User login
More Is Less
A 64-year-old man presented with a 2-month history of a nonproductive cough, weight loss, and subjective fevers. He had no chest pain, hemoptysis, or shortness of breath. He also described worsening anorexia and a 15-pound weight loss over the previous 3 months. He had no arthralgias, myalgias, abdominal pain, nausea, emesis, or diarrhea.
Two weeks prior to his presentation, he was diagnosed with pneumonia and given a 5-day course of azithromycin. His symptoms did not improve, so he presented to the emergency room.
He had not been seen regularly by a physician in decades and had no known medical conditions. He did not take any medications. He immigrated from China 3 years prior and lived with his wife in California. He had a 30 pack-year smoking history. He drank a shot glass of liquor daily and denied any drug use.
Weight loss might result from inflammatory disorders like cancer or noninflammatory causes such as decreased oral intake (eg, diminished appetite) or malabsorption (eg, celiac disease). However, his fevers suggest inflammation, which usually reflects an underlying infection, cancer, or autoimmune process. While chronic cough typically results from upper airway cough syndrome (allergic or nonallergic rhinitis), gastroesophageal reflux disease, or asthma, it can also point to pathology of the lung, which may be intrinsic (bronchiectasis) or extrinsic (mediastinal mass). The duration of 2 months makes a typical infectious process like pneumococcal pneumonia unlikely. Atypical infections such as tuberculosis, melioidosis, and talaromycosis are possible given his immigration from East Asia, and coccidioidomycosis given his residence in California. He might have undiagnosed medical conditions, such as diabetes, that could be relevant to his current presentation and classify him as immunocompromised. His smoking history prompts consideration of lung cancer.
His temperature was 36.5 oC, heart rate 70 beats per minute, blood pressure 118/66 mm Hg, respiratory rate 16 breaths per minute, oxygen saturation 98% on room air, and body mass index 23 kg/m2. He was in no acute distress. The findings from the cardiac, lung, abdominal, and neurological exams were normal.
Skin examination found a fixed, symmetric, 5-cm, firm nodule at top of sternum (Figure 1A). In addition, he had two 1-cm, mobile, firm, subcutaneous nodules, one on his anterior left chest and another underneath his right axilla. He also had two 2-cm, erythematous, tender nodules on his left anterior forearm and a 1-cm nodule with a central black plug on the dorsal surface of his right hand (Figure 1B). He did not have any edema.

The white blood cell count was 10,500/mm3 (42% neutrophils, 37% lymphocytes, 16.4% monocytes, and 2.9% eosinophils), hemoglobin was 12.2 g/dL with a mean corpuscular volume of 91 fL, and the platelet count was 441,000/mm3. Basic metabolic panel, aminotransferase, bilirubin, and alkaline phosphatase were within reference ranges. Serum albumin was 3.1 g/dL. Serum total protein was elevated at 8.8 g/dL. Serum calcium was 9.0 mg/dL. Urinalysis results were normal.
The slightly low albumin, mildly elevated platelet count, monocytosis, and normocytic anemia suggest inflammation, although monocytosis might represent a hematologic malignancy like chronic myelomonocytic leukemia (CMML). His subjective fevers and weight loss further corroborate underlying inflammation. What is driving the inflammation? There are two localizing findings: cough and nodular skin lesions.
His lack of dyspnea and normal oxygen saturation, respiratory rate, and lung exam make an extrapulmonary cause of cough such as lymphadenopathy or mediastinal infection possible. The number of nodular skin lesions, wide-spread distribution, and appearance (eg, erythematous, tender) point to either a primary cutaneous disease with systemic manifestations (eg, cutaneous lymphoma) or a systemic disease with cutaneous features (eg, sarcoidosis).
Three categories—inflammatory, infectious, and neoplastic—account for most nodular skin lesions. Usually microscopic evaluation is necessary for definitive diagnosis, though epidemiology, associated symptoms, and characteristics of the nodules help prioritize the differential diagnosis. Tender nodules might reflect a panniculitis; erythema nodosum is the most common type, and while this classically develops on the anterior shins, it may also occur on the forearm. His immigration from China prompts consideration of tuberculosis and cutaneous leishmaniasis. Coccidioidomycosis can lead to inflammation and nodular skin lesions. Other infections such as nontuberculous mycobacteria, nocardiosis, and cryptococcosis may cause disseminated infection with pulmonary and skin manifestations. His smoking puts him at risk of lung cancer, which rarely results in metastatic subcutaneous infiltrates.
A chest radiograph demonstrated a prominent density in the right paratracheal region of the mediastinum with adjacent streaky opacities. A computed tomography scan of the chest with intravenous contrast demonstrated centrilobular emphysematous changes and revealed a 2.6 × 4.7-cm necrotic mass in the anterior chest wall with erosion into the manubrium, a 3.8 × 2.1-cm centrally necrotic soft-tissue mass in the right hilum, a 5-mm left upper-lobe noncalcified solid pulmonary nodule, and prominent subcarinal, paratracheal, hilar, and bilateral supraclavicular lymphadenopathy (Figure 2).

Flow cytometry of the peripheral blood did not demonstrate a lymphoproliferative disorder. Blood smear demonstrated normal red blood cell, white blood cell, and platelet morphology. HIV antibody was negative. Hemoglobin A1c was 6.1%. Smear microscopy for acid-fast bacilli (AFB) was negative and sputum AFB samples were sent for culture. Bacterial, fungal, and AFB blood cultures were collected and pending.
Causes of necrotizing pneumonia include liquid (eg, lymphoma) and solid (eg, squamous cell carcinoma) cancers, infections, and noninfectious inflammatory processes such as granulomatosis with polyangiitis (GPA). Given his subacute presentation and extrapulmonary cutaneous manifestations, consideration of mycobacteria, fungi (eg, Coccidioides, Aspergillus, and Cryptococcus), and filamentous bacteria (eg, Nocardia and Actinomyces) is prioritized among the myriad of infections that can cause a lung cavity. His smoking history and centrilobular emphysematous changes are highly suggestive of chronic obstructive pulmonary disease, which puts him at increased risk of bacterial colonization and recurrent pulmonary infections. Tuberculosis is still possible despite three negative AFB-sputa smears given the sensitivity of smear microscopy (with three specimens) is roughly 70% in an immunocompetent host.
The lymphadenopathy likely reflects spread from the necrotic lung mass. The frequency of non-Hodgkin lymphoma increases with age. The results of the peripheral flow cytometry do not exclude the possibility of an aggressive lymphoma with pulmonary and cutaneous manifestations.
The erosive property of the chest wall mass makes an autoimmune process like GPA unlikely. An aggressive and disseminated infection or cancer is most likely. A pathologic process that originated in the lung and then spread to the lymph nodes and skin is more likely than a disorder which started in the skin. It would be unlikely for a primary cutaneous disorder to cause such a well-defined necrotic lung mass. Lung cancer rarely metastasizes to the skin and, instead, preferentially involves the chest. Ultimately, ascertaining what the patient experienced first (ie, respiratory or cutaneous symptoms) will determine where the pathology originated.
Computed tomography scan of the abdomen and pelvis with intravenous contrast demonstrated multiple ill-defined lytic lesions in the pelvis, including a 12-mm lesion of the left sacral ala and multiple subcentimeter lesions in the medial left iliac bone and superior right acetabulum. In addition, there were two 1-cm, rim-enhancing, hypodense nodules in the subcutaneous fat of the right flank at the level of L5 and the left lower quadrant, respectively. There was also a 2.2 × 1.9-cm faintly rim-enhancing hypodensity within the left iliopsoas muscle belly.
These imaging findings further corroborate a widely metastatic process probably originating in the lung and spreading to the lymph nodes, skin, muscles, and bones. The characterization of lesions as lytic as opposed to blastic is less helpful because many diseases can cause both. It does prompt consideration of multiple myeloma; however, multiple myeloma less commonly manifests with extramedullary plasmacytomas and is less likely given his normal renal function and calcium level. Bone lesions lessen the likelihood of GPA, and his necrotic lung mass makes sarcoidosis unlikely. Atypical infections and cancers are the prime suspect of his multisystemic disease.
There are no data yet to suggest a weakened immune system, which would increase his risk for atypical infections. His chronic lung disease, identified on imaging, is a risk factor for nocardiosis. This gram-positive, weakly acid-fast bacterium can involve any organ, although lung, brain, and skin are most commonly involved. Disseminated nocardiosis can result from a pulmonary or cutaneous site of origin. Mycobacteria; Actinomyces; dimorphic fungi like Histoplasma, Coccidioides, and Blastomyces; and molds such as Aspergillus can also cause disseminated disease with pulmonary, cutaneous, and musculoskeletal manifestations.
While metastases to muscle itself are rare, they can occur with primary lung cancers. Primary lung cancer with extrapulmonary features is feasible. Squamous cell lung cancer is the most likely to cavitate, although it rarely spreads to the skin. An aggressive lymphoma like diffuse large B-cell lymphoma or cutaneous T-cell lymphoma (higher occurrence in Asians) might also explain his constellation of findings. If culture data remain negative, then biopsy of the chest wall mass might be the safest and highest-yield target.
On hospital day 2, the patient developed new-onset severe neck pain. Magnetic resonance imaging of the cervical, thoracic, and lumbar spine revealed multilevel, bony, lytic lesions with notable cortical breakthrough of the C2 and C3 vertebrae into the prevertebral space, as well as epidural extension and paraspinal soft-tissue extension of the thoracic and lumbar vertebral lesions (Figure 3).

On hospital day 3, the patient reported increased tenderness in his skin nodules with one on his left forearm spontaneously draining purulent fluid. Repeat complete blood count demonstrated a white blood cell count of 12,600/mm3 (45% neutrophils, 43% lymphocytes, 8.4% monocytes, and 4.3% eosinophils), hemoglobin of 16 g/dL, and platelet count of 355,000/mm3.
The erosion into the manubrium and cortical destruction of the cervical spine attests to the aggressiveness of the underlying disease process. Noncutaneous lymphoma and lung cancer are unlikely to have such prominent skin findings; the visceral pathology, necrotizing lung mass, and bone lesions make cutaneous lymphoma less likely. At this point, a disseminated infectious process is most likely. Leading considerations based on his emigration from China and residence in California are tuberculosis and coccidioidomycosis, respectively. Tuberculous spondylitis most commonly involves the lower thoracic and upper lumbar region, and less commonly the cervical spine. His three negative AFB sputa samples further reduce its posttest probability. Ultimately microbiologic data are needed to distinguish between a disseminated fungal process, like coccidioidomycosis, or tuberculosis.
Given the concern for malignancy, a fine needle aspiration of the left supraclavicular lymph node was pursued. This revealed fungal microorganisms morphologically compatible with Coccidioides spp. with a background of necrotizing granulomas and acute inflammation. Fungal blood cultures grew Coccidioides immitis. AFB blood cultures were discontinued due to overgrowth of mold. The Coccidioides immitis antibody immunodiffusion titer was positive at 1:256.
During the remainder of the hospitalization, the patient was treated with oral fluconazole 800 mg daily. The patient underwent surgical debridement of the manubrium. In addition, given the concern for cervical spine instability, neurosurgery recommended follow-up with interval imaging. Since his discharge from the hospital, the patient continues to take oral fluconazole with resolution of his cutaneous lesions and respiratory symptoms. His titers have incrementally decreased from 1:256 to 1:16 after 8 months of treatment.
COMMENTARY
This elderly gentleman from China presented with subacute symptoms and was found to have numerous cutaneous nodules, lymphadenopathy, and diffuse osseous lesions. This multisystem illness posed a diagnostic challenge, forcing our discussant to search for a disease process that could lead to such varied findings. Ultimately, epidemiologic and clinical clues suggested a diagnosis of disseminated coccidioidomycosis, which was later confirmed on lymph node biopsy.
Coccidioides species are important fungal pathogens in the Western Hemisphere. This organism exhibits dimorphism, existing as mycelia (with arthroconidia) in soil and spherules in tissues. Coccidioides spp are endemic to the Southwestern United States, particularly California’s central valley and parts of Arizona; it additionally remains an important pathogen in Mexico, Central America, and South America.1 Newer epidemiologic studies have raised concerns that the incidence of coccidioidomycosis is increasing and that its geographic range may be more extensive than previously appreciated, with it now being found as far north as Washington state.2
Coccidioidal infection can take several forms. One-half to two-thirds of infections may be asymptomatic.3 Clinically significant infections can include an acute self-limiting respiratory illness, pulmonary nodules and cavities, chronic fibrocavitary pneumonia, and infections with extrapulmonary dissemination. Early respiratory infection is often indistinguishable from typical community-acquired pneumonia (10%-15% of pneumonia in endemic areas) but can be associated with certain suggestive features, such as erythema nodosum, erythema multiforme, prominent arthralgias (ie, “desert rheumatism”), and a peripheral eosinophilia.4,5
Extrapulmonary dissemination is rare and most commonly associated with immunocompromising states.6 However, individuals of African or Filipino ancestry also appear to be at increased risk for disseminated disease, which led to a California court decision that excluded African American inmates from state prisons located in Coccidioides endemic areas.7 The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the central nervous system (CNS).6 CNS disease has a predilection to manifest as a chronic basilar meningitis, most often complicated by hydrocephalus, vasculitic infarction, and spinal arachnoiditis.8
Cutaneous manifestations of coccidioidomycosis can occur as immunologic phenomenon associated with pulmonary disease or represent skin and soft tissue foci of disseminated infection.9 In primary pulmonary infection, skin findings can range from a nonspecific exanthem to erythema nodosum and erythema multiforme, which are thought to represent hypersensitivity responses. In contrast, Coccidioides spp can infect the skin either through direct inoculation (as in primary cutaneous coccidioidomycosis) or via hematogenous dissemination.9,10 A variety of lesions have been described, with painless nodules being the most frequently encountered morphotype in one study.11,12 On histopathologic examination, these lesions often have features of granulomatous dermatitis, eosinophilic infiltration, gummatous necrosis, microabscesses, or perivascular inflammation.13
Another common and highly morbid site of extrapulmonary dissemination is the musculoskeletal system. Bone and joint coccidioidomycosis most frequently affect the axial skeleton, although peripheral skeletal structures and joints can also be involved.6,12 Vertebral coccidioidomycosis is associated with significant morbidity. A study describing the magnetic resonance imaging findings of patients with vertebral coccidioidomycosis found that Coccidioides spp appeared to have a predilection for the thoracic vertebrae (in up to 80% of the study’s cohort).14 Skip lesions with noncontiguously involved vertebrae occurred in roughly half of patients, highlighting the usefulness of imaging the total spine in suspected cases.
The diagnosis of coccidioidomycosis is often established through serologic testing or by isolation of Coccidioides spp. on histopathology or culture. Obtaining sputum or tissue may be difficult, so clinicians often rely on noninvasive diagnostic tests such as coccidioidal antigen and serologies by enzyme immunoassays, immunodiffusion, and complement fixation. Enzyme immunoassays IgM and IgG results are positive early in the disease process and need to be confirmed with immunodiffusion or complement fixation testing. Complement fixation IgG is additionally useful to monitor disease activity over time and can help inform risk of disseminated disease.15 The gold standard of diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation either by direct visualization of a spherule or growth in fungal cultures.16 Polymerase chain reaction testing of sputum samples is an emerging diagnostic technique that has been found to have similar sensitivity rates to fungal culture.17
Treatment decisions in coccidioidomycosis are complex and vary by site of infection, immune status of the host, and extent of disease.16 While uncomplicated primary pulmonary infections can often be managed with observation alone, prolonged medical therapy with azole antifungals is often recommended for complicated pulmonary infections, symptomatic cavitary disease, and virtually all forms of extrapulmonary disease. Intravenous liposomal amphotericin is often used as initial therapy in immunosuppressed individuals, pregnant women, and those with extensive disease. CNS disease represents a particularly challenging treatment scenario and requires lifelong azole therapy.8,16
The patient in this case initially presented with vague inflammatory symptoms, with each aliquot revealing further evidence of a metastatic disease process. Such multisystem presentations are diagnostically challenging and force clinicians to reach for some feature around which to build their differential diagnosis. It is with this in mind that we are often taught to “localize the lesion” in order to focus our search for a unifying diagnosis. Yet, in this case, the sheer number of disease foci ultimately helped the discussant to narrow the range of diagnostic possibilities because only a limited number of conditions could present with such widespread, multisystem manifestations. Therefore, this case serves as a reminder that, sometimes in clinical reasoning, “more is less.”
KEY TEACHING POINTS
- Coccidioidomycosis is a fungal infection that can present with pulmonary or extrapulmonary disease. Risk of extrapulmonary dissemination is greatest among immunocompromised individuals and those of African or Filipino ancestry.3,7
- The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the CNS.6
- While serologic testing can be diagnostically useful, the gold standard for diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation with direct visualization of a spherule or growth in fungal cultures.16
1. Benedict K, McCotter OZ, Brady S, et al. Surveillance for Coccidioidomycosis - United States, 2011-2017. MMWR Surveill Summ. 2019;68(No. SS-7):1-15. http://dx.doi.org/10.15585/mmwr.ss6807a1
2. McCotter OZ, Benedict K, Engelthaler DM, et al. Update on the epidemiology of coccidioidomycosis in the United States. Med Mycol. 2019;57(Suppl 1):S30-s40. https://doi.org/10.1093/mmy/myy095
3. Galgiani JN, Ampel NM, Blair JE, et al. Coccidioidomycosis. Clin Infect Dis. 2005;41(9):1217-1223. https://doi.org/10.1086/496991
4. Chang DC, Anderson S, Wannemuehler K, et al. Testing for coccidioidomycosis among patients with community-acquired pneumonia. Emerg Infect Dis. 2008;14(7):1053-1059. https://doi.org/10.3201/eid1407.070832
5. Saubolle MA, McKellar PP, Sussland D. Epidemiologic, clinical, and diagnostic aspects of coccidioidomycosis. J Clin Microbiol. 2007;45(1):26-30. https://doi.org/10.1128/jcm.02230-06
6. Adam RD, Elliott SP, Taljanovic MS. The spectrum and presentation of disseminated coccidioidomycosis. Am J Med. 2009;122(8):770-777. https://doi.org/10.1016/j.amjmed.2008.12.024
7. Wheeler C, Lucas KD, Mohle-Boetani JC. Rates and risk factors for Coccidioidomycosis among prison inmates, California, USA, 2011. Emerg Infect Dis. 2015;21(1):70-75. https://doi.org/10.3201/eid2101.140836
8. Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis. 2006;42(1):103-107. https://doi.org/10.1086/497596
9. Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections. Ann N Y Acad Sci. 2007;1111:411-421. https://doi.org/10.1196/annals.1406.010
10. Chang A, Tung RC, McGillis TS, Bergfeld WF, Taylor JS. Primary cutaneous coccidioidomycosis. J Am Acad Dermatol. 2003;49(5):944-949. https://doi.org/10.1016/s0190-9622(03)00462-6
11. Quimby SR, Connolly SM, Winkelmann RK, Smilack JD. Clinicopathologic spectrum of specific cutaneous lesions of disseminated coccidioidomycosis. J Am Acad Dermatol. 1992;26(1):79-85. https://doi.org/10.1016/0190-9622(92)70011-4
12. Crum NF, Lederman ER, Stafford CM, Parrish JS, Wallace MR. Coccidioidomycosis: a descriptive survey of a reemerging disease. clinical characteristics and current controversies. Medicine (Baltimore). 2004;83(3):149-175. https://doi.org/10.1097/01.md.0000126762.91040.fd
13. Carpenter JB, Feldman JS, Leyva WH, DiCaudo DJ. Clinical and pathologic characteristics of disseminated cutaneous coccidioidomycosis. J Am Acad Dermatol. 2010;62(5):831-837. https://doi.org/10.1016/j.jaad.2008.07.031
14. Crete RN, Gallmann W, Karis JP, Ross J. Spinal coccidioidomycosis: MR imaging findings in 41 patients. AJNR Am J Neuroradiol. 2018;39(11):2148-2153. https://doi.org/10.3174/ajnr.a5818
15. McHardy IH, Dinh BN, Waldman S, et al. Coccidioidomycosis complement fixation titer trends in the age of antifungals. J Clin Microbiol. 2018;56(12):e01318-18. https://doi.org/10.1128/jcm.01318-18
16. Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious Diseases Society of America (IDSA) clinical practice guideline for the treatment of coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112-e146. https://doi.org/10.1093/cid/ciw360
17. Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia. 2010;170(5):345-351. https://doi.org/10.1007/s11046-010-9327-0
A 64-year-old man presented with a 2-month history of a nonproductive cough, weight loss, and subjective fevers. He had no chest pain, hemoptysis, or shortness of breath. He also described worsening anorexia and a 15-pound weight loss over the previous 3 months. He had no arthralgias, myalgias, abdominal pain, nausea, emesis, or diarrhea.
Two weeks prior to his presentation, he was diagnosed with pneumonia and given a 5-day course of azithromycin. His symptoms did not improve, so he presented to the emergency room.
He had not been seen regularly by a physician in decades and had no known medical conditions. He did not take any medications. He immigrated from China 3 years prior and lived with his wife in California. He had a 30 pack-year smoking history. He drank a shot glass of liquor daily and denied any drug use.
Weight loss might result from inflammatory disorders like cancer or noninflammatory causes such as decreased oral intake (eg, diminished appetite) or malabsorption (eg, celiac disease). However, his fevers suggest inflammation, which usually reflects an underlying infection, cancer, or autoimmune process. While chronic cough typically results from upper airway cough syndrome (allergic or nonallergic rhinitis), gastroesophageal reflux disease, or asthma, it can also point to pathology of the lung, which may be intrinsic (bronchiectasis) or extrinsic (mediastinal mass). The duration of 2 months makes a typical infectious process like pneumococcal pneumonia unlikely. Atypical infections such as tuberculosis, melioidosis, and talaromycosis are possible given his immigration from East Asia, and coccidioidomycosis given his residence in California. He might have undiagnosed medical conditions, such as diabetes, that could be relevant to his current presentation and classify him as immunocompromised. His smoking history prompts consideration of lung cancer.
His temperature was 36.5 oC, heart rate 70 beats per minute, blood pressure 118/66 mm Hg, respiratory rate 16 breaths per minute, oxygen saturation 98% on room air, and body mass index 23 kg/m2. He was in no acute distress. The findings from the cardiac, lung, abdominal, and neurological exams were normal.
Skin examination found a fixed, symmetric, 5-cm, firm nodule at top of sternum (Figure 1A). In addition, he had two 1-cm, mobile, firm, subcutaneous nodules, one on his anterior left chest and another underneath his right axilla. He also had two 2-cm, erythematous, tender nodules on his left anterior forearm and a 1-cm nodule with a central black plug on the dorsal surface of his right hand (Figure 1B). He did not have any edema.
The white blood cell count was 10,500/mm3 (42% neutrophils, 37% lymphocytes, 16.4% monocytes, and 2.9% eosinophils), hemoglobin was 12.2 g/dL with a mean corpuscular volume of 91 fL, and the platelet count was 441,000/mm3. Basic metabolic panel, aminotransferase, bilirubin, and alkaline phosphatase were within reference ranges. Serum albumin was 3.1 g/dL. Serum total protein was elevated at 8.8 g/dL. Serum calcium was 9.0 mg/dL. Urinalysis results were normal.
The slightly low albumin, mildly elevated platelet count, monocytosis, and normocytic anemia suggest inflammation, although monocytosis might represent a hematologic malignancy like chronic myelomonocytic leukemia (CMML). His subjective fevers and weight loss further corroborate underlying inflammation. What is driving the inflammation? There are two localizing findings: cough and nodular skin lesions.
His lack of dyspnea and normal oxygen saturation, respiratory rate, and lung exam make an extrapulmonary cause of cough such as lymphadenopathy or mediastinal infection possible. The number of nodular skin lesions, wide-spread distribution, and appearance (eg, erythematous, tender) point to either a primary cutaneous disease with systemic manifestations (eg, cutaneous lymphoma) or a systemic disease with cutaneous features (eg, sarcoidosis).
Three categories—inflammatory, infectious, and neoplastic—account for most nodular skin lesions. Usually microscopic evaluation is necessary for definitive diagnosis, though epidemiology, associated symptoms, and characteristics of the nodules help prioritize the differential diagnosis. Tender nodules might reflect a panniculitis; erythema nodosum is the most common type, and while this classically develops on the anterior shins, it may also occur on the forearm. His immigration from China prompts consideration of tuberculosis and cutaneous leishmaniasis. Coccidioidomycosis can lead to inflammation and nodular skin lesions. Other infections such as nontuberculous mycobacteria, nocardiosis, and cryptococcosis may cause disseminated infection with pulmonary and skin manifestations. His smoking puts him at risk of lung cancer, which rarely results in metastatic subcutaneous infiltrates.
A chest radiograph demonstrated a prominent density in the right paratracheal region of the mediastinum with adjacent streaky opacities. A computed tomography scan of the chest with intravenous contrast demonstrated centrilobular emphysematous changes and revealed a 2.6 × 4.7-cm necrotic mass in the anterior chest wall with erosion into the manubrium, a 3.8 × 2.1-cm centrally necrotic soft-tissue mass in the right hilum, a 5-mm left upper-lobe noncalcified solid pulmonary nodule, and prominent subcarinal, paratracheal, hilar, and bilateral supraclavicular lymphadenopathy (Figure 2).
Flow cytometry of the peripheral blood did not demonstrate a lymphoproliferative disorder. Blood smear demonstrated normal red blood cell, white blood cell, and platelet morphology. HIV antibody was negative. Hemoglobin A1c was 6.1%. Smear microscopy for acid-fast bacilli (AFB) was negative and sputum AFB samples were sent for culture. Bacterial, fungal, and AFB blood cultures were collected and pending.
Causes of necrotizing pneumonia include liquid (eg, lymphoma) and solid (eg, squamous cell carcinoma) cancers, infections, and noninfectious inflammatory processes such as granulomatosis with polyangiitis (GPA). Given his subacute presentation and extrapulmonary cutaneous manifestations, consideration of mycobacteria, fungi (eg, Coccidioides, Aspergillus, and Cryptococcus), and filamentous bacteria (eg, Nocardia and Actinomyces) is prioritized among the myriad of infections that can cause a lung cavity. His smoking history and centrilobular emphysematous changes are highly suggestive of chronic obstructive pulmonary disease, which puts him at increased risk of bacterial colonization and recurrent pulmonary infections. Tuberculosis is still possible despite three negative AFB-sputa smears given the sensitivity of smear microscopy (with three specimens) is roughly 70% in an immunocompetent host.
The lymphadenopathy likely reflects spread from the necrotic lung mass. The frequency of non-Hodgkin lymphoma increases with age. The results of the peripheral flow cytometry do not exclude the possibility of an aggressive lymphoma with pulmonary and cutaneous manifestations.
The erosive property of the chest wall mass makes an autoimmune process like GPA unlikely. An aggressive and disseminated infection or cancer is most likely. A pathologic process that originated in the lung and then spread to the lymph nodes and skin is more likely than a disorder which started in the skin. It would be unlikely for a primary cutaneous disorder to cause such a well-defined necrotic lung mass. Lung cancer rarely metastasizes to the skin and, instead, preferentially involves the chest. Ultimately, ascertaining what the patient experienced first (ie, respiratory or cutaneous symptoms) will determine where the pathology originated.
Computed tomography scan of the abdomen and pelvis with intravenous contrast demonstrated multiple ill-defined lytic lesions in the pelvis, including a 12-mm lesion of the left sacral ala and multiple subcentimeter lesions in the medial left iliac bone and superior right acetabulum. In addition, there were two 1-cm, rim-enhancing, hypodense nodules in the subcutaneous fat of the right flank at the level of L5 and the left lower quadrant, respectively. There was also a 2.2 × 1.9-cm faintly rim-enhancing hypodensity within the left iliopsoas muscle belly.
These imaging findings further corroborate a widely metastatic process probably originating in the lung and spreading to the lymph nodes, skin, muscles, and bones. The characterization of lesions as lytic as opposed to blastic is less helpful because many diseases can cause both. It does prompt consideration of multiple myeloma; however, multiple myeloma less commonly manifests with extramedullary plasmacytomas and is less likely given his normal renal function and calcium level. Bone lesions lessen the likelihood of GPA, and his necrotic lung mass makes sarcoidosis unlikely. Atypical infections and cancers are the prime suspect of his multisystemic disease.
There are no data yet to suggest a weakened immune system, which would increase his risk for atypical infections. His chronic lung disease, identified on imaging, is a risk factor for nocardiosis. This gram-positive, weakly acid-fast bacterium can involve any organ, although lung, brain, and skin are most commonly involved. Disseminated nocardiosis can result from a pulmonary or cutaneous site of origin. Mycobacteria; Actinomyces; dimorphic fungi like Histoplasma, Coccidioides, and Blastomyces; and molds such as Aspergillus can also cause disseminated disease with pulmonary, cutaneous, and musculoskeletal manifestations.
While metastases to muscle itself are rare, they can occur with primary lung cancers. Primary lung cancer with extrapulmonary features is feasible. Squamous cell lung cancer is the most likely to cavitate, although it rarely spreads to the skin. An aggressive lymphoma like diffuse large B-cell lymphoma or cutaneous T-cell lymphoma (higher occurrence in Asians) might also explain his constellation of findings. If culture data remain negative, then biopsy of the chest wall mass might be the safest and highest-yield target.
On hospital day 2, the patient developed new-onset severe neck pain. Magnetic resonance imaging of the cervical, thoracic, and lumbar spine revealed multilevel, bony, lytic lesions with notable cortical breakthrough of the C2 and C3 vertebrae into the prevertebral space, as well as epidural extension and paraspinal soft-tissue extension of the thoracic and lumbar vertebral lesions (Figure 3).
On hospital day 3, the patient reported increased tenderness in his skin nodules with one on his left forearm spontaneously draining purulent fluid. Repeat complete blood count demonstrated a white blood cell count of 12,600/mm3 (45% neutrophils, 43% lymphocytes, 8.4% monocytes, and 4.3% eosinophils), hemoglobin of 16 g/dL, and platelet count of 355,000/mm3.
The erosion into the manubrium and cortical destruction of the cervical spine attests to the aggressiveness of the underlying disease process. Noncutaneous lymphoma and lung cancer are unlikely to have such prominent skin findings; the visceral pathology, necrotizing lung mass, and bone lesions make cutaneous lymphoma less likely. At this point, a disseminated infectious process is most likely. Leading considerations based on his emigration from China and residence in California are tuberculosis and coccidioidomycosis, respectively. Tuberculous spondylitis most commonly involves the lower thoracic and upper lumbar region, and less commonly the cervical spine. His three negative AFB sputa samples further reduce its posttest probability. Ultimately microbiologic data are needed to distinguish between a disseminated fungal process, like coccidioidomycosis, or tuberculosis.
Given the concern for malignancy, a fine needle aspiration of the left supraclavicular lymph node was pursued. This revealed fungal microorganisms morphologically compatible with Coccidioides spp. with a background of necrotizing granulomas and acute inflammation. Fungal blood cultures grew Coccidioides immitis. AFB blood cultures were discontinued due to overgrowth of mold. The Coccidioides immitis antibody immunodiffusion titer was positive at 1:256.
During the remainder of the hospitalization, the patient was treated with oral fluconazole 800 mg daily. The patient underwent surgical debridement of the manubrium. In addition, given the concern for cervical spine instability, neurosurgery recommended follow-up with interval imaging. Since his discharge from the hospital, the patient continues to take oral fluconazole with resolution of his cutaneous lesions and respiratory symptoms. His titers have incrementally decreased from 1:256 to 1:16 after 8 months of treatment.
COMMENTARY
This elderly gentleman from China presented with subacute symptoms and was found to have numerous cutaneous nodules, lymphadenopathy, and diffuse osseous lesions. This multisystem illness posed a diagnostic challenge, forcing our discussant to search for a disease process that could lead to such varied findings. Ultimately, epidemiologic and clinical clues suggested a diagnosis of disseminated coccidioidomycosis, which was later confirmed on lymph node biopsy.
Coccidioides species are important fungal pathogens in the Western Hemisphere. This organism exhibits dimorphism, existing as mycelia (with arthroconidia) in soil and spherules in tissues. Coccidioides spp are endemic to the Southwestern United States, particularly California’s central valley and parts of Arizona; it additionally remains an important pathogen in Mexico, Central America, and South America.1 Newer epidemiologic studies have raised concerns that the incidence of coccidioidomycosis is increasing and that its geographic range may be more extensive than previously appreciated, with it now being found as far north as Washington state.2
Coccidioidal infection can take several forms. One-half to two-thirds of infections may be asymptomatic.3 Clinically significant infections can include an acute self-limiting respiratory illness, pulmonary nodules and cavities, chronic fibrocavitary pneumonia, and infections with extrapulmonary dissemination. Early respiratory infection is often indistinguishable from typical community-acquired pneumonia (10%-15% of pneumonia in endemic areas) but can be associated with certain suggestive features, such as erythema nodosum, erythema multiforme, prominent arthralgias (ie, “desert rheumatism”), and a peripheral eosinophilia.4,5
Extrapulmonary dissemination is rare and most commonly associated with immunocompromising states.6 However, individuals of African or Filipino ancestry also appear to be at increased risk for disseminated disease, which led to a California court decision that excluded African American inmates from state prisons located in Coccidioides endemic areas.7 The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the central nervous system (CNS).6 CNS disease has a predilection to manifest as a chronic basilar meningitis, most often complicated by hydrocephalus, vasculitic infarction, and spinal arachnoiditis.8
Cutaneous manifestations of coccidioidomycosis can occur as immunologic phenomenon associated with pulmonary disease or represent skin and soft tissue foci of disseminated infection.9 In primary pulmonary infection, skin findings can range from a nonspecific exanthem to erythema nodosum and erythema multiforme, which are thought to represent hypersensitivity responses. In contrast, Coccidioides spp can infect the skin either through direct inoculation (as in primary cutaneous coccidioidomycosis) or via hematogenous dissemination.9,10 A variety of lesions have been described, with painless nodules being the most frequently encountered morphotype in one study.11,12 On histopathologic examination, these lesions often have features of granulomatous dermatitis, eosinophilic infiltration, gummatous necrosis, microabscesses, or perivascular inflammation.13
Another common and highly morbid site of extrapulmonary dissemination is the musculoskeletal system. Bone and joint coccidioidomycosis most frequently affect the axial skeleton, although peripheral skeletal structures and joints can also be involved.6,12 Vertebral coccidioidomycosis is associated with significant morbidity. A study describing the magnetic resonance imaging findings of patients with vertebral coccidioidomycosis found that Coccidioides spp appeared to have a predilection for the thoracic vertebrae (in up to 80% of the study’s cohort).14 Skip lesions with noncontiguously involved vertebrae occurred in roughly half of patients, highlighting the usefulness of imaging the total spine in suspected cases.
The diagnosis of coccidioidomycosis is often established through serologic testing or by isolation of Coccidioides spp. on histopathology or culture. Obtaining sputum or tissue may be difficult, so clinicians often rely on noninvasive diagnostic tests such as coccidioidal antigen and serologies by enzyme immunoassays, immunodiffusion, and complement fixation. Enzyme immunoassays IgM and IgG results are positive early in the disease process and need to be confirmed with immunodiffusion or complement fixation testing. Complement fixation IgG is additionally useful to monitor disease activity over time and can help inform risk of disseminated disease.15 The gold standard of diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation either by direct visualization of a spherule or growth in fungal cultures.16 Polymerase chain reaction testing of sputum samples is an emerging diagnostic technique that has been found to have similar sensitivity rates to fungal culture.17
Treatment decisions in coccidioidomycosis are complex and vary by site of infection, immune status of the host, and extent of disease.16 While uncomplicated primary pulmonary infections can often be managed with observation alone, prolonged medical therapy with azole antifungals is often recommended for complicated pulmonary infections, symptomatic cavitary disease, and virtually all forms of extrapulmonary disease. Intravenous liposomal amphotericin is often used as initial therapy in immunosuppressed individuals, pregnant women, and those with extensive disease. CNS disease represents a particularly challenging treatment scenario and requires lifelong azole therapy.8,16
The patient in this case initially presented with vague inflammatory symptoms, with each aliquot revealing further evidence of a metastatic disease process. Such multisystem presentations are diagnostically challenging and force clinicians to reach for some feature around which to build their differential diagnosis. It is with this in mind that we are often taught to “localize the lesion” in order to focus our search for a unifying diagnosis. Yet, in this case, the sheer number of disease foci ultimately helped the discussant to narrow the range of diagnostic possibilities because only a limited number of conditions could present with such widespread, multisystem manifestations. Therefore, this case serves as a reminder that, sometimes in clinical reasoning, “more is less.”
KEY TEACHING POINTS
- Coccidioidomycosis is a fungal infection that can present with pulmonary or extrapulmonary disease. Risk of extrapulmonary dissemination is greatest among immunocompromised individuals and those of African or Filipino ancestry.3,7
- The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the CNS.6
- While serologic testing can be diagnostically useful, the gold standard for diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation with direct visualization of a spherule or growth in fungal cultures.16
A 64-year-old man presented with a 2-month history of a nonproductive cough, weight loss, and subjective fevers. He had no chest pain, hemoptysis, or shortness of breath. He also described worsening anorexia and a 15-pound weight loss over the previous 3 months. He had no arthralgias, myalgias, abdominal pain, nausea, emesis, or diarrhea.
Two weeks prior to his presentation, he was diagnosed with pneumonia and given a 5-day course of azithromycin. His symptoms did not improve, so he presented to the emergency room.
He had not been seen regularly by a physician in decades and had no known medical conditions. He did not take any medications. He immigrated from China 3 years prior and lived with his wife in California. He had a 30 pack-year smoking history. He drank a shot glass of liquor daily and denied any drug use.
Weight loss might result from inflammatory disorders like cancer or noninflammatory causes such as decreased oral intake (eg, diminished appetite) or malabsorption (eg, celiac disease). However, his fevers suggest inflammation, which usually reflects an underlying infection, cancer, or autoimmune process. While chronic cough typically results from upper airway cough syndrome (allergic or nonallergic rhinitis), gastroesophageal reflux disease, or asthma, it can also point to pathology of the lung, which may be intrinsic (bronchiectasis) or extrinsic (mediastinal mass). The duration of 2 months makes a typical infectious process like pneumococcal pneumonia unlikely. Atypical infections such as tuberculosis, melioidosis, and talaromycosis are possible given his immigration from East Asia, and coccidioidomycosis given his residence in California. He might have undiagnosed medical conditions, such as diabetes, that could be relevant to his current presentation and classify him as immunocompromised. His smoking history prompts consideration of lung cancer.
His temperature was 36.5 oC, heart rate 70 beats per minute, blood pressure 118/66 mm Hg, respiratory rate 16 breaths per minute, oxygen saturation 98% on room air, and body mass index 23 kg/m2. He was in no acute distress. The findings from the cardiac, lung, abdominal, and neurological exams were normal.
Skin examination found a fixed, symmetric, 5-cm, firm nodule at top of sternum (Figure 1A). In addition, he had two 1-cm, mobile, firm, subcutaneous nodules, one on his anterior left chest and another underneath his right axilla. He also had two 2-cm, erythematous, tender nodules on his left anterior forearm and a 1-cm nodule with a central black plug on the dorsal surface of his right hand (Figure 1B). He did not have any edema.
The white blood cell count was 10,500/mm3 (42% neutrophils, 37% lymphocytes, 16.4% monocytes, and 2.9% eosinophils), hemoglobin was 12.2 g/dL with a mean corpuscular volume of 91 fL, and the platelet count was 441,000/mm3. Basic metabolic panel, aminotransferase, bilirubin, and alkaline phosphatase were within reference ranges. Serum albumin was 3.1 g/dL. Serum total protein was elevated at 8.8 g/dL. Serum calcium was 9.0 mg/dL. Urinalysis results were normal.
The slightly low albumin, mildly elevated platelet count, monocytosis, and normocytic anemia suggest inflammation, although monocytosis might represent a hematologic malignancy like chronic myelomonocytic leukemia (CMML). His subjective fevers and weight loss further corroborate underlying inflammation. What is driving the inflammation? There are two localizing findings: cough and nodular skin lesions.
His lack of dyspnea and normal oxygen saturation, respiratory rate, and lung exam make an extrapulmonary cause of cough such as lymphadenopathy or mediastinal infection possible. The number of nodular skin lesions, wide-spread distribution, and appearance (eg, erythematous, tender) point to either a primary cutaneous disease with systemic manifestations (eg, cutaneous lymphoma) or a systemic disease with cutaneous features (eg, sarcoidosis).
Three categories—inflammatory, infectious, and neoplastic—account for most nodular skin lesions. Usually microscopic evaluation is necessary for definitive diagnosis, though epidemiology, associated symptoms, and characteristics of the nodules help prioritize the differential diagnosis. Tender nodules might reflect a panniculitis; erythema nodosum is the most common type, and while this classically develops on the anterior shins, it may also occur on the forearm. His immigration from China prompts consideration of tuberculosis and cutaneous leishmaniasis. Coccidioidomycosis can lead to inflammation and nodular skin lesions. Other infections such as nontuberculous mycobacteria, nocardiosis, and cryptococcosis may cause disseminated infection with pulmonary and skin manifestations. His smoking puts him at risk of lung cancer, which rarely results in metastatic subcutaneous infiltrates.
A chest radiograph demonstrated a prominent density in the right paratracheal region of the mediastinum with adjacent streaky opacities. A computed tomography scan of the chest with intravenous contrast demonstrated centrilobular emphysematous changes and revealed a 2.6 × 4.7-cm necrotic mass in the anterior chest wall with erosion into the manubrium, a 3.8 × 2.1-cm centrally necrotic soft-tissue mass in the right hilum, a 5-mm left upper-lobe noncalcified solid pulmonary nodule, and prominent subcarinal, paratracheal, hilar, and bilateral supraclavicular lymphadenopathy (Figure 2).
Flow cytometry of the peripheral blood did not demonstrate a lymphoproliferative disorder. Blood smear demonstrated normal red blood cell, white blood cell, and platelet morphology. HIV antibody was negative. Hemoglobin A1c was 6.1%. Smear microscopy for acid-fast bacilli (AFB) was negative and sputum AFB samples were sent for culture. Bacterial, fungal, and AFB blood cultures were collected and pending.
Causes of necrotizing pneumonia include liquid (eg, lymphoma) and solid (eg, squamous cell carcinoma) cancers, infections, and noninfectious inflammatory processes such as granulomatosis with polyangiitis (GPA). Given his subacute presentation and extrapulmonary cutaneous manifestations, consideration of mycobacteria, fungi (eg, Coccidioides, Aspergillus, and Cryptococcus), and filamentous bacteria (eg, Nocardia and Actinomyces) is prioritized among the myriad of infections that can cause a lung cavity. His smoking history and centrilobular emphysematous changes are highly suggestive of chronic obstructive pulmonary disease, which puts him at increased risk of bacterial colonization and recurrent pulmonary infections. Tuberculosis is still possible despite three negative AFB-sputa smears given the sensitivity of smear microscopy (with three specimens) is roughly 70% in an immunocompetent host.
The lymphadenopathy likely reflects spread from the necrotic lung mass. The frequency of non-Hodgkin lymphoma increases with age. The results of the peripheral flow cytometry do not exclude the possibility of an aggressive lymphoma with pulmonary and cutaneous manifestations.
The erosive property of the chest wall mass makes an autoimmune process like GPA unlikely. An aggressive and disseminated infection or cancer is most likely. A pathologic process that originated in the lung and then spread to the lymph nodes and skin is more likely than a disorder which started in the skin. It would be unlikely for a primary cutaneous disorder to cause such a well-defined necrotic lung mass. Lung cancer rarely metastasizes to the skin and, instead, preferentially involves the chest. Ultimately, ascertaining what the patient experienced first (ie, respiratory or cutaneous symptoms) will determine where the pathology originated.
Computed tomography scan of the abdomen and pelvis with intravenous contrast demonstrated multiple ill-defined lytic lesions in the pelvis, including a 12-mm lesion of the left sacral ala and multiple subcentimeter lesions in the medial left iliac bone and superior right acetabulum. In addition, there were two 1-cm, rim-enhancing, hypodense nodules in the subcutaneous fat of the right flank at the level of L5 and the left lower quadrant, respectively. There was also a 2.2 × 1.9-cm faintly rim-enhancing hypodensity within the left iliopsoas muscle belly.
These imaging findings further corroborate a widely metastatic process probably originating in the lung and spreading to the lymph nodes, skin, muscles, and bones. The characterization of lesions as lytic as opposed to blastic is less helpful because many diseases can cause both. It does prompt consideration of multiple myeloma; however, multiple myeloma less commonly manifests with extramedullary plasmacytomas and is less likely given his normal renal function and calcium level. Bone lesions lessen the likelihood of GPA, and his necrotic lung mass makes sarcoidosis unlikely. Atypical infections and cancers are the prime suspect of his multisystemic disease.
There are no data yet to suggest a weakened immune system, which would increase his risk for atypical infections. His chronic lung disease, identified on imaging, is a risk factor for nocardiosis. This gram-positive, weakly acid-fast bacterium can involve any organ, although lung, brain, and skin are most commonly involved. Disseminated nocardiosis can result from a pulmonary or cutaneous site of origin. Mycobacteria; Actinomyces; dimorphic fungi like Histoplasma, Coccidioides, and Blastomyces; and molds such as Aspergillus can also cause disseminated disease with pulmonary, cutaneous, and musculoskeletal manifestations.
While metastases to muscle itself are rare, they can occur with primary lung cancers. Primary lung cancer with extrapulmonary features is feasible. Squamous cell lung cancer is the most likely to cavitate, although it rarely spreads to the skin. An aggressive lymphoma like diffuse large B-cell lymphoma or cutaneous T-cell lymphoma (higher occurrence in Asians) might also explain his constellation of findings. If culture data remain negative, then biopsy of the chest wall mass might be the safest and highest-yield target.
On hospital day 2, the patient developed new-onset severe neck pain. Magnetic resonance imaging of the cervical, thoracic, and lumbar spine revealed multilevel, bony, lytic lesions with notable cortical breakthrough of the C2 and C3 vertebrae into the prevertebral space, as well as epidural extension and paraspinal soft-tissue extension of the thoracic and lumbar vertebral lesions (Figure 3).
On hospital day 3, the patient reported increased tenderness in his skin nodules with one on his left forearm spontaneously draining purulent fluid. Repeat complete blood count demonstrated a white blood cell count of 12,600/mm3 (45% neutrophils, 43% lymphocytes, 8.4% monocytes, and 4.3% eosinophils), hemoglobin of 16 g/dL, and platelet count of 355,000/mm3.
The erosion into the manubrium and cortical destruction of the cervical spine attests to the aggressiveness of the underlying disease process. Noncutaneous lymphoma and lung cancer are unlikely to have such prominent skin findings; the visceral pathology, necrotizing lung mass, and bone lesions make cutaneous lymphoma less likely. At this point, a disseminated infectious process is most likely. Leading considerations based on his emigration from China and residence in California are tuberculosis and coccidioidomycosis, respectively. Tuberculous spondylitis most commonly involves the lower thoracic and upper lumbar region, and less commonly the cervical spine. His three negative AFB sputa samples further reduce its posttest probability. Ultimately microbiologic data are needed to distinguish between a disseminated fungal process, like coccidioidomycosis, or tuberculosis.
Given the concern for malignancy, a fine needle aspiration of the left supraclavicular lymph node was pursued. This revealed fungal microorganisms morphologically compatible with Coccidioides spp. with a background of necrotizing granulomas and acute inflammation. Fungal blood cultures grew Coccidioides immitis. AFB blood cultures were discontinued due to overgrowth of mold. The Coccidioides immitis antibody immunodiffusion titer was positive at 1:256.
During the remainder of the hospitalization, the patient was treated with oral fluconazole 800 mg daily. The patient underwent surgical debridement of the manubrium. In addition, given the concern for cervical spine instability, neurosurgery recommended follow-up with interval imaging. Since his discharge from the hospital, the patient continues to take oral fluconazole with resolution of his cutaneous lesions and respiratory symptoms. His titers have incrementally decreased from 1:256 to 1:16 after 8 months of treatment.
COMMENTARY
This elderly gentleman from China presented with subacute symptoms and was found to have numerous cutaneous nodules, lymphadenopathy, and diffuse osseous lesions. This multisystem illness posed a diagnostic challenge, forcing our discussant to search for a disease process that could lead to such varied findings. Ultimately, epidemiologic and clinical clues suggested a diagnosis of disseminated coccidioidomycosis, which was later confirmed on lymph node biopsy.
Coccidioides species are important fungal pathogens in the Western Hemisphere. This organism exhibits dimorphism, existing as mycelia (with arthroconidia) in soil and spherules in tissues. Coccidioides spp are endemic to the Southwestern United States, particularly California’s central valley and parts of Arizona; it additionally remains an important pathogen in Mexico, Central America, and South America.1 Newer epidemiologic studies have raised concerns that the incidence of coccidioidomycosis is increasing and that its geographic range may be more extensive than previously appreciated, with it now being found as far north as Washington state.2
Coccidioidal infection can take several forms. One-half to two-thirds of infections may be asymptomatic.3 Clinically significant infections can include an acute self-limiting respiratory illness, pulmonary nodules and cavities, chronic fibrocavitary pneumonia, and infections with extrapulmonary dissemination. Early respiratory infection is often indistinguishable from typical community-acquired pneumonia (10%-15% of pneumonia in endemic areas) but can be associated with certain suggestive features, such as erythema nodosum, erythema multiforme, prominent arthralgias (ie, “desert rheumatism”), and a peripheral eosinophilia.4,5
Extrapulmonary dissemination is rare and most commonly associated with immunocompromising states.6 However, individuals of African or Filipino ancestry also appear to be at increased risk for disseminated disease, which led to a California court decision that excluded African American inmates from state prisons located in Coccidioides endemic areas.7 The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the central nervous system (CNS).6 CNS disease has a predilection to manifest as a chronic basilar meningitis, most often complicated by hydrocephalus, vasculitic infarction, and spinal arachnoiditis.8
Cutaneous manifestations of coccidioidomycosis can occur as immunologic phenomenon associated with pulmonary disease or represent skin and soft tissue foci of disseminated infection.9 In primary pulmonary infection, skin findings can range from a nonspecific exanthem to erythema nodosum and erythema multiforme, which are thought to represent hypersensitivity responses. In contrast, Coccidioides spp can infect the skin either through direct inoculation (as in primary cutaneous coccidioidomycosis) or via hematogenous dissemination.9,10 A variety of lesions have been described, with painless nodules being the most frequently encountered morphotype in one study.11,12 On histopathologic examination, these lesions often have features of granulomatous dermatitis, eosinophilic infiltration, gummatous necrosis, microabscesses, or perivascular inflammation.13
Another common and highly morbid site of extrapulmonary dissemination is the musculoskeletal system. Bone and joint coccidioidomycosis most frequently affect the axial skeleton, although peripheral skeletal structures and joints can also be involved.6,12 Vertebral coccidioidomycosis is associated with significant morbidity. A study describing the magnetic resonance imaging findings of patients with vertebral coccidioidomycosis found that Coccidioides spp appeared to have a predilection for the thoracic vertebrae (in up to 80% of the study’s cohort).14 Skip lesions with noncontiguously involved vertebrae occurred in roughly half of patients, highlighting the usefulness of imaging the total spine in suspected cases.
The diagnosis of coccidioidomycosis is often established through serologic testing or by isolation of Coccidioides spp. on histopathology or culture. Obtaining sputum or tissue may be difficult, so clinicians often rely on noninvasive diagnostic tests such as coccidioidal antigen and serologies by enzyme immunoassays, immunodiffusion, and complement fixation. Enzyme immunoassays IgM and IgG results are positive early in the disease process and need to be confirmed with immunodiffusion or complement fixation testing. Complement fixation IgG is additionally useful to monitor disease activity over time and can help inform risk of disseminated disease.15 The gold standard of diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation either by direct visualization of a spherule or growth in fungal cultures.16 Polymerase chain reaction testing of sputum samples is an emerging diagnostic technique that has been found to have similar sensitivity rates to fungal culture.17
Treatment decisions in coccidioidomycosis are complex and vary by site of infection, immune status of the host, and extent of disease.16 While uncomplicated primary pulmonary infections can often be managed with observation alone, prolonged medical therapy with azole antifungals is often recommended for complicated pulmonary infections, symptomatic cavitary disease, and virtually all forms of extrapulmonary disease. Intravenous liposomal amphotericin is often used as initial therapy in immunosuppressed individuals, pregnant women, and those with extensive disease. CNS disease represents a particularly challenging treatment scenario and requires lifelong azole therapy.8,16
The patient in this case initially presented with vague inflammatory symptoms, with each aliquot revealing further evidence of a metastatic disease process. Such multisystem presentations are diagnostically challenging and force clinicians to reach for some feature around which to build their differential diagnosis. It is with this in mind that we are often taught to “localize the lesion” in order to focus our search for a unifying diagnosis. Yet, in this case, the sheer number of disease foci ultimately helped the discussant to narrow the range of diagnostic possibilities because only a limited number of conditions could present with such widespread, multisystem manifestations. Therefore, this case serves as a reminder that, sometimes in clinical reasoning, “more is less.”
KEY TEACHING POINTS
- Coccidioidomycosis is a fungal infection that can present with pulmonary or extrapulmonary disease. Risk of extrapulmonary dissemination is greatest among immunocompromised individuals and those of African or Filipino ancestry.3,7
- The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the CNS.6
- While serologic testing can be diagnostically useful, the gold standard for diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation with direct visualization of a spherule or growth in fungal cultures.16
1. Benedict K, McCotter OZ, Brady S, et al. Surveillance for Coccidioidomycosis - United States, 2011-2017. MMWR Surveill Summ. 2019;68(No. SS-7):1-15. http://dx.doi.org/10.15585/mmwr.ss6807a1
2. McCotter OZ, Benedict K, Engelthaler DM, et al. Update on the epidemiology of coccidioidomycosis in the United States. Med Mycol. 2019;57(Suppl 1):S30-s40. https://doi.org/10.1093/mmy/myy095
3. Galgiani JN, Ampel NM, Blair JE, et al. Coccidioidomycosis. Clin Infect Dis. 2005;41(9):1217-1223. https://doi.org/10.1086/496991
4. Chang DC, Anderson S, Wannemuehler K, et al. Testing for coccidioidomycosis among patients with community-acquired pneumonia. Emerg Infect Dis. 2008;14(7):1053-1059. https://doi.org/10.3201/eid1407.070832
5. Saubolle MA, McKellar PP, Sussland D. Epidemiologic, clinical, and diagnostic aspects of coccidioidomycosis. J Clin Microbiol. 2007;45(1):26-30. https://doi.org/10.1128/jcm.02230-06
6. Adam RD, Elliott SP, Taljanovic MS. The spectrum and presentation of disseminated coccidioidomycosis. Am J Med. 2009;122(8):770-777. https://doi.org/10.1016/j.amjmed.2008.12.024
7. Wheeler C, Lucas KD, Mohle-Boetani JC. Rates and risk factors for Coccidioidomycosis among prison inmates, California, USA, 2011. Emerg Infect Dis. 2015;21(1):70-75. https://doi.org/10.3201/eid2101.140836
8. Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis. 2006;42(1):103-107. https://doi.org/10.1086/497596
9. Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections. Ann N Y Acad Sci. 2007;1111:411-421. https://doi.org/10.1196/annals.1406.010
10. Chang A, Tung RC, McGillis TS, Bergfeld WF, Taylor JS. Primary cutaneous coccidioidomycosis. J Am Acad Dermatol. 2003;49(5):944-949. https://doi.org/10.1016/s0190-9622(03)00462-6
11. Quimby SR, Connolly SM, Winkelmann RK, Smilack JD. Clinicopathologic spectrum of specific cutaneous lesions of disseminated coccidioidomycosis. J Am Acad Dermatol. 1992;26(1):79-85. https://doi.org/10.1016/0190-9622(92)70011-4
12. Crum NF, Lederman ER, Stafford CM, Parrish JS, Wallace MR. Coccidioidomycosis: a descriptive survey of a reemerging disease. clinical characteristics and current controversies. Medicine (Baltimore). 2004;83(3):149-175. https://doi.org/10.1097/01.md.0000126762.91040.fd
13. Carpenter JB, Feldman JS, Leyva WH, DiCaudo DJ. Clinical and pathologic characteristics of disseminated cutaneous coccidioidomycosis. J Am Acad Dermatol. 2010;62(5):831-837. https://doi.org/10.1016/j.jaad.2008.07.031
14. Crete RN, Gallmann W, Karis JP, Ross J. Spinal coccidioidomycosis: MR imaging findings in 41 patients. AJNR Am J Neuroradiol. 2018;39(11):2148-2153. https://doi.org/10.3174/ajnr.a5818
15. McHardy IH, Dinh BN, Waldman S, et al. Coccidioidomycosis complement fixation titer trends in the age of antifungals. J Clin Microbiol. 2018;56(12):e01318-18. https://doi.org/10.1128/jcm.01318-18
16. Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious Diseases Society of America (IDSA) clinical practice guideline for the treatment of coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112-e146. https://doi.org/10.1093/cid/ciw360
17. Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia. 2010;170(5):345-351. https://doi.org/10.1007/s11046-010-9327-0
1. Benedict K, McCotter OZ, Brady S, et al. Surveillance for Coccidioidomycosis - United States, 2011-2017. MMWR Surveill Summ. 2019;68(No. SS-7):1-15. http://dx.doi.org/10.15585/mmwr.ss6807a1
2. McCotter OZ, Benedict K, Engelthaler DM, et al. Update on the epidemiology of coccidioidomycosis in the United States. Med Mycol. 2019;57(Suppl 1):S30-s40. https://doi.org/10.1093/mmy/myy095
3. Galgiani JN, Ampel NM, Blair JE, et al. Coccidioidomycosis. Clin Infect Dis. 2005;41(9):1217-1223. https://doi.org/10.1086/496991
4. Chang DC, Anderson S, Wannemuehler K, et al. Testing for coccidioidomycosis among patients with community-acquired pneumonia. Emerg Infect Dis. 2008;14(7):1053-1059. https://doi.org/10.3201/eid1407.070832
5. Saubolle MA, McKellar PP, Sussland D. Epidemiologic, clinical, and diagnostic aspects of coccidioidomycosis. J Clin Microbiol. 2007;45(1):26-30. https://doi.org/10.1128/jcm.02230-06
6. Adam RD, Elliott SP, Taljanovic MS. The spectrum and presentation of disseminated coccidioidomycosis. Am J Med. 2009;122(8):770-777. https://doi.org/10.1016/j.amjmed.2008.12.024
7. Wheeler C, Lucas KD, Mohle-Boetani JC. Rates and risk factors for Coccidioidomycosis among prison inmates, California, USA, 2011. Emerg Infect Dis. 2015;21(1):70-75. https://doi.org/10.3201/eid2101.140836
8. Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis. 2006;42(1):103-107. https://doi.org/10.1086/497596
9. Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections. Ann N Y Acad Sci. 2007;1111:411-421. https://doi.org/10.1196/annals.1406.010
10. Chang A, Tung RC, McGillis TS, Bergfeld WF, Taylor JS. Primary cutaneous coccidioidomycosis. J Am Acad Dermatol. 2003;49(5):944-949. https://doi.org/10.1016/s0190-9622(03)00462-6
11. Quimby SR, Connolly SM, Winkelmann RK, Smilack JD. Clinicopathologic spectrum of specific cutaneous lesions of disseminated coccidioidomycosis. J Am Acad Dermatol. 1992;26(1):79-85. https://doi.org/10.1016/0190-9622(92)70011-4
12. Crum NF, Lederman ER, Stafford CM, Parrish JS, Wallace MR. Coccidioidomycosis: a descriptive survey of a reemerging disease. clinical characteristics and current controversies. Medicine (Baltimore). 2004;83(3):149-175. https://doi.org/10.1097/01.md.0000126762.91040.fd
13. Carpenter JB, Feldman JS, Leyva WH, DiCaudo DJ. Clinical and pathologic characteristics of disseminated cutaneous coccidioidomycosis. J Am Acad Dermatol. 2010;62(5):831-837. https://doi.org/10.1016/j.jaad.2008.07.031
14. Crete RN, Gallmann W, Karis JP, Ross J. Spinal coccidioidomycosis: MR imaging findings in 41 patients. AJNR Am J Neuroradiol. 2018;39(11):2148-2153. https://doi.org/10.3174/ajnr.a5818
15. McHardy IH, Dinh BN, Waldman S, et al. Coccidioidomycosis complement fixation titer trends in the age of antifungals. J Clin Microbiol. 2018;56(12):e01318-18. https://doi.org/10.1128/jcm.01318-18
16. Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious Diseases Society of America (IDSA) clinical practice guideline for the treatment of coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112-e146. https://doi.org/10.1093/cid/ciw360
17. Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia. 2010;170(5):345-351. https://doi.org/10.1007/s11046-010-9327-0
© 2020 Society of Hospital Medicine
Left Out in the Cold
A previously healthy 4-year-old boy presented to his pediatrician for nasal congestion, left ear pain, and intermittent fevers, which he’d been experiencing for 2 days. His exam was consistent with acute otitis media. Cefdinir was prescribed given a rash allergy to amoxicillin. His fever, congestion, and otalgia improved the next day.
Three days later he developed abdominal pain, fever, and labored breathing; his mother brought him to the emergency department (ED). His temperature was 38.0 °C, heart rate 141 beats per minute, blood pressure 117/71 mm Hg, respiratory rate 22 breaths per minute; he had oxygen saturation of 96% on ambient air. Despite mild accessory muscle use, he appeared comfortable and interactive. His left tympanic membrane was bulging without erythema. His neck was supple and mucous membranes moist. He had neither cervical lymphadenopathy nor conjunctival pallor. The cardiopulmonary exam was normal except for tachycardia. His abdomen was soft and not distended without organomegaly or tenderness.
Upper respiratory tract symptoms are commonly encountered in pediatrics and most often result from self-limited viral processes. Evaluation of a child with upper respiratory tract symptoms aims to identify serious causes like meningitis, as well as assessing the need for antimicrobial therapy. Supportive management is often appropriate in otitis media. His new, more concerning symptoms portend either a progression of the original process causing his upper respiratory tract symptoms or a separate etiology. It is key to determine which signs and symptoms are associated with the primary process and which are compensatory or secondary. If he were to be more ill appearing, for example, it is possible that his respiratory distress may be related to an underlying systemic illness rather than a primary lung process. Respiratory distress, abdominal pain, and fever could be a result of sepsis from an intrabdominal process such as ruptured appendicitis, intussusception, or malrotation with volvulus. Other causes of sepsis, such as meningitis or severe mastoiditis, both rare complications of otitis media, should be considered, although he does not appear severely ill. Acute myelogenous leukemia or other malignancies and illnesses associated with immunodeficiency can present with sepsis and chloromas in the middle ear that can be misconstrued as otitis media.
A chest radiograph demonstrated left lower lobe patchy opacities concerning for pneumonia. Rapid respiratory syncytial virus and influenza antigen test results were negative. Laboratory testing for general bloodwork was not obtained. He was administered a single dose of intramuscular ceftriaxone, prescribed a 5-day course of azithromycin, and discharged home. The child’s breathing gradually improved, but he continued to have subjective fevers. Two days later, he developed dark red urine. His mother brought him back to the outpatient clinic.
At the time of the ED visit, a diagnosis of community-acquired pneumonia was plausible given fever, mildly increased work of breathing, and an opacification on chest radiography. Most community-acquired pneumonia is caused by viruses; common bacterial causes for his age include Streptococcus pneumoniae and Moraxella catarrhalis. The first-line treatment for uncomplicated community-acquired pneumonia in children is amoxicillin, but this was appropriately avoided given his allergy.
The persistent fevers are surprising. The improvement in breathing corresponds to the treatment (and resolution) of community-acquired pneumonia. However, the development of dark urine does not. Red urine—in the absence of ingested pigments (such as those found in beets)—usually results from hematuria, hemoglobinuria, or myoglobinuria. Gross hematuria can originate from the kidneys to the urethral meatus. Abdominal masses, kidney trauma, or underlying kidney disease may all present with gross hematuria (or microscopic hematuria, seen only on urinalysis). The urine should be examined for the presence of heme, protein, and for evidence of infection; microscopy should be performed to examine for cellular casts and dysmorphic red cells. Tests of renal function, a comprehensive metabolic panel, evaluation of hematologic indexes, and assessments of inflammatory markers should be performed.
The child lived with his parents and had no siblings. He experienced no physical trauma, and there was no family history of kidney disease or hematuria. His father had a persistent cough and fever for 1 month, but recovered around the time the patient began to experience his initial symptoms. This was the patient’s third diagnosis of pneumonia. He had not traveled and was up to date with immunizations. He attended day care.
The fact that this is not the first episode of “pneumonia” raises important possibilities. The most likely one is that the child has had multiple viral infections; however, he could have an underlying primary immunodeficiency (PI) that predisposes him to recurrent infections. More severe PIs often present with recurrent sepsis, bacteremia, and failure to thrive, none of which were present in this case. Less severe PIs (such as selective IgA deficiency) could be possible. Another possibility is that these recurrent episodes of pneumonia are a relapsing and remitting noninfectious process, such as an antineutrophil cytoplasmic antibodies–associated vasculitis or anti–glomerular basement membrane disease. The patient’s father’s recent prolonged respiratory symptoms may be suggestive of pertussis or a “walking pneumonia” potentially caused by Mycoplasma or another atypical bacterium.
His temperature was 36.9 °C, heart rate 107 beats per minute, blood pressure was 106/67 mm Hg, and respiratory rate was 24 breaths per minute with oxygen saturation of 100% on ambient air. He was well appearing. His mucous membranes were moist, and oropharynx was clear. He had scleral icterus. The cardiopulmonary exam was normal. He had no significant lymphadenopathy, hepatosplenomegaly, or rashes.
The finding of jaundice is an important diagnostic pivot point, especially when combined with hematuria. The next step is determining if the jaundice is resulting from unconjugated or conjugated hyperbilirubinemia; the former most often stems from hemolysis or impairment in conjugation, while the latter results from intrahepatic or extrahepatic biliary defects. Tests for hepatobiliary injury including evaluations of alanine and aspartate aminotransferases and alkaline phosphatase, as well as for hepatic function such as tests of coagulation, should be performed.
The patient was referred to the ED and admitted for further evaluation. A complete blood count revealed a white blood cell (WBC) count of 10,700/µL (61% polymorphonuclear neutrophils, 30% lymphocytes, 5% monocytes, 3% eosinophils, 1% basophils), hemoglobin count was 10.3 g/dL (reticulocyte 2% with absolute reticulocyte count 58,400/μL), and platelet count was 265,000/µL. Components of the basic metabolic panel were within reference ranges except for a mildly elevated blood urea nitrogen level of 14 mg/dL with normal creatinine level of 0.3 mg/dL. Total protein was 6.7 g/dL (reference range, 6.4-8.3) and albumin 3.9 g/dL (reference range, 3.4-4.8). Alkaline phosphatase level was 188 U/L (reference range, 44-147), aspartate aminotransferase level 76 U/L (reference range, 0-40), and alanine aminotransferase level 12 U/L (reference range, 7-40). Total bilirubin level was 2.4 mg/dL (reference range, less than 1.5) with direct bilirubin level of 0.4 mg/dL. His C-reactive protein level was 1.5 mg/mL (reference range, 0-0.75). Creatinine kinase (CK) level was 2,550 U/L (reference range, 2-198). International Normalized Ratio (INR) was 1.0. Urinalysis was notable for 2+ proteinuria, large hemoglobin pigment, and 6 red blood cells per high power field (reference range, 0-4).
His blood urea nitrogen is elevated, reflecting either prerenal azotemia or increased absorption of nitrogenous products. Unconjugated hyperbilirubinemia may result from impaired hepatic bilirubin uptake (such as in heart failure or portosystemic shunts), impaired bilirubin conjugation (resulting from genetic conditions or drugs), or excess bilirubin production (such as in hemolysis); his anemia and lack of other evidence of hepatic dysfunction point to hemolysis as the etiology. The reticulocyte production index is approximately 1%, which suggests that an increase in erythrocyte generation is present but inadequate. This, however, does not mean that an erythrocyte production abnormality is present since reticulocytosis can be delayed in many cases of acute hemolytic anemia. It is also possible that the same hemolytic process is affecting mature and immature erythrocytes. A peripheral blood smear should be reviewed for evidence of intravascular hemolysis and testing for autoimmune hemolysis should be performed. Notably, his white blood cell and platelet counts are preserved, which makes a bone marrow–involved malignancy or infiltrative process less likely. The alkaline phosphatase elevation may result from either intrahepatic or extrahepatic biliopathy; bone damage is also possible. The elevation of aspartate aminotransferase, CK, and potassium, along with marked urinary heme pigment, may indicate muscle damage; the most common myositis in children is benign acute childhood myositis resulting from viral infection. However, the moderate level of CK elevation seen in this case is nonspecific and can result from many different etiologies. A metabolic myopathy, such as carnitine palmitoyltransferase II deficiency, can be made worse by metabolic stress and result in rhabdomyolysis; the presentations of inborn errors of metabolism are varied and a planned-out, stepwise approach in evaluation is fundamental.
Lactic acid dehydrogenase (LDH) level was 1,457 U/L (reference range, 140-280), and haptoglobin level was less than 6 mg/dL (reference range, 30-200). Peripheral blood smear demonstrated occasional atypical, reactive-appearing lymphocytes with red cell clumping and agglutination, as well as rare target, burr, and fragmented red cells. Test results for urine myoglobin were negative. Results of urine culture were negative. No blood culture was collected.
The elevated LDH, decreased haptoglobin, and findings on the peripheral blood smear confirm hemolysis. The clumping of erythrocytes can be artifactual in the preparation of peripheral smears, but when considered in the context of hemolysis, may be clinically important. Clumping of erythrocytes on the peripheral smear indicates the binding of a protein to antigens on the erythrocyte membrane; when this occurs below body temperature, this is consistent with the presence of a “cold agglutinin,” usually an IgM antibody directed at erythrocyte surface antigens that causes agglutination and destruction, especially in cooler areas of the body. This is a well-known complication of Mycoplasma pneumoniae infections as well as Epstein-Barr virus (EBV) infections; it may also occur with lymphoid malignancies or autoimmune disease.
Direct Coombs IgG test findings were negative, direct Coombs C3 test was positive, and direct Coombs polyspecific test was positive. M pneumoniae IgG antibody level was 1.4 mg/dL (reference ranges: <0.9, negative; 0.91-1.09, equivocal; >1.1, positive); M pneumoniae IgM level was 529 U/mL (reference range: <770, negative). EBV capsid IgM and IgG levels were undetectable. EBV nuclear antigen IgG level was also undetectable. EBV viral load was fewer than 10 copies/mL. Antinuclear antibodies (ANA) level was negative. General IgE and IgM levels were normal, at 11 and 81 mg/dL, respectively. Repeat complete blood count showed WBC of 7,800/µL, hemoglobin of 8.7 g/dL, and platelet count of 341,000/µL. The patient’s hemoglobin remained stable during hospitalization.
This directed testing is helpful in further classifying the patient’s hemolytic anemia. Autoimmune hemolytic anemias are classified into warm antibody–mediated, cold antibody–mediated, and mixed-type forms; drug-induced and alloimmune hemolytic anemias also occur. In addition, both systemic lupus erythematosus and antiphospholipid antibody syndrome can have hemolytic anemia with variable Coombs testing results; neither fit well in this case. The absence of red blood cell–directed IgG antibodies substantially decreases the likelihood of warm antibody–mediated hemolytic anemia. In cold antibody–mediated hemolytic anemia, antibodies bind to the erythrocyte membrane and then adhere to complement C3, which leads to both intravascular and extravascular hemolysis. Important types of cold antibody–mediated hemolytic anemia in children are primary and secondary cold agglutinin disease, along with paroxysmal cold hemoglobinuria. The Donath-Landsteiner test can be helpful in differentiating these conditions. Antibodies to Mycoplasma may be delayed in response to acute infection, and a child who is reinfected may only produce IgG antibodies. Given the patient’s clinical stability and previous health, the most likely diagnosis is Mycoplasma-induced cold antibody–mediated hemolytic anemia. It may be helpful to check convalescent titers to Mycoplasma in 2 to 4 weeks.
Donath-Landsteiner (D-L) antibody test results were positive. Medication-derived hemolytic anemia testing was conducted, but the presence of positive D-L antibody makes the test results inconclusive. This ultimately led to a diagnosis of paroxysmal cold hemoglobinuria (PCH), presumably triggered by a viral syndrome. Convalescent titers to Mycoplasma were not checked given clinical improvement. Because the patient’s hemoglobin was stable during hospitalization, he was not treated with steroids. His parents were counseled on avoiding cold temperatures for several days. Within 1 month, his hemoglobin had recovered without further evidence of hemolysis.
DISCUSSION
Hemolytic anemia refers to the accelerated destruction of red blood cells and can be further classified as acquired or hereditary.1 Hereditary conditions causing hemolytic anemia include enzymopathies (eg, glucose-6-phosphate dehydrogenase deficiency), hemoglobinopathies (eg, sickle cell disease), and membrane abnormalities (eg, hereditary spherocytosis). Acquired pathologies include microangiopathic hemolytic anemia (MAHA), anemias directly caused by certain infections such as malaria, and immune-mediated (Coombs-positive) hemolytic anemias.
MAHA can sometimes be life-threatening and is therefore important to identify quickly. In the right clinical context, such processes may be rapidly recognized by the presence of schistocytes on blood smear in addition to an elevated serum LDH level. Schistocytes suggest mechanical destruction of erythrocytes in the vasculature, the hallmark of MAHA. Important MAHAs include thrombocytopenic purpura, hemolytic-uremic syndrome, and disseminated intravascular coagulation. Though this patient did have a mildly elevated LDH, MAHA was less likely because there were no schistocytes on the blood smear.
Autoimmune hemolytic anemias (AIHAs) are another important subset of acquired hemolytic anemias. AIHAs occur when there is antibody-mediated destruction of erythrocytes. The direct Coombs test evaluates for antibody- or complement-coated erythrocytes. After administration of anti-IgG and anti-C3 serum, the test evaluates for agglutination of the red cells caused by attached antibodies or complement. Coombs-positive AIHA can also be categorized by the temperature of agglutination. “Warm” hemolysis often involves IgG autoantibodies (ie, warm agglutinins), while “cold” antibodies, usually IgM autoantibodies, bind at colder temperatures (0-4 °C) and activate complements, including C3. In this patient, the Coombs C3 was positive while the Coombs IgG was negative, which is more suggestive of a cold complement–mediated pathway.
Cold AIHA can be further categorized into primary cold agglutinin disease, secondary cold agglutinin disease, and PCH. Primary cold agglutinin disease is an autoimmune disorder that mostly occurs in adults. Secondary cold AIHA can often be triggered by bacterial infection (commonly M pneumoniae) or viruses including EBV, measles, and mumps.2 Medications, including penicillin and cephalosporins, can also be implicated. Secondary cold AIHA is also linked with autoimmune diseases, such as systemic lupus erythematosus and lymphoproliferative disorders. PCH can be identified with the unique presence of a specific autoantibody (ie, D-L autoantibody) that agglutinates at cold temperatures but dissociates on subsequent rewarming.3 Complement remains affixed and activates hemolysis.
The D-L antibody responsible for PCH is an IgG antibody to the P-antigen present on the erythrocyte surface. Since the Coombs test is conducted at normal temperature, it will be positive for the affixed complement but not for IgG. The underlying mechanism for PCH was proposed by Julius Donath, MD, and Karl Landsteiner, MD, in 1904 and is considered to be the first description of autoimmune disease being precipitated by antibodies.4 The D-L antibody test itself is uncommonly performed and somewhat difficult to interpret, particularly in adults, and may lead to false-negative results.5
PCH is an acquired, cold AIHA more common to children6,7 and may account for up to 33% of pediatric AIHA cases.8 Typical presentation is after an upper respiratory tract illness; however, the trigger is often not identified. Implicated triggers include a number of viruses.9 Clinical presentation includes findings of intravascular hemolysis similar to those in our patient. The pathogenic IgG autoantibody is polyclonal and is likely formed because of immune stimulation, which is consistent with the predominance of nonmalignant triggers of this disease process.10 Hemolysis and associated symptoms are often exacerbated with cold exposure; both typically resolve within 2 weeks. In recurrent cases, which are a minority, immunosuppression may be considered.10
PCH remains an often-understated cause of hemolytic anemia particularly in children. Lacking obvious pathognomonic clinical symptoms, it may be overlooked for other forms of AIHA or MAHA. However, with a structured approach to evaluation, as with this patient who had hematuria and jaundice, early diagnosis can prevent an unnecessarily extensive workup and can provide reassurance to patient and parents. By understanding the basic categories of hemolytic anemia, the relevant blood testing available, and interpretation of Coombs test results, clinicians can ensure that PCH is a diagnosis that is not left out in the cold.
KEY TEACHING POINTS
- Examination for schistocytes on a blood smear can help identify life-threatening causes of hemolytic anemia.
- Characterization of cold AIHA includes defining the underlying etiology as primary cold agglutinin disease, secondary cold agglutinin disease, or PCH.
- PCH is a cold AIHA that is an underrecognized cause of hemolytic anemia in children. The diagnosis of PCH is made by testing for the presence of the D-L antibody.
1. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
2. Djaldetti M. Paroxysmal cold hemoglobinuria. CRC Crit Rev Clin Lab Sci. 1978;9(1):49-83. https://doi.org/10.3109/10408367809150915
3. Levine P, Celano MJ, Falkowski F. The specificity of the antibody in paroxysmal cold hemoglobinuria (P.C.H.). Transfusion. 1963;3(4):278-280. https://doi.org/10.1111/j.1537-2995.1963.tb04643.x
4. Donath J, Landsteiner K. Uber Paroxysmale Hamoglobinurie. Munch Med Wochenschr. 1904;51:1590-1593
5. Zeller MP, Arnold DM, Al Habsi K, et al. Paroxysmal cold hemoglobinuria: a difficult diagnosis in adult patients. Transfusion. 2017;57(1):137-143. https://doi.org/10.1111/trf.13888
6. Göttsche B, Salama A, Mueller-Eckhardt C. Donath-Landsteiner autoimmune hemolytic anemia in children. a study of 22 cases. Vox Sang. 1990;58(4):281-286. https://doi.org/10.1111/j.1423-0410.1990.tb05000.x
7. Sokol RJ, Booker DJ, Stamps R. Erythropoiesis: paroxysmal cold haemoglobinuria: a clinico-pathological study of patients with a positive Donath-Landsteiner test. Hematology. 1999;4(2):137-164. https://doi.org/10.1080/10245332.1999.11746439
8. Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008;22(1):1-15. https://doi.org/10.1016/j.blre.2007.08.002
9. Leibrandt R, Angelino K, Vizel-Schwartz M, Shapira I. Paroxysmal cold hemoglobinuria in an adult with respiratory syncytial virus. Case Rep Hematol. 2018;2018:1-3. https://doi.org/10.1155/2018/7586719
10. Gertz MA. Management of cold haemolytic syndrome. Br J Haematol. 2007;138(4):422-429. https://doi.org/10.1111/j.1365-2141.2007.06664.x
A previously healthy 4-year-old boy presented to his pediatrician for nasal congestion, left ear pain, and intermittent fevers, which he’d been experiencing for 2 days. His exam was consistent with acute otitis media. Cefdinir was prescribed given a rash allergy to amoxicillin. His fever, congestion, and otalgia improved the next day.
Three days later he developed abdominal pain, fever, and labored breathing; his mother brought him to the emergency department (ED). His temperature was 38.0 °C, heart rate 141 beats per minute, blood pressure 117/71 mm Hg, respiratory rate 22 breaths per minute; he had oxygen saturation of 96% on ambient air. Despite mild accessory muscle use, he appeared comfortable and interactive. His left tympanic membrane was bulging without erythema. His neck was supple and mucous membranes moist. He had neither cervical lymphadenopathy nor conjunctival pallor. The cardiopulmonary exam was normal except for tachycardia. His abdomen was soft and not distended without organomegaly or tenderness.
Upper respiratory tract symptoms are commonly encountered in pediatrics and most often result from self-limited viral processes. Evaluation of a child with upper respiratory tract symptoms aims to identify serious causes like meningitis, as well as assessing the need for antimicrobial therapy. Supportive management is often appropriate in otitis media. His new, more concerning symptoms portend either a progression of the original process causing his upper respiratory tract symptoms or a separate etiology. It is key to determine which signs and symptoms are associated with the primary process and which are compensatory or secondary. If he were to be more ill appearing, for example, it is possible that his respiratory distress may be related to an underlying systemic illness rather than a primary lung process. Respiratory distress, abdominal pain, and fever could be a result of sepsis from an intrabdominal process such as ruptured appendicitis, intussusception, or malrotation with volvulus. Other causes of sepsis, such as meningitis or severe mastoiditis, both rare complications of otitis media, should be considered, although he does not appear severely ill. Acute myelogenous leukemia or other malignancies and illnesses associated with immunodeficiency can present with sepsis and chloromas in the middle ear that can be misconstrued as otitis media.
A chest radiograph demonstrated left lower lobe patchy opacities concerning for pneumonia. Rapid respiratory syncytial virus and influenza antigen test results were negative. Laboratory testing for general bloodwork was not obtained. He was administered a single dose of intramuscular ceftriaxone, prescribed a 5-day course of azithromycin, and discharged home. The child’s breathing gradually improved, but he continued to have subjective fevers. Two days later, he developed dark red urine. His mother brought him back to the outpatient clinic.
At the time of the ED visit, a diagnosis of community-acquired pneumonia was plausible given fever, mildly increased work of breathing, and an opacification on chest radiography. Most community-acquired pneumonia is caused by viruses; common bacterial causes for his age include Streptococcus pneumoniae and Moraxella catarrhalis. The first-line treatment for uncomplicated community-acquired pneumonia in children is amoxicillin, but this was appropriately avoided given his allergy.
The persistent fevers are surprising. The improvement in breathing corresponds to the treatment (and resolution) of community-acquired pneumonia. However, the development of dark urine does not. Red urine—in the absence of ingested pigments (such as those found in beets)—usually results from hematuria, hemoglobinuria, or myoglobinuria. Gross hematuria can originate from the kidneys to the urethral meatus. Abdominal masses, kidney trauma, or underlying kidney disease may all present with gross hematuria (or microscopic hematuria, seen only on urinalysis). The urine should be examined for the presence of heme, protein, and for evidence of infection; microscopy should be performed to examine for cellular casts and dysmorphic red cells. Tests of renal function, a comprehensive metabolic panel, evaluation of hematologic indexes, and assessments of inflammatory markers should be performed.
The child lived with his parents and had no siblings. He experienced no physical trauma, and there was no family history of kidney disease or hematuria. His father had a persistent cough and fever for 1 month, but recovered around the time the patient began to experience his initial symptoms. This was the patient’s third diagnosis of pneumonia. He had not traveled and was up to date with immunizations. He attended day care.
The fact that this is not the first episode of “pneumonia” raises important possibilities. The most likely one is that the child has had multiple viral infections; however, he could have an underlying primary immunodeficiency (PI) that predisposes him to recurrent infections. More severe PIs often present with recurrent sepsis, bacteremia, and failure to thrive, none of which were present in this case. Less severe PIs (such as selective IgA deficiency) could be possible. Another possibility is that these recurrent episodes of pneumonia are a relapsing and remitting noninfectious process, such as an antineutrophil cytoplasmic antibodies–associated vasculitis or anti–glomerular basement membrane disease. The patient’s father’s recent prolonged respiratory symptoms may be suggestive of pertussis or a “walking pneumonia” potentially caused by Mycoplasma or another atypical bacterium.
His temperature was 36.9 °C, heart rate 107 beats per minute, blood pressure was 106/67 mm Hg, and respiratory rate was 24 breaths per minute with oxygen saturation of 100% on ambient air. He was well appearing. His mucous membranes were moist, and oropharynx was clear. He had scleral icterus. The cardiopulmonary exam was normal. He had no significant lymphadenopathy, hepatosplenomegaly, or rashes.
The finding of jaundice is an important diagnostic pivot point, especially when combined with hematuria. The next step is determining if the jaundice is resulting from unconjugated or conjugated hyperbilirubinemia; the former most often stems from hemolysis or impairment in conjugation, while the latter results from intrahepatic or extrahepatic biliary defects. Tests for hepatobiliary injury including evaluations of alanine and aspartate aminotransferases and alkaline phosphatase, as well as for hepatic function such as tests of coagulation, should be performed.
The patient was referred to the ED and admitted for further evaluation. A complete blood count revealed a white blood cell (WBC) count of 10,700/µL (61% polymorphonuclear neutrophils, 30% lymphocytes, 5% monocytes, 3% eosinophils, 1% basophils), hemoglobin count was 10.3 g/dL (reticulocyte 2% with absolute reticulocyte count 58,400/μL), and platelet count was 265,000/µL. Components of the basic metabolic panel were within reference ranges except for a mildly elevated blood urea nitrogen level of 14 mg/dL with normal creatinine level of 0.3 mg/dL. Total protein was 6.7 g/dL (reference range, 6.4-8.3) and albumin 3.9 g/dL (reference range, 3.4-4.8). Alkaline phosphatase level was 188 U/L (reference range, 44-147), aspartate aminotransferase level 76 U/L (reference range, 0-40), and alanine aminotransferase level 12 U/L (reference range, 7-40). Total bilirubin level was 2.4 mg/dL (reference range, less than 1.5) with direct bilirubin level of 0.4 mg/dL. His C-reactive protein level was 1.5 mg/mL (reference range, 0-0.75). Creatinine kinase (CK) level was 2,550 U/L (reference range, 2-198). International Normalized Ratio (INR) was 1.0. Urinalysis was notable for 2+ proteinuria, large hemoglobin pigment, and 6 red blood cells per high power field (reference range, 0-4).
His blood urea nitrogen is elevated, reflecting either prerenal azotemia or increased absorption of nitrogenous products. Unconjugated hyperbilirubinemia may result from impaired hepatic bilirubin uptake (such as in heart failure or portosystemic shunts), impaired bilirubin conjugation (resulting from genetic conditions or drugs), or excess bilirubin production (such as in hemolysis); his anemia and lack of other evidence of hepatic dysfunction point to hemolysis as the etiology. The reticulocyte production index is approximately 1%, which suggests that an increase in erythrocyte generation is present but inadequate. This, however, does not mean that an erythrocyte production abnormality is present since reticulocytosis can be delayed in many cases of acute hemolytic anemia. It is also possible that the same hemolytic process is affecting mature and immature erythrocytes. A peripheral blood smear should be reviewed for evidence of intravascular hemolysis and testing for autoimmune hemolysis should be performed. Notably, his white blood cell and platelet counts are preserved, which makes a bone marrow–involved malignancy or infiltrative process less likely. The alkaline phosphatase elevation may result from either intrahepatic or extrahepatic biliopathy; bone damage is also possible. The elevation of aspartate aminotransferase, CK, and potassium, along with marked urinary heme pigment, may indicate muscle damage; the most common myositis in children is benign acute childhood myositis resulting from viral infection. However, the moderate level of CK elevation seen in this case is nonspecific and can result from many different etiologies. A metabolic myopathy, such as carnitine palmitoyltransferase II deficiency, can be made worse by metabolic stress and result in rhabdomyolysis; the presentations of inborn errors of metabolism are varied and a planned-out, stepwise approach in evaluation is fundamental.
Lactic acid dehydrogenase (LDH) level was 1,457 U/L (reference range, 140-280), and haptoglobin level was less than 6 mg/dL (reference range, 30-200). Peripheral blood smear demonstrated occasional atypical, reactive-appearing lymphocytes with red cell clumping and agglutination, as well as rare target, burr, and fragmented red cells. Test results for urine myoglobin were negative. Results of urine culture were negative. No blood culture was collected.
The elevated LDH, decreased haptoglobin, and findings on the peripheral blood smear confirm hemolysis. The clumping of erythrocytes can be artifactual in the preparation of peripheral smears, but when considered in the context of hemolysis, may be clinically important. Clumping of erythrocytes on the peripheral smear indicates the binding of a protein to antigens on the erythrocyte membrane; when this occurs below body temperature, this is consistent with the presence of a “cold agglutinin,” usually an IgM antibody directed at erythrocyte surface antigens that causes agglutination and destruction, especially in cooler areas of the body. This is a well-known complication of Mycoplasma pneumoniae infections as well as Epstein-Barr virus (EBV) infections; it may also occur with lymphoid malignancies or autoimmune disease.
Direct Coombs IgG test findings were negative, direct Coombs C3 test was positive, and direct Coombs polyspecific test was positive. M pneumoniae IgG antibody level was 1.4 mg/dL (reference ranges: <0.9, negative; 0.91-1.09, equivocal; >1.1, positive); M pneumoniae IgM level was 529 U/mL (reference range: <770, negative). EBV capsid IgM and IgG levels were undetectable. EBV nuclear antigen IgG level was also undetectable. EBV viral load was fewer than 10 copies/mL. Antinuclear antibodies (ANA) level was negative. General IgE and IgM levels were normal, at 11 and 81 mg/dL, respectively. Repeat complete blood count showed WBC of 7,800/µL, hemoglobin of 8.7 g/dL, and platelet count of 341,000/µL. The patient’s hemoglobin remained stable during hospitalization.
This directed testing is helpful in further classifying the patient’s hemolytic anemia. Autoimmune hemolytic anemias are classified into warm antibody–mediated, cold antibody–mediated, and mixed-type forms; drug-induced and alloimmune hemolytic anemias also occur. In addition, both systemic lupus erythematosus and antiphospholipid antibody syndrome can have hemolytic anemia with variable Coombs testing results; neither fit well in this case. The absence of red blood cell–directed IgG antibodies substantially decreases the likelihood of warm antibody–mediated hemolytic anemia. In cold antibody–mediated hemolytic anemia, antibodies bind to the erythrocyte membrane and then adhere to complement C3, which leads to both intravascular and extravascular hemolysis. Important types of cold antibody–mediated hemolytic anemia in children are primary and secondary cold agglutinin disease, along with paroxysmal cold hemoglobinuria. The Donath-Landsteiner test can be helpful in differentiating these conditions. Antibodies to Mycoplasma may be delayed in response to acute infection, and a child who is reinfected may only produce IgG antibodies. Given the patient’s clinical stability and previous health, the most likely diagnosis is Mycoplasma-induced cold antibody–mediated hemolytic anemia. It may be helpful to check convalescent titers to Mycoplasma in 2 to 4 weeks.
Donath-Landsteiner (D-L) antibody test results were positive. Medication-derived hemolytic anemia testing was conducted, but the presence of positive D-L antibody makes the test results inconclusive. This ultimately led to a diagnosis of paroxysmal cold hemoglobinuria (PCH), presumably triggered by a viral syndrome. Convalescent titers to Mycoplasma were not checked given clinical improvement. Because the patient’s hemoglobin was stable during hospitalization, he was not treated with steroids. His parents were counseled on avoiding cold temperatures for several days. Within 1 month, his hemoglobin had recovered without further evidence of hemolysis.
DISCUSSION
Hemolytic anemia refers to the accelerated destruction of red blood cells and can be further classified as acquired or hereditary.1 Hereditary conditions causing hemolytic anemia include enzymopathies (eg, glucose-6-phosphate dehydrogenase deficiency), hemoglobinopathies (eg, sickle cell disease), and membrane abnormalities (eg, hereditary spherocytosis). Acquired pathologies include microangiopathic hemolytic anemia (MAHA), anemias directly caused by certain infections such as malaria, and immune-mediated (Coombs-positive) hemolytic anemias.
MAHA can sometimes be life-threatening and is therefore important to identify quickly. In the right clinical context, such processes may be rapidly recognized by the presence of schistocytes on blood smear in addition to an elevated serum LDH level. Schistocytes suggest mechanical destruction of erythrocytes in the vasculature, the hallmark of MAHA. Important MAHAs include thrombocytopenic purpura, hemolytic-uremic syndrome, and disseminated intravascular coagulation. Though this patient did have a mildly elevated LDH, MAHA was less likely because there were no schistocytes on the blood smear.
Autoimmune hemolytic anemias (AIHAs) are another important subset of acquired hemolytic anemias. AIHAs occur when there is antibody-mediated destruction of erythrocytes. The direct Coombs test evaluates for antibody- or complement-coated erythrocytes. After administration of anti-IgG and anti-C3 serum, the test evaluates for agglutination of the red cells caused by attached antibodies or complement. Coombs-positive AIHA can also be categorized by the temperature of agglutination. “Warm” hemolysis often involves IgG autoantibodies (ie, warm agglutinins), while “cold” antibodies, usually IgM autoantibodies, bind at colder temperatures (0-4 °C) and activate complements, including C3. In this patient, the Coombs C3 was positive while the Coombs IgG was negative, which is more suggestive of a cold complement–mediated pathway.
Cold AIHA can be further categorized into primary cold agglutinin disease, secondary cold agglutinin disease, and PCH. Primary cold agglutinin disease is an autoimmune disorder that mostly occurs in adults. Secondary cold AIHA can often be triggered by bacterial infection (commonly M pneumoniae) or viruses including EBV, measles, and mumps.2 Medications, including penicillin and cephalosporins, can also be implicated. Secondary cold AIHA is also linked with autoimmune diseases, such as systemic lupus erythematosus and lymphoproliferative disorders. PCH can be identified with the unique presence of a specific autoantibody (ie, D-L autoantibody) that agglutinates at cold temperatures but dissociates on subsequent rewarming.3 Complement remains affixed and activates hemolysis.
The D-L antibody responsible for PCH is an IgG antibody to the P-antigen present on the erythrocyte surface. Since the Coombs test is conducted at normal temperature, it will be positive for the affixed complement but not for IgG. The underlying mechanism for PCH was proposed by Julius Donath, MD, and Karl Landsteiner, MD, in 1904 and is considered to be the first description of autoimmune disease being precipitated by antibodies.4 The D-L antibody test itself is uncommonly performed and somewhat difficult to interpret, particularly in adults, and may lead to false-negative results.5
PCH is an acquired, cold AIHA more common to children6,7 and may account for up to 33% of pediatric AIHA cases.8 Typical presentation is after an upper respiratory tract illness; however, the trigger is often not identified. Implicated triggers include a number of viruses.9 Clinical presentation includes findings of intravascular hemolysis similar to those in our patient. The pathogenic IgG autoantibody is polyclonal and is likely formed because of immune stimulation, which is consistent with the predominance of nonmalignant triggers of this disease process.10 Hemolysis and associated symptoms are often exacerbated with cold exposure; both typically resolve within 2 weeks. In recurrent cases, which are a minority, immunosuppression may be considered.10
PCH remains an often-understated cause of hemolytic anemia particularly in children. Lacking obvious pathognomonic clinical symptoms, it may be overlooked for other forms of AIHA or MAHA. However, with a structured approach to evaluation, as with this patient who had hematuria and jaundice, early diagnosis can prevent an unnecessarily extensive workup and can provide reassurance to patient and parents. By understanding the basic categories of hemolytic anemia, the relevant blood testing available, and interpretation of Coombs test results, clinicians can ensure that PCH is a diagnosis that is not left out in the cold.
KEY TEACHING POINTS
- Examination for schistocytes on a blood smear can help identify life-threatening causes of hemolytic anemia.
- Characterization of cold AIHA includes defining the underlying etiology as primary cold agglutinin disease, secondary cold agglutinin disease, or PCH.
- PCH is a cold AIHA that is an underrecognized cause of hemolytic anemia in children. The diagnosis of PCH is made by testing for the presence of the D-L antibody.
A previously healthy 4-year-old boy presented to his pediatrician for nasal congestion, left ear pain, and intermittent fevers, which he’d been experiencing for 2 days. His exam was consistent with acute otitis media. Cefdinir was prescribed given a rash allergy to amoxicillin. His fever, congestion, and otalgia improved the next day.
Three days later he developed abdominal pain, fever, and labored breathing; his mother brought him to the emergency department (ED). His temperature was 38.0 °C, heart rate 141 beats per minute, blood pressure 117/71 mm Hg, respiratory rate 22 breaths per minute; he had oxygen saturation of 96% on ambient air. Despite mild accessory muscle use, he appeared comfortable and interactive. His left tympanic membrane was bulging without erythema. His neck was supple and mucous membranes moist. He had neither cervical lymphadenopathy nor conjunctival pallor. The cardiopulmonary exam was normal except for tachycardia. His abdomen was soft and not distended without organomegaly or tenderness.
Upper respiratory tract symptoms are commonly encountered in pediatrics and most often result from self-limited viral processes. Evaluation of a child with upper respiratory tract symptoms aims to identify serious causes like meningitis, as well as assessing the need for antimicrobial therapy. Supportive management is often appropriate in otitis media. His new, more concerning symptoms portend either a progression of the original process causing his upper respiratory tract symptoms or a separate etiology. It is key to determine which signs and symptoms are associated with the primary process and which are compensatory or secondary. If he were to be more ill appearing, for example, it is possible that his respiratory distress may be related to an underlying systemic illness rather than a primary lung process. Respiratory distress, abdominal pain, and fever could be a result of sepsis from an intrabdominal process such as ruptured appendicitis, intussusception, or malrotation with volvulus. Other causes of sepsis, such as meningitis or severe mastoiditis, both rare complications of otitis media, should be considered, although he does not appear severely ill. Acute myelogenous leukemia or other malignancies and illnesses associated with immunodeficiency can present with sepsis and chloromas in the middle ear that can be misconstrued as otitis media.
A chest radiograph demonstrated left lower lobe patchy opacities concerning for pneumonia. Rapid respiratory syncytial virus and influenza antigen test results were negative. Laboratory testing for general bloodwork was not obtained. He was administered a single dose of intramuscular ceftriaxone, prescribed a 5-day course of azithromycin, and discharged home. The child’s breathing gradually improved, but he continued to have subjective fevers. Two days later, he developed dark red urine. His mother brought him back to the outpatient clinic.
At the time of the ED visit, a diagnosis of community-acquired pneumonia was plausible given fever, mildly increased work of breathing, and an opacification on chest radiography. Most community-acquired pneumonia is caused by viruses; common bacterial causes for his age include Streptococcus pneumoniae and Moraxella catarrhalis. The first-line treatment for uncomplicated community-acquired pneumonia in children is amoxicillin, but this was appropriately avoided given his allergy.
The persistent fevers are surprising. The improvement in breathing corresponds to the treatment (and resolution) of community-acquired pneumonia. However, the development of dark urine does not. Red urine—in the absence of ingested pigments (such as those found in beets)—usually results from hematuria, hemoglobinuria, or myoglobinuria. Gross hematuria can originate from the kidneys to the urethral meatus. Abdominal masses, kidney trauma, or underlying kidney disease may all present with gross hematuria (or microscopic hematuria, seen only on urinalysis). The urine should be examined for the presence of heme, protein, and for evidence of infection; microscopy should be performed to examine for cellular casts and dysmorphic red cells. Tests of renal function, a comprehensive metabolic panel, evaluation of hematologic indexes, and assessments of inflammatory markers should be performed.
The child lived with his parents and had no siblings. He experienced no physical trauma, and there was no family history of kidney disease or hematuria. His father had a persistent cough and fever for 1 month, but recovered around the time the patient began to experience his initial symptoms. This was the patient’s third diagnosis of pneumonia. He had not traveled and was up to date with immunizations. He attended day care.
The fact that this is not the first episode of “pneumonia” raises important possibilities. The most likely one is that the child has had multiple viral infections; however, he could have an underlying primary immunodeficiency (PI) that predisposes him to recurrent infections. More severe PIs often present with recurrent sepsis, bacteremia, and failure to thrive, none of which were present in this case. Less severe PIs (such as selective IgA deficiency) could be possible. Another possibility is that these recurrent episodes of pneumonia are a relapsing and remitting noninfectious process, such as an antineutrophil cytoplasmic antibodies–associated vasculitis or anti–glomerular basement membrane disease. The patient’s father’s recent prolonged respiratory symptoms may be suggestive of pertussis or a “walking pneumonia” potentially caused by Mycoplasma or another atypical bacterium.
His temperature was 36.9 °C, heart rate 107 beats per minute, blood pressure was 106/67 mm Hg, and respiratory rate was 24 breaths per minute with oxygen saturation of 100% on ambient air. He was well appearing. His mucous membranes were moist, and oropharynx was clear. He had scleral icterus. The cardiopulmonary exam was normal. He had no significant lymphadenopathy, hepatosplenomegaly, or rashes.
The finding of jaundice is an important diagnostic pivot point, especially when combined with hematuria. The next step is determining if the jaundice is resulting from unconjugated or conjugated hyperbilirubinemia; the former most often stems from hemolysis or impairment in conjugation, while the latter results from intrahepatic or extrahepatic biliary defects. Tests for hepatobiliary injury including evaluations of alanine and aspartate aminotransferases and alkaline phosphatase, as well as for hepatic function such as tests of coagulation, should be performed.
The patient was referred to the ED and admitted for further evaluation. A complete blood count revealed a white blood cell (WBC) count of 10,700/µL (61% polymorphonuclear neutrophils, 30% lymphocytes, 5% monocytes, 3% eosinophils, 1% basophils), hemoglobin count was 10.3 g/dL (reticulocyte 2% with absolute reticulocyte count 58,400/μL), and platelet count was 265,000/µL. Components of the basic metabolic panel were within reference ranges except for a mildly elevated blood urea nitrogen level of 14 mg/dL with normal creatinine level of 0.3 mg/dL. Total protein was 6.7 g/dL (reference range, 6.4-8.3) and albumin 3.9 g/dL (reference range, 3.4-4.8). Alkaline phosphatase level was 188 U/L (reference range, 44-147), aspartate aminotransferase level 76 U/L (reference range, 0-40), and alanine aminotransferase level 12 U/L (reference range, 7-40). Total bilirubin level was 2.4 mg/dL (reference range, less than 1.5) with direct bilirubin level of 0.4 mg/dL. His C-reactive protein level was 1.5 mg/mL (reference range, 0-0.75). Creatinine kinase (CK) level was 2,550 U/L (reference range, 2-198). International Normalized Ratio (INR) was 1.0. Urinalysis was notable for 2+ proteinuria, large hemoglobin pigment, and 6 red blood cells per high power field (reference range, 0-4).
His blood urea nitrogen is elevated, reflecting either prerenal azotemia or increased absorption of nitrogenous products. Unconjugated hyperbilirubinemia may result from impaired hepatic bilirubin uptake (such as in heart failure or portosystemic shunts), impaired bilirubin conjugation (resulting from genetic conditions or drugs), or excess bilirubin production (such as in hemolysis); his anemia and lack of other evidence of hepatic dysfunction point to hemolysis as the etiology. The reticulocyte production index is approximately 1%, which suggests that an increase in erythrocyte generation is present but inadequate. This, however, does not mean that an erythrocyte production abnormality is present since reticulocytosis can be delayed in many cases of acute hemolytic anemia. It is also possible that the same hemolytic process is affecting mature and immature erythrocytes. A peripheral blood smear should be reviewed for evidence of intravascular hemolysis and testing for autoimmune hemolysis should be performed. Notably, his white blood cell and platelet counts are preserved, which makes a bone marrow–involved malignancy or infiltrative process less likely. The alkaline phosphatase elevation may result from either intrahepatic or extrahepatic biliopathy; bone damage is also possible. The elevation of aspartate aminotransferase, CK, and potassium, along with marked urinary heme pigment, may indicate muscle damage; the most common myositis in children is benign acute childhood myositis resulting from viral infection. However, the moderate level of CK elevation seen in this case is nonspecific and can result from many different etiologies. A metabolic myopathy, such as carnitine palmitoyltransferase II deficiency, can be made worse by metabolic stress and result in rhabdomyolysis; the presentations of inborn errors of metabolism are varied and a planned-out, stepwise approach in evaluation is fundamental.
Lactic acid dehydrogenase (LDH) level was 1,457 U/L (reference range, 140-280), and haptoglobin level was less than 6 mg/dL (reference range, 30-200). Peripheral blood smear demonstrated occasional atypical, reactive-appearing lymphocytes with red cell clumping and agglutination, as well as rare target, burr, and fragmented red cells. Test results for urine myoglobin were negative. Results of urine culture were negative. No blood culture was collected.
The elevated LDH, decreased haptoglobin, and findings on the peripheral blood smear confirm hemolysis. The clumping of erythrocytes can be artifactual in the preparation of peripheral smears, but when considered in the context of hemolysis, may be clinically important. Clumping of erythrocytes on the peripheral smear indicates the binding of a protein to antigens on the erythrocyte membrane; when this occurs below body temperature, this is consistent with the presence of a “cold agglutinin,” usually an IgM antibody directed at erythrocyte surface antigens that causes agglutination and destruction, especially in cooler areas of the body. This is a well-known complication of Mycoplasma pneumoniae infections as well as Epstein-Barr virus (EBV) infections; it may also occur with lymphoid malignancies or autoimmune disease.
Direct Coombs IgG test findings were negative, direct Coombs C3 test was positive, and direct Coombs polyspecific test was positive. M pneumoniae IgG antibody level was 1.4 mg/dL (reference ranges: <0.9, negative; 0.91-1.09, equivocal; >1.1, positive); M pneumoniae IgM level was 529 U/mL (reference range: <770, negative). EBV capsid IgM and IgG levels were undetectable. EBV nuclear antigen IgG level was also undetectable. EBV viral load was fewer than 10 copies/mL. Antinuclear antibodies (ANA) level was negative. General IgE and IgM levels were normal, at 11 and 81 mg/dL, respectively. Repeat complete blood count showed WBC of 7,800/µL, hemoglobin of 8.7 g/dL, and platelet count of 341,000/µL. The patient’s hemoglobin remained stable during hospitalization.
This directed testing is helpful in further classifying the patient’s hemolytic anemia. Autoimmune hemolytic anemias are classified into warm antibody–mediated, cold antibody–mediated, and mixed-type forms; drug-induced and alloimmune hemolytic anemias also occur. In addition, both systemic lupus erythematosus and antiphospholipid antibody syndrome can have hemolytic anemia with variable Coombs testing results; neither fit well in this case. The absence of red blood cell–directed IgG antibodies substantially decreases the likelihood of warm antibody–mediated hemolytic anemia. In cold antibody–mediated hemolytic anemia, antibodies bind to the erythrocyte membrane and then adhere to complement C3, which leads to both intravascular and extravascular hemolysis. Important types of cold antibody–mediated hemolytic anemia in children are primary and secondary cold agglutinin disease, along with paroxysmal cold hemoglobinuria. The Donath-Landsteiner test can be helpful in differentiating these conditions. Antibodies to Mycoplasma may be delayed in response to acute infection, and a child who is reinfected may only produce IgG antibodies. Given the patient’s clinical stability and previous health, the most likely diagnosis is Mycoplasma-induced cold antibody–mediated hemolytic anemia. It may be helpful to check convalescent titers to Mycoplasma in 2 to 4 weeks.
Donath-Landsteiner (D-L) antibody test results were positive. Medication-derived hemolytic anemia testing was conducted, but the presence of positive D-L antibody makes the test results inconclusive. This ultimately led to a diagnosis of paroxysmal cold hemoglobinuria (PCH), presumably triggered by a viral syndrome. Convalescent titers to Mycoplasma were not checked given clinical improvement. Because the patient’s hemoglobin was stable during hospitalization, he was not treated with steroids. His parents were counseled on avoiding cold temperatures for several days. Within 1 month, his hemoglobin had recovered without further evidence of hemolysis.
DISCUSSION
Hemolytic anemia refers to the accelerated destruction of red blood cells and can be further classified as acquired or hereditary.1 Hereditary conditions causing hemolytic anemia include enzymopathies (eg, glucose-6-phosphate dehydrogenase deficiency), hemoglobinopathies (eg, sickle cell disease), and membrane abnormalities (eg, hereditary spherocytosis). Acquired pathologies include microangiopathic hemolytic anemia (MAHA), anemias directly caused by certain infections such as malaria, and immune-mediated (Coombs-positive) hemolytic anemias.
MAHA can sometimes be life-threatening and is therefore important to identify quickly. In the right clinical context, such processes may be rapidly recognized by the presence of schistocytes on blood smear in addition to an elevated serum LDH level. Schistocytes suggest mechanical destruction of erythrocytes in the vasculature, the hallmark of MAHA. Important MAHAs include thrombocytopenic purpura, hemolytic-uremic syndrome, and disseminated intravascular coagulation. Though this patient did have a mildly elevated LDH, MAHA was less likely because there were no schistocytes on the blood smear.
Autoimmune hemolytic anemias (AIHAs) are another important subset of acquired hemolytic anemias. AIHAs occur when there is antibody-mediated destruction of erythrocytes. The direct Coombs test evaluates for antibody- or complement-coated erythrocytes. After administration of anti-IgG and anti-C3 serum, the test evaluates for agglutination of the red cells caused by attached antibodies or complement. Coombs-positive AIHA can also be categorized by the temperature of agglutination. “Warm” hemolysis often involves IgG autoantibodies (ie, warm agglutinins), while “cold” antibodies, usually IgM autoantibodies, bind at colder temperatures (0-4 °C) and activate complements, including C3. In this patient, the Coombs C3 was positive while the Coombs IgG was negative, which is more suggestive of a cold complement–mediated pathway.
Cold AIHA can be further categorized into primary cold agglutinin disease, secondary cold agglutinin disease, and PCH. Primary cold agglutinin disease is an autoimmune disorder that mostly occurs in adults. Secondary cold AIHA can often be triggered by bacterial infection (commonly M pneumoniae) or viruses including EBV, measles, and mumps.2 Medications, including penicillin and cephalosporins, can also be implicated. Secondary cold AIHA is also linked with autoimmune diseases, such as systemic lupus erythematosus and lymphoproliferative disorders. PCH can be identified with the unique presence of a specific autoantibody (ie, D-L autoantibody) that agglutinates at cold temperatures but dissociates on subsequent rewarming.3 Complement remains affixed and activates hemolysis.
The D-L antibody responsible for PCH is an IgG antibody to the P-antigen present on the erythrocyte surface. Since the Coombs test is conducted at normal temperature, it will be positive for the affixed complement but not for IgG. The underlying mechanism for PCH was proposed by Julius Donath, MD, and Karl Landsteiner, MD, in 1904 and is considered to be the first description of autoimmune disease being precipitated by antibodies.4 The D-L antibody test itself is uncommonly performed and somewhat difficult to interpret, particularly in adults, and may lead to false-negative results.5
PCH is an acquired, cold AIHA more common to children6,7 and may account for up to 33% of pediatric AIHA cases.8 Typical presentation is after an upper respiratory tract illness; however, the trigger is often not identified. Implicated triggers include a number of viruses.9 Clinical presentation includes findings of intravascular hemolysis similar to those in our patient. The pathogenic IgG autoantibody is polyclonal and is likely formed because of immune stimulation, which is consistent with the predominance of nonmalignant triggers of this disease process.10 Hemolysis and associated symptoms are often exacerbated with cold exposure; both typically resolve within 2 weeks. In recurrent cases, which are a minority, immunosuppression may be considered.10
PCH remains an often-understated cause of hemolytic anemia particularly in children. Lacking obvious pathognomonic clinical symptoms, it may be overlooked for other forms of AIHA or MAHA. However, with a structured approach to evaluation, as with this patient who had hematuria and jaundice, early diagnosis can prevent an unnecessarily extensive workup and can provide reassurance to patient and parents. By understanding the basic categories of hemolytic anemia, the relevant blood testing available, and interpretation of Coombs test results, clinicians can ensure that PCH is a diagnosis that is not left out in the cold.
KEY TEACHING POINTS
- Examination for schistocytes on a blood smear can help identify life-threatening causes of hemolytic anemia.
- Characterization of cold AIHA includes defining the underlying etiology as primary cold agglutinin disease, secondary cold agglutinin disease, or PCH.
- PCH is a cold AIHA that is an underrecognized cause of hemolytic anemia in children. The diagnosis of PCH is made by testing for the presence of the D-L antibody.
1. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
2. Djaldetti M. Paroxysmal cold hemoglobinuria. CRC Crit Rev Clin Lab Sci. 1978;9(1):49-83. https://doi.org/10.3109/10408367809150915
3. Levine P, Celano MJ, Falkowski F. The specificity of the antibody in paroxysmal cold hemoglobinuria (P.C.H.). Transfusion. 1963;3(4):278-280. https://doi.org/10.1111/j.1537-2995.1963.tb04643.x
4. Donath J, Landsteiner K. Uber Paroxysmale Hamoglobinurie. Munch Med Wochenschr. 1904;51:1590-1593
5. Zeller MP, Arnold DM, Al Habsi K, et al. Paroxysmal cold hemoglobinuria: a difficult diagnosis in adult patients. Transfusion. 2017;57(1):137-143. https://doi.org/10.1111/trf.13888
6. Göttsche B, Salama A, Mueller-Eckhardt C. Donath-Landsteiner autoimmune hemolytic anemia in children. a study of 22 cases. Vox Sang. 1990;58(4):281-286. https://doi.org/10.1111/j.1423-0410.1990.tb05000.x
7. Sokol RJ, Booker DJ, Stamps R. Erythropoiesis: paroxysmal cold haemoglobinuria: a clinico-pathological study of patients with a positive Donath-Landsteiner test. Hematology. 1999;4(2):137-164. https://doi.org/10.1080/10245332.1999.11746439
8. Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008;22(1):1-15. https://doi.org/10.1016/j.blre.2007.08.002
9. Leibrandt R, Angelino K, Vizel-Schwartz M, Shapira I. Paroxysmal cold hemoglobinuria in an adult with respiratory syncytial virus. Case Rep Hematol. 2018;2018:1-3. https://doi.org/10.1155/2018/7586719
10. Gertz MA. Management of cold haemolytic syndrome. Br J Haematol. 2007;138(4):422-429. https://doi.org/10.1111/j.1365-2141.2007.06664.x
1. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
2. Djaldetti M. Paroxysmal cold hemoglobinuria. CRC Crit Rev Clin Lab Sci. 1978;9(1):49-83. https://doi.org/10.3109/10408367809150915
3. Levine P, Celano MJ, Falkowski F. The specificity of the antibody in paroxysmal cold hemoglobinuria (P.C.H.). Transfusion. 1963;3(4):278-280. https://doi.org/10.1111/j.1537-2995.1963.tb04643.x
4. Donath J, Landsteiner K. Uber Paroxysmale Hamoglobinurie. Munch Med Wochenschr. 1904;51:1590-1593
5. Zeller MP, Arnold DM, Al Habsi K, et al. Paroxysmal cold hemoglobinuria: a difficult diagnosis in adult patients. Transfusion. 2017;57(1):137-143. https://doi.org/10.1111/trf.13888
6. Göttsche B, Salama A, Mueller-Eckhardt C. Donath-Landsteiner autoimmune hemolytic anemia in children. a study of 22 cases. Vox Sang. 1990;58(4):281-286. https://doi.org/10.1111/j.1423-0410.1990.tb05000.x
7. Sokol RJ, Booker DJ, Stamps R. Erythropoiesis: paroxysmal cold haemoglobinuria: a clinico-pathological study of patients with a positive Donath-Landsteiner test. Hematology. 1999;4(2):137-164. https://doi.org/10.1080/10245332.1999.11746439
8. Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008;22(1):1-15. https://doi.org/10.1016/j.blre.2007.08.002
9. Leibrandt R, Angelino K, Vizel-Schwartz M, Shapira I. Paroxysmal cold hemoglobinuria in an adult with respiratory syncytial virus. Case Rep Hematol. 2018;2018:1-3. https://doi.org/10.1155/2018/7586719
10. Gertz MA. Management of cold haemolytic syndrome. Br J Haematol. 2007;138(4):422-429. https://doi.org/10.1111/j.1365-2141.2007.06664.x
© 2020 Society of Hospital Medicine
A Fiery Pivot
A 62-year-old man with metastatic non–small cell lung cancer (NSCLC) presented to the Emergency Department with 3 days of progressive generalized weakness, anorexia, and nonbloody diarrhea. He denied fever, chills, nausea, vomiting, cough, shortness of breath, or abdominal pain. He had no sick contacts.
One diagnostic approach for patients with cancer who present with new symptoms is to consider diagnoses both related and unrelated to the cancer. Cancer-related diagnoses can include the broad categories of complications related to the tumor itself (such as mass effect), paraneoplastic phenomena, or treatment-related complications (such as infection from immunosuppression or chemotherapy toxicity).
For this patient with metastatic NSCLC, weakness, anorexia, and diarrhea are unlikely to be related to mass effect unless the patient has peritoneal metastases (an uncommon complication of NSCLC) with carcinomatosis-associated diarrhea.
Paraneoplastic phenomena, such as hypercalcemia or hyponatremia from the syndrome of inappropriate antidiuretic hormone (SIADH), are common with NSCLC and could both lead to weakness and anorexia. Hematologic consequences of NSCLC (or its treatment) include anemia, thrombosis, and thrombotic microangiopathy (TMA), though diarrhea, in the absence of abdominal pain or hematochezia, would be unexpected.
Weakness, anorexia, and diarrhea may also be symptoms of chemotherapy toxicity or an infection resulting from immunosuppression. It would be important to know what specific treatment the patient has received. Chemotherapy commonly causes neutropenia and predisposes to rapidly progressive infections, while immunotherapies have other toxicities. Diarrhea is a common toxicity of the checkpoint inhibitors and anaplastic lymphoma kinase (ALK) inhibitors that are frequently used to treat metastatic NSCLC. Checkpoint inhibitors also are known to cause a wide range of autoimmune phenomena including colitis.
Finally, the patient’s symptoms may be unrelated to the cancer. Weakness, anorexia, and nonbloody diarrhea could be signs of viral or bacterial gastroenteritis or Clostridioides difficile colitis particularly with frequent healthcare contact or antimicrobial use.
Two days prior, he had been diagnosed with nonsevere Clostridioides difficile colitis in an acute care clinic. He was started on oral metronidazole, but his diarrhea worsened over the next day and was accompanied by weakness and anorexia. Additional past medical history included untreated hepatitis C infection, chronic kidney disease stage 3, seizure disorder, and left lung NSCLC (adenocarcinoma). The lung cancer was diagnosed 8 months prior when he had presented with hemoptysis and 3 months of progressive constitutional symptoms. Imaging at that time revealed metastases to the contralateral lung and regional lymph nodes, as well as vertebrae, ribs, and pelvis. He had no abdominal metastases. He was initially treated with carboplatin and paclitaxel. After a partial response to initial chemotherapy, he developed peripheral neuropathy and was switched to gemcitabine 12 weeks ago. He received five cycles of gemcitabine over 10 weeks. He was last administered gemcitabine 2 weeks prior. He had not received any additional chemotherapy or immunotherapy. He had a 40 pack-year history of smoking, but quit when diagnosed with cancer. He did not drink alcohol. He had no recent travel or sick contacts. He was not on any medications. He was homeless but staying with family members in the area. Additional review of systems was negative for recent bleeding, bruising, hemoptysis, melena, hematochezia, or hematuria.
Recent treatment with gemcitabine could contribute to the presentation in a number of ways. First, gemcitabine is associated with myelosuppression and neutropenia that could predispose him to infectious colitis. Second, gemcitabine is known to cause anemia, anorexia, diarrhea, and fatigue. Third, gemcitabine may also cause renal injury that can contribute to worsening anemia. He may be at greater risk of anemia and renal toxicity because of preexisting chronic kidney disease. Finally, gemcitabine can rarely cause TMA with characteristics that mimic the hemolytic-uremic syndrome with microangiopathic hemolytic anemia, mild thrombocytopenia, and severe acute kidney injury (AKI).

In addition, worsening infectious colitis could certainly explain his presenting symptoms. At this point, local mass effect seems unlikely despite his metastatic disease. Lastly, it should be noted that, in an immunosuppressed cancer patient, multiple problems could be present at the same time. Laboratory testing should evaluate for hypercalcemia, SIADH, hematologic indexes, and renal function. If initial laboratory evaluation is unrevealing, abdominal imaging may be needed to assess for carcinomatosis, complications from colitis, typhlitis, abscess, or perforation.

On physical examination, the patient appeared fatigued. His temperature was 36.8°C, blood pressure 158/72 mm Hg, pulse 88 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation was 96% while breathing ambient air. There was neither scleral icterus nor conjunctival injection but he had mild conjunctival pallor. Cardiovascular and lung examinations were normal. Abdominal exam revealed normal bowel sounds without tenderness or organomegaly. He had no supraclavicular, axillary, or inguinal lymphadenopathy. He was alert and oriented. Cranial nerves II through XII were intact. He had decreased muscle bulk in his extremities without focal weakness. Gait and reflexes were not tested.
Initial laboratory testing revealed a white blood cell count of 5.5 K/mm3, hemoglobin of 5 g/dL (hemoglobin 1 month prior was 10.1 g/dL), and platelet count of 20 K/mm3 (platelet count 1 month prior was 246 K/mm3). Creatinine was 3.9 mg/dL (compared with a baseline of 1.8 mg/dL), and blood urea nitrogen was 39 mg/dL. His sodium was 137 mEq/L, potassium 4.2 mEq/L, chloride 105 mEq/L, bicarbonate 22 mEq/L, and thyroid stimulating hormone 0.9 mU/L. His total protein was 4.9 g/dL, albumin 2.1 g/dL, alkaline phosphatase 60 IU/L, alanine aminotransferase 17 IU/L, aspartate aminotransferase 60 IU/L, direct bilirubin 0.2 mg/dL, and total bilirubin 0.5 mg/dL. A chest x-ray showed no infiltrates.
The patient’s laboratory tests reveal several important new findings, including severe acute on chronic anemia, acute thrombocytopenia, and AKI, without clinical evidence of acute blood loss. These changes could be parts of a syndrome or multiple independent disorders. The most urgent priority is to evaluate for TMAs, many of which are fatal if not diagnosed and treated expeditiously. This includes thrombotic thrombocytopenic purpura (TTP), disseminated intravascular hemolysis (DIC), and atypical hemolytic uremic syndrome (aHUS). A manual review of a peripheral blood smear is required to evaluate for fragmented red blood cells (schistocytes). Thereafter, ancillary testing to confirm intravascular hemolysis would include measuring free plasma hemoglobin and lactate dehydrogenase (LDH). Additionally, in intravascular hemolysis, haptoglobin should be depleted and urinalysis should show heme-positive urine without RBCs. In this case the patient’s normal bilirubin studies argue against hemolysis; however, elevated bilirubin is variably present in hemolytic anemias depending on the liver’s ability to conjugate and excrete bilirubin, the relative degree of RBC turnover, and type of hemolysis. Patients with intravascular hemolysis lose hemoglobin directly into the urine leaving relatively little hemoglobin to be incorporated into bile once it has reached the reticuloendothelial system. This results in relatively normal bilirubin levels. More specific indicators of intravascular hemolysis include pink colored plasma on visual inspection (commonly done in the blood bank as part of assessing for hemolytic transfusion reactions), measuring plasma free hemoglobin, or by detecting hemoglobin in the urine.
If microangiopathic hemolytic anemia (MAHA) is excluded, then other causes of these laboratory abnormalities should be considered. Bleeding is the most common cause for anemia, and thrombocytopenia predisposes patients to bleeding. However, there is no evidence of bleeding in this patient, and such a rapid acute anemia is unlikely to be caused by occult blood loss alone. Concurrent anemia and thrombocytopenia could be evidence of bone marrow toxicity from chemotherapy or neoplastic infiltration. With marrow infiltration, there are typically signs on the peripheral smear of leukoerythroblastosis, with circulating nucleated red blood cells and early myeloid forms. Concurrent immune thrombocytopenia (ITP) and autoimmune hemolytic anemia (AIHA), or Evans’ Syndrome, should also be considered. AIHA would be suggested by spherocytes on the peripheral smear, elevated LDH and a positive direct antibody test (DAT).

Regarding the AKI, the patient has diarrhea, which could lead to prerenal azotemia and acute tubular necrosis. A formal urinalysis would evaluate for prerenal and intrinsic kidney disease. TMA can cause intrinsic kidney injury with a benign urinary sediment. The blood urea nitrogen-to-creatinine ratio is not elevated, but in a patient with malnutrition this may not indicate prerenal azotemia. In summary, to differentiate potential TMAs from other causes, the patient needs a blood smear, coagulation studies, and an evaluation for hemolysis, including a urinalysis for free heme and any evidence of intrinsic kidney disease.
Urinalysis showed amber-colored, dilute urine with no white blood cells, red blood cells, protein, or casts. It was positive for blood and negative for bilirubin and hemosiderin. LDH was 1,382 IU/L (reference range 135-225 IU/L), and haptoglobin was unmeasurably low. His ferritin was 2,267 ng/mL, serum iron was 57 mcg/dL, total iron-binding capacity was 241 mcg/dL, and transferrin was 162 mcg/dL. Reticulocyte count was 6% (reticulocyte index of 0.86). Vitamin B12 level was normal. DAT was negative; INR and aPTT were normal. Fibrinogen was 287 mg/dL (reference range 200-400 mg/dL), and D-dimer was 5,095 ng/mL (reference range 0-229 ng/mL).
The urinalysis shows no active sediment to suggest vasculitis or glomerulonephritis. The kidney injury could be the result of renal toxicity from free hemoglobin or as part of TMA caused by microvascular thrombosis. The dilute urine makes prerenal azotemia less likely.
There is clearly acute intravascular hemolysis occurring as evidenced by hemoglobinuria, very high LDH, and undetectable serum haptoglobin. The hemolysis is acute because chronic intravascular hemolysis would lead to positive urine hemosiderin via deposition in the renal tubules. Autoimmune hemolytic anemia is much less likely, but not ruled out, by a negative DAT.
This syndrome can be further refined from acute anemia to acute anemia with likely nonimmune intravascular hemolysis, acute thrombocytopenia, and AKI with hemoglobinuria and a bland urinary sediment. At this point, intravascular hemolysis and kidney injury could be part of a unifying diagnosis. However, this does not account for the patient’s thrombocytopenia, and TMA remains the best explanation for the constellation of findings. Review of the peripheral blood smear is urgent because evidence of MAHA would prompt urgent plasma exchange based on presumptive diagnosis of acquired TTP to later be confirmed with ADAMTS13 activity testing. Most TMAs are treated with supportive care only; TTP and aHUS have specific interventions that change the natural history of the disease (plasma exchange and anticomplement therapy, respectively). Given both the deadly natural history and opportunity to intervene with plasma exchange, patients with TMA should be treated with urgent plasma exchange until ADAMTS13 deficiency is confirmed or refuted. One TMA that can be excluded at this point is DIC. DIC in its acute and chronic forms nearly universally causes MAHA, thrombocytopenia, and consumptive coagulopathy including hypofibrinogenemia.
If MAHA is excluded, then other causes of intravascular hemolysis should be considered, along with causes of thrombocytopenia that might be occurring concurrently. Intravascular hemolysis can be further differentiated by etiologies primarily related to the RBC or whether the RBC is the innocent bystander amidst a systemic illness. RBC disorders include syndromes affecting RBC fragility like hereditary spherocytosis or RBC enzymopathies (G6PD deficiency), but these do not cause thrombocytopenia. One exception is an acquired membrane defect, paroxysmal nocturnal hemoglobinuria (PNH), in which RBCs and other blood cells become susceptible to complement-mediated lysis. Testing for PNH by peripheral blood flow cytometry should be considered if the blood film lacks schistocytes. Systemic disorders that cause intravascular hemolysis include severe burns (heat damage to RBCs), RBC trauma from “march hemoglobinuria” or mechanical heart valves, immune (antibody-mediated) hemolysis from Rh immune globulin administration, cold agglutinin disease or ABO mismatched transfusion, and infections including the intraerythrocyte parasites malaria, Bartonellosis, and Babesiosis, as well as organisms that induce RBC fragility such as Leishmaniasis, Clostridium perfringens, and Haemophilus influenzae B.
On review of additional history, the patient had not recently received blood products. He had received heparin during prior hospitalizations, but had no prior history of thrombosis. He had no history of tick exposure. Peripheral blood smear was obtained and reviewed by a
The blood smear helps narrow the differential further. The lack of schistocytes makes TMA far less likely and so plasma exchange is not urgently indicated. The differential still includes drug-induced TMA (gemcitabine being a well-known cause for TMA) and cancer-associated TMA could still cause these findings, but plasma exchange does not improve outcomes. Acquired (immune) TTP is very unlikely unless the patient did not improve with supportive care or developed neurologic symptoms. Similarly, atypical (complement-driven) HUS would only be considered if renal failure did not improve with supportive care.
The blood smear does show a surprising finding of pyropoikilocytosis. Pyropoikilocytosis refers to changes in RBC shape (poikilocytosis) typically seen with thermal injury or rare RBC membrane structural defects. Hereditary pyropoikilocytosis, a very rare disease, is characterized by chronic hyperproliferative, compensated anemia, and occasional hemolytic crises. These crises are associated with splenomegaly, reticulocytosis, and elevated bilirubin with jaundice. As the patient has no history of similar episodes, the blood smear changes are not due to a hereditary cause and obviously not due to thermal injury (ie, severe burns). Pyropoikilocytosis has been rarely reported in drug-induced TMA and in severe bacterial bloodstream infections (most commonly Gram-negative bacilli). This patient has received gemcitabine (a known cause of drug-induced TMA) and has a recently diagnosed infection (C difficile colitis), either of which could be linked to this rare blood smear finding. Both of these syndromes would be treated with supportive care plus avoidance of future gemcitabine.
Transfusion of packed RBCs is indicated given his profound anemia and symptoms of fatigue. One should obtain further testing for cold agglutinins, PNH, and echocardiography to exclude endocarditis. If he were to become critically ill, anuric, or encephalopathic, then one could consider plasma exchange for treatment of TMA and hemoglobin-mediated AKI. Pyropoikilocytosis should be considered the result of drug-induced TMA, severe C difficile colitis, or an occult infection.
The patient was transfused packed RBCs. Because of a concern for an acute TMA such as TTP, both a hematopathologist and the consulting hematology/oncology team reviewed the peripheral blood morphology emergently. He was given aggressive fluid resuscitation and received 3 L of IV lactated ringers’ solution. An echocardiogram did not show valvular abnormalities. A renal biopsy was contraindicated because of the severe thrombocytopenia.
Given the recently confirmed C difficile colitis along with the findings of pyropoikilocytosis on the peripheral smear, toxin-mediated intravascular hemolysis from systemic C difficile infection became the leading diagnosis. Positing that the C difficile colitis was inadequately treated with oral metronidazole, aggressive treatment for C difficile was initiated with oral vancomycin in addition to intravenous metronidazole. Intravenous metronidazole was included given his elevated creatinine, presence of severe colitis on imaging, and concern he may be at risk for translocation of colonic C difficile or exotoxin into the bloodstream.
Over the course of the next 3 days, the patient’s platelet count normalized and his hemoglobin, creatinine, and symptoms of fatigue improved. Blood cultures remained negative. The patient’s rapid improvement with antibiotics supported our final diagnosis of toxin-mediated hemolysis caused by a systemic C difficile infection. On follow-up testing after hospital discharge, hemoglobin had returned to prior baseline and there was no recurrent hemolysis. Gemcitabine was considered to be a possible cause of his hemolytic anemia and was not continued in further treatment for his NSCLC.
COMMENTARY
When evaluating patients with cancer who present with fatigue, hospitalists should consider a broad list of potential causes. The differential should include etiologies directly related to the malignancy, paraneoplastic phenomena, treatment-related complications, and diseases unrelated to cancer. In addition, as the number of medications used for cancer proliferates, hospitalists must take a detailed history of the agents used and be aware of major side effects. Using this information, hospitalists may undertake a targeted approach to diagnostics while searching for a cause of fatigue.
When lab testing reveals profound anemia, hospitalists must consider syndromes that may require emergent management. Anemia can be caused by decreased RBC production, and acute anemia in the absence of clear blood loss suggests hemolysis. Moreover, the combination of elevated LDH and low haptoglobin is quite specific of hemolytic anemia.1,2 Once hemolytic anemia is identified, DIC and TMA syndromes (such as TTP) need to be considered. The combination of hemolytic anemia and AKI may indicate a medical emergency and should prompt hospitalists to obtain an urgent peripheral blood smear to help narrow the differential.3
The absence of schistocytes on a blood smear does not rule out TTP or HUS, but does argue strongly against these diagnoses.4,5 Of note, consultation with a hematopathologist and hematology subspecialist should be done to ensure appropriate and timely review of the peripheral blood smear.
In this case, the blood smear led to a very rare finding of pyropoikilocytosis. The unexpected result should prompt a broader review of the medical history particularly as it relates to the patient’s broader symptoms and laboratory abnormalities. Acquired pyropoikilocytosis is a very specific finding known to be associated only with hyperthermal injury (seen in burn patients), drug-induced TMA, and bacterial bloodstream infections, mainly Gram-negative toxins and Clostridioidal infections.6-8 In this case, both drug-induced TMA and C difficile infection were considered.
Gemcitabine-induced TMA can occur with either short or long term use of the medication and can be difficult to distinguish from TTP. While both TTP and gemcitabine-induced TMA can cause thrombocytopenia, hemolytic anemia, and schistocytes on a blood smear, the latter causes acute kidney injury more frequently than TTP. In addition, gemcitabine-induced TMA may not lead to severe decrease in ADAMTS13 activity. A kidney biopsy could confirm drug-induced TMA but was contraindicated in this case because of the thrombocytopenia. Gemcitabine should not be restarted if this side effect is suspected.
Given the continued rise in C difficile incidence, hospitalists should be aware that C difficile infection can cause extraintestinal illness.9,10 Although uncommon, these extraintestinal complications are associated with high risk of mortality and frequently occur in those with a history of intestinal injury or inflammation and a concomitant bloodstream infection.10 Regarding the possibility of C difficile contributing to hemolysis in this case, the patient’s low blood counts and hemolysis improved concomitantly with more aggressive treatment of C difficile infection. Although his blood cultures were sterile, C difficile is notoriously difficult to culture. Prior case reports have associated C difficile with intravascular hemolysis, which leads to the possibility that the patient did have a very rare manifestation of this unfortunately common infection.11
This case provides an excellent example of a diagnostic pivot point
KEY TEACHING POINTS
- In evaluating symptomatic cancer patients, providers must consider sequelae of the tumor, paraneoplastic phenomena, and treatment-related complications.
- Hemolytic anemia may represent a life-threatening emergency particularly when accompanied by AKI and requires urgent peripheral blood smear evaluation.
- Acquired pyropoikilocytosis is a specific finding known to be associated only with thermal injury, drug-induced TMA, and bacterial toxin–mediated hemolysis.
Disclosures
The authors have nothing to disclose.
1. Weinzierl EP, Arber DA. The differential diagnosis and bone marrow evaluation of new-onset pancytopenia. Am J Clin Pathol. 2013:139(1):9-29. https://doi.org/10.1309/AJCP50AEEYGREWUZ.
2. Marchand A, Galen RS, Van Lente F. The predictive value of serum haptoglobin in hemolytic disease. JAMA.1980;243(19):1909-1911. https://doi:10.1001/jama.1980.03300450023014.
3. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
4. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836-2846. https://doi.org/10.1182/blood-2016-10-709857.
5. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856. https://doi.org/10.1182/blood-2016-11-709865.
6. Baar S, Arrowsmith DJ. Thermal damage to red cells. J Clin Path. 1970;23(7):572-576. https://doi.org/10.1136/jcp.23.7.572.
7. Meinders AJ, Dijkstra I. Massive hemolysis and erythrophagocytosis in severe sepsis. Blood. 2014;124(6):841. https://doi.org/10.1182/blood-2014-04-565663.
8. McIlwaine K, Leach MT. Clostridium perfringens septicaemia. Br J Haematol. 2013;163(5):549. https://doi.org/10.1111/bjh.12551.
9. Evans CT, Safdar N. Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis. 2015;60 (Supp 2):S66-71. https://doi.org/10.1093/cid/civ140.
10. Gupta A, Patel R, Baddour LM, Pardi DS, Khanna S. Extraintestinal Clostridium difficile infections: a single-center experience. Mayo Clin Proc. 2014;89(11):1525-36. https://doi.org/10.1016/j.mayocp.2014.07.012.
11. Alvarado AS, Brodsky SV, Nadasdy T, Singh N. Hemolytic uremic syndrome associated with Clostridium difficile infection. Clin Nephrol. 2014;81(4):302-6. https://doi.org/10.5414/CN107691.
A 62-year-old man with metastatic non–small cell lung cancer (NSCLC) presented to the Emergency Department with 3 days of progressive generalized weakness, anorexia, and nonbloody diarrhea. He denied fever, chills, nausea, vomiting, cough, shortness of breath, or abdominal pain. He had no sick contacts.
One diagnostic approach for patients with cancer who present with new symptoms is to consider diagnoses both related and unrelated to the cancer. Cancer-related diagnoses can include the broad categories of complications related to the tumor itself (such as mass effect), paraneoplastic phenomena, or treatment-related complications (such as infection from immunosuppression or chemotherapy toxicity).
For this patient with metastatic NSCLC, weakness, anorexia, and diarrhea are unlikely to be related to mass effect unless the patient has peritoneal metastases (an uncommon complication of NSCLC) with carcinomatosis-associated diarrhea.
Paraneoplastic phenomena, such as hypercalcemia or hyponatremia from the syndrome of inappropriate antidiuretic hormone (SIADH), are common with NSCLC and could both lead to weakness and anorexia. Hematologic consequences of NSCLC (or its treatment) include anemia, thrombosis, and thrombotic microangiopathy (TMA), though diarrhea, in the absence of abdominal pain or hematochezia, would be unexpected.
Weakness, anorexia, and diarrhea may also be symptoms of chemotherapy toxicity or an infection resulting from immunosuppression. It would be important to know what specific treatment the patient has received. Chemotherapy commonly causes neutropenia and predisposes to rapidly progressive infections, while immunotherapies have other toxicities. Diarrhea is a common toxicity of the checkpoint inhibitors and anaplastic lymphoma kinase (ALK) inhibitors that are frequently used to treat metastatic NSCLC. Checkpoint inhibitors also are known to cause a wide range of autoimmune phenomena including colitis.
Finally, the patient’s symptoms may be unrelated to the cancer. Weakness, anorexia, and nonbloody diarrhea could be signs of viral or bacterial gastroenteritis or Clostridioides difficile colitis particularly with frequent healthcare contact or antimicrobial use.
Two days prior, he had been diagnosed with nonsevere Clostridioides difficile colitis in an acute care clinic. He was started on oral metronidazole, but his diarrhea worsened over the next day and was accompanied by weakness and anorexia. Additional past medical history included untreated hepatitis C infection, chronic kidney disease stage 3, seizure disorder, and left lung NSCLC (adenocarcinoma). The lung cancer was diagnosed 8 months prior when he had presented with hemoptysis and 3 months of progressive constitutional symptoms. Imaging at that time revealed metastases to the contralateral lung and regional lymph nodes, as well as vertebrae, ribs, and pelvis. He had no abdominal metastases. He was initially treated with carboplatin and paclitaxel. After a partial response to initial chemotherapy, he developed peripheral neuropathy and was switched to gemcitabine 12 weeks ago. He received five cycles of gemcitabine over 10 weeks. He was last administered gemcitabine 2 weeks prior. He had not received any additional chemotherapy or immunotherapy. He had a 40 pack-year history of smoking, but quit when diagnosed with cancer. He did not drink alcohol. He had no recent travel or sick contacts. He was not on any medications. He was homeless but staying with family members in the area. Additional review of systems was negative for recent bleeding, bruising, hemoptysis, melena, hematochezia, or hematuria.
Recent treatment with gemcitabine could contribute to the presentation in a number of ways. First, gemcitabine is associated with myelosuppression and neutropenia that could predispose him to infectious colitis. Second, gemcitabine is known to cause anemia, anorexia, diarrhea, and fatigue. Third, gemcitabine may also cause renal injury that can contribute to worsening anemia. He may be at greater risk of anemia and renal toxicity because of preexisting chronic kidney disease. Finally, gemcitabine can rarely cause TMA with characteristics that mimic the hemolytic-uremic syndrome with microangiopathic hemolytic anemia, mild thrombocytopenia, and severe acute kidney injury (AKI).
In addition, worsening infectious colitis could certainly explain his presenting symptoms. At this point, local mass effect seems unlikely despite his metastatic disease. Lastly, it should be noted that, in an immunosuppressed cancer patient, multiple problems could be present at the same time. Laboratory testing should evaluate for hypercalcemia, SIADH, hematologic indexes, and renal function. If initial laboratory evaluation is unrevealing, abdominal imaging may be needed to assess for carcinomatosis, complications from colitis, typhlitis, abscess, or perforation.
On physical examination, the patient appeared fatigued. His temperature was 36.8°C, blood pressure 158/72 mm Hg, pulse 88 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation was 96% while breathing ambient air. There was neither scleral icterus nor conjunctival injection but he had mild conjunctival pallor. Cardiovascular and lung examinations were normal. Abdominal exam revealed normal bowel sounds without tenderness or organomegaly. He had no supraclavicular, axillary, or inguinal lymphadenopathy. He was alert and oriented. Cranial nerves II through XII were intact. He had decreased muscle bulk in his extremities without focal weakness. Gait and reflexes were not tested.
Initial laboratory testing revealed a white blood cell count of 5.5 K/mm3, hemoglobin of 5 g/dL (hemoglobin 1 month prior was 10.1 g/dL), and platelet count of 20 K/mm3 (platelet count 1 month prior was 246 K/mm3). Creatinine was 3.9 mg/dL (compared with a baseline of 1.8 mg/dL), and blood urea nitrogen was 39 mg/dL. His sodium was 137 mEq/L, potassium 4.2 mEq/L, chloride 105 mEq/L, bicarbonate 22 mEq/L, and thyroid stimulating hormone 0.9 mU/L. His total protein was 4.9 g/dL, albumin 2.1 g/dL, alkaline phosphatase 60 IU/L, alanine aminotransferase 17 IU/L, aspartate aminotransferase 60 IU/L, direct bilirubin 0.2 mg/dL, and total bilirubin 0.5 mg/dL. A chest x-ray showed no infiltrates.
The patient’s laboratory tests reveal several important new findings, including severe acute on chronic anemia, acute thrombocytopenia, and AKI, without clinical evidence of acute blood loss. These changes could be parts of a syndrome or multiple independent disorders. The most urgent priority is to evaluate for TMAs, many of which are fatal if not diagnosed and treated expeditiously. This includes thrombotic thrombocytopenic purpura (TTP), disseminated intravascular hemolysis (DIC), and atypical hemolytic uremic syndrome (aHUS). A manual review of a peripheral blood smear is required to evaluate for fragmented red blood cells (schistocytes). Thereafter, ancillary testing to confirm intravascular hemolysis would include measuring free plasma hemoglobin and lactate dehydrogenase (LDH). Additionally, in intravascular hemolysis, haptoglobin should be depleted and urinalysis should show heme-positive urine without RBCs. In this case the patient’s normal bilirubin studies argue against hemolysis; however, elevated bilirubin is variably present in hemolytic anemias depending on the liver’s ability to conjugate and excrete bilirubin, the relative degree of RBC turnover, and type of hemolysis. Patients with intravascular hemolysis lose hemoglobin directly into the urine leaving relatively little hemoglobin to be incorporated into bile once it has reached the reticuloendothelial system. This results in relatively normal bilirubin levels. More specific indicators of intravascular hemolysis include pink colored plasma on visual inspection (commonly done in the blood bank as part of assessing for hemolytic transfusion reactions), measuring plasma free hemoglobin, or by detecting hemoglobin in the urine.
If microangiopathic hemolytic anemia (MAHA) is excluded, then other causes of these laboratory abnormalities should be considered. Bleeding is the most common cause for anemia, and thrombocytopenia predisposes patients to bleeding. However, there is no evidence of bleeding in this patient, and such a rapid acute anemia is unlikely to be caused by occult blood loss alone. Concurrent anemia and thrombocytopenia could be evidence of bone marrow toxicity from chemotherapy or neoplastic infiltration. With marrow infiltration, there are typically signs on the peripheral smear of leukoerythroblastosis, with circulating nucleated red blood cells and early myeloid forms. Concurrent immune thrombocytopenia (ITP) and autoimmune hemolytic anemia (AIHA), or Evans’ Syndrome, should also be considered. AIHA would be suggested by spherocytes on the peripheral smear, elevated LDH and a positive direct antibody test (DAT).
Regarding the AKI, the patient has diarrhea, which could lead to prerenal azotemia and acute tubular necrosis. A formal urinalysis would evaluate for prerenal and intrinsic kidney disease. TMA can cause intrinsic kidney injury with a benign urinary sediment. The blood urea nitrogen-to-creatinine ratio is not elevated, but in a patient with malnutrition this may not indicate prerenal azotemia. In summary, to differentiate potential TMAs from other causes, the patient needs a blood smear, coagulation studies, and an evaluation for hemolysis, including a urinalysis for free heme and any evidence of intrinsic kidney disease.
Urinalysis showed amber-colored, dilute urine with no white blood cells, red blood cells, protein, or casts. It was positive for blood and negative for bilirubin and hemosiderin. LDH was 1,382 IU/L (reference range 135-225 IU/L), and haptoglobin was unmeasurably low. His ferritin was 2,267 ng/mL, serum iron was 57 mcg/dL, total iron-binding capacity was 241 mcg/dL, and transferrin was 162 mcg/dL. Reticulocyte count was 6% (reticulocyte index of 0.86). Vitamin B12 level was normal. DAT was negative; INR and aPTT were normal. Fibrinogen was 287 mg/dL (reference range 200-400 mg/dL), and D-dimer was 5,095 ng/mL (reference range 0-229 ng/mL).
The urinalysis shows no active sediment to suggest vasculitis or glomerulonephritis. The kidney injury could be the result of renal toxicity from free hemoglobin or as part of TMA caused by microvascular thrombosis. The dilute urine makes prerenal azotemia less likely.
There is clearly acute intravascular hemolysis occurring as evidenced by hemoglobinuria, very high LDH, and undetectable serum haptoglobin. The hemolysis is acute because chronic intravascular hemolysis would lead to positive urine hemosiderin via deposition in the renal tubules. Autoimmune hemolytic anemia is much less likely, but not ruled out, by a negative DAT.
This syndrome can be further refined from acute anemia to acute anemia with likely nonimmune intravascular hemolysis, acute thrombocytopenia, and AKI with hemoglobinuria and a bland urinary sediment. At this point, intravascular hemolysis and kidney injury could be part of a unifying diagnosis. However, this does not account for the patient’s thrombocytopenia, and TMA remains the best explanation for the constellation of findings. Review of the peripheral blood smear is urgent because evidence of MAHA would prompt urgent plasma exchange based on presumptive diagnosis of acquired TTP to later be confirmed with ADAMTS13 activity testing. Most TMAs are treated with supportive care only; TTP and aHUS have specific interventions that change the natural history of the disease (plasma exchange and anticomplement therapy, respectively). Given both the deadly natural history and opportunity to intervene with plasma exchange, patients with TMA should be treated with urgent plasma exchange until ADAMTS13 deficiency is confirmed or refuted. One TMA that can be excluded at this point is DIC. DIC in its acute and chronic forms nearly universally causes MAHA, thrombocytopenia, and consumptive coagulopathy including hypofibrinogenemia.
If MAHA is excluded, then other causes of intravascular hemolysis should be considered, along with causes of thrombocytopenia that might be occurring concurrently. Intravascular hemolysis can be further differentiated by etiologies primarily related to the RBC or whether the RBC is the innocent bystander amidst a systemic illness. RBC disorders include syndromes affecting RBC fragility like hereditary spherocytosis or RBC enzymopathies (G6PD deficiency), but these do not cause thrombocytopenia. One exception is an acquired membrane defect, paroxysmal nocturnal hemoglobinuria (PNH), in which RBCs and other blood cells become susceptible to complement-mediated lysis. Testing for PNH by peripheral blood flow cytometry should be considered if the blood film lacks schistocytes. Systemic disorders that cause intravascular hemolysis include severe burns (heat damage to RBCs), RBC trauma from “march hemoglobinuria” or mechanical heart valves, immune (antibody-mediated) hemolysis from Rh immune globulin administration, cold agglutinin disease or ABO mismatched transfusion, and infections including the intraerythrocyte parasites malaria, Bartonellosis, and Babesiosis, as well as organisms that induce RBC fragility such as Leishmaniasis, Clostridium perfringens, and Haemophilus influenzae B.
On review of additional history, the patient had not recently received blood products. He had received heparin during prior hospitalizations, but had no prior history of thrombosis. He had no history of tick exposure. Peripheral blood smear was obtained and reviewed by a
The blood smear helps narrow the differential further. The lack of schistocytes makes TMA far less likely and so plasma exchange is not urgently indicated. The differential still includes drug-induced TMA (gemcitabine being a well-known cause for TMA) and cancer-associated TMA could still cause these findings, but plasma exchange does not improve outcomes. Acquired (immune) TTP is very unlikely unless the patient did not improve with supportive care or developed neurologic symptoms. Similarly, atypical (complement-driven) HUS would only be considered if renal failure did not improve with supportive care.
The blood smear does show a surprising finding of pyropoikilocytosis. Pyropoikilocytosis refers to changes in RBC shape (poikilocytosis) typically seen with thermal injury or rare RBC membrane structural defects. Hereditary pyropoikilocytosis, a very rare disease, is characterized by chronic hyperproliferative, compensated anemia, and occasional hemolytic crises. These crises are associated with splenomegaly, reticulocytosis, and elevated bilirubin with jaundice. As the patient has no history of similar episodes, the blood smear changes are not due to a hereditary cause and obviously not due to thermal injury (ie, severe burns). Pyropoikilocytosis has been rarely reported in drug-induced TMA and in severe bacterial bloodstream infections (most commonly Gram-negative bacilli). This patient has received gemcitabine (a known cause of drug-induced TMA) and has a recently diagnosed infection (C difficile colitis), either of which could be linked to this rare blood smear finding. Both of these syndromes would be treated with supportive care plus avoidance of future gemcitabine.
Transfusion of packed RBCs is indicated given his profound anemia and symptoms of fatigue. One should obtain further testing for cold agglutinins, PNH, and echocardiography to exclude endocarditis. If he were to become critically ill, anuric, or encephalopathic, then one could consider plasma exchange for treatment of TMA and hemoglobin-mediated AKI. Pyropoikilocytosis should be considered the result of drug-induced TMA, severe C difficile colitis, or an occult infection.
The patient was transfused packed RBCs. Because of a concern for an acute TMA such as TTP, both a hematopathologist and the consulting hematology/oncology team reviewed the peripheral blood morphology emergently. He was given aggressive fluid resuscitation and received 3 L of IV lactated ringers’ solution. An echocardiogram did not show valvular abnormalities. A renal biopsy was contraindicated because of the severe thrombocytopenia.
Given the recently confirmed C difficile colitis along with the findings of pyropoikilocytosis on the peripheral smear, toxin-mediated intravascular hemolysis from systemic C difficile infection became the leading diagnosis. Positing that the C difficile colitis was inadequately treated with oral metronidazole, aggressive treatment for C difficile was initiated with oral vancomycin in addition to intravenous metronidazole. Intravenous metronidazole was included given his elevated creatinine, presence of severe colitis on imaging, and concern he may be at risk for translocation of colonic C difficile or exotoxin into the bloodstream.
Over the course of the next 3 days, the patient’s platelet count normalized and his hemoglobin, creatinine, and symptoms of fatigue improved. Blood cultures remained negative. The patient’s rapid improvement with antibiotics supported our final diagnosis of toxin-mediated hemolysis caused by a systemic C difficile infection. On follow-up testing after hospital discharge, hemoglobin had returned to prior baseline and there was no recurrent hemolysis. Gemcitabine was considered to be a possible cause of his hemolytic anemia and was not continued in further treatment for his NSCLC.
COMMENTARY
When evaluating patients with cancer who present with fatigue, hospitalists should consider a broad list of potential causes. The differential should include etiologies directly related to the malignancy, paraneoplastic phenomena, treatment-related complications, and diseases unrelated to cancer. In addition, as the number of medications used for cancer proliferates, hospitalists must take a detailed history of the agents used and be aware of major side effects. Using this information, hospitalists may undertake a targeted approach to diagnostics while searching for a cause of fatigue.
When lab testing reveals profound anemia, hospitalists must consider syndromes that may require emergent management. Anemia can be caused by decreased RBC production, and acute anemia in the absence of clear blood loss suggests hemolysis. Moreover, the combination of elevated LDH and low haptoglobin is quite specific of hemolytic anemia.1,2 Once hemolytic anemia is identified, DIC and TMA syndromes (such as TTP) need to be considered. The combination of hemolytic anemia and AKI may indicate a medical emergency and should prompt hospitalists to obtain an urgent peripheral blood smear to help narrow the differential.3
The absence of schistocytes on a blood smear does not rule out TTP or HUS, but does argue strongly against these diagnoses.4,5 Of note, consultation with a hematopathologist and hematology subspecialist should be done to ensure appropriate and timely review of the peripheral blood smear.
In this case, the blood smear led to a very rare finding of pyropoikilocytosis. The unexpected result should prompt a broader review of the medical history particularly as it relates to the patient’s broader symptoms and laboratory abnormalities. Acquired pyropoikilocytosis is a very specific finding known to be associated only with hyperthermal injury (seen in burn patients), drug-induced TMA, and bacterial bloodstream infections, mainly Gram-negative toxins and Clostridioidal infections.6-8 In this case, both drug-induced TMA and C difficile infection were considered.
Gemcitabine-induced TMA can occur with either short or long term use of the medication and can be difficult to distinguish from TTP. While both TTP and gemcitabine-induced TMA can cause thrombocytopenia, hemolytic anemia, and schistocytes on a blood smear, the latter causes acute kidney injury more frequently than TTP. In addition, gemcitabine-induced TMA may not lead to severe decrease in ADAMTS13 activity. A kidney biopsy could confirm drug-induced TMA but was contraindicated in this case because of the thrombocytopenia. Gemcitabine should not be restarted if this side effect is suspected.
Given the continued rise in C difficile incidence, hospitalists should be aware that C difficile infection can cause extraintestinal illness.9,10 Although uncommon, these extraintestinal complications are associated with high risk of mortality and frequently occur in those with a history of intestinal injury or inflammation and a concomitant bloodstream infection.10 Regarding the possibility of C difficile contributing to hemolysis in this case, the patient’s low blood counts and hemolysis improved concomitantly with more aggressive treatment of C difficile infection. Although his blood cultures were sterile, C difficile is notoriously difficult to culture. Prior case reports have associated C difficile with intravascular hemolysis, which leads to the possibility that the patient did have a very rare manifestation of this unfortunately common infection.11
This case provides an excellent example of a diagnostic pivot point
KEY TEACHING POINTS
- In evaluating symptomatic cancer patients, providers must consider sequelae of the tumor, paraneoplastic phenomena, and treatment-related complications.
- Hemolytic anemia may represent a life-threatening emergency particularly when accompanied by AKI and requires urgent peripheral blood smear evaluation.
- Acquired pyropoikilocytosis is a specific finding known to be associated only with thermal injury, drug-induced TMA, and bacterial toxin–mediated hemolysis.
Disclosures
The authors have nothing to disclose.
A 62-year-old man with metastatic non–small cell lung cancer (NSCLC) presented to the Emergency Department with 3 days of progressive generalized weakness, anorexia, and nonbloody diarrhea. He denied fever, chills, nausea, vomiting, cough, shortness of breath, or abdominal pain. He had no sick contacts.
One diagnostic approach for patients with cancer who present with new symptoms is to consider diagnoses both related and unrelated to the cancer. Cancer-related diagnoses can include the broad categories of complications related to the tumor itself (such as mass effect), paraneoplastic phenomena, or treatment-related complications (such as infection from immunosuppression or chemotherapy toxicity).
For this patient with metastatic NSCLC, weakness, anorexia, and diarrhea are unlikely to be related to mass effect unless the patient has peritoneal metastases (an uncommon complication of NSCLC) with carcinomatosis-associated diarrhea.
Paraneoplastic phenomena, such as hypercalcemia or hyponatremia from the syndrome of inappropriate antidiuretic hormone (SIADH), are common with NSCLC and could both lead to weakness and anorexia. Hematologic consequences of NSCLC (or its treatment) include anemia, thrombosis, and thrombotic microangiopathy (TMA), though diarrhea, in the absence of abdominal pain or hematochezia, would be unexpected.
Weakness, anorexia, and diarrhea may also be symptoms of chemotherapy toxicity or an infection resulting from immunosuppression. It would be important to know what specific treatment the patient has received. Chemotherapy commonly causes neutropenia and predisposes to rapidly progressive infections, while immunotherapies have other toxicities. Diarrhea is a common toxicity of the checkpoint inhibitors and anaplastic lymphoma kinase (ALK) inhibitors that are frequently used to treat metastatic NSCLC. Checkpoint inhibitors also are known to cause a wide range of autoimmune phenomena including colitis.
Finally, the patient’s symptoms may be unrelated to the cancer. Weakness, anorexia, and nonbloody diarrhea could be signs of viral or bacterial gastroenteritis or Clostridioides difficile colitis particularly with frequent healthcare contact or antimicrobial use.
Two days prior, he had been diagnosed with nonsevere Clostridioides difficile colitis in an acute care clinic. He was started on oral metronidazole, but his diarrhea worsened over the next day and was accompanied by weakness and anorexia. Additional past medical history included untreated hepatitis C infection, chronic kidney disease stage 3, seizure disorder, and left lung NSCLC (adenocarcinoma). The lung cancer was diagnosed 8 months prior when he had presented with hemoptysis and 3 months of progressive constitutional symptoms. Imaging at that time revealed metastases to the contralateral lung and regional lymph nodes, as well as vertebrae, ribs, and pelvis. He had no abdominal metastases. He was initially treated with carboplatin and paclitaxel. After a partial response to initial chemotherapy, he developed peripheral neuropathy and was switched to gemcitabine 12 weeks ago. He received five cycles of gemcitabine over 10 weeks. He was last administered gemcitabine 2 weeks prior. He had not received any additional chemotherapy or immunotherapy. He had a 40 pack-year history of smoking, but quit when diagnosed with cancer. He did not drink alcohol. He had no recent travel or sick contacts. He was not on any medications. He was homeless but staying with family members in the area. Additional review of systems was negative for recent bleeding, bruising, hemoptysis, melena, hematochezia, or hematuria.
Recent treatment with gemcitabine could contribute to the presentation in a number of ways. First, gemcitabine is associated with myelosuppression and neutropenia that could predispose him to infectious colitis. Second, gemcitabine is known to cause anemia, anorexia, diarrhea, and fatigue. Third, gemcitabine may also cause renal injury that can contribute to worsening anemia. He may be at greater risk of anemia and renal toxicity because of preexisting chronic kidney disease. Finally, gemcitabine can rarely cause TMA with characteristics that mimic the hemolytic-uremic syndrome with microangiopathic hemolytic anemia, mild thrombocytopenia, and severe acute kidney injury (AKI).
In addition, worsening infectious colitis could certainly explain his presenting symptoms. At this point, local mass effect seems unlikely despite his metastatic disease. Lastly, it should be noted that, in an immunosuppressed cancer patient, multiple problems could be present at the same time. Laboratory testing should evaluate for hypercalcemia, SIADH, hematologic indexes, and renal function. If initial laboratory evaluation is unrevealing, abdominal imaging may be needed to assess for carcinomatosis, complications from colitis, typhlitis, abscess, or perforation.
On physical examination, the patient appeared fatigued. His temperature was 36.8°C, blood pressure 158/72 mm Hg, pulse 88 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation was 96% while breathing ambient air. There was neither scleral icterus nor conjunctival injection but he had mild conjunctival pallor. Cardiovascular and lung examinations were normal. Abdominal exam revealed normal bowel sounds without tenderness or organomegaly. He had no supraclavicular, axillary, or inguinal lymphadenopathy. He was alert and oriented. Cranial nerves II through XII were intact. He had decreased muscle bulk in his extremities without focal weakness. Gait and reflexes were not tested.
Initial laboratory testing revealed a white blood cell count of 5.5 K/mm3, hemoglobin of 5 g/dL (hemoglobin 1 month prior was 10.1 g/dL), and platelet count of 20 K/mm3 (platelet count 1 month prior was 246 K/mm3). Creatinine was 3.9 mg/dL (compared with a baseline of 1.8 mg/dL), and blood urea nitrogen was 39 mg/dL. His sodium was 137 mEq/L, potassium 4.2 mEq/L, chloride 105 mEq/L, bicarbonate 22 mEq/L, and thyroid stimulating hormone 0.9 mU/L. His total protein was 4.9 g/dL, albumin 2.1 g/dL, alkaline phosphatase 60 IU/L, alanine aminotransferase 17 IU/L, aspartate aminotransferase 60 IU/L, direct bilirubin 0.2 mg/dL, and total bilirubin 0.5 mg/dL. A chest x-ray showed no infiltrates.
The patient’s laboratory tests reveal several important new findings, including severe acute on chronic anemia, acute thrombocytopenia, and AKI, without clinical evidence of acute blood loss. These changes could be parts of a syndrome or multiple independent disorders. The most urgent priority is to evaluate for TMAs, many of which are fatal if not diagnosed and treated expeditiously. This includes thrombotic thrombocytopenic purpura (TTP), disseminated intravascular hemolysis (DIC), and atypical hemolytic uremic syndrome (aHUS). A manual review of a peripheral blood smear is required to evaluate for fragmented red blood cells (schistocytes). Thereafter, ancillary testing to confirm intravascular hemolysis would include measuring free plasma hemoglobin and lactate dehydrogenase (LDH). Additionally, in intravascular hemolysis, haptoglobin should be depleted and urinalysis should show heme-positive urine without RBCs. In this case the patient’s normal bilirubin studies argue against hemolysis; however, elevated bilirubin is variably present in hemolytic anemias depending on the liver’s ability to conjugate and excrete bilirubin, the relative degree of RBC turnover, and type of hemolysis. Patients with intravascular hemolysis lose hemoglobin directly into the urine leaving relatively little hemoglobin to be incorporated into bile once it has reached the reticuloendothelial system. This results in relatively normal bilirubin levels. More specific indicators of intravascular hemolysis include pink colored plasma on visual inspection (commonly done in the blood bank as part of assessing for hemolytic transfusion reactions), measuring plasma free hemoglobin, or by detecting hemoglobin in the urine.
If microangiopathic hemolytic anemia (MAHA) is excluded, then other causes of these laboratory abnormalities should be considered. Bleeding is the most common cause for anemia, and thrombocytopenia predisposes patients to bleeding. However, there is no evidence of bleeding in this patient, and such a rapid acute anemia is unlikely to be caused by occult blood loss alone. Concurrent anemia and thrombocytopenia could be evidence of bone marrow toxicity from chemotherapy or neoplastic infiltration. With marrow infiltration, there are typically signs on the peripheral smear of leukoerythroblastosis, with circulating nucleated red blood cells and early myeloid forms. Concurrent immune thrombocytopenia (ITP) and autoimmune hemolytic anemia (AIHA), or Evans’ Syndrome, should also be considered. AIHA would be suggested by spherocytes on the peripheral smear, elevated LDH and a positive direct antibody test (DAT).
Regarding the AKI, the patient has diarrhea, which could lead to prerenal azotemia and acute tubular necrosis. A formal urinalysis would evaluate for prerenal and intrinsic kidney disease. TMA can cause intrinsic kidney injury with a benign urinary sediment. The blood urea nitrogen-to-creatinine ratio is not elevated, but in a patient with malnutrition this may not indicate prerenal azotemia. In summary, to differentiate potential TMAs from other causes, the patient needs a blood smear, coagulation studies, and an evaluation for hemolysis, including a urinalysis for free heme and any evidence of intrinsic kidney disease.
Urinalysis showed amber-colored, dilute urine with no white blood cells, red blood cells, protein, or casts. It was positive for blood and negative for bilirubin and hemosiderin. LDH was 1,382 IU/L (reference range 135-225 IU/L), and haptoglobin was unmeasurably low. His ferritin was 2,267 ng/mL, serum iron was 57 mcg/dL, total iron-binding capacity was 241 mcg/dL, and transferrin was 162 mcg/dL. Reticulocyte count was 6% (reticulocyte index of 0.86). Vitamin B12 level was normal. DAT was negative; INR and aPTT were normal. Fibrinogen was 287 mg/dL (reference range 200-400 mg/dL), and D-dimer was 5,095 ng/mL (reference range 0-229 ng/mL).
The urinalysis shows no active sediment to suggest vasculitis or glomerulonephritis. The kidney injury could be the result of renal toxicity from free hemoglobin or as part of TMA caused by microvascular thrombosis. The dilute urine makes prerenal azotemia less likely.
There is clearly acute intravascular hemolysis occurring as evidenced by hemoglobinuria, very high LDH, and undetectable serum haptoglobin. The hemolysis is acute because chronic intravascular hemolysis would lead to positive urine hemosiderin via deposition in the renal tubules. Autoimmune hemolytic anemia is much less likely, but not ruled out, by a negative DAT.
This syndrome can be further refined from acute anemia to acute anemia with likely nonimmune intravascular hemolysis, acute thrombocytopenia, and AKI with hemoglobinuria and a bland urinary sediment. At this point, intravascular hemolysis and kidney injury could be part of a unifying diagnosis. However, this does not account for the patient’s thrombocytopenia, and TMA remains the best explanation for the constellation of findings. Review of the peripheral blood smear is urgent because evidence of MAHA would prompt urgent plasma exchange based on presumptive diagnosis of acquired TTP to later be confirmed with ADAMTS13 activity testing. Most TMAs are treated with supportive care only; TTP and aHUS have specific interventions that change the natural history of the disease (plasma exchange and anticomplement therapy, respectively). Given both the deadly natural history and opportunity to intervene with plasma exchange, patients with TMA should be treated with urgent plasma exchange until ADAMTS13 deficiency is confirmed or refuted. One TMA that can be excluded at this point is DIC. DIC in its acute and chronic forms nearly universally causes MAHA, thrombocytopenia, and consumptive coagulopathy including hypofibrinogenemia.
If MAHA is excluded, then other causes of intravascular hemolysis should be considered, along with causes of thrombocytopenia that might be occurring concurrently. Intravascular hemolysis can be further differentiated by etiologies primarily related to the RBC or whether the RBC is the innocent bystander amidst a systemic illness. RBC disorders include syndromes affecting RBC fragility like hereditary spherocytosis or RBC enzymopathies (G6PD deficiency), but these do not cause thrombocytopenia. One exception is an acquired membrane defect, paroxysmal nocturnal hemoglobinuria (PNH), in which RBCs and other blood cells become susceptible to complement-mediated lysis. Testing for PNH by peripheral blood flow cytometry should be considered if the blood film lacks schistocytes. Systemic disorders that cause intravascular hemolysis include severe burns (heat damage to RBCs), RBC trauma from “march hemoglobinuria” or mechanical heart valves, immune (antibody-mediated) hemolysis from Rh immune globulin administration, cold agglutinin disease or ABO mismatched transfusion, and infections including the intraerythrocyte parasites malaria, Bartonellosis, and Babesiosis, as well as organisms that induce RBC fragility such as Leishmaniasis, Clostridium perfringens, and Haemophilus influenzae B.
On review of additional history, the patient had not recently received blood products. He had received heparin during prior hospitalizations, but had no prior history of thrombosis. He had no history of tick exposure. Peripheral blood smear was obtained and reviewed by a
The blood smear helps narrow the differential further. The lack of schistocytes makes TMA far less likely and so plasma exchange is not urgently indicated. The differential still includes drug-induced TMA (gemcitabine being a well-known cause for TMA) and cancer-associated TMA could still cause these findings, but plasma exchange does not improve outcomes. Acquired (immune) TTP is very unlikely unless the patient did not improve with supportive care or developed neurologic symptoms. Similarly, atypical (complement-driven) HUS would only be considered if renal failure did not improve with supportive care.
The blood smear does show a surprising finding of pyropoikilocytosis. Pyropoikilocytosis refers to changes in RBC shape (poikilocytosis) typically seen with thermal injury or rare RBC membrane structural defects. Hereditary pyropoikilocytosis, a very rare disease, is characterized by chronic hyperproliferative, compensated anemia, and occasional hemolytic crises. These crises are associated with splenomegaly, reticulocytosis, and elevated bilirubin with jaundice. As the patient has no history of similar episodes, the blood smear changes are not due to a hereditary cause and obviously not due to thermal injury (ie, severe burns). Pyropoikilocytosis has been rarely reported in drug-induced TMA and in severe bacterial bloodstream infections (most commonly Gram-negative bacilli). This patient has received gemcitabine (a known cause of drug-induced TMA) and has a recently diagnosed infection (C difficile colitis), either of which could be linked to this rare blood smear finding. Both of these syndromes would be treated with supportive care plus avoidance of future gemcitabine.
Transfusion of packed RBCs is indicated given his profound anemia and symptoms of fatigue. One should obtain further testing for cold agglutinins, PNH, and echocardiography to exclude endocarditis. If he were to become critically ill, anuric, or encephalopathic, then one could consider plasma exchange for treatment of TMA and hemoglobin-mediated AKI. Pyropoikilocytosis should be considered the result of drug-induced TMA, severe C difficile colitis, or an occult infection.
The patient was transfused packed RBCs. Because of a concern for an acute TMA such as TTP, both a hematopathologist and the consulting hematology/oncology team reviewed the peripheral blood morphology emergently. He was given aggressive fluid resuscitation and received 3 L of IV lactated ringers’ solution. An echocardiogram did not show valvular abnormalities. A renal biopsy was contraindicated because of the severe thrombocytopenia.
Given the recently confirmed C difficile colitis along with the findings of pyropoikilocytosis on the peripheral smear, toxin-mediated intravascular hemolysis from systemic C difficile infection became the leading diagnosis. Positing that the C difficile colitis was inadequately treated with oral metronidazole, aggressive treatment for C difficile was initiated with oral vancomycin in addition to intravenous metronidazole. Intravenous metronidazole was included given his elevated creatinine, presence of severe colitis on imaging, and concern he may be at risk for translocation of colonic C difficile or exotoxin into the bloodstream.
Over the course of the next 3 days, the patient’s platelet count normalized and his hemoglobin, creatinine, and symptoms of fatigue improved. Blood cultures remained negative. The patient’s rapid improvement with antibiotics supported our final diagnosis of toxin-mediated hemolysis caused by a systemic C difficile infection. On follow-up testing after hospital discharge, hemoglobin had returned to prior baseline and there was no recurrent hemolysis. Gemcitabine was considered to be a possible cause of his hemolytic anemia and was not continued in further treatment for his NSCLC.
COMMENTARY
When evaluating patients with cancer who present with fatigue, hospitalists should consider a broad list of potential causes. The differential should include etiologies directly related to the malignancy, paraneoplastic phenomena, treatment-related complications, and diseases unrelated to cancer. In addition, as the number of medications used for cancer proliferates, hospitalists must take a detailed history of the agents used and be aware of major side effects. Using this information, hospitalists may undertake a targeted approach to diagnostics while searching for a cause of fatigue.
When lab testing reveals profound anemia, hospitalists must consider syndromes that may require emergent management. Anemia can be caused by decreased RBC production, and acute anemia in the absence of clear blood loss suggests hemolysis. Moreover, the combination of elevated LDH and low haptoglobin is quite specific of hemolytic anemia.1,2 Once hemolytic anemia is identified, DIC and TMA syndromes (such as TTP) need to be considered. The combination of hemolytic anemia and AKI may indicate a medical emergency and should prompt hospitalists to obtain an urgent peripheral blood smear to help narrow the differential.3
The absence of schistocytes on a blood smear does not rule out TTP or HUS, but does argue strongly against these diagnoses.4,5 Of note, consultation with a hematopathologist and hematology subspecialist should be done to ensure appropriate and timely review of the peripheral blood smear.
In this case, the blood smear led to a very rare finding of pyropoikilocytosis. The unexpected result should prompt a broader review of the medical history particularly as it relates to the patient’s broader symptoms and laboratory abnormalities. Acquired pyropoikilocytosis is a very specific finding known to be associated only with hyperthermal injury (seen in burn patients), drug-induced TMA, and bacterial bloodstream infections, mainly Gram-negative toxins and Clostridioidal infections.6-8 In this case, both drug-induced TMA and C difficile infection were considered.
Gemcitabine-induced TMA can occur with either short or long term use of the medication and can be difficult to distinguish from TTP. While both TTP and gemcitabine-induced TMA can cause thrombocytopenia, hemolytic anemia, and schistocytes on a blood smear, the latter causes acute kidney injury more frequently than TTP. In addition, gemcitabine-induced TMA may not lead to severe decrease in ADAMTS13 activity. A kidney biopsy could confirm drug-induced TMA but was contraindicated in this case because of the thrombocytopenia. Gemcitabine should not be restarted if this side effect is suspected.
Given the continued rise in C difficile incidence, hospitalists should be aware that C difficile infection can cause extraintestinal illness.9,10 Although uncommon, these extraintestinal complications are associated with high risk of mortality and frequently occur in those with a history of intestinal injury or inflammation and a concomitant bloodstream infection.10 Regarding the possibility of C difficile contributing to hemolysis in this case, the patient’s low blood counts and hemolysis improved concomitantly with more aggressive treatment of C difficile infection. Although his blood cultures were sterile, C difficile is notoriously difficult to culture. Prior case reports have associated C difficile with intravascular hemolysis, which leads to the possibility that the patient did have a very rare manifestation of this unfortunately common infection.11
This case provides an excellent example of a diagnostic pivot point
KEY TEACHING POINTS
- In evaluating symptomatic cancer patients, providers must consider sequelae of the tumor, paraneoplastic phenomena, and treatment-related complications.
- Hemolytic anemia may represent a life-threatening emergency particularly when accompanied by AKI and requires urgent peripheral blood smear evaluation.
- Acquired pyropoikilocytosis is a specific finding known to be associated only with thermal injury, drug-induced TMA, and bacterial toxin–mediated hemolysis.
Disclosures
The authors have nothing to disclose.
1. Weinzierl EP, Arber DA. The differential diagnosis and bone marrow evaluation of new-onset pancytopenia. Am J Clin Pathol. 2013:139(1):9-29. https://doi.org/10.1309/AJCP50AEEYGREWUZ.
2. Marchand A, Galen RS, Van Lente F. The predictive value of serum haptoglobin in hemolytic disease. JAMA.1980;243(19):1909-1911. https://doi:10.1001/jama.1980.03300450023014.
3. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
4. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836-2846. https://doi.org/10.1182/blood-2016-10-709857.
5. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856. https://doi.org/10.1182/blood-2016-11-709865.
6. Baar S, Arrowsmith DJ. Thermal damage to red cells. J Clin Path. 1970;23(7):572-576. https://doi.org/10.1136/jcp.23.7.572.
7. Meinders AJ, Dijkstra I. Massive hemolysis and erythrophagocytosis in severe sepsis. Blood. 2014;124(6):841. https://doi.org/10.1182/blood-2014-04-565663.
8. McIlwaine K, Leach MT. Clostridium perfringens septicaemia. Br J Haematol. 2013;163(5):549. https://doi.org/10.1111/bjh.12551.
9. Evans CT, Safdar N. Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis. 2015;60 (Supp 2):S66-71. https://doi.org/10.1093/cid/civ140.
10. Gupta A, Patel R, Baddour LM, Pardi DS, Khanna S. Extraintestinal Clostridium difficile infections: a single-center experience. Mayo Clin Proc. 2014;89(11):1525-36. https://doi.org/10.1016/j.mayocp.2014.07.012.
11. Alvarado AS, Brodsky SV, Nadasdy T, Singh N. Hemolytic uremic syndrome associated with Clostridium difficile infection. Clin Nephrol. 2014;81(4):302-6. https://doi.org/10.5414/CN107691.
1. Weinzierl EP, Arber DA. The differential diagnosis and bone marrow evaluation of new-onset pancytopenia. Am J Clin Pathol. 2013:139(1):9-29. https://doi.org/10.1309/AJCP50AEEYGREWUZ.
2. Marchand A, Galen RS, Van Lente F. The predictive value of serum haptoglobin in hemolytic disease. JAMA.1980;243(19):1909-1911. https://doi:10.1001/jama.1980.03300450023014.
3. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
4. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836-2846. https://doi.org/10.1182/blood-2016-10-709857.
5. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856. https://doi.org/10.1182/blood-2016-11-709865.
6. Baar S, Arrowsmith DJ. Thermal damage to red cells. J Clin Path. 1970;23(7):572-576. https://doi.org/10.1136/jcp.23.7.572.
7. Meinders AJ, Dijkstra I. Massive hemolysis and erythrophagocytosis in severe sepsis. Blood. 2014;124(6):841. https://doi.org/10.1182/blood-2014-04-565663.
8. McIlwaine K, Leach MT. Clostridium perfringens septicaemia. Br J Haematol. 2013;163(5):549. https://doi.org/10.1111/bjh.12551.
9. Evans CT, Safdar N. Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis. 2015;60 (Supp 2):S66-71. https://doi.org/10.1093/cid/civ140.
10. Gupta A, Patel R, Baddour LM, Pardi DS, Khanna S. Extraintestinal Clostridium difficile infections: a single-center experience. Mayo Clin Proc. 2014;89(11):1525-36. https://doi.org/10.1016/j.mayocp.2014.07.012.
11. Alvarado AS, Brodsky SV, Nadasdy T, Singh N. Hemolytic uremic syndrome associated with Clostridium difficile infection. Clin Nephrol. 2014;81(4):302-6. https://doi.org/10.5414/CN107691.
© 2020 Society of Hospital Medicine
A Jaw-Dropping Diagnosis
A 73-year-old man presented to primary care for an annual examination. Four days prior, he noted right-sided sharp jaw pain such that he could not open his mouth nor chew solid food; it radiated from the right mandible to the ipsilateral temple. He also noted bilateral aching hip pain for several years that increased in severity in the prior 2 months. He reported an intentional weight loss of 9 kg over the past year, achieved through dietary modification. He denied fever, chills, and visual disturbance.
Acute onset of unilateral jaw pain that is worsened by chewing is a feature consistent with a temporomandibular disorder (TMD). TMD consists of musculoskeletal and neuromuscular conditions that affect the temporomandibular joints (TMJs), masticatory muscles, and associated tissues. Common symptoms of TMD include facial or ear pain, temporal headache, and TMJ dysfunction or discomfort. In addition to TMD, craniofacial pain has many possible etiologies such as dental pathology, neuralgias, sinus and otologic disorders, headache and migraine disorders, infections, rheumatologic conditions, and neoplasms.
Systemic etiologies for this patient’s symptoms are a consideration given his age and concomitant worsening of chronic hip pain. Rheumatologic conditions such as giant cell arteritis (GCA) and polymyalgia rheumatica (PMR) are more common in adults older than 50 years of age and cause headache, jaw claudication, and pelvic girdle pain. Rarely, hematologic malignancies (eg, lymphoma), solid tumor metastases (eg, breast cancer, melanoma), and primary tumors of the head and neck (eg, nasopharyngeal carcinoma) can involve the mandible, TMJ, or parotid gland and result in symptoms of TMD.
Medical history was notable for hypertension and type 2 diabetes mellitus complicated by peripheral neuropathy. He smoked one pack of cigarettes daily for 40 years but quit 15 years prior. He drank 4 ounces of vodka each night.
On examination, temperature was 36.5°C, heart rate 92 beats per minute, blood pressure 127/60 mmHg, respiratory rate 12 breaths per minute, oxygen saturation 98% on ambient air, and weight 118 kg. Extraocular movements were intact, pupils were equal and reactive to light and accommodation, and there were no visual field deficits. Nondilated funduscopic examination revealed normal blood vessels, optic disc, and optic cup-to-disc ratio. Dentition was good with pink gingiva. Bilateral temples were nontender. There was normal range of motion and strength in the shoulders, hips, and lower extremities with no tenderness over the trochanters. Patellar and ankle reflexes were present and symmetric bilaterally. He had no rashes or ecchymoses.
The history of smoking, especially with concomitant alcohol intake, is a risk factor for head and neck cancer, and these malignancies can lead to facial pain. While the normal oral cavity exam argues against localized oral and dental causes of the patient’s symptoms, direct fiberoptic endoscopy should be considered. The neck should be examined for lymphadenopathy. Normal vital signs point away from severe infection. The lack of findings in the head and musculoskeletal regions does not exclude systemic etiologies such as rheumatologic conditions or neoplasm. Complete blood cell count and markers of inflammation including erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) levels should be obtained. Hip and pelvic radiographs should be obtained to evaluate for hip osteoarthritis, fractures, or osseous lesions.
The appointment occurred during evening hours and the patient declined further evaluation until the following morning, at which time laboratory studies revealed normal serum levels of electrolytes, blood urea nitrogen, and creatinine. White blood cell (WBC) count was 6,800/mm3 with an immature granulocyte ratio of 1.8% (normal, 0.0-0.5%), hemoglobin 13.2 g/dL, and platelet count 163,000/mm3. ESR was 118 mm/hr (normal, 0-15 mm/hr) and CRP was 1.5 mg/dL (normal, 0-0.75 mg/dL). Radiographs of the hips and pelvis showed osteoarthritis of the bilateral hip joints and degenerative disc disease of the lower lumbar spine.
Granulocytosis may occur in response to infection, rheumatologic conditions, and hematologic malignancies such as chronic myelogenous leukemia. While infectious etiologies (eg, abscess, osteomyelitis) are the most common cause of an extremely elevated ESR level, this patient does not have other signs or symptoms of infection such as fever or leukocytosis. Therefore, other common causes for an extremely elevated ESR level should be considered, including malignancy (eg, multiple myeloma, lymphoma, metastatic solid tumor) and autoimmune conditions (eg, rheumatoid arthritis, vasculitis). While multiple myeloma is the most common malignant etiology for extremely elevated ESR, the patient lacks signs of this condition such as anemia, elevated creatinine, or osteolytic lesions on radiographic imaging. Osteoarthritis identified on the radiographs may contribute to the patient’s hip pain but would not explain the patient’s jaw pain, weight loss, granulocytosis, and elevated ESR. These findings, taken together with the patient’s age, are most suggestive of GCA with possible coexisting PMR. Temporal artery biopsy should be obtained as it is the gold standard test for diagnosing GCA.
The patient was contacted by telephone that same day with laboratory test results. During the call, he endorsed increased jaw and temple pain. He was advised to proceed to the emergency department (ED) for timely evaluation and treatment.
Because GCA was being considered, ophthalmology performed an ocular examination in the ED, which demonstrated no signs of optic nerve or retinal ischemia. Computed tomography (CT) scan of the head and neck with intravenous contrast revealed no abscess or soft tissue abnormalities. Right temporal artery biopsy was performed.
The normal ocular examination does not exclude GCA, and temporal artery biopsy is appropriate. The mainstay of treatment for GCA is high-dose systemic glucocorticoids, which should not be withheld while awaiting biopsy results since ophthalmic artery inflammation may occur and threaten vision.
While GCA remains the leading diagnosis, malignant etiologies warrant further consideration because they are a common cause of extreme ESR elevation, particularly among older patients. The patient’s cancer screening history should be reviewed. The normal CT scan of the head and neck reduces the likelihood of localized solid tumor etiologies; however, additional CT imaging of the chest, abdomen, and pelvis is warranted to evaluate for metastatic solid tumors or lymphoma.
A 10-day course of prednisone 60 mg daily was prescribed for empiric treatment of GCA. The patient was discharged home with follow-up scheduled in rheumatology and primary care clinics. Pain in the jaw and temple resolved within several days.
Two weeks later, he presented to the rheumatology clinic. He noted 1 week of lower right back pain described as dull, aching, radiating to the lateral right hip, and occurring when transitioning from sitting to standing. He had no leg numbness, weakness, or change in bowel habits. Bladder habits were also unchanged, although he reported chronic urinary frequency and occasional incontinence. He reported further weight loss, this time an unintentional loss of 9 kg. He noted frequent sweating but no fever.
He reported a normal colonoscopy within the prior 5 years. Because these records were not available for review, a fecal immunochemical test was obtained and negative for hemoglobin. He had previously declined prostate cancer screening.
The resolution of jaw and temple pain with prednisone supports the presumed diagnosis of GCA. Up to half of patients with GCA may also have PMR, which can cause aching and stiffness in the arms, hips, and lumbar region, and pain may be abrupt in onset. However, PMR-related pain would be expected to improve rather than develop or worsen in the setting of high-dose glucocorticoid use. Therefore, other causes of acute-onset back pain must be considered.
While localized musculoskeletal etiologies such as lumbar muscle strain, radiculopathy, and vertebral compression fracture are possible, co-occurrence of unintentional weight loss and diaphoresis with elevated inflammatory markers suggests a systemic etiology. A neoplastic process with bony metastasis is possible. The reportedly normal colonoscopy and the negative fecal immunochemical test make colorectal cancer less likely. Inflammatory conditions such as ankylosing spondylitis and rheumatoid arthritis are also possible. Ankylosing spondylitis usually presents at a much younger age, however, and axial skeletal involvement in rheumatoid arthritis often involves the cervical spine and is usually seen after longstanding disease. Additionally, the hallmark of inflammatory back pain is morning stiffness which the patient does not endorse. Nonetheless, additional laboratory testing should include antinuclear antibody, rheumatoid factor, and anti-cyclic citrullinated peptide (anti-CCP) antibody. Vertebral osteomyelitis remains on the differential diagnosis, and repeat WBC count and inflammatory markers should be assessed. Lumbosacral radiographs should be obtained to rule out fracture.
Physical examination in the rheumatology clinic revealed a temperature of 37.0°C, heart rate 100 beats per minute, blood pressure 146/72 mmHg, respiratory rate 12 breaths per minute, and oxygen saturation 98% on ambient air. Weight was 109 kg. He was pale and diaphoretic. There was diffuse tenderness to palpation of the right-sided lumbar paraspinal muscles. Straight leg raise was negative bilaterally. Patellar reflexes and gait were normal.
Blood chemistries, renal function, and aminotransferase levels were normal. WBC count was 7,100/mm3, hemoglobin 8.0 g/dL, mean corpuscular volume 88.9 fL, platelet count 128,000/mm3, ESR 66 mm/hr, CRP 0.57 mg/dL, alkaline phosphatase 438 IU/L (normal, 30-130 IU/L), and thyroid-stimulating hormone 0.925 mU/L (normal, 0.34-5.60 mU/L). Testing for antinuclear antibodies, rheumatoid factor, and anti-CCP antibody was unremarkable. Prostate-specific antigen (PSA) level was 2.2 ng/mL (normal, 0-4 ng/mL). Urinalysis was unremarkable. Antibodies to hepatitis C and Treponema pallidum were negative. Interferon gamma release assay was negative.
Findings of new onset anemia and thrombocytopenia, in combination with elevated ESR and alkaline phosphatase level, are concerning for disseminated intravascular coagulation (DIC) and microangiopathic hemolytic anemia (MAHA), bone marrow infiltration of a metastatic neoplasm, or ineffective hematopoiesis caused by myelodysplastic syndromes or myelofibrosis.
Laboratory evaluation should include iron studies, lactate dehydrogenase (LDH), haptoglobin, fibrinogen, D-dimer, reticulocyte count, and peripheral blood smear to assess for hemolysis and erythrocyte morphology. Advanced imaging with lumbosacral magnetic resonance imaging (MRI) should be obtained to evaluate for focal etiologies of back pain such as disc herniation, abscess, marrow infiltration, and infarction.
Additional laboratory studies revealed a gamma-glutamyl transferase level of 49 IU/L (normal, 8-56 IU/L), LDH 288 IU/L (normal, 98-192 IU/L), haptoglobin 495 mg/dL (normal, 32-240 mg/dL), fibrinogen >700 mg/dL (normal, 225-550 mg/dL), D-dimer 693 ng/mL (normal, 200-250 ng/mL), serum iron 57 mcg/dL (normal, 33-150 mcg/dL), total iron binding capacity 286 mcg/dL (normal, 250-450 mcg/dL), ferritin 1,012 ng/mL (normal, 17.9-464 ng/mL), and reticulocyte count 2.9% (normal, 0.5-2.5%). Coagulation studies and serum protein electrophoresis were normal. Erythropoietin level was 109 mIU/mL (normal, 4.0-20.0 mIU/mL). Peripheral blood smear demonstrated moderate anemia with 8% nucleated erythrocytes per white blood cell (normal, 0%) and no circulating blasts.
MRI of the thoracolumbar spine and pelvis revealed diffusely abnormal bone marrow signal with multiple superimposed focal and poorly defined enhancing lesions along the lumbar spine marrow, sacrum, and bilateral iliac bones (Figure 1). Positron emission tomography/computed tomography (PET/CT) scan showed no scintigraphic evidence of metabolically active neoplastic, paraneoplastic, or inflammatory disorder.

The elevated haptoglobin, normal coagulation studies, and absence of fragmented erythrocytes on peripheral smear exclude an intravascular hemolytic process. The patient’s lower than expected reticulocyte count for the degree of anemia, elevated erythropoietin, and nucleated erythrocytes constitute a pattern that can be seen with bone marrow infiltration. There are no circulating blasts, making leukemia less likely. A solid organ tumor with bone metastases may cause enhancing lesions on MRI since this form of imaging is more sensitive than radiography for detecting skeletal malignancies. The negative PET/CT, however, does not reveal a primary tumor. Myelofibrosis is an infiltrative myeloproliferative disorder associated with nonspecific laboratory abnormalities, bone pain, weight loss, and night sweats that could cause diffuse MRI bone marrow signal alterations with normal PET/CT findings. However, myelofibrosis would not typically cause a significantly elevated ESR, and thus would be an unlikely cause for this patient’s presentation.
Given the constellation of symptoms, hematologic abnormalities, and bone marrow infiltration on imaging, hematology should be consulted to perform a bone marrow biopsy to assist with definitive diagnosis.
Bone marrow biopsy demonstrated metastatic adenocarcinoma consistent with prostatic origin (Figure 2). Bone scan demonstrated widespread osteoblastic metastases, which included the skull and temporal regions. These lesions were thought to be the cause of the patient’s original presenting symptom of jaw pain.

The patient was started on androgen deprivation therapy, initially with degarelix and subsequently leuprolide shots and abiraterone with prednisone. PSA was 0.08 ng/mL after 3 months of androgen deprivation therapy. His back and hip pain slowly improved.
DISCUSSION
Prostate cancer is the most common cancer in men with one out of every nine men diagnosed in his lifetime.1 While most men initially present with localized, curable disease,1 4% present with metastatic disease, an incidence that has been increasing since 2004.2 Despite available treatments, metastatic prostate cancer has a poor prognosis, with an average overall survival of approximately 5 years.3
Prostate cancer can be challenging to diagnose. Men with prostate cancer are commonly asymptomatic. Rarely, patients may present with hematuria, bony pain caused by metastasis, or obstructive urinary symptoms like hesitancy or incomplete bladder emptying. Our patient presented with jaw pain, which was ultimately attributed to osteoblastic lesions of the skull. Additionally, his history of urinary frequency and incontinence may have been clues to his underlying diagnosis of prostate cancer.
Prostate cancer screening remains highly nuanced and relies on shared decision-making between patients and healthcare providers. Clinical practice guidelines for early detection of prostate cancer recommend individualized PSA-based serologic screening.4,5 Specifically, the United States Preventive Services Task Force recommends screening men aged 55 to 69 years who desire screening and understand the potential harms associated with a positive test result. These harms may include psychological distress and complications from prostate biopsy (eg, pain or infection) or prostate cancer treatment (eg, erectile, urinary, and/or bowel dysfunction).4-6 The decision to screen can be guided by individuals’ risk factors including African American race, family history, and older age.
While our patient elected not to undergo routine prostate cancer screening, a PSA level was obtained during his diagnostic evaluation and highlights the limitations of PSA-based screening. A PSA level ≤4.0 ng/mL has 21% sensitivity and 91% specificity for detecting prostate cancer.7 PSA levels above 4.0 ng/mL warrant repeat testing and, if persistently elevated, referral to urology for possible prostate biopsy. PSA levels often correlate with burden of disease, and patients with PSA levels >20 ng/mL are referred for CT imaging to evaluate for metastatic disease.8 PSA’s poor sensitivity was underscored in a study by Thompson et al who evaluated the incidence of prostate cancer in men participating in the Prostate Cancer Prevention Trial with PSA levels of <4 ng/mL.9 In this study, 15% of men diagnosed with prostate cancer never had a PSA level >4 ng/mL.9 While most of the cancers in this study were low grade and may have been clinically insignificant, 15% demonstrated histologic signs of at least intermediate-risk disease. Our patient’s PSA level of 2.2 ng/mL was below the threshold that triggers additional evaluation even though he had widely metastatic prostate cancer.
Our patient’s severe jaw and temple pain, weight loss, and progressive hip pain were concerning for GCA. This vasculitis of large- and medium-sized arteries predominantly affects older adults with greatest incidence among those 70 years of age and older.10 Symptoms occur because of cranial artery inflammation and may include headache, visual disturbance, erythema or tenderness of the temporal artery, and jaw claudication. Extracranial inflammation may affect the thoracic aorta and its branches and rarely the abdominal aorta and lower limb arteries. Pelvic girdle pain more typically results from associated PMR. Patients may also note systemic symptoms such as fever, weight loss, and fatigue.
Prompt diagnostic testing is important when considering GCA. Most patients with GCA have ESR levels greater than 40 mm/hr.11 ESR is a laboratory test that measures the vertical distance erythrocytes travel in a column of blood over 1 hour; in the setting of inflammation, cells form clumps and travel more quickly than individual cells, resulting in a higher value. While moderate elevations in ESR may occur without an identifiable cause, extreme ESR levels—those above 100 mm/hr, as observed in our patient—are highly suggestive of certain serious conditions, including infection, malignancy, and autoimmune disease such as GCA.12,13 Temporal artery biopsy is the gold standard test to diagnose GCA. However, because of noncontiguous inflammation of the temporal artery, biopsies may be falsely negative. Thus, sampling of the contralateral temporal artery may be warranted if suspicion remains high.
As was the case for our patient, PET/CT is not reliable for diagnosing prostate cancer. In contrast to other malignancies (eg, lymphoma, lung cancer), prostate cancer typically does not display increased glucose metabolism. Moreover, the close proximity of the bladder and prostate can interfere with imaging interpretation because the fluorodeoxyglucose (FDG) tracer is excreted in the urine.14 The reported sensitivity of PET/CT for the diagnosis of prostate cancer ranges from 17%-65%.15,16 In a small study of men with metastatic prostate cancer, only 18% of bony metastases were FDG avid, and there was no correlation between FDG avidity and PSA level.15 Notably, although PET/CT includes CT imaging, this CT is used to map anatomic landmarks and is not separately interpreted by the radiologist. Thus, even if evidence of prostate cancer was apparent on traditional CT, it may be overlooked on PET/CT.
Several important points regarding diagnostic testing are raised by this case. First, PSA-based screening for prostate cancer may be falsely negative, even in the setting of widely metastatic disease. Second, extreme ESR elevation is a marker for serious underlying disease and warrants a thorough diagnostic evaluation. Finally, PET/CT has limited diagnostic utility in evaluating metastatic prostate cancer because of the normal rates of glucose metabolism. Our patient initially presented with jaw pain, yet his progressive physical symptoms and laboratory abnormalities prompted an evaluation which ultimately revealed the jaw-dropping diagnosis of PSA-negative, metastatic prostate cancer.
KEY TEACHING POINTS
- ESR levels greater than 100 mm/hr are highly suggestive of certain serious conditions including infection, autoimmune disease, and malignancy.
- PSA-based screening for prostate cancer can result in false negative test results. In one study, 15% of men diagnosed with prostate cancer never had a PSA level greater than 4 ng/mL (ie, the level at which repeat laboratory testing and/or referral to urology for possible prostate biopsy is advisable).
- PET/CT has limited diagnostic utility in evaluating metastatic prostate cancer, because prostate cancer cells typically demonstrate normal glucose metabolism.
Disclosures
Drs Griauzde, Northway, Yentz, and Houchens have nothing to disclose. Dr Saint reports personal fees from ISMIE Mutual Insurance Company during the conduct of the study, as well as personal fees from Jvion and Doximity outside the submitted work.
1. Prostate Cancer - Cancer Stat Facts. SEER. https://seer.cancer.gov/statfacts/html/prost.html. Accessed October 23, 2018.
2. Li J, Siegel DA, King JB. Stage-specific incidence rates and trends of prostate cancer by age, race, and ethnicity, United States, 2004-2014. Ann Epidemiol. 2018;28(5):328-330. https://doi.org/10.1016/j.annepidem.2018.03.001.
3. Sweeney CJ, Chen YH, Carducci M, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med. 2015;373(8):737-746. https://doi.org/10.1056/NEJMoa1503747.
4. US Preventive Services Task Force. Final Recommendation Statement: Prostate Cancer: Screening. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/prostate-cancer-screening1. Accessed August 8, 2018.
5. American Urological Association. http://www.auanet.org/guidelines/prostate-cancer-early-detection. Accessed August 8, 2018.
6. American Cancer Society. American Cancer Society Recommendations for Prostate Cancer Early Detection. https://www.cancer.org/cancer/prostate-cancer/early-detection/acs-recommendations.html. Accessed August 8, 2018.
7. Wolf AM, Wender RC, Etzioni RB, et al. American Cancer Society guideline for the early detection of prostate cancer: update 2010. CA Cancer J Clin. 2010;60(2):70-98. https://doi.org/10.3322/caac.20066.
8. Mohler JL, Lee RJ, Antonarakis ES, Higano CS, Richey S. NCCN Guidelines Index Table of Contents. Prostate Cancer. 2018:151.
9. Thompson IM, Pauler DK, Goodman PJ, et al. Prevalence of prostate cancer among men with a prostate-specific antigen level ≤4.0 ng per milliliter. N Engl J Med. 2004;350(22):2239-2246. https://doi.org/10.1056/NEJMoa031918.
10. Pioro MH. Primary care vasculitis: Polymyalgia rheumatica and giant cell arteritis. Prim Care. 2018;45(2):305-323. https://doi.org/10.1016/j.pop.2018.02.007.
11. Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurrence in a population-based study. Arthritis Rheum. 2001;45(2):140-145. https://doi.org/10.1002/1529-0131(200104)45:2<140::AID-ANR166>3.0.CO;2-2
12. Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60(5):1443-1450.
13. Daniels LM, Tosh PK, Fiala JA, Schleck CD, Mandrekar JN, Beckman TJ. Extremely elevated erythrocyte sedimentation rates: Associations with patients’ diagnoses, demographic dharacteristics, and comorbidities. Mayo Clin Proc. 2017;92(11):1636-1643. https://doi.org/10.1016/j.mayocp.2017.07.018.
14. Powles T, Murray I, Brock C, Oliver T, Avril N. Molecular positron emission tomography and PET/CT imaging in urological malignancies. Eur Urol. 2007;51(6):1511-1521. http://doi.org/10.1016/j.eururo.2007.01.061.
15. Yeh SDJ, Imbriaco M, Larson SM, et al. Detection of bony metastases of androgen-independent prostate cancer by PET-FDG. Nucl Med Biol. 1996;23(6):693-697. https://doi.org/10.1016/0969-8051(96)00044-3.
16. Perera M, Papa N, Christidis D, et al. Sensitivity, specificity, and predictors of positive 68ga-prostate-specific membrane antigen positron emission tomography in advanced prostate cancer: a systematic review and meta-analysis. Eur Urol. 2016;70(6):926-937. https://doi.org/10.1016/j.eururo.2016.06.021.
A 73-year-old man presented to primary care for an annual examination. Four days prior, he noted right-sided sharp jaw pain such that he could not open his mouth nor chew solid food; it radiated from the right mandible to the ipsilateral temple. He also noted bilateral aching hip pain for several years that increased in severity in the prior 2 months. He reported an intentional weight loss of 9 kg over the past year, achieved through dietary modification. He denied fever, chills, and visual disturbance.
Acute onset of unilateral jaw pain that is worsened by chewing is a feature consistent with a temporomandibular disorder (TMD). TMD consists of musculoskeletal and neuromuscular conditions that affect the temporomandibular joints (TMJs), masticatory muscles, and associated tissues. Common symptoms of TMD include facial or ear pain, temporal headache, and TMJ dysfunction or discomfort. In addition to TMD, craniofacial pain has many possible etiologies such as dental pathology, neuralgias, sinus and otologic disorders, headache and migraine disorders, infections, rheumatologic conditions, and neoplasms.
Systemic etiologies for this patient’s symptoms are a consideration given his age and concomitant worsening of chronic hip pain. Rheumatologic conditions such as giant cell arteritis (GCA) and polymyalgia rheumatica (PMR) are more common in adults older than 50 years of age and cause headache, jaw claudication, and pelvic girdle pain. Rarely, hematologic malignancies (eg, lymphoma), solid tumor metastases (eg, breast cancer, melanoma), and primary tumors of the head and neck (eg, nasopharyngeal carcinoma) can involve the mandible, TMJ, or parotid gland and result in symptoms of TMD.
Medical history was notable for hypertension and type 2 diabetes mellitus complicated by peripheral neuropathy. He smoked one pack of cigarettes daily for 40 years but quit 15 years prior. He drank 4 ounces of vodka each night.
On examination, temperature was 36.5°C, heart rate 92 beats per minute, blood pressure 127/60 mmHg, respiratory rate 12 breaths per minute, oxygen saturation 98% on ambient air, and weight 118 kg. Extraocular movements were intact, pupils were equal and reactive to light and accommodation, and there were no visual field deficits. Nondilated funduscopic examination revealed normal blood vessels, optic disc, and optic cup-to-disc ratio. Dentition was good with pink gingiva. Bilateral temples were nontender. There was normal range of motion and strength in the shoulders, hips, and lower extremities with no tenderness over the trochanters. Patellar and ankle reflexes were present and symmetric bilaterally. He had no rashes or ecchymoses.
The history of smoking, especially with concomitant alcohol intake, is a risk factor for head and neck cancer, and these malignancies can lead to facial pain. While the normal oral cavity exam argues against localized oral and dental causes of the patient’s symptoms, direct fiberoptic endoscopy should be considered. The neck should be examined for lymphadenopathy. Normal vital signs point away from severe infection. The lack of findings in the head and musculoskeletal regions does not exclude systemic etiologies such as rheumatologic conditions or neoplasm. Complete blood cell count and markers of inflammation including erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) levels should be obtained. Hip and pelvic radiographs should be obtained to evaluate for hip osteoarthritis, fractures, or osseous lesions.
The appointment occurred during evening hours and the patient declined further evaluation until the following morning, at which time laboratory studies revealed normal serum levels of electrolytes, blood urea nitrogen, and creatinine. White blood cell (WBC) count was 6,800/mm3 with an immature granulocyte ratio of 1.8% (normal, 0.0-0.5%), hemoglobin 13.2 g/dL, and platelet count 163,000/mm3. ESR was 118 mm/hr (normal, 0-15 mm/hr) and CRP was 1.5 mg/dL (normal, 0-0.75 mg/dL). Radiographs of the hips and pelvis showed osteoarthritis of the bilateral hip joints and degenerative disc disease of the lower lumbar spine.
Granulocytosis may occur in response to infection, rheumatologic conditions, and hematologic malignancies such as chronic myelogenous leukemia. While infectious etiologies (eg, abscess, osteomyelitis) are the most common cause of an extremely elevated ESR level, this patient does not have other signs or symptoms of infection such as fever or leukocytosis. Therefore, other common causes for an extremely elevated ESR level should be considered, including malignancy (eg, multiple myeloma, lymphoma, metastatic solid tumor) and autoimmune conditions (eg, rheumatoid arthritis, vasculitis). While multiple myeloma is the most common malignant etiology for extremely elevated ESR, the patient lacks signs of this condition such as anemia, elevated creatinine, or osteolytic lesions on radiographic imaging. Osteoarthritis identified on the radiographs may contribute to the patient’s hip pain but would not explain the patient’s jaw pain, weight loss, granulocytosis, and elevated ESR. These findings, taken together with the patient’s age, are most suggestive of GCA with possible coexisting PMR. Temporal artery biopsy should be obtained as it is the gold standard test for diagnosing GCA.
The patient was contacted by telephone that same day with laboratory test results. During the call, he endorsed increased jaw and temple pain. He was advised to proceed to the emergency department (ED) for timely evaluation and treatment.
Because GCA was being considered, ophthalmology performed an ocular examination in the ED, which demonstrated no signs of optic nerve or retinal ischemia. Computed tomography (CT) scan of the head and neck with intravenous contrast revealed no abscess or soft tissue abnormalities. Right temporal artery biopsy was performed.
The normal ocular examination does not exclude GCA, and temporal artery biopsy is appropriate. The mainstay of treatment for GCA is high-dose systemic glucocorticoids, which should not be withheld while awaiting biopsy results since ophthalmic artery inflammation may occur and threaten vision.
While GCA remains the leading diagnosis, malignant etiologies warrant further consideration because they are a common cause of extreme ESR elevation, particularly among older patients. The patient’s cancer screening history should be reviewed. The normal CT scan of the head and neck reduces the likelihood of localized solid tumor etiologies; however, additional CT imaging of the chest, abdomen, and pelvis is warranted to evaluate for metastatic solid tumors or lymphoma.
A 10-day course of prednisone 60 mg daily was prescribed for empiric treatment of GCA. The patient was discharged home with follow-up scheduled in rheumatology and primary care clinics. Pain in the jaw and temple resolved within several days.
Two weeks later, he presented to the rheumatology clinic. He noted 1 week of lower right back pain described as dull, aching, radiating to the lateral right hip, and occurring when transitioning from sitting to standing. He had no leg numbness, weakness, or change in bowel habits. Bladder habits were also unchanged, although he reported chronic urinary frequency and occasional incontinence. He reported further weight loss, this time an unintentional loss of 9 kg. He noted frequent sweating but no fever.
He reported a normal colonoscopy within the prior 5 years. Because these records were not available for review, a fecal immunochemical test was obtained and negative for hemoglobin. He had previously declined prostate cancer screening.
The resolution of jaw and temple pain with prednisone supports the presumed diagnosis of GCA. Up to half of patients with GCA may also have PMR, which can cause aching and stiffness in the arms, hips, and lumbar region, and pain may be abrupt in onset. However, PMR-related pain would be expected to improve rather than develop or worsen in the setting of high-dose glucocorticoid use. Therefore, other causes of acute-onset back pain must be considered.
While localized musculoskeletal etiologies such as lumbar muscle strain, radiculopathy, and vertebral compression fracture are possible, co-occurrence of unintentional weight loss and diaphoresis with elevated inflammatory markers suggests a systemic etiology. A neoplastic process with bony metastasis is possible. The reportedly normal colonoscopy and the negative fecal immunochemical test make colorectal cancer less likely. Inflammatory conditions such as ankylosing spondylitis and rheumatoid arthritis are also possible. Ankylosing spondylitis usually presents at a much younger age, however, and axial skeletal involvement in rheumatoid arthritis often involves the cervical spine and is usually seen after longstanding disease. Additionally, the hallmark of inflammatory back pain is morning stiffness which the patient does not endorse. Nonetheless, additional laboratory testing should include antinuclear antibody, rheumatoid factor, and anti-cyclic citrullinated peptide (anti-CCP) antibody. Vertebral osteomyelitis remains on the differential diagnosis, and repeat WBC count and inflammatory markers should be assessed. Lumbosacral radiographs should be obtained to rule out fracture.
Physical examination in the rheumatology clinic revealed a temperature of 37.0°C, heart rate 100 beats per minute, blood pressure 146/72 mmHg, respiratory rate 12 breaths per minute, and oxygen saturation 98% on ambient air. Weight was 109 kg. He was pale and diaphoretic. There was diffuse tenderness to palpation of the right-sided lumbar paraspinal muscles. Straight leg raise was negative bilaterally. Patellar reflexes and gait were normal.
Blood chemistries, renal function, and aminotransferase levels were normal. WBC count was 7,100/mm3, hemoglobin 8.0 g/dL, mean corpuscular volume 88.9 fL, platelet count 128,000/mm3, ESR 66 mm/hr, CRP 0.57 mg/dL, alkaline phosphatase 438 IU/L (normal, 30-130 IU/L), and thyroid-stimulating hormone 0.925 mU/L (normal, 0.34-5.60 mU/L). Testing for antinuclear antibodies, rheumatoid factor, and anti-CCP antibody was unremarkable. Prostate-specific antigen (PSA) level was 2.2 ng/mL (normal, 0-4 ng/mL). Urinalysis was unremarkable. Antibodies to hepatitis C and Treponema pallidum were negative. Interferon gamma release assay was negative.
Findings of new onset anemia and thrombocytopenia, in combination with elevated ESR and alkaline phosphatase level, are concerning for disseminated intravascular coagulation (DIC) and microangiopathic hemolytic anemia (MAHA), bone marrow infiltration of a metastatic neoplasm, or ineffective hematopoiesis caused by myelodysplastic syndromes or myelofibrosis.
Laboratory evaluation should include iron studies, lactate dehydrogenase (LDH), haptoglobin, fibrinogen, D-dimer, reticulocyte count, and peripheral blood smear to assess for hemolysis and erythrocyte morphology. Advanced imaging with lumbosacral magnetic resonance imaging (MRI) should be obtained to evaluate for focal etiologies of back pain such as disc herniation, abscess, marrow infiltration, and infarction.
Additional laboratory studies revealed a gamma-glutamyl transferase level of 49 IU/L (normal, 8-56 IU/L), LDH 288 IU/L (normal, 98-192 IU/L), haptoglobin 495 mg/dL (normal, 32-240 mg/dL), fibrinogen >700 mg/dL (normal, 225-550 mg/dL), D-dimer 693 ng/mL (normal, 200-250 ng/mL), serum iron 57 mcg/dL (normal, 33-150 mcg/dL), total iron binding capacity 286 mcg/dL (normal, 250-450 mcg/dL), ferritin 1,012 ng/mL (normal, 17.9-464 ng/mL), and reticulocyte count 2.9% (normal, 0.5-2.5%). Coagulation studies and serum protein electrophoresis were normal. Erythropoietin level was 109 mIU/mL (normal, 4.0-20.0 mIU/mL). Peripheral blood smear demonstrated moderate anemia with 8% nucleated erythrocytes per white blood cell (normal, 0%) and no circulating blasts.
MRI of the thoracolumbar spine and pelvis revealed diffusely abnormal bone marrow signal with multiple superimposed focal and poorly defined enhancing lesions along the lumbar spine marrow, sacrum, and bilateral iliac bones (Figure 1). Positron emission tomography/computed tomography (PET/CT) scan showed no scintigraphic evidence of metabolically active neoplastic, paraneoplastic, or inflammatory disorder.
The elevated haptoglobin, normal coagulation studies, and absence of fragmented erythrocytes on peripheral smear exclude an intravascular hemolytic process. The patient’s lower than expected reticulocyte count for the degree of anemia, elevated erythropoietin, and nucleated erythrocytes constitute a pattern that can be seen with bone marrow infiltration. There are no circulating blasts, making leukemia less likely. A solid organ tumor with bone metastases may cause enhancing lesions on MRI since this form of imaging is more sensitive than radiography for detecting skeletal malignancies. The negative PET/CT, however, does not reveal a primary tumor. Myelofibrosis is an infiltrative myeloproliferative disorder associated with nonspecific laboratory abnormalities, bone pain, weight loss, and night sweats that could cause diffuse MRI bone marrow signal alterations with normal PET/CT findings. However, myelofibrosis would not typically cause a significantly elevated ESR, and thus would be an unlikely cause for this patient’s presentation.
Given the constellation of symptoms, hematologic abnormalities, and bone marrow infiltration on imaging, hematology should be consulted to perform a bone marrow biopsy to assist with definitive diagnosis.
Bone marrow biopsy demonstrated metastatic adenocarcinoma consistent with prostatic origin (Figure 2). Bone scan demonstrated widespread osteoblastic metastases, which included the skull and temporal regions. These lesions were thought to be the cause of the patient’s original presenting symptom of jaw pain.
The patient was started on androgen deprivation therapy, initially with degarelix and subsequently leuprolide shots and abiraterone with prednisone. PSA was 0.08 ng/mL after 3 months of androgen deprivation therapy. His back and hip pain slowly improved.
DISCUSSION
Prostate cancer is the most common cancer in men with one out of every nine men diagnosed in his lifetime.1 While most men initially present with localized, curable disease,1 4% present with metastatic disease, an incidence that has been increasing since 2004.2 Despite available treatments, metastatic prostate cancer has a poor prognosis, with an average overall survival of approximately 5 years.3
Prostate cancer can be challenging to diagnose. Men with prostate cancer are commonly asymptomatic. Rarely, patients may present with hematuria, bony pain caused by metastasis, or obstructive urinary symptoms like hesitancy or incomplete bladder emptying. Our patient presented with jaw pain, which was ultimately attributed to osteoblastic lesions of the skull. Additionally, his history of urinary frequency and incontinence may have been clues to his underlying diagnosis of prostate cancer.
Prostate cancer screening remains highly nuanced and relies on shared decision-making between patients and healthcare providers. Clinical practice guidelines for early detection of prostate cancer recommend individualized PSA-based serologic screening.4,5 Specifically, the United States Preventive Services Task Force recommends screening men aged 55 to 69 years who desire screening and understand the potential harms associated with a positive test result. These harms may include psychological distress and complications from prostate biopsy (eg, pain or infection) or prostate cancer treatment (eg, erectile, urinary, and/or bowel dysfunction).4-6 The decision to screen can be guided by individuals’ risk factors including African American race, family history, and older age.
While our patient elected not to undergo routine prostate cancer screening, a PSA level was obtained during his diagnostic evaluation and highlights the limitations of PSA-based screening. A PSA level ≤4.0 ng/mL has 21% sensitivity and 91% specificity for detecting prostate cancer.7 PSA levels above 4.0 ng/mL warrant repeat testing and, if persistently elevated, referral to urology for possible prostate biopsy. PSA levels often correlate with burden of disease, and patients with PSA levels >20 ng/mL are referred for CT imaging to evaluate for metastatic disease.8 PSA’s poor sensitivity was underscored in a study by Thompson et al who evaluated the incidence of prostate cancer in men participating in the Prostate Cancer Prevention Trial with PSA levels of <4 ng/mL.9 In this study, 15% of men diagnosed with prostate cancer never had a PSA level >4 ng/mL.9 While most of the cancers in this study were low grade and may have been clinically insignificant, 15% demonstrated histologic signs of at least intermediate-risk disease. Our patient’s PSA level of 2.2 ng/mL was below the threshold that triggers additional evaluation even though he had widely metastatic prostate cancer.
Our patient’s severe jaw and temple pain, weight loss, and progressive hip pain were concerning for GCA. This vasculitis of large- and medium-sized arteries predominantly affects older adults with greatest incidence among those 70 years of age and older.10 Symptoms occur because of cranial artery inflammation and may include headache, visual disturbance, erythema or tenderness of the temporal artery, and jaw claudication. Extracranial inflammation may affect the thoracic aorta and its branches and rarely the abdominal aorta and lower limb arteries. Pelvic girdle pain more typically results from associated PMR. Patients may also note systemic symptoms such as fever, weight loss, and fatigue.
Prompt diagnostic testing is important when considering GCA. Most patients with GCA have ESR levels greater than 40 mm/hr.11 ESR is a laboratory test that measures the vertical distance erythrocytes travel in a column of blood over 1 hour; in the setting of inflammation, cells form clumps and travel more quickly than individual cells, resulting in a higher value. While moderate elevations in ESR may occur without an identifiable cause, extreme ESR levels—those above 100 mm/hr, as observed in our patient—are highly suggestive of certain serious conditions, including infection, malignancy, and autoimmune disease such as GCA.12,13 Temporal artery biopsy is the gold standard test to diagnose GCA. However, because of noncontiguous inflammation of the temporal artery, biopsies may be falsely negative. Thus, sampling of the contralateral temporal artery may be warranted if suspicion remains high.
As was the case for our patient, PET/CT is not reliable for diagnosing prostate cancer. In contrast to other malignancies (eg, lymphoma, lung cancer), prostate cancer typically does not display increased glucose metabolism. Moreover, the close proximity of the bladder and prostate can interfere with imaging interpretation because the fluorodeoxyglucose (FDG) tracer is excreted in the urine.14 The reported sensitivity of PET/CT for the diagnosis of prostate cancer ranges from 17%-65%.15,16 In a small study of men with metastatic prostate cancer, only 18% of bony metastases were FDG avid, and there was no correlation between FDG avidity and PSA level.15 Notably, although PET/CT includes CT imaging, this CT is used to map anatomic landmarks and is not separately interpreted by the radiologist. Thus, even if evidence of prostate cancer was apparent on traditional CT, it may be overlooked on PET/CT.
Several important points regarding diagnostic testing are raised by this case. First, PSA-based screening for prostate cancer may be falsely negative, even in the setting of widely metastatic disease. Second, extreme ESR elevation is a marker for serious underlying disease and warrants a thorough diagnostic evaluation. Finally, PET/CT has limited diagnostic utility in evaluating metastatic prostate cancer because of the normal rates of glucose metabolism. Our patient initially presented with jaw pain, yet his progressive physical symptoms and laboratory abnormalities prompted an evaluation which ultimately revealed the jaw-dropping diagnosis of PSA-negative, metastatic prostate cancer.
KEY TEACHING POINTS
- ESR levels greater than 100 mm/hr are highly suggestive of certain serious conditions including infection, autoimmune disease, and malignancy.
- PSA-based screening for prostate cancer can result in false negative test results. In one study, 15% of men diagnosed with prostate cancer never had a PSA level greater than 4 ng/mL (ie, the level at which repeat laboratory testing and/or referral to urology for possible prostate biopsy is advisable).
- PET/CT has limited diagnostic utility in evaluating metastatic prostate cancer, because prostate cancer cells typically demonstrate normal glucose metabolism.
Disclosures
Drs Griauzde, Northway, Yentz, and Houchens have nothing to disclose. Dr Saint reports personal fees from ISMIE Mutual Insurance Company during the conduct of the study, as well as personal fees from Jvion and Doximity outside the submitted work.
A 73-year-old man presented to primary care for an annual examination. Four days prior, he noted right-sided sharp jaw pain such that he could not open his mouth nor chew solid food; it radiated from the right mandible to the ipsilateral temple. He also noted bilateral aching hip pain for several years that increased in severity in the prior 2 months. He reported an intentional weight loss of 9 kg over the past year, achieved through dietary modification. He denied fever, chills, and visual disturbance.
Acute onset of unilateral jaw pain that is worsened by chewing is a feature consistent with a temporomandibular disorder (TMD). TMD consists of musculoskeletal and neuromuscular conditions that affect the temporomandibular joints (TMJs), masticatory muscles, and associated tissues. Common symptoms of TMD include facial or ear pain, temporal headache, and TMJ dysfunction or discomfort. In addition to TMD, craniofacial pain has many possible etiologies such as dental pathology, neuralgias, sinus and otologic disorders, headache and migraine disorders, infections, rheumatologic conditions, and neoplasms.
Systemic etiologies for this patient’s symptoms are a consideration given his age and concomitant worsening of chronic hip pain. Rheumatologic conditions such as giant cell arteritis (GCA) and polymyalgia rheumatica (PMR) are more common in adults older than 50 years of age and cause headache, jaw claudication, and pelvic girdle pain. Rarely, hematologic malignancies (eg, lymphoma), solid tumor metastases (eg, breast cancer, melanoma), and primary tumors of the head and neck (eg, nasopharyngeal carcinoma) can involve the mandible, TMJ, or parotid gland and result in symptoms of TMD.
Medical history was notable for hypertension and type 2 diabetes mellitus complicated by peripheral neuropathy. He smoked one pack of cigarettes daily for 40 years but quit 15 years prior. He drank 4 ounces of vodka each night.
On examination, temperature was 36.5°C, heart rate 92 beats per minute, blood pressure 127/60 mmHg, respiratory rate 12 breaths per minute, oxygen saturation 98% on ambient air, and weight 118 kg. Extraocular movements were intact, pupils were equal and reactive to light and accommodation, and there were no visual field deficits. Nondilated funduscopic examination revealed normal blood vessels, optic disc, and optic cup-to-disc ratio. Dentition was good with pink gingiva. Bilateral temples were nontender. There was normal range of motion and strength in the shoulders, hips, and lower extremities with no tenderness over the trochanters. Patellar and ankle reflexes were present and symmetric bilaterally. He had no rashes or ecchymoses.
The history of smoking, especially with concomitant alcohol intake, is a risk factor for head and neck cancer, and these malignancies can lead to facial pain. While the normal oral cavity exam argues against localized oral and dental causes of the patient’s symptoms, direct fiberoptic endoscopy should be considered. The neck should be examined for lymphadenopathy. Normal vital signs point away from severe infection. The lack of findings in the head and musculoskeletal regions does not exclude systemic etiologies such as rheumatologic conditions or neoplasm. Complete blood cell count and markers of inflammation including erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) levels should be obtained. Hip and pelvic radiographs should be obtained to evaluate for hip osteoarthritis, fractures, or osseous lesions.
The appointment occurred during evening hours and the patient declined further evaluation until the following morning, at which time laboratory studies revealed normal serum levels of electrolytes, blood urea nitrogen, and creatinine. White blood cell (WBC) count was 6,800/mm3 with an immature granulocyte ratio of 1.8% (normal, 0.0-0.5%), hemoglobin 13.2 g/dL, and platelet count 163,000/mm3. ESR was 118 mm/hr (normal, 0-15 mm/hr) and CRP was 1.5 mg/dL (normal, 0-0.75 mg/dL). Radiographs of the hips and pelvis showed osteoarthritis of the bilateral hip joints and degenerative disc disease of the lower lumbar spine.
Granulocytosis may occur in response to infection, rheumatologic conditions, and hematologic malignancies such as chronic myelogenous leukemia. While infectious etiologies (eg, abscess, osteomyelitis) are the most common cause of an extremely elevated ESR level, this patient does not have other signs or symptoms of infection such as fever or leukocytosis. Therefore, other common causes for an extremely elevated ESR level should be considered, including malignancy (eg, multiple myeloma, lymphoma, metastatic solid tumor) and autoimmune conditions (eg, rheumatoid arthritis, vasculitis). While multiple myeloma is the most common malignant etiology for extremely elevated ESR, the patient lacks signs of this condition such as anemia, elevated creatinine, or osteolytic lesions on radiographic imaging. Osteoarthritis identified on the radiographs may contribute to the patient’s hip pain but would not explain the patient’s jaw pain, weight loss, granulocytosis, and elevated ESR. These findings, taken together with the patient’s age, are most suggestive of GCA with possible coexisting PMR. Temporal artery biopsy should be obtained as it is the gold standard test for diagnosing GCA.
The patient was contacted by telephone that same day with laboratory test results. During the call, he endorsed increased jaw and temple pain. He was advised to proceed to the emergency department (ED) for timely evaluation and treatment.
Because GCA was being considered, ophthalmology performed an ocular examination in the ED, which demonstrated no signs of optic nerve or retinal ischemia. Computed tomography (CT) scan of the head and neck with intravenous contrast revealed no abscess or soft tissue abnormalities. Right temporal artery biopsy was performed.
The normal ocular examination does not exclude GCA, and temporal artery biopsy is appropriate. The mainstay of treatment for GCA is high-dose systemic glucocorticoids, which should not be withheld while awaiting biopsy results since ophthalmic artery inflammation may occur and threaten vision.
While GCA remains the leading diagnosis, malignant etiologies warrant further consideration because they are a common cause of extreme ESR elevation, particularly among older patients. The patient’s cancer screening history should be reviewed. The normal CT scan of the head and neck reduces the likelihood of localized solid tumor etiologies; however, additional CT imaging of the chest, abdomen, and pelvis is warranted to evaluate for metastatic solid tumors or lymphoma.
A 10-day course of prednisone 60 mg daily was prescribed for empiric treatment of GCA. The patient was discharged home with follow-up scheduled in rheumatology and primary care clinics. Pain in the jaw and temple resolved within several days.
Two weeks later, he presented to the rheumatology clinic. He noted 1 week of lower right back pain described as dull, aching, radiating to the lateral right hip, and occurring when transitioning from sitting to standing. He had no leg numbness, weakness, or change in bowel habits. Bladder habits were also unchanged, although he reported chronic urinary frequency and occasional incontinence. He reported further weight loss, this time an unintentional loss of 9 kg. He noted frequent sweating but no fever.
He reported a normal colonoscopy within the prior 5 years. Because these records were not available for review, a fecal immunochemical test was obtained and negative for hemoglobin. He had previously declined prostate cancer screening.
The resolution of jaw and temple pain with prednisone supports the presumed diagnosis of GCA. Up to half of patients with GCA may also have PMR, which can cause aching and stiffness in the arms, hips, and lumbar region, and pain may be abrupt in onset. However, PMR-related pain would be expected to improve rather than develop or worsen in the setting of high-dose glucocorticoid use. Therefore, other causes of acute-onset back pain must be considered.
While localized musculoskeletal etiologies such as lumbar muscle strain, radiculopathy, and vertebral compression fracture are possible, co-occurrence of unintentional weight loss and diaphoresis with elevated inflammatory markers suggests a systemic etiology. A neoplastic process with bony metastasis is possible. The reportedly normal colonoscopy and the negative fecal immunochemical test make colorectal cancer less likely. Inflammatory conditions such as ankylosing spondylitis and rheumatoid arthritis are also possible. Ankylosing spondylitis usually presents at a much younger age, however, and axial skeletal involvement in rheumatoid arthritis often involves the cervical spine and is usually seen after longstanding disease. Additionally, the hallmark of inflammatory back pain is morning stiffness which the patient does not endorse. Nonetheless, additional laboratory testing should include antinuclear antibody, rheumatoid factor, and anti-cyclic citrullinated peptide (anti-CCP) antibody. Vertebral osteomyelitis remains on the differential diagnosis, and repeat WBC count and inflammatory markers should be assessed. Lumbosacral radiographs should be obtained to rule out fracture.
Physical examination in the rheumatology clinic revealed a temperature of 37.0°C, heart rate 100 beats per minute, blood pressure 146/72 mmHg, respiratory rate 12 breaths per minute, and oxygen saturation 98% on ambient air. Weight was 109 kg. He was pale and diaphoretic. There was diffuse tenderness to palpation of the right-sided lumbar paraspinal muscles. Straight leg raise was negative bilaterally. Patellar reflexes and gait were normal.
Blood chemistries, renal function, and aminotransferase levels were normal. WBC count was 7,100/mm3, hemoglobin 8.0 g/dL, mean corpuscular volume 88.9 fL, platelet count 128,000/mm3, ESR 66 mm/hr, CRP 0.57 mg/dL, alkaline phosphatase 438 IU/L (normal, 30-130 IU/L), and thyroid-stimulating hormone 0.925 mU/L (normal, 0.34-5.60 mU/L). Testing for antinuclear antibodies, rheumatoid factor, and anti-CCP antibody was unremarkable. Prostate-specific antigen (PSA) level was 2.2 ng/mL (normal, 0-4 ng/mL). Urinalysis was unremarkable. Antibodies to hepatitis C and Treponema pallidum were negative. Interferon gamma release assay was negative.
Findings of new onset anemia and thrombocytopenia, in combination with elevated ESR and alkaline phosphatase level, are concerning for disseminated intravascular coagulation (DIC) and microangiopathic hemolytic anemia (MAHA), bone marrow infiltration of a metastatic neoplasm, or ineffective hematopoiesis caused by myelodysplastic syndromes or myelofibrosis.
Laboratory evaluation should include iron studies, lactate dehydrogenase (LDH), haptoglobin, fibrinogen, D-dimer, reticulocyte count, and peripheral blood smear to assess for hemolysis and erythrocyte morphology. Advanced imaging with lumbosacral magnetic resonance imaging (MRI) should be obtained to evaluate for focal etiologies of back pain such as disc herniation, abscess, marrow infiltration, and infarction.
Additional laboratory studies revealed a gamma-glutamyl transferase level of 49 IU/L (normal, 8-56 IU/L), LDH 288 IU/L (normal, 98-192 IU/L), haptoglobin 495 mg/dL (normal, 32-240 mg/dL), fibrinogen >700 mg/dL (normal, 225-550 mg/dL), D-dimer 693 ng/mL (normal, 200-250 ng/mL), serum iron 57 mcg/dL (normal, 33-150 mcg/dL), total iron binding capacity 286 mcg/dL (normal, 250-450 mcg/dL), ferritin 1,012 ng/mL (normal, 17.9-464 ng/mL), and reticulocyte count 2.9% (normal, 0.5-2.5%). Coagulation studies and serum protein electrophoresis were normal. Erythropoietin level was 109 mIU/mL (normal, 4.0-20.0 mIU/mL). Peripheral blood smear demonstrated moderate anemia with 8% nucleated erythrocytes per white blood cell (normal, 0%) and no circulating blasts.
MRI of the thoracolumbar spine and pelvis revealed diffusely abnormal bone marrow signal with multiple superimposed focal and poorly defined enhancing lesions along the lumbar spine marrow, sacrum, and bilateral iliac bones (Figure 1). Positron emission tomography/computed tomography (PET/CT) scan showed no scintigraphic evidence of metabolically active neoplastic, paraneoplastic, or inflammatory disorder.
The elevated haptoglobin, normal coagulation studies, and absence of fragmented erythrocytes on peripheral smear exclude an intravascular hemolytic process. The patient’s lower than expected reticulocyte count for the degree of anemia, elevated erythropoietin, and nucleated erythrocytes constitute a pattern that can be seen with bone marrow infiltration. There are no circulating blasts, making leukemia less likely. A solid organ tumor with bone metastases may cause enhancing lesions on MRI since this form of imaging is more sensitive than radiography for detecting skeletal malignancies. The negative PET/CT, however, does not reveal a primary tumor. Myelofibrosis is an infiltrative myeloproliferative disorder associated with nonspecific laboratory abnormalities, bone pain, weight loss, and night sweats that could cause diffuse MRI bone marrow signal alterations with normal PET/CT findings. However, myelofibrosis would not typically cause a significantly elevated ESR, and thus would be an unlikely cause for this patient’s presentation.
Given the constellation of symptoms, hematologic abnormalities, and bone marrow infiltration on imaging, hematology should be consulted to perform a bone marrow biopsy to assist with definitive diagnosis.
Bone marrow biopsy demonstrated metastatic adenocarcinoma consistent with prostatic origin (Figure 2). Bone scan demonstrated widespread osteoblastic metastases, which included the skull and temporal regions. These lesions were thought to be the cause of the patient’s original presenting symptom of jaw pain.
The patient was started on androgen deprivation therapy, initially with degarelix and subsequently leuprolide shots and abiraterone with prednisone. PSA was 0.08 ng/mL after 3 months of androgen deprivation therapy. His back and hip pain slowly improved.
DISCUSSION
Prostate cancer is the most common cancer in men with one out of every nine men diagnosed in his lifetime.1 While most men initially present with localized, curable disease,1 4% present with metastatic disease, an incidence that has been increasing since 2004.2 Despite available treatments, metastatic prostate cancer has a poor prognosis, with an average overall survival of approximately 5 years.3
Prostate cancer can be challenging to diagnose. Men with prostate cancer are commonly asymptomatic. Rarely, patients may present with hematuria, bony pain caused by metastasis, or obstructive urinary symptoms like hesitancy or incomplete bladder emptying. Our patient presented with jaw pain, which was ultimately attributed to osteoblastic lesions of the skull. Additionally, his history of urinary frequency and incontinence may have been clues to his underlying diagnosis of prostate cancer.
Prostate cancer screening remains highly nuanced and relies on shared decision-making between patients and healthcare providers. Clinical practice guidelines for early detection of prostate cancer recommend individualized PSA-based serologic screening.4,5 Specifically, the United States Preventive Services Task Force recommends screening men aged 55 to 69 years who desire screening and understand the potential harms associated with a positive test result. These harms may include psychological distress and complications from prostate biopsy (eg, pain or infection) or prostate cancer treatment (eg, erectile, urinary, and/or bowel dysfunction).4-6 The decision to screen can be guided by individuals’ risk factors including African American race, family history, and older age.
While our patient elected not to undergo routine prostate cancer screening, a PSA level was obtained during his diagnostic evaluation and highlights the limitations of PSA-based screening. A PSA level ≤4.0 ng/mL has 21% sensitivity and 91% specificity for detecting prostate cancer.7 PSA levels above 4.0 ng/mL warrant repeat testing and, if persistently elevated, referral to urology for possible prostate biopsy. PSA levels often correlate with burden of disease, and patients with PSA levels >20 ng/mL are referred for CT imaging to evaluate for metastatic disease.8 PSA’s poor sensitivity was underscored in a study by Thompson et al who evaluated the incidence of prostate cancer in men participating in the Prostate Cancer Prevention Trial with PSA levels of <4 ng/mL.9 In this study, 15% of men diagnosed with prostate cancer never had a PSA level >4 ng/mL.9 While most of the cancers in this study were low grade and may have been clinically insignificant, 15% demonstrated histologic signs of at least intermediate-risk disease. Our patient’s PSA level of 2.2 ng/mL was below the threshold that triggers additional evaluation even though he had widely metastatic prostate cancer.
Our patient’s severe jaw and temple pain, weight loss, and progressive hip pain were concerning for GCA. This vasculitis of large- and medium-sized arteries predominantly affects older adults with greatest incidence among those 70 years of age and older.10 Symptoms occur because of cranial artery inflammation and may include headache, visual disturbance, erythema or tenderness of the temporal artery, and jaw claudication. Extracranial inflammation may affect the thoracic aorta and its branches and rarely the abdominal aorta and lower limb arteries. Pelvic girdle pain more typically results from associated PMR. Patients may also note systemic symptoms such as fever, weight loss, and fatigue.
Prompt diagnostic testing is important when considering GCA. Most patients with GCA have ESR levels greater than 40 mm/hr.11 ESR is a laboratory test that measures the vertical distance erythrocytes travel in a column of blood over 1 hour; in the setting of inflammation, cells form clumps and travel more quickly than individual cells, resulting in a higher value. While moderate elevations in ESR may occur without an identifiable cause, extreme ESR levels—those above 100 mm/hr, as observed in our patient—are highly suggestive of certain serious conditions, including infection, malignancy, and autoimmune disease such as GCA.12,13 Temporal artery biopsy is the gold standard test to diagnose GCA. However, because of noncontiguous inflammation of the temporal artery, biopsies may be falsely negative. Thus, sampling of the contralateral temporal artery may be warranted if suspicion remains high.
As was the case for our patient, PET/CT is not reliable for diagnosing prostate cancer. In contrast to other malignancies (eg, lymphoma, lung cancer), prostate cancer typically does not display increased glucose metabolism. Moreover, the close proximity of the bladder and prostate can interfere with imaging interpretation because the fluorodeoxyglucose (FDG) tracer is excreted in the urine.14 The reported sensitivity of PET/CT for the diagnosis of prostate cancer ranges from 17%-65%.15,16 In a small study of men with metastatic prostate cancer, only 18% of bony metastases were FDG avid, and there was no correlation between FDG avidity and PSA level.15 Notably, although PET/CT includes CT imaging, this CT is used to map anatomic landmarks and is not separately interpreted by the radiologist. Thus, even if evidence of prostate cancer was apparent on traditional CT, it may be overlooked on PET/CT.
Several important points regarding diagnostic testing are raised by this case. First, PSA-based screening for prostate cancer may be falsely negative, even in the setting of widely metastatic disease. Second, extreme ESR elevation is a marker for serious underlying disease and warrants a thorough diagnostic evaluation. Finally, PET/CT has limited diagnostic utility in evaluating metastatic prostate cancer because of the normal rates of glucose metabolism. Our patient initially presented with jaw pain, yet his progressive physical symptoms and laboratory abnormalities prompted an evaluation which ultimately revealed the jaw-dropping diagnosis of PSA-negative, metastatic prostate cancer.
KEY TEACHING POINTS
- ESR levels greater than 100 mm/hr are highly suggestive of certain serious conditions including infection, autoimmune disease, and malignancy.
- PSA-based screening for prostate cancer can result in false negative test results. In one study, 15% of men diagnosed with prostate cancer never had a PSA level greater than 4 ng/mL (ie, the level at which repeat laboratory testing and/or referral to urology for possible prostate biopsy is advisable).
- PET/CT has limited diagnostic utility in evaluating metastatic prostate cancer, because prostate cancer cells typically demonstrate normal glucose metabolism.
Disclosures
Drs Griauzde, Northway, Yentz, and Houchens have nothing to disclose. Dr Saint reports personal fees from ISMIE Mutual Insurance Company during the conduct of the study, as well as personal fees from Jvion and Doximity outside the submitted work.
1. Prostate Cancer - Cancer Stat Facts. SEER. https://seer.cancer.gov/statfacts/html/prost.html. Accessed October 23, 2018.
2. Li J, Siegel DA, King JB. Stage-specific incidence rates and trends of prostate cancer by age, race, and ethnicity, United States, 2004-2014. Ann Epidemiol. 2018;28(5):328-330. https://doi.org/10.1016/j.annepidem.2018.03.001.
3. Sweeney CJ, Chen YH, Carducci M, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med. 2015;373(8):737-746. https://doi.org/10.1056/NEJMoa1503747.
4. US Preventive Services Task Force. Final Recommendation Statement: Prostate Cancer: Screening. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/prostate-cancer-screening1. Accessed August 8, 2018.
5. American Urological Association. http://www.auanet.org/guidelines/prostate-cancer-early-detection. Accessed August 8, 2018.
6. American Cancer Society. American Cancer Society Recommendations for Prostate Cancer Early Detection. https://www.cancer.org/cancer/prostate-cancer/early-detection/acs-recommendations.html. Accessed August 8, 2018.
7. Wolf AM, Wender RC, Etzioni RB, et al. American Cancer Society guideline for the early detection of prostate cancer: update 2010. CA Cancer J Clin. 2010;60(2):70-98. https://doi.org/10.3322/caac.20066.
8. Mohler JL, Lee RJ, Antonarakis ES, Higano CS, Richey S. NCCN Guidelines Index Table of Contents. Prostate Cancer. 2018:151.
9. Thompson IM, Pauler DK, Goodman PJ, et al. Prevalence of prostate cancer among men with a prostate-specific antigen level ≤4.0 ng per milliliter. N Engl J Med. 2004;350(22):2239-2246. https://doi.org/10.1056/NEJMoa031918.
10. Pioro MH. Primary care vasculitis: Polymyalgia rheumatica and giant cell arteritis. Prim Care. 2018;45(2):305-323. https://doi.org/10.1016/j.pop.2018.02.007.
11. Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurrence in a population-based study. Arthritis Rheum. 2001;45(2):140-145. https://doi.org/10.1002/1529-0131(200104)45:2<140::AID-ANR166>3.0.CO;2-2
12. Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60(5):1443-1450.
13. Daniels LM, Tosh PK, Fiala JA, Schleck CD, Mandrekar JN, Beckman TJ. Extremely elevated erythrocyte sedimentation rates: Associations with patients’ diagnoses, demographic dharacteristics, and comorbidities. Mayo Clin Proc. 2017;92(11):1636-1643. https://doi.org/10.1016/j.mayocp.2017.07.018.
14. Powles T, Murray I, Brock C, Oliver T, Avril N. Molecular positron emission tomography and PET/CT imaging in urological malignancies. Eur Urol. 2007;51(6):1511-1521. http://doi.org/10.1016/j.eururo.2007.01.061.
15. Yeh SDJ, Imbriaco M, Larson SM, et al. Detection of bony metastases of androgen-independent prostate cancer by PET-FDG. Nucl Med Biol. 1996;23(6):693-697. https://doi.org/10.1016/0969-8051(96)00044-3.
16. Perera M, Papa N, Christidis D, et al. Sensitivity, specificity, and predictors of positive 68ga-prostate-specific membrane antigen positron emission tomography in advanced prostate cancer: a systematic review and meta-analysis. Eur Urol. 2016;70(6):926-937. https://doi.org/10.1016/j.eururo.2016.06.021.
1. Prostate Cancer - Cancer Stat Facts. SEER. https://seer.cancer.gov/statfacts/html/prost.html. Accessed October 23, 2018.
2. Li J, Siegel DA, King JB. Stage-specific incidence rates and trends of prostate cancer by age, race, and ethnicity, United States, 2004-2014. Ann Epidemiol. 2018;28(5):328-330. https://doi.org/10.1016/j.annepidem.2018.03.001.
3. Sweeney CJ, Chen YH, Carducci M, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med. 2015;373(8):737-746. https://doi.org/10.1056/NEJMoa1503747.
4. US Preventive Services Task Force. Final Recommendation Statement: Prostate Cancer: Screening. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/prostate-cancer-screening1. Accessed August 8, 2018.
5. American Urological Association. http://www.auanet.org/guidelines/prostate-cancer-early-detection. Accessed August 8, 2018.
6. American Cancer Society. American Cancer Society Recommendations for Prostate Cancer Early Detection. https://www.cancer.org/cancer/prostate-cancer/early-detection/acs-recommendations.html. Accessed August 8, 2018.
7. Wolf AM, Wender RC, Etzioni RB, et al. American Cancer Society guideline for the early detection of prostate cancer: update 2010. CA Cancer J Clin. 2010;60(2):70-98. https://doi.org/10.3322/caac.20066.
8. Mohler JL, Lee RJ, Antonarakis ES, Higano CS, Richey S. NCCN Guidelines Index Table of Contents. Prostate Cancer. 2018:151.
9. Thompson IM, Pauler DK, Goodman PJ, et al. Prevalence of prostate cancer among men with a prostate-specific antigen level ≤4.0 ng per milliliter. N Engl J Med. 2004;350(22):2239-2246. https://doi.org/10.1056/NEJMoa031918.
10. Pioro MH. Primary care vasculitis: Polymyalgia rheumatica and giant cell arteritis. Prim Care. 2018;45(2):305-323. https://doi.org/10.1016/j.pop.2018.02.007.
11. Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurrence in a population-based study. Arthritis Rheum. 2001;45(2):140-145. https://doi.org/10.1002/1529-0131(200104)45:2<140::AID-ANR166>3.0.CO;2-2
12. Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60(5):1443-1450.
13. Daniels LM, Tosh PK, Fiala JA, Schleck CD, Mandrekar JN, Beckman TJ. Extremely elevated erythrocyte sedimentation rates: Associations with patients’ diagnoses, demographic dharacteristics, and comorbidities. Mayo Clin Proc. 2017;92(11):1636-1643. https://doi.org/10.1016/j.mayocp.2017.07.018.
14. Powles T, Murray I, Brock C, Oliver T, Avril N. Molecular positron emission tomography and PET/CT imaging in urological malignancies. Eur Urol. 2007;51(6):1511-1521. http://doi.org/10.1016/j.eururo.2007.01.061.
15. Yeh SDJ, Imbriaco M, Larson SM, et al. Detection of bony metastases of androgen-independent prostate cancer by PET-FDG. Nucl Med Biol. 1996;23(6):693-697. https://doi.org/10.1016/0969-8051(96)00044-3.
16. Perera M, Papa N, Christidis D, et al. Sensitivity, specificity, and predictors of positive 68ga-prostate-specific membrane antigen positron emission tomography in advanced prostate cancer: a systematic review and meta-analysis. Eur Urol. 2016;70(6):926-937. https://doi.org/10.1016/j.eururo.2016.06.021.
© 2020 Society of Hospital Medicine
A Traumatic Traveler
A 19-year-old man with Duchenne Muscular Dystrophy (DMD) presented to the Emergency Department (ED) for left knee pain after ejection from his motorized wheelchair at a low velocity. In the ED, he developed increasing respiratory distress.
When addressing a new problem in a patient with a chronic condition, it is crucial to first understand the chronic condition and then consider whether the presenting symptoms relate to that condition or stem from an unrelated inciting event.
Patients with DMD are at risk of pulmonary complications relating to their underlying disease. For instance, dysphagia and ineffective cough can predispose them to recurrent aspiration pneumonitis and/or pneumonia, whereas decreased lung compliance (from scoliosis, atelectasis, and/or pulmonary fibrosis) and respiratory muscle weakness can progress to ventilatory failure. In addition, patients with DMD are at risk for pulmonary thromboembolism in the setting of immobility. Patients with DMD may also develop congestive heart failure resulting from myocardial fibrosis and nonischemic cardiomyopathy.
The ejection from his wheelchair signals potential trauma-associated conditions that could explain his respiratory distress. Respiratory complications of blunt thoracic trauma include pulmonary contusion, pneumothorax, flail chest (resulting from fractured ribs), and acute respiratory distress syndrome (ARDS). Lower extremity injury can result in venous thrombosis and pulmonary thromboembolism. While classically associated with long bone fractures, fat embolism syndrome (FES) may rarely occur with rib fractures and soft-tissue trauma. Respiratory compromise may also result from cervical spinal cord injury or severe anemia from trauma-associated hemorrhage.
Additional past medical history included growth hormone deficiency, migraine headaches, osteoporosis secondary to chronic steroid use, cardiac fibrosis of the inferolateral wall and septum with a baseline left ventricular ejection fraction of 65%, and atrial fibrillation. His medications included calcium carbonate, vitamin D, omeprazole, lisinopril, metoprolol, prednisone, escitalopram, and testosterone. Physical examination revealed an ill-appearing obese man in respiratory distress. Temperature was 37.3°C, heart rate was 102 beats per minute (bpm), blood pressure was 110/74 mm Hg. His oxygen saturation was 93% with a respiratory rate of 25 breaths per minute while breathing ambient air. His lung sounds were clear, and his heart was without murmur. The left knee was diffusely tender to palpation without specific point tenderness. Strength was 2/5 with flexion and extension at the bilateral knees and hips and 3/5 flexion and extension at the bilateral elbows. He reported this level of weakness was his baseline. Radiographs revealed a minimally displaced Salter Harris II fracture (fracture line through the metaphysis and growth plate) of the left distal femur. His fracture was splinted early in his ED course. During his ED evaluation, the patient had acute worsening of tachycardia to 130 bpm, increased respiratory rate of 34 breaths per minute, and hypoxemia with an oxygen saturation of 83% on ambient air. He was placed on 3 L/min of oxygen via nasal cannula with improvement in his oxygen saturation to 90%. A chest radiograph was unremarkable, without evidence of pneumothorax, effusion, or pneumonia. The patient was admitted to the hospital.
The acute onset of tachypnea, tachycardia, and hypoxia, accompanied by a clear lung exam and normal chest radiograph, increases the likelihood of a pulmonary embolism. Obesity, testosterone therapy, and trauma increase his susceptibility to venous thromboembolism, while a distal femur fracture increases his risk for FES. Acute pulmonary aspiration often presents with initially absent or subtle radiographic findings. An arterial blood gas analysis would determine the presence and extent of an alveolar-arterial (A-a) gradient; a normal A-a gradient is seen in hypoventilation, while an elevated A-a gradient is seen in conditions affecting gas exchange, including pulmonary emboli and alveolar filling processes. His hypoxemia only partially corrects with supplemental oxygen, raising the possibility of capillary or anatomic shunting. Capillary shunting may occur with atelectasis, aspiration/pneumonia and pulmonary edema, whereas anatomic shunting can be intra-cardiac (eg, patent foramen ovale or septic defect) or intrapulmonary (eg, arteriovenous malformations). Patients with pulmonary emboli may also develop right-to-left shunting because of increased pulmonary vascular resistance, although hypoxemia with pulmonary emboli largely relates to ventilation/perfusion mismatch and decreased level of mixed venous blood oxygen (PvO2).
This patient’s complex medical history warrants a broadened differential with consideration of his cardiac history, including myocardial fibrosis and arrhythmia, and the impact of exposure to steroids on his immune and musculoskeletal systems. He has a history of atrial fibrillation, and an electrocardiogram is warranted to determine the underlying rhythm. Prolonged periods of rapid ventricular response may lead to tachycardia-induced cardiomyopathy. Myocardial fibrosis may progress despite use of angiotensin-converting enzyme inhibitors and is associated with systolic and/or diastolic dysfunction, although neither the examination findings provided nor the chest radiograph are suggestive of decompensated heart failure. Chronic exposure to corticosteroids (used in DMD to improve muscle strength and function) may predispose to numerous infectious and metabolic complications. Up to 10%-15% of patients with Pneumocystis jirovecii pneumonia may present with a normal chest radiograph. Acute adrenal insufficiency can present with tachycardia, weakness, and respiratory distress, so recent prednisone dose changes or interruptions should be assessed.
The patient’s respiratory status worsened. In light of his complex medical history, he was transferred to a children’s hospital for a higher level of care with a presumptive diagnosis of aspiration pneumonia. Upon reassessment at the new facility, the patient reported an ongoing and severe headache since his initial injury. NSAIDs had been given prior to transfer. His exam continued to be significant for tachycardia, tachypnea, and hypoxemia. His cardiac and lung examinations were otherwise normal. A comprehensive metabolic panel, procalcitonin, complete blood count with differential, and lactate were normal; his C-reactive protein (CRP) was 46.8 mg/dL (Normal <8 mg/dL). A computed tomography (CT) angiogram of the chest revealed small multifocal nodular ground-glass opacities, especially in the lower lobes, concerning for microatelectasis, multifocal pneumonia, or aspiration pneumonia. After consultation with pediatric pulmonology consultants, antimicrobials were held during the initial phase of work-up.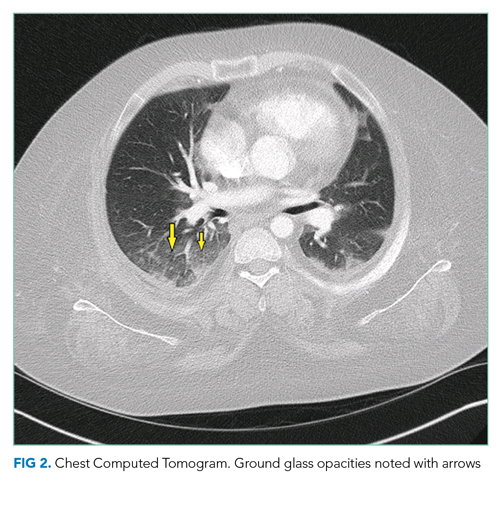
His headache may reflect a migraine, although further characterization and assessment for the presence and extent of head or neck trauma is warranted. Headache following trauma warrants consideration of cerebral contusion, diffuse axonal injury, intracranial hemorrhage, and carotid or vertebral artery dissection. Screening for concussion should also be performed. Hypoxemia may increase cerebral blood flow and raise intracranial pressure, resulting in headache.
CRP elevation is nonspecific and signals the presence of focal or systemic inflammation and is often elevated to a milder extent in obese patients with DMD. While normal procalcitonin argues against bacterial pneumonia, the precise level can be informative, and serial procalcitonin values may be more helpful than a single value. Although antecedent respiratory symptoms were not mentioned, viral or fungal pneumonia can present insidiously. An occult malignancy may be incidentally discovered when patients present for unrelated issues, although this and other sources of elevated CRP (eg, exacerbation of an autoimmune disease or drug reaction) remain less likely given the acuity of his presentation. Acute pulmonary embolism may be associated with a systemic inflammatory response and elevation in CRP.
In addition to the radiographic differential diagnosis already presented, the appearance of multifocal opacifications with hypoxemia raises the possibility of pulmonary infarcts or noncardiogenic pulmonary edema.
On hospital day 2, the patient continued to complain of “the worst headache of his life” as well as blurry vision and seeing “dark spots.” His headache did not improve with NSAIDs. A noncontrast CT scan of the head was normal. Neurology was consulted. Given his symptoms, history of migraines, stable neurological examination, and normal head CT, he was diagnosed with migraines and administered fluids, prochlorperazine, diphenhydramine, ondansetron, and NSAIDs. His headache continued and he continued to require supplemental oxygen.
The combination of hypoxemia, severe headache, and vision changes remains consistent with systemic emboli caused by thromboembolism or fat embolism. Headache assessment must also involve screening for “red flags,” which include sudden onset, antecedent head trauma, systemic illness (eg, fever or meningismus), focal neurologic findings, papilledema, changes with position or Valsalva, and immunosuppression. Although primary headache syndromes (eg, migraines or tension and cluster headache) may be triggered in the setting of trauma and systemic illness, “the worst headache of my life” is a concerning symptom that warrants urgent attention. While this invokes the possibility of a subarachnoid hemorrhage (SAH), headache severity is nonspecific, and rapid onset (ie, thunderclap headache) would be more suggestive. After 6 hours of symptoms, the sensitivity of head CT for detecting SAH declines, and lumbar puncture would be warranted to evaluate for xanthochromia.
His blurry vision and dark spots require testing of visual acuity and visual fields, as well as fundoscopic examination to assess for embolic phenomena or papilledema. Migraine is classically associated with “positive” or scintillating scotomata, although dark spots may occur. The presence of horizontal diplopia would indicate a cranial nerve VI palsy, which can occur with increased intracranial pressure. Visual-field cuts may also present as blurry vision, and monocular vs binocular deficits signal whether the issue is anterior or involving/posterior to the optic chiasm, respectively. Magnetic resonance imaging (MRI) may reveal the presence or sequelae of cerebral emboli (eg, fat emboli), including vasogenic edema.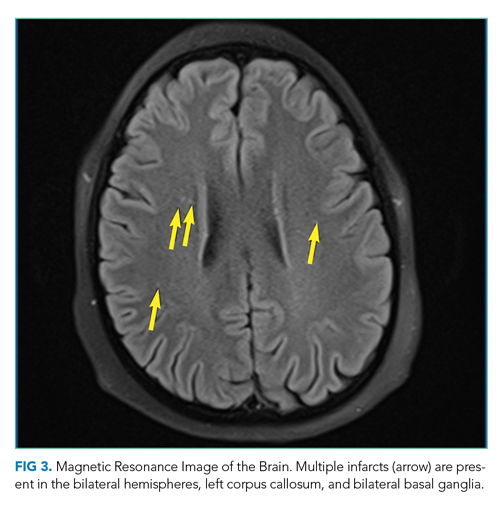
Dilated fundus examination revealed Purtscher retinopathy: bilateral cotton-wool spots and larger areas of retinal whitening (Purtscher flecken).
Typical findings of Purtscher retinopathy include Purtscher flecken, cotton-wool spots, retinal hemorrhage, and optic disc edema. Purtscher retinopathy is classically associated with severe head trauma. Without associated head trauma, the term “Purtscher-like retinopathy” is used. Conditions that can cause Purtscher-like retinopathy include pancreatitis, vasculitis, microangiopathy, chronic renal failure, and systemic embolization. The most likely source of systemic embolization remains fat emboli stemming from his femur fracture. Treatment of FES is largely supportive.
The possibility of fat emboli had been repeatedly raised by the patient’s mother since admission. While providers had considered this a possibility, it was discounted early on because of the minor nature of the patient’s orthopedic trauma, the lack of clear radiographic evidence for pulmonary emboli on chest CT, and the normal head CT. The findings on the ophthalmologist’s fundoscopic examination led the primary team to reconsider FES, along with thromboemboli and pancreatitis. Lipase was normal. MRI of the brain with contrast revealed >20 microinfarcts in the bilateral hemispheres, left corpus callosum, and bilateral basal ganglia. The CT angiogram of the chest was rereviewed; the pediatric radiologists suggested that microinfarcts could explain the patchy small ground glass opacities seen in the lungs. A transthoracic echocardiogram and electrocardiogram were normal. The diagnosis of FES was made, and the patient was started on aspirin and enoxaparin prophylaxis. His headache and respiratory status improved, and he was discharged home with close follow-up.
DISCUSSION
FES is a rare complication associated with long bone fractures and orthopedic manipulation.1,2 The exact mechanism of fat emboli production is unknown, but two theories prevail. The mechanical theory states that an outside mechanical source causes bone marrow contents or adipose tissue contents to be dislodged into the circulation where they travel through the venous circulation to become embedded in the lungs.1,2 These fragments may also migrate to the arterial circulation, through a patent foramen ovale or intrapulmonary shunts, leading to end organ damage.1,2 The biochemical theory suggests that fat emboli in the venous circulation precipitate an inflammatory and prothrombotic cascade that triggers fibrin production, platelet aggregation, and release of free fatty acids into the circulation, predisposing patients to develop multifocal systemic emboli.1
Although the classic triad in FES includes respiratory symptoms, rash, and CNS symptoms, all three findings are only present in 1%-29% of cases.1,2 Respiratory abnormalities, ranging from tachypnea and dyspnea to ARDS and hypoxic respiratory failure, occur in up to 75% of patients with FES.1 Central nervous system (CNS) complications, including headache, confusion, coma, seizures, and death caused by cerebral ischemia, occur in up to 86% of patients.1,2 Petechiae may occur in 20%-60% of patients and are usually located on nondependent regions of the body such as the head, neck, and chest.
Diagnosis of FES is largely clinical and requires a high index of suspicion and elimination of other conditions, including pulmonary thromboembolism, diffuse intravascular coagulation, and sepsis. The CNS complications must be differentiated from CNS infection, stroke, migraine, benign intracranial hypertension, and intracranial hemorrhage. There is no gold standard test for diagnosis. The Gurd and Wilson criteria, modified Gurd criteria, and Schonfeld’s criteria (Table) are commonly used but have not been clinically validated.1,3-5 These use a combination of clinical signs of respiratory distress, neurological symptoms, petechial rash, and various other diagnostic factors. When patients have risk factors, such as trauma, surgery, or predisposing conditions (eg, mobility-limiting neuromuscular disorders) and signs and symptoms potentially consistent with FES, the diagnostic evaluation should include arterial blood gas analysis, complete blood count, chest radiographs, and coagulation studies. Ophthalmological exam to evaluate for cotton-wool spots, brain MRI to detect cerebral emboli, and CT pulmonary angiogram to assess for pulmonary infarcts may help to eliminate other diagnoses and/or confirm FES.
Diagnosis of uncommon conditions that present with nonspecific symptoms, like FES, can be challenging because the symptoms may overlap with many other possible diagnoses. This challenge is further exacerbated in patients with significant medical complexity, as with the patient discussed here. Specifically, this patient had multiple plausible explanations for CNS symptoms and respiratory symptoms. It was ultimately the visual symptoms that began to link his symptoms together into a unifying syndrome and the ophthalmologic examination that prompted confirmatory imaging. It is important to continually revisit and revise the differential diagnosis in patients with medical complexity and avoid the competing temptations to abandon the search for a unifying diagnosis and attribute all symptoms to a patient’s underlying condition.
Treatment of FES is largely supportive with close monitoring of neurological status and providing supplemental oxygen as needed. Corticosteroids have been suggested to help prevent FES in patients with long bone fractures, but there is no evidence to suggest they are helpful once FES is diagnosed.2 There is conflicting evidence for the efficacy of heparin or low-molecular-weight heparin as treatment in FES.2,6 After discussions with consulting physician teams, the patient, and his family, enoxaparin and aspirin were started for this patient in light of his tenuous condition in order to decrease the risk of further embolic complications.
Patients with DMD and other neuromuscular disorders likely have a greater propensity to develop FES even after minor trauma.1,6 This is believed to be caused by patients becoming nonambulatory early in life and receiving substantial corticosteroid therapy, which can lead to osteopenia and fatty replacement of the bone marrow.1,6 This population is also often obese by the second or third decade of life, which contributes to their already increased propensity to fall.1,6
To our knowledge, this patient is 1 of 18 reported cases of FES after trauma in DMD patients. Two-thirds of these cases occurred when an unrestrained patient fell from their wheelchair. The other cases occurred while walking, during physical therapy, and during assisted transfers.6-12 In these cases, FES had a guarded prognosis, with 7 of the 18 patients dying and 1 of the patients remaining in a persistent vegetative state.8,9 While caution is warranted in generalizing these findings, given the small number of reported cases and likely publication bias,education of caregivers and patients on use of restraints and safe transfers is paramount to limit the risk of trauma.
TEACHING POINTS
- FES is a rare condition that most commonly manifests with respiratory, neurological, and cutaneous findings.
- Patients with Duchenne’s Muscular Dystrophy are likely at increased risk for FES even with minor trauma; this makes wheelchair restraints and safe transfers fundamental.
- Patients with medical complexity and their caregivers are key members of the diagnostic team.
1. Fukumoto LE, Fukumoto KD. Fat embolism syndrome. Nurs Clin North Am. 2018;53(3):335-347. https://doi.org/10.1016/j.cnur.2018.04.003.
2. Scarpino M, Lanzo G, Lolli F, Grippo A. From the diagnosis to the therapeutic management: Cerebral fat embolism, a clinical challenge. Int J Gen Med. 2019;2019(12):39-48. https://doi.org/10.2147/IJGM.S177407.
A 19-year-old man with Duchenne Muscular Dystrophy (DMD) presented to the Emergency Department (ED) for left knee pain after ejection from his motorized wheelchair at a low velocity. In the ED, he developed increasing respiratory distress.
When addressing a new problem in a patient with a chronic condition, it is crucial to first understand the chronic condition and then consider whether the presenting symptoms relate to that condition or stem from an unrelated inciting event.
Patients with DMD are at risk of pulmonary complications relating to their underlying disease. For instance, dysphagia and ineffective cough can predispose them to recurrent aspiration pneumonitis and/or pneumonia, whereas decreased lung compliance (from scoliosis, atelectasis, and/or pulmonary fibrosis) and respiratory muscle weakness can progress to ventilatory failure. In addition, patients with DMD are at risk for pulmonary thromboembolism in the setting of immobility. Patients with DMD may also develop congestive heart failure resulting from myocardial fibrosis and nonischemic cardiomyopathy.
The ejection from his wheelchair signals potential trauma-associated conditions that could explain his respiratory distress. Respiratory complications of blunt thoracic trauma include pulmonary contusion, pneumothorax, flail chest (resulting from fractured ribs), and acute respiratory distress syndrome (ARDS). Lower extremity injury can result in venous thrombosis and pulmonary thromboembolism. While classically associated with long bone fractures, fat embolism syndrome (FES) may rarely occur with rib fractures and soft-tissue trauma. Respiratory compromise may also result from cervical spinal cord injury or severe anemia from trauma-associated hemorrhage.
Additional past medical history included growth hormone deficiency, migraine headaches, osteoporosis secondary to chronic steroid use, cardiac fibrosis of the inferolateral wall and septum with a baseline left ventricular ejection fraction of 65%, and atrial fibrillation. His medications included calcium carbonate, vitamin D, omeprazole, lisinopril, metoprolol, prednisone, escitalopram, and testosterone. Physical examination revealed an ill-appearing obese man in respiratory distress. Temperature was 37.3°C, heart rate was 102 beats per minute (bpm), blood pressure was 110/74 mm Hg. His oxygen saturation was 93% with a respiratory rate of 25 breaths per minute while breathing ambient air. His lung sounds were clear, and his heart was without murmur. The left knee was diffusely tender to palpation without specific point tenderness. Strength was 2/5 with flexion and extension at the bilateral knees and hips and 3/5 flexion and extension at the bilateral elbows. He reported this level of weakness was his baseline. Radiographs revealed a minimally displaced Salter Harris II fracture (fracture line through the metaphysis and growth plate) of the left distal femur. His fracture was splinted early in his ED course. During his ED evaluation, the patient had acute worsening of tachycardia to 130 bpm, increased respiratory rate of 34 breaths per minute, and hypoxemia with an oxygen saturation of 83% on ambient air. He was placed on 3 L/min of oxygen via nasal cannula with improvement in his oxygen saturation to 90%. A chest radiograph was unremarkable, without evidence of pneumothorax, effusion, or pneumonia. The patient was admitted to the hospital.
The acute onset of tachypnea, tachycardia, and hypoxia, accompanied by a clear lung exam and normal chest radiograph, increases the likelihood of a pulmonary embolism. Obesity, testosterone therapy, and trauma increase his susceptibility to venous thromboembolism, while a distal femur fracture increases his risk for FES. Acute pulmonary aspiration often presents with initially absent or subtle radiographic findings. An arterial blood gas analysis would determine the presence and extent of an alveolar-arterial (A-a) gradient; a normal A-a gradient is seen in hypoventilation, while an elevated A-a gradient is seen in conditions affecting gas exchange, including pulmonary emboli and alveolar filling processes. His hypoxemia only partially corrects with supplemental oxygen, raising the possibility of capillary or anatomic shunting. Capillary shunting may occur with atelectasis, aspiration/pneumonia and pulmonary edema, whereas anatomic shunting can be intra-cardiac (eg, patent foramen ovale or septic defect) or intrapulmonary (eg, arteriovenous malformations). Patients with pulmonary emboli may also develop right-to-left shunting because of increased pulmonary vascular resistance, although hypoxemia with pulmonary emboli largely relates to ventilation/perfusion mismatch and decreased level of mixed venous blood oxygen (PvO2).
This patient’s complex medical history warrants a broadened differential with consideration of his cardiac history, including myocardial fibrosis and arrhythmia, and the impact of exposure to steroids on his immune and musculoskeletal systems. He has a history of atrial fibrillation, and an electrocardiogram is warranted to determine the underlying rhythm. Prolonged periods of rapid ventricular response may lead to tachycardia-induced cardiomyopathy. Myocardial fibrosis may progress despite use of angiotensin-converting enzyme inhibitors and is associated with systolic and/or diastolic dysfunction, although neither the examination findings provided nor the chest radiograph are suggestive of decompensated heart failure. Chronic exposure to corticosteroids (used in DMD to improve muscle strength and function) may predispose to numerous infectious and metabolic complications. Up to 10%-15% of patients with Pneumocystis jirovecii pneumonia may present with a normal chest radiograph. Acute adrenal insufficiency can present with tachycardia, weakness, and respiratory distress, so recent prednisone dose changes or interruptions should be assessed.
The patient’s respiratory status worsened. In light of his complex medical history, he was transferred to a children’s hospital for a higher level of care with a presumptive diagnosis of aspiration pneumonia. Upon reassessment at the new facility, the patient reported an ongoing and severe headache since his initial injury. NSAIDs had been given prior to transfer. His exam continued to be significant for tachycardia, tachypnea, and hypoxemia. His cardiac and lung examinations were otherwise normal. A comprehensive metabolic panel, procalcitonin, complete blood count with differential, and lactate were normal; his C-reactive protein (CRP) was 46.8 mg/dL (Normal <8 mg/dL). A computed tomography (CT) angiogram of the chest revealed small multifocal nodular ground-glass opacities, especially in the lower lobes, concerning for microatelectasis, multifocal pneumonia, or aspiration pneumonia. After consultation with pediatric pulmonology consultants, antimicrobials were held during the initial phase of work-up.
His headache may reflect a migraine, although further characterization and assessment for the presence and extent of head or neck trauma is warranted. Headache following trauma warrants consideration of cerebral contusion, diffuse axonal injury, intracranial hemorrhage, and carotid or vertebral artery dissection. Screening for concussion should also be performed. Hypoxemia may increase cerebral blood flow and raise intracranial pressure, resulting in headache.
CRP elevation is nonspecific and signals the presence of focal or systemic inflammation and is often elevated to a milder extent in obese patients with DMD. While normal procalcitonin argues against bacterial pneumonia, the precise level can be informative, and serial procalcitonin values may be more helpful than a single value. Although antecedent respiratory symptoms were not mentioned, viral or fungal pneumonia can present insidiously. An occult malignancy may be incidentally discovered when patients present for unrelated issues, although this and other sources of elevated CRP (eg, exacerbation of an autoimmune disease or drug reaction) remain less likely given the acuity of his presentation. Acute pulmonary embolism may be associated with a systemic inflammatory response and elevation in CRP.
In addition to the radiographic differential diagnosis already presented, the appearance of multifocal opacifications with hypoxemia raises the possibility of pulmonary infarcts or noncardiogenic pulmonary edema.
On hospital day 2, the patient continued to complain of “the worst headache of his life” as well as blurry vision and seeing “dark spots.” His headache did not improve with NSAIDs. A noncontrast CT scan of the head was normal. Neurology was consulted. Given his symptoms, history of migraines, stable neurological examination, and normal head CT, he was diagnosed with migraines and administered fluids, prochlorperazine, diphenhydramine, ondansetron, and NSAIDs. His headache continued and he continued to require supplemental oxygen.
The combination of hypoxemia, severe headache, and vision changes remains consistent with systemic emboli caused by thromboembolism or fat embolism. Headache assessment must also involve screening for “red flags,” which include sudden onset, antecedent head trauma, systemic illness (eg, fever or meningismus), focal neurologic findings, papilledema, changes with position or Valsalva, and immunosuppression. Although primary headache syndromes (eg, migraines or tension and cluster headache) may be triggered in the setting of trauma and systemic illness, “the worst headache of my life” is a concerning symptom that warrants urgent attention. While this invokes the possibility of a subarachnoid hemorrhage (SAH), headache severity is nonspecific, and rapid onset (ie, thunderclap headache) would be more suggestive. After 6 hours of symptoms, the sensitivity of head CT for detecting SAH declines, and lumbar puncture would be warranted to evaluate for xanthochromia.
His blurry vision and dark spots require testing of visual acuity and visual fields, as well as fundoscopic examination to assess for embolic phenomena or papilledema. Migraine is classically associated with “positive” or scintillating scotomata, although dark spots may occur. The presence of horizontal diplopia would indicate a cranial nerve VI palsy, which can occur with increased intracranial pressure. Visual-field cuts may also present as blurry vision, and monocular vs binocular deficits signal whether the issue is anterior or involving/posterior to the optic chiasm, respectively. Magnetic resonance imaging (MRI) may reveal the presence or sequelae of cerebral emboli (eg, fat emboli), including vasogenic edema.
Dilated fundus examination revealed Purtscher retinopathy: bilateral cotton-wool spots and larger areas of retinal whitening (Purtscher flecken).
Typical findings of Purtscher retinopathy include Purtscher flecken, cotton-wool spots, retinal hemorrhage, and optic disc edema. Purtscher retinopathy is classically associated with severe head trauma. Without associated head trauma, the term “Purtscher-like retinopathy” is used. Conditions that can cause Purtscher-like retinopathy include pancreatitis, vasculitis, microangiopathy, chronic renal failure, and systemic embolization. The most likely source of systemic embolization remains fat emboli stemming from his femur fracture. Treatment of FES is largely supportive.
The possibility of fat emboli had been repeatedly raised by the patient’s mother since admission. While providers had considered this a possibility, it was discounted early on because of the minor nature of the patient’s orthopedic trauma, the lack of clear radiographic evidence for pulmonary emboli on chest CT, and the normal head CT. The findings on the ophthalmologist’s fundoscopic examination led the primary team to reconsider FES, along with thromboemboli and pancreatitis. Lipase was normal. MRI of the brain with contrast revealed >20 microinfarcts in the bilateral hemispheres, left corpus callosum, and bilateral basal ganglia. The CT angiogram of the chest was rereviewed; the pediatric radiologists suggested that microinfarcts could explain the patchy small ground glass opacities seen in the lungs. A transthoracic echocardiogram and electrocardiogram were normal. The diagnosis of FES was made, and the patient was started on aspirin and enoxaparin prophylaxis. His headache and respiratory status improved, and he was discharged home with close follow-up.
DISCUSSION
FES is a rare complication associated with long bone fractures and orthopedic manipulation.1,2 The exact mechanism of fat emboli production is unknown, but two theories prevail. The mechanical theory states that an outside mechanical source causes bone marrow contents or adipose tissue contents to be dislodged into the circulation where they travel through the venous circulation to become embedded in the lungs.1,2 These fragments may also migrate to the arterial circulation, through a patent foramen ovale or intrapulmonary shunts, leading to end organ damage.1,2 The biochemical theory suggests that fat emboli in the venous circulation precipitate an inflammatory and prothrombotic cascade that triggers fibrin production, platelet aggregation, and release of free fatty acids into the circulation, predisposing patients to develop multifocal systemic emboli.1
Although the classic triad in FES includes respiratory symptoms, rash, and CNS symptoms, all three findings are only present in 1%-29% of cases.1,2 Respiratory abnormalities, ranging from tachypnea and dyspnea to ARDS and hypoxic respiratory failure, occur in up to 75% of patients with FES.1 Central nervous system (CNS) complications, including headache, confusion, coma, seizures, and death caused by cerebral ischemia, occur in up to 86% of patients.1,2 Petechiae may occur in 20%-60% of patients and are usually located on nondependent regions of the body such as the head, neck, and chest.
Diagnosis of FES is largely clinical and requires a high index of suspicion and elimination of other conditions, including pulmonary thromboembolism, diffuse intravascular coagulation, and sepsis. The CNS complications must be differentiated from CNS infection, stroke, migraine, benign intracranial hypertension, and intracranial hemorrhage. There is no gold standard test for diagnosis. The Gurd and Wilson criteria, modified Gurd criteria, and Schonfeld’s criteria (Table) are commonly used but have not been clinically validated.1,3-5 These use a combination of clinical signs of respiratory distress, neurological symptoms, petechial rash, and various other diagnostic factors. When patients have risk factors, such as trauma, surgery, or predisposing conditions (eg, mobility-limiting neuromuscular disorders) and signs and symptoms potentially consistent with FES, the diagnostic evaluation should include arterial blood gas analysis, complete blood count, chest radiographs, and coagulation studies. Ophthalmological exam to evaluate for cotton-wool spots, brain MRI to detect cerebral emboli, and CT pulmonary angiogram to assess for pulmonary infarcts may help to eliminate other diagnoses and/or confirm FES.
Diagnosis of uncommon conditions that present with nonspecific symptoms, like FES, can be challenging because the symptoms may overlap with many other possible diagnoses. This challenge is further exacerbated in patients with significant medical complexity, as with the patient discussed here. Specifically, this patient had multiple plausible explanations for CNS symptoms and respiratory symptoms. It was ultimately the visual symptoms that began to link his symptoms together into a unifying syndrome and the ophthalmologic examination that prompted confirmatory imaging. It is important to continually revisit and revise the differential diagnosis in patients with medical complexity and avoid the competing temptations to abandon the search for a unifying diagnosis and attribute all symptoms to a patient’s underlying condition.
Treatment of FES is largely supportive with close monitoring of neurological status and providing supplemental oxygen as needed. Corticosteroids have been suggested to help prevent FES in patients with long bone fractures, but there is no evidence to suggest they are helpful once FES is diagnosed.2 There is conflicting evidence for the efficacy of heparin or low-molecular-weight heparin as treatment in FES.2,6 After discussions with consulting physician teams, the patient, and his family, enoxaparin and aspirin were started for this patient in light of his tenuous condition in order to decrease the risk of further embolic complications.
Patients with DMD and other neuromuscular disorders likely have a greater propensity to develop FES even after minor trauma.1,6 This is believed to be caused by patients becoming nonambulatory early in life and receiving substantial corticosteroid therapy, which can lead to osteopenia and fatty replacement of the bone marrow.1,6 This population is also often obese by the second or third decade of life, which contributes to their already increased propensity to fall.1,6
To our knowledge, this patient is 1 of 18 reported cases of FES after trauma in DMD patients. Two-thirds of these cases occurred when an unrestrained patient fell from their wheelchair. The other cases occurred while walking, during physical therapy, and during assisted transfers.6-12 In these cases, FES had a guarded prognosis, with 7 of the 18 patients dying and 1 of the patients remaining in a persistent vegetative state.8,9 While caution is warranted in generalizing these findings, given the small number of reported cases and likely publication bias,education of caregivers and patients on use of restraints and safe transfers is paramount to limit the risk of trauma.
TEACHING POINTS
- FES is a rare condition that most commonly manifests with respiratory, neurological, and cutaneous findings.
- Patients with Duchenne’s Muscular Dystrophy are likely at increased risk for FES even with minor trauma; this makes wheelchair restraints and safe transfers fundamental.
- Patients with medical complexity and their caregivers are key members of the diagnostic team.
A 19-year-old man with Duchenne Muscular Dystrophy (DMD) presented to the Emergency Department (ED) for left knee pain after ejection from his motorized wheelchair at a low velocity. In the ED, he developed increasing respiratory distress.
When addressing a new problem in a patient with a chronic condition, it is crucial to first understand the chronic condition and then consider whether the presenting symptoms relate to that condition or stem from an unrelated inciting event.
Patients with DMD are at risk of pulmonary complications relating to their underlying disease. For instance, dysphagia and ineffective cough can predispose them to recurrent aspiration pneumonitis and/or pneumonia, whereas decreased lung compliance (from scoliosis, atelectasis, and/or pulmonary fibrosis) and respiratory muscle weakness can progress to ventilatory failure. In addition, patients with DMD are at risk for pulmonary thromboembolism in the setting of immobility. Patients with DMD may also develop congestive heart failure resulting from myocardial fibrosis and nonischemic cardiomyopathy.
The ejection from his wheelchair signals potential trauma-associated conditions that could explain his respiratory distress. Respiratory complications of blunt thoracic trauma include pulmonary contusion, pneumothorax, flail chest (resulting from fractured ribs), and acute respiratory distress syndrome (ARDS). Lower extremity injury can result in venous thrombosis and pulmonary thromboembolism. While classically associated with long bone fractures, fat embolism syndrome (FES) may rarely occur with rib fractures and soft-tissue trauma. Respiratory compromise may also result from cervical spinal cord injury or severe anemia from trauma-associated hemorrhage.
Additional past medical history included growth hormone deficiency, migraine headaches, osteoporosis secondary to chronic steroid use, cardiac fibrosis of the inferolateral wall and septum with a baseline left ventricular ejection fraction of 65%, and atrial fibrillation. His medications included calcium carbonate, vitamin D, omeprazole, lisinopril, metoprolol, prednisone, escitalopram, and testosterone. Physical examination revealed an ill-appearing obese man in respiratory distress. Temperature was 37.3°C, heart rate was 102 beats per minute (bpm), blood pressure was 110/74 mm Hg. His oxygen saturation was 93% with a respiratory rate of 25 breaths per minute while breathing ambient air. His lung sounds were clear, and his heart was without murmur. The left knee was diffusely tender to palpation without specific point tenderness. Strength was 2/5 with flexion and extension at the bilateral knees and hips and 3/5 flexion and extension at the bilateral elbows. He reported this level of weakness was his baseline. Radiographs revealed a minimally displaced Salter Harris II fracture (fracture line through the metaphysis and growth plate) of the left distal femur. His fracture was splinted early in his ED course. During his ED evaluation, the patient had acute worsening of tachycardia to 130 bpm, increased respiratory rate of 34 breaths per minute, and hypoxemia with an oxygen saturation of 83% on ambient air. He was placed on 3 L/min of oxygen via nasal cannula with improvement in his oxygen saturation to 90%. A chest radiograph was unremarkable, without evidence of pneumothorax, effusion, or pneumonia. The patient was admitted to the hospital.
The acute onset of tachypnea, tachycardia, and hypoxia, accompanied by a clear lung exam and normal chest radiograph, increases the likelihood of a pulmonary embolism. Obesity, testosterone therapy, and trauma increase his susceptibility to venous thromboembolism, while a distal femur fracture increases his risk for FES. Acute pulmonary aspiration often presents with initially absent or subtle radiographic findings. An arterial blood gas analysis would determine the presence and extent of an alveolar-arterial (A-a) gradient; a normal A-a gradient is seen in hypoventilation, while an elevated A-a gradient is seen in conditions affecting gas exchange, including pulmonary emboli and alveolar filling processes. His hypoxemia only partially corrects with supplemental oxygen, raising the possibility of capillary or anatomic shunting. Capillary shunting may occur with atelectasis, aspiration/pneumonia and pulmonary edema, whereas anatomic shunting can be intra-cardiac (eg, patent foramen ovale or septic defect) or intrapulmonary (eg, arteriovenous malformations). Patients with pulmonary emboli may also develop right-to-left shunting because of increased pulmonary vascular resistance, although hypoxemia with pulmonary emboli largely relates to ventilation/perfusion mismatch and decreased level of mixed venous blood oxygen (PvO2).
This patient’s complex medical history warrants a broadened differential with consideration of his cardiac history, including myocardial fibrosis and arrhythmia, and the impact of exposure to steroids on his immune and musculoskeletal systems. He has a history of atrial fibrillation, and an electrocardiogram is warranted to determine the underlying rhythm. Prolonged periods of rapid ventricular response may lead to tachycardia-induced cardiomyopathy. Myocardial fibrosis may progress despite use of angiotensin-converting enzyme inhibitors and is associated with systolic and/or diastolic dysfunction, although neither the examination findings provided nor the chest radiograph are suggestive of decompensated heart failure. Chronic exposure to corticosteroids (used in DMD to improve muscle strength and function) may predispose to numerous infectious and metabolic complications. Up to 10%-15% of patients with Pneumocystis jirovecii pneumonia may present with a normal chest radiograph. Acute adrenal insufficiency can present with tachycardia, weakness, and respiratory distress, so recent prednisone dose changes or interruptions should be assessed.
The patient’s respiratory status worsened. In light of his complex medical history, he was transferred to a children’s hospital for a higher level of care with a presumptive diagnosis of aspiration pneumonia. Upon reassessment at the new facility, the patient reported an ongoing and severe headache since his initial injury. NSAIDs had been given prior to transfer. His exam continued to be significant for tachycardia, tachypnea, and hypoxemia. His cardiac and lung examinations were otherwise normal. A comprehensive metabolic panel, procalcitonin, complete blood count with differential, and lactate were normal; his C-reactive protein (CRP) was 46.8 mg/dL (Normal <8 mg/dL). A computed tomography (CT) angiogram of the chest revealed small multifocal nodular ground-glass opacities, especially in the lower lobes, concerning for microatelectasis, multifocal pneumonia, or aspiration pneumonia. After consultation with pediatric pulmonology consultants, antimicrobials were held during the initial phase of work-up.
His headache may reflect a migraine, although further characterization and assessment for the presence and extent of head or neck trauma is warranted. Headache following trauma warrants consideration of cerebral contusion, diffuse axonal injury, intracranial hemorrhage, and carotid or vertebral artery dissection. Screening for concussion should also be performed. Hypoxemia may increase cerebral blood flow and raise intracranial pressure, resulting in headache.
CRP elevation is nonspecific and signals the presence of focal or systemic inflammation and is often elevated to a milder extent in obese patients with DMD. While normal procalcitonin argues against bacterial pneumonia, the precise level can be informative, and serial procalcitonin values may be more helpful than a single value. Although antecedent respiratory symptoms were not mentioned, viral or fungal pneumonia can present insidiously. An occult malignancy may be incidentally discovered when patients present for unrelated issues, although this and other sources of elevated CRP (eg, exacerbation of an autoimmune disease or drug reaction) remain less likely given the acuity of his presentation. Acute pulmonary embolism may be associated with a systemic inflammatory response and elevation in CRP.
In addition to the radiographic differential diagnosis already presented, the appearance of multifocal opacifications with hypoxemia raises the possibility of pulmonary infarcts or noncardiogenic pulmonary edema.
On hospital day 2, the patient continued to complain of “the worst headache of his life” as well as blurry vision and seeing “dark spots.” His headache did not improve with NSAIDs. A noncontrast CT scan of the head was normal. Neurology was consulted. Given his symptoms, history of migraines, stable neurological examination, and normal head CT, he was diagnosed with migraines and administered fluids, prochlorperazine, diphenhydramine, ondansetron, and NSAIDs. His headache continued and he continued to require supplemental oxygen.
The combination of hypoxemia, severe headache, and vision changes remains consistent with systemic emboli caused by thromboembolism or fat embolism. Headache assessment must also involve screening for “red flags,” which include sudden onset, antecedent head trauma, systemic illness (eg, fever or meningismus), focal neurologic findings, papilledema, changes with position or Valsalva, and immunosuppression. Although primary headache syndromes (eg, migraines or tension and cluster headache) may be triggered in the setting of trauma and systemic illness, “the worst headache of my life” is a concerning symptom that warrants urgent attention. While this invokes the possibility of a subarachnoid hemorrhage (SAH), headache severity is nonspecific, and rapid onset (ie, thunderclap headache) would be more suggestive. After 6 hours of symptoms, the sensitivity of head CT for detecting SAH declines, and lumbar puncture would be warranted to evaluate for xanthochromia.
His blurry vision and dark spots require testing of visual acuity and visual fields, as well as fundoscopic examination to assess for embolic phenomena or papilledema. Migraine is classically associated with “positive” or scintillating scotomata, although dark spots may occur. The presence of horizontal diplopia would indicate a cranial nerve VI palsy, which can occur with increased intracranial pressure. Visual-field cuts may also present as blurry vision, and monocular vs binocular deficits signal whether the issue is anterior or involving/posterior to the optic chiasm, respectively. Magnetic resonance imaging (MRI) may reveal the presence or sequelae of cerebral emboli (eg, fat emboli), including vasogenic edema.
Dilated fundus examination revealed Purtscher retinopathy: bilateral cotton-wool spots and larger areas of retinal whitening (Purtscher flecken).
Typical findings of Purtscher retinopathy include Purtscher flecken, cotton-wool spots, retinal hemorrhage, and optic disc edema. Purtscher retinopathy is classically associated with severe head trauma. Without associated head trauma, the term “Purtscher-like retinopathy” is used. Conditions that can cause Purtscher-like retinopathy include pancreatitis, vasculitis, microangiopathy, chronic renal failure, and systemic embolization. The most likely source of systemic embolization remains fat emboli stemming from his femur fracture. Treatment of FES is largely supportive.
The possibility of fat emboli had been repeatedly raised by the patient’s mother since admission. While providers had considered this a possibility, it was discounted early on because of the minor nature of the patient’s orthopedic trauma, the lack of clear radiographic evidence for pulmonary emboli on chest CT, and the normal head CT. The findings on the ophthalmologist’s fundoscopic examination led the primary team to reconsider FES, along with thromboemboli and pancreatitis. Lipase was normal. MRI of the brain with contrast revealed >20 microinfarcts in the bilateral hemispheres, left corpus callosum, and bilateral basal ganglia. The CT angiogram of the chest was rereviewed; the pediatric radiologists suggested that microinfarcts could explain the patchy small ground glass opacities seen in the lungs. A transthoracic echocardiogram and electrocardiogram were normal. The diagnosis of FES was made, and the patient was started on aspirin and enoxaparin prophylaxis. His headache and respiratory status improved, and he was discharged home with close follow-up.
DISCUSSION
FES is a rare complication associated with long bone fractures and orthopedic manipulation.1,2 The exact mechanism of fat emboli production is unknown, but two theories prevail. The mechanical theory states that an outside mechanical source causes bone marrow contents or adipose tissue contents to be dislodged into the circulation where they travel through the venous circulation to become embedded in the lungs.1,2 These fragments may also migrate to the arterial circulation, through a patent foramen ovale or intrapulmonary shunts, leading to end organ damage.1,2 The biochemical theory suggests that fat emboli in the venous circulation precipitate an inflammatory and prothrombotic cascade that triggers fibrin production, platelet aggregation, and release of free fatty acids into the circulation, predisposing patients to develop multifocal systemic emboli.1
Although the classic triad in FES includes respiratory symptoms, rash, and CNS symptoms, all three findings are only present in 1%-29% of cases.1,2 Respiratory abnormalities, ranging from tachypnea and dyspnea to ARDS and hypoxic respiratory failure, occur in up to 75% of patients with FES.1 Central nervous system (CNS) complications, including headache, confusion, coma, seizures, and death caused by cerebral ischemia, occur in up to 86% of patients.1,2 Petechiae may occur in 20%-60% of patients and are usually located on nondependent regions of the body such as the head, neck, and chest.
Diagnosis of FES is largely clinical and requires a high index of suspicion and elimination of other conditions, including pulmonary thromboembolism, diffuse intravascular coagulation, and sepsis. The CNS complications must be differentiated from CNS infection, stroke, migraine, benign intracranial hypertension, and intracranial hemorrhage. There is no gold standard test for diagnosis. The Gurd and Wilson criteria, modified Gurd criteria, and Schonfeld’s criteria (Table) are commonly used but have not been clinically validated.1,3-5 These use a combination of clinical signs of respiratory distress, neurological symptoms, petechial rash, and various other diagnostic factors. When patients have risk factors, such as trauma, surgery, or predisposing conditions (eg, mobility-limiting neuromuscular disorders) and signs and symptoms potentially consistent with FES, the diagnostic evaluation should include arterial blood gas analysis, complete blood count, chest radiographs, and coagulation studies. Ophthalmological exam to evaluate for cotton-wool spots, brain MRI to detect cerebral emboli, and CT pulmonary angiogram to assess for pulmonary infarcts may help to eliminate other diagnoses and/or confirm FES.
Diagnosis of uncommon conditions that present with nonspecific symptoms, like FES, can be challenging because the symptoms may overlap with many other possible diagnoses. This challenge is further exacerbated in patients with significant medical complexity, as with the patient discussed here. Specifically, this patient had multiple plausible explanations for CNS symptoms and respiratory symptoms. It was ultimately the visual symptoms that began to link his symptoms together into a unifying syndrome and the ophthalmologic examination that prompted confirmatory imaging. It is important to continually revisit and revise the differential diagnosis in patients with medical complexity and avoid the competing temptations to abandon the search for a unifying diagnosis and attribute all symptoms to a patient’s underlying condition.
Treatment of FES is largely supportive with close monitoring of neurological status and providing supplemental oxygen as needed. Corticosteroids have been suggested to help prevent FES in patients with long bone fractures, but there is no evidence to suggest they are helpful once FES is diagnosed.2 There is conflicting evidence for the efficacy of heparin or low-molecular-weight heparin as treatment in FES.2,6 After discussions with consulting physician teams, the patient, and his family, enoxaparin and aspirin were started for this patient in light of his tenuous condition in order to decrease the risk of further embolic complications.
Patients with DMD and other neuromuscular disorders likely have a greater propensity to develop FES even after minor trauma.1,6 This is believed to be caused by patients becoming nonambulatory early in life and receiving substantial corticosteroid therapy, which can lead to osteopenia and fatty replacement of the bone marrow.1,6 This population is also often obese by the second or third decade of life, which contributes to their already increased propensity to fall.1,6
To our knowledge, this patient is 1 of 18 reported cases of FES after trauma in DMD patients. Two-thirds of these cases occurred when an unrestrained patient fell from their wheelchair. The other cases occurred while walking, during physical therapy, and during assisted transfers.6-12 In these cases, FES had a guarded prognosis, with 7 of the 18 patients dying and 1 of the patients remaining in a persistent vegetative state.8,9 While caution is warranted in generalizing these findings, given the small number of reported cases and likely publication bias,education of caregivers and patients on use of restraints and safe transfers is paramount to limit the risk of trauma.
TEACHING POINTS
- FES is a rare condition that most commonly manifests with respiratory, neurological, and cutaneous findings.
- Patients with Duchenne’s Muscular Dystrophy are likely at increased risk for FES even with minor trauma; this makes wheelchair restraints and safe transfers fundamental.
- Patients with medical complexity and their caregivers are key members of the diagnostic team.
1. Fukumoto LE, Fukumoto KD. Fat embolism syndrome. Nurs Clin North Am. 2018;53(3):335-347. https://doi.org/10.1016/j.cnur.2018.04.003.
2. Scarpino M, Lanzo G, Lolli F, Grippo A. From the diagnosis to the therapeutic management: Cerebral fat embolism, a clinical challenge. Int J Gen Med. 2019;2019(12):39-48. https://doi.org/10.2147/IJGM.S177407.
1. Fukumoto LE, Fukumoto KD. Fat embolism syndrome. Nurs Clin North Am. 2018;53(3):335-347. https://doi.org/10.1016/j.cnur.2018.04.003.
2. Scarpino M, Lanzo G, Lolli F, Grippo A. From the diagnosis to the therapeutic management: Cerebral fat embolism, a clinical challenge. Int J Gen Med. 2019;2019(12):39-48. https://doi.org/10.2147/IJGM.S177407.
© 2020 Society of Hospital Medicine
Hindsight Is 20/20
A 38-year-old woman presented to her primary care clinic with 3 weeks of progressive numbness and tingling sensation, which began in both hands and then progressed to involv
As with all neurological complaints, localization of the process will often inform a more specific differential diagnosis. If both sensory and motor findings are present, both central and peripheral nerve processes deserve consideration. The onset of paresthesia in the hands, rapid progression to the trunk, and unilateral leg weakness would be inconsistent with a length-dependent peripheral neuropathy. The distribution of complaints and the sacral sparing suggests a myelopathic process involving the cervical region rather than a cauda equina or conus lesions. In an otherwise healthy person of this age and gender, an inflammatory demyelinating disease affecting the cord including multiple sclerosis (MS) would be a strong consideration, although metabolic, vascular, infectious, compressive, or neoplastic disease of the spinal cord could also present with similar subacute onset and pattern of deficits.
Her medical history included morbid obesity, dry eyes, depression, iron deficiency anemia requiring recurrent intravenous replenishment, and abnormal uterine bleeding. Her surgical history included gastric band placement 7 years earlier with removal 5 years later due to persistent gastroesophageal reflux disease, dysphagia, nausea, and vomiting. The gastric band removal was complicated by chronic abdominal pain. Her medications consisted of duloxetine, intermittent iron infusions, artificial tears, loratadine, and pregabalin. She was sexually active with her husband. She consumed alcohol occasionally but did not smoke tobacco or use illicit drugs.
On exam, her temperature was 36.6°C (97.8°F), blood pressure 132/84 mm Hg, and heart rate 85 beats per minute. Body mass index was 39.5 kg/m2. The cardiac, pulmonary, and skin examinations were normal. The abdomen was soft with diffuse tenderness to palpation without rebound or guarding. Examination of cranial nerves 2-12 was normal. Cognition, strength, proprioception, deep tendon reflexes, and light touch were all normal. Her gait was normal, and the Romberg test was negative.
The normal neurologic exam is reassuring but imperfectly sensitive and does not eliminate the possibility of underlying neuropathology. Bariatric surgery may result in an array of nutritional deficiencies such as vitamin E, B12, and copper, which can cause myelopathy and/or neuropathy. However, these abnormalities occur less frequently with gastric banding procedures. If her dry eyes are part of the sicca syndrome, an underlying autoimmune diathesis may be present. Her unexplained chronic abdominal pain prompts considering nonmenstrual causes of iron deficiency anemia, such as celiac disease. Bariatric surgery may contribute to iron deficiency through impaired iron absorption. Her stable weight and lack of diarrhea argue against Crohn’s or celiac disease. Iron deficiency predisposes individuals to pica, most commonly described with ice chip ingestion. If lead pica had occurred, abdominal and neurological symptoms could result. Nevertheless, the abdominal pain is nonspecific, and its occurrence after gastric band removal makes its link to her neurologic syndrome unclear. An initial evaluation would include basic metabolic panel, complete blood count with differential, erythrocyte sedimentation rate, C-reactive protein (CRP), thyroid-stimulating hormone, vitamin B12, and copper levels.
A basic metabolic panel was normal. The white cell count was 5,710 per cubic millimeter, hemoglobin level 12.2 g per deciliter, mean corpuscular volume 85.2 fl, and platelet count 279,000 per cubic millimeter. The serum ferritin level was 18 ng per milliliter (normal range, 13-150), iron 28 µg per deciliter (normal range, 50-170), total iron-binding capacity 364 µg per deciliter (normal range, 250-450), and iron saturation 8% (normal range, 20-55). The vitamin B12 level was 621 pg per milliliter (normal range, 232-1,245) and thyroid-stimulating hormone level 1.87 units per milliliter (normal range, 0.50-4.50). Electrolyte and aminotransferase levels were within normal limits. CRP was 1.0 mg per deciliter (normal range, <0.5) and erythrocyte sedimentation rate 33 millimeters per hour (normal range, 4-25). Hepatitis C and HIV antibodies were nonreactive.
The ongoing iron deficiency despite parenteral iron replacement raises the question of ongoing gastrointestinal or genitourinary blood loss. While the level of vitamin B12 in the serum may be misleadingly normal with cobalamin deficiency, a methylmalonic acid level is indicated to evaluate whether tissue stores are depleted. Copper levels are warranted given the prior bariatric surgery. The mild elevations of inflammatory markers are nonspecific but reduce the likelihood of a highly inflammatory process to account for the neurological and abdominal symptoms.
At her 3-month follow-up visit, she noted that the paresthesia had improved and was now limited to her bilateral lower extremities. During the same clinic visit, she experienced a 45-minute episode of ascending left upper extremity numbness. Her physical examination revealed normal strength and reflexes. She had diminished response to pinprick in both legs to the knees and in both hands to the wrists. Vibration sense was diminished in the bilateral lower extremities.
A glycosylated hemoglobin (HbA1c) level was 6.2%. Methylmalonic acid was 69 nmol per liter (normal range, 45-325). Antibodies to Borrelia burgdorferi and Treponema pallidum were absent. Impaired glucose metabolism was the leading diagnosis for her polyneuropathy, and it was recommended that she undergo an oral glucose tolerance test. Electromyography was not performed.
The neurological symptoms are now chronic, and importantly, the patient has developed sensory deficits on neurological examination, suggesting worsening of the underlying process. While the paresthesia is now limited to a “stocking/glove” distribution consistent with distal sensory polyneuropathy, there should still be a concern for spinal cord pathology given that the HbA1c level of 6.2 would not explain her initial distribution of symptoms. Myelopathy may mimic peripheral nerve disease if, for example, there is involvement of the dorsal columns leading to sensory deficits of vibration and proprioception. Additionally, the transient episode of upper extremity numbness raises the question of sensory nerve root involvement (ie, sensory radiculopathy). Unexplained abdominal pain could possibly represent the involvement of other nerve roots innervating the abdominal wall. The patient’s episode of focal arm numbness recalls the lancinating radicular pain of tabes dorsalis; however, the negative specific treponemal antibody test excludes neurosyphilis.
The differential diagnosis going forward will be strongly conditioned by the localization of the neurological lesion(s). To differentiate between myelopathy, radiculopathy, and peripheral neuropathy, I would perform nerve conduction studies, magnetic resonance imaging (MRI) of the spinal cord, and cerebrospinal fluid analysis.
The patient began taking a multivitamin, and after weeks her paresthesia had resolved. One month later, she developed an intermittent, throbbing left-sided headache and pain behind the left eye that was worsened with ocular movement. She then noted decreased visual acuity in her left eye that progressed the following month. She denied photophobia, flashers, or floaters.
In the emergency department, visual acuity was 20/25 in her right eye; in the left eye she was only able to count fingers. Extraocular movements of both eyes were normal as was her right pupillary reflex. Red desaturation and a relative afferent papillary defect were present in the left eye. Fundoscopic exam demonstrated left optic disc swelling. The remainder of her cranial nerves were normal. She had pronation of the left upper extremity and mild right finger-to-nose dysmetria. Muscle tone, strength, sensation, and deep tendon reflexes were normal.
The improvement in the sensory symptoms was unlikely to be related to the nutritional intervention and provides a clue to an underlying waxing and waning illness. That interpretation is supported by the subsequent development of new visual symptoms and signs, which point to optic nerve pathology. Optic neuropathy has a broad differential diagnosis that includes ischemic, metabolic, toxic, and compressive causes. Eye pain, swelling of the optic disc, and prominent impairment of color vision all point to the more specific syndrome of optic neuritis caused by infections (including both Treponema pallidum and Borrelia species), systemic autoimmune diseases (systemic lupus erythematosus or Sjogren’s syndrome), and central nervous system (CNS) demyelinating diseases. Of these, inflammatory demyelinating processes would be the likeliest explanation of intermittent and improving neurologic findings.
With relapsing symptoms and findings that are separate in distribution and time, two diagnoses become most likely, and both of these are most often diagnosed in young women. MS is common, and optic neuritis occurs in more than 50% of patients over the course of illness. Neuromyelitis optica spectrum disorder (NMOSD) is a rare condition that can exist in isolation or be associated with other autoimmune illnesses. While these entities are difficult to differentiate clinically, neuroimaging that demonstrates extensive intracerebral demyelinating lesions and cerebrospinal fluid with oligoclonal bands favor MS, whereas extensive, predominant spinal cord involvement is suggestive of NMOSD. Approximately 70% of NMO patients harbor an antibody directed against the aquaporin-4 channel, and these antibodies are not seen in patients with MS. A milder NMO-like disorder has also been associated with antimyelin oligodendrocyte antibodies (MOG).
Testing for antinuclear antibodies, anti–double-stranded DNA, anti-Ro (SSA), and anti-La (SSB) antibodies was negative. The level of C3 was 162 mg per deciliter (normal range 81-157) and C4 38 (normal range 13-39). T-spot testing for latent tuberculosis was negative.
There is no serological evidence of active systemic lupus erythematosus or Sjogren’s syndrome. The pretest probability of CNS tuberculosis was low in light of her presenting complaints, relatively protracted course, and overall clinical stability without antituberculous therapy. Tests for latent tuberculosis infection have significant limitations of both sensitivity and specificity for the diagnosis of active disease.
Optical coherence tomography showed optic disc edema in the left eye only. MRI of the head with contrast revealed abnormal signal intensity involving the posterior aspect of the pons, right middle cerebellar peduncle, anterior left temporal lobe, bilateral periventricular white matter, subcortical white matter of the frontal lobes bilaterally, and medulla with abnormal signal and enhancement of the left optic nerve (Figure, Panel A). MRI of the cervical and thoracic spine demonstrated multifocal demyelinating lesions at C3, C4, C7, T4, T5, T7, and T8 (Figure, Panel B). The lesions were not longitudinally extensive. There was no significant postcontrast enhancement to suggest active demyelination.
The cerebrospinal fluid analysis revealed glucose of 105 mg per deciliter and a total protein of 26.1 mg per deciliter. In the fourth tube, there were 20 red cells per cubic and four white cells with a differential of 62% neutrophils, 35% lymphocytes, and 3% monocytes. Epstein-Barr and herpes simplex virus DNA were negative. A Venereal Disease Research Laboratory test was negative. Multiple oligoclonal IgG bands were identified only in the cerebrospinal fluid. Aquaporin-4 IgG and MOG antibodies were negative.
In addition to the expected finding of enhancement of the optic nerve, MRI demonstrated numerous multifocal white matter lesions throughout the cerebrum, brainstem, and spinal cord. Many of the lesions were in “silent” areas, which is not directly attributable to specific symptoms, but several did correlate with the subtler deficits of weakness and dysmetria that were noted on examination. Although such lesions may be seen with a diverse group of systemic diseases including adrenal leukodystrophy, sarcoidosis, Behcet’s, cerebral lupus, and vasculitis, primary CNS inflammatory demyelinating diseases are much more likely. The extensive distribution of demyelination argues against NMOSD. The negative aquaporin-4 and MOG assays support this conclusion. Not all multifocal CNS demyelination is caused by MS and can be seen in posterior reversible encephalopathy syndrome, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, and adult polyglucosan body disease. Osmotic demyelination is increasingly being recognized as a process that can be more widespread rather than just being limited to the pons. Viral infections of the CNS such as the JC virus (PML) may also provoke multifocal demyelination. Acute disseminated encephalomyelitis is most often seen during childhood, usually after vaccination or after an infectious prodrome. The tempo of the progression of these other diseases tends to be much more rapid than this woman’s course, and often, the neurological deficits are more profound and debilitating. The clinical presentation of sensory-predominant myelopathy, followed by optic neuritis, absence of systemic inflammatory signs or laboratory markers, exclusion of other relevant diseases, multifocal white matter lesions on imaging, minimal pleocytosis, and presence of oligoclonal bands in cerebrospinal fluid, all point to a diagnosis of relapsing-remitting MS.
The patient was diagnosed with MS. She was admitted to the neurology service and treated with 1,000 mg IV methylprednisolone for 3 days with a prompt improvement in her vision. She was started on natalizumab without a relapse of symptoms over the past year.
COMMENTARY
Multiple sclerosis is a chronic demyelinating disease of the CNS.1 The diagnosis of MS has classically been based upon compatible clinical and radiographic evidence of pathology that is disseminated in space and time. Patients typically present with an initial clinically isolated syndrome—involving changes in vision, sensation, strength, mobility, or cognition—for which there is radiographic evidence of demyelination.2 A diagnosis of clinically definite MS is then often made based on a subsequent relapse of symptoms.3
An interval from initial symptoms has been central to the diagnosis of MS (“lesions disseminated in time”). However, recent evidence questions this diagnostic paradigm, and a more rapid diagnosis of MS has been recommended. This recommendation is reflected in the updated McDonald criteria, according to which, if a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.4 The importance of such early diagnosis has been supported by numerous studies that have demonstrated improved clinical outcomes with early therapy.5-7
Despite the McDonald criteria, delays in definitive diagnosis are common in MS. Patients with MS in Spain were found to experience a 2-year delay from the first onset of symptoms to diagnosis.8 In this cohort, patients exhibited delays in presenting to a healthcare provider, as well as delays in diagnosis with an average time from seeing an initial provider to diagnosis of 6 months. When patients who were referred for a demyelinating episode were surveyed, over a third reported a prior suggestive event.9 The time from the first suggestive episode to referral to a neurologist for a recognized demyelinating event was 46 months. Other studies have shown that delays in diagnosis are especially common in younger patients, those with primary progressive MS, and those with comorbid disease.10,11
Misapplication of an MS diagnosis also occurs frequently. In one case series, such misapplication was found most often in cases involving migraine, fibromyalgia, psychogenic disorders, and NMOSD.12 NMOSD is distinguished from MS by the presence of typical brain and spine findings on MRI.13 Antibodies to aquaporin-4 are highly specific and moderately sensitive for the disease.14 It is important to distinguish NMOSD from MS as certain disease-modifying drugs used for MS might actually exacerbate NMOSD.15 A lesion that traverses over three or more contiguous vertebral segments with predominant involvement of central gray matter (ie, longitudinally extensive transverse myelitis) on MRI is the most distinct finding of NMOSD. In contrast, similar to our patient, short and often multiple lesions are demonstrated on spinal cord MRI in patients with MS. Sensitive and specific findings of brain MRI in patients with MS include the presence of lateral ventricle and inferior temporal lobe lesion, Dawson’s fingers, central vein sign, or an S-shaped U-fiber lesion. In NMOSD, brain MRI might reveal periependymal lesions surrounding the ventricular system.
This case highlights the diagnostic challenges related to presentations of a waxing and waning neurological process. At the time of the second evaluation, the presentation was interpreted as a length-dependent polyneuropathy due to glucose intolerance. Our patient’s relatively normal HbA1c, subacute onset of neuropathic symptoms (ie, <4 weeks), sensory and motor complaints, and onset in the upper extremities suggested an alternative diagnosis to prediabetes. Once the patient presented with optic neuritis, the cause of the initial symptoms was obvious, but then, hindsight is 20/20.
TEACHING POINTS
- Early treatment of MS results in improved clinical outcomes.
- Delays in the definitive diagnosis of MS are common, especially in younger patients, those with primary progressive MS, and those with comorbid disease.
- If a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis of MS can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.
Acknowledgments
The authors wish to thank Rabih Geha, MD, and Gurpreet Dhaliwal, MD, for providing feedback on an earlier version of this manuscript.
1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378:169-180. https://doi.org/10.1056/NEJMra140148.
2. Brownlee WJ, Hardy TA, Fazekas F, Miller DH. Diagnosis of multiple sclerosis: progress and challenges. Lancet. 2017;389(10076):1336-1346. https://doi.org/10.1016/S0140-6736(16)30959-X.
3. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622-1636. https://doi.org/10.1016/S0140-6736(18)30481-1.
4. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173. https://doi.org/10.1016/S1474-4422(17)30470-2.
5. Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017;389(10076):1347-1356. https://doi.org/10.1016/S0140-6736(16)32388-1.
6. Freedman MS, Comi G, De Stefano N, et al. Moving toward earlier treatment of multiple sclerosis: Findings from a decade of clinical trials and implications for clinical practice. Mult Scler Relat Disord. 2014;3(2):147-155. https://doi.org/10.1016/j.msard.2013.07.001.
7. Harding K, Williams O, Willis M, et al. Clinical outcomes of escalation vs early intensive disease-modifying therapy in patients with multiple sclerosis. JAMA Neurol. 2019;76(5):536-541. https://doi.org/10.1001/jamaneurol.2018.4905.
8. Fernández O, Fernández V, Arbizu T, et al. Characteristics of multiple sclerosis at onset and delay of diagnosis and treatment in Spain (the Novo Study). J Neurol. 257(9):1500-1507. https://doi.org/10.1007/s00415-010-5560-1.
9. Gout O, Lebrun-Frenay C, Labauge P, et al. Prior suggestive symptoms in one-third of patients consulting for a “first” demyelinating event. J Neurol Neurosurg Psychiatry 2011;82(3):323-325. https://doi.org/10.1136/jnnp.2008.166421.
10. Kingwell E, Leung A, Roger E, et al. Factors associated with delay to medical recognition in two Canadian multiple sclerosis cohorts. J Neurol Sci. 2010(1-2);292:57-62. https://doi.org/10.1016/j.jns.2010.02.007.
11. Marrie RA, Horwitz R, Cutter G, Tyry T, Campagnolo D, Vollmer T. Comorbidity delays diagnosis and increases disability at diagnosis in MS. Neurology. 2009;72(2):117-124. https://doi.org/10.1212/01.wnl.0000333252.78173.5f.
12. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: A multicenter study. Neurology. 2016;87(13):1393-1399. https://doi.org/10.1212/WNL.0000000000003152.
13. Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: An international update. Neurology. 2015;84(11):1165-1173. https://doi.org/10.1212/WNL.0000000000001367.
14. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189. https://doi.org/10.1212/WNL.0000000000001729.
15. Jacob A, Hutchinson M, Elsone L, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012;79(10):1065-1066. https://doi.org/10.1212/WNL.0b013e31826845fe.
A 38-year-old woman presented to her primary care clinic with 3 weeks of progressive numbness and tingling sensation, which began in both hands and then progressed to involv
As with all neurological complaints, localization of the process will often inform a more specific differential diagnosis. If both sensory and motor findings are present, both central and peripheral nerve processes deserve consideration. The onset of paresthesia in the hands, rapid progression to the trunk, and unilateral leg weakness would be inconsistent with a length-dependent peripheral neuropathy. The distribution of complaints and the sacral sparing suggests a myelopathic process involving the cervical region rather than a cauda equina or conus lesions. In an otherwise healthy person of this age and gender, an inflammatory demyelinating disease affecting the cord including multiple sclerosis (MS) would be a strong consideration, although metabolic, vascular, infectious, compressive, or neoplastic disease of the spinal cord could also present with similar subacute onset and pattern of deficits.
Her medical history included morbid obesity, dry eyes, depression, iron deficiency anemia requiring recurrent intravenous replenishment, and abnormal uterine bleeding. Her surgical history included gastric band placement 7 years earlier with removal 5 years later due to persistent gastroesophageal reflux disease, dysphagia, nausea, and vomiting. The gastric band removal was complicated by chronic abdominal pain. Her medications consisted of duloxetine, intermittent iron infusions, artificial tears, loratadine, and pregabalin. She was sexually active with her husband. She consumed alcohol occasionally but did not smoke tobacco or use illicit drugs.
On exam, her temperature was 36.6°C (97.8°F), blood pressure 132/84 mm Hg, and heart rate 85 beats per minute. Body mass index was 39.5 kg/m2. The cardiac, pulmonary, and skin examinations were normal. The abdomen was soft with diffuse tenderness to palpation without rebound or guarding. Examination of cranial nerves 2-12 was normal. Cognition, strength, proprioception, deep tendon reflexes, and light touch were all normal. Her gait was normal, and the Romberg test was negative.
The normal neurologic exam is reassuring but imperfectly sensitive and does not eliminate the possibility of underlying neuropathology. Bariatric surgery may result in an array of nutritional deficiencies such as vitamin E, B12, and copper, which can cause myelopathy and/or neuropathy. However, these abnormalities occur less frequently with gastric banding procedures. If her dry eyes are part of the sicca syndrome, an underlying autoimmune diathesis may be present. Her unexplained chronic abdominal pain prompts considering nonmenstrual causes of iron deficiency anemia, such as celiac disease. Bariatric surgery may contribute to iron deficiency through impaired iron absorption. Her stable weight and lack of diarrhea argue against Crohn’s or celiac disease. Iron deficiency predisposes individuals to pica, most commonly described with ice chip ingestion. If lead pica had occurred, abdominal and neurological symptoms could result. Nevertheless, the abdominal pain is nonspecific, and its occurrence after gastric band removal makes its link to her neurologic syndrome unclear. An initial evaluation would include basic metabolic panel, complete blood count with differential, erythrocyte sedimentation rate, C-reactive protein (CRP), thyroid-stimulating hormone, vitamin B12, and copper levels.
A basic metabolic panel was normal. The white cell count was 5,710 per cubic millimeter, hemoglobin level 12.2 g per deciliter, mean corpuscular volume 85.2 fl, and platelet count 279,000 per cubic millimeter. The serum ferritin level was 18 ng per milliliter (normal range, 13-150), iron 28 µg per deciliter (normal range, 50-170), total iron-binding capacity 364 µg per deciliter (normal range, 250-450), and iron saturation 8% (normal range, 20-55). The vitamin B12 level was 621 pg per milliliter (normal range, 232-1,245) and thyroid-stimulating hormone level 1.87 units per milliliter (normal range, 0.50-4.50). Electrolyte and aminotransferase levels were within normal limits. CRP was 1.0 mg per deciliter (normal range, <0.5) and erythrocyte sedimentation rate 33 millimeters per hour (normal range, 4-25). Hepatitis C and HIV antibodies were nonreactive.
The ongoing iron deficiency despite parenteral iron replacement raises the question of ongoing gastrointestinal or genitourinary blood loss. While the level of vitamin B12 in the serum may be misleadingly normal with cobalamin deficiency, a methylmalonic acid level is indicated to evaluate whether tissue stores are depleted. Copper levels are warranted given the prior bariatric surgery. The mild elevations of inflammatory markers are nonspecific but reduce the likelihood of a highly inflammatory process to account for the neurological and abdominal symptoms.
At her 3-month follow-up visit, she noted that the paresthesia had improved and was now limited to her bilateral lower extremities. During the same clinic visit, she experienced a 45-minute episode of ascending left upper extremity numbness. Her physical examination revealed normal strength and reflexes. She had diminished response to pinprick in both legs to the knees and in both hands to the wrists. Vibration sense was diminished in the bilateral lower extremities.
A glycosylated hemoglobin (HbA1c) level was 6.2%. Methylmalonic acid was 69 nmol per liter (normal range, 45-325). Antibodies to Borrelia burgdorferi and Treponema pallidum were absent. Impaired glucose metabolism was the leading diagnosis for her polyneuropathy, and it was recommended that she undergo an oral glucose tolerance test. Electromyography was not performed.
The neurological symptoms are now chronic, and importantly, the patient has developed sensory deficits on neurological examination, suggesting worsening of the underlying process. While the paresthesia is now limited to a “stocking/glove” distribution consistent with distal sensory polyneuropathy, there should still be a concern for spinal cord pathology given that the HbA1c level of 6.2 would not explain her initial distribution of symptoms. Myelopathy may mimic peripheral nerve disease if, for example, there is involvement of the dorsal columns leading to sensory deficits of vibration and proprioception. Additionally, the transient episode of upper extremity numbness raises the question of sensory nerve root involvement (ie, sensory radiculopathy). Unexplained abdominal pain could possibly represent the involvement of other nerve roots innervating the abdominal wall. The patient’s episode of focal arm numbness recalls the lancinating radicular pain of tabes dorsalis; however, the negative specific treponemal antibody test excludes neurosyphilis.
The differential diagnosis going forward will be strongly conditioned by the localization of the neurological lesion(s). To differentiate between myelopathy, radiculopathy, and peripheral neuropathy, I would perform nerve conduction studies, magnetic resonance imaging (MRI) of the spinal cord, and cerebrospinal fluid analysis.
The patient began taking a multivitamin, and after weeks her paresthesia had resolved. One month later, she developed an intermittent, throbbing left-sided headache and pain behind the left eye that was worsened with ocular movement. She then noted decreased visual acuity in her left eye that progressed the following month. She denied photophobia, flashers, or floaters.
In the emergency department, visual acuity was 20/25 in her right eye; in the left eye she was only able to count fingers. Extraocular movements of both eyes were normal as was her right pupillary reflex. Red desaturation and a relative afferent papillary defect were present in the left eye. Fundoscopic exam demonstrated left optic disc swelling. The remainder of her cranial nerves were normal. She had pronation of the left upper extremity and mild right finger-to-nose dysmetria. Muscle tone, strength, sensation, and deep tendon reflexes were normal.
The improvement in the sensory symptoms was unlikely to be related to the nutritional intervention and provides a clue to an underlying waxing and waning illness. That interpretation is supported by the subsequent development of new visual symptoms and signs, which point to optic nerve pathology. Optic neuropathy has a broad differential diagnosis that includes ischemic, metabolic, toxic, and compressive causes. Eye pain, swelling of the optic disc, and prominent impairment of color vision all point to the more specific syndrome of optic neuritis caused by infections (including both Treponema pallidum and Borrelia species), systemic autoimmune diseases (systemic lupus erythematosus or Sjogren’s syndrome), and central nervous system (CNS) demyelinating diseases. Of these, inflammatory demyelinating processes would be the likeliest explanation of intermittent and improving neurologic findings.
With relapsing symptoms and findings that are separate in distribution and time, two diagnoses become most likely, and both of these are most often diagnosed in young women. MS is common, and optic neuritis occurs in more than 50% of patients over the course of illness. Neuromyelitis optica spectrum disorder (NMOSD) is a rare condition that can exist in isolation or be associated with other autoimmune illnesses. While these entities are difficult to differentiate clinically, neuroimaging that demonstrates extensive intracerebral demyelinating lesions and cerebrospinal fluid with oligoclonal bands favor MS, whereas extensive, predominant spinal cord involvement is suggestive of NMOSD. Approximately 70% of NMO patients harbor an antibody directed against the aquaporin-4 channel, and these antibodies are not seen in patients with MS. A milder NMO-like disorder has also been associated with antimyelin oligodendrocyte antibodies (MOG).
Testing for antinuclear antibodies, anti–double-stranded DNA, anti-Ro (SSA), and anti-La (SSB) antibodies was negative. The level of C3 was 162 mg per deciliter (normal range 81-157) and C4 38 (normal range 13-39). T-spot testing for latent tuberculosis was negative.
There is no serological evidence of active systemic lupus erythematosus or Sjogren’s syndrome. The pretest probability of CNS tuberculosis was low in light of her presenting complaints, relatively protracted course, and overall clinical stability without antituberculous therapy. Tests for latent tuberculosis infection have significant limitations of both sensitivity and specificity for the diagnosis of active disease.
Optical coherence tomography showed optic disc edema in the left eye only. MRI of the head with contrast revealed abnormal signal intensity involving the posterior aspect of the pons, right middle cerebellar peduncle, anterior left temporal lobe, bilateral periventricular white matter, subcortical white matter of the frontal lobes bilaterally, and medulla with abnormal signal and enhancement of the left optic nerve (Figure, Panel A). MRI of the cervical and thoracic spine demonstrated multifocal demyelinating lesions at C3, C4, C7, T4, T5, T7, and T8 (Figure, Panel B). The lesions were not longitudinally extensive. There was no significant postcontrast enhancement to suggest active demyelination.
The cerebrospinal fluid analysis revealed glucose of 105 mg per deciliter and a total protein of 26.1 mg per deciliter. In the fourth tube, there were 20 red cells per cubic and four white cells with a differential of 62% neutrophils, 35% lymphocytes, and 3% monocytes. Epstein-Barr and herpes simplex virus DNA were negative. A Venereal Disease Research Laboratory test was negative. Multiple oligoclonal IgG bands were identified only in the cerebrospinal fluid. Aquaporin-4 IgG and MOG antibodies were negative.
In addition to the expected finding of enhancement of the optic nerve, MRI demonstrated numerous multifocal white matter lesions throughout the cerebrum, brainstem, and spinal cord. Many of the lesions were in “silent” areas, which is not directly attributable to specific symptoms, but several did correlate with the subtler deficits of weakness and dysmetria that were noted on examination. Although such lesions may be seen with a diverse group of systemic diseases including adrenal leukodystrophy, sarcoidosis, Behcet’s, cerebral lupus, and vasculitis, primary CNS inflammatory demyelinating diseases are much more likely. The extensive distribution of demyelination argues against NMOSD. The negative aquaporin-4 and MOG assays support this conclusion. Not all multifocal CNS demyelination is caused by MS and can be seen in posterior reversible encephalopathy syndrome, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, and adult polyglucosan body disease. Osmotic demyelination is increasingly being recognized as a process that can be more widespread rather than just being limited to the pons. Viral infections of the CNS such as the JC virus (PML) may also provoke multifocal demyelination. Acute disseminated encephalomyelitis is most often seen during childhood, usually after vaccination or after an infectious prodrome. The tempo of the progression of these other diseases tends to be much more rapid than this woman’s course, and often, the neurological deficits are more profound and debilitating. The clinical presentation of sensory-predominant myelopathy, followed by optic neuritis, absence of systemic inflammatory signs or laboratory markers, exclusion of other relevant diseases, multifocal white matter lesions on imaging, minimal pleocytosis, and presence of oligoclonal bands in cerebrospinal fluid, all point to a diagnosis of relapsing-remitting MS.
The patient was diagnosed with MS. She was admitted to the neurology service and treated with 1,000 mg IV methylprednisolone for 3 days with a prompt improvement in her vision. She was started on natalizumab without a relapse of symptoms over the past year.
COMMENTARY
Multiple sclerosis is a chronic demyelinating disease of the CNS.1 The diagnosis of MS has classically been based upon compatible clinical and radiographic evidence of pathology that is disseminated in space and time. Patients typically present with an initial clinically isolated syndrome—involving changes in vision, sensation, strength, mobility, or cognition—for which there is radiographic evidence of demyelination.2 A diagnosis of clinically definite MS is then often made based on a subsequent relapse of symptoms.3
An interval from initial symptoms has been central to the diagnosis of MS (“lesions disseminated in time”). However, recent evidence questions this diagnostic paradigm, and a more rapid diagnosis of MS has been recommended. This recommendation is reflected in the updated McDonald criteria, according to which, if a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.4 The importance of such early diagnosis has been supported by numerous studies that have demonstrated improved clinical outcomes with early therapy.5-7
Despite the McDonald criteria, delays in definitive diagnosis are common in MS. Patients with MS in Spain were found to experience a 2-year delay from the first onset of symptoms to diagnosis.8 In this cohort, patients exhibited delays in presenting to a healthcare provider, as well as delays in diagnosis with an average time from seeing an initial provider to diagnosis of 6 months. When patients who were referred for a demyelinating episode were surveyed, over a third reported a prior suggestive event.9 The time from the first suggestive episode to referral to a neurologist for a recognized demyelinating event was 46 months. Other studies have shown that delays in diagnosis are especially common in younger patients, those with primary progressive MS, and those with comorbid disease.10,11
Misapplication of an MS diagnosis also occurs frequently. In one case series, such misapplication was found most often in cases involving migraine, fibromyalgia, psychogenic disorders, and NMOSD.12 NMOSD is distinguished from MS by the presence of typical brain and spine findings on MRI.13 Antibodies to aquaporin-4 are highly specific and moderately sensitive for the disease.14 It is important to distinguish NMOSD from MS as certain disease-modifying drugs used for MS might actually exacerbate NMOSD.15 A lesion that traverses over three or more contiguous vertebral segments with predominant involvement of central gray matter (ie, longitudinally extensive transverse myelitis) on MRI is the most distinct finding of NMOSD. In contrast, similar to our patient, short and often multiple lesions are demonstrated on spinal cord MRI in patients with MS. Sensitive and specific findings of brain MRI in patients with MS include the presence of lateral ventricle and inferior temporal lobe lesion, Dawson’s fingers, central vein sign, or an S-shaped U-fiber lesion. In NMOSD, brain MRI might reveal periependymal lesions surrounding the ventricular system.
This case highlights the diagnostic challenges related to presentations of a waxing and waning neurological process. At the time of the second evaluation, the presentation was interpreted as a length-dependent polyneuropathy due to glucose intolerance. Our patient’s relatively normal HbA1c, subacute onset of neuropathic symptoms (ie, <4 weeks), sensory and motor complaints, and onset in the upper extremities suggested an alternative diagnosis to prediabetes. Once the patient presented with optic neuritis, the cause of the initial symptoms was obvious, but then, hindsight is 20/20.
TEACHING POINTS
- Early treatment of MS results in improved clinical outcomes.
- Delays in the definitive diagnosis of MS are common, especially in younger patients, those with primary progressive MS, and those with comorbid disease.
- If a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis of MS can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.
Acknowledgments
The authors wish to thank Rabih Geha, MD, and Gurpreet Dhaliwal, MD, for providing feedback on an earlier version of this manuscript.
A 38-year-old woman presented to her primary care clinic with 3 weeks of progressive numbness and tingling sensation, which began in both hands and then progressed to involv
As with all neurological complaints, localization of the process will often inform a more specific differential diagnosis. If both sensory and motor findings are present, both central and peripheral nerve processes deserve consideration. The onset of paresthesia in the hands, rapid progression to the trunk, and unilateral leg weakness would be inconsistent with a length-dependent peripheral neuropathy. The distribution of complaints and the sacral sparing suggests a myelopathic process involving the cervical region rather than a cauda equina or conus lesions. In an otherwise healthy person of this age and gender, an inflammatory demyelinating disease affecting the cord including multiple sclerosis (MS) would be a strong consideration, although metabolic, vascular, infectious, compressive, or neoplastic disease of the spinal cord could also present with similar subacute onset and pattern of deficits.
Her medical history included morbid obesity, dry eyes, depression, iron deficiency anemia requiring recurrent intravenous replenishment, and abnormal uterine bleeding. Her surgical history included gastric band placement 7 years earlier with removal 5 years later due to persistent gastroesophageal reflux disease, dysphagia, nausea, and vomiting. The gastric band removal was complicated by chronic abdominal pain. Her medications consisted of duloxetine, intermittent iron infusions, artificial tears, loratadine, and pregabalin. She was sexually active with her husband. She consumed alcohol occasionally but did not smoke tobacco or use illicit drugs.
On exam, her temperature was 36.6°C (97.8°F), blood pressure 132/84 mm Hg, and heart rate 85 beats per minute. Body mass index was 39.5 kg/m2. The cardiac, pulmonary, and skin examinations were normal. The abdomen was soft with diffuse tenderness to palpation without rebound or guarding. Examination of cranial nerves 2-12 was normal. Cognition, strength, proprioception, deep tendon reflexes, and light touch were all normal. Her gait was normal, and the Romberg test was negative.
The normal neurologic exam is reassuring but imperfectly sensitive and does not eliminate the possibility of underlying neuropathology. Bariatric surgery may result in an array of nutritional deficiencies such as vitamin E, B12, and copper, which can cause myelopathy and/or neuropathy. However, these abnormalities occur less frequently with gastric banding procedures. If her dry eyes are part of the sicca syndrome, an underlying autoimmune diathesis may be present. Her unexplained chronic abdominal pain prompts considering nonmenstrual causes of iron deficiency anemia, such as celiac disease. Bariatric surgery may contribute to iron deficiency through impaired iron absorption. Her stable weight and lack of diarrhea argue against Crohn’s or celiac disease. Iron deficiency predisposes individuals to pica, most commonly described with ice chip ingestion. If lead pica had occurred, abdominal and neurological symptoms could result. Nevertheless, the abdominal pain is nonspecific, and its occurrence after gastric band removal makes its link to her neurologic syndrome unclear. An initial evaluation would include basic metabolic panel, complete blood count with differential, erythrocyte sedimentation rate, C-reactive protein (CRP), thyroid-stimulating hormone, vitamin B12, and copper levels.
A basic metabolic panel was normal. The white cell count was 5,710 per cubic millimeter, hemoglobin level 12.2 g per deciliter, mean corpuscular volume 85.2 fl, and platelet count 279,000 per cubic millimeter. The serum ferritin level was 18 ng per milliliter (normal range, 13-150), iron 28 µg per deciliter (normal range, 50-170), total iron-binding capacity 364 µg per deciliter (normal range, 250-450), and iron saturation 8% (normal range, 20-55). The vitamin B12 level was 621 pg per milliliter (normal range, 232-1,245) and thyroid-stimulating hormone level 1.87 units per milliliter (normal range, 0.50-4.50). Electrolyte and aminotransferase levels were within normal limits. CRP was 1.0 mg per deciliter (normal range, <0.5) and erythrocyte sedimentation rate 33 millimeters per hour (normal range, 4-25). Hepatitis C and HIV antibodies were nonreactive.
The ongoing iron deficiency despite parenteral iron replacement raises the question of ongoing gastrointestinal or genitourinary blood loss. While the level of vitamin B12 in the serum may be misleadingly normal with cobalamin deficiency, a methylmalonic acid level is indicated to evaluate whether tissue stores are depleted. Copper levels are warranted given the prior bariatric surgery. The mild elevations of inflammatory markers are nonspecific but reduce the likelihood of a highly inflammatory process to account for the neurological and abdominal symptoms.
At her 3-month follow-up visit, she noted that the paresthesia had improved and was now limited to her bilateral lower extremities. During the same clinic visit, she experienced a 45-minute episode of ascending left upper extremity numbness. Her physical examination revealed normal strength and reflexes. She had diminished response to pinprick in both legs to the knees and in both hands to the wrists. Vibration sense was diminished in the bilateral lower extremities.
A glycosylated hemoglobin (HbA1c) level was 6.2%. Methylmalonic acid was 69 nmol per liter (normal range, 45-325). Antibodies to Borrelia burgdorferi and Treponema pallidum were absent. Impaired glucose metabolism was the leading diagnosis for her polyneuropathy, and it was recommended that she undergo an oral glucose tolerance test. Electromyography was not performed.
The neurological symptoms are now chronic, and importantly, the patient has developed sensory deficits on neurological examination, suggesting worsening of the underlying process. While the paresthesia is now limited to a “stocking/glove” distribution consistent with distal sensory polyneuropathy, there should still be a concern for spinal cord pathology given that the HbA1c level of 6.2 would not explain her initial distribution of symptoms. Myelopathy may mimic peripheral nerve disease if, for example, there is involvement of the dorsal columns leading to sensory deficits of vibration and proprioception. Additionally, the transient episode of upper extremity numbness raises the question of sensory nerve root involvement (ie, sensory radiculopathy). Unexplained abdominal pain could possibly represent the involvement of other nerve roots innervating the abdominal wall. The patient’s episode of focal arm numbness recalls the lancinating radicular pain of tabes dorsalis; however, the negative specific treponemal antibody test excludes neurosyphilis.
The differential diagnosis going forward will be strongly conditioned by the localization of the neurological lesion(s). To differentiate between myelopathy, radiculopathy, and peripheral neuropathy, I would perform nerve conduction studies, magnetic resonance imaging (MRI) of the spinal cord, and cerebrospinal fluid analysis.
The patient began taking a multivitamin, and after weeks her paresthesia had resolved. One month later, she developed an intermittent, throbbing left-sided headache and pain behind the left eye that was worsened with ocular movement. She then noted decreased visual acuity in her left eye that progressed the following month. She denied photophobia, flashers, or floaters.
In the emergency department, visual acuity was 20/25 in her right eye; in the left eye she was only able to count fingers. Extraocular movements of both eyes were normal as was her right pupillary reflex. Red desaturation and a relative afferent papillary defect were present in the left eye. Fundoscopic exam demonstrated left optic disc swelling. The remainder of her cranial nerves were normal. She had pronation of the left upper extremity and mild right finger-to-nose dysmetria. Muscle tone, strength, sensation, and deep tendon reflexes were normal.
The improvement in the sensory symptoms was unlikely to be related to the nutritional intervention and provides a clue to an underlying waxing and waning illness. That interpretation is supported by the subsequent development of new visual symptoms and signs, which point to optic nerve pathology. Optic neuropathy has a broad differential diagnosis that includes ischemic, metabolic, toxic, and compressive causes. Eye pain, swelling of the optic disc, and prominent impairment of color vision all point to the more specific syndrome of optic neuritis caused by infections (including both Treponema pallidum and Borrelia species), systemic autoimmune diseases (systemic lupus erythematosus or Sjogren’s syndrome), and central nervous system (CNS) demyelinating diseases. Of these, inflammatory demyelinating processes would be the likeliest explanation of intermittent and improving neurologic findings.
With relapsing symptoms and findings that are separate in distribution and time, two diagnoses become most likely, and both of these are most often diagnosed in young women. MS is common, and optic neuritis occurs in more than 50% of patients over the course of illness. Neuromyelitis optica spectrum disorder (NMOSD) is a rare condition that can exist in isolation or be associated with other autoimmune illnesses. While these entities are difficult to differentiate clinically, neuroimaging that demonstrates extensive intracerebral demyelinating lesions and cerebrospinal fluid with oligoclonal bands favor MS, whereas extensive, predominant spinal cord involvement is suggestive of NMOSD. Approximately 70% of NMO patients harbor an antibody directed against the aquaporin-4 channel, and these antibodies are not seen in patients with MS. A milder NMO-like disorder has also been associated with antimyelin oligodendrocyte antibodies (MOG).
Testing for antinuclear antibodies, anti–double-stranded DNA, anti-Ro (SSA), and anti-La (SSB) antibodies was negative. The level of C3 was 162 mg per deciliter (normal range 81-157) and C4 38 (normal range 13-39). T-spot testing for latent tuberculosis was negative.
There is no serological evidence of active systemic lupus erythematosus or Sjogren’s syndrome. The pretest probability of CNS tuberculosis was low in light of her presenting complaints, relatively protracted course, and overall clinical stability without antituberculous therapy. Tests for latent tuberculosis infection have significant limitations of both sensitivity and specificity for the diagnosis of active disease.
Optical coherence tomography showed optic disc edema in the left eye only. MRI of the head with contrast revealed abnormal signal intensity involving the posterior aspect of the pons, right middle cerebellar peduncle, anterior left temporal lobe, bilateral periventricular white matter, subcortical white matter of the frontal lobes bilaterally, and medulla with abnormal signal and enhancement of the left optic nerve (Figure, Panel A). MRI of the cervical and thoracic spine demonstrated multifocal demyelinating lesions at C3, C4, C7, T4, T5, T7, and T8 (Figure, Panel B). The lesions were not longitudinally extensive. There was no significant postcontrast enhancement to suggest active demyelination.
The cerebrospinal fluid analysis revealed glucose of 105 mg per deciliter and a total protein of 26.1 mg per deciliter. In the fourth tube, there were 20 red cells per cubic and four white cells with a differential of 62% neutrophils, 35% lymphocytes, and 3% monocytes. Epstein-Barr and herpes simplex virus DNA were negative. A Venereal Disease Research Laboratory test was negative. Multiple oligoclonal IgG bands were identified only in the cerebrospinal fluid. Aquaporin-4 IgG and MOG antibodies were negative.
In addition to the expected finding of enhancement of the optic nerve, MRI demonstrated numerous multifocal white matter lesions throughout the cerebrum, brainstem, and spinal cord. Many of the lesions were in “silent” areas, which is not directly attributable to specific symptoms, but several did correlate with the subtler deficits of weakness and dysmetria that were noted on examination. Although such lesions may be seen with a diverse group of systemic diseases including adrenal leukodystrophy, sarcoidosis, Behcet’s, cerebral lupus, and vasculitis, primary CNS inflammatory demyelinating diseases are much more likely. The extensive distribution of demyelination argues against NMOSD. The negative aquaporin-4 and MOG assays support this conclusion. Not all multifocal CNS demyelination is caused by MS and can be seen in posterior reversible encephalopathy syndrome, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, and adult polyglucosan body disease. Osmotic demyelination is increasingly being recognized as a process that can be more widespread rather than just being limited to the pons. Viral infections of the CNS such as the JC virus (PML) may also provoke multifocal demyelination. Acute disseminated encephalomyelitis is most often seen during childhood, usually after vaccination or after an infectious prodrome. The tempo of the progression of these other diseases tends to be much more rapid than this woman’s course, and often, the neurological deficits are more profound and debilitating. The clinical presentation of sensory-predominant myelopathy, followed by optic neuritis, absence of systemic inflammatory signs or laboratory markers, exclusion of other relevant diseases, multifocal white matter lesions on imaging, minimal pleocytosis, and presence of oligoclonal bands in cerebrospinal fluid, all point to a diagnosis of relapsing-remitting MS.
The patient was diagnosed with MS. She was admitted to the neurology service and treated with 1,000 mg IV methylprednisolone for 3 days with a prompt improvement in her vision. She was started on natalizumab without a relapse of symptoms over the past year.
COMMENTARY
Multiple sclerosis is a chronic demyelinating disease of the CNS.1 The diagnosis of MS has classically been based upon compatible clinical and radiographic evidence of pathology that is disseminated in space and time. Patients typically present with an initial clinically isolated syndrome—involving changes in vision, sensation, strength, mobility, or cognition—for which there is radiographic evidence of demyelination.2 A diagnosis of clinically definite MS is then often made based on a subsequent relapse of symptoms.3
An interval from initial symptoms has been central to the diagnosis of MS (“lesions disseminated in time”). However, recent evidence questions this diagnostic paradigm, and a more rapid diagnosis of MS has been recommended. This recommendation is reflected in the updated McDonald criteria, according to which, if a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.4 The importance of such early diagnosis has been supported by numerous studies that have demonstrated improved clinical outcomes with early therapy.5-7
Despite the McDonald criteria, delays in definitive diagnosis are common in MS. Patients with MS in Spain were found to experience a 2-year delay from the first onset of symptoms to diagnosis.8 In this cohort, patients exhibited delays in presenting to a healthcare provider, as well as delays in diagnosis with an average time from seeing an initial provider to diagnosis of 6 months. When patients who were referred for a demyelinating episode were surveyed, over a third reported a prior suggestive event.9 The time from the first suggestive episode to referral to a neurologist for a recognized demyelinating event was 46 months. Other studies have shown that delays in diagnosis are especially common in younger patients, those with primary progressive MS, and those with comorbid disease.10,11
Misapplication of an MS diagnosis also occurs frequently. In one case series, such misapplication was found most often in cases involving migraine, fibromyalgia, psychogenic disorders, and NMOSD.12 NMOSD is distinguished from MS by the presence of typical brain and spine findings on MRI.13 Antibodies to aquaporin-4 are highly specific and moderately sensitive for the disease.14 It is important to distinguish NMOSD from MS as certain disease-modifying drugs used for MS might actually exacerbate NMOSD.15 A lesion that traverses over three or more contiguous vertebral segments with predominant involvement of central gray matter (ie, longitudinally extensive transverse myelitis) on MRI is the most distinct finding of NMOSD. In contrast, similar to our patient, short and often multiple lesions are demonstrated on spinal cord MRI in patients with MS. Sensitive and specific findings of brain MRI in patients with MS include the presence of lateral ventricle and inferior temporal lobe lesion, Dawson’s fingers, central vein sign, or an S-shaped U-fiber lesion. In NMOSD, brain MRI might reveal periependymal lesions surrounding the ventricular system.
This case highlights the diagnostic challenges related to presentations of a waxing and waning neurological process. At the time of the second evaluation, the presentation was interpreted as a length-dependent polyneuropathy due to glucose intolerance. Our patient’s relatively normal HbA1c, subacute onset of neuropathic symptoms (ie, <4 weeks), sensory and motor complaints, and onset in the upper extremities suggested an alternative diagnosis to prediabetes. Once the patient presented with optic neuritis, the cause of the initial symptoms was obvious, but then, hindsight is 20/20.
TEACHING POINTS
- Early treatment of MS results in improved clinical outcomes.
- Delays in the definitive diagnosis of MS are common, especially in younger patients, those with primary progressive MS, and those with comorbid disease.
- If a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis of MS can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.
Acknowledgments
The authors wish to thank Rabih Geha, MD, and Gurpreet Dhaliwal, MD, for providing feedback on an earlier version of this manuscript.
1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378:169-180. https://doi.org/10.1056/NEJMra140148.
2. Brownlee WJ, Hardy TA, Fazekas F, Miller DH. Diagnosis of multiple sclerosis: progress and challenges. Lancet. 2017;389(10076):1336-1346. https://doi.org/10.1016/S0140-6736(16)30959-X.
3. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622-1636. https://doi.org/10.1016/S0140-6736(18)30481-1.
4. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173. https://doi.org/10.1016/S1474-4422(17)30470-2.
5. Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017;389(10076):1347-1356. https://doi.org/10.1016/S0140-6736(16)32388-1.
6. Freedman MS, Comi G, De Stefano N, et al. Moving toward earlier treatment of multiple sclerosis: Findings from a decade of clinical trials and implications for clinical practice. Mult Scler Relat Disord. 2014;3(2):147-155. https://doi.org/10.1016/j.msard.2013.07.001.
7. Harding K, Williams O, Willis M, et al. Clinical outcomes of escalation vs early intensive disease-modifying therapy in patients with multiple sclerosis. JAMA Neurol. 2019;76(5):536-541. https://doi.org/10.1001/jamaneurol.2018.4905.
8. Fernández O, Fernández V, Arbizu T, et al. Characteristics of multiple sclerosis at onset and delay of diagnosis and treatment in Spain (the Novo Study). J Neurol. 257(9):1500-1507. https://doi.org/10.1007/s00415-010-5560-1.
9. Gout O, Lebrun-Frenay C, Labauge P, et al. Prior suggestive symptoms in one-third of patients consulting for a “first” demyelinating event. J Neurol Neurosurg Psychiatry 2011;82(3):323-325. https://doi.org/10.1136/jnnp.2008.166421.
10. Kingwell E, Leung A, Roger E, et al. Factors associated with delay to medical recognition in two Canadian multiple sclerosis cohorts. J Neurol Sci. 2010(1-2);292:57-62. https://doi.org/10.1016/j.jns.2010.02.007.
11. Marrie RA, Horwitz R, Cutter G, Tyry T, Campagnolo D, Vollmer T. Comorbidity delays diagnosis and increases disability at diagnosis in MS. Neurology. 2009;72(2):117-124. https://doi.org/10.1212/01.wnl.0000333252.78173.5f.
12. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: A multicenter study. Neurology. 2016;87(13):1393-1399. https://doi.org/10.1212/WNL.0000000000003152.
13. Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: An international update. Neurology. 2015;84(11):1165-1173. https://doi.org/10.1212/WNL.0000000000001367.
14. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189. https://doi.org/10.1212/WNL.0000000000001729.
15. Jacob A, Hutchinson M, Elsone L, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012;79(10):1065-1066. https://doi.org/10.1212/WNL.0b013e31826845fe.
1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378:169-180. https://doi.org/10.1056/NEJMra140148.
2. Brownlee WJ, Hardy TA, Fazekas F, Miller DH. Diagnosis of multiple sclerosis: progress and challenges. Lancet. 2017;389(10076):1336-1346. https://doi.org/10.1016/S0140-6736(16)30959-X.
3. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622-1636. https://doi.org/10.1016/S0140-6736(18)30481-1.
4. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173. https://doi.org/10.1016/S1474-4422(17)30470-2.
5. Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017;389(10076):1347-1356. https://doi.org/10.1016/S0140-6736(16)32388-1.
6. Freedman MS, Comi G, De Stefano N, et al. Moving toward earlier treatment of multiple sclerosis: Findings from a decade of clinical trials and implications for clinical practice. Mult Scler Relat Disord. 2014;3(2):147-155. https://doi.org/10.1016/j.msard.2013.07.001.
7. Harding K, Williams O, Willis M, et al. Clinical outcomes of escalation vs early intensive disease-modifying therapy in patients with multiple sclerosis. JAMA Neurol. 2019;76(5):536-541. https://doi.org/10.1001/jamaneurol.2018.4905.
8. Fernández O, Fernández V, Arbizu T, et al. Characteristics of multiple sclerosis at onset and delay of diagnosis and treatment in Spain (the Novo Study). J Neurol. 257(9):1500-1507. https://doi.org/10.1007/s00415-010-5560-1.
9. Gout O, Lebrun-Frenay C, Labauge P, et al. Prior suggestive symptoms in one-third of patients consulting for a “first” demyelinating event. J Neurol Neurosurg Psychiatry 2011;82(3):323-325. https://doi.org/10.1136/jnnp.2008.166421.
10. Kingwell E, Leung A, Roger E, et al. Factors associated with delay to medical recognition in two Canadian multiple sclerosis cohorts. J Neurol Sci. 2010(1-2);292:57-62. https://doi.org/10.1016/j.jns.2010.02.007.
11. Marrie RA, Horwitz R, Cutter G, Tyry T, Campagnolo D, Vollmer T. Comorbidity delays diagnosis and increases disability at diagnosis in MS. Neurology. 2009;72(2):117-124. https://doi.org/10.1212/01.wnl.0000333252.78173.5f.
12. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: A multicenter study. Neurology. 2016;87(13):1393-1399. https://doi.org/10.1212/WNL.0000000000003152.
13. Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: An international update. Neurology. 2015;84(11):1165-1173. https://doi.org/10.1212/WNL.0000000000001367.
14. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189. https://doi.org/10.1212/WNL.0000000000001729.
15. Jacob A, Hutchinson M, Elsone L, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012;79(10):1065-1066. https://doi.org/10.1212/WNL.0b013e31826845fe.
© 2020 Society of Hospital Medicine