User login
Painful Losses
A 58‐year‐old man presented to the emergency department with a 1‐month history of progressive, severe left hip pain that had become unbearable. The pain was constant and significantly worse with weight‐bearing, and the patient was now confined to bed. He denied back pain, falls, or trauma.
Although hip pain is a common complaint and a frequent manifestation of chronic degenerative joint disease, the debilitating and subacute nature of the pain suggests a potentially more serious underlying cause. Patients and even clinicians may refer to hip pain when the actual symptoms are periarticular, often presenting over the trochanter laterally, or muscular, presenting as posterior pain. The true hip joint is located in the anterior hip and groin area and often causes symptoms that radiate to the buttock. Pain can also be referred to the hip area from the spine, pelvis, or retroperitoneum, so it is crucial not to restrict the differential diagnosis to hip pathology.
Key diagnostic considerations include (1) inflammatory conditions such as trochanteric bursitis or gout; (2) bacterial infection of the hip joint, adjacent bone, or a nearby structure; (3) benign nerve compression (such as meralgia paresthetica); and (4) tumor (particularly myeloma or metastatic disease to the bone, but also potentially a pelvic or spinal mass with nerve compression). Polymyalgia rheumatica and other systemic rheumatologic complaints are a consideration, but because a single joint is involved, these conditions are less likely. The hip would be an unusual location for a first gout flare, and the duration of symptoms would be unusually long for gout. Avascular necrosis should be considered if the patient has received glucocorticoids for his previously diagnosed rheumatologic disease. If the patient is anticoagulated, consideration of spontaneous hematoma is reasonable, but usually this would present over a course of days, not weeks. The absence of trauma makes a fracture of the hip or pelvis less likely, and the insidious progression of symptoms makes a pathologic fracture less likely.
The patient reported 6 months of worsening proximal upper and lower extremity myalgia and weakness, with arthralgia of the hips and shoulders. The weakness was most notable in his proximal lower extremities, although he had remained ambulatory until the hip pain became limiting. He maintained normal use of his arms. The patient denied current rash but noted photosensitivity and a mild facial rash several months earlier. He described having transient mouth sores intermittently for several years. He denied fever, chills, night sweats, weight loss, dyspnea, recent travel, and outdoor exposures. Several months previously, he had been evaluated for these symptoms at another institution and given the diagnoses of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). At that time, he had initiated treatment with weekly dosing of methotrexate and etanercept.
The patient's medical history was also notable for hypertension, Graves' disease treated previously with radioiodine ablation, quiescent ulcerative colitis, and depression. Current medications included methotrexate, etanercept, levothyroxine, enalapril, hydrochlorothiazide, fluoxetine, ibuprofen, and oxycodone‐acetaminophen. He denied tobacco, alcohol, and recreational drug use.
Weakness occurring in the proximal lower extremities is the classic distribution for polymyositis and dermatomyositis. In contrast to polymyalgia rheumatica, dermatomyositis and polymyositis do not generally feature severe muscle pain, but they can be associated with a painful polyarthritis. Oral ulcers, photosensitivity, and facial rash are consistent with SLE, but dermatomyositis can also lead to a symmetrical erythema of the eyelids (commonly referred to as a heliotrope rash, named after the flower bearing that name) and sometimes can be associated with photosensitivity. Oral ulcers, particularly the painful ones known as canker sores, are extraordinarily common in the general population, and patients and providers may miss the mucosal lesions of SLE because they are usually painless. As methotrexate and etanercept are immunosuppressive, opportunistic pathogens such as typical or atypical mycobacteria and disseminated fungal infections should be considered, with special attention to the possibility of infection in or near the left hip. Given that SLE and RA rarely coexist, it would be helpful to seek outside medical records to know what the prior serologic evaluation entailed, but it is unlikely that this presentation is a manifestation of a diffuse connective tissue process.
Physical examination should focus on the features of dermatomyositis including heliotrope rash, truncal erythema, and papules over the knuckles (Gottron's papules); objective proximal muscle weakness in the shoulder and hip girdle; and findings that might suggest antisynthetase syndrome such as hyperkeratotic mechanic hand palmar and digital changes, and interstitial crackles on lung exam. If necrotic skin lesions are found, this would raise concern for a disseminated infection. The joints should be examined for inflammation and effusions.
His temperature was 36.6C, heart rate 74 beats per minute, blood pressure 134/76 mm Hg, respiratory rate 16 breaths per minute, and O2 saturation 97% on room air. He was obese but did not have moon facies or a buffalo hump. There were no rashes or mucosal lesions. Active and passive motion of his left hip joint elicited pain with both flexion/extension and internal/external rotation. Muscle strength was limited by pain in the left hip flexors and extenders, but was 5/5 in all other muscle groups. Palpation of the proximal muscles of his arms and legs did not elicit pain. His extremities were without edema, and examination of his shoulders, elbows, wrists, hands, knees, ankles, and feet did not reveal any erythema, synovial thickening, effusion, or deformity. Examination of the heart, chest, and abdomen was normal.
Given the reassuring strength examination, the absence of rashes or skin lesions, and the reassuring joint exam aside from the left hip, a focal infectious, inflammatory, or malignant process seems most likely. The pain with range of motion of the hip does not definitively localize the pathology to the hip joint, because pathology of the nearby structures can lead to pain when the hip is moved. Laboratory evaluation should include a complete blood count to screen for evidence of infection or marrow suppression, complete metabolic panel, and creatine kinase. The history of ulcerative colitis raises the possibility of an enthesitis (inflammation of tendons or ligaments) occurring near the hip. Enthesitis is sometimes a feature of the seronegative spondyloarthropathy‐associated conditions and can occur in the absence of sacroiliitis or spondyloarthropathy.
The patient's myalgias and arthralgias had recently been evaluated in the rheumatology clinic. Laboratory evaluation from that visit was remarkable only for an antinuclear antibody (ANA) test that was positive at a titer of 1:320 in a homogeneous pattern, creatine phosphokinase 366 IU/L (normal range [NR] 38240), and alkaline phosphatase 203 IU/L (NR 30130). All of the following labs from that visit were within normal ranges: cyclic citrullinated peptide, rheumatoid factor, antidouble stranded DNA, aldolase, complement levels, serum and urine protein electrophoresis, thyroglobulin antibody, thyroid microsomal antibody, thyroid‐stimulating hormone, erythrocyte sedimentation rate (10 mm/h), and C‐reactive protein (0.3 mg/dL).
The patient was admitted to the hospital. Initial blood test results on admission included sodium 139 mEq/L, potassium 3.9 mEq/L, chloride 105 mEq/L, bicarbonate 27 mEq/L, urea nitrogen 16 mg/dL, creatinine 0.6 mg/dL, glucose 85 mg/dL, calcium 9.2 mg/dL (NR 8.810.3), phosphate 1.3 mg/dL (NR 2.74.6), albumin 4.7 g/dL (NR 3.54.9), and alkaline phosphatase 195 IU/L (NR 30130). The remainder of a comprehensive metabolic profile, complete blood count with differential, and coagulation studies were all normal.
The homogeneous ANA titer of 1:320 is high enough to raise eyebrows, but is nonspecific for lupus and other ANA‐associated rheumatologic conditions, and may be a red herring, particularly given the low likelihood of a systemic inflammatory process explaining this new focal hip pain. The alkaline phosphatase is only mildly elevated and could be of bone or liver origin. The reassuringly low inflammatory markers are potentially helpful, because if checked now and substantially increased from the prior outpatient visit, they would be suggestive of a new inflammatory process. However, this would not point to a specific cause of inflammation.
Given the focality of the symptoms, imaging is warranted. As opposed to plain films, contrast‐enhanced computed tomography (CT) of the pelvis or magnetic resonance imaging (MRI) may be an efficient first step, because there is low suspicion for fracture and high suspicion for an inflammatory, neoplastic, or infectious process. MRI is more expensive and usually cannot be obtained as rapidly as CT. There is a chance that CT imaging alone will provide enough information to guide the next diagnostic steps, such as aspiration of an abscess or joint, or biopsy of a suspicious lesion. However, for soft tissue lesions and many bone lesions, including osteomyelitis, MRI offers better delineation of pathology.
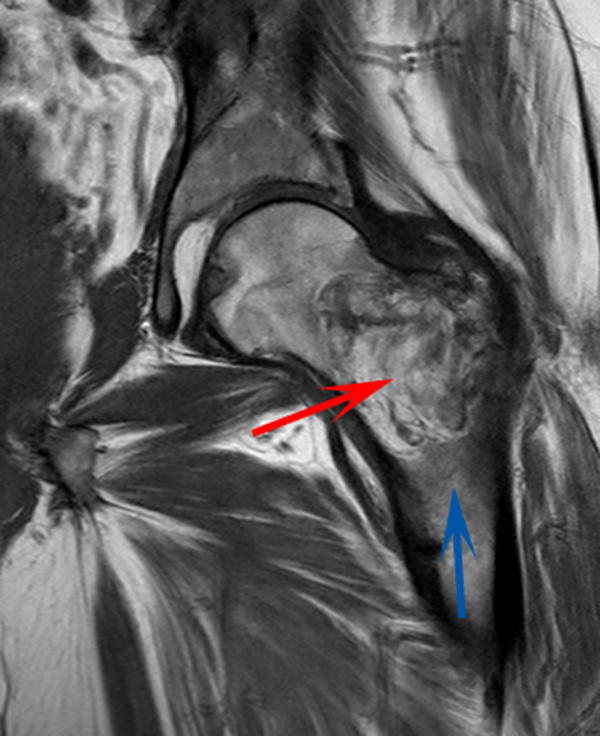
CT scan of the left femur demonstrated a large lytic lesion in the femoral neck that contained macroscopic fat and had an aggressive appearance with significant thinning of the cortex. MRI confirmed these findings and demonstrated a nondisplaced subtrochanteric femur fracture in the proximity of the lesion (Figure 1). Contrast‐enhanced CT scans of the thorax, abdomen, and pelvis revealed no other neoplastic lesions. Prostate‐specific antigen level was normal. The patient's significant hypophosphatemia persisted, with levels dropping to as low as 0.9 mg/dL despite aggressive oral phosphate replacement.
Although hypophosphatemia is often a nonspecific finding in hospitalized patients and is usually of little clinical importance, profound hypophosphatemia that is refractory to supplementation suggests an underlying metabolic disorder. Phosphate levels less than 1.0 mg/dL, particularly if prolonged, can lead to decreased adenosine triphosphate production and subsequent weakness of respiratory and cardiac muscles. Parathyroid hormone (PTH) excess and production of parathyroid hormone‐related protein (PTHrP) by a malignancy can cause profound hypophosphatemia, but are generally associated with hypercalcemia, a finding not seen in this case. Occasionally, tumors can lead to renal phosphate wasting via nonPTH‐related mechanisms. The best characterized example of this is the paraneoplastic syndrome oncogenic osteomalacia caused by tumor production of a fibroblast growth factor. Tumors that lead to this syndrome are usually benign mesenchymal tumors. This patient's tumor may be of this type, causing local destruction and metabolic disturbance. The next step would be consultation with orthopedic surgery for resection of the tumor and total hip arthroplasty with aggressive perioperative repletion of phosphate. Assessment of intact PTH, ionized calcium, 24‐hour urinary phosphate excretion, and even PTHrP levels may help to rule out other etiologies of hypophosphatemia, but given that surgery is needed regardless, it might be reasonable to proceed to the operating room without these diagnostics. If the phosphate levels return to normal postoperatively, then the diagnosis is clear and no further metabolic testing is needed.
PTH level was 47 pg/mL (NR 1065), 25‐hydroxyvitamin D level was 25 ng/mL (NR 2580), and 1,25‐dihydroxyvitamin D level was 18 pg/mL (NR 1872). Urinalysis was normal without proteinuria or glucosuria. A 24‐hour urine collection contained 1936 mg of phosphate (NR 4001200). The ratio of maximum rate of renal tubular reabsorption of phosphate to glomerular filtration rate (TmP/GFR) was 1.3 mg/dL (NR 2.44.2). Tissue obtained by CT‐guided needle biopsy of the femoral mass was consistent with a benign spindle cell neoplasm.
With normal calcium levels, the PTH level is appropriate, and hyperparathyroidism is excluded. The levels of 25‐hydroxyvitamin D and 1‐25‐dihydroxyvitamin D are not low enough to suggest that vitamin D deficiency is driving the impressive hypophosphatemia. What is impressive is the phosphate wasting demonstrated by the 24‐hour urine collection, consistent with paraneoplastic overproduction of fibroblast growth factor 23 (FGF23) by the benign bone tumor. Overproduction of this protein can be detected by blood tests or staining of the tumor specimen, but surgery should be performed as soon as possible independent of any further test results. Once the tumor is resected, phosphate metabolism should normalize.
FGF23 level was 266 RU/mL (NR < 180). The patient was diagnosed with tumor‐induced osteomalacia (TIO). He underwent complete resection of the femoral tumor as well as open reduction and internal fixation of the fracture. After surgery, his symptoms of pain and subjective muscle weakness improved, his serum phosphate level normalized, his need for phosphate supplementation resolved, and his blood levels of FGF23 decreased into the normal range (111 RU/mL). The rapid improvement of his symptoms after surgery suggested that they were related to TIO, and not manifestations of SLE or RA. His immunosuppressant medications were discontinued. Surgical pathology demonstrated a heterogeneous tumor consisting of sheets of uniform spindle cells interspersed with mature adipose tissue. This was diagnosed descriptively as a benign spindle cell and lipomatous neoplasm without further classification. Two months later, the patient was ambulating without pain, and muscle strength was subjectively normal.
DISCUSSION
TIO is a rare paraneoplastic syndrome affecting phosphate and vitamin D metabolism, leading to hypophosphatemia and osteomalacia.[1] TIO is caused by the inappropriate tumor secretion of the phosphatonin hormone, FGF23.
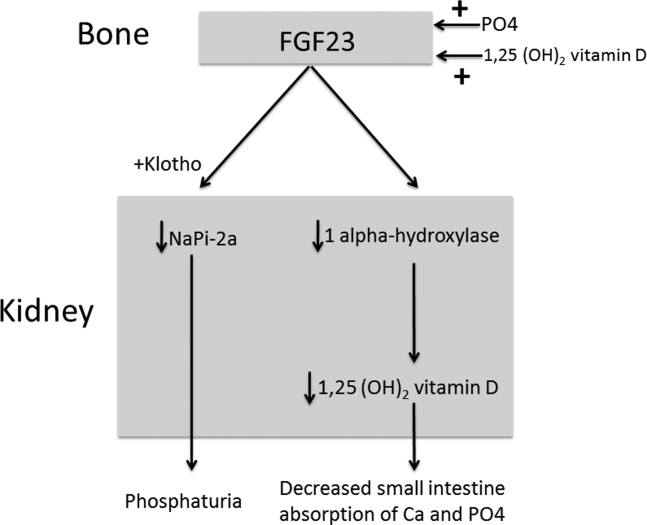
The normal physiology of FGF23 is illustrated in Figure 2. Osteocytes appear to be the primary source of FGF23, but the regulation of FGF23 production is not completely understood. FGF23 production may be influenced by several factors, including 1,25 dihydroxyvitamin D levels, and serum phosphate and PTH concentrations. This hormone binds to the FGF receptor and its coreceptor, Klotho,[2] causing 2 major physiological effects. First, it decreases the expression of the sodium‐phosphate cotransporters in the renal proximal tubular cells,[3, 4] resulting in increased tubular phosphate wasting. This effect appears to be partly PTH dependent.[5] Second, it has effects on vitamin D metabolism, decreasing renal production of activated vitamin D.[3, 4, 6]
In overproduction states, the elevated FGF23 leads to chronically low serum phosphate levels (with renal phosphate wasting) and the clinical syndrome of osteomalacia, manifested by bone pain, fractures, and deformities. Hypophosphatemia can also lead to painful proximal myopathy, cardiorespiratory dysfunction, and a spectrum of neuropsychiatric findings. The clinical findings in TIO are similar to those seen in genetic diseases in which hypophosphatemia results from the same mechanism.[3, 4]
In this case, measurement of the serum phosphate level was important in reaching the diagnosis. Although hypophosphatemia in the hospitalized patient is often easily explained, severe or persistent hypophosphatemia requires a focused evaluation. Causes of hypophosphatemia are categorized in Table 1.[7, 8, 9] In patients with hypophosphatemia that is not explained by the clinical situation (eg, osmotic diuresis, insulin treatment, refeeding syndrome, postparathyroidectomy, chronic diarrhea), measurement of serum calcium, PTH, and 25‐hydroxyvitamin D are used to investigate possible primary or secondary hyperparathyroidism. In addition, low‐normal or low serum 1,25‐dihydroxyvitamin D with normal PTH, normal 25‐hydroxyvitamin D stores, and normal renal function are clues to the presence of TIO. Urine phosphate wasting can be measured by collecting a 24‐hour urine sample. Calculation of the TmP/GFR (a measure of the maximum tubular resorption of phosphate relative to the glomerular filtration rate), as described by the nomogram of Walton and Bijvoet, may improve the accuracy of this assessment and confirm a renal source of the hypophosphatemia.[10]
|
| Internal redistribution |
| Insulin or catecholamine effect (including that related to refeeding syndrome, and infusion of glucose or TPN) |
| Acute respiratory alkalosis |
| Accelerated bone formation or rapid cell proliferation (eg, hungry bone syndrome, leukemic blast crisis, erythropoietin, or granulocyte colony stimulating factor administration) |
| Decreased absorption |
| Poor intake (including that seen in alcoholism*) |
| Vitamin D deficiency |
| Gastrointestinal losses (eg, chronic diarrhea) |
| Malabsorption (eg, phosphate‐binding antacids) |
| Urinary losses |
| Osmotic diuresis (eg, poorly controlled diabetes, acetazolamide) or volume expansion |
| Other diuretics: thiazides, indapamide |
| Hyperparathyroidism |
| Primary |
| Secondary (including vitamin D or calcium deficiency) |
| Parathyroid hormone‐related peptide |
| Renal tubular disease |
| Medications (eg, ethanol,* high‐dose glucocorticoids, cisplatin, bisphosphonates, estrogens, imatinib, acyclovir) |
| Fanconi syndrome |
| Medications inducing Fanconi syndrome: tenofovir, cidofovir, adefovir, aminoglycosides, ifosfamide, tetracyclines, valproic acid |
| Other (eg, postrenal transplant) |
| Excessive phosphatonin hormone activity (eg, hereditary syndromes [rickets], tumor‐induced osteomalacia) |
| Multifactorial causes |
| Alcoholism* |
| Acetaminophen toxicity |
| Parenteral iron administration |
The patient presented here had inappropriate urinary phosphate losses, and laboratory testing ruled out primary and secondary hyperparathyroidism and Fanconi syndrome. The patient was not taking medications known to cause tubular phosphate wasting. The patient's age and clinical history made hereditary syndromes unlikely. Therefore, the urinary phosphate wasting had to be related to an acquired defect in phosphate metabolism. The diagnostic characteristics of TIO are summarized in Table 2.
|
| Patients may present with symptoms of osteomalacia (eg, bone pain, fractures), hypophosphatemia (eg, proximal myopathy), and/or neoplasm. |
| Hypophosphatemia with urinary phosphate wasting. |
| Serum calcium level is usually normal. |
| Serum 1,25‐dihydroxyvitamin D level is usually low or low‐normal. |
| Parathyroid hormone is usually normal. |
| Plasma FGF23 level is elevated. |
| A neoplasm with the appropriate histology is identified, although the osteomalacia syndrome may precede identification of the tumor, which may be occult. |
| The syndrome resolves after complete resection of the tumor. |
The presence of a known neoplasm makes the diagnosis of TIO considerably easier. However, osteomalacia often precedes the tumor diagnosis. In these cases, the discovery of this clinical syndrome necessitates a search for the tumor. The tumors can be small, occult, and often located in the extremities. In addition to standard cross‐sectional imaging, specialized diagnostic modalities can be helpful in localizing culprit tumors. These include F‐18 flourodeoxyglucose positron emission tomography with computed tomography, 111‐Indium octreotide single photon emission CT/CT, 68‐Gallium‐DOTA‐octreotide positron emission tomography with computed tomography, and even selective venous sampling for FGF23 levels.[1, 11] The octreotide tests capitalize on the fact that culprit tumors often express somatostatin receptors.
TIO is most often associated with mesenchymal tumors of the bone or soft tissue. It has also been reported in association with several malignancies (small cell carcinoma, hematologic malignancies, prostate cancer), and with polyostotic fibrous dysplasia, neurofibromatosis, and the epidermal nevus syndrome. The mesenchymal tumors are heterogeneous in appearance and can be variably classified as hemangiopericytomas, hemangiomas, sarcomas, ossifying fibromas, granulomas, giant cell tumors, or osteoblastomas.[1] However, 1 review suggests that most of these tumors actually represent a distinct but heterogeneous, under‐recognized entity that is best classified as a phosphaturic mesenchymal tumor.[11]
TIO is only cured by complete resection of the tumor.[1] Local recurrences have been described, as have rare occurrences of metastatic disease.[1, 12] Medical treatment can be used to normalize serum phosphate levels in patients who are unable to be cured by surgery. The goal is to bring serum phosphate into the low‐normal range via phosphate supplementation (typically 13 g/day of elemental phosphorus is required) and treatment with either calcitriol or alfacalcidol. Due to the inhibition of 1,25‐dihydroxyvitamin D activation in TIO, relatively large doses of calcitriol are needed. A reasonable starting dose of calcitriol is 1.5 g/day, and most patients require 15 to 60 ng/kg per day. Because PTH action is involved in FGF23‐mediated hypophosphatemia, suppression of PTH may also be useful in these patients.[13]
This patient presented with a painful femoral tumor in the setting of muscle and joint pain that had been erroneously attributed to connective tissue disease. However, recognition and thorough evaluation of the patient's hypophosphatemia led to a unifying diagnosis of TIO. This diagnosis altered the surgical approach (emphasizing complete resection to eradicate the FGF23 production) and helped alleviate the patient's painful losses of phosphate.
TEACHING POINTS
- Hypophosphatemia, especially if severe or persistent, should not be dismissed as an unimportant laboratory finding. A focused evaluation should be performed to determine the etiology.
- In patients with unexplained hypophosphatemia, the measurement of serum calcium, parathyroid hormone, and vitamin D levels can identify primary or secondary hyperparathyroidism.
- The differential diagnosis of hypophosphatemia is narrowed if there is clinical evidence of inappropriate urinary phosphate wasting (ie, urinary phosphate levels remain high, despite low serum levels).
- TIO is a rare paraneoplastic syndrome caused by FGF23, a phosphatonin hormone that causes renal phosphate wasting, hypophosphatemia, and osteomalacia.
Disclosure: Nothing to report.
- , , , . Tumor‐induced osteomalacia. Endocr Relat Cancer. 2011;18:R53–R77.
- . The FGF23‐Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619.
- , . Genetic disorders of renal phosphate transport. N Engl J Med. 2010;362:2399–2409.
- . The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. 2012;30:1–9.
- , , , , . FGF‐23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492.
- , , , et al. FGF‐23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435.
- , . Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101.
- , , . In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: Elsevier; 2011:1237–1304.
- , , . Medication‐induced hypophosphatemia: a review. QJM. 2010;103:449–459.
- , . Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–310.
- , , , et al. Improving diagnosis of tumor‐induced osteomalacia with gallium‐68 DOTATATE PET/CT. J Clin Endocrinol Metab. 2013; 98:687–694.
- , , , et al. Most osteomalacia‐associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
- , , , , , . Cinacalcet in the management of tumor‐induced osteomalacia. J Bone Miner Res. 2007;22:931–937.
A 58‐year‐old man presented to the emergency department with a 1‐month history of progressive, severe left hip pain that had become unbearable. The pain was constant and significantly worse with weight‐bearing, and the patient was now confined to bed. He denied back pain, falls, or trauma.
Although hip pain is a common complaint and a frequent manifestation of chronic degenerative joint disease, the debilitating and subacute nature of the pain suggests a potentially more serious underlying cause. Patients and even clinicians may refer to hip pain when the actual symptoms are periarticular, often presenting over the trochanter laterally, or muscular, presenting as posterior pain. The true hip joint is located in the anterior hip and groin area and often causes symptoms that radiate to the buttock. Pain can also be referred to the hip area from the spine, pelvis, or retroperitoneum, so it is crucial not to restrict the differential diagnosis to hip pathology.
Key diagnostic considerations include (1) inflammatory conditions such as trochanteric bursitis or gout; (2) bacterial infection of the hip joint, adjacent bone, or a nearby structure; (3) benign nerve compression (such as meralgia paresthetica); and (4) tumor (particularly myeloma or metastatic disease to the bone, but also potentially a pelvic or spinal mass with nerve compression). Polymyalgia rheumatica and other systemic rheumatologic complaints are a consideration, but because a single joint is involved, these conditions are less likely. The hip would be an unusual location for a first gout flare, and the duration of symptoms would be unusually long for gout. Avascular necrosis should be considered if the patient has received glucocorticoids for his previously diagnosed rheumatologic disease. If the patient is anticoagulated, consideration of spontaneous hematoma is reasonable, but usually this would present over a course of days, not weeks. The absence of trauma makes a fracture of the hip or pelvis less likely, and the insidious progression of symptoms makes a pathologic fracture less likely.
The patient reported 6 months of worsening proximal upper and lower extremity myalgia and weakness, with arthralgia of the hips and shoulders. The weakness was most notable in his proximal lower extremities, although he had remained ambulatory until the hip pain became limiting. He maintained normal use of his arms. The patient denied current rash but noted photosensitivity and a mild facial rash several months earlier. He described having transient mouth sores intermittently for several years. He denied fever, chills, night sweats, weight loss, dyspnea, recent travel, and outdoor exposures. Several months previously, he had been evaluated for these symptoms at another institution and given the diagnoses of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). At that time, he had initiated treatment with weekly dosing of methotrexate and etanercept.
The patient's medical history was also notable for hypertension, Graves' disease treated previously with radioiodine ablation, quiescent ulcerative colitis, and depression. Current medications included methotrexate, etanercept, levothyroxine, enalapril, hydrochlorothiazide, fluoxetine, ibuprofen, and oxycodone‐acetaminophen. He denied tobacco, alcohol, and recreational drug use.
Weakness occurring in the proximal lower extremities is the classic distribution for polymyositis and dermatomyositis. In contrast to polymyalgia rheumatica, dermatomyositis and polymyositis do not generally feature severe muscle pain, but they can be associated with a painful polyarthritis. Oral ulcers, photosensitivity, and facial rash are consistent with SLE, but dermatomyositis can also lead to a symmetrical erythema of the eyelids (commonly referred to as a heliotrope rash, named after the flower bearing that name) and sometimes can be associated with photosensitivity. Oral ulcers, particularly the painful ones known as canker sores, are extraordinarily common in the general population, and patients and providers may miss the mucosal lesions of SLE because they are usually painless. As methotrexate and etanercept are immunosuppressive, opportunistic pathogens such as typical or atypical mycobacteria and disseminated fungal infections should be considered, with special attention to the possibility of infection in or near the left hip. Given that SLE and RA rarely coexist, it would be helpful to seek outside medical records to know what the prior serologic evaluation entailed, but it is unlikely that this presentation is a manifestation of a diffuse connective tissue process.
Physical examination should focus on the features of dermatomyositis including heliotrope rash, truncal erythema, and papules over the knuckles (Gottron's papules); objective proximal muscle weakness in the shoulder and hip girdle; and findings that might suggest antisynthetase syndrome such as hyperkeratotic mechanic hand palmar and digital changes, and interstitial crackles on lung exam. If necrotic skin lesions are found, this would raise concern for a disseminated infection. The joints should be examined for inflammation and effusions.
His temperature was 36.6C, heart rate 74 beats per minute, blood pressure 134/76 mm Hg, respiratory rate 16 breaths per minute, and O2 saturation 97% on room air. He was obese but did not have moon facies or a buffalo hump. There were no rashes or mucosal lesions. Active and passive motion of his left hip joint elicited pain with both flexion/extension and internal/external rotation. Muscle strength was limited by pain in the left hip flexors and extenders, but was 5/5 in all other muscle groups. Palpation of the proximal muscles of his arms and legs did not elicit pain. His extremities were without edema, and examination of his shoulders, elbows, wrists, hands, knees, ankles, and feet did not reveal any erythema, synovial thickening, effusion, or deformity. Examination of the heart, chest, and abdomen was normal.
Given the reassuring strength examination, the absence of rashes or skin lesions, and the reassuring joint exam aside from the left hip, a focal infectious, inflammatory, or malignant process seems most likely. The pain with range of motion of the hip does not definitively localize the pathology to the hip joint, because pathology of the nearby structures can lead to pain when the hip is moved. Laboratory evaluation should include a complete blood count to screen for evidence of infection or marrow suppression, complete metabolic panel, and creatine kinase. The history of ulcerative colitis raises the possibility of an enthesitis (inflammation of tendons or ligaments) occurring near the hip. Enthesitis is sometimes a feature of the seronegative spondyloarthropathy‐associated conditions and can occur in the absence of sacroiliitis or spondyloarthropathy.
The patient's myalgias and arthralgias had recently been evaluated in the rheumatology clinic. Laboratory evaluation from that visit was remarkable only for an antinuclear antibody (ANA) test that was positive at a titer of 1:320 in a homogeneous pattern, creatine phosphokinase 366 IU/L (normal range [NR] 38240), and alkaline phosphatase 203 IU/L (NR 30130). All of the following labs from that visit were within normal ranges: cyclic citrullinated peptide, rheumatoid factor, antidouble stranded DNA, aldolase, complement levels, serum and urine protein electrophoresis, thyroglobulin antibody, thyroid microsomal antibody, thyroid‐stimulating hormone, erythrocyte sedimentation rate (10 mm/h), and C‐reactive protein (0.3 mg/dL).
The patient was admitted to the hospital. Initial blood test results on admission included sodium 139 mEq/L, potassium 3.9 mEq/L, chloride 105 mEq/L, bicarbonate 27 mEq/L, urea nitrogen 16 mg/dL, creatinine 0.6 mg/dL, glucose 85 mg/dL, calcium 9.2 mg/dL (NR 8.810.3), phosphate 1.3 mg/dL (NR 2.74.6), albumin 4.7 g/dL (NR 3.54.9), and alkaline phosphatase 195 IU/L (NR 30130). The remainder of a comprehensive metabolic profile, complete blood count with differential, and coagulation studies were all normal.
The homogeneous ANA titer of 1:320 is high enough to raise eyebrows, but is nonspecific for lupus and other ANA‐associated rheumatologic conditions, and may be a red herring, particularly given the low likelihood of a systemic inflammatory process explaining this new focal hip pain. The alkaline phosphatase is only mildly elevated and could be of bone or liver origin. The reassuringly low inflammatory markers are potentially helpful, because if checked now and substantially increased from the prior outpatient visit, they would be suggestive of a new inflammatory process. However, this would not point to a specific cause of inflammation.
Given the focality of the symptoms, imaging is warranted. As opposed to plain films, contrast‐enhanced computed tomography (CT) of the pelvis or magnetic resonance imaging (MRI) may be an efficient first step, because there is low suspicion for fracture and high suspicion for an inflammatory, neoplastic, or infectious process. MRI is more expensive and usually cannot be obtained as rapidly as CT. There is a chance that CT imaging alone will provide enough information to guide the next diagnostic steps, such as aspiration of an abscess or joint, or biopsy of a suspicious lesion. However, for soft tissue lesions and many bone lesions, including osteomyelitis, MRI offers better delineation of pathology.
CT scan of the left femur demonstrated a large lytic lesion in the femoral neck that contained macroscopic fat and had an aggressive appearance with significant thinning of the cortex. MRI confirmed these findings and demonstrated a nondisplaced subtrochanteric femur fracture in the proximity of the lesion (Figure 1). Contrast‐enhanced CT scans of the thorax, abdomen, and pelvis revealed no other neoplastic lesions. Prostate‐specific antigen level was normal. The patient's significant hypophosphatemia persisted, with levels dropping to as low as 0.9 mg/dL despite aggressive oral phosphate replacement.
Although hypophosphatemia is often a nonspecific finding in hospitalized patients and is usually of little clinical importance, profound hypophosphatemia that is refractory to supplementation suggests an underlying metabolic disorder. Phosphate levels less than 1.0 mg/dL, particularly if prolonged, can lead to decreased adenosine triphosphate production and subsequent weakness of respiratory and cardiac muscles. Parathyroid hormone (PTH) excess and production of parathyroid hormone‐related protein (PTHrP) by a malignancy can cause profound hypophosphatemia, but are generally associated with hypercalcemia, a finding not seen in this case. Occasionally, tumors can lead to renal phosphate wasting via nonPTH‐related mechanisms. The best characterized example of this is the paraneoplastic syndrome oncogenic osteomalacia caused by tumor production of a fibroblast growth factor. Tumors that lead to this syndrome are usually benign mesenchymal tumors. This patient's tumor may be of this type, causing local destruction and metabolic disturbance. The next step would be consultation with orthopedic surgery for resection of the tumor and total hip arthroplasty with aggressive perioperative repletion of phosphate. Assessment of intact PTH, ionized calcium, 24‐hour urinary phosphate excretion, and even PTHrP levels may help to rule out other etiologies of hypophosphatemia, but given that surgery is needed regardless, it might be reasonable to proceed to the operating room without these diagnostics. If the phosphate levels return to normal postoperatively, then the diagnosis is clear and no further metabolic testing is needed.
PTH level was 47 pg/mL (NR 1065), 25‐hydroxyvitamin D level was 25 ng/mL (NR 2580), and 1,25‐dihydroxyvitamin D level was 18 pg/mL (NR 1872). Urinalysis was normal without proteinuria or glucosuria. A 24‐hour urine collection contained 1936 mg of phosphate (NR 4001200). The ratio of maximum rate of renal tubular reabsorption of phosphate to glomerular filtration rate (TmP/GFR) was 1.3 mg/dL (NR 2.44.2). Tissue obtained by CT‐guided needle biopsy of the femoral mass was consistent with a benign spindle cell neoplasm.
With normal calcium levels, the PTH level is appropriate, and hyperparathyroidism is excluded. The levels of 25‐hydroxyvitamin D and 1‐25‐dihydroxyvitamin D are not low enough to suggest that vitamin D deficiency is driving the impressive hypophosphatemia. What is impressive is the phosphate wasting demonstrated by the 24‐hour urine collection, consistent with paraneoplastic overproduction of fibroblast growth factor 23 (FGF23) by the benign bone tumor. Overproduction of this protein can be detected by blood tests or staining of the tumor specimen, but surgery should be performed as soon as possible independent of any further test results. Once the tumor is resected, phosphate metabolism should normalize.
FGF23 level was 266 RU/mL (NR < 180). The patient was diagnosed with tumor‐induced osteomalacia (TIO). He underwent complete resection of the femoral tumor as well as open reduction and internal fixation of the fracture. After surgery, his symptoms of pain and subjective muscle weakness improved, his serum phosphate level normalized, his need for phosphate supplementation resolved, and his blood levels of FGF23 decreased into the normal range (111 RU/mL). The rapid improvement of his symptoms after surgery suggested that they were related to TIO, and not manifestations of SLE or RA. His immunosuppressant medications were discontinued. Surgical pathology demonstrated a heterogeneous tumor consisting of sheets of uniform spindle cells interspersed with mature adipose tissue. This was diagnosed descriptively as a benign spindle cell and lipomatous neoplasm without further classification. Two months later, the patient was ambulating without pain, and muscle strength was subjectively normal.
DISCUSSION
TIO is a rare paraneoplastic syndrome affecting phosphate and vitamin D metabolism, leading to hypophosphatemia and osteomalacia.[1] TIO is caused by the inappropriate tumor secretion of the phosphatonin hormone, FGF23.
The normal physiology of FGF23 is illustrated in Figure 2. Osteocytes appear to be the primary source of FGF23, but the regulation of FGF23 production is not completely understood. FGF23 production may be influenced by several factors, including 1,25 dihydroxyvitamin D levels, and serum phosphate and PTH concentrations. This hormone binds to the FGF receptor and its coreceptor, Klotho,[2] causing 2 major physiological effects. First, it decreases the expression of the sodium‐phosphate cotransporters in the renal proximal tubular cells,[3, 4] resulting in increased tubular phosphate wasting. This effect appears to be partly PTH dependent.[5] Second, it has effects on vitamin D metabolism, decreasing renal production of activated vitamin D.[3, 4, 6]
In overproduction states, the elevated FGF23 leads to chronically low serum phosphate levels (with renal phosphate wasting) and the clinical syndrome of osteomalacia, manifested by bone pain, fractures, and deformities. Hypophosphatemia can also lead to painful proximal myopathy, cardiorespiratory dysfunction, and a spectrum of neuropsychiatric findings. The clinical findings in TIO are similar to those seen in genetic diseases in which hypophosphatemia results from the same mechanism.[3, 4]
In this case, measurement of the serum phosphate level was important in reaching the diagnosis. Although hypophosphatemia in the hospitalized patient is often easily explained, severe or persistent hypophosphatemia requires a focused evaluation. Causes of hypophosphatemia are categorized in Table 1.[7, 8, 9] In patients with hypophosphatemia that is not explained by the clinical situation (eg, osmotic diuresis, insulin treatment, refeeding syndrome, postparathyroidectomy, chronic diarrhea), measurement of serum calcium, PTH, and 25‐hydroxyvitamin D are used to investigate possible primary or secondary hyperparathyroidism. In addition, low‐normal or low serum 1,25‐dihydroxyvitamin D with normal PTH, normal 25‐hydroxyvitamin D stores, and normal renal function are clues to the presence of TIO. Urine phosphate wasting can be measured by collecting a 24‐hour urine sample. Calculation of the TmP/GFR (a measure of the maximum tubular resorption of phosphate relative to the glomerular filtration rate), as described by the nomogram of Walton and Bijvoet, may improve the accuracy of this assessment and confirm a renal source of the hypophosphatemia.[10]
|
| Internal redistribution |
| Insulin or catecholamine effect (including that related to refeeding syndrome, and infusion of glucose or TPN) |
| Acute respiratory alkalosis |
| Accelerated bone formation or rapid cell proliferation (eg, hungry bone syndrome, leukemic blast crisis, erythropoietin, or granulocyte colony stimulating factor administration) |
| Decreased absorption |
| Poor intake (including that seen in alcoholism*) |
| Vitamin D deficiency |
| Gastrointestinal losses (eg, chronic diarrhea) |
| Malabsorption (eg, phosphate‐binding antacids) |
| Urinary losses |
| Osmotic diuresis (eg, poorly controlled diabetes, acetazolamide) or volume expansion |
| Other diuretics: thiazides, indapamide |
| Hyperparathyroidism |
| Primary |
| Secondary (including vitamin D or calcium deficiency) |
| Parathyroid hormone‐related peptide |
| Renal tubular disease |
| Medications (eg, ethanol,* high‐dose glucocorticoids, cisplatin, bisphosphonates, estrogens, imatinib, acyclovir) |
| Fanconi syndrome |
| Medications inducing Fanconi syndrome: tenofovir, cidofovir, adefovir, aminoglycosides, ifosfamide, tetracyclines, valproic acid |
| Other (eg, postrenal transplant) |
| Excessive phosphatonin hormone activity (eg, hereditary syndromes [rickets], tumor‐induced osteomalacia) |
| Multifactorial causes |
| Alcoholism* |
| Acetaminophen toxicity |
| Parenteral iron administration |
The patient presented here had inappropriate urinary phosphate losses, and laboratory testing ruled out primary and secondary hyperparathyroidism and Fanconi syndrome. The patient was not taking medications known to cause tubular phosphate wasting. The patient's age and clinical history made hereditary syndromes unlikely. Therefore, the urinary phosphate wasting had to be related to an acquired defect in phosphate metabolism. The diagnostic characteristics of TIO are summarized in Table 2.
|
| Patients may present with symptoms of osteomalacia (eg, bone pain, fractures), hypophosphatemia (eg, proximal myopathy), and/or neoplasm. |
| Hypophosphatemia with urinary phosphate wasting. |
| Serum calcium level is usually normal. |
| Serum 1,25‐dihydroxyvitamin D level is usually low or low‐normal. |
| Parathyroid hormone is usually normal. |
| Plasma FGF23 level is elevated. |
| A neoplasm with the appropriate histology is identified, although the osteomalacia syndrome may precede identification of the tumor, which may be occult. |
| The syndrome resolves after complete resection of the tumor. |
The presence of a known neoplasm makes the diagnosis of TIO considerably easier. However, osteomalacia often precedes the tumor diagnosis. In these cases, the discovery of this clinical syndrome necessitates a search for the tumor. The tumors can be small, occult, and often located in the extremities. In addition to standard cross‐sectional imaging, specialized diagnostic modalities can be helpful in localizing culprit tumors. These include F‐18 flourodeoxyglucose positron emission tomography with computed tomography, 111‐Indium octreotide single photon emission CT/CT, 68‐Gallium‐DOTA‐octreotide positron emission tomography with computed tomography, and even selective venous sampling for FGF23 levels.[1, 11] The octreotide tests capitalize on the fact that culprit tumors often express somatostatin receptors.
TIO is most often associated with mesenchymal tumors of the bone or soft tissue. It has also been reported in association with several malignancies (small cell carcinoma, hematologic malignancies, prostate cancer), and with polyostotic fibrous dysplasia, neurofibromatosis, and the epidermal nevus syndrome. The mesenchymal tumors are heterogeneous in appearance and can be variably classified as hemangiopericytomas, hemangiomas, sarcomas, ossifying fibromas, granulomas, giant cell tumors, or osteoblastomas.[1] However, 1 review suggests that most of these tumors actually represent a distinct but heterogeneous, under‐recognized entity that is best classified as a phosphaturic mesenchymal tumor.[11]
TIO is only cured by complete resection of the tumor.[1] Local recurrences have been described, as have rare occurrences of metastatic disease.[1, 12] Medical treatment can be used to normalize serum phosphate levels in patients who are unable to be cured by surgery. The goal is to bring serum phosphate into the low‐normal range via phosphate supplementation (typically 13 g/day of elemental phosphorus is required) and treatment with either calcitriol or alfacalcidol. Due to the inhibition of 1,25‐dihydroxyvitamin D activation in TIO, relatively large doses of calcitriol are needed. A reasonable starting dose of calcitriol is 1.5 g/day, and most patients require 15 to 60 ng/kg per day. Because PTH action is involved in FGF23‐mediated hypophosphatemia, suppression of PTH may also be useful in these patients.[13]
This patient presented with a painful femoral tumor in the setting of muscle and joint pain that had been erroneously attributed to connective tissue disease. However, recognition and thorough evaluation of the patient's hypophosphatemia led to a unifying diagnosis of TIO. This diagnosis altered the surgical approach (emphasizing complete resection to eradicate the FGF23 production) and helped alleviate the patient's painful losses of phosphate.
TEACHING POINTS
- Hypophosphatemia, especially if severe or persistent, should not be dismissed as an unimportant laboratory finding. A focused evaluation should be performed to determine the etiology.
- In patients with unexplained hypophosphatemia, the measurement of serum calcium, parathyroid hormone, and vitamin D levels can identify primary or secondary hyperparathyroidism.
- The differential diagnosis of hypophosphatemia is narrowed if there is clinical evidence of inappropriate urinary phosphate wasting (ie, urinary phosphate levels remain high, despite low serum levels).
- TIO is a rare paraneoplastic syndrome caused by FGF23, a phosphatonin hormone that causes renal phosphate wasting, hypophosphatemia, and osteomalacia.
Disclosure: Nothing to report.
A 58‐year‐old man presented to the emergency department with a 1‐month history of progressive, severe left hip pain that had become unbearable. The pain was constant and significantly worse with weight‐bearing, and the patient was now confined to bed. He denied back pain, falls, or trauma.
Although hip pain is a common complaint and a frequent manifestation of chronic degenerative joint disease, the debilitating and subacute nature of the pain suggests a potentially more serious underlying cause. Patients and even clinicians may refer to hip pain when the actual symptoms are periarticular, often presenting over the trochanter laterally, or muscular, presenting as posterior pain. The true hip joint is located in the anterior hip and groin area and often causes symptoms that radiate to the buttock. Pain can also be referred to the hip area from the spine, pelvis, or retroperitoneum, so it is crucial not to restrict the differential diagnosis to hip pathology.
Key diagnostic considerations include (1) inflammatory conditions such as trochanteric bursitis or gout; (2) bacterial infection of the hip joint, adjacent bone, or a nearby structure; (3) benign nerve compression (such as meralgia paresthetica); and (4) tumor (particularly myeloma or metastatic disease to the bone, but also potentially a pelvic or spinal mass with nerve compression). Polymyalgia rheumatica and other systemic rheumatologic complaints are a consideration, but because a single joint is involved, these conditions are less likely. The hip would be an unusual location for a first gout flare, and the duration of symptoms would be unusually long for gout. Avascular necrosis should be considered if the patient has received glucocorticoids for his previously diagnosed rheumatologic disease. If the patient is anticoagulated, consideration of spontaneous hematoma is reasonable, but usually this would present over a course of days, not weeks. The absence of trauma makes a fracture of the hip or pelvis less likely, and the insidious progression of symptoms makes a pathologic fracture less likely.
The patient reported 6 months of worsening proximal upper and lower extremity myalgia and weakness, with arthralgia of the hips and shoulders. The weakness was most notable in his proximal lower extremities, although he had remained ambulatory until the hip pain became limiting. He maintained normal use of his arms. The patient denied current rash but noted photosensitivity and a mild facial rash several months earlier. He described having transient mouth sores intermittently for several years. He denied fever, chills, night sweats, weight loss, dyspnea, recent travel, and outdoor exposures. Several months previously, he had been evaluated for these symptoms at another institution and given the diagnoses of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). At that time, he had initiated treatment with weekly dosing of methotrexate and etanercept.
The patient's medical history was also notable for hypertension, Graves' disease treated previously with radioiodine ablation, quiescent ulcerative colitis, and depression. Current medications included methotrexate, etanercept, levothyroxine, enalapril, hydrochlorothiazide, fluoxetine, ibuprofen, and oxycodone‐acetaminophen. He denied tobacco, alcohol, and recreational drug use.
Weakness occurring in the proximal lower extremities is the classic distribution for polymyositis and dermatomyositis. In contrast to polymyalgia rheumatica, dermatomyositis and polymyositis do not generally feature severe muscle pain, but they can be associated with a painful polyarthritis. Oral ulcers, photosensitivity, and facial rash are consistent with SLE, but dermatomyositis can also lead to a symmetrical erythema of the eyelids (commonly referred to as a heliotrope rash, named after the flower bearing that name) and sometimes can be associated with photosensitivity. Oral ulcers, particularly the painful ones known as canker sores, are extraordinarily common in the general population, and patients and providers may miss the mucosal lesions of SLE because they are usually painless. As methotrexate and etanercept are immunosuppressive, opportunistic pathogens such as typical or atypical mycobacteria and disseminated fungal infections should be considered, with special attention to the possibility of infection in or near the left hip. Given that SLE and RA rarely coexist, it would be helpful to seek outside medical records to know what the prior serologic evaluation entailed, but it is unlikely that this presentation is a manifestation of a diffuse connective tissue process.
Physical examination should focus on the features of dermatomyositis including heliotrope rash, truncal erythema, and papules over the knuckles (Gottron's papules); objective proximal muscle weakness in the shoulder and hip girdle; and findings that might suggest antisynthetase syndrome such as hyperkeratotic mechanic hand palmar and digital changes, and interstitial crackles on lung exam. If necrotic skin lesions are found, this would raise concern for a disseminated infection. The joints should be examined for inflammation and effusions.
His temperature was 36.6C, heart rate 74 beats per minute, blood pressure 134/76 mm Hg, respiratory rate 16 breaths per minute, and O2 saturation 97% on room air. He was obese but did not have moon facies or a buffalo hump. There were no rashes or mucosal lesions. Active and passive motion of his left hip joint elicited pain with both flexion/extension and internal/external rotation. Muscle strength was limited by pain in the left hip flexors and extenders, but was 5/5 in all other muscle groups. Palpation of the proximal muscles of his arms and legs did not elicit pain. His extremities were without edema, and examination of his shoulders, elbows, wrists, hands, knees, ankles, and feet did not reveal any erythema, synovial thickening, effusion, or deformity. Examination of the heart, chest, and abdomen was normal.
Given the reassuring strength examination, the absence of rashes or skin lesions, and the reassuring joint exam aside from the left hip, a focal infectious, inflammatory, or malignant process seems most likely. The pain with range of motion of the hip does not definitively localize the pathology to the hip joint, because pathology of the nearby structures can lead to pain when the hip is moved. Laboratory evaluation should include a complete blood count to screen for evidence of infection or marrow suppression, complete metabolic panel, and creatine kinase. The history of ulcerative colitis raises the possibility of an enthesitis (inflammation of tendons or ligaments) occurring near the hip. Enthesitis is sometimes a feature of the seronegative spondyloarthropathy‐associated conditions and can occur in the absence of sacroiliitis or spondyloarthropathy.
The patient's myalgias and arthralgias had recently been evaluated in the rheumatology clinic. Laboratory evaluation from that visit was remarkable only for an antinuclear antibody (ANA) test that was positive at a titer of 1:320 in a homogeneous pattern, creatine phosphokinase 366 IU/L (normal range [NR] 38240), and alkaline phosphatase 203 IU/L (NR 30130). All of the following labs from that visit were within normal ranges: cyclic citrullinated peptide, rheumatoid factor, antidouble stranded DNA, aldolase, complement levels, serum and urine protein electrophoresis, thyroglobulin antibody, thyroid microsomal antibody, thyroid‐stimulating hormone, erythrocyte sedimentation rate (10 mm/h), and C‐reactive protein (0.3 mg/dL).
The patient was admitted to the hospital. Initial blood test results on admission included sodium 139 mEq/L, potassium 3.9 mEq/L, chloride 105 mEq/L, bicarbonate 27 mEq/L, urea nitrogen 16 mg/dL, creatinine 0.6 mg/dL, glucose 85 mg/dL, calcium 9.2 mg/dL (NR 8.810.3), phosphate 1.3 mg/dL (NR 2.74.6), albumin 4.7 g/dL (NR 3.54.9), and alkaline phosphatase 195 IU/L (NR 30130). The remainder of a comprehensive metabolic profile, complete blood count with differential, and coagulation studies were all normal.
The homogeneous ANA titer of 1:320 is high enough to raise eyebrows, but is nonspecific for lupus and other ANA‐associated rheumatologic conditions, and may be a red herring, particularly given the low likelihood of a systemic inflammatory process explaining this new focal hip pain. The alkaline phosphatase is only mildly elevated and could be of bone or liver origin. The reassuringly low inflammatory markers are potentially helpful, because if checked now and substantially increased from the prior outpatient visit, they would be suggestive of a new inflammatory process. However, this would not point to a specific cause of inflammation.
Given the focality of the symptoms, imaging is warranted. As opposed to plain films, contrast‐enhanced computed tomography (CT) of the pelvis or magnetic resonance imaging (MRI) may be an efficient first step, because there is low suspicion for fracture and high suspicion for an inflammatory, neoplastic, or infectious process. MRI is more expensive and usually cannot be obtained as rapidly as CT. There is a chance that CT imaging alone will provide enough information to guide the next diagnostic steps, such as aspiration of an abscess or joint, or biopsy of a suspicious lesion. However, for soft tissue lesions and many bone lesions, including osteomyelitis, MRI offers better delineation of pathology.
CT scan of the left femur demonstrated a large lytic lesion in the femoral neck that contained macroscopic fat and had an aggressive appearance with significant thinning of the cortex. MRI confirmed these findings and demonstrated a nondisplaced subtrochanteric femur fracture in the proximity of the lesion (Figure 1). Contrast‐enhanced CT scans of the thorax, abdomen, and pelvis revealed no other neoplastic lesions. Prostate‐specific antigen level was normal. The patient's significant hypophosphatemia persisted, with levels dropping to as low as 0.9 mg/dL despite aggressive oral phosphate replacement.
Although hypophosphatemia is often a nonspecific finding in hospitalized patients and is usually of little clinical importance, profound hypophosphatemia that is refractory to supplementation suggests an underlying metabolic disorder. Phosphate levels less than 1.0 mg/dL, particularly if prolonged, can lead to decreased adenosine triphosphate production and subsequent weakness of respiratory and cardiac muscles. Parathyroid hormone (PTH) excess and production of parathyroid hormone‐related protein (PTHrP) by a malignancy can cause profound hypophosphatemia, but are generally associated with hypercalcemia, a finding not seen in this case. Occasionally, tumors can lead to renal phosphate wasting via nonPTH‐related mechanisms. The best characterized example of this is the paraneoplastic syndrome oncogenic osteomalacia caused by tumor production of a fibroblast growth factor. Tumors that lead to this syndrome are usually benign mesenchymal tumors. This patient's tumor may be of this type, causing local destruction and metabolic disturbance. The next step would be consultation with orthopedic surgery for resection of the tumor and total hip arthroplasty with aggressive perioperative repletion of phosphate. Assessment of intact PTH, ionized calcium, 24‐hour urinary phosphate excretion, and even PTHrP levels may help to rule out other etiologies of hypophosphatemia, but given that surgery is needed regardless, it might be reasonable to proceed to the operating room without these diagnostics. If the phosphate levels return to normal postoperatively, then the diagnosis is clear and no further metabolic testing is needed.
PTH level was 47 pg/mL (NR 1065), 25‐hydroxyvitamin D level was 25 ng/mL (NR 2580), and 1,25‐dihydroxyvitamin D level was 18 pg/mL (NR 1872). Urinalysis was normal without proteinuria or glucosuria. A 24‐hour urine collection contained 1936 mg of phosphate (NR 4001200). The ratio of maximum rate of renal tubular reabsorption of phosphate to glomerular filtration rate (TmP/GFR) was 1.3 mg/dL (NR 2.44.2). Tissue obtained by CT‐guided needle biopsy of the femoral mass was consistent with a benign spindle cell neoplasm.
With normal calcium levels, the PTH level is appropriate, and hyperparathyroidism is excluded. The levels of 25‐hydroxyvitamin D and 1‐25‐dihydroxyvitamin D are not low enough to suggest that vitamin D deficiency is driving the impressive hypophosphatemia. What is impressive is the phosphate wasting demonstrated by the 24‐hour urine collection, consistent with paraneoplastic overproduction of fibroblast growth factor 23 (FGF23) by the benign bone tumor. Overproduction of this protein can be detected by blood tests or staining of the tumor specimen, but surgery should be performed as soon as possible independent of any further test results. Once the tumor is resected, phosphate metabolism should normalize.
FGF23 level was 266 RU/mL (NR < 180). The patient was diagnosed with tumor‐induced osteomalacia (TIO). He underwent complete resection of the femoral tumor as well as open reduction and internal fixation of the fracture. After surgery, his symptoms of pain and subjective muscle weakness improved, his serum phosphate level normalized, his need for phosphate supplementation resolved, and his blood levels of FGF23 decreased into the normal range (111 RU/mL). The rapid improvement of his symptoms after surgery suggested that they were related to TIO, and not manifestations of SLE or RA. His immunosuppressant medications were discontinued. Surgical pathology demonstrated a heterogeneous tumor consisting of sheets of uniform spindle cells interspersed with mature adipose tissue. This was diagnosed descriptively as a benign spindle cell and lipomatous neoplasm without further classification. Two months later, the patient was ambulating without pain, and muscle strength was subjectively normal.
DISCUSSION
TIO is a rare paraneoplastic syndrome affecting phosphate and vitamin D metabolism, leading to hypophosphatemia and osteomalacia.[1] TIO is caused by the inappropriate tumor secretion of the phosphatonin hormone, FGF23.
The normal physiology of FGF23 is illustrated in Figure 2. Osteocytes appear to be the primary source of FGF23, but the regulation of FGF23 production is not completely understood. FGF23 production may be influenced by several factors, including 1,25 dihydroxyvitamin D levels, and serum phosphate and PTH concentrations. This hormone binds to the FGF receptor and its coreceptor, Klotho,[2] causing 2 major physiological effects. First, it decreases the expression of the sodium‐phosphate cotransporters in the renal proximal tubular cells,[3, 4] resulting in increased tubular phosphate wasting. This effect appears to be partly PTH dependent.[5] Second, it has effects on vitamin D metabolism, decreasing renal production of activated vitamin D.[3, 4, 6]
In overproduction states, the elevated FGF23 leads to chronically low serum phosphate levels (with renal phosphate wasting) and the clinical syndrome of osteomalacia, manifested by bone pain, fractures, and deformities. Hypophosphatemia can also lead to painful proximal myopathy, cardiorespiratory dysfunction, and a spectrum of neuropsychiatric findings. The clinical findings in TIO are similar to those seen in genetic diseases in which hypophosphatemia results from the same mechanism.[3, 4]
In this case, measurement of the serum phosphate level was important in reaching the diagnosis. Although hypophosphatemia in the hospitalized patient is often easily explained, severe or persistent hypophosphatemia requires a focused evaluation. Causes of hypophosphatemia are categorized in Table 1.[7, 8, 9] In patients with hypophosphatemia that is not explained by the clinical situation (eg, osmotic diuresis, insulin treatment, refeeding syndrome, postparathyroidectomy, chronic diarrhea), measurement of serum calcium, PTH, and 25‐hydroxyvitamin D are used to investigate possible primary or secondary hyperparathyroidism. In addition, low‐normal or low serum 1,25‐dihydroxyvitamin D with normal PTH, normal 25‐hydroxyvitamin D stores, and normal renal function are clues to the presence of TIO. Urine phosphate wasting can be measured by collecting a 24‐hour urine sample. Calculation of the TmP/GFR (a measure of the maximum tubular resorption of phosphate relative to the glomerular filtration rate), as described by the nomogram of Walton and Bijvoet, may improve the accuracy of this assessment and confirm a renal source of the hypophosphatemia.[10]
|
| Internal redistribution |
| Insulin or catecholamine effect (including that related to refeeding syndrome, and infusion of glucose or TPN) |
| Acute respiratory alkalosis |
| Accelerated bone formation or rapid cell proliferation (eg, hungry bone syndrome, leukemic blast crisis, erythropoietin, or granulocyte colony stimulating factor administration) |
| Decreased absorption |
| Poor intake (including that seen in alcoholism*) |
| Vitamin D deficiency |
| Gastrointestinal losses (eg, chronic diarrhea) |
| Malabsorption (eg, phosphate‐binding antacids) |
| Urinary losses |
| Osmotic diuresis (eg, poorly controlled diabetes, acetazolamide) or volume expansion |
| Other diuretics: thiazides, indapamide |
| Hyperparathyroidism |
| Primary |
| Secondary (including vitamin D or calcium deficiency) |
| Parathyroid hormone‐related peptide |
| Renal tubular disease |
| Medications (eg, ethanol,* high‐dose glucocorticoids, cisplatin, bisphosphonates, estrogens, imatinib, acyclovir) |
| Fanconi syndrome |
| Medications inducing Fanconi syndrome: tenofovir, cidofovir, adefovir, aminoglycosides, ifosfamide, tetracyclines, valproic acid |
| Other (eg, postrenal transplant) |
| Excessive phosphatonin hormone activity (eg, hereditary syndromes [rickets], tumor‐induced osteomalacia) |
| Multifactorial causes |
| Alcoholism* |
| Acetaminophen toxicity |
| Parenteral iron administration |
The patient presented here had inappropriate urinary phosphate losses, and laboratory testing ruled out primary and secondary hyperparathyroidism and Fanconi syndrome. The patient was not taking medications known to cause tubular phosphate wasting. The patient's age and clinical history made hereditary syndromes unlikely. Therefore, the urinary phosphate wasting had to be related to an acquired defect in phosphate metabolism. The diagnostic characteristics of TIO are summarized in Table 2.
|
| Patients may present with symptoms of osteomalacia (eg, bone pain, fractures), hypophosphatemia (eg, proximal myopathy), and/or neoplasm. |
| Hypophosphatemia with urinary phosphate wasting. |
| Serum calcium level is usually normal. |
| Serum 1,25‐dihydroxyvitamin D level is usually low or low‐normal. |
| Parathyroid hormone is usually normal. |
| Plasma FGF23 level is elevated. |
| A neoplasm with the appropriate histology is identified, although the osteomalacia syndrome may precede identification of the tumor, which may be occult. |
| The syndrome resolves after complete resection of the tumor. |
The presence of a known neoplasm makes the diagnosis of TIO considerably easier. However, osteomalacia often precedes the tumor diagnosis. In these cases, the discovery of this clinical syndrome necessitates a search for the tumor. The tumors can be small, occult, and often located in the extremities. In addition to standard cross‐sectional imaging, specialized diagnostic modalities can be helpful in localizing culprit tumors. These include F‐18 flourodeoxyglucose positron emission tomography with computed tomography, 111‐Indium octreotide single photon emission CT/CT, 68‐Gallium‐DOTA‐octreotide positron emission tomography with computed tomography, and even selective venous sampling for FGF23 levels.[1, 11] The octreotide tests capitalize on the fact that culprit tumors often express somatostatin receptors.
TIO is most often associated with mesenchymal tumors of the bone or soft tissue. It has also been reported in association with several malignancies (small cell carcinoma, hematologic malignancies, prostate cancer), and with polyostotic fibrous dysplasia, neurofibromatosis, and the epidermal nevus syndrome. The mesenchymal tumors are heterogeneous in appearance and can be variably classified as hemangiopericytomas, hemangiomas, sarcomas, ossifying fibromas, granulomas, giant cell tumors, or osteoblastomas.[1] However, 1 review suggests that most of these tumors actually represent a distinct but heterogeneous, under‐recognized entity that is best classified as a phosphaturic mesenchymal tumor.[11]
TIO is only cured by complete resection of the tumor.[1] Local recurrences have been described, as have rare occurrences of metastatic disease.[1, 12] Medical treatment can be used to normalize serum phosphate levels in patients who are unable to be cured by surgery. The goal is to bring serum phosphate into the low‐normal range via phosphate supplementation (typically 13 g/day of elemental phosphorus is required) and treatment with either calcitriol or alfacalcidol. Due to the inhibition of 1,25‐dihydroxyvitamin D activation in TIO, relatively large doses of calcitriol are needed. A reasonable starting dose of calcitriol is 1.5 g/day, and most patients require 15 to 60 ng/kg per day. Because PTH action is involved in FGF23‐mediated hypophosphatemia, suppression of PTH may also be useful in these patients.[13]
This patient presented with a painful femoral tumor in the setting of muscle and joint pain that had been erroneously attributed to connective tissue disease. However, recognition and thorough evaluation of the patient's hypophosphatemia led to a unifying diagnosis of TIO. This diagnosis altered the surgical approach (emphasizing complete resection to eradicate the FGF23 production) and helped alleviate the patient's painful losses of phosphate.
TEACHING POINTS
- Hypophosphatemia, especially if severe or persistent, should not be dismissed as an unimportant laboratory finding. A focused evaluation should be performed to determine the etiology.
- In patients with unexplained hypophosphatemia, the measurement of serum calcium, parathyroid hormone, and vitamin D levels can identify primary or secondary hyperparathyroidism.
- The differential diagnosis of hypophosphatemia is narrowed if there is clinical evidence of inappropriate urinary phosphate wasting (ie, urinary phosphate levels remain high, despite low serum levels).
- TIO is a rare paraneoplastic syndrome caused by FGF23, a phosphatonin hormone that causes renal phosphate wasting, hypophosphatemia, and osteomalacia.
Disclosure: Nothing to report.
- , , , . Tumor‐induced osteomalacia. Endocr Relat Cancer. 2011;18:R53–R77.
- . The FGF23‐Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619.
- , . Genetic disorders of renal phosphate transport. N Engl J Med. 2010;362:2399–2409.
- . The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. 2012;30:1–9.
- , , , , . FGF‐23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492.
- , , , et al. FGF‐23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435.
- , . Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101.
- , , . In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: Elsevier; 2011:1237–1304.
- , , . Medication‐induced hypophosphatemia: a review. QJM. 2010;103:449–459.
- , . Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–310.
- , , , et al. Improving diagnosis of tumor‐induced osteomalacia with gallium‐68 DOTATATE PET/CT. J Clin Endocrinol Metab. 2013; 98:687–694.
- , , , et al. Most osteomalacia‐associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
- , , , , , . Cinacalcet in the management of tumor‐induced osteomalacia. J Bone Miner Res. 2007;22:931–937.
- , , , . Tumor‐induced osteomalacia. Endocr Relat Cancer. 2011;18:R53–R77.
- . The FGF23‐Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619.
- , . Genetic disorders of renal phosphate transport. N Engl J Med. 2010;362:2399–2409.
- . The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. 2012;30:1–9.
- , , , , . FGF‐23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492.
- , , , et al. FGF‐23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435.
- , . Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101.
- , , . In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: Elsevier; 2011:1237–1304.
- , , . Medication‐induced hypophosphatemia: a review. QJM. 2010;103:449–459.
- , . Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–310.
- , , , et al. Improving diagnosis of tumor‐induced osteomalacia with gallium‐68 DOTATATE PET/CT. J Clin Endocrinol Metab. 2013; 98:687–694.
- , , , et al. Most osteomalacia‐associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
- , , , , , . Cinacalcet in the management of tumor‐induced osteomalacia. J Bone Miner Res. 2007;22:931–937.
Should Patients Who Develop Postoperative Atrial Fibrillation Start Anticoagulation?
Case
A 66-year-old man with diabetes mellitus type 2 and hypertension underwent left total knee replacement. Several hours after surgery, the patient developed atrial fibrillation (AF). He was asymptomatic, and reversible causes of AF were ruled out. Approximately 18 hours later, he spontaneously reverted back to sinus rhythm. Should this patient, who has no known prior history of AF and a CHA2DS2-VASc score of 3, be started on anticoagulation?
Background

Hospitalists are commonly consulted for evaluation and management of postoperative atrial fibrillation (POAF). The incidence of new-onset AF associated with non-cardiac surgery is approximately 2% and may be more frequent in an elderly population.1 The increased adrenergic tone associated with surgery is thought to elicit AF in some patients. POAF has also been associated with positive fluid balance, electrolyte abnormalities, and hypoxemia.2 Some of these patients will spontaneously revert back to sinus rhythm after these issues are reversed. Others will go on to develop chronic or paroxysmal AF that persists indefinitely. It is also likely that some patients with POAF, in fact, already had asymptomatic AF that was simply undetected prior to hospitalization.
Hospitalists are faced with the difficult task of determining which patients with POAF will benefit from either short-term or long-term anticoagulation. This has not been well studied in postsurgical patients, in contrast to medical patients in whom stroke risk from AF has been very well-characterized. The decision may be further complicated by bleeding risk (associated with either some surgeries or with patient-dependent factors).3
It is worth noting that following major cardiac or thoracic surgery, POAF is common; the incidence ranges from 10% to 60%. In these cases, POAF may be triggered by transient atrial ischemia or by postoperative inflammation and may have a different natural history from POAF in non-cardiac surgery patients in terms of both reversibility and stroke risk. More retrospective data are available regarding cardiothoracic surgery patients.
Previous American Heart Association (AHA) and American College of Cardiology (ACC) guidelines stated that POAF lasting longer than 48 hours warranted anticoagulation. This recommendation was removed from the newest update. The 2014 updated AHA/ACC guidelines are less absolute and now state only that “it is reasonable to administer antithrombotic medication in patients who developed postoperative AF, as recommended for nonsurgical patients” (Level of Evidence: B) in regard to cardiothoracic surgery.4
There is no specific recommendation regarding POAF for non-cardiac surgery patients. The current guidelines are likely purposefully vague due to the lack of direct evidence. The following is a review of the existing literature and a suggested approach to anticoagulation in POAF.
Review
How common is postoperative atrial fibrillation? New-onset AF during hospitalization is known to occur in association with many acute conditions including surgery, infection, and myocardial infarction. About half of the cases of in-hospital new-onset AF are associated with surgery. AF is more commonly seen in surgery that involves the thoracic cavity and cardiac structures. In a cross-sectional epidemiologic study of 22 million patients in California, 20.8% of patients undergoing cardiac surgery developed POAF compared with only 1.3% of patients undergoing non-cardiac surgery.5 A smaller study of non-cardiac surgery patients found a 30-day POAF incidence of 0.37%.2

It is not clear that all of the increase in stroke risk is a direct effect of POAF. Indeed, in a retrospective analysis of almost 3,000 CABG patients, 1.1% suffered a stroke during their hospital stay. Fewer than half of those had a cardiac rhythm other than sinus rhythm. In the 15 stroke patients who developed POAF, nine presented with stroke symptoms prior to the first episode of AF.9 The authors suggest that aggressive anticoagulation for POAF would not have prevented most of these events.
Furthermore, the rate of in-hospital stroke after non-cardiac surgery is probably much lower, though it has not been as well studied. These data raise some questions as to the benefit of anticoagulation in the immediate postoperative period, though it is difficult to draw firm conclusions without randomized data.
What about non-cardiac surgery? There is less evidence available for patients undergoing non-cardiac surgery, but the few studies that do exist also point to higher stroke risk in patients with POAF. A large population-based study using ICD codes found that the one-year risk of stroke for patients with POAF after non-cardiac surgery was 1.47% compared to 0.36% in non-cardiac surgery patients without POAF (P<0.001). Based on these data, the long-term stroke risk after POAF in non-cardiac surgery patients is similar to that of medical AF patients with a CHA2DS2-VASc score of 2. The authors of this study suggest that transient POAF after non-cardiac surgery may carry a long-term stroke risk similar to any other AF diagnosis.10 However, this study design is subject to significant ascertainment bias (i.e., they may have unintentionally captured some patients with preexisting or prolonged AF), and further research is needed to better delineate this risk.
Does increased stroke risk translate into increased mortality? In a retrospective study of 17,000 patients, El-Chami et al found that POAF after CABG was associated with decreased survival after one year (90% versus 96%) and 10 years (55% versus 70%).11 However, those patients who develop POAF may be sicker overall.
Another study showed that death due to stroke occurred in 4.2% of POAF patients compared to 0.2% of non-POAF patients in a five-year period.12 Based on these studies, POAF is likely associated with increased mortality, but there may be other unaccounted variables. Nevertheless, the increased mortality associated with POAF in these populations is similar to that seen for non-surgical population-based studies13 and provides support that those with newly diagnosed AF in the post-surgical setting should at least be followed closely to assess for recurrence.
What is a patient’s risk of developing atrial fibrillation later in life? When we choose to anticoagulate patients with POAF, we then have to determine whether they should be committed to long-term anticoagulation. It is thought that many cases of POAF are transient; however, some patients will go on to have persistent or paroxysmal AF after discharge.

In another study of about 300 CABG patients, about 20% of patients with POAF also went on to develop post-discharge AF, defined as symptomatic AF that led to medical evaluation. As in the previous study, it is likely that there were undetected episodes of AF.14 Thus, in cardiothoracic surgery patients, some but not all of whom develop POAF have recurrent or ongoing AF. For this reason, if anticoagulation is started, it may be reasonable to stop anticoagulation after weeks or months if ongoing AF is not apparent.
What is the risk of postoperative bleeding if anticoagulation is started? Any decision about the benefits of anticoagulation must be weighed against the risks, most notably the risk of serious or life-threatening bleeding. This risk may be heightened in the immediate perioperative period. Discussions should always take place with our surgical colleagues about type of surgery, intraoperative complications, and postoperative risk of bleeding.
Anticoagulation, if indicated, should not be started until postoperative bleeding risk is deemed appropriately low. That said, the 2015 BRIDGE trial (looking at the benefits and risks of “bridging” patients before surgery) provides some peripheral but meaningful information about postoperative bleeding risk. In this study, patients with preexisting AF who underwent low-bleeding-risk surgery and were bridged on day one after surgery with therapeutic doses of unfractionated or low-molecular-weight heparin had a significantly higher risk of postoperative bleeding compared to non-bridged patients, with a number needed to harm of 50.15 It may be reasonable—and likely safer—to wait a couple days to start anticoagulation for patients with POAF.
What is the expert’s opinion? We asked one of our cardiac electrophysiologists what her approach is to this situation. In general, if a patient has a low stroke risk and is in AF for fewer than 24 hours, it is reasonable to defer anticoagulation and follow as an outpatient. Regardless of risk, if AF is sustained for more than 24 hours, we recommend at least four weeks of anticoagulation and close outpatient follow-up, which should include a period of ambulatory monitoring to determine the need for continued anticoagulation. We also recommend considering what comprises the patient’s stroke risk.
For example, if the CHA2DS2-VASc score is 2 but the points come from being a female with coronary artery disease, we would consider forgoing anticoagulation but arranging for an outpatient cardiac monitor with cardiology follow-up. If the patient has a history of stroke or TIA, we recommend continuing anticoagulation indefinitely.
Back to the Case
Given our patient’s episode of POAF lasted fewer than 24 hours, it would be reasonable to hold off starting anticoagulation, but he should be followed as an outpatient with ambulatory monitoring at a minimum, monitoring for recurrence. If he were to develop recurrent AF, then he would warrant anticoagulation based on an annual stroke risk of 3.2% as determined by a CHA2DS2-VASc score of 3.
Bottom Line
Our strategy is as follows: If a patient has a low stroke risk (i.e., CHA2DS2-VASc score <2) and is in AF for fewer than 24 hours, anticoagulation is not started, but outpatient follow-up is arranged to monitor symptoms. Regardless of stroke risk, if a patient is in AF for more than 24 hours, we initiate and continue anticoagulation for a minimum of four weeks and arrange outpatient follow-up with a period of ambulatory monitoring to determine need for continued anticoagulation. If a patient has a high stroke risk (CHA2DS2-VASc >2) or if their risk factors include a history of stroke or TIA, anticoagulation is started and continued indefinitely. Risk-benefit discussion is held with the patient, especially with regard to bleeding risk, prior to anticoagulation initiation. If the individual patient’s situation presents further nuance, we ask for the assistance of our cardiology or cardiac electrophysiology colleagues.
Final Thought
None of the mentioned studies investigated or included newer oral anticoagulants. Risk-benefit ratios may change (potentially considerably) with these agents. Further study is needed. We expect, in due time, studies will look at the question of POAF in regard to newer anticoagulant agents, and perhaps then our decision making will change. TH
Dr. Evavold is a resident in the hospitalist training program, while Dr. Lessing and Dr. Merritt are hospitalists in the Department of Internal Medicine at the University of Colorado. Dr. Tzou is a cardiologist in the section of electrophysiology at the University of Colorado.
References:
- POISE Study Group, Devereaux PJ, Yang H, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet. 2008;371(9627):1839-1847. doi:10.1016/s0140-6736(08)60601-7.
- Christians K, Wu B, Quebbeman E, Brasel K. Postoperative atrial fibrillation in noncardiothoracic surgical patients. Am J Surg. 2001;182(6):713-715. doi:10.1016/s0002-9610(01)00799-1.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138(5):1093-1100. doi:10.1378/chest.10-0134.
- Fleisher L, Beckman J, Brown K, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 2002 guidelines on perioperative cardiovascular evaluation for noncardiac surgery). Circulation. 2007;116(17):e418-e500. doi:10.1161/circulationaha.107.185699.
- Walkey A, Benjamin E, Lubitz S. New-onset atrial fibrillation during hospitalization. J Am Coll Cardiol. 2014;64(22):2432-2433. doi:10.1016/j.jacc.2014.09.034.
- Creswell L, Schuessler R, Rosenbloom M, Cox J. Hazards of postoperative atrial arrhythmias. Ann Thorac Surg. 1993;56(3):539-549. doi:10.1016/0003-4975(93)90894-n.
- Almassi G, Schowalter T, Nicolosi A, et al. Atrial fibrillation after cardiac surgery: a major morbid event? Ann Surg. 1997;226(4):501-513.
- Horwich P, Buth K, Légaré J. New onset postoperative atrial fibrillation is associated with a long-term risk for stroke and death following cardiac surgery. J Card Surg. 2013;28(1):8-13. doi:10.1111/jocs.12033.
- Kollar A, Lick S, Vasquez K, Conti V. Relationship of atrial fibrillation and stroke after coronary artery bypass graft surgery: when is anticoagulation indicated? Ann Thorac Surg. 2006;82(2):515-523. doi:10.1016/j.athoracsur.2006.03.037.
- Gialdini G, Nearing K, Bhave P, et al. Perioperative atrial fibrillation and the long-term risk of ischemic stroke. JAMA. 2014;312(6):616. doi:10.1001/jama.2014.9143.
- El-Chami M, Kilgo P, Thourani V, et al. New-onset atrial fibrillation predicts long-term mortality after coronary artery bypass graft. J Am Coll Cardiol. 2010;55(13):1370-1376. doi:10.1016/j.jacc.2009.10.058.
- Ahlsson A, Fengsrud E, Bodin L, Englund A. Postoperative atrial fibrillation in patients undergoing aortocoronary bypass surgery carries an eightfold risk of future atrial fibrillation and a doubled cardiovascular mortality. Euro J Cardiothorac Surg. 2010;37(6):1353-1359. doi:10.1016/j.ejcts.2009.12.033.
- Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98(10):946-952.
- Antonelli D, Peres D, Freedberg N, Feldman A, Rosenfeld T. Incidence of postdischarge symptomatic paroxysmal atrial fibrillation in patients who underwent coronary artery bypass graft: long-term follow-up. Pacing Clin Electrophysiol. 2004;27(3):365-367. doi:10.1111/j.1540-8159.2004.00443.x.
- Douketis J, Spyropoulos A, Kaatz S, et al. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med. 2015;373(9):823-833. doi:10.1056/nejmoa1501035.
Case
A 66-year-old man with diabetes mellitus type 2 and hypertension underwent left total knee replacement. Several hours after surgery, the patient developed atrial fibrillation (AF). He was asymptomatic, and reversible causes of AF were ruled out. Approximately 18 hours later, he spontaneously reverted back to sinus rhythm. Should this patient, who has no known prior history of AF and a CHA2DS2-VASc score of 3, be started on anticoagulation?
Background
Hospitalists are commonly consulted for evaluation and management of postoperative atrial fibrillation (POAF). The incidence of new-onset AF associated with non-cardiac surgery is approximately 2% and may be more frequent in an elderly population.1 The increased adrenergic tone associated with surgery is thought to elicit AF in some patients. POAF has also been associated with positive fluid balance, electrolyte abnormalities, and hypoxemia.2 Some of these patients will spontaneously revert back to sinus rhythm after these issues are reversed. Others will go on to develop chronic or paroxysmal AF that persists indefinitely. It is also likely that some patients with POAF, in fact, already had asymptomatic AF that was simply undetected prior to hospitalization.
Hospitalists are faced with the difficult task of determining which patients with POAF will benefit from either short-term or long-term anticoagulation. This has not been well studied in postsurgical patients, in contrast to medical patients in whom stroke risk from AF has been very well-characterized. The decision may be further complicated by bleeding risk (associated with either some surgeries or with patient-dependent factors).3
It is worth noting that following major cardiac or thoracic surgery, POAF is common; the incidence ranges from 10% to 60%. In these cases, POAF may be triggered by transient atrial ischemia or by postoperative inflammation and may have a different natural history from POAF in non-cardiac surgery patients in terms of both reversibility and stroke risk. More retrospective data are available regarding cardiothoracic surgery patients.
Previous American Heart Association (AHA) and American College of Cardiology (ACC) guidelines stated that POAF lasting longer than 48 hours warranted anticoagulation. This recommendation was removed from the newest update. The 2014 updated AHA/ACC guidelines are less absolute and now state only that “it is reasonable to administer antithrombotic medication in patients who developed postoperative AF, as recommended for nonsurgical patients” (Level of Evidence: B) in regard to cardiothoracic surgery.4
There is no specific recommendation regarding POAF for non-cardiac surgery patients. The current guidelines are likely purposefully vague due to the lack of direct evidence. The following is a review of the existing literature and a suggested approach to anticoagulation in POAF.
Review
How common is postoperative atrial fibrillation? New-onset AF during hospitalization is known to occur in association with many acute conditions including surgery, infection, and myocardial infarction. About half of the cases of in-hospital new-onset AF are associated with surgery. AF is more commonly seen in surgery that involves the thoracic cavity and cardiac structures. In a cross-sectional epidemiologic study of 22 million patients in California, 20.8% of patients undergoing cardiac surgery developed POAF compared with only 1.3% of patients undergoing non-cardiac surgery.5 A smaller study of non-cardiac surgery patients found a 30-day POAF incidence of 0.37%.2
It is not clear that all of the increase in stroke risk is a direct effect of POAF. Indeed, in a retrospective analysis of almost 3,000 CABG patients, 1.1% suffered a stroke during their hospital stay. Fewer than half of those had a cardiac rhythm other than sinus rhythm. In the 15 stroke patients who developed POAF, nine presented with stroke symptoms prior to the first episode of AF.9 The authors suggest that aggressive anticoagulation for POAF would not have prevented most of these events.
Furthermore, the rate of in-hospital stroke after non-cardiac surgery is probably much lower, though it has not been as well studied. These data raise some questions as to the benefit of anticoagulation in the immediate postoperative period, though it is difficult to draw firm conclusions without randomized data.
What about non-cardiac surgery? There is less evidence available for patients undergoing non-cardiac surgery, but the few studies that do exist also point to higher stroke risk in patients with POAF. A large population-based study using ICD codes found that the one-year risk of stroke for patients with POAF after non-cardiac surgery was 1.47% compared to 0.36% in non-cardiac surgery patients without POAF (P<0.001). Based on these data, the long-term stroke risk after POAF in non-cardiac surgery patients is similar to that of medical AF patients with a CHA2DS2-VASc score of 2. The authors of this study suggest that transient POAF after non-cardiac surgery may carry a long-term stroke risk similar to any other AF diagnosis.10 However, this study design is subject to significant ascertainment bias (i.e., they may have unintentionally captured some patients with preexisting or prolonged AF), and further research is needed to better delineate this risk.
Does increased stroke risk translate into increased mortality? In a retrospective study of 17,000 patients, El-Chami et al found that POAF after CABG was associated with decreased survival after one year (90% versus 96%) and 10 years (55% versus 70%).11 However, those patients who develop POAF may be sicker overall.
Another study showed that death due to stroke occurred in 4.2% of POAF patients compared to 0.2% of non-POAF patients in a five-year period.12 Based on these studies, POAF is likely associated with increased mortality, but there may be other unaccounted variables. Nevertheless, the increased mortality associated with POAF in these populations is similar to that seen for non-surgical population-based studies13 and provides support that those with newly diagnosed AF in the post-surgical setting should at least be followed closely to assess for recurrence.
What is a patient’s risk of developing atrial fibrillation later in life? When we choose to anticoagulate patients with POAF, we then have to determine whether they should be committed to long-term anticoagulation. It is thought that many cases of POAF are transient; however, some patients will go on to have persistent or paroxysmal AF after discharge.
In another study of about 300 CABG patients, about 20% of patients with POAF also went on to develop post-discharge AF, defined as symptomatic AF that led to medical evaluation. As in the previous study, it is likely that there were undetected episodes of AF.14 Thus, in cardiothoracic surgery patients, some but not all of whom develop POAF have recurrent or ongoing AF. For this reason, if anticoagulation is started, it may be reasonable to stop anticoagulation after weeks or months if ongoing AF is not apparent.
What is the risk of postoperative bleeding if anticoagulation is started? Any decision about the benefits of anticoagulation must be weighed against the risks, most notably the risk of serious or life-threatening bleeding. This risk may be heightened in the immediate perioperative period. Discussions should always take place with our surgical colleagues about type of surgery, intraoperative complications, and postoperative risk of bleeding.
Anticoagulation, if indicated, should not be started until postoperative bleeding risk is deemed appropriately low. That said, the 2015 BRIDGE trial (looking at the benefits and risks of “bridging” patients before surgery) provides some peripheral but meaningful information about postoperative bleeding risk. In this study, patients with preexisting AF who underwent low-bleeding-risk surgery and were bridged on day one after surgery with therapeutic doses of unfractionated or low-molecular-weight heparin had a significantly higher risk of postoperative bleeding compared to non-bridged patients, with a number needed to harm of 50.15 It may be reasonable—and likely safer—to wait a couple days to start anticoagulation for patients with POAF.
What is the expert’s opinion? We asked one of our cardiac electrophysiologists what her approach is to this situation. In general, if a patient has a low stroke risk and is in AF for fewer than 24 hours, it is reasonable to defer anticoagulation and follow as an outpatient. Regardless of risk, if AF is sustained for more than 24 hours, we recommend at least four weeks of anticoagulation and close outpatient follow-up, which should include a period of ambulatory monitoring to determine the need for continued anticoagulation. We also recommend considering what comprises the patient’s stroke risk.
For example, if the CHA2DS2-VASc score is 2 but the points come from being a female with coronary artery disease, we would consider forgoing anticoagulation but arranging for an outpatient cardiac monitor with cardiology follow-up. If the patient has a history of stroke or TIA, we recommend continuing anticoagulation indefinitely.
Back to the Case
Given our patient’s episode of POAF lasted fewer than 24 hours, it would be reasonable to hold off starting anticoagulation, but he should be followed as an outpatient with ambulatory monitoring at a minimum, monitoring for recurrence. If he were to develop recurrent AF, then he would warrant anticoagulation based on an annual stroke risk of 3.2% as determined by a CHA2DS2-VASc score of 3.
Bottom Line
Our strategy is as follows: If a patient has a low stroke risk (i.e., CHA2DS2-VASc score <2) and is in AF for fewer than 24 hours, anticoagulation is not started, but outpatient follow-up is arranged to monitor symptoms. Regardless of stroke risk, if a patient is in AF for more than 24 hours, we initiate and continue anticoagulation for a minimum of four weeks and arrange outpatient follow-up with a period of ambulatory monitoring to determine need for continued anticoagulation. If a patient has a high stroke risk (CHA2DS2-VASc >2) or if their risk factors include a history of stroke or TIA, anticoagulation is started and continued indefinitely. Risk-benefit discussion is held with the patient, especially with regard to bleeding risk, prior to anticoagulation initiation. If the individual patient’s situation presents further nuance, we ask for the assistance of our cardiology or cardiac electrophysiology colleagues.
Final Thought
None of the mentioned studies investigated or included newer oral anticoagulants. Risk-benefit ratios may change (potentially considerably) with these agents. Further study is needed. We expect, in due time, studies will look at the question of POAF in regard to newer anticoagulant agents, and perhaps then our decision making will change. TH
Dr. Evavold is a resident in the hospitalist training program, while Dr. Lessing and Dr. Merritt are hospitalists in the Department of Internal Medicine at the University of Colorado. Dr. Tzou is a cardiologist in the section of electrophysiology at the University of Colorado.
References:
- POISE Study Group, Devereaux PJ, Yang H, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet. 2008;371(9627):1839-1847. doi:10.1016/s0140-6736(08)60601-7.
- Christians K, Wu B, Quebbeman E, Brasel K. Postoperative atrial fibrillation in noncardiothoracic surgical patients. Am J Surg. 2001;182(6):713-715. doi:10.1016/s0002-9610(01)00799-1.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138(5):1093-1100. doi:10.1378/chest.10-0134.
- Fleisher L, Beckman J, Brown K, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 2002 guidelines on perioperative cardiovascular evaluation for noncardiac surgery). Circulation. 2007;116(17):e418-e500. doi:10.1161/circulationaha.107.185699.
- Walkey A, Benjamin E, Lubitz S. New-onset atrial fibrillation during hospitalization. J Am Coll Cardiol. 2014;64(22):2432-2433. doi:10.1016/j.jacc.2014.09.034.
- Creswell L, Schuessler R, Rosenbloom M, Cox J. Hazards of postoperative atrial arrhythmias. Ann Thorac Surg. 1993;56(3):539-549. doi:10.1016/0003-4975(93)90894-n.
- Almassi G, Schowalter T, Nicolosi A, et al. Atrial fibrillation after cardiac surgery: a major morbid event? Ann Surg. 1997;226(4):501-513.
- Horwich P, Buth K, Légaré J. New onset postoperative atrial fibrillation is associated with a long-term risk for stroke and death following cardiac surgery. J Card Surg. 2013;28(1):8-13. doi:10.1111/jocs.12033.
- Kollar A, Lick S, Vasquez K, Conti V. Relationship of atrial fibrillation and stroke after coronary artery bypass graft surgery: when is anticoagulation indicated? Ann Thorac Surg. 2006;82(2):515-523. doi:10.1016/j.athoracsur.2006.03.037.
- Gialdini G, Nearing K, Bhave P, et al. Perioperative atrial fibrillation and the long-term risk of ischemic stroke. JAMA. 2014;312(6):616. doi:10.1001/jama.2014.9143.
- El-Chami M, Kilgo P, Thourani V, et al. New-onset atrial fibrillation predicts long-term mortality after coronary artery bypass graft. J Am Coll Cardiol. 2010;55(13):1370-1376. doi:10.1016/j.jacc.2009.10.058.
- Ahlsson A, Fengsrud E, Bodin L, Englund A. Postoperative atrial fibrillation in patients undergoing aortocoronary bypass surgery carries an eightfold risk of future atrial fibrillation and a doubled cardiovascular mortality. Euro J Cardiothorac Surg. 2010;37(6):1353-1359. doi:10.1016/j.ejcts.2009.12.033.
- Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98(10):946-952.
- Antonelli D, Peres D, Freedberg N, Feldman A, Rosenfeld T. Incidence of postdischarge symptomatic paroxysmal atrial fibrillation in patients who underwent coronary artery bypass graft: long-term follow-up. Pacing Clin Electrophysiol. 2004;27(3):365-367. doi:10.1111/j.1540-8159.2004.00443.x.
- Douketis J, Spyropoulos A, Kaatz S, et al. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med. 2015;373(9):823-833. doi:10.1056/nejmoa1501035.
Case
A 66-year-old man with diabetes mellitus type 2 and hypertension underwent left total knee replacement. Several hours after surgery, the patient developed atrial fibrillation (AF). He was asymptomatic, and reversible causes of AF were ruled out. Approximately 18 hours later, he spontaneously reverted back to sinus rhythm. Should this patient, who has no known prior history of AF and a CHA2DS2-VASc score of 3, be started on anticoagulation?
Background
Hospitalists are commonly consulted for evaluation and management of postoperative atrial fibrillation (POAF). The incidence of new-onset AF associated with non-cardiac surgery is approximately 2% and may be more frequent in an elderly population.1 The increased adrenergic tone associated with surgery is thought to elicit AF in some patients. POAF has also been associated with positive fluid balance, electrolyte abnormalities, and hypoxemia.2 Some of these patients will spontaneously revert back to sinus rhythm after these issues are reversed. Others will go on to develop chronic or paroxysmal AF that persists indefinitely. It is also likely that some patients with POAF, in fact, already had asymptomatic AF that was simply undetected prior to hospitalization.
Hospitalists are faced with the difficult task of determining which patients with POAF will benefit from either short-term or long-term anticoagulation. This has not been well studied in postsurgical patients, in contrast to medical patients in whom stroke risk from AF has been very well-characterized. The decision may be further complicated by bleeding risk (associated with either some surgeries or with patient-dependent factors).3
It is worth noting that following major cardiac or thoracic surgery, POAF is common; the incidence ranges from 10% to 60%. In these cases, POAF may be triggered by transient atrial ischemia or by postoperative inflammation and may have a different natural history from POAF in non-cardiac surgery patients in terms of both reversibility and stroke risk. More retrospective data are available regarding cardiothoracic surgery patients.
Previous American Heart Association (AHA) and American College of Cardiology (ACC) guidelines stated that POAF lasting longer than 48 hours warranted anticoagulation. This recommendation was removed from the newest update. The 2014 updated AHA/ACC guidelines are less absolute and now state only that “it is reasonable to administer antithrombotic medication in patients who developed postoperative AF, as recommended for nonsurgical patients” (Level of Evidence: B) in regard to cardiothoracic surgery.4
There is no specific recommendation regarding POAF for non-cardiac surgery patients. The current guidelines are likely purposefully vague due to the lack of direct evidence. The following is a review of the existing literature and a suggested approach to anticoagulation in POAF.
Review
How common is postoperative atrial fibrillation? New-onset AF during hospitalization is known to occur in association with many acute conditions including surgery, infection, and myocardial infarction. About half of the cases of in-hospital new-onset AF are associated with surgery. AF is more commonly seen in surgery that involves the thoracic cavity and cardiac structures. In a cross-sectional epidemiologic study of 22 million patients in California, 20.8% of patients undergoing cardiac surgery developed POAF compared with only 1.3% of patients undergoing non-cardiac surgery.5 A smaller study of non-cardiac surgery patients found a 30-day POAF incidence of 0.37%.2
It is not clear that all of the increase in stroke risk is a direct effect of POAF. Indeed, in a retrospective analysis of almost 3,000 CABG patients, 1.1% suffered a stroke during their hospital stay. Fewer than half of those had a cardiac rhythm other than sinus rhythm. In the 15 stroke patients who developed POAF, nine presented with stroke symptoms prior to the first episode of AF.9 The authors suggest that aggressive anticoagulation for POAF would not have prevented most of these events.
Furthermore, the rate of in-hospital stroke after non-cardiac surgery is probably much lower, though it has not been as well studied. These data raise some questions as to the benefit of anticoagulation in the immediate postoperative period, though it is difficult to draw firm conclusions without randomized data.
What about non-cardiac surgery? There is less evidence available for patients undergoing non-cardiac surgery, but the few studies that do exist also point to higher stroke risk in patients with POAF. A large population-based study using ICD codes found that the one-year risk of stroke for patients with POAF after non-cardiac surgery was 1.47% compared to 0.36% in non-cardiac surgery patients without POAF (P<0.001). Based on these data, the long-term stroke risk after POAF in non-cardiac surgery patients is similar to that of medical AF patients with a CHA2DS2-VASc score of 2. The authors of this study suggest that transient POAF after non-cardiac surgery may carry a long-term stroke risk similar to any other AF diagnosis.10 However, this study design is subject to significant ascertainment bias (i.e., they may have unintentionally captured some patients with preexisting or prolonged AF), and further research is needed to better delineate this risk.
Does increased stroke risk translate into increased mortality? In a retrospective study of 17,000 patients, El-Chami et al found that POAF after CABG was associated with decreased survival after one year (90% versus 96%) and 10 years (55% versus 70%).11 However, those patients who develop POAF may be sicker overall.
Another study showed that death due to stroke occurred in 4.2% of POAF patients compared to 0.2% of non-POAF patients in a five-year period.12 Based on these studies, POAF is likely associated with increased mortality, but there may be other unaccounted variables. Nevertheless, the increased mortality associated with POAF in these populations is similar to that seen for non-surgical population-based studies13 and provides support that those with newly diagnosed AF in the post-surgical setting should at least be followed closely to assess for recurrence.
What is a patient’s risk of developing atrial fibrillation later in life? When we choose to anticoagulate patients with POAF, we then have to determine whether they should be committed to long-term anticoagulation. It is thought that many cases of POAF are transient; however, some patients will go on to have persistent or paroxysmal AF after discharge.
In another study of about 300 CABG patients, about 20% of patients with POAF also went on to develop post-discharge AF, defined as symptomatic AF that led to medical evaluation. As in the previous study, it is likely that there were undetected episodes of AF.14 Thus, in cardiothoracic surgery patients, some but not all of whom develop POAF have recurrent or ongoing AF. For this reason, if anticoagulation is started, it may be reasonable to stop anticoagulation after weeks or months if ongoing AF is not apparent.
What is the risk of postoperative bleeding if anticoagulation is started? Any decision about the benefits of anticoagulation must be weighed against the risks, most notably the risk of serious or life-threatening bleeding. This risk may be heightened in the immediate perioperative period. Discussions should always take place with our surgical colleagues about type of surgery, intraoperative complications, and postoperative risk of bleeding.
Anticoagulation, if indicated, should not be started until postoperative bleeding risk is deemed appropriately low. That said, the 2015 BRIDGE trial (looking at the benefits and risks of “bridging” patients before surgery) provides some peripheral but meaningful information about postoperative bleeding risk. In this study, patients with preexisting AF who underwent low-bleeding-risk surgery and were bridged on day one after surgery with therapeutic doses of unfractionated or low-molecular-weight heparin had a significantly higher risk of postoperative bleeding compared to non-bridged patients, with a number needed to harm of 50.15 It may be reasonable—and likely safer—to wait a couple days to start anticoagulation for patients with POAF.
What is the expert’s opinion? We asked one of our cardiac electrophysiologists what her approach is to this situation. In general, if a patient has a low stroke risk and is in AF for fewer than 24 hours, it is reasonable to defer anticoagulation and follow as an outpatient. Regardless of risk, if AF is sustained for more than 24 hours, we recommend at least four weeks of anticoagulation and close outpatient follow-up, which should include a period of ambulatory monitoring to determine the need for continued anticoagulation. We also recommend considering what comprises the patient’s stroke risk.
For example, if the CHA2DS2-VASc score is 2 but the points come from being a female with coronary artery disease, we would consider forgoing anticoagulation but arranging for an outpatient cardiac monitor with cardiology follow-up. If the patient has a history of stroke or TIA, we recommend continuing anticoagulation indefinitely.
Back to the Case
Given our patient’s episode of POAF lasted fewer than 24 hours, it would be reasonable to hold off starting anticoagulation, but he should be followed as an outpatient with ambulatory monitoring at a minimum, monitoring for recurrence. If he were to develop recurrent AF, then he would warrant anticoagulation based on an annual stroke risk of 3.2% as determined by a CHA2DS2-VASc score of 3.
Bottom Line
Our strategy is as follows: If a patient has a low stroke risk (i.e., CHA2DS2-VASc score <2) and is in AF for fewer than 24 hours, anticoagulation is not started, but outpatient follow-up is arranged to monitor symptoms. Regardless of stroke risk, if a patient is in AF for more than 24 hours, we initiate and continue anticoagulation for a minimum of four weeks and arrange outpatient follow-up with a period of ambulatory monitoring to determine need for continued anticoagulation. If a patient has a high stroke risk (CHA2DS2-VASc >2) or if their risk factors include a history of stroke or TIA, anticoagulation is started and continued indefinitely. Risk-benefit discussion is held with the patient, especially with regard to bleeding risk, prior to anticoagulation initiation. If the individual patient’s situation presents further nuance, we ask for the assistance of our cardiology or cardiac electrophysiology colleagues.
Final Thought
None of the mentioned studies investigated or included newer oral anticoagulants. Risk-benefit ratios may change (potentially considerably) with these agents. Further study is needed. We expect, in due time, studies will look at the question of POAF in regard to newer anticoagulant agents, and perhaps then our decision making will change. TH
Dr. Evavold is a resident in the hospitalist training program, while Dr. Lessing and Dr. Merritt are hospitalists in the Department of Internal Medicine at the University of Colorado. Dr. Tzou is a cardiologist in the section of electrophysiology at the University of Colorado.
References:
- POISE Study Group, Devereaux PJ, Yang H, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet. 2008;371(9627):1839-1847. doi:10.1016/s0140-6736(08)60601-7.
- Christians K, Wu B, Quebbeman E, Brasel K. Postoperative atrial fibrillation in noncardiothoracic surgical patients. Am J Surg. 2001;182(6):713-715. doi:10.1016/s0002-9610(01)00799-1.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138(5):1093-1100. doi:10.1378/chest.10-0134.
- Fleisher L, Beckman J, Brown K, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 2002 guidelines on perioperative cardiovascular evaluation for noncardiac surgery). Circulation. 2007;116(17):e418-e500. doi:10.1161/circulationaha.107.185699.
- Walkey A, Benjamin E, Lubitz S. New-onset atrial fibrillation during hospitalization. J Am Coll Cardiol. 2014;64(22):2432-2433. doi:10.1016/j.jacc.2014.09.034.
- Creswell L, Schuessler R, Rosenbloom M, Cox J. Hazards of postoperative atrial arrhythmias. Ann Thorac Surg. 1993;56(3):539-549. doi:10.1016/0003-4975(93)90894-n.
- Almassi G, Schowalter T, Nicolosi A, et al. Atrial fibrillation after cardiac surgery: a major morbid event? Ann Surg. 1997;226(4):501-513.
- Horwich P, Buth K, Légaré J. New onset postoperative atrial fibrillation is associated with a long-term risk for stroke and death following cardiac surgery. J Card Surg. 2013;28(1):8-13. doi:10.1111/jocs.12033.
- Kollar A, Lick S, Vasquez K, Conti V. Relationship of atrial fibrillation and stroke after coronary artery bypass graft surgery: when is anticoagulation indicated? Ann Thorac Surg. 2006;82(2):515-523. doi:10.1016/j.athoracsur.2006.03.037.
- Gialdini G, Nearing K, Bhave P, et al. Perioperative atrial fibrillation and the long-term risk of ischemic stroke. JAMA. 2014;312(6):616. doi:10.1001/jama.2014.9143.
- El-Chami M, Kilgo P, Thourani V, et al. New-onset atrial fibrillation predicts long-term mortality after coronary artery bypass graft. J Am Coll Cardiol. 2010;55(13):1370-1376. doi:10.1016/j.jacc.2009.10.058.
- Ahlsson A, Fengsrud E, Bodin L, Englund A. Postoperative atrial fibrillation in patients undergoing aortocoronary bypass surgery carries an eightfold risk of future atrial fibrillation and a doubled cardiovascular mortality. Euro J Cardiothorac Surg. 2010;37(6):1353-1359. doi:10.1016/j.ejcts.2009.12.033.
- Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98(10):946-952.
- Antonelli D, Peres D, Freedberg N, Feldman A, Rosenfeld T. Incidence of postdischarge symptomatic paroxysmal atrial fibrillation in patients who underwent coronary artery bypass graft: long-term follow-up. Pacing Clin Electrophysiol. 2004;27(3):365-367. doi:10.1111/j.1540-8159.2004.00443.x.
- Douketis J, Spyropoulos A, Kaatz S, et al. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med. 2015;373(9):823-833. doi:10.1056/nejmoa1501035.

When Should Harm-Reduction Strategies Be Used for Inpatients with Opioid Misuse?
Case
A 33-year-old male with a history of opioid overdose and opioid use disorder is admitted with IV heroin use complicated by injection site cellulitis. He is started on antibiotics with improvement in his cellulitis; however, his hospitalization is complicated by acute opioid withdrawal. Despite his history of opioid overdose and opioid use disorder, he has never seen a substance use disorder specialist nor received any education or treatment for his addiction. He reports that he will stop using illicit drugs but declines any further addiction treatment.
What strategies can be employed to reduce his risk of future harm from opioid misuse?
Background
Over the past decade, the U.S. has experienced a rapid increase in the rates of opioid prescriptions and opioid misuse.1 Consequently, the number of ED visits and hospitalizations for opioid-related complications has also increased.2 Many complications result from the practice of injection drug use (IDU), which predisposes individuals to serious blood-borne viral infections such as human immunodeficiency virus (HIV) and hepatitis C virus (HCV) as well as bacterial infections such as infective endocarditis. In addition, individuals who misuse opioids are at risk of death related to opioid overdose. In 2013, there were more than 24,000 deaths in the U.S. due to opioid overdose (see Figure 1).3
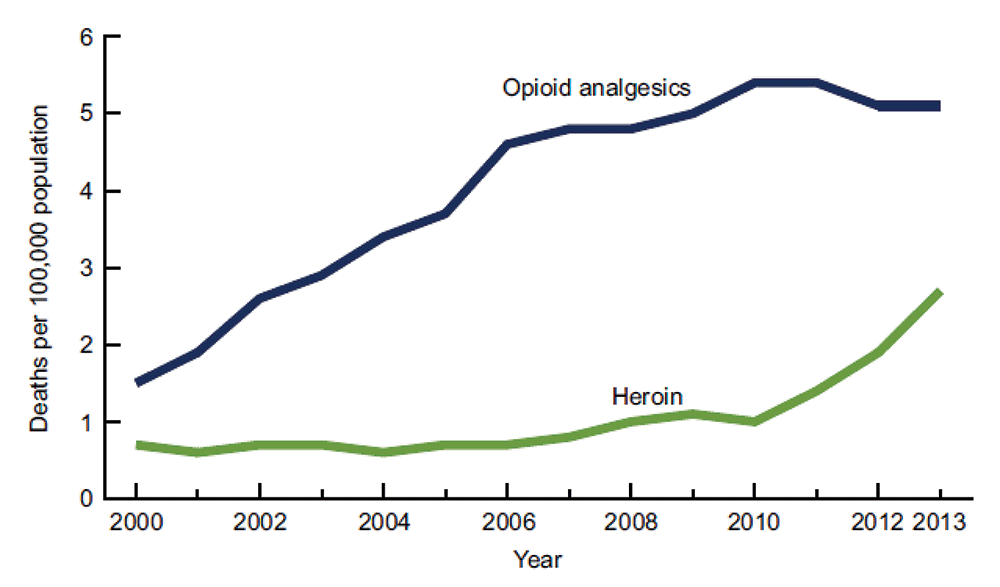
In response to the opioid epidemic, there have been a number of local, state, and federal public health initiatives to monitor and secure the opioid drug supply, improve treatment resources, and promulgate harm-reduction interventions. At a more individual level, hospitalists have an important role to play in combating the opioid epidemic. As frontline providers, hospitalists have access to hospitalized individuals with opioid misuse who may not otherwise be exposed to the healthcare system. Therefore, inpatient hospitalizations serve as a unique and important opportunity to engage individuals in the management of their addiction.
There are a number of interventions that hospitalists and substance use disorder specialists can pursue. Psychiatric evaluation and initiation of medication-assisted treatment often aim to aid patients in abstaining from further opioid misuse. However, many individuals with opioid use disorder are not ready for treatment or experience relapses of opioid misuse despite treatment. Given this, a secondary goal is to reduce any harm that may result from opioid misuse. This is done through the implementation of harm-reduction strategies. These strategies include teaching safe injection practices, facilitating the use of syringe exchange programs, and providing opioid overdose education and naloxone distribution.
Overview of Data
Safe Injection Education. People who inject drugs are at risk for viral, bacterial, and fungal infections. These infections are often the result of nonsterile injection and may be minimized by the utilization of safe injection practices. In order to educate people who inject drugs on safe injection practices, the hospitalist must first understand the process involved in injecting drugs. In Table 1, the process of injecting heroin is outlined (of note, other illicit drugs can be injected, and processes may vary).4

As evidenced by Table 1, the process of sterile injection can be complicated, especially for an individual who may be withdrawing from opioids. Table 1 is also optimistic in that it recommends new and sterile products be used with every injection. If new and sterile equipment is not available, another option is to clean the equipment after every use, which can be done by using bleach and water. This may mitigate the risk of viral, bacterial, and fungal infections. However, the risk is still present, so users should not share or use another individual’s equipment even if it has been cleaned. Due to the risk of viral, bacterial, and fungal infections, all hospitalized individuals who inject drugs should receive education on safe injection practices.
Syringe Exchange Programs. IDU accounts for up to 15% of all new HIV infections and is the primary risk factor for the transmission of HCV.5 These infections occur when people inject using equipment contaminated with blood that contains HIV and/or HCV. Given this, if people who inject drugs could access and consistently use sterile syringes and other injection paraphernalia, the risk of transmitting blood-borne infections would be dramatically reduced. This is the concept behind syringe exchange programs (also known as needle exchange programs), which serve to increase access to sterile syringes while removing contaminated or used syringes from the community.
There is compelling evidence that syringe exchange programs decrease the rate of HIV transmission and likely reduce the rate of HCV transmission as well.6 In addition, syringe exchange programs often provide other beneficial services, such as counseling, testing, and prevention efforts for HIV, HCV, and sexually transmitted infections; distribution of condoms; and referrals to treatment services for substance use disorder.5
Unfortunately, in the U.S., restrictive state laws and lack of funding limit the number of established syringe exchange programs. According to the North American Syringe Exchange Network, there are only 226 programs in 33 states and the District of Columbia. Hospitalists and social workers should be aware of available local resources, including syringe exchange programs, and distribute this information to hospitalized individuals who inject drugs.
Opioid Overdose Education and Naloxone Distribution. Syringe exchange programs and safe injection education aim to reduce harm by decreasing the transmission of infections; however, they do not address the problem of deaths related to opioid overdose. The primary harm-reduction strategy used to address deaths related to opioid overdose in the U.S is opioid overdose education and naloxone distribution (OEND). Naloxone is an opioid antagonist that reverses the respiratory depression and decreased consciousness caused by opioids. The OEND strategy involves educating first responders— including individuals and friends and family of individuals who use opioids—to recognize the signs of an opioid overdose, seek help, provide rescue breathing, administer naloxone, and stay with the individual until emergency medical services arrive.7 This strategy has been observed to decrease rates of death related to opioid overdose.7
Given the evolving opioid epidemic and effectiveness of the OEND strategy, it is not surprising that the number of local opioid overdose prevention programs adopting OEND has risen dramatically. As of 2014, there were 140 organizations, with 644 local sites providing naloxone in 29 states and the District of Columbia. These organizations have distributed 152,000 naloxone kits and have reported more than 26,000 reversals.8 Certainly, OEND has prevented morbidity and mortality in some of these patients.
The adoption of OEND can be performed by individual prescribers as well. Naloxone is U.S. FDA-approved for the treatment of opioid overdose, and thus the liability to prescribers is similar to that of other FDA-approved drugs. However, the distribution of naloxone to third parties, such as friends and family of individuals with opioid misuse, is more complex and regulated by state laws. Many states have created liability protection for naloxone prescription to third parties. Individual state laws and additional information can be found at prescribetoprevent.org.
Hospitalists should provide opioid overdose education to all individuals with opioid misuse and friends and family of individuals with opioid misuse. In addition, hospitalists should prescribe naloxone to individuals with opioid misuse and, in states where the law allows, distribute naloxone to friends and family of individuals with opioid misuse as well.
Controversies. In general, opioid use disorder treatment providers; public health officials; and local, state, and federal government agencies have increasingly embraced harm-reduction strategies. However, some feel that harm-reduction strategies are misguided or even detrimental due to concern that they implicitly condone or enable the use of illicit substances. There have been a number of studies to evaluate the potential unintended consequences of harm-reduction strategies, and overwhelmingly, these have been either neutral or have shown the benefit of harm-reduction interventions. At this point, there is no good evidence to prevent the widespread adoption of harm-reduction strategies for hospitalists.
Back to the Case
The case involves an individual who has already had at least two complications of his IV heroin use, including cellulitis and opioid overdose. Ideally, this individual would be willing to see an addiction specialist and start medication-assisted treatment. Unfortunately, he is unwilling to be further evaluated by a specialist at this time. Regardless, he remains at risk of future complications, and it is the hospitalist’s responsibility to intervene with a goal of reducing future harm that may result from his IV heroin use.
The hospitalist in this case advises the patient to abstain from heroin and IDU, encourages him to seek treatment for his opioid use disorder, and gives him resources for linkage to care if he becomes interested. In addition, the hospitalist educates the patient on safe injection practices and provides a list of local syringe exchange programs to decrease future risk of viral, bacterial, and fungal infections. Furthermore, the hospitalist provides opioid overdose education and distributes naloxone to the patient, along with friends and family of the patient, to reduce the risk of death related to opioid overdose.
Bottom Line
Hospitalists should utilize harm-reduction interventions in individuals hospitalized with opioid misuse. TH
Dr. Theisen-Toupal is a hospitalist at the Veterans Affairs Medical Center and assistant professor of medicine at the George Washington University School of Medicine & Health Sciences, both in Washington, D.C.
References
- Vital signs: overdoses of prescription opioid pain relievers—United States, 1999–2008. Morbidity and Mortality Weekly Report. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6043a4.htm. Published November 4, 2011.
- Drug abuse warning network, 2011: national estimates of drug-related emergency department visits. Substance Abuse and Mental Health Services Administration website. Available at: http://www.samhsa.gov/data/2k13/DAWN2k11ED/DAWN2k11ED.htm#5. Accessed July 29, 2015.
- Hedergaard H, Chen LH, Warner M. Drug-poisoning deaths involving heroin: United States, 2000–2013. National Center for Health Statistics Data Brief. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/nchs/data/databriefs/db190.htm. Published March 2015.
- Getting off right: a safety manual for injection drug users. Harm Reduction Coalition website. Available at: http://harmreduction.org/wp-content/uploads/2011/12/getting-off-right.pdf.
- Syringe exchange programs—United States, 2008. Morbidity and Mortality Weekly Report. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5945a4.htm/Syringe-Exchange-Programs-United-States-2008. Published November 19, 2010.
- Wodak A, Conney A. Effectiveness of sterile needle and syringe programming in reducing HIV/AIDS among injecting drug users. World Health Organization website. Available at: http://apps.who.int/iris/bitstream/10665/43107/1/9241591641.pdf. Published 2004.
- Walley AY, Xuan Z, Hackman HH, et al. Opioid overdose rates and implementation of overdose education and nasal naloxone distribution in Massachusetts: interrupted time series analysis. BMJ. 2013;346:f174.
- Wheeler E, Jones TS, Gilbert MK, Davidson PJ. Opioid overdose prevention programs providing naloxone to laypersons—United States, 2014. Morbidity and Mortality Weekly Report. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6423a2.htm. Published June 19, 2015.
Case
A 33-year-old male with a history of opioid overdose and opioid use disorder is admitted with IV heroin use complicated by injection site cellulitis. He is started on antibiotics with improvement in his cellulitis; however, his hospitalization is complicated by acute opioid withdrawal. Despite his history of opioid overdose and opioid use disorder, he has never seen a substance use disorder specialist nor received any education or treatment for his addiction. He reports that he will stop using illicit drugs but declines any further addiction treatment.
What strategies can be employed to reduce his risk of future harm from opioid misuse?
Background
Over the past decade, the U.S. has experienced a rapid increase in the rates of opioid prescriptions and opioid misuse.1 Consequently, the number of ED visits and hospitalizations for opioid-related complications has also increased.2 Many complications result from the practice of injection drug use (IDU), which predisposes individuals to serious blood-borne viral infections such as human immunodeficiency virus (HIV) and hepatitis C virus (HCV) as well as bacterial infections such as infective endocarditis. In addition, individuals who misuse opioids are at risk of death related to opioid overdose. In 2013, there were more than 24,000 deaths in the U.S. due to opioid overdose (see Figure 1).3
In response to the opioid epidemic, there have been a number of local, state, and federal public health initiatives to monitor and secure the opioid drug supply, improve treatment resources, and promulgate harm-reduction interventions. At a more individual level, hospitalists have an important role to play in combating the opioid epidemic. As frontline providers, hospitalists have access to hospitalized individuals with opioid misuse who may not otherwise be exposed to the healthcare system. Therefore, inpatient hospitalizations serve as a unique and important opportunity to engage individuals in the management of their addiction.
There are a number of interventions that hospitalists and substance use disorder specialists can pursue. Psychiatric evaluation and initiation of medication-assisted treatment often aim to aid patients in abstaining from further opioid misuse. However, many individuals with opioid use disorder are not ready for treatment or experience relapses of opioid misuse despite treatment. Given this, a secondary goal is to reduce any harm that may result from opioid misuse. This is done through the implementation of harm-reduction strategies. These strategies include teaching safe injection practices, facilitating the use of syringe exchange programs, and providing opioid overdose education and naloxone distribution.
Overview of Data
Safe Injection Education. People who inject drugs are at risk for viral, bacterial, and fungal infections. These infections are often the result of nonsterile injection and may be minimized by the utilization of safe injection practices. In order to educate people who inject drugs on safe injection practices, the hospitalist must first understand the process involved in injecting drugs. In Table 1, the process of injecting heroin is outlined (of note, other illicit drugs can be injected, and processes may vary).4
As evidenced by Table 1, the process of sterile injection can be complicated, especially for an individual who may be withdrawing from opioids. Table 1 is also optimistic in that it recommends new and sterile products be used with every injection. If new and sterile equipment is not available, another option is to clean the equipment after every use, which can be done by using bleach and water. This may mitigate the risk of viral, bacterial, and fungal infections. However, the risk is still present, so users should not share or use another individual’s equipment even if it has been cleaned. Due to the risk of viral, bacterial, and fungal infections, all hospitalized individuals who inject drugs should receive education on safe injection practices.
Syringe Exchange Programs. IDU accounts for up to 15% of all new HIV infections and is the primary risk factor for the transmission of HCV.5 These infections occur when people inject using equipment contaminated with blood that contains HIV and/or HCV. Given this, if people who inject drugs could access and consistently use sterile syringes and other injection paraphernalia, the risk of transmitting blood-borne infections would be dramatically reduced. This is the concept behind syringe exchange programs (also known as needle exchange programs), which serve to increase access to sterile syringes while removing contaminated or used syringes from the community.
There is compelling evidence that syringe exchange programs decrease the rate of HIV transmission and likely reduce the rate of HCV transmission as well.6 In addition, syringe exchange programs often provide other beneficial services, such as counseling, testing, and prevention efforts for HIV, HCV, and sexually transmitted infections; distribution of condoms; and referrals to treatment services for substance use disorder.5
Unfortunately, in the U.S., restrictive state laws and lack of funding limit the number of established syringe exchange programs. According to the North American Syringe Exchange Network, there are only 226 programs in 33 states and the District of Columbia. Hospitalists and social workers should be aware of available local resources, including syringe exchange programs, and distribute this information to hospitalized individuals who inject drugs.
Opioid Overdose Education and Naloxone Distribution. Syringe exchange programs and safe injection education aim to reduce harm by decreasing the transmission of infections; however, they do not address the problem of deaths related to opioid overdose. The primary harm-reduction strategy used to address deaths related to opioid overdose in the U.S is opioid overdose education and naloxone distribution (OEND). Naloxone is an opioid antagonist that reverses the respiratory depression and decreased consciousness caused by opioids. The OEND strategy involves educating first responders— including individuals and friends and family of individuals who use opioids—to recognize the signs of an opioid overdose, seek help, provide rescue breathing, administer naloxone, and stay with the individual until emergency medical services arrive.7 This strategy has been observed to decrease rates of death related to opioid overdose.7
Given the evolving opioid epidemic and effectiveness of the OEND strategy, it is not surprising that the number of local opioid overdose prevention programs adopting OEND has risen dramatically. As of 2014, there were 140 organizations, with 644 local sites providing naloxone in 29 states and the District of Columbia. These organizations have distributed 152,000 naloxone kits and have reported more than 26,000 reversals.8 Certainly, OEND has prevented morbidity and mortality in some of these patients.
The adoption of OEND can be performed by individual prescribers as well. Naloxone is U.S. FDA-approved for the treatment of opioid overdose, and thus the liability to prescribers is similar to that of other FDA-approved drugs. However, the distribution of naloxone to third parties, such as friends and family of individuals with opioid misuse, is more complex and regulated by state laws. Many states have created liability protection for naloxone prescription to third parties. Individual state laws and additional information can be found at prescribetoprevent.org.
Hospitalists should provide opioid overdose education to all individuals with opioid misuse and friends and family of individuals with opioid misuse. In addition, hospitalists should prescribe naloxone to individuals with opioid misuse and, in states where the law allows, distribute naloxone to friends and family of individuals with opioid misuse as well.
Controversies. In general, opioid use disorder treatment providers; public health officials; and local, state, and federal government agencies have increasingly embraced harm-reduction strategies. However, some feel that harm-reduction strategies are misguided or even detrimental due to concern that they implicitly condone or enable the use of illicit substances. There have been a number of studies to evaluate the potential unintended consequences of harm-reduction strategies, and overwhelmingly, these have been either neutral or have shown the benefit of harm-reduction interventions. At this point, there is no good evidence to prevent the widespread adoption of harm-reduction strategies for hospitalists.
Back to the Case
The case involves an individual who has already had at least two complications of his IV heroin use, including cellulitis and opioid overdose. Ideally, this individual would be willing to see an addiction specialist and start medication-assisted treatment. Unfortunately, he is unwilling to be further evaluated by a specialist at this time. Regardless, he remains at risk of future complications, and it is the hospitalist’s responsibility to intervene with a goal of reducing future harm that may result from his IV heroin use.
The hospitalist in this case advises the patient to abstain from heroin and IDU, encourages him to seek treatment for his opioid use disorder, and gives him resources for linkage to care if he becomes interested. In addition, the hospitalist educates the patient on safe injection practices and provides a list of local syringe exchange programs to decrease future risk of viral, bacterial, and fungal infections. Furthermore, the hospitalist provides opioid overdose education and distributes naloxone to the patient, along with friends and family of the patient, to reduce the risk of death related to opioid overdose.
Bottom Line
Hospitalists should utilize harm-reduction interventions in individuals hospitalized with opioid misuse. TH
Dr. Theisen-Toupal is a hospitalist at the Veterans Affairs Medical Center and assistant professor of medicine at the George Washington University School of Medicine & Health Sciences, both in Washington, D.C.
References
- Vital signs: overdoses of prescription opioid pain relievers—United States, 1999–2008. Morbidity and Mortality Weekly Report. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6043a4.htm. Published November 4, 2011.
- Drug abuse warning network, 2011: national estimates of drug-related emergency department visits. Substance Abuse and Mental Health Services Administration website. Available at: http://www.samhsa.gov/data/2k13/DAWN2k11ED/DAWN2k11ED.htm#5. Accessed July 29, 2015.
- Hedergaard H, Chen LH, Warner M. Drug-poisoning deaths involving heroin: United States, 2000–2013. National Center for Health Statistics Data Brief. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/nchs/data/databriefs/db190.htm. Published March 2015.
- Getting off right: a safety manual for injection drug users. Harm Reduction Coalition website. Available at: http://harmreduction.org/wp-content/uploads/2011/12/getting-off-right.pdf.
- Syringe exchange programs—United States, 2008. Morbidity and Mortality Weekly Report. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5945a4.htm/Syringe-Exchange-Programs-United-States-2008. Published November 19, 2010.
- Wodak A, Conney A. Effectiveness of sterile needle and syringe programming in reducing HIV/AIDS among injecting drug users. World Health Organization website. Available at: http://apps.who.int/iris/bitstream/10665/43107/1/9241591641.pdf. Published 2004.
- Walley AY, Xuan Z, Hackman HH, et al. Opioid overdose rates and implementation of overdose education and nasal naloxone distribution in Massachusetts: interrupted time series analysis. BMJ. 2013;346:f174.
- Wheeler E, Jones TS, Gilbert MK, Davidson PJ. Opioid overdose prevention programs providing naloxone to laypersons—United States, 2014. Morbidity and Mortality Weekly Report. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6423a2.htm. Published June 19, 2015.
Case
A 33-year-old male with a history of opioid overdose and opioid use disorder is admitted with IV heroin use complicated by injection site cellulitis. He is started on antibiotics with improvement in his cellulitis; however, his hospitalization is complicated by acute opioid withdrawal. Despite his history of opioid overdose and opioid use disorder, he has never seen a substance use disorder specialist nor received any education or treatment for his addiction. He reports that he will stop using illicit drugs but declines any further addiction treatment.
What strategies can be employed to reduce his risk of future harm from opioid misuse?
Background
Over the past decade, the U.S. has experienced a rapid increase in the rates of opioid prescriptions and opioid misuse.1 Consequently, the number of ED visits and hospitalizations for opioid-related complications has also increased.2 Many complications result from the practice of injection drug use (IDU), which predisposes individuals to serious blood-borne viral infections such as human immunodeficiency virus (HIV) and hepatitis C virus (HCV) as well as bacterial infections such as infective endocarditis. In addition, individuals who misuse opioids are at risk of death related to opioid overdose. In 2013, there were more than 24,000 deaths in the U.S. due to opioid overdose (see Figure 1).3
In response to the opioid epidemic, there have been a number of local, state, and federal public health initiatives to monitor and secure the opioid drug supply, improve treatment resources, and promulgate harm-reduction interventions. At a more individual level, hospitalists have an important role to play in combating the opioid epidemic. As frontline providers, hospitalists have access to hospitalized individuals with opioid misuse who may not otherwise be exposed to the healthcare system. Therefore, inpatient hospitalizations serve as a unique and important opportunity to engage individuals in the management of their addiction.
There are a number of interventions that hospitalists and substance use disorder specialists can pursue. Psychiatric evaluation and initiation of medication-assisted treatment often aim to aid patients in abstaining from further opioid misuse. However, many individuals with opioid use disorder are not ready for treatment or experience relapses of opioid misuse despite treatment. Given this, a secondary goal is to reduce any harm that may result from opioid misuse. This is done through the implementation of harm-reduction strategies. These strategies include teaching safe injection practices, facilitating the use of syringe exchange programs, and providing opioid overdose education and naloxone distribution.
Overview of Data
Safe Injection Education. People who inject drugs are at risk for viral, bacterial, and fungal infections. These infections are often the result of nonsterile injection and may be minimized by the utilization of safe injection practices. In order to educate people who inject drugs on safe injection practices, the hospitalist must first understand the process involved in injecting drugs. In Table 1, the process of injecting heroin is outlined (of note, other illicit drugs can be injected, and processes may vary).4
As evidenced by Table 1, the process of sterile injection can be complicated, especially for an individual who may be withdrawing from opioids. Table 1 is also optimistic in that it recommends new and sterile products be used with every injection. If new and sterile equipment is not available, another option is to clean the equipment after every use, which can be done by using bleach and water. This may mitigate the risk of viral, bacterial, and fungal infections. However, the risk is still present, so users should not share or use another individual’s equipment even if it has been cleaned. Due to the risk of viral, bacterial, and fungal infections, all hospitalized individuals who inject drugs should receive education on safe injection practices.
Syringe Exchange Programs. IDU accounts for up to 15% of all new HIV infections and is the primary risk factor for the transmission of HCV.5 These infections occur when people inject using equipment contaminated with blood that contains HIV and/or HCV. Given this, if people who inject drugs could access and consistently use sterile syringes and other injection paraphernalia, the risk of transmitting blood-borne infections would be dramatically reduced. This is the concept behind syringe exchange programs (also known as needle exchange programs), which serve to increase access to sterile syringes while removing contaminated or used syringes from the community.
There is compelling evidence that syringe exchange programs decrease the rate of HIV transmission and likely reduce the rate of HCV transmission as well.6 In addition, syringe exchange programs often provide other beneficial services, such as counseling, testing, and prevention efforts for HIV, HCV, and sexually transmitted infections; distribution of condoms; and referrals to treatment services for substance use disorder.5
Unfortunately, in the U.S., restrictive state laws and lack of funding limit the number of established syringe exchange programs. According to the North American Syringe Exchange Network, there are only 226 programs in 33 states and the District of Columbia. Hospitalists and social workers should be aware of available local resources, including syringe exchange programs, and distribute this information to hospitalized individuals who inject drugs.
Opioid Overdose Education and Naloxone Distribution. Syringe exchange programs and safe injection education aim to reduce harm by decreasing the transmission of infections; however, they do not address the problem of deaths related to opioid overdose. The primary harm-reduction strategy used to address deaths related to opioid overdose in the U.S is opioid overdose education and naloxone distribution (OEND). Naloxone is an opioid antagonist that reverses the respiratory depression and decreased consciousness caused by opioids. The OEND strategy involves educating first responders— including individuals and friends and family of individuals who use opioids—to recognize the signs of an opioid overdose, seek help, provide rescue breathing, administer naloxone, and stay with the individual until emergency medical services arrive.7 This strategy has been observed to decrease rates of death related to opioid overdose.7
Given the evolving opioid epidemic and effectiveness of the OEND strategy, it is not surprising that the number of local opioid overdose prevention programs adopting OEND has risen dramatically. As of 2014, there were 140 organizations, with 644 local sites providing naloxone in 29 states and the District of Columbia. These organizations have distributed 152,000 naloxone kits and have reported more than 26,000 reversals.8 Certainly, OEND has prevented morbidity and mortality in some of these patients.
The adoption of OEND can be performed by individual prescribers as well. Naloxone is U.S. FDA-approved for the treatment of opioid overdose, and thus the liability to prescribers is similar to that of other FDA-approved drugs. However, the distribution of naloxone to third parties, such as friends and family of individuals with opioid misuse, is more complex and regulated by state laws. Many states have created liability protection for naloxone prescription to third parties. Individual state laws and additional information can be found at prescribetoprevent.org.
Hospitalists should provide opioid overdose education to all individuals with opioid misuse and friends and family of individuals with opioid misuse. In addition, hospitalists should prescribe naloxone to individuals with opioid misuse and, in states where the law allows, distribute naloxone to friends and family of individuals with opioid misuse as well.
Controversies. In general, opioid use disorder treatment providers; public health officials; and local, state, and federal government agencies have increasingly embraced harm-reduction strategies. However, some feel that harm-reduction strategies are misguided or even detrimental due to concern that they implicitly condone or enable the use of illicit substances. There have been a number of studies to evaluate the potential unintended consequences of harm-reduction strategies, and overwhelmingly, these have been either neutral or have shown the benefit of harm-reduction interventions. At this point, there is no good evidence to prevent the widespread adoption of harm-reduction strategies for hospitalists.
Back to the Case
The case involves an individual who has already had at least two complications of his IV heroin use, including cellulitis and opioid overdose. Ideally, this individual would be willing to see an addiction specialist and start medication-assisted treatment. Unfortunately, he is unwilling to be further evaluated by a specialist at this time. Regardless, he remains at risk of future complications, and it is the hospitalist’s responsibility to intervene with a goal of reducing future harm that may result from his IV heroin use.
The hospitalist in this case advises the patient to abstain from heroin and IDU, encourages him to seek treatment for his opioid use disorder, and gives him resources for linkage to care if he becomes interested. In addition, the hospitalist educates the patient on safe injection practices and provides a list of local syringe exchange programs to decrease future risk of viral, bacterial, and fungal infections. Furthermore, the hospitalist provides opioid overdose education and distributes naloxone to the patient, along with friends and family of the patient, to reduce the risk of death related to opioid overdose.
Bottom Line
Hospitalists should utilize harm-reduction interventions in individuals hospitalized with opioid misuse. TH
Dr. Theisen-Toupal is a hospitalist at the Veterans Affairs Medical Center and assistant professor of medicine at the George Washington University School of Medicine & Health Sciences, both in Washington, D.C.
References
- Vital signs: overdoses of prescription opioid pain relievers—United States, 1999–2008. Morbidity and Mortality Weekly Report. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6043a4.htm. Published November 4, 2011.
- Drug abuse warning network, 2011: national estimates of drug-related emergency department visits. Substance Abuse and Mental Health Services Administration website. Available at: http://www.samhsa.gov/data/2k13/DAWN2k11ED/DAWN2k11ED.htm#5. Accessed July 29, 2015.
- Hedergaard H, Chen LH, Warner M. Drug-poisoning deaths involving heroin: United States, 2000–2013. National Center for Health Statistics Data Brief. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/nchs/data/databriefs/db190.htm. Published March 2015.
- Getting off right: a safety manual for injection drug users. Harm Reduction Coalition website. Available at: http://harmreduction.org/wp-content/uploads/2011/12/getting-off-right.pdf.
- Syringe exchange programs—United States, 2008. Morbidity and Mortality Weekly Report. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5945a4.htm/Syringe-Exchange-Programs-United-States-2008. Published November 19, 2010.
- Wodak A, Conney A. Effectiveness of sterile needle and syringe programming in reducing HIV/AIDS among injecting drug users. World Health Organization website. Available at: http://apps.who.int/iris/bitstream/10665/43107/1/9241591641.pdf. Published 2004.
- Walley AY, Xuan Z, Hackman HH, et al. Opioid overdose rates and implementation of overdose education and nasal naloxone distribution in Massachusetts: interrupted time series analysis. BMJ. 2013;346:f174.
- Wheeler E, Jones TS, Gilbert MK, Davidson PJ. Opioid overdose prevention programs providing naloxone to laypersons—United States, 2014. Morbidity and Mortality Weekly Report. Centers for Disease Control and Prevention website. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6423a2.htm. Published June 19, 2015.

QUIZ: Which Strategy Should Hospitalists Employ to Reduce the Risk of Opioid Misuse?
[WpProQuiz 6]
[WpProQuiz_toplist 6]
[WpProQuiz 6]
[WpProQuiz_toplist 6]
[WpProQuiz 6]
[WpProQuiz_toplist 6]
Caught red‐handed
A previously healthy 58‐year‐old man presented to a community hospital's emergency department 1 day after the sudden onset of a severe headache, fever, diffuse abdominal pain, nausea, vomiting, and disorientation. The patient had a history of allergic rhinitis and his only medication was a daily multivitamin.
Key features of this patient's presentation include the abrupt onset of severe headache, disorientation, fever, and abdominal pain. The list of entities likely to make a previously healthy individual this ill this quickly is typically circumscribed. His presentation raises the possibility of bacterial meningitis (including Listeria, given his age), viral encephalitis, or other extraneural etiologies of sepsis. Noninfectious explanations seem much less likely given the rapid tempo of illness.
His proclivity for gardening and apparent tick exposure raise the question of tick‐borne illnesses. This would constitute a rather explosive onset for any of these; however, babesiosis, Rocky Mountain spotted fever (RMSF), ehrlichiosis, and anaplasmosis could present this abruptly, with dog exposure linked to RMSF.
The potential causes of fever and rash are myriad, although the severity and acuity of this patient's illness narrow the differential considerably, likely to an infectious cause. Diagnoses that typically include a generalized exanthem involving the palms and soles are meningococcal meningitis, overwhelming Staphylococcus aureus sepsis, RMSF (realizing that this disease is not common in the upper Midwest), and toxic shock syndrome. The rash described is not the classic and/or fully developed rash typical of any of these; subsequent evolution to a petechial appearance would lend further support to the first 3 diagnoses. Ehrlichiosis is still a possibility, although the palm and sole involvement would be unusual. The presence of a rash makes anaplasmosis very unlikely, although not entirely excluded. The finding of modest splenomegaly does not help further distinguish between these possibilities.
Empiric antimicrobials should be immediately administered after blood cultures, a complete blood count, and coagulation studies are obtained. Doxycycline would be appropriate to treat the possible tick‐borne diseases already mentioned, whereas antimicrobials appropriate to cover community‐acquired bacterial meningitis in a 58‐year‐old (ie, vancomycin, ampicillin, and a third‐generation cephalosporin) should also be empirically administered. Given the patient's altered mentation, a brain computed tomography (CT) should be urgently obtained. Provided this did not show evidence of increased intracranial pressure and that coagulation studies and a platelet count did not suggest a contraindication, a lumbar puncture should then be performed promptly. The patient should be placed in droplet precautions until meningococcal disease is excluded. Although most patients with bacterial meningitis will exhibit meningismus, a substantial minority will not.
These laboratory results do not significantly affect the differential diagnosis. Although nonspecific, moderate thrombocytopenia and modest elevation of hepatic transaminases are typical for tick‐borne diseases, whereas leukocytosis is somewhat atypical for these entities. Marked elevation of the C‐reactive protein with a less striking increase in the erythrocyte sedimentation rate, along with significant hypoalbuminemia, are commonly encountered early in the course of critical infectious illnesses. The elevated troponin likely reflects severe sepsis and demand ischemia, and is associated with a less favorable prognosis; an electrocardiogram and serial cardiac biomarkers are appropriate to help exclude an acute coronary syndrome. As already noted, blood cultures need to be obtained and a lumbar puncture should be performed, provided this can be safely accomplished.
Results of the lumbar puncture exclude bacterial meningitis as the explanation of this patient's illness; the mildly elevated protein is nonspecific. These studies do not otherwise change the differential diagnosis.
Supporting data for a diagnosis of pneumonia, such as pulmonary infiltrates or supplemental oxygen requirement, are lacking. Given his critical illness, broad spectrum antimicrobial coverage is indicated, and as a primary central nervous system (CNS) infection now appears unlikely, piperacillin/tazobactam (which does not have adequate CNS penetration) and vancomycin are reasonable. Empiric treatment for RMSF is appropriate, and should have been initiated earlier in the patient's course, despite the upper Midwest being out of the typical range for this disease. Doxycycline will also provide excellent coverage for ehrlichiosis and anaplasmosis.
Given the patient's deterioration, it is important to stop and reconsider the differential diagnosis in an attempt to avoid anchoring bias and premature closure. The patient's illness is almost certainly infectious in nature, and the differential is not substantially altered by the most recent information. A skin biopsy should be performed in an attempt to secure the diagnosis.
The patient's overall course, including rapid onset of severe illness and especially the apparent dramatic response to doxycycline, make tick‐borne illness very likely. Completing a course of doxycycline is certainly appropriate, typically for 7 to 14 days. The acute serologies drawn prior to discharge may well reveal the causative agent, but convalescent serology should also be obtained at the time of an outpatient follow‐up visit as immunoglobulin G has a delayed rise. Without hyponatremia or respiratory symptoms, Legionella seems unlikely.
The appearance of late desquamation of the palms and soles is an unexpected and important sign. Desquamation in this pattern following an illness of this nature strongly suggests a diagnosis of staphylococcal toxic shock syndrome (TSS), and in conjunction with the negative serologies, argues that tick‐borne disease is unlikely. The list of other entities that might lead to desquamation in this setting is very short, namely adult Kawasaki disease and drug reaction. The former seems reasonably excluded based on details of the case, whereas a doxycycline‐related drug reaction, although not entirely implausible, seems quite unlikely as this medication was started after the onset of the initial rash. This patient most likely had staphylococcal TSS secondary to a minor and unappreciated skin lesion.
DISCUSSION
TSS is a systemic illness resulting in multiorgan dysfunction.[1] Infection by S aureus or Streptococcus pyogenes causes TSS by stimulating maladaptive T‐cell proliferation and cytokine release resulting in shock.[1, 2] A definitive diagnosis requires fever, a diffuse macular erythematous rash (often resembling a sunburn), with subsequent desquamation, hypotension, and involvement of at least 3 organ systems. Blood cultures, cerebrospinal cultures, and serologies for other organisms should be negative; although Staphylococcus and Streptococcus species may be isolated, they frequently are not (Table 1).[3]
| Diagnostic Criteria* | This Case |
|---|---|
| |
| Fever: Temperature 102.0F | Fever: 105.3F on admission |
| Rash: Diffuse macular erythroderma | Diffuse morbilliform rash with progression to confluent erythroderma |
| Desquamation of rash: occurs 12 weeks following rash onset | Desquamation 12 days after discharge |
| Hypotension: SBP 90 mm Hg for adults | Intermittent |
| Multisystem involvement, 3 of the following: | 4 organ systems definitively involved |
| GI: vomiting or diarrhea at disease onset | Vomiting and abdominal pain |
| Muscular: severe myalgias, or creatine phosphokinase >2 times the upper limit of normal | |
| Mucous membranes: vaginal, oropharyngeal, or conjunctival hyperemia | |
| Renal: BUN or Cr >2 times the upper limit of normal, or pyuria without evidence of infection | |
| Hepatic: total bilirubin, AST, or ALT levels >2 times the upper limit of normal | AST and ALT peaked at 128IU/L and 94 IU/L |
| Hematologic: platelets <100,000/mm3 | Platelet nadir of 80,000/mm3 |
| CNS: disorientation or altered consciousness without focal neurologic signs | Disorientation and somnolence |
| Probable case: 4 out of 5 clinical criteria present | |
| Confirmed case: 5 out of 5 clinical criteria present, or patient dies before desquamation can occur | |
A rare cause of shock, TSS is most associated with a surge of menstruation‐related cases linked to tampon use in young women in the 1980s.[4] However, in Centers for Disease Control and Prevention (CDC) surveillance between 1987 and 1996, only 59% of the 1069 cases identified were noted to be menstruation‐related, as compared to nearly 80% of all cases earlier in the decade.[4, 5] Today, the syndrome is more likely to present after musculoskeletal and cutaneous trauma, oropharyngeal infections, surgical procedures, and device implantation.[1, 6] Despite the disease's evolving epidemiology, the illness script used by physicians likely continues to focus on young women as the primary at risk population for TSS, causing physicians to neglect the diagnosis in other populations.[1, 6, 7, 8, 9] Given this change in risk factors, it is imperative that clinicians rewrite their scripts and recognize the early signs of TSS in all patients to enable quick and effective treatment.
In addition to its shifting epidemiology and rarity, the diagnosis of TSS vexes clinicians for several reasons. First, TSS cannot be quickly and definitively diagnosed because 2 diagnostic criteria cannot be fulfilled during the acute illness. The disease's hallmarka desquamative rashoccurs only if the patient survives.[3] Serologies often take weeks to return, further delaying diagnosis. During this period of diagnostic delay, the illness has usually already resolved or resulted in death. In addition, the presenting symptoms of rash, fever, and shock are nonspecific. Alternative etiologies include meningococcal meningitis, which can also present dramatically as with this patient; RMSF, which can occasionally have a fulminant presentation; bacterial sepsis, usually from Staphylococcus or Streptococcus species; acute viral syndromes; and severe drug reactions.[6, 10, 11, 12] Palmoplantar desquamation, as in this case, can further narrow the differential as this presentation is uncommon but characteristic of TSS, RMSF, and secondary syphilis.[11] Other diagnostic clues offered by the pattern of the rash may be limited by physician discomfort with diagnosing and describing rashes. Because of this lack of a definitive diagnostic test in the acute setting, it is imperative that the clinician include TSS in the differential of fever, shock, and rash, as mortality from TSS can exceed 20% in patients who are untreated.[13]
Treatment of TSS is straightforward once considered and includes the administration of antibiotics that cover both Staphylococcus and Streptococcus species, in addition to aggressive hydration and supportive care.[14] The final critical detail in this case was the appropriate arrangement of follow‐up. Given the patient's drastic improvement, the complicated process of arranging follow‐up for a transferred patient, and the current model where the hospitalists providing inpatient care do not typically follow their patients in clinic, patients such as these can easily be lost to follow‐up. Had this occurred, the desquamation would have been missed, and the patient's diagnosis would have been incomplete.
This patient was eventually diagnosed with TSS by fulfilling all 5 CDC criteria (Table 1).[3] He made a full recovery, likely aided by the administration of broad‐spectrum antibiotics (followed by doxycycline, which provided community‐acquired methicillin‐resistant S aureus coverage) and his lack of serious comorbidities. This case should serve as a reminder to hospitalists that with a discerning eye, a careful assessment of the clinical facts, and appropriate follow‐up, perhaps the next case of TSS can be caught red‐handed.
KEY POINTS
- When presented with a patient with fever, rash, and shock, hospitalists should consider meningococcal meningitis, RMSF bacterial sepsis, acute viral illness, severe drug reaction, and TSS.
- TSS, caused by S aureus or S pyogenes, is no longer predominantly associated with tampon use. Postsurgical infection and cutaneous trauma have become important present‐day risk factors.
- The initial presentation of TSS is nonspecific. Definitive diagnosis requires proper follow‐up, allowing time for infectious serologies to return negative and for the disease's hallmark desquamation to occur.
Disclosure
Nothing to report.
- . Toxic shock syndrome: major advances in pathogenesis, but not treatment. Crit Care Clin. 2013;29:651–675.
- . The toxic shock syndromes. Infect Dis Clin North Am. 1996;10(4):727–746.
- Centers for Disease Control and Prevention. National Notifiable Diseases Surveillance System. Toxic shock syndrome (other than Streptococcal) (TSS) 2011 Case Definition. Available at: http://wwwn.cdc.gov/nndss/conditions/toxic‐shock‐syndrome‐other‐than‐streptococcal/case‐definition/2011. Accessed June 4, 2015.
- Centers for Disease Control and Prevention. Update: toxic‐shock syndrome—United States. MMWR Morb Mortal Wkly Rep. 1983;32(30):398–400.
- , , , , , . Toxic shock syndrome in the United States: surveillance update, 1979–1996. Emerg Infect Dis. 1999;5(6):807–810.
- . Fever and rash. Infect Dis Clin North Am. 1996;10(1):101–110.
- , , , et al. Staphylococcal toxic shock syndrome 2000–2006: epidemiology, clinical features, and molecular characteristics. PLoS One. 2011;6(8):e22997.
- , , , et al. Toxic‐shock syndrome in menstruating women: association with tampon use and staphylococcus aureus and clinical features in 52 cases. N Engl J Med. 1980;303(25):1436–1442.
- , , , . Toxic‐shock syndrome—epidemiologic features, recurrence, risk factors, and prevention. N Engl J Med. 1980;303:1429–1435.
- , . Evaluating the febrile patient with a rash. Am Fam Physician. 2000;62(4):804–816.
- . Toxic shock syndrome: broadening the differential diagnosis. J Am Board Fam Pract. 2001;14(2):131–136.
- , , , . Spatial clustering by disease severity among reported Rocky Mountain spotted fever cases in the United States, 2001–2005. Am J Trop Med Hyg. 2009;80(1):72–77.
- , , , et al. One in five mortality in non‐menstrual toxic shock syndrome versus no mortality in menstrual cases in a balanced French series of 55 cases. Eur J Clin Microbio Infect Dis. 2008;27(1):37–43.
- , . Gram‐positive toxic shock syndromes. Lancet Infect Dis. 2009;9(5):281–290.
A previously healthy 58‐year‐old man presented to a community hospital's emergency department 1 day after the sudden onset of a severe headache, fever, diffuse abdominal pain, nausea, vomiting, and disorientation. The patient had a history of allergic rhinitis and his only medication was a daily multivitamin.
Key features of this patient's presentation include the abrupt onset of severe headache, disorientation, fever, and abdominal pain. The list of entities likely to make a previously healthy individual this ill this quickly is typically circumscribed. His presentation raises the possibility of bacterial meningitis (including Listeria, given his age), viral encephalitis, or other extraneural etiologies of sepsis. Noninfectious explanations seem much less likely given the rapid tempo of illness.
His proclivity for gardening and apparent tick exposure raise the question of tick‐borne illnesses. This would constitute a rather explosive onset for any of these; however, babesiosis, Rocky Mountain spotted fever (RMSF), ehrlichiosis, and anaplasmosis could present this abruptly, with dog exposure linked to RMSF.
The potential causes of fever and rash are myriad, although the severity and acuity of this patient's illness narrow the differential considerably, likely to an infectious cause. Diagnoses that typically include a generalized exanthem involving the palms and soles are meningococcal meningitis, overwhelming Staphylococcus aureus sepsis, RMSF (realizing that this disease is not common in the upper Midwest), and toxic shock syndrome. The rash described is not the classic and/or fully developed rash typical of any of these; subsequent evolution to a petechial appearance would lend further support to the first 3 diagnoses. Ehrlichiosis is still a possibility, although the palm and sole involvement would be unusual. The presence of a rash makes anaplasmosis very unlikely, although not entirely excluded. The finding of modest splenomegaly does not help further distinguish between these possibilities.
Empiric antimicrobials should be immediately administered after blood cultures, a complete blood count, and coagulation studies are obtained. Doxycycline would be appropriate to treat the possible tick‐borne diseases already mentioned, whereas antimicrobials appropriate to cover community‐acquired bacterial meningitis in a 58‐year‐old (ie, vancomycin, ampicillin, and a third‐generation cephalosporin) should also be empirically administered. Given the patient's altered mentation, a brain computed tomography (CT) should be urgently obtained. Provided this did not show evidence of increased intracranial pressure and that coagulation studies and a platelet count did not suggest a contraindication, a lumbar puncture should then be performed promptly. The patient should be placed in droplet precautions until meningococcal disease is excluded. Although most patients with bacterial meningitis will exhibit meningismus, a substantial minority will not.
These laboratory results do not significantly affect the differential diagnosis. Although nonspecific, moderate thrombocytopenia and modest elevation of hepatic transaminases are typical for tick‐borne diseases, whereas leukocytosis is somewhat atypical for these entities. Marked elevation of the C‐reactive protein with a less striking increase in the erythrocyte sedimentation rate, along with significant hypoalbuminemia, are commonly encountered early in the course of critical infectious illnesses. The elevated troponin likely reflects severe sepsis and demand ischemia, and is associated with a less favorable prognosis; an electrocardiogram and serial cardiac biomarkers are appropriate to help exclude an acute coronary syndrome. As already noted, blood cultures need to be obtained and a lumbar puncture should be performed, provided this can be safely accomplished.
Results of the lumbar puncture exclude bacterial meningitis as the explanation of this patient's illness; the mildly elevated protein is nonspecific. These studies do not otherwise change the differential diagnosis.
Supporting data for a diagnosis of pneumonia, such as pulmonary infiltrates or supplemental oxygen requirement, are lacking. Given his critical illness, broad spectrum antimicrobial coverage is indicated, and as a primary central nervous system (CNS) infection now appears unlikely, piperacillin/tazobactam (which does not have adequate CNS penetration) and vancomycin are reasonable. Empiric treatment for RMSF is appropriate, and should have been initiated earlier in the patient's course, despite the upper Midwest being out of the typical range for this disease. Doxycycline will also provide excellent coverage for ehrlichiosis and anaplasmosis.
Given the patient's deterioration, it is important to stop and reconsider the differential diagnosis in an attempt to avoid anchoring bias and premature closure. The patient's illness is almost certainly infectious in nature, and the differential is not substantially altered by the most recent information. A skin biopsy should be performed in an attempt to secure the diagnosis.
The patient's overall course, including rapid onset of severe illness and especially the apparent dramatic response to doxycycline, make tick‐borne illness very likely. Completing a course of doxycycline is certainly appropriate, typically for 7 to 14 days. The acute serologies drawn prior to discharge may well reveal the causative agent, but convalescent serology should also be obtained at the time of an outpatient follow‐up visit as immunoglobulin G has a delayed rise. Without hyponatremia or respiratory symptoms, Legionella seems unlikely.
The appearance of late desquamation of the palms and soles is an unexpected and important sign. Desquamation in this pattern following an illness of this nature strongly suggests a diagnosis of staphylococcal toxic shock syndrome (TSS), and in conjunction with the negative serologies, argues that tick‐borne disease is unlikely. The list of other entities that might lead to desquamation in this setting is very short, namely adult Kawasaki disease and drug reaction. The former seems reasonably excluded based on details of the case, whereas a doxycycline‐related drug reaction, although not entirely implausible, seems quite unlikely as this medication was started after the onset of the initial rash. This patient most likely had staphylococcal TSS secondary to a minor and unappreciated skin lesion.
DISCUSSION
TSS is a systemic illness resulting in multiorgan dysfunction.[1] Infection by S aureus or Streptococcus pyogenes causes TSS by stimulating maladaptive T‐cell proliferation and cytokine release resulting in shock.[1, 2] A definitive diagnosis requires fever, a diffuse macular erythematous rash (often resembling a sunburn), with subsequent desquamation, hypotension, and involvement of at least 3 organ systems. Blood cultures, cerebrospinal cultures, and serologies for other organisms should be negative; although Staphylococcus and Streptococcus species may be isolated, they frequently are not (Table 1).[3]
| Diagnostic Criteria* | This Case |
|---|---|
| |
| Fever: Temperature 102.0F | Fever: 105.3F on admission |
| Rash: Diffuse macular erythroderma | Diffuse morbilliform rash with progression to confluent erythroderma |
| Desquamation of rash: occurs 12 weeks following rash onset | Desquamation 12 days after discharge |
| Hypotension: SBP 90 mm Hg for adults | Intermittent |
| Multisystem involvement, 3 of the following: | 4 organ systems definitively involved |
| GI: vomiting or diarrhea at disease onset | Vomiting and abdominal pain |
| Muscular: severe myalgias, or creatine phosphokinase >2 times the upper limit of normal | |
| Mucous membranes: vaginal, oropharyngeal, or conjunctival hyperemia | |
| Renal: BUN or Cr >2 times the upper limit of normal, or pyuria without evidence of infection | |
| Hepatic: total bilirubin, AST, or ALT levels >2 times the upper limit of normal | AST and ALT peaked at 128IU/L and 94 IU/L |
| Hematologic: platelets <100,000/mm3 | Platelet nadir of 80,000/mm3 |
| CNS: disorientation or altered consciousness without focal neurologic signs | Disorientation and somnolence |
| Probable case: 4 out of 5 clinical criteria present | |
| Confirmed case: 5 out of 5 clinical criteria present, or patient dies before desquamation can occur | |
A rare cause of shock, TSS is most associated with a surge of menstruation‐related cases linked to tampon use in young women in the 1980s.[4] However, in Centers for Disease Control and Prevention (CDC) surveillance between 1987 and 1996, only 59% of the 1069 cases identified were noted to be menstruation‐related, as compared to nearly 80% of all cases earlier in the decade.[4, 5] Today, the syndrome is more likely to present after musculoskeletal and cutaneous trauma, oropharyngeal infections, surgical procedures, and device implantation.[1, 6] Despite the disease's evolving epidemiology, the illness script used by physicians likely continues to focus on young women as the primary at risk population for TSS, causing physicians to neglect the diagnosis in other populations.[1, 6, 7, 8, 9] Given this change in risk factors, it is imperative that clinicians rewrite their scripts and recognize the early signs of TSS in all patients to enable quick and effective treatment.
In addition to its shifting epidemiology and rarity, the diagnosis of TSS vexes clinicians for several reasons. First, TSS cannot be quickly and definitively diagnosed because 2 diagnostic criteria cannot be fulfilled during the acute illness. The disease's hallmarka desquamative rashoccurs only if the patient survives.[3] Serologies often take weeks to return, further delaying diagnosis. During this period of diagnostic delay, the illness has usually already resolved or resulted in death. In addition, the presenting symptoms of rash, fever, and shock are nonspecific. Alternative etiologies include meningococcal meningitis, which can also present dramatically as with this patient; RMSF, which can occasionally have a fulminant presentation; bacterial sepsis, usually from Staphylococcus or Streptococcus species; acute viral syndromes; and severe drug reactions.[6, 10, 11, 12] Palmoplantar desquamation, as in this case, can further narrow the differential as this presentation is uncommon but characteristic of TSS, RMSF, and secondary syphilis.[11] Other diagnostic clues offered by the pattern of the rash may be limited by physician discomfort with diagnosing and describing rashes. Because of this lack of a definitive diagnostic test in the acute setting, it is imperative that the clinician include TSS in the differential of fever, shock, and rash, as mortality from TSS can exceed 20% in patients who are untreated.[13]
Treatment of TSS is straightforward once considered and includes the administration of antibiotics that cover both Staphylococcus and Streptococcus species, in addition to aggressive hydration and supportive care.[14] The final critical detail in this case was the appropriate arrangement of follow‐up. Given the patient's drastic improvement, the complicated process of arranging follow‐up for a transferred patient, and the current model where the hospitalists providing inpatient care do not typically follow their patients in clinic, patients such as these can easily be lost to follow‐up. Had this occurred, the desquamation would have been missed, and the patient's diagnosis would have been incomplete.
This patient was eventually diagnosed with TSS by fulfilling all 5 CDC criteria (Table 1).[3] He made a full recovery, likely aided by the administration of broad‐spectrum antibiotics (followed by doxycycline, which provided community‐acquired methicillin‐resistant S aureus coverage) and his lack of serious comorbidities. This case should serve as a reminder to hospitalists that with a discerning eye, a careful assessment of the clinical facts, and appropriate follow‐up, perhaps the next case of TSS can be caught red‐handed.
KEY POINTS
- When presented with a patient with fever, rash, and shock, hospitalists should consider meningococcal meningitis, RMSF bacterial sepsis, acute viral illness, severe drug reaction, and TSS.
- TSS, caused by S aureus or S pyogenes, is no longer predominantly associated with tampon use. Postsurgical infection and cutaneous trauma have become important present‐day risk factors.
- The initial presentation of TSS is nonspecific. Definitive diagnosis requires proper follow‐up, allowing time for infectious serologies to return negative and for the disease's hallmark desquamation to occur.
Disclosure
Nothing to report.
A previously healthy 58‐year‐old man presented to a community hospital's emergency department 1 day after the sudden onset of a severe headache, fever, diffuse abdominal pain, nausea, vomiting, and disorientation. The patient had a history of allergic rhinitis and his only medication was a daily multivitamin.
Key features of this patient's presentation include the abrupt onset of severe headache, disorientation, fever, and abdominal pain. The list of entities likely to make a previously healthy individual this ill this quickly is typically circumscribed. His presentation raises the possibility of bacterial meningitis (including Listeria, given his age), viral encephalitis, or other extraneural etiologies of sepsis. Noninfectious explanations seem much less likely given the rapid tempo of illness.
His proclivity for gardening and apparent tick exposure raise the question of tick‐borne illnesses. This would constitute a rather explosive onset for any of these; however, babesiosis, Rocky Mountain spotted fever (RMSF), ehrlichiosis, and anaplasmosis could present this abruptly, with dog exposure linked to RMSF.
The potential causes of fever and rash are myriad, although the severity and acuity of this patient's illness narrow the differential considerably, likely to an infectious cause. Diagnoses that typically include a generalized exanthem involving the palms and soles are meningococcal meningitis, overwhelming Staphylococcus aureus sepsis, RMSF (realizing that this disease is not common in the upper Midwest), and toxic shock syndrome. The rash described is not the classic and/or fully developed rash typical of any of these; subsequent evolution to a petechial appearance would lend further support to the first 3 diagnoses. Ehrlichiosis is still a possibility, although the palm and sole involvement would be unusual. The presence of a rash makes anaplasmosis very unlikely, although not entirely excluded. The finding of modest splenomegaly does not help further distinguish between these possibilities.
Empiric antimicrobials should be immediately administered after blood cultures, a complete blood count, and coagulation studies are obtained. Doxycycline would be appropriate to treat the possible tick‐borne diseases already mentioned, whereas antimicrobials appropriate to cover community‐acquired bacterial meningitis in a 58‐year‐old (ie, vancomycin, ampicillin, and a third‐generation cephalosporin) should also be empirically administered. Given the patient's altered mentation, a brain computed tomography (CT) should be urgently obtained. Provided this did not show evidence of increased intracranial pressure and that coagulation studies and a platelet count did not suggest a contraindication, a lumbar puncture should then be performed promptly. The patient should be placed in droplet precautions until meningococcal disease is excluded. Although most patients with bacterial meningitis will exhibit meningismus, a substantial minority will not.
These laboratory results do not significantly affect the differential diagnosis. Although nonspecific, moderate thrombocytopenia and modest elevation of hepatic transaminases are typical for tick‐borne diseases, whereas leukocytosis is somewhat atypical for these entities. Marked elevation of the C‐reactive protein with a less striking increase in the erythrocyte sedimentation rate, along with significant hypoalbuminemia, are commonly encountered early in the course of critical infectious illnesses. The elevated troponin likely reflects severe sepsis and demand ischemia, and is associated with a less favorable prognosis; an electrocardiogram and serial cardiac biomarkers are appropriate to help exclude an acute coronary syndrome. As already noted, blood cultures need to be obtained and a lumbar puncture should be performed, provided this can be safely accomplished.
Results of the lumbar puncture exclude bacterial meningitis as the explanation of this patient's illness; the mildly elevated protein is nonspecific. These studies do not otherwise change the differential diagnosis.
Supporting data for a diagnosis of pneumonia, such as pulmonary infiltrates or supplemental oxygen requirement, are lacking. Given his critical illness, broad spectrum antimicrobial coverage is indicated, and as a primary central nervous system (CNS) infection now appears unlikely, piperacillin/tazobactam (which does not have adequate CNS penetration) and vancomycin are reasonable. Empiric treatment for RMSF is appropriate, and should have been initiated earlier in the patient's course, despite the upper Midwest being out of the typical range for this disease. Doxycycline will also provide excellent coverage for ehrlichiosis and anaplasmosis.
Given the patient's deterioration, it is important to stop and reconsider the differential diagnosis in an attempt to avoid anchoring bias and premature closure. The patient's illness is almost certainly infectious in nature, and the differential is not substantially altered by the most recent information. A skin biopsy should be performed in an attempt to secure the diagnosis.
The patient's overall course, including rapid onset of severe illness and especially the apparent dramatic response to doxycycline, make tick‐borne illness very likely. Completing a course of doxycycline is certainly appropriate, typically for 7 to 14 days. The acute serologies drawn prior to discharge may well reveal the causative agent, but convalescent serology should also be obtained at the time of an outpatient follow‐up visit as immunoglobulin G has a delayed rise. Without hyponatremia or respiratory symptoms, Legionella seems unlikely.
The appearance of late desquamation of the palms and soles is an unexpected and important sign. Desquamation in this pattern following an illness of this nature strongly suggests a diagnosis of staphylococcal toxic shock syndrome (TSS), and in conjunction with the negative serologies, argues that tick‐borne disease is unlikely. The list of other entities that might lead to desquamation in this setting is very short, namely adult Kawasaki disease and drug reaction. The former seems reasonably excluded based on details of the case, whereas a doxycycline‐related drug reaction, although not entirely implausible, seems quite unlikely as this medication was started after the onset of the initial rash. This patient most likely had staphylococcal TSS secondary to a minor and unappreciated skin lesion.
DISCUSSION
TSS is a systemic illness resulting in multiorgan dysfunction.[1] Infection by S aureus or Streptococcus pyogenes causes TSS by stimulating maladaptive T‐cell proliferation and cytokine release resulting in shock.[1, 2] A definitive diagnosis requires fever, a diffuse macular erythematous rash (often resembling a sunburn), with subsequent desquamation, hypotension, and involvement of at least 3 organ systems. Blood cultures, cerebrospinal cultures, and serologies for other organisms should be negative; although Staphylococcus and Streptococcus species may be isolated, they frequently are not (Table 1).[3]
| Diagnostic Criteria* | This Case |
|---|---|
| |
| Fever: Temperature 102.0F | Fever: 105.3F on admission |
| Rash: Diffuse macular erythroderma | Diffuse morbilliform rash with progression to confluent erythroderma |
| Desquamation of rash: occurs 12 weeks following rash onset | Desquamation 12 days after discharge |
| Hypotension: SBP 90 mm Hg for adults | Intermittent |
| Multisystem involvement, 3 of the following: | 4 organ systems definitively involved |
| GI: vomiting or diarrhea at disease onset | Vomiting and abdominal pain |
| Muscular: severe myalgias, or creatine phosphokinase >2 times the upper limit of normal | |
| Mucous membranes: vaginal, oropharyngeal, or conjunctival hyperemia | |
| Renal: BUN or Cr >2 times the upper limit of normal, or pyuria without evidence of infection | |
| Hepatic: total bilirubin, AST, or ALT levels >2 times the upper limit of normal | AST and ALT peaked at 128IU/L and 94 IU/L |
| Hematologic: platelets <100,000/mm3 | Platelet nadir of 80,000/mm3 |
| CNS: disorientation or altered consciousness without focal neurologic signs | Disorientation and somnolence |
| Probable case: 4 out of 5 clinical criteria present | |
| Confirmed case: 5 out of 5 clinical criteria present, or patient dies before desquamation can occur | |
A rare cause of shock, TSS is most associated with a surge of menstruation‐related cases linked to tampon use in young women in the 1980s.[4] However, in Centers for Disease Control and Prevention (CDC) surveillance between 1987 and 1996, only 59% of the 1069 cases identified were noted to be menstruation‐related, as compared to nearly 80% of all cases earlier in the decade.[4, 5] Today, the syndrome is more likely to present after musculoskeletal and cutaneous trauma, oropharyngeal infections, surgical procedures, and device implantation.[1, 6] Despite the disease's evolving epidemiology, the illness script used by physicians likely continues to focus on young women as the primary at risk population for TSS, causing physicians to neglect the diagnosis in other populations.[1, 6, 7, 8, 9] Given this change in risk factors, it is imperative that clinicians rewrite their scripts and recognize the early signs of TSS in all patients to enable quick and effective treatment.
In addition to its shifting epidemiology and rarity, the diagnosis of TSS vexes clinicians for several reasons. First, TSS cannot be quickly and definitively diagnosed because 2 diagnostic criteria cannot be fulfilled during the acute illness. The disease's hallmarka desquamative rashoccurs only if the patient survives.[3] Serologies often take weeks to return, further delaying diagnosis. During this period of diagnostic delay, the illness has usually already resolved or resulted in death. In addition, the presenting symptoms of rash, fever, and shock are nonspecific. Alternative etiologies include meningococcal meningitis, which can also present dramatically as with this patient; RMSF, which can occasionally have a fulminant presentation; bacterial sepsis, usually from Staphylococcus or Streptococcus species; acute viral syndromes; and severe drug reactions.[6, 10, 11, 12] Palmoplantar desquamation, as in this case, can further narrow the differential as this presentation is uncommon but characteristic of TSS, RMSF, and secondary syphilis.[11] Other diagnostic clues offered by the pattern of the rash may be limited by physician discomfort with diagnosing and describing rashes. Because of this lack of a definitive diagnostic test in the acute setting, it is imperative that the clinician include TSS in the differential of fever, shock, and rash, as mortality from TSS can exceed 20% in patients who are untreated.[13]
Treatment of TSS is straightforward once considered and includes the administration of antibiotics that cover both Staphylococcus and Streptococcus species, in addition to aggressive hydration and supportive care.[14] The final critical detail in this case was the appropriate arrangement of follow‐up. Given the patient's drastic improvement, the complicated process of arranging follow‐up for a transferred patient, and the current model where the hospitalists providing inpatient care do not typically follow their patients in clinic, patients such as these can easily be lost to follow‐up. Had this occurred, the desquamation would have been missed, and the patient's diagnosis would have been incomplete.
This patient was eventually diagnosed with TSS by fulfilling all 5 CDC criteria (Table 1).[3] He made a full recovery, likely aided by the administration of broad‐spectrum antibiotics (followed by doxycycline, which provided community‐acquired methicillin‐resistant S aureus coverage) and his lack of serious comorbidities. This case should serve as a reminder to hospitalists that with a discerning eye, a careful assessment of the clinical facts, and appropriate follow‐up, perhaps the next case of TSS can be caught red‐handed.
KEY POINTS
- When presented with a patient with fever, rash, and shock, hospitalists should consider meningococcal meningitis, RMSF bacterial sepsis, acute viral illness, severe drug reaction, and TSS.
- TSS, caused by S aureus or S pyogenes, is no longer predominantly associated with tampon use. Postsurgical infection and cutaneous trauma have become important present‐day risk factors.
- The initial presentation of TSS is nonspecific. Definitive diagnosis requires proper follow‐up, allowing time for infectious serologies to return negative and for the disease's hallmark desquamation to occur.
Disclosure
Nothing to report.
- . Toxic shock syndrome: major advances in pathogenesis, but not treatment. Crit Care Clin. 2013;29:651–675.
- . The toxic shock syndromes. Infect Dis Clin North Am. 1996;10(4):727–746.
- Centers for Disease Control and Prevention. National Notifiable Diseases Surveillance System. Toxic shock syndrome (other than Streptococcal) (TSS) 2011 Case Definition. Available at: http://wwwn.cdc.gov/nndss/conditions/toxic‐shock‐syndrome‐other‐than‐streptococcal/case‐definition/2011. Accessed June 4, 2015.
- Centers for Disease Control and Prevention. Update: toxic‐shock syndrome—United States. MMWR Morb Mortal Wkly Rep. 1983;32(30):398–400.
- , , , , , . Toxic shock syndrome in the United States: surveillance update, 1979–1996. Emerg Infect Dis. 1999;5(6):807–810.
- . Fever and rash. Infect Dis Clin North Am. 1996;10(1):101–110.
- , , , et al. Staphylococcal toxic shock syndrome 2000–2006: epidemiology, clinical features, and molecular characteristics. PLoS One. 2011;6(8):e22997.
- , , , et al. Toxic‐shock syndrome in menstruating women: association with tampon use and staphylococcus aureus and clinical features in 52 cases. N Engl J Med. 1980;303(25):1436–1442.
- , , , . Toxic‐shock syndrome—epidemiologic features, recurrence, risk factors, and prevention. N Engl J Med. 1980;303:1429–1435.
- , . Evaluating the febrile patient with a rash. Am Fam Physician. 2000;62(4):804–816.
- . Toxic shock syndrome: broadening the differential diagnosis. J Am Board Fam Pract. 2001;14(2):131–136.
- , , , . Spatial clustering by disease severity among reported Rocky Mountain spotted fever cases in the United States, 2001–2005. Am J Trop Med Hyg. 2009;80(1):72–77.
- , , , et al. One in five mortality in non‐menstrual toxic shock syndrome versus no mortality in menstrual cases in a balanced French series of 55 cases. Eur J Clin Microbio Infect Dis. 2008;27(1):37–43.
- , . Gram‐positive toxic shock syndromes. Lancet Infect Dis. 2009;9(5):281–290.
- . Toxic shock syndrome: major advances in pathogenesis, but not treatment. Crit Care Clin. 2013;29:651–675.
- . The toxic shock syndromes. Infect Dis Clin North Am. 1996;10(4):727–746.
- Centers for Disease Control and Prevention. National Notifiable Diseases Surveillance System. Toxic shock syndrome (other than Streptococcal) (TSS) 2011 Case Definition. Available at: http://wwwn.cdc.gov/nndss/conditions/toxic‐shock‐syndrome‐other‐than‐streptococcal/case‐definition/2011. Accessed June 4, 2015.
- Centers for Disease Control and Prevention. Update: toxic‐shock syndrome—United States. MMWR Morb Mortal Wkly Rep. 1983;32(30):398–400.
- , , , , , . Toxic shock syndrome in the United States: surveillance update, 1979–1996. Emerg Infect Dis. 1999;5(6):807–810.
- . Fever and rash. Infect Dis Clin North Am. 1996;10(1):101–110.
- , , , et al. Staphylococcal toxic shock syndrome 2000–2006: epidemiology, clinical features, and molecular characteristics. PLoS One. 2011;6(8):e22997.
- , , , et al. Toxic‐shock syndrome in menstruating women: association with tampon use and staphylococcus aureus and clinical features in 52 cases. N Engl J Med. 1980;303(25):1436–1442.
- , , , . Toxic‐shock syndrome—epidemiologic features, recurrence, risk factors, and prevention. N Engl J Med. 1980;303:1429–1435.
- , . Evaluating the febrile patient with a rash. Am Fam Physician. 2000;62(4):804–816.
- . Toxic shock syndrome: broadening the differential diagnosis. J Am Board Fam Pract. 2001;14(2):131–136.
- , , , . Spatial clustering by disease severity among reported Rocky Mountain spotted fever cases in the United States, 2001–2005. Am J Trop Med Hyg. 2009;80(1):72–77.
- , , , et al. One in five mortality in non‐menstrual toxic shock syndrome versus no mortality in menstrual cases in a balanced French series of 55 cases. Eur J Clin Microbio Infect Dis. 2008;27(1):37–43.
- , . Gram‐positive toxic shock syndromes. Lancet Infect Dis. 2009;9(5):281–290.
Another Spin
A previously healthy 11‐year‐old boy presented to the emergency department after referral from his pediatrician for 1 week of fevers. Seven days prior to admission he developed a fever to 40.8C, vomiting, and mild left knee pain. The vomiting resolved within 2 days. Five days prior to admission he developed a pruritic, pinpoint rash over his abdomen that resolved within 24 hours. He also developed red, cracked lips, redness of his tongue, redness surrounding his eyes, and slight swelling of his hands. Three days prior to admission his pediatrician noted a 1‐cm anterior cervical lymph node. His fevers occurred throughout each of the prior 7 days without a discernible pattern, and his mild knee pain persisted at the time of presentation.
This preteen has had high fevers for 1 week associated with arthralgia, pruritic rash, emesis, and oral mucosal erythema. His rash, lip and tongue erythema, and swollen hands are classic features of Kawasaki disease (KD), but he lacks the other characteristic physical examination findings. The diagnosis of KD requires fever for at least 5 days accompanied by 4 of the following 5 signs: polymorphous rash, oral mucous membrane changes, peripheral extremity changes such as swelling or skin desquamation, bilateral bulbar conjunctival injection, and cervical lymphadenopathy >1.5 cm in diameter. Children meeting fewer than 4 of these criteria may have an incomplete form of KD.
Because most patients with KD (80%) are under 5 years old, alternative diagnoses such as autoimmune illnesses or a hypersensitivity reaction should be considered. Travel, medication, and animal exposure histories may reveal clues to an infectious or drug‐induced etiology of his fever. Immunization status should be assessed, as measles is also associated with fever, rash, and mucosal changes. Arthralgia or arthritis may occur in KD, but these findings suggest the need to entertain other possibilities, including bone or joint infection, infective endocarditis, inflammatory bowel disease, juvenile idiopathic arthritis (JIA), or systemic lupus erythematosus (SLE).
The child's only past medical history was an episode of croup as an infant. There was no family history of autoimmune diseases. He was not taking any medications and had no known allergies. His immunizations were up to date, including measles, mumps, rubella, and varicella. He lived with his parents and his dog. He swam in fresh water during a trip to Maine 2 months earlier. Neither he nor his family recalled a tick bite. He had no exposure to raw meat or unpasteurized dairy products.
The travel to New England raises the possibility of Lyme disease, although a 2‐month interval between exposure and a high, prolonged fever would be very unusual. Knee arthralgia or arthritis is common in children with late‐stage Lyme disease, but can also be seen in early‐disseminated disease. The prior description of the rash is not suggestive of erythema chronicum migrans, which is seen in early‐stage Lyme disease.
C‐reactive protein (CRP) was 189 mg/L (normal <6.3 mg/L). An echocardiogram was normal. Intravenous immunoglobulin (IVIG) was administered for presumed KD, with immediate improvement of the periorbital erythema, tongue redness, and hand swelling. He was discharged the next day on aspirin with cardiology clinic follow‐up.
Improvement after IVIG supports the diagnosis of KD. It is typical to discharge KD patients from the hospital when they have been afebrile for 24 hours or when the CRP level has declined by approximately 50%.
Over the next 48 hours he felt unwell with high‐grade fevers, continued left knee pain, and new left hip pain. He was readmitted to the hospital. His temperature was 39.4C, respiratory rate was 22 breaths per minute, heart rate was 122 beats per minute, blood pressure was 103/50 mm Hg, and oxygen saturation was 100% while breathing ambient air. He appeared mildly uncomfortable. His conjunctivae were normal. His lips were dry, red, and cracked, and his tongue was red with prominent papillae. His neck was supple without lymphadenopathy. His lungs were clear to auscultation. His heart exam was without murmurs. His abdomen was soft, and the liver and spleen were not enlarged. He had no swelling or erythema of his joints; however, he experienced pain with range of motion of his left knee, and tenderness and restricted range of motion of his left hip. His neurologic exam was normal. There were no rashes.
He has persistent fever, tachycardia, and tachypnea, now without features of KD except oral mucosal changes including prominent tongue papillae consistent with a strawberry tongue. Continued or recurrent fever may suggest persistent KD with ongoing inflammation or the need to search for an alternative diagnoses. An echocardiogram should be repeated, as the coronary artery abnormalities in KD can evolve rapidly, particularly when inflammation persists. Additional findings may include decreased left ventricular function, mitral regurgitation, or pericardial effusion. A second dose of IVIG is necessary to control fever and inflammation in about 15% of patients with KD, although in this case IVIG should be withheld pending further evaluation.
Arthralgia occurs commonly in KD, whereas frank arthritis is less typical. Polyarticular or oligoarticular arthritis involving small or large joints (especially knee or ankle) affects 5% to 10% of patients. The severity of findings in his left hip warrants consideration of septic arthritis with pain referred to the knee; pelvic or femoral osteomyelitis; psoas abscess; or pyomyositis. Following basic lab tests, imaging of the left hip region is indicated.
Laboratory evaluation revealed: white blood cell (WBC) count 10,000/L (absolute neutrophil count 8,460/L, absolute lymphocyte count 530/L), hemoglobin 10.6 g/dL, platelet count 208,000/L, serum sodium 130 mmol/L, serum potassium 3.3 mmol/L, serum urea nitrogen 11 mg/dL, serum creatinine 0.54 mg/dL, aspartate transaminase (AST) 26 U/L, alanine transaminase (ALT) 31 U/L, albumin 1.7 g/dL, erythrocyte sedimentation rate (ESR) > 100 mm/h, and CRP 263 mg/L. No blast cells were seen on peripheral blood smear.
Hypoalbuminemia and markedly elevated inflammatory markers indicate an inflammatory condition that has been active for more than a week. Assessing ESR after IVIG therapy is not useful because exogenous globulins increase the ESR; however, CRP is useful to monitor inflammation and remains elevated here.
Incomplete KD is still possible. Hyponatremia, hypoalbuminemia, and anemia are all features of persistent KD, and have been utilized in several clinical scoring systems in Japan to identify KD patients at increased risk for developing coronary complications. A neoplastic process cannot be excluded, but does not appear likely based on the acuity of his presentation and peripheral blood smear review.
Upon readmission he received a second dose of 2 g/kg IVIG. He remained on aspirin and continued to have fevers. A repeat echocardiogram was normal. He had worsening pain in his left knee and hip with difficulty straightening his left leg. Physical examination was notable for tenderness to palpation over his left hip joint, refusal to bear weight, and resistance to passive range of motion. On hospital day 2, an ultrasound of his left hip and knee revealed a complex left hip effusion and small left knee effusion.
KD becomes less likely in the presence of persistent fevers after IVIG and a repeatedly normal echocardiogram. Worsening left leg symptoms including impaired hip extension with a complex hip effusion suggests an infectious process in or adjacent to the left hip, such as septic arthritis, myositis, or osteomyelitis of the pelvis or proximal femur. A complex hip effusion is less likely to be present with arthritis related to JIA or SLE. The patient needs an emergent hip aspiration and possibly magnetic resonance imaging (MRI) to evaluate adjacent structures.
Arthrotomy and open drainage of his left hip revealed purulent fluid with a WBC count of 49,000/L with 89% neutrophils and 2% lymphocytes. Gram stain was negative. A left knee aspirate demonstrated straw‐colored synovial fluid (which was not sent for cell counts). Bacterial, fungal, and acid‐fast bacilli cultures were requested from hip and knee aspirates. Intravenous ceftriaxone and vancomycin were administered.
The most likely organism in pediatric pyogenic arthritis is Staphylococcus aureus, but there is a long list of other potential pathogens, including Streptococcus pyogenes (group A streptococcus) and Streptococcus pneumoniae. Most pediatric patients with acute pyogenic arthritis have synovial fluid WBC counts in excess of 75,000 to 100,000/L. The protracted course and the initial lack of hip symptoms raise the possibility of a primary osteomyelitis of the femur (particularly the intracapsular portion of the femoral neck or head) or of the acetabulum, with subsequent extension into the hip joint. Pyogenic myositis involving muscle groups adjacent to the hip would be unlikely to spread into the hip space, but can lead to synovial irritation, characterized by sterile joint fluid and WBC counts that fall short of the usual numbers seen in septic arthritis. The blood supply to the femoral head can become compromised with prolonged inflammation and increased intracapsular pressure, resulting in aseptic necrosis.
All cultures from his hip and knee aspirations were sterile. He continued to have daily fevers and persistent tachycardia while receiving intravenous ceftriaxone and vancomycin. Additional testing was notable for: antinuclear antibody (ANA) 1:80, anti‐streptolysin O (ASO) titer 344 IU (normal <150 IU), AST and ALT within normal limits, ferritin 568 ng/mL (normal <322 ng/mL), and lactate dehydrogenase (LDH) 212 units/L (normal <257 units/L). Abdominal ultrasound revealed borderline hepatosplenomegaly. An ophthalmologic examination was normal.
On postoperative day 4 he developed left upper thigh swelling. An MRI showed rim‐enhancing juxta‐articular complex fluid collections surrounding the left femur with decreased marrow enhancement of the left proximal femur (Figure 1).
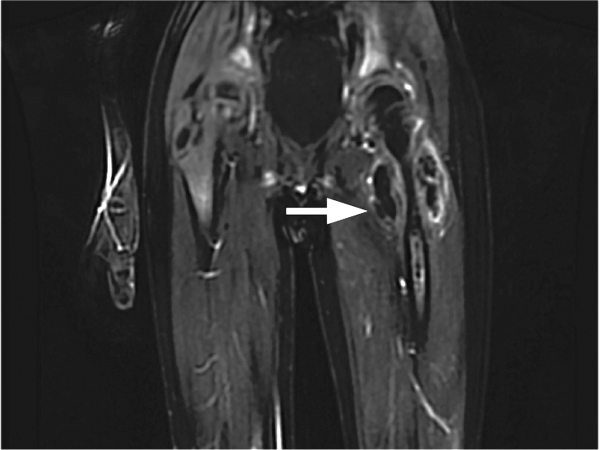
The limited rheumatologic evaluation is unrevealing; the ANA result is nondiagnostic and the ASO titer is normal for age. Laboratories generally report adult normal values for streptococcal antibodies regardless of the patient's age; children from ages 7 to 12 years are at their life peak frequency of group A streptococcal pharyngitis and typically have higher normal values of streptococcal antibodies, including ASO (up to about 480640 IU). The moderately elevated ferritin level is most likely an acute phase reactant and not high enough to suggest macrophage activation syndrome, which is unlikely with the normal AST, ALT, and LDH levels, the absence of significant splenomegaly, and the lack of cytopenias. Continued fever with progressive left upper thigh swelling point to osteomyelitis of the proximal femur, which may have initially ruptured into the hip and then infiltrated the femoral cortex and spread infection into the adjacent soft tissues. Surgical debridement is indicated.
The relative prevalence of methicillin‐sensitive S aureus (MSSA) and methicillin‐resistant S aureus vary widely with geography. MSSA strains are more likely to be highly toxigenic. The elaboration of 1 or more extracellular toxins could account for the patient's initial symptoms.
The patient was brought back to the operating room for drainage of the juxta‐articular fluid collections and a biopsy of his femur. The fluid collections were grossly purulent. His intraoperative cultures were positive for MSSA. The bone biopsy revealed necrotic tissue, acute inflammation, and bacterial colonies, consistent with acute osteomyelitis. Further testing of his S aureus isolate was positive for staphylococcal enterotoxin B. He completed a 4‐week course of oral clindamycin with subsequent normalization of his hip exam and inflammatory markers. At a follow‐up visit the patient was feeling better, but had developed skin peeling on the lateral aspects of his feet consistent with late sequelae of toxin‐mediated disease (Figure 2). Three months after discharge the patient had returned to his baseline activity level and remained asymptomatic.
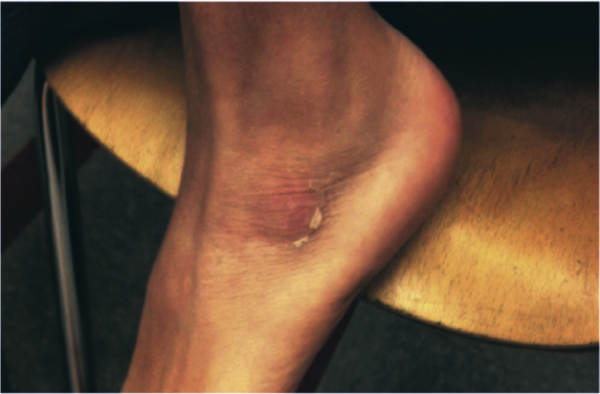
COMMENTARY
The patient presented with a constellation of symptoms that was initially mistaken for incomplete KD until focal progression of his symptoms exposed an underlying femoral osteomyelitis with periarticular abscess formation. Bacterial cultures and subsequent toxin assay revealed an enterotoxin B‐producing strain of S aureus.
Certain staphylococcal strains secrete superantigens that may lead to the development of a systemic toxin‐mediated syndrome. Toxins elaborated by S aureus include toxic shock syndrome toxin‐1 (TSST‐1) and enterotoxins, which have been implicated in menstrual and nonmenstrual toxic shock syndromes.[1, 2] Enterotoxin B is a staphylococcal superantigen found in most strains of the USA400 clonal group, and has been frequently associated with skin and soft tissue infections.[3] Enterotoxin B production has been reported in nearly half of S aureus isolates from skin, soft tissue, and bone infections.[4]
Staphylococcal and streptococcal toxin‐mediated diseases can mimic vasculitis, systemic juvenile idiopathic arthritis, viral infections, and Stevens‐Johnson syndrome. Glossitis in toxin‐mediated syndromes manifests with a swollen, red tongue with overlying enlarged papillae, giving the appearance of a strawberry. Although pediatric providers often equate strawberry tongue, conjunctival injection, rash, and erythematous lips with KD, these findings are also seen in toxin‐mediated diseases, such as scarlet fever or staphylococcal toxic shock syndrome. Enterotoxin B mediated staphylococcal disease masquerading as KD has been reported in 2 cases: a 7‐month‐old boy with multifocal S aureus osteomyelitis and a 5‐year‐old boy with S aureus bacteremia. Both staphylococcal isolates produced enterotoxin B but were negative for other staphylococcus‐related toxins including TSST‐1.[5]
The Institute of Medicine (IOM) recently released its report Improving Diagnosis in Health Care, highlighting the under‐recognized quality and safety issue of diagnostic error.[6] The report uses the following broad and patient‐centered definition of diagnostic error: the failure to (a) establish an accurate and timely explanation of the patient's health problem(s) or (b) communicate that explanation to the patient. The IOM's conceptual model of diagnosis emphasizes the iterative nature of the diagnostic process, including the importance of generating a working diagnosis, gathering and incorporating new information in the reassessment of that diagnosis, and integrating treatment response into the formulation of the final diagnosis (Figure 3).

Even though the patient initially had several features consistent with KD, the increasing number of atypical features could have prompted the clinical team to reconsider their working diagnosis. The patient's age was atypical for KD, he had progressive knee and hip arthritis, and his fevers persisted after IVIG. An expanded differential should have included toxic shock syndrome; the resolution of conjunctival and mucosal injection and edema after IVIG may have been the result of antibodies in the IVIG preparation with neutralizing activity against superantigens. This antitoxin activity has established a role for IVIG in the management of staphylococcal toxic shock syndrome.[7] Ultimately, his imaging and surgical drainage revealed a focal staphylococcal toxin‐producing infectious source from which his fevers, rash, and mucosal and extremity changes emanated. This case reminds us that the more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should take it for another spin around the diagnostic wheel in search of a more suitable alternative.
KEY LEARNING POINTS
- Staphylococcal toxin‐mediated disease may mimic KD, with common features including strawberry tongue, oral and conjunctival injection, and skin desquamation.
- Improvement after treatment with IVIG is characteristic but not diagnostic of KD, and may be seen in toxin‐mediated disease.
- KD may present with arthralgia or arthritis, but severe joint abnormalities warrant consideration of infectious and other autoimmune conditions.
- The more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should gather, interpret, and integrate new information in search of a more suitable alternative.
Disclosure: Nothing to report.
- . Staphylococcal enterotoxin B and toxic shock syndrome toxin‐1 are significantly associated with non‐menstrual TSS. Lancet. 1986;1:1149–1150.
- , , , , . Toxic shock syndrome caused by a strain of staphylococcus aureus that produces enterotoxin C but not toxic shock syndrome toxin‐1. Am J Dis Child. 1989;143 (7):848–849.
- , , , , , . Staphylococcus aureus isolates encode variant staphylococcal enterotoxin B proteins that are diverse in superantigenicity and lethality. PLoS One. 2012;7(7):e41157.
- , , , et al. Variability of antibiotic susceptibility and toxin production of Staphylococcal aureus stains isolated from skin, soft tissue, and bone related infections. BMC Microbiol. 2013;13:188.
- , , , , . Kawasaki syndrome‐like illness associated with infection caused by enterotoxin B‐secreting Staphylococcus aureus. Clin Microbiol Rev. 2013;26:422–447.
- National Academies of Sciences, Engineering, and Medicine. Improving Diagnosis in Health Care. Washington, DC: The National Academies Press; 2015.
- , , , et al. Intravenous immunoglobulin G therapy in streptococcal toxic shock syndrome: a European randomized, double‐blind, placebo‐controlled trial. Clin Infect Dis. 2003;37(3):333–340.
A previously healthy 11‐year‐old boy presented to the emergency department after referral from his pediatrician for 1 week of fevers. Seven days prior to admission he developed a fever to 40.8C, vomiting, and mild left knee pain. The vomiting resolved within 2 days. Five days prior to admission he developed a pruritic, pinpoint rash over his abdomen that resolved within 24 hours. He also developed red, cracked lips, redness of his tongue, redness surrounding his eyes, and slight swelling of his hands. Three days prior to admission his pediatrician noted a 1‐cm anterior cervical lymph node. His fevers occurred throughout each of the prior 7 days without a discernible pattern, and his mild knee pain persisted at the time of presentation.
This preteen has had high fevers for 1 week associated with arthralgia, pruritic rash, emesis, and oral mucosal erythema. His rash, lip and tongue erythema, and swollen hands are classic features of Kawasaki disease (KD), but he lacks the other characteristic physical examination findings. The diagnosis of KD requires fever for at least 5 days accompanied by 4 of the following 5 signs: polymorphous rash, oral mucous membrane changes, peripheral extremity changes such as swelling or skin desquamation, bilateral bulbar conjunctival injection, and cervical lymphadenopathy >1.5 cm in diameter. Children meeting fewer than 4 of these criteria may have an incomplete form of KD.
Because most patients with KD (80%) are under 5 years old, alternative diagnoses such as autoimmune illnesses or a hypersensitivity reaction should be considered. Travel, medication, and animal exposure histories may reveal clues to an infectious or drug‐induced etiology of his fever. Immunization status should be assessed, as measles is also associated with fever, rash, and mucosal changes. Arthralgia or arthritis may occur in KD, but these findings suggest the need to entertain other possibilities, including bone or joint infection, infective endocarditis, inflammatory bowel disease, juvenile idiopathic arthritis (JIA), or systemic lupus erythematosus (SLE).
The child's only past medical history was an episode of croup as an infant. There was no family history of autoimmune diseases. He was not taking any medications and had no known allergies. His immunizations were up to date, including measles, mumps, rubella, and varicella. He lived with his parents and his dog. He swam in fresh water during a trip to Maine 2 months earlier. Neither he nor his family recalled a tick bite. He had no exposure to raw meat or unpasteurized dairy products.
The travel to New England raises the possibility of Lyme disease, although a 2‐month interval between exposure and a high, prolonged fever would be very unusual. Knee arthralgia or arthritis is common in children with late‐stage Lyme disease, but can also be seen in early‐disseminated disease. The prior description of the rash is not suggestive of erythema chronicum migrans, which is seen in early‐stage Lyme disease.
C‐reactive protein (CRP) was 189 mg/L (normal <6.3 mg/L). An echocardiogram was normal. Intravenous immunoglobulin (IVIG) was administered for presumed KD, with immediate improvement of the periorbital erythema, tongue redness, and hand swelling. He was discharged the next day on aspirin with cardiology clinic follow‐up.
Improvement after IVIG supports the diagnosis of KD. It is typical to discharge KD patients from the hospital when they have been afebrile for 24 hours or when the CRP level has declined by approximately 50%.
Over the next 48 hours he felt unwell with high‐grade fevers, continued left knee pain, and new left hip pain. He was readmitted to the hospital. His temperature was 39.4C, respiratory rate was 22 breaths per minute, heart rate was 122 beats per minute, blood pressure was 103/50 mm Hg, and oxygen saturation was 100% while breathing ambient air. He appeared mildly uncomfortable. His conjunctivae were normal. His lips were dry, red, and cracked, and his tongue was red with prominent papillae. His neck was supple without lymphadenopathy. His lungs were clear to auscultation. His heart exam was without murmurs. His abdomen was soft, and the liver and spleen were not enlarged. He had no swelling or erythema of his joints; however, he experienced pain with range of motion of his left knee, and tenderness and restricted range of motion of his left hip. His neurologic exam was normal. There were no rashes.
He has persistent fever, tachycardia, and tachypnea, now without features of KD except oral mucosal changes including prominent tongue papillae consistent with a strawberry tongue. Continued or recurrent fever may suggest persistent KD with ongoing inflammation or the need to search for an alternative diagnoses. An echocardiogram should be repeated, as the coronary artery abnormalities in KD can evolve rapidly, particularly when inflammation persists. Additional findings may include decreased left ventricular function, mitral regurgitation, or pericardial effusion. A second dose of IVIG is necessary to control fever and inflammation in about 15% of patients with KD, although in this case IVIG should be withheld pending further evaluation.
Arthralgia occurs commonly in KD, whereas frank arthritis is less typical. Polyarticular or oligoarticular arthritis involving small or large joints (especially knee or ankle) affects 5% to 10% of patients. The severity of findings in his left hip warrants consideration of septic arthritis with pain referred to the knee; pelvic or femoral osteomyelitis; psoas abscess; or pyomyositis. Following basic lab tests, imaging of the left hip region is indicated.
Laboratory evaluation revealed: white blood cell (WBC) count 10,000/L (absolute neutrophil count 8,460/L, absolute lymphocyte count 530/L), hemoglobin 10.6 g/dL, platelet count 208,000/L, serum sodium 130 mmol/L, serum potassium 3.3 mmol/L, serum urea nitrogen 11 mg/dL, serum creatinine 0.54 mg/dL, aspartate transaminase (AST) 26 U/L, alanine transaminase (ALT) 31 U/L, albumin 1.7 g/dL, erythrocyte sedimentation rate (ESR) > 100 mm/h, and CRP 263 mg/L. No blast cells were seen on peripheral blood smear.
Hypoalbuminemia and markedly elevated inflammatory markers indicate an inflammatory condition that has been active for more than a week. Assessing ESR after IVIG therapy is not useful because exogenous globulins increase the ESR; however, CRP is useful to monitor inflammation and remains elevated here.
Incomplete KD is still possible. Hyponatremia, hypoalbuminemia, and anemia are all features of persistent KD, and have been utilized in several clinical scoring systems in Japan to identify KD patients at increased risk for developing coronary complications. A neoplastic process cannot be excluded, but does not appear likely based on the acuity of his presentation and peripheral blood smear review.
Upon readmission he received a second dose of 2 g/kg IVIG. He remained on aspirin and continued to have fevers. A repeat echocardiogram was normal. He had worsening pain in his left knee and hip with difficulty straightening his left leg. Physical examination was notable for tenderness to palpation over his left hip joint, refusal to bear weight, and resistance to passive range of motion. On hospital day 2, an ultrasound of his left hip and knee revealed a complex left hip effusion and small left knee effusion.
KD becomes less likely in the presence of persistent fevers after IVIG and a repeatedly normal echocardiogram. Worsening left leg symptoms including impaired hip extension with a complex hip effusion suggests an infectious process in or adjacent to the left hip, such as septic arthritis, myositis, or osteomyelitis of the pelvis or proximal femur. A complex hip effusion is less likely to be present with arthritis related to JIA or SLE. The patient needs an emergent hip aspiration and possibly magnetic resonance imaging (MRI) to evaluate adjacent structures.
Arthrotomy and open drainage of his left hip revealed purulent fluid with a WBC count of 49,000/L with 89% neutrophils and 2% lymphocytes. Gram stain was negative. A left knee aspirate demonstrated straw‐colored synovial fluid (which was not sent for cell counts). Bacterial, fungal, and acid‐fast bacilli cultures were requested from hip and knee aspirates. Intravenous ceftriaxone and vancomycin were administered.
The most likely organism in pediatric pyogenic arthritis is Staphylococcus aureus, but there is a long list of other potential pathogens, including Streptococcus pyogenes (group A streptococcus) and Streptococcus pneumoniae. Most pediatric patients with acute pyogenic arthritis have synovial fluid WBC counts in excess of 75,000 to 100,000/L. The protracted course and the initial lack of hip symptoms raise the possibility of a primary osteomyelitis of the femur (particularly the intracapsular portion of the femoral neck or head) or of the acetabulum, with subsequent extension into the hip joint. Pyogenic myositis involving muscle groups adjacent to the hip would be unlikely to spread into the hip space, but can lead to synovial irritation, characterized by sterile joint fluid and WBC counts that fall short of the usual numbers seen in septic arthritis. The blood supply to the femoral head can become compromised with prolonged inflammation and increased intracapsular pressure, resulting in aseptic necrosis.
All cultures from his hip and knee aspirations were sterile. He continued to have daily fevers and persistent tachycardia while receiving intravenous ceftriaxone and vancomycin. Additional testing was notable for: antinuclear antibody (ANA) 1:80, anti‐streptolysin O (ASO) titer 344 IU (normal <150 IU), AST and ALT within normal limits, ferritin 568 ng/mL (normal <322 ng/mL), and lactate dehydrogenase (LDH) 212 units/L (normal <257 units/L). Abdominal ultrasound revealed borderline hepatosplenomegaly. An ophthalmologic examination was normal.
On postoperative day 4 he developed left upper thigh swelling. An MRI showed rim‐enhancing juxta‐articular complex fluid collections surrounding the left femur with decreased marrow enhancement of the left proximal femur (Figure 1).
The limited rheumatologic evaluation is unrevealing; the ANA result is nondiagnostic and the ASO titer is normal for age. Laboratories generally report adult normal values for streptococcal antibodies regardless of the patient's age; children from ages 7 to 12 years are at their life peak frequency of group A streptococcal pharyngitis and typically have higher normal values of streptococcal antibodies, including ASO (up to about 480640 IU). The moderately elevated ferritin level is most likely an acute phase reactant and not high enough to suggest macrophage activation syndrome, which is unlikely with the normal AST, ALT, and LDH levels, the absence of significant splenomegaly, and the lack of cytopenias. Continued fever with progressive left upper thigh swelling point to osteomyelitis of the proximal femur, which may have initially ruptured into the hip and then infiltrated the femoral cortex and spread infection into the adjacent soft tissues. Surgical debridement is indicated.
The relative prevalence of methicillin‐sensitive S aureus (MSSA) and methicillin‐resistant S aureus vary widely with geography. MSSA strains are more likely to be highly toxigenic. The elaboration of 1 or more extracellular toxins could account for the patient's initial symptoms.
The patient was brought back to the operating room for drainage of the juxta‐articular fluid collections and a biopsy of his femur. The fluid collections were grossly purulent. His intraoperative cultures were positive for MSSA. The bone biopsy revealed necrotic tissue, acute inflammation, and bacterial colonies, consistent with acute osteomyelitis. Further testing of his S aureus isolate was positive for staphylococcal enterotoxin B. He completed a 4‐week course of oral clindamycin with subsequent normalization of his hip exam and inflammatory markers. At a follow‐up visit the patient was feeling better, but had developed skin peeling on the lateral aspects of his feet consistent with late sequelae of toxin‐mediated disease (Figure 2). Three months after discharge the patient had returned to his baseline activity level and remained asymptomatic.
COMMENTARY
The patient presented with a constellation of symptoms that was initially mistaken for incomplete KD until focal progression of his symptoms exposed an underlying femoral osteomyelitis with periarticular abscess formation. Bacterial cultures and subsequent toxin assay revealed an enterotoxin B‐producing strain of S aureus.
Certain staphylococcal strains secrete superantigens that may lead to the development of a systemic toxin‐mediated syndrome. Toxins elaborated by S aureus include toxic shock syndrome toxin‐1 (TSST‐1) and enterotoxins, which have been implicated in menstrual and nonmenstrual toxic shock syndromes.[1, 2] Enterotoxin B is a staphylococcal superantigen found in most strains of the USA400 clonal group, and has been frequently associated with skin and soft tissue infections.[3] Enterotoxin B production has been reported in nearly half of S aureus isolates from skin, soft tissue, and bone infections.[4]
Staphylococcal and streptococcal toxin‐mediated diseases can mimic vasculitis, systemic juvenile idiopathic arthritis, viral infections, and Stevens‐Johnson syndrome. Glossitis in toxin‐mediated syndromes manifests with a swollen, red tongue with overlying enlarged papillae, giving the appearance of a strawberry. Although pediatric providers often equate strawberry tongue, conjunctival injection, rash, and erythematous lips with KD, these findings are also seen in toxin‐mediated diseases, such as scarlet fever or staphylococcal toxic shock syndrome. Enterotoxin B mediated staphylococcal disease masquerading as KD has been reported in 2 cases: a 7‐month‐old boy with multifocal S aureus osteomyelitis and a 5‐year‐old boy with S aureus bacteremia. Both staphylococcal isolates produced enterotoxin B but were negative for other staphylococcus‐related toxins including TSST‐1.[5]
The Institute of Medicine (IOM) recently released its report Improving Diagnosis in Health Care, highlighting the under‐recognized quality and safety issue of diagnostic error.[6] The report uses the following broad and patient‐centered definition of diagnostic error: the failure to (a) establish an accurate and timely explanation of the patient's health problem(s) or (b) communicate that explanation to the patient. The IOM's conceptual model of diagnosis emphasizes the iterative nature of the diagnostic process, including the importance of generating a working diagnosis, gathering and incorporating new information in the reassessment of that diagnosis, and integrating treatment response into the formulation of the final diagnosis (Figure 3).
Even though the patient initially had several features consistent with KD, the increasing number of atypical features could have prompted the clinical team to reconsider their working diagnosis. The patient's age was atypical for KD, he had progressive knee and hip arthritis, and his fevers persisted after IVIG. An expanded differential should have included toxic shock syndrome; the resolution of conjunctival and mucosal injection and edema after IVIG may have been the result of antibodies in the IVIG preparation with neutralizing activity against superantigens. This antitoxin activity has established a role for IVIG in the management of staphylococcal toxic shock syndrome.[7] Ultimately, his imaging and surgical drainage revealed a focal staphylococcal toxin‐producing infectious source from which his fevers, rash, and mucosal and extremity changes emanated. This case reminds us that the more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should take it for another spin around the diagnostic wheel in search of a more suitable alternative.
KEY LEARNING POINTS
- Staphylococcal toxin‐mediated disease may mimic KD, with common features including strawberry tongue, oral and conjunctival injection, and skin desquamation.
- Improvement after treatment with IVIG is characteristic but not diagnostic of KD, and may be seen in toxin‐mediated disease.
- KD may present with arthralgia or arthritis, but severe joint abnormalities warrant consideration of infectious and other autoimmune conditions.
- The more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should gather, interpret, and integrate new information in search of a more suitable alternative.
Disclosure: Nothing to report.
A previously healthy 11‐year‐old boy presented to the emergency department after referral from his pediatrician for 1 week of fevers. Seven days prior to admission he developed a fever to 40.8C, vomiting, and mild left knee pain. The vomiting resolved within 2 days. Five days prior to admission he developed a pruritic, pinpoint rash over his abdomen that resolved within 24 hours. He also developed red, cracked lips, redness of his tongue, redness surrounding his eyes, and slight swelling of his hands. Three days prior to admission his pediatrician noted a 1‐cm anterior cervical lymph node. His fevers occurred throughout each of the prior 7 days without a discernible pattern, and his mild knee pain persisted at the time of presentation.
This preteen has had high fevers for 1 week associated with arthralgia, pruritic rash, emesis, and oral mucosal erythema. His rash, lip and tongue erythema, and swollen hands are classic features of Kawasaki disease (KD), but he lacks the other characteristic physical examination findings. The diagnosis of KD requires fever for at least 5 days accompanied by 4 of the following 5 signs: polymorphous rash, oral mucous membrane changes, peripheral extremity changes such as swelling or skin desquamation, bilateral bulbar conjunctival injection, and cervical lymphadenopathy >1.5 cm in diameter. Children meeting fewer than 4 of these criteria may have an incomplete form of KD.
Because most patients with KD (80%) are under 5 years old, alternative diagnoses such as autoimmune illnesses or a hypersensitivity reaction should be considered. Travel, medication, and animal exposure histories may reveal clues to an infectious or drug‐induced etiology of his fever. Immunization status should be assessed, as measles is also associated with fever, rash, and mucosal changes. Arthralgia or arthritis may occur in KD, but these findings suggest the need to entertain other possibilities, including bone or joint infection, infective endocarditis, inflammatory bowel disease, juvenile idiopathic arthritis (JIA), or systemic lupus erythematosus (SLE).
The child's only past medical history was an episode of croup as an infant. There was no family history of autoimmune diseases. He was not taking any medications and had no known allergies. His immunizations were up to date, including measles, mumps, rubella, and varicella. He lived with his parents and his dog. He swam in fresh water during a trip to Maine 2 months earlier. Neither he nor his family recalled a tick bite. He had no exposure to raw meat or unpasteurized dairy products.
The travel to New England raises the possibility of Lyme disease, although a 2‐month interval between exposure and a high, prolonged fever would be very unusual. Knee arthralgia or arthritis is common in children with late‐stage Lyme disease, but can also be seen in early‐disseminated disease. The prior description of the rash is not suggestive of erythema chronicum migrans, which is seen in early‐stage Lyme disease.
C‐reactive protein (CRP) was 189 mg/L (normal <6.3 mg/L). An echocardiogram was normal. Intravenous immunoglobulin (IVIG) was administered for presumed KD, with immediate improvement of the periorbital erythema, tongue redness, and hand swelling. He was discharged the next day on aspirin with cardiology clinic follow‐up.
Improvement after IVIG supports the diagnosis of KD. It is typical to discharge KD patients from the hospital when they have been afebrile for 24 hours or when the CRP level has declined by approximately 50%.
Over the next 48 hours he felt unwell with high‐grade fevers, continued left knee pain, and new left hip pain. He was readmitted to the hospital. His temperature was 39.4C, respiratory rate was 22 breaths per minute, heart rate was 122 beats per minute, blood pressure was 103/50 mm Hg, and oxygen saturation was 100% while breathing ambient air. He appeared mildly uncomfortable. His conjunctivae were normal. His lips were dry, red, and cracked, and his tongue was red with prominent papillae. His neck was supple without lymphadenopathy. His lungs were clear to auscultation. His heart exam was without murmurs. His abdomen was soft, and the liver and spleen were not enlarged. He had no swelling or erythema of his joints; however, he experienced pain with range of motion of his left knee, and tenderness and restricted range of motion of his left hip. His neurologic exam was normal. There were no rashes.
He has persistent fever, tachycardia, and tachypnea, now without features of KD except oral mucosal changes including prominent tongue papillae consistent with a strawberry tongue. Continued or recurrent fever may suggest persistent KD with ongoing inflammation or the need to search for an alternative diagnoses. An echocardiogram should be repeated, as the coronary artery abnormalities in KD can evolve rapidly, particularly when inflammation persists. Additional findings may include decreased left ventricular function, mitral regurgitation, or pericardial effusion. A second dose of IVIG is necessary to control fever and inflammation in about 15% of patients with KD, although in this case IVIG should be withheld pending further evaluation.
Arthralgia occurs commonly in KD, whereas frank arthritis is less typical. Polyarticular or oligoarticular arthritis involving small or large joints (especially knee or ankle) affects 5% to 10% of patients. The severity of findings in his left hip warrants consideration of septic arthritis with pain referred to the knee; pelvic or femoral osteomyelitis; psoas abscess; or pyomyositis. Following basic lab tests, imaging of the left hip region is indicated.
Laboratory evaluation revealed: white blood cell (WBC) count 10,000/L (absolute neutrophil count 8,460/L, absolute lymphocyte count 530/L), hemoglobin 10.6 g/dL, platelet count 208,000/L, serum sodium 130 mmol/L, serum potassium 3.3 mmol/L, serum urea nitrogen 11 mg/dL, serum creatinine 0.54 mg/dL, aspartate transaminase (AST) 26 U/L, alanine transaminase (ALT) 31 U/L, albumin 1.7 g/dL, erythrocyte sedimentation rate (ESR) > 100 mm/h, and CRP 263 mg/L. No blast cells were seen on peripheral blood smear.
Hypoalbuminemia and markedly elevated inflammatory markers indicate an inflammatory condition that has been active for more than a week. Assessing ESR after IVIG therapy is not useful because exogenous globulins increase the ESR; however, CRP is useful to monitor inflammation and remains elevated here.
Incomplete KD is still possible. Hyponatremia, hypoalbuminemia, and anemia are all features of persistent KD, and have been utilized in several clinical scoring systems in Japan to identify KD patients at increased risk for developing coronary complications. A neoplastic process cannot be excluded, but does not appear likely based on the acuity of his presentation and peripheral blood smear review.
Upon readmission he received a second dose of 2 g/kg IVIG. He remained on aspirin and continued to have fevers. A repeat echocardiogram was normal. He had worsening pain in his left knee and hip with difficulty straightening his left leg. Physical examination was notable for tenderness to palpation over his left hip joint, refusal to bear weight, and resistance to passive range of motion. On hospital day 2, an ultrasound of his left hip and knee revealed a complex left hip effusion and small left knee effusion.
KD becomes less likely in the presence of persistent fevers after IVIG and a repeatedly normal echocardiogram. Worsening left leg symptoms including impaired hip extension with a complex hip effusion suggests an infectious process in or adjacent to the left hip, such as septic arthritis, myositis, or osteomyelitis of the pelvis or proximal femur. A complex hip effusion is less likely to be present with arthritis related to JIA or SLE. The patient needs an emergent hip aspiration and possibly magnetic resonance imaging (MRI) to evaluate adjacent structures.
Arthrotomy and open drainage of his left hip revealed purulent fluid with a WBC count of 49,000/L with 89% neutrophils and 2% lymphocytes. Gram stain was negative. A left knee aspirate demonstrated straw‐colored synovial fluid (which was not sent for cell counts). Bacterial, fungal, and acid‐fast bacilli cultures were requested from hip and knee aspirates. Intravenous ceftriaxone and vancomycin were administered.
The most likely organism in pediatric pyogenic arthritis is Staphylococcus aureus, but there is a long list of other potential pathogens, including Streptococcus pyogenes (group A streptococcus) and Streptococcus pneumoniae. Most pediatric patients with acute pyogenic arthritis have synovial fluid WBC counts in excess of 75,000 to 100,000/L. The protracted course and the initial lack of hip symptoms raise the possibility of a primary osteomyelitis of the femur (particularly the intracapsular portion of the femoral neck or head) or of the acetabulum, with subsequent extension into the hip joint. Pyogenic myositis involving muscle groups adjacent to the hip would be unlikely to spread into the hip space, but can lead to synovial irritation, characterized by sterile joint fluid and WBC counts that fall short of the usual numbers seen in septic arthritis. The blood supply to the femoral head can become compromised with prolonged inflammation and increased intracapsular pressure, resulting in aseptic necrosis.
All cultures from his hip and knee aspirations were sterile. He continued to have daily fevers and persistent tachycardia while receiving intravenous ceftriaxone and vancomycin. Additional testing was notable for: antinuclear antibody (ANA) 1:80, anti‐streptolysin O (ASO) titer 344 IU (normal <150 IU), AST and ALT within normal limits, ferritin 568 ng/mL (normal <322 ng/mL), and lactate dehydrogenase (LDH) 212 units/L (normal <257 units/L). Abdominal ultrasound revealed borderline hepatosplenomegaly. An ophthalmologic examination was normal.
On postoperative day 4 he developed left upper thigh swelling. An MRI showed rim‐enhancing juxta‐articular complex fluid collections surrounding the left femur with decreased marrow enhancement of the left proximal femur (Figure 1).
The limited rheumatologic evaluation is unrevealing; the ANA result is nondiagnostic and the ASO titer is normal for age. Laboratories generally report adult normal values for streptococcal antibodies regardless of the patient's age; children from ages 7 to 12 years are at their life peak frequency of group A streptococcal pharyngitis and typically have higher normal values of streptococcal antibodies, including ASO (up to about 480640 IU). The moderately elevated ferritin level is most likely an acute phase reactant and not high enough to suggest macrophage activation syndrome, which is unlikely with the normal AST, ALT, and LDH levels, the absence of significant splenomegaly, and the lack of cytopenias. Continued fever with progressive left upper thigh swelling point to osteomyelitis of the proximal femur, which may have initially ruptured into the hip and then infiltrated the femoral cortex and spread infection into the adjacent soft tissues. Surgical debridement is indicated.
The relative prevalence of methicillin‐sensitive S aureus (MSSA) and methicillin‐resistant S aureus vary widely with geography. MSSA strains are more likely to be highly toxigenic. The elaboration of 1 or more extracellular toxins could account for the patient's initial symptoms.
The patient was brought back to the operating room for drainage of the juxta‐articular fluid collections and a biopsy of his femur. The fluid collections were grossly purulent. His intraoperative cultures were positive for MSSA. The bone biopsy revealed necrotic tissue, acute inflammation, and bacterial colonies, consistent with acute osteomyelitis. Further testing of his S aureus isolate was positive for staphylococcal enterotoxin B. He completed a 4‐week course of oral clindamycin with subsequent normalization of his hip exam and inflammatory markers. At a follow‐up visit the patient was feeling better, but had developed skin peeling on the lateral aspects of his feet consistent with late sequelae of toxin‐mediated disease (Figure 2). Three months after discharge the patient had returned to his baseline activity level and remained asymptomatic.
COMMENTARY
The patient presented with a constellation of symptoms that was initially mistaken for incomplete KD until focal progression of his symptoms exposed an underlying femoral osteomyelitis with periarticular abscess formation. Bacterial cultures and subsequent toxin assay revealed an enterotoxin B‐producing strain of S aureus.
Certain staphylococcal strains secrete superantigens that may lead to the development of a systemic toxin‐mediated syndrome. Toxins elaborated by S aureus include toxic shock syndrome toxin‐1 (TSST‐1) and enterotoxins, which have been implicated in menstrual and nonmenstrual toxic shock syndromes.[1, 2] Enterotoxin B is a staphylococcal superantigen found in most strains of the USA400 clonal group, and has been frequently associated with skin and soft tissue infections.[3] Enterotoxin B production has been reported in nearly half of S aureus isolates from skin, soft tissue, and bone infections.[4]
Staphylococcal and streptococcal toxin‐mediated diseases can mimic vasculitis, systemic juvenile idiopathic arthritis, viral infections, and Stevens‐Johnson syndrome. Glossitis in toxin‐mediated syndromes manifests with a swollen, red tongue with overlying enlarged papillae, giving the appearance of a strawberry. Although pediatric providers often equate strawberry tongue, conjunctival injection, rash, and erythematous lips with KD, these findings are also seen in toxin‐mediated diseases, such as scarlet fever or staphylococcal toxic shock syndrome. Enterotoxin B mediated staphylococcal disease masquerading as KD has been reported in 2 cases: a 7‐month‐old boy with multifocal S aureus osteomyelitis and a 5‐year‐old boy with S aureus bacteremia. Both staphylococcal isolates produced enterotoxin B but were negative for other staphylococcus‐related toxins including TSST‐1.[5]
The Institute of Medicine (IOM) recently released its report Improving Diagnosis in Health Care, highlighting the under‐recognized quality and safety issue of diagnostic error.[6] The report uses the following broad and patient‐centered definition of diagnostic error: the failure to (a) establish an accurate and timely explanation of the patient's health problem(s) or (b) communicate that explanation to the patient. The IOM's conceptual model of diagnosis emphasizes the iterative nature of the diagnostic process, including the importance of generating a working diagnosis, gathering and incorporating new information in the reassessment of that diagnosis, and integrating treatment response into the formulation of the final diagnosis (Figure 3).
Even though the patient initially had several features consistent with KD, the increasing number of atypical features could have prompted the clinical team to reconsider their working diagnosis. The patient's age was atypical for KD, he had progressive knee and hip arthritis, and his fevers persisted after IVIG. An expanded differential should have included toxic shock syndrome; the resolution of conjunctival and mucosal injection and edema after IVIG may have been the result of antibodies in the IVIG preparation with neutralizing activity against superantigens. This antitoxin activity has established a role for IVIG in the management of staphylococcal toxic shock syndrome.[7] Ultimately, his imaging and surgical drainage revealed a focal staphylococcal toxin‐producing infectious source from which his fevers, rash, and mucosal and extremity changes emanated. This case reminds us that the more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should take it for another spin around the diagnostic wheel in search of a more suitable alternative.
KEY LEARNING POINTS
- Staphylococcal toxin‐mediated disease may mimic KD, with common features including strawberry tongue, oral and conjunctival injection, and skin desquamation.
- Improvement after treatment with IVIG is characteristic but not diagnostic of KD, and may be seen in toxin‐mediated disease.
- KD may present with arthralgia or arthritis, but severe joint abnormalities warrant consideration of infectious and other autoimmune conditions.
- The more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should gather, interpret, and integrate new information in search of a more suitable alternative.
Disclosure: Nothing to report.
- . Staphylococcal enterotoxin B and toxic shock syndrome toxin‐1 are significantly associated with non‐menstrual TSS. Lancet. 1986;1:1149–1150.
- , , , , . Toxic shock syndrome caused by a strain of staphylococcus aureus that produces enterotoxin C but not toxic shock syndrome toxin‐1. Am J Dis Child. 1989;143 (7):848–849.
- , , , , , . Staphylococcus aureus isolates encode variant staphylococcal enterotoxin B proteins that are diverse in superantigenicity and lethality. PLoS One. 2012;7(7):e41157.
- , , , et al. Variability of antibiotic susceptibility and toxin production of Staphylococcal aureus stains isolated from skin, soft tissue, and bone related infections. BMC Microbiol. 2013;13:188.
- , , , , . Kawasaki syndrome‐like illness associated with infection caused by enterotoxin B‐secreting Staphylococcus aureus. Clin Microbiol Rev. 2013;26:422–447.
- National Academies of Sciences, Engineering, and Medicine. Improving Diagnosis in Health Care. Washington, DC: The National Academies Press; 2015.
- , , , et al. Intravenous immunoglobulin G therapy in streptococcal toxic shock syndrome: a European randomized, double‐blind, placebo‐controlled trial. Clin Infect Dis. 2003;37(3):333–340.
- . Staphylococcal enterotoxin B and toxic shock syndrome toxin‐1 are significantly associated with non‐menstrual TSS. Lancet. 1986;1:1149–1150.
- , , , , . Toxic shock syndrome caused by a strain of staphylococcus aureus that produces enterotoxin C but not toxic shock syndrome toxin‐1. Am J Dis Child. 1989;143 (7):848–849.
- , , , , , . Staphylococcus aureus isolates encode variant staphylococcal enterotoxin B proteins that are diverse in superantigenicity and lethality. PLoS One. 2012;7(7):e41157.
- , , , et al. Variability of antibiotic susceptibility and toxin production of Staphylococcal aureus stains isolated from skin, soft tissue, and bone related infections. BMC Microbiol. 2013;13:188.
- , , , , . Kawasaki syndrome‐like illness associated with infection caused by enterotoxin B‐secreting Staphylococcus aureus. Clin Microbiol Rev. 2013;26:422–447.
- National Academies of Sciences, Engineering, and Medicine. Improving Diagnosis in Health Care. Washington, DC: The National Academies Press; 2015.
- , , , et al. Intravenous immunoglobulin G therapy in streptococcal toxic shock syndrome: a European randomized, double‐blind, placebo‐controlled trial. Clin Infect Dis. 2003;37(3):333–340.
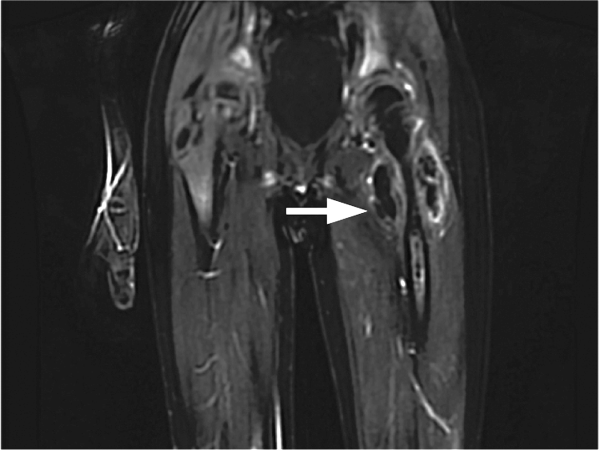
Should a Patient Who Requests Alcohol Detoxification Be Admitted or Treated as Outpatient?
Case
A 42-year-old man with a history of posttraumatic stress disorder (PTSD), hypertension, and alcohol use disorder (AUD) presents to the ED requesting alcohol detoxification. He has had six admissions in the last six months for alcohol detoxification. Two years ago, the patient had a documented alcohol withdrawal seizure. His last drink was eight hours ago, and he currently drinks a liter of vodka a day. On exam, his pulse rate is 126 bpm, and his blood pressure is 162/91 mm Hg. He appears anxious and has bilateral hand tremors. His serum ethanol level is 388.6 mg/dL.
Overview
DSM-5 integrated alcohol abuse and alcohol dependence that were previously classified in DSM-IV into AUDs with mild, moderate, and severe subclassifications. AUDs are the most serious substance abuse problem in the U.S. In the general population, the lifetime prevalence of alcohol abuse is 17.8% and of alcohol dependence is 12.5%.1–3 One study estimates that 24% of adult patients brought to the ED by ambulance suffer from alcoholism, and approximately 10% to 32% of hospitalized medical patients have an AUD.4–8 Patients who stop drinking will develop alcohol withdrawal as early as six hours after their last drink (see Figure 1). The majority of patients at risk of alcohol withdrawal syndrome (AWS) will develop only minor uncomplicated symptoms, but up to 20% will develop symptoms associated with complicated AWS, including withdrawal seizures and delirium tremens (DT).9 It is not entirely clear why some individuals suffer from more severe withdrawal symptoms than others, but genetic predisposition may play a role.10
DT is a syndrome characterized by agitation, disorientation, hallucinations, and autonomic instability (tachycardia, hypertension, hyperthermia, and diaphoresis) in the setting of acute reduction or abstinence from alcohol and is associated with a mortality rate as high as 20%.11 Complicated AWS is associated with increased in-hospital morbidity and mortality, longer lengths of stay, inflated costs of care, increased burden and frustration of nursing and medical staff, and worse cognitive functioning.9 In 80% of cases, the symptoms of uncomplicated alcohol withdrawal do not require aggressive medical intervention and usually disappear within two to seven days of the last drink.12 Physicians making triage decisions for patients who present to the ED in need of detoxification face a difficult dilemma concerning inpatient versus outpatient treatment.

Review of the Data
The literature on both inpatient and outpatient management and treatment of AWS is well-described. Currently, there are no guidelines or consensus on whether to admit patients with alcohol abuse syndromes to the hospital when the request for detoxification is made. Admission should be considered for all patients experiencing alcohol withdrawal who present to the ED.13 Patients with mild AWS may be discharged if they do not require admission for an additional medical condition, but patients experiencing moderate to severe withdrawal require admission for monitoring and treatment. Many physicians use a simple assessment of past history of DT and pulse rate, which may be easily evaluated in clinical settings, to readily identify patients who are at high risk of developing DT during an alcohol dependence period.14
Since 1978, the Clinical Institute Withdrawal Assessment for Alcohol (CIWA) has been consistently used for both monitoring patients with alcohol withdrawal and for making an initial assessment. CIWA-Ar was developed as a revised scale and is frequently used to monitor the severity of ongoing alcohol withdrawal and the response to treatment for the clinical care of patients in alcohol withdrawal (see Figure 2). CIWA-Ar was not developed to identify patients at risk for AWS but is frequently used to determine if patients require admission to the hospital for detoxification.15 Patients with CIWA-Ar scores > 15 require inpatient detoxification. Patients with scores between 8 and 15 should be admitted if they have a history of prior seizures or DT but could otherwise be considered for outpatient detoxification. Patients with scores < 8, which are considered mild alcohol withdrawal, can likely be safely treated as outpatients unless they have a history of DT or alcohol withdrawal seizures.16 Because symptoms of severe alcohol withdrawal are often not present for more than six hours after the patient’s last drink, or often longer, CIWA-Ar is limited and does not identify patients who are otherwise at high risk for complicated withdrawal. A protocol was developed incorporating the patient’s history of alcohol withdrawal seizure, DT, and the CIWA to evaluate the outcome of outpatient versus inpatient detoxification.16

The most promising tool to screen patients for AWS was developed recently by researchers at Stanford University in Stanford, Calif., using an extensive systematic literature search to identify evidence-based clinical factors associated with the development of AWS.15 The Prediction of Alcohol Withdrawal Severity Scale (PAWSS) was subsequently constructed from 10 items correlating with complicated AWS (see Figure 3). When using a PAWSS score cutoff of ≥ 4, the predictive value of identifying a patient who is at risk for complicated withdrawal is significantly increased to 93.1%. This tool has only been used in medically ill patients but could be extrapolated for use in patients who present to an acute-care setting requesting inpatient detoxification.

Patients presenting to the ED with alcohol withdrawal seizures have been shown to have an associated 35% risk of progression to DT when found to have a low platelet count, low blood pyridoxine, and a high blood level of homocysteine. In another retrospective cohort study in Hepatology, three clinical features were identified to be associated with an increased risk for DT: alcohol dependence, a prior history of DT, and a higher pulse rate at admission (> 100 bpm).14
Instructions for the assessment of the patient who requests detoxification are as follows:
- A patient whose last drink of alcohol was more than five days ago and who shows no signs of withdrawal is unlikely to develop significant withdrawal symptoms and does not require inpatient detoxification.
- Other medical and psychiatric conditions should be evaluated for admission including alcohol use disorder complications.
- Calculate CIWA-Ar score:
Scores < 8 may not need detoxification; consider calculating PAWSS score.
Scores of 8 to 15 without symptoms of DT or seizures can be treated as an outpatient detoxification if no contraindication.
Scores of ≥ 15 should be admitted to the hospital.
- Calculate PAWSS score:
Scores ≥ 4 suggest high risk for moderate to severe complicated AWS, and admission should be considered.
Scores < 4 suggest lower risk for complicated AWS, and outpatient treatment should be considered if patients do not have a medical or surgical diagnosis requiring admission.
Back to the Case
At the time of his presentation, the patient was beginning to show signs of early withdrawal symptoms, including tremor and tachycardia, despite having an elevated blood alcohol level. This patient had a PAWSS score of 6, placing him at increased risk of complicated AWS, and a CIWA-Ar score of 13. He was subsequently admitted to the hospital, and symptom-triggered therapy for treatment of his alcohol withdrawal was used. The patient’s CIWA-Ar score peaked at 21 some 24 hours after his last drink. The patient otherwise had an uncomplicated four-day hospital course due to persistent nausea.
Bottom Line
Hospitalists unsure of which patients should be admitted for alcohol detoxification can use the PAWSS tool and an initial CIWA-Ar score to help determine a patient’s risk for developing complicated AWS. TH
Dr. Velasquez and Dr. Kornsawad are assistant professors and hospitalists at the University of Texas Health Science Center at San Antonio. Dr. Velasquez also serves as assistant professor and hospitalist at the South Texas Veterans Health Care System serving the San Antonio area.
References
- Grant BF, Stinson FS, Dawson DA, et al. Prevalence and co-occurrence of substance use disorder and independent mood and anxiety disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2004;61(8):807-816.
- Lieber CS. Medical disorders of alcoholism. N Engl J Med. 1995;333(16):1058-1065.
- Hasin SD, Stinson SF, Ogburn E, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830-842.
- Whiteman PJ, Hoffman RS, Goldfrank LR. Alcoholism in the emergency department: an epidemiologic study. Acad Emerg Med. 2000;7(1):14-20.
- Nielson SD, Storgarrd H, Moesgarrd F, Gluud C. Prevalence of alcohol problems among adult somatic in-patients of a Copenhagen hospital. Alcohol Alcohol. 1994;29(5):583-590.
- Smothers BA, Yahr HT, Ruhl CE. Detection of alcohol use disorders in general hospital admissions in the United States. Arch Intern Med. 2004;164(7):749-756.
- Dolman JM, Hawkes ND. Combining the audit questionnaire and biochemical markers to assess alcohol use and risk of alcohol withdrawal in medical inpatients. Alcohol Alcohol. 2005;40(6):515-519.
- Doering-Silveira J, Fidalgo TM, Nascimento CL, et al. Assessing alcohol dependence in hospitalized patients. Int J Environ Res Public Health. 2014;11(6):5783-5791.
- Maldonado JR, Sher Y, Das S, et al. Prospective validation study of the prediction of alcohol withdrawal severity scale (PAWSS) in medically ill inpatients: a new scale for the prediction of complicated alcohol withdrawal syndrome. Alcohol Alcohol. 2015;50(5):509-518.
- Saitz R, O’Malley SS. Pharmacotherapies for alcohol abuse. Withdrawal and treatment. Med Clin North Am. 1997;81(4):881-907.
- Turner RC, Lichstein PR, Pedan Jr JG, Busher JT, Waivers LE. Alcohol withdrawal syndromes: a review of pathophysiology, clinical presentation, and treatment. J Gen Intern Med. 1989;4(5):432-444.
- Schuckit MA. Alcohol-use disorders. Lancet. 2009;373(9662):492-501.
- Stehman CR, Mycyk MB. A rational approach to the treatment of alcohol withdrawal in the ED. Am J Emerg Med. 2013;31(4):734-742.
- Lee JH, Jang MK, Lee JY, et al. Clinical predictors for delirium tremens in alcohol dependence. J Gastroenterol Hepatol. 2005;20(12):1833-1837.
- Maldonado JR, Sher Y, Ashouri JF, et al. The “prediction of alcohol withdrawal severity scale” (PAWSS): systematic literature review and pilot study of a new scale for the prediction of complicated alcohol withdrawal syndrome. Alcohol. 2014;48(4):375-390.
- Stephens JR, Liles AE, Dancel R, Gilchrist M, Kirsch J, DeWalt DA. Who needs inpatient detox? Development and implementation of a hospitalist protocol for the evaluation of patients for alcohol detoxification. J Gen Intern Med. 2014;29(4):587-593.
Case
A 42-year-old man with a history of posttraumatic stress disorder (PTSD), hypertension, and alcohol use disorder (AUD) presents to the ED requesting alcohol detoxification. He has had six admissions in the last six months for alcohol detoxification. Two years ago, the patient had a documented alcohol withdrawal seizure. His last drink was eight hours ago, and he currently drinks a liter of vodka a day. On exam, his pulse rate is 126 bpm, and his blood pressure is 162/91 mm Hg. He appears anxious and has bilateral hand tremors. His serum ethanol level is 388.6 mg/dL.
Overview
DSM-5 integrated alcohol abuse and alcohol dependence that were previously classified in DSM-IV into AUDs with mild, moderate, and severe subclassifications. AUDs are the most serious substance abuse problem in the U.S. In the general population, the lifetime prevalence of alcohol abuse is 17.8% and of alcohol dependence is 12.5%.1–3 One study estimates that 24% of adult patients brought to the ED by ambulance suffer from alcoholism, and approximately 10% to 32% of hospitalized medical patients have an AUD.4–8 Patients who stop drinking will develop alcohol withdrawal as early as six hours after their last drink (see Figure 1). The majority of patients at risk of alcohol withdrawal syndrome (AWS) will develop only minor uncomplicated symptoms, but up to 20% will develop symptoms associated with complicated AWS, including withdrawal seizures and delirium tremens (DT).9 It is not entirely clear why some individuals suffer from more severe withdrawal symptoms than others, but genetic predisposition may play a role.10
DT is a syndrome characterized by agitation, disorientation, hallucinations, and autonomic instability (tachycardia, hypertension, hyperthermia, and diaphoresis) in the setting of acute reduction or abstinence from alcohol and is associated with a mortality rate as high as 20%.11 Complicated AWS is associated with increased in-hospital morbidity and mortality, longer lengths of stay, inflated costs of care, increased burden and frustration of nursing and medical staff, and worse cognitive functioning.9 In 80% of cases, the symptoms of uncomplicated alcohol withdrawal do not require aggressive medical intervention and usually disappear within two to seven days of the last drink.12 Physicians making triage decisions for patients who present to the ED in need of detoxification face a difficult dilemma concerning inpatient versus outpatient treatment.
Review of the Data
The literature on both inpatient and outpatient management and treatment of AWS is well-described. Currently, there are no guidelines or consensus on whether to admit patients with alcohol abuse syndromes to the hospital when the request for detoxification is made. Admission should be considered for all patients experiencing alcohol withdrawal who present to the ED.13 Patients with mild AWS may be discharged if they do not require admission for an additional medical condition, but patients experiencing moderate to severe withdrawal require admission for monitoring and treatment. Many physicians use a simple assessment of past history of DT and pulse rate, which may be easily evaluated in clinical settings, to readily identify patients who are at high risk of developing DT during an alcohol dependence period.14
Since 1978, the Clinical Institute Withdrawal Assessment for Alcohol (CIWA) has been consistently used for both monitoring patients with alcohol withdrawal and for making an initial assessment. CIWA-Ar was developed as a revised scale and is frequently used to monitor the severity of ongoing alcohol withdrawal and the response to treatment for the clinical care of patients in alcohol withdrawal (see Figure 2). CIWA-Ar was not developed to identify patients at risk for AWS but is frequently used to determine if patients require admission to the hospital for detoxification.15 Patients with CIWA-Ar scores > 15 require inpatient detoxification. Patients with scores between 8 and 15 should be admitted if they have a history of prior seizures or DT but could otherwise be considered for outpatient detoxification. Patients with scores < 8, which are considered mild alcohol withdrawal, can likely be safely treated as outpatients unless they have a history of DT or alcohol withdrawal seizures.16 Because symptoms of severe alcohol withdrawal are often not present for more than six hours after the patient’s last drink, or often longer, CIWA-Ar is limited and does not identify patients who are otherwise at high risk for complicated withdrawal. A protocol was developed incorporating the patient’s history of alcohol withdrawal seizure, DT, and the CIWA to evaluate the outcome of outpatient versus inpatient detoxification.16
The most promising tool to screen patients for AWS was developed recently by researchers at Stanford University in Stanford, Calif., using an extensive systematic literature search to identify evidence-based clinical factors associated with the development of AWS.15 The Prediction of Alcohol Withdrawal Severity Scale (PAWSS) was subsequently constructed from 10 items correlating with complicated AWS (see Figure 3). When using a PAWSS score cutoff of ≥ 4, the predictive value of identifying a patient who is at risk for complicated withdrawal is significantly increased to 93.1%. This tool has only been used in medically ill patients but could be extrapolated for use in patients who present to an acute-care setting requesting inpatient detoxification.
Patients presenting to the ED with alcohol withdrawal seizures have been shown to have an associated 35% risk of progression to DT when found to have a low platelet count, low blood pyridoxine, and a high blood level of homocysteine. In another retrospective cohort study in Hepatology, three clinical features were identified to be associated with an increased risk for DT: alcohol dependence, a prior history of DT, and a higher pulse rate at admission (> 100 bpm).14
Instructions for the assessment of the patient who requests detoxification are as follows:
- A patient whose last drink of alcohol was more than five days ago and who shows no signs of withdrawal is unlikely to develop significant withdrawal symptoms and does not require inpatient detoxification.
- Other medical and psychiatric conditions should be evaluated for admission including alcohol use disorder complications.
- Calculate CIWA-Ar score:
Scores < 8 may not need detoxification; consider calculating PAWSS score.
Scores of 8 to 15 without symptoms of DT or seizures can be treated as an outpatient detoxification if no contraindication.
Scores of ≥ 15 should be admitted to the hospital.
- Calculate PAWSS score:
Scores ≥ 4 suggest high risk for moderate to severe complicated AWS, and admission should be considered.
Scores < 4 suggest lower risk for complicated AWS, and outpatient treatment should be considered if patients do not have a medical or surgical diagnosis requiring admission.
Back to the Case
At the time of his presentation, the patient was beginning to show signs of early withdrawal symptoms, including tremor and tachycardia, despite having an elevated blood alcohol level. This patient had a PAWSS score of 6, placing him at increased risk of complicated AWS, and a CIWA-Ar score of 13. He was subsequently admitted to the hospital, and symptom-triggered therapy for treatment of his alcohol withdrawal was used. The patient’s CIWA-Ar score peaked at 21 some 24 hours after his last drink. The patient otherwise had an uncomplicated four-day hospital course due to persistent nausea.
Bottom Line
Hospitalists unsure of which patients should be admitted for alcohol detoxification can use the PAWSS tool and an initial CIWA-Ar score to help determine a patient’s risk for developing complicated AWS. TH
Dr. Velasquez and Dr. Kornsawad are assistant professors and hospitalists at the University of Texas Health Science Center at San Antonio. Dr. Velasquez also serves as assistant professor and hospitalist at the South Texas Veterans Health Care System serving the San Antonio area.
References
- Grant BF, Stinson FS, Dawson DA, et al. Prevalence and co-occurrence of substance use disorder and independent mood and anxiety disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2004;61(8):807-816.
- Lieber CS. Medical disorders of alcoholism. N Engl J Med. 1995;333(16):1058-1065.
- Hasin SD, Stinson SF, Ogburn E, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830-842.
- Whiteman PJ, Hoffman RS, Goldfrank LR. Alcoholism in the emergency department: an epidemiologic study. Acad Emerg Med. 2000;7(1):14-20.
- Nielson SD, Storgarrd H, Moesgarrd F, Gluud C. Prevalence of alcohol problems among adult somatic in-patients of a Copenhagen hospital. Alcohol Alcohol. 1994;29(5):583-590.
- Smothers BA, Yahr HT, Ruhl CE. Detection of alcohol use disorders in general hospital admissions in the United States. Arch Intern Med. 2004;164(7):749-756.
- Dolman JM, Hawkes ND. Combining the audit questionnaire and biochemical markers to assess alcohol use and risk of alcohol withdrawal in medical inpatients. Alcohol Alcohol. 2005;40(6):515-519.
- Doering-Silveira J, Fidalgo TM, Nascimento CL, et al. Assessing alcohol dependence in hospitalized patients. Int J Environ Res Public Health. 2014;11(6):5783-5791.
- Maldonado JR, Sher Y, Das S, et al. Prospective validation study of the prediction of alcohol withdrawal severity scale (PAWSS) in medically ill inpatients: a new scale for the prediction of complicated alcohol withdrawal syndrome. Alcohol Alcohol. 2015;50(5):509-518.
- Saitz R, O’Malley SS. Pharmacotherapies for alcohol abuse. Withdrawal and treatment. Med Clin North Am. 1997;81(4):881-907.
- Turner RC, Lichstein PR, Pedan Jr JG, Busher JT, Waivers LE. Alcohol withdrawal syndromes: a review of pathophysiology, clinical presentation, and treatment. J Gen Intern Med. 1989;4(5):432-444.
- Schuckit MA. Alcohol-use disorders. Lancet. 2009;373(9662):492-501.
- Stehman CR, Mycyk MB. A rational approach to the treatment of alcohol withdrawal in the ED. Am J Emerg Med. 2013;31(4):734-742.
- Lee JH, Jang MK, Lee JY, et al. Clinical predictors for delirium tremens in alcohol dependence. J Gastroenterol Hepatol. 2005;20(12):1833-1837.
- Maldonado JR, Sher Y, Ashouri JF, et al. The “prediction of alcohol withdrawal severity scale” (PAWSS): systematic literature review and pilot study of a new scale for the prediction of complicated alcohol withdrawal syndrome. Alcohol. 2014;48(4):375-390.
- Stephens JR, Liles AE, Dancel R, Gilchrist M, Kirsch J, DeWalt DA. Who needs inpatient detox? Development and implementation of a hospitalist protocol for the evaluation of patients for alcohol detoxification. J Gen Intern Med. 2014;29(4):587-593.
Case
A 42-year-old man with a history of posttraumatic stress disorder (PTSD), hypertension, and alcohol use disorder (AUD) presents to the ED requesting alcohol detoxification. He has had six admissions in the last six months for alcohol detoxification. Two years ago, the patient had a documented alcohol withdrawal seizure. His last drink was eight hours ago, and he currently drinks a liter of vodka a day. On exam, his pulse rate is 126 bpm, and his blood pressure is 162/91 mm Hg. He appears anxious and has bilateral hand tremors. His serum ethanol level is 388.6 mg/dL.
Overview
DSM-5 integrated alcohol abuse and alcohol dependence that were previously classified in DSM-IV into AUDs with mild, moderate, and severe subclassifications. AUDs are the most serious substance abuse problem in the U.S. In the general population, the lifetime prevalence of alcohol abuse is 17.8% and of alcohol dependence is 12.5%.1–3 One study estimates that 24% of adult patients brought to the ED by ambulance suffer from alcoholism, and approximately 10% to 32% of hospitalized medical patients have an AUD.4–8 Patients who stop drinking will develop alcohol withdrawal as early as six hours after their last drink (see Figure 1). The majority of patients at risk of alcohol withdrawal syndrome (AWS) will develop only minor uncomplicated symptoms, but up to 20% will develop symptoms associated with complicated AWS, including withdrawal seizures and delirium tremens (DT).9 It is not entirely clear why some individuals suffer from more severe withdrawal symptoms than others, but genetic predisposition may play a role.10
DT is a syndrome characterized by agitation, disorientation, hallucinations, and autonomic instability (tachycardia, hypertension, hyperthermia, and diaphoresis) in the setting of acute reduction or abstinence from alcohol and is associated with a mortality rate as high as 20%.11 Complicated AWS is associated with increased in-hospital morbidity and mortality, longer lengths of stay, inflated costs of care, increased burden and frustration of nursing and medical staff, and worse cognitive functioning.9 In 80% of cases, the symptoms of uncomplicated alcohol withdrawal do not require aggressive medical intervention and usually disappear within two to seven days of the last drink.12 Physicians making triage decisions for patients who present to the ED in need of detoxification face a difficult dilemma concerning inpatient versus outpatient treatment.
Review of the Data
The literature on both inpatient and outpatient management and treatment of AWS is well-described. Currently, there are no guidelines or consensus on whether to admit patients with alcohol abuse syndromes to the hospital when the request for detoxification is made. Admission should be considered for all patients experiencing alcohol withdrawal who present to the ED.13 Patients with mild AWS may be discharged if they do not require admission for an additional medical condition, but patients experiencing moderate to severe withdrawal require admission for monitoring and treatment. Many physicians use a simple assessment of past history of DT and pulse rate, which may be easily evaluated in clinical settings, to readily identify patients who are at high risk of developing DT during an alcohol dependence period.14
Since 1978, the Clinical Institute Withdrawal Assessment for Alcohol (CIWA) has been consistently used for both monitoring patients with alcohol withdrawal and for making an initial assessment. CIWA-Ar was developed as a revised scale and is frequently used to monitor the severity of ongoing alcohol withdrawal and the response to treatment for the clinical care of patients in alcohol withdrawal (see Figure 2). CIWA-Ar was not developed to identify patients at risk for AWS but is frequently used to determine if patients require admission to the hospital for detoxification.15 Patients with CIWA-Ar scores > 15 require inpatient detoxification. Patients with scores between 8 and 15 should be admitted if they have a history of prior seizures or DT but could otherwise be considered for outpatient detoxification. Patients with scores < 8, which are considered mild alcohol withdrawal, can likely be safely treated as outpatients unless they have a history of DT or alcohol withdrawal seizures.16 Because symptoms of severe alcohol withdrawal are often not present for more than six hours after the patient’s last drink, or often longer, CIWA-Ar is limited and does not identify patients who are otherwise at high risk for complicated withdrawal. A protocol was developed incorporating the patient’s history of alcohol withdrawal seizure, DT, and the CIWA to evaluate the outcome of outpatient versus inpatient detoxification.16
The most promising tool to screen patients for AWS was developed recently by researchers at Stanford University in Stanford, Calif., using an extensive systematic literature search to identify evidence-based clinical factors associated with the development of AWS.15 The Prediction of Alcohol Withdrawal Severity Scale (PAWSS) was subsequently constructed from 10 items correlating with complicated AWS (see Figure 3). When using a PAWSS score cutoff of ≥ 4, the predictive value of identifying a patient who is at risk for complicated withdrawal is significantly increased to 93.1%. This tool has only been used in medically ill patients but could be extrapolated for use in patients who present to an acute-care setting requesting inpatient detoxification.
Patients presenting to the ED with alcohol withdrawal seizures have been shown to have an associated 35% risk of progression to DT when found to have a low platelet count, low blood pyridoxine, and a high blood level of homocysteine. In another retrospective cohort study in Hepatology, three clinical features were identified to be associated with an increased risk for DT: alcohol dependence, a prior history of DT, and a higher pulse rate at admission (> 100 bpm).14
Instructions for the assessment of the patient who requests detoxification are as follows:
- A patient whose last drink of alcohol was more than five days ago and who shows no signs of withdrawal is unlikely to develop significant withdrawal symptoms and does not require inpatient detoxification.
- Other medical and psychiatric conditions should be evaluated for admission including alcohol use disorder complications.
- Calculate CIWA-Ar score:
Scores < 8 may not need detoxification; consider calculating PAWSS score.
Scores of 8 to 15 without symptoms of DT or seizures can be treated as an outpatient detoxification if no contraindication.
Scores of ≥ 15 should be admitted to the hospital.
- Calculate PAWSS score:
Scores ≥ 4 suggest high risk for moderate to severe complicated AWS, and admission should be considered.
Scores < 4 suggest lower risk for complicated AWS, and outpatient treatment should be considered if patients do not have a medical or surgical diagnosis requiring admission.
Back to the Case
At the time of his presentation, the patient was beginning to show signs of early withdrawal symptoms, including tremor and tachycardia, despite having an elevated blood alcohol level. This patient had a PAWSS score of 6, placing him at increased risk of complicated AWS, and a CIWA-Ar score of 13. He was subsequently admitted to the hospital, and symptom-triggered therapy for treatment of his alcohol withdrawal was used. The patient’s CIWA-Ar score peaked at 21 some 24 hours after his last drink. The patient otherwise had an uncomplicated four-day hospital course due to persistent nausea.
Bottom Line
Hospitalists unsure of which patients should be admitted for alcohol detoxification can use the PAWSS tool and an initial CIWA-Ar score to help determine a patient’s risk for developing complicated AWS. TH
Dr. Velasquez and Dr. Kornsawad are assistant professors and hospitalists at the University of Texas Health Science Center at San Antonio. Dr. Velasquez also serves as assistant professor and hospitalist at the South Texas Veterans Health Care System serving the San Antonio area.
References
- Grant BF, Stinson FS, Dawson DA, et al. Prevalence and co-occurrence of substance use disorder and independent mood and anxiety disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2004;61(8):807-816.
- Lieber CS. Medical disorders of alcoholism. N Engl J Med. 1995;333(16):1058-1065.
- Hasin SD, Stinson SF, Ogburn E, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830-842.
- Whiteman PJ, Hoffman RS, Goldfrank LR. Alcoholism in the emergency department: an epidemiologic study. Acad Emerg Med. 2000;7(1):14-20.
- Nielson SD, Storgarrd H, Moesgarrd F, Gluud C. Prevalence of alcohol problems among adult somatic in-patients of a Copenhagen hospital. Alcohol Alcohol. 1994;29(5):583-590.
- Smothers BA, Yahr HT, Ruhl CE. Detection of alcohol use disorders in general hospital admissions in the United States. Arch Intern Med. 2004;164(7):749-756.
- Dolman JM, Hawkes ND. Combining the audit questionnaire and biochemical markers to assess alcohol use and risk of alcohol withdrawal in medical inpatients. Alcohol Alcohol. 2005;40(6):515-519.
- Doering-Silveira J, Fidalgo TM, Nascimento CL, et al. Assessing alcohol dependence in hospitalized patients. Int J Environ Res Public Health. 2014;11(6):5783-5791.
- Maldonado JR, Sher Y, Das S, et al. Prospective validation study of the prediction of alcohol withdrawal severity scale (PAWSS) in medically ill inpatients: a new scale for the prediction of complicated alcohol withdrawal syndrome. Alcohol Alcohol. 2015;50(5):509-518.
- Saitz R, O’Malley SS. Pharmacotherapies for alcohol abuse. Withdrawal and treatment. Med Clin North Am. 1997;81(4):881-907.
- Turner RC, Lichstein PR, Pedan Jr JG, Busher JT, Waivers LE. Alcohol withdrawal syndromes: a review of pathophysiology, clinical presentation, and treatment. J Gen Intern Med. 1989;4(5):432-444.
- Schuckit MA. Alcohol-use disorders. Lancet. 2009;373(9662):492-501.
- Stehman CR, Mycyk MB. A rational approach to the treatment of alcohol withdrawal in the ED. Am J Emerg Med. 2013;31(4):734-742.
- Lee JH, Jang MK, Lee JY, et al. Clinical predictors for delirium tremens in alcohol dependence. J Gastroenterol Hepatol. 2005;20(12):1833-1837.
- Maldonado JR, Sher Y, Ashouri JF, et al. The “prediction of alcohol withdrawal severity scale” (PAWSS): systematic literature review and pilot study of a new scale for the prediction of complicated alcohol withdrawal syndrome. Alcohol. 2014;48(4):375-390.
- Stephens JR, Liles AE, Dancel R, Gilchrist M, Kirsch J, DeWalt DA. Who needs inpatient detox? Development and implementation of a hospitalist protocol for the evaluation of patients for alcohol detoxification. J Gen Intern Med. 2014;29(4):587-593.
Betting the Farm
A 65‐year‐old man with a 6‐month history of diabetes mellitus presented to the emergency department in May with 1 week of fevers, headaches, myalgia, polydipsia, and polyuria.
The patient presents with symptoms suggestive of uncontrolled diabetes and infection. The broad diagnostic categories include acute infection, an emerging chronic process aggravating his diabetes, or a noninfectious mimic such as autoimmune disease or lymphoproliferative disease. New onset headache in an older patient is concerning. Although it may be attributed to fever and dehydration, primary central nervous system processes such as meningitis or encephalitis must be considered. At this stage, a detailed exposure history, including travel, food, pets, hobbies, and sick contacts as well as occupation and national origins is needed. This patient presented in May, making illnesses that peak in other seasons such as influenza and West Nile fever less likely.
He had no other medical problems except diabetes. He was not taking any medications; he had been started on glipizide but had stopped taking it 1 month prior. He denied fever, cough, chest pain, palpitations, abdominal pain, nausea, vomiting, dysuria, focal weakness, visual changes, or photophobia. He was born in Mexico and emigrated at the age of 25 years. Two months prior to presentation he visited a cattle farm in Mexico; he denied any direct contact with farm animals or dairy products. He denied ill contacts, pets, known tuberculosis exposures, and sexual partners other than his wife.
The history of recent travel to Mexico with a visit to a farm raises concerns about zoonoses. The endemic zoonoses that should be considered include parasitic (toxoplasmosis), fungal (coccidiodomycosis), and bacterial (brucellosis, Q fever, leptospirosis, tularemia, salmonellosis) infections. Nonzoonotic granulomatous infections such as cytomegalovirus (CMV) and Epstein‐Barr virus (EBV), mycobacteria, fungi (histoplasmosis, blastomycosis, cryptococcosis, aspergillosis), and bacteria (actinomycosis) should also be considered.
On examination, he was an elderly Hispanic male who appeared ill but in no acute distress. He was overweight, with a BMI of 29. His temperature was 39C, pulse 66 beats/minute, blood pressure 108/68 mm Hg, respiratory rate 18 per minute, and oxygen saturation was 96% on room air. There were no ulcerations, exudates, or erythema in the oropharynx. There was no sinus tenderness or lymphadenopathy. Cardiac examination revealed normal heart sounds with no murmurs. Respiratory examination demonstrated clear lungs. His abdomen was soft and nontender, whereas the liver and spleen were not palpable. There was no nuchal rigidity, and his mental status was normal. There were no cranial nerve deficits or weakness in his extremities. There was no skin rash or peripheral stigmata of infectious endocarditis. Genitourinary examination revealed no ulcerations, inguinal lymphadenopathy, or urethral discharge. There was no tenderness, warmth, or erythema on examination of all joints.
The physical exam is notable for temperaturepulse dissociation. Heart rate should increase by about 10 beats/minute for every 1‐degree increase in Fahrenheit temperature. The infectious causes of temperaturepulse dissociation are largely intracellular pathogens such as Salmonella, Coxiella, Chlamydia, Leptospira, Legionella, Francisella, Mycoplasma, and dengue virus. This patient is at increased risk for infection by any of these pathogens based on his recent travel to Mexico. Drug fever is the most common noninfectious cause of temperaturepulse dissociation, but this patient took no medications. At this point, a complete blood count and differential, urinalysis, blood cultures, chest x‐ray, and electrocardiogram should be ordered. Testing for human immunodeficiency virus (HIV) is appropriate, as up to 50% of patients with newly diagnosed HIV have no acknowledged risk factors. Serological studies for the aforementioned pathogens may be indicated depending on the results of these initial diagnostic tests.
Serum sodium concentration was 122 mEq/L, potassium 4.0 mEq/L, chloride 88 mEq/L, bicarbonate 14 mEq/L, blood urea nitrogen 17 mg/dL, creatinine 0.7 mg/dL, glucose 402 mg/dL, and calcium 8.5 mg/dL. Total protein was 5.4 g/dL, albumin 2.9 g/dL, total bilirubin 0.9 mg/dL, direct bilirubin 0.4 mg/dL, alkaline phosphatase 126 U/L (normal 53128), gamma‐glutamyl transferase 264 U/L (normal 360), aspartate aminotransferase 51 U/L (normal 840), alanine aminotransferase 62 U/L (normal 556), and lactate dehydrogenase 248 U/L (normal 85210). The white blood cell (WBC) count was 6800 mm3 (51% band forms, 38% segmented neutrophils, 6% monocytes, 5% lymphocytes). The hemoglobin was 15.7 g/dL, with mean corpuscular volume (MCV) of 102 fL and platelet count 59,000/mm3. Peripheral‐blood smear showed occasional macrocytes. Prothrombin time was 13.6 seconds and partial thromboplastin time was 34.5 seconds. C‐reactive protein was 11.8 mg/dL. Urinalysis revealed 80 mg of ketones per deciliter, no cells, and nitrite was negative. Hemoglobin A1c was 13%, and HIV antibody testing was negative.
Elevated circulating bands and thrombocytopenia suggest infection; however, bone marrow infiltration by infectious or neoplastic process is also possible and should be investigated. The increased gamma‐glutamyl transferase, alkaline phosphatase, and mild increases in transaminases suggest hepatic pathology. The combination of unexplained fever, hyponatremia, thrombocytopenia, elevated liver enzymes, and travel to Mexico mandates investigation for infectious diseases that often involve both the bone marrow and liver such as Brucella, Coxiella, and fungal infections such as histoplasmosis. Autoimmune diseases such as systemic lupus erythematosus and malignancy should also be considered. Blood cultures should be incubated beyond the usual 5 days because of the slower growth of Brucella or Salmonella typhi. An HIV viral load should be obtained to evaluate for acute retroviral syndrome. Serologic tests for Rickettsia, Coccidiodes, and hepatitis A, B, and C viruses should be obtained. Urine should be tested for Histoplasma and Legionella antigens. Abdominal imaging should be obtained to evaluate for hepatobiliary disease, occult intra‐abdominal abscess, or malignancy. Because the patient has unexplained fever and headache, imaging of the central nervous system and lumbar puncture are warranted.
His diabetic ketoacidosis (DKA) was treated with intravenous fluids and insulin. Lumbar puncture and cerebrospinal fluid (CSF) analysis revealed opening pressure of 18 cm H20 (normal 1025), cell count WBC 3/L (normal 05), red blood cell 204/L (normal 0), CSF protein 25 mg/dL (normal 2050), and glucose 68 mg/dL (normal 5070). Blood cultures showed no growth. HIV RNA was undetectable. Hepatitis C antibody was negative, and hepatitis A and B serologies were not consistent with an acute infection. Serum ferritin was 1147 ng/mL. Histoplasma and Legionella urine antigen tests were negative. CMV, EBV, and herpes simplex virus DNA were not detected in blood samples. Anti‐neutrophil antibody, anti‐mitochondrial antibody and anti‐neutrophil cytoplasmic antibodies were undetectable. Anti‐smooth muscle antibody was positive at a titer of 1:80. Transthoracic echocardiogram revealed normal heart valves without vegetations. A chest radiograph was normal. Brain computed tomography (CT) revealed atrophic frontal lobes. CT of his chest, abdomen, and pelvis demonstrated focal inflammatory changes of a loop of distal small bowel with surrounding fluid collection, suggesting small bowel diverticulitis. There were no pulmonary infiltrates noted, and the remainder of the CT was unremarkable.
Because the patient remains ill and additional serological test results will take time to return, a key consideration at this point is empiric treatment while awaiting test results. The CSF examination was normal. A history of travel including animal and tick exposures should be reevaluated. The timing of the trip to Mexico was outside the usual incubation period for many pathogens except for Coxiella or Brucella, and empiric therapy for both would be appropriate. The abdominal CT suggests small bowel diverticulitis, which is a rare clinical entity.
The benign abdominal examination suggests the finding is incidental. However, there are several infections that may involve the distal small bowel and proximal colon, such as yersiniosis, salmonellosis, tuberculosis, actinomycosis, histoplasmosis, and noninfectious processes including Crohn's disease and neoplasia. The absence of diarrhea or hematochezia makes yersiniosis, salmonellosis, and Crohn's disease unlikely. Histoplasmosis is unlikely given the negative urine antigen. Evaluation for neoplasia of the distal small bowel requires histologic examination. A colonoscopy with random biopsies of the colon and terminal ileum is the next step if other tests are unrevealing.
The patient was empirically treated for small bowel diverticulitis with ceftriaxone and metronidazole. Because of continued daily fevers as high as 39C, his therapy was changed to vancomycin and piperacillin‐tazobactam to cover methicillin‐resistant Staphylococcus aureus and resistant gram‐negative bacilli. The patient developed new scleral icterus on hospital day 6; the remainder of his examination was unchanged. Serum sodium concentration was 127 mEq/L, potassium 2.7 mEq/L, phosphorus 1.3 mg/dL, magnesium 1.6 mg/dL, total bilirubin 5.6 mg/dL, direct bilirubin 3.6 mg/dL, alkaline phosphatase 193 U/L, gamma‐glutamyl transferase 300 U/L, aspartate aminotransferase 91 U/L, alanine aminotransferase 52 U/L. Brucella serology was negative.
His liver enzymes remain elevated with new onset jaundice consistent with hepatitis and intrahepatic cholestasis. His persistent hypophosphatemia, hypokalemia, and hypomagnesaemia well after resolution of diabetic ketoacidosis suggests acute tubulointerstitial dysfunction, which may be a complication of empiric antibiotic treatment or renal involvement by his underlying condition. Additional blood cultures, and tissue examination and culture are the next appropriate steps. Liver or bone marrow biopsy may suggest a diagnosis that can be confirmed by tissue culture or immunohistochemistry. Histologic findings such as fibrin ringed granulomas, caseating or noncaseating granulomas, or lymphomatous infiltration may suggest Coxiella (Q fever), tuberculosis, or lymphoma respectively. Because a liver biopsy is invasive and usually provides less tissue for culture, bone marrow examination should be obtained first.
A gallium 67 scan showed nonhomogenous increased uptake in both lungs and kidneys, consistent with interstitial nephritis and bilateral pneumonia. Serum protein electrophoresis demonstrated a monoclonal immunoglobulin (Ig)G lambda band with a kappa/lambda ratio of 0.9 (normal 1.42.8). Bone marrow biopsy showed normal hematopoiesis; no plasma or malignant cells, granulomas, or evidence of hemophagocytosis; and fungal and mycobacterial stains and cultures were negative. Colonoscopy revealed normal‐appearing mucosa. Histologic examination and culture of random biopsies from the colon and terminal ileum were negative for fungi, viruses, and mycobacteria. An ultrasound‐guided liver biopsy revealed numerous noncaseating granulomas formed of histiocytes and neutrophils with occasional fibrin rings. Fungal, viral, and mycobacterial stains and cultures were negative. The patient's fever resolved after 14 days, and he was discharged home without a diagnosis and close outpatient follow‐up.
The hepatic granulomas with fibrin rings are highly suggestive of Q fever, although ring granulomas may be seen in tuberculosis, typhoid fever, lymphoma, drug reactions, sarcoidosis, and CMV infections. Competing diagnoses such as CMV have been excluded by negative serology. Microscopic examination, tissue staining, and culture from liver and bone marrow biopsies were negative for S typhi, mycobacteria, and lymphoma. Gallium scan findings are generally nonspecific and of little utility in cases such as this. The kidney involvement correlates with the biochemical evidence of tubulointerstitial dysfunction; pulmonary involvement may reflect subclinical pulmonary infection with Coxiella. Given the normal bone marrow biopsy, the monoclonal gammopathy is of undetermined significance. The positive anti‐smooth muscle antibody can be related to Q fever. Anti‐smooth muscle antibodies frequently occur in Q fever, especially in those patients with hepatitis. Given the history of exposure to cattle, unexplained fever with temperaturepulse dissociation and liver biopsy findings, Q fever is the most likely diagnosis and empiric treatment with doxycycline is warranted.
Results of serology for Coxiella burnetii sent during admission were returned after the patient's discharge. C burnetii phase I IgG and IgM antibody titers were positive (1:512 each). C burnetii phase II IgG and IgM titers also were positive (1:1024 each). The patient was seen within a week and started on doxycycline 100 mg twice daily for 2 weeks for acute Q fever. His symptoms improved; hyponatremia, liver function tests, and thrombocytopenia normalized after treatment.
DISCUSSION
Q fever was first described in 1937 as a febrile illness affecting Australian slaughterhouse workers.[1] The Q in Q fever stands for query and reflected the initial uncertainty surrounding the underlying cause of the illness. The causative organism, C burnetti, is an obligate intracellular bacterium that resides within macrophage lysosomes. It can be found in the urine, feces, milk, placenta, and amniotic fluid of ungulates (cattle, sheep, and other ruminants), and other animals such as domestic cats and dogs. C burnetii is transmitted via inhalation, ingestion, occupational, or common source exposures, and in 1 case report by person‐to‐person sexual transmission.[2] In addition to slaughterhouse workers, pregnant women and immunosuppressed patients are more susceptible to developing Q fever.[3] For patients with suspected Q fever, a detailed occupational history, including specific job duties and potential exposure to animal products, is imperative.
Q fever has both acute and chronic presentations, which are differentiated based on the clinical illness and serologies. The symptoms of acute Q fever are nonspecific and may include influenza‐like illness, fever, pneumonia, and hepatitis. It presents less commonly with hemolytic anemia, interstitial nephritis, monoclonal gammopathy, or aseptic meningitis.[4, 5, 6, 7] Symptoms typically begin between 1 and 3 weeks after animal exposure and may persist for several months. Chronic Q fever occurs when unrecognized or untreated infection persists for greater than 6 months. It commonly presents with culture‐negative endocarditis, although infected aneurysms, osteomyelitis, or other distant sites of infection may also occur.
C burnetti is present in 2 antigenic forms that can be assessed by serology. Phase I is the more virulent, infectious form of C burnetti, which transitions to the avirulent phase II form during laboratory handling. In acute Q fever, phase II serologies are typically elevated out of proportion to phase I serologies, whereas this pattern is reversed in chronic Q fever. The diagnostic gold standard of acute Q fever is a 4‐fold rise in phase II antibody titers taken 3 to 6 weeks apart.[8] Histologic examination of affected organs can support a diagnosis of Q fever. The presence of ringed granulomas on liver or bone marrow biopsy specimens is highly suggestive, but not pathognomonic, of Q fever.[9]
Q fever is highly susceptible to several classes of antibiotics. For acute Q fever, doxycycline and tetracycline are typically used, with fluoroquinolones and chloramphenicol as alternatives.[8, 10] Patients with chronic Q fever should be treated with doxycycline and hydroxychloroquine. The addition of hydroxychloroquine alkalinizes the macrophage lysosome and enhances bacterial eradication.[8] For patients with acute Q fever, physicians should determine the risk of progression to chronic Q fever because closer monitoring is necessary. Patients with valvular heart lesions, immunosuppression, and pregnant women are at elevated risk of chronic Q fever. Trimethoprim/sulfamethoxazole can be used in place of doxycycline in pregnant women, as doxycycline and fluoroquinolones are contraindicated in pregnancy.[8]
This patient presented with a nonspecific febrile illness. Although the treating clinicians obtained a history of exposure to cattle early in his course, both the diagnosis and treatment were delayed. There are several possible explanations for the delay. First, although Q fever is a relatively common zoonosis, it remains an uncommon diagnosis, particularly among hospitalized patients. As a result, clinicians often focus on more common conditions. In this case, typical infections, malignancies, and inflammatory diseases were considered more likely. Second, the patient presented with hepatitis, an uncommon presentation of Q fever. Classical clinical reasoning suggests that atypical presentations of common diseases will occur more frequently than typical presentations of uncommon diseases. This case presented with an atypical presentation of an uncommon disease. The resultant lower pretest probability further dissuaded the patient's physicians from consideration of Q fever. Third, the finding of small bowel diverticulitis was a potential distractor. In patients with nonspecific febrile illnesses, it is common for physicians to anchor on any abnormal findings. In this case, the small bowel diverticulitis led to antibiotic treatment that was ineffective against C burnetti.
There were several clues to the diagnosis of Q fever in this patient's presentation. First, the pulsetemperature dissociation suggested infection with an intracellular pathogen. Hospitalists should recognize this association and be mindful of this often‐subtle clinical finding when faced with diagnostic uncertainty. Second, the patient was exposed to cattle prior to the onset of his illness. The fact that he did not have a direct exposure to animals underscores the infectivity of C burnetti. Finally, elevated alkaline phosphatase and transaminases were suggestive of an infiltrative disease; in the setting of a nonspecific febrile illness, Q fever was an important diagnostic consideration.
The key treatment decision in this case was the initiation and choice of antibiotics. Because of this patient's history of exposure to cattle and lack of a compelling alternative diagnosis, empiric treatment with doxycycline would have been appropriate. Hospitalists must weigh the potential benefit of early treatment of Q fever against the risks associated with antibiotic overuse. In patients presenting with a febrile illness after ungulate exposure, the decision to bet the farm with empiric doxycycline therapy may lead to clinical improvement, obviating a more invasive or extensive diagnostic evaluation.
TEACHING POINTS
- Acute Q fever typically presents 2 to 3 weeks after ungulate exposure with a febrile illness, pneumonia, and granulomatous hepatitis.
- Pulsetemperature dissociation is suggestive of infection by intracellular pathogens such as Coxiella, Salmonella, Leptospira, Legionella, and Mycoplasma.
- Clinicians should consider empiric doxycycline therapy in patients with suspected zoonosis (eg, Q fever, brucellosis, anaplasmosis, leptospirosis, Rocky Mountain spotted fever) while awaiting confirmatory tests, as improvement may obviate invasive testing.
Disclosure: Nothing to report.
- . “Q” fever, a new fever entity: clinical features, diagnosis, and laboratory investigation. Rev Infect Dis. 1983;5(4):790–800.
- , , , . Q fever: a biological weapon in your backyard. Lancet Infect Dis. 2003;3(11):709–721.
- , , , . Role of sex, age, previous valve lesion, and pregnancy in the clinical expression and outcome of Q fever after a large outbreak. Clin Infect Dis. 2007;15:44(2):232–237.
- , , , , . Unusual manifestations of acute Q fever: autoimmune hemolytic anemia and tubulointerstitial nephritis. Ann of Clin Microbiol Antimicrob. 2012;11:14.
- , , . Q fever. Lancet. 2006;367(9511):679–688.
- , , , , , . Transitory monoclonal gammopathy and acute Q fever. Enferm Infecc Microbiol Clin. 1995;13(7):442.
- , . Q fever. Clin Microbiol Rev. 1999;12(4):518–553.
- , , , et al. Diagnosis and management of Q fever—United States, 2013: recommendations from CDC and the Q Fever Working Group. MMWR Recomm Rep. 2013;62(RR‐03):1–30.
- , , , et al. Hepatic fibrin‐ring granulomas: a clinicopathologic study of 23 patients. Hum Pathol. 1991;22(6):607–613.
- , , . Travel‐associated zoonotic bacterial diseases. Curr Opin Infect Dis. 2011;24(5):457–463.
A 65‐year‐old man with a 6‐month history of diabetes mellitus presented to the emergency department in May with 1 week of fevers, headaches, myalgia, polydipsia, and polyuria.
The patient presents with symptoms suggestive of uncontrolled diabetes and infection. The broad diagnostic categories include acute infection, an emerging chronic process aggravating his diabetes, or a noninfectious mimic such as autoimmune disease or lymphoproliferative disease. New onset headache in an older patient is concerning. Although it may be attributed to fever and dehydration, primary central nervous system processes such as meningitis or encephalitis must be considered. At this stage, a detailed exposure history, including travel, food, pets, hobbies, and sick contacts as well as occupation and national origins is needed. This patient presented in May, making illnesses that peak in other seasons such as influenza and West Nile fever less likely.
He had no other medical problems except diabetes. He was not taking any medications; he had been started on glipizide but had stopped taking it 1 month prior. He denied fever, cough, chest pain, palpitations, abdominal pain, nausea, vomiting, dysuria, focal weakness, visual changes, or photophobia. He was born in Mexico and emigrated at the age of 25 years. Two months prior to presentation he visited a cattle farm in Mexico; he denied any direct contact with farm animals or dairy products. He denied ill contacts, pets, known tuberculosis exposures, and sexual partners other than his wife.
The history of recent travel to Mexico with a visit to a farm raises concerns about zoonoses. The endemic zoonoses that should be considered include parasitic (toxoplasmosis), fungal (coccidiodomycosis), and bacterial (brucellosis, Q fever, leptospirosis, tularemia, salmonellosis) infections. Nonzoonotic granulomatous infections such as cytomegalovirus (CMV) and Epstein‐Barr virus (EBV), mycobacteria, fungi (histoplasmosis, blastomycosis, cryptococcosis, aspergillosis), and bacteria (actinomycosis) should also be considered.
On examination, he was an elderly Hispanic male who appeared ill but in no acute distress. He was overweight, with a BMI of 29. His temperature was 39C, pulse 66 beats/minute, blood pressure 108/68 mm Hg, respiratory rate 18 per minute, and oxygen saturation was 96% on room air. There were no ulcerations, exudates, or erythema in the oropharynx. There was no sinus tenderness or lymphadenopathy. Cardiac examination revealed normal heart sounds with no murmurs. Respiratory examination demonstrated clear lungs. His abdomen was soft and nontender, whereas the liver and spleen were not palpable. There was no nuchal rigidity, and his mental status was normal. There were no cranial nerve deficits or weakness in his extremities. There was no skin rash or peripheral stigmata of infectious endocarditis. Genitourinary examination revealed no ulcerations, inguinal lymphadenopathy, or urethral discharge. There was no tenderness, warmth, or erythema on examination of all joints.
The physical exam is notable for temperaturepulse dissociation. Heart rate should increase by about 10 beats/minute for every 1‐degree increase in Fahrenheit temperature. The infectious causes of temperaturepulse dissociation are largely intracellular pathogens such as Salmonella, Coxiella, Chlamydia, Leptospira, Legionella, Francisella, Mycoplasma, and dengue virus. This patient is at increased risk for infection by any of these pathogens based on his recent travel to Mexico. Drug fever is the most common noninfectious cause of temperaturepulse dissociation, but this patient took no medications. At this point, a complete blood count and differential, urinalysis, blood cultures, chest x‐ray, and electrocardiogram should be ordered. Testing for human immunodeficiency virus (HIV) is appropriate, as up to 50% of patients with newly diagnosed HIV have no acknowledged risk factors. Serological studies for the aforementioned pathogens may be indicated depending on the results of these initial diagnostic tests.
Serum sodium concentration was 122 mEq/L, potassium 4.0 mEq/L, chloride 88 mEq/L, bicarbonate 14 mEq/L, blood urea nitrogen 17 mg/dL, creatinine 0.7 mg/dL, glucose 402 mg/dL, and calcium 8.5 mg/dL. Total protein was 5.4 g/dL, albumin 2.9 g/dL, total bilirubin 0.9 mg/dL, direct bilirubin 0.4 mg/dL, alkaline phosphatase 126 U/L (normal 53128), gamma‐glutamyl transferase 264 U/L (normal 360), aspartate aminotransferase 51 U/L (normal 840), alanine aminotransferase 62 U/L (normal 556), and lactate dehydrogenase 248 U/L (normal 85210). The white blood cell (WBC) count was 6800 mm3 (51% band forms, 38% segmented neutrophils, 6% monocytes, 5% lymphocytes). The hemoglobin was 15.7 g/dL, with mean corpuscular volume (MCV) of 102 fL and platelet count 59,000/mm3. Peripheral‐blood smear showed occasional macrocytes. Prothrombin time was 13.6 seconds and partial thromboplastin time was 34.5 seconds. C‐reactive protein was 11.8 mg/dL. Urinalysis revealed 80 mg of ketones per deciliter, no cells, and nitrite was negative. Hemoglobin A1c was 13%, and HIV antibody testing was negative.
Elevated circulating bands and thrombocytopenia suggest infection; however, bone marrow infiltration by infectious or neoplastic process is also possible and should be investigated. The increased gamma‐glutamyl transferase, alkaline phosphatase, and mild increases in transaminases suggest hepatic pathology. The combination of unexplained fever, hyponatremia, thrombocytopenia, elevated liver enzymes, and travel to Mexico mandates investigation for infectious diseases that often involve both the bone marrow and liver such as Brucella, Coxiella, and fungal infections such as histoplasmosis. Autoimmune diseases such as systemic lupus erythematosus and malignancy should also be considered. Blood cultures should be incubated beyond the usual 5 days because of the slower growth of Brucella or Salmonella typhi. An HIV viral load should be obtained to evaluate for acute retroviral syndrome. Serologic tests for Rickettsia, Coccidiodes, and hepatitis A, B, and C viruses should be obtained. Urine should be tested for Histoplasma and Legionella antigens. Abdominal imaging should be obtained to evaluate for hepatobiliary disease, occult intra‐abdominal abscess, or malignancy. Because the patient has unexplained fever and headache, imaging of the central nervous system and lumbar puncture are warranted.
His diabetic ketoacidosis (DKA) was treated with intravenous fluids and insulin. Lumbar puncture and cerebrospinal fluid (CSF) analysis revealed opening pressure of 18 cm H20 (normal 1025), cell count WBC 3/L (normal 05), red blood cell 204/L (normal 0), CSF protein 25 mg/dL (normal 2050), and glucose 68 mg/dL (normal 5070). Blood cultures showed no growth. HIV RNA was undetectable. Hepatitis C antibody was negative, and hepatitis A and B serologies were not consistent with an acute infection. Serum ferritin was 1147 ng/mL. Histoplasma and Legionella urine antigen tests were negative. CMV, EBV, and herpes simplex virus DNA were not detected in blood samples. Anti‐neutrophil antibody, anti‐mitochondrial antibody and anti‐neutrophil cytoplasmic antibodies were undetectable. Anti‐smooth muscle antibody was positive at a titer of 1:80. Transthoracic echocardiogram revealed normal heart valves without vegetations. A chest radiograph was normal. Brain computed tomography (CT) revealed atrophic frontal lobes. CT of his chest, abdomen, and pelvis demonstrated focal inflammatory changes of a loop of distal small bowel with surrounding fluid collection, suggesting small bowel diverticulitis. There were no pulmonary infiltrates noted, and the remainder of the CT was unremarkable.
Because the patient remains ill and additional serological test results will take time to return, a key consideration at this point is empiric treatment while awaiting test results. The CSF examination was normal. A history of travel including animal and tick exposures should be reevaluated. The timing of the trip to Mexico was outside the usual incubation period for many pathogens except for Coxiella or Brucella, and empiric therapy for both would be appropriate. The abdominal CT suggests small bowel diverticulitis, which is a rare clinical entity.
The benign abdominal examination suggests the finding is incidental. However, there are several infections that may involve the distal small bowel and proximal colon, such as yersiniosis, salmonellosis, tuberculosis, actinomycosis, histoplasmosis, and noninfectious processes including Crohn's disease and neoplasia. The absence of diarrhea or hematochezia makes yersiniosis, salmonellosis, and Crohn's disease unlikely. Histoplasmosis is unlikely given the negative urine antigen. Evaluation for neoplasia of the distal small bowel requires histologic examination. A colonoscopy with random biopsies of the colon and terminal ileum is the next step if other tests are unrevealing.
The patient was empirically treated for small bowel diverticulitis with ceftriaxone and metronidazole. Because of continued daily fevers as high as 39C, his therapy was changed to vancomycin and piperacillin‐tazobactam to cover methicillin‐resistant Staphylococcus aureus and resistant gram‐negative bacilli. The patient developed new scleral icterus on hospital day 6; the remainder of his examination was unchanged. Serum sodium concentration was 127 mEq/L, potassium 2.7 mEq/L, phosphorus 1.3 mg/dL, magnesium 1.6 mg/dL, total bilirubin 5.6 mg/dL, direct bilirubin 3.6 mg/dL, alkaline phosphatase 193 U/L, gamma‐glutamyl transferase 300 U/L, aspartate aminotransferase 91 U/L, alanine aminotransferase 52 U/L. Brucella serology was negative.
His liver enzymes remain elevated with new onset jaundice consistent with hepatitis and intrahepatic cholestasis. His persistent hypophosphatemia, hypokalemia, and hypomagnesaemia well after resolution of diabetic ketoacidosis suggests acute tubulointerstitial dysfunction, which may be a complication of empiric antibiotic treatment or renal involvement by his underlying condition. Additional blood cultures, and tissue examination and culture are the next appropriate steps. Liver or bone marrow biopsy may suggest a diagnosis that can be confirmed by tissue culture or immunohistochemistry. Histologic findings such as fibrin ringed granulomas, caseating or noncaseating granulomas, or lymphomatous infiltration may suggest Coxiella (Q fever), tuberculosis, or lymphoma respectively. Because a liver biopsy is invasive and usually provides less tissue for culture, bone marrow examination should be obtained first.
A gallium 67 scan showed nonhomogenous increased uptake in both lungs and kidneys, consistent with interstitial nephritis and bilateral pneumonia. Serum protein electrophoresis demonstrated a monoclonal immunoglobulin (Ig)G lambda band with a kappa/lambda ratio of 0.9 (normal 1.42.8). Bone marrow biopsy showed normal hematopoiesis; no plasma or malignant cells, granulomas, or evidence of hemophagocytosis; and fungal and mycobacterial stains and cultures were negative. Colonoscopy revealed normal‐appearing mucosa. Histologic examination and culture of random biopsies from the colon and terminal ileum were negative for fungi, viruses, and mycobacteria. An ultrasound‐guided liver biopsy revealed numerous noncaseating granulomas formed of histiocytes and neutrophils with occasional fibrin rings. Fungal, viral, and mycobacterial stains and cultures were negative. The patient's fever resolved after 14 days, and he was discharged home without a diagnosis and close outpatient follow‐up.
The hepatic granulomas with fibrin rings are highly suggestive of Q fever, although ring granulomas may be seen in tuberculosis, typhoid fever, lymphoma, drug reactions, sarcoidosis, and CMV infections. Competing diagnoses such as CMV have been excluded by negative serology. Microscopic examination, tissue staining, and culture from liver and bone marrow biopsies were negative for S typhi, mycobacteria, and lymphoma. Gallium scan findings are generally nonspecific and of little utility in cases such as this. The kidney involvement correlates with the biochemical evidence of tubulointerstitial dysfunction; pulmonary involvement may reflect subclinical pulmonary infection with Coxiella. Given the normal bone marrow biopsy, the monoclonal gammopathy is of undetermined significance. The positive anti‐smooth muscle antibody can be related to Q fever. Anti‐smooth muscle antibodies frequently occur in Q fever, especially in those patients with hepatitis. Given the history of exposure to cattle, unexplained fever with temperaturepulse dissociation and liver biopsy findings, Q fever is the most likely diagnosis and empiric treatment with doxycycline is warranted.
Results of serology for Coxiella burnetii sent during admission were returned after the patient's discharge. C burnetii phase I IgG and IgM antibody titers were positive (1:512 each). C burnetii phase II IgG and IgM titers also were positive (1:1024 each). The patient was seen within a week and started on doxycycline 100 mg twice daily for 2 weeks for acute Q fever. His symptoms improved; hyponatremia, liver function tests, and thrombocytopenia normalized after treatment.
DISCUSSION
Q fever was first described in 1937 as a febrile illness affecting Australian slaughterhouse workers.[1] The Q in Q fever stands for query and reflected the initial uncertainty surrounding the underlying cause of the illness. The causative organism, C burnetti, is an obligate intracellular bacterium that resides within macrophage lysosomes. It can be found in the urine, feces, milk, placenta, and amniotic fluid of ungulates (cattle, sheep, and other ruminants), and other animals such as domestic cats and dogs. C burnetii is transmitted via inhalation, ingestion, occupational, or common source exposures, and in 1 case report by person‐to‐person sexual transmission.[2] In addition to slaughterhouse workers, pregnant women and immunosuppressed patients are more susceptible to developing Q fever.[3] For patients with suspected Q fever, a detailed occupational history, including specific job duties and potential exposure to animal products, is imperative.
Q fever has both acute and chronic presentations, which are differentiated based on the clinical illness and serologies. The symptoms of acute Q fever are nonspecific and may include influenza‐like illness, fever, pneumonia, and hepatitis. It presents less commonly with hemolytic anemia, interstitial nephritis, monoclonal gammopathy, or aseptic meningitis.[4, 5, 6, 7] Symptoms typically begin between 1 and 3 weeks after animal exposure and may persist for several months. Chronic Q fever occurs when unrecognized or untreated infection persists for greater than 6 months. It commonly presents with culture‐negative endocarditis, although infected aneurysms, osteomyelitis, or other distant sites of infection may also occur.
C burnetti is present in 2 antigenic forms that can be assessed by serology. Phase I is the more virulent, infectious form of C burnetti, which transitions to the avirulent phase II form during laboratory handling. In acute Q fever, phase II serologies are typically elevated out of proportion to phase I serologies, whereas this pattern is reversed in chronic Q fever. The diagnostic gold standard of acute Q fever is a 4‐fold rise in phase II antibody titers taken 3 to 6 weeks apart.[8] Histologic examination of affected organs can support a diagnosis of Q fever. The presence of ringed granulomas on liver or bone marrow biopsy specimens is highly suggestive, but not pathognomonic, of Q fever.[9]
Q fever is highly susceptible to several classes of antibiotics. For acute Q fever, doxycycline and tetracycline are typically used, with fluoroquinolones and chloramphenicol as alternatives.[8, 10] Patients with chronic Q fever should be treated with doxycycline and hydroxychloroquine. The addition of hydroxychloroquine alkalinizes the macrophage lysosome and enhances bacterial eradication.[8] For patients with acute Q fever, physicians should determine the risk of progression to chronic Q fever because closer monitoring is necessary. Patients with valvular heart lesions, immunosuppression, and pregnant women are at elevated risk of chronic Q fever. Trimethoprim/sulfamethoxazole can be used in place of doxycycline in pregnant women, as doxycycline and fluoroquinolones are contraindicated in pregnancy.[8]
This patient presented with a nonspecific febrile illness. Although the treating clinicians obtained a history of exposure to cattle early in his course, both the diagnosis and treatment were delayed. There are several possible explanations for the delay. First, although Q fever is a relatively common zoonosis, it remains an uncommon diagnosis, particularly among hospitalized patients. As a result, clinicians often focus on more common conditions. In this case, typical infections, malignancies, and inflammatory diseases were considered more likely. Second, the patient presented with hepatitis, an uncommon presentation of Q fever. Classical clinical reasoning suggests that atypical presentations of common diseases will occur more frequently than typical presentations of uncommon diseases. This case presented with an atypical presentation of an uncommon disease. The resultant lower pretest probability further dissuaded the patient's physicians from consideration of Q fever. Third, the finding of small bowel diverticulitis was a potential distractor. In patients with nonspecific febrile illnesses, it is common for physicians to anchor on any abnormal findings. In this case, the small bowel diverticulitis led to antibiotic treatment that was ineffective against C burnetti.
There were several clues to the diagnosis of Q fever in this patient's presentation. First, the pulsetemperature dissociation suggested infection with an intracellular pathogen. Hospitalists should recognize this association and be mindful of this often‐subtle clinical finding when faced with diagnostic uncertainty. Second, the patient was exposed to cattle prior to the onset of his illness. The fact that he did not have a direct exposure to animals underscores the infectivity of C burnetti. Finally, elevated alkaline phosphatase and transaminases were suggestive of an infiltrative disease; in the setting of a nonspecific febrile illness, Q fever was an important diagnostic consideration.
The key treatment decision in this case was the initiation and choice of antibiotics. Because of this patient's history of exposure to cattle and lack of a compelling alternative diagnosis, empiric treatment with doxycycline would have been appropriate. Hospitalists must weigh the potential benefit of early treatment of Q fever against the risks associated with antibiotic overuse. In patients presenting with a febrile illness after ungulate exposure, the decision to bet the farm with empiric doxycycline therapy may lead to clinical improvement, obviating a more invasive or extensive diagnostic evaluation.
TEACHING POINTS
- Acute Q fever typically presents 2 to 3 weeks after ungulate exposure with a febrile illness, pneumonia, and granulomatous hepatitis.
- Pulsetemperature dissociation is suggestive of infection by intracellular pathogens such as Coxiella, Salmonella, Leptospira, Legionella, and Mycoplasma.
- Clinicians should consider empiric doxycycline therapy in patients with suspected zoonosis (eg, Q fever, brucellosis, anaplasmosis, leptospirosis, Rocky Mountain spotted fever) while awaiting confirmatory tests, as improvement may obviate invasive testing.
Disclosure: Nothing to report.
A 65‐year‐old man with a 6‐month history of diabetes mellitus presented to the emergency department in May with 1 week of fevers, headaches, myalgia, polydipsia, and polyuria.
The patient presents with symptoms suggestive of uncontrolled diabetes and infection. The broad diagnostic categories include acute infection, an emerging chronic process aggravating his diabetes, or a noninfectious mimic such as autoimmune disease or lymphoproliferative disease. New onset headache in an older patient is concerning. Although it may be attributed to fever and dehydration, primary central nervous system processes such as meningitis or encephalitis must be considered. At this stage, a detailed exposure history, including travel, food, pets, hobbies, and sick contacts as well as occupation and national origins is needed. This patient presented in May, making illnesses that peak in other seasons such as influenza and West Nile fever less likely.
He had no other medical problems except diabetes. He was not taking any medications; he had been started on glipizide but had stopped taking it 1 month prior. He denied fever, cough, chest pain, palpitations, abdominal pain, nausea, vomiting, dysuria, focal weakness, visual changes, or photophobia. He was born in Mexico and emigrated at the age of 25 years. Two months prior to presentation he visited a cattle farm in Mexico; he denied any direct contact with farm animals or dairy products. He denied ill contacts, pets, known tuberculosis exposures, and sexual partners other than his wife.
The history of recent travel to Mexico with a visit to a farm raises concerns about zoonoses. The endemic zoonoses that should be considered include parasitic (toxoplasmosis), fungal (coccidiodomycosis), and bacterial (brucellosis, Q fever, leptospirosis, tularemia, salmonellosis) infections. Nonzoonotic granulomatous infections such as cytomegalovirus (CMV) and Epstein‐Barr virus (EBV), mycobacteria, fungi (histoplasmosis, blastomycosis, cryptococcosis, aspergillosis), and bacteria (actinomycosis) should also be considered.
On examination, he was an elderly Hispanic male who appeared ill but in no acute distress. He was overweight, with a BMI of 29. His temperature was 39C, pulse 66 beats/minute, blood pressure 108/68 mm Hg, respiratory rate 18 per minute, and oxygen saturation was 96% on room air. There were no ulcerations, exudates, or erythema in the oropharynx. There was no sinus tenderness or lymphadenopathy. Cardiac examination revealed normal heart sounds with no murmurs. Respiratory examination demonstrated clear lungs. His abdomen was soft and nontender, whereas the liver and spleen were not palpable. There was no nuchal rigidity, and his mental status was normal. There were no cranial nerve deficits or weakness in his extremities. There was no skin rash or peripheral stigmata of infectious endocarditis. Genitourinary examination revealed no ulcerations, inguinal lymphadenopathy, or urethral discharge. There was no tenderness, warmth, or erythema on examination of all joints.
The physical exam is notable for temperaturepulse dissociation. Heart rate should increase by about 10 beats/minute for every 1‐degree increase in Fahrenheit temperature. The infectious causes of temperaturepulse dissociation are largely intracellular pathogens such as Salmonella, Coxiella, Chlamydia, Leptospira, Legionella, Francisella, Mycoplasma, and dengue virus. This patient is at increased risk for infection by any of these pathogens based on his recent travel to Mexico. Drug fever is the most common noninfectious cause of temperaturepulse dissociation, but this patient took no medications. At this point, a complete blood count and differential, urinalysis, blood cultures, chest x‐ray, and electrocardiogram should be ordered. Testing for human immunodeficiency virus (HIV) is appropriate, as up to 50% of patients with newly diagnosed HIV have no acknowledged risk factors. Serological studies for the aforementioned pathogens may be indicated depending on the results of these initial diagnostic tests.
Serum sodium concentration was 122 mEq/L, potassium 4.0 mEq/L, chloride 88 mEq/L, bicarbonate 14 mEq/L, blood urea nitrogen 17 mg/dL, creatinine 0.7 mg/dL, glucose 402 mg/dL, and calcium 8.5 mg/dL. Total protein was 5.4 g/dL, albumin 2.9 g/dL, total bilirubin 0.9 mg/dL, direct bilirubin 0.4 mg/dL, alkaline phosphatase 126 U/L (normal 53128), gamma‐glutamyl transferase 264 U/L (normal 360), aspartate aminotransferase 51 U/L (normal 840), alanine aminotransferase 62 U/L (normal 556), and lactate dehydrogenase 248 U/L (normal 85210). The white blood cell (WBC) count was 6800 mm3 (51% band forms, 38% segmented neutrophils, 6% monocytes, 5% lymphocytes). The hemoglobin was 15.7 g/dL, with mean corpuscular volume (MCV) of 102 fL and platelet count 59,000/mm3. Peripheral‐blood smear showed occasional macrocytes. Prothrombin time was 13.6 seconds and partial thromboplastin time was 34.5 seconds. C‐reactive protein was 11.8 mg/dL. Urinalysis revealed 80 mg of ketones per deciliter, no cells, and nitrite was negative. Hemoglobin A1c was 13%, and HIV antibody testing was negative.
Elevated circulating bands and thrombocytopenia suggest infection; however, bone marrow infiltration by infectious or neoplastic process is also possible and should be investigated. The increased gamma‐glutamyl transferase, alkaline phosphatase, and mild increases in transaminases suggest hepatic pathology. The combination of unexplained fever, hyponatremia, thrombocytopenia, elevated liver enzymes, and travel to Mexico mandates investigation for infectious diseases that often involve both the bone marrow and liver such as Brucella, Coxiella, and fungal infections such as histoplasmosis. Autoimmune diseases such as systemic lupus erythematosus and malignancy should also be considered. Blood cultures should be incubated beyond the usual 5 days because of the slower growth of Brucella or Salmonella typhi. An HIV viral load should be obtained to evaluate for acute retroviral syndrome. Serologic tests for Rickettsia, Coccidiodes, and hepatitis A, B, and C viruses should be obtained. Urine should be tested for Histoplasma and Legionella antigens. Abdominal imaging should be obtained to evaluate for hepatobiliary disease, occult intra‐abdominal abscess, or malignancy. Because the patient has unexplained fever and headache, imaging of the central nervous system and lumbar puncture are warranted.
His diabetic ketoacidosis (DKA) was treated with intravenous fluids and insulin. Lumbar puncture and cerebrospinal fluid (CSF) analysis revealed opening pressure of 18 cm H20 (normal 1025), cell count WBC 3/L (normal 05), red blood cell 204/L (normal 0), CSF protein 25 mg/dL (normal 2050), and glucose 68 mg/dL (normal 5070). Blood cultures showed no growth. HIV RNA was undetectable. Hepatitis C antibody was negative, and hepatitis A and B serologies were not consistent with an acute infection. Serum ferritin was 1147 ng/mL. Histoplasma and Legionella urine antigen tests were negative. CMV, EBV, and herpes simplex virus DNA were not detected in blood samples. Anti‐neutrophil antibody, anti‐mitochondrial antibody and anti‐neutrophil cytoplasmic antibodies were undetectable. Anti‐smooth muscle antibody was positive at a titer of 1:80. Transthoracic echocardiogram revealed normal heart valves without vegetations. A chest radiograph was normal. Brain computed tomography (CT) revealed atrophic frontal lobes. CT of his chest, abdomen, and pelvis demonstrated focal inflammatory changes of a loop of distal small bowel with surrounding fluid collection, suggesting small bowel diverticulitis. There were no pulmonary infiltrates noted, and the remainder of the CT was unremarkable.
Because the patient remains ill and additional serological test results will take time to return, a key consideration at this point is empiric treatment while awaiting test results. The CSF examination was normal. A history of travel including animal and tick exposures should be reevaluated. The timing of the trip to Mexico was outside the usual incubation period for many pathogens except for Coxiella or Brucella, and empiric therapy for both would be appropriate. The abdominal CT suggests small bowel diverticulitis, which is a rare clinical entity.
The benign abdominal examination suggests the finding is incidental. However, there are several infections that may involve the distal small bowel and proximal colon, such as yersiniosis, salmonellosis, tuberculosis, actinomycosis, histoplasmosis, and noninfectious processes including Crohn's disease and neoplasia. The absence of diarrhea or hematochezia makes yersiniosis, salmonellosis, and Crohn's disease unlikely. Histoplasmosis is unlikely given the negative urine antigen. Evaluation for neoplasia of the distal small bowel requires histologic examination. A colonoscopy with random biopsies of the colon and terminal ileum is the next step if other tests are unrevealing.
The patient was empirically treated for small bowel diverticulitis with ceftriaxone and metronidazole. Because of continued daily fevers as high as 39C, his therapy was changed to vancomycin and piperacillin‐tazobactam to cover methicillin‐resistant Staphylococcus aureus and resistant gram‐negative bacilli. The patient developed new scleral icterus on hospital day 6; the remainder of his examination was unchanged. Serum sodium concentration was 127 mEq/L, potassium 2.7 mEq/L, phosphorus 1.3 mg/dL, magnesium 1.6 mg/dL, total bilirubin 5.6 mg/dL, direct bilirubin 3.6 mg/dL, alkaline phosphatase 193 U/L, gamma‐glutamyl transferase 300 U/L, aspartate aminotransferase 91 U/L, alanine aminotransferase 52 U/L. Brucella serology was negative.
His liver enzymes remain elevated with new onset jaundice consistent with hepatitis and intrahepatic cholestasis. His persistent hypophosphatemia, hypokalemia, and hypomagnesaemia well after resolution of diabetic ketoacidosis suggests acute tubulointerstitial dysfunction, which may be a complication of empiric antibiotic treatment or renal involvement by his underlying condition. Additional blood cultures, and tissue examination and culture are the next appropriate steps. Liver or bone marrow biopsy may suggest a diagnosis that can be confirmed by tissue culture or immunohistochemistry. Histologic findings such as fibrin ringed granulomas, caseating or noncaseating granulomas, or lymphomatous infiltration may suggest Coxiella (Q fever), tuberculosis, or lymphoma respectively. Because a liver biopsy is invasive and usually provides less tissue for culture, bone marrow examination should be obtained first.
A gallium 67 scan showed nonhomogenous increased uptake in both lungs and kidneys, consistent with interstitial nephritis and bilateral pneumonia. Serum protein electrophoresis demonstrated a monoclonal immunoglobulin (Ig)G lambda band with a kappa/lambda ratio of 0.9 (normal 1.42.8). Bone marrow biopsy showed normal hematopoiesis; no plasma or malignant cells, granulomas, or evidence of hemophagocytosis; and fungal and mycobacterial stains and cultures were negative. Colonoscopy revealed normal‐appearing mucosa. Histologic examination and culture of random biopsies from the colon and terminal ileum were negative for fungi, viruses, and mycobacteria. An ultrasound‐guided liver biopsy revealed numerous noncaseating granulomas formed of histiocytes and neutrophils with occasional fibrin rings. Fungal, viral, and mycobacterial stains and cultures were negative. The patient's fever resolved after 14 days, and he was discharged home without a diagnosis and close outpatient follow‐up.
The hepatic granulomas with fibrin rings are highly suggestive of Q fever, although ring granulomas may be seen in tuberculosis, typhoid fever, lymphoma, drug reactions, sarcoidosis, and CMV infections. Competing diagnoses such as CMV have been excluded by negative serology. Microscopic examination, tissue staining, and culture from liver and bone marrow biopsies were negative for S typhi, mycobacteria, and lymphoma. Gallium scan findings are generally nonspecific and of little utility in cases such as this. The kidney involvement correlates with the biochemical evidence of tubulointerstitial dysfunction; pulmonary involvement may reflect subclinical pulmonary infection with Coxiella. Given the normal bone marrow biopsy, the monoclonal gammopathy is of undetermined significance. The positive anti‐smooth muscle antibody can be related to Q fever. Anti‐smooth muscle antibodies frequently occur in Q fever, especially in those patients with hepatitis. Given the history of exposure to cattle, unexplained fever with temperaturepulse dissociation and liver biopsy findings, Q fever is the most likely diagnosis and empiric treatment with doxycycline is warranted.
Results of serology for Coxiella burnetii sent during admission were returned after the patient's discharge. C burnetii phase I IgG and IgM antibody titers were positive (1:512 each). C burnetii phase II IgG and IgM titers also were positive (1:1024 each). The patient was seen within a week and started on doxycycline 100 mg twice daily for 2 weeks for acute Q fever. His symptoms improved; hyponatremia, liver function tests, and thrombocytopenia normalized after treatment.
DISCUSSION
Q fever was first described in 1937 as a febrile illness affecting Australian slaughterhouse workers.[1] The Q in Q fever stands for query and reflected the initial uncertainty surrounding the underlying cause of the illness. The causative organism, C burnetti, is an obligate intracellular bacterium that resides within macrophage lysosomes. It can be found in the urine, feces, milk, placenta, and amniotic fluid of ungulates (cattle, sheep, and other ruminants), and other animals such as domestic cats and dogs. C burnetii is transmitted via inhalation, ingestion, occupational, or common source exposures, and in 1 case report by person‐to‐person sexual transmission.[2] In addition to slaughterhouse workers, pregnant women and immunosuppressed patients are more susceptible to developing Q fever.[3] For patients with suspected Q fever, a detailed occupational history, including specific job duties and potential exposure to animal products, is imperative.
Q fever has both acute and chronic presentations, which are differentiated based on the clinical illness and serologies. The symptoms of acute Q fever are nonspecific and may include influenza‐like illness, fever, pneumonia, and hepatitis. It presents less commonly with hemolytic anemia, interstitial nephritis, monoclonal gammopathy, or aseptic meningitis.[4, 5, 6, 7] Symptoms typically begin between 1 and 3 weeks after animal exposure and may persist for several months. Chronic Q fever occurs when unrecognized or untreated infection persists for greater than 6 months. It commonly presents with culture‐negative endocarditis, although infected aneurysms, osteomyelitis, or other distant sites of infection may also occur.
C burnetti is present in 2 antigenic forms that can be assessed by serology. Phase I is the more virulent, infectious form of C burnetti, which transitions to the avirulent phase II form during laboratory handling. In acute Q fever, phase II serologies are typically elevated out of proportion to phase I serologies, whereas this pattern is reversed in chronic Q fever. The diagnostic gold standard of acute Q fever is a 4‐fold rise in phase II antibody titers taken 3 to 6 weeks apart.[8] Histologic examination of affected organs can support a diagnosis of Q fever. The presence of ringed granulomas on liver or bone marrow biopsy specimens is highly suggestive, but not pathognomonic, of Q fever.[9]
Q fever is highly susceptible to several classes of antibiotics. For acute Q fever, doxycycline and tetracycline are typically used, with fluoroquinolones and chloramphenicol as alternatives.[8, 10] Patients with chronic Q fever should be treated with doxycycline and hydroxychloroquine. The addition of hydroxychloroquine alkalinizes the macrophage lysosome and enhances bacterial eradication.[8] For patients with acute Q fever, physicians should determine the risk of progression to chronic Q fever because closer monitoring is necessary. Patients with valvular heart lesions, immunosuppression, and pregnant women are at elevated risk of chronic Q fever. Trimethoprim/sulfamethoxazole can be used in place of doxycycline in pregnant women, as doxycycline and fluoroquinolones are contraindicated in pregnancy.[8]
This patient presented with a nonspecific febrile illness. Although the treating clinicians obtained a history of exposure to cattle early in his course, both the diagnosis and treatment were delayed. There are several possible explanations for the delay. First, although Q fever is a relatively common zoonosis, it remains an uncommon diagnosis, particularly among hospitalized patients. As a result, clinicians often focus on more common conditions. In this case, typical infections, malignancies, and inflammatory diseases were considered more likely. Second, the patient presented with hepatitis, an uncommon presentation of Q fever. Classical clinical reasoning suggests that atypical presentations of common diseases will occur more frequently than typical presentations of uncommon diseases. This case presented with an atypical presentation of an uncommon disease. The resultant lower pretest probability further dissuaded the patient's physicians from consideration of Q fever. Third, the finding of small bowel diverticulitis was a potential distractor. In patients with nonspecific febrile illnesses, it is common for physicians to anchor on any abnormal findings. In this case, the small bowel diverticulitis led to antibiotic treatment that was ineffective against C burnetti.
There were several clues to the diagnosis of Q fever in this patient's presentation. First, the pulsetemperature dissociation suggested infection with an intracellular pathogen. Hospitalists should recognize this association and be mindful of this often‐subtle clinical finding when faced with diagnostic uncertainty. Second, the patient was exposed to cattle prior to the onset of his illness. The fact that he did not have a direct exposure to animals underscores the infectivity of C burnetti. Finally, elevated alkaline phosphatase and transaminases were suggestive of an infiltrative disease; in the setting of a nonspecific febrile illness, Q fever was an important diagnostic consideration.
The key treatment decision in this case was the initiation and choice of antibiotics. Because of this patient's history of exposure to cattle and lack of a compelling alternative diagnosis, empiric treatment with doxycycline would have been appropriate. Hospitalists must weigh the potential benefit of early treatment of Q fever against the risks associated with antibiotic overuse. In patients presenting with a febrile illness after ungulate exposure, the decision to bet the farm with empiric doxycycline therapy may lead to clinical improvement, obviating a more invasive or extensive diagnostic evaluation.
TEACHING POINTS
- Acute Q fever typically presents 2 to 3 weeks after ungulate exposure with a febrile illness, pneumonia, and granulomatous hepatitis.
- Pulsetemperature dissociation is suggestive of infection by intracellular pathogens such as Coxiella, Salmonella, Leptospira, Legionella, and Mycoplasma.
- Clinicians should consider empiric doxycycline therapy in patients with suspected zoonosis (eg, Q fever, brucellosis, anaplasmosis, leptospirosis, Rocky Mountain spotted fever) while awaiting confirmatory tests, as improvement may obviate invasive testing.
Disclosure: Nothing to report.
- . “Q” fever, a new fever entity: clinical features, diagnosis, and laboratory investigation. Rev Infect Dis. 1983;5(4):790–800.
- , , , . Q fever: a biological weapon in your backyard. Lancet Infect Dis. 2003;3(11):709–721.
- , , , . Role of sex, age, previous valve lesion, and pregnancy in the clinical expression and outcome of Q fever after a large outbreak. Clin Infect Dis. 2007;15:44(2):232–237.
- , , , , . Unusual manifestations of acute Q fever: autoimmune hemolytic anemia and tubulointerstitial nephritis. Ann of Clin Microbiol Antimicrob. 2012;11:14.
- , , . Q fever. Lancet. 2006;367(9511):679–688.
- , , , , , . Transitory monoclonal gammopathy and acute Q fever. Enferm Infecc Microbiol Clin. 1995;13(7):442.
- , . Q fever. Clin Microbiol Rev. 1999;12(4):518–553.
- , , , et al. Diagnosis and management of Q fever—United States, 2013: recommendations from CDC and the Q Fever Working Group. MMWR Recomm Rep. 2013;62(RR‐03):1–30.
- , , , et al. Hepatic fibrin‐ring granulomas: a clinicopathologic study of 23 patients. Hum Pathol. 1991;22(6):607–613.
- , , . Travel‐associated zoonotic bacterial diseases. Curr Opin Infect Dis. 2011;24(5):457–463.
- . “Q” fever, a new fever entity: clinical features, diagnosis, and laboratory investigation. Rev Infect Dis. 1983;5(4):790–800.
- , , , . Q fever: a biological weapon in your backyard. Lancet Infect Dis. 2003;3(11):709–721.
- , , , . Role of sex, age, previous valve lesion, and pregnancy in the clinical expression and outcome of Q fever after a large outbreak. Clin Infect Dis. 2007;15:44(2):232–237.
- , , , , . Unusual manifestations of acute Q fever: autoimmune hemolytic anemia and tubulointerstitial nephritis. Ann of Clin Microbiol Antimicrob. 2012;11:14.
- , , . Q fever. Lancet. 2006;367(9511):679–688.
- , , , , , . Transitory monoclonal gammopathy and acute Q fever. Enferm Infecc Microbiol Clin. 1995;13(7):442.
- , . Q fever. Clin Microbiol Rev. 1999;12(4):518–553.
- , , , et al. Diagnosis and management of Q fever—United States, 2013: recommendations from CDC and the Q Fever Working Group. MMWR Recomm Rep. 2013;62(RR‐03):1–30.
- , , , et al. Hepatic fibrin‐ring granulomas: a clinicopathologic study of 23 patients. Hum Pathol. 1991;22(6):607–613.
- , , . Travel‐associated zoonotic bacterial diseases. Curr Opin Infect Dis. 2011;24(5):457–463.
What Are Best Practices for Patients Discharged against Medical Advice?
Case No. 1
A 41-year-old woman with a history of asthma presents to the emergency department (ED) with shortness of breath and wheezing. She is diagnosed with a mild asthma exacerbation. After three albuterol nebulizer treatments, she still has wheezing on physical examination but appears comfortable and has no oxygen requirement. She has a primary medical doctor at the hospital and follows up with her regularly.

The hospitalist recommends that she stay in the hospital for further treatment, but the patient says she has a nebulizer machine at home and asks to be discharged. In addition, she is worried about her frail elderly mother, for whom she is the primary caretaker. The hospitalist acknowledges her concerns but continues to recommend that she remain in the hospital for additional care and monitoring. She becomes visibly upset and insists that she must return home. She asks for prescriptions for albuterol and prednisone and is discharged against medical advice (AMA).
Case No. 2
A 52-year-old man with a history of hypertension and diabetes presents to the ED with left foot pain. He frequently presents with this complaint but often leaves AMA before treatment is completed. He has no known physical address or telephone number and has no known outpatient healthcare providers. Physical examination reveals several ulcers on the dorsum of the foot, one with purulent drainage, and generalized lower extremity pallor. His left leg is cool to the touch, and vascular surgery is consulted for suspected limb-threatening ischemia; IV antibiotics are started for suspected osteomyelitis.
During the interview, he states that he wishes to leave the hospital because he has “things to take care of.” The hospitalist recommends that he remain in the hospital for limb-preserving surgery and antibiotics. He then explains that he is homeless and needs to return to his shelter to keep his bed. He is able to articulate the risks of premature discharge and the medical concerns, and it is determined that he has the capacity to participate in discharge planning. The hospitalist therefore discharges him AMA.
Background
AMA discharges represent 1%–2% of all inpatient discharges.¹,² Despite being a small percentage of total discharges, these patients have disproportionately high healthcare costs. One study reported that healthcare costs among these patients were 56% higher than expected.² Furthermore, AMA patients suffer higher than expected rates of morbidity, mortality, and hospital readmission.
For example, in one case-control study in an urban teaching hospital, patients discharged AMA from the general medicine service had a 21% 15-day readmission rate compared to a 3% readmission rate among age, gender, and diagnosis-matched controls.3,4,5
Additionally, history of AMA discharge appears to confer risk of increased future utilization of healthcare resources. In a cohort study of hospital admissions among HIV-infected patients with high rates of intravenous drug abuse, patients discharged AMA (13% of the cohort) were not only more likely to be readmitted within 30 days for a related diagnosis (odds ratio = 5.0) but also were more likely to have increased length of stay during the year following the index admission.6
These studies highlight the barriers to safe and effective transitions of care for this vulnerable population and demonstrate the increased burden that this population places on the health system.
Several retrospective studies have identified psychosocial and demographic risk factors associated with AMA discharge. These include younger age, male sex, substance abuse, lack of a primary care physician or health insurance, and history of previous AMA discharge.1,3,7,8 Insurance status is also associated with AMA discharge, with increased odds of AMA discharge among Medicare and Medicaid patients and patients without health insurance.9,10
Of note, one study found that race did not act as an independent predictor of AMA discharge when adjusted for age, gender, and socioeconomic factors.11
The AMA population is clinically heterogeneous. Among patients with pneumonia, for example, Saitz et al showed that a patient’s documented clinical severity did not independently predict AMA discharge, suggesting that there is great clinical heterogeneity even among AMA patients with similar admission diagnoses.12
These studies highlight the clinical and demographic heterogeneity within this population, suggesting that patients discharged AMA require individualized attention from hospitalists and other healthcare providers.
Patients describe numerous motivations for leaving the hospital prematurely, including needing to pick up public-assistance checks, personal financial issues, and familial obligations.13 Interestingly, in the cohort of HIV patients referenced above, discharge on the day welfare checks were distributed was an independent predictor of AMA discharge.6 In focus groups composed of patients discharged AMA and their treating nurses and physicians, several themes were identified as potential contributors to AMA discharge, including drug addiction, pain management issues, external obligations, wait time, the physician’s bedside manner, being in a teaching hospital, and communication issues.14
Clearly, patients have a diversity of reasons for requesting to be discharged AMA, and further research is necessary to define clear and potentially modifiable risk factors.
Discussion
The clinical scenarios outlined above present two patients with very different clinical presentations and outpatient support systems as well as demonstrate the great variability in clinical risk at the time of discharge AMA. These examples emphasize the importance of an individualized approach to care for each patient.
In Case No. 1, the patient is admitted with a mild asthma exacerbation with persistent bronchospasm, though she clinically appears well and has reliable follow-up. In contrast, in Case No. 2, the patient has life-threatening disease and no established primary care physician or mechanism for outpatient care. These examples demonstrate extremes on the clinical and psychosocial spectrum of patients requesting an “early” discharge and suggest that no two patients at risk of AMA discharge are the same. Patient 1 could likely be safely managed at home with close outpatient follow-up, while Patient 2 presents a high-risk scenario with very few safe outpatient treatment options.
We suggest that an individualized approach be taken for each patient, with attention to both clinical and psychosocial risk. In clinically low-risk cases (e.g., Case No. 1), an approach that prioritizes shared decision making and coordination with the outpatient care team may be preferable to an AMA discharge, particularly given the often adversarial nature of the later.2 In such cases, a collaborative approach may provide greater opportunity for harm reduction, provision of appropriate prescriptions, and follow-up appointments. In clinically high-risk patients such as Case No. 2, however, premature discharge is clearly inappropriate. Even in such clinically high-risk cases, however, we argue that a collaborative strategy aimed at identifying and addressing the patient’s psychosocial concerns is appropriate, as such an approach promotes shared decision making, builds trust between the patient and the care team, and therefore may facilitate improved follow-up at the time of discharge. Research is needed to formally assess the optimal approach for this patient population, including impact on rates of AMA discharge and the quality of post-discharge follow-up.
At present, the decision to classify a discharge as AMA falls solely on the treating provider, and we suspect that there is great variability in practice patterns, particularly as there are few established professional society practice guidelines regarding this difficult issue. As with all discharges from the hospital, the burden falls on the provider to engage the patient in shared decision making and ensure that the patient has the capacity to understand the risks and benefits of the proposed treatment plan. It is in this spirit that simply “filling out an AMA form” does not provide legal protection to a physician who does not adequately explain the full risks and benefits of refusal of inpatient treatment.2,15
We propose that a high-quality AMA discharge be defined as a discharge in which the patient is informed of the clinical team’s determination that further hospitalization is required but elects to leave the hospital, and it includes a clear discussion of the risks of outpatient treatment, a determination of capacity, and an exploration of safe alternative care plans that could satisfy both the patient’s medical and social needs. This definition places the burden on hospitalists and other providers to fully explore the motivations behind a patient’s request to leave the hospital and treats psychosocial motivators for premature discharge as variables in the complex risk-benefit analysis that underlies the informed consent discussion prior to AMA discharge.
Furthermore, AMA discharge does not obviate a physician’s responsibility to advocate for a patient’s well-being, and therefore an AMA discharge should be accompanied by reasonable efforts to coordinate a patient’s ongoing outpatient care. Of note, this approach is consistent with previous reviews and attempts to balance the physician’s duty to honor a patient’s autonomy with the responsibility to protect the patient from harm.2,16
Conclusion
Patients discharged AMA are a diverse population at markedly increased risk of morbidity, readmissions, and subsequent healthcare cost. We argue that in all cases of a potential premature discharge, a collaborative and patient-centered approach is crucial. Such an approach allows the provider to identify and address the patient’s concerns regarding further inpatient care, to explore possible safe outpatient treatment options, to document patient capacity, and to provide appropriate harm-reduction measures such as prescriptions.
Further research into the current practice patterns of hospitalists and other providers is necessary to allow for the formulation and adoption of best practices and implementation of appropriate harm-reduction strategies. TH
Dr. Tummalapalli is an internal medicine resident in the department of medicine at Icahn School of Medicine at Mount Sinai in New York City. Dr. Goodman is a hospitalist in the division of hospital medicine, department of medicine, at the Icahn School of Medicine at Mount Sinai.
References
- Aliyu ZY. Discharge against medical advice: sociodemographic, clinical and financial perspectives. Int J Clin Pract. 2002;56(5):325-327.
- Kahle CH, Rubio ML, Santos RA. Discharges against medical advice: considerations for the hospitalist and the patient. Hospital Medicine Clinics. 2015;4(3):421-429.
- Baptist AP, Warrier I, Arora R, Ager J, Massanari RM. Hospitalized patients with asthma who leave against medical advice: characteristics, reasons, and outcomes. J Allergy Clin Immunol. 2007;119(4):924-929.
- Hwang SW, Li J, Gupta R, Chien V, Martin RE. What happens to patients who leave hospital against medical advice? CMAJ. 2003;168(4):417-420.
- Glasgow JM, Vaughn-Sarrazin MV, Kaboli PJ. Leaving against medical advice (AMA): risk of 30-day mortality and hospital readmission. J Gen Int Med. 2010;25(9):926-929.
- Anis AH, Sun H, Guh DP, Palepu A, Schechter MT, O’Shaughnessy MV. Leaving hospital against medical advice among HIV-positive patients. CMAJ. 2002;167(6):633-637.
- Jeremiah J, O’Sullivan P, Stein MD. Who leaves against medical advice? J Gen Int Med. 1995; 10(7);403-405.
- O’Hara D, Hart W, McDonald I. Leaving hospital against medical advice. J Qual Clin Pract.1996;16(3):157-164.
- Ibrahim SA, Kwoh CK, Krishnan E. Factors associated with patients who leave acute-care hospitals against medical advice. Am J Public Health. 2007;97(12): 2204-2208.
- Weingart SN, Davis RB, Phillips RS. Patients discharged against medical advice from a general medicine service. J Gen Intern Med. 1998;13(8):568-571.
- Franks P, Meldrum S, Fiscella K. Discharges against medical advice: are race/ethnicity predictors? J Gen Intern Med. 2006;21(9):955-960.
- Saitz R, Ghali WA, Moskowitz MA. Characteristics of patients with pneumonia who are discharged from hospitals against medical advice. Am J Med. 1999;107(5):507-509.
- Alfandre, DJ. “I’m going home”: discharges against medical advice. Mayo Clin Proc. 2009;84(3):255-260.
- Onukwugha E, Saunders E, Mullins CD, Pradel FG, Zuckerman M, Weir MR. Reasons for discharges against medical advice: a qualitative study. Qual Saf Health Care. 2010;19(5):420-424. doi: 10.1136/qshc.2009.036269.
- Battenfeld v. Gregory, 589 A.2d 1059, 1061 (N.J. Super. Ct. App. Div. 1991).
- Berger J. Discharge against medical advice: ethical considerations and professional obligations. J Hosp Med. 2008;3(5):403-408.
Case No. 1
A 41-year-old woman with a history of asthma presents to the emergency department (ED) with shortness of breath and wheezing. She is diagnosed with a mild asthma exacerbation. After three albuterol nebulizer treatments, she still has wheezing on physical examination but appears comfortable and has no oxygen requirement. She has a primary medical doctor at the hospital and follows up with her regularly.
The hospitalist recommends that she stay in the hospital for further treatment, but the patient says she has a nebulizer machine at home and asks to be discharged. In addition, she is worried about her frail elderly mother, for whom she is the primary caretaker. The hospitalist acknowledges her concerns but continues to recommend that she remain in the hospital for additional care and monitoring. She becomes visibly upset and insists that she must return home. She asks for prescriptions for albuterol and prednisone and is discharged against medical advice (AMA).
Case No. 2
A 52-year-old man with a history of hypertension and diabetes presents to the ED with left foot pain. He frequently presents with this complaint but often leaves AMA before treatment is completed. He has no known physical address or telephone number and has no known outpatient healthcare providers. Physical examination reveals several ulcers on the dorsum of the foot, one with purulent drainage, and generalized lower extremity pallor. His left leg is cool to the touch, and vascular surgery is consulted for suspected limb-threatening ischemia; IV antibiotics are started for suspected osteomyelitis.
During the interview, he states that he wishes to leave the hospital because he has “things to take care of.” The hospitalist recommends that he remain in the hospital for limb-preserving surgery and antibiotics. He then explains that he is homeless and needs to return to his shelter to keep his bed. He is able to articulate the risks of premature discharge and the medical concerns, and it is determined that he has the capacity to participate in discharge planning. The hospitalist therefore discharges him AMA.
Background
AMA discharges represent 1%–2% of all inpatient discharges.¹,² Despite being a small percentage of total discharges, these patients have disproportionately high healthcare costs. One study reported that healthcare costs among these patients were 56% higher than expected.² Furthermore, AMA patients suffer higher than expected rates of morbidity, mortality, and hospital readmission.
For example, in one case-control study in an urban teaching hospital, patients discharged AMA from the general medicine service had a 21% 15-day readmission rate compared to a 3% readmission rate among age, gender, and diagnosis-matched controls.3,4,5
Additionally, history of AMA discharge appears to confer risk of increased future utilization of healthcare resources. In a cohort study of hospital admissions among HIV-infected patients with high rates of intravenous drug abuse, patients discharged AMA (13% of the cohort) were not only more likely to be readmitted within 30 days for a related diagnosis (odds ratio = 5.0) but also were more likely to have increased length of stay during the year following the index admission.6
These studies highlight the barriers to safe and effective transitions of care for this vulnerable population and demonstrate the increased burden that this population places on the health system.
Several retrospective studies have identified psychosocial and demographic risk factors associated with AMA discharge. These include younger age, male sex, substance abuse, lack of a primary care physician or health insurance, and history of previous AMA discharge.1,3,7,8 Insurance status is also associated with AMA discharge, with increased odds of AMA discharge among Medicare and Medicaid patients and patients without health insurance.9,10
Of note, one study found that race did not act as an independent predictor of AMA discharge when adjusted for age, gender, and socioeconomic factors.11
The AMA population is clinically heterogeneous. Among patients with pneumonia, for example, Saitz et al showed that a patient’s documented clinical severity did not independently predict AMA discharge, suggesting that there is great clinical heterogeneity even among AMA patients with similar admission diagnoses.12
These studies highlight the clinical and demographic heterogeneity within this population, suggesting that patients discharged AMA require individualized attention from hospitalists and other healthcare providers.
Patients describe numerous motivations for leaving the hospital prematurely, including needing to pick up public-assistance checks, personal financial issues, and familial obligations.13 Interestingly, in the cohort of HIV patients referenced above, discharge on the day welfare checks were distributed was an independent predictor of AMA discharge.6 In focus groups composed of patients discharged AMA and their treating nurses and physicians, several themes were identified as potential contributors to AMA discharge, including drug addiction, pain management issues, external obligations, wait time, the physician’s bedside manner, being in a teaching hospital, and communication issues.14
Clearly, patients have a diversity of reasons for requesting to be discharged AMA, and further research is necessary to define clear and potentially modifiable risk factors.
Discussion
The clinical scenarios outlined above present two patients with very different clinical presentations and outpatient support systems as well as demonstrate the great variability in clinical risk at the time of discharge AMA. These examples emphasize the importance of an individualized approach to care for each patient.
In Case No. 1, the patient is admitted with a mild asthma exacerbation with persistent bronchospasm, though she clinically appears well and has reliable follow-up. In contrast, in Case No. 2, the patient has life-threatening disease and no established primary care physician or mechanism for outpatient care. These examples demonstrate extremes on the clinical and psychosocial spectrum of patients requesting an “early” discharge and suggest that no two patients at risk of AMA discharge are the same. Patient 1 could likely be safely managed at home with close outpatient follow-up, while Patient 2 presents a high-risk scenario with very few safe outpatient treatment options.
We suggest that an individualized approach be taken for each patient, with attention to both clinical and psychosocial risk. In clinically low-risk cases (e.g., Case No. 1), an approach that prioritizes shared decision making and coordination with the outpatient care team may be preferable to an AMA discharge, particularly given the often adversarial nature of the later.2 In such cases, a collaborative approach may provide greater opportunity for harm reduction, provision of appropriate prescriptions, and follow-up appointments. In clinically high-risk patients such as Case No. 2, however, premature discharge is clearly inappropriate. Even in such clinically high-risk cases, however, we argue that a collaborative strategy aimed at identifying and addressing the patient’s psychosocial concerns is appropriate, as such an approach promotes shared decision making, builds trust between the patient and the care team, and therefore may facilitate improved follow-up at the time of discharge. Research is needed to formally assess the optimal approach for this patient population, including impact on rates of AMA discharge and the quality of post-discharge follow-up.
At present, the decision to classify a discharge as AMA falls solely on the treating provider, and we suspect that there is great variability in practice patterns, particularly as there are few established professional society practice guidelines regarding this difficult issue. As with all discharges from the hospital, the burden falls on the provider to engage the patient in shared decision making and ensure that the patient has the capacity to understand the risks and benefits of the proposed treatment plan. It is in this spirit that simply “filling out an AMA form” does not provide legal protection to a physician who does not adequately explain the full risks and benefits of refusal of inpatient treatment.2,15
We propose that a high-quality AMA discharge be defined as a discharge in which the patient is informed of the clinical team’s determination that further hospitalization is required but elects to leave the hospital, and it includes a clear discussion of the risks of outpatient treatment, a determination of capacity, and an exploration of safe alternative care plans that could satisfy both the patient’s medical and social needs. This definition places the burden on hospitalists and other providers to fully explore the motivations behind a patient’s request to leave the hospital and treats psychosocial motivators for premature discharge as variables in the complex risk-benefit analysis that underlies the informed consent discussion prior to AMA discharge.
Furthermore, AMA discharge does not obviate a physician’s responsibility to advocate for a patient’s well-being, and therefore an AMA discharge should be accompanied by reasonable efforts to coordinate a patient’s ongoing outpatient care. Of note, this approach is consistent with previous reviews and attempts to balance the physician’s duty to honor a patient’s autonomy with the responsibility to protect the patient from harm.2,16
Conclusion
Patients discharged AMA are a diverse population at markedly increased risk of morbidity, readmissions, and subsequent healthcare cost. We argue that in all cases of a potential premature discharge, a collaborative and patient-centered approach is crucial. Such an approach allows the provider to identify and address the patient’s concerns regarding further inpatient care, to explore possible safe outpatient treatment options, to document patient capacity, and to provide appropriate harm-reduction measures such as prescriptions.
Further research into the current practice patterns of hospitalists and other providers is necessary to allow for the formulation and adoption of best practices and implementation of appropriate harm-reduction strategies. TH
Dr. Tummalapalli is an internal medicine resident in the department of medicine at Icahn School of Medicine at Mount Sinai in New York City. Dr. Goodman is a hospitalist in the division of hospital medicine, department of medicine, at the Icahn School of Medicine at Mount Sinai.
References
- Aliyu ZY. Discharge against medical advice: sociodemographic, clinical and financial perspectives. Int J Clin Pract. 2002;56(5):325-327.
- Kahle CH, Rubio ML, Santos RA. Discharges against medical advice: considerations for the hospitalist and the patient. Hospital Medicine Clinics. 2015;4(3):421-429.
- Baptist AP, Warrier I, Arora R, Ager J, Massanari RM. Hospitalized patients with asthma who leave against medical advice: characteristics, reasons, and outcomes. J Allergy Clin Immunol. 2007;119(4):924-929.
- Hwang SW, Li J, Gupta R, Chien V, Martin RE. What happens to patients who leave hospital against medical advice? CMAJ. 2003;168(4):417-420.
- Glasgow JM, Vaughn-Sarrazin MV, Kaboli PJ. Leaving against medical advice (AMA): risk of 30-day mortality and hospital readmission. J Gen Int Med. 2010;25(9):926-929.
- Anis AH, Sun H, Guh DP, Palepu A, Schechter MT, O’Shaughnessy MV. Leaving hospital against medical advice among HIV-positive patients. CMAJ. 2002;167(6):633-637.
- Jeremiah J, O’Sullivan P, Stein MD. Who leaves against medical advice? J Gen Int Med. 1995; 10(7);403-405.
- O’Hara D, Hart W, McDonald I. Leaving hospital against medical advice. J Qual Clin Pract.1996;16(3):157-164.
- Ibrahim SA, Kwoh CK, Krishnan E. Factors associated with patients who leave acute-care hospitals against medical advice. Am J Public Health. 2007;97(12): 2204-2208.
- Weingart SN, Davis RB, Phillips RS. Patients discharged against medical advice from a general medicine service. J Gen Intern Med. 1998;13(8):568-571.
- Franks P, Meldrum S, Fiscella K. Discharges against medical advice: are race/ethnicity predictors? J Gen Intern Med. 2006;21(9):955-960.
- Saitz R, Ghali WA, Moskowitz MA. Characteristics of patients with pneumonia who are discharged from hospitals against medical advice. Am J Med. 1999;107(5):507-509.
- Alfandre, DJ. “I’m going home”: discharges against medical advice. Mayo Clin Proc. 2009;84(3):255-260.
- Onukwugha E, Saunders E, Mullins CD, Pradel FG, Zuckerman M, Weir MR. Reasons for discharges against medical advice: a qualitative study. Qual Saf Health Care. 2010;19(5):420-424. doi: 10.1136/qshc.2009.036269.
- Battenfeld v. Gregory, 589 A.2d 1059, 1061 (N.J. Super. Ct. App. Div. 1991).
- Berger J. Discharge against medical advice: ethical considerations and professional obligations. J Hosp Med. 2008;3(5):403-408.
Case No. 1
A 41-year-old woman with a history of asthma presents to the emergency department (ED) with shortness of breath and wheezing. She is diagnosed with a mild asthma exacerbation. After three albuterol nebulizer treatments, she still has wheezing on physical examination but appears comfortable and has no oxygen requirement. She has a primary medical doctor at the hospital and follows up with her regularly.
The hospitalist recommends that she stay in the hospital for further treatment, but the patient says she has a nebulizer machine at home and asks to be discharged. In addition, she is worried about her frail elderly mother, for whom she is the primary caretaker. The hospitalist acknowledges her concerns but continues to recommend that she remain in the hospital for additional care and monitoring. She becomes visibly upset and insists that she must return home. She asks for prescriptions for albuterol and prednisone and is discharged against medical advice (AMA).
Case No. 2
A 52-year-old man with a history of hypertension and diabetes presents to the ED with left foot pain. He frequently presents with this complaint but often leaves AMA before treatment is completed. He has no known physical address or telephone number and has no known outpatient healthcare providers. Physical examination reveals several ulcers on the dorsum of the foot, one with purulent drainage, and generalized lower extremity pallor. His left leg is cool to the touch, and vascular surgery is consulted for suspected limb-threatening ischemia; IV antibiotics are started for suspected osteomyelitis.
During the interview, he states that he wishes to leave the hospital because he has “things to take care of.” The hospitalist recommends that he remain in the hospital for limb-preserving surgery and antibiotics. He then explains that he is homeless and needs to return to his shelter to keep his bed. He is able to articulate the risks of premature discharge and the medical concerns, and it is determined that he has the capacity to participate in discharge planning. The hospitalist therefore discharges him AMA.
Background
AMA discharges represent 1%–2% of all inpatient discharges.¹,² Despite being a small percentage of total discharges, these patients have disproportionately high healthcare costs. One study reported that healthcare costs among these patients were 56% higher than expected.² Furthermore, AMA patients suffer higher than expected rates of morbidity, mortality, and hospital readmission.
For example, in one case-control study in an urban teaching hospital, patients discharged AMA from the general medicine service had a 21% 15-day readmission rate compared to a 3% readmission rate among age, gender, and diagnosis-matched controls.3,4,5
Additionally, history of AMA discharge appears to confer risk of increased future utilization of healthcare resources. In a cohort study of hospital admissions among HIV-infected patients with high rates of intravenous drug abuse, patients discharged AMA (13% of the cohort) were not only more likely to be readmitted within 30 days for a related diagnosis (odds ratio = 5.0) but also were more likely to have increased length of stay during the year following the index admission.6
These studies highlight the barriers to safe and effective transitions of care for this vulnerable population and demonstrate the increased burden that this population places on the health system.
Several retrospective studies have identified psychosocial and demographic risk factors associated with AMA discharge. These include younger age, male sex, substance abuse, lack of a primary care physician or health insurance, and history of previous AMA discharge.1,3,7,8 Insurance status is also associated with AMA discharge, with increased odds of AMA discharge among Medicare and Medicaid patients and patients without health insurance.9,10
Of note, one study found that race did not act as an independent predictor of AMA discharge when adjusted for age, gender, and socioeconomic factors.11
The AMA population is clinically heterogeneous. Among patients with pneumonia, for example, Saitz et al showed that a patient’s documented clinical severity did not independently predict AMA discharge, suggesting that there is great clinical heterogeneity even among AMA patients with similar admission diagnoses.12
These studies highlight the clinical and demographic heterogeneity within this population, suggesting that patients discharged AMA require individualized attention from hospitalists and other healthcare providers.
Patients describe numerous motivations for leaving the hospital prematurely, including needing to pick up public-assistance checks, personal financial issues, and familial obligations.13 Interestingly, in the cohort of HIV patients referenced above, discharge on the day welfare checks were distributed was an independent predictor of AMA discharge.6 In focus groups composed of patients discharged AMA and their treating nurses and physicians, several themes were identified as potential contributors to AMA discharge, including drug addiction, pain management issues, external obligations, wait time, the physician’s bedside manner, being in a teaching hospital, and communication issues.14
Clearly, patients have a diversity of reasons for requesting to be discharged AMA, and further research is necessary to define clear and potentially modifiable risk factors.
Discussion
The clinical scenarios outlined above present two patients with very different clinical presentations and outpatient support systems as well as demonstrate the great variability in clinical risk at the time of discharge AMA. These examples emphasize the importance of an individualized approach to care for each patient.
In Case No. 1, the patient is admitted with a mild asthma exacerbation with persistent bronchospasm, though she clinically appears well and has reliable follow-up. In contrast, in Case No. 2, the patient has life-threatening disease and no established primary care physician or mechanism for outpatient care. These examples demonstrate extremes on the clinical and psychosocial spectrum of patients requesting an “early” discharge and suggest that no two patients at risk of AMA discharge are the same. Patient 1 could likely be safely managed at home with close outpatient follow-up, while Patient 2 presents a high-risk scenario with very few safe outpatient treatment options.
We suggest that an individualized approach be taken for each patient, with attention to both clinical and psychosocial risk. In clinically low-risk cases (e.g., Case No. 1), an approach that prioritizes shared decision making and coordination with the outpatient care team may be preferable to an AMA discharge, particularly given the often adversarial nature of the later.2 In such cases, a collaborative approach may provide greater opportunity for harm reduction, provision of appropriate prescriptions, and follow-up appointments. In clinically high-risk patients such as Case No. 2, however, premature discharge is clearly inappropriate. Even in such clinically high-risk cases, however, we argue that a collaborative strategy aimed at identifying and addressing the patient’s psychosocial concerns is appropriate, as such an approach promotes shared decision making, builds trust between the patient and the care team, and therefore may facilitate improved follow-up at the time of discharge. Research is needed to formally assess the optimal approach for this patient population, including impact on rates of AMA discharge and the quality of post-discharge follow-up.
At present, the decision to classify a discharge as AMA falls solely on the treating provider, and we suspect that there is great variability in practice patterns, particularly as there are few established professional society practice guidelines regarding this difficult issue. As with all discharges from the hospital, the burden falls on the provider to engage the patient in shared decision making and ensure that the patient has the capacity to understand the risks and benefits of the proposed treatment plan. It is in this spirit that simply “filling out an AMA form” does not provide legal protection to a physician who does not adequately explain the full risks and benefits of refusal of inpatient treatment.2,15
We propose that a high-quality AMA discharge be defined as a discharge in which the patient is informed of the clinical team’s determination that further hospitalization is required but elects to leave the hospital, and it includes a clear discussion of the risks of outpatient treatment, a determination of capacity, and an exploration of safe alternative care plans that could satisfy both the patient’s medical and social needs. This definition places the burden on hospitalists and other providers to fully explore the motivations behind a patient’s request to leave the hospital and treats psychosocial motivators for premature discharge as variables in the complex risk-benefit analysis that underlies the informed consent discussion prior to AMA discharge.
Furthermore, AMA discharge does not obviate a physician’s responsibility to advocate for a patient’s well-being, and therefore an AMA discharge should be accompanied by reasonable efforts to coordinate a patient’s ongoing outpatient care. Of note, this approach is consistent with previous reviews and attempts to balance the physician’s duty to honor a patient’s autonomy with the responsibility to protect the patient from harm.2,16
Conclusion
Patients discharged AMA are a diverse population at markedly increased risk of morbidity, readmissions, and subsequent healthcare cost. We argue that in all cases of a potential premature discharge, a collaborative and patient-centered approach is crucial. Such an approach allows the provider to identify and address the patient’s concerns regarding further inpatient care, to explore possible safe outpatient treatment options, to document patient capacity, and to provide appropriate harm-reduction measures such as prescriptions.
Further research into the current practice patterns of hospitalists and other providers is necessary to allow for the formulation and adoption of best practices and implementation of appropriate harm-reduction strategies. TH
Dr. Tummalapalli is an internal medicine resident in the department of medicine at Icahn School of Medicine at Mount Sinai in New York City. Dr. Goodman is a hospitalist in the division of hospital medicine, department of medicine, at the Icahn School of Medicine at Mount Sinai.
References
- Aliyu ZY. Discharge against medical advice: sociodemographic, clinical and financial perspectives. Int J Clin Pract. 2002;56(5):325-327.
- Kahle CH, Rubio ML, Santos RA. Discharges against medical advice: considerations for the hospitalist and the patient. Hospital Medicine Clinics. 2015;4(3):421-429.
- Baptist AP, Warrier I, Arora R, Ager J, Massanari RM. Hospitalized patients with asthma who leave against medical advice: characteristics, reasons, and outcomes. J Allergy Clin Immunol. 2007;119(4):924-929.
- Hwang SW, Li J, Gupta R, Chien V, Martin RE. What happens to patients who leave hospital against medical advice? CMAJ. 2003;168(4):417-420.
- Glasgow JM, Vaughn-Sarrazin MV, Kaboli PJ. Leaving against medical advice (AMA): risk of 30-day mortality and hospital readmission. J Gen Int Med. 2010;25(9):926-929.
- Anis AH, Sun H, Guh DP, Palepu A, Schechter MT, O’Shaughnessy MV. Leaving hospital against medical advice among HIV-positive patients. CMAJ. 2002;167(6):633-637.
- Jeremiah J, O’Sullivan P, Stein MD. Who leaves against medical advice? J Gen Int Med. 1995; 10(7);403-405.
- O’Hara D, Hart W, McDonald I. Leaving hospital against medical advice. J Qual Clin Pract.1996;16(3):157-164.
- Ibrahim SA, Kwoh CK, Krishnan E. Factors associated with patients who leave acute-care hospitals against medical advice. Am J Public Health. 2007;97(12): 2204-2208.
- Weingart SN, Davis RB, Phillips RS. Patients discharged against medical advice from a general medicine service. J Gen Intern Med. 1998;13(8):568-571.
- Franks P, Meldrum S, Fiscella K. Discharges against medical advice: are race/ethnicity predictors? J Gen Intern Med. 2006;21(9):955-960.
- Saitz R, Ghali WA, Moskowitz MA. Characteristics of patients with pneumonia who are discharged from hospitals against medical advice. Am J Med. 1999;107(5):507-509.
- Alfandre, DJ. “I’m going home”: discharges against medical advice. Mayo Clin Proc. 2009;84(3):255-260.
- Onukwugha E, Saunders E, Mullins CD, Pradel FG, Zuckerman M, Weir MR. Reasons for discharges against medical advice: a qualitative study. Qual Saf Health Care. 2010;19(5):420-424. doi: 10.1136/qshc.2009.036269.
- Battenfeld v. Gregory, 589 A.2d 1059, 1061 (N.J. Super. Ct. App. Div. 1991).
- Berger J. Discharge against medical advice: ethical considerations and professional obligations. J Hosp Med. 2008;3(5):403-408.
Breakdown
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.