User login
ASCO addresses needs of SGMs with cancer

The American Society of Clinical Oncology (ASCO) has issued recommendations addressing the needs of sexual and gender minority (SGM) populations with cancer.
The recommendations are designed to focus attention on the challenges facing the SGM community—including discrimination and greater risk of anxiety and depression, resulting in disparate care—and concrete steps that can help minimize health disparities among SGM individuals.
The recommendations were published in a policy statement in the Journal of Clinical Oncology.
“Sexual and gender minorities face unique challenges related to cancer risk, discrimination, and other psychosocial issues,” said ASCO President Daniel F. Hayes, MD.
“Compounding these challenges is the fact that providers may have a lack of knowledge and sensitivity about the health risks and health needs facing their SGM patients.”
SGMs include individuals who are lesbian, gay, bisexual, transgender, and intersex (also referred to as those with differences in sex development).
ASCO’s policy statement notes that SGM populations bear a disproportionate cancer burden stemming from several factors, including:
- Lower rates of cancer screening, in part due to lower rates of insurance coverage, exclusion from traditional screening campaigns, and previous experience of discrimination in the healthcare system
- A hesitancy on the part of SGM patients to disclose their sexual orientation to providers due to a fear of stigmatization, which can create additional barriers to care.
ASCO’s statement calls for a coordinated effort to address health disparities among SGM populations, including:
- Increased patient access to culturally competent support services
- Expanded cancer prevention education for SGM individuals
- Robust policies prohibiting discrimination
- Adequate insurance coverage to meet the needs of SGM individuals affected by cancer
- Inclusion of SGM status as a required data element in cancer registries and clinical trials
- Increased focus on SGM populations in cancer research.
“Our objective was to raise awareness among oncology providers, patients, policy makers, and other stakeholders about the cancer care needs of SGM populations and the barriers that SGM individuals face in getting the highest-quality care,” said Jennifer J. Griggs, MD, lead author of the policy statement and a professor at the University of Michigan in Ann Arbor.
“To address these barriers, a coordinated effort is needed to enhance education for patients and providers, to improve outreach and support, and to encourage productive policy and legislative action.” ![]()
The American Society of Clinical Oncology (ASCO) has issued recommendations addressing the needs of sexual and gender minority (SGM) populations with cancer.
The recommendations are designed to focus attention on the challenges facing the SGM community—including discrimination and greater risk of anxiety and depression, resulting in disparate care—and concrete steps that can help minimize health disparities among SGM individuals.
The recommendations were published in a policy statement in the Journal of Clinical Oncology.
“Sexual and gender minorities face unique challenges related to cancer risk, discrimination, and other psychosocial issues,” said ASCO President Daniel F. Hayes, MD.
“Compounding these challenges is the fact that providers may have a lack of knowledge and sensitivity about the health risks and health needs facing their SGM patients.”
SGMs include individuals who are lesbian, gay, bisexual, transgender, and intersex (also referred to as those with differences in sex development).
ASCO’s policy statement notes that SGM populations bear a disproportionate cancer burden stemming from several factors, including:
- Lower rates of cancer screening, in part due to lower rates of insurance coverage, exclusion from traditional screening campaigns, and previous experience of discrimination in the healthcare system
- A hesitancy on the part of SGM patients to disclose their sexual orientation to providers due to a fear of stigmatization, which can create additional barriers to care.
ASCO’s statement calls for a coordinated effort to address health disparities among SGM populations, including:
- Increased patient access to culturally competent support services
- Expanded cancer prevention education for SGM individuals
- Robust policies prohibiting discrimination
- Adequate insurance coverage to meet the needs of SGM individuals affected by cancer
- Inclusion of SGM status as a required data element in cancer registries and clinical trials
- Increased focus on SGM populations in cancer research.
“Our objective was to raise awareness among oncology providers, patients, policy makers, and other stakeholders about the cancer care needs of SGM populations and the barriers that SGM individuals face in getting the highest-quality care,” said Jennifer J. Griggs, MD, lead author of the policy statement and a professor at the University of Michigan in Ann Arbor.
“To address these barriers, a coordinated effort is needed to enhance education for patients and providers, to improve outreach and support, and to encourage productive policy and legislative action.” ![]()
The American Society of Clinical Oncology (ASCO) has issued recommendations addressing the needs of sexual and gender minority (SGM) populations with cancer.
The recommendations are designed to focus attention on the challenges facing the SGM community—including discrimination and greater risk of anxiety and depression, resulting in disparate care—and concrete steps that can help minimize health disparities among SGM individuals.
The recommendations were published in a policy statement in the Journal of Clinical Oncology.
“Sexual and gender minorities face unique challenges related to cancer risk, discrimination, and other psychosocial issues,” said ASCO President Daniel F. Hayes, MD.
“Compounding these challenges is the fact that providers may have a lack of knowledge and sensitivity about the health risks and health needs facing their SGM patients.”
SGMs include individuals who are lesbian, gay, bisexual, transgender, and intersex (also referred to as those with differences in sex development).
ASCO’s policy statement notes that SGM populations bear a disproportionate cancer burden stemming from several factors, including:
- Lower rates of cancer screening, in part due to lower rates of insurance coverage, exclusion from traditional screening campaigns, and previous experience of discrimination in the healthcare system
- A hesitancy on the part of SGM patients to disclose their sexual orientation to providers due to a fear of stigmatization, which can create additional barriers to care.
ASCO’s statement calls for a coordinated effort to address health disparities among SGM populations, including:
- Increased patient access to culturally competent support services
- Expanded cancer prevention education for SGM individuals
- Robust policies prohibiting discrimination
- Adequate insurance coverage to meet the needs of SGM individuals affected by cancer
- Inclusion of SGM status as a required data element in cancer registries and clinical trials
- Increased focus on SGM populations in cancer research.
“Our objective was to raise awareness among oncology providers, patients, policy makers, and other stakeholders about the cancer care needs of SGM populations and the barriers that SGM individuals face in getting the highest-quality care,” said Jennifer J. Griggs, MD, lead author of the policy statement and a professor at the University of Michigan in Ann Arbor.
“To address these barriers, a coordinated effort is needed to enhance education for patients and providers, to improve outreach and support, and to encourage productive policy and legislative action.” ![]()
Report shows increase in blood cancer incidence and survival

A report on cancer in the US suggests the incidence of leukemia and myeloma has been on the rise in recent years, but the incidence of non-Hodgkin lymphoma (NHL) has been on the decline.
Meanwhile, annual death rates for leukemia and NHL have decreased, and annual death rates for myeloma have decreased in men but not in women.
Furthermore, patients with leukemia, NHL, and myeloma have seen a substantial improvement in 5-year survival rates in recent years relative to patients in the late 1970s.
These findings are part of the Annual Report to the Nation on the Status of Cancer, 1975-2014, which has been published in the Journal of the National Cancer Institute.
This report is released each year, but the current edition includes a special section focused on survival.
“While trends in death rates are the most commonly used measure to assess progress against cancer, survival trends are also an important measure to evaluate progress in improvement of cancer outcomes,” said Ahmedin Jemal, DVM, PhD, of the American Cancer Society.
“We last included a special section on cancer survival in 2004, and, as we found then, survival improved over time for almost all cancers at every stage of diagnosis.”
For the current report, researchers calculated the 5-year average annual percent changes (AAPCs) for 2009 to 2013 for cancer incidence and for 2010 to 2014 for cancer mortality.
Cancer incidence (2009-2013)
In women, the AAPC increased 1.5% for leukemia (P<0.05), decreased 0.5% for NHL (P<0.05), and increased 2.2% for myeloma (P<0.05).
In men, the AAPC increased 1.7% for leukemia (P<0.05), decreased 0.2% for NHL, and increased 2.8% for myeloma (P<0.05).
Cancer mortality (2010-2014)
In women, the AAPC decreased 1.2% for leukemia (P<0.05), decreased 2.2% for NHL (P<0.05), and increased 0.5% for myeloma.
In men, the AAPC decreased 1.0% for leukemia (P<0.05), decreased 2.0% for NHL (P<0.05), and decreased 0.9% for myeloma (P<0.05).
5-year survival
The researchers compared 5-year relative survival for cancers diagnosed from 1975 to 1977 and those diagnosed from 2006 to 2012.
The absolute percentage change over time (for both sexes combined) was 26.1% for NHL, 25.7% for myeloma, and 28.5% for leukemia.
Five-year survival for patients diagnosed in 1975-1977 was 46.5% for NHL, 24.6% for myeloma, and 34.2% for leukemia.
Five-year survival for patients diagnosed in 2006-2012 was 72.6% for NHL, 50.2% for myeloma, and 62.7% for leukemia. ![]()
A report on cancer in the US suggests the incidence of leukemia and myeloma has been on the rise in recent years, but the incidence of non-Hodgkin lymphoma (NHL) has been on the decline.
Meanwhile, annual death rates for leukemia and NHL have decreased, and annual death rates for myeloma have decreased in men but not in women.
Furthermore, patients with leukemia, NHL, and myeloma have seen a substantial improvement in 5-year survival rates in recent years relative to patients in the late 1970s.
These findings are part of the Annual Report to the Nation on the Status of Cancer, 1975-2014, which has been published in the Journal of the National Cancer Institute.
This report is released each year, but the current edition includes a special section focused on survival.
“While trends in death rates are the most commonly used measure to assess progress against cancer, survival trends are also an important measure to evaluate progress in improvement of cancer outcomes,” said Ahmedin Jemal, DVM, PhD, of the American Cancer Society.
“We last included a special section on cancer survival in 2004, and, as we found then, survival improved over time for almost all cancers at every stage of diagnosis.”
For the current report, researchers calculated the 5-year average annual percent changes (AAPCs) for 2009 to 2013 for cancer incidence and for 2010 to 2014 for cancer mortality.
Cancer incidence (2009-2013)
In women, the AAPC increased 1.5% for leukemia (P<0.05), decreased 0.5% for NHL (P<0.05), and increased 2.2% for myeloma (P<0.05).
In men, the AAPC increased 1.7% for leukemia (P<0.05), decreased 0.2% for NHL, and increased 2.8% for myeloma (P<0.05).
Cancer mortality (2010-2014)
In women, the AAPC decreased 1.2% for leukemia (P<0.05), decreased 2.2% for NHL (P<0.05), and increased 0.5% for myeloma.
In men, the AAPC decreased 1.0% for leukemia (P<0.05), decreased 2.0% for NHL (P<0.05), and decreased 0.9% for myeloma (P<0.05).
5-year survival
The researchers compared 5-year relative survival for cancers diagnosed from 1975 to 1977 and those diagnosed from 2006 to 2012.
The absolute percentage change over time (for both sexes combined) was 26.1% for NHL, 25.7% for myeloma, and 28.5% for leukemia.
Five-year survival for patients diagnosed in 1975-1977 was 46.5% for NHL, 24.6% for myeloma, and 34.2% for leukemia.
Five-year survival for patients diagnosed in 2006-2012 was 72.6% for NHL, 50.2% for myeloma, and 62.7% for leukemia. ![]()
A report on cancer in the US suggests the incidence of leukemia and myeloma has been on the rise in recent years, but the incidence of non-Hodgkin lymphoma (NHL) has been on the decline.
Meanwhile, annual death rates for leukemia and NHL have decreased, and annual death rates for myeloma have decreased in men but not in women.
Furthermore, patients with leukemia, NHL, and myeloma have seen a substantial improvement in 5-year survival rates in recent years relative to patients in the late 1970s.
These findings are part of the Annual Report to the Nation on the Status of Cancer, 1975-2014, which has been published in the Journal of the National Cancer Institute.
This report is released each year, but the current edition includes a special section focused on survival.
“While trends in death rates are the most commonly used measure to assess progress against cancer, survival trends are also an important measure to evaluate progress in improvement of cancer outcomes,” said Ahmedin Jemal, DVM, PhD, of the American Cancer Society.
“We last included a special section on cancer survival in 2004, and, as we found then, survival improved over time for almost all cancers at every stage of diagnosis.”
For the current report, researchers calculated the 5-year average annual percent changes (AAPCs) for 2009 to 2013 for cancer incidence and for 2010 to 2014 for cancer mortality.
Cancer incidence (2009-2013)
In women, the AAPC increased 1.5% for leukemia (P<0.05), decreased 0.5% for NHL (P<0.05), and increased 2.2% for myeloma (P<0.05).
In men, the AAPC increased 1.7% for leukemia (P<0.05), decreased 0.2% for NHL, and increased 2.8% for myeloma (P<0.05).
Cancer mortality (2010-2014)
In women, the AAPC decreased 1.2% for leukemia (P<0.05), decreased 2.2% for NHL (P<0.05), and increased 0.5% for myeloma.
In men, the AAPC decreased 1.0% for leukemia (P<0.05), decreased 2.0% for NHL (P<0.05), and decreased 0.9% for myeloma (P<0.05).
5-year survival
The researchers compared 5-year relative survival for cancers diagnosed from 1975 to 1977 and those diagnosed from 2006 to 2012.
The absolute percentage change over time (for both sexes combined) was 26.1% for NHL, 25.7% for myeloma, and 28.5% for leukemia.
Five-year survival for patients diagnosed in 1975-1977 was 46.5% for NHL, 24.6% for myeloma, and 34.2% for leukemia.
Five-year survival for patients diagnosed in 2006-2012 was 72.6% for NHL, 50.2% for myeloma, and 62.7% for leukemia. ![]()
Demystifying the diagnosis and classification of lymphoma: a guide to the hematopathologist’s galaxy
Lymphomas constitute a very heterogeneous group of neoplasms with diverse clinical presentations, prognoses, and responses to therapy. Approximately 80,500 new cases of lymphoma are expected to be diagnosed in the United States in 2017, of which about one quarter will lead to the death of the patient.1 Perhaps more so than any other group of neoplasms, the diagnosis of lymphoma involves the integration of a multiplicity of clinical, histologic and immunophenotypic findings and, on occasion, cytogenetic and molecular results as well. An accurate diagnosis of lymphoma, usually rendered by hematopathologists, allows hematologists/oncologists to treat patients appropriately. Herein we will describe a simplified approach to the diagnosis and classification of lymphomas (Figure 1).
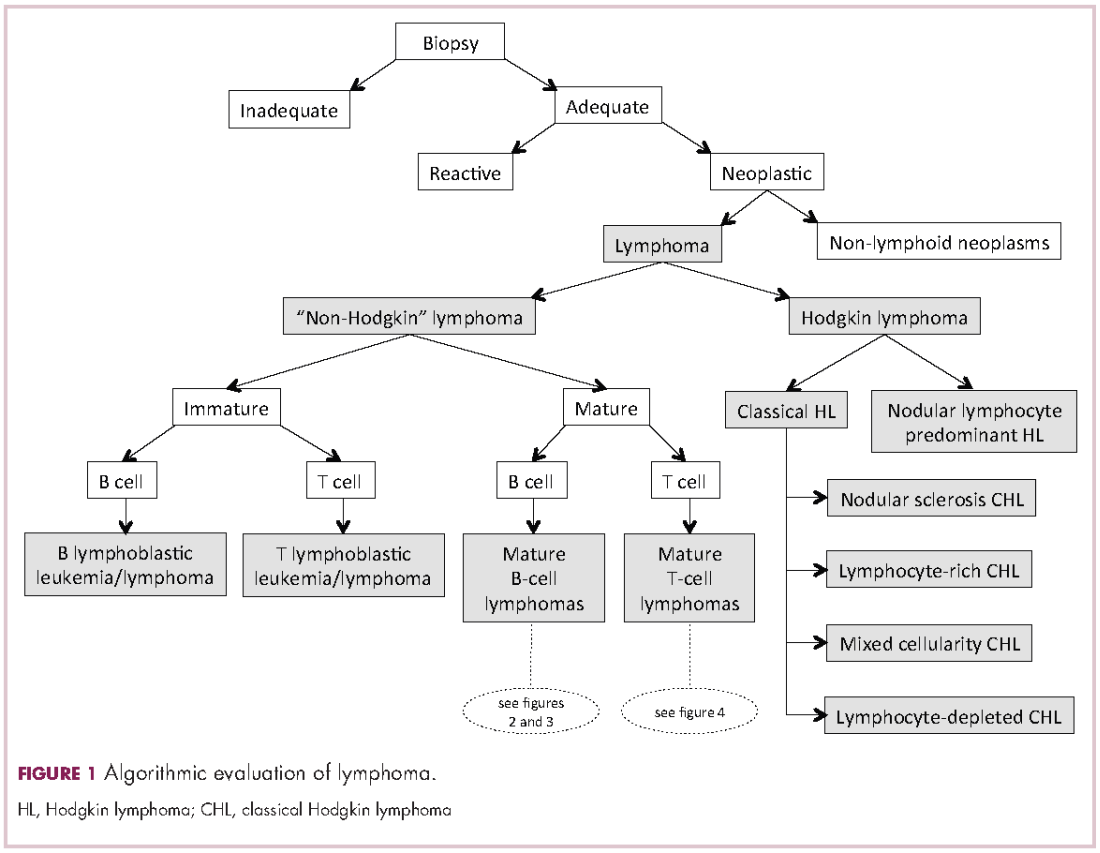
Lymphoma classification
Lymphomas are clonal neoplasms characterized by the expansion of abnormal lymphoid cells that may develop in any organ but commonly involve lymph nodes. The fourth edition of the World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid tissues, published in 2008, is the official and most current guideline used for diagnosis of lymphoid neoplasms.2 The WHO scheme classifies lymphomas according to the type of cell from which they are derived (mature and immature B cells, T cells, or natural killer (NK) cells, findings determined by their morphology and immunophenotype) and their clinical, cytogenetic, and/or molecular features. This official classification is currently being updated3 and is expected to be published in full in 2017, at which time it is anticipated to include definitions for more than 70 distinct neoplasms.
Lymphomas are broadly and informally classified as Hodgkin lymphomas (HLs) and non-Hodgkin lymphomas (NHLs), based on the differences these two groups show in their clinical presentation, treatment, prognosis, and proportion of neoplastic cells, among others. NHLs are by far the most common type of lymphomas, accounting for approximately 90% of all new cases of lymphoma in the United States and 70% worldwide.1,2 NHLs are a very heterogeneous group of B-, T-, or NK-cell neoplasms that, in turn, can also be informally subclassified as low-grade (or indolent) or high-grade (or aggressive) according to their predicted clinical behavior. HLs are comparatively rare, less heterogeneous, uniformly of B-cell origin and, in the case of classical Hodgkin lymphoma, highly curable.1,2 It is beyond the scope of this manuscript to outline the features of each of the >70 specific entities, but the reader is referred elsewhere for more detail and encouraged to become familiarized with the complexity, challenges, and beauty of lymphoma diagnosis.2,3
Biopsy procedure
A correct diagnosis begins with an adequate biopsy procedure. It is essential that biopsy specimens for lymphoma evaluation be submitted fresh and unfixed, because some crucial analyses such as flow cytometry or conventional cytogenetics can only be performed on fresh tissue. Indeed, it is important for the hematologist/oncologist and/or surgeon and/or interventional radiologist to converse with the hematopathologist prior to and even during some procedures to ensure the correct processing of the specimen. Also, it is important to limit the compression of the specimen and the excessive use of cauterization during the biopsy procedure, both of which cause artifacts that may render impossible the interpretation of the histopathologic findings.
Given that the diagnosis of lymphoma is based not only on the cytologic details of the lymphoma cells but also on the architectural pattern with which they infiltrate an organ, the larger the biopsy specimen, the easier it will be for a hematopathologist to identify the pattern. In addition, excisional biopsies frequently contain more diagnostic tissue than needle core biopsies and this provides pathologists with the option to submit tissue fragments for ancillary tests that require unfixed tissue as noted above. Needle core biopsies of lymph nodes are increasingly being used because of their association with fewer complications and lower cost than excisional biopsies. However, needle core biopsies provide only a glimpse of the pattern of infiltration and may not be completely representative of the architecture. Therefore, excisional lymph node biopsies of lymph nodes are preferred over needle core biopsies, recognizing that in the setting of deeply seated lymph nodes, needle core biopsies may be the only or the best surgical option.
Clinical presentation
Accurate diagnosis of lymphoma cannot take place in a vacuum. The hematopathologist’s initial approach to the diagnosis of lymphoid processes in tissue biopsies should begin with a thorough review of the clinical history, although some pathology laboratories may not have immediate access to this information. The hematopathologist should evaluate factors such as age, gender, location of the tumor, symptomatology, medications, serology, and prior history of malignancy, immunosuppression or immunodeficiency in every case. Other important but frequently omitted parts of the clinical history are the patient’s occupation, history of exposure to animals, and the presence of tattoos, which may be associated with certain reactive lymphadenopathies.
Histomorphologic evaluation
Despite the plethora of new and increasingly sophisticated tools, histologic and morphologic analysis still remains the cornerstone of diagnosis in hematopathology. However, for the characterization of an increasing number of reactive and neoplastic lymphoid processes, hematopathologists may also require immunophenotypic, molecular, and cytogenetic tests for an accurate diagnosis. Upon review of the clinical information, a microscopic evaluation of the tissue submitted for processing by the histology laboratory will be performed. The results of concurrent flow cytometric evaluation (performed on fresh unfixed material) should also be available in most if not all cases before the H&E-stained slides are available for review. Upon receipt of H&E-stained slides, the hematopathologist will evaluate the quality of the submitted specimen, since many diagnostic difficulties stem from suboptimal techniques related to the biopsy procedure, fixation, processing, cutting, or staining (Figure 1). If deemed suitable for accurate diagnosis, a search for signs of preservation or disruption of the organ that was biopsied will follow. The identification of certain morphologic patterns aids the hematopathologist in answering the first question: “what organ is this and is this consistent with what is indicated on the requisition?” This is usually immediately followed by “is this sufficient and adequate material for a diagnosis?” and “is there any normal architecture?” If the architecture is not normal, “is this alteration due to a reactive or a neoplastic process?” If neoplastic, “is it lymphoma or a non-hematolymphoid neoplasm?”
Both reactive and neoplastic processes have variably unique morphologic features that if properly recognized, guide the subsequent testing. However, some reactive and neoplastic processes can present with overlapping features, and even after extensive immunophenotypic evaluation and the performance of ancillary studies, it may not be possible to conclusively determine its nature. If the lymph node architecture is altered or effaced, the predominant pattern of infiltration (eg, nodular, diffuse, interfollicular, intrasinusoidal) and the degree of alteration of the normal architecture is evaluated, usually at low magnification. When the presence of an infiltrate is recognized, its components must be characterized. If the infiltrate is composed of a homogeneous expansion of lymphoid cells that disrupts or replaces the normal lymphoid architecture, a lymphoma will be suspected or diagnosed. The pattern of distribution of the cells along with their individual morphologic characteristics (ie, size, nuclear shape, chromatin configuration, nucleoli, amount and hue of cytoplasm) are key factors for the diagnosis and classification of the lymphoma that will guide subsequent testing. The immunophenotypic analysis (by immunohistochemistry, flow cytometry or a combination of both) may confirm the reactive or neoplastic nature of the process, and its subclassification. B-cell lymphomas, in particular have variable and distinctive histologic features: as a diffuse infiltrate of large mature lymphoid cells (eg, diffuse large B-cell lymphoma), an expansion of immature lymphoid cells (lymphoblastic lymphoma), and a nodular infiltrate of small, intermediate and/or mature large B cells (eg, follicular lymphoma).

Mature T-cell lymphomas may display similar histologic, features but they can be quite heterogeneous with an infiltrate composed of one predominant cell type or a mixture of small, medium-sized, and large atypical lymphoid cells (on occasion with abundant clear cytoplasm) and a variable number of eosinophils, plasma cells, macrophages (including granulomas), and B cells. HLs most commonly efface the lymph node architecture with a nodular or diffuse infiltrate variably composed of reactive lymphocytes, granulocytes, macrophages, and plasma cells and usually a minority of large neoplastic cells (Hodgkin/Reed-Sternberg cells and/or lymphocyte predominant cells).
Once the H&E-stained slides are evaluated and a diagnosis of lymphoma is suspected or established, the hematopathologist will attempt to determine whether it has mature or immature features, and whether low- or high-grade morphologic characteristics are present. The maturity of lymphoid cells is generally determined by the nature of the chromatin, which if “fine” and homogeneous (with or without a conspicuous nucleolus) will usually, but not always, be considered immature, whereas clumped, vesicular or hyperchromatic chromatin is generally, but not always, associated with maturity. If the chromatin displays immature features, the differential diagnosis will mainly include B- and T-lymphoblastic lymphomas, but also blastoid variants of mature neoplasm such as mantle cell lymphoma, and follicular lymphoma, as well as high-grade B-cell lymphomas. Features associated with low-grade lymphomas (eg, follicular lymphoma, small lymphocytic lymphoma/chronic lymphocytic leukemia, marginal zone lymphoma, lymphoplasmacytic lymphoma) include small cell morphology, mature chromatin, absence of a significant number of mitoses or apoptotic cells, and a low proliferation index as shown by immunohistochemistry for Ki67. High-grade lymphomas, such as lymphoblastic lymphoma, Burkitt lymphoma, or certain large B-cell lymphomas tend to show opposite features, and some of the mature entities are frequently associated with MYC rearrangements. Of note, immature lymphomas tend to be clinically high grade, but not all clinically high-grade lymphomas are immature. Conversely, the majority of low-grade lymphomas are usually mature.
Immunophenotypic evaluation
Immunophenotypic evaluation is essential because the lineage of lymphoma cells cannot be determined by morphology alone. The immunophenotype is the combination of proteins/markers (eg, CD20, CD3, TdT) expressed by cells. Usually, it is evaluated by immunohistochemistry and/or flow cytometry, which help determine the proportion of lymphoid cells that express a certain marker and its location and intensity within the cells. While immunohistochemistry is normally performed on formalin-fixed and paraffin-embedded tissue, flow cytometry can be evaluated only on fresh unfixed tissue. Flow cytometry has the advantage over immunohistochemistry of being faster and better at simultaneously identifying coexpression of multiple markers on multiple cell populations. However, certain markers can only be evaluated by immunohistochemistry.
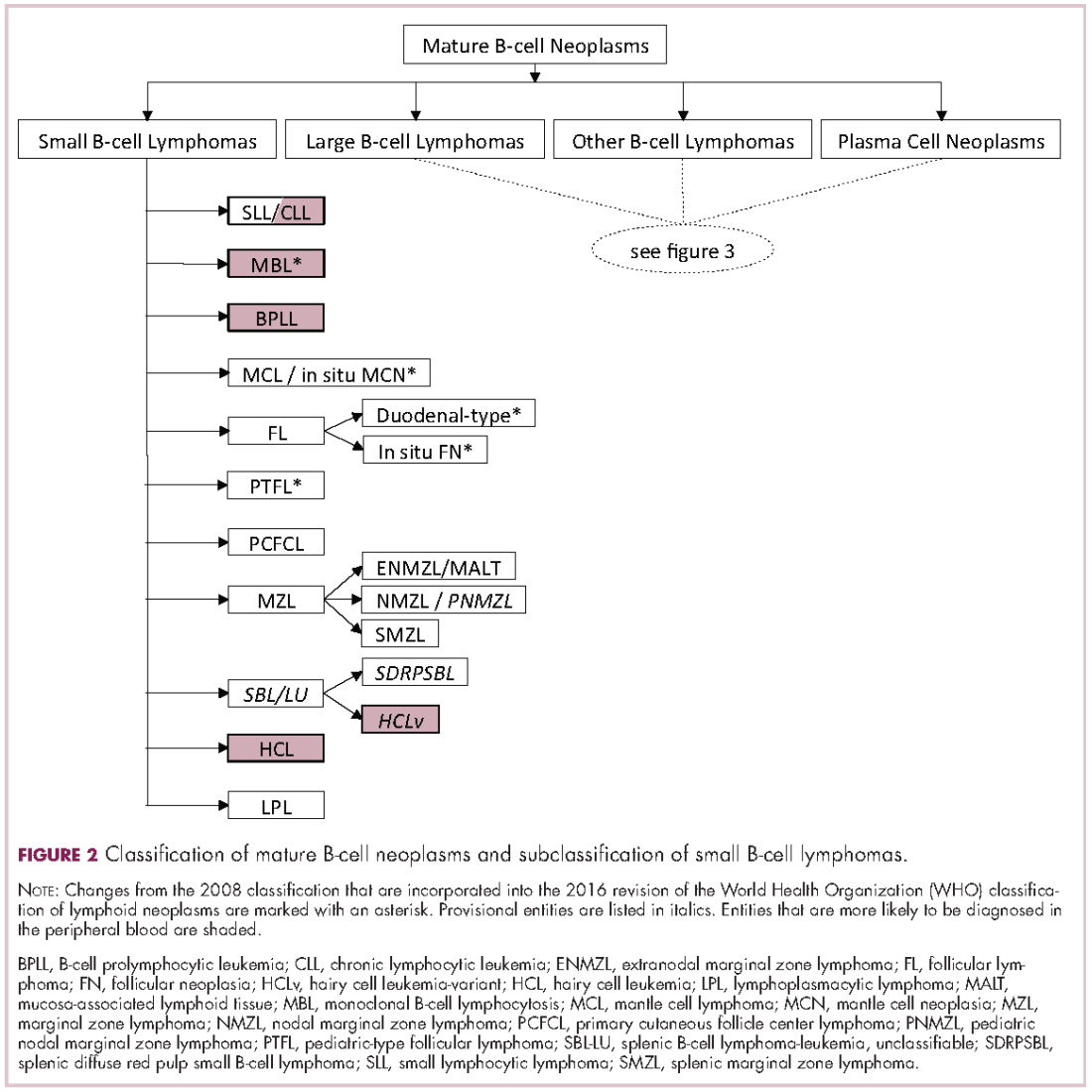
The immunophenotypic analysis will in most cases reveal whether the lymphomas is of B-, T- or NK-cell origin, and whether a lymphoma subtype associated immunophenotype is present. Typical pan B-cell antigens include PAX5, CD19, and CD79a (CD20 is less broadly expressed throughout B-cell differentiation, although it is usually evident in most mature B-cell lymphomas), and typical pan T-cell antigens include CD2, CD5, and CD7. The immature or mature nature of a lymphoma can also be confirmed by evaluation of the immunophenotype. Immature lymphomas commonly express one or more of TdT, CD10, or CD34; T-lymphoblastic lymphoma cells may also coexpress CD1a. The majority of NHLs and all HLs are derived from (or reflect) B cells at different stages of maturation. Mature B-cell lymphomas are the most common type of lymphoma and typically, but not always, express pan B-cell markers as well as surface membrane immunoglobulin, with the latter also most useful in assessing clonality via a determination of light chain restriction. Some mature B-cell lymphomas tend to acquire markers that are either never physiologically expressed by normal mature B cells (eg, cyclin D1 in mantle cell lymphoma, or BCL2 in germinal center B cells in follicular lymphoma) or only expressed in a minor fraction (eg, CD5 that is characteristically expressed in small lymphocytic and mantle cell lymphoma). The most common mature B-cell lymphomas include diffuse large B-cell lymphoma, follicular lymphoma, small lymphocytic lymphoma, mantle cell lymphoma, marginal zone lymphoma, Burkitt lymphoma, and lymphoplasmacytic lymphoma (Figures 2 and 3). Classical HLs are also lymphomas of B-cell origin that demonstrate diminished preservation of their B-cell immunophenotype (as evidenced by the dim expression of PAX5 but absence of most other pan B-cell antigens), expression of CD30, variable expression of CD15, and loss of CD45 (Figure 1). In contrast, nodular lymphocyte predominant HL shows a preserved B-cell immunophenotypic program and expression of CD45, typically without CD30 and CD15. Of note, the evaluation of the immunophenotype of the neoplastic cells in HL is routinely assessed by immunohistochemistry because most flow cytometry laboratories cannot reliably detect and characterize the low numbers of these cells.
Mature T-cell lymphomas generally express one or more T-cell markers, and tend to display a T-helper (CD4-positive) or cytotoxic (CD8-positive) immunophenotype and may show loss of markers expressed by most normal T-cells (eg, CD5, CD7; Figure 4). However, a subset of them may express markers not commonly detected in normal T cells, such as ALK. NK-cell lymphomas lack surface CD3 (expressing only cytoplasmic CD3) and CD5 but express some pan T-cell antigens (such as CD2 and CD7) as well as CD16 and/or CD56.
Patients with primary or acquired immune dysfunction are at risk for development of lymphoma and other less clearly defined lymphoproliferative disorders, the majority of which are associated with infection of the lymphoid cells with Epstein-Barr virus (EBV). Therefore, evaluation with chromogenic in situ hybridization for an EBV-encoded early RNA (EBER1) is routinely performed in these cases; it is thus essential that the hematopathologist be informed of the altered immune system of the patient. If lymphoma develops, they may be morphologically similar to those that appear in immunocompetent patients, which specifically in the post-transplant setting are known as monomorphic post-transplant lymphoproliferative disorders (PTLD). If the PTLD does not meet the criteria for any of the recognized types of lymphoma, it may be best characterized as a polymorphic PTLD.
Once the lineage (B-, T-, or NK-cell) of the mature lymphoma has been established, the sum (and on occasion the gestalt) of the clinical, morphologic, immunophenotypic and other findings will be considered for the subclassification of the neoplasm.
Cytogenetic and molecular evaluation
If the morphologic and immunophenotypic analysis is inconclusive or nondiagnostic, then molecular and/or cytogenetic testing may further aid in the characterization of the process. Some of available molecular tests include analyses for the rearrangements of the variable region of the immunoglobulin (IG) or T-cell receptor (TCR) genes and for mutations on specific genes. The identification of specific mutations not only confirms the clonal nature of the process but, on occasion, it may also help subclassify the lymphoma, whereas IG or TCR rearrangement studies are used to establish whether a lymphoid expansion is polyclonal or monoclonal. The molecular findings should not be evaluated in isolation, because not all monoclonal rearrangements are diagnostic of lymphoma, and not all lymphomas will show a monoclonal rearrangement. Other methodologies that can aid in the identification of a clonal process or specific genetic abnormalities include metaphase cytogenetics (karyotyping) and fluorescence in situ hybridization (FISH). If any cytogenetic abnormalities are found in sufficient numbers (and constitutional abnormalities are excluded), their identification indicates the presence of a clonal process. Also, some cytogenetic abnormalities are characteristic of certain lymphomas. However, they may be neither 100% diagnostically sensitive nor diagnostically specific, for example, the hallmark t(14;18)/IGH-BCL2 is not present in all follicular lymphomas and not all lymphomas with this translocation are follicular lymphomas. Whereas FISH is generally performed on a minimum of 200 cells, compared with typically 20 metaphase by “conventional” karyotyping, and is therefore considered to have higher analytical sensitivity, it evaluates only for the presence or absence of the abnormality being investigated with a given set of probes, and therefore other abnormalities, if present, will not be identified. The value of FISH cytogenetic studies is perhaps best illustrated in the need to diagnose double hit lymphomas, amongst other scenarios. The detection of certain mutations can aid in the diagnosis of certain lymphomas, such as MYD88 in lymphoplasmacytic lymphoma, prognosis of others, such as in follicular lymphoma and identify pathways that may be precisely therapeutically targeted.
Final remarks
The diagnosis of lymphoma can be complex and usually requires the hematopathologist to integrate multiple parameters. The classification of lymphomas is not static, and new entities or variants are continuously described, and the facets of well-known ones refined. While such changes are often to the chagrin of hematologists/oncologists and hematopathologists alike, we should embrace the incorporation of nascent and typically cool data into our practice, as more therapeutically relevant entities are molded.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017 ;67(1):7-30.
2. Swerdlow SH, Campo E, Harris NL, et al, eds. WHO classification of tumours of haematopoietic and lymphoid tissues. In: Bosman FT, Jaffe ES, Lakhani SR, Ohgaki H, eds. World Health Organization Classification of Tumours. Lyon, France: IARC; 2008.
3. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016 ;127(20):2375-2390.
Lymphomas constitute a very heterogeneous group of neoplasms with diverse clinical presentations, prognoses, and responses to therapy. Approximately 80,500 new cases of lymphoma are expected to be diagnosed in the United States in 2017, of which about one quarter will lead to the death of the patient.1 Perhaps more so than any other group of neoplasms, the diagnosis of lymphoma involves the integration of a multiplicity of clinical, histologic and immunophenotypic findings and, on occasion, cytogenetic and molecular results as well. An accurate diagnosis of lymphoma, usually rendered by hematopathologists, allows hematologists/oncologists to treat patients appropriately. Herein we will describe a simplified approach to the diagnosis and classification of lymphomas (Figure 1).
Lymphoma classification
Lymphomas are clonal neoplasms characterized by the expansion of abnormal lymphoid cells that may develop in any organ but commonly involve lymph nodes. The fourth edition of the World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid tissues, published in 2008, is the official and most current guideline used for diagnosis of lymphoid neoplasms.2 The WHO scheme classifies lymphomas according to the type of cell from which they are derived (mature and immature B cells, T cells, or natural killer (NK) cells, findings determined by their morphology and immunophenotype) and their clinical, cytogenetic, and/or molecular features. This official classification is currently being updated3 and is expected to be published in full in 2017, at which time it is anticipated to include definitions for more than 70 distinct neoplasms.
Lymphomas are broadly and informally classified as Hodgkin lymphomas (HLs) and non-Hodgkin lymphomas (NHLs), based on the differences these two groups show in their clinical presentation, treatment, prognosis, and proportion of neoplastic cells, among others. NHLs are by far the most common type of lymphomas, accounting for approximately 90% of all new cases of lymphoma in the United States and 70% worldwide.1,2 NHLs are a very heterogeneous group of B-, T-, or NK-cell neoplasms that, in turn, can also be informally subclassified as low-grade (or indolent) or high-grade (or aggressive) according to their predicted clinical behavior. HLs are comparatively rare, less heterogeneous, uniformly of B-cell origin and, in the case of classical Hodgkin lymphoma, highly curable.1,2 It is beyond the scope of this manuscript to outline the features of each of the >70 specific entities, but the reader is referred elsewhere for more detail and encouraged to become familiarized with the complexity, challenges, and beauty of lymphoma diagnosis.2,3
Biopsy procedure
A correct diagnosis begins with an adequate biopsy procedure. It is essential that biopsy specimens for lymphoma evaluation be submitted fresh and unfixed, because some crucial analyses such as flow cytometry or conventional cytogenetics can only be performed on fresh tissue. Indeed, it is important for the hematologist/oncologist and/or surgeon and/or interventional radiologist to converse with the hematopathologist prior to and even during some procedures to ensure the correct processing of the specimen. Also, it is important to limit the compression of the specimen and the excessive use of cauterization during the biopsy procedure, both of which cause artifacts that may render impossible the interpretation of the histopathologic findings.
Given that the diagnosis of lymphoma is based not only on the cytologic details of the lymphoma cells but also on the architectural pattern with which they infiltrate an organ, the larger the biopsy specimen, the easier it will be for a hematopathologist to identify the pattern. In addition, excisional biopsies frequently contain more diagnostic tissue than needle core biopsies and this provides pathologists with the option to submit tissue fragments for ancillary tests that require unfixed tissue as noted above. Needle core biopsies of lymph nodes are increasingly being used because of their association with fewer complications and lower cost than excisional biopsies. However, needle core biopsies provide only a glimpse of the pattern of infiltration and may not be completely representative of the architecture. Therefore, excisional lymph node biopsies of lymph nodes are preferred over needle core biopsies, recognizing that in the setting of deeply seated lymph nodes, needle core biopsies may be the only or the best surgical option.
Clinical presentation
Accurate diagnosis of lymphoma cannot take place in a vacuum. The hematopathologist’s initial approach to the diagnosis of lymphoid processes in tissue biopsies should begin with a thorough review of the clinical history, although some pathology laboratories may not have immediate access to this information. The hematopathologist should evaluate factors such as age, gender, location of the tumor, symptomatology, medications, serology, and prior history of malignancy, immunosuppression or immunodeficiency in every case. Other important but frequently omitted parts of the clinical history are the patient’s occupation, history of exposure to animals, and the presence of tattoos, which may be associated with certain reactive lymphadenopathies.
Histomorphologic evaluation
Despite the plethora of new and increasingly sophisticated tools, histologic and morphologic analysis still remains the cornerstone of diagnosis in hematopathology. However, for the characterization of an increasing number of reactive and neoplastic lymphoid processes, hematopathologists may also require immunophenotypic, molecular, and cytogenetic tests for an accurate diagnosis. Upon review of the clinical information, a microscopic evaluation of the tissue submitted for processing by the histology laboratory will be performed. The results of concurrent flow cytometric evaluation (performed on fresh unfixed material) should also be available in most if not all cases before the H&E-stained slides are available for review. Upon receipt of H&E-stained slides, the hematopathologist will evaluate the quality of the submitted specimen, since many diagnostic difficulties stem from suboptimal techniques related to the biopsy procedure, fixation, processing, cutting, or staining (Figure 1). If deemed suitable for accurate diagnosis, a search for signs of preservation or disruption of the organ that was biopsied will follow. The identification of certain morphologic patterns aids the hematopathologist in answering the first question: “what organ is this and is this consistent with what is indicated on the requisition?” This is usually immediately followed by “is this sufficient and adequate material for a diagnosis?” and “is there any normal architecture?” If the architecture is not normal, “is this alteration due to a reactive or a neoplastic process?” If neoplastic, “is it lymphoma or a non-hematolymphoid neoplasm?”
Both reactive and neoplastic processes have variably unique morphologic features that if properly recognized, guide the subsequent testing. However, some reactive and neoplastic processes can present with overlapping features, and even after extensive immunophenotypic evaluation and the performance of ancillary studies, it may not be possible to conclusively determine its nature. If the lymph node architecture is altered or effaced, the predominant pattern of infiltration (eg, nodular, diffuse, interfollicular, intrasinusoidal) and the degree of alteration of the normal architecture is evaluated, usually at low magnification. When the presence of an infiltrate is recognized, its components must be characterized. If the infiltrate is composed of a homogeneous expansion of lymphoid cells that disrupts or replaces the normal lymphoid architecture, a lymphoma will be suspected or diagnosed. The pattern of distribution of the cells along with their individual morphologic characteristics (ie, size, nuclear shape, chromatin configuration, nucleoli, amount and hue of cytoplasm) are key factors for the diagnosis and classification of the lymphoma that will guide subsequent testing. The immunophenotypic analysis (by immunohistochemistry, flow cytometry or a combination of both) may confirm the reactive or neoplastic nature of the process, and its subclassification. B-cell lymphomas, in particular have variable and distinctive histologic features: as a diffuse infiltrate of large mature lymphoid cells (eg, diffuse large B-cell lymphoma), an expansion of immature lymphoid cells (lymphoblastic lymphoma), and a nodular infiltrate of small, intermediate and/or mature large B cells (eg, follicular lymphoma).
Mature T-cell lymphomas may display similar histologic, features but they can be quite heterogeneous with an infiltrate composed of one predominant cell type or a mixture of small, medium-sized, and large atypical lymphoid cells (on occasion with abundant clear cytoplasm) and a variable number of eosinophils, plasma cells, macrophages (including granulomas), and B cells. HLs most commonly efface the lymph node architecture with a nodular or diffuse infiltrate variably composed of reactive lymphocytes, granulocytes, macrophages, and plasma cells and usually a minority of large neoplastic cells (Hodgkin/Reed-Sternberg cells and/or lymphocyte predominant cells).
Once the H&E-stained slides are evaluated and a diagnosis of lymphoma is suspected or established, the hematopathologist will attempt to determine whether it has mature or immature features, and whether low- or high-grade morphologic characteristics are present. The maturity of lymphoid cells is generally determined by the nature of the chromatin, which if “fine” and homogeneous (with or without a conspicuous nucleolus) will usually, but not always, be considered immature, whereas clumped, vesicular or hyperchromatic chromatin is generally, but not always, associated with maturity. If the chromatin displays immature features, the differential diagnosis will mainly include B- and T-lymphoblastic lymphomas, but also blastoid variants of mature neoplasm such as mantle cell lymphoma, and follicular lymphoma, as well as high-grade B-cell lymphomas. Features associated with low-grade lymphomas (eg, follicular lymphoma, small lymphocytic lymphoma/chronic lymphocytic leukemia, marginal zone lymphoma, lymphoplasmacytic lymphoma) include small cell morphology, mature chromatin, absence of a significant number of mitoses or apoptotic cells, and a low proliferation index as shown by immunohistochemistry for Ki67. High-grade lymphomas, such as lymphoblastic lymphoma, Burkitt lymphoma, or certain large B-cell lymphomas tend to show opposite features, and some of the mature entities are frequently associated with MYC rearrangements. Of note, immature lymphomas tend to be clinically high grade, but not all clinically high-grade lymphomas are immature. Conversely, the majority of low-grade lymphomas are usually mature.
Immunophenotypic evaluation
Immunophenotypic evaluation is essential because the lineage of lymphoma cells cannot be determined by morphology alone. The immunophenotype is the combination of proteins/markers (eg, CD20, CD3, TdT) expressed by cells. Usually, it is evaluated by immunohistochemistry and/or flow cytometry, which help determine the proportion of lymphoid cells that express a certain marker and its location and intensity within the cells. While immunohistochemistry is normally performed on formalin-fixed and paraffin-embedded tissue, flow cytometry can be evaluated only on fresh unfixed tissue. Flow cytometry has the advantage over immunohistochemistry of being faster and better at simultaneously identifying coexpression of multiple markers on multiple cell populations. However, certain markers can only be evaluated by immunohistochemistry.
The immunophenotypic analysis will in most cases reveal whether the lymphomas is of B-, T- or NK-cell origin, and whether a lymphoma subtype associated immunophenotype is present. Typical pan B-cell antigens include PAX5, CD19, and CD79a (CD20 is less broadly expressed throughout B-cell differentiation, although it is usually evident in most mature B-cell lymphomas), and typical pan T-cell antigens include CD2, CD5, and CD7. The immature or mature nature of a lymphoma can also be confirmed by evaluation of the immunophenotype. Immature lymphomas commonly express one or more of TdT, CD10, or CD34; T-lymphoblastic lymphoma cells may also coexpress CD1a. The majority of NHLs and all HLs are derived from (or reflect) B cells at different stages of maturation. Mature B-cell lymphomas are the most common type of lymphoma and typically, but not always, express pan B-cell markers as well as surface membrane immunoglobulin, with the latter also most useful in assessing clonality via a determination of light chain restriction. Some mature B-cell lymphomas tend to acquire markers that are either never physiologically expressed by normal mature B cells (eg, cyclin D1 in mantle cell lymphoma, or BCL2 in germinal center B cells in follicular lymphoma) or only expressed in a minor fraction (eg, CD5 that is characteristically expressed in small lymphocytic and mantle cell lymphoma). The most common mature B-cell lymphomas include diffuse large B-cell lymphoma, follicular lymphoma, small lymphocytic lymphoma, mantle cell lymphoma, marginal zone lymphoma, Burkitt lymphoma, and lymphoplasmacytic lymphoma (Figures 2 and 3). Classical HLs are also lymphomas of B-cell origin that demonstrate diminished preservation of their B-cell immunophenotype (as evidenced by the dim expression of PAX5 but absence of most other pan B-cell antigens), expression of CD30, variable expression of CD15, and loss of CD45 (Figure 1). In contrast, nodular lymphocyte predominant HL shows a preserved B-cell immunophenotypic program and expression of CD45, typically without CD30 and CD15. Of note, the evaluation of the immunophenotype of the neoplastic cells in HL is routinely assessed by immunohistochemistry because most flow cytometry laboratories cannot reliably detect and characterize the low numbers of these cells.
Mature T-cell lymphomas generally express one or more T-cell markers, and tend to display a T-helper (CD4-positive) or cytotoxic (CD8-positive) immunophenotype and may show loss of markers expressed by most normal T-cells (eg, CD5, CD7; Figure 4). However, a subset of them may express markers not commonly detected in normal T cells, such as ALK. NK-cell lymphomas lack surface CD3 (expressing only cytoplasmic CD3) and CD5 but express some pan T-cell antigens (such as CD2 and CD7) as well as CD16 and/or CD56.
Patients with primary or acquired immune dysfunction are at risk for development of lymphoma and other less clearly defined lymphoproliferative disorders, the majority of which are associated with infection of the lymphoid cells with Epstein-Barr virus (EBV). Therefore, evaluation with chromogenic in situ hybridization for an EBV-encoded early RNA (EBER1) is routinely performed in these cases; it is thus essential that the hematopathologist be informed of the altered immune system of the patient. If lymphoma develops, they may be morphologically similar to those that appear in immunocompetent patients, which specifically in the post-transplant setting are known as monomorphic post-transplant lymphoproliferative disorders (PTLD). If the PTLD does not meet the criteria for any of the recognized types of lymphoma, it may be best characterized as a polymorphic PTLD.
Once the lineage (B-, T-, or NK-cell) of the mature lymphoma has been established, the sum (and on occasion the gestalt) of the clinical, morphologic, immunophenotypic and other findings will be considered for the subclassification of the neoplasm.
Cytogenetic and molecular evaluation
If the morphologic and immunophenotypic analysis is inconclusive or nondiagnostic, then molecular and/or cytogenetic testing may further aid in the characterization of the process. Some of available molecular tests include analyses for the rearrangements of the variable region of the immunoglobulin (IG) or T-cell receptor (TCR) genes and for mutations on specific genes. The identification of specific mutations not only confirms the clonal nature of the process but, on occasion, it may also help subclassify the lymphoma, whereas IG or TCR rearrangement studies are used to establish whether a lymphoid expansion is polyclonal or monoclonal. The molecular findings should not be evaluated in isolation, because not all monoclonal rearrangements are diagnostic of lymphoma, and not all lymphomas will show a monoclonal rearrangement. Other methodologies that can aid in the identification of a clonal process or specific genetic abnormalities include metaphase cytogenetics (karyotyping) and fluorescence in situ hybridization (FISH). If any cytogenetic abnormalities are found in sufficient numbers (and constitutional abnormalities are excluded), their identification indicates the presence of a clonal process. Also, some cytogenetic abnormalities are characteristic of certain lymphomas. However, they may be neither 100% diagnostically sensitive nor diagnostically specific, for example, the hallmark t(14;18)/IGH-BCL2 is not present in all follicular lymphomas and not all lymphomas with this translocation are follicular lymphomas. Whereas FISH is generally performed on a minimum of 200 cells, compared with typically 20 metaphase by “conventional” karyotyping, and is therefore considered to have higher analytical sensitivity, it evaluates only for the presence or absence of the abnormality being investigated with a given set of probes, and therefore other abnormalities, if present, will not be identified. The value of FISH cytogenetic studies is perhaps best illustrated in the need to diagnose double hit lymphomas, amongst other scenarios. The detection of certain mutations can aid in the diagnosis of certain lymphomas, such as MYD88 in lymphoplasmacytic lymphoma, prognosis of others, such as in follicular lymphoma and identify pathways that may be precisely therapeutically targeted.
Final remarks
The diagnosis of lymphoma can be complex and usually requires the hematopathologist to integrate multiple parameters. The classification of lymphomas is not static, and new entities or variants are continuously described, and the facets of well-known ones refined. While such changes are often to the chagrin of hematologists/oncologists and hematopathologists alike, we should embrace the incorporation of nascent and typically cool data into our practice, as more therapeutically relevant entities are molded.
Lymphomas constitute a very heterogeneous group of neoplasms with diverse clinical presentations, prognoses, and responses to therapy. Approximately 80,500 new cases of lymphoma are expected to be diagnosed in the United States in 2017, of which about one quarter will lead to the death of the patient.1 Perhaps more so than any other group of neoplasms, the diagnosis of lymphoma involves the integration of a multiplicity of clinical, histologic and immunophenotypic findings and, on occasion, cytogenetic and molecular results as well. An accurate diagnosis of lymphoma, usually rendered by hematopathologists, allows hematologists/oncologists to treat patients appropriately. Herein we will describe a simplified approach to the diagnosis and classification of lymphomas (Figure 1).
Lymphoma classification
Lymphomas are clonal neoplasms characterized by the expansion of abnormal lymphoid cells that may develop in any organ but commonly involve lymph nodes. The fourth edition of the World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid tissues, published in 2008, is the official and most current guideline used for diagnosis of lymphoid neoplasms.2 The WHO scheme classifies lymphomas according to the type of cell from which they are derived (mature and immature B cells, T cells, or natural killer (NK) cells, findings determined by their morphology and immunophenotype) and their clinical, cytogenetic, and/or molecular features. This official classification is currently being updated3 and is expected to be published in full in 2017, at which time it is anticipated to include definitions for more than 70 distinct neoplasms.
Lymphomas are broadly and informally classified as Hodgkin lymphomas (HLs) and non-Hodgkin lymphomas (NHLs), based on the differences these two groups show in their clinical presentation, treatment, prognosis, and proportion of neoplastic cells, among others. NHLs are by far the most common type of lymphomas, accounting for approximately 90% of all new cases of lymphoma in the United States and 70% worldwide.1,2 NHLs are a very heterogeneous group of B-, T-, or NK-cell neoplasms that, in turn, can also be informally subclassified as low-grade (or indolent) or high-grade (or aggressive) according to their predicted clinical behavior. HLs are comparatively rare, less heterogeneous, uniformly of B-cell origin and, in the case of classical Hodgkin lymphoma, highly curable.1,2 It is beyond the scope of this manuscript to outline the features of each of the >70 specific entities, but the reader is referred elsewhere for more detail and encouraged to become familiarized with the complexity, challenges, and beauty of lymphoma diagnosis.2,3
Biopsy procedure
A correct diagnosis begins with an adequate biopsy procedure. It is essential that biopsy specimens for lymphoma evaluation be submitted fresh and unfixed, because some crucial analyses such as flow cytometry or conventional cytogenetics can only be performed on fresh tissue. Indeed, it is important for the hematologist/oncologist and/or surgeon and/or interventional radiologist to converse with the hematopathologist prior to and even during some procedures to ensure the correct processing of the specimen. Also, it is important to limit the compression of the specimen and the excessive use of cauterization during the biopsy procedure, both of which cause artifacts that may render impossible the interpretation of the histopathologic findings.
Given that the diagnosis of lymphoma is based not only on the cytologic details of the lymphoma cells but also on the architectural pattern with which they infiltrate an organ, the larger the biopsy specimen, the easier it will be for a hematopathologist to identify the pattern. In addition, excisional biopsies frequently contain more diagnostic tissue than needle core biopsies and this provides pathologists with the option to submit tissue fragments for ancillary tests that require unfixed tissue as noted above. Needle core biopsies of lymph nodes are increasingly being used because of their association with fewer complications and lower cost than excisional biopsies. However, needle core biopsies provide only a glimpse of the pattern of infiltration and may not be completely representative of the architecture. Therefore, excisional lymph node biopsies of lymph nodes are preferred over needle core biopsies, recognizing that in the setting of deeply seated lymph nodes, needle core biopsies may be the only or the best surgical option.
Clinical presentation
Accurate diagnosis of lymphoma cannot take place in a vacuum. The hematopathologist’s initial approach to the diagnosis of lymphoid processes in tissue biopsies should begin with a thorough review of the clinical history, although some pathology laboratories may not have immediate access to this information. The hematopathologist should evaluate factors such as age, gender, location of the tumor, symptomatology, medications, serology, and prior history of malignancy, immunosuppression or immunodeficiency in every case. Other important but frequently omitted parts of the clinical history are the patient’s occupation, history of exposure to animals, and the presence of tattoos, which may be associated with certain reactive lymphadenopathies.
Histomorphologic evaluation
Despite the plethora of new and increasingly sophisticated tools, histologic and morphologic analysis still remains the cornerstone of diagnosis in hematopathology. However, for the characterization of an increasing number of reactive and neoplastic lymphoid processes, hematopathologists may also require immunophenotypic, molecular, and cytogenetic tests for an accurate diagnosis. Upon review of the clinical information, a microscopic evaluation of the tissue submitted for processing by the histology laboratory will be performed. The results of concurrent flow cytometric evaluation (performed on fresh unfixed material) should also be available in most if not all cases before the H&E-stained slides are available for review. Upon receipt of H&E-stained slides, the hematopathologist will evaluate the quality of the submitted specimen, since many diagnostic difficulties stem from suboptimal techniques related to the biopsy procedure, fixation, processing, cutting, or staining (Figure 1). If deemed suitable for accurate diagnosis, a search for signs of preservation or disruption of the organ that was biopsied will follow. The identification of certain morphologic patterns aids the hematopathologist in answering the first question: “what organ is this and is this consistent with what is indicated on the requisition?” This is usually immediately followed by “is this sufficient and adequate material for a diagnosis?” and “is there any normal architecture?” If the architecture is not normal, “is this alteration due to a reactive or a neoplastic process?” If neoplastic, “is it lymphoma or a non-hematolymphoid neoplasm?”
Both reactive and neoplastic processes have variably unique morphologic features that if properly recognized, guide the subsequent testing. However, some reactive and neoplastic processes can present with overlapping features, and even after extensive immunophenotypic evaluation and the performance of ancillary studies, it may not be possible to conclusively determine its nature. If the lymph node architecture is altered or effaced, the predominant pattern of infiltration (eg, nodular, diffuse, interfollicular, intrasinusoidal) and the degree of alteration of the normal architecture is evaluated, usually at low magnification. When the presence of an infiltrate is recognized, its components must be characterized. If the infiltrate is composed of a homogeneous expansion of lymphoid cells that disrupts or replaces the normal lymphoid architecture, a lymphoma will be suspected or diagnosed. The pattern of distribution of the cells along with their individual morphologic characteristics (ie, size, nuclear shape, chromatin configuration, nucleoli, amount and hue of cytoplasm) are key factors for the diagnosis and classification of the lymphoma that will guide subsequent testing. The immunophenotypic analysis (by immunohistochemistry, flow cytometry or a combination of both) may confirm the reactive or neoplastic nature of the process, and its subclassification. B-cell lymphomas, in particular have variable and distinctive histologic features: as a diffuse infiltrate of large mature lymphoid cells (eg, diffuse large B-cell lymphoma), an expansion of immature lymphoid cells (lymphoblastic lymphoma), and a nodular infiltrate of small, intermediate and/or mature large B cells (eg, follicular lymphoma).
Mature T-cell lymphomas may display similar histologic, features but they can be quite heterogeneous with an infiltrate composed of one predominant cell type or a mixture of small, medium-sized, and large atypical lymphoid cells (on occasion with abundant clear cytoplasm) and a variable number of eosinophils, plasma cells, macrophages (including granulomas), and B cells. HLs most commonly efface the lymph node architecture with a nodular or diffuse infiltrate variably composed of reactive lymphocytes, granulocytes, macrophages, and plasma cells and usually a minority of large neoplastic cells (Hodgkin/Reed-Sternberg cells and/or lymphocyte predominant cells).
Once the H&E-stained slides are evaluated and a diagnosis of lymphoma is suspected or established, the hematopathologist will attempt to determine whether it has mature or immature features, and whether low- or high-grade morphologic characteristics are present. The maturity of lymphoid cells is generally determined by the nature of the chromatin, which if “fine” and homogeneous (with or without a conspicuous nucleolus) will usually, but not always, be considered immature, whereas clumped, vesicular or hyperchromatic chromatin is generally, but not always, associated with maturity. If the chromatin displays immature features, the differential diagnosis will mainly include B- and T-lymphoblastic lymphomas, but also blastoid variants of mature neoplasm such as mantle cell lymphoma, and follicular lymphoma, as well as high-grade B-cell lymphomas. Features associated with low-grade lymphomas (eg, follicular lymphoma, small lymphocytic lymphoma/chronic lymphocytic leukemia, marginal zone lymphoma, lymphoplasmacytic lymphoma) include small cell morphology, mature chromatin, absence of a significant number of mitoses or apoptotic cells, and a low proliferation index as shown by immunohistochemistry for Ki67. High-grade lymphomas, such as lymphoblastic lymphoma, Burkitt lymphoma, or certain large B-cell lymphomas tend to show opposite features, and some of the mature entities are frequently associated with MYC rearrangements. Of note, immature lymphomas tend to be clinically high grade, but not all clinically high-grade lymphomas are immature. Conversely, the majority of low-grade lymphomas are usually mature.
Immunophenotypic evaluation
Immunophenotypic evaluation is essential because the lineage of lymphoma cells cannot be determined by morphology alone. The immunophenotype is the combination of proteins/markers (eg, CD20, CD3, TdT) expressed by cells. Usually, it is evaluated by immunohistochemistry and/or flow cytometry, which help determine the proportion of lymphoid cells that express a certain marker and its location and intensity within the cells. While immunohistochemistry is normally performed on formalin-fixed and paraffin-embedded tissue, flow cytometry can be evaluated only on fresh unfixed tissue. Flow cytometry has the advantage over immunohistochemistry of being faster and better at simultaneously identifying coexpression of multiple markers on multiple cell populations. However, certain markers can only be evaluated by immunohistochemistry.
The immunophenotypic analysis will in most cases reveal whether the lymphomas is of B-, T- or NK-cell origin, and whether a lymphoma subtype associated immunophenotype is present. Typical pan B-cell antigens include PAX5, CD19, and CD79a (CD20 is less broadly expressed throughout B-cell differentiation, although it is usually evident in most mature B-cell lymphomas), and typical pan T-cell antigens include CD2, CD5, and CD7. The immature or mature nature of a lymphoma can also be confirmed by evaluation of the immunophenotype. Immature lymphomas commonly express one or more of TdT, CD10, or CD34; T-lymphoblastic lymphoma cells may also coexpress CD1a. The majority of NHLs and all HLs are derived from (or reflect) B cells at different stages of maturation. Mature B-cell lymphomas are the most common type of lymphoma and typically, but not always, express pan B-cell markers as well as surface membrane immunoglobulin, with the latter also most useful in assessing clonality via a determination of light chain restriction. Some mature B-cell lymphomas tend to acquire markers that are either never physiologically expressed by normal mature B cells (eg, cyclin D1 in mantle cell lymphoma, or BCL2 in germinal center B cells in follicular lymphoma) or only expressed in a minor fraction (eg, CD5 that is characteristically expressed in small lymphocytic and mantle cell lymphoma). The most common mature B-cell lymphomas include diffuse large B-cell lymphoma, follicular lymphoma, small lymphocytic lymphoma, mantle cell lymphoma, marginal zone lymphoma, Burkitt lymphoma, and lymphoplasmacytic lymphoma (Figures 2 and 3). Classical HLs are also lymphomas of B-cell origin that demonstrate diminished preservation of their B-cell immunophenotype (as evidenced by the dim expression of PAX5 but absence of most other pan B-cell antigens), expression of CD30, variable expression of CD15, and loss of CD45 (Figure 1). In contrast, nodular lymphocyte predominant HL shows a preserved B-cell immunophenotypic program and expression of CD45, typically without CD30 and CD15. Of note, the evaluation of the immunophenotype of the neoplastic cells in HL is routinely assessed by immunohistochemistry because most flow cytometry laboratories cannot reliably detect and characterize the low numbers of these cells.
Mature T-cell lymphomas generally express one or more T-cell markers, and tend to display a T-helper (CD4-positive) or cytotoxic (CD8-positive) immunophenotype and may show loss of markers expressed by most normal T-cells (eg, CD5, CD7; Figure 4). However, a subset of them may express markers not commonly detected in normal T cells, such as ALK. NK-cell lymphomas lack surface CD3 (expressing only cytoplasmic CD3) and CD5 but express some pan T-cell antigens (such as CD2 and CD7) as well as CD16 and/or CD56.
Patients with primary or acquired immune dysfunction are at risk for development of lymphoma and other less clearly defined lymphoproliferative disorders, the majority of which are associated with infection of the lymphoid cells with Epstein-Barr virus (EBV). Therefore, evaluation with chromogenic in situ hybridization for an EBV-encoded early RNA (EBER1) is routinely performed in these cases; it is thus essential that the hematopathologist be informed of the altered immune system of the patient. If lymphoma develops, they may be morphologically similar to those that appear in immunocompetent patients, which specifically in the post-transplant setting are known as monomorphic post-transplant lymphoproliferative disorders (PTLD). If the PTLD does not meet the criteria for any of the recognized types of lymphoma, it may be best characterized as a polymorphic PTLD.
Once the lineage (B-, T-, or NK-cell) of the mature lymphoma has been established, the sum (and on occasion the gestalt) of the clinical, morphologic, immunophenotypic and other findings will be considered for the subclassification of the neoplasm.
Cytogenetic and molecular evaluation
If the morphologic and immunophenotypic analysis is inconclusive or nondiagnostic, then molecular and/or cytogenetic testing may further aid in the characterization of the process. Some of available molecular tests include analyses for the rearrangements of the variable region of the immunoglobulin (IG) or T-cell receptor (TCR) genes and for mutations on specific genes. The identification of specific mutations not only confirms the clonal nature of the process but, on occasion, it may also help subclassify the lymphoma, whereas IG or TCR rearrangement studies are used to establish whether a lymphoid expansion is polyclonal or monoclonal. The molecular findings should not be evaluated in isolation, because not all monoclonal rearrangements are diagnostic of lymphoma, and not all lymphomas will show a monoclonal rearrangement. Other methodologies that can aid in the identification of a clonal process or specific genetic abnormalities include metaphase cytogenetics (karyotyping) and fluorescence in situ hybridization (FISH). If any cytogenetic abnormalities are found in sufficient numbers (and constitutional abnormalities are excluded), their identification indicates the presence of a clonal process. Also, some cytogenetic abnormalities are characteristic of certain lymphomas. However, they may be neither 100% diagnostically sensitive nor diagnostically specific, for example, the hallmark t(14;18)/IGH-BCL2 is not present in all follicular lymphomas and not all lymphomas with this translocation are follicular lymphomas. Whereas FISH is generally performed on a minimum of 200 cells, compared with typically 20 metaphase by “conventional” karyotyping, and is therefore considered to have higher analytical sensitivity, it evaluates only for the presence or absence of the abnormality being investigated with a given set of probes, and therefore other abnormalities, if present, will not be identified. The value of FISH cytogenetic studies is perhaps best illustrated in the need to diagnose double hit lymphomas, amongst other scenarios. The detection of certain mutations can aid in the diagnosis of certain lymphomas, such as MYD88 in lymphoplasmacytic lymphoma, prognosis of others, such as in follicular lymphoma and identify pathways that may be precisely therapeutically targeted.
Final remarks
The diagnosis of lymphoma can be complex and usually requires the hematopathologist to integrate multiple parameters. The classification of lymphomas is not static, and new entities or variants are continuously described, and the facets of well-known ones refined. While such changes are often to the chagrin of hematologists/oncologists and hematopathologists alike, we should embrace the incorporation of nascent and typically cool data into our practice, as more therapeutically relevant entities are molded.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017 ;67(1):7-30.
2. Swerdlow SH, Campo E, Harris NL, et al, eds. WHO classification of tumours of haematopoietic and lymphoid tissues. In: Bosman FT, Jaffe ES, Lakhani SR, Ohgaki H, eds. World Health Organization Classification of Tumours. Lyon, France: IARC; 2008.
3. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016 ;127(20):2375-2390.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017 ;67(1):7-30.
2. Swerdlow SH, Campo E, Harris NL, et al, eds. WHO classification of tumours of haematopoietic and lymphoid tissues. In: Bosman FT, Jaffe ES, Lakhani SR, Ohgaki H, eds. World Health Organization Classification of Tumours. Lyon, France: IARC; 2008.
3. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016 ;127(20):2375-2390.
FDA lifts partial clinical hold for some selinexor trials

The US Food and Drug Administration (FDA) has lifted the partial clinical hold on trials of selinexor (KPT-330) in patients with hematologic malignancies.
The partial hold, which was announced on March 10, was placed on all trials of the drug, including those in patients with solid tumor malignancies.
The hold meant that no new patients could be enrolled in selinexor trials.
Patients who were already enrolled and had stable disease or better could remain on selinexor therapy.
Now, the FDA has lifted the hold on trials of patients with hematologic malignancies, so new patients can be enrolled in these trials and begin receiving selinexor.
The FDA had placed the hold due to a lack of information in the investigator’s brochure, including an incomplete list of serious adverse events associated with selinexor.
Karyopharm Therapeutics Inc., the company developing selinexor, noted that the hold was not the result of patient deaths or any new information regarding the safety profile of selinexor.
In response to the hold, Karyopharm amended the investigator’s brochure, updated informed consent documents, and submitted the documents to the FDA.
“The Karyopharm team worked diligently to update and submit the required documents to the FDA, which allowed the hematology division to expeditiously remove the partial clinical hold,” said Michael G. Kauffman, MD, PhD, chief executive officer of Karyopharm.
“We anticipate that the solid tumor divisions will follow suit shortly. Patient enrollment is again underway in our hematologic oncology studies. Our previously disclosed enrollment rates and timelines for both ongoing and planned trials are not expected to be materially impacted.”
About selinexor
Selinexor is a first-in-class, oral, selective inhibitor of nuclear export compound. The drug functions by inhibiting the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which subsequently reinitiates and amplifies their tumor suppressor function. This is thought to prompt apoptosis in cancer cells while largely sparing normal cells.
To date, more than 1900 patients have been treated with selinexor.
The drug is currently being evaluated in clinical trials across multiple cancer indications, including in acute myeloid leukemia (SOPRA), in multiple myeloma in combination with low-dose dexamethasone (STORM) and backbone therapies (STOMP), as well as in diffuse large B-cell lymphoma (SADAL).
The US Food and Drug Administration (FDA) has lifted the partial clinical hold on trials of selinexor (KPT-330) in patients with hematologic malignancies.
The partial hold, which was announced on March 10, was placed on all trials of the drug, including those in patients with solid tumor malignancies.
The hold meant that no new patients could be enrolled in selinexor trials.
Patients who were already enrolled and had stable disease or better could remain on selinexor therapy.
Now, the FDA has lifted the hold on trials of patients with hematologic malignancies, so new patients can be enrolled in these trials and begin receiving selinexor.
The FDA had placed the hold due to a lack of information in the investigator’s brochure, including an incomplete list of serious adverse events associated with selinexor.
Karyopharm Therapeutics Inc., the company developing selinexor, noted that the hold was not the result of patient deaths or any new information regarding the safety profile of selinexor.
In response to the hold, Karyopharm amended the investigator’s brochure, updated informed consent documents, and submitted the documents to the FDA.
“The Karyopharm team worked diligently to update and submit the required documents to the FDA, which allowed the hematology division to expeditiously remove the partial clinical hold,” said Michael G. Kauffman, MD, PhD, chief executive officer of Karyopharm.
“We anticipate that the solid tumor divisions will follow suit shortly. Patient enrollment is again underway in our hematologic oncology studies. Our previously disclosed enrollment rates and timelines for both ongoing and planned trials are not expected to be materially impacted.”
About selinexor
Selinexor is a first-in-class, oral, selective inhibitor of nuclear export compound. The drug functions by inhibiting the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which subsequently reinitiates and amplifies their tumor suppressor function. This is thought to prompt apoptosis in cancer cells while largely sparing normal cells.
To date, more than 1900 patients have been treated with selinexor.
The drug is currently being evaluated in clinical trials across multiple cancer indications, including in acute myeloid leukemia (SOPRA), in multiple myeloma in combination with low-dose dexamethasone (STORM) and backbone therapies (STOMP), as well as in diffuse large B-cell lymphoma (SADAL).
The US Food and Drug Administration (FDA) has lifted the partial clinical hold on trials of selinexor (KPT-330) in patients with hematologic malignancies.
The partial hold, which was announced on March 10, was placed on all trials of the drug, including those in patients with solid tumor malignancies.
The hold meant that no new patients could be enrolled in selinexor trials.
Patients who were already enrolled and had stable disease or better could remain on selinexor therapy.
Now, the FDA has lifted the hold on trials of patients with hematologic malignancies, so new patients can be enrolled in these trials and begin receiving selinexor.
The FDA had placed the hold due to a lack of information in the investigator’s brochure, including an incomplete list of serious adverse events associated with selinexor.
Karyopharm Therapeutics Inc., the company developing selinexor, noted that the hold was not the result of patient deaths or any new information regarding the safety profile of selinexor.
In response to the hold, Karyopharm amended the investigator’s brochure, updated informed consent documents, and submitted the documents to the FDA.
“The Karyopharm team worked diligently to update and submit the required documents to the FDA, which allowed the hematology division to expeditiously remove the partial clinical hold,” said Michael G. Kauffman, MD, PhD, chief executive officer of Karyopharm.
“We anticipate that the solid tumor divisions will follow suit shortly. Patient enrollment is again underway in our hematologic oncology studies. Our previously disclosed enrollment rates and timelines for both ongoing and planned trials are not expected to be materially impacted.”
About selinexor
Selinexor is a first-in-class, oral, selective inhibitor of nuclear export compound. The drug functions by inhibiting the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which subsequently reinitiates and amplifies their tumor suppressor function. This is thought to prompt apoptosis in cancer cells while largely sparing normal cells.
To date, more than 1900 patients have been treated with selinexor.
The drug is currently being evaluated in clinical trials across multiple cancer indications, including in acute myeloid leukemia (SOPRA), in multiple myeloma in combination with low-dose dexamethasone (STORM) and backbone therapies (STOMP), as well as in diffuse large B-cell lymphoma (SADAL).
Phase 2 study of daratumumab in NHL won’t proceed

The phase 2 CARINA study of daratumumab in non-Hodgkin lymphoma (NHL) will not proceed to stage 2, according to Genmab A/S and Janssen Biotech, Inc.
In this study, researchers have been investigating daratumumab monotherapy in patients with relapsed or refractory follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL), or mantle cell lymphoma (MCL).
Researchers planned to enroll up to 210 patients in this trial in 2 stages. Stage 1 was designed to provide a preliminary assessment of activity.
The goal of stage 2 was to further evaluate the safety and efficacy of daratumumab in the 3 patient groups.
Stage 2 will not proceed because a data review showed the FL and DLBCL cohorts did not reach the predefined futility thresholds, which were overall response rates of 50% and 30%, respectively. In the MCL cohort, the overall response rate was not evaluable due to slow recruitment.
The decision regarding this study has no impact on other ongoing or planned studies with daratumumab.
“While we hoped that daratumumab as a monotherapy could potentially provide a new treatment option in NHL patients with a high unmet medical need, the preliminary activity profile seen was not sufficient for the study to continue,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
“Daratumumab is still being investigated in a number of indications, including multiple myeloma and other hematological cancers, such as NK/T-cell lymphoma and myelodysplastic syndrome, as well as in solid tumors.”
About daratumumab
Daratumumab is a human IgG1k monoclonal antibody that binds to the CD38 molecule.
In the US, daratumumab is approved for use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least 1 prior therapy.
Daratumumab monotherapy is approved in the US for patients with multiple myeloma who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab. ![]()
The phase 2 CARINA study of daratumumab in non-Hodgkin lymphoma (NHL) will not proceed to stage 2, according to Genmab A/S and Janssen Biotech, Inc.
In this study, researchers have been investigating daratumumab monotherapy in patients with relapsed or refractory follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL), or mantle cell lymphoma (MCL).
Researchers planned to enroll up to 210 patients in this trial in 2 stages. Stage 1 was designed to provide a preliminary assessment of activity.
The goal of stage 2 was to further evaluate the safety and efficacy of daratumumab in the 3 patient groups.
Stage 2 will not proceed because a data review showed the FL and DLBCL cohorts did not reach the predefined futility thresholds, which were overall response rates of 50% and 30%, respectively. In the MCL cohort, the overall response rate was not evaluable due to slow recruitment.
The decision regarding this study has no impact on other ongoing or planned studies with daratumumab.
“While we hoped that daratumumab as a monotherapy could potentially provide a new treatment option in NHL patients with a high unmet medical need, the preliminary activity profile seen was not sufficient for the study to continue,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
“Daratumumab is still being investigated in a number of indications, including multiple myeloma and other hematological cancers, such as NK/T-cell lymphoma and myelodysplastic syndrome, as well as in solid tumors.”
About daratumumab
Daratumumab is a human IgG1k monoclonal antibody that binds to the CD38 molecule.
In the US, daratumumab is approved for use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least 1 prior therapy.
Daratumumab monotherapy is approved in the US for patients with multiple myeloma who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab. ![]()
The phase 2 CARINA study of daratumumab in non-Hodgkin lymphoma (NHL) will not proceed to stage 2, according to Genmab A/S and Janssen Biotech, Inc.
In this study, researchers have been investigating daratumumab monotherapy in patients with relapsed or refractory follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL), or mantle cell lymphoma (MCL).
Researchers planned to enroll up to 210 patients in this trial in 2 stages. Stage 1 was designed to provide a preliminary assessment of activity.
The goal of stage 2 was to further evaluate the safety and efficacy of daratumumab in the 3 patient groups.
Stage 2 will not proceed because a data review showed the FL and DLBCL cohorts did not reach the predefined futility thresholds, which were overall response rates of 50% and 30%, respectively. In the MCL cohort, the overall response rate was not evaluable due to slow recruitment.
The decision regarding this study has no impact on other ongoing or planned studies with daratumumab.
“While we hoped that daratumumab as a monotherapy could potentially provide a new treatment option in NHL patients with a high unmet medical need, the preliminary activity profile seen was not sufficient for the study to continue,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
“Daratumumab is still being investigated in a number of indications, including multiple myeloma and other hematological cancers, such as NK/T-cell lymphoma and myelodysplastic syndrome, as well as in solid tumors.”
About daratumumab
Daratumumab is a human IgG1k monoclonal antibody that binds to the CD38 molecule.
In the US, daratumumab is approved for use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least 1 prior therapy.
Daratumumab monotherapy is approved in the US for patients with multiple myeloma who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab. ![]()
NCCN launches radiation therapy resource

The National Comprehensive Cancer Network® (NCCN®) recently launched the NCCN Radiation Therapy Compendium™, which provides a single access point for NCCN recommendations pertaining to radiation therapy.
The compendium provides guidance on all radiation therapy modalities recommended within NCCN guidelines, including intensity modulated radiation therapy, intra-operative radiation therapy, stereotactic radiosurgery/stereotactic body radiotherapy/stereotactic ablative radiotherapy, image-guided radiotherapy, low dose-rate brachytherapy/high dose-rate brachytherapy, radioisotope, and particle therapy.
“As a single source for all radiation therapy recommendations within the NCCN guidelines, the compendium benefits patients with cancer by assisting providers and payers in making evidence-based treatment and coverage decisions,” said Robert W. Carlson, MD, chief executive officer of NCCN.
The NCCN Radiation Therapy Compendium™ includes recommendations for the following 24 cancer types:
Acute myeloid leukemia
Anal cancer
B-cell lymphomas
Bladder cancer
Breast cancer
Chronic lymphocytic leukemia/small lymphoblastic lymphoma
Colon cancer
Hepatobiliary cancers
Kidney cancer
Malignant pleural mesothelioma
Melanoma
Multiple myeloma
Neuroendocrine tumors
Non-small cell lung cancer
Occult primary cancer
Pancreatic adenocarcinoma
Penile cancer
Primary cutaneous B-cell lymphomas
Prostate cancer
Rectal cancer
Small cell lung cancer
Soft tissue sarcoma
T-cell lymphomas
Testicular cancer
NCCN said additional cancer types will be published on a rolling basis over the coming months. ![]()
The National Comprehensive Cancer Network® (NCCN®) recently launched the NCCN Radiation Therapy Compendium™, which provides a single access point for NCCN recommendations pertaining to radiation therapy.
The compendium provides guidance on all radiation therapy modalities recommended within NCCN guidelines, including intensity modulated radiation therapy, intra-operative radiation therapy, stereotactic radiosurgery/stereotactic body radiotherapy/stereotactic ablative radiotherapy, image-guided radiotherapy, low dose-rate brachytherapy/high dose-rate brachytherapy, radioisotope, and particle therapy.
“As a single source for all radiation therapy recommendations within the NCCN guidelines, the compendium benefits patients with cancer by assisting providers and payers in making evidence-based treatment and coverage decisions,” said Robert W. Carlson, MD, chief executive officer of NCCN.
The NCCN Radiation Therapy Compendium™ includes recommendations for the following 24 cancer types:
Acute myeloid leukemia
Anal cancer
B-cell lymphomas
Bladder cancer
Breast cancer
Chronic lymphocytic leukemia/small lymphoblastic lymphoma
Colon cancer
Hepatobiliary cancers
Kidney cancer
Malignant pleural mesothelioma
Melanoma
Multiple myeloma
Neuroendocrine tumors
Non-small cell lung cancer
Occult primary cancer
Pancreatic adenocarcinoma
Penile cancer
Primary cutaneous B-cell lymphomas
Prostate cancer
Rectal cancer
Small cell lung cancer
Soft tissue sarcoma
T-cell lymphomas
Testicular cancer
NCCN said additional cancer types will be published on a rolling basis over the coming months. ![]()
The National Comprehensive Cancer Network® (NCCN®) recently launched the NCCN Radiation Therapy Compendium™, which provides a single access point for NCCN recommendations pertaining to radiation therapy.
The compendium provides guidance on all radiation therapy modalities recommended within NCCN guidelines, including intensity modulated radiation therapy, intra-operative radiation therapy, stereotactic radiosurgery/stereotactic body radiotherapy/stereotactic ablative radiotherapy, image-guided radiotherapy, low dose-rate brachytherapy/high dose-rate brachytherapy, radioisotope, and particle therapy.
“As a single source for all radiation therapy recommendations within the NCCN guidelines, the compendium benefits patients with cancer by assisting providers and payers in making evidence-based treatment and coverage decisions,” said Robert W. Carlson, MD, chief executive officer of NCCN.
The NCCN Radiation Therapy Compendium™ includes recommendations for the following 24 cancer types:
Acute myeloid leukemia
Anal cancer
B-cell lymphomas
Bladder cancer
Breast cancer
Chronic lymphocytic leukemia/small lymphoblastic lymphoma
Colon cancer
Hepatobiliary cancers
Kidney cancer
Malignant pleural mesothelioma
Melanoma
Multiple myeloma
Neuroendocrine tumors
Non-small cell lung cancer
Occult primary cancer
Pancreatic adenocarcinoma
Penile cancer
Primary cutaneous B-cell lymphomas
Prostate cancer
Rectal cancer
Small cell lung cancer
Soft tissue sarcoma
T-cell lymphomas
Testicular cancer
NCCN said additional cancer types will be published on a rolling basis over the coming months. ![]()
CHMP recommends drug for relapsed/refractory cHL

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for the anti-PD-1 therapy pembrolizumab (Keytruda) as a treatment for patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
The recommendation pertains specifically to adults with cHL who have failed autologous hematopoietic stem cell transplant (auto-HSCT) and treatment with brentuximab vedotin (BV) or adults with cHL who are transplant-ineligible and have failed treatment with BV.
The CHMP’s recommendation will be reviewed by the European Commission, which is expected to make a decision about the drug in the second quarter of 2017.
Pembrolizumab is already approved for use in the European Union as a treatment for melanoma and non-small-cell lung cancer.
The CHMP’s positive opinion of pembrolizumab for cHL was based on data from the KEYNOTE-087 and KEYNOTE-013 trials. Results from both trials were presented at ASH 2016 (abstract 1107 and abstract 1108).
KEYNOTE-087
KEYNOTE-087 is a phase 2 trial in which researchers evaluated pembrolizumab (a 200 mg fixed dose every 3 weeks) in patients with relapsed or refractory cHL across 3 cohorts:
- Cohort 1: Patients who progressed after auto-HSCT and subsequent treatment with BV
- Cohort 2: Patients who failed salvage chemotherapy, were ineligible for a transplant, and progressed after BV
- Cohort 3: Patients who progressed after auto-HSCT and did not receive BV after transplant.
Across all 210 enrolled patients, the overall response rate (ORR) was 69.0%, and the complete response (CR) rate was 22.4%.
In Cohort 1 (n=69), the ORR was 73.9%. The CR rate was 21.7%, the partial response (PR) rate was 52.2%, 15.9% of patients had stable disease (SD), and 7.2% progressed. In 82.2% of responders, the response lasted 6 months or more.
In Cohort 2 (n=81), the ORR was 64.2%. The CR rate was 24.7%, the PR rate was 39.5%, 12.3% of patients had SD, and 21.0% progressed. In 70.0% of responders, the response lasted 6 months or more.
In Cohort 3 (n=60), the ORR was 70.0%. Twenty percent of patients had a CR, 50.0% had a PR, 16.7% had SD, and 13.3% progressed. In 75.6% of responders, the response lasted 6 months or more.
Results also included an analysis of patients with primary refractory disease (n=73), which was defined as failure to achieve CR or PR with first-line treatment. In this patient population, the ORR was 79.5%.
An ORR of 67.8% was reported in patients who relapsed after 3 or more lines of prior therapy (99/146).
The most common treatment-related adverse events (AEs) were hypothyroidism (12.4%), pyrexia (10.5%), fatigue (9.0%), rash (7.6%), diarrhea (7.1%), headache (6.2%), nausea (5.7%), cough (5.7%), and neutropenia (5.2%).
The most common grade 3/4 treatment-related AEs were neutropenia (2.4%), diarrhea (1.0%), and dyspnea (1.0%). Immune-mediated AEs included pneumonitis (2.9%), hyperthyroidism (2.9%), colitis (1.0%), and myositis (1.0%).
There were 9 discontinuations because of treatment-related AEs and no treatment-related deaths.
KEYNOTE-013
KEYNOTE-013 is a phase 1b trial that has enrolled 31 patients with relapsed or refractory cHL who failed auto-HSCT and subsequent BV or who were transplant-ineligible.
Patients received pembrolizumab at 10 mg/kg every 2 weeks. The median duration of follow-up was 29 months.
The ORR was 58%. Nineteen percent of patients achieved a CR, 39% had a PR, and 23% had SD.
The median duration of response had not been reached at last follow-up (range, 0.0+ to 26.1+ months), and 70% of responding patients had a response lasting 12 months or more.
The median progression-free survival (PFS) was 11.4 months (range, 4.9-27.8 months). The six-month PFS rate was 66%, and the 12-month PFS rate was 48%.
The median overall survival was not reached. Six-month and 12-month overall survival rates were 100% and 87%, respectively.
The most common treatment-related AEs were diarrhea (19%), hypothyroidism (13%), pneumonitis (13%), nausea (13%), fatigue (10%), and dyspnea (10%).
The most common grade 3/4 treatment-related AEs were colitis (3%), axillary pain (3%), AST increase (3%), joint swelling (3%), nephrotic syndrome back pain (3%), and dyspnea (3%).
AEs leading to discontinuation were nephrotic syndrome (grade 3), interstitial lung disease (grade 2), and pneumonitis (grade 2). There were no treatment-related deaths. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for the anti-PD-1 therapy pembrolizumab (Keytruda) as a treatment for patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
The recommendation pertains specifically to adults with cHL who have failed autologous hematopoietic stem cell transplant (auto-HSCT) and treatment with brentuximab vedotin (BV) or adults with cHL who are transplant-ineligible and have failed treatment with BV.
The CHMP’s recommendation will be reviewed by the European Commission, which is expected to make a decision about the drug in the second quarter of 2017.
Pembrolizumab is already approved for use in the European Union as a treatment for melanoma and non-small-cell lung cancer.
The CHMP’s positive opinion of pembrolizumab for cHL was based on data from the KEYNOTE-087 and KEYNOTE-013 trials. Results from both trials were presented at ASH 2016 (abstract 1107 and abstract 1108).
KEYNOTE-087
KEYNOTE-087 is a phase 2 trial in which researchers evaluated pembrolizumab (a 200 mg fixed dose every 3 weeks) in patients with relapsed or refractory cHL across 3 cohorts:
- Cohort 1: Patients who progressed after auto-HSCT and subsequent treatment with BV
- Cohort 2: Patients who failed salvage chemotherapy, were ineligible for a transplant, and progressed after BV
- Cohort 3: Patients who progressed after auto-HSCT and did not receive BV after transplant.
Across all 210 enrolled patients, the overall response rate (ORR) was 69.0%, and the complete response (CR) rate was 22.4%.
In Cohort 1 (n=69), the ORR was 73.9%. The CR rate was 21.7%, the partial response (PR) rate was 52.2%, 15.9% of patients had stable disease (SD), and 7.2% progressed. In 82.2% of responders, the response lasted 6 months or more.
In Cohort 2 (n=81), the ORR was 64.2%. The CR rate was 24.7%, the PR rate was 39.5%, 12.3% of patients had SD, and 21.0% progressed. In 70.0% of responders, the response lasted 6 months or more.
In Cohort 3 (n=60), the ORR was 70.0%. Twenty percent of patients had a CR, 50.0% had a PR, 16.7% had SD, and 13.3% progressed. In 75.6% of responders, the response lasted 6 months or more.
Results also included an analysis of patients with primary refractory disease (n=73), which was defined as failure to achieve CR or PR with first-line treatment. In this patient population, the ORR was 79.5%.
An ORR of 67.8% was reported in patients who relapsed after 3 or more lines of prior therapy (99/146).
The most common treatment-related adverse events (AEs) were hypothyroidism (12.4%), pyrexia (10.5%), fatigue (9.0%), rash (7.6%), diarrhea (7.1%), headache (6.2%), nausea (5.7%), cough (5.7%), and neutropenia (5.2%).
The most common grade 3/4 treatment-related AEs were neutropenia (2.4%), diarrhea (1.0%), and dyspnea (1.0%). Immune-mediated AEs included pneumonitis (2.9%), hyperthyroidism (2.9%), colitis (1.0%), and myositis (1.0%).
There were 9 discontinuations because of treatment-related AEs and no treatment-related deaths.
KEYNOTE-013
KEYNOTE-013 is a phase 1b trial that has enrolled 31 patients with relapsed or refractory cHL who failed auto-HSCT and subsequent BV or who were transplant-ineligible.
Patients received pembrolizumab at 10 mg/kg every 2 weeks. The median duration of follow-up was 29 months.
The ORR was 58%. Nineteen percent of patients achieved a CR, 39% had a PR, and 23% had SD.
The median duration of response had not been reached at last follow-up (range, 0.0+ to 26.1+ months), and 70% of responding patients had a response lasting 12 months or more.
The median progression-free survival (PFS) was 11.4 months (range, 4.9-27.8 months). The six-month PFS rate was 66%, and the 12-month PFS rate was 48%.
The median overall survival was not reached. Six-month and 12-month overall survival rates were 100% and 87%, respectively.
The most common treatment-related AEs were diarrhea (19%), hypothyroidism (13%), pneumonitis (13%), nausea (13%), fatigue (10%), and dyspnea (10%).
The most common grade 3/4 treatment-related AEs were colitis (3%), axillary pain (3%), AST increase (3%), joint swelling (3%), nephrotic syndrome back pain (3%), and dyspnea (3%).
AEs leading to discontinuation were nephrotic syndrome (grade 3), interstitial lung disease (grade 2), and pneumonitis (grade 2). There were no treatment-related deaths. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for the anti-PD-1 therapy pembrolizumab (Keytruda) as a treatment for patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
The recommendation pertains specifically to adults with cHL who have failed autologous hematopoietic stem cell transplant (auto-HSCT) and treatment with brentuximab vedotin (BV) or adults with cHL who are transplant-ineligible and have failed treatment with BV.
The CHMP’s recommendation will be reviewed by the European Commission, which is expected to make a decision about the drug in the second quarter of 2017.
Pembrolizumab is already approved for use in the European Union as a treatment for melanoma and non-small-cell lung cancer.
The CHMP’s positive opinion of pembrolizumab for cHL was based on data from the KEYNOTE-087 and KEYNOTE-013 trials. Results from both trials were presented at ASH 2016 (abstract 1107 and abstract 1108).
KEYNOTE-087
KEYNOTE-087 is a phase 2 trial in which researchers evaluated pembrolizumab (a 200 mg fixed dose every 3 weeks) in patients with relapsed or refractory cHL across 3 cohorts:
- Cohort 1: Patients who progressed after auto-HSCT and subsequent treatment with BV
- Cohort 2: Patients who failed salvage chemotherapy, were ineligible for a transplant, and progressed after BV
- Cohort 3: Patients who progressed after auto-HSCT and did not receive BV after transplant.
Across all 210 enrolled patients, the overall response rate (ORR) was 69.0%, and the complete response (CR) rate was 22.4%.
In Cohort 1 (n=69), the ORR was 73.9%. The CR rate was 21.7%, the partial response (PR) rate was 52.2%, 15.9% of patients had stable disease (SD), and 7.2% progressed. In 82.2% of responders, the response lasted 6 months or more.
In Cohort 2 (n=81), the ORR was 64.2%. The CR rate was 24.7%, the PR rate was 39.5%, 12.3% of patients had SD, and 21.0% progressed. In 70.0% of responders, the response lasted 6 months or more.
In Cohort 3 (n=60), the ORR was 70.0%. Twenty percent of patients had a CR, 50.0% had a PR, 16.7% had SD, and 13.3% progressed. In 75.6% of responders, the response lasted 6 months or more.
Results also included an analysis of patients with primary refractory disease (n=73), which was defined as failure to achieve CR or PR with first-line treatment. In this patient population, the ORR was 79.5%.
An ORR of 67.8% was reported in patients who relapsed after 3 or more lines of prior therapy (99/146).
The most common treatment-related adverse events (AEs) were hypothyroidism (12.4%), pyrexia (10.5%), fatigue (9.0%), rash (7.6%), diarrhea (7.1%), headache (6.2%), nausea (5.7%), cough (5.7%), and neutropenia (5.2%).
The most common grade 3/4 treatment-related AEs were neutropenia (2.4%), diarrhea (1.0%), and dyspnea (1.0%). Immune-mediated AEs included pneumonitis (2.9%), hyperthyroidism (2.9%), colitis (1.0%), and myositis (1.0%).
There were 9 discontinuations because of treatment-related AEs and no treatment-related deaths.
KEYNOTE-013
KEYNOTE-013 is a phase 1b trial that has enrolled 31 patients with relapsed or refractory cHL who failed auto-HSCT and subsequent BV or who were transplant-ineligible.
Patients received pembrolizumab at 10 mg/kg every 2 weeks. The median duration of follow-up was 29 months.
The ORR was 58%. Nineteen percent of patients achieved a CR, 39% had a PR, and 23% had SD.
The median duration of response had not been reached at last follow-up (range, 0.0+ to 26.1+ months), and 70% of responding patients had a response lasting 12 months or more.
The median progression-free survival (PFS) was 11.4 months (range, 4.9-27.8 months). The six-month PFS rate was 66%, and the 12-month PFS rate was 48%.
The median overall survival was not reached. Six-month and 12-month overall survival rates were 100% and 87%, respectively.
The most common treatment-related AEs were diarrhea (19%), hypothyroidism (13%), pneumonitis (13%), nausea (13%), fatigue (10%), and dyspnea (10%).
The most common grade 3/4 treatment-related AEs were colitis (3%), axillary pain (3%), AST increase (3%), joint swelling (3%), nephrotic syndrome back pain (3%), and dyspnea (3%).
AEs leading to discontinuation were nephrotic syndrome (grade 3), interstitial lung disease (grade 2), and pneumonitis (grade 2). There were no treatment-related deaths. ![]()
Most blood cancer mutations due to DNA replication errors

A new study supports the idea that most cancer-driving mutations are a result of DNA replication errors, not heredity or lifestyle/environmental factors.
For all 32 cancer types studied, researchers found that 66% of driver mutations resulted from DNA replication errors, 29% could be attributed to lifestyle or environmental factors, and the remaining 5% were inherited.
In hematologic malignancies, the percentage of mutations caused by DNA replication errors was even higher—70% in Hodgkin lymphoma, 85% in leukemias, 96% in non-Hodgkin lymphomas, and 99% in myeloma.
Cristian Tomasetti, PhD, of Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues reported these findings in Science.
“It is well-known that we must avoid environmental factors such as smoking to decrease our risk of getting cancer, but it is not as well-known that each time a normal cell divides and copies its DNA to produce 2 new cells, it makes multiple mistakes,” Dr Tomasetti said.
“These copying mistakes are a potent source of cancer mutations that, historically, have been scientifically undervalued, and this new work provides the first estimate of the fraction of mutations caused by these mistakes.”
In 2015, Dr Tomasetti and his colleagues reported that DNA replication errors could explain why certain cancers occur more often than others in the US.
The current study builds upon that research but includes additional cancers and encompasses an international population.
The researchers first studied the relationship between the number of normal stem cell divisions and the risk of 17 cancer types in 69 countries representing 4.8 billion people, or more than half of the world’s population.
The team said they observed a strong correlation between cancer incidence and normal stem cell divisions in all countries, regardless of their environment.
Next, the researchers set out to determine the percentage of driver mutations caused by DNA replication errors in 32 cancer types. The team developed a mathematical model using DNA sequencing data from The Cancer Genome Atlas and epidemiologic data from the Cancer Research UK database.
According to the researchers, it generally takes 2 or more critical mutations for cancer to occur. In an individual, these mutations can be due to random DNA replication errors, the environment, or inherited genes.
Knowing this, the researchers used their mathematical model to show, for example, that when critical mutations in leukemia are added together, 85.2% of them are due to random DNA replication errors, 14.3% to environmental factors, and 0.5% to heredity.
In Hodgkin lymphoma, 69.5% are due to DNA replication errors, 30% to environmental factors, and 0.5% to heredity. In non-Hodgkin lymphoma, 95.6% are due to random DNA replication errors, 3.9% to environmental factors, and 0.5% to heredity.
In myeloma, 99.3% are due to DNA replication errors, 0.2% to environmental factors, and 0.5% to heredity.
Dr Tomasetti said these random DNA replication errors will only get more important as aging populations continue to grow, prolonging the opportunity for cells to make more and more errors.
“We need to continue to encourage people to avoid environmental agents and lifestyles that increase their risk of developing cancer mutations,” said study author Bert Vogelstein, MD, of The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University.
“However, many people will still develop cancers due to these random DNA copying errors, and better methods to detect all cancers earlier, while they are still curable, are urgently needed.” ![]()
A new study supports the idea that most cancer-driving mutations are a result of DNA replication errors, not heredity or lifestyle/environmental factors.
For all 32 cancer types studied, researchers found that 66% of driver mutations resulted from DNA replication errors, 29% could be attributed to lifestyle or environmental factors, and the remaining 5% were inherited.
In hematologic malignancies, the percentage of mutations caused by DNA replication errors was even higher—70% in Hodgkin lymphoma, 85% in leukemias, 96% in non-Hodgkin lymphomas, and 99% in myeloma.
Cristian Tomasetti, PhD, of Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues reported these findings in Science.
“It is well-known that we must avoid environmental factors such as smoking to decrease our risk of getting cancer, but it is not as well-known that each time a normal cell divides and copies its DNA to produce 2 new cells, it makes multiple mistakes,” Dr Tomasetti said.
“These copying mistakes are a potent source of cancer mutations that, historically, have been scientifically undervalued, and this new work provides the first estimate of the fraction of mutations caused by these mistakes.”
In 2015, Dr Tomasetti and his colleagues reported that DNA replication errors could explain why certain cancers occur more often than others in the US.
The current study builds upon that research but includes additional cancers and encompasses an international population.
The researchers first studied the relationship between the number of normal stem cell divisions and the risk of 17 cancer types in 69 countries representing 4.8 billion people, or more than half of the world’s population.
The team said they observed a strong correlation between cancer incidence and normal stem cell divisions in all countries, regardless of their environment.
Next, the researchers set out to determine the percentage of driver mutations caused by DNA replication errors in 32 cancer types. The team developed a mathematical model using DNA sequencing data from The Cancer Genome Atlas and epidemiologic data from the Cancer Research UK database.
According to the researchers, it generally takes 2 or more critical mutations for cancer to occur. In an individual, these mutations can be due to random DNA replication errors, the environment, or inherited genes.
Knowing this, the researchers used their mathematical model to show, for example, that when critical mutations in leukemia are added together, 85.2% of them are due to random DNA replication errors, 14.3% to environmental factors, and 0.5% to heredity.
In Hodgkin lymphoma, 69.5% are due to DNA replication errors, 30% to environmental factors, and 0.5% to heredity. In non-Hodgkin lymphoma, 95.6% are due to random DNA replication errors, 3.9% to environmental factors, and 0.5% to heredity.
In myeloma, 99.3% are due to DNA replication errors, 0.2% to environmental factors, and 0.5% to heredity.
Dr Tomasetti said these random DNA replication errors will only get more important as aging populations continue to grow, prolonging the opportunity for cells to make more and more errors.
“We need to continue to encourage people to avoid environmental agents and lifestyles that increase their risk of developing cancer mutations,” said study author Bert Vogelstein, MD, of The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University.
“However, many people will still develop cancers due to these random DNA copying errors, and better methods to detect all cancers earlier, while they are still curable, are urgently needed.” ![]()
A new study supports the idea that most cancer-driving mutations are a result of DNA replication errors, not heredity or lifestyle/environmental factors.
For all 32 cancer types studied, researchers found that 66% of driver mutations resulted from DNA replication errors, 29% could be attributed to lifestyle or environmental factors, and the remaining 5% were inherited.
In hematologic malignancies, the percentage of mutations caused by DNA replication errors was even higher—70% in Hodgkin lymphoma, 85% in leukemias, 96% in non-Hodgkin lymphomas, and 99% in myeloma.
Cristian Tomasetti, PhD, of Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues reported these findings in Science.
“It is well-known that we must avoid environmental factors such as smoking to decrease our risk of getting cancer, but it is not as well-known that each time a normal cell divides and copies its DNA to produce 2 new cells, it makes multiple mistakes,” Dr Tomasetti said.
“These copying mistakes are a potent source of cancer mutations that, historically, have been scientifically undervalued, and this new work provides the first estimate of the fraction of mutations caused by these mistakes.”
In 2015, Dr Tomasetti and his colleagues reported that DNA replication errors could explain why certain cancers occur more often than others in the US.
The current study builds upon that research but includes additional cancers and encompasses an international population.
The researchers first studied the relationship between the number of normal stem cell divisions and the risk of 17 cancer types in 69 countries representing 4.8 billion people, or more than half of the world’s population.
The team said they observed a strong correlation between cancer incidence and normal stem cell divisions in all countries, regardless of their environment.
Next, the researchers set out to determine the percentage of driver mutations caused by DNA replication errors in 32 cancer types. The team developed a mathematical model using DNA sequencing data from The Cancer Genome Atlas and epidemiologic data from the Cancer Research UK database.
According to the researchers, it generally takes 2 or more critical mutations for cancer to occur. In an individual, these mutations can be due to random DNA replication errors, the environment, or inherited genes.
Knowing this, the researchers used their mathematical model to show, for example, that when critical mutations in leukemia are added together, 85.2% of them are due to random DNA replication errors, 14.3% to environmental factors, and 0.5% to heredity.
In Hodgkin lymphoma, 69.5% are due to DNA replication errors, 30% to environmental factors, and 0.5% to heredity. In non-Hodgkin lymphoma, 95.6% are due to random DNA replication errors, 3.9% to environmental factors, and 0.5% to heredity.
In myeloma, 99.3% are due to DNA replication errors, 0.2% to environmental factors, and 0.5% to heredity.
Dr Tomasetti said these random DNA replication errors will only get more important as aging populations continue to grow, prolonging the opportunity for cells to make more and more errors.
“We need to continue to encourage people to avoid environmental agents and lifestyles that increase their risk of developing cancer mutations,” said study author Bert Vogelstein, MD, of The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University.
“However, many people will still develop cancers due to these random DNA copying errors, and better methods to detect all cancers earlier, while they are still curable, are urgently needed.” ![]()
Preterm births more common in cancer survivors

Women diagnosed with cancer during their childbearing years have an increased risk of preterm births, according to research published in JAMA Oncology.
The study showed that cancer survivors were more likely than women who never had cancer to give birth prematurely, have underweight babies, and undergo cesarean section deliveries.
The researchers said women diagnosed with cancer during pregnancy may be delivering early in order to start their cancer treatment, but that does not fully explain these findings.
The team also detected an increased risk of preterm delivery in women who had already received cancer treatment.
“We found that women were more likely to deliver preterm if they’ve been treated for cancer overall, with greater risks for women who had chemotherapy,” said study author Hazel B. Nichols, PhD, of University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“While we believe these findings are something women should be aware of, we still have a lot of work to do to understand why this risk is becoming apparent and whether or not the children who are born preterm to these women go on to develop any health concerns.”
Dr Nichols and her colleagues analyzed data on 2598 births to female adolescent and young adult cancer survivors (ages 15 to 39) and 12,990 births to women without a cancer diagnosis.
Among cancer survivors, there was a significantly increased prevalence of preterm birth (prevalence ratio [PR]=1.52), low birth weight (PR=1.59), and cesarean delivery (PR=1.08), compared to women without a cancer diagnosis.
Timing of diagnosis and cancer type
When the researchers broke the data down by cancer diagnosis, they found a higher risk of preterm birth and low birth weight for women with lymphoma as well as breast and gynecologic cancers.
The PR for preterm birth was 1.59 for Hodgkin lymphoma, 1.98 for breast cancer, 2.11 for non-Hodgkin lymphoma, and 2.58 for gynecologic cancer. The PR for low birth weight was 1.59 for breast cancer, 2.41 for non-Hodgkin lymphoma, and 2.74 for gynecologic cancer.
The researchers found an increased risk of adverse birth outcomes among women who were diagnosed with cancer while pregnant and before pregnancy.
Among women diagnosed while pregnant, the PR was 2.97 for preterm birth, 2.82 for low birth weight, 1.21 for cesarean delivery, and 1.90 for low Apgar score. Among women diagnosed before pregnancy, the PR was 1.23 for preterm birth and 1.36 for low birth weight.
Role of treatment
Compared to women without a cancer diagnosis, cancer survivors who received chemotherapy but no radiation were more likely to have preterm births (PR=2.11), infants with low birth weight (PR=2.36), and cesarean deliveries (PR=1.16).
There was no significant increase in adverse birth outcomes among cancer survivors who received radiation but not chemotherapy.
Among the cancer survivors, women who received chemotherapy without radiation were more likely to have preterm births (PR=2.12), infants with low birth weight (PR=2.13), and infants who were small for their gestational age (PR=1.43) when compared to women treated with surgery only.
Dr Nichols said the role of treatment is an area of possible future research.
“We’d like to get better information about the types of chemotherapy women receive,” she said. “Chemotherapy is a very broad category, and the agents have very different effects on the body. In the future, we’d like to get more detailed information on the types of drugs that were involved in treatment.” ![]()
Women diagnosed with cancer during their childbearing years have an increased risk of preterm births, according to research published in JAMA Oncology.
The study showed that cancer survivors were more likely than women who never had cancer to give birth prematurely, have underweight babies, and undergo cesarean section deliveries.
The researchers said women diagnosed with cancer during pregnancy may be delivering early in order to start their cancer treatment, but that does not fully explain these findings.
The team also detected an increased risk of preterm delivery in women who had already received cancer treatment.
“We found that women were more likely to deliver preterm if they’ve been treated for cancer overall, with greater risks for women who had chemotherapy,” said study author Hazel B. Nichols, PhD, of University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“While we believe these findings are something women should be aware of, we still have a lot of work to do to understand why this risk is becoming apparent and whether or not the children who are born preterm to these women go on to develop any health concerns.”
Dr Nichols and her colleagues analyzed data on 2598 births to female adolescent and young adult cancer survivors (ages 15 to 39) and 12,990 births to women without a cancer diagnosis.
Among cancer survivors, there was a significantly increased prevalence of preterm birth (prevalence ratio [PR]=1.52), low birth weight (PR=1.59), and cesarean delivery (PR=1.08), compared to women without a cancer diagnosis.
Timing of diagnosis and cancer type
When the researchers broke the data down by cancer diagnosis, they found a higher risk of preterm birth and low birth weight for women with lymphoma as well as breast and gynecologic cancers.
The PR for preterm birth was 1.59 for Hodgkin lymphoma, 1.98 for breast cancer, 2.11 for non-Hodgkin lymphoma, and 2.58 for gynecologic cancer. The PR for low birth weight was 1.59 for breast cancer, 2.41 for non-Hodgkin lymphoma, and 2.74 for gynecologic cancer.
The researchers found an increased risk of adverse birth outcomes among women who were diagnosed with cancer while pregnant and before pregnancy.
Among women diagnosed while pregnant, the PR was 2.97 for preterm birth, 2.82 for low birth weight, 1.21 for cesarean delivery, and 1.90 for low Apgar score. Among women diagnosed before pregnancy, the PR was 1.23 for preterm birth and 1.36 for low birth weight.
Role of treatment
Compared to women without a cancer diagnosis, cancer survivors who received chemotherapy but no radiation were more likely to have preterm births (PR=2.11), infants with low birth weight (PR=2.36), and cesarean deliveries (PR=1.16).
There was no significant increase in adverse birth outcomes among cancer survivors who received radiation but not chemotherapy.
Among the cancer survivors, women who received chemotherapy without radiation were more likely to have preterm births (PR=2.12), infants with low birth weight (PR=2.13), and infants who were small for their gestational age (PR=1.43) when compared to women treated with surgery only.
Dr Nichols said the role of treatment is an area of possible future research.
“We’d like to get better information about the types of chemotherapy women receive,” she said. “Chemotherapy is a very broad category, and the agents have very different effects on the body. In the future, we’d like to get more detailed information on the types of drugs that were involved in treatment.” ![]()
Women diagnosed with cancer during their childbearing years have an increased risk of preterm births, according to research published in JAMA Oncology.
The study showed that cancer survivors were more likely than women who never had cancer to give birth prematurely, have underweight babies, and undergo cesarean section deliveries.
The researchers said women diagnosed with cancer during pregnancy may be delivering early in order to start their cancer treatment, but that does not fully explain these findings.
The team also detected an increased risk of preterm delivery in women who had already received cancer treatment.
“We found that women were more likely to deliver preterm if they’ve been treated for cancer overall, with greater risks for women who had chemotherapy,” said study author Hazel B. Nichols, PhD, of University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“While we believe these findings are something women should be aware of, we still have a lot of work to do to understand why this risk is becoming apparent and whether or not the children who are born preterm to these women go on to develop any health concerns.”
Dr Nichols and her colleagues analyzed data on 2598 births to female adolescent and young adult cancer survivors (ages 15 to 39) and 12,990 births to women without a cancer diagnosis.
Among cancer survivors, there was a significantly increased prevalence of preterm birth (prevalence ratio [PR]=1.52), low birth weight (PR=1.59), and cesarean delivery (PR=1.08), compared to women without a cancer diagnosis.
Timing of diagnosis and cancer type
When the researchers broke the data down by cancer diagnosis, they found a higher risk of preterm birth and low birth weight for women with lymphoma as well as breast and gynecologic cancers.
The PR for preterm birth was 1.59 for Hodgkin lymphoma, 1.98 for breast cancer, 2.11 for non-Hodgkin lymphoma, and 2.58 for gynecologic cancer. The PR for low birth weight was 1.59 for breast cancer, 2.41 for non-Hodgkin lymphoma, and 2.74 for gynecologic cancer.
The researchers found an increased risk of adverse birth outcomes among women who were diagnosed with cancer while pregnant and before pregnancy.
Among women diagnosed while pregnant, the PR was 2.97 for preterm birth, 2.82 for low birth weight, 1.21 for cesarean delivery, and 1.90 for low Apgar score. Among women diagnosed before pregnancy, the PR was 1.23 for preterm birth and 1.36 for low birth weight.
Role of treatment
Compared to women without a cancer diagnosis, cancer survivors who received chemotherapy but no radiation were more likely to have preterm births (PR=2.11), infants with low birth weight (PR=2.36), and cesarean deliveries (PR=1.16).
There was no significant increase in adverse birth outcomes among cancer survivors who received radiation but not chemotherapy.
Among the cancer survivors, women who received chemotherapy without radiation were more likely to have preterm births (PR=2.12), infants with low birth weight (PR=2.13), and infants who were small for their gestational age (PR=1.43) when compared to women treated with surgery only.
Dr Nichols said the role of treatment is an area of possible future research.
“We’d like to get better information about the types of chemotherapy women receive,” she said. “Chemotherapy is a very broad category, and the agents have very different effects on the body. In the future, we’d like to get more detailed information on the types of drugs that were involved in treatment.” ![]()
ASCO reports progress, challenges in cancer care

The US cancer care delivery system is undergoing changes to better meet the needs of cancer patients, but persistent hurdles threaten to slow progress, according to the American Society of Clinical Oncology (ASCO).
ASCO’s “The State of Cancer Care in America, 2017” report describes areas of progress, including new approaches for cancer diagnosis and treatment, improved data sharing to drive innovation, and an increased focus on value-based healthcare.
However, the report also suggests that access and affordability challenges, along with increased practice burdens, continue to pose barriers to high-value, high-quality cancer care.
The report was published in the Journal of Oncology Practice.
Challenges
The report notes that the US population is growing rapidly, changing demographically, and living longer. And all of these factors contribute to a record number of cancer cases/survivors.
It has been estimated that the number of cancer survivors in the US will grow from 15.5 million to 20.3 million by 2026.
Unfortunately, the report says, cancer care is unaffordable for many patients, even those with health insurance.
And significant health disparities persist that are independent of insurance status. Socioeconomic status, geography, and race/ethnicity all impact patient health outcomes.
The report also suggests that oncology practices are facing increased administrative burdens that divert time and resources from their patients.
Progress
Despite the aforementioned challenges, the report paints an optimistic vision about the future of cancer care and highlights activity in the past year aimed at improving care.
For instance, the Food and Drug Administration approved 5 new anticancer therapies, expanded the use of 13, and approved several diagnostic tests in 2016.
In addition, overall cancer incidence and mortality rates were lower in 2016 than in previous decades.
“Since 1991, we’ve been able to save 2.1 million lives because of significant advances in prevention, diagnosis, and treatment—something unimaginable even a decade ago,” said ASCO President Daniel F. Hayes, MD.
“But there’s still more work to be done to ensure that every patient with cancer, no matter who they are or where they live, has access to high-quality, high-value cancer care.”
The report includes a list of recommendations that, ASCO believes, could help bring the US closer to achieving that goal. ![]()
The US cancer care delivery system is undergoing changes to better meet the needs of cancer patients, but persistent hurdles threaten to slow progress, according to the American Society of Clinical Oncology (ASCO).
ASCO’s “The State of Cancer Care in America, 2017” report describes areas of progress, including new approaches for cancer diagnosis and treatment, improved data sharing to drive innovation, and an increased focus on value-based healthcare.
However, the report also suggests that access and affordability challenges, along with increased practice burdens, continue to pose barriers to high-value, high-quality cancer care.
The report was published in the Journal of Oncology Practice.
Challenges
The report notes that the US population is growing rapidly, changing demographically, and living longer. And all of these factors contribute to a record number of cancer cases/survivors.
It has been estimated that the number of cancer survivors in the US will grow from 15.5 million to 20.3 million by 2026.
Unfortunately, the report says, cancer care is unaffordable for many patients, even those with health insurance.
And significant health disparities persist that are independent of insurance status. Socioeconomic status, geography, and race/ethnicity all impact patient health outcomes.
The report also suggests that oncology practices are facing increased administrative burdens that divert time and resources from their patients.
Progress
Despite the aforementioned challenges, the report paints an optimistic vision about the future of cancer care and highlights activity in the past year aimed at improving care.
For instance, the Food and Drug Administration approved 5 new anticancer therapies, expanded the use of 13, and approved several diagnostic tests in 2016.
In addition, overall cancer incidence and mortality rates were lower in 2016 than in previous decades.
“Since 1991, we’ve been able to save 2.1 million lives because of significant advances in prevention, diagnosis, and treatment—something unimaginable even a decade ago,” said ASCO President Daniel F. Hayes, MD.
“But there’s still more work to be done to ensure that every patient with cancer, no matter who they are or where they live, has access to high-quality, high-value cancer care.”
The report includes a list of recommendations that, ASCO believes, could help bring the US closer to achieving that goal. ![]()
The US cancer care delivery system is undergoing changes to better meet the needs of cancer patients, but persistent hurdles threaten to slow progress, according to the American Society of Clinical Oncology (ASCO).
ASCO’s “The State of Cancer Care in America, 2017” report describes areas of progress, including new approaches for cancer diagnosis and treatment, improved data sharing to drive innovation, and an increased focus on value-based healthcare.
However, the report also suggests that access and affordability challenges, along with increased practice burdens, continue to pose barriers to high-value, high-quality cancer care.
The report was published in the Journal of Oncology Practice.
Challenges
The report notes that the US population is growing rapidly, changing demographically, and living longer. And all of these factors contribute to a record number of cancer cases/survivors.
It has been estimated that the number of cancer survivors in the US will grow from 15.5 million to 20.3 million by 2026.
Unfortunately, the report says, cancer care is unaffordable for many patients, even those with health insurance.
And significant health disparities persist that are independent of insurance status. Socioeconomic status, geography, and race/ethnicity all impact patient health outcomes.
The report also suggests that oncology practices are facing increased administrative burdens that divert time and resources from their patients.
Progress
Despite the aforementioned challenges, the report paints an optimistic vision about the future of cancer care and highlights activity in the past year aimed at improving care.
For instance, the Food and Drug Administration approved 5 new anticancer therapies, expanded the use of 13, and approved several diagnostic tests in 2016.
In addition, overall cancer incidence and mortality rates were lower in 2016 than in previous decades.
“Since 1991, we’ve been able to save 2.1 million lives because of significant advances in prevention, diagnosis, and treatment—something unimaginable even a decade ago,” said ASCO President Daniel F. Hayes, MD.
“But there’s still more work to be done to ensure that every patient with cancer, no matter who they are or where they live, has access to high-quality, high-value cancer care.”
The report includes a list of recommendations that, ASCO believes, could help bring the US closer to achieving that goal.