User login
G-CSF could prevent infertility in cancer patients

Granulocyte colony-stimulating factor (G-CSF) could prevent infertility in male cancer patients, according to preclinical research published in Reproductive Biology and Endocrinology.
Researchers said they found that G-CSF protects spermatogenesis after alkylating chemotherapy by stimulating the proliferation of surviving spermatogonia.
The team also found evidence to suggest that G-CSF may be useful as a fertility-restoring treatment.
The researchers have been pursuing initiatives to restore fertility in men who have lost their ability to have children as a result of cancer treatments they received as children.
While working on methods to restart sperm production, the team discovered a link between G-CSF and the absence of normal damage to reproductive ability.
“We were using G-CSF to prevent infections in our research experiments,” said study author Brian Hermann, PhD, of The University of Texas at San Antonio.
“It turned out that the drug also had the unexpected impact of guarding against male infertility.”
To test the fertility-related impact of G-CSF, the researchers treated male mice with G-CSF before and/or after treatment with busulfan.
The team then evaluated effects on spermatogenesis in these mice and control mice that only received busulfan.
G-CSF had a protective effect on spermatogenesis that was stable for at least 19 weeks after chemotherapy.
And mice treated with G-CSF for 4 days after busulfan showed modestly enhanced spermatogenic recovery compared to controls.
The researchers said these results suggest G-CSF promotes spermatogonial proliferation, leading to enhanced spermatogenic regeneration from surviving spermatogonial stem cells. ![]()
Granulocyte colony-stimulating factor (G-CSF) could prevent infertility in male cancer patients, according to preclinical research published in Reproductive Biology and Endocrinology.
Researchers said they found that G-CSF protects spermatogenesis after alkylating chemotherapy by stimulating the proliferation of surviving spermatogonia.
The team also found evidence to suggest that G-CSF may be useful as a fertility-restoring treatment.
The researchers have been pursuing initiatives to restore fertility in men who have lost their ability to have children as a result of cancer treatments they received as children.
While working on methods to restart sperm production, the team discovered a link between G-CSF and the absence of normal damage to reproductive ability.
“We were using G-CSF to prevent infections in our research experiments,” said study author Brian Hermann, PhD, of The University of Texas at San Antonio.
“It turned out that the drug also had the unexpected impact of guarding against male infertility.”
To test the fertility-related impact of G-CSF, the researchers treated male mice with G-CSF before and/or after treatment with busulfan.
The team then evaluated effects on spermatogenesis in these mice and control mice that only received busulfan.
G-CSF had a protective effect on spermatogenesis that was stable for at least 19 weeks after chemotherapy.
And mice treated with G-CSF for 4 days after busulfan showed modestly enhanced spermatogenic recovery compared to controls.
The researchers said these results suggest G-CSF promotes spermatogonial proliferation, leading to enhanced spermatogenic regeneration from surviving spermatogonial stem cells. ![]()
Granulocyte colony-stimulating factor (G-CSF) could prevent infertility in male cancer patients, according to preclinical research published in Reproductive Biology and Endocrinology.
Researchers said they found that G-CSF protects spermatogenesis after alkylating chemotherapy by stimulating the proliferation of surviving spermatogonia.
The team also found evidence to suggest that G-CSF may be useful as a fertility-restoring treatment.
The researchers have been pursuing initiatives to restore fertility in men who have lost their ability to have children as a result of cancer treatments they received as children.
While working on methods to restart sperm production, the team discovered a link between G-CSF and the absence of normal damage to reproductive ability.
“We were using G-CSF to prevent infections in our research experiments,” said study author Brian Hermann, PhD, of The University of Texas at San Antonio.
“It turned out that the drug also had the unexpected impact of guarding against male infertility.”
To test the fertility-related impact of G-CSF, the researchers treated male mice with G-CSF before and/or after treatment with busulfan.
The team then evaluated effects on spermatogenesis in these mice and control mice that only received busulfan.
G-CSF had a protective effect on spermatogenesis that was stable for at least 19 weeks after chemotherapy.
And mice treated with G-CSF for 4 days after busulfan showed modestly enhanced spermatogenic recovery compared to controls.
The researchers said these results suggest G-CSF promotes spermatogonial proliferation, leading to enhanced spermatogenic regeneration from surviving spermatogonial stem cells. ![]()
In CLL, specific mutation is key to ibrutinib resistance
Acquired BTKC481S and PLCG2 mutations led to ibrutinib resistance in chronic lymphocytic leukemia (CLL), investigators reported online in the Journal of Clinical Oncology.
These mutations preceded 85% of clinical relapses, appearing a median of 9.3 months beforehand, Jennifer A. Woyach, MD, and her associates from the Ohio State University, Columbus, concluded from a retrospective study of 308 patients. In a separate prospective study of 112 patients, acquired BTKC481S mutation and clonal expansion preceded all eight cases of relapse, they said. “Relapse of CLL after ibrutinib is an issue of increasing clinical significance,” they concluded. “We show that mutations in Bruton tyrosine kinase (BTK) and PLCG2 appear early and have the potential to be used as a biomarker for future relapse, suggesting an opportunity for intervention.”
Ibrutinib has transformed the CLL treatment landscape, but patients face poor outcomes if they relapse with Richter transformation or develop progressive disease. Past work has linked ibrutinib resistance to acquired mutations in BTK at the binding site of ibrutinib and in PLCG2 located just downstream. But the scope of ibrutinib resistance in CLL and key mutational players were unknown (J Clin Oncol. 2017. doi: 10.1200/JCO.2016.70.2282).
To fill that gap, the researchers retrospectively analyzed data from four sequential ibrutinib CLL trials at the Ohio State University. The separate prospective analysis involved analyzing the entire BTK and PLCG2 coding regions every 3 months.
In the retrospective study, patients had received a median of 3 and up to 16 prior therapies. Given the median follow-up period of 3.4 years, about 19% of patients experienced clinical relapse within 4 years of starting ibrutinib, the researchers estimated (95% confidence interval, 14%-24%). Deep sequencing by Ion Torrent (Life Technologies) identified mutations in BTKC481S and/or PLCG2, in 40 of 47 (85%) relapses. In 31 cases, BTKC481S was the sole mutation. Mutational burdens varied among patients, but generally correlated with CLL progression in peripheral blood versus primarily nodal relapse.
At baseline, 172 (58%) of retrospective study participants had complex cytogenetics, 52% had del(13q), 40% had del(17p), and 21% had MYC abnormality. Median age was 65 years (range, 26-91 years) and 70% of patients were female. Multivariable analyses linked transformation to complex karyotype (hazard ratio, 5.0; 95% CI, 1.5-16.5) and MYC abnormality (HR, 2.5; 95% CI, 1.0-4.7), and linked progressive CLL to age younger than 65 years, complex karyotype, and del(17)(p13.1).
Richter transformation usually occurred within 2 years of starting ibrutinib and had a cumulative 4-year incidence of 10%, the investigators also reported. Patients survived a median of only 3.9 months after stopping ibrutinib because of transformation. The cumulative rate of progressive CLL was higher (19.1%), but early progression was rare, and patients who stopped ibrutinib because of progression survived longer (median, 22.7 months).
In the prospective study, all eight patients with BTKC481S who had not yet clinically relapsed nonetheless had increasing frequency of this mutation over time, the investigators reported. Together, the findings confirm BTK and PLCG2 mutations as the key players in CLL resistance to ibrutinib, they stated. Perhaps most importantly, they reveal “a prolonged period of asymptomatic clonal expression” in CLL that precedes clinical relapse and provides a window of opportunity to target these cells with novel therapies in clinical trials, they wrote.
Given that ibrutinib was approved for use in relapsed CLL only 2 years ago, “We are likely just starting to see the first emergence of relapse in the community setting,” the researchers concluded. “Enhanced knowledge of both the molecular and clinical mechanisms of relapse may allow for strategic alterations in monitoring and management that could change the natural history of ibrutinib resistance.”
Funding sources included the D. Warren Brown Foundation, Mr. and Mrs. Michael Thomas, the Four Winds Foundation, the Leukemia and Lymphoma Society, Pelotonia, and the National Cancer Institute. Pharmacyclics also provided partial support. Dr. Woyach disclosed ties to Janssen, Acerta Pharma, Karyopharm Therapeutics, and MorphoSys, and a provisional patent related to C481S detection.
Acquired BTKC481S and PLCG2 mutations led to ibrutinib resistance in chronic lymphocytic leukemia (CLL), investigators reported online in the Journal of Clinical Oncology.
These mutations preceded 85% of clinical relapses, appearing a median of 9.3 months beforehand, Jennifer A. Woyach, MD, and her associates from the Ohio State University, Columbus, concluded from a retrospective study of 308 patients. In a separate prospective study of 112 patients, acquired BTKC481S mutation and clonal expansion preceded all eight cases of relapse, they said. “Relapse of CLL after ibrutinib is an issue of increasing clinical significance,” they concluded. “We show that mutations in Bruton tyrosine kinase (BTK) and PLCG2 appear early and have the potential to be used as a biomarker for future relapse, suggesting an opportunity for intervention.”
Ibrutinib has transformed the CLL treatment landscape, but patients face poor outcomes if they relapse with Richter transformation or develop progressive disease. Past work has linked ibrutinib resistance to acquired mutations in BTK at the binding site of ibrutinib and in PLCG2 located just downstream. But the scope of ibrutinib resistance in CLL and key mutational players were unknown (J Clin Oncol. 2017. doi: 10.1200/JCO.2016.70.2282).
To fill that gap, the researchers retrospectively analyzed data from four sequential ibrutinib CLL trials at the Ohio State University. The separate prospective analysis involved analyzing the entire BTK and PLCG2 coding regions every 3 months.
In the retrospective study, patients had received a median of 3 and up to 16 prior therapies. Given the median follow-up period of 3.4 years, about 19% of patients experienced clinical relapse within 4 years of starting ibrutinib, the researchers estimated (95% confidence interval, 14%-24%). Deep sequencing by Ion Torrent (Life Technologies) identified mutations in BTKC481S and/or PLCG2, in 40 of 47 (85%) relapses. In 31 cases, BTKC481S was the sole mutation. Mutational burdens varied among patients, but generally correlated with CLL progression in peripheral blood versus primarily nodal relapse.
At baseline, 172 (58%) of retrospective study participants had complex cytogenetics, 52% had del(13q), 40% had del(17p), and 21% had MYC abnormality. Median age was 65 years (range, 26-91 years) and 70% of patients were female. Multivariable analyses linked transformation to complex karyotype (hazard ratio, 5.0; 95% CI, 1.5-16.5) and MYC abnormality (HR, 2.5; 95% CI, 1.0-4.7), and linked progressive CLL to age younger than 65 years, complex karyotype, and del(17)(p13.1).
Richter transformation usually occurred within 2 years of starting ibrutinib and had a cumulative 4-year incidence of 10%, the investigators also reported. Patients survived a median of only 3.9 months after stopping ibrutinib because of transformation. The cumulative rate of progressive CLL was higher (19.1%), but early progression was rare, and patients who stopped ibrutinib because of progression survived longer (median, 22.7 months).
In the prospective study, all eight patients with BTKC481S who had not yet clinically relapsed nonetheless had increasing frequency of this mutation over time, the investigators reported. Together, the findings confirm BTK and PLCG2 mutations as the key players in CLL resistance to ibrutinib, they stated. Perhaps most importantly, they reveal “a prolonged period of asymptomatic clonal expression” in CLL that precedes clinical relapse and provides a window of opportunity to target these cells with novel therapies in clinical trials, they wrote.
Given that ibrutinib was approved for use in relapsed CLL only 2 years ago, “We are likely just starting to see the first emergence of relapse in the community setting,” the researchers concluded. “Enhanced knowledge of both the molecular and clinical mechanisms of relapse may allow for strategic alterations in monitoring and management that could change the natural history of ibrutinib resistance.”
Funding sources included the D. Warren Brown Foundation, Mr. and Mrs. Michael Thomas, the Four Winds Foundation, the Leukemia and Lymphoma Society, Pelotonia, and the National Cancer Institute. Pharmacyclics also provided partial support. Dr. Woyach disclosed ties to Janssen, Acerta Pharma, Karyopharm Therapeutics, and MorphoSys, and a provisional patent related to C481S detection.
Acquired BTKC481S and PLCG2 mutations led to ibrutinib resistance in chronic lymphocytic leukemia (CLL), investigators reported online in the Journal of Clinical Oncology.
These mutations preceded 85% of clinical relapses, appearing a median of 9.3 months beforehand, Jennifer A. Woyach, MD, and her associates from the Ohio State University, Columbus, concluded from a retrospective study of 308 patients. In a separate prospective study of 112 patients, acquired BTKC481S mutation and clonal expansion preceded all eight cases of relapse, they said. “Relapse of CLL after ibrutinib is an issue of increasing clinical significance,” they concluded. “We show that mutations in Bruton tyrosine kinase (BTK) and PLCG2 appear early and have the potential to be used as a biomarker for future relapse, suggesting an opportunity for intervention.”
Ibrutinib has transformed the CLL treatment landscape, but patients face poor outcomes if they relapse with Richter transformation or develop progressive disease. Past work has linked ibrutinib resistance to acquired mutations in BTK at the binding site of ibrutinib and in PLCG2 located just downstream. But the scope of ibrutinib resistance in CLL and key mutational players were unknown (J Clin Oncol. 2017. doi: 10.1200/JCO.2016.70.2282).
To fill that gap, the researchers retrospectively analyzed data from four sequential ibrutinib CLL trials at the Ohio State University. The separate prospective analysis involved analyzing the entire BTK and PLCG2 coding regions every 3 months.
In the retrospective study, patients had received a median of 3 and up to 16 prior therapies. Given the median follow-up period of 3.4 years, about 19% of patients experienced clinical relapse within 4 years of starting ibrutinib, the researchers estimated (95% confidence interval, 14%-24%). Deep sequencing by Ion Torrent (Life Technologies) identified mutations in BTKC481S and/or PLCG2, in 40 of 47 (85%) relapses. In 31 cases, BTKC481S was the sole mutation. Mutational burdens varied among patients, but generally correlated with CLL progression in peripheral blood versus primarily nodal relapse.
At baseline, 172 (58%) of retrospective study participants had complex cytogenetics, 52% had del(13q), 40% had del(17p), and 21% had MYC abnormality. Median age was 65 years (range, 26-91 years) and 70% of patients were female. Multivariable analyses linked transformation to complex karyotype (hazard ratio, 5.0; 95% CI, 1.5-16.5) and MYC abnormality (HR, 2.5; 95% CI, 1.0-4.7), and linked progressive CLL to age younger than 65 years, complex karyotype, and del(17)(p13.1).
Richter transformation usually occurred within 2 years of starting ibrutinib and had a cumulative 4-year incidence of 10%, the investigators also reported. Patients survived a median of only 3.9 months after stopping ibrutinib because of transformation. The cumulative rate of progressive CLL was higher (19.1%), but early progression was rare, and patients who stopped ibrutinib because of progression survived longer (median, 22.7 months).
In the prospective study, all eight patients with BTKC481S who had not yet clinically relapsed nonetheless had increasing frequency of this mutation over time, the investigators reported. Together, the findings confirm BTK and PLCG2 mutations as the key players in CLL resistance to ibrutinib, they stated. Perhaps most importantly, they reveal “a prolonged period of asymptomatic clonal expression” in CLL that precedes clinical relapse and provides a window of opportunity to target these cells with novel therapies in clinical trials, they wrote.
Given that ibrutinib was approved for use in relapsed CLL only 2 years ago, “We are likely just starting to see the first emergence of relapse in the community setting,” the researchers concluded. “Enhanced knowledge of both the molecular and clinical mechanisms of relapse may allow for strategic alterations in monitoring and management that could change the natural history of ibrutinib resistance.”
Funding sources included the D. Warren Brown Foundation, Mr. and Mrs. Michael Thomas, the Four Winds Foundation, the Leukemia and Lymphoma Society, Pelotonia, and the National Cancer Institute. Pharmacyclics also provided partial support. Dr. Woyach disclosed ties to Janssen, Acerta Pharma, Karyopharm Therapeutics, and MorphoSys, and a provisional patent related to C481S detection.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Acquired mutations in BTKC481S and PLCG2 predict ibrutinib resistance in chronic lymphocytic leukemia.
Major finding: These mutations appeared a median of 9.3 months before clinical relapse in 85% of cases. In a separate study, all eight CLL patients who relapsed on ibrutinib had previously developed the BTKC481S mutation with clonal expansion.
Data source: A retrospective analysis of 308 CLL patients from four ibrutinib trials, and a separate prospective study of 118 CLL patients.
Disclosures: Funding sources included the D. Warren Brown Foundation, Mr. and Mrs. Michael Thomas, the Four Winds Foundation, the Leukemia and Lymphoma Society, Pelotonia, and the National Cancer Institute. Pharmacyclics also provided partial support. Dr. Woyach disclosed ties to Janssen, Acerta Pharma, Karyopharm Therapeutics, and MorphoSys, and a provisional patent related to C481S detection.
Team creates online database of cancer mutations

Image by Spencer Phillips
Researchers have developed an online “knowledgebase” called CIViC, an open access resource for collecting and interpreting information from scientific publications on cancer genetics.
“CIViC” stands for Clinical Interpretations of Variants in Cancer, and the researchers liken it to a Wikipedia of cancer genetics.
Anyone can create an account and contribute information. That information is then curated by editors and moderators who are considered experts in the field.
The researchers described the resource in Nature Genetics.
“It’s relatively easy now to sequence the DNA of tumors—to gather the raw information—but there’s a big interpretation problem,” said study author Obi L. Griffith, PhD, of Washington University School of Medicine in St Louis, Missouri.
“What do these hundreds or thousands of mutations mean for this patient? There are a lot of studies being done to answer these questions. But oncologists trying to interpret the raw data are faced with an overwhelming task of plumbing the literature, reading papers, trying to understand what the latest studies tell them about these mutations and how they may or may not be important.”
The CIViC knowledgebase is an attempt to solve this problem. The researchers said this is one of many efforts to collect and interpret such information, but, to their knowledge, CIViC is the only one that is entirely open access. Anyone is free to contribute and use the content as well as the source code.
“We are committed to keeping this resource open and available to anyone who wants to contribute or make use of the information,” said Malachi Griffith, PhD, of Washington University School of Medicine.
“We would like it to be a community exercise and public resource. The information is in the public domain. There are no restrictions on its use, academic or commercial.”
Though anyone can submit a new piece of information or suggest edits to existing data, at least 2 independent contributors must agree that the new information should be incorporated, and 1 of those users must be an “expert editor.”
Expert editors are not permitted to approve their own submissions. Information on the CIViC website provides details about how new users may be promoted to expert editors and administrators.
To date, the site has seen over 17,500 users from academic institutions, governmental organizations, and commercial entities around the world.
Since CIViC’s launch, 59 users have volunteered their time to contribute their knowledge to CIViC, including descriptions of the clinical relevance of 732 mutations from 285 genes for 203 types of cancer, all gleaned from reviewing 1090 scientific and medical publications.
Despite the fact that there are many groups attempting to collect and interpret genomic variants in cancer, the researchers said the sheer volume of information has resulted in relatively little overlap in data gathered so far.
“While we believe this is the only such open access knowledgebase, there are other large research centers with similar resources,” Malachi Griffith said. “We did an analysis to compare the big ones.”
“Even though we all have access to the same published literature, if you look at the overlap of the information mined by each of these resources, it’s remarkably small. We’re all approaching the same problem, and, just by chance—and probably because of the amount of information out there—we haven’t duplicated our efforts very much yet.”
Obi and Malachi Griffith said finding a way to combine these resources is the primary goal of an international group they are helping lead called the Variant Interpretation for Cancer Consortium, which is a part of the Global Alliance for Genomics and Health (GA4GH).
“We’re just scratching the surface of the potential this holds for precision medicine,” Obi Griffith said. “There’s a lot of work to do.” ![]()
Image by Spencer Phillips
Researchers have developed an online “knowledgebase” called CIViC, an open access resource for collecting and interpreting information from scientific publications on cancer genetics.
“CIViC” stands for Clinical Interpretations of Variants in Cancer, and the researchers liken it to a Wikipedia of cancer genetics.
Anyone can create an account and contribute information. That information is then curated by editors and moderators who are considered experts in the field.
The researchers described the resource in Nature Genetics.
“It’s relatively easy now to sequence the DNA of tumors—to gather the raw information—but there’s a big interpretation problem,” said study author Obi L. Griffith, PhD, of Washington University School of Medicine in St Louis, Missouri.
“What do these hundreds or thousands of mutations mean for this patient? There are a lot of studies being done to answer these questions. But oncologists trying to interpret the raw data are faced with an overwhelming task of plumbing the literature, reading papers, trying to understand what the latest studies tell them about these mutations and how they may or may not be important.”
The CIViC knowledgebase is an attempt to solve this problem. The researchers said this is one of many efforts to collect and interpret such information, but, to their knowledge, CIViC is the only one that is entirely open access. Anyone is free to contribute and use the content as well as the source code.
“We are committed to keeping this resource open and available to anyone who wants to contribute or make use of the information,” said Malachi Griffith, PhD, of Washington University School of Medicine.
“We would like it to be a community exercise and public resource. The information is in the public domain. There are no restrictions on its use, academic or commercial.”
Though anyone can submit a new piece of information or suggest edits to existing data, at least 2 independent contributors must agree that the new information should be incorporated, and 1 of those users must be an “expert editor.”
Expert editors are not permitted to approve their own submissions. Information on the CIViC website provides details about how new users may be promoted to expert editors and administrators.
To date, the site has seen over 17,500 users from academic institutions, governmental organizations, and commercial entities around the world.
Since CIViC’s launch, 59 users have volunteered their time to contribute their knowledge to CIViC, including descriptions of the clinical relevance of 732 mutations from 285 genes for 203 types of cancer, all gleaned from reviewing 1090 scientific and medical publications.
Despite the fact that there are many groups attempting to collect and interpret genomic variants in cancer, the researchers said the sheer volume of information has resulted in relatively little overlap in data gathered so far.
“While we believe this is the only such open access knowledgebase, there are other large research centers with similar resources,” Malachi Griffith said. “We did an analysis to compare the big ones.”
“Even though we all have access to the same published literature, if you look at the overlap of the information mined by each of these resources, it’s remarkably small. We’re all approaching the same problem, and, just by chance—and probably because of the amount of information out there—we haven’t duplicated our efforts very much yet.”
Obi and Malachi Griffith said finding a way to combine these resources is the primary goal of an international group they are helping lead called the Variant Interpretation for Cancer Consortium, which is a part of the Global Alliance for Genomics and Health (GA4GH).
“We’re just scratching the surface of the potential this holds for precision medicine,” Obi Griffith said. “There’s a lot of work to do.” ![]()
Image by Spencer Phillips
Researchers have developed an online “knowledgebase” called CIViC, an open access resource for collecting and interpreting information from scientific publications on cancer genetics.
“CIViC” stands for Clinical Interpretations of Variants in Cancer, and the researchers liken it to a Wikipedia of cancer genetics.
Anyone can create an account and contribute information. That information is then curated by editors and moderators who are considered experts in the field.
The researchers described the resource in Nature Genetics.
“It’s relatively easy now to sequence the DNA of tumors—to gather the raw information—but there’s a big interpretation problem,” said study author Obi L. Griffith, PhD, of Washington University School of Medicine in St Louis, Missouri.
“What do these hundreds or thousands of mutations mean for this patient? There are a lot of studies being done to answer these questions. But oncologists trying to interpret the raw data are faced with an overwhelming task of plumbing the literature, reading papers, trying to understand what the latest studies tell them about these mutations and how they may or may not be important.”
The CIViC knowledgebase is an attempt to solve this problem. The researchers said this is one of many efforts to collect and interpret such information, but, to their knowledge, CIViC is the only one that is entirely open access. Anyone is free to contribute and use the content as well as the source code.
“We are committed to keeping this resource open and available to anyone who wants to contribute or make use of the information,” said Malachi Griffith, PhD, of Washington University School of Medicine.
“We would like it to be a community exercise and public resource. The information is in the public domain. There are no restrictions on its use, academic or commercial.”
Though anyone can submit a new piece of information or suggest edits to existing data, at least 2 independent contributors must agree that the new information should be incorporated, and 1 of those users must be an “expert editor.”
Expert editors are not permitted to approve their own submissions. Information on the CIViC website provides details about how new users may be promoted to expert editors and administrators.
To date, the site has seen over 17,500 users from academic institutions, governmental organizations, and commercial entities around the world.
Since CIViC’s launch, 59 users have volunteered their time to contribute their knowledge to CIViC, including descriptions of the clinical relevance of 732 mutations from 285 genes for 203 types of cancer, all gleaned from reviewing 1090 scientific and medical publications.
Despite the fact that there are many groups attempting to collect and interpret genomic variants in cancer, the researchers said the sheer volume of information has resulted in relatively little overlap in data gathered so far.
“While we believe this is the only such open access knowledgebase, there are other large research centers with similar resources,” Malachi Griffith said. “We did an analysis to compare the big ones.”
“Even though we all have access to the same published literature, if you look at the overlap of the information mined by each of these resources, it’s remarkably small. We’re all approaching the same problem, and, just by chance—and probably because of the amount of information out there—we haven’t duplicated our efforts very much yet.”
Obi and Malachi Griffith said finding a way to combine these resources is the primary goal of an international group they are helping lead called the Variant Interpretation for Cancer Consortium, which is a part of the Global Alliance for Genomics and Health (GA4GH).
“We’re just scratching the surface of the potential this holds for precision medicine,” Obi Griffith said. “There’s a lot of work to do.” ![]()
Group identifies ‘essential’ genes in AML
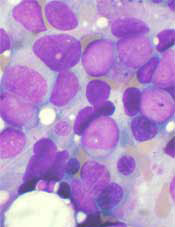
CRISPR-based genetic screens have revealed essential genes—those required for cellular proliferation and survival—in 14 acute myeloid leukemia (AML) cell lines, according to investigators.
By combining this information with the existing genomic information on these cell lines, the investigators believe they have identified vulnerabilities that could potentially be exploited with new therapies.
The group described their research in Cell.
A major aspect of their study focused on the genes and protein pathways connected to the Ras oncogene, which is the most commonly mutated oncogene in human cancers and plays a role in AML.
“For the most part, the mutant Ras protein itself has been considered to be ‘undruggable,’” said study author Tim Wang, a doctoral student at the Massachusetts Institute of Technology in Cambridge.
“An alternative approach has been to find other genes that Ras-mutant cancers rely on with the hope that one of them may be druggable. Unfortunately, such ‘Ras-synthetic-lethal’ genes have been difficult to identify.”
Using CRISPR-based screens, the investigators were able to gauge the impact of individually knocking out each of the 18,000 protein-coding genes in the human genome.
“This process rapidly enabled us to identify the short list of genes that were selectively required in only the Ras-mutant cells,” explained study author David Sabatini, MD, PhD, of the Massachusetts Institute of Technology.
He and his colleagues found that the enzymes catalyzing the latter steps of the Ras processing pathway, Rce1 and Icmt, displayed synthetic lethality with oncogenic Ras, which suggests they might be therapeutic targets in AML and other cancers driven by oncogenic Ras.
The investigators also said their findings provide further support for the central role of MAPK signaling in Ras-driven cancers, suggest PREX1 and the Rac pathway are critical regulators of MAPK pathway activation, and indicate that c-Raf could be a therapeutic target in cancers driven by oncogenic Ras.
In addition to defining the Ras-specific gene essentiality network, the investigators said they were able to determine the function of previously unstudied genes.
The team started by focusing on genes that were essential in some of the AML cell lines but dispensable for others. For each of these genes, the investigators sifted through their data to find others that showed a matching pattern of essentiality, with the idea that all of them had similar functions.
Indeed, this analysis revealed gene groups that were already known to act together and uncovered novel associations between genes that were not known to be related or had been previously unstudied.
“What’s particularly exciting about this work is that we have just begun to scratch the surface with our method,” Wang concluded. “By applying it broadly, we could reveal a huge amount of information about the functional organization of human genes and their roles in many diseases.” ![]()
CRISPR-based genetic screens have revealed essential genes—those required for cellular proliferation and survival—in 14 acute myeloid leukemia (AML) cell lines, according to investigators.
By combining this information with the existing genomic information on these cell lines, the investigators believe they have identified vulnerabilities that could potentially be exploited with new therapies.
The group described their research in Cell.
A major aspect of their study focused on the genes and protein pathways connected to the Ras oncogene, which is the most commonly mutated oncogene in human cancers and plays a role in AML.
“For the most part, the mutant Ras protein itself has been considered to be ‘undruggable,’” said study author Tim Wang, a doctoral student at the Massachusetts Institute of Technology in Cambridge.
“An alternative approach has been to find other genes that Ras-mutant cancers rely on with the hope that one of them may be druggable. Unfortunately, such ‘Ras-synthetic-lethal’ genes have been difficult to identify.”
Using CRISPR-based screens, the investigators were able to gauge the impact of individually knocking out each of the 18,000 protein-coding genes in the human genome.
“This process rapidly enabled us to identify the short list of genes that were selectively required in only the Ras-mutant cells,” explained study author David Sabatini, MD, PhD, of the Massachusetts Institute of Technology.
He and his colleagues found that the enzymes catalyzing the latter steps of the Ras processing pathway, Rce1 and Icmt, displayed synthetic lethality with oncogenic Ras, which suggests they might be therapeutic targets in AML and other cancers driven by oncogenic Ras.
The investigators also said their findings provide further support for the central role of MAPK signaling in Ras-driven cancers, suggest PREX1 and the Rac pathway are critical regulators of MAPK pathway activation, and indicate that c-Raf could be a therapeutic target in cancers driven by oncogenic Ras.
In addition to defining the Ras-specific gene essentiality network, the investigators said they were able to determine the function of previously unstudied genes.
The team started by focusing on genes that were essential in some of the AML cell lines but dispensable for others. For each of these genes, the investigators sifted through their data to find others that showed a matching pattern of essentiality, with the idea that all of them had similar functions.
Indeed, this analysis revealed gene groups that were already known to act together and uncovered novel associations between genes that were not known to be related or had been previously unstudied.
“What’s particularly exciting about this work is that we have just begun to scratch the surface with our method,” Wang concluded. “By applying it broadly, we could reveal a huge amount of information about the functional organization of human genes and their roles in many diseases.” ![]()
CRISPR-based genetic screens have revealed essential genes—those required for cellular proliferation and survival—in 14 acute myeloid leukemia (AML) cell lines, according to investigators.
By combining this information with the existing genomic information on these cell lines, the investigators believe they have identified vulnerabilities that could potentially be exploited with new therapies.
The group described their research in Cell.
A major aspect of their study focused on the genes and protein pathways connected to the Ras oncogene, which is the most commonly mutated oncogene in human cancers and plays a role in AML.
“For the most part, the mutant Ras protein itself has been considered to be ‘undruggable,’” said study author Tim Wang, a doctoral student at the Massachusetts Institute of Technology in Cambridge.
“An alternative approach has been to find other genes that Ras-mutant cancers rely on with the hope that one of them may be druggable. Unfortunately, such ‘Ras-synthetic-lethal’ genes have been difficult to identify.”
Using CRISPR-based screens, the investigators were able to gauge the impact of individually knocking out each of the 18,000 protein-coding genes in the human genome.
“This process rapidly enabled us to identify the short list of genes that were selectively required in only the Ras-mutant cells,” explained study author David Sabatini, MD, PhD, of the Massachusetts Institute of Technology.
He and his colleagues found that the enzymes catalyzing the latter steps of the Ras processing pathway, Rce1 and Icmt, displayed synthetic lethality with oncogenic Ras, which suggests they might be therapeutic targets in AML and other cancers driven by oncogenic Ras.
The investigators also said their findings provide further support for the central role of MAPK signaling in Ras-driven cancers, suggest PREX1 and the Rac pathway are critical regulators of MAPK pathway activation, and indicate that c-Raf could be a therapeutic target in cancers driven by oncogenic Ras.
In addition to defining the Ras-specific gene essentiality network, the investigators said they were able to determine the function of previously unstudied genes.
The team started by focusing on genes that were essential in some of the AML cell lines but dispensable for others. For each of these genes, the investigators sifted through their data to find others that showed a matching pattern of essentiality, with the idea that all of them had similar functions.
Indeed, this analysis revealed gene groups that were already known to act together and uncovered novel associations between genes that were not known to be related or had been previously unstudied.
“What’s particularly exciting about this work is that we have just begun to scratch the surface with our method,” Wang concluded. “By applying it broadly, we could reveal a huge amount of information about the functional organization of human genes and their roles in many diseases.” ![]()
Computer predicts remission, relapse in AML

Photo by Darren Baker
Researchers say they have developed the first computer machine-learning model to accurately predict which patients diagnosed with acute
myeloid leukemia (AML) will go into remission after treatment and which will relapse.
“It’s pretty straightforward to teach a computer to recognize AML, once you develop a robust algorithm, and, in previous work, we did it with almost 100% accuracy,” said study author Murat Dundar, PhD of Indiana University-Purdue University Indianapolis.
“What was challenging was to go beyond that work and teach the computer to accurately predict the direction of change in disease progression in AML patients, interpreting new data to predict the unknown: which new AML patients will go into remission and which will relapse.”
Dr Dundar and his colleagues described this work in IEEE Transactions on Biomedical Engineering.
The researchers said they modeled data from multiple flow cytometry samples to identify functionally distinct cell populations and their local realizations. Each sample was characterized by the proportions of recovered cell populations, which were used to predict the direction of change in disease progression for each AML patient.
“As the input, our computational system employs data from flow cytometry, a widely utilized technology that can rapidly provide detailed characteristics of single cells in samples such as blood or bone marrow,” explained study author Bartek Rajwa, PhD, of Purdue University in Lafayette, Indiana.
“Traditionally, the results of flow cytometry analyses are evaluated by highly trained human experts rather than by machine-learning algorithms. But computers are often better at extracting knowledge from complex data than humans are.”
The researchers used 200 diseased and non-diseased immunophenotypic panels for training and tested
the computational system with samples collected at multiple time points from 36 additional AML patients.
The system was able to predict remission with 100% accuracy (26 of 26 cases) and relapse with 90% accuracy (9 of 10 cases).
“Machine learning is not about modeling data,” Dr Dundar noted. “It’s about extracting knowledge from the data you have so you can build a powerful, intuitive tool that can make predictions about future data that the computer has not previously seen. The machine is learning, not memorizing, and that’s what we did.” ![]()
Photo by Darren Baker
Researchers say they have developed the first computer machine-learning model to accurately predict which patients diagnosed with acute
myeloid leukemia (AML) will go into remission after treatment and which will relapse.
“It’s pretty straightforward to teach a computer to recognize AML, once you develop a robust algorithm, and, in previous work, we did it with almost 100% accuracy,” said study author Murat Dundar, PhD of Indiana University-Purdue University Indianapolis.
“What was challenging was to go beyond that work and teach the computer to accurately predict the direction of change in disease progression in AML patients, interpreting new data to predict the unknown: which new AML patients will go into remission and which will relapse.”
Dr Dundar and his colleagues described this work in IEEE Transactions on Biomedical Engineering.
The researchers said they modeled data from multiple flow cytometry samples to identify functionally distinct cell populations and their local realizations. Each sample was characterized by the proportions of recovered cell populations, which were used to predict the direction of change in disease progression for each AML patient.
“As the input, our computational system employs data from flow cytometry, a widely utilized technology that can rapidly provide detailed characteristics of single cells in samples such as blood or bone marrow,” explained study author Bartek Rajwa, PhD, of Purdue University in Lafayette, Indiana.
“Traditionally, the results of flow cytometry analyses are evaluated by highly trained human experts rather than by machine-learning algorithms. But computers are often better at extracting knowledge from complex data than humans are.”
The researchers used 200 diseased and non-diseased immunophenotypic panels for training and tested
the computational system with samples collected at multiple time points from 36 additional AML patients.
The system was able to predict remission with 100% accuracy (26 of 26 cases) and relapse with 90% accuracy (9 of 10 cases).
“Machine learning is not about modeling data,” Dr Dundar noted. “It’s about extracting knowledge from the data you have so you can build a powerful, intuitive tool that can make predictions about future data that the computer has not previously seen. The machine is learning, not memorizing, and that’s what we did.” ![]()
Photo by Darren Baker
Researchers say they have developed the first computer machine-learning model to accurately predict which patients diagnosed with acute
myeloid leukemia (AML) will go into remission after treatment and which will relapse.
“It’s pretty straightforward to teach a computer to recognize AML, once you develop a robust algorithm, and, in previous work, we did it with almost 100% accuracy,” said study author Murat Dundar, PhD of Indiana University-Purdue University Indianapolis.
“What was challenging was to go beyond that work and teach the computer to accurately predict the direction of change in disease progression in AML patients, interpreting new data to predict the unknown: which new AML patients will go into remission and which will relapse.”
Dr Dundar and his colleagues described this work in IEEE Transactions on Biomedical Engineering.
The researchers said they modeled data from multiple flow cytometry samples to identify functionally distinct cell populations and their local realizations. Each sample was characterized by the proportions of recovered cell populations, which were used to predict the direction of change in disease progression for each AML patient.
“As the input, our computational system employs data from flow cytometry, a widely utilized technology that can rapidly provide detailed characteristics of single cells in samples such as blood or bone marrow,” explained study author Bartek Rajwa, PhD, of Purdue University in Lafayette, Indiana.
“Traditionally, the results of flow cytometry analyses are evaluated by highly trained human experts rather than by machine-learning algorithms. But computers are often better at extracting knowledge from complex data than humans are.”
The researchers used 200 diseased and non-diseased immunophenotypic panels for training and tested
the computational system with samples collected at multiple time points from 36 additional AML patients.
The system was able to predict remission with 100% accuracy (26 of 26 cases) and relapse with 90% accuracy (9 of 10 cases).
“Machine learning is not about modeling data,” Dr Dundar noted. “It’s about extracting knowledge from the data you have so you can build a powerful, intuitive tool that can make predictions about future data that the computer has not previously seen. The machine is learning, not memorizing, and that’s what we did.” ![]()
Genetic profiling can guide HSCT in MDS, team says

Genetic profiling can be used to determine which patients with myelodysplastic syndrome (MDS) are likely to benefit from allogeneic hematopoietic stem cell transplant (HSCT), according to research published in NEJM.
Targeted sequencing of 129 genes revealed mutations that, after adjustment for clinical variables, were associated with shorter survival and/or relapse after HSCT.
Patients with mutations in TP53, JAK2, and the RAS pathway tended to have worse outcomes after HSCT than patients without such mutations.
“Although donor stem cell transplantation is the only curative therapy for MDS, many patients die after transplantation, largely due to relapse of the disease or complications relating to the transplant itself,” said study author R. Coleman Lindsley, MD, PhD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
“As physicians, one of our major challenges is to be able to predict which patients are most likely to benefit from a transplant. Improving our ability to identify patients who are most likely to have a relapse or to experience life-threatening complications from a transplant could lead to better pre-transplant therapies and strategies for preventing relapse.”
Researchers have long known that specific genetic mutations are closely related to the course MDS takes. With this study, Dr Lindsley and his colleagues sought to discover whether mutations can be used to predict how patients will fare following allogeneic HSCT.
The team analyzed blood samples from 1514 MDS patients, performing targeted sequencing of 129 genes. The genes were selected based on their known or suspected involvement in the pathogenesis of myeloid cancers or bone marrow failure syndromes.
Dr Lindsley and his colleagues then evaluated the association between mutations and HSCT outcomes, including overall survival, relapse, and death without relapse.
After adjusting for significant clinical variables, the researchers found that having mutated TP53 was significantly associated with shorter survival and shorter time to relapse after HSCT (P<0.001 for both comparisons). This was true whether patients received standard conditioning or reduced-intensity conditioning.
In patients age 40 and older who did not have TP53 mutations, mutations in RAS pathway genes (P=0.004) or JAK2 (P=0.001) were significantly associated with shorter survival.
The shorter survival in patients with mutated RAS pathway genes was due to a higher risk of relapse, while the shorter survival in patients with JAK2 mutations was due to a higher risk of death without relapse.
In contrast to TP53 mutations, the adverse effect of RAS mutations on survival and risk of relapse was evident only in patients who received reduced-intensity conditioning (P<0.001). This suggests these patients may benefit from higher intensity conditioning regimens, the researchers said.
This study also yielded insights about the biology of MDS in specific groups of patients.
For example, the researchers found that 4% of MDS patients between the ages of 18 and 40 had mutations associated with Shwachman-Diamond syndrome (in the SBDS gene), but most of them had not previously been diagnosed with the syndrome.
In each case, the patients had acquired a TP53 mutation, suggesting not only how MDS develops in patients with Schwachman-Diamond syndrome but also what underlies their poor prognosis after HSCT.
The researchers also analyzed patients with therapy-related MDS. The team found that TP53 mutations and mutations in PPM1D, a gene that regulates TP53 function, were far more common in these patients than in those with primary MDS (15% and 3%, respectively, P<0.001).
“In deciding whether a stem cell transplant is appropriate for a patient with MDS, it’s always necessary to balance the potential benefit with the risk of complications,” Dr Lindsley noted.
“Our findings offer physicians a guide—based on the genetic profile of the disease and certain clinical factors—to identifying patients for whom a transplant is appropriate, and the intensity of treatment most likely to be effective.” ![]()
Genetic profiling can be used to determine which patients with myelodysplastic syndrome (MDS) are likely to benefit from allogeneic hematopoietic stem cell transplant (HSCT), according to research published in NEJM.
Targeted sequencing of 129 genes revealed mutations that, after adjustment for clinical variables, were associated with shorter survival and/or relapse after HSCT.
Patients with mutations in TP53, JAK2, and the RAS pathway tended to have worse outcomes after HSCT than patients without such mutations.
“Although donor stem cell transplantation is the only curative therapy for MDS, many patients die after transplantation, largely due to relapse of the disease or complications relating to the transplant itself,” said study author R. Coleman Lindsley, MD, PhD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
“As physicians, one of our major challenges is to be able to predict which patients are most likely to benefit from a transplant. Improving our ability to identify patients who are most likely to have a relapse or to experience life-threatening complications from a transplant could lead to better pre-transplant therapies and strategies for preventing relapse.”
Researchers have long known that specific genetic mutations are closely related to the course MDS takes. With this study, Dr Lindsley and his colleagues sought to discover whether mutations can be used to predict how patients will fare following allogeneic HSCT.
The team analyzed blood samples from 1514 MDS patients, performing targeted sequencing of 129 genes. The genes were selected based on their known or suspected involvement in the pathogenesis of myeloid cancers or bone marrow failure syndromes.
Dr Lindsley and his colleagues then evaluated the association between mutations and HSCT outcomes, including overall survival, relapse, and death without relapse.
After adjusting for significant clinical variables, the researchers found that having mutated TP53 was significantly associated with shorter survival and shorter time to relapse after HSCT (P<0.001 for both comparisons). This was true whether patients received standard conditioning or reduced-intensity conditioning.
In patients age 40 and older who did not have TP53 mutations, mutations in RAS pathway genes (P=0.004) or JAK2 (P=0.001) were significantly associated with shorter survival.
The shorter survival in patients with mutated RAS pathway genes was due to a higher risk of relapse, while the shorter survival in patients with JAK2 mutations was due to a higher risk of death without relapse.
In contrast to TP53 mutations, the adverse effect of RAS mutations on survival and risk of relapse was evident only in patients who received reduced-intensity conditioning (P<0.001). This suggests these patients may benefit from higher intensity conditioning regimens, the researchers said.
This study also yielded insights about the biology of MDS in specific groups of patients.
For example, the researchers found that 4% of MDS patients between the ages of 18 and 40 had mutations associated with Shwachman-Diamond syndrome (in the SBDS gene), but most of them had not previously been diagnosed with the syndrome.
In each case, the patients had acquired a TP53 mutation, suggesting not only how MDS develops in patients with Schwachman-Diamond syndrome but also what underlies their poor prognosis after HSCT.
The researchers also analyzed patients with therapy-related MDS. The team found that TP53 mutations and mutations in PPM1D, a gene that regulates TP53 function, were far more common in these patients than in those with primary MDS (15% and 3%, respectively, P<0.001).
“In deciding whether a stem cell transplant is appropriate for a patient with MDS, it’s always necessary to balance the potential benefit with the risk of complications,” Dr Lindsley noted.
“Our findings offer physicians a guide—based on the genetic profile of the disease and certain clinical factors—to identifying patients for whom a transplant is appropriate, and the intensity of treatment most likely to be effective.” ![]()
Genetic profiling can be used to determine which patients with myelodysplastic syndrome (MDS) are likely to benefit from allogeneic hematopoietic stem cell transplant (HSCT), according to research published in NEJM.
Targeted sequencing of 129 genes revealed mutations that, after adjustment for clinical variables, were associated with shorter survival and/or relapse after HSCT.
Patients with mutations in TP53, JAK2, and the RAS pathway tended to have worse outcomes after HSCT than patients without such mutations.
“Although donor stem cell transplantation is the only curative therapy for MDS, many patients die after transplantation, largely due to relapse of the disease or complications relating to the transplant itself,” said study author R. Coleman Lindsley, MD, PhD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
“As physicians, one of our major challenges is to be able to predict which patients are most likely to benefit from a transplant. Improving our ability to identify patients who are most likely to have a relapse or to experience life-threatening complications from a transplant could lead to better pre-transplant therapies and strategies for preventing relapse.”
Researchers have long known that specific genetic mutations are closely related to the course MDS takes. With this study, Dr Lindsley and his colleagues sought to discover whether mutations can be used to predict how patients will fare following allogeneic HSCT.
The team analyzed blood samples from 1514 MDS patients, performing targeted sequencing of 129 genes. The genes were selected based on their known or suspected involvement in the pathogenesis of myeloid cancers or bone marrow failure syndromes.
Dr Lindsley and his colleagues then evaluated the association between mutations and HSCT outcomes, including overall survival, relapse, and death without relapse.
After adjusting for significant clinical variables, the researchers found that having mutated TP53 was significantly associated with shorter survival and shorter time to relapse after HSCT (P<0.001 for both comparisons). This was true whether patients received standard conditioning or reduced-intensity conditioning.
In patients age 40 and older who did not have TP53 mutations, mutations in RAS pathway genes (P=0.004) or JAK2 (P=0.001) were significantly associated with shorter survival.
The shorter survival in patients with mutated RAS pathway genes was due to a higher risk of relapse, while the shorter survival in patients with JAK2 mutations was due to a higher risk of death without relapse.
In contrast to TP53 mutations, the adverse effect of RAS mutations on survival and risk of relapse was evident only in patients who received reduced-intensity conditioning (P<0.001). This suggests these patients may benefit from higher intensity conditioning regimens, the researchers said.
This study also yielded insights about the biology of MDS in specific groups of patients.
For example, the researchers found that 4% of MDS patients between the ages of 18 and 40 had mutations associated with Shwachman-Diamond syndrome (in the SBDS gene), but most of them had not previously been diagnosed with the syndrome.
In each case, the patients had acquired a TP53 mutation, suggesting not only how MDS develops in patients with Schwachman-Diamond syndrome but also what underlies their poor prognosis after HSCT.
The researchers also analyzed patients with therapy-related MDS. The team found that TP53 mutations and mutations in PPM1D, a gene that regulates TP53 function, were far more common in these patients than in those with primary MDS (15% and 3%, respectively, P<0.001).
“In deciding whether a stem cell transplant is appropriate for a patient with MDS, it’s always necessary to balance the potential benefit with the risk of complications,” Dr Lindsley noted.
“Our findings offer physicians a guide—based on the genetic profile of the disease and certain clinical factors—to identifying patients for whom a transplant is appropriate, and the intensity of treatment most likely to be effective.” ![]()
In active CLL with deletion 17p, consider trial enrollment
NEW YORK – Outside of clinical trials, therapy for early stage chronic lymphocytic leukemia in patients with deletion of the short arm of chromosome 17 (del[17]p) and/or mutation of the tumor suppressor gene TP53 requires the presence of active disease, according to Neil E. Kay, MD.
“Right now, we would propose that patients with del[17]p should have additional prognostic work-up. It’s very important to know if they are unmutated or mutated for the IgVH gene,” he said, adding that stimulated karyotype is also important to perform in those with del[17]p.
“The median overall survivorship in many phase II and phase III trials appears to be around 2 to 3 years,” he said.
Those with a chromosome 17 (del[17]p) and/or mutation of the tumor suppressor gene TP53 who receive chemoimmunotherapy very rarely achieve a complete response, or if they do they have a short duration of response, he added.
In treated patients, 17p deletion and p53 mutation are the most common abnormalities acquired during the course of the disease.
“Unfortunately there appears to be a selection pressure, and [in treated patients] the incidence of 17p and the p53 mutation has been reported up to 23%-44%. No one understands completely the biology of this, but it may be that subclones are present and expand, or that new mutations occur due to selection pressure of [chemoimmunotherapy] and the overgrowth of these subclones,” he said.
Importantly, not all patients with del[17]p or p53 mutation have bad outcomes; there are patients with indolent disease, he noted, adding that various criteria have been shown to help identify which patients are at risk for poor outcomes and to classify them according to risk. In general, lower-risk patients have mutated immunoglobulin heavy chain variable gene status, early stage disease, younger age, good performance status, and normal serum lactate dehydrogenase. These criteria could be used to identify low-risk patients who can be followed, he said.
Fluorescence in situ hybridization (FISH) evaluation of patients is a useful tool when there is no access to sequencing and other tests, Dr. Kay said, describing a recent multinational CLL Research Consortium study of nearly 1,600 patients (Br J Haematol. 2016 Apr;173[1]:105-13).
In that study, he and his colleagues found that patients with less than 50% 17p did not have such poor outcomes, but at 50%-plus they did much worse in terms of time to first treatment.
Based on the available data, Dr. Kay said that treatment is unnecessary in asymptomatic patients, except, perhaps, in high-risk patients identified using recently published risk models, for whom clinical trial enrollment may be considered.
“We do advocate having a discussion about allogeneic stem cell transplant since this may still be the only curative approach,” he added.
In patients with del[17]p and/or p53 mutation who have progressive disease, Dr. Kay said his take on the available data is that patients should first be categorized by age, then by whether they are fit or frail, and finally by whether or not they have del[17]p. Those under age 70 years without del[17]p and who have a mutated IgVH status should be considered for a clinical trial, and are also good candidates for chemoimmunotherapy. If they do have del[17]p or p53 mutation, consider clinical trial enrollment or treatment with ibrutinib, he said.
Fit patients in a complete response can be referred for transplant evaluation, but while the other treatments can be considered in frail patients or those aged 70 years or older, transplant is not advised, he added.
For relapsed or refractory patients, FISH testing should be performed or repeated, because such patients are at high risk of progression to develop del[17]p or mutation, he noted.
Those who are asymptomatic can be observed or enrolled in a clinical trial, and those who are symptomatic can be enrolled in a clinical trial or treated with various novel agents, including ibrutinib, idelalisib/rituximab, venetoclax, or combination therapies with methylpred–anti-CD20, or alemtuzumab with or without rituximab. Referral for transplant may be warranted in these patients if they are fit.
“Progressive CLL patients with 17p deletion/p53 mutations are much less likely to do well with chemoimmunotherapy, and novel inhibitors are effective, but we still need to enhance complete response rates and minimal residual disease-negative status for these high-risk patients,” he said.
Dr. Kay reported consulting for or receiving grant/research support from Acerta, Celgene, Gilead, Infinity, MorphoSys, Pharmacyclics, and Tolero.
NEW YORK – Outside of clinical trials, therapy for early stage chronic lymphocytic leukemia in patients with deletion of the short arm of chromosome 17 (del[17]p) and/or mutation of the tumor suppressor gene TP53 requires the presence of active disease, according to Neil E. Kay, MD.
“Right now, we would propose that patients with del[17]p should have additional prognostic work-up. It’s very important to know if they are unmutated or mutated for the IgVH gene,” he said, adding that stimulated karyotype is also important to perform in those with del[17]p.
“The median overall survivorship in many phase II and phase III trials appears to be around 2 to 3 years,” he said.
Those with a chromosome 17 (del[17]p) and/or mutation of the tumor suppressor gene TP53 who receive chemoimmunotherapy very rarely achieve a complete response, or if they do they have a short duration of response, he added.
In treated patients, 17p deletion and p53 mutation are the most common abnormalities acquired during the course of the disease.
“Unfortunately there appears to be a selection pressure, and [in treated patients] the incidence of 17p and the p53 mutation has been reported up to 23%-44%. No one understands completely the biology of this, but it may be that subclones are present and expand, or that new mutations occur due to selection pressure of [chemoimmunotherapy] and the overgrowth of these subclones,” he said.
Importantly, not all patients with del[17]p or p53 mutation have bad outcomes; there are patients with indolent disease, he noted, adding that various criteria have been shown to help identify which patients are at risk for poor outcomes and to classify them according to risk. In general, lower-risk patients have mutated immunoglobulin heavy chain variable gene status, early stage disease, younger age, good performance status, and normal serum lactate dehydrogenase. These criteria could be used to identify low-risk patients who can be followed, he said.
Fluorescence in situ hybridization (FISH) evaluation of patients is a useful tool when there is no access to sequencing and other tests, Dr. Kay said, describing a recent multinational CLL Research Consortium study of nearly 1,600 patients (Br J Haematol. 2016 Apr;173[1]:105-13).
In that study, he and his colleagues found that patients with less than 50% 17p did not have such poor outcomes, but at 50%-plus they did much worse in terms of time to first treatment.
Based on the available data, Dr. Kay said that treatment is unnecessary in asymptomatic patients, except, perhaps, in high-risk patients identified using recently published risk models, for whom clinical trial enrollment may be considered.
“We do advocate having a discussion about allogeneic stem cell transplant since this may still be the only curative approach,” he added.
In patients with del[17]p and/or p53 mutation who have progressive disease, Dr. Kay said his take on the available data is that patients should first be categorized by age, then by whether they are fit or frail, and finally by whether or not they have del[17]p. Those under age 70 years without del[17]p and who have a mutated IgVH status should be considered for a clinical trial, and are also good candidates for chemoimmunotherapy. If they do have del[17]p or p53 mutation, consider clinical trial enrollment or treatment with ibrutinib, he said.
Fit patients in a complete response can be referred for transplant evaluation, but while the other treatments can be considered in frail patients or those aged 70 years or older, transplant is not advised, he added.
For relapsed or refractory patients, FISH testing should be performed or repeated, because such patients are at high risk of progression to develop del[17]p or mutation, he noted.
Those who are asymptomatic can be observed or enrolled in a clinical trial, and those who are symptomatic can be enrolled in a clinical trial or treated with various novel agents, including ibrutinib, idelalisib/rituximab, venetoclax, or combination therapies with methylpred–anti-CD20, or alemtuzumab with or without rituximab. Referral for transplant may be warranted in these patients if they are fit.
“Progressive CLL patients with 17p deletion/p53 mutations are much less likely to do well with chemoimmunotherapy, and novel inhibitors are effective, but we still need to enhance complete response rates and minimal residual disease-negative status for these high-risk patients,” he said.
Dr. Kay reported consulting for or receiving grant/research support from Acerta, Celgene, Gilead, Infinity, MorphoSys, Pharmacyclics, and Tolero.
NEW YORK – Outside of clinical trials, therapy for early stage chronic lymphocytic leukemia in patients with deletion of the short arm of chromosome 17 (del[17]p) and/or mutation of the tumor suppressor gene TP53 requires the presence of active disease, according to Neil E. Kay, MD.
“Right now, we would propose that patients with del[17]p should have additional prognostic work-up. It’s very important to know if they are unmutated or mutated for the IgVH gene,” he said, adding that stimulated karyotype is also important to perform in those with del[17]p.
“The median overall survivorship in many phase II and phase III trials appears to be around 2 to 3 years,” he said.
Those with a chromosome 17 (del[17]p) and/or mutation of the tumor suppressor gene TP53 who receive chemoimmunotherapy very rarely achieve a complete response, or if they do they have a short duration of response, he added.
In treated patients, 17p deletion and p53 mutation are the most common abnormalities acquired during the course of the disease.
“Unfortunately there appears to be a selection pressure, and [in treated patients] the incidence of 17p and the p53 mutation has been reported up to 23%-44%. No one understands completely the biology of this, but it may be that subclones are present and expand, or that new mutations occur due to selection pressure of [chemoimmunotherapy] and the overgrowth of these subclones,” he said.
Importantly, not all patients with del[17]p or p53 mutation have bad outcomes; there are patients with indolent disease, he noted, adding that various criteria have been shown to help identify which patients are at risk for poor outcomes and to classify them according to risk. In general, lower-risk patients have mutated immunoglobulin heavy chain variable gene status, early stage disease, younger age, good performance status, and normal serum lactate dehydrogenase. These criteria could be used to identify low-risk patients who can be followed, he said.
Fluorescence in situ hybridization (FISH) evaluation of patients is a useful tool when there is no access to sequencing and other tests, Dr. Kay said, describing a recent multinational CLL Research Consortium study of nearly 1,600 patients (Br J Haematol. 2016 Apr;173[1]:105-13).
In that study, he and his colleagues found that patients with less than 50% 17p did not have such poor outcomes, but at 50%-plus they did much worse in terms of time to first treatment.
Based on the available data, Dr. Kay said that treatment is unnecessary in asymptomatic patients, except, perhaps, in high-risk patients identified using recently published risk models, for whom clinical trial enrollment may be considered.
“We do advocate having a discussion about allogeneic stem cell transplant since this may still be the only curative approach,” he added.
In patients with del[17]p and/or p53 mutation who have progressive disease, Dr. Kay said his take on the available data is that patients should first be categorized by age, then by whether they are fit or frail, and finally by whether or not they have del[17]p. Those under age 70 years without del[17]p and who have a mutated IgVH status should be considered for a clinical trial, and are also good candidates for chemoimmunotherapy. If they do have del[17]p or p53 mutation, consider clinical trial enrollment or treatment with ibrutinib, he said.
Fit patients in a complete response can be referred for transplant evaluation, but while the other treatments can be considered in frail patients or those aged 70 years or older, transplant is not advised, he added.
For relapsed or refractory patients, FISH testing should be performed or repeated, because such patients are at high risk of progression to develop del[17]p or mutation, he noted.
Those who are asymptomatic can be observed or enrolled in a clinical trial, and those who are symptomatic can be enrolled in a clinical trial or treated with various novel agents, including ibrutinib, idelalisib/rituximab, venetoclax, or combination therapies with methylpred–anti-CD20, or alemtuzumab with or without rituximab. Referral for transplant may be warranted in these patients if they are fit.
“Progressive CLL patients with 17p deletion/p53 mutations are much less likely to do well with chemoimmunotherapy, and novel inhibitors are effective, but we still need to enhance complete response rates and minimal residual disease-negative status for these high-risk patients,” he said.
Dr. Kay reported consulting for or receiving grant/research support from Acerta, Celgene, Gilead, Infinity, MorphoSys, Pharmacyclics, and Tolero.
EXPERT ANALYSIS FROM LYMPHOMA & MYELOMA
Investigators report new risk loci for CLL

Investigators say they have identified 9 new risk loci for chronic lymphocytic leukemia (CLL).
The team says the research, published in Nature Communications, provides additional evidence for genetic susceptibility to CLL and sheds new light on the biological basis of CLL development.
They also believe their findings could aid the development of new drugs for CLL or help in selecting existing therapies for CLL
patients.
“We knew people were more likely to develop chronic lymphocytic leukemia if someone in their family had suffered from the disease, but our new research takes a big step towards explaining the underlying genetics,” said study author Richard Houlston, MD, PhD, of The Institute of Cancer Research in London, UK.
“CLL is essentially a disease of the immune system, and it’s fascinating that so many of the new genetic variants we have uncovered seem to directly affect the behavior of white blood cells and their ability to fight disease. Understanding the genetics of CLL can point us towards new treatments for the disease and help us to use existing targeted drugs more effectively.”
For this study, Dr Houlston and his colleagues analyzed data from 8 studies involving a total of 6200 CLL patients and 17,598 controls.
From this, the team identified 9 CLL risk loci:
- 1p36.11 (rs34676223, P=5.04 × 10−13)
- 1q42.13 (rs41271473, P=1.06 × 10−10)
- 4q24 (rs71597109, P=1.37 × 10−10)
- 4q35.1 (rs57214277, P=3.69 × 10−8)
- 6p21.31 (rs3800461, P=1.97 × 10−8)
- 11q23.2 (rs61904987, P=2.64 × 10−11)
- 18q21.1 (rs1036935, P=3.27 × 10−8)
- 19p13.3 (rs7254272, P=4.67 × 10−8)
- 22q13.33 (rs140522, P=2.70 × 10−9).
The investigators noted that the 4q24 association marked by rs71597109 maps to intron 1 of the gene encoding BANK1 (B-cell scaffold protein with ankyrin repeats 1). BANK1 is only ever activated in B cells and is linked to the autoimmune disease lupus.
The team also pointed out that the 19p13.3 association marked by rs7254272 maps 2.5 kb 5′ to ZBTB7A (zinc finger and BTB domain-containing protein 7a), which is a master regulator of B versus T lymphoid fate. So errors in ZBTB7A could lead to too many B cells in the bloodstream and bone marrow.
And rs140522 maps to 22q13.33, which has been linked to the development of multiple sclerosis. The investigators noted that this region of linkage disequilibrium contains 4 genes. One of them, NCAPH2 (non-SMC condensin II complex subunit H2), is differentially expressed in CLL and normal B cells.
“This fascinating study makes a link between genetic variants in the immune system and the development of leukemia and implicates regions of DNA which are also involved in autoimmune diseases,” said Paul Workman, PhD, chief executive and president of The Institute of Cancer Research, who was not involved in this research.
“The findings could point us towards new ways of treating leukemia or better ways of using existing treatments—potentially including immunotherapies.” ![]()
Investigators say they have identified 9 new risk loci for chronic lymphocytic leukemia (CLL).
The team says the research, published in Nature Communications, provides additional evidence for genetic susceptibility to CLL and sheds new light on the biological basis of CLL development.
They also believe their findings could aid the development of new drugs for CLL or help in selecting existing therapies for CLL
patients.
“We knew people were more likely to develop chronic lymphocytic leukemia if someone in their family had suffered from the disease, but our new research takes a big step towards explaining the underlying genetics,” said study author Richard Houlston, MD, PhD, of The Institute of Cancer Research in London, UK.
“CLL is essentially a disease of the immune system, and it’s fascinating that so many of the new genetic variants we have uncovered seem to directly affect the behavior of white blood cells and their ability to fight disease. Understanding the genetics of CLL can point us towards new treatments for the disease and help us to use existing targeted drugs more effectively.”
For this study, Dr Houlston and his colleagues analyzed data from 8 studies involving a total of 6200 CLL patients and 17,598 controls.
From this, the team identified 9 CLL risk loci:
- 1p36.11 (rs34676223, P=5.04 × 10−13)
- 1q42.13 (rs41271473, P=1.06 × 10−10)
- 4q24 (rs71597109, P=1.37 × 10−10)
- 4q35.1 (rs57214277, P=3.69 × 10−8)
- 6p21.31 (rs3800461, P=1.97 × 10−8)
- 11q23.2 (rs61904987, P=2.64 × 10−11)
- 18q21.1 (rs1036935, P=3.27 × 10−8)
- 19p13.3 (rs7254272, P=4.67 × 10−8)
- 22q13.33 (rs140522, P=2.70 × 10−9).
The investigators noted that the 4q24 association marked by rs71597109 maps to intron 1 of the gene encoding BANK1 (B-cell scaffold protein with ankyrin repeats 1). BANK1 is only ever activated in B cells and is linked to the autoimmune disease lupus.
The team also pointed out that the 19p13.3 association marked by rs7254272 maps 2.5 kb 5′ to ZBTB7A (zinc finger and BTB domain-containing protein 7a), which is a master regulator of B versus T lymphoid fate. So errors in ZBTB7A could lead to too many B cells in the bloodstream and bone marrow.
And rs140522 maps to 22q13.33, which has been linked to the development of multiple sclerosis. The investigators noted that this region of linkage disequilibrium contains 4 genes. One of them, NCAPH2 (non-SMC condensin II complex subunit H2), is differentially expressed in CLL and normal B cells.
“This fascinating study makes a link between genetic variants in the immune system and the development of leukemia and implicates regions of DNA which are also involved in autoimmune diseases,” said Paul Workman, PhD, chief executive and president of The Institute of Cancer Research, who was not involved in this research.
“The findings could point us towards new ways of treating leukemia or better ways of using existing treatments—potentially including immunotherapies.” ![]()
Investigators say they have identified 9 new risk loci for chronic lymphocytic leukemia (CLL).
The team says the research, published in Nature Communications, provides additional evidence for genetic susceptibility to CLL and sheds new light on the biological basis of CLL development.
They also believe their findings could aid the development of new drugs for CLL or help in selecting existing therapies for CLL
patients.
“We knew people were more likely to develop chronic lymphocytic leukemia if someone in their family had suffered from the disease, but our new research takes a big step towards explaining the underlying genetics,” said study author Richard Houlston, MD, PhD, of The Institute of Cancer Research in London, UK.
“CLL is essentially a disease of the immune system, and it’s fascinating that so many of the new genetic variants we have uncovered seem to directly affect the behavior of white blood cells and their ability to fight disease. Understanding the genetics of CLL can point us towards new treatments for the disease and help us to use existing targeted drugs more effectively.”
For this study, Dr Houlston and his colleagues analyzed data from 8 studies involving a total of 6200 CLL patients and 17,598 controls.
From this, the team identified 9 CLL risk loci:
- 1p36.11 (rs34676223, P=5.04 × 10−13)
- 1q42.13 (rs41271473, P=1.06 × 10−10)
- 4q24 (rs71597109, P=1.37 × 10−10)
- 4q35.1 (rs57214277, P=3.69 × 10−8)
- 6p21.31 (rs3800461, P=1.97 × 10−8)
- 11q23.2 (rs61904987, P=2.64 × 10−11)
- 18q21.1 (rs1036935, P=3.27 × 10−8)
- 19p13.3 (rs7254272, P=4.67 × 10−8)
- 22q13.33 (rs140522, P=2.70 × 10−9).
The investigators noted that the 4q24 association marked by rs71597109 maps to intron 1 of the gene encoding BANK1 (B-cell scaffold protein with ankyrin repeats 1). BANK1 is only ever activated in B cells and is linked to the autoimmune disease lupus.
The team also pointed out that the 19p13.3 association marked by rs7254272 maps 2.5 kb 5′ to ZBTB7A (zinc finger and BTB domain-containing protein 7a), which is a master regulator of B versus T lymphoid fate. So errors in ZBTB7A could lead to too many B cells in the bloodstream and bone marrow.
And rs140522 maps to 22q13.33, which has been linked to the development of multiple sclerosis. The investigators noted that this region of linkage disequilibrium contains 4 genes. One of them, NCAPH2 (non-SMC condensin II complex subunit H2), is differentially expressed in CLL and normal B cells.
“This fascinating study makes a link between genetic variants in the immune system and the development of leukemia and implicates regions of DNA which are also involved in autoimmune diseases,” said Paul Workman, PhD, chief executive and president of The Institute of Cancer Research, who was not involved in this research.
“The findings could point us towards new ways of treating leukemia or better ways of using existing treatments—potentially including immunotherapies.” ![]()
Why CLL may go chemo free
NEW YORK – The approach to treating chronic lymphocytic leukemia (CLL) is evolving, and while chemoimmunotherapy remains a reasonable initial option in some cases, a “chemo-free” approach is also a very real possibility, according to Bruce D. Cheson, MD.
A number of studies showing survival benefits with targeted therapies vs. chemoimmunotherapy (CIT) regimens have been completed, including studies that look specifically at outcomes by mutation status and other factors. One example – a likely game changer – is the ALLIANCE trial, a randomized phase III study of bendamustine plus rituximab vs. ibrutinib plus rituximab vs. ibrutinib alone in untreated CLL patients aged 65 years or older, Dr. Cheson of Georgetown University Hospital, Washington, said at an international congress on hematologic malignancies.
“I think [the ALLIANCE] trial has the possibility of totally changing how we treat patients with CLL, even though it was done in older patients,” he said, noting that the study is completed, but final results are pending adequate follow-up.
Based on the available data, he suggests a treatment paradigm for untreated CLL patients who require therapy that begins with consideration of patient age, comorbidities, functional status, and fluorescence in situ hybridization (FISH).
While clinical trial enrollment is preferable, those who are CIT eligible based on age and comorbidities can be treated with bendamustine/rituximab (BR), fludarabine/cyclophosphamide/rituximab (FCR), or ibrutinib.
“I think BR and FCR are both reasonable options, although the latter primarily for young, IGHV-mutated patients, and certainly ibrutinib remains an option for this patient population,” he said.
For those not eligible for CIT, options include ibrutinib and chlorambucil/obinutuzumab. For those with deletion 17p, ibrutinib is the standard for frontline therapy. And for those who are frail, ibrutinib is a good option.
“Some might use an anti-CD20, but as a single agent, it’s not what I would prefer,” Dr. Cheson said, explaining that response rates with such agents are low and tend to lack durability.
CIT remains a reasonable initial option for those patients who are mutated, but with the prolonged progression-free survival seen with ibrutinib in several trials, he predicted that will change over time.
“The role of targeted approaches is a subject of discussion. It takes the most time in my clinic of any discussion I have. [Patients ask] ‘Should I get ibrutinib? Should I get chemoimmunotherapy?’ ” he said. “One needs to take into account patient age and comorbidities, the fact that with CIT you are six [treatments] and done vs. indefinite therapy [with ibrutinib]. There is cost and there is compliance that one needs to consider.”
As for relapsed/refractory CLL, the role of CIT is particularly diminished in the wake of trials such as HELIOS and RESONATE, showing survival benefits with ibrutinib, others showing survival benefits with rituximab/idelalisib (R-idelalisib), and trials showing better results with venetoclax non-CIT regimens than would be expected with CIT regimens. As with treatment-naive CLL patients, age, comorbidities, functional status, and FISH should be considered in those with previously treated CLL who require therapy, and if clinical trial enrollment is not possible, treatment options depend on certain patient characteristics.
“For patients who had a long first response to chemoimmunotherapy, I would still use ibrutinib, and in select patients, R-idelalisib,” he said.
If they had a long response with BR, or FCR, retreatment with those can be considered as well, he noted, adding, “But I don’t see the point when the results with kinase inhibitors are at least as good as, if not better than one would expect with CIT in this context.”
In patients who had short first response, ibrutinib and R-idelalisib are the best options. For those with deletion 17p, the best options are ibrutinib or venetoclax, and possibly R-idelalisib. For frail patients, options include ibrutinib, R-idelalisib, or anti-CD20, although, as with untreated patients, the latter is his least favorite option because of the increased risk of toxicity in this population, he said.
“I think the role of chemoimmunotherapy in CLL is vanishing, and a chemo-free world for CLL patients is a reality,” he said.
Dr. Cheson reported consulting for Acerta, Celgene Pharmacyclics, and Roche-Genentech.
NEW YORK – The approach to treating chronic lymphocytic leukemia (CLL) is evolving, and while chemoimmunotherapy remains a reasonable initial option in some cases, a “chemo-free” approach is also a very real possibility, according to Bruce D. Cheson, MD.
A number of studies showing survival benefits with targeted therapies vs. chemoimmunotherapy (CIT) regimens have been completed, including studies that look specifically at outcomes by mutation status and other factors. One example – a likely game changer – is the ALLIANCE trial, a randomized phase III study of bendamustine plus rituximab vs. ibrutinib plus rituximab vs. ibrutinib alone in untreated CLL patients aged 65 years or older, Dr. Cheson of Georgetown University Hospital, Washington, said at an international congress on hematologic malignancies.
“I think [the ALLIANCE] trial has the possibility of totally changing how we treat patients with CLL, even though it was done in older patients,” he said, noting that the study is completed, but final results are pending adequate follow-up.
Based on the available data, he suggests a treatment paradigm for untreated CLL patients who require therapy that begins with consideration of patient age, comorbidities, functional status, and fluorescence in situ hybridization (FISH).
While clinical trial enrollment is preferable, those who are CIT eligible based on age and comorbidities can be treated with bendamustine/rituximab (BR), fludarabine/cyclophosphamide/rituximab (FCR), or ibrutinib.
“I think BR and FCR are both reasonable options, although the latter primarily for young, IGHV-mutated patients, and certainly ibrutinib remains an option for this patient population,” he said.
For those not eligible for CIT, options include ibrutinib and chlorambucil/obinutuzumab. For those with deletion 17p, ibrutinib is the standard for frontline therapy. And for those who are frail, ibrutinib is a good option.
“Some might use an anti-CD20, but as a single agent, it’s not what I would prefer,” Dr. Cheson said, explaining that response rates with such agents are low and tend to lack durability.
CIT remains a reasonable initial option for those patients who are mutated, but with the prolonged progression-free survival seen with ibrutinib in several trials, he predicted that will change over time.
“The role of targeted approaches is a subject of discussion. It takes the most time in my clinic of any discussion I have. [Patients ask] ‘Should I get ibrutinib? Should I get chemoimmunotherapy?’ ” he said. “One needs to take into account patient age and comorbidities, the fact that with CIT you are six [treatments] and done vs. indefinite therapy [with ibrutinib]. There is cost and there is compliance that one needs to consider.”
As for relapsed/refractory CLL, the role of CIT is particularly diminished in the wake of trials such as HELIOS and RESONATE, showing survival benefits with ibrutinib, others showing survival benefits with rituximab/idelalisib (R-idelalisib), and trials showing better results with venetoclax non-CIT regimens than would be expected with CIT regimens. As with treatment-naive CLL patients, age, comorbidities, functional status, and FISH should be considered in those with previously treated CLL who require therapy, and if clinical trial enrollment is not possible, treatment options depend on certain patient characteristics.
“For patients who had a long first response to chemoimmunotherapy, I would still use ibrutinib, and in select patients, R-idelalisib,” he said.
If they had a long response with BR, or FCR, retreatment with those can be considered as well, he noted, adding, “But I don’t see the point when the results with kinase inhibitors are at least as good as, if not better than one would expect with CIT in this context.”
In patients who had short first response, ibrutinib and R-idelalisib are the best options. For those with deletion 17p, the best options are ibrutinib or venetoclax, and possibly R-idelalisib. For frail patients, options include ibrutinib, R-idelalisib, or anti-CD20, although, as with untreated patients, the latter is his least favorite option because of the increased risk of toxicity in this population, he said.
“I think the role of chemoimmunotherapy in CLL is vanishing, and a chemo-free world for CLL patients is a reality,” he said.
Dr. Cheson reported consulting for Acerta, Celgene Pharmacyclics, and Roche-Genentech.
NEW YORK – The approach to treating chronic lymphocytic leukemia (CLL) is evolving, and while chemoimmunotherapy remains a reasonable initial option in some cases, a “chemo-free” approach is also a very real possibility, according to Bruce D. Cheson, MD.
A number of studies showing survival benefits with targeted therapies vs. chemoimmunotherapy (CIT) regimens have been completed, including studies that look specifically at outcomes by mutation status and other factors. One example – a likely game changer – is the ALLIANCE trial, a randomized phase III study of bendamustine plus rituximab vs. ibrutinib plus rituximab vs. ibrutinib alone in untreated CLL patients aged 65 years or older, Dr. Cheson of Georgetown University Hospital, Washington, said at an international congress on hematologic malignancies.
“I think [the ALLIANCE] trial has the possibility of totally changing how we treat patients with CLL, even though it was done in older patients,” he said, noting that the study is completed, but final results are pending adequate follow-up.
Based on the available data, he suggests a treatment paradigm for untreated CLL patients who require therapy that begins with consideration of patient age, comorbidities, functional status, and fluorescence in situ hybridization (FISH).
While clinical trial enrollment is preferable, those who are CIT eligible based on age and comorbidities can be treated with bendamustine/rituximab (BR), fludarabine/cyclophosphamide/rituximab (FCR), or ibrutinib.
“I think BR and FCR are both reasonable options, although the latter primarily for young, IGHV-mutated patients, and certainly ibrutinib remains an option for this patient population,” he said.
For those not eligible for CIT, options include ibrutinib and chlorambucil/obinutuzumab. For those with deletion 17p, ibrutinib is the standard for frontline therapy. And for those who are frail, ibrutinib is a good option.
“Some might use an anti-CD20, but as a single agent, it’s not what I would prefer,” Dr. Cheson said, explaining that response rates with such agents are low and tend to lack durability.
CIT remains a reasonable initial option for those patients who are mutated, but with the prolonged progression-free survival seen with ibrutinib in several trials, he predicted that will change over time.
“The role of targeted approaches is a subject of discussion. It takes the most time in my clinic of any discussion I have. [Patients ask] ‘Should I get ibrutinib? Should I get chemoimmunotherapy?’ ” he said. “One needs to take into account patient age and comorbidities, the fact that with CIT you are six [treatments] and done vs. indefinite therapy [with ibrutinib]. There is cost and there is compliance that one needs to consider.”
As for relapsed/refractory CLL, the role of CIT is particularly diminished in the wake of trials such as HELIOS and RESONATE, showing survival benefits with ibrutinib, others showing survival benefits with rituximab/idelalisib (R-idelalisib), and trials showing better results with venetoclax non-CIT regimens than would be expected with CIT regimens. As with treatment-naive CLL patients, age, comorbidities, functional status, and FISH should be considered in those with previously treated CLL who require therapy, and if clinical trial enrollment is not possible, treatment options depend on certain patient characteristics.
“For patients who had a long first response to chemoimmunotherapy, I would still use ibrutinib, and in select patients, R-idelalisib,” he said.
If they had a long response with BR, or FCR, retreatment with those can be considered as well, he noted, adding, “But I don’t see the point when the results with kinase inhibitors are at least as good as, if not better than one would expect with CIT in this context.”
In patients who had short first response, ibrutinib and R-idelalisib are the best options. For those with deletion 17p, the best options are ibrutinib or venetoclax, and possibly R-idelalisib. For frail patients, options include ibrutinib, R-idelalisib, or anti-CD20, although, as with untreated patients, the latter is his least favorite option because of the increased risk of toxicity in this population, he said.
“I think the role of chemoimmunotherapy in CLL is vanishing, and a chemo-free world for CLL patients is a reality,” he said.
Dr. Cheson reported consulting for Acerta, Celgene Pharmacyclics, and Roche-Genentech.
MDS gene mutations predict response to HSCT
Genetic mutations in blood samples may predict outcomes and guide treatment for patients of all ages who have myelodysplastic syndrome and are undergoing hematopoietic stem-cell transplantation, according to a report published online Feb. 8 in the New England Journal of Medicine.
Allogeneic hematopoietic stem-cell transplantation is the only potentially curative therapy currently available for myelodysplastic syndrome (MDS), but mortality due to relapse and to transplant-related complications is high. “Predicting which patients are most likely to benefit from transplantation is thus a central challenge,” and identifying patients most likely to relapse could help clinicians refine conditioning regimens and relapse-prevention strategies, said R. Coleman Lindsley, MD, PhD, of the division of hematological malignancies, Dana-Farber Cancer Institute, Boston, and his associates.
The analyses included targeted sequencing of 129 genes known or suspected to be involved in the pathogenesis of myeloid cancers or syndromes related to bone marrow failure. Approximately 80% of the study participants were found to have at least one such driver mutation, with a median of two mutations per patient.
Mutations in the TP53 gene turned out to be the single most powerful predictor of survival after transplantation, independent of factors such as patient age, performance status, and hematologic variables. Moreover, intensive (myeloablative) conditioning regimens did not attenuate this effect, “a finding that is consistent with clinical and experimental evidence showing TP53 mutation-mediated chemoresistance,” Dr. Lindsley and his associates said.
“Our data suggest that escalating the intensity of the conditioning regimen in order to improve outcomes in patients with TP53-mutated MDS will not be successful. ... These patients, who have an exceptionally high risk of relapse-related death after transplantation, should be considered for investigative approaches to conditioning or new relapse-prevention strategies after transplantation,” they added.
Among patients over age 40, mutations in the RAS pathway were associated with a significantly elevated risk of early relapse – an outcome that might be ameliorated by more intensive conditioning. “RAS-pathway mutations may thus reflect the presence of low-volume but biologically transformed disease that, without adequate cytoreduction before transplantation, outpaces the development of effective graft-versus-leukemia activity,” the investigators said.
However, this association between RAS mutations and relapse was not seen in patients younger than age 40 years, they noted.
Conversely, JAK2 mutations were associated with a higher rate of death without relapse but not a higher rate of relapse. And this association was not affected by conditioning intensity. Although the mechanism of such an effect is not yet known, early death without relapse may be driven by factors that are susceptible to targeting by JAK2 inhibitors. In addition, minimizing treatment toxicity should be the focus of treatment in patients who carry JAK2 mutations, since their poor survival rate is driven by deaths unrelated to relapse, Dr. Lindsley and his associates said.
Mutations in the PPM1D gene, especially when accompanied by TP53 mutations, were strongly associated with previous exposure to leukemogenic therapies. “PPM1D encodes a serine-threonine protein phosphatase that regulates the cellular response to environmental stress, in part by means of inhibition of TP53 activity, which suggests that TP53 and PPM1D mutations represent convergent mechanisms of clonal survival in the context of leukemogenic exposures,” the investigators said.
“Our results... provide strong genetic evidence of the role of PPM1D mutations in the pathogenesis of therapy-related myelodysplastic syndromes.”
Mutations in the SBDS gene, which has been linked to Shwachman-Diamond syndrome, were “unexpectedly common” in young-adult patients and were associated with a poor prognosis. (Shwachman-Diamond syndrome is a rare congenital syndrome of bone-marrow failure.) This finding suggests that early stem-cell transplantation should be considered for patients who have this disorder, since transplantation after full-blown MDS develops “may not offer long-term benefit.”
Genetic mutations in blood samples may predict outcomes and guide treatment for patients of all ages who have myelodysplastic syndrome and are undergoing hematopoietic stem-cell transplantation, according to a report published online Feb. 8 in the New England Journal of Medicine.
Allogeneic hematopoietic stem-cell transplantation is the only potentially curative therapy currently available for myelodysplastic syndrome (MDS), but mortality due to relapse and to transplant-related complications is high. “Predicting which patients are most likely to benefit from transplantation is thus a central challenge,” and identifying patients most likely to relapse could help clinicians refine conditioning regimens and relapse-prevention strategies, said R. Coleman Lindsley, MD, PhD, of the division of hematological malignancies, Dana-Farber Cancer Institute, Boston, and his associates.
The analyses included targeted sequencing of 129 genes known or suspected to be involved in the pathogenesis of myeloid cancers or syndromes related to bone marrow failure. Approximately 80% of the study participants were found to have at least one such driver mutation, with a median of two mutations per patient.
Mutations in the TP53 gene turned out to be the single most powerful predictor of survival after transplantation, independent of factors such as patient age, performance status, and hematologic variables. Moreover, intensive (myeloablative) conditioning regimens did not attenuate this effect, “a finding that is consistent with clinical and experimental evidence showing TP53 mutation-mediated chemoresistance,” Dr. Lindsley and his associates said.
“Our data suggest that escalating the intensity of the conditioning regimen in order to improve outcomes in patients with TP53-mutated MDS will not be successful. ... These patients, who have an exceptionally high risk of relapse-related death after transplantation, should be considered for investigative approaches to conditioning or new relapse-prevention strategies after transplantation,” they added.
Among patients over age 40, mutations in the RAS pathway were associated with a significantly elevated risk of early relapse – an outcome that might be ameliorated by more intensive conditioning. “RAS-pathway mutations may thus reflect the presence of low-volume but biologically transformed disease that, without adequate cytoreduction before transplantation, outpaces the development of effective graft-versus-leukemia activity,” the investigators said.
However, this association between RAS mutations and relapse was not seen in patients younger than age 40 years, they noted.
Conversely, JAK2 mutations were associated with a higher rate of death without relapse but not a higher rate of relapse. And this association was not affected by conditioning intensity. Although the mechanism of such an effect is not yet known, early death without relapse may be driven by factors that are susceptible to targeting by JAK2 inhibitors. In addition, minimizing treatment toxicity should be the focus of treatment in patients who carry JAK2 mutations, since their poor survival rate is driven by deaths unrelated to relapse, Dr. Lindsley and his associates said.
Mutations in the PPM1D gene, especially when accompanied by TP53 mutations, were strongly associated with previous exposure to leukemogenic therapies. “PPM1D encodes a serine-threonine protein phosphatase that regulates the cellular response to environmental stress, in part by means of inhibition of TP53 activity, which suggests that TP53 and PPM1D mutations represent convergent mechanisms of clonal survival in the context of leukemogenic exposures,” the investigators said.
“Our results... provide strong genetic evidence of the role of PPM1D mutations in the pathogenesis of therapy-related myelodysplastic syndromes.”
Mutations in the SBDS gene, which has been linked to Shwachman-Diamond syndrome, were “unexpectedly common” in young-adult patients and were associated with a poor prognosis. (Shwachman-Diamond syndrome is a rare congenital syndrome of bone-marrow failure.) This finding suggests that early stem-cell transplantation should be considered for patients who have this disorder, since transplantation after full-blown MDS develops “may not offer long-term benefit.”
Genetic mutations in blood samples may predict outcomes and guide treatment for patients of all ages who have myelodysplastic syndrome and are undergoing hematopoietic stem-cell transplantation, according to a report published online Feb. 8 in the New England Journal of Medicine.
Allogeneic hematopoietic stem-cell transplantation is the only potentially curative therapy currently available for myelodysplastic syndrome (MDS), but mortality due to relapse and to transplant-related complications is high. “Predicting which patients are most likely to benefit from transplantation is thus a central challenge,” and identifying patients most likely to relapse could help clinicians refine conditioning regimens and relapse-prevention strategies, said R. Coleman Lindsley, MD, PhD, of the division of hematological malignancies, Dana-Farber Cancer Institute, Boston, and his associates.
The analyses included targeted sequencing of 129 genes known or suspected to be involved in the pathogenesis of myeloid cancers or syndromes related to bone marrow failure. Approximately 80% of the study participants were found to have at least one such driver mutation, with a median of two mutations per patient.
Mutations in the TP53 gene turned out to be the single most powerful predictor of survival after transplantation, independent of factors such as patient age, performance status, and hematologic variables. Moreover, intensive (myeloablative) conditioning regimens did not attenuate this effect, “a finding that is consistent with clinical and experimental evidence showing TP53 mutation-mediated chemoresistance,” Dr. Lindsley and his associates said.
“Our data suggest that escalating the intensity of the conditioning regimen in order to improve outcomes in patients with TP53-mutated MDS will not be successful. ... These patients, who have an exceptionally high risk of relapse-related death after transplantation, should be considered for investigative approaches to conditioning or new relapse-prevention strategies after transplantation,” they added.
Among patients over age 40, mutations in the RAS pathway were associated with a significantly elevated risk of early relapse – an outcome that might be ameliorated by more intensive conditioning. “RAS-pathway mutations may thus reflect the presence of low-volume but biologically transformed disease that, without adequate cytoreduction before transplantation, outpaces the development of effective graft-versus-leukemia activity,” the investigators said.
However, this association between RAS mutations and relapse was not seen in patients younger than age 40 years, they noted.
Conversely, JAK2 mutations were associated with a higher rate of death without relapse but not a higher rate of relapse. And this association was not affected by conditioning intensity. Although the mechanism of such an effect is not yet known, early death without relapse may be driven by factors that are susceptible to targeting by JAK2 inhibitors. In addition, minimizing treatment toxicity should be the focus of treatment in patients who carry JAK2 mutations, since their poor survival rate is driven by deaths unrelated to relapse, Dr. Lindsley and his associates said.
Mutations in the PPM1D gene, especially when accompanied by TP53 mutations, were strongly associated with previous exposure to leukemogenic therapies. “PPM1D encodes a serine-threonine protein phosphatase that regulates the cellular response to environmental stress, in part by means of inhibition of TP53 activity, which suggests that TP53 and PPM1D mutations represent convergent mechanisms of clonal survival in the context of leukemogenic exposures,” the investigators said.
“Our results... provide strong genetic evidence of the role of PPM1D mutations in the pathogenesis of therapy-related myelodysplastic syndromes.”
Mutations in the SBDS gene, which has been linked to Shwachman-Diamond syndrome, were “unexpectedly common” in young-adult patients and were associated with a poor prognosis. (Shwachman-Diamond syndrome is a rare congenital syndrome of bone-marrow failure.) This finding suggests that early stem-cell transplantation should be considered for patients who have this disorder, since transplantation after full-blown MDS develops “may not offer long-term benefit.”
Key clinical point: Genetic mutations in blood samples may predict outcomes and guide treatment for patients of all ages who have myelodysplastic syndrome and are undergoing hematopoietic stem-cell transplantation.
Key numerical finding: Approximately 80% of the study participants were found to have at least one driver mutation, with a median of two such mutations per patient.
Data source: Targeted mutational analyses of banked blood samples from 1,514 patients treated at 130 transplantation centers in the U.S. and Germany.
Disclosures: This study was supported by the Edward P. Evans Foundation, the Harvard Catalyst Program, the National Marrow Donor Program, the National Institutes of Health, and the Leukemia and Lymphoma Society. Dr. Lindsley reported ties to Takeda, and one of his associates reported ties to Celgene, Genoptix, and H3 Biomedicine.