User login
Molecule is active against MYC-driven malignancies

Credit: Ed Uthman
A small molecule can disrupt the interactions between MYC and its binding partner MAX in MYC-driven cancers, according to research published in PNAS.
The molecule, KJ-Pyr-9, inhibited MYC-induced oncogenic transformation in cell culture but had little to no effect on the oncogenic activity of several unrelated oncoproteins.
KJ-Pyr-9 preferentially interfered with proliferation in a range of cells that overexpressed MYC, including leukemia and lymphoma cells.
In vivo, the molecule inhibited the growth of MYC-amplified human cancer cells.
“We finally hit a home run with this—maybe a grand slam,” said study author Kim Janda, PhD, of The Scripps Research Institute in La Jolla, California.
For years, MYC has challenged researchers seeking to disrupt its activity in cancer cells.
“At room temperature or body temperature, MYC without any binding partners is random and constantly shifting,” said study author Jonathan Ross Hart, PhD, also of The Scripps Research Institute. “It’s like a piece of spaghetti.”
So instead of designing a compound to target the structure of MYC, the researchers tested a range of compounds from a library to see if any could disrupt the interactions between MYC and other proteins important in cell proliferation. One did—the small molecule KJ-Pyr-9.
To further investigate, the researchers ran tests in a variety of cell lines, including chronic myeloid leukemia, acute lymphoblastic leukemia, acute myeloid leukemia, Burkitt lymphoma, and solid tumors. And they tested KJ-Pyr-9 in mouse models of breast cancer.
The experiments showed that MYC-dependent cells die if treated with KJ-Pyr-9. In fact, a dose of KJ-Pyr-9 made it seem as if MYC was not present at all.
When mice with MYC-dependent tumors received KJ-Pyr-9, the tumors showed no growth after 31 days, compared with significant tumor growth in untreated mice.
Dr Janda said he hopes further research will reveal exactly how KJ-Pyr-9 interacts with MYC and how the compound can more effectively reach tumor cells. ![]()
Credit: Ed Uthman
A small molecule can disrupt the interactions between MYC and its binding partner MAX in MYC-driven cancers, according to research published in PNAS.
The molecule, KJ-Pyr-9, inhibited MYC-induced oncogenic transformation in cell culture but had little to no effect on the oncogenic activity of several unrelated oncoproteins.
KJ-Pyr-9 preferentially interfered with proliferation in a range of cells that overexpressed MYC, including leukemia and lymphoma cells.
In vivo, the molecule inhibited the growth of MYC-amplified human cancer cells.
“We finally hit a home run with this—maybe a grand slam,” said study author Kim Janda, PhD, of The Scripps Research Institute in La Jolla, California.
For years, MYC has challenged researchers seeking to disrupt its activity in cancer cells.
“At room temperature or body temperature, MYC without any binding partners is random and constantly shifting,” said study author Jonathan Ross Hart, PhD, also of The Scripps Research Institute. “It’s like a piece of spaghetti.”
So instead of designing a compound to target the structure of MYC, the researchers tested a range of compounds from a library to see if any could disrupt the interactions between MYC and other proteins important in cell proliferation. One did—the small molecule KJ-Pyr-9.
To further investigate, the researchers ran tests in a variety of cell lines, including chronic myeloid leukemia, acute lymphoblastic leukemia, acute myeloid leukemia, Burkitt lymphoma, and solid tumors. And they tested KJ-Pyr-9 in mouse models of breast cancer.
The experiments showed that MYC-dependent cells die if treated with KJ-Pyr-9. In fact, a dose of KJ-Pyr-9 made it seem as if MYC was not present at all.
When mice with MYC-dependent tumors received KJ-Pyr-9, the tumors showed no growth after 31 days, compared with significant tumor growth in untreated mice.
Dr Janda said he hopes further research will reveal exactly how KJ-Pyr-9 interacts with MYC and how the compound can more effectively reach tumor cells. ![]()
Credit: Ed Uthman
A small molecule can disrupt the interactions between MYC and its binding partner MAX in MYC-driven cancers, according to research published in PNAS.
The molecule, KJ-Pyr-9, inhibited MYC-induced oncogenic transformation in cell culture but had little to no effect on the oncogenic activity of several unrelated oncoproteins.
KJ-Pyr-9 preferentially interfered with proliferation in a range of cells that overexpressed MYC, including leukemia and lymphoma cells.
In vivo, the molecule inhibited the growth of MYC-amplified human cancer cells.
“We finally hit a home run with this—maybe a grand slam,” said study author Kim Janda, PhD, of The Scripps Research Institute in La Jolla, California.
For years, MYC has challenged researchers seeking to disrupt its activity in cancer cells.
“At room temperature or body temperature, MYC without any binding partners is random and constantly shifting,” said study author Jonathan Ross Hart, PhD, also of The Scripps Research Institute. “It’s like a piece of spaghetti.”
So instead of designing a compound to target the structure of MYC, the researchers tested a range of compounds from a library to see if any could disrupt the interactions between MYC and other proteins important in cell proliferation. One did—the small molecule KJ-Pyr-9.
To further investigate, the researchers ran tests in a variety of cell lines, including chronic myeloid leukemia, acute lymphoblastic leukemia, acute myeloid leukemia, Burkitt lymphoma, and solid tumors. And they tested KJ-Pyr-9 in mouse models of breast cancer.
The experiments showed that MYC-dependent cells die if treated with KJ-Pyr-9. In fact, a dose of KJ-Pyr-9 made it seem as if MYC was not present at all.
When mice with MYC-dependent tumors received KJ-Pyr-9, the tumors showed no growth after 31 days, compared with significant tumor growth in untreated mice.
Dr Janda said he hopes further research will reveal exactly how KJ-Pyr-9 interacts with MYC and how the compound can more effectively reach tumor cells. ![]()
Signaling Pathways and Novel Inhibitors in Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia (CLL) is a common hematological malignancy in the U.S. with 15,000 new patients diagnosed each year.1 This leukemia is frequently diagnosed in veterans since it is more commonly seen in an elderly male population. The disease is characterized by a slow accumulation of mature B cells that are functionally incompetent and resist apoptosis. CLL has an indolent clinical course, but about 60% to 70% of patients require treatment. The disease also runs a variable course, and a number of genetic abnormalities and prognostic markers have been defined to subclassify CLL patients and prognosticate.2-4 This article reviews important CLL signaling pathways and novel therapeutic agents in this leukemia.
Signaling Pathways
B-Cell Receptor Signaling
The B-cell receptor (BCR) signaling is the major signaling pathway in CLL, because it defines clinical, biologic, and prognostic characteristics of the disease.5 The BCR is composed of a surface transmembrane immunoglobulin that binds the antigen with CD79 alpha and beta chains. The activation of BCR results in the formation of a signaling complex or signalosome, which includes Lyn, Syk, BTK, and ZAP-70, among other components that assemble with other adaptor proteins (Figure). This assembly of proteins occurs on the cytoplasmic tails of immunoglobulin chains on regions called immunoreceptor tyrosine-based motifs (ITAMs).
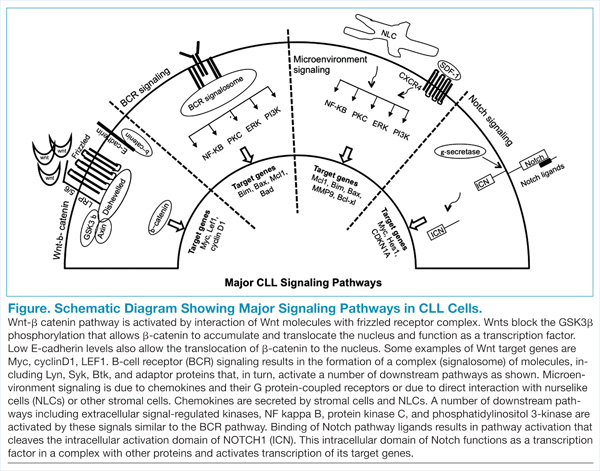
With the assembly of this signaling complex, BCR stimulates a number of downstream pathways, such as phosphatidylinositol 3-kinase (PI3K), protein kinase B (Akt), protein kinase C, nuclear factor-κB (NFκB), and extracellular signal-regulated kinases (ERKs) (Figure). Activation of these pathways results in cell proliferation, resistance to apoptosis, increased cell motility and migration. Recent studies have identified additional novel components of this signaling complex, including a guanine nucleotide exchange factor (GEF) RASGRF1. This GEF is activated by BCR signaling and, in turn, stimulates the ERK pathway by increasing the production of active GTP-bound Ras.6
The ability of BCR to activate a number of downstream signaling pathways makes it a highly relevant and investigated pathway in this leukemia. Inhibitors have been developed and/or identified against a number of signalosome components to block the BCR signaling.7 Syk and Lyn are Src kinases, and their phosphorylation is one of the initial events of BCR signaling. Syk is overexpressed in CLL specimens, and Syk inhibitors (R406 and P505-15, also known as PRT062607) have shown activity in CLL.8,9 Dasatinib is a Src inhibitor that also shows activity in CLL specimens and is being studied in combination with chemotherapy drugs in refractory CLL patients.10
BTK, a component of the BCR signalosome, is required for BCR function, and loss of its function is seen in X-linked agammaglobulinemia. PCI-32765 (ibrutinib) is an oral BTK inhibitor that irreversibly inactivates this kinase and has been approved for clinical use in CLL patients.11,12 Another signaling pathway activated by BCR is the PI3K, and a promising inhibitor (CAL-101) blocks its activity in CLL specimens.13 Investigative work has identified that the delta isoform of PI3K p110 is highly expressed in B cells and lymphocytes.14 This is a catalytic subunit of a class I PI3K with a role in BCR signaling. A selective inhibitor GS-1101 (CAL-101) is able to block PI3K signaling in CLL specimens and inhibits Akt phosphorylation and other downstream effectors along with induction of apoptosis.15 The clinical data with BTK and PI3K inhibitors will be discussed later in this review.
CLL and the Microenvironment
Interactions between CLL cells and the microenvironment allow CLL cells to thrive in certain niche environments.16,17 Interaction mainly occurs via bone marrow stromal cells and nurselike cells (NLCs), which evolve from monocytes (Figure). These interactions can be divided into 2 groups. First, CLL cell growth is supported by a number of chemokine receptor-ligand interactions. CXCR4 is the receptor for CXCL12 (SDF-1) that stimulates chemotaxis and tissue homing. Another chemokine is CXCL13, which acts via its receptor CXCR5 and is involved in chemotaxis and activation of other kinases. Second, NLCs also support CLL cells by expressing TNF family members BAFF and APRIL, which interact with their receptors and activate the NFκB pathway.
Leukemic cells also express VLA-4 integrins, which further their support adhesion to the stromal cells and predict for an aggressive phenotype. Specific inhibitors that block the stimulation by chemokines and cytokines are not yet available; however, one can envision that this class of inhibitors will decrease the chemoresistance of leukemic cells and will be used in conjunction with other chemotherapy agents. Interestingly, inhibitors that block BCR-mediated signaling (BTK and PI3K inhibitors) also inhibit signaling via the microenvironment and chemokines.
Wnt-β-catenin Pathway
Wnt signaling affects developmental pathways, and its aberrant activation has major oncogenic effects as well. This pathway is activated in CLL as these leukemic cells express high levels of Wnt and frizzled along with epigenetic downregulation of Wnt pathway antagonist genes, including secreted frizzled-related protein (SFRP) family members and WIF1 (Figure).18-20 The binding of Wnts to their cognate receptors results in inhibition of GSK3β phosphorylation and stabilization of β-catenin, which then translocates to the nucleus and interacts with lymphoid-enhancing (LEF) and T-cell transcription factors to activate transcription of Wnt-target genes. Lack of E-cadherin expression in CLL cells also results in an increase in translocation of β-catenin and upregulation of the Wnt pathway.20
Wnt-target genes include Myc, LEF, cyclinD1, COX-2, and MMP. Gene expression profiling from our laboratory and other groups have identified the overexpression of these wnt-target genes and support this pathway activation in CLL cells.20 This is a promising signalling pathway and an active area of research for developing inhibitors that will have a growth inhibitory effect on CLL leukemic cells. GSK3b inhibitors and other drugs that re-express epigenetically silenced Wnt antagonist genes have been shown to inhibit this pathway activity in CLL cells in vitro.
Notch Pathway Activation
High-throughput exome sequencing has identified recurring mutations in a number of genes, including NOTCH1.21 Analysis of additional CLL patients confirmed activating NOTCH1 mutations in 10% to 15% of CLL patients and were also associated with poor outcome.22 This pathway is activated by ligands such as Jagged and Delta-like, which interact with the Notch receptor, which is then cleaved by γ-secretases. The cleaved intracellular domain of the NOTCH1 receptor in combination with other factors activates transcription of target genes, including Myc and HES1 (Figure). Besides the mutations that generate a truncated protein or may stabilize the pathway, the Notch pathway is also constitutively active in CLL specimens.23 Notch stimulation increases activity of prosurvival pathways and genes such as NFκB that resist apoptotic signals. The pathway can be inhibited by γ-secretase inhibitors (GSIs), which reduce the levels of cleaved NOTCH1 protein and downregulated Notch target genes. This pathway is also able to modulate the microenvironment stimuli as the GSIs inhibit responses to chemokines such as CXCL12 and inhibit migration and invasion.24
Newer Theraputic Agents
Work on signaling mechanisms paid dividends in CLL with the recent development of 2 inhibitors. Ibrutinib (BTK inhibitor) and idelalisib (PI3K inhibitor) are being studied in clinical trials, and both drugs block the BCR and microenvironment signaling pathways, thereby inhibiting the growth of CLL cells.
BTK Inhibitor: Ibrutinib
The activity of BTK is critical for a number of CLL signaling pathways, and it is a component of the initial signaling complex or signalosome that is formed with BCR signaling. Studies have shown that inhibiting this kinase blocks a number of pathways, including ERK, NFκB, and others. The drug ibrutinib blocks this kinase by forming a covalent bond and inhibiting its enzyme activity. This orally bioavailable drug showed activity in phase 1 trials in different B-cell malignancies.25 In a phase 2 study, high-risk CLL patients were given 2 different doses of this inhibitor, and the overall response rate was 71% with an overall survival at 26 months of 83%.11 Responses were seen in all patients irrespective of clinical and genetic risk factors. Based on these findings, the drug was approved for clinical use in patients with relapsed or refractory disease. Recently, there are data on the use of this drug as frontline therapy in elderly patients, and the drug was well tolerated.26 There are additional ongoing trials to compare this drug with other agents, including chlorambucil (in chemotherapy-naïve patients) and ofatumumab (in relapsed or refractory patients).
PI3 Kinase p110 Delta Inhibitor: Idelalisib
The crucial finding for the development of this inhibitor was the over-expression of the delta isoform of PI3K p110 in B-cell malignancies.14 The drug CAL-101 selectively inhibits this constitutively active isoform and induces apoptosis in a number of B-cell malignancies.15,27 In the phase 1 trial, this inhibitor was evaluated in relapsed/refractory patients at multiple dose levels.28 There was inhibition of PI3K signaling with an overall response rate of 72%, and a partial response rate of 39% was observed in CLL patients. This was followed by a randomized, placebo-controlled phase 3 study in which patients with myelosuppression, decreased renal function, or other illnesses were treated with either rituximab alone or with rituximab and idelalisib.29
At the time of reporting, the median progression-free survival (PFS) was 5.5 months in the placebo arm and was not reached in the idelalisib arm. Overall response rates were higher in the idelalisib group (81% vs 13%) with similar toxicity profiles in the 2 groups. This drug is now being extensively studied in combination with bendamustine and other anti-CD20 antibodies in clinical trials.
A unique toxicity observed with both these inhibitors is the initial lymphocytosis. In the case of ibrutinib, this was seen in a majority of patients (77%) and at the same time there was a response in the nodal disease, implying a redistribution of leukemic cells from the tissues to the peripheral blood.30
A potential explanation is that these drugs inhibit signaling via chemokines and other components of the microenvironment and by inhibiting the homing signals, allows leukemic cells to move out of their niche areas. This was analyzed in a recent study that compared clinical and biochemical parameters of patients who had a complete or partial response with ibrutinib compared with a “partial response except for lymphocytosis.”30 Patients with “partial response except for lymphocytosis” were found to have favorable prognostic factors, and the persisting leukemic cells were not clonally different from the original cells. The progression free survival of patients with “partial response except for lymphocytosis” was also similar to the subgroup with no prolonged lymphocytosis.
Discussion
Several therapeutic agents with novel mechanisms of action are effective in killing the CLL leukemic cells, and a number of targeted agents are currently in the pipeline. The next challenge for treating CLL will be the proper integration of these novel targeted agents with the traditional chemotherapy and chemoimmunotherapy approaches. Let us consider CLL patients in different clinical settings. First, a patient aged 60 years who is otherwise healthy will be treated with possibly all the available chemotherapy and chemoimmunotherapy options, as well as the newer targeted agents. In this clinical setting sequencing of therapy is not a major concern. On the other hand, a patient aged 70 years who is already refractory to multiple lines of therapy is a good candidate for these newer drugs.
The more controversial use of these targeted agents will be in an older patient with some comorbidities and newly diagnosed CLL. In this clinical setting, should one go with traditional chemotherapy/chemoimmunotherapy approaches or consider newer targeted agents? These issues are now being addressed in clinical trials, and with acceptable toxicity profiles these newer drugs will move to the frontline setting.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect an endorsement by or opinion of Federal Practitioner, Frontline Medical Communications, the U.S. Air Force, the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drug combinations–including indications, contraindications, warnings, and adverse effects–before administering pharmacologic therapy to patients.
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9-29.
2. Döhner H, Stilgenbauer S, Döhner K, Bentz M, Lichter P. Chromosome aberrations in B-cell chronic lymphocytic leukemia: reassessment based on molecular cytogenetic analysis. J Mol Med. 1999;77(2):266-281.
3. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848-1854.
4. Chen L, Widhopf G, Huynh L, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2002;100(13):4609-4614.
5. Wickremasinghe RG, Prentice AG, Steele AJ. Aberrantly activated anti-apoptotic signalling mechanisms in chronic lymphocytic leukaemia cells: Clues to the identification of novel therapeutic targets. Br J Haematol. 2011;153(5):545-556.
6. Liao W, Jordaan G, Coriaty N, Sharma S. Amplification of B cell receptor-Erk signaling by Rasgrf-1 overexpression in chronic lymphocytic leukemia [published online ahead of print April 2, 2014]. Leuk Lymphoma. doi: 10.3109/10428194.2014898759.
7. Burger JA. Inhibiting B-cell receptor signaling pathways in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2012;7(1):26-33.
8. Buchner M, Fuchs S, Prinz G, et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009;69(13):5424-5432.
9. Spurgeon SE, Coffey G, Fletcher LB, et al. The selective SYK inhibitor P505-15 (PRT062607) inhibits B cell signaling and function in vitro and in vivo and augments the activity of fludarabine in chronic lymphocytic leukemia. J Pharmacol Exp Ther. 2013;344(2):378-387.
10. Veldurthy A, Patz M, Hagist S, et al. The kinase inhibitor dasatinib induces apoptosis in chronic lymphocytic leukemia cells in vitro with preference for a subgroup of patients with unmutated IgVH genes. Blood. 2008;112(4):1443-1452.
11. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia [published correction appears in N Engl J Med. 2014;370(8):786]. N Engl J Med. 2013;369(1):32-42.
12. Cheng S, Ma J, Guo A, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia. 2014;28(3):649-657.
13. Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591-594.
14. Chantry D, Vojtek A, Kashishian A, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997;272(31):19236-19241.
15. Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3’-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603-3612.
16. Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood. 2009;114(16):3367-3375.
17. ten Hacken E, Burger JA. Molecular pathways: targeting the microenvironment in chronic lymphocytic leukemia—focus on the B-cell receptor. Clin Cancer Res. 2014;20(3):548-556.
18. Gandhirajan RK, Poll-Wolbeck SJ, Gehrke I, Kreuzer KA. Wnt/b-catenin/LEF-1 signaling in chronic lymphocytic leukemia (CLL): a target for current and potential therapeutic options. Curr Cancer Drug Targets. 2010;10(7):716-727.
19. Gutierrez A, Jr, Tschumper RC, Wu X, et al. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood. 2010;116(16):2975-2983.
20. Jordaan G, Liao W, Sharma S. E-cadherin gene re-expression in chronic lymphocytic leukemia cells by HDAC inhibitors. BMC Cancer. 2013;13:88.
21. Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101-105.
22. Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389-1401.
23. Rosati E, Sabatini R, Rampino G, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113(4):856-865.
24. López-Guerra M, Xargay-Torrent S, Rosich L, et al. The g-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells [published online ahead of print April 30, 2014]. Leukemia. doi: 10.1038/leu.2014.143.
25. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88-94.
26. O’Brien S, Furman RR, Coutre SE, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: An open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15(1):48-58.
27. Brown JR, Byrd JC, Coutre SE, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110∂, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390-3397.
28. Brown JR, Furman RR, Flinn I, et al. Final results of a phase I study of idelalisib (GS-1101) a selective inhibitor of PI3K∂, in patients with relapsed or refractory CLL. J Clin Oncol. 2013;31:Absract 7003.
29. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007.
30. Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2013;123(12):1810-1817.
Chronic lymphocytic leukemia (CLL) is a common hematological malignancy in the U.S. with 15,000 new patients diagnosed each year.1 This leukemia is frequently diagnosed in veterans since it is more commonly seen in an elderly male population. The disease is characterized by a slow accumulation of mature B cells that are functionally incompetent and resist apoptosis. CLL has an indolent clinical course, but about 60% to 70% of patients require treatment. The disease also runs a variable course, and a number of genetic abnormalities and prognostic markers have been defined to subclassify CLL patients and prognosticate.2-4 This article reviews important CLL signaling pathways and novel therapeutic agents in this leukemia.
Signaling Pathways
B-Cell Receptor Signaling
The B-cell receptor (BCR) signaling is the major signaling pathway in CLL, because it defines clinical, biologic, and prognostic characteristics of the disease.5 The BCR is composed of a surface transmembrane immunoglobulin that binds the antigen with CD79 alpha and beta chains. The activation of BCR results in the formation of a signaling complex or signalosome, which includes Lyn, Syk, BTK, and ZAP-70, among other components that assemble with other adaptor proteins (Figure). This assembly of proteins occurs on the cytoplasmic tails of immunoglobulin chains on regions called immunoreceptor tyrosine-based motifs (ITAMs).
With the assembly of this signaling complex, BCR stimulates a number of downstream pathways, such as phosphatidylinositol 3-kinase (PI3K), protein kinase B (Akt), protein kinase C, nuclear factor-κB (NFκB), and extracellular signal-regulated kinases (ERKs) (Figure). Activation of these pathways results in cell proliferation, resistance to apoptosis, increased cell motility and migration. Recent studies have identified additional novel components of this signaling complex, including a guanine nucleotide exchange factor (GEF) RASGRF1. This GEF is activated by BCR signaling and, in turn, stimulates the ERK pathway by increasing the production of active GTP-bound Ras.6
The ability of BCR to activate a number of downstream signaling pathways makes it a highly relevant and investigated pathway in this leukemia. Inhibitors have been developed and/or identified against a number of signalosome components to block the BCR signaling.7 Syk and Lyn are Src kinases, and their phosphorylation is one of the initial events of BCR signaling. Syk is overexpressed in CLL specimens, and Syk inhibitors (R406 and P505-15, also known as PRT062607) have shown activity in CLL.8,9 Dasatinib is a Src inhibitor that also shows activity in CLL specimens and is being studied in combination with chemotherapy drugs in refractory CLL patients.10
BTK, a component of the BCR signalosome, is required for BCR function, and loss of its function is seen in X-linked agammaglobulinemia. PCI-32765 (ibrutinib) is an oral BTK inhibitor that irreversibly inactivates this kinase and has been approved for clinical use in CLL patients.11,12 Another signaling pathway activated by BCR is the PI3K, and a promising inhibitor (CAL-101) blocks its activity in CLL specimens.13 Investigative work has identified that the delta isoform of PI3K p110 is highly expressed in B cells and lymphocytes.14 This is a catalytic subunit of a class I PI3K with a role in BCR signaling. A selective inhibitor GS-1101 (CAL-101) is able to block PI3K signaling in CLL specimens and inhibits Akt phosphorylation and other downstream effectors along with induction of apoptosis.15 The clinical data with BTK and PI3K inhibitors will be discussed later in this review.
CLL and the Microenvironment
Interactions between CLL cells and the microenvironment allow CLL cells to thrive in certain niche environments.16,17 Interaction mainly occurs via bone marrow stromal cells and nurselike cells (NLCs), which evolve from monocytes (Figure). These interactions can be divided into 2 groups. First, CLL cell growth is supported by a number of chemokine receptor-ligand interactions. CXCR4 is the receptor for CXCL12 (SDF-1) that stimulates chemotaxis and tissue homing. Another chemokine is CXCL13, which acts via its receptor CXCR5 and is involved in chemotaxis and activation of other kinases. Second, NLCs also support CLL cells by expressing TNF family members BAFF and APRIL, which interact with their receptors and activate the NFκB pathway.
Leukemic cells also express VLA-4 integrins, which further their support adhesion to the stromal cells and predict for an aggressive phenotype. Specific inhibitors that block the stimulation by chemokines and cytokines are not yet available; however, one can envision that this class of inhibitors will decrease the chemoresistance of leukemic cells and will be used in conjunction with other chemotherapy agents. Interestingly, inhibitors that block BCR-mediated signaling (BTK and PI3K inhibitors) also inhibit signaling via the microenvironment and chemokines.
Wnt-β-catenin Pathway
Wnt signaling affects developmental pathways, and its aberrant activation has major oncogenic effects as well. This pathway is activated in CLL as these leukemic cells express high levels of Wnt and frizzled along with epigenetic downregulation of Wnt pathway antagonist genes, including secreted frizzled-related protein (SFRP) family members and WIF1 (Figure).18-20 The binding of Wnts to their cognate receptors results in inhibition of GSK3β phosphorylation and stabilization of β-catenin, which then translocates to the nucleus and interacts with lymphoid-enhancing (LEF) and T-cell transcription factors to activate transcription of Wnt-target genes. Lack of E-cadherin expression in CLL cells also results in an increase in translocation of β-catenin and upregulation of the Wnt pathway.20
Wnt-target genes include Myc, LEF, cyclinD1, COX-2, and MMP. Gene expression profiling from our laboratory and other groups have identified the overexpression of these wnt-target genes and support this pathway activation in CLL cells.20 This is a promising signalling pathway and an active area of research for developing inhibitors that will have a growth inhibitory effect on CLL leukemic cells. GSK3b inhibitors and other drugs that re-express epigenetically silenced Wnt antagonist genes have been shown to inhibit this pathway activity in CLL cells in vitro.
Notch Pathway Activation
High-throughput exome sequencing has identified recurring mutations in a number of genes, including NOTCH1.21 Analysis of additional CLL patients confirmed activating NOTCH1 mutations in 10% to 15% of CLL patients and were also associated with poor outcome.22 This pathway is activated by ligands such as Jagged and Delta-like, which interact with the Notch receptor, which is then cleaved by γ-secretases. The cleaved intracellular domain of the NOTCH1 receptor in combination with other factors activates transcription of target genes, including Myc and HES1 (Figure). Besides the mutations that generate a truncated protein or may stabilize the pathway, the Notch pathway is also constitutively active in CLL specimens.23 Notch stimulation increases activity of prosurvival pathways and genes such as NFκB that resist apoptotic signals. The pathway can be inhibited by γ-secretase inhibitors (GSIs), which reduce the levels of cleaved NOTCH1 protein and downregulated Notch target genes. This pathway is also able to modulate the microenvironment stimuli as the GSIs inhibit responses to chemokines such as CXCL12 and inhibit migration and invasion.24
Newer Theraputic Agents
Work on signaling mechanisms paid dividends in CLL with the recent development of 2 inhibitors. Ibrutinib (BTK inhibitor) and idelalisib (PI3K inhibitor) are being studied in clinical trials, and both drugs block the BCR and microenvironment signaling pathways, thereby inhibiting the growth of CLL cells.
BTK Inhibitor: Ibrutinib
The activity of BTK is critical for a number of CLL signaling pathways, and it is a component of the initial signaling complex or signalosome that is formed with BCR signaling. Studies have shown that inhibiting this kinase blocks a number of pathways, including ERK, NFκB, and others. The drug ibrutinib blocks this kinase by forming a covalent bond and inhibiting its enzyme activity. This orally bioavailable drug showed activity in phase 1 trials in different B-cell malignancies.25 In a phase 2 study, high-risk CLL patients were given 2 different doses of this inhibitor, and the overall response rate was 71% with an overall survival at 26 months of 83%.11 Responses were seen in all patients irrespective of clinical and genetic risk factors. Based on these findings, the drug was approved for clinical use in patients with relapsed or refractory disease. Recently, there are data on the use of this drug as frontline therapy in elderly patients, and the drug was well tolerated.26 There are additional ongoing trials to compare this drug with other agents, including chlorambucil (in chemotherapy-naïve patients) and ofatumumab (in relapsed or refractory patients).
PI3 Kinase p110 Delta Inhibitor: Idelalisib
The crucial finding for the development of this inhibitor was the over-expression of the delta isoform of PI3K p110 in B-cell malignancies.14 The drug CAL-101 selectively inhibits this constitutively active isoform and induces apoptosis in a number of B-cell malignancies.15,27 In the phase 1 trial, this inhibitor was evaluated in relapsed/refractory patients at multiple dose levels.28 There was inhibition of PI3K signaling with an overall response rate of 72%, and a partial response rate of 39% was observed in CLL patients. This was followed by a randomized, placebo-controlled phase 3 study in which patients with myelosuppression, decreased renal function, or other illnesses were treated with either rituximab alone or with rituximab and idelalisib.29
At the time of reporting, the median progression-free survival (PFS) was 5.5 months in the placebo arm and was not reached in the idelalisib arm. Overall response rates were higher in the idelalisib group (81% vs 13%) with similar toxicity profiles in the 2 groups. This drug is now being extensively studied in combination with bendamustine and other anti-CD20 antibodies in clinical trials.
A unique toxicity observed with both these inhibitors is the initial lymphocytosis. In the case of ibrutinib, this was seen in a majority of patients (77%) and at the same time there was a response in the nodal disease, implying a redistribution of leukemic cells from the tissues to the peripheral blood.30
A potential explanation is that these drugs inhibit signaling via chemokines and other components of the microenvironment and by inhibiting the homing signals, allows leukemic cells to move out of their niche areas. This was analyzed in a recent study that compared clinical and biochemical parameters of patients who had a complete or partial response with ibrutinib compared with a “partial response except for lymphocytosis.”30 Patients with “partial response except for lymphocytosis” were found to have favorable prognostic factors, and the persisting leukemic cells were not clonally different from the original cells. The progression free survival of patients with “partial response except for lymphocytosis” was also similar to the subgroup with no prolonged lymphocytosis.
Discussion
Several therapeutic agents with novel mechanisms of action are effective in killing the CLL leukemic cells, and a number of targeted agents are currently in the pipeline. The next challenge for treating CLL will be the proper integration of these novel targeted agents with the traditional chemotherapy and chemoimmunotherapy approaches. Let us consider CLL patients in different clinical settings. First, a patient aged 60 years who is otherwise healthy will be treated with possibly all the available chemotherapy and chemoimmunotherapy options, as well as the newer targeted agents. In this clinical setting sequencing of therapy is not a major concern. On the other hand, a patient aged 70 years who is already refractory to multiple lines of therapy is a good candidate for these newer drugs.
The more controversial use of these targeted agents will be in an older patient with some comorbidities and newly diagnosed CLL. In this clinical setting, should one go with traditional chemotherapy/chemoimmunotherapy approaches or consider newer targeted agents? These issues are now being addressed in clinical trials, and with acceptable toxicity profiles these newer drugs will move to the frontline setting.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect an endorsement by or opinion of Federal Practitioner, Frontline Medical Communications, the U.S. Air Force, the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drug combinations–including indications, contraindications, warnings, and adverse effects–before administering pharmacologic therapy to patients.
Chronic lymphocytic leukemia (CLL) is a common hematological malignancy in the U.S. with 15,000 new patients diagnosed each year.1 This leukemia is frequently diagnosed in veterans since it is more commonly seen in an elderly male population. The disease is characterized by a slow accumulation of mature B cells that are functionally incompetent and resist apoptosis. CLL has an indolent clinical course, but about 60% to 70% of patients require treatment. The disease also runs a variable course, and a number of genetic abnormalities and prognostic markers have been defined to subclassify CLL patients and prognosticate.2-4 This article reviews important CLL signaling pathways and novel therapeutic agents in this leukemia.
Signaling Pathways
B-Cell Receptor Signaling
The B-cell receptor (BCR) signaling is the major signaling pathway in CLL, because it defines clinical, biologic, and prognostic characteristics of the disease.5 The BCR is composed of a surface transmembrane immunoglobulin that binds the antigen with CD79 alpha and beta chains. The activation of BCR results in the formation of a signaling complex or signalosome, which includes Lyn, Syk, BTK, and ZAP-70, among other components that assemble with other adaptor proteins (Figure). This assembly of proteins occurs on the cytoplasmic tails of immunoglobulin chains on regions called immunoreceptor tyrosine-based motifs (ITAMs).
With the assembly of this signaling complex, BCR stimulates a number of downstream pathways, such as phosphatidylinositol 3-kinase (PI3K), protein kinase B (Akt), protein kinase C, nuclear factor-κB (NFκB), and extracellular signal-regulated kinases (ERKs) (Figure). Activation of these pathways results in cell proliferation, resistance to apoptosis, increased cell motility and migration. Recent studies have identified additional novel components of this signaling complex, including a guanine nucleotide exchange factor (GEF) RASGRF1. This GEF is activated by BCR signaling and, in turn, stimulates the ERK pathway by increasing the production of active GTP-bound Ras.6
The ability of BCR to activate a number of downstream signaling pathways makes it a highly relevant and investigated pathway in this leukemia. Inhibitors have been developed and/or identified against a number of signalosome components to block the BCR signaling.7 Syk and Lyn are Src kinases, and their phosphorylation is one of the initial events of BCR signaling. Syk is overexpressed in CLL specimens, and Syk inhibitors (R406 and P505-15, also known as PRT062607) have shown activity in CLL.8,9 Dasatinib is a Src inhibitor that also shows activity in CLL specimens and is being studied in combination with chemotherapy drugs in refractory CLL patients.10
BTK, a component of the BCR signalosome, is required for BCR function, and loss of its function is seen in X-linked agammaglobulinemia. PCI-32765 (ibrutinib) is an oral BTK inhibitor that irreversibly inactivates this kinase and has been approved for clinical use in CLL patients.11,12 Another signaling pathway activated by BCR is the PI3K, and a promising inhibitor (CAL-101) blocks its activity in CLL specimens.13 Investigative work has identified that the delta isoform of PI3K p110 is highly expressed in B cells and lymphocytes.14 This is a catalytic subunit of a class I PI3K with a role in BCR signaling. A selective inhibitor GS-1101 (CAL-101) is able to block PI3K signaling in CLL specimens and inhibits Akt phosphorylation and other downstream effectors along with induction of apoptosis.15 The clinical data with BTK and PI3K inhibitors will be discussed later in this review.
CLL and the Microenvironment
Interactions between CLL cells and the microenvironment allow CLL cells to thrive in certain niche environments.16,17 Interaction mainly occurs via bone marrow stromal cells and nurselike cells (NLCs), which evolve from monocytes (Figure). These interactions can be divided into 2 groups. First, CLL cell growth is supported by a number of chemokine receptor-ligand interactions. CXCR4 is the receptor for CXCL12 (SDF-1) that stimulates chemotaxis and tissue homing. Another chemokine is CXCL13, which acts via its receptor CXCR5 and is involved in chemotaxis and activation of other kinases. Second, NLCs also support CLL cells by expressing TNF family members BAFF and APRIL, which interact with their receptors and activate the NFκB pathway.
Leukemic cells also express VLA-4 integrins, which further their support adhesion to the stromal cells and predict for an aggressive phenotype. Specific inhibitors that block the stimulation by chemokines and cytokines are not yet available; however, one can envision that this class of inhibitors will decrease the chemoresistance of leukemic cells and will be used in conjunction with other chemotherapy agents. Interestingly, inhibitors that block BCR-mediated signaling (BTK and PI3K inhibitors) also inhibit signaling via the microenvironment and chemokines.
Wnt-β-catenin Pathway
Wnt signaling affects developmental pathways, and its aberrant activation has major oncogenic effects as well. This pathway is activated in CLL as these leukemic cells express high levels of Wnt and frizzled along with epigenetic downregulation of Wnt pathway antagonist genes, including secreted frizzled-related protein (SFRP) family members and WIF1 (Figure).18-20 The binding of Wnts to their cognate receptors results in inhibition of GSK3β phosphorylation and stabilization of β-catenin, which then translocates to the nucleus and interacts with lymphoid-enhancing (LEF) and T-cell transcription factors to activate transcription of Wnt-target genes. Lack of E-cadherin expression in CLL cells also results in an increase in translocation of β-catenin and upregulation of the Wnt pathway.20
Wnt-target genes include Myc, LEF, cyclinD1, COX-2, and MMP. Gene expression profiling from our laboratory and other groups have identified the overexpression of these wnt-target genes and support this pathway activation in CLL cells.20 This is a promising signalling pathway and an active area of research for developing inhibitors that will have a growth inhibitory effect on CLL leukemic cells. GSK3b inhibitors and other drugs that re-express epigenetically silenced Wnt antagonist genes have been shown to inhibit this pathway activity in CLL cells in vitro.
Notch Pathway Activation
High-throughput exome sequencing has identified recurring mutations in a number of genes, including NOTCH1.21 Analysis of additional CLL patients confirmed activating NOTCH1 mutations in 10% to 15% of CLL patients and were also associated with poor outcome.22 This pathway is activated by ligands such as Jagged and Delta-like, which interact with the Notch receptor, which is then cleaved by γ-secretases. The cleaved intracellular domain of the NOTCH1 receptor in combination with other factors activates transcription of target genes, including Myc and HES1 (Figure). Besides the mutations that generate a truncated protein or may stabilize the pathway, the Notch pathway is also constitutively active in CLL specimens.23 Notch stimulation increases activity of prosurvival pathways and genes such as NFκB that resist apoptotic signals. The pathway can be inhibited by γ-secretase inhibitors (GSIs), which reduce the levels of cleaved NOTCH1 protein and downregulated Notch target genes. This pathway is also able to modulate the microenvironment stimuli as the GSIs inhibit responses to chemokines such as CXCL12 and inhibit migration and invasion.24
Newer Theraputic Agents
Work on signaling mechanisms paid dividends in CLL with the recent development of 2 inhibitors. Ibrutinib (BTK inhibitor) and idelalisib (PI3K inhibitor) are being studied in clinical trials, and both drugs block the BCR and microenvironment signaling pathways, thereby inhibiting the growth of CLL cells.
BTK Inhibitor: Ibrutinib
The activity of BTK is critical for a number of CLL signaling pathways, and it is a component of the initial signaling complex or signalosome that is formed with BCR signaling. Studies have shown that inhibiting this kinase blocks a number of pathways, including ERK, NFκB, and others. The drug ibrutinib blocks this kinase by forming a covalent bond and inhibiting its enzyme activity. This orally bioavailable drug showed activity in phase 1 trials in different B-cell malignancies.25 In a phase 2 study, high-risk CLL patients were given 2 different doses of this inhibitor, and the overall response rate was 71% with an overall survival at 26 months of 83%.11 Responses were seen in all patients irrespective of clinical and genetic risk factors. Based on these findings, the drug was approved for clinical use in patients with relapsed or refractory disease. Recently, there are data on the use of this drug as frontline therapy in elderly patients, and the drug was well tolerated.26 There are additional ongoing trials to compare this drug with other agents, including chlorambucil (in chemotherapy-naïve patients) and ofatumumab (in relapsed or refractory patients).
PI3 Kinase p110 Delta Inhibitor: Idelalisib
The crucial finding for the development of this inhibitor was the over-expression of the delta isoform of PI3K p110 in B-cell malignancies.14 The drug CAL-101 selectively inhibits this constitutively active isoform and induces apoptosis in a number of B-cell malignancies.15,27 In the phase 1 trial, this inhibitor was evaluated in relapsed/refractory patients at multiple dose levels.28 There was inhibition of PI3K signaling with an overall response rate of 72%, and a partial response rate of 39% was observed in CLL patients. This was followed by a randomized, placebo-controlled phase 3 study in which patients with myelosuppression, decreased renal function, or other illnesses were treated with either rituximab alone or with rituximab and idelalisib.29
At the time of reporting, the median progression-free survival (PFS) was 5.5 months in the placebo arm and was not reached in the idelalisib arm. Overall response rates were higher in the idelalisib group (81% vs 13%) with similar toxicity profiles in the 2 groups. This drug is now being extensively studied in combination with bendamustine and other anti-CD20 antibodies in clinical trials.
A unique toxicity observed with both these inhibitors is the initial lymphocytosis. In the case of ibrutinib, this was seen in a majority of patients (77%) and at the same time there was a response in the nodal disease, implying a redistribution of leukemic cells from the tissues to the peripheral blood.30
A potential explanation is that these drugs inhibit signaling via chemokines and other components of the microenvironment and by inhibiting the homing signals, allows leukemic cells to move out of their niche areas. This was analyzed in a recent study that compared clinical and biochemical parameters of patients who had a complete or partial response with ibrutinib compared with a “partial response except for lymphocytosis.”30 Patients with “partial response except for lymphocytosis” were found to have favorable prognostic factors, and the persisting leukemic cells were not clonally different from the original cells. The progression free survival of patients with “partial response except for lymphocytosis” was also similar to the subgroup with no prolonged lymphocytosis.
Discussion
Several therapeutic agents with novel mechanisms of action are effective in killing the CLL leukemic cells, and a number of targeted agents are currently in the pipeline. The next challenge for treating CLL will be the proper integration of these novel targeted agents with the traditional chemotherapy and chemoimmunotherapy approaches. Let us consider CLL patients in different clinical settings. First, a patient aged 60 years who is otherwise healthy will be treated with possibly all the available chemotherapy and chemoimmunotherapy options, as well as the newer targeted agents. In this clinical setting sequencing of therapy is not a major concern. On the other hand, a patient aged 70 years who is already refractory to multiple lines of therapy is a good candidate for these newer drugs.
The more controversial use of these targeted agents will be in an older patient with some comorbidities and newly diagnosed CLL. In this clinical setting, should one go with traditional chemotherapy/chemoimmunotherapy approaches or consider newer targeted agents? These issues are now being addressed in clinical trials, and with acceptable toxicity profiles these newer drugs will move to the frontline setting.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect an endorsement by or opinion of Federal Practitioner, Frontline Medical Communications, the U.S. Air Force, the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drug combinations–including indications, contraindications, warnings, and adverse effects–before administering pharmacologic therapy to patients.
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9-29.
2. Döhner H, Stilgenbauer S, Döhner K, Bentz M, Lichter P. Chromosome aberrations in B-cell chronic lymphocytic leukemia: reassessment based on molecular cytogenetic analysis. J Mol Med. 1999;77(2):266-281.
3. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848-1854.
4. Chen L, Widhopf G, Huynh L, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2002;100(13):4609-4614.
5. Wickremasinghe RG, Prentice AG, Steele AJ. Aberrantly activated anti-apoptotic signalling mechanisms in chronic lymphocytic leukaemia cells: Clues to the identification of novel therapeutic targets. Br J Haematol. 2011;153(5):545-556.
6. Liao W, Jordaan G, Coriaty N, Sharma S. Amplification of B cell receptor-Erk signaling by Rasgrf-1 overexpression in chronic lymphocytic leukemia [published online ahead of print April 2, 2014]. Leuk Lymphoma. doi: 10.3109/10428194.2014898759.
7. Burger JA. Inhibiting B-cell receptor signaling pathways in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2012;7(1):26-33.
8. Buchner M, Fuchs S, Prinz G, et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009;69(13):5424-5432.
9. Spurgeon SE, Coffey G, Fletcher LB, et al. The selective SYK inhibitor P505-15 (PRT062607) inhibits B cell signaling and function in vitro and in vivo and augments the activity of fludarabine in chronic lymphocytic leukemia. J Pharmacol Exp Ther. 2013;344(2):378-387.
10. Veldurthy A, Patz M, Hagist S, et al. The kinase inhibitor dasatinib induces apoptosis in chronic lymphocytic leukemia cells in vitro with preference for a subgroup of patients with unmutated IgVH genes. Blood. 2008;112(4):1443-1452.
11. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia [published correction appears in N Engl J Med. 2014;370(8):786]. N Engl J Med. 2013;369(1):32-42.
12. Cheng S, Ma J, Guo A, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia. 2014;28(3):649-657.
13. Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591-594.
14. Chantry D, Vojtek A, Kashishian A, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997;272(31):19236-19241.
15. Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3’-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603-3612.
16. Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood. 2009;114(16):3367-3375.
17. ten Hacken E, Burger JA. Molecular pathways: targeting the microenvironment in chronic lymphocytic leukemia—focus on the B-cell receptor. Clin Cancer Res. 2014;20(3):548-556.
18. Gandhirajan RK, Poll-Wolbeck SJ, Gehrke I, Kreuzer KA. Wnt/b-catenin/LEF-1 signaling in chronic lymphocytic leukemia (CLL): a target for current and potential therapeutic options. Curr Cancer Drug Targets. 2010;10(7):716-727.
19. Gutierrez A, Jr, Tschumper RC, Wu X, et al. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood. 2010;116(16):2975-2983.
20. Jordaan G, Liao W, Sharma S. E-cadherin gene re-expression in chronic lymphocytic leukemia cells by HDAC inhibitors. BMC Cancer. 2013;13:88.
21. Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101-105.
22. Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389-1401.
23. Rosati E, Sabatini R, Rampino G, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113(4):856-865.
24. López-Guerra M, Xargay-Torrent S, Rosich L, et al. The g-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells [published online ahead of print April 30, 2014]. Leukemia. doi: 10.1038/leu.2014.143.
25. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88-94.
26. O’Brien S, Furman RR, Coutre SE, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: An open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15(1):48-58.
27. Brown JR, Byrd JC, Coutre SE, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110∂, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390-3397.
28. Brown JR, Furman RR, Flinn I, et al. Final results of a phase I study of idelalisib (GS-1101) a selective inhibitor of PI3K∂, in patients with relapsed or refractory CLL. J Clin Oncol. 2013;31:Absract 7003.
29. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007.
30. Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2013;123(12):1810-1817.
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9-29.
2. Döhner H, Stilgenbauer S, Döhner K, Bentz M, Lichter P. Chromosome aberrations in B-cell chronic lymphocytic leukemia: reassessment based on molecular cytogenetic analysis. J Mol Med. 1999;77(2):266-281.
3. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848-1854.
4. Chen L, Widhopf G, Huynh L, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2002;100(13):4609-4614.
5. Wickremasinghe RG, Prentice AG, Steele AJ. Aberrantly activated anti-apoptotic signalling mechanisms in chronic lymphocytic leukaemia cells: Clues to the identification of novel therapeutic targets. Br J Haematol. 2011;153(5):545-556.
6. Liao W, Jordaan G, Coriaty N, Sharma S. Amplification of B cell receptor-Erk signaling by Rasgrf-1 overexpression in chronic lymphocytic leukemia [published online ahead of print April 2, 2014]. Leuk Lymphoma. doi: 10.3109/10428194.2014898759.
7. Burger JA. Inhibiting B-cell receptor signaling pathways in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2012;7(1):26-33.
8. Buchner M, Fuchs S, Prinz G, et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009;69(13):5424-5432.
9. Spurgeon SE, Coffey G, Fletcher LB, et al. The selective SYK inhibitor P505-15 (PRT062607) inhibits B cell signaling and function in vitro and in vivo and augments the activity of fludarabine in chronic lymphocytic leukemia. J Pharmacol Exp Ther. 2013;344(2):378-387.
10. Veldurthy A, Patz M, Hagist S, et al. The kinase inhibitor dasatinib induces apoptosis in chronic lymphocytic leukemia cells in vitro with preference for a subgroup of patients with unmutated IgVH genes. Blood. 2008;112(4):1443-1452.
11. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia [published correction appears in N Engl J Med. 2014;370(8):786]. N Engl J Med. 2013;369(1):32-42.
12. Cheng S, Ma J, Guo A, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia. 2014;28(3):649-657.
13. Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591-594.
14. Chantry D, Vojtek A, Kashishian A, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997;272(31):19236-19241.
15. Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3’-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603-3612.
16. Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood. 2009;114(16):3367-3375.
17. ten Hacken E, Burger JA. Molecular pathways: targeting the microenvironment in chronic lymphocytic leukemia—focus on the B-cell receptor. Clin Cancer Res. 2014;20(3):548-556.
18. Gandhirajan RK, Poll-Wolbeck SJ, Gehrke I, Kreuzer KA. Wnt/b-catenin/LEF-1 signaling in chronic lymphocytic leukemia (CLL): a target for current and potential therapeutic options. Curr Cancer Drug Targets. 2010;10(7):716-727.
19. Gutierrez A, Jr, Tschumper RC, Wu X, et al. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood. 2010;116(16):2975-2983.
20. Jordaan G, Liao W, Sharma S. E-cadherin gene re-expression in chronic lymphocytic leukemia cells by HDAC inhibitors. BMC Cancer. 2013;13:88.
21. Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101-105.
22. Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389-1401.
23. Rosati E, Sabatini R, Rampino G, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113(4):856-865.
24. López-Guerra M, Xargay-Torrent S, Rosich L, et al. The g-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells [published online ahead of print April 30, 2014]. Leukemia. doi: 10.1038/leu.2014.143.
25. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88-94.
26. O’Brien S, Furman RR, Coutre SE, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: An open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15(1):48-58.
27. Brown JR, Byrd JC, Coutre SE, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110∂, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390-3397.
28. Brown JR, Furman RR, Flinn I, et al. Final results of a phase I study of idelalisib (GS-1101) a selective inhibitor of PI3K∂, in patients with relapsed or refractory CLL. J Clin Oncol. 2013;31:Absract 7003.
29. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007.
30. Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2013;123(12):1810-1817.
Blast Phase Chronic Myelogenous Leukemia
Chronic myelogenous leukemia (CML) is caused by the constitutively active BCR-ABL fusion protein that results from t(9;22), the Philadelphia (Ph+) chromosome. Chronic myelogenous leukemia typically evolves through 3 clinical phases: an indolent chronic phase, an accelerated phase, and a terminal blast phase analogous to acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL). Fortunately, today more than 80% of patients are diagnosed in the chronic phase of the disease.1
Before the development of the tyrosine kinase inhibitor (TKI) imatinib, > 20% of the patients with chronic phase CML progressed to the blast phase every year.2 Based on data from 8 years of follow-up with imatinib therapy, the rate of progression to the advanced phases of CML is about 1% per year, with freedom from progression at 92%.3 For the majority of patients with chronic phase CML, due to advances in treatment, the disease does not affect mortality.
For those who progress to the terminal blast phase of CML, survival is typically measured in months unless allogeneic stem cell transplant (allo-SCT) is an option. This article will review one of the major remaining problems in CML: how to manage blast phase CML.
Definition and Diagnosis
Defining blast phase CML can be confusing, because different criteria have been proposed, none of which are biologically based. The most widely used definition is set forth by the European LeukemiaNet, recommending 30% blasts in the blood or bone marrow or the presence of extramedullary disease.1 Clinically, blast phase CML may present with constitutional symptoms, bone pain, or symptoms related to cytopenias (fatigue, dyspnea, bleeding, infections).
Diagnostic workup should include a complete blood cell count (CBC) with differential, bone marrow analysis with conventional cytogenetics, flow cytometry to determine whether the blast phase is of myeloid or lymphoid origin, and molecular mutational analysis of the BCR-ABL tyrosine kinase domain to help guide the choice of TKI. If age and performance status are favorable, a donor search for allo-SCT should be started promptly.
Chronic myelogenous leukemia cells that contain the BCR-ABL kinase protein are genetically unstable.4,5 Additional cytogenetic aberrations (ACAs) are seen in up to 80% of those with blast phase CML and are the most consistent predictor of blast transformation in those with chronic phase CML.6 Chromosomal changes are broken down into the nonrandom, “major route” ACAs (trisomy 8, additional Ph+ chromosome, isochromosome 17q, trisomy 19), considered likely to be involved in the evolution of CML, and the more random “minor route” ACAs, which may denote nothing more than the instability of the genome.5,7 Mutations of the BCR-ABL tyrosine kinase domain are also seen in the majority of those in blast phase CML and, depending on the specific mutation, can negatively predict the response to certain TKI therapies.4
Prognosis
The single most important prognostic indicator for patients with CML remains the length of response to initial BCR-ABL–specific TKI therapy. Only a very small minority of patients are refractory to TKIs from the beginning; these are the patients with the worst prognosis.8 If the response to treatment seems inadequate, then the health care professional should first verify with the patient that he or she is taking the medicine as prescribed.1 Lack of adherence continues to be the most common reason for less-than-ideal outcomes or fluctuations in response and plays a critical role in treatment with TKI therapy, with worse outcomes when < 90% of doses are taken.9
Other features associated with a poor prognosis include cytogenetic clonal evolution, > 50% blasts, and/or extramedullary disease.7,10,11 At the time of imatinib failure, detection of mutations of the BCR-ABL tyrosine kinase domain correlates to worse 4-year event-free survival.12 Showing the instability of the genome in CML, patients who harbor mutations of the BCR-ABL domain have a higher likelihood of relapse associated with further mutations and, therefore, potentially further TKI resistance.13 Once CML has progressed to the blast phase, life expectancy is, on average, less than a year.11
Treatment Strategy
Currently, the most effective treatment strategy in blast phase CML is to prevent the transformation from chronic phase from ever occurring. Management of blast phase CML depends on 2 factors: (1) previous therapies; and (2) type of blast phase—myeloid or lymphoid. The goal of treatment is to knock the disease back to a clinical remission and/or a chronic phase for a long enough period to get the patient to allo-SCT if age, performance status, and suitable donor allow for it.
Using single-agent imatinib for blast phase CML has been tried in patients who have never been on TKI therapy before. Hematologic responses were seen in the majority of patients, but any form of cytogenetic response was seen in fewer than 20% of patients. Median overall survival, although better than with previous conventional chemotherapies, was still measured in months.6 A patient with blast phase CML who has never been on BCR-ABL–specific TKIs is very rare now; at a minimum, the patient has usually tried at least 1 TKI previously.
If blast phase CML develops while a patient is taking imatinib, treatment with a second-generation TKIs—nilotinib or dasatinib— should be attempted if the BCR-ABL tyrosine kinase domain analysis shows no resistant mutations.14 Both nilotinib and dasatinib have been tried as single agents in patients with imatinib-refractory CML or who are unable to tolerate imatinib.15,16 Cytogenetic response rates were 2 to 4 times higher for these agents than for imatinib when used in blast phase CML.
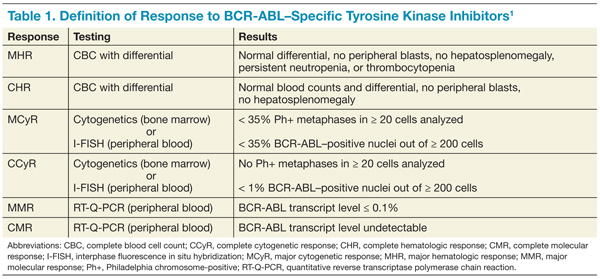
Table 1 reviews the common definitions of response, including cytogenetic response, to TKIs in CML. The pattern of response is usually very predictable: First, a hematologic response is seen, then a cytogenetic response, and finally, a hoped-for molecular response. Interestingly, in these studies, not all patients with blast phase CML who experienced a cytogenetic response had a hematologic response. This makes CBCs less reliable for assessing response and other peripheral blood tests, such as the interphase fluorescence in situ hybridization (I-FISH) test or the quantitative reverse transcriptase polymerase chain reaction (RT-Q-PCR) test, more important. Unfortunately, this improved cytogenetic response in blast phase CML did not translate to long-term survival advantage; median survival with these second- generation TKIs was still less than a year without transplant. If the T315I mutation is present, then clinical trials involving ponatinib or one of the newest non–FDA-approved TKIs should be considered.
Recent data involving ponatinib suggest similar response and survival rates to nilotinib and dasatinib, but this was in more heavily-pretreated CML patients who had resistance to, or unacceptable adverse effects from the second-generation TKIs or who had the BCR-ABL T315I mutation.17
In late 2013, ponatinib was voluntarily suspended from marketing and sales by its manufacturer due to a worrisome rate of serious arterial thromboembolic events reported in clinical trials and in postmarketing experience. However, the FDA reintroduced ponatinib in 2014 once additional safety measures were put in place, such as changes to the black box warning and review of the risk of arterial and venous thrombosis and occlusions.18
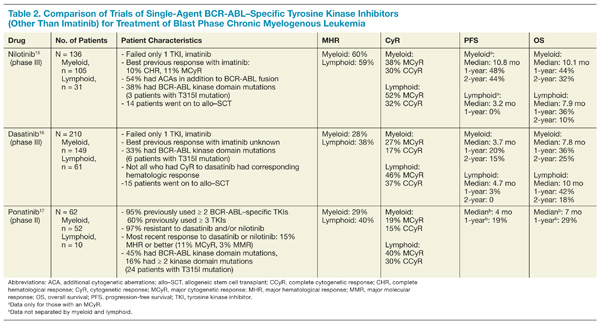
Table 2 compares the results between these newer TKIs in blast phase CML. If the patient can tolerate it, a combination of TKI with AML or ALL-type induction chemotherapy, preferably in a clinical trial setting, provides the best opportunity to return the patient to the chronic phase. If this is achieved, then allo-SCT represents the best chance for sustained remission or cure; allo-SCT was standard first-line therapy prior to the advent of BCR-ABL–specific TKIs. Tyrosine kinase inhibitor exposure prior to allo-SCT does not seem to affect transplantation outcomes.19 Allo-SCT while still in blast phase is discouraged because of its high risks with minimal benefit; disease-free survival rates are <10%.19 Although no scientific data support this, maintenance TKI posttransplantation seems logical, with monitoring of BCR-ABL transcript levels every 3 months.
Conclusion
With the advent of TKI therapy, the overall prognosis of CML has changed drastically. Unfortunately, the success seen with these novel agents in the chronic phase of CML has not translated into success in the blast phase of CML. Therefore, the best way to manage blast phase CML is to prevent this transformation from ever happening. The deeper and more rapid the cytogenetic and molecular response after TKI initiation, the better the long-term outcome for the patient.
If the patient progresses though TKI therapy, then combining a different TKI with a conventional induction chemotherapy regimen for acute leukemia should be tried; the goal is to achieve a remission that lasts long enough for the patient to be able to undergo allo-SCT. If the patient is not a candidate for allo-SCT, then the prognosis is extremely poor, and clinical trials with best supportive care should be considered.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Baccarani M, Pileri S, Steegmann JL, Muller M, Soverini S, Dreyling M; ESMO Guidelines Working Group. Chronic myeloid leukemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(7):vii72-vii77.
2. Sokal JE. Evaluation of survival data for chronic myelocytic leukemia. Am J Hematol. 1976;1(4):493-500.
3. Deininger M, O’Brien SG, Guilhot F, et al. International randomized study of interferon vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood (ASH Annual Meeting Abstracts). 2009;114(22):abstract 1126.
4. Fabarius A, Leitner A, Hochhaus A, et al, Schweizerische Arbeitsgemeinschaft für Klinische Krebsforschung (SAKK) and the German CML Study Group. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood. 2011;118(26):6760-6768.
5. Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol. 2002;107(2):76-94.
6. Hehlmann R. How I treat CML blast crisis. Blood. 2012;120(4):737-747.
7. Jabbour EJ, Hughes TP, Cortes JE, Kantarjian HM, Hochhaus A. Potential mechanisms of disease progression and management of advanced-phase chronic myeloid leukemia [published online ahead of print November 12, 2013]. Leuk Lymphoma. doi:10.3109/10428194.2013.845883.
8. Jabbour E, Kantarjian H, O’Brien S, et al. The achievement of an early complete cytogenetic response is a major determinant for outcome in patients with early chronic phase chronic myeloid leukemia treated with tyrosine kinase inhibitors. Blood. 2011;118(17):4541-4546.
9. Marin D, Bazeos A, Mahon FX, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol. 2010;28(14):2381-2388.
10. Cervantes F, Rozman M, Rosell J, Urbano-Ispizua A, Montserrat E, Rozman C. A study of prognostic factors in blast crisis of Philadelphia chromosome-positive chronic myelogenous leukemia. Br J Haematol. 1990;76(1):27-32.
11. Wadhwa J, Szydlo RM, Apperley JF, et al. Factors affecting duration of survival after onset of blastic transformation of chronic myeloid leukemia. Blood. 2002;99(7):2304-2309.
12. Quintas-Cardama A, Kantarjian H, O’Brien S, et al. Outcome of patients with chronic myeloid leukemia with multiple ABL1 kinase domain mutations receiving tyrosine kinase inhibitor therapy. Haematologica. 2011;96(6):918-921.
13. Soverini S, Gnani A, Colarossi S, et al. Philadelphia-positive patients who already harbor imatinib-resistant BCR-ABL kinase domain mutations have a higher likelihood of developing additional mutations associated with resistance to second- or third-line tyrosine kinase inhibitors. Blood. 2009;114(10):2168-2171.
14. Soverini S, Hochhaus A, Nicolini FE, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118(5):1208-1215.
15. Giles FJ, Kantarjian HM, le Coutre PD, et al. Nilotinib is effective in imatinib-resistant or -intolerant patients with chronic myeloid leukemia in blastic phase. Leukemia. 2012;26(5):959-962.
16. Saglio G, Hochhaus A, Goh YT, et al. Dasatinib in imatinib-resistant or imatinib-intolerant chronic myeloid leukemia in blast phase after 2 years of follow-up in a phase 3 study: efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer. 2010;116(16):3852-3861.
17. Cortes JE, Kim D-W, Pinilla-Ibarz J, et al; PACE Investigators. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783-1796.
18. Food and Drug Administration. FDA Drug Safety Communication: FDA requires multiple new safety measures for leukemia drug Iclusig; company expected to resume marketing. U.S. Food and Drug Administration Website. http://www.fda.gov/drugs/drugsafety/ucm379554.htm. Updated December 20, 2013. Accessed June 13, 2014.
19. Khoury HJ, Kukreja M, Goldman JM, et al. Prognostic factors for outcomes in allogeneic transplantation for CML in the imatinib era: a CIBMTR analysis. Bone Marrow Transplant. 2012;47(6):810-816.
Chronic myelogenous leukemia (CML) is caused by the constitutively active BCR-ABL fusion protein that results from t(9;22), the Philadelphia (Ph+) chromosome. Chronic myelogenous leukemia typically evolves through 3 clinical phases: an indolent chronic phase, an accelerated phase, and a terminal blast phase analogous to acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL). Fortunately, today more than 80% of patients are diagnosed in the chronic phase of the disease.1
Before the development of the tyrosine kinase inhibitor (TKI) imatinib, > 20% of the patients with chronic phase CML progressed to the blast phase every year.2 Based on data from 8 years of follow-up with imatinib therapy, the rate of progression to the advanced phases of CML is about 1% per year, with freedom from progression at 92%.3 For the majority of patients with chronic phase CML, due to advances in treatment, the disease does not affect mortality.
For those who progress to the terminal blast phase of CML, survival is typically measured in months unless allogeneic stem cell transplant (allo-SCT) is an option. This article will review one of the major remaining problems in CML: how to manage blast phase CML.
Definition and Diagnosis
Defining blast phase CML can be confusing, because different criteria have been proposed, none of which are biologically based. The most widely used definition is set forth by the European LeukemiaNet, recommending 30% blasts in the blood or bone marrow or the presence of extramedullary disease.1 Clinically, blast phase CML may present with constitutional symptoms, bone pain, or symptoms related to cytopenias (fatigue, dyspnea, bleeding, infections).
Diagnostic workup should include a complete blood cell count (CBC) with differential, bone marrow analysis with conventional cytogenetics, flow cytometry to determine whether the blast phase is of myeloid or lymphoid origin, and molecular mutational analysis of the BCR-ABL tyrosine kinase domain to help guide the choice of TKI. If age and performance status are favorable, a donor search for allo-SCT should be started promptly.
Chronic myelogenous leukemia cells that contain the BCR-ABL kinase protein are genetically unstable.4,5 Additional cytogenetic aberrations (ACAs) are seen in up to 80% of those with blast phase CML and are the most consistent predictor of blast transformation in those with chronic phase CML.6 Chromosomal changes are broken down into the nonrandom, “major route” ACAs (trisomy 8, additional Ph+ chromosome, isochromosome 17q, trisomy 19), considered likely to be involved in the evolution of CML, and the more random “minor route” ACAs, which may denote nothing more than the instability of the genome.5,7 Mutations of the BCR-ABL tyrosine kinase domain are also seen in the majority of those in blast phase CML and, depending on the specific mutation, can negatively predict the response to certain TKI therapies.4
Prognosis
The single most important prognostic indicator for patients with CML remains the length of response to initial BCR-ABL–specific TKI therapy. Only a very small minority of patients are refractory to TKIs from the beginning; these are the patients with the worst prognosis.8 If the response to treatment seems inadequate, then the health care professional should first verify with the patient that he or she is taking the medicine as prescribed.1 Lack of adherence continues to be the most common reason for less-than-ideal outcomes or fluctuations in response and plays a critical role in treatment with TKI therapy, with worse outcomes when < 90% of doses are taken.9
Other features associated with a poor prognosis include cytogenetic clonal evolution, > 50% blasts, and/or extramedullary disease.7,10,11 At the time of imatinib failure, detection of mutations of the BCR-ABL tyrosine kinase domain correlates to worse 4-year event-free survival.12 Showing the instability of the genome in CML, patients who harbor mutations of the BCR-ABL domain have a higher likelihood of relapse associated with further mutations and, therefore, potentially further TKI resistance.13 Once CML has progressed to the blast phase, life expectancy is, on average, less than a year.11
Treatment Strategy
Currently, the most effective treatment strategy in blast phase CML is to prevent the transformation from chronic phase from ever occurring. Management of blast phase CML depends on 2 factors: (1) previous therapies; and (2) type of blast phase—myeloid or lymphoid. The goal of treatment is to knock the disease back to a clinical remission and/or a chronic phase for a long enough period to get the patient to allo-SCT if age, performance status, and suitable donor allow for it.
Using single-agent imatinib for blast phase CML has been tried in patients who have never been on TKI therapy before. Hematologic responses were seen in the majority of patients, but any form of cytogenetic response was seen in fewer than 20% of patients. Median overall survival, although better than with previous conventional chemotherapies, was still measured in months.6 A patient with blast phase CML who has never been on BCR-ABL–specific TKIs is very rare now; at a minimum, the patient has usually tried at least 1 TKI previously.
If blast phase CML develops while a patient is taking imatinib, treatment with a second-generation TKIs—nilotinib or dasatinib— should be attempted if the BCR-ABL tyrosine kinase domain analysis shows no resistant mutations.14 Both nilotinib and dasatinib have been tried as single agents in patients with imatinib-refractory CML or who are unable to tolerate imatinib.15,16 Cytogenetic response rates were 2 to 4 times higher for these agents than for imatinib when used in blast phase CML.
Table 1 reviews the common definitions of response, including cytogenetic response, to TKIs in CML. The pattern of response is usually very predictable: First, a hematologic response is seen, then a cytogenetic response, and finally, a hoped-for molecular response. Interestingly, in these studies, not all patients with blast phase CML who experienced a cytogenetic response had a hematologic response. This makes CBCs less reliable for assessing response and other peripheral blood tests, such as the interphase fluorescence in situ hybridization (I-FISH) test or the quantitative reverse transcriptase polymerase chain reaction (RT-Q-PCR) test, more important. Unfortunately, this improved cytogenetic response in blast phase CML did not translate to long-term survival advantage; median survival with these second- generation TKIs was still less than a year without transplant. If the T315I mutation is present, then clinical trials involving ponatinib or one of the newest non–FDA-approved TKIs should be considered.
Recent data involving ponatinib suggest similar response and survival rates to nilotinib and dasatinib, but this was in more heavily-pretreated CML patients who had resistance to, or unacceptable adverse effects from the second-generation TKIs or who had the BCR-ABL T315I mutation.17
In late 2013, ponatinib was voluntarily suspended from marketing and sales by its manufacturer due to a worrisome rate of serious arterial thromboembolic events reported in clinical trials and in postmarketing experience. However, the FDA reintroduced ponatinib in 2014 once additional safety measures were put in place, such as changes to the black box warning and review of the risk of arterial and venous thrombosis and occlusions.18
Table 2 compares the results between these newer TKIs in blast phase CML. If the patient can tolerate it, a combination of TKI with AML or ALL-type induction chemotherapy, preferably in a clinical trial setting, provides the best opportunity to return the patient to the chronic phase. If this is achieved, then allo-SCT represents the best chance for sustained remission or cure; allo-SCT was standard first-line therapy prior to the advent of BCR-ABL–specific TKIs. Tyrosine kinase inhibitor exposure prior to allo-SCT does not seem to affect transplantation outcomes.19 Allo-SCT while still in blast phase is discouraged because of its high risks with minimal benefit; disease-free survival rates are <10%.19 Although no scientific data support this, maintenance TKI posttransplantation seems logical, with monitoring of BCR-ABL transcript levels every 3 months.
Conclusion
With the advent of TKI therapy, the overall prognosis of CML has changed drastically. Unfortunately, the success seen with these novel agents in the chronic phase of CML has not translated into success in the blast phase of CML. Therefore, the best way to manage blast phase CML is to prevent this transformation from ever happening. The deeper and more rapid the cytogenetic and molecular response after TKI initiation, the better the long-term outcome for the patient.
If the patient progresses though TKI therapy, then combining a different TKI with a conventional induction chemotherapy regimen for acute leukemia should be tried; the goal is to achieve a remission that lasts long enough for the patient to be able to undergo allo-SCT. If the patient is not a candidate for allo-SCT, then the prognosis is extremely poor, and clinical trials with best supportive care should be considered.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Chronic myelogenous leukemia (CML) is caused by the constitutively active BCR-ABL fusion protein that results from t(9;22), the Philadelphia (Ph+) chromosome. Chronic myelogenous leukemia typically evolves through 3 clinical phases: an indolent chronic phase, an accelerated phase, and a terminal blast phase analogous to acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL). Fortunately, today more than 80% of patients are diagnosed in the chronic phase of the disease.1
Before the development of the tyrosine kinase inhibitor (TKI) imatinib, > 20% of the patients with chronic phase CML progressed to the blast phase every year.2 Based on data from 8 years of follow-up with imatinib therapy, the rate of progression to the advanced phases of CML is about 1% per year, with freedom from progression at 92%.3 For the majority of patients with chronic phase CML, due to advances in treatment, the disease does not affect mortality.
For those who progress to the terminal blast phase of CML, survival is typically measured in months unless allogeneic stem cell transplant (allo-SCT) is an option. This article will review one of the major remaining problems in CML: how to manage blast phase CML.
Definition and Diagnosis
Defining blast phase CML can be confusing, because different criteria have been proposed, none of which are biologically based. The most widely used definition is set forth by the European LeukemiaNet, recommending 30% blasts in the blood or bone marrow or the presence of extramedullary disease.1 Clinically, blast phase CML may present with constitutional symptoms, bone pain, or symptoms related to cytopenias (fatigue, dyspnea, bleeding, infections).
Diagnostic workup should include a complete blood cell count (CBC) with differential, bone marrow analysis with conventional cytogenetics, flow cytometry to determine whether the blast phase is of myeloid or lymphoid origin, and molecular mutational analysis of the BCR-ABL tyrosine kinase domain to help guide the choice of TKI. If age and performance status are favorable, a donor search for allo-SCT should be started promptly.
Chronic myelogenous leukemia cells that contain the BCR-ABL kinase protein are genetically unstable.4,5 Additional cytogenetic aberrations (ACAs) are seen in up to 80% of those with blast phase CML and are the most consistent predictor of blast transformation in those with chronic phase CML.6 Chromosomal changes are broken down into the nonrandom, “major route” ACAs (trisomy 8, additional Ph+ chromosome, isochromosome 17q, trisomy 19), considered likely to be involved in the evolution of CML, and the more random “minor route” ACAs, which may denote nothing more than the instability of the genome.5,7 Mutations of the BCR-ABL tyrosine kinase domain are also seen in the majority of those in blast phase CML and, depending on the specific mutation, can negatively predict the response to certain TKI therapies.4
Prognosis
The single most important prognostic indicator for patients with CML remains the length of response to initial BCR-ABL–specific TKI therapy. Only a very small minority of patients are refractory to TKIs from the beginning; these are the patients with the worst prognosis.8 If the response to treatment seems inadequate, then the health care professional should first verify with the patient that he or she is taking the medicine as prescribed.1 Lack of adherence continues to be the most common reason for less-than-ideal outcomes or fluctuations in response and plays a critical role in treatment with TKI therapy, with worse outcomes when < 90% of doses are taken.9
Other features associated with a poor prognosis include cytogenetic clonal evolution, > 50% blasts, and/or extramedullary disease.7,10,11 At the time of imatinib failure, detection of mutations of the BCR-ABL tyrosine kinase domain correlates to worse 4-year event-free survival.12 Showing the instability of the genome in CML, patients who harbor mutations of the BCR-ABL domain have a higher likelihood of relapse associated with further mutations and, therefore, potentially further TKI resistance.13 Once CML has progressed to the blast phase, life expectancy is, on average, less than a year.11
Treatment Strategy
Currently, the most effective treatment strategy in blast phase CML is to prevent the transformation from chronic phase from ever occurring. Management of blast phase CML depends on 2 factors: (1) previous therapies; and (2) type of blast phase—myeloid or lymphoid. The goal of treatment is to knock the disease back to a clinical remission and/or a chronic phase for a long enough period to get the patient to allo-SCT if age, performance status, and suitable donor allow for it.
Using single-agent imatinib for blast phase CML has been tried in patients who have never been on TKI therapy before. Hematologic responses were seen in the majority of patients, but any form of cytogenetic response was seen in fewer than 20% of patients. Median overall survival, although better than with previous conventional chemotherapies, was still measured in months.6 A patient with blast phase CML who has never been on BCR-ABL–specific TKIs is very rare now; at a minimum, the patient has usually tried at least 1 TKI previously.
If blast phase CML develops while a patient is taking imatinib, treatment with a second-generation TKIs—nilotinib or dasatinib— should be attempted if the BCR-ABL tyrosine kinase domain analysis shows no resistant mutations.14 Both nilotinib and dasatinib have been tried as single agents in patients with imatinib-refractory CML or who are unable to tolerate imatinib.15,16 Cytogenetic response rates were 2 to 4 times higher for these agents than for imatinib when used in blast phase CML.
Table 1 reviews the common definitions of response, including cytogenetic response, to TKIs in CML. The pattern of response is usually very predictable: First, a hematologic response is seen, then a cytogenetic response, and finally, a hoped-for molecular response. Interestingly, in these studies, not all patients with blast phase CML who experienced a cytogenetic response had a hematologic response. This makes CBCs less reliable for assessing response and other peripheral blood tests, such as the interphase fluorescence in situ hybridization (I-FISH) test or the quantitative reverse transcriptase polymerase chain reaction (RT-Q-PCR) test, more important. Unfortunately, this improved cytogenetic response in blast phase CML did not translate to long-term survival advantage; median survival with these second- generation TKIs was still less than a year without transplant. If the T315I mutation is present, then clinical trials involving ponatinib or one of the newest non–FDA-approved TKIs should be considered.
Recent data involving ponatinib suggest similar response and survival rates to nilotinib and dasatinib, but this was in more heavily-pretreated CML patients who had resistance to, or unacceptable adverse effects from the second-generation TKIs or who had the BCR-ABL T315I mutation.17
In late 2013, ponatinib was voluntarily suspended from marketing and sales by its manufacturer due to a worrisome rate of serious arterial thromboembolic events reported in clinical trials and in postmarketing experience. However, the FDA reintroduced ponatinib in 2014 once additional safety measures were put in place, such as changes to the black box warning and review of the risk of arterial and venous thrombosis and occlusions.18
Table 2 compares the results between these newer TKIs in blast phase CML. If the patient can tolerate it, a combination of TKI with AML or ALL-type induction chemotherapy, preferably in a clinical trial setting, provides the best opportunity to return the patient to the chronic phase. If this is achieved, then allo-SCT represents the best chance for sustained remission or cure; allo-SCT was standard first-line therapy prior to the advent of BCR-ABL–specific TKIs. Tyrosine kinase inhibitor exposure prior to allo-SCT does not seem to affect transplantation outcomes.19 Allo-SCT while still in blast phase is discouraged because of its high risks with minimal benefit; disease-free survival rates are <10%.19 Although no scientific data support this, maintenance TKI posttransplantation seems logical, with monitoring of BCR-ABL transcript levels every 3 months.
Conclusion
With the advent of TKI therapy, the overall prognosis of CML has changed drastically. Unfortunately, the success seen with these novel agents in the chronic phase of CML has not translated into success in the blast phase of CML. Therefore, the best way to manage blast phase CML is to prevent this transformation from ever happening. The deeper and more rapid the cytogenetic and molecular response after TKI initiation, the better the long-term outcome for the patient.
If the patient progresses though TKI therapy, then combining a different TKI with a conventional induction chemotherapy regimen for acute leukemia should be tried; the goal is to achieve a remission that lasts long enough for the patient to be able to undergo allo-SCT. If the patient is not a candidate for allo-SCT, then the prognosis is extremely poor, and clinical trials with best supportive care should be considered.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Baccarani M, Pileri S, Steegmann JL, Muller M, Soverini S, Dreyling M; ESMO Guidelines Working Group. Chronic myeloid leukemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(7):vii72-vii77.
2. Sokal JE. Evaluation of survival data for chronic myelocytic leukemia. Am J Hematol. 1976;1(4):493-500.
3. Deininger M, O’Brien SG, Guilhot F, et al. International randomized study of interferon vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood (ASH Annual Meeting Abstracts). 2009;114(22):abstract 1126.
4. Fabarius A, Leitner A, Hochhaus A, et al, Schweizerische Arbeitsgemeinschaft für Klinische Krebsforschung (SAKK) and the German CML Study Group. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood. 2011;118(26):6760-6768.
5. Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol. 2002;107(2):76-94.
6. Hehlmann R. How I treat CML blast crisis. Blood. 2012;120(4):737-747.
7. Jabbour EJ, Hughes TP, Cortes JE, Kantarjian HM, Hochhaus A. Potential mechanisms of disease progression and management of advanced-phase chronic myeloid leukemia [published online ahead of print November 12, 2013]. Leuk Lymphoma. doi:10.3109/10428194.2013.845883.
8. Jabbour E, Kantarjian H, O’Brien S, et al. The achievement of an early complete cytogenetic response is a major determinant for outcome in patients with early chronic phase chronic myeloid leukemia treated with tyrosine kinase inhibitors. Blood. 2011;118(17):4541-4546.
9. Marin D, Bazeos A, Mahon FX, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol. 2010;28(14):2381-2388.
10. Cervantes F, Rozman M, Rosell J, Urbano-Ispizua A, Montserrat E, Rozman C. A study of prognostic factors in blast crisis of Philadelphia chromosome-positive chronic myelogenous leukemia. Br J Haematol. 1990;76(1):27-32.
11. Wadhwa J, Szydlo RM, Apperley JF, et al. Factors affecting duration of survival after onset of blastic transformation of chronic myeloid leukemia. Blood. 2002;99(7):2304-2309.
12. Quintas-Cardama A, Kantarjian H, O’Brien S, et al. Outcome of patients with chronic myeloid leukemia with multiple ABL1 kinase domain mutations receiving tyrosine kinase inhibitor therapy. Haematologica. 2011;96(6):918-921.
13. Soverini S, Gnani A, Colarossi S, et al. Philadelphia-positive patients who already harbor imatinib-resistant BCR-ABL kinase domain mutations have a higher likelihood of developing additional mutations associated with resistance to second- or third-line tyrosine kinase inhibitors. Blood. 2009;114(10):2168-2171.
14. Soverini S, Hochhaus A, Nicolini FE, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118(5):1208-1215.
15. Giles FJ, Kantarjian HM, le Coutre PD, et al. Nilotinib is effective in imatinib-resistant or -intolerant patients with chronic myeloid leukemia in blastic phase. Leukemia. 2012;26(5):959-962.
16. Saglio G, Hochhaus A, Goh YT, et al. Dasatinib in imatinib-resistant or imatinib-intolerant chronic myeloid leukemia in blast phase after 2 years of follow-up in a phase 3 study: efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer. 2010;116(16):3852-3861.
17. Cortes JE, Kim D-W, Pinilla-Ibarz J, et al; PACE Investigators. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783-1796.
18. Food and Drug Administration. FDA Drug Safety Communication: FDA requires multiple new safety measures for leukemia drug Iclusig; company expected to resume marketing. U.S. Food and Drug Administration Website. http://www.fda.gov/drugs/drugsafety/ucm379554.htm. Updated December 20, 2013. Accessed June 13, 2014.
19. Khoury HJ, Kukreja M, Goldman JM, et al. Prognostic factors for outcomes in allogeneic transplantation for CML in the imatinib era: a CIBMTR analysis. Bone Marrow Transplant. 2012;47(6):810-816.
1. Baccarani M, Pileri S, Steegmann JL, Muller M, Soverini S, Dreyling M; ESMO Guidelines Working Group. Chronic myeloid leukemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(7):vii72-vii77.
2. Sokal JE. Evaluation of survival data for chronic myelocytic leukemia. Am J Hematol. 1976;1(4):493-500.
3. Deininger M, O’Brien SG, Guilhot F, et al. International randomized study of interferon vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood (ASH Annual Meeting Abstracts). 2009;114(22):abstract 1126.
4. Fabarius A, Leitner A, Hochhaus A, et al, Schweizerische Arbeitsgemeinschaft für Klinische Krebsforschung (SAKK) and the German CML Study Group. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood. 2011;118(26):6760-6768.
5. Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol. 2002;107(2):76-94.
6. Hehlmann R. How I treat CML blast crisis. Blood. 2012;120(4):737-747.
7. Jabbour EJ, Hughes TP, Cortes JE, Kantarjian HM, Hochhaus A. Potential mechanisms of disease progression and management of advanced-phase chronic myeloid leukemia [published online ahead of print November 12, 2013]. Leuk Lymphoma. doi:10.3109/10428194.2013.845883.
8. Jabbour E, Kantarjian H, O’Brien S, et al. The achievement of an early complete cytogenetic response is a major determinant for outcome in patients with early chronic phase chronic myeloid leukemia treated with tyrosine kinase inhibitors. Blood. 2011;118(17):4541-4546.
9. Marin D, Bazeos A, Mahon FX, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol. 2010;28(14):2381-2388.
10. Cervantes F, Rozman M, Rosell J, Urbano-Ispizua A, Montserrat E, Rozman C. A study of prognostic factors in blast crisis of Philadelphia chromosome-positive chronic myelogenous leukemia. Br J Haematol. 1990;76(1):27-32.
11. Wadhwa J, Szydlo RM, Apperley JF, et al. Factors affecting duration of survival after onset of blastic transformation of chronic myeloid leukemia. Blood. 2002;99(7):2304-2309.
12. Quintas-Cardama A, Kantarjian H, O’Brien S, et al. Outcome of patients with chronic myeloid leukemia with multiple ABL1 kinase domain mutations receiving tyrosine kinase inhibitor therapy. Haematologica. 2011;96(6):918-921.
13. Soverini S, Gnani A, Colarossi S, et al. Philadelphia-positive patients who already harbor imatinib-resistant BCR-ABL kinase domain mutations have a higher likelihood of developing additional mutations associated with resistance to second- or third-line tyrosine kinase inhibitors. Blood. 2009;114(10):2168-2171.
14. Soverini S, Hochhaus A, Nicolini FE, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118(5):1208-1215.
15. Giles FJ, Kantarjian HM, le Coutre PD, et al. Nilotinib is effective in imatinib-resistant or -intolerant patients with chronic myeloid leukemia in blastic phase. Leukemia. 2012;26(5):959-962.
16. Saglio G, Hochhaus A, Goh YT, et al. Dasatinib in imatinib-resistant or imatinib-intolerant chronic myeloid leukemia in blast phase after 2 years of follow-up in a phase 3 study: efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer. 2010;116(16):3852-3861.
17. Cortes JE, Kim D-W, Pinilla-Ibarz J, et al; PACE Investigators. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783-1796.
18. Food and Drug Administration. FDA Drug Safety Communication: FDA requires multiple new safety measures for leukemia drug Iclusig; company expected to resume marketing. U.S. Food and Drug Administration Website. http://www.fda.gov/drugs/drugsafety/ucm379554.htm. Updated December 20, 2013. Accessed June 13, 2014.
19. Khoury HJ, Kukreja M, Goldman JM, et al. Prognostic factors for outcomes in allogeneic transplantation for CML in the imatinib era: a CIBMTR analysis. Bone Marrow Transplant. 2012;47(6):810-816.
Study reveals potential targets for MYC-dependent cancers

Credit: Juha Klefstrom
New research suggests the MYC protein drives cell growth by inhibiting a handful of genes involved in DNA packaging and cell death.
The study showed that MYC works through a microRNA to suppress the genes’ expression.
This marks the first time that a subset of MYC-controlled genes have been identified as critical players in the protein’s cancer-causing function, and it points to new therapeutic targets for MYC-dependent cancers.
“This is a different way of thinking about the roles of microRNA and chromatin packaging in cancer,” said Dean Felsher, MD, PhD, of the Stanford University School of Medicine in California.
“We were very surprised to learn that the overexpression of one microRNA can mimic the cancerous effect of MYC.”
Dr Felsher and his colleagues reported this discovery in Cancer Cell.
The team noted that MYC overexpression has been known to prompt an increase in the levels of a microRNA family called miR-17-92.
“People have known for several years that MYC regulates the expression of microRNAs,” Dr Felsher said. “But it wasn’t clear how this was related to MYC’s oncogenic function.”
To gain some insight, Dr Felsher and his colleagues analyzed MYC-dependent cancer cells in vitro and in vivo.
The cells in which miR-17-92 expression was locked in the “on” position kept dividing even when MYC expression was blocked. This suggested that MYC works through the microRNA family to exert its cancer-causing effects.
The researchers then looked for an overlap among genes affected by MYC overexpression and those affected by miR-17-92. There were about 401 genes whose expression was either increased or suppressed by both MYC and miR-17-92.
The team chose to focus on genes that were suppressed because these genes exhibited, on average, many more binding sites for the microRNAs. They further narrowed their panel down to 15 genes regulated by more than one miR-17-92 binding site.
Of these genes, 5 stood out. Four of them—Sin3b, Hbp1, Suv420h1, and Btg1—encode proteins known to regulate chromatin packaging.
These 4 proteins affect cell proliferation and senescence by regulating gene accessibility within the chromatin. They had never before been identified as MYC or miR-17-92 targets.
The fifth gene encodes the apoptotic protein Bim. Previous research suggested that Bim expression is affected by miR-17-92.
All 5 of the proteins are known to affect either cellular proliferation, entry into senescence, or apoptosis, in part by granting or prohibiting access to genes in tightly packaged stretches of DNA in the chromatin.
“MYC is still a general amplifier of gene transcription and expression,” Dr Felsher said. “But our study shows that the maintenance of the cancerous state relies on a more focused mechanism.”
Lastly, the researchers showed that suppressing the expression of the 5 target genes, effectively mimicking MYC overexpression, partially mitigates the effect of MYC deactivation.
Up to 30% of MYC-dependent cancer cells in culture continued to grow—compared to 11% of control cells—in the absence of MYC expression. And tumors in mice either failed to regress or recurred within a few weeks.
“One of the biggest unanswered questions in oncology is how oncogenes cause cancer, and whether you can replace an oncogene with another gene product,” Dr Felsher said.
“These experiments begin to reveal how MYC affects the self-renewal decisions of cells. They may also help us target those aspects of MYC overexpression that contribute to the cancer phenotype.” ![]()
Credit: Juha Klefstrom
New research suggests the MYC protein drives cell growth by inhibiting a handful of genes involved in DNA packaging and cell death.
The study showed that MYC works through a microRNA to suppress the genes’ expression.
This marks the first time that a subset of MYC-controlled genes have been identified as critical players in the protein’s cancer-causing function, and it points to new therapeutic targets for MYC-dependent cancers.
“This is a different way of thinking about the roles of microRNA and chromatin packaging in cancer,” said Dean Felsher, MD, PhD, of the Stanford University School of Medicine in California.
“We were very surprised to learn that the overexpression of one microRNA can mimic the cancerous effect of MYC.”
Dr Felsher and his colleagues reported this discovery in Cancer Cell.
The team noted that MYC overexpression has been known to prompt an increase in the levels of a microRNA family called miR-17-92.
“People have known for several years that MYC regulates the expression of microRNAs,” Dr Felsher said. “But it wasn’t clear how this was related to MYC’s oncogenic function.”
To gain some insight, Dr Felsher and his colleagues analyzed MYC-dependent cancer cells in vitro and in vivo.
The cells in which miR-17-92 expression was locked in the “on” position kept dividing even when MYC expression was blocked. This suggested that MYC works through the microRNA family to exert its cancer-causing effects.
The researchers then looked for an overlap among genes affected by MYC overexpression and those affected by miR-17-92. There were about 401 genes whose expression was either increased or suppressed by both MYC and miR-17-92.
The team chose to focus on genes that were suppressed because these genes exhibited, on average, many more binding sites for the microRNAs. They further narrowed their panel down to 15 genes regulated by more than one miR-17-92 binding site.
Of these genes, 5 stood out. Four of them—Sin3b, Hbp1, Suv420h1, and Btg1—encode proteins known to regulate chromatin packaging.
These 4 proteins affect cell proliferation and senescence by regulating gene accessibility within the chromatin. They had never before been identified as MYC or miR-17-92 targets.
The fifth gene encodes the apoptotic protein Bim. Previous research suggested that Bim expression is affected by miR-17-92.
All 5 of the proteins are known to affect either cellular proliferation, entry into senescence, or apoptosis, in part by granting or prohibiting access to genes in tightly packaged stretches of DNA in the chromatin.
“MYC is still a general amplifier of gene transcription and expression,” Dr Felsher said. “But our study shows that the maintenance of the cancerous state relies on a more focused mechanism.”
Lastly, the researchers showed that suppressing the expression of the 5 target genes, effectively mimicking MYC overexpression, partially mitigates the effect of MYC deactivation.
Up to 30% of MYC-dependent cancer cells in culture continued to grow—compared to 11% of control cells—in the absence of MYC expression. And tumors in mice either failed to regress or recurred within a few weeks.
“One of the biggest unanswered questions in oncology is how oncogenes cause cancer, and whether you can replace an oncogene with another gene product,” Dr Felsher said.
“These experiments begin to reveal how MYC affects the self-renewal decisions of cells. They may also help us target those aspects of MYC overexpression that contribute to the cancer phenotype.” ![]()
Credit: Juha Klefstrom
New research suggests the MYC protein drives cell growth by inhibiting a handful of genes involved in DNA packaging and cell death.
The study showed that MYC works through a microRNA to suppress the genes’ expression.
This marks the first time that a subset of MYC-controlled genes have been identified as critical players in the protein’s cancer-causing function, and it points to new therapeutic targets for MYC-dependent cancers.
“This is a different way of thinking about the roles of microRNA and chromatin packaging in cancer,” said Dean Felsher, MD, PhD, of the Stanford University School of Medicine in California.
“We were very surprised to learn that the overexpression of one microRNA can mimic the cancerous effect of MYC.”
Dr Felsher and his colleagues reported this discovery in Cancer Cell.
The team noted that MYC overexpression has been known to prompt an increase in the levels of a microRNA family called miR-17-92.
“People have known for several years that MYC regulates the expression of microRNAs,” Dr Felsher said. “But it wasn’t clear how this was related to MYC’s oncogenic function.”
To gain some insight, Dr Felsher and his colleagues analyzed MYC-dependent cancer cells in vitro and in vivo.
The cells in which miR-17-92 expression was locked in the “on” position kept dividing even when MYC expression was blocked. This suggested that MYC works through the microRNA family to exert its cancer-causing effects.
The researchers then looked for an overlap among genes affected by MYC overexpression and those affected by miR-17-92. There were about 401 genes whose expression was either increased or suppressed by both MYC and miR-17-92.
The team chose to focus on genes that were suppressed because these genes exhibited, on average, many more binding sites for the microRNAs. They further narrowed their panel down to 15 genes regulated by more than one miR-17-92 binding site.
Of these genes, 5 stood out. Four of them—Sin3b, Hbp1, Suv420h1, and Btg1—encode proteins known to regulate chromatin packaging.
These 4 proteins affect cell proliferation and senescence by regulating gene accessibility within the chromatin. They had never before been identified as MYC or miR-17-92 targets.
The fifth gene encodes the apoptotic protein Bim. Previous research suggested that Bim expression is affected by miR-17-92.
All 5 of the proteins are known to affect either cellular proliferation, entry into senescence, or apoptosis, in part by granting or prohibiting access to genes in tightly packaged stretches of DNA in the chromatin.
“MYC is still a general amplifier of gene transcription and expression,” Dr Felsher said. “But our study shows that the maintenance of the cancerous state relies on a more focused mechanism.”
Lastly, the researchers showed that suppressing the expression of the 5 target genes, effectively mimicking MYC overexpression, partially mitigates the effect of MYC deactivation.
Up to 30% of MYC-dependent cancer cells in culture continued to grow—compared to 11% of control cells—in the absence of MYC expression. And tumors in mice either failed to regress or recurred within a few weeks.
“One of the biggest unanswered questions in oncology is how oncogenes cause cancer, and whether you can replace an oncogene with another gene product,” Dr Felsher said.
“These experiments begin to reveal how MYC affects the self-renewal decisions of cells. They may also help us target those aspects of MYC overexpression that contribute to the cancer phenotype.” ![]()
Method could speed up cancer diagnosis

Credit: NIGMS
A new technique could enable faster diagnosis of cancer and various prenatal conditions, according to a paper published in Proceedings of the National Academy of Sciences.
The method, known as convex lens-induced confinement (CLIC), allows researchers to load long strands of DNA into a tunable, nanoscale imaging chamber in ways that maintain their structural identity and under conditions that are similar to those in the human body.
CLIC lets researchers map large genomes rapidly and identify specific gene sequences from single cells with single-molecule resolution, a process that is critical to diagnosing diseases like cancer.
“Current practices of genomic analysis typically require tens of thousands of cells worth of genomic material to obtain the information we need, but this new approach works with single cells,” said study author Rob Sladek, MD, of McGill University in Montreal, Canada.
“CLIC will allow researchers to avoid having to spend time stitching together maps of entire genomes, as we do under current techniques, and promises to make genomic analysis a much simpler and more efficient process.”
The CLIC imaging chamber can sit on top of a standard inverted fluorescence microscope, and strands of DNA can be loaded into the chamber from above, which allows the strands to maintain their integrity.
Existing tools used for genomic analysis rely on side-loading DNA under pressure into nanochannels in the imaging chamber. This breaks the DNA molecules into small pieces, making it a challenge to reconstruct the genome.
CLIC, on the other hand, is “like squeezing many soft spaghetti noodles into long, narrow tubes without breaking them,” according to study author Sabrina Leslie, PhD, also of McGill University.
“Once these long strands of DNA are gently squeezed down into nanochannels from a nanoscale bath above, they become effectively rigid, which means that we can map positions along uniformly stretched strands of DNA, while holding them still,” she said.
“This means diagnostics can be performed quickly, one cell at a time, which is critical for diagnosing many prenatal conditions and the onset of cancer.” ![]()
Credit: NIGMS
A new technique could enable faster diagnosis of cancer and various prenatal conditions, according to a paper published in Proceedings of the National Academy of Sciences.
The method, known as convex lens-induced confinement (CLIC), allows researchers to load long strands of DNA into a tunable, nanoscale imaging chamber in ways that maintain their structural identity and under conditions that are similar to those in the human body.
CLIC lets researchers map large genomes rapidly and identify specific gene sequences from single cells with single-molecule resolution, a process that is critical to diagnosing diseases like cancer.
“Current practices of genomic analysis typically require tens of thousands of cells worth of genomic material to obtain the information we need, but this new approach works with single cells,” said study author Rob Sladek, MD, of McGill University in Montreal, Canada.
“CLIC will allow researchers to avoid having to spend time stitching together maps of entire genomes, as we do under current techniques, and promises to make genomic analysis a much simpler and more efficient process.”
The CLIC imaging chamber can sit on top of a standard inverted fluorescence microscope, and strands of DNA can be loaded into the chamber from above, which allows the strands to maintain their integrity.
Existing tools used for genomic analysis rely on side-loading DNA under pressure into nanochannels in the imaging chamber. This breaks the DNA molecules into small pieces, making it a challenge to reconstruct the genome.
CLIC, on the other hand, is “like squeezing many soft spaghetti noodles into long, narrow tubes without breaking them,” according to study author Sabrina Leslie, PhD, also of McGill University.
“Once these long strands of DNA are gently squeezed down into nanochannels from a nanoscale bath above, they become effectively rigid, which means that we can map positions along uniformly stretched strands of DNA, while holding them still,” she said.
“This means diagnostics can be performed quickly, one cell at a time, which is critical for diagnosing many prenatal conditions and the onset of cancer.” ![]()
Credit: NIGMS
A new technique could enable faster diagnosis of cancer and various prenatal conditions, according to a paper published in Proceedings of the National Academy of Sciences.
The method, known as convex lens-induced confinement (CLIC), allows researchers to load long strands of DNA into a tunable, nanoscale imaging chamber in ways that maintain their structural identity and under conditions that are similar to those in the human body.
CLIC lets researchers map large genomes rapidly and identify specific gene sequences from single cells with single-molecule resolution, a process that is critical to diagnosing diseases like cancer.
“Current practices of genomic analysis typically require tens of thousands of cells worth of genomic material to obtain the information we need, but this new approach works with single cells,” said study author Rob Sladek, MD, of McGill University in Montreal, Canada.
“CLIC will allow researchers to avoid having to spend time stitching together maps of entire genomes, as we do under current techniques, and promises to make genomic analysis a much simpler and more efficient process.”
The CLIC imaging chamber can sit on top of a standard inverted fluorescence microscope, and strands of DNA can be loaded into the chamber from above, which allows the strands to maintain their integrity.
Existing tools used for genomic analysis rely on side-loading DNA under pressure into nanochannels in the imaging chamber. This breaks the DNA molecules into small pieces, making it a challenge to reconstruct the genome.
CLIC, on the other hand, is “like squeezing many soft spaghetti noodles into long, narrow tubes without breaking them,” according to study author Sabrina Leslie, PhD, also of McGill University.
“Once these long strands of DNA are gently squeezed down into nanochannels from a nanoscale bath above, they become effectively rigid, which means that we can map positions along uniformly stretched strands of DNA, while holding them still,” she said.
“This means diagnostics can be performed quickly, one cell at a time, which is critical for diagnosing many prenatal conditions and the onset of cancer.” ![]()
Study challenges traditional cancer classification

a tumor sample in a test tube
Credit: Rhoda Baer
Defining cancers by molecular criteria rather than their tissue of origin can provide patients with more accurate diagnoses, researchers have reported in Cell.
The group analyzed the molecular characteristics of more than 3500 samples of 12 different cancers and reclassified them according to the new information.
For 5 of the cancer types, including acute myeloid leukemia (AML), the molecular classification largely matched the tissue-of-origin classification.
For the remaining malignancies, that was not the case.
“This genomic study not only challenges our existing system of classifying cancers based on tissue type, but also provides a massive new data resource for further exploration, as well as a comprehensive list of the molecular features distinguishing each of the newly described cancer classes,” said study author Christopher Benz, MD, of the University of California, San Francisco.
The researchers said each molecular subtype they identified may reflect tumors arising from distinct cell types. For example, the data showed a marked difference between cancers of epithelial and non-epithelial origins.
“We think the subtypes reflect, primarily, the cell of origin,” said study author Joshua Stuart, PhD, of the University of California, Santa Cruz.
“Another factor is the nature of the genomic lesion, and third is the microenvironment of the cell and how surrounding cells influence it. We are disentangling the signals from these different factors so we can gauge each one for its prognostic power.”
Identifying molecular subtypes
The researchers performed an integrative analysis using 5 genome-wide platforms and 1 proteomic platform on 3527 specimens from 12 cancer types.
This included AML, glioblastoma multiforme, serous ovarian carcinoma, colon and rectal adenocarcinomas, lung squamous cell carcinoma, breast cancer, endometrial cancer, renal cell carcinoma, bladder urothelial adenocarcinoma, lung adenocarcinoma, and head and neck squamous cell carcinoma.
The group’s analyses allowed them to classify these cancer types into 11 major cellular/molecular subtypes. Two of the initial 13 subtypes (numbers 11 and 12) were eliminated from further analysis because they included fewer than 10 samples.
Five of the classification types—C5-renal cell carcinoma, C6-endometrial cancer, C9-serous ovarian carcinoma, C10-glioblastoma multiforme, and C13-AML—showed near 1-to-1 relationships with the tissue site of origin. However, there were a few cases of reclassification here and there, such as a case of breast cancer that fell in the AML subtype.
Another subtype stayed pretty true to its tissues of origin. C7-colon adenocarcinoma/rectal adenocarcinoma was composed mainly of colon and rectal adenocarcinomas but also included a case of endometrial cancer.
The C1-lung adenocarcinoma-enriched subtype was predominantly composed of non-small cell lung adenocarcinoma samples. But it also included cases of bladder cancer, breast cancer, colon adenocarcinoma, glioblastoma multiforme, head and neck squamous cell carcinoma, renal cell carcinoma, lung squamous cell carcinoma , serous ovarian carcinoma, and endometrial cancer.
The C2-squamous-like subtype consisted largely of head and neck squamous cell carcinoma and lung squamous cell carcinoma but also included bladder urothelial adenocarcinoma and breast cancer.
Breast cancers were further divided into the C3-breast cancer/luminal subtype and the C4-breast cancer/basal subtype. The C4 subtype also included lung adenocarcinoma and lung squamous cell carcinoma.
The researchers noted that breast cancers were present in 7 of the subtype classifications. And while this study confirmed known differences between the subtypes of breast cancer, the team was surprised to discover that basal-like breast cancers actually constitute their own cancer class.
“Even though these basal-like cancers arise in the breast, on the molecular level, they have more in common with ovarian cancers and cancers of squamous-cell origin than with other subtypes of breast cancer,” said study author Christina Yau, PhD, of the University of California, San Francisco.
Like breast cancers, bladder cancers were present in 7 of the subtype classifications. There were 1 or 2 cases in C5, C10, C11, and C12. But most bladder cancer samples fell into 1 of 3 categories: C1-lung adenocarcinoma-enriched, C2-squamous-like, and C8-bladder urothelial adenocarcinoma.
Although the C8-bladder urothelial adenocarcinoma subtype consisted largely of bladder cancer, it also included breast cancer, head and neck squamous cell carcinoma, lung adenocarcinoma, and lung squamous cell carcinoma.
These findings may help explain why patients with bladder cancer “often respond very differently when treated with the same systemic therapy for their seemingly identical cancer type,” Dr Benz said.
In fact, the researchers found the bladder cancers that clustered with other tumor types had a worse prognosis.
Next steps
The researchers noted that follow-up studies are needed to validate these findings, but this analysis lays the groundwork for classifying tumors into molecularly defined subtypes. The new classification scheme could be used to enroll patients in clinical trials and could lead to different treatment options based on molecular subtypes.
“We can now say what the telltale signatures of the subtypes are, so you can classify a patient’s tumor just based on the gene expression data, or just based on mutation data, if that’s what you have,” Dr Stuart said. “Having a molecular map like this could help get a patient into the right clinical trial.”
The researchers believe the percentage of tumors that should be reclassified based on molecular signatures is likely to grow as more samples and tumor types are analyzed. This study suggested that 1 in 10 cancers could be reclassified in clinically meaningful ways, but the researchers said their next analysis will include 21 tumor types instead of 12.
“We’re just appreciating the tip of the iceberg when considering the potential of this multiplatform type of genomic analysis,” Dr Benz said. “It could be that as many as 30% or 50% of cancers need to be reclassified.”
The data sets and results from this study have been made available to other researchers through the Synapse website. ![]()
a tumor sample in a test tube
Credit: Rhoda Baer
Defining cancers by molecular criteria rather than their tissue of origin can provide patients with more accurate diagnoses, researchers have reported in Cell.
The group analyzed the molecular characteristics of more than 3500 samples of 12 different cancers and reclassified them according to the new information.
For 5 of the cancer types, including acute myeloid leukemia (AML), the molecular classification largely matched the tissue-of-origin classification.
For the remaining malignancies, that was not the case.
“This genomic study not only challenges our existing system of classifying cancers based on tissue type, but also provides a massive new data resource for further exploration, as well as a comprehensive list of the molecular features distinguishing each of the newly described cancer classes,” said study author Christopher Benz, MD, of the University of California, San Francisco.
The researchers said each molecular subtype they identified may reflect tumors arising from distinct cell types. For example, the data showed a marked difference between cancers of epithelial and non-epithelial origins.
“We think the subtypes reflect, primarily, the cell of origin,” said study author Joshua Stuart, PhD, of the University of California, Santa Cruz.
“Another factor is the nature of the genomic lesion, and third is the microenvironment of the cell and how surrounding cells influence it. We are disentangling the signals from these different factors so we can gauge each one for its prognostic power.”
Identifying molecular subtypes
The researchers performed an integrative analysis using 5 genome-wide platforms and 1 proteomic platform on 3527 specimens from 12 cancer types.
This included AML, glioblastoma multiforme, serous ovarian carcinoma, colon and rectal adenocarcinomas, lung squamous cell carcinoma, breast cancer, endometrial cancer, renal cell carcinoma, bladder urothelial adenocarcinoma, lung adenocarcinoma, and head and neck squamous cell carcinoma.
The group’s analyses allowed them to classify these cancer types into 11 major cellular/molecular subtypes. Two of the initial 13 subtypes (numbers 11 and 12) were eliminated from further analysis because they included fewer than 10 samples.
Five of the classification types—C5-renal cell carcinoma, C6-endometrial cancer, C9-serous ovarian carcinoma, C10-glioblastoma multiforme, and C13-AML—showed near 1-to-1 relationships with the tissue site of origin. However, there were a few cases of reclassification here and there, such as a case of breast cancer that fell in the AML subtype.
Another subtype stayed pretty true to its tissues of origin. C7-colon adenocarcinoma/rectal adenocarcinoma was composed mainly of colon and rectal adenocarcinomas but also included a case of endometrial cancer.
The C1-lung adenocarcinoma-enriched subtype was predominantly composed of non-small cell lung adenocarcinoma samples. But it also included cases of bladder cancer, breast cancer, colon adenocarcinoma, glioblastoma multiforme, head and neck squamous cell carcinoma, renal cell carcinoma, lung squamous cell carcinoma , serous ovarian carcinoma, and endometrial cancer.
The C2-squamous-like subtype consisted largely of head and neck squamous cell carcinoma and lung squamous cell carcinoma but also included bladder urothelial adenocarcinoma and breast cancer.
Breast cancers were further divided into the C3-breast cancer/luminal subtype and the C4-breast cancer/basal subtype. The C4 subtype also included lung adenocarcinoma and lung squamous cell carcinoma.
The researchers noted that breast cancers were present in 7 of the subtype classifications. And while this study confirmed known differences between the subtypes of breast cancer, the team was surprised to discover that basal-like breast cancers actually constitute their own cancer class.
“Even though these basal-like cancers arise in the breast, on the molecular level, they have more in common with ovarian cancers and cancers of squamous-cell origin than with other subtypes of breast cancer,” said study author Christina Yau, PhD, of the University of California, San Francisco.
Like breast cancers, bladder cancers were present in 7 of the subtype classifications. There were 1 or 2 cases in C5, C10, C11, and C12. But most bladder cancer samples fell into 1 of 3 categories: C1-lung adenocarcinoma-enriched, C2-squamous-like, and C8-bladder urothelial adenocarcinoma.
Although the C8-bladder urothelial adenocarcinoma subtype consisted largely of bladder cancer, it also included breast cancer, head and neck squamous cell carcinoma, lung adenocarcinoma, and lung squamous cell carcinoma.
These findings may help explain why patients with bladder cancer “often respond very differently when treated with the same systemic therapy for their seemingly identical cancer type,” Dr Benz said.
In fact, the researchers found the bladder cancers that clustered with other tumor types had a worse prognosis.
Next steps
The researchers noted that follow-up studies are needed to validate these findings, but this analysis lays the groundwork for classifying tumors into molecularly defined subtypes. The new classification scheme could be used to enroll patients in clinical trials and could lead to different treatment options based on molecular subtypes.
“We can now say what the telltale signatures of the subtypes are, so you can classify a patient’s tumor just based on the gene expression data, or just based on mutation data, if that’s what you have,” Dr Stuart said. “Having a molecular map like this could help get a patient into the right clinical trial.”
The researchers believe the percentage of tumors that should be reclassified based on molecular signatures is likely to grow as more samples and tumor types are analyzed. This study suggested that 1 in 10 cancers could be reclassified in clinically meaningful ways, but the researchers said their next analysis will include 21 tumor types instead of 12.
“We’re just appreciating the tip of the iceberg when considering the potential of this multiplatform type of genomic analysis,” Dr Benz said. “It could be that as many as 30% or 50% of cancers need to be reclassified.”
The data sets and results from this study have been made available to other researchers through the Synapse website. ![]()
a tumor sample in a test tube
Credit: Rhoda Baer
Defining cancers by molecular criteria rather than their tissue of origin can provide patients with more accurate diagnoses, researchers have reported in Cell.
The group analyzed the molecular characteristics of more than 3500 samples of 12 different cancers and reclassified them according to the new information.
For 5 of the cancer types, including acute myeloid leukemia (AML), the molecular classification largely matched the tissue-of-origin classification.
For the remaining malignancies, that was not the case.
“This genomic study not only challenges our existing system of classifying cancers based on tissue type, but also provides a massive new data resource for further exploration, as well as a comprehensive list of the molecular features distinguishing each of the newly described cancer classes,” said study author Christopher Benz, MD, of the University of California, San Francisco.
The researchers said each molecular subtype they identified may reflect tumors arising from distinct cell types. For example, the data showed a marked difference between cancers of epithelial and non-epithelial origins.
“We think the subtypes reflect, primarily, the cell of origin,” said study author Joshua Stuart, PhD, of the University of California, Santa Cruz.
“Another factor is the nature of the genomic lesion, and third is the microenvironment of the cell and how surrounding cells influence it. We are disentangling the signals from these different factors so we can gauge each one for its prognostic power.”
Identifying molecular subtypes
The researchers performed an integrative analysis using 5 genome-wide platforms and 1 proteomic platform on 3527 specimens from 12 cancer types.
This included AML, glioblastoma multiforme, serous ovarian carcinoma, colon and rectal adenocarcinomas, lung squamous cell carcinoma, breast cancer, endometrial cancer, renal cell carcinoma, bladder urothelial adenocarcinoma, lung adenocarcinoma, and head and neck squamous cell carcinoma.
The group’s analyses allowed them to classify these cancer types into 11 major cellular/molecular subtypes. Two of the initial 13 subtypes (numbers 11 and 12) were eliminated from further analysis because they included fewer than 10 samples.
Five of the classification types—C5-renal cell carcinoma, C6-endometrial cancer, C9-serous ovarian carcinoma, C10-glioblastoma multiforme, and C13-AML—showed near 1-to-1 relationships with the tissue site of origin. However, there were a few cases of reclassification here and there, such as a case of breast cancer that fell in the AML subtype.
Another subtype stayed pretty true to its tissues of origin. C7-colon adenocarcinoma/rectal adenocarcinoma was composed mainly of colon and rectal adenocarcinomas but also included a case of endometrial cancer.
The C1-lung adenocarcinoma-enriched subtype was predominantly composed of non-small cell lung adenocarcinoma samples. But it also included cases of bladder cancer, breast cancer, colon adenocarcinoma, glioblastoma multiforme, head and neck squamous cell carcinoma, renal cell carcinoma, lung squamous cell carcinoma , serous ovarian carcinoma, and endometrial cancer.
The C2-squamous-like subtype consisted largely of head and neck squamous cell carcinoma and lung squamous cell carcinoma but also included bladder urothelial adenocarcinoma and breast cancer.
Breast cancers were further divided into the C3-breast cancer/luminal subtype and the C4-breast cancer/basal subtype. The C4 subtype also included lung adenocarcinoma and lung squamous cell carcinoma.
The researchers noted that breast cancers were present in 7 of the subtype classifications. And while this study confirmed known differences between the subtypes of breast cancer, the team was surprised to discover that basal-like breast cancers actually constitute their own cancer class.
“Even though these basal-like cancers arise in the breast, on the molecular level, they have more in common with ovarian cancers and cancers of squamous-cell origin than with other subtypes of breast cancer,” said study author Christina Yau, PhD, of the University of California, San Francisco.
Like breast cancers, bladder cancers were present in 7 of the subtype classifications. There were 1 or 2 cases in C5, C10, C11, and C12. But most bladder cancer samples fell into 1 of 3 categories: C1-lung adenocarcinoma-enriched, C2-squamous-like, and C8-bladder urothelial adenocarcinoma.
Although the C8-bladder urothelial adenocarcinoma subtype consisted largely of bladder cancer, it also included breast cancer, head and neck squamous cell carcinoma, lung adenocarcinoma, and lung squamous cell carcinoma.
These findings may help explain why patients with bladder cancer “often respond very differently when treated with the same systemic therapy for their seemingly identical cancer type,” Dr Benz said.
In fact, the researchers found the bladder cancers that clustered with other tumor types had a worse prognosis.
Next steps
The researchers noted that follow-up studies are needed to validate these findings, but this analysis lays the groundwork for classifying tumors into molecularly defined subtypes. The new classification scheme could be used to enroll patients in clinical trials and could lead to different treatment options based on molecular subtypes.
“We can now say what the telltale signatures of the subtypes are, so you can classify a patient’s tumor just based on the gene expression data, or just based on mutation data, if that’s what you have,” Dr Stuart said. “Having a molecular map like this could help get a patient into the right clinical trial.”
The researchers believe the percentage of tumors that should be reclassified based on molecular signatures is likely to grow as more samples and tumor types are analyzed. This study suggested that 1 in 10 cancers could be reclassified in clinically meaningful ways, but the researchers said their next analysis will include 21 tumor types instead of 12.
“We’re just appreciating the tip of the iceberg when considering the potential of this multiplatform type of genomic analysis,” Dr Benz said. “It could be that as many as 30% or 50% of cancers need to be reclassified.”
The data sets and results from this study have been made available to other researchers through the Synapse website. ![]()
Gene plays crucial role in cancer development, team says
Credit: Beth A. Sullivan
New research suggests DNA ligase 3 is crucial for the evolutionary processes that drive cancer.
“We have identified a gene that, as cells age, seems to regulate whether the cells become cancerous or not,” said Eric A. Hendrickson, PhD, of the University of Minnesota in Minneapolis.
“This gene has never been identified before in this role, so this makes it a potentially very important therapeutic target.”
Dr Hendrickson and his colleagues recounted this discovery in Cell Reports.
The researchers noted that short, dysfunctional telomeres can fuse, thereby generating dicentric chromosomes and initiating breakage-fusion-bridge cycles. The cells that manage to escape the subsequent crisis have genomic rearrangements that drive clonal evolution and malignant progression.
The team wanted to determine exactly what allows these malignant cells to escape telomere-driven crisis and avoid death.
To find out, the group disabled certain genes in human cells and then studied the impact this had on telomere fusion.
They found that cells escaped death when ligase 3 was active but not when its action, which appears to promote fusion within like chromosomes rather than between different chromosomes, was inhibited.
“Telomere dysfunction has been identified in many human cancers,” said study author Duncan Baird, PhD, of Cardiff University in the UK.
“And, as we have shown previously, short telomeres can predict the outcome of patients with [chronic lymphocytic leukemia] and probably many other tumor types. Thus, the discovery that ligase 3 is required for this process is fundamentally important.”
This research was made possible by a chance meeting between Dr Baird and Dr Hendrickson at an international conference. The pair discovered they were both looking at the role of ligase 3 in cancer and decided to collaborate.
“The collaboration paid off, as we were able to uncover something that neither one of us could have done on our own,” Dr Hendrickson said.
Additional studies are already underway. The researchers are investigating the discovery that the reliance on ligase 3 appears to be dependent upon the activity of another key DNA repair gene, p53.
“Since p53 is the most commonly mutated gene in human cancer, it now behooves us to discover how these two genes are interacting and to see if we can’t use that information to develop synergistic treatment modalities,” Dr Hendrickson concluded. ![]()
Credit: Beth A. Sullivan
New research suggests DNA ligase 3 is crucial for the evolutionary processes that drive cancer.
“We have identified a gene that, as cells age, seems to regulate whether the cells become cancerous or not,” said Eric A. Hendrickson, PhD, of the University of Minnesota in Minneapolis.
“This gene has never been identified before in this role, so this makes it a potentially very important therapeutic target.”
Dr Hendrickson and his colleagues recounted this discovery in Cell Reports.
The researchers noted that short, dysfunctional telomeres can fuse, thereby generating dicentric chromosomes and initiating breakage-fusion-bridge cycles. The cells that manage to escape the subsequent crisis have genomic rearrangements that drive clonal evolution and malignant progression.
The team wanted to determine exactly what allows these malignant cells to escape telomere-driven crisis and avoid death.
To find out, the group disabled certain genes in human cells and then studied the impact this had on telomere fusion.
They found that cells escaped death when ligase 3 was active but not when its action, which appears to promote fusion within like chromosomes rather than between different chromosomes, was inhibited.
“Telomere dysfunction has been identified in many human cancers,” said study author Duncan Baird, PhD, of Cardiff University in the UK.
“And, as we have shown previously, short telomeres can predict the outcome of patients with [chronic lymphocytic leukemia] and probably many other tumor types. Thus, the discovery that ligase 3 is required for this process is fundamentally important.”
This research was made possible by a chance meeting between Dr Baird and Dr Hendrickson at an international conference. The pair discovered they were both looking at the role of ligase 3 in cancer and decided to collaborate.
“The collaboration paid off, as we were able to uncover something that neither one of us could have done on our own,” Dr Hendrickson said.
Additional studies are already underway. The researchers are investigating the discovery that the reliance on ligase 3 appears to be dependent upon the activity of another key DNA repair gene, p53.
“Since p53 is the most commonly mutated gene in human cancer, it now behooves us to discover how these two genes are interacting and to see if we can’t use that information to develop synergistic treatment modalities,” Dr Hendrickson concluded. ![]()
Credit: Beth A. Sullivan
New research suggests DNA ligase 3 is crucial for the evolutionary processes that drive cancer.
“We have identified a gene that, as cells age, seems to regulate whether the cells become cancerous or not,” said Eric A. Hendrickson, PhD, of the University of Minnesota in Minneapolis.
“This gene has never been identified before in this role, so this makes it a potentially very important therapeutic target.”
Dr Hendrickson and his colleagues recounted this discovery in Cell Reports.
The researchers noted that short, dysfunctional telomeres can fuse, thereby generating dicentric chromosomes and initiating breakage-fusion-bridge cycles. The cells that manage to escape the subsequent crisis have genomic rearrangements that drive clonal evolution and malignant progression.
The team wanted to determine exactly what allows these malignant cells to escape telomere-driven crisis and avoid death.
To find out, the group disabled certain genes in human cells and then studied the impact this had on telomere fusion.
They found that cells escaped death when ligase 3 was active but not when its action, which appears to promote fusion within like chromosomes rather than between different chromosomes, was inhibited.
“Telomere dysfunction has been identified in many human cancers,” said study author Duncan Baird, PhD, of Cardiff University in the UK.
“And, as we have shown previously, short telomeres can predict the outcome of patients with [chronic lymphocytic leukemia] and probably many other tumor types. Thus, the discovery that ligase 3 is required for this process is fundamentally important.”
This research was made possible by a chance meeting between Dr Baird and Dr Hendrickson at an international conference. The pair discovered they were both looking at the role of ligase 3 in cancer and decided to collaborate.
“The collaboration paid off, as we were able to uncover something that neither one of us could have done on our own,” Dr Hendrickson said.
Additional studies are already underway. The researchers are investigating the discovery that the reliance on ligase 3 appears to be dependent upon the activity of another key DNA repair gene, p53.
“Since p53 is the most commonly mutated gene in human cancer, it now behooves us to discover how these two genes are interacting and to see if we can’t use that information to develop synergistic treatment modalities,” Dr Hendrickson concluded. ![]()
Drugs can increase risk of MDS and AML
A class of immunosuppressive agents appear to increase the risk of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) in patients with inflammatory bowel disease (IBD).
In an observational study of more than 19,000 IBD patients, past exposure to the agents—thiopurines—increased the risk of developing AML or MDS nearly 7-fold, when compared to the general population.
However, the absolute risk to an individual patient was about 1 in 10,000.
The researchers reported these results in Clinical Gastroenterology and Hepatology.
Thiopurines are an established treatment for IBD patients, but the drugs are also used to prevent rejection after a kidney transplant, to treat rheumatoid arthritis, as maintenance therapy for acute lymphocytic leukemia, and to induce remission in patients with AML.
Previous research showed that long-term use of thiopurines can increase a person’s risk of developing lymphoma.
“In order to make appropriate, informed decisions about thiopurines, patients and providers need to be well-educated about the risks and benefits of this treatment,” said study author Laurent Peyrin-Biroulet, MD, PhD, of the University Hospital of Nancy-Brabois in France.
“According to our research, the risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. However, it was increased amongst those taking thiopurines. We hope these findings encourage other researchers to investigate more about the drug and its potentially harmful effects.”
The researchers analyzed 19,486 patients who were enrolled in the Cancers Et Surrisque Associé aux Maladies inflammatoires intestinales En France study from May 2004 through June 2005.
At study entry, 10,810 patients had never received thiopurines, 2810 patients had discontinued such drugs, and 5866 patients were still receiving them.
After 3 years of follow up, 5 patients were diagnosed with incident myeloid disorders—2 with AML and 3 with MDS. Four of these patients had been exposed to thiopurines—1 with ongoing treatment and 3 with past exposure.
The risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. The standardized incidence ratio (SIR) was 1.80.
Similarly, the risk of myeloid disorders was not increased among IBD patients still receiving thiopurine treatment. The SIR was 1.54.
However, patients with prior exposure to thiopurines did have a significantly increased risk of myeloid disorders, with an SIR of 6.98.
The researchers noted that, although these findings provide evidence of a connection between thiopurines and myeloid disorders in IBD patients, the absolute risk to an individual patient was low.
So it seems the link between thiopurines and myeloid disorders remains complex. And physicians must balance the risk against the known benefits of thiopurines in the management of IBD.
The American Gastroenterological Association has developed a guideline-based clinical decision support tool to help providers determine when to use thiopurines in these patients. ![]()
A class of immunosuppressive agents appear to increase the risk of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) in patients with inflammatory bowel disease (IBD).
In an observational study of more than 19,000 IBD patients, past exposure to the agents—thiopurines—increased the risk of developing AML or MDS nearly 7-fold, when compared to the general population.
However, the absolute risk to an individual patient was about 1 in 10,000.
The researchers reported these results in Clinical Gastroenterology and Hepatology.
Thiopurines are an established treatment for IBD patients, but the drugs are also used to prevent rejection after a kidney transplant, to treat rheumatoid arthritis, as maintenance therapy for acute lymphocytic leukemia, and to induce remission in patients with AML.
Previous research showed that long-term use of thiopurines can increase a person’s risk of developing lymphoma.
“In order to make appropriate, informed decisions about thiopurines, patients and providers need to be well-educated about the risks and benefits of this treatment,” said study author Laurent Peyrin-Biroulet, MD, PhD, of the University Hospital of Nancy-Brabois in France.
“According to our research, the risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. However, it was increased amongst those taking thiopurines. We hope these findings encourage other researchers to investigate more about the drug and its potentially harmful effects.”
The researchers analyzed 19,486 patients who were enrolled in the Cancers Et Surrisque Associé aux Maladies inflammatoires intestinales En France study from May 2004 through June 2005.
At study entry, 10,810 patients had never received thiopurines, 2810 patients had discontinued such drugs, and 5866 patients were still receiving them.
After 3 years of follow up, 5 patients were diagnosed with incident myeloid disorders—2 with AML and 3 with MDS. Four of these patients had been exposed to thiopurines—1 with ongoing treatment and 3 with past exposure.
The risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. The standardized incidence ratio (SIR) was 1.80.
Similarly, the risk of myeloid disorders was not increased among IBD patients still receiving thiopurine treatment. The SIR was 1.54.
However, patients with prior exposure to thiopurines did have a significantly increased risk of myeloid disorders, with an SIR of 6.98.
The researchers noted that, although these findings provide evidence of a connection between thiopurines and myeloid disorders in IBD patients, the absolute risk to an individual patient was low.
So it seems the link between thiopurines and myeloid disorders remains complex. And physicians must balance the risk against the known benefits of thiopurines in the management of IBD.
The American Gastroenterological Association has developed a guideline-based clinical decision support tool to help providers determine when to use thiopurines in these patients. ![]()
A class of immunosuppressive agents appear to increase the risk of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) in patients with inflammatory bowel disease (IBD).
In an observational study of more than 19,000 IBD patients, past exposure to the agents—thiopurines—increased the risk of developing AML or MDS nearly 7-fold, when compared to the general population.
However, the absolute risk to an individual patient was about 1 in 10,000.
The researchers reported these results in Clinical Gastroenterology and Hepatology.
Thiopurines are an established treatment for IBD patients, but the drugs are also used to prevent rejection after a kidney transplant, to treat rheumatoid arthritis, as maintenance therapy for acute lymphocytic leukemia, and to induce remission in patients with AML.
Previous research showed that long-term use of thiopurines can increase a person’s risk of developing lymphoma.
“In order to make appropriate, informed decisions about thiopurines, patients and providers need to be well-educated about the risks and benefits of this treatment,” said study author Laurent Peyrin-Biroulet, MD, PhD, of the University Hospital of Nancy-Brabois in France.
“According to our research, the risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. However, it was increased amongst those taking thiopurines. We hope these findings encourage other researchers to investigate more about the drug and its potentially harmful effects.”
The researchers analyzed 19,486 patients who were enrolled in the Cancers Et Surrisque Associé aux Maladies inflammatoires intestinales En France study from May 2004 through June 2005.
At study entry, 10,810 patients had never received thiopurines, 2810 patients had discontinued such drugs, and 5866 patients were still receiving them.
After 3 years of follow up, 5 patients were diagnosed with incident myeloid disorders—2 with AML and 3 with MDS. Four of these patients had been exposed to thiopurines—1 with ongoing treatment and 3 with past exposure.
The risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. The standardized incidence ratio (SIR) was 1.80.
Similarly, the risk of myeloid disorders was not increased among IBD patients still receiving thiopurine treatment. The SIR was 1.54.
However, patients with prior exposure to thiopurines did have a significantly increased risk of myeloid disorders, with an SIR of 6.98.
The researchers noted that, although these findings provide evidence of a connection between thiopurines and myeloid disorders in IBD patients, the absolute risk to an individual patient was low.
So it seems the link between thiopurines and myeloid disorders remains complex. And physicians must balance the risk against the known benefits of thiopurines in the management of IBD.
The American Gastroenterological Association has developed a guideline-based clinical decision support tool to help providers determine when to use thiopurines in these patients. ![]()
Ofatumumab prompts fatal reaction in CLL patient

Credit: Bill Branson
Health Canada and GlaxoSmithKline (GSK) have reported a fatal infusion reaction in a patient receiving the monoclonal antibody ofatumumab (Arzerra) to treat
chronic lymphocytic leukemia (CLL).
The patient had no known history of cardiac disease.
Ofatumumab’s product monograph is being updated to include a warning about the potential for fatal infusion reactions.
In Canada, ofatumumab is approved to treat patients with CLL that is refractory to fludarabine and alemtuzumab.
The drug received this marketing authorization with conditions, pending the results of trials to verify its clinical benefit.
In light of the fatal infusion reaction, GSK and Health Canada are reminding healthcare professionals that ofatumumab should be administered under the supervision of a physician experienced in the use of cancer therapy. The drug should be given in an environment where facilities to adequately monitor and treat infusion reactions are available.
Prior to infusion, patients should always receive the appropriate premedication, as outlined in the product label. However, serious infusion reactions may occur despite premedication.
If you suspect a severe infusion reaction, stop the infusion immediately and provide symptomatic treatment. Signs and symptoms of an infusion reaction may include swelling of the face or mouth, fever, chills, difficulty breathing, tightness of the chest and/or throat, light headedness, nausea, diarrhea, and rash.
These symptoms can occur during or shortly after the infusion, predominantly with the first 2 infusions. So ensure patients are closely monitored, especially those with heart conditions. And inform patients about the risk of fatal infusion reactions associated with ofatumumab.
GSK has sent a letter to healthcare professionals detailing the risk of fatal infusion reactions. The information is also available on the Canadian website of GSK and the Health Canada website.
Any case of serious hypersensitivity, infusion reactions, or other serious or unexpected side effects in patients receiving ofatumumab should be reported to GSK or Health Canada.
Ofatumumab is also known to pose a risk of hepatitis B virus reactivation, progressive multifocal leukoencephalopathy, serious and/or fatal cardiovascular events, and serious and/or fatal infections (bacterial, fungal, and viral).
Ofatumumab recently received approval in the European Union to be used in combination with chlorambucil or bendamustine for untreated CLL patients who are not eligible for fludarabine-based therapy. The drug previously received conditional approval in Europe as monotherapy to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Ofatumumab is approved for both of these indications in the US as well. ![]()
Credit: Bill Branson
Health Canada and GlaxoSmithKline (GSK) have reported a fatal infusion reaction in a patient receiving the monoclonal antibody ofatumumab (Arzerra) to treat
chronic lymphocytic leukemia (CLL).
The patient had no known history of cardiac disease.
Ofatumumab’s product monograph is being updated to include a warning about the potential for fatal infusion reactions.
In Canada, ofatumumab is approved to treat patients with CLL that is refractory to fludarabine and alemtuzumab.
The drug received this marketing authorization with conditions, pending the results of trials to verify its clinical benefit.
In light of the fatal infusion reaction, GSK and Health Canada are reminding healthcare professionals that ofatumumab should be administered under the supervision of a physician experienced in the use of cancer therapy. The drug should be given in an environment where facilities to adequately monitor and treat infusion reactions are available.
Prior to infusion, patients should always receive the appropriate premedication, as outlined in the product label. However, serious infusion reactions may occur despite premedication.
If you suspect a severe infusion reaction, stop the infusion immediately and provide symptomatic treatment. Signs and symptoms of an infusion reaction may include swelling of the face or mouth, fever, chills, difficulty breathing, tightness of the chest and/or throat, light headedness, nausea, diarrhea, and rash.
These symptoms can occur during or shortly after the infusion, predominantly with the first 2 infusions. So ensure patients are closely monitored, especially those with heart conditions. And inform patients about the risk of fatal infusion reactions associated with ofatumumab.
GSK has sent a letter to healthcare professionals detailing the risk of fatal infusion reactions. The information is also available on the Canadian website of GSK and the Health Canada website.
Any case of serious hypersensitivity, infusion reactions, or other serious or unexpected side effects in patients receiving ofatumumab should be reported to GSK or Health Canada.
Ofatumumab is also known to pose a risk of hepatitis B virus reactivation, progressive multifocal leukoencephalopathy, serious and/or fatal cardiovascular events, and serious and/or fatal infections (bacterial, fungal, and viral).
Ofatumumab recently received approval in the European Union to be used in combination with chlorambucil or bendamustine for untreated CLL patients who are not eligible for fludarabine-based therapy. The drug previously received conditional approval in Europe as monotherapy to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Ofatumumab is approved for both of these indications in the US as well. ![]()
Credit: Bill Branson
Health Canada and GlaxoSmithKline (GSK) have reported a fatal infusion reaction in a patient receiving the monoclonal antibody ofatumumab (Arzerra) to treat
chronic lymphocytic leukemia (CLL).
The patient had no known history of cardiac disease.
Ofatumumab’s product monograph is being updated to include a warning about the potential for fatal infusion reactions.
In Canada, ofatumumab is approved to treat patients with CLL that is refractory to fludarabine and alemtuzumab.
The drug received this marketing authorization with conditions, pending the results of trials to verify its clinical benefit.
In light of the fatal infusion reaction, GSK and Health Canada are reminding healthcare professionals that ofatumumab should be administered under the supervision of a physician experienced in the use of cancer therapy. The drug should be given in an environment where facilities to adequately monitor and treat infusion reactions are available.
Prior to infusion, patients should always receive the appropriate premedication, as outlined in the product label. However, serious infusion reactions may occur despite premedication.
If you suspect a severe infusion reaction, stop the infusion immediately and provide symptomatic treatment. Signs and symptoms of an infusion reaction may include swelling of the face or mouth, fever, chills, difficulty breathing, tightness of the chest and/or throat, light headedness, nausea, diarrhea, and rash.
These symptoms can occur during or shortly after the infusion, predominantly with the first 2 infusions. So ensure patients are closely monitored, especially those with heart conditions. And inform patients about the risk of fatal infusion reactions associated with ofatumumab.
GSK has sent a letter to healthcare professionals detailing the risk of fatal infusion reactions. The information is also available on the Canadian website of GSK and the Health Canada website.
Any case of serious hypersensitivity, infusion reactions, or other serious or unexpected side effects in patients receiving ofatumumab should be reported to GSK or Health Canada.
Ofatumumab is also known to pose a risk of hepatitis B virus reactivation, progressive multifocal leukoencephalopathy, serious and/or fatal cardiovascular events, and serious and/or fatal infections (bacterial, fungal, and viral).
Ofatumumab recently received approval in the European Union to be used in combination with chlorambucil or bendamustine for untreated CLL patients who are not eligible for fludarabine-based therapy. The drug previously received conditional approval in Europe as monotherapy to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Ofatumumab is approved for both of these indications in the US as well.
CHMP recommends antifungal agent
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for intravenous (IV) posaconazole (Noxafil), an antifungal agent.
If the European Commission affirms the CHMP opinion, IV posaconazole will be authorized for use in the European Union, Iceland, Liechtenstein, and Norway.
The commission previously granted marketing authorization for posaconazole delayed-release tablets and oral suspension.
Posaconazole is used to prevent invasive fungal infections in severely immunocompromised patients, such as hematopoietic stem cell transplant recipients with graft-vs-host disease or patients with hematologic malignancies and prolonged neutropenia from chemotherapy.
The drug is also used to treat fungal diseases—invasive aspergillosis, fusariosis, chromoblastomycosis, mycetoma, and coccidioidomycosis—when other antifungal agents—amphotericin B, itraconazole, or fluconazole—cannot be tolerated or have failed.
And posaconazole oral suspension is used as a first-line treatment for thrush, a fungal infection of the mouth and throat due to Candida.
Posaconazole injection is administered with a loading dose of 300 mg twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by IV infusion over approximately 90 minutes.
Once combined with a mixture of IV solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2°-8° C (36°-46° F).
The safety and effectiveness of IV posaconazole in patients younger than 18 years has not been established. IV posaconazole should not be used in pediatric patients because of non-clinical safety concerns.
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
Patients with known hypersensitivity to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
Hepatic reactions have been reported as well. This includes mild to moderate elevations in ALT, AST, alkaline phosphatase, total bilirubin, and/or clinical hepatitis. Monitoring or discontinuation may be necessary in patients with hepatic reactions to posaconazole.
IV posaconazole should be avoided in patients with moderate or severe renal impairment (estimated glomerular filtration rate <50 mL/min), unless an assessment of the benefit/risk to the patient justifies the use of posaconazole.
In clinical trials, the adverse events associated with IV posaconazole were generally similar to those in trials of posaconazole oral suspension. The most frequently reported events were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
IV posaconazole is under development by MSD (known as Merck in the US and Canada).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for intravenous (IV) posaconazole (Noxafil), an antifungal agent.
If the European Commission affirms the CHMP opinion, IV posaconazole will be authorized for use in the European Union, Iceland, Liechtenstein, and Norway.
The commission previously granted marketing authorization for posaconazole delayed-release tablets and oral suspension.
Posaconazole is used to prevent invasive fungal infections in severely immunocompromised patients, such as hematopoietic stem cell transplant recipients with graft-vs-host disease or patients with hematologic malignancies and prolonged neutropenia from chemotherapy.
The drug is also used to treat fungal diseases—invasive aspergillosis, fusariosis, chromoblastomycosis, mycetoma, and coccidioidomycosis—when other antifungal agents—amphotericin B, itraconazole, or fluconazole—cannot be tolerated or have failed.
And posaconazole oral suspension is used as a first-line treatment for thrush, a fungal infection of the mouth and throat due to Candida.
Posaconazole injection is administered with a loading dose of 300 mg twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by IV infusion over approximately 90 minutes.
Once combined with a mixture of IV solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2°-8° C (36°-46° F).
The safety and effectiveness of IV posaconazole in patients younger than 18 years has not been established. IV posaconazole should not be used in pediatric patients because of non-clinical safety concerns.
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
Patients with known hypersensitivity to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
Hepatic reactions have been reported as well. This includes mild to moderate elevations in ALT, AST, alkaline phosphatase, total bilirubin, and/or clinical hepatitis. Monitoring or discontinuation may be necessary in patients with hepatic reactions to posaconazole.
IV posaconazole should be avoided in patients with moderate or severe renal impairment (estimated glomerular filtration rate <50 mL/min), unless an assessment of the benefit/risk to the patient justifies the use of posaconazole.
In clinical trials, the adverse events associated with IV posaconazole were generally similar to those in trials of posaconazole oral suspension. The most frequently reported events were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
IV posaconazole is under development by MSD (known as Merck in the US and Canada).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for intravenous (IV) posaconazole (Noxafil), an antifungal agent.
If the European Commission affirms the CHMP opinion, IV posaconazole will be authorized for use in the European Union, Iceland, Liechtenstein, and Norway.
The commission previously granted marketing authorization for posaconazole delayed-release tablets and oral suspension.
Posaconazole is used to prevent invasive fungal infections in severely immunocompromised patients, such as hematopoietic stem cell transplant recipients with graft-vs-host disease or patients with hematologic malignancies and prolonged neutropenia from chemotherapy.
The drug is also used to treat fungal diseases—invasive aspergillosis, fusariosis, chromoblastomycosis, mycetoma, and coccidioidomycosis—when other antifungal agents—amphotericin B, itraconazole, or fluconazole—cannot be tolerated or have failed.
And posaconazole oral suspension is used as a first-line treatment for thrush, a fungal infection of the mouth and throat due to Candida.
Posaconazole injection is administered with a loading dose of 300 mg twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by IV infusion over approximately 90 minutes.
Once combined with a mixture of IV solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2°-8° C (36°-46° F).
The safety and effectiveness of IV posaconazole in patients younger than 18 years has not been established. IV posaconazole should not be used in pediatric patients because of non-clinical safety concerns.
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
Patients with known hypersensitivity to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
Hepatic reactions have been reported as well. This includes mild to moderate elevations in ALT, AST, alkaline phosphatase, total bilirubin, and/or clinical hepatitis. Monitoring or discontinuation may be necessary in patients with hepatic reactions to posaconazole.
IV posaconazole should be avoided in patients with moderate or severe renal impairment (estimated glomerular filtration rate <50 mL/min), unless an assessment of the benefit/risk to the patient justifies the use of posaconazole.
In clinical trials, the adverse events associated with IV posaconazole were generally similar to those in trials of posaconazole oral suspension. The most frequently reported events were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
IV posaconazole is under development by MSD (known as Merck in the US and Canada).