User login
A shocking diagnosis
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 75-year-old man was brought by ambulance to the emergency department (ED) after the acute onset of palpitations, lightheadedness, and confusion. His medical history, provided by his wife, included osteoarthritis and remote cholecystectomy. He was not a smoker but drank 2 to 4 cans of beer daily. His medications were aspirin 162 mg daily and naproxen as needed. There was no history of bruising, diarrhea, melena, or bleeding.
Palpitations may represent an arrhythmia arising from an ischemic or alcoholic cardiomyopathy. Mental status changes usually have metabolic, infectious, structural (eg, hemorrhage, tumor), or toxic causes. Lightheadedness and confusion could occur with arrhythmia-associated cerebral hypoperfusion or a seizure. Daily alcohol use could cause confusion through acute intoxication, thiamine or B12 deficiency, repeated head trauma, or liver failure.
The patient’s systolic blood pressure (BP) was 60 mm Hg, heart rate (HR) was 120 beats per minute (bpm), and oral temperature was 98.4°F. Rousing him was difficult. There were no localizing neurologic abnormalities, and the rest of the physical examination findings were normal. Point-of-care blood glucose level was 155 mg/dL. Blood cultures were obtained and broad-spectrum antibiotics initiated. After fluid resuscitation, BP improved to 116/87 mm Hg, HR fell to 105 bpm, and the patient became alert and oriented. He denied chest pain, fever, or diaphoresis.
The patient’s improvement with intravenous (IV) fluids makes cardiogenic shock unlikely but does not exclude an underlying compensated cardiomyopathy that may be predisposing to arrhythmia. Hypotension, tachycardia, and somnolence may represent sepsis, but the near normalization of vital signs and mental status shortly after administration of IV fluids, the normal temperature, and the absence of localizing signs of infection favor withholding additional antibiotics. Other causes of hypotension are hypovolemia, medication effects, adrenal insufficiency, anaphylaxis, and autonomic insufficiency. There was no reported nausea, vomiting, diarrhea, bleeding, polyuria, or impaired oral intake to support hypovolemia, though the response to IV fluids suggests hypovolemia may still be playing a role.
White blood cell (WBC) count was 15,450/µL with a normal differential; hemoglobin level was 15.8 g/dL; and platelet count was 176,000/µL. Electrolytes, liver function tests, cardiac enzymes, and urinalysis were normal. Electrocardiogram showed sinus tachycardia with premature atrial complexes and no ST-segment abnormalities. Radiograph of the chest and computed tomography scan of the head were normal. Echocardiogram showed moderate left ventricular hypertrophy with a normal ejection fraction and no valvular abnormalities. Exercise nuclear cardiac stress test was negative for ischemia. Blood cultures were sterile. The patient quickly became asymptomatic and remained so during his 3-day hospitalization. There were no arrhythmias on telemetry. The patient was discharged with follow-up scheduled with his primary care physician.
The nonlocalizing history and physical examination findings, normal chest radiograph and urinalysis, absence of fevers, negative blood cultures, and quick recovery make infection unlikely, despite the moderate leukocytosis. Conditions that present with acute and transient hypotension and altered mental status include arrhythmias, seizures, and reactions to drugs or toxins. Given the cardiac test results, a chronic cardiomyopathy seems unlikely, but arrhythmia is still possible. Continuous outpatient monitoring is required to assess the palpitations and the frequency of the premature atrial complexes.
Two days after discharge, the patient suddenly became diaphoretic and lost consciousness while walking to the bathroom. He was taken to the ED, where his BP was 90/60 mm Hg and HR was 108 bpm. Family members reported that he had appeared flushed during the syncopal episode, showed no seizure activity, and been unconscious for 15 to 20 minutes. The patient denied chest pain, dyspnea, fever, bowel or bladder incontinence, focal weakness, slurred speech, visual changes, nausea or vomiting either before or after the episode. Physical examination revealed a tongue laceration and facial erythema; all other findings were normal. In the ED, there was an asymptomatic 7-beat run of nonsustained ventricular tachycardia, and the hypotension resolved after fluid resuscitation. The patient now reported 2 similar syncopal episodes in the past. The first occurred in a restaurant 6 years earlier, and the second occurred 3 years later, at which time he was hospitalized and no etiology was found.
The loss of consciousness is attributable to cerebral hypoperfusion. Hypotension has 3 principal categories: hypovolemic, cardiogenic, and distributive. With syncopal episodes recurring over several years, hypovolemia seems unlikely. Given the palpitations and ventricular tachycardia, it is reasonable to suspect a cardiogenic cause. Although his heart appears to be structurally normal on echocardiogram, genetic, electrophysiologic, or magnetic resonance imaging (MRI) testing will occasionally reveal an unsuspected substrate for arrhythmia.
The recurring yet self-limited nature, diaphoresis, flushing, and facial erythema suggest a non-sepsis distributive cause of hypotension. It is possible the patient is recurrently exposed to a toxin (eg, alcohol) that causes both flushing and dehydration. Flushing disorders include carcinoid syndrome, pheochromocytoma, drug reaction with eosinophilia and systemic symptoms (DRESS), and mastocytosis. Carcinoid syndrome is characterized by bronchospasm and diarrhea and, in some cases, right-sided valvulopathy, all of which are absent in this patient. Pheochromocytoma is associated with orthostasis, but patients typically are hypertensive at baseline. DRESS, which may arise from nonsteroidal anti-inflammatory drug (NSAID) or aspirin use, can cause facial erythema and swelling but is also characterized by liver, renal, and hematologic abnormalities, none of which was demonstrated. Furthermore, DRESS typically does not cause hypotension. Mastocytosis can manifest as isolated or recurrent anaphylaxis.
It is important to investigate antecedents of these syncopal episodes. If the earlier episodes were food-related—one occurred at a restaurant—then deglutition syncope (syncope precipitated by swallowing) should be considered. If an NSAID or aspirin was ingested before each episode, then medication hypersensitivity or mast cell degranulation (which can be triggered by these medications) should be further examined. Loss of consciousness lasting 20 minutes without causing any neurologic sequelae is unusual for most causes of recurrent syncope. This feature raises the possibility that a toxin or mediator might still be present in the patient’s system.
Serial cardiac enzymes and electrocardiogram were normal. A tilt-table study was negative. The cortisol response to ACTH (cosyntropin) stimulation was normal. The level of serum tryptase, drawn 2 days after syncope, was 18.4 ng/dL (normal, <11.5 ng/dL). Computed tomography scan of chest and abdomen was negative for pulmonary embolism but showed a 1.4×1.3-cm hypervascular lesion in the tail of pancreas. The following neuroendocrine tests were within normal limits: serum and urine catecholamines; urine 5-hydroxyindoleacetic acid (5-HIAA); and serum chromogranin A, insulin, serotonin, vasoactive intestinal polypeptide (VIP), and somatostatin (Table 1). The patient remained asymptomatic during his hospital stay and was discharged home with appointments for cardiology follow-up and endoscopic ultrasound-guided biopsy of the pancreatic mass.

Pheochromocytoma is unlikely with normal serum and urine catecholamine levels and normal adrenal images. The differential diagnosis for a pancreatic mass includes pancreatic carcinoma, lymphoma, cystic neoplasm, and neuroendocrine tumor. All markers of neuroendocrine excess are normal, though elevations can be episodic. The normal 5-HIAA level makes carcinoid syndrome unlikely. VIPomas are associated with flushing, but the absence of profound and protracted diarrhea makes a VIPoma unlikely.
As hypoglycemia from a pancreatic insulinoma is plausible as a cause of episodic loss of consciousness lasting 15 minutes or more, it is important to inquire if giving food or drink helped resolve previous episodes. The normal insulin level reported here is of limited value, because it is the combination of insulin and C-peptide levels at time of hypoglycemia that is diagnostic. The normal glucose level recorded during one of the earlier episodes and the hypotension argue against hypoglycemia.
The elevated tryptase level is an indicator of mast cell degranulation. Tryptase levels are transiently elevated during the initial 2 to 4 hours after an anaphylactic episode and then normalize. An elevated level many hours or days later is considered a sign of mast cell excess. Although there is no evidence of the multi-organ disease (eg, cytopenia, bone disease, hepatosplenomegaly) seen in patients with a high systemic burden of mast cells, mast cell disorders exist on a spectrum. There may be a focal excess of mast cells confined to one organ or an isolated mass.
The same day as discharge, the patient’s wife drove them to the grocery store. He remained in the car while she shopped. When she returned, she found him confused and minimally responsive with subsequent brief loss of consciousness. He was taken to an ED, where he was flushed and hypotensive (systolic BP, 60 mm Hg) and tachycardic. Other examination findings were normal. After fluid resuscitation he became alert and oriented. WBC count was 20,850/μL with 89% neutrophils, hemoglobin level was 14.6 g/dL, and platelet count was 168,000/μL. Serum lactate level was 3.7 mmol/L (normal, <2.3 mmol/L). Chest radiograph was normal. He was treated with broad-spectrum antibiotic therapy and admitted to the hospital. Blood and urine cultures were sterile. Fine-needle aspiration of the pancreatic mass demonstrated nonspecific inflammation. Four days after admission (3 days after pancreatic mass biopsy) the patient developed palpitations, felt unwell, and had marked flushing of the face and trunk, with concomitant BP of 90/50 mm Hg and HR of 140 bpm.
The salient features of this case are recurrent hypotension, tachycardia, and flushing. Autonomic insufficiency, to which elderly patients are prone, causes hemodynamic perturbations but rarely flushing. The patient does not have diabetes mellitus, Parkinson disease, or another condition that puts him at risk for dysautonomia. Pancreatic neuroendocrine tumors secrete mediators that lead to vasodilation and hypotension but are unlikely given the clinical and biochemical data.
The patient’s symptoms are consistent with anaphylaxis, though prototypical immunoglobulin E (IgE)–mediated anaphylaxis is usually accompanied by urticaria, angioedema, and wheezing, which have been absent during his presentations. There are no clear food, pharmacologic, or environmental precipitants.
Recurrent anaphylaxis can be a manifestation of mast cell excess (eg, cutaneous or systemic mastocytosis). A markedly elevated tryptase level during an anaphylactic episode is consistent with mastocytosis or IgE-mediated anaphylaxis. An elevated baseline tryptase level days after an anaphylactic episode signals increased mast cell burden. There may be a reservoir of mast cells in the bone marrow. Alternatively, the hypervascular pancreatic mass may be a mastocytoma or a mast cell sarcoma (missed because of inadequate sampling or staining).
The lactic acidosis likely reflects global tissue hypoperfusion from vasodilatory hypotension. The leukocytosis may reflect WBC mobilization secondary to endogenous corticosteroids and catecholamines in response to hypotension or may be a direct response to the release of mast cell–derived mediators of inflammation.
The patient was treated with diphenhydramine and ranitidine. Serum tryptase level was 46.8 ng/mL (normal, <11.5 ng/mL), and 24-hour urine histamine level was 95 µ g/dL (normal, <60 µ g/dL). Bone marrow biopsy results showed multifocal dense infiltrative aggregates of mast cells (>15 cells/aggregate), which were confirmed by CD117 (Kit) and tryptase positivity (Figure). Mutation analysis for Kit Asp816Val, which is present in 80% to 90% of patients with mastocytosis, was positive. He fulfilled the 2008 World Health Organization criteria for systemic mastocytosis (Table 2). Prednisone, histamine inhibitors, and montelukast were prescribed. Six months later, magnetic resonance imaging of the abdomen showed no change in the pancreatic mass, which was now characterized as a possible splenule. The patient had no additional episodes of flushing or syncope over 2 years.


DISCUSSION
Cardiovascular collapse (hypotension, tachycardia, syncope) in an elderly patient prompts clinicians to focus on life-threatening conditions, such as acute coronary syndrome, pulmonary embolus, arrhythmia, and sepsis. Each of these diagnoses was considered early in the course of this patient’s presentations, but each was deemed unlikely as it became apparent that the episodes were self-limited and recurrent over years. Incorporating flushing into the diagnostic problem representation allowed the clinicians to focus on a subset of causes of hypotension.
Flushing disorders may be classified by whether they are mediated by the autonomic nervous system (wet flushes, because they are usually accompanied by diaphoresis) or by exogenous or endogenous vasoactive substances (dry flushes).1 Autonomic nervous system flushing is triggered by emotions, fever, exercise, perimenopause (hot flashes), and neurologic conditions (eg, Parkinson disease, spinal cord injury, multiple sclerosis). Vasoactive flushing precipitants include drugs (eg, niacin); alcohol (secondary to cutaneous vasodilation, or acetaldehyde particularly in people with insufficient acetaldehyde dehydrogenase activity)2; foods that contain capsaicin, tyramine, sulfites, or histamine (eg, eating improperly handled fish can cause scombroid poisoning); and anaphylaxis. Rare causes of vasoactive flushing include carcinoid syndrome, pheochromocytoma, medullary thyroid carcinoma, VIPoma, and mastocytosis.2
Mastocytosis is a rare clonal disorder characterized by the accumulation of abnormal mast cells in the skin (cutaneous mastocytosis), in multiple organs (systemic mastocytosis), or in a solid tumor (mastocytoma). Urticaria pigmentosa is the most common form of cutaneous mastocytosis; it is seen more often in children than in adults and typically is associated with a maculopapular rash and dermatographism. Systemic mastocytosis is the most common form of the disorder in adults.3 Symptoms are related to mast cell infiltration or mast cell mediator–related effects, which range from itching, flushing, and diarrhea to hypotension and anaphylaxis. Other manifestations are fatigue, urticaria pigmentosa, osteoporosis, hepatosplenomegaly, bone pain, cytopenias, and lymphadenopathy.4
Systemic mastocytosis can occur at any age and should be considered in patients with recurrent unexplained flushing, syncope, or hypotension. Eighty percent to 90% of patients with systemic mastocytosis have a mutation in Kit,5 a transmembrane tyrosine kinase that is the receptor for stem cell factor. The Asp816Val mutation leads to increased proliferation and reduced apoptosis of mast cells.3,6,7 Proposed diagnostic algorithms8-11 involve measurement of serum tryptase levels and examination of bone marrow. Bone marrow biopsy and testing for the Asp816Val
The primary goals of treatment are managing mast cell–mediated symptoms and, in advanced cases, achieving cytoreduction. Alcohol can trigger mast cell degranulation in indolent systemic mastocytosis and should be avoided. Mast cell–mediated symptoms are managed with histamine blockers, leukotriene antagonists, and mast cell stabilizers.12 Targeted therapy with tyrosine kinase inhibitors (eg, imatinib) in patients with transmembrane Kit mutation (eg, Phe522Cys, Lys509Ile) associated with systemic mastocytosis has had promising results.13,14 However, this patient’s Asp816Val mutation is in the Kit catalytic domain, not the transmembrane region, and therefore would not be expected to respond to imatinib. A recent open-label trial of the multikinase inhibitor midostaurin demonstrated resolution of organ damage, reduced bone marrow burden, and lowered serum tryptase levels in patients with advanced systemic mastocytosis.15 Interferon, cladribine, and high-dose corticosteroids are prescribed in patients for whom other therapies have been ineffective.8
The differential diagnosis is broad for both hypotension and for flushing, but the differential diagnosis for recurrent hypotension and flushing is limited. Recognizing that flushing was an essential feature of this patient’s hypotensive condition, and not an epiphenomenon of syncope, allowed the clinicians to focus on the overlap and make a shocking diagnosis.
Acknowledgment
The authors thank David Bosler, MD (Cleveland Clinic) for interpreting the pathology image.
Disclosure
Nothing to report.
1. Wilkin JK. The red face: flushing disorders. Clin Dermatol. 1993;11(2):211-223. PubMed
2. Izikson L, English JC 3rd, Zirwas MJ. The flushing patient: differential diagnosis, workup, and treatment. J Am Acad Dermatol. 2006;55(2):193-208. PubMed
3. Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37(6):435-453. PubMed
4. Hermans MA, Rietveld MJ, van Laar JA, et al. Systemic mastocytosis: a cohort study on clinical characteristics of 136 patients in a large tertiary centre. Eur J Intern Med. 2016;30:25-30. PubMed
5. Kristensen T, Vestergaard H, Bindslev-Jensen C, Møller MB, Broesby-Olsen S; Mastocytosis Centre, Odense University Hospital (MastOUH). Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am J Hematol. 2014;89(5):493-498. PubMed
6. Verstovsek S. Advanced systemic mastocytosis: the impact of KIT mutations in diagnosis, treatment, and progression. Eur J Haematol. 2013;90(2):89-98. PubMed
7. Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108(7):2366-2372. PubMed
8. Pardanani A. Systemic mastocytosis in adults: 2015 update on diagnosis, risk stratification, and management. Am J Hematol. 2015;90(3):250-262. PubMed
9. Valent P, Aberer E, Beham-Schmid C, et al. Guidelines and diagnostic algorithm for patients with suspected systemic mastocytosis: a proposal of the Austrian Competence Network (AUCNM). Am J Blood Res. 2013;3(2):174-180. PubMed
10. Valent P, Escribano L, Broesby-Olsen S, et al; European Competence Network on Mastocytosis. Proposed diagnostic algorithm for patients with suspected mastocytosis: a proposal of the European Competence Network on Mastocytosis. Allergy. 2014;69(10):1267-1274. PubMed
11. Akin C, Soto D, Brittain E, et al. Tryptase haplotype in mastocytosis: relationship to disease variant and diagnostic utility of total tryptase levels. Clin Immunol. 2007;123(3):268-271. PubMed
12. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(19):1885-1886. PubMed
13. Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103(8):3222-3225. PubMed
14. Zhang LY, Smith ML, Schultheis B, et al. A novel K509I mutation of KIT identified in familial mastocytosis—in vitro and in vivo responsiveness to imatinib therapy. Leuk Res. 2006;30(4):373-378. PubMed
15. Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374(26):2530-2541. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 75-year-old man was brought by ambulance to the emergency department (ED) after the acute onset of palpitations, lightheadedness, and confusion. His medical history, provided by his wife, included osteoarthritis and remote cholecystectomy. He was not a smoker but drank 2 to 4 cans of beer daily. His medications were aspirin 162 mg daily and naproxen as needed. There was no history of bruising, diarrhea, melena, or bleeding.
Palpitations may represent an arrhythmia arising from an ischemic or alcoholic cardiomyopathy. Mental status changes usually have metabolic, infectious, structural (eg, hemorrhage, tumor), or toxic causes. Lightheadedness and confusion could occur with arrhythmia-associated cerebral hypoperfusion or a seizure. Daily alcohol use could cause confusion through acute intoxication, thiamine or B12 deficiency, repeated head trauma, or liver failure.
The patient’s systolic blood pressure (BP) was 60 mm Hg, heart rate (HR) was 120 beats per minute (bpm), and oral temperature was 98.4°F. Rousing him was difficult. There were no localizing neurologic abnormalities, and the rest of the physical examination findings were normal. Point-of-care blood glucose level was 155 mg/dL. Blood cultures were obtained and broad-spectrum antibiotics initiated. After fluid resuscitation, BP improved to 116/87 mm Hg, HR fell to 105 bpm, and the patient became alert and oriented. He denied chest pain, fever, or diaphoresis.
The patient’s improvement with intravenous (IV) fluids makes cardiogenic shock unlikely but does not exclude an underlying compensated cardiomyopathy that may be predisposing to arrhythmia. Hypotension, tachycardia, and somnolence may represent sepsis, but the near normalization of vital signs and mental status shortly after administration of IV fluids, the normal temperature, and the absence of localizing signs of infection favor withholding additional antibiotics. Other causes of hypotension are hypovolemia, medication effects, adrenal insufficiency, anaphylaxis, and autonomic insufficiency. There was no reported nausea, vomiting, diarrhea, bleeding, polyuria, or impaired oral intake to support hypovolemia, though the response to IV fluids suggests hypovolemia may still be playing a role.
White blood cell (WBC) count was 15,450/µL with a normal differential; hemoglobin level was 15.8 g/dL; and platelet count was 176,000/µL. Electrolytes, liver function tests, cardiac enzymes, and urinalysis were normal. Electrocardiogram showed sinus tachycardia with premature atrial complexes and no ST-segment abnormalities. Radiograph of the chest and computed tomography scan of the head were normal. Echocardiogram showed moderate left ventricular hypertrophy with a normal ejection fraction and no valvular abnormalities. Exercise nuclear cardiac stress test was negative for ischemia. Blood cultures were sterile. The patient quickly became asymptomatic and remained so during his 3-day hospitalization. There were no arrhythmias on telemetry. The patient was discharged with follow-up scheduled with his primary care physician.
The nonlocalizing history and physical examination findings, normal chest radiograph and urinalysis, absence of fevers, negative blood cultures, and quick recovery make infection unlikely, despite the moderate leukocytosis. Conditions that present with acute and transient hypotension and altered mental status include arrhythmias, seizures, and reactions to drugs or toxins. Given the cardiac test results, a chronic cardiomyopathy seems unlikely, but arrhythmia is still possible. Continuous outpatient monitoring is required to assess the palpitations and the frequency of the premature atrial complexes.
Two days after discharge, the patient suddenly became diaphoretic and lost consciousness while walking to the bathroom. He was taken to the ED, where his BP was 90/60 mm Hg and HR was 108 bpm. Family members reported that he had appeared flushed during the syncopal episode, showed no seizure activity, and been unconscious for 15 to 20 minutes. The patient denied chest pain, dyspnea, fever, bowel or bladder incontinence, focal weakness, slurred speech, visual changes, nausea or vomiting either before or after the episode. Physical examination revealed a tongue laceration and facial erythema; all other findings were normal. In the ED, there was an asymptomatic 7-beat run of nonsustained ventricular tachycardia, and the hypotension resolved after fluid resuscitation. The patient now reported 2 similar syncopal episodes in the past. The first occurred in a restaurant 6 years earlier, and the second occurred 3 years later, at which time he was hospitalized and no etiology was found.
The loss of consciousness is attributable to cerebral hypoperfusion. Hypotension has 3 principal categories: hypovolemic, cardiogenic, and distributive. With syncopal episodes recurring over several years, hypovolemia seems unlikely. Given the palpitations and ventricular tachycardia, it is reasonable to suspect a cardiogenic cause. Although his heart appears to be structurally normal on echocardiogram, genetic, electrophysiologic, or magnetic resonance imaging (MRI) testing will occasionally reveal an unsuspected substrate for arrhythmia.
The recurring yet self-limited nature, diaphoresis, flushing, and facial erythema suggest a non-sepsis distributive cause of hypotension. It is possible the patient is recurrently exposed to a toxin (eg, alcohol) that causes both flushing and dehydration. Flushing disorders include carcinoid syndrome, pheochromocytoma, drug reaction with eosinophilia and systemic symptoms (DRESS), and mastocytosis. Carcinoid syndrome is characterized by bronchospasm and diarrhea and, in some cases, right-sided valvulopathy, all of which are absent in this patient. Pheochromocytoma is associated with orthostasis, but patients typically are hypertensive at baseline. DRESS, which may arise from nonsteroidal anti-inflammatory drug (NSAID) or aspirin use, can cause facial erythema and swelling but is also characterized by liver, renal, and hematologic abnormalities, none of which was demonstrated. Furthermore, DRESS typically does not cause hypotension. Mastocytosis can manifest as isolated or recurrent anaphylaxis.
It is important to investigate antecedents of these syncopal episodes. If the earlier episodes were food-related—one occurred at a restaurant—then deglutition syncope (syncope precipitated by swallowing) should be considered. If an NSAID or aspirin was ingested before each episode, then medication hypersensitivity or mast cell degranulation (which can be triggered by these medications) should be further examined. Loss of consciousness lasting 20 minutes without causing any neurologic sequelae is unusual for most causes of recurrent syncope. This feature raises the possibility that a toxin or mediator might still be present in the patient’s system.
Serial cardiac enzymes and electrocardiogram were normal. A tilt-table study was negative. The cortisol response to ACTH (cosyntropin) stimulation was normal. The level of serum tryptase, drawn 2 days after syncope, was 18.4 ng/dL (normal, <11.5 ng/dL). Computed tomography scan of chest and abdomen was negative for pulmonary embolism but showed a 1.4×1.3-cm hypervascular lesion in the tail of pancreas. The following neuroendocrine tests were within normal limits: serum and urine catecholamines; urine 5-hydroxyindoleacetic acid (5-HIAA); and serum chromogranin A, insulin, serotonin, vasoactive intestinal polypeptide (VIP), and somatostatin (Table 1). The patient remained asymptomatic during his hospital stay and was discharged home with appointments for cardiology follow-up and endoscopic ultrasound-guided biopsy of the pancreatic mass.
Pheochromocytoma is unlikely with normal serum and urine catecholamine levels and normal adrenal images. The differential diagnosis for a pancreatic mass includes pancreatic carcinoma, lymphoma, cystic neoplasm, and neuroendocrine tumor. All markers of neuroendocrine excess are normal, though elevations can be episodic. The normal 5-HIAA level makes carcinoid syndrome unlikely. VIPomas are associated with flushing, but the absence of profound and protracted diarrhea makes a VIPoma unlikely.
As hypoglycemia from a pancreatic insulinoma is plausible as a cause of episodic loss of consciousness lasting 15 minutes or more, it is important to inquire if giving food or drink helped resolve previous episodes. The normal insulin level reported here is of limited value, because it is the combination of insulin and C-peptide levels at time of hypoglycemia that is diagnostic. The normal glucose level recorded during one of the earlier episodes and the hypotension argue against hypoglycemia.
The elevated tryptase level is an indicator of mast cell degranulation. Tryptase levels are transiently elevated during the initial 2 to 4 hours after an anaphylactic episode and then normalize. An elevated level many hours or days later is considered a sign of mast cell excess. Although there is no evidence of the multi-organ disease (eg, cytopenia, bone disease, hepatosplenomegaly) seen in patients with a high systemic burden of mast cells, mast cell disorders exist on a spectrum. There may be a focal excess of mast cells confined to one organ or an isolated mass.
The same day as discharge, the patient’s wife drove them to the grocery store. He remained in the car while she shopped. When she returned, she found him confused and minimally responsive with subsequent brief loss of consciousness. He was taken to an ED, where he was flushed and hypotensive (systolic BP, 60 mm Hg) and tachycardic. Other examination findings were normal. After fluid resuscitation he became alert and oriented. WBC count was 20,850/μL with 89% neutrophils, hemoglobin level was 14.6 g/dL, and platelet count was 168,000/μL. Serum lactate level was 3.7 mmol/L (normal, <2.3 mmol/L). Chest radiograph was normal. He was treated with broad-spectrum antibiotic therapy and admitted to the hospital. Blood and urine cultures were sterile. Fine-needle aspiration of the pancreatic mass demonstrated nonspecific inflammation. Four days after admission (3 days after pancreatic mass biopsy) the patient developed palpitations, felt unwell, and had marked flushing of the face and trunk, with concomitant BP of 90/50 mm Hg and HR of 140 bpm.
The salient features of this case are recurrent hypotension, tachycardia, and flushing. Autonomic insufficiency, to which elderly patients are prone, causes hemodynamic perturbations but rarely flushing. The patient does not have diabetes mellitus, Parkinson disease, or another condition that puts him at risk for dysautonomia. Pancreatic neuroendocrine tumors secrete mediators that lead to vasodilation and hypotension but are unlikely given the clinical and biochemical data.
The patient’s symptoms are consistent with anaphylaxis, though prototypical immunoglobulin E (IgE)–mediated anaphylaxis is usually accompanied by urticaria, angioedema, and wheezing, which have been absent during his presentations. There are no clear food, pharmacologic, or environmental precipitants.
Recurrent anaphylaxis can be a manifestation of mast cell excess (eg, cutaneous or systemic mastocytosis). A markedly elevated tryptase level during an anaphylactic episode is consistent with mastocytosis or IgE-mediated anaphylaxis. An elevated baseline tryptase level days after an anaphylactic episode signals increased mast cell burden. There may be a reservoir of mast cells in the bone marrow. Alternatively, the hypervascular pancreatic mass may be a mastocytoma or a mast cell sarcoma (missed because of inadequate sampling or staining).
The lactic acidosis likely reflects global tissue hypoperfusion from vasodilatory hypotension. The leukocytosis may reflect WBC mobilization secondary to endogenous corticosteroids and catecholamines in response to hypotension or may be a direct response to the release of mast cell–derived mediators of inflammation.
The patient was treated with diphenhydramine and ranitidine. Serum tryptase level was 46.8 ng/mL (normal, <11.5 ng/mL), and 24-hour urine histamine level was 95 µ g/dL (normal, <60 µ g/dL). Bone marrow biopsy results showed multifocal dense infiltrative aggregates of mast cells (>15 cells/aggregate), which were confirmed by CD117 (Kit) and tryptase positivity (Figure). Mutation analysis for Kit Asp816Val, which is present in 80% to 90% of patients with mastocytosis, was positive. He fulfilled the 2008 World Health Organization criteria for systemic mastocytosis (Table 2). Prednisone, histamine inhibitors, and montelukast were prescribed. Six months later, magnetic resonance imaging of the abdomen showed no change in the pancreatic mass, which was now characterized as a possible splenule. The patient had no additional episodes of flushing or syncope over 2 years.
DISCUSSION
Cardiovascular collapse (hypotension, tachycardia, syncope) in an elderly patient prompts clinicians to focus on life-threatening conditions, such as acute coronary syndrome, pulmonary embolus, arrhythmia, and sepsis. Each of these diagnoses was considered early in the course of this patient’s presentations, but each was deemed unlikely as it became apparent that the episodes were self-limited and recurrent over years. Incorporating flushing into the diagnostic problem representation allowed the clinicians to focus on a subset of causes of hypotension.
Flushing disorders may be classified by whether they are mediated by the autonomic nervous system (wet flushes, because they are usually accompanied by diaphoresis) or by exogenous or endogenous vasoactive substances (dry flushes).1 Autonomic nervous system flushing is triggered by emotions, fever, exercise, perimenopause (hot flashes), and neurologic conditions (eg, Parkinson disease, spinal cord injury, multiple sclerosis). Vasoactive flushing precipitants include drugs (eg, niacin); alcohol (secondary to cutaneous vasodilation, or acetaldehyde particularly in people with insufficient acetaldehyde dehydrogenase activity)2; foods that contain capsaicin, tyramine, sulfites, or histamine (eg, eating improperly handled fish can cause scombroid poisoning); and anaphylaxis. Rare causes of vasoactive flushing include carcinoid syndrome, pheochromocytoma, medullary thyroid carcinoma, VIPoma, and mastocytosis.2
Mastocytosis is a rare clonal disorder characterized by the accumulation of abnormal mast cells in the skin (cutaneous mastocytosis), in multiple organs (systemic mastocytosis), or in a solid tumor (mastocytoma). Urticaria pigmentosa is the most common form of cutaneous mastocytosis; it is seen more often in children than in adults and typically is associated with a maculopapular rash and dermatographism. Systemic mastocytosis is the most common form of the disorder in adults.3 Symptoms are related to mast cell infiltration or mast cell mediator–related effects, which range from itching, flushing, and diarrhea to hypotension and anaphylaxis. Other manifestations are fatigue, urticaria pigmentosa, osteoporosis, hepatosplenomegaly, bone pain, cytopenias, and lymphadenopathy.4
Systemic mastocytosis can occur at any age and should be considered in patients with recurrent unexplained flushing, syncope, or hypotension. Eighty percent to 90% of patients with systemic mastocytosis have a mutation in Kit,5 a transmembrane tyrosine kinase that is the receptor for stem cell factor. The Asp816Val mutation leads to increased proliferation and reduced apoptosis of mast cells.3,6,7 Proposed diagnostic algorithms8-11 involve measurement of serum tryptase levels and examination of bone marrow. Bone marrow biopsy and testing for the Asp816Val
The primary goals of treatment are managing mast cell–mediated symptoms and, in advanced cases, achieving cytoreduction. Alcohol can trigger mast cell degranulation in indolent systemic mastocytosis and should be avoided. Mast cell–mediated symptoms are managed with histamine blockers, leukotriene antagonists, and mast cell stabilizers.12 Targeted therapy with tyrosine kinase inhibitors (eg, imatinib) in patients with transmembrane Kit mutation (eg, Phe522Cys, Lys509Ile) associated with systemic mastocytosis has had promising results.13,14 However, this patient’s Asp816Val mutation is in the Kit catalytic domain, not the transmembrane region, and therefore would not be expected to respond to imatinib. A recent open-label trial of the multikinase inhibitor midostaurin demonstrated resolution of organ damage, reduced bone marrow burden, and lowered serum tryptase levels in patients with advanced systemic mastocytosis.15 Interferon, cladribine, and high-dose corticosteroids are prescribed in patients for whom other therapies have been ineffective.8
The differential diagnosis is broad for both hypotension and for flushing, but the differential diagnosis for recurrent hypotension and flushing is limited. Recognizing that flushing was an essential feature of this patient’s hypotensive condition, and not an epiphenomenon of syncope, allowed the clinicians to focus on the overlap and make a shocking diagnosis.
Acknowledgment
The authors thank David Bosler, MD (Cleveland Clinic) for interpreting the pathology image.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 75-year-old man was brought by ambulance to the emergency department (ED) after the acute onset of palpitations, lightheadedness, and confusion. His medical history, provided by his wife, included osteoarthritis and remote cholecystectomy. He was not a smoker but drank 2 to 4 cans of beer daily. His medications were aspirin 162 mg daily and naproxen as needed. There was no history of bruising, diarrhea, melena, or bleeding.
Palpitations may represent an arrhythmia arising from an ischemic or alcoholic cardiomyopathy. Mental status changes usually have metabolic, infectious, structural (eg, hemorrhage, tumor), or toxic causes. Lightheadedness and confusion could occur with arrhythmia-associated cerebral hypoperfusion or a seizure. Daily alcohol use could cause confusion through acute intoxication, thiamine or B12 deficiency, repeated head trauma, or liver failure.
The patient’s systolic blood pressure (BP) was 60 mm Hg, heart rate (HR) was 120 beats per minute (bpm), and oral temperature was 98.4°F. Rousing him was difficult. There were no localizing neurologic abnormalities, and the rest of the physical examination findings were normal. Point-of-care blood glucose level was 155 mg/dL. Blood cultures were obtained and broad-spectrum antibiotics initiated. After fluid resuscitation, BP improved to 116/87 mm Hg, HR fell to 105 bpm, and the patient became alert and oriented. He denied chest pain, fever, or diaphoresis.
The patient’s improvement with intravenous (IV) fluids makes cardiogenic shock unlikely but does not exclude an underlying compensated cardiomyopathy that may be predisposing to arrhythmia. Hypotension, tachycardia, and somnolence may represent sepsis, but the near normalization of vital signs and mental status shortly after administration of IV fluids, the normal temperature, and the absence of localizing signs of infection favor withholding additional antibiotics. Other causes of hypotension are hypovolemia, medication effects, adrenal insufficiency, anaphylaxis, and autonomic insufficiency. There was no reported nausea, vomiting, diarrhea, bleeding, polyuria, or impaired oral intake to support hypovolemia, though the response to IV fluids suggests hypovolemia may still be playing a role.
White blood cell (WBC) count was 15,450/µL with a normal differential; hemoglobin level was 15.8 g/dL; and platelet count was 176,000/µL. Electrolytes, liver function tests, cardiac enzymes, and urinalysis were normal. Electrocardiogram showed sinus tachycardia with premature atrial complexes and no ST-segment abnormalities. Radiograph of the chest and computed tomography scan of the head were normal. Echocardiogram showed moderate left ventricular hypertrophy with a normal ejection fraction and no valvular abnormalities. Exercise nuclear cardiac stress test was negative for ischemia. Blood cultures were sterile. The patient quickly became asymptomatic and remained so during his 3-day hospitalization. There were no arrhythmias on telemetry. The patient was discharged with follow-up scheduled with his primary care physician.
The nonlocalizing history and physical examination findings, normal chest radiograph and urinalysis, absence of fevers, negative blood cultures, and quick recovery make infection unlikely, despite the moderate leukocytosis. Conditions that present with acute and transient hypotension and altered mental status include arrhythmias, seizures, and reactions to drugs or toxins. Given the cardiac test results, a chronic cardiomyopathy seems unlikely, but arrhythmia is still possible. Continuous outpatient monitoring is required to assess the palpitations and the frequency of the premature atrial complexes.
Two days after discharge, the patient suddenly became diaphoretic and lost consciousness while walking to the bathroom. He was taken to the ED, where his BP was 90/60 mm Hg and HR was 108 bpm. Family members reported that he had appeared flushed during the syncopal episode, showed no seizure activity, and been unconscious for 15 to 20 minutes. The patient denied chest pain, dyspnea, fever, bowel or bladder incontinence, focal weakness, slurred speech, visual changes, nausea or vomiting either before or after the episode. Physical examination revealed a tongue laceration and facial erythema; all other findings were normal. In the ED, there was an asymptomatic 7-beat run of nonsustained ventricular tachycardia, and the hypotension resolved after fluid resuscitation. The patient now reported 2 similar syncopal episodes in the past. The first occurred in a restaurant 6 years earlier, and the second occurred 3 years later, at which time he was hospitalized and no etiology was found.
The loss of consciousness is attributable to cerebral hypoperfusion. Hypotension has 3 principal categories: hypovolemic, cardiogenic, and distributive. With syncopal episodes recurring over several years, hypovolemia seems unlikely. Given the palpitations and ventricular tachycardia, it is reasonable to suspect a cardiogenic cause. Although his heart appears to be structurally normal on echocardiogram, genetic, electrophysiologic, or magnetic resonance imaging (MRI) testing will occasionally reveal an unsuspected substrate for arrhythmia.
The recurring yet self-limited nature, diaphoresis, flushing, and facial erythema suggest a non-sepsis distributive cause of hypotension. It is possible the patient is recurrently exposed to a toxin (eg, alcohol) that causes both flushing and dehydration. Flushing disorders include carcinoid syndrome, pheochromocytoma, drug reaction with eosinophilia and systemic symptoms (DRESS), and mastocytosis. Carcinoid syndrome is characterized by bronchospasm and diarrhea and, in some cases, right-sided valvulopathy, all of which are absent in this patient. Pheochromocytoma is associated with orthostasis, but patients typically are hypertensive at baseline. DRESS, which may arise from nonsteroidal anti-inflammatory drug (NSAID) or aspirin use, can cause facial erythema and swelling but is also characterized by liver, renal, and hematologic abnormalities, none of which was demonstrated. Furthermore, DRESS typically does not cause hypotension. Mastocytosis can manifest as isolated or recurrent anaphylaxis.
It is important to investigate antecedents of these syncopal episodes. If the earlier episodes were food-related—one occurred at a restaurant—then deglutition syncope (syncope precipitated by swallowing) should be considered. If an NSAID or aspirin was ingested before each episode, then medication hypersensitivity or mast cell degranulation (which can be triggered by these medications) should be further examined. Loss of consciousness lasting 20 minutes without causing any neurologic sequelae is unusual for most causes of recurrent syncope. This feature raises the possibility that a toxin or mediator might still be present in the patient’s system.
Serial cardiac enzymes and electrocardiogram were normal. A tilt-table study was negative. The cortisol response to ACTH (cosyntropin) stimulation was normal. The level of serum tryptase, drawn 2 days after syncope, was 18.4 ng/dL (normal, <11.5 ng/dL). Computed tomography scan of chest and abdomen was negative for pulmonary embolism but showed a 1.4×1.3-cm hypervascular lesion in the tail of pancreas. The following neuroendocrine tests were within normal limits: serum and urine catecholamines; urine 5-hydroxyindoleacetic acid (5-HIAA); and serum chromogranin A, insulin, serotonin, vasoactive intestinal polypeptide (VIP), and somatostatin (Table 1). The patient remained asymptomatic during his hospital stay and was discharged home with appointments for cardiology follow-up and endoscopic ultrasound-guided biopsy of the pancreatic mass.
Pheochromocytoma is unlikely with normal serum and urine catecholamine levels and normal adrenal images. The differential diagnosis for a pancreatic mass includes pancreatic carcinoma, lymphoma, cystic neoplasm, and neuroendocrine tumor. All markers of neuroendocrine excess are normal, though elevations can be episodic. The normal 5-HIAA level makes carcinoid syndrome unlikely. VIPomas are associated with flushing, but the absence of profound and protracted diarrhea makes a VIPoma unlikely.
As hypoglycemia from a pancreatic insulinoma is plausible as a cause of episodic loss of consciousness lasting 15 minutes or more, it is important to inquire if giving food or drink helped resolve previous episodes. The normal insulin level reported here is of limited value, because it is the combination of insulin and C-peptide levels at time of hypoglycemia that is diagnostic. The normal glucose level recorded during one of the earlier episodes and the hypotension argue against hypoglycemia.
The elevated tryptase level is an indicator of mast cell degranulation. Tryptase levels are transiently elevated during the initial 2 to 4 hours after an anaphylactic episode and then normalize. An elevated level many hours or days later is considered a sign of mast cell excess. Although there is no evidence of the multi-organ disease (eg, cytopenia, bone disease, hepatosplenomegaly) seen in patients with a high systemic burden of mast cells, mast cell disorders exist on a spectrum. There may be a focal excess of mast cells confined to one organ or an isolated mass.
The same day as discharge, the patient’s wife drove them to the grocery store. He remained in the car while she shopped. When she returned, she found him confused and minimally responsive with subsequent brief loss of consciousness. He was taken to an ED, where he was flushed and hypotensive (systolic BP, 60 mm Hg) and tachycardic. Other examination findings were normal. After fluid resuscitation he became alert and oriented. WBC count was 20,850/μL with 89% neutrophils, hemoglobin level was 14.6 g/dL, and platelet count was 168,000/μL. Serum lactate level was 3.7 mmol/L (normal, <2.3 mmol/L). Chest radiograph was normal. He was treated with broad-spectrum antibiotic therapy and admitted to the hospital. Blood and urine cultures were sterile. Fine-needle aspiration of the pancreatic mass demonstrated nonspecific inflammation. Four days after admission (3 days after pancreatic mass biopsy) the patient developed palpitations, felt unwell, and had marked flushing of the face and trunk, with concomitant BP of 90/50 mm Hg and HR of 140 bpm.
The salient features of this case are recurrent hypotension, tachycardia, and flushing. Autonomic insufficiency, to which elderly patients are prone, causes hemodynamic perturbations but rarely flushing. The patient does not have diabetes mellitus, Parkinson disease, or another condition that puts him at risk for dysautonomia. Pancreatic neuroendocrine tumors secrete mediators that lead to vasodilation and hypotension but are unlikely given the clinical and biochemical data.
The patient’s symptoms are consistent with anaphylaxis, though prototypical immunoglobulin E (IgE)–mediated anaphylaxis is usually accompanied by urticaria, angioedema, and wheezing, which have been absent during his presentations. There are no clear food, pharmacologic, or environmental precipitants.
Recurrent anaphylaxis can be a manifestation of mast cell excess (eg, cutaneous or systemic mastocytosis). A markedly elevated tryptase level during an anaphylactic episode is consistent with mastocytosis or IgE-mediated anaphylaxis. An elevated baseline tryptase level days after an anaphylactic episode signals increased mast cell burden. There may be a reservoir of mast cells in the bone marrow. Alternatively, the hypervascular pancreatic mass may be a mastocytoma or a mast cell sarcoma (missed because of inadequate sampling or staining).
The lactic acidosis likely reflects global tissue hypoperfusion from vasodilatory hypotension. The leukocytosis may reflect WBC mobilization secondary to endogenous corticosteroids and catecholamines in response to hypotension or may be a direct response to the release of mast cell–derived mediators of inflammation.
The patient was treated with diphenhydramine and ranitidine. Serum tryptase level was 46.8 ng/mL (normal, <11.5 ng/mL), and 24-hour urine histamine level was 95 µ g/dL (normal, <60 µ g/dL). Bone marrow biopsy results showed multifocal dense infiltrative aggregates of mast cells (>15 cells/aggregate), which were confirmed by CD117 (Kit) and tryptase positivity (Figure). Mutation analysis for Kit Asp816Val, which is present in 80% to 90% of patients with mastocytosis, was positive. He fulfilled the 2008 World Health Organization criteria for systemic mastocytosis (Table 2). Prednisone, histamine inhibitors, and montelukast were prescribed. Six months later, magnetic resonance imaging of the abdomen showed no change in the pancreatic mass, which was now characterized as a possible splenule. The patient had no additional episodes of flushing or syncope over 2 years.
DISCUSSION
Cardiovascular collapse (hypotension, tachycardia, syncope) in an elderly patient prompts clinicians to focus on life-threatening conditions, such as acute coronary syndrome, pulmonary embolus, arrhythmia, and sepsis. Each of these diagnoses was considered early in the course of this patient’s presentations, but each was deemed unlikely as it became apparent that the episodes were self-limited and recurrent over years. Incorporating flushing into the diagnostic problem representation allowed the clinicians to focus on a subset of causes of hypotension.
Flushing disorders may be classified by whether they are mediated by the autonomic nervous system (wet flushes, because they are usually accompanied by diaphoresis) or by exogenous or endogenous vasoactive substances (dry flushes).1 Autonomic nervous system flushing is triggered by emotions, fever, exercise, perimenopause (hot flashes), and neurologic conditions (eg, Parkinson disease, spinal cord injury, multiple sclerosis). Vasoactive flushing precipitants include drugs (eg, niacin); alcohol (secondary to cutaneous vasodilation, or acetaldehyde particularly in people with insufficient acetaldehyde dehydrogenase activity)2; foods that contain capsaicin, tyramine, sulfites, or histamine (eg, eating improperly handled fish can cause scombroid poisoning); and anaphylaxis. Rare causes of vasoactive flushing include carcinoid syndrome, pheochromocytoma, medullary thyroid carcinoma, VIPoma, and mastocytosis.2
Mastocytosis is a rare clonal disorder characterized by the accumulation of abnormal mast cells in the skin (cutaneous mastocytosis), in multiple organs (systemic mastocytosis), or in a solid tumor (mastocytoma). Urticaria pigmentosa is the most common form of cutaneous mastocytosis; it is seen more often in children than in adults and typically is associated with a maculopapular rash and dermatographism. Systemic mastocytosis is the most common form of the disorder in adults.3 Symptoms are related to mast cell infiltration or mast cell mediator–related effects, which range from itching, flushing, and diarrhea to hypotension and anaphylaxis. Other manifestations are fatigue, urticaria pigmentosa, osteoporosis, hepatosplenomegaly, bone pain, cytopenias, and lymphadenopathy.4
Systemic mastocytosis can occur at any age and should be considered in patients with recurrent unexplained flushing, syncope, or hypotension. Eighty percent to 90% of patients with systemic mastocytosis have a mutation in Kit,5 a transmembrane tyrosine kinase that is the receptor for stem cell factor. The Asp816Val mutation leads to increased proliferation and reduced apoptosis of mast cells.3,6,7 Proposed diagnostic algorithms8-11 involve measurement of serum tryptase levels and examination of bone marrow. Bone marrow biopsy and testing for the Asp816Val
The primary goals of treatment are managing mast cell–mediated symptoms and, in advanced cases, achieving cytoreduction. Alcohol can trigger mast cell degranulation in indolent systemic mastocytosis and should be avoided. Mast cell–mediated symptoms are managed with histamine blockers, leukotriene antagonists, and mast cell stabilizers.12 Targeted therapy with tyrosine kinase inhibitors (eg, imatinib) in patients with transmembrane Kit mutation (eg, Phe522Cys, Lys509Ile) associated with systemic mastocytosis has had promising results.13,14 However, this patient’s Asp816Val mutation is in the Kit catalytic domain, not the transmembrane region, and therefore would not be expected to respond to imatinib. A recent open-label trial of the multikinase inhibitor midostaurin demonstrated resolution of organ damage, reduced bone marrow burden, and lowered serum tryptase levels in patients with advanced systemic mastocytosis.15 Interferon, cladribine, and high-dose corticosteroids are prescribed in patients for whom other therapies have been ineffective.8
The differential diagnosis is broad for both hypotension and for flushing, but the differential diagnosis for recurrent hypotension and flushing is limited. Recognizing that flushing was an essential feature of this patient’s hypotensive condition, and not an epiphenomenon of syncope, allowed the clinicians to focus on the overlap and make a shocking diagnosis.
Acknowledgment
The authors thank David Bosler, MD (Cleveland Clinic) for interpreting the pathology image.
Disclosure
Nothing to report.
1. Wilkin JK. The red face: flushing disorders. Clin Dermatol. 1993;11(2):211-223. PubMed
2. Izikson L, English JC 3rd, Zirwas MJ. The flushing patient: differential diagnosis, workup, and treatment. J Am Acad Dermatol. 2006;55(2):193-208. PubMed
3. Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37(6):435-453. PubMed
4. Hermans MA, Rietveld MJ, van Laar JA, et al. Systemic mastocytosis: a cohort study on clinical characteristics of 136 patients in a large tertiary centre. Eur J Intern Med. 2016;30:25-30. PubMed
5. Kristensen T, Vestergaard H, Bindslev-Jensen C, Møller MB, Broesby-Olsen S; Mastocytosis Centre, Odense University Hospital (MastOUH). Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am J Hematol. 2014;89(5):493-498. PubMed
6. Verstovsek S. Advanced systemic mastocytosis: the impact of KIT mutations in diagnosis, treatment, and progression. Eur J Haematol. 2013;90(2):89-98. PubMed
7. Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108(7):2366-2372. PubMed
8. Pardanani A. Systemic mastocytosis in adults: 2015 update on diagnosis, risk stratification, and management. Am J Hematol. 2015;90(3):250-262. PubMed
9. Valent P, Aberer E, Beham-Schmid C, et al. Guidelines and diagnostic algorithm for patients with suspected systemic mastocytosis: a proposal of the Austrian Competence Network (AUCNM). Am J Blood Res. 2013;3(2):174-180. PubMed
10. Valent P, Escribano L, Broesby-Olsen S, et al; European Competence Network on Mastocytosis. Proposed diagnostic algorithm for patients with suspected mastocytosis: a proposal of the European Competence Network on Mastocytosis. Allergy. 2014;69(10):1267-1274. PubMed
11. Akin C, Soto D, Brittain E, et al. Tryptase haplotype in mastocytosis: relationship to disease variant and diagnostic utility of total tryptase levels. Clin Immunol. 2007;123(3):268-271. PubMed
12. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(19):1885-1886. PubMed
13. Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103(8):3222-3225. PubMed
14. Zhang LY, Smith ML, Schultheis B, et al. A novel K509I mutation of KIT identified in familial mastocytosis—in vitro and in vivo responsiveness to imatinib therapy. Leuk Res. 2006;30(4):373-378. PubMed
15. Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374(26):2530-2541. PubMed
1. Wilkin JK. The red face: flushing disorders. Clin Dermatol. 1993;11(2):211-223. PubMed
2. Izikson L, English JC 3rd, Zirwas MJ. The flushing patient: differential diagnosis, workup, and treatment. J Am Acad Dermatol. 2006;55(2):193-208. PubMed
3. Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37(6):435-453. PubMed
4. Hermans MA, Rietveld MJ, van Laar JA, et al. Systemic mastocytosis: a cohort study on clinical characteristics of 136 patients in a large tertiary centre. Eur J Intern Med. 2016;30:25-30. PubMed
5. Kristensen T, Vestergaard H, Bindslev-Jensen C, Møller MB, Broesby-Olsen S; Mastocytosis Centre, Odense University Hospital (MastOUH). Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am J Hematol. 2014;89(5):493-498. PubMed
6. Verstovsek S. Advanced systemic mastocytosis: the impact of KIT mutations in diagnosis, treatment, and progression. Eur J Haematol. 2013;90(2):89-98. PubMed
7. Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108(7):2366-2372. PubMed
8. Pardanani A. Systemic mastocytosis in adults: 2015 update on diagnosis, risk stratification, and management. Am J Hematol. 2015;90(3):250-262. PubMed
9. Valent P, Aberer E, Beham-Schmid C, et al. Guidelines and diagnostic algorithm for patients with suspected systemic mastocytosis: a proposal of the Austrian Competence Network (AUCNM). Am J Blood Res. 2013;3(2):174-180. PubMed
10. Valent P, Escribano L, Broesby-Olsen S, et al; European Competence Network on Mastocytosis. Proposed diagnostic algorithm for patients with suspected mastocytosis: a proposal of the European Competence Network on Mastocytosis. Allergy. 2014;69(10):1267-1274. PubMed
11. Akin C, Soto D, Brittain E, et al. Tryptase haplotype in mastocytosis: relationship to disease variant and diagnostic utility of total tryptase levels. Clin Immunol. 2007;123(3):268-271. PubMed
12. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(19):1885-1886. PubMed
13. Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103(8):3222-3225. PubMed
14. Zhang LY, Smith ML, Schultheis B, et al. A novel K509I mutation of KIT identified in familial mastocytosis—in vitro and in vivo responsiveness to imatinib therapy. Leuk Res. 2006;30(4):373-378. PubMed
15. Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374(26):2530-2541. PubMed
© 2017 Society of Hospital Medicine
Getting Warmer
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 3-month-old otherwise healthy, immunized female presented to clinic with 2 days of intermittent low-grade fevers (maximum, 100º F), decreased oral intake, and sleepiness. Her pediatrician noted a faint, maculopapular rash on her trunk and extremities with mild conjunctival injection bilaterally that appeared that day, according to her mother. The infant otherwise appeared alert, well-hydrated, and without respiratory distress. She had no history of sick contacts or recent travel. She was prescribed amoxicillin for empiric treatment of a possible bacterial sinusitis or pharyngitis, despite a negative rapid strep antigen test.
At this age, multiple conditions can cause rashes. Given that this is early in the course of illness, without focal symptoms but with low-grade fevers, the initial differential diagnosis is broad and would include infectious, rheumatologic, and hematologic-oncologic etiologies, although the latter would be less likely. While the patient’s mother reports decreased oral intake, the fact that the patient is alert and appears hydrated is encouraging, suggesting time to observe and see if other symptoms present that may assist in elucidating the cause. The history of increased sleepiness warrants further investigation of meningeal signs, which would point to a central nervous system infection.
While streptococcal infection is possible, it would be uncommon at this age. The patient would have a higher fever and focal infection, and the rash does not appear consistent unless it was described as “sandpaper” in feel and appearance. A negative rapid strep test, while not sensitive, further supports this impression. A low-grade fever and rash would be consistent with a viral syndrome and, given the conjunctival injection, adenovirus, cytomegalovirus, rhinovirus, and Epstein Barr virus (EBV) are possibilities. Without ocular discharge, bacterial conjunctivitis would be unlikely. Another consideration would be Kawasaki disease, though it would be too early to diagnose this condition since at least 5 days of fever are required. Next steps include a detailed physical examination, looking for other focal signs such as swelling or desquamation of hands and feet, lymphadenopathy, strawberry tongue, and mucositis. Rather than empirically starting antibiotics, it would be more reasonable to observe her with close outpatient follow-up. The patient’s family should be instructed to monitor for additional and/or worsening symptoms, further decreased oral intake, signs of dehydration, or changes in alertness.
At home, the patient completed 5 doses of amoxicillin but continued to be febrile (maximum, 102.6º F). She was taken to a local emergency department on day 6 of her illness. She had worsening conjunctival injection and progression of the rash, involving the palms and soles. She was noted to have edema of hands and feet without desquamation (Figure 1). She had no oral mucous membrane changes and no cervical lymphadenopathy. Cerebrospinal fluid (CSF) was unremarkable, and empiric treatment with intravenous (IV) ceftriaxone was initiated. Complete blood count was notable for a white blood cell (WBC) count of 18.9 k/μL (normal range, 6.0-17.0); hemoglobin, 7.6 g/dL (normal range, 10-13); mean corpuscular volume, 84 (normal range, 74-108); and platelet count, 105 k/μL (normal range, 150-400). A peripheral blood smear revealed no abnormal cells. C-reactive protein (CRP) was elevated at 6.5 mg/dL (normal range, 0.0-0.6). She was admitted for further management.

Infection remains on the differential diagnosis given the elevated WBC count. Since the patient has completed a reasonable course of antibiotics, a bacterial infection would be less likely but not fully excluded. The cultures obtained would be helpful if they become positive, but given that the patient has been on antibiotics, a negative culture may represent partial sterilization and would not rule out infection. A viral infection continues to be high on the differential, but one would expect that symptoms and fever would have begun to abate. The normal peripheral blood smear makes a hematologic disorder less likely.
Kawasaki disease has risen on the differential with 5 days of fever surpassing 102º F. She has 3 of 5 primary clinical criteria, including conjunctival injection, rash, and edema of the hands and feet. Desquamation of the peripheral extremities would not be expected until the convalescent phase. A diagnosis of typical Kawasaki disease would require a fourth criterion, either oral mucous membrane changes or cervical lymphadenopathy. She meets the criteria for atypical or incomplete Kawasaki disease, which requires only fever for at least 5 days, elevated CRP, and 2 or 3 additional clinical criteria. She also meets supplemental laboratory criteria with an elevated WBC count greater than 15,000/μL, normocytic and normochromic anemia for age, and elevated CRP. Urinalysis positive for pyuria or serum albumin less than 3 g/dL would lend further support but is not necessary. Fever of 7 or more days in a child less than 6 months old without other explanation would also increase the likelihood of incomplete Kawasaki disease. Admission to the hospital, treatment with IV immunoglobulin (IVIg), and echocardiography to evaluate for typical cardiac involvement (eg, aneurysms, coronary arteritis, and pericardial effusion) are the appropriate next steps.
The patient was diagnosed with atypical Kawasaki disease. A transthoracic echocardiogram was normal on admission. On day 7 of her illness, she was treated with 1 dose of IVIg at 2 g/kg and high-dose aspirin at 100 mg/kg per day in divided doses. Despite this treatment, she continued to be febrile and was given a second dose of IVIg on day 9. Her fevers persisted.
In Kawasaki disease, persistent fever is concerning for long-term sequelae, including coronary artery aneurysms. Continued treatment is reasonable. After 2 doses of IVIg with a cumulative dose of 4 g/kg, it is prudent to switch therapy to IV methylprednisolone 30mg/kg with repeated doses as needed for up to 3 days should her fevers persist.
Her blood culture was negative. EBV serology, enterovirus polymerase chain reaction, and viral cultures were negative. Chest radiography on day 9 was normal. Abdominal ultrasonography on day 10 showed hydrops of the gallbladder.
The patient was started on IV corticosteroids on day 11 with resolution of her fevers and improvement in her rash. A repeat echocardiogram revealed new findings of dilated left main, left anterior descending, and right coronary arteries. On day 13, a steroid wean was attempted because she had remained afebrile for more than 48 hours, but the wean was halted due to recurrence of fevers and rash. Her high-dose aspirin was reduced to 81 mg PO daily on day 14, and she was started on enoxaparin injections.
It is unusual for Kawasaki disease not to respond to 2 doses of IVIg, followed by corticosteroids. As such, the differential diagnosis must be revisited. The findings of coronary artery dilation, prolonged fever, and rash corroborate the diagnosis of Kawasaki disease, although this could be an atypical presentation of another vasculitis. Systemic onset juvenile idiopathic arthritis usually affects children at 2 to 5 years old and is, therefore, less likely. Henoch-Schönlein purpura manifests with a rash but is often associated with diarrhea. There does not appear to be objective evidence of polyarteritis nodosa, although biopsy or angiography would be required to make this diagnosis. Hydrops of the gallbladder is an over-distention of the organ filled with watery or mucoid content. While hydrops can be noninflammatory and seen in gallstone disease, it can also occur in vasculitides. Despite the reassuring serologies, false negative results are possible. Thus, these viral infections are not eliminated, but they are less likely. Given the echocardiogram findings and continued concern for atypical Kawasaki disease, high-dose aspirin should be continued. It is reasonable to consider rheumatology consultation for assessment and recommendations as to length of steroid treatment and/or alternative interventions.
Pediatric cardiology was consulted. Repeat echocardiogram on day 16 showed an increase in the size of her coronary artery aneurysms, and her fevers persisted. Computed tomography scan of the abdomen and pelvis with contrast, obtained to further evaluate for a source of infection, was unremarkable.
The patient was transferred to a tertiary care institution on day 19, at which time she remained on aspirin, enoxaparin, and oral corticosteroids. On arrival, her temperature was 101.3º F, heart rate 225 beats per minute, and respiratory rate 57 breaths per minute. She was fussy with bilateral conjunctivitis and a maculopapular rash involving palms, soles, and right infraorbital region. Laboratory studies were significant for a WBC count of 30.3 k/μL; hemoglobin, 10.9 g/dL; platelets, 106 k/μL; and CRP, 8.3 mg/dL.
Pediatric rheumatology was consulted on day 20. The patient was treated with 3 days IV pulse-dose methylprednisolone at 30 mg/kg daily. Her fevers resolved, although her CRP level remained elevated. She was treated with 1 dose of infliximab 10 mg/kg IV on day 24, followed by 1 dose of anakinra 15 mg subcutaneously on day 27 due to persistently elevated CRP.
The symptoms and diagnostic evaluation remain most consistent with atypical Kawasaki disease. Her tachycardia and tachypnea are likely driven by her fever and fussiness, and should be followed closely. The elevated WBC is likely a consequence of the steroids and demargination of neutrophils. The elevated and increasing CRP is a marker of acute inflammation. The adage “treat the patient, not the numbers” comes to mind, because it is reassuring that the patient’s overall clinical picture seems to be improving with resolution of her fevers. However, further discussion with the pediatric rheumatology consultant is prudent, specifically regarding the significance of the persistently elevated CRP, refinement of the differential diagnosis including the potential for other vasculitides and appropriate evaluation of such, as well as recommendations for further treatment.
The patient was noted to have ongoing fevers. Based on reports of success with cyclophosphamide in refractory Kawasaki disease, she was treated with 2 doses at 60 mg IV per dose starting on day 28. Her CRP level decreased. Cardiology and rheumatology consultants recommended magnetic resonance imaging/magnetic resonance angiography of the chest, abdomen, and pelvis with and without contrast. These studies revealed dilation of the axillary and brachial arteries (Figure 2).

The response to cyclophosphamide confirms an autoimmune/inflammatory process. The imaging results and pattern are most consistent with either Kawasaki disease or polyarteritis nodosa. Therefore, rheumatology’s input will be invaluable with regard to which diagnosis is most likely, additional diagnostic testing, and appropriate medical regimen and follow-up plans.
Systemic extracoronary vascular inflammation on imaging and the refractory nature of the patient’s disease process, despite appropriate treatment for Kawasaki disease, led to the diagnosis of childhood polyarteritis nodosa (PAN). The patient was discharged home and closely followed in rheumatology clinic. Her most recent outpatient visit 1 year after the initial onset of her illness showed no further fevers or rashes, normal inflammatory markers, and stabilization of her coronary aneurysms on daily maintenance azathioprine.
DISCUSSION
Fever with an accompanying rash is a common issue in children. The extensive differential diagnosis includes infectious diseases, rheumatologic disorders, and medication reactions (Table 1). A thorough history and physical examination are essential in guiding the physician toward the proper diagnosis and management. Important information includes patient age, season, associated symptoms, exposure to sick contacts, travel history, host immune status, and immunization history. Fever duration and pattern must be elicited, as should features of the rash, including temporal relationship to the fever, distribution, progression, and morphology.1

When unexplained fever persists for 5 days or more in the pediatric patient, the diagnosis of KD must be suspected. KD is an acute, febrile, primary systemic vasculitis affecting small- and medium-sized vessels, with a predilection for coronary arteries.2 KD affects younger children, with approximately 85% of cases occurring in children under 5 years old. KD has a higher incidence in Asian populations, suggesting a possible genetic predisposition.3 The etiology of KD is not well understood, but infection and immune dysregulation have been proposed as contributing factors. KD is the leading cause of acquired heart disease in developed countries.2
The diagnosis of KD is made clinically (Table 2). Atypical KD is considered in patients with at least 5 days of fever but only 2 or 3 clinical criteria. Supportive laboratory findings include elevated inflammatory markers, anemia, neutrophilia, abnormal plasma lipids, low albumin, sterile pyuria, CSF pleocytosis, and elevated serum transaminases. Two-dimensional echocardiography should be performed in all children with definite or suspected KD at the time of diagnosis, 1 to 2 weeks later, and 6 weeks following discharge for evaluation of the coronary arteries, left ventricular function, and valve function. The American Heart Association recommends follow-up echocardiography at 1 year in children without coronary vessel involvement.4

Treatment is aimed at minimizing inflammation and coronary artery involvement, and should be initiated promptly.5 Therapy includes a single infusion of high-dose IVIg and aspirin;6,7 the latter is initially provided at high anti-inflammatory doses, followed by lower antithrombotic doses once fever and laboratory markers have resolved.2 Aspirin can be discontinued if there is no evidence of coronary involvement at the 6-week follow-up echocardiogram.5 A second dose of IVIg is given within 48 hours for refractory cases, defined as persistent fever following the first dose of IVIg.4 Fifteen percent of children have refractory illness, and refractory KD is associated with a higher risk of coronary artery lesions.5 Additional agents that suppress immune activation and cytokine secretion contributing to KD pathogenesis have been studied. Corticosteroids inhibit phospholipase A, an enzyme required for production of inflammatory markers.8 Infliximab, a tumor necrosis factor-alpha inhibitor, has been shown to reduce duration of fever and length of hospital stay.8,9 Anakinra, an interleukin-1 receptor antagonist, has been shown to decrease fever duration and prevent progression of vascular injury in cases of refractory KD.10 There is, however, a lack of sufficient evidence and consensus on best practice.8-10
If inflammation, evidenced by fever, elevated inflammatory markers (such as erythrocyte sedimentation rate, CRP), or vessel involvement on imaging, persists or worsens despite standard therapy, physicians should seek alternative diagnoses. This patient’s extracoronary vascular inflammation and favorable response only to cyclophosphamide led to the diagnosis of systemic PAN. Like KD, PAN is a multi-system vasculitis affecting small- and medium-sized vessels. Unlike KD, PAN is rarely seen in children.11 Historically, PAN was thought to represent an extreme fatal end of the KD spectrum. Today, PAN is accepted as a separate entity. Clinical features and histological findings often overlap with KD, creating a diagnostic dilemma for providers.12
At the onset of illness, clinical features of systemic PAN may include recurrent fever, weight loss, and myalgia, with gradual progression to multi-organ system involvement. Laboratory assessment reveals elevated inflammatory markers and leukocytosis. Thrombocytosis, anemia, proteinuria, and hematuria may be present. A positive antineutrophil cytoplasmic antibody is rare in PAN and should raise suspicion for a microscopic polyangiitis, which is distinguished from PAN by small vessel involvement only. When compared to KD, cardiac vessel involvement in PAN is more variable.11 Diagnostic criteria for childhood PAN are listed in Table 2.13
Treatment of PAN is aimed at inducing remission with high-dose steroids and cyclophosphamide. Maintenance of remission is achieved using low-dose steroids and azathioprine.11 Total duration of treatment averages 2 to 3 years, with a minimum of 18 months.14 Plasma exchange has been used in severe, life-threatening cases.11 Prognosis for children with PAN is more favorable compared to adults with PAN, in whom the mortality rate is as high as 20% to 30%, even with aggressive treatment. In 1 multicenter study of childhood and adolescent PAN, overall mortality was 1.1%.15
This patient initially presented with findings consistent with KD. As her inflammatory markers remained elevated and fevers persisted, her physicians appropriately reconsidered the etiology of her symptoms, thereby “getting warmer” in the search for the correct diagnosis of systemic PAN, a rare disease and a separate entity from KD. Recognizing the overlapping and distinct clinical features of each entity can promote more timely and appropriate selection of therapy, thereby minimizing clinical manifestations and complications associated with each vasculitis.
KEY TEACHING POINTS
- KD and childhood PAN are disseminated vasculitides affecting small- and medium-sized vessels. Although they are distinct entities, KD and PAN exhibit overlapping clinical and pathological features that make appropriate diagnosis and treatment challenging.
- In cases of refractory KD, alternative diagnoses should be considered.
- Recognizing the individual features of both entities is imperative because treatment differs: KD is treated with high-dose aspirin and IVIg; corticosteroids and immunosuppressive agents are used to treat PAN.
Disclosure
Nothing to report.
1. McKinnon HD Jr, Howard T. Evaluating the febrile patient with a rash. Am Fam Physician. 2000;62:804-816. PubMed
2. Dimitriades V, Brown AG, Gedalia A. Kawasaki disease: pathophysiology, clinical manifestations, and management. Curr Rheumatol Rep. 2014;16:423. PubMed
3. Callinan L, Holman RC, Vugia DJ, Schonberger LB, Belay ED. Kawasaki disease hospitalization rate among children younger than 5 years of age in California, 2003-2010. Pediatr Infect Dis J. 2014;33:781-783. PubMed
4. Newburger JW, Takahashi M, Gerber MA, Gewirtz MH, Tani LY, Burns JC, et al. Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association; American Academy of Pediatrics. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747-2771. PubMed
5. Son M, Newburger JW. Kawasaki disease. Pediatr Rev. 2013;34:151-61. PubMed
6. Newberger JW, Takahasi M, Beiser AS, et al. A single intravenous infusion of gammaglobulin as compared with four infusions in treatment of acute Kawasaki syndrome. N Engl J Med. 1991;324:1633-1639. PubMed
7. Dajani AS, Taubert KA, Gerber MA, et al. Diagnosis and therapy of Kawasaki disease in children. Circulation. 1993;87:1776-1780. PubMed
8. Saneeymehri S, Baker K, So TY. Overview of pharmacological treatment options for pediatric patients with refractory Kawasaki disease. J Pediatr Pharmacol Ther. 2015;20:163-177. PubMed
9. Brogan R, Eleftheriou D, Gnanapragasam J, Klein NJ, Brogan PA. Infliximab for the treatment of intravenous immunoglobulin resistant Kawasaki disease complicated by coronary artery aneurysms: a case report. Pediatr Rheumatol Online J. 2009;7:3. PubMed
10. Cohen S, Tacke CE, Straver B, Meijer N, Kuipers IM, Kuijpers TW. A child with severe relapsing Kawasaki disease rescued by IL-1 receptor blockade and extracorporeal membrane oxygenation. Ann Rheum Dis. 2012;71:2059-2061. PubMed
11. Kelly A, Tizard E. Vasculitis in children. Paediatrics and Child Health. 2010;20:65-72.
12. Yamazaki-Nakashimada MA, Espinosa-Lopez M, Hernandez-Bautista V, Espinosa-Padilla S, Espinosa-Rosales F. Catastrophic Kawasaki disease or juvenile polyarteritis nodosa? Semin Arthritis Rheum. 2006;35:349-354. PubMed
13. Ozen S, Pistorio A, Iusan SM, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis. 2010;69:798-806. PubMed
14. Eleftheriou D, Brogan PA. Vasculitis in children. Best Pract Res Clin Rheumatol. 2009;23:309-323. PubMed
15. Ozen S, Anton J, Arisoy N, et al. Juvenile polyarteritis: results of a multicenter survey of 110 children. J Pediatr. 2004;145:517-522. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 3-month-old otherwise healthy, immunized female presented to clinic with 2 days of intermittent low-grade fevers (maximum, 100º F), decreased oral intake, and sleepiness. Her pediatrician noted a faint, maculopapular rash on her trunk and extremities with mild conjunctival injection bilaterally that appeared that day, according to her mother. The infant otherwise appeared alert, well-hydrated, and without respiratory distress. She had no history of sick contacts or recent travel. She was prescribed amoxicillin for empiric treatment of a possible bacterial sinusitis or pharyngitis, despite a negative rapid strep antigen test.
At this age, multiple conditions can cause rashes. Given that this is early in the course of illness, without focal symptoms but with low-grade fevers, the initial differential diagnosis is broad and would include infectious, rheumatologic, and hematologic-oncologic etiologies, although the latter would be less likely. While the patient’s mother reports decreased oral intake, the fact that the patient is alert and appears hydrated is encouraging, suggesting time to observe and see if other symptoms present that may assist in elucidating the cause. The history of increased sleepiness warrants further investigation of meningeal signs, which would point to a central nervous system infection.
While streptococcal infection is possible, it would be uncommon at this age. The patient would have a higher fever and focal infection, and the rash does not appear consistent unless it was described as “sandpaper” in feel and appearance. A negative rapid strep test, while not sensitive, further supports this impression. A low-grade fever and rash would be consistent with a viral syndrome and, given the conjunctival injection, adenovirus, cytomegalovirus, rhinovirus, and Epstein Barr virus (EBV) are possibilities. Without ocular discharge, bacterial conjunctivitis would be unlikely. Another consideration would be Kawasaki disease, though it would be too early to diagnose this condition since at least 5 days of fever are required. Next steps include a detailed physical examination, looking for other focal signs such as swelling or desquamation of hands and feet, lymphadenopathy, strawberry tongue, and mucositis. Rather than empirically starting antibiotics, it would be more reasonable to observe her with close outpatient follow-up. The patient’s family should be instructed to monitor for additional and/or worsening symptoms, further decreased oral intake, signs of dehydration, or changes in alertness.
At home, the patient completed 5 doses of amoxicillin but continued to be febrile (maximum, 102.6º F). She was taken to a local emergency department on day 6 of her illness. She had worsening conjunctival injection and progression of the rash, involving the palms and soles. She was noted to have edema of hands and feet without desquamation (Figure 1). She had no oral mucous membrane changes and no cervical lymphadenopathy. Cerebrospinal fluid (CSF) was unremarkable, and empiric treatment with intravenous (IV) ceftriaxone was initiated. Complete blood count was notable for a white blood cell (WBC) count of 18.9 k/μL (normal range, 6.0-17.0); hemoglobin, 7.6 g/dL (normal range, 10-13); mean corpuscular volume, 84 (normal range, 74-108); and platelet count, 105 k/μL (normal range, 150-400). A peripheral blood smear revealed no abnormal cells. C-reactive protein (CRP) was elevated at 6.5 mg/dL (normal range, 0.0-0.6). She was admitted for further management.
Infection remains on the differential diagnosis given the elevated WBC count. Since the patient has completed a reasonable course of antibiotics, a bacterial infection would be less likely but not fully excluded. The cultures obtained would be helpful if they become positive, but given that the patient has been on antibiotics, a negative culture may represent partial sterilization and would not rule out infection. A viral infection continues to be high on the differential, but one would expect that symptoms and fever would have begun to abate. The normal peripheral blood smear makes a hematologic disorder less likely.
Kawasaki disease has risen on the differential with 5 days of fever surpassing 102º F. She has 3 of 5 primary clinical criteria, including conjunctival injection, rash, and edema of the hands and feet. Desquamation of the peripheral extremities would not be expected until the convalescent phase. A diagnosis of typical Kawasaki disease would require a fourth criterion, either oral mucous membrane changes or cervical lymphadenopathy. She meets the criteria for atypical or incomplete Kawasaki disease, which requires only fever for at least 5 days, elevated CRP, and 2 or 3 additional clinical criteria. She also meets supplemental laboratory criteria with an elevated WBC count greater than 15,000/μL, normocytic and normochromic anemia for age, and elevated CRP. Urinalysis positive for pyuria or serum albumin less than 3 g/dL would lend further support but is not necessary. Fever of 7 or more days in a child less than 6 months old without other explanation would also increase the likelihood of incomplete Kawasaki disease. Admission to the hospital, treatment with IV immunoglobulin (IVIg), and echocardiography to evaluate for typical cardiac involvement (eg, aneurysms, coronary arteritis, and pericardial effusion) are the appropriate next steps.
The patient was diagnosed with atypical Kawasaki disease. A transthoracic echocardiogram was normal on admission. On day 7 of her illness, she was treated with 1 dose of IVIg at 2 g/kg and high-dose aspirin at 100 mg/kg per day in divided doses. Despite this treatment, she continued to be febrile and was given a second dose of IVIg on day 9. Her fevers persisted.
In Kawasaki disease, persistent fever is concerning for long-term sequelae, including coronary artery aneurysms. Continued treatment is reasonable. After 2 doses of IVIg with a cumulative dose of 4 g/kg, it is prudent to switch therapy to IV methylprednisolone 30mg/kg with repeated doses as needed for up to 3 days should her fevers persist.
Her blood culture was negative. EBV serology, enterovirus polymerase chain reaction, and viral cultures were negative. Chest radiography on day 9 was normal. Abdominal ultrasonography on day 10 showed hydrops of the gallbladder.
The patient was started on IV corticosteroids on day 11 with resolution of her fevers and improvement in her rash. A repeat echocardiogram revealed new findings of dilated left main, left anterior descending, and right coronary arteries. On day 13, a steroid wean was attempted because she had remained afebrile for more than 48 hours, but the wean was halted due to recurrence of fevers and rash. Her high-dose aspirin was reduced to 81 mg PO daily on day 14, and she was started on enoxaparin injections.
It is unusual for Kawasaki disease not to respond to 2 doses of IVIg, followed by corticosteroids. As such, the differential diagnosis must be revisited. The findings of coronary artery dilation, prolonged fever, and rash corroborate the diagnosis of Kawasaki disease, although this could be an atypical presentation of another vasculitis. Systemic onset juvenile idiopathic arthritis usually affects children at 2 to 5 years old and is, therefore, less likely. Henoch-Schönlein purpura manifests with a rash but is often associated with diarrhea. There does not appear to be objective evidence of polyarteritis nodosa, although biopsy or angiography would be required to make this diagnosis. Hydrops of the gallbladder is an over-distention of the organ filled with watery or mucoid content. While hydrops can be noninflammatory and seen in gallstone disease, it can also occur in vasculitides. Despite the reassuring serologies, false negative results are possible. Thus, these viral infections are not eliminated, but they are less likely. Given the echocardiogram findings and continued concern for atypical Kawasaki disease, high-dose aspirin should be continued. It is reasonable to consider rheumatology consultation for assessment and recommendations as to length of steroid treatment and/or alternative interventions.
Pediatric cardiology was consulted. Repeat echocardiogram on day 16 showed an increase in the size of her coronary artery aneurysms, and her fevers persisted. Computed tomography scan of the abdomen and pelvis with contrast, obtained to further evaluate for a source of infection, was unremarkable.
The patient was transferred to a tertiary care institution on day 19, at which time she remained on aspirin, enoxaparin, and oral corticosteroids. On arrival, her temperature was 101.3º F, heart rate 225 beats per minute, and respiratory rate 57 breaths per minute. She was fussy with bilateral conjunctivitis and a maculopapular rash involving palms, soles, and right infraorbital region. Laboratory studies were significant for a WBC count of 30.3 k/μL; hemoglobin, 10.9 g/dL; platelets, 106 k/μL; and CRP, 8.3 mg/dL.
Pediatric rheumatology was consulted on day 20. The patient was treated with 3 days IV pulse-dose methylprednisolone at 30 mg/kg daily. Her fevers resolved, although her CRP level remained elevated. She was treated with 1 dose of infliximab 10 mg/kg IV on day 24, followed by 1 dose of anakinra 15 mg subcutaneously on day 27 due to persistently elevated CRP.
The symptoms and diagnostic evaluation remain most consistent with atypical Kawasaki disease. Her tachycardia and tachypnea are likely driven by her fever and fussiness, and should be followed closely. The elevated WBC is likely a consequence of the steroids and demargination of neutrophils. The elevated and increasing CRP is a marker of acute inflammation. The adage “treat the patient, not the numbers” comes to mind, because it is reassuring that the patient’s overall clinical picture seems to be improving with resolution of her fevers. However, further discussion with the pediatric rheumatology consultant is prudent, specifically regarding the significance of the persistently elevated CRP, refinement of the differential diagnosis including the potential for other vasculitides and appropriate evaluation of such, as well as recommendations for further treatment.
The patient was noted to have ongoing fevers. Based on reports of success with cyclophosphamide in refractory Kawasaki disease, she was treated with 2 doses at 60 mg IV per dose starting on day 28. Her CRP level decreased. Cardiology and rheumatology consultants recommended magnetic resonance imaging/magnetic resonance angiography of the chest, abdomen, and pelvis with and without contrast. These studies revealed dilation of the axillary and brachial arteries (Figure 2).
The response to cyclophosphamide confirms an autoimmune/inflammatory process. The imaging results and pattern are most consistent with either Kawasaki disease or polyarteritis nodosa. Therefore, rheumatology’s input will be invaluable with regard to which diagnosis is most likely, additional diagnostic testing, and appropriate medical regimen and follow-up plans.
Systemic extracoronary vascular inflammation on imaging and the refractory nature of the patient’s disease process, despite appropriate treatment for Kawasaki disease, led to the diagnosis of childhood polyarteritis nodosa (PAN). The patient was discharged home and closely followed in rheumatology clinic. Her most recent outpatient visit 1 year after the initial onset of her illness showed no further fevers or rashes, normal inflammatory markers, and stabilization of her coronary aneurysms on daily maintenance azathioprine.
DISCUSSION
Fever with an accompanying rash is a common issue in children. The extensive differential diagnosis includes infectious diseases, rheumatologic disorders, and medication reactions (Table 1). A thorough history and physical examination are essential in guiding the physician toward the proper diagnosis and management. Important information includes patient age, season, associated symptoms, exposure to sick contacts, travel history, host immune status, and immunization history. Fever duration and pattern must be elicited, as should features of the rash, including temporal relationship to the fever, distribution, progression, and morphology.1
When unexplained fever persists for 5 days or more in the pediatric patient, the diagnosis of KD must be suspected. KD is an acute, febrile, primary systemic vasculitis affecting small- and medium-sized vessels, with a predilection for coronary arteries.2 KD affects younger children, with approximately 85% of cases occurring in children under 5 years old. KD has a higher incidence in Asian populations, suggesting a possible genetic predisposition.3 The etiology of KD is not well understood, but infection and immune dysregulation have been proposed as contributing factors. KD is the leading cause of acquired heart disease in developed countries.2
The diagnosis of KD is made clinically (Table 2). Atypical KD is considered in patients with at least 5 days of fever but only 2 or 3 clinical criteria. Supportive laboratory findings include elevated inflammatory markers, anemia, neutrophilia, abnormal plasma lipids, low albumin, sterile pyuria, CSF pleocytosis, and elevated serum transaminases. Two-dimensional echocardiography should be performed in all children with definite or suspected KD at the time of diagnosis, 1 to 2 weeks later, and 6 weeks following discharge for evaluation of the coronary arteries, left ventricular function, and valve function. The American Heart Association recommends follow-up echocardiography at 1 year in children without coronary vessel involvement.4
Treatment is aimed at minimizing inflammation and coronary artery involvement, and should be initiated promptly.5 Therapy includes a single infusion of high-dose IVIg and aspirin;6,7 the latter is initially provided at high anti-inflammatory doses, followed by lower antithrombotic doses once fever and laboratory markers have resolved.2 Aspirin can be discontinued if there is no evidence of coronary involvement at the 6-week follow-up echocardiogram.5 A second dose of IVIg is given within 48 hours for refractory cases, defined as persistent fever following the first dose of IVIg.4 Fifteen percent of children have refractory illness, and refractory KD is associated with a higher risk of coronary artery lesions.5 Additional agents that suppress immune activation and cytokine secretion contributing to KD pathogenesis have been studied. Corticosteroids inhibit phospholipase A, an enzyme required for production of inflammatory markers.8 Infliximab, a tumor necrosis factor-alpha inhibitor, has been shown to reduce duration of fever and length of hospital stay.8,9 Anakinra, an interleukin-1 receptor antagonist, has been shown to decrease fever duration and prevent progression of vascular injury in cases of refractory KD.10 There is, however, a lack of sufficient evidence and consensus on best practice.8-10
If inflammation, evidenced by fever, elevated inflammatory markers (such as erythrocyte sedimentation rate, CRP), or vessel involvement on imaging, persists or worsens despite standard therapy, physicians should seek alternative diagnoses. This patient’s extracoronary vascular inflammation and favorable response only to cyclophosphamide led to the diagnosis of systemic PAN. Like KD, PAN is a multi-system vasculitis affecting small- and medium-sized vessels. Unlike KD, PAN is rarely seen in children.11 Historically, PAN was thought to represent an extreme fatal end of the KD spectrum. Today, PAN is accepted as a separate entity. Clinical features and histological findings often overlap with KD, creating a diagnostic dilemma for providers.12
At the onset of illness, clinical features of systemic PAN may include recurrent fever, weight loss, and myalgia, with gradual progression to multi-organ system involvement. Laboratory assessment reveals elevated inflammatory markers and leukocytosis. Thrombocytosis, anemia, proteinuria, and hematuria may be present. A positive antineutrophil cytoplasmic antibody is rare in PAN and should raise suspicion for a microscopic polyangiitis, which is distinguished from PAN by small vessel involvement only. When compared to KD, cardiac vessel involvement in PAN is more variable.11 Diagnostic criteria for childhood PAN are listed in Table 2.13
Treatment of PAN is aimed at inducing remission with high-dose steroids and cyclophosphamide. Maintenance of remission is achieved using low-dose steroids and azathioprine.11 Total duration of treatment averages 2 to 3 years, with a minimum of 18 months.14 Plasma exchange has been used in severe, life-threatening cases.11 Prognosis for children with PAN is more favorable compared to adults with PAN, in whom the mortality rate is as high as 20% to 30%, even with aggressive treatment. In 1 multicenter study of childhood and adolescent PAN, overall mortality was 1.1%.15
This patient initially presented with findings consistent with KD. As her inflammatory markers remained elevated and fevers persisted, her physicians appropriately reconsidered the etiology of her symptoms, thereby “getting warmer” in the search for the correct diagnosis of systemic PAN, a rare disease and a separate entity from KD. Recognizing the overlapping and distinct clinical features of each entity can promote more timely and appropriate selection of therapy, thereby minimizing clinical manifestations and complications associated with each vasculitis.
KEY TEACHING POINTS
- KD and childhood PAN are disseminated vasculitides affecting small- and medium-sized vessels. Although they are distinct entities, KD and PAN exhibit overlapping clinical and pathological features that make appropriate diagnosis and treatment challenging.
- In cases of refractory KD, alternative diagnoses should be considered.
- Recognizing the individual features of both entities is imperative because treatment differs: KD is treated with high-dose aspirin and IVIg; corticosteroids and immunosuppressive agents are used to treat PAN.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 3-month-old otherwise healthy, immunized female presented to clinic with 2 days of intermittent low-grade fevers (maximum, 100º F), decreased oral intake, and sleepiness. Her pediatrician noted a faint, maculopapular rash on her trunk and extremities with mild conjunctival injection bilaterally that appeared that day, according to her mother. The infant otherwise appeared alert, well-hydrated, and without respiratory distress. She had no history of sick contacts or recent travel. She was prescribed amoxicillin for empiric treatment of a possible bacterial sinusitis or pharyngitis, despite a negative rapid strep antigen test.
At this age, multiple conditions can cause rashes. Given that this is early in the course of illness, without focal symptoms but with low-grade fevers, the initial differential diagnosis is broad and would include infectious, rheumatologic, and hematologic-oncologic etiologies, although the latter would be less likely. While the patient’s mother reports decreased oral intake, the fact that the patient is alert and appears hydrated is encouraging, suggesting time to observe and see if other symptoms present that may assist in elucidating the cause. The history of increased sleepiness warrants further investigation of meningeal signs, which would point to a central nervous system infection.
While streptococcal infection is possible, it would be uncommon at this age. The patient would have a higher fever and focal infection, and the rash does not appear consistent unless it was described as “sandpaper” in feel and appearance. A negative rapid strep test, while not sensitive, further supports this impression. A low-grade fever and rash would be consistent with a viral syndrome and, given the conjunctival injection, adenovirus, cytomegalovirus, rhinovirus, and Epstein Barr virus (EBV) are possibilities. Without ocular discharge, bacterial conjunctivitis would be unlikely. Another consideration would be Kawasaki disease, though it would be too early to diagnose this condition since at least 5 days of fever are required. Next steps include a detailed physical examination, looking for other focal signs such as swelling or desquamation of hands and feet, lymphadenopathy, strawberry tongue, and mucositis. Rather than empirically starting antibiotics, it would be more reasonable to observe her with close outpatient follow-up. The patient’s family should be instructed to monitor for additional and/or worsening symptoms, further decreased oral intake, signs of dehydration, or changes in alertness.
At home, the patient completed 5 doses of amoxicillin but continued to be febrile (maximum, 102.6º F). She was taken to a local emergency department on day 6 of her illness. She had worsening conjunctival injection and progression of the rash, involving the palms and soles. She was noted to have edema of hands and feet without desquamation (Figure 1). She had no oral mucous membrane changes and no cervical lymphadenopathy. Cerebrospinal fluid (CSF) was unremarkable, and empiric treatment with intravenous (IV) ceftriaxone was initiated. Complete blood count was notable for a white blood cell (WBC) count of 18.9 k/μL (normal range, 6.0-17.0); hemoglobin, 7.6 g/dL (normal range, 10-13); mean corpuscular volume, 84 (normal range, 74-108); and platelet count, 105 k/μL (normal range, 150-400). A peripheral blood smear revealed no abnormal cells. C-reactive protein (CRP) was elevated at 6.5 mg/dL (normal range, 0.0-0.6). She was admitted for further management.
Infection remains on the differential diagnosis given the elevated WBC count. Since the patient has completed a reasonable course of antibiotics, a bacterial infection would be less likely but not fully excluded. The cultures obtained would be helpful if they become positive, but given that the patient has been on antibiotics, a negative culture may represent partial sterilization and would not rule out infection. A viral infection continues to be high on the differential, but one would expect that symptoms and fever would have begun to abate. The normal peripheral blood smear makes a hematologic disorder less likely.
Kawasaki disease has risen on the differential with 5 days of fever surpassing 102º F. She has 3 of 5 primary clinical criteria, including conjunctival injection, rash, and edema of the hands and feet. Desquamation of the peripheral extremities would not be expected until the convalescent phase. A diagnosis of typical Kawasaki disease would require a fourth criterion, either oral mucous membrane changes or cervical lymphadenopathy. She meets the criteria for atypical or incomplete Kawasaki disease, which requires only fever for at least 5 days, elevated CRP, and 2 or 3 additional clinical criteria. She also meets supplemental laboratory criteria with an elevated WBC count greater than 15,000/μL, normocytic and normochromic anemia for age, and elevated CRP. Urinalysis positive for pyuria or serum albumin less than 3 g/dL would lend further support but is not necessary. Fever of 7 or more days in a child less than 6 months old without other explanation would also increase the likelihood of incomplete Kawasaki disease. Admission to the hospital, treatment with IV immunoglobulin (IVIg), and echocardiography to evaluate for typical cardiac involvement (eg, aneurysms, coronary arteritis, and pericardial effusion) are the appropriate next steps.
The patient was diagnosed with atypical Kawasaki disease. A transthoracic echocardiogram was normal on admission. On day 7 of her illness, she was treated with 1 dose of IVIg at 2 g/kg and high-dose aspirin at 100 mg/kg per day in divided doses. Despite this treatment, she continued to be febrile and was given a second dose of IVIg on day 9. Her fevers persisted.
In Kawasaki disease, persistent fever is concerning for long-term sequelae, including coronary artery aneurysms. Continued treatment is reasonable. After 2 doses of IVIg with a cumulative dose of 4 g/kg, it is prudent to switch therapy to IV methylprednisolone 30mg/kg with repeated doses as needed for up to 3 days should her fevers persist.
Her blood culture was negative. EBV serology, enterovirus polymerase chain reaction, and viral cultures were negative. Chest radiography on day 9 was normal. Abdominal ultrasonography on day 10 showed hydrops of the gallbladder.
The patient was started on IV corticosteroids on day 11 with resolution of her fevers and improvement in her rash. A repeat echocardiogram revealed new findings of dilated left main, left anterior descending, and right coronary arteries. On day 13, a steroid wean was attempted because she had remained afebrile for more than 48 hours, but the wean was halted due to recurrence of fevers and rash. Her high-dose aspirin was reduced to 81 mg PO daily on day 14, and she was started on enoxaparin injections.
It is unusual for Kawasaki disease not to respond to 2 doses of IVIg, followed by corticosteroids. As such, the differential diagnosis must be revisited. The findings of coronary artery dilation, prolonged fever, and rash corroborate the diagnosis of Kawasaki disease, although this could be an atypical presentation of another vasculitis. Systemic onset juvenile idiopathic arthritis usually affects children at 2 to 5 years old and is, therefore, less likely. Henoch-Schönlein purpura manifests with a rash but is often associated with diarrhea. There does not appear to be objective evidence of polyarteritis nodosa, although biopsy or angiography would be required to make this diagnosis. Hydrops of the gallbladder is an over-distention of the organ filled with watery or mucoid content. While hydrops can be noninflammatory and seen in gallstone disease, it can also occur in vasculitides. Despite the reassuring serologies, false negative results are possible. Thus, these viral infections are not eliminated, but they are less likely. Given the echocardiogram findings and continued concern for atypical Kawasaki disease, high-dose aspirin should be continued. It is reasonable to consider rheumatology consultation for assessment and recommendations as to length of steroid treatment and/or alternative interventions.
Pediatric cardiology was consulted. Repeat echocardiogram on day 16 showed an increase in the size of her coronary artery aneurysms, and her fevers persisted. Computed tomography scan of the abdomen and pelvis with contrast, obtained to further evaluate for a source of infection, was unremarkable.
The patient was transferred to a tertiary care institution on day 19, at which time she remained on aspirin, enoxaparin, and oral corticosteroids. On arrival, her temperature was 101.3º F, heart rate 225 beats per minute, and respiratory rate 57 breaths per minute. She was fussy with bilateral conjunctivitis and a maculopapular rash involving palms, soles, and right infraorbital region. Laboratory studies were significant for a WBC count of 30.3 k/μL; hemoglobin, 10.9 g/dL; platelets, 106 k/μL; and CRP, 8.3 mg/dL.
Pediatric rheumatology was consulted on day 20. The patient was treated with 3 days IV pulse-dose methylprednisolone at 30 mg/kg daily. Her fevers resolved, although her CRP level remained elevated. She was treated with 1 dose of infliximab 10 mg/kg IV on day 24, followed by 1 dose of anakinra 15 mg subcutaneously on day 27 due to persistently elevated CRP.
The symptoms and diagnostic evaluation remain most consistent with atypical Kawasaki disease. Her tachycardia and tachypnea are likely driven by her fever and fussiness, and should be followed closely. The elevated WBC is likely a consequence of the steroids and demargination of neutrophils. The elevated and increasing CRP is a marker of acute inflammation. The adage “treat the patient, not the numbers” comes to mind, because it is reassuring that the patient’s overall clinical picture seems to be improving with resolution of her fevers. However, further discussion with the pediatric rheumatology consultant is prudent, specifically regarding the significance of the persistently elevated CRP, refinement of the differential diagnosis including the potential for other vasculitides and appropriate evaluation of such, as well as recommendations for further treatment.
The patient was noted to have ongoing fevers. Based on reports of success with cyclophosphamide in refractory Kawasaki disease, she was treated with 2 doses at 60 mg IV per dose starting on day 28. Her CRP level decreased. Cardiology and rheumatology consultants recommended magnetic resonance imaging/magnetic resonance angiography of the chest, abdomen, and pelvis with and without contrast. These studies revealed dilation of the axillary and brachial arteries (Figure 2).
The response to cyclophosphamide confirms an autoimmune/inflammatory process. The imaging results and pattern are most consistent with either Kawasaki disease or polyarteritis nodosa. Therefore, rheumatology’s input will be invaluable with regard to which diagnosis is most likely, additional diagnostic testing, and appropriate medical regimen and follow-up plans.
Systemic extracoronary vascular inflammation on imaging and the refractory nature of the patient’s disease process, despite appropriate treatment for Kawasaki disease, led to the diagnosis of childhood polyarteritis nodosa (PAN). The patient was discharged home and closely followed in rheumatology clinic. Her most recent outpatient visit 1 year after the initial onset of her illness showed no further fevers or rashes, normal inflammatory markers, and stabilization of her coronary aneurysms on daily maintenance azathioprine.
DISCUSSION
Fever with an accompanying rash is a common issue in children. The extensive differential diagnosis includes infectious diseases, rheumatologic disorders, and medication reactions (Table 1). A thorough history and physical examination are essential in guiding the physician toward the proper diagnosis and management. Important information includes patient age, season, associated symptoms, exposure to sick contacts, travel history, host immune status, and immunization history. Fever duration and pattern must be elicited, as should features of the rash, including temporal relationship to the fever, distribution, progression, and morphology.1
When unexplained fever persists for 5 days or more in the pediatric patient, the diagnosis of KD must be suspected. KD is an acute, febrile, primary systemic vasculitis affecting small- and medium-sized vessels, with a predilection for coronary arteries.2 KD affects younger children, with approximately 85% of cases occurring in children under 5 years old. KD has a higher incidence in Asian populations, suggesting a possible genetic predisposition.3 The etiology of KD is not well understood, but infection and immune dysregulation have been proposed as contributing factors. KD is the leading cause of acquired heart disease in developed countries.2
The diagnosis of KD is made clinically (Table 2). Atypical KD is considered in patients with at least 5 days of fever but only 2 or 3 clinical criteria. Supportive laboratory findings include elevated inflammatory markers, anemia, neutrophilia, abnormal plasma lipids, low albumin, sterile pyuria, CSF pleocytosis, and elevated serum transaminases. Two-dimensional echocardiography should be performed in all children with definite or suspected KD at the time of diagnosis, 1 to 2 weeks later, and 6 weeks following discharge for evaluation of the coronary arteries, left ventricular function, and valve function. The American Heart Association recommends follow-up echocardiography at 1 year in children without coronary vessel involvement.4
Treatment is aimed at minimizing inflammation and coronary artery involvement, and should be initiated promptly.5 Therapy includes a single infusion of high-dose IVIg and aspirin;6,7 the latter is initially provided at high anti-inflammatory doses, followed by lower antithrombotic doses once fever and laboratory markers have resolved.2 Aspirin can be discontinued if there is no evidence of coronary involvement at the 6-week follow-up echocardiogram.5 A second dose of IVIg is given within 48 hours for refractory cases, defined as persistent fever following the first dose of IVIg.4 Fifteen percent of children have refractory illness, and refractory KD is associated with a higher risk of coronary artery lesions.5 Additional agents that suppress immune activation and cytokine secretion contributing to KD pathogenesis have been studied. Corticosteroids inhibit phospholipase A, an enzyme required for production of inflammatory markers.8 Infliximab, a tumor necrosis factor-alpha inhibitor, has been shown to reduce duration of fever and length of hospital stay.8,9 Anakinra, an interleukin-1 receptor antagonist, has been shown to decrease fever duration and prevent progression of vascular injury in cases of refractory KD.10 There is, however, a lack of sufficient evidence and consensus on best practice.8-10
If inflammation, evidenced by fever, elevated inflammatory markers (such as erythrocyte sedimentation rate, CRP), or vessel involvement on imaging, persists or worsens despite standard therapy, physicians should seek alternative diagnoses. This patient’s extracoronary vascular inflammation and favorable response only to cyclophosphamide led to the diagnosis of systemic PAN. Like KD, PAN is a multi-system vasculitis affecting small- and medium-sized vessels. Unlike KD, PAN is rarely seen in children.11 Historically, PAN was thought to represent an extreme fatal end of the KD spectrum. Today, PAN is accepted as a separate entity. Clinical features and histological findings often overlap with KD, creating a diagnostic dilemma for providers.12
At the onset of illness, clinical features of systemic PAN may include recurrent fever, weight loss, and myalgia, with gradual progression to multi-organ system involvement. Laboratory assessment reveals elevated inflammatory markers and leukocytosis. Thrombocytosis, anemia, proteinuria, and hematuria may be present. A positive antineutrophil cytoplasmic antibody is rare in PAN and should raise suspicion for a microscopic polyangiitis, which is distinguished from PAN by small vessel involvement only. When compared to KD, cardiac vessel involvement in PAN is more variable.11 Diagnostic criteria for childhood PAN are listed in Table 2.13
Treatment of PAN is aimed at inducing remission with high-dose steroids and cyclophosphamide. Maintenance of remission is achieved using low-dose steroids and azathioprine.11 Total duration of treatment averages 2 to 3 years, with a minimum of 18 months.14 Plasma exchange has been used in severe, life-threatening cases.11 Prognosis for children with PAN is more favorable compared to adults with PAN, in whom the mortality rate is as high as 20% to 30%, even with aggressive treatment. In 1 multicenter study of childhood and adolescent PAN, overall mortality was 1.1%.15
This patient initially presented with findings consistent with KD. As her inflammatory markers remained elevated and fevers persisted, her physicians appropriately reconsidered the etiology of her symptoms, thereby “getting warmer” in the search for the correct diagnosis of systemic PAN, a rare disease and a separate entity from KD. Recognizing the overlapping and distinct clinical features of each entity can promote more timely and appropriate selection of therapy, thereby minimizing clinical manifestations and complications associated with each vasculitis.
KEY TEACHING POINTS
- KD and childhood PAN are disseminated vasculitides affecting small- and medium-sized vessels. Although they are distinct entities, KD and PAN exhibit overlapping clinical and pathological features that make appropriate diagnosis and treatment challenging.
- In cases of refractory KD, alternative diagnoses should be considered.
- Recognizing the individual features of both entities is imperative because treatment differs: KD is treated with high-dose aspirin and IVIg; corticosteroids and immunosuppressive agents are used to treat PAN.
Disclosure
Nothing to report.
1. McKinnon HD Jr, Howard T. Evaluating the febrile patient with a rash. Am Fam Physician. 2000;62:804-816. PubMed
2. Dimitriades V, Brown AG, Gedalia A. Kawasaki disease: pathophysiology, clinical manifestations, and management. Curr Rheumatol Rep. 2014;16:423. PubMed
3. Callinan L, Holman RC, Vugia DJ, Schonberger LB, Belay ED. Kawasaki disease hospitalization rate among children younger than 5 years of age in California, 2003-2010. Pediatr Infect Dis J. 2014;33:781-783. PubMed
4. Newburger JW, Takahashi M, Gerber MA, Gewirtz MH, Tani LY, Burns JC, et al. Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association; American Academy of Pediatrics. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747-2771. PubMed
5. Son M, Newburger JW. Kawasaki disease. Pediatr Rev. 2013;34:151-61. PubMed
6. Newberger JW, Takahasi M, Beiser AS, et al. A single intravenous infusion of gammaglobulin as compared with four infusions in treatment of acute Kawasaki syndrome. N Engl J Med. 1991;324:1633-1639. PubMed
7. Dajani AS, Taubert KA, Gerber MA, et al. Diagnosis and therapy of Kawasaki disease in children. Circulation. 1993;87:1776-1780. PubMed
8. Saneeymehri S, Baker K, So TY. Overview of pharmacological treatment options for pediatric patients with refractory Kawasaki disease. J Pediatr Pharmacol Ther. 2015;20:163-177. PubMed
9. Brogan R, Eleftheriou D, Gnanapragasam J, Klein NJ, Brogan PA. Infliximab for the treatment of intravenous immunoglobulin resistant Kawasaki disease complicated by coronary artery aneurysms: a case report. Pediatr Rheumatol Online J. 2009;7:3. PubMed
10. Cohen S, Tacke CE, Straver B, Meijer N, Kuipers IM, Kuijpers TW. A child with severe relapsing Kawasaki disease rescued by IL-1 receptor blockade and extracorporeal membrane oxygenation. Ann Rheum Dis. 2012;71:2059-2061. PubMed
11. Kelly A, Tizard E. Vasculitis in children. Paediatrics and Child Health. 2010;20:65-72.
12. Yamazaki-Nakashimada MA, Espinosa-Lopez M, Hernandez-Bautista V, Espinosa-Padilla S, Espinosa-Rosales F. Catastrophic Kawasaki disease or juvenile polyarteritis nodosa? Semin Arthritis Rheum. 2006;35:349-354. PubMed
13. Ozen S, Pistorio A, Iusan SM, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis. 2010;69:798-806. PubMed
14. Eleftheriou D, Brogan PA. Vasculitis in children. Best Pract Res Clin Rheumatol. 2009;23:309-323. PubMed
15. Ozen S, Anton J, Arisoy N, et al. Juvenile polyarteritis: results of a multicenter survey of 110 children. J Pediatr. 2004;145:517-522. PubMed
1. McKinnon HD Jr, Howard T. Evaluating the febrile patient with a rash. Am Fam Physician. 2000;62:804-816. PubMed
2. Dimitriades V, Brown AG, Gedalia A. Kawasaki disease: pathophysiology, clinical manifestations, and management. Curr Rheumatol Rep. 2014;16:423. PubMed
3. Callinan L, Holman RC, Vugia DJ, Schonberger LB, Belay ED. Kawasaki disease hospitalization rate among children younger than 5 years of age in California, 2003-2010. Pediatr Infect Dis J. 2014;33:781-783. PubMed
4. Newburger JW, Takahashi M, Gerber MA, Gewirtz MH, Tani LY, Burns JC, et al. Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association; American Academy of Pediatrics. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747-2771. PubMed
5. Son M, Newburger JW. Kawasaki disease. Pediatr Rev. 2013;34:151-61. PubMed
6. Newberger JW, Takahasi M, Beiser AS, et al. A single intravenous infusion of gammaglobulin as compared with four infusions in treatment of acute Kawasaki syndrome. N Engl J Med. 1991;324:1633-1639. PubMed
7. Dajani AS, Taubert KA, Gerber MA, et al. Diagnosis and therapy of Kawasaki disease in children. Circulation. 1993;87:1776-1780. PubMed
8. Saneeymehri S, Baker K, So TY. Overview of pharmacological treatment options for pediatric patients with refractory Kawasaki disease. J Pediatr Pharmacol Ther. 2015;20:163-177. PubMed
9. Brogan R, Eleftheriou D, Gnanapragasam J, Klein NJ, Brogan PA. Infliximab for the treatment of intravenous immunoglobulin resistant Kawasaki disease complicated by coronary artery aneurysms: a case report. Pediatr Rheumatol Online J. 2009;7:3. PubMed
10. Cohen S, Tacke CE, Straver B, Meijer N, Kuipers IM, Kuijpers TW. A child with severe relapsing Kawasaki disease rescued by IL-1 receptor blockade and extracorporeal membrane oxygenation. Ann Rheum Dis. 2012;71:2059-2061. PubMed
11. Kelly A, Tizard E. Vasculitis in children. Paediatrics and Child Health. 2010;20:65-72.
12. Yamazaki-Nakashimada MA, Espinosa-Lopez M, Hernandez-Bautista V, Espinosa-Padilla S, Espinosa-Rosales F. Catastrophic Kawasaki disease or juvenile polyarteritis nodosa? Semin Arthritis Rheum. 2006;35:349-354. PubMed
13. Ozen S, Pistorio A, Iusan SM, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis. 2010;69:798-806. PubMed
14. Eleftheriou D, Brogan PA. Vasculitis in children. Best Pract Res Clin Rheumatol. 2009;23:309-323. PubMed
15. Ozen S, Anton J, Arisoy N, et al. Juvenile polyarteritis: results of a multicenter survey of 110 children. J Pediatr. 2004;145:517-522. PubMed
© 2017 Society of Hospital Medicine
How Should Hospitalists Manage Elderly Patients with Dysphagia?
The Case
A 74-year-old man with Alzheimer’s dementia presents with urinary tract infection (UTI), hypovolemia, and hypernatremia. He also has chronic dysphagia with a history of aspiration pneumonia and has been on thickened liquids at home for the past five months. As his infection is treated, he improves and requests water to drink.
Background

The diagnosis of dysphagia is clinical, and assessments from patients and family are often sufficient. The optimal test to assess the severity of dysphagia is a bedside swallow evaluation using small amounts of water.1 Video-assisted fluoroscopic examinations can identify problem areas within the oropharynx and esophagus and may help determine the etiology of dysphagia.
What evidence supports various treatment options for dysphagia?
Access to Water
Water is a thin liquid with low viscosity, which allows for rapid transit through the oropharynx. In debilitated and elderly patients, thin liquids easily reach the epiglottis and enter the trachea before pharyngeal muscles compensate. As such, access to water and other thin liquids is often restricted in patients suspected to have dysphagia.4
However, allowing access to water improves patient satisfaction, reduces the development of dehydration, and does not increase the incidence of AP. Bedside therapy interventions such as correct positioning and chin-tuck and sipping technique as well as attention to oral hygiene are recommended prior to more noxious options such as thickened liquids.1 The Frazier water protocol may help provide logistical guidance for facilities interested in improving access to water for patients with dysphagia.
Liquid Modification
Many clinicians manage dysphagia through restricting access to all thin liquids. In the hospital setting where video fluoroscopy and speech therapy are readily available, clinicians frequently employ the use of modified diets with thickened liquids in order to minimize the risk of aspiration despite the lack of high-quality evidence supporting liquid modification.2 Patients associate thickened liquids and restricted diets with a reduction in quality of life. Compliance studies have shown that only a minority of patients are compliant with thickened liquids at five days. In addition, thickening liquids has not been shown to decrease the risk of AP nor improve nutritional status, and it may actually cause harm by increasing the risk of dehydration and UTI.4
Tube Feeding
In patients with severe dysphagia in whom conservative management is not feasible or has failed, maintaining adequate nutrition can be a challenge. There are encouraging data with nutritionally enriching and modifying the texture of solid foods.1 Alternative methods of enteral nutrition delivery are often also considered. The most common vehicles of delivery are nasogastric tubes, post-pyloric feeding tubes, and percutaneous endoscopic gastrostomy (PEG) tubes. In theory, bypassing the pharynx and esophagus could result in fewer aspiration events and less AP.3 However, nasogastric, post-pyloric, or PEG feeding does not decrease the risk of AP. For patients with advanced dementia, there have been no randomized trials demonstrating an improvement in mortality with tube feeds.4 Tube feeding also carries with it a slight procedural risk and a high incidence of associated diarrhea, plus is associated with electrolyte derangements such as hypernatremia. The decision to pursue tube feeding should be weighed heavily in every patient and is highly influenced by the etiology and anticipated duration of dysphagia.
Selective Digestive Decontamination
Selective digestive decontamination (SDD) is a protocol-based treatment that aims to eradicate potentially pathogenic gut flora, particularly aerobic gram-negatives, in critically ill patients to reduce the impact of aspiration events. The utilization of SDD and the available literature center firmly on critically ill and ventilated patients. Subsequent studies have demonstrated recolonization after protocol cessation, and long-term effects are currently undefined.5 Until it can be studied in broader populations and proven to have clinical benefit, employing SSD in non-critically ill patients with dysphagia remains unsupported.
Multimodal Approach
Many rehabilitation centers incorporate a therapist-driven swallowing treatment program. Evidence suggests patient and family counseling alone may not be effective, so these programs variably incorporate diet/liquid modification, strengthening exercises, sensory processing techniques, and even neuromuscular electrical stimulation for muscle building.1 Accordingly, these programs are resource-intensive.
Management
Dysphagia remains a significant clinical problem for hospitalized patients. The existing literature and practice guidelines generally support a “less is more” approach. Though liquid/diet modification is common practice, it is not based in solid evidence and may contribute to unnecessary tube feeding. The best current evidence supports allowing access to water and ice chips. The ideal management plan for each patient will differ and should incorporate patient and family preferences in a multidisciplinary approach.
Back to the Case
Our patient requests water. He coughs after drinking during a bedside swallow evaluation. The risks of potential aspiration and AP are explained, and he expresses his understanding. He reiterates his choice to be allowed access to water as it is important to his quality of life. The speech therapy team is consulted and provides instruction on chin-tuck positioning, oral care, and timing water between meals rather than while eating food. He does well for the remainder of the hospital stay, and by time of discharge, his electrolytes are corrected, and he is much more comfortable being allowed to drink water. He is discharged home and encouraged to continue with these conservative measures.
Bottom Line
Evidence to support many common interventions for dysphagia is lacking; patients with dysphagia are best managed via a multidisciplinary, multimodal approach that provides access to water whenever possible. TH
Vijay G. Paryani, MD, is an internal medicine resident in the department of internal medicine at the University of Kentucky. Joseph R. Sweigart, MD, is a hospitalist and assistant professor of hospital medicine in the division of hospital medicine at the University of Kentucky. Laura C. Fanucchi, MD, is a hospitalist and assistant professor of hospital medicine in the division of hospital medicine at the University of Kentucky.
References
- Karagiannis MJ, Chivers L, Karagiannis TC. Effects of oral intake of water in patients with oropharyngeal dysphagia. BMC Geriatr. 2011;11(2):9.
- Foley N, Teasell R, Salter K, Kruger E, Martino R. Dysphagia treatment post stroke: a systematic review of randomized controlled trials. Age Ageing. 2008;37(3):258-264.
- Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med. 2001;344(9):665-671.
- Loeb MB, Becker M, Eady A, Walker-Dilks C. Interventions to prevent aspiration pneumonia in older adults: a systematic review. J Am Geriatr Soc. 2003;51(7):1018-1022.
- Gosney M, Martin MV, Wright AE. The role of selective decontamination of the digestive tract in acute stroke. Age Ageing 2006;35(1):42-47.
The Case
A 74-year-old man with Alzheimer’s dementia presents with urinary tract infection (UTI), hypovolemia, and hypernatremia. He also has chronic dysphagia with a history of aspiration pneumonia and has been on thickened liquids at home for the past five months. As his infection is treated, he improves and requests water to drink.
Background
The diagnosis of dysphagia is clinical, and assessments from patients and family are often sufficient. The optimal test to assess the severity of dysphagia is a bedside swallow evaluation using small amounts of water.1 Video-assisted fluoroscopic examinations can identify problem areas within the oropharynx and esophagus and may help determine the etiology of dysphagia.
What evidence supports various treatment options for dysphagia?
Access to Water
Water is a thin liquid with low viscosity, which allows for rapid transit through the oropharynx. In debilitated and elderly patients, thin liquids easily reach the epiglottis and enter the trachea before pharyngeal muscles compensate. As such, access to water and other thin liquids is often restricted in patients suspected to have dysphagia.4
However, allowing access to water improves patient satisfaction, reduces the development of dehydration, and does not increase the incidence of AP. Bedside therapy interventions such as correct positioning and chin-tuck and sipping technique as well as attention to oral hygiene are recommended prior to more noxious options such as thickened liquids.1 The Frazier water protocol may help provide logistical guidance for facilities interested in improving access to water for patients with dysphagia.
Liquid Modification
Many clinicians manage dysphagia through restricting access to all thin liquids. In the hospital setting where video fluoroscopy and speech therapy are readily available, clinicians frequently employ the use of modified diets with thickened liquids in order to minimize the risk of aspiration despite the lack of high-quality evidence supporting liquid modification.2 Patients associate thickened liquids and restricted diets with a reduction in quality of life. Compliance studies have shown that only a minority of patients are compliant with thickened liquids at five days. In addition, thickening liquids has not been shown to decrease the risk of AP nor improve nutritional status, and it may actually cause harm by increasing the risk of dehydration and UTI.4
Tube Feeding
In patients with severe dysphagia in whom conservative management is not feasible or has failed, maintaining adequate nutrition can be a challenge. There are encouraging data with nutritionally enriching and modifying the texture of solid foods.1 Alternative methods of enteral nutrition delivery are often also considered. The most common vehicles of delivery are nasogastric tubes, post-pyloric feeding tubes, and percutaneous endoscopic gastrostomy (PEG) tubes. In theory, bypassing the pharynx and esophagus could result in fewer aspiration events and less AP.3 However, nasogastric, post-pyloric, or PEG feeding does not decrease the risk of AP. For patients with advanced dementia, there have been no randomized trials demonstrating an improvement in mortality with tube feeds.4 Tube feeding also carries with it a slight procedural risk and a high incidence of associated diarrhea, plus is associated with electrolyte derangements such as hypernatremia. The decision to pursue tube feeding should be weighed heavily in every patient and is highly influenced by the etiology and anticipated duration of dysphagia.
Selective Digestive Decontamination
Selective digestive decontamination (SDD) is a protocol-based treatment that aims to eradicate potentially pathogenic gut flora, particularly aerobic gram-negatives, in critically ill patients to reduce the impact of aspiration events. The utilization of SDD and the available literature center firmly on critically ill and ventilated patients. Subsequent studies have demonstrated recolonization after protocol cessation, and long-term effects are currently undefined.5 Until it can be studied in broader populations and proven to have clinical benefit, employing SSD in non-critically ill patients with dysphagia remains unsupported.
Multimodal Approach
Many rehabilitation centers incorporate a therapist-driven swallowing treatment program. Evidence suggests patient and family counseling alone may not be effective, so these programs variably incorporate diet/liquid modification, strengthening exercises, sensory processing techniques, and even neuromuscular electrical stimulation for muscle building.1 Accordingly, these programs are resource-intensive.
Management
Dysphagia remains a significant clinical problem for hospitalized patients. The existing literature and practice guidelines generally support a “less is more” approach. Though liquid/diet modification is common practice, it is not based in solid evidence and may contribute to unnecessary tube feeding. The best current evidence supports allowing access to water and ice chips. The ideal management plan for each patient will differ and should incorporate patient and family preferences in a multidisciplinary approach.
Back to the Case
Our patient requests water. He coughs after drinking during a bedside swallow evaluation. The risks of potential aspiration and AP are explained, and he expresses his understanding. He reiterates his choice to be allowed access to water as it is important to his quality of life. The speech therapy team is consulted and provides instruction on chin-tuck positioning, oral care, and timing water between meals rather than while eating food. He does well for the remainder of the hospital stay, and by time of discharge, his electrolytes are corrected, and he is much more comfortable being allowed to drink water. He is discharged home and encouraged to continue with these conservative measures.
Bottom Line
Evidence to support many common interventions for dysphagia is lacking; patients with dysphagia are best managed via a multidisciplinary, multimodal approach that provides access to water whenever possible. TH
Vijay G. Paryani, MD, is an internal medicine resident in the department of internal medicine at the University of Kentucky. Joseph R. Sweigart, MD, is a hospitalist and assistant professor of hospital medicine in the division of hospital medicine at the University of Kentucky. Laura C. Fanucchi, MD, is a hospitalist and assistant professor of hospital medicine in the division of hospital medicine at the University of Kentucky.
References
- Karagiannis MJ, Chivers L, Karagiannis TC. Effects of oral intake of water in patients with oropharyngeal dysphagia. BMC Geriatr. 2011;11(2):9.
- Foley N, Teasell R, Salter K, Kruger E, Martino R. Dysphagia treatment post stroke: a systematic review of randomized controlled trials. Age Ageing. 2008;37(3):258-264.
- Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med. 2001;344(9):665-671.
- Loeb MB, Becker M, Eady A, Walker-Dilks C. Interventions to prevent aspiration pneumonia in older adults: a systematic review. J Am Geriatr Soc. 2003;51(7):1018-1022.
- Gosney M, Martin MV, Wright AE. The role of selective decontamination of the digestive tract in acute stroke. Age Ageing 2006;35(1):42-47.
The Case
A 74-year-old man with Alzheimer’s dementia presents with urinary tract infection (UTI), hypovolemia, and hypernatremia. He also has chronic dysphagia with a history of aspiration pneumonia and has been on thickened liquids at home for the past five months. As his infection is treated, he improves and requests water to drink.
Background
The diagnosis of dysphagia is clinical, and assessments from patients and family are often sufficient. The optimal test to assess the severity of dysphagia is a bedside swallow evaluation using small amounts of water.1 Video-assisted fluoroscopic examinations can identify problem areas within the oropharynx and esophagus and may help determine the etiology of dysphagia.
What evidence supports various treatment options for dysphagia?
Access to Water
Water is a thin liquid with low viscosity, which allows for rapid transit through the oropharynx. In debilitated and elderly patients, thin liquids easily reach the epiglottis and enter the trachea before pharyngeal muscles compensate. As such, access to water and other thin liquids is often restricted in patients suspected to have dysphagia.4
However, allowing access to water improves patient satisfaction, reduces the development of dehydration, and does not increase the incidence of AP. Bedside therapy interventions such as correct positioning and chin-tuck and sipping technique as well as attention to oral hygiene are recommended prior to more noxious options such as thickened liquids.1 The Frazier water protocol may help provide logistical guidance for facilities interested in improving access to water for patients with dysphagia.
Liquid Modification
Many clinicians manage dysphagia through restricting access to all thin liquids. In the hospital setting where video fluoroscopy and speech therapy are readily available, clinicians frequently employ the use of modified diets with thickened liquids in order to minimize the risk of aspiration despite the lack of high-quality evidence supporting liquid modification.2 Patients associate thickened liquids and restricted diets with a reduction in quality of life. Compliance studies have shown that only a minority of patients are compliant with thickened liquids at five days. In addition, thickening liquids has not been shown to decrease the risk of AP nor improve nutritional status, and it may actually cause harm by increasing the risk of dehydration and UTI.4
Tube Feeding
In patients with severe dysphagia in whom conservative management is not feasible or has failed, maintaining adequate nutrition can be a challenge. There are encouraging data with nutritionally enriching and modifying the texture of solid foods.1 Alternative methods of enteral nutrition delivery are often also considered. The most common vehicles of delivery are nasogastric tubes, post-pyloric feeding tubes, and percutaneous endoscopic gastrostomy (PEG) tubes. In theory, bypassing the pharynx and esophagus could result in fewer aspiration events and less AP.3 However, nasogastric, post-pyloric, or PEG feeding does not decrease the risk of AP. For patients with advanced dementia, there have been no randomized trials demonstrating an improvement in mortality with tube feeds.4 Tube feeding also carries with it a slight procedural risk and a high incidence of associated diarrhea, plus is associated with electrolyte derangements such as hypernatremia. The decision to pursue tube feeding should be weighed heavily in every patient and is highly influenced by the etiology and anticipated duration of dysphagia.
Selective Digestive Decontamination
Selective digestive decontamination (SDD) is a protocol-based treatment that aims to eradicate potentially pathogenic gut flora, particularly aerobic gram-negatives, in critically ill patients to reduce the impact of aspiration events. The utilization of SDD and the available literature center firmly on critically ill and ventilated patients. Subsequent studies have demonstrated recolonization after protocol cessation, and long-term effects are currently undefined.5 Until it can be studied in broader populations and proven to have clinical benefit, employing SSD in non-critically ill patients with dysphagia remains unsupported.
Multimodal Approach
Many rehabilitation centers incorporate a therapist-driven swallowing treatment program. Evidence suggests patient and family counseling alone may not be effective, so these programs variably incorporate diet/liquid modification, strengthening exercises, sensory processing techniques, and even neuromuscular electrical stimulation for muscle building.1 Accordingly, these programs are resource-intensive.
Management
Dysphagia remains a significant clinical problem for hospitalized patients. The existing literature and practice guidelines generally support a “less is more” approach. Though liquid/diet modification is common practice, it is not based in solid evidence and may contribute to unnecessary tube feeding. The best current evidence supports allowing access to water and ice chips. The ideal management plan for each patient will differ and should incorporate patient and family preferences in a multidisciplinary approach.
Back to the Case
Our patient requests water. He coughs after drinking during a bedside swallow evaluation. The risks of potential aspiration and AP are explained, and he expresses his understanding. He reiterates his choice to be allowed access to water as it is important to his quality of life. The speech therapy team is consulted and provides instruction on chin-tuck positioning, oral care, and timing water between meals rather than while eating food. He does well for the remainder of the hospital stay, and by time of discharge, his electrolytes are corrected, and he is much more comfortable being allowed to drink water. He is discharged home and encouraged to continue with these conservative measures.
Bottom Line
Evidence to support many common interventions for dysphagia is lacking; patients with dysphagia are best managed via a multidisciplinary, multimodal approach that provides access to water whenever possible. TH
Vijay G. Paryani, MD, is an internal medicine resident in the department of internal medicine at the University of Kentucky. Joseph R. Sweigart, MD, is a hospitalist and assistant professor of hospital medicine in the division of hospital medicine at the University of Kentucky. Laura C. Fanucchi, MD, is a hospitalist and assistant professor of hospital medicine in the division of hospital medicine at the University of Kentucky.
References
- Karagiannis MJ, Chivers L, Karagiannis TC. Effects of oral intake of water in patients with oropharyngeal dysphagia. BMC Geriatr. 2011;11(2):9.
- Foley N, Teasell R, Salter K, Kruger E, Martino R. Dysphagia treatment post stroke: a systematic review of randomized controlled trials. Age Ageing. 2008;37(3):258-264.
- Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med. 2001;344(9):665-671.
- Loeb MB, Becker M, Eady A, Walker-Dilks C. Interventions to prevent aspiration pneumonia in older adults: a systematic review. J Am Geriatr Soc. 2003;51(7):1018-1022.
- Gosney M, Martin MV, Wright AE. The role of selective decontamination of the digestive tract in acute stroke. Age Ageing 2006;35(1):42-47.
The Missing Element
A 57‐year‐old man presented to an emergency department with 1 month of progressive, bilateral lower extremity pain and weakness.
The first step in evaluating weakness is to determine whether it is objective (ie, decreased muscle strength due to pathology along the neuromuscular axis) or subjective. The sensation of weakness without loss of muscle strength may result from a debilitating chronic disease (eg, congestive heart failure, anemia, or chronic obstructive pulmonary disease). In patients with true lower extremity weakness it is prudent to assess for a myelopathy with a focused history and exam that includes assessment of bowel or bladder impairment and anal reflex. The presence of pain along with weakness might suggest disease of the muscle itself. A myopathy may arise from an infectious (eg, influenza), inflammatory (eg, polymyositis), endocrine (eg, hypothyroidism), or drug‐related (eg, statin) process.
The patient described 1 month of generalized weakness and pain in his lower extremities, which had worsened progressively to the point where ambulation was difficult. He was able to rise from a seated position using his arms for assistance, but had difficulty balancing in a standing position without assistance. The pain also involved both of his knees and increased with weight bearing. He also complained of bilateral lower extremity numbness and paresthesias, which had been migrating proximally from his toes over several months. He denied any recent trauma to his legs or back.
These symmetrical, distal sensory deficits favor a peripheral neuropathy over a myopathy, with neuropathic pain and arthralgia causing his impaired ability to ambulate or remain standing. In polyneuropathy, the type of nerve involvement (sensory vs motor) and pathology (axonal vs demyelinating) helps prioritize the differential. In developed countries, the most common causes of polyneuropathy are diabetes mellitus and alcohol. However, the tempo of his disease broadens the possibilities to include acute inflammatory demyelinating polyneuropathy, paraneoplastic syndrome (eg, monoclonal gammopathy), an autoimmune process (eg, rheumatoid arthritis, vasculitis), and heavy metal toxicity such as lead poisoning.
He had no history of chronic medical illness or hospitalizations and took no medications. His social history was notable for a history of alcohol abuse. For the past several years, he had only been drinking 1 to 2 beers daily due to cost, but had a history of more significant alcohol abuse in the distant past. He smoked 1 pack of tobacco per day, and denied illicit drug use. He denied any sexual activity or recent travel. He lived in a van, and had been homeless for over 10 years.
His socioeconomic status adds a layer of complexity to the case. Human immunodeficiency virus and hepatitis C virus (HCV) are more prevalent in the homeless and are associated with polyneuropathy. His lack of funds may drive him to drink illegally distilled alcohol, which can cause polyneuropathy through lead or arsenic toxicity. Excessive smoking could be linked to a peripheral neuropathy through a paraneoplastic syndrome (eg, small cell lung cancer).
Alcohol causes polyneuropathy through toxic effects on nerves and may be playing a role in his polyneuropathy, but the rapid pace and severity suggests an additional process. Alcoholism can be associated with deficiency of various B vitamins, such as thiamine, pyridoxine, and cobalamin, which can cause polyneuropathy. In alcoholics who are hospitalized, thiamine should be administered prior to glucose to decrease risk of Wernicke encephalopathy.
His temperature was 38.0C, heart rate 93 beats/min, blood pressure 121/60 mm Hg, respiratory rate 14/min, with an oxygen saturation of 97% on ambient air. He appeared cachectic and disheveled. He had moist mucous membranes, poor dentition with missing teeth, and no mucosal bleeding or oropharyngeal erythema. His cardiac exam revealed no murmurs, rubs, or gallops. His lungs were clear. His abdominal exam was benign, without masses or tenderness. His skin exam (Figure 1) was notable for nonpalpable petechiae on his anterior shins and thighs up to his buttocks. His extremity exam was significant for diffuse tenderness to light palpation on both lower extremities, a large indurated tender ecchymosis 15 15 cm behind the right knee, and another ecchymosis 6 8 cm behind the left knee. His dorsalis pedis and anterior tibialis pulses were appreciated by Doppler but not by palpation. He had decreased sensation to light touch of his bilateral feet to his ankles. Strength exam was challenging to assess secondary to posterior leg pain, but he demonstrated 4/5 strength of his hip flexors, quadriceps, and plantar flexors of the foot. His upper extremity strength and sensory exam were normal. Examination of the cranial nerves was normal. He had 2+ patellar and Achilles reflexes. Gait could not be adequately assessed.

Petechiae manifest as a nonblanchable rash caused by extravasated red blood cells. Common etiologies include quantitative or qualitative platelet defects, disseminated intravascular coagulopathy, trauma, and vasculitis. Cirrhosis from alcohol leading to thrombocytopenia and petechial rash is unlikely given no other stigmata of liver disease such as jaundice, spider angiomata, caput medusae, or palmar erythema. Less common causes include nutritional deficiency and light chain (AL) amyloidosis, which could explain both the neuropathy and rash.
The constellation of fever and petechial rash can represent a life‐threatening systemic process. Infectious agents that require immediate consideration with fever and petechiae include Neisseria meningitidis (meningococcemia), Rickettsia rickettsii (Rocky Mountain spotted fever), Staphylococcus, and Streptococcus. However, his normal blood pressure, dependent distribution of rash, and neuropathy make a severe bacterial infection less likely. Thrombotic thrombocytopenic purpura is possible and should prompt assessment of platelets, peripheral blood smear, and lactate dehydrogenase. Among vasculitides, the polyneuropathy, fever, and dependent distribution of petechial rash prioritize a small‐to‐medium vessel vasculitis, where the pathophysiology involves inflammation of dermal vessels and vasa nervorum (blood supply of nerves). Examples include HCV‐related cryoglobulinemic vasculitis, polyarteritis nodosa (PAN), and antineutrophilic cytoplasmic antibody (ANCA)associated vasculitis. However, ANCA‐associated vasculitis is less likely without upper or lower respiratory symptoms. Henoch‐Schonlein purpura may explain the rash but is more common in children and is not associated with neuropathy.
Posterior knee ecchymosis, in absence of trauma, raises suspicion for a ruptured Baker's cyst. However, the bilateral involvement and lack of calf manifestations makes this unlikely. The location raises concern for hemarthrosis, so a more likely explanation would be coagulopathy (eg, an acquired factor inhibitor) or a collagen defect. In developed countries, a commonly overlooked category of diseasenutritional deficiencywarrants serious consideration in alcoholics. Vitamin C deficiency (scurvy) may cause a petechial rash and ecchymosis from perifollicular hemorrhage and impaired collagen synthesis, respectively. Scurvy can masquerade as small vessel vasculitis because of its associated petechial rash. The neuropathy might be explained by concomitant thiamine or cobalamin deficiency. It is important to obtain a thorough dietary history and assess vibration and proprioception, which may be impaired from pathology of the dorsal column in cobalamin deficiency. The low‐grade fever may be a red herring, but if it becomes significant would be difficult to explain with nutritional deficiency.
In summary, a judicious evaluation for infection is mandatory, but the leading diagnoses are a small‐to‐medium vessel vasculitis (PAN or HCV‐related cryoglobulinemia), deficiency of multiple vitamins, and AL amyloidosis.
Initial labs showed white blood cell count 7800/L, hematocrit 39.2%, and platelet count of 251,000/L. Serum chemistry demonstrated a sodium of 131 mEq/L, potassium 4.7 mEq/L, chloride 93 mEq/L, bicarbonate 23 mEq/L, blood urea nitrogen 8 mg/dL, and creatinine 0.8 mg/dL. His aminotransferases, albumin, alkaline phosphatase, and coagulation studies were within normal limits. Urinalysis was remarkable for 2+ urobilinogen, 1+ ketones, and a bland sediment. Urine toxicology screen was negative.
His white blood cell count is normal, so with a heart rate of 93 beats/minute, he barely meets a single criterion of systemic inflammatory response syndrome (SIRS). The lack of SIRS and normal platelet, albumin, white blood cell, and red blood cell counts significantly reduces the likelihood of an infectious or inflammatory process. Without any clinical or biochemical evidence of HCV infection, HCV‐associated cryoglobulinemia is less likely. A normal creatinine might overestimate renal function in setting of decreased protein intake and muscle mass; nevertheless, the bland urine sediment further lowers probability of PAN and ANCA‐associated vasculitides. The normal platelet count and coagulation studies suggest either a qualitative platelet defect (eg, acquired von Willebrand disease) or impaired vessel integrity (eg, collagen defect) to explain the petechial rash. The urine ketones likely represent alcohol and/or starvation‐related ketosis. These data reduce the probability of infection and vasculitis, and prioritize vitamin deficiency and AL amyloidosis. Antibiotic therapy is not appropriate, given the absence of SIRS and subacute course. His presentation likely prompted a wide variety of tests, but most relevant would be a dietary history, cobalamin and vitamin C levels, serum free light chains, and skin biopsy. Biopsy of the rash would allow assessment for vasculitis and AL amyloidosis. The former is marked by inflammatory infiltrate of vessels, and the latter by perivascular invasion with amyloid fibrils. If the dietary history was consistent with ascorbic acid deficiency (scurvy), in addition to thiamine, he should be empirically treated with vitamin C. Patients with scurvy demonstrate rapid clinical improvement with treatment.
C‐reactive protein (CRP) was 47.9 mg/L and erythrocyte sedimentation rate (ESR) was 44 mm/hr. Human immunodeficiency antibody screen was negative. Anti‐nuclear antibodies and anti‐nuclear cytoplasmic antibody panel were negative. Computed tomography angiogram (CTA) of the lower extremities demonstrated severe stenosis of the left superficial femoral artery and severe stenosis of the right posterior tibial artery. Ankle‐brachial indices were 0.83 on the right side and 0.72 on the left, indicating mild to moderate arterial disease.
ESR and CRP are nonspecific markers of inflammation. Their elevation does not prioritize malignancy, autoimmunity, or infection. ANCA might be negative in commonly ANCA‐associated vasculitides such as eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis, and granulomatosis with polyangiitis. However, the lack of respiratory and renal involvement in addition to the negative ANCA panel make such diagnoses unlikely. CTA of the patient's legs showed significant peripheral artery disease (PAD). This is unlikely to be the cause of his presentation; PAD should not cause petechiae, and his pain is disproportionate to the severity of the vascular disease reported. The additional information leaves the differential unchanged.
A dermatologist was consulted. She described and photographed a perifollicular distribution of the lower extremity petechiae with associated perifollicular hyperkeratosis and retained curled hairs (Figure 2).
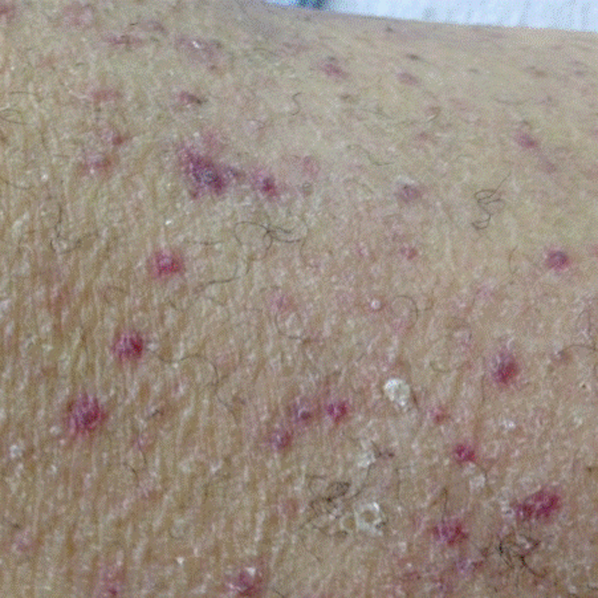
The described rash is specific for scurvy. His homelessness and alcohol intake likely made him vulnerable to ascorbic acid deficiency from lack of access to fruits and vegetables. Measurement of vitamin C level is unnecessary as the pretest probability for scurvy is very high. More relevant than a vitamin C level or skin biopsy is empiric treatment with ascorbic acid; as mentioned, patients with scurvy respond rapidly to vitamin C therapy. Given the neuropathy, he should be assessed for concomitant thiamine and/or cobalamin deficiency. His peripheral arterial disease is unlikely to be related.
His ascorbic acid level was 0.0 mg/dL (reference range, 0.22.0 mg/dL). Further history was obtained, and the patient reported exclusively eating frozen hamburgers and burritos for almost 1 year. He believed he had not had a fruit or vegetable in over 10 years. He was started on 1000 mg daily of ascorbic acid. By hospital day 2, his rash had mostly resolved and he was able to stand with some support. The patient was seen by his primary care physician 3 weeks after diagnosis, with his exercise tolerance nearly back to baseline. His rash had entirely resolved.
DISCUSSION
Unlike other mammals, humans do not have the ability to convert glucose to vitamin C and thus require an exogenous source, such as fruits and vegetables. The oft‐cited observation of scurvy in sailors during long journeys in the 18th century is a classic example of clinical disease due to vitamin C deficiency.[1] Once replete, body stores of vitamin C are usually sufficient to last over 6 months of deprivation. In some patients, symptoms of deficiency may appear within 3 months.[2] The patient in this report likely suffered years of vitamin C deficiency, resulting in the significant manifestations of scurvy reported here.
Vitamin C is a water‐soluble vitamin necessary for the biosynthesis of collagen, L‐carnitine, and neurotransmitters.[3] With deficiency, the resulting impairment in the formation of collagen affects blood vessel integrity and results in perivascular edema and erythrocyte extravasation. Clinically, this leads to hemorrhagic manifestations (eg, periosteal hemorrhage and perifollicular petechiae) and poor wound healing. Corkscrew hairs result because of vitamin C's role in disulfide bonding during hair formation. Woody edema and dyspnea are thought to be a consequence of leaky capillaries.[4]
Scurvy is still a significant cause of morbidity in at‐risk populations in the United States. Several populations have been identified as high risk for vitamin C deficiency, including the elderly, persons who live alone, alcoholics, smokers, individuals of low socioeconomic status, patients on hemodialysis, and those with psychiatric disease.[5] Specifically, the high oxidative stress associated with smoking, the history of alcohol abuse, and homelessness put this patient at an especially high risk.[6] Those with oxidative stressors have been postulated to require up to 125 mg/d of vitamin C compared to 60 to 90 mg/d of those without the same risks.[7] In a national health and nutrition survey in the United States in 2004, the prevalence of vitamin C deficiency as defined by a serum level <0.2 mg/dL was noted in 7.1% of those surveyed.[8] This study also noted a significantly higher prevalence of deficiency in smokers and individuals with low socioeconomic status.
Scurvy is a clinical diagnosis based on clinical features and dietary history. Severe manifestations of scurvy may happen quickly after the initial presentation, making early diagnosis especially important.[2] These include anemia, bone pain, ocular hemorrhage, cerebral hemorrhage, and hemopericardium.[2, 4] If needed, laboratory diagnosis can be made by demonstrating a serum ascorbic acid level <0.2 mg/dL. However, the level may be normal if the patient has had recent intake of vitamin C. In that scenario, the leukocyte vitamin C concentration may be a more accurate measure of the body stores as leukocyte levels change more slowly.[4] Biopsy of skin lesions is not necessary for the diagnosis and typically show a dilated hair follicle with keratin plugging and perifollicular hemorrhage.[9] Given the lack of adverse effects, treatment with vitamin C supplementation should begin immediately, even with low suspicion of scurvy, and response can serve as further clinical evidence and render laboratory testing unnecessary.
In this patient, the diagnosis was challenging for several reasons. The presentation was concerning for vasculitis given the dependent petechiae and elevated inflammatory markers. However, in scurvy, the petechiae are perifollicular and associated with hyperkeratosis, as opposed to the palpable purpura often described in vasculitis. Further, marked elevations in ESR and CRP have also been reported in scurvy.[10] The initial concern for vasculitis and clinician discomfort with a diagnosis based solely on a rash delayed the diagnosis. The complaint of polyneuropathy also seemed inconsistent with scurvy. Very rarely, scurvy may cause a neuropathy by hemorrhage into the nerve sheath, as seen in a case of bilateral femoral neuropathy.[11] Most likely, this patient had an underlying vitamin B deficiency explaining his polyneuropathy. Unfortunately, the patient was lost to follow‐up after his postdischarge visit with his primary physician and was not tested for other concomitant vitamin deficiencies.
Scurvy is very responsive to even small doses of vitamin C supplementation. For rapid recovery, doses ranging from 100 mg 3 times daily to 1000 mg daily of oral vitamin C are recommended for at least 1 month. Resolution of symptoms will begin within 24 hours, and complete recovery should occur by 3 months.[4] Scurvy is a classic example of how nutritional deficiencies can have a myriad of presentations and may mimic other systemic diseases. Clinicians who recall these manifestations and carefully assess patients for nutritional risks may be able to quickly identify the missing element (or elements) in a patient's diet, and initiate treatment that is often rapidly effective.
KEY TEACHING POINTS
- Vitamin C deficiency initially presents with classic dermatological findings of perifollicular petechiae with associated hyperkeratosis and corkscrew hairs.
- Scurvy is a clinical diagnosis based on history and presentation. Vitamin C serum level may not accurately reflect body stores, and a leukocyte vitamin C level may be obtained. The diagnosis may also be confirmed with observed response to vitamin C supplementation.
- Scurvy should be suspected in high‐risk populations, especially the marginally housed, the elderly, alcoholics, and smokers.
- Clinicians should screen patients with scurvy for other nutritional deficiencies including thiamine, folate, B12, and vitamin D levels.
Disclosures: Nothing to report.
- . The discovery of vitamin C. Ann Nutr Metab. 2012;61(3):259–264.
- , , , , . Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24(4):432–443.
- . New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314(14):892–902.
- , . Adult scurvy. J Am Acad Dermatol. 1999;41(6):895–910.
- , . Be vigilant for scurvy in high‐risk groups. Practitioner. 2012;256(1755):23–5, 3.
- . Estimating ascorbic acid requirements for cigarette smokers. Ann N Y Acad Sci. 1993;686:335–346.
- , , . Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health. 2004;94(5):870–875.
- , , , . Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90(5):1252–1263.
- , , . Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol. 2003;44(1):48–51.
- , . Rheumatic manifestations of scurvy: a report of three recent cases in a major urban center and a review. Semin Arthritis Rheum. 2011;41(2):286–290.
- . Femoral neuropathy in scurvy. N Engl J Med. 1969;281(23):1292–1293.
A 57‐year‐old man presented to an emergency department with 1 month of progressive, bilateral lower extremity pain and weakness.
The first step in evaluating weakness is to determine whether it is objective (ie, decreased muscle strength due to pathology along the neuromuscular axis) or subjective. The sensation of weakness without loss of muscle strength may result from a debilitating chronic disease (eg, congestive heart failure, anemia, or chronic obstructive pulmonary disease). In patients with true lower extremity weakness it is prudent to assess for a myelopathy with a focused history and exam that includes assessment of bowel or bladder impairment and anal reflex. The presence of pain along with weakness might suggest disease of the muscle itself. A myopathy may arise from an infectious (eg, influenza), inflammatory (eg, polymyositis), endocrine (eg, hypothyroidism), or drug‐related (eg, statin) process.
The patient described 1 month of generalized weakness and pain in his lower extremities, which had worsened progressively to the point where ambulation was difficult. He was able to rise from a seated position using his arms for assistance, but had difficulty balancing in a standing position without assistance. The pain also involved both of his knees and increased with weight bearing. He also complained of bilateral lower extremity numbness and paresthesias, which had been migrating proximally from his toes over several months. He denied any recent trauma to his legs or back.
These symmetrical, distal sensory deficits favor a peripheral neuropathy over a myopathy, with neuropathic pain and arthralgia causing his impaired ability to ambulate or remain standing. In polyneuropathy, the type of nerve involvement (sensory vs motor) and pathology (axonal vs demyelinating) helps prioritize the differential. In developed countries, the most common causes of polyneuropathy are diabetes mellitus and alcohol. However, the tempo of his disease broadens the possibilities to include acute inflammatory demyelinating polyneuropathy, paraneoplastic syndrome (eg, monoclonal gammopathy), an autoimmune process (eg, rheumatoid arthritis, vasculitis), and heavy metal toxicity such as lead poisoning.
He had no history of chronic medical illness or hospitalizations and took no medications. His social history was notable for a history of alcohol abuse. For the past several years, he had only been drinking 1 to 2 beers daily due to cost, but had a history of more significant alcohol abuse in the distant past. He smoked 1 pack of tobacco per day, and denied illicit drug use. He denied any sexual activity or recent travel. He lived in a van, and had been homeless for over 10 years.
His socioeconomic status adds a layer of complexity to the case. Human immunodeficiency virus and hepatitis C virus (HCV) are more prevalent in the homeless and are associated with polyneuropathy. His lack of funds may drive him to drink illegally distilled alcohol, which can cause polyneuropathy through lead or arsenic toxicity. Excessive smoking could be linked to a peripheral neuropathy through a paraneoplastic syndrome (eg, small cell lung cancer).
Alcohol causes polyneuropathy through toxic effects on nerves and may be playing a role in his polyneuropathy, but the rapid pace and severity suggests an additional process. Alcoholism can be associated with deficiency of various B vitamins, such as thiamine, pyridoxine, and cobalamin, which can cause polyneuropathy. In alcoholics who are hospitalized, thiamine should be administered prior to glucose to decrease risk of Wernicke encephalopathy.
His temperature was 38.0C, heart rate 93 beats/min, blood pressure 121/60 mm Hg, respiratory rate 14/min, with an oxygen saturation of 97% on ambient air. He appeared cachectic and disheveled. He had moist mucous membranes, poor dentition with missing teeth, and no mucosal bleeding or oropharyngeal erythema. His cardiac exam revealed no murmurs, rubs, or gallops. His lungs were clear. His abdominal exam was benign, without masses or tenderness. His skin exam (Figure 1) was notable for nonpalpable petechiae on his anterior shins and thighs up to his buttocks. His extremity exam was significant for diffuse tenderness to light palpation on both lower extremities, a large indurated tender ecchymosis 15 15 cm behind the right knee, and another ecchymosis 6 8 cm behind the left knee. His dorsalis pedis and anterior tibialis pulses were appreciated by Doppler but not by palpation. He had decreased sensation to light touch of his bilateral feet to his ankles. Strength exam was challenging to assess secondary to posterior leg pain, but he demonstrated 4/5 strength of his hip flexors, quadriceps, and plantar flexors of the foot. His upper extremity strength and sensory exam were normal. Examination of the cranial nerves was normal. He had 2+ patellar and Achilles reflexes. Gait could not be adequately assessed.
Petechiae manifest as a nonblanchable rash caused by extravasated red blood cells. Common etiologies include quantitative or qualitative platelet defects, disseminated intravascular coagulopathy, trauma, and vasculitis. Cirrhosis from alcohol leading to thrombocytopenia and petechial rash is unlikely given no other stigmata of liver disease such as jaundice, spider angiomata, caput medusae, or palmar erythema. Less common causes include nutritional deficiency and light chain (AL) amyloidosis, which could explain both the neuropathy and rash.
The constellation of fever and petechial rash can represent a life‐threatening systemic process. Infectious agents that require immediate consideration with fever and petechiae include Neisseria meningitidis (meningococcemia), Rickettsia rickettsii (Rocky Mountain spotted fever), Staphylococcus, and Streptococcus. However, his normal blood pressure, dependent distribution of rash, and neuropathy make a severe bacterial infection less likely. Thrombotic thrombocytopenic purpura is possible and should prompt assessment of platelets, peripheral blood smear, and lactate dehydrogenase. Among vasculitides, the polyneuropathy, fever, and dependent distribution of petechial rash prioritize a small‐to‐medium vessel vasculitis, where the pathophysiology involves inflammation of dermal vessels and vasa nervorum (blood supply of nerves). Examples include HCV‐related cryoglobulinemic vasculitis, polyarteritis nodosa (PAN), and antineutrophilic cytoplasmic antibody (ANCA)associated vasculitis. However, ANCA‐associated vasculitis is less likely without upper or lower respiratory symptoms. Henoch‐Schonlein purpura may explain the rash but is more common in children and is not associated with neuropathy.
Posterior knee ecchymosis, in absence of trauma, raises suspicion for a ruptured Baker's cyst. However, the bilateral involvement and lack of calf manifestations makes this unlikely. The location raises concern for hemarthrosis, so a more likely explanation would be coagulopathy (eg, an acquired factor inhibitor) or a collagen defect. In developed countries, a commonly overlooked category of diseasenutritional deficiencywarrants serious consideration in alcoholics. Vitamin C deficiency (scurvy) may cause a petechial rash and ecchymosis from perifollicular hemorrhage and impaired collagen synthesis, respectively. Scurvy can masquerade as small vessel vasculitis because of its associated petechial rash. The neuropathy might be explained by concomitant thiamine or cobalamin deficiency. It is important to obtain a thorough dietary history and assess vibration and proprioception, which may be impaired from pathology of the dorsal column in cobalamin deficiency. The low‐grade fever may be a red herring, but if it becomes significant would be difficult to explain with nutritional deficiency.
In summary, a judicious evaluation for infection is mandatory, but the leading diagnoses are a small‐to‐medium vessel vasculitis (PAN or HCV‐related cryoglobulinemia), deficiency of multiple vitamins, and AL amyloidosis.
Initial labs showed white blood cell count 7800/L, hematocrit 39.2%, and platelet count of 251,000/L. Serum chemistry demonstrated a sodium of 131 mEq/L, potassium 4.7 mEq/L, chloride 93 mEq/L, bicarbonate 23 mEq/L, blood urea nitrogen 8 mg/dL, and creatinine 0.8 mg/dL. His aminotransferases, albumin, alkaline phosphatase, and coagulation studies were within normal limits. Urinalysis was remarkable for 2+ urobilinogen, 1+ ketones, and a bland sediment. Urine toxicology screen was negative.
His white blood cell count is normal, so with a heart rate of 93 beats/minute, he barely meets a single criterion of systemic inflammatory response syndrome (SIRS). The lack of SIRS and normal platelet, albumin, white blood cell, and red blood cell counts significantly reduces the likelihood of an infectious or inflammatory process. Without any clinical or biochemical evidence of HCV infection, HCV‐associated cryoglobulinemia is less likely. A normal creatinine might overestimate renal function in setting of decreased protein intake and muscle mass; nevertheless, the bland urine sediment further lowers probability of PAN and ANCA‐associated vasculitides. The normal platelet count and coagulation studies suggest either a qualitative platelet defect (eg, acquired von Willebrand disease) or impaired vessel integrity (eg, collagen defect) to explain the petechial rash. The urine ketones likely represent alcohol and/or starvation‐related ketosis. These data reduce the probability of infection and vasculitis, and prioritize vitamin deficiency and AL amyloidosis. Antibiotic therapy is not appropriate, given the absence of SIRS and subacute course. His presentation likely prompted a wide variety of tests, but most relevant would be a dietary history, cobalamin and vitamin C levels, serum free light chains, and skin biopsy. Biopsy of the rash would allow assessment for vasculitis and AL amyloidosis. The former is marked by inflammatory infiltrate of vessels, and the latter by perivascular invasion with amyloid fibrils. If the dietary history was consistent with ascorbic acid deficiency (scurvy), in addition to thiamine, he should be empirically treated with vitamin C. Patients with scurvy demonstrate rapid clinical improvement with treatment.
C‐reactive protein (CRP) was 47.9 mg/L and erythrocyte sedimentation rate (ESR) was 44 mm/hr. Human immunodeficiency antibody screen was negative. Anti‐nuclear antibodies and anti‐nuclear cytoplasmic antibody panel were negative. Computed tomography angiogram (CTA) of the lower extremities demonstrated severe stenosis of the left superficial femoral artery and severe stenosis of the right posterior tibial artery. Ankle‐brachial indices were 0.83 on the right side and 0.72 on the left, indicating mild to moderate arterial disease.
ESR and CRP are nonspecific markers of inflammation. Their elevation does not prioritize malignancy, autoimmunity, or infection. ANCA might be negative in commonly ANCA‐associated vasculitides such as eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis, and granulomatosis with polyangiitis. However, the lack of respiratory and renal involvement in addition to the negative ANCA panel make such diagnoses unlikely. CTA of the patient's legs showed significant peripheral artery disease (PAD). This is unlikely to be the cause of his presentation; PAD should not cause petechiae, and his pain is disproportionate to the severity of the vascular disease reported. The additional information leaves the differential unchanged.
A dermatologist was consulted. She described and photographed a perifollicular distribution of the lower extremity petechiae with associated perifollicular hyperkeratosis and retained curled hairs (Figure 2).
The described rash is specific for scurvy. His homelessness and alcohol intake likely made him vulnerable to ascorbic acid deficiency from lack of access to fruits and vegetables. Measurement of vitamin C level is unnecessary as the pretest probability for scurvy is very high. More relevant than a vitamin C level or skin biopsy is empiric treatment with ascorbic acid; as mentioned, patients with scurvy respond rapidly to vitamin C therapy. Given the neuropathy, he should be assessed for concomitant thiamine and/or cobalamin deficiency. His peripheral arterial disease is unlikely to be related.
His ascorbic acid level was 0.0 mg/dL (reference range, 0.22.0 mg/dL). Further history was obtained, and the patient reported exclusively eating frozen hamburgers and burritos for almost 1 year. He believed he had not had a fruit or vegetable in over 10 years. He was started on 1000 mg daily of ascorbic acid. By hospital day 2, his rash had mostly resolved and he was able to stand with some support. The patient was seen by his primary care physician 3 weeks after diagnosis, with his exercise tolerance nearly back to baseline. His rash had entirely resolved.
DISCUSSION
Unlike other mammals, humans do not have the ability to convert glucose to vitamin C and thus require an exogenous source, such as fruits and vegetables. The oft‐cited observation of scurvy in sailors during long journeys in the 18th century is a classic example of clinical disease due to vitamin C deficiency.[1] Once replete, body stores of vitamin C are usually sufficient to last over 6 months of deprivation. In some patients, symptoms of deficiency may appear within 3 months.[2] The patient in this report likely suffered years of vitamin C deficiency, resulting in the significant manifestations of scurvy reported here.
Vitamin C is a water‐soluble vitamin necessary for the biosynthesis of collagen, L‐carnitine, and neurotransmitters.[3] With deficiency, the resulting impairment in the formation of collagen affects blood vessel integrity and results in perivascular edema and erythrocyte extravasation. Clinically, this leads to hemorrhagic manifestations (eg, periosteal hemorrhage and perifollicular petechiae) and poor wound healing. Corkscrew hairs result because of vitamin C's role in disulfide bonding during hair formation. Woody edema and dyspnea are thought to be a consequence of leaky capillaries.[4]
Scurvy is still a significant cause of morbidity in at‐risk populations in the United States. Several populations have been identified as high risk for vitamin C deficiency, including the elderly, persons who live alone, alcoholics, smokers, individuals of low socioeconomic status, patients on hemodialysis, and those with psychiatric disease.[5] Specifically, the high oxidative stress associated with smoking, the history of alcohol abuse, and homelessness put this patient at an especially high risk.[6] Those with oxidative stressors have been postulated to require up to 125 mg/d of vitamin C compared to 60 to 90 mg/d of those without the same risks.[7] In a national health and nutrition survey in the United States in 2004, the prevalence of vitamin C deficiency as defined by a serum level <0.2 mg/dL was noted in 7.1% of those surveyed.[8] This study also noted a significantly higher prevalence of deficiency in smokers and individuals with low socioeconomic status.
Scurvy is a clinical diagnosis based on clinical features and dietary history. Severe manifestations of scurvy may happen quickly after the initial presentation, making early diagnosis especially important.[2] These include anemia, bone pain, ocular hemorrhage, cerebral hemorrhage, and hemopericardium.[2, 4] If needed, laboratory diagnosis can be made by demonstrating a serum ascorbic acid level <0.2 mg/dL. However, the level may be normal if the patient has had recent intake of vitamin C. In that scenario, the leukocyte vitamin C concentration may be a more accurate measure of the body stores as leukocyte levels change more slowly.[4] Biopsy of skin lesions is not necessary for the diagnosis and typically show a dilated hair follicle with keratin plugging and perifollicular hemorrhage.[9] Given the lack of adverse effects, treatment with vitamin C supplementation should begin immediately, even with low suspicion of scurvy, and response can serve as further clinical evidence and render laboratory testing unnecessary.
In this patient, the diagnosis was challenging for several reasons. The presentation was concerning for vasculitis given the dependent petechiae and elevated inflammatory markers. However, in scurvy, the petechiae are perifollicular and associated with hyperkeratosis, as opposed to the palpable purpura often described in vasculitis. Further, marked elevations in ESR and CRP have also been reported in scurvy.[10] The initial concern for vasculitis and clinician discomfort with a diagnosis based solely on a rash delayed the diagnosis. The complaint of polyneuropathy also seemed inconsistent with scurvy. Very rarely, scurvy may cause a neuropathy by hemorrhage into the nerve sheath, as seen in a case of bilateral femoral neuropathy.[11] Most likely, this patient had an underlying vitamin B deficiency explaining his polyneuropathy. Unfortunately, the patient was lost to follow‐up after his postdischarge visit with his primary physician and was not tested for other concomitant vitamin deficiencies.
Scurvy is very responsive to even small doses of vitamin C supplementation. For rapid recovery, doses ranging from 100 mg 3 times daily to 1000 mg daily of oral vitamin C are recommended for at least 1 month. Resolution of symptoms will begin within 24 hours, and complete recovery should occur by 3 months.[4] Scurvy is a classic example of how nutritional deficiencies can have a myriad of presentations and may mimic other systemic diseases. Clinicians who recall these manifestations and carefully assess patients for nutritional risks may be able to quickly identify the missing element (or elements) in a patient's diet, and initiate treatment that is often rapidly effective.
KEY TEACHING POINTS
- Vitamin C deficiency initially presents with classic dermatological findings of perifollicular petechiae with associated hyperkeratosis and corkscrew hairs.
- Scurvy is a clinical diagnosis based on history and presentation. Vitamin C serum level may not accurately reflect body stores, and a leukocyte vitamin C level may be obtained. The diagnosis may also be confirmed with observed response to vitamin C supplementation.
- Scurvy should be suspected in high‐risk populations, especially the marginally housed, the elderly, alcoholics, and smokers.
- Clinicians should screen patients with scurvy for other nutritional deficiencies including thiamine, folate, B12, and vitamin D levels.
Disclosures: Nothing to report.
A 57‐year‐old man presented to an emergency department with 1 month of progressive, bilateral lower extremity pain and weakness.
The first step in evaluating weakness is to determine whether it is objective (ie, decreased muscle strength due to pathology along the neuromuscular axis) or subjective. The sensation of weakness without loss of muscle strength may result from a debilitating chronic disease (eg, congestive heart failure, anemia, or chronic obstructive pulmonary disease). In patients with true lower extremity weakness it is prudent to assess for a myelopathy with a focused history and exam that includes assessment of bowel or bladder impairment and anal reflex. The presence of pain along with weakness might suggest disease of the muscle itself. A myopathy may arise from an infectious (eg, influenza), inflammatory (eg, polymyositis), endocrine (eg, hypothyroidism), or drug‐related (eg, statin) process.
The patient described 1 month of generalized weakness and pain in his lower extremities, which had worsened progressively to the point where ambulation was difficult. He was able to rise from a seated position using his arms for assistance, but had difficulty balancing in a standing position without assistance. The pain also involved both of his knees and increased with weight bearing. He also complained of bilateral lower extremity numbness and paresthesias, which had been migrating proximally from his toes over several months. He denied any recent trauma to his legs or back.
These symmetrical, distal sensory deficits favor a peripheral neuropathy over a myopathy, with neuropathic pain and arthralgia causing his impaired ability to ambulate or remain standing. In polyneuropathy, the type of nerve involvement (sensory vs motor) and pathology (axonal vs demyelinating) helps prioritize the differential. In developed countries, the most common causes of polyneuropathy are diabetes mellitus and alcohol. However, the tempo of his disease broadens the possibilities to include acute inflammatory demyelinating polyneuropathy, paraneoplastic syndrome (eg, monoclonal gammopathy), an autoimmune process (eg, rheumatoid arthritis, vasculitis), and heavy metal toxicity such as lead poisoning.
He had no history of chronic medical illness or hospitalizations and took no medications. His social history was notable for a history of alcohol abuse. For the past several years, he had only been drinking 1 to 2 beers daily due to cost, but had a history of more significant alcohol abuse in the distant past. He smoked 1 pack of tobacco per day, and denied illicit drug use. He denied any sexual activity or recent travel. He lived in a van, and had been homeless for over 10 years.
His socioeconomic status adds a layer of complexity to the case. Human immunodeficiency virus and hepatitis C virus (HCV) are more prevalent in the homeless and are associated with polyneuropathy. His lack of funds may drive him to drink illegally distilled alcohol, which can cause polyneuropathy through lead or arsenic toxicity. Excessive smoking could be linked to a peripheral neuropathy through a paraneoplastic syndrome (eg, small cell lung cancer).
Alcohol causes polyneuropathy through toxic effects on nerves and may be playing a role in his polyneuropathy, but the rapid pace and severity suggests an additional process. Alcoholism can be associated with deficiency of various B vitamins, such as thiamine, pyridoxine, and cobalamin, which can cause polyneuropathy. In alcoholics who are hospitalized, thiamine should be administered prior to glucose to decrease risk of Wernicke encephalopathy.
His temperature was 38.0C, heart rate 93 beats/min, blood pressure 121/60 mm Hg, respiratory rate 14/min, with an oxygen saturation of 97% on ambient air. He appeared cachectic and disheveled. He had moist mucous membranes, poor dentition with missing teeth, and no mucosal bleeding or oropharyngeal erythema. His cardiac exam revealed no murmurs, rubs, or gallops. His lungs were clear. His abdominal exam was benign, without masses or tenderness. His skin exam (Figure 1) was notable for nonpalpable petechiae on his anterior shins and thighs up to his buttocks. His extremity exam was significant for diffuse tenderness to light palpation on both lower extremities, a large indurated tender ecchymosis 15 15 cm behind the right knee, and another ecchymosis 6 8 cm behind the left knee. His dorsalis pedis and anterior tibialis pulses were appreciated by Doppler but not by palpation. He had decreased sensation to light touch of his bilateral feet to his ankles. Strength exam was challenging to assess secondary to posterior leg pain, but he demonstrated 4/5 strength of his hip flexors, quadriceps, and plantar flexors of the foot. His upper extremity strength and sensory exam were normal. Examination of the cranial nerves was normal. He had 2+ patellar and Achilles reflexes. Gait could not be adequately assessed.
Petechiae manifest as a nonblanchable rash caused by extravasated red blood cells. Common etiologies include quantitative or qualitative platelet defects, disseminated intravascular coagulopathy, trauma, and vasculitis. Cirrhosis from alcohol leading to thrombocytopenia and petechial rash is unlikely given no other stigmata of liver disease such as jaundice, spider angiomata, caput medusae, or palmar erythema. Less common causes include nutritional deficiency and light chain (AL) amyloidosis, which could explain both the neuropathy and rash.
The constellation of fever and petechial rash can represent a life‐threatening systemic process. Infectious agents that require immediate consideration with fever and petechiae include Neisseria meningitidis (meningococcemia), Rickettsia rickettsii (Rocky Mountain spotted fever), Staphylococcus, and Streptococcus. However, his normal blood pressure, dependent distribution of rash, and neuropathy make a severe bacterial infection less likely. Thrombotic thrombocytopenic purpura is possible and should prompt assessment of platelets, peripheral blood smear, and lactate dehydrogenase. Among vasculitides, the polyneuropathy, fever, and dependent distribution of petechial rash prioritize a small‐to‐medium vessel vasculitis, where the pathophysiology involves inflammation of dermal vessels and vasa nervorum (blood supply of nerves). Examples include HCV‐related cryoglobulinemic vasculitis, polyarteritis nodosa (PAN), and antineutrophilic cytoplasmic antibody (ANCA)associated vasculitis. However, ANCA‐associated vasculitis is less likely without upper or lower respiratory symptoms. Henoch‐Schonlein purpura may explain the rash but is more common in children and is not associated with neuropathy.
Posterior knee ecchymosis, in absence of trauma, raises suspicion for a ruptured Baker's cyst. However, the bilateral involvement and lack of calf manifestations makes this unlikely. The location raises concern for hemarthrosis, so a more likely explanation would be coagulopathy (eg, an acquired factor inhibitor) or a collagen defect. In developed countries, a commonly overlooked category of diseasenutritional deficiencywarrants serious consideration in alcoholics. Vitamin C deficiency (scurvy) may cause a petechial rash and ecchymosis from perifollicular hemorrhage and impaired collagen synthesis, respectively. Scurvy can masquerade as small vessel vasculitis because of its associated petechial rash. The neuropathy might be explained by concomitant thiamine or cobalamin deficiency. It is important to obtain a thorough dietary history and assess vibration and proprioception, which may be impaired from pathology of the dorsal column in cobalamin deficiency. The low‐grade fever may be a red herring, but if it becomes significant would be difficult to explain with nutritional deficiency.
In summary, a judicious evaluation for infection is mandatory, but the leading diagnoses are a small‐to‐medium vessel vasculitis (PAN or HCV‐related cryoglobulinemia), deficiency of multiple vitamins, and AL amyloidosis.
Initial labs showed white blood cell count 7800/L, hematocrit 39.2%, and platelet count of 251,000/L. Serum chemistry demonstrated a sodium of 131 mEq/L, potassium 4.7 mEq/L, chloride 93 mEq/L, bicarbonate 23 mEq/L, blood urea nitrogen 8 mg/dL, and creatinine 0.8 mg/dL. His aminotransferases, albumin, alkaline phosphatase, and coagulation studies were within normal limits. Urinalysis was remarkable for 2+ urobilinogen, 1+ ketones, and a bland sediment. Urine toxicology screen was negative.
His white blood cell count is normal, so with a heart rate of 93 beats/minute, he barely meets a single criterion of systemic inflammatory response syndrome (SIRS). The lack of SIRS and normal platelet, albumin, white blood cell, and red blood cell counts significantly reduces the likelihood of an infectious or inflammatory process. Without any clinical or biochemical evidence of HCV infection, HCV‐associated cryoglobulinemia is less likely. A normal creatinine might overestimate renal function in setting of decreased protein intake and muscle mass; nevertheless, the bland urine sediment further lowers probability of PAN and ANCA‐associated vasculitides. The normal platelet count and coagulation studies suggest either a qualitative platelet defect (eg, acquired von Willebrand disease) or impaired vessel integrity (eg, collagen defect) to explain the petechial rash. The urine ketones likely represent alcohol and/or starvation‐related ketosis. These data reduce the probability of infection and vasculitis, and prioritize vitamin deficiency and AL amyloidosis. Antibiotic therapy is not appropriate, given the absence of SIRS and subacute course. His presentation likely prompted a wide variety of tests, but most relevant would be a dietary history, cobalamin and vitamin C levels, serum free light chains, and skin biopsy. Biopsy of the rash would allow assessment for vasculitis and AL amyloidosis. The former is marked by inflammatory infiltrate of vessels, and the latter by perivascular invasion with amyloid fibrils. If the dietary history was consistent with ascorbic acid deficiency (scurvy), in addition to thiamine, he should be empirically treated with vitamin C. Patients with scurvy demonstrate rapid clinical improvement with treatment.
C‐reactive protein (CRP) was 47.9 mg/L and erythrocyte sedimentation rate (ESR) was 44 mm/hr. Human immunodeficiency antibody screen was negative. Anti‐nuclear antibodies and anti‐nuclear cytoplasmic antibody panel were negative. Computed tomography angiogram (CTA) of the lower extremities demonstrated severe stenosis of the left superficial femoral artery and severe stenosis of the right posterior tibial artery. Ankle‐brachial indices were 0.83 on the right side and 0.72 on the left, indicating mild to moderate arterial disease.
ESR and CRP are nonspecific markers of inflammation. Their elevation does not prioritize malignancy, autoimmunity, or infection. ANCA might be negative in commonly ANCA‐associated vasculitides such as eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis, and granulomatosis with polyangiitis. However, the lack of respiratory and renal involvement in addition to the negative ANCA panel make such diagnoses unlikely. CTA of the patient's legs showed significant peripheral artery disease (PAD). This is unlikely to be the cause of his presentation; PAD should not cause petechiae, and his pain is disproportionate to the severity of the vascular disease reported. The additional information leaves the differential unchanged.
A dermatologist was consulted. She described and photographed a perifollicular distribution of the lower extremity petechiae with associated perifollicular hyperkeratosis and retained curled hairs (Figure 2).
The described rash is specific for scurvy. His homelessness and alcohol intake likely made him vulnerable to ascorbic acid deficiency from lack of access to fruits and vegetables. Measurement of vitamin C level is unnecessary as the pretest probability for scurvy is very high. More relevant than a vitamin C level or skin biopsy is empiric treatment with ascorbic acid; as mentioned, patients with scurvy respond rapidly to vitamin C therapy. Given the neuropathy, he should be assessed for concomitant thiamine and/or cobalamin deficiency. His peripheral arterial disease is unlikely to be related.
His ascorbic acid level was 0.0 mg/dL (reference range, 0.22.0 mg/dL). Further history was obtained, and the patient reported exclusively eating frozen hamburgers and burritos for almost 1 year. He believed he had not had a fruit or vegetable in over 10 years. He was started on 1000 mg daily of ascorbic acid. By hospital day 2, his rash had mostly resolved and he was able to stand with some support. The patient was seen by his primary care physician 3 weeks after diagnosis, with his exercise tolerance nearly back to baseline. His rash had entirely resolved.
DISCUSSION
Unlike other mammals, humans do not have the ability to convert glucose to vitamin C and thus require an exogenous source, such as fruits and vegetables. The oft‐cited observation of scurvy in sailors during long journeys in the 18th century is a classic example of clinical disease due to vitamin C deficiency.[1] Once replete, body stores of vitamin C are usually sufficient to last over 6 months of deprivation. In some patients, symptoms of deficiency may appear within 3 months.[2] The patient in this report likely suffered years of vitamin C deficiency, resulting in the significant manifestations of scurvy reported here.
Vitamin C is a water‐soluble vitamin necessary for the biosynthesis of collagen, L‐carnitine, and neurotransmitters.[3] With deficiency, the resulting impairment in the formation of collagen affects blood vessel integrity and results in perivascular edema and erythrocyte extravasation. Clinically, this leads to hemorrhagic manifestations (eg, periosteal hemorrhage and perifollicular petechiae) and poor wound healing. Corkscrew hairs result because of vitamin C's role in disulfide bonding during hair formation. Woody edema and dyspnea are thought to be a consequence of leaky capillaries.[4]
Scurvy is still a significant cause of morbidity in at‐risk populations in the United States. Several populations have been identified as high risk for vitamin C deficiency, including the elderly, persons who live alone, alcoholics, smokers, individuals of low socioeconomic status, patients on hemodialysis, and those with psychiatric disease.[5] Specifically, the high oxidative stress associated with smoking, the history of alcohol abuse, and homelessness put this patient at an especially high risk.[6] Those with oxidative stressors have been postulated to require up to 125 mg/d of vitamin C compared to 60 to 90 mg/d of those without the same risks.[7] In a national health and nutrition survey in the United States in 2004, the prevalence of vitamin C deficiency as defined by a serum level <0.2 mg/dL was noted in 7.1% of those surveyed.[8] This study also noted a significantly higher prevalence of deficiency in smokers and individuals with low socioeconomic status.
Scurvy is a clinical diagnosis based on clinical features and dietary history. Severe manifestations of scurvy may happen quickly after the initial presentation, making early diagnosis especially important.[2] These include anemia, bone pain, ocular hemorrhage, cerebral hemorrhage, and hemopericardium.[2, 4] If needed, laboratory diagnosis can be made by demonstrating a serum ascorbic acid level <0.2 mg/dL. However, the level may be normal if the patient has had recent intake of vitamin C. In that scenario, the leukocyte vitamin C concentration may be a more accurate measure of the body stores as leukocyte levels change more slowly.[4] Biopsy of skin lesions is not necessary for the diagnosis and typically show a dilated hair follicle with keratin plugging and perifollicular hemorrhage.[9] Given the lack of adverse effects, treatment with vitamin C supplementation should begin immediately, even with low suspicion of scurvy, and response can serve as further clinical evidence and render laboratory testing unnecessary.
In this patient, the diagnosis was challenging for several reasons. The presentation was concerning for vasculitis given the dependent petechiae and elevated inflammatory markers. However, in scurvy, the petechiae are perifollicular and associated with hyperkeratosis, as opposed to the palpable purpura often described in vasculitis. Further, marked elevations in ESR and CRP have also been reported in scurvy.[10] The initial concern for vasculitis and clinician discomfort with a diagnosis based solely on a rash delayed the diagnosis. The complaint of polyneuropathy also seemed inconsistent with scurvy. Very rarely, scurvy may cause a neuropathy by hemorrhage into the nerve sheath, as seen in a case of bilateral femoral neuropathy.[11] Most likely, this patient had an underlying vitamin B deficiency explaining his polyneuropathy. Unfortunately, the patient was lost to follow‐up after his postdischarge visit with his primary physician and was not tested for other concomitant vitamin deficiencies.
Scurvy is very responsive to even small doses of vitamin C supplementation. For rapid recovery, doses ranging from 100 mg 3 times daily to 1000 mg daily of oral vitamin C are recommended for at least 1 month. Resolution of symptoms will begin within 24 hours, and complete recovery should occur by 3 months.[4] Scurvy is a classic example of how nutritional deficiencies can have a myriad of presentations and may mimic other systemic diseases. Clinicians who recall these manifestations and carefully assess patients for nutritional risks may be able to quickly identify the missing element (or elements) in a patient's diet, and initiate treatment that is often rapidly effective.
KEY TEACHING POINTS
- Vitamin C deficiency initially presents with classic dermatological findings of perifollicular petechiae with associated hyperkeratosis and corkscrew hairs.
- Scurvy is a clinical diagnosis based on history and presentation. Vitamin C serum level may not accurately reflect body stores, and a leukocyte vitamin C level may be obtained. The diagnosis may also be confirmed with observed response to vitamin C supplementation.
- Scurvy should be suspected in high‐risk populations, especially the marginally housed, the elderly, alcoholics, and smokers.
- Clinicians should screen patients with scurvy for other nutritional deficiencies including thiamine, folate, B12, and vitamin D levels.
Disclosures: Nothing to report.
- . The discovery of vitamin C. Ann Nutr Metab. 2012;61(3):259–264.
- , , , , . Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24(4):432–443.
- . New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314(14):892–902.
- , . Adult scurvy. J Am Acad Dermatol. 1999;41(6):895–910.
- , . Be vigilant for scurvy in high‐risk groups. Practitioner. 2012;256(1755):23–5, 3.
- . Estimating ascorbic acid requirements for cigarette smokers. Ann N Y Acad Sci. 1993;686:335–346.
- , , . Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health. 2004;94(5):870–875.
- , , , . Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90(5):1252–1263.
- , , . Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol. 2003;44(1):48–51.
- , . Rheumatic manifestations of scurvy: a report of three recent cases in a major urban center and a review. Semin Arthritis Rheum. 2011;41(2):286–290.
- . Femoral neuropathy in scurvy. N Engl J Med. 1969;281(23):1292–1293.
- . The discovery of vitamin C. Ann Nutr Metab. 2012;61(3):259–264.
- , , , , . Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24(4):432–443.
- . New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314(14):892–902.
- , . Adult scurvy. J Am Acad Dermatol. 1999;41(6):895–910.
- , . Be vigilant for scurvy in high‐risk groups. Practitioner. 2012;256(1755):23–5, 3.
- . Estimating ascorbic acid requirements for cigarette smokers. Ann N Y Acad Sci. 1993;686:335–346.
- , , . Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health. 2004;94(5):870–875.
- , , , . Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90(5):1252–1263.
- , , . Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol. 2003;44(1):48–51.
- , . Rheumatic manifestations of scurvy: a report of three recent cases in a major urban center and a review. Semin Arthritis Rheum. 2011;41(2):286–290.
- . Femoral neuropathy in scurvy. N Engl J Med. 1969;281(23):1292–1293.
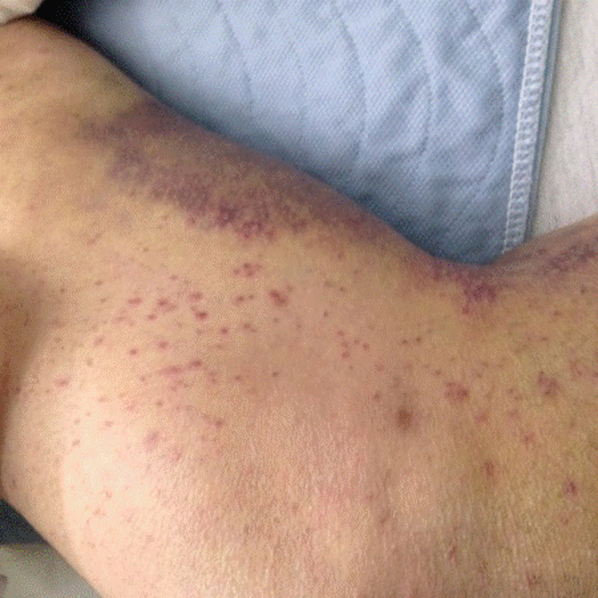
How Should a Hospitalized Patient with Newly Diagnosed Cirrhosis Be Evaluated and Managed?
The Case
A 50-year-old man with no known medical history presents with two months of increasing abdominal distension. Exam is notable for scleral icterus, telangiectasias on the upper chest, abdominal distention with a positive fluid wave, and bilateral pitting lower-extremity edema. An abdominal ultrasound shows large ascites and a nodular liver consistent with cirrhosis. How should this patient with newly diagnosed cirrhosis be evaluated and managed?
Background
Cirrhosis is a leading cause of death among people ages 25–64 and associated with a mortality rate of 11.5 per 100,000 people.1 In 2010, 101,000 people were discharged from the hospital with chronic liver disease and cirrhosis as the first-listed diagnosis.2 Given the myriad etiologies and the asymptomatic nature of many of these conditions, hospitalists frequently encounter patients presenting with advanced disease.
Evaluation

The gold standard for diagnosis is liver biopsy, although this is now usually reserved for atypical cases or where the etiology of cirrhosis is unclear. Alcohol and viral hepatitis (B and C) are the most common causes of chronic liver disease, with nonalcoholic steatohepatitis (NASH) increasing in prevalence. Other less common etiologies and characteristic test findings are listed in Figure 2.
Recently, the Centers for Disease Control and Prevention (CDC) recommended that adults born between 1945 and 1965 receive one-time testing for hepatitis C virus (HCV) infection, regardless of other risk factors, given the higher prevalence in this birth cohort and the introduction of newer oral treatments that achieve sustained virologic response.3
Management
The three classic complications of cirrhosis that will typically prompt inpatient admission are volume overload/ascites, gastrointestinal variceal bleeding, and hepatic encephalopathy.
Volume overload/ascites. Ascites is the most common major complication of cirrhosis, with roughly 50% of patients with asymptomatic cirrhosis developing ascites within 10 years.4 Ascites development portends a poor prognosis, with a mortality of 15% within one year and 44% within five years of diagnosis.4 Patients presenting with new-onset ascites should have a diagnostic paracentesis performed to determine the etiology and evaluate for infection.
Ascitic fluid should be sent for an albumin level and a cell count with differential. A serum-ascites albumin gradient (SAAG) of greater than or equal to 1.1 g/dL is consistent with portal hypertension and cirrhosis, while values less than 1.1 g/dL suggest a non-cirrhotic cause, such as infection or malignancy. Due to the high prevalence of spontaneous bacterial peritonitis (SBP) in hospitalized patients, fluid should also be immediately inoculated in aerobic and anaerobic culture bottles at the bedside, as this has been shown to improve the yield compared to inoculation of culture bottles in the laboratory. Other testing (such as cytology for the evaluation of malignancy) should only be performed if there is significant concern for a particular disease since the vast majority of cases are secondary to uncomplicated cirrhosis.4
In patients with a large amount of ascites and related symptoms (eg, abdominal pain, shortness of breath), therapeutic paracentesis should be performed. Although there is controversy over the need for routine albumin administration, guidelines currently recommend the infusion of 6–8 g of albumin per liter of ascites removed for paracentesis volumes of greater than 4–5 liters.4
No data support the routine administration of fresh frozen plasma (FFP) or platelets prior to paracentesis. Although significant complications of paracentesis (including bowel perforation and hemorrhage) may occur, these are exceedingly rare. Ultrasonography can be used to decrease risks and identify suitable pockets of fluid to tap, even when fluid is not obvious on physical exam alone.5
For patients with significant edema or ascites that is due to portal hypertension (SAAG >1.1 g/dL), the first-line therapy is sodium restriction to less than 2,000 mg/day. Consulting a nutritionist may be beneficial for patient education.
For patients with significant natriuresis (>78 mmol daily urine sodium excretion), dietary restriction alone can manage fluid retention. Most patients (85%–90%), however, require diuretics to increase sodium output. Single-agent spironolactone is more efficacious than single-agent furosemide, but diuresis is improved when both agents are used.4 A dosing regimen of once-daily 40 mg furosemide and 100 mg spironolactone is the recommended starting regimen to promote diuresis while maintaining normokalemia. Due to the long half-life of spironolactone, the dose can be increased every three to five days if needed for diuresis.4
Gastroesophageal variceal bleeding. Approximately 50% of patients with cirrhosis have gastroesophageal varices as a consequence of portal hypertension, with prevalence increasing in those with more severe disease.6 As many patients with cirrhosis have advanced disease at the time of diagnosis, it is recommended that patients be referred for endoscopic screening when diagnosed.6 Nonselective beta-blockers decrease the risk of bleeding in patients with known varices but should not be initiated empirically in all patients with cirrhosis given significant side effects, including worsening of ascites.
There is increasing evidence that there is a “window” period for beta-blocker use in cirrhosis with the window opening after the diagnosis of varices and the window closing at advanced stages of disease (marked by an episode of spontaneous bacterial peritonitis, refractory ascites, or hepatorenal syndrome, for example).7
Hepatic encephalopathy. Hepatic encephalopathy (HE) is another complication of portal hypertension and is seen in 10%–14% of patients at the time of cirrhosis diagnosis.8 Overt HE is estimated to occur in 30%–40% of patients with cirrhosis at some point during their disease course, and more subtle forms (minimal or covert HE) are seen in up to 80%.8 HE can cause numerous neurologic and psychiatric issues including personality changes, poor memory, sleep-wake disturbances, and alterations in consciousness.
In patients with an episode of encephalopathy, precipitating factors should be evaluated. Typical precipitants include infections, bleeding, electrolyte disorders, and constipation. Ammonia levels are frequently drawn as part of the evaluation of hepatic encephalopathy, but elevated levels do not significantly change diagnostic probabilities or add prognostic information.8 A low ammonia level, on the other hand, may be useful in lowering the probability of hepatic encephalopathy in a patient with altered mental status of unknown etiology.8
Routine primary prophylaxis of HE in all patients with cirrhosis is not currently recommended. Treatment is only recommended in patients with overt HE, with secondary prophylaxis administered following an episode due to the high risk for recurrence.
Other Issues
VTE prophylaxis. Although patients with cirrhosis are often presumed to be “auto-anticoagulated” due to an elevated international normalized ratio (INR), they experience thrombotic complications during hospitalization at the same rate or higher than patients with other chronic illnesses.9 Unfortunately, studies examining venous thromboembolism (VTE) prophylaxis in hospitalized patients have generally excluded cirrhotics. Therefore, risks/benefits of prophylaxis need to be considered on an individual basis, taking into account the presence of varices (if known), platelet count, and other VTE risk factors.
Drugs to avoid. As detailed above, nonselective beta-blockers should be avoided when outside the “window” period of benefit. Patients with cirrhosis should be counseled to avoid nonsteroidal anti-inflammatory drugs (NSAIDs) due to an increased risk of bleeding and renal dysfunction. ACE inhibitors (ACE-Is) and angiotensin-receptor blockers (ARBs) can also precipitate renal dysfunction and should generally be avoided unless strongly indicated for another diagnosis.
There is conflicting evidence with regard to whether the use of proton-pump inhibitors (PPIs) in cirrhotics increases the risk of SBP.10,11 Nevertheless, it is prudent to reevaluate the need for PPIs in patients with cirrhosis to determine where a true indication exists.
Post-hospitalization care. Patients with a new diagnosis of cirrhosis require screening for esophageal varices and hepatocellular carcinoma (HCC), with frequency of subsequent testing based on initial results. They should also be immunized against hepatitis A (HAV) and hepatitis B (HBV), if not already immune. Specific treatments are available for many causes of cirrhosis, including new antiviral agents against hepatitis C (HCV), and liver transplantation is an option for select patients. Given the complexity of subsequent diagnostic and treatment options, patients with new cirrhosis should be referred to a gastroenterologist or hepatologist, if possible.
Back to the Case
The patient is hospitalized, and a large-volume paracentesis is performed. Four liters are removed without the administration of albumin. Ascitic fluid analysis reveals a SAAG of greater than 1.1 g/dL and a polymorphonuclear cell count of 50 cell/mm3, suggesting ascites due to portal hypertension and ruling out infection. Nutrition is consulted and educates the patient on a restricted-sodium diet. Furosemide is started at 40 mg daily; spironolactone is started at 100 mg daily. Initial workup and serologies demonstrate active HCV infection (HCV RNA positive), with immunity to HBV due to vaccination. HAV vaccination is administered given lack of seropositivity. The patient is screened for alcohol and found not to drink alcohol. By the time of discharge, the patient is experiencing daily 0.5 kg weight loss due to diuretics and has stable renal function. The patient is referred to outpatient gastroenterology for gastroesophageal variceal screening and consideration of HCV treatment and/or liver transplantation.
Bottom Line
Workup and management of cirrhosis should focus on revealing the underlying etiology, managing complications, and discharging patients with a comprehensive follow-up plan. TH
Dr. Sehgal and Dr. Hanson are hospitalists in the division of hospital medicine at the University of Texas Health Science Center at San Antonio and the South Texas Veterans Health Care System.
References
- Heron M. Deaths: leading causes for 2012. Natl Vital Stat Rep. 2015;64(10):1-93.
- Chronic liver disease and cirrhosis. Centers for Disease Control and Prevention website. Accessed March 17, 2016.
- Smith BD, Morgan RL, Beckett GA, Falck-Ytter Y, Holtzman D, Ward JW. Hepatitis C virus testing of persons born during 1945-1965: recommendations from the Centers for Disease Control and Prevention. Ann Intern Med. 2012;157(11):817-822. doi:10.7326/0003-4819-157-9-201211060-00529.
- Runyon BA, AASLD. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology. 2013;57(4):1651-1653. doi:10.1002/hep.26359.
- Udell JA, Wang CS, Tinmouth J, et al. Does this patient with liver disease have cirrhosis? JAMA. 2012;307(8):832-842. doi:10.1001/jama.2012.186.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W, Practice Guidelines Committee of the American Association for the Study of Liver Diseases, Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46(3):922-938. doi:10.1002/hep.21907.
- Mandorfer M, Bota S, Schwabl P, et al. Nonselective β blockers increase risk for hepatorenal syndrome and death in patients with cirrhosis and spontaneous bacterial peritonitis. Gastroenterology. 2014;146(7):1680-90.e1. doi:10.1053/j.gastro.2014.03.005.
- Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology. 2014;60(2):715-735. doi:10.1002/hep.27210.
- Khoury T, Ayman AR, Cohen J, Daher S, Shmuel C, Mizrahi M. The complex role of anticoagulation in cirrhosis: an updated review of where we are and where we are going. Digestion. 2016;93(2):149-159. doi:10.1159/000442877.
- Terg R, Casciato P, Garbe C, et al. Proton pump inhibitor therapy does not increase the incidence of spontaneous bacterial peritonitis in cirrhosis: a multicenter prospective study. J Hepatol. 2015;62(5):1056-1060. doi:10.1016/j.jhep.2014.11.036.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol. 2013;28(2):235-242. doi:10.1111/jgh.12065.
Key Points
- Cirrhosis has many etiologies, and new diagnoses require further investigation as to the underlying etiology.
- Initial management should focus on evaluation and treatment of complications, including ascites, esophageal varices, and hepatic encephalopathy.
- A diagnostic paracentesis, salt restriction, and a nutrition consult are the initial therapies for ascites although most patients will also require diuretics to increase sodium excretion.
- Once stabilized, the cirrhotic patient will require specialty care for possible liver biopsy (if etiology remains unclear), treatment (eg, HCV antivirals), and/or referral for liver transplantation.
The Case
A 50-year-old man with no known medical history presents with two months of increasing abdominal distension. Exam is notable for scleral icterus, telangiectasias on the upper chest, abdominal distention with a positive fluid wave, and bilateral pitting lower-extremity edema. An abdominal ultrasound shows large ascites and a nodular liver consistent with cirrhosis. How should this patient with newly diagnosed cirrhosis be evaluated and managed?
Background
Cirrhosis is a leading cause of death among people ages 25–64 and associated with a mortality rate of 11.5 per 100,000 people.1 In 2010, 101,000 people were discharged from the hospital with chronic liver disease and cirrhosis as the first-listed diagnosis.2 Given the myriad etiologies and the asymptomatic nature of many of these conditions, hospitalists frequently encounter patients presenting with advanced disease.
Evaluation
The gold standard for diagnosis is liver biopsy, although this is now usually reserved for atypical cases or where the etiology of cirrhosis is unclear. Alcohol and viral hepatitis (B and C) are the most common causes of chronic liver disease, with nonalcoholic steatohepatitis (NASH) increasing in prevalence. Other less common etiologies and characteristic test findings are listed in Figure 2.
Recently, the Centers for Disease Control and Prevention (CDC) recommended that adults born between 1945 and 1965 receive one-time testing for hepatitis C virus (HCV) infection, regardless of other risk factors, given the higher prevalence in this birth cohort and the introduction of newer oral treatments that achieve sustained virologic response.3
Management
The three classic complications of cirrhosis that will typically prompt inpatient admission are volume overload/ascites, gastrointestinal variceal bleeding, and hepatic encephalopathy.
Volume overload/ascites. Ascites is the most common major complication of cirrhosis, with roughly 50% of patients with asymptomatic cirrhosis developing ascites within 10 years.4 Ascites development portends a poor prognosis, with a mortality of 15% within one year and 44% within five years of diagnosis.4 Patients presenting with new-onset ascites should have a diagnostic paracentesis performed to determine the etiology and evaluate for infection.
Ascitic fluid should be sent for an albumin level and a cell count with differential. A serum-ascites albumin gradient (SAAG) of greater than or equal to 1.1 g/dL is consistent with portal hypertension and cirrhosis, while values less than 1.1 g/dL suggest a non-cirrhotic cause, such as infection or malignancy. Due to the high prevalence of spontaneous bacterial peritonitis (SBP) in hospitalized patients, fluid should also be immediately inoculated in aerobic and anaerobic culture bottles at the bedside, as this has been shown to improve the yield compared to inoculation of culture bottles in the laboratory. Other testing (such as cytology for the evaluation of malignancy) should only be performed if there is significant concern for a particular disease since the vast majority of cases are secondary to uncomplicated cirrhosis.4
In patients with a large amount of ascites and related symptoms (eg, abdominal pain, shortness of breath), therapeutic paracentesis should be performed. Although there is controversy over the need for routine albumin administration, guidelines currently recommend the infusion of 6–8 g of albumin per liter of ascites removed for paracentesis volumes of greater than 4–5 liters.4
No data support the routine administration of fresh frozen plasma (FFP) or platelets prior to paracentesis. Although significant complications of paracentesis (including bowel perforation and hemorrhage) may occur, these are exceedingly rare. Ultrasonography can be used to decrease risks and identify suitable pockets of fluid to tap, even when fluid is not obvious on physical exam alone.5
For patients with significant edema or ascites that is due to portal hypertension (SAAG >1.1 g/dL), the first-line therapy is sodium restriction to less than 2,000 mg/day. Consulting a nutritionist may be beneficial for patient education.
For patients with significant natriuresis (>78 mmol daily urine sodium excretion), dietary restriction alone can manage fluid retention. Most patients (85%–90%), however, require diuretics to increase sodium output. Single-agent spironolactone is more efficacious than single-agent furosemide, but diuresis is improved when both agents are used.4 A dosing regimen of once-daily 40 mg furosemide and 100 mg spironolactone is the recommended starting regimen to promote diuresis while maintaining normokalemia. Due to the long half-life of spironolactone, the dose can be increased every three to five days if needed for diuresis.4
Gastroesophageal variceal bleeding. Approximately 50% of patients with cirrhosis have gastroesophageal varices as a consequence of portal hypertension, with prevalence increasing in those with more severe disease.6 As many patients with cirrhosis have advanced disease at the time of diagnosis, it is recommended that patients be referred for endoscopic screening when diagnosed.6 Nonselective beta-blockers decrease the risk of bleeding in patients with known varices but should not be initiated empirically in all patients with cirrhosis given significant side effects, including worsening of ascites.
There is increasing evidence that there is a “window” period for beta-blocker use in cirrhosis with the window opening after the diagnosis of varices and the window closing at advanced stages of disease (marked by an episode of spontaneous bacterial peritonitis, refractory ascites, or hepatorenal syndrome, for example).7
Hepatic encephalopathy. Hepatic encephalopathy (HE) is another complication of portal hypertension and is seen in 10%–14% of patients at the time of cirrhosis diagnosis.8 Overt HE is estimated to occur in 30%–40% of patients with cirrhosis at some point during their disease course, and more subtle forms (minimal or covert HE) are seen in up to 80%.8 HE can cause numerous neurologic and psychiatric issues including personality changes, poor memory, sleep-wake disturbances, and alterations in consciousness.
In patients with an episode of encephalopathy, precipitating factors should be evaluated. Typical precipitants include infections, bleeding, electrolyte disorders, and constipation. Ammonia levels are frequently drawn as part of the evaluation of hepatic encephalopathy, but elevated levels do not significantly change diagnostic probabilities or add prognostic information.8 A low ammonia level, on the other hand, may be useful in lowering the probability of hepatic encephalopathy in a patient with altered mental status of unknown etiology.8
Routine primary prophylaxis of HE in all patients with cirrhosis is not currently recommended. Treatment is only recommended in patients with overt HE, with secondary prophylaxis administered following an episode due to the high risk for recurrence.
Other Issues
VTE prophylaxis. Although patients with cirrhosis are often presumed to be “auto-anticoagulated” due to an elevated international normalized ratio (INR), they experience thrombotic complications during hospitalization at the same rate or higher than patients with other chronic illnesses.9 Unfortunately, studies examining venous thromboembolism (VTE) prophylaxis in hospitalized patients have generally excluded cirrhotics. Therefore, risks/benefits of prophylaxis need to be considered on an individual basis, taking into account the presence of varices (if known), platelet count, and other VTE risk factors.
Drugs to avoid. As detailed above, nonselective beta-blockers should be avoided when outside the “window” period of benefit. Patients with cirrhosis should be counseled to avoid nonsteroidal anti-inflammatory drugs (NSAIDs) due to an increased risk of bleeding and renal dysfunction. ACE inhibitors (ACE-Is) and angiotensin-receptor blockers (ARBs) can also precipitate renal dysfunction and should generally be avoided unless strongly indicated for another diagnosis.
There is conflicting evidence with regard to whether the use of proton-pump inhibitors (PPIs) in cirrhotics increases the risk of SBP.10,11 Nevertheless, it is prudent to reevaluate the need for PPIs in patients with cirrhosis to determine where a true indication exists.
Post-hospitalization care. Patients with a new diagnosis of cirrhosis require screening for esophageal varices and hepatocellular carcinoma (HCC), with frequency of subsequent testing based on initial results. They should also be immunized against hepatitis A (HAV) and hepatitis B (HBV), if not already immune. Specific treatments are available for many causes of cirrhosis, including new antiviral agents against hepatitis C (HCV), and liver transplantation is an option for select patients. Given the complexity of subsequent diagnostic and treatment options, patients with new cirrhosis should be referred to a gastroenterologist or hepatologist, if possible.
Back to the Case
The patient is hospitalized, and a large-volume paracentesis is performed. Four liters are removed without the administration of albumin. Ascitic fluid analysis reveals a SAAG of greater than 1.1 g/dL and a polymorphonuclear cell count of 50 cell/mm3, suggesting ascites due to portal hypertension and ruling out infection. Nutrition is consulted and educates the patient on a restricted-sodium diet. Furosemide is started at 40 mg daily; spironolactone is started at 100 mg daily. Initial workup and serologies demonstrate active HCV infection (HCV RNA positive), with immunity to HBV due to vaccination. HAV vaccination is administered given lack of seropositivity. The patient is screened for alcohol and found not to drink alcohol. By the time of discharge, the patient is experiencing daily 0.5 kg weight loss due to diuretics and has stable renal function. The patient is referred to outpatient gastroenterology for gastroesophageal variceal screening and consideration of HCV treatment and/or liver transplantation.
Bottom Line
Workup and management of cirrhosis should focus on revealing the underlying etiology, managing complications, and discharging patients with a comprehensive follow-up plan. TH
Dr. Sehgal and Dr. Hanson are hospitalists in the division of hospital medicine at the University of Texas Health Science Center at San Antonio and the South Texas Veterans Health Care System.
References
- Heron M. Deaths: leading causes for 2012. Natl Vital Stat Rep. 2015;64(10):1-93.
- Chronic liver disease and cirrhosis. Centers for Disease Control and Prevention website. Accessed March 17, 2016.
- Smith BD, Morgan RL, Beckett GA, Falck-Ytter Y, Holtzman D, Ward JW. Hepatitis C virus testing of persons born during 1945-1965: recommendations from the Centers for Disease Control and Prevention. Ann Intern Med. 2012;157(11):817-822. doi:10.7326/0003-4819-157-9-201211060-00529.
- Runyon BA, AASLD. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology. 2013;57(4):1651-1653. doi:10.1002/hep.26359.
- Udell JA, Wang CS, Tinmouth J, et al. Does this patient with liver disease have cirrhosis? JAMA. 2012;307(8):832-842. doi:10.1001/jama.2012.186.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W, Practice Guidelines Committee of the American Association for the Study of Liver Diseases, Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46(3):922-938. doi:10.1002/hep.21907.
- Mandorfer M, Bota S, Schwabl P, et al. Nonselective β blockers increase risk for hepatorenal syndrome and death in patients with cirrhosis and spontaneous bacterial peritonitis. Gastroenterology. 2014;146(7):1680-90.e1. doi:10.1053/j.gastro.2014.03.005.
- Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology. 2014;60(2):715-735. doi:10.1002/hep.27210.
- Khoury T, Ayman AR, Cohen J, Daher S, Shmuel C, Mizrahi M. The complex role of anticoagulation in cirrhosis: an updated review of where we are and where we are going. Digestion. 2016;93(2):149-159. doi:10.1159/000442877.
- Terg R, Casciato P, Garbe C, et al. Proton pump inhibitor therapy does not increase the incidence of spontaneous bacterial peritonitis in cirrhosis: a multicenter prospective study. J Hepatol. 2015;62(5):1056-1060. doi:10.1016/j.jhep.2014.11.036.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol. 2013;28(2):235-242. doi:10.1111/jgh.12065.
Key Points
- Cirrhosis has many etiologies, and new diagnoses require further investigation as to the underlying etiology.
- Initial management should focus on evaluation and treatment of complications, including ascites, esophageal varices, and hepatic encephalopathy.
- A diagnostic paracentesis, salt restriction, and a nutrition consult are the initial therapies for ascites although most patients will also require diuretics to increase sodium excretion.
- Once stabilized, the cirrhotic patient will require specialty care for possible liver biopsy (if etiology remains unclear), treatment (eg, HCV antivirals), and/or referral for liver transplantation.
The Case
A 50-year-old man with no known medical history presents with two months of increasing abdominal distension. Exam is notable for scleral icterus, telangiectasias on the upper chest, abdominal distention with a positive fluid wave, and bilateral pitting lower-extremity edema. An abdominal ultrasound shows large ascites and a nodular liver consistent with cirrhosis. How should this patient with newly diagnosed cirrhosis be evaluated and managed?
Background
Cirrhosis is a leading cause of death among people ages 25–64 and associated with a mortality rate of 11.5 per 100,000 people.1 In 2010, 101,000 people were discharged from the hospital with chronic liver disease and cirrhosis as the first-listed diagnosis.2 Given the myriad etiologies and the asymptomatic nature of many of these conditions, hospitalists frequently encounter patients presenting with advanced disease.
Evaluation
The gold standard for diagnosis is liver biopsy, although this is now usually reserved for atypical cases or where the etiology of cirrhosis is unclear. Alcohol and viral hepatitis (B and C) are the most common causes of chronic liver disease, with nonalcoholic steatohepatitis (NASH) increasing in prevalence. Other less common etiologies and characteristic test findings are listed in Figure 2.
Recently, the Centers for Disease Control and Prevention (CDC) recommended that adults born between 1945 and 1965 receive one-time testing for hepatitis C virus (HCV) infection, regardless of other risk factors, given the higher prevalence in this birth cohort and the introduction of newer oral treatments that achieve sustained virologic response.3
Management
The three classic complications of cirrhosis that will typically prompt inpatient admission are volume overload/ascites, gastrointestinal variceal bleeding, and hepatic encephalopathy.
Volume overload/ascites. Ascites is the most common major complication of cirrhosis, with roughly 50% of patients with asymptomatic cirrhosis developing ascites within 10 years.4 Ascites development portends a poor prognosis, with a mortality of 15% within one year and 44% within five years of diagnosis.4 Patients presenting with new-onset ascites should have a diagnostic paracentesis performed to determine the etiology and evaluate for infection.
Ascitic fluid should be sent for an albumin level and a cell count with differential. A serum-ascites albumin gradient (SAAG) of greater than or equal to 1.1 g/dL is consistent with portal hypertension and cirrhosis, while values less than 1.1 g/dL suggest a non-cirrhotic cause, such as infection or malignancy. Due to the high prevalence of spontaneous bacterial peritonitis (SBP) in hospitalized patients, fluid should also be immediately inoculated in aerobic and anaerobic culture bottles at the bedside, as this has been shown to improve the yield compared to inoculation of culture bottles in the laboratory. Other testing (such as cytology for the evaluation of malignancy) should only be performed if there is significant concern for a particular disease since the vast majority of cases are secondary to uncomplicated cirrhosis.4
In patients with a large amount of ascites and related symptoms (eg, abdominal pain, shortness of breath), therapeutic paracentesis should be performed. Although there is controversy over the need for routine albumin administration, guidelines currently recommend the infusion of 6–8 g of albumin per liter of ascites removed for paracentesis volumes of greater than 4–5 liters.4
No data support the routine administration of fresh frozen plasma (FFP) or platelets prior to paracentesis. Although significant complications of paracentesis (including bowel perforation and hemorrhage) may occur, these are exceedingly rare. Ultrasonography can be used to decrease risks and identify suitable pockets of fluid to tap, even when fluid is not obvious on physical exam alone.5
For patients with significant edema or ascites that is due to portal hypertension (SAAG >1.1 g/dL), the first-line therapy is sodium restriction to less than 2,000 mg/day. Consulting a nutritionist may be beneficial for patient education.
For patients with significant natriuresis (>78 mmol daily urine sodium excretion), dietary restriction alone can manage fluid retention. Most patients (85%–90%), however, require diuretics to increase sodium output. Single-agent spironolactone is more efficacious than single-agent furosemide, but diuresis is improved when both agents are used.4 A dosing regimen of once-daily 40 mg furosemide and 100 mg spironolactone is the recommended starting regimen to promote diuresis while maintaining normokalemia. Due to the long half-life of spironolactone, the dose can be increased every three to five days if needed for diuresis.4
Gastroesophageal variceal bleeding. Approximately 50% of patients with cirrhosis have gastroesophageal varices as a consequence of portal hypertension, with prevalence increasing in those with more severe disease.6 As many patients with cirrhosis have advanced disease at the time of diagnosis, it is recommended that patients be referred for endoscopic screening when diagnosed.6 Nonselective beta-blockers decrease the risk of bleeding in patients with known varices but should not be initiated empirically in all patients with cirrhosis given significant side effects, including worsening of ascites.
There is increasing evidence that there is a “window” period for beta-blocker use in cirrhosis with the window opening after the diagnosis of varices and the window closing at advanced stages of disease (marked by an episode of spontaneous bacterial peritonitis, refractory ascites, or hepatorenal syndrome, for example).7
Hepatic encephalopathy. Hepatic encephalopathy (HE) is another complication of portal hypertension and is seen in 10%–14% of patients at the time of cirrhosis diagnosis.8 Overt HE is estimated to occur in 30%–40% of patients with cirrhosis at some point during their disease course, and more subtle forms (minimal or covert HE) are seen in up to 80%.8 HE can cause numerous neurologic and psychiatric issues including personality changes, poor memory, sleep-wake disturbances, and alterations in consciousness.
In patients with an episode of encephalopathy, precipitating factors should be evaluated. Typical precipitants include infections, bleeding, electrolyte disorders, and constipation. Ammonia levels are frequently drawn as part of the evaluation of hepatic encephalopathy, but elevated levels do not significantly change diagnostic probabilities or add prognostic information.8 A low ammonia level, on the other hand, may be useful in lowering the probability of hepatic encephalopathy in a patient with altered mental status of unknown etiology.8
Routine primary prophylaxis of HE in all patients with cirrhosis is not currently recommended. Treatment is only recommended in patients with overt HE, with secondary prophylaxis administered following an episode due to the high risk for recurrence.
Other Issues
VTE prophylaxis. Although patients with cirrhosis are often presumed to be “auto-anticoagulated” due to an elevated international normalized ratio (INR), they experience thrombotic complications during hospitalization at the same rate or higher than patients with other chronic illnesses.9 Unfortunately, studies examining venous thromboembolism (VTE) prophylaxis in hospitalized patients have generally excluded cirrhotics. Therefore, risks/benefits of prophylaxis need to be considered on an individual basis, taking into account the presence of varices (if known), platelet count, and other VTE risk factors.
Drugs to avoid. As detailed above, nonselective beta-blockers should be avoided when outside the “window” period of benefit. Patients with cirrhosis should be counseled to avoid nonsteroidal anti-inflammatory drugs (NSAIDs) due to an increased risk of bleeding and renal dysfunction. ACE inhibitors (ACE-Is) and angiotensin-receptor blockers (ARBs) can also precipitate renal dysfunction and should generally be avoided unless strongly indicated for another diagnosis.
There is conflicting evidence with regard to whether the use of proton-pump inhibitors (PPIs) in cirrhotics increases the risk of SBP.10,11 Nevertheless, it is prudent to reevaluate the need for PPIs in patients with cirrhosis to determine where a true indication exists.
Post-hospitalization care. Patients with a new diagnosis of cirrhosis require screening for esophageal varices and hepatocellular carcinoma (HCC), with frequency of subsequent testing based on initial results. They should also be immunized against hepatitis A (HAV) and hepatitis B (HBV), if not already immune. Specific treatments are available for many causes of cirrhosis, including new antiviral agents against hepatitis C (HCV), and liver transplantation is an option for select patients. Given the complexity of subsequent diagnostic and treatment options, patients with new cirrhosis should be referred to a gastroenterologist or hepatologist, if possible.
Back to the Case
The patient is hospitalized, and a large-volume paracentesis is performed. Four liters are removed without the administration of albumin. Ascitic fluid analysis reveals a SAAG of greater than 1.1 g/dL and a polymorphonuclear cell count of 50 cell/mm3, suggesting ascites due to portal hypertension and ruling out infection. Nutrition is consulted and educates the patient on a restricted-sodium diet. Furosemide is started at 40 mg daily; spironolactone is started at 100 mg daily. Initial workup and serologies demonstrate active HCV infection (HCV RNA positive), with immunity to HBV due to vaccination. HAV vaccination is administered given lack of seropositivity. The patient is screened for alcohol and found not to drink alcohol. By the time of discharge, the patient is experiencing daily 0.5 kg weight loss due to diuretics and has stable renal function. The patient is referred to outpatient gastroenterology for gastroesophageal variceal screening and consideration of HCV treatment and/or liver transplantation.
Bottom Line
Workup and management of cirrhosis should focus on revealing the underlying etiology, managing complications, and discharging patients with a comprehensive follow-up plan. TH
Dr. Sehgal and Dr. Hanson are hospitalists in the division of hospital medicine at the University of Texas Health Science Center at San Antonio and the South Texas Veterans Health Care System.
References
- Heron M. Deaths: leading causes for 2012. Natl Vital Stat Rep. 2015;64(10):1-93.
- Chronic liver disease and cirrhosis. Centers for Disease Control and Prevention website. Accessed March 17, 2016.
- Smith BD, Morgan RL, Beckett GA, Falck-Ytter Y, Holtzman D, Ward JW. Hepatitis C virus testing of persons born during 1945-1965: recommendations from the Centers for Disease Control and Prevention. Ann Intern Med. 2012;157(11):817-822. doi:10.7326/0003-4819-157-9-201211060-00529.
- Runyon BA, AASLD. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology. 2013;57(4):1651-1653. doi:10.1002/hep.26359.
- Udell JA, Wang CS, Tinmouth J, et al. Does this patient with liver disease have cirrhosis? JAMA. 2012;307(8):832-842. doi:10.1001/jama.2012.186.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W, Practice Guidelines Committee of the American Association for the Study of Liver Diseases, Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46(3):922-938. doi:10.1002/hep.21907.
- Mandorfer M, Bota S, Schwabl P, et al. Nonselective β blockers increase risk for hepatorenal syndrome and death in patients with cirrhosis and spontaneous bacterial peritonitis. Gastroenterology. 2014;146(7):1680-90.e1. doi:10.1053/j.gastro.2014.03.005.
- Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology. 2014;60(2):715-735. doi:10.1002/hep.27210.
- Khoury T, Ayman AR, Cohen J, Daher S, Shmuel C, Mizrahi M. The complex role of anticoagulation in cirrhosis: an updated review of where we are and where we are going. Digestion. 2016;93(2):149-159. doi:10.1159/000442877.
- Terg R, Casciato P, Garbe C, et al. Proton pump inhibitor therapy does not increase the incidence of spontaneous bacterial peritonitis in cirrhosis: a multicenter prospective study. J Hepatol. 2015;62(5):1056-1060. doi:10.1016/j.jhep.2014.11.036.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol. 2013;28(2):235-242. doi:10.1111/jgh.12065.
Key Points
- Cirrhosis has many etiologies, and new diagnoses require further investigation as to the underlying etiology.
- Initial management should focus on evaluation and treatment of complications, including ascites, esophageal varices, and hepatic encephalopathy.
- A diagnostic paracentesis, salt restriction, and a nutrition consult are the initial therapies for ascites although most patients will also require diuretics to increase sodium excretion.
- Once stabilized, the cirrhotic patient will require specialty care for possible liver biopsy (if etiology remains unclear), treatment (eg, HCV antivirals), and/or referral for liver transplantation.
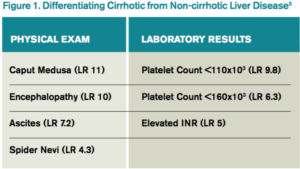
Atrial Fibrillation Linked with Greater Alcohol Access
NEW YORK - Greater access to alcohol is linked with more atrial fibrillation but less myocardial infarction and congestive heart failure, researchers report.
Dr. Gregory M. Marcus, from the Division of Cardiology at the University of California, San Francisco, and colleagues conducted an observational cohort study of differences in health outcomes based on alcohol sales laws by county in Texas.
All patients were residents of Texas, 21 years old or older, and were admitted to hospitals in Texas between 2005 and 2010. More than 1 million patients were included in the analysis.
Of the counties, 47 were wet (no restrictions on the sale of alcohol) and 29 were dry (prohibition of alcohol sales). Seven of them changed from dry to wet during the study period.
The main cardiovascular outcomes were atrial fibrillation, acute myocardial infarction, and congestive heart failure.
After multivariable adjustment, wet county residents had a greater prevalence (odds ratio 1.05, p=0.007) and incidence (HR 1.07, p=0.014) of atrial fibrillation.
Prevalence of myocardial infarction was lower (OR 0.83, p<0.001), as was its incidence (HR 0.91, p=0.019). Prevalence of congestive heart failure was also lower (OR 0.87, p<0.001).
In the seven dry counties that changed their status to wet, the post-conversion interval (from dry to wet county status) was associated with greater odds of hospitalization for atrial fibrillation (OR 1.07, p=0.001) and congestive heart failure (OR 1.07, p<0.001).
The researchers found no difference in acute myocardial infarction (OR 0.99, p=0.746).
"Cardiovascular disease is the most common cause of death worldwide, and alcohol is the most widely consumed drug in the United States," said Dr. Rory Brett Weiner, a cardiologist at Massachusetts General Hospital in Boston.
He said that studying the impact of alcohol intake on incident cardiovascular disease is important for its public health implications.
"Differences in laws affecting access to alcohol are associated with changes in health outcomes, both harmful and protective," said Dr. Marcus, "but the study's findings shouldn't be used to change any specific legislation."
Dr. Weiner agreed. "The study design minimizes confounders commonly seen in prior research that relied on self-report," he said, "but it still doesn't provide conclusive evidence with regard to the use of alcohol and incident cardiovascular disease."
Dr. Weiner said that the study did not contain information on the level of individual alcohol exposure, and therefore, the impact of the 'dose' of alcohol on cardiovascular outcomes could not be ascertained.
"Based on the question at hand -- the impact of alcohol -- it's unlikely that a randomized controlled study will ever be performed," he said, "so analyses like the current one are important."
According to Dr. Marcus, "We still don't understand the mechanisms underlying the relationship between alcohol and cardiovascular disease, and have a long way to go to achieve the sort of personalized medicine needed to figure out how to counsel an individual patient on their particular "prescribed" amount, if any, of alcohol."
The National Institute on Alcohol Abuse and Alcoholism supported this research. Dr. Marcus reported research support from Medtronic and Pfizer and equity interest in InCarda.
SOURCE: http://bit.ly/1tlx1cx
BMJ 2016
NEW YORK - Greater access to alcohol is linked with more atrial fibrillation but less myocardial infarction and congestive heart failure, researchers report.
Dr. Gregory M. Marcus, from the Division of Cardiology at the University of California, San Francisco, and colleagues conducted an observational cohort study of differences in health outcomes based on alcohol sales laws by county in Texas.
All patients were residents of Texas, 21 years old or older, and were admitted to hospitals in Texas between 2005 and 2010. More than 1 million patients were included in the analysis.
Of the counties, 47 were wet (no restrictions on the sale of alcohol) and 29 were dry (prohibition of alcohol sales). Seven of them changed from dry to wet during the study period.
The main cardiovascular outcomes were atrial fibrillation, acute myocardial infarction, and congestive heart failure.
After multivariable adjustment, wet county residents had a greater prevalence (odds ratio 1.05, p=0.007) and incidence (HR 1.07, p=0.014) of atrial fibrillation.
Prevalence of myocardial infarction was lower (OR 0.83, p<0.001), as was its incidence (HR 0.91, p=0.019). Prevalence of congestive heart failure was also lower (OR 0.87, p<0.001).
In the seven dry counties that changed their status to wet, the post-conversion interval (from dry to wet county status) was associated with greater odds of hospitalization for atrial fibrillation (OR 1.07, p=0.001) and congestive heart failure (OR 1.07, p<0.001).
The researchers found no difference in acute myocardial infarction (OR 0.99, p=0.746).
"Cardiovascular disease is the most common cause of death worldwide, and alcohol is the most widely consumed drug in the United States," said Dr. Rory Brett Weiner, a cardiologist at Massachusetts General Hospital in Boston.
He said that studying the impact of alcohol intake on incident cardiovascular disease is important for its public health implications.
"Differences in laws affecting access to alcohol are associated with changes in health outcomes, both harmful and protective," said Dr. Marcus, "but the study's findings shouldn't be used to change any specific legislation."
Dr. Weiner agreed. "The study design minimizes confounders commonly seen in prior research that relied on self-report," he said, "but it still doesn't provide conclusive evidence with regard to the use of alcohol and incident cardiovascular disease."
Dr. Weiner said that the study did not contain information on the level of individual alcohol exposure, and therefore, the impact of the 'dose' of alcohol on cardiovascular outcomes could not be ascertained.
"Based on the question at hand -- the impact of alcohol -- it's unlikely that a randomized controlled study will ever be performed," he said, "so analyses like the current one are important."
According to Dr. Marcus, "We still don't understand the mechanisms underlying the relationship between alcohol and cardiovascular disease, and have a long way to go to achieve the sort of personalized medicine needed to figure out how to counsel an individual patient on their particular "prescribed" amount, if any, of alcohol."
The National Institute on Alcohol Abuse and Alcoholism supported this research. Dr. Marcus reported research support from Medtronic and Pfizer and equity interest in InCarda.
SOURCE: http://bit.ly/1tlx1cx
BMJ 2016
NEW YORK - Greater access to alcohol is linked with more atrial fibrillation but less myocardial infarction and congestive heart failure, researchers report.
Dr. Gregory M. Marcus, from the Division of Cardiology at the University of California, San Francisco, and colleagues conducted an observational cohort study of differences in health outcomes based on alcohol sales laws by county in Texas.
All patients were residents of Texas, 21 years old or older, and were admitted to hospitals in Texas between 2005 and 2010. More than 1 million patients were included in the analysis.
Of the counties, 47 were wet (no restrictions on the sale of alcohol) and 29 were dry (prohibition of alcohol sales). Seven of them changed from dry to wet during the study period.
The main cardiovascular outcomes were atrial fibrillation, acute myocardial infarction, and congestive heart failure.
After multivariable adjustment, wet county residents had a greater prevalence (odds ratio 1.05, p=0.007) and incidence (HR 1.07, p=0.014) of atrial fibrillation.
Prevalence of myocardial infarction was lower (OR 0.83, p<0.001), as was its incidence (HR 0.91, p=0.019). Prevalence of congestive heart failure was also lower (OR 0.87, p<0.001).
In the seven dry counties that changed their status to wet, the post-conversion interval (from dry to wet county status) was associated with greater odds of hospitalization for atrial fibrillation (OR 1.07, p=0.001) and congestive heart failure (OR 1.07, p<0.001).
The researchers found no difference in acute myocardial infarction (OR 0.99, p=0.746).
"Cardiovascular disease is the most common cause of death worldwide, and alcohol is the most widely consumed drug in the United States," said Dr. Rory Brett Weiner, a cardiologist at Massachusetts General Hospital in Boston.
He said that studying the impact of alcohol intake on incident cardiovascular disease is important for its public health implications.
"Differences in laws affecting access to alcohol are associated with changes in health outcomes, both harmful and protective," said Dr. Marcus, "but the study's findings shouldn't be used to change any specific legislation."
Dr. Weiner agreed. "The study design minimizes confounders commonly seen in prior research that relied on self-report," he said, "but it still doesn't provide conclusive evidence with regard to the use of alcohol and incident cardiovascular disease."
Dr. Weiner said that the study did not contain information on the level of individual alcohol exposure, and therefore, the impact of the 'dose' of alcohol on cardiovascular outcomes could not be ascertained.
"Based on the question at hand -- the impact of alcohol -- it's unlikely that a randomized controlled study will ever be performed," he said, "so analyses like the current one are important."
According to Dr. Marcus, "We still don't understand the mechanisms underlying the relationship between alcohol and cardiovascular disease, and have a long way to go to achieve the sort of personalized medicine needed to figure out how to counsel an individual patient on their particular "prescribed" amount, if any, of alcohol."
The National Institute on Alcohol Abuse and Alcoholism supported this research. Dr. Marcus reported research support from Medtronic and Pfizer and equity interest in InCarda.
SOURCE: http://bit.ly/1tlx1cx
BMJ 2016
CT Scans Reliable Determinants of Blunt Trauma
NEW YORK - CT scans identify all clinically significant cervical spine injuries in intoxicated patients with blunt trauma, according to a new study.
"I don't think any of the results were particularly surprising to any of us who regularly do trauma care, but what I do think is remarkable about them is that they dispel several long-held myths about the c-spine, intoxicated patients, and the clearance process," Dr. Matthew J. Martin from Legacy Emanuel Medical Center, Portland, Oregon told Reuters Health.
"I think it again confirms that modern CT scan is highly reliable for identifying significant c-spine injuries, but also that the majority of so called 'intoxicated' patients are examinable enough to determine whether the collar can be removed (when combined with the CT scan)," he said.
Up to half of trauma patients are intoxicated, making clearance of the cervical spine a commonly encountered dilemma with both medical and medicolegal implications. Most guidelines indicate that the cervical spine should not be cleared in such patients, resulting in prolonged immobilization or additional imaging even in the face of a normal CT scan.
Dr. Martin's team examined cervical spine clearance practices for intoxicated trauma patients, examined the reliability of cervical spine CT scans for identifying clinically significant injuries (CSIs), and looked for CSIs that might have been missed by CT scans.
Among 1,429 patients who had an alcohol or drug screen performed, 44.2% were intoxicated, the researchers report in JAMA Surgery, online June 15.
Cervical spine injuries were identified in 11.3% of the sober group, 8.1% of the alcohol-intoxicated group, and 12.0% of the drug-intoxicated group.
CT scans yielded negative predictive values of 99.2% for all injuries and 99.8% for unstable injuries. There were five false-negative CT scans, including four central cord syndromes without associated fractures and one potentially unstable injury in a drug-intoxicated patient who presented with clear quadriplegia on examination.
Half of the intoxicated patients were admitted with continued cervical spine immobilization only on the basis of their intoxication. There were no missed CSIs in this group, and all patients were discharged without evidence of an injury or neurologic deficit. They underwent cervical spine immobilization for an average of 15.1 hours, about four times the average time to cervical spine clearance among sober patients (3.7 hours).
"The finding of how long we are keeping these patients in a c-collar based solely on intoxication should raise some eyebrows, and identifies an easy target for process improvement," Dr. Martin said.
"Cervical collars and immobilization are not therapeutic for the vast majority of c-spine injuries; they are really only to prevent inadvertent motion of an unstable c-spine injury," Dr. Martin said. "This is exceedingly rare in a patient who presents with no gross motor deficit, and a high quality CT scan will identify these unstable injuries very reliably. In addition, there are multiple adverse effects of prolonged immobilization, and even of getting an MRI."
"When these are factored in, I think the risk:benefit analysis falls squarely on the side of early clearance based on CT scan," he concluded.
"A key point is that this should be done by experts who are familiar with not only the global concept (the collar can be removed with a negative CT scan), but also the finer points where you could potentially cause harm, or where you should not remove the collar," Dr. Martin added. "This is where a very clear written protocol comes into play and reduces variation or errors that could cause patient harm."
"The results of this study suggest that it is unnecessary to delay cervical spine clearance until intoxicated patients are sober or until magnetic resonance imaging is performed," write Dr. Olubode A. Olufajo and Dr. Ali Salim from Brigham and Women's Hospital, Boston, in a related editorial. "However, caution must be taken in making conclusions based on these data."
"Although the authors conducted the study at an institution with high-quality CT technology and well-trained radiologists, they still recorded a false-negative CT report consistent with a misread," they note. "With the higher potential for this nature of error in lower-resourced settings, it becomes important to compare the costs and benefits of early removal of cervical collars."
They wonder, "With our knowledge that intoxicated patients form up to half of the population of trauma patients, is it really safe to risk irreversible injuries in 1% of the population to save a few hours in cervical clearance times?"
Dr. Stephen Asha from the University of New South Wales in Sydney, Australia, who has reported on various aspects of cervical spine imaging, told Reuters Health by email, "I think this study confirms what clinical experience as well as much of the more recent studies on cervical spine CT scanning tells us, which is that if there is nothing abnormal detected on a new generation, multi-slice CT, then the neck can be cleared."
"Of course there were a few missed injuries, but this needs to be put into context: no one just does a test in isolation, it is always combined with a clinical assessment, and a consideration the mechanism of injury," said Dr. Asha, who was not involved in the new work. "In this case there were five injuries not apparent on the CT scan, but all had obvious spinal cord injury on clinical examination before the CT was done, so these injuries were never going to be missed in a real clinical setting."
"MRI use should be carefully considered because the problem with MRI is that it can be over-sensitive, demonstrating abnormal signal suggesting ligamentous injury in patient who simply have a ligamentous 'strain,'" Dr. Asha explained. "The false-positive results then lead to further periods of inappropriate immobilization and testing, with the accompanying costs, inconvenience, and complications."
"In patients in whom the clinical assessment raises no concerns for injury, then a normal CT should herald the end of investigations," he said. "MRI should be reserved for those where the clinical assessment is abnormal or where the CT is abnormal and further evaluation for ligamentous or spinal injury is required."
Dr. Asha concluded, "If the clinical exam is not concerning and the CT is normal, then clear the neck."
SOURCE: http://bit.ly/28MxHxA and http://bit.ly/28MxHO5
JAMA Surg 2016.
NEW YORK - CT scans identify all clinically significant cervical spine injuries in intoxicated patients with blunt trauma, according to a new study.
"I don't think any of the results were particularly surprising to any of us who regularly do trauma care, but what I do think is remarkable about them is that they dispel several long-held myths about the c-spine, intoxicated patients, and the clearance process," Dr. Matthew J. Martin from Legacy Emanuel Medical Center, Portland, Oregon told Reuters Health.
"I think it again confirms that modern CT scan is highly reliable for identifying significant c-spine injuries, but also that the majority of so called 'intoxicated' patients are examinable enough to determine whether the collar can be removed (when combined with the CT scan)," he said.
Up to half of trauma patients are intoxicated, making clearance of the cervical spine a commonly encountered dilemma with both medical and medicolegal implications. Most guidelines indicate that the cervical spine should not be cleared in such patients, resulting in prolonged immobilization or additional imaging even in the face of a normal CT scan.
Dr. Martin's team examined cervical spine clearance practices for intoxicated trauma patients, examined the reliability of cervical spine CT scans for identifying clinically significant injuries (CSIs), and looked for CSIs that might have been missed by CT scans.
Among 1,429 patients who had an alcohol or drug screen performed, 44.2% were intoxicated, the researchers report in JAMA Surgery, online June 15.
Cervical spine injuries were identified in 11.3% of the sober group, 8.1% of the alcohol-intoxicated group, and 12.0% of the drug-intoxicated group.
CT scans yielded negative predictive values of 99.2% for all injuries and 99.8% for unstable injuries. There were five false-negative CT scans, including four central cord syndromes without associated fractures and one potentially unstable injury in a drug-intoxicated patient who presented with clear quadriplegia on examination.
Half of the intoxicated patients were admitted with continued cervical spine immobilization only on the basis of their intoxication. There were no missed CSIs in this group, and all patients were discharged without evidence of an injury or neurologic deficit. They underwent cervical spine immobilization for an average of 15.1 hours, about four times the average time to cervical spine clearance among sober patients (3.7 hours).
"The finding of how long we are keeping these patients in a c-collar based solely on intoxication should raise some eyebrows, and identifies an easy target for process improvement," Dr. Martin said.
"Cervical collars and immobilization are not therapeutic for the vast majority of c-spine injuries; they are really only to prevent inadvertent motion of an unstable c-spine injury," Dr. Martin said. "This is exceedingly rare in a patient who presents with no gross motor deficit, and a high quality CT scan will identify these unstable injuries very reliably. In addition, there are multiple adverse effects of prolonged immobilization, and even of getting an MRI."
"When these are factored in, I think the risk:benefit analysis falls squarely on the side of early clearance based on CT scan," he concluded.
"A key point is that this should be done by experts who are familiar with not only the global concept (the collar can be removed with a negative CT scan), but also the finer points where you could potentially cause harm, or where you should not remove the collar," Dr. Martin added. "This is where a very clear written protocol comes into play and reduces variation or errors that could cause patient harm."
"The results of this study suggest that it is unnecessary to delay cervical spine clearance until intoxicated patients are sober or until magnetic resonance imaging is performed," write Dr. Olubode A. Olufajo and Dr. Ali Salim from Brigham and Women's Hospital, Boston, in a related editorial. "However, caution must be taken in making conclusions based on these data."
"Although the authors conducted the study at an institution with high-quality CT technology and well-trained radiologists, they still recorded a false-negative CT report consistent with a misread," they note. "With the higher potential for this nature of error in lower-resourced settings, it becomes important to compare the costs and benefits of early removal of cervical collars."
They wonder, "With our knowledge that intoxicated patients form up to half of the population of trauma patients, is it really safe to risk irreversible injuries in 1% of the population to save a few hours in cervical clearance times?"
Dr. Stephen Asha from the University of New South Wales in Sydney, Australia, who has reported on various aspects of cervical spine imaging, told Reuters Health by email, "I think this study confirms what clinical experience as well as much of the more recent studies on cervical spine CT scanning tells us, which is that if there is nothing abnormal detected on a new generation, multi-slice CT, then the neck can be cleared."
"Of course there were a few missed injuries, but this needs to be put into context: no one just does a test in isolation, it is always combined with a clinical assessment, and a consideration the mechanism of injury," said Dr. Asha, who was not involved in the new work. "In this case there were five injuries not apparent on the CT scan, but all had obvious spinal cord injury on clinical examination before the CT was done, so these injuries were never going to be missed in a real clinical setting."
"MRI use should be carefully considered because the problem with MRI is that it can be over-sensitive, demonstrating abnormal signal suggesting ligamentous injury in patient who simply have a ligamentous 'strain,'" Dr. Asha explained. "The false-positive results then lead to further periods of inappropriate immobilization and testing, with the accompanying costs, inconvenience, and complications."
"In patients in whom the clinical assessment raises no concerns for injury, then a normal CT should herald the end of investigations," he said. "MRI should be reserved for those where the clinical assessment is abnormal or where the CT is abnormal and further evaluation for ligamentous or spinal injury is required."
Dr. Asha concluded, "If the clinical exam is not concerning and the CT is normal, then clear the neck."
SOURCE: http://bit.ly/28MxHxA and http://bit.ly/28MxHO5
JAMA Surg 2016.
NEW YORK - CT scans identify all clinically significant cervical spine injuries in intoxicated patients with blunt trauma, according to a new study.
"I don't think any of the results were particularly surprising to any of us who regularly do trauma care, but what I do think is remarkable about them is that they dispel several long-held myths about the c-spine, intoxicated patients, and the clearance process," Dr. Matthew J. Martin from Legacy Emanuel Medical Center, Portland, Oregon told Reuters Health.
"I think it again confirms that modern CT scan is highly reliable for identifying significant c-spine injuries, but also that the majority of so called 'intoxicated' patients are examinable enough to determine whether the collar can be removed (when combined with the CT scan)," he said.
Up to half of trauma patients are intoxicated, making clearance of the cervical spine a commonly encountered dilemma with both medical and medicolegal implications. Most guidelines indicate that the cervical spine should not be cleared in such patients, resulting in prolonged immobilization or additional imaging even in the face of a normal CT scan.
Dr. Martin's team examined cervical spine clearance practices for intoxicated trauma patients, examined the reliability of cervical spine CT scans for identifying clinically significant injuries (CSIs), and looked for CSIs that might have been missed by CT scans.
Among 1,429 patients who had an alcohol or drug screen performed, 44.2% were intoxicated, the researchers report in JAMA Surgery, online June 15.
Cervical spine injuries were identified in 11.3% of the sober group, 8.1% of the alcohol-intoxicated group, and 12.0% of the drug-intoxicated group.
CT scans yielded negative predictive values of 99.2% for all injuries and 99.8% for unstable injuries. There were five false-negative CT scans, including four central cord syndromes without associated fractures and one potentially unstable injury in a drug-intoxicated patient who presented with clear quadriplegia on examination.
Half of the intoxicated patients were admitted with continued cervical spine immobilization only on the basis of their intoxication. There were no missed CSIs in this group, and all patients were discharged without evidence of an injury or neurologic deficit. They underwent cervical spine immobilization for an average of 15.1 hours, about four times the average time to cervical spine clearance among sober patients (3.7 hours).
"The finding of how long we are keeping these patients in a c-collar based solely on intoxication should raise some eyebrows, and identifies an easy target for process improvement," Dr. Martin said.
"Cervical collars and immobilization are not therapeutic for the vast majority of c-spine injuries; they are really only to prevent inadvertent motion of an unstable c-spine injury," Dr. Martin said. "This is exceedingly rare in a patient who presents with no gross motor deficit, and a high quality CT scan will identify these unstable injuries very reliably. In addition, there are multiple adverse effects of prolonged immobilization, and even of getting an MRI."
"When these are factored in, I think the risk:benefit analysis falls squarely on the side of early clearance based on CT scan," he concluded.
"A key point is that this should be done by experts who are familiar with not only the global concept (the collar can be removed with a negative CT scan), but also the finer points where you could potentially cause harm, or where you should not remove the collar," Dr. Martin added. "This is where a very clear written protocol comes into play and reduces variation or errors that could cause patient harm."
"The results of this study suggest that it is unnecessary to delay cervical spine clearance until intoxicated patients are sober or until magnetic resonance imaging is performed," write Dr. Olubode A. Olufajo and Dr. Ali Salim from Brigham and Women's Hospital, Boston, in a related editorial. "However, caution must be taken in making conclusions based on these data."
"Although the authors conducted the study at an institution with high-quality CT technology and well-trained radiologists, they still recorded a false-negative CT report consistent with a misread," they note. "With the higher potential for this nature of error in lower-resourced settings, it becomes important to compare the costs and benefits of early removal of cervical collars."
They wonder, "With our knowledge that intoxicated patients form up to half of the population of trauma patients, is it really safe to risk irreversible injuries in 1% of the population to save a few hours in cervical clearance times?"
Dr. Stephen Asha from the University of New South Wales in Sydney, Australia, who has reported on various aspects of cervical spine imaging, told Reuters Health by email, "I think this study confirms what clinical experience as well as much of the more recent studies on cervical spine CT scanning tells us, which is that if there is nothing abnormal detected on a new generation, multi-slice CT, then the neck can be cleared."
"Of course there were a few missed injuries, but this needs to be put into context: no one just does a test in isolation, it is always combined with a clinical assessment, and a consideration the mechanism of injury," said Dr. Asha, who was not involved in the new work. "In this case there were five injuries not apparent on the CT scan, but all had obvious spinal cord injury on clinical examination before the CT was done, so these injuries were never going to be missed in a real clinical setting."
"MRI use should be carefully considered because the problem with MRI is that it can be over-sensitive, demonstrating abnormal signal suggesting ligamentous injury in patient who simply have a ligamentous 'strain,'" Dr. Asha explained. "The false-positive results then lead to further periods of inappropriate immobilization and testing, with the accompanying costs, inconvenience, and complications."
"In patients in whom the clinical assessment raises no concerns for injury, then a normal CT should herald the end of investigations," he said. "MRI should be reserved for those where the clinical assessment is abnormal or where the CT is abnormal and further evaluation for ligamentous or spinal injury is required."
Dr. Asha concluded, "If the clinical exam is not concerning and the CT is normal, then clear the neck."
SOURCE: http://bit.ly/28MxHxA and http://bit.ly/28MxHO5
JAMA Surg 2016.
Is It Safe to Discharge a Patient with IDU History, PICC for Outpatient Antimicrobial Therapy?
Case
A 42-year-old female with a history of intravenous (IV) drug use presents with severe neck pain, gait instability, and bilateral C5 motor weakness. A cervical MRI shows inflammation consistent with infection of her cervical spine at C5 and C6 and significant boney destruction. The patient undergoes kyphoplasty and debridement of her cervical spine. Operative cultures are significant for Pseudomonas aeruginosa. Infectious disease consultants recommend parenteral ceftriaxone for six weeks. The patient has no insurance, and efforts to obtain long-term placement are unsuccessful. The patient states that her last use of IV drugs was three months ago, and she insists that she will abstain from illicit IV drug abuse going forward.
Background
Outpatient parenteral antibiotic treatment (OPAT) has proven to be a cost-effective and relatively safe treatment option for most patients.1 For these reasons, it has been encouraged for use among a wide a variety of clinical situations. Intravenous drug users (IDUs) are often underinsured and have few options other than costly treatment in an inpatient acute-care facility.
A history of illicit injection drug use frequently raises questions about the appropriateness of OPAT. Some of our most vulnerable patients are those who abuse illicit drugs. Due to psychiatric, social, and financial factors, their ability to adequately transition to outpatient care may be limited. They are often underinsured, and appropriate options for inpatient post-acute care may not exist. Hospitalists often feel pressure to discharge these patients despite the lack of optimal follow-up care, and they must weigh the risks and benefits in each case.
The enrollment of IDUs into an OPAT service using a peripherally inserted central catheter (PICC) is controversial and often avoided. No clear-cut guidelines concerning the use of OPAT in IDUs by national medical societies exist.2 Consultants are often reluctant to recommend options that deviate from the typical standard of inpatient or directly observed care. The obvious risk is that a PICC line provides easy and tempting access to veins for continued drug abuse. In addition, there is an increased risk of infection and/or thrombosis if the PICC is abused.3
The safety and efficacy of PICC line use for OPAT in IDUs are unknown, and studies addressing these issues are limited. In one study at the National University Hospital of Singapore, 29 IDU patients received OPAT without complications.4 Patients were closely monitored, including by use of a tamper-proof security seal on the PICC. Infective endocarditis was the primary diagnosis in 42% of the cases studied. There were no deaths or cases of PICC abuse reported. In another abstract presentation, 39 IDU patients at Henry Ford Health System in Detroit were discharged to outpatient therapy with a PICC line and demonstrated a high cure rate (73.3%). Nine patients were lost to follow-up.5
No studies have compared OPAT therapy to inpatient therapy in IDU patients.
Back to the Case
Despite multiple attempts and due to financial considerations, no long-term care facility is able to admit the patient for therapy. The frequency of required antibiotics makes outpatient therapy in an infusion center problematic. The primary service is reluctant to discharge the patient home with a PICC line in place due to the potential of abuse and complications. A “Goals of Care” committee, consisting of several physicians from multiple specialties, legal counsel, and case management, is convened to review the case. The committee concludes that, in this particular case, it would be a reasonable option to discharge the patient to home with a PICC line in place to complete OPAT. A patient agreement document is drafted; it describes the complications of PICC line abuse and stipulates that the patient agrees to drug testing throughout the duration of her treatment. A similar agreement is required by the home infusion company. Both documents are signed by the patient, and she is subsequently discharged home.
Bottom Line
Our strategy is to deal with each of these cases as unique situations because no policies, procedures, protocols, or guidelines currently exist. One of the guiding principles should be, despite financial pressures, that the primary focus is on appropriate care of this vulnerable population. A type of “Goals of Care” committee (or organizational equivalent) can be utilized to offer assistance in decision making. Unfortunately, the safety and efficacy of OPAT in IDU patients are uncertain, and there is a lack of studies to support definitive protocols. In select cases, OPAT in IDU patients may be considered, but signed consent of the risks and the patient’s responsibilities concerning OPAT should be clearly documented in the medical record by the discharging team. TH
Dr. Conrad is a hospitalist with Ochsner Health System in New Orleans.
References
- Tice AD, Hoaglund PA, Nolet B, McKinnon PS, Mozaffari E. Cost perspectives for outpatient intravenous antimicrobial therapy. Pharmacotherapy. 2002;22(2, pt 2):63S-70S.
- Tice AD, Rehm SJ, Dalovisio JR, et al. Practice guidelines for outpatient parenteral antimicrobial therapy. IDSA guidelines. Clin Infect Dis. 2004;38(12):1651-1672.
- Chemaly R, de Parres JB, Rehm SJ, et al. Venous thrombosis associated with peripherally inserted central catheters: a retrospective analysis of the Cleveland Clinic experience. Clin Infect Dis. 2002;34(9):1179-1183.
- Ho J, Archuleta S, Sulaiman Z, Fisher D. Safe and successful treatment of intravenous drug users with a peripherally inserted central catheter in an outpatient parenteral antibiotic treatment service. J Antimicrobial Chemotherapy. 2010;65(12):2641-2644.
- Papalekas E, Patel N, Neph A, Moreno D, Zervos M, Reyes K. Outpatient parenteral antimicrobial therapy (OPAT) in intravenous drug users (IVDUs): epidemiology and outcomes. Oral abstract presented at: IDWeek; October 2014; Philadelphia.
Case
A 42-year-old female with a history of intravenous (IV) drug use presents with severe neck pain, gait instability, and bilateral C5 motor weakness. A cervical MRI shows inflammation consistent with infection of her cervical spine at C5 and C6 and significant boney destruction. The patient undergoes kyphoplasty and debridement of her cervical spine. Operative cultures are significant for Pseudomonas aeruginosa. Infectious disease consultants recommend parenteral ceftriaxone for six weeks. The patient has no insurance, and efforts to obtain long-term placement are unsuccessful. The patient states that her last use of IV drugs was three months ago, and she insists that she will abstain from illicit IV drug abuse going forward.
Background
Outpatient parenteral antibiotic treatment (OPAT) has proven to be a cost-effective and relatively safe treatment option for most patients.1 For these reasons, it has been encouraged for use among a wide a variety of clinical situations. Intravenous drug users (IDUs) are often underinsured and have few options other than costly treatment in an inpatient acute-care facility.
A history of illicit injection drug use frequently raises questions about the appropriateness of OPAT. Some of our most vulnerable patients are those who abuse illicit drugs. Due to psychiatric, social, and financial factors, their ability to adequately transition to outpatient care may be limited. They are often underinsured, and appropriate options for inpatient post-acute care may not exist. Hospitalists often feel pressure to discharge these patients despite the lack of optimal follow-up care, and they must weigh the risks and benefits in each case.
The enrollment of IDUs into an OPAT service using a peripherally inserted central catheter (PICC) is controversial and often avoided. No clear-cut guidelines concerning the use of OPAT in IDUs by national medical societies exist.2 Consultants are often reluctant to recommend options that deviate from the typical standard of inpatient or directly observed care. The obvious risk is that a PICC line provides easy and tempting access to veins for continued drug abuse. In addition, there is an increased risk of infection and/or thrombosis if the PICC is abused.3
The safety and efficacy of PICC line use for OPAT in IDUs are unknown, and studies addressing these issues are limited. In one study at the National University Hospital of Singapore, 29 IDU patients received OPAT without complications.4 Patients were closely monitored, including by use of a tamper-proof security seal on the PICC. Infective endocarditis was the primary diagnosis in 42% of the cases studied. There were no deaths or cases of PICC abuse reported. In another abstract presentation, 39 IDU patients at Henry Ford Health System in Detroit were discharged to outpatient therapy with a PICC line and demonstrated a high cure rate (73.3%). Nine patients were lost to follow-up.5
No studies have compared OPAT therapy to inpatient therapy in IDU patients.
Back to the Case
Despite multiple attempts and due to financial considerations, no long-term care facility is able to admit the patient for therapy. The frequency of required antibiotics makes outpatient therapy in an infusion center problematic. The primary service is reluctant to discharge the patient home with a PICC line in place due to the potential of abuse and complications. A “Goals of Care” committee, consisting of several physicians from multiple specialties, legal counsel, and case management, is convened to review the case. The committee concludes that, in this particular case, it would be a reasonable option to discharge the patient to home with a PICC line in place to complete OPAT. A patient agreement document is drafted; it describes the complications of PICC line abuse and stipulates that the patient agrees to drug testing throughout the duration of her treatment. A similar agreement is required by the home infusion company. Both documents are signed by the patient, and she is subsequently discharged home.
Bottom Line
Our strategy is to deal with each of these cases as unique situations because no policies, procedures, protocols, or guidelines currently exist. One of the guiding principles should be, despite financial pressures, that the primary focus is on appropriate care of this vulnerable population. A type of “Goals of Care” committee (or organizational equivalent) can be utilized to offer assistance in decision making. Unfortunately, the safety and efficacy of OPAT in IDU patients are uncertain, and there is a lack of studies to support definitive protocols. In select cases, OPAT in IDU patients may be considered, but signed consent of the risks and the patient’s responsibilities concerning OPAT should be clearly documented in the medical record by the discharging team. TH
Dr. Conrad is a hospitalist with Ochsner Health System in New Orleans.
References
- Tice AD, Hoaglund PA, Nolet B, McKinnon PS, Mozaffari E. Cost perspectives for outpatient intravenous antimicrobial therapy. Pharmacotherapy. 2002;22(2, pt 2):63S-70S.
- Tice AD, Rehm SJ, Dalovisio JR, et al. Practice guidelines for outpatient parenteral antimicrobial therapy. IDSA guidelines. Clin Infect Dis. 2004;38(12):1651-1672.
- Chemaly R, de Parres JB, Rehm SJ, et al. Venous thrombosis associated with peripherally inserted central catheters: a retrospective analysis of the Cleveland Clinic experience. Clin Infect Dis. 2002;34(9):1179-1183.
- Ho J, Archuleta S, Sulaiman Z, Fisher D. Safe and successful treatment of intravenous drug users with a peripherally inserted central catheter in an outpatient parenteral antibiotic treatment service. J Antimicrobial Chemotherapy. 2010;65(12):2641-2644.
- Papalekas E, Patel N, Neph A, Moreno D, Zervos M, Reyes K. Outpatient parenteral antimicrobial therapy (OPAT) in intravenous drug users (IVDUs): epidemiology and outcomes. Oral abstract presented at: IDWeek; October 2014; Philadelphia.
Case
A 42-year-old female with a history of intravenous (IV) drug use presents with severe neck pain, gait instability, and bilateral C5 motor weakness. A cervical MRI shows inflammation consistent with infection of her cervical spine at C5 and C6 and significant boney destruction. The patient undergoes kyphoplasty and debridement of her cervical spine. Operative cultures are significant for Pseudomonas aeruginosa. Infectious disease consultants recommend parenteral ceftriaxone for six weeks. The patient has no insurance, and efforts to obtain long-term placement are unsuccessful. The patient states that her last use of IV drugs was three months ago, and she insists that she will abstain from illicit IV drug abuse going forward.
Background
Outpatient parenteral antibiotic treatment (OPAT) has proven to be a cost-effective and relatively safe treatment option for most patients.1 For these reasons, it has been encouraged for use among a wide a variety of clinical situations. Intravenous drug users (IDUs) are often underinsured and have few options other than costly treatment in an inpatient acute-care facility.
A history of illicit injection drug use frequently raises questions about the appropriateness of OPAT. Some of our most vulnerable patients are those who abuse illicit drugs. Due to psychiatric, social, and financial factors, their ability to adequately transition to outpatient care may be limited. They are often underinsured, and appropriate options for inpatient post-acute care may not exist. Hospitalists often feel pressure to discharge these patients despite the lack of optimal follow-up care, and they must weigh the risks and benefits in each case.
The enrollment of IDUs into an OPAT service using a peripherally inserted central catheter (PICC) is controversial and often avoided. No clear-cut guidelines concerning the use of OPAT in IDUs by national medical societies exist.2 Consultants are often reluctant to recommend options that deviate from the typical standard of inpatient or directly observed care. The obvious risk is that a PICC line provides easy and tempting access to veins for continued drug abuse. In addition, there is an increased risk of infection and/or thrombosis if the PICC is abused.3
The safety and efficacy of PICC line use for OPAT in IDUs are unknown, and studies addressing these issues are limited. In one study at the National University Hospital of Singapore, 29 IDU patients received OPAT without complications.4 Patients were closely monitored, including by use of a tamper-proof security seal on the PICC. Infective endocarditis was the primary diagnosis in 42% of the cases studied. There were no deaths or cases of PICC abuse reported. In another abstract presentation, 39 IDU patients at Henry Ford Health System in Detroit were discharged to outpatient therapy with a PICC line and demonstrated a high cure rate (73.3%). Nine patients were lost to follow-up.5
No studies have compared OPAT therapy to inpatient therapy in IDU patients.
Back to the Case
Despite multiple attempts and due to financial considerations, no long-term care facility is able to admit the patient for therapy. The frequency of required antibiotics makes outpatient therapy in an infusion center problematic. The primary service is reluctant to discharge the patient home with a PICC line in place due to the potential of abuse and complications. A “Goals of Care” committee, consisting of several physicians from multiple specialties, legal counsel, and case management, is convened to review the case. The committee concludes that, in this particular case, it would be a reasonable option to discharge the patient to home with a PICC line in place to complete OPAT. A patient agreement document is drafted; it describes the complications of PICC line abuse and stipulates that the patient agrees to drug testing throughout the duration of her treatment. A similar agreement is required by the home infusion company. Both documents are signed by the patient, and she is subsequently discharged home.
Bottom Line
Our strategy is to deal with each of these cases as unique situations because no policies, procedures, protocols, or guidelines currently exist. One of the guiding principles should be, despite financial pressures, that the primary focus is on appropriate care of this vulnerable population. A type of “Goals of Care” committee (or organizational equivalent) can be utilized to offer assistance in decision making. Unfortunately, the safety and efficacy of OPAT in IDU patients are uncertain, and there is a lack of studies to support definitive protocols. In select cases, OPAT in IDU patients may be considered, but signed consent of the risks and the patient’s responsibilities concerning OPAT should be clearly documented in the medical record by the discharging team. TH
Dr. Conrad is a hospitalist with Ochsner Health System in New Orleans.
References
- Tice AD, Hoaglund PA, Nolet B, McKinnon PS, Mozaffari E. Cost perspectives for outpatient intravenous antimicrobial therapy. Pharmacotherapy. 2002;22(2, pt 2):63S-70S.
- Tice AD, Rehm SJ, Dalovisio JR, et al. Practice guidelines for outpatient parenteral antimicrobial therapy. IDSA guidelines. Clin Infect Dis. 2004;38(12):1651-1672.
- Chemaly R, de Parres JB, Rehm SJ, et al. Venous thrombosis associated with peripherally inserted central catheters: a retrospective analysis of the Cleveland Clinic experience. Clin Infect Dis. 2002;34(9):1179-1183.
- Ho J, Archuleta S, Sulaiman Z, Fisher D. Safe and successful treatment of intravenous drug users with a peripherally inserted central catheter in an outpatient parenteral antibiotic treatment service. J Antimicrobial Chemotherapy. 2010;65(12):2641-2644.
- Papalekas E, Patel N, Neph A, Moreno D, Zervos M, Reyes K. Outpatient parenteral antimicrobial therapy (OPAT) in intravenous drug users (IVDUs): epidemiology and outcomes. Oral abstract presented at: IDWeek; October 2014; Philadelphia.
Unnerving
A 73‐year‐old African American man presented to his primary care physician's office concerned about several years of muscle cramps throughout his body as if his nerves were jumping and 1 month of bilateral arm weakness.
For the past 10 years, he had experienced intermittent cramping in his calves and thighs, described as a slow tightening of the muscles associated with mild pain. Initially, the cramps lasted less than 5 minutes, occurred every few days at various times of the day, and might awaken him from sleep. They happened more often following periods of inactivity and on occasion would resolve after playing golf. In recent weeks, the sensations became more frequent, more diffuse, and lasted up to several hours. He described them as a shivering. They began to affect his biceps, pectorals, deltoids, forearms, back, and calves, and would occur unrelated to activity or inactivity. He denied sensory disturbances, facial twitching or facial weakness, diplopia, dysarthria, dysphagia, dyspnea, changes in bowel or bladder function, unexplained lapses of consciousness, fevers, or weight loss.
Long‐standing cramping is nonspecific and may reflect transient electrolyte derangements or muscle overuse. However, the more recent change in frequency, duration, and quality of these sensations, along with the reported weakness, raises concern for a process involving the peripheral nervous system. It will be important to differentiate cramping from other abnormal movements such as fasciculations, tremor, or myoclonus, and to determine whether there is objective weakness on the neurological examination.
His past medical history was significant for coronary artery disease with an ST‐segment elevation myocardial infarction several years prior, which was treated with a drug‐eluting stent. He was also diagnosed with essential thrombocythemia at the time of his myocardial infarction and tested positive for the JAK2 mutation. He was treated for several years with hydroxyurea following his diagnosis of essential thrombocythemia. Hydroxyurea had been discontinued 6 months prior due to cytopenias. The remainder of his history was significant for hypertension, chronic kidney disease stage 3, and prediabetes.
Medications were clopidogrel, atorvastatin, metoprolol, lisinopril, and hydrochlorothiazide. He did not use tobacco nor consume alcohol or illicit drugs, and he drank caffeine only occasionally. He had no family history of neurologic disorders.
Apart from his use of statins, which often affect muscles (and less commonly the nerves), the past medical history provides minimal additional insights into the cause of his symptoms. If weakness is detected on physical exam, the next step would be to distinguish upper (central) from a lower motor neuron (peripheral) localization. A diffuse problem involving all 4 limbs is generally more likely to arise from a disorder of a lower motor neuron (LMN) structure (anterior horn cell, nerve, neuromuscular junction, or muscle). To explain bilateral symptoms of the upper and lower limbs, an upper motor neuron (UMN) disease would have to affect the bilateral brain or cervical cord, a somewhat less likely possibility given the cramps described. It would also be quite unusual to have weakness of central nervous system origin without sensory deficits.
On physical examination, the patient was well‐appearing and in no apparent distress. Temperature was 98.1, blood pressure 134/84, pulse 110 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation was 100% while breathing ambient air. There was no lymphadenopathy. Lung, heart, abdominal, and skin exams were unremarkable. He was alert and oriented. His speech was without dysarthria. Examinations of the cranial nerves were intact. No tongue atrophy or fasciculations were noted. No pooling of secretions was appreciated in the oropharynx. Examination of the musculature revealed normal tone, strength, and bulk. However, there were diffuse fasciculations present, most prominent in the bilateral biceps, pectorals, deltoids, forearms, upper back, and calves. Sensation to light touch, temperature, and vibration were intact. Babinski's sign was absent, and deep tendon reflexes were normal, except at the ankles where they were reduced. Coordination and gait were normal.
The exam is notable for diffuse fasciculations, defined as spontaneous local involuntary muscle contraction and relaxation, which is often visible. Benign fasciculations are extremely common, with up to 70% of otherwise healthy adults experiencing them, and may be brought on by physical exertion. Men experience these benign fasciculations more frequently than women, and they can occur at any age and persist throughout life. Fasciculations may point to LMN disease, usually localizing to the anterior horn cell (for instance in amyotrophic lateral sclerosis [ALS]), muscle, or nerve disorders (including diffuse polyneuropathy). The presence of fasciculations in patients without other complaints and an otherwise normal physical examination supports benign fasciculations. The presence of neurologic deficits, however, such as weakness or reflex loss, is worrisome for another etiology. The absence of sensory changes makes anterior horn cell disease or myopathy most likely, as pure motor neuropathies are uncommon.
The fasciculations in this patient are most prominent in the proximal muscles, which may indicate a primary muscle disorder. Myopathies are typically characterized by diffuse symmetric weakness that is more proximal than distal, with no changes in sensation or deep tendon reflexes. One muscle disease characterized by fasciculations and cramping is periodic paralysis, which is often associated with potassium abnormalities or thyroid dysfunction caused by specific channelopathies. However, patients with this disorder typically present with episodic crises in contrast to the constant symptoms in this case.
Given the accelerated tempo of this patient's symptoms, further diagnostic evaluation should include basic laboratory testing including electrolytes, creatinine kinase, and thyrotropin. If these initial tests fail to reveal an etiology, the next study of choice would be an electromyography (EMG) and nerve conduction study (NCS), which can definitively localize the disorder to and within the peripheral nervous system. Unlike in UMN disease, in which neuroimaging is crucial, imaging is unlikely to be informative in patients with an LMN disorder.
Results of a complete blood count demonstrated a platelet count of 590,000/L (normal 140400 K/L), but was otherwise normal. Sodium, potassium, magnesium, and calcium levels were normal as were alanine aminotransferase, thyrotropin, and urinalysis. The hemoglobin A1c was 6.4%. Serum creatinine kinase was 157 U/L (normal<170 U/L), and the serum creatinine was 1.7mg/dL, similar to previous results. The erythrocyte sedimentation rate was 58mm/h (normal 020mm/h). Magnetic resonance imaging (MRI) (Figure 1) of the cervical spine demonstrated diffuse disc desiccation, and multilevel spondylosis most prominent at C4C5 and C5C6, with severe central canal stenosis and neural foraminal narrowing.
The routine laboratory tests do not point to an obvious cause of this man's symptoms. As expected, the MRI findings do not explain diffuse fascinations in all limbs with no sensory disturbance. Further evaluation should include EMG and NCS.
NCS of the median and ulnar nerves demonstrated minimally reduced conduction velocities and markedly prolonged latencies. Sensory responses were absent. No conduction block was detected. EMG demonstrated fasciculations of the right extensor digitorum and first dorsal interossei, as well as decreased amplitude and decreased recruitment of motor units.
Abnormalities on the EMG testing can reflect either a primary muscle disorder or muscle derangement resulting from disease of the nerves. NCSs are important to differentiate these 2 possibilities. This patient's NCS indicates that the primary process is a diffuse motor and sensory neuropathy, not myopathy. The lack of sensory findings on physical examination emphasizes the ability of electrodiagnostic testing to extend the clinical neurologic examination in some cases. Markedly prolonged latencies and more modest reduction in amplitude support a motor and sensory neuropathy mostly due to demyelination, rather than axonal loss, and that it is more severe in the lower limbs.
Among the demyelinating neuropathies, acute inflammatory demyelinating neuropathy (Guillain‐Barr syndrome), is the most commonly recognized. The prolonged time course of this patient's illness excludes this possibility. Chronic inflammatory demyelinating polyneuropathy is also very unlikely in the absence of conduction block on NCS. Demyelinating neuropathies may also result from antibody‐mediated nerve injury. The serum paraprotein most commonly involved is immunoglobulin M (IgM), as is detected in neuropathy due to antibodies to myelin‐associated glycoprotein (anti‐MAG) neuropathy. Another variant is the ganglioside monosialic acid antibody (anti‐GM1) associated with a rare disease called multifocal motor neuropathy (MMN), an important condition to recognize because symptoms of this illness may mimic the presentation of ALS, often with fasciculations and weakness. Unlike ALS, MMN is very responsive to treatment. Other antibody‐mediated neuropathies are much rarer. In this patient, MMN is unlikely because sensory nerves are affected in addition to motor nerves.
Because NCSs also indicate some axonal loss, it would be reasonable to screen for vitamin deficiencies, human immunodeficiency virus, and viral hepatitis. The pattern here is more symmetric and confluent than would be expected if he had mononeuritic multiplex from vasculitis.
Vitamin E level was normal. Vitamin B12 level was 323 pg/mL (normal>200 pg/mL), methylmalonate 0.3mol/L (normal 00.3mol/L). Antibodies to human immunodeficiency virus and surface antibody and antigen to hepatitis B were not detected. Cryoglobulins, anti‐nuclear antibody, and antibodies to myeloperoxidase and proteinase 3 were not detected. Serum antibodies to tissue transglutaminase and Borrelia burgdorferi were not detectable. Serum protein electrophoresis demonstrated 2 small spikes in the gamma region. Quantitative serum immunoglobulin levels were normal except for IgM, which was elevated at 1.7g/dL (normal<0.19g/dL). Serum free light chains showed a kappa component of 43.7mg/L (normal 319mg/L), a lambda component of 13.8mg/L (normal 526mg/L), and a kappa/lambda ratio of 3.17mg/L (normal 0.261.65mg/L).
The differential for a symmetric demyelinating neuropathy is quite narrow, and tests for vasculitis, celiac disease, and Lyme disease are not necessary. To pursue the cause of the elevated IgM, specific serum testing should be obtained for anti‐MAG antibodies. Many cases of anti‐MAG neuropathy are associated with an underlying lymphoproliferative disorder. As such, additional imaging to identify occult lymphoma is warranted.
Anti‐GM1 and asialoganglioside were not detectable. The anti‐MAG IgM titer was >102,400 (normal<1:1600). Abdominal ultrasound showed normal sized kidneys with normal cortical echogenicity and no splenomegaly. Computed tomography with contrast was not performed due to chronic kidney disease.
Treatment for anti‐MAG neuropathy is evolving rapidly as our understanding of the entity improves. Cyclophosphamide, intravenous immune globulin, and plasmapheresis have been the traditional treatments, but in the past decade, favorable experiences with rituximab have led some to try this medication earlier in the course. Prognosis can be favorable in many patients.
Over the next 2 months he continued to have fasciculations. He developed progressive generalized weakness, an unsteady gait, required a walker for mobility, and began to have trouble with his activities of daily living. His cognition remained intact. There was no pooling of secretions. Serial neurologic examinations demonstrated persistent fasciculations, progressive atrophy, most notably in the intrinsic hand muscles and legs, and progressive weakness of all limbs, worse in the distal muscle groups. Deep tendon reflexes remained preserved, except at the ankles where they were absent. Sensory exam showed stocking diminution to temperature up to his knees and elbows. Romberg sign was present, and he could not walk without support. He was started on rituximab, and after 4 weeks his condition continued to deteriorate.
The response to rituximab may be delayed. Alternatively, his disease may have an underlying cause such as occult lymphoma not yet identified, which would require treatment to control the neuropathy. Because of the potential association between lymphoma in some patients with anti‐MAG neuropathy, and because he is not responding to immunotherapy, whole body imaging with positron emission tomography and bone marrow biopsy should be performed.
CD19 levels indicated an appropriate B cell response to rituximab, but the anti‐MAG titer remained elevated at >102,400. He received additional doses of rituximab, but continued to decline. A bone marrow biopsy was considered, but the patient opted to forgo the procedure. After several months of rituximab, he developed mild dysarthria and dysphagia and was hospitalized for plasma exchange. After 5 sessions of plasma exchange, he showed no improvement and was discharged to a rehabilitation facility. Over the ensuing months, he became restricted to wheel chair or bed and eventually opted for comfort measures. He died after an aspiration pneumonia 15 months after his initial visit to his physician. Permission for an autopsy was not granted.
COMMENTARY
When encountering patients with involuntary muscle movements, hospitalists must recognize potential serious underlying disorders and implement a cost‐effective evaluation strategy. Fasciculations are a common finding that represent involuntary discharges of a motor unit, with a wide array of causes including radiculopathies, neuropathies, metabolic disturbances, and motor neuron diseases.[1, 2] Useful clues might point to a probable cause, such as a statin‐induced myopathy in patients with concomitant myalgias, or hypokalemia in patients on loop diuretics. Confinement of fasciculations to specific anatomic structures may be useful, as in carpal tunnel syndrome, where fasciculations would only be expected distal to median nerve compression. Features such as sensory loss, muscle atrophy, or abnormal reflexes should alert the clinician to a possible neurologic lesion.
Although fasciculations rarely reflect serious underlying pathology, the presence of neurologic deficits, such as muscle weakness, abnormal reflexes, or sensory loss, should prompt further investigation.[3] Because fasciculations typically point to an abnormality of LMN structures, a reasonable approach is to measure serum electrolytes, creatinine kinase, and thyrotropin to evaluate for myopathy. If these tests are unrevealing, the next step would be to perform EMG and NCS to help localize the lesion among the LMN structures. Muscle localization could then be pursued with muscle biopsy. Alternatively, when electrodiagnostic testing indicates peripheral nerve pathology, further evaluation is guided by the type of neuropathy: demyelinating, axonal, or mixed. If electrodiagnostic and clinical findings are unrevealing, the patient is diagnosed with benign fasciculations.
Demyelinating neuropathy, as seen in our patient, is relevant to hospitalists for several reasons. First, the list of diagnostic possibilities is narrow, allowing hospitalists to forgo many unnecessary laboratory tests and brain MRI. Second, unlike many axonal neuropathies, demyelinating neuropathies are potentially reversible if recognized early and promptly treated. Third, demyelinating neuropathy may involve the diaphragm, necessitating vigilance for neuromuscular respiratory failure. Finally, hospitalists need to be aware that some demyelinating neuropathies are associated with underlying malignancy, and identifying and treating the primary cancer may be critical to ameliorating the neuropathy.[4, 5, 6, 7, 8]
IgM paraproteinemia, with or without an underlying malignancy, is 1 type of demyelinating neuropathy that is potentially reversible with early treatment. The typical patient is exemplified by the case presented in this report: an older man who experiences symmetric, gradually worsening sensory disturbances and ataxia over months to years.[9] Motor deficits may progress more rapidly, prompting patients to seek hospital care.[7, 10] The hallmark of NCS in anti‐MAG disease is a demyelinating pattern with a predominance of distal abnormalities including marked prolongation of distal motor latencies and reductions in conduction velocities and sensory action potentials.[9] Findings of areflexia or conduction block should prompt consideration of other etiologies, such as acute or chronic inflammatory demyelinating polyneuropathy.
For unclear reasons, IgM is more likely than other immunoglobulins to cause neuropathy. Although IgM accounts for only 17% of monoclonal gammopathies, IgM is detected in 50% to 70% of patients who have both monoclonal gammopathy and peripheral neuropathy.[11] Approximately half of the patients with IgM‐associated neuropathy produce antibodies to MAG.[11, 12] Several lines of evidence have firmly established the causative role of anti‐MAG antibodies.[13]
Because the majority of patients with anti‐MAG neuropathy will have no malignant source of IgM paraprotein identified, it is unclear how extensively to search for occult malignancy. A reasonable approach is to perform a bone marrow biopsy to distinguish underlying IgM monoclonal gammopathy of undetermined significance from Waldenstrom's macroglobulinemia.[14] Bone marrow analysis may also detect B‐cell lymphoma, primary amyloidosis, chronic lymphocytic leukemia, and hairy cell leukemia, which have been described in cases of anti‐MAG syndrome.[4, 5, 6, 7, 8] There are no reports of anti‐MAG neuropathy linked to either essential thrombocythemia nor hydroxyurea use.
The goals of treatment in anti‐MAG neuropathy are to deplete monoclonal B cells and to reduce antibody levels. Although it is reported that approximately half of patients will improve with some form of immunotherapy, a Cochrane review of randomized controlled trials of treatments for anti‐MAG neuropathy (including plasma exchange, intravenous immunoglobulin [IVIG], rituximab, corticosteroids, and chemotherapy) concluded that evidence is lacking to recommend 1 treatment over another.[15] European guidelines suggest deferring therapy unless progressive or severe neuropathy is present, in which case IVIG, plasma exchange, or rituximab may be tried.[14] In patients with underlying malignancy, treatment of the hematologic disorder may improve the neuropathy.[4, 8]
Although fasciculations and peripheral neuropathy typically present in outpatient settings, they can be harbingers of more dire diagnoses that prompt patients to seek hospitalization. A sequential and cost‐effective approach can allow the astute hospitalist to pinpoint the diagnosis in what might otherwise be an unnerving case.
KEY TEACHING POINTS
- Fasciculations are extremely common and usually benign, but may indicate a more serious neurologic process, especially when accompanied by weakness or other neurologic symptoms.
- Localizing neurologic deficits to upper motor neuron or lower motor neuron structures guides further evaluation.
- Central nervous system imaging is not indicated in demyelinating neuropathies.
- Bone marrow biopsy and cross‐sectional imaging to evaluate for malignancy should be considered in patients with anti‐MAG neuropathy who fail to improve despite therapy.
- , . Fasciculations: what do we know of their significance? J Neurol Sci. 1997;152(suppl 1):S43–S48.
- , . Muscle fasciculations in a healthy population. Arch Neurol. 1963;9:363–367.
- . Muscle pain, fatigue, and fasciculations. Neurol Clin. 1997;15(3):697–709.
- , , , et al. Monocytoid B cell lymphoma associated with antibodies to myelin‐associated glycoprotein and sulphated glucuronyl paragloboside. Acta Haematol. 2001;106(3):130–132.
- . Anti‐myelin‐associated glycoprotein peripheral neuropathy as the only presentation of low grade lymphoma: a case report. Cases J. 2009;2(1):6370–6373.
- , , , et al. Antibodies to myelin‐associated glycoprotein (anti‐Mag) in IgM amyloidosis may influence expression of neuropathy in rare patients. Muscle Nerve. 2008;37(4):490–495.
- , , , et al. Heterogeneity of polyneuropathy associated with anti‐MAG antibodies. J Immunol Res. 2015;2015(3):450391–450399.
- , , , et al. Hairy cell leukaemia complicated by anti‐MAG paraproteinemic demyelinating neuropathy: resolution of neurological syndrome after cladribrine treatment. Leuk Res. 2007;31(6):873–876.
- , , , , , . Neuropathy associated with “benign” anti‐myelin‐associated glycoprotein IgM gammopathy: clinical, immunological, neurophysiological pathological findings and response to treatment in 33 cases. J Neurol. 1996;243(1):34–43.
- , , , et al. IgM MGUS anti‐MAG neuropathy with predominant muscle weakness and extensive muscle atrophy. Muscle Nerve. 2010;42(3):433–435.
- , , , et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–1369.
- , . Review of peripheral neuropathy in plasma cell disorders. Hematol Oncol. 2008;26(2):55–65.
- , . Effector mechanisms in anti‐MAG antibody‐mediated and other demyelinating neuropathies. J Neurol Sci. 2004;220(1–2):127–129.
- Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. J Peripher Nerv Syst. 2010;15(3):185–195.
- , . Immunotherapy for IgM anti‐myelin‐associated glycoprotein paraprotein‐associated peripheral neuropathies. Cochrane Database Syst Rev. 2012;5:CD002827.
A 73‐year‐old African American man presented to his primary care physician's office concerned about several years of muscle cramps throughout his body as if his nerves were jumping and 1 month of bilateral arm weakness.
For the past 10 years, he had experienced intermittent cramping in his calves and thighs, described as a slow tightening of the muscles associated with mild pain. Initially, the cramps lasted less than 5 minutes, occurred every few days at various times of the day, and might awaken him from sleep. They happened more often following periods of inactivity and on occasion would resolve after playing golf. In recent weeks, the sensations became more frequent, more diffuse, and lasted up to several hours. He described them as a shivering. They began to affect his biceps, pectorals, deltoids, forearms, back, and calves, and would occur unrelated to activity or inactivity. He denied sensory disturbances, facial twitching or facial weakness, diplopia, dysarthria, dysphagia, dyspnea, changes in bowel or bladder function, unexplained lapses of consciousness, fevers, or weight loss.
Long‐standing cramping is nonspecific and may reflect transient electrolyte derangements or muscle overuse. However, the more recent change in frequency, duration, and quality of these sensations, along with the reported weakness, raises concern for a process involving the peripheral nervous system. It will be important to differentiate cramping from other abnormal movements such as fasciculations, tremor, or myoclonus, and to determine whether there is objective weakness on the neurological examination.
His past medical history was significant for coronary artery disease with an ST‐segment elevation myocardial infarction several years prior, which was treated with a drug‐eluting stent. He was also diagnosed with essential thrombocythemia at the time of his myocardial infarction and tested positive for the JAK2 mutation. He was treated for several years with hydroxyurea following his diagnosis of essential thrombocythemia. Hydroxyurea had been discontinued 6 months prior due to cytopenias. The remainder of his history was significant for hypertension, chronic kidney disease stage 3, and prediabetes.
Medications were clopidogrel, atorvastatin, metoprolol, lisinopril, and hydrochlorothiazide. He did not use tobacco nor consume alcohol or illicit drugs, and he drank caffeine only occasionally. He had no family history of neurologic disorders.
Apart from his use of statins, which often affect muscles (and less commonly the nerves), the past medical history provides minimal additional insights into the cause of his symptoms. If weakness is detected on physical exam, the next step would be to distinguish upper (central) from a lower motor neuron (peripheral) localization. A diffuse problem involving all 4 limbs is generally more likely to arise from a disorder of a lower motor neuron (LMN) structure (anterior horn cell, nerve, neuromuscular junction, or muscle). To explain bilateral symptoms of the upper and lower limbs, an upper motor neuron (UMN) disease would have to affect the bilateral brain or cervical cord, a somewhat less likely possibility given the cramps described. It would also be quite unusual to have weakness of central nervous system origin without sensory deficits.
On physical examination, the patient was well‐appearing and in no apparent distress. Temperature was 98.1, blood pressure 134/84, pulse 110 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation was 100% while breathing ambient air. There was no lymphadenopathy. Lung, heart, abdominal, and skin exams were unremarkable. He was alert and oriented. His speech was without dysarthria. Examinations of the cranial nerves were intact. No tongue atrophy or fasciculations were noted. No pooling of secretions was appreciated in the oropharynx. Examination of the musculature revealed normal tone, strength, and bulk. However, there were diffuse fasciculations present, most prominent in the bilateral biceps, pectorals, deltoids, forearms, upper back, and calves. Sensation to light touch, temperature, and vibration were intact. Babinski's sign was absent, and deep tendon reflexes were normal, except at the ankles where they were reduced. Coordination and gait were normal.
The exam is notable for diffuse fasciculations, defined as spontaneous local involuntary muscle contraction and relaxation, which is often visible. Benign fasciculations are extremely common, with up to 70% of otherwise healthy adults experiencing them, and may be brought on by physical exertion. Men experience these benign fasciculations more frequently than women, and they can occur at any age and persist throughout life. Fasciculations may point to LMN disease, usually localizing to the anterior horn cell (for instance in amyotrophic lateral sclerosis [ALS]), muscle, or nerve disorders (including diffuse polyneuropathy). The presence of fasciculations in patients without other complaints and an otherwise normal physical examination supports benign fasciculations. The presence of neurologic deficits, however, such as weakness or reflex loss, is worrisome for another etiology. The absence of sensory changes makes anterior horn cell disease or myopathy most likely, as pure motor neuropathies are uncommon.
The fasciculations in this patient are most prominent in the proximal muscles, which may indicate a primary muscle disorder. Myopathies are typically characterized by diffuse symmetric weakness that is more proximal than distal, with no changes in sensation or deep tendon reflexes. One muscle disease characterized by fasciculations and cramping is periodic paralysis, which is often associated with potassium abnormalities or thyroid dysfunction caused by specific channelopathies. However, patients with this disorder typically present with episodic crises in contrast to the constant symptoms in this case.
Given the accelerated tempo of this patient's symptoms, further diagnostic evaluation should include basic laboratory testing including electrolytes, creatinine kinase, and thyrotropin. If these initial tests fail to reveal an etiology, the next study of choice would be an electromyography (EMG) and nerve conduction study (NCS), which can definitively localize the disorder to and within the peripheral nervous system. Unlike in UMN disease, in which neuroimaging is crucial, imaging is unlikely to be informative in patients with an LMN disorder.
Results of a complete blood count demonstrated a platelet count of 590,000/L (normal 140400 K/L), but was otherwise normal. Sodium, potassium, magnesium, and calcium levels were normal as were alanine aminotransferase, thyrotropin, and urinalysis. The hemoglobin A1c was 6.4%. Serum creatinine kinase was 157 U/L (normal<170 U/L), and the serum creatinine was 1.7mg/dL, similar to previous results. The erythrocyte sedimentation rate was 58mm/h (normal 020mm/h). Magnetic resonance imaging (MRI) (Figure 1) of the cervical spine demonstrated diffuse disc desiccation, and multilevel spondylosis most prominent at C4C5 and C5C6, with severe central canal stenosis and neural foraminal narrowing.
The routine laboratory tests do not point to an obvious cause of this man's symptoms. As expected, the MRI findings do not explain diffuse fascinations in all limbs with no sensory disturbance. Further evaluation should include EMG and NCS.
NCS of the median and ulnar nerves demonstrated minimally reduced conduction velocities and markedly prolonged latencies. Sensory responses were absent. No conduction block was detected. EMG demonstrated fasciculations of the right extensor digitorum and first dorsal interossei, as well as decreased amplitude and decreased recruitment of motor units.
Abnormalities on the EMG testing can reflect either a primary muscle disorder or muscle derangement resulting from disease of the nerves. NCSs are important to differentiate these 2 possibilities. This patient's NCS indicates that the primary process is a diffuse motor and sensory neuropathy, not myopathy. The lack of sensory findings on physical examination emphasizes the ability of electrodiagnostic testing to extend the clinical neurologic examination in some cases. Markedly prolonged latencies and more modest reduction in amplitude support a motor and sensory neuropathy mostly due to demyelination, rather than axonal loss, and that it is more severe in the lower limbs.
Among the demyelinating neuropathies, acute inflammatory demyelinating neuropathy (Guillain‐Barr syndrome), is the most commonly recognized. The prolonged time course of this patient's illness excludes this possibility. Chronic inflammatory demyelinating polyneuropathy is also very unlikely in the absence of conduction block on NCS. Demyelinating neuropathies may also result from antibody‐mediated nerve injury. The serum paraprotein most commonly involved is immunoglobulin M (IgM), as is detected in neuropathy due to antibodies to myelin‐associated glycoprotein (anti‐MAG) neuropathy. Another variant is the ganglioside monosialic acid antibody (anti‐GM1) associated with a rare disease called multifocal motor neuropathy (MMN), an important condition to recognize because symptoms of this illness may mimic the presentation of ALS, often with fasciculations and weakness. Unlike ALS, MMN is very responsive to treatment. Other antibody‐mediated neuropathies are much rarer. In this patient, MMN is unlikely because sensory nerves are affected in addition to motor nerves.
Because NCSs also indicate some axonal loss, it would be reasonable to screen for vitamin deficiencies, human immunodeficiency virus, and viral hepatitis. The pattern here is more symmetric and confluent than would be expected if he had mononeuritic multiplex from vasculitis.
Vitamin E level was normal. Vitamin B12 level was 323 pg/mL (normal>200 pg/mL), methylmalonate 0.3mol/L (normal 00.3mol/L). Antibodies to human immunodeficiency virus and surface antibody and antigen to hepatitis B were not detected. Cryoglobulins, anti‐nuclear antibody, and antibodies to myeloperoxidase and proteinase 3 were not detected. Serum antibodies to tissue transglutaminase and Borrelia burgdorferi were not detectable. Serum protein electrophoresis demonstrated 2 small spikes in the gamma region. Quantitative serum immunoglobulin levels were normal except for IgM, which was elevated at 1.7g/dL (normal<0.19g/dL). Serum free light chains showed a kappa component of 43.7mg/L (normal 319mg/L), a lambda component of 13.8mg/L (normal 526mg/L), and a kappa/lambda ratio of 3.17mg/L (normal 0.261.65mg/L).
The differential for a symmetric demyelinating neuropathy is quite narrow, and tests for vasculitis, celiac disease, and Lyme disease are not necessary. To pursue the cause of the elevated IgM, specific serum testing should be obtained for anti‐MAG antibodies. Many cases of anti‐MAG neuropathy are associated with an underlying lymphoproliferative disorder. As such, additional imaging to identify occult lymphoma is warranted.
Anti‐GM1 and asialoganglioside were not detectable. The anti‐MAG IgM titer was >102,400 (normal<1:1600). Abdominal ultrasound showed normal sized kidneys with normal cortical echogenicity and no splenomegaly. Computed tomography with contrast was not performed due to chronic kidney disease.
Treatment for anti‐MAG neuropathy is evolving rapidly as our understanding of the entity improves. Cyclophosphamide, intravenous immune globulin, and plasmapheresis have been the traditional treatments, but in the past decade, favorable experiences with rituximab have led some to try this medication earlier in the course. Prognosis can be favorable in many patients.
Over the next 2 months he continued to have fasciculations. He developed progressive generalized weakness, an unsteady gait, required a walker for mobility, and began to have trouble with his activities of daily living. His cognition remained intact. There was no pooling of secretions. Serial neurologic examinations demonstrated persistent fasciculations, progressive atrophy, most notably in the intrinsic hand muscles and legs, and progressive weakness of all limbs, worse in the distal muscle groups. Deep tendon reflexes remained preserved, except at the ankles where they were absent. Sensory exam showed stocking diminution to temperature up to his knees and elbows. Romberg sign was present, and he could not walk without support. He was started on rituximab, and after 4 weeks his condition continued to deteriorate.
The response to rituximab may be delayed. Alternatively, his disease may have an underlying cause such as occult lymphoma not yet identified, which would require treatment to control the neuropathy. Because of the potential association between lymphoma in some patients with anti‐MAG neuropathy, and because he is not responding to immunotherapy, whole body imaging with positron emission tomography and bone marrow biopsy should be performed.
CD19 levels indicated an appropriate B cell response to rituximab, but the anti‐MAG titer remained elevated at >102,400. He received additional doses of rituximab, but continued to decline. A bone marrow biopsy was considered, but the patient opted to forgo the procedure. After several months of rituximab, he developed mild dysarthria and dysphagia and was hospitalized for plasma exchange. After 5 sessions of plasma exchange, he showed no improvement and was discharged to a rehabilitation facility. Over the ensuing months, he became restricted to wheel chair or bed and eventually opted for comfort measures. He died after an aspiration pneumonia 15 months after his initial visit to his physician. Permission for an autopsy was not granted.
COMMENTARY
When encountering patients with involuntary muscle movements, hospitalists must recognize potential serious underlying disorders and implement a cost‐effective evaluation strategy. Fasciculations are a common finding that represent involuntary discharges of a motor unit, with a wide array of causes including radiculopathies, neuropathies, metabolic disturbances, and motor neuron diseases.[1, 2] Useful clues might point to a probable cause, such as a statin‐induced myopathy in patients with concomitant myalgias, or hypokalemia in patients on loop diuretics. Confinement of fasciculations to specific anatomic structures may be useful, as in carpal tunnel syndrome, where fasciculations would only be expected distal to median nerve compression. Features such as sensory loss, muscle atrophy, or abnormal reflexes should alert the clinician to a possible neurologic lesion.
Although fasciculations rarely reflect serious underlying pathology, the presence of neurologic deficits, such as muscle weakness, abnormal reflexes, or sensory loss, should prompt further investigation.[3] Because fasciculations typically point to an abnormality of LMN structures, a reasonable approach is to measure serum electrolytes, creatinine kinase, and thyrotropin to evaluate for myopathy. If these tests are unrevealing, the next step would be to perform EMG and NCS to help localize the lesion among the LMN structures. Muscle localization could then be pursued with muscle biopsy. Alternatively, when electrodiagnostic testing indicates peripheral nerve pathology, further evaluation is guided by the type of neuropathy: demyelinating, axonal, or mixed. If electrodiagnostic and clinical findings are unrevealing, the patient is diagnosed with benign fasciculations.
Demyelinating neuropathy, as seen in our patient, is relevant to hospitalists for several reasons. First, the list of diagnostic possibilities is narrow, allowing hospitalists to forgo many unnecessary laboratory tests and brain MRI. Second, unlike many axonal neuropathies, demyelinating neuropathies are potentially reversible if recognized early and promptly treated. Third, demyelinating neuropathy may involve the diaphragm, necessitating vigilance for neuromuscular respiratory failure. Finally, hospitalists need to be aware that some demyelinating neuropathies are associated with underlying malignancy, and identifying and treating the primary cancer may be critical to ameliorating the neuropathy.[4, 5, 6, 7, 8]
IgM paraproteinemia, with or without an underlying malignancy, is 1 type of demyelinating neuropathy that is potentially reversible with early treatment. The typical patient is exemplified by the case presented in this report: an older man who experiences symmetric, gradually worsening sensory disturbances and ataxia over months to years.[9] Motor deficits may progress more rapidly, prompting patients to seek hospital care.[7, 10] The hallmark of NCS in anti‐MAG disease is a demyelinating pattern with a predominance of distal abnormalities including marked prolongation of distal motor latencies and reductions in conduction velocities and sensory action potentials.[9] Findings of areflexia or conduction block should prompt consideration of other etiologies, such as acute or chronic inflammatory demyelinating polyneuropathy.
For unclear reasons, IgM is more likely than other immunoglobulins to cause neuropathy. Although IgM accounts for only 17% of monoclonal gammopathies, IgM is detected in 50% to 70% of patients who have both monoclonal gammopathy and peripheral neuropathy.[11] Approximately half of the patients with IgM‐associated neuropathy produce antibodies to MAG.[11, 12] Several lines of evidence have firmly established the causative role of anti‐MAG antibodies.[13]
Because the majority of patients with anti‐MAG neuropathy will have no malignant source of IgM paraprotein identified, it is unclear how extensively to search for occult malignancy. A reasonable approach is to perform a bone marrow biopsy to distinguish underlying IgM monoclonal gammopathy of undetermined significance from Waldenstrom's macroglobulinemia.[14] Bone marrow analysis may also detect B‐cell lymphoma, primary amyloidosis, chronic lymphocytic leukemia, and hairy cell leukemia, which have been described in cases of anti‐MAG syndrome.[4, 5, 6, 7, 8] There are no reports of anti‐MAG neuropathy linked to either essential thrombocythemia nor hydroxyurea use.
The goals of treatment in anti‐MAG neuropathy are to deplete monoclonal B cells and to reduce antibody levels. Although it is reported that approximately half of patients will improve with some form of immunotherapy, a Cochrane review of randomized controlled trials of treatments for anti‐MAG neuropathy (including plasma exchange, intravenous immunoglobulin [IVIG], rituximab, corticosteroids, and chemotherapy) concluded that evidence is lacking to recommend 1 treatment over another.[15] European guidelines suggest deferring therapy unless progressive or severe neuropathy is present, in which case IVIG, plasma exchange, or rituximab may be tried.[14] In patients with underlying malignancy, treatment of the hematologic disorder may improve the neuropathy.[4, 8]
Although fasciculations and peripheral neuropathy typically present in outpatient settings, they can be harbingers of more dire diagnoses that prompt patients to seek hospitalization. A sequential and cost‐effective approach can allow the astute hospitalist to pinpoint the diagnosis in what might otherwise be an unnerving case.
KEY TEACHING POINTS
- Fasciculations are extremely common and usually benign, but may indicate a more serious neurologic process, especially when accompanied by weakness or other neurologic symptoms.
- Localizing neurologic deficits to upper motor neuron or lower motor neuron structures guides further evaluation.
- Central nervous system imaging is not indicated in demyelinating neuropathies.
- Bone marrow biopsy and cross‐sectional imaging to evaluate for malignancy should be considered in patients with anti‐MAG neuropathy who fail to improve despite therapy.
A 73‐year‐old African American man presented to his primary care physician's office concerned about several years of muscle cramps throughout his body as if his nerves were jumping and 1 month of bilateral arm weakness.
For the past 10 years, he had experienced intermittent cramping in his calves and thighs, described as a slow tightening of the muscles associated with mild pain. Initially, the cramps lasted less than 5 minutes, occurred every few days at various times of the day, and might awaken him from sleep. They happened more often following periods of inactivity and on occasion would resolve after playing golf. In recent weeks, the sensations became more frequent, more diffuse, and lasted up to several hours. He described them as a shivering. They began to affect his biceps, pectorals, deltoids, forearms, back, and calves, and would occur unrelated to activity or inactivity. He denied sensory disturbances, facial twitching or facial weakness, diplopia, dysarthria, dysphagia, dyspnea, changes in bowel or bladder function, unexplained lapses of consciousness, fevers, or weight loss.
Long‐standing cramping is nonspecific and may reflect transient electrolyte derangements or muscle overuse. However, the more recent change in frequency, duration, and quality of these sensations, along with the reported weakness, raises concern for a process involving the peripheral nervous system. It will be important to differentiate cramping from other abnormal movements such as fasciculations, tremor, or myoclonus, and to determine whether there is objective weakness on the neurological examination.
His past medical history was significant for coronary artery disease with an ST‐segment elevation myocardial infarction several years prior, which was treated with a drug‐eluting stent. He was also diagnosed with essential thrombocythemia at the time of his myocardial infarction and tested positive for the JAK2 mutation. He was treated for several years with hydroxyurea following his diagnosis of essential thrombocythemia. Hydroxyurea had been discontinued 6 months prior due to cytopenias. The remainder of his history was significant for hypertension, chronic kidney disease stage 3, and prediabetes.
Medications were clopidogrel, atorvastatin, metoprolol, lisinopril, and hydrochlorothiazide. He did not use tobacco nor consume alcohol or illicit drugs, and he drank caffeine only occasionally. He had no family history of neurologic disorders.
Apart from his use of statins, which often affect muscles (and less commonly the nerves), the past medical history provides minimal additional insights into the cause of his symptoms. If weakness is detected on physical exam, the next step would be to distinguish upper (central) from a lower motor neuron (peripheral) localization. A diffuse problem involving all 4 limbs is generally more likely to arise from a disorder of a lower motor neuron (LMN) structure (anterior horn cell, nerve, neuromuscular junction, or muscle). To explain bilateral symptoms of the upper and lower limbs, an upper motor neuron (UMN) disease would have to affect the bilateral brain or cervical cord, a somewhat less likely possibility given the cramps described. It would also be quite unusual to have weakness of central nervous system origin without sensory deficits.
On physical examination, the patient was well‐appearing and in no apparent distress. Temperature was 98.1, blood pressure 134/84, pulse 110 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation was 100% while breathing ambient air. There was no lymphadenopathy. Lung, heart, abdominal, and skin exams were unremarkable. He was alert and oriented. His speech was without dysarthria. Examinations of the cranial nerves were intact. No tongue atrophy or fasciculations were noted. No pooling of secretions was appreciated in the oropharynx. Examination of the musculature revealed normal tone, strength, and bulk. However, there were diffuse fasciculations present, most prominent in the bilateral biceps, pectorals, deltoids, forearms, upper back, and calves. Sensation to light touch, temperature, and vibration were intact. Babinski's sign was absent, and deep tendon reflexes were normal, except at the ankles where they were reduced. Coordination and gait were normal.
The exam is notable for diffuse fasciculations, defined as spontaneous local involuntary muscle contraction and relaxation, which is often visible. Benign fasciculations are extremely common, with up to 70% of otherwise healthy adults experiencing them, and may be brought on by physical exertion. Men experience these benign fasciculations more frequently than women, and they can occur at any age and persist throughout life. Fasciculations may point to LMN disease, usually localizing to the anterior horn cell (for instance in amyotrophic lateral sclerosis [ALS]), muscle, or nerve disorders (including diffuse polyneuropathy). The presence of fasciculations in patients without other complaints and an otherwise normal physical examination supports benign fasciculations. The presence of neurologic deficits, however, such as weakness or reflex loss, is worrisome for another etiology. The absence of sensory changes makes anterior horn cell disease or myopathy most likely, as pure motor neuropathies are uncommon.
The fasciculations in this patient are most prominent in the proximal muscles, which may indicate a primary muscle disorder. Myopathies are typically characterized by diffuse symmetric weakness that is more proximal than distal, with no changes in sensation or deep tendon reflexes. One muscle disease characterized by fasciculations and cramping is periodic paralysis, which is often associated with potassium abnormalities or thyroid dysfunction caused by specific channelopathies. However, patients with this disorder typically present with episodic crises in contrast to the constant symptoms in this case.
Given the accelerated tempo of this patient's symptoms, further diagnostic evaluation should include basic laboratory testing including electrolytes, creatinine kinase, and thyrotropin. If these initial tests fail to reveal an etiology, the next study of choice would be an electromyography (EMG) and nerve conduction study (NCS), which can definitively localize the disorder to and within the peripheral nervous system. Unlike in UMN disease, in which neuroimaging is crucial, imaging is unlikely to be informative in patients with an LMN disorder.
Results of a complete blood count demonstrated a platelet count of 590,000/L (normal 140400 K/L), but was otherwise normal. Sodium, potassium, magnesium, and calcium levels were normal as were alanine aminotransferase, thyrotropin, and urinalysis. The hemoglobin A1c was 6.4%. Serum creatinine kinase was 157 U/L (normal<170 U/L), and the serum creatinine was 1.7mg/dL, similar to previous results. The erythrocyte sedimentation rate was 58mm/h (normal 020mm/h). Magnetic resonance imaging (MRI) (Figure 1) of the cervical spine demonstrated diffuse disc desiccation, and multilevel spondylosis most prominent at C4C5 and C5C6, with severe central canal stenosis and neural foraminal narrowing.
The routine laboratory tests do not point to an obvious cause of this man's symptoms. As expected, the MRI findings do not explain diffuse fascinations in all limbs with no sensory disturbance. Further evaluation should include EMG and NCS.
NCS of the median and ulnar nerves demonstrated minimally reduced conduction velocities and markedly prolonged latencies. Sensory responses were absent. No conduction block was detected. EMG demonstrated fasciculations of the right extensor digitorum and first dorsal interossei, as well as decreased amplitude and decreased recruitment of motor units.
Abnormalities on the EMG testing can reflect either a primary muscle disorder or muscle derangement resulting from disease of the nerves. NCSs are important to differentiate these 2 possibilities. This patient's NCS indicates that the primary process is a diffuse motor and sensory neuropathy, not myopathy. The lack of sensory findings on physical examination emphasizes the ability of electrodiagnostic testing to extend the clinical neurologic examination in some cases. Markedly prolonged latencies and more modest reduction in amplitude support a motor and sensory neuropathy mostly due to demyelination, rather than axonal loss, and that it is more severe in the lower limbs.
Among the demyelinating neuropathies, acute inflammatory demyelinating neuropathy (Guillain‐Barr syndrome), is the most commonly recognized. The prolonged time course of this patient's illness excludes this possibility. Chronic inflammatory demyelinating polyneuropathy is also very unlikely in the absence of conduction block on NCS. Demyelinating neuropathies may also result from antibody‐mediated nerve injury. The serum paraprotein most commonly involved is immunoglobulin M (IgM), as is detected in neuropathy due to antibodies to myelin‐associated glycoprotein (anti‐MAG) neuropathy. Another variant is the ganglioside monosialic acid antibody (anti‐GM1) associated with a rare disease called multifocal motor neuropathy (MMN), an important condition to recognize because symptoms of this illness may mimic the presentation of ALS, often with fasciculations and weakness. Unlike ALS, MMN is very responsive to treatment. Other antibody‐mediated neuropathies are much rarer. In this patient, MMN is unlikely because sensory nerves are affected in addition to motor nerves.
Because NCSs also indicate some axonal loss, it would be reasonable to screen for vitamin deficiencies, human immunodeficiency virus, and viral hepatitis. The pattern here is more symmetric and confluent than would be expected if he had mononeuritic multiplex from vasculitis.
Vitamin E level was normal. Vitamin B12 level was 323 pg/mL (normal>200 pg/mL), methylmalonate 0.3mol/L (normal 00.3mol/L). Antibodies to human immunodeficiency virus and surface antibody and antigen to hepatitis B were not detected. Cryoglobulins, anti‐nuclear antibody, and antibodies to myeloperoxidase and proteinase 3 were not detected. Serum antibodies to tissue transglutaminase and Borrelia burgdorferi were not detectable. Serum protein electrophoresis demonstrated 2 small spikes in the gamma region. Quantitative serum immunoglobulin levels were normal except for IgM, which was elevated at 1.7g/dL (normal<0.19g/dL). Serum free light chains showed a kappa component of 43.7mg/L (normal 319mg/L), a lambda component of 13.8mg/L (normal 526mg/L), and a kappa/lambda ratio of 3.17mg/L (normal 0.261.65mg/L).
The differential for a symmetric demyelinating neuropathy is quite narrow, and tests for vasculitis, celiac disease, and Lyme disease are not necessary. To pursue the cause of the elevated IgM, specific serum testing should be obtained for anti‐MAG antibodies. Many cases of anti‐MAG neuropathy are associated with an underlying lymphoproliferative disorder. As such, additional imaging to identify occult lymphoma is warranted.
Anti‐GM1 and asialoganglioside were not detectable. The anti‐MAG IgM titer was >102,400 (normal<1:1600). Abdominal ultrasound showed normal sized kidneys with normal cortical echogenicity and no splenomegaly. Computed tomography with contrast was not performed due to chronic kidney disease.
Treatment for anti‐MAG neuropathy is evolving rapidly as our understanding of the entity improves. Cyclophosphamide, intravenous immune globulin, and plasmapheresis have been the traditional treatments, but in the past decade, favorable experiences with rituximab have led some to try this medication earlier in the course. Prognosis can be favorable in many patients.
Over the next 2 months he continued to have fasciculations. He developed progressive generalized weakness, an unsteady gait, required a walker for mobility, and began to have trouble with his activities of daily living. His cognition remained intact. There was no pooling of secretions. Serial neurologic examinations demonstrated persistent fasciculations, progressive atrophy, most notably in the intrinsic hand muscles and legs, and progressive weakness of all limbs, worse in the distal muscle groups. Deep tendon reflexes remained preserved, except at the ankles where they were absent. Sensory exam showed stocking diminution to temperature up to his knees and elbows. Romberg sign was present, and he could not walk without support. He was started on rituximab, and after 4 weeks his condition continued to deteriorate.
The response to rituximab may be delayed. Alternatively, his disease may have an underlying cause such as occult lymphoma not yet identified, which would require treatment to control the neuropathy. Because of the potential association between lymphoma in some patients with anti‐MAG neuropathy, and because he is not responding to immunotherapy, whole body imaging with positron emission tomography and bone marrow biopsy should be performed.
CD19 levels indicated an appropriate B cell response to rituximab, but the anti‐MAG titer remained elevated at >102,400. He received additional doses of rituximab, but continued to decline. A bone marrow biopsy was considered, but the patient opted to forgo the procedure. After several months of rituximab, he developed mild dysarthria and dysphagia and was hospitalized for plasma exchange. After 5 sessions of plasma exchange, he showed no improvement and was discharged to a rehabilitation facility. Over the ensuing months, he became restricted to wheel chair or bed and eventually opted for comfort measures. He died after an aspiration pneumonia 15 months after his initial visit to his physician. Permission for an autopsy was not granted.
COMMENTARY
When encountering patients with involuntary muscle movements, hospitalists must recognize potential serious underlying disorders and implement a cost‐effective evaluation strategy. Fasciculations are a common finding that represent involuntary discharges of a motor unit, with a wide array of causes including radiculopathies, neuropathies, metabolic disturbances, and motor neuron diseases.[1, 2] Useful clues might point to a probable cause, such as a statin‐induced myopathy in patients with concomitant myalgias, or hypokalemia in patients on loop diuretics. Confinement of fasciculations to specific anatomic structures may be useful, as in carpal tunnel syndrome, where fasciculations would only be expected distal to median nerve compression. Features such as sensory loss, muscle atrophy, or abnormal reflexes should alert the clinician to a possible neurologic lesion.
Although fasciculations rarely reflect serious underlying pathology, the presence of neurologic deficits, such as muscle weakness, abnormal reflexes, or sensory loss, should prompt further investigation.[3] Because fasciculations typically point to an abnormality of LMN structures, a reasonable approach is to measure serum electrolytes, creatinine kinase, and thyrotropin to evaluate for myopathy. If these tests are unrevealing, the next step would be to perform EMG and NCS to help localize the lesion among the LMN structures. Muscle localization could then be pursued with muscle biopsy. Alternatively, when electrodiagnostic testing indicates peripheral nerve pathology, further evaluation is guided by the type of neuropathy: demyelinating, axonal, or mixed. If electrodiagnostic and clinical findings are unrevealing, the patient is diagnosed with benign fasciculations.
Demyelinating neuropathy, as seen in our patient, is relevant to hospitalists for several reasons. First, the list of diagnostic possibilities is narrow, allowing hospitalists to forgo many unnecessary laboratory tests and brain MRI. Second, unlike many axonal neuropathies, demyelinating neuropathies are potentially reversible if recognized early and promptly treated. Third, demyelinating neuropathy may involve the diaphragm, necessitating vigilance for neuromuscular respiratory failure. Finally, hospitalists need to be aware that some demyelinating neuropathies are associated with underlying malignancy, and identifying and treating the primary cancer may be critical to ameliorating the neuropathy.[4, 5, 6, 7, 8]
IgM paraproteinemia, with or without an underlying malignancy, is 1 type of demyelinating neuropathy that is potentially reversible with early treatment. The typical patient is exemplified by the case presented in this report: an older man who experiences symmetric, gradually worsening sensory disturbances and ataxia over months to years.[9] Motor deficits may progress more rapidly, prompting patients to seek hospital care.[7, 10] The hallmark of NCS in anti‐MAG disease is a demyelinating pattern with a predominance of distal abnormalities including marked prolongation of distal motor latencies and reductions in conduction velocities and sensory action potentials.[9] Findings of areflexia or conduction block should prompt consideration of other etiologies, such as acute or chronic inflammatory demyelinating polyneuropathy.
For unclear reasons, IgM is more likely than other immunoglobulins to cause neuropathy. Although IgM accounts for only 17% of monoclonal gammopathies, IgM is detected in 50% to 70% of patients who have both monoclonal gammopathy and peripheral neuropathy.[11] Approximately half of the patients with IgM‐associated neuropathy produce antibodies to MAG.[11, 12] Several lines of evidence have firmly established the causative role of anti‐MAG antibodies.[13]
Because the majority of patients with anti‐MAG neuropathy will have no malignant source of IgM paraprotein identified, it is unclear how extensively to search for occult malignancy. A reasonable approach is to perform a bone marrow biopsy to distinguish underlying IgM monoclonal gammopathy of undetermined significance from Waldenstrom's macroglobulinemia.[14] Bone marrow analysis may also detect B‐cell lymphoma, primary amyloidosis, chronic lymphocytic leukemia, and hairy cell leukemia, which have been described in cases of anti‐MAG syndrome.[4, 5, 6, 7, 8] There are no reports of anti‐MAG neuropathy linked to either essential thrombocythemia nor hydroxyurea use.
The goals of treatment in anti‐MAG neuropathy are to deplete monoclonal B cells and to reduce antibody levels. Although it is reported that approximately half of patients will improve with some form of immunotherapy, a Cochrane review of randomized controlled trials of treatments for anti‐MAG neuropathy (including plasma exchange, intravenous immunoglobulin [IVIG], rituximab, corticosteroids, and chemotherapy) concluded that evidence is lacking to recommend 1 treatment over another.[15] European guidelines suggest deferring therapy unless progressive or severe neuropathy is present, in which case IVIG, plasma exchange, or rituximab may be tried.[14] In patients with underlying malignancy, treatment of the hematologic disorder may improve the neuropathy.[4, 8]
Although fasciculations and peripheral neuropathy typically present in outpatient settings, they can be harbingers of more dire diagnoses that prompt patients to seek hospitalization. A sequential and cost‐effective approach can allow the astute hospitalist to pinpoint the diagnosis in what might otherwise be an unnerving case.
KEY TEACHING POINTS
- Fasciculations are extremely common and usually benign, but may indicate a more serious neurologic process, especially when accompanied by weakness or other neurologic symptoms.
- Localizing neurologic deficits to upper motor neuron or lower motor neuron structures guides further evaluation.
- Central nervous system imaging is not indicated in demyelinating neuropathies.
- Bone marrow biopsy and cross‐sectional imaging to evaluate for malignancy should be considered in patients with anti‐MAG neuropathy who fail to improve despite therapy.
- , . Fasciculations: what do we know of their significance? J Neurol Sci. 1997;152(suppl 1):S43–S48.
- , . Muscle fasciculations in a healthy population. Arch Neurol. 1963;9:363–367.
- . Muscle pain, fatigue, and fasciculations. Neurol Clin. 1997;15(3):697–709.
- , , , et al. Monocytoid B cell lymphoma associated with antibodies to myelin‐associated glycoprotein and sulphated glucuronyl paragloboside. Acta Haematol. 2001;106(3):130–132.
- . Anti‐myelin‐associated glycoprotein peripheral neuropathy as the only presentation of low grade lymphoma: a case report. Cases J. 2009;2(1):6370–6373.
- , , , et al. Antibodies to myelin‐associated glycoprotein (anti‐Mag) in IgM amyloidosis may influence expression of neuropathy in rare patients. Muscle Nerve. 2008;37(4):490–495.
- , , , et al. Heterogeneity of polyneuropathy associated with anti‐MAG antibodies. J Immunol Res. 2015;2015(3):450391–450399.
- , , , et al. Hairy cell leukaemia complicated by anti‐MAG paraproteinemic demyelinating neuropathy: resolution of neurological syndrome after cladribrine treatment. Leuk Res. 2007;31(6):873–876.
- , , , , , . Neuropathy associated with “benign” anti‐myelin‐associated glycoprotein IgM gammopathy: clinical, immunological, neurophysiological pathological findings and response to treatment in 33 cases. J Neurol. 1996;243(1):34–43.
- , , , et al. IgM MGUS anti‐MAG neuropathy with predominant muscle weakness and extensive muscle atrophy. Muscle Nerve. 2010;42(3):433–435.
- , , , et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–1369.
- , . Review of peripheral neuropathy in plasma cell disorders. Hematol Oncol. 2008;26(2):55–65.
- , . Effector mechanisms in anti‐MAG antibody‐mediated and other demyelinating neuropathies. J Neurol Sci. 2004;220(1–2):127–129.
- Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. J Peripher Nerv Syst. 2010;15(3):185–195.
- , . Immunotherapy for IgM anti‐myelin‐associated glycoprotein paraprotein‐associated peripheral neuropathies. Cochrane Database Syst Rev. 2012;5:CD002827.
- , . Fasciculations: what do we know of their significance? J Neurol Sci. 1997;152(suppl 1):S43–S48.
- , . Muscle fasciculations in a healthy population. Arch Neurol. 1963;9:363–367.
- . Muscle pain, fatigue, and fasciculations. Neurol Clin. 1997;15(3):697–709.
- , , , et al. Monocytoid B cell lymphoma associated with antibodies to myelin‐associated glycoprotein and sulphated glucuronyl paragloboside. Acta Haematol. 2001;106(3):130–132.
- . Anti‐myelin‐associated glycoprotein peripheral neuropathy as the only presentation of low grade lymphoma: a case report. Cases J. 2009;2(1):6370–6373.
- , , , et al. Antibodies to myelin‐associated glycoprotein (anti‐Mag) in IgM amyloidosis may influence expression of neuropathy in rare patients. Muscle Nerve. 2008;37(4):490–495.
- , , , et al. Heterogeneity of polyneuropathy associated with anti‐MAG antibodies. J Immunol Res. 2015;2015(3):450391–450399.
- , , , et al. Hairy cell leukaemia complicated by anti‐MAG paraproteinemic demyelinating neuropathy: resolution of neurological syndrome after cladribrine treatment. Leuk Res. 2007;31(6):873–876.
- , , , , , . Neuropathy associated with “benign” anti‐myelin‐associated glycoprotein IgM gammopathy: clinical, immunological, neurophysiological pathological findings and response to treatment in 33 cases. J Neurol. 1996;243(1):34–43.
- , , , et al. IgM MGUS anti‐MAG neuropathy with predominant muscle weakness and extensive muscle atrophy. Muscle Nerve. 2010;42(3):433–435.
- , , , et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–1369.
- , . Review of peripheral neuropathy in plasma cell disorders. Hematol Oncol. 2008;26(2):55–65.
- , . Effector mechanisms in anti‐MAG antibody‐mediated and other demyelinating neuropathies. J Neurol Sci. 2004;220(1–2):127–129.
- Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. J Peripher Nerv Syst. 2010;15(3):185–195.
- , . Immunotherapy for IgM anti‐myelin‐associated glycoprotein paraprotein‐associated peripheral neuropathies. Cochrane Database Syst Rev. 2012;5:CD002827.