User login
Team targets fructose to treat AML

Photo from the University
of Hawaii Cancer Center
Researchers say they have discovered a novel treatment strategy for acute myeloid leukemia (AML)—inhibiting fructose utilization.
The team discovered that AML cells are prone to using fructose for energy, and increased fructose utilization predicts poor treatment outcomes in AML patients.
The researchers also found that 2,5-anhydro-D-mannitol (2,5-AM) could inhibit fructose use in AML cells and therefore hinder their growth.
Wei Jia, PhD, of the University of Hawaii Cancer Center in Honolulu, and his colleagues reported these findings in Cancer Cell.
The researchers said their findings highlight the unique ability of AML cells to switch their energy supply from glucose to fructose, when glucose is in short supply.
Fructose is the second most abundant blood sugar in the body and is used as a glucose alternative by AML cells to retain energy. After the switch, AML cells begin to multiply faster.
The study revealed a potential treatment by stopping the glucose transporter GLUT5. This restricts the AML energy supply and effectively slows AML growth.
To target GLUT5, the researchers used 2,5-AM, a fructose analog with high affinity for GLUT5.
The team tested 2,5-AM in AML cells with enhanced fructose utilization and found the drug significantly suppressed fructose-induced proliferation, colony growth, and migration in the absence of glucose or when glucose levels were low.
The researchers tested 2,5-AM in 4 different AML cell lines and found the drug suppressed fructose-induced cell proliferation in a dose-dependent manner in all of the cell lines under glucose-limiting conditions. However, 2,5-AM had little effect on glucose-induced cell proliferation.
The team tested 2,5-AM in normal monocytes as well. They said the drug had a negligible effect on glucose-induced cell growth.
“Our normal cells hardly rely on fructose for growth,” Dr Jia noted. “This makes the fructose transport in cancer cells an attractive drug target.”
Finally, the researchers tested 2,5-AM in combination with ara-C in the 4 AML cell lines. They observed a synergistic effect between the drugs in all cell lines in the absence of glucose or when glucose levels were low.
“We are in the process of developing a GLUT5 inhibitor, thus cutting the cancer cells’ energy source and eventually killing them,” Dr Jia said. “The new GLUT5 inhibitor can potentially be used alone or in addition to the current chemotherapy drugs to enhance anticancer effects.” ![]()
Photo from the University
of Hawaii Cancer Center
Researchers say they have discovered a novel treatment strategy for acute myeloid leukemia (AML)—inhibiting fructose utilization.
The team discovered that AML cells are prone to using fructose for energy, and increased fructose utilization predicts poor treatment outcomes in AML patients.
The researchers also found that 2,5-anhydro-D-mannitol (2,5-AM) could inhibit fructose use in AML cells and therefore hinder their growth.
Wei Jia, PhD, of the University of Hawaii Cancer Center in Honolulu, and his colleagues reported these findings in Cancer Cell.
The researchers said their findings highlight the unique ability of AML cells to switch their energy supply from glucose to fructose, when glucose is in short supply.
Fructose is the second most abundant blood sugar in the body and is used as a glucose alternative by AML cells to retain energy. After the switch, AML cells begin to multiply faster.
The study revealed a potential treatment by stopping the glucose transporter GLUT5. This restricts the AML energy supply and effectively slows AML growth.
To target GLUT5, the researchers used 2,5-AM, a fructose analog with high affinity for GLUT5.
The team tested 2,5-AM in AML cells with enhanced fructose utilization and found the drug significantly suppressed fructose-induced proliferation, colony growth, and migration in the absence of glucose or when glucose levels were low.
The researchers tested 2,5-AM in 4 different AML cell lines and found the drug suppressed fructose-induced cell proliferation in a dose-dependent manner in all of the cell lines under glucose-limiting conditions. However, 2,5-AM had little effect on glucose-induced cell proliferation.
The team tested 2,5-AM in normal monocytes as well. They said the drug had a negligible effect on glucose-induced cell growth.
“Our normal cells hardly rely on fructose for growth,” Dr Jia noted. “This makes the fructose transport in cancer cells an attractive drug target.”
Finally, the researchers tested 2,5-AM in combination with ara-C in the 4 AML cell lines. They observed a synergistic effect between the drugs in all cell lines in the absence of glucose or when glucose levels were low.
“We are in the process of developing a GLUT5 inhibitor, thus cutting the cancer cells’ energy source and eventually killing them,” Dr Jia said. “The new GLUT5 inhibitor can potentially be used alone or in addition to the current chemotherapy drugs to enhance anticancer effects.” ![]()
Photo from the University
of Hawaii Cancer Center
Researchers say they have discovered a novel treatment strategy for acute myeloid leukemia (AML)—inhibiting fructose utilization.
The team discovered that AML cells are prone to using fructose for energy, and increased fructose utilization predicts poor treatment outcomes in AML patients.
The researchers also found that 2,5-anhydro-D-mannitol (2,5-AM) could inhibit fructose use in AML cells and therefore hinder their growth.
Wei Jia, PhD, of the University of Hawaii Cancer Center in Honolulu, and his colleagues reported these findings in Cancer Cell.
The researchers said their findings highlight the unique ability of AML cells to switch their energy supply from glucose to fructose, when glucose is in short supply.
Fructose is the second most abundant blood sugar in the body and is used as a glucose alternative by AML cells to retain energy. After the switch, AML cells begin to multiply faster.
The study revealed a potential treatment by stopping the glucose transporter GLUT5. This restricts the AML energy supply and effectively slows AML growth.
To target GLUT5, the researchers used 2,5-AM, a fructose analog with high affinity for GLUT5.
The team tested 2,5-AM in AML cells with enhanced fructose utilization and found the drug significantly suppressed fructose-induced proliferation, colony growth, and migration in the absence of glucose or when glucose levels were low.
The researchers tested 2,5-AM in 4 different AML cell lines and found the drug suppressed fructose-induced cell proliferation in a dose-dependent manner in all of the cell lines under glucose-limiting conditions. However, 2,5-AM had little effect on glucose-induced cell proliferation.
The team tested 2,5-AM in normal monocytes as well. They said the drug had a negligible effect on glucose-induced cell growth.
“Our normal cells hardly rely on fructose for growth,” Dr Jia noted. “This makes the fructose transport in cancer cells an attractive drug target.”
Finally, the researchers tested 2,5-AM in combination with ara-C in the 4 AML cell lines. They observed a synergistic effect between the drugs in all cell lines in the absence of glucose or when glucose levels were low.
“We are in the process of developing a GLUT5 inhibitor, thus cutting the cancer cells’ energy source and eventually killing them,” Dr Jia said. “The new GLUT5 inhibitor can potentially be used alone or in addition to the current chemotherapy drugs to enhance anticancer effects.” ![]()
Targeting CD98 to treat AML
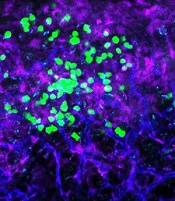
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
Two-drug combination targets LSCs in CML

Image by Difu Wu
Targeting a pair of transcription factors might improve the treatment of chronic myeloid leukemia (CML), according to researchers.
The team found that p53 and c-MYC have “defining roles” in the survival of leukemia stem cells (LSCs) in CML.
And by targeting these transcription factors with a pair of investigational drugs, the researchers were able to kill LSCs.
The team described this work in Nature.
“This collaborative study combined proteomics, transcriptomics, and systems biology to identify a novel, precision medicine-based approach for eradicating leukemic stem cells,” said study author Tony Whetton, PhD, of the University of Manchester in the UK.
Dr Whetton and his colleagues first discovered that p53 and c-MYC are “central hubs” in a CML network of deregulated proteins. The team also found that CML cells express increased c-MYC and decreased p53 levels.
So the researchers theorized that simultaneously activating p53 and inhibiting c-MYC could be a method for treating CML.
To that end, the team tested 2 drugs—RITA (or NSC652287), which binds p53 and blocks its degradation, and CPI-203, a BET inhibitor that hinders transcription by disrupting chromatin-dependent signal transduction.
The researchers found that CPI-203 successfully downregulated c-MYC but also reduced p53, while RITA increased p53.
Treating CML CD34+ cells with RITA or CPI-203 for 72 hours reduced cell viability and induced significant apoptosis, the team said. Combining the drugs enhanced these effects.
The researchers also found evidence to suggest that c-MYC inhibition induces differentiation of CML CD34+ cells. The team said that labelling with the cell-division tracker carboxyfluorescein succinimidyl ester (CFSE) and CD34 antibody showed that, as CML cells divided in the presence of CPI-203, there was a clear and rapid loss of CD34 expression that was not seen in the presence of RITA.
The researchers did not observe any differences in the effects of RITA and CPI-203 when they were tested in CML CD34+ cells pretreated with imatinib.
Furthermore, RITA and CPI-203, either alone or in combination, had no significant effects on normal CD34+ cells when tested at lower concentrations. However, when CPI-203 was used alone at higher concentrations (2 or 5 μ M) or with RITA at the highest concentrations tested (RITA at 25 nM, CPI-203 at 5 μ M), apoptosis did occur.
In CML cells, the researchers observed “significant apoptosis” with all concentrations of CPI-203 and RITA tested.
The team also exposed CML LSCs, defined as either CFSEmax or CD34+CD38− cells, to CPI-203 and RITA as well as a pair of tyrosine kinase inhibitors.
The CFSEmax population persisted despite 5 days of treatment with dasatinib or nilotinib, but the cells were “significantly reduced” after 5 days of treatment with CPI-203 alone and in combination with RITA.
Similarly, 72 hours of treatment with RITA with CPI-203 eliminated residual CD34+CD38− cells.
The researchers also assessed LSC engraftment after treatment with RITA and/or CPI-203, as well as dasatinib. They exposed CML CD34+ cells to the drugs for 48 hours before transplanting the cells into sublethally irradiated NSG mice.
The team said dasatinib had no significant effect on NSG-repopulating CML LSCs. However, RITA, CPI-203, and the drugs in combination reduced engraftment, as indicated by decreased CD45+, CD34+, CD33+, CD11b+, CD19+ and CD14+ cells. ![]()
Image by Difu Wu
Targeting a pair of transcription factors might improve the treatment of chronic myeloid leukemia (CML), according to researchers.
The team found that p53 and c-MYC have “defining roles” in the survival of leukemia stem cells (LSCs) in CML.
And by targeting these transcription factors with a pair of investigational drugs, the researchers were able to kill LSCs.
The team described this work in Nature.
“This collaborative study combined proteomics, transcriptomics, and systems biology to identify a novel, precision medicine-based approach for eradicating leukemic stem cells,” said study author Tony Whetton, PhD, of the University of Manchester in the UK.
Dr Whetton and his colleagues first discovered that p53 and c-MYC are “central hubs” in a CML network of deregulated proteins. The team also found that CML cells express increased c-MYC and decreased p53 levels.
So the researchers theorized that simultaneously activating p53 and inhibiting c-MYC could be a method for treating CML.
To that end, the team tested 2 drugs—RITA (or NSC652287), which binds p53 and blocks its degradation, and CPI-203, a BET inhibitor that hinders transcription by disrupting chromatin-dependent signal transduction.
The researchers found that CPI-203 successfully downregulated c-MYC but also reduced p53, while RITA increased p53.
Treating CML CD34+ cells with RITA or CPI-203 for 72 hours reduced cell viability and induced significant apoptosis, the team said. Combining the drugs enhanced these effects.
The researchers also found evidence to suggest that c-MYC inhibition induces differentiation of CML CD34+ cells. The team said that labelling with the cell-division tracker carboxyfluorescein succinimidyl ester (CFSE) and CD34 antibody showed that, as CML cells divided in the presence of CPI-203, there was a clear and rapid loss of CD34 expression that was not seen in the presence of RITA.
The researchers did not observe any differences in the effects of RITA and CPI-203 when they were tested in CML CD34+ cells pretreated with imatinib.
Furthermore, RITA and CPI-203, either alone or in combination, had no significant effects on normal CD34+ cells when tested at lower concentrations. However, when CPI-203 was used alone at higher concentrations (2 or 5 μ M) or with RITA at the highest concentrations tested (RITA at 25 nM, CPI-203 at 5 μ M), apoptosis did occur.
In CML cells, the researchers observed “significant apoptosis” with all concentrations of CPI-203 and RITA tested.
The team also exposed CML LSCs, defined as either CFSEmax or CD34+CD38− cells, to CPI-203 and RITA as well as a pair of tyrosine kinase inhibitors.
The CFSEmax population persisted despite 5 days of treatment with dasatinib or nilotinib, but the cells were “significantly reduced” after 5 days of treatment with CPI-203 alone and in combination with RITA.
Similarly, 72 hours of treatment with RITA with CPI-203 eliminated residual CD34+CD38− cells.
The researchers also assessed LSC engraftment after treatment with RITA and/or CPI-203, as well as dasatinib. They exposed CML CD34+ cells to the drugs for 48 hours before transplanting the cells into sublethally irradiated NSG mice.
The team said dasatinib had no significant effect on NSG-repopulating CML LSCs. However, RITA, CPI-203, and the drugs in combination reduced engraftment, as indicated by decreased CD45+, CD34+, CD33+, CD11b+, CD19+ and CD14+ cells. ![]()
Image by Difu Wu
Targeting a pair of transcription factors might improve the treatment of chronic myeloid leukemia (CML), according to researchers.
The team found that p53 and c-MYC have “defining roles” in the survival of leukemia stem cells (LSCs) in CML.
And by targeting these transcription factors with a pair of investigational drugs, the researchers were able to kill LSCs.
The team described this work in Nature.
“This collaborative study combined proteomics, transcriptomics, and systems biology to identify a novel, precision medicine-based approach for eradicating leukemic stem cells,” said study author Tony Whetton, PhD, of the University of Manchester in the UK.
Dr Whetton and his colleagues first discovered that p53 and c-MYC are “central hubs” in a CML network of deregulated proteins. The team also found that CML cells express increased c-MYC and decreased p53 levels.
So the researchers theorized that simultaneously activating p53 and inhibiting c-MYC could be a method for treating CML.
To that end, the team tested 2 drugs—RITA (or NSC652287), which binds p53 and blocks its degradation, and CPI-203, a BET inhibitor that hinders transcription by disrupting chromatin-dependent signal transduction.
The researchers found that CPI-203 successfully downregulated c-MYC but also reduced p53, while RITA increased p53.
Treating CML CD34+ cells with RITA or CPI-203 for 72 hours reduced cell viability and induced significant apoptosis, the team said. Combining the drugs enhanced these effects.
The researchers also found evidence to suggest that c-MYC inhibition induces differentiation of CML CD34+ cells. The team said that labelling with the cell-division tracker carboxyfluorescein succinimidyl ester (CFSE) and CD34 antibody showed that, as CML cells divided in the presence of CPI-203, there was a clear and rapid loss of CD34 expression that was not seen in the presence of RITA.
The researchers did not observe any differences in the effects of RITA and CPI-203 when they were tested in CML CD34+ cells pretreated with imatinib.
Furthermore, RITA and CPI-203, either alone or in combination, had no significant effects on normal CD34+ cells when tested at lower concentrations. However, when CPI-203 was used alone at higher concentrations (2 or 5 μ M) or with RITA at the highest concentrations tested (RITA at 25 nM, CPI-203 at 5 μ M), apoptosis did occur.
In CML cells, the researchers observed “significant apoptosis” with all concentrations of CPI-203 and RITA tested.
The team also exposed CML LSCs, defined as either CFSEmax or CD34+CD38− cells, to CPI-203 and RITA as well as a pair of tyrosine kinase inhibitors.
The CFSEmax population persisted despite 5 days of treatment with dasatinib or nilotinib, but the cells were “significantly reduced” after 5 days of treatment with CPI-203 alone and in combination with RITA.
Similarly, 72 hours of treatment with RITA with CPI-203 eliminated residual CD34+CD38− cells.
The researchers also assessed LSC engraftment after treatment with RITA and/or CPI-203, as well as dasatinib. They exposed CML CD34+ cells to the drugs for 48 hours before transplanting the cells into sublethally irradiated NSG mice.
The team said dasatinib had no significant effect on NSG-repopulating CML LSCs. However, RITA, CPI-203, and the drugs in combination reduced engraftment, as indicated by decreased CD45+, CD34+, CD33+, CD11b+, CD19+ and CD14+ cells. ![]()
Team maps genomic landscape of CBF-AML

whole-genome sequencing
Photo courtesy of the US
Food and Drug Administration
Whole-genome and whole-exome sequencing has provided new insight into the pathogenesis and development of core-binding factor acute myeloid leukemia (CBF-AML), according to researchers.
The team said their work has revealed “dramatic” differences in the genomic landscape of CBF-AMLs that contribute to the diversity of this disease.
The researchers reported their findings in Nature Genetics.
“We set out to understand the genetic variations that contribute to the development of CBF-AML using whole-exome and whole-genome sequencing,” said study author Jeffery Klco, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our goal was to define a detailed mutational landscape to understand better the genetic changes that contribute to disease.”
Dr Klco and his colleagues sequenced samples from 87 children and 78 adults with CBF-AML. Eighty-five of the patients had the RUNX1-RUNX1T1 subtype, and 80 had the CBFB-MYH11 subtype.
Development and relapse
The researchers identified several genes with mutations that may contribute to CBF-AML development, including CCND2, DHX15, ASXL2, ZBTB7A, and MGA.
“Many of the mutations we identified interfered with molecular signaling or epigenetic factors,” said study author Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital.
“Some of the mutations, like ASXL2, are epigenetic regulators that modify the local state of chromatin,” Dr Klco noted. “Others, like ZBTB7A, appear to act like tumor suppressors.”
The researchers also compared mutations present at diagnosis and relapse in an attempt to understand how CBF-AML changes over time.
Their results suggested that KMT2C mutations are associated with relapse. Of the 4 patients in this study who had KMT2C mutations, 3 relapsed in less than 12 months, and the fourth had residual disease after a course of remission-induction therapy.
Similarities and differences
The researchers found a similar mutational landscape in adults and children with CBF-AML but differences between patients with the RUNX1-RUNX1T1 and CBFB-MYH11 subtypes.
NRAS was the most frequently mutated gene in CBF-AMLs, but NRAS mutations were more common in CBFB-MYH11 AML than RUNX1-RUNX1T1 AML. The same was true for mutations in NF1 and WT1.
Patients with both subtypes of CBF-AML had mutations in NRAS, KIT, NF1, WT1, FLT3, KRAS, MGA, TTN, CCND2, KDM6A, PHIP, TET2, HCN1, KMT2C, and SETD2.
But only patients with CBFB-MYH11 AML had mutations in PTPN11.
Only patients with RUNX1-RUNX1T1 AML had mutations in ASXL2, ZBTB7A, EZH2, SMC1A, DHX15, RAD21, CBL, DNM2, CSF3R, GIGYF2, SMC3, and ZNF687.
The researchers said their findings suggest a range of mutations may play roles in CBF-AML, but additional research is needed to confirm their precise function in the disease.
Further studies are already underway to fully evaluate the contributions of the different genes as well as the roles of the newly identified genetic alterations in CBF-AML. ![]()
whole-genome sequencing
Photo courtesy of the US
Food and Drug Administration
Whole-genome and whole-exome sequencing has provided new insight into the pathogenesis and development of core-binding factor acute myeloid leukemia (CBF-AML), according to researchers.
The team said their work has revealed “dramatic” differences in the genomic landscape of CBF-AMLs that contribute to the diversity of this disease.
The researchers reported their findings in Nature Genetics.
“We set out to understand the genetic variations that contribute to the development of CBF-AML using whole-exome and whole-genome sequencing,” said study author Jeffery Klco, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our goal was to define a detailed mutational landscape to understand better the genetic changes that contribute to disease.”
Dr Klco and his colleagues sequenced samples from 87 children and 78 adults with CBF-AML. Eighty-five of the patients had the RUNX1-RUNX1T1 subtype, and 80 had the CBFB-MYH11 subtype.
Development and relapse
The researchers identified several genes with mutations that may contribute to CBF-AML development, including CCND2, DHX15, ASXL2, ZBTB7A, and MGA.
“Many of the mutations we identified interfered with molecular signaling or epigenetic factors,” said study author Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital.
“Some of the mutations, like ASXL2, are epigenetic regulators that modify the local state of chromatin,” Dr Klco noted. “Others, like ZBTB7A, appear to act like tumor suppressors.”
The researchers also compared mutations present at diagnosis and relapse in an attempt to understand how CBF-AML changes over time.
Their results suggested that KMT2C mutations are associated with relapse. Of the 4 patients in this study who had KMT2C mutations, 3 relapsed in less than 12 months, and the fourth had residual disease after a course of remission-induction therapy.
Similarities and differences
The researchers found a similar mutational landscape in adults and children with CBF-AML but differences between patients with the RUNX1-RUNX1T1 and CBFB-MYH11 subtypes.
NRAS was the most frequently mutated gene in CBF-AMLs, but NRAS mutations were more common in CBFB-MYH11 AML than RUNX1-RUNX1T1 AML. The same was true for mutations in NF1 and WT1.
Patients with both subtypes of CBF-AML had mutations in NRAS, KIT, NF1, WT1, FLT3, KRAS, MGA, TTN, CCND2, KDM6A, PHIP, TET2, HCN1, KMT2C, and SETD2.
But only patients with CBFB-MYH11 AML had mutations in PTPN11.
Only patients with RUNX1-RUNX1T1 AML had mutations in ASXL2, ZBTB7A, EZH2, SMC1A, DHX15, RAD21, CBL, DNM2, CSF3R, GIGYF2, SMC3, and ZNF687.
The researchers said their findings suggest a range of mutations may play roles in CBF-AML, but additional research is needed to confirm their precise function in the disease.
Further studies are already underway to fully evaluate the contributions of the different genes as well as the roles of the newly identified genetic alterations in CBF-AML. ![]()
whole-genome sequencing
Photo courtesy of the US
Food and Drug Administration
Whole-genome and whole-exome sequencing has provided new insight into the pathogenesis and development of core-binding factor acute myeloid leukemia (CBF-AML), according to researchers.
The team said their work has revealed “dramatic” differences in the genomic landscape of CBF-AMLs that contribute to the diversity of this disease.
The researchers reported their findings in Nature Genetics.
“We set out to understand the genetic variations that contribute to the development of CBF-AML using whole-exome and whole-genome sequencing,” said study author Jeffery Klco, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our goal was to define a detailed mutational landscape to understand better the genetic changes that contribute to disease.”
Dr Klco and his colleagues sequenced samples from 87 children and 78 adults with CBF-AML. Eighty-five of the patients had the RUNX1-RUNX1T1 subtype, and 80 had the CBFB-MYH11 subtype.
Development and relapse
The researchers identified several genes with mutations that may contribute to CBF-AML development, including CCND2, DHX15, ASXL2, ZBTB7A, and MGA.
“Many of the mutations we identified interfered with molecular signaling or epigenetic factors,” said study author Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital.
“Some of the mutations, like ASXL2, are epigenetic regulators that modify the local state of chromatin,” Dr Klco noted. “Others, like ZBTB7A, appear to act like tumor suppressors.”
The researchers also compared mutations present at diagnosis and relapse in an attempt to understand how CBF-AML changes over time.
Their results suggested that KMT2C mutations are associated with relapse. Of the 4 patients in this study who had KMT2C mutations, 3 relapsed in less than 12 months, and the fourth had residual disease after a course of remission-induction therapy.
Similarities and differences
The researchers found a similar mutational landscape in adults and children with CBF-AML but differences between patients with the RUNX1-RUNX1T1 and CBFB-MYH11 subtypes.
NRAS was the most frequently mutated gene in CBF-AMLs, but NRAS mutations were more common in CBFB-MYH11 AML than RUNX1-RUNX1T1 AML. The same was true for mutations in NF1 and WT1.
Patients with both subtypes of CBF-AML had mutations in NRAS, KIT, NF1, WT1, FLT3, KRAS, MGA, TTN, CCND2, KDM6A, PHIP, TET2, HCN1, KMT2C, and SETD2.
But only patients with CBFB-MYH11 AML had mutations in PTPN11.
Only patients with RUNX1-RUNX1T1 AML had mutations in ASXL2, ZBTB7A, EZH2, SMC1A, DHX15, RAD21, CBL, DNM2, CSF3R, GIGYF2, SMC3, and ZNF687.
The researchers said their findings suggest a range of mutations may play roles in CBF-AML, but additional research is needed to confirm their precise function in the disease.
Further studies are already underway to fully evaluate the contributions of the different genes as well as the roles of the newly identified genetic alterations in CBF-AML. ![]()
Combo shows promise for treating T-ALL
acute lymphoblastic leukemia
Image by Hind Medyou
Preclinical research suggests that 2 investigational drugs synergize to kill T-cell acute lymphoblastic leukemia (T-ALL) cells while having a minimal impact on normal blood cells.
Both drugs—the CK2 inhibitor CX-4945 and the BET inhibitor JQ1—have already been tested as single agents in clinical trials of hematologic malignancies and solid tumors.
However, the effects of the drugs in combination were not known until now.
“Previous studies provided us a rationale to test the combination of CX-4945 and JQ1 on refractory/relapsed T-cell leukemia,” said Hui Feng, MD, PhD, of Boston University School of Medicine in Massachusetts.
“Our findings suggest that the combination treatment of CX-4945 and JQ1 could be an effective strategy to target refractory/relapsed T-cell leukemia.”
Dr Feng and her colleagues reported these findings in Haematologica.
The researchers noted that targeting MYC-mediated transcriptional programs using JQ1 produces anti-leukemic activity in vitro and in vivo. However, global repression of transcription is likely to cause toxicities.
Therefore, the team theorized that finding drugs that synergize with JQ1 might allow them to reduce the dose of JQ1 and therefore decrease the risk of toxicity while enhancing the efficacy of treatment.
For this, the researchers looked to CX-4945, which is currently being investigated in clinical trials of breast cancer and multiple myeloma.
The team said CX-4945 has been shown to significantly reduce the growth and survival of human T-ALL cells on its own.
In a series of experiments, Dr Feng and her colleagues were able to show that CX-4945 destabilizes NOTCH1 and synergizes with JQ1 to induce apoptosis in human T-ALL cells.
The researchers also assessed the effects of JQ1 and CX-4946, alone and in combination, on normal peripheral blood monocytes (PBMs).
PBMs proved less sensitive than ALL-SIL T-ALL cells to each drug alone and to the drugs in combination. In fact, the combination had an antagonistic effect in PBMs.
Dr Feng and her colleagues said this research suggests JQ1 and CX-4946 in combination may be a feasible treatment option for relapsed/refractory T-ALL and other cancers involving CK2 and NOTCH1/MYC. ![]()
acute lymphoblastic leukemia
Image by Hind Medyou
Preclinical research suggests that 2 investigational drugs synergize to kill T-cell acute lymphoblastic leukemia (T-ALL) cells while having a minimal impact on normal blood cells.
Both drugs—the CK2 inhibitor CX-4945 and the BET inhibitor JQ1—have already been tested as single agents in clinical trials of hematologic malignancies and solid tumors.
However, the effects of the drugs in combination were not known until now.
“Previous studies provided us a rationale to test the combination of CX-4945 and JQ1 on refractory/relapsed T-cell leukemia,” said Hui Feng, MD, PhD, of Boston University School of Medicine in Massachusetts.
“Our findings suggest that the combination treatment of CX-4945 and JQ1 could be an effective strategy to target refractory/relapsed T-cell leukemia.”
Dr Feng and her colleagues reported these findings in Haematologica.
The researchers noted that targeting MYC-mediated transcriptional programs using JQ1 produces anti-leukemic activity in vitro and in vivo. However, global repression of transcription is likely to cause toxicities.
Therefore, the team theorized that finding drugs that synergize with JQ1 might allow them to reduce the dose of JQ1 and therefore decrease the risk of toxicity while enhancing the efficacy of treatment.
For this, the researchers looked to CX-4945, which is currently being investigated in clinical trials of breast cancer and multiple myeloma.
The team said CX-4945 has been shown to significantly reduce the growth and survival of human T-ALL cells on its own.
In a series of experiments, Dr Feng and her colleagues were able to show that CX-4945 destabilizes NOTCH1 and synergizes with JQ1 to induce apoptosis in human T-ALL cells.
The researchers also assessed the effects of JQ1 and CX-4946, alone and in combination, on normal peripheral blood monocytes (PBMs).
PBMs proved less sensitive than ALL-SIL T-ALL cells to each drug alone and to the drugs in combination. In fact, the combination had an antagonistic effect in PBMs.
Dr Feng and her colleagues said this research suggests JQ1 and CX-4946 in combination may be a feasible treatment option for relapsed/refractory T-ALL and other cancers involving CK2 and NOTCH1/MYC. ![]()
acute lymphoblastic leukemia
Image by Hind Medyou
Preclinical research suggests that 2 investigational drugs synergize to kill T-cell acute lymphoblastic leukemia (T-ALL) cells while having a minimal impact on normal blood cells.
Both drugs—the CK2 inhibitor CX-4945 and the BET inhibitor JQ1—have already been tested as single agents in clinical trials of hematologic malignancies and solid tumors.
However, the effects of the drugs in combination were not known until now.
“Previous studies provided us a rationale to test the combination of CX-4945 and JQ1 on refractory/relapsed T-cell leukemia,” said Hui Feng, MD, PhD, of Boston University School of Medicine in Massachusetts.
“Our findings suggest that the combination treatment of CX-4945 and JQ1 could be an effective strategy to target refractory/relapsed T-cell leukemia.”
Dr Feng and her colleagues reported these findings in Haematologica.
The researchers noted that targeting MYC-mediated transcriptional programs using JQ1 produces anti-leukemic activity in vitro and in vivo. However, global repression of transcription is likely to cause toxicities.
Therefore, the team theorized that finding drugs that synergize with JQ1 might allow them to reduce the dose of JQ1 and therefore decrease the risk of toxicity while enhancing the efficacy of treatment.
For this, the researchers looked to CX-4945, which is currently being investigated in clinical trials of breast cancer and multiple myeloma.
The team said CX-4945 has been shown to significantly reduce the growth and survival of human T-ALL cells on its own.
In a series of experiments, Dr Feng and her colleagues were able to show that CX-4945 destabilizes NOTCH1 and synergizes with JQ1 to induce apoptosis in human T-ALL cells.
The researchers also assessed the effects of JQ1 and CX-4946, alone and in combination, on normal peripheral blood monocytes (PBMs).
PBMs proved less sensitive than ALL-SIL T-ALL cells to each drug alone and to the drugs in combination. In fact, the combination had an antagonistic effect in PBMs.
Dr Feng and her colleagues said this research suggests JQ1 and CX-4946 in combination may be a feasible treatment option for relapsed/refractory T-ALL and other cancers involving CK2 and NOTCH1/MYC. ![]()
Explaining the development of MPNs, leukemia
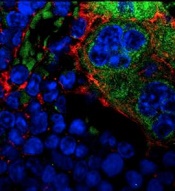
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation. ![]()
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation. ![]()
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation. ![]()
Genetic screening for CLL premature, speaker says

Photo courtesy of the
National Institute
of General Medical Science
NEW YORK—Research has shown that family history is a strong risk factor for developing chronic lymphocytic leukemia (CLL).
First-degree relatives have an 8.5-fold risk of getting CLL and an increased risk of other lymphoproliferative disorders, according to a study published in 2009.
However, despite the strong evidence of a genetic contribution, one expert believes it’s premature to bring genetic testing into the clinic for screening in CLL.
“At this time, we do not recommend genetic screening,” said Susan Slager, PhD, of the Mayo Clinic in Rochester, Minnesota.
“There’s no known relationship between the inherited variants and treatment response,” she explained, and the relatively low incidence of CLL argues against active screening in affected families at present.
Dr Slager discussed genetic and non-genetic factors associated with CLL and the clinical implications of these factors at Lymphoma & Myeloma 2016.
Demographic risk factors
Dr Slager noted that age, gender, and race are risk factors for CLL.
Individuals aged 65 to 74 have the highest incidence of CLL, at 28%, while the risk is almost non-existent for those under age 20, she said.
There is a higher incidence of CLL in males than in females, and the reason for this gender disparity is unknown.
There is a higher incidence of CLL in Caucasians than Asians, for both males and females.
“Again, it’s unknown why there’s this variability in incidence in CLL,” Dr Slager said. “Obviously, age, sex, and race—these are things you can’t modify. You’re stuck with them.”
However, several studies have been undertaken to look at some of the potentially modifiable factors associated with CLL.
Beyond demographic factors
The International Lymphoma Epidemiology Consortium, known as InterLymph, was initiated in 2001 to evaluate the association of risk factors in CLL. Study centers are located primarily in North America and Europe, with one in Australia.
In one of the larger InterLymph studies, investigators evaluated risk factors—lifestyle exposure, reproductive history, medical history, occupational exposures, farming exposure, and family history—in 2440 CLL patients and 15,186 controls.
The investigators found that sun exposure and atopy—allergies, asthma, eczema, and hay fever—have a protective effect in CLL, while serological hepatitis C virus (HCV) infections, farming exposure, and family history carry an increased risk of CLL.
This confirmed an earlier study conducted in New South Wales, Australia, that had uncovered an inverse association between sun exposure and non-Hodgkin lymphoma (NHL) risk, which fell significantly with increasing recreational sun exposure.
Medical history
Another earlier study from New South Wales revealed a 20% reduction in the risk of NHL for any specific allergy.
However, the investigators of the large, more recent study observed little to no evidence of reduced risk for asthma and eczema.
The underlying biology for atopy or allergies is a hyper-immune system, Dr Slager explained.
“So if you have a hyper-immune system, then we hypothesize that you have protection against CLL,” she said.
Another medical exposure investigators analyzed that impacts CLL risk is HCV. People infected with HCV have an increased risk of CLL, perhaps due to chronic antigen stimulation or possibly disruption of the T-cell function.
Height is also associated with CLL. CLL risk increases with greater height. The concept is that taller individuals have increased exposure to growth hormones that possibly result in cell proliferation.
Another hypothesis supporting the height association is that people of shorter stature experience more infections, which could result in a stronger immune system. And a stronger immune system perhaps protects against NHL.
Occupational exposures
Investigators consistently observed a 20% increased risk of CLL for people living or working on a farm.
Animal farmers, as opposed to crop farmers, experienced some protection. However, the sample size was too small to be conclusive, with only 29 people across all studies being animal farmers.
Among other occupations evaluated, hairdressers also had an increased risk of CLL, although this too was based on a small sample size.
Family history
One of the strongest risk factors for CLL is family history.
Using population-based registry data from Sweden, investigators found that people with a first-degree relative with CLL have an 8.5-fold risk of CLL.
They also have an elevated risk of other lymphoproliferative disorders, including NHL (1.9-fold risk), Waldenström’s macroglobulinemia (4.0-fold risk), hairy cell leukemia (3.3-fold risk), and follicular lymphoma (1.6-fold risk).
GWAS in CLL
Investigators conducted genome-wide association studies (GWAS) to determine what is driving the familial risk.
Dr Slager described these studies as an agnostic approach that looks across the entire genome to determine which regions are associated with a trait of interest.
Typically, many markers are genotyped—somewhere between half a million to 5 million markers—and each is looked at individually with respect to CLL, she said.
Unrelated cases and controls are included in the studies.
The first GWAS study identifying susceptibility loci for CLL was published in 2008. Subsequently, more studies were published with increasing sample sizes—more cases, more controls, and more genetic variants identified.
In the largest meta-analysis for CLL to date (Slager and Houlston et al, not yet published), investigators analyzed 4400 CLL cases and 13,000 controls.
They identified 9 more inherited variances with CLL, for a total of 43 identified to date.
The genes involved follow an apoptosis pathway, the telomere length pathway, and the B-cell lymphocyte development pathway.
“We have to remember, though, that these are non-causal,” Dr Slager cautioned. “We are just identifying the region in the genome that’s associated with CLL . . . . So now we have to dig deeper in these relationships to understand what’s going on.”
Using the identified CLL single-nucleotide polymorphisms, the investigators computed a polygenic risk score. CLL cases in the highest quintile had 2.7-fold increased risk of CLL.
However, the most common GWAS variants explain only 17% of the genetic heritability of CLL, which suggests that more loci are yet to be identified, Dr Slager clarified.
She went on to say that CLL incidence varies by ethnicity. Caucasians have a very high rate of CLL, while Asians have a very low rate. And African Americans have an incidence rate between those of Caucasians and Asians.
Investigators have hypothesized that the differences in incidence are based on the distinct genetic variants that are associated with the ethnicities.
For example, 4 of the variants with more than 20% frequency in Caucasians are quite rare in Chinese individuals and are also quite uncommon in African Americans, with frequencies less than 10%.
Dr Slager suggested that conducting these kinds of studies in Asians and African Americans will take a large sample size and most likely require an international consortium to bring enough CLL cases together.
Impact on clinical practice
Because of the strong genetic risk, patients with CLL naturally want to know about their offspring and their siblings, Dr Slager has found.
Patients who have monoclonal B-cell lymphocytosis (MBL), which is a precursor to CLL, pose the biggest quandary.
MBL is detected in about 5% of people over age 40. However, it’s detected in about 15% to 18% of people with a first-degree relative with CLL.
“These are individuals who have lymphocytosis,” Dr Slager said. “They come to your clinic and have an elevated blood cell count, flow cytometry. [So] you screen them for MBL, and these individuals who have more than 500 cells per microliter, they are the ones who progress to CLL, at 1% per year.”
Individuals who don’t have the elevated blood counts do have the clonal cells, Dr Slager noted.
“They just don’t have the expansion,” she said. “The progression of these individuals to CLL is still yet to be determined.”
For these reasons, Dr Slager believes “it’s still premature to bring genetic testing into clinical practice.”
Future directions include bringing together the non-environmental issues and the inherited issues to create a model that will accurately predict the risk of CLL. ![]()
Photo courtesy of the
National Institute
of General Medical Science
NEW YORK—Research has shown that family history is a strong risk factor for developing chronic lymphocytic leukemia (CLL).
First-degree relatives have an 8.5-fold risk of getting CLL and an increased risk of other lymphoproliferative disorders, according to a study published in 2009.
However, despite the strong evidence of a genetic contribution, one expert believes it’s premature to bring genetic testing into the clinic for screening in CLL.
“At this time, we do not recommend genetic screening,” said Susan Slager, PhD, of the Mayo Clinic in Rochester, Minnesota.
“There’s no known relationship between the inherited variants and treatment response,” she explained, and the relatively low incidence of CLL argues against active screening in affected families at present.
Dr Slager discussed genetic and non-genetic factors associated with CLL and the clinical implications of these factors at Lymphoma & Myeloma 2016.
Demographic risk factors
Dr Slager noted that age, gender, and race are risk factors for CLL.
Individuals aged 65 to 74 have the highest incidence of CLL, at 28%, while the risk is almost non-existent for those under age 20, she said.
There is a higher incidence of CLL in males than in females, and the reason for this gender disparity is unknown.
There is a higher incidence of CLL in Caucasians than Asians, for both males and females.
“Again, it’s unknown why there’s this variability in incidence in CLL,” Dr Slager said. “Obviously, age, sex, and race—these are things you can’t modify. You’re stuck with them.”
However, several studies have been undertaken to look at some of the potentially modifiable factors associated with CLL.
Beyond demographic factors
The International Lymphoma Epidemiology Consortium, known as InterLymph, was initiated in 2001 to evaluate the association of risk factors in CLL. Study centers are located primarily in North America and Europe, with one in Australia.
In one of the larger InterLymph studies, investigators evaluated risk factors—lifestyle exposure, reproductive history, medical history, occupational exposures, farming exposure, and family history—in 2440 CLL patients and 15,186 controls.
The investigators found that sun exposure and atopy—allergies, asthma, eczema, and hay fever—have a protective effect in CLL, while serological hepatitis C virus (HCV) infections, farming exposure, and family history carry an increased risk of CLL.
This confirmed an earlier study conducted in New South Wales, Australia, that had uncovered an inverse association between sun exposure and non-Hodgkin lymphoma (NHL) risk, which fell significantly with increasing recreational sun exposure.
Medical history
Another earlier study from New South Wales revealed a 20% reduction in the risk of NHL for any specific allergy.
However, the investigators of the large, more recent study observed little to no evidence of reduced risk for asthma and eczema.
The underlying biology for atopy or allergies is a hyper-immune system, Dr Slager explained.
“So if you have a hyper-immune system, then we hypothesize that you have protection against CLL,” she said.
Another medical exposure investigators analyzed that impacts CLL risk is HCV. People infected with HCV have an increased risk of CLL, perhaps due to chronic antigen stimulation or possibly disruption of the T-cell function.
Height is also associated with CLL. CLL risk increases with greater height. The concept is that taller individuals have increased exposure to growth hormones that possibly result in cell proliferation.
Another hypothesis supporting the height association is that people of shorter stature experience more infections, which could result in a stronger immune system. And a stronger immune system perhaps protects against NHL.
Occupational exposures
Investigators consistently observed a 20% increased risk of CLL for people living or working on a farm.
Animal farmers, as opposed to crop farmers, experienced some protection. However, the sample size was too small to be conclusive, with only 29 people across all studies being animal farmers.
Among other occupations evaluated, hairdressers also had an increased risk of CLL, although this too was based on a small sample size.
Family history
One of the strongest risk factors for CLL is family history.
Using population-based registry data from Sweden, investigators found that people with a first-degree relative with CLL have an 8.5-fold risk of CLL.
They also have an elevated risk of other lymphoproliferative disorders, including NHL (1.9-fold risk), Waldenström’s macroglobulinemia (4.0-fold risk), hairy cell leukemia (3.3-fold risk), and follicular lymphoma (1.6-fold risk).
GWAS in CLL
Investigators conducted genome-wide association studies (GWAS) to determine what is driving the familial risk.
Dr Slager described these studies as an agnostic approach that looks across the entire genome to determine which regions are associated with a trait of interest.
Typically, many markers are genotyped—somewhere between half a million to 5 million markers—and each is looked at individually with respect to CLL, she said.
Unrelated cases and controls are included in the studies.
The first GWAS study identifying susceptibility loci for CLL was published in 2008. Subsequently, more studies were published with increasing sample sizes—more cases, more controls, and more genetic variants identified.
In the largest meta-analysis for CLL to date (Slager and Houlston et al, not yet published), investigators analyzed 4400 CLL cases and 13,000 controls.
They identified 9 more inherited variances with CLL, for a total of 43 identified to date.
The genes involved follow an apoptosis pathway, the telomere length pathway, and the B-cell lymphocyte development pathway.
“We have to remember, though, that these are non-causal,” Dr Slager cautioned. “We are just identifying the region in the genome that’s associated with CLL . . . . So now we have to dig deeper in these relationships to understand what’s going on.”
Using the identified CLL single-nucleotide polymorphisms, the investigators computed a polygenic risk score. CLL cases in the highest quintile had 2.7-fold increased risk of CLL.
However, the most common GWAS variants explain only 17% of the genetic heritability of CLL, which suggests that more loci are yet to be identified, Dr Slager clarified.
She went on to say that CLL incidence varies by ethnicity. Caucasians have a very high rate of CLL, while Asians have a very low rate. And African Americans have an incidence rate between those of Caucasians and Asians.
Investigators have hypothesized that the differences in incidence are based on the distinct genetic variants that are associated with the ethnicities.
For example, 4 of the variants with more than 20% frequency in Caucasians are quite rare in Chinese individuals and are also quite uncommon in African Americans, with frequencies less than 10%.
Dr Slager suggested that conducting these kinds of studies in Asians and African Americans will take a large sample size and most likely require an international consortium to bring enough CLL cases together.
Impact on clinical practice
Because of the strong genetic risk, patients with CLL naturally want to know about their offspring and their siblings, Dr Slager has found.
Patients who have monoclonal B-cell lymphocytosis (MBL), which is a precursor to CLL, pose the biggest quandary.
MBL is detected in about 5% of people over age 40. However, it’s detected in about 15% to 18% of people with a first-degree relative with CLL.
“These are individuals who have lymphocytosis,” Dr Slager said. “They come to your clinic and have an elevated blood cell count, flow cytometry. [So] you screen them for MBL, and these individuals who have more than 500 cells per microliter, they are the ones who progress to CLL, at 1% per year.”
Individuals who don’t have the elevated blood counts do have the clonal cells, Dr Slager noted.
“They just don’t have the expansion,” she said. “The progression of these individuals to CLL is still yet to be determined.”
For these reasons, Dr Slager believes “it’s still premature to bring genetic testing into clinical practice.”
Future directions include bringing together the non-environmental issues and the inherited issues to create a model that will accurately predict the risk of CLL. ![]()
Photo courtesy of the
National Institute
of General Medical Science
NEW YORK—Research has shown that family history is a strong risk factor for developing chronic lymphocytic leukemia (CLL).
First-degree relatives have an 8.5-fold risk of getting CLL and an increased risk of other lymphoproliferative disorders, according to a study published in 2009.
However, despite the strong evidence of a genetic contribution, one expert believes it’s premature to bring genetic testing into the clinic for screening in CLL.
“At this time, we do not recommend genetic screening,” said Susan Slager, PhD, of the Mayo Clinic in Rochester, Minnesota.
“There’s no known relationship between the inherited variants and treatment response,” she explained, and the relatively low incidence of CLL argues against active screening in affected families at present.
Dr Slager discussed genetic and non-genetic factors associated with CLL and the clinical implications of these factors at Lymphoma & Myeloma 2016.
Demographic risk factors
Dr Slager noted that age, gender, and race are risk factors for CLL.
Individuals aged 65 to 74 have the highest incidence of CLL, at 28%, while the risk is almost non-existent for those under age 20, she said.
There is a higher incidence of CLL in males than in females, and the reason for this gender disparity is unknown.
There is a higher incidence of CLL in Caucasians than Asians, for both males and females.
“Again, it’s unknown why there’s this variability in incidence in CLL,” Dr Slager said. “Obviously, age, sex, and race—these are things you can’t modify. You’re stuck with them.”
However, several studies have been undertaken to look at some of the potentially modifiable factors associated with CLL.
Beyond demographic factors
The International Lymphoma Epidemiology Consortium, known as InterLymph, was initiated in 2001 to evaluate the association of risk factors in CLL. Study centers are located primarily in North America and Europe, with one in Australia.
In one of the larger InterLymph studies, investigators evaluated risk factors—lifestyle exposure, reproductive history, medical history, occupational exposures, farming exposure, and family history—in 2440 CLL patients and 15,186 controls.
The investigators found that sun exposure and atopy—allergies, asthma, eczema, and hay fever—have a protective effect in CLL, while serological hepatitis C virus (HCV) infections, farming exposure, and family history carry an increased risk of CLL.
This confirmed an earlier study conducted in New South Wales, Australia, that had uncovered an inverse association between sun exposure and non-Hodgkin lymphoma (NHL) risk, which fell significantly with increasing recreational sun exposure.
Medical history
Another earlier study from New South Wales revealed a 20% reduction in the risk of NHL for any specific allergy.
However, the investigators of the large, more recent study observed little to no evidence of reduced risk for asthma and eczema.
The underlying biology for atopy or allergies is a hyper-immune system, Dr Slager explained.
“So if you have a hyper-immune system, then we hypothesize that you have protection against CLL,” she said.
Another medical exposure investigators analyzed that impacts CLL risk is HCV. People infected with HCV have an increased risk of CLL, perhaps due to chronic antigen stimulation or possibly disruption of the T-cell function.
Height is also associated with CLL. CLL risk increases with greater height. The concept is that taller individuals have increased exposure to growth hormones that possibly result in cell proliferation.
Another hypothesis supporting the height association is that people of shorter stature experience more infections, which could result in a stronger immune system. And a stronger immune system perhaps protects against NHL.
Occupational exposures
Investigators consistently observed a 20% increased risk of CLL for people living or working on a farm.
Animal farmers, as opposed to crop farmers, experienced some protection. However, the sample size was too small to be conclusive, with only 29 people across all studies being animal farmers.
Among other occupations evaluated, hairdressers also had an increased risk of CLL, although this too was based on a small sample size.
Family history
One of the strongest risk factors for CLL is family history.
Using population-based registry data from Sweden, investigators found that people with a first-degree relative with CLL have an 8.5-fold risk of CLL.
They also have an elevated risk of other lymphoproliferative disorders, including NHL (1.9-fold risk), Waldenström’s macroglobulinemia (4.0-fold risk), hairy cell leukemia (3.3-fold risk), and follicular lymphoma (1.6-fold risk).
GWAS in CLL
Investigators conducted genome-wide association studies (GWAS) to determine what is driving the familial risk.
Dr Slager described these studies as an agnostic approach that looks across the entire genome to determine which regions are associated with a trait of interest.
Typically, many markers are genotyped—somewhere between half a million to 5 million markers—and each is looked at individually with respect to CLL, she said.
Unrelated cases and controls are included in the studies.
The first GWAS study identifying susceptibility loci for CLL was published in 2008. Subsequently, more studies were published with increasing sample sizes—more cases, more controls, and more genetic variants identified.
In the largest meta-analysis for CLL to date (Slager and Houlston et al, not yet published), investigators analyzed 4400 CLL cases and 13,000 controls.
They identified 9 more inherited variances with CLL, for a total of 43 identified to date.
The genes involved follow an apoptosis pathway, the telomere length pathway, and the B-cell lymphocyte development pathway.
“We have to remember, though, that these are non-causal,” Dr Slager cautioned. “We are just identifying the region in the genome that’s associated with CLL . . . . So now we have to dig deeper in these relationships to understand what’s going on.”
Using the identified CLL single-nucleotide polymorphisms, the investigators computed a polygenic risk score. CLL cases in the highest quintile had 2.7-fold increased risk of CLL.
However, the most common GWAS variants explain only 17% of the genetic heritability of CLL, which suggests that more loci are yet to be identified, Dr Slager clarified.
She went on to say that CLL incidence varies by ethnicity. Caucasians have a very high rate of CLL, while Asians have a very low rate. And African Americans have an incidence rate between those of Caucasians and Asians.
Investigators have hypothesized that the differences in incidence are based on the distinct genetic variants that are associated with the ethnicities.
For example, 4 of the variants with more than 20% frequency in Caucasians are quite rare in Chinese individuals and are also quite uncommon in African Americans, with frequencies less than 10%.
Dr Slager suggested that conducting these kinds of studies in Asians and African Americans will take a large sample size and most likely require an international consortium to bring enough CLL cases together.
Impact on clinical practice
Because of the strong genetic risk, patients with CLL naturally want to know about their offspring and their siblings, Dr Slager has found.
Patients who have monoclonal B-cell lymphocytosis (MBL), which is a precursor to CLL, pose the biggest quandary.
MBL is detected in about 5% of people over age 40. However, it’s detected in about 15% to 18% of people with a first-degree relative with CLL.
“These are individuals who have lymphocytosis,” Dr Slager said. “They come to your clinic and have an elevated blood cell count, flow cytometry. [So] you screen them for MBL, and these individuals who have more than 500 cells per microliter, they are the ones who progress to CLL, at 1% per year.”
Individuals who don’t have the elevated blood counts do have the clonal cells, Dr Slager noted.
“They just don’t have the expansion,” she said. “The progression of these individuals to CLL is still yet to be determined.”
For these reasons, Dr Slager believes “it’s still premature to bring genetic testing into clinical practice.”
Future directions include bringing together the non-environmental issues and the inherited issues to create a model that will accurately predict the risk of CLL.
Androgen improved survival in elderly AML patients
Elderly patients with acute myeloid leukemia (AML) generally have a poor prognosis, but maintenance therapy with an androgen, norethandrolone, significantly improved survival in this population, investigators report in the Journal of Clinical Oncology.
The 5-year disease-free survival was almost double in patients who received norethandrolone (31.2% vs 16.2%), and event-free survival (EFS) and overall survival (OS) were also markedly improved.
A number of hypotheses could explain why norethandrolone improves outcomes in this population. “Because androgen supplementation was initiated when the tumoral mass was decreased, it is possible that norethandrolone decreased the proliferation of remaining blast cells and/or their genetic instability in restoring proper telomere length,” wrote Arnaud Pigneux, MD, PhD, of the Centre Hospitalier Universitaire, Bordeaux (France), and his coauthors. “In addition, as in aplastic anemia, a beneficial effect could be exerted by androgens on normal hematopoietic cells recovering after the end of the postinduction aplastic phase,” they said (J Clin Oncol. 2016 Oct 17. doi: 10.1200/JCO.2016.67.6213)
The majority of patients with AML are older than 60 years of age at the time of their diagnosis, and the prognosis is particularly poor for a number of reasons, including a greater intolerance to intensive chemotherapies and the presence of comorbidities. In addition, older patients also frequently present with unfavorable prognostic features, including multidrug resistant phenotypes or poor-risk cytogenetics.
In this study, Dr. Pigneux and his team enrolled 330 patients with AML de novo or secondary to chemotherapy or radiotherapy, who were 60 years of age or older. Induction therapy of idarubicin 8 mg/m2 on days 1 to 5, cytarabine 100 mg/m2 on days 1 to 7, and lomustine 200 mg/m2 on day 1 was administered.
Those who achieved complete or partial remission received six reinduction courses, alternating idarubicin and cytarabine, and a regimen of methotrexate and mercaptopurine. The cohort was then randomized to receive norethandrolone 10 or 20 mg/day (arm A) according to body weight, or no norethandrolone (arm B) for a 2-year maintenance therapy regimen.
Disease-free survival (DFS) was significantly improved in patients who received androgen therapy, but there was no difference between groups in patients with a high WBC count.
The 5-year DFS was 39.2% for arm A versus 15.1% in arm B for patients with a low WBC count, and 14.3% in arm A versus 20.8% in arm B for patients with a high WBC count.
From the time of inclusion, the 5-year EFS and OS were 21.5% (95% confidence interval, 15.5%-28.1%) and 26.3% (95% CI, 19.7%-33.2%), respectively, in arm A versus 12.9% (95% CI, 8.3%-18.5%) and 17.2% (95% CI, 11.9%-23.4%), respectively, in arm B.
For patients with unfavorable cytogenetics (n = 78), outcomes were better if they received norethandrolone.
The 5-year DFS for this subset was 15.79% in arm A versus 7.69% in arm B. For patients with low- or intermediate-risk cytogenetics, the 5-year DFS was 39.73% in arm A versus 19.72% in arm B.
No funding source was disclosed. Dr. Pigneux has disclosed consulting or advisory roles with Amgen, Celgene, MSD, and Novartis, and disclosed receiving funding for travel and accommodations expenses from Amgen and Pfizer. Several of the authors have disclosed relationships with industry.
Elderly patients with acute myeloid leukemia (AML) generally have a poor prognosis, but maintenance therapy with an androgen, norethandrolone, significantly improved survival in this population, investigators report in the Journal of Clinical Oncology.
The 5-year disease-free survival was almost double in patients who received norethandrolone (31.2% vs 16.2%), and event-free survival (EFS) and overall survival (OS) were also markedly improved.
A number of hypotheses could explain why norethandrolone improves outcomes in this population. “Because androgen supplementation was initiated when the tumoral mass was decreased, it is possible that norethandrolone decreased the proliferation of remaining blast cells and/or their genetic instability in restoring proper telomere length,” wrote Arnaud Pigneux, MD, PhD, of the Centre Hospitalier Universitaire, Bordeaux (France), and his coauthors. “In addition, as in aplastic anemia, a beneficial effect could be exerted by androgens on normal hematopoietic cells recovering after the end of the postinduction aplastic phase,” they said (J Clin Oncol. 2016 Oct 17. doi: 10.1200/JCO.2016.67.6213)
The majority of patients with AML are older than 60 years of age at the time of their diagnosis, and the prognosis is particularly poor for a number of reasons, including a greater intolerance to intensive chemotherapies and the presence of comorbidities. In addition, older patients also frequently present with unfavorable prognostic features, including multidrug resistant phenotypes or poor-risk cytogenetics.
In this study, Dr. Pigneux and his team enrolled 330 patients with AML de novo or secondary to chemotherapy or radiotherapy, who were 60 years of age or older. Induction therapy of idarubicin 8 mg/m2 on days 1 to 5, cytarabine 100 mg/m2 on days 1 to 7, and lomustine 200 mg/m2 on day 1 was administered.
Those who achieved complete or partial remission received six reinduction courses, alternating idarubicin and cytarabine, and a regimen of methotrexate and mercaptopurine. The cohort was then randomized to receive norethandrolone 10 or 20 mg/day (arm A) according to body weight, or no norethandrolone (arm B) for a 2-year maintenance therapy regimen.
Disease-free survival (DFS) was significantly improved in patients who received androgen therapy, but there was no difference between groups in patients with a high WBC count.
The 5-year DFS was 39.2% for arm A versus 15.1% in arm B for patients with a low WBC count, and 14.3% in arm A versus 20.8% in arm B for patients with a high WBC count.
From the time of inclusion, the 5-year EFS and OS were 21.5% (95% confidence interval, 15.5%-28.1%) and 26.3% (95% CI, 19.7%-33.2%), respectively, in arm A versus 12.9% (95% CI, 8.3%-18.5%) and 17.2% (95% CI, 11.9%-23.4%), respectively, in arm B.
For patients with unfavorable cytogenetics (n = 78), outcomes were better if they received norethandrolone.
The 5-year DFS for this subset was 15.79% in arm A versus 7.69% in arm B. For patients with low- or intermediate-risk cytogenetics, the 5-year DFS was 39.73% in arm A versus 19.72% in arm B.
No funding source was disclosed. Dr. Pigneux has disclosed consulting or advisory roles with Amgen, Celgene, MSD, and Novartis, and disclosed receiving funding for travel and accommodations expenses from Amgen and Pfizer. Several of the authors have disclosed relationships with industry.
Elderly patients with acute myeloid leukemia (AML) generally have a poor prognosis, but maintenance therapy with an androgen, norethandrolone, significantly improved survival in this population, investigators report in the Journal of Clinical Oncology.
The 5-year disease-free survival was almost double in patients who received norethandrolone (31.2% vs 16.2%), and event-free survival (EFS) and overall survival (OS) were also markedly improved.
A number of hypotheses could explain why norethandrolone improves outcomes in this population. “Because androgen supplementation was initiated when the tumoral mass was decreased, it is possible that norethandrolone decreased the proliferation of remaining blast cells and/or their genetic instability in restoring proper telomere length,” wrote Arnaud Pigneux, MD, PhD, of the Centre Hospitalier Universitaire, Bordeaux (France), and his coauthors. “In addition, as in aplastic anemia, a beneficial effect could be exerted by androgens on normal hematopoietic cells recovering after the end of the postinduction aplastic phase,” they said (J Clin Oncol. 2016 Oct 17. doi: 10.1200/JCO.2016.67.6213)
The majority of patients with AML are older than 60 years of age at the time of their diagnosis, and the prognosis is particularly poor for a number of reasons, including a greater intolerance to intensive chemotherapies and the presence of comorbidities. In addition, older patients also frequently present with unfavorable prognostic features, including multidrug resistant phenotypes or poor-risk cytogenetics.
In this study, Dr. Pigneux and his team enrolled 330 patients with AML de novo or secondary to chemotherapy or radiotherapy, who were 60 years of age or older. Induction therapy of idarubicin 8 mg/m2 on days 1 to 5, cytarabine 100 mg/m2 on days 1 to 7, and lomustine 200 mg/m2 on day 1 was administered.
Those who achieved complete or partial remission received six reinduction courses, alternating idarubicin and cytarabine, and a regimen of methotrexate and mercaptopurine. The cohort was then randomized to receive norethandrolone 10 or 20 mg/day (arm A) according to body weight, or no norethandrolone (arm B) for a 2-year maintenance therapy regimen.
Disease-free survival (DFS) was significantly improved in patients who received androgen therapy, but there was no difference between groups in patients with a high WBC count.
The 5-year DFS was 39.2% for arm A versus 15.1% in arm B for patients with a low WBC count, and 14.3% in arm A versus 20.8% in arm B for patients with a high WBC count.
From the time of inclusion, the 5-year EFS and OS were 21.5% (95% confidence interval, 15.5%-28.1%) and 26.3% (95% CI, 19.7%-33.2%), respectively, in arm A versus 12.9% (95% CI, 8.3%-18.5%) and 17.2% (95% CI, 11.9%-23.4%), respectively, in arm B.
For patients with unfavorable cytogenetics (n = 78), outcomes were better if they received norethandrolone.
The 5-year DFS for this subset was 15.79% in arm A versus 7.69% in arm B. For patients with low- or intermediate-risk cytogenetics, the 5-year DFS was 39.73% in arm A versus 19.72% in arm B.
No funding source was disclosed. Dr. Pigneux has disclosed consulting or advisory roles with Amgen, Celgene, MSD, and Novartis, and disclosed receiving funding for travel and accommodations expenses from Amgen and Pfizer. Several of the authors have disclosed relationships with industry.
Key clinical point: Maintenance therapy with norethandrolone improved outcomes in elderly patients with acute myeloid leukemia.
Major finding: 5-year disease-free survival was 31.2% (for the norethandrolone group) vs. 16.2% and event-free survival was 21.5% and 12.9%, respectively.
Data source: Prospective, randomized, multicenter, open-label phase III study that included 330 patients with AML.
Disclosures: No funding source was disclosed. Dr. Pigneux has disclosed consulting or advisory roles with Amgen, Celgene, MSD, and Novartis, and disclosed receiving funding for travel and accommodations expenses from Amgen and Pfizer. Several of the authors have also disclosed relationships with industry.
Team identifies genetic hallmarks of B-ALL subtype

Photo courtesy of St. Jude
Children’s Research Hospital
Researchers say they have uncovered a unique paradigm of transcription factor deregulation in B-precursor acute lymphoblastic leukemia (B-ALL).
The team found that deregulation of the homeobox transcription factor gene DUX4 and the ETS transcription factor gene ERG is a hallmark of a subtype of B-ALL that may comprise up to 8% of B-ALL cases.
The researchers reported these findings in Nature Genetics.
“Our work is motivated by a lack of information on the genetic basis of many B-ALL cases,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We discovered a distinct gene pattern in blood samples from some patients in our study and wanted to determine the underlying molecular events behind this signal.”
The researchers studied a group of 1913 B-ALL patients (including children, adolescents, and young adults) to understand the genetic basis of the disease.
Microarray and transcriptome sequencing revealed that 7.6% of these patients had the distinctive genetic profile the researchers wanted to characterize further.
“Our work revealed that, in this type of B-ALL, there is a sequence of molecular events that involves the interplay of 2 transcription factors,” Dr Mullighan said.
The team observed rearrangement of the gene DUX4 in all cases of this subtype of B-ALL, which resulted in high-level expression of DUX4. DUX4 was shown to bind to the ERG gene, leading to deregulated expression of ERG.
The deregulation of ERG compromised the function of ERG either by deleting part of the gene or by expressing another form of ERG—ERGalt. In both cases, loss of activity was observed for the ERG transcription factor, which led to leukemia.
“These results underscore that there is still more to be learned about the genetic changes in ALL, and that this knowledge can help refine treatment for patients,” said study author Stephen Hunger, MD, of the Children’s Hospital of Philadelphia in Pennsylvania.
The researchers hope identification of the relationships between the 2 transcription factors will lead to new diagnostic tests for patients. DUX4/ERG ALL is linked to favorable outcomes even when other detrimental genetic mutations are present.
Currently, only transcriptome or genome sequencing helps identify the DUX4 rearrangements. The researchers say other detection methods, such as fluorescence hybridization or karyotyping, are not sufficient to recognize genetic changes to DUX4.
Photo courtesy of St. Jude
Children’s Research Hospital
Researchers say they have uncovered a unique paradigm of transcription factor deregulation in B-precursor acute lymphoblastic leukemia (B-ALL).
The team found that deregulation of the homeobox transcription factor gene DUX4 and the ETS transcription factor gene ERG is a hallmark of a subtype of B-ALL that may comprise up to 8% of B-ALL cases.
The researchers reported these findings in Nature Genetics.
“Our work is motivated by a lack of information on the genetic basis of many B-ALL cases,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We discovered a distinct gene pattern in blood samples from some patients in our study and wanted to determine the underlying molecular events behind this signal.”
The researchers studied a group of 1913 B-ALL patients (including children, adolescents, and young adults) to understand the genetic basis of the disease.
Microarray and transcriptome sequencing revealed that 7.6% of these patients had the distinctive genetic profile the researchers wanted to characterize further.
“Our work revealed that, in this type of B-ALL, there is a sequence of molecular events that involves the interplay of 2 transcription factors,” Dr Mullighan said.
The team observed rearrangement of the gene DUX4 in all cases of this subtype of B-ALL, which resulted in high-level expression of DUX4. DUX4 was shown to bind to the ERG gene, leading to deregulated expression of ERG.
The deregulation of ERG compromised the function of ERG either by deleting part of the gene or by expressing another form of ERG—ERGalt. In both cases, loss of activity was observed for the ERG transcription factor, which led to leukemia.
“These results underscore that there is still more to be learned about the genetic changes in ALL, and that this knowledge can help refine treatment for patients,” said study author Stephen Hunger, MD, of the Children’s Hospital of Philadelphia in Pennsylvania.
The researchers hope identification of the relationships between the 2 transcription factors will lead to new diagnostic tests for patients. DUX4/ERG ALL is linked to favorable outcomes even when other detrimental genetic mutations are present.
Currently, only transcriptome or genome sequencing helps identify the DUX4 rearrangements. The researchers say other detection methods, such as fluorescence hybridization or karyotyping, are not sufficient to recognize genetic changes to DUX4.
Photo courtesy of St. Jude
Children’s Research Hospital
Researchers say they have uncovered a unique paradigm of transcription factor deregulation in B-precursor acute lymphoblastic leukemia (B-ALL).
The team found that deregulation of the homeobox transcription factor gene DUX4 and the ETS transcription factor gene ERG is a hallmark of a subtype of B-ALL that may comprise up to 8% of B-ALL cases.
The researchers reported these findings in Nature Genetics.
“Our work is motivated by a lack of information on the genetic basis of many B-ALL cases,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We discovered a distinct gene pattern in blood samples from some patients in our study and wanted to determine the underlying molecular events behind this signal.”
The researchers studied a group of 1913 B-ALL patients (including children, adolescents, and young adults) to understand the genetic basis of the disease.
Microarray and transcriptome sequencing revealed that 7.6% of these patients had the distinctive genetic profile the researchers wanted to characterize further.
“Our work revealed that, in this type of B-ALL, there is a sequence of molecular events that involves the interplay of 2 transcription factors,” Dr Mullighan said.
The team observed rearrangement of the gene DUX4 in all cases of this subtype of B-ALL, which resulted in high-level expression of DUX4. DUX4 was shown to bind to the ERG gene, leading to deregulated expression of ERG.
The deregulation of ERG compromised the function of ERG either by deleting part of the gene or by expressing another form of ERG—ERGalt. In both cases, loss of activity was observed for the ERG transcription factor, which led to leukemia.
“These results underscore that there is still more to be learned about the genetic changes in ALL, and that this knowledge can help refine treatment for patients,” said study author Stephen Hunger, MD, of the Children’s Hospital of Philadelphia in Pennsylvania.
The researchers hope identification of the relationships between the 2 transcription factors will lead to new diagnostic tests for patients. DUX4/ERG ALL is linked to favorable outcomes even when other detrimental genetic mutations are present.
Currently, only transcriptome or genome sequencing helps identify the DUX4 rearrangements. The researchers say other detection methods, such as fluorescence hybridization or karyotyping, are not sufficient to recognize genetic changes to DUX4.
Compound could treat a range of blood cancers

Image by Ed Uthman
A new compound has shown promise for treating hematologic malignancies and other cancers, according to preclinical research published in Nature.
The compound, known as S63845, targets the BCL2 family protein MCL1.
Investigators said their research on S63845 provides the first clear evidence that inhibiting MCL1 is effective in targeting several cancer types, including leukemia, lymphoma, and multiple myeloma (MM).
“MCL1 is important for many cancers because it is a pro-survival protein that allows the cancerous cells to evade the process of programmed cell death that normally removes cancer cells from the body,” said study author Guillaume Lessene, PhD, of the Walter and Eliza Hall Institute in Melbourne, Australia.
“Extensive studies performed in a variety of cancer models have shown that S63845 potently targets cancer cells dependent on MCL1 for their survival.”
About S63845
Dr Lessene and his colleagues said S63845 binds with high affinity to the BH3-binding groove of MCL1. And the compound kills MCL1-dependent cancer cells by activating the BAX/BAK-dependent mitochondrial apoptotic pathway.
In solid tumors, S63845 often wasn’t effective enough on its own. However, when the compound was combined with various kinase inhibitors, it induced a “potent cytotoxic response” in breast cancer, lung cancer, and melanoma cells.
In hematologic malignancies, S63845 proved effective when given alone.
Myeloma
The investigators said 17 of 25 MM cell lines tested were highly sensitive to S63845 (IC50 < 0.1 μ M), 6 cell lines were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 2 cell lines were insensitive (IC50 > 1 μ M).
The team also administered S63845 (at 25 mg/kg) to mice with MM. Seven of 8 mice had complete regression at 100 days after treatment.
Lymphoma
The investigators tested S63845 in 11 cell lines representative of human lymphomas. Five were highly sensitive to the compound (IC50 < 0.1 μ M), 3 were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 3 were insensitive (IC50 > 1 μ M).
The team also tested S63845 in 7 c-MYC-driven human Burkitt lymphoma cell lines and found the compound exhibited “potent cytotoxic activity” in all of them.
The investigators then tested S63845 in a c-MYC-driven mouse lymphoma model. They noted that both tumor cells and normal tissues express mouse MCL1 protein in this model.
Treatment with S63845 (25 mg/kg) for 5 consecutive days cured 70% of these mice, and the investigators said there were no evident side effects in normal tissues.
Leukemia
The investigators tested S63845 in 5 chronic myeloid leukemia cell lines, and none of them were sensitive to the compound.
However, the team also tested S63845 in 8 acute myeloid leukemia (AML) cell lines, and all of them were sensitive to the compound (IC50 4–233 nM).
When S63845 was given to mice with AML (25 mg/kg), 6 of the 8 mice achieved complete remission after 80 days.
The investigators also tested S63845 in 25 freshly derived samples from patients with AML. The team said these samples displayed a wide range of responses to S63845.
The most sensitive samples required 100- to 1000-fold less drug (to induce apoptosis) than the resistant samples or normal CD34+ progenitor cells.
Development/funding
S63845 was discovered through a collaboration between 2 pharmaceutical companies—Servier, which is headquartered in France, and Vernalis (R&D), which is based in the UK.
“[C]linical development of a MCL1 inhibitor should be launched in the near future,” said Olivier Geneste, director of oncology research at Servier.
The current research was supported through a collaboration with Servier and through funding from the National Health and Medical Research Council of Australia, the Leukemia and Lymphoma Society, Cancer Council Victoria, the Kay Kendall Leukemia Fund, Victorian Cancer Agency, Australian Cancer Research Foundation, the Victorian Government Operational Infrastructure Scheme, and the estate of Anthony Redstone.
Image by Ed Uthman
A new compound has shown promise for treating hematologic malignancies and other cancers, according to preclinical research published in Nature.
The compound, known as S63845, targets the BCL2 family protein MCL1.
Investigators said their research on S63845 provides the first clear evidence that inhibiting MCL1 is effective in targeting several cancer types, including leukemia, lymphoma, and multiple myeloma (MM).
“MCL1 is important for many cancers because it is a pro-survival protein that allows the cancerous cells to evade the process of programmed cell death that normally removes cancer cells from the body,” said study author Guillaume Lessene, PhD, of the Walter and Eliza Hall Institute in Melbourne, Australia.
“Extensive studies performed in a variety of cancer models have shown that S63845 potently targets cancer cells dependent on MCL1 for their survival.”
About S63845
Dr Lessene and his colleagues said S63845 binds with high affinity to the BH3-binding groove of MCL1. And the compound kills MCL1-dependent cancer cells by activating the BAX/BAK-dependent mitochondrial apoptotic pathway.
In solid tumors, S63845 often wasn’t effective enough on its own. However, when the compound was combined with various kinase inhibitors, it induced a “potent cytotoxic response” in breast cancer, lung cancer, and melanoma cells.
In hematologic malignancies, S63845 proved effective when given alone.
Myeloma
The investigators said 17 of 25 MM cell lines tested were highly sensitive to S63845 (IC50 < 0.1 μ M), 6 cell lines were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 2 cell lines were insensitive (IC50 > 1 μ M).
The team also administered S63845 (at 25 mg/kg) to mice with MM. Seven of 8 mice had complete regression at 100 days after treatment.
Lymphoma
The investigators tested S63845 in 11 cell lines representative of human lymphomas. Five were highly sensitive to the compound (IC50 < 0.1 μ M), 3 were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 3 were insensitive (IC50 > 1 μ M).
The team also tested S63845 in 7 c-MYC-driven human Burkitt lymphoma cell lines and found the compound exhibited “potent cytotoxic activity” in all of them.
The investigators then tested S63845 in a c-MYC-driven mouse lymphoma model. They noted that both tumor cells and normal tissues express mouse MCL1 protein in this model.
Treatment with S63845 (25 mg/kg) for 5 consecutive days cured 70% of these mice, and the investigators said there were no evident side effects in normal tissues.
Leukemia
The investigators tested S63845 in 5 chronic myeloid leukemia cell lines, and none of them were sensitive to the compound.
However, the team also tested S63845 in 8 acute myeloid leukemia (AML) cell lines, and all of them were sensitive to the compound (IC50 4–233 nM).
When S63845 was given to mice with AML (25 mg/kg), 6 of the 8 mice achieved complete remission after 80 days.
The investigators also tested S63845 in 25 freshly derived samples from patients with AML. The team said these samples displayed a wide range of responses to S63845.
The most sensitive samples required 100- to 1000-fold less drug (to induce apoptosis) than the resistant samples or normal CD34+ progenitor cells.
Development/funding
S63845 was discovered through a collaboration between 2 pharmaceutical companies—Servier, which is headquartered in France, and Vernalis (R&D), which is based in the UK.
“[C]linical development of a MCL1 inhibitor should be launched in the near future,” said Olivier Geneste, director of oncology research at Servier.
The current research was supported through a collaboration with Servier and through funding from the National Health and Medical Research Council of Australia, the Leukemia and Lymphoma Society, Cancer Council Victoria, the Kay Kendall Leukemia Fund, Victorian Cancer Agency, Australian Cancer Research Foundation, the Victorian Government Operational Infrastructure Scheme, and the estate of Anthony Redstone.
Image by Ed Uthman
A new compound has shown promise for treating hematologic malignancies and other cancers, according to preclinical research published in Nature.
The compound, known as S63845, targets the BCL2 family protein MCL1.
Investigators said their research on S63845 provides the first clear evidence that inhibiting MCL1 is effective in targeting several cancer types, including leukemia, lymphoma, and multiple myeloma (MM).
“MCL1 is important for many cancers because it is a pro-survival protein that allows the cancerous cells to evade the process of programmed cell death that normally removes cancer cells from the body,” said study author Guillaume Lessene, PhD, of the Walter and Eliza Hall Institute in Melbourne, Australia.
“Extensive studies performed in a variety of cancer models have shown that S63845 potently targets cancer cells dependent on MCL1 for their survival.”
About S63845
Dr Lessene and his colleagues said S63845 binds with high affinity to the BH3-binding groove of MCL1. And the compound kills MCL1-dependent cancer cells by activating the BAX/BAK-dependent mitochondrial apoptotic pathway.
In solid tumors, S63845 often wasn’t effective enough on its own. However, when the compound was combined with various kinase inhibitors, it induced a “potent cytotoxic response” in breast cancer, lung cancer, and melanoma cells.
In hematologic malignancies, S63845 proved effective when given alone.
Myeloma
The investigators said 17 of 25 MM cell lines tested were highly sensitive to S63845 (IC50 < 0.1 μ M), 6 cell lines were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 2 cell lines were insensitive (IC50 > 1 μ M).
The team also administered S63845 (at 25 mg/kg) to mice with MM. Seven of 8 mice had complete regression at 100 days after treatment.
Lymphoma
The investigators tested S63845 in 11 cell lines representative of human lymphomas. Five were highly sensitive to the compound (IC50 < 0.1 μ M), 3 were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 3 were insensitive (IC50 > 1 μ M).
The team also tested S63845 in 7 c-MYC-driven human Burkitt lymphoma cell lines and found the compound exhibited “potent cytotoxic activity” in all of them.
The investigators then tested S63845 in a c-MYC-driven mouse lymphoma model. They noted that both tumor cells and normal tissues express mouse MCL1 protein in this model.
Treatment with S63845 (25 mg/kg) for 5 consecutive days cured 70% of these mice, and the investigators said there were no evident side effects in normal tissues.
Leukemia
The investigators tested S63845 in 5 chronic myeloid leukemia cell lines, and none of them were sensitive to the compound.
However, the team also tested S63845 in 8 acute myeloid leukemia (AML) cell lines, and all of them were sensitive to the compound (IC50 4–233 nM).
When S63845 was given to mice with AML (25 mg/kg), 6 of the 8 mice achieved complete remission after 80 days.
The investigators also tested S63845 in 25 freshly derived samples from patients with AML. The team said these samples displayed a wide range of responses to S63845.
The most sensitive samples required 100- to 1000-fold less drug (to induce apoptosis) than the resistant samples or normal CD34+ progenitor cells.
Development/funding
S63845 was discovered through a collaboration between 2 pharmaceutical companies—Servier, which is headquartered in France, and Vernalis (R&D), which is based in the UK.
“[C]linical development of a MCL1 inhibitor should be launched in the near future,” said Olivier Geneste, director of oncology research at Servier.
The current research was supported through a collaboration with Servier and through funding from the National Health and Medical Research Council of Australia, the Leukemia and Lymphoma Society, Cancer Council Victoria, the Kay Kendall Leukemia Fund, Victorian Cancer Agency, Australian Cancer Research Foundation, the Victorian Government Operational Infrastructure Scheme, and the estate of Anthony Redstone.