User login
Pediatric Dermatology Consult - May 2017
BY LAWRENCE F. EICHENFIELD, MD, and JENNA BOROK
Nevus sebaceous (NS) is considered a subtype of an epidermal nevus, a benign hamartoma that includes an excess or deficiency of structural elements of the skin, such as epidermis, sebaceous, and apocrine glands.1
Nevus sebaceous, also known as nevus sebaceous of Jadassohn, was first described in 1895 by Josef Jadassohn as a nevoid growth composed predominantly of sebaceous glands.2 Sebaceous glands are found everywhere on the body where hair is found and are located adjacent to hair follicles with ducts, through which sebum flows.3
NS clinically appears as a waxy, yellowish-orange to pink, hairless plaque, ranging from 1 cm to over 10 cm in size and usually located on the scalp, face, neck, or trunk.1 When the lesions are linear, they typically follow Blaschko lines.1 The lesions change over time. An infant’s lesion will be slightly raised and can be hardly noticeable. If the lesion is located on the scalp, it will remain hairless. During later childhood, the nevus may thicken. The lesions may become thicker and verrucous during adolescence.
Histologically, NS are benign hamartomas with epidermal, follicular, sebaceous, and apocrine elements.4 The malformation is primarily within the individual folliculosebaceous units.1 An infantile lesion will deviate little from normal skin as the follicular units are so small.1 During childhood, microscopically, it is easier to see misshapen follicles and thickened epidermis.1 During adolescence, the histological pattern is similar to that of an epidermal nevi, which includes acanthosis and fibroplasia of the papillary dermis.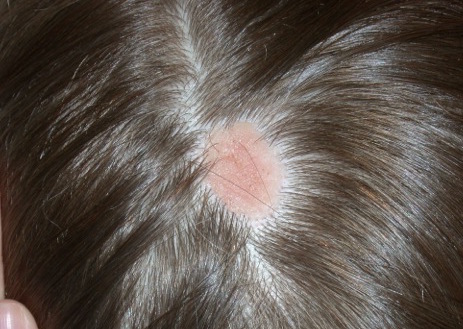
There are now several lines of evidence showing that nevus sebaceous is caused by genetic postzygotic mosaic mutations in the mitogen–activated protein kinase (MAPK) pathway, which specifically activate ras mutations including H ras and K ras genes.5-7 Since NS is caused by a somatic mosaicism mutation, there are a several syndromic findings associated with NS depending on the timing of the mutations during development and whether single versus multiple tissues are affected.8 Specifically, NS has been associated with Schimmelpenning-Feuerstein-Mims syndrome that includes NS, and skeletal, ocular, and neurologic abnormalities.1,7 A study found that cutaneous skeletal hypophosphatemia syndrome, manifested as NS and vitamin D–resistant rickets, has identical ras mutations in both skin and bone tissues, providing further support that these syndromic findings are a result of a multilineage somatic ras mutation.8
Diagnosis
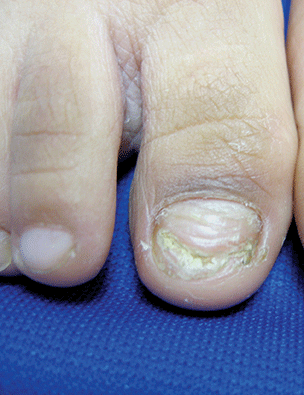
NS is typically diagnosed clinically, based on the presence of a circumscribed, thin, yellow-orange, oval, round, or linear plaque, usually on the scalp or face. During infancy or the first stage, the lesions remain stable. In the second stage, during puberty, the lesions thicken and become verrucous or nodular because of changes in sebaceous gland activity that are driven by hormones. In the third stage of their natural course, benign and malignant epithelial neoplasms can develop, including trichoblastoma, syringocystadenoma papilliferum, sebaceous epithelioma, basal cell carcinoma, and trichilemmoma.9
The associated syndrome, nevus sebaceous syndrome, also known as Schimmelpenning syndrome, has more extensive cutaneous lesions along Blaschko lines and presents with CNS, ocular, or skeletal defects. The CNS abnormalities include mental retardation, seizures, and hemimegalencephaly.1 Therefore, a thorough neurologic and ophthalmologic examination should be performed in patients with suspected nevus sebaceous syndrome.
Aplasia cutis congenita is the absence of the skin at birth that presents with an erosion or deep ulceration to a scar or a defect that is covered with an epidermal membrane.1 Some aplasia cutis may present as a smooth, hairless surface at birth, making differentiation from nevus sebaceous difficult. The pinkish, orange to yellow hue of NS may help differentiate these entities. Juvenile xanthogranuloma is a fairly common histiocytosis and is the most common histiocytic disease of childhood.1 It is a benign proliferation of dermal dendrocytes, and it presents in infants as many red to yellow papules or a few nodules on the head and neck. Many lesions even remain undetected.1
Syringocystadenoma papilliferum is a benign neoplasm with apocrine differentiation and presents as a papule or plaque on the head and neck. It can be associated with nevus sebaceous.1
Treatment
The definitive treatment is an excision. The necessity and timing of excision of these lesions is still under debate.9 Secondary neoplasms do arise from the nevus sebaceous, although the incidence is low pre puberty.1 It has been estimated that 16% of cases develop benign tumors and that 8% of cases develop malignant tumors.9 The majority of malignant lesions are basal cell carcinomas, and a large recent study found that only 1% of patients had malignancies.10 While most of these tumors develop in adulthood, there have been reports of malignancies in children.10
An additional reason to excise NS is that, over time, they grow more verrucous, become inflamed, bleed with trauma, and may be unappealing cosmetically.1 Some experts recommend earlier excision in childhood, especially with larger lesions or facial lesions where minimizing deformity is important and with the possibility of less noticeable scarring.9 The prophylactic removal of NS remains controversial, and there is a lack of consensus among experts about the timing of excision. It is recommended that each lesion be evaluated on a case-by-case basis.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Borok is a medical student at the University of California, Los Angeles. Dr. Eichenfield and Ms. Borok said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. “Dermatology.” 3rd ed. (St Louis, Mo: Elsevier, 2012).
2. Arch Dermatol Res. 1895. doi: 10.1007/BF01842810.
3. J Invest Dermatol. 2004 Jul;123(1):1-12.
4. J Cutan Pathol. 1984;11(5):396-414.
5. J Invest Dermatol. 2013 Mar;133(3):827-30.
6. J Invest Dermatol. 2013 Mar;133(3):597-600.
7. Nat Genet. 2012 Jun 10;44(7):783-7.
8. JAAD. 2016 Aug;75(2):420-7.
9. Pediatr Dermatol. 2012 Jan-Feb;29(1):15-23.
10. Pediatr Dermatol. 2009 Nov-Dec;26(6):676-81.
BY LAWRENCE F. EICHENFIELD, MD, and JENNA BOROK
Nevus sebaceous (NS) is considered a subtype of an epidermal nevus, a benign hamartoma that includes an excess or deficiency of structural elements of the skin, such as epidermis, sebaceous, and apocrine glands.1
Nevus sebaceous, also known as nevus sebaceous of Jadassohn, was first described in 1895 by Josef Jadassohn as a nevoid growth composed predominantly of sebaceous glands.2 Sebaceous glands are found everywhere on the body where hair is found and are located adjacent to hair follicles with ducts, through which sebum flows.3
NS clinically appears as a waxy, yellowish-orange to pink, hairless plaque, ranging from 1 cm to over 10 cm in size and usually located on the scalp, face, neck, or trunk.1 When the lesions are linear, they typically follow Blaschko lines.1 The lesions change over time. An infant’s lesion will be slightly raised and can be hardly noticeable. If the lesion is located on the scalp, it will remain hairless. During later childhood, the nevus may thicken. The lesions may become thicker and verrucous during adolescence.
Histologically, NS are benign hamartomas with epidermal, follicular, sebaceous, and apocrine elements.4 The malformation is primarily within the individual folliculosebaceous units.1 An infantile lesion will deviate little from normal skin as the follicular units are so small.1 During childhood, microscopically, it is easier to see misshapen follicles and thickened epidermis.1 During adolescence, the histological pattern is similar to that of an epidermal nevi, which includes acanthosis and fibroplasia of the papillary dermis.
There are now several lines of evidence showing that nevus sebaceous is caused by genetic postzygotic mosaic mutations in the mitogen–activated protein kinase (MAPK) pathway, which specifically activate ras mutations including H ras and K ras genes.5-7 Since NS is caused by a somatic mosaicism mutation, there are a several syndromic findings associated with NS depending on the timing of the mutations during development and whether single versus multiple tissues are affected.8 Specifically, NS has been associated with Schimmelpenning-Feuerstein-Mims syndrome that includes NS, and skeletal, ocular, and neurologic abnormalities.1,7 A study found that cutaneous skeletal hypophosphatemia syndrome, manifested as NS and vitamin D–resistant rickets, has identical ras mutations in both skin and bone tissues, providing further support that these syndromic findings are a result of a multilineage somatic ras mutation.8
Diagnosis
NS is typically diagnosed clinically, based on the presence of a circumscribed, thin, yellow-orange, oval, round, or linear plaque, usually on the scalp or face. During infancy or the first stage, the lesions remain stable. In the second stage, during puberty, the lesions thicken and become verrucous or nodular because of changes in sebaceous gland activity that are driven by hormones. In the third stage of their natural course, benign and malignant epithelial neoplasms can develop, including trichoblastoma, syringocystadenoma papilliferum, sebaceous epithelioma, basal cell carcinoma, and trichilemmoma.9
The associated syndrome, nevus sebaceous syndrome, also known as Schimmelpenning syndrome, has more extensive cutaneous lesions along Blaschko lines and presents with CNS, ocular, or skeletal defects. The CNS abnormalities include mental retardation, seizures, and hemimegalencephaly.1 Therefore, a thorough neurologic and ophthalmologic examination should be performed in patients with suspected nevus sebaceous syndrome.
Aplasia cutis congenita is the absence of the skin at birth that presents with an erosion or deep ulceration to a scar or a defect that is covered with an epidermal membrane.1 Some aplasia cutis may present as a smooth, hairless surface at birth, making differentiation from nevus sebaceous difficult. The pinkish, orange to yellow hue of NS may help differentiate these entities. Juvenile xanthogranuloma is a fairly common histiocytosis and is the most common histiocytic disease of childhood.1 It is a benign proliferation of dermal dendrocytes, and it presents in infants as many red to yellow papules or a few nodules on the head and neck. Many lesions even remain undetected.1
Syringocystadenoma papilliferum is a benign neoplasm with apocrine differentiation and presents as a papule or plaque on the head and neck. It can be associated with nevus sebaceous.1
Treatment
The definitive treatment is an excision. The necessity and timing of excision of these lesions is still under debate.9 Secondary neoplasms do arise from the nevus sebaceous, although the incidence is low pre puberty.1 It has been estimated that 16% of cases develop benign tumors and that 8% of cases develop malignant tumors.9 The majority of malignant lesions are basal cell carcinomas, and a large recent study found that only 1% of patients had malignancies.10 While most of these tumors develop in adulthood, there have been reports of malignancies in children.10
An additional reason to excise NS is that, over time, they grow more verrucous, become inflamed, bleed with trauma, and may be unappealing cosmetically.1 Some experts recommend earlier excision in childhood, especially with larger lesions or facial lesions where minimizing deformity is important and with the possibility of less noticeable scarring.9 The prophylactic removal of NS remains controversial, and there is a lack of consensus among experts about the timing of excision. It is recommended that each lesion be evaluated on a case-by-case basis.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Borok is a medical student at the University of California, Los Angeles. Dr. Eichenfield and Ms. Borok said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. “Dermatology.” 3rd ed. (St Louis, Mo: Elsevier, 2012).
2. Arch Dermatol Res. 1895. doi: 10.1007/BF01842810.
3. J Invest Dermatol. 2004 Jul;123(1):1-12.
4. J Cutan Pathol. 1984;11(5):396-414.
5. J Invest Dermatol. 2013 Mar;133(3):827-30.
6. J Invest Dermatol. 2013 Mar;133(3):597-600.
7. Nat Genet. 2012 Jun 10;44(7):783-7.
8. JAAD. 2016 Aug;75(2):420-7.
9. Pediatr Dermatol. 2012 Jan-Feb;29(1):15-23.
10. Pediatr Dermatol. 2009 Nov-Dec;26(6):676-81.
BY LAWRENCE F. EICHENFIELD, MD, and JENNA BOROK
Nevus sebaceous (NS) is considered a subtype of an epidermal nevus, a benign hamartoma that includes an excess or deficiency of structural elements of the skin, such as epidermis, sebaceous, and apocrine glands.1
Nevus sebaceous, also known as nevus sebaceous of Jadassohn, was first described in 1895 by Josef Jadassohn as a nevoid growth composed predominantly of sebaceous glands.2 Sebaceous glands are found everywhere on the body where hair is found and are located adjacent to hair follicles with ducts, through which sebum flows.3
NS clinically appears as a waxy, yellowish-orange to pink, hairless plaque, ranging from 1 cm to over 10 cm in size and usually located on the scalp, face, neck, or trunk.1 When the lesions are linear, they typically follow Blaschko lines.1 The lesions change over time. An infant’s lesion will be slightly raised and can be hardly noticeable. If the lesion is located on the scalp, it will remain hairless. During later childhood, the nevus may thicken. The lesions may become thicker and verrucous during adolescence.
Histologically, NS are benign hamartomas with epidermal, follicular, sebaceous, and apocrine elements.4 The malformation is primarily within the individual folliculosebaceous units.1 An infantile lesion will deviate little from normal skin as the follicular units are so small.1 During childhood, microscopically, it is easier to see misshapen follicles and thickened epidermis.1 During adolescence, the histological pattern is similar to that of an epidermal nevi, which includes acanthosis and fibroplasia of the papillary dermis.
There are now several lines of evidence showing that nevus sebaceous is caused by genetic postzygotic mosaic mutations in the mitogen–activated protein kinase (MAPK) pathway, which specifically activate ras mutations including H ras and K ras genes.5-7 Since NS is caused by a somatic mosaicism mutation, there are a several syndromic findings associated with NS depending on the timing of the mutations during development and whether single versus multiple tissues are affected.8 Specifically, NS has been associated with Schimmelpenning-Feuerstein-Mims syndrome that includes NS, and skeletal, ocular, and neurologic abnormalities.1,7 A study found that cutaneous skeletal hypophosphatemia syndrome, manifested as NS and vitamin D–resistant rickets, has identical ras mutations in both skin and bone tissues, providing further support that these syndromic findings are a result of a multilineage somatic ras mutation.8
Diagnosis
NS is typically diagnosed clinically, based on the presence of a circumscribed, thin, yellow-orange, oval, round, or linear plaque, usually on the scalp or face. During infancy or the first stage, the lesions remain stable. In the second stage, during puberty, the lesions thicken and become verrucous or nodular because of changes in sebaceous gland activity that are driven by hormones. In the third stage of their natural course, benign and malignant epithelial neoplasms can develop, including trichoblastoma, syringocystadenoma papilliferum, sebaceous epithelioma, basal cell carcinoma, and trichilemmoma.9
The associated syndrome, nevus sebaceous syndrome, also known as Schimmelpenning syndrome, has more extensive cutaneous lesions along Blaschko lines and presents with CNS, ocular, or skeletal defects. The CNS abnormalities include mental retardation, seizures, and hemimegalencephaly.1 Therefore, a thorough neurologic and ophthalmologic examination should be performed in patients with suspected nevus sebaceous syndrome.
Aplasia cutis congenita is the absence of the skin at birth that presents with an erosion or deep ulceration to a scar or a defect that is covered with an epidermal membrane.1 Some aplasia cutis may present as a smooth, hairless surface at birth, making differentiation from nevus sebaceous difficult. The pinkish, orange to yellow hue of NS may help differentiate these entities. Juvenile xanthogranuloma is a fairly common histiocytosis and is the most common histiocytic disease of childhood.1 It is a benign proliferation of dermal dendrocytes, and it presents in infants as many red to yellow papules or a few nodules on the head and neck. Many lesions even remain undetected.1
Syringocystadenoma papilliferum is a benign neoplasm with apocrine differentiation and presents as a papule or plaque on the head and neck. It can be associated with nevus sebaceous.1
Treatment
The definitive treatment is an excision. The necessity and timing of excision of these lesions is still under debate.9 Secondary neoplasms do arise from the nevus sebaceous, although the incidence is low pre puberty.1 It has been estimated that 16% of cases develop benign tumors and that 8% of cases develop malignant tumors.9 The majority of malignant lesions are basal cell carcinomas, and a large recent study found that only 1% of patients had malignancies.10 While most of these tumors develop in adulthood, there have been reports of malignancies in children.10
An additional reason to excise NS is that, over time, they grow more verrucous, become inflamed, bleed with trauma, and may be unappealing cosmetically.1 Some experts recommend earlier excision in childhood, especially with larger lesions or facial lesions where minimizing deformity is important and with the possibility of less noticeable scarring.9 The prophylactic removal of NS remains controversial, and there is a lack of consensus among experts about the timing of excision. It is recommended that each lesion be evaluated on a case-by-case basis.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Borok is a medical student at the University of California, Los Angeles. Dr. Eichenfield and Ms. Borok said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. “Dermatology.” 3rd ed. (St Louis, Mo: Elsevier, 2012).
2. Arch Dermatol Res. 1895. doi: 10.1007/BF01842810.
3. J Invest Dermatol. 2004 Jul;123(1):1-12.
4. J Cutan Pathol. 1984;11(5):396-414.
5. J Invest Dermatol. 2013 Mar;133(3):827-30.
6. J Invest Dermatol. 2013 Mar;133(3):597-600.
7. Nat Genet. 2012 Jun 10;44(7):783-7.
8. JAAD. 2016 Aug;75(2):420-7.
9. Pediatr Dermatol. 2012 Jan-Feb;29(1):15-23.
10. Pediatr Dermatol. 2009 Nov-Dec;26(6):676-81.
A 4-year-old boy presents with a yellow-orange lesion on his scalp. His mother states that it was present at birth and does not seem to bother him. The mother also complains that he is not growing hair over the bump area on his head. The bump has grown proportionally with the patient since birth.
He is otherwise healthy and has no other bumps like this one on his body. He was born at term with an unremarkable perinatal history.
During the physical exam, you find a yellow-orange, smooth, hairless plaque on the scalp. The plaque is 3 cm in size and is well circumscribed. The infant’s general physical, skin exam, and neurological exam are normal. He has no skeletal defects.
Pediatric Dermatology Consult - April 2017
BY JEREMY UDKOFF AND CATALINA MATIZ, MD
Juvenile xanthogranuloma (JXG), a non–Langerhans cell histiocytosis, is a common pediatric tumor that most commonly presents either at birth, in infants, or in young children – with the majority of cases occurring before 2 years. There is a male predominance with a 50% increased prevalence for solitary lesion disease and a 12 times higher prevalence in multilesion disease.1 Few lesions are concerning, and spontaneous regression of cutaneous lesions over the subsequent 1-5 years is a hallmark feature of JXG.2
Clinically, the JXGs are initially smooth, pinkish papules that may enlarge to 1-cm nodules and become yellowish in appearance before resolving to become atrophic macules or patches.2,3 JXGs are firm but rubbery and may become scaly and/or ulcerate as the lesion progresses.2 The JXGs most frequently occur superficially on the scalp and flexural areas of the upper extremities but infrequently present in the subcutaneous soft tissue, central nervous system, liver/spleen, eye/orbit, oropharynx, and muscle tissue.3,4 A well-described and concerning extracutaneous manifestations of JXG is ocular involvement and may be associated with bleeding into the anterior chamber of the eye. Despite this potentially disabling complication, screening for ocular involvement is recommended only in patients under age 2 years and in those with multiple skin lesions.5
The etiology of JXG is largely unknown. However, an association between neurofibromatosis type 1 (NF1) and the development of JXG and other diseases such as juvenile myelomonocytic leukemia, previously called juvenile chronic myelomonocytic leukemia, exists. It was thought that patients with NF1 and JXGs had a higher risk to develop juvenile myelomonocytic leukemia.6 However, a recent study showed that JXG alone does not appear to confer an increased risk for developing malignancy in children with NF1.6,7
Differential diagnosis and work-up
The clinical differential diagnosis for JXG includes dermatofibromas, Langerhans cell histiocytosis, mastocytosis, Spitz nevus, hemangioendothelioma, and other xanthomas. Because of the concerning nature of these look-alikes, equivocal cases should be referred to a pediatric dermatologist.
Although, altered laboratory values may be seen with systemic JXG with solid organ involvement, there are no systemic tests that can be used to determine if a cutaneous lesion is JXG. Thus, biopsy is the gold standard diagnostic for confirmatory testing. As a histiocytic disorder, JXG will display various macrophages on histologic examination. Additionally, one may observe a dense dermal infiltrate of vacuolated cells, along with wreathlike giant cells (Touton cells) and eosinophils.3 Although these Touton cells are thought to be pathognomonic of JXG, early lesions may lack these cells.8 Thus, their absence does not exclude the diagnosis of JXG. These are more serious cases, and the work-up conducted depends upon the organ system(s) involved. Systemic disease occurs in approximately 5% of patients.
Treatment and prognosis
Clinical monitoring is the only therapy required if there are only a few cutaneous JXGs present. However, systemic JXG is a concerning disease and various chemotherapy regimens have been recommended.3 Additionally, the use of a vinca alkaloid in conjunction with a steroid is associated with better outcomes than either of these agents alone.9 As a word of caution, in one study of 12 patients who received therapeutic systemic chemotherapy or radiation therapy to the brain, eye, skin, or heart, the patients had long-term disabilities and 2 patients died of their disease.4 In another study, children with systemic JXG, again, had a poor prognosis: Despite courses of multiagent chemotherapy, 2 of 17 patients died.9
Despite the poor results associated with systemic JXG, the vast majority of JXG patients have localized disease, which is associated with an excellent prognosis. The majority of these lesions spontaneously regress.
In conclusion, JXG is typically a benign, cutaneous disease. It presents in infants and children and involutes over a 1-5 year period. Lesions that are not classic for JXG should be referred to a pediatric dermatologist, and biopsy is the gold standard diagnosis. Most manifestations of JXG do not require therapy. However, systemic JXG may be difficult to treat and is associated with poor outcomes.
Dr. Catalina Matiz is assistant professor of dermatology at Rady Children’s Hospital-San Diego, associated with the University of California, San Diego. Jeremy Udkoff is a medical student at the university. Neither Dr. Matiz nor Mr. Udkoff have relevant financial disclosures.
References
1. Am J Surg Pathol. 2005 Jan;29(1):21-8.
2. Int J Dermatol. 2015 Oct;54(10):1109-23.
4. J Pediatr. 1996 Aug;129(2):227-37.
5. J Am Acad Dermatol. 1996 Mar;34(3):445-9.
6. J Am Acad Dermatol. 2017 Feb 8. pii: S0190-9622(16)31196-3.
7. Pediatr Dermatol. 2004 Mar-Apr;21(2):97-101.
BY JEREMY UDKOFF AND CATALINA MATIZ, MD
Juvenile xanthogranuloma (JXG), a non–Langerhans cell histiocytosis, is a common pediatric tumor that most commonly presents either at birth, in infants, or in young children – with the majority of cases occurring before 2 years. There is a male predominance with a 50% increased prevalence for solitary lesion disease and a 12 times higher prevalence in multilesion disease.1 Few lesions are concerning, and spontaneous regression of cutaneous lesions over the subsequent 1-5 years is a hallmark feature of JXG.2
Clinically, the JXGs are initially smooth, pinkish papules that may enlarge to 1-cm nodules and become yellowish in appearance before resolving to become atrophic macules or patches.2,3 JXGs are firm but rubbery and may become scaly and/or ulcerate as the lesion progresses.2 The JXGs most frequently occur superficially on the scalp and flexural areas of the upper extremities but infrequently present in the subcutaneous soft tissue, central nervous system, liver/spleen, eye/orbit, oropharynx, and muscle tissue.3,4 A well-described and concerning extracutaneous manifestations of JXG is ocular involvement and may be associated with bleeding into the anterior chamber of the eye. Despite this potentially disabling complication, screening for ocular involvement is recommended only in patients under age 2 years and in those with multiple skin lesions.5
The etiology of JXG is largely unknown. However, an association between neurofibromatosis type 1 (NF1) and the development of JXG and other diseases such as juvenile myelomonocytic leukemia, previously called juvenile chronic myelomonocytic leukemia, exists. It was thought that patients with NF1 and JXGs had a higher risk to develop juvenile myelomonocytic leukemia.6 However, a recent study showed that JXG alone does not appear to confer an increased risk for developing malignancy in children with NF1.6,7
Differential diagnosis and work-up
The clinical differential diagnosis for JXG includes dermatofibromas, Langerhans cell histiocytosis, mastocytosis, Spitz nevus, hemangioendothelioma, and other xanthomas. Because of the concerning nature of these look-alikes, equivocal cases should be referred to a pediatric dermatologist.
Although, altered laboratory values may be seen with systemic JXG with solid organ involvement, there are no systemic tests that can be used to determine if a cutaneous lesion is JXG. Thus, biopsy is the gold standard diagnostic for confirmatory testing. As a histiocytic disorder, JXG will display various macrophages on histologic examination. Additionally, one may observe a dense dermal infiltrate of vacuolated cells, along with wreathlike giant cells (Touton cells) and eosinophils.3 Although these Touton cells are thought to be pathognomonic of JXG, early lesions may lack these cells.8 Thus, their absence does not exclude the diagnosis of JXG. These are more serious cases, and the work-up conducted depends upon the organ system(s) involved. Systemic disease occurs in approximately 5% of patients.
Treatment and prognosis
Clinical monitoring is the only therapy required if there are only a few cutaneous JXGs present. However, systemic JXG is a concerning disease and various chemotherapy regimens have been recommended.3 Additionally, the use of a vinca alkaloid in conjunction with a steroid is associated with better outcomes than either of these agents alone.9 As a word of caution, in one study of 12 patients who received therapeutic systemic chemotherapy or radiation therapy to the brain, eye, skin, or heart, the patients had long-term disabilities and 2 patients died of their disease.4 In another study, children with systemic JXG, again, had a poor prognosis: Despite courses of multiagent chemotherapy, 2 of 17 patients died.9
Despite the poor results associated with systemic JXG, the vast majority of JXG patients have localized disease, which is associated with an excellent prognosis. The majority of these lesions spontaneously regress.
In conclusion, JXG is typically a benign, cutaneous disease. It presents in infants and children and involutes over a 1-5 year period. Lesions that are not classic for JXG should be referred to a pediatric dermatologist, and biopsy is the gold standard diagnosis. Most manifestations of JXG do not require therapy. However, systemic JXG may be difficult to treat and is associated with poor outcomes.
Dr. Catalina Matiz is assistant professor of dermatology at Rady Children’s Hospital-San Diego, associated with the University of California, San Diego. Jeremy Udkoff is a medical student at the university. Neither Dr. Matiz nor Mr. Udkoff have relevant financial disclosures.
References
1. Am J Surg Pathol. 2005 Jan;29(1):21-8.
2. Int J Dermatol. 2015 Oct;54(10):1109-23.
4. J Pediatr. 1996 Aug;129(2):227-37.
5. J Am Acad Dermatol. 1996 Mar;34(3):445-9.
6. J Am Acad Dermatol. 2017 Feb 8. pii: S0190-9622(16)31196-3.
7. Pediatr Dermatol. 2004 Mar-Apr;21(2):97-101.
BY JEREMY UDKOFF AND CATALINA MATIZ, MD
Juvenile xanthogranuloma (JXG), a non–Langerhans cell histiocytosis, is a common pediatric tumor that most commonly presents either at birth, in infants, or in young children – with the majority of cases occurring before 2 years. There is a male predominance with a 50% increased prevalence for solitary lesion disease and a 12 times higher prevalence in multilesion disease.1 Few lesions are concerning, and spontaneous regression of cutaneous lesions over the subsequent 1-5 years is a hallmark feature of JXG.2
Clinically, the JXGs are initially smooth, pinkish papules that may enlarge to 1-cm nodules and become yellowish in appearance before resolving to become atrophic macules or patches.2,3 JXGs are firm but rubbery and may become scaly and/or ulcerate as the lesion progresses.2 The JXGs most frequently occur superficially on the scalp and flexural areas of the upper extremities but infrequently present in the subcutaneous soft tissue, central nervous system, liver/spleen, eye/orbit, oropharynx, and muscle tissue.3,4 A well-described and concerning extracutaneous manifestations of JXG is ocular involvement and may be associated with bleeding into the anterior chamber of the eye. Despite this potentially disabling complication, screening for ocular involvement is recommended only in patients under age 2 years and in those with multiple skin lesions.5
The etiology of JXG is largely unknown. However, an association between neurofibromatosis type 1 (NF1) and the development of JXG and other diseases such as juvenile myelomonocytic leukemia, previously called juvenile chronic myelomonocytic leukemia, exists. It was thought that patients with NF1 and JXGs had a higher risk to develop juvenile myelomonocytic leukemia.6 However, a recent study showed that JXG alone does not appear to confer an increased risk for developing malignancy in children with NF1.6,7
Differential diagnosis and work-up
The clinical differential diagnosis for JXG includes dermatofibromas, Langerhans cell histiocytosis, mastocytosis, Spitz nevus, hemangioendothelioma, and other xanthomas. Because of the concerning nature of these look-alikes, equivocal cases should be referred to a pediatric dermatologist.
Although, altered laboratory values may be seen with systemic JXG with solid organ involvement, there are no systemic tests that can be used to determine if a cutaneous lesion is JXG. Thus, biopsy is the gold standard diagnostic for confirmatory testing. As a histiocytic disorder, JXG will display various macrophages on histologic examination. Additionally, one may observe a dense dermal infiltrate of vacuolated cells, along with wreathlike giant cells (Touton cells) and eosinophils.3 Although these Touton cells are thought to be pathognomonic of JXG, early lesions may lack these cells.8 Thus, their absence does not exclude the diagnosis of JXG. These are more serious cases, and the work-up conducted depends upon the organ system(s) involved. Systemic disease occurs in approximately 5% of patients.
Treatment and prognosis
Clinical monitoring is the only therapy required if there are only a few cutaneous JXGs present. However, systemic JXG is a concerning disease and various chemotherapy regimens have been recommended.3 Additionally, the use of a vinca alkaloid in conjunction with a steroid is associated with better outcomes than either of these agents alone.9 As a word of caution, in one study of 12 patients who received therapeutic systemic chemotherapy or radiation therapy to the brain, eye, skin, or heart, the patients had long-term disabilities and 2 patients died of their disease.4 In another study, children with systemic JXG, again, had a poor prognosis: Despite courses of multiagent chemotherapy, 2 of 17 patients died.9
Despite the poor results associated with systemic JXG, the vast majority of JXG patients have localized disease, which is associated with an excellent prognosis. The majority of these lesions spontaneously regress.
In conclusion, JXG is typically a benign, cutaneous disease. It presents in infants and children and involutes over a 1-5 year period. Lesions that are not classic for JXG should be referred to a pediatric dermatologist, and biopsy is the gold standard diagnosis. Most manifestations of JXG do not require therapy. However, systemic JXG may be difficult to treat and is associated with poor outcomes.
Dr. Catalina Matiz is assistant professor of dermatology at Rady Children’s Hospital-San Diego, associated with the University of California, San Diego. Jeremy Udkoff is a medical student at the university. Neither Dr. Matiz nor Mr. Udkoff have relevant financial disclosures.
References
1. Am J Surg Pathol. 2005 Jan;29(1):21-8.
2. Int J Dermatol. 2015 Oct;54(10):1109-23.
4. J Pediatr. 1996 Aug;129(2):227-37.
5. J Am Acad Dermatol. 1996 Mar;34(3):445-9.
6. J Am Acad Dermatol. 2017 Feb 8. pii: S0190-9622(16)31196-3.
7. Pediatr Dermatol. 2004 Mar-Apr;21(2):97-101.
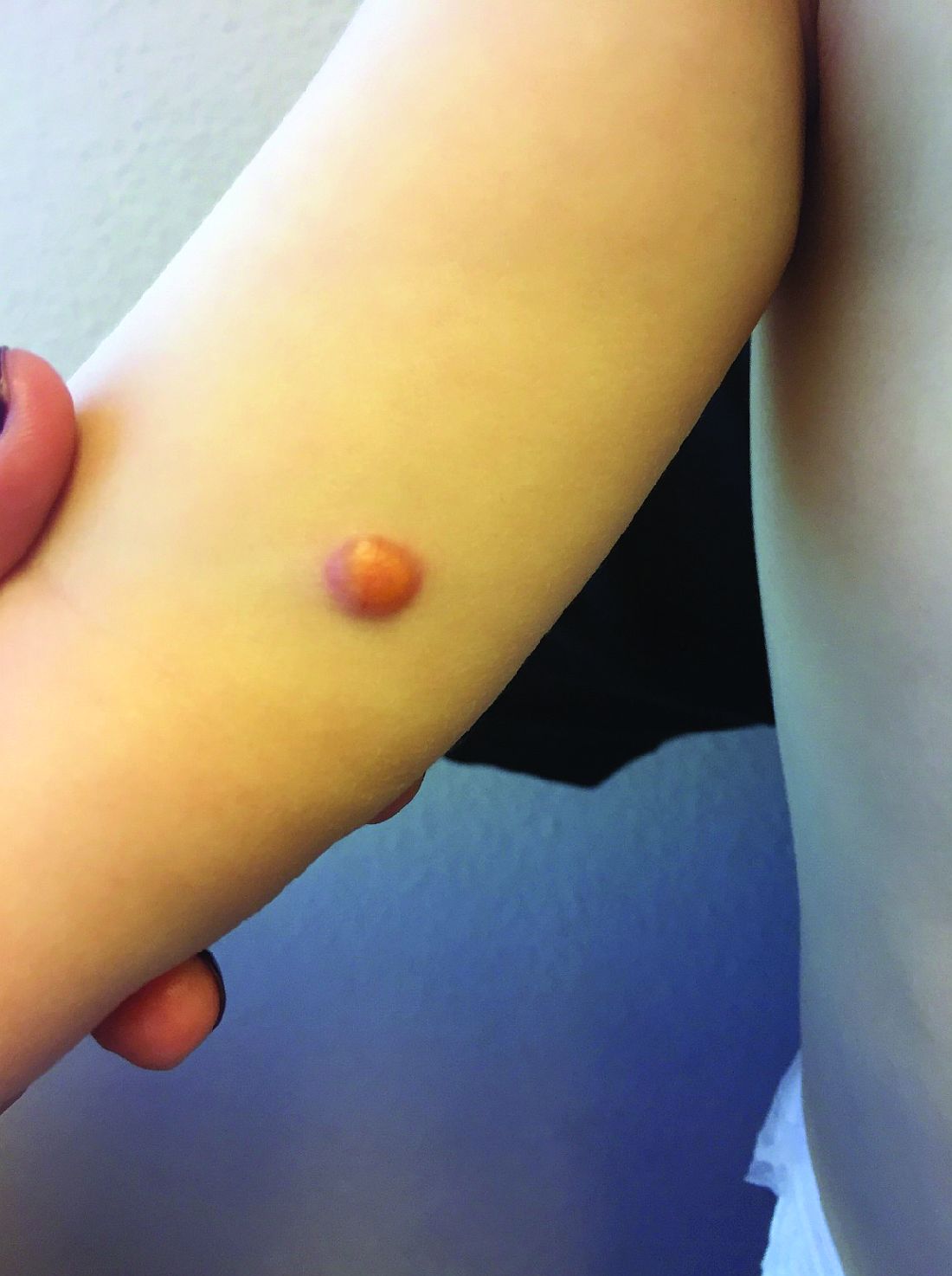
One month later, the patient returned to our clinic with an urgent concern. The patient accidentally scraped the lesion, and it bled heavily for 15 minutes before subsiding. The mother stated that the lesion grew rapidly since the prior visit and became painful. On repeat physical examination the lesion was a 1-cm fungiform, yellow to pink nodule, with central ulceration.
Pediatric Dermatology Consult - March 2017
By Jusleen Ahluwalia, MD, and Lawrence F. Eichenfield, MD
Subcutaneous fat necrosis of the newborn
The clinical history and morphology of our patient’s cutaneous manifestation is highly suggestive of subcutaneous fat necrosis of the newborn (SFN). SFN is a self-limited, lobular form of panniculitis that typically affects newborns at term or post term until the first 6 weeks of life.1 Delivery complications resulting in perinatal stress – including perinatal hypothermia, hypoxia, and birth trauma – have been associated with the development of SFN.2 Other risk factors include maternal disorders during pregnancy, such as diabetes, hypertension, exposure to tobacco, and thrombotic events.1,2 SFN has been noted after therapeutic hypothermia that is utilized to minimize neurologic effects of neonatal hypoxic ischemic encephalopathy.1
It has been hypothesized that SFN follows hypoxic injury to fat caused by local trauma, while in some cases it is proposed that SFN results from an imbalance of saturated and monounsaturated fats leading to crystallization at certain temperatures in the neonatal subcutis.1
The clinical presentation of SFN is characterized by the development of one to several erythematous violaceous subcutaneous nodules and plaques that can evolve into firm calcifications, and may be tender to palpation. They are characteristically located on the shoulders, back, and upper limbs. Spontaneous regression has been observed without scarring within 2-5 months; however, cutaneous atrophy of the affected areas can follow recovery from the condition.1-3
Hypercalcemia is a potentially fatal complication of SFN that infrequently occurs in a subset of patients. Its risk increases with the extent of perinatal injury and degree of fat necrosis.1 Several studies have documented a prevalence of hypercalcemia in 36%-56% of infants with SFN; however, most of these cases were mild and conservatively managed.2,4 Hypercalcemia may manifest without symptoms or present with vomiting, irritability, and seizures.1 Nephrocalcinosis can complicate high calcium levels and is detected by abdominal ultrasonography. Other complications that can accompany the development of SFN include thrombocytopenia, hyperglycemia, and hypertriglyceridemia, although this relationship is controversial as these conditions can be attributed to other neonatal disease or maternal factors.5
Differential diagnosis
Although SFN is generally a transient, self-limited condition, recognition of SFN is critical to monitor and avoid metabolic alterations associated with SFN. Sclerema neonatorum is another rare condition characterized by diffuse hardening of the skin affecting infants up to 4 months of age with severe underlying disease and systemic symptoms.1
Deep soft tissue infections, such as cellulitis, are usually accompanied with fever and other signs of infection.1 Mechanical trauma may induce firm, subcutaneous nodules in areas where fat is adjacent to bone and should be considered in any infant or child with subcutaneous nodules over areas prone to injury.1 Sudden withdrawal of systemic steroids can cause subcutaneous nodules typically located on cheeks, arms, and trunk.1
Management
Most infants with SFN are managed conservatively.1 Recently proposed guidelines for the management of SFN include weekly monitoring of calcium levels until 1 month of age and monthly until 6 months of age or after resolution of the cutaneous lesion, and more frequently if hypercalcemia is documented. Platelet count and creatinine, glucose, and triglyceride levels also should be assessed.5 At-risk infants should be evaluated for nephrocalcinosis with abdominal ultrasonagraphy.1,6 Treatment of hypercalcemia can consist of modification of diet with low levels of calcium and vitamin D, intravenous saline, calcium-wasting diuretics, or occasionally corticosteroids. Bisphosphonates also have been reported to successfully treat hypercalcemia in the setting of SFN.1,6
References
1. Disorders of the subcutaneous tissue, in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Saunders, 2015, p. 443-55).
2. Br J Dermatol. 2007 Apr;156(4):709-15.
3. An Bras Dermatol. 2013 Nov-Dec;88(6 Suppl 1):154-7.
4. Pediatr Dermatol. 1999 Sep-Oct;16(5):384-7.
5. Pediatr Dermatol. 2016 Nov;33(6):e353-5.
6. Arch Dis Child Fetal Neonatal Ed. 2014 Sep;99(5):F419-21.
Dr. Ahluwalia and Dr. Eichenfield are in the division of pediatric and adolescent dermatology, Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics, University of California, San Diego. They said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
By Jusleen Ahluwalia, MD, and Lawrence F. Eichenfield, MD
Subcutaneous fat necrosis of the newborn
The clinical history and morphology of our patient’s cutaneous manifestation is highly suggestive of subcutaneous fat necrosis of the newborn (SFN). SFN is a self-limited, lobular form of panniculitis that typically affects newborns at term or post term until the first 6 weeks of life.1 Delivery complications resulting in perinatal stress – including perinatal hypothermia, hypoxia, and birth trauma – have been associated with the development of SFN.2 Other risk factors include maternal disorders during pregnancy, such as diabetes, hypertension, exposure to tobacco, and thrombotic events.1,2 SFN has been noted after therapeutic hypothermia that is utilized to minimize neurologic effects of neonatal hypoxic ischemic encephalopathy.1
It has been hypothesized that SFN follows hypoxic injury to fat caused by local trauma, while in some cases it is proposed that SFN results from an imbalance of saturated and monounsaturated fats leading to crystallization at certain temperatures in the neonatal subcutis.1
The clinical presentation of SFN is characterized by the development of one to several erythematous violaceous subcutaneous nodules and plaques that can evolve into firm calcifications, and may be tender to palpation. They are characteristically located on the shoulders, back, and upper limbs. Spontaneous regression has been observed without scarring within 2-5 months; however, cutaneous atrophy of the affected areas can follow recovery from the condition.1-3
Hypercalcemia is a potentially fatal complication of SFN that infrequently occurs in a subset of patients. Its risk increases with the extent of perinatal injury and degree of fat necrosis.1 Several studies have documented a prevalence of hypercalcemia in 36%-56% of infants with SFN; however, most of these cases were mild and conservatively managed.2,4 Hypercalcemia may manifest without symptoms or present with vomiting, irritability, and seizures.1 Nephrocalcinosis can complicate high calcium levels and is detected by abdominal ultrasonography. Other complications that can accompany the development of SFN include thrombocytopenia, hyperglycemia, and hypertriglyceridemia, although this relationship is controversial as these conditions can be attributed to other neonatal disease or maternal factors.5
Differential diagnosis
Although SFN is generally a transient, self-limited condition, recognition of SFN is critical to monitor and avoid metabolic alterations associated with SFN. Sclerema neonatorum is another rare condition characterized by diffuse hardening of the skin affecting infants up to 4 months of age with severe underlying disease and systemic symptoms.1
Deep soft tissue infections, such as cellulitis, are usually accompanied with fever and other signs of infection.1 Mechanical trauma may induce firm, subcutaneous nodules in areas where fat is adjacent to bone and should be considered in any infant or child with subcutaneous nodules over areas prone to injury.1 Sudden withdrawal of systemic steroids can cause subcutaneous nodules typically located on cheeks, arms, and trunk.1
Management
Most infants with SFN are managed conservatively.1 Recently proposed guidelines for the management of SFN include weekly monitoring of calcium levels until 1 month of age and monthly until 6 months of age or after resolution of the cutaneous lesion, and more frequently if hypercalcemia is documented. Platelet count and creatinine, glucose, and triglyceride levels also should be assessed.5 At-risk infants should be evaluated for nephrocalcinosis with abdominal ultrasonagraphy.1,6 Treatment of hypercalcemia can consist of modification of diet with low levels of calcium and vitamin D, intravenous saline, calcium-wasting diuretics, or occasionally corticosteroids. Bisphosphonates also have been reported to successfully treat hypercalcemia in the setting of SFN.1,6
References
1. Disorders of the subcutaneous tissue, in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Saunders, 2015, p. 443-55).
2. Br J Dermatol. 2007 Apr;156(4):709-15.
3. An Bras Dermatol. 2013 Nov-Dec;88(6 Suppl 1):154-7.
4. Pediatr Dermatol. 1999 Sep-Oct;16(5):384-7.
5. Pediatr Dermatol. 2016 Nov;33(6):e353-5.
6. Arch Dis Child Fetal Neonatal Ed. 2014 Sep;99(5):F419-21.
Dr. Ahluwalia and Dr. Eichenfield are in the division of pediatric and adolescent dermatology, Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics, University of California, San Diego. They said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
By Jusleen Ahluwalia, MD, and Lawrence F. Eichenfield, MD
Subcutaneous fat necrosis of the newborn
The clinical history and morphology of our patient’s cutaneous manifestation is highly suggestive of subcutaneous fat necrosis of the newborn (SFN). SFN is a self-limited, lobular form of panniculitis that typically affects newborns at term or post term until the first 6 weeks of life.1 Delivery complications resulting in perinatal stress – including perinatal hypothermia, hypoxia, and birth trauma – have been associated with the development of SFN.2 Other risk factors include maternal disorders during pregnancy, such as diabetes, hypertension, exposure to tobacco, and thrombotic events.1,2 SFN has been noted after therapeutic hypothermia that is utilized to minimize neurologic effects of neonatal hypoxic ischemic encephalopathy.1
It has been hypothesized that SFN follows hypoxic injury to fat caused by local trauma, while in some cases it is proposed that SFN results from an imbalance of saturated and monounsaturated fats leading to crystallization at certain temperatures in the neonatal subcutis.1
The clinical presentation of SFN is characterized by the development of one to several erythematous violaceous subcutaneous nodules and plaques that can evolve into firm calcifications, and may be tender to palpation. They are characteristically located on the shoulders, back, and upper limbs. Spontaneous regression has been observed without scarring within 2-5 months; however, cutaneous atrophy of the affected areas can follow recovery from the condition.1-3
Hypercalcemia is a potentially fatal complication of SFN that infrequently occurs in a subset of patients. Its risk increases with the extent of perinatal injury and degree of fat necrosis.1 Several studies have documented a prevalence of hypercalcemia in 36%-56% of infants with SFN; however, most of these cases were mild and conservatively managed.2,4 Hypercalcemia may manifest without symptoms or present with vomiting, irritability, and seizures.1 Nephrocalcinosis can complicate high calcium levels and is detected by abdominal ultrasonography. Other complications that can accompany the development of SFN include thrombocytopenia, hyperglycemia, and hypertriglyceridemia, although this relationship is controversial as these conditions can be attributed to other neonatal disease or maternal factors.5
Differential diagnosis
Although SFN is generally a transient, self-limited condition, recognition of SFN is critical to monitor and avoid metabolic alterations associated with SFN. Sclerema neonatorum is another rare condition characterized by diffuse hardening of the skin affecting infants up to 4 months of age with severe underlying disease and systemic symptoms.1
Deep soft tissue infections, such as cellulitis, are usually accompanied with fever and other signs of infection.1 Mechanical trauma may induce firm, subcutaneous nodules in areas where fat is adjacent to bone and should be considered in any infant or child with subcutaneous nodules over areas prone to injury.1 Sudden withdrawal of systemic steroids can cause subcutaneous nodules typically located on cheeks, arms, and trunk.1
Management
Most infants with SFN are managed conservatively.1 Recently proposed guidelines for the management of SFN include weekly monitoring of calcium levels until 1 month of age and monthly until 6 months of age or after resolution of the cutaneous lesion, and more frequently if hypercalcemia is documented. Platelet count and creatinine, glucose, and triglyceride levels also should be assessed.5 At-risk infants should be evaluated for nephrocalcinosis with abdominal ultrasonagraphy.1,6 Treatment of hypercalcemia can consist of modification of diet with low levels of calcium and vitamin D, intravenous saline, calcium-wasting diuretics, or occasionally corticosteroids. Bisphosphonates also have been reported to successfully treat hypercalcemia in the setting of SFN.1,6
References
1. Disorders of the subcutaneous tissue, in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Saunders, 2015, p. 443-55).
2. Br J Dermatol. 2007 Apr;156(4):709-15.
3. An Bras Dermatol. 2013 Nov-Dec;88(6 Suppl 1):154-7.
4. Pediatr Dermatol. 1999 Sep-Oct;16(5):384-7.
5. Pediatr Dermatol. 2016 Nov;33(6):e353-5.
6. Arch Dis Child Fetal Neonatal Ed. 2014 Sep;99(5):F419-21.
Dr. Ahluwalia and Dr. Eichenfield are in the division of pediatric and adolescent dermatology, Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics, University of California, San Diego. They said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
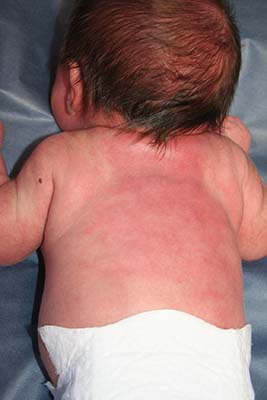
An 8.8 pound boy was born to a 29-year-old healthy mother at full term by emergency cesarean section secondary to fetal distress. Apgar scores were 4 and 6 at 1 and 5 minutes, respectively. Pregnancy was otherwise uncomplicated. The infant’s postnatal course was complicated by sepsis requiring intravenous antibiotics. The infant was discharged home after 1 week of hospitalization. At 3 weeks of life, the infant developed a red, firm, woody area on the back that did not appear painful and did not appear to spread in 2 days of observation. There was no fever. He was referred to the dermatology clinic for evaluation.
Physical exam showed an afebrile, well-appearing infant with an 11.5-cm by 7-cm red, indurated, ill-defined plaque overlying his back. The area was nontender and nonfluctuant. Complete blood count, comprehensive metabolic panel, and inflammatory markers were within normal limits.
Pediatric Dermatology Consult - February 2017
Diagnosing CARP
BY CATALINA MATIZ, MD, AND ANDREA WALDMAN, MD
Frontline Medical News
The distribution and morphology of our patient’s cutaneous eruption was highly suggestive of confluent and reticulated papillomatosis (CARP) of Gougerot and Carteaud, a rare dermatologic disorder characterized by epidermal changes (hyperkeratosis, scaling) and hyperpigmentation. The lesions classically begin as brown or red hyperpigmented hyperkeratotic or verrucous papules that spread centrifugally, coalescing into confluent plaques with reticulation at the periphery.1 CARP also may manifest as shiny papules or atrophic macules.1 Reports of scaly papules with rippling also are documented in the literature.2 Characteristically, the lesions frequently involve the inframammary region and the posterior trunk. Other less common sites of presentation include the abdomen, neck, axilla, face, and shoulders.1
Pathogenically, CARP results from disordered keratinization of the epidermis and increased melanosomes within the stratum corneum. The condition is chronic in nature and often intermittent, with a remitting-relapsing course. CARP is limited to cutaneous involvement, with no systemic manifestations. The lesions are typically asymptomatic; however, some individuals complain of mild pruritus.1 Primarily seen in adolescent patients, this dermatitis afflicts females twice as often as males, and occurs in all races.1
Several hypotheses concerning the etiology of CARP have been suggested in the literature; however, the definitive pathophysiology remains unknown. Suggested causes include defective keratinization, and infectious, endocrine, genetic, and environmental etiologies.3 In light of its clinical similarity to tinea versicolor and occasional clinical response to antifungal therapies, Malassezia has been postulated as a potential etiology for CARP. Potassium hydroxide examinations of the skin for fungal species are negative in most patients, and thus, this suggestion remains highly controversial.1 More convincing infectious culprits implicated include bacterial species such as Rhodococcus and Dietzia, papillomatosis previously isolated from skin scrapings of CARP patients. This is further supported by the successful treatment of CARP with antibiotic therapy.4 A highly controversial association between CARP and endocrine abnormalities such as insulin resistance and thyroid disease is well documented in the literature. Acanthosis nigricans and obesity may present concurrently with CARP as well; however, in most cases this association does not occur.3
The diagnosis of CARP is primarily clinical, formulated based on the presence of cutaneous lesions with the characteristic distribution and morphology. Biopsy and histopathology often are utilized to exclude other diagnoses. Davis et al. previously proposed the following five diagnostic criteria for CARP: involvement of the upper trunk and neck, clinical findings of scaly brown macules and patches (at least a portion of which appear reticulated and papillomatous), negative fungal staining of lesions, lack of response to antifungal therapy, and excellent response to minocycline therapy.5 These diagnostic criteria were validated by a retrospective analysis of CARP patients in Singapore.6
An extensive variety of cutaneous conditions bearing a resemblance to CARP were considered in the differential diagnosis, including acanthosis nigricans (AN), tinea versicolor, Darier disease, terra firma-forme dermatosis, prurigo pigmentosa, flagellate dermatosis, and dyskeratosis congenita.1 A common challenge for practitioners is distinguishing CARP from similar dermatoses, particularly AN. Differentiating AN from CARP cannot be accomplished based on the history of insulin resistance or increased body mass index alone, as these comorbidities may coexist with CARP.3 The clinical presence of reticulation peripherally, combined with a positive response to minocycline or azithromycin therapy, distinguish CARP from AN in most cases.
Other disorders commonly presenting in a similar distribution to CARP were further excluded based on the morphologic appearance of the lesions and further testing. Tinea versicolor was unlikely, because of the lack of response to antifungal agents and the absence of organisms on KOH examination. Furthermore, the morphologic characteristics of our patient’s lesions were inconsistent with the typical appearance of tinea versicolor – hypopigmented or hyperpigmented patches with overlying scale. Reticulated hyperpigmentation without papules or plaques may occur in dyskeratosis congenita.1 If the patient presents with reticulated hyperpigmentation in addition to pruritus, prurigo pigmentosa may be considered in the differential.4 The presentation of slightly verrucous “dirtlike” plaques refractory to cleansing with soap and water alone, should prompt the consideration of terra firma-forme dermatosis. Terra firma-forme dermatosis, a benign disorder of keratinization, may be confirmed at the bedside upon the removal of the plaques with a 70% alcohol swab.7 Flagellate erythema, characterized by the presence of linear streaks of erythema or brown pigmentation, often presents in a similar distribution to CARP. This condition occurs mostly in individuals treated with bleomycin or other chemotherapeutic agents, and spontaneously resolves.1 Darier disease classically presents as hyperkeratotic papules coalescing into plaques in seborrheic areas, and may manifest in the same distribution as CARP. Palmoplantar pits and nail changes (blue-red striations and distal V-shaped nicking) may be present in the former.1
A variety of treatments for CARP are documented in the literature, including antibiotic therapy with minocycline or azithromycin, topical salicylic acid, urea or lactic acid–containing emollients, and oral or topical retinoids.1,3 A substantial proportion of patients respond to oral minocycline (100 mg twice daily). At the time of initial evaluation, our patient was prescribed a 6-week course of systemic minocycline (100 mg twice daily), with subsequent improvement.
References
1. Am J Clin Dermatol. 2006;7(5):305-13.
2. Postepy Dermatol Alergol. 2014 Oct;31(5):335-7.
4. Br J Dermatol. 2006 Feb;154(2):287-93.
5. Ann Dermatol. 2014 Jun;26(3):409-10.
6. Am J Clin Dermatol. 2015 Apr;16(2):131-6.
7. Clinical Experiment Dermatol. 2012;37(4):446-7.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego, University of California, San Diego. Dr. Waldman is a clinical research fellow at the hospital. Dr. Matiz and Dr. Waldman said they had no relevant financial disclosures.
Diagnosing CARP
BY CATALINA MATIZ, MD, AND ANDREA WALDMAN, MD
Frontline Medical News
The distribution and morphology of our patient’s cutaneous eruption was highly suggestive of confluent and reticulated papillomatosis (CARP) of Gougerot and Carteaud, a rare dermatologic disorder characterized by epidermal changes (hyperkeratosis, scaling) and hyperpigmentation. The lesions classically begin as brown or red hyperpigmented hyperkeratotic or verrucous papules that spread centrifugally, coalescing into confluent plaques with reticulation at the periphery.1 CARP also may manifest as shiny papules or atrophic macules.1 Reports of scaly papules with rippling also are documented in the literature.2 Characteristically, the lesions frequently involve the inframammary region and the posterior trunk. Other less common sites of presentation include the abdomen, neck, axilla, face, and shoulders.1
Pathogenically, CARP results from disordered keratinization of the epidermis and increased melanosomes within the stratum corneum. The condition is chronic in nature and often intermittent, with a remitting-relapsing course. CARP is limited to cutaneous involvement, with no systemic manifestations. The lesions are typically asymptomatic; however, some individuals complain of mild pruritus.1 Primarily seen in adolescent patients, this dermatitis afflicts females twice as often as males, and occurs in all races.1
Several hypotheses concerning the etiology of CARP have been suggested in the literature; however, the definitive pathophysiology remains unknown. Suggested causes include defective keratinization, and infectious, endocrine, genetic, and environmental etiologies.3 In light of its clinical similarity to tinea versicolor and occasional clinical response to antifungal therapies, Malassezia has been postulated as a potential etiology for CARP. Potassium hydroxide examinations of the skin for fungal species are negative in most patients, and thus, this suggestion remains highly controversial.1 More convincing infectious culprits implicated include bacterial species such as Rhodococcus and Dietzia, papillomatosis previously isolated from skin scrapings of CARP patients. This is further supported by the successful treatment of CARP with antibiotic therapy.4 A highly controversial association between CARP and endocrine abnormalities such as insulin resistance and thyroid disease is well documented in the literature. Acanthosis nigricans and obesity may present concurrently with CARP as well; however, in most cases this association does not occur.3
The diagnosis of CARP is primarily clinical, formulated based on the presence of cutaneous lesions with the characteristic distribution and morphology. Biopsy and histopathology often are utilized to exclude other diagnoses. Davis et al. previously proposed the following five diagnostic criteria for CARP: involvement of the upper trunk and neck, clinical findings of scaly brown macules and patches (at least a portion of which appear reticulated and papillomatous), negative fungal staining of lesions, lack of response to antifungal therapy, and excellent response to minocycline therapy.5 These diagnostic criteria were validated by a retrospective analysis of CARP patients in Singapore.6
An extensive variety of cutaneous conditions bearing a resemblance to CARP were considered in the differential diagnosis, including acanthosis nigricans (AN), tinea versicolor, Darier disease, terra firma-forme dermatosis, prurigo pigmentosa, flagellate dermatosis, and dyskeratosis congenita.1 A common challenge for practitioners is distinguishing CARP from similar dermatoses, particularly AN. Differentiating AN from CARP cannot be accomplished based on the history of insulin resistance or increased body mass index alone, as these comorbidities may coexist with CARP.3 The clinical presence of reticulation peripherally, combined with a positive response to minocycline or azithromycin therapy, distinguish CARP from AN in most cases.
Other disorders commonly presenting in a similar distribution to CARP were further excluded based on the morphologic appearance of the lesions and further testing. Tinea versicolor was unlikely, because of the lack of response to antifungal agents and the absence of organisms on KOH examination. Furthermore, the morphologic characteristics of our patient’s lesions were inconsistent with the typical appearance of tinea versicolor – hypopigmented or hyperpigmented patches with overlying scale. Reticulated hyperpigmentation without papules or plaques may occur in dyskeratosis congenita.1 If the patient presents with reticulated hyperpigmentation in addition to pruritus, prurigo pigmentosa may be considered in the differential.4 The presentation of slightly verrucous “dirtlike” plaques refractory to cleansing with soap and water alone, should prompt the consideration of terra firma-forme dermatosis. Terra firma-forme dermatosis, a benign disorder of keratinization, may be confirmed at the bedside upon the removal of the plaques with a 70% alcohol swab.7 Flagellate erythema, characterized by the presence of linear streaks of erythema or brown pigmentation, often presents in a similar distribution to CARP. This condition occurs mostly in individuals treated with bleomycin or other chemotherapeutic agents, and spontaneously resolves.1 Darier disease classically presents as hyperkeratotic papules coalescing into plaques in seborrheic areas, and may manifest in the same distribution as CARP. Palmoplantar pits and nail changes (blue-red striations and distal V-shaped nicking) may be present in the former.1
A variety of treatments for CARP are documented in the literature, including antibiotic therapy with minocycline or azithromycin, topical salicylic acid, urea or lactic acid–containing emollients, and oral or topical retinoids.1,3 A substantial proportion of patients respond to oral minocycline (100 mg twice daily). At the time of initial evaluation, our patient was prescribed a 6-week course of systemic minocycline (100 mg twice daily), with subsequent improvement.
References
1. Am J Clin Dermatol. 2006;7(5):305-13.
2. Postepy Dermatol Alergol. 2014 Oct;31(5):335-7.
4. Br J Dermatol. 2006 Feb;154(2):287-93.
5. Ann Dermatol. 2014 Jun;26(3):409-10.
6. Am J Clin Dermatol. 2015 Apr;16(2):131-6.
7. Clinical Experiment Dermatol. 2012;37(4):446-7.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego, University of California, San Diego. Dr. Waldman is a clinical research fellow at the hospital. Dr. Matiz and Dr. Waldman said they had no relevant financial disclosures.
Diagnosing CARP
BY CATALINA MATIZ, MD, AND ANDREA WALDMAN, MD
Frontline Medical News
The distribution and morphology of our patient’s cutaneous eruption was highly suggestive of confluent and reticulated papillomatosis (CARP) of Gougerot and Carteaud, a rare dermatologic disorder characterized by epidermal changes (hyperkeratosis, scaling) and hyperpigmentation. The lesions classically begin as brown or red hyperpigmented hyperkeratotic or verrucous papules that spread centrifugally, coalescing into confluent plaques with reticulation at the periphery.1 CARP also may manifest as shiny papules or atrophic macules.1 Reports of scaly papules with rippling also are documented in the literature.2 Characteristically, the lesions frequently involve the inframammary region and the posterior trunk. Other less common sites of presentation include the abdomen, neck, axilla, face, and shoulders.1
Pathogenically, CARP results from disordered keratinization of the epidermis and increased melanosomes within the stratum corneum. The condition is chronic in nature and often intermittent, with a remitting-relapsing course. CARP is limited to cutaneous involvement, with no systemic manifestations. The lesions are typically asymptomatic; however, some individuals complain of mild pruritus.1 Primarily seen in adolescent patients, this dermatitis afflicts females twice as often as males, and occurs in all races.1
Several hypotheses concerning the etiology of CARP have been suggested in the literature; however, the definitive pathophysiology remains unknown. Suggested causes include defective keratinization, and infectious, endocrine, genetic, and environmental etiologies.3 In light of its clinical similarity to tinea versicolor and occasional clinical response to antifungal therapies, Malassezia has been postulated as a potential etiology for CARP. Potassium hydroxide examinations of the skin for fungal species are negative in most patients, and thus, this suggestion remains highly controversial.1 More convincing infectious culprits implicated include bacterial species such as Rhodococcus and Dietzia, papillomatosis previously isolated from skin scrapings of CARP patients. This is further supported by the successful treatment of CARP with antibiotic therapy.4 A highly controversial association between CARP and endocrine abnormalities such as insulin resistance and thyroid disease is well documented in the literature. Acanthosis nigricans and obesity may present concurrently with CARP as well; however, in most cases this association does not occur.3
The diagnosis of CARP is primarily clinical, formulated based on the presence of cutaneous lesions with the characteristic distribution and morphology. Biopsy and histopathology often are utilized to exclude other diagnoses. Davis et al. previously proposed the following five diagnostic criteria for CARP: involvement of the upper trunk and neck, clinical findings of scaly brown macules and patches (at least a portion of which appear reticulated and papillomatous), negative fungal staining of lesions, lack of response to antifungal therapy, and excellent response to minocycline therapy.5 These diagnostic criteria were validated by a retrospective analysis of CARP patients in Singapore.6
An extensive variety of cutaneous conditions bearing a resemblance to CARP were considered in the differential diagnosis, including acanthosis nigricans (AN), tinea versicolor, Darier disease, terra firma-forme dermatosis, prurigo pigmentosa, flagellate dermatosis, and dyskeratosis congenita.1 A common challenge for practitioners is distinguishing CARP from similar dermatoses, particularly AN. Differentiating AN from CARP cannot be accomplished based on the history of insulin resistance or increased body mass index alone, as these comorbidities may coexist with CARP.3 The clinical presence of reticulation peripherally, combined with a positive response to minocycline or azithromycin therapy, distinguish CARP from AN in most cases.
Other disorders commonly presenting in a similar distribution to CARP were further excluded based on the morphologic appearance of the lesions and further testing. Tinea versicolor was unlikely, because of the lack of response to antifungal agents and the absence of organisms on KOH examination. Furthermore, the morphologic characteristics of our patient’s lesions were inconsistent with the typical appearance of tinea versicolor – hypopigmented or hyperpigmented patches with overlying scale. Reticulated hyperpigmentation without papules or plaques may occur in dyskeratosis congenita.1 If the patient presents with reticulated hyperpigmentation in addition to pruritus, prurigo pigmentosa may be considered in the differential.4 The presentation of slightly verrucous “dirtlike” plaques refractory to cleansing with soap and water alone, should prompt the consideration of terra firma-forme dermatosis. Terra firma-forme dermatosis, a benign disorder of keratinization, may be confirmed at the bedside upon the removal of the plaques with a 70% alcohol swab.7 Flagellate erythema, characterized by the presence of linear streaks of erythema or brown pigmentation, often presents in a similar distribution to CARP. This condition occurs mostly in individuals treated with bleomycin or other chemotherapeutic agents, and spontaneously resolves.1 Darier disease classically presents as hyperkeratotic papules coalescing into plaques in seborrheic areas, and may manifest in the same distribution as CARP. Palmoplantar pits and nail changes (blue-red striations and distal V-shaped nicking) may be present in the former.1
A variety of treatments for CARP are documented in the literature, including antibiotic therapy with minocycline or azithromycin, topical salicylic acid, urea or lactic acid–containing emollients, and oral or topical retinoids.1,3 A substantial proportion of patients respond to oral minocycline (100 mg twice daily). At the time of initial evaluation, our patient was prescribed a 6-week course of systemic minocycline (100 mg twice daily), with subsequent improvement.
References
1. Am J Clin Dermatol. 2006;7(5):305-13.
2. Postepy Dermatol Alergol. 2014 Oct;31(5):335-7.
4. Br J Dermatol. 2006 Feb;154(2):287-93.
5. Ann Dermatol. 2014 Jun;26(3):409-10.
6. Am J Clin Dermatol. 2015 Apr;16(2):131-6.
7. Clinical Experiment Dermatol. 2012;37(4):446-7.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego, University of California, San Diego. Dr. Waldman is a clinical research fellow at the hospital. Dr. Matiz and Dr. Waldman said they had no relevant financial disclosures.

Clinical presentation
A 14-year-old previously healthy female presents to the dermatology clinic for evaluation of a persistent dermatitis on the trunk and abdomen. The patient reports the development of two distinct lesions covering the back and abdomen 6 months prior to initial presentation. The patient denies pruritus, pain, burning, or xerosis. Shortly after the lesions erupted, the pediatrician diagnosed her with tinea versicolor. The patient’s rash failed to improve over the following 4 months, despite the intermittent use of clotrimazole 1% cream and ketoconazole 2% cream. The patient denies any recent travel or pets residing in the home. The family history is noncontributory.
Physical exam
The patient is a well-appearing nonobese teenage female who is in no acute distress, with normal affect. On skin examination, there are confluent tan-brown papules coalescing into plaques involving the back and abdomen, with a reticulated appearance around the periphery of the plaques. There are no other lesions present on the cutaneous surface. The patient is afebrile, and vital signs are within normal limits.
Vascular anomalies often misdiagnosed amidst confusion
CHICAGO – Thanks to convoluted terminology, not to mention confusion in the literature, physicians have been known to frequently misdiagnose vascular malformations as hemangiomas, but an evolving understanding of their differences may lead to more precise diagnoses, according to a report at a symposium on vascular surgery sponsored by Northwestern University.
“Historically there has been a great deal of confusion in the literature when it comes to the nomenclature used to describe vascular anomalies,” said Naiem Nassiri, MD, of Robert Wood Johnson Medical School, New Brunswick, N.J. He pointed out that the term hemangioma “or derivatives thereof” – cavernous hemangioma, cavernous angioma, lymphangioma and cystic hygroma – are “absolute misnomers and continue to be misused and applied almost haphazardly to any anomalous vascular lesion.”

He cited reports that 71% of vascular anomalies have been improperly called hemangiomas, 69% have initially been diagnosed incorrectly, and 21% received the wrong treatment (Pediatr Dermatol. 2008;25[1]:7-12; Plast Reconstr Surg. 2011:127[1]:347-51). “Erroneous terminology has prognostic as well as diagnostic and therapeutic implications, and these can actually be quite devastating for the patient, not only clinically and physically but psychologically as well,” Dr. Nassiri said.
Using the International Society for the Study of Vascular Anomalies classification for hemangiomas and vascular malformations can help physicians make the differential diagnosis, Dr. Nassiri said. Hemangiomas are neoplastic lesions of infancy, though not always congenital, with a finite growth phase, whereas vascular malformations (VMs) are nonneoplastic, congenital lesions that can appear at any age and do not regress spontaneously, he said.
Infantile hemangiomas typically appear as the classic strawberry birthmark in children, whereas VMs tend to appear later in life. “They require some environmental trigger, such as trauma, activity, or changes in the hormonal milieu to manifest onset,” he said of VMs.
Simply put, VMs fall into three broad categories: slow-flow malformations, which include lymphatic and venous malformations; high-flow arteriovenous malformations (AVMs) and fistulas; and congenital mixed syndromes, which can include combinations thereof.
Dr. Nassiri noted that contrast-enhanced MRI is the standard imaging modality for diagnosis of VMs, and can differentiate between slow-flow and high-flow lesions. However, vascular specialists must be vigilant in ordering imaging for slow-flow lesions. “Orders can be changed to MR venography, and I’ve had patients who’ve gone decades with multiple MR venograms and no one can figure out what’s going on as no identifiable lesion is readily detected,” he said. “MR venograms are fantastic for detecting truncular blood flow where there typically are no anomalies in the vast majority of patients with isolated venous malformations, but on contrast-enhanced MRI these convoluted cluster of anomalous veins light up like Christmas trees.”
Lymphatic malformations affect the head and neck more so than the extremities, trunk or viscera, and are prone to infection and bleeding. “You can think of these as fluid-filled balloons, and the goal of treatment is fairly simple: You want to puncture the balloon and drain the fluid inside so as to obtain maximum wall collapse,” Dr. Nassiri said. Infusion of a sclerosant causes an inflammatory reaction leading to fibrosis, which then prevents balloon re-expansion. Surgical excision is best used as a secondary adjunct.
Venous malformations, comprising about 80% of all VMs, typically present as soft, spongy blue or purple compressible masses with associated pain that worsens with exertion, Dr. Nassiri said. “The most dangerous thing that is often overlooked, even by some of the physicians that treat these on a regular basis, is localized intravascular coagulopathy, which if left untreated can progress to fulminant disseminated intravascular coagulopathy,” he said. This tends to occur more in the more widespread varieties of venous malformations.
A common misnomer associated with venous malformations in adults is “liver hemangioma,” owing to the confusing nomenclature, Dr. Nassiri said. “When interrogated angiographically,” he said, “what is often labeled as a hepatic hemangioma is in fact a venous malformation. Natural history of the two entities is completely different.”
Dr. Nassiri described congenital high-flow AVMs as “convoluted networks of blood vessels with poorly differentiated endothelial cells that have neither a venous nor an arterial designation; this entity, otherwise known as a nidus, sits between the feeding arteries and the draining veins.” Treatment aims to eliminate the flow within that nidus.
Super-selective microcatheterization is the best option for nidus access and embolization using liquid embolic agents, preferably those that polymerize when infused. “This is probably the most potent angiogenic entity I’ve ever seen,” Dr. Nassiri said of the nidus.
“It’s like a low-pressure sump and it will recruit collaterals vigorously, so you have to eliminate that nidus.” A variety of different embolic agents, some off label, may be used for high flow AVMs.
For congenital mixed syndromes, the same diagnostic and therapeutic concepts hold true depending on the type of VM involved. Dr. Nassiri advised a multidisciplinary approach, and noted that early trials have investigated the use of sirolimus in severe, life-threatening cases (Br J Clin Pharmacol. 2016;82[5]:1171-9. doi: 10.1111/bcp.13022).
Dr. Nassiri disclosed serving on the speakers bureaus for Boston Scientific, Penumbra, and Merritt Medical, and is a consultant to Merritt Medical.
CHICAGO – Thanks to convoluted terminology, not to mention confusion in the literature, physicians have been known to frequently misdiagnose vascular malformations as hemangiomas, but an evolving understanding of their differences may lead to more precise diagnoses, according to a report at a symposium on vascular surgery sponsored by Northwestern University.
“Historically there has been a great deal of confusion in the literature when it comes to the nomenclature used to describe vascular anomalies,” said Naiem Nassiri, MD, of Robert Wood Johnson Medical School, New Brunswick, N.J. He pointed out that the term hemangioma “or derivatives thereof” – cavernous hemangioma, cavernous angioma, lymphangioma and cystic hygroma – are “absolute misnomers and continue to be misused and applied almost haphazardly to any anomalous vascular lesion.”
He cited reports that 71% of vascular anomalies have been improperly called hemangiomas, 69% have initially been diagnosed incorrectly, and 21% received the wrong treatment (Pediatr Dermatol. 2008;25[1]:7-12; Plast Reconstr Surg. 2011:127[1]:347-51). “Erroneous terminology has prognostic as well as diagnostic and therapeutic implications, and these can actually be quite devastating for the patient, not only clinically and physically but psychologically as well,” Dr. Nassiri said.
Using the International Society for the Study of Vascular Anomalies classification for hemangiomas and vascular malformations can help physicians make the differential diagnosis, Dr. Nassiri said. Hemangiomas are neoplastic lesions of infancy, though not always congenital, with a finite growth phase, whereas vascular malformations (VMs) are nonneoplastic, congenital lesions that can appear at any age and do not regress spontaneously, he said.
Infantile hemangiomas typically appear as the classic strawberry birthmark in children, whereas VMs tend to appear later in life. “They require some environmental trigger, such as trauma, activity, or changes in the hormonal milieu to manifest onset,” he said of VMs.
Simply put, VMs fall into three broad categories: slow-flow malformations, which include lymphatic and venous malformations; high-flow arteriovenous malformations (AVMs) and fistulas; and congenital mixed syndromes, which can include combinations thereof.
Dr. Nassiri noted that contrast-enhanced MRI is the standard imaging modality for diagnosis of VMs, and can differentiate between slow-flow and high-flow lesions. However, vascular specialists must be vigilant in ordering imaging for slow-flow lesions. “Orders can be changed to MR venography, and I’ve had patients who’ve gone decades with multiple MR venograms and no one can figure out what’s going on as no identifiable lesion is readily detected,” he said. “MR venograms are fantastic for detecting truncular blood flow where there typically are no anomalies in the vast majority of patients with isolated venous malformations, but on contrast-enhanced MRI these convoluted cluster of anomalous veins light up like Christmas trees.”
Lymphatic malformations affect the head and neck more so than the extremities, trunk or viscera, and are prone to infection and bleeding. “You can think of these as fluid-filled balloons, and the goal of treatment is fairly simple: You want to puncture the balloon and drain the fluid inside so as to obtain maximum wall collapse,” Dr. Nassiri said. Infusion of a sclerosant causes an inflammatory reaction leading to fibrosis, which then prevents balloon re-expansion. Surgical excision is best used as a secondary adjunct.
Venous malformations, comprising about 80% of all VMs, typically present as soft, spongy blue or purple compressible masses with associated pain that worsens with exertion, Dr. Nassiri said. “The most dangerous thing that is often overlooked, even by some of the physicians that treat these on a regular basis, is localized intravascular coagulopathy, which if left untreated can progress to fulminant disseminated intravascular coagulopathy,” he said. This tends to occur more in the more widespread varieties of venous malformations.
A common misnomer associated with venous malformations in adults is “liver hemangioma,” owing to the confusing nomenclature, Dr. Nassiri said. “When interrogated angiographically,” he said, “what is often labeled as a hepatic hemangioma is in fact a venous malformation. Natural history of the two entities is completely different.”
Dr. Nassiri described congenital high-flow AVMs as “convoluted networks of blood vessels with poorly differentiated endothelial cells that have neither a venous nor an arterial designation; this entity, otherwise known as a nidus, sits between the feeding arteries and the draining veins.” Treatment aims to eliminate the flow within that nidus.
Super-selective microcatheterization is the best option for nidus access and embolization using liquid embolic agents, preferably those that polymerize when infused. “This is probably the most potent angiogenic entity I’ve ever seen,” Dr. Nassiri said of the nidus.
“It’s like a low-pressure sump and it will recruit collaterals vigorously, so you have to eliminate that nidus.” A variety of different embolic agents, some off label, may be used for high flow AVMs.
For congenital mixed syndromes, the same diagnostic and therapeutic concepts hold true depending on the type of VM involved. Dr. Nassiri advised a multidisciplinary approach, and noted that early trials have investigated the use of sirolimus in severe, life-threatening cases (Br J Clin Pharmacol. 2016;82[5]:1171-9. doi: 10.1111/bcp.13022).
Dr. Nassiri disclosed serving on the speakers bureaus for Boston Scientific, Penumbra, and Merritt Medical, and is a consultant to Merritt Medical.
CHICAGO – Thanks to convoluted terminology, not to mention confusion in the literature, physicians have been known to frequently misdiagnose vascular malformations as hemangiomas, but an evolving understanding of their differences may lead to more precise diagnoses, according to a report at a symposium on vascular surgery sponsored by Northwestern University.
“Historically there has been a great deal of confusion in the literature when it comes to the nomenclature used to describe vascular anomalies,” said Naiem Nassiri, MD, of Robert Wood Johnson Medical School, New Brunswick, N.J. He pointed out that the term hemangioma “or derivatives thereof” – cavernous hemangioma, cavernous angioma, lymphangioma and cystic hygroma – are “absolute misnomers and continue to be misused and applied almost haphazardly to any anomalous vascular lesion.”
He cited reports that 71% of vascular anomalies have been improperly called hemangiomas, 69% have initially been diagnosed incorrectly, and 21% received the wrong treatment (Pediatr Dermatol. 2008;25[1]:7-12; Plast Reconstr Surg. 2011:127[1]:347-51). “Erroneous terminology has prognostic as well as diagnostic and therapeutic implications, and these can actually be quite devastating for the patient, not only clinically and physically but psychologically as well,” Dr. Nassiri said.
Using the International Society for the Study of Vascular Anomalies classification for hemangiomas and vascular malformations can help physicians make the differential diagnosis, Dr. Nassiri said. Hemangiomas are neoplastic lesions of infancy, though not always congenital, with a finite growth phase, whereas vascular malformations (VMs) are nonneoplastic, congenital lesions that can appear at any age and do not regress spontaneously, he said.
Infantile hemangiomas typically appear as the classic strawberry birthmark in children, whereas VMs tend to appear later in life. “They require some environmental trigger, such as trauma, activity, or changes in the hormonal milieu to manifest onset,” he said of VMs.
Simply put, VMs fall into three broad categories: slow-flow malformations, which include lymphatic and venous malformations; high-flow arteriovenous malformations (AVMs) and fistulas; and congenital mixed syndromes, which can include combinations thereof.
Dr. Nassiri noted that contrast-enhanced MRI is the standard imaging modality for diagnosis of VMs, and can differentiate between slow-flow and high-flow lesions. However, vascular specialists must be vigilant in ordering imaging for slow-flow lesions. “Orders can be changed to MR venography, and I’ve had patients who’ve gone decades with multiple MR venograms and no one can figure out what’s going on as no identifiable lesion is readily detected,” he said. “MR venograms are fantastic for detecting truncular blood flow where there typically are no anomalies in the vast majority of patients with isolated venous malformations, but on contrast-enhanced MRI these convoluted cluster of anomalous veins light up like Christmas trees.”
Lymphatic malformations affect the head and neck more so than the extremities, trunk or viscera, and are prone to infection and bleeding. “You can think of these as fluid-filled balloons, and the goal of treatment is fairly simple: You want to puncture the balloon and drain the fluid inside so as to obtain maximum wall collapse,” Dr. Nassiri said. Infusion of a sclerosant causes an inflammatory reaction leading to fibrosis, which then prevents balloon re-expansion. Surgical excision is best used as a secondary adjunct.
Venous malformations, comprising about 80% of all VMs, typically present as soft, spongy blue or purple compressible masses with associated pain that worsens with exertion, Dr. Nassiri said. “The most dangerous thing that is often overlooked, even by some of the physicians that treat these on a regular basis, is localized intravascular coagulopathy, which if left untreated can progress to fulminant disseminated intravascular coagulopathy,” he said. This tends to occur more in the more widespread varieties of venous malformations.
A common misnomer associated with venous malformations in adults is “liver hemangioma,” owing to the confusing nomenclature, Dr. Nassiri said. “When interrogated angiographically,” he said, “what is often labeled as a hepatic hemangioma is in fact a venous malformation. Natural history of the two entities is completely different.”
Dr. Nassiri described congenital high-flow AVMs as “convoluted networks of blood vessels with poorly differentiated endothelial cells that have neither a venous nor an arterial designation; this entity, otherwise known as a nidus, sits between the feeding arteries and the draining veins.” Treatment aims to eliminate the flow within that nidus.
Super-selective microcatheterization is the best option for nidus access and embolization using liquid embolic agents, preferably those that polymerize when infused. “This is probably the most potent angiogenic entity I’ve ever seen,” Dr. Nassiri said of the nidus.
“It’s like a low-pressure sump and it will recruit collaterals vigorously, so you have to eliminate that nidus.” A variety of different embolic agents, some off label, may be used for high flow AVMs.
For congenital mixed syndromes, the same diagnostic and therapeutic concepts hold true depending on the type of VM involved. Dr. Nassiri advised a multidisciplinary approach, and noted that early trials have investigated the use of sirolimus in severe, life-threatening cases (Br J Clin Pharmacol. 2016;82[5]:1171-9. doi: 10.1111/bcp.13022).
Dr. Nassiri disclosed serving on the speakers bureaus for Boston Scientific, Penumbra, and Merritt Medical, and is a consultant to Merritt Medical.
AT THE NORTHWESTERN VASCULAR SYMPOSIUM
Key clinical point:
Major finding: Use of imaging and a clearer understanding of the lack of neoplastic activity are key to more precisely diagnosing vascular malformations.
Data source: Review of literature and center experience.
Disclosure: Dr. Nassiri disclosed serving on the speakers bureaus for Boston Scientific, Penumbra, and Merritt Medical, and is a consultant to Merritt Medical.
Pediatric Dermatology Consult - January 2017
Scabies
BY JENNA BOROK AND LAWRENCE EICHENFIELD, MD
Frontline Medical News
The scabies mite or the “itch mite” was discovered in 1687 as the first identifiable microorganism that caused human disease.1 Scabies is an infection of the epidermis with the mite, Sarcoptes scabiei variety hominis (S. scabiei) affecting approximately 300 million people worldwide.2 It is a common disease, especially among school-aged children and is particularly rampant in areas of poor sanitation and overcrowding.2,3
S. scabiei live in and on human skin where the impregnated female mite burrows into the stratum corneum and lays two to three eggs daily for as long as 30 days.3,4 The egg becomes a larva, which leaves the burrow and molts into a nymph, and it continues to molt into a mature mite.3 Mating then occurs during the mite stage. After mating, the male mite dies and the female completes the life cycle by burrowing back into the stratum corneum; this process takes about 2 weeks.3,4
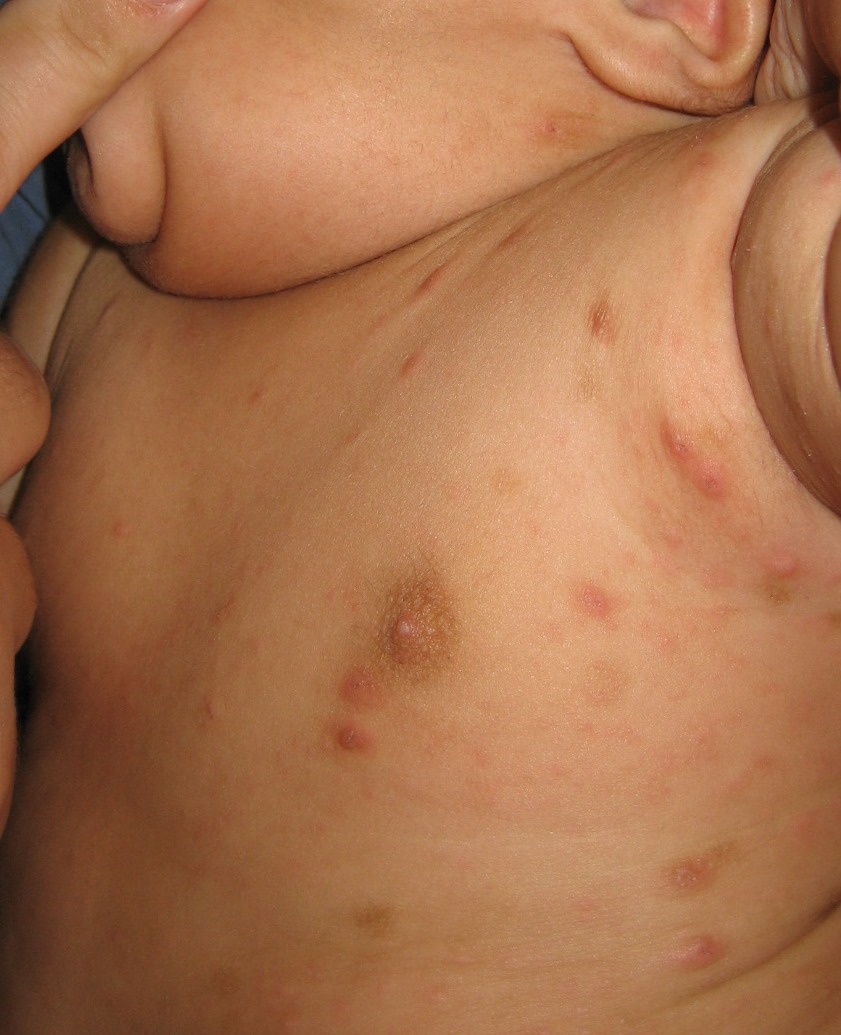
Diagnosis
The diagnosis is usually based on strong clinical suspicion. The chief complaint is often a generalized itching rash that is often worse at nighttime.3,6 Small inflammatory papules are the main physical exam finding, and they usually are widely distributed, the favored locations including the finger webs, wrists, elbows, axillae, girdle area, and feet.3 In addition, male genitalia oftentimes are involved, and small itching papules on the penis should be considered scabies until proven otherwise.3 The head almost always is spared except in infants, who may present with pustules on the palms and soles of the feet and vesicles or lesions on the neck and face.7
The pathognomonic exam finding is the mite’s burrow, which appears as a 2-5 mm white, superficial, threadlike line. Upon close inspection, a tiny black speck often can be seen at the end of the burrow, which represents the adult mite.3 The presence of mites, eggs, or feces also is diagnostic and is accomplished by a skin scraping with a No. 15 blade.3 A positive scraping is diagnostic, although a negative scraping does not rule out the condition. (We explain this to our patients by saying “If you call someone’s home phone and they answer the phone, you know they are home. If you call and there is no answer, it could be that they aren’t home, or they are home and just not answering.”) A biopsy usually is not required, but may provide a diagnosis when scabies is unsuspected.3
The differential for red papules that itch in children includes atopic dermatitis, impetigo, papular urticaria, contact dermatitis, and other infestations, including bites from mosquitoes, fleas, and bed bugs. Atopic dermatitis (AD) can present as eczematous, erythematous papular lesions with oozing in flexural areas similar to areas affected by scabies. However, there are no burrows seen in AD, and the lesions are not in the interdigital web spaces. Impetigo has erythematous vesiculopustular lesions that form honey-colored crusts. Papular urticaria may be a hypersensitivity reaction to another insect, and the urticarial lesions are usually on the exposed parts of extremities. Unlike scabies, there are no burrows and usually no symptomatic family members. Contact dermatitis can present with vesicular, bullous, and sometimes papular erythematous lesions. It occurs after exposure to an allergen and often has well-demarcated borders with geometric shapes.
Treatment
Few randomized control or head-to-head trials exist on scabies treatment.2 First-line treatment is permethrin 5% cream, which is applied to the entire body surface from the neck down in children, but includes the face and scalp in infants.8,9 It is applied at bedtime and is washed off in the morning, and a second application is recommended after 7 days.3,8 It can be used in infants 2 months of age and older, and in pregnant females.3 Household contacts should be treated too, and those who are asymptomatic only require one application of permethrin.3 Permethrin is effective and more cost effective than ivermectin. Oral ivermectin has been used to treat scabies with a dose of 0.2 mg/kg and then repeated 2 weeks later, but the safety in children under 15 kg has not been determined.9 Lindane is an alternative option and is not recommended as first-line therapy because of its toxicity. It only should be used if the patient cannot tolerate or failed the previously mentioned therapies, with particular concern in children less than 50 kg.8,9 Infants and young children should be treated with permethrin and not with lindane.8 Clothes and bed linens can be decontaminated by machine-washing at a hot temperature.3,8 Topical corticosteroids and oral antihistamines can be helpful post-treatment to minimize pruritus and secondary eczematous changes.9
References
1. Int J Dermatol. 1998 Aug;37(8):625-30.
3. Lookingbill and Marks’ Principles of Dermatology, 5th edition (Philadelphia: Elsevier, 2013).
4. BMJ. 2005 Sep 17;331(7517):619-22.
5. Br Med J. 1941 Sep 20;2(4211):405-6.
6. Lancet. 2006 May 27;367(9524):1767-74.
7. Am J Clin Dermatol. 2002;3(1):9-18.
8. MMWR June 5, 2015 / 64(RR3);1-137.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Borok is a medical student at the University of California, Los Angeles. Dr. Eichenfield and Ms. Borok said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
Scabies
BY JENNA BOROK AND LAWRENCE EICHENFIELD, MD
Frontline Medical News
The scabies mite or the “itch mite” was discovered in 1687 as the first identifiable microorganism that caused human disease.1 Scabies is an infection of the epidermis with the mite, Sarcoptes scabiei variety hominis (S. scabiei) affecting approximately 300 million people worldwide.2 It is a common disease, especially among school-aged children and is particularly rampant in areas of poor sanitation and overcrowding.2,3
S. scabiei live in and on human skin where the impregnated female mite burrows into the stratum corneum and lays two to three eggs daily for as long as 30 days.3,4 The egg becomes a larva, which leaves the burrow and molts into a nymph, and it continues to molt into a mature mite.3 Mating then occurs during the mite stage. After mating, the male mite dies and the female completes the life cycle by burrowing back into the stratum corneum; this process takes about 2 weeks.3,4
Diagnosis
The diagnosis is usually based on strong clinical suspicion. The chief complaint is often a generalized itching rash that is often worse at nighttime.3,6 Small inflammatory papules are the main physical exam finding, and they usually are widely distributed, the favored locations including the finger webs, wrists, elbows, axillae, girdle area, and feet.3 In addition, male genitalia oftentimes are involved, and small itching papules on the penis should be considered scabies until proven otherwise.3 The head almost always is spared except in infants, who may present with pustules on the palms and soles of the feet and vesicles or lesions on the neck and face.7
The pathognomonic exam finding is the mite’s burrow, which appears as a 2-5 mm white, superficial, threadlike line. Upon close inspection, a tiny black speck often can be seen at the end of the burrow, which represents the adult mite.3 The presence of mites, eggs, or feces also is diagnostic and is accomplished by a skin scraping with a No. 15 blade.3 A positive scraping is diagnostic, although a negative scraping does not rule out the condition. (We explain this to our patients by saying “If you call someone’s home phone and they answer the phone, you know they are home. If you call and there is no answer, it could be that they aren’t home, or they are home and just not answering.”) A biopsy usually is not required, but may provide a diagnosis when scabies is unsuspected.3
The differential for red papules that itch in children includes atopic dermatitis, impetigo, papular urticaria, contact dermatitis, and other infestations, including bites from mosquitoes, fleas, and bed bugs. Atopic dermatitis (AD) can present as eczematous, erythematous papular lesions with oozing in flexural areas similar to areas affected by scabies. However, there are no burrows seen in AD, and the lesions are not in the interdigital web spaces. Impetigo has erythematous vesiculopustular lesions that form honey-colored crusts. Papular urticaria may be a hypersensitivity reaction to another insect, and the urticarial lesions are usually on the exposed parts of extremities. Unlike scabies, there are no burrows and usually no symptomatic family members. Contact dermatitis can present with vesicular, bullous, and sometimes papular erythematous lesions. It occurs after exposure to an allergen and often has well-demarcated borders with geometric shapes.
Treatment
Few randomized control or head-to-head trials exist on scabies treatment.2 First-line treatment is permethrin 5% cream, which is applied to the entire body surface from the neck down in children, but includes the face and scalp in infants.8,9 It is applied at bedtime and is washed off in the morning, and a second application is recommended after 7 days.3,8 It can be used in infants 2 months of age and older, and in pregnant females.3 Household contacts should be treated too, and those who are asymptomatic only require one application of permethrin.3 Permethrin is effective and more cost effective than ivermectin. Oral ivermectin has been used to treat scabies with a dose of 0.2 mg/kg and then repeated 2 weeks later, but the safety in children under 15 kg has not been determined.9 Lindane is an alternative option and is not recommended as first-line therapy because of its toxicity. It only should be used if the patient cannot tolerate or failed the previously mentioned therapies, with particular concern in children less than 50 kg.8,9 Infants and young children should be treated with permethrin and not with lindane.8 Clothes and bed linens can be decontaminated by machine-washing at a hot temperature.3,8 Topical corticosteroids and oral antihistamines can be helpful post-treatment to minimize pruritus and secondary eczematous changes.9
References
1. Int J Dermatol. 1998 Aug;37(8):625-30.
3. Lookingbill and Marks’ Principles of Dermatology, 5th edition (Philadelphia: Elsevier, 2013).
4. BMJ. 2005 Sep 17;331(7517):619-22.
5. Br Med J. 1941 Sep 20;2(4211):405-6.
6. Lancet. 2006 May 27;367(9524):1767-74.
7. Am J Clin Dermatol. 2002;3(1):9-18.
8. MMWR June 5, 2015 / 64(RR3);1-137.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Borok is a medical student at the University of California, Los Angeles. Dr. Eichenfield and Ms. Borok said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
Scabies
BY JENNA BOROK AND LAWRENCE EICHENFIELD, MD
Frontline Medical News
The scabies mite or the “itch mite” was discovered in 1687 as the first identifiable microorganism that caused human disease.1 Scabies is an infection of the epidermis with the mite, Sarcoptes scabiei variety hominis (S. scabiei) affecting approximately 300 million people worldwide.2 It is a common disease, especially among school-aged children and is particularly rampant in areas of poor sanitation and overcrowding.2,3
S. scabiei live in and on human skin where the impregnated female mite burrows into the stratum corneum and lays two to three eggs daily for as long as 30 days.3,4 The egg becomes a larva, which leaves the burrow and molts into a nymph, and it continues to molt into a mature mite.3 Mating then occurs during the mite stage. After mating, the male mite dies and the female completes the life cycle by burrowing back into the stratum corneum; this process takes about 2 weeks.3,4
Diagnosis
The diagnosis is usually based on strong clinical suspicion. The chief complaint is often a generalized itching rash that is often worse at nighttime.3,6 Small inflammatory papules are the main physical exam finding, and they usually are widely distributed, the favored locations including the finger webs, wrists, elbows, axillae, girdle area, and feet.3 In addition, male genitalia oftentimes are involved, and small itching papules on the penis should be considered scabies until proven otherwise.3 The head almost always is spared except in infants, who may present with pustules on the palms and soles of the feet and vesicles or lesions on the neck and face.7
The pathognomonic exam finding is the mite’s burrow, which appears as a 2-5 mm white, superficial, threadlike line. Upon close inspection, a tiny black speck often can be seen at the end of the burrow, which represents the adult mite.3 The presence of mites, eggs, or feces also is diagnostic and is accomplished by a skin scraping with a No. 15 blade.3 A positive scraping is diagnostic, although a negative scraping does not rule out the condition. (We explain this to our patients by saying “If you call someone’s home phone and they answer the phone, you know they are home. If you call and there is no answer, it could be that they aren’t home, or they are home and just not answering.”) A biopsy usually is not required, but may provide a diagnosis when scabies is unsuspected.3
The differential for red papules that itch in children includes atopic dermatitis, impetigo, papular urticaria, contact dermatitis, and other infestations, including bites from mosquitoes, fleas, and bed bugs. Atopic dermatitis (AD) can present as eczematous, erythematous papular lesions with oozing in flexural areas similar to areas affected by scabies. However, there are no burrows seen in AD, and the lesions are not in the interdigital web spaces. Impetigo has erythematous vesiculopustular lesions that form honey-colored crusts. Papular urticaria may be a hypersensitivity reaction to another insect, and the urticarial lesions are usually on the exposed parts of extremities. Unlike scabies, there are no burrows and usually no symptomatic family members. Contact dermatitis can present with vesicular, bullous, and sometimes papular erythematous lesions. It occurs after exposure to an allergen and often has well-demarcated borders with geometric shapes.
Treatment
Few randomized control or head-to-head trials exist on scabies treatment.2 First-line treatment is permethrin 5% cream, which is applied to the entire body surface from the neck down in children, but includes the face and scalp in infants.8,9 It is applied at bedtime and is washed off in the morning, and a second application is recommended after 7 days.3,8 It can be used in infants 2 months of age and older, and in pregnant females.3 Household contacts should be treated too, and those who are asymptomatic only require one application of permethrin.3 Permethrin is effective and more cost effective than ivermectin. Oral ivermectin has been used to treat scabies with a dose of 0.2 mg/kg and then repeated 2 weeks later, but the safety in children under 15 kg has not been determined.9 Lindane is an alternative option and is not recommended as first-line therapy because of its toxicity. It only should be used if the patient cannot tolerate or failed the previously mentioned therapies, with particular concern in children less than 50 kg.8,9 Infants and young children should be treated with permethrin and not with lindane.8 Clothes and bed linens can be decontaminated by machine-washing at a hot temperature.3,8 Topical corticosteroids and oral antihistamines can be helpful post-treatment to minimize pruritus and secondary eczematous changes.9
References
1. Int J Dermatol. 1998 Aug;37(8):625-30.
3. Lookingbill and Marks’ Principles of Dermatology, 5th edition (Philadelphia: Elsevier, 2013).
4. BMJ. 2005 Sep 17;331(7517):619-22.
5. Br Med J. 1941 Sep 20;2(4211):405-6.
6. Lancet. 2006 May 27;367(9524):1767-74.
7. Am J Clin Dermatol. 2002;3(1):9-18.
8. MMWR June 5, 2015 / 64(RR3);1-137.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Borok is a medical student at the University of California, Los Angeles. Dr. Eichenfield and Ms. Borok said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
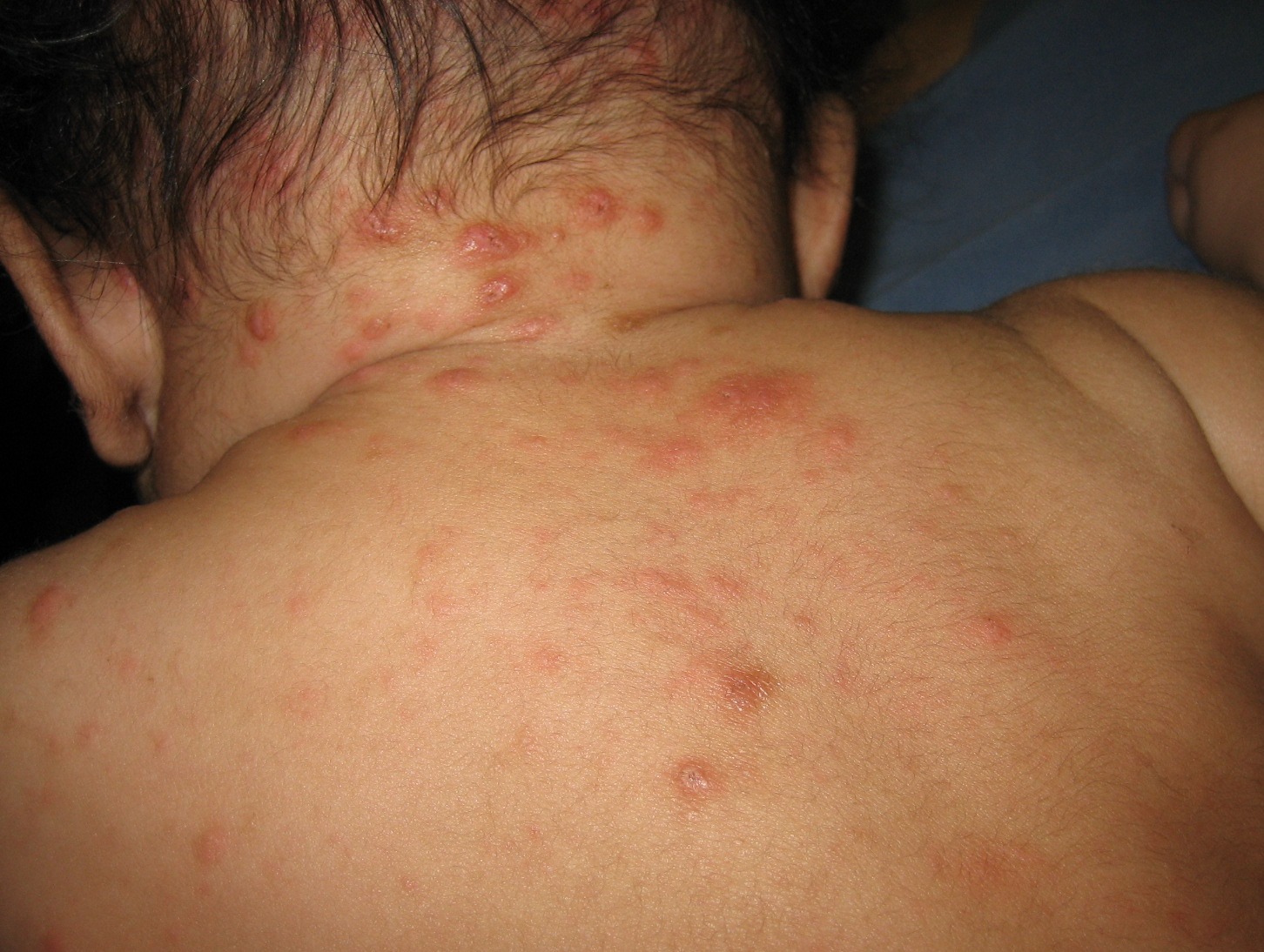
A 3-month-old boy presents to his physician for evaluation of a diffusely itchy rash. The rash started about 1 month ago and has been getting progressively worse; it is now very pruritic. The rash is diffuse, but includes the face, trunk, hands, and feet.
He is otherwise healthy and has no history of eczema or infections.
There are no animals at home. The infant was born at term with an unremarkable perinatal history.
Pediatric Dermatology Consult - December 2016
Urticaria pigmentosa
BY CATALINA MATIZ, MD, AND JEREMY UDKOFF
Frontline Medical News
Maculopapular cutaneous mastocytosis (MPCM), colloquially known as urticaria pigmentosa, typically presents as multiple brown lesions on the skin and accounts for the majority of pediatric cutaneous mastocytosis.1 The category of cutaneous mastocytosis also includes diffuse cutaneous mastocytosis (characterized by generalized erythema and thickened skin), solitary mastocytoma (present as a solitary brown or red lesion), and telangiectasia macularis eruptiva perstans (diffuse telangiectatic macules on the skin, usually in adult patients).
Cutaneous manifestations of mastocytosis are caused by collections of mast cells and the local release of their mediators into the skin. Some of these patients can present with pruritus and flushing, and rarely with diarrhea, abdominal pain, or heart palpitations. Alternatively, systemic mastocytosis, where mast cells invade extracutaneous organs such as the bone marrow or the gastrointestinal tract, may present with or without associated skin lesions. These patients usually have more severe systemic symptoms such as fever, weight loss, epigastric pain, neuropsychiatric symptoms, or bone pain.2,3

The pathogenesis of mastocytosis revolves around abnormal mast cell proliferation. These mast cells then accumulate in organs such as the skin. Mechanical stimulations result in a release of cell mediators, such as histamine, eicosanoids, and cytokines, that causes the associated symptoms. Recent studies have shown c-KIT mutations to be present in the majority of mastocytosis cases, and these mutations are thought to contribute to disease progression.6
Differential diagnosis
Because of its varied presentation, the differential diagnosis for MPCM includes café au lait macules, lentigines, nevi, lichen planus, pseudolymphoma, diffuse juvenile xanthogranuloma, and Langerhans cell histiocytosis. Although these diseases may include macules and papules that look similar to MPCM, the Darier’s sign is an extremely powerful tool in differentiating these diseases as it is almost always present in mastocytosis. When there is a history of blistering lesions, the differential diagnosis should include epidermolysis bullosa, herpes simplex, bullous arthropod bites, bullous impetigo, linear IgA dermatosis, and rarely autoimmune bullous conditions. Equivocal cases should be referred to a pediatric dermatologist for evaluation, as conditions such as Langerhans cell histiocytosis and epidermolysis bullosa are usually associated with poor outcomes.
Work-up and treatment
In cases where the lesions are not classic or there is not a clear Darier’s sign, a skin biopsy with special staining, such as Giemsa or toluidine blue, is recommended to help clarify the diagnosis and rule out other worrisome conditions as mentioned above. If the child presents with systemic symptoms, performing a complete blood count with differential, tryptase level, and organ-specific testing based on the symptoms is recommended.
Serum tryptase is a biomarker for mast cell degranulation. It is typically normal in pediatric patients with MPCM but may be positive if there are symptoms of systemic disease such as fever, weight loss, bone pain, epigastric pain, headaches, or anaphylaxis. Similarly, the risk of anaphylactic reactions correlates with the serum tryptase level.4,7 If the tryptase level is elevated, a bone marrow biopsy should be performed to rule out more severe forms of systemic disease.
Potent topical corticosteroids, such as fluocinonide, are typically used to the treat cutaneous lesions when they are inflamed. Other supportive treatments used in MPCM usually revolve around controlling the cutaneous symptoms as there is no cure for the disease. H1 and H2 antihistamines may be used alone or together to control itching and are thought to decrease flushing and blistering associated with MPCM. Other possible treatments include topical cromolyn sodium and leukotriene antagonists. Mastocytoma lesions that are increasing in size or are very symptomatic despite the treatments listed above may benefit from local immunosuppressants, ultraviolet radiation and psoralen application, or excision.8
Lifestyle modifications also are recommended for patients with mastocytosis. They are advised to avoid foods, medications, and environmental triggers that can cause mast cell degranulation. These triggers include aspirin, nonsteroidal anti-inflammatory agents and narcotics, topical polymyxin B sulfate, heat, friction, and certain systemic anesthetic agents.9
References
1. Br J Dermatol. 2015;172(3):642-51.
2. Clin Pediatr. (Phila) 2008;47(8):757-61.
3. “Mastocytosis,” in Dermatology, 3rd ed. (China: Elsevier, 2012).
4. J Allergy Clin Immunol. 2016;137(1):35-45.
5. J Allergy Clin Immunol. 2015 Dec;136(6):1581-90.e1-e3.
6. J Invest Dermatol. 2010;130(3):804-15.
7. Pediatr Dermatol. 2014;31(3):271-5.
8. Eur J Clin Invest. 2007;37(6):435-53.
9. “Histiocytosis and Malignant Skin Diseases,” in Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence, 5th ed. (China: Elsevier, 2016).
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Jeremy Udkoff is a medical student at the university.
Urticaria pigmentosa
BY CATALINA MATIZ, MD, AND JEREMY UDKOFF
Frontline Medical News
Maculopapular cutaneous mastocytosis (MPCM), colloquially known as urticaria pigmentosa, typically presents as multiple brown lesions on the skin and accounts for the majority of pediatric cutaneous mastocytosis.1 The category of cutaneous mastocytosis also includes diffuse cutaneous mastocytosis (characterized by generalized erythema and thickened skin), solitary mastocytoma (present as a solitary brown or red lesion), and telangiectasia macularis eruptiva perstans (diffuse telangiectatic macules on the skin, usually in adult patients).
Cutaneous manifestations of mastocytosis are caused by collections of mast cells and the local release of their mediators into the skin. Some of these patients can present with pruritus and flushing, and rarely with diarrhea, abdominal pain, or heart palpitations. Alternatively, systemic mastocytosis, where mast cells invade extracutaneous organs such as the bone marrow or the gastrointestinal tract, may present with or without associated skin lesions. These patients usually have more severe systemic symptoms such as fever, weight loss, epigastric pain, neuropsychiatric symptoms, or bone pain.2,3
The pathogenesis of mastocytosis revolves around abnormal mast cell proliferation. These mast cells then accumulate in organs such as the skin. Mechanical stimulations result in a release of cell mediators, such as histamine, eicosanoids, and cytokines, that causes the associated symptoms. Recent studies have shown c-KIT mutations to be present in the majority of mastocytosis cases, and these mutations are thought to contribute to disease progression.6
Differential diagnosis
Because of its varied presentation, the differential diagnosis for MPCM includes café au lait macules, lentigines, nevi, lichen planus, pseudolymphoma, diffuse juvenile xanthogranuloma, and Langerhans cell histiocytosis. Although these diseases may include macules and papules that look similar to MPCM, the Darier’s sign is an extremely powerful tool in differentiating these diseases as it is almost always present in mastocytosis. When there is a history of blistering lesions, the differential diagnosis should include epidermolysis bullosa, herpes simplex, bullous arthropod bites, bullous impetigo, linear IgA dermatosis, and rarely autoimmune bullous conditions. Equivocal cases should be referred to a pediatric dermatologist for evaluation, as conditions such as Langerhans cell histiocytosis and epidermolysis bullosa are usually associated with poor outcomes.
Work-up and treatment
In cases where the lesions are not classic or there is not a clear Darier’s sign, a skin biopsy with special staining, such as Giemsa or toluidine blue, is recommended to help clarify the diagnosis and rule out other worrisome conditions as mentioned above. If the child presents with systemic symptoms, performing a complete blood count with differential, tryptase level, and organ-specific testing based on the symptoms is recommended.
Serum tryptase is a biomarker for mast cell degranulation. It is typically normal in pediatric patients with MPCM but may be positive if there are symptoms of systemic disease such as fever, weight loss, bone pain, epigastric pain, headaches, or anaphylaxis. Similarly, the risk of anaphylactic reactions correlates with the serum tryptase level.4,7 If the tryptase level is elevated, a bone marrow biopsy should be performed to rule out more severe forms of systemic disease.
Potent topical corticosteroids, such as fluocinonide, are typically used to the treat cutaneous lesions when they are inflamed. Other supportive treatments used in MPCM usually revolve around controlling the cutaneous symptoms as there is no cure for the disease. H1 and H2 antihistamines may be used alone or together to control itching and are thought to decrease flushing and blistering associated with MPCM. Other possible treatments include topical cromolyn sodium and leukotriene antagonists. Mastocytoma lesions that are increasing in size or are very symptomatic despite the treatments listed above may benefit from local immunosuppressants, ultraviolet radiation and psoralen application, or excision.8
Lifestyle modifications also are recommended for patients with mastocytosis. They are advised to avoid foods, medications, and environmental triggers that can cause mast cell degranulation. These triggers include aspirin, nonsteroidal anti-inflammatory agents and narcotics, topical polymyxin B sulfate, heat, friction, and certain systemic anesthetic agents.9
References
1. Br J Dermatol. 2015;172(3):642-51.
2. Clin Pediatr. (Phila) 2008;47(8):757-61.
3. “Mastocytosis,” in Dermatology, 3rd ed. (China: Elsevier, 2012).
4. J Allergy Clin Immunol. 2016;137(1):35-45.
5. J Allergy Clin Immunol. 2015 Dec;136(6):1581-90.e1-e3.
6. J Invest Dermatol. 2010;130(3):804-15.
7. Pediatr Dermatol. 2014;31(3):271-5.
8. Eur J Clin Invest. 2007;37(6):435-53.
9. “Histiocytosis and Malignant Skin Diseases,” in Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence, 5th ed. (China: Elsevier, 2016).
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Jeremy Udkoff is a medical student at the university.
Urticaria pigmentosa
BY CATALINA MATIZ, MD, AND JEREMY UDKOFF
Frontline Medical News
Maculopapular cutaneous mastocytosis (MPCM), colloquially known as urticaria pigmentosa, typically presents as multiple brown lesions on the skin and accounts for the majority of pediatric cutaneous mastocytosis.1 The category of cutaneous mastocytosis also includes diffuse cutaneous mastocytosis (characterized by generalized erythema and thickened skin), solitary mastocytoma (present as a solitary brown or red lesion), and telangiectasia macularis eruptiva perstans (diffuse telangiectatic macules on the skin, usually in adult patients).
Cutaneous manifestations of mastocytosis are caused by collections of mast cells and the local release of their mediators into the skin. Some of these patients can present with pruritus and flushing, and rarely with diarrhea, abdominal pain, or heart palpitations. Alternatively, systemic mastocytosis, where mast cells invade extracutaneous organs such as the bone marrow or the gastrointestinal tract, may present with or without associated skin lesions. These patients usually have more severe systemic symptoms such as fever, weight loss, epigastric pain, neuropsychiatric symptoms, or bone pain.2,3
The pathogenesis of mastocytosis revolves around abnormal mast cell proliferation. These mast cells then accumulate in organs such as the skin. Mechanical stimulations result in a release of cell mediators, such as histamine, eicosanoids, and cytokines, that causes the associated symptoms. Recent studies have shown c-KIT mutations to be present in the majority of mastocytosis cases, and these mutations are thought to contribute to disease progression.6
Differential diagnosis
Because of its varied presentation, the differential diagnosis for MPCM includes café au lait macules, lentigines, nevi, lichen planus, pseudolymphoma, diffuse juvenile xanthogranuloma, and Langerhans cell histiocytosis. Although these diseases may include macules and papules that look similar to MPCM, the Darier’s sign is an extremely powerful tool in differentiating these diseases as it is almost always present in mastocytosis. When there is a history of blistering lesions, the differential diagnosis should include epidermolysis bullosa, herpes simplex, bullous arthropod bites, bullous impetigo, linear IgA dermatosis, and rarely autoimmune bullous conditions. Equivocal cases should be referred to a pediatric dermatologist for evaluation, as conditions such as Langerhans cell histiocytosis and epidermolysis bullosa are usually associated with poor outcomes.
Work-up and treatment
In cases where the lesions are not classic or there is not a clear Darier’s sign, a skin biopsy with special staining, such as Giemsa or toluidine blue, is recommended to help clarify the diagnosis and rule out other worrisome conditions as mentioned above. If the child presents with systemic symptoms, performing a complete blood count with differential, tryptase level, and organ-specific testing based on the symptoms is recommended.
Serum tryptase is a biomarker for mast cell degranulation. It is typically normal in pediatric patients with MPCM but may be positive if there are symptoms of systemic disease such as fever, weight loss, bone pain, epigastric pain, headaches, or anaphylaxis. Similarly, the risk of anaphylactic reactions correlates with the serum tryptase level.4,7 If the tryptase level is elevated, a bone marrow biopsy should be performed to rule out more severe forms of systemic disease.
Potent topical corticosteroids, such as fluocinonide, are typically used to the treat cutaneous lesions when they are inflamed. Other supportive treatments used in MPCM usually revolve around controlling the cutaneous symptoms as there is no cure for the disease. H1 and H2 antihistamines may be used alone or together to control itching and are thought to decrease flushing and blistering associated with MPCM. Other possible treatments include topical cromolyn sodium and leukotriene antagonists. Mastocytoma lesions that are increasing in size or are very symptomatic despite the treatments listed above may benefit from local immunosuppressants, ultraviolet radiation and psoralen application, or excision.8
Lifestyle modifications also are recommended for patients with mastocytosis. They are advised to avoid foods, medications, and environmental triggers that can cause mast cell degranulation. These triggers include aspirin, nonsteroidal anti-inflammatory agents and narcotics, topical polymyxin B sulfate, heat, friction, and certain systemic anesthetic agents.9
References
1. Br J Dermatol. 2015;172(3):642-51.
2. Clin Pediatr. (Phila) 2008;47(8):757-61.
3. “Mastocytosis,” in Dermatology, 3rd ed. (China: Elsevier, 2012).
4. J Allergy Clin Immunol. 2016;137(1):35-45.
5. J Allergy Clin Immunol. 2015 Dec;136(6):1581-90.e1-e3.
6. J Invest Dermatol. 2010;130(3):804-15.
7. Pediatr Dermatol. 2014;31(3):271-5.
8. Eur J Clin Invest. 2007;37(6):435-53.
9. “Histiocytosis and Malignant Skin Diseases,” in Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence, 5th ed. (China: Elsevier, 2016).
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Jeremy Udkoff is a medical student at the university.

An 18-month-old girl was referred to our pediatric dermatology clinic for an evaluation of multiple brown macules all over her body. The lesions had been present for about 1 year and were not increasing in size, but she was getting new lesions on her face and scalp. They were not symptomatic and did not seem to bother the patient. The mother noted that some lesions may become red with changes in the weather. She denied symptoms such as wheezing, flushing, or diarrhea. No other family members were affected, and the patient was not taking any medications. She had no family history of neurofibromatosis or other genetic conditions. The physical exam was notable for many scattered, 3- to 5-mm, polymorphic, light brown to orange macules and papules. One of the lesions reddened and wheeled after we stroked it. The patient had no lymphadenopathy or hepatosplenomegaly.
Pediatric Dermatology Consult - November 2016
BY JUSLEEN AHLUWALIA, MD, AND LAWRENCE F. EICHENFIELD, MD
The presence of an initial tense bulla on the vertex scalp suggests a diagnosis of aplasia cutis congenita. Given the palpable quality of the lesion, magnetic resonance imaging and angiogram (MRI/MRA) of the head with gadolinium enhancement was performed, revealing a 1.5-cm mass underlying the scalp defect, without calvarial deformations. The mass was excised with advancement flap closure. Histologic evaluation of the excised specimen revealed polypoid tissue with unremarkable epidermis and hypocellular dermis with loose connective tissue, lacking adnexal structures. Neural and glial tissue were not identified.
Aplasia cutis congenita (ACC) is a term used to describe congenital absence or defects of the skin.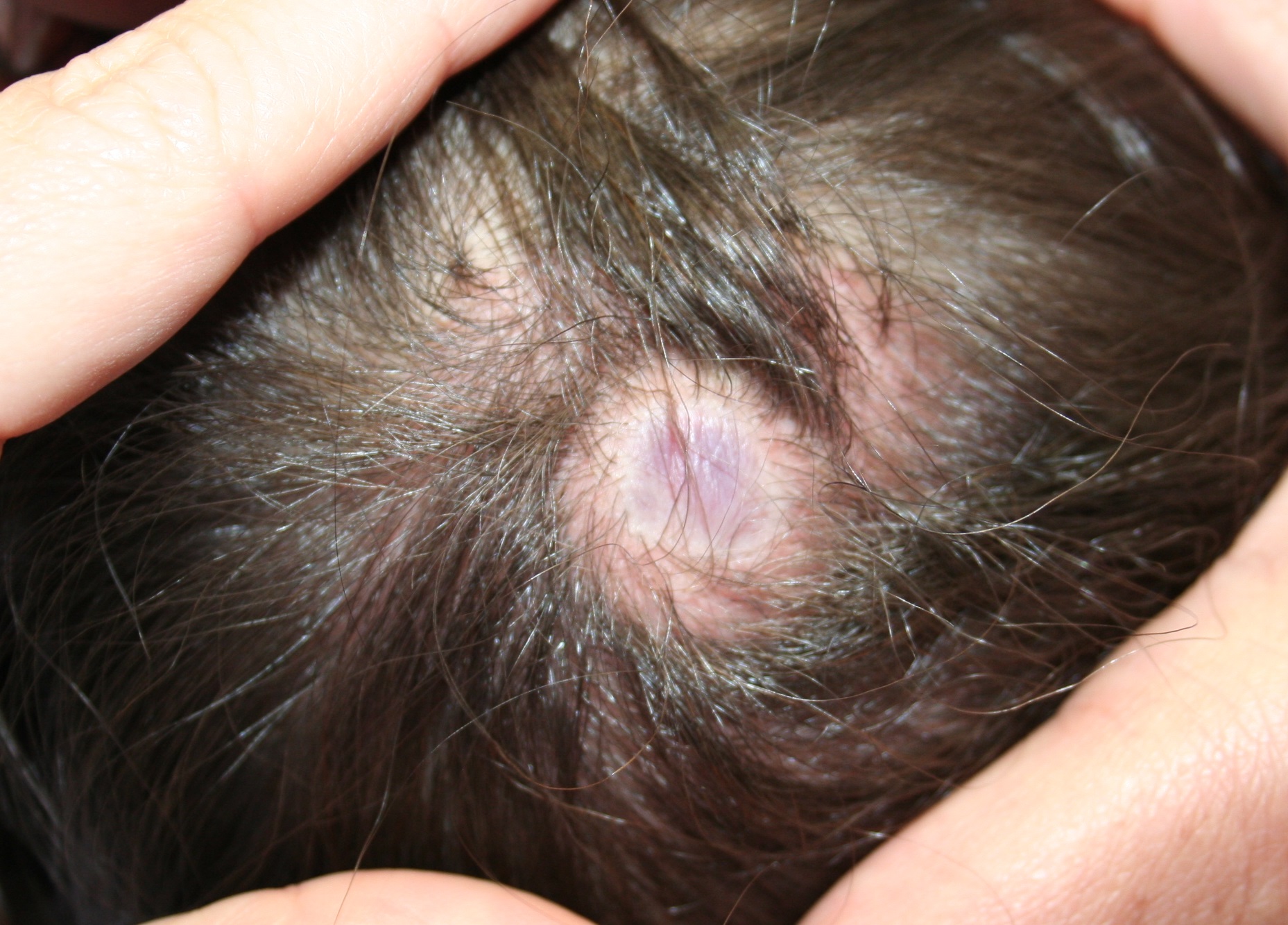
According to its developmental stage in utero, scalp ACC may present at birth as a deep ulceration, superficial erosion, or a healed, alopecic scar.2 Rarely, some defects may have a cystic or bullous component that may transform into a smooth, atrophic scar with an overlying translucent membrane. This clinical subtype has been described as the “bullous” or “membranous” variant, depending on the stage of bulla maturation.4-8 Our patient displays features demonstrative of the membranous variant. Most commonly, membranous aplasia cutis is an isolated defect and, if there is no palpable component, does not require imaging.2 These usually heal spontaneously, including those with small bone defects. Larger lesions of AC (greater than 3 cm) with large bone defects require urgent imaging.2 Papules or nodules around aplasia cutis may be associated with more significant neural tube anomalies, including congenital ectopic meningeal tissue, also termed atretic or rudimentary meningoceles.7 These may be associated with defects of the skull or tracts with intracranial connections.7 Hair collars, a collarette of coarser hair surrounding bullous or membranous variant of AC, may be indicative of defective neural tube closure.4-8 MRI of the head is recommended in cases of bullous or membranous scalp ACC with a palpable nodule to evaluate for intracranial connection and ectopic neural tissue, as performed in our patient.2 Because imaging of our patient was concerning for ectopic neural tissue within the defect, excision was recommended with histologic examination for neural or glial tissue.
Small scalp ACC lesions can heal by secondary intention with supportive wound care, whereas larger defects may require bone or skin grafting.2 Cautious monitoring of the defect will help prevent the serious sequelae rarely associated with large scalp ACC, including hemorrhage, infection, sagittal sinus thrombosis, and hydrocephalus.2 Generally, most defects heal well within months and the resultant scars become relatively unnoticeable. Those that are apparent can be surgically reconstructed.2
Although most infants with ACC are otherwise healthy, several anomalies have been noted to be associated with the presence of ACC, and thus a multisystem evaluation is recommended during the initial visit. These anomalies include limb abnormalities, epidermal nevi, epidermolysis bullosa, chromosomal abnormalities, such as trisomy 13, ectodermal dysplasias, or other malformation syndromes, including Adams-Oliver syndrome.2,8 Around 30 years ago, Frieden et al. stratified ACC into nine different groups based on the location and presence of these associations.9
Differentiation of scalp ACC from other conditions may be necessary. HSV usually appears as grouped vesicles that can evolve to appear punched out.2 Iatrogenic trauma can produce erosions at the site of scalp electrode placement or forceps use, but is less likely to occur over the vertex scalp.2 Congenital triangular alopecia usually presents as an alopecic, lancet-shaped pattern on the frontal temporal scalp without evidence of scarring. Nevus sebaceous characteristically presents as a solitary yellow-orange, mammilated plaque, but may mimic an erosion given its slightly pink appearance in the neonate.2
References
1. Developmental abnormalities, in “Neonatal and infant dermatology,” 3rd ed. (Philadelphia: Saunders, 2015).
2. Dermatol Ther. 2013 Nov-Dec;26(6):439-44.
3. Br J Plast Surg. 2002 Sep;55(6):530-2.
4. J Am Acad Dermatol. 2003 May;48(5 Suppl):S95-8.
5. J Ultrasound Med. 2009 Oct;28(10):1393-6.
6. Arch Dermatol. 1995 Dec;131(12):1427-31.
7. Pediatr Dermatol. 2015 Mar-Apr;32(2):161-70.
8. Indian J Dermatol. 2011 May;56(3):337-8.
9. J Am Acad Dermatol. 1986 Apr;14(4):646-60.
Dr. Ahluwalia is with the division of pediatric and adolescent dermatology, Rady Children’s Hospital, San Diego. Dr. Eichenfield is in the departments of dermatology and pediatrics, University of California, San Diego.
BY JUSLEEN AHLUWALIA, MD, AND LAWRENCE F. EICHENFIELD, MD
The presence of an initial tense bulla on the vertex scalp suggests a diagnosis of aplasia cutis congenita. Given the palpable quality of the lesion, magnetic resonance imaging and angiogram (MRI/MRA) of the head with gadolinium enhancement was performed, revealing a 1.5-cm mass underlying the scalp defect, without calvarial deformations. The mass was excised with advancement flap closure. Histologic evaluation of the excised specimen revealed polypoid tissue with unremarkable epidermis and hypocellular dermis with loose connective tissue, lacking adnexal structures. Neural and glial tissue were not identified.
Aplasia cutis congenita (ACC) is a term used to describe congenital absence or defects of the skin.
According to its developmental stage in utero, scalp ACC may present at birth as a deep ulceration, superficial erosion, or a healed, alopecic scar.2 Rarely, some defects may have a cystic or bullous component that may transform into a smooth, atrophic scar with an overlying translucent membrane. This clinical subtype has been described as the “bullous” or “membranous” variant, depending on the stage of bulla maturation.4-8 Our patient displays features demonstrative of the membranous variant. Most commonly, membranous aplasia cutis is an isolated defect and, if there is no palpable component, does not require imaging.2 These usually heal spontaneously, including those with small bone defects. Larger lesions of AC (greater than 3 cm) with large bone defects require urgent imaging.2 Papules or nodules around aplasia cutis may be associated with more significant neural tube anomalies, including congenital ectopic meningeal tissue, also termed atretic or rudimentary meningoceles.7 These may be associated with defects of the skull or tracts with intracranial connections.7 Hair collars, a collarette of coarser hair surrounding bullous or membranous variant of AC, may be indicative of defective neural tube closure.4-8 MRI of the head is recommended in cases of bullous or membranous scalp ACC with a palpable nodule to evaluate for intracranial connection and ectopic neural tissue, as performed in our patient.2 Because imaging of our patient was concerning for ectopic neural tissue within the defect, excision was recommended with histologic examination for neural or glial tissue.
Small scalp ACC lesions can heal by secondary intention with supportive wound care, whereas larger defects may require bone or skin grafting.2 Cautious monitoring of the defect will help prevent the serious sequelae rarely associated with large scalp ACC, including hemorrhage, infection, sagittal sinus thrombosis, and hydrocephalus.2 Generally, most defects heal well within months and the resultant scars become relatively unnoticeable. Those that are apparent can be surgically reconstructed.2
Although most infants with ACC are otherwise healthy, several anomalies have been noted to be associated with the presence of ACC, and thus a multisystem evaluation is recommended during the initial visit. These anomalies include limb abnormalities, epidermal nevi, epidermolysis bullosa, chromosomal abnormalities, such as trisomy 13, ectodermal dysplasias, or other malformation syndromes, including Adams-Oliver syndrome.2,8 Around 30 years ago, Frieden et al. stratified ACC into nine different groups based on the location and presence of these associations.9
Differentiation of scalp ACC from other conditions may be necessary. HSV usually appears as grouped vesicles that can evolve to appear punched out.2 Iatrogenic trauma can produce erosions at the site of scalp electrode placement or forceps use, but is less likely to occur over the vertex scalp.2 Congenital triangular alopecia usually presents as an alopecic, lancet-shaped pattern on the frontal temporal scalp without evidence of scarring. Nevus sebaceous characteristically presents as a solitary yellow-orange, mammilated plaque, but may mimic an erosion given its slightly pink appearance in the neonate.2
References
1. Developmental abnormalities, in “Neonatal and infant dermatology,” 3rd ed. (Philadelphia: Saunders, 2015).
2. Dermatol Ther. 2013 Nov-Dec;26(6):439-44.
3. Br J Plast Surg. 2002 Sep;55(6):530-2.
4. J Am Acad Dermatol. 2003 May;48(5 Suppl):S95-8.
5. J Ultrasound Med. 2009 Oct;28(10):1393-6.
6. Arch Dermatol. 1995 Dec;131(12):1427-31.
7. Pediatr Dermatol. 2015 Mar-Apr;32(2):161-70.
8. Indian J Dermatol. 2011 May;56(3):337-8.
9. J Am Acad Dermatol. 1986 Apr;14(4):646-60.
Dr. Ahluwalia is with the division of pediatric and adolescent dermatology, Rady Children’s Hospital, San Diego. Dr. Eichenfield is in the departments of dermatology and pediatrics, University of California, San Diego.
BY JUSLEEN AHLUWALIA, MD, AND LAWRENCE F. EICHENFIELD, MD
The presence of an initial tense bulla on the vertex scalp suggests a diagnosis of aplasia cutis congenita. Given the palpable quality of the lesion, magnetic resonance imaging and angiogram (MRI/MRA) of the head with gadolinium enhancement was performed, revealing a 1.5-cm mass underlying the scalp defect, without calvarial deformations. The mass was excised with advancement flap closure. Histologic evaluation of the excised specimen revealed polypoid tissue with unremarkable epidermis and hypocellular dermis with loose connective tissue, lacking adnexal structures. Neural and glial tissue were not identified.
Aplasia cutis congenita (ACC) is a term used to describe congenital absence or defects of the skin.
According to its developmental stage in utero, scalp ACC may present at birth as a deep ulceration, superficial erosion, or a healed, alopecic scar.2 Rarely, some defects may have a cystic or bullous component that may transform into a smooth, atrophic scar with an overlying translucent membrane. This clinical subtype has been described as the “bullous” or “membranous” variant, depending on the stage of bulla maturation.4-8 Our patient displays features demonstrative of the membranous variant. Most commonly, membranous aplasia cutis is an isolated defect and, if there is no palpable component, does not require imaging.2 These usually heal spontaneously, including those with small bone defects. Larger lesions of AC (greater than 3 cm) with large bone defects require urgent imaging.2 Papules or nodules around aplasia cutis may be associated with more significant neural tube anomalies, including congenital ectopic meningeal tissue, also termed atretic or rudimentary meningoceles.7 These may be associated with defects of the skull or tracts with intracranial connections.7 Hair collars, a collarette of coarser hair surrounding bullous or membranous variant of AC, may be indicative of defective neural tube closure.4-8 MRI of the head is recommended in cases of bullous or membranous scalp ACC with a palpable nodule to evaluate for intracranial connection and ectopic neural tissue, as performed in our patient.2 Because imaging of our patient was concerning for ectopic neural tissue within the defect, excision was recommended with histologic examination for neural or glial tissue.
Small scalp ACC lesions can heal by secondary intention with supportive wound care, whereas larger defects may require bone or skin grafting.2 Cautious monitoring of the defect will help prevent the serious sequelae rarely associated with large scalp ACC, including hemorrhage, infection, sagittal sinus thrombosis, and hydrocephalus.2 Generally, most defects heal well within months and the resultant scars become relatively unnoticeable. Those that are apparent can be surgically reconstructed.2
Although most infants with ACC are otherwise healthy, several anomalies have been noted to be associated with the presence of ACC, and thus a multisystem evaluation is recommended during the initial visit. These anomalies include limb abnormalities, epidermal nevi, epidermolysis bullosa, chromosomal abnormalities, such as trisomy 13, ectodermal dysplasias, or other malformation syndromes, including Adams-Oliver syndrome.2,8 Around 30 years ago, Frieden et al. stratified ACC into nine different groups based on the location and presence of these associations.9
Differentiation of scalp ACC from other conditions may be necessary. HSV usually appears as grouped vesicles that can evolve to appear punched out.2 Iatrogenic trauma can produce erosions at the site of scalp electrode placement or forceps use, but is less likely to occur over the vertex scalp.2 Congenital triangular alopecia usually presents as an alopecic, lancet-shaped pattern on the frontal temporal scalp without evidence of scarring. Nevus sebaceous characteristically presents as a solitary yellow-orange, mammilated plaque, but may mimic an erosion given its slightly pink appearance in the neonate.2
References
1. Developmental abnormalities, in “Neonatal and infant dermatology,” 3rd ed. (Philadelphia: Saunders, 2015).
2. Dermatol Ther. 2013 Nov-Dec;26(6):439-44.
3. Br J Plast Surg. 2002 Sep;55(6):530-2.
4. J Am Acad Dermatol. 2003 May;48(5 Suppl):S95-8.
5. J Ultrasound Med. 2009 Oct;28(10):1393-6.
6. Arch Dermatol. 1995 Dec;131(12):1427-31.
7. Pediatr Dermatol. 2015 Mar-Apr;32(2):161-70.
8. Indian J Dermatol. 2011 May;56(3):337-8.
9. J Am Acad Dermatol. 1986 Apr;14(4):646-60.
Dr. Ahluwalia is with the division of pediatric and adolescent dermatology, Rady Children’s Hospital, San Diego. Dr. Eichenfield is in the departments of dermatology and pediatrics, University of California, San Diego.
A 9-month-old previously healthy female, born at term, presents to the dermatology clinic for evaluation of a bluish nodule on the posterior vertex of the scalp (Figure 1). Initially at birth, the area was described as a hairless, tense blister that flattened over several weeks. The hairless nodule has persisted without symptoms. The family denies any history of trauma, including the use of forceps or scalp electrodes during labor. Family history was noncontributory. Review of systems was otherwise negative.
On examination, the patient is a well-developed, active female with a 2-cm, bluish nodule with an overlying atrophic, glistening membrane on the posterior parietal scalp, lateral to the midline. The lesion is surrounded by a ring of subtly coarse, terminal hair. There is no evidence of bleeding or ulceration, and no scalp defects were palpable. The lesion was unchanged with crying. The remainder of the physical examination was normal.
Pediatric Dermatology Consult - October 2016
BY CATALINA MATIZ, MD, AND ANDREA WALDMAN, MD
At the time of initial evaluation, the patient was prescribed a 2-week course of topical triamcinolone 0.1% ointment. Subsequently, the cutaneous lesions clinically improved, but did not completely resolve. Mother had no recollection of infant contact with metal, plastic, or additional topical emollients or medications. On further investigation, the patient’s mother revealed the recent conversion to a different brand of diaper. The diapers had a blue and green polka dot design corresponding with the distribution of our patient’s cutaneous lesions, highly suggestive of allergic contact dermatitis (ACD).
Diaper dermatitis represents a common complaint peaking at 9-12 months of age, estimated to occur in 25% of infants in the primary care office setting.1 Irritant contact dermatitis (ICD) predominates as the most common etiology of diaper dermatitis. ICD represents a nonimmunologic response, triggered by maceration and breakdown of the cutaneous barrier, directly resulting from chronic exposure to urine and feces.1 In contrast, ACD characterizes an immunologic reaction to specific offending allergen(s).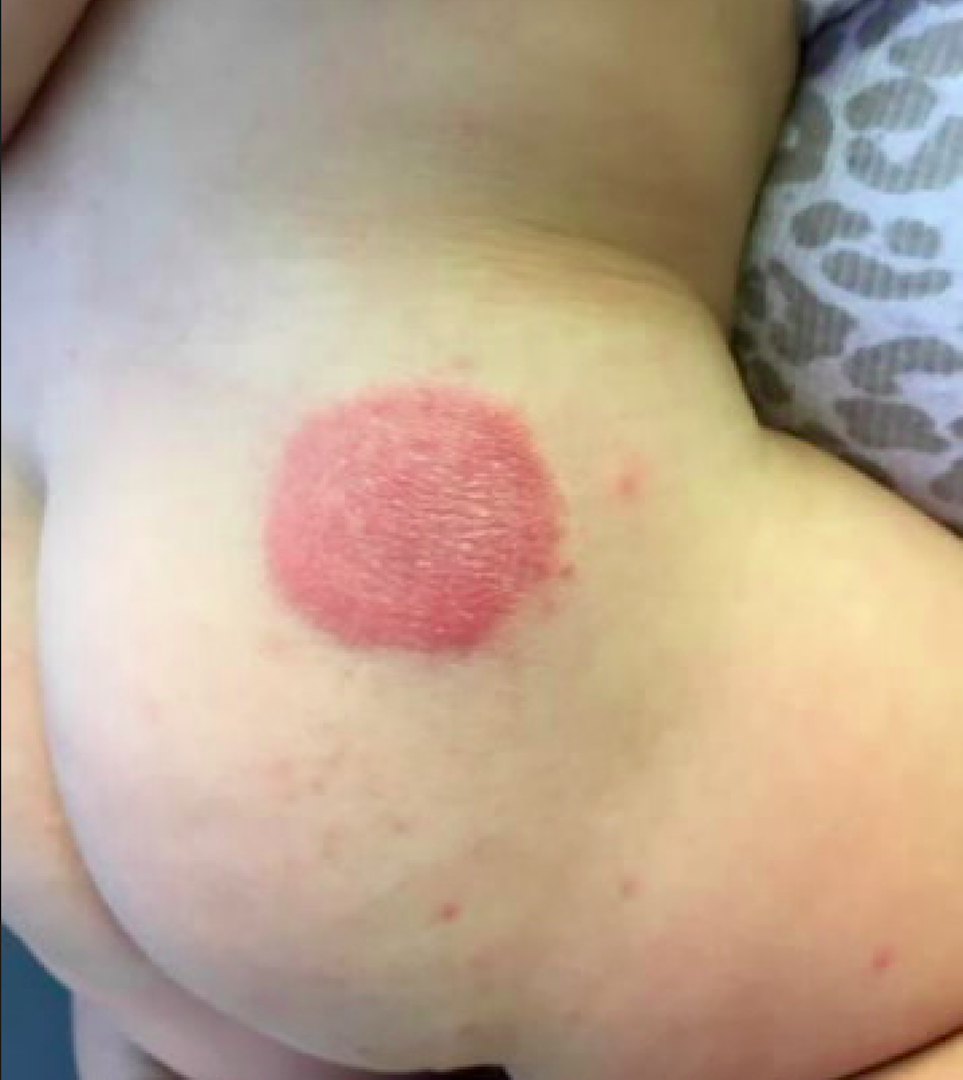
ACD can arise in the diaper area following contact with dyes, fragrances, or other constituents of disposable diapers, wet wipes, diaper creams, or medications. Alberta et al. initially described ACD in several infants, resulting from blue, green, and pink dyes present in various disposable diapers. The dermatitis promptly resolved upon switching to dye-free diapers.2 Furthermore, topical products applied to the diaper region such as barrier creams and wet wipes may trigger allergic sensitization. Sensitization to an allergen can occur after few exposures to years after introduction. Following the initial contact reaction, re-exposure to the offending allergen can elicit a quicker cutaneous response. Case reports have detailed cases of ACD presenting in neonates as early as 1 week of age.3
If ACD is suspected as an etiologic factor in diaper dermatitis, providers should consider constituents present in topical formulations, wet wipes, and components of the diaper itself. A specific example includes mercaptobenzothiazole, a rubber additive commonly found in elastic bands of disposable diapers.2 The most common potential allergens discovered in a recent review of 63 diaper wipes, 41 topical diaper preparations, and 3 top-selling diaper brands included botanical extracts (i.e. aloe vera), alpha-tocopherol, fragrances, propylene glycol, parabens, iodopropynyl butylcarbamate, and lanolin.4
ACD may be difficult to diagnose. Obtaining a thorough clinical history is essential to diagnosis. ACD is often distinguished by its distinct cutaneous morphology, with initial lesions commonly characterized by sharply demarcated erythema associated with vesiculation and edema. The dermatitis progressively evolves to a mixed presentation of predominantly eczematous papules and plaques with crusting, scaling, and lichenification. Another clue suggestive of ACD is cutaneous involvement limited to areas of allergen contact, such as the geometric plaques corresponding to areas of skin exposure to blue and green dye in our patient.1
Differential diagnosis
In contrast to ACD, ICD commonly erupts in the perianal region in the presence of feces, especially during diarrheal episodes. Common clinical manifestations include skin maceration, erosions, erythema, papulation, and scaling. More severe presentations of irritant dermatitis include Jacquet’s diaper dermatitis, perianal pseudoverrucous papules and nodules and granuloma gluteale infantum.1 These cutaneous findings also may develop in older patients with irritated peristomal skin.5
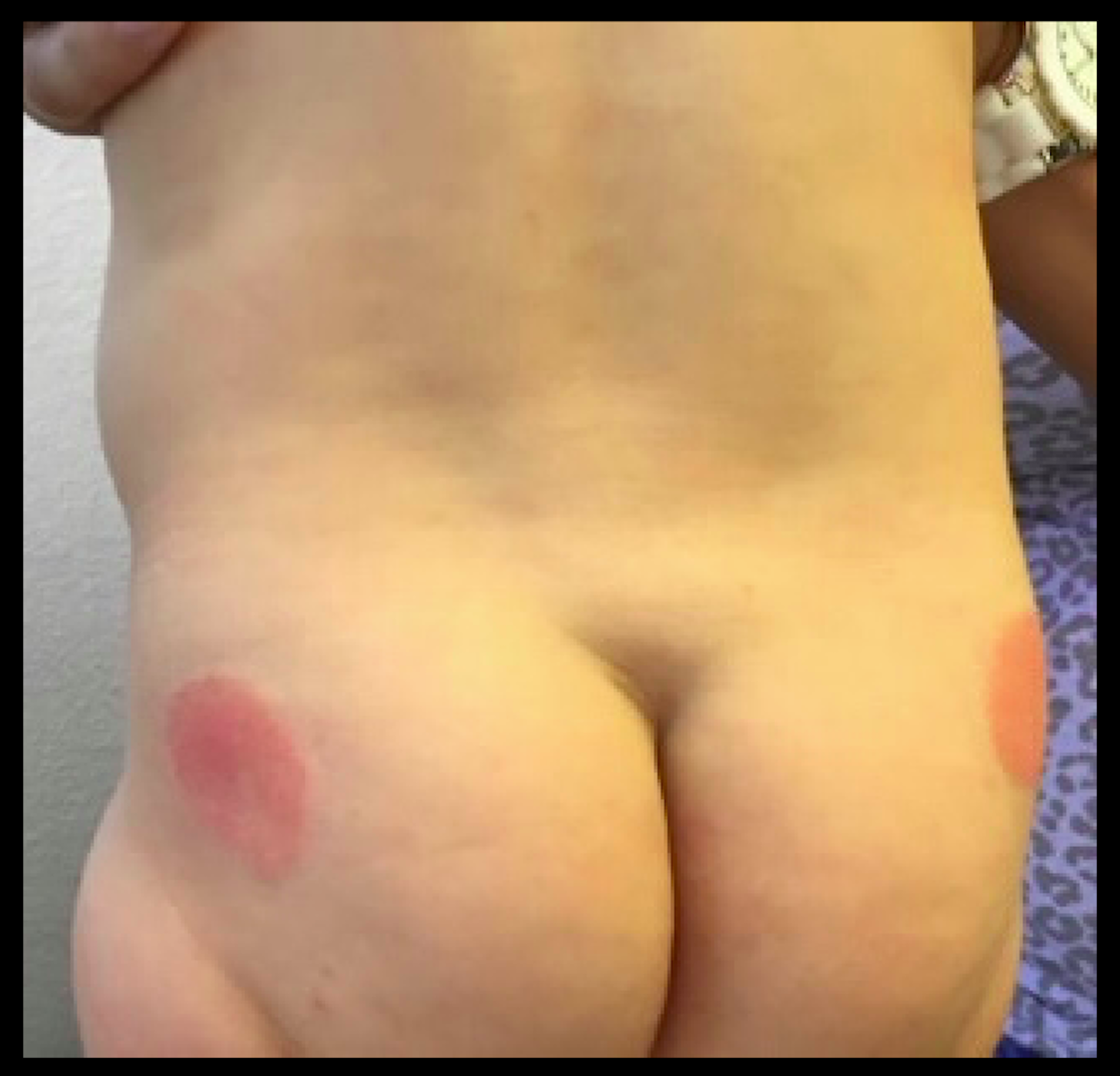
Inflammatory dermatoses such as psoriasis and seborrheic dermatitis also may erupt in the diaper region. Psoriasis commonly involves the gluteal cleft and inguinal folds, appearing as well-demarcated, pink plaques with thin white scale.5 Persistence of plaques despite topical corticosteroid application and lack of pruritus further delineates this disorder. Family history of psoriasis may provide an additional clue to the diagnosis of psoriasis. Similarly, seborrheic dermatitis may present with diaper involvement characterized by well-demarcated erythematous plaques with minimal or absent greasy yellow scale. This dermatitis commonly erupts in infants and neonates less than 6 weeks old and resolves by 6-9 months of age.1
Langerhans cell histiocytosis (LCH) represents a serious condition important to consider in the differential diagnosis of persistent recalcitrant diaper dermatitis. Often confused with seborrheic dermatitis, the diaper dermatitis of LCH may be differentiated by petechiae, purpura, and/or ulceration and atrophy of the inguinal folds.5
Management
Avoidance of the allergen constitutes the gold standard of treatment for ACD. Initial treatment involves adequate-potency topical corticosteroids; for the diaper area, low- to mid-potency corticosteroids are recommended. Changing to dye-free diapers and/or avoiding contact with the suspected allergen also is advised. Epicutaneous patch testing is recommended if the dermatitis fails to improve despite removal of the suspected offending agent.5
References
1. Pediatr Dermatol. 2014 Nov;31 Suppl 1:19-24.
2. Pediatrics. 2005 Sep;116(3):e450-2.
3. Cutis. 1994 Nov;54(5):300-2.
4. Dermatitis. 2016 May-Jun;27(3):110-8.
5. “Neonatal and Infant Dermatology,” 3rd edition (Philadelphia: Elsevier, 2015, p. 251)
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Dr. Waldman is a clinical research fellow at the hospital. Email them at pdnews@frontlinemedcom.com.
BY CATALINA MATIZ, MD, AND ANDREA WALDMAN, MD
At the time of initial evaluation, the patient was prescribed a 2-week course of topical triamcinolone 0.1% ointment. Subsequently, the cutaneous lesions clinically improved, but did not completely resolve. Mother had no recollection of infant contact with metal, plastic, or additional topical emollients or medications. On further investigation, the patient’s mother revealed the recent conversion to a different brand of diaper. The diapers had a blue and green polka dot design corresponding with the distribution of our patient’s cutaneous lesions, highly suggestive of allergic contact dermatitis (ACD).
Diaper dermatitis represents a common complaint peaking at 9-12 months of age, estimated to occur in 25% of infants in the primary care office setting.1 Irritant contact dermatitis (ICD) predominates as the most common etiology of diaper dermatitis. ICD represents a nonimmunologic response, triggered by maceration and breakdown of the cutaneous barrier, directly resulting from chronic exposure to urine and feces.1 In contrast, ACD characterizes an immunologic reaction to specific offending allergen(s).
ACD can arise in the diaper area following contact with dyes, fragrances, or other constituents of disposable diapers, wet wipes, diaper creams, or medications. Alberta et al. initially described ACD in several infants, resulting from blue, green, and pink dyes present in various disposable diapers. The dermatitis promptly resolved upon switching to dye-free diapers.2 Furthermore, topical products applied to the diaper region such as barrier creams and wet wipes may trigger allergic sensitization. Sensitization to an allergen can occur after few exposures to years after introduction. Following the initial contact reaction, re-exposure to the offending allergen can elicit a quicker cutaneous response. Case reports have detailed cases of ACD presenting in neonates as early as 1 week of age.3
If ACD is suspected as an etiologic factor in diaper dermatitis, providers should consider constituents present in topical formulations, wet wipes, and components of the diaper itself. A specific example includes mercaptobenzothiazole, a rubber additive commonly found in elastic bands of disposable diapers.2 The most common potential allergens discovered in a recent review of 63 diaper wipes, 41 topical diaper preparations, and 3 top-selling diaper brands included botanical extracts (i.e. aloe vera), alpha-tocopherol, fragrances, propylene glycol, parabens, iodopropynyl butylcarbamate, and lanolin.4
ACD may be difficult to diagnose. Obtaining a thorough clinical history is essential to diagnosis. ACD is often distinguished by its distinct cutaneous morphology, with initial lesions commonly characterized by sharply demarcated erythema associated with vesiculation and edema. The dermatitis progressively evolves to a mixed presentation of predominantly eczematous papules and plaques with crusting, scaling, and lichenification. Another clue suggestive of ACD is cutaneous involvement limited to areas of allergen contact, such as the geometric plaques corresponding to areas of skin exposure to blue and green dye in our patient.1
Differential diagnosis
In contrast to ACD, ICD commonly erupts in the perianal region in the presence of feces, especially during diarrheal episodes. Common clinical manifestations include skin maceration, erosions, erythema, papulation, and scaling. More severe presentations of irritant dermatitis include Jacquet’s diaper dermatitis, perianal pseudoverrucous papules and nodules and granuloma gluteale infantum.1 These cutaneous findings also may develop in older patients with irritated peristomal skin.5
Inflammatory dermatoses such as psoriasis and seborrheic dermatitis also may erupt in the diaper region. Psoriasis commonly involves the gluteal cleft and inguinal folds, appearing as well-demarcated, pink plaques with thin white scale.5 Persistence of plaques despite topical corticosteroid application and lack of pruritus further delineates this disorder. Family history of psoriasis may provide an additional clue to the diagnosis of psoriasis. Similarly, seborrheic dermatitis may present with diaper involvement characterized by well-demarcated erythematous plaques with minimal or absent greasy yellow scale. This dermatitis commonly erupts in infants and neonates less than 6 weeks old and resolves by 6-9 months of age.1
Langerhans cell histiocytosis (LCH) represents a serious condition important to consider in the differential diagnosis of persistent recalcitrant diaper dermatitis. Often confused with seborrheic dermatitis, the diaper dermatitis of LCH may be differentiated by petechiae, purpura, and/or ulceration and atrophy of the inguinal folds.5
Management
Avoidance of the allergen constitutes the gold standard of treatment for ACD. Initial treatment involves adequate-potency topical corticosteroids; for the diaper area, low- to mid-potency corticosteroids are recommended. Changing to dye-free diapers and/or avoiding contact with the suspected allergen also is advised. Epicutaneous patch testing is recommended if the dermatitis fails to improve despite removal of the suspected offending agent.5
References
1. Pediatr Dermatol. 2014 Nov;31 Suppl 1:19-24.
2. Pediatrics. 2005 Sep;116(3):e450-2.
3. Cutis. 1994 Nov;54(5):300-2.
4. Dermatitis. 2016 May-Jun;27(3):110-8.
5. “Neonatal and Infant Dermatology,” 3rd edition (Philadelphia: Elsevier, 2015, p. 251)
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Dr. Waldman is a clinical research fellow at the hospital. Email them at pdnews@frontlinemedcom.com.
BY CATALINA MATIZ, MD, AND ANDREA WALDMAN, MD
At the time of initial evaluation, the patient was prescribed a 2-week course of topical triamcinolone 0.1% ointment. Subsequently, the cutaneous lesions clinically improved, but did not completely resolve. Mother had no recollection of infant contact with metal, plastic, or additional topical emollients or medications. On further investigation, the patient’s mother revealed the recent conversion to a different brand of diaper. The diapers had a blue and green polka dot design corresponding with the distribution of our patient’s cutaneous lesions, highly suggestive of allergic contact dermatitis (ACD).
Diaper dermatitis represents a common complaint peaking at 9-12 months of age, estimated to occur in 25% of infants in the primary care office setting.1 Irritant contact dermatitis (ICD) predominates as the most common etiology of diaper dermatitis. ICD represents a nonimmunologic response, triggered by maceration and breakdown of the cutaneous barrier, directly resulting from chronic exposure to urine and feces.1 In contrast, ACD characterizes an immunologic reaction to specific offending allergen(s).
ACD can arise in the diaper area following contact with dyes, fragrances, or other constituents of disposable diapers, wet wipes, diaper creams, or medications. Alberta et al. initially described ACD in several infants, resulting from blue, green, and pink dyes present in various disposable diapers. The dermatitis promptly resolved upon switching to dye-free diapers.2 Furthermore, topical products applied to the diaper region such as barrier creams and wet wipes may trigger allergic sensitization. Sensitization to an allergen can occur after few exposures to years after introduction. Following the initial contact reaction, re-exposure to the offending allergen can elicit a quicker cutaneous response. Case reports have detailed cases of ACD presenting in neonates as early as 1 week of age.3
If ACD is suspected as an etiologic factor in diaper dermatitis, providers should consider constituents present in topical formulations, wet wipes, and components of the diaper itself. A specific example includes mercaptobenzothiazole, a rubber additive commonly found in elastic bands of disposable diapers.2 The most common potential allergens discovered in a recent review of 63 diaper wipes, 41 topical diaper preparations, and 3 top-selling diaper brands included botanical extracts (i.e. aloe vera), alpha-tocopherol, fragrances, propylene glycol, parabens, iodopropynyl butylcarbamate, and lanolin.4
ACD may be difficult to diagnose. Obtaining a thorough clinical history is essential to diagnosis. ACD is often distinguished by its distinct cutaneous morphology, with initial lesions commonly characterized by sharply demarcated erythema associated with vesiculation and edema. The dermatitis progressively evolves to a mixed presentation of predominantly eczematous papules and plaques with crusting, scaling, and lichenification. Another clue suggestive of ACD is cutaneous involvement limited to areas of allergen contact, such as the geometric plaques corresponding to areas of skin exposure to blue and green dye in our patient.1
Differential diagnosis
In contrast to ACD, ICD commonly erupts in the perianal region in the presence of feces, especially during diarrheal episodes. Common clinical manifestations include skin maceration, erosions, erythema, papulation, and scaling. More severe presentations of irritant dermatitis include Jacquet’s diaper dermatitis, perianal pseudoverrucous papules and nodules and granuloma gluteale infantum.1 These cutaneous findings also may develop in older patients with irritated peristomal skin.5
Inflammatory dermatoses such as psoriasis and seborrheic dermatitis also may erupt in the diaper region. Psoriasis commonly involves the gluteal cleft and inguinal folds, appearing as well-demarcated, pink plaques with thin white scale.5 Persistence of plaques despite topical corticosteroid application and lack of pruritus further delineates this disorder. Family history of psoriasis may provide an additional clue to the diagnosis of psoriasis. Similarly, seborrheic dermatitis may present with diaper involvement characterized by well-demarcated erythematous plaques with minimal or absent greasy yellow scale. This dermatitis commonly erupts in infants and neonates less than 6 weeks old and resolves by 6-9 months of age.1
Langerhans cell histiocytosis (LCH) represents a serious condition important to consider in the differential diagnosis of persistent recalcitrant diaper dermatitis. Often confused with seborrheic dermatitis, the diaper dermatitis of LCH may be differentiated by petechiae, purpura, and/or ulceration and atrophy of the inguinal folds.5
Management
Avoidance of the allergen constitutes the gold standard of treatment for ACD. Initial treatment involves adequate-potency topical corticosteroids; for the diaper area, low- to mid-potency corticosteroids are recommended. Changing to dye-free diapers and/or avoiding contact with the suspected allergen also is advised. Epicutaneous patch testing is recommended if the dermatitis fails to improve despite removal of the suspected offending agent.5
References
1. Pediatr Dermatol. 2014 Nov;31 Suppl 1:19-24.
2. Pediatrics. 2005 Sep;116(3):e450-2.
3. Cutis. 1994 Nov;54(5):300-2.
4. Dermatitis. 2016 May-Jun;27(3):110-8.
5. “Neonatal and Infant Dermatology,” 3rd edition (Philadelphia: Elsevier, 2015, p. 251)
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Dr. Waldman is a clinical research fellow at the hospital. Email them at pdnews@frontlinemedcom.com.
A 10-month-old previously healthy male presents to the dermatology clinic for evaluation of a persistent annular rash. The mother reports the development of two distinct symmetrical circular lesions on the upper lateral buttocks bilaterally 7 weeks earlier. Shortly after the lesions erupted, the pediatrician diagnosed him with tinea corporis. The patient’s rash markedly worsened over the following 3 weeks, despite the use of clotrimazole 1% cream and miconazole 2% cream. Patient’s mother denies any recent travel or pets residing in the home. Other than a history of mild atopic dermatitis in the mother, the family history is noncontributory.
The patient is a well-appearing infant who is attentive and in no acute distress. On skin examination there are two symmetric well-demarcated annular erythematous eczematous plaques on the upper lateral buttocks. The annular shape presents as geometric circles. There are no other lesions present on the cutaneous surface. Patient is afebrile and vital signs are within normal limits.
Pediatric Dermatology Consult - September 2016
Onychomycosis
Onychomycosis is a nail infection caused by a variety of fungi, including dermatophytes, yeasts, and nondermatophyte molds. Tinea unguium refers specifically to nail infections caused by dermatophytes, which are the most common cause of onychomycosis; they cause 82% of cases in the U.S. hospital population.1
Onychomycosis is more prevalent in adults than in the pediatric population.2 A recent study showed that 3.22%, 0.40%, and 0.37% of adults have culture-proven dermatophyte, yeast, and nondermatophyte mold onychomycosis, respectively, while in the pediatric population, 0.14% have dermatophyte and 0.09% have yeast toenail onychomycosis.2
The likely reason for lower prevalence of onychomycosis in the pediatric population is the faster growth rate of pediatric nails, the smaller surface area susceptible to infection, and the absence of cumulative trauma and tinea pedis.2 Distal lateral subungual onychomycosis is the most common clinical pattern of onychomycosis.2
The diagnosis usually is based on strong clinical suspicion, but laboratory evidence to support a clinical diagnosis is ideal. Patients may be evaluated with a fungal culture, potassium hydroxide (KOH) preparation, or histologic evaluation of the nail clippings with periodic acid-Schiff (PAS) staining. A KOH preparation is highly specific for onychomycosis, but sensitivity depends on the specimen obtained. The histopathology of the nail clippings sample treated with a PAS stain demonstrates fungal elements and is the most sensitive test, but it does not identify the species.3 It takes a few days for the results and costs more than a KOH preparation.
Differential diagnosis
The differential for nail dystrophy in children includes trauma, fungal infection, congenital dystrophies, psoriasis, and lichen planus.4
Trauma can result in similar changes to onychomycosis, such as distal onycholysis. Unlike the most common type of onychomycosis, there is rarely distal thickening of the nail. Usually the morphology, history of trauma, and culture can be used to differentiate the two.
Congenital dystrophies often include diseases that have other clinical manifestations. Children exhibited nail alterations in diseases such as dystrophic epidermolysis bullosa, focal dermal hypoplasia, Turner syndrome, and Down syndrome.4
"Twenty-nail dystrophy," also known as trachyonychia, presents with longitudinal ridges, lost of luster, sandpaper-like rough appearance, and pitting. While the cause is not known, it may be associated with lichen planus, psoriasis, alopecia areata, and atopic dermatitis.
Nail psoriasis can present similarly to onychomycosis with subungual hyperkeratosis and onycholysis. However, distinguishing features for nail psoriasis include pitting, nail bed salmon patches (areas of yellow or pink discoloration), or "oil drop" discoloration, and other systemic findings such as cutaneous or joint findings.
Lichen planus is an inflammatory condition of unknown etiology that also can present with onycholysis with or without subungual hyperkeratosis when it involves the nail matrix. Its clinical characteristics include longitudinal ridging, nail plate thinning, and longitudinal fissuring.
Onychomadesis is the proximal separation of the nail plate from the nail matrix and bed. It is caused by temporary arrest of the nail matrix activity associated with a variety of systemic illnesses or drug exposure, and presents with "peeling" or shedding of the nail from the proximal portion of the plate. It has been noticed commonly with hand, foot, and mouth disease in children.
Etiology
The term dermatophytosis describes infections caused by members of the genera Microsporum, Trichophyton, and Epidermophyton. Trichophyton rubrum is the most common dermatophyte to cause onychomycosis.1 Risk factors for developing onychomycosis include older age, tinea pedis, psoriasis, diabetes, immunodeficiency, genetic predisposition, swimming, and living with family members who have onychomycosis.5 Tinea pedis is a major risk factor for the development of onychomycosis, with concurrent rates of the two diseases reported as high as 47%.6 Candida species may cause onychomycosis, while nondermatophyte molds (such as Acremonium, Alternaria, Aspergillus, Fusarium, Scytalidium, and Scopulariopsis species) are rarely true pathogens in immunocompetent children.2
Treatment
Onychomycosis may cause physical discomfort and pain, and may increase the risk for developing bacterial cellulitis, especially in patients with tinea pedis.7 Treatment options can include observation, if there is minimal discomfort, oral systemic antifungal medications, topical antifungal medications, and physical interventions.
While there is no systemic antifungal approved by the Food and Drug Administration for use in children, several systemic antifungals may be utilized off-label. Oral terbinafine or itraconazole are the most effective in achieving cure, with griseofulvin next most effective and fluconazole less so.8 The safety and effectiveness of these medications in children have not been established. With the exception of one case of ataxia with the use of itraconazole, adverse events for terbinafine and itraconazole treatment in children are limited to reports listed in the prescribing information and include: gastrointestinal side effects, urticaria, hepatotoxicity, neutropenia, thrombocytopenia, and cytochrome P450 enzyme inhibition.8 Terbinafine dosing for children is based on studies for tinea capitis and determined by weight: 10-20 kg, 62.5 mg/day; 20-40 kg, 125 mg/day; greater than 40 kg, 250 mg/day for 6 weeks.9 FDA prescribing information suggests a baseline liver function panel prior to initiation of the drug, but there are no recommendations on serial lab monitoring. Dosing of itraconazole in the pediatric population is not as well established.9
Topical antifungal agents can be used in pediatric nail infections that do not involve the nail matrix (lunula). Pediatric nails grow faster than adult nails and children have a thinner nail plate, which may allow better penetration of the drug, making children more likely to respond better to topical treatment.10 Topical therapy options for onychomycosis include ciclopirox and amorolfine nail lacquers, and bifonazole-urea; these require application for prolonged periods of time. Friedlander et al. showed that children with onychomycosis without nail matrix treated with ciclopirox 8% over 32 weeks had a 90% mycologic cure rate.11 Recently, new topical treatments (efinaconazole and tavaborole) became available for treatment of onychomycosis in adults and appear to be more effective.12,13 The data for these treatments in pediatric onychomycosis are being gathered, and the results will provide insight into the efficacy of these new formulations in the pediatric population.
Behavioral measures that may reduce risk of onychomycosis include: keeping feet cool and dry, wearing shoes in public areas, and avoidance of shared, unsterilized nail manicure equipment.5
References
- J Eur Acad Dermatol Venereol. 2014 Nov;28(11):1480-91.
- J Eur Acad Dermatol Venereol. 2015 Jun;29(6):1039-44.
- J Eur Acad Dermatol Venereol. 2011 Feb;25(2):235-7.
- Pediatric Dermatology 2001 Mar;18:107-9.
- J Drugs Dermatol. 2015 Oct;14(10 Suppl):s32-4.
- J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):395-402.
- Dermatology. 2004;209(4):301-7.
- Pediatr Dermatol. 2013 May-Jun;30(3):294-302.
- Tinea Pedis and Tinea Unguium, in "Red Book: 2015 Report of the Committee on Infectious Diseases, 30th Edition (Elk Grove Village, IL: American Academy of Pediatrics, 2015; 784-6).
- Am J Clin Dermatol. 2014 Dec;15(6):489-502.
- Pediatr Dermatol. 2013 May-Jun;30(3):316-22.
- J Am Acad Dermatol. 2015 Jul;73(1):62-9.
- J Am Acad Dermatol. 2013 Apr;68(4):600-8.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Mr. Ginsberg is a research associate at the hospital. Dr. Matiz and Mr. Ginsberg said they have no relevant financial disclosures.
Onychomycosis
Onychomycosis is a nail infection caused by a variety of fungi, including dermatophytes, yeasts, and nondermatophyte molds. Tinea unguium refers specifically to nail infections caused by dermatophytes, which are the most common cause of onychomycosis; they cause 82% of cases in the U.S. hospital population.1
Onychomycosis is more prevalent in adults than in the pediatric population.2 A recent study showed that 3.22%, 0.40%, and 0.37% of adults have culture-proven dermatophyte, yeast, and nondermatophyte mold onychomycosis, respectively, while in the pediatric population, 0.14% have dermatophyte and 0.09% have yeast toenail onychomycosis.2
The likely reason for lower prevalence of onychomycosis in the pediatric population is the faster growth rate of pediatric nails, the smaller surface area susceptible to infection, and the absence of cumulative trauma and tinea pedis.2 Distal lateral subungual onychomycosis is the most common clinical pattern of onychomycosis.2
The diagnosis usually is based on strong clinical suspicion, but laboratory evidence to support a clinical diagnosis is ideal. Patients may be evaluated with a fungal culture, potassium hydroxide (KOH) preparation, or histologic evaluation of the nail clippings with periodic acid-Schiff (PAS) staining. A KOH preparation is highly specific for onychomycosis, but sensitivity depends on the specimen obtained. The histopathology of the nail clippings sample treated with a PAS stain demonstrates fungal elements and is the most sensitive test, but it does not identify the species.3 It takes a few days for the results and costs more than a KOH preparation.
Differential diagnosis
The differential for nail dystrophy in children includes trauma, fungal infection, congenital dystrophies, psoriasis, and lichen planus.4
Trauma can result in similar changes to onychomycosis, such as distal onycholysis. Unlike the most common type of onychomycosis, there is rarely distal thickening of the nail. Usually the morphology, history of trauma, and culture can be used to differentiate the two.
Congenital dystrophies often include diseases that have other clinical manifestations. Children exhibited nail alterations in diseases such as dystrophic epidermolysis bullosa, focal dermal hypoplasia, Turner syndrome, and Down syndrome.4
"Twenty-nail dystrophy," also known as trachyonychia, presents with longitudinal ridges, lost of luster, sandpaper-like rough appearance, and pitting. While the cause is not known, it may be associated with lichen planus, psoriasis, alopecia areata, and atopic dermatitis.
Nail psoriasis can present similarly to onychomycosis with subungual hyperkeratosis and onycholysis. However, distinguishing features for nail psoriasis include pitting, nail bed salmon patches (areas of yellow or pink discoloration), or "oil drop" discoloration, and other systemic findings such as cutaneous or joint findings.
Lichen planus is an inflammatory condition of unknown etiology that also can present with onycholysis with or without subungual hyperkeratosis when it involves the nail matrix. Its clinical characteristics include longitudinal ridging, nail plate thinning, and longitudinal fissuring.
Onychomadesis is the proximal separation of the nail plate from the nail matrix and bed. It is caused by temporary arrest of the nail matrix activity associated with a variety of systemic illnesses or drug exposure, and presents with "peeling" or shedding of the nail from the proximal portion of the plate. It has been noticed commonly with hand, foot, and mouth disease in children.
Etiology
The term dermatophytosis describes infections caused by members of the genera Microsporum, Trichophyton, and Epidermophyton. Trichophyton rubrum is the most common dermatophyte to cause onychomycosis.1 Risk factors for developing onychomycosis include older age, tinea pedis, psoriasis, diabetes, immunodeficiency, genetic predisposition, swimming, and living with family members who have onychomycosis.5 Tinea pedis is a major risk factor for the development of onychomycosis, with concurrent rates of the two diseases reported as high as 47%.6 Candida species may cause onychomycosis, while nondermatophyte molds (such as Acremonium, Alternaria, Aspergillus, Fusarium, Scytalidium, and Scopulariopsis species) are rarely true pathogens in immunocompetent children.2
Treatment
Onychomycosis may cause physical discomfort and pain, and may increase the risk for developing bacterial cellulitis, especially in patients with tinea pedis.7 Treatment options can include observation, if there is minimal discomfort, oral systemic antifungal medications, topical antifungal medications, and physical interventions.
While there is no systemic antifungal approved by the Food and Drug Administration for use in children, several systemic antifungals may be utilized off-label. Oral terbinafine or itraconazole are the most effective in achieving cure, with griseofulvin next most effective and fluconazole less so.8 The safety and effectiveness of these medications in children have not been established. With the exception of one case of ataxia with the use of itraconazole, adverse events for terbinafine and itraconazole treatment in children are limited to reports listed in the prescribing information and include: gastrointestinal side effects, urticaria, hepatotoxicity, neutropenia, thrombocytopenia, and cytochrome P450 enzyme inhibition.8 Terbinafine dosing for children is based on studies for tinea capitis and determined by weight: 10-20 kg, 62.5 mg/day; 20-40 kg, 125 mg/day; greater than 40 kg, 250 mg/day for 6 weeks.9 FDA prescribing information suggests a baseline liver function panel prior to initiation of the drug, but there are no recommendations on serial lab monitoring. Dosing of itraconazole in the pediatric population is not as well established.9
Topical antifungal agents can be used in pediatric nail infections that do not involve the nail matrix (lunula). Pediatric nails grow faster than adult nails and children have a thinner nail plate, which may allow better penetration of the drug, making children more likely to respond better to topical treatment.10 Topical therapy options for onychomycosis include ciclopirox and amorolfine nail lacquers, and bifonazole-urea; these require application for prolonged periods of time. Friedlander et al. showed that children with onychomycosis without nail matrix treated with ciclopirox 8% over 32 weeks had a 90% mycologic cure rate.11 Recently, new topical treatments (efinaconazole and tavaborole) became available for treatment of onychomycosis in adults and appear to be more effective.12,13 The data for these treatments in pediatric onychomycosis are being gathered, and the results will provide insight into the efficacy of these new formulations in the pediatric population.
Behavioral measures that may reduce risk of onychomycosis include: keeping feet cool and dry, wearing shoes in public areas, and avoidance of shared, unsterilized nail manicure equipment.5
References
- J Eur Acad Dermatol Venereol. 2014 Nov;28(11):1480-91.
- J Eur Acad Dermatol Venereol. 2015 Jun;29(6):1039-44.
- J Eur Acad Dermatol Venereol. 2011 Feb;25(2):235-7.
- Pediatric Dermatology 2001 Mar;18:107-9.
- J Drugs Dermatol. 2015 Oct;14(10 Suppl):s32-4.
- J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):395-402.
- Dermatology. 2004;209(4):301-7.
- Pediatr Dermatol. 2013 May-Jun;30(3):294-302.
- Tinea Pedis and Tinea Unguium, in "Red Book: 2015 Report of the Committee on Infectious Diseases, 30th Edition (Elk Grove Village, IL: American Academy of Pediatrics, 2015; 784-6).
- Am J Clin Dermatol. 2014 Dec;15(6):489-502.
- Pediatr Dermatol. 2013 May-Jun;30(3):316-22.
- J Am Acad Dermatol. 2015 Jul;73(1):62-9.
- J Am Acad Dermatol. 2013 Apr;68(4):600-8.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Mr. Ginsberg is a research associate at the hospital. Dr. Matiz and Mr. Ginsberg said they have no relevant financial disclosures.
Onychomycosis
Onychomycosis is a nail infection caused by a variety of fungi, including dermatophytes, yeasts, and nondermatophyte molds. Tinea unguium refers specifically to nail infections caused by dermatophytes, which are the most common cause of onychomycosis; they cause 82% of cases in the U.S. hospital population.1
Onychomycosis is more prevalent in adults than in the pediatric population.2 A recent study showed that 3.22%, 0.40%, and 0.37% of adults have culture-proven dermatophyte, yeast, and nondermatophyte mold onychomycosis, respectively, while in the pediatric population, 0.14% have dermatophyte and 0.09% have yeast toenail onychomycosis.2
The likely reason for lower prevalence of onychomycosis in the pediatric population is the faster growth rate of pediatric nails, the smaller surface area susceptible to infection, and the absence of cumulative trauma and tinea pedis.2 Distal lateral subungual onychomycosis is the most common clinical pattern of onychomycosis.2
The diagnosis usually is based on strong clinical suspicion, but laboratory evidence to support a clinical diagnosis is ideal. Patients may be evaluated with a fungal culture, potassium hydroxide (KOH) preparation, or histologic evaluation of the nail clippings with periodic acid-Schiff (PAS) staining. A KOH preparation is highly specific for onychomycosis, but sensitivity depends on the specimen obtained. The histopathology of the nail clippings sample treated with a PAS stain demonstrates fungal elements and is the most sensitive test, but it does not identify the species.3 It takes a few days for the results and costs more than a KOH preparation.
Differential diagnosis
The differential for nail dystrophy in children includes trauma, fungal infection, congenital dystrophies, psoriasis, and lichen planus.4
Trauma can result in similar changes to onychomycosis, such as distal onycholysis. Unlike the most common type of onychomycosis, there is rarely distal thickening of the nail. Usually the morphology, history of trauma, and culture can be used to differentiate the two.
Congenital dystrophies often include diseases that have other clinical manifestations. Children exhibited nail alterations in diseases such as dystrophic epidermolysis bullosa, focal dermal hypoplasia, Turner syndrome, and Down syndrome.4
"Twenty-nail dystrophy," also known as trachyonychia, presents with longitudinal ridges, lost of luster, sandpaper-like rough appearance, and pitting. While the cause is not known, it may be associated with lichen planus, psoriasis, alopecia areata, and atopic dermatitis.
Nail psoriasis can present similarly to onychomycosis with subungual hyperkeratosis and onycholysis. However, distinguishing features for nail psoriasis include pitting, nail bed salmon patches (areas of yellow or pink discoloration), or "oil drop" discoloration, and other systemic findings such as cutaneous or joint findings.
Lichen planus is an inflammatory condition of unknown etiology that also can present with onycholysis with or without subungual hyperkeratosis when it involves the nail matrix. Its clinical characteristics include longitudinal ridging, nail plate thinning, and longitudinal fissuring.
Onychomadesis is the proximal separation of the nail plate from the nail matrix and bed. It is caused by temporary arrest of the nail matrix activity associated with a variety of systemic illnesses or drug exposure, and presents with "peeling" or shedding of the nail from the proximal portion of the plate. It has been noticed commonly with hand, foot, and mouth disease in children.
Etiology
The term dermatophytosis describes infections caused by members of the genera Microsporum, Trichophyton, and Epidermophyton. Trichophyton rubrum is the most common dermatophyte to cause onychomycosis.1 Risk factors for developing onychomycosis include older age, tinea pedis, psoriasis, diabetes, immunodeficiency, genetic predisposition, swimming, and living with family members who have onychomycosis.5 Tinea pedis is a major risk factor for the development of onychomycosis, with concurrent rates of the two diseases reported as high as 47%.6 Candida species may cause onychomycosis, while nondermatophyte molds (such as Acremonium, Alternaria, Aspergillus, Fusarium, Scytalidium, and Scopulariopsis species) are rarely true pathogens in immunocompetent children.2
Treatment
Onychomycosis may cause physical discomfort and pain, and may increase the risk for developing bacterial cellulitis, especially in patients with tinea pedis.7 Treatment options can include observation, if there is minimal discomfort, oral systemic antifungal medications, topical antifungal medications, and physical interventions.
While there is no systemic antifungal approved by the Food and Drug Administration for use in children, several systemic antifungals may be utilized off-label. Oral terbinafine or itraconazole are the most effective in achieving cure, with griseofulvin next most effective and fluconazole less so.8 The safety and effectiveness of these medications in children have not been established. With the exception of one case of ataxia with the use of itraconazole, adverse events for terbinafine and itraconazole treatment in children are limited to reports listed in the prescribing information and include: gastrointestinal side effects, urticaria, hepatotoxicity, neutropenia, thrombocytopenia, and cytochrome P450 enzyme inhibition.8 Terbinafine dosing for children is based on studies for tinea capitis and determined by weight: 10-20 kg, 62.5 mg/day; 20-40 kg, 125 mg/day; greater than 40 kg, 250 mg/day for 6 weeks.9 FDA prescribing information suggests a baseline liver function panel prior to initiation of the drug, but there are no recommendations on serial lab monitoring. Dosing of itraconazole in the pediatric population is not as well established.9
Topical antifungal agents can be used in pediatric nail infections that do not involve the nail matrix (lunula). Pediatric nails grow faster than adult nails and children have a thinner nail plate, which may allow better penetration of the drug, making children more likely to respond better to topical treatment.10 Topical therapy options for onychomycosis include ciclopirox and amorolfine nail lacquers, and bifonazole-urea; these require application for prolonged periods of time. Friedlander et al. showed that children with onychomycosis without nail matrix treated with ciclopirox 8% over 32 weeks had a 90% mycologic cure rate.11 Recently, new topical treatments (efinaconazole and tavaborole) became available for treatment of onychomycosis in adults and appear to be more effective.12,13 The data for these treatments in pediatric onychomycosis are being gathered, and the results will provide insight into the efficacy of these new formulations in the pediatric population.
Behavioral measures that may reduce risk of onychomycosis include: keeping feet cool and dry, wearing shoes in public areas, and avoidance of shared, unsterilized nail manicure equipment.5
References
- J Eur Acad Dermatol Venereol. 2014 Nov;28(11):1480-91.
- J Eur Acad Dermatol Venereol. 2015 Jun;29(6):1039-44.
- J Eur Acad Dermatol Venereol. 2011 Feb;25(2):235-7.
- Pediatric Dermatology 2001 Mar;18:107-9.
- J Drugs Dermatol. 2015 Oct;14(10 Suppl):s32-4.
- J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):395-402.
- Dermatology. 2004;209(4):301-7.
- Pediatr Dermatol. 2013 May-Jun;30(3):294-302.
- Tinea Pedis and Tinea Unguium, in "Red Book: 2015 Report of the Committee on Infectious Diseases, 30th Edition (Elk Grove Village, IL: American Academy of Pediatrics, 2015; 784-6).
- Am J Clin Dermatol. 2014 Dec;15(6):489-502.
- Pediatr Dermatol. 2013 May-Jun;30(3):316-22.
- J Am Acad Dermatol. 2015 Jul;73(1):62-9.
- J Am Acad Dermatol. 2013 Apr;68(4):600-8.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Mr. Ginsberg is a research associate at the hospital. Dr. Matiz and Mr. Ginsberg said they have no relevant financial disclosures.
A 5-year-old boy presents to his physician for evaluation of "toenail issues" for at least 1 year. The family has noticed changes of the his right great toenail, which they thought might be due to "tight shoes," stating that the boy has been growing out of his shoes quickly. In the last 6 months, his mother has noted a "crumbly" nail with yellow discoloration. There has been no prior treatment, although his parents now are replacing his sneakers more regularly to allow him "room to grow." He has no history of toe swelling or pain. He is otherwise healthy, and he has no history of psoriasis or eczema. He has had no significant viral infections, although some children in his school did have hand, foot, and mouth disease several months ago. His mother states that her husband has athlete's foot, which has been treated with "creams and sprays." Physical exam The toenail of the right foot great toe has thickening of the distal part of the nail, with onycholysis (separation of the nail plate from the nail bed), yellow discoloration, and subungual debris. The right toe shows some chronic dystrophy. Other toenails appear normal, and the skin of the feet is otherwise unremarkable.