User login
Midostaurin improves survival in new AML
Adding the multitargeted kinase inhibitor midostaurin to standard chemotherapy led to significantly longer overall and event-free survival, compared with placebo and standard chemotherapy in newly diagnosed acute myeloid leukemia (AML) patients with FLT3 gene mutations, according to phase III trial results published in the New England Journal of Medicine.*
About 30% of AML patients have mutations to the FLT3 gene – with three-quarters of those internal tandem duplication (ITD) mutations, which involves duplication of between 3 and 100 amino acids in the juxtamembrane region. These mutations are linked with a high relapse rate and poor prognosis, especially when there is a high ratio of these mutations to wild-type FLT3. About 8% of patients with newly diagnosed AML have an FLT3 point mutation in the tyrosine kinase domain (TKD), but the effect of these on prognosis isn’t clear.
In the trial, called RATIFY and conducted at 225 sites in 17 countries, 360 patients were randomized to the midostaurin group and 357* to placebo, and they were treated from 2008 to 2013. In all, 29.8% of patients were “ITD high,” meaning their ITD FLT3 mutation to wild-type FLT3 ratio was higher than 0.7, and 47.6% were “ITD low,” with a mutation-to-wild-type FLT3 ratio of 0.5 to 0.7. A total of 22.6% of patients had TKD mutations.
Patients received standard induction chemotherapy, with daunorubicine and cytarabine, and on days 8 through 21 either 50 mg of midostaurin or placebo orally twice a day. Patients were given an identical second cycle of induction therapy, with midostaurin or placebo, if they showed definitive clinically significant residual leukemia after the first induction treatment.
Those who achieved complete remission after induction were given 4, 28-day cycles of consolidation treatment, with midostaurin or placebo on days 8 through 21. If they stayed in remission after that, they were given maintenance of 12, 28-day cycles of midostaurin or placebo.
They were not required to receive hematopoetic stem cell transplantation (HSCT), but it was performed at investigator discretion.
Midostaurin improved survival but not rates of complete remission as defined in the trial protocol, researchers reported.
The hazard ratio for death in the midostaurin group was 0.78 (95% CI, 0.63 to 0.96; one-sided P = .0009). The 4-year overall survival rate was 51.4% for the midostaurin group and 44.3% for the placebo group. Midostaurin was shown to benefit all mutation subgroups, but with no greater benefit in one group than another.
Patients in the midostaurin group had a 21.6% lower likelihood of having an event, defined as failure to achieve protocol-defined complete remission, relapse or death without relapse.
There was no significant difference between the groups in complete remission, which under protocol had to occur by day 60.
HSCT was performed in 57% of patients – during the first complete remission in 28.1% of the midostaurin group and in 22.7% during the first complete remission in the placebo group. For those who were transplanted after the first complete remission, no treatment effect was seen.
Researchers noted that there was a therapeutic benefit even among patients with ITD mutations but with a low allelic burden, in whom the disease might be due largely to mutations other than FLT3.
“It is possible that the benefit of midostaurin, which is a multitargeted kinase inhibitor, might lie beyond its ability to inhibit FLT3,” possibly through inhibition of KIT, researchers said.
They also noted that as the trial went on, more and more investigators decided to treat patients with hematopoietic stem cell transplantation, based on newly reported data elsewhere. Since midostaurin was discontinued at the time of transplant, that could have limited exposure to the drug and limited its effect.
*CORRECTION 7/5/2017: An earlier version of this article misstated the number of patients in the placebo group as well as where the study originally appeared.
Adding the multitargeted kinase inhibitor midostaurin to standard chemotherapy led to significantly longer overall and event-free survival, compared with placebo and standard chemotherapy in newly diagnosed acute myeloid leukemia (AML) patients with FLT3 gene mutations, according to phase III trial results published in the New England Journal of Medicine.*
About 30% of AML patients have mutations to the FLT3 gene – with three-quarters of those internal tandem duplication (ITD) mutations, which involves duplication of between 3 and 100 amino acids in the juxtamembrane region. These mutations are linked with a high relapse rate and poor prognosis, especially when there is a high ratio of these mutations to wild-type FLT3. About 8% of patients with newly diagnosed AML have an FLT3 point mutation in the tyrosine kinase domain (TKD), but the effect of these on prognosis isn’t clear.
In the trial, called RATIFY and conducted at 225 sites in 17 countries, 360 patients were randomized to the midostaurin group and 357* to placebo, and they were treated from 2008 to 2013. In all, 29.8% of patients were “ITD high,” meaning their ITD FLT3 mutation to wild-type FLT3 ratio was higher than 0.7, and 47.6% were “ITD low,” with a mutation-to-wild-type FLT3 ratio of 0.5 to 0.7. A total of 22.6% of patients had TKD mutations.
Patients received standard induction chemotherapy, with daunorubicine and cytarabine, and on days 8 through 21 either 50 mg of midostaurin or placebo orally twice a day. Patients were given an identical second cycle of induction therapy, with midostaurin or placebo, if they showed definitive clinically significant residual leukemia after the first induction treatment.
Those who achieved complete remission after induction were given 4, 28-day cycles of consolidation treatment, with midostaurin or placebo on days 8 through 21. If they stayed in remission after that, they were given maintenance of 12, 28-day cycles of midostaurin or placebo.
They were not required to receive hematopoetic stem cell transplantation (HSCT), but it was performed at investigator discretion.
Midostaurin improved survival but not rates of complete remission as defined in the trial protocol, researchers reported.
The hazard ratio for death in the midostaurin group was 0.78 (95% CI, 0.63 to 0.96; one-sided P = .0009). The 4-year overall survival rate was 51.4% for the midostaurin group and 44.3% for the placebo group. Midostaurin was shown to benefit all mutation subgroups, but with no greater benefit in one group than another.
Patients in the midostaurin group had a 21.6% lower likelihood of having an event, defined as failure to achieve protocol-defined complete remission, relapse or death without relapse.
There was no significant difference between the groups in complete remission, which under protocol had to occur by day 60.
HSCT was performed in 57% of patients – during the first complete remission in 28.1% of the midostaurin group and in 22.7% during the first complete remission in the placebo group. For those who were transplanted after the first complete remission, no treatment effect was seen.
Researchers noted that there was a therapeutic benefit even among patients with ITD mutations but with a low allelic burden, in whom the disease might be due largely to mutations other than FLT3.
“It is possible that the benefit of midostaurin, which is a multitargeted kinase inhibitor, might lie beyond its ability to inhibit FLT3,” possibly through inhibition of KIT, researchers said.
They also noted that as the trial went on, more and more investigators decided to treat patients with hematopoietic stem cell transplantation, based on newly reported data elsewhere. Since midostaurin was discontinued at the time of transplant, that could have limited exposure to the drug and limited its effect.
*CORRECTION 7/5/2017: An earlier version of this article misstated the number of patients in the placebo group as well as where the study originally appeared.
Adding the multitargeted kinase inhibitor midostaurin to standard chemotherapy led to significantly longer overall and event-free survival, compared with placebo and standard chemotherapy in newly diagnosed acute myeloid leukemia (AML) patients with FLT3 gene mutations, according to phase III trial results published in the New England Journal of Medicine.*
About 30% of AML patients have mutations to the FLT3 gene – with three-quarters of those internal tandem duplication (ITD) mutations, which involves duplication of between 3 and 100 amino acids in the juxtamembrane region. These mutations are linked with a high relapse rate and poor prognosis, especially when there is a high ratio of these mutations to wild-type FLT3. About 8% of patients with newly diagnosed AML have an FLT3 point mutation in the tyrosine kinase domain (TKD), but the effect of these on prognosis isn’t clear.
In the trial, called RATIFY and conducted at 225 sites in 17 countries, 360 patients were randomized to the midostaurin group and 357* to placebo, and they were treated from 2008 to 2013. In all, 29.8% of patients were “ITD high,” meaning their ITD FLT3 mutation to wild-type FLT3 ratio was higher than 0.7, and 47.6% were “ITD low,” with a mutation-to-wild-type FLT3 ratio of 0.5 to 0.7. A total of 22.6% of patients had TKD mutations.
Patients received standard induction chemotherapy, with daunorubicine and cytarabine, and on days 8 through 21 either 50 mg of midostaurin or placebo orally twice a day. Patients were given an identical second cycle of induction therapy, with midostaurin or placebo, if they showed definitive clinically significant residual leukemia after the first induction treatment.
Those who achieved complete remission after induction were given 4, 28-day cycles of consolidation treatment, with midostaurin or placebo on days 8 through 21. If they stayed in remission after that, they were given maintenance of 12, 28-day cycles of midostaurin or placebo.
They were not required to receive hematopoetic stem cell transplantation (HSCT), but it was performed at investigator discretion.
Midostaurin improved survival but not rates of complete remission as defined in the trial protocol, researchers reported.
The hazard ratio for death in the midostaurin group was 0.78 (95% CI, 0.63 to 0.96; one-sided P = .0009). The 4-year overall survival rate was 51.4% for the midostaurin group and 44.3% for the placebo group. Midostaurin was shown to benefit all mutation subgroups, but with no greater benefit in one group than another.
Patients in the midostaurin group had a 21.6% lower likelihood of having an event, defined as failure to achieve protocol-defined complete remission, relapse or death without relapse.
There was no significant difference between the groups in complete remission, which under protocol had to occur by day 60.
HSCT was performed in 57% of patients – during the first complete remission in 28.1% of the midostaurin group and in 22.7% during the first complete remission in the placebo group. For those who were transplanted after the first complete remission, no treatment effect was seen.
Researchers noted that there was a therapeutic benefit even among patients with ITD mutations but with a low allelic burden, in whom the disease might be due largely to mutations other than FLT3.
“It is possible that the benefit of midostaurin, which is a multitargeted kinase inhibitor, might lie beyond its ability to inhibit FLT3,” possibly through inhibition of KIT, researchers said.
They also noted that as the trial went on, more and more investigators decided to treat patients with hematopoietic stem cell transplantation, based on newly reported data elsewhere. Since midostaurin was discontinued at the time of transplant, that could have limited exposure to the drug and limited its effect.
*CORRECTION 7/5/2017: An earlier version of this article misstated the number of patients in the placebo group as well as where the study originally appeared.
FROM NEJM
Key clinical point: Multitargeted kinase inhibitor midostaurin combined with standard chemotherapy improved survival in newly diagnosed acute myeloid leukemia patients.
Major finding: The 4-year overall survival rate was 51.4% for the midostaurin group and 44.3% for the placebo group. Midostaurin was shown to benefit all mutation subgroups — internal tandem mutations and point mutations in the tyrosine kinase domain – but with no greater benefit in one group than another.
Data source: A multicenter, multinational, randomized, double-blind, placebo-controlled trial.
Disclosures: The trial was funded by the National Cancer Institute and Novartis. Researchers reported receiving personal fees from Novartis and other companies.
Gilteritinib shows safety, efficacy in relapsed/refractory AML
Gilteritinib, a tyrosine kinase inhibitor, had a generally favorable safety profile and inhibited FLT3 in a population enriched with relapsed/refractory acute myeloid leukemia (AML) patients who had the target mutations, based on results of a phase I/II trial.
The findings represent a step forward in treatment of AML with FLT3 inhibition, according to Alexander E. Perl, MD, of the University of Pennsylvania Abramson Comprehensive Cancer Center, Philadelphia, and his colleagues in the trial (NCT02014558), which is sponsored by Astellas Pharma Global Development.
Gilteritinib at 120 mg/day is being tested in phase III trials and in combination with chemotherapy regimens.
Initial entrants in the FLT3 inhibitor class had poor bioavailability, lacked potency and kinase specificity, and had low rates of response. While newer FLT3 inhibitors have had more potent effects, the proportions of patients who have responded have varied and their responses have often been transient, with resistance emerging within a few weeks of treatment.
Gilteritinib is attractive because it has in vitro activity against FLT3 internal tandem duplication mutations and tyrosine kinase domain mutations.
In the first-in-human, single-arm, open-label study — conducted at centers in the United States, Germany, France, and Italy — 252 patients were given one of seven gilteritinib doses, from 20 to 450 mg per day, either as part of a cohort to assess dose escalation or to expand a given dose.
FTL3 mutations were not required for study enrollment, but researchers did require 10 or more patients with confirmed FLT3 mutations to be enrolled in each of the dose expansion groups. Because they found that patients with the mutations were responding so much better than those with wild-type FLT3, they expanded the 120-mg and 200-mg dose cohorts to include only those with FLT3 mutations. In the end, 162 of 252 treated patients had internal tandem duplication mutations, 12 had codon D835 mutations, and 15 had both.
The most common grade 3 or 4 adverse events, regardless of relation to treatment, were neutropenia, seen in 39%, anemia (24%), thrombocytopenia (13%), sepsis (11%), and pneumonia (11%).
Commonly reported treatment-related adverse events were diarrhea (37%), anemia (34%), fatigue (33%), elevated aspartate aminotransferase (26%), and elevated alanine aminotransferase (19%).
Serious adverse events seen in at least 5% of patients included febrile neutropenia (39%; five cases of which were related to the treatment), progressive disease (17%), sepsis (14%; two of which were related to treatment), and pneumonia (11%), and acute renal failure (10%; five related to treatment), the researchers reported in The Lancet Oncology (doi: 10.1016/S1470-2045(17)30416-3).
Seven deaths were judged to be possibly or probably related to treatment, seen in the 20-mg, 80-mg, 120-mg, and 200-mg groups.
Of the 249 patients with data allowing a full analysis, 100 (40%) achieved a response, with 8% achieving a complete remission, 4% a complete remission with incomplete platelet recovery, 18% a complete remission with incomplete hematologic recovery, and 10% a partial remission.
At least 90% of the FLT3 inhibition was seen by the eighth day of treatment among those getting at least the 80-mg dose.
Median overall survival was 25 weeks, and leukemia-free survival will be reported in future data analyses, researchers said.
Only 19% of the patients with FLT3 mutations underwent a hematopoetic stem cell transplant after treatment, which was attributed in part to prior hematopioetic stem cell transplant and the advanced age of many of the patients. Among the patients who subsequently had transplants, the results did not have much effect. Median survival was 47 weeks for those with mutations who had an overall response to gilteritinib and had a transplant after treatment, compared to 42 weeks for those with mutations and an overall response but didn’t go on to transplant.
“Because gilteritinib as a single agent is likely to have limited curative capacity, even when used early in the disease course,” researchers wrote, “studies that integrate gilteritnib into frontline chemotherapy regimens are underway.”
Study authors reported receiving fees, grants, or nonfinancial support from Astellas, the sponsor of the trial, and other pharmaceutical companies.
Gilteritinib, a tyrosine kinase inhibitor, had a generally favorable safety profile and inhibited FLT3 in a population enriched with relapsed/refractory acute myeloid leukemia (AML) patients who had the target mutations, based on results of a phase I/II trial.
The findings represent a step forward in treatment of AML with FLT3 inhibition, according to Alexander E. Perl, MD, of the University of Pennsylvania Abramson Comprehensive Cancer Center, Philadelphia, and his colleagues in the trial (NCT02014558), which is sponsored by Astellas Pharma Global Development.
Gilteritinib at 120 mg/day is being tested in phase III trials and in combination with chemotherapy regimens.
Initial entrants in the FLT3 inhibitor class had poor bioavailability, lacked potency and kinase specificity, and had low rates of response. While newer FLT3 inhibitors have had more potent effects, the proportions of patients who have responded have varied and their responses have often been transient, with resistance emerging within a few weeks of treatment.
Gilteritinib is attractive because it has in vitro activity against FLT3 internal tandem duplication mutations and tyrosine kinase domain mutations.
In the first-in-human, single-arm, open-label study — conducted at centers in the United States, Germany, France, and Italy — 252 patients were given one of seven gilteritinib doses, from 20 to 450 mg per day, either as part of a cohort to assess dose escalation or to expand a given dose.
FTL3 mutations were not required for study enrollment, but researchers did require 10 or more patients with confirmed FLT3 mutations to be enrolled in each of the dose expansion groups. Because they found that patients with the mutations were responding so much better than those with wild-type FLT3, they expanded the 120-mg and 200-mg dose cohorts to include only those with FLT3 mutations. In the end, 162 of 252 treated patients had internal tandem duplication mutations, 12 had codon D835 mutations, and 15 had both.
The most common grade 3 or 4 adverse events, regardless of relation to treatment, were neutropenia, seen in 39%, anemia (24%), thrombocytopenia (13%), sepsis (11%), and pneumonia (11%).
Commonly reported treatment-related adverse events were diarrhea (37%), anemia (34%), fatigue (33%), elevated aspartate aminotransferase (26%), and elevated alanine aminotransferase (19%).
Serious adverse events seen in at least 5% of patients included febrile neutropenia (39%; five cases of which were related to the treatment), progressive disease (17%), sepsis (14%; two of which were related to treatment), and pneumonia (11%), and acute renal failure (10%; five related to treatment), the researchers reported in The Lancet Oncology (doi: 10.1016/S1470-2045(17)30416-3).
Seven deaths were judged to be possibly or probably related to treatment, seen in the 20-mg, 80-mg, 120-mg, and 200-mg groups.
Of the 249 patients with data allowing a full analysis, 100 (40%) achieved a response, with 8% achieving a complete remission, 4% a complete remission with incomplete platelet recovery, 18% a complete remission with incomplete hematologic recovery, and 10% a partial remission.
At least 90% of the FLT3 inhibition was seen by the eighth day of treatment among those getting at least the 80-mg dose.
Median overall survival was 25 weeks, and leukemia-free survival will be reported in future data analyses, researchers said.
Only 19% of the patients with FLT3 mutations underwent a hematopoetic stem cell transplant after treatment, which was attributed in part to prior hematopioetic stem cell transplant and the advanced age of many of the patients. Among the patients who subsequently had transplants, the results did not have much effect. Median survival was 47 weeks for those with mutations who had an overall response to gilteritinib and had a transplant after treatment, compared to 42 weeks for those with mutations and an overall response but didn’t go on to transplant.
“Because gilteritinib as a single agent is likely to have limited curative capacity, even when used early in the disease course,” researchers wrote, “studies that integrate gilteritnib into frontline chemotherapy regimens are underway.”
Study authors reported receiving fees, grants, or nonfinancial support from Astellas, the sponsor of the trial, and other pharmaceutical companies.
Gilteritinib, a tyrosine kinase inhibitor, had a generally favorable safety profile and inhibited FLT3 in a population enriched with relapsed/refractory acute myeloid leukemia (AML) patients who had the target mutations, based on results of a phase I/II trial.
The findings represent a step forward in treatment of AML with FLT3 inhibition, according to Alexander E. Perl, MD, of the University of Pennsylvania Abramson Comprehensive Cancer Center, Philadelphia, and his colleagues in the trial (NCT02014558), which is sponsored by Astellas Pharma Global Development.
Gilteritinib at 120 mg/day is being tested in phase III trials and in combination with chemotherapy regimens.
Initial entrants in the FLT3 inhibitor class had poor bioavailability, lacked potency and kinase specificity, and had low rates of response. While newer FLT3 inhibitors have had more potent effects, the proportions of patients who have responded have varied and their responses have often been transient, with resistance emerging within a few weeks of treatment.
Gilteritinib is attractive because it has in vitro activity against FLT3 internal tandem duplication mutations and tyrosine kinase domain mutations.
In the first-in-human, single-arm, open-label study — conducted at centers in the United States, Germany, France, and Italy — 252 patients were given one of seven gilteritinib doses, from 20 to 450 mg per day, either as part of a cohort to assess dose escalation or to expand a given dose.
FTL3 mutations were not required for study enrollment, but researchers did require 10 or more patients with confirmed FLT3 mutations to be enrolled in each of the dose expansion groups. Because they found that patients with the mutations were responding so much better than those with wild-type FLT3, they expanded the 120-mg and 200-mg dose cohorts to include only those with FLT3 mutations. In the end, 162 of 252 treated patients had internal tandem duplication mutations, 12 had codon D835 mutations, and 15 had both.
The most common grade 3 or 4 adverse events, regardless of relation to treatment, were neutropenia, seen in 39%, anemia (24%), thrombocytopenia (13%), sepsis (11%), and pneumonia (11%).
Commonly reported treatment-related adverse events were diarrhea (37%), anemia (34%), fatigue (33%), elevated aspartate aminotransferase (26%), and elevated alanine aminotransferase (19%).
Serious adverse events seen in at least 5% of patients included febrile neutropenia (39%; five cases of which were related to the treatment), progressive disease (17%), sepsis (14%; two of which were related to treatment), and pneumonia (11%), and acute renal failure (10%; five related to treatment), the researchers reported in The Lancet Oncology (doi: 10.1016/S1470-2045(17)30416-3).
Seven deaths were judged to be possibly or probably related to treatment, seen in the 20-mg, 80-mg, 120-mg, and 200-mg groups.
Of the 249 patients with data allowing a full analysis, 100 (40%) achieved a response, with 8% achieving a complete remission, 4% a complete remission with incomplete platelet recovery, 18% a complete remission with incomplete hematologic recovery, and 10% a partial remission.
At least 90% of the FLT3 inhibition was seen by the eighth day of treatment among those getting at least the 80-mg dose.
Median overall survival was 25 weeks, and leukemia-free survival will be reported in future data analyses, researchers said.
Only 19% of the patients with FLT3 mutations underwent a hematopoetic stem cell transplant after treatment, which was attributed in part to prior hematopioetic stem cell transplant and the advanced age of many of the patients. Among the patients who subsequently had transplants, the results did not have much effect. Median survival was 47 weeks for those with mutations who had an overall response to gilteritinib and had a transplant after treatment, compared to 42 weeks for those with mutations and an overall response but didn’t go on to transplant.
“Because gilteritinib as a single agent is likely to have limited curative capacity, even when used early in the disease course,” researchers wrote, “studies that integrate gilteritnib into frontline chemotherapy regimens are underway.”
Study authors reported receiving fees, grants, or nonfinancial support from Astellas, the sponsor of the trial, and other pharmaceutical companies.
FROM THE LANCET ONCOLOGY
Key clinical point: The highly selective tyrosine kinase inhibitor gilternitinib was generally safe and elicited responses in relapsed-refractory AML patients.
Major finding: Of the 249 patients with data allowing a full analysis, 100 (40%) achieved a response, with a median overall survival of 25 weeks.
Data source: Multicenter, single-arm, open-label study in Europe and the United States.
Disclosures: Astellas Pharma funded the study, and study authors reported receiving fees, grants or nonfinancial support from Astellas and other pharmaceutical companies.
Antibody from AML survivor may prove therapeutic
CHICAGO – A therapeutic target and possibly a treatment for acute myeloid leukemia and myelodysplastic syndrome may lie in the immortalized B cells of a patient whose acute myeloid leukemia was cured after allogeneic stem cell transplantation.
A B cell clone isolated from this patient makes a hypermutated immunoglobulin G1 antibody that binds leukemic blasts of all World Health Organization 2008 AML and myelodysplastic syndrome (MDS) types, based on cells obtained from 60 AML or MDS patients, but does not target healthy cells and lymphoid tissue, Mette D. Hazenberg, MD, PhD, reported at the annual meeting of the American Society of Clinical Oncology.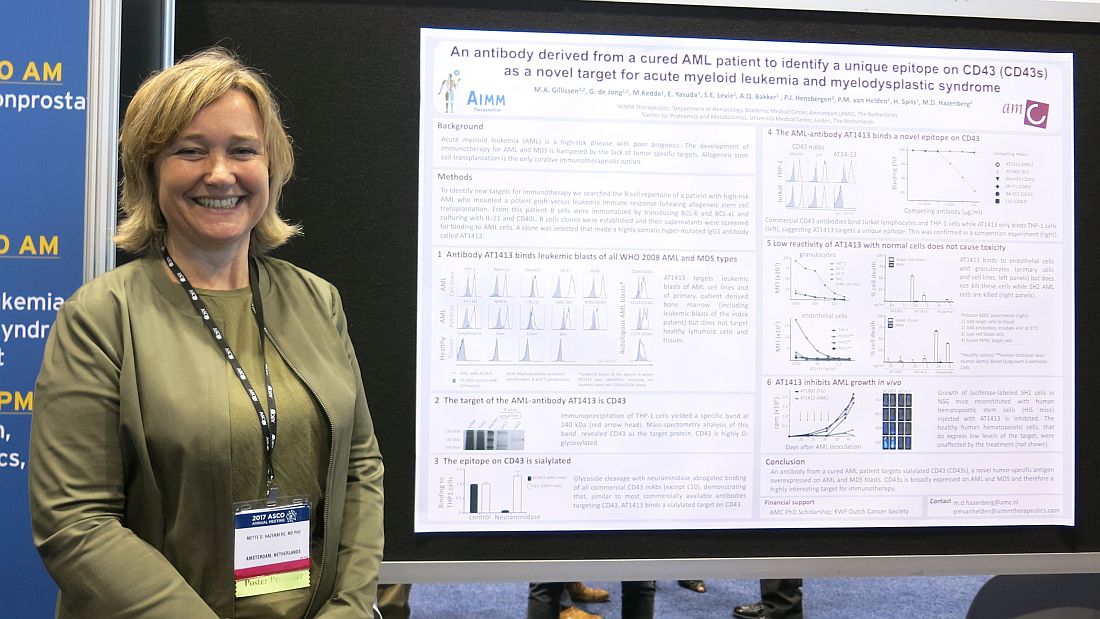
“CD43 is broadly expressed on AML and MDS and, therefore, is a highly interesting target for immunotherapy,” said Dr. Hazenberg of AIMM Therapeutics and Academic Medical Center, Amsterdam.
The growth of luciferase-labeled AML cells expressing CD43s was inhibited in highly immunodeficient NOD scid-gamma mice that were reconstituted with human hematopoietic stem cells injected with AT1413. Healthy human hematopoietic cells, which express low levels of the target, were not affected by the treatment.
Next steps include further in vivo preclinical studies, according to Dr. Hazenberg.
AIMM Therapeutics is a biotech company comprising a joint venture between Immpact and the Academic Medical Center (AMC) at the University of Amsterdam. The study was supported by an AMC PhD scholarship and the KWF Dutch Cancer Society.
mdales@frontlinemedcom.com
On Twitter @maryjodales
CHICAGO – A therapeutic target and possibly a treatment for acute myeloid leukemia and myelodysplastic syndrome may lie in the immortalized B cells of a patient whose acute myeloid leukemia was cured after allogeneic stem cell transplantation.
A B cell clone isolated from this patient makes a hypermutated immunoglobulin G1 antibody that binds leukemic blasts of all World Health Organization 2008 AML and myelodysplastic syndrome (MDS) types, based on cells obtained from 60 AML or MDS patients, but does not target healthy cells and lymphoid tissue, Mette D. Hazenberg, MD, PhD, reported at the annual meeting of the American Society of Clinical Oncology.
“CD43 is broadly expressed on AML and MDS and, therefore, is a highly interesting target for immunotherapy,” said Dr. Hazenberg of AIMM Therapeutics and Academic Medical Center, Amsterdam.
The growth of luciferase-labeled AML cells expressing CD43s was inhibited in highly immunodeficient NOD scid-gamma mice that were reconstituted with human hematopoietic stem cells injected with AT1413. Healthy human hematopoietic cells, which express low levels of the target, were not affected by the treatment.
Next steps include further in vivo preclinical studies, according to Dr. Hazenberg.
AIMM Therapeutics is a biotech company comprising a joint venture between Immpact and the Academic Medical Center (AMC) at the University of Amsterdam. The study was supported by an AMC PhD scholarship and the KWF Dutch Cancer Society.
mdales@frontlinemedcom.com
On Twitter @maryjodales
CHICAGO – A therapeutic target and possibly a treatment for acute myeloid leukemia and myelodysplastic syndrome may lie in the immortalized B cells of a patient whose acute myeloid leukemia was cured after allogeneic stem cell transplantation.
A B cell clone isolated from this patient makes a hypermutated immunoglobulin G1 antibody that binds leukemic blasts of all World Health Organization 2008 AML and myelodysplastic syndrome (MDS) types, based on cells obtained from 60 AML or MDS patients, but does not target healthy cells and lymphoid tissue, Mette D. Hazenberg, MD, PhD, reported at the annual meeting of the American Society of Clinical Oncology.
“CD43 is broadly expressed on AML and MDS and, therefore, is a highly interesting target for immunotherapy,” said Dr. Hazenberg of AIMM Therapeutics and Academic Medical Center, Amsterdam.
The growth of luciferase-labeled AML cells expressing CD43s was inhibited in highly immunodeficient NOD scid-gamma mice that were reconstituted with human hematopoietic stem cells injected with AT1413. Healthy human hematopoietic cells, which express low levels of the target, were not affected by the treatment.
Next steps include further in vivo preclinical studies, according to Dr. Hazenberg.
AIMM Therapeutics is a biotech company comprising a joint venture between Immpact and the Academic Medical Center (AMC) at the University of Amsterdam. The study was supported by an AMC PhD scholarship and the KWF Dutch Cancer Society.
mdales@frontlinemedcom.com
On Twitter @maryjodales
AT ASCO 2017
Key clinical point:
Major finding: The growth of luciferase-labeled SH2 cells was inhibited in highly immunodeficient NSG (NOD scid-gamma) mice that were reconstituted with human hematopoietic stem cells injected with AT1413.
Data source: Cellular studies and studies in severely immunodeficient mice.
Disclosures: Dr. Hazenberg is with AIMM Therapeutics and Academic Medical Center, Amsterdam. AIMM Therapeutics is a biotech company comprising a joint venture between Immpact and the Academic Medical Center (AMC) at the University of Amsterdam. The study was supported by an AMC PhD scholarship and the KWF Dutch Cancer Society.
Company discontinues phase 3 trial of vadastuximab talirine in AML

Update as of June 21: The US Food and Drug Administration (FDA) has placed the Investigational New Drug (IND) application for vadastuximab talirine on hold. No clinical trial may resume under the IND until the FDA lifts the clinical hold.
On the advice of the Independent Data Monitoring Committee, Seattle Genetics is discontinuing the phase 3 CASCADE clinical trial of vadastuximab talirine as frontline treatment in older patients with acute myeloid leukemia (AML).
The company is also suspending patient enrollment and treatment in all its vadastuximab trials, including the ongoing phase 1/2 trial in frontline high-risk myelodysplastic syndromes (MDS).
In December last year, the US Food and Drug Administration (FDA) had placed the trials of vadastuximab on full and partial clinical holds due to the potential risk of hepatotoxicity.
The FDA lifted the hold in March of this year. However, concerns regarding a higher rate of deaths, including fatal infections but not liver toxicity, in the vadastuximab arm compared to control prompted the company to discontinue the phase 3 trial.
Vadastuximab talirene is an antibody-drug conjugate (ADC) targeted to CD33, which is expressed on most AML and MDS blasts. The ADC technology links anti-cancer compounds with targeting antibodies to precisely kill cancer cells and spare healthy ones.
Seattle Genetics’ ADC for Hodgkin lymphoma, brentuximab vedotin, was granted accelerated approval by the FDA in 2011.
The CASCADE trial was evaluating vadastuximab in combination with the hypomethylating agents (HMAs) azacytidine or decitabine compared to an HMA alone in older patients with newly diagnosed AML.
In addition to the MDS trial, the company is stopping enrollment onto the trial of vadastuximab in combination with 7+3 chemotherapy in newly diagnosed, younger AML patients and vadastuximab given prior to or after allogeneic hematopoietic stem cell transplant in AML patients.
Calling the decision “disappointing and unexpected,” Clay Siegall, PhD, president and CEO of Seattle Genetics, said, “Patient safety is our highest priority, and we will closely review the data and evaluate next steps.” ![]()
Update as of June 21: The US Food and Drug Administration (FDA) has placed the Investigational New Drug (IND) application for vadastuximab talirine on hold. No clinical trial may resume under the IND until the FDA lifts the clinical hold.
On the advice of the Independent Data Monitoring Committee, Seattle Genetics is discontinuing the phase 3 CASCADE clinical trial of vadastuximab talirine as frontline treatment in older patients with acute myeloid leukemia (AML).
The company is also suspending patient enrollment and treatment in all its vadastuximab trials, including the ongoing phase 1/2 trial in frontline high-risk myelodysplastic syndromes (MDS).
In December last year, the US Food and Drug Administration (FDA) had placed the trials of vadastuximab on full and partial clinical holds due to the potential risk of hepatotoxicity.
The FDA lifted the hold in March of this year. However, concerns regarding a higher rate of deaths, including fatal infections but not liver toxicity, in the vadastuximab arm compared to control prompted the company to discontinue the phase 3 trial.
Vadastuximab talirene is an antibody-drug conjugate (ADC) targeted to CD33, which is expressed on most AML and MDS blasts. The ADC technology links anti-cancer compounds with targeting antibodies to precisely kill cancer cells and spare healthy ones.
Seattle Genetics’ ADC for Hodgkin lymphoma, brentuximab vedotin, was granted accelerated approval by the FDA in 2011.
The CASCADE trial was evaluating vadastuximab in combination with the hypomethylating agents (HMAs) azacytidine or decitabine compared to an HMA alone in older patients with newly diagnosed AML.
In addition to the MDS trial, the company is stopping enrollment onto the trial of vadastuximab in combination with 7+3 chemotherapy in newly diagnosed, younger AML patients and vadastuximab given prior to or after allogeneic hematopoietic stem cell transplant in AML patients.
Calling the decision “disappointing and unexpected,” Clay Siegall, PhD, president and CEO of Seattle Genetics, said, “Patient safety is our highest priority, and we will closely review the data and evaluate next steps.” ![]()
Update as of June 21: The US Food and Drug Administration (FDA) has placed the Investigational New Drug (IND) application for vadastuximab talirine on hold. No clinical trial may resume under the IND until the FDA lifts the clinical hold.
On the advice of the Independent Data Monitoring Committee, Seattle Genetics is discontinuing the phase 3 CASCADE clinical trial of vadastuximab talirine as frontline treatment in older patients with acute myeloid leukemia (AML).
The company is also suspending patient enrollment and treatment in all its vadastuximab trials, including the ongoing phase 1/2 trial in frontline high-risk myelodysplastic syndromes (MDS).
In December last year, the US Food and Drug Administration (FDA) had placed the trials of vadastuximab on full and partial clinical holds due to the potential risk of hepatotoxicity.
The FDA lifted the hold in March of this year. However, concerns regarding a higher rate of deaths, including fatal infections but not liver toxicity, in the vadastuximab arm compared to control prompted the company to discontinue the phase 3 trial.
Vadastuximab talirene is an antibody-drug conjugate (ADC) targeted to CD33, which is expressed on most AML and MDS blasts. The ADC technology links anti-cancer compounds with targeting antibodies to precisely kill cancer cells and spare healthy ones.
Seattle Genetics’ ADC for Hodgkin lymphoma, brentuximab vedotin, was granted accelerated approval by the FDA in 2011.
The CASCADE trial was evaluating vadastuximab in combination with the hypomethylating agents (HMAs) azacytidine or decitabine compared to an HMA alone in older patients with newly diagnosed AML.
In addition to the MDS trial, the company is stopping enrollment onto the trial of vadastuximab in combination with 7+3 chemotherapy in newly diagnosed, younger AML patients and vadastuximab given prior to or after allogeneic hematopoietic stem cell transplant in AML patients.
Calling the decision “disappointing and unexpected,” Clay Siegall, PhD, president and CEO of Seattle Genetics, said, “Patient safety is our highest priority, and we will closely review the data and evaluate next steps.” ![]()
‘Admirable’ overall survival attainable in AML with enasidenib

CHICAGO—The experimental mutant IDH2 (mIDH2) inhibitor enasidenib has produced “admirable” overall survival in patients with mIDH2 relapsed or refractory acute myeloid leukemia (AML), according to Eytan M. Stein, MD, an investigator on the phase 1 dose escalation and expansion study.
Patients who achieved a complete remission (CR) had a median overall survival (OS) of 19.7 months and non-CR responders, 13.8 months.
“I really want to make the point,” Dr Stein said, “this is a group of patients that are highly refractory, either refractory to induction chemotherapy, refractory to standard of care approaches for patients who are unable to get induction chemotherapy, so refractory to hypomethylating agents or low-dose cytarabine.”
Mutations in IDH2 occur in approximately 12% of AML patients.
Dr Stein explained that the mutant protein converts alpha ketoglutarate to beta hydroxyglutarate (2-HG). And increased levels of intracellular 2-HG lead to methylation changes in the cell that cause a block in myeloid differentiation.
Enasidenib, also known as AG-221, is a selective, oral, potent inhibitor of the mIDH2 enzyyme.
Dr Stein, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the results during the ASCO 2017 Annual Meeting (abstract 7004).
The clinical and translational papers were published simultaneously in Blood.
Study design
The phase 1/2 study had a large dose-escalation component, with 113 patients enrolled. Patients had to have an advanced hematologic malignancy with an IDH2 mutation.
Patients received cumulative daily doses of 50 mg – 650 mg of enasidenib in continuous 28-day cycles.
Four expansion arms were added, with 126 patients.
Two expansion arms were in relapsed/refractory AML patients: one in patients 60 years or older or any age if they had relapsed after bone marrow transplant (BMT), and the other in patients younger than 60 excluding those relapsed after BMT.
The other 2 expansion arms were in untreated AML patients and in patients with any hematologic malignancy ineligible for the other arms.
Dr Stein presented results for the relapsed/refractory AML patients in the dose escalation and expansion phases of the study.
The key endpoints were safety, tolerability, maximum tolerated dose (MTD), and dose-limiting toxicities; response rates as assessed by the local investigator according to IWG criteria; and assessment of clinical activity.
Dr Stein noted the phase 2 study is now completely accrued (n=91) and the recommended enasidenib dose is 100 mg/day in relapsed/refractory AML.
The MTD was not reached at doses up to 650 mg/day.
Baseline characteristics
Median age of all 239 phase 1 patients was 70 years (range, 19-100), 57% were male, and almost all patients had intermediate- or poor-risk disease.
The investigators were also interested in the co-occurring mutations in patients on screening and whether there were differences between patients with mIDH2 at R172 and R140.
Seventy-five percent of the patients (n=179) had R140 and 24% had R172 (n=57).
There was a statistically significant difference in the number of co-occurring mutations in the R140 and R172 patients, with the R140 patients having a higher co-mutation burden compared with the R172 patients, (P=0.020).
The most frequent mutations co-occurring in R140 patients were SRSF2, followed by, in descending order of frequency, DNMT3A, RUNX1, ASXL1, and 24 others.
SFSR2 does not occur in R172 patients. DNMT3A was the most frequently co-occurring mutation in R172, followed by ASXL1, BCOR, NRAS, RUNX1, KMT2A, KRAS, and STAG2.
Safety
The most common treatment-emergent adverse events (TEAE) that occurred in 20% or more of all patients of any grade included nausea (46%), hyperbilirubinemia (45%), diarrhea and fatigue (40% each), decreased appetite (38%), vomiting (32%), dyspnea (31%), cough (29%), pyrexia and febrile neutropenia (28% each), thrombocytopenia, anemia, constipation, hypokalemia, and peripheral edema (27% each), pneumonia (21%), and hyperuricemia (20%).
The only 2 grade 3/4 TEAEs that rose above the level of 5% were hyperbilirubinemia (12%) and thrombocytopenia (6%).
“The hyperbilirubinemia, as I’ve mentioned in a number of meetings before this,” Dr Stein clarified, “is one that occurs because the enzyme is an off-target effect of inhibiting the UGT1A1 enzyme, which conjugates bilirubin.”
“So a patient who goes on this study who has a defect in bilirubin conjugation because they have Gilbert’s disease, they will have a higher level of bilirubin compared to a patient who doesn’t have Gilbert’s disease. This does not appear to have any clinical sequelae. You’ll also notice AST, ALT, alkaline phosphatase or any liver failure is not on this [TEAE] list.”
Response
The overall response rate for the patients who received enasidenib 100 mg/day was 38.5% (42/109) and for all doses 40.3% (71/176).
The true CR rate was 20.2% (100 mg/day) and 19.3% for all doses.
An additional 20% achieved a CR with incomplete hematologic recovery, CR with incomplete platelet recovery, partial response (PR), and morphologic leukemia-free state with either 100 mg enasidenib daily or all doses.
“Time to first response is not immediate,” Dr Stein pointed out. “It takes a median of 1.9 months to get there, and the time to complete remission takes even longer, a median of 3.7 months in the 100-mg experience, 3.8 months in all doses, to get to that best response.”
“I think the clinical importance of this is,” he added, “for a patient that one might have who is on this drug, it is important to keep them on the drug for a prolonged period of time so that they have the opportunity to have that response.”
Hematologic parameters also improved gradually.
Increases in platelet count, absolute neutrophil count, and hemoglobin level did not rise exponentially upon administration of study drug, but rather they slowly rose, “again getting to this point, that the drug takes time to work,” Dr Stein emphasized.
Patients in CR had very high transfusion independence rates, “which is what I would expect,” Dr Stein said. “If you are in complete remission, you should be transfusion independent.”
“What’s a little bit more interesting, though,” Dr Stein added, “is those patients who are non-CR responders. [I]n those patients who have responded but have less than a complete remission, 50% of them are independent of red cell transfusions and 50% of them are independent of platelet transfusions.”
Survival
The CR data and transfusion independence data translated into a median OS in these relapsed and refractory AML patients of 9.3 months.
And about 10% - 15% of the patients had prolonged survival up to 2 years and longer on the single agent.
Analysis of OS by best response revealed that for patients with a CR, “they really have an admirable overall survival of 19.7 months, almost 20 months,” Dr Stein said.
Patients who had a non-CR response had a median OS of 13.8 months, and non-responders had a median OS of 7.0 months.
And there was a qualitative improvement in response over time: the number of patients with CRs and PRs increased, while the number with stable disease decreased.
“Again, I think getting at the point it takes time for these responses to occur,” Dr Stein iterated.
Over the course of therapy, some responders had a differentiation of myeloblasts, so that by cycle 3, the marrow looked largely normal.
The investigators did not observe any morphological evidence of cytotoxicity or cellular aplasia.
But they did observe myeloid differentiation using FISH.
Trisomy 8 that was evident at the time of screening in responders’ myeloblasts, persisted in the promyelocytes and mature granulocyte population, and was no longer evident in the lymphoid compartment.
Baseline 2-HG levels and mIDH2 variant allele frequency were similar for responding and non-responding patients.
The investigators believe that differentiation of myeloblsts, not cytotoxicity, may drive the clinical efficacy of enasidenib.
A phase 3 trial of enasidenib monotherapy versus conventional care regimens is underway in older patients with late-stage AML, and phase 1/2 studies of enasidenib combinations are ongoing in newly diagnosed AML patients.
Enasidenib, which also has efficacy in myelodysplastic syndromes, has been granted priority review for relapsed/refractory AML by the US Food and Drug Administration. ![]()
CHICAGO—The experimental mutant IDH2 (mIDH2) inhibitor enasidenib has produced “admirable” overall survival in patients with mIDH2 relapsed or refractory acute myeloid leukemia (AML), according to Eytan M. Stein, MD, an investigator on the phase 1 dose escalation and expansion study.
Patients who achieved a complete remission (CR) had a median overall survival (OS) of 19.7 months and non-CR responders, 13.8 months.
“I really want to make the point,” Dr Stein said, “this is a group of patients that are highly refractory, either refractory to induction chemotherapy, refractory to standard of care approaches for patients who are unable to get induction chemotherapy, so refractory to hypomethylating agents or low-dose cytarabine.”
Mutations in IDH2 occur in approximately 12% of AML patients.
Dr Stein explained that the mutant protein converts alpha ketoglutarate to beta hydroxyglutarate (2-HG). And increased levels of intracellular 2-HG lead to methylation changes in the cell that cause a block in myeloid differentiation.
Enasidenib, also known as AG-221, is a selective, oral, potent inhibitor of the mIDH2 enzyyme.
Dr Stein, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the results during the ASCO 2017 Annual Meeting (abstract 7004).
The clinical and translational papers were published simultaneously in Blood.
Study design
The phase 1/2 study had a large dose-escalation component, with 113 patients enrolled. Patients had to have an advanced hematologic malignancy with an IDH2 mutation.
Patients received cumulative daily doses of 50 mg – 650 mg of enasidenib in continuous 28-day cycles.
Four expansion arms were added, with 126 patients.
Two expansion arms were in relapsed/refractory AML patients: one in patients 60 years or older or any age if they had relapsed after bone marrow transplant (BMT), and the other in patients younger than 60 excluding those relapsed after BMT.
The other 2 expansion arms were in untreated AML patients and in patients with any hematologic malignancy ineligible for the other arms.
Dr Stein presented results for the relapsed/refractory AML patients in the dose escalation and expansion phases of the study.
The key endpoints were safety, tolerability, maximum tolerated dose (MTD), and dose-limiting toxicities; response rates as assessed by the local investigator according to IWG criteria; and assessment of clinical activity.
Dr Stein noted the phase 2 study is now completely accrued (n=91) and the recommended enasidenib dose is 100 mg/day in relapsed/refractory AML.
The MTD was not reached at doses up to 650 mg/day.
Baseline characteristics
Median age of all 239 phase 1 patients was 70 years (range, 19-100), 57% were male, and almost all patients had intermediate- or poor-risk disease.
The investigators were also interested in the co-occurring mutations in patients on screening and whether there were differences between patients with mIDH2 at R172 and R140.
Seventy-five percent of the patients (n=179) had R140 and 24% had R172 (n=57).
There was a statistically significant difference in the number of co-occurring mutations in the R140 and R172 patients, with the R140 patients having a higher co-mutation burden compared with the R172 patients, (P=0.020).
The most frequent mutations co-occurring in R140 patients were SRSF2, followed by, in descending order of frequency, DNMT3A, RUNX1, ASXL1, and 24 others.
SFSR2 does not occur in R172 patients. DNMT3A was the most frequently co-occurring mutation in R172, followed by ASXL1, BCOR, NRAS, RUNX1, KMT2A, KRAS, and STAG2.
Safety
The most common treatment-emergent adverse events (TEAE) that occurred in 20% or more of all patients of any grade included nausea (46%), hyperbilirubinemia (45%), diarrhea and fatigue (40% each), decreased appetite (38%), vomiting (32%), dyspnea (31%), cough (29%), pyrexia and febrile neutropenia (28% each), thrombocytopenia, anemia, constipation, hypokalemia, and peripheral edema (27% each), pneumonia (21%), and hyperuricemia (20%).
The only 2 grade 3/4 TEAEs that rose above the level of 5% were hyperbilirubinemia (12%) and thrombocytopenia (6%).
“The hyperbilirubinemia, as I’ve mentioned in a number of meetings before this,” Dr Stein clarified, “is one that occurs because the enzyme is an off-target effect of inhibiting the UGT1A1 enzyme, which conjugates bilirubin.”
“So a patient who goes on this study who has a defect in bilirubin conjugation because they have Gilbert’s disease, they will have a higher level of bilirubin compared to a patient who doesn’t have Gilbert’s disease. This does not appear to have any clinical sequelae. You’ll also notice AST, ALT, alkaline phosphatase or any liver failure is not on this [TEAE] list.”
Response
The overall response rate for the patients who received enasidenib 100 mg/day was 38.5% (42/109) and for all doses 40.3% (71/176).
The true CR rate was 20.2% (100 mg/day) and 19.3% for all doses.
An additional 20% achieved a CR with incomplete hematologic recovery, CR with incomplete platelet recovery, partial response (PR), and morphologic leukemia-free state with either 100 mg enasidenib daily or all doses.
“Time to first response is not immediate,” Dr Stein pointed out. “It takes a median of 1.9 months to get there, and the time to complete remission takes even longer, a median of 3.7 months in the 100-mg experience, 3.8 months in all doses, to get to that best response.”
“I think the clinical importance of this is,” he added, “for a patient that one might have who is on this drug, it is important to keep them on the drug for a prolonged period of time so that they have the opportunity to have that response.”
Hematologic parameters also improved gradually.
Increases in platelet count, absolute neutrophil count, and hemoglobin level did not rise exponentially upon administration of study drug, but rather they slowly rose, “again getting to this point, that the drug takes time to work,” Dr Stein emphasized.
Patients in CR had very high transfusion independence rates, “which is what I would expect,” Dr Stein said. “If you are in complete remission, you should be transfusion independent.”
“What’s a little bit more interesting, though,” Dr Stein added, “is those patients who are non-CR responders. [I]n those patients who have responded but have less than a complete remission, 50% of them are independent of red cell transfusions and 50% of them are independent of platelet transfusions.”
Survival
The CR data and transfusion independence data translated into a median OS in these relapsed and refractory AML patients of 9.3 months.
And about 10% - 15% of the patients had prolonged survival up to 2 years and longer on the single agent.
Analysis of OS by best response revealed that for patients with a CR, “they really have an admirable overall survival of 19.7 months, almost 20 months,” Dr Stein said.
Patients who had a non-CR response had a median OS of 13.8 months, and non-responders had a median OS of 7.0 months.
And there was a qualitative improvement in response over time: the number of patients with CRs and PRs increased, while the number with stable disease decreased.
“Again, I think getting at the point it takes time for these responses to occur,” Dr Stein iterated.
Over the course of therapy, some responders had a differentiation of myeloblasts, so that by cycle 3, the marrow looked largely normal.
The investigators did not observe any morphological evidence of cytotoxicity or cellular aplasia.
But they did observe myeloid differentiation using FISH.
Trisomy 8 that was evident at the time of screening in responders’ myeloblasts, persisted in the promyelocytes and mature granulocyte population, and was no longer evident in the lymphoid compartment.
Baseline 2-HG levels and mIDH2 variant allele frequency were similar for responding and non-responding patients.
The investigators believe that differentiation of myeloblsts, not cytotoxicity, may drive the clinical efficacy of enasidenib.
A phase 3 trial of enasidenib monotherapy versus conventional care regimens is underway in older patients with late-stage AML, and phase 1/2 studies of enasidenib combinations are ongoing in newly diagnosed AML patients.
Enasidenib, which also has efficacy in myelodysplastic syndromes, has been granted priority review for relapsed/refractory AML by the US Food and Drug Administration. ![]()
CHICAGO—The experimental mutant IDH2 (mIDH2) inhibitor enasidenib has produced “admirable” overall survival in patients with mIDH2 relapsed or refractory acute myeloid leukemia (AML), according to Eytan M. Stein, MD, an investigator on the phase 1 dose escalation and expansion study.
Patients who achieved a complete remission (CR) had a median overall survival (OS) of 19.7 months and non-CR responders, 13.8 months.
“I really want to make the point,” Dr Stein said, “this is a group of patients that are highly refractory, either refractory to induction chemotherapy, refractory to standard of care approaches for patients who are unable to get induction chemotherapy, so refractory to hypomethylating agents or low-dose cytarabine.”
Mutations in IDH2 occur in approximately 12% of AML patients.
Dr Stein explained that the mutant protein converts alpha ketoglutarate to beta hydroxyglutarate (2-HG). And increased levels of intracellular 2-HG lead to methylation changes in the cell that cause a block in myeloid differentiation.
Enasidenib, also known as AG-221, is a selective, oral, potent inhibitor of the mIDH2 enzyyme.
Dr Stein, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the results during the ASCO 2017 Annual Meeting (abstract 7004).
The clinical and translational papers were published simultaneously in Blood.
Study design
The phase 1/2 study had a large dose-escalation component, with 113 patients enrolled. Patients had to have an advanced hematologic malignancy with an IDH2 mutation.
Patients received cumulative daily doses of 50 mg – 650 mg of enasidenib in continuous 28-day cycles.
Four expansion arms were added, with 126 patients.
Two expansion arms were in relapsed/refractory AML patients: one in patients 60 years or older or any age if they had relapsed after bone marrow transplant (BMT), and the other in patients younger than 60 excluding those relapsed after BMT.
The other 2 expansion arms were in untreated AML patients and in patients with any hematologic malignancy ineligible for the other arms.
Dr Stein presented results for the relapsed/refractory AML patients in the dose escalation and expansion phases of the study.
The key endpoints were safety, tolerability, maximum tolerated dose (MTD), and dose-limiting toxicities; response rates as assessed by the local investigator according to IWG criteria; and assessment of clinical activity.
Dr Stein noted the phase 2 study is now completely accrued (n=91) and the recommended enasidenib dose is 100 mg/day in relapsed/refractory AML.
The MTD was not reached at doses up to 650 mg/day.
Baseline characteristics
Median age of all 239 phase 1 patients was 70 years (range, 19-100), 57% were male, and almost all patients had intermediate- or poor-risk disease.
The investigators were also interested in the co-occurring mutations in patients on screening and whether there were differences between patients with mIDH2 at R172 and R140.
Seventy-five percent of the patients (n=179) had R140 and 24% had R172 (n=57).
There was a statistically significant difference in the number of co-occurring mutations in the R140 and R172 patients, with the R140 patients having a higher co-mutation burden compared with the R172 patients, (P=0.020).
The most frequent mutations co-occurring in R140 patients were SRSF2, followed by, in descending order of frequency, DNMT3A, RUNX1, ASXL1, and 24 others.
SFSR2 does not occur in R172 patients. DNMT3A was the most frequently co-occurring mutation in R172, followed by ASXL1, BCOR, NRAS, RUNX1, KMT2A, KRAS, and STAG2.
Safety
The most common treatment-emergent adverse events (TEAE) that occurred in 20% or more of all patients of any grade included nausea (46%), hyperbilirubinemia (45%), diarrhea and fatigue (40% each), decreased appetite (38%), vomiting (32%), dyspnea (31%), cough (29%), pyrexia and febrile neutropenia (28% each), thrombocytopenia, anemia, constipation, hypokalemia, and peripheral edema (27% each), pneumonia (21%), and hyperuricemia (20%).
The only 2 grade 3/4 TEAEs that rose above the level of 5% were hyperbilirubinemia (12%) and thrombocytopenia (6%).
“The hyperbilirubinemia, as I’ve mentioned in a number of meetings before this,” Dr Stein clarified, “is one that occurs because the enzyme is an off-target effect of inhibiting the UGT1A1 enzyme, which conjugates bilirubin.”
“So a patient who goes on this study who has a defect in bilirubin conjugation because they have Gilbert’s disease, they will have a higher level of bilirubin compared to a patient who doesn’t have Gilbert’s disease. This does not appear to have any clinical sequelae. You’ll also notice AST, ALT, alkaline phosphatase or any liver failure is not on this [TEAE] list.”
Response
The overall response rate for the patients who received enasidenib 100 mg/day was 38.5% (42/109) and for all doses 40.3% (71/176).
The true CR rate was 20.2% (100 mg/day) and 19.3% for all doses.
An additional 20% achieved a CR with incomplete hematologic recovery, CR with incomplete platelet recovery, partial response (PR), and morphologic leukemia-free state with either 100 mg enasidenib daily or all doses.
“Time to first response is not immediate,” Dr Stein pointed out. “It takes a median of 1.9 months to get there, and the time to complete remission takes even longer, a median of 3.7 months in the 100-mg experience, 3.8 months in all doses, to get to that best response.”
“I think the clinical importance of this is,” he added, “for a patient that one might have who is on this drug, it is important to keep them on the drug for a prolonged period of time so that they have the opportunity to have that response.”
Hematologic parameters also improved gradually.
Increases in platelet count, absolute neutrophil count, and hemoglobin level did not rise exponentially upon administration of study drug, but rather they slowly rose, “again getting to this point, that the drug takes time to work,” Dr Stein emphasized.
Patients in CR had very high transfusion independence rates, “which is what I would expect,” Dr Stein said. “If you are in complete remission, you should be transfusion independent.”
“What’s a little bit more interesting, though,” Dr Stein added, “is those patients who are non-CR responders. [I]n those patients who have responded but have less than a complete remission, 50% of them are independent of red cell transfusions and 50% of them are independent of platelet transfusions.”
Survival
The CR data and transfusion independence data translated into a median OS in these relapsed and refractory AML patients of 9.3 months.
And about 10% - 15% of the patients had prolonged survival up to 2 years and longer on the single agent.
Analysis of OS by best response revealed that for patients with a CR, “they really have an admirable overall survival of 19.7 months, almost 20 months,” Dr Stein said.
Patients who had a non-CR response had a median OS of 13.8 months, and non-responders had a median OS of 7.0 months.
And there was a qualitative improvement in response over time: the number of patients with CRs and PRs increased, while the number with stable disease decreased.
“Again, I think getting at the point it takes time for these responses to occur,” Dr Stein iterated.
Over the course of therapy, some responders had a differentiation of myeloblasts, so that by cycle 3, the marrow looked largely normal.
The investigators did not observe any morphological evidence of cytotoxicity or cellular aplasia.
But they did observe myeloid differentiation using FISH.
Trisomy 8 that was evident at the time of screening in responders’ myeloblasts, persisted in the promyelocytes and mature granulocyte population, and was no longer evident in the lymphoid compartment.
Baseline 2-HG levels and mIDH2 variant allele frequency were similar for responding and non-responding patients.
The investigators believe that differentiation of myeloblsts, not cytotoxicity, may drive the clinical efficacy of enasidenib.
A phase 3 trial of enasidenib monotherapy versus conventional care regimens is underway in older patients with late-stage AML, and phase 1/2 studies of enasidenib combinations are ongoing in newly diagnosed AML patients.
Enasidenib, which also has efficacy in myelodysplastic syndromes, has been granted priority review for relapsed/refractory AML by the US Food and Drug Administration. ![]()
Deep molecular responses achievable in AML pts treated with gilteritinib

CHICAGO—Next generation sequencing (NGS) has shown that the FLT3 inhibitor gilteritinib can produce deep molecular responses in a subset of patients with acute myeloid leukemia (AML), according to new research.
Gilteritinib is a highly selective FLT3/AXL inhibitor that is active against FLT3-ITD and FLT3-D835 mutations, but minimal residual disease (MRD) had not systematically been assessed previously in AML patients treated with potent FLT3 inhibitors.
Investigators believed that MRD evaluation in these patients could serve as a useful marker of FLT3 inhibitor efficacy. They therefore conducted an exploratory analysis of a subset of AML patients treated with gilteritinib on the Chrysalis study.
Jessica K. Altman, MD, of the Robert H. Lurie Cancer Center of Northwestern University in Chicago, Illinois, presented the findings at the ASCO 2017 Annual Meeting (abstract 7003).
Chrysalis study: Efficacy and survival
The phase 1/2 Chrysalis study examined the tolerability and antileukemic activity of once daily gilteritinib in a FLT3-ITD-enriched relapsed/refractory AML population of approximately 250 patients.
Overall, gilteritinib was well tolerated and had consistency and potent FLT3 inhibition at doses of >80 mg/day.
The maximum tolerated dose was 300 mg/day. Dose-limiting toxicities were diarrhea and liver function abnormalities.
The greatest overall response rate was 52% and the longest median overall survival (OS) duration was 31 weeks, observed in patients at doses >80 mg/day.
The composite complete remission (CR) rate, comprised of CR, CR with incomplete count recovery (Cri), and CR with incomplete platelet recovery (CRp), was 41%.
The median OS was 31 weeks, and median duration of response 20 weeks.
“Survival probabilities demonstrated that the overall survival for patients who received 80 mg of gilteritinib was higher than those who received less than 80 mg,” Dr Altman said.
Molecular response assessment
Dr Altman then presented the molecular response assessment.
The investigators included all FLT3-ITD mutated patients enrolled in the gilteritinib 120 and 200 mg/day dose cohorts and had bone marrow aspirates available at baseline and at 1 or more additional time points.
“The group I’m reporting on,” Dr Altman explained, “comprises 51% of all FLT3-ITD mutated patients treated at these 2 dose levels.”
FLT3-ITD and total FLT3 alleles were quantified by a novel NGS assay using an Illumina® sequencing platform. Read depth of at least 100,000 reads per sample were implemented.
“Evaluation of MRD was exploratory and it was not prespecified in the study,” Dr Altman noted.
Hence, the investigators defined a molecular response as an ITD signal ratio—FLT3-ITD : FLT3 total—of <10-2.
They defined major molecular response (MMR) as an ITD signal ratio of <10-3, and negative MRD status as <10-4.
Patient characteristics
Baseline characteristics of the 80 patients in the MRD analysis group were similar to those of the entire Chrysalis study population.
Median age was 61 years (range, 23 – 86) and the patients were heavily pretreated: 35% had 3 or more prior lines of AML therapy, and 28% had received a prior FLT3 inhibitor. About a third had prior allogeneic hematopoietic stem cell transplant.
Molecular response
Median OS in this cohort was 32.6 weeks, very similar to the entire study population.
Twenty patients (25%) achieved a molecular response, 18 (23%) an MMR, and 13 (16%) were MRD negative.
The median time to achieve a minimum ITD signal ratio was 8.2 weeks (range, 3.7 – 64).
And molecular response correlated with improved OS.
“The 20 patients who achieved a molecular response had a median overall survival of 59.6 weeks,” Dr Altman said, “which is statistically significantly different and I think clinically different than those who did not attain a molecular response.”
Patients who did not achieve a molecular response had a median overall survival of 28.4 weeks.
“As you could predict,” she added, “the molecular response was greater in those who attained a complete remission than those who had a CRp or Cri.”
Investigators observed similar results in patients who achieved an MMR, using the the cutoff point of 10-3.
“When we stratified by MRD negative status,” she said, “which was an ITD signal ratio of 10-4 or better, there’s clear separation of the Kaplan Meier curves for OS in this cohort again.”
Dr Altman pointed out that this was the first clinical trial to demonstrate that patients with AML treated with a FLT3 inhibitor can attain a molecular response.
“Also, and importantly, there is now a sensitive and specific assay for the detection of minimal residual disease in FLT3-ITD mutated patients and it has the potential to be widely adopted across trials and in clinical practice.”
MRD is prospectively being evaluated in 2 gilteritinib phase 3 maintenance studies.
The trial was sponsored by Astellas Pharma Global Development, Inc. ![]()
CHICAGO—Next generation sequencing (NGS) has shown that the FLT3 inhibitor gilteritinib can produce deep molecular responses in a subset of patients with acute myeloid leukemia (AML), according to new research.
Gilteritinib is a highly selective FLT3/AXL inhibitor that is active against FLT3-ITD and FLT3-D835 mutations, but minimal residual disease (MRD) had not systematically been assessed previously in AML patients treated with potent FLT3 inhibitors.
Investigators believed that MRD evaluation in these patients could serve as a useful marker of FLT3 inhibitor efficacy. They therefore conducted an exploratory analysis of a subset of AML patients treated with gilteritinib on the Chrysalis study.
Jessica K. Altman, MD, of the Robert H. Lurie Cancer Center of Northwestern University in Chicago, Illinois, presented the findings at the ASCO 2017 Annual Meeting (abstract 7003).
Chrysalis study: Efficacy and survival
The phase 1/2 Chrysalis study examined the tolerability and antileukemic activity of once daily gilteritinib in a FLT3-ITD-enriched relapsed/refractory AML population of approximately 250 patients.
Overall, gilteritinib was well tolerated and had consistency and potent FLT3 inhibition at doses of >80 mg/day.
The maximum tolerated dose was 300 mg/day. Dose-limiting toxicities were diarrhea and liver function abnormalities.
The greatest overall response rate was 52% and the longest median overall survival (OS) duration was 31 weeks, observed in patients at doses >80 mg/day.
The composite complete remission (CR) rate, comprised of CR, CR with incomplete count recovery (Cri), and CR with incomplete platelet recovery (CRp), was 41%.
The median OS was 31 weeks, and median duration of response 20 weeks.
“Survival probabilities demonstrated that the overall survival for patients who received 80 mg of gilteritinib was higher than those who received less than 80 mg,” Dr Altman said.
Molecular response assessment
Dr Altman then presented the molecular response assessment.
The investigators included all FLT3-ITD mutated patients enrolled in the gilteritinib 120 and 200 mg/day dose cohorts and had bone marrow aspirates available at baseline and at 1 or more additional time points.
“The group I’m reporting on,” Dr Altman explained, “comprises 51% of all FLT3-ITD mutated patients treated at these 2 dose levels.”
FLT3-ITD and total FLT3 alleles were quantified by a novel NGS assay using an Illumina® sequencing platform. Read depth of at least 100,000 reads per sample were implemented.
“Evaluation of MRD was exploratory and it was not prespecified in the study,” Dr Altman noted.
Hence, the investigators defined a molecular response as an ITD signal ratio—FLT3-ITD : FLT3 total—of <10-2.
They defined major molecular response (MMR) as an ITD signal ratio of <10-3, and negative MRD status as <10-4.
Patient characteristics
Baseline characteristics of the 80 patients in the MRD analysis group were similar to those of the entire Chrysalis study population.
Median age was 61 years (range, 23 – 86) and the patients were heavily pretreated: 35% had 3 or more prior lines of AML therapy, and 28% had received a prior FLT3 inhibitor. About a third had prior allogeneic hematopoietic stem cell transplant.
Molecular response
Median OS in this cohort was 32.6 weeks, very similar to the entire study population.
Twenty patients (25%) achieved a molecular response, 18 (23%) an MMR, and 13 (16%) were MRD negative.
The median time to achieve a minimum ITD signal ratio was 8.2 weeks (range, 3.7 – 64).
And molecular response correlated with improved OS.
“The 20 patients who achieved a molecular response had a median overall survival of 59.6 weeks,” Dr Altman said, “which is statistically significantly different and I think clinically different than those who did not attain a molecular response.”
Patients who did not achieve a molecular response had a median overall survival of 28.4 weeks.
“As you could predict,” she added, “the molecular response was greater in those who attained a complete remission than those who had a CRp or Cri.”
Investigators observed similar results in patients who achieved an MMR, using the the cutoff point of 10-3.
“When we stratified by MRD negative status,” she said, “which was an ITD signal ratio of 10-4 or better, there’s clear separation of the Kaplan Meier curves for OS in this cohort again.”
Dr Altman pointed out that this was the first clinical trial to demonstrate that patients with AML treated with a FLT3 inhibitor can attain a molecular response.
“Also, and importantly, there is now a sensitive and specific assay for the detection of minimal residual disease in FLT3-ITD mutated patients and it has the potential to be widely adopted across trials and in clinical practice.”
MRD is prospectively being evaluated in 2 gilteritinib phase 3 maintenance studies.
The trial was sponsored by Astellas Pharma Global Development, Inc. ![]()
CHICAGO—Next generation sequencing (NGS) has shown that the FLT3 inhibitor gilteritinib can produce deep molecular responses in a subset of patients with acute myeloid leukemia (AML), according to new research.
Gilteritinib is a highly selective FLT3/AXL inhibitor that is active against FLT3-ITD and FLT3-D835 mutations, but minimal residual disease (MRD) had not systematically been assessed previously in AML patients treated with potent FLT3 inhibitors.
Investigators believed that MRD evaluation in these patients could serve as a useful marker of FLT3 inhibitor efficacy. They therefore conducted an exploratory analysis of a subset of AML patients treated with gilteritinib on the Chrysalis study.
Jessica K. Altman, MD, of the Robert H. Lurie Cancer Center of Northwestern University in Chicago, Illinois, presented the findings at the ASCO 2017 Annual Meeting (abstract 7003).
Chrysalis study: Efficacy and survival
The phase 1/2 Chrysalis study examined the tolerability and antileukemic activity of once daily gilteritinib in a FLT3-ITD-enriched relapsed/refractory AML population of approximately 250 patients.
Overall, gilteritinib was well tolerated and had consistency and potent FLT3 inhibition at doses of >80 mg/day.
The maximum tolerated dose was 300 mg/day. Dose-limiting toxicities were diarrhea and liver function abnormalities.
The greatest overall response rate was 52% and the longest median overall survival (OS) duration was 31 weeks, observed in patients at doses >80 mg/day.
The composite complete remission (CR) rate, comprised of CR, CR with incomplete count recovery (Cri), and CR with incomplete platelet recovery (CRp), was 41%.
The median OS was 31 weeks, and median duration of response 20 weeks.
“Survival probabilities demonstrated that the overall survival for patients who received 80 mg of gilteritinib was higher than those who received less than 80 mg,” Dr Altman said.
Molecular response assessment
Dr Altman then presented the molecular response assessment.
The investigators included all FLT3-ITD mutated patients enrolled in the gilteritinib 120 and 200 mg/day dose cohorts and had bone marrow aspirates available at baseline and at 1 or more additional time points.
“The group I’m reporting on,” Dr Altman explained, “comprises 51% of all FLT3-ITD mutated patients treated at these 2 dose levels.”
FLT3-ITD and total FLT3 alleles were quantified by a novel NGS assay using an Illumina® sequencing platform. Read depth of at least 100,000 reads per sample were implemented.
“Evaluation of MRD was exploratory and it was not prespecified in the study,” Dr Altman noted.
Hence, the investigators defined a molecular response as an ITD signal ratio—FLT3-ITD : FLT3 total—of <10-2.
They defined major molecular response (MMR) as an ITD signal ratio of <10-3, and negative MRD status as <10-4.
Patient characteristics
Baseline characteristics of the 80 patients in the MRD analysis group were similar to those of the entire Chrysalis study population.
Median age was 61 years (range, 23 – 86) and the patients were heavily pretreated: 35% had 3 or more prior lines of AML therapy, and 28% had received a prior FLT3 inhibitor. About a third had prior allogeneic hematopoietic stem cell transplant.
Molecular response
Median OS in this cohort was 32.6 weeks, very similar to the entire study population.
Twenty patients (25%) achieved a molecular response, 18 (23%) an MMR, and 13 (16%) were MRD negative.
The median time to achieve a minimum ITD signal ratio was 8.2 weeks (range, 3.7 – 64).
And molecular response correlated with improved OS.
“The 20 patients who achieved a molecular response had a median overall survival of 59.6 weeks,” Dr Altman said, “which is statistically significantly different and I think clinically different than those who did not attain a molecular response.”
Patients who did not achieve a molecular response had a median overall survival of 28.4 weeks.
“As you could predict,” she added, “the molecular response was greater in those who attained a complete remission than those who had a CRp or Cri.”
Investigators observed similar results in patients who achieved an MMR, using the the cutoff point of 10-3.
“When we stratified by MRD negative status,” she said, “which was an ITD signal ratio of 10-4 or better, there’s clear separation of the Kaplan Meier curves for OS in this cohort again.”
Dr Altman pointed out that this was the first clinical trial to demonstrate that patients with AML treated with a FLT3 inhibitor can attain a molecular response.
“Also, and importantly, there is now a sensitive and specific assay for the detection of minimal residual disease in FLT3-ITD mutated patients and it has the potential to be widely adopted across trials and in clinical practice.”
MRD is prospectively being evaluated in 2 gilteritinib phase 3 maintenance studies.
The trial was sponsored by Astellas Pharma Global Development, Inc. ![]()
Majority of AML patients do not receive recommended molecular genetic testing

CHICAGO—While 67% of newly diagnosed patients with acute myeloid leukemia (AML) receive some genetic testing, only 9% receive all 7 of the genetic tests recommended by the National Comprehensive Cancer Network (NCCN), according to new research.
The data comes from the CONNECT MDS/AML Disease Registry, which collects treatment and outcome statistics from 86 sites across the United States, both academic medical centers and community settings. The findings reflect data gathered from 2013 to 2016.
Previously, data on genetic testing in AML patients came primarily from clinical trials, where adherence to guidelines is very high. The current analysis evaluated adherence to genetic testing guidelines in AML treated outside the clinic trial setting.
Daniel A. Pollyea, MD, of the University of Colorado Comprehensive Cancer Center in Aurora, presented the findings on behalf of the CONNECT MDS/AML Disease Registry Scientific Steering Committee (abstract 7022).
The CONNECT registry is a US prospective, observational cohort study of patients with newly diagnosed AML or myelodysplastic syndromes (MDS) aged 55 years or older. Enrollment is ongoing and study clinicians make all clinical decisions.
NCCN guidelines recommend testing AML patients for NPM1, FLT3-ITD, CEBPA, IDH1, IDH2, DNMT3A, and KIT mutations.
“We now know a tremendous amount about the genetic underpinnings of the disease,” Dr Pollyea said.
“We can test for these genetic changes in the clinic to see what’s making a patient’s disease tick. And often there are targeted therapies that can be matched with these genetic changes," he added. "But there’s a disconnect between what can be done, what should be done, and what is being done.”
The current analysis evaluated genetic testing in 259 AML patients, 173 (67%) of whom had some genetic testing.
The likelihood of patients getting tested varied by type of treatment center, age, karyotype, and insurance.
Patients treated at academic medical centers had higher rates of testing than those treated at community clinics (76% and 62%, respectively), P= 0.018
Patients younger than 65 years more often were tested than older patients (83% and 60%, respectively), P= 0.0003.
Patients with non-Medicare insurance were more often tested than those with Medicare (74% and 61%, respectively), P= .025.
And patients with normal karyotype were more often tested than those with abnormal karyotype (77% and 59%, respectively), P= 0.006.
Of the 173 patients who had some genetic testing, only 15 (9%) had all the molecular tests recommended by NCCN.
Of the 7 recommended tests, NPM1 (77%) and FLT3-ITD (76%) were most often reported and DNMT3A least often (16%).
Dr Pollyea attributed the lack of adherence to the guidelines in part to willingness of insurance companies to pay for testing.
He also suggested the guidelines themselves might need adjustment, given the low adherence rate.
Nevertheless, he affirmed the guidelines are well founded. “I think the guidelines are pretty solid and, in my opinion, I would say they don’t go far enough in recommending genetic testing.”
“We’re in our infancy with this testing, and even earlier than infancy in seeing how we’re doing on testing. But now with this registry we at least have the infrastructure available to ask these kinds of questions,” Dr Pollyea added. ![]()
CHICAGO—While 67% of newly diagnosed patients with acute myeloid leukemia (AML) receive some genetic testing, only 9% receive all 7 of the genetic tests recommended by the National Comprehensive Cancer Network (NCCN), according to new research.
The data comes from the CONNECT MDS/AML Disease Registry, which collects treatment and outcome statistics from 86 sites across the United States, both academic medical centers and community settings. The findings reflect data gathered from 2013 to 2016.
Previously, data on genetic testing in AML patients came primarily from clinical trials, where adherence to guidelines is very high. The current analysis evaluated adherence to genetic testing guidelines in AML treated outside the clinic trial setting.
Daniel A. Pollyea, MD, of the University of Colorado Comprehensive Cancer Center in Aurora, presented the findings on behalf of the CONNECT MDS/AML Disease Registry Scientific Steering Committee (abstract 7022).
The CONNECT registry is a US prospective, observational cohort study of patients with newly diagnosed AML or myelodysplastic syndromes (MDS) aged 55 years or older. Enrollment is ongoing and study clinicians make all clinical decisions.
NCCN guidelines recommend testing AML patients for NPM1, FLT3-ITD, CEBPA, IDH1, IDH2, DNMT3A, and KIT mutations.
“We now know a tremendous amount about the genetic underpinnings of the disease,” Dr Pollyea said.
“We can test for these genetic changes in the clinic to see what’s making a patient’s disease tick. And often there are targeted therapies that can be matched with these genetic changes," he added. "But there’s a disconnect between what can be done, what should be done, and what is being done.”
The current analysis evaluated genetic testing in 259 AML patients, 173 (67%) of whom had some genetic testing.
The likelihood of patients getting tested varied by type of treatment center, age, karyotype, and insurance.
Patients treated at academic medical centers had higher rates of testing than those treated at community clinics (76% and 62%, respectively), P= 0.018
Patients younger than 65 years more often were tested than older patients (83% and 60%, respectively), P= 0.0003.
Patients with non-Medicare insurance were more often tested than those with Medicare (74% and 61%, respectively), P= .025.
And patients with normal karyotype were more often tested than those with abnormal karyotype (77% and 59%, respectively), P= 0.006.
Of the 173 patients who had some genetic testing, only 15 (9%) had all the molecular tests recommended by NCCN.
Of the 7 recommended tests, NPM1 (77%) and FLT3-ITD (76%) were most often reported and DNMT3A least often (16%).
Dr Pollyea attributed the lack of adherence to the guidelines in part to willingness of insurance companies to pay for testing.
He also suggested the guidelines themselves might need adjustment, given the low adherence rate.
Nevertheless, he affirmed the guidelines are well founded. “I think the guidelines are pretty solid and, in my opinion, I would say they don’t go far enough in recommending genetic testing.”
“We’re in our infancy with this testing, and even earlier than infancy in seeing how we’re doing on testing. But now with this registry we at least have the infrastructure available to ask these kinds of questions,” Dr Pollyea added. ![]()
CHICAGO—While 67% of newly diagnosed patients with acute myeloid leukemia (AML) receive some genetic testing, only 9% receive all 7 of the genetic tests recommended by the National Comprehensive Cancer Network (NCCN), according to new research.
The data comes from the CONNECT MDS/AML Disease Registry, which collects treatment and outcome statistics from 86 sites across the United States, both academic medical centers and community settings. The findings reflect data gathered from 2013 to 2016.
Previously, data on genetic testing in AML patients came primarily from clinical trials, where adherence to guidelines is very high. The current analysis evaluated adherence to genetic testing guidelines in AML treated outside the clinic trial setting.
Daniel A. Pollyea, MD, of the University of Colorado Comprehensive Cancer Center in Aurora, presented the findings on behalf of the CONNECT MDS/AML Disease Registry Scientific Steering Committee (abstract 7022).
The CONNECT registry is a US prospective, observational cohort study of patients with newly diagnosed AML or myelodysplastic syndromes (MDS) aged 55 years or older. Enrollment is ongoing and study clinicians make all clinical decisions.
NCCN guidelines recommend testing AML patients for NPM1, FLT3-ITD, CEBPA, IDH1, IDH2, DNMT3A, and KIT mutations.
“We now know a tremendous amount about the genetic underpinnings of the disease,” Dr Pollyea said.
“We can test for these genetic changes in the clinic to see what’s making a patient’s disease tick. And often there are targeted therapies that can be matched with these genetic changes," he added. "But there’s a disconnect between what can be done, what should be done, and what is being done.”
The current analysis evaluated genetic testing in 259 AML patients, 173 (67%) of whom had some genetic testing.
The likelihood of patients getting tested varied by type of treatment center, age, karyotype, and insurance.
Patients treated at academic medical centers had higher rates of testing than those treated at community clinics (76% and 62%, respectively), P= 0.018
Patients younger than 65 years more often were tested than older patients (83% and 60%, respectively), P= 0.0003.
Patients with non-Medicare insurance were more often tested than those with Medicare (74% and 61%, respectively), P= .025.
And patients with normal karyotype were more often tested than those with abnormal karyotype (77% and 59%, respectively), P= 0.006.
Of the 173 patients who had some genetic testing, only 15 (9%) had all the molecular tests recommended by NCCN.
Of the 7 recommended tests, NPM1 (77%) and FLT3-ITD (76%) were most often reported and DNMT3A least often (16%).
Dr Pollyea attributed the lack of adherence to the guidelines in part to willingness of insurance companies to pay for testing.
He also suggested the guidelines themselves might need adjustment, given the low adherence rate.
Nevertheless, he affirmed the guidelines are well founded. “I think the guidelines are pretty solid and, in my opinion, I would say they don’t go far enough in recommending genetic testing.”
“We’re in our infancy with this testing, and even earlier than infancy in seeing how we’re doing on testing. But now with this registry we at least have the infrastructure available to ask these kinds of questions,” Dr Pollyea added. ![]()
Differences emerge in new guidelines for managing FN in kids

A multidisciplinary, international panel of experts has updated earlier clinical practice guidelines on managing fever and neutropenia (FN) in children with cancer and in those undergoing hematopoietic stem cell transplantation (HSCT). And while most of the recommendations remained unchanged from the 2012 guidelines, a few key differences emerged. The changes included addition of a 4th generation cephalosporin for empirical antifungal therapy and refinements in risk stratification for invasive fungal disease (IFD), among others.
The new guidelines were published by The International Pediatric Fever and Neutropenia Guideline Panel in the Journal of Clinical Oncology.
The recommendations were organized into 3 major sections: initial presentation, ongoing management, and empirical antifungal therapy. The guidelines panel followed procedures previously validated for creating evidence-based guidelines and used the Appraisal of Guidelines for Research & Evaluation II instrument as a framework.
For the initial presentation of FN, the panel increased the quality of evidence from low to moderate in the recommendation to obtain peripheral blood cultures concurrent with central venous catheter cultures.
In the treatment of FN, the panel added a 4th-generation cephalosporin as empirical therapy in high-risk FN.
The panel refined the IFD risk factors and decreased the quality of evidence from moderate to low. Children with acute myeloid leukemia (AML), high-risk acute lymphoblastic leukemia (ALL), relapsed acute leukemia, those undergoing allogeneic HSCT, those with prolonged neutropenia, and those receiving high-dose corticosteroids are at high risk of IFD. All others should be categorized as IFD low risk.
The panel suggested serum galactomannan not be used to guide empirical antifungal management for prolonged FN lasting 96 hours or more in high-risk IFD patients. GM does not rule out non-Aspergillus molds, and therefore high negative values provide less useful predictions. Previously, the use of galactomannan was a weak recommendation.
The panel added a new recommendation against using fungal polymerase chain reaction (PCR) testing in blood. They explained PCR testing provides poor positive predictive values and negative predictive values are not sufficiently high to be clinically useful. Also, PCR testing is not yet standardized.
Another new recommendation is the addition of imaging of the abdomen in patients without localizing signs or symptoms. Even though the ideal imaging modality is not known, ultrasound is readily available, not associated with radiation exposure, and usually does not require sedation. For these reasons, the panel said it is preferable to computed tomography or magnetic resonance imaging.
The panel also changed a previously weak recommendation to administer empirical therapy for IFD low-risk patients with prolonged FN to a weak recommendation against administering therapy for these patients.
The panel's recommendations and their rationale can be found in the JCO article.
The guidelines update was supported by meeting grants from the Canadian Institutes of Health Research and the Garron Comprehensive Cancer Centre. ![]()
A multidisciplinary, international panel of experts has updated earlier clinical practice guidelines on managing fever and neutropenia (FN) in children with cancer and in those undergoing hematopoietic stem cell transplantation (HSCT). And while most of the recommendations remained unchanged from the 2012 guidelines, a few key differences emerged. The changes included addition of a 4th generation cephalosporin for empirical antifungal therapy and refinements in risk stratification for invasive fungal disease (IFD), among others.
The new guidelines were published by The International Pediatric Fever and Neutropenia Guideline Panel in the Journal of Clinical Oncology.
The recommendations were organized into 3 major sections: initial presentation, ongoing management, and empirical antifungal therapy. The guidelines panel followed procedures previously validated for creating evidence-based guidelines and used the Appraisal of Guidelines for Research & Evaluation II instrument as a framework.
For the initial presentation of FN, the panel increased the quality of evidence from low to moderate in the recommendation to obtain peripheral blood cultures concurrent with central venous catheter cultures.
In the treatment of FN, the panel added a 4th-generation cephalosporin as empirical therapy in high-risk FN.
The panel refined the IFD risk factors and decreased the quality of evidence from moderate to low. Children with acute myeloid leukemia (AML), high-risk acute lymphoblastic leukemia (ALL), relapsed acute leukemia, those undergoing allogeneic HSCT, those with prolonged neutropenia, and those receiving high-dose corticosteroids are at high risk of IFD. All others should be categorized as IFD low risk.
The panel suggested serum galactomannan not be used to guide empirical antifungal management for prolonged FN lasting 96 hours or more in high-risk IFD patients. GM does not rule out non-Aspergillus molds, and therefore high negative values provide less useful predictions. Previously, the use of galactomannan was a weak recommendation.
The panel added a new recommendation against using fungal polymerase chain reaction (PCR) testing in blood. They explained PCR testing provides poor positive predictive values and negative predictive values are not sufficiently high to be clinically useful. Also, PCR testing is not yet standardized.
Another new recommendation is the addition of imaging of the abdomen in patients without localizing signs or symptoms. Even though the ideal imaging modality is not known, ultrasound is readily available, not associated with radiation exposure, and usually does not require sedation. For these reasons, the panel said it is preferable to computed tomography or magnetic resonance imaging.
The panel also changed a previously weak recommendation to administer empirical therapy for IFD low-risk patients with prolonged FN to a weak recommendation against administering therapy for these patients.
The panel's recommendations and their rationale can be found in the JCO article.
The guidelines update was supported by meeting grants from the Canadian Institutes of Health Research and the Garron Comprehensive Cancer Centre. ![]()
A multidisciplinary, international panel of experts has updated earlier clinical practice guidelines on managing fever and neutropenia (FN) in children with cancer and in those undergoing hematopoietic stem cell transplantation (HSCT). And while most of the recommendations remained unchanged from the 2012 guidelines, a few key differences emerged. The changes included addition of a 4th generation cephalosporin for empirical antifungal therapy and refinements in risk stratification for invasive fungal disease (IFD), among others.
The new guidelines were published by The International Pediatric Fever and Neutropenia Guideline Panel in the Journal of Clinical Oncology.
The recommendations were organized into 3 major sections: initial presentation, ongoing management, and empirical antifungal therapy. The guidelines panel followed procedures previously validated for creating evidence-based guidelines and used the Appraisal of Guidelines for Research & Evaluation II instrument as a framework.
For the initial presentation of FN, the panel increased the quality of evidence from low to moderate in the recommendation to obtain peripheral blood cultures concurrent with central venous catheter cultures.
In the treatment of FN, the panel added a 4th-generation cephalosporin as empirical therapy in high-risk FN.
The panel refined the IFD risk factors and decreased the quality of evidence from moderate to low. Children with acute myeloid leukemia (AML), high-risk acute lymphoblastic leukemia (ALL), relapsed acute leukemia, those undergoing allogeneic HSCT, those with prolonged neutropenia, and those receiving high-dose corticosteroids are at high risk of IFD. All others should be categorized as IFD low risk.
The panel suggested serum galactomannan not be used to guide empirical antifungal management for prolonged FN lasting 96 hours or more in high-risk IFD patients. GM does not rule out non-Aspergillus molds, and therefore high negative values provide less useful predictions. Previously, the use of galactomannan was a weak recommendation.
The panel added a new recommendation against using fungal polymerase chain reaction (PCR) testing in blood. They explained PCR testing provides poor positive predictive values and negative predictive values are not sufficiently high to be clinically useful. Also, PCR testing is not yet standardized.
Another new recommendation is the addition of imaging of the abdomen in patients without localizing signs or symptoms. Even though the ideal imaging modality is not known, ultrasound is readily available, not associated with radiation exposure, and usually does not require sedation. For these reasons, the panel said it is preferable to computed tomography or magnetic resonance imaging.
The panel also changed a previously weak recommendation to administer empirical therapy for IFD low-risk patients with prolonged FN to a weak recommendation against administering therapy for these patients.
The panel's recommendations and their rationale can be found in the JCO article.
The guidelines update was supported by meeting grants from the Canadian Institutes of Health Research and the Garron Comprehensive Cancer Centre. ![]()
EC grants drug orphan designation for AML

The European Commission (EC) has granted orphan designation to GMI-1271 for the treatment of acute myeloid leukemia (AML).
GMI-1271 is an E-selectin antagonist being developed by GlycoMimetics, Inc.
The product also has orphan designation, fast track designation, and breakthrough therapy designation in the US.
GMI-1271 is currently being evaluated in a phase 1/2 trial of patients with relapsed or refractory AML and patients age 60 and older with newly diagnosed AML.
The patients are receiving GM-1271 in combination with chemotherapy. The relapsed/refractory group is receiving mitoxantrone, etoposide, and cytarabine. The newly diagnosed patients are receiving cytarabine and idarubicin (7+3).
GlycoMimetics plans to present data from this trial at the 2017 American Society of Clinical Oncology (ASCO) Annual Meeting as abstracts 2520 and 2560.
The company also plans to present the research at the 22nd Congress of the European Hematology Association (EHA) as abstracts P547 and P203.
About orphan designation
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure.
The European Medicines Agency adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the EC for a final decision. The EC typically makes a decision within 30 days of that submission. ![]()
The European Commission (EC) has granted orphan designation to GMI-1271 for the treatment of acute myeloid leukemia (AML).
GMI-1271 is an E-selectin antagonist being developed by GlycoMimetics, Inc.
The product also has orphan designation, fast track designation, and breakthrough therapy designation in the US.
GMI-1271 is currently being evaluated in a phase 1/2 trial of patients with relapsed or refractory AML and patients age 60 and older with newly diagnosed AML.
The patients are receiving GM-1271 in combination with chemotherapy. The relapsed/refractory group is receiving mitoxantrone, etoposide, and cytarabine. The newly diagnosed patients are receiving cytarabine and idarubicin (7+3).
GlycoMimetics plans to present data from this trial at the 2017 American Society of Clinical Oncology (ASCO) Annual Meeting as abstracts 2520 and 2560.
The company also plans to present the research at the 22nd Congress of the European Hematology Association (EHA) as abstracts P547 and P203.
About orphan designation
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure.
The European Medicines Agency adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the EC for a final decision. The EC typically makes a decision within 30 days of that submission. ![]()
The European Commission (EC) has granted orphan designation to GMI-1271 for the treatment of acute myeloid leukemia (AML).
GMI-1271 is an E-selectin antagonist being developed by GlycoMimetics, Inc.
The product also has orphan designation, fast track designation, and breakthrough therapy designation in the US.
GMI-1271 is currently being evaluated in a phase 1/2 trial of patients with relapsed or refractory AML and patients age 60 and older with newly diagnosed AML.
The patients are receiving GM-1271 in combination with chemotherapy. The relapsed/refractory group is receiving mitoxantrone, etoposide, and cytarabine. The newly diagnosed patients are receiving cytarabine and idarubicin (7+3).
GlycoMimetics plans to present data from this trial at the 2017 American Society of Clinical Oncology (ASCO) Annual Meeting as abstracts 2520 and 2560.
The company also plans to present the research at the 22nd Congress of the European Hematology Association (EHA) as abstracts P547 and P203.
About orphan designation
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure.
The European Medicines Agency adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the EC for a final decision. The EC typically makes a decision within 30 days of that submission. ![]()
EMA recommends orphan designation for AML drug
The European Medicines Agency (EMA) has recommended orphan designation for Actimab-A, a product intended to treat patients with newly diagnosed acute myeloid leukemia (AML) who are over the age of 60 and are ineligible for standard induction therapy.
Actimab-A targets CD33, a protein expressed on the surface of AML cells, via the monoclonal antibody, HuM195, which carries the cytotoxic radioisotope actinium-225 to the AML cells.
Actinium Pharmaceuticals, Inc., the company developing Actimab-A, is testing the drug in a phase 2 trial.
Results from a phase 1 trial of the drug were presented at the 2016 ASH Annual Meeting.
At that time, researchers reported results in 18 patients who had been newly diagnosed with AML and were age 60 and older. Their median age was 77 (range, 68-87).
The patients received Actimab-A in combination with low-dose cytarabine. Actimab-A was given at 0.5 μCi/kg/fraction (n=3), 1 μCi/kg/fraction (n=6), 1.5 μCi/kg/fraction (n=3), or 2 μCi/kg/fraction (n=6).
Two patients experienced dose-limiting toxicities. Both had grade 4 thrombocytopenia with marrow aplasia for more than 6 weeks after therapy. One patient was in the 1 µCi/kg/fraction cohort, and the other was in the 2 µCi/kg/fraction cohort.
The maximum-tolerated dose was not reached, but 2 µCi/kg/fraction was chosen as the phase 2 dose.
Grade 3/4 toxicities included neutropenia (n=5), thrombocytopenia (n=9), febrile neutropenia (n=6), pneumonia (n=5), other infections (n=3), atrial fibrillation/syncope (n=1), transient creatinine increase (n=1), generalized fatigue (n=1), hypokalemia (n=1), mucositis (n=1), and rectal hemorrhage (n=1).
Twenty-eight percent of patients (5/18) had objective responses to treatment. Two patients achieved a complete response (CR), 1 had a CR with incomplete platelet recovery, and 2 had a CR with incomplete marrow recovery.
The median duration of response was 9.1 months (range, 4.1-16.9).
About orphan designation
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The European Commission typically makes a decision within 30 days. ![]()
The European Medicines Agency (EMA) has recommended orphan designation for Actimab-A, a product intended to treat patients with newly diagnosed acute myeloid leukemia (AML) who are over the age of 60 and are ineligible for standard induction therapy.
Actimab-A targets CD33, a protein expressed on the surface of AML cells, via the monoclonal antibody, HuM195, which carries the cytotoxic radioisotope actinium-225 to the AML cells.
Actinium Pharmaceuticals, Inc., the company developing Actimab-A, is testing the drug in a phase 2 trial.
Results from a phase 1 trial of the drug were presented at the 2016 ASH Annual Meeting.
At that time, researchers reported results in 18 patients who had been newly diagnosed with AML and were age 60 and older. Their median age was 77 (range, 68-87).
The patients received Actimab-A in combination with low-dose cytarabine. Actimab-A was given at 0.5 μCi/kg/fraction (n=3), 1 μCi/kg/fraction (n=6), 1.5 μCi/kg/fraction (n=3), or 2 μCi/kg/fraction (n=6).
Two patients experienced dose-limiting toxicities. Both had grade 4 thrombocytopenia with marrow aplasia for more than 6 weeks after therapy. One patient was in the 1 µCi/kg/fraction cohort, and the other was in the 2 µCi/kg/fraction cohort.
The maximum-tolerated dose was not reached, but 2 µCi/kg/fraction was chosen as the phase 2 dose.
Grade 3/4 toxicities included neutropenia (n=5), thrombocytopenia (n=9), febrile neutropenia (n=6), pneumonia (n=5), other infections (n=3), atrial fibrillation/syncope (n=1), transient creatinine increase (n=1), generalized fatigue (n=1), hypokalemia (n=1), mucositis (n=1), and rectal hemorrhage (n=1).
Twenty-eight percent of patients (5/18) had objective responses to treatment. Two patients achieved a complete response (CR), 1 had a CR with incomplete platelet recovery, and 2 had a CR with incomplete marrow recovery.
The median duration of response was 9.1 months (range, 4.1-16.9).
About orphan designation
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The European Commission typically makes a decision within 30 days. ![]()
The European Medicines Agency (EMA) has recommended orphan designation for Actimab-A, a product intended to treat patients with newly diagnosed acute myeloid leukemia (AML) who are over the age of 60 and are ineligible for standard induction therapy.
Actimab-A targets CD33, a protein expressed on the surface of AML cells, via the monoclonal antibody, HuM195, which carries the cytotoxic radioisotope actinium-225 to the AML cells.
Actinium Pharmaceuticals, Inc., the company developing Actimab-A, is testing the drug in a phase 2 trial.
Results from a phase 1 trial of the drug were presented at the 2016 ASH Annual Meeting.
At that time, researchers reported results in 18 patients who had been newly diagnosed with AML and were age 60 and older. Their median age was 77 (range, 68-87).
The patients received Actimab-A in combination with low-dose cytarabine. Actimab-A was given at 0.5 μCi/kg/fraction (n=3), 1 μCi/kg/fraction (n=6), 1.5 μCi/kg/fraction (n=3), or 2 μCi/kg/fraction (n=6).
Two patients experienced dose-limiting toxicities. Both had grade 4 thrombocytopenia with marrow aplasia for more than 6 weeks after therapy. One patient was in the 1 µCi/kg/fraction cohort, and the other was in the 2 µCi/kg/fraction cohort.
The maximum-tolerated dose was not reached, but 2 µCi/kg/fraction was chosen as the phase 2 dose.
Grade 3/4 toxicities included neutropenia (n=5), thrombocytopenia (n=9), febrile neutropenia (n=6), pneumonia (n=5), other infections (n=3), atrial fibrillation/syncope (n=1), transient creatinine increase (n=1), generalized fatigue (n=1), hypokalemia (n=1), mucositis (n=1), and rectal hemorrhage (n=1).
Twenty-eight percent of patients (5/18) had objective responses to treatment. Two patients achieved a complete response (CR), 1 had a CR with incomplete platelet recovery, and 2 had a CR with incomplete marrow recovery.
The median duration of response was 9.1 months (range, 4.1-16.9).
About orphan designation
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The European Commission typically makes a decision within 30 days. ![]()