User login
CAR T-cell trial placed on hold again

Once again, the phase 2 ROCKET trial has been placed on clinical hold due to patient deaths.
In this trial, researchers are testing the chimeric antigen receptor (CAR) T-cell therapy JCAR015 in adults with relapsed or refractory B-cell acute lymphoblastic leukemia.
Juno Therapeutics, Inc. voluntarily put the trial on hold after 2 more patients suffered cerebral edema and died.
A total of 5 patients have died of cerebral edema in this trial.
Juno has notified the US Food and Drug Administration (FDA) of the latest clinical hold on the ROCKET trial and is working with the agency and the company’s data and safety monitoring board to determine next steps.
The ROCKET trial was previously placed on clinical hold in July, after 3 patients died of cerebral edema. The FDA lifted the hold less than a week later, allowing the trial to continue with a revised protocol.
Juno had theorized the deaths were likely a result of adding fludarabine to the conditioning regimen.
Patients enrolled in ROCKET initially received conditioning with cyclophosphamide alone, but researchers later decided to add fludarabine in the hopes of increasing efficacy. Previous trials of 2 other CAR T-cell therapies, JCAR014 and JCAR017, had suggested that adding fludarabine to conditioning could increase efficacy.
However, in the ROCKET trial, the addition of fludarabine was associated with an increase in the incidence of severe neurotoxicity and the 3 deaths from cerebral edema.
Juno said that, although other factors may have contributed to the deaths, fludarabine was the most likely culprit. So the company revised the trial protocol, and the FDA allowed ROCKET to continue with a conditioning regimen consisting of cyclophosphamide alone.
Since that time, 12 patients have been treated on the ROCKET trial. Two patients who were treated the week of November 14 developed cerebral edema and died on November 22 and 23, respectively.
In a conference call, Juno’s Chief Medical Officer Mark Gilbert, MD, said the etiology of cerebral edema is multi-factorial, and Juno will need more time to draw even preliminary conclusions about what factors contributed to the cases of cerebral edema in ROCKET.
Right now, the company is assessing data from the cases and the trial and is evaluating its options regarding the JCAR015 program.
Juno’s President and CEO Hans Bishop said the options for JCAR015 going forward include continuing the ROCKET trial with a modified protocol, beginning a new study of JCAR015, and terminating the JCAR015 development program.
Bishop said the company expects to provide an update on the status of ROCKET and JCAR015 in the next few weeks.
Juno’s other trials and plans for its other CD19-directed CAR T-cell product candidates are not affected by the issues with ROCKET and JCAR015.
ROCKET is not the first trial of JCAR015 to be placed on hold. The phase 1 trial of the therapy was placed on clinical hold in 2014, after 2 patients died of cytokine release syndrome.
That hold was lifted following changes to enrollment criteria and dosing. Results from this trial were presented at ASCO 2015 and ASCO 2016.
Once again, the phase 2 ROCKET trial has been placed on clinical hold due to patient deaths.
In this trial, researchers are testing the chimeric antigen receptor (CAR) T-cell therapy JCAR015 in adults with relapsed or refractory B-cell acute lymphoblastic leukemia.
Juno Therapeutics, Inc. voluntarily put the trial on hold after 2 more patients suffered cerebral edema and died.
A total of 5 patients have died of cerebral edema in this trial.
Juno has notified the US Food and Drug Administration (FDA) of the latest clinical hold on the ROCKET trial and is working with the agency and the company’s data and safety monitoring board to determine next steps.
The ROCKET trial was previously placed on clinical hold in July, after 3 patients died of cerebral edema. The FDA lifted the hold less than a week later, allowing the trial to continue with a revised protocol.
Juno had theorized the deaths were likely a result of adding fludarabine to the conditioning regimen.
Patients enrolled in ROCKET initially received conditioning with cyclophosphamide alone, but researchers later decided to add fludarabine in the hopes of increasing efficacy. Previous trials of 2 other CAR T-cell therapies, JCAR014 and JCAR017, had suggested that adding fludarabine to conditioning could increase efficacy.
However, in the ROCKET trial, the addition of fludarabine was associated with an increase in the incidence of severe neurotoxicity and the 3 deaths from cerebral edema.
Juno said that, although other factors may have contributed to the deaths, fludarabine was the most likely culprit. So the company revised the trial protocol, and the FDA allowed ROCKET to continue with a conditioning regimen consisting of cyclophosphamide alone.
Since that time, 12 patients have been treated on the ROCKET trial. Two patients who were treated the week of November 14 developed cerebral edema and died on November 22 and 23, respectively.
In a conference call, Juno’s Chief Medical Officer Mark Gilbert, MD, said the etiology of cerebral edema is multi-factorial, and Juno will need more time to draw even preliminary conclusions about what factors contributed to the cases of cerebral edema in ROCKET.
Right now, the company is assessing data from the cases and the trial and is evaluating its options regarding the JCAR015 program.
Juno’s President and CEO Hans Bishop said the options for JCAR015 going forward include continuing the ROCKET trial with a modified protocol, beginning a new study of JCAR015, and terminating the JCAR015 development program.
Bishop said the company expects to provide an update on the status of ROCKET and JCAR015 in the next few weeks.
Juno’s other trials and plans for its other CD19-directed CAR T-cell product candidates are not affected by the issues with ROCKET and JCAR015.
ROCKET is not the first trial of JCAR015 to be placed on hold. The phase 1 trial of the therapy was placed on clinical hold in 2014, after 2 patients died of cytokine release syndrome.
That hold was lifted following changes to enrollment criteria and dosing. Results from this trial were presented at ASCO 2015 and ASCO 2016.
Once again, the phase 2 ROCKET trial has been placed on clinical hold due to patient deaths.
In this trial, researchers are testing the chimeric antigen receptor (CAR) T-cell therapy JCAR015 in adults with relapsed or refractory B-cell acute lymphoblastic leukemia.
Juno Therapeutics, Inc. voluntarily put the trial on hold after 2 more patients suffered cerebral edema and died.
A total of 5 patients have died of cerebral edema in this trial.
Juno has notified the US Food and Drug Administration (FDA) of the latest clinical hold on the ROCKET trial and is working with the agency and the company’s data and safety monitoring board to determine next steps.
The ROCKET trial was previously placed on clinical hold in July, after 3 patients died of cerebral edema. The FDA lifted the hold less than a week later, allowing the trial to continue with a revised protocol.
Juno had theorized the deaths were likely a result of adding fludarabine to the conditioning regimen.
Patients enrolled in ROCKET initially received conditioning with cyclophosphamide alone, but researchers later decided to add fludarabine in the hopes of increasing efficacy. Previous trials of 2 other CAR T-cell therapies, JCAR014 and JCAR017, had suggested that adding fludarabine to conditioning could increase efficacy.
However, in the ROCKET trial, the addition of fludarabine was associated with an increase in the incidence of severe neurotoxicity and the 3 deaths from cerebral edema.
Juno said that, although other factors may have contributed to the deaths, fludarabine was the most likely culprit. So the company revised the trial protocol, and the FDA allowed ROCKET to continue with a conditioning regimen consisting of cyclophosphamide alone.
Since that time, 12 patients have been treated on the ROCKET trial. Two patients who were treated the week of November 14 developed cerebral edema and died on November 22 and 23, respectively.
In a conference call, Juno’s Chief Medical Officer Mark Gilbert, MD, said the etiology of cerebral edema is multi-factorial, and Juno will need more time to draw even preliminary conclusions about what factors contributed to the cases of cerebral edema in ROCKET.
Right now, the company is assessing data from the cases and the trial and is evaluating its options regarding the JCAR015 program.
Juno’s President and CEO Hans Bishop said the options for JCAR015 going forward include continuing the ROCKET trial with a modified protocol, beginning a new study of JCAR015, and terminating the JCAR015 development program.
Bishop said the company expects to provide an update on the status of ROCKET and JCAR015 in the next few weeks.
Juno’s other trials and plans for its other CD19-directed CAR T-cell product candidates are not affected by the issues with ROCKET and JCAR015.
ROCKET is not the first trial of JCAR015 to be placed on hold. The phase 1 trial of the therapy was placed on clinical hold in 2014, after 2 patients died of cytokine release syndrome.
That hold was lifted following changes to enrollment criteria and dosing. Results from this trial were presented at ASCO 2015 and ASCO 2016.
Ph-like ALL is highly prevalent in adults
Philadelphia chromosome–like acute lymphoblastic leukemia, a high-risk subtype of childhood ALL, accounts for more than 24% of ALL in adults and is associated with poor outcome, according to gene-expression profiling in nearly 800 adults with B-cell ALL.
In the 798 subjects aged 21-86 years, Philadelphia chromosome–like (Ph-like) ALL accounted for 27.9% of ALL cases in those aged 21-39 years, 20.4% of cases in those aged 40-59 years, and 24% of cases in those aged 60-86 years. The overall 5-year event-free survival rate was inferior in those with Ph-like ALL vs. non–Ph-like ALL (22.5% vs. 49.3%), as was the 5-year overall survival (23.8% vs. 52.4%). Increasing age also was associated with inferior outcomes: 5-year event-free survival was 40.4%, 29.8%, and 18.9% in the age groups, respectively, and 5-year overall survival was 45.2%, 35.1%, and 28.4% in the groups, respectively, Kathryn G. Roberts, Ph.D., of St. Jude Children’s Research Hospital in Memphis and her colleagues reported online ahead of print (J Clin Oncol. 2016 Nov 21. doi: 10.1200/JCO.2016.69.0073).
Ph-like ALL in children is characterized by kinase-activating alterations amenable to treatment with tyrosine kinase inhibitors, but the prevalence in adults was unclear.
“These findings warrant the development of clinical trials in adults that assess the efficacy of TKIs, similar to those that are being established for pediatric ALL,” Dr. Roberts and her associates wrote.
This study was supported by grants and other awards, some to individual authors, from the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital; Stand Up to Cancer, St. Baldrick’s Foundation, the American Society of Hematology, the Lady Tata Memorial Trust, the Leukemia Research Foundation, the National Cancer Institute, and the National Institutes of Health. Dr. Roberts reported that she is the inventor on a pending patent application related to gene-expression signatures for detection of underlying Philadelphia chromosome-live events and therapeutic targeting in leukemia.
Philadelphia chromosome–like acute lymphoblastic leukemia, a high-risk subtype of childhood ALL, accounts for more than 24% of ALL in adults and is associated with poor outcome, according to gene-expression profiling in nearly 800 adults with B-cell ALL.
In the 798 subjects aged 21-86 years, Philadelphia chromosome–like (Ph-like) ALL accounted for 27.9% of ALL cases in those aged 21-39 years, 20.4% of cases in those aged 40-59 years, and 24% of cases in those aged 60-86 years. The overall 5-year event-free survival rate was inferior in those with Ph-like ALL vs. non–Ph-like ALL (22.5% vs. 49.3%), as was the 5-year overall survival (23.8% vs. 52.4%). Increasing age also was associated with inferior outcomes: 5-year event-free survival was 40.4%, 29.8%, and 18.9% in the age groups, respectively, and 5-year overall survival was 45.2%, 35.1%, and 28.4% in the groups, respectively, Kathryn G. Roberts, Ph.D., of St. Jude Children’s Research Hospital in Memphis and her colleagues reported online ahead of print (J Clin Oncol. 2016 Nov 21. doi: 10.1200/JCO.2016.69.0073).
Ph-like ALL in children is characterized by kinase-activating alterations amenable to treatment with tyrosine kinase inhibitors, but the prevalence in adults was unclear.
“These findings warrant the development of clinical trials in adults that assess the efficacy of TKIs, similar to those that are being established for pediatric ALL,” Dr. Roberts and her associates wrote.
This study was supported by grants and other awards, some to individual authors, from the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital; Stand Up to Cancer, St. Baldrick’s Foundation, the American Society of Hematology, the Lady Tata Memorial Trust, the Leukemia Research Foundation, the National Cancer Institute, and the National Institutes of Health. Dr. Roberts reported that she is the inventor on a pending patent application related to gene-expression signatures for detection of underlying Philadelphia chromosome-live events and therapeutic targeting in leukemia.
Philadelphia chromosome–like acute lymphoblastic leukemia, a high-risk subtype of childhood ALL, accounts for more than 24% of ALL in adults and is associated with poor outcome, according to gene-expression profiling in nearly 800 adults with B-cell ALL.
In the 798 subjects aged 21-86 years, Philadelphia chromosome–like (Ph-like) ALL accounted for 27.9% of ALL cases in those aged 21-39 years, 20.4% of cases in those aged 40-59 years, and 24% of cases in those aged 60-86 years. The overall 5-year event-free survival rate was inferior in those with Ph-like ALL vs. non–Ph-like ALL (22.5% vs. 49.3%), as was the 5-year overall survival (23.8% vs. 52.4%). Increasing age also was associated with inferior outcomes: 5-year event-free survival was 40.4%, 29.8%, and 18.9% in the age groups, respectively, and 5-year overall survival was 45.2%, 35.1%, and 28.4% in the groups, respectively, Kathryn G. Roberts, Ph.D., of St. Jude Children’s Research Hospital in Memphis and her colleagues reported online ahead of print (J Clin Oncol. 2016 Nov 21. doi: 10.1200/JCO.2016.69.0073).
Ph-like ALL in children is characterized by kinase-activating alterations amenable to treatment with tyrosine kinase inhibitors, but the prevalence in adults was unclear.
“These findings warrant the development of clinical trials in adults that assess the efficacy of TKIs, similar to those that are being established for pediatric ALL,” Dr. Roberts and her associates wrote.
This study was supported by grants and other awards, some to individual authors, from the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital; Stand Up to Cancer, St. Baldrick’s Foundation, the American Society of Hematology, the Lady Tata Memorial Trust, the Leukemia Research Foundation, the National Cancer Institute, and the National Institutes of Health. Dr. Roberts reported that she is the inventor on a pending patent application related to gene-expression signatures for detection of underlying Philadelphia chromosome-live events and therapeutic targeting in leukemia.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point:
Major finding: Ph-like ALL accounted for 27.9% of ALL cases in adults aged 21-39 years, 20.4% of cases in those aged 40-59 years, and 24% of cases in those aged 60-86 years.
Data source: Gene-expression profiling of 798 patients.
Disclosures: This study was supported by grants and other awards, some to individual authors, from the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital; Stand Up to Cancer, St. Baldrick’s Foundation, the American Society of Hematology, the Lady Tata Memorial Trust, the Leukemia Research Foundation, the National Cancer Institute, and the National Institutes of Health. Dr. Roberts reported that she is the inventor on a pending patent application related to gene-expression signatures for detection of underlying Philadelphia chromosome–like events and therapeutic targeting in leukemia.
ALL subtype ‘highly prevalent’ in adults

Photo courtesy of St. Jude
Children’s Research Hospital
Researchers have found evidence to suggest that a high-risk subtype of acute lymphoblastic leukemia (ALL) is “highly prevalent” in adults with ALL.
In a study of nearly 800 adults with ALL, roughly a quarter of the patients had Philadelphia chromosome-like (Ph-like) ALL.
Patients with Ph-like ALL had inferior overall survival (OS) and event-free survival (EFS), but most of them also had kinase-activating alterations that suggest they might respond well to tyrosine kinase inhibitors.
The researchers reported these findings in the Journal of Clinical Oncology.
“This study establishes that a large percentage of adults with ALL have this high-risk subtype,” said study author Charles Mullighan, MD, MBBS, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The finding provides a compelling reason to identify those with Ph-like ALL and move forward with clinical trials of these targeted therapies in combination with current chemotherapeutic regimens.”
This study builds on previous research, which suggested that Ph-like ALL becomes more common with age, is associated with poor prognosis, and is characterized by genomic alterations that appear to make patients responsive to treatment with tyrosine kinase inhibitors.
“Our 2014 findings that the prevalence of Ph-like ALL increased with age and was particularly common in young adults generated tremendous interest because adult ALL is difficult to treat,” Dr Mullighan said. “In this study, we determined that the prevalence remains high across the age spectrum of adults with ALL.”
Prevalence and outcomes
The study included 798 adults who were between the ages of 21 and 86 when diagnosed with ALL. A total of 194 patients (24%) had Ph-like ALL.
The incidence of Ph-like ALL was 27.9% among young adults (ages 21 to 39), 20.4% in adults (ages 40 to 59), and 24.0% in older adults (ages 60 to 86).
Patients with Ph-like ALL had significantly inferior 5-year OS compared to patients with non-Ph-like ALL—23.8% and 52.4%, respectively (P<0.001). The same was true for 5-year EFS—22.5% and 49.3%, respectively (P<0.001).
Among Ph-like ALL patients, the 5-year EFS rates were 40.4% for young adults, 29.8% for adults, and 18.9% for older adults. The 5-year OS rates were 45.2%, 35.1%, and 16.2%, respectively.
Genomic analysis
The researchers performed genomic analysis of 180 of the Ph-like ALL cases and found that 88% had kinase-activating alterations.
This included CRLF2 rearrangements in 51% of cases, JAK2 or EPOR rearrangements in 12.4%, ABL class fusions in 9.8%, other JAK-STAT sequence mutations in 7.2%, other kinase alterations in 4.1%, and Ras pathway mutations in 3.6%.
“Our comprehensive sequencing showed that Ph-like ALL in adults is the most genetically diverse subtype of leukemia that has been described,” said study author Kathryn Roberts, PhD, of St. Jude.
“Cumulatively, more than 50 different chromosomal rearrangements involving 15 different kinases and cytokine receptors have been identified. In this study, we identified 11 chromosomal rearrangements that are new to Ph-like ALL.”
These 11 rearrangements are FIP1L1-PDGFRA, SNX29-PDGFRB, SMU1-JAK2, ZNF340-JAK2, THADA-EPOR, SMARCA4-TYK2, ZNF340-TYK2, ZYMM2-FLT3, DNTT-BLNK, TMEM2-PTK2B, and KANK1-CBL1.
The diversity of kinase-activating alterations in Ph-like ALL has important clinical implications, said study author Hagop Kantarjian, MD, of the University of Texas MD Anderson Cancer Center in Houston.
“It is important that we now identify patients with Ph-like ALL at diagnosis to provide optimal treatment with targeted agents,” he said.
The findings also highlight the importance of centralized comprehensive genomic sequencing for patients, said study author Elisabeth Paietta, PhD, of the Montefiore Health System and Albert Einstein College of Medicine in Bronx, New York.
“Lymphoblasts from almost half of the patients with Ph-like ALL harbor a genomic rearrangement of CRLF2, which can be detected by flow cytometry using an antibody to CRLF2,” Dr Paietta said. “This is very important as it allows a quick characterization of this Ph-like ALL subtype prior to any detailed sequencing.” ![]()
Photo courtesy of St. Jude
Children’s Research Hospital
Researchers have found evidence to suggest that a high-risk subtype of acute lymphoblastic leukemia (ALL) is “highly prevalent” in adults with ALL.
In a study of nearly 800 adults with ALL, roughly a quarter of the patients had Philadelphia chromosome-like (Ph-like) ALL.
Patients with Ph-like ALL had inferior overall survival (OS) and event-free survival (EFS), but most of them also had kinase-activating alterations that suggest they might respond well to tyrosine kinase inhibitors.
The researchers reported these findings in the Journal of Clinical Oncology.
“This study establishes that a large percentage of adults with ALL have this high-risk subtype,” said study author Charles Mullighan, MD, MBBS, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The finding provides a compelling reason to identify those with Ph-like ALL and move forward with clinical trials of these targeted therapies in combination with current chemotherapeutic regimens.”
This study builds on previous research, which suggested that Ph-like ALL becomes more common with age, is associated with poor prognosis, and is characterized by genomic alterations that appear to make patients responsive to treatment with tyrosine kinase inhibitors.
“Our 2014 findings that the prevalence of Ph-like ALL increased with age and was particularly common in young adults generated tremendous interest because adult ALL is difficult to treat,” Dr Mullighan said. “In this study, we determined that the prevalence remains high across the age spectrum of adults with ALL.”
Prevalence and outcomes
The study included 798 adults who were between the ages of 21 and 86 when diagnosed with ALL. A total of 194 patients (24%) had Ph-like ALL.
The incidence of Ph-like ALL was 27.9% among young adults (ages 21 to 39), 20.4% in adults (ages 40 to 59), and 24.0% in older adults (ages 60 to 86).
Patients with Ph-like ALL had significantly inferior 5-year OS compared to patients with non-Ph-like ALL—23.8% and 52.4%, respectively (P<0.001). The same was true for 5-year EFS—22.5% and 49.3%, respectively (P<0.001).
Among Ph-like ALL patients, the 5-year EFS rates were 40.4% for young adults, 29.8% for adults, and 18.9% for older adults. The 5-year OS rates were 45.2%, 35.1%, and 16.2%, respectively.
Genomic analysis
The researchers performed genomic analysis of 180 of the Ph-like ALL cases and found that 88% had kinase-activating alterations.
This included CRLF2 rearrangements in 51% of cases, JAK2 or EPOR rearrangements in 12.4%, ABL class fusions in 9.8%, other JAK-STAT sequence mutations in 7.2%, other kinase alterations in 4.1%, and Ras pathway mutations in 3.6%.
“Our comprehensive sequencing showed that Ph-like ALL in adults is the most genetically diverse subtype of leukemia that has been described,” said study author Kathryn Roberts, PhD, of St. Jude.
“Cumulatively, more than 50 different chromosomal rearrangements involving 15 different kinases and cytokine receptors have been identified. In this study, we identified 11 chromosomal rearrangements that are new to Ph-like ALL.”
These 11 rearrangements are FIP1L1-PDGFRA, SNX29-PDGFRB, SMU1-JAK2, ZNF340-JAK2, THADA-EPOR, SMARCA4-TYK2, ZNF340-TYK2, ZYMM2-FLT3, DNTT-BLNK, TMEM2-PTK2B, and KANK1-CBL1.
The diversity of kinase-activating alterations in Ph-like ALL has important clinical implications, said study author Hagop Kantarjian, MD, of the University of Texas MD Anderson Cancer Center in Houston.
“It is important that we now identify patients with Ph-like ALL at diagnosis to provide optimal treatment with targeted agents,” he said.
The findings also highlight the importance of centralized comprehensive genomic sequencing for patients, said study author Elisabeth Paietta, PhD, of the Montefiore Health System and Albert Einstein College of Medicine in Bronx, New York.
“Lymphoblasts from almost half of the patients with Ph-like ALL harbor a genomic rearrangement of CRLF2, which can be detected by flow cytometry using an antibody to CRLF2,” Dr Paietta said. “This is very important as it allows a quick characterization of this Ph-like ALL subtype prior to any detailed sequencing.” ![]()
Photo courtesy of St. Jude
Children’s Research Hospital
Researchers have found evidence to suggest that a high-risk subtype of acute lymphoblastic leukemia (ALL) is “highly prevalent” in adults with ALL.
In a study of nearly 800 adults with ALL, roughly a quarter of the patients had Philadelphia chromosome-like (Ph-like) ALL.
Patients with Ph-like ALL had inferior overall survival (OS) and event-free survival (EFS), but most of them also had kinase-activating alterations that suggest they might respond well to tyrosine kinase inhibitors.
The researchers reported these findings in the Journal of Clinical Oncology.
“This study establishes that a large percentage of adults with ALL have this high-risk subtype,” said study author Charles Mullighan, MD, MBBS, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The finding provides a compelling reason to identify those with Ph-like ALL and move forward with clinical trials of these targeted therapies in combination with current chemotherapeutic regimens.”
This study builds on previous research, which suggested that Ph-like ALL becomes more common with age, is associated with poor prognosis, and is characterized by genomic alterations that appear to make patients responsive to treatment with tyrosine kinase inhibitors.
“Our 2014 findings that the prevalence of Ph-like ALL increased with age and was particularly common in young adults generated tremendous interest because adult ALL is difficult to treat,” Dr Mullighan said. “In this study, we determined that the prevalence remains high across the age spectrum of adults with ALL.”
Prevalence and outcomes
The study included 798 adults who were between the ages of 21 and 86 when diagnosed with ALL. A total of 194 patients (24%) had Ph-like ALL.
The incidence of Ph-like ALL was 27.9% among young adults (ages 21 to 39), 20.4% in adults (ages 40 to 59), and 24.0% in older adults (ages 60 to 86).
Patients with Ph-like ALL had significantly inferior 5-year OS compared to patients with non-Ph-like ALL—23.8% and 52.4%, respectively (P<0.001). The same was true for 5-year EFS—22.5% and 49.3%, respectively (P<0.001).
Among Ph-like ALL patients, the 5-year EFS rates were 40.4% for young adults, 29.8% for adults, and 18.9% for older adults. The 5-year OS rates were 45.2%, 35.1%, and 16.2%, respectively.
Genomic analysis
The researchers performed genomic analysis of 180 of the Ph-like ALL cases and found that 88% had kinase-activating alterations.
This included CRLF2 rearrangements in 51% of cases, JAK2 or EPOR rearrangements in 12.4%, ABL class fusions in 9.8%, other JAK-STAT sequence mutations in 7.2%, other kinase alterations in 4.1%, and Ras pathway mutations in 3.6%.
“Our comprehensive sequencing showed that Ph-like ALL in adults is the most genetically diverse subtype of leukemia that has been described,” said study author Kathryn Roberts, PhD, of St. Jude.
“Cumulatively, more than 50 different chromosomal rearrangements involving 15 different kinases and cytokine receptors have been identified. In this study, we identified 11 chromosomal rearrangements that are new to Ph-like ALL.”
These 11 rearrangements are FIP1L1-PDGFRA, SNX29-PDGFRB, SMU1-JAK2, ZNF340-JAK2, THADA-EPOR, SMARCA4-TYK2, ZNF340-TYK2, ZYMM2-FLT3, DNTT-BLNK, TMEM2-PTK2B, and KANK1-CBL1.
The diversity of kinase-activating alterations in Ph-like ALL has important clinical implications, said study author Hagop Kantarjian, MD, of the University of Texas MD Anderson Cancer Center in Houston.
“It is important that we now identify patients with Ph-like ALL at diagnosis to provide optimal treatment with targeted agents,” he said.
The findings also highlight the importance of centralized comprehensive genomic sequencing for patients, said study author Elisabeth Paietta, PhD, of the Montefiore Health System and Albert Einstein College of Medicine in Bronx, New York.
“Lymphoblasts from almost half of the patients with Ph-like ALL harbor a genomic rearrangement of CRLF2, which can be detected by flow cytometry using an antibody to CRLF2,” Dr Paietta said. “This is very important as it allows a quick characterization of this Ph-like ALL subtype prior to any detailed sequencing.” ![]()
Team develops model of common infant ALL

Photo by Petr Kratochvil
After trying for nearly 2 decades, researchers have created a mouse model of t(4;11) pro-B acute lymphoblastic
leukemia (ALL).
The team said this model, described in Cancer Cell, mimics the human disease phenotypically and molecularly.
This type of ALL, which is common in infants, results from the translocation t(4;11)(q21;q23), which fuses the mixed-lineage leukemia (MLL) gene on chromosome 11 to the ALL-1 fused gene on chromosome 4 (AF4).
“For 20 years, scientists have repeatedly tried and consistently failed to make a model of MLL-AF4 pro-B acute lymphoblastic leukemia,” said study author Michael Thirman, MD, of the University of Chicago in Illinois.
“Even though we understood the basic genetic flaw, no one had been able create a mouse model that mimicked the human disease, which is crucial for evaluating potential therapies.”
That frustrated many researchers, who shifted their focus to test alternative hypotheses on the causes of t(4;11) pro-B ALL or refocused their laboratories to study different aspects of the disease.
However, Dr Thirman and his colleagues began working on this problem “years ago,” he said, and stayed with it.
The team identified 2 hurdles. The first was a problem with the retrovirus used to insert the MLL-AF4 fusion gene into mouse cells.
“We soon discovered that the virus wasn’t working,” Dr Thirman explained. “We knew that certain parts of human DNA can decrease viral titers. So we switched from the human version of AF4 to the mouse version, Af4, which is slightly different. This increased viral titers 30-fold.”
That worked, but it led to the second hurdle. The mice injected with virus transporting MLL-Af4 developed leukemia, but it was acute myeloid leukemia.
So the researchers inserted the fused MLL-Af4 gene into human CD34 cells, which were derived from cord blood or peripheral blood from volunteer donors.
The team then transferred those cells to mice, and, this time, the mice developed t(4;11) pro-B ALL.
The researchers said this model “fully recapitulates the immunophenotypic and molecular aspects” of human t(4;11) pro-B ALL and will therefore be “a valuable tool” for studying the disease. ![]()
Photo by Petr Kratochvil
After trying for nearly 2 decades, researchers have created a mouse model of t(4;11) pro-B acute lymphoblastic
leukemia (ALL).
The team said this model, described in Cancer Cell, mimics the human disease phenotypically and molecularly.
This type of ALL, which is common in infants, results from the translocation t(4;11)(q21;q23), which fuses the mixed-lineage leukemia (MLL) gene on chromosome 11 to the ALL-1 fused gene on chromosome 4 (AF4).
“For 20 years, scientists have repeatedly tried and consistently failed to make a model of MLL-AF4 pro-B acute lymphoblastic leukemia,” said study author Michael Thirman, MD, of the University of Chicago in Illinois.
“Even though we understood the basic genetic flaw, no one had been able create a mouse model that mimicked the human disease, which is crucial for evaluating potential therapies.”
That frustrated many researchers, who shifted their focus to test alternative hypotheses on the causes of t(4;11) pro-B ALL or refocused their laboratories to study different aspects of the disease.
However, Dr Thirman and his colleagues began working on this problem “years ago,” he said, and stayed with it.
The team identified 2 hurdles. The first was a problem with the retrovirus used to insert the MLL-AF4 fusion gene into mouse cells.
“We soon discovered that the virus wasn’t working,” Dr Thirman explained. “We knew that certain parts of human DNA can decrease viral titers. So we switched from the human version of AF4 to the mouse version, Af4, which is slightly different. This increased viral titers 30-fold.”
That worked, but it led to the second hurdle. The mice injected with virus transporting MLL-Af4 developed leukemia, but it was acute myeloid leukemia.
So the researchers inserted the fused MLL-Af4 gene into human CD34 cells, which were derived from cord blood or peripheral blood from volunteer donors.
The team then transferred those cells to mice, and, this time, the mice developed t(4;11) pro-B ALL.
The researchers said this model “fully recapitulates the immunophenotypic and molecular aspects” of human t(4;11) pro-B ALL and will therefore be “a valuable tool” for studying the disease. ![]()
Photo by Petr Kratochvil
After trying for nearly 2 decades, researchers have created a mouse model of t(4;11) pro-B acute lymphoblastic
leukemia (ALL).
The team said this model, described in Cancer Cell, mimics the human disease phenotypically and molecularly.
This type of ALL, which is common in infants, results from the translocation t(4;11)(q21;q23), which fuses the mixed-lineage leukemia (MLL) gene on chromosome 11 to the ALL-1 fused gene on chromosome 4 (AF4).
“For 20 years, scientists have repeatedly tried and consistently failed to make a model of MLL-AF4 pro-B acute lymphoblastic leukemia,” said study author Michael Thirman, MD, of the University of Chicago in Illinois.
“Even though we understood the basic genetic flaw, no one had been able create a mouse model that mimicked the human disease, which is crucial for evaluating potential therapies.”
That frustrated many researchers, who shifted their focus to test alternative hypotheses on the causes of t(4;11) pro-B ALL or refocused their laboratories to study different aspects of the disease.
However, Dr Thirman and his colleagues began working on this problem “years ago,” he said, and stayed with it.
The team identified 2 hurdles. The first was a problem with the retrovirus used to insert the MLL-AF4 fusion gene into mouse cells.
“We soon discovered that the virus wasn’t working,” Dr Thirman explained. “We knew that certain parts of human DNA can decrease viral titers. So we switched from the human version of AF4 to the mouse version, Af4, which is slightly different. This increased viral titers 30-fold.”
That worked, but it led to the second hurdle. The mice injected with virus transporting MLL-Af4 developed leukemia, but it was acute myeloid leukemia.
So the researchers inserted the fused MLL-Af4 gene into human CD34 cells, which were derived from cord blood or peripheral blood from volunteer donors.
The team then transferred those cells to mice, and, this time, the mice developed t(4;11) pro-B ALL.
The researchers said this model “fully recapitulates the immunophenotypic and molecular aspects” of human t(4;11) pro-B ALL and will therefore be “a valuable tool” for studying the disease. ![]()
Company withdraws application for eryaspase in ALL
ERYTECH Pharma has announced its decision to withdraw the European marketing authorization application (MAA) for eryaspase (GRASPA®) as a treatment for acute lymphoblastic leukemia (ALL).
The European Medicines Agency’s (EMA’s) Committee for Medicinal Products for Human Use (CHMP) asked for additional data on eryaspase, but ERYTECH said the time allowed by the EMA’s approval process is not sufficient to provide the data requested.
Therefore, the company decided to withdraw the MAA and resubmit it next year.
About eryaspase
Eryaspase consists of L-asparaginase encapsulated inside donor-derived red blood cells. These enzyme-loaded red blood cells function as bioreactors to eliminate circulating asparagine and “starve” cancer cells, thereby inducing their death.
Research has suggested this delivery system provides improved pharmacodynamics, protecting L-asparaginase from circulating proteolytic enzymes and preventing early liver or renal clearance.
The system also appears to reduce the risk of adverse events compared to native L-asparaginase.
The EMA and the US Food and Drug Administration have granted orphan drug designations for eryaspase for the treatment of ALL, acute myeloid
leukemia, and pancreatic cancer.
About the MAA
ERYTECH submitted an MAA for eryaspase in September 2015, based on positive results from a phase 2/3 study in which researchers compared eryaspase and native L-asparaginase in patients with relapsed and refractory ALL.
One year later (September 2016), the company received the CHMP’s Day 180 List of Outstanding Issues, which highlighted the need for additional data.
Specifically, the CHMP asked for data regarding the comparability between the old and new form of asparaginase encapsulated in eryaspase and the development of a new immunogenicity assay, as well as the pharmacodynamic effects of eryaspase.
Given the fact that the generation of these data will require more time than allowed in the EMA’s approval procedures, ERYTECH has notified the CHMP of the withdrawal of the MAA.
The company intends to resubmit the MAA in mid-2017, as soon as the newly generated data are available.
ERYTECH stressed that there have been no safety issues with eryaspase, and the withdrawal of this MAA will not affect any ongoing trials.
“We are committed to pursuing regulatory approval for GRASPA and intend to work closely with our investigators and advisors to generate the additional information requested and to resubmit an MAA next year,” said Iman El-Hariry, chief medical officer of ERYTECH.
“We believe we have generated strong clinical data in our different programs of eryaspase, and we continue to execute our plans towards making the product available to patients with aggressive forms of cancer, such as acute lymphoblastic leukemia, acute myeloid leukemia, and pancreatic cancer,” added Gil Beyen, ERYTECH’s chairman and chief executive officer. ![]()
ERYTECH Pharma has announced its decision to withdraw the European marketing authorization application (MAA) for eryaspase (GRASPA®) as a treatment for acute lymphoblastic leukemia (ALL).
The European Medicines Agency’s (EMA’s) Committee for Medicinal Products for Human Use (CHMP) asked for additional data on eryaspase, but ERYTECH said the time allowed by the EMA’s approval process is not sufficient to provide the data requested.
Therefore, the company decided to withdraw the MAA and resubmit it next year.
About eryaspase
Eryaspase consists of L-asparaginase encapsulated inside donor-derived red blood cells. These enzyme-loaded red blood cells function as bioreactors to eliminate circulating asparagine and “starve” cancer cells, thereby inducing their death.
Research has suggested this delivery system provides improved pharmacodynamics, protecting L-asparaginase from circulating proteolytic enzymes and preventing early liver or renal clearance.
The system also appears to reduce the risk of adverse events compared to native L-asparaginase.
The EMA and the US Food and Drug Administration have granted orphan drug designations for eryaspase for the treatment of ALL, acute myeloid
leukemia, and pancreatic cancer.
About the MAA
ERYTECH submitted an MAA for eryaspase in September 2015, based on positive results from a phase 2/3 study in which researchers compared eryaspase and native L-asparaginase in patients with relapsed and refractory ALL.
One year later (September 2016), the company received the CHMP’s Day 180 List of Outstanding Issues, which highlighted the need for additional data.
Specifically, the CHMP asked for data regarding the comparability between the old and new form of asparaginase encapsulated in eryaspase and the development of a new immunogenicity assay, as well as the pharmacodynamic effects of eryaspase.
Given the fact that the generation of these data will require more time than allowed in the EMA’s approval procedures, ERYTECH has notified the CHMP of the withdrawal of the MAA.
The company intends to resubmit the MAA in mid-2017, as soon as the newly generated data are available.
ERYTECH stressed that there have been no safety issues with eryaspase, and the withdrawal of this MAA will not affect any ongoing trials.
“We are committed to pursuing regulatory approval for GRASPA and intend to work closely with our investigators and advisors to generate the additional information requested and to resubmit an MAA next year,” said Iman El-Hariry, chief medical officer of ERYTECH.
“We believe we have generated strong clinical data in our different programs of eryaspase, and we continue to execute our plans towards making the product available to patients with aggressive forms of cancer, such as acute lymphoblastic leukemia, acute myeloid leukemia, and pancreatic cancer,” added Gil Beyen, ERYTECH’s chairman and chief executive officer. ![]()
ERYTECH Pharma has announced its decision to withdraw the European marketing authorization application (MAA) for eryaspase (GRASPA®) as a treatment for acute lymphoblastic leukemia (ALL).
The European Medicines Agency’s (EMA’s) Committee for Medicinal Products for Human Use (CHMP) asked for additional data on eryaspase, but ERYTECH said the time allowed by the EMA’s approval process is not sufficient to provide the data requested.
Therefore, the company decided to withdraw the MAA and resubmit it next year.
About eryaspase
Eryaspase consists of L-asparaginase encapsulated inside donor-derived red blood cells. These enzyme-loaded red blood cells function as bioreactors to eliminate circulating asparagine and “starve” cancer cells, thereby inducing their death.
Research has suggested this delivery system provides improved pharmacodynamics, protecting L-asparaginase from circulating proteolytic enzymes and preventing early liver or renal clearance.
The system also appears to reduce the risk of adverse events compared to native L-asparaginase.
The EMA and the US Food and Drug Administration have granted orphan drug designations for eryaspase for the treatment of ALL, acute myeloid
leukemia, and pancreatic cancer.
About the MAA
ERYTECH submitted an MAA for eryaspase in September 2015, based on positive results from a phase 2/3 study in which researchers compared eryaspase and native L-asparaginase in patients with relapsed and refractory ALL.
One year later (September 2016), the company received the CHMP’s Day 180 List of Outstanding Issues, which highlighted the need for additional data.
Specifically, the CHMP asked for data regarding the comparability between the old and new form of asparaginase encapsulated in eryaspase and the development of a new immunogenicity assay, as well as the pharmacodynamic effects of eryaspase.
Given the fact that the generation of these data will require more time than allowed in the EMA’s approval procedures, ERYTECH has notified the CHMP of the withdrawal of the MAA.
The company intends to resubmit the MAA in mid-2017, as soon as the newly generated data are available.
ERYTECH stressed that there have been no safety issues with eryaspase, and the withdrawal of this MAA will not affect any ongoing trials.
“We are committed to pursuing regulatory approval for GRASPA and intend to work closely with our investigators and advisors to generate the additional information requested and to resubmit an MAA next year,” said Iman El-Hariry, chief medical officer of ERYTECH.
“We believe we have generated strong clinical data in our different programs of eryaspase, and we continue to execute our plans towards making the product available to patients with aggressive forms of cancer, such as acute lymphoblastic leukemia, acute myeloid leukemia, and pancreatic cancer,” added Gil Beyen, ERYTECH’s chairman and chief executive officer. ![]()
Panobinostat might treat high-risk ALL subtype
Photo courtesy of Novartis
Researchers say they have identified a high-risk subtype of acute lymphoblastic leukemia (ALL) that may respond to treatment with the histone deacetylase (HDAC) inhibitor panobinostat.
The ALL subtype is characterized by chromosomal rearrangements that involve the MEF2D gene and 1 of 6 partner genes, most often BCL9.
The researchers described this subtype, known as MEF2D-rearranged ALL, in Nature Communications.
“MEF2D is a transcription factor that switches on expression of other genes during normal development,” said study author Charles Mullighan, MD, MBBS, of the St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We found that MEF2D chromosomal rearrangements disrupt expression of those genes and create a vulnerability to at least one targeted therapy, the drug panobinostat.”
Dr Mullighan and his colleagues performed genomic analyses on samples from a total of 1724 children, adolescents, and adults with ALL. This revealed 52 patients with MEF2D rearrangements.
MEF2D-rearranged ALL
The researchers calculated that MEF2D-rearranged ALL accounted for 5.3% of the ALL cases whose genetic basis was unknown.
The team also noted that MEF2D-rearranged ALL occurred most frequently in adolescents. Although, overall, ALL occurs most often in children between 3 and 5 years old, the average patient with MEF2D-rearranged ALL was 14.
In addition, MEF2D-rearranged ALL was associated with reduced survival when compared to some other ALL subtypes. The 5-year cancer-free survival for MEF2D-rearranged ALL patients was 71.6%.
The researchers also found that a fusion protein resulting from the MEF2D rearrangement led to sustained growth of mouse cells when compared to wild-type MEF2D or other proteins.
“That indicates the MEF2D fusion is a key step in transforming a normal white blood cell with a finite lifespan into a leukemic cell that is immortal,” Dr Mullighan said.
Role for panobinostat
MEF2D-rearranged leukemic cells produced high levels of HDAC9, which is targeted by panobinostat.
The researchers tested panobinostat in the lab and found the drug stopped proliferation of human MEF2D-rearranged leukemic cells.
Dr Mullighan said MEF2D-rearranged leukemic cells were exquisitely sensitive to panobinostat, which suggested the drug might function in a more targeted manner against cells with the rearrangement.
“If further testing of panobinostat, either alone or in combination therapy, confirms the anti-proliferative activity, that would lay the foundation for a clinical trial in patients, particularly patients with high-risk disease or those who have relapsed,” he said. ![]()
Photo courtesy of Novartis
Researchers say they have identified a high-risk subtype of acute lymphoblastic leukemia (ALL) that may respond to treatment with the histone deacetylase (HDAC) inhibitor panobinostat.
The ALL subtype is characterized by chromosomal rearrangements that involve the MEF2D gene and 1 of 6 partner genes, most often BCL9.
The researchers described this subtype, known as MEF2D-rearranged ALL, in Nature Communications.
“MEF2D is a transcription factor that switches on expression of other genes during normal development,” said study author Charles Mullighan, MD, MBBS, of the St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We found that MEF2D chromosomal rearrangements disrupt expression of those genes and create a vulnerability to at least one targeted therapy, the drug panobinostat.”
Dr Mullighan and his colleagues performed genomic analyses on samples from a total of 1724 children, adolescents, and adults with ALL. This revealed 52 patients with MEF2D rearrangements.
MEF2D-rearranged ALL
The researchers calculated that MEF2D-rearranged ALL accounted for 5.3% of the ALL cases whose genetic basis was unknown.
The team also noted that MEF2D-rearranged ALL occurred most frequently in adolescents. Although, overall, ALL occurs most often in children between 3 and 5 years old, the average patient with MEF2D-rearranged ALL was 14.
In addition, MEF2D-rearranged ALL was associated with reduced survival when compared to some other ALL subtypes. The 5-year cancer-free survival for MEF2D-rearranged ALL patients was 71.6%.
The researchers also found that a fusion protein resulting from the MEF2D rearrangement led to sustained growth of mouse cells when compared to wild-type MEF2D or other proteins.
“That indicates the MEF2D fusion is a key step in transforming a normal white blood cell with a finite lifespan into a leukemic cell that is immortal,” Dr Mullighan said.
Role for panobinostat
MEF2D-rearranged leukemic cells produced high levels of HDAC9, which is targeted by panobinostat.
The researchers tested panobinostat in the lab and found the drug stopped proliferation of human MEF2D-rearranged leukemic cells.
Dr Mullighan said MEF2D-rearranged leukemic cells were exquisitely sensitive to panobinostat, which suggested the drug might function in a more targeted manner against cells with the rearrangement.
“If further testing of panobinostat, either alone or in combination therapy, confirms the anti-proliferative activity, that would lay the foundation for a clinical trial in patients, particularly patients with high-risk disease or those who have relapsed,” he said. ![]()
Photo courtesy of Novartis
Researchers say they have identified a high-risk subtype of acute lymphoblastic leukemia (ALL) that may respond to treatment with the histone deacetylase (HDAC) inhibitor panobinostat.
The ALL subtype is characterized by chromosomal rearrangements that involve the MEF2D gene and 1 of 6 partner genes, most often BCL9.
The researchers described this subtype, known as MEF2D-rearranged ALL, in Nature Communications.
“MEF2D is a transcription factor that switches on expression of other genes during normal development,” said study author Charles Mullighan, MD, MBBS, of the St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We found that MEF2D chromosomal rearrangements disrupt expression of those genes and create a vulnerability to at least one targeted therapy, the drug panobinostat.”
Dr Mullighan and his colleagues performed genomic analyses on samples from a total of 1724 children, adolescents, and adults with ALL. This revealed 52 patients with MEF2D rearrangements.
MEF2D-rearranged ALL
The researchers calculated that MEF2D-rearranged ALL accounted for 5.3% of the ALL cases whose genetic basis was unknown.
The team also noted that MEF2D-rearranged ALL occurred most frequently in adolescents. Although, overall, ALL occurs most often in children between 3 and 5 years old, the average patient with MEF2D-rearranged ALL was 14.
In addition, MEF2D-rearranged ALL was associated with reduced survival when compared to some other ALL subtypes. The 5-year cancer-free survival for MEF2D-rearranged ALL patients was 71.6%.
The researchers also found that a fusion protein resulting from the MEF2D rearrangement led to sustained growth of mouse cells when compared to wild-type MEF2D or other proteins.
“That indicates the MEF2D fusion is a key step in transforming a normal white blood cell with a finite lifespan into a leukemic cell that is immortal,” Dr Mullighan said.
Role for panobinostat
MEF2D-rearranged leukemic cells produced high levels of HDAC9, which is targeted by panobinostat.
The researchers tested panobinostat in the lab and found the drug stopped proliferation of human MEF2D-rearranged leukemic cells.
Dr Mullighan said MEF2D-rearranged leukemic cells were exquisitely sensitive to panobinostat, which suggested the drug might function in a more targeted manner against cells with the rearrangement.
“If further testing of panobinostat, either alone or in combination therapy, confirms the anti-proliferative activity, that would lay the foundation for a clinical trial in patients, particularly patients with high-risk disease or those who have relapsed,” he said. ![]()
Combo shows promise for treating T-ALL
acute lymphoblastic leukemia
Image by Hind Medyou
Preclinical research suggests that 2 investigational drugs synergize to kill T-cell acute lymphoblastic leukemia (T-ALL) cells while having a minimal impact on normal blood cells.
Both drugs—the CK2 inhibitor CX-4945 and the BET inhibitor JQ1—have already been tested as single agents in clinical trials of hematologic malignancies and solid tumors.
However, the effects of the drugs in combination were not known until now.
“Previous studies provided us a rationale to test the combination of CX-4945 and JQ1 on refractory/relapsed T-cell leukemia,” said Hui Feng, MD, PhD, of Boston University School of Medicine in Massachusetts.
“Our findings suggest that the combination treatment of CX-4945 and JQ1 could be an effective strategy to target refractory/relapsed T-cell leukemia.”
Dr Feng and her colleagues reported these findings in Haematologica.
The researchers noted that targeting MYC-mediated transcriptional programs using JQ1 produces anti-leukemic activity in vitro and in vivo. However, global repression of transcription is likely to cause toxicities.
Therefore, the team theorized that finding drugs that synergize with JQ1 might allow them to reduce the dose of JQ1 and therefore decrease the risk of toxicity while enhancing the efficacy of treatment.
For this, the researchers looked to CX-4945, which is currently being investigated in clinical trials of breast cancer and multiple myeloma.
The team said CX-4945 has been shown to significantly reduce the growth and survival of human T-ALL cells on its own.
In a series of experiments, Dr Feng and her colleagues were able to show that CX-4945 destabilizes NOTCH1 and synergizes with JQ1 to induce apoptosis in human T-ALL cells.
The researchers also assessed the effects of JQ1 and CX-4946, alone and in combination, on normal peripheral blood monocytes (PBMs).
PBMs proved less sensitive than ALL-SIL T-ALL cells to each drug alone and to the drugs in combination. In fact, the combination had an antagonistic effect in PBMs.
Dr Feng and her colleagues said this research suggests JQ1 and CX-4946 in combination may be a feasible treatment option for relapsed/refractory T-ALL and other cancers involving CK2 and NOTCH1/MYC. ![]()
acute lymphoblastic leukemia
Image by Hind Medyou
Preclinical research suggests that 2 investigational drugs synergize to kill T-cell acute lymphoblastic leukemia (T-ALL) cells while having a minimal impact on normal blood cells.
Both drugs—the CK2 inhibitor CX-4945 and the BET inhibitor JQ1—have already been tested as single agents in clinical trials of hematologic malignancies and solid tumors.
However, the effects of the drugs in combination were not known until now.
“Previous studies provided us a rationale to test the combination of CX-4945 and JQ1 on refractory/relapsed T-cell leukemia,” said Hui Feng, MD, PhD, of Boston University School of Medicine in Massachusetts.
“Our findings suggest that the combination treatment of CX-4945 and JQ1 could be an effective strategy to target refractory/relapsed T-cell leukemia.”
Dr Feng and her colleagues reported these findings in Haematologica.
The researchers noted that targeting MYC-mediated transcriptional programs using JQ1 produces anti-leukemic activity in vitro and in vivo. However, global repression of transcription is likely to cause toxicities.
Therefore, the team theorized that finding drugs that synergize with JQ1 might allow them to reduce the dose of JQ1 and therefore decrease the risk of toxicity while enhancing the efficacy of treatment.
For this, the researchers looked to CX-4945, which is currently being investigated in clinical trials of breast cancer and multiple myeloma.
The team said CX-4945 has been shown to significantly reduce the growth and survival of human T-ALL cells on its own.
In a series of experiments, Dr Feng and her colleagues were able to show that CX-4945 destabilizes NOTCH1 and synergizes with JQ1 to induce apoptosis in human T-ALL cells.
The researchers also assessed the effects of JQ1 and CX-4946, alone and in combination, on normal peripheral blood monocytes (PBMs).
PBMs proved less sensitive than ALL-SIL T-ALL cells to each drug alone and to the drugs in combination. In fact, the combination had an antagonistic effect in PBMs.
Dr Feng and her colleagues said this research suggests JQ1 and CX-4946 in combination may be a feasible treatment option for relapsed/refractory T-ALL and other cancers involving CK2 and NOTCH1/MYC. ![]()
acute lymphoblastic leukemia
Image by Hind Medyou
Preclinical research suggests that 2 investigational drugs synergize to kill T-cell acute lymphoblastic leukemia (T-ALL) cells while having a minimal impact on normal blood cells.
Both drugs—the CK2 inhibitor CX-4945 and the BET inhibitor JQ1—have already been tested as single agents in clinical trials of hematologic malignancies and solid tumors.
However, the effects of the drugs in combination were not known until now.
“Previous studies provided us a rationale to test the combination of CX-4945 and JQ1 on refractory/relapsed T-cell leukemia,” said Hui Feng, MD, PhD, of Boston University School of Medicine in Massachusetts.
“Our findings suggest that the combination treatment of CX-4945 and JQ1 could be an effective strategy to target refractory/relapsed T-cell leukemia.”
Dr Feng and her colleagues reported these findings in Haematologica.
The researchers noted that targeting MYC-mediated transcriptional programs using JQ1 produces anti-leukemic activity in vitro and in vivo. However, global repression of transcription is likely to cause toxicities.
Therefore, the team theorized that finding drugs that synergize with JQ1 might allow them to reduce the dose of JQ1 and therefore decrease the risk of toxicity while enhancing the efficacy of treatment.
For this, the researchers looked to CX-4945, which is currently being investigated in clinical trials of breast cancer and multiple myeloma.
The team said CX-4945 has been shown to significantly reduce the growth and survival of human T-ALL cells on its own.
In a series of experiments, Dr Feng and her colleagues were able to show that CX-4945 destabilizes NOTCH1 and synergizes with JQ1 to induce apoptosis in human T-ALL cells.
The researchers also assessed the effects of JQ1 and CX-4946, alone and in combination, on normal peripheral blood monocytes (PBMs).
PBMs proved less sensitive than ALL-SIL T-ALL cells to each drug alone and to the drugs in combination. In fact, the combination had an antagonistic effect in PBMs.
Dr Feng and her colleagues said this research suggests JQ1 and CX-4946 in combination may be a feasible treatment option for relapsed/refractory T-ALL and other cancers involving CK2 and NOTCH1/MYC. ![]()
Team identifies genetic hallmarks of B-ALL subtype
Photo courtesy of St. Jude
Children’s Research Hospital
Researchers say they have uncovered a unique paradigm of transcription factor deregulation in B-precursor acute lymphoblastic leukemia (B-ALL).
The team found that deregulation of the homeobox transcription factor gene DUX4 and the ETS transcription factor gene ERG is a hallmark of a subtype of B-ALL that may comprise up to 8% of B-ALL cases.
The researchers reported these findings in Nature Genetics.
“Our work is motivated by a lack of information on the genetic basis of many B-ALL cases,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We discovered a distinct gene pattern in blood samples from some patients in our study and wanted to determine the underlying molecular events behind this signal.”
The researchers studied a group of 1913 B-ALL patients (including children, adolescents, and young adults) to understand the genetic basis of the disease.
Microarray and transcriptome sequencing revealed that 7.6% of these patients had the distinctive genetic profile the researchers wanted to characterize further.
“Our work revealed that, in this type of B-ALL, there is a sequence of molecular events that involves the interplay of 2 transcription factors,” Dr Mullighan said.
The team observed rearrangement of the gene DUX4 in all cases of this subtype of B-ALL, which resulted in high-level expression of DUX4. DUX4 was shown to bind to the ERG gene, leading to deregulated expression of ERG.
The deregulation of ERG compromised the function of ERG either by deleting part of the gene or by expressing another form of ERG—ERGalt. In both cases, loss of activity was observed for the ERG transcription factor, which led to leukemia.
“These results underscore that there is still more to be learned about the genetic changes in ALL, and that this knowledge can help refine treatment for patients,” said study author Stephen Hunger, MD, of the Children’s Hospital of Philadelphia in Pennsylvania.
The researchers hope identification of the relationships between the 2 transcription factors will lead to new diagnostic tests for patients. DUX4/ERG ALL is linked to favorable outcomes even when other detrimental genetic mutations are present.
Currently, only transcriptome or genome sequencing helps identify the DUX4 rearrangements. The researchers say other detection methods, such as fluorescence hybridization or karyotyping, are not sufficient to recognize genetic changes to DUX4. ![]()
Photo courtesy of St. Jude
Children’s Research Hospital
Researchers say they have uncovered a unique paradigm of transcription factor deregulation in B-precursor acute lymphoblastic leukemia (B-ALL).
The team found that deregulation of the homeobox transcription factor gene DUX4 and the ETS transcription factor gene ERG is a hallmark of a subtype of B-ALL that may comprise up to 8% of B-ALL cases.
The researchers reported these findings in Nature Genetics.
“Our work is motivated by a lack of information on the genetic basis of many B-ALL cases,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We discovered a distinct gene pattern in blood samples from some patients in our study and wanted to determine the underlying molecular events behind this signal.”
The researchers studied a group of 1913 B-ALL patients (including children, adolescents, and young adults) to understand the genetic basis of the disease.
Microarray and transcriptome sequencing revealed that 7.6% of these patients had the distinctive genetic profile the researchers wanted to characterize further.
“Our work revealed that, in this type of B-ALL, there is a sequence of molecular events that involves the interplay of 2 transcription factors,” Dr Mullighan said.
The team observed rearrangement of the gene DUX4 in all cases of this subtype of B-ALL, which resulted in high-level expression of DUX4. DUX4 was shown to bind to the ERG gene, leading to deregulated expression of ERG.
The deregulation of ERG compromised the function of ERG either by deleting part of the gene or by expressing another form of ERG—ERGalt. In both cases, loss of activity was observed for the ERG transcription factor, which led to leukemia.
“These results underscore that there is still more to be learned about the genetic changes in ALL, and that this knowledge can help refine treatment for patients,” said study author Stephen Hunger, MD, of the Children’s Hospital of Philadelphia in Pennsylvania.
The researchers hope identification of the relationships between the 2 transcription factors will lead to new diagnostic tests for patients. DUX4/ERG ALL is linked to favorable outcomes even when other detrimental genetic mutations are present.
Currently, only transcriptome or genome sequencing helps identify the DUX4 rearrangements. The researchers say other detection methods, such as fluorescence hybridization or karyotyping, are not sufficient to recognize genetic changes to DUX4. ![]()
Photo courtesy of St. Jude
Children’s Research Hospital
Researchers say they have uncovered a unique paradigm of transcription factor deregulation in B-precursor acute lymphoblastic leukemia (B-ALL).
The team found that deregulation of the homeobox transcription factor gene DUX4 and the ETS transcription factor gene ERG is a hallmark of a subtype of B-ALL that may comprise up to 8% of B-ALL cases.
The researchers reported these findings in Nature Genetics.
“Our work is motivated by a lack of information on the genetic basis of many B-ALL cases,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We discovered a distinct gene pattern in blood samples from some patients in our study and wanted to determine the underlying molecular events behind this signal.”
The researchers studied a group of 1913 B-ALL patients (including children, adolescents, and young adults) to understand the genetic basis of the disease.
Microarray and transcriptome sequencing revealed that 7.6% of these patients had the distinctive genetic profile the researchers wanted to characterize further.
“Our work revealed that, in this type of B-ALL, there is a sequence of molecular events that involves the interplay of 2 transcription factors,” Dr Mullighan said.
The team observed rearrangement of the gene DUX4 in all cases of this subtype of B-ALL, which resulted in high-level expression of DUX4. DUX4 was shown to bind to the ERG gene, leading to deregulated expression of ERG.
The deregulation of ERG compromised the function of ERG either by deleting part of the gene or by expressing another form of ERG—ERGalt. In both cases, loss of activity was observed for the ERG transcription factor, which led to leukemia.
“These results underscore that there is still more to be learned about the genetic changes in ALL, and that this knowledge can help refine treatment for patients,” said study author Stephen Hunger, MD, of the Children’s Hospital of Philadelphia in Pennsylvania.
The researchers hope identification of the relationships between the 2 transcription factors will lead to new diagnostic tests for patients. DUX4/ERG ALL is linked to favorable outcomes even when other detrimental genetic mutations are present.
Currently, only transcriptome or genome sequencing helps identify the DUX4 rearrangements. The researchers say other detection methods, such as fluorescence hybridization or karyotyping, are not sufficient to recognize genetic changes to DUX4. ![]()
Legislators question price of leukemia drug
Photo from Business Wire
A pair of US legislators are questioning why ARIAD Pharmaceuticals, Inc. has increased the price of its leukemia drug Iclusig (ponatinib) by more than $80,000 over the last several years.
ARIAD raised the price of Iclusig 4 times in 2016. The drug now costs nearly $199,000 a year.
Senator Bernie Sanders (Vermont) and Congressman Elijah Cummings (Maryland) sent a letter to ARIAD last week requesting information about these price increases.
Cummings and Sanders are also investigating whether ARIAD took additional steps to boost profits by discontinuing sales of certain dosages and quantities of Iclusig in order to charge patients and insurers more in exchange for less medicine.
“These outrageous sales tactics indicate that ARIAD is more concerned with its profit than with its patients,” Sanders and Cummings wrote in the letter.
The US Food and Drug Administration (FDA) approved Iclusig in December 2012 to treat chronic myeloid leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL).
In late 2013, the FDA suspended sales and clinical trials of the drug due to reports of serious adverse events.
The FDA allowed ARIAD to resume selling Iclusig in December 2013, but only to CML/ALL patients who cannot tolerate, or whose disease is resistant to, other tyrosine kinase inhibitors.
“Despite this new evidence showing the drug posed a far greater safety risk to patients than was known when the drug came on the market, ARIAD nonetheless raised the price of Iclusig several times over the subsequent 4 years,” Sanders and Cummings wrote.
“In the interest of patients and taxpayers, we are interested in learning more about the impact that the escalating price and restrictions on product availability have had.”
ARIAD has released a statement acknowledging Cummings’ and Sanders’ letter and defending its decisions to increase the price of Iclusig.
The company said it “makes significant investments in research and development (R&D) to advance breakthrough treatments” for patients with rare cancers.
In fact, ARIAD has invested more than $1.3 billion in R&D and accumulated losses of approximately $1.4 billion, which have not been recovered. In 2015, ARIAD generated $119 million in total revenue and invested $171 million in R&D.
The company said it intends to respond to Cummings’ and Sanders’ request for information. ![]()
Photo from Business Wire
A pair of US legislators are questioning why ARIAD Pharmaceuticals, Inc. has increased the price of its leukemia drug Iclusig (ponatinib) by more than $80,000 over the last several years.
ARIAD raised the price of Iclusig 4 times in 2016. The drug now costs nearly $199,000 a year.
Senator Bernie Sanders (Vermont) and Congressman Elijah Cummings (Maryland) sent a letter to ARIAD last week requesting information about these price increases.
Cummings and Sanders are also investigating whether ARIAD took additional steps to boost profits by discontinuing sales of certain dosages and quantities of Iclusig in order to charge patients and insurers more in exchange for less medicine.
“These outrageous sales tactics indicate that ARIAD is more concerned with its profit than with its patients,” Sanders and Cummings wrote in the letter.
The US Food and Drug Administration (FDA) approved Iclusig in December 2012 to treat chronic myeloid leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL).
In late 2013, the FDA suspended sales and clinical trials of the drug due to reports of serious adverse events.
The FDA allowed ARIAD to resume selling Iclusig in December 2013, but only to CML/ALL patients who cannot tolerate, or whose disease is resistant to, other tyrosine kinase inhibitors.
“Despite this new evidence showing the drug posed a far greater safety risk to patients than was known when the drug came on the market, ARIAD nonetheless raised the price of Iclusig several times over the subsequent 4 years,” Sanders and Cummings wrote.
“In the interest of patients and taxpayers, we are interested in learning more about the impact that the escalating price and restrictions on product availability have had.”
ARIAD has released a statement acknowledging Cummings’ and Sanders’ letter and defending its decisions to increase the price of Iclusig.
The company said it “makes significant investments in research and development (R&D) to advance breakthrough treatments” for patients with rare cancers.
In fact, ARIAD has invested more than $1.3 billion in R&D and accumulated losses of approximately $1.4 billion, which have not been recovered. In 2015, ARIAD generated $119 million in total revenue and invested $171 million in R&D.
The company said it intends to respond to Cummings’ and Sanders’ request for information. ![]()
Photo from Business Wire
A pair of US legislators are questioning why ARIAD Pharmaceuticals, Inc. has increased the price of its leukemia drug Iclusig (ponatinib) by more than $80,000 over the last several years.
ARIAD raised the price of Iclusig 4 times in 2016. The drug now costs nearly $199,000 a year.
Senator Bernie Sanders (Vermont) and Congressman Elijah Cummings (Maryland) sent a letter to ARIAD last week requesting information about these price increases.
Cummings and Sanders are also investigating whether ARIAD took additional steps to boost profits by discontinuing sales of certain dosages and quantities of Iclusig in order to charge patients and insurers more in exchange for less medicine.
“These outrageous sales tactics indicate that ARIAD is more concerned with its profit than with its patients,” Sanders and Cummings wrote in the letter.
The US Food and Drug Administration (FDA) approved Iclusig in December 2012 to treat chronic myeloid leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL).
In late 2013, the FDA suspended sales and clinical trials of the drug due to reports of serious adverse events.
The FDA allowed ARIAD to resume selling Iclusig in December 2013, but only to CML/ALL patients who cannot tolerate, or whose disease is resistant to, other tyrosine kinase inhibitors.
“Despite this new evidence showing the drug posed a far greater safety risk to patients than was known when the drug came on the market, ARIAD nonetheless raised the price of Iclusig several times over the subsequent 4 years,” Sanders and Cummings wrote.
“In the interest of patients and taxpayers, we are interested in learning more about the impact that the escalating price and restrictions on product availability have had.”
ARIAD has released a statement acknowledging Cummings’ and Sanders’ letter and defending its decisions to increase the price of Iclusig.
The company said it “makes significant investments in research and development (R&D) to advance breakthrough treatments” for patients with rare cancers.
In fact, ARIAD has invested more than $1.3 billion in R&D and accumulated losses of approximately $1.4 billion, which have not been recovered. In 2015, ARIAD generated $119 million in total revenue and invested $171 million in R&D.
The company said it intends to respond to Cummings’ and Sanders’ request for information.
Findings could aid treatment of resistant T-ALL
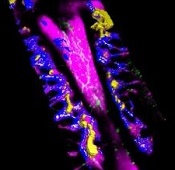
(blue and purple) invaded
by leukemia cells (yellow).
Image courtesy of Edwin
Hawkins, Delfim Duarte,
and Imperial College London
Preclinical research has shed light on how certain leukemia cells survive treatment and could pave the way for better therapeutic targeting of these resistant cells.
Researchers have speculated that some leukemia cells survive treatment by hiding out in specific niches in the bone marrow.
Results of the new research, conducted in mouse models and human samples of T-cell acute lymphoblastic leukemia (T-ALL), contradict that theory.
The experiments showed that resistant T-ALL cells move rapidly through the bone marrow before, during, and after treatment, interacting with—and sometimes killing—healthy cells.
“We expected the cells that survived treatment to be sat in particular niches, but, instead, they are very active throughout the bone marrow,” said Cristina Lo Celso, PhD, of Imperial College London in the UK.
“We now know that it would be ineffective to target particular niches in the bone marrow to tackle treatment-resistant leukemia. Now that we know that the cells don’t hide, we can explore why that is and how their movement helps them to survive. Ultimately, we want to find out whether we can stop the movement and whether this could kill the treatment-resistant cells.”
Dr Lo Celso and her colleagues described these findings in a letter to Nature.
The researchers used intravital microscopy to track the movement of T-ALL cells in mice—before, during, and after treatment. Treatment consisted of dexamethasone alone, vincristine alone, or combination dexamethasone, vincristine, and L-asparaginase.
The team found that T-ALL cells moved around rapidly, not showing any preference for bone marrow subcompartments. The cells’ behavior was consistent over time—from the earliest bone marrow seeding through to treatment response and resistance.
However, the researchers noted that surviving T-ALL cells were “highly migratory” and travelled at significantly faster speeds than early infiltrating cells. In addition, resistant T-ALL cells were still capable of undergoing division at times when other T-ALL cells were dying.
The team suggested that the act of moving may help T-ALL cells to survive, possibly through short-lived interactions with other cells.
This theory was supported by the discovery that T-ALL cells actively attack osteoblasts. The researchers noted that osteoblasts are associated with hematopoietic fitness. So the loss of osteoblasts may contribute to the loss of healthy hematopoiesis observed in leukemia patients.
The team believes this insight could aid the development of treatments to safeguard the production of healthy blood cells in T-ALL patients.
“Our study supports the idea that, at least in this leukemia, new therapies should target the cancer cells themselves instead of the surrounding normal stromal cells to better eradicate the disease,” said study author Delfim Duarte, MD, a PhD student at Imperial College London.
“Our work also suggests that protecting normal stromal bone cells from the attack of leukemia cells can have wide implications in the support of healthy blood cell production,” said Edwin Hawkins, PhD, of the Walter and Eliza Hall Institute of Medical Research in Melbourne, Victoria, Australia.
“Keeping blood cell levels up would prevent anemia, infection, and bleeding.”
(blue and purple) invaded
by leukemia cells (yellow).
Image courtesy of Edwin
Hawkins, Delfim Duarte,
and Imperial College London
Preclinical research has shed light on how certain leukemia cells survive treatment and could pave the way for better therapeutic targeting of these resistant cells.
Researchers have speculated that some leukemia cells survive treatment by hiding out in specific niches in the bone marrow.
Results of the new research, conducted in mouse models and human samples of T-cell acute lymphoblastic leukemia (T-ALL), contradict that theory.
The experiments showed that resistant T-ALL cells move rapidly through the bone marrow before, during, and after treatment, interacting with—and sometimes killing—healthy cells.
“We expected the cells that survived treatment to be sat in particular niches, but, instead, they are very active throughout the bone marrow,” said Cristina Lo Celso, PhD, of Imperial College London in the UK.
“We now know that it would be ineffective to target particular niches in the bone marrow to tackle treatment-resistant leukemia. Now that we know that the cells don’t hide, we can explore why that is and how their movement helps them to survive. Ultimately, we want to find out whether we can stop the movement and whether this could kill the treatment-resistant cells.”
Dr Lo Celso and her colleagues described these findings in a letter to Nature.
The researchers used intravital microscopy to track the movement of T-ALL cells in mice—before, during, and after treatment. Treatment consisted of dexamethasone alone, vincristine alone, or combination dexamethasone, vincristine, and L-asparaginase.
The team found that T-ALL cells moved around rapidly, not showing any preference for bone marrow subcompartments. The cells’ behavior was consistent over time—from the earliest bone marrow seeding through to treatment response and resistance.
However, the researchers noted that surviving T-ALL cells were “highly migratory” and travelled at significantly faster speeds than early infiltrating cells. In addition, resistant T-ALL cells were still capable of undergoing division at times when other T-ALL cells were dying.
The team suggested that the act of moving may help T-ALL cells to survive, possibly through short-lived interactions with other cells.
This theory was supported by the discovery that T-ALL cells actively attack osteoblasts. The researchers noted that osteoblasts are associated with hematopoietic fitness. So the loss of osteoblasts may contribute to the loss of healthy hematopoiesis observed in leukemia patients.
The team believes this insight could aid the development of treatments to safeguard the production of healthy blood cells in T-ALL patients.
“Our study supports the idea that, at least in this leukemia, new therapies should target the cancer cells themselves instead of the surrounding normal stromal cells to better eradicate the disease,” said study author Delfim Duarte, MD, a PhD student at Imperial College London.
“Our work also suggests that protecting normal stromal bone cells from the attack of leukemia cells can have wide implications in the support of healthy blood cell production,” said Edwin Hawkins, PhD, of the Walter and Eliza Hall Institute of Medical Research in Melbourne, Victoria, Australia.
“Keeping blood cell levels up would prevent anemia, infection, and bleeding.”
(blue and purple) invaded
by leukemia cells (yellow).
Image courtesy of Edwin
Hawkins, Delfim Duarte,
and Imperial College London
Preclinical research has shed light on how certain leukemia cells survive treatment and could pave the way for better therapeutic targeting of these resistant cells.
Researchers have speculated that some leukemia cells survive treatment by hiding out in specific niches in the bone marrow.
Results of the new research, conducted in mouse models and human samples of T-cell acute lymphoblastic leukemia (T-ALL), contradict that theory.
The experiments showed that resistant T-ALL cells move rapidly through the bone marrow before, during, and after treatment, interacting with—and sometimes killing—healthy cells.
“We expected the cells that survived treatment to be sat in particular niches, but, instead, they are very active throughout the bone marrow,” said Cristina Lo Celso, PhD, of Imperial College London in the UK.
“We now know that it would be ineffective to target particular niches in the bone marrow to tackle treatment-resistant leukemia. Now that we know that the cells don’t hide, we can explore why that is and how their movement helps them to survive. Ultimately, we want to find out whether we can stop the movement and whether this could kill the treatment-resistant cells.”
Dr Lo Celso and her colleagues described these findings in a letter to Nature.
The researchers used intravital microscopy to track the movement of T-ALL cells in mice—before, during, and after treatment. Treatment consisted of dexamethasone alone, vincristine alone, or combination dexamethasone, vincristine, and L-asparaginase.
The team found that T-ALL cells moved around rapidly, not showing any preference for bone marrow subcompartments. The cells’ behavior was consistent over time—from the earliest bone marrow seeding through to treatment response and resistance.
However, the researchers noted that surviving T-ALL cells were “highly migratory” and travelled at significantly faster speeds than early infiltrating cells. In addition, resistant T-ALL cells were still capable of undergoing division at times when other T-ALL cells were dying.
The team suggested that the act of moving may help T-ALL cells to survive, possibly through short-lived interactions with other cells.
This theory was supported by the discovery that T-ALL cells actively attack osteoblasts. The researchers noted that osteoblasts are associated with hematopoietic fitness. So the loss of osteoblasts may contribute to the loss of healthy hematopoiesis observed in leukemia patients.
The team believes this insight could aid the development of treatments to safeguard the production of healthy blood cells in T-ALL patients.
“Our study supports the idea that, at least in this leukemia, new therapies should target the cancer cells themselves instead of the surrounding normal stromal cells to better eradicate the disease,” said study author Delfim Duarte, MD, a PhD student at Imperial College London.
“Our work also suggests that protecting normal stromal bone cells from the attack of leukemia cells can have wide implications in the support of healthy blood cell production,” said Edwin Hawkins, PhD, of the Walter and Eliza Hall Institute of Medical Research in Melbourne, Victoria, Australia.
“Keeping blood cell levels up would prevent anemia, infection, and bleeding.”