User login
Anifrolumab shows promise in refractory discoid lupus erythematosus
a small retrospective study reports.
DLE, the most common form of chronic cutaneous lupus erythematosus, can permanently scar and disfigure patients, and traditional treatments such as antimalarials, steroid-sparing immunosuppressive agents, thalidomide, retinoids, and lenalidomide don’t consistently improve refractory DLE, the authors noted.
“All patients demonstrated significant improvement in symptomatology and disease activity within 2 months of initiating anifrolumab,” lead study author Katharina Shaw, MD, of the department of dermatology of Brigham and Women’s Hospital and Harvard Medical School, both in Boston, and colleagues wrote in a research letter published in JAMA Dermatology. “These early results highlight the potential for anifrolumab to be a viable therapeutic option for patients with DLE, particularly those with severe or recalcitrant disease.”
The Food and Drug Administration approved anifrolumab (Saphnelo), a human monoclonal antibody targeting type 1 interferon receptor subunit 1, in 2021 for adults with moderate to severe systemic lupus erythematosus, but it has not been approved for the treatment of DLE.
Dr. Shaw and colleagues queried the medical records from Brigham and Women’s Hospital and Massachusetts General Hospital, Boston, to find all cases of DLE based on biopsy, expert opinion, or both from January 2000 to October 2022.
The researchers identified eight female patients who had received anifrolumab for at least 8 weeks. The women were aged between 19 and 75 years (median, 42.5 years), and all had DLE recalcitrant to standard therapies and had been treated with hydroxychloroquine and between 1 and 10 other drugs, most commonly methotrexate and mycophenolate mofetil (MMF).
The authors looked for improvements in patient-reported symptoms and Cutaneous Lupus Erythematosus Disease Area and Severity Index scores, including CLASI A (activity) score 0-70, and CLASI-D (damage) score 0-56.
All patients showed significantly improved symptoms and disease activity within 2 months of their first infusion of the treatment. The mean decrease and mean percentage decrease in CLASI-A scores were 17.1 and 65.1%, respectively. The mean decrease and mean percentage decrease in CLASI-D scores were 0.5 and 2.9%, respectively.
The rapid clinical improvements with anifrolumab, compared with improvements with traditional medications, were striking, the authors wrote. “Given the risk for permanent scarring, dyspigmentation, and alopecia with poorly controlled DLE, the importance of rapidly mitigating disease activity cannot be overemphasized.”
They acknowledged that the results are limited by the study’s small sample size and retrospective design, and they recommend larger related prospective studies.

Asked to comment on the results, Kaveh Ardalan, MD, MS, assistant professor of pediatrics in the division of pediatric rheumatology at Duke University, Durham, N.C., said that finding new DLE therapeutics is important because of the huge impact of uncontrolled DLE on patients’ quality of life, body image, and social roles.
Dr. Ardalan noted that he sees DLE in his pediatric patients, “either as an isolated finding or in the context of systemic lupus erythematosus. Anifrolumab is not approved by the FDA to treat DLE or children.
“Randomized controlled trials, including the TULIP-1 and TULIP-2 studies of anifrolumab in systemic lupus, have indicated that lupus skin manifestations can improve in patients who receive anifrolumab,” said Dr. Ardalan, who was not involved in the study. “And we know that type I interferons are major drivers of cutaneous disease activity in patients with lupus, so targeting that mechanism with anifrolumab makes biological sense.”
The authors’ use of the validated CLASI classification system to quantify disease activity and damage over time, and their determination of the length of time for the drug to take effect are strengths of the study, he added.
Funding information was not provided. Two authors reported financial relationships with Pfizer, which does not manufacture anifrolumab. Dr. Ardalan reported no conflicts of interest with the study.
a small retrospective study reports.
DLE, the most common form of chronic cutaneous lupus erythematosus, can permanently scar and disfigure patients, and traditional treatments such as antimalarials, steroid-sparing immunosuppressive agents, thalidomide, retinoids, and lenalidomide don’t consistently improve refractory DLE, the authors noted.
“All patients demonstrated significant improvement in symptomatology and disease activity within 2 months of initiating anifrolumab,” lead study author Katharina Shaw, MD, of the department of dermatology of Brigham and Women’s Hospital and Harvard Medical School, both in Boston, and colleagues wrote in a research letter published in JAMA Dermatology. “These early results highlight the potential for anifrolumab to be a viable therapeutic option for patients with DLE, particularly those with severe or recalcitrant disease.”
The Food and Drug Administration approved anifrolumab (Saphnelo), a human monoclonal antibody targeting type 1 interferon receptor subunit 1, in 2021 for adults with moderate to severe systemic lupus erythematosus, but it has not been approved for the treatment of DLE.
Dr. Shaw and colleagues queried the medical records from Brigham and Women’s Hospital and Massachusetts General Hospital, Boston, to find all cases of DLE based on biopsy, expert opinion, or both from January 2000 to October 2022.
The researchers identified eight female patients who had received anifrolumab for at least 8 weeks. The women were aged between 19 and 75 years (median, 42.5 years), and all had DLE recalcitrant to standard therapies and had been treated with hydroxychloroquine and between 1 and 10 other drugs, most commonly methotrexate and mycophenolate mofetil (MMF).
The authors looked for improvements in patient-reported symptoms and Cutaneous Lupus Erythematosus Disease Area and Severity Index scores, including CLASI A (activity) score 0-70, and CLASI-D (damage) score 0-56.
All patients showed significantly improved symptoms and disease activity within 2 months of their first infusion of the treatment. The mean decrease and mean percentage decrease in CLASI-A scores were 17.1 and 65.1%, respectively. The mean decrease and mean percentage decrease in CLASI-D scores were 0.5 and 2.9%, respectively.
The rapid clinical improvements with anifrolumab, compared with improvements with traditional medications, were striking, the authors wrote. “Given the risk for permanent scarring, dyspigmentation, and alopecia with poorly controlled DLE, the importance of rapidly mitigating disease activity cannot be overemphasized.”
They acknowledged that the results are limited by the study’s small sample size and retrospective design, and they recommend larger related prospective studies.
Asked to comment on the results, Kaveh Ardalan, MD, MS, assistant professor of pediatrics in the division of pediatric rheumatology at Duke University, Durham, N.C., said that finding new DLE therapeutics is important because of the huge impact of uncontrolled DLE on patients’ quality of life, body image, and social roles.
Dr. Ardalan noted that he sees DLE in his pediatric patients, “either as an isolated finding or in the context of systemic lupus erythematosus. Anifrolumab is not approved by the FDA to treat DLE or children.
“Randomized controlled trials, including the TULIP-1 and TULIP-2 studies of anifrolumab in systemic lupus, have indicated that lupus skin manifestations can improve in patients who receive anifrolumab,” said Dr. Ardalan, who was not involved in the study. “And we know that type I interferons are major drivers of cutaneous disease activity in patients with lupus, so targeting that mechanism with anifrolumab makes biological sense.”
The authors’ use of the validated CLASI classification system to quantify disease activity and damage over time, and their determination of the length of time for the drug to take effect are strengths of the study, he added.
Funding information was not provided. Two authors reported financial relationships with Pfizer, which does not manufacture anifrolumab. Dr. Ardalan reported no conflicts of interest with the study.
a small retrospective study reports.
DLE, the most common form of chronic cutaneous lupus erythematosus, can permanently scar and disfigure patients, and traditional treatments such as antimalarials, steroid-sparing immunosuppressive agents, thalidomide, retinoids, and lenalidomide don’t consistently improve refractory DLE, the authors noted.
“All patients demonstrated significant improvement in symptomatology and disease activity within 2 months of initiating anifrolumab,” lead study author Katharina Shaw, MD, of the department of dermatology of Brigham and Women’s Hospital and Harvard Medical School, both in Boston, and colleagues wrote in a research letter published in JAMA Dermatology. “These early results highlight the potential for anifrolumab to be a viable therapeutic option for patients with DLE, particularly those with severe or recalcitrant disease.”
The Food and Drug Administration approved anifrolumab (Saphnelo), a human monoclonal antibody targeting type 1 interferon receptor subunit 1, in 2021 for adults with moderate to severe systemic lupus erythematosus, but it has not been approved for the treatment of DLE.
Dr. Shaw and colleagues queried the medical records from Brigham and Women’s Hospital and Massachusetts General Hospital, Boston, to find all cases of DLE based on biopsy, expert opinion, or both from January 2000 to October 2022.
The researchers identified eight female patients who had received anifrolumab for at least 8 weeks. The women were aged between 19 and 75 years (median, 42.5 years), and all had DLE recalcitrant to standard therapies and had been treated with hydroxychloroquine and between 1 and 10 other drugs, most commonly methotrexate and mycophenolate mofetil (MMF).
The authors looked for improvements in patient-reported symptoms and Cutaneous Lupus Erythematosus Disease Area and Severity Index scores, including CLASI A (activity) score 0-70, and CLASI-D (damage) score 0-56.
All patients showed significantly improved symptoms and disease activity within 2 months of their first infusion of the treatment. The mean decrease and mean percentage decrease in CLASI-A scores were 17.1 and 65.1%, respectively. The mean decrease and mean percentage decrease in CLASI-D scores were 0.5 and 2.9%, respectively.
The rapid clinical improvements with anifrolumab, compared with improvements with traditional medications, were striking, the authors wrote. “Given the risk for permanent scarring, dyspigmentation, and alopecia with poorly controlled DLE, the importance of rapidly mitigating disease activity cannot be overemphasized.”
They acknowledged that the results are limited by the study’s small sample size and retrospective design, and they recommend larger related prospective studies.
Asked to comment on the results, Kaveh Ardalan, MD, MS, assistant professor of pediatrics in the division of pediatric rheumatology at Duke University, Durham, N.C., said that finding new DLE therapeutics is important because of the huge impact of uncontrolled DLE on patients’ quality of life, body image, and social roles.
Dr. Ardalan noted that he sees DLE in his pediatric patients, “either as an isolated finding or in the context of systemic lupus erythematosus. Anifrolumab is not approved by the FDA to treat DLE or children.
“Randomized controlled trials, including the TULIP-1 and TULIP-2 studies of anifrolumab in systemic lupus, have indicated that lupus skin manifestations can improve in patients who receive anifrolumab,” said Dr. Ardalan, who was not involved in the study. “And we know that type I interferons are major drivers of cutaneous disease activity in patients with lupus, so targeting that mechanism with anifrolumab makes biological sense.”
The authors’ use of the validated CLASI classification system to quantify disease activity and damage over time, and their determination of the length of time for the drug to take effect are strengths of the study, he added.
Funding information was not provided. Two authors reported financial relationships with Pfizer, which does not manufacture anifrolumab. Dr. Ardalan reported no conflicts of interest with the study.
FROM JAMA DERMATOLOGY
New data forecast more oral PDE4 inhibitors for psoriasis
NEW ORLEANS – according to results of a phase 2 clinical trial presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
The phase 2b data, which are prompting a phase 3 trial, suggest that the drug, called orismilast, “is a potential new addition to the psoriasis armamentarium,” reported Lars E. French, MD, professor and chair, department of dermatology, Ludwig Maximilian University of Munich (Germany).

At the same session, findings from another study supported off-label use of oral roflumilast (Daliresp and generic), a PDE4 inhibitor approved for severe chronic obstructive pulmonary disease (COPD). The only PDE4 inhibitors with an indication for psoriasis are roflumilast, approved as a cream (Zoryve), and apremilast (Otezla), approved as an oral therapy.
Phase 2 study of orismilast
In the orismilast trial, Dr. French attributed the efficacy observed to the potency of orismilast on the B and D subtypes of PDE4 associated with inflammation. One clue is that these specific subtypes are overly expressed in the skin of patients with either psoriasis or atopic dermatitis.
“When compared to apremilast, orismilast is at least two to fivefold more potent on all PDE4 isoforms and up to 39 times more potent on some of the PDE4 B and D isoforms,” said Dr. French, referring to preclinical findings in human whole blood and blood cells and in a mouse model of chronic inflammation.
The efficacy of orismilast in an immediate-release oral formulation was previously demonstrated in a recently published phase 2a trial, but the newest study tested a modified-release formulation of orismilast to test its potential to improve tolerability.
In the study, 202 adult patients with moderate to severe psoriasis (Psoriasis Area Severity Index [PASI] score ≥ 12) were randomly assigned to one of three doses of orismilast or to placebo. Each of the three doses – 20 mg, 30 mg, or 40 mg – were administered twice daily. The primary endpoint was change in PASI score at 16 weeks. Secondary endpoints included PASI 75 responses (signifying 75% clearance) and safety.
Relative to placebo, which was associated with a PASI improvement of 17%, all three of the tested orismilast doses were superior in a dose-dependent manner. The rates of response were 53%, 61%, and 64% for the 20-mg, 30-mg, and 40-mg twice-daily doses, respectively.
The PASI improvements were rapid, Dr. French said. At 4 weeks, PASI scores climbed from baseline by nearly 40% for those on all orismilast doses, which was more than double the improvement in the placebo group.
In the intention-to-treat analysis with missing data counted as nonresponders, the proportion of patients reaching PASI-75 scores at 16 weeks were 39%, 49%, 45%, and 17%, in the 20-mg, 30-mg, 40-mg, and placebo groups, respectively. The proportion of patients experiencing complete or near-complete skin clearance defined by a PASI 90 were 24%, 22%, 28%, and 8%, respectively.
The side-effect profile was consistent with other PDE4 inhibitors. The most common adverse events included gastrointestinal complaints, such as diarrhea and nausea, as well as headache and dizziness. But the majority of these events were of low grade, and they were largely confined to the first 4 weeks of treatment, which is a pattern reported with other PDE4 inhibitors in psoriasis and other chronic inflammatory diseases, such as COPD, according to Dr. French.
“There were no discontinuations for a treatment-related adverse event in the arms receiving either the 20-mg or the 30-mg doses,” Dr. French reported. There were only two serious adverse events, and neither were considered by trial investigators to be related to orismilast.
Based on the limited therapeutic gain but greater risk for adverse events on the 40-mg twice-daily dose, “the question is now whether to move forward with the 20-mg or the 30-mg dose,” said Dr. French, who said planning of a phase 3 trial is underway.
Phase 2 study of roflumilast
However, this was not the only set of data on an oral PDE4 inhibitor presented as a late-breaker at the AAD meeting. For clinicians looking for a more immediate and less expensive alternative to apremilast, another study indicated that off-label use of oral roflumilast is an option.
In an investigator-initiated, multicenter, double-blind, placebo-controlled trial conducted in Denmark, the rate of response to oral roflumilast at 24 weeks, including the clear or almost clear response, was on the same general order of magnitude as that seen in the orismilast study, reported Alexander Egeberg, MD, PhD, professor of dermatology, University of Copenhagen.
“At 24 weeks, 21.7% had achieved a PASI 90, and 8.7% achieved a PASI 100,” Dr. Egeberg said.
Oral roflumilast has been available for the treatment of COPD for more than 10 years and is now available in a generic formulation. This study was conducted independent of any pharmaceutical company involvement, and the high rate of response and low risk of adverse events suggests that patients can benefit from a PDE4 inhibitor in a very low-cost form.
“Generic oral roflumilast is cheaper than a Starbucks coffee,” Dr. Egeberg said.
In this trial, 46 patients were randomly assigned to placebo or to the COPD-approved roflumilast dose of 500 mcg once daily. The primary endpoint was change in PASI scores from baseline to week 12, which Dr. Egeberg pointed out is a shorter time frame than the 16 weeks more typical of psoriasis treatment studies.
At week 12, the median improvement in PASI was 34.8% in the roflumilast group versus 0% in the placebo group. Patients were then followed for an additional 12 weeks, but those randomized to placebo were switched to the active treatment. By week 24, the switch patients had largely caught up to those initiated on roflumilast for median PASI improvement (39.1% vs. 43.5%).
Similar to orismilast, roflumilast “was generally well tolerated,” Dr. Egeberg said. The adverse events were consistent with those associated with PDE4 inhibitors in previous trials, whether in psoriasis or COPD. There was only one serious adverse event, and it was not considered treatment related. Discontinuations for adverse events “were very low.”
In a population with a relatively high rate of smoking, Dr. Egeberg further reported, lung function was improved, a remark initially interpreted as a joke by some attending the presentation. However, Dr. Egeberg confirmed that lung function was monitored, and objective improvements were recorded.
By Danish law, the investigators were required to inform the manufacturers of roflumilast. Despite the results of this study, he is not aware of any plans to seek an indication for roflumilast in psoriasis, but he noted that the drug is readily available at a low price.
For those willing to offer this therapy off label, “you can start using it tomorrow if you’d like,” he said.
Dr. French reports financial relationships with Almirall, Amgen, Biotest, Galderma, Janssen Cilag, Leo Pharma, Pincell, Regeneron, UCB, and UNION Therapeutics, which provided funding for this trial. Dr. Egeberg reports financial relationships with Eli Lilly, Galderma, Janssen-Cilag, Novartis, and Pfizer.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – according to results of a phase 2 clinical trial presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
The phase 2b data, which are prompting a phase 3 trial, suggest that the drug, called orismilast, “is a potential new addition to the psoriasis armamentarium,” reported Lars E. French, MD, professor and chair, department of dermatology, Ludwig Maximilian University of Munich (Germany).
At the same session, findings from another study supported off-label use of oral roflumilast (Daliresp and generic), a PDE4 inhibitor approved for severe chronic obstructive pulmonary disease (COPD). The only PDE4 inhibitors with an indication for psoriasis are roflumilast, approved as a cream (Zoryve), and apremilast (Otezla), approved as an oral therapy.
Phase 2 study of orismilast
In the orismilast trial, Dr. French attributed the efficacy observed to the potency of orismilast on the B and D subtypes of PDE4 associated with inflammation. One clue is that these specific subtypes are overly expressed in the skin of patients with either psoriasis or atopic dermatitis.
“When compared to apremilast, orismilast is at least two to fivefold more potent on all PDE4 isoforms and up to 39 times more potent on some of the PDE4 B and D isoforms,” said Dr. French, referring to preclinical findings in human whole blood and blood cells and in a mouse model of chronic inflammation.
The efficacy of orismilast in an immediate-release oral formulation was previously demonstrated in a recently published phase 2a trial, but the newest study tested a modified-release formulation of orismilast to test its potential to improve tolerability.
In the study, 202 adult patients with moderate to severe psoriasis (Psoriasis Area Severity Index [PASI] score ≥ 12) were randomly assigned to one of three doses of orismilast or to placebo. Each of the three doses – 20 mg, 30 mg, or 40 mg – were administered twice daily. The primary endpoint was change in PASI score at 16 weeks. Secondary endpoints included PASI 75 responses (signifying 75% clearance) and safety.
Relative to placebo, which was associated with a PASI improvement of 17%, all three of the tested orismilast doses were superior in a dose-dependent manner. The rates of response were 53%, 61%, and 64% for the 20-mg, 30-mg, and 40-mg twice-daily doses, respectively.
The PASI improvements were rapid, Dr. French said. At 4 weeks, PASI scores climbed from baseline by nearly 40% for those on all orismilast doses, which was more than double the improvement in the placebo group.
In the intention-to-treat analysis with missing data counted as nonresponders, the proportion of patients reaching PASI-75 scores at 16 weeks were 39%, 49%, 45%, and 17%, in the 20-mg, 30-mg, 40-mg, and placebo groups, respectively. The proportion of patients experiencing complete or near-complete skin clearance defined by a PASI 90 were 24%, 22%, 28%, and 8%, respectively.
The side-effect profile was consistent with other PDE4 inhibitors. The most common adverse events included gastrointestinal complaints, such as diarrhea and nausea, as well as headache and dizziness. But the majority of these events were of low grade, and they were largely confined to the first 4 weeks of treatment, which is a pattern reported with other PDE4 inhibitors in psoriasis and other chronic inflammatory diseases, such as COPD, according to Dr. French.
“There were no discontinuations for a treatment-related adverse event in the arms receiving either the 20-mg or the 30-mg doses,” Dr. French reported. There were only two serious adverse events, and neither were considered by trial investigators to be related to orismilast.
Based on the limited therapeutic gain but greater risk for adverse events on the 40-mg twice-daily dose, “the question is now whether to move forward with the 20-mg or the 30-mg dose,” said Dr. French, who said planning of a phase 3 trial is underway.
Phase 2 study of roflumilast
However, this was not the only set of data on an oral PDE4 inhibitor presented as a late-breaker at the AAD meeting. For clinicians looking for a more immediate and less expensive alternative to apremilast, another study indicated that off-label use of oral roflumilast is an option.
In an investigator-initiated, multicenter, double-blind, placebo-controlled trial conducted in Denmark, the rate of response to oral roflumilast at 24 weeks, including the clear or almost clear response, was on the same general order of magnitude as that seen in the orismilast study, reported Alexander Egeberg, MD, PhD, professor of dermatology, University of Copenhagen.
“At 24 weeks, 21.7% had achieved a PASI 90, and 8.7% achieved a PASI 100,” Dr. Egeberg said.
Oral roflumilast has been available for the treatment of COPD for more than 10 years and is now available in a generic formulation. This study was conducted independent of any pharmaceutical company involvement, and the high rate of response and low risk of adverse events suggests that patients can benefit from a PDE4 inhibitor in a very low-cost form.
“Generic oral roflumilast is cheaper than a Starbucks coffee,” Dr. Egeberg said.
In this trial, 46 patients were randomly assigned to placebo or to the COPD-approved roflumilast dose of 500 mcg once daily. The primary endpoint was change in PASI scores from baseline to week 12, which Dr. Egeberg pointed out is a shorter time frame than the 16 weeks more typical of psoriasis treatment studies.
At week 12, the median improvement in PASI was 34.8% in the roflumilast group versus 0% in the placebo group. Patients were then followed for an additional 12 weeks, but those randomized to placebo were switched to the active treatment. By week 24, the switch patients had largely caught up to those initiated on roflumilast for median PASI improvement (39.1% vs. 43.5%).
Similar to orismilast, roflumilast “was generally well tolerated,” Dr. Egeberg said. The adverse events were consistent with those associated with PDE4 inhibitors in previous trials, whether in psoriasis or COPD. There was only one serious adverse event, and it was not considered treatment related. Discontinuations for adverse events “were very low.”
In a population with a relatively high rate of smoking, Dr. Egeberg further reported, lung function was improved, a remark initially interpreted as a joke by some attending the presentation. However, Dr. Egeberg confirmed that lung function was monitored, and objective improvements were recorded.
By Danish law, the investigators were required to inform the manufacturers of roflumilast. Despite the results of this study, he is not aware of any plans to seek an indication for roflumilast in psoriasis, but he noted that the drug is readily available at a low price.
For those willing to offer this therapy off label, “you can start using it tomorrow if you’d like,” he said.
Dr. French reports financial relationships with Almirall, Amgen, Biotest, Galderma, Janssen Cilag, Leo Pharma, Pincell, Regeneron, UCB, and UNION Therapeutics, which provided funding for this trial. Dr. Egeberg reports financial relationships with Eli Lilly, Galderma, Janssen-Cilag, Novartis, and Pfizer.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – according to results of a phase 2 clinical trial presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
The phase 2b data, which are prompting a phase 3 trial, suggest that the drug, called orismilast, “is a potential new addition to the psoriasis armamentarium,” reported Lars E. French, MD, professor and chair, department of dermatology, Ludwig Maximilian University of Munich (Germany).
At the same session, findings from another study supported off-label use of oral roflumilast (Daliresp and generic), a PDE4 inhibitor approved for severe chronic obstructive pulmonary disease (COPD). The only PDE4 inhibitors with an indication for psoriasis are roflumilast, approved as a cream (Zoryve), and apremilast (Otezla), approved as an oral therapy.
Phase 2 study of orismilast
In the orismilast trial, Dr. French attributed the efficacy observed to the potency of orismilast on the B and D subtypes of PDE4 associated with inflammation. One clue is that these specific subtypes are overly expressed in the skin of patients with either psoriasis or atopic dermatitis.
“When compared to apremilast, orismilast is at least two to fivefold more potent on all PDE4 isoforms and up to 39 times more potent on some of the PDE4 B and D isoforms,” said Dr. French, referring to preclinical findings in human whole blood and blood cells and in a mouse model of chronic inflammation.
The efficacy of orismilast in an immediate-release oral formulation was previously demonstrated in a recently published phase 2a trial, but the newest study tested a modified-release formulation of orismilast to test its potential to improve tolerability.
In the study, 202 adult patients with moderate to severe psoriasis (Psoriasis Area Severity Index [PASI] score ≥ 12) were randomly assigned to one of three doses of orismilast or to placebo. Each of the three doses – 20 mg, 30 mg, or 40 mg – were administered twice daily. The primary endpoint was change in PASI score at 16 weeks. Secondary endpoints included PASI 75 responses (signifying 75% clearance) and safety.
Relative to placebo, which was associated with a PASI improvement of 17%, all three of the tested orismilast doses were superior in a dose-dependent manner. The rates of response were 53%, 61%, and 64% for the 20-mg, 30-mg, and 40-mg twice-daily doses, respectively.
The PASI improvements were rapid, Dr. French said. At 4 weeks, PASI scores climbed from baseline by nearly 40% for those on all orismilast doses, which was more than double the improvement in the placebo group.
In the intention-to-treat analysis with missing data counted as nonresponders, the proportion of patients reaching PASI-75 scores at 16 weeks were 39%, 49%, 45%, and 17%, in the 20-mg, 30-mg, 40-mg, and placebo groups, respectively. The proportion of patients experiencing complete or near-complete skin clearance defined by a PASI 90 were 24%, 22%, 28%, and 8%, respectively.
The side-effect profile was consistent with other PDE4 inhibitors. The most common adverse events included gastrointestinal complaints, such as diarrhea and nausea, as well as headache and dizziness. But the majority of these events were of low grade, and they were largely confined to the first 4 weeks of treatment, which is a pattern reported with other PDE4 inhibitors in psoriasis and other chronic inflammatory diseases, such as COPD, according to Dr. French.
“There were no discontinuations for a treatment-related adverse event in the arms receiving either the 20-mg or the 30-mg doses,” Dr. French reported. There were only two serious adverse events, and neither were considered by trial investigators to be related to orismilast.
Based on the limited therapeutic gain but greater risk for adverse events on the 40-mg twice-daily dose, “the question is now whether to move forward with the 20-mg or the 30-mg dose,” said Dr. French, who said planning of a phase 3 trial is underway.
Phase 2 study of roflumilast
However, this was not the only set of data on an oral PDE4 inhibitor presented as a late-breaker at the AAD meeting. For clinicians looking for a more immediate and less expensive alternative to apremilast, another study indicated that off-label use of oral roflumilast is an option.
In an investigator-initiated, multicenter, double-blind, placebo-controlled trial conducted in Denmark, the rate of response to oral roflumilast at 24 weeks, including the clear or almost clear response, was on the same general order of magnitude as that seen in the orismilast study, reported Alexander Egeberg, MD, PhD, professor of dermatology, University of Copenhagen.
“At 24 weeks, 21.7% had achieved a PASI 90, and 8.7% achieved a PASI 100,” Dr. Egeberg said.
Oral roflumilast has been available for the treatment of COPD for more than 10 years and is now available in a generic formulation. This study was conducted independent of any pharmaceutical company involvement, and the high rate of response and low risk of adverse events suggests that patients can benefit from a PDE4 inhibitor in a very low-cost form.
“Generic oral roflumilast is cheaper than a Starbucks coffee,” Dr. Egeberg said.
In this trial, 46 patients were randomly assigned to placebo or to the COPD-approved roflumilast dose of 500 mcg once daily. The primary endpoint was change in PASI scores from baseline to week 12, which Dr. Egeberg pointed out is a shorter time frame than the 16 weeks more typical of psoriasis treatment studies.
At week 12, the median improvement in PASI was 34.8% in the roflumilast group versus 0% in the placebo group. Patients were then followed for an additional 12 weeks, but those randomized to placebo were switched to the active treatment. By week 24, the switch patients had largely caught up to those initiated on roflumilast for median PASI improvement (39.1% vs. 43.5%).
Similar to orismilast, roflumilast “was generally well tolerated,” Dr. Egeberg said. The adverse events were consistent with those associated with PDE4 inhibitors in previous trials, whether in psoriasis or COPD. There was only one serious adverse event, and it was not considered treatment related. Discontinuations for adverse events “were very low.”
In a population with a relatively high rate of smoking, Dr. Egeberg further reported, lung function was improved, a remark initially interpreted as a joke by some attending the presentation. However, Dr. Egeberg confirmed that lung function was monitored, and objective improvements were recorded.
By Danish law, the investigators were required to inform the manufacturers of roflumilast. Despite the results of this study, he is not aware of any plans to seek an indication for roflumilast in psoriasis, but he noted that the drug is readily available at a low price.
For those willing to offer this therapy off label, “you can start using it tomorrow if you’d like,” he said.
Dr. French reports financial relationships with Almirall, Amgen, Biotest, Galderma, Janssen Cilag, Leo Pharma, Pincell, Regeneron, UCB, and UNION Therapeutics, which provided funding for this trial. Dr. Egeberg reports financial relationships with Eli Lilly, Galderma, Janssen-Cilag, Novartis, and Pfizer.
A version of this article first appeared on Medscape.com.
AT AAD 2023
New JAK inhibitor study data confirm benefit in alopecia areata
from clinical trials of two drugs presented at a late-breaker research session at the annual meeting of the American Academy of Dermatology.
Based on phase 3 studies that document robust hair growth in about one third of patients, deuruxolitinib (CTP-543), an inhibitor of the JAK1 and JAK2 enzymes, has the potential to become the second JAK inhibitor available for the treatment of alopecia areata. If approved, it will join baricitinib (Olumiant), which received U.S. approval almost 1 year ago.
In his talk on THRIVE-AA2, a phase 3 trial of the investigational medicine deuruxolitinib, the principal investigator, Brett A. King, MD, PhD, displayed several before-and-after photos and said, “The photos tell the whole story. This is why there is so much excitement about these drugs.”
THRIVE-AA2 was the second of two phase 3 studies of deuruxolitinib. King was a principal investigator for both pivotal trials, called THRIVE-AA1 and THRIVE AA-2. He characterized the results of the two THRIVE trials as “comparable.”
Dr. King also was a principal investigator for the trials with baricitinib, called BRAVE-AA1 and BRAVE AA-2, which were published last year in the New England Journal of Medicine. The trials for both drugs had similar designs and endpoints.
Deuruxolitinib and the THRIVE studies
In the THRIVE-AA2 trial, 517 adult patients were enrolled with moderate to severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of ≥ 50%, which signifies a hair loss of at least 50%. Like THRIVE-AA1, patients participated at treatment centers in North America and Europe. About two-thirds were female. The mean age was 39 years. The majority of patients had complete or near complete hair loss at baseline.
“Many of these patients are the ones we have historically characterized as having alopecia totalis or universalis,” Dr. King said.
Participating patients were randomly assigned to 8 mg deuruxolitinib twice daily, 12 mg deuruxolitinib twice daily, or placebo. The primary endpoint was a SALT score of ≤ 20% at week 24.
At 24 weeks, almost no patients in the placebo group (1%) vs. 33% and 38% in the 8 mg and 12 mg twice-daily groups, respectively, met the primary endpoint. Each active treatment group was highly significant vs. placebo.
Of the responders, the majority achieved complete or near complete hair growth as defined by a SALT score of ≤ 10%, Dr. King reported.
Based on a graph that showed a relatively steep climb over the entire 24-week study period, deuruxolitinib “had a really fast onset of action,” Dr. King said. By week 8, which was the time of the first assessment, both doses of deuruxolitinib were superior to placebo.
The majority of patients had complete or significant loss of eyebrows and eye lashes at baseline, but more than two-thirds of these patients had regrowth by week 24, Dr. King said. Again, no significant regrowth was observed in the placebo arm.
On the Satisfaction of Hair Patient Reported Outcomes (SPRO), more than half of patients on both doses reported being satisfied or very satisfied with the improvement when evaluated at 24 weeks.
“The patient satisfaction overshot what one would expect by looking at the SALT scores, but a lot of subjects were at the precipice of the primary endpoint, sitting on SALT scores of 21, 25, or 30,” Dr. King said.
High participation in extension trial
More than 90% of the patients assigned to deuruxolitinib completed the trial and have entered an open-label extension (OLE). Dr. King credited the substantial rates of hair growth and the low rate of significant adverse events for the high rate of transition to OLE. Those who experienced the response were motivated to maintain it.
“This is a devastating disease. Patients want to get better,” Dr. King said.
There were no serious treatment-emergent adverse events associated with deuruxolitinib, including no thromboembolic events or other off-target events that have been reported previously with other JAK inhibitors in other disease states, such as rheumatoid arthritis. Although some adverse events, such as nasopharyngitis, were observed more often in those taking deuruxolitinib than placebo, there were “very few” discontinuations because of an adverse event, he said.
The data of THRIVE-AA2 are wholly compatible with the previously reported 706-patient THRIVE-AA1, according to Dr. King. In THRIVE-AA1, the primary endpoint of SALT ≤ 20% was reached by 29.6%, 41.5%, and 0.8% of the 8 mg, 12 mg, and placebo groups, respectively. Patient satisfaction scores, safety, and tolerability were also similar, according to Dr. King.
The experience with deuruxolitinib in the THRIVE-AA phase 3 program is similar to the experience with baricitinib in the BRAVE-AA trials. Although they cannot be compared directly because of potential differences between study populations, the 4-mg dose of baricitinib also achieved SALT score ≤ 20 in about 35% of patients, he said. The proportion was lower in the 2-mg group but was also superior to the placebo group.
“JAK inhibitors are changing the paradigm of alopecia areata,” Dr. King said. Responding to a question about payers reluctant to reimburse therapies for a “cosmetic” condition, Dr. King added that the effective treatments are “changing the landscape of how we think about this disease.” Dr. King believes these kinds of data show that “we are literally transforming lives forever.”
Baricitinib and the BRAVE studies
When baricitinib received regulatory approval for alopecia areata last year, it was not just the first JAK inhibitor approved for this disease, but the first systemic therapy of any kind, according to Maryanne Senna, MD, an assistant professor of dermatology at Harvard Medical School, Boston, and the director of the Lahey Hair Loss Center of Excellence, Burlington, Mass. Dr. Senna was a clinical investigator of BRAVE-AA1, as well as of THRIVE-AA2.
Providing an update on the BRAVE-AA program, Dr. Senna reported 104-week data that appear to support the idea of a life-changing benefit from JAK inhibitor therapy. This is because the effects appear durable.
In the data she presented at the AAD, responders and mixed responders at 52 weeks were followed to 104 weeks. Mixed responders were defined as those without a SALT response of ≤ 20 at week 52 but who had achieved this degree of hair regrowth at some earlier point.
Of the responders, 90% maintained their response at 104 weeks. In addition, many of the mixed responders and patients with a partial response but who never achieved a SALT score ≤ 20% gained additional hair growth, including complete or near complete hair growth, when maintained on treatment over the 2 years of follow-up.
“The follow-up suggests that, if you keep patients on treatment, you can get many of them to a meaningful response,” she said.
Meanwhile, “there have been no new safety signals,” Dr. Senna said. She based this statement not only of the 104-week data but on follow-up of up to 3.6 years among patients who have remained on treatment after participating in previous studies.
According to Dr. Senna, the off-target events that have been reported previously in other diseases with other JAK inhibitors, such as major adverse cardiovascular events and thromboembolic events, have not so far been observed in the BRAVE-AA phase 3 program.
Baricitinib, much like all but one of the JAK inhibitors with dermatologic indications, carries a black box warning that lists multiple risks for drugs in this class, based on a rheumatoid arthritis study.
The Food and Drug Administration has granted deuruxolitinib Breakthrough Therapy designation for the treatment of adult patients with moderate to severe alopecia areata and Fast Track designation for the treatment of alopecia areata, according to its manufacturer Concert Pharmaceuticals.
Dr. King reports financial relationships with more than 15 pharmaceutical companies, including Concert Pharmaceuticals, which provided the funding for the THRIVE-AA trial program, and for Eli Lilly, which provided funding for the BRAVE-AA trial program. Dr. Senna reports financial relationships with Arena pharmaceuticals, Follica, and both Concert Pharmaceuticals and Eli Lilly.
A version of this article originally appeared on Medscape.com.
from clinical trials of two drugs presented at a late-breaker research session at the annual meeting of the American Academy of Dermatology.
Based on phase 3 studies that document robust hair growth in about one third of patients, deuruxolitinib (CTP-543), an inhibitor of the JAK1 and JAK2 enzymes, has the potential to become the second JAK inhibitor available for the treatment of alopecia areata. If approved, it will join baricitinib (Olumiant), which received U.S. approval almost 1 year ago.
In his talk on THRIVE-AA2, a phase 3 trial of the investigational medicine deuruxolitinib, the principal investigator, Brett A. King, MD, PhD, displayed several before-and-after photos and said, “The photos tell the whole story. This is why there is so much excitement about these drugs.”
THRIVE-AA2 was the second of two phase 3 studies of deuruxolitinib. King was a principal investigator for both pivotal trials, called THRIVE-AA1 and THRIVE AA-2. He characterized the results of the two THRIVE trials as “comparable.”
Dr. King also was a principal investigator for the trials with baricitinib, called BRAVE-AA1 and BRAVE AA-2, which were published last year in the New England Journal of Medicine. The trials for both drugs had similar designs and endpoints.
Deuruxolitinib and the THRIVE studies
In the THRIVE-AA2 trial, 517 adult patients were enrolled with moderate to severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of ≥ 50%, which signifies a hair loss of at least 50%. Like THRIVE-AA1, patients participated at treatment centers in North America and Europe. About two-thirds were female. The mean age was 39 years. The majority of patients had complete or near complete hair loss at baseline.
“Many of these patients are the ones we have historically characterized as having alopecia totalis or universalis,” Dr. King said.
Participating patients were randomly assigned to 8 mg deuruxolitinib twice daily, 12 mg deuruxolitinib twice daily, or placebo. The primary endpoint was a SALT score of ≤ 20% at week 24.
At 24 weeks, almost no patients in the placebo group (1%) vs. 33% and 38% in the 8 mg and 12 mg twice-daily groups, respectively, met the primary endpoint. Each active treatment group was highly significant vs. placebo.
Of the responders, the majority achieved complete or near complete hair growth as defined by a SALT score of ≤ 10%, Dr. King reported.
Based on a graph that showed a relatively steep climb over the entire 24-week study period, deuruxolitinib “had a really fast onset of action,” Dr. King said. By week 8, which was the time of the first assessment, both doses of deuruxolitinib were superior to placebo.
The majority of patients had complete or significant loss of eyebrows and eye lashes at baseline, but more than two-thirds of these patients had regrowth by week 24, Dr. King said. Again, no significant regrowth was observed in the placebo arm.
On the Satisfaction of Hair Patient Reported Outcomes (SPRO), more than half of patients on both doses reported being satisfied or very satisfied with the improvement when evaluated at 24 weeks.
“The patient satisfaction overshot what one would expect by looking at the SALT scores, but a lot of subjects were at the precipice of the primary endpoint, sitting on SALT scores of 21, 25, or 30,” Dr. King said.
High participation in extension trial
More than 90% of the patients assigned to deuruxolitinib completed the trial and have entered an open-label extension (OLE). Dr. King credited the substantial rates of hair growth and the low rate of significant adverse events for the high rate of transition to OLE. Those who experienced the response were motivated to maintain it.
“This is a devastating disease. Patients want to get better,” Dr. King said.
There were no serious treatment-emergent adverse events associated with deuruxolitinib, including no thromboembolic events or other off-target events that have been reported previously with other JAK inhibitors in other disease states, such as rheumatoid arthritis. Although some adverse events, such as nasopharyngitis, were observed more often in those taking deuruxolitinib than placebo, there were “very few” discontinuations because of an adverse event, he said.
The data of THRIVE-AA2 are wholly compatible with the previously reported 706-patient THRIVE-AA1, according to Dr. King. In THRIVE-AA1, the primary endpoint of SALT ≤ 20% was reached by 29.6%, 41.5%, and 0.8% of the 8 mg, 12 mg, and placebo groups, respectively. Patient satisfaction scores, safety, and tolerability were also similar, according to Dr. King.
The experience with deuruxolitinib in the THRIVE-AA phase 3 program is similar to the experience with baricitinib in the BRAVE-AA trials. Although they cannot be compared directly because of potential differences between study populations, the 4-mg dose of baricitinib also achieved SALT score ≤ 20 in about 35% of patients, he said. The proportion was lower in the 2-mg group but was also superior to the placebo group.
“JAK inhibitors are changing the paradigm of alopecia areata,” Dr. King said. Responding to a question about payers reluctant to reimburse therapies for a “cosmetic” condition, Dr. King added that the effective treatments are “changing the landscape of how we think about this disease.” Dr. King believes these kinds of data show that “we are literally transforming lives forever.”
Baricitinib and the BRAVE studies
When baricitinib received regulatory approval for alopecia areata last year, it was not just the first JAK inhibitor approved for this disease, but the first systemic therapy of any kind, according to Maryanne Senna, MD, an assistant professor of dermatology at Harvard Medical School, Boston, and the director of the Lahey Hair Loss Center of Excellence, Burlington, Mass. Dr. Senna was a clinical investigator of BRAVE-AA1, as well as of THRIVE-AA2.
Providing an update on the BRAVE-AA program, Dr. Senna reported 104-week data that appear to support the idea of a life-changing benefit from JAK inhibitor therapy. This is because the effects appear durable.
In the data she presented at the AAD, responders and mixed responders at 52 weeks were followed to 104 weeks. Mixed responders were defined as those without a SALT response of ≤ 20 at week 52 but who had achieved this degree of hair regrowth at some earlier point.
Of the responders, 90% maintained their response at 104 weeks. In addition, many of the mixed responders and patients with a partial response but who never achieved a SALT score ≤ 20% gained additional hair growth, including complete or near complete hair growth, when maintained on treatment over the 2 years of follow-up.
“The follow-up suggests that, if you keep patients on treatment, you can get many of them to a meaningful response,” she said.
Meanwhile, “there have been no new safety signals,” Dr. Senna said. She based this statement not only of the 104-week data but on follow-up of up to 3.6 years among patients who have remained on treatment after participating in previous studies.
According to Dr. Senna, the off-target events that have been reported previously in other diseases with other JAK inhibitors, such as major adverse cardiovascular events and thromboembolic events, have not so far been observed in the BRAVE-AA phase 3 program.
Baricitinib, much like all but one of the JAK inhibitors with dermatologic indications, carries a black box warning that lists multiple risks for drugs in this class, based on a rheumatoid arthritis study.
The Food and Drug Administration has granted deuruxolitinib Breakthrough Therapy designation for the treatment of adult patients with moderate to severe alopecia areata and Fast Track designation for the treatment of alopecia areata, according to its manufacturer Concert Pharmaceuticals.
Dr. King reports financial relationships with more than 15 pharmaceutical companies, including Concert Pharmaceuticals, which provided the funding for the THRIVE-AA trial program, and for Eli Lilly, which provided funding for the BRAVE-AA trial program. Dr. Senna reports financial relationships with Arena pharmaceuticals, Follica, and both Concert Pharmaceuticals and Eli Lilly.
A version of this article originally appeared on Medscape.com.
from clinical trials of two drugs presented at a late-breaker research session at the annual meeting of the American Academy of Dermatology.
Based on phase 3 studies that document robust hair growth in about one third of patients, deuruxolitinib (CTP-543), an inhibitor of the JAK1 and JAK2 enzymes, has the potential to become the second JAK inhibitor available for the treatment of alopecia areata. If approved, it will join baricitinib (Olumiant), which received U.S. approval almost 1 year ago.
In his talk on THRIVE-AA2, a phase 3 trial of the investigational medicine deuruxolitinib, the principal investigator, Brett A. King, MD, PhD, displayed several before-and-after photos and said, “The photos tell the whole story. This is why there is so much excitement about these drugs.”
THRIVE-AA2 was the second of two phase 3 studies of deuruxolitinib. King was a principal investigator for both pivotal trials, called THRIVE-AA1 and THRIVE AA-2. He characterized the results of the two THRIVE trials as “comparable.”
Dr. King also was a principal investigator for the trials with baricitinib, called BRAVE-AA1 and BRAVE AA-2, which were published last year in the New England Journal of Medicine. The trials for both drugs had similar designs and endpoints.
Deuruxolitinib and the THRIVE studies
In the THRIVE-AA2 trial, 517 adult patients were enrolled with moderate to severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of ≥ 50%, which signifies a hair loss of at least 50%. Like THRIVE-AA1, patients participated at treatment centers in North America and Europe. About two-thirds were female. The mean age was 39 years. The majority of patients had complete or near complete hair loss at baseline.
“Many of these patients are the ones we have historically characterized as having alopecia totalis or universalis,” Dr. King said.
Participating patients were randomly assigned to 8 mg deuruxolitinib twice daily, 12 mg deuruxolitinib twice daily, or placebo. The primary endpoint was a SALT score of ≤ 20% at week 24.
At 24 weeks, almost no patients in the placebo group (1%) vs. 33% and 38% in the 8 mg and 12 mg twice-daily groups, respectively, met the primary endpoint. Each active treatment group was highly significant vs. placebo.
Of the responders, the majority achieved complete or near complete hair growth as defined by a SALT score of ≤ 10%, Dr. King reported.
Based on a graph that showed a relatively steep climb over the entire 24-week study period, deuruxolitinib “had a really fast onset of action,” Dr. King said. By week 8, which was the time of the first assessment, both doses of deuruxolitinib were superior to placebo.
The majority of patients had complete or significant loss of eyebrows and eye lashes at baseline, but more than two-thirds of these patients had regrowth by week 24, Dr. King said. Again, no significant regrowth was observed in the placebo arm.
On the Satisfaction of Hair Patient Reported Outcomes (SPRO), more than half of patients on both doses reported being satisfied or very satisfied with the improvement when evaluated at 24 weeks.
“The patient satisfaction overshot what one would expect by looking at the SALT scores, but a lot of subjects were at the precipice of the primary endpoint, sitting on SALT scores of 21, 25, or 30,” Dr. King said.
High participation in extension trial
More than 90% of the patients assigned to deuruxolitinib completed the trial and have entered an open-label extension (OLE). Dr. King credited the substantial rates of hair growth and the low rate of significant adverse events for the high rate of transition to OLE. Those who experienced the response were motivated to maintain it.
“This is a devastating disease. Patients want to get better,” Dr. King said.
There were no serious treatment-emergent adverse events associated with deuruxolitinib, including no thromboembolic events or other off-target events that have been reported previously with other JAK inhibitors in other disease states, such as rheumatoid arthritis. Although some adverse events, such as nasopharyngitis, were observed more often in those taking deuruxolitinib than placebo, there were “very few” discontinuations because of an adverse event, he said.
The data of THRIVE-AA2 are wholly compatible with the previously reported 706-patient THRIVE-AA1, according to Dr. King. In THRIVE-AA1, the primary endpoint of SALT ≤ 20% was reached by 29.6%, 41.5%, and 0.8% of the 8 mg, 12 mg, and placebo groups, respectively. Patient satisfaction scores, safety, and tolerability were also similar, according to Dr. King.
The experience with deuruxolitinib in the THRIVE-AA phase 3 program is similar to the experience with baricitinib in the BRAVE-AA trials. Although they cannot be compared directly because of potential differences between study populations, the 4-mg dose of baricitinib also achieved SALT score ≤ 20 in about 35% of patients, he said. The proportion was lower in the 2-mg group but was also superior to the placebo group.
“JAK inhibitors are changing the paradigm of alopecia areata,” Dr. King said. Responding to a question about payers reluctant to reimburse therapies for a “cosmetic” condition, Dr. King added that the effective treatments are “changing the landscape of how we think about this disease.” Dr. King believes these kinds of data show that “we are literally transforming lives forever.”
Baricitinib and the BRAVE studies
When baricitinib received regulatory approval for alopecia areata last year, it was not just the first JAK inhibitor approved for this disease, but the first systemic therapy of any kind, according to Maryanne Senna, MD, an assistant professor of dermatology at Harvard Medical School, Boston, and the director of the Lahey Hair Loss Center of Excellence, Burlington, Mass. Dr. Senna was a clinical investigator of BRAVE-AA1, as well as of THRIVE-AA2.
Providing an update on the BRAVE-AA program, Dr. Senna reported 104-week data that appear to support the idea of a life-changing benefit from JAK inhibitor therapy. This is because the effects appear durable.
In the data she presented at the AAD, responders and mixed responders at 52 weeks were followed to 104 weeks. Mixed responders were defined as those without a SALT response of ≤ 20 at week 52 but who had achieved this degree of hair regrowth at some earlier point.
Of the responders, 90% maintained their response at 104 weeks. In addition, many of the mixed responders and patients with a partial response but who never achieved a SALT score ≤ 20% gained additional hair growth, including complete or near complete hair growth, when maintained on treatment over the 2 years of follow-up.
“The follow-up suggests that, if you keep patients on treatment, you can get many of them to a meaningful response,” she said.
Meanwhile, “there have been no new safety signals,” Dr. Senna said. She based this statement not only of the 104-week data but on follow-up of up to 3.6 years among patients who have remained on treatment after participating in previous studies.
According to Dr. Senna, the off-target events that have been reported previously in other diseases with other JAK inhibitors, such as major adverse cardiovascular events and thromboembolic events, have not so far been observed in the BRAVE-AA phase 3 program.
Baricitinib, much like all but one of the JAK inhibitors with dermatologic indications, carries a black box warning that lists multiple risks for drugs in this class, based on a rheumatoid arthritis study.
The Food and Drug Administration has granted deuruxolitinib Breakthrough Therapy designation for the treatment of adult patients with moderate to severe alopecia areata and Fast Track designation for the treatment of alopecia areata, according to its manufacturer Concert Pharmaceuticals.
Dr. King reports financial relationships with more than 15 pharmaceutical companies, including Concert Pharmaceuticals, which provided the funding for the THRIVE-AA trial program, and for Eli Lilly, which provided funding for the BRAVE-AA trial program. Dr. Senna reports financial relationships with Arena pharmaceuticals, Follica, and both Concert Pharmaceuticals and Eli Lilly.
A version of this article originally appeared on Medscape.com.
AT AAD 2023
Pilot study evaluates sensitive skin burden in persons of color
NEW ORLEANS – .
Respondents also reported high rates of reactions to skin care products marketed for sensitive skin, and most said they had visited a dermatologist about their condition.
Those are among the key findings of a pilot study designed to assess the prevalence, symptom burden, and behaviors of self-identified persons of color with sensitive skin, which senior author Adam Friedman, MD, and colleagues defined as a subjective syndrome of cutaneous hyperreactivity to otherwise innocuous stimuli. “Improved understanding of sensitive skin is essential, and we encourage additional research into pathophysiology and creating a consensus definition for sensitive skin,” Dr. Friedman, professor and chair of dermatology at George Washington University, Washington, said in an interview in advance of the annual meeting of the American Academy of Dermatology, where the study was presented during an e-poster session. The findings were also reported online in JAAD International.
In May of 2022, Dr. Friedman, first author Erika McCormick, a 4th-year medical student at George Washington University, and colleagues invited individuals attending a community health fair in an undeserved area of Washington, to complete the Sensitive Scale-10 (SS-10) and to answer other questions after receiving a brief education about sensitive skin. Of the 58 respondents, 78% were female, and 86% self-identified as a person of color.
“Our study population predominantly self-identified as Black, which only represents one piece of those who would be characterized as persons of color,” Dr. Friedman said. “That said, improved representation of both our study population, and furthermore persons of color, in all aspects of dermatology research is crucial to at a minimum ensure generalizability of findings to the U.S. population, and research on sensitive skin is but one component of this.”
Nearly two-thirds of all respondents (63.8%) reported having an underlying skin condition, most commonly acne (21%), eczema (17%), and rosacea (6%). More than half (57%) reported sensitive skin, 27% of whom reported no other skin disease. Individuals with sensitive skin had higher mean SS-10 scores, compared with those with nonsensitive skin (14.61 vs. 4.32; P = .002) and burning was the main symptom among those with sensitive skin (56%), followed by itch (50%), redness (39%), dryness (39%) and pain (17%).
Compared with those who did not meet criteria for sensitive skin, those who did were more likely to report a personal history of allergy (56.25% vs. 8.33%; P = .0002) and were nearly seven times more likely to have seen a dermatologist about their concerns (odds ratio, 6.857; P = .0012).
In other findings limited to respondents with sensitive skin, 72% who reported reactions to general consumer skin care products also reported reacting to products marketed for sensitive skin, and 94% reported reactivity to at least one trigger, most commonly extreme temperatures (34%), stress (34%), sweat (33%), sun exposure (29%), and diet (28%). “We were particularly surprised by the high rates of reactivity to skin care products designed for and marketed to those suffering with sensitive skin,” Ms. McCormick told this news organization. “Importantly, there is currently no federal or legal standard regulating ingredients in products marketed for sensitive skin, and many products lack testing in sensitive skin specifically. Our data suggest an opportunity for improvement of sensitive skin care.”
She acknowledged certain limitations of the study, including its small sample size. “Reconducting this survey in a larger population will help validate our findings,” she said.
The research was supported by two independent research grants from Galderma: one supporting Ms. McCormick with a Sensitive Skin Research Fellowship and the other a Sensitive Skin Research Acceleration Fund. Dr. Friedman reported having no relevant disclosures.
NEW ORLEANS – .
Respondents also reported high rates of reactions to skin care products marketed for sensitive skin, and most said they had visited a dermatologist about their condition.
Those are among the key findings of a pilot study designed to assess the prevalence, symptom burden, and behaviors of self-identified persons of color with sensitive skin, which senior author Adam Friedman, MD, and colleagues defined as a subjective syndrome of cutaneous hyperreactivity to otherwise innocuous stimuli. “Improved understanding of sensitive skin is essential, and we encourage additional research into pathophysiology and creating a consensus definition for sensitive skin,” Dr. Friedman, professor and chair of dermatology at George Washington University, Washington, said in an interview in advance of the annual meeting of the American Academy of Dermatology, where the study was presented during an e-poster session. The findings were also reported online in JAAD International.
In May of 2022, Dr. Friedman, first author Erika McCormick, a 4th-year medical student at George Washington University, and colleagues invited individuals attending a community health fair in an undeserved area of Washington, to complete the Sensitive Scale-10 (SS-10) and to answer other questions after receiving a brief education about sensitive skin. Of the 58 respondents, 78% were female, and 86% self-identified as a person of color.
“Our study population predominantly self-identified as Black, which only represents one piece of those who would be characterized as persons of color,” Dr. Friedman said. “That said, improved representation of both our study population, and furthermore persons of color, in all aspects of dermatology research is crucial to at a minimum ensure generalizability of findings to the U.S. population, and research on sensitive skin is but one component of this.”
Nearly two-thirds of all respondents (63.8%) reported having an underlying skin condition, most commonly acne (21%), eczema (17%), and rosacea (6%). More than half (57%) reported sensitive skin, 27% of whom reported no other skin disease. Individuals with sensitive skin had higher mean SS-10 scores, compared with those with nonsensitive skin (14.61 vs. 4.32; P = .002) and burning was the main symptom among those with sensitive skin (56%), followed by itch (50%), redness (39%), dryness (39%) and pain (17%).
Compared with those who did not meet criteria for sensitive skin, those who did were more likely to report a personal history of allergy (56.25% vs. 8.33%; P = .0002) and were nearly seven times more likely to have seen a dermatologist about their concerns (odds ratio, 6.857; P = .0012).
In other findings limited to respondents with sensitive skin, 72% who reported reactions to general consumer skin care products also reported reacting to products marketed for sensitive skin, and 94% reported reactivity to at least one trigger, most commonly extreme temperatures (34%), stress (34%), sweat (33%), sun exposure (29%), and diet (28%). “We were particularly surprised by the high rates of reactivity to skin care products designed for and marketed to those suffering with sensitive skin,” Ms. McCormick told this news organization. “Importantly, there is currently no federal or legal standard regulating ingredients in products marketed for sensitive skin, and many products lack testing in sensitive skin specifically. Our data suggest an opportunity for improvement of sensitive skin care.”
She acknowledged certain limitations of the study, including its small sample size. “Reconducting this survey in a larger population will help validate our findings,” she said.
The research was supported by two independent research grants from Galderma: one supporting Ms. McCormick with a Sensitive Skin Research Fellowship and the other a Sensitive Skin Research Acceleration Fund. Dr. Friedman reported having no relevant disclosures.
NEW ORLEANS – .
Respondents also reported high rates of reactions to skin care products marketed for sensitive skin, and most said they had visited a dermatologist about their condition.
Those are among the key findings of a pilot study designed to assess the prevalence, symptom burden, and behaviors of self-identified persons of color with sensitive skin, which senior author Adam Friedman, MD, and colleagues defined as a subjective syndrome of cutaneous hyperreactivity to otherwise innocuous stimuli. “Improved understanding of sensitive skin is essential, and we encourage additional research into pathophysiology and creating a consensus definition for sensitive skin,” Dr. Friedman, professor and chair of dermatology at George Washington University, Washington, said in an interview in advance of the annual meeting of the American Academy of Dermatology, where the study was presented during an e-poster session. The findings were also reported online in JAAD International.
In May of 2022, Dr. Friedman, first author Erika McCormick, a 4th-year medical student at George Washington University, and colleagues invited individuals attending a community health fair in an undeserved area of Washington, to complete the Sensitive Scale-10 (SS-10) and to answer other questions after receiving a brief education about sensitive skin. Of the 58 respondents, 78% were female, and 86% self-identified as a person of color.
“Our study population predominantly self-identified as Black, which only represents one piece of those who would be characterized as persons of color,” Dr. Friedman said. “That said, improved representation of both our study population, and furthermore persons of color, in all aspects of dermatology research is crucial to at a minimum ensure generalizability of findings to the U.S. population, and research on sensitive skin is but one component of this.”
Nearly two-thirds of all respondents (63.8%) reported having an underlying skin condition, most commonly acne (21%), eczema (17%), and rosacea (6%). More than half (57%) reported sensitive skin, 27% of whom reported no other skin disease. Individuals with sensitive skin had higher mean SS-10 scores, compared with those with nonsensitive skin (14.61 vs. 4.32; P = .002) and burning was the main symptom among those with sensitive skin (56%), followed by itch (50%), redness (39%), dryness (39%) and pain (17%).
Compared with those who did not meet criteria for sensitive skin, those who did were more likely to report a personal history of allergy (56.25% vs. 8.33%; P = .0002) and were nearly seven times more likely to have seen a dermatologist about their concerns (odds ratio, 6.857; P = .0012).
In other findings limited to respondents with sensitive skin, 72% who reported reactions to general consumer skin care products also reported reacting to products marketed for sensitive skin, and 94% reported reactivity to at least one trigger, most commonly extreme temperatures (34%), stress (34%), sweat (33%), sun exposure (29%), and diet (28%). “We were particularly surprised by the high rates of reactivity to skin care products designed for and marketed to those suffering with sensitive skin,” Ms. McCormick told this news organization. “Importantly, there is currently no federal or legal standard regulating ingredients in products marketed for sensitive skin, and many products lack testing in sensitive skin specifically. Our data suggest an opportunity for improvement of sensitive skin care.”
She acknowledged certain limitations of the study, including its small sample size. “Reconducting this survey in a larger population will help validate our findings,” she said.
The research was supported by two independent research grants from Galderma: one supporting Ms. McCormick with a Sensitive Skin Research Fellowship and the other a Sensitive Skin Research Acceleration Fund. Dr. Friedman reported having no relevant disclosures.
AT AAD 2023
Expert shares her tips for diagnosing, treating onychomycosis
NEW ORLEANS – .
“The PAS [periodic acid-Schiff] stain is very popular because it can identify the presence or absence of fungal elements, but a fungal culture will identify the organism living in the nail,” Dr. Elewski, professor and chair of dermatology at the University of Alabama, Birmingham, said at the annual meeting of the American Academy of Dermatology. “You also could do a PCR to identify the organism, with or without a KOH or PAS stain. It is often helpful to know what organism is causing the infection.”

While waiting for lab results, there are three clinical clues to look for – the first being that an infection likely resides in the toenail. “You almost never see dermatophyte onychomycosis in the fingernails without it being in the toenails, too,” Dr. Elewski said.
The presence of tinea pedis is a second clinical clue. “Sometimes it’s subtle, so I will ask the patient, ‘Have you been treating yourself for athlete’s foot?’ If they say ‘no, I’ve never had it,’ put down on your list that it’s unlikely they have onychomycosis. How is the fungus going to jump from the floor into the nail without taking a little vacation on the bottom of the foot? It just isn’t going to happen.”
The presence of dermatophytoma is the third clinical clue. “These are dermatophyte abscesses encased in a biofilm, and they’re really hard to treat,” she said.
Treatments
Clinicians typically turn to one of three oral drugs for treating onychomycosis: terbinafine, itraconazole, and fluconazole, Dr. Elewski noted. Referring to terbinafine as “the gold standard,” she said that she typically writes a prescription for 90 250-mg pills. “When I give terbinafine, I often do baseline liver profiling, depending on the patient’s age, their state of health, their comorbidities, and other medications they’re taking,” she said. “If they’re 18 years old and otherwise healthy, I probably don’t.” While she generally prescribes 90 pills, she added, “keep in mind that 90 pills are not going to cure everybody. I see the patient 4 months later because the drug should stay in the nail for 30 days or more at therapeutic levels after you take that 90-day course.”
Another option is itraconazole, which can be taken at a dose of 200 mg a day for 12 weeks, or at a pulse dose, where patients take 400 mg every day for 1 week, 1 week a month, for 4 consecutive months. “I’ll often do a baseline liver profile with itraconazole, too,” Dr. Elewski said. “I don’t think you have to, but it makes sense if it’s feasible for you. Decide that based on each patient.”
Itraconazole can’t be given concomitantly with statins because of the potential for rhabdomyolysis. For patients taking statins, she consults with their physicians to make sure it’s safe to stop the statin a couple of days before and after their scheduled pulse dose of itraconazole. “This involves 1 week per month of taking itraconazole without the statin,” she said. “Or they could stop statins for the time you treat, if cleared by their doctor.”
As for fluconazole, Dr. Elewski usually prescribes 200 mg once or twice per week until the nail is normal. She offers patients the mnemonic for “Fungal Fridays” or Toesdays” as a way for them to remember which day to take the fluconazole.
According to data in the package inserts, rates of complete and mycologic cures are 38% and 70% for terbinafine, respectively, 14% and 54% for itraconazole, and 37% to 48% and 47% to 62% for fluconazole. “These cures are not 100% based on the standard course [of the drug],” Dr. Elewski noted. “I don’t use the standard course. I believe in treating to terminate. You want to kill the fungus.”
Resistant dermatophytes ‘are coming’
Halting treatment with an oral drug at a particular time point instead of when the nail is fungal-free likely contributes to resistant strains, she added, noting that she has at least two dozen patients in her practice with dermatophyte resistance documented in labs. “We need to be antifungal stewards, because resistant dermatophytes are coming to us,” she said. “They’re here already, and we don’t want it to be endemic in the U.S.”
In a published study from 2020, researchers from India enrolled 200 patients with relapsing tinea corporis, tinea cruris, and tinea faciei and allocated 50 each to treatment with either fluconazole, griseofulvin, itraconazole, or terbinafine. At week 4, all treatment arms had cure rates of less than 8%. At week 8, the cure rates were 42% for fluconazole, 16% for griseofulvin, 28% for terbinafine, and 66% for itraconazole.
Based in part on these study findings, Dr. Elewski said that she has become more aggressive in her therapeutic approach, including treating some of her patients on terbinafine for a minimum of 6 months. “If that’s not enough, I keep treating,” she said. “But, patients may not respond to terbinafine; we see resistance. So, itraconazole may be our best drug going forward for treating onychomycosis. You just have to watch out for side effects of itraconazole, mainly drug-drug interactions.”
Dr. Elewski reported having no relevant financial disclosures related to her presentation.
NEW ORLEANS – .
“The PAS [periodic acid-Schiff] stain is very popular because it can identify the presence or absence of fungal elements, but a fungal culture will identify the organism living in the nail,” Dr. Elewski, professor and chair of dermatology at the University of Alabama, Birmingham, said at the annual meeting of the American Academy of Dermatology. “You also could do a PCR to identify the organism, with or without a KOH or PAS stain. It is often helpful to know what organism is causing the infection.”
While waiting for lab results, there are three clinical clues to look for – the first being that an infection likely resides in the toenail. “You almost never see dermatophyte onychomycosis in the fingernails without it being in the toenails, too,” Dr. Elewski said.
The presence of tinea pedis is a second clinical clue. “Sometimes it’s subtle, so I will ask the patient, ‘Have you been treating yourself for athlete’s foot?’ If they say ‘no, I’ve never had it,’ put down on your list that it’s unlikely they have onychomycosis. How is the fungus going to jump from the floor into the nail without taking a little vacation on the bottom of the foot? It just isn’t going to happen.”
The presence of dermatophytoma is the third clinical clue. “These are dermatophyte abscesses encased in a biofilm, and they’re really hard to treat,” she said.
Treatments
Clinicians typically turn to one of three oral drugs for treating onychomycosis: terbinafine, itraconazole, and fluconazole, Dr. Elewski noted. Referring to terbinafine as “the gold standard,” she said that she typically writes a prescription for 90 250-mg pills. “When I give terbinafine, I often do baseline liver profiling, depending on the patient’s age, their state of health, their comorbidities, and other medications they’re taking,” she said. “If they’re 18 years old and otherwise healthy, I probably don’t.” While she generally prescribes 90 pills, she added, “keep in mind that 90 pills are not going to cure everybody. I see the patient 4 months later because the drug should stay in the nail for 30 days or more at therapeutic levels after you take that 90-day course.”
Another option is itraconazole, which can be taken at a dose of 200 mg a day for 12 weeks, or at a pulse dose, where patients take 400 mg every day for 1 week, 1 week a month, for 4 consecutive months. “I’ll often do a baseline liver profile with itraconazole, too,” Dr. Elewski said. “I don’t think you have to, but it makes sense if it’s feasible for you. Decide that based on each patient.”
Itraconazole can’t be given concomitantly with statins because of the potential for rhabdomyolysis. For patients taking statins, she consults with their physicians to make sure it’s safe to stop the statin a couple of days before and after their scheduled pulse dose of itraconazole. “This involves 1 week per month of taking itraconazole without the statin,” she said. “Or they could stop statins for the time you treat, if cleared by their doctor.”
As for fluconazole, Dr. Elewski usually prescribes 200 mg once or twice per week until the nail is normal. She offers patients the mnemonic for “Fungal Fridays” or Toesdays” as a way for them to remember which day to take the fluconazole.
According to data in the package inserts, rates of complete and mycologic cures are 38% and 70% for terbinafine, respectively, 14% and 54% for itraconazole, and 37% to 48% and 47% to 62% for fluconazole. “These cures are not 100% based on the standard course [of the drug],” Dr. Elewski noted. “I don’t use the standard course. I believe in treating to terminate. You want to kill the fungus.”
Resistant dermatophytes ‘are coming’
Halting treatment with an oral drug at a particular time point instead of when the nail is fungal-free likely contributes to resistant strains, she added, noting that she has at least two dozen patients in her practice with dermatophyte resistance documented in labs. “We need to be antifungal stewards, because resistant dermatophytes are coming to us,” she said. “They’re here already, and we don’t want it to be endemic in the U.S.”
In a published study from 2020, researchers from India enrolled 200 patients with relapsing tinea corporis, tinea cruris, and tinea faciei and allocated 50 each to treatment with either fluconazole, griseofulvin, itraconazole, or terbinafine. At week 4, all treatment arms had cure rates of less than 8%. At week 8, the cure rates were 42% for fluconazole, 16% for griseofulvin, 28% for terbinafine, and 66% for itraconazole.
Based in part on these study findings, Dr. Elewski said that she has become more aggressive in her therapeutic approach, including treating some of her patients on terbinafine for a minimum of 6 months. “If that’s not enough, I keep treating,” she said. “But, patients may not respond to terbinafine; we see resistance. So, itraconazole may be our best drug going forward for treating onychomycosis. You just have to watch out for side effects of itraconazole, mainly drug-drug interactions.”
Dr. Elewski reported having no relevant financial disclosures related to her presentation.
NEW ORLEANS – .
“The PAS [periodic acid-Schiff] stain is very popular because it can identify the presence or absence of fungal elements, but a fungal culture will identify the organism living in the nail,” Dr. Elewski, professor and chair of dermatology at the University of Alabama, Birmingham, said at the annual meeting of the American Academy of Dermatology. “You also could do a PCR to identify the organism, with or without a KOH or PAS stain. It is often helpful to know what organism is causing the infection.”
While waiting for lab results, there are three clinical clues to look for – the first being that an infection likely resides in the toenail. “You almost never see dermatophyte onychomycosis in the fingernails without it being in the toenails, too,” Dr. Elewski said.
The presence of tinea pedis is a second clinical clue. “Sometimes it’s subtle, so I will ask the patient, ‘Have you been treating yourself for athlete’s foot?’ If they say ‘no, I’ve never had it,’ put down on your list that it’s unlikely they have onychomycosis. How is the fungus going to jump from the floor into the nail without taking a little vacation on the bottom of the foot? It just isn’t going to happen.”
The presence of dermatophytoma is the third clinical clue. “These are dermatophyte abscesses encased in a biofilm, and they’re really hard to treat,” she said.
Treatments
Clinicians typically turn to one of three oral drugs for treating onychomycosis: terbinafine, itraconazole, and fluconazole, Dr. Elewski noted. Referring to terbinafine as “the gold standard,” she said that she typically writes a prescription for 90 250-mg pills. “When I give terbinafine, I often do baseline liver profiling, depending on the patient’s age, their state of health, their comorbidities, and other medications they’re taking,” she said. “If they’re 18 years old and otherwise healthy, I probably don’t.” While she generally prescribes 90 pills, she added, “keep in mind that 90 pills are not going to cure everybody. I see the patient 4 months later because the drug should stay in the nail for 30 days or more at therapeutic levels after you take that 90-day course.”
Another option is itraconazole, which can be taken at a dose of 200 mg a day for 12 weeks, or at a pulse dose, where patients take 400 mg every day for 1 week, 1 week a month, for 4 consecutive months. “I’ll often do a baseline liver profile with itraconazole, too,” Dr. Elewski said. “I don’t think you have to, but it makes sense if it’s feasible for you. Decide that based on each patient.”
Itraconazole can’t be given concomitantly with statins because of the potential for rhabdomyolysis. For patients taking statins, she consults with their physicians to make sure it’s safe to stop the statin a couple of days before and after their scheduled pulse dose of itraconazole. “This involves 1 week per month of taking itraconazole without the statin,” she said. “Or they could stop statins for the time you treat, if cleared by their doctor.”
As for fluconazole, Dr. Elewski usually prescribes 200 mg once or twice per week until the nail is normal. She offers patients the mnemonic for “Fungal Fridays” or Toesdays” as a way for them to remember which day to take the fluconazole.
According to data in the package inserts, rates of complete and mycologic cures are 38% and 70% for terbinafine, respectively, 14% and 54% for itraconazole, and 37% to 48% and 47% to 62% for fluconazole. “These cures are not 100% based on the standard course [of the drug],” Dr. Elewski noted. “I don’t use the standard course. I believe in treating to terminate. You want to kill the fungus.”
Resistant dermatophytes ‘are coming’
Halting treatment with an oral drug at a particular time point instead of when the nail is fungal-free likely contributes to resistant strains, she added, noting that she has at least two dozen patients in her practice with dermatophyte resistance documented in labs. “We need to be antifungal stewards, because resistant dermatophytes are coming to us,” she said. “They’re here already, and we don’t want it to be endemic in the U.S.”
In a published study from 2020, researchers from India enrolled 200 patients with relapsing tinea corporis, tinea cruris, and tinea faciei and allocated 50 each to treatment with either fluconazole, griseofulvin, itraconazole, or terbinafine. At week 4, all treatment arms had cure rates of less than 8%. At week 8, the cure rates were 42% for fluconazole, 16% for griseofulvin, 28% for terbinafine, and 66% for itraconazole.
Based in part on these study findings, Dr. Elewski said that she has become more aggressive in her therapeutic approach, including treating some of her patients on terbinafine for a minimum of 6 months. “If that’s not enough, I keep treating,” she said. “But, patients may not respond to terbinafine; we see resistance. So, itraconazole may be our best drug going forward for treating onychomycosis. You just have to watch out for side effects of itraconazole, mainly drug-drug interactions.”
Dr. Elewski reported having no relevant financial disclosures related to her presentation.
AT AAD 2023
Rash and edema

Lab work was ordered and came back within normal range, except for an elevated white blood cell count (19,700/mm3; reference range, 4500-13,500/mm3). His mild systemic symptoms, skin lesions without blistering or necrosis, acral edema, and the absence of lymphadenopathy pointed to a diagnosis of urticaria multiforme.
Urticaria multiforme, also called acute annular urticaria or acute urticarial hypersensitivity syndrome, is a histamine-mediated hypersensitivity reaction characterized by transient annular, polycyclic, urticarial lesions with central ecchymosis. It typically manifests in children ages 4 months to 4 years and begins with small erythematous macules, papules, and plaques that progress to large blanchable wheals with dusky blue centers.1-3 Lesions are usually located on the face, trunk, and extremities and are often pruritic (60%-94%).1-3 The rash generally lasts 2 to 12 days.1,3
Patients often report a preceding viral illness, otitis media, recent use of antibiotics, or recent immunizations. Patients also often have associated facial or acral edema (72%).1 Children with significant edema of the feet may find walking difficult, which should not be confused with arthritis or arthralgias.
The diagnosis is made clinically and should not require a skin biopsy or extensive laboratory testing. When performed, laboratory studies—including CBC, erythrocyte sedimentation rate, C-reactive protein, and urinalysis—are routinely normal. The differential diagnosis in this case included erythema multiforme, Henoch-Schönlein purpura, serum sickness-like reaction, and urticarial vasculitis.1,2,4
Treatment consists of discontinuing any offending agent (if suspected) and using systemic H1 or H2 antihistamines for symptom relief. Systemic steroids should only be given in refractory cases.
Our patient’s amoxicillin was discontinued, and he was started on a 14-day course of cetirizine 5 mg bid and hydroxyzine 10 mg at bedtime. He was also started on triamcinolone 0.1% cream to be applied twice daily for 1 week. During his 3-day hospital stay, his fever resolved, and his rash and edema improved.
During an outpatient follow-up visit with a pediatric dermatologist 2 weeks after discharge, the patient’s rash was still present and dermatographism was noted. In light of this, his parents were instructed to continue giving the cetirizine and hydroxyzine once daily for an additional 2 weeks and to return as needed.
This case was adapted from: Cook JS, Angles A, Morley E. Urticaria and edema in a 2-year-old boy. J Fam Pract. 2021;70:353-355.
Photos courtesy of Jeffrey S. Cook, MD, FAAFP
1. Shah KN, Honig PJ, Yan AC. “Urticaria multiforme”: a case series and review of acute annular urticarial hypersensitivity syndromes in children. Pediatrics. 2007;119:e1177-e1183. doi: 10.1542/peds.2006-1553
2. Emer JJ, Bernardo SG, Kovalerchik O, et al. Urticaria multiforme. J Clin Aesthet Dermatol. 2013;6:34-39.
3. Starnes L, Patel T, Skinner RB. Urticaria multiforme – a case report. Pediatr Dermatol. 2011; 28:436-438. doi: 10.1111/j.1525-1470.2011.01311.x
4. Reamy BV, Williams PM, Lindsay TJ. Henoch-Schönlein purpura. Am Fam Physician. 2009;80:697-704.
Lab work was ordered and came back within normal range, except for an elevated white blood cell count (19,700/mm3; reference range, 4500-13,500/mm3). His mild systemic symptoms, skin lesions without blistering or necrosis, acral edema, and the absence of lymphadenopathy pointed to a diagnosis of urticaria multiforme.
Urticaria multiforme, also called acute annular urticaria or acute urticarial hypersensitivity syndrome, is a histamine-mediated hypersensitivity reaction characterized by transient annular, polycyclic, urticarial lesions with central ecchymosis. It typically manifests in children ages 4 months to 4 years and begins with small erythematous macules, papules, and plaques that progress to large blanchable wheals with dusky blue centers.1-3 Lesions are usually located on the face, trunk, and extremities and are often pruritic (60%-94%).1-3 The rash generally lasts 2 to 12 days.1,3
Patients often report a preceding viral illness, otitis media, recent use of antibiotics, or recent immunizations. Patients also often have associated facial or acral edema (72%).1 Children with significant edema of the feet may find walking difficult, which should not be confused with arthritis or arthralgias.
The diagnosis is made clinically and should not require a skin biopsy or extensive laboratory testing. When performed, laboratory studies—including CBC, erythrocyte sedimentation rate, C-reactive protein, and urinalysis—are routinely normal. The differential diagnosis in this case included erythema multiforme, Henoch-Schönlein purpura, serum sickness-like reaction, and urticarial vasculitis.1,2,4
Treatment consists of discontinuing any offending agent (if suspected) and using systemic H1 or H2 antihistamines for symptom relief. Systemic steroids should only be given in refractory cases.
Our patient’s amoxicillin was discontinued, and he was started on a 14-day course of cetirizine 5 mg bid and hydroxyzine 10 mg at bedtime. He was also started on triamcinolone 0.1% cream to be applied twice daily for 1 week. During his 3-day hospital stay, his fever resolved, and his rash and edema improved.
During an outpatient follow-up visit with a pediatric dermatologist 2 weeks after discharge, the patient’s rash was still present and dermatographism was noted. In light of this, his parents were instructed to continue giving the cetirizine and hydroxyzine once daily for an additional 2 weeks and to return as needed.
This case was adapted from: Cook JS, Angles A, Morley E. Urticaria and edema in a 2-year-old boy. J Fam Pract. 2021;70:353-355.
Photos courtesy of Jeffrey S. Cook, MD, FAAFP
Lab work was ordered and came back within normal range, except for an elevated white blood cell count (19,700/mm3; reference range, 4500-13,500/mm3). His mild systemic symptoms, skin lesions without blistering or necrosis, acral edema, and the absence of lymphadenopathy pointed to a diagnosis of urticaria multiforme.
Urticaria multiforme, also called acute annular urticaria or acute urticarial hypersensitivity syndrome, is a histamine-mediated hypersensitivity reaction characterized by transient annular, polycyclic, urticarial lesions with central ecchymosis. It typically manifests in children ages 4 months to 4 years and begins with small erythematous macules, papules, and plaques that progress to large blanchable wheals with dusky blue centers.1-3 Lesions are usually located on the face, trunk, and extremities and are often pruritic (60%-94%).1-3 The rash generally lasts 2 to 12 days.1,3
Patients often report a preceding viral illness, otitis media, recent use of antibiotics, or recent immunizations. Patients also often have associated facial or acral edema (72%).1 Children with significant edema of the feet may find walking difficult, which should not be confused with arthritis or arthralgias.
The diagnosis is made clinically and should not require a skin biopsy or extensive laboratory testing. When performed, laboratory studies—including CBC, erythrocyte sedimentation rate, C-reactive protein, and urinalysis—are routinely normal. The differential diagnosis in this case included erythema multiforme, Henoch-Schönlein purpura, serum sickness-like reaction, and urticarial vasculitis.1,2,4
Treatment consists of discontinuing any offending agent (if suspected) and using systemic H1 or H2 antihistamines for symptom relief. Systemic steroids should only be given in refractory cases.
Our patient’s amoxicillin was discontinued, and he was started on a 14-day course of cetirizine 5 mg bid and hydroxyzine 10 mg at bedtime. He was also started on triamcinolone 0.1% cream to be applied twice daily for 1 week. During his 3-day hospital stay, his fever resolved, and his rash and edema improved.
During an outpatient follow-up visit with a pediatric dermatologist 2 weeks after discharge, the patient’s rash was still present and dermatographism was noted. In light of this, his parents were instructed to continue giving the cetirizine and hydroxyzine once daily for an additional 2 weeks and to return as needed.
This case was adapted from: Cook JS, Angles A, Morley E. Urticaria and edema in a 2-year-old boy. J Fam Pract. 2021;70:353-355.
Photos courtesy of Jeffrey S. Cook, MD, FAAFP
1. Shah KN, Honig PJ, Yan AC. “Urticaria multiforme”: a case series and review of acute annular urticarial hypersensitivity syndromes in children. Pediatrics. 2007;119:e1177-e1183. doi: 10.1542/peds.2006-1553
2. Emer JJ, Bernardo SG, Kovalerchik O, et al. Urticaria multiforme. J Clin Aesthet Dermatol. 2013;6:34-39.
3. Starnes L, Patel T, Skinner RB. Urticaria multiforme – a case report. Pediatr Dermatol. 2011; 28:436-438. doi: 10.1111/j.1525-1470.2011.01311.x
4. Reamy BV, Williams PM, Lindsay TJ. Henoch-Schönlein purpura. Am Fam Physician. 2009;80:697-704.
1. Shah KN, Honig PJ, Yan AC. “Urticaria multiforme”: a case series and review of acute annular urticarial hypersensitivity syndromes in children. Pediatrics. 2007;119:e1177-e1183. doi: 10.1542/peds.2006-1553
2. Emer JJ, Bernardo SG, Kovalerchik O, et al. Urticaria multiforme. J Clin Aesthet Dermatol. 2013;6:34-39.
3. Starnes L, Patel T, Skinner RB. Urticaria multiforme – a case report. Pediatr Dermatol. 2011; 28:436-438. doi: 10.1111/j.1525-1470.2011.01311.x
4. Reamy BV, Williams PM, Lindsay TJ. Henoch-Schönlein purpura. Am Fam Physician. 2009;80:697-704.

Cases of potentially deadly fungus jump 200%: CDC
prompting the Centers for Disease Control and Prevention to issue a warning to health care facilities about the rising threat.
C. auris is a yeast that spreads easily from touching it on a surface like a countertop. It can also spread from person to person. It isn’t a threat to healthy people, but people in hospitals and nursing homes are at a heightened risk because they might have weakened immune systems or be using invasive medical devices that can introduce the fungus inside their bodies. When C. auris progresses to causing an infection that reaches the brain, blood, or lungs, more than one in three people die.
The worrying increase was detailed in the journal Annals of Internal Medicine. In 2021, cases reached a count of 3,270 with an active infection, and 7,413 cases showed the fungus was present but hadn’t caused an infection. Infection counts were up 95% over the previous year, and the fungus showed up on screenings three times as often. The number of cases resistant to medication also tripled.
The CDC called the figures “alarming,” noting that the fungus was only detected in the United States in 2016.
“The timing of this increase and findings from public health investigations suggest C. auris spread may have worsened due to strain on health care and public health systems during the COVID-19 pandemic,” the CDC explained in a news release.
Another potential reason for the jump could be that screening for C. auris has simply increased and it’s being found more often because it’s being looked for more often. But researchers believe that, even with the increase in testing, the reported counts are underestimated. That’s because even though screening has increased, health care providers still aren’t looking for the presence of the fungus as often as the CDC would like.
“The rapid rise and geographic spread of cases is concerning and emphasizes the need for continued surveillance, expanded lab capacity, quicker diagnostic tests, and adherence to proven infection prevention and control,” said study author Meghan Lyman, MD, a CDC epidemiologist in Atlanta, in a statement.
Cases of C. auris continued to rise in 2022, the CDC said. A map on the agency’s website of reported cases from 2022 shows it was found in more than half of U.S. states, with the highest counts occurring in California, Florida, Illinois, Nevada, New York, and Texas. The fungus is a problem worldwide and is listed among the most threatening treatment-resistant fungi by the World Health Organization.
The study authors concluded that screening capacity for the fungus needs to be expanded nationwide so that when C. auris is detected, measures can be taken to prevent its spread.
A version of this article originally appeared on WebMD.com.
prompting the Centers for Disease Control and Prevention to issue a warning to health care facilities about the rising threat.
C. auris is a yeast that spreads easily from touching it on a surface like a countertop. It can also spread from person to person. It isn’t a threat to healthy people, but people in hospitals and nursing homes are at a heightened risk because they might have weakened immune systems or be using invasive medical devices that can introduce the fungus inside their bodies. When C. auris progresses to causing an infection that reaches the brain, blood, or lungs, more than one in three people die.
The worrying increase was detailed in the journal Annals of Internal Medicine. In 2021, cases reached a count of 3,270 with an active infection, and 7,413 cases showed the fungus was present but hadn’t caused an infection. Infection counts were up 95% over the previous year, and the fungus showed up on screenings three times as often. The number of cases resistant to medication also tripled.
The CDC called the figures “alarming,” noting that the fungus was only detected in the United States in 2016.
“The timing of this increase and findings from public health investigations suggest C. auris spread may have worsened due to strain on health care and public health systems during the COVID-19 pandemic,” the CDC explained in a news release.
Another potential reason for the jump could be that screening for C. auris has simply increased and it’s being found more often because it’s being looked for more often. But researchers believe that, even with the increase in testing, the reported counts are underestimated. That’s because even though screening has increased, health care providers still aren’t looking for the presence of the fungus as often as the CDC would like.
“The rapid rise and geographic spread of cases is concerning and emphasizes the need for continued surveillance, expanded lab capacity, quicker diagnostic tests, and adherence to proven infection prevention and control,” said study author Meghan Lyman, MD, a CDC epidemiologist in Atlanta, in a statement.
Cases of C. auris continued to rise in 2022, the CDC said. A map on the agency’s website of reported cases from 2022 shows it was found in more than half of U.S. states, with the highest counts occurring in California, Florida, Illinois, Nevada, New York, and Texas. The fungus is a problem worldwide and is listed among the most threatening treatment-resistant fungi by the World Health Organization.
The study authors concluded that screening capacity for the fungus needs to be expanded nationwide so that when C. auris is detected, measures can be taken to prevent its spread.
A version of this article originally appeared on WebMD.com.
prompting the Centers for Disease Control and Prevention to issue a warning to health care facilities about the rising threat.
C. auris is a yeast that spreads easily from touching it on a surface like a countertop. It can also spread from person to person. It isn’t a threat to healthy people, but people in hospitals and nursing homes are at a heightened risk because they might have weakened immune systems or be using invasive medical devices that can introduce the fungus inside their bodies. When C. auris progresses to causing an infection that reaches the brain, blood, or lungs, more than one in three people die.
The worrying increase was detailed in the journal Annals of Internal Medicine. In 2021, cases reached a count of 3,270 with an active infection, and 7,413 cases showed the fungus was present but hadn’t caused an infection. Infection counts were up 95% over the previous year, and the fungus showed up on screenings three times as often. The number of cases resistant to medication also tripled.
The CDC called the figures “alarming,” noting that the fungus was only detected in the United States in 2016.
“The timing of this increase and findings from public health investigations suggest C. auris spread may have worsened due to strain on health care and public health systems during the COVID-19 pandemic,” the CDC explained in a news release.
Another potential reason for the jump could be that screening for C. auris has simply increased and it’s being found more often because it’s being looked for more often. But researchers believe that, even with the increase in testing, the reported counts are underestimated. That’s because even though screening has increased, health care providers still aren’t looking for the presence of the fungus as often as the CDC would like.
“The rapid rise and geographic spread of cases is concerning and emphasizes the need for continued surveillance, expanded lab capacity, quicker diagnostic tests, and adherence to proven infection prevention and control,” said study author Meghan Lyman, MD, a CDC epidemiologist in Atlanta, in a statement.
Cases of C. auris continued to rise in 2022, the CDC said. A map on the agency’s website of reported cases from 2022 shows it was found in more than half of U.S. states, with the highest counts occurring in California, Florida, Illinois, Nevada, New York, and Texas. The fungus is a problem worldwide and is listed among the most threatening treatment-resistant fungi by the World Health Organization.
The study authors concluded that screening capacity for the fungus needs to be expanded nationwide so that when C. auris is detected, measures can be taken to prevent its spread.
A version of this article originally appeared on WebMD.com.
Phase 3 prurigo nodularis trial shows positive results for nemolizumab
NEW ORLEANS – demonstrated.
Nemolizumab is a first-in-class investigational monoclonal antibody directed against the interleukin-31 receptor alpha that blocks signaling from IL-31. “From prior studies we know that it modulates pruritus, but also alters keratinocyte differentiation, inflammation, and fibrosis,” one of the investigators, Shawn G. Kwatra, MD, of the department of dermatology, Johns Hopkins University, Baltimore, said during a late-breaking research session at the annual meeting of the American Academy of Dermatology.

OLYMPIA 2 was a phase 3, multicenter, double-blind study in adults with PN presenting with 20 or more nodules, and Investigator’s Global Assessment (IGA) score of 3 or more, and the Peak Pruritus Numerical Rating Scale (PP-NRS) score of 7 or more. Exclusion criteria included chronic pruritus resulting from an active condition other than PN, such as neuropathic and psychogenic pruritus and active atopic dermatitis. In addition, the use of topical steroids, considered a rescue therapy, was not allowed in the trial, Dr. Kwatra said.
After an initial screening period, 274 patients at 73 sites in nine countries were randomized 2:1 either to the nemolizumab monotherapy or placebo. Following an initial 60-mg subcutaneous dose, patients received 30 mg or 60 mg (depending on their baseline weight) every 4 weeks for 16 weeks. The primary endpoint was the proportion of patients with a 4-point or greater improvement in the PP-NRS from baseline at week 16 and the proportion of patients with IGA success at week 16.
Selected key secondary endpoints included the proportion of patients with a 4 point or greater improvement from baseline in the PP-NRS at week 4, the Sleep Disturbance Numerical Rating Scale at week 4, and the SD-NRS at week 16. Safety endpoints included the incidence and severity of all adverse events.
Of the 274 patients randomized, 183 received nemolizumab and 91 received placebo. A total of 174 patients in the nemolizumab group completed the study, compared with 88 in the placebo group. The mean age of study participants was 53 years, 61% were women, 79% were White, 14% were Asian, and the rest were from other racial groups. More than half (57%) had IGA category 3 disease (moderate) and the remainder had IGA category 4 disease (severe); 63% had 20-100 lesions, and the remainder had more than 100. About one-third of study enrollees (32%) had a history of atopy.
Primary, secondary endpoint results
Dr. Kwatra reported that 56.3% of the patients in the nemolizumab group achieved a 4-point or greater improvement in the PP-NRS at week 16, compared with 20.9% of those in the placebo group (P < .0001), while 37.7% of those in the nemolizumab group achieved IGA success at week 16, compared with 11% of those in the placebo group (P < .0001).
As for secondary endpoints, 41% of patients in the nemolizumab group achieved a 4-point or greater improvement in PP-NRS at week 4, compared with 7.7% of those in the placebo group (P < .0001); and 37.2% of patients in the nemolizumab group achieved a 4-point or greater improvement in SD-NRS at week 4, compared with 9.9% of those in the placebo group (P < .0001). Almost 52% of patients in the nemolizumab group achieved a 4-point or greater improvement in SD-NRS at week 16, compared with 20.9% of those in the placebo group (P < .0001); and 9.8% of those in the nemolizumab group achieved IGA success at week 4, compared with 1.1% of those in the placebo group (P < .0074).
Adverse events
Treatment-emergent adverse events occurred in 61.2% of subjects in the nemolizumab group, compared with 52.7% of those in the placebo group. “There were no imbalances overall, [including] no injection-related reactions in either group,” Dr. Kwatra said. There was one case of newly diagnosed asthma in the placebo arm, and none in the treatment arm.
The researchers observed a slightly increased onset of atopic dermatitis in the treatment arm, compared with the placebo arm (5.5% vs. 0%). “Seven out of those 10 patients actually had a history of atopic dermatitis or high IgE [levels] and they were mostly managed with topical steroids without study drug discontinuation,” Dr. Kwatra added. Neurodermatitis, or worsening of PN, occurred in 3.8% of patients in the nemolizumab group, compared with 11% of those in the placebo group.
“The results of this study extend the efficacy and safety findings from the phase 2 study of nemolizumab in patients with PN,” Dr. Kwatra concluded. “I think they also help to usher in a new era of PN [treatment] in prime time.”
Kenneth B. Gordon, MD, who chairs the department of dermatology at the Medical College of Wisconsin, Milwaukee, and was asked to comment on the study, was impressed with nemolizumab’s propensity for blocking IL-31. “To be able to treat PN effectively by simply blocking the itch and not having a significant inflammatory function is really interesting,” he said in an interview at the meeting. If approved, nemolizumab “gives us another treatment option for a disease that is really debilitating. It’s very promising and we hope [the drug] will be available to us in the near future.”
Nemolizumab is being developed by Galderma. According to a press release from the company, nemolizumab was granted Breakthrough Therapy designation by the Food and Drug Administration in December 2019 for the treatment of pruritus associated with PN, a status that was reconfirmed in February 2023.
Dr. Kwatra disclosed that he is an advisory board member/consultant for Galderma, AbbVie, Amgen, Arcutis, ASLAN Pharmaceuticals, Cara Therapeutics, Castle Biosciences, Celldex, Incyte, Johnson and Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi. Dr. Gordon disclosed that he is a consultant to, an investigator for, and/or a member of the advisory board for several pharmaceutical companies, but not Galderma.
NEW ORLEANS – demonstrated.
Nemolizumab is a first-in-class investigational monoclonal antibody directed against the interleukin-31 receptor alpha that blocks signaling from IL-31. “From prior studies we know that it modulates pruritus, but also alters keratinocyte differentiation, inflammation, and fibrosis,” one of the investigators, Shawn G. Kwatra, MD, of the department of dermatology, Johns Hopkins University, Baltimore, said during a late-breaking research session at the annual meeting of the American Academy of Dermatology.
OLYMPIA 2 was a phase 3, multicenter, double-blind study in adults with PN presenting with 20 or more nodules, and Investigator’s Global Assessment (IGA) score of 3 or more, and the Peak Pruritus Numerical Rating Scale (PP-NRS) score of 7 or more. Exclusion criteria included chronic pruritus resulting from an active condition other than PN, such as neuropathic and psychogenic pruritus and active atopic dermatitis. In addition, the use of topical steroids, considered a rescue therapy, was not allowed in the trial, Dr. Kwatra said.
After an initial screening period, 274 patients at 73 sites in nine countries were randomized 2:1 either to the nemolizumab monotherapy or placebo. Following an initial 60-mg subcutaneous dose, patients received 30 mg or 60 mg (depending on their baseline weight) every 4 weeks for 16 weeks. The primary endpoint was the proportion of patients with a 4-point or greater improvement in the PP-NRS from baseline at week 16 and the proportion of patients with IGA success at week 16.
Selected key secondary endpoints included the proportion of patients with a 4 point or greater improvement from baseline in the PP-NRS at week 4, the Sleep Disturbance Numerical Rating Scale at week 4, and the SD-NRS at week 16. Safety endpoints included the incidence and severity of all adverse events.
Of the 274 patients randomized, 183 received nemolizumab and 91 received placebo. A total of 174 patients in the nemolizumab group completed the study, compared with 88 in the placebo group. The mean age of study participants was 53 years, 61% were women, 79% were White, 14% were Asian, and the rest were from other racial groups. More than half (57%) had IGA category 3 disease (moderate) and the remainder had IGA category 4 disease (severe); 63% had 20-100 lesions, and the remainder had more than 100. About one-third of study enrollees (32%) had a history of atopy.
Primary, secondary endpoint results
Dr. Kwatra reported that 56.3% of the patients in the nemolizumab group achieved a 4-point or greater improvement in the PP-NRS at week 16, compared with 20.9% of those in the placebo group (P < .0001), while 37.7% of those in the nemolizumab group achieved IGA success at week 16, compared with 11% of those in the placebo group (P < .0001).
As for secondary endpoints, 41% of patients in the nemolizumab group achieved a 4-point or greater improvement in PP-NRS at week 4, compared with 7.7% of those in the placebo group (P < .0001); and 37.2% of patients in the nemolizumab group achieved a 4-point or greater improvement in SD-NRS at week 4, compared with 9.9% of those in the placebo group (P < .0001). Almost 52% of patients in the nemolizumab group achieved a 4-point or greater improvement in SD-NRS at week 16, compared with 20.9% of those in the placebo group (P < .0001); and 9.8% of those in the nemolizumab group achieved IGA success at week 4, compared with 1.1% of those in the placebo group (P < .0074).
Adverse events
Treatment-emergent adverse events occurred in 61.2% of subjects in the nemolizumab group, compared with 52.7% of those in the placebo group. “There were no imbalances overall, [including] no injection-related reactions in either group,” Dr. Kwatra said. There was one case of newly diagnosed asthma in the placebo arm, and none in the treatment arm.
The researchers observed a slightly increased onset of atopic dermatitis in the treatment arm, compared with the placebo arm (5.5% vs. 0%). “Seven out of those 10 patients actually had a history of atopic dermatitis or high IgE [levels] and they were mostly managed with topical steroids without study drug discontinuation,” Dr. Kwatra added. Neurodermatitis, or worsening of PN, occurred in 3.8% of patients in the nemolizumab group, compared with 11% of those in the placebo group.
“The results of this study extend the efficacy and safety findings from the phase 2 study of nemolizumab in patients with PN,” Dr. Kwatra concluded. “I think they also help to usher in a new era of PN [treatment] in prime time.”
Kenneth B. Gordon, MD, who chairs the department of dermatology at the Medical College of Wisconsin, Milwaukee, and was asked to comment on the study, was impressed with nemolizumab’s propensity for blocking IL-31. “To be able to treat PN effectively by simply blocking the itch and not having a significant inflammatory function is really interesting,” he said in an interview at the meeting. If approved, nemolizumab “gives us another treatment option for a disease that is really debilitating. It’s very promising and we hope [the drug] will be available to us in the near future.”
Nemolizumab is being developed by Galderma. According to a press release from the company, nemolizumab was granted Breakthrough Therapy designation by the Food and Drug Administration in December 2019 for the treatment of pruritus associated with PN, a status that was reconfirmed in February 2023.
Dr. Kwatra disclosed that he is an advisory board member/consultant for Galderma, AbbVie, Amgen, Arcutis, ASLAN Pharmaceuticals, Cara Therapeutics, Castle Biosciences, Celldex, Incyte, Johnson and Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi. Dr. Gordon disclosed that he is a consultant to, an investigator for, and/or a member of the advisory board for several pharmaceutical companies, but not Galderma.
NEW ORLEANS – demonstrated.
Nemolizumab is a first-in-class investigational monoclonal antibody directed against the interleukin-31 receptor alpha that blocks signaling from IL-31. “From prior studies we know that it modulates pruritus, but also alters keratinocyte differentiation, inflammation, and fibrosis,” one of the investigators, Shawn G. Kwatra, MD, of the department of dermatology, Johns Hopkins University, Baltimore, said during a late-breaking research session at the annual meeting of the American Academy of Dermatology.
OLYMPIA 2 was a phase 3, multicenter, double-blind study in adults with PN presenting with 20 or more nodules, and Investigator’s Global Assessment (IGA) score of 3 or more, and the Peak Pruritus Numerical Rating Scale (PP-NRS) score of 7 or more. Exclusion criteria included chronic pruritus resulting from an active condition other than PN, such as neuropathic and psychogenic pruritus and active atopic dermatitis. In addition, the use of topical steroids, considered a rescue therapy, was not allowed in the trial, Dr. Kwatra said.
After an initial screening period, 274 patients at 73 sites in nine countries were randomized 2:1 either to the nemolizumab monotherapy or placebo. Following an initial 60-mg subcutaneous dose, patients received 30 mg or 60 mg (depending on their baseline weight) every 4 weeks for 16 weeks. The primary endpoint was the proportion of patients with a 4-point or greater improvement in the PP-NRS from baseline at week 16 and the proportion of patients with IGA success at week 16.
Selected key secondary endpoints included the proportion of patients with a 4 point or greater improvement from baseline in the PP-NRS at week 4, the Sleep Disturbance Numerical Rating Scale at week 4, and the SD-NRS at week 16. Safety endpoints included the incidence and severity of all adverse events.
Of the 274 patients randomized, 183 received nemolizumab and 91 received placebo. A total of 174 patients in the nemolizumab group completed the study, compared with 88 in the placebo group. The mean age of study participants was 53 years, 61% were women, 79% were White, 14% were Asian, and the rest were from other racial groups. More than half (57%) had IGA category 3 disease (moderate) and the remainder had IGA category 4 disease (severe); 63% had 20-100 lesions, and the remainder had more than 100. About one-third of study enrollees (32%) had a history of atopy.
Primary, secondary endpoint results
Dr. Kwatra reported that 56.3% of the patients in the nemolizumab group achieved a 4-point or greater improvement in the PP-NRS at week 16, compared with 20.9% of those in the placebo group (P < .0001), while 37.7% of those in the nemolizumab group achieved IGA success at week 16, compared with 11% of those in the placebo group (P < .0001).
As for secondary endpoints, 41% of patients in the nemolizumab group achieved a 4-point or greater improvement in PP-NRS at week 4, compared with 7.7% of those in the placebo group (P < .0001); and 37.2% of patients in the nemolizumab group achieved a 4-point or greater improvement in SD-NRS at week 4, compared with 9.9% of those in the placebo group (P < .0001). Almost 52% of patients in the nemolizumab group achieved a 4-point or greater improvement in SD-NRS at week 16, compared with 20.9% of those in the placebo group (P < .0001); and 9.8% of those in the nemolizumab group achieved IGA success at week 4, compared with 1.1% of those in the placebo group (P < .0074).
Adverse events
Treatment-emergent adverse events occurred in 61.2% of subjects in the nemolizumab group, compared with 52.7% of those in the placebo group. “There were no imbalances overall, [including] no injection-related reactions in either group,” Dr. Kwatra said. There was one case of newly diagnosed asthma in the placebo arm, and none in the treatment arm.
The researchers observed a slightly increased onset of atopic dermatitis in the treatment arm, compared with the placebo arm (5.5% vs. 0%). “Seven out of those 10 patients actually had a history of atopic dermatitis or high IgE [levels] and they were mostly managed with topical steroids without study drug discontinuation,” Dr. Kwatra added. Neurodermatitis, or worsening of PN, occurred in 3.8% of patients in the nemolizumab group, compared with 11% of those in the placebo group.
“The results of this study extend the efficacy and safety findings from the phase 2 study of nemolizumab in patients with PN,” Dr. Kwatra concluded. “I think they also help to usher in a new era of PN [treatment] in prime time.”
Kenneth B. Gordon, MD, who chairs the department of dermatology at the Medical College of Wisconsin, Milwaukee, and was asked to comment on the study, was impressed with nemolizumab’s propensity for blocking IL-31. “To be able to treat PN effectively by simply blocking the itch and not having a significant inflammatory function is really interesting,” he said in an interview at the meeting. If approved, nemolizumab “gives us another treatment option for a disease that is really debilitating. It’s very promising and we hope [the drug] will be available to us in the near future.”
Nemolizumab is being developed by Galderma. According to a press release from the company, nemolizumab was granted Breakthrough Therapy designation by the Food and Drug Administration in December 2019 for the treatment of pruritus associated with PN, a status that was reconfirmed in February 2023.
Dr. Kwatra disclosed that he is an advisory board member/consultant for Galderma, AbbVie, Amgen, Arcutis, ASLAN Pharmaceuticals, Cara Therapeutics, Castle Biosciences, Celldex, Incyte, Johnson and Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi. Dr. Gordon disclosed that he is a consultant to, an investigator for, and/or a member of the advisory board for several pharmaceutical companies, but not Galderma.
AT AAD 2023
Blisters on arms and legs

This patient was given a diagnosis of bullous pemphigoid. Although there were a number of clues that pointed to this diagnosis, confirming that this was the case required 2 biopsies and a blood draw. (More on this in a bit.)
Although rare and potentially lethal, bullous pemphigoid is the most common autoimmune blistering disease in the elderly. Patients present with tense bullae over limited or widespread areas of the skin. The pathogenesis includes development of autoimmune antibodies that target important proteins (BP180 and BP230) that bind basal epidermal keratinocytes to the dermis. When weakened by inflammation at these sites, the skin delaminates at the dermal-epidermal junction, while the cells of the epidermis continue to bind to each other. This leads to itching, hive-like wheals, and tense fluid-filled bullae.
The differential diagnosis of an acute or semi-acute bullous disease includes bullous pemphigoid, IgA pemphigoid, linear IgA bullous dermatosis, epidermolysis bullosa acquisita, and Senear-Usher syndrome. In this case, the large tense bullae suggested bullous pemphigoid over the other diagnoses.
Initial diagnosis requires 2 biopsies be performed: One at the edge of a bulla for a standard pathologic exam to identify the skin level at which the bulla is forming, and another biopsy of skin near the site of inflammation (5-10 mm away) to be sent for direct immunofluorescence (DIF) in Michel’s medium or Zeus medium. In bullous pemphigoid, the separation is at the dermal-epidermal junction, and IgG and C3 are found in the DIF in the same location. There are a couple ways to differentiate this disorder from epidermolysis bullosa acquisita—a similar blistering disorder in which autoantibodies attack collagen at the dermal-epidermal junction. A common approach is to send a patient’s serum for indirect immunofluorescence. This is done because it is impossible to distinguish between the 2 clinically.
While bullous pemphigoid has historically been treated with high-dose prednisone, it is more common now to treat with whole-body topical clobetasol and oral doxycycline 100 mg twice a day to avoid the adverse effects of the prednisone. Other immunosuppressive options, such as mycophenolate mofetil and cyclosporine, can provide the potency of prednisone with a more favorable long-term safety profile. Rituximab infusions are another very powerful and durable option in refractory or severe cases.1
This patient was treated with topical clobetasol and doxycycline 100 mg twice a day, but he had incomplete clearance after 2 to 3 weeks. At that point, mycophenolate mofetil was added to the regimen and was titrated up to 1000 mg twice daily. When clearance occurred, the clobetasol was discontinued and the mycophenolate mofetil was titrated down to 250 mg/d; the patient continues to maintain clearance at this dose. He continues on doxycycline 100 mg bid.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Ruggiero A, Megna M, Villani A, et al. Strategies to improve outcomes of bullous pemphigoid: a comprehensive review of clinical presentations, diagnosis, and patients' assessment. Clin Cosmet Investig Dermatol. 2022;15:661-673. doi:10.2147/CCID.S267573
This patient was given a diagnosis of bullous pemphigoid. Although there were a number of clues that pointed to this diagnosis, confirming that this was the case required 2 biopsies and a blood draw. (More on this in a bit.)
Although rare and potentially lethal, bullous pemphigoid is the most common autoimmune blistering disease in the elderly. Patients present with tense bullae over limited or widespread areas of the skin. The pathogenesis includes development of autoimmune antibodies that target important proteins (BP180 and BP230) that bind basal epidermal keratinocytes to the dermis. When weakened by inflammation at these sites, the skin delaminates at the dermal-epidermal junction, while the cells of the epidermis continue to bind to each other. This leads to itching, hive-like wheals, and tense fluid-filled bullae.
The differential diagnosis of an acute or semi-acute bullous disease includes bullous pemphigoid, IgA pemphigoid, linear IgA bullous dermatosis, epidermolysis bullosa acquisita, and Senear-Usher syndrome. In this case, the large tense bullae suggested bullous pemphigoid over the other diagnoses.
Initial diagnosis requires 2 biopsies be performed: One at the edge of a bulla for a standard pathologic exam to identify the skin level at which the bulla is forming, and another biopsy of skin near the site of inflammation (5-10 mm away) to be sent for direct immunofluorescence (DIF) in Michel’s medium or Zeus medium. In bullous pemphigoid, the separation is at the dermal-epidermal junction, and IgG and C3 are found in the DIF in the same location. There are a couple ways to differentiate this disorder from epidermolysis bullosa acquisita—a similar blistering disorder in which autoantibodies attack collagen at the dermal-epidermal junction. A common approach is to send a patient’s serum for indirect immunofluorescence. This is done because it is impossible to distinguish between the 2 clinically.
While bullous pemphigoid has historically been treated with high-dose prednisone, it is more common now to treat with whole-body topical clobetasol and oral doxycycline 100 mg twice a day to avoid the adverse effects of the prednisone. Other immunosuppressive options, such as mycophenolate mofetil and cyclosporine, can provide the potency of prednisone with a more favorable long-term safety profile. Rituximab infusions are another very powerful and durable option in refractory or severe cases.1
This patient was treated with topical clobetasol and doxycycline 100 mg twice a day, but he had incomplete clearance after 2 to 3 weeks. At that point, mycophenolate mofetil was added to the regimen and was titrated up to 1000 mg twice daily. When clearance occurred, the clobetasol was discontinued and the mycophenolate mofetil was titrated down to 250 mg/d; the patient continues to maintain clearance at this dose. He continues on doxycycline 100 mg bid.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
This patient was given a diagnosis of bullous pemphigoid. Although there were a number of clues that pointed to this diagnosis, confirming that this was the case required 2 biopsies and a blood draw. (More on this in a bit.)
Although rare and potentially lethal, bullous pemphigoid is the most common autoimmune blistering disease in the elderly. Patients present with tense bullae over limited or widespread areas of the skin. The pathogenesis includes development of autoimmune antibodies that target important proteins (BP180 and BP230) that bind basal epidermal keratinocytes to the dermis. When weakened by inflammation at these sites, the skin delaminates at the dermal-epidermal junction, while the cells of the epidermis continue to bind to each other. This leads to itching, hive-like wheals, and tense fluid-filled bullae.
The differential diagnosis of an acute or semi-acute bullous disease includes bullous pemphigoid, IgA pemphigoid, linear IgA bullous dermatosis, epidermolysis bullosa acquisita, and Senear-Usher syndrome. In this case, the large tense bullae suggested bullous pemphigoid over the other diagnoses.
Initial diagnosis requires 2 biopsies be performed: One at the edge of a bulla for a standard pathologic exam to identify the skin level at which the bulla is forming, and another biopsy of skin near the site of inflammation (5-10 mm away) to be sent for direct immunofluorescence (DIF) in Michel’s medium or Zeus medium. In bullous pemphigoid, the separation is at the dermal-epidermal junction, and IgG and C3 are found in the DIF in the same location. There are a couple ways to differentiate this disorder from epidermolysis bullosa acquisita—a similar blistering disorder in which autoantibodies attack collagen at the dermal-epidermal junction. A common approach is to send a patient’s serum for indirect immunofluorescence. This is done because it is impossible to distinguish between the 2 clinically.
While bullous pemphigoid has historically been treated with high-dose prednisone, it is more common now to treat with whole-body topical clobetasol and oral doxycycline 100 mg twice a day to avoid the adverse effects of the prednisone. Other immunosuppressive options, such as mycophenolate mofetil and cyclosporine, can provide the potency of prednisone with a more favorable long-term safety profile. Rituximab infusions are another very powerful and durable option in refractory or severe cases.1
This patient was treated with topical clobetasol and doxycycline 100 mg twice a day, but he had incomplete clearance after 2 to 3 weeks. At that point, mycophenolate mofetil was added to the regimen and was titrated up to 1000 mg twice daily. When clearance occurred, the clobetasol was discontinued and the mycophenolate mofetil was titrated down to 250 mg/d; the patient continues to maintain clearance at this dose. He continues on doxycycline 100 mg bid.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Ruggiero A, Megna M, Villani A, et al. Strategies to improve outcomes of bullous pemphigoid: a comprehensive review of clinical presentations, diagnosis, and patients' assessment. Clin Cosmet Investig Dermatol. 2022;15:661-673. doi:10.2147/CCID.S267573
1. Ruggiero A, Megna M, Villani A, et al. Strategies to improve outcomes of bullous pemphigoid: a comprehensive review of clinical presentations, diagnosis, and patients' assessment. Clin Cosmet Investig Dermatol. 2022;15:661-673. doi:10.2147/CCID.S267573

Novel therapy shows promise for treating skin-predominant dermatomyositis
NEW ORLEANS – in a double-blind, placebo-controlled phase 2 trial, according to results presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
“These findings support the inhibition of IFN-beta as a promising therapeutic strategy in skin-predominant disease,” said principal investigator Aaron Mangold, MD, associate professor of dermatology, Mayo Clinic, Scottsdale, Ariz.

Dermatomyositis, a rare autoimmune inflammatory condition that typically involves both skeletal muscles and skin, is a challenging disease with a diverse set of potential complications.
Immunosuppressive and immunomodulatory agents are used with mixed success for myositis, but skin manifestations, which include papular eruptions, heliotrope rash, photoerythema, burning, and pruritus, are often the most troublesome and the most difficult to control. Treatment options other than immunomodulators that target cutaneous involvement – which include steroids, emollients, and photoprotection – are generally modestly effective, according to Dr. Mangold.
Targeting an elevated cytokine
Interest in IFN-beta, which is elevated in the blood of individuals with dermatomyositis, was triggered by evidence that this cytokine plays an important role in driving the skin inflammation, Dr. Mangold explained.
“The blood concentrations of IFN-beta are positively correlated with cutaneous disease activity and severity,” he said.
The study drug, currently known as PF-06823859 (Dazukibart), “is a potent, selective humanized IgG1-neutralizing antibody directed at IFN-beta,” Dr. Mangold said. A dose-ranging phase 1 study published 2 years ago provided evidence of acceptable pharmacokinetics and safety in healthy individuals to support treatment studies for disorders associated with elevated IFN-beta levels. In addition to dermatomyositis, this includes systemic lupus erythematosus.
In this phase 2 trial, patients whose condition was not improved by at least one standard-care therapy for skin manifestations of dermatomyositis were eligible if they had moderate to severe disease as measured with the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI), according to Dr. Mangold. During the study, patients were allowed to remain on a disease modifying antirheumatic drug and/or prednisone if they had been on stable doses and did not change the dose.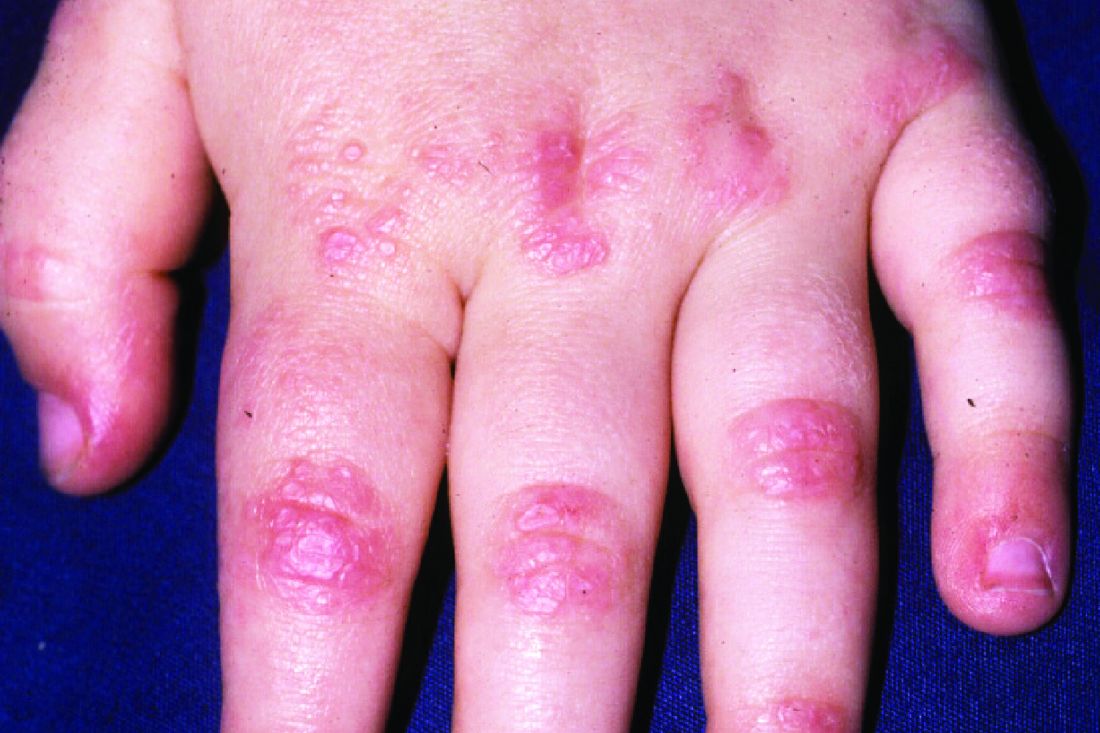
After a screening run-in, the trial had two blinded stages. In stage 1, 30 patients were randomly assigned either to 600 mg of PF-06823859 or to placebo, both administered intravenously every 4 weeks. A second cohort of 25 patients was randomly assigned in stage 2 to placebo, 150 mg of PF-06823859, or 600 mg of PF-06823859. The primary endpoint assessed at 12 weeks was a greater than 5-point reduction in CDASI score or greater than 40% CDASI improvement from baseline.
Both endpoints are associated with a clinically meaningful response in regard to an improved quality of life, Dr. Mangold noted.
Both doses better than placebo
In results from the stage 1 portion, the mean reduction in CDASI at 12 weeks after three doses of the assigned therapy was 18.8 points in the active-treatment group versus 3.9 points in the placebo group. In pooled data from stage 1 and 2, the reductions were 16.6 points, 19.2 points, and 2.9 points for the 150-mg, 600-mg, and placebo arms, respectively. Both doses achieved a highly significant advantage over placebo.
For both stages and doses, the response curves of the active-treatment groups and the placebo group diverged almost immediately. By 4 weeks, both measures of CDASI reductions on active therapy were significantly improved relative to placebo, and the response curves had a consistent downward slope through the end of the 12-week study, Dr. Mangold reported.
The majority of patients responded by either of the primary endpoint criteria. For a CDASI reduction of greater than 5 points, the response rates were 100% and 96% for the 150-mg and 600-mg doses of PF-06823859, respectively. The placebo response was 35.7%. For the CDASI reduction of greater than 40%, the rates were 80%, 82.1%, and 7.1% for the 150-mg, 600-mg, and placebo arms, respectively.
“There were no major safety concerns. Most of the treatment-emergent adverse events were mild, and adverse events did not have a relationship to dose,” Dr. Mangold said. Notably, there were no cases of herpes zoster, and infections of any kind were low in all study groups.
A phase 3 study is being planned with the 600-mg dose, according to Dr. Mangold, but he acknowledged that regulatory authorities have generally required endpoints for both cutaneous and muscle manifestations in previous trials of therapies for dermatomyositis.
It is not yet certain that “there will be a carve-out for skin,” he said in answer to a question about investigations moving forward. So far, studies have been focused on skin response. However, a meaningful degree of benefit against muscle involvement, which has not yet been well studied, has not been ruled out.
Even though this is a phase 2 trial with small numbers, it was controlled and blinded, and the potential of an inhibitor of IFN-beta to control the skin manifestations of dermatomyositis “is kind of a big deal,” said Paul Nghiem, MD, PhD, professor of dermatology, University of Washington, Seattle.
“There is definitely an unmet need for better therapies to control the skin involvement,” Dr. Nghiem said.
Hensin Tsao, MD, PhD, clinical director of the Melanoma and Pigmented Lesion Center at Massachusetts General Hospital, Boston, agreed. Like Dr. Nghiem, Dr. Tsao was a panelist during the late-breaker session where the study was presented, and he was impressed by the data.
“This is something that is definitely newsworthy,” Dr. Tsao said.
Dr. Mangold reports financial relationships with Actelion, Amgen, Corbus, Eli Lilly, Incyte, miRagen, Novartis, Regeneron, Solagenix, Sun Pharmaceuticals, Teva, and Pfizer, which provided funding for this trial. Both Dr. Nghiem and Dr. Tsao reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – in a double-blind, placebo-controlled phase 2 trial, according to results presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
“These findings support the inhibition of IFN-beta as a promising therapeutic strategy in skin-predominant disease,” said principal investigator Aaron Mangold, MD, associate professor of dermatology, Mayo Clinic, Scottsdale, Ariz.
Dermatomyositis, a rare autoimmune inflammatory condition that typically involves both skeletal muscles and skin, is a challenging disease with a diverse set of potential complications.
Immunosuppressive and immunomodulatory agents are used with mixed success for myositis, but skin manifestations, which include papular eruptions, heliotrope rash, photoerythema, burning, and pruritus, are often the most troublesome and the most difficult to control. Treatment options other than immunomodulators that target cutaneous involvement – which include steroids, emollients, and photoprotection – are generally modestly effective, according to Dr. Mangold.
Targeting an elevated cytokine
Interest in IFN-beta, which is elevated in the blood of individuals with dermatomyositis, was triggered by evidence that this cytokine plays an important role in driving the skin inflammation, Dr. Mangold explained.
“The blood concentrations of IFN-beta are positively correlated with cutaneous disease activity and severity,” he said.
The study drug, currently known as PF-06823859 (Dazukibart), “is a potent, selective humanized IgG1-neutralizing antibody directed at IFN-beta,” Dr. Mangold said. A dose-ranging phase 1 study published 2 years ago provided evidence of acceptable pharmacokinetics and safety in healthy individuals to support treatment studies for disorders associated with elevated IFN-beta levels. In addition to dermatomyositis, this includes systemic lupus erythematosus.
In this phase 2 trial, patients whose condition was not improved by at least one standard-care therapy for skin manifestations of dermatomyositis were eligible if they had moderate to severe disease as measured with the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI), according to Dr. Mangold. During the study, patients were allowed to remain on a disease modifying antirheumatic drug and/or prednisone if they had been on stable doses and did not change the dose.
After a screening run-in, the trial had two blinded stages. In stage 1, 30 patients were randomly assigned either to 600 mg of PF-06823859 or to placebo, both administered intravenously every 4 weeks. A second cohort of 25 patients was randomly assigned in stage 2 to placebo, 150 mg of PF-06823859, or 600 mg of PF-06823859. The primary endpoint assessed at 12 weeks was a greater than 5-point reduction in CDASI score or greater than 40% CDASI improvement from baseline.
Both endpoints are associated with a clinically meaningful response in regard to an improved quality of life, Dr. Mangold noted.
Both doses better than placebo
In results from the stage 1 portion, the mean reduction in CDASI at 12 weeks after three doses of the assigned therapy was 18.8 points in the active-treatment group versus 3.9 points in the placebo group. In pooled data from stage 1 and 2, the reductions were 16.6 points, 19.2 points, and 2.9 points for the 150-mg, 600-mg, and placebo arms, respectively. Both doses achieved a highly significant advantage over placebo.
For both stages and doses, the response curves of the active-treatment groups and the placebo group diverged almost immediately. By 4 weeks, both measures of CDASI reductions on active therapy were significantly improved relative to placebo, and the response curves had a consistent downward slope through the end of the 12-week study, Dr. Mangold reported.
The majority of patients responded by either of the primary endpoint criteria. For a CDASI reduction of greater than 5 points, the response rates were 100% and 96% for the 150-mg and 600-mg doses of PF-06823859, respectively. The placebo response was 35.7%. For the CDASI reduction of greater than 40%, the rates were 80%, 82.1%, and 7.1% for the 150-mg, 600-mg, and placebo arms, respectively.
“There were no major safety concerns. Most of the treatment-emergent adverse events were mild, and adverse events did not have a relationship to dose,” Dr. Mangold said. Notably, there were no cases of herpes zoster, and infections of any kind were low in all study groups.
A phase 3 study is being planned with the 600-mg dose, according to Dr. Mangold, but he acknowledged that regulatory authorities have generally required endpoints for both cutaneous and muscle manifestations in previous trials of therapies for dermatomyositis.
It is not yet certain that “there will be a carve-out for skin,” he said in answer to a question about investigations moving forward. So far, studies have been focused on skin response. However, a meaningful degree of benefit against muscle involvement, which has not yet been well studied, has not been ruled out.
Even though this is a phase 2 trial with small numbers, it was controlled and blinded, and the potential of an inhibitor of IFN-beta to control the skin manifestations of dermatomyositis “is kind of a big deal,” said Paul Nghiem, MD, PhD, professor of dermatology, University of Washington, Seattle.
“There is definitely an unmet need for better therapies to control the skin involvement,” Dr. Nghiem said.
Hensin Tsao, MD, PhD, clinical director of the Melanoma and Pigmented Lesion Center at Massachusetts General Hospital, Boston, agreed. Like Dr. Nghiem, Dr. Tsao was a panelist during the late-breaker session where the study was presented, and he was impressed by the data.
“This is something that is definitely newsworthy,” Dr. Tsao said.
Dr. Mangold reports financial relationships with Actelion, Amgen, Corbus, Eli Lilly, Incyte, miRagen, Novartis, Regeneron, Solagenix, Sun Pharmaceuticals, Teva, and Pfizer, which provided funding for this trial. Both Dr. Nghiem and Dr. Tsao reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – in a double-blind, placebo-controlled phase 2 trial, according to results presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
“These findings support the inhibition of IFN-beta as a promising therapeutic strategy in skin-predominant disease,” said principal investigator Aaron Mangold, MD, associate professor of dermatology, Mayo Clinic, Scottsdale, Ariz.
Dermatomyositis, a rare autoimmune inflammatory condition that typically involves both skeletal muscles and skin, is a challenging disease with a diverse set of potential complications.
Immunosuppressive and immunomodulatory agents are used with mixed success for myositis, but skin manifestations, which include papular eruptions, heliotrope rash, photoerythema, burning, and pruritus, are often the most troublesome and the most difficult to control. Treatment options other than immunomodulators that target cutaneous involvement – which include steroids, emollients, and photoprotection – are generally modestly effective, according to Dr. Mangold.
Targeting an elevated cytokine
Interest in IFN-beta, which is elevated in the blood of individuals with dermatomyositis, was triggered by evidence that this cytokine plays an important role in driving the skin inflammation, Dr. Mangold explained.
“The blood concentrations of IFN-beta are positively correlated with cutaneous disease activity and severity,” he said.
The study drug, currently known as PF-06823859 (Dazukibart), “is a potent, selective humanized IgG1-neutralizing antibody directed at IFN-beta,” Dr. Mangold said. A dose-ranging phase 1 study published 2 years ago provided evidence of acceptable pharmacokinetics and safety in healthy individuals to support treatment studies for disorders associated with elevated IFN-beta levels. In addition to dermatomyositis, this includes systemic lupus erythematosus.
In this phase 2 trial, patients whose condition was not improved by at least one standard-care therapy for skin manifestations of dermatomyositis were eligible if they had moderate to severe disease as measured with the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI), according to Dr. Mangold. During the study, patients were allowed to remain on a disease modifying antirheumatic drug and/or prednisone if they had been on stable doses and did not change the dose.
After a screening run-in, the trial had two blinded stages. In stage 1, 30 patients were randomly assigned either to 600 mg of PF-06823859 or to placebo, both administered intravenously every 4 weeks. A second cohort of 25 patients was randomly assigned in stage 2 to placebo, 150 mg of PF-06823859, or 600 mg of PF-06823859. The primary endpoint assessed at 12 weeks was a greater than 5-point reduction in CDASI score or greater than 40% CDASI improvement from baseline.
Both endpoints are associated with a clinically meaningful response in regard to an improved quality of life, Dr. Mangold noted.
Both doses better than placebo
In results from the stage 1 portion, the mean reduction in CDASI at 12 weeks after three doses of the assigned therapy was 18.8 points in the active-treatment group versus 3.9 points in the placebo group. In pooled data from stage 1 and 2, the reductions were 16.6 points, 19.2 points, and 2.9 points for the 150-mg, 600-mg, and placebo arms, respectively. Both doses achieved a highly significant advantage over placebo.
For both stages and doses, the response curves of the active-treatment groups and the placebo group diverged almost immediately. By 4 weeks, both measures of CDASI reductions on active therapy were significantly improved relative to placebo, and the response curves had a consistent downward slope through the end of the 12-week study, Dr. Mangold reported.
The majority of patients responded by either of the primary endpoint criteria. For a CDASI reduction of greater than 5 points, the response rates were 100% and 96% for the 150-mg and 600-mg doses of PF-06823859, respectively. The placebo response was 35.7%. For the CDASI reduction of greater than 40%, the rates were 80%, 82.1%, and 7.1% for the 150-mg, 600-mg, and placebo arms, respectively.
“There were no major safety concerns. Most of the treatment-emergent adverse events were mild, and adverse events did not have a relationship to dose,” Dr. Mangold said. Notably, there were no cases of herpes zoster, and infections of any kind were low in all study groups.
A phase 3 study is being planned with the 600-mg dose, according to Dr. Mangold, but he acknowledged that regulatory authorities have generally required endpoints for both cutaneous and muscle manifestations in previous trials of therapies for dermatomyositis.
It is not yet certain that “there will be a carve-out for skin,” he said in answer to a question about investigations moving forward. So far, studies have been focused on skin response. However, a meaningful degree of benefit against muscle involvement, which has not yet been well studied, has not been ruled out.
Even though this is a phase 2 trial with small numbers, it was controlled and blinded, and the potential of an inhibitor of IFN-beta to control the skin manifestations of dermatomyositis “is kind of a big deal,” said Paul Nghiem, MD, PhD, professor of dermatology, University of Washington, Seattle.
“There is definitely an unmet need for better therapies to control the skin involvement,” Dr. Nghiem said.
Hensin Tsao, MD, PhD, clinical director of the Melanoma and Pigmented Lesion Center at Massachusetts General Hospital, Boston, agreed. Like Dr. Nghiem, Dr. Tsao was a panelist during the late-breaker session where the study was presented, and he was impressed by the data.
“This is something that is definitely newsworthy,” Dr. Tsao said.
Dr. Mangold reports financial relationships with Actelion, Amgen, Corbus, Eli Lilly, Incyte, miRagen, Novartis, Regeneron, Solagenix, Sun Pharmaceuticals, Teva, and Pfizer, which provided funding for this trial. Both Dr. Nghiem and Dr. Tsao reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT AAD 2023