User login
We must counsel against heat-not-burn cigarettes
Tobacco companies are marketing a new version of cigarettes dubbed heat-not-burn (HNB) cigarettes.1,2 Offered as a “modified-risk tobacco product,” HNB cigarettes utilize a lithium battery-powered heating element and are available all over the world.1,2 Like conventional smokes, they contain tobacco, but deliver nicotine by heating leaves at 350° C rather than burning them at 600° C.1-3 Heating the tobacco produces an inhalable aerosol with tobacco flavor and nicotine, without smoke. These HNB cigarettes are also different from e-cigarettes that aerosolize a liquid.
Tobacco companies contend that HNB cigarettes are safer than smoking tobacco.1 Consumers inhale a heated tobacco aerosol that reportedly contains less nicotine and fewer toxicities; yet, HNB are not independently substantiated as being healthier, nor proven safe.1-5 Thermal decomposition, rather than combustion, may afford a less dangerous nicotine consumption; however, HNB aerosols deliver many of the same dangerous compounds as traditional cigarettes, including carbon monoxide, tar, and aromatic hydrocarbons.2-6 Despite possible harm reduction in the short-run, long-term safety remains unconfirmed.
Safety in passive environmental inhalations is not established.2 HNB cigarettes are contraindicated during pregnancy and/or lactation. Nicotine is provided in addictive quantities, enough to foster continued dependence. Exposure to HNB products can promote longer-term usage or lead to smoking traditional tobacco cigarettes. There is also an increased risk to non-smokers of exposure to HNB aerosols. Additionally, lithium batteries have been known to burn or explode. HNB devices may even lead to privacy concerns due micro-controller chips contained within that harvest information. These chips could inform manufacturers about device usage.7
Tobacco is a global health hazard and smoking is the number one preventable cause of disease.1,5,8 Global smoking prevalence is nearing 19%.9 There are concerns about dual use, rather than HNB cigarettes alone as a substitute for conventional smoking. The ultimate hope is to abstain from all tobacco and nicotine. Although HNB inhalations contain fewer toxic chemicals than by smoking, evidence regarding mitigation of tobacco-related diseases is inconclusive.10
Physicians have an obligation to minimize tobacco and nicotine-related hazards. Ongoing research and clinical exposure might better document the health impact of HNB cigarettes. Until the risks and benefits of HNB cigarettes are confirmed, health care professionals would be wise to counsel against their use.
Diksha Mohanty, MD; Steven Lippmann, MD
Louisville, Ky
1. Combustible cigarettes kill millions a year. Can Big Tobacco save them? The Economist Web site. https://www.economist.com/business/2017/12/19/combustible-cigarettes-kill-millions-a-year-can-big-tobacco-save-them. Accessed November 9, 2018.
2. Auer R, Concha-Lozano N, Jacot-Sadowski I, et al. Heat-not-burn tobacco cigarettes: smoke by any other name. JAMA Intern Med. 2017;177:1050-1052.
3. Caputi TL. Industry watch: heat-not-burn tobacco products are about to reach their boiling point. Tob Control. 2016;26:609-610.
4. Jenssen BP, Walley SC, McGrath-Morrow SA. Heat-not-burn tobacco products: Tobacco industry claims no substitute for science. Pediatrics. 2018;141:e20172383.
5. Levy DT, Cummings KM, Villanti AC, et al. A framework for evaluating the public health impact of e-cigarettes and other vaporized nicotine products. Addiction. 2017;112:8-17.
6. Bekki K, Inaba Y, Uchiyama S, et al. Comparison of chemicals in mainstream smoke in heat-not-burn tobacco and combustion cigarettes. J UOEH, 2017;39:201-207.
7. Lasseter T, Wilson D, Wilson T, et al. Philip Morris device knows a lot about your smoking habit. Reuters. https://www.reuters.com/investigates/special-report/tobacco-iqos-device. Accessed November 9, 2018.
8. Carter BD, Abnet CC, Feskanich D, et al. Smoking and mortality — beyond established causes. New Engl J Med. 2015;372:631-640.
9. World Health Organization. WHO global report on trends in tobacco smoking 2000-2025 - First edition. http://www.who.int/tobacco/publications/surveillance/reportontrendstobaccosmoking/en/index4.html. Accessed November 9, 2018.
10. U.S. Food & Drug Administration. CTPConnect—September 2017. https://www.fda.gov/TobaccoProducts/NewsEvents/ucm576895.htm. Updated June 14, 2018. Accessed Nov ember 9, 2018.
Tobacco companies are marketing a new version of cigarettes dubbed heat-not-burn (HNB) cigarettes.1,2 Offered as a “modified-risk tobacco product,” HNB cigarettes utilize a lithium battery-powered heating element and are available all over the world.1,2 Like conventional smokes, they contain tobacco, but deliver nicotine by heating leaves at 350° C rather than burning them at 600° C.1-3 Heating the tobacco produces an inhalable aerosol with tobacco flavor and nicotine, without smoke. These HNB cigarettes are also different from e-cigarettes that aerosolize a liquid.
Tobacco companies contend that HNB cigarettes are safer than smoking tobacco.1 Consumers inhale a heated tobacco aerosol that reportedly contains less nicotine and fewer toxicities; yet, HNB are not independently substantiated as being healthier, nor proven safe.1-5 Thermal decomposition, rather than combustion, may afford a less dangerous nicotine consumption; however, HNB aerosols deliver many of the same dangerous compounds as traditional cigarettes, including carbon monoxide, tar, and aromatic hydrocarbons.2-6 Despite possible harm reduction in the short-run, long-term safety remains unconfirmed.
Safety in passive environmental inhalations is not established.2 HNB cigarettes are contraindicated during pregnancy and/or lactation. Nicotine is provided in addictive quantities, enough to foster continued dependence. Exposure to HNB products can promote longer-term usage or lead to smoking traditional tobacco cigarettes. There is also an increased risk to non-smokers of exposure to HNB aerosols. Additionally, lithium batteries have been known to burn or explode. HNB devices may even lead to privacy concerns due micro-controller chips contained within that harvest information. These chips could inform manufacturers about device usage.7
Tobacco is a global health hazard and smoking is the number one preventable cause of disease.1,5,8 Global smoking prevalence is nearing 19%.9 There are concerns about dual use, rather than HNB cigarettes alone as a substitute for conventional smoking. The ultimate hope is to abstain from all tobacco and nicotine. Although HNB inhalations contain fewer toxic chemicals than by smoking, evidence regarding mitigation of tobacco-related diseases is inconclusive.10
Physicians have an obligation to minimize tobacco and nicotine-related hazards. Ongoing research and clinical exposure might better document the health impact of HNB cigarettes. Until the risks and benefits of HNB cigarettes are confirmed, health care professionals would be wise to counsel against their use.
Diksha Mohanty, MD; Steven Lippmann, MD
Louisville, Ky
Tobacco companies are marketing a new version of cigarettes dubbed heat-not-burn (HNB) cigarettes.1,2 Offered as a “modified-risk tobacco product,” HNB cigarettes utilize a lithium battery-powered heating element and are available all over the world.1,2 Like conventional smokes, they contain tobacco, but deliver nicotine by heating leaves at 350° C rather than burning them at 600° C.1-3 Heating the tobacco produces an inhalable aerosol with tobacco flavor and nicotine, without smoke. These HNB cigarettes are also different from e-cigarettes that aerosolize a liquid.
Tobacco companies contend that HNB cigarettes are safer than smoking tobacco.1 Consumers inhale a heated tobacco aerosol that reportedly contains less nicotine and fewer toxicities; yet, HNB are not independently substantiated as being healthier, nor proven safe.1-5 Thermal decomposition, rather than combustion, may afford a less dangerous nicotine consumption; however, HNB aerosols deliver many of the same dangerous compounds as traditional cigarettes, including carbon monoxide, tar, and aromatic hydrocarbons.2-6 Despite possible harm reduction in the short-run, long-term safety remains unconfirmed.
Safety in passive environmental inhalations is not established.2 HNB cigarettes are contraindicated during pregnancy and/or lactation. Nicotine is provided in addictive quantities, enough to foster continued dependence. Exposure to HNB products can promote longer-term usage or lead to smoking traditional tobacco cigarettes. There is also an increased risk to non-smokers of exposure to HNB aerosols. Additionally, lithium batteries have been known to burn or explode. HNB devices may even lead to privacy concerns due micro-controller chips contained within that harvest information. These chips could inform manufacturers about device usage.7
Tobacco is a global health hazard and smoking is the number one preventable cause of disease.1,5,8 Global smoking prevalence is nearing 19%.9 There are concerns about dual use, rather than HNB cigarettes alone as a substitute for conventional smoking. The ultimate hope is to abstain from all tobacco and nicotine. Although HNB inhalations contain fewer toxic chemicals than by smoking, evidence regarding mitigation of tobacco-related diseases is inconclusive.10
Physicians have an obligation to minimize tobacco and nicotine-related hazards. Ongoing research and clinical exposure might better document the health impact of HNB cigarettes. Until the risks and benefits of HNB cigarettes are confirmed, health care professionals would be wise to counsel against their use.
Diksha Mohanty, MD; Steven Lippmann, MD
Louisville, Ky
1. Combustible cigarettes kill millions a year. Can Big Tobacco save them? The Economist Web site. https://www.economist.com/business/2017/12/19/combustible-cigarettes-kill-millions-a-year-can-big-tobacco-save-them. Accessed November 9, 2018.
2. Auer R, Concha-Lozano N, Jacot-Sadowski I, et al. Heat-not-burn tobacco cigarettes: smoke by any other name. JAMA Intern Med. 2017;177:1050-1052.
3. Caputi TL. Industry watch: heat-not-burn tobacco products are about to reach their boiling point. Tob Control. 2016;26:609-610.
4. Jenssen BP, Walley SC, McGrath-Morrow SA. Heat-not-burn tobacco products: Tobacco industry claims no substitute for science. Pediatrics. 2018;141:e20172383.
5. Levy DT, Cummings KM, Villanti AC, et al. A framework for evaluating the public health impact of e-cigarettes and other vaporized nicotine products. Addiction. 2017;112:8-17.
6. Bekki K, Inaba Y, Uchiyama S, et al. Comparison of chemicals in mainstream smoke in heat-not-burn tobacco and combustion cigarettes. J UOEH, 2017;39:201-207.
7. Lasseter T, Wilson D, Wilson T, et al. Philip Morris device knows a lot about your smoking habit. Reuters. https://www.reuters.com/investigates/special-report/tobacco-iqos-device. Accessed November 9, 2018.
8. Carter BD, Abnet CC, Feskanich D, et al. Smoking and mortality — beyond established causes. New Engl J Med. 2015;372:631-640.
9. World Health Organization. WHO global report on trends in tobacco smoking 2000-2025 - First edition. http://www.who.int/tobacco/publications/surveillance/reportontrendstobaccosmoking/en/index4.html. Accessed November 9, 2018.
10. U.S. Food & Drug Administration. CTPConnect—September 2017. https://www.fda.gov/TobaccoProducts/NewsEvents/ucm576895.htm. Updated June 14, 2018. Accessed Nov ember 9, 2018.
1. Combustible cigarettes kill millions a year. Can Big Tobacco save them? The Economist Web site. https://www.economist.com/business/2017/12/19/combustible-cigarettes-kill-millions-a-year-can-big-tobacco-save-them. Accessed November 9, 2018.
2. Auer R, Concha-Lozano N, Jacot-Sadowski I, et al. Heat-not-burn tobacco cigarettes: smoke by any other name. JAMA Intern Med. 2017;177:1050-1052.
3. Caputi TL. Industry watch: heat-not-burn tobacco products are about to reach their boiling point. Tob Control. 2016;26:609-610.
4. Jenssen BP, Walley SC, McGrath-Morrow SA. Heat-not-burn tobacco products: Tobacco industry claims no substitute for science. Pediatrics. 2018;141:e20172383.
5. Levy DT, Cummings KM, Villanti AC, et al. A framework for evaluating the public health impact of e-cigarettes and other vaporized nicotine products. Addiction. 2017;112:8-17.
6. Bekki K, Inaba Y, Uchiyama S, et al. Comparison of chemicals in mainstream smoke in heat-not-burn tobacco and combustion cigarettes. J UOEH, 2017;39:201-207.
7. Lasseter T, Wilson D, Wilson T, et al. Philip Morris device knows a lot about your smoking habit. Reuters. https://www.reuters.com/investigates/special-report/tobacco-iqos-device. Accessed November 9, 2018.
8. Carter BD, Abnet CC, Feskanich D, et al. Smoking and mortality — beyond established causes. New Engl J Med. 2015;372:631-640.
9. World Health Organization. WHO global report on trends in tobacco smoking 2000-2025 - First edition. http://www.who.int/tobacco/publications/surveillance/reportontrendstobaccosmoking/en/index4.html. Accessed November 9, 2018.
10. U.S. Food & Drug Administration. CTPConnect—September 2017. https://www.fda.gov/TobaccoProducts/NewsEvents/ucm576895.htm. Updated June 14, 2018. Accessed Nov ember 9, 2018.
Dehydration in terminal illness: Which path forward?
CASE 1
A 94-year-old white woman, who had been in excellent health (other than pernicious anemia, treated with monthly cyanocobalamin injections), suddenly developed gastrointestinal distress 2 weeks earlier. A work-up performed by her physician revealed advanced pancreatic cancer.
Over the next 2 weeks, she experienced pain and nausea. A left-sided fistula developed externally at her flank that drained feces and induced considerable discomfort. An indwelling drain was placed, which provided some relief, but the patient’s dyspepsia, pain, and nausea escalated.
One month into her disease course, an oncologist reported on her potential treatment options and prognosis. Her life expectancy was about 3 months without treatment. This could be extended by 1 to 2 months with extensive surgical and chemotherapeutic interventions, but would further diminish her quality of life. The patient declined further treatment.
Her clinical status declined, and her quality of life significantly deteriorated. At 3 months, she felt life had lost meaning and was not worth living. She began asking for a morphine overdose, stating a desire to end her life.
After several discussions with the oncologist, one of the patient’s adult children suggested that her mother stop eating and drinking in order to diminish discomfort and hasten her demise. This plan was adopted, and the patient declined food and drank only enough to swish for oral comfort.
CASE 2
An 83-year-old woman with advanced Parkinson’s disease had become increasingly disabled. Her gait and motor skills were dramatically and progressively compromised. Pharmacotherapy yielded only transient improvement and considerable adverse effects of choreiform hyperkinesia and hallucinations, which were troublesome and embarrassing. Her social, physical, and personal well-being declined to the point that she was placed in a nursing home.
Despite this help, worsening parkinsonism progressively diminished her physical capacity. She became largely bedridden and developed decubitus ulcerations, especially at the coccyx, which produced severe pain and distress.
Continue to: The confluence of pain...
The confluence of pain, bedfastness, constipation, and social isolation yielded a loss of interest and joy in life. The patient required assistance with almost every aspect of daily life, including eating. As the illness progressed, she prayed at night that God would “take her.” Each morning, she spoke of disappointment upon reawakening. She overtly expressed her lack of desire to live to her family. Medical interventions were increasingly ineffective.
After repeated family and physician discussions had focused on her death wishes, one adult daughter recommended her mother stop eating and drinking; her food intake was already minimal. Although she did not endorse this plan verbally, the patient’s oral intake significantly diminished. Within 2 weeks, her physical state had declined, and she died one night during sleep.
Adequate hydration is stressed in physician education and practice. A conventional expectation to normalize fluid balance is important to restore health and improve well-being. In addition to being good medical practice, it can also show patients (and their families) that we care about their well-being.1-3
Treating dehydration in individuals with terminal illness is controversial from both medical and ethical standpoints. While the natural tendency of physicians is to restore full hydration to their patients, in select cases of imminent death, being fully hydrated may prolong discomfort.1,2 Emphasis in this population should be consistently placed on improving comfort care and quality of life, rather than prolonging life or delaying death.3-5
Continue to: A multifactorial, patient-based decision
A multifactorial, patient-based decision
Years ago, before the advent of hospitalizing people with terminal illnesses, dying at home amongst loved ones was believed to be peaceful. Nevertheless, questions arise about the practical vs ethical approach to caring for patients with terminal illness.2 Sometimes it is difficult to find a balance between potential health care benefits and the burdens induced by medical, legal, moral, and/or social pressures. Our medical communities and the general population uphold preserving dignity at the end of life, which is supported by organizations such as Compassion & Choices (a nonprofit group that seeks to improve and expand options for end of life care; https://www.compassionandchoices.org).
Allowing for voluntary, patient-determined dehydration in those with terminal illness can offer greater comfort than maintaining the physiologic degrees of fluid balance. There are 3 key considerations to bear in mind:
- Hydration is usually a standard part of quality medical care.1
- Selectively allowing dehydration in patients who are dying can facilitate comfort.1-5
- Dehydration may be a deliberate strategy to hasten death.6
When is dehydration appropriate?
Hydration is not favored whenever doing so may increase discomfort and prolong pain without meaningful life.3 In people with terminal illness, hydration may reduce quality of life.7
The data support dehydration in certain patients. A randomized controlled trial involving 129 patients receiving hospice care compared parenteral hydration with placebo, documenting that rehydration did not improve symptoms or quality of life; there was no significant difference between patients who were hydrated and those who were dehydrated.7 In fact, dehydration may even yield greater patient comfort.8
Case reports, retrospective chart reviews, and testimonials from health care professionals have reported that being less hydrated can diminish nausea, vomiting, diarrhea, ascites, edema, and urinary or bowel incontinence, with less skin breakdown.8 Hydration, on the other hand, may exacerbate dyspnea, coughing, and choking, increasing the need for suctioning.
Continue to: A component of palliative care
A component of palliative care. When death is imminent, palliation becomes key. Pain may be more manageable with less fluids, an important goal for this population.6,8 Dehydration is associated with an accumulation of opioids throughout body fluid volumes, which may decrease pain, consciousness, and/or agony.2 Pharmacotherapies might also have greater efficacy in a dehydrated patient.9 In addition, tissue shrinkage might mitigate pain from tumors, especially those in confined spaces.8
Hospice care and palliative medicine confirm that routine hydration is not always advisable; allowing for dehydration is a conventional practice, especially in older adults with terminal illness.7 However, do not deny access to liquids if a patient wants them, and never force unwanted fluids by any route.8 Facilitate oral care in the form of swishing fluids, elective drinking, or providing mouth lubrication for any patients selectively allowed to become dehydrated.3,8
The role of the physician in decision-making
Patients with terminal illness sometimes do not want fluids and may actively decline food and drink.10 This can be emotionally distressing for family members and/or caregivers to witness. Physicians can address this concern by compassionately explaining: “I know you are concerned that your relative is not eating or drinking, but there is no indication that hydration or parenteral feeding will improve function or quality of life.”10 This can generate a discussion between physicians and families by acknowledging concerns, relieving distress, and leading to what is ultimately best for the patient.
Implications for practice: Individualized autonomy
Physicians must identify patients who wish to die by purposely becoming dehydrated and uphold the important physician obligation to hydrate those with a recoverable illness. Allowing for a moderate degree of dehydration might provide greater comfort in select people with terminal illness. Some individuals for whom life has lost meaning may choose dehydration as a means to hasten their departure.4-6 Allowing individualized autonomy over life and death choices is part of a physician’s obligation to their patients. It can be difficult for caregivers, but it is medically indicated to comply with a patient’s desire for comfort when death is imminent.
Providing palliation as a priority over treatment is sometimes challenging, but comfort care takes preference and is always coordinated with the person’s own wishes. Facilitating dehydration removes assisted-suicide issues or requests and thus affords everyone involved more emotional comfort. An advantage of this method is that a decisional patient maintains full control over the direction of their choices and helps preserve dignity during the end of life.
CORRESPONDENCE
Steven Lippmann, MD, Department of Psychiatry, University of Louisville School of Medicine, 401 East Chestnut Street, Suite 610, Louisville, KY 40202; sblipp01@louisville.edu
1. Burge FI. Dehydration and provision of fluids in palliative care. What is the evidence? Can Fam Physician. 1996;42:2383-2388.
2. Printz LA. Is withholding hydration a valid comfort measure in the terminally ill? Geriatrics. 1988;43:84-88.
3. Lippmann S. Palliative dehydration. Prim Care Companion CNS Disord. 2015;17: doi: 10.4088/PCC.15101797.
4. Bernat JL, Gert B, Mogielnicki RP. Patient refusal of hydration and nutrition: an alternative to physician-assisted suicide or voluntary active euthanasia. Arch Intern Med. 1993;153:2723-2728.
5. Sullivan RJ. Accepting death without artificial nutrition or hydration. J Gen Intern Med.1993;8:220-224.
6. Miller FG, Meier DE. Voluntary death: a comparison of terminal dehydration and physician-assisted suicide. Ann Intern Med. 1998;128:559-562.
7. Bruera E, Hui D, Dalal S, et al. Parenteral hydration in patients with advanced cancer: a multicenter, double-blind, placebo-controlled randomized trial. J Clin Oncol. 2013;31:111-118.
8. Forrow L, Smith HS. Pain management in end of life: palliative care. In: Warfield CA, Bajwa ZH, ed. Principles and Practice of Pain Management. 2nd ed. New York, NY: McGraw-Hill; 2004.
9. Zerwekh JV. The dehydration question. Nursing. 1983;13:47-51.
10. Bailey F, Harman S. Palliative care: The last hours and days of life. www.uptodate.com. September, 2016. Accessed on September 11, 2018.
CASE 1
A 94-year-old white woman, who had been in excellent health (other than pernicious anemia, treated with monthly cyanocobalamin injections), suddenly developed gastrointestinal distress 2 weeks earlier. A work-up performed by her physician revealed advanced pancreatic cancer.
Over the next 2 weeks, she experienced pain and nausea. A left-sided fistula developed externally at her flank that drained feces and induced considerable discomfort. An indwelling drain was placed, which provided some relief, but the patient’s dyspepsia, pain, and nausea escalated.
One month into her disease course, an oncologist reported on her potential treatment options and prognosis. Her life expectancy was about 3 months without treatment. This could be extended by 1 to 2 months with extensive surgical and chemotherapeutic interventions, but would further diminish her quality of life. The patient declined further treatment.
Her clinical status declined, and her quality of life significantly deteriorated. At 3 months, she felt life had lost meaning and was not worth living. She began asking for a morphine overdose, stating a desire to end her life.
After several discussions with the oncologist, one of the patient’s adult children suggested that her mother stop eating and drinking in order to diminish discomfort and hasten her demise. This plan was adopted, and the patient declined food and drank only enough to swish for oral comfort.
CASE 2
An 83-year-old woman with advanced Parkinson’s disease had become increasingly disabled. Her gait and motor skills were dramatically and progressively compromised. Pharmacotherapy yielded only transient improvement and considerable adverse effects of choreiform hyperkinesia and hallucinations, which were troublesome and embarrassing. Her social, physical, and personal well-being declined to the point that she was placed in a nursing home.
Despite this help, worsening parkinsonism progressively diminished her physical capacity. She became largely bedridden and developed decubitus ulcerations, especially at the coccyx, which produced severe pain and distress.
Continue to: The confluence of pain...
The confluence of pain, bedfastness, constipation, and social isolation yielded a loss of interest and joy in life. The patient required assistance with almost every aspect of daily life, including eating. As the illness progressed, she prayed at night that God would “take her.” Each morning, she spoke of disappointment upon reawakening. She overtly expressed her lack of desire to live to her family. Medical interventions were increasingly ineffective.
After repeated family and physician discussions had focused on her death wishes, one adult daughter recommended her mother stop eating and drinking; her food intake was already minimal. Although she did not endorse this plan verbally, the patient’s oral intake significantly diminished. Within 2 weeks, her physical state had declined, and she died one night during sleep.
Adequate hydration is stressed in physician education and practice. A conventional expectation to normalize fluid balance is important to restore health and improve well-being. In addition to being good medical practice, it can also show patients (and their families) that we care about their well-being.1-3
Treating dehydration in individuals with terminal illness is controversial from both medical and ethical standpoints. While the natural tendency of physicians is to restore full hydration to their patients, in select cases of imminent death, being fully hydrated may prolong discomfort.1,2 Emphasis in this population should be consistently placed on improving comfort care and quality of life, rather than prolonging life or delaying death.3-5
Continue to: A multifactorial, patient-based decision
A multifactorial, patient-based decision
Years ago, before the advent of hospitalizing people with terminal illnesses, dying at home amongst loved ones was believed to be peaceful. Nevertheless, questions arise about the practical vs ethical approach to caring for patients with terminal illness.2 Sometimes it is difficult to find a balance between potential health care benefits and the burdens induced by medical, legal, moral, and/or social pressures. Our medical communities and the general population uphold preserving dignity at the end of life, which is supported by organizations such as Compassion & Choices (a nonprofit group that seeks to improve and expand options for end of life care; https://www.compassionandchoices.org).
Allowing for voluntary, patient-determined dehydration in those with terminal illness can offer greater comfort than maintaining the physiologic degrees of fluid balance. There are 3 key considerations to bear in mind:
- Hydration is usually a standard part of quality medical care.1
- Selectively allowing dehydration in patients who are dying can facilitate comfort.1-5
- Dehydration may be a deliberate strategy to hasten death.6
When is dehydration appropriate?
Hydration is not favored whenever doing so may increase discomfort and prolong pain without meaningful life.3 In people with terminal illness, hydration may reduce quality of life.7
The data support dehydration in certain patients. A randomized controlled trial involving 129 patients receiving hospice care compared parenteral hydration with placebo, documenting that rehydration did not improve symptoms or quality of life; there was no significant difference between patients who were hydrated and those who were dehydrated.7 In fact, dehydration may even yield greater patient comfort.8
Case reports, retrospective chart reviews, and testimonials from health care professionals have reported that being less hydrated can diminish nausea, vomiting, diarrhea, ascites, edema, and urinary or bowel incontinence, with less skin breakdown.8 Hydration, on the other hand, may exacerbate dyspnea, coughing, and choking, increasing the need for suctioning.
Continue to: A component of palliative care
A component of palliative care. When death is imminent, palliation becomes key. Pain may be more manageable with less fluids, an important goal for this population.6,8 Dehydration is associated with an accumulation of opioids throughout body fluid volumes, which may decrease pain, consciousness, and/or agony.2 Pharmacotherapies might also have greater efficacy in a dehydrated patient.9 In addition, tissue shrinkage might mitigate pain from tumors, especially those in confined spaces.8
Hospice care and palliative medicine confirm that routine hydration is not always advisable; allowing for dehydration is a conventional practice, especially in older adults with terminal illness.7 However, do not deny access to liquids if a patient wants them, and never force unwanted fluids by any route.8 Facilitate oral care in the form of swishing fluids, elective drinking, or providing mouth lubrication for any patients selectively allowed to become dehydrated.3,8
The role of the physician in decision-making
Patients with terminal illness sometimes do not want fluids and may actively decline food and drink.10 This can be emotionally distressing for family members and/or caregivers to witness. Physicians can address this concern by compassionately explaining: “I know you are concerned that your relative is not eating or drinking, but there is no indication that hydration or parenteral feeding will improve function or quality of life.”10 This can generate a discussion between physicians and families by acknowledging concerns, relieving distress, and leading to what is ultimately best for the patient.
Implications for practice: Individualized autonomy
Physicians must identify patients who wish to die by purposely becoming dehydrated and uphold the important physician obligation to hydrate those with a recoverable illness. Allowing for a moderate degree of dehydration might provide greater comfort in select people with terminal illness. Some individuals for whom life has lost meaning may choose dehydration as a means to hasten their departure.4-6 Allowing individualized autonomy over life and death choices is part of a physician’s obligation to their patients. It can be difficult for caregivers, but it is medically indicated to comply with a patient’s desire for comfort when death is imminent.
Providing palliation as a priority over treatment is sometimes challenging, but comfort care takes preference and is always coordinated with the person’s own wishes. Facilitating dehydration removes assisted-suicide issues or requests and thus affords everyone involved more emotional comfort. An advantage of this method is that a decisional patient maintains full control over the direction of their choices and helps preserve dignity during the end of life.
CORRESPONDENCE
Steven Lippmann, MD, Department of Psychiatry, University of Louisville School of Medicine, 401 East Chestnut Street, Suite 610, Louisville, KY 40202; sblipp01@louisville.edu
CASE 1
A 94-year-old white woman, who had been in excellent health (other than pernicious anemia, treated with monthly cyanocobalamin injections), suddenly developed gastrointestinal distress 2 weeks earlier. A work-up performed by her physician revealed advanced pancreatic cancer.
Over the next 2 weeks, she experienced pain and nausea. A left-sided fistula developed externally at her flank that drained feces and induced considerable discomfort. An indwelling drain was placed, which provided some relief, but the patient’s dyspepsia, pain, and nausea escalated.
One month into her disease course, an oncologist reported on her potential treatment options and prognosis. Her life expectancy was about 3 months without treatment. This could be extended by 1 to 2 months with extensive surgical and chemotherapeutic interventions, but would further diminish her quality of life. The patient declined further treatment.
Her clinical status declined, and her quality of life significantly deteriorated. At 3 months, she felt life had lost meaning and was not worth living. She began asking for a morphine overdose, stating a desire to end her life.
After several discussions with the oncologist, one of the patient’s adult children suggested that her mother stop eating and drinking in order to diminish discomfort and hasten her demise. This plan was adopted, and the patient declined food and drank only enough to swish for oral comfort.
CASE 2
An 83-year-old woman with advanced Parkinson’s disease had become increasingly disabled. Her gait and motor skills were dramatically and progressively compromised. Pharmacotherapy yielded only transient improvement and considerable adverse effects of choreiform hyperkinesia and hallucinations, which were troublesome and embarrassing. Her social, physical, and personal well-being declined to the point that she was placed in a nursing home.
Despite this help, worsening parkinsonism progressively diminished her physical capacity. She became largely bedridden and developed decubitus ulcerations, especially at the coccyx, which produced severe pain and distress.
Continue to: The confluence of pain...
The confluence of pain, bedfastness, constipation, and social isolation yielded a loss of interest and joy in life. The patient required assistance with almost every aspect of daily life, including eating. As the illness progressed, she prayed at night that God would “take her.” Each morning, she spoke of disappointment upon reawakening. She overtly expressed her lack of desire to live to her family. Medical interventions were increasingly ineffective.
After repeated family and physician discussions had focused on her death wishes, one adult daughter recommended her mother stop eating and drinking; her food intake was already minimal. Although she did not endorse this plan verbally, the patient’s oral intake significantly diminished. Within 2 weeks, her physical state had declined, and she died one night during sleep.
Adequate hydration is stressed in physician education and practice. A conventional expectation to normalize fluid balance is important to restore health and improve well-being. In addition to being good medical practice, it can also show patients (and their families) that we care about their well-being.1-3
Treating dehydration in individuals with terminal illness is controversial from both medical and ethical standpoints. While the natural tendency of physicians is to restore full hydration to their patients, in select cases of imminent death, being fully hydrated may prolong discomfort.1,2 Emphasis in this population should be consistently placed on improving comfort care and quality of life, rather than prolonging life or delaying death.3-5
Continue to: A multifactorial, patient-based decision
A multifactorial, patient-based decision
Years ago, before the advent of hospitalizing people with terminal illnesses, dying at home amongst loved ones was believed to be peaceful. Nevertheless, questions arise about the practical vs ethical approach to caring for patients with terminal illness.2 Sometimes it is difficult to find a balance between potential health care benefits and the burdens induced by medical, legal, moral, and/or social pressures. Our medical communities and the general population uphold preserving dignity at the end of life, which is supported by organizations such as Compassion & Choices (a nonprofit group that seeks to improve and expand options for end of life care; https://www.compassionandchoices.org).
Allowing for voluntary, patient-determined dehydration in those with terminal illness can offer greater comfort than maintaining the physiologic degrees of fluid balance. There are 3 key considerations to bear in mind:
- Hydration is usually a standard part of quality medical care.1
- Selectively allowing dehydration in patients who are dying can facilitate comfort.1-5
- Dehydration may be a deliberate strategy to hasten death.6
When is dehydration appropriate?
Hydration is not favored whenever doing so may increase discomfort and prolong pain without meaningful life.3 In people with terminal illness, hydration may reduce quality of life.7
The data support dehydration in certain patients. A randomized controlled trial involving 129 patients receiving hospice care compared parenteral hydration with placebo, documenting that rehydration did not improve symptoms or quality of life; there was no significant difference between patients who were hydrated and those who were dehydrated.7 In fact, dehydration may even yield greater patient comfort.8
Case reports, retrospective chart reviews, and testimonials from health care professionals have reported that being less hydrated can diminish nausea, vomiting, diarrhea, ascites, edema, and urinary or bowel incontinence, with less skin breakdown.8 Hydration, on the other hand, may exacerbate dyspnea, coughing, and choking, increasing the need for suctioning.
Continue to: A component of palliative care
A component of palliative care. When death is imminent, palliation becomes key. Pain may be more manageable with less fluids, an important goal for this population.6,8 Dehydration is associated with an accumulation of opioids throughout body fluid volumes, which may decrease pain, consciousness, and/or agony.2 Pharmacotherapies might also have greater efficacy in a dehydrated patient.9 In addition, tissue shrinkage might mitigate pain from tumors, especially those in confined spaces.8
Hospice care and palliative medicine confirm that routine hydration is not always advisable; allowing for dehydration is a conventional practice, especially in older adults with terminal illness.7 However, do not deny access to liquids if a patient wants them, and never force unwanted fluids by any route.8 Facilitate oral care in the form of swishing fluids, elective drinking, or providing mouth lubrication for any patients selectively allowed to become dehydrated.3,8
The role of the physician in decision-making
Patients with terminal illness sometimes do not want fluids and may actively decline food and drink.10 This can be emotionally distressing for family members and/or caregivers to witness. Physicians can address this concern by compassionately explaining: “I know you are concerned that your relative is not eating or drinking, but there is no indication that hydration or parenteral feeding will improve function or quality of life.”10 This can generate a discussion between physicians and families by acknowledging concerns, relieving distress, and leading to what is ultimately best for the patient.
Implications for practice: Individualized autonomy
Physicians must identify patients who wish to die by purposely becoming dehydrated and uphold the important physician obligation to hydrate those with a recoverable illness. Allowing for a moderate degree of dehydration might provide greater comfort in select people with terminal illness. Some individuals for whom life has lost meaning may choose dehydration as a means to hasten their departure.4-6 Allowing individualized autonomy over life and death choices is part of a physician’s obligation to their patients. It can be difficult for caregivers, but it is medically indicated to comply with a patient’s desire for comfort when death is imminent.
Providing palliation as a priority over treatment is sometimes challenging, but comfort care takes preference and is always coordinated with the person’s own wishes. Facilitating dehydration removes assisted-suicide issues or requests and thus affords everyone involved more emotional comfort. An advantage of this method is that a decisional patient maintains full control over the direction of their choices and helps preserve dignity during the end of life.
CORRESPONDENCE
Steven Lippmann, MD, Department of Psychiatry, University of Louisville School of Medicine, 401 East Chestnut Street, Suite 610, Louisville, KY 40202; sblipp01@louisville.edu
1. Burge FI. Dehydration and provision of fluids in palliative care. What is the evidence? Can Fam Physician. 1996;42:2383-2388.
2. Printz LA. Is withholding hydration a valid comfort measure in the terminally ill? Geriatrics. 1988;43:84-88.
3. Lippmann S. Palliative dehydration. Prim Care Companion CNS Disord. 2015;17: doi: 10.4088/PCC.15101797.
4. Bernat JL, Gert B, Mogielnicki RP. Patient refusal of hydration and nutrition: an alternative to physician-assisted suicide or voluntary active euthanasia. Arch Intern Med. 1993;153:2723-2728.
5. Sullivan RJ. Accepting death without artificial nutrition or hydration. J Gen Intern Med.1993;8:220-224.
6. Miller FG, Meier DE. Voluntary death: a comparison of terminal dehydration and physician-assisted suicide. Ann Intern Med. 1998;128:559-562.
7. Bruera E, Hui D, Dalal S, et al. Parenteral hydration in patients with advanced cancer: a multicenter, double-blind, placebo-controlled randomized trial. J Clin Oncol. 2013;31:111-118.
8. Forrow L, Smith HS. Pain management in end of life: palliative care. In: Warfield CA, Bajwa ZH, ed. Principles and Practice of Pain Management. 2nd ed. New York, NY: McGraw-Hill; 2004.
9. Zerwekh JV. The dehydration question. Nursing. 1983;13:47-51.
10. Bailey F, Harman S. Palliative care: The last hours and days of life. www.uptodate.com. September, 2016. Accessed on September 11, 2018.
1. Burge FI. Dehydration and provision of fluids in palliative care. What is the evidence? Can Fam Physician. 1996;42:2383-2388.
2. Printz LA. Is withholding hydration a valid comfort measure in the terminally ill? Geriatrics. 1988;43:84-88.
3. Lippmann S. Palliative dehydration. Prim Care Companion CNS Disord. 2015;17: doi: 10.4088/PCC.15101797.
4. Bernat JL, Gert B, Mogielnicki RP. Patient refusal of hydration and nutrition: an alternative to physician-assisted suicide or voluntary active euthanasia. Arch Intern Med. 1993;153:2723-2728.
5. Sullivan RJ. Accepting death without artificial nutrition or hydration. J Gen Intern Med.1993;8:220-224.
6. Miller FG, Meier DE. Voluntary death: a comparison of terminal dehydration and physician-assisted suicide. Ann Intern Med. 1998;128:559-562.
7. Bruera E, Hui D, Dalal S, et al. Parenteral hydration in patients with advanced cancer: a multicenter, double-blind, placebo-controlled randomized trial. J Clin Oncol. 2013;31:111-118.
8. Forrow L, Smith HS. Pain management in end of life: palliative care. In: Warfield CA, Bajwa ZH, ed. Principles and Practice of Pain Management. 2nd ed. New York, NY: McGraw-Hill; 2004.
9. Zerwekh JV. The dehydration question. Nursing. 1983;13:47-51.
10. Bailey F, Harman S. Palliative care: The last hours and days of life. www.uptodate.com. September, 2016. Accessed on September 11, 2018.

Risks vs Benefits for SGLT2 Inhibitor Medications
Diabetes mellitus (DM) is a metabolic disorder affecting about 5% to 13% of the population in the US.1 Since 1552, the earliest record of a person with DM, many treatment advances have been made.2Sodium-glucose cotransporter 2 (SGLT2) inhibitors are one of the newest antidiabetic pharmaceuticals on the market. The SGLT2 inhibitor drugs include canagliflozin, dapagliflozin, empagliflozin, ipragliflozin, and tofogliflozin; however, only canagliflozin, dapagliflozin, and empagliflozin have been approved by the US Food and Drug Administration (FDA). These pharmaceuticals promote glycosuria via the kidneys and enhance sugar excretion from the body. Along with lifestyle changes and self-care measures, such as healthful eating and increased physical activity, SGLT2 inhibitor pharmaceuticals provide antidiabetic efficacy by facilitating normoglycemia and minimizing vascular pathology.
Although SGLT2 inhibitor pharmaceuticals are newly introduced into the market, their discovery dates to 1835.3 Phlorizin, a nonselective SGLT inhibitor, was first isolated by French chemists from the bark of an apple tree.4 Phlorizin inhibits SGLT1 mostly in small intestinal cells, and SGLT2 similarly affects the kidney.4 Renal SGLT2 is the primary therapeutic target. Canagliflozin was the first pharmaceutical SGLT2 inhibitor approved by the FDA in 2013. Dapagliflozin’s FDA approval followed in 2013 and empagliflozin in 2014.5
Mechanism Of Action
In healthy individuals, tubular glucose is absorbed, resulting in no urinary glucose excretion. Sodium-glucose cotransporters 1 and 2 contribute to the renal absorption of glucose. A SGLT2 is responsible for 90% of the glucose reuptake in the segment 1 of the proximal tubule, while SGLT 1 is accountable for the remaining 10%.3 Unlike other antidiabetic medications, which act by increasing insulin secretion or improving insulin sensitivity for the receptors, SGLT2 inhibitor drugs prevent the reuptake of glucose into the bloodstream. This selective action spares the inhibition of SGLT1 present in other tissues, avoiding gastrointestinal effects.6
Benefits
The SGLT2 inhibitor action is focused on renal excretion of glucose and is independent of insulin action. 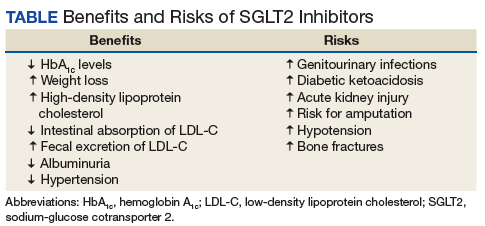
Hemoglobin A1c Levels
Canagliflozin, dapagliflozin, and empagliflozin reduce hemoglobin A1c (HbA1c) levels.5 Inagaki and colleagues found significant reductions in HbA1c and weight gain with > 100 mg canagliflozin compared with that of placebo when used for 12 weeks.7 In a study where 2.5-mg, 5-mg, and 10-mg dapagliflozin was compared with placebo, the mean HbA1c change from the baseline was -0.23% with placebo; -0.58% at 2.5 mg; -0.77% at 5 mg; and -0.89% at 10 mg.8 Empagliflozin was more effective in reducing HbA1c levels than was sitagliptin.9 When patients were treated with 10-mg empagliflozin, 25-mg empagliflozin, and sitagliptin, HbA1c levels dropped -1.44%, -1.43%, and -1.04%, respectively.9
Cholesterol
Sodium-glucose cotransporter 2 inhibitors have the beneficial effect of reducing vascular disease risk factors.10,11 A study by Hayashi and colleagues found that dapagliflozin decreases harmful atherogenic small, low-density lipoprotein-cholesterol (LDL-C), increases less atherogenic large, buoyant LDL-C, and increases high-density, lipoprotein-2 cholesterol (HDL-2C).10 Empagliflozin, however, can cause a small dose-dependent increase in HDL-C and LDL-C.11 Although there is an increase in serum LDL-C concentrations, empagliflozin can induce a decrease in intestinal absorption of cholesterol, thus promoting fecal excretion of LDL-C and macrophage-derived cholesterol.11
Weight Loss
A study by Weber and colleagues found that the SGLT2 inhibitor dapagloflozin lead to a reduction in body weight from -1.0 kg to -0.3 kg compared with placebo.12 Cefalu and colleagues found that daily prescribing of 100 mg and/or 300 mg of canagliflozin evidenced dose-dependent loss of weight.13 Neeland and colleagues found that empagliflozin utilization resulted in less adiposity indices in 3,300 subjects.14
Albuminuria
Sodium-glucose cotransporter 2 inhibitors have a reno-protective role in patients with type 2 DM (T2DM). In those receiving renin-angiotensin blockers with T2DM and hypertension, dapagliflozin decreased their albuminuria.15 Canagliflozin has a similar potential.16 Empagliflozin reduced the urine albumin-creatinine ratio in patients with macro- or micro-albuminuria, supporting a direct renal effect by SGLT2 inhibitors.17
Systolic Blood Pressure
Sodium-glucose cotransporter 2 inhibitors can have beneficial effects on physiologic vascular outcomes. In patients with T2DM and hypertension, dapagliflozin reduced mean systolic blood pressure (SBP) compared with placebo: -7.3 mm Hg vs -10.4 mm Hg, respectively.12 Prescribing canagliflozin treatment at 100 mg or 300 mg reduced SBP (-4.3 mm Hg and -5.0 mm Hg, respectively, vs placebo at -0.3 mm Hg).18 Subjects taking empagliflozin 10 mg or 25 mg exhibited an adjusted mean BP change from baseline of -4.60 mm Hg and -5.47mm Hg, respectively, whereas placebo induced a -0.67 mm Hg decline.19
Risks
Nausea, fatigue, polyuria, polydipsia, and xerostomia are common SGLT2 AEs. Use of SGLT2 inhibitors can induce certain other more serious AEs as well.
Increased Risk for Amputations
The Canagliflozin Cardiovascular Assessment Study (CANVAS) and the Canagliflozin Cardiovascular Assessment Study-Renal (CANVAS-R) documented that canagliflozin doubled the incidence of leg and foot amputations in research participants compared with placebo (6.3 vs 3.4 per 1,000 patient-years).16 Therefore, canagliflozin should be prescribed with caution in persons with a prior history of foot ulceration, neuropathy, and/or vascular diseases.20
Acute Renal Injury
The mechanism of kidney damage by SGLT2 inhibitor drugs is not completely understood. About 100 patients experienced renal failure after the intake of SGLT2 inhibitor drugs.21 Among them, more than half reported symptom onset within a month of starting the medication, and their symptoms improved after discontinuing the SGLT2 medication. As a result, the FDA issued a warning to monitor renal function before initiating and during such pharmacotherapy.21
Ketoacidosis
Sodium-glucose cotransporter 2 inhibitors might lead to elevated ketone body levels22 and euglycemic ketoacidosis;23 however, this risk reportedly is negligible.24 Use of SGLT2 inhibitors is not recommended for patients evidencing the presence of precipitating factors like acute gastroenteritis or insulin pump failure.25
Genitourinary Infections
About 10% to 15% of women taking SGLT2 inhibitor medications developed urinary tract infections and vulvovaginitis.26 This could be because of a glycosuria effect caused by SGLT2 inhibitors.27
Hypotension
Sodium-glucose cotransporter 2 inhibitors cause contraction of intravascular volume. Therefore, patients taking SGLT2 inhibitors are at risk for hypotension, leading to dizziness and potentially dangerous falls. Patients already taking volume-depleting medications, such as diuretics, should be advised to use this group of medications with caution and report these AEs.28
Bone Fractures
A clinical trial revealed that SGLT2 inhibitors, such as canagliflozin, decrease bone mineral density possibly leading to bone fractures.29 Bone fractures occurred in about 1.5% of cases of patients taking 100 mg and 300 mg of canagliflozin compared with a 1.1% fracture rate among the placebo group.29
Conclusion
Since the FDA approval of SGLT2 inhibitor medications, their usage has increased. The American Diabetes Association first recommends nonpharmacologic approaches, such as diet modification, exercise, and weight loss for patients diagnosed with DM, followed by a medicinal intervention with metformin if required. Sodium-glucose cotransporter 2 inhibitors are suggested as an additional medication in dual or triple pharmacotherapies when metformin alone fails to achieve normoglycemia.
Prior to starting a patient on SGLT2 inhibitor medication, clinicians should monitor hydration adequacy, check bone density, review the patient’s cardiac profile, and assess hepatic and renal function. Prescribing SGLT2 inhibitors should be restricted if the patient has a history of type 1 DM, ketosisprone T2DM, and in those with a glomerular filtration rate of < 60 mL/min. Considering the preexisting medical conditions of the patient and monitoring the blood glucose levels, renal function, and volume status at every visit should minimize risks and enhance the benefits of prescribing this new medication class.
1. Li C, Balluz LS, Okoro CA, et al; Centers for Disease Control and Prevention. Surveillance of certain health behaviors and condition among states and selected local areas—Behavioral Risk Factor Surveillance System, United States, 2009. MMWR Surveill Summ. 2011;60(9):1-250.
2. Loriaux DL. Diabetes and the ebers papyrus: 1552 BC. Endocrinologist. 2006;16(2):55-56.
3. Malhotra A, Kudyar S, Gupta AK, Kudyar RP, Malhotra P. Sodium glucose cotransporter inhibitors—a new class of old drugs. Int J Appl Basic Med Res. 2015;5(3):161-163.
4. Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21(1):31-38.
5. Mosley JF II, Smith L, Everton E, Fellner C. Sodium-glucose linked transporter 2 (SGLT2) inhibitors in the management of type-2 diabetes: a drug class overview. PT. 2015;40(7):451-462.
6. Bays H. Sodium glucose cotransporter type 2 (SGLT2) inhibitors: targeting the kidney to improve glycemic control in diabetes mellitus. Diabetes Ther. 2013;4(2):195-220.
7. Inagaki N, Kondo K, Yoshinari T, Maruyama N, Susuta Y, Kuki H. Efficacy and safety of canagliflozin in Japanese patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, 12-week study. Diabetes Obes Metab. 2013;15(12):1136-1145.
8. Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010;33(10):2217-2224.
9. Roden M, Weng J, Eilbracht J, et al; EMPA-REG MONO trial investigators. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2013;1(3):208-219.
10. Hayashi T, Fukui T, Nakanishi N, et al. Dapagliflozin decreases small dense low-density lipoprotein-cholesterol and increases high-density lipoprotein 2-cholesterol in patients with type 2 diabetes: comparison with sitagliptin. Cardiovasc Diabetology. 2017;16:8.
11. Tsimihodimos V, Filippatos TD, Elisaf MS. Effects of sodium-glucose cotransporter 2 inhibitors on metabolism: unanswered questions and controversies. Expert Opin Drug Metab Toxicol. 2017;13(4):399-408.
12. Weber MA, Mansfield TA, Alessi F, Iqbal N, Parikh S, Ptaszynska A. Effects of dapagliflozin on blood pressure in hypertensive diabetic patients on renin–angiotensin system blockade. Blood Press. 2016;25(2):93-103.
13. Cefalu WT, Stenlöf K, Leiter LA, et al. Effects of canagliflozin on body weight and relationship to HbA1c and blood pressure changes in patients with type 2 diabetes. Diabetologia. 2015;58(6):1183-1187.
14. Neeland IJ, McGuire DK, Chilton R, et al. Empagliflozin reduces body weight and indices of adipose distribution in patients with type 2 diabetes mellitus. Diab Vasc Dis Res. 2016;13(2):119-126.
15. Heerspink HJ, Johnsson E, Gause-Nilsson I, Cain VA, Sjöström CD. Dapagliflozin reduces albuminuria in patients with diabetes and hypertension receiving renin-angiotensin blockers. Diabetes Obes Metab. 2016;18(6):590-597.
16. Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377(7): 644-657.
17. Cherney D, Lund SS, Perkins BA, et al. The effect of sodium glucose cotransporter 2 inhibition with empagliflozin on microalbuminuria and macroalbuminuria in patients with type 2 diabetes. Diabetologia. 2016;59(9):1860-1870.
18. Pfeifer M, Townsend RR, Davies MJ, Vijapurkar U, Ren J. Effects of canagliflozin, a sodium glucose cotransporter 2 inhibitor, on blood pressure and markers of arterial stiffness in patients with type 2 diabetes mellitus: a post hoc analysis. Cardiovasc Diabetol. 2017;16(1):29.
19. Tikkanen I, Narko K, Zeller C, et al; EMPA-REG BP Investigators. Empagliflozin reduces blood pressure in patients with type 2 diabetes and hypertension. Diabetes Care. 2015;38(3):420-428.
20. Boulton AJM, Armstrong DG, Albert SF, et al. Comprehensive foot examination and risk assessment: a report of the task force of the foot care interest group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31(8):1679-1685.
21. Hahn K, Ejaz AA, Kanbay M, Lanaspa MA, Johnson RJ. Acute kidney injury from SGLT2 inhibitors: potential mechanisms. Nat Rev Nephrol. 2016;12(12):711-712.
22. Taylor SI, Blau JE, Rother KI. SGLT2 inhibitors may predispose to ketoacidosis. J Clin Endocrinol Metab. 2015;100(8):2849-2852.
23. Ogawa W, Sakaguchi K. Euglycemic diabetic ketoacidosis induced by SGLT2 inhibitors: possible mechanism and contributing factors. J Diabetes Investig. 2016;7(2):135-138.
24. Monami M, Nreu B, Zannoni S, Lualdi C, Mannucci E. Effects of SGLT-2 inhibitors on diabetic ketoacidosis: a meta-analysis of randomised controlled trials. Diabetes Res Clin Pract. 2017;130:53-60.
25. Burke KR, Schumacher CA, Harpe SE. SGLT2 inhibitors: a systematic review of diabetic ketoacidosis and related risk factors in the primary literature. Pharmacotherapy. 2017;37(2):187-194.
26. Liu J, Li L, Li S, et al. Effects of SGLT2 inhibitors on UTIs and genital infections in type 2 diabetes mellitus: a systematic review and meta-analysis. Sci Rep. 2017;7(1):2824.
27. Chaplin S. SGLT2 inhibitors and risk of genitourinary infections. Prescriber. 2016;27(12):26-30.
28. Weir MR, Januszewicz A, Gilbert RE, et al. Effect of canagliflozin on blood pressure and adverse events related to osmotic diuresis and reduced intravascular volume in patients with type 2 diabetes mellitus. J Clin Hypertens (Greenwich). 2014;16(12):875-882.
29. Watts NB, Bilezkian JP, Usiskin K, et al. Effects of canagliflozin on fracture risk in patients with type 2 diabetes mellietus. J Clin Endocrinol Metab. 2016;101(1):157-166.
Diabetes mellitus (DM) is a metabolic disorder affecting about 5% to 13% of the population in the US.1 Since 1552, the earliest record of a person with DM, many treatment advances have been made.2Sodium-glucose cotransporter 2 (SGLT2) inhibitors are one of the newest antidiabetic pharmaceuticals on the market. The SGLT2 inhibitor drugs include canagliflozin, dapagliflozin, empagliflozin, ipragliflozin, and tofogliflozin; however, only canagliflozin, dapagliflozin, and empagliflozin have been approved by the US Food and Drug Administration (FDA). These pharmaceuticals promote glycosuria via the kidneys and enhance sugar excretion from the body. Along with lifestyle changes and self-care measures, such as healthful eating and increased physical activity, SGLT2 inhibitor pharmaceuticals provide antidiabetic efficacy by facilitating normoglycemia and minimizing vascular pathology.
Although SGLT2 inhibitor pharmaceuticals are newly introduced into the market, their discovery dates to 1835.3 Phlorizin, a nonselective SGLT inhibitor, was first isolated by French chemists from the bark of an apple tree.4 Phlorizin inhibits SGLT1 mostly in small intestinal cells, and SGLT2 similarly affects the kidney.4 Renal SGLT2 is the primary therapeutic target. Canagliflozin was the first pharmaceutical SGLT2 inhibitor approved by the FDA in 2013. Dapagliflozin’s FDA approval followed in 2013 and empagliflozin in 2014.5
Mechanism Of Action
In healthy individuals, tubular glucose is absorbed, resulting in no urinary glucose excretion. Sodium-glucose cotransporters 1 and 2 contribute to the renal absorption of glucose. A SGLT2 is responsible for 90% of the glucose reuptake in the segment 1 of the proximal tubule, while SGLT 1 is accountable for the remaining 10%.3 Unlike other antidiabetic medications, which act by increasing insulin secretion or improving insulin sensitivity for the receptors, SGLT2 inhibitor drugs prevent the reuptake of glucose into the bloodstream. This selective action spares the inhibition of SGLT1 present in other tissues, avoiding gastrointestinal effects.6
Benefits
The SGLT2 inhibitor action is focused on renal excretion of glucose and is independent of insulin action.
Hemoglobin A1c Levels
Canagliflozin, dapagliflozin, and empagliflozin reduce hemoglobin A1c (HbA1c) levels.5 Inagaki and colleagues found significant reductions in HbA1c and weight gain with > 100 mg canagliflozin compared with that of placebo when used for 12 weeks.7 In a study where 2.5-mg, 5-mg, and 10-mg dapagliflozin was compared with placebo, the mean HbA1c change from the baseline was -0.23% with placebo; -0.58% at 2.5 mg; -0.77% at 5 mg; and -0.89% at 10 mg.8 Empagliflozin was more effective in reducing HbA1c levels than was sitagliptin.9 When patients were treated with 10-mg empagliflozin, 25-mg empagliflozin, and sitagliptin, HbA1c levels dropped -1.44%, -1.43%, and -1.04%, respectively.9
Cholesterol
Sodium-glucose cotransporter 2 inhibitors have the beneficial effect of reducing vascular disease risk factors.10,11 A study by Hayashi and colleagues found that dapagliflozin decreases harmful atherogenic small, low-density lipoprotein-cholesterol (LDL-C), increases less atherogenic large, buoyant LDL-C, and increases high-density, lipoprotein-2 cholesterol (HDL-2C).10 Empagliflozin, however, can cause a small dose-dependent increase in HDL-C and LDL-C.11 Although there is an increase in serum LDL-C concentrations, empagliflozin can induce a decrease in intestinal absorption of cholesterol, thus promoting fecal excretion of LDL-C and macrophage-derived cholesterol.11
Weight Loss
A study by Weber and colleagues found that the SGLT2 inhibitor dapagloflozin lead to a reduction in body weight from -1.0 kg to -0.3 kg compared with placebo.12 Cefalu and colleagues found that daily prescribing of 100 mg and/or 300 mg of canagliflozin evidenced dose-dependent loss of weight.13 Neeland and colleagues found that empagliflozin utilization resulted in less adiposity indices in 3,300 subjects.14
Albuminuria
Sodium-glucose cotransporter 2 inhibitors have a reno-protective role in patients with type 2 DM (T2DM). In those receiving renin-angiotensin blockers with T2DM and hypertension, dapagliflozin decreased their albuminuria.15 Canagliflozin has a similar potential.16 Empagliflozin reduced the urine albumin-creatinine ratio in patients with macro- or micro-albuminuria, supporting a direct renal effect by SGLT2 inhibitors.17
Systolic Blood Pressure
Sodium-glucose cotransporter 2 inhibitors can have beneficial effects on physiologic vascular outcomes. In patients with T2DM and hypertension, dapagliflozin reduced mean systolic blood pressure (SBP) compared with placebo: -7.3 mm Hg vs -10.4 mm Hg, respectively.12 Prescribing canagliflozin treatment at 100 mg or 300 mg reduced SBP (-4.3 mm Hg and -5.0 mm Hg, respectively, vs placebo at -0.3 mm Hg).18 Subjects taking empagliflozin 10 mg or 25 mg exhibited an adjusted mean BP change from baseline of -4.60 mm Hg and -5.47mm Hg, respectively, whereas placebo induced a -0.67 mm Hg decline.19
Risks
Nausea, fatigue, polyuria, polydipsia, and xerostomia are common SGLT2 AEs. Use of SGLT2 inhibitors can induce certain other more serious AEs as well.
Increased Risk for Amputations
The Canagliflozin Cardiovascular Assessment Study (CANVAS) and the Canagliflozin Cardiovascular Assessment Study-Renal (CANVAS-R) documented that canagliflozin doubled the incidence of leg and foot amputations in research participants compared with placebo (6.3 vs 3.4 per 1,000 patient-years).16 Therefore, canagliflozin should be prescribed with caution in persons with a prior history of foot ulceration, neuropathy, and/or vascular diseases.20
Acute Renal Injury
The mechanism of kidney damage by SGLT2 inhibitor drugs is not completely understood. About 100 patients experienced renal failure after the intake of SGLT2 inhibitor drugs.21 Among them, more than half reported symptom onset within a month of starting the medication, and their symptoms improved after discontinuing the SGLT2 medication. As a result, the FDA issued a warning to monitor renal function before initiating and during such pharmacotherapy.21
Ketoacidosis
Sodium-glucose cotransporter 2 inhibitors might lead to elevated ketone body levels22 and euglycemic ketoacidosis;23 however, this risk reportedly is negligible.24 Use of SGLT2 inhibitors is not recommended for patients evidencing the presence of precipitating factors like acute gastroenteritis or insulin pump failure.25
Genitourinary Infections
About 10% to 15% of women taking SGLT2 inhibitor medications developed urinary tract infections and vulvovaginitis.26 This could be because of a glycosuria effect caused by SGLT2 inhibitors.27
Hypotension
Sodium-glucose cotransporter 2 inhibitors cause contraction of intravascular volume. Therefore, patients taking SGLT2 inhibitors are at risk for hypotension, leading to dizziness and potentially dangerous falls. Patients already taking volume-depleting medications, such as diuretics, should be advised to use this group of medications with caution and report these AEs.28
Bone Fractures
A clinical trial revealed that SGLT2 inhibitors, such as canagliflozin, decrease bone mineral density possibly leading to bone fractures.29 Bone fractures occurred in about 1.5% of cases of patients taking 100 mg and 300 mg of canagliflozin compared with a 1.1% fracture rate among the placebo group.29
Conclusion
Since the FDA approval of SGLT2 inhibitor medications, their usage has increased. The American Diabetes Association first recommends nonpharmacologic approaches, such as diet modification, exercise, and weight loss for patients diagnosed with DM, followed by a medicinal intervention with metformin if required. Sodium-glucose cotransporter 2 inhibitors are suggested as an additional medication in dual or triple pharmacotherapies when metformin alone fails to achieve normoglycemia.
Prior to starting a patient on SGLT2 inhibitor medication, clinicians should monitor hydration adequacy, check bone density, review the patient’s cardiac profile, and assess hepatic and renal function. Prescribing SGLT2 inhibitors should be restricted if the patient has a history of type 1 DM, ketosisprone T2DM, and in those with a glomerular filtration rate of < 60 mL/min. Considering the preexisting medical conditions of the patient and monitoring the blood glucose levels, renal function, and volume status at every visit should minimize risks and enhance the benefits of prescribing this new medication class.
Diabetes mellitus (DM) is a metabolic disorder affecting about 5% to 13% of the population in the US.1 Since 1552, the earliest record of a person with DM, many treatment advances have been made.2Sodium-glucose cotransporter 2 (SGLT2) inhibitors are one of the newest antidiabetic pharmaceuticals on the market. The SGLT2 inhibitor drugs include canagliflozin, dapagliflozin, empagliflozin, ipragliflozin, and tofogliflozin; however, only canagliflozin, dapagliflozin, and empagliflozin have been approved by the US Food and Drug Administration (FDA). These pharmaceuticals promote glycosuria via the kidneys and enhance sugar excretion from the body. Along with lifestyle changes and self-care measures, such as healthful eating and increased physical activity, SGLT2 inhibitor pharmaceuticals provide antidiabetic efficacy by facilitating normoglycemia and minimizing vascular pathology.
Although SGLT2 inhibitor pharmaceuticals are newly introduced into the market, their discovery dates to 1835.3 Phlorizin, a nonselective SGLT inhibitor, was first isolated by French chemists from the bark of an apple tree.4 Phlorizin inhibits SGLT1 mostly in small intestinal cells, and SGLT2 similarly affects the kidney.4 Renal SGLT2 is the primary therapeutic target. Canagliflozin was the first pharmaceutical SGLT2 inhibitor approved by the FDA in 2013. Dapagliflozin’s FDA approval followed in 2013 and empagliflozin in 2014.5
Mechanism Of Action
In healthy individuals, tubular glucose is absorbed, resulting in no urinary glucose excretion. Sodium-glucose cotransporters 1 and 2 contribute to the renal absorption of glucose. A SGLT2 is responsible for 90% of the glucose reuptake in the segment 1 of the proximal tubule, while SGLT 1 is accountable for the remaining 10%.3 Unlike other antidiabetic medications, which act by increasing insulin secretion or improving insulin sensitivity for the receptors, SGLT2 inhibitor drugs prevent the reuptake of glucose into the bloodstream. This selective action spares the inhibition of SGLT1 present in other tissues, avoiding gastrointestinal effects.6
Benefits
The SGLT2 inhibitor action is focused on renal excretion of glucose and is independent of insulin action.
Hemoglobin A1c Levels
Canagliflozin, dapagliflozin, and empagliflozin reduce hemoglobin A1c (HbA1c) levels.5 Inagaki and colleagues found significant reductions in HbA1c and weight gain with > 100 mg canagliflozin compared with that of placebo when used for 12 weeks.7 In a study where 2.5-mg, 5-mg, and 10-mg dapagliflozin was compared with placebo, the mean HbA1c change from the baseline was -0.23% with placebo; -0.58% at 2.5 mg; -0.77% at 5 mg; and -0.89% at 10 mg.8 Empagliflozin was more effective in reducing HbA1c levels than was sitagliptin.9 When patients were treated with 10-mg empagliflozin, 25-mg empagliflozin, and sitagliptin, HbA1c levels dropped -1.44%, -1.43%, and -1.04%, respectively.9
Cholesterol
Sodium-glucose cotransporter 2 inhibitors have the beneficial effect of reducing vascular disease risk factors.10,11 A study by Hayashi and colleagues found that dapagliflozin decreases harmful atherogenic small, low-density lipoprotein-cholesterol (LDL-C), increases less atherogenic large, buoyant LDL-C, and increases high-density, lipoprotein-2 cholesterol (HDL-2C).10 Empagliflozin, however, can cause a small dose-dependent increase in HDL-C and LDL-C.11 Although there is an increase in serum LDL-C concentrations, empagliflozin can induce a decrease in intestinal absorption of cholesterol, thus promoting fecal excretion of LDL-C and macrophage-derived cholesterol.11
Weight Loss
A study by Weber and colleagues found that the SGLT2 inhibitor dapagloflozin lead to a reduction in body weight from -1.0 kg to -0.3 kg compared with placebo.12 Cefalu and colleagues found that daily prescribing of 100 mg and/or 300 mg of canagliflozin evidenced dose-dependent loss of weight.13 Neeland and colleagues found that empagliflozin utilization resulted in less adiposity indices in 3,300 subjects.14
Albuminuria
Sodium-glucose cotransporter 2 inhibitors have a reno-protective role in patients with type 2 DM (T2DM). In those receiving renin-angiotensin blockers with T2DM and hypertension, dapagliflozin decreased their albuminuria.15 Canagliflozin has a similar potential.16 Empagliflozin reduced the urine albumin-creatinine ratio in patients with macro- or micro-albuminuria, supporting a direct renal effect by SGLT2 inhibitors.17
Systolic Blood Pressure
Sodium-glucose cotransporter 2 inhibitors can have beneficial effects on physiologic vascular outcomes. In patients with T2DM and hypertension, dapagliflozin reduced mean systolic blood pressure (SBP) compared with placebo: -7.3 mm Hg vs -10.4 mm Hg, respectively.12 Prescribing canagliflozin treatment at 100 mg or 300 mg reduced SBP (-4.3 mm Hg and -5.0 mm Hg, respectively, vs placebo at -0.3 mm Hg).18 Subjects taking empagliflozin 10 mg or 25 mg exhibited an adjusted mean BP change from baseline of -4.60 mm Hg and -5.47mm Hg, respectively, whereas placebo induced a -0.67 mm Hg decline.19
Risks
Nausea, fatigue, polyuria, polydipsia, and xerostomia are common SGLT2 AEs. Use of SGLT2 inhibitors can induce certain other more serious AEs as well.
Increased Risk for Amputations
The Canagliflozin Cardiovascular Assessment Study (CANVAS) and the Canagliflozin Cardiovascular Assessment Study-Renal (CANVAS-R) documented that canagliflozin doubled the incidence of leg and foot amputations in research participants compared with placebo (6.3 vs 3.4 per 1,000 patient-years).16 Therefore, canagliflozin should be prescribed with caution in persons with a prior history of foot ulceration, neuropathy, and/or vascular diseases.20
Acute Renal Injury
The mechanism of kidney damage by SGLT2 inhibitor drugs is not completely understood. About 100 patients experienced renal failure after the intake of SGLT2 inhibitor drugs.21 Among them, more than half reported symptom onset within a month of starting the medication, and their symptoms improved after discontinuing the SGLT2 medication. As a result, the FDA issued a warning to monitor renal function before initiating and during such pharmacotherapy.21
Ketoacidosis
Sodium-glucose cotransporter 2 inhibitors might lead to elevated ketone body levels22 and euglycemic ketoacidosis;23 however, this risk reportedly is negligible.24 Use of SGLT2 inhibitors is not recommended for patients evidencing the presence of precipitating factors like acute gastroenteritis or insulin pump failure.25
Genitourinary Infections
About 10% to 15% of women taking SGLT2 inhibitor medications developed urinary tract infections and vulvovaginitis.26 This could be because of a glycosuria effect caused by SGLT2 inhibitors.27
Hypotension
Sodium-glucose cotransporter 2 inhibitors cause contraction of intravascular volume. Therefore, patients taking SGLT2 inhibitors are at risk for hypotension, leading to dizziness and potentially dangerous falls. Patients already taking volume-depleting medications, such as diuretics, should be advised to use this group of medications with caution and report these AEs.28
Bone Fractures
A clinical trial revealed that SGLT2 inhibitors, such as canagliflozin, decrease bone mineral density possibly leading to bone fractures.29 Bone fractures occurred in about 1.5% of cases of patients taking 100 mg and 300 mg of canagliflozin compared with a 1.1% fracture rate among the placebo group.29
Conclusion
Since the FDA approval of SGLT2 inhibitor medications, their usage has increased. The American Diabetes Association first recommends nonpharmacologic approaches, such as diet modification, exercise, and weight loss for patients diagnosed with DM, followed by a medicinal intervention with metformin if required. Sodium-glucose cotransporter 2 inhibitors are suggested as an additional medication in dual or triple pharmacotherapies when metformin alone fails to achieve normoglycemia.
Prior to starting a patient on SGLT2 inhibitor medication, clinicians should monitor hydration adequacy, check bone density, review the patient’s cardiac profile, and assess hepatic and renal function. Prescribing SGLT2 inhibitors should be restricted if the patient has a history of type 1 DM, ketosisprone T2DM, and in those with a glomerular filtration rate of < 60 mL/min. Considering the preexisting medical conditions of the patient and monitoring the blood glucose levels, renal function, and volume status at every visit should minimize risks and enhance the benefits of prescribing this new medication class.
1. Li C, Balluz LS, Okoro CA, et al; Centers for Disease Control and Prevention. Surveillance of certain health behaviors and condition among states and selected local areas—Behavioral Risk Factor Surveillance System, United States, 2009. MMWR Surveill Summ. 2011;60(9):1-250.
2. Loriaux DL. Diabetes and the ebers papyrus: 1552 BC. Endocrinologist. 2006;16(2):55-56.
3. Malhotra A, Kudyar S, Gupta AK, Kudyar RP, Malhotra P. Sodium glucose cotransporter inhibitors—a new class of old drugs. Int J Appl Basic Med Res. 2015;5(3):161-163.
4. Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21(1):31-38.
5. Mosley JF II, Smith L, Everton E, Fellner C. Sodium-glucose linked transporter 2 (SGLT2) inhibitors in the management of type-2 diabetes: a drug class overview. PT. 2015;40(7):451-462.
6. Bays H. Sodium glucose cotransporter type 2 (SGLT2) inhibitors: targeting the kidney to improve glycemic control in diabetes mellitus. Diabetes Ther. 2013;4(2):195-220.
7. Inagaki N, Kondo K, Yoshinari T, Maruyama N, Susuta Y, Kuki H. Efficacy and safety of canagliflozin in Japanese patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, 12-week study. Diabetes Obes Metab. 2013;15(12):1136-1145.
8. Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010;33(10):2217-2224.
9. Roden M, Weng J, Eilbracht J, et al; EMPA-REG MONO trial investigators. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2013;1(3):208-219.
10. Hayashi T, Fukui T, Nakanishi N, et al. Dapagliflozin decreases small dense low-density lipoprotein-cholesterol and increases high-density lipoprotein 2-cholesterol in patients with type 2 diabetes: comparison with sitagliptin. Cardiovasc Diabetology. 2017;16:8.
11. Tsimihodimos V, Filippatos TD, Elisaf MS. Effects of sodium-glucose cotransporter 2 inhibitors on metabolism: unanswered questions and controversies. Expert Opin Drug Metab Toxicol. 2017;13(4):399-408.
12. Weber MA, Mansfield TA, Alessi F, Iqbal N, Parikh S, Ptaszynska A. Effects of dapagliflozin on blood pressure in hypertensive diabetic patients on renin–angiotensin system blockade. Blood Press. 2016;25(2):93-103.
13. Cefalu WT, Stenlöf K, Leiter LA, et al. Effects of canagliflozin on body weight and relationship to HbA1c and blood pressure changes in patients with type 2 diabetes. Diabetologia. 2015;58(6):1183-1187.
14. Neeland IJ, McGuire DK, Chilton R, et al. Empagliflozin reduces body weight and indices of adipose distribution in patients with type 2 diabetes mellitus. Diab Vasc Dis Res. 2016;13(2):119-126.
15. Heerspink HJ, Johnsson E, Gause-Nilsson I, Cain VA, Sjöström CD. Dapagliflozin reduces albuminuria in patients with diabetes and hypertension receiving renin-angiotensin blockers. Diabetes Obes Metab. 2016;18(6):590-597.
16. Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377(7): 644-657.
17. Cherney D, Lund SS, Perkins BA, et al. The effect of sodium glucose cotransporter 2 inhibition with empagliflozin on microalbuminuria and macroalbuminuria in patients with type 2 diabetes. Diabetologia. 2016;59(9):1860-1870.
18. Pfeifer M, Townsend RR, Davies MJ, Vijapurkar U, Ren J. Effects of canagliflozin, a sodium glucose cotransporter 2 inhibitor, on blood pressure and markers of arterial stiffness in patients with type 2 diabetes mellitus: a post hoc analysis. Cardiovasc Diabetol. 2017;16(1):29.
19. Tikkanen I, Narko K, Zeller C, et al; EMPA-REG BP Investigators. Empagliflozin reduces blood pressure in patients with type 2 diabetes and hypertension. Diabetes Care. 2015;38(3):420-428.
20. Boulton AJM, Armstrong DG, Albert SF, et al. Comprehensive foot examination and risk assessment: a report of the task force of the foot care interest group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31(8):1679-1685.
21. Hahn K, Ejaz AA, Kanbay M, Lanaspa MA, Johnson RJ. Acute kidney injury from SGLT2 inhibitors: potential mechanisms. Nat Rev Nephrol. 2016;12(12):711-712.
22. Taylor SI, Blau JE, Rother KI. SGLT2 inhibitors may predispose to ketoacidosis. J Clin Endocrinol Metab. 2015;100(8):2849-2852.
23. Ogawa W, Sakaguchi K. Euglycemic diabetic ketoacidosis induced by SGLT2 inhibitors: possible mechanism and contributing factors. J Diabetes Investig. 2016;7(2):135-138.
24. Monami M, Nreu B, Zannoni S, Lualdi C, Mannucci E. Effects of SGLT-2 inhibitors on diabetic ketoacidosis: a meta-analysis of randomised controlled trials. Diabetes Res Clin Pract. 2017;130:53-60.
25. Burke KR, Schumacher CA, Harpe SE. SGLT2 inhibitors: a systematic review of diabetic ketoacidosis and related risk factors in the primary literature. Pharmacotherapy. 2017;37(2):187-194.
26. Liu J, Li L, Li S, et al. Effects of SGLT2 inhibitors on UTIs and genital infections in type 2 diabetes mellitus: a systematic review and meta-analysis. Sci Rep. 2017;7(1):2824.
27. Chaplin S. SGLT2 inhibitors and risk of genitourinary infections. Prescriber. 2016;27(12):26-30.
28. Weir MR, Januszewicz A, Gilbert RE, et al. Effect of canagliflozin on blood pressure and adverse events related to osmotic diuresis and reduced intravascular volume in patients with type 2 diabetes mellitus. J Clin Hypertens (Greenwich). 2014;16(12):875-882.
29. Watts NB, Bilezkian JP, Usiskin K, et al. Effects of canagliflozin on fracture risk in patients with type 2 diabetes mellietus. J Clin Endocrinol Metab. 2016;101(1):157-166.
1. Li C, Balluz LS, Okoro CA, et al; Centers for Disease Control and Prevention. Surveillance of certain health behaviors and condition among states and selected local areas—Behavioral Risk Factor Surveillance System, United States, 2009. MMWR Surveill Summ. 2011;60(9):1-250.
2. Loriaux DL. Diabetes and the ebers papyrus: 1552 BC. Endocrinologist. 2006;16(2):55-56.
3. Malhotra A, Kudyar S, Gupta AK, Kudyar RP, Malhotra P. Sodium glucose cotransporter inhibitors—a new class of old drugs. Int J Appl Basic Med Res. 2015;5(3):161-163.
4. Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21(1):31-38.
5. Mosley JF II, Smith L, Everton E, Fellner C. Sodium-glucose linked transporter 2 (SGLT2) inhibitors in the management of type-2 diabetes: a drug class overview. PT. 2015;40(7):451-462.
6. Bays H. Sodium glucose cotransporter type 2 (SGLT2) inhibitors: targeting the kidney to improve glycemic control in diabetes mellitus. Diabetes Ther. 2013;4(2):195-220.
7. Inagaki N, Kondo K, Yoshinari T, Maruyama N, Susuta Y, Kuki H. Efficacy and safety of canagliflozin in Japanese patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, 12-week study. Diabetes Obes Metab. 2013;15(12):1136-1145.
8. Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010;33(10):2217-2224.
9. Roden M, Weng J, Eilbracht J, et al; EMPA-REG MONO trial investigators. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2013;1(3):208-219.
10. Hayashi T, Fukui T, Nakanishi N, et al. Dapagliflozin decreases small dense low-density lipoprotein-cholesterol and increases high-density lipoprotein 2-cholesterol in patients with type 2 diabetes: comparison with sitagliptin. Cardiovasc Diabetology. 2017;16:8.
11. Tsimihodimos V, Filippatos TD, Elisaf MS. Effects of sodium-glucose cotransporter 2 inhibitors on metabolism: unanswered questions and controversies. Expert Opin Drug Metab Toxicol. 2017;13(4):399-408.
12. Weber MA, Mansfield TA, Alessi F, Iqbal N, Parikh S, Ptaszynska A. Effects of dapagliflozin on blood pressure in hypertensive diabetic patients on renin–angiotensin system blockade. Blood Press. 2016;25(2):93-103.
13. Cefalu WT, Stenlöf K, Leiter LA, et al. Effects of canagliflozin on body weight and relationship to HbA1c and blood pressure changes in patients with type 2 diabetes. Diabetologia. 2015;58(6):1183-1187.
14. Neeland IJ, McGuire DK, Chilton R, et al. Empagliflozin reduces body weight and indices of adipose distribution in patients with type 2 diabetes mellitus. Diab Vasc Dis Res. 2016;13(2):119-126.
15. Heerspink HJ, Johnsson E, Gause-Nilsson I, Cain VA, Sjöström CD. Dapagliflozin reduces albuminuria in patients with diabetes and hypertension receiving renin-angiotensin blockers. Diabetes Obes Metab. 2016;18(6):590-597.
16. Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377(7): 644-657.
17. Cherney D, Lund SS, Perkins BA, et al. The effect of sodium glucose cotransporter 2 inhibition with empagliflozin on microalbuminuria and macroalbuminuria in patients with type 2 diabetes. Diabetologia. 2016;59(9):1860-1870.
18. Pfeifer M, Townsend RR, Davies MJ, Vijapurkar U, Ren J. Effects of canagliflozin, a sodium glucose cotransporter 2 inhibitor, on blood pressure and markers of arterial stiffness in patients with type 2 diabetes mellitus: a post hoc analysis. Cardiovasc Diabetol. 2017;16(1):29.
19. Tikkanen I, Narko K, Zeller C, et al; EMPA-REG BP Investigators. Empagliflozin reduces blood pressure in patients with type 2 diabetes and hypertension. Diabetes Care. 2015;38(3):420-428.
20. Boulton AJM, Armstrong DG, Albert SF, et al. Comprehensive foot examination and risk assessment: a report of the task force of the foot care interest group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31(8):1679-1685.
21. Hahn K, Ejaz AA, Kanbay M, Lanaspa MA, Johnson RJ. Acute kidney injury from SGLT2 inhibitors: potential mechanisms. Nat Rev Nephrol. 2016;12(12):711-712.
22. Taylor SI, Blau JE, Rother KI. SGLT2 inhibitors may predispose to ketoacidosis. J Clin Endocrinol Metab. 2015;100(8):2849-2852.
23. Ogawa W, Sakaguchi K. Euglycemic diabetic ketoacidosis induced by SGLT2 inhibitors: possible mechanism and contributing factors. J Diabetes Investig. 2016;7(2):135-138.
24. Monami M, Nreu B, Zannoni S, Lualdi C, Mannucci E. Effects of SGLT-2 inhibitors on diabetic ketoacidosis: a meta-analysis of randomised controlled trials. Diabetes Res Clin Pract. 2017;130:53-60.
25. Burke KR, Schumacher CA, Harpe SE. SGLT2 inhibitors: a systematic review of diabetic ketoacidosis and related risk factors in the primary literature. Pharmacotherapy. 2017;37(2):187-194.
26. Liu J, Li L, Li S, et al. Effects of SGLT2 inhibitors on UTIs and genital infections in type 2 diabetes mellitus: a systematic review and meta-analysis. Sci Rep. 2017;7(1):2824.
27. Chaplin S. SGLT2 inhibitors and risk of genitourinary infections. Prescriber. 2016;27(12):26-30.
28. Weir MR, Januszewicz A, Gilbert RE, et al. Effect of canagliflozin on blood pressure and adverse events related to osmotic diuresis and reduced intravascular volume in patients with type 2 diabetes mellitus. J Clin Hypertens (Greenwich). 2014;16(12):875-882.
29. Watts NB, Bilezkian JP, Usiskin K, et al. Effects of canagliflozin on fracture risk in patients with type 2 diabetes mellietus. J Clin Endocrinol Metab. 2016;101(1):157-166.
The naloxone option
More than 64,000 people in the United States died of drug overdoses in 2016.1 Of those overdose deaths, more than 34,000 were related to the use of natural (eg, codeine, morphine); synthetic (eg, fentanyl); and semisynthetic (eg, oxycodone, hydrocodone) opioids.1 The number of drug-overdose fatalities (driven largely by opioids) has increased so dramatically in recent years that drug overdose is now the leading cause of intentional and unintentional injury-related death in the United States.2 Furthermore, opioid use is increasing among college students, with many injecting these agents.3 Those injecting (as opposed to other routes of delivery) have the highest death rate.4
The Department of Health and Human Services has identified 3 important issues to address with regard to the opioid epidemic: prescriber education, community naloxone access, and better interventions (such as naloxone overdose-reversal take-home kits) for people with opioid use disorders and/or a history of overdoses.5 (For more on overdose reversal kits, see “What FPs need to know about naloxone kits,” a 3-in-3 video.) With these goals in mind, we provide the following review of naloxone dosing and postoverdose treatment.
Steps FPs can take to reverse the overdose
Opioids act on delta, kappa, and mu receptors in the brain to produce analgesic effects,6 but, in large quantities, their mu receptor activity can cause fatal respiratory depression.7 Some of the most commonly abused opioids are heroin and the prescription opioids fentanyl, oxycodone, and hydrocodone.8
People who have overdosed on opioids generally present with evidence of obtundation, miosis, and difficulty breathing. Respiratory failure is the most common cause of death.9 Hypothermia, compartment syndrome, rhabdomyolysis, renal failure, and acute pulmonary edema are less common complications. Overdoses and these medical issues can potentially be reversed and/or mitigated by naloxone administration.10,11
Naloxone and its routes of administration. Naloxone is the agent of choice in overdose situations.12 It works as an antagonist of the delta, kappa, and mu receptors,6,13 has a rapid onset of action, and is associated with minimal adverse effects.14
Naloxone can be administered via the intravenous (IV), intranasal, intramuscular, subcutaneous, intraosseous, or endotracheal routes.6 Although IV administration has been the most common and is still generally preferred in the hospital setting, the intranasal route has gained favor, partly because it can be difficult to establish an IV in IV drug users and partly because it is easier for nonmedical people to administer.6
In addition, the nasal mucosa has an abundant blood supply resulting in rapid absorption. The drug reaches the systemic circulation quickly and avoids first-pass hepatic metabolism.6 Intranasal route absorption is enhanced by deep inhalation and patient cooperation, but it can still be effective in an unconscious patient. Response time is nearly the same as that with IV administration (both act within 1-2 mins).6
Naloxone has a short duration of action (shorter than that of some opiates), and its duration of action is influenced by the pharmacology and toxicity of the overdose drug.15 The serum half-life in adults ranges from 30 to 81 mins, and clinical impact varies from minutes to an hour.15 Thus, even if a patient initially improves after administration, close observation is mandatory due to the frequent need for repeat naloxone dosing.
Adverse effects. Naloxone is considered safe, with relatively few adverse effects and doesn’t have any effects on someone who isn’t experiencing an opioid overdose or currently on opioids.15 The only downside is that naloxone administration to an opioid-dependent person often precipitates an acute withdrawal event, characterized by global pain, agitation, generalized distress, and gastrointestinal complaints, including vomiting and diarrhea. Although withdrawal is not life-threatening, it can cause great discomfort.
Getting a handle on naloxone dosing
The starting dose of naloxone used to be 0.04 mg, but this was later increased to 0.4 mg. The advent and high overdose lethality of more potent drugs like fentanyl and carfentanil has made low-dose naloxone less effective.12
Currently, 1 mg is often the initial recommendation, but doses of 2 to 4 mg are not uncommon, and multiple administrations or continuous IV administration are frequently needed to reverse severe toxicities, such as those involving fentanyl or longer-action opioids like methadone. Anyone exhibiting difficulty breathing mandates a starting naloxone dose of at least 1 to 2 mg.12,16 In addition to breathing, additional doses are indicated clinically by medical parameters such as vital signs, ocular pupil diameter, and/or alertness.6
Intranasal administration often utilizes up to 4 mg of naloxone in one nostril, followed by a titrated additional administration in the other nostril. In life-threatening circumstances, especially those in which a patient is exhibiting respiratory depression, a much larger quantity of naloxone—up to 10 mg—may be administered by trained medical personnel.12,16 In the end, all dosing varies and must be individualized to the patient’s signs and symptoms. Those who have overdosed require prolonged monitoring to treat potential complications.
Emergency assistance and transport. Because of the dangers that can result from opioid toxicities, any hint or evidence of physiologic compromise merits a 911 call for emergency medical assistance and transport to a hospital emergency department (ED). Hospitalization is at the physician’s discretion.
Expanding the availability of naloxone in the community
The availability of naloxone overdose-reversal kits is growing among hospitals, other types of health care facilities, first responders, medical offices, and the general public. Distributing the kits to opioid users and their families has wide support but remains controversial (more on this in a bit).
Support even includes that from the current US Surgeon General, Jerome Adams, MD, MPH, who noted in a statement on April 5, 2018, the lifesaving success of opioid-overdose reversal naloxone kits by medical personnel, first responders, and other people. As a result, he formally recommended that more Americans keep such kits available in order to be able to quickly diminish opioid toxicities.17,18 His advice was especially directed toward people at risk for an opioid overdose or anyone associated with opioid drug users.
Prehospital management of overdoses is ideally managed by emergency medical service (EMS) personnel,10 but even nonmedical people can safely administer naloxone. About 10,000 overdose cases were documented to have been reversed by nonmedical providers between 1996 and 2010.10 Many states have laws limiting the civil and criminal liability for naloxone administrators. New Mexico was the first state to legally allow naloxone administration by individuals without a prescription.7 Pharmacists often participate in efforts to counter opioid drug overdose deaths by offering naloxone administration kits, along with training about techniques of use, to people filling opioid prescriptions and to household members and/or other individuals in the social support network of an opioid user.6 Some physicians co-prescribe naloxone to patients along with opioid therapies during long-term pain management. Such dual prescribing is encouraged by many clinics.19 This method has decreased opioid overdose deaths in North Carolina,20 in its army base at Fort Bragg,19 and in California.21
The issue of “risk compensation”
To those who say that having naloxone available to users of opioids or those in their social network promotes even riskier behavior resulting in increased overdoses, research points to just the opposite. A nonrandomized study that examined co-prescribing naloxone to patients on chronic opioid therapy for non-cancer-related pain, documented fewer opioid-related ED visits following use by prescribers and patients at community health centers.22 Other research has demonstrated a reduced number of community-level opioid overdose deaths once opioid overdose education and community naloxone distribution were implemented.23,24
After the overdose: Getting patients into treatment
After reversing initial toxicities, a protracted period of assessment is required to assure patient safety. Beyond prolonged observation after an overdose, it is critical to recommend and provide long-term substance abuse therapies. Simply reversing the overdose is not medically sufficient, even if postoverdose patients refuse such treatment referrals. The fact that many of these people subsequently die is evidence of the importance of adhering to a formal, long-term chemical dependence intervention program.
Persistent diligence is usually needed to convince a patient who has recovered from an acute drug overdose event to accept a treatment referral. Some EDs institute special teams to facilitate such referrals, using a multidisciplinary approach, including substance abuse counselors and social workers. Referral agencies are also sometimes included to aid patient acceptance and retention in drug abuse treatment interventions. (See "Resources" below for more information.)
SIDEBAR
Resources
- The Centers for Disease Control and Prevention’s Guideline for Prescribing Opioids for Chronic Pain. Available at: https://www.cdc.gov/drugoverdose/prescribing/guideline.html.
- National Institute on Drug Abuse. Available at: https://www.drugabuse.gov.
- Substance Abuse and Mental Health Services Administration. Available at: https://www.samhsa.gov/find-help/national-helpline.
- Your state’s prescription drug monitoring program. Available at: https://www.cdc.gov/drugoverdose/pdmp/states.html.
CORRESPONDENCE
Steven Lippmann, MD, 401 East Chestnut Street, Suite 610, Louisville, KY 40202; Steven.lippmann@louisville.edu.
1. National Institute on Drug Abuse. Overdose death rates. Revised September 2017. Available at: https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates. Accessed April 11, 2018.
2. Xu J, Murphy SL, Kochanek KD, et al. Deaths: final data for 2013. Nat Vital Stat Syst. 2016;64:1-119.
3. McCabe SE, West BT, Teter CJ, et al. Trends in medical use, diversion, and nonmedical use of prescription medications among college students from 2003 to 2013: connecting the dots. Addict Behav. 2014;39:1176-1182.
4. Green TC, Heimer R, Grau LE. Distinguishing signs of opioid overdose and indication for naloxone: an evaluation of six overdose training and naloxone distribution programs in the United States. Addiction. 2008;103:979-989.
5. US Department of Health and Human Services. HHS takes strong steps to address opioid-drug related overdose, death and dependence. March 26, 2015. Available at: http://wayback.archive-it.org/3926/20170127185704/https://www.hhs.gov/about/news/2015/03/26/hhs-takes-strong-steps-to-address-opioid-drug-related-overdose-death-and-dependence.html. Accessed April 16, 2018.
6. Robinson A, Wermeling DP. Intranasal naloxone administration for treatment of opioid overdose. Am J Health Syst Pharm. 2014;71:2129-2135.
7. Doyon S, Aks SE, Schaeffer S. Expanding access to naloxone in the United States. J Med Toxicol. 2014;10:431-434.
8. National Institute on Drug Abuse. Which classes of prescription drugs are commonly misused? Available at: https://www.drugabuse.gov/publications/research-reports/misuse-prescription-drugs/which-classes-prescription-drugs-are-commonly-misused. Accessed April 16, 2018.
9. Boom M, Niesters M, Sarton E, et al. Non-analgesic effects of opioids: opioid-induced respiratory depression. Curr Pharm Des. 2012;18:5994-6004.
10. Weaver L, Palombi L, Bastianelli KMS. Naloxone administration for opioid overdose reversal in the prehospital setting: implications for pharmacists. J Pharm Pract. 2018;31:91-98.
11. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367:146-155.
12. Jordan MR, Morrisonponce D. Naloxone. StatPearls. Available at: https://www.ncbi.nlm.nih.gov/books/NBK441910/. Accessed September 1, 2017.
13. Wilkerson RG, Kim HK, Windsor TA, et al. The opioid epidemic in the United States. Emerg Med Clin North Am. 2016;34:e1-e23.
14. Jeffery RM, Dickinson L, Ng ND, et al. Naloxone administration for suspected opioid overdose: an expanded scope of practice by a basic life support collegiate-based emergency medical services agency. J Am Coll Health. 2017;65:212-216.
15. Drugs.com. Naloxone. Available at: https://www.drugs.com/pro/naloxone.html. Accessed April 16, 2018.
16. Prabhu A, Abaid B, Naik S, et al. Naloxone for opioid overdoses. Internet and Psychiatry 2017. Available at: https://www.internetandpsychiatry.com/wp/editorials/naloxone-for-opioid-overdoses/. Accessed September 19, 2017.
17. HHS.gov. Surgeon General releases advisory on naloxone, an opioid overdose-reversing drug. Available at: https://www.hhs.gov/about/news/2018/04/05/surgeon-general-releases-advisory-on-naloxone-an-opioid-overdose-reversing-drug.html. Accessed April 16, 2018.
18. US Department of Health and Human Services. Surgeongeneral.gov. Surgeon General’s advisory on naloxone and opioid overdose. Available at: https://www.surgeongeneral.gov/priorities/opioid-overdose-prevention/naloxone-advisory.html. Accessed April 16, 2018.
19. Behar E, Rowe C, Santos GM, et al. Acceptability of naloxone co-prescription among primary care providers treating patients on long-term opioid therapy for pain. J Gen Intern Med. 2017;32:291-295.
20. Albert S, Brason FW 2nd, Sanford CK, et al. Project Lazarus: community‐based overdose prevention in rural North Carolina. Pain Med. 2011;12:S77-S85.
21. Rowe C, Santos GM, Vittinghoff E, et al. Predictors of participant engagement and naloxone utilization in a community‐based naloxone distribution program. Addiction. 2015;110:1301-1310.
22. Coffin PO, Behar E, Rowe C, et al. Nonrandomized intervention study of naloxone coprescription for primary care patients receiving long-term opioid therapy for pain. Ann Intern Med. 2016;165:245-252.
23. Walley AY, Xuan Z, Hackman HH, et al. Opioid overdose rates and implementation of overdose education and nasal naloxone distribution in Massachusetts: interrupted time series analysis. BMJ. 2013;346:f174.
24. Bird SM, McAuley A, Perry S, et al. Effectiveness of Scotland’s National Naloxone Programme for reducing opioid-related deaths: a before (2006-10) versus after (2011-13) comparison. Addiction. 2016;111:883-891.
More than 64,000 people in the United States died of drug overdoses in 2016.1 Of those overdose deaths, more than 34,000 were related to the use of natural (eg, codeine, morphine); synthetic (eg, fentanyl); and semisynthetic (eg, oxycodone, hydrocodone) opioids.1 The number of drug-overdose fatalities (driven largely by opioids) has increased so dramatically in recent years that drug overdose is now the leading cause of intentional and unintentional injury-related death in the United States.2 Furthermore, opioid use is increasing among college students, with many injecting these agents.3 Those injecting (as opposed to other routes of delivery) have the highest death rate.4
The Department of Health and Human Services has identified 3 important issues to address with regard to the opioid epidemic: prescriber education, community naloxone access, and better interventions (such as naloxone overdose-reversal take-home kits) for people with opioid use disorders and/or a history of overdoses.5 (For more on overdose reversal kits, see “What FPs need to know about naloxone kits,” a 3-in-3 video.) With these goals in mind, we provide the following review of naloxone dosing and postoverdose treatment.
Steps FPs can take to reverse the overdose
Opioids act on delta, kappa, and mu receptors in the brain to produce analgesic effects,6 but, in large quantities, their mu receptor activity can cause fatal respiratory depression.7 Some of the most commonly abused opioids are heroin and the prescription opioids fentanyl, oxycodone, and hydrocodone.8
People who have overdosed on opioids generally present with evidence of obtundation, miosis, and difficulty breathing. Respiratory failure is the most common cause of death.9 Hypothermia, compartment syndrome, rhabdomyolysis, renal failure, and acute pulmonary edema are less common complications. Overdoses and these medical issues can potentially be reversed and/or mitigated by naloxone administration.10,11
Naloxone and its routes of administration. Naloxone is the agent of choice in overdose situations.12 It works as an antagonist of the delta, kappa, and mu receptors,6,13 has a rapid onset of action, and is associated with minimal adverse effects.14
Naloxone can be administered via the intravenous (IV), intranasal, intramuscular, subcutaneous, intraosseous, or endotracheal routes.6 Although IV administration has been the most common and is still generally preferred in the hospital setting, the intranasal route has gained favor, partly because it can be difficult to establish an IV in IV drug users and partly because it is easier for nonmedical people to administer.6
In addition, the nasal mucosa has an abundant blood supply resulting in rapid absorption. The drug reaches the systemic circulation quickly and avoids first-pass hepatic metabolism.6 Intranasal route absorption is enhanced by deep inhalation and patient cooperation, but it can still be effective in an unconscious patient. Response time is nearly the same as that with IV administration (both act within 1-2 mins).6
Naloxone has a short duration of action (shorter than that of some opiates), and its duration of action is influenced by the pharmacology and toxicity of the overdose drug.15 The serum half-life in adults ranges from 30 to 81 mins, and clinical impact varies from minutes to an hour.15 Thus, even if a patient initially improves after administration, close observation is mandatory due to the frequent need for repeat naloxone dosing.
Adverse effects. Naloxone is considered safe, with relatively few adverse effects and doesn’t have any effects on someone who isn’t experiencing an opioid overdose or currently on opioids.15 The only downside is that naloxone administration to an opioid-dependent person often precipitates an acute withdrawal event, characterized by global pain, agitation, generalized distress, and gastrointestinal complaints, including vomiting and diarrhea. Although withdrawal is not life-threatening, it can cause great discomfort.
Getting a handle on naloxone dosing
The starting dose of naloxone used to be 0.04 mg, but this was later increased to 0.4 mg. The advent and high overdose lethality of more potent drugs like fentanyl and carfentanil has made low-dose naloxone less effective.12
Currently, 1 mg is often the initial recommendation, but doses of 2 to 4 mg are not uncommon, and multiple administrations or continuous IV administration are frequently needed to reverse severe toxicities, such as those involving fentanyl or longer-action opioids like methadone. Anyone exhibiting difficulty breathing mandates a starting naloxone dose of at least 1 to 2 mg.12,16 In addition to breathing, additional doses are indicated clinically by medical parameters such as vital signs, ocular pupil diameter, and/or alertness.6
Intranasal administration often utilizes up to 4 mg of naloxone in one nostril, followed by a titrated additional administration in the other nostril. In life-threatening circumstances, especially those in which a patient is exhibiting respiratory depression, a much larger quantity of naloxone—up to 10 mg—may be administered by trained medical personnel.12,16 In the end, all dosing varies and must be individualized to the patient’s signs and symptoms. Those who have overdosed require prolonged monitoring to treat potential complications.
Emergency assistance and transport. Because of the dangers that can result from opioid toxicities, any hint or evidence of physiologic compromise merits a 911 call for emergency medical assistance and transport to a hospital emergency department (ED). Hospitalization is at the physician’s discretion.
Expanding the availability of naloxone in the community
The availability of naloxone overdose-reversal kits is growing among hospitals, other types of health care facilities, first responders, medical offices, and the general public. Distributing the kits to opioid users and their families has wide support but remains controversial (more on this in a bit).
Support even includes that from the current US Surgeon General, Jerome Adams, MD, MPH, who noted in a statement on April 5, 2018, the lifesaving success of opioid-overdose reversal naloxone kits by medical personnel, first responders, and other people. As a result, he formally recommended that more Americans keep such kits available in order to be able to quickly diminish opioid toxicities.17,18 His advice was especially directed toward people at risk for an opioid overdose or anyone associated with opioid drug users.
Prehospital management of overdoses is ideally managed by emergency medical service (EMS) personnel,10 but even nonmedical people can safely administer naloxone. About 10,000 overdose cases were documented to have been reversed by nonmedical providers between 1996 and 2010.10 Many states have laws limiting the civil and criminal liability for naloxone administrators. New Mexico was the first state to legally allow naloxone administration by individuals without a prescription.7 Pharmacists often participate in efforts to counter opioid drug overdose deaths by offering naloxone administration kits, along with training about techniques of use, to people filling opioid prescriptions and to household members and/or other individuals in the social support network of an opioid user.6 Some physicians co-prescribe naloxone to patients along with opioid therapies during long-term pain management. Such dual prescribing is encouraged by many clinics.19 This method has decreased opioid overdose deaths in North Carolina,20 in its army base at Fort Bragg,19 and in California.21
The issue of “risk compensation”
To those who say that having naloxone available to users of opioids or those in their social network promotes even riskier behavior resulting in increased overdoses, research points to just the opposite. A nonrandomized study that examined co-prescribing naloxone to patients on chronic opioid therapy for non-cancer-related pain, documented fewer opioid-related ED visits following use by prescribers and patients at community health centers.22 Other research has demonstrated a reduced number of community-level opioid overdose deaths once opioid overdose education and community naloxone distribution were implemented.23,24
After the overdose: Getting patients into treatment
After reversing initial toxicities, a protracted period of assessment is required to assure patient safety. Beyond prolonged observation after an overdose, it is critical to recommend and provide long-term substance abuse therapies. Simply reversing the overdose is not medically sufficient, even if postoverdose patients refuse such treatment referrals. The fact that many of these people subsequently die is evidence of the importance of adhering to a formal, long-term chemical dependence intervention program.
Persistent diligence is usually needed to convince a patient who has recovered from an acute drug overdose event to accept a treatment referral. Some EDs institute special teams to facilitate such referrals, using a multidisciplinary approach, including substance abuse counselors and social workers. Referral agencies are also sometimes included to aid patient acceptance and retention in drug abuse treatment interventions. (See "Resources" below for more information.)
SIDEBAR
Resources
- The Centers for Disease Control and Prevention’s Guideline for Prescribing Opioids for Chronic Pain. Available at: https://www.cdc.gov/drugoverdose/prescribing/guideline.html.
- National Institute on Drug Abuse. Available at: https://www.drugabuse.gov.
- Substance Abuse and Mental Health Services Administration. Available at: https://www.samhsa.gov/find-help/national-helpline.
- Your state’s prescription drug monitoring program. Available at: https://www.cdc.gov/drugoverdose/pdmp/states.html.
CORRESPONDENCE
Steven Lippmann, MD, 401 East Chestnut Street, Suite 610, Louisville, KY 40202; Steven.lippmann@louisville.edu.
More than 64,000 people in the United States died of drug overdoses in 2016.1 Of those overdose deaths, more than 34,000 were related to the use of natural (eg, codeine, morphine); synthetic (eg, fentanyl); and semisynthetic (eg, oxycodone, hydrocodone) opioids.1 The number of drug-overdose fatalities (driven largely by opioids) has increased so dramatically in recent years that drug overdose is now the leading cause of intentional and unintentional injury-related death in the United States.2 Furthermore, opioid use is increasing among college students, with many injecting these agents.3 Those injecting (as opposed to other routes of delivery) have the highest death rate.4
The Department of Health and Human Services has identified 3 important issues to address with regard to the opioid epidemic: prescriber education, community naloxone access, and better interventions (such as naloxone overdose-reversal take-home kits) for people with opioid use disorders and/or a history of overdoses.5 (For more on overdose reversal kits, see “What FPs need to know about naloxone kits,” a 3-in-3 video.) With these goals in mind, we provide the following review of naloxone dosing and postoverdose treatment.
Steps FPs can take to reverse the overdose
Opioids act on delta, kappa, and mu receptors in the brain to produce analgesic effects,6 but, in large quantities, their mu receptor activity can cause fatal respiratory depression.7 Some of the most commonly abused opioids are heroin and the prescription opioids fentanyl, oxycodone, and hydrocodone.8
People who have overdosed on opioids generally present with evidence of obtundation, miosis, and difficulty breathing. Respiratory failure is the most common cause of death.9 Hypothermia, compartment syndrome, rhabdomyolysis, renal failure, and acute pulmonary edema are less common complications. Overdoses and these medical issues can potentially be reversed and/or mitigated by naloxone administration.10,11
Naloxone and its routes of administration. Naloxone is the agent of choice in overdose situations.12 It works as an antagonist of the delta, kappa, and mu receptors,6,13 has a rapid onset of action, and is associated with minimal adverse effects.14
Naloxone can be administered via the intravenous (IV), intranasal, intramuscular, subcutaneous, intraosseous, or endotracheal routes.6 Although IV administration has been the most common and is still generally preferred in the hospital setting, the intranasal route has gained favor, partly because it can be difficult to establish an IV in IV drug users and partly because it is easier for nonmedical people to administer.6
In addition, the nasal mucosa has an abundant blood supply resulting in rapid absorption. The drug reaches the systemic circulation quickly and avoids first-pass hepatic metabolism.6 Intranasal route absorption is enhanced by deep inhalation and patient cooperation, but it can still be effective in an unconscious patient. Response time is nearly the same as that with IV administration (both act within 1-2 mins).6
Naloxone has a short duration of action (shorter than that of some opiates), and its duration of action is influenced by the pharmacology and toxicity of the overdose drug.15 The serum half-life in adults ranges from 30 to 81 mins, and clinical impact varies from minutes to an hour.15 Thus, even if a patient initially improves after administration, close observation is mandatory due to the frequent need for repeat naloxone dosing.
Adverse effects. Naloxone is considered safe, with relatively few adverse effects and doesn’t have any effects on someone who isn’t experiencing an opioid overdose or currently on opioids.15 The only downside is that naloxone administration to an opioid-dependent person often precipitates an acute withdrawal event, characterized by global pain, agitation, generalized distress, and gastrointestinal complaints, including vomiting and diarrhea. Although withdrawal is not life-threatening, it can cause great discomfort.
Getting a handle on naloxone dosing
The starting dose of naloxone used to be 0.04 mg, but this was later increased to 0.4 mg. The advent and high overdose lethality of more potent drugs like fentanyl and carfentanil has made low-dose naloxone less effective.12
Currently, 1 mg is often the initial recommendation, but doses of 2 to 4 mg are not uncommon, and multiple administrations or continuous IV administration are frequently needed to reverse severe toxicities, such as those involving fentanyl or longer-action opioids like methadone. Anyone exhibiting difficulty breathing mandates a starting naloxone dose of at least 1 to 2 mg.12,16 In addition to breathing, additional doses are indicated clinically by medical parameters such as vital signs, ocular pupil diameter, and/or alertness.6
Intranasal administration often utilizes up to 4 mg of naloxone in one nostril, followed by a titrated additional administration in the other nostril. In life-threatening circumstances, especially those in which a patient is exhibiting respiratory depression, a much larger quantity of naloxone—up to 10 mg—may be administered by trained medical personnel.12,16 In the end, all dosing varies and must be individualized to the patient’s signs and symptoms. Those who have overdosed require prolonged monitoring to treat potential complications.
Emergency assistance and transport. Because of the dangers that can result from opioid toxicities, any hint or evidence of physiologic compromise merits a 911 call for emergency medical assistance and transport to a hospital emergency department (ED). Hospitalization is at the physician’s discretion.
Expanding the availability of naloxone in the community
The availability of naloxone overdose-reversal kits is growing among hospitals, other types of health care facilities, first responders, medical offices, and the general public. Distributing the kits to opioid users and their families has wide support but remains controversial (more on this in a bit).
Support even includes that from the current US Surgeon General, Jerome Adams, MD, MPH, who noted in a statement on April 5, 2018, the lifesaving success of opioid-overdose reversal naloxone kits by medical personnel, first responders, and other people. As a result, he formally recommended that more Americans keep such kits available in order to be able to quickly diminish opioid toxicities.17,18 His advice was especially directed toward people at risk for an opioid overdose or anyone associated with opioid drug users.
Prehospital management of overdoses is ideally managed by emergency medical service (EMS) personnel,10 but even nonmedical people can safely administer naloxone. About 10,000 overdose cases were documented to have been reversed by nonmedical providers between 1996 and 2010.10 Many states have laws limiting the civil and criminal liability for naloxone administrators. New Mexico was the first state to legally allow naloxone administration by individuals without a prescription.7 Pharmacists often participate in efforts to counter opioid drug overdose deaths by offering naloxone administration kits, along with training about techniques of use, to people filling opioid prescriptions and to household members and/or other individuals in the social support network of an opioid user.6 Some physicians co-prescribe naloxone to patients along with opioid therapies during long-term pain management. Such dual prescribing is encouraged by many clinics.19 This method has decreased opioid overdose deaths in North Carolina,20 in its army base at Fort Bragg,19 and in California.21
The issue of “risk compensation”
To those who say that having naloxone available to users of opioids or those in their social network promotes even riskier behavior resulting in increased overdoses, research points to just the opposite. A nonrandomized study that examined co-prescribing naloxone to patients on chronic opioid therapy for non-cancer-related pain, documented fewer opioid-related ED visits following use by prescribers and patients at community health centers.22 Other research has demonstrated a reduced number of community-level opioid overdose deaths once opioid overdose education and community naloxone distribution were implemented.23,24
After the overdose: Getting patients into treatment
After reversing initial toxicities, a protracted period of assessment is required to assure patient safety. Beyond prolonged observation after an overdose, it is critical to recommend and provide long-term substance abuse therapies. Simply reversing the overdose is not medically sufficient, even if postoverdose patients refuse such treatment referrals. The fact that many of these people subsequently die is evidence of the importance of adhering to a formal, long-term chemical dependence intervention program.
Persistent diligence is usually needed to convince a patient who has recovered from an acute drug overdose event to accept a treatment referral. Some EDs institute special teams to facilitate such referrals, using a multidisciplinary approach, including substance abuse counselors and social workers. Referral agencies are also sometimes included to aid patient acceptance and retention in drug abuse treatment interventions. (See "Resources" below for more information.)
SIDEBAR
Resources
- The Centers for Disease Control and Prevention’s Guideline for Prescribing Opioids for Chronic Pain. Available at: https://www.cdc.gov/drugoverdose/prescribing/guideline.html.
- National Institute on Drug Abuse. Available at: https://www.drugabuse.gov.
- Substance Abuse and Mental Health Services Administration. Available at: https://www.samhsa.gov/find-help/national-helpline.
- Your state’s prescription drug monitoring program. Available at: https://www.cdc.gov/drugoverdose/pdmp/states.html.
CORRESPONDENCE
Steven Lippmann, MD, 401 East Chestnut Street, Suite 610, Louisville, KY 40202; Steven.lippmann@louisville.edu.
1. National Institute on Drug Abuse. Overdose death rates. Revised September 2017. Available at: https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates. Accessed April 11, 2018.
2. Xu J, Murphy SL, Kochanek KD, et al. Deaths: final data for 2013. Nat Vital Stat Syst. 2016;64:1-119.
3. McCabe SE, West BT, Teter CJ, et al. Trends in medical use, diversion, and nonmedical use of prescription medications among college students from 2003 to 2013: connecting the dots. Addict Behav. 2014;39:1176-1182.
4. Green TC, Heimer R, Grau LE. Distinguishing signs of opioid overdose and indication for naloxone: an evaluation of six overdose training and naloxone distribution programs in the United States. Addiction. 2008;103:979-989.
5. US Department of Health and Human Services. HHS takes strong steps to address opioid-drug related overdose, death and dependence. March 26, 2015. Available at: http://wayback.archive-it.org/3926/20170127185704/https://www.hhs.gov/about/news/2015/03/26/hhs-takes-strong-steps-to-address-opioid-drug-related-overdose-death-and-dependence.html. Accessed April 16, 2018.
6. Robinson A, Wermeling DP. Intranasal naloxone administration for treatment of opioid overdose. Am J Health Syst Pharm. 2014;71:2129-2135.
7. Doyon S, Aks SE, Schaeffer S. Expanding access to naloxone in the United States. J Med Toxicol. 2014;10:431-434.
8. National Institute on Drug Abuse. Which classes of prescription drugs are commonly misused? Available at: https://www.drugabuse.gov/publications/research-reports/misuse-prescription-drugs/which-classes-prescription-drugs-are-commonly-misused. Accessed April 16, 2018.
9. Boom M, Niesters M, Sarton E, et al. Non-analgesic effects of opioids: opioid-induced respiratory depression. Curr Pharm Des. 2012;18:5994-6004.
10. Weaver L, Palombi L, Bastianelli KMS. Naloxone administration for opioid overdose reversal in the prehospital setting: implications for pharmacists. J Pharm Pract. 2018;31:91-98.
11. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367:146-155.
12. Jordan MR, Morrisonponce D. Naloxone. StatPearls. Available at: https://www.ncbi.nlm.nih.gov/books/NBK441910/. Accessed September 1, 2017.
13. Wilkerson RG, Kim HK, Windsor TA, et al. The opioid epidemic in the United States. Emerg Med Clin North Am. 2016;34:e1-e23.
14. Jeffery RM, Dickinson L, Ng ND, et al. Naloxone administration for suspected opioid overdose: an expanded scope of practice by a basic life support collegiate-based emergency medical services agency. J Am Coll Health. 2017;65:212-216.
15. Drugs.com. Naloxone. Available at: https://www.drugs.com/pro/naloxone.html. Accessed April 16, 2018.
16. Prabhu A, Abaid B, Naik S, et al. Naloxone for opioid overdoses. Internet and Psychiatry 2017. Available at: https://www.internetandpsychiatry.com/wp/editorials/naloxone-for-opioid-overdoses/. Accessed September 19, 2017.
17. HHS.gov. Surgeon General releases advisory on naloxone, an opioid overdose-reversing drug. Available at: https://www.hhs.gov/about/news/2018/04/05/surgeon-general-releases-advisory-on-naloxone-an-opioid-overdose-reversing-drug.html. Accessed April 16, 2018.
18. US Department of Health and Human Services. Surgeongeneral.gov. Surgeon General’s advisory on naloxone and opioid overdose. Available at: https://www.surgeongeneral.gov/priorities/opioid-overdose-prevention/naloxone-advisory.html. Accessed April 16, 2018.
19. Behar E, Rowe C, Santos GM, et al. Acceptability of naloxone co-prescription among primary care providers treating patients on long-term opioid therapy for pain. J Gen Intern Med. 2017;32:291-295.
20. Albert S, Brason FW 2nd, Sanford CK, et al. Project Lazarus: community‐based overdose prevention in rural North Carolina. Pain Med. 2011;12:S77-S85.
21. Rowe C, Santos GM, Vittinghoff E, et al. Predictors of participant engagement and naloxone utilization in a community‐based naloxone distribution program. Addiction. 2015;110:1301-1310.
22. Coffin PO, Behar E, Rowe C, et al. Nonrandomized intervention study of naloxone coprescription for primary care patients receiving long-term opioid therapy for pain. Ann Intern Med. 2016;165:245-252.
23. Walley AY, Xuan Z, Hackman HH, et al. Opioid overdose rates and implementation of overdose education and nasal naloxone distribution in Massachusetts: interrupted time series analysis. BMJ. 2013;346:f174.
24. Bird SM, McAuley A, Perry S, et al. Effectiveness of Scotland’s National Naloxone Programme for reducing opioid-related deaths: a before (2006-10) versus after (2011-13) comparison. Addiction. 2016;111:883-891.
1. National Institute on Drug Abuse. Overdose death rates. Revised September 2017. Available at: https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates. Accessed April 11, 2018.
2. Xu J, Murphy SL, Kochanek KD, et al. Deaths: final data for 2013. Nat Vital Stat Syst. 2016;64:1-119.
3. McCabe SE, West BT, Teter CJ, et al. Trends in medical use, diversion, and nonmedical use of prescription medications among college students from 2003 to 2013: connecting the dots. Addict Behav. 2014;39:1176-1182.
4. Green TC, Heimer R, Grau LE. Distinguishing signs of opioid overdose and indication for naloxone: an evaluation of six overdose training and naloxone distribution programs in the United States. Addiction. 2008;103:979-989.
5. US Department of Health and Human Services. HHS takes strong steps to address opioid-drug related overdose, death and dependence. March 26, 2015. Available at: http://wayback.archive-it.org/3926/20170127185704/https://www.hhs.gov/about/news/2015/03/26/hhs-takes-strong-steps-to-address-opioid-drug-related-overdose-death-and-dependence.html. Accessed April 16, 2018.
6. Robinson A, Wermeling DP. Intranasal naloxone administration for treatment of opioid overdose. Am J Health Syst Pharm. 2014;71:2129-2135.
7. Doyon S, Aks SE, Schaeffer S. Expanding access to naloxone in the United States. J Med Toxicol. 2014;10:431-434.
8. National Institute on Drug Abuse. Which classes of prescription drugs are commonly misused? Available at: https://www.drugabuse.gov/publications/research-reports/misuse-prescription-drugs/which-classes-prescription-drugs-are-commonly-misused. Accessed April 16, 2018.
9. Boom M, Niesters M, Sarton E, et al. Non-analgesic effects of opioids: opioid-induced respiratory depression. Curr Pharm Des. 2012;18:5994-6004.
10. Weaver L, Palombi L, Bastianelli KMS. Naloxone administration for opioid overdose reversal in the prehospital setting: implications for pharmacists. J Pharm Pract. 2018;31:91-98.
11. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367:146-155.
12. Jordan MR, Morrisonponce D. Naloxone. StatPearls. Available at: https://www.ncbi.nlm.nih.gov/books/NBK441910/. Accessed September 1, 2017.
13. Wilkerson RG, Kim HK, Windsor TA, et al. The opioid epidemic in the United States. Emerg Med Clin North Am. 2016;34:e1-e23.
14. Jeffery RM, Dickinson L, Ng ND, et al. Naloxone administration for suspected opioid overdose: an expanded scope of practice by a basic life support collegiate-based emergency medical services agency. J Am Coll Health. 2017;65:212-216.
15. Drugs.com. Naloxone. Available at: https://www.drugs.com/pro/naloxone.html. Accessed April 16, 2018.
16. Prabhu A, Abaid B, Naik S, et al. Naloxone for opioid overdoses. Internet and Psychiatry 2017. Available at: https://www.internetandpsychiatry.com/wp/editorials/naloxone-for-opioid-overdoses/. Accessed September 19, 2017.
17. HHS.gov. Surgeon General releases advisory on naloxone, an opioid overdose-reversing drug. Available at: https://www.hhs.gov/about/news/2018/04/05/surgeon-general-releases-advisory-on-naloxone-an-opioid-overdose-reversing-drug.html. Accessed April 16, 2018.
18. US Department of Health and Human Services. Surgeongeneral.gov. Surgeon General’s advisory on naloxone and opioid overdose. Available at: https://www.surgeongeneral.gov/priorities/opioid-overdose-prevention/naloxone-advisory.html. Accessed April 16, 2018.
19. Behar E, Rowe C, Santos GM, et al. Acceptability of naloxone co-prescription among primary care providers treating patients on long-term opioid therapy for pain. J Gen Intern Med. 2017;32:291-295.
20. Albert S, Brason FW 2nd, Sanford CK, et al. Project Lazarus: community‐based overdose prevention in rural North Carolina. Pain Med. 2011;12:S77-S85.
21. Rowe C, Santos GM, Vittinghoff E, et al. Predictors of participant engagement and naloxone utilization in a community‐based naloxone distribution program. Addiction. 2015;110:1301-1310.
22. Coffin PO, Behar E, Rowe C, et al. Nonrandomized intervention study of naloxone coprescription for primary care patients receiving long-term opioid therapy for pain. Ann Intern Med. 2016;165:245-252.
23. Walley AY, Xuan Z, Hackman HH, et al. Opioid overdose rates and implementation of overdose education and nasal naloxone distribution in Massachusetts: interrupted time series analysis. BMJ. 2013;346:f174.
24. Bird SM, McAuley A, Perry S, et al. Effectiveness of Scotland’s National Naloxone Programme for reducing opioid-related deaths: a before (2006-10) versus after (2011-13) comparison. Addiction. 2016;111:883-891.
From The Journal of Family Practice | 2018;67(5):288-290,292.
Depression and Bipolar Disorders in Patients With Alcohol Use Disorders (FULL)
Co-occurrence of depression and substance abuse often poses diagnostic and therapeutic challenges. This article reviews the prevalence, clinical considerations, and treatment of depression coexisting with alcohol use disorders (AUDs).
Prevalence
Mood and substance use disorders (SUDs) are very common with an estimated lifetime prevalence in the U.S. of 17% for major depression, 4% for bipolar I and II disorders, 13% for alcohol abuse, and 5% for alcohol dependence.1 Almost all of the associations between disorders of mood or anxiety and drug use were positive and statistically significant in the National Epidemiologic Survey, on Alcohol and Related Conditions (NESARC), which included 43,093 noninstitutionalized patients.2
There is a reciprocal relationship between depression and alcoholism. Epidemiologic Catchment Area Survey results indicated that baseline symptoms of depression or alcohol abuse increased the risk of developing alcohol dependence or depression.3 The risk of developing depression were elevated among people with increasing levels of alcohol-induced debility. Conversely, the presence of depressive symptoms increased the chance of developing alcohol dependence. The association between alcohol dependence and depression may be attributable to the depressive effects of ethanol; depression often remits with sobriety. Psychosocial consequences of problem drinking also may contribute to affective illnesses.
Alcohol dependence poses a major depression risk that contributes to higher rates of alcohol use. In people with ethanol dependence, the prevalence of major depressive disorder (MDD) is 21%.4 People who are alcohol dependent are 4 times more likely than are nondependents to have MDD. Forty-one percent of people who seek treatment for current alcohol abuse have a mood disorder.
The NESARC survey revealed strong associations between depression, substance use, and other psychopathologies.5 Compared with MDD alone, SUD combined with MDD conferred high vulnerability to additional psychopathology, depressive episodes that were more severe and greater in number, and more suicide attempts.
Depression Clincal Considerations
Depression linked to recent alcohol abuse may not respond well to an antidepressant drug beyond what is achieved with ethanol abstinence. In one study, depressive symptoms were assessed over the course of alcohol-related hospitalizations.6 Depression was evident in 42% of patients 48 hours after admission, but only 6% remained clinically depressed by week 4 of hospitalization. Therefore, in the treatment of patients hospitalized for alcohol detoxification, it is common to observe them for 1 month before considering antidepressant medication. Mood likely will improve without pharmacotherapy.
However, delaying treatment for depression while a patient is hospitalized for alcohol detoxification presents some difficulties. Many patients do not remain sober during the first month after detoxification. One study found that 65% of patients imbibed alcohol within 2 weeks after discharge.7 Furthermore, 50% relapsed into heavy drinking during the same period. More than 25% of patients who used alcohol and were diagnosed with substance-induced depression at baseline were reclassified with MDD the next year.8
Careful clinical assessment is needed after alcohol detoxification. Depression that persists during ethanol abstinence predisposes a patient to relapse into heavy drinking. Therefore, failure to treat depression after alcohol detoxification poses considerable risk.9 A study of the effect of depression on the return to drinking among patients with alcohol dependence found that depression at entry into inpatient treatment for alcohol dependence predicted a shorter time to first drink.9 The prognosis for a drinking relapse was worse no matter whether the depression came first or was triggered by the alcohol. Depression does not predict drinking outcomes, but it is associated with a more rapid relapse to ethanol consumption.
Similarly, inpatients with premorbid or substanceinduced depression were more likely to meet the criteria for drug dependence during outpatient follow-up.10 In addition, patients who developed depression during the first 26 weeks after hospitalization were 3 times more likely than those without depression to relapse into drug dependence during follow-up.
Alcohol dependence may hasten the progression of depression. A study on the prognostic effect of alcoholism on the 10-year course of depression found a deleterious influence of current alcoholism after recovery from depression.11 Patients with MDD were more likely to transition from being ill to improving if either they were forgoing alcohol or had never abused it. Another investigation verified that alcohol and drug dependence increased perceptions of affective symptomatology.12
Substance-induced depression also increases the risk for suicide. In 602 patients with substance dependence, depression was classified as occurring before dependence, during abstinence, or during substance use.13 Depression increased the risk for suicide in 34% of patients
who had already attempted suicide at least once. Compared with depression absent substance abuse, depression preceding substance use was associated with high vulnerability to additional psychopathology, depressive episodes that were more severe and greater in number, and more suicide attempts. Substance dependence predicted severity of suicidal intent, and abstinence predicted number of attempts.
Psychiatric hospitalizations often involve patients with a history of suicidal thinking or behavior and substance-induced depression. Clinicians can make reliable assessments of the degree to which a presenting psychiatric syndrome is substance-induced.14 These patients require addiction treatment, including outpatient addiction services capable of caring for suicidal persons. These individuals also are more likely to be homeless, unemployed, and uncooperative.15
Taking a psychiatric history and making a detailed inquiry into potential suicidal behavior, recent substance abuse, and current mood symptoms are warranted in persons with depression and/or SUD. Close follow-up is especially important for depressed patients likely to relapse into alcoholism soon after hospital discharge. Failure to recognize MDD or a bipolar disorder in such a patient may result in more relapses, recurrence of mood episodes, and elevated risk of completing suicide.16
Bipolar Clinical Considerations
There is a lack of clarity regarding the effect of moderate-to-excessive alcohol use on the course of bipolar disorders. There is a negative effect on patients with alcohol-induced bipolar depression. In a study of group therapy patients with bipolar disorder co-occurring with substance dependence, data indicated that number of days of alcohol use predicted development of depression a month later.17 These findings were associated with heavy alcohol consumption. In these patients, substantial drinking increased the risk of a depressive episode. In another study, comorbid SUDs were correlated with suboptimal treatment compliance.18 The authors of a 1998 literature review concluded that comorbid SUD makes bipolar symptoms more severe.19
A number of studies have failed to confirm a negative effect of alcohol on bipolar depression.20 There were no differences in 1-year course and outcome between bipolar patients with different alcohol use levels (abstinence, incidental use, moderate abuse, excessive consumption). Other investigators concluded that SUDs were not associated with slower recovery from depression but could contribute to a higher risk of switching to a manic, mixed, or hypomanic state.21
Substance use disorders are associated with increased suicidal behavior in people with a bipolar disorder. The risk of attempted suicide is about double for these patients relative to bipolar patients who do not abuse alcohol.22 Of those who abuse drugs, 14% to 16% complete suicide.23
Psychotherapy
Reportedly, integrated cognitive behavioral therapy (CBT) provided better substance abuse outcomes compared with 12-step programs.24 There also was less substance abuse within the year after CBT. Integrated psychosocial treatment for patients with a mood disorder and substance abuse should involve simultaneous treatment of the 2 conditions. A sequential approach addresses the primary concern and subsequently treats the comorbid disorder, whereas a parallel approach manages both at the same time but in different surroundings. In both approaches, conflicting therapeutic ideologies are a potential difficulty. Given the multiple treatment locations and separate appointments, scheduling problems are an additional difficulty. Coexisting illnesses also are important to consider in the clinical treatment for bipolar patients. As with individual treatments, group therapies take either a sequential approach (more acute disorder treated first) or a parallel approach (disorders treated simultaneously but in separate settings).
Integrated group therapy (IGT) considers patients as having a single diagnosis, focuses on commonalities between relapse and recovery, and reviews the relationship between both conditions. One study compared IGT and treatment as usual in subjects with comorbid bipolar and AUD.25 The IGT group evidenced fewer days of alcohol use. Other research compared IGT with group drug treatment and found that IGT subjects were more likely to remain abstinent.26 This type of psychotherapy showed promise in a meta-analysis of integrated treatment in patients with depression and SUDs.26
Compared with placebo, sertraline/CBT combined treatment reduced alcohol consumption on drinking days.27 This combination was effective in reducing depression, especially in females.
Acceptance and commitment therapy (ACT) combines mindfulness and behavioral change to increase psychological flexibility. The goal in ACT is for patients to become more accepting of their unpleasant feelings. In a study of alcohol abusers with affective disorders, those treated with ACT, compared with controls, had higher abstinence rates and lower depression scores.28
Phamacotherapy and Bipolar Disorder
Even when bipolar symptoms were resolved with use of mood-stabilizing medications, usually some alcohol use continued, though no association was found between bipolar disorder and AUDs.29 With patients’ illness severity and ethanol consumption rated weekly over 7 years, no temporal correlation was found between drinking alcohol and bipolar symptoms.
Similarly, in a study, relief of depressive bipolar symptoms did not result in less frequent alcohol relapse.30 One hundred fifteen outpatients with bipolar disorder and AUD were randomly assigned to either 12 weeks of quetiapine therapy or placebo. Patients in the quetiapine group experienced significant improvement in mood, but sobriety was not enhanced.
Two studies indicated trends of reduced drinking with use of prescribed alcohol-deterrent drugs. An investigation that compared naltrexone with placebo did not reach statistical significance, but naltrexone was reasonably effective in reducing alcohol consumption and craving.31 A report on patients with bipolar disorder treated with acamprosate also did not identify any significant differences in alcohol drinking prognosis.32 Nevertheless, acamprosate was well tolerated and seemed to confer some clinical benefit.
There is a paucity of research focused on patients with bipolar disorder and substance dependence.33 In one trial, patients with bipolar disorder and a diagnosis of alcohol dependence were randomly assigned to receive either valproate or placebo.34 Valproate therapy decreased the number of heavy consumption days and drinks per drinking day in these patients. In a study of 362 patients with bipolar disorder and alcohol dependence treated with lithium or valproic acid, there was no change in drinking days despite adding quetiapine to the regimen.35
Pharmacotherapy and Depression
Lithium is not effective for patients with MDD and AUD. Lithium treatment for depressed patients with alcohol dependence did not improve abstinence, alcohol-related hospitalizations, or severity of either condition.36
Aripiprazole is an antipsychotic that partially agonizes dopamine receptors. Dopamine implicates reward circuitry and has a role in AUDs. Aripiprazole was used as an adjunctive intervention in a randomized trial of 35 patients with comorbid alcohol dependence and depression.37 There was less depression in both the aripiprazole plus escitalopram group and the escitalopram group. Imaging showed a change in activity in the left cingulate gyrus in the patients with comorbid alcohol dependence and MDD. The action of aripiprazole may be mediated through the anterior cingulate cortex.
Research on patients with alcohol dependence treated with fluoxetine found decreased Hamilton Depression Rating Scale (HDRS) scores but no change in alcohol consumption.38
Sertraline diminishes depressive symptoms in abstinent alcoholics. In one study, depressed, recently abstinent alcohol users were randomly assigned to receive sertraline 100 mg daily or placebo.39 Significant improvement was noted in HDRS and Beck Depression Inventory scores at 3- and 6-week intervals.
Citalopram was studied in patients randomly assigned to receive citalopram or placebo for alcohol abuse or dependence.40 Patients in the citalopram group had more days of drinking and showed little change in frequency of alcohol consumption. There was no improvement in depression severity in the citalopram group relative to the placebo group. Citalopram also has been studied in combination with naltrexone.41 Patients with depression and alcohol dependence were randomly assigned to receive either citalopram or placebo, as well as naltrexone. There were no significant differences in depression severity or drinking outcomes.
Treating depression with selective serotonin reuptake inhibitors (SSRIs) had variable results. Most SSRIs improve depression severity but largely have no effect on drinking outcomes.
Antidepressants
A meta-analysis on the efficacy of antidepressant medications in treating patients with depression and substance abuse revealed that the antidepressants had a somewhat advantageous effect.42 That finding was supported by the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study.43 About 33% of patients with citalopramtreated major depression endorsed concurrent SUD symptoms, 19% reported diagnosable alcohol use, 6% had other drug abuses, and 5% exhibited both alcohol and drug use. The groups did not differ in time needed to attain a better mood or in rate of response to citalopram.
Patients with citalopram-treated MDD and alcohol or drug abuse responded about as well as those without an SUD. However, those with alcohol and/or drug abuse had reduced rates of remission, and their remission was delayed, as compared with those without alcohol or drug abuse. There were more suicide attempts and psychiatric hospitalizations among the cohort with drug abuse.
Selective serotonin reuptake inhibitors have a reported safety advantage in treating patients with a history of excessive alcohol intake.44 Another advantage is that SSRIs are seldom abused and seldom lower seizure thresholds significantly. Deleterious effects of alcohol on motor skills or cognition are not potentiated. Adverse effects are usually mild, and overdoses are rarely dangerous.
Antidepressant medication decreased depression and diminished the amount of drinking in patients with depression who use alcohol.45 In controlled research of patients with comorbid depression and alcohol dependence, fluoxetine reduced the severity of these conditions. Substantial reductions in depressive symptoms occurred during detoxification and washout in both groups. There was a strong relationship between depression and drinking among people with depression and AUD.
Desipramine can produce similar results, with positive antidepressant drug effects on depression and drinking. Therefore, pharmacotherapy is indicated for patients with depression who abuse ethanol. Research found that alcohol-dependent patients with depression responded to desipramine.46 Desipramine yielded prolonged abstinence in patients with depression who were using alcohol but not in alcohol users without depression.
A study of imipramine use in actively drinking outpatients found decreased alcohol consumption only for those whose depression responded to treatment.47 However, there was no influence on drinking outcome. Patients whose mood improved reported decreased alcohol consumption after imipramine therapy.
Conslusion
People with co-occurring depression and alcohol dependence are optimally treated with pharmacotherapies that address each condition. One investigation randomly assigned alcohol-dependent patients with depression to 14 weeks of treatment with sertraline 200 mg/d, naltrexone 100 mg/d, a combination of the drugs, or placebo.48 The combination treatment produced the best rate of abstinence before a heavy drinking relapse. Also, fewer patients tended to be depressed in the final weeks of treatment when prescribed the combined regimen. Pharmacotherapy is the best approach for both depression and AUDs.
Click here to read the digital edition.
1. Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Conway KP, Compton W, Stinson FS, Grant BF. Lifetime comorbidity of DSM-IV mood and anxiety disorders and specific drug use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2006;67(2):247-257.
3. Gilman SE, Abraham HD. A longitudinal study of the order of onset of alcohol dependence and major depression. Drug Alcohol Depend. 2001;63(3):277-286.
4. Kessler RC, Crum RM, Warner LA, Nelson CB, Schulenberg J, Anthony JC. Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
5. Blanco C, Alegría AA, Liu SM, et al. Differences among major depressive disorder with and without co-occurring substance use disorders and substance-induced depressive disorder: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2012;73(6):865-873.
6. Brown SA, Schuckit MA. Changes in depression among abstinent alcoholics. J Stud Alcohol. 1988;49(5):412-417.
7. Kiefer F, Jahn H, Tarnaske T, et al. Comparing and combining naltrexone and acamprosate in relapse prevention of alcoholism: a double-blind, placebo-controlled study. Arch Gen Psychiatry. 2003;60(1):92-99.
8. Ramsey SE, Kahler CW, Read JP, Stuart GL, Brown RA. Discriminating between substance-induced and independent depressive episodes in alcohol-dependent patients. J Stud Alcohol. 2004;65(5):672-676.
9. Greenfield SF, Weiss RD, Muenz LR, et al. The effect of depression on return to drinking: a prospective study. Arch Gen Psychiatry. 1998;55(3):259-265.
10. Hasin D, Liu X, Nunes E, McCloud S, Samet S, Endicott J. Effects of major depression on remission and relapse of substance dependence. Arch Gen Psychiatry. 2002;59(4):375-380.
11. Mueller TI, Lavori PW, Martin B, et al. Prognostic effect of the variable course of alcoholism on the 10-year course of depression. Am J Psychiatry. 1994;151(5):701-706.
12. Agosti V, Levin FR. The effects of alcohol and drug dependence on the course of depression. Am J Addict. 2006;15(1):71-75.
13. Aharonovich E, Liu X, Nunes E, Hasin DS. Suicide attempts in substance abusers: effects of major depression in relation to substance use disorders. Am J Psychiatry. 2002;159(9):1600-1602.
14. Ries RK, Demirsoy A, Russo JE, Barrett J, Roy-Byrne PP. Reliability and clinical utility of DSM-IV substance-induced psychiatric disorders in acute psychiatric inpatients. Am J Addict. 2001;10(4):308-318.
15. Ries RK, Yuodelis-Flores C, Comtois KA, Roy-Byrne PP, Russo JE. Substanceinduced suicidal admissions to an acute psychiatric service: characteristics and outcomes. J Subst Abuse Treat. 2008;34(1):72-79.
16. Toliver BK, Anton RF. Assessment and treatment of mood disorders in the context of substance abuse. Dialogues Clin Neurosci. 2015;17(2):181-190.
17. Jaffee WB, Griffin ML, Gallop R, et al. Depression precipitated by alcohol use in patients with co-occurring bipolar and substance use disorders. J Clin Psychiatry. 2009;70(2):171-176.
18. Manwani SG, Szilagyi KA, Zablotsky B, Hennen J, Griffin ML, Weiss RD. Adherence to pharmacotherapy in bipolar disorder patients with and without co-occurring substance use disorders. J Clin Psychiatry. 2007;68(8):1172-1176.
19. Tohen M, Greenfield SF, Weiss RD, Zarate CA Jr, Vagge LM. The effect of comorbid substance disorders on the course of bipolar disorder: a review. Harv Rev Psychiatry. 1998;6(3):133-141.
20. van Zaane J, van den Brink W, Draisma S, Smit JH, Nolen WA. The effect of moderate and excessive alcohol use on the course and outcome of patients with bipolar disorders: a prospective cohort study. J Clin Psychiatry. 2010;71(7):885-893.
21. Ostacher MJ, Perlis RH, Nierenberg AA, et al; STEP-BD Investigators. Impact of substance use disorders on recovery from episodes of depression in bipolar disorder patients: prospective data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). Am J Psychiatry. 2010;167(3):289-297.
22. Oquendo MA, Currier D, Liu SM, Hasin DS, Grant BF, Blanco C. Increased risk for suicidal behavior in comorbid bipolar disorder and alcohol use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). J Clin Psychiatry. 2010;71(7):902-909.
23. Yoon YH, Chen CM, Yi HY, Moss HB. Effect of comorbid alcohol and drug use disorders on premature death of unipolar and bipolar decedents in the United States, 1999 to 2006. Compr Psychiatry. 2011;52(5):453-464.
24. Lydecker KP, Tate SR, Cummins KM, McQuaid J, Granholm E, Brown SA. Clinical outcomes of an integrated treatment for depression and substance use disorders. Psychol Addict Behav. 2010;24(3):453-465.
25. Weiss RD, Griffin ML, Kolodziej ME, et al. A randomized trial of integrated group therapy versus group drug counseling for patients with bipolar disorder and substance dependence. Am J Psychiatry. 2007;164(1):100-107.
26. Hesse M. Integrated psychological treatment for substance use and co-morbid anxiety or depression vs. treatment for substance use alone. A systematic review of the published literature. BMC Psychiatry. 2009;9:6.
27. Moak DH, Anton RF, Latham PK, Voronin KE, Waid RL, Durazo-Arvizu R. Sertraline and cognitive behavioral therapy for depressed alcoholics: results of a placebo-controlled trial. J Clin Psychopharmacol. 2003;23(6):553-562.
28. Thekiso TB, Murphy P, Milnes J, Lambe K, Curtin A, Farren CK. Acceptance and commitment therapy in the treatment of alcohol use disorder and comorbid affective disorder: a pilot matched control trial. Behav Ther. 2015;46(6):717-728.
29. Fleck DE, Amdt S, Delbello MP, Strakowski SM. Concurrent tracking of alcohol use and bipolar disorder symptoms. Bipolar Disord. 2006:8(4):338-344.
30. Brown ES, Gaza M, Carmody TJ. A randomized, double-blind, placebo-controlled add-on trial of quetiapine in outpatients with bipolar disorder and alcohol use disorders. J Clin Psychiatry. 2008;69(5):701-705.
31. Brown ES, Carmody TJ, Schmitz JM, et al. A randomized, double-blind, placebocontrolled pilot study of naltrexone in outpatients with bipolar disorder and alcohol dependence. Alcohol Clin Exp Res. 2009;33(11):1863-1869.
32. Tolliver BK, Desantis SM, Brown DG, Prisciandaro JJ, Brady KT. A randomized, double-blind, placebo-controlled clinical trial of acamprosate in alcoholdependent individuals with bipolar disorder: a preliminary report. Bipolar Disord. 2012;14(1):54-63.
33. Pettinati HM, O’Brien CP, Dundon WD. Current status of co-occurring mood and substance use disorders: a new therapeutic target. Am J Psychiatry. 2013;170(1):23-30.
34. Salloum IM, Cornelius JR, Daley DC, Kirisci L, Himmelhoch JM, Thase ME. Efficacy of valproate maintenance in patients with bipolar disorder and alcoholism: a double-blind placebo-controlled study. Arch Gen Psychiatry. 2005;62(1):37-45.
35. Farren CK, Hill KP, Weiss RD. Bipolar disorder and alcohol use disorder: a review. Curr Psychiatry Rep. 2012;14(6):659-666.
36. Dorus W, Ostrow DG, Anton R, et al. Lithium treatment of depressed and nondepressed alcoholics. JAMA. 1989;262(12):1646-1652.
37. Han DH, Kim SM, Choi JE, Min KJ, Renshaw PF. Adjunctive aripiprazole therapy with escitalopram in patients with co-morbid major depressive disorder and alcohol dependence: clinical and neuroimaging evidence. J Psychopharmacol. 2013;27(3):282-291.
38. Kranzler HR, Burleson JA, Korner P, et al. Placebo-controlled trial of fluoxetine as an adjunct to relapse prevention in alcoholics. Am J Psychiatry. 1995;152(3):391-397.
39. Roy A. Placebo-controlled study of sertraline in depressed recently abstinent alcoholics. Biol Psychiatry. 1998;44(7):633-637.
40. Charney DA, Heath LM, Zikos E, Palacios-Boix J, Gill KJ. Poorer drinking outcomes with citalopram treatment for alcohol dependence: a randomized, doubleblind, placebo-controlled trial. Alcohol Clin Exp Res. 2015;39(9):1756-1765.
41. Adamson SJ, Sellman JD, Foulds JA, et al. A randomized trial of combined citalopram and naltrexone for non-abstinent outpatients with co-occurring alcohol dependence and major depression. J Clin Psychopharmacol. 2015;35(2):143-149.
42. Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
43. Davis LL, Wisniewski SR, Howland RH, et al. Does comorbid substance use disorder impair recovery from major depression with SSRI treatment? An analysis of the STAR*D level one treatment outcomes. Drug Alcohol Depend. 2010;107(2-3):161-170.
44. Pettinati HM. The use of selective reuptake inhibitors in treating alcoholic subtypes. J Clin Psychiatry. 2001;62(suppl 20):26-31.
45. Cornelius JR, Salloum IM, Ehler JG, et al. Fluoxetine in depressed alcoholics. A double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1997;54(8):700-705.
46. Mason BJ, Kocsis JH, Ritvo EC, Cutler RB. A double-blind, placebo-controlled trial of desipramine for primary alcohol dependence stratified on the presence of absence of major depression. JAMA. 1996;275(10):761-767.
47. McGrath PJ, Nunes EV, Stewart JW, et al. Imipramine treatment of alcoholics with primary depression: a placebo-controlled clinical trial. Arch Gen Psychiatry. 1996;53(3):232-240.
48. Pettinati HM, Oslin DW, Kampman KM, et al. A double-blind, placebo-controlled trial combining sertraline and naltrexone for treating co-occurring depression and alcohol dependence. Am J Psychiatry. 2010;167(6):668-675.
Co-occurrence of depression and substance abuse often poses diagnostic and therapeutic challenges. This article reviews the prevalence, clinical considerations, and treatment of depression coexisting with alcohol use disorders (AUDs).
Prevalence
Mood and substance use disorders (SUDs) are very common with an estimated lifetime prevalence in the U.S. of 17% for major depression, 4% for bipolar I and II disorders, 13% for alcohol abuse, and 5% for alcohol dependence.1 Almost all of the associations between disorders of mood or anxiety and drug use were positive and statistically significant in the National Epidemiologic Survey, on Alcohol and Related Conditions (NESARC), which included 43,093 noninstitutionalized patients.2
There is a reciprocal relationship between depression and alcoholism. Epidemiologic Catchment Area Survey results indicated that baseline symptoms of depression or alcohol abuse increased the risk of developing alcohol dependence or depression.3 The risk of developing depression were elevated among people with increasing levels of alcohol-induced debility. Conversely, the presence of depressive symptoms increased the chance of developing alcohol dependence. The association between alcohol dependence and depression may be attributable to the depressive effects of ethanol; depression often remits with sobriety. Psychosocial consequences of problem drinking also may contribute to affective illnesses.
Alcohol dependence poses a major depression risk that contributes to higher rates of alcohol use. In people with ethanol dependence, the prevalence of major depressive disorder (MDD) is 21%.4 People who are alcohol dependent are 4 times more likely than are nondependents to have MDD. Forty-one percent of people who seek treatment for current alcohol abuse have a mood disorder.
The NESARC survey revealed strong associations between depression, substance use, and other psychopathologies.5 Compared with MDD alone, SUD combined with MDD conferred high vulnerability to additional psychopathology, depressive episodes that were more severe and greater in number, and more suicide attempts.
Depression Clincal Considerations
Depression linked to recent alcohol abuse may not respond well to an antidepressant drug beyond what is achieved with ethanol abstinence. In one study, depressive symptoms were assessed over the course of alcohol-related hospitalizations.6 Depression was evident in 42% of patients 48 hours after admission, but only 6% remained clinically depressed by week 4 of hospitalization. Therefore, in the treatment of patients hospitalized for alcohol detoxification, it is common to observe them for 1 month before considering antidepressant medication. Mood likely will improve without pharmacotherapy.
However, delaying treatment for depression while a patient is hospitalized for alcohol detoxification presents some difficulties. Many patients do not remain sober during the first month after detoxification. One study found that 65% of patients imbibed alcohol within 2 weeks after discharge.7 Furthermore, 50% relapsed into heavy drinking during the same period. More than 25% of patients who used alcohol and were diagnosed with substance-induced depression at baseline were reclassified with MDD the next year.8
Careful clinical assessment is needed after alcohol detoxification. Depression that persists during ethanol abstinence predisposes a patient to relapse into heavy drinking. Therefore, failure to treat depression after alcohol detoxification poses considerable risk.9 A study of the effect of depression on the return to drinking among patients with alcohol dependence found that depression at entry into inpatient treatment for alcohol dependence predicted a shorter time to first drink.9 The prognosis for a drinking relapse was worse no matter whether the depression came first or was triggered by the alcohol. Depression does not predict drinking outcomes, but it is associated with a more rapid relapse to ethanol consumption.
Similarly, inpatients with premorbid or substanceinduced depression were more likely to meet the criteria for drug dependence during outpatient follow-up.10 In addition, patients who developed depression during the first 26 weeks after hospitalization were 3 times more likely than those without depression to relapse into drug dependence during follow-up.
Alcohol dependence may hasten the progression of depression. A study on the prognostic effect of alcoholism on the 10-year course of depression found a deleterious influence of current alcoholism after recovery from depression.11 Patients with MDD were more likely to transition from being ill to improving if either they were forgoing alcohol or had never abused it. Another investigation verified that alcohol and drug dependence increased perceptions of affective symptomatology.12
Substance-induced depression also increases the risk for suicide. In 602 patients with substance dependence, depression was classified as occurring before dependence, during abstinence, or during substance use.13 Depression increased the risk for suicide in 34% of patients
who had already attempted suicide at least once. Compared with depression absent substance abuse, depression preceding substance use was associated with high vulnerability to additional psychopathology, depressive episodes that were more severe and greater in number, and more suicide attempts. Substance dependence predicted severity of suicidal intent, and abstinence predicted number of attempts.
Psychiatric hospitalizations often involve patients with a history of suicidal thinking or behavior and substance-induced depression. Clinicians can make reliable assessments of the degree to which a presenting psychiatric syndrome is substance-induced.14 These patients require addiction treatment, including outpatient addiction services capable of caring for suicidal persons. These individuals also are more likely to be homeless, unemployed, and uncooperative.15
Taking a psychiatric history and making a detailed inquiry into potential suicidal behavior, recent substance abuse, and current mood symptoms are warranted in persons with depression and/or SUD. Close follow-up is especially important for depressed patients likely to relapse into alcoholism soon after hospital discharge. Failure to recognize MDD or a bipolar disorder in such a patient may result in more relapses, recurrence of mood episodes, and elevated risk of completing suicide.16
Bipolar Clinical Considerations
There is a lack of clarity regarding the effect of moderate-to-excessive alcohol use on the course of bipolar disorders. There is a negative effect on patients with alcohol-induced bipolar depression. In a study of group therapy patients with bipolar disorder co-occurring with substance dependence, data indicated that number of days of alcohol use predicted development of depression a month later.17 These findings were associated with heavy alcohol consumption. In these patients, substantial drinking increased the risk of a depressive episode. In another study, comorbid SUDs were correlated with suboptimal treatment compliance.18 The authors of a 1998 literature review concluded that comorbid SUD makes bipolar symptoms more severe.19
A number of studies have failed to confirm a negative effect of alcohol on bipolar depression.20 There were no differences in 1-year course and outcome between bipolar patients with different alcohol use levels (abstinence, incidental use, moderate abuse, excessive consumption). Other investigators concluded that SUDs were not associated with slower recovery from depression but could contribute to a higher risk of switching to a manic, mixed, or hypomanic state.21
Substance use disorders are associated with increased suicidal behavior in people with a bipolar disorder. The risk of attempted suicide is about double for these patients relative to bipolar patients who do not abuse alcohol.22 Of those who abuse drugs, 14% to 16% complete suicide.23
Psychotherapy
Reportedly, integrated cognitive behavioral therapy (CBT) provided better substance abuse outcomes compared with 12-step programs.24 There also was less substance abuse within the year after CBT. Integrated psychosocial treatment for patients with a mood disorder and substance abuse should involve simultaneous treatment of the 2 conditions. A sequential approach addresses the primary concern and subsequently treats the comorbid disorder, whereas a parallel approach manages both at the same time but in different surroundings. In both approaches, conflicting therapeutic ideologies are a potential difficulty. Given the multiple treatment locations and separate appointments, scheduling problems are an additional difficulty. Coexisting illnesses also are important to consider in the clinical treatment for bipolar patients. As with individual treatments, group therapies take either a sequential approach (more acute disorder treated first) or a parallel approach (disorders treated simultaneously but in separate settings).
Integrated group therapy (IGT) considers patients as having a single diagnosis, focuses on commonalities between relapse and recovery, and reviews the relationship between both conditions. One study compared IGT and treatment as usual in subjects with comorbid bipolar and AUD.25 The IGT group evidenced fewer days of alcohol use. Other research compared IGT with group drug treatment and found that IGT subjects were more likely to remain abstinent.26 This type of psychotherapy showed promise in a meta-analysis of integrated treatment in patients with depression and SUDs.26
Compared with placebo, sertraline/CBT combined treatment reduced alcohol consumption on drinking days.27 This combination was effective in reducing depression, especially in females.
Acceptance and commitment therapy (ACT) combines mindfulness and behavioral change to increase psychological flexibility. The goal in ACT is for patients to become more accepting of their unpleasant feelings. In a study of alcohol abusers with affective disorders, those treated with ACT, compared with controls, had higher abstinence rates and lower depression scores.28
Phamacotherapy and Bipolar Disorder
Even when bipolar symptoms were resolved with use of mood-stabilizing medications, usually some alcohol use continued, though no association was found between bipolar disorder and AUDs.29 With patients’ illness severity and ethanol consumption rated weekly over 7 years, no temporal correlation was found between drinking alcohol and bipolar symptoms.
Similarly, in a study, relief of depressive bipolar symptoms did not result in less frequent alcohol relapse.30 One hundred fifteen outpatients with bipolar disorder and AUD were randomly assigned to either 12 weeks of quetiapine therapy or placebo. Patients in the quetiapine group experienced significant improvement in mood, but sobriety was not enhanced.
Two studies indicated trends of reduced drinking with use of prescribed alcohol-deterrent drugs. An investigation that compared naltrexone with placebo did not reach statistical significance, but naltrexone was reasonably effective in reducing alcohol consumption and craving.31 A report on patients with bipolar disorder treated with acamprosate also did not identify any significant differences in alcohol drinking prognosis.32 Nevertheless, acamprosate was well tolerated and seemed to confer some clinical benefit.
There is a paucity of research focused on patients with bipolar disorder and substance dependence.33 In one trial, patients with bipolar disorder and a diagnosis of alcohol dependence were randomly assigned to receive either valproate or placebo.34 Valproate therapy decreased the number of heavy consumption days and drinks per drinking day in these patients. In a study of 362 patients with bipolar disorder and alcohol dependence treated with lithium or valproic acid, there was no change in drinking days despite adding quetiapine to the regimen.35
Pharmacotherapy and Depression
Lithium is not effective for patients with MDD and AUD. Lithium treatment for depressed patients with alcohol dependence did not improve abstinence, alcohol-related hospitalizations, or severity of either condition.36
Aripiprazole is an antipsychotic that partially agonizes dopamine receptors. Dopamine implicates reward circuitry and has a role in AUDs. Aripiprazole was used as an adjunctive intervention in a randomized trial of 35 patients with comorbid alcohol dependence and depression.37 There was less depression in both the aripiprazole plus escitalopram group and the escitalopram group. Imaging showed a change in activity in the left cingulate gyrus in the patients with comorbid alcohol dependence and MDD. The action of aripiprazole may be mediated through the anterior cingulate cortex.
Research on patients with alcohol dependence treated with fluoxetine found decreased Hamilton Depression Rating Scale (HDRS) scores but no change in alcohol consumption.38
Sertraline diminishes depressive symptoms in abstinent alcoholics. In one study, depressed, recently abstinent alcohol users were randomly assigned to receive sertraline 100 mg daily or placebo.39 Significant improvement was noted in HDRS and Beck Depression Inventory scores at 3- and 6-week intervals.
Citalopram was studied in patients randomly assigned to receive citalopram or placebo for alcohol abuse or dependence.40 Patients in the citalopram group had more days of drinking and showed little change in frequency of alcohol consumption. There was no improvement in depression severity in the citalopram group relative to the placebo group. Citalopram also has been studied in combination with naltrexone.41 Patients with depression and alcohol dependence were randomly assigned to receive either citalopram or placebo, as well as naltrexone. There were no significant differences in depression severity or drinking outcomes.
Treating depression with selective serotonin reuptake inhibitors (SSRIs) had variable results. Most SSRIs improve depression severity but largely have no effect on drinking outcomes.
Antidepressants
A meta-analysis on the efficacy of antidepressant medications in treating patients with depression and substance abuse revealed that the antidepressants had a somewhat advantageous effect.42 That finding was supported by the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study.43 About 33% of patients with citalopramtreated major depression endorsed concurrent SUD symptoms, 19% reported diagnosable alcohol use, 6% had other drug abuses, and 5% exhibited both alcohol and drug use. The groups did not differ in time needed to attain a better mood or in rate of response to citalopram.
Patients with citalopram-treated MDD and alcohol or drug abuse responded about as well as those without an SUD. However, those with alcohol and/or drug abuse had reduced rates of remission, and their remission was delayed, as compared with those without alcohol or drug abuse. There were more suicide attempts and psychiatric hospitalizations among the cohort with drug abuse.
Selective serotonin reuptake inhibitors have a reported safety advantage in treating patients with a history of excessive alcohol intake.44 Another advantage is that SSRIs are seldom abused and seldom lower seizure thresholds significantly. Deleterious effects of alcohol on motor skills or cognition are not potentiated. Adverse effects are usually mild, and overdoses are rarely dangerous.
Antidepressant medication decreased depression and diminished the amount of drinking in patients with depression who use alcohol.45 In controlled research of patients with comorbid depression and alcohol dependence, fluoxetine reduced the severity of these conditions. Substantial reductions in depressive symptoms occurred during detoxification and washout in both groups. There was a strong relationship between depression and drinking among people with depression and AUD.
Desipramine can produce similar results, with positive antidepressant drug effects on depression and drinking. Therefore, pharmacotherapy is indicated for patients with depression who abuse ethanol. Research found that alcohol-dependent patients with depression responded to desipramine.46 Desipramine yielded prolonged abstinence in patients with depression who were using alcohol but not in alcohol users without depression.
A study of imipramine use in actively drinking outpatients found decreased alcohol consumption only for those whose depression responded to treatment.47 However, there was no influence on drinking outcome. Patients whose mood improved reported decreased alcohol consumption after imipramine therapy.
Conslusion
People with co-occurring depression and alcohol dependence are optimally treated with pharmacotherapies that address each condition. One investigation randomly assigned alcohol-dependent patients with depression to 14 weeks of treatment with sertraline 200 mg/d, naltrexone 100 mg/d, a combination of the drugs, or placebo.48 The combination treatment produced the best rate of abstinence before a heavy drinking relapse. Also, fewer patients tended to be depressed in the final weeks of treatment when prescribed the combined regimen. Pharmacotherapy is the best approach for both depression and AUDs.
Click here to read the digital edition.
Co-occurrence of depression and substance abuse often poses diagnostic and therapeutic challenges. This article reviews the prevalence, clinical considerations, and treatment of depression coexisting with alcohol use disorders (AUDs).
Prevalence
Mood and substance use disorders (SUDs) are very common with an estimated lifetime prevalence in the U.S. of 17% for major depression, 4% for bipolar I and II disorders, 13% for alcohol abuse, and 5% for alcohol dependence.1 Almost all of the associations between disorders of mood or anxiety and drug use were positive and statistically significant in the National Epidemiologic Survey, on Alcohol and Related Conditions (NESARC), which included 43,093 noninstitutionalized patients.2
There is a reciprocal relationship between depression and alcoholism. Epidemiologic Catchment Area Survey results indicated that baseline symptoms of depression or alcohol abuse increased the risk of developing alcohol dependence or depression.3 The risk of developing depression were elevated among people with increasing levels of alcohol-induced debility. Conversely, the presence of depressive symptoms increased the chance of developing alcohol dependence. The association between alcohol dependence and depression may be attributable to the depressive effects of ethanol; depression often remits with sobriety. Psychosocial consequences of problem drinking also may contribute to affective illnesses.
Alcohol dependence poses a major depression risk that contributes to higher rates of alcohol use. In people with ethanol dependence, the prevalence of major depressive disorder (MDD) is 21%.4 People who are alcohol dependent are 4 times more likely than are nondependents to have MDD. Forty-one percent of people who seek treatment for current alcohol abuse have a mood disorder.
The NESARC survey revealed strong associations between depression, substance use, and other psychopathologies.5 Compared with MDD alone, SUD combined with MDD conferred high vulnerability to additional psychopathology, depressive episodes that were more severe and greater in number, and more suicide attempts.
Depression Clincal Considerations
Depression linked to recent alcohol abuse may not respond well to an antidepressant drug beyond what is achieved with ethanol abstinence. In one study, depressive symptoms were assessed over the course of alcohol-related hospitalizations.6 Depression was evident in 42% of patients 48 hours after admission, but only 6% remained clinically depressed by week 4 of hospitalization. Therefore, in the treatment of patients hospitalized for alcohol detoxification, it is common to observe them for 1 month before considering antidepressant medication. Mood likely will improve without pharmacotherapy.
However, delaying treatment for depression while a patient is hospitalized for alcohol detoxification presents some difficulties. Many patients do not remain sober during the first month after detoxification. One study found that 65% of patients imbibed alcohol within 2 weeks after discharge.7 Furthermore, 50% relapsed into heavy drinking during the same period. More than 25% of patients who used alcohol and were diagnosed with substance-induced depression at baseline were reclassified with MDD the next year.8
Careful clinical assessment is needed after alcohol detoxification. Depression that persists during ethanol abstinence predisposes a patient to relapse into heavy drinking. Therefore, failure to treat depression after alcohol detoxification poses considerable risk.9 A study of the effect of depression on the return to drinking among patients with alcohol dependence found that depression at entry into inpatient treatment for alcohol dependence predicted a shorter time to first drink.9 The prognosis for a drinking relapse was worse no matter whether the depression came first or was triggered by the alcohol. Depression does not predict drinking outcomes, but it is associated with a more rapid relapse to ethanol consumption.
Similarly, inpatients with premorbid or substanceinduced depression were more likely to meet the criteria for drug dependence during outpatient follow-up.10 In addition, patients who developed depression during the first 26 weeks after hospitalization were 3 times more likely than those without depression to relapse into drug dependence during follow-up.
Alcohol dependence may hasten the progression of depression. A study on the prognostic effect of alcoholism on the 10-year course of depression found a deleterious influence of current alcoholism after recovery from depression.11 Patients with MDD were more likely to transition from being ill to improving if either they were forgoing alcohol or had never abused it. Another investigation verified that alcohol and drug dependence increased perceptions of affective symptomatology.12
Substance-induced depression also increases the risk for suicide. In 602 patients with substance dependence, depression was classified as occurring before dependence, during abstinence, or during substance use.13 Depression increased the risk for suicide in 34% of patients
who had already attempted suicide at least once. Compared with depression absent substance abuse, depression preceding substance use was associated with high vulnerability to additional psychopathology, depressive episodes that were more severe and greater in number, and more suicide attempts. Substance dependence predicted severity of suicidal intent, and abstinence predicted number of attempts.
Psychiatric hospitalizations often involve patients with a history of suicidal thinking or behavior and substance-induced depression. Clinicians can make reliable assessments of the degree to which a presenting psychiatric syndrome is substance-induced.14 These patients require addiction treatment, including outpatient addiction services capable of caring for suicidal persons. These individuals also are more likely to be homeless, unemployed, and uncooperative.15
Taking a psychiatric history and making a detailed inquiry into potential suicidal behavior, recent substance abuse, and current mood symptoms are warranted in persons with depression and/or SUD. Close follow-up is especially important for depressed patients likely to relapse into alcoholism soon after hospital discharge. Failure to recognize MDD or a bipolar disorder in such a patient may result in more relapses, recurrence of mood episodes, and elevated risk of completing suicide.16
Bipolar Clinical Considerations
There is a lack of clarity regarding the effect of moderate-to-excessive alcohol use on the course of bipolar disorders. There is a negative effect on patients with alcohol-induced bipolar depression. In a study of group therapy patients with bipolar disorder co-occurring with substance dependence, data indicated that number of days of alcohol use predicted development of depression a month later.17 These findings were associated with heavy alcohol consumption. In these patients, substantial drinking increased the risk of a depressive episode. In another study, comorbid SUDs were correlated with suboptimal treatment compliance.18 The authors of a 1998 literature review concluded that comorbid SUD makes bipolar symptoms more severe.19
A number of studies have failed to confirm a negative effect of alcohol on bipolar depression.20 There were no differences in 1-year course and outcome between bipolar patients with different alcohol use levels (abstinence, incidental use, moderate abuse, excessive consumption). Other investigators concluded that SUDs were not associated with slower recovery from depression but could contribute to a higher risk of switching to a manic, mixed, or hypomanic state.21
Substance use disorders are associated with increased suicidal behavior in people with a bipolar disorder. The risk of attempted suicide is about double for these patients relative to bipolar patients who do not abuse alcohol.22 Of those who abuse drugs, 14% to 16% complete suicide.23
Psychotherapy
Reportedly, integrated cognitive behavioral therapy (CBT) provided better substance abuse outcomes compared with 12-step programs.24 There also was less substance abuse within the year after CBT. Integrated psychosocial treatment for patients with a mood disorder and substance abuse should involve simultaneous treatment of the 2 conditions. A sequential approach addresses the primary concern and subsequently treats the comorbid disorder, whereas a parallel approach manages both at the same time but in different surroundings. In both approaches, conflicting therapeutic ideologies are a potential difficulty. Given the multiple treatment locations and separate appointments, scheduling problems are an additional difficulty. Coexisting illnesses also are important to consider in the clinical treatment for bipolar patients. As with individual treatments, group therapies take either a sequential approach (more acute disorder treated first) or a parallel approach (disorders treated simultaneously but in separate settings).
Integrated group therapy (IGT) considers patients as having a single diagnosis, focuses on commonalities between relapse and recovery, and reviews the relationship between both conditions. One study compared IGT and treatment as usual in subjects with comorbid bipolar and AUD.25 The IGT group evidenced fewer days of alcohol use. Other research compared IGT with group drug treatment and found that IGT subjects were more likely to remain abstinent.26 This type of psychotherapy showed promise in a meta-analysis of integrated treatment in patients with depression and SUDs.26
Compared with placebo, sertraline/CBT combined treatment reduced alcohol consumption on drinking days.27 This combination was effective in reducing depression, especially in females.
Acceptance and commitment therapy (ACT) combines mindfulness and behavioral change to increase psychological flexibility. The goal in ACT is for patients to become more accepting of their unpleasant feelings. In a study of alcohol abusers with affective disorders, those treated with ACT, compared with controls, had higher abstinence rates and lower depression scores.28
Phamacotherapy and Bipolar Disorder
Even when bipolar symptoms were resolved with use of mood-stabilizing medications, usually some alcohol use continued, though no association was found between bipolar disorder and AUDs.29 With patients’ illness severity and ethanol consumption rated weekly over 7 years, no temporal correlation was found between drinking alcohol and bipolar symptoms.
Similarly, in a study, relief of depressive bipolar symptoms did not result in less frequent alcohol relapse.30 One hundred fifteen outpatients with bipolar disorder and AUD were randomly assigned to either 12 weeks of quetiapine therapy or placebo. Patients in the quetiapine group experienced significant improvement in mood, but sobriety was not enhanced.
Two studies indicated trends of reduced drinking with use of prescribed alcohol-deterrent drugs. An investigation that compared naltrexone with placebo did not reach statistical significance, but naltrexone was reasonably effective in reducing alcohol consumption and craving.31 A report on patients with bipolar disorder treated with acamprosate also did not identify any significant differences in alcohol drinking prognosis.32 Nevertheless, acamprosate was well tolerated and seemed to confer some clinical benefit.
There is a paucity of research focused on patients with bipolar disorder and substance dependence.33 In one trial, patients with bipolar disorder and a diagnosis of alcohol dependence were randomly assigned to receive either valproate or placebo.34 Valproate therapy decreased the number of heavy consumption days and drinks per drinking day in these patients. In a study of 362 patients with bipolar disorder and alcohol dependence treated with lithium or valproic acid, there was no change in drinking days despite adding quetiapine to the regimen.35
Pharmacotherapy and Depression
Lithium is not effective for patients with MDD and AUD. Lithium treatment for depressed patients with alcohol dependence did not improve abstinence, alcohol-related hospitalizations, or severity of either condition.36
Aripiprazole is an antipsychotic that partially agonizes dopamine receptors. Dopamine implicates reward circuitry and has a role in AUDs. Aripiprazole was used as an adjunctive intervention in a randomized trial of 35 patients with comorbid alcohol dependence and depression.37 There was less depression in both the aripiprazole plus escitalopram group and the escitalopram group. Imaging showed a change in activity in the left cingulate gyrus in the patients with comorbid alcohol dependence and MDD. The action of aripiprazole may be mediated through the anterior cingulate cortex.
Research on patients with alcohol dependence treated with fluoxetine found decreased Hamilton Depression Rating Scale (HDRS) scores but no change in alcohol consumption.38
Sertraline diminishes depressive symptoms in abstinent alcoholics. In one study, depressed, recently abstinent alcohol users were randomly assigned to receive sertraline 100 mg daily or placebo.39 Significant improvement was noted in HDRS and Beck Depression Inventory scores at 3- and 6-week intervals.
Citalopram was studied in patients randomly assigned to receive citalopram or placebo for alcohol abuse or dependence.40 Patients in the citalopram group had more days of drinking and showed little change in frequency of alcohol consumption. There was no improvement in depression severity in the citalopram group relative to the placebo group. Citalopram also has been studied in combination with naltrexone.41 Patients with depression and alcohol dependence were randomly assigned to receive either citalopram or placebo, as well as naltrexone. There were no significant differences in depression severity or drinking outcomes.
Treating depression with selective serotonin reuptake inhibitors (SSRIs) had variable results. Most SSRIs improve depression severity but largely have no effect on drinking outcomes.
Antidepressants
A meta-analysis on the efficacy of antidepressant medications in treating patients with depression and substance abuse revealed that the antidepressants had a somewhat advantageous effect.42 That finding was supported by the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study.43 About 33% of patients with citalopramtreated major depression endorsed concurrent SUD symptoms, 19% reported diagnosable alcohol use, 6% had other drug abuses, and 5% exhibited both alcohol and drug use. The groups did not differ in time needed to attain a better mood or in rate of response to citalopram.
Patients with citalopram-treated MDD and alcohol or drug abuse responded about as well as those without an SUD. However, those with alcohol and/or drug abuse had reduced rates of remission, and their remission was delayed, as compared with those without alcohol or drug abuse. There were more suicide attempts and psychiatric hospitalizations among the cohort with drug abuse.
Selective serotonin reuptake inhibitors have a reported safety advantage in treating patients with a history of excessive alcohol intake.44 Another advantage is that SSRIs are seldom abused and seldom lower seizure thresholds significantly. Deleterious effects of alcohol on motor skills or cognition are not potentiated. Adverse effects are usually mild, and overdoses are rarely dangerous.
Antidepressant medication decreased depression and diminished the amount of drinking in patients with depression who use alcohol.45 In controlled research of patients with comorbid depression and alcohol dependence, fluoxetine reduced the severity of these conditions. Substantial reductions in depressive symptoms occurred during detoxification and washout in both groups. There was a strong relationship between depression and drinking among people with depression and AUD.
Desipramine can produce similar results, with positive antidepressant drug effects on depression and drinking. Therefore, pharmacotherapy is indicated for patients with depression who abuse ethanol. Research found that alcohol-dependent patients with depression responded to desipramine.46 Desipramine yielded prolonged abstinence in patients with depression who were using alcohol but not in alcohol users without depression.
A study of imipramine use in actively drinking outpatients found decreased alcohol consumption only for those whose depression responded to treatment.47 However, there was no influence on drinking outcome. Patients whose mood improved reported decreased alcohol consumption after imipramine therapy.
Conslusion
People with co-occurring depression and alcohol dependence are optimally treated with pharmacotherapies that address each condition. One investigation randomly assigned alcohol-dependent patients with depression to 14 weeks of treatment with sertraline 200 mg/d, naltrexone 100 mg/d, a combination of the drugs, or placebo.48 The combination treatment produced the best rate of abstinence before a heavy drinking relapse. Also, fewer patients tended to be depressed in the final weeks of treatment when prescribed the combined regimen. Pharmacotherapy is the best approach for both depression and AUDs.
Click here to read the digital edition.
1. Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Conway KP, Compton W, Stinson FS, Grant BF. Lifetime comorbidity of DSM-IV mood and anxiety disorders and specific drug use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2006;67(2):247-257.
3. Gilman SE, Abraham HD. A longitudinal study of the order of onset of alcohol dependence and major depression. Drug Alcohol Depend. 2001;63(3):277-286.
4. Kessler RC, Crum RM, Warner LA, Nelson CB, Schulenberg J, Anthony JC. Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
5. Blanco C, Alegría AA, Liu SM, et al. Differences among major depressive disorder with and without co-occurring substance use disorders and substance-induced depressive disorder: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2012;73(6):865-873.
6. Brown SA, Schuckit MA. Changes in depression among abstinent alcoholics. J Stud Alcohol. 1988;49(5):412-417.
7. Kiefer F, Jahn H, Tarnaske T, et al. Comparing and combining naltrexone and acamprosate in relapse prevention of alcoholism: a double-blind, placebo-controlled study. Arch Gen Psychiatry. 2003;60(1):92-99.
8. Ramsey SE, Kahler CW, Read JP, Stuart GL, Brown RA. Discriminating between substance-induced and independent depressive episodes in alcohol-dependent patients. J Stud Alcohol. 2004;65(5):672-676.
9. Greenfield SF, Weiss RD, Muenz LR, et al. The effect of depression on return to drinking: a prospective study. Arch Gen Psychiatry. 1998;55(3):259-265.
10. Hasin D, Liu X, Nunes E, McCloud S, Samet S, Endicott J. Effects of major depression on remission and relapse of substance dependence. Arch Gen Psychiatry. 2002;59(4):375-380.
11. Mueller TI, Lavori PW, Martin B, et al. Prognostic effect of the variable course of alcoholism on the 10-year course of depression. Am J Psychiatry. 1994;151(5):701-706.
12. Agosti V, Levin FR. The effects of alcohol and drug dependence on the course of depression. Am J Addict. 2006;15(1):71-75.
13. Aharonovich E, Liu X, Nunes E, Hasin DS. Suicide attempts in substance abusers: effects of major depression in relation to substance use disorders. Am J Psychiatry. 2002;159(9):1600-1602.
14. Ries RK, Demirsoy A, Russo JE, Barrett J, Roy-Byrne PP. Reliability and clinical utility of DSM-IV substance-induced psychiatric disorders in acute psychiatric inpatients. Am J Addict. 2001;10(4):308-318.
15. Ries RK, Yuodelis-Flores C, Comtois KA, Roy-Byrne PP, Russo JE. Substanceinduced suicidal admissions to an acute psychiatric service: characteristics and outcomes. J Subst Abuse Treat. 2008;34(1):72-79.
16. Toliver BK, Anton RF. Assessment and treatment of mood disorders in the context of substance abuse. Dialogues Clin Neurosci. 2015;17(2):181-190.
17. Jaffee WB, Griffin ML, Gallop R, et al. Depression precipitated by alcohol use in patients with co-occurring bipolar and substance use disorders. J Clin Psychiatry. 2009;70(2):171-176.
18. Manwani SG, Szilagyi KA, Zablotsky B, Hennen J, Griffin ML, Weiss RD. Adherence to pharmacotherapy in bipolar disorder patients with and without co-occurring substance use disorders. J Clin Psychiatry. 2007;68(8):1172-1176.
19. Tohen M, Greenfield SF, Weiss RD, Zarate CA Jr, Vagge LM. The effect of comorbid substance disorders on the course of bipolar disorder: a review. Harv Rev Psychiatry. 1998;6(3):133-141.
20. van Zaane J, van den Brink W, Draisma S, Smit JH, Nolen WA. The effect of moderate and excessive alcohol use on the course and outcome of patients with bipolar disorders: a prospective cohort study. J Clin Psychiatry. 2010;71(7):885-893.
21. Ostacher MJ, Perlis RH, Nierenberg AA, et al; STEP-BD Investigators. Impact of substance use disorders on recovery from episodes of depression in bipolar disorder patients: prospective data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). Am J Psychiatry. 2010;167(3):289-297.
22. Oquendo MA, Currier D, Liu SM, Hasin DS, Grant BF, Blanco C. Increased risk for suicidal behavior in comorbid bipolar disorder and alcohol use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). J Clin Psychiatry. 2010;71(7):902-909.
23. Yoon YH, Chen CM, Yi HY, Moss HB. Effect of comorbid alcohol and drug use disorders on premature death of unipolar and bipolar decedents in the United States, 1999 to 2006. Compr Psychiatry. 2011;52(5):453-464.
24. Lydecker KP, Tate SR, Cummins KM, McQuaid J, Granholm E, Brown SA. Clinical outcomes of an integrated treatment for depression and substance use disorders. Psychol Addict Behav. 2010;24(3):453-465.
25. Weiss RD, Griffin ML, Kolodziej ME, et al. A randomized trial of integrated group therapy versus group drug counseling for patients with bipolar disorder and substance dependence. Am J Psychiatry. 2007;164(1):100-107.
26. Hesse M. Integrated psychological treatment for substance use and co-morbid anxiety or depression vs. treatment for substance use alone. A systematic review of the published literature. BMC Psychiatry. 2009;9:6.
27. Moak DH, Anton RF, Latham PK, Voronin KE, Waid RL, Durazo-Arvizu R. Sertraline and cognitive behavioral therapy for depressed alcoholics: results of a placebo-controlled trial. J Clin Psychopharmacol. 2003;23(6):553-562.
28. Thekiso TB, Murphy P, Milnes J, Lambe K, Curtin A, Farren CK. Acceptance and commitment therapy in the treatment of alcohol use disorder and comorbid affective disorder: a pilot matched control trial. Behav Ther. 2015;46(6):717-728.
29. Fleck DE, Amdt S, Delbello MP, Strakowski SM. Concurrent tracking of alcohol use and bipolar disorder symptoms. Bipolar Disord. 2006:8(4):338-344.
30. Brown ES, Gaza M, Carmody TJ. A randomized, double-blind, placebo-controlled add-on trial of quetiapine in outpatients with bipolar disorder and alcohol use disorders. J Clin Psychiatry. 2008;69(5):701-705.
31. Brown ES, Carmody TJ, Schmitz JM, et al. A randomized, double-blind, placebocontrolled pilot study of naltrexone in outpatients with bipolar disorder and alcohol dependence. Alcohol Clin Exp Res. 2009;33(11):1863-1869.
32. Tolliver BK, Desantis SM, Brown DG, Prisciandaro JJ, Brady KT. A randomized, double-blind, placebo-controlled clinical trial of acamprosate in alcoholdependent individuals with bipolar disorder: a preliminary report. Bipolar Disord. 2012;14(1):54-63.
33. Pettinati HM, O’Brien CP, Dundon WD. Current status of co-occurring mood and substance use disorders: a new therapeutic target. Am J Psychiatry. 2013;170(1):23-30.
34. Salloum IM, Cornelius JR, Daley DC, Kirisci L, Himmelhoch JM, Thase ME. Efficacy of valproate maintenance in patients with bipolar disorder and alcoholism: a double-blind placebo-controlled study. Arch Gen Psychiatry. 2005;62(1):37-45.
35. Farren CK, Hill KP, Weiss RD. Bipolar disorder and alcohol use disorder: a review. Curr Psychiatry Rep. 2012;14(6):659-666.
36. Dorus W, Ostrow DG, Anton R, et al. Lithium treatment of depressed and nondepressed alcoholics. JAMA. 1989;262(12):1646-1652.
37. Han DH, Kim SM, Choi JE, Min KJ, Renshaw PF. Adjunctive aripiprazole therapy with escitalopram in patients with co-morbid major depressive disorder and alcohol dependence: clinical and neuroimaging evidence. J Psychopharmacol. 2013;27(3):282-291.
38. Kranzler HR, Burleson JA, Korner P, et al. Placebo-controlled trial of fluoxetine as an adjunct to relapse prevention in alcoholics. Am J Psychiatry. 1995;152(3):391-397.
39. Roy A. Placebo-controlled study of sertraline in depressed recently abstinent alcoholics. Biol Psychiatry. 1998;44(7):633-637.
40. Charney DA, Heath LM, Zikos E, Palacios-Boix J, Gill KJ. Poorer drinking outcomes with citalopram treatment for alcohol dependence: a randomized, doubleblind, placebo-controlled trial. Alcohol Clin Exp Res. 2015;39(9):1756-1765.
41. Adamson SJ, Sellman JD, Foulds JA, et al. A randomized trial of combined citalopram and naltrexone for non-abstinent outpatients with co-occurring alcohol dependence and major depression. J Clin Psychopharmacol. 2015;35(2):143-149.
42. Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
43. Davis LL, Wisniewski SR, Howland RH, et al. Does comorbid substance use disorder impair recovery from major depression with SSRI treatment? An analysis of the STAR*D level one treatment outcomes. Drug Alcohol Depend. 2010;107(2-3):161-170.
44. Pettinati HM. The use of selective reuptake inhibitors in treating alcoholic subtypes. J Clin Psychiatry. 2001;62(suppl 20):26-31.
45. Cornelius JR, Salloum IM, Ehler JG, et al. Fluoxetine in depressed alcoholics. A double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1997;54(8):700-705.
46. Mason BJ, Kocsis JH, Ritvo EC, Cutler RB. A double-blind, placebo-controlled trial of desipramine for primary alcohol dependence stratified on the presence of absence of major depression. JAMA. 1996;275(10):761-767.
47. McGrath PJ, Nunes EV, Stewart JW, et al. Imipramine treatment of alcoholics with primary depression: a placebo-controlled clinical trial. Arch Gen Psychiatry. 1996;53(3):232-240.
48. Pettinati HM, Oslin DW, Kampman KM, et al. A double-blind, placebo-controlled trial combining sertraline and naltrexone for treating co-occurring depression and alcohol dependence. Am J Psychiatry. 2010;167(6):668-675.
1. Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Conway KP, Compton W, Stinson FS, Grant BF. Lifetime comorbidity of DSM-IV mood and anxiety disorders and specific drug use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2006;67(2):247-257.
3. Gilman SE, Abraham HD. A longitudinal study of the order of onset of alcohol dependence and major depression. Drug Alcohol Depend. 2001;63(3):277-286.
4. Kessler RC, Crum RM, Warner LA, Nelson CB, Schulenberg J, Anthony JC. Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
5. Blanco C, Alegría AA, Liu SM, et al. Differences among major depressive disorder with and without co-occurring substance use disorders and substance-induced depressive disorder: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2012;73(6):865-873.
6. Brown SA, Schuckit MA. Changes in depression among abstinent alcoholics. J Stud Alcohol. 1988;49(5):412-417.
7. Kiefer F, Jahn H, Tarnaske T, et al. Comparing and combining naltrexone and acamprosate in relapse prevention of alcoholism: a double-blind, placebo-controlled study. Arch Gen Psychiatry. 2003;60(1):92-99.
8. Ramsey SE, Kahler CW, Read JP, Stuart GL, Brown RA. Discriminating between substance-induced and independent depressive episodes in alcohol-dependent patients. J Stud Alcohol. 2004;65(5):672-676.
9. Greenfield SF, Weiss RD, Muenz LR, et al. The effect of depression on return to drinking: a prospective study. Arch Gen Psychiatry. 1998;55(3):259-265.
10. Hasin D, Liu X, Nunes E, McCloud S, Samet S, Endicott J. Effects of major depression on remission and relapse of substance dependence. Arch Gen Psychiatry. 2002;59(4):375-380.
11. Mueller TI, Lavori PW, Martin B, et al. Prognostic effect of the variable course of alcoholism on the 10-year course of depression. Am J Psychiatry. 1994;151(5):701-706.
12. Agosti V, Levin FR. The effects of alcohol and drug dependence on the course of depression. Am J Addict. 2006;15(1):71-75.
13. Aharonovich E, Liu X, Nunes E, Hasin DS. Suicide attempts in substance abusers: effects of major depression in relation to substance use disorders. Am J Psychiatry. 2002;159(9):1600-1602.
14. Ries RK, Demirsoy A, Russo JE, Barrett J, Roy-Byrne PP. Reliability and clinical utility of DSM-IV substance-induced psychiatric disorders in acute psychiatric inpatients. Am J Addict. 2001;10(4):308-318.
15. Ries RK, Yuodelis-Flores C, Comtois KA, Roy-Byrne PP, Russo JE. Substanceinduced suicidal admissions to an acute psychiatric service: characteristics and outcomes. J Subst Abuse Treat. 2008;34(1):72-79.
16. Toliver BK, Anton RF. Assessment and treatment of mood disorders in the context of substance abuse. Dialogues Clin Neurosci. 2015;17(2):181-190.
17. Jaffee WB, Griffin ML, Gallop R, et al. Depression precipitated by alcohol use in patients with co-occurring bipolar and substance use disorders. J Clin Psychiatry. 2009;70(2):171-176.
18. Manwani SG, Szilagyi KA, Zablotsky B, Hennen J, Griffin ML, Weiss RD. Adherence to pharmacotherapy in bipolar disorder patients with and without co-occurring substance use disorders. J Clin Psychiatry. 2007;68(8):1172-1176.
19. Tohen M, Greenfield SF, Weiss RD, Zarate CA Jr, Vagge LM. The effect of comorbid substance disorders on the course of bipolar disorder: a review. Harv Rev Psychiatry. 1998;6(3):133-141.
20. van Zaane J, van den Brink W, Draisma S, Smit JH, Nolen WA. The effect of moderate and excessive alcohol use on the course and outcome of patients with bipolar disorders: a prospective cohort study. J Clin Psychiatry. 2010;71(7):885-893.
21. Ostacher MJ, Perlis RH, Nierenberg AA, et al; STEP-BD Investigators. Impact of substance use disorders on recovery from episodes of depression in bipolar disorder patients: prospective data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). Am J Psychiatry. 2010;167(3):289-297.
22. Oquendo MA, Currier D, Liu SM, Hasin DS, Grant BF, Blanco C. Increased risk for suicidal behavior in comorbid bipolar disorder and alcohol use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). J Clin Psychiatry. 2010;71(7):902-909.
23. Yoon YH, Chen CM, Yi HY, Moss HB. Effect of comorbid alcohol and drug use disorders on premature death of unipolar and bipolar decedents in the United States, 1999 to 2006. Compr Psychiatry. 2011;52(5):453-464.
24. Lydecker KP, Tate SR, Cummins KM, McQuaid J, Granholm E, Brown SA. Clinical outcomes of an integrated treatment for depression and substance use disorders. Psychol Addict Behav. 2010;24(3):453-465.
25. Weiss RD, Griffin ML, Kolodziej ME, et al. A randomized trial of integrated group therapy versus group drug counseling for patients with bipolar disorder and substance dependence. Am J Psychiatry. 2007;164(1):100-107.
26. Hesse M. Integrated psychological treatment for substance use and co-morbid anxiety or depression vs. treatment for substance use alone. A systematic review of the published literature. BMC Psychiatry. 2009;9:6.
27. Moak DH, Anton RF, Latham PK, Voronin KE, Waid RL, Durazo-Arvizu R. Sertraline and cognitive behavioral therapy for depressed alcoholics: results of a placebo-controlled trial. J Clin Psychopharmacol. 2003;23(6):553-562.
28. Thekiso TB, Murphy P, Milnes J, Lambe K, Curtin A, Farren CK. Acceptance and commitment therapy in the treatment of alcohol use disorder and comorbid affective disorder: a pilot matched control trial. Behav Ther. 2015;46(6):717-728.
29. Fleck DE, Amdt S, Delbello MP, Strakowski SM. Concurrent tracking of alcohol use and bipolar disorder symptoms. Bipolar Disord. 2006:8(4):338-344.
30. Brown ES, Gaza M, Carmody TJ. A randomized, double-blind, placebo-controlled add-on trial of quetiapine in outpatients with bipolar disorder and alcohol use disorders. J Clin Psychiatry. 2008;69(5):701-705.
31. Brown ES, Carmody TJ, Schmitz JM, et al. A randomized, double-blind, placebocontrolled pilot study of naltrexone in outpatients with bipolar disorder and alcohol dependence. Alcohol Clin Exp Res. 2009;33(11):1863-1869.
32. Tolliver BK, Desantis SM, Brown DG, Prisciandaro JJ, Brady KT. A randomized, double-blind, placebo-controlled clinical trial of acamprosate in alcoholdependent individuals with bipolar disorder: a preliminary report. Bipolar Disord. 2012;14(1):54-63.
33. Pettinati HM, O’Brien CP, Dundon WD. Current status of co-occurring mood and substance use disorders: a new therapeutic target. Am J Psychiatry. 2013;170(1):23-30.
34. Salloum IM, Cornelius JR, Daley DC, Kirisci L, Himmelhoch JM, Thase ME. Efficacy of valproate maintenance in patients with bipolar disorder and alcoholism: a double-blind placebo-controlled study. Arch Gen Psychiatry. 2005;62(1):37-45.
35. Farren CK, Hill KP, Weiss RD. Bipolar disorder and alcohol use disorder: a review. Curr Psychiatry Rep. 2012;14(6):659-666.
36. Dorus W, Ostrow DG, Anton R, et al. Lithium treatment of depressed and nondepressed alcoholics. JAMA. 1989;262(12):1646-1652.
37. Han DH, Kim SM, Choi JE, Min KJ, Renshaw PF. Adjunctive aripiprazole therapy with escitalopram in patients with co-morbid major depressive disorder and alcohol dependence: clinical and neuroimaging evidence. J Psychopharmacol. 2013;27(3):282-291.
38. Kranzler HR, Burleson JA, Korner P, et al. Placebo-controlled trial of fluoxetine as an adjunct to relapse prevention in alcoholics. Am J Psychiatry. 1995;152(3):391-397.
39. Roy A. Placebo-controlled study of sertraline in depressed recently abstinent alcoholics. Biol Psychiatry. 1998;44(7):633-637.
40. Charney DA, Heath LM, Zikos E, Palacios-Boix J, Gill KJ. Poorer drinking outcomes with citalopram treatment for alcohol dependence: a randomized, doubleblind, placebo-controlled trial. Alcohol Clin Exp Res. 2015;39(9):1756-1765.
41. Adamson SJ, Sellman JD, Foulds JA, et al. A randomized trial of combined citalopram and naltrexone for non-abstinent outpatients with co-occurring alcohol dependence and major depression. J Clin Psychopharmacol. 2015;35(2):143-149.
42. Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
43. Davis LL, Wisniewski SR, Howland RH, et al. Does comorbid substance use disorder impair recovery from major depression with SSRI treatment? An analysis of the STAR*D level one treatment outcomes. Drug Alcohol Depend. 2010;107(2-3):161-170.
44. Pettinati HM. The use of selective reuptake inhibitors in treating alcoholic subtypes. J Clin Psychiatry. 2001;62(suppl 20):26-31.
45. Cornelius JR, Salloum IM, Ehler JG, et al. Fluoxetine in depressed alcoholics. A double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1997;54(8):700-705.
46. Mason BJ, Kocsis JH, Ritvo EC, Cutler RB. A double-blind, placebo-controlled trial of desipramine for primary alcohol dependence stratified on the presence of absence of major depression. JAMA. 1996;275(10):761-767.
47. McGrath PJ, Nunes EV, Stewart JW, et al. Imipramine treatment of alcoholics with primary depression: a placebo-controlled clinical trial. Arch Gen Psychiatry. 1996;53(3):232-240.
48. Pettinati HM, Oslin DW, Kampman KM, et al. A double-blind, placebo-controlled trial combining sertraline and naltrexone for treating co-occurring depression and alcohol dependence. Am J Psychiatry. 2010;167(6):668-675.
A 95-year-old man with treatment-resistant depression
CASE Depressed, avoidant
Mr. R, age 95, has a history of recurrent major depressive disorder. He presents to the emergency department with depressive symptoms that began 6 weeks ago. His symptoms include depressed mood, hopelessness, anhedonia, anxiety, and insomnia. Co-occurring anorexia nervosa has resulted in a 20-lb weight loss. He denies suicidal ideation.
A mental status examination reveals profound psychomotor agitation, dysphoric mood, tearfulness, and mood-congruent delusions. Mr. R’s Mini-Mental State Examination (MMSE) score is 14/30; his Hamilton Depression Rating Scale (HAM-D) score is 21, indicating severe depression (19 to 22). However, the examiner feels that these scores may not reflect an accurate assessment because Mr. R gave flippant responses and did not cooperate during the interview. Physical examination is unremarkable. Previous medication trials included buspirone, escitalopram, and risperidone; none of these medications successfully alleviated his depressive symptoms.
On admission, Mr. R is given oral mirtazapine, 15 mg/d, and quetiapine, 25 mg/d, to target depressive mood, insomnia, and weight loss. Urgent intervention is indicated because his depressive symptoms are profoundly causing failure to thrive and are compromising his physical health. Mr. R’s deterioration concerns the physician team. Because of a history of failed pharmacotherapy trials, the team reassesses Mr. R’s treatment options.
[polldaddy:9903171]
The authors’ observations
The physician team recommends that Mr. R undergo ECT to obtain rapid relief from his depressive symptoms. After discussion of the potential risks and benefits, Mr. R agrees to this treatment. Quetiapine is discontinued prior to initiating ECT to avoid unnecessary medications; mirtazapine is continued.
Mr. R’s lack of response to previous antidepressants and significant deterioration were concerning. The physicians wanted to avoid higher-dose medications because of the risk of falls or somnolence. Their clinical experience and the literature supporting ECT for patients of Mr. R’s age lead them to select ECT as the most appropriate therapeutic option.
ECT has no absolute contraindications.1 The rate of ECT use in the United States has fluctuated over time because of factors unrelated to the efficacy and availability of ECT or alternative treatments.2 This form of intervention is also somewhat stigmatized.
Some psychiatrists are reluctant to prescribe ECT for geriatric patients because of concerns of potential neurocognitive or medical complications and risks during anesthesia. However, in the United States, older patients with depression are more likely to be treated with ECT than their younger counterparts.3 ECT usually induces greater immediate efficacy than antidepressants.4
Evidence supports using ECT in older patients
Multiple studies have found that ECT is a rapid, safe, and efficacious intervention for treating older persons with depression. Patients age >60 who receive ECT plus pharmacotherapy have lower HAM-D scores than those receiving pharmacotherapy alone.5 Overall, the rates of remission for depression range from 50% to 70%; yet geriatric patients who receive only ECT have response rates around 90%.6 Older age, presence of psychotic symptoms, and shorter duration of illness can predict a rapidly positive ECT response.7
When treated with ECT, older patients, including those age >85, have fewer subsequent episodes of depression compared with those who receive pharmacotherapy alone.1 Older individuals with physical illness or cognitive impairment respond to and tolerate ECT much like younger patients.6 Older patients receiving ECT may experience less cognitive decline than younger ones.7 Those in their ninth decade of life with treatment-resistant depression, psychotic features, and post-stroke depression often respond robustly with improvement following ECT.8
Remission rates also depend on the technique of administration. Interactions between electrode placement and stimulus parameter dosage affect efficacy and adverse effects.9 Right-sided, unilateral ECT induces less cognitive dysfunction compared with bilateral electrode placement,9 but bilateral ECT is more clinically effective.10 However, the efficacy of right-sided ECT is more dose-sensitive, and some data suggest that suboptimal response is due to insufficient stimulus dosages.11 One double-blind randomized controlled trial documented that when using a high-dose stimulus parameter, unilateral ECT is as effective as bilateral ECT.12 When there is a suboptimal response to unilateral ECT, bilateral ECT might be beneficial.12,13 For preventing relapse in older patients, increasing the interval between ECT treatments is more effective than stopping ECT abruptly.13
[polldaddy:9903172]

Indications of ECT

ECT is indicated for patients with severe depression, mania, and other conditions (Table).14 The most common indication for ECT in older persons is a history of treatment-resistant depression, with melancholia, psychosis, or suicidal ideations.1-6,12 There are also age-related and clinical factors to consider with ECT. This treatment provides a safe, rapid remission for patients age >65, even after adjusting for somatic conditions, duration of illness, medication resistance, or case severity.15 Compared with younger patients, older adults may not tolerate antidepressants as well because of age-related pharmacokinetic alterations, including increased sensitivity to anticholinergic and/or hypotensive effects.1
Factors that favor ECT include a previous good response to it; patient preference; and an indication for rapid intervention, such as suicidality, catatonia, dehydration, malnutrition, or a suboptimal result from pharmacotherapy.3 Mortality among individuals age >85 who receive ECT reportedly is lower than that among their counterparts who receive alternative treatments.16 ECT has been administered safely and effectively in patients with comorbid medical illnesses such as stroke, cerebral aneurysm, cardiovascular disease with ischemia or arrhythmia, dementia, and osteoporosis.17
Neurocognitive effects
Reports on the effects of ECT on neurocognitive functioning have varied. In some studies, performance improved or did not change in severely depressed older patients who received ECT.18,19 In older people who receive ECT, MMSE scores often return to baseline by the end of treatment.20 There often is only mild transient cognitive impairment in patients with late-life depression who receive ECT. Areas of concern include attention span, orientation, and speed of mental processing.20 Physicians should conduct cognitive tests before, during, and after ECT sessions to monitor their patient’s mental status.20
Cognitive stability can be maintained by administering ECT twice a week; applying right-sided, unilateral electrode placement; and using short, ultra-brief stimulus pulse width parameters.21 Cognitive impairment induced by ECT is not associated with age in geriatric patients with depression.22 Older adults who experienced longer postictal reorientation time periods have better outcomes than others who reach orientation faster; their intellectual impairment returned to baseline.20 Falling is another complication associated with ECT. A longitudinal cohort study found the incident of falls among patients receiving ECT was 13%.22 Risk factors for falls during a course of ECT include the number of treatments and the presence of coexisting Parkinson’s disease.23
OUTCOME Improvement
Mr. R receives 8 sessions of right-sided, unilateral ECT with an individualized dosage titration method. Treatments are completed with a stimulus intensity at 6 times seizure threshold, with an ultra-brief pulse width at 0.3 milliseconds. Mr. R’s mood and affect begin to improve after 3 ECT sessions. His MMSE score increases to 28/30 (Figure). His clinical improvement is progressively sustained; he develops an increasingly jovial attitude and experiences less anxiety. Mr. R’s confidence, appetite, and sleep also improve. There are no complications with treatment, and Mr. R has no complaints. After 8 ECT sessions, Mr. R has no affective symptoms and does not experience any cognitive impairment.
The authors’ observations
Depression among older people is a growing public health concern. It is a leading cause of disability, and often leads to nursing home placement.24 ECT is a safe, effective treatment for late-life depression, but is underutilized in patients age >75 because of concerns for cognitive impairment.6 However, there is evidence that response rates to ECT are higher in patients ages 45 to 85, compared with young individuals ages 18 to 45.25 ECT is a viable intervention for older depressed patients, particularly for those who do not tolerate or fail to respond to pharmacotherapy. Many of these patients are at risk for drug-induced toxicities or interactions or suicide.1
1. Kerner N, Prudic J. Current electroconvulsive therapy practice and research in the geriatric population. Neuropsychiatry (London). 2014;4(1):33-54.
2. Dombrovski AY, Mulsant BH. The evidence for electroconvulsive therapy (ECT) in the treatment of severe late-life depression. ECT: the preferred treatment for severe depression in late life. Int Psychogeriatr. 2007;19(1):10-14,27-35; discussion 24-26.
3. Olfson M, Marcus S, Sackeim HA, et al. Use of ECT for the inpatient treatment of recurrent major depression. Am J Psychiatry. 1998;155(1):22-29.
4. Salzman C, Wong E, Wright BC. Drug and ECT treatment of depression in the elderly, 1996-2001: a literature review. Biol Psychiatry. 2002;52(3):265-284.
5. Kellner CH, Husain MM, Knapp RG, et al; CORE/PRIDE Work Group. A novel strategy for continuation ect in geriatric depression: phase 2 of the PRIDE study. A
6. Tew JD Jr, Mulsant BH, Haskett RF, et al. Acute efficacy of ECT in the treatment of major depression in the old-old. Am J Psychiatry. 1999;156(12):1865-1870.
7. Dombrovski AY, Mulsant BH, Haskett RF, et al. Predictors of remission after electroconvulsive therapy in unipolar major depression. J Clin Psychiatry. 2005;66(8):1043-1049.
8. Charles K. UpToDate. Unipolar major depression in adults: indications for efficacy of electroconvulsive therapy (ECT). https://www.uptodate.com/contents/unipolar-major-depression-in-adults-indications-for-and-efficacy-of-electroconvulsive-therapy-ect. Updated May 16, 2017. Accessed November 26, 2017.
9. Sackeim HA, Prudic J, Devanand DP, et al. A prospective, randomized, double-blind comparison of bilateral and right unilateral electroconvulsive therapy at different stimulus intensities. Arch Gen Psychiatry. 2000;57(5):425-434.
10. UK ECT Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet. 2003;361(9360):799-808.
11. Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med. 2007;357(19):1939-1945.
12. Stoppe A, Louzã M, Rosa M, et al. Fixed high dose electroconvulsive therapy in elderly with depression: a double-blind, randomized comparison of efficacy and tolerability between unilateral and bilateral electrode placement. J ECT. 2006;22(2):92-99.
13. Geduldig ET, Kellner CH. Electroconvulsive therapy in the elderly: new findings in geriatric depression. Curr Psychiatry Rep. 2016;18(4):40.
14. Practice guideline for the treatment of patients with major depressive disorder (revision). American Psychiatric Association. Am J Psychiatry. 2000;157(suppl 4):1-45.
15. Rhebergen D, Huisman A, Bouckaert F, et al. Older age is associated with rapid remission of depression after electroconvulsive therapy: a latent class growth analysis. Am J Geriatr Psychiatry. 2015;23(3):274-282.
16. Philibert RA, Richards L, Lynch CF, et al. Effect of ECT on mortality and clinical outcome in geriatric unipolar depression. J Clin Psychiatry. 1995;56(9):390-394.
17. Tomac TA, Rummans TA, Pileggi TS, et al. Safety and efficacy of electroconvulsive therapy in patients over age 85. Am J Geriatr Psychiatry. 1997;5(2):126-130.
18. Verwijk E, Comijs HC, Kok RM, et al. Short and long-term neurocognitive functioning after electroconvulsive therapy in depressed elderly: a prospective naturalistic study. Int Psychogeriatr. 2014;26(2):315-324.
19. Flint AJ, Gagnon N. Effective use of electroconvulsive therapy in late life depression. Can J Psychiatry. 2002;47(8):734-741.
20. Bjolseth TM, Engedal K, Benth JS, et al. Speed of recovery from disorientation may predict the treatment outcome of electroconvulsive therapy (ECT) in elderly patients with major depression. J Affect Disord. 2016;190:178-186.
21. Sackeim HA, Prudic J, Nobler MS, et al. Ultra-brief pulse ECT and the affective and cognitive consequences of ECT. J ECT. 2001;17(1):77.
22. Bjolseth TM, Engedal K, Benth JS, et al. Baseline cognitive function does not predict the treatment outcome of electroconvulsive therapy (ECT) in late-life depression. J Affect Disord. 2015;185:67-75.
23. de Carle AJ, Kohn R. Electroconvulsive therapy and falls in the elderly. J ECT. 2000;16(3):252-257.
24. Hoover DR, Siegel M, Lucas J, et al. Depression in the first year of stay for elderly long-term nursing home residents in the USA. Int Psychogeriatr. 2010;22:1161.
25. O’Connor MK, Knapp R, Husain M, et al. The influence of age on the response of major depression to electroconvulsive therapy: a C.O.R.E. Report. Am J Geriatr Psychiatry. 2001; 9:382.
CASE Depressed, avoidant
Mr. R, age 95, has a history of recurrent major depressive disorder. He presents to the emergency department with depressive symptoms that began 6 weeks ago. His symptoms include depressed mood, hopelessness, anhedonia, anxiety, and insomnia. Co-occurring anorexia nervosa has resulted in a 20-lb weight loss. He denies suicidal ideation.
A mental status examination reveals profound psychomotor agitation, dysphoric mood, tearfulness, and mood-congruent delusions. Mr. R’s Mini-Mental State Examination (MMSE) score is 14/30; his Hamilton Depression Rating Scale (HAM-D) score is 21, indicating severe depression (19 to 22). However, the examiner feels that these scores may not reflect an accurate assessment because Mr. R gave flippant responses and did not cooperate during the interview. Physical examination is unremarkable. Previous medication trials included buspirone, escitalopram, and risperidone; none of these medications successfully alleviated his depressive symptoms.
On admission, Mr. R is given oral mirtazapine, 15 mg/d, and quetiapine, 25 mg/d, to target depressive mood, insomnia, and weight loss. Urgent intervention is indicated because his depressive symptoms are profoundly causing failure to thrive and are compromising his physical health. Mr. R’s deterioration concerns the physician team. Because of a history of failed pharmacotherapy trials, the team reassesses Mr. R’s treatment options.
[polldaddy:9903171]
The authors’ observations
The physician team recommends that Mr. R undergo ECT to obtain rapid relief from his depressive symptoms. After discussion of the potential risks and benefits, Mr. R agrees to this treatment. Quetiapine is discontinued prior to initiating ECT to avoid unnecessary medications; mirtazapine is continued.
Mr. R’s lack of response to previous antidepressants and significant deterioration were concerning. The physicians wanted to avoid higher-dose medications because of the risk of falls or somnolence. Their clinical experience and the literature supporting ECT for patients of Mr. R’s age lead them to select ECT as the most appropriate therapeutic option.
ECT has no absolute contraindications.1 The rate of ECT use in the United States has fluctuated over time because of factors unrelated to the efficacy and availability of ECT or alternative treatments.2 This form of intervention is also somewhat stigmatized.
Some psychiatrists are reluctant to prescribe ECT for geriatric patients because of concerns of potential neurocognitive or medical complications and risks during anesthesia. However, in the United States, older patients with depression are more likely to be treated with ECT than their younger counterparts.3 ECT usually induces greater immediate efficacy than antidepressants.4
Evidence supports using ECT in older patients
Multiple studies have found that ECT is a rapid, safe, and efficacious intervention for treating older persons with depression. Patients age >60 who receive ECT plus pharmacotherapy have lower HAM-D scores than those receiving pharmacotherapy alone.5 Overall, the rates of remission for depression range from 50% to 70%; yet geriatric patients who receive only ECT have response rates around 90%.6 Older age, presence of psychotic symptoms, and shorter duration of illness can predict a rapidly positive ECT response.7
When treated with ECT, older patients, including those age >85, have fewer subsequent episodes of depression compared with those who receive pharmacotherapy alone.1 Older individuals with physical illness or cognitive impairment respond to and tolerate ECT much like younger patients.6 Older patients receiving ECT may experience less cognitive decline than younger ones.7 Those in their ninth decade of life with treatment-resistant depression, psychotic features, and post-stroke depression often respond robustly with improvement following ECT.8
Remission rates also depend on the technique of administration. Interactions between electrode placement and stimulus parameter dosage affect efficacy and adverse effects.9 Right-sided, unilateral ECT induces less cognitive dysfunction compared with bilateral electrode placement,9 but bilateral ECT is more clinically effective.10 However, the efficacy of right-sided ECT is more dose-sensitive, and some data suggest that suboptimal response is due to insufficient stimulus dosages.11 One double-blind randomized controlled trial documented that when using a high-dose stimulus parameter, unilateral ECT is as effective as bilateral ECT.12 When there is a suboptimal response to unilateral ECT, bilateral ECT might be beneficial.12,13 For preventing relapse in older patients, increasing the interval between ECT treatments is more effective than stopping ECT abruptly.13
[polldaddy:9903172]
Indications of ECT
ECT is indicated for patients with severe depression, mania, and other conditions (Table).14 The most common indication for ECT in older persons is a history of treatment-resistant depression, with melancholia, psychosis, or suicidal ideations.1-6,12 There are also age-related and clinical factors to consider with ECT. This treatment provides a safe, rapid remission for patients age >65, even after adjusting for somatic conditions, duration of illness, medication resistance, or case severity.15 Compared with younger patients, older adults may not tolerate antidepressants as well because of age-related pharmacokinetic alterations, including increased sensitivity to anticholinergic and/or hypotensive effects.1
Factors that favor ECT include a previous good response to it; patient preference; and an indication for rapid intervention, such as suicidality, catatonia, dehydration, malnutrition, or a suboptimal result from pharmacotherapy.3 Mortality among individuals age >85 who receive ECT reportedly is lower than that among their counterparts who receive alternative treatments.16 ECT has been administered safely and effectively in patients with comorbid medical illnesses such as stroke, cerebral aneurysm, cardiovascular disease with ischemia or arrhythmia, dementia, and osteoporosis.17
Neurocognitive effects
Reports on the effects of ECT on neurocognitive functioning have varied. In some studies, performance improved or did not change in severely depressed older patients who received ECT.18,19 In older people who receive ECT, MMSE scores often return to baseline by the end of treatment.20 There often is only mild transient cognitive impairment in patients with late-life depression who receive ECT. Areas of concern include attention span, orientation, and speed of mental processing.20 Physicians should conduct cognitive tests before, during, and after ECT sessions to monitor their patient’s mental status.20
Cognitive stability can be maintained by administering ECT twice a week; applying right-sided, unilateral electrode placement; and using short, ultra-brief stimulus pulse width parameters.21 Cognitive impairment induced by ECT is not associated with age in geriatric patients with depression.22 Older adults who experienced longer postictal reorientation time periods have better outcomes than others who reach orientation faster; their intellectual impairment returned to baseline.20 Falling is another complication associated with ECT. A longitudinal cohort study found the incident of falls among patients receiving ECT was 13%.22 Risk factors for falls during a course of ECT include the number of treatments and the presence of coexisting Parkinson’s disease.23
OUTCOME Improvement
Mr. R receives 8 sessions of right-sided, unilateral ECT with an individualized dosage titration method. Treatments are completed with a stimulus intensity at 6 times seizure threshold, with an ultra-brief pulse width at 0.3 milliseconds. Mr. R’s mood and affect begin to improve after 3 ECT sessions. His MMSE score increases to 28/30 (Figure). His clinical improvement is progressively sustained; he develops an increasingly jovial attitude and experiences less anxiety. Mr. R’s confidence, appetite, and sleep also improve. There are no complications with treatment, and Mr. R has no complaints. After 8 ECT sessions, Mr. R has no affective symptoms and does not experience any cognitive impairment.
The authors’ observations
Depression among older people is a growing public health concern. It is a leading cause of disability, and often leads to nursing home placement.24 ECT is a safe, effective treatment for late-life depression, but is underutilized in patients age >75 because of concerns for cognitive impairment.6 However, there is evidence that response rates to ECT are higher in patients ages 45 to 85, compared with young individuals ages 18 to 45.25 ECT is a viable intervention for older depressed patients, particularly for those who do not tolerate or fail to respond to pharmacotherapy. Many of these patients are at risk for drug-induced toxicities or interactions or suicide.1
CASE Depressed, avoidant
Mr. R, age 95, has a history of recurrent major depressive disorder. He presents to the emergency department with depressive symptoms that began 6 weeks ago. His symptoms include depressed mood, hopelessness, anhedonia, anxiety, and insomnia. Co-occurring anorexia nervosa has resulted in a 20-lb weight loss. He denies suicidal ideation.
A mental status examination reveals profound psychomotor agitation, dysphoric mood, tearfulness, and mood-congruent delusions. Mr. R’s Mini-Mental State Examination (MMSE) score is 14/30; his Hamilton Depression Rating Scale (HAM-D) score is 21, indicating severe depression (19 to 22). However, the examiner feels that these scores may not reflect an accurate assessment because Mr. R gave flippant responses and did not cooperate during the interview. Physical examination is unremarkable. Previous medication trials included buspirone, escitalopram, and risperidone; none of these medications successfully alleviated his depressive symptoms.
On admission, Mr. R is given oral mirtazapine, 15 mg/d, and quetiapine, 25 mg/d, to target depressive mood, insomnia, and weight loss. Urgent intervention is indicated because his depressive symptoms are profoundly causing failure to thrive and are compromising his physical health. Mr. R’s deterioration concerns the physician team. Because of a history of failed pharmacotherapy trials, the team reassesses Mr. R’s treatment options.
[polldaddy:9903171]
The authors’ observations
The physician team recommends that Mr. R undergo ECT to obtain rapid relief from his depressive symptoms. After discussion of the potential risks and benefits, Mr. R agrees to this treatment. Quetiapine is discontinued prior to initiating ECT to avoid unnecessary medications; mirtazapine is continued.
Mr. R’s lack of response to previous antidepressants and significant deterioration were concerning. The physicians wanted to avoid higher-dose medications because of the risk of falls or somnolence. Their clinical experience and the literature supporting ECT for patients of Mr. R’s age lead them to select ECT as the most appropriate therapeutic option.
ECT has no absolute contraindications.1 The rate of ECT use in the United States has fluctuated over time because of factors unrelated to the efficacy and availability of ECT or alternative treatments.2 This form of intervention is also somewhat stigmatized.
Some psychiatrists are reluctant to prescribe ECT for geriatric patients because of concerns of potential neurocognitive or medical complications and risks during anesthesia. However, in the United States, older patients with depression are more likely to be treated with ECT than their younger counterparts.3 ECT usually induces greater immediate efficacy than antidepressants.4
Evidence supports using ECT in older patients
Multiple studies have found that ECT is a rapid, safe, and efficacious intervention for treating older persons with depression. Patients age >60 who receive ECT plus pharmacotherapy have lower HAM-D scores than those receiving pharmacotherapy alone.5 Overall, the rates of remission for depression range from 50% to 70%; yet geriatric patients who receive only ECT have response rates around 90%.6 Older age, presence of psychotic symptoms, and shorter duration of illness can predict a rapidly positive ECT response.7
When treated with ECT, older patients, including those age >85, have fewer subsequent episodes of depression compared with those who receive pharmacotherapy alone.1 Older individuals with physical illness or cognitive impairment respond to and tolerate ECT much like younger patients.6 Older patients receiving ECT may experience less cognitive decline than younger ones.7 Those in their ninth decade of life with treatment-resistant depression, psychotic features, and post-stroke depression often respond robustly with improvement following ECT.8
Remission rates also depend on the technique of administration. Interactions between electrode placement and stimulus parameter dosage affect efficacy and adverse effects.9 Right-sided, unilateral ECT induces less cognitive dysfunction compared with bilateral electrode placement,9 but bilateral ECT is more clinically effective.10 However, the efficacy of right-sided ECT is more dose-sensitive, and some data suggest that suboptimal response is due to insufficient stimulus dosages.11 One double-blind randomized controlled trial documented that when using a high-dose stimulus parameter, unilateral ECT is as effective as bilateral ECT.12 When there is a suboptimal response to unilateral ECT, bilateral ECT might be beneficial.12,13 For preventing relapse in older patients, increasing the interval between ECT treatments is more effective than stopping ECT abruptly.13
[polldaddy:9903172]
Indications of ECT
ECT is indicated for patients with severe depression, mania, and other conditions (Table).14 The most common indication for ECT in older persons is a history of treatment-resistant depression, with melancholia, psychosis, or suicidal ideations.1-6,12 There are also age-related and clinical factors to consider with ECT. This treatment provides a safe, rapid remission for patients age >65, even after adjusting for somatic conditions, duration of illness, medication resistance, or case severity.15 Compared with younger patients, older adults may not tolerate antidepressants as well because of age-related pharmacokinetic alterations, including increased sensitivity to anticholinergic and/or hypotensive effects.1
Factors that favor ECT include a previous good response to it; patient preference; and an indication for rapid intervention, such as suicidality, catatonia, dehydration, malnutrition, or a suboptimal result from pharmacotherapy.3 Mortality among individuals age >85 who receive ECT reportedly is lower than that among their counterparts who receive alternative treatments.16 ECT has been administered safely and effectively in patients with comorbid medical illnesses such as stroke, cerebral aneurysm, cardiovascular disease with ischemia or arrhythmia, dementia, and osteoporosis.17
Neurocognitive effects
Reports on the effects of ECT on neurocognitive functioning have varied. In some studies, performance improved or did not change in severely depressed older patients who received ECT.18,19 In older people who receive ECT, MMSE scores often return to baseline by the end of treatment.20 There often is only mild transient cognitive impairment in patients with late-life depression who receive ECT. Areas of concern include attention span, orientation, and speed of mental processing.20 Physicians should conduct cognitive tests before, during, and after ECT sessions to monitor their patient’s mental status.20
Cognitive stability can be maintained by administering ECT twice a week; applying right-sided, unilateral electrode placement; and using short, ultra-brief stimulus pulse width parameters.21 Cognitive impairment induced by ECT is not associated with age in geriatric patients with depression.22 Older adults who experienced longer postictal reorientation time periods have better outcomes than others who reach orientation faster; their intellectual impairment returned to baseline.20 Falling is another complication associated with ECT. A longitudinal cohort study found the incident of falls among patients receiving ECT was 13%.22 Risk factors for falls during a course of ECT include the number of treatments and the presence of coexisting Parkinson’s disease.23
OUTCOME Improvement
Mr. R receives 8 sessions of right-sided, unilateral ECT with an individualized dosage titration method. Treatments are completed with a stimulus intensity at 6 times seizure threshold, with an ultra-brief pulse width at 0.3 milliseconds. Mr. R’s mood and affect begin to improve after 3 ECT sessions. His MMSE score increases to 28/30 (Figure). His clinical improvement is progressively sustained; he develops an increasingly jovial attitude and experiences less anxiety. Mr. R’s confidence, appetite, and sleep also improve. There are no complications with treatment, and Mr. R has no complaints. After 8 ECT sessions, Mr. R has no affective symptoms and does not experience any cognitive impairment.
The authors’ observations
Depression among older people is a growing public health concern. It is a leading cause of disability, and often leads to nursing home placement.24 ECT is a safe, effective treatment for late-life depression, but is underutilized in patients age >75 because of concerns for cognitive impairment.6 However, there is evidence that response rates to ECT are higher in patients ages 45 to 85, compared with young individuals ages 18 to 45.25 ECT is a viable intervention for older depressed patients, particularly for those who do not tolerate or fail to respond to pharmacotherapy. Many of these patients are at risk for drug-induced toxicities or interactions or suicide.1
1. Kerner N, Prudic J. Current electroconvulsive therapy practice and research in the geriatric population. Neuropsychiatry (London). 2014;4(1):33-54.
2. Dombrovski AY, Mulsant BH. The evidence for electroconvulsive therapy (ECT) in the treatment of severe late-life depression. ECT: the preferred treatment for severe depression in late life. Int Psychogeriatr. 2007;19(1):10-14,27-35; discussion 24-26.
3. Olfson M, Marcus S, Sackeim HA, et al. Use of ECT for the inpatient treatment of recurrent major depression. Am J Psychiatry. 1998;155(1):22-29.
4. Salzman C, Wong E, Wright BC. Drug and ECT treatment of depression in the elderly, 1996-2001: a literature review. Biol Psychiatry. 2002;52(3):265-284.
5. Kellner CH, Husain MM, Knapp RG, et al; CORE/PRIDE Work Group. A novel strategy for continuation ect in geriatric depression: phase 2 of the PRIDE study. A
6. Tew JD Jr, Mulsant BH, Haskett RF, et al. Acute efficacy of ECT in the treatment of major depression in the old-old. Am J Psychiatry. 1999;156(12):1865-1870.
7. Dombrovski AY, Mulsant BH, Haskett RF, et al. Predictors of remission after electroconvulsive therapy in unipolar major depression. J Clin Psychiatry. 2005;66(8):1043-1049.
8. Charles K. UpToDate. Unipolar major depression in adults: indications for efficacy of electroconvulsive therapy (ECT). https://www.uptodate.com/contents/unipolar-major-depression-in-adults-indications-for-and-efficacy-of-electroconvulsive-therapy-ect. Updated May 16, 2017. Accessed November 26, 2017.
9. Sackeim HA, Prudic J, Devanand DP, et al. A prospective, randomized, double-blind comparison of bilateral and right unilateral electroconvulsive therapy at different stimulus intensities. Arch Gen Psychiatry. 2000;57(5):425-434.
10. UK ECT Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet. 2003;361(9360):799-808.
11. Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med. 2007;357(19):1939-1945.
12. Stoppe A, Louzã M, Rosa M, et al. Fixed high dose electroconvulsive therapy in elderly with depression: a double-blind, randomized comparison of efficacy and tolerability between unilateral and bilateral electrode placement. J ECT. 2006;22(2):92-99.
13. Geduldig ET, Kellner CH. Electroconvulsive therapy in the elderly: new findings in geriatric depression. Curr Psychiatry Rep. 2016;18(4):40.
14. Practice guideline for the treatment of patients with major depressive disorder (revision). American Psychiatric Association. Am J Psychiatry. 2000;157(suppl 4):1-45.
15. Rhebergen D, Huisman A, Bouckaert F, et al. Older age is associated with rapid remission of depression after electroconvulsive therapy: a latent class growth analysis. Am J Geriatr Psychiatry. 2015;23(3):274-282.
16. Philibert RA, Richards L, Lynch CF, et al. Effect of ECT on mortality and clinical outcome in geriatric unipolar depression. J Clin Psychiatry. 1995;56(9):390-394.
17. Tomac TA, Rummans TA, Pileggi TS, et al. Safety and efficacy of electroconvulsive therapy in patients over age 85. Am J Geriatr Psychiatry. 1997;5(2):126-130.
18. Verwijk E, Comijs HC, Kok RM, et al. Short and long-term neurocognitive functioning after electroconvulsive therapy in depressed elderly: a prospective naturalistic study. Int Psychogeriatr. 2014;26(2):315-324.
19. Flint AJ, Gagnon N. Effective use of electroconvulsive therapy in late life depression. Can J Psychiatry. 2002;47(8):734-741.
20. Bjolseth TM, Engedal K, Benth JS, et al. Speed of recovery from disorientation may predict the treatment outcome of electroconvulsive therapy (ECT) in elderly patients with major depression. J Affect Disord. 2016;190:178-186.
21. Sackeim HA, Prudic J, Nobler MS, et al. Ultra-brief pulse ECT and the affective and cognitive consequences of ECT. J ECT. 2001;17(1):77.
22. Bjolseth TM, Engedal K, Benth JS, et al. Baseline cognitive function does not predict the treatment outcome of electroconvulsive therapy (ECT) in late-life depression. J Affect Disord. 2015;185:67-75.
23. de Carle AJ, Kohn R. Electroconvulsive therapy and falls in the elderly. J ECT. 2000;16(3):252-257.
24. Hoover DR, Siegel M, Lucas J, et al. Depression in the first year of stay for elderly long-term nursing home residents in the USA. Int Psychogeriatr. 2010;22:1161.
25. O’Connor MK, Knapp R, Husain M, et al. The influence of age on the response of major depression to electroconvulsive therapy: a C.O.R.E. Report. Am J Geriatr Psychiatry. 2001; 9:382.
1. Kerner N, Prudic J. Current electroconvulsive therapy practice and research in the geriatric population. Neuropsychiatry (London). 2014;4(1):33-54.
2. Dombrovski AY, Mulsant BH. The evidence for electroconvulsive therapy (ECT) in the treatment of severe late-life depression. ECT: the preferred treatment for severe depression in late life. Int Psychogeriatr. 2007;19(1):10-14,27-35; discussion 24-26.
3. Olfson M, Marcus S, Sackeim HA, et al. Use of ECT for the inpatient treatment of recurrent major depression. Am J Psychiatry. 1998;155(1):22-29.
4. Salzman C, Wong E, Wright BC. Drug and ECT treatment of depression in the elderly, 1996-2001: a literature review. Biol Psychiatry. 2002;52(3):265-284.
5. Kellner CH, Husain MM, Knapp RG, et al; CORE/PRIDE Work Group. A novel strategy for continuation ect in geriatric depression: phase 2 of the PRIDE study. A
6. Tew JD Jr, Mulsant BH, Haskett RF, et al. Acute efficacy of ECT in the treatment of major depression in the old-old. Am J Psychiatry. 1999;156(12):1865-1870.
7. Dombrovski AY, Mulsant BH, Haskett RF, et al. Predictors of remission after electroconvulsive therapy in unipolar major depression. J Clin Psychiatry. 2005;66(8):1043-1049.
8. Charles K. UpToDate. Unipolar major depression in adults: indications for efficacy of electroconvulsive therapy (ECT). https://www.uptodate.com/contents/unipolar-major-depression-in-adults-indications-for-and-efficacy-of-electroconvulsive-therapy-ect. Updated May 16, 2017. Accessed November 26, 2017.
9. Sackeim HA, Prudic J, Devanand DP, et al. A prospective, randomized, double-blind comparison of bilateral and right unilateral electroconvulsive therapy at different stimulus intensities. Arch Gen Psychiatry. 2000;57(5):425-434.
10. UK ECT Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet. 2003;361(9360):799-808.
11. Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med. 2007;357(19):1939-1945.
12. Stoppe A, Louzã M, Rosa M, et al. Fixed high dose electroconvulsive therapy in elderly with depression: a double-blind, randomized comparison of efficacy and tolerability between unilateral and bilateral electrode placement. J ECT. 2006;22(2):92-99.
13. Geduldig ET, Kellner CH. Electroconvulsive therapy in the elderly: new findings in geriatric depression. Curr Psychiatry Rep. 2016;18(4):40.
14. Practice guideline for the treatment of patients with major depressive disorder (revision). American Psychiatric Association. Am J Psychiatry. 2000;157(suppl 4):1-45.
15. Rhebergen D, Huisman A, Bouckaert F, et al. Older age is associated with rapid remission of depression after electroconvulsive therapy: a latent class growth analysis. Am J Geriatr Psychiatry. 2015;23(3):274-282.
16. Philibert RA, Richards L, Lynch CF, et al. Effect of ECT on mortality and clinical outcome in geriatric unipolar depression. J Clin Psychiatry. 1995;56(9):390-394.
17. Tomac TA, Rummans TA, Pileggi TS, et al. Safety and efficacy of electroconvulsive therapy in patients over age 85. Am J Geriatr Psychiatry. 1997;5(2):126-130.
18. Verwijk E, Comijs HC, Kok RM, et al. Short and long-term neurocognitive functioning after electroconvulsive therapy in depressed elderly: a prospective naturalistic study. Int Psychogeriatr. 2014;26(2):315-324.
19. Flint AJ, Gagnon N. Effective use of electroconvulsive therapy in late life depression. Can J Psychiatry. 2002;47(8):734-741.
20. Bjolseth TM, Engedal K, Benth JS, et al. Speed of recovery from disorientation may predict the treatment outcome of electroconvulsive therapy (ECT) in elderly patients with major depression. J Affect Disord. 2016;190:178-186.
21. Sackeim HA, Prudic J, Nobler MS, et al. Ultra-brief pulse ECT and the affective and cognitive consequences of ECT. J ECT. 2001;17(1):77.
22. Bjolseth TM, Engedal K, Benth JS, et al. Baseline cognitive function does not predict the treatment outcome of electroconvulsive therapy (ECT) in late-life depression. J Affect Disord. 2015;185:67-75.
23. de Carle AJ, Kohn R. Electroconvulsive therapy and falls in the elderly. J ECT. 2000;16(3):252-257.
24. Hoover DR, Siegel M, Lucas J, et al. Depression in the first year of stay for elderly long-term nursing home residents in the USA. Int Psychogeriatr. 2010;22:1161.
25. O’Connor MK, Knapp R, Husain M, et al. The influence of age on the response of major depression to electroconvulsive therapy: a C.O.R.E. Report. Am J Geriatr Psychiatry. 2001; 9:382.
Vaping marijuana?
Cannavaping—the inhalation of a cannabis-containing aerosol, created by a battery-driven, heated atomizer in e-cigarettes or similar devices1—is touted as a less expensive and safer alternative to smoking marijuana. It’s also gaining in popularity.2 One study of Connecticut high school students found that 5.4% had used e-cigarettes to vaporize cannabis.3 But what do we know about this new way to get high?
We know that those who wish to cannavape can easily obtain e-cigarettes from gas stations and tobacco shops. They then have to obtain a cartridge, filled with either hash oil or tetrahydrocannabinol-infused wax, to attach to the e-cigarette. These cartridges are available for purchase in states that have legalized the sale of marijuana. They also find their way into states where the sale of marijuana is not legal, and are purchased illegally for the purpose of cannavaping.
And while cannavaping does appear to reduce the cost of smoking marijuana,4 it has not been widely researched, nor determined to be safe.5
In fact, although marijuana has several important therapeutic and medicinal purposes, cannavaping the substance can result in medical concerns.6 The vaping aerosols of some compounds can induce lung pathology and may be carcinogenic, since they often contain a number of dangerous toxins.4
Chronic marijuana use can increase the likelihood of motor vehicles accidents, cognitive impairment, psychoses, and demotivation.4 It may predispose certain individuals to use other drugs and tobacco products and could increase the consumption of marijuana.4,5 Increased consumption could have a detrimental effect on intellect and behavior when used chronically—especially in youngsters, whose nervous systems are not yet fully matured.7-9
Because cannavaping has potentially deleterious effects, more regulations on the manufacture, distribution, access, and use are indicated—at least until research sheds more light on issues surrounding this practice.
Steven Lippman, MD; Devina Singh, MD
Louisville, KY
1. Varlet V, Concha-Lozano N, Berthlet A, et al. Drug vaping applied to cannabis: is “cannavaping” a therapeutic alternative to marijuana? Sci Rep. 2016;6:25599.
2. Giroud C, de Cesare M, Berthet A, et al. E-cigarettes: a review of new trends in cannabis use. Int J Environ Res Public Health. 2015;12:9988-10008.
3. Morean ME, Kong G, Camenga DR, et al. High school students’ use of electronic cigarettes to vaporize cannabis. Pediatrics. 2015;136:611-616.
4. Budney AJ, Sargent JD, Lee DC. Vaping cannabis (marijuana): parallel concerns to e-cigs? Addiction. 2015;110:1699-1704.
5. Cox B. Can the research community respond adequately to the health risks of vaping? Addiction. 2015;110:1709-1709.
6. Rong C, Lee Y, Carmona NE, et al. Cannabidiol in medical marijuana: research vistas and potential opportunities. Pharmacol Res. 2017;121:213-218.
7. Schweinsburg AD, Brown SA, Tapert SF. The influence of marijuana use on neurocognitive functioning in adolescents. Curr Drug Abuse Rev. 2008;1:99-111.
8. Meier MH, Caspi A, Ambler A, et al. Persistent cannabis users show neuropsychological decline from childhood to midlife. Proc Natl Acad Sci USA. 2012;109:E2657-2664.
9. Castellanos-Ryan N, Pingault J, Parent S, et al. Adolescent cannabis use, change in neurocognitive function, and high-school graduation: a longitudinal study from early adolescence to young adulthood. Dev Psychopathol . 2017;29:1253-1266.
Cannavaping—the inhalation of a cannabis-containing aerosol, created by a battery-driven, heated atomizer in e-cigarettes or similar devices1—is touted as a less expensive and safer alternative to smoking marijuana. It’s also gaining in popularity.2 One study of Connecticut high school students found that 5.4% had used e-cigarettes to vaporize cannabis.3 But what do we know about this new way to get high?
We know that those who wish to cannavape can easily obtain e-cigarettes from gas stations and tobacco shops. They then have to obtain a cartridge, filled with either hash oil or tetrahydrocannabinol-infused wax, to attach to the e-cigarette. These cartridges are available for purchase in states that have legalized the sale of marijuana. They also find their way into states where the sale of marijuana is not legal, and are purchased illegally for the purpose of cannavaping.
And while cannavaping does appear to reduce the cost of smoking marijuana,4 it has not been widely researched, nor determined to be safe.5
In fact, although marijuana has several important therapeutic and medicinal purposes, cannavaping the substance can result in medical concerns.6 The vaping aerosols of some compounds can induce lung pathology and may be carcinogenic, since they often contain a number of dangerous toxins.4
Chronic marijuana use can increase the likelihood of motor vehicles accidents, cognitive impairment, psychoses, and demotivation.4 It may predispose certain individuals to use other drugs and tobacco products and could increase the consumption of marijuana.4,5 Increased consumption could have a detrimental effect on intellect and behavior when used chronically—especially in youngsters, whose nervous systems are not yet fully matured.7-9
Because cannavaping has potentially deleterious effects, more regulations on the manufacture, distribution, access, and use are indicated—at least until research sheds more light on issues surrounding this practice.
Steven Lippman, MD; Devina Singh, MD
Louisville, KY
Cannavaping—the inhalation of a cannabis-containing aerosol, created by a battery-driven, heated atomizer in e-cigarettes or similar devices1—is touted as a less expensive and safer alternative to smoking marijuana. It’s also gaining in popularity.2 One study of Connecticut high school students found that 5.4% had used e-cigarettes to vaporize cannabis.3 But what do we know about this new way to get high?
We know that those who wish to cannavape can easily obtain e-cigarettes from gas stations and tobacco shops. They then have to obtain a cartridge, filled with either hash oil or tetrahydrocannabinol-infused wax, to attach to the e-cigarette. These cartridges are available for purchase in states that have legalized the sale of marijuana. They also find their way into states where the sale of marijuana is not legal, and are purchased illegally for the purpose of cannavaping.
And while cannavaping does appear to reduce the cost of smoking marijuana,4 it has not been widely researched, nor determined to be safe.5
In fact, although marijuana has several important therapeutic and medicinal purposes, cannavaping the substance can result in medical concerns.6 The vaping aerosols of some compounds can induce lung pathology and may be carcinogenic, since they often contain a number of dangerous toxins.4
Chronic marijuana use can increase the likelihood of motor vehicles accidents, cognitive impairment, psychoses, and demotivation.4 It may predispose certain individuals to use other drugs and tobacco products and could increase the consumption of marijuana.4,5 Increased consumption could have a detrimental effect on intellect and behavior when used chronically—especially in youngsters, whose nervous systems are not yet fully matured.7-9
Because cannavaping has potentially deleterious effects, more regulations on the manufacture, distribution, access, and use are indicated—at least until research sheds more light on issues surrounding this practice.
Steven Lippman, MD; Devina Singh, MD
Louisville, KY
1. Varlet V, Concha-Lozano N, Berthlet A, et al. Drug vaping applied to cannabis: is “cannavaping” a therapeutic alternative to marijuana? Sci Rep. 2016;6:25599.
2. Giroud C, de Cesare M, Berthet A, et al. E-cigarettes: a review of new trends in cannabis use. Int J Environ Res Public Health. 2015;12:9988-10008.
3. Morean ME, Kong G, Camenga DR, et al. High school students’ use of electronic cigarettes to vaporize cannabis. Pediatrics. 2015;136:611-616.
4. Budney AJ, Sargent JD, Lee DC. Vaping cannabis (marijuana): parallel concerns to e-cigs? Addiction. 2015;110:1699-1704.
5. Cox B. Can the research community respond adequately to the health risks of vaping? Addiction. 2015;110:1709-1709.
6. Rong C, Lee Y, Carmona NE, et al. Cannabidiol in medical marijuana: research vistas and potential opportunities. Pharmacol Res. 2017;121:213-218.
7. Schweinsburg AD, Brown SA, Tapert SF. The influence of marijuana use on neurocognitive functioning in adolescents. Curr Drug Abuse Rev. 2008;1:99-111.
8. Meier MH, Caspi A, Ambler A, et al. Persistent cannabis users show neuropsychological decline from childhood to midlife. Proc Natl Acad Sci USA. 2012;109:E2657-2664.
9. Castellanos-Ryan N, Pingault J, Parent S, et al. Adolescent cannabis use, change in neurocognitive function, and high-school graduation: a longitudinal study from early adolescence to young adulthood. Dev Psychopathol . 2017;29:1253-1266.
1. Varlet V, Concha-Lozano N, Berthlet A, et al. Drug vaping applied to cannabis: is “cannavaping” a therapeutic alternative to marijuana? Sci Rep. 2016;6:25599.
2. Giroud C, de Cesare M, Berthet A, et al. E-cigarettes: a review of new trends in cannabis use. Int J Environ Res Public Health. 2015;12:9988-10008.
3. Morean ME, Kong G, Camenga DR, et al. High school students’ use of electronic cigarettes to vaporize cannabis. Pediatrics. 2015;136:611-616.
4. Budney AJ, Sargent JD, Lee DC. Vaping cannabis (marijuana): parallel concerns to e-cigs? Addiction. 2015;110:1699-1704.
5. Cox B. Can the research community respond adequately to the health risks of vaping? Addiction. 2015;110:1709-1709.
6. Rong C, Lee Y, Carmona NE, et al. Cannabidiol in medical marijuana: research vistas and potential opportunities. Pharmacol Res. 2017;121:213-218.
7. Schweinsburg AD, Brown SA, Tapert SF. The influence of marijuana use on neurocognitive functioning in adolescents. Curr Drug Abuse Rev. 2008;1:99-111.
8. Meier MH, Caspi A, Ambler A, et al. Persistent cannabis users show neuropsychological decline from childhood to midlife. Proc Natl Acad Sci USA. 2012;109:E2657-2664.
9. Castellanos-Ryan N, Pingault J, Parent S, et al. Adolescent cannabis use, change in neurocognitive function, and high-school graduation: a longitudinal study from early adolescence to young adulthood. Dev Psychopathol . 2017;29:1253-1266.
A Review of Psychostimulants for Adults With Depression
Depression is a common condition that significantly impairs social and occupational functioning. Many patients do not respond to first-line pharmacotherapies and are considered to have treatment-resistant depression (TRD). These patients may benefit from augmentation of their antidepressant to reduce depression. Multiple medications have demonstrated various degrees of efficacy for augmentation, including psychostimulants. This article reviews studies of psychostimulants as augmentation agents for TRD and discusses risks, offers advice, and makes recommendations for clinicians who prescribe stimulants.
Background
Major depressive disorder (MDD) is a common psychiatric condition that significantly impairs quality of life.1 It is a recurrent illness, averaging 2 relapses per decade. The probability of recurrence increases with the number of depressive episodes.2,3 A patient who experiences major depressive episodes alternating with euthymia has unipolar depression; whereas one who experiences major depressive episodes alternating with episodes of mania or hypomania has bipolar depression.4
Despite adequate dose and duration of pharmacotherapy, many individuals with unipolar or bipolar depression do not achieve and sustain remission.5 Remission rates decrease and relapse rates increase with subsequent failed antidepressant trials.6 It is difficult to identify factors that predict treatment resistance, but one review of antidepressant studies found that patients who did not demonstrate a response within 3 weeks of medication initiation were less likely to respond after a longer duration.7
Treatment-resistant depression is commonly, but not universally, defined as lack of response after trials of 2 or more antidepressants with different mechanisms of action for sufficient duration.5 This definition will be used here as well. Other definitions have proposed stages of TRD, but these require further study to evaluate their reliability and predictive utility.8 Due to lack of consensus regarding the definition of TRD, it is not possible to determine the exact prevalence of TRD.
Patients with TRD may benefit from augmentation of their medication regimen. Augmentation with lithium has yielded conflicting results, and its efficacy with newer antidepressants is not well studied.9-12 Triiodothyronine, buspirone, and pindolol have demonstrated some efficacy when added to serotonin reuptake inhibitors (SRIs).10,12,13 Second-generation antipsychotic drugs, antidepressant drug combinations, omega-3 fatty acids, S-adenosyl methionine (SAMe), and L-methylfolate have demonstrated some efficacy in some studies as well.12,14-23 In patients with depression who have not responded to these strategies, psychostimulant augmentation may be appropriate.
Methods
A literature search was conducted following an algorithmic approach in the MEDLINE/PubMed database for studies in English from January 1985 to August 2014 of stimulants as augmenting agents for depression, using the Medical Subject Headings stimulant, depression, and augmentation, combined with an AND operator. The search was limited to adult humans and excluded case reports and letters, to identify studies with stronger evidence. Also excluded were studies using caffeine (to augment electroconvulsive therapy for depression) and pemoline as the sole augmenting stimulant as well as studies of patients with comorbid mental health diagnoses and studies that initiated stimulants and antidepressants simultaneously to assess antidepressant response.
This review organized results by stimulant rather than by depression type, even though some studies used > 1 stimulant or recruited patients with different types of depression. Although prevalence, prognosis, and monotherapy differ for unipolar and bipolar depression, psychostimulants target similar symptoms, despite augmenting different monotherapies in unipolar and bipolar depression. Therefore, no distinction is made between assessing studies of stimulants for unipolar and bipolar depression.
Results
A total of 70 articles were identified, and 31 studies met inclusion criteria (Figure). Of the studies included, 12 were double-blind, placebo-controlled (DBPC) trials and 19 were retrospective chart reviews or open studies. Most studies evaluated depression, using validated scales, such as the Hamilton Depression Rating Scale, Montgomery-Asberg Depression Rating Scale, Clinical Global Impressions of Severity, Inventory of Depressive Symptoms, Carroll Depression Rating Scale, Global Assessment of Functioning, Quick Inventory of Depressive Symptomatology, or the Psychiatric Symptom Assessment Scale. Study details are provided in Tables 1 to 4.
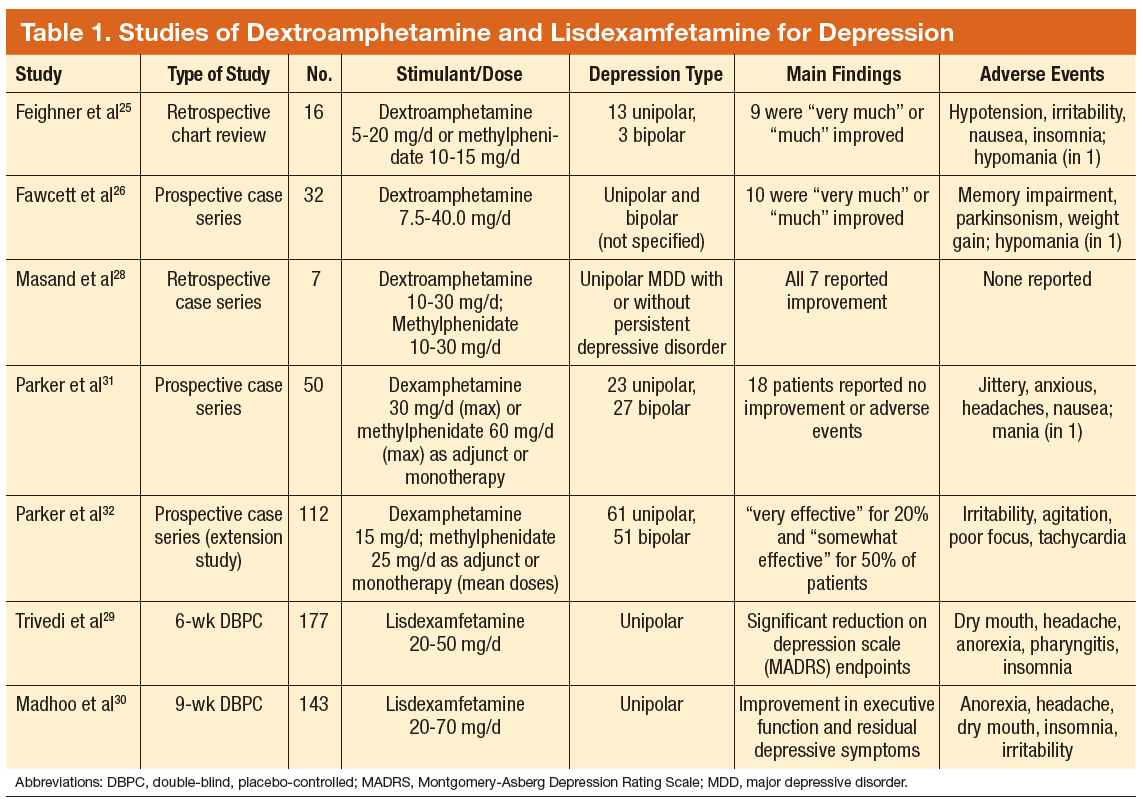
Dextroamphetamine and Methylphenidate
Dextroamphetamine and methylphenidate are indicated for the treatment of attention-deficit/hyperactivity disorder (ADHD) and exert their effects by inhibiting uptake of norepinephrine and dopamine.24 In one chart review, patients received dextroamphetamine or methylphenidate augmentation of monoamine oxidase inhibitors (MAOIs) alone or with concurrent tricyclic antidepressants; the majority reported decreased depression.25 In an openlabel trial, dextroamphetamine was titrated to efficacy in patients who were receiving an MAOI with or without pemoline.26 Nearly 80% of patients reported long-lasting improvement in depression. In an open-label trial, all patients reported decreased depression when methylphenidate was added to SRIs; however, no scales were used.27
In a case series, patients with both major depression and persistent depressive disorder (dysthymia) experienced a substantial, quick, and sustained response to dextroamphetamine or methylphenidate augmentation.28 Addition of lisdexamfetamine significantly reduced depressive symptoms in individuals with inadequate response to escitalopram.29 Patients with full or partial remission of depression noted improved executive function and residual depressive symptoms after lisdexamfetamine was added to SRI monotherapy.30 In a trial in which patients received dexamphetamine or methylphenidate as monotherapy or augmentation, 30% to 34% of patients reported mood improvement, but 36% reported no improvement.31 In an extension study, low-dose psychostimulants quickly diminished melancholia.32
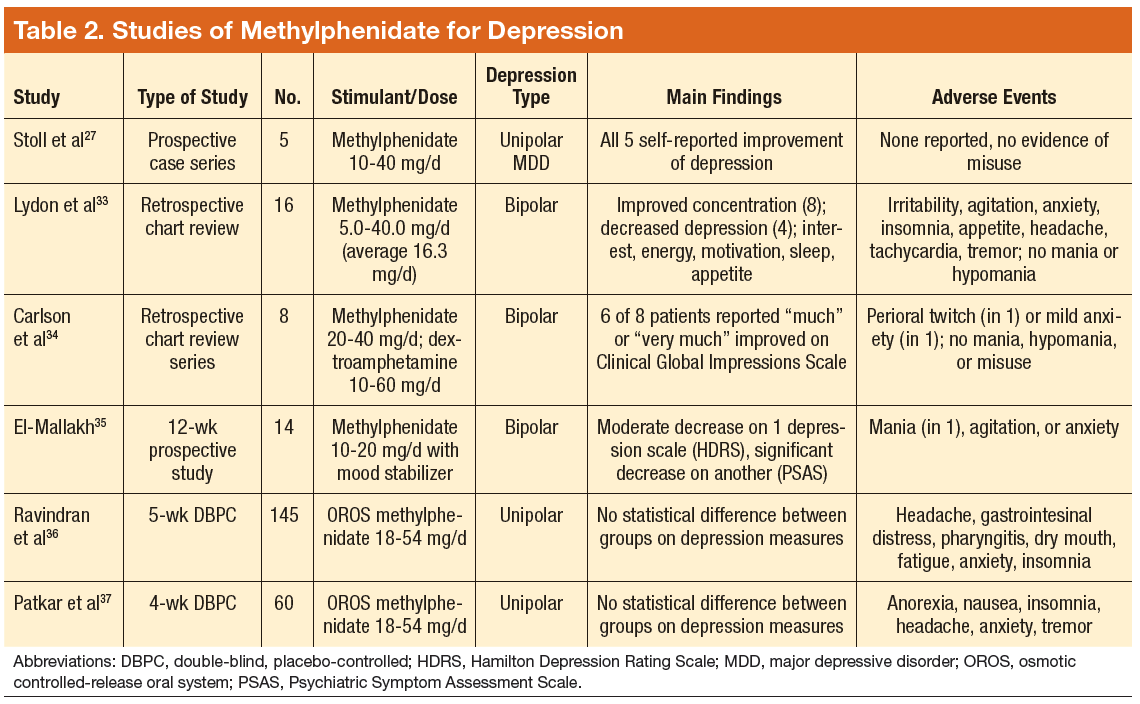
Methylphenidate was safe and effective in patients with bipolar depression receiving treatment for 1 to 5 years; 44% evidenced significant improvement.33 When offered to patients with bipolar depression, patients receiving methylphenidate or dextroamphetamine reported less depression or sedation and did not develop tolerance, mania, or misuse.34 A case series concluded that methylphenidate addition to mood stabilizers was generally effective and safe.35
However, not all preparations of methylphenidate have demonstrated efficacy. In one study, osmotic controlledrelease oral system (OROS) methylphenidate improved apathy and fatigue but not overall depression.36 Although OROS methylphenidate similarly failed to demonstrate statistically significant efficacy in another study, more responders were documented in the treatment group.37
Although this review focuses on stimulants as augmenting agents in patients with depression, it is worth noting the limited number of studies evaluating stimulants’ effect on depression in patients with traumatic brain injury. This observation is of concern, as these conditions are frequently comorbid in returning veterans. One study noted that methylphenidate was an effective monotherapy for depression; whereas another study found that methylphenidate monotherapy reduced depression as well as sertraline, was better tolerated, and improved fatigue and cognition.38,39
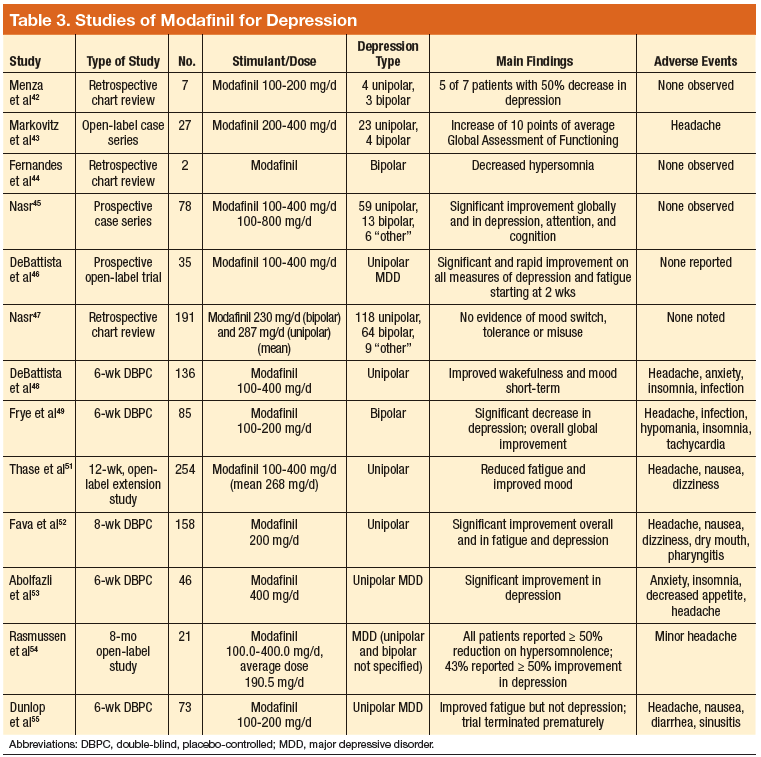
Modafinil and Armodafinil
Modafinil and armodafinil (the R-enantiomer of modafinil) are indicated for improving wakefulness in individuals with narcolepsy, obstructive sleep apnea, and shift work sleep disorder by modulating glutamate, gamma amino-butyric acid, and histamine.40,41 Although they increase extracellular dopamine concentrations, they do not cause an increase in dopamine release and may have less misuse potential than that of dextroamphetamine and methylphenidate.40,41 In a study of 7 patients with unipolar or bipolar depression, all patients achieved full or partial remission with minimal adverse effects (AEs).42 In a prospective study, 41% of patients reported only mild depression or full remission with modafinil augmentation.43
Multiple trials and a pooled analysis noted decreased depression and fatigue and improved cognition in patients receiving modafinil augmentation compared with mood stabilizers or antidepressants.44-49 Modafinil is a useful adjunct for partial responders to SRIs, resulting in rapid mood improvement and decreased fatigue.50-54 However, in one study, modafinil did not demonstrate efficacy compared with placebo. This result was attributed to premature study termination after 2 modafinil-treated patients developed suicidal ideation.55 A post hoc analysis found no difference in frequency of suicidal ideation between groups.
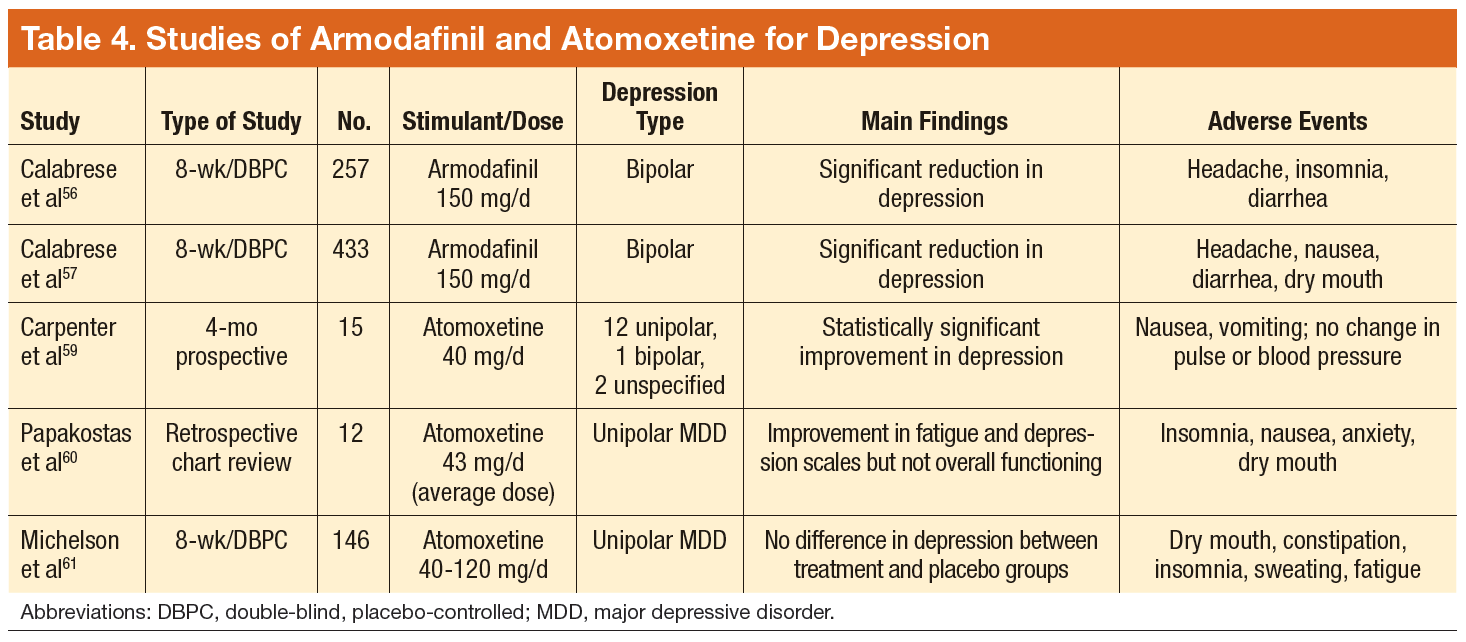
Two DBPC studies evaluated armodafinil in patients with bipolar depression. In both studies it was added to a mood-stabilizing agent (lithium, valproate, aripiprazole, olanzapine, lamotrigine, risperidone, or ziprasidone), and patients receiving armodafinil reported significant reductions
in depression.56,57
Atomoxetine
Atomoxetine is a norepinephrine reuptake inhibitor indicated for the treatment of ADHD and is considered to have no misuse potential due to lack of dopamine modulation.58 In one study, 15 patients received atomoxetine added to their antidepressant, and 60% experienced significant symptom reduction.59 A chart review noted decreases in fatigue and depression when atomoxetine was added to an SRI, mirtazapine, or amitriptyline.60 However, in a DBPC trial, atomoxetine did not lead to significant changes in depression.61
Discussion
There is a limited amount of high-quality evidence to support psychostimulant augmentation, as noted by the relatively few DBPC trials, most of short duration. The evidence supports their efficacy primarily for unipolar depression, as 14 studies evaluated patients with unipolar depression, whereas only 7 studies evaluated patients with bipolar depression. The remaining studies recruited patients with both depression types. Collectively, modafinil and armodafinil have the most evidence in DBPC trials.
There are relatively few DBPC trials with high power and sufficient duration for dextroamphetamine and methylphenidate preparations. This discovery is surprising, considering the duration that these medications have been available. However, several chart reviews and open-label trials provided some evidence to support their use in patients without a history of substance misuse or cardiac conditions.62 Osmotic controlled- release oral system methylphenidate seems to be ineffective, and the efficacy of atomoxetine for augmentation
is uncertain.
Precautions
Prescribing physicians who offer stimulants should consider potential AEs, such as psychosis, anorexia, anxiety, insomnia, mood changes (eg, anger), misuse, addiction, mania, and cardiovascular problems. Psychostimulants have been implicated in precipitating psychosis.63,64 However, in a 12-month study of 250 adults with ADHD, 73 reported AEs, and only 31 discontinued the stimulant. Adverse effects leading to discontinuation included mood instability (n = 7), agitation (n = 6), irritability (n = 4), or decreased appetite (n = 4).65
Although associated with the risks of anorexia and insomnia in patients with ADHD, methylphenidate rapidly improved daytime sleepiness and mood, and—paradoxically—appetite and nighttime sleep in medically ill elderly patients with depression.66 Misuse or abuse of methylphenidate and dextroamphetamine were noted in 23% of patients referred for substance misuse.67 Nonetheless, little evidence exists that these drugs possess significant misuse potential in patients taking them as prescribed. As a prodrug, lisdexamfetamine is hypothesized to have less abuse potential compared with dextroamphetamine and methylphenidate, but it carries the same prescribing and monitoring precautions.68 Risks related to stimulant usage extend to manic symptoms.69 Patients with bipolar disorder should not receive stimulants if they have a history of stimulant-induced mania, rapid cycling, or psychosis.70
Long-term cardiovascular safety data exist for dextroamphetamine and methylphenidate but are limited or unavailable for modafinil, armodafinil, and atomoxetine. A retrospective cohort study found no significant increase in the number of cardiac events in patients receiving dextroamphetamine,
methylphenidate, or atomoxetine for an average of 1 year compared with controls.71 Another cohort study of > 44,000 patients found that initiation of
methylphenidate was associated with increased risk of sudden death or arrhythmia, but the risk was attributed to an unmeasured confounding factor, as the authors found a negative correlation between methylphenidate dose and all cardiovascular events.72
Recent practice guidelines recommend that before prescribing stimulants, clinicians should perform a physical examination (including heart and lung auscultation), obtain vital signs and height and weight, and request an electrocardiogram in case of abnormal findings on a cardiovascular examination or in case of a personal or family history of heart disease. Before offering atomoxetine, clinicians should evaluate the patient for a history of liver disease (and check liver function studies in case of a positive history). Clinicians should also assess risk of self-harm prior to initiating psychostimulant therapy.73 Throughout treatment, clinicians should evaluate the patient for changes in blood pressure, pulse, weight or mood, as well as the development of dependence or misuse. Urine toxicology testing is recommended for dextroamphetamine and methylphenidate to screen for adherence and diversion.
Limitations
Using only PubMed and MEDLINE databases limited the search to articles published in English after 1985, excluding letters and case reports to identify studies with higher evidence (the studies were not weighted based on study design). In addition, the studies had certain limitations. These include a limited number of DBPC trials, most were of short duration. It is also difficult to compare studies due to various rating scales used and concurrent
medication regimens of study subjects. These limitations raise questions surrounding the long-term efficacy of stimulants, and there is no consensus for how long a stimulant should be continued if beneficial. Longer, higherpowered, DBPC trials are warranted to determine longterm efficacy and safety of stimulant augmentation.62
Conclusion
For patients with depression who have not responded to other augmentation strategies, psychostimulants may be offered to improve mood, energy, and concentration. For clinicians considering stimulant augmentation, modafinil and armodafinil are reasonable choices given their efficacy in double-blind, placebo-controlled trials and lower risk of misuse. Dextroamphetamine (particularly lisdexamphetamine) and methylphenidate may be appropriate for patients who have not benefited from or tolerated modafinil or armodafinil, provided these patients do not have a medical history of cardiac disease or current substance use.
Osmotic controlled-release oral system methylphenidate seems to be ineffective as an augmenting agent. The efficacy of atomoxetine for augmentation is questionable, but atomoxetine could be offered if other stimulants were contraindicated, ineffective, or poorly tolerated. Both OROS methylphenidate and atomoxetine should be evaluated in additional trials before they can be recommended as augmentation therapies. Certain psychostimulants may be appropriate and reasonable adjunctive pharmacotherapies for patients with unipolar or bipolar depression who have failed other augmentation strategies, for patients who have significant fatigue or cognitive complaints, or for elderly patients with melancholic or somatic features of depression.
Acknowledgements
The authors thank Maureen Humphrey-Shelton and Kathy Thomas for their help in obtaining references.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distribution of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Solomon DA, Keller MB, Leon AC, et al. Multiple recurrences of major depressive disorder. Am J Psychiatry. 2000;157(2):229-233.
3. Katon WJ, Fan MY, Lin EH, Unützer J. Depressive symptom deterioration in a large
primary care-based elderly cohort. Am J Geriatr Psychiatry. 2006;14(3):246-254.
4. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
5. McIntyre RS, Filteau M-J, Martin L, et al. Treatment-resistant depression: Definitions, review of the evidence, and algorithmic approach. J Affect Disord. 2014;156:1-7.
6. Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush AJ. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Focus. 2012;10(4):510-517.
7. Kudlow PA, Cha DS, McIntyre RS. Predicting treatment response in major depressive disorder: The impact of early symptomatic improvement. Can J Psychiatry. 2012;57(12):782-788.
8. Ruhé HG, van Rooijen G, Spijker J, Peeters FP, Schene AH. Staging methods for treatment resistant depression. A systematic review. J Affect Disord. 2012;137(1-3):35-45.
9. Bauer M, Dopfmer S. Lithium augmentation treatment-resistant depression: Metaanalysis of placebo-controlled studies. J Clin Psychopharmacol. 1999;19(5):427-434.
10. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: A STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530.
11. Nierenberg AA, Papakostas GI, Petersen T, et al. Lithium augmentation of nortriptyline
for subjects resistant to multiple antidepressants. J Clin Psychopharmacol. 2003;23(1):92-95.
12. Connolly KR, Thase ME. If at first you don’t succeed: A review of the evidence for antidepressant augmentation, combination, and switching strategies. Drugs. 2011;71(1):43-64.
13. Trivedi MH, Fava M, Wisniewski SR, et al; STAR*D Study Team. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354(12):1243-1252.
14. Papakostas GI, Shelton RC, Smith J, Fava M. Augmentation of antidepressants with atypical antipsychotic medications for treatment resistant major depressive disorder: A meta-analysis. J Clin Psychiatry. 2007;68(6):826-831.
15. Mahmoud RA, Pandina GJ, Turkoz I, et al. Risperidone for treatment-refractory major depressive disorder: A randomized trial. Ann Intern Med. 2007;147(9):593-602.
16. Barbee JG, Conrad EJ, Jamhour NJ. The effectiveness of olanzapine, risperidone, quetiapine, and ziprasidone as augmentation agents in treatment resistant depressive disorder. J Clin Psychiatry. 2004;65(7):975-981.
17. Fatemi SH, Emamian ES, Kist DA. Venlafaxine and bupropion combination therapy in a case of treatment-resistant depression. Ann Pharmacother.1999;33(6):701-703.
18. Carpenter LL, Yasman S, Price LH. A double-blind, placebo-controlled study of antidepressant augmentation with mirtazapine. Biol Psychiatry. 2002;51(2):183-188.
19. Hannan N, Hamzah Z, Akinpeloye HO, Meagher D. Venlafaxine-mirtazapine combination therapy in the treatment of persistent depressive illness. J Psychopharmacol. 2007;21(2):161-164.
20. McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: A STAR*D report. Am J Psychiatry. 2006;163(9):1531-1541.
21. Blier P, Ward HE, Tremblay P, Laberge L, Hébert C, Bergeron R. Combination of antidepressant medications from treatment initiation for major depressive disorder: A double-blind randomized study. Am J Psychiatry. 2010;167(3):281-288.
22. Papakostas GI, Mischoulon D, Shyu I, Alpert JE, Fava M. S-adenosyl methionine (SAMe) augmentation of serotonin reuptake inhibitors for antidepressant nonresponders with major depressive disorder: A double blind randomized clinical trial. Am J Psychiatry. 2010;167(8):942-948.
23. Papakostas GI, Shelton RC, Zajecka JM, et al. L-methylfolate as adjunctive therapy
for SSRI-resistant major depression: Results of two randomized, double-blind,
parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
24. Korston TR. Drugs of abuse. In: Katzung BG, ed. Basic and Clinical Pharmacology. 9th ed. New York, NY: McGraw-Hill; 2004:521-523.
25. Feighner JP, Herbstein J, Damlouji N. Combined MAOI, TCA, and direct stimulant therapy of treatment-resistant depression. J Clin Psychiatry. 1985;46(6):206-209.
26. Fawcett J, Kravitz HM, Zajecka JM, Schaff MR. CNS stimulant potentiation of monoamine oxidase inhibitors in treatment-refractory depression. J Clin Psychopharmacol. 1991;11(2):127-132.
27. Stoll AL, Pillay SS, Diamond L, Workum SB, Cole JO. Methylphenidate augmentation of serotonin selective reuptake inhibitors: A case series. J Clin Psychiatry. 1996;57(2):72-76.
28. Masand PS, Anand VS, Tanquary JF. Psychostimulant augmentation of second generation antidepressants: A case series. Depress Anxiety. 1998;7(2):89-91.
29. Trivedi MH, Cutler AJ, Richards C, et al. A randomized control trial of the efficacy and safety of lisdesxamfetamine dimesylate as augmentation therapy in adults with residual symptoms of major depressive disorder after treatment with escitalopram. J Clin Psychiatry. 2013;74(8):802-809.
30. Madhoo M, Keefe RS, Roth RM, et al. Lisdexamfetamine dimesylate augmentation in adults with persistent executive dysfunction after partial or full remission of major depressive disorder. Neuropsychopharmacology. 2014;39(6):1388-1398.
31. Parker G, Brotchie H. Do the old psychostimulant drugs have a role in managing treatment-resistant depression. Acta Psychiatr Scand. 2010;121(4):308-314.
32. Parker G, Brotchie H, McClure G, Fletcher K. Psychostimulants for managing unipolar and bipolar treatment-resistant melancholic depression: A medium term evaluation of cost benefits. J Affect Disord. 2013;151(1):360-364.
33. Lydon E, El-Mallakh RS. Naturalistic long-term use of methylphenidate in bipolar disorder. J Clin Psychopharmacol. 2006;26(5):516-518.
34. Carlson PJ, Merlock MC, Suppes T. Adjunctive stimulant use in patients with bipolar disorder: Treatment of residual depression and sedation. Bipolar Disord. 2004;6(5):416-420.
35. El-Mallakh RS. An open study of methylphenidate in bipolar depression. Bipolar Disord. 2000;2(1):56-59.
36. Ravindran AV, Kennedy SH, O’Donovan MC, Fallu A, Camacho F, Binder CE. Osmotic-release oral system methylphenidate augmentation of antidepressant monotherapy in major depressive disorder: Results of a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2008;69(1):87-94.
37. Patkar AA, Masand PS, Pae CU, et al. A randomized, double-blind, placebocontrolled
trial of augmentation with an extended release formulation of methylphenidate in outpatients with treatment-resistant depression. J Clin Psychopharmacol. 2006;26(6):653-656.
38. Lee H, Kim SW, Kim JM, Shin IS, Yang SJ, Yoon JS Comparing effects of methylphenidate, sertraline, and placebo on neuropsychiatric sequelae in patients with
traumatic brain injury. Hum Psychopharmacol. 2005;20(2):97-104.
39. Gualtieri CT, Evans RW. Stimulant treatment for the neurobehavioural sequelae of traumatic brain injury. Brain Inj. 1988;2(4):273-290.
40. Provigil [package insert]. North Wales, PA: Cephalon Inc; 2015.
41. Nuvigil [package insert]. Frazer, PA: Cephalon, Inc; 2013.
42. Menza MA, Kaufman KR, Castellanos A. Modafinil augmentation of antidepressant treatment in depression. J Clin Psychiatry. 2000;61(5):378-381.
43. Markovitz PJ, Wagner S. An open-label trial of modafinil augmentation in patients with partial response to antidepressant therapy. J Clin Psychopharmacol. 2003;23(2):207-209.
44. Fernandes PP, Petty F. Modafinil for remitted bipolar depression with hypersomnia. Ann Pharmacother. 2003;37(12):1807-1809.
45. Nasr S. Modafinil as adjunctive therapy in depressed outpatients. Ann Clin Psychiatry. 2004;16(3):133-138.
46. DeBattista C, Lembke A, Solvason HB, Ghebremichael R, Poirier J. A prospective trial of modafinil as an adjunctive treatment of major depression. J Clin Psychopharmacol. 2004;24(1):87-90.
47. Nasr S, Wendt B, Steiner K. Absence of mood switch with and tolerance to modafinil: A replication study from a large private practice. J Affect Disord. 2006;95(1-3):111-114.
48. DeBattista C, Doghramji K, Menza MA, Rosenthal MH, Fieve RR; Modafinil in Depression Study Group. Adjunct modafinil for the short-term treatment of fatigue and sleepiness in patients with major depressive disorder: A preliminary doubleblind, placebo-controlled study. J Clin Psychiatry. 2003;64(9):1057-1064.
49. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry. 2007;164(8):1242-1249.
50. Fava M, Thase ME, DeBattista C, Doghramji K, Arora S, Hughes RJ. Modafinil augmentation of selective serotonin reuptake inhibitor therapy in MDD partial responders with persistent fatigue and sleepiness. Ann Clin Psychiatry. 2007;19(3):153-159.
51. Thase ME, Fava M, DeBattista C, Arora S, Hughes RJ. Modafinil augmentation of SSRI therapy in patients with major depressive disorder and excessive sleepiness and fatigue: A 12-week, open-label, extension study. CNS Spectr. 2006;11(2):93-102.
52. Fava M, Thase ME, DeBattista C. A multicenter, placebo-controlled study of modafinil augmentation in partial responders to selective serotonin reuptake inhibitors with persistent fatigue and sleepiness. J Clin Psychiatry. 2005;66(1):85-93.
53. Abolfazli R, Hosseini M, Ghanizadeh A, et al. Double-blind randomized parallelgroup clinical trial of efficacy of the combination fluoxetine plus modafinil versus fluoxetine plus placebo in the treatment of major depression. Depress Anxiety. 2011;28(4):297-302.
54. Rasmussen NA, Schrøder P, Olsen LR, Brødsgaard M, Undén M, Bech P. Modafinil augmentation in depressed patients with partial response to antidepressants: A pilot study on self-reported symptoms covered by the Major Depression Inventory (MDI) and the Symptom Checklist (SCL-92). Nord J Psychiatry. 2005;59(3):173-178.
55. Dunlop BW, Crits-Christoph P, Evans DL, et al. Coadministration of modafinil and a selective serotonin reuptake inhibitor from the initiation of treatment of major depressive disorder with fatigue and sleepiness: A double-blind, placebocontrolled study. J Clin Psychopharmacol. 2007;27(6):614-619.
56. Calabrese JR, Ketter TA, Youakim JM, Tiller JM, Yang R, Frye MA. Adjunctive armodafinil
for major depressive episodes associated with bipolar I disorder: A randomized multicenter, double-blind, placebo-controlled, proof-of-concept study. J Clin Psychiatry. 2010;71(10):1363-1370.
57. Calabrese JR, Frye MA, Yang R, Ketter TA; Armodafinil Treatment Trial Study Network. Efficacy and safety of adjunctive armodafinil in adults with major depressive episodes associated with bipolar I disorder: A randomized, double-blind, placebo-controlled, multicenter trial. J Clin Psychiatry. 2014;75(10):1054-1061.
58. Strattera [package insert]. Indianapolis, IN. Lilly; 2015.
59. Carpenter LL, Milosavljevic N, Schecter JM, Tyrka AR, Price LH. Augmentation with open-label atomoxetine for partial or nonresponse to antidepressants. J Clin Psychiatry. 2005;66(10):1234-1238.
60. Papakostas GI, Petersen TJ, Burns AM, Fava M. Adjunctive atomoxetine for residual
fatigue in major depressive disorder. J Psychiatr Res. 2006;40(4):370-373.
61. Michelson D, Adler LA, Amsterdam JD, et al. Addition of atomoxetine for depression
incompletely responsive to sertraline: A randomized, double-blind, placebocontrolled study. J Clin Psychiatry. 2007;68(4):582-587.
62. Corp SA, Gitlin MJ, Altshuler LL. A review of the use of stimulants and stimulant alternatives in treating bipolar depression and major depressive disorder. J Clin Psychiatry. 2014;75(9):1010-1018.
63. Kraemer M, Uekermann J, Wiltfang J, Kis B. Methylphenidate-induced psychosis in adult attention-deficit/hyperactivity disorder: Report of 3 new cases and review of the literature. Clin Neuropharmacol. 2010;33(4):204-206.
64. Berman SM, Kuczenski R, McCracken JT, London ED. Potential adverse effects of amphetamine treatment on brain and behavior: A review. Mol Psychiatry. 2009;14(2):123-142.
65. Fredriksen M, Dahl AA, Martinsen EW, Klungsøyr O, Haavik J, Peleikis DE. Effectiveness of one-year pharmacological treatment of adult attention-deficit/hyperactivity disorder (ADHD): An open-label prospective study of time in treatment, dose, side-effects and comorbidity. Eur Neuropsychopharmacol. 2014;24(12):1873-1874.
66. Hardy SE. Methylphenidate for the treatment of depressive symptoms, including fatigue and apathy, in medically ill older adults and terminally ill adults. Am J Geriatr Pharmacother. 2009;7(1):34-59.
67. Williams RJ, Goodale LA, Shay-Fiddler MA, Gloster SP, Chang SY. Methylphenidate and dextroamphetamine abuse in substance-abusing adolescents. Am J Addict. 2004;13(4):381-389.
68. Madaan V, Kolli V, Bestha DP, Shah MJ. Update on optimal use of lisdexamfetamine in the treatment of ADHD. Neuropsychiatr Dis Treat. 2013;9:977-983.
69. Ross RG. Psychotic and manic-like symptoms during stimulant treatment of attention deficit hyperactivity disorder. Am J Psychiatry. 2006;163(7):1149-1152.
70. Dell’Osso B, Ketter TA. Use of adjunctive stimulants in adult bipolar depression. Int J Neuropsychopharmacol. 2013;16(1):55-68.
71. Habel LA, Cooper WO, Sox CM, et al. ADHD medications and risk of serious cardiovascular events in young and middle-aged adults. JAMA. 2011;306(24):2673-2683.
72. Schelleman H, Bilker WB, Kimmel SE, et al. Methylphenidate and risk of serious cardiovascular events in adults. Am J Psychiatry. 2012;169(2):178-185.
73. Bolea-Alamañac B, Nutt DJ, Adamou M, et al; British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological management of attention deficit hyperactivity disorder: Update on recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(3):179-203.
74. Moher D, Liberati A, Tetzlaff J, Altman DG; The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009;6(6):e1000097.
Depression is a common condition that significantly impairs social and occupational functioning. Many patients do not respond to first-line pharmacotherapies and are considered to have treatment-resistant depression (TRD). These patients may benefit from augmentation of their antidepressant to reduce depression. Multiple medications have demonstrated various degrees of efficacy for augmentation, including psychostimulants. This article reviews studies of psychostimulants as augmentation agents for TRD and discusses risks, offers advice, and makes recommendations for clinicians who prescribe stimulants.
Background
Major depressive disorder (MDD) is a common psychiatric condition that significantly impairs quality of life.1 It is a recurrent illness, averaging 2 relapses per decade. The probability of recurrence increases with the number of depressive episodes.2,3 A patient who experiences major depressive episodes alternating with euthymia has unipolar depression; whereas one who experiences major depressive episodes alternating with episodes of mania or hypomania has bipolar depression.4
Despite adequate dose and duration of pharmacotherapy, many individuals with unipolar or bipolar depression do not achieve and sustain remission.5 Remission rates decrease and relapse rates increase with subsequent failed antidepressant trials.6 It is difficult to identify factors that predict treatment resistance, but one review of antidepressant studies found that patients who did not demonstrate a response within 3 weeks of medication initiation were less likely to respond after a longer duration.7
Treatment-resistant depression is commonly, but not universally, defined as lack of response after trials of 2 or more antidepressants with different mechanisms of action for sufficient duration.5 This definition will be used here as well. Other definitions have proposed stages of TRD, but these require further study to evaluate their reliability and predictive utility.8 Due to lack of consensus regarding the definition of TRD, it is not possible to determine the exact prevalence of TRD.
Patients with TRD may benefit from augmentation of their medication regimen. Augmentation with lithium has yielded conflicting results, and its efficacy with newer antidepressants is not well studied.9-12 Triiodothyronine, buspirone, and pindolol have demonstrated some efficacy when added to serotonin reuptake inhibitors (SRIs).10,12,13 Second-generation antipsychotic drugs, antidepressant drug combinations, omega-3 fatty acids, S-adenosyl methionine (SAMe), and L-methylfolate have demonstrated some efficacy in some studies as well.12,14-23 In patients with depression who have not responded to these strategies, psychostimulant augmentation may be appropriate.
Methods
A literature search was conducted following an algorithmic approach in the MEDLINE/PubMed database for studies in English from January 1985 to August 2014 of stimulants as augmenting agents for depression, using the Medical Subject Headings stimulant, depression, and augmentation, combined with an AND operator. The search was limited to adult humans and excluded case reports and letters, to identify studies with stronger evidence. Also excluded were studies using caffeine (to augment electroconvulsive therapy for depression) and pemoline as the sole augmenting stimulant as well as studies of patients with comorbid mental health diagnoses and studies that initiated stimulants and antidepressants simultaneously to assess antidepressant response.
This review organized results by stimulant rather than by depression type, even though some studies used > 1 stimulant or recruited patients with different types of depression. Although prevalence, prognosis, and monotherapy differ for unipolar and bipolar depression, psychostimulants target similar symptoms, despite augmenting different monotherapies in unipolar and bipolar depression. Therefore, no distinction is made between assessing studies of stimulants for unipolar and bipolar depression.
Results
A total of 70 articles were identified, and 31 studies met inclusion criteria (Figure). Of the studies included, 12 were double-blind, placebo-controlled (DBPC) trials and 19 were retrospective chart reviews or open studies. Most studies evaluated depression, using validated scales, such as the Hamilton Depression Rating Scale, Montgomery-Asberg Depression Rating Scale, Clinical Global Impressions of Severity, Inventory of Depressive Symptoms, Carroll Depression Rating Scale, Global Assessment of Functioning, Quick Inventory of Depressive Symptomatology, or the Psychiatric Symptom Assessment Scale. Study details are provided in Tables 1 to 4.
Dextroamphetamine and Methylphenidate
Dextroamphetamine and methylphenidate are indicated for the treatment of attention-deficit/hyperactivity disorder (ADHD) and exert their effects by inhibiting uptake of norepinephrine and dopamine.24 In one chart review, patients received dextroamphetamine or methylphenidate augmentation of monoamine oxidase inhibitors (MAOIs) alone or with concurrent tricyclic antidepressants; the majority reported decreased depression.25 In an openlabel trial, dextroamphetamine was titrated to efficacy in patients who were receiving an MAOI with or without pemoline.26 Nearly 80% of patients reported long-lasting improvement in depression. In an open-label trial, all patients reported decreased depression when methylphenidate was added to SRIs; however, no scales were used.27
In a case series, patients with both major depression and persistent depressive disorder (dysthymia) experienced a substantial, quick, and sustained response to dextroamphetamine or methylphenidate augmentation.28 Addition of lisdexamfetamine significantly reduced depressive symptoms in individuals with inadequate response to escitalopram.29 Patients with full or partial remission of depression noted improved executive function and residual depressive symptoms after lisdexamfetamine was added to SRI monotherapy.30 In a trial in which patients received dexamphetamine or methylphenidate as monotherapy or augmentation, 30% to 34% of patients reported mood improvement, but 36% reported no improvement.31 In an extension study, low-dose psychostimulants quickly diminished melancholia.32
Methylphenidate was safe and effective in patients with bipolar depression receiving treatment for 1 to 5 years; 44% evidenced significant improvement.33 When offered to patients with bipolar depression, patients receiving methylphenidate or dextroamphetamine reported less depression or sedation and did not develop tolerance, mania, or misuse.34 A case series concluded that methylphenidate addition to mood stabilizers was generally effective and safe.35
However, not all preparations of methylphenidate have demonstrated efficacy. In one study, osmotic controlledrelease oral system (OROS) methylphenidate improved apathy and fatigue but not overall depression.36 Although OROS methylphenidate similarly failed to demonstrate statistically significant efficacy in another study, more responders were documented in the treatment group.37
Although this review focuses on stimulants as augmenting agents in patients with depression, it is worth noting the limited number of studies evaluating stimulants’ effect on depression in patients with traumatic brain injury. This observation is of concern, as these conditions are frequently comorbid in returning veterans. One study noted that methylphenidate was an effective monotherapy for depression; whereas another study found that methylphenidate monotherapy reduced depression as well as sertraline, was better tolerated, and improved fatigue and cognition.38,39
Modafinil and Armodafinil
Modafinil and armodafinil (the R-enantiomer of modafinil) are indicated for improving wakefulness in individuals with narcolepsy, obstructive sleep apnea, and shift work sleep disorder by modulating glutamate, gamma amino-butyric acid, and histamine.40,41 Although they increase extracellular dopamine concentrations, they do not cause an increase in dopamine release and may have less misuse potential than that of dextroamphetamine and methylphenidate.40,41 In a study of 7 patients with unipolar or bipolar depression, all patients achieved full or partial remission with minimal adverse effects (AEs).42 In a prospective study, 41% of patients reported only mild depression or full remission with modafinil augmentation.43
Multiple trials and a pooled analysis noted decreased depression and fatigue and improved cognition in patients receiving modafinil augmentation compared with mood stabilizers or antidepressants.44-49 Modafinil is a useful adjunct for partial responders to SRIs, resulting in rapid mood improvement and decreased fatigue.50-54 However, in one study, modafinil did not demonstrate efficacy compared with placebo. This result was attributed to premature study termination after 2 modafinil-treated patients developed suicidal ideation.55 A post hoc analysis found no difference in frequency of suicidal ideation between groups.
Two DBPC studies evaluated armodafinil in patients with bipolar depression. In both studies it was added to a mood-stabilizing agent (lithium, valproate, aripiprazole, olanzapine, lamotrigine, risperidone, or ziprasidone), and patients receiving armodafinil reported significant reductions
in depression.56,57
Atomoxetine
Atomoxetine is a norepinephrine reuptake inhibitor indicated for the treatment of ADHD and is considered to have no misuse potential due to lack of dopamine modulation.58 In one study, 15 patients received atomoxetine added to their antidepressant, and 60% experienced significant symptom reduction.59 A chart review noted decreases in fatigue and depression when atomoxetine was added to an SRI, mirtazapine, or amitriptyline.60 However, in a DBPC trial, atomoxetine did not lead to significant changes in depression.61
Discussion
There is a limited amount of high-quality evidence to support psychostimulant augmentation, as noted by the relatively few DBPC trials, most of short duration. The evidence supports their efficacy primarily for unipolar depression, as 14 studies evaluated patients with unipolar depression, whereas only 7 studies evaluated patients with bipolar depression. The remaining studies recruited patients with both depression types. Collectively, modafinil and armodafinil have the most evidence in DBPC trials.
There are relatively few DBPC trials with high power and sufficient duration for dextroamphetamine and methylphenidate preparations. This discovery is surprising, considering the duration that these medications have been available. However, several chart reviews and open-label trials provided some evidence to support their use in patients without a history of substance misuse or cardiac conditions.62 Osmotic controlled- release oral system methylphenidate seems to be ineffective, and the efficacy of atomoxetine for augmentation
is uncertain.
Precautions
Prescribing physicians who offer stimulants should consider potential AEs, such as psychosis, anorexia, anxiety, insomnia, mood changes (eg, anger), misuse, addiction, mania, and cardiovascular problems. Psychostimulants have been implicated in precipitating psychosis.63,64 However, in a 12-month study of 250 adults with ADHD, 73 reported AEs, and only 31 discontinued the stimulant. Adverse effects leading to discontinuation included mood instability (n = 7), agitation (n = 6), irritability (n = 4), or decreased appetite (n = 4).65
Although associated with the risks of anorexia and insomnia in patients with ADHD, methylphenidate rapidly improved daytime sleepiness and mood, and—paradoxically—appetite and nighttime sleep in medically ill elderly patients with depression.66 Misuse or abuse of methylphenidate and dextroamphetamine were noted in 23% of patients referred for substance misuse.67 Nonetheless, little evidence exists that these drugs possess significant misuse potential in patients taking them as prescribed. As a prodrug, lisdexamfetamine is hypothesized to have less abuse potential compared with dextroamphetamine and methylphenidate, but it carries the same prescribing and monitoring precautions.68 Risks related to stimulant usage extend to manic symptoms.69 Patients with bipolar disorder should not receive stimulants if they have a history of stimulant-induced mania, rapid cycling, or psychosis.70
Long-term cardiovascular safety data exist for dextroamphetamine and methylphenidate but are limited or unavailable for modafinil, armodafinil, and atomoxetine. A retrospective cohort study found no significant increase in the number of cardiac events in patients receiving dextroamphetamine,
methylphenidate, or atomoxetine for an average of 1 year compared with controls.71 Another cohort study of > 44,000 patients found that initiation of
methylphenidate was associated with increased risk of sudden death or arrhythmia, but the risk was attributed to an unmeasured confounding factor, as the authors found a negative correlation between methylphenidate dose and all cardiovascular events.72
Recent practice guidelines recommend that before prescribing stimulants, clinicians should perform a physical examination (including heart and lung auscultation), obtain vital signs and height and weight, and request an electrocardiogram in case of abnormal findings on a cardiovascular examination or in case of a personal or family history of heart disease. Before offering atomoxetine, clinicians should evaluate the patient for a history of liver disease (and check liver function studies in case of a positive history). Clinicians should also assess risk of self-harm prior to initiating psychostimulant therapy.73 Throughout treatment, clinicians should evaluate the patient for changes in blood pressure, pulse, weight or mood, as well as the development of dependence or misuse. Urine toxicology testing is recommended for dextroamphetamine and methylphenidate to screen for adherence and diversion.
Limitations
Using only PubMed and MEDLINE databases limited the search to articles published in English after 1985, excluding letters and case reports to identify studies with higher evidence (the studies were not weighted based on study design). In addition, the studies had certain limitations. These include a limited number of DBPC trials, most were of short duration. It is also difficult to compare studies due to various rating scales used and concurrent
medication regimens of study subjects. These limitations raise questions surrounding the long-term efficacy of stimulants, and there is no consensus for how long a stimulant should be continued if beneficial. Longer, higherpowered, DBPC trials are warranted to determine longterm efficacy and safety of stimulant augmentation.62
Conclusion
For patients with depression who have not responded to other augmentation strategies, psychostimulants may be offered to improve mood, energy, and concentration. For clinicians considering stimulant augmentation, modafinil and armodafinil are reasonable choices given their efficacy in double-blind, placebo-controlled trials and lower risk of misuse. Dextroamphetamine (particularly lisdexamphetamine) and methylphenidate may be appropriate for patients who have not benefited from or tolerated modafinil or armodafinil, provided these patients do not have a medical history of cardiac disease or current substance use.
Osmotic controlled-release oral system methylphenidate seems to be ineffective as an augmenting agent. The efficacy of atomoxetine for augmentation is questionable, but atomoxetine could be offered if other stimulants were contraindicated, ineffective, or poorly tolerated. Both OROS methylphenidate and atomoxetine should be evaluated in additional trials before they can be recommended as augmentation therapies. Certain psychostimulants may be appropriate and reasonable adjunctive pharmacotherapies for patients with unipolar or bipolar depression who have failed other augmentation strategies, for patients who have significant fatigue or cognitive complaints, or for elderly patients with melancholic or somatic features of depression.
Acknowledgements
The authors thank Maureen Humphrey-Shelton and Kathy Thomas for their help in obtaining references.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Depression is a common condition that significantly impairs social and occupational functioning. Many patients do not respond to first-line pharmacotherapies and are considered to have treatment-resistant depression (TRD). These patients may benefit from augmentation of their antidepressant to reduce depression. Multiple medications have demonstrated various degrees of efficacy for augmentation, including psychostimulants. This article reviews studies of psychostimulants as augmentation agents for TRD and discusses risks, offers advice, and makes recommendations for clinicians who prescribe stimulants.
Background
Major depressive disorder (MDD) is a common psychiatric condition that significantly impairs quality of life.1 It is a recurrent illness, averaging 2 relapses per decade. The probability of recurrence increases with the number of depressive episodes.2,3 A patient who experiences major depressive episodes alternating with euthymia has unipolar depression; whereas one who experiences major depressive episodes alternating with episodes of mania or hypomania has bipolar depression.4
Despite adequate dose and duration of pharmacotherapy, many individuals with unipolar or bipolar depression do not achieve and sustain remission.5 Remission rates decrease and relapse rates increase with subsequent failed antidepressant trials.6 It is difficult to identify factors that predict treatment resistance, but one review of antidepressant studies found that patients who did not demonstrate a response within 3 weeks of medication initiation were less likely to respond after a longer duration.7
Treatment-resistant depression is commonly, but not universally, defined as lack of response after trials of 2 or more antidepressants with different mechanisms of action for sufficient duration.5 This definition will be used here as well. Other definitions have proposed stages of TRD, but these require further study to evaluate their reliability and predictive utility.8 Due to lack of consensus regarding the definition of TRD, it is not possible to determine the exact prevalence of TRD.
Patients with TRD may benefit from augmentation of their medication regimen. Augmentation with lithium has yielded conflicting results, and its efficacy with newer antidepressants is not well studied.9-12 Triiodothyronine, buspirone, and pindolol have demonstrated some efficacy when added to serotonin reuptake inhibitors (SRIs).10,12,13 Second-generation antipsychotic drugs, antidepressant drug combinations, omega-3 fatty acids, S-adenosyl methionine (SAMe), and L-methylfolate have demonstrated some efficacy in some studies as well.12,14-23 In patients with depression who have not responded to these strategies, psychostimulant augmentation may be appropriate.
Methods
A literature search was conducted following an algorithmic approach in the MEDLINE/PubMed database for studies in English from January 1985 to August 2014 of stimulants as augmenting agents for depression, using the Medical Subject Headings stimulant, depression, and augmentation, combined with an AND operator. The search was limited to adult humans and excluded case reports and letters, to identify studies with stronger evidence. Also excluded were studies using caffeine (to augment electroconvulsive therapy for depression) and pemoline as the sole augmenting stimulant as well as studies of patients with comorbid mental health diagnoses and studies that initiated stimulants and antidepressants simultaneously to assess antidepressant response.
This review organized results by stimulant rather than by depression type, even though some studies used > 1 stimulant or recruited patients with different types of depression. Although prevalence, prognosis, and monotherapy differ for unipolar and bipolar depression, psychostimulants target similar symptoms, despite augmenting different monotherapies in unipolar and bipolar depression. Therefore, no distinction is made between assessing studies of stimulants for unipolar and bipolar depression.
Results
A total of 70 articles were identified, and 31 studies met inclusion criteria (Figure). Of the studies included, 12 were double-blind, placebo-controlled (DBPC) trials and 19 were retrospective chart reviews or open studies. Most studies evaluated depression, using validated scales, such as the Hamilton Depression Rating Scale, Montgomery-Asberg Depression Rating Scale, Clinical Global Impressions of Severity, Inventory of Depressive Symptoms, Carroll Depression Rating Scale, Global Assessment of Functioning, Quick Inventory of Depressive Symptomatology, or the Psychiatric Symptom Assessment Scale. Study details are provided in Tables 1 to 4.
Dextroamphetamine and Methylphenidate
Dextroamphetamine and methylphenidate are indicated for the treatment of attention-deficit/hyperactivity disorder (ADHD) and exert their effects by inhibiting uptake of norepinephrine and dopamine.24 In one chart review, patients received dextroamphetamine or methylphenidate augmentation of monoamine oxidase inhibitors (MAOIs) alone or with concurrent tricyclic antidepressants; the majority reported decreased depression.25 In an openlabel trial, dextroamphetamine was titrated to efficacy in patients who were receiving an MAOI with or without pemoline.26 Nearly 80% of patients reported long-lasting improvement in depression. In an open-label trial, all patients reported decreased depression when methylphenidate was added to SRIs; however, no scales were used.27
In a case series, patients with both major depression and persistent depressive disorder (dysthymia) experienced a substantial, quick, and sustained response to dextroamphetamine or methylphenidate augmentation.28 Addition of lisdexamfetamine significantly reduced depressive symptoms in individuals with inadequate response to escitalopram.29 Patients with full or partial remission of depression noted improved executive function and residual depressive symptoms after lisdexamfetamine was added to SRI monotherapy.30 In a trial in which patients received dexamphetamine or methylphenidate as monotherapy or augmentation, 30% to 34% of patients reported mood improvement, but 36% reported no improvement.31 In an extension study, low-dose psychostimulants quickly diminished melancholia.32
Methylphenidate was safe and effective in patients with bipolar depression receiving treatment for 1 to 5 years; 44% evidenced significant improvement.33 When offered to patients with bipolar depression, patients receiving methylphenidate or dextroamphetamine reported less depression or sedation and did not develop tolerance, mania, or misuse.34 A case series concluded that methylphenidate addition to mood stabilizers was generally effective and safe.35
However, not all preparations of methylphenidate have demonstrated efficacy. In one study, osmotic controlledrelease oral system (OROS) methylphenidate improved apathy and fatigue but not overall depression.36 Although OROS methylphenidate similarly failed to demonstrate statistically significant efficacy in another study, more responders were documented in the treatment group.37
Although this review focuses on stimulants as augmenting agents in patients with depression, it is worth noting the limited number of studies evaluating stimulants’ effect on depression in patients with traumatic brain injury. This observation is of concern, as these conditions are frequently comorbid in returning veterans. One study noted that methylphenidate was an effective monotherapy for depression; whereas another study found that methylphenidate monotherapy reduced depression as well as sertraline, was better tolerated, and improved fatigue and cognition.38,39
Modafinil and Armodafinil
Modafinil and armodafinil (the R-enantiomer of modafinil) are indicated for improving wakefulness in individuals with narcolepsy, obstructive sleep apnea, and shift work sleep disorder by modulating glutamate, gamma amino-butyric acid, and histamine.40,41 Although they increase extracellular dopamine concentrations, they do not cause an increase in dopamine release and may have less misuse potential than that of dextroamphetamine and methylphenidate.40,41 In a study of 7 patients with unipolar or bipolar depression, all patients achieved full or partial remission with minimal adverse effects (AEs).42 In a prospective study, 41% of patients reported only mild depression or full remission with modafinil augmentation.43
Multiple trials and a pooled analysis noted decreased depression and fatigue and improved cognition in patients receiving modafinil augmentation compared with mood stabilizers or antidepressants.44-49 Modafinil is a useful adjunct for partial responders to SRIs, resulting in rapid mood improvement and decreased fatigue.50-54 However, in one study, modafinil did not demonstrate efficacy compared with placebo. This result was attributed to premature study termination after 2 modafinil-treated patients developed suicidal ideation.55 A post hoc analysis found no difference in frequency of suicidal ideation between groups.
Two DBPC studies evaluated armodafinil in patients with bipolar depression. In both studies it was added to a mood-stabilizing agent (lithium, valproate, aripiprazole, olanzapine, lamotrigine, risperidone, or ziprasidone), and patients receiving armodafinil reported significant reductions
in depression.56,57
Atomoxetine
Atomoxetine is a norepinephrine reuptake inhibitor indicated for the treatment of ADHD and is considered to have no misuse potential due to lack of dopamine modulation.58 In one study, 15 patients received atomoxetine added to their antidepressant, and 60% experienced significant symptom reduction.59 A chart review noted decreases in fatigue and depression when atomoxetine was added to an SRI, mirtazapine, or amitriptyline.60 However, in a DBPC trial, atomoxetine did not lead to significant changes in depression.61
Discussion
There is a limited amount of high-quality evidence to support psychostimulant augmentation, as noted by the relatively few DBPC trials, most of short duration. The evidence supports their efficacy primarily for unipolar depression, as 14 studies evaluated patients with unipolar depression, whereas only 7 studies evaluated patients with bipolar depression. The remaining studies recruited patients with both depression types. Collectively, modafinil and armodafinil have the most evidence in DBPC trials.
There are relatively few DBPC trials with high power and sufficient duration for dextroamphetamine and methylphenidate preparations. This discovery is surprising, considering the duration that these medications have been available. However, several chart reviews and open-label trials provided some evidence to support their use in patients without a history of substance misuse or cardiac conditions.62 Osmotic controlled- release oral system methylphenidate seems to be ineffective, and the efficacy of atomoxetine for augmentation
is uncertain.
Precautions
Prescribing physicians who offer stimulants should consider potential AEs, such as psychosis, anorexia, anxiety, insomnia, mood changes (eg, anger), misuse, addiction, mania, and cardiovascular problems. Psychostimulants have been implicated in precipitating psychosis.63,64 However, in a 12-month study of 250 adults with ADHD, 73 reported AEs, and only 31 discontinued the stimulant. Adverse effects leading to discontinuation included mood instability (n = 7), agitation (n = 6), irritability (n = 4), or decreased appetite (n = 4).65
Although associated with the risks of anorexia and insomnia in patients with ADHD, methylphenidate rapidly improved daytime sleepiness and mood, and—paradoxically—appetite and nighttime sleep in medically ill elderly patients with depression.66 Misuse or abuse of methylphenidate and dextroamphetamine were noted in 23% of patients referred for substance misuse.67 Nonetheless, little evidence exists that these drugs possess significant misuse potential in patients taking them as prescribed. As a prodrug, lisdexamfetamine is hypothesized to have less abuse potential compared with dextroamphetamine and methylphenidate, but it carries the same prescribing and monitoring precautions.68 Risks related to stimulant usage extend to manic symptoms.69 Patients with bipolar disorder should not receive stimulants if they have a history of stimulant-induced mania, rapid cycling, or psychosis.70
Long-term cardiovascular safety data exist for dextroamphetamine and methylphenidate but are limited or unavailable for modafinil, armodafinil, and atomoxetine. A retrospective cohort study found no significant increase in the number of cardiac events in patients receiving dextroamphetamine,
methylphenidate, or atomoxetine for an average of 1 year compared with controls.71 Another cohort study of > 44,000 patients found that initiation of
methylphenidate was associated with increased risk of sudden death or arrhythmia, but the risk was attributed to an unmeasured confounding factor, as the authors found a negative correlation between methylphenidate dose and all cardiovascular events.72
Recent practice guidelines recommend that before prescribing stimulants, clinicians should perform a physical examination (including heart and lung auscultation), obtain vital signs and height and weight, and request an electrocardiogram in case of abnormal findings on a cardiovascular examination or in case of a personal or family history of heart disease. Before offering atomoxetine, clinicians should evaluate the patient for a history of liver disease (and check liver function studies in case of a positive history). Clinicians should also assess risk of self-harm prior to initiating psychostimulant therapy.73 Throughout treatment, clinicians should evaluate the patient for changes in blood pressure, pulse, weight or mood, as well as the development of dependence or misuse. Urine toxicology testing is recommended for dextroamphetamine and methylphenidate to screen for adherence and diversion.
Limitations
Using only PubMed and MEDLINE databases limited the search to articles published in English after 1985, excluding letters and case reports to identify studies with higher evidence (the studies were not weighted based on study design). In addition, the studies had certain limitations. These include a limited number of DBPC trials, most were of short duration. It is also difficult to compare studies due to various rating scales used and concurrent
medication regimens of study subjects. These limitations raise questions surrounding the long-term efficacy of stimulants, and there is no consensus for how long a stimulant should be continued if beneficial. Longer, higherpowered, DBPC trials are warranted to determine longterm efficacy and safety of stimulant augmentation.62
Conclusion
For patients with depression who have not responded to other augmentation strategies, psychostimulants may be offered to improve mood, energy, and concentration. For clinicians considering stimulant augmentation, modafinil and armodafinil are reasonable choices given their efficacy in double-blind, placebo-controlled trials and lower risk of misuse. Dextroamphetamine (particularly lisdexamphetamine) and methylphenidate may be appropriate for patients who have not benefited from or tolerated modafinil or armodafinil, provided these patients do not have a medical history of cardiac disease or current substance use.
Osmotic controlled-release oral system methylphenidate seems to be ineffective as an augmenting agent. The efficacy of atomoxetine for augmentation is questionable, but atomoxetine could be offered if other stimulants were contraindicated, ineffective, or poorly tolerated. Both OROS methylphenidate and atomoxetine should be evaluated in additional trials before they can be recommended as augmentation therapies. Certain psychostimulants may be appropriate and reasonable adjunctive pharmacotherapies for patients with unipolar or bipolar depression who have failed other augmentation strategies, for patients who have significant fatigue or cognitive complaints, or for elderly patients with melancholic or somatic features of depression.
Acknowledgements
The authors thank Maureen Humphrey-Shelton and Kathy Thomas for their help in obtaining references.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distribution of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Solomon DA, Keller MB, Leon AC, et al. Multiple recurrences of major depressive disorder. Am J Psychiatry. 2000;157(2):229-233.
3. Katon WJ, Fan MY, Lin EH, Unützer J. Depressive symptom deterioration in a large
primary care-based elderly cohort. Am J Geriatr Psychiatry. 2006;14(3):246-254.
4. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
5. McIntyre RS, Filteau M-J, Martin L, et al. Treatment-resistant depression: Definitions, review of the evidence, and algorithmic approach. J Affect Disord. 2014;156:1-7.
6. Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush AJ. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Focus. 2012;10(4):510-517.
7. Kudlow PA, Cha DS, McIntyre RS. Predicting treatment response in major depressive disorder: The impact of early symptomatic improvement. Can J Psychiatry. 2012;57(12):782-788.
8. Ruhé HG, van Rooijen G, Spijker J, Peeters FP, Schene AH. Staging methods for treatment resistant depression. A systematic review. J Affect Disord. 2012;137(1-3):35-45.
9. Bauer M, Dopfmer S. Lithium augmentation treatment-resistant depression: Metaanalysis of placebo-controlled studies. J Clin Psychopharmacol. 1999;19(5):427-434.
10. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: A STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530.
11. Nierenberg AA, Papakostas GI, Petersen T, et al. Lithium augmentation of nortriptyline
for subjects resistant to multiple antidepressants. J Clin Psychopharmacol. 2003;23(1):92-95.
12. Connolly KR, Thase ME. If at first you don’t succeed: A review of the evidence for antidepressant augmentation, combination, and switching strategies. Drugs. 2011;71(1):43-64.
13. Trivedi MH, Fava M, Wisniewski SR, et al; STAR*D Study Team. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354(12):1243-1252.
14. Papakostas GI, Shelton RC, Smith J, Fava M. Augmentation of antidepressants with atypical antipsychotic medications for treatment resistant major depressive disorder: A meta-analysis. J Clin Psychiatry. 2007;68(6):826-831.
15. Mahmoud RA, Pandina GJ, Turkoz I, et al. Risperidone for treatment-refractory major depressive disorder: A randomized trial. Ann Intern Med. 2007;147(9):593-602.
16. Barbee JG, Conrad EJ, Jamhour NJ. The effectiveness of olanzapine, risperidone, quetiapine, and ziprasidone as augmentation agents in treatment resistant depressive disorder. J Clin Psychiatry. 2004;65(7):975-981.
17. Fatemi SH, Emamian ES, Kist DA. Venlafaxine and bupropion combination therapy in a case of treatment-resistant depression. Ann Pharmacother.1999;33(6):701-703.
18. Carpenter LL, Yasman S, Price LH. A double-blind, placebo-controlled study of antidepressant augmentation with mirtazapine. Biol Psychiatry. 2002;51(2):183-188.
19. Hannan N, Hamzah Z, Akinpeloye HO, Meagher D. Venlafaxine-mirtazapine combination therapy in the treatment of persistent depressive illness. J Psychopharmacol. 2007;21(2):161-164.
20. McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: A STAR*D report. Am J Psychiatry. 2006;163(9):1531-1541.
21. Blier P, Ward HE, Tremblay P, Laberge L, Hébert C, Bergeron R. Combination of antidepressant medications from treatment initiation for major depressive disorder: A double-blind randomized study. Am J Psychiatry. 2010;167(3):281-288.
22. Papakostas GI, Mischoulon D, Shyu I, Alpert JE, Fava M. S-adenosyl methionine (SAMe) augmentation of serotonin reuptake inhibitors for antidepressant nonresponders with major depressive disorder: A double blind randomized clinical trial. Am J Psychiatry. 2010;167(8):942-948.
23. Papakostas GI, Shelton RC, Zajecka JM, et al. L-methylfolate as adjunctive therapy
for SSRI-resistant major depression: Results of two randomized, double-blind,
parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
24. Korston TR. Drugs of abuse. In: Katzung BG, ed. Basic and Clinical Pharmacology. 9th ed. New York, NY: McGraw-Hill; 2004:521-523.
25. Feighner JP, Herbstein J, Damlouji N. Combined MAOI, TCA, and direct stimulant therapy of treatment-resistant depression. J Clin Psychiatry. 1985;46(6):206-209.
26. Fawcett J, Kravitz HM, Zajecka JM, Schaff MR. CNS stimulant potentiation of monoamine oxidase inhibitors in treatment-refractory depression. J Clin Psychopharmacol. 1991;11(2):127-132.
27. Stoll AL, Pillay SS, Diamond L, Workum SB, Cole JO. Methylphenidate augmentation of serotonin selective reuptake inhibitors: A case series. J Clin Psychiatry. 1996;57(2):72-76.
28. Masand PS, Anand VS, Tanquary JF. Psychostimulant augmentation of second generation antidepressants: A case series. Depress Anxiety. 1998;7(2):89-91.
29. Trivedi MH, Cutler AJ, Richards C, et al. A randomized control trial of the efficacy and safety of lisdesxamfetamine dimesylate as augmentation therapy in adults with residual symptoms of major depressive disorder after treatment with escitalopram. J Clin Psychiatry. 2013;74(8):802-809.
30. Madhoo M, Keefe RS, Roth RM, et al. Lisdexamfetamine dimesylate augmentation in adults with persistent executive dysfunction after partial or full remission of major depressive disorder. Neuropsychopharmacology. 2014;39(6):1388-1398.
31. Parker G, Brotchie H. Do the old psychostimulant drugs have a role in managing treatment-resistant depression. Acta Psychiatr Scand. 2010;121(4):308-314.
32. Parker G, Brotchie H, McClure G, Fletcher K. Psychostimulants for managing unipolar and bipolar treatment-resistant melancholic depression: A medium term evaluation of cost benefits. J Affect Disord. 2013;151(1):360-364.
33. Lydon E, El-Mallakh RS. Naturalistic long-term use of methylphenidate in bipolar disorder. J Clin Psychopharmacol. 2006;26(5):516-518.
34. Carlson PJ, Merlock MC, Suppes T. Adjunctive stimulant use in patients with bipolar disorder: Treatment of residual depression and sedation. Bipolar Disord. 2004;6(5):416-420.
35. El-Mallakh RS. An open study of methylphenidate in bipolar depression. Bipolar Disord. 2000;2(1):56-59.
36. Ravindran AV, Kennedy SH, O’Donovan MC, Fallu A, Camacho F, Binder CE. Osmotic-release oral system methylphenidate augmentation of antidepressant monotherapy in major depressive disorder: Results of a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2008;69(1):87-94.
37. Patkar AA, Masand PS, Pae CU, et al. A randomized, double-blind, placebocontrolled
trial of augmentation with an extended release formulation of methylphenidate in outpatients with treatment-resistant depression. J Clin Psychopharmacol. 2006;26(6):653-656.
38. Lee H, Kim SW, Kim JM, Shin IS, Yang SJ, Yoon JS Comparing effects of methylphenidate, sertraline, and placebo on neuropsychiatric sequelae in patients with
traumatic brain injury. Hum Psychopharmacol. 2005;20(2):97-104.
39. Gualtieri CT, Evans RW. Stimulant treatment for the neurobehavioural sequelae of traumatic brain injury. Brain Inj. 1988;2(4):273-290.
40. Provigil [package insert]. North Wales, PA: Cephalon Inc; 2015.
41. Nuvigil [package insert]. Frazer, PA: Cephalon, Inc; 2013.
42. Menza MA, Kaufman KR, Castellanos A. Modafinil augmentation of antidepressant treatment in depression. J Clin Psychiatry. 2000;61(5):378-381.
43. Markovitz PJ, Wagner S. An open-label trial of modafinil augmentation in patients with partial response to antidepressant therapy. J Clin Psychopharmacol. 2003;23(2):207-209.
44. Fernandes PP, Petty F. Modafinil for remitted bipolar depression with hypersomnia. Ann Pharmacother. 2003;37(12):1807-1809.
45. Nasr S. Modafinil as adjunctive therapy in depressed outpatients. Ann Clin Psychiatry. 2004;16(3):133-138.
46. DeBattista C, Lembke A, Solvason HB, Ghebremichael R, Poirier J. A prospective trial of modafinil as an adjunctive treatment of major depression. J Clin Psychopharmacol. 2004;24(1):87-90.
47. Nasr S, Wendt B, Steiner K. Absence of mood switch with and tolerance to modafinil: A replication study from a large private practice. J Affect Disord. 2006;95(1-3):111-114.
48. DeBattista C, Doghramji K, Menza MA, Rosenthal MH, Fieve RR; Modafinil in Depression Study Group. Adjunct modafinil for the short-term treatment of fatigue and sleepiness in patients with major depressive disorder: A preliminary doubleblind, placebo-controlled study. J Clin Psychiatry. 2003;64(9):1057-1064.
49. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry. 2007;164(8):1242-1249.
50. Fava M, Thase ME, DeBattista C, Doghramji K, Arora S, Hughes RJ. Modafinil augmentation of selective serotonin reuptake inhibitor therapy in MDD partial responders with persistent fatigue and sleepiness. Ann Clin Psychiatry. 2007;19(3):153-159.
51. Thase ME, Fava M, DeBattista C, Arora S, Hughes RJ. Modafinil augmentation of SSRI therapy in patients with major depressive disorder and excessive sleepiness and fatigue: A 12-week, open-label, extension study. CNS Spectr. 2006;11(2):93-102.
52. Fava M, Thase ME, DeBattista C. A multicenter, placebo-controlled study of modafinil augmentation in partial responders to selective serotonin reuptake inhibitors with persistent fatigue and sleepiness. J Clin Psychiatry. 2005;66(1):85-93.
53. Abolfazli R, Hosseini M, Ghanizadeh A, et al. Double-blind randomized parallelgroup clinical trial of efficacy of the combination fluoxetine plus modafinil versus fluoxetine plus placebo in the treatment of major depression. Depress Anxiety. 2011;28(4):297-302.
54. Rasmussen NA, Schrøder P, Olsen LR, Brødsgaard M, Undén M, Bech P. Modafinil augmentation in depressed patients with partial response to antidepressants: A pilot study on self-reported symptoms covered by the Major Depression Inventory (MDI) and the Symptom Checklist (SCL-92). Nord J Psychiatry. 2005;59(3):173-178.
55. Dunlop BW, Crits-Christoph P, Evans DL, et al. Coadministration of modafinil and a selective serotonin reuptake inhibitor from the initiation of treatment of major depressive disorder with fatigue and sleepiness: A double-blind, placebocontrolled study. J Clin Psychopharmacol. 2007;27(6):614-619.
56. Calabrese JR, Ketter TA, Youakim JM, Tiller JM, Yang R, Frye MA. Adjunctive armodafinil
for major depressive episodes associated with bipolar I disorder: A randomized multicenter, double-blind, placebo-controlled, proof-of-concept study. J Clin Psychiatry. 2010;71(10):1363-1370.
57. Calabrese JR, Frye MA, Yang R, Ketter TA; Armodafinil Treatment Trial Study Network. Efficacy and safety of adjunctive armodafinil in adults with major depressive episodes associated with bipolar I disorder: A randomized, double-blind, placebo-controlled, multicenter trial. J Clin Psychiatry. 2014;75(10):1054-1061.
58. Strattera [package insert]. Indianapolis, IN. Lilly; 2015.
59. Carpenter LL, Milosavljevic N, Schecter JM, Tyrka AR, Price LH. Augmentation with open-label atomoxetine for partial or nonresponse to antidepressants. J Clin Psychiatry. 2005;66(10):1234-1238.
60. Papakostas GI, Petersen TJ, Burns AM, Fava M. Adjunctive atomoxetine for residual
fatigue in major depressive disorder. J Psychiatr Res. 2006;40(4):370-373.
61. Michelson D, Adler LA, Amsterdam JD, et al. Addition of atomoxetine for depression
incompletely responsive to sertraline: A randomized, double-blind, placebocontrolled study. J Clin Psychiatry. 2007;68(4):582-587.
62. Corp SA, Gitlin MJ, Altshuler LL. A review of the use of stimulants and stimulant alternatives in treating bipolar depression and major depressive disorder. J Clin Psychiatry. 2014;75(9):1010-1018.
63. Kraemer M, Uekermann J, Wiltfang J, Kis B. Methylphenidate-induced psychosis in adult attention-deficit/hyperactivity disorder: Report of 3 new cases and review of the literature. Clin Neuropharmacol. 2010;33(4):204-206.
64. Berman SM, Kuczenski R, McCracken JT, London ED. Potential adverse effects of amphetamine treatment on brain and behavior: A review. Mol Psychiatry. 2009;14(2):123-142.
65. Fredriksen M, Dahl AA, Martinsen EW, Klungsøyr O, Haavik J, Peleikis DE. Effectiveness of one-year pharmacological treatment of adult attention-deficit/hyperactivity disorder (ADHD): An open-label prospective study of time in treatment, dose, side-effects and comorbidity. Eur Neuropsychopharmacol. 2014;24(12):1873-1874.
66. Hardy SE. Methylphenidate for the treatment of depressive symptoms, including fatigue and apathy, in medically ill older adults and terminally ill adults. Am J Geriatr Pharmacother. 2009;7(1):34-59.
67. Williams RJ, Goodale LA, Shay-Fiddler MA, Gloster SP, Chang SY. Methylphenidate and dextroamphetamine abuse in substance-abusing adolescents. Am J Addict. 2004;13(4):381-389.
68. Madaan V, Kolli V, Bestha DP, Shah MJ. Update on optimal use of lisdexamfetamine in the treatment of ADHD. Neuropsychiatr Dis Treat. 2013;9:977-983.
69. Ross RG. Psychotic and manic-like symptoms during stimulant treatment of attention deficit hyperactivity disorder. Am J Psychiatry. 2006;163(7):1149-1152.
70. Dell’Osso B, Ketter TA. Use of adjunctive stimulants in adult bipolar depression. Int J Neuropsychopharmacol. 2013;16(1):55-68.
71. Habel LA, Cooper WO, Sox CM, et al. ADHD medications and risk of serious cardiovascular events in young and middle-aged adults. JAMA. 2011;306(24):2673-2683.
72. Schelleman H, Bilker WB, Kimmel SE, et al. Methylphenidate and risk of serious cardiovascular events in adults. Am J Psychiatry. 2012;169(2):178-185.
73. Bolea-Alamañac B, Nutt DJ, Adamou M, et al; British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological management of attention deficit hyperactivity disorder: Update on recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(3):179-203.
74. Moher D, Liberati A, Tetzlaff J, Altman DG; The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009;6(6):e1000097.
1. Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distribution of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Solomon DA, Keller MB, Leon AC, et al. Multiple recurrences of major depressive disorder. Am J Psychiatry. 2000;157(2):229-233.
3. Katon WJ, Fan MY, Lin EH, Unützer J. Depressive symptom deterioration in a large
primary care-based elderly cohort. Am J Geriatr Psychiatry. 2006;14(3):246-254.
4. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
5. McIntyre RS, Filteau M-J, Martin L, et al. Treatment-resistant depression: Definitions, review of the evidence, and algorithmic approach. J Affect Disord. 2014;156:1-7.
6. Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush AJ. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Focus. 2012;10(4):510-517.
7. Kudlow PA, Cha DS, McIntyre RS. Predicting treatment response in major depressive disorder: The impact of early symptomatic improvement. Can J Psychiatry. 2012;57(12):782-788.
8. Ruhé HG, van Rooijen G, Spijker J, Peeters FP, Schene AH. Staging methods for treatment resistant depression. A systematic review. J Affect Disord. 2012;137(1-3):35-45.
9. Bauer M, Dopfmer S. Lithium augmentation treatment-resistant depression: Metaanalysis of placebo-controlled studies. J Clin Psychopharmacol. 1999;19(5):427-434.
10. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: A STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530.
11. Nierenberg AA, Papakostas GI, Petersen T, et al. Lithium augmentation of nortriptyline
for subjects resistant to multiple antidepressants. J Clin Psychopharmacol. 2003;23(1):92-95.
12. Connolly KR, Thase ME. If at first you don’t succeed: A review of the evidence for antidepressant augmentation, combination, and switching strategies. Drugs. 2011;71(1):43-64.
13. Trivedi MH, Fava M, Wisniewski SR, et al; STAR*D Study Team. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354(12):1243-1252.
14. Papakostas GI, Shelton RC, Smith J, Fava M. Augmentation of antidepressants with atypical antipsychotic medications for treatment resistant major depressive disorder: A meta-analysis. J Clin Psychiatry. 2007;68(6):826-831.
15. Mahmoud RA, Pandina GJ, Turkoz I, et al. Risperidone for treatment-refractory major depressive disorder: A randomized trial. Ann Intern Med. 2007;147(9):593-602.
16. Barbee JG, Conrad EJ, Jamhour NJ. The effectiveness of olanzapine, risperidone, quetiapine, and ziprasidone as augmentation agents in treatment resistant depressive disorder. J Clin Psychiatry. 2004;65(7):975-981.
17. Fatemi SH, Emamian ES, Kist DA. Venlafaxine and bupropion combination therapy in a case of treatment-resistant depression. Ann Pharmacother.1999;33(6):701-703.
18. Carpenter LL, Yasman S, Price LH. A double-blind, placebo-controlled study of antidepressant augmentation with mirtazapine. Biol Psychiatry. 2002;51(2):183-188.
19. Hannan N, Hamzah Z, Akinpeloye HO, Meagher D. Venlafaxine-mirtazapine combination therapy in the treatment of persistent depressive illness. J Psychopharmacol. 2007;21(2):161-164.
20. McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: A STAR*D report. Am J Psychiatry. 2006;163(9):1531-1541.
21. Blier P, Ward HE, Tremblay P, Laberge L, Hébert C, Bergeron R. Combination of antidepressant medications from treatment initiation for major depressive disorder: A double-blind randomized study. Am J Psychiatry. 2010;167(3):281-288.
22. Papakostas GI, Mischoulon D, Shyu I, Alpert JE, Fava M. S-adenosyl methionine (SAMe) augmentation of serotonin reuptake inhibitors for antidepressant nonresponders with major depressive disorder: A double blind randomized clinical trial. Am J Psychiatry. 2010;167(8):942-948.
23. Papakostas GI, Shelton RC, Zajecka JM, et al. L-methylfolate as adjunctive therapy
for SSRI-resistant major depression: Results of two randomized, double-blind,
parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
24. Korston TR. Drugs of abuse. In: Katzung BG, ed. Basic and Clinical Pharmacology. 9th ed. New York, NY: McGraw-Hill; 2004:521-523.
25. Feighner JP, Herbstein J, Damlouji N. Combined MAOI, TCA, and direct stimulant therapy of treatment-resistant depression. J Clin Psychiatry. 1985;46(6):206-209.
26. Fawcett J, Kravitz HM, Zajecka JM, Schaff MR. CNS stimulant potentiation of monoamine oxidase inhibitors in treatment-refractory depression. J Clin Psychopharmacol. 1991;11(2):127-132.
27. Stoll AL, Pillay SS, Diamond L, Workum SB, Cole JO. Methylphenidate augmentation of serotonin selective reuptake inhibitors: A case series. J Clin Psychiatry. 1996;57(2):72-76.
28. Masand PS, Anand VS, Tanquary JF. Psychostimulant augmentation of second generation antidepressants: A case series. Depress Anxiety. 1998;7(2):89-91.
29. Trivedi MH, Cutler AJ, Richards C, et al. A randomized control trial of the efficacy and safety of lisdesxamfetamine dimesylate as augmentation therapy in adults with residual symptoms of major depressive disorder after treatment with escitalopram. J Clin Psychiatry. 2013;74(8):802-809.
30. Madhoo M, Keefe RS, Roth RM, et al. Lisdexamfetamine dimesylate augmentation in adults with persistent executive dysfunction after partial or full remission of major depressive disorder. Neuropsychopharmacology. 2014;39(6):1388-1398.
31. Parker G, Brotchie H. Do the old psychostimulant drugs have a role in managing treatment-resistant depression. Acta Psychiatr Scand. 2010;121(4):308-314.
32. Parker G, Brotchie H, McClure G, Fletcher K. Psychostimulants for managing unipolar and bipolar treatment-resistant melancholic depression: A medium term evaluation of cost benefits. J Affect Disord. 2013;151(1):360-364.
33. Lydon E, El-Mallakh RS. Naturalistic long-term use of methylphenidate in bipolar disorder. J Clin Psychopharmacol. 2006;26(5):516-518.
34. Carlson PJ, Merlock MC, Suppes T. Adjunctive stimulant use in patients with bipolar disorder: Treatment of residual depression and sedation. Bipolar Disord. 2004;6(5):416-420.
35. El-Mallakh RS. An open study of methylphenidate in bipolar depression. Bipolar Disord. 2000;2(1):56-59.
36. Ravindran AV, Kennedy SH, O’Donovan MC, Fallu A, Camacho F, Binder CE. Osmotic-release oral system methylphenidate augmentation of antidepressant monotherapy in major depressive disorder: Results of a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2008;69(1):87-94.
37. Patkar AA, Masand PS, Pae CU, et al. A randomized, double-blind, placebocontrolled
trial of augmentation with an extended release formulation of methylphenidate in outpatients with treatment-resistant depression. J Clin Psychopharmacol. 2006;26(6):653-656.
38. Lee H, Kim SW, Kim JM, Shin IS, Yang SJ, Yoon JS Comparing effects of methylphenidate, sertraline, and placebo on neuropsychiatric sequelae in patients with
traumatic brain injury. Hum Psychopharmacol. 2005;20(2):97-104.
39. Gualtieri CT, Evans RW. Stimulant treatment for the neurobehavioural sequelae of traumatic brain injury. Brain Inj. 1988;2(4):273-290.
40. Provigil [package insert]. North Wales, PA: Cephalon Inc; 2015.
41. Nuvigil [package insert]. Frazer, PA: Cephalon, Inc; 2013.
42. Menza MA, Kaufman KR, Castellanos A. Modafinil augmentation of antidepressant treatment in depression. J Clin Psychiatry. 2000;61(5):378-381.
43. Markovitz PJ, Wagner S. An open-label trial of modafinil augmentation in patients with partial response to antidepressant therapy. J Clin Psychopharmacol. 2003;23(2):207-209.
44. Fernandes PP, Petty F. Modafinil for remitted bipolar depression with hypersomnia. Ann Pharmacother. 2003;37(12):1807-1809.
45. Nasr S. Modafinil as adjunctive therapy in depressed outpatients. Ann Clin Psychiatry. 2004;16(3):133-138.
46. DeBattista C, Lembke A, Solvason HB, Ghebremichael R, Poirier J. A prospective trial of modafinil as an adjunctive treatment of major depression. J Clin Psychopharmacol. 2004;24(1):87-90.
47. Nasr S, Wendt B, Steiner K. Absence of mood switch with and tolerance to modafinil: A replication study from a large private practice. J Affect Disord. 2006;95(1-3):111-114.
48. DeBattista C, Doghramji K, Menza MA, Rosenthal MH, Fieve RR; Modafinil in Depression Study Group. Adjunct modafinil for the short-term treatment of fatigue and sleepiness in patients with major depressive disorder: A preliminary doubleblind, placebo-controlled study. J Clin Psychiatry. 2003;64(9):1057-1064.
49. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry. 2007;164(8):1242-1249.
50. Fava M, Thase ME, DeBattista C, Doghramji K, Arora S, Hughes RJ. Modafinil augmentation of selective serotonin reuptake inhibitor therapy in MDD partial responders with persistent fatigue and sleepiness. Ann Clin Psychiatry. 2007;19(3):153-159.
51. Thase ME, Fava M, DeBattista C, Arora S, Hughes RJ. Modafinil augmentation of SSRI therapy in patients with major depressive disorder and excessive sleepiness and fatigue: A 12-week, open-label, extension study. CNS Spectr. 2006;11(2):93-102.
52. Fava M, Thase ME, DeBattista C. A multicenter, placebo-controlled study of modafinil augmentation in partial responders to selective serotonin reuptake inhibitors with persistent fatigue and sleepiness. J Clin Psychiatry. 2005;66(1):85-93.
53. Abolfazli R, Hosseini M, Ghanizadeh A, et al. Double-blind randomized parallelgroup clinical trial of efficacy of the combination fluoxetine plus modafinil versus fluoxetine plus placebo in the treatment of major depression. Depress Anxiety. 2011;28(4):297-302.
54. Rasmussen NA, Schrøder P, Olsen LR, Brødsgaard M, Undén M, Bech P. Modafinil augmentation in depressed patients with partial response to antidepressants: A pilot study on self-reported symptoms covered by the Major Depression Inventory (MDI) and the Symptom Checklist (SCL-92). Nord J Psychiatry. 2005;59(3):173-178.
55. Dunlop BW, Crits-Christoph P, Evans DL, et al. Coadministration of modafinil and a selective serotonin reuptake inhibitor from the initiation of treatment of major depressive disorder with fatigue and sleepiness: A double-blind, placebocontrolled study. J Clin Psychopharmacol. 2007;27(6):614-619.
56. Calabrese JR, Ketter TA, Youakim JM, Tiller JM, Yang R, Frye MA. Adjunctive armodafinil
for major depressive episodes associated with bipolar I disorder: A randomized multicenter, double-blind, placebo-controlled, proof-of-concept study. J Clin Psychiatry. 2010;71(10):1363-1370.
57. Calabrese JR, Frye MA, Yang R, Ketter TA; Armodafinil Treatment Trial Study Network. Efficacy and safety of adjunctive armodafinil in adults with major depressive episodes associated with bipolar I disorder: A randomized, double-blind, placebo-controlled, multicenter trial. J Clin Psychiatry. 2014;75(10):1054-1061.
58. Strattera [package insert]. Indianapolis, IN. Lilly; 2015.
59. Carpenter LL, Milosavljevic N, Schecter JM, Tyrka AR, Price LH. Augmentation with open-label atomoxetine for partial or nonresponse to antidepressants. J Clin Psychiatry. 2005;66(10):1234-1238.
60. Papakostas GI, Petersen TJ, Burns AM, Fava M. Adjunctive atomoxetine for residual
fatigue in major depressive disorder. J Psychiatr Res. 2006;40(4):370-373.
61. Michelson D, Adler LA, Amsterdam JD, et al. Addition of atomoxetine for depression
incompletely responsive to sertraline: A randomized, double-blind, placebocontrolled study. J Clin Psychiatry. 2007;68(4):582-587.
62. Corp SA, Gitlin MJ, Altshuler LL. A review of the use of stimulants and stimulant alternatives in treating bipolar depression and major depressive disorder. J Clin Psychiatry. 2014;75(9):1010-1018.
63. Kraemer M, Uekermann J, Wiltfang J, Kis B. Methylphenidate-induced psychosis in adult attention-deficit/hyperactivity disorder: Report of 3 new cases and review of the literature. Clin Neuropharmacol. 2010;33(4):204-206.
64. Berman SM, Kuczenski R, McCracken JT, London ED. Potential adverse effects of amphetamine treatment on brain and behavior: A review. Mol Psychiatry. 2009;14(2):123-142.
65. Fredriksen M, Dahl AA, Martinsen EW, Klungsøyr O, Haavik J, Peleikis DE. Effectiveness of one-year pharmacological treatment of adult attention-deficit/hyperactivity disorder (ADHD): An open-label prospective study of time in treatment, dose, side-effects and comorbidity. Eur Neuropsychopharmacol. 2014;24(12):1873-1874.
66. Hardy SE. Methylphenidate for the treatment of depressive symptoms, including fatigue and apathy, in medically ill older adults and terminally ill adults. Am J Geriatr Pharmacother. 2009;7(1):34-59.
67. Williams RJ, Goodale LA, Shay-Fiddler MA, Gloster SP, Chang SY. Methylphenidate and dextroamphetamine abuse in substance-abusing adolescents. Am J Addict. 2004;13(4):381-389.
68. Madaan V, Kolli V, Bestha DP, Shah MJ. Update on optimal use of lisdexamfetamine in the treatment of ADHD. Neuropsychiatr Dis Treat. 2013;9:977-983.
69. Ross RG. Psychotic and manic-like symptoms during stimulant treatment of attention deficit hyperactivity disorder. Am J Psychiatry. 2006;163(7):1149-1152.
70. Dell’Osso B, Ketter TA. Use of adjunctive stimulants in adult bipolar depression. Int J Neuropsychopharmacol. 2013;16(1):55-68.
71. Habel LA, Cooper WO, Sox CM, et al. ADHD medications and risk of serious cardiovascular events in young and middle-aged adults. JAMA. 2011;306(24):2673-2683.
72. Schelleman H, Bilker WB, Kimmel SE, et al. Methylphenidate and risk of serious cardiovascular events in adults. Am J Psychiatry. 2012;169(2):178-185.
73. Bolea-Alamañac B, Nutt DJ, Adamou M, et al; British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological management of attention deficit hyperactivity disorder: Update on recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(3):179-203.
74. Moher D, Liberati A, Tetzlaff J, Altman DG; The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009;6(6):e1000097.
What can we do about the Zika virus in the United States?
Since Florida has seen several new cases of local mosquito-borne infection, controlling and preventing Zika infection has great urgency. Zika virus involves an arthropod-borne infection transmitted by Aedes aegypti and Aedes albopictus mosquitoes. Other modes of transmission include the maternal-fetal route, any sexual contact, blood transfusions, organ or tissue transplantation, and laboratory exposure.1
The first case of Zika infection in the United States and its territories occurred through international travel. According to the Centers for Disease Control and Prevention, as of October 12, 2016, there were 3807 travel-associated cases of Zika infection in the United States and 84 instances in its territories.2 As for local transmission, there were 128 people evidencing a Zika infection in the United States and 25,871 in US territories.2 Regions between Texas and Florida are at high risk because Aedes mosquitoes primarily inhabit the gulf coast.3 Many cases have occurred despite repellent use and eradication efforts, possibly due to resistance acquired by these mosquitoes.1
Control measures include using insect repellents, aerial spraying of insecticides, eliminating mosquito breeding sites, covering water tanks, and using mosquito nets or door and window screens. Infection during pregnancy is the greatest concern because of congenital anomalies (including microcephaly) that negatively affect brain development.4
Before a possible conception or any sexual contact, women exposed to Zika—with or without symptoms—must wait at least 8 weeks; men with or without symptoms should abstain for 6 months.4 Individuals should avoid traveling to areas with Zika infestation, wear long-sleeved clothing treated with permethrin, and minimize outside exposure, especially in evening hours.4
The World Health Organization is utilizing genetically modified mosquitoes to diminish Aedes populations; trials conducted in affected areas of Brazil revealed that the number of Aedes mosquitoes was reduced by 90%.5 This method of mosquito control is currently being studied in the United States.6 Vaccinations to prevent Zika infection are also under investigation.
Physicians should educate patients regarding the clinical manifestations and complications of Zika virus infection; people need to know that the Zika virus can be sexually transmitted. Doctors should also counsel patients to curtail travel to areas that have Zika infestations, or to at least wear protective clothing while in such areas to minimize mosquito bite risk. Educating travelers about appropriate postponement of sexual contact after any exposure to the Zika virus is also essential.4
Hema Madhuri Mekala, MD
Priyanga Jayakumar, MD
Rajashekar Reddy Yeruva, MD
Steven Lippmann, MD
Louisville, KY
1. Centers for Disease Control and Prevention. Zika virus: Transmission & risks. Available at: http://www.cdc.gov/zika/transmission/index.html. Accessed October 14, 2016.
2. Centers for Disease Control and Prevention. Zika virus: Case counts in the US. Available at: http://www.cdc.gov/zika/geo/united-states.html. Accessed October 14, 2016.
3. Castro L, Chen X, Dimitrov NB, et al. The University of Texas at Austin. Texas Arbovirus Risk. 2015. Available at: http://hdl.handle.net/2152/31934. Accessed October 14, 2016.
4. Centers for Disease Control and Prevention. Zika virus: Zika is in your area: What to do. Available at: http://www.cdc.gov/zika/intheus/what-to-do.html. Accessed October 14, 2016.
5. FL KEYS NEWS. Available at: http://www.flkeysnews.com/opinion/opn-columns-blogs/article83328707.html. Accessed October 14, 2016.
6. Ernst KC, Haenchen S, Dickinson K, et al. Awareness and support of release of genetically modified “sterile” mosquitoes, Key West, Florida, USA. Emerg Infect Dis. 2015;21:320-324.
Since Florida has seen several new cases of local mosquito-borne infection, controlling and preventing Zika infection has great urgency. Zika virus involves an arthropod-borne infection transmitted by Aedes aegypti and Aedes albopictus mosquitoes. Other modes of transmission include the maternal-fetal route, any sexual contact, blood transfusions, organ or tissue transplantation, and laboratory exposure.1
The first case of Zika infection in the United States and its territories occurred through international travel. According to the Centers for Disease Control and Prevention, as of October 12, 2016, there were 3807 travel-associated cases of Zika infection in the United States and 84 instances in its territories.2 As for local transmission, there were 128 people evidencing a Zika infection in the United States and 25,871 in US territories.2 Regions between Texas and Florida are at high risk because Aedes mosquitoes primarily inhabit the gulf coast.3 Many cases have occurred despite repellent use and eradication efforts, possibly due to resistance acquired by these mosquitoes.1
Control measures include using insect repellents, aerial spraying of insecticides, eliminating mosquito breeding sites, covering water tanks, and using mosquito nets or door and window screens. Infection during pregnancy is the greatest concern because of congenital anomalies (including microcephaly) that negatively affect brain development.4
Before a possible conception or any sexual contact, women exposed to Zika—with or without symptoms—must wait at least 8 weeks; men with or without symptoms should abstain for 6 months.4 Individuals should avoid traveling to areas with Zika infestation, wear long-sleeved clothing treated with permethrin, and minimize outside exposure, especially in evening hours.4
The World Health Organization is utilizing genetically modified mosquitoes to diminish Aedes populations; trials conducted in affected areas of Brazil revealed that the number of Aedes mosquitoes was reduced by 90%.5 This method of mosquito control is currently being studied in the United States.6 Vaccinations to prevent Zika infection are also under investigation.
Physicians should educate patients regarding the clinical manifestations and complications of Zika virus infection; people need to know that the Zika virus can be sexually transmitted. Doctors should also counsel patients to curtail travel to areas that have Zika infestations, or to at least wear protective clothing while in such areas to minimize mosquito bite risk. Educating travelers about appropriate postponement of sexual contact after any exposure to the Zika virus is also essential.4
Hema Madhuri Mekala, MD
Priyanga Jayakumar, MD
Rajashekar Reddy Yeruva, MD
Steven Lippmann, MD
Louisville, KY
Since Florida has seen several new cases of local mosquito-borne infection, controlling and preventing Zika infection has great urgency. Zika virus involves an arthropod-borne infection transmitted by Aedes aegypti and Aedes albopictus mosquitoes. Other modes of transmission include the maternal-fetal route, any sexual contact, blood transfusions, organ or tissue transplantation, and laboratory exposure.1
The first case of Zika infection in the United States and its territories occurred through international travel. According to the Centers for Disease Control and Prevention, as of October 12, 2016, there were 3807 travel-associated cases of Zika infection in the United States and 84 instances in its territories.2 As for local transmission, there were 128 people evidencing a Zika infection in the United States and 25,871 in US territories.2 Regions between Texas and Florida are at high risk because Aedes mosquitoes primarily inhabit the gulf coast.3 Many cases have occurred despite repellent use and eradication efforts, possibly due to resistance acquired by these mosquitoes.1
Control measures include using insect repellents, aerial spraying of insecticides, eliminating mosquito breeding sites, covering water tanks, and using mosquito nets or door and window screens. Infection during pregnancy is the greatest concern because of congenital anomalies (including microcephaly) that negatively affect brain development.4
Before a possible conception or any sexual contact, women exposed to Zika—with or without symptoms—must wait at least 8 weeks; men with or without symptoms should abstain for 6 months.4 Individuals should avoid traveling to areas with Zika infestation, wear long-sleeved clothing treated with permethrin, and minimize outside exposure, especially in evening hours.4
The World Health Organization is utilizing genetically modified mosquitoes to diminish Aedes populations; trials conducted in affected areas of Brazil revealed that the number of Aedes mosquitoes was reduced by 90%.5 This method of mosquito control is currently being studied in the United States.6 Vaccinations to prevent Zika infection are also under investigation.
Physicians should educate patients regarding the clinical manifestations and complications of Zika virus infection; people need to know that the Zika virus can be sexually transmitted. Doctors should also counsel patients to curtail travel to areas that have Zika infestations, or to at least wear protective clothing while in such areas to minimize mosquito bite risk. Educating travelers about appropriate postponement of sexual contact after any exposure to the Zika virus is also essential.4
Hema Madhuri Mekala, MD
Priyanga Jayakumar, MD
Rajashekar Reddy Yeruva, MD
Steven Lippmann, MD
Louisville, KY
1. Centers for Disease Control and Prevention. Zika virus: Transmission & risks. Available at: http://www.cdc.gov/zika/transmission/index.html. Accessed October 14, 2016.
2. Centers for Disease Control and Prevention. Zika virus: Case counts in the US. Available at: http://www.cdc.gov/zika/geo/united-states.html. Accessed October 14, 2016.
3. Castro L, Chen X, Dimitrov NB, et al. The University of Texas at Austin. Texas Arbovirus Risk. 2015. Available at: http://hdl.handle.net/2152/31934. Accessed October 14, 2016.
4. Centers for Disease Control and Prevention. Zika virus: Zika is in your area: What to do. Available at: http://www.cdc.gov/zika/intheus/what-to-do.html. Accessed October 14, 2016.
5. FL KEYS NEWS. Available at: http://www.flkeysnews.com/opinion/opn-columns-blogs/article83328707.html. Accessed October 14, 2016.
6. Ernst KC, Haenchen S, Dickinson K, et al. Awareness and support of release of genetically modified “sterile” mosquitoes, Key West, Florida, USA. Emerg Infect Dis. 2015;21:320-324.
1. Centers for Disease Control and Prevention. Zika virus: Transmission & risks. Available at: http://www.cdc.gov/zika/transmission/index.html. Accessed October 14, 2016.
2. Centers for Disease Control and Prevention. Zika virus: Case counts in the US. Available at: http://www.cdc.gov/zika/geo/united-states.html. Accessed October 14, 2016.
3. Castro L, Chen X, Dimitrov NB, et al. The University of Texas at Austin. Texas Arbovirus Risk. 2015. Available at: http://hdl.handle.net/2152/31934. Accessed October 14, 2016.
4. Centers for Disease Control and Prevention. Zika virus: Zika is in your area: What to do. Available at: http://www.cdc.gov/zika/intheus/what-to-do.html. Accessed October 14, 2016.
5. FL KEYS NEWS. Available at: http://www.flkeysnews.com/opinion/opn-columns-blogs/article83328707.html. Accessed October 14, 2016.
6. Ernst KC, Haenchen S, Dickinson K, et al. Awareness and support of release of genetically modified “sterile” mosquitoes, Key West, Florida, USA. Emerg Infect Dis. 2015;21:320-324.
The future of ketamine in psychiatry
Ketamine, a high-affinity, noncompetitive N-methyl-
How ketamine works
Water- and lipid-soluble, ketamine is available in oral, topical, IM, and IV forms. Plasma concentrations reach maximum levels minutes after IV infusion; 5 to 15 minutes after IM administration; and 30 minutes after oral ingestion.1 The duration of action is as long as 2 hours after IM injection, and 4 to 6 hours orally. Metabolites are eliminated in urine.
Ketamine, co-prescribed with stimulants and some antidepressant drugs, can induce unwanted effects, such as increased blood pressure. Auditory and visual hallucinations are reported occasionally, especially in patients receiving a high dosage or in those with alcohol dependence.1 Hypertension, tachycardia, cardiac arrhythmia, and pain at injection site are the most common adverse effects.
Some advantages over ECT in treating depression
The efficacy of electroconvulsive therapy (ECT) in alleviating depression depends on seizure duration. Compared with methohexital, an anesthetic used for ECT, ketamine offers some advantages:
- increased ictal time
- augmented mid-ictal slow-wave amplitude
- shortened post-treatment re-orientation time
- less cognitive dysfunction.2
Uses for ketamine
Treatment-resistant depression. The glutamatergic system is implicated in depression.2,3 Ketamine works in patients with treatment-resistant depression by blocking glutamate NMDA receptors and increasing the activity of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, resulting in a rapid, sustained antidepressant effect. Response to ketamine occurs within 2 hours and lasts approximately 1 week.
Bipolar and unipolar depression. Ketamine has rapid antidepressant properties in unipolar and bipolar depression. It is most beneficial in people with a family history of alcohol dependence, because similar glutamatergic system alterations might be involved in the pathophysiology of both disorders.3,4 An antidepressant effect has been reported as soon as 40 minutes after ketamine infusions.3
Suicide prevention. A single sub-anesthetic IV dose of ketamine rapidly diminishes acute suicidal ideation.1 This effect can be maintained through repeated ketamine infusions, episodically on a clinically derived basis. The exact duration and period between ketamine readministrations are not fully established. A variety of clinical-, patient-, and circumstance-related factors, history, response, and physician preferences alter such patterns, in an individualized way. This is also a promising means to reduce hospitalizations and at least mitigate the severity of depressive patient presentations.
Anesthesia and analgesia. Because ketamine induces anesthesia with minimal effect on respiratory function, it could be used in patients with pulmonary conditions.5 Ketamine can provide analgesia during brief operative and diagnostic procedures; because of its hypertensive actions, it is useful in trauma patients with hypotension.A low dose of ketamine effectively diminishes the discomfort of complex regional pain and other pain syndromes.
Abuse potential
There is documented risk of ketamine abuse. It may create psychedelic effects that some people find pleasurable, such as sedation, disinhibition, and altered perceptions.6 There also may be a component of physiological dependence.6
Conclusion
Ketamine’s rapid antidepressant effect results could be beneficial when used in severely depressed and suicidal patients. Given the potential risks of ketamine, safety considerations will determine whether this drug is successful as a therapy for people with a mood disorder.
Further research about ketamine usage including pain management and affective disorders is anticipated.7 Investigations substantiating relative safety and clinical trials are still on-going.8
Related Resources
• Nichols SD, Bishop J. Is the evidence compelling for using ketamine to treat resistant depression? Current Psychiatry. 2015;15(5):48-51.
• National Institute of Mental Health. Highlight: ketamine: a new (and faster) path to treating depression. www.nimh.nih.gov/about/strategic-planning-reports/highlights/highlight-ketamine-a-new-and-faster-path-to-treatingdepression.shtml.
1. Sinner B, Graf BM. Ketamine. Handb Exp Pharmacol. 2008;(128):313-333.
2. Krystal AD, Dean MD, Weiner RD, et al. ECT stimulus intensity: are present ECT devices too limited? Am J Psychiatry. 2000;157(6):963-967.
3. Phelps LE, Brutsche N, Moral JR, et al. Family history of alcohol dependence and initial antidepressant response to an N-methyl-D-aspartate antagonist. Biol Psychiatry. 2009;65:181-184.
4. Nery FG, Stanley JA, Chen HH, et al. Bipolar disorder comorbid with alcoholism: a 1H magnetic resonance spectroscopy study. J Psychiatry Res. 2010;44(5):278-285.
5. Meller, ST. Ketamine: relief from chronic pain through actions at the NMDA receptor. Pain. 1996;68(2-3):435-436.
6. Sassano-Higgins S, Baron D, Juarez G, et al. A review of ketamine abuse and diversion. Depress Anxiety. 2016;33(8):718-727.
7. Jafarinia M, Afarideh M, Tafakhori A, et al. Efficacy and safety of oral ketamine versus diclofenac to alleviate mild to moderate depression in chronic pain patients: A double-blind, randomized, controlled trial. J Affect Disord. 2016;204:1-8.
8. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.
Ketamine, a high-affinity, noncompetitive N-methyl-
How ketamine works
Water- and lipid-soluble, ketamine is available in oral, topical, IM, and IV forms. Plasma concentrations reach maximum levels minutes after IV infusion; 5 to 15 minutes after IM administration; and 30 minutes after oral ingestion.1 The duration of action is as long as 2 hours after IM injection, and 4 to 6 hours orally. Metabolites are eliminated in urine.
Ketamine, co-prescribed with stimulants and some antidepressant drugs, can induce unwanted effects, such as increased blood pressure. Auditory and visual hallucinations are reported occasionally, especially in patients receiving a high dosage or in those with alcohol dependence.1 Hypertension, tachycardia, cardiac arrhythmia, and pain at injection site are the most common adverse effects.
Some advantages over ECT in treating depression
The efficacy of electroconvulsive therapy (ECT) in alleviating depression depends on seizure duration. Compared with methohexital, an anesthetic used for ECT, ketamine offers some advantages:
- increased ictal time
- augmented mid-ictal slow-wave amplitude
- shortened post-treatment re-orientation time
- less cognitive dysfunction.2
Uses for ketamine
Treatment-resistant depression. The glutamatergic system is implicated in depression.2,3 Ketamine works in patients with treatment-resistant depression by blocking glutamate NMDA receptors and increasing the activity of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, resulting in a rapid, sustained antidepressant effect. Response to ketamine occurs within 2 hours and lasts approximately 1 week.
Bipolar and unipolar depression. Ketamine has rapid antidepressant properties in unipolar and bipolar depression. It is most beneficial in people with a family history of alcohol dependence, because similar glutamatergic system alterations might be involved in the pathophysiology of both disorders.3,4 An antidepressant effect has been reported as soon as 40 minutes after ketamine infusions.3
Suicide prevention. A single sub-anesthetic IV dose of ketamine rapidly diminishes acute suicidal ideation.1 This effect can be maintained through repeated ketamine infusions, episodically on a clinically derived basis. The exact duration and period between ketamine readministrations are not fully established. A variety of clinical-, patient-, and circumstance-related factors, history, response, and physician preferences alter such patterns, in an individualized way. This is also a promising means to reduce hospitalizations and at least mitigate the severity of depressive patient presentations.
Anesthesia and analgesia. Because ketamine induces anesthesia with minimal effect on respiratory function, it could be used in patients with pulmonary conditions.5 Ketamine can provide analgesia during brief operative and diagnostic procedures; because of its hypertensive actions, it is useful in trauma patients with hypotension.A low dose of ketamine effectively diminishes the discomfort of complex regional pain and other pain syndromes.
Abuse potential
There is documented risk of ketamine abuse. It may create psychedelic effects that some people find pleasurable, such as sedation, disinhibition, and altered perceptions.6 There also may be a component of physiological dependence.6
Conclusion
Ketamine’s rapid antidepressant effect results could be beneficial when used in severely depressed and suicidal patients. Given the potential risks of ketamine, safety considerations will determine whether this drug is successful as a therapy for people with a mood disorder.
Further research about ketamine usage including pain management and affective disorders is anticipated.7 Investigations substantiating relative safety and clinical trials are still on-going.8
Related Resources
• Nichols SD, Bishop J. Is the evidence compelling for using ketamine to treat resistant depression? Current Psychiatry. 2015;15(5):48-51.
• National Institute of Mental Health. Highlight: ketamine: a new (and faster) path to treating depression. www.nimh.nih.gov/about/strategic-planning-reports/highlights/highlight-ketamine-a-new-and-faster-path-to-treatingdepression.shtml.
Ketamine, a high-affinity, noncompetitive N-methyl-
How ketamine works
Water- and lipid-soluble, ketamine is available in oral, topical, IM, and IV forms. Plasma concentrations reach maximum levels minutes after IV infusion; 5 to 15 minutes after IM administration; and 30 minutes after oral ingestion.1 The duration of action is as long as 2 hours after IM injection, and 4 to 6 hours orally. Metabolites are eliminated in urine.
Ketamine, co-prescribed with stimulants and some antidepressant drugs, can induce unwanted effects, such as increased blood pressure. Auditory and visual hallucinations are reported occasionally, especially in patients receiving a high dosage or in those with alcohol dependence.1 Hypertension, tachycardia, cardiac arrhythmia, and pain at injection site are the most common adverse effects.
Some advantages over ECT in treating depression
The efficacy of electroconvulsive therapy (ECT) in alleviating depression depends on seizure duration. Compared with methohexital, an anesthetic used for ECT, ketamine offers some advantages:
- increased ictal time
- augmented mid-ictal slow-wave amplitude
- shortened post-treatment re-orientation time
- less cognitive dysfunction.2
Uses for ketamine
Treatment-resistant depression. The glutamatergic system is implicated in depression.2,3 Ketamine works in patients with treatment-resistant depression by blocking glutamate NMDA receptors and increasing the activity of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, resulting in a rapid, sustained antidepressant effect. Response to ketamine occurs within 2 hours and lasts approximately 1 week.
Bipolar and unipolar depression. Ketamine has rapid antidepressant properties in unipolar and bipolar depression. It is most beneficial in people with a family history of alcohol dependence, because similar glutamatergic system alterations might be involved in the pathophysiology of both disorders.3,4 An antidepressant effect has been reported as soon as 40 minutes after ketamine infusions.3
Suicide prevention. A single sub-anesthetic IV dose of ketamine rapidly diminishes acute suicidal ideation.1 This effect can be maintained through repeated ketamine infusions, episodically on a clinically derived basis. The exact duration and period between ketamine readministrations are not fully established. A variety of clinical-, patient-, and circumstance-related factors, history, response, and physician preferences alter such patterns, in an individualized way. This is also a promising means to reduce hospitalizations and at least mitigate the severity of depressive patient presentations.
Anesthesia and analgesia. Because ketamine induces anesthesia with minimal effect on respiratory function, it could be used in patients with pulmonary conditions.5 Ketamine can provide analgesia during brief operative and diagnostic procedures; because of its hypertensive actions, it is useful in trauma patients with hypotension.A low dose of ketamine effectively diminishes the discomfort of complex regional pain and other pain syndromes.
Abuse potential
There is documented risk of ketamine abuse. It may create psychedelic effects that some people find pleasurable, such as sedation, disinhibition, and altered perceptions.6 There also may be a component of physiological dependence.6
Conclusion
Ketamine’s rapid antidepressant effect results could be beneficial when used in severely depressed and suicidal patients. Given the potential risks of ketamine, safety considerations will determine whether this drug is successful as a therapy for people with a mood disorder.
Further research about ketamine usage including pain management and affective disorders is anticipated.7 Investigations substantiating relative safety and clinical trials are still on-going.8
Related Resources
• Nichols SD, Bishop J. Is the evidence compelling for using ketamine to treat resistant depression? Current Psychiatry. 2015;15(5):48-51.
• National Institute of Mental Health. Highlight: ketamine: a new (and faster) path to treating depression. www.nimh.nih.gov/about/strategic-planning-reports/highlights/highlight-ketamine-a-new-and-faster-path-to-treatingdepression.shtml.
1. Sinner B, Graf BM. Ketamine. Handb Exp Pharmacol. 2008;(128):313-333.
2. Krystal AD, Dean MD, Weiner RD, et al. ECT stimulus intensity: are present ECT devices too limited? Am J Psychiatry. 2000;157(6):963-967.
3. Phelps LE, Brutsche N, Moral JR, et al. Family history of alcohol dependence and initial antidepressant response to an N-methyl-D-aspartate antagonist. Biol Psychiatry. 2009;65:181-184.
4. Nery FG, Stanley JA, Chen HH, et al. Bipolar disorder comorbid with alcoholism: a 1H magnetic resonance spectroscopy study. J Psychiatry Res. 2010;44(5):278-285.
5. Meller, ST. Ketamine: relief from chronic pain through actions at the NMDA receptor. Pain. 1996;68(2-3):435-436.
6. Sassano-Higgins S, Baron D, Juarez G, et al. A review of ketamine abuse and diversion. Depress Anxiety. 2016;33(8):718-727.
7. Jafarinia M, Afarideh M, Tafakhori A, et al. Efficacy and safety of oral ketamine versus diclofenac to alleviate mild to moderate depression in chronic pain patients: A double-blind, randomized, controlled trial. J Affect Disord. 2016;204:1-8.
8. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.
1. Sinner B, Graf BM. Ketamine. Handb Exp Pharmacol. 2008;(128):313-333.
2. Krystal AD, Dean MD, Weiner RD, et al. ECT stimulus intensity: are present ECT devices too limited? Am J Psychiatry. 2000;157(6):963-967.
3. Phelps LE, Brutsche N, Moral JR, et al. Family history of alcohol dependence and initial antidepressant response to an N-methyl-D-aspartate antagonist. Biol Psychiatry. 2009;65:181-184.
4. Nery FG, Stanley JA, Chen HH, et al. Bipolar disorder comorbid with alcoholism: a 1H magnetic resonance spectroscopy study. J Psychiatry Res. 2010;44(5):278-285.
5. Meller, ST. Ketamine: relief from chronic pain through actions at the NMDA receptor. Pain. 1996;68(2-3):435-436.
6. Sassano-Higgins S, Baron D, Juarez G, et al. A review of ketamine abuse and diversion. Depress Anxiety. 2016;33(8):718-727.
7. Jafarinia M, Afarideh M, Tafakhori A, et al. Efficacy and safety of oral ketamine versus diclofenac to alleviate mild to moderate depression in chronic pain patients: A double-blind, randomized, controlled trial. J Affect Disord. 2016;204:1-8.
8. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.