in San Francisco, was among those voting not to continue the 19-day lockout.
“I think this places an unduly high burden physically and psychologically on our patients. It seems arbitrary,” he said. “Likely we will miss some pregnancies; we are missing some already. But the burden is not matched by the benefit.”
The second question concerned patients who cannot become pregnant, and it asked when REMS should require that the prescriber document counseling the patient in the iPLEDGE system. The current requirement is monthly.
Listed options and the number of votes for each were:
- Only with the first prescription as part of patient enrollment (10)
- Monthly (1)
- Every 120 days (6)
- Some other frequency (5)
For this question too, while the members largely agreed the current monthly requirement is too burdensome, there was little agreement on what the most appropriate interval should be.
Lack of data
On both questions, several advisory committee members cited a lack of data on which they could base their decision.
On the documentation question, Megha Tollefson, MD, professor of dermatology at the Mayo Clinic, Rochester, Minn., said she voted for the fourth option (some other frequency) with the thought of yearly attestation.
“As a part of this, providers have to provide monthly counseling,” Dr. Tollefson said. “This is just a documentation requirement in the iPLEDGE system. I think most prescribers do document their monthly counseling in their own medical records. I would say it would be okay not to redocument that in iPLEDGE.”
The two votes came at the end of the second day of a joint meeting of the FDA’s Drug Safety and Risk Management Advisory Committee and Dermatologic and Ophthalmic Drugs Advisory Committee in which experts addressed ways to improve the iPLEDGE REMS for isotretinoin. A transition to a new platform for the iPLEDGE program caused chaos after its rollout at the end of 2021, resulting in extensive delays and denial of prescriptions.
The committees sought to balance reducing burden with maintaining safety and preventing fetal exposures to isotretinoin.
They were also tasked with discussing other REMS requirements without taking a vote on each topic.
Among those topics was whether home pregnancy tests, allowed during the COVID-19 public health emergency, should continue to be allowed. Most who spoke to the issue agreed that home tests should continue in an effort to increase access and decrease burden. Members suggested safeguards against falsified results that have been documented, including assigning names and barcodes to the test results and uploading the verification to the iPLEDGE website.
The advisory committees also discussed recommendations to encourage more participation in the iPLEDGE Pregnancy Registry.
The advisory committees’ recommendations to the FDA are nonbinding, but the FDA generally follows the recommendations of advisory panels.
A version of this article first appeared on Medscape.com.
At a joint meeting of a drug for severe, nodular acne that is highly teratogenic.
The first vote was on whether to continue the 19-day lockout period for patients who can become pregnant and do not pick up their first prescription of isotretinoin within the 7-day prescription window. Those patients currently have to wait 19 days to get their second pregnancy test and receive the medication.
Most (17) of the 22 voting members voted not to continue the 19-day period; 4 voted to keep it; and 1 abstained. But there was no consensus on when the second pregnancy test should occur if the 19-day lockout is changed.
Ken Katz, MD, MSc, a dermatologist at Kaiser Permanente in San Francisco, was among those voting not to continue the 19-day lockout.
“I think this places an unduly high burden physically and psychologically on our patients. It seems arbitrary,” he said. “Likely we will miss some pregnancies; we are missing some already. But the burden is not matched by the benefit.”
The second question concerned patients who cannot become pregnant, and it asked when REMS should require that the prescriber document counseling the patient in the iPLEDGE system. The current requirement is monthly.
Listed options and the number of votes for each were:
- Only with the first prescription as part of patient enrollment (10)
- Monthly (1)
- Every 120 days (6)
- Some other frequency (5)
For this question too, while the members largely agreed the current monthly requirement is too burdensome, there was little agreement on what the most appropriate interval should be.
Lack of data
On both questions, several advisory committee members cited a lack of data on which they could base their decision.
On the documentation question, Megha Tollefson, MD, professor of dermatology at the Mayo Clinic, Rochester, Minn., said she voted for the fourth option (some other frequency) with the thought of yearly attestation.
“As a part of this, providers have to provide monthly counseling,” Dr. Tollefson said. “This is just a documentation requirement in the iPLEDGE system. I think most prescribers do document their monthly counseling in their own medical records. I would say it would be okay not to redocument that in iPLEDGE.”
The two votes came at the end of the second day of a joint meeting of the FDA’s Drug Safety and Risk Management Advisory Committee and Dermatologic and Ophthalmic Drugs Advisory Committee in which experts addressed ways to improve the iPLEDGE REMS for isotretinoin. A transition to a new platform for the iPLEDGE program caused chaos after its rollout at the end of 2021, resulting in extensive delays and denial of prescriptions.
The committees sought to balance reducing burden with maintaining safety and preventing fetal exposures to isotretinoin.
They were also tasked with discussing other REMS requirements without taking a vote on each topic.
Among those topics was whether home pregnancy tests, allowed during the COVID-19 public health emergency, should continue to be allowed. Most who spoke to the issue agreed that home tests should continue in an effort to increase access and decrease burden. Members suggested safeguards against falsified results that have been documented, including assigning names and barcodes to the test results and uploading the verification to the iPLEDGE website.
The advisory committees also discussed recommendations to encourage more participation in the iPLEDGE Pregnancy Registry.
The advisory committees’ recommendations to the FDA are nonbinding, but the FDA generally follows the recommendations of advisory panels.
A version of this article first appeared on Medscape.com.
At a joint meeting of a drug for severe, nodular acne that is highly teratogenic.
The first vote was on whether to continue the 19-day lockout period for patients who can become pregnant and do not pick up their first prescription of isotretinoin within the 7-day prescription window. Those patients currently have to wait 19 days to get their second pregnancy test and receive the medication.
Most (17) of the 22 voting members voted not to continue the 19-day period; 4 voted to keep it; and 1 abstained. But there was no consensus on when the second pregnancy test should occur if the 19-day lockout is changed.
Ken Katz, MD, MSc, a dermatologist at Kaiser Permanente in San Francisco, was among those voting not to continue the 19-day lockout.
“I think this places an unduly high burden physically and psychologically on our patients. It seems arbitrary,” he said. “Likely we will miss some pregnancies; we are missing some already. But the burden is not matched by the benefit.”
The second question concerned patients who cannot become pregnant, and it asked when REMS should require that the prescriber document counseling the patient in the iPLEDGE system. The current requirement is monthly.
Listed options and the number of votes for each were:
- Only with the first prescription as part of patient enrollment (10)
- Monthly (1)
- Every 120 days (6)
- Some other frequency (5)
For this question too, while the members largely agreed the current monthly requirement is too burdensome, there was little agreement on what the most appropriate interval should be.
Lack of data
On both questions, several advisory committee members cited a lack of data on which they could base their decision.
On the documentation question, Megha Tollefson, MD, professor of dermatology at the Mayo Clinic, Rochester, Minn., said she voted for the fourth option (some other frequency) with the thought of yearly attestation.
“As a part of this, providers have to provide monthly counseling,” Dr. Tollefson said. “This is just a documentation requirement in the iPLEDGE system. I think most prescribers do document their monthly counseling in their own medical records. I would say it would be okay not to redocument that in iPLEDGE.”
The two votes came at the end of the second day of a joint meeting of the FDA’s Drug Safety and Risk Management Advisory Committee and Dermatologic and Ophthalmic Drugs Advisory Committee in which experts addressed ways to improve the iPLEDGE REMS for isotretinoin. A transition to a new platform for the iPLEDGE program caused chaos after its rollout at the end of 2021, resulting in extensive delays and denial of prescriptions.
The committees sought to balance reducing burden with maintaining safety and preventing fetal exposures to isotretinoin.
They were also tasked with discussing other REMS requirements without taking a vote on each topic.
Among those topics was whether home pregnancy tests, allowed during the COVID-19 public health emergency, should continue to be allowed. Most who spoke to the issue agreed that home tests should continue in an effort to increase access and decrease burden. Members suggested safeguards against falsified results that have been documented, including assigning names and barcodes to the test results and uploading the verification to the iPLEDGE website.
The advisory committees also discussed recommendations to encourage more participation in the iPLEDGE Pregnancy Registry.
The advisory committees’ recommendations to the FDA are nonbinding, but the FDA generally follows the recommendations of advisory panels.
A version of this article first appeared on Medscape.com.
Limited treatment options exist for brittle nail syndrome
NEW ORLEANS – .
“The mainstay of treatment is irritant avoidance and moisturization,” Shari R. Lipner, MD, PhD, associate professor of clinical dermatology and director of the nail division at Weill Cornell Medicine, New York, said at the annual meeting of the American Academy of Dermatology. “This works well if patients are religious about doing it.”

Brittle nail syndrome affects about 20% of adults, she said, and is more common in females, particularly those older than age 50. Most cases are idiopathic, but some are secondary to dermatologic diseases including nail psoriasis and nail lichen planus, and systemic diseases such as hyperthyroidism and hypothyroidism. They are more common in patients in certain occupations such as carpentry. “The pathogenesis is poorly understood but is thought to be due to weakened intercellular keratinocyte bridges, decreased cholesterol sulphate in the nail plate, and reduced water content in the nail plate,” Dr. Lipner said.
Key clinical findings include onychoschizia (peeling of the nail plate), onychorrhexis (an increase in the longitudinal ridges and furrows, sometimes leading to splitting), and superficial granulation of keratin. Treatment involves general measures. “You want to treat the underlying cause and recommend that the patient avoid water and irritant exposure,” she said. Her general instructions for affected patients are to wear latex gloves for wet work and cotton gloves for dry work, avoid triclosan-based hand sanitizers, avoid nail cosmetics, minimize nail trauma, and foster moisturization.“It’s important to give these instructions verbally and in written form,” she said. “In our practice, we designed a QR code that links to our patient handout.”
According to Dr. Lipner, the promotion of vitamins and supplements such as biotin, vitamin D, amino acids, and chromium for treating brittle nail syndrome is rampant on the Internet and on social media, but no rigorously designed clinical trials have shown efficacy for any of them. “Very few people are deficient in biotin, except for those with inherited enzyme deficiencies,” and most people “can get all the biotin they need from a regular diet,” she said.
The initial rationale for using biotin for nails comes from the veterinary literature, she continued. In the 1940s, chickens with biotin deficiency developed fissures in their feet and parrot-like beaks. In the 1970s, pigs with biotin deficiency developed friable hooves, which was corrected with biotin supplementation. “By the 1980s it was standard practice to supplement the feet of pigs with biotin,” she said.
In a human trial from 1989, German researchers enrolled 71 patients with brittle nail syndrome who took oral biotin, 2.5 mg daily. Of the 45 patients evaluated, 41 (91%) showed improvement in firmness and hardness of the fingernails over the course of 5.5 months, but there was no good control group, Dr. Lipner said. In a follow-up study, the same German researchers used scanning electron microscopy to evaluate 22 patients with brittle nails who took oral biotin 2.5 mg daily and compared them with 10 patients with normal nails who did not take biotin. They found a 25% increase in nail plate thickness in the biotin group and onychoschizia resolved in 50% of patients who received biotin. “But again, there was no good control group,” Dr. Lipner said.
In a third study on the topic, researchers surveyed 46 patients who presented with onychorrhexis and/or onychoschizia on clinical exam and took 2.5 mg of biotin daily. Of the 35 survey respondents, 63% subjectively reported improvement in their nails at a mean of 2 months. “This is where we are today: There have been studies of only 80 patients that were done 25 years ago,” Dr. Lipner said. “That’s all of our evidence for biotin for the treatment of brittle nail syndrome.”
FDA warning about biotin
Additional cause for concern, she continued, is the safety communication issued by the FDA in 2017, stating that the use of biotin may interfere with certain lab tests such as thyroid tests and cardiac enzymes, in some cases leading to death. The safety communication was updated in 2019.
In 2018, Dr. Lipner and colleagues administered an anonymous survey to 447 patients at their clinic asking about their use of biotin supplements. Of the 447 patients, 34% reported current use of biotin. Among biotin users, 7% were aware of the FDA warning, 29% of respondents reported that it was recommended by either a primary care physician or a dermatologist, and 56% underwent laboratory testing while taking biotin. “It’s our duty to warn our patients about the evidence for biotin for treating brittle nails, and about this interference on laboratory tests,” Dr. Lipner said.
Other treatment options for brittle nail syndrome include two lacquers that are available by prescription. One contains hydroxypropyl chitosan, Equisetum arvense, and methylsulphonylmethane; the other contains 16% poly-ureaurethane, but has not been well studied. “These products can be very expensive if not covered by insurance,” Dr. Lipner said.
As an alternative, she recommends Nail Tek CITRA 2 Nail Strengthener, which is available for less than $10 from Walmart and other retailers.
Cyclosporine emulsion also has been studied for brittle nail syndrome, but results to date have been underwhelming. Dr. Lipner and colleagues are exploring the effect of platelet rich plasma for treating brittle nails on the premise that it will improve nail growth and promote healing, in a 16-week trial that has enrolled 10 patients and includes both a Physician Global Improvement Assessment (PGIA) and a Physician Global Assessment (PGA) score. “Our data is being analyzed by three independent nail experts, and we hope to report the findings next year,” she said.
Dr. Lipner reported having no disclosures relevant to her presentation.
NEW ORLEANS – .
“The mainstay of treatment is irritant avoidance and moisturization,” Shari R. Lipner, MD, PhD, associate professor of clinical dermatology and director of the nail division at Weill Cornell Medicine, New York, said at the annual meeting of the American Academy of Dermatology. “This works well if patients are religious about doing it.”
Brittle nail syndrome affects about 20% of adults, she said, and is more common in females, particularly those older than age 50. Most cases are idiopathic, but some are secondary to dermatologic diseases including nail psoriasis and nail lichen planus, and systemic diseases such as hyperthyroidism and hypothyroidism. They are more common in patients in certain occupations such as carpentry. “The pathogenesis is poorly understood but is thought to be due to weakened intercellular keratinocyte bridges, decreased cholesterol sulphate in the nail plate, and reduced water content in the nail plate,” Dr. Lipner said.
Key clinical findings include onychoschizia (peeling of the nail plate), onychorrhexis (an increase in the longitudinal ridges and furrows, sometimes leading to splitting), and superficial granulation of keratin. Treatment involves general measures. “You want to treat the underlying cause and recommend that the patient avoid water and irritant exposure,” she said. Her general instructions for affected patients are to wear latex gloves for wet work and cotton gloves for dry work, avoid triclosan-based hand sanitizers, avoid nail cosmetics, minimize nail trauma, and foster moisturization.“It’s important to give these instructions verbally and in written form,” she said. “In our practice, we designed a QR code that links to our patient handout.”
According to Dr. Lipner, the promotion of vitamins and supplements such as biotin, vitamin D, amino acids, and chromium for treating brittle nail syndrome is rampant on the Internet and on social media, but no rigorously designed clinical trials have shown efficacy for any of them. “Very few people are deficient in biotin, except for those with inherited enzyme deficiencies,” and most people “can get all the biotin they need from a regular diet,” she said.
The initial rationale for using biotin for nails comes from the veterinary literature, she continued. In the 1940s, chickens with biotin deficiency developed fissures in their feet and parrot-like beaks. In the 1970s, pigs with biotin deficiency developed friable hooves, which was corrected with biotin supplementation. “By the 1980s it was standard practice to supplement the feet of pigs with biotin,” she said.
In a human trial from 1989, German researchers enrolled 71 patients with brittle nail syndrome who took oral biotin, 2.5 mg daily. Of the 45 patients evaluated, 41 (91%) showed improvement in firmness and hardness of the fingernails over the course of 5.5 months, but there was no good control group, Dr. Lipner said. In a follow-up study, the same German researchers used scanning electron microscopy to evaluate 22 patients with brittle nails who took oral biotin 2.5 mg daily and compared them with 10 patients with normal nails who did not take biotin. They found a 25% increase in nail plate thickness in the biotin group and onychoschizia resolved in 50% of patients who received biotin. “But again, there was no good control group,” Dr. Lipner said.
In a third study on the topic, researchers surveyed 46 patients who presented with onychorrhexis and/or onychoschizia on clinical exam and took 2.5 mg of biotin daily. Of the 35 survey respondents, 63% subjectively reported improvement in their nails at a mean of 2 months. “This is where we are today: There have been studies of only 80 patients that were done 25 years ago,” Dr. Lipner said. “That’s all of our evidence for biotin for the treatment of brittle nail syndrome.”
FDA warning about biotin
Additional cause for concern, she continued, is the safety communication issued by the FDA in 2017, stating that the use of biotin may interfere with certain lab tests such as thyroid tests and cardiac enzymes, in some cases leading to death. The safety communication was updated in 2019.
In 2018, Dr. Lipner and colleagues administered an anonymous survey to 447 patients at their clinic asking about their use of biotin supplements. Of the 447 patients, 34% reported current use of biotin. Among biotin users, 7% were aware of the FDA warning, 29% of respondents reported that it was recommended by either a primary care physician or a dermatologist, and 56% underwent laboratory testing while taking biotin. “It’s our duty to warn our patients about the evidence for biotin for treating brittle nails, and about this interference on laboratory tests,” Dr. Lipner said.
Other treatment options for brittle nail syndrome include two lacquers that are available by prescription. One contains hydroxypropyl chitosan, Equisetum arvense, and methylsulphonylmethane; the other contains 16% poly-ureaurethane, but has not been well studied. “These products can be very expensive if not covered by insurance,” Dr. Lipner said.
As an alternative, she recommends Nail Tek CITRA 2 Nail Strengthener, which is available for less than $10 from Walmart and other retailers.
Cyclosporine emulsion also has been studied for brittle nail syndrome, but results to date have been underwhelming. Dr. Lipner and colleagues are exploring the effect of platelet rich plasma for treating brittle nails on the premise that it will improve nail growth and promote healing, in a 16-week trial that has enrolled 10 patients and includes both a Physician Global Improvement Assessment (PGIA) and a Physician Global Assessment (PGA) score. “Our data is being analyzed by three independent nail experts, and we hope to report the findings next year,” she said.
Dr. Lipner reported having no disclosures relevant to her presentation.
NEW ORLEANS – .
“The mainstay of treatment is irritant avoidance and moisturization,” Shari R. Lipner, MD, PhD, associate professor of clinical dermatology and director of the nail division at Weill Cornell Medicine, New York, said at the annual meeting of the American Academy of Dermatology. “This works well if patients are religious about doing it.”
Brittle nail syndrome affects about 20% of adults, she said, and is more common in females, particularly those older than age 50. Most cases are idiopathic, but some are secondary to dermatologic diseases including nail psoriasis and nail lichen planus, and systemic diseases such as hyperthyroidism and hypothyroidism. They are more common in patients in certain occupations such as carpentry. “The pathogenesis is poorly understood but is thought to be due to weakened intercellular keratinocyte bridges, decreased cholesterol sulphate in the nail plate, and reduced water content in the nail plate,” Dr. Lipner said.
Key clinical findings include onychoschizia (peeling of the nail plate), onychorrhexis (an increase in the longitudinal ridges and furrows, sometimes leading to splitting), and superficial granulation of keratin. Treatment involves general measures. “You want to treat the underlying cause and recommend that the patient avoid water and irritant exposure,” she said. Her general instructions for affected patients are to wear latex gloves for wet work and cotton gloves for dry work, avoid triclosan-based hand sanitizers, avoid nail cosmetics, minimize nail trauma, and foster moisturization.“It’s important to give these instructions verbally and in written form,” she said. “In our practice, we designed a QR code that links to our patient handout.”
According to Dr. Lipner, the promotion of vitamins and supplements such as biotin, vitamin D, amino acids, and chromium for treating brittle nail syndrome is rampant on the Internet and on social media, but no rigorously designed clinical trials have shown efficacy for any of them. “Very few people are deficient in biotin, except for those with inherited enzyme deficiencies,” and most people “can get all the biotin they need from a regular diet,” she said.
The initial rationale for using biotin for nails comes from the veterinary literature, she continued. In the 1940s, chickens with biotin deficiency developed fissures in their feet and parrot-like beaks. In the 1970s, pigs with biotin deficiency developed friable hooves, which was corrected with biotin supplementation. “By the 1980s it was standard practice to supplement the feet of pigs with biotin,” she said.
In a human trial from 1989, German researchers enrolled 71 patients with brittle nail syndrome who took oral biotin, 2.5 mg daily. Of the 45 patients evaluated, 41 (91%) showed improvement in firmness and hardness of the fingernails over the course of 5.5 months, but there was no good control group, Dr. Lipner said. In a follow-up study, the same German researchers used scanning electron microscopy to evaluate 22 patients with brittle nails who took oral biotin 2.5 mg daily and compared them with 10 patients with normal nails who did not take biotin. They found a 25% increase in nail plate thickness in the biotin group and onychoschizia resolved in 50% of patients who received biotin. “But again, there was no good control group,” Dr. Lipner said.
In a third study on the topic, researchers surveyed 46 patients who presented with onychorrhexis and/or onychoschizia on clinical exam and took 2.5 mg of biotin daily. Of the 35 survey respondents, 63% subjectively reported improvement in their nails at a mean of 2 months. “This is where we are today: There have been studies of only 80 patients that were done 25 years ago,” Dr. Lipner said. “That’s all of our evidence for biotin for the treatment of brittle nail syndrome.”
FDA warning about biotin
Additional cause for concern, she continued, is the safety communication issued by the FDA in 2017, stating that the use of biotin may interfere with certain lab tests such as thyroid tests and cardiac enzymes, in some cases leading to death. The safety communication was updated in 2019.
In 2018, Dr. Lipner and colleagues administered an anonymous survey to 447 patients at their clinic asking about their use of biotin supplements. Of the 447 patients, 34% reported current use of biotin. Among biotin users, 7% were aware of the FDA warning, 29% of respondents reported that it was recommended by either a primary care physician or a dermatologist, and 56% underwent laboratory testing while taking biotin. “It’s our duty to warn our patients about the evidence for biotin for treating brittle nails, and about this interference on laboratory tests,” Dr. Lipner said.
Other treatment options for brittle nail syndrome include two lacquers that are available by prescription. One contains hydroxypropyl chitosan, Equisetum arvense, and methylsulphonylmethane; the other contains 16% poly-ureaurethane, but has not been well studied. “These products can be very expensive if not covered by insurance,” Dr. Lipner said.
As an alternative, she recommends Nail Tek CITRA 2 Nail Strengthener, which is available for less than $10 from Walmart and other retailers.
Cyclosporine emulsion also has been studied for brittle nail syndrome, but results to date have been underwhelming. Dr. Lipner and colleagues are exploring the effect of platelet rich plasma for treating brittle nails on the premise that it will improve nail growth and promote healing, in a 16-week trial that has enrolled 10 patients and includes both a Physician Global Improvement Assessment (PGIA) and a Physician Global Assessment (PGA) score. “Our data is being analyzed by three independent nail experts, and we hope to report the findings next year,” she said.
Dr. Lipner reported having no disclosures relevant to her presentation.
AT AAD 2023
FDA Advisory panels consider easing isotretinoin requirements
for patients, pharmacies, and prescribers.
Isotretinoin, previously called Accutane, is marketed as Absorica, Absorica LD, Claravis, Amnesteem, Myorisan, and Zenatane.
In a joint meeting of the FDA’s Drug Safety and Risk Management Advisory Committee and Dermatologic and Ophthalmic Drugs Advisory Committee, experts addressed ways to improve the modified iPLEDGE Risk Evaluation and Mitigation Strategy (iPLEDGE REMS) for isotretinoin that caused chaos after its rollout at the end of 2021.
In January 2022, problems were multiplying with the program for clinicians, pharmacists, and patients, causing extensive delays and prescription denials. In response, the FDA said it would continue to meet with the Isotretinoin Products Manufacturers Group (IPMG) to resolve problems.
March 28 was the first day of a 2-day meeting addressing what can be done to reduce burden with the iPLEDGE REMS while maintaining safety and preventing fetal exposure to the drug.
Key areas of concern
The meeting focused on several key areas.
The 19-day lockout period
The lockout is a current restriction for patients who can become pregnant and do not pick up their first prescription of isotretinoin within the specified 7-day prescription window. Currently, those who miss the window must wait 19 days from the date of the first pregnancy test to take an additional pregnancy test to be eligible to receive the drug.
Lindsey Crist, PharmD, a risk management analyst for the FDA, who presented the FDA review committee’s analysis, acknowledged that the lockout period causes delays in treatment and adds frustration and costs.
She said it’s important to remember that the lockout applies only to the first prescription. “It’s intended as an additional layer of screening to detect pregnancy,” she said.
“At least 12 pregnancies have been identified during the 19-day lockout from March 2017–September of 2022,” she noted.
The FDA is looking to the advisory committee to provide recommendations on whether the lockout period should be changed.
Home testing
During the pandemic, iPLEDGE rules have been relaxed from having a pregnancy test done only at a Clinical Laboratory Improvement Amendments–certified laboratory and home pregnancy tests have been allowed. The question now is whether home tests should continue to be allowed.
Ms. Crist said that the FDA’s review committee recommends ending the allowance of home tests, citing insufficient data on use and the discovery of instances of falsification of pregnancy tests.
“One study at an academic medical center reviewed the medical records of 89 patients who used home pregnancy tests while taking isotretinoin during the public health emergency. It found that 15.7% submitted falsified pregnancy test results,” she said.
Ms. Crist added, however, that the review committee recommends allowing the tests to be done in a provider’s office as an alternative.
Documenting counseling patients who cannot get pregnant
Currently, this documentation must be done monthly, primarily to counsel patients against drug sharing or giving blood. Proposed changes include extending the intervals for attestation or eliminating it to reduce burden on clinicians.
IPMG representative Gregory Wedin, PharmD, pharmacovigilance and risk management director for Upsher-Smith Laboratories, said, “while we cannot support eliminating or extending the confirmation interval to a year, the [iPLEDGE] sponsors are agreeable [to] a 120-day confirmation interval.”
He said that while extending to 120 days would reduce burden on prescribers, it comes with risk in reducing oversight by a certified iPLEDGE prescriber and potentially increasing the risk for drug sharing.
“A patient may be more likely to share their drug with another person the further along with therapy they get as their condition improves,” Mr. Wedin said.
On March 29, the panel will hear more recommendations for and against modifications to iPLEDGE REMS and will vote on select modifications at the end of the meeting.
A version of this article first appeared on Medscape.com.
for patients, pharmacies, and prescribers.
Isotretinoin, previously called Accutane, is marketed as Absorica, Absorica LD, Claravis, Amnesteem, Myorisan, and Zenatane.
In a joint meeting of the FDA’s Drug Safety and Risk Management Advisory Committee and Dermatologic and Ophthalmic Drugs Advisory Committee, experts addressed ways to improve the modified iPLEDGE Risk Evaluation and Mitigation Strategy (iPLEDGE REMS) for isotretinoin that caused chaos after its rollout at the end of 2021.
In January 2022, problems were multiplying with the program for clinicians, pharmacists, and patients, causing extensive delays and prescription denials. In response, the FDA said it would continue to meet with the Isotretinoin Products Manufacturers Group (IPMG) to resolve problems.
March 28 was the first day of a 2-day meeting addressing what can be done to reduce burden with the iPLEDGE REMS while maintaining safety and preventing fetal exposure to the drug.
Key areas of concern
The meeting focused on several key areas.
The 19-day lockout period
The lockout is a current restriction for patients who can become pregnant and do not pick up their first prescription of isotretinoin within the specified 7-day prescription window. Currently, those who miss the window must wait 19 days from the date of the first pregnancy test to take an additional pregnancy test to be eligible to receive the drug.
Lindsey Crist, PharmD, a risk management analyst for the FDA, who presented the FDA review committee’s analysis, acknowledged that the lockout period causes delays in treatment and adds frustration and costs.
She said it’s important to remember that the lockout applies only to the first prescription. “It’s intended as an additional layer of screening to detect pregnancy,” she said.
“At least 12 pregnancies have been identified during the 19-day lockout from March 2017–September of 2022,” she noted.
The FDA is looking to the advisory committee to provide recommendations on whether the lockout period should be changed.
Home testing
During the pandemic, iPLEDGE rules have been relaxed from having a pregnancy test done only at a Clinical Laboratory Improvement Amendments–certified laboratory and home pregnancy tests have been allowed. The question now is whether home tests should continue to be allowed.
Ms. Crist said that the FDA’s review committee recommends ending the allowance of home tests, citing insufficient data on use and the discovery of instances of falsification of pregnancy tests.
“One study at an academic medical center reviewed the medical records of 89 patients who used home pregnancy tests while taking isotretinoin during the public health emergency. It found that 15.7% submitted falsified pregnancy test results,” she said.
Ms. Crist added, however, that the review committee recommends allowing the tests to be done in a provider’s office as an alternative.
Documenting counseling patients who cannot get pregnant
Currently, this documentation must be done monthly, primarily to counsel patients against drug sharing or giving blood. Proposed changes include extending the intervals for attestation or eliminating it to reduce burden on clinicians.
IPMG representative Gregory Wedin, PharmD, pharmacovigilance and risk management director for Upsher-Smith Laboratories, said, “while we cannot support eliminating or extending the confirmation interval to a year, the [iPLEDGE] sponsors are agreeable [to] a 120-day confirmation interval.”
He said that while extending to 120 days would reduce burden on prescribers, it comes with risk in reducing oversight by a certified iPLEDGE prescriber and potentially increasing the risk for drug sharing.
“A patient may be more likely to share their drug with another person the further along with therapy they get as their condition improves,” Mr. Wedin said.
On March 29, the panel will hear more recommendations for and against modifications to iPLEDGE REMS and will vote on select modifications at the end of the meeting.
A version of this article first appeared on Medscape.com.
for patients, pharmacies, and prescribers.
Isotretinoin, previously called Accutane, is marketed as Absorica, Absorica LD, Claravis, Amnesteem, Myorisan, and Zenatane.
In a joint meeting of the FDA’s Drug Safety and Risk Management Advisory Committee and Dermatologic and Ophthalmic Drugs Advisory Committee, experts addressed ways to improve the modified iPLEDGE Risk Evaluation and Mitigation Strategy (iPLEDGE REMS) for isotretinoin that caused chaos after its rollout at the end of 2021.
In January 2022, problems were multiplying with the program for clinicians, pharmacists, and patients, causing extensive delays and prescription denials. In response, the FDA said it would continue to meet with the Isotretinoin Products Manufacturers Group (IPMG) to resolve problems.
March 28 was the first day of a 2-day meeting addressing what can be done to reduce burden with the iPLEDGE REMS while maintaining safety and preventing fetal exposure to the drug.
Key areas of concern
The meeting focused on several key areas.
The 19-day lockout period
The lockout is a current restriction for patients who can become pregnant and do not pick up their first prescription of isotretinoin within the specified 7-day prescription window. Currently, those who miss the window must wait 19 days from the date of the first pregnancy test to take an additional pregnancy test to be eligible to receive the drug.
Lindsey Crist, PharmD, a risk management analyst for the FDA, who presented the FDA review committee’s analysis, acknowledged that the lockout period causes delays in treatment and adds frustration and costs.
She said it’s important to remember that the lockout applies only to the first prescription. “It’s intended as an additional layer of screening to detect pregnancy,” she said.
“At least 12 pregnancies have been identified during the 19-day lockout from March 2017–September of 2022,” she noted.
The FDA is looking to the advisory committee to provide recommendations on whether the lockout period should be changed.
Home testing
During the pandemic, iPLEDGE rules have been relaxed from having a pregnancy test done only at a Clinical Laboratory Improvement Amendments–certified laboratory and home pregnancy tests have been allowed. The question now is whether home tests should continue to be allowed.
Ms. Crist said that the FDA’s review committee recommends ending the allowance of home tests, citing insufficient data on use and the discovery of instances of falsification of pregnancy tests.
“One study at an academic medical center reviewed the medical records of 89 patients who used home pregnancy tests while taking isotretinoin during the public health emergency. It found that 15.7% submitted falsified pregnancy test results,” she said.
Ms. Crist added, however, that the review committee recommends allowing the tests to be done in a provider’s office as an alternative.
Documenting counseling patients who cannot get pregnant
Currently, this documentation must be done monthly, primarily to counsel patients against drug sharing or giving blood. Proposed changes include extending the intervals for attestation or eliminating it to reduce burden on clinicians.
IPMG representative Gregory Wedin, PharmD, pharmacovigilance and risk management director for Upsher-Smith Laboratories, said, “while we cannot support eliminating or extending the confirmation interval to a year, the [iPLEDGE] sponsors are agreeable [to] a 120-day confirmation interval.”
He said that while extending to 120 days would reduce burden on prescribers, it comes with risk in reducing oversight by a certified iPLEDGE prescriber and potentially increasing the risk for drug sharing.
“A patient may be more likely to share their drug with another person the further along with therapy they get as their condition improves,” Mr. Wedin said.
On March 29, the panel will hear more recommendations for and against modifications to iPLEDGE REMS and will vote on select modifications at the end of the meeting.
A version of this article first appeared on Medscape.com.
JAK inhibitor ivarmacitinib shows efficacy for atopic dermatitis in a pivotal trial
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
AT AAD 2023
Topical delgocitinib shows promise for chronic hand eczema, pivotal trial shows
NEW ORLEANS – , compared with those who received vehicle cream, results from a pivotal phase 3 trial showed.
“Chronic hand eczema is the most frequent chronic inflammatory disorder affecting the hands,” Robert Bissonnette, MD, a dermatologist who is founder and CEO of Innovaderm Research, said at the annual meeting of the American Academy of Dermatology, where the study was presented during a late-breaking research session. “It’s associated with pain, pruritus, and has a huge impact on quality of life,” and results with current topical treatments are often unsatisfactory, he noted.
Delgocitinib is an investigational topical pan-JAK inhibitor that inhibits activation of the JAK-STAT pathway and targets key mediators of chronic hand eczema. In a phase 2b dose-ranging trial, twice-daily treatment with delgocitinib cream demonstrated significantly greater efficacy, compared with the cream vehicle, and was well tolerated in adults with mild to severe chronic hand eczema.
For the phase 3 study, known as DELTA 1, researchers randomized 487 adults with moderate to severe chronic hand eczema to receive twice-daily applications of delgocitinib cream 20 mg/g or cream vehicle for 16 weeks. After week 16, patients had the option to enter a long-term extension trial, which is currently ongoing. DELTA 1 was limited to adults with a diagnosis of chronic hand eczema defined as hand eczema that had persisted for more than 3 months or had returned more than twice within the past 12 months; an Investigator’s Global Assessment for chronic hand eczema (IGA-CHE) score of 3 (moderate) or 4 (severe); a weekly average Hand Eczema Symptom Diary (HESD) itch score of 4 or more points, and a medical history of inadequate response to topical corticosteroids within the past 12 months or for whom treatment with topical corticosteroids was not medically advisable.
The IGA-CHE scale used in the trial was new, “where, in order to be almost clear, the only sign that could be present on the skin was barely perceptible erythema,” Dr. Bissonnette said. He noted that he has used many IGA scales over the more than 25 years he has been involved with clinical trials, and “this was the first that used a scale with a bar so high.” Key secondary endpoints include a 75% and 90% improvement in Hand Eczema Severity Index (HECSI) from baseline at week 16 and a 4-point or greater improvement in the Dermatology Life Quality Index (DLQI) from baseline at week 16.
The median age of patients was 44 years, 88% were White, 4% were Asian, 1% were Black, and the remainder were from other racial groups. One-third of patients (33%) had severe hand eczema based on their IGA-CHE score, the median HECSI was 65 (in line with severe disease), and the median DLQI was 12. As for previous chronic hand eczema treatments, 19% had undergone phototherapy, 14% had tried oral retinoids, and 12% had tried oral corticosteroids.
In the study, a greater proportion of delgocitinib-treated patients achieved the primary endpoint of IGA-CHE 0/1, compared with the cream vehicle group at week 4 (15.4% vs. 4.9%; P < .001); week 8 (22.8% vs. 10.5%; P = .001), and week 16 (19.7% vs. 9.9%; P = .006). “As early as week 2, there is a separation between cream and vehicle,” Dr. Bissonnette said. When reviewing the results and the patients in the trial, he said that, in his personal opinion, “I don’t think this is uniquely representative of the efficacy of the drug,” because of the IGA scale that was used, which set such a high bar for efficacy.
As for secondary endpoints, a greater proportion of delgocitinib-treated patients than those in the vehicle group achieved a HESCI-75 (49.2% vs. 23.5%), a HECSI-90 (29.5% vs. 12.3%), and a 4-point or greater improvement on the DLQI (74.4% vs 50%; P < .001 for all endpoints).
Delgocitinib had a similar safety profile as the vehicle over 16 weeks, with no difference between the delgocitinib and vehicle arms in the proportion of patients who had adverse events (45.2% vs. 50.6%, respectively) and serious adverse events (1.8% vs. 1.9%). The most common adverse events (defined as 5% or greater in any treatment group) during the study were COVID-19 infections and nasopharyngitis; rates were comparable in the two arms.
Raj Chovatiya, MD, PhD, a dermatologist who directs the Center for Eczema and Itch at Northwestern University, Chicago, who was asked to comment on the study, said that chronic hand eczema can be functionally limiting for many patients. “Given its focal symptoms but multifaceted immunopathogenesis, topical JAK inhibition represents a rational strategy for targeted treatment,” Dr. Chovatiya told this news organization. He was not an investigator in the trial.

“In the phase 3 DELTA 1 study, topical delgocitinib cream was superior to vehicle control with nearly one out of five patients achieving clear or almost clear skin, with no difference in total adverse events between groups. While both comparative and long-term data would be helpful to better assess how delgocitinib cream stacks up against common topical anti-inflammatories and how it may be used for a chronic condition that typically requires ongoing treatment, these findings move us closer to a potential first-in-class approved therapy for chronic hand eczema.”
Dr. Bissonnette disclosed that he served as a consultant and investigator for the developer of delgocitinib, LEO Pharma, on this study. He has also received grants and research funding from many other pharmaceutical companies. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for several pharmaceutical companies, including LEO Pharma.
NEW ORLEANS – , compared with those who received vehicle cream, results from a pivotal phase 3 trial showed.
“Chronic hand eczema is the most frequent chronic inflammatory disorder affecting the hands,” Robert Bissonnette, MD, a dermatologist who is founder and CEO of Innovaderm Research, said at the annual meeting of the American Academy of Dermatology, where the study was presented during a late-breaking research session. “It’s associated with pain, pruritus, and has a huge impact on quality of life,” and results with current topical treatments are often unsatisfactory, he noted.
Delgocitinib is an investigational topical pan-JAK inhibitor that inhibits activation of the JAK-STAT pathway and targets key mediators of chronic hand eczema. In a phase 2b dose-ranging trial, twice-daily treatment with delgocitinib cream demonstrated significantly greater efficacy, compared with the cream vehicle, and was well tolerated in adults with mild to severe chronic hand eczema.
For the phase 3 study, known as DELTA 1, researchers randomized 487 adults with moderate to severe chronic hand eczema to receive twice-daily applications of delgocitinib cream 20 mg/g or cream vehicle for 16 weeks. After week 16, patients had the option to enter a long-term extension trial, which is currently ongoing. DELTA 1 was limited to adults with a diagnosis of chronic hand eczema defined as hand eczema that had persisted for more than 3 months or had returned more than twice within the past 12 months; an Investigator’s Global Assessment for chronic hand eczema (IGA-CHE) score of 3 (moderate) or 4 (severe); a weekly average Hand Eczema Symptom Diary (HESD) itch score of 4 or more points, and a medical history of inadequate response to topical corticosteroids within the past 12 months or for whom treatment with topical corticosteroids was not medically advisable.
The IGA-CHE scale used in the trial was new, “where, in order to be almost clear, the only sign that could be present on the skin was barely perceptible erythema,” Dr. Bissonnette said. He noted that he has used many IGA scales over the more than 25 years he has been involved with clinical trials, and “this was the first that used a scale with a bar so high.” Key secondary endpoints include a 75% and 90% improvement in Hand Eczema Severity Index (HECSI) from baseline at week 16 and a 4-point or greater improvement in the Dermatology Life Quality Index (DLQI) from baseline at week 16.
The median age of patients was 44 years, 88% were White, 4% were Asian, 1% were Black, and the remainder were from other racial groups. One-third of patients (33%) had severe hand eczema based on their IGA-CHE score, the median HECSI was 65 (in line with severe disease), and the median DLQI was 12. As for previous chronic hand eczema treatments, 19% had undergone phototherapy, 14% had tried oral retinoids, and 12% had tried oral corticosteroids.
In the study, a greater proportion of delgocitinib-treated patients achieved the primary endpoint of IGA-CHE 0/1, compared with the cream vehicle group at week 4 (15.4% vs. 4.9%; P < .001); week 8 (22.8% vs. 10.5%; P = .001), and week 16 (19.7% vs. 9.9%; P = .006). “As early as week 2, there is a separation between cream and vehicle,” Dr. Bissonnette said. When reviewing the results and the patients in the trial, he said that, in his personal opinion, “I don’t think this is uniquely representative of the efficacy of the drug,” because of the IGA scale that was used, which set such a high bar for efficacy.
As for secondary endpoints, a greater proportion of delgocitinib-treated patients than those in the vehicle group achieved a HESCI-75 (49.2% vs. 23.5%), a HECSI-90 (29.5% vs. 12.3%), and a 4-point or greater improvement on the DLQI (74.4% vs 50%; P < .001 for all endpoints).
Delgocitinib had a similar safety profile as the vehicle over 16 weeks, with no difference between the delgocitinib and vehicle arms in the proportion of patients who had adverse events (45.2% vs. 50.6%, respectively) and serious adverse events (1.8% vs. 1.9%). The most common adverse events (defined as 5% or greater in any treatment group) during the study were COVID-19 infections and nasopharyngitis; rates were comparable in the two arms.
Raj Chovatiya, MD, PhD, a dermatologist who directs the Center for Eczema and Itch at Northwestern University, Chicago, who was asked to comment on the study, said that chronic hand eczema can be functionally limiting for many patients. “Given its focal symptoms but multifaceted immunopathogenesis, topical JAK inhibition represents a rational strategy for targeted treatment,” Dr. Chovatiya told this news organization. He was not an investigator in the trial.
“In the phase 3 DELTA 1 study, topical delgocitinib cream was superior to vehicle control with nearly one out of five patients achieving clear or almost clear skin, with no difference in total adverse events between groups. While both comparative and long-term data would be helpful to better assess how delgocitinib cream stacks up against common topical anti-inflammatories and how it may be used for a chronic condition that typically requires ongoing treatment, these findings move us closer to a potential first-in-class approved therapy for chronic hand eczema.”
Dr. Bissonnette disclosed that he served as a consultant and investigator for the developer of delgocitinib, LEO Pharma, on this study. He has also received grants and research funding from many other pharmaceutical companies. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for several pharmaceutical companies, including LEO Pharma.
NEW ORLEANS – , compared with those who received vehicle cream, results from a pivotal phase 3 trial showed.
“Chronic hand eczema is the most frequent chronic inflammatory disorder affecting the hands,” Robert Bissonnette, MD, a dermatologist who is founder and CEO of Innovaderm Research, said at the annual meeting of the American Academy of Dermatology, where the study was presented during a late-breaking research session. “It’s associated with pain, pruritus, and has a huge impact on quality of life,” and results with current topical treatments are often unsatisfactory, he noted.
Delgocitinib is an investigational topical pan-JAK inhibitor that inhibits activation of the JAK-STAT pathway and targets key mediators of chronic hand eczema. In a phase 2b dose-ranging trial, twice-daily treatment with delgocitinib cream demonstrated significantly greater efficacy, compared with the cream vehicle, and was well tolerated in adults with mild to severe chronic hand eczema.
For the phase 3 study, known as DELTA 1, researchers randomized 487 adults with moderate to severe chronic hand eczema to receive twice-daily applications of delgocitinib cream 20 mg/g or cream vehicle for 16 weeks. After week 16, patients had the option to enter a long-term extension trial, which is currently ongoing. DELTA 1 was limited to adults with a diagnosis of chronic hand eczema defined as hand eczema that had persisted for more than 3 months or had returned more than twice within the past 12 months; an Investigator’s Global Assessment for chronic hand eczema (IGA-CHE) score of 3 (moderate) or 4 (severe); a weekly average Hand Eczema Symptom Diary (HESD) itch score of 4 or more points, and a medical history of inadequate response to topical corticosteroids within the past 12 months or for whom treatment with topical corticosteroids was not medically advisable.
The IGA-CHE scale used in the trial was new, “where, in order to be almost clear, the only sign that could be present on the skin was barely perceptible erythema,” Dr. Bissonnette said. He noted that he has used many IGA scales over the more than 25 years he has been involved with clinical trials, and “this was the first that used a scale with a bar so high.” Key secondary endpoints include a 75% and 90% improvement in Hand Eczema Severity Index (HECSI) from baseline at week 16 and a 4-point or greater improvement in the Dermatology Life Quality Index (DLQI) from baseline at week 16.
The median age of patients was 44 years, 88% were White, 4% were Asian, 1% were Black, and the remainder were from other racial groups. One-third of patients (33%) had severe hand eczema based on their IGA-CHE score, the median HECSI was 65 (in line with severe disease), and the median DLQI was 12. As for previous chronic hand eczema treatments, 19% had undergone phototherapy, 14% had tried oral retinoids, and 12% had tried oral corticosteroids.
In the study, a greater proportion of delgocitinib-treated patients achieved the primary endpoint of IGA-CHE 0/1, compared with the cream vehicle group at week 4 (15.4% vs. 4.9%; P < .001); week 8 (22.8% vs. 10.5%; P = .001), and week 16 (19.7% vs. 9.9%; P = .006). “As early as week 2, there is a separation between cream and vehicle,” Dr. Bissonnette said. When reviewing the results and the patients in the trial, he said that, in his personal opinion, “I don’t think this is uniquely representative of the efficacy of the drug,” because of the IGA scale that was used, which set such a high bar for efficacy.
As for secondary endpoints, a greater proportion of delgocitinib-treated patients than those in the vehicle group achieved a HESCI-75 (49.2% vs. 23.5%), a HECSI-90 (29.5% vs. 12.3%), and a 4-point or greater improvement on the DLQI (74.4% vs 50%; P < .001 for all endpoints).
Delgocitinib had a similar safety profile as the vehicle over 16 weeks, with no difference between the delgocitinib and vehicle arms in the proportion of patients who had adverse events (45.2% vs. 50.6%, respectively) and serious adverse events (1.8% vs. 1.9%). The most common adverse events (defined as 5% or greater in any treatment group) during the study were COVID-19 infections and nasopharyngitis; rates were comparable in the two arms.
Raj Chovatiya, MD, PhD, a dermatologist who directs the Center for Eczema and Itch at Northwestern University, Chicago, who was asked to comment on the study, said that chronic hand eczema can be functionally limiting for many patients. “Given its focal symptoms but multifaceted immunopathogenesis, topical JAK inhibition represents a rational strategy for targeted treatment,” Dr. Chovatiya told this news organization. He was not an investigator in the trial.
“In the phase 3 DELTA 1 study, topical delgocitinib cream was superior to vehicle control with nearly one out of five patients achieving clear or almost clear skin, with no difference in total adverse events between groups. While both comparative and long-term data would be helpful to better assess how delgocitinib cream stacks up against common topical anti-inflammatories and how it may be used for a chronic condition that typically requires ongoing treatment, these findings move us closer to a potential first-in-class approved therapy for chronic hand eczema.”
Dr. Bissonnette disclosed that he served as a consultant and investigator for the developer of delgocitinib, LEO Pharma, on this study. He has also received grants and research funding from many other pharmaceutical companies. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for several pharmaceutical companies, including LEO Pharma.
AT AAD 2023
Air pollution may be causing eczema
The finding points scientists toward how to better treat the skin ailment. There are now more than three times as many eczema cases as there were in the 1970s, and it now affects as many as 20% of children and 10% of adults.
“I think these authors are spot-on in recognizing that the incidence of allergic conditions is increasing concurrently with how different pollutants are increasing in our environment,” said Denver-based pediatric allergist and immunologist Jessica Hui, MD, according to NBC News. “We’re finally understanding more about why people are getting eczema.”
Some people get eczema because of genetics, but the new research built on the previous understanding of how chemicals called diisocyanates can trigger the eczema symptoms of severe itching, skin redness, and oozing or painful rashes. An experiment on mice showed that exposure to a specific part of diisocyanates, called isocyanates, disrupted oil production that the skin needs to stay healthy.
Researchers at the National Institutes of Health “found that when bacteria that live on healthy skin are exposed to isocyanate, they must adapt to survive,” the agency summarized in a news release. “When they adapt, these bacteria shift their metabolism away from making the lipids, or oils, that skin needs to stay healthy. This finding suggests that eczema may be treatable by replacing the modified skin bacteria with healthy bacteria.”
The study was published in the journal Science Advances.
The chemicals also trigger a message to the brain that causes skin inflammation and itching, lead researcher Ian Myles, MD, told NBC News. Dr. Myles is also chief of the Epithelial Research Unit in the National Institute of Allergy and Infectious Diseases Laboratory of Clinical Immunology and Microbiology.
“So much of this is out of our control. I mean, you can’t shut the highways down,” he said of the environmental sources.
Previous research that explored attempting to restore healthy skin bacteria called Roseomonas mucosa to treat eczema symptoms had mixed results. The NIH says it has made the bacteria available “for commercial, nontherapeutic development ... as a potentially beneficial probiotic.”
A version of this article first appeared on WebMD.com.
The finding points scientists toward how to better treat the skin ailment. There are now more than three times as many eczema cases as there were in the 1970s, and it now affects as many as 20% of children and 10% of adults.
“I think these authors are spot-on in recognizing that the incidence of allergic conditions is increasing concurrently with how different pollutants are increasing in our environment,” said Denver-based pediatric allergist and immunologist Jessica Hui, MD, according to NBC News. “We’re finally understanding more about why people are getting eczema.”
Some people get eczema because of genetics, but the new research built on the previous understanding of how chemicals called diisocyanates can trigger the eczema symptoms of severe itching, skin redness, and oozing or painful rashes. An experiment on mice showed that exposure to a specific part of diisocyanates, called isocyanates, disrupted oil production that the skin needs to stay healthy.
Researchers at the National Institutes of Health “found that when bacteria that live on healthy skin are exposed to isocyanate, they must adapt to survive,” the agency summarized in a news release. “When they adapt, these bacteria shift their metabolism away from making the lipids, or oils, that skin needs to stay healthy. This finding suggests that eczema may be treatable by replacing the modified skin bacteria with healthy bacteria.”
The study was published in the journal Science Advances.
The chemicals also trigger a message to the brain that causes skin inflammation and itching, lead researcher Ian Myles, MD, told NBC News. Dr. Myles is also chief of the Epithelial Research Unit in the National Institute of Allergy and Infectious Diseases Laboratory of Clinical Immunology and Microbiology.
“So much of this is out of our control. I mean, you can’t shut the highways down,” he said of the environmental sources.
Previous research that explored attempting to restore healthy skin bacteria called Roseomonas mucosa to treat eczema symptoms had mixed results. The NIH says it has made the bacteria available “for commercial, nontherapeutic development ... as a potentially beneficial probiotic.”
A version of this article first appeared on WebMD.com.
The finding points scientists toward how to better treat the skin ailment. There are now more than three times as many eczema cases as there were in the 1970s, and it now affects as many as 20% of children and 10% of adults.
“I think these authors are spot-on in recognizing that the incidence of allergic conditions is increasing concurrently with how different pollutants are increasing in our environment,” said Denver-based pediatric allergist and immunologist Jessica Hui, MD, according to NBC News. “We’re finally understanding more about why people are getting eczema.”
Some people get eczema because of genetics, but the new research built on the previous understanding of how chemicals called diisocyanates can trigger the eczema symptoms of severe itching, skin redness, and oozing or painful rashes. An experiment on mice showed that exposure to a specific part of diisocyanates, called isocyanates, disrupted oil production that the skin needs to stay healthy.
Researchers at the National Institutes of Health “found that when bacteria that live on healthy skin are exposed to isocyanate, they must adapt to survive,” the agency summarized in a news release. “When they adapt, these bacteria shift their metabolism away from making the lipids, or oils, that skin needs to stay healthy. This finding suggests that eczema may be treatable by replacing the modified skin bacteria with healthy bacteria.”
The study was published in the journal Science Advances.
The chemicals also trigger a message to the brain that causes skin inflammation and itching, lead researcher Ian Myles, MD, told NBC News. Dr. Myles is also chief of the Epithelial Research Unit in the National Institute of Allergy and Infectious Diseases Laboratory of Clinical Immunology and Microbiology.
“So much of this is out of our control. I mean, you can’t shut the highways down,” he said of the environmental sources.
Previous research that explored attempting to restore healthy skin bacteria called Roseomonas mucosa to treat eczema symptoms had mixed results. The NIH says it has made the bacteria available “for commercial, nontherapeutic development ... as a potentially beneficial probiotic.”
A version of this article first appeared on WebMD.com.
FROM SCIENCE ADVANCES
FDA approves new formulation of Hyrimoz adalimumab biosimilar
The Food and Drug Administration has approved a citrate-free, 100 mg/mL formulation of the biosimilar adalimumab-adaz (Hyrimoz), according to a statement from manufacturer Sandoz.
Hyrimoz, a tumor necrosis factor (TNF) blocker that is biosimilar to its reference product Humira, was approved by the FDA in 2018 at a concentration of 50 mg/mL for rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. The high-concentration formula is indicated for these same conditions.
Sandoz said that it intends to launch the citrate-free formulation in the United States on July 1. It will be one of up to nine other adalimumab biosimilars that are expected to launch in July. On January 31, Amjevita (adalimumab-atto) became the first adalimumab biosimilar to launch in the United States.
The current label for Hyrimoz contains a black box warning emphasizing certain risks, notably the increased risk for serious infections, such as tuberculosis or sepsis, and an increased risk of malignancy, particularly lymphomas.
Adverse effects associated with Hyrimoz with an incidence greater than 10% include upper respiratory infections and sinusitis, injection-site reactions, headache, and rash.
The approval for the high-concentration formulation was based on data from a phase 1 pharmacokinetics bridging study that compared Hyrimoz 50 mg/mL and citrate-free Hyrimoz 100 mg/mL.
“This study met all of the primary objectives, demonstrating comparable pharmacokinetics and showing similar safety and immunogenicity of the Hyrimoz 50 mg/mL and Hyrimoz [100 mg/mL],” according to Sandoz, a division of Novartis.
The approval for Hyrimoz 50 mg/mL in 2018 was based on preclinical and clinical research comparing Hyrimoz and Humira. In a phase 3 trial published in the British Journal of Dermatology, which included adults with clinically stable but active moderate to severe chronic plaque psoriasis, Hyrimoz and Humira showed a similar percentage of patients met the primary endpoint of a 75% reduction or more in Psoriasis Area and Severity Index (PASI 75) score at 16 weeks, compared with baseline (66.8% and 65%, respectively).
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved a citrate-free, 100 mg/mL formulation of the biosimilar adalimumab-adaz (Hyrimoz), according to a statement from manufacturer Sandoz.
Hyrimoz, a tumor necrosis factor (TNF) blocker that is biosimilar to its reference product Humira, was approved by the FDA in 2018 at a concentration of 50 mg/mL for rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. The high-concentration formula is indicated for these same conditions.
Sandoz said that it intends to launch the citrate-free formulation in the United States on July 1. It will be one of up to nine other adalimumab biosimilars that are expected to launch in July. On January 31, Amjevita (adalimumab-atto) became the first adalimumab biosimilar to launch in the United States.
The current label for Hyrimoz contains a black box warning emphasizing certain risks, notably the increased risk for serious infections, such as tuberculosis or sepsis, and an increased risk of malignancy, particularly lymphomas.
Adverse effects associated with Hyrimoz with an incidence greater than 10% include upper respiratory infections and sinusitis, injection-site reactions, headache, and rash.
The approval for the high-concentration formulation was based on data from a phase 1 pharmacokinetics bridging study that compared Hyrimoz 50 mg/mL and citrate-free Hyrimoz 100 mg/mL.
“This study met all of the primary objectives, demonstrating comparable pharmacokinetics and showing similar safety and immunogenicity of the Hyrimoz 50 mg/mL and Hyrimoz [100 mg/mL],” according to Sandoz, a division of Novartis.
The approval for Hyrimoz 50 mg/mL in 2018 was based on preclinical and clinical research comparing Hyrimoz and Humira. In a phase 3 trial published in the British Journal of Dermatology, which included adults with clinically stable but active moderate to severe chronic plaque psoriasis, Hyrimoz and Humira showed a similar percentage of patients met the primary endpoint of a 75% reduction or more in Psoriasis Area and Severity Index (PASI 75) score at 16 weeks, compared with baseline (66.8% and 65%, respectively).
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved a citrate-free, 100 mg/mL formulation of the biosimilar adalimumab-adaz (Hyrimoz), according to a statement from manufacturer Sandoz.
Hyrimoz, a tumor necrosis factor (TNF) blocker that is biosimilar to its reference product Humira, was approved by the FDA in 2018 at a concentration of 50 mg/mL for rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. The high-concentration formula is indicated for these same conditions.
Sandoz said that it intends to launch the citrate-free formulation in the United States on July 1. It will be one of up to nine other adalimumab biosimilars that are expected to launch in July. On January 31, Amjevita (adalimumab-atto) became the first adalimumab biosimilar to launch in the United States.
The current label for Hyrimoz contains a black box warning emphasizing certain risks, notably the increased risk for serious infections, such as tuberculosis or sepsis, and an increased risk of malignancy, particularly lymphomas.
Adverse effects associated with Hyrimoz with an incidence greater than 10% include upper respiratory infections and sinusitis, injection-site reactions, headache, and rash.
The approval for the high-concentration formulation was based on data from a phase 1 pharmacokinetics bridging study that compared Hyrimoz 50 mg/mL and citrate-free Hyrimoz 100 mg/mL.
“This study met all of the primary objectives, demonstrating comparable pharmacokinetics and showing similar safety and immunogenicity of the Hyrimoz 50 mg/mL and Hyrimoz [100 mg/mL],” according to Sandoz, a division of Novartis.
The approval for Hyrimoz 50 mg/mL in 2018 was based on preclinical and clinical research comparing Hyrimoz and Humira. In a phase 3 trial published in the British Journal of Dermatology, which included adults with clinically stable but active moderate to severe chronic plaque psoriasis, Hyrimoz and Humira showed a similar percentage of patients met the primary endpoint of a 75% reduction or more in Psoriasis Area and Severity Index (PASI 75) score at 16 weeks, compared with baseline (66.8% and 65%, respectively).
A version of this article originally appeared on Medscape.com.