CCJM delivers practical clinical articles relevant to internists, cardiologists, endocrinologists, and other specialists, all written by known experts.
Correction: Anemia, leukocytosis, abdominal pain, flushing, and bone and skin lesion
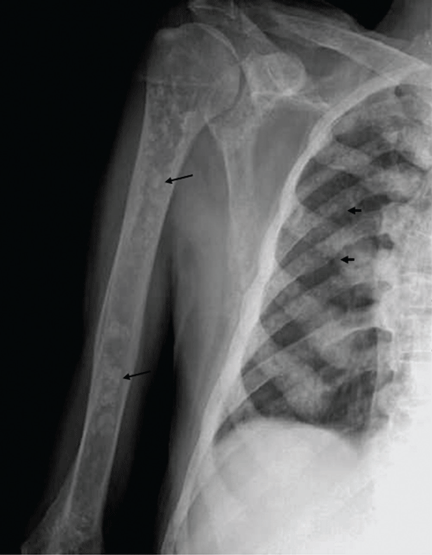
In the June 2012 issue, on page 384 of the Clinical Picture article by Álvarez-Twose et al (Álvarez-Twose I, Vañó-Galván S, Sanchez-Muñoz L, Fernandez-Zapardiel S, Escribano L. The Clinical Picture: anemia, leukocytosis, abdominal pain, flushing, and bone and skin lesions. Cleve Clin J Med 2012; 79:384–386), Dr. Alvarez-Twose’s first name was spelled incorrectly. The correct spelling is Iván. This error has been corrected in the online version.
In the June 2012 issue, on page 384 of the Clinical Picture article by Álvarez-Twose et al (Álvarez-Twose I, Vañó-Galván S, Sanchez-Muñoz L, Fernandez-Zapardiel S, Escribano L. The Clinical Picture: anemia, leukocytosis, abdominal pain, flushing, and bone and skin lesions. Cleve Clin J Med 2012; 79:384–386), Dr. Alvarez-Twose’s first name was spelled incorrectly. The correct spelling is Iván. This error has been corrected in the online version.
In the June 2012 issue, on page 384 of the Clinical Picture article by Álvarez-Twose et al (Álvarez-Twose I, Vañó-Galván S, Sanchez-Muñoz L, Fernandez-Zapardiel S, Escribano L. The Clinical Picture: anemia, leukocytosis, abdominal pain, flushing, and bone and skin lesions. Cleve Clin J Med 2012; 79:384–386), Dr. Alvarez-Twose’s first name was spelled incorrectly. The correct spelling is Iván. This error has been corrected in the online version.
Everyone should avoid overexposure to the sun’s rays. But the desire for the “perfect tan,” the belief that a tan enables one to spend more time in the sun, and a lack of awareness about the dangers of ultraviolet (UV) radiation are factors that contribute to UV-induced skin damage and to an increased risk of skin cancer. Physicians need to be prepared to counsel patients on why and how to avoid damaging UV radiation.
Some measures are straightforward, such as wearing protective clothing, limiting sun exposure during the peak daylight hours, and avoiding tanning booths. The issue of which sunscreen to use can be more difficult, given the quantity of sunscreen products and the confusing claims made on product labels.
In this article, we review UV radiation, the consequences of increased exposure to different parts of the UV spectrum, tanning, and the fundamentals of sunscreens. We also briefly review current guidelines from professional organizations and rulings on sunscreen products by the US Food and Drug Administration (FDA).
FACTORS AFFECTING UV EXPOSURE
UV radiation from the sun is strongest between 10:00 am and 4:00 pm at equatorial latitudes and during summer months.1 Certain wavelengths of UV radiation have long been known to contribute to skin cancer in humans: the wavelengths considered most damaging are those from 320 to 400 nm, referred to as UV-A, and from 290 to 320 nm, referred to as UV-B.1,2 The UV spectrum also includes UV-C and other subdivisions, but in this article we are mainly concerned with UV-A and UV-B. From 90% to 95% of UV radiation that reaches the earth’s surface is UV-A, and most of the rest is UV-B.
The different wavelengths of UV-A and UV-B have different effects on the skin. Much of the shorter-wavelength UV-B radiation is scattered by the atmospheric ozone layer, by clouds, by air pollution, and by glass; on the other hand, UV-B rays are the main cause of sunburn in humans. The longer-wavelength UV-A radiation penetrates more deeply into the skin and so may have greater destructive potential.1,3
The daily UV index
The daily UV index of the US National Weather Service and the US Environmental Protection Agency (EPA) (www.epa.gov/sunwise/uvindex.html) offers a direct measurement of the level of UV radiation on a scale of 1 (low) to 11+ (extremely high). The higher the number, the greater the risk of sunburn for a fair-skinned person, even after allowing for cloud cover.
UV EXPOSURE RISKS ARE WELL KNOWN
The American Cancer Society has estimated that the annual incidence of nonmelanoma skin cancer is greater than 2 million, and the incidence of melanoma is from 65,000 to 70,000.4 The incidence of all types of skin cancer has been increasing for the last 30 years.4,5
Exposure to UV radiation is the major environmental risk factor for nonmelanoma skin cancer.6 It is also believed to be a major risk factor for melanoma; although definitive evidence is still lacking, research is beginning to uncover mechanisms linking UV-related gene damage to melanoma.7
UV LIGHT’S EFFECTS ON THE SKIN
The effects of UV light on the skin can be immediate (eg, erythema) and long-term (eg, photoaging, immunosuppression, carcinogenicity).1
Sunburn
Excessive UV damage creates a biochemical milieu that manifests grossly on the skin as a “sunburn.” Excessive UV exposure is damaging regardless of whether a sunburn occurs. Intensive intermittent UV exposure in childhood and teen years leading to blistering sunburn is a risk factor for basal cell carcinoma and malignant melanoma, whereas excessive chronic cumulative exposure is a risk factor for squamous cell carcinoma. In addition, both types of exposure can lead to photoaging.
Sunburn is noticeable 3 to 4 hours after exposure, peaking at around 24 hours.
Photoaging
A long-term effect of UV exposure is photoaging. Although how photoaging occurs is unclear, studies suggest that UV-A contributes more to photoaging, while UV-B contributes to burning, which results in extracellular matrix degradation and dysregulation of collagen metabolism. These changes in matrix and collagen may cause wrinkles and loss of skin turgor; increases in vascular growth factors may induce telangiectasia. All of these effects are characteristic of photoaging.8,9
Immunosuppression, sun exposure, cancer
Profound systemic immunosuppression, such as in organ transplantation patients, can lead to an increased risk of skin cancer, as evidenced by the frequent development of nonmelanoma skin cancers in patients who have undergone organ transplantation, with reported incidence rates of 21% to 50%.6,10
But sun exposure itself can also cause both local and systemic immunosuppression depending on the area of exposure and the dosage of UV radiation. The immunosuppressive and carcinogenic effects of UV light on the skin are complex, involving a variety of cell types, including antigen-presenting cells, lymphocytes, and cytokines. UV radiation can cause dysregulation of antigen-presenting cells such as Langerhans cells and dermal dendritic cells, which in turn can activate regulatory T cells to suppress the immune system. UV radiation can also induce keratinocytes to produce immunosuppressive cytokines that inhibit the production of a number of “repair cytokines” that fix UV-induced DNA damage. The repair cytokines can mitigate UV-induced immunosuppression.6,11 These effects can suppress the induction of local, systemic, and memory immunity.
Both UV-A and UV-B interact to enhance UV-induced immunosuppression, and this can occur even at doses that do not cause erythema.12 Profound immunosuppression—whether UV-induced or due to HIV infection or immunosuppressive drugs—can lead to an increased risk of skin cancer, as evidenced by the frequent development of nonmelanoma skin cancers in patients who have undergone organ transplantation, with reported incidence rates of 21% to 50%.6,10
Animal studies linking UV-B exposure to skin cancer found that UV-B energy is directly absorbed by DNA, resulting in the formation of cyclobutane pyrimidine dimers and pyrimidine-pyrimidone photoproducts in the DNA, which block replication and transcription.6 The resulting mutations specifically occur in the tumor suppressor gene p53, and these mutations have been linked to squamous cell carcinoma.13,14
UV-A light has also been reported to induce cyclobutane dimers, but via an indirect mechanism, since DNA does not directly absorb UV-A. Dimers induced by UV-A light are apparently cleared at a slower rate than those induced by UV-B, suggesting that UV-A may have a greater potential for carcinogenesis.15 UV-A light can also directly induce carcinogenesis through reactive oxygen species that cause tumorogenic modified bases in the DNA. These modified bases can be misread, leading to decreased DNA integrity.6
WHAT IS TANNING?
UV radiation produces darkening of the skin, or tanning. UV exposure results in both immediate and persistent pigment darkening. Immediate pigment darkening, which is visible and transient, occurs within seconds of UV exposure as a result of the formation of reactive oxygen species and photooxidation of preexisting melanin, and it resolves in a couple of hours. Persistent pigment darkening results from photooxidation and redistribution of preexisting melanin, occurring 2 to 24 hours after sun exposure. Neither type of pigment darkening protects the skin, since no new melanin is produced.16,17
UV-B rays can induce skin erythema, edema, and sunburn, followed by skin desquamation and tanning. Its effects can be seen immediately, but typically the erythema reaches its peak 24 hours later.1
“Delayed tanning” is an adaptive response seen about 3 days after sun exposure and is caused by increased melanocyte activity and new melanin formation in response to UV-B; this effect is considered mildly photoprotective, with a sun protection factor (SPF) of 3. In other words, there is a tiny bit of truth to the common belief that a tan that develops a few days after sun exposure (delayed tanning) can provide a small increase in protection from sunburn. However, the real health concern is not only sunburn, but increased cancer risk and photoaging from UV exposure.
INDOOR TANNING
Every year, nearly 28 million Americans use a sunbed or a sunlamp, and 2.3 million of them are teenagers.18,19 Every day in the United States more than 1 million people use an indoor tanning device.20 Nearly 70% of those who use tanning devices are white women ages 16 to 29.21
Tanning is big business. In 2010, there were 20,000 tanning salons in the United States, and the number of health clubs and spas with tanning beds was between 15,000 and 20,000. In 2010, the tanning industry generated an estimated $4.7 billion in revenue.22
In their search for the perfect tan, people receive very large doses of UV light, and most tanning lamps emit 95% to 99% of their light as UV-A. In fact, the typical sunlamp user can receive an annual dose of UV-A that is 0.3 to 1.2 times the average annual cumulative dose received from sun exposure (7,700 kJ/m2).11 A typical customer of a tanning salon in the course of 20 sessions is exposed to up to 1.2 times the average normal annual exposure from sunlight. Also, for a frequent tanner, the exposure can increase to 4.7 times the average normal annual exposure and up to 12 times the exposure if using high-pressure sunlamps.11 Indoor tanners not only receive large doses of a known carcinogen, but the body’s pigmentary responses to a sunlamp’s UV-A (immediate and persistent pigment darkening) do not protect it from sunburn, cancer-inducing DNA damage, immunosuppression, or photoaging.
Additionally, even though tanning bed lamps only emit 1% to 5% of their light in the UV-B spectrum, one can still receive a very large dose of UV-B radiation with enough exposure.
The American Academy of Dermatology opposes indoor tanning and supports a ban on the nonmedical production and sale of indoor tanning devices. The World Health Organization classifies tanning lamps as carcinogenic and advises minors to avoid indoor tanning.23
SUNSCREEN PROTECTION
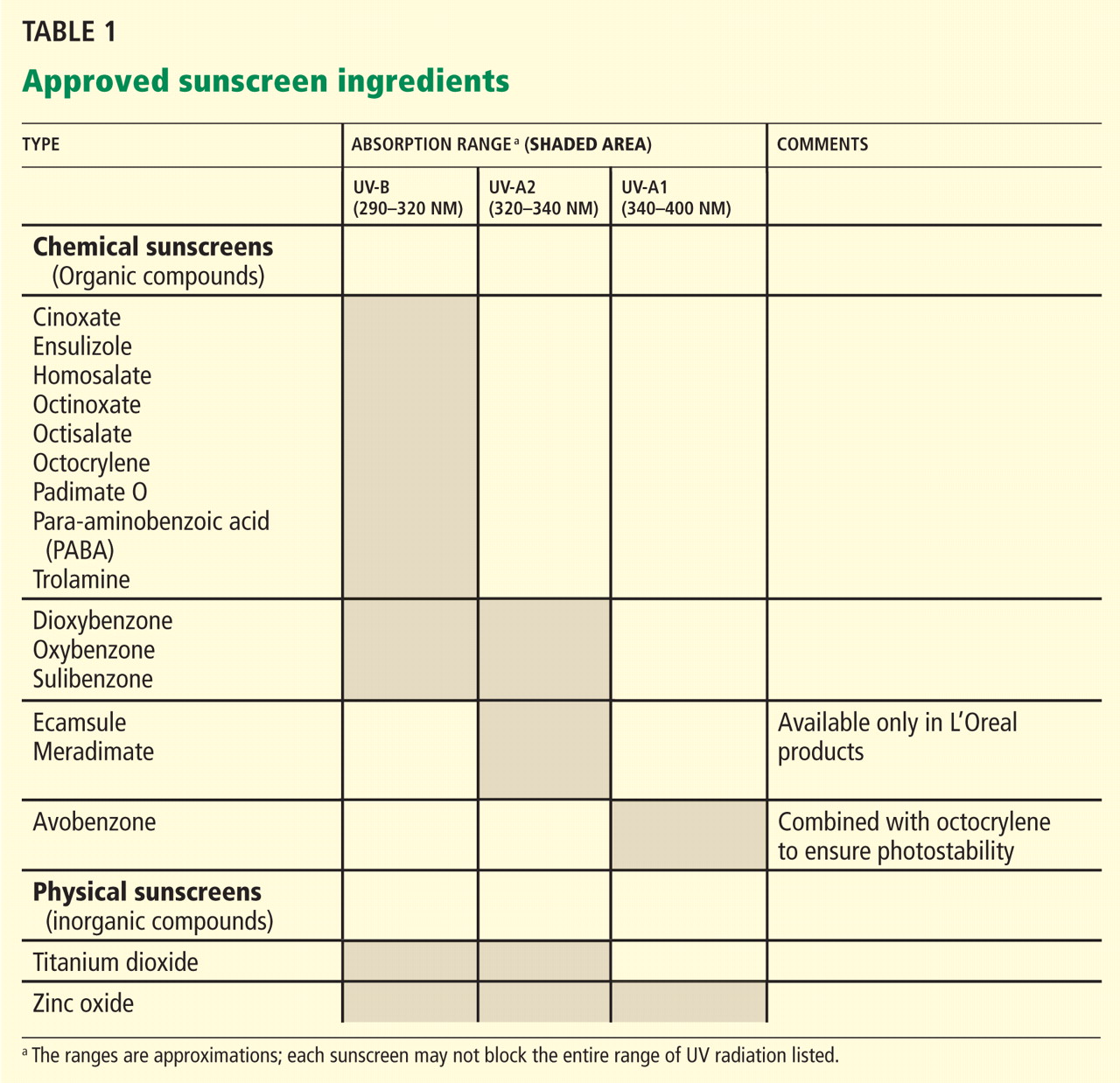
Sunscreen products must contain an active sunscreen ingredient that absorbs radiation in the range of 290 to 400 nm. In “physical” sunscreens, the ingredient is an inorganic compound with particles that physically block out UV radiation; in “chemical” sunscreens, the ingredient is an organic compound that absorbs UV radiation.
Most organic UV filters absorb UV-B radiation, and a few act in the UV-A2 range (320–340 nm). Only one FDA-approved organic sunscreen, avobenzone, protects against UV-A1 (340–400 nm).
Inorganic compounds function by physically reflecting and scattering UV radiation from a film of inert metal particles, ie, in a manner similar to protective clothing.24 Two FDA-approved inorganic sunscreens—titanium dioxide and zinc oxide—provide UV-A and UV-B protection. Zinc oxide and the non-micronized form of titanium dioxide provide UV-A1 and UV-A2 protection.
Inorganic sunscreens have a thick consistency and tend to clump. Advances in nanoparticle technology have improved their consistency,25 but micronized titanium dioxide does not provide UV-A1 protection.
The FDA regulates the active ingredients in sunscreen products, determines the methods of testing them, and dictates labelling requirements.
CATEGORIES OF SUNSCREENS
Sunscreens are categorized according to their SPF,26 UV-A protection,27,28 substantivity, and stability.29
Understanding the ‘sun protection factor’
SPF is a laboratory measure of sunscreen efficacy and is defined as the amount of UV radiation required to produce a sunburn on protected skin relative to that of unprotected skin. Since SPF assessment is based on erythema, it is mainly a measure of UV-B exposure, not UV-A exposure.
Contrary to popular belief, the SPF of a product is not related to the duration of UV exposure.30 Also, the relationship between SPF and UV-B protection is not linear: a sunscreen with an SPF of 15 can filter 94% of UV-B radiation, whereas an SPF of 30 provides greater than 97% protection at an equal UV-B dosage. UV radiation dosage depends on both the duration of exposure and the intensity of the UV radiation. Thus, a sunscreen with twice the SPF does not necessarily mean one can stay out in the sun twice as long before developing a sunburn.
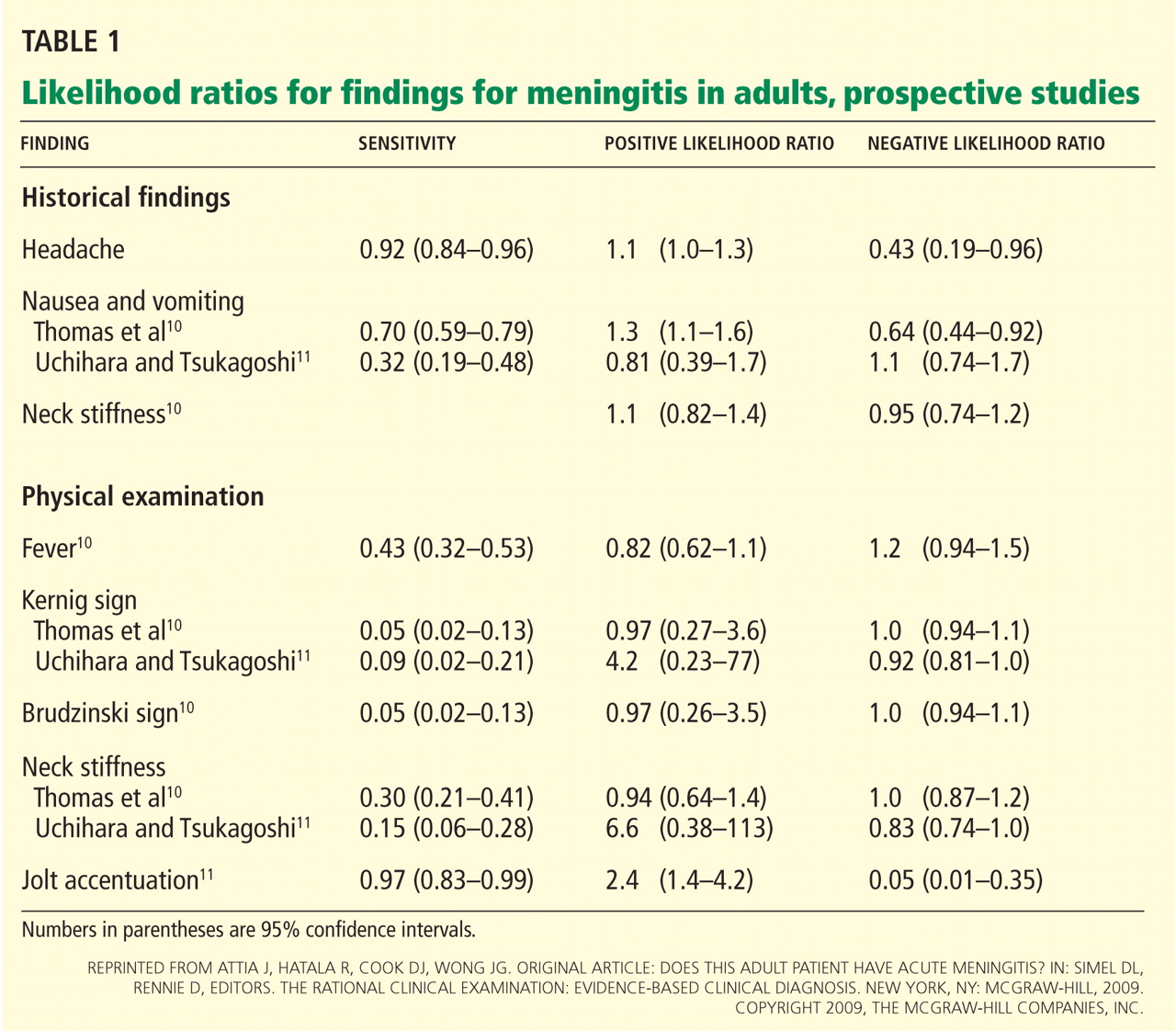
The FDA has established acceptable sunscreen filters and their maximal concentrations for over-the-counter sunscreens.31 The FDA approval of ecamsule (Mexoryl SX) in 2006 brought the total number of sunscreens to 17 (Table 1).1
Ability to block UV-A radiation
As UV-A causes significant immunosuppression and is the major type of UV radiation reaching Earth, a systematic and repeatable method of measuring a sunscreen’s ability to block UV-A light is necessary.
For each sunscreen, laboratory testing generates a curve of the absorbance within the UV spectrum. The area under this curve is calculated, and a “critical wavelength” is defined as the wavelength where the area under the absorbance curve up to that value is 90% of the total area under the curve. A sunscreen with “broad-spectrum” UV-A protection is one for which the critical wavelength is greater than or equal to 370 nm. The critical wavelength measures the breadth of UV-A absorbance by a sunscreen and must be used in combination with the SPF value to provide a complete assessment of UV protection.27,28,32,33
Substantivity
Substantivity is a sunscreen’s ability to remain effective under adverse conditions such as exposure to water and sweat. A water-resistant product maintains the indicated protection after 40 minutes of water immersion, whereas a very-water-resistant (formerly called “waterproof”) product maintains the indicated protection after 80 minutes of water immersion.27,28,32,33
Stability
The stability of the sunscreen is important for long-lasting protection with continuous exposure to UV light, in particular to prevent photodegradation. The FDA has established maximum levels of each filter allowed in the sunscreen. Several filters can be combined to achieve a high SPF level, to provide broadspectrum UV-A and UV-B protection, and to prevent photodegradation. For example, octocrylene prevents the degradation of the photosensitive compound avobenzone, whereas ecamsule has been combined with avobenzone and octocrylene to provide broad-spectrum UV-A and UV-B protection. Ecamsule is currently patent-protected by L’Oreal and is found only in products produced by it and its subsidiaries.
SUNSCREEN USES AND ABUSES
Sunscreen use generally falls into three categories: daily use, short-term use (eg, for an activity involving increased sun exposure, such as outdoor exercise or work), and use for preventing sunburn during tan acquisition, ie, to increase the time of UV radiation exposure.
Most published studies report on the effects of daily sunscreen protection or on cutaneous immune responses to sunscreen use. However, the use of sunscreens to enhance tan acquisition and to increase sun exposure duration is an abuse of the product and can actually increase the risk of skin cancer. A common misperception is that sunscreens decrease the risk of burning and allow people to increase their exposure to UV radiation. This results in increased exposure to UV-A and thus increases the risk of skin cancers and facilitates photoaging.34
In 2003, Baron et al35 published a randomized trial evaluating the protective effects of UV-B sunscreens (SPF 15) and UV-A/UV-B sunscreens (SPF 15) against UV radiation, using contact hypersensitivity as a model for immunosuppression. The study involved 211 volunteers ages 18 to 59. Measuring skinfold thickness vs total UV dose to calculate an immune protection factor, they reported that the UV-A/UV-B sunscreens had a greater average immune protection factor than the UV-B sunscreen. They concluded that though both types of sunscreen can protect against immunosuppression, the addition of a UV-A filter provides greater protection against immunosuppression.35
A French study36 in 104 volunteers examined the immunoprotective effects of sunscreens with equal SPF but differing levels of UV-A protection after UV exposure, and used delayed-type hypersensitivity as a model for cutaneous immune response. Broader UV-A protection yielded smaller reductions in delayed-type hypersensitivity after UV exposure, leading to the conclusion that UV-A contributes greatly to cutaneous immunosuppression and that UV-A filters can mitigate some of these effects.36
Sunscreens and photoaging
Only a few clinical studies have examined the effects of sunscreen use on photoaging.
In 1995, a randomized, double-blind, placebo-controlled trial involving 53 adults with previously diagnosed with actinic keratosis or skin cancer, or both, showed that those who applied a UV-A/UV-B sunscreen over a 24-month period had less solar elastosis on biopsy compared with controls.37
In 2008, a French study of 12 volunteers showed that broad-spectrum UV protection prevented histologic changes attributed to 6 weeks of chronic UV exposure. The control group exhibited structural and molecular evidence of UV damage (eg, epidermal thickening, decreased procollagen expression, higher lysozyme-to-elastin ratio), whereas chronic use of a broad-spectrum sunscreen either minimized or abrogated these findings.12
Evidence also suggests that broad-spectrum sunscreens can prevent damage from suberythemal doses of UV. A study published in 200738 investigated whether broad-spectrum sunscreen use affects the development of genetic and cellular markers of UV damage after daily suberythemal UV exposure. It reported that unprotected individuals exhibited more thymine dimers, higher p53 expression, and loss of Langerhans cells compared with protected individuals.38
Similarly, a study published in 201012 assessed cellular and molecular markers of photodamage after 19 daily suberythemal UV exposures with or without a broad-spectrum, low-SPF (SPF 8) sunscreen and found that consistent sunscreen use resulted in fewer p53-positive cells, a lower lysozyme-to-elastin ratio, a decreased number and size of melanocytes, and an increased number of Langerhans cells.
Thus, evidence supports the idea that consistent use of a broad-spectrum sunscreen can protect against photodamage, even at doses that do not cause erythema.12
Sunscreens and squamous cell carcinoma
Several large trials provide appreciable evidence that sunscreen is effective in preventing squamous cell carcinoma.
A randomized, controlled, 7-month trial in Australia of a broad-spectrum sunscreen with an SPF of 17 noted a dose-dependent reduction in the development of new actinic keratosis.39 Another randomized, controlled trial from Australia showed a 40% reduction in the development of squamous cell carcinoma over a 4.5-year period in participants who applied a broad-spectrum SPF-16 sunscreen 3 to 4 days per week vs discretionary use.40 Follow-up data at 8 years showed that daily sunscreen users continued to have a 40% lower incidence rate of squamous cell carcinoma than controls.41
Sunscreens and basal cell carcinoma
Although sunscreens appear to be effective in preventing actinic keratosis and squamous cell carcinoma, the evidence that they also prevent basal cell carcinoma and melanoma has been inconclusive.
Sunscreens and melanoma
Using a high number of nevi as a surrogate measure of the risk of developing melanoma, a randomized controlled trial of a broad-spectrum SPF-30 sunscreen in Canadian children over a 3-year period showed a slight decrease in the number of new nevi compared with controls. However, this effect was seen only in children with freckles.42
In a large European study of white school-age children, sunscreen use was associated with an increased number of nevi compared with the use of clothing, which prevented new nevi.43
A large meta-analysis of 18 case-controlled studies failed to show a protective association of sunscreen use with melanoma.44 Postulated confounding factors in earlier studies included older sunscreen formulations with no UV-A protection, low SPF, and limited substantivity. In many cases, sunscreen users exposed themselves to higher doses of UV because of the perceived decreased risk of burning with sunscreen use. This is especially the case when sun exposure was intentional to acquire a tan.34 Individuals who burn easily or may have had a family history of melanoma tended to use more sunscreen, thus creating another confounder. Finally, extrapolation of results from data performed in different geographic latitudes may not be appropriate.
Recently, Green et al45 published a study using the same cohort from a previous study of sunscreens and nonmelanoma skin cancer to examine new primary melanomas as a secondary outcome. They reported that, during the 5-year trial period and during the 10-year follow-up, fewer participants in the intervention group developed primary melanoma compared with the control group (11 vs 21). They concluded that regular applications of a broad-spectrum SPF-16 sunscreen in white adults ages 25 to 75 can decrease the incidence of melanoma.45 The study had serious limitations: the authors admitted that the results were marginally statistically significant; intervention sites of sunscreen application were chosen for nonmelanoma skin cancer and excluded the trunk and lower extremities, where melanomas often occur; and the entire body was analyzed for melanomas, not just the intervention site.46 Thus, despite providing some of the first evidence supporting sunscreen’s ability to prevent melanoma, these results are controversial and are by no means conclusive.
HOW TO USE SUNSCREEN
The American Academy of Dermatology guidelines47 recommend daily, year-round use of a broad-spectrum, water-resistant sunscreen with an SPF of at least 30, regardless of age or skin type. Cloud cover and windows block UV-B but not UV-A. Additionally, 80% of UV light can pass through cloud cover, while 25% is reflected by sand and 80% by snow. Thus, sunscreen should be used daily throughout the year.
Sunscreen should be applied to exposed dry skin 15 to 30 minutes before sun exposure, paying particular attention to common areas of nonmelanoma skin cancer, such as the face, ears, hands, arms, and lips. The standard amount of sunscreen used in SPF testing is 2 mg/cm2, which is difficult to translate into real use; most people apply only 25% to 50% of the recommended amount of sunscreen.48 According to the guidelines, 1 oz of sunscreen—2 tablespoons, or enough to fill a shot glass—is enough to cover sun-exposed parts of the adult body. Sunscreen should be reapplied every 2 hours or after swimming or heavy perspiration; many water-resistant sunscreens lose effectiveness after 40 minutes in the water.
Despite the protective effects of sunscreen, the following are still recommended:
Seek shade or avoid exposure between 10:00 am and 4:00 pm, ie, when the sun’s rays are strongest
Take caution around water, sand, and snow, which reflect UV radiation
Wear protective clothing such as long-sleeved shirts, pants, sunglasses, and wide-brimmed hats
Do not use tanning beds
Do not use sunscreens to increase the time of UV exposure.
SPECIAL CONSIDERATIONS: INFANTS
Infants and toddlers are at higher risk of UV damage and skin cancer. Structurally, children’s skin is thinner than that of adults and has lower melanin concentrations. Thus, UV penetrates more deeply into skin that is less able to absorb UV radiation. Animal studies suggest that the skin of children, especially infants, is immunologically immature and less able to respond to UV damage than adult skin. Therefore, extra care must be taken to protect children from UV exposure.49
The American Academy of Pediatrics recommends that infants under 6 months of age should be kept out of direct sunlight whenever possible. A broad-spectrum, water-resistant sunscreen with an SPF of at least 30 should be applied to skin that is not protected by clothing or shade (eg, face, hands, neck).50
NEW FDA GUIDELINES AND OTHER PROPOSED CHANGES
Figure 1. New US Food and Drug Administration (FDA) labeling standards include separately delineating “broad-spectrum” and sun protection factor (SPF) information in an equal font size. The claim “water-resistant” must be specified with a time, ie, 40 or 80 minutes. The “drug facts” box on the back of the product must include usage directions, guidelines for sun protection, and other FDA-required statements.In June 2011, the FDA released a new set of testing and labeling requirements for sunscreens (Figure 1)51 and proposed further modifications to the rules for manufacturing sunscreen products. Manufacturers must comply with these new rules within 12 months of the date of release (at least by June 17, 2012). Manufacturers with annual sales of less than $25,000 were given 24 months to comply.
The FDA’s SPF labeling requirements remained unchanged; however, the FDA instituted new regulations regarding UV-A protection. Sunscreens that qualify as broad-spectrum are to be labeled as such, indicating that they protect against radiation in the entire UV spectrum. Products that are “broad-spectrum SPF ≥ 15” can now include the following statement in the “drug facts” part of the label: “If used as directed with other sun protection measures, decreases the risk of skin cancer and early skin aging caused by the sun.”
The FDA now requires sunscreens that are not broad-spectrum or that have an SPF less than 15 to include the following alert: “Spending time in the sun increases your risk of skin cancer and early skin aging.”33 These products can only claim protection from sunburn with the statement: “This product has been shown only to prevent sunburn, not skin cancer or early skin aging.”27,28,32,33
In terms of water resistance, the FDA now bans the terms “sunblock,” “waterproof,” or “sweatproof,” as these claims cannot be substantiated. Instead, the label on the front of the package can only read either “water resistant (40 minutes)” or “water resistant (80 minutes).” Also, sunscreens may no longer claim to provide “instant protection,” nor can they claim to maintain efficacy for more than 2 hours without reapplication.27,28,32,33
Some sunscreen products have been labeled with SPF values exceeding 100. The FDA decided that because there is insufficient evidence of clinical benefit for such SPFs, sunscreen product labels may claim a maximum SPF value of “50+.”28,52
The FDA now also specifies approved formulations for sunscreen products. Oils, lotions, creams, gels, butters, pastes, and ointments are acceptable, and this applies to all products that contain sunscreens, including cosmetics. Wipes, towelettes, powders, body washes, and shampoos are not acceptable as sunscreen products. The FDA now considers the popular spray form as potentially acceptable; a final decision awaits the results of further testing.28,53
Editor’s note: As this paper was being sent to press, the US Food and Drug Administration announced that sunscreen manufacturers would have an additional 6 months to comply with the new labeling rules for sunscreens. The new deadline is December 2012. Smaller companies have until December 2013 to implement the labeling changes.
References
Kullavanijaya P, Lim HW. Photoprotection. J Am Acad Dermatol2005; 52:937–958.
Sivamani RK, Ghiya M, Maibach HI. Shedding light on sunscreens and their labels: testing policies need to match actual use. Am J Prev Med2010; 38:679–681.
Miyamura Y, Coelho SG, Schlenz K, et al. The deceptive nature of UVA tanning versus the modest protective effects of UVB tanning on human skin. Pigment Cell Melanoma Res2011; 24:136–147.
Wang Y, Digiovanna JJ, Stern JB, et al. Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc Natl Acad Sci U S A2009; 106:6279–6284.
Yano K, Kadoya K, Kajiya K, Hong YK, Detmar M. Ultraviolet B irradiation of human skin induces an angiogenic switch that is mediated by upregulation of vascular endothelial growth factor and by downregulation of thrombospondin-1. Br J Dermatol2005; 152:115–121.
Rabe JH, Mamelak AJ, McElgunn PJ, Morison WL, Sauder DN. Photoaging: mechanisms and repair. J Am Acad Dermatol2006; 55:1–19.
Damian DL, Patterson CR, Stapelberg M, Park J, Barnetson RS, Halliday GM. UV radiation-induced immunosuppression is greater in men and prevented by topical nicotinamide. J Invest Dermatol2008; 128:447–454.
Miller SA, Hamilton SL, Wester UG, Cyr WH. An analysis of UVA emissions from sunlamps and the potential importance for melanoma. Photochem Photobiol1998; 68:63–70.
Seité S, Fourtanier AM. The benefit of daily photoprotection. J Am Acad Dermatol2008; 58(5 suppl 2):S160–166.
Besaratinia A, Synold TW, Chen HH, et al. DNA lesions induced by UV A1 and B radiation in human cells: comparative analyses in the overall genome and in the p53 tumor suppressor gene. Proc Natl Acad Sci U S A2005; 102:10058–10063.
May P, May E. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene1999; 18:7621–7636.
Mouret S, Baudouin C, Charveron M, Favier A, Cadet J, Douki T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc Natl Acad Sci USA2006; 103:13765–13770.
Wolber R, Schlenz K, Wakamatsu K, et al. Pigmentation effects of solar-simulated radiation as compared with UVA and UVB radiation. Pigment Cell Melanoma Res2008; 21:487–491.
Miyamura Y, Coelho SG, Wolber R, et al. Regulation of human skin pigmentation and responses to ultraviolet radiation. Pigment Cell Res2007; 20:2–13.
Kwon HT, Mayer JA, Walker KK, Yu H, Lewis EC, Belch GE. Promotion of frequent tanning sessions by indoor tanning facilities: two studies. J Am Acad Dermatol2002; 46:700–705.
Dellavalle RP, Parker ER, Cersonsky N, et al. Youth access laws: in the dark at the tanning parlor?Arch Dermatol2003; 139:443–448.
Whitmore SE, Morison WL, Potten CS, Chadwick C. Tanning salon exposure and molecular alterations. J Am Acad Dermatol2001; 44:775–780.
Swerdlow AJ, Weinstock MA. Do tanning lamps cause melanoma? An epidemiologic assessment. J Am Acad Dermatol1998; 38:89–98.
IBISWorld. Tanning salons in the US: Market research report NAICS 81219c. www.ibisworld.com. Accesssed May 9, 2012.
Wang SQ, Lim HW. Current status of the sunscreen regulation in the United States: 2011 Food and Drug Administration’s final rule on labeling and effectiveness testing. J Am Acad Dermatol2011; 65:863–869.
Food and Drug Administration (FDA). Labeling and effectiveness testing; sunscreen drug products for over-the-counter human use (final rule). Federal Register2011. http://www.gpo.gov/fdsys/pkg/FR-2011-06-17/pdf/2011-14766.pdf. Accessed May 9, 2012.
Scherschun L, Lim HW. Photoprotection by sunscreens. Am J Clin Dermatol2001; 2:131–134.
US Food and Drug Administration (FDA). FDA Press Release. FDA announces changes to better inform consumers about sunscreen: new rules give consumers more information to help reduce the risk of skin cancer, early aging. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm258940.htm. Accessed May 9, 2012.
Autier P. Sunscreen abuse for intentional sun exposure. Br J Dermatol2009; 161(suppl 3):40–45.
Baron ED, Fourtanier A, Compan D, Medaisko C, Cooper KD, Stevens SR. High ultraviolet A protection affords greater immune protection confirming that ultraviolet A contributes to photoimmunosuppression in humans. J Invest Dermatol2003; 121:869–875.
Moyal DD, Fourtanier AM. Broad-spectrum sunscreens provide better protection from solar ultraviolet-simulated radiation and natural sunlight-induced immunosuppression in human beings. J Am Acad Dermatol2008; 58(suppl 2):S149–S154.
Boyd AS, Naylor M, Cameron GS, Pearse AD, Gaskell SA, Neldner KH. The effects of chronic sunscreen use on the histologic changes of dermatoheliosis. J Am Acad Dermatol1995; 33:941–946.
Young AR, Orchard GE, Harrison GI, Klock JL. The detrimental effects of daily sub-erythemal exposure on human skin in vivo can be prevented by a daily-care broad-spectrum sunscreen. J Invest Dermatol2007; 127:975–978.
Thompson SC, Jolley D, Marks R. Reduction of solar keratoses by regular sunscreen use. N Engl J Med1993; 329:1147–1151.
Green A, Williams G, Neale R, et al. Daily sunscreen application and betacarotene supplementation in prevention of basal-cell and squamous-cell carcinomas of the skin: a randomised controlled trial. Lancet1999; 354:723–729.
van der Pols JC, Williams GM, Pandeya N, Logan V, Green AC. Prolonged prevention of squamous cell carcinoma of the skin by regular sunscreen use. Cancer Epidemiol Biomarkers Prev2006; 15:2546–2548.
Gallagher RP, Rivers JK, Lee TK, Bajdik CD, McLean DI, Coldman AJ. Broad-spectrum sunscreen use and the development of new nevi in white children: a randomized controlled trial. JAMA2000; 283:2955–2960.
Autier P, Doré JF, Cattaruzza MS, et al. Sunscreen use, wearing clothes, and number of nevi in 6- to 7-year-old European children. European Organization for Research and Treatment of Cancer Melanoma Cooperative Group. J Natl Cancer Inst1998; 90:1873–1880.
Dennis LK, Beane Freeman LE, VanBeek MJ. Sunscreen use and the risk for melanoma: a quantitative review. Ann Intern Med2003; 139:966–978.
Green AC, Williams GM, Logan V, Strutton GM. Reduced melanoma after regular sunscreen use: randomized trial follow-up. J Clin Oncol2011; 29:257–263.
Goldenhersh MA, Koslowsky M. Increased melanoma after regular sunscreen use?J Clin Oncol2011; 29:e557–e558.
Neale R, Williams G, Green A. Application patterns among participants randomized to daily sunscreen use in a skin cancer prevention trial. Arch Dermatol2002; 138:1319–1325.
Paller AS, Hawk JL, Honig P, et al. New insights about infant and toddler skin: implications for sun protection. Pediatrics2011; 128:92–102.
Food and Drug Administration (FDA). Revised effectiveness determination; Sunscreen drug products for over-the-counter human use (proposed rule.) Federal Register 2011. http://69.175.53.6/register/2011/jun/17/2011-14769.pdf. Accessed May 9, 2012.
Food and Drug Administration (FDA). Sunscreen drug products for over-the-counter human use: Request for data and information regarding dosage forms (advance notice of proposed rulemaking), Federal Register 2011). http://69.175.53.6/register/2011/jun/17/2011-14768.pdf. Accessed May 9, 2012.
Everyone should avoid overexposure to the sun’s rays. But the desire for the “perfect tan,” the belief that a tan enables one to spend more time in the sun, and a lack of awareness about the dangers of ultraviolet (UV) radiation are factors that contribute to UV-induced skin damage and to an increased risk of skin cancer. Physicians need to be prepared to counsel patients on why and how to avoid damaging UV radiation.
Some measures are straightforward, such as wearing protective clothing, limiting sun exposure during the peak daylight hours, and avoiding tanning booths. The issue of which sunscreen to use can be more difficult, given the quantity of sunscreen products and the confusing claims made on product labels.
In this article, we review UV radiation, the consequences of increased exposure to different parts of the UV spectrum, tanning, and the fundamentals of sunscreens. We also briefly review current guidelines from professional organizations and rulings on sunscreen products by the US Food and Drug Administration (FDA).
FACTORS AFFECTING UV EXPOSURE
UV radiation from the sun is strongest between 10:00 am and 4:00 pm at equatorial latitudes and during summer months.1 Certain wavelengths of UV radiation have long been known to contribute to skin cancer in humans: the wavelengths considered most damaging are those from 320 to 400 nm, referred to as UV-A, and from 290 to 320 nm, referred to as UV-B.1,2 The UV spectrum also includes UV-C and other subdivisions, but in this article we are mainly concerned with UV-A and UV-B. From 90% to 95% of UV radiation that reaches the earth’s surface is UV-A, and most of the rest is UV-B.
The different wavelengths of UV-A and UV-B have different effects on the skin. Much of the shorter-wavelength UV-B radiation is scattered by the atmospheric ozone layer, by clouds, by air pollution, and by glass; on the other hand, UV-B rays are the main cause of sunburn in humans. The longer-wavelength UV-A radiation penetrates more deeply into the skin and so may have greater destructive potential.1,3
The daily UV index
The daily UV index of the US National Weather Service and the US Environmental Protection Agency (EPA) (www.epa.gov/sunwise/uvindex.html) offers a direct measurement of the level of UV radiation on a scale of 1 (low) to 11+ (extremely high). The higher the number, the greater the risk of sunburn for a fair-skinned person, even after allowing for cloud cover.
UV EXPOSURE RISKS ARE WELL KNOWN
The American Cancer Society has estimated that the annual incidence of nonmelanoma skin cancer is greater than 2 million, and the incidence of melanoma is from 65,000 to 70,000.4 The incidence of all types of skin cancer has been increasing for the last 30 years.4,5
Exposure to UV radiation is the major environmental risk factor for nonmelanoma skin cancer.6 It is also believed to be a major risk factor for melanoma; although definitive evidence is still lacking, research is beginning to uncover mechanisms linking UV-related gene damage to melanoma.7
UV LIGHT’S EFFECTS ON THE SKIN
The effects of UV light on the skin can be immediate (eg, erythema) and long-term (eg, photoaging, immunosuppression, carcinogenicity).1
Sunburn
Excessive UV damage creates a biochemical milieu that manifests grossly on the skin as a “sunburn.” Excessive UV exposure is damaging regardless of whether a sunburn occurs. Intensive intermittent UV exposure in childhood and teen years leading to blistering sunburn is a risk factor for basal cell carcinoma and malignant melanoma, whereas excessive chronic cumulative exposure is a risk factor for squamous cell carcinoma. In addition, both types of exposure can lead to photoaging.
Sunburn is noticeable 3 to 4 hours after exposure, peaking at around 24 hours.
Photoaging
A long-term effect of UV exposure is photoaging. Although how photoaging occurs is unclear, studies suggest that UV-A contributes more to photoaging, while UV-B contributes to burning, which results in extracellular matrix degradation and dysregulation of collagen metabolism. These changes in matrix and collagen may cause wrinkles and loss of skin turgor; increases in vascular growth factors may induce telangiectasia. All of these effects are characteristic of photoaging.8,9
Immunosuppression, sun exposure, cancer
Profound systemic immunosuppression, such as in organ transplantation patients, can lead to an increased risk of skin cancer, as evidenced by the frequent development of nonmelanoma skin cancers in patients who have undergone organ transplantation, with reported incidence rates of 21% to 50%.6,10
But sun exposure itself can also cause both local and systemic immunosuppression depending on the area of exposure and the dosage of UV radiation. The immunosuppressive and carcinogenic effects of UV light on the skin are complex, involving a variety of cell types, including antigen-presenting cells, lymphocytes, and cytokines. UV radiation can cause dysregulation of antigen-presenting cells such as Langerhans cells and dermal dendritic cells, which in turn can activate regulatory T cells to suppress the immune system. UV radiation can also induce keratinocytes to produce immunosuppressive cytokines that inhibit the production of a number of “repair cytokines” that fix UV-induced DNA damage. The repair cytokines can mitigate UV-induced immunosuppression.6,11 These effects can suppress the induction of local, systemic, and memory immunity.
Both UV-A and UV-B interact to enhance UV-induced immunosuppression, and this can occur even at doses that do not cause erythema.12 Profound immunosuppression—whether UV-induced or due to HIV infection or immunosuppressive drugs—can lead to an increased risk of skin cancer, as evidenced by the frequent development of nonmelanoma skin cancers in patients who have undergone organ transplantation, with reported incidence rates of 21% to 50%.6,10
Animal studies linking UV-B exposure to skin cancer found that UV-B energy is directly absorbed by DNA, resulting in the formation of cyclobutane pyrimidine dimers and pyrimidine-pyrimidone photoproducts in the DNA, which block replication and transcription.6 The resulting mutations specifically occur in the tumor suppressor gene p53, and these mutations have been linked to squamous cell carcinoma.13,14
UV-A light has also been reported to induce cyclobutane dimers, but via an indirect mechanism, since DNA does not directly absorb UV-A. Dimers induced by UV-A light are apparently cleared at a slower rate than those induced by UV-B, suggesting that UV-A may have a greater potential for carcinogenesis.15 UV-A light can also directly induce carcinogenesis through reactive oxygen species that cause tumorogenic modified bases in the DNA. These modified bases can be misread, leading to decreased DNA integrity.6
WHAT IS TANNING?
UV radiation produces darkening of the skin, or tanning. UV exposure results in both immediate and persistent pigment darkening. Immediate pigment darkening, which is visible and transient, occurs within seconds of UV exposure as a result of the formation of reactive oxygen species and photooxidation of preexisting melanin, and it resolves in a couple of hours. Persistent pigment darkening results from photooxidation and redistribution of preexisting melanin, occurring 2 to 24 hours after sun exposure. Neither type of pigment darkening protects the skin, since no new melanin is produced.16,17
UV-B rays can induce skin erythema, edema, and sunburn, followed by skin desquamation and tanning. Its effects can be seen immediately, but typically the erythema reaches its peak 24 hours later.1
“Delayed tanning” is an adaptive response seen about 3 days after sun exposure and is caused by increased melanocyte activity and new melanin formation in response to UV-B; this effect is considered mildly photoprotective, with a sun protection factor (SPF) of 3. In other words, there is a tiny bit of truth to the common belief that a tan that develops a few days after sun exposure (delayed tanning) can provide a small increase in protection from sunburn. However, the real health concern is not only sunburn, but increased cancer risk and photoaging from UV exposure.
INDOOR TANNING
Every year, nearly 28 million Americans use a sunbed or a sunlamp, and 2.3 million of them are teenagers.18,19 Every day in the United States more than 1 million people use an indoor tanning device.20 Nearly 70% of those who use tanning devices are white women ages 16 to 29.21
Tanning is big business. In 2010, there were 20,000 tanning salons in the United States, and the number of health clubs and spas with tanning beds was between 15,000 and 20,000. In 2010, the tanning industry generated an estimated $4.7 billion in revenue.22
In their search for the perfect tan, people receive very large doses of UV light, and most tanning lamps emit 95% to 99% of their light as UV-A. In fact, the typical sunlamp user can receive an annual dose of UV-A that is 0.3 to 1.2 times the average annual cumulative dose received from sun exposure (7,700 kJ/m2).11 A typical customer of a tanning salon in the course of 20 sessions is exposed to up to 1.2 times the average normal annual exposure from sunlight. Also, for a frequent tanner, the exposure can increase to 4.7 times the average normal annual exposure and up to 12 times the exposure if using high-pressure sunlamps.11 Indoor tanners not only receive large doses of a known carcinogen, but the body’s pigmentary responses to a sunlamp’s UV-A (immediate and persistent pigment darkening) do not protect it from sunburn, cancer-inducing DNA damage, immunosuppression, or photoaging.
Additionally, even though tanning bed lamps only emit 1% to 5% of their light in the UV-B spectrum, one can still receive a very large dose of UV-B radiation with enough exposure.
The American Academy of Dermatology opposes indoor tanning and supports a ban on the nonmedical production and sale of indoor tanning devices. The World Health Organization classifies tanning lamps as carcinogenic and advises minors to avoid indoor tanning.23
SUNSCREEN PROTECTION
Sunscreen products must contain an active sunscreen ingredient that absorbs radiation in the range of 290 to 400 nm. In “physical” sunscreens, the ingredient is an inorganic compound with particles that physically block out UV radiation; in “chemical” sunscreens, the ingredient is an organic compound that absorbs UV radiation.
Most organic UV filters absorb UV-B radiation, and a few act in the UV-A2 range (320–340 nm). Only one FDA-approved organic sunscreen, avobenzone, protects against UV-A1 (340–400 nm).
Inorganic compounds function by physically reflecting and scattering UV radiation from a film of inert metal particles, ie, in a manner similar to protective clothing.24 Two FDA-approved inorganic sunscreens—titanium dioxide and zinc oxide—provide UV-A and UV-B protection. Zinc oxide and the non-micronized form of titanium dioxide provide UV-A1 and UV-A2 protection.
Inorganic sunscreens have a thick consistency and tend to clump. Advances in nanoparticle technology have improved their consistency,25 but micronized titanium dioxide does not provide UV-A1 protection.
The FDA regulates the active ingredients in sunscreen products, determines the methods of testing them, and dictates labelling requirements.
CATEGORIES OF SUNSCREENS
Sunscreens are categorized according to their SPF,26 UV-A protection,27,28 substantivity, and stability.29
Understanding the ‘sun protection factor’
SPF is a laboratory measure of sunscreen efficacy and is defined as the amount of UV radiation required to produce a sunburn on protected skin relative to that of unprotected skin. Since SPF assessment is based on erythema, it is mainly a measure of UV-B exposure, not UV-A exposure.
Contrary to popular belief, the SPF of a product is not related to the duration of UV exposure.30 Also, the relationship between SPF and UV-B protection is not linear: a sunscreen with an SPF of 15 can filter 94% of UV-B radiation, whereas an SPF of 30 provides greater than 97% protection at an equal UV-B dosage. UV radiation dosage depends on both the duration of exposure and the intensity of the UV radiation. Thus, a sunscreen with twice the SPF does not necessarily mean one can stay out in the sun twice as long before developing a sunburn.
The FDA has established acceptable sunscreen filters and their maximal concentrations for over-the-counter sunscreens.31 The FDA approval of ecamsule (Mexoryl SX) in 2006 brought the total number of sunscreens to 17 (Table 1).1
Ability to block UV-A radiation
As UV-A causes significant immunosuppression and is the major type of UV radiation reaching Earth, a systematic and repeatable method of measuring a sunscreen’s ability to block UV-A light is necessary.
For each sunscreen, laboratory testing generates a curve of the absorbance within the UV spectrum. The area under this curve is calculated, and a “critical wavelength” is defined as the wavelength where the area under the absorbance curve up to that value is 90% of the total area under the curve. A sunscreen with “broad-spectrum” UV-A protection is one for which the critical wavelength is greater than or equal to 370 nm. The critical wavelength measures the breadth of UV-A absorbance by a sunscreen and must be used in combination with the SPF value to provide a complete assessment of UV protection.27,28,32,33
Substantivity
Substantivity is a sunscreen’s ability to remain effective under adverse conditions such as exposure to water and sweat. A water-resistant product maintains the indicated protection after 40 minutes of water immersion, whereas a very-water-resistant (formerly called “waterproof”) product maintains the indicated protection after 80 minutes of water immersion.27,28,32,33
Stability
The stability of the sunscreen is important for long-lasting protection with continuous exposure to UV light, in particular to prevent photodegradation. The FDA has established maximum levels of each filter allowed in the sunscreen. Several filters can be combined to achieve a high SPF level, to provide broadspectrum UV-A and UV-B protection, and to prevent photodegradation. For example, octocrylene prevents the degradation of the photosensitive compound avobenzone, whereas ecamsule has been combined with avobenzone and octocrylene to provide broad-spectrum UV-A and UV-B protection. Ecamsule is currently patent-protected by L’Oreal and is found only in products produced by it and its subsidiaries.
SUNSCREEN USES AND ABUSES
Sunscreen use generally falls into three categories: daily use, short-term use (eg, for an activity involving increased sun exposure, such as outdoor exercise or work), and use for preventing sunburn during tan acquisition, ie, to increase the time of UV radiation exposure.
Most published studies report on the effects of daily sunscreen protection or on cutaneous immune responses to sunscreen use. However, the use of sunscreens to enhance tan acquisition and to increase sun exposure duration is an abuse of the product and can actually increase the risk of skin cancer. A common misperception is that sunscreens decrease the risk of burning and allow people to increase their exposure to UV radiation. This results in increased exposure to UV-A and thus increases the risk of skin cancers and facilitates photoaging.34
In 2003, Baron et al35 published a randomized trial evaluating the protective effects of UV-B sunscreens (SPF 15) and UV-A/UV-B sunscreens (SPF 15) against UV radiation, using contact hypersensitivity as a model for immunosuppression. The study involved 211 volunteers ages 18 to 59. Measuring skinfold thickness vs total UV dose to calculate an immune protection factor, they reported that the UV-A/UV-B sunscreens had a greater average immune protection factor than the UV-B sunscreen. They concluded that though both types of sunscreen can protect against immunosuppression, the addition of a UV-A filter provides greater protection against immunosuppression.35
A French study36 in 104 volunteers examined the immunoprotective effects of sunscreens with equal SPF but differing levels of UV-A protection after UV exposure, and used delayed-type hypersensitivity as a model for cutaneous immune response. Broader UV-A protection yielded smaller reductions in delayed-type hypersensitivity after UV exposure, leading to the conclusion that UV-A contributes greatly to cutaneous immunosuppression and that UV-A filters can mitigate some of these effects.36
Sunscreens and photoaging
Only a few clinical studies have examined the effects of sunscreen use on photoaging.
In 1995, a randomized, double-blind, placebo-controlled trial involving 53 adults with previously diagnosed with actinic keratosis or skin cancer, or both, showed that those who applied a UV-A/UV-B sunscreen over a 24-month period had less solar elastosis on biopsy compared with controls.37
In 2008, a French study of 12 volunteers showed that broad-spectrum UV protection prevented histologic changes attributed to 6 weeks of chronic UV exposure. The control group exhibited structural and molecular evidence of UV damage (eg, epidermal thickening, decreased procollagen expression, higher lysozyme-to-elastin ratio), whereas chronic use of a broad-spectrum sunscreen either minimized or abrogated these findings.12
Evidence also suggests that broad-spectrum sunscreens can prevent damage from suberythemal doses of UV. A study published in 200738 investigated whether broad-spectrum sunscreen use affects the development of genetic and cellular markers of UV damage after daily suberythemal UV exposure. It reported that unprotected individuals exhibited more thymine dimers, higher p53 expression, and loss of Langerhans cells compared with protected individuals.38
Similarly, a study published in 201012 assessed cellular and molecular markers of photodamage after 19 daily suberythemal UV exposures with or without a broad-spectrum, low-SPF (SPF 8) sunscreen and found that consistent sunscreen use resulted in fewer p53-positive cells, a lower lysozyme-to-elastin ratio, a decreased number and size of melanocytes, and an increased number of Langerhans cells.
Thus, evidence supports the idea that consistent use of a broad-spectrum sunscreen can protect against photodamage, even at doses that do not cause erythema.12
Sunscreens and squamous cell carcinoma
Several large trials provide appreciable evidence that sunscreen is effective in preventing squamous cell carcinoma.
A randomized, controlled, 7-month trial in Australia of a broad-spectrum sunscreen with an SPF of 17 noted a dose-dependent reduction in the development of new actinic keratosis.39 Another randomized, controlled trial from Australia showed a 40% reduction in the development of squamous cell carcinoma over a 4.5-year period in participants who applied a broad-spectrum SPF-16 sunscreen 3 to 4 days per week vs discretionary use.40 Follow-up data at 8 years showed that daily sunscreen users continued to have a 40% lower incidence rate of squamous cell carcinoma than controls.41
Sunscreens and basal cell carcinoma
Although sunscreens appear to be effective in preventing actinic keratosis and squamous cell carcinoma, the evidence that they also prevent basal cell carcinoma and melanoma has been inconclusive.
Sunscreens and melanoma
Using a high number of nevi as a surrogate measure of the risk of developing melanoma, a randomized controlled trial of a broad-spectrum SPF-30 sunscreen in Canadian children over a 3-year period showed a slight decrease in the number of new nevi compared with controls. However, this effect was seen only in children with freckles.42
In a large European study of white school-age children, sunscreen use was associated with an increased number of nevi compared with the use of clothing, which prevented new nevi.43
A large meta-analysis of 18 case-controlled studies failed to show a protective association of sunscreen use with melanoma.44 Postulated confounding factors in earlier studies included older sunscreen formulations with no UV-A protection, low SPF, and limited substantivity. In many cases, sunscreen users exposed themselves to higher doses of UV because of the perceived decreased risk of burning with sunscreen use. This is especially the case when sun exposure was intentional to acquire a tan.34 Individuals who burn easily or may have had a family history of melanoma tended to use more sunscreen, thus creating another confounder. Finally, extrapolation of results from data performed in different geographic latitudes may not be appropriate.
Recently, Green et al45 published a study using the same cohort from a previous study of sunscreens and nonmelanoma skin cancer to examine new primary melanomas as a secondary outcome. They reported that, during the 5-year trial period and during the 10-year follow-up, fewer participants in the intervention group developed primary melanoma compared with the control group (11 vs 21). They concluded that regular applications of a broad-spectrum SPF-16 sunscreen in white adults ages 25 to 75 can decrease the incidence of melanoma.45 The study had serious limitations: the authors admitted that the results were marginally statistically significant; intervention sites of sunscreen application were chosen for nonmelanoma skin cancer and excluded the trunk and lower extremities, where melanomas often occur; and the entire body was analyzed for melanomas, not just the intervention site.46 Thus, despite providing some of the first evidence supporting sunscreen’s ability to prevent melanoma, these results are controversial and are by no means conclusive.
HOW TO USE SUNSCREEN
The American Academy of Dermatology guidelines47 recommend daily, year-round use of a broad-spectrum, water-resistant sunscreen with an SPF of at least 30, regardless of age or skin type. Cloud cover and windows block UV-B but not UV-A. Additionally, 80% of UV light can pass through cloud cover, while 25% is reflected by sand and 80% by snow. Thus, sunscreen should be used daily throughout the year.
Sunscreen should be applied to exposed dry skin 15 to 30 minutes before sun exposure, paying particular attention to common areas of nonmelanoma skin cancer, such as the face, ears, hands, arms, and lips. The standard amount of sunscreen used in SPF testing is 2 mg/cm2, which is difficult to translate into real use; most people apply only 25% to 50% of the recommended amount of sunscreen.48 According to the guidelines, 1 oz of sunscreen—2 tablespoons, or enough to fill a shot glass—is enough to cover sun-exposed parts of the adult body. Sunscreen should be reapplied every 2 hours or after swimming or heavy perspiration; many water-resistant sunscreens lose effectiveness after 40 minutes in the water.
Despite the protective effects of sunscreen, the following are still recommended:
Seek shade or avoid exposure between 10:00 am and 4:00 pm, ie, when the sun’s rays are strongest
Take caution around water, sand, and snow, which reflect UV radiation
Wear protective clothing such as long-sleeved shirts, pants, sunglasses, and wide-brimmed hats
Do not use tanning beds
Do not use sunscreens to increase the time of UV exposure.
SPECIAL CONSIDERATIONS: INFANTS
Infants and toddlers are at higher risk of UV damage and skin cancer. Structurally, children’s skin is thinner than that of adults and has lower melanin concentrations. Thus, UV penetrates more deeply into skin that is less able to absorb UV radiation. Animal studies suggest that the skin of children, especially infants, is immunologically immature and less able to respond to UV damage than adult skin. Therefore, extra care must be taken to protect children from UV exposure.49
The American Academy of Pediatrics recommends that infants under 6 months of age should be kept out of direct sunlight whenever possible. A broad-spectrum, water-resistant sunscreen with an SPF of at least 30 should be applied to skin that is not protected by clothing or shade (eg, face, hands, neck).50
NEW FDA GUIDELINES AND OTHER PROPOSED CHANGES
Figure 1. New US Food and Drug Administration (FDA) labeling standards include separately delineating “broad-spectrum” and sun protection factor (SPF) information in an equal font size. The claim “water-resistant” must be specified with a time, ie, 40 or 80 minutes. The “drug facts” box on the back of the product must include usage directions, guidelines for sun protection, and other FDA-required statements.In June 2011, the FDA released a new set of testing and labeling requirements for sunscreens (Figure 1)51 and proposed further modifications to the rules for manufacturing sunscreen products. Manufacturers must comply with these new rules within 12 months of the date of release (at least by June 17, 2012). Manufacturers with annual sales of less than $25,000 were given 24 months to comply.
The FDA’s SPF labeling requirements remained unchanged; however, the FDA instituted new regulations regarding UV-A protection. Sunscreens that qualify as broad-spectrum are to be labeled as such, indicating that they protect against radiation in the entire UV spectrum. Products that are “broad-spectrum SPF ≥ 15” can now include the following statement in the “drug facts” part of the label: “If used as directed with other sun protection measures, decreases the risk of skin cancer and early skin aging caused by the sun.”
The FDA now requires sunscreens that are not broad-spectrum or that have an SPF less than 15 to include the following alert: “Spending time in the sun increases your risk of skin cancer and early skin aging.”33 These products can only claim protection from sunburn with the statement: “This product has been shown only to prevent sunburn, not skin cancer or early skin aging.”27,28,32,33
In terms of water resistance, the FDA now bans the terms “sunblock,” “waterproof,” or “sweatproof,” as these claims cannot be substantiated. Instead, the label on the front of the package can only read either “water resistant (40 minutes)” or “water resistant (80 minutes).” Also, sunscreens may no longer claim to provide “instant protection,” nor can they claim to maintain efficacy for more than 2 hours without reapplication.27,28,32,33
Some sunscreen products have been labeled with SPF values exceeding 100. The FDA decided that because there is insufficient evidence of clinical benefit for such SPFs, sunscreen product labels may claim a maximum SPF value of “50+.”28,52
The FDA now also specifies approved formulations for sunscreen products. Oils, lotions, creams, gels, butters, pastes, and ointments are acceptable, and this applies to all products that contain sunscreens, including cosmetics. Wipes, towelettes, powders, body washes, and shampoos are not acceptable as sunscreen products. The FDA now considers the popular spray form as potentially acceptable; a final decision awaits the results of further testing.28,53
Editor’s note: As this paper was being sent to press, the US Food and Drug Administration announced that sunscreen manufacturers would have an additional 6 months to comply with the new labeling rules for sunscreens. The new deadline is December 2012. Smaller companies have until December 2013 to implement the labeling changes.
Everyone should avoid overexposure to the sun’s rays. But the desire for the “perfect tan,” the belief that a tan enables one to spend more time in the sun, and a lack of awareness about the dangers of ultraviolet (UV) radiation are factors that contribute to UV-induced skin damage and to an increased risk of skin cancer. Physicians need to be prepared to counsel patients on why and how to avoid damaging UV radiation.
Some measures are straightforward, such as wearing protective clothing, limiting sun exposure during the peak daylight hours, and avoiding tanning booths. The issue of which sunscreen to use can be more difficult, given the quantity of sunscreen products and the confusing claims made on product labels.
In this article, we review UV radiation, the consequences of increased exposure to different parts of the UV spectrum, tanning, and the fundamentals of sunscreens. We also briefly review current guidelines from professional organizations and rulings on sunscreen products by the US Food and Drug Administration (FDA).
FACTORS AFFECTING UV EXPOSURE
UV radiation from the sun is strongest between 10:00 am and 4:00 pm at equatorial latitudes and during summer months.1 Certain wavelengths of UV radiation have long been known to contribute to skin cancer in humans: the wavelengths considered most damaging are those from 320 to 400 nm, referred to as UV-A, and from 290 to 320 nm, referred to as UV-B.1,2 The UV spectrum also includes UV-C and other subdivisions, but in this article we are mainly concerned with UV-A and UV-B. From 90% to 95% of UV radiation that reaches the earth’s surface is UV-A, and most of the rest is UV-B.
The different wavelengths of UV-A and UV-B have different effects on the skin. Much of the shorter-wavelength UV-B radiation is scattered by the atmospheric ozone layer, by clouds, by air pollution, and by glass; on the other hand, UV-B rays are the main cause of sunburn in humans. The longer-wavelength UV-A radiation penetrates more deeply into the skin and so may have greater destructive potential.1,3
The daily UV index
The daily UV index of the US National Weather Service and the US Environmental Protection Agency (EPA) (www.epa.gov/sunwise/uvindex.html) offers a direct measurement of the level of UV radiation on a scale of 1 (low) to 11+ (extremely high). The higher the number, the greater the risk of sunburn for a fair-skinned person, even after allowing for cloud cover.
UV EXPOSURE RISKS ARE WELL KNOWN
The American Cancer Society has estimated that the annual incidence of nonmelanoma skin cancer is greater than 2 million, and the incidence of melanoma is from 65,000 to 70,000.4 The incidence of all types of skin cancer has been increasing for the last 30 years.4,5
Exposure to UV radiation is the major environmental risk factor for nonmelanoma skin cancer.6 It is also believed to be a major risk factor for melanoma; although definitive evidence is still lacking, research is beginning to uncover mechanisms linking UV-related gene damage to melanoma.7
UV LIGHT’S EFFECTS ON THE SKIN
The effects of UV light on the skin can be immediate (eg, erythema) and long-term (eg, photoaging, immunosuppression, carcinogenicity).1
Sunburn
Excessive UV damage creates a biochemical milieu that manifests grossly on the skin as a “sunburn.” Excessive UV exposure is damaging regardless of whether a sunburn occurs. Intensive intermittent UV exposure in childhood and teen years leading to blistering sunburn is a risk factor for basal cell carcinoma and malignant melanoma, whereas excessive chronic cumulative exposure is a risk factor for squamous cell carcinoma. In addition, both types of exposure can lead to photoaging.
Sunburn is noticeable 3 to 4 hours after exposure, peaking at around 24 hours.
Photoaging
A long-term effect of UV exposure is photoaging. Although how photoaging occurs is unclear, studies suggest that UV-A contributes more to photoaging, while UV-B contributes to burning, which results in extracellular matrix degradation and dysregulation of collagen metabolism. These changes in matrix and collagen may cause wrinkles and loss of skin turgor; increases in vascular growth factors may induce telangiectasia. All of these effects are characteristic of photoaging.8,9
Immunosuppression, sun exposure, cancer
Profound systemic immunosuppression, such as in organ transplantation patients, can lead to an increased risk of skin cancer, as evidenced by the frequent development of nonmelanoma skin cancers in patients who have undergone organ transplantation, with reported incidence rates of 21% to 50%.6,10
But sun exposure itself can also cause both local and systemic immunosuppression depending on the area of exposure and the dosage of UV radiation. The immunosuppressive and carcinogenic effects of UV light on the skin are complex, involving a variety of cell types, including antigen-presenting cells, lymphocytes, and cytokines. UV radiation can cause dysregulation of antigen-presenting cells such as Langerhans cells and dermal dendritic cells, which in turn can activate regulatory T cells to suppress the immune system. UV radiation can also induce keratinocytes to produce immunosuppressive cytokines that inhibit the production of a number of “repair cytokines” that fix UV-induced DNA damage. The repair cytokines can mitigate UV-induced immunosuppression.6,11 These effects can suppress the induction of local, systemic, and memory immunity.
Both UV-A and UV-B interact to enhance UV-induced immunosuppression, and this can occur even at doses that do not cause erythema.12 Profound immunosuppression—whether UV-induced or due to HIV infection or immunosuppressive drugs—can lead to an increased risk of skin cancer, as evidenced by the frequent development of nonmelanoma skin cancers in patients who have undergone organ transplantation, with reported incidence rates of 21% to 50%.6,10
Animal studies linking UV-B exposure to skin cancer found that UV-B energy is directly absorbed by DNA, resulting in the formation of cyclobutane pyrimidine dimers and pyrimidine-pyrimidone photoproducts in the DNA, which block replication and transcription.6 The resulting mutations specifically occur in the tumor suppressor gene p53, and these mutations have been linked to squamous cell carcinoma.13,14
UV-A light has also been reported to induce cyclobutane dimers, but via an indirect mechanism, since DNA does not directly absorb UV-A. Dimers induced by UV-A light are apparently cleared at a slower rate than those induced by UV-B, suggesting that UV-A may have a greater potential for carcinogenesis.15 UV-A light can also directly induce carcinogenesis through reactive oxygen species that cause tumorogenic modified bases in the DNA. These modified bases can be misread, leading to decreased DNA integrity.6
WHAT IS TANNING?
UV radiation produces darkening of the skin, or tanning. UV exposure results in both immediate and persistent pigment darkening. Immediate pigment darkening, which is visible and transient, occurs within seconds of UV exposure as a result of the formation of reactive oxygen species and photooxidation of preexisting melanin, and it resolves in a couple of hours. Persistent pigment darkening results from photooxidation and redistribution of preexisting melanin, occurring 2 to 24 hours after sun exposure. Neither type of pigment darkening protects the skin, since no new melanin is produced.16,17
UV-B rays can induce skin erythema, edema, and sunburn, followed by skin desquamation and tanning. Its effects can be seen immediately, but typically the erythema reaches its peak 24 hours later.1
“Delayed tanning” is an adaptive response seen about 3 days after sun exposure and is caused by increased melanocyte activity and new melanin formation in response to UV-B; this effect is considered mildly photoprotective, with a sun protection factor (SPF) of 3. In other words, there is a tiny bit of truth to the common belief that a tan that develops a few days after sun exposure (delayed tanning) can provide a small increase in protection from sunburn. However, the real health concern is not only sunburn, but increased cancer risk and photoaging from UV exposure.
INDOOR TANNING
Every year, nearly 28 million Americans use a sunbed or a sunlamp, and 2.3 million of them are teenagers.18,19 Every day in the United States more than 1 million people use an indoor tanning device.20 Nearly 70% of those who use tanning devices are white women ages 16 to 29.21
Tanning is big business. In 2010, there were 20,000 tanning salons in the United States, and the number of health clubs and spas with tanning beds was between 15,000 and 20,000. In 2010, the tanning industry generated an estimated $4.7 billion in revenue.22
In their search for the perfect tan, people receive very large doses of UV light, and most tanning lamps emit 95% to 99% of their light as UV-A. In fact, the typical sunlamp user can receive an annual dose of UV-A that is 0.3 to 1.2 times the average annual cumulative dose received from sun exposure (7,700 kJ/m2).11 A typical customer of a tanning salon in the course of 20 sessions is exposed to up to 1.2 times the average normal annual exposure from sunlight. Also, for a frequent tanner, the exposure can increase to 4.7 times the average normal annual exposure and up to 12 times the exposure if using high-pressure sunlamps.11 Indoor tanners not only receive large doses of a known carcinogen, but the body’s pigmentary responses to a sunlamp’s UV-A (immediate and persistent pigment darkening) do not protect it from sunburn, cancer-inducing DNA damage, immunosuppression, or photoaging.
Additionally, even though tanning bed lamps only emit 1% to 5% of their light in the UV-B spectrum, one can still receive a very large dose of UV-B radiation with enough exposure.
The American Academy of Dermatology opposes indoor tanning and supports a ban on the nonmedical production and sale of indoor tanning devices. The World Health Organization classifies tanning lamps as carcinogenic and advises minors to avoid indoor tanning.23
SUNSCREEN PROTECTION
Sunscreen products must contain an active sunscreen ingredient that absorbs radiation in the range of 290 to 400 nm. In “physical” sunscreens, the ingredient is an inorganic compound with particles that physically block out UV radiation; in “chemical” sunscreens, the ingredient is an organic compound that absorbs UV radiation.
Most organic UV filters absorb UV-B radiation, and a few act in the UV-A2 range (320–340 nm). Only one FDA-approved organic sunscreen, avobenzone, protects against UV-A1 (340–400 nm).
Inorganic compounds function by physically reflecting and scattering UV radiation from a film of inert metal particles, ie, in a manner similar to protective clothing.24 Two FDA-approved inorganic sunscreens—titanium dioxide and zinc oxide—provide UV-A and UV-B protection. Zinc oxide and the non-micronized form of titanium dioxide provide UV-A1 and UV-A2 protection.
Inorganic sunscreens have a thick consistency and tend to clump. Advances in nanoparticle technology have improved their consistency,25 but micronized titanium dioxide does not provide UV-A1 protection.
The FDA regulates the active ingredients in sunscreen products, determines the methods of testing them, and dictates labelling requirements.
CATEGORIES OF SUNSCREENS
Sunscreens are categorized according to their SPF,26 UV-A protection,27,28 substantivity, and stability.29
Understanding the ‘sun protection factor’
SPF is a laboratory measure of sunscreen efficacy and is defined as the amount of UV radiation required to produce a sunburn on protected skin relative to that of unprotected skin. Since SPF assessment is based on erythema, it is mainly a measure of UV-B exposure, not UV-A exposure.
Contrary to popular belief, the SPF of a product is not related to the duration of UV exposure.30 Also, the relationship between SPF and UV-B protection is not linear: a sunscreen with an SPF of 15 can filter 94% of UV-B radiation, whereas an SPF of 30 provides greater than 97% protection at an equal UV-B dosage. UV radiation dosage depends on both the duration of exposure and the intensity of the UV radiation. Thus, a sunscreen with twice the SPF does not necessarily mean one can stay out in the sun twice as long before developing a sunburn.
The FDA has established acceptable sunscreen filters and their maximal concentrations for over-the-counter sunscreens.31 The FDA approval of ecamsule (Mexoryl SX) in 2006 brought the total number of sunscreens to 17 (Table 1).1
Ability to block UV-A radiation
As UV-A causes significant immunosuppression and is the major type of UV radiation reaching Earth, a systematic and repeatable method of measuring a sunscreen’s ability to block UV-A light is necessary.
For each sunscreen, laboratory testing generates a curve of the absorbance within the UV spectrum. The area under this curve is calculated, and a “critical wavelength” is defined as the wavelength where the area under the absorbance curve up to that value is 90% of the total area under the curve. A sunscreen with “broad-spectrum” UV-A protection is one for which the critical wavelength is greater than or equal to 370 nm. The critical wavelength measures the breadth of UV-A absorbance by a sunscreen and must be used in combination with the SPF value to provide a complete assessment of UV protection.27,28,32,33
Substantivity
Substantivity is a sunscreen’s ability to remain effective under adverse conditions such as exposure to water and sweat. A water-resistant product maintains the indicated protection after 40 minutes of water immersion, whereas a very-water-resistant (formerly called “waterproof”) product maintains the indicated protection after 80 minutes of water immersion.27,28,32,33
Stability
The stability of the sunscreen is important for long-lasting protection with continuous exposure to UV light, in particular to prevent photodegradation. The FDA has established maximum levels of each filter allowed in the sunscreen. Several filters can be combined to achieve a high SPF level, to provide broadspectrum UV-A and UV-B protection, and to prevent photodegradation. For example, octocrylene prevents the degradation of the photosensitive compound avobenzone, whereas ecamsule has been combined with avobenzone and octocrylene to provide broad-spectrum UV-A and UV-B protection. Ecamsule is currently patent-protected by L’Oreal and is found only in products produced by it and its subsidiaries.
SUNSCREEN USES AND ABUSES
Sunscreen use generally falls into three categories: daily use, short-term use (eg, for an activity involving increased sun exposure, such as outdoor exercise or work), and use for preventing sunburn during tan acquisition, ie, to increase the time of UV radiation exposure.
Most published studies report on the effects of daily sunscreen protection or on cutaneous immune responses to sunscreen use. However, the use of sunscreens to enhance tan acquisition and to increase sun exposure duration is an abuse of the product and can actually increase the risk of skin cancer. A common misperception is that sunscreens decrease the risk of burning and allow people to increase their exposure to UV radiation. This results in increased exposure to UV-A and thus increases the risk of skin cancers and facilitates photoaging.34
In 2003, Baron et al35 published a randomized trial evaluating the protective effects of UV-B sunscreens (SPF 15) and UV-A/UV-B sunscreens (SPF 15) against UV radiation, using contact hypersensitivity as a model for immunosuppression. The study involved 211 volunteers ages 18 to 59. Measuring skinfold thickness vs total UV dose to calculate an immune protection factor, they reported that the UV-A/UV-B sunscreens had a greater average immune protection factor than the UV-B sunscreen. They concluded that though both types of sunscreen can protect against immunosuppression, the addition of a UV-A filter provides greater protection against immunosuppression.35
A French study36 in 104 volunteers examined the immunoprotective effects of sunscreens with equal SPF but differing levels of UV-A protection after UV exposure, and used delayed-type hypersensitivity as a model for cutaneous immune response. Broader UV-A protection yielded smaller reductions in delayed-type hypersensitivity after UV exposure, leading to the conclusion that UV-A contributes greatly to cutaneous immunosuppression and that UV-A filters can mitigate some of these effects.36
Sunscreens and photoaging
Only a few clinical studies have examined the effects of sunscreen use on photoaging.
In 1995, a randomized, double-blind, placebo-controlled trial involving 53 adults with previously diagnosed with actinic keratosis or skin cancer, or both, showed that those who applied a UV-A/UV-B sunscreen over a 24-month period had less solar elastosis on biopsy compared with controls.37
In 2008, a French study of 12 volunteers showed that broad-spectrum UV protection prevented histologic changes attributed to 6 weeks of chronic UV exposure. The control group exhibited structural and molecular evidence of UV damage (eg, epidermal thickening, decreased procollagen expression, higher lysozyme-to-elastin ratio), whereas chronic use of a broad-spectrum sunscreen either minimized or abrogated these findings.12
Evidence also suggests that broad-spectrum sunscreens can prevent damage from suberythemal doses of UV. A study published in 200738 investigated whether broad-spectrum sunscreen use affects the development of genetic and cellular markers of UV damage after daily suberythemal UV exposure. It reported that unprotected individuals exhibited more thymine dimers, higher p53 expression, and loss of Langerhans cells compared with protected individuals.38
Similarly, a study published in 201012 assessed cellular and molecular markers of photodamage after 19 daily suberythemal UV exposures with or without a broad-spectrum, low-SPF (SPF 8) sunscreen and found that consistent sunscreen use resulted in fewer p53-positive cells, a lower lysozyme-to-elastin ratio, a decreased number and size of melanocytes, and an increased number of Langerhans cells.
Thus, evidence supports the idea that consistent use of a broad-spectrum sunscreen can protect against photodamage, even at doses that do not cause erythema.12
Sunscreens and squamous cell carcinoma
Several large trials provide appreciable evidence that sunscreen is effective in preventing squamous cell carcinoma.
A randomized, controlled, 7-month trial in Australia of a broad-spectrum sunscreen with an SPF of 17 noted a dose-dependent reduction in the development of new actinic keratosis.39 Another randomized, controlled trial from Australia showed a 40% reduction in the development of squamous cell carcinoma over a 4.5-year period in participants who applied a broad-spectrum SPF-16 sunscreen 3 to 4 days per week vs discretionary use.40 Follow-up data at 8 years showed that daily sunscreen users continued to have a 40% lower incidence rate of squamous cell carcinoma than controls.41
Sunscreens and basal cell carcinoma
Although sunscreens appear to be effective in preventing actinic keratosis and squamous cell carcinoma, the evidence that they also prevent basal cell carcinoma and melanoma has been inconclusive.
Sunscreens and melanoma
Using a high number of nevi as a surrogate measure of the risk of developing melanoma, a randomized controlled trial of a broad-spectrum SPF-30 sunscreen in Canadian children over a 3-year period showed a slight decrease in the number of new nevi compared with controls. However, this effect was seen only in children with freckles.42
In a large European study of white school-age children, sunscreen use was associated with an increased number of nevi compared with the use of clothing, which prevented new nevi.43
A large meta-analysis of 18 case-controlled studies failed to show a protective association of sunscreen use with melanoma.44 Postulated confounding factors in earlier studies included older sunscreen formulations with no UV-A protection, low SPF, and limited substantivity. In many cases, sunscreen users exposed themselves to higher doses of UV because of the perceived decreased risk of burning with sunscreen use. This is especially the case when sun exposure was intentional to acquire a tan.34 Individuals who burn easily or may have had a family history of melanoma tended to use more sunscreen, thus creating another confounder. Finally, extrapolation of results from data performed in different geographic latitudes may not be appropriate.
Recently, Green et al45 published a study using the same cohort from a previous study of sunscreens and nonmelanoma skin cancer to examine new primary melanomas as a secondary outcome. They reported that, during the 5-year trial period and during the 10-year follow-up, fewer participants in the intervention group developed primary melanoma compared with the control group (11 vs 21). They concluded that regular applications of a broad-spectrum SPF-16 sunscreen in white adults ages 25 to 75 can decrease the incidence of melanoma.45 The study had serious limitations: the authors admitted that the results were marginally statistically significant; intervention sites of sunscreen application were chosen for nonmelanoma skin cancer and excluded the trunk and lower extremities, where melanomas often occur; and the entire body was analyzed for melanomas, not just the intervention site.46 Thus, despite providing some of the first evidence supporting sunscreen’s ability to prevent melanoma, these results are controversial and are by no means conclusive.
HOW TO USE SUNSCREEN
The American Academy of Dermatology guidelines47 recommend daily, year-round use of a broad-spectrum, water-resistant sunscreen with an SPF of at least 30, regardless of age or skin type. Cloud cover and windows block UV-B but not UV-A. Additionally, 80% of UV light can pass through cloud cover, while 25% is reflected by sand and 80% by snow. Thus, sunscreen should be used daily throughout the year.
Sunscreen should be applied to exposed dry skin 15 to 30 minutes before sun exposure, paying particular attention to common areas of nonmelanoma skin cancer, such as the face, ears, hands, arms, and lips. The standard amount of sunscreen used in SPF testing is 2 mg/cm2, which is difficult to translate into real use; most people apply only 25% to 50% of the recommended amount of sunscreen.48 According to the guidelines, 1 oz of sunscreen—2 tablespoons, or enough to fill a shot glass—is enough to cover sun-exposed parts of the adult body. Sunscreen should be reapplied every 2 hours or after swimming or heavy perspiration; many water-resistant sunscreens lose effectiveness after 40 minutes in the water.
Despite the protective effects of sunscreen, the following are still recommended:
Seek shade or avoid exposure between 10:00 am and 4:00 pm, ie, when the sun’s rays are strongest
Take caution around water, sand, and snow, which reflect UV radiation
Wear protective clothing such as long-sleeved shirts, pants, sunglasses, and wide-brimmed hats
Do not use tanning beds
Do not use sunscreens to increase the time of UV exposure.
SPECIAL CONSIDERATIONS: INFANTS
Infants and toddlers are at higher risk of UV damage and skin cancer. Structurally, children’s skin is thinner than that of adults and has lower melanin concentrations. Thus, UV penetrates more deeply into skin that is less able to absorb UV radiation. Animal studies suggest that the skin of children, especially infants, is immunologically immature and less able to respond to UV damage than adult skin. Therefore, extra care must be taken to protect children from UV exposure.49
The American Academy of Pediatrics recommends that infants under 6 months of age should be kept out of direct sunlight whenever possible. A broad-spectrum, water-resistant sunscreen with an SPF of at least 30 should be applied to skin that is not protected by clothing or shade (eg, face, hands, neck).50
NEW FDA GUIDELINES AND OTHER PROPOSED CHANGES
Figure 1. New US Food and Drug Administration (FDA) labeling standards include separately delineating “broad-spectrum” and sun protection factor (SPF) information in an equal font size. The claim “water-resistant” must be specified with a time, ie, 40 or 80 minutes. The “drug facts” box on the back of the product must include usage directions, guidelines for sun protection, and other FDA-required statements.In June 2011, the FDA released a new set of testing and labeling requirements for sunscreens (Figure 1)51 and proposed further modifications to the rules for manufacturing sunscreen products. Manufacturers must comply with these new rules within 12 months of the date of release (at least by June 17, 2012). Manufacturers with annual sales of less than $25,000 were given 24 months to comply.
The FDA’s SPF labeling requirements remained unchanged; however, the FDA instituted new regulations regarding UV-A protection. Sunscreens that qualify as broad-spectrum are to be labeled as such, indicating that they protect against radiation in the entire UV spectrum. Products that are “broad-spectrum SPF ≥ 15” can now include the following statement in the “drug facts” part of the label: “If used as directed with other sun protection measures, decreases the risk of skin cancer and early skin aging caused by the sun.”
The FDA now requires sunscreens that are not broad-spectrum or that have an SPF less than 15 to include the following alert: “Spending time in the sun increases your risk of skin cancer and early skin aging.”33 These products can only claim protection from sunburn with the statement: “This product has been shown only to prevent sunburn, not skin cancer or early skin aging.”27,28,32,33
In terms of water resistance, the FDA now bans the terms “sunblock,” “waterproof,” or “sweatproof,” as these claims cannot be substantiated. Instead, the label on the front of the package can only read either “water resistant (40 minutes)” or “water resistant (80 minutes).” Also, sunscreens may no longer claim to provide “instant protection,” nor can they claim to maintain efficacy for more than 2 hours without reapplication.27,28,32,33
Some sunscreen products have been labeled with SPF values exceeding 100. The FDA decided that because there is insufficient evidence of clinical benefit for such SPFs, sunscreen product labels may claim a maximum SPF value of “50+.”28,52
The FDA now also specifies approved formulations for sunscreen products. Oils, lotions, creams, gels, butters, pastes, and ointments are acceptable, and this applies to all products that contain sunscreens, including cosmetics. Wipes, towelettes, powders, body washes, and shampoos are not acceptable as sunscreen products. The FDA now considers the popular spray form as potentially acceptable; a final decision awaits the results of further testing.28,53
Editor’s note: As this paper was being sent to press, the US Food and Drug Administration announced that sunscreen manufacturers would have an additional 6 months to comply with the new labeling rules for sunscreens. The new deadline is December 2012. Smaller companies have until December 2013 to implement the labeling changes.
References
Kullavanijaya P, Lim HW. Photoprotection. J Am Acad Dermatol2005; 52:937–958.
Sivamani RK, Ghiya M, Maibach HI. Shedding light on sunscreens and their labels: testing policies need to match actual use. Am J Prev Med2010; 38:679–681.
Miyamura Y, Coelho SG, Schlenz K, et al. The deceptive nature of UVA tanning versus the modest protective effects of UVB tanning on human skin. Pigment Cell Melanoma Res2011; 24:136–147.
Wang Y, Digiovanna JJ, Stern JB, et al. Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc Natl Acad Sci U S A2009; 106:6279–6284.
Yano K, Kadoya K, Kajiya K, Hong YK, Detmar M. Ultraviolet B irradiation of human skin induces an angiogenic switch that is mediated by upregulation of vascular endothelial growth factor and by downregulation of thrombospondin-1. Br J Dermatol2005; 152:115–121.
Rabe JH, Mamelak AJ, McElgunn PJ, Morison WL, Sauder DN. Photoaging: mechanisms and repair. J Am Acad Dermatol2006; 55:1–19.
Damian DL, Patterson CR, Stapelberg M, Park J, Barnetson RS, Halliday GM. UV radiation-induced immunosuppression is greater in men and prevented by topical nicotinamide. J Invest Dermatol2008; 128:447–454.
Miller SA, Hamilton SL, Wester UG, Cyr WH. An analysis of UVA emissions from sunlamps and the potential importance for melanoma. Photochem Photobiol1998; 68:63–70.
Seité S, Fourtanier AM. The benefit of daily photoprotection. J Am Acad Dermatol2008; 58(5 suppl 2):S160–166.
Besaratinia A, Synold TW, Chen HH, et al. DNA lesions induced by UV A1 and B radiation in human cells: comparative analyses in the overall genome and in the p53 tumor suppressor gene. Proc Natl Acad Sci U S A2005; 102:10058–10063.
May P, May E. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene1999; 18:7621–7636.
Mouret S, Baudouin C, Charveron M, Favier A, Cadet J, Douki T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc Natl Acad Sci USA2006; 103:13765–13770.
Wolber R, Schlenz K, Wakamatsu K, et al. Pigmentation effects of solar-simulated radiation as compared with UVA and UVB radiation. Pigment Cell Melanoma Res2008; 21:487–491.
Miyamura Y, Coelho SG, Wolber R, et al. Regulation of human skin pigmentation and responses to ultraviolet radiation. Pigment Cell Res2007; 20:2–13.
Kwon HT, Mayer JA, Walker KK, Yu H, Lewis EC, Belch GE. Promotion of frequent tanning sessions by indoor tanning facilities: two studies. J Am Acad Dermatol2002; 46:700–705.
Dellavalle RP, Parker ER, Cersonsky N, et al. Youth access laws: in the dark at the tanning parlor?Arch Dermatol2003; 139:443–448.
Whitmore SE, Morison WL, Potten CS, Chadwick C. Tanning salon exposure and molecular alterations. J Am Acad Dermatol2001; 44:775–780.
Swerdlow AJ, Weinstock MA. Do tanning lamps cause melanoma? An epidemiologic assessment. J Am Acad Dermatol1998; 38:89–98.
IBISWorld. Tanning salons in the US: Market research report NAICS 81219c. www.ibisworld.com. Accesssed May 9, 2012.
Wang SQ, Lim HW. Current status of the sunscreen regulation in the United States: 2011 Food and Drug Administration’s final rule on labeling and effectiveness testing. J Am Acad Dermatol2011; 65:863–869.
Food and Drug Administration (FDA). Labeling and effectiveness testing; sunscreen drug products for over-the-counter human use (final rule). Federal Register2011. http://www.gpo.gov/fdsys/pkg/FR-2011-06-17/pdf/2011-14766.pdf. Accessed May 9, 2012.
Scherschun L, Lim HW. Photoprotection by sunscreens. Am J Clin Dermatol2001; 2:131–134.
US Food and Drug Administration (FDA). FDA Press Release. FDA announces changes to better inform consumers about sunscreen: new rules give consumers more information to help reduce the risk of skin cancer, early aging. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm258940.htm. Accessed May 9, 2012.
Autier P. Sunscreen abuse for intentional sun exposure. Br J Dermatol2009; 161(suppl 3):40–45.
Baron ED, Fourtanier A, Compan D, Medaisko C, Cooper KD, Stevens SR. High ultraviolet A protection affords greater immune protection confirming that ultraviolet A contributes to photoimmunosuppression in humans. J Invest Dermatol2003; 121:869–875.
Moyal DD, Fourtanier AM. Broad-spectrum sunscreens provide better protection from solar ultraviolet-simulated radiation and natural sunlight-induced immunosuppression in human beings. J Am Acad Dermatol2008; 58(suppl 2):S149–S154.
Boyd AS, Naylor M, Cameron GS, Pearse AD, Gaskell SA, Neldner KH. The effects of chronic sunscreen use on the histologic changes of dermatoheliosis. J Am Acad Dermatol1995; 33:941–946.
Young AR, Orchard GE, Harrison GI, Klock JL. The detrimental effects of daily sub-erythemal exposure on human skin in vivo can be prevented by a daily-care broad-spectrum sunscreen. J Invest Dermatol2007; 127:975–978.
Thompson SC, Jolley D, Marks R. Reduction of solar keratoses by regular sunscreen use. N Engl J Med1993; 329:1147–1151.
Green A, Williams G, Neale R, et al. Daily sunscreen application and betacarotene supplementation in prevention of basal-cell and squamous-cell carcinomas of the skin: a randomised controlled trial. Lancet1999; 354:723–729.
van der Pols JC, Williams GM, Pandeya N, Logan V, Green AC. Prolonged prevention of squamous cell carcinoma of the skin by regular sunscreen use. Cancer Epidemiol Biomarkers Prev2006; 15:2546–2548.
Gallagher RP, Rivers JK, Lee TK, Bajdik CD, McLean DI, Coldman AJ. Broad-spectrum sunscreen use and the development of new nevi in white children: a randomized controlled trial. JAMA2000; 283:2955–2960.
Autier P, Doré JF, Cattaruzza MS, et al. Sunscreen use, wearing clothes, and number of nevi in 6- to 7-year-old European children. European Organization for Research and Treatment of Cancer Melanoma Cooperative Group. J Natl Cancer Inst1998; 90:1873–1880.
Dennis LK, Beane Freeman LE, VanBeek MJ. Sunscreen use and the risk for melanoma: a quantitative review. Ann Intern Med2003; 139:966–978.
Green AC, Williams GM, Logan V, Strutton GM. Reduced melanoma after regular sunscreen use: randomized trial follow-up. J Clin Oncol2011; 29:257–263.
Goldenhersh MA, Koslowsky M. Increased melanoma after regular sunscreen use?J Clin Oncol2011; 29:e557–e558.
Neale R, Williams G, Green A. Application patterns among participants randomized to daily sunscreen use in a skin cancer prevention trial. Arch Dermatol2002; 138:1319–1325.
Paller AS, Hawk JL, Honig P, et al. New insights about infant and toddler skin: implications for sun protection. Pediatrics2011; 128:92–102.
Food and Drug Administration (FDA). Revised effectiveness determination; Sunscreen drug products for over-the-counter human use (proposed rule.) Federal Register 2011. http://69.175.53.6/register/2011/jun/17/2011-14769.pdf. Accessed May 9, 2012.
Food and Drug Administration (FDA). Sunscreen drug products for over-the-counter human use: Request for data and information regarding dosage forms (advance notice of proposed rulemaking), Federal Register 2011). http://69.175.53.6/register/2011/jun/17/2011-14768.pdf. Accessed May 9, 2012.
References
Kullavanijaya P, Lim HW. Photoprotection. J Am Acad Dermatol2005; 52:937–958.
Sivamani RK, Ghiya M, Maibach HI. Shedding light on sunscreens and their labels: testing policies need to match actual use. Am J Prev Med2010; 38:679–681.
Miyamura Y, Coelho SG, Schlenz K, et al. The deceptive nature of UVA tanning versus the modest protective effects of UVB tanning on human skin. Pigment Cell Melanoma Res2011; 24:136–147.
Wang Y, Digiovanna JJ, Stern JB, et al. Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc Natl Acad Sci U S A2009; 106:6279–6284.
Yano K, Kadoya K, Kajiya K, Hong YK, Detmar M. Ultraviolet B irradiation of human skin induces an angiogenic switch that is mediated by upregulation of vascular endothelial growth factor and by downregulation of thrombospondin-1. Br J Dermatol2005; 152:115–121.
Rabe JH, Mamelak AJ, McElgunn PJ, Morison WL, Sauder DN. Photoaging: mechanisms and repair. J Am Acad Dermatol2006; 55:1–19.
Damian DL, Patterson CR, Stapelberg M, Park J, Barnetson RS, Halliday GM. UV radiation-induced immunosuppression is greater in men and prevented by topical nicotinamide. J Invest Dermatol2008; 128:447–454.
Miller SA, Hamilton SL, Wester UG, Cyr WH. An analysis of UVA emissions from sunlamps and the potential importance for melanoma. Photochem Photobiol1998; 68:63–70.
Seité S, Fourtanier AM. The benefit of daily photoprotection. J Am Acad Dermatol2008; 58(5 suppl 2):S160–166.
Besaratinia A, Synold TW, Chen HH, et al. DNA lesions induced by UV A1 and B radiation in human cells: comparative analyses in the overall genome and in the p53 tumor suppressor gene. Proc Natl Acad Sci U S A2005; 102:10058–10063.
May P, May E. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene1999; 18:7621–7636.
Mouret S, Baudouin C, Charveron M, Favier A, Cadet J, Douki T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc Natl Acad Sci USA2006; 103:13765–13770.
Wolber R, Schlenz K, Wakamatsu K, et al. Pigmentation effects of solar-simulated radiation as compared with UVA and UVB radiation. Pigment Cell Melanoma Res2008; 21:487–491.
Miyamura Y, Coelho SG, Wolber R, et al. Regulation of human skin pigmentation and responses to ultraviolet radiation. Pigment Cell Res2007; 20:2–13.
Kwon HT, Mayer JA, Walker KK, Yu H, Lewis EC, Belch GE. Promotion of frequent tanning sessions by indoor tanning facilities: two studies. J Am Acad Dermatol2002; 46:700–705.
Dellavalle RP, Parker ER, Cersonsky N, et al. Youth access laws: in the dark at the tanning parlor?Arch Dermatol2003; 139:443–448.
Whitmore SE, Morison WL, Potten CS, Chadwick C. Tanning salon exposure and molecular alterations. J Am Acad Dermatol2001; 44:775–780.
Swerdlow AJ, Weinstock MA. Do tanning lamps cause melanoma? An epidemiologic assessment. J Am Acad Dermatol1998; 38:89–98.
IBISWorld. Tanning salons in the US: Market research report NAICS 81219c. www.ibisworld.com. Accesssed May 9, 2012.
Wang SQ, Lim HW. Current status of the sunscreen regulation in the United States: 2011 Food and Drug Administration’s final rule on labeling and effectiveness testing. J Am Acad Dermatol2011; 65:863–869.
Food and Drug Administration (FDA). Labeling and effectiveness testing; sunscreen drug products for over-the-counter human use (final rule). Federal Register2011. http://www.gpo.gov/fdsys/pkg/FR-2011-06-17/pdf/2011-14766.pdf. Accessed May 9, 2012.
Scherschun L, Lim HW. Photoprotection by sunscreens. Am J Clin Dermatol2001; 2:131–134.
US Food and Drug Administration (FDA). FDA Press Release. FDA announces changes to better inform consumers about sunscreen: new rules give consumers more information to help reduce the risk of skin cancer, early aging. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm258940.htm. Accessed May 9, 2012.
Autier P. Sunscreen abuse for intentional sun exposure. Br J Dermatol2009; 161(suppl 3):40–45.
Baron ED, Fourtanier A, Compan D, Medaisko C, Cooper KD, Stevens SR. High ultraviolet A protection affords greater immune protection confirming that ultraviolet A contributes to photoimmunosuppression in humans. J Invest Dermatol2003; 121:869–875.
Moyal DD, Fourtanier AM. Broad-spectrum sunscreens provide better protection from solar ultraviolet-simulated radiation and natural sunlight-induced immunosuppression in human beings. J Am Acad Dermatol2008; 58(suppl 2):S149–S154.
Boyd AS, Naylor M, Cameron GS, Pearse AD, Gaskell SA, Neldner KH. The effects of chronic sunscreen use on the histologic changes of dermatoheliosis. J Am Acad Dermatol1995; 33:941–946.
Young AR, Orchard GE, Harrison GI, Klock JL. The detrimental effects of daily sub-erythemal exposure on human skin in vivo can be prevented by a daily-care broad-spectrum sunscreen. J Invest Dermatol2007; 127:975–978.
Thompson SC, Jolley D, Marks R. Reduction of solar keratoses by regular sunscreen use. N Engl J Med1993; 329:1147–1151.
Green A, Williams G, Neale R, et al. Daily sunscreen application and betacarotene supplementation in prevention of basal-cell and squamous-cell carcinomas of the skin: a randomised controlled trial. Lancet1999; 354:723–729.
van der Pols JC, Williams GM, Pandeya N, Logan V, Green AC. Prolonged prevention of squamous cell carcinoma of the skin by regular sunscreen use. Cancer Epidemiol Biomarkers Prev2006; 15:2546–2548.
Gallagher RP, Rivers JK, Lee TK, Bajdik CD, McLean DI, Coldman AJ. Broad-spectrum sunscreen use and the development of new nevi in white children: a randomized controlled trial. JAMA2000; 283:2955–2960.
Autier P, Doré JF, Cattaruzza MS, et al. Sunscreen use, wearing clothes, and number of nevi in 6- to 7-year-old European children. European Organization for Research and Treatment of Cancer Melanoma Cooperative Group. J Natl Cancer Inst1998; 90:1873–1880.
Dennis LK, Beane Freeman LE, VanBeek MJ. Sunscreen use and the risk for melanoma: a quantitative review. Ann Intern Med2003; 139:966–978.
Green AC, Williams GM, Logan V, Strutton GM. Reduced melanoma after regular sunscreen use: randomized trial follow-up. J Clin Oncol2011; 29:257–263.
Goldenhersh MA, Koslowsky M. Increased melanoma after regular sunscreen use?J Clin Oncol2011; 29:e557–e558.
Neale R, Williams G, Green A. Application patterns among participants randomized to daily sunscreen use in a skin cancer prevention trial. Arch Dermatol2002; 138:1319–1325.
Paller AS, Hawk JL, Honig P, et al. New insights about infant and toddler skin: implications for sun protection. Pediatrics2011; 128:92–102.
Food and Drug Administration (FDA). Revised effectiveness determination; Sunscreen drug products for over-the-counter human use (proposed rule.) Federal Register 2011. http://69.175.53.6/register/2011/jun/17/2011-14769.pdf. Accessed May 9, 2012.
Food and Drug Administration (FDA). Sunscreen drug products for over-the-counter human use: Request for data and information regarding dosage forms (advance notice of proposed rulemaking), Federal Register 2011). http://69.175.53.6/register/2011/jun/17/2011-14768.pdf. Accessed May 9, 2012.
Despite the known risks, nearly 28 million Americans use a sunbed or a sunlamp every year, and 70% of those are white women ages 16 to 29.
Sunscreens have been a source of confusion in their labeling and their sun protection factor ratings. Revised FDA labeling requirements may help clinicians provide useful guidance to patients.
The American Academy of Dermatology supports a ban on the nonmedical production and sale of indoor tanning devices.
Recommendations to prevent UV damage include minimizing sun exposure during peak daylight hours, wearing clothing such as long-sleeve shirts, wide-brimmed hats, and sunglasses, and application of a broad-spectrum sunscreen with UV-A protection. Infants less than 6 months of age require additional protective measures.
Overexposure to the sun’s ultraviolet (UV) radiation—whether accidental or to acquire a tan—raises your risk of skin cancer and sun-related aging of the skin. Here are some basic steps to take to avoid UV damage:
Seek shade or minimize your sun exposure between 10 am and 4 pm, when the sun’s rays are the most intense. Wear protective clothing such as a wide-brimmed hat and long sleeves. Wear UV-blocking sunglasses. Whenever possible, keep children under 6 months old out of direct sunlight.
Apply sunscreen to minimize UV damage when out in the sun. Choose a sunscreen that is broad-spectrum and water-resistant and with a sun protection factor—or SPF—of at least 30. This can be used all year long, regardless of your age or whether or not you are “fair-skinned.”
Apply sunscreen to exposed dry skin 10–15 minutes before going out into the sun. The amount to use for an adult is about 2 tablespoons. Wear sunscreen even when the sky is overcast, as UV rays penetrate clouds and windows (unless the windows are treated to block UV light). Also, take cover and use sunscreen around water, sand, and snow: they reflect UV radiation.
Sunscreen is only effective for a limited time. Be sure to reapply sunscreen every 2 hours, or even sooner if you have been swimming or sweating heavily (eg, playing tennis or volleyball).
Don’t abuse sunscreen lotions. Don’t apply sunscreen so that you can stay longer in the sun—for example, in order to tan. They are not intended to let you stay in the sun for long periods. Long exposure to the sun’s damaging UV rays increases your risk of skin cancer and photoaging.
Do not use tanning beds. They expose the skin to much larger amounts of damaging UV light than normal sun exposure. The American Dermatologic Association supports a ban on tanning beds, and the World Health Organization classifies them as carcinogenic.
This information is provided by your physician and the Cleveland Clinic Journal of Medicine. It is not designed to replace a physician’s medical assessment and judgment.
This page may be reproduced noncommercially to share with patients. Any other reproduction is subject to Cleveland Clinic Journal of Medicine approval. Bulk color reprints are available by calling 216-444-2661.
For patient information on hundreds of health topics, see the Patient Education and Health Information web site, www.clevelandclinic.org/health
Overexposure to the sun’s ultraviolet (UV) radiation—whether accidental or to acquire a tan—raises your risk of skin cancer and sun-related aging of the skin. Here are some basic steps to take to avoid UV damage:
Seek shade or minimize your sun exposure between 10 am and 4 pm, when the sun’s rays are the most intense. Wear protective clothing such as a wide-brimmed hat and long sleeves. Wear UV-blocking sunglasses. Whenever possible, keep children under 6 months old out of direct sunlight.
Apply sunscreen to minimize UV damage when out in the sun. Choose a sunscreen that is broad-spectrum and water-resistant and with a sun protection factor—or SPF—of at least 30. This can be used all year long, regardless of your age or whether or not you are “fair-skinned.”
Apply sunscreen to exposed dry skin 10–15 minutes before going out into the sun. The amount to use for an adult is about 2 tablespoons. Wear sunscreen even when the sky is overcast, as UV rays penetrate clouds and windows (unless the windows are treated to block UV light). Also, take cover and use sunscreen around water, sand, and snow: they reflect UV radiation.
Sunscreen is only effective for a limited time. Be sure to reapply sunscreen every 2 hours, or even sooner if you have been swimming or sweating heavily (eg, playing tennis or volleyball).
Don’t abuse sunscreen lotions. Don’t apply sunscreen so that you can stay longer in the sun—for example, in order to tan. They are not intended to let you stay in the sun for long periods. Long exposure to the sun’s damaging UV rays increases your risk of skin cancer and photoaging.
Do not use tanning beds. They expose the skin to much larger amounts of damaging UV light than normal sun exposure. The American Dermatologic Association supports a ban on tanning beds, and the World Health Organization classifies them as carcinogenic.
This information is provided by your physician and the Cleveland Clinic Journal of Medicine. It is not designed to replace a physician’s medical assessment and judgment.
This page may be reproduced noncommercially to share with patients. Any other reproduction is subject to Cleveland Clinic Journal of Medicine approval. Bulk color reprints are available by calling 216-444-2661.
For patient information on hundreds of health topics, see the Patient Education and Health Information web site, www.clevelandclinic.org/health
Overexposure to the sun’s ultraviolet (UV) radiation—whether accidental or to acquire a tan—raises your risk of skin cancer and sun-related aging of the skin. Here are some basic steps to take to avoid UV damage:
Seek shade or minimize your sun exposure between 10 am and 4 pm, when the sun’s rays are the most intense. Wear protective clothing such as a wide-brimmed hat and long sleeves. Wear UV-blocking sunglasses. Whenever possible, keep children under 6 months old out of direct sunlight.
Apply sunscreen to minimize UV damage when out in the sun. Choose a sunscreen that is broad-spectrum and water-resistant and with a sun protection factor—or SPF—of at least 30. This can be used all year long, regardless of your age or whether or not you are “fair-skinned.”
Apply sunscreen to exposed dry skin 10–15 minutes before going out into the sun. The amount to use for an adult is about 2 tablespoons. Wear sunscreen even when the sky is overcast, as UV rays penetrate clouds and windows (unless the windows are treated to block UV light). Also, take cover and use sunscreen around water, sand, and snow: they reflect UV radiation.
Sunscreen is only effective for a limited time. Be sure to reapply sunscreen every 2 hours, or even sooner if you have been swimming or sweating heavily (eg, playing tennis or volleyball).
Don’t abuse sunscreen lotions. Don’t apply sunscreen so that you can stay longer in the sun—for example, in order to tan. They are not intended to let you stay in the sun for long periods. Long exposure to the sun’s damaging UV rays increases your risk of skin cancer and photoaging.
Do not use tanning beds. They expose the skin to much larger amounts of damaging UV light than normal sun exposure. The American Dermatologic Association supports a ban on tanning beds, and the World Health Organization classifies them as carcinogenic.
This information is provided by your physician and the Cleveland Clinic Journal of Medicine. It is not designed to replace a physician’s medical assessment and judgment.
This page may be reproduced noncommercially to share with patients. Any other reproduction is subject to Cleveland Clinic Journal of Medicine approval. Bulk color reprints are available by calling 216-444-2661.
For patient information on hundreds of health topics, see the Patient Education and Health Information web site, www.clevelandclinic.org/health
A 64-year-old man with diabetes and hypertension presented with a 2-day history of sudden onset of acute pain and cyanosis on the sole of his right foot 4 days after undergoing cardiac catheterization and coronary angiography.
Figure1. Macular violaceous connecting rings in a net-like pattern compatible with livedo reticularis on the foot.The physical examination revealed macular, violaceous, connecting rings in a net-like pattern that blanched with pressure and disappeared when the foot was elevated, a presentation compatible with livedo reticularis (Figure 1). Laboratory testing (complete blood cell count, biochemistry panel, coagulation test, and C-reactive protein test) was notable only for eosinophilia.
A few days later, the patient returned with abdominal pain, diarrhea, and acute renal injury with urinary eosinophils (7% of the white blood cells in the urine) and proteinuria.
Q: Which is the most likely diagnosis?
Infective endocarditis
Pernio (chilblain)
Cholesterol crystal embolism
Cutaneous small-vessel vasculitis
A: Cholesterol crystal embolism is the correct diagnosis.
Infective endocarditis is an infection of the endocardium, but systemic signs may be present, including cutaneous lesions such as Osler nodes (painful papules on the tips of the fingers and toes) and Janeway lesions (painless macules on the palms and soles). Histologic staining of skin biopsy specimens often shows vasculitis, occasionally with a positive Gram stain. Severe renal injury is not common, and the timing of the acute illness and skin lesion fits better with an acute embolic phenomenon.
Pernio is a form of cold injury, localized in peripheral parts of the body and occurring after exposure to cold temperatures in damp conditions. It usually manifests bilaterally as painful erythematous or purple lesions on the acral areas of the hands and feet, nose, ears, and, rarely, the thighs and buttocks. Pernio most commonly affects women between 20 and 40 years of age. It can be idiopathic or associated with a systemic disease such as systemic lupus erythematosus or Sjögren syndrome.
Cutaneous small-vessel vasculitis is a heterogeneous group of disorders with inflammation and damage of the blood vessels; it may be limited to the skin or it may involve multiple systems. Palpable or nonpalpable purpura and ulceration are common clinical findings, and histologic study shows an inflammatory infiltrate of vessel walls, fibrinoid necrosis, thrombosis, and extravasation of red blood cells.
While this patient’s problems are not consistent with small-vessel vasculitis, the single skin lesion, the timing after the catheterization, and the urinary eosinophils are best explained by cholesterol embolization.
CHOLESTEROL CRYSTAL EMBOLISM
Cholesterol crystal embolism is commonly iatrogenic, a complication of mechanical damage to the arterial walls from vascular surgery or invasive percutaneous procedures. Material dislodged from atheromatous arterial plaques can occlude the small vessels of the feet, producing this syndrome.
The onset of the clinical disease is often delayed for days to weeks after an angiographic procedure.1 Spontaneous hemorrhage, disruption of plaque, or drug therapy with an anticoagulant or a fibrinolytic can precipitate embolization of cholesterol crystals. The source of the emboli is atheromatous plaque in major blood vessels, particularly the abdominal aorta.
Many organs and systems can be affected
These emboli can affect many organs and systems: eg, the kidneys (causing hypertension and acute renal failure), the muscles (causing myalgias), gastrointestinal organs (causing bleeding, abdominal pain, and bowel infarction), the lungs (causing acute respiratory distress syndrome), the eyes (causing Hollenhorst plaques in retinal arteries), and the central nervous system (causing stroke, confusion, and delirium). Cardiac or central nervous system involvement is associated with a high risk of death.
After angiography, clinical manifestations of cholesterol embolization have been reported in 0.06% to 1.4% of patients,2,3 although the finding of cholesterol emboli is more common in autopsy studies.4
Recognizing skin signs is the key
Cutaneous abnormalities are usually the earliest and often the only clinical manifestation of this syndrome. Findings on the lower limbs include blue toes, cutaneous nodules, and livedo reticularis, affecting the feet and legs and sometimes extending upward to the trunk. Other findings include digital infarcts, ulceration, gangrene, purpura, cyanosis, and splinter hemorrhages in the nail bed.
Figure 2. Skin biopsy showed needle-shaped clefts within the lumen of blood vessels, ie, dissolved cholesterol crystals obstructing small arteries.
In our patient, microscopic study of skin biopsy specimens showed needle-shaped clefts within the lumen of blood vessels—ie, dissolved cholesterol crystals obstructing small arteries (Figure 2).
Biopsy studies of skin lesions are positive in a high percentage of cases, showing dissolved cholesterol crystals within the lumen of blood vessels, especially in the small to large arteries and arterioles of the deep dermis or subcutaneous fat. Deep biopsies and carefully examination are necessary, as emboli tend to be patchily distributed. Early recognition of cutaneous clinical findings is essential to establish the proper diagnosis and treatment.
The diagnostic triad of this disease includes blue toe syndrome, acute or subacute renal failure, and a temporal relation with an inciting event (particularly angiography). But despite these diagnostic criteria,2 the diagnosis is often based on a combination of signs and symptoms specific to end-organ damage and a systemic inflammatory response.3
Histologic confirmation is considered essential to the diagnosis of cholesterol crystal embolism, and as the skin is the most accessible site, skin biopsy provides the best sample for histologic diagnosis.5
Postprocedural embolism of a blood clot, vasculitis, and infective endocarditis are the most important differential diagnoses.6,7
Treatment is supportive, preventive
Treatment is mainly supportive with hemodynamic monitoring, nutritional and metabolic support, mechanical ventilation, and dialysis if necessary. The underlying atherosclerotic disease should be treated aggressively. Prevention of another episode involves modification of traditional risk factors such as serum cholesterol, diabetes, hypertension, and smoking. Additional vascular surgery procedures should be avoided, as they can induce new episodes.
References
Donohue KG, Saap L, Falanga V. Cholesterol crystal embolization: an atherosclerotic disease with frequent and varied cutaneous manifestations. J Eur Acad Dermatol Venereol2003; 17:504–511.
Fukumoto Y, Tsutsui H, Tsuchihashi M, Masumoto A, Takeshita A; Cholesterol Embolism Study (CHEST) Investigators. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol2003; 42:211–216.
Johnson LW, Esente P, Giambartolomei A, et al. Peripheral vascular complications of coronary angioplasty by the femoral and brachial techniques. Cathet Cardiovasc Diagn1994; 31:165–172.
Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation2010; 122:631–641.
Jucgla A, Moreso F, Muniesa C, Moreno A, Vidaller A. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol2006; 55:786–793.
Maki T, Izumi C, Miyake M, et al. Cholesterol embolism after cardiac catheterization mimicking infective endocarditis. Intern Med2005; 44:1060–1063.
Salvador Arias-Santiago, MD, PhD Department of Dermatology, San Cecilio University Hospital, Department of Dermatology, Baza General Hospital, and Department of Histology, School of Medicine, Granada University, Granada, Spain
Jose Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Victor Carriel, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Jacinto Orgaz-Molina, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Miguel Gonález-Andrades, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Agustín Buendía-Eisman, MD, PhD Department of Dermatology, School of Medicine, Granada University, Granada, Spain
Miguel Alaminos, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Oloriz 16, Granada 18012, Spain; e-mail salvadorarias@hotmail.es
Salvador Arias-Santiago, MD, PhD Department of Dermatology, San Cecilio University Hospital, Department of Dermatology, Baza General Hospital, and Department of Histology, School of Medicine, Granada University, Granada, Spain
Jose Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Victor Carriel, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Jacinto Orgaz-Molina, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Miguel Gonález-Andrades, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Agustín Buendía-Eisman, MD, PhD Department of Dermatology, School of Medicine, Granada University, Granada, Spain
Miguel Alaminos, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Oloriz 16, Granada 18012, Spain; e-mail salvadorarias@hotmail.es
Author and Disclosure Information
Salvador Arias-Santiago, MD, PhD Department of Dermatology, San Cecilio University Hospital, Department of Dermatology, Baza General Hospital, and Department of Histology, School of Medicine, Granada University, Granada, Spain
Jose Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Victor Carriel, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Jacinto Orgaz-Molina, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Miguel Gonález-Andrades, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Agustín Buendía-Eisman, MD, PhD Department of Dermatology, School of Medicine, Granada University, Granada, Spain
Miguel Alaminos, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Oloriz 16, Granada 18012, Spain; e-mail salvadorarias@hotmail.es
A 64-year-old man with diabetes and hypertension presented with a 2-day history of sudden onset of acute pain and cyanosis on the sole of his right foot 4 days after undergoing cardiac catheterization and coronary angiography.
Figure1. Macular violaceous connecting rings in a net-like pattern compatible with livedo reticularis on the foot.The physical examination revealed macular, violaceous, connecting rings in a net-like pattern that blanched with pressure and disappeared when the foot was elevated, a presentation compatible with livedo reticularis (Figure 1). Laboratory testing (complete blood cell count, biochemistry panel, coagulation test, and C-reactive protein test) was notable only for eosinophilia.
A few days later, the patient returned with abdominal pain, diarrhea, and acute renal injury with urinary eosinophils (7% of the white blood cells in the urine) and proteinuria.
Q: Which is the most likely diagnosis?
Infective endocarditis
Pernio (chilblain)
Cholesterol crystal embolism
Cutaneous small-vessel vasculitis
A: Cholesterol crystal embolism is the correct diagnosis.
Infective endocarditis is an infection of the endocardium, but systemic signs may be present, including cutaneous lesions such as Osler nodes (painful papules on the tips of the fingers and toes) and Janeway lesions (painless macules on the palms and soles). Histologic staining of skin biopsy specimens often shows vasculitis, occasionally with a positive Gram stain. Severe renal injury is not common, and the timing of the acute illness and skin lesion fits better with an acute embolic phenomenon.
Pernio is a form of cold injury, localized in peripheral parts of the body and occurring after exposure to cold temperatures in damp conditions. It usually manifests bilaterally as painful erythematous or purple lesions on the acral areas of the hands and feet, nose, ears, and, rarely, the thighs and buttocks. Pernio most commonly affects women between 20 and 40 years of age. It can be idiopathic or associated with a systemic disease such as systemic lupus erythematosus or Sjögren syndrome.
Cutaneous small-vessel vasculitis is a heterogeneous group of disorders with inflammation and damage of the blood vessels; it may be limited to the skin or it may involve multiple systems. Palpable or nonpalpable purpura and ulceration are common clinical findings, and histologic study shows an inflammatory infiltrate of vessel walls, fibrinoid necrosis, thrombosis, and extravasation of red blood cells.
While this patient’s problems are not consistent with small-vessel vasculitis, the single skin lesion, the timing after the catheterization, and the urinary eosinophils are best explained by cholesterol embolization.
CHOLESTEROL CRYSTAL EMBOLISM
Cholesterol crystal embolism is commonly iatrogenic, a complication of mechanical damage to the arterial walls from vascular surgery or invasive percutaneous procedures. Material dislodged from atheromatous arterial plaques can occlude the small vessels of the feet, producing this syndrome.
The onset of the clinical disease is often delayed for days to weeks after an angiographic procedure.1 Spontaneous hemorrhage, disruption of plaque, or drug therapy with an anticoagulant or a fibrinolytic can precipitate embolization of cholesterol crystals. The source of the emboli is atheromatous plaque in major blood vessels, particularly the abdominal aorta.
Many organs and systems can be affected
These emboli can affect many organs and systems: eg, the kidneys (causing hypertension and acute renal failure), the muscles (causing myalgias), gastrointestinal organs (causing bleeding, abdominal pain, and bowel infarction), the lungs (causing acute respiratory distress syndrome), the eyes (causing Hollenhorst plaques in retinal arteries), and the central nervous system (causing stroke, confusion, and delirium). Cardiac or central nervous system involvement is associated with a high risk of death.
After angiography, clinical manifestations of cholesterol embolization have been reported in 0.06% to 1.4% of patients,2,3 although the finding of cholesterol emboli is more common in autopsy studies.4
Recognizing skin signs is the key
Cutaneous abnormalities are usually the earliest and often the only clinical manifestation of this syndrome. Findings on the lower limbs include blue toes, cutaneous nodules, and livedo reticularis, affecting the feet and legs and sometimes extending upward to the trunk. Other findings include digital infarcts, ulceration, gangrene, purpura, cyanosis, and splinter hemorrhages in the nail bed.
Figure 2. Skin biopsy showed needle-shaped clefts within the lumen of blood vessels, ie, dissolved cholesterol crystals obstructing small arteries.
In our patient, microscopic study of skin biopsy specimens showed needle-shaped clefts within the lumen of blood vessels—ie, dissolved cholesterol crystals obstructing small arteries (Figure 2).
Biopsy studies of skin lesions are positive in a high percentage of cases, showing dissolved cholesterol crystals within the lumen of blood vessels, especially in the small to large arteries and arterioles of the deep dermis or subcutaneous fat. Deep biopsies and carefully examination are necessary, as emboli tend to be patchily distributed. Early recognition of cutaneous clinical findings is essential to establish the proper diagnosis and treatment.
The diagnostic triad of this disease includes blue toe syndrome, acute or subacute renal failure, and a temporal relation with an inciting event (particularly angiography). But despite these diagnostic criteria,2 the diagnosis is often based on a combination of signs and symptoms specific to end-organ damage and a systemic inflammatory response.3
Histologic confirmation is considered essential to the diagnosis of cholesterol crystal embolism, and as the skin is the most accessible site, skin biopsy provides the best sample for histologic diagnosis.5
Postprocedural embolism of a blood clot, vasculitis, and infective endocarditis are the most important differential diagnoses.6,7
Treatment is supportive, preventive
Treatment is mainly supportive with hemodynamic monitoring, nutritional and metabolic support, mechanical ventilation, and dialysis if necessary. The underlying atherosclerotic disease should be treated aggressively. Prevention of another episode involves modification of traditional risk factors such as serum cholesterol, diabetes, hypertension, and smoking. Additional vascular surgery procedures should be avoided, as they can induce new episodes.
A 64-year-old man with diabetes and hypertension presented with a 2-day history of sudden onset of acute pain and cyanosis on the sole of his right foot 4 days after undergoing cardiac catheterization and coronary angiography.
Figure1. Macular violaceous connecting rings in a net-like pattern compatible with livedo reticularis on the foot.The physical examination revealed macular, violaceous, connecting rings in a net-like pattern that blanched with pressure and disappeared when the foot was elevated, a presentation compatible with livedo reticularis (Figure 1). Laboratory testing (complete blood cell count, biochemistry panel, coagulation test, and C-reactive protein test) was notable only for eosinophilia.
A few days later, the patient returned with abdominal pain, diarrhea, and acute renal injury with urinary eosinophils (7% of the white blood cells in the urine) and proteinuria.
Q: Which is the most likely diagnosis?
Infective endocarditis
Pernio (chilblain)
Cholesterol crystal embolism
Cutaneous small-vessel vasculitis
A: Cholesterol crystal embolism is the correct diagnosis.
Infective endocarditis is an infection of the endocardium, but systemic signs may be present, including cutaneous lesions such as Osler nodes (painful papules on the tips of the fingers and toes) and Janeway lesions (painless macules on the palms and soles). Histologic staining of skin biopsy specimens often shows vasculitis, occasionally with a positive Gram stain. Severe renal injury is not common, and the timing of the acute illness and skin lesion fits better with an acute embolic phenomenon.
Pernio is a form of cold injury, localized in peripheral parts of the body and occurring after exposure to cold temperatures in damp conditions. It usually manifests bilaterally as painful erythematous or purple lesions on the acral areas of the hands and feet, nose, ears, and, rarely, the thighs and buttocks. Pernio most commonly affects women between 20 and 40 years of age. It can be idiopathic or associated with a systemic disease such as systemic lupus erythematosus or Sjögren syndrome.
Cutaneous small-vessel vasculitis is a heterogeneous group of disorders with inflammation and damage of the blood vessels; it may be limited to the skin or it may involve multiple systems. Palpable or nonpalpable purpura and ulceration are common clinical findings, and histologic study shows an inflammatory infiltrate of vessel walls, fibrinoid necrosis, thrombosis, and extravasation of red blood cells.
While this patient’s problems are not consistent with small-vessel vasculitis, the single skin lesion, the timing after the catheterization, and the urinary eosinophils are best explained by cholesterol embolization.
CHOLESTEROL CRYSTAL EMBOLISM
Cholesterol crystal embolism is commonly iatrogenic, a complication of mechanical damage to the arterial walls from vascular surgery or invasive percutaneous procedures. Material dislodged from atheromatous arterial plaques can occlude the small vessels of the feet, producing this syndrome.
The onset of the clinical disease is often delayed for days to weeks after an angiographic procedure.1 Spontaneous hemorrhage, disruption of plaque, or drug therapy with an anticoagulant or a fibrinolytic can precipitate embolization of cholesterol crystals. The source of the emboli is atheromatous plaque in major blood vessels, particularly the abdominal aorta.
Many organs and systems can be affected
These emboli can affect many organs and systems: eg, the kidneys (causing hypertension and acute renal failure), the muscles (causing myalgias), gastrointestinal organs (causing bleeding, abdominal pain, and bowel infarction), the lungs (causing acute respiratory distress syndrome), the eyes (causing Hollenhorst plaques in retinal arteries), and the central nervous system (causing stroke, confusion, and delirium). Cardiac or central nervous system involvement is associated with a high risk of death.
After angiography, clinical manifestations of cholesterol embolization have been reported in 0.06% to 1.4% of patients,2,3 although the finding of cholesterol emboli is more common in autopsy studies.4
Recognizing skin signs is the key
Cutaneous abnormalities are usually the earliest and often the only clinical manifestation of this syndrome. Findings on the lower limbs include blue toes, cutaneous nodules, and livedo reticularis, affecting the feet and legs and sometimes extending upward to the trunk. Other findings include digital infarcts, ulceration, gangrene, purpura, cyanosis, and splinter hemorrhages in the nail bed.
Figure 2. Skin biopsy showed needle-shaped clefts within the lumen of blood vessels, ie, dissolved cholesterol crystals obstructing small arteries.
In our patient, microscopic study of skin biopsy specimens showed needle-shaped clefts within the lumen of blood vessels—ie, dissolved cholesterol crystals obstructing small arteries (Figure 2).
Biopsy studies of skin lesions are positive in a high percentage of cases, showing dissolved cholesterol crystals within the lumen of blood vessels, especially in the small to large arteries and arterioles of the deep dermis or subcutaneous fat. Deep biopsies and carefully examination are necessary, as emboli tend to be patchily distributed. Early recognition of cutaneous clinical findings is essential to establish the proper diagnosis and treatment.
The diagnostic triad of this disease includes blue toe syndrome, acute or subacute renal failure, and a temporal relation with an inciting event (particularly angiography). But despite these diagnostic criteria,2 the diagnosis is often based on a combination of signs and symptoms specific to end-organ damage and a systemic inflammatory response.3
Histologic confirmation is considered essential to the diagnosis of cholesterol crystal embolism, and as the skin is the most accessible site, skin biopsy provides the best sample for histologic diagnosis.5
Postprocedural embolism of a blood clot, vasculitis, and infective endocarditis are the most important differential diagnoses.6,7
Treatment is supportive, preventive
Treatment is mainly supportive with hemodynamic monitoring, nutritional and metabolic support, mechanical ventilation, and dialysis if necessary. The underlying atherosclerotic disease should be treated aggressively. Prevention of another episode involves modification of traditional risk factors such as serum cholesterol, diabetes, hypertension, and smoking. Additional vascular surgery procedures should be avoided, as they can induce new episodes.
References
Donohue KG, Saap L, Falanga V. Cholesterol crystal embolization: an atherosclerotic disease with frequent and varied cutaneous manifestations. J Eur Acad Dermatol Venereol2003; 17:504–511.
Fukumoto Y, Tsutsui H, Tsuchihashi M, Masumoto A, Takeshita A; Cholesterol Embolism Study (CHEST) Investigators. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol2003; 42:211–216.
Johnson LW, Esente P, Giambartolomei A, et al. Peripheral vascular complications of coronary angioplasty by the femoral and brachial techniques. Cathet Cardiovasc Diagn1994; 31:165–172.
Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation2010; 122:631–641.
Jucgla A, Moreso F, Muniesa C, Moreno A, Vidaller A. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol2006; 55:786–793.
Maki T, Izumi C, Miyake M, et al. Cholesterol embolism after cardiac catheterization mimicking infective endocarditis. Intern Med2005; 44:1060–1063.
Donohue KG, Saap L, Falanga V. Cholesterol crystal embolization: an atherosclerotic disease with frequent and varied cutaneous manifestations. J Eur Acad Dermatol Venereol2003; 17:504–511.
Fukumoto Y, Tsutsui H, Tsuchihashi M, Masumoto A, Takeshita A; Cholesterol Embolism Study (CHEST) Investigators. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol2003; 42:211–216.
Johnson LW, Esente P, Giambartolomei A, et al. Peripheral vascular complications of coronary angioplasty by the femoral and brachial techniques. Cathet Cardiovasc Diagn1994; 31:165–172.
Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation2010; 122:631–641.
Jucgla A, Moreso F, Muniesa C, Moreno A, Vidaller A. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol2006; 55:786–793.
Maki T, Izumi C, Miyake M, et al. Cholesterol embolism after cardiac catheterization mimicking infective endocarditis. Intern Med2005; 44:1060–1063.
A 60-year-old man presented with progressive swelling of his face and neck, which had begun 2 weeks earlier. He denied any headache, lightheadedness, blurry vision, syncope, or change in his cognitive or memory function. A review of symptoms was unremarkable.
The patient had hypertension and end-stage renal disease, for which he was receiving hemodialysis via a catheter tunneled into his right internal jugular vein. He had undergone multiple unsuccessful attempts to create an arteriovenous fistula over the previous 2 years.
Figure 1. Swollen face, congested conjunctivae, and multiple dilated tortuous veins on the chest and abdominal walls.On physical examination, his vital signs were normal. Swelling of the face and fullness in his neck with bilateral congested conjunctivae were noted. His jugular venous pressure was elevated at 10 cm. Multiple dilated tortuous veins were noticed on his upper chest and across his abdominal wall (Figure 1). The rest of the examination was unremarkable.
Doppler ultrasonography revealed chronic thrombosis and reverse flow in the right internal jugular vein and reverse flow in the right subclavian vein. These findings were consistent with central venous thrombosis and superior vena cava (SVC) syndrome.
Figure 2. Multiple collateral veins in the upper chest (black arrows, left panel), stenosis of the superior vena cava, and the intraluminal catheter in place (right panel).Computed tomography (CT) of the chest showed significant stenosis of the right innominate vein and SVC due to thrombosis. Numerous collateral veins were seen in the neck and across both shoulders (Figure 2).
Diagnosis: SVC syndrome secondary to intravascular thrombosis related to his central venous dialysis catheter.
SVC SYNDROME
The SVC is the major drainage vessel for venous blood from the head, neck, upper extremities, and upper thorax. Obstruction to its flow increases venous pressure, which results in interstitial edema and retrograde collateral flow.1
More than 80% of cases of SVC syndrome are caused by malignant lung tumors and lymphoma.
Nonmalignant causes include mediastinal fibrosis; vascular diseases (eg, aortic aneurysm, large-vessel vasculitis); infections such as histoplasmosis, tuberculosis, syphilis, and actinomycosis; benign mediastinal tumors such as teratoma, cystic hygroma, thymoma, and dermoid cyst; and thrombosis from central venous catheters, pacemaker leads, and guidewires.2–6 A recent report suggests that benign causes may now account for up to 40% of cases as a result of a rise in the use of indwelling central venous catheters and cardiac pacemakers during the past 2 decades, resulting in a higher incidence of SVC thrombosis.7
An obstructed SVC initiates collateral venous return to the heart from the upper half of the body through different pathways. The most important pathway is the azygos venous system, which includes the azygos vein. Occlusion of the SVC at the level of the azygos vein contributes to the appearance of collateral veins on the chest and abdominal walls, and venous blood flows via these collaterals into the inferior vena cava.1,8,9
Different presentations
The diagnosis of SVC syndrome is often made on clinical grounds alone, ie, the combination of the clinical presentation and, often, a thoracic malignancy or contributing factors such as a central catheter.1
With slowly progressive obstruction of the SVC, the most common presenting symptoms include swelling of the face, neck, and both arms. On the other hand, adequate collateral drainage may develop,1 and patients may have minimal symptoms.
However, a rapid onset of SVC syndrome in the absence of collateral circulation will cause a more dramatic and life-threatening presentation, often with neurologic and respiratory sequelae such as cerebral and laryngeal edema and respiratory embarrassment, which were not present in our patient’s case.1,10–15 These serious complications are rare and are considered an acute emergency. In these cases, special attention to airway, breathing, and circulation (the “ABCs”) is essential, and endovascular repairs and stenting or open surgical reconstruction and alternate approaches for renal replacement therapy should be considered.1,12,13,15
CT is diagnostic and provides accurate information about the location of the obstruction and about other critical surrounding structures such as the lungs, mediastinum, and adjacent blood vessels.1,7,10,11 Our patient’s CT scan confirmed a significant stenosis of the SVC due to thrombosis, with no compression coming from the lungs or mediastinal structures.
Thrombolytic therapy in acute cases
In cases of acute thrombosis (with symptom onset less than 2 days previously), thrombolytic therapy followed by anticoagulation is recommended and may both cause the symptoms to regress within several days and allow the central catheter to be kept in.16 However, thrombolytic therapy is less effective in chronic thrombosis (with onset of symptoms more than 10 days previously).16
Vascular or surgical intervention is often needed to treat SVC syndrome related to dialysis access.
Most experts recommend anticoagulation after thrombosis to prevent disease progression and recurrence, although the benefit of either short-term or long-term anticoagulation therapy for this syndrome is unclear.16
Recommended treatments for cancer-related SVC syndrome include chemotherapy and radiation to shrink the tumor that is causing the obstruction. Tissue diagnosis is often necessary to direct treatment decisions.1 However, percutaneous angioplasty and the use of intravenous stents are becoming increasingly common and are simple, safe, and effective in rapidly relieving SVC syndrome caused by malignant diseases.1 A bypass of the SVC may be indicated in some cases.1 Adjunctive therapies include diuretics, corticosteroids, thrombolytics, anticoagulation, and elevating the head of the patient’s bed.1
CASE CONTINUED
Our patient was started on heparin intravenously for 7 days and long-term oral anticoagulant therapy with warfarin (Coumadin) to continue as long as the catheter was in place, with a target international normalized ratio between 2 and 2.5. He required no other interventions, and his dialysis catheter remained functioning. He was monitored in the hospital for 2 weeks, during which his symptoms gradually improved, with noticeable resolution of his facial swelling.
He was discharged home to continue on an oral anticoagulant and was then followed to monitor for a reappearance of the symptoms (which would force the removal of the catheter), and to pursue possible percutaneous angioplasty, stenting, or surgical reconstruction of the SVC if needed.
References
Wilson LD, Detterbeck FC, Yahalom J. Clinical practice. Superior vena cava syndrome with malignant causes. N Engl J Med2007; 356:1862–1869.
Parish JM, Marschke RF, Dines DE, Lee RE. Etiologic considerations in superior vena cava syndrome. Mayo Clin Proc1981; 56:407–413.
Markman M. Diagnosis and management of superior vena cava syndrome. Cleve Clin J Med1999; 66:59–61.
Khanna S, Sniderman K, Simons M, Besley M, Uldall R. Superior vena cava stenosis associated with hemodialysis catheters. Am J Kidney Dis1993; 21:278–281.
Bertrand M, Presant CA, Klein L, Scott E. Iatrogenic superior vena cava syndrome. A new entity. Cancer1984; 54:376–378.
Rice TW, Rodriguez RM, Light RW. The superior vena cava syndrome: clinical characteristics and evolving etiology. Medicine (Baltimore)2006; 85:37–42.
Plekker D, Ellis T, Irusen EM, Bolliger CT, Diacon AH. Clinical and radiological grading of superior vena cava obstruction. Respiration2008; 76:69–75.
Sheth S, Ebert MD, Fishman EK. Superior vena cava obstruction evaluation with MDCT. AJR Am J Roentgenol2010; 194:W336–W346.
DeMichele A, Glick J. Cancer-related emergencies. In:Lenhard R, Osteen R, Gansler T, eds. Clinical Oncology. Atlanta, GA: American Cancer Society; 2001:733–764.
Chen JC, Bongard F, Klein SR. A contemporary perspective on superior vena cava syndrome. Am J Surg1990; 160:207–211.
Sheikh MA, Fernandez BB, Gray BH, Graham LM, Carman TL. Endovascular stenting of nonmalignant superior vena cava syndrome. Catheter Cardiovasc Interv2005; 65:405–411.
Flinterman LE, Van Der Meer FJ, Rosendaal FR, Doggen CJ. Current perspective of venous thrombosis in the upper extremity. J Thromb Haemost2008; 6:1262–1266.
Greenberg S, Kosinski R, Daniels J. Treatment of superior vena cava thrombosis with recombinant tissue type plasminogen activator. Chest1991; 99:1298–1301.
Molhem A, Sabry A, Bawadekji H, Al Saran K. Superior vena cava syndrome in hemodialysis patient. Saudi J Kidney Dis Transpl2011; 22:381–386.
Akoglu H, Yilmaz R, Peynircioglu B, et al. A rare complication of hemodialysis catheters: superior vena cava syndrome. Hemodial Int2007; 11:385–391.
Khaldoon Shaheen, MD Department of Medicine, Case Western Reserve University/St. Vincent Charity Medical Center, Cleveland, OH
M. Chadi Alraies, MD, FACP Clinical Assistant Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Staff, Department of Hospital Medicine, Cleveland Clinic
Address: Khaldoon Shaheen, MD, Department of Internal Medicine, St. Vincent Charity Medical Center, 2351 East 22nd Street, Cleveland, OH 44115; e-mail Khaldoon.Shaheen@stvincentcharity.com
Khaldoon Shaheen, MD Department of Medicine, Case Western Reserve University/St. Vincent Charity Medical Center, Cleveland, OH
M. Chadi Alraies, MD, FACP Clinical Assistant Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Staff, Department of Hospital Medicine, Cleveland Clinic
Address: Khaldoon Shaheen, MD, Department of Internal Medicine, St. Vincent Charity Medical Center, 2351 East 22nd Street, Cleveland, OH 44115; e-mail Khaldoon.Shaheen@stvincentcharity.com
Author and Disclosure Information
Khaldoon Shaheen, MD Department of Medicine, Case Western Reserve University/St. Vincent Charity Medical Center, Cleveland, OH
M. Chadi Alraies, MD, FACP Clinical Assistant Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Staff, Department of Hospital Medicine, Cleveland Clinic
Address: Khaldoon Shaheen, MD, Department of Internal Medicine, St. Vincent Charity Medical Center, 2351 East 22nd Street, Cleveland, OH 44115; e-mail Khaldoon.Shaheen@stvincentcharity.com
A 60-year-old man presented with progressive swelling of his face and neck, which had begun 2 weeks earlier. He denied any headache, lightheadedness, blurry vision, syncope, or change in his cognitive or memory function. A review of symptoms was unremarkable.
The patient had hypertension and end-stage renal disease, for which he was receiving hemodialysis via a catheter tunneled into his right internal jugular vein. He had undergone multiple unsuccessful attempts to create an arteriovenous fistula over the previous 2 years.
Figure 1. Swollen face, congested conjunctivae, and multiple dilated tortuous veins on the chest and abdominal walls.On physical examination, his vital signs were normal. Swelling of the face and fullness in his neck with bilateral congested conjunctivae were noted. His jugular venous pressure was elevated at 10 cm. Multiple dilated tortuous veins were noticed on his upper chest and across his abdominal wall (Figure 1). The rest of the examination was unremarkable.
Doppler ultrasonography revealed chronic thrombosis and reverse flow in the right internal jugular vein and reverse flow in the right subclavian vein. These findings were consistent with central venous thrombosis and superior vena cava (SVC) syndrome.
Figure 2. Multiple collateral veins in the upper chest (black arrows, left panel), stenosis of the superior vena cava, and the intraluminal catheter in place (right panel).Computed tomography (CT) of the chest showed significant stenosis of the right innominate vein and SVC due to thrombosis. Numerous collateral veins were seen in the neck and across both shoulders (Figure 2).
Diagnosis: SVC syndrome secondary to intravascular thrombosis related to his central venous dialysis catheter.
SVC SYNDROME
The SVC is the major drainage vessel for venous blood from the head, neck, upper extremities, and upper thorax. Obstruction to its flow increases venous pressure, which results in interstitial edema and retrograde collateral flow.1
More than 80% of cases of SVC syndrome are caused by malignant lung tumors and lymphoma.
Nonmalignant causes include mediastinal fibrosis; vascular diseases (eg, aortic aneurysm, large-vessel vasculitis); infections such as histoplasmosis, tuberculosis, syphilis, and actinomycosis; benign mediastinal tumors such as teratoma, cystic hygroma, thymoma, and dermoid cyst; and thrombosis from central venous catheters, pacemaker leads, and guidewires.2–6 A recent report suggests that benign causes may now account for up to 40% of cases as a result of a rise in the use of indwelling central venous catheters and cardiac pacemakers during the past 2 decades, resulting in a higher incidence of SVC thrombosis.7
An obstructed SVC initiates collateral venous return to the heart from the upper half of the body through different pathways. The most important pathway is the azygos venous system, which includes the azygos vein. Occlusion of the SVC at the level of the azygos vein contributes to the appearance of collateral veins on the chest and abdominal walls, and venous blood flows via these collaterals into the inferior vena cava.1,8,9
Different presentations
The diagnosis of SVC syndrome is often made on clinical grounds alone, ie, the combination of the clinical presentation and, often, a thoracic malignancy or contributing factors such as a central catheter.1
With slowly progressive obstruction of the SVC, the most common presenting symptoms include swelling of the face, neck, and both arms. On the other hand, adequate collateral drainage may develop,1 and patients may have minimal symptoms.
However, a rapid onset of SVC syndrome in the absence of collateral circulation will cause a more dramatic and life-threatening presentation, often with neurologic and respiratory sequelae such as cerebral and laryngeal edema and respiratory embarrassment, which were not present in our patient’s case.1,10–15 These serious complications are rare and are considered an acute emergency. In these cases, special attention to airway, breathing, and circulation (the “ABCs”) is essential, and endovascular repairs and stenting or open surgical reconstruction and alternate approaches for renal replacement therapy should be considered.1,12,13,15
CT is diagnostic and provides accurate information about the location of the obstruction and about other critical surrounding structures such as the lungs, mediastinum, and adjacent blood vessels.1,7,10,11 Our patient’s CT scan confirmed a significant stenosis of the SVC due to thrombosis, with no compression coming from the lungs or mediastinal structures.
Thrombolytic therapy in acute cases
In cases of acute thrombosis (with symptom onset less than 2 days previously), thrombolytic therapy followed by anticoagulation is recommended and may both cause the symptoms to regress within several days and allow the central catheter to be kept in.16 However, thrombolytic therapy is less effective in chronic thrombosis (with onset of symptoms more than 10 days previously).16
Vascular or surgical intervention is often needed to treat SVC syndrome related to dialysis access.
Most experts recommend anticoagulation after thrombosis to prevent disease progression and recurrence, although the benefit of either short-term or long-term anticoagulation therapy for this syndrome is unclear.16
Recommended treatments for cancer-related SVC syndrome include chemotherapy and radiation to shrink the tumor that is causing the obstruction. Tissue diagnosis is often necessary to direct treatment decisions.1 However, percutaneous angioplasty and the use of intravenous stents are becoming increasingly common and are simple, safe, and effective in rapidly relieving SVC syndrome caused by malignant diseases.1 A bypass of the SVC may be indicated in some cases.1 Adjunctive therapies include diuretics, corticosteroids, thrombolytics, anticoagulation, and elevating the head of the patient’s bed.1
CASE CONTINUED
Our patient was started on heparin intravenously for 7 days and long-term oral anticoagulant therapy with warfarin (Coumadin) to continue as long as the catheter was in place, with a target international normalized ratio between 2 and 2.5. He required no other interventions, and his dialysis catheter remained functioning. He was monitored in the hospital for 2 weeks, during which his symptoms gradually improved, with noticeable resolution of his facial swelling.
He was discharged home to continue on an oral anticoagulant and was then followed to monitor for a reappearance of the symptoms (which would force the removal of the catheter), and to pursue possible percutaneous angioplasty, stenting, or surgical reconstruction of the SVC if needed.
A 60-year-old man presented with progressive swelling of his face and neck, which had begun 2 weeks earlier. He denied any headache, lightheadedness, blurry vision, syncope, or change in his cognitive or memory function. A review of symptoms was unremarkable.
The patient had hypertension and end-stage renal disease, for which he was receiving hemodialysis via a catheter tunneled into his right internal jugular vein. He had undergone multiple unsuccessful attempts to create an arteriovenous fistula over the previous 2 years.
Figure 1. Swollen face, congested conjunctivae, and multiple dilated tortuous veins on the chest and abdominal walls.On physical examination, his vital signs were normal. Swelling of the face and fullness in his neck with bilateral congested conjunctivae were noted. His jugular venous pressure was elevated at 10 cm. Multiple dilated tortuous veins were noticed on his upper chest and across his abdominal wall (Figure 1). The rest of the examination was unremarkable.
Doppler ultrasonography revealed chronic thrombosis and reverse flow in the right internal jugular vein and reverse flow in the right subclavian vein. These findings were consistent with central venous thrombosis and superior vena cava (SVC) syndrome.
Figure 2. Multiple collateral veins in the upper chest (black arrows, left panel), stenosis of the superior vena cava, and the intraluminal catheter in place (right panel).Computed tomography (CT) of the chest showed significant stenosis of the right innominate vein and SVC due to thrombosis. Numerous collateral veins were seen in the neck and across both shoulders (Figure 2).
Diagnosis: SVC syndrome secondary to intravascular thrombosis related to his central venous dialysis catheter.
SVC SYNDROME
The SVC is the major drainage vessel for venous blood from the head, neck, upper extremities, and upper thorax. Obstruction to its flow increases venous pressure, which results in interstitial edema and retrograde collateral flow.1
More than 80% of cases of SVC syndrome are caused by malignant lung tumors and lymphoma.
Nonmalignant causes include mediastinal fibrosis; vascular diseases (eg, aortic aneurysm, large-vessel vasculitis); infections such as histoplasmosis, tuberculosis, syphilis, and actinomycosis; benign mediastinal tumors such as teratoma, cystic hygroma, thymoma, and dermoid cyst; and thrombosis from central venous catheters, pacemaker leads, and guidewires.2–6 A recent report suggests that benign causes may now account for up to 40% of cases as a result of a rise in the use of indwelling central venous catheters and cardiac pacemakers during the past 2 decades, resulting in a higher incidence of SVC thrombosis.7
An obstructed SVC initiates collateral venous return to the heart from the upper half of the body through different pathways. The most important pathway is the azygos venous system, which includes the azygos vein. Occlusion of the SVC at the level of the azygos vein contributes to the appearance of collateral veins on the chest and abdominal walls, and venous blood flows via these collaterals into the inferior vena cava.1,8,9
Different presentations
The diagnosis of SVC syndrome is often made on clinical grounds alone, ie, the combination of the clinical presentation and, often, a thoracic malignancy or contributing factors such as a central catheter.1
With slowly progressive obstruction of the SVC, the most common presenting symptoms include swelling of the face, neck, and both arms. On the other hand, adequate collateral drainage may develop,1 and patients may have minimal symptoms.
However, a rapid onset of SVC syndrome in the absence of collateral circulation will cause a more dramatic and life-threatening presentation, often with neurologic and respiratory sequelae such as cerebral and laryngeal edema and respiratory embarrassment, which were not present in our patient’s case.1,10–15 These serious complications are rare and are considered an acute emergency. In these cases, special attention to airway, breathing, and circulation (the “ABCs”) is essential, and endovascular repairs and stenting or open surgical reconstruction and alternate approaches for renal replacement therapy should be considered.1,12,13,15
CT is diagnostic and provides accurate information about the location of the obstruction and about other critical surrounding structures such as the lungs, mediastinum, and adjacent blood vessels.1,7,10,11 Our patient’s CT scan confirmed a significant stenosis of the SVC due to thrombosis, with no compression coming from the lungs or mediastinal structures.
Thrombolytic therapy in acute cases
In cases of acute thrombosis (with symptom onset less than 2 days previously), thrombolytic therapy followed by anticoagulation is recommended and may both cause the symptoms to regress within several days and allow the central catheter to be kept in.16 However, thrombolytic therapy is less effective in chronic thrombosis (with onset of symptoms more than 10 days previously).16
Vascular or surgical intervention is often needed to treat SVC syndrome related to dialysis access.
Most experts recommend anticoagulation after thrombosis to prevent disease progression and recurrence, although the benefit of either short-term or long-term anticoagulation therapy for this syndrome is unclear.16
Recommended treatments for cancer-related SVC syndrome include chemotherapy and radiation to shrink the tumor that is causing the obstruction. Tissue diagnosis is often necessary to direct treatment decisions.1 However, percutaneous angioplasty and the use of intravenous stents are becoming increasingly common and are simple, safe, and effective in rapidly relieving SVC syndrome caused by malignant diseases.1 A bypass of the SVC may be indicated in some cases.1 Adjunctive therapies include diuretics, corticosteroids, thrombolytics, anticoagulation, and elevating the head of the patient’s bed.1
CASE CONTINUED
Our patient was started on heparin intravenously for 7 days and long-term oral anticoagulant therapy with warfarin (Coumadin) to continue as long as the catheter was in place, with a target international normalized ratio between 2 and 2.5. He required no other interventions, and his dialysis catheter remained functioning. He was monitored in the hospital for 2 weeks, during which his symptoms gradually improved, with noticeable resolution of his facial swelling.
He was discharged home to continue on an oral anticoagulant and was then followed to monitor for a reappearance of the symptoms (which would force the removal of the catheter), and to pursue possible percutaneous angioplasty, stenting, or surgical reconstruction of the SVC if needed.
References
Wilson LD, Detterbeck FC, Yahalom J. Clinical practice. Superior vena cava syndrome with malignant causes. N Engl J Med2007; 356:1862–1869.
Parish JM, Marschke RF, Dines DE, Lee RE. Etiologic considerations in superior vena cava syndrome. Mayo Clin Proc1981; 56:407–413.
Markman M. Diagnosis and management of superior vena cava syndrome. Cleve Clin J Med1999; 66:59–61.
Khanna S, Sniderman K, Simons M, Besley M, Uldall R. Superior vena cava stenosis associated with hemodialysis catheters. Am J Kidney Dis1993; 21:278–281.
Bertrand M, Presant CA, Klein L, Scott E. Iatrogenic superior vena cava syndrome. A new entity. Cancer1984; 54:376–378.
Rice TW, Rodriguez RM, Light RW. The superior vena cava syndrome: clinical characteristics and evolving etiology. Medicine (Baltimore)2006; 85:37–42.
Plekker D, Ellis T, Irusen EM, Bolliger CT, Diacon AH. Clinical and radiological grading of superior vena cava obstruction. Respiration2008; 76:69–75.
Sheth S, Ebert MD, Fishman EK. Superior vena cava obstruction evaluation with MDCT. AJR Am J Roentgenol2010; 194:W336–W346.
DeMichele A, Glick J. Cancer-related emergencies. In:Lenhard R, Osteen R, Gansler T, eds. Clinical Oncology. Atlanta, GA: American Cancer Society; 2001:733–764.
Chen JC, Bongard F, Klein SR. A contemporary perspective on superior vena cava syndrome. Am J Surg1990; 160:207–211.
Sheikh MA, Fernandez BB, Gray BH, Graham LM, Carman TL. Endovascular stenting of nonmalignant superior vena cava syndrome. Catheter Cardiovasc Interv2005; 65:405–411.
Flinterman LE, Van Der Meer FJ, Rosendaal FR, Doggen CJ. Current perspective of venous thrombosis in the upper extremity. J Thromb Haemost2008; 6:1262–1266.
Greenberg S, Kosinski R, Daniels J. Treatment of superior vena cava thrombosis with recombinant tissue type plasminogen activator. Chest1991; 99:1298–1301.
Molhem A, Sabry A, Bawadekji H, Al Saran K. Superior vena cava syndrome in hemodialysis patient. Saudi J Kidney Dis Transpl2011; 22:381–386.
Akoglu H, Yilmaz R, Peynircioglu B, et al. A rare complication of hemodialysis catheters: superior vena cava syndrome. Hemodial Int2007; 11:385–391.
References
Wilson LD, Detterbeck FC, Yahalom J. Clinical practice. Superior vena cava syndrome with malignant causes. N Engl J Med2007; 356:1862–1869.
Parish JM, Marschke RF, Dines DE, Lee RE. Etiologic considerations in superior vena cava syndrome. Mayo Clin Proc1981; 56:407–413.
Markman M. Diagnosis and management of superior vena cava syndrome. Cleve Clin J Med1999; 66:59–61.
Khanna S, Sniderman K, Simons M, Besley M, Uldall R. Superior vena cava stenosis associated with hemodialysis catheters. Am J Kidney Dis1993; 21:278–281.
Bertrand M, Presant CA, Klein L, Scott E. Iatrogenic superior vena cava syndrome. A new entity. Cancer1984; 54:376–378.
Rice TW, Rodriguez RM, Light RW. The superior vena cava syndrome: clinical characteristics and evolving etiology. Medicine (Baltimore)2006; 85:37–42.
Plekker D, Ellis T, Irusen EM, Bolliger CT, Diacon AH. Clinical and radiological grading of superior vena cava obstruction. Respiration2008; 76:69–75.
Sheth S, Ebert MD, Fishman EK. Superior vena cava obstruction evaluation with MDCT. AJR Am J Roentgenol2010; 194:W336–W346.
DeMichele A, Glick J. Cancer-related emergencies. In:Lenhard R, Osteen R, Gansler T, eds. Clinical Oncology. Atlanta, GA: American Cancer Society; 2001:733–764.
Chen JC, Bongard F, Klein SR. A contemporary perspective on superior vena cava syndrome. Am J Surg1990; 160:207–211.
Sheikh MA, Fernandez BB, Gray BH, Graham LM, Carman TL. Endovascular stenting of nonmalignant superior vena cava syndrome. Catheter Cardiovasc Interv2005; 65:405–411.
Flinterman LE, Van Der Meer FJ, Rosendaal FR, Doggen CJ. Current perspective of venous thrombosis in the upper extremity. J Thromb Haemost2008; 6:1262–1266.
Greenberg S, Kosinski R, Daniels J. Treatment of superior vena cava thrombosis with recombinant tissue type plasminogen activator. Chest1991; 99:1298–1301.
Molhem A, Sabry A, Bawadekji H, Al Saran K. Superior vena cava syndrome in hemodialysis patient. Saudi J Kidney Dis Transpl2011; 22:381–386.
Akoglu H, Yilmaz R, Peynircioglu B, et al. A rare complication of hemodialysis catheters: superior vena cava syndrome. Hemodial Int2007; 11:385–391.
A 75-year-old man with type 2 diabetes and hypothyroidism underwent bilateral total knee replacement at our hospital.
His functional capacity had been moderately limited by knee pain, but he could easily climb one flight of stairs without symptoms. His medications at that time included levothyroxine (Synthroid) and metformin (Glucophage). He had no known cardiac or pulmonary disease. The preoperative evaluation, including laboratory tests and electrocardiography, was within normal limits.
Spinal anesthesia was used for surgery, and he was given 2 mg of midazolam (Versed) intravenously for sedation. No additional sedation was given. He was given oxygen via nasal cannula at 2 L/min.
All vital signs were stable at the start of the procedure. However, about halfway through, when the thigh tourniquet was released, his oxygen saturation dropped abruptly from 100% to 92%. All other vital signs remained stable, and he was asymptomatic, was oriented to person, time, and place, was conversing freely, and was in no distress. The oxygen flow was increased to 6 L/min, his oxygen saturation improved, and the procedure was then completed as planned.
At the conclusion of the surgery, before the patient was transported to the postanesthesia care unit (PACU) and while his oxygen flow rate was still 6 L/min, his oxygen saturation again dropped to 92%. A simple face mask was placed, and the oxygen flow rate was increased to 10 L/min. His oxygen saturation stayed low, near 90%.
Bleeding during surgery had been nominal. He had received 2 L of lactated Ringer’s solution and 500 mL of hetastarch (Hextend) during surgery. He continued to be asymptomatic in the PACU.
1.What is the most likely cause of oxygen desaturation during bilateral total knee arthroplasty?
Fat embolism
Intraoperative pneumonia
Venous thromboembolism with pulmonary embolism
Acute myocardial infarction
Acute pulmonary edema
Excessive sedation
The differential diagnosis of oxygen desaturation during orthopedic procedures is listed in Table 1.
Fat embolism is the most likely cause, particularly given the greater fatty embolic load that occurs with bilateral total knee arthroplasty than with unilateral total knee arthroplasty.
At what point the maximal showering of fat emboli occurs is not known. Fat may be released into the circulation with pressurization of the medullary canal during surgery or with manipulation of a fracture. The emboli may collect in the leg veins and then be released in a shower when the thigh tourniquet is released. Vasoactive mediators and methylmethacrylate cement released into the circulatory system after tourniquet deflation may also cause vasodilation, hypotension, and increased dead-space ventilation, resulting in hypoxia and a drop in end-tidal CO2.
Pneumonia during surgery is rare without an apparent aspiration event.
Venous thromboembolism is possible but is more likely later in the postoperative period after major orthopedic surgery.
Acute myocardial infarction could present with hypoxia, particularly in a diabetic patient, who may not experience chest pain. However, intraoperative electrocardiographic changes would likely be seen. If myocardial infarction is suspected, postoperative serial electrocardiograms and measuring troponin and cardiac enzyme levels aid in the diagnosis.
Acute pulmonary edema is possible but not as highly suspected, as the patient had no history of congestive heart failure and received an appropriate amount of fluid for this type of surgery.
Excessive sedation could cause hypoventilation and, thus, oxygen desaturatation. However, this patient’s oxygen desaturatation began more than an hour after the midazolam was given. Midazolam is a short-acting benzodiazepine. It is unlikely that the patient would show signs of hypoventilation and oversedation an hour after the drug was given. Our patient also did not show any signs of excessive sedation, as he was awake and conversing during the surgery.
Fat emboli vs fat embolism syndrome
Fat embolism is the presence of fat drops within the systemic and pulmonary microcirculation, with or without clinical sequelae.1 Fat embolism syndrome, on the other hand, is defined as injury to and dysfunction of one or more organs as a result of the embolization of fat, usually within 24 hours of injury or orthopedic surgery.2
Fat embolism syndrome is an unpredictable condition with a varied presentation. Fat droplets are thought to embolize via the venous circulation into the pulmonary arteries, occluding small blood vessels in the lung. However, they also get into the arterial circulation and occlude arteries in the brain, kidney, heart, and liver (more on this phenomenon below).
Fat embolism is reported to originate primarily from fractures of the femur, tibia, and pelvis.2,3 As many as 90% of trauma patients have been shown to have evidence of fat embolism on autopsy.4 However, only a small number of patients develop the classic fat embolism syndrome,2,3,5 Why some develop the syndrome and others do not is still unknown.
Orthopedic procedures associated with fat embolization include knee arthroplasty and hip arthroplasty, particularly if it involves intramedullary manipulation or medullary fixation.6 It has also been reported during spinal procedures in which pedicular screws are used.7 The syndrome occurs in 0.25% to 30% of patients following multiple fractures and in 0.1% to 12% of patients during or following knee or hip arthroplasty.
One study8 showed evidence of fat on transesophageal echocardiography in 88% of patients undergoing medullary reaming of lower-extremity fractures and hip hemiarthroplasty. Blood sampling from the right atrium confirmed that fat was responsible for the echocardiographic abnormalities. The study also showed that the severity of the embolic showering correlated with the severity of hypoxia and the decrease in end-tidal CO2.8
CASE CONTINUED
On arrival at the PACU, our patient’s oxygen saturation was 94% while he was breathing oxygen via a simple face mask at a flow rate of 10 L/min. His heart rate was 60 bpm, blood pressure 110/60 mm Hg, and temperature 37.5°C (96.3°F). Chest sounds were normal on auscultation.
However, 3 hours later, his mental status rapidly deteriorated. He was oriented only to person, and he was drowsy. He had escalating respiratory distress with a rapid respiratory rate and decreasing oxygen saturation. At this point, auscultation of his chest wall revealed bilateral crackles and rales.
He was promptly intubated. Profuse fluid and secretions were noted to be coming from his lungs, filling the endotracheal tube. Arterial blood gas measurement showed a pH of 7.22, Pao2 64 mm Hg, and Paco2 56 mm Hg on 100% fraction of inspired oxygen, with no increased anion gap.
2.Which consequence of fat embolism is most likely at this time in this patient?
Coexisting sepsis
Fat embolism syndrome
Acute cardioembolic stroke
Anaphylaxis
Fat embolism syndrome should be highly suspected in this patient. As mentioned, it can affect many different organs. It is the most serious condition resulting from fat embolization after surgery or trauma.
Sepsis was unlikely in our patient, since he presented for his surgery in good health and with no preexisting signs or symptoms of infection. Acute cardioembolic stroke could have caused the neurologic signs, but this would not necessarily explain the coexisting hypoxia. An anaphylactic reaction to drugs or surgical cement would most likely present intraoperatively, shortly after exposure occurred, rather than several hours after surgery.
How common is fat embolism syndrome?
The occurrence rate of fat embolism syndrome has been reported to be 0.25% to 30% after multiple fractures and 0.1% to 12% after knee and hip joint surgery, with a mortality rate of 13% to 36%.2,9–14 The rate of occurrence after unilateral total knee joint replacement has been reported to be 1.8% to 5%, and 4% to 12% after bilateral total knee replacement.15–19
The syndrome is relatively more common with traumatic fractures of the lower extremities. However, it has also been reported with liposuction, total parenteral nutrition, bone marrow harvest and transplantation, burns, and acute pancreatitis, to mention a few.10
The broad range of reported incidence rates can be attributed to the fact that many studies were in patients with multiple trauma, whose concomitant injuries may have made it difficult to clearly define the contribution of fat embolism syndrome to the overall rates of morbidity and mortality. Also, different studies used different criteria to define the syndrome.
How does fat embolism syndrome occur?
Two hypotheses for how this syndrome occurs were proposed nearly a century ago.20,21
The “mechanical” theory is that fat emboli are formed as a result of trauma and disruption of adipose tissue and other cells in the bone marrow. Increases in intramedullary pressure force the fat emboli through damaged medullary venous channels in the bone and into the circulation of the lower extremities. This embolization of fat causes an initial mechanical pulmonary obstruction. Mechanical obstruction by fat emboli in the pulmonary system leads to increased pulmonary pressures and an increase in right heart outflow pressure. The right heart becomes strained, leading to a decreased right-sided cardiac output. As a result, the left heart filling pressures diminish and hypotension ensues.20
The “biochemical” theory, on the other hand, is that chylomicrons within the vascular system are modified and their stability is compromised as a result of stress. These traumatized chylomicrons then coalesce to form droplets of fat that accumulate in the pulmonary circulation and produce a mechanical obstruction. This would explain why nontraumatic, nonorthopedic insults can produce this syndrome.
Autopsy studies show that there is little correlation between the presence and quantity of intravascular fat and the severity of clinical symptoms, thus implying that the syndrome is caused by more than just mechanical obstruction. The biochemical theory postulates that fat globules within the circulatory system then cause the release of lipase from the pulmonary alveolar cells, which then hydrolyses the fat into free fatty acids. These free fatty acids cause an inflammatory reaction, complementmediated leukocyte aggregation, chemotoxin release, and subsequently endothelial damage. These vasoactive substances damage type 2 pneumocytes and lead to an increased permeability of the pulmonary capillary beds. Acute respiratory distress syndrome (ARDS) may ensue. Disseminated intravascular coagulation may occur as a result of the formation of microthrombi involving lipids, platelets, and fibrin.21,22
Embolization of fat to the central nervous system can occur as fat globules cross into the systemic circulation via a patent foramen ovale, an atrioventricular shunt, or the pulmonary capillaries. This can then result in cerebral ischemia.23
Although patent foramen ovale may seem the most direct route for cerebral embolization, the neurologic impairment and signs of cerebral emboli in fat embolism syndrome may occur in the absence of patent foramen ovale.24,25 The fat globules may actually go through the lung capillaries, being flexible and forced through by increased pulmonary pressure.
But whether the cause of fat embolism syndrome is occlusion by globules, the release of biochemical mediators, or a combination of both is unknown. Both mechanisms are likely responsible. We can only suspect that the degree of fat load and intrinsic metabolic differences between individuals account for the variation in susceptibility.
FAT EMBOLI AFFECT THE LUNGS, SKIN, AND BRAIN
3.Where on the body is the rash associated with fat embolism syndrome usually seen?
Face
Near a site of fracture or surgery
Chest, axilla, conjunctiva
Distal extremities
Petechiae are part of the classic presenting triad of fat embolism syndrome, which also includes pulmonary and cerebral dysfunction.
Petechiae usually appear on the 2nd to 4th day after injury.26 They are usually found across the chest, the anterior axillary folds, and the neck, as well as on the oral mucosa and the conjunctiva. The rash is caused by occlusion of dermal capillaries by fat, which increases their fragility.10
Pulmonary changes usually begin with tachypnea, dyspnea, and a drop in oxygen saturation, leading to generalized hypoxia. Respiratory symptoms are present in 100% of cases.2 Respiratory symptoms can acutely develop with the sudden manipulation of a fracture, reaming of bone, or release of a limb tourniquet.27
Cerebral dysfunction can be variable, from anxiety and confusion to seizures and coma. The neurologic signs are typically diffuse; however, focal symptoms such as hemiplegia or aphasia can occasionally occur. Neurologic signs are present in 80% of cases.2,28
Body systems affected by fat embolism syndrome are summarized in Table 2.
4.How many hours after injury does fat embolism syndrome typically manifest?
1 to 2 hours
6 to 12 hours
12 to 20 hours
24 to 48 hours
72 to 84 hours
Most patients develop signs and symptoms 24 to 48 hours after injury. Patients presenting earlier than 12 hours usually have a more fulminant course.29
The time between fat embolization and the development of fat embolism syndrome is thought to be related to the time required for the metabolic conversion of fat to free fatty acids.30 We suspect that the early desaturation seen in our patient was the result of a heavy showering of fat intraoperatively. However, this could only be concluded after we had ruled out other causes of acute hypoxia and hypotension.
Fat embolism syndrome is a diagnosis of exclusion and is based on clinical criteria. No specific sign, symptom, or test is pathognomonic. It may often be confused with other conditions such as systemic inflammatory response syndrome or sepsis. However, the triad of respiratory and neurologic symptoms and petechiae coupled with the clinical picture of recent trauma or orthopedic surgery almost assures the diagnosis.
Fat embolism syndrome can range from subclinical to fulminating, with the more fulminating course attributable to a huge load of fat emboli, which leads to acute cor pulmonale.
The diagnostic criteria established by Gurd and Wilson13 are widely accepted and include major, minor, and laboratory criteria (Table 3). According to their criteria, the diagnosis of fat embolism syndrome requires the presence of one major feature plus four minor features plus fat macroglobulinemia. Major signs appear in 60% of patients within 24 hours and in 85% of patients within 48 hours.13
Variations on these diagnostic guidelines require two major criteria, one major and three minor criteria, two major and two minor criteria, and one major and two minor criteria.31 Other authors, perceiving these criteria to be insensitive, have focused on other factors, including hypoxemia by arterial blood gas monitoring.12,32 Lindeque at al12 thus included arterial blood gas analysis in their criteria (Table 4). However, their criteria have been criticized for focusing only on the pulmonary system, and many of these features may be present in patients with ARDS with a cause other than fat embolization, such as burns, septicemia, aspiration, and multiple transfusions.
Schonfeld et al32 created a fat embolism index to diagnose fat embolism syndrome; a score greater than 5 indicates that the syndrome is likely (Table 5).32
Regardless of the criteria used, one must have a high index of suspicion for fat embolization syndrome in patients undergoing orthopedic procedures, particularly hip and knee surgery, and in patients with fractures, especially fractures of the femur, tibia, or pelvis and multiple, concomitant fractures.
CASE CONTINUED
Our patient was given furosemide (Lasix) empirically for diuresis and to improve oxygenation. However, his oxygen saturation remained low.
Chest radiography 4 hours after surgery showed bilateral pulmonary infiltrates. Serial electrocardiography showed no acute changes. Levels of cardiac enzymes and troponins were normal. Transthoracic echocardiography showed no left ventricular dysfunction, a normal right ventricle, and no evidence of valvular lesions. Urine and blood fat stains were negative, but the sputum stain was positive for copious extracellular fat. The patient became comatose 5 hours postoperatively. Computed tomography of the brain was normal. He was transferred to the surgical intensive care unit.
The clinical course was marked by hemodynamic instability requiring norepinephrine (Levophed) and vasopressin (Pitressin) for hypotension. Right ventricular filling pressures via central venous pressure monitoring showed no evidence of hypovolemia. The hemoglobin concentration and the hematocrit were stable, with no evidence of acute or ongoing bleeding. Blood, urine, and sputum cultures remained negative. Acute myocardial infarction was ruled out by serial electrocardiography, cardiac enzyme testing, and troponin testing.
Figure 1. Magnetic resonance imaging on postoperative day 2 showed multiple hyperintense areas, consistent with emboli.
Magnetic resonance imaging (MRI) of the brain on postoperative day 2 showed foci of acute ischemia suggestive of embolic phenomena consistent with fat embolism syndrome(Figure 1). Transthoracic echocardiography was repeated but again showed no evidence of a patent foramen ovale. Electroencephalography on postoperative day 4 showed severe, diffuse encephalopathy. There was no petechial skin rash. Other laboratory studies showed progressive thrombocytopenia with a platelet count of 53 × 199/L on postoperative day 3.
TESTS THAT AID THE CLINICAL DIAGNOSIS
Although no single laboratory test is pathognomonic for fat embolism syndrome, several tests may help raise suspicion of it, especially in the setting of fracture or an orthopedic surgical procedure.
Arterial blood gases must be measured. A Pao2 of less than 60 mm Hg with no other obvious lung pathology in an orthopedic surgery patient is highly suspicious.12 An alveolar-arterial gradient of greater than 100 mm Hg may further increase suspicion.
Tests for fat. The blood and urine may be examined for fat, although positive findings are not specific for fat embolism syndrome.33 Fat in the urine indicates the occurrence of massive fat embolism, but this is not always accompanied by the syndrome.34 Gurd and Wilson13 found fat globules larger than 8 μm circulating in the serum in all documented cases. They stated that, even though the relationship of large fat globules to the pathogenesis of the clinical picture remains obscure, the demonstration of their presence can be helpful in the diagnosis.13
Also, samples obtained with bronchoalveolar lavage may be examined for fat. The macrophages may be stained for fat using the oil red O stain. Again, this is a nonspecific marker, as fat-stained macrophages are seen in trauma patients,35 but the finding has a very high negative predictive value.36 Anemia, thrombocytopenia, hypofibrinogenemia, an elevated lipase level, and a high erythrocyte sedimentation rate may be found in fat embolism syndrome.13
Chest radiography may show bilateral infiltrates, as in ARDS, but this is not diagnostic for fat embolism syndrome.
Electrocardiography may show changes in ST and T waves and signs of right heart strain.
Transesophageal echocardiography may show increased right heart and pulmonary artery pressures.
Computed tomography is often negative,37,38 but T2-weighted MRI is useful in the diagnosis of cerebral fat embolism syndrome, as it can show intracerebral microinfarcts as early as 4 hours after the onset of neurologic symptoms, and these findings correlate well with the clinical severity of brain injury.
Diffusion-weighted MRI may enhance the sensitivity and specificity of the neuroradiologic diagnosis. Diffusion-weighted MRI typically shows multiple nonconfluent areas of high-intensity signals or bright spots on a dark background, known as a “starfield pattern.” This pattern has been suggested to be pathognomonic of acute cerebral microinfarction. The abnormalities presumably reflect foci of cytotoxic edema that develops immediately, unlike vasogenic edema, seen in T2-weighted images, which may take up to several days to develop. Although these images are not necessarily specific for fat emboli, they are useful in helping make the diagnosis. Thus, diffusionweighted MRI should be done if fat embolism syndrome is suspected.38,39
CASE CONCLUDED
The patient’s course in the intensive care unit was further complicated by gastrointestinal bleeding and renal failure. His neurologic status did not improve. Repeated MRI of the brain showed evolving bilateral watershed infarction throughout the cortices. The neurologic consult service diagnosed the patient as having severe encephalopathy with a very poor prognosis. The decision was made to withdraw care. He was placed under palliative care and died on postoperative day 22.
DRUG TREATMENT OF FAT EMBOLISM SYNDROME
5.Which of the following drugs has been proven to be effective in treating fat embolism syndrome?
Intravenous ethanol
Steroids
Heparin
Dextran
Aspirin
None of the above
None of the above has been proven to be effective in treating this disorder. The management is largely supportive. Thus, prevention, early diagnosis, and symptom management are vital.
Pulmonary and hemodynamic support are the cornerstones of successful treatment. Aggressive respiratory support is often needed. Management of acute lung injury and ARDS focuses on achieving acceptable gas exchange while preventing ventilator-associated lung injury. Intravascular volume must be supported. Inotropes and pulmonary vasodilators may be required to maintain hemodynamics. Exacerbation of central nervous system ischemia from hypotension or hypoxia should be avoided.
If the thrombocytopenia leads to clinical bleeding, platelet transfusions may be warranted.
Supportive care should include prophylaxis of deep venous thrombosis and of gastrointestinal bleeding, and maintenance of nutrition.40 Patients who receive supportive care generally have a favorable outcome, with a mortality rate of less than 10%.28
Drug studies have been inconclusive
Drugs suggested in the treatment of fat embolism syndrome include heparin, aspirin, dextran, hypertonic glucose, and alcohol, but the results have been inconclusive.3,11,23,40–43
Heparin stimulates lipase activity, consequently decreasing the concentration of circulating fat globules. However, the increase in levels of free fatty acids may actually worsen the clinical picture. For this reason, and because of anticoagulation concerns and evidence of increased mortality rates, heparin is now contraindicated in the treatment of fat embolism syndrome.2,41,43
Alcohol. Patients with a higher blood alcohol level at the time of injury have been reported to have a lower incidence of fat embolism syndrome. Alcohol inhibits lipase, suppressing the rise of free fatty acids. In experimental studies, the incidence of fat embolism syndrome was lower when the blood alcohol level was maintained at 20 mg/dL. However, no prospective randomized trial has been done to determine the clinical efficacy of ethanol as a treatment for this condition.5,42
Dextran has been advocated, owing to its ability to improve small-vessel perfusion, but bleeding risk and acute renal failure associated with this drug have limited its use.5
N-acetylcysteine has been shown to attenuate fat-induced lung injury in a study of rats with induced fat embolism syndrome.44
Corticosteroid treatment for this condition is controversial. Studies in patients with femoral and tibial fractures show that steroids reduce the incidence of fat embolism syndrome when given prophylactically, and those treated with steroids had a higher Pao2 than controls. Doses of methylprednisolone in these studies ranged between 9 mg/kg to 90 mg/kg. A drawback of these studies is their small number of patients.12,32,45,46
A meta-analysis47 of randomized trials of corticosteroids to prevent fat embolism syndrome in patients with long-bone fractures identified 104 such studies. Only 7 of the 104 were considered adequate. In 389 patients with long-bone fractures, prophylactic corticosteroids reduced the risk of fat embolism syndrome by 78% (95% confidence interval 43%–92%) and corticosteroids also significantly reduced the risk of hypoxia with no difference in rates of infection or death. However, the overall quality of the trials was poor, and the authors of the meta-analysis concluded that more study is needed before corticosteroids could be formally recommended.47
There is no evidence that steroids improve the overall clinical course of already established fat embolism syndrome.12,32,45 The dosing and optimal timing of administration have also not been established. High doses pose a risk of septic complications, which may be devastating for the posttrauma or postoperative patient.
References
Akhtar S. Fat embolism. Anesthesiol Clin2009; 27:533–550.
Filomeno LT, Carelli CR, Silva NC, Filho TE, Amatuzzi MM. Fat embolism: a review for current orthopaedics practice. Acta Ortop Bras2005; 13:196–208.
ten Duis HJ. The fat embolism syndrome. Injury1997; 28:77–85.
Peltier LF. Fat embolism. A current concept. Clin Orthop Relat Res1969; 66:241–253.
Gossling HR, Pellegrini VD. Fat embolism syndrome: a review of the pathophysiology and physiological basis of treatment. Clin Orthop Relat Res1982; 165:68–82.
Papagelopoulos PJ, Apostolou CD, Karachalios TS, Themistocleous GS, Giannakopoulos CK, Ioannidis TT. Pulmonary fat embolism after total hip and total knee arthroplasty. Orthopedics2003; 26:523–527.
Takahashi S, Kitagawa H, Ishii T. Intraoperative pulmonary embolism during spinal instrumentation surgery. A prospective study using transoesophageal echocardiography. J Bone Joint Surg Br2003; 85:90–94.
Christie J, Robinson CM, Pell AC, McBirnie J, Burnett R. Transcardiac echocardiography during invasive intramedullary procedures. J Bone Joint Surg Br1995; 77:450–455.
Robert JH, Hoffmeyer P, Broquet PE, Cerutti P, Vasey H. Fat embolism syndrome. Orthop Rev1993; 22:567–571.
Mellor A, Soni N. Fat embolism. Anaesthesia2001; 56:145–154.
Taviloglu K, Yanar H. Fat embolism syndrome. Surg Today2007; 37:5–8.
Lindeque BG, Schoeman HS, Dommisse GF, Boeyens MC, Vlok AL. Fat embolism and the fat embolism syndrome. A double-blind therapeutic study. J Bone Joint Surg Br1987; 69:128–131.
Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br1974; 56B:408–416.
Ganong RB. Fat emboli syndrome in isolated fractures of the tibia and femur. Clin Orthop Relat Res1993; 291:208–214.
Djelouah I, Lefèvre G, Ozier Y, Rosencher N, Tallet F. Fat embolism in orthopedic surgery: role of bone marrow fatty acid. Anesth Analg1997; 85:441–443.
Barre J, Lepouse C, Segal P. Embolism and intramedullary femoral surgery. Rev Chir Orthop Reparatrice Appar Mot1997; 83:9–21.
Kim YH. Incidence of fat embolism syndrome after cemented or cementless bilateral simultaneous and unilateral total knee arthroplasty. J Arthroplasty2001; 16:730–739.
Dorr LD, Merkel C, Mellman MF, Klein I. Fat emboli in bilateral total knee arthroplasty. Predictive factors for neurologic manifestations. Clin Orthop Relat Res1989; 248:112–118.
Jankiewicz JJ, Sculco TP, Ranawat CS, Behr C, Tarrentino S. Onestage versus 2-stage bilateral total knee arthroplasty. Clin Orthop Relat Res1994; 309:94–101.
Gauss H. The pathology of fat embolism. Arch Surg1924; 9:593–605.
Lehman EP, Moore RM. Fat embolism, including experimental production without trauma. Arch Surg1927; 14:621–662.
Johnson MJ, Lucas GL. Fat embolism syndrome. Orthopedics1996; 19:41–48.
Benson KT. Diagnosis and treatment of fat embolism syndrome. Anesthesiology Rev1993; 20:165–170.
Colonna DM, Kilgus D, Brown W, Challa V, Stump DA, Moody DM. Acute brain fat embolization occurring after total hip arthroplasty in the absence of a patent foramen ovale. Anesthesiology2002; 96:1027–1029.
Byrick RJ, Mullen JB, Mazer CD, Guest CB. Transpulmonary systemic fat embolism. Studies in mongrel dogs after cemented arthroplasty. Am J Respir Crit Care Med1994; 150:1416–1422.
Benestad G. Drei Fälle von Fettembolie mit punktförmigen Blutungen in der Haut. Deutsche Ztschr f Chir1911; 112:192.
Fulde GW, Harrison P. Fat embolism—a review. Arch Emerg Med1991; 8:233–239.
Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg1997; 132:435–439.
King EG, Wagner WW, Ashbaugh DG, Latham LP, Halsey DR. Alterations in pulmonary microanatomy after fat embolism. In vivo observations via thoracic window of the oleic acid-embolized canine lung. Chest1971; 59:524–530.
Schonfeld SA, Ploysongsang Y, DiLisio R, et al. Fat embolism prophylaxis with corticosteroids. A prospective study in high-risk patients. Ann Intern Med1983; 99:438–443.
Capan LM, Miller SM, Patel KP. Fat embolism. Anesthesiol Clin North Am1993; 11:25–54.
Reider E, Sherman Y, Weiss Y, Liebergall M, Pizov R. Alveolar macrophages fat stain in early diagnosis of fat embolism syndrome. Isr J Med Sci1997; 33:654–658.
Aoki N, Soma K, Shindo M, Kurosawa T, Ohwada T. Evaluation of potential fat emboli during placement of intramedullary nails after orthopedic fractures. Chest1998; 113:178–181.
Stoeger A, Daniaux M, Felber S, Stockhammer G, Aichner F, zur Nedden D. MRI findings in cerebral fat embolism. Eur Radiol1998; 8:1590–1593.
Takahashi M, Suzuki R, Osakabe Y, et al. Magnetic resonance imaging findings in cerebral fat embolism: correlation with clinical manifestations. J Trauma1999; 46:324–327.
Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke2001; 32:2942–2944.
Sage RH, Tudor RW. Treatment of fat embolism with heparin. Br Med J1958; 1:1160–1161.
Myers R, Taljaard JJ. Blood alcohol and fat embolism syndrome. J Bone Joint Surg Am1977; 59:878–880.
Denman EE, Cairnes CS, Holmes CM. Case of severe fat embolism treated by intermittent positive-pressure respiration. Br Med J1964; 2:101–102.
Liu DD, Kao SJ, Chen HI. N-Acetylcysteine attenuates acute lung injury induced by fat embolism. Crit Care Med2008; 36:565–571.
Kallenbach J, Lewis M, Zaltzman M, Feldman C, Orford A, Zwi S. ‘Low-dose’ corticosteroid prophylaxis against fat embolism. J Trauma1987; 27:1173–1176.
Stoltenberg JJ, Gustilo RB. The use of methylprednisolone and hypertonic glucose in the prophylaxis of fat embolism syndrome. Clin Orthop Relat Res1979; 143:211–221.
Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg2009; 52:386–393.
Uusula A. Galway, MD Department of General Anesthesiology, Cleveland Clinic, and Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
David Gugliotti, MD, FACP, SFHM Department of Hospital Medicine, Cleveland Clinic
Address: Ursula A. Galway, MD, Department of General Anesthesiology, E31, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail galwayu@ccf.org
Uusula A. Galway, MD Department of General Anesthesiology, Cleveland Clinic, and Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
David Gugliotti, MD, FACP, SFHM Department of Hospital Medicine, Cleveland Clinic
Address: Ursula A. Galway, MD, Department of General Anesthesiology, E31, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail galwayu@ccf.org
Author and Disclosure Information
Uusula A. Galway, MD Department of General Anesthesiology, Cleveland Clinic, and Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
David Gugliotti, MD, FACP, SFHM Department of Hospital Medicine, Cleveland Clinic
Address: Ursula A. Galway, MD, Department of General Anesthesiology, E31, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail galwayu@ccf.org
A 75-year-old man with type 2 diabetes and hypothyroidism underwent bilateral total knee replacement at our hospital.
His functional capacity had been moderately limited by knee pain, but he could easily climb one flight of stairs without symptoms. His medications at that time included levothyroxine (Synthroid) and metformin (Glucophage). He had no known cardiac or pulmonary disease. The preoperative evaluation, including laboratory tests and electrocardiography, was within normal limits.
Spinal anesthesia was used for surgery, and he was given 2 mg of midazolam (Versed) intravenously for sedation. No additional sedation was given. He was given oxygen via nasal cannula at 2 L/min.
All vital signs were stable at the start of the procedure. However, about halfway through, when the thigh tourniquet was released, his oxygen saturation dropped abruptly from 100% to 92%. All other vital signs remained stable, and he was asymptomatic, was oriented to person, time, and place, was conversing freely, and was in no distress. The oxygen flow was increased to 6 L/min, his oxygen saturation improved, and the procedure was then completed as planned.
At the conclusion of the surgery, before the patient was transported to the postanesthesia care unit (PACU) and while his oxygen flow rate was still 6 L/min, his oxygen saturation again dropped to 92%. A simple face mask was placed, and the oxygen flow rate was increased to 10 L/min. His oxygen saturation stayed low, near 90%.
Bleeding during surgery had been nominal. He had received 2 L of lactated Ringer’s solution and 500 mL of hetastarch (Hextend) during surgery. He continued to be asymptomatic in the PACU.
1.What is the most likely cause of oxygen desaturation during bilateral total knee arthroplasty?
Fat embolism
Intraoperative pneumonia
Venous thromboembolism with pulmonary embolism
Acute myocardial infarction
Acute pulmonary edema
Excessive sedation
The differential diagnosis of oxygen desaturation during orthopedic procedures is listed in Table 1.
Fat embolism is the most likely cause, particularly given the greater fatty embolic load that occurs with bilateral total knee arthroplasty than with unilateral total knee arthroplasty.
At what point the maximal showering of fat emboli occurs is not known. Fat may be released into the circulation with pressurization of the medullary canal during surgery or with manipulation of a fracture. The emboli may collect in the leg veins and then be released in a shower when the thigh tourniquet is released. Vasoactive mediators and methylmethacrylate cement released into the circulatory system after tourniquet deflation may also cause vasodilation, hypotension, and increased dead-space ventilation, resulting in hypoxia and a drop in end-tidal CO2.
Pneumonia during surgery is rare without an apparent aspiration event.
Venous thromboembolism is possible but is more likely later in the postoperative period after major orthopedic surgery.
Acute myocardial infarction could present with hypoxia, particularly in a diabetic patient, who may not experience chest pain. However, intraoperative electrocardiographic changes would likely be seen. If myocardial infarction is suspected, postoperative serial electrocardiograms and measuring troponin and cardiac enzyme levels aid in the diagnosis.
Acute pulmonary edema is possible but not as highly suspected, as the patient had no history of congestive heart failure and received an appropriate amount of fluid for this type of surgery.
Excessive sedation could cause hypoventilation and, thus, oxygen desaturatation. However, this patient’s oxygen desaturatation began more than an hour after the midazolam was given. Midazolam is a short-acting benzodiazepine. It is unlikely that the patient would show signs of hypoventilation and oversedation an hour after the drug was given. Our patient also did not show any signs of excessive sedation, as he was awake and conversing during the surgery.
Fat emboli vs fat embolism syndrome
Fat embolism is the presence of fat drops within the systemic and pulmonary microcirculation, with or without clinical sequelae.1 Fat embolism syndrome, on the other hand, is defined as injury to and dysfunction of one or more organs as a result of the embolization of fat, usually within 24 hours of injury or orthopedic surgery.2
Fat embolism syndrome is an unpredictable condition with a varied presentation. Fat droplets are thought to embolize via the venous circulation into the pulmonary arteries, occluding small blood vessels in the lung. However, they also get into the arterial circulation and occlude arteries in the brain, kidney, heart, and liver (more on this phenomenon below).
Fat embolism is reported to originate primarily from fractures of the femur, tibia, and pelvis.2,3 As many as 90% of trauma patients have been shown to have evidence of fat embolism on autopsy.4 However, only a small number of patients develop the classic fat embolism syndrome,2,3,5 Why some develop the syndrome and others do not is still unknown.
Orthopedic procedures associated with fat embolization include knee arthroplasty and hip arthroplasty, particularly if it involves intramedullary manipulation or medullary fixation.6 It has also been reported during spinal procedures in which pedicular screws are used.7 The syndrome occurs in 0.25% to 30% of patients following multiple fractures and in 0.1% to 12% of patients during or following knee or hip arthroplasty.
One study8 showed evidence of fat on transesophageal echocardiography in 88% of patients undergoing medullary reaming of lower-extremity fractures and hip hemiarthroplasty. Blood sampling from the right atrium confirmed that fat was responsible for the echocardiographic abnormalities. The study also showed that the severity of the embolic showering correlated with the severity of hypoxia and the decrease in end-tidal CO2.8
CASE CONTINUED
On arrival at the PACU, our patient’s oxygen saturation was 94% while he was breathing oxygen via a simple face mask at a flow rate of 10 L/min. His heart rate was 60 bpm, blood pressure 110/60 mm Hg, and temperature 37.5°C (96.3°F). Chest sounds were normal on auscultation.
However, 3 hours later, his mental status rapidly deteriorated. He was oriented only to person, and he was drowsy. He had escalating respiratory distress with a rapid respiratory rate and decreasing oxygen saturation. At this point, auscultation of his chest wall revealed bilateral crackles and rales.
He was promptly intubated. Profuse fluid and secretions were noted to be coming from his lungs, filling the endotracheal tube. Arterial blood gas measurement showed a pH of 7.22, Pao2 64 mm Hg, and Paco2 56 mm Hg on 100% fraction of inspired oxygen, with no increased anion gap.
2.Which consequence of fat embolism is most likely at this time in this patient?
Coexisting sepsis
Fat embolism syndrome
Acute cardioembolic stroke
Anaphylaxis
Fat embolism syndrome should be highly suspected in this patient. As mentioned, it can affect many different organs. It is the most serious condition resulting from fat embolization after surgery or trauma.
Sepsis was unlikely in our patient, since he presented for his surgery in good health and with no preexisting signs or symptoms of infection. Acute cardioembolic stroke could have caused the neurologic signs, but this would not necessarily explain the coexisting hypoxia. An anaphylactic reaction to drugs or surgical cement would most likely present intraoperatively, shortly after exposure occurred, rather than several hours after surgery.
How common is fat embolism syndrome?
The occurrence rate of fat embolism syndrome has been reported to be 0.25% to 30% after multiple fractures and 0.1% to 12% after knee and hip joint surgery, with a mortality rate of 13% to 36%.2,9–14 The rate of occurrence after unilateral total knee joint replacement has been reported to be 1.8% to 5%, and 4% to 12% after bilateral total knee replacement.15–19
The syndrome is relatively more common with traumatic fractures of the lower extremities. However, it has also been reported with liposuction, total parenteral nutrition, bone marrow harvest and transplantation, burns, and acute pancreatitis, to mention a few.10
The broad range of reported incidence rates can be attributed to the fact that many studies were in patients with multiple trauma, whose concomitant injuries may have made it difficult to clearly define the contribution of fat embolism syndrome to the overall rates of morbidity and mortality. Also, different studies used different criteria to define the syndrome.
How does fat embolism syndrome occur?
Two hypotheses for how this syndrome occurs were proposed nearly a century ago.20,21
The “mechanical” theory is that fat emboli are formed as a result of trauma and disruption of adipose tissue and other cells in the bone marrow. Increases in intramedullary pressure force the fat emboli through damaged medullary venous channels in the bone and into the circulation of the lower extremities. This embolization of fat causes an initial mechanical pulmonary obstruction. Mechanical obstruction by fat emboli in the pulmonary system leads to increased pulmonary pressures and an increase in right heart outflow pressure. The right heart becomes strained, leading to a decreased right-sided cardiac output. As a result, the left heart filling pressures diminish and hypotension ensues.20
The “biochemical” theory, on the other hand, is that chylomicrons within the vascular system are modified and their stability is compromised as a result of stress. These traumatized chylomicrons then coalesce to form droplets of fat that accumulate in the pulmonary circulation and produce a mechanical obstruction. This would explain why nontraumatic, nonorthopedic insults can produce this syndrome.
Autopsy studies show that there is little correlation between the presence and quantity of intravascular fat and the severity of clinical symptoms, thus implying that the syndrome is caused by more than just mechanical obstruction. The biochemical theory postulates that fat globules within the circulatory system then cause the release of lipase from the pulmonary alveolar cells, which then hydrolyses the fat into free fatty acids. These free fatty acids cause an inflammatory reaction, complementmediated leukocyte aggregation, chemotoxin release, and subsequently endothelial damage. These vasoactive substances damage type 2 pneumocytes and lead to an increased permeability of the pulmonary capillary beds. Acute respiratory distress syndrome (ARDS) may ensue. Disseminated intravascular coagulation may occur as a result of the formation of microthrombi involving lipids, platelets, and fibrin.21,22
Embolization of fat to the central nervous system can occur as fat globules cross into the systemic circulation via a patent foramen ovale, an atrioventricular shunt, or the pulmonary capillaries. This can then result in cerebral ischemia.23
Although patent foramen ovale may seem the most direct route for cerebral embolization, the neurologic impairment and signs of cerebral emboli in fat embolism syndrome may occur in the absence of patent foramen ovale.24,25 The fat globules may actually go through the lung capillaries, being flexible and forced through by increased pulmonary pressure.
But whether the cause of fat embolism syndrome is occlusion by globules, the release of biochemical mediators, or a combination of both is unknown. Both mechanisms are likely responsible. We can only suspect that the degree of fat load and intrinsic metabolic differences between individuals account for the variation in susceptibility.
FAT EMBOLI AFFECT THE LUNGS, SKIN, AND BRAIN
3.Where on the body is the rash associated with fat embolism syndrome usually seen?
Face
Near a site of fracture or surgery
Chest, axilla, conjunctiva
Distal extremities
Petechiae are part of the classic presenting triad of fat embolism syndrome, which also includes pulmonary and cerebral dysfunction.
Petechiae usually appear on the 2nd to 4th day after injury.26 They are usually found across the chest, the anterior axillary folds, and the neck, as well as on the oral mucosa and the conjunctiva. The rash is caused by occlusion of dermal capillaries by fat, which increases their fragility.10
Pulmonary changes usually begin with tachypnea, dyspnea, and a drop in oxygen saturation, leading to generalized hypoxia. Respiratory symptoms are present in 100% of cases.2 Respiratory symptoms can acutely develop with the sudden manipulation of a fracture, reaming of bone, or release of a limb tourniquet.27
Cerebral dysfunction can be variable, from anxiety and confusion to seizures and coma. The neurologic signs are typically diffuse; however, focal symptoms such as hemiplegia or aphasia can occasionally occur. Neurologic signs are present in 80% of cases.2,28
Body systems affected by fat embolism syndrome are summarized in Table 2.
4.How many hours after injury does fat embolism syndrome typically manifest?
1 to 2 hours
6 to 12 hours
12 to 20 hours
24 to 48 hours
72 to 84 hours
Most patients develop signs and symptoms 24 to 48 hours after injury. Patients presenting earlier than 12 hours usually have a more fulminant course.29
The time between fat embolization and the development of fat embolism syndrome is thought to be related to the time required for the metabolic conversion of fat to free fatty acids.30 We suspect that the early desaturation seen in our patient was the result of a heavy showering of fat intraoperatively. However, this could only be concluded after we had ruled out other causes of acute hypoxia and hypotension.
Fat embolism syndrome is a diagnosis of exclusion and is based on clinical criteria. No specific sign, symptom, or test is pathognomonic. It may often be confused with other conditions such as systemic inflammatory response syndrome or sepsis. However, the triad of respiratory and neurologic symptoms and petechiae coupled with the clinical picture of recent trauma or orthopedic surgery almost assures the diagnosis.
Fat embolism syndrome can range from subclinical to fulminating, with the more fulminating course attributable to a huge load of fat emboli, which leads to acute cor pulmonale.
The diagnostic criteria established by Gurd and Wilson13 are widely accepted and include major, minor, and laboratory criteria (Table 3). According to their criteria, the diagnosis of fat embolism syndrome requires the presence of one major feature plus four minor features plus fat macroglobulinemia. Major signs appear in 60% of patients within 24 hours and in 85% of patients within 48 hours.13
Variations on these diagnostic guidelines require two major criteria, one major and three minor criteria, two major and two minor criteria, and one major and two minor criteria.31 Other authors, perceiving these criteria to be insensitive, have focused on other factors, including hypoxemia by arterial blood gas monitoring.12,32 Lindeque at al12 thus included arterial blood gas analysis in their criteria (Table 4). However, their criteria have been criticized for focusing only on the pulmonary system, and many of these features may be present in patients with ARDS with a cause other than fat embolization, such as burns, septicemia, aspiration, and multiple transfusions.
Schonfeld et al32 created a fat embolism index to diagnose fat embolism syndrome; a score greater than 5 indicates that the syndrome is likely (Table 5).32
Regardless of the criteria used, one must have a high index of suspicion for fat embolization syndrome in patients undergoing orthopedic procedures, particularly hip and knee surgery, and in patients with fractures, especially fractures of the femur, tibia, or pelvis and multiple, concomitant fractures.
CASE CONTINUED
Our patient was given furosemide (Lasix) empirically for diuresis and to improve oxygenation. However, his oxygen saturation remained low.
Chest radiography 4 hours after surgery showed bilateral pulmonary infiltrates. Serial electrocardiography showed no acute changes. Levels of cardiac enzymes and troponins were normal. Transthoracic echocardiography showed no left ventricular dysfunction, a normal right ventricle, and no evidence of valvular lesions. Urine and blood fat stains were negative, but the sputum stain was positive for copious extracellular fat. The patient became comatose 5 hours postoperatively. Computed tomography of the brain was normal. He was transferred to the surgical intensive care unit.
The clinical course was marked by hemodynamic instability requiring norepinephrine (Levophed) and vasopressin (Pitressin) for hypotension. Right ventricular filling pressures via central venous pressure monitoring showed no evidence of hypovolemia. The hemoglobin concentration and the hematocrit were stable, with no evidence of acute or ongoing bleeding. Blood, urine, and sputum cultures remained negative. Acute myocardial infarction was ruled out by serial electrocardiography, cardiac enzyme testing, and troponin testing.
Figure 1. Magnetic resonance imaging on postoperative day 2 showed multiple hyperintense areas, consistent with emboli.
Magnetic resonance imaging (MRI) of the brain on postoperative day 2 showed foci of acute ischemia suggestive of embolic phenomena consistent with fat embolism syndrome(Figure 1). Transthoracic echocardiography was repeated but again showed no evidence of a patent foramen ovale. Electroencephalography on postoperative day 4 showed severe, diffuse encephalopathy. There was no petechial skin rash. Other laboratory studies showed progressive thrombocytopenia with a platelet count of 53 × 199/L on postoperative day 3.
TESTS THAT AID THE CLINICAL DIAGNOSIS
Although no single laboratory test is pathognomonic for fat embolism syndrome, several tests may help raise suspicion of it, especially in the setting of fracture or an orthopedic surgical procedure.
Arterial blood gases must be measured. A Pao2 of less than 60 mm Hg with no other obvious lung pathology in an orthopedic surgery patient is highly suspicious.12 An alveolar-arterial gradient of greater than 100 mm Hg may further increase suspicion.
Tests for fat. The blood and urine may be examined for fat, although positive findings are not specific for fat embolism syndrome.33 Fat in the urine indicates the occurrence of massive fat embolism, but this is not always accompanied by the syndrome.34 Gurd and Wilson13 found fat globules larger than 8 μm circulating in the serum in all documented cases. They stated that, even though the relationship of large fat globules to the pathogenesis of the clinical picture remains obscure, the demonstration of their presence can be helpful in the diagnosis.13
Also, samples obtained with bronchoalveolar lavage may be examined for fat. The macrophages may be stained for fat using the oil red O stain. Again, this is a nonspecific marker, as fat-stained macrophages are seen in trauma patients,35 but the finding has a very high negative predictive value.36 Anemia, thrombocytopenia, hypofibrinogenemia, an elevated lipase level, and a high erythrocyte sedimentation rate may be found in fat embolism syndrome.13
Chest radiography may show bilateral infiltrates, as in ARDS, but this is not diagnostic for fat embolism syndrome.
Electrocardiography may show changes in ST and T waves and signs of right heart strain.
Transesophageal echocardiography may show increased right heart and pulmonary artery pressures.
Computed tomography is often negative,37,38 but T2-weighted MRI is useful in the diagnosis of cerebral fat embolism syndrome, as it can show intracerebral microinfarcts as early as 4 hours after the onset of neurologic symptoms, and these findings correlate well with the clinical severity of brain injury.
Diffusion-weighted MRI may enhance the sensitivity and specificity of the neuroradiologic diagnosis. Diffusion-weighted MRI typically shows multiple nonconfluent areas of high-intensity signals or bright spots on a dark background, known as a “starfield pattern.” This pattern has been suggested to be pathognomonic of acute cerebral microinfarction. The abnormalities presumably reflect foci of cytotoxic edema that develops immediately, unlike vasogenic edema, seen in T2-weighted images, which may take up to several days to develop. Although these images are not necessarily specific for fat emboli, they are useful in helping make the diagnosis. Thus, diffusionweighted MRI should be done if fat embolism syndrome is suspected.38,39
CASE CONCLUDED
The patient’s course in the intensive care unit was further complicated by gastrointestinal bleeding and renal failure. His neurologic status did not improve. Repeated MRI of the brain showed evolving bilateral watershed infarction throughout the cortices. The neurologic consult service diagnosed the patient as having severe encephalopathy with a very poor prognosis. The decision was made to withdraw care. He was placed under palliative care and died on postoperative day 22.
DRUG TREATMENT OF FAT EMBOLISM SYNDROME
5.Which of the following drugs has been proven to be effective in treating fat embolism syndrome?
Intravenous ethanol
Steroids
Heparin
Dextran
Aspirin
None of the above
None of the above has been proven to be effective in treating this disorder. The management is largely supportive. Thus, prevention, early diagnosis, and symptom management are vital.
Pulmonary and hemodynamic support are the cornerstones of successful treatment. Aggressive respiratory support is often needed. Management of acute lung injury and ARDS focuses on achieving acceptable gas exchange while preventing ventilator-associated lung injury. Intravascular volume must be supported. Inotropes and pulmonary vasodilators may be required to maintain hemodynamics. Exacerbation of central nervous system ischemia from hypotension or hypoxia should be avoided.
If the thrombocytopenia leads to clinical bleeding, platelet transfusions may be warranted.
Supportive care should include prophylaxis of deep venous thrombosis and of gastrointestinal bleeding, and maintenance of nutrition.40 Patients who receive supportive care generally have a favorable outcome, with a mortality rate of less than 10%.28
Drug studies have been inconclusive
Drugs suggested in the treatment of fat embolism syndrome include heparin, aspirin, dextran, hypertonic glucose, and alcohol, but the results have been inconclusive.3,11,23,40–43
Heparin stimulates lipase activity, consequently decreasing the concentration of circulating fat globules. However, the increase in levels of free fatty acids may actually worsen the clinical picture. For this reason, and because of anticoagulation concerns and evidence of increased mortality rates, heparin is now contraindicated in the treatment of fat embolism syndrome.2,41,43
Alcohol. Patients with a higher blood alcohol level at the time of injury have been reported to have a lower incidence of fat embolism syndrome. Alcohol inhibits lipase, suppressing the rise of free fatty acids. In experimental studies, the incidence of fat embolism syndrome was lower when the blood alcohol level was maintained at 20 mg/dL. However, no prospective randomized trial has been done to determine the clinical efficacy of ethanol as a treatment for this condition.5,42
Dextran has been advocated, owing to its ability to improve small-vessel perfusion, but bleeding risk and acute renal failure associated with this drug have limited its use.5
N-acetylcysteine has been shown to attenuate fat-induced lung injury in a study of rats with induced fat embolism syndrome.44
Corticosteroid treatment for this condition is controversial. Studies in patients with femoral and tibial fractures show that steroids reduce the incidence of fat embolism syndrome when given prophylactically, and those treated with steroids had a higher Pao2 than controls. Doses of methylprednisolone in these studies ranged between 9 mg/kg to 90 mg/kg. A drawback of these studies is their small number of patients.12,32,45,46
A meta-analysis47 of randomized trials of corticosteroids to prevent fat embolism syndrome in patients with long-bone fractures identified 104 such studies. Only 7 of the 104 were considered adequate. In 389 patients with long-bone fractures, prophylactic corticosteroids reduced the risk of fat embolism syndrome by 78% (95% confidence interval 43%–92%) and corticosteroids also significantly reduced the risk of hypoxia with no difference in rates of infection or death. However, the overall quality of the trials was poor, and the authors of the meta-analysis concluded that more study is needed before corticosteroids could be formally recommended.47
There is no evidence that steroids improve the overall clinical course of already established fat embolism syndrome.12,32,45 The dosing and optimal timing of administration have also not been established. High doses pose a risk of septic complications, which may be devastating for the posttrauma or postoperative patient.
A 75-year-old man with type 2 diabetes and hypothyroidism underwent bilateral total knee replacement at our hospital.
His functional capacity had been moderately limited by knee pain, but he could easily climb one flight of stairs without symptoms. His medications at that time included levothyroxine (Synthroid) and metformin (Glucophage). He had no known cardiac or pulmonary disease. The preoperative evaluation, including laboratory tests and electrocardiography, was within normal limits.
Spinal anesthesia was used for surgery, and he was given 2 mg of midazolam (Versed) intravenously for sedation. No additional sedation was given. He was given oxygen via nasal cannula at 2 L/min.
All vital signs were stable at the start of the procedure. However, about halfway through, when the thigh tourniquet was released, his oxygen saturation dropped abruptly from 100% to 92%. All other vital signs remained stable, and he was asymptomatic, was oriented to person, time, and place, was conversing freely, and was in no distress. The oxygen flow was increased to 6 L/min, his oxygen saturation improved, and the procedure was then completed as planned.
At the conclusion of the surgery, before the patient was transported to the postanesthesia care unit (PACU) and while his oxygen flow rate was still 6 L/min, his oxygen saturation again dropped to 92%. A simple face mask was placed, and the oxygen flow rate was increased to 10 L/min. His oxygen saturation stayed low, near 90%.
Bleeding during surgery had been nominal. He had received 2 L of lactated Ringer’s solution and 500 mL of hetastarch (Hextend) during surgery. He continued to be asymptomatic in the PACU.
1.What is the most likely cause of oxygen desaturation during bilateral total knee arthroplasty?
Fat embolism
Intraoperative pneumonia
Venous thromboembolism with pulmonary embolism
Acute myocardial infarction
Acute pulmonary edema
Excessive sedation
The differential diagnosis of oxygen desaturation during orthopedic procedures is listed in Table 1.
Fat embolism is the most likely cause, particularly given the greater fatty embolic load that occurs with bilateral total knee arthroplasty than with unilateral total knee arthroplasty.
At what point the maximal showering of fat emboli occurs is not known. Fat may be released into the circulation with pressurization of the medullary canal during surgery or with manipulation of a fracture. The emboli may collect in the leg veins and then be released in a shower when the thigh tourniquet is released. Vasoactive mediators and methylmethacrylate cement released into the circulatory system after tourniquet deflation may also cause vasodilation, hypotension, and increased dead-space ventilation, resulting in hypoxia and a drop in end-tidal CO2.
Pneumonia during surgery is rare without an apparent aspiration event.
Venous thromboembolism is possible but is more likely later in the postoperative period after major orthopedic surgery.
Acute myocardial infarction could present with hypoxia, particularly in a diabetic patient, who may not experience chest pain. However, intraoperative electrocardiographic changes would likely be seen. If myocardial infarction is suspected, postoperative serial electrocardiograms and measuring troponin and cardiac enzyme levels aid in the diagnosis.
Acute pulmonary edema is possible but not as highly suspected, as the patient had no history of congestive heart failure and received an appropriate amount of fluid for this type of surgery.
Excessive sedation could cause hypoventilation and, thus, oxygen desaturatation. However, this patient’s oxygen desaturatation began more than an hour after the midazolam was given. Midazolam is a short-acting benzodiazepine. It is unlikely that the patient would show signs of hypoventilation and oversedation an hour after the drug was given. Our patient also did not show any signs of excessive sedation, as he was awake and conversing during the surgery.
Fat emboli vs fat embolism syndrome
Fat embolism is the presence of fat drops within the systemic and pulmonary microcirculation, with or without clinical sequelae.1 Fat embolism syndrome, on the other hand, is defined as injury to and dysfunction of one or more organs as a result of the embolization of fat, usually within 24 hours of injury or orthopedic surgery.2
Fat embolism syndrome is an unpredictable condition with a varied presentation. Fat droplets are thought to embolize via the venous circulation into the pulmonary arteries, occluding small blood vessels in the lung. However, they also get into the arterial circulation and occlude arteries in the brain, kidney, heart, and liver (more on this phenomenon below).
Fat embolism is reported to originate primarily from fractures of the femur, tibia, and pelvis.2,3 As many as 90% of trauma patients have been shown to have evidence of fat embolism on autopsy.4 However, only a small number of patients develop the classic fat embolism syndrome,2,3,5 Why some develop the syndrome and others do not is still unknown.
Orthopedic procedures associated with fat embolization include knee arthroplasty and hip arthroplasty, particularly if it involves intramedullary manipulation or medullary fixation.6 It has also been reported during spinal procedures in which pedicular screws are used.7 The syndrome occurs in 0.25% to 30% of patients following multiple fractures and in 0.1% to 12% of patients during or following knee or hip arthroplasty.
One study8 showed evidence of fat on transesophageal echocardiography in 88% of patients undergoing medullary reaming of lower-extremity fractures and hip hemiarthroplasty. Blood sampling from the right atrium confirmed that fat was responsible for the echocardiographic abnormalities. The study also showed that the severity of the embolic showering correlated with the severity of hypoxia and the decrease in end-tidal CO2.8
CASE CONTINUED
On arrival at the PACU, our patient’s oxygen saturation was 94% while he was breathing oxygen via a simple face mask at a flow rate of 10 L/min. His heart rate was 60 bpm, blood pressure 110/60 mm Hg, and temperature 37.5°C (96.3°F). Chest sounds were normal on auscultation.
However, 3 hours later, his mental status rapidly deteriorated. He was oriented only to person, and he was drowsy. He had escalating respiratory distress with a rapid respiratory rate and decreasing oxygen saturation. At this point, auscultation of his chest wall revealed bilateral crackles and rales.
He was promptly intubated. Profuse fluid and secretions were noted to be coming from his lungs, filling the endotracheal tube. Arterial blood gas measurement showed a pH of 7.22, Pao2 64 mm Hg, and Paco2 56 mm Hg on 100% fraction of inspired oxygen, with no increased anion gap.
2.Which consequence of fat embolism is most likely at this time in this patient?
Coexisting sepsis
Fat embolism syndrome
Acute cardioembolic stroke
Anaphylaxis
Fat embolism syndrome should be highly suspected in this patient. As mentioned, it can affect many different organs. It is the most serious condition resulting from fat embolization after surgery or trauma.
Sepsis was unlikely in our patient, since he presented for his surgery in good health and with no preexisting signs or symptoms of infection. Acute cardioembolic stroke could have caused the neurologic signs, but this would not necessarily explain the coexisting hypoxia. An anaphylactic reaction to drugs or surgical cement would most likely present intraoperatively, shortly after exposure occurred, rather than several hours after surgery.
How common is fat embolism syndrome?
The occurrence rate of fat embolism syndrome has been reported to be 0.25% to 30% after multiple fractures and 0.1% to 12% after knee and hip joint surgery, with a mortality rate of 13% to 36%.2,9–14 The rate of occurrence after unilateral total knee joint replacement has been reported to be 1.8% to 5%, and 4% to 12% after bilateral total knee replacement.15–19
The syndrome is relatively more common with traumatic fractures of the lower extremities. However, it has also been reported with liposuction, total parenteral nutrition, bone marrow harvest and transplantation, burns, and acute pancreatitis, to mention a few.10
The broad range of reported incidence rates can be attributed to the fact that many studies were in patients with multiple trauma, whose concomitant injuries may have made it difficult to clearly define the contribution of fat embolism syndrome to the overall rates of morbidity and mortality. Also, different studies used different criteria to define the syndrome.
How does fat embolism syndrome occur?
Two hypotheses for how this syndrome occurs were proposed nearly a century ago.20,21
The “mechanical” theory is that fat emboli are formed as a result of trauma and disruption of adipose tissue and other cells in the bone marrow. Increases in intramedullary pressure force the fat emboli through damaged medullary venous channels in the bone and into the circulation of the lower extremities. This embolization of fat causes an initial mechanical pulmonary obstruction. Mechanical obstruction by fat emboli in the pulmonary system leads to increased pulmonary pressures and an increase in right heart outflow pressure. The right heart becomes strained, leading to a decreased right-sided cardiac output. As a result, the left heart filling pressures diminish and hypotension ensues.20
The “biochemical” theory, on the other hand, is that chylomicrons within the vascular system are modified and their stability is compromised as a result of stress. These traumatized chylomicrons then coalesce to form droplets of fat that accumulate in the pulmonary circulation and produce a mechanical obstruction. This would explain why nontraumatic, nonorthopedic insults can produce this syndrome.
Autopsy studies show that there is little correlation between the presence and quantity of intravascular fat and the severity of clinical symptoms, thus implying that the syndrome is caused by more than just mechanical obstruction. The biochemical theory postulates that fat globules within the circulatory system then cause the release of lipase from the pulmonary alveolar cells, which then hydrolyses the fat into free fatty acids. These free fatty acids cause an inflammatory reaction, complementmediated leukocyte aggregation, chemotoxin release, and subsequently endothelial damage. These vasoactive substances damage type 2 pneumocytes and lead to an increased permeability of the pulmonary capillary beds. Acute respiratory distress syndrome (ARDS) may ensue. Disseminated intravascular coagulation may occur as a result of the formation of microthrombi involving lipids, platelets, and fibrin.21,22
Embolization of fat to the central nervous system can occur as fat globules cross into the systemic circulation via a patent foramen ovale, an atrioventricular shunt, or the pulmonary capillaries. This can then result in cerebral ischemia.23
Although patent foramen ovale may seem the most direct route for cerebral embolization, the neurologic impairment and signs of cerebral emboli in fat embolism syndrome may occur in the absence of patent foramen ovale.24,25 The fat globules may actually go through the lung capillaries, being flexible and forced through by increased pulmonary pressure.
But whether the cause of fat embolism syndrome is occlusion by globules, the release of biochemical mediators, or a combination of both is unknown. Both mechanisms are likely responsible. We can only suspect that the degree of fat load and intrinsic metabolic differences between individuals account for the variation in susceptibility.
FAT EMBOLI AFFECT THE LUNGS, SKIN, AND BRAIN
3.Where on the body is the rash associated with fat embolism syndrome usually seen?
Face
Near a site of fracture or surgery
Chest, axilla, conjunctiva
Distal extremities
Petechiae are part of the classic presenting triad of fat embolism syndrome, which also includes pulmonary and cerebral dysfunction.
Petechiae usually appear on the 2nd to 4th day after injury.26 They are usually found across the chest, the anterior axillary folds, and the neck, as well as on the oral mucosa and the conjunctiva. The rash is caused by occlusion of dermal capillaries by fat, which increases their fragility.10
Pulmonary changes usually begin with tachypnea, dyspnea, and a drop in oxygen saturation, leading to generalized hypoxia. Respiratory symptoms are present in 100% of cases.2 Respiratory symptoms can acutely develop with the sudden manipulation of a fracture, reaming of bone, or release of a limb tourniquet.27
Cerebral dysfunction can be variable, from anxiety and confusion to seizures and coma. The neurologic signs are typically diffuse; however, focal symptoms such as hemiplegia or aphasia can occasionally occur. Neurologic signs are present in 80% of cases.2,28
Body systems affected by fat embolism syndrome are summarized in Table 2.
4.How many hours after injury does fat embolism syndrome typically manifest?
1 to 2 hours
6 to 12 hours
12 to 20 hours
24 to 48 hours
72 to 84 hours
Most patients develop signs and symptoms 24 to 48 hours after injury. Patients presenting earlier than 12 hours usually have a more fulminant course.29
The time between fat embolization and the development of fat embolism syndrome is thought to be related to the time required for the metabolic conversion of fat to free fatty acids.30 We suspect that the early desaturation seen in our patient was the result of a heavy showering of fat intraoperatively. However, this could only be concluded after we had ruled out other causes of acute hypoxia and hypotension.
Fat embolism syndrome is a diagnosis of exclusion and is based on clinical criteria. No specific sign, symptom, or test is pathognomonic. It may often be confused with other conditions such as systemic inflammatory response syndrome or sepsis. However, the triad of respiratory and neurologic symptoms and petechiae coupled with the clinical picture of recent trauma or orthopedic surgery almost assures the diagnosis.
Fat embolism syndrome can range from subclinical to fulminating, with the more fulminating course attributable to a huge load of fat emboli, which leads to acute cor pulmonale.
The diagnostic criteria established by Gurd and Wilson13 are widely accepted and include major, minor, and laboratory criteria (Table 3). According to their criteria, the diagnosis of fat embolism syndrome requires the presence of one major feature plus four minor features plus fat macroglobulinemia. Major signs appear in 60% of patients within 24 hours and in 85% of patients within 48 hours.13
Variations on these diagnostic guidelines require two major criteria, one major and three minor criteria, two major and two minor criteria, and one major and two minor criteria.31 Other authors, perceiving these criteria to be insensitive, have focused on other factors, including hypoxemia by arterial blood gas monitoring.12,32 Lindeque at al12 thus included arterial blood gas analysis in their criteria (Table 4). However, their criteria have been criticized for focusing only on the pulmonary system, and many of these features may be present in patients with ARDS with a cause other than fat embolization, such as burns, septicemia, aspiration, and multiple transfusions.
Schonfeld et al32 created a fat embolism index to diagnose fat embolism syndrome; a score greater than 5 indicates that the syndrome is likely (Table 5).32
Regardless of the criteria used, one must have a high index of suspicion for fat embolization syndrome in patients undergoing orthopedic procedures, particularly hip and knee surgery, and in patients with fractures, especially fractures of the femur, tibia, or pelvis and multiple, concomitant fractures.
CASE CONTINUED
Our patient was given furosemide (Lasix) empirically for diuresis and to improve oxygenation. However, his oxygen saturation remained low.
Chest radiography 4 hours after surgery showed bilateral pulmonary infiltrates. Serial electrocardiography showed no acute changes. Levels of cardiac enzymes and troponins were normal. Transthoracic echocardiography showed no left ventricular dysfunction, a normal right ventricle, and no evidence of valvular lesions. Urine and blood fat stains were negative, but the sputum stain was positive for copious extracellular fat. The patient became comatose 5 hours postoperatively. Computed tomography of the brain was normal. He was transferred to the surgical intensive care unit.
The clinical course was marked by hemodynamic instability requiring norepinephrine (Levophed) and vasopressin (Pitressin) for hypotension. Right ventricular filling pressures via central venous pressure monitoring showed no evidence of hypovolemia. The hemoglobin concentration and the hematocrit were stable, with no evidence of acute or ongoing bleeding. Blood, urine, and sputum cultures remained negative. Acute myocardial infarction was ruled out by serial electrocardiography, cardiac enzyme testing, and troponin testing.
Figure 1. Magnetic resonance imaging on postoperative day 2 showed multiple hyperintense areas, consistent with emboli.
Magnetic resonance imaging (MRI) of the brain on postoperative day 2 showed foci of acute ischemia suggestive of embolic phenomena consistent with fat embolism syndrome(Figure 1). Transthoracic echocardiography was repeated but again showed no evidence of a patent foramen ovale. Electroencephalography on postoperative day 4 showed severe, diffuse encephalopathy. There was no petechial skin rash. Other laboratory studies showed progressive thrombocytopenia with a platelet count of 53 × 199/L on postoperative day 3.
TESTS THAT AID THE CLINICAL DIAGNOSIS
Although no single laboratory test is pathognomonic for fat embolism syndrome, several tests may help raise suspicion of it, especially in the setting of fracture or an orthopedic surgical procedure.
Arterial blood gases must be measured. A Pao2 of less than 60 mm Hg with no other obvious lung pathology in an orthopedic surgery patient is highly suspicious.12 An alveolar-arterial gradient of greater than 100 mm Hg may further increase suspicion.
Tests for fat. The blood and urine may be examined for fat, although positive findings are not specific for fat embolism syndrome.33 Fat in the urine indicates the occurrence of massive fat embolism, but this is not always accompanied by the syndrome.34 Gurd and Wilson13 found fat globules larger than 8 μm circulating in the serum in all documented cases. They stated that, even though the relationship of large fat globules to the pathogenesis of the clinical picture remains obscure, the demonstration of their presence can be helpful in the diagnosis.13
Also, samples obtained with bronchoalveolar lavage may be examined for fat. The macrophages may be stained for fat using the oil red O stain. Again, this is a nonspecific marker, as fat-stained macrophages are seen in trauma patients,35 but the finding has a very high negative predictive value.36 Anemia, thrombocytopenia, hypofibrinogenemia, an elevated lipase level, and a high erythrocyte sedimentation rate may be found in fat embolism syndrome.13
Chest radiography may show bilateral infiltrates, as in ARDS, but this is not diagnostic for fat embolism syndrome.
Electrocardiography may show changes in ST and T waves and signs of right heart strain.
Transesophageal echocardiography may show increased right heart and pulmonary artery pressures.
Computed tomography is often negative,37,38 but T2-weighted MRI is useful in the diagnosis of cerebral fat embolism syndrome, as it can show intracerebral microinfarcts as early as 4 hours after the onset of neurologic symptoms, and these findings correlate well with the clinical severity of brain injury.
Diffusion-weighted MRI may enhance the sensitivity and specificity of the neuroradiologic diagnosis. Diffusion-weighted MRI typically shows multiple nonconfluent areas of high-intensity signals or bright spots on a dark background, known as a “starfield pattern.” This pattern has been suggested to be pathognomonic of acute cerebral microinfarction. The abnormalities presumably reflect foci of cytotoxic edema that develops immediately, unlike vasogenic edema, seen in T2-weighted images, which may take up to several days to develop. Although these images are not necessarily specific for fat emboli, they are useful in helping make the diagnosis. Thus, diffusionweighted MRI should be done if fat embolism syndrome is suspected.38,39
CASE CONCLUDED
The patient’s course in the intensive care unit was further complicated by gastrointestinal bleeding and renal failure. His neurologic status did not improve. Repeated MRI of the brain showed evolving bilateral watershed infarction throughout the cortices. The neurologic consult service diagnosed the patient as having severe encephalopathy with a very poor prognosis. The decision was made to withdraw care. He was placed under palliative care and died on postoperative day 22.
DRUG TREATMENT OF FAT EMBOLISM SYNDROME
5.Which of the following drugs has been proven to be effective in treating fat embolism syndrome?
Intravenous ethanol
Steroids
Heparin
Dextran
Aspirin
None of the above
None of the above has been proven to be effective in treating this disorder. The management is largely supportive. Thus, prevention, early diagnosis, and symptom management are vital.
Pulmonary and hemodynamic support are the cornerstones of successful treatment. Aggressive respiratory support is often needed. Management of acute lung injury and ARDS focuses on achieving acceptable gas exchange while preventing ventilator-associated lung injury. Intravascular volume must be supported. Inotropes and pulmonary vasodilators may be required to maintain hemodynamics. Exacerbation of central nervous system ischemia from hypotension or hypoxia should be avoided.
If the thrombocytopenia leads to clinical bleeding, platelet transfusions may be warranted.
Supportive care should include prophylaxis of deep venous thrombosis and of gastrointestinal bleeding, and maintenance of nutrition.40 Patients who receive supportive care generally have a favorable outcome, with a mortality rate of less than 10%.28
Drug studies have been inconclusive
Drugs suggested in the treatment of fat embolism syndrome include heparin, aspirin, dextran, hypertonic glucose, and alcohol, but the results have been inconclusive.3,11,23,40–43
Heparin stimulates lipase activity, consequently decreasing the concentration of circulating fat globules. However, the increase in levels of free fatty acids may actually worsen the clinical picture. For this reason, and because of anticoagulation concerns and evidence of increased mortality rates, heparin is now contraindicated in the treatment of fat embolism syndrome.2,41,43
Alcohol. Patients with a higher blood alcohol level at the time of injury have been reported to have a lower incidence of fat embolism syndrome. Alcohol inhibits lipase, suppressing the rise of free fatty acids. In experimental studies, the incidence of fat embolism syndrome was lower when the blood alcohol level was maintained at 20 mg/dL. However, no prospective randomized trial has been done to determine the clinical efficacy of ethanol as a treatment for this condition.5,42
Dextran has been advocated, owing to its ability to improve small-vessel perfusion, but bleeding risk and acute renal failure associated with this drug have limited its use.5
N-acetylcysteine has been shown to attenuate fat-induced lung injury in a study of rats with induced fat embolism syndrome.44
Corticosteroid treatment for this condition is controversial. Studies in patients with femoral and tibial fractures show that steroids reduce the incidence of fat embolism syndrome when given prophylactically, and those treated with steroids had a higher Pao2 than controls. Doses of methylprednisolone in these studies ranged between 9 mg/kg to 90 mg/kg. A drawback of these studies is their small number of patients.12,32,45,46
A meta-analysis47 of randomized trials of corticosteroids to prevent fat embolism syndrome in patients with long-bone fractures identified 104 such studies. Only 7 of the 104 were considered adequate. In 389 patients with long-bone fractures, prophylactic corticosteroids reduced the risk of fat embolism syndrome by 78% (95% confidence interval 43%–92%) and corticosteroids also significantly reduced the risk of hypoxia with no difference in rates of infection or death. However, the overall quality of the trials was poor, and the authors of the meta-analysis concluded that more study is needed before corticosteroids could be formally recommended.47
There is no evidence that steroids improve the overall clinical course of already established fat embolism syndrome.12,32,45 The dosing and optimal timing of administration have also not been established. High doses pose a risk of septic complications, which may be devastating for the posttrauma or postoperative patient.
References
Akhtar S. Fat embolism. Anesthesiol Clin2009; 27:533–550.
Filomeno LT, Carelli CR, Silva NC, Filho TE, Amatuzzi MM. Fat embolism: a review for current orthopaedics practice. Acta Ortop Bras2005; 13:196–208.
ten Duis HJ. The fat embolism syndrome. Injury1997; 28:77–85.
Peltier LF. Fat embolism. A current concept. Clin Orthop Relat Res1969; 66:241–253.
Gossling HR, Pellegrini VD. Fat embolism syndrome: a review of the pathophysiology and physiological basis of treatment. Clin Orthop Relat Res1982; 165:68–82.
Papagelopoulos PJ, Apostolou CD, Karachalios TS, Themistocleous GS, Giannakopoulos CK, Ioannidis TT. Pulmonary fat embolism after total hip and total knee arthroplasty. Orthopedics2003; 26:523–527.
Takahashi S, Kitagawa H, Ishii T. Intraoperative pulmonary embolism during spinal instrumentation surgery. A prospective study using transoesophageal echocardiography. J Bone Joint Surg Br2003; 85:90–94.
Christie J, Robinson CM, Pell AC, McBirnie J, Burnett R. Transcardiac echocardiography during invasive intramedullary procedures. J Bone Joint Surg Br1995; 77:450–455.
Robert JH, Hoffmeyer P, Broquet PE, Cerutti P, Vasey H. Fat embolism syndrome. Orthop Rev1993; 22:567–571.
Mellor A, Soni N. Fat embolism. Anaesthesia2001; 56:145–154.
Taviloglu K, Yanar H. Fat embolism syndrome. Surg Today2007; 37:5–8.
Lindeque BG, Schoeman HS, Dommisse GF, Boeyens MC, Vlok AL. Fat embolism and the fat embolism syndrome. A double-blind therapeutic study. J Bone Joint Surg Br1987; 69:128–131.
Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br1974; 56B:408–416.
Ganong RB. Fat emboli syndrome in isolated fractures of the tibia and femur. Clin Orthop Relat Res1993; 291:208–214.
Djelouah I, Lefèvre G, Ozier Y, Rosencher N, Tallet F. Fat embolism in orthopedic surgery: role of bone marrow fatty acid. Anesth Analg1997; 85:441–443.
Barre J, Lepouse C, Segal P. Embolism and intramedullary femoral surgery. Rev Chir Orthop Reparatrice Appar Mot1997; 83:9–21.
Kim YH. Incidence of fat embolism syndrome after cemented or cementless bilateral simultaneous and unilateral total knee arthroplasty. J Arthroplasty2001; 16:730–739.
Dorr LD, Merkel C, Mellman MF, Klein I. Fat emboli in bilateral total knee arthroplasty. Predictive factors for neurologic manifestations. Clin Orthop Relat Res1989; 248:112–118.
Jankiewicz JJ, Sculco TP, Ranawat CS, Behr C, Tarrentino S. Onestage versus 2-stage bilateral total knee arthroplasty. Clin Orthop Relat Res1994; 309:94–101.
Gauss H. The pathology of fat embolism. Arch Surg1924; 9:593–605.
Lehman EP, Moore RM. Fat embolism, including experimental production without trauma. Arch Surg1927; 14:621–662.
Johnson MJ, Lucas GL. Fat embolism syndrome. Orthopedics1996; 19:41–48.
Benson KT. Diagnosis and treatment of fat embolism syndrome. Anesthesiology Rev1993; 20:165–170.
Colonna DM, Kilgus D, Brown W, Challa V, Stump DA, Moody DM. Acute brain fat embolization occurring after total hip arthroplasty in the absence of a patent foramen ovale. Anesthesiology2002; 96:1027–1029.
Byrick RJ, Mullen JB, Mazer CD, Guest CB. Transpulmonary systemic fat embolism. Studies in mongrel dogs after cemented arthroplasty. Am J Respir Crit Care Med1994; 150:1416–1422.
Benestad G. Drei Fälle von Fettembolie mit punktförmigen Blutungen in der Haut. Deutsche Ztschr f Chir1911; 112:192.
Fulde GW, Harrison P. Fat embolism—a review. Arch Emerg Med1991; 8:233–239.
Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg1997; 132:435–439.
King EG, Wagner WW, Ashbaugh DG, Latham LP, Halsey DR. Alterations in pulmonary microanatomy after fat embolism. In vivo observations via thoracic window of the oleic acid-embolized canine lung. Chest1971; 59:524–530.
Schonfeld SA, Ploysongsang Y, DiLisio R, et al. Fat embolism prophylaxis with corticosteroids. A prospective study in high-risk patients. Ann Intern Med1983; 99:438–443.
Capan LM, Miller SM, Patel KP. Fat embolism. Anesthesiol Clin North Am1993; 11:25–54.
Reider E, Sherman Y, Weiss Y, Liebergall M, Pizov R. Alveolar macrophages fat stain in early diagnosis of fat embolism syndrome. Isr J Med Sci1997; 33:654–658.
Aoki N, Soma K, Shindo M, Kurosawa T, Ohwada T. Evaluation of potential fat emboli during placement of intramedullary nails after orthopedic fractures. Chest1998; 113:178–181.
Stoeger A, Daniaux M, Felber S, Stockhammer G, Aichner F, zur Nedden D. MRI findings in cerebral fat embolism. Eur Radiol1998; 8:1590–1593.
Takahashi M, Suzuki R, Osakabe Y, et al. Magnetic resonance imaging findings in cerebral fat embolism: correlation with clinical manifestations. J Trauma1999; 46:324–327.
Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke2001; 32:2942–2944.
Sage RH, Tudor RW. Treatment of fat embolism with heparin. Br Med J1958; 1:1160–1161.
Myers R, Taljaard JJ. Blood alcohol and fat embolism syndrome. J Bone Joint Surg Am1977; 59:878–880.
Denman EE, Cairnes CS, Holmes CM. Case of severe fat embolism treated by intermittent positive-pressure respiration. Br Med J1964; 2:101–102.
Liu DD, Kao SJ, Chen HI. N-Acetylcysteine attenuates acute lung injury induced by fat embolism. Crit Care Med2008; 36:565–571.
Kallenbach J, Lewis M, Zaltzman M, Feldman C, Orford A, Zwi S. ‘Low-dose’ corticosteroid prophylaxis against fat embolism. J Trauma1987; 27:1173–1176.
Stoltenberg JJ, Gustilo RB. The use of methylprednisolone and hypertonic glucose in the prophylaxis of fat embolism syndrome. Clin Orthop Relat Res1979; 143:211–221.
Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg2009; 52:386–393.
References
Akhtar S. Fat embolism. Anesthesiol Clin2009; 27:533–550.
Filomeno LT, Carelli CR, Silva NC, Filho TE, Amatuzzi MM. Fat embolism: a review for current orthopaedics practice. Acta Ortop Bras2005; 13:196–208.
ten Duis HJ. The fat embolism syndrome. Injury1997; 28:77–85.
Peltier LF. Fat embolism. A current concept. Clin Orthop Relat Res1969; 66:241–253.
Gossling HR, Pellegrini VD. Fat embolism syndrome: a review of the pathophysiology and physiological basis of treatment. Clin Orthop Relat Res1982; 165:68–82.
Papagelopoulos PJ, Apostolou CD, Karachalios TS, Themistocleous GS, Giannakopoulos CK, Ioannidis TT. Pulmonary fat embolism after total hip and total knee arthroplasty. Orthopedics2003; 26:523–527.
Takahashi S, Kitagawa H, Ishii T. Intraoperative pulmonary embolism during spinal instrumentation surgery. A prospective study using transoesophageal echocardiography. J Bone Joint Surg Br2003; 85:90–94.
Christie J, Robinson CM, Pell AC, McBirnie J, Burnett R. Transcardiac echocardiography during invasive intramedullary procedures. J Bone Joint Surg Br1995; 77:450–455.
Robert JH, Hoffmeyer P, Broquet PE, Cerutti P, Vasey H. Fat embolism syndrome. Orthop Rev1993; 22:567–571.
Mellor A, Soni N. Fat embolism. Anaesthesia2001; 56:145–154.
Taviloglu K, Yanar H. Fat embolism syndrome. Surg Today2007; 37:5–8.
Lindeque BG, Schoeman HS, Dommisse GF, Boeyens MC, Vlok AL. Fat embolism and the fat embolism syndrome. A double-blind therapeutic study. J Bone Joint Surg Br1987; 69:128–131.
Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br1974; 56B:408–416.
Ganong RB. Fat emboli syndrome in isolated fractures of the tibia and femur. Clin Orthop Relat Res1993; 291:208–214.
Djelouah I, Lefèvre G, Ozier Y, Rosencher N, Tallet F. Fat embolism in orthopedic surgery: role of bone marrow fatty acid. Anesth Analg1997; 85:441–443.
Barre J, Lepouse C, Segal P. Embolism and intramedullary femoral surgery. Rev Chir Orthop Reparatrice Appar Mot1997; 83:9–21.
Kim YH. Incidence of fat embolism syndrome after cemented or cementless bilateral simultaneous and unilateral total knee arthroplasty. J Arthroplasty2001; 16:730–739.
Dorr LD, Merkel C, Mellman MF, Klein I. Fat emboli in bilateral total knee arthroplasty. Predictive factors for neurologic manifestations. Clin Orthop Relat Res1989; 248:112–118.
Jankiewicz JJ, Sculco TP, Ranawat CS, Behr C, Tarrentino S. Onestage versus 2-stage bilateral total knee arthroplasty. Clin Orthop Relat Res1994; 309:94–101.
Gauss H. The pathology of fat embolism. Arch Surg1924; 9:593–605.
Lehman EP, Moore RM. Fat embolism, including experimental production without trauma. Arch Surg1927; 14:621–662.
Johnson MJ, Lucas GL. Fat embolism syndrome. Orthopedics1996; 19:41–48.
Benson KT. Diagnosis and treatment of fat embolism syndrome. Anesthesiology Rev1993; 20:165–170.
Colonna DM, Kilgus D, Brown W, Challa V, Stump DA, Moody DM. Acute brain fat embolization occurring after total hip arthroplasty in the absence of a patent foramen ovale. Anesthesiology2002; 96:1027–1029.
Byrick RJ, Mullen JB, Mazer CD, Guest CB. Transpulmonary systemic fat embolism. Studies in mongrel dogs after cemented arthroplasty. Am J Respir Crit Care Med1994; 150:1416–1422.
Benestad G. Drei Fälle von Fettembolie mit punktförmigen Blutungen in der Haut. Deutsche Ztschr f Chir1911; 112:192.
Fulde GW, Harrison P. Fat embolism—a review. Arch Emerg Med1991; 8:233–239.
Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg1997; 132:435–439.
King EG, Wagner WW, Ashbaugh DG, Latham LP, Halsey DR. Alterations in pulmonary microanatomy after fat embolism. In vivo observations via thoracic window of the oleic acid-embolized canine lung. Chest1971; 59:524–530.
Schonfeld SA, Ploysongsang Y, DiLisio R, et al. Fat embolism prophylaxis with corticosteroids. A prospective study in high-risk patients. Ann Intern Med1983; 99:438–443.
Capan LM, Miller SM, Patel KP. Fat embolism. Anesthesiol Clin North Am1993; 11:25–54.
Reider E, Sherman Y, Weiss Y, Liebergall M, Pizov R. Alveolar macrophages fat stain in early diagnosis of fat embolism syndrome. Isr J Med Sci1997; 33:654–658.
Aoki N, Soma K, Shindo M, Kurosawa T, Ohwada T. Evaluation of potential fat emboli during placement of intramedullary nails after orthopedic fractures. Chest1998; 113:178–181.
Stoeger A, Daniaux M, Felber S, Stockhammer G, Aichner F, zur Nedden D. MRI findings in cerebral fat embolism. Eur Radiol1998; 8:1590–1593.
Takahashi M, Suzuki R, Osakabe Y, et al. Magnetic resonance imaging findings in cerebral fat embolism: correlation with clinical manifestations. J Trauma1999; 46:324–327.
Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke2001; 32:2942–2944.
Sage RH, Tudor RW. Treatment of fat embolism with heparin. Br Med J1958; 1:1160–1161.
Myers R, Taljaard JJ. Blood alcohol and fat embolism syndrome. J Bone Joint Surg Am1977; 59:878–880.
Denman EE, Cairnes CS, Holmes CM. Case of severe fat embolism treated by intermittent positive-pressure respiration. Br Med J1964; 2:101–102.
Liu DD, Kao SJ, Chen HI. N-Acetylcysteine attenuates acute lung injury induced by fat embolism. Crit Care Med2008; 36:565–571.
Kallenbach J, Lewis M, Zaltzman M, Feldman C, Orford A, Zwi S. ‘Low-dose’ corticosteroid prophylaxis against fat embolism. J Trauma1987; 27:1173–1176.
Stoltenberg JJ, Gustilo RB. The use of methylprednisolone and hypertonic glucose in the prophylaxis of fat embolism syndrome. Clin Orthop Relat Res1979; 143:211–221.
Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg2009; 52:386–393.
Although the incidence and rates of morbidity and death from acute community-acquired bacterial meningitis have dramatically declined, probably as a result of vaccination and better antimicrobial and adjuvant therapy, the disease still has a high toll. From 10% to 20% of people who contract it in the United States still die of it.1,2
In the United States, meningitis from all causes accounts for about 72,000 hospitalizations and up to $1.2 billion in hospital costs annually.3 However, the incidence of bacterial meningitis has declined from 3 to 5 per 100,000 per year a few decades ago to 1.3 to 2 per 100,000 per year currently.2 In less-developed countries, rates are much higher.
In the early 1900s in the United States, the death rate from bacterial meningitis was 80% to 100%. The use of intrathecal equine meningococcal antiserum during the first decades of the 1900s dramatically reduced the rate of death from meningococcal meningitis. With the advent of antimicrobial drugs in the 1930s and 1940s, the death rate from bacterial meningitis further declined.
The organisms that cause community-acquired bacterial meningitis differ somewhat by geographic region and by age. In a recent paper based on surveillance data, in the United States, from 1998 to 2007, the most common cause of bacterial meningitis among adults was Streptococcus pneumoniae. Among young adults, Neisseria meningitidis is nearly as common as S pneumoniae. The incidence of Listeria infections increases with age in adults.2
The epidemiologic features of bacterial meningitis have changed dramatically over the past decades with the advent of the Haemophilus influenzae vaccine. In 1986, about half the cases of acute bacterial meningitis were caused by H influenzae, but a decade later the incidence of H influenzae meningitis had been reduced by 94%.4
Meningitis is inflammation of the pia and arachnoid (the inner two layers of the meninges). Acute community-acquired meningitis can develop within hours to days and can be viral or bacterial. Viral meningitis usually has a good prognosis, whereas bacterial meningitis is associated with significant rates of morbidity and death, so it is critical to recognize and differentiate them promptly.
PATHOGENESIS
Most cases of community-acquired bacterial meningitis begin with colonization of the nasopharyngeal mucosa. In certain individuals this leads to mucosal invasion and bacteremia. Not all organisms that cause bacteremia are capable of breaching the blood-cerebrospinal fluid barrier to enter the subarachnoid space to cause meningitis. Very few organisms have this capacity, but N meningitidis and S pneumoniae do.5
Some patients are at higher risk of meningitis because of an abnormal communication between the nasopharynx and the subarachnoid space due either to trauma or a congenital anatomic abnormality. The organisms in these instances can directly spread from the nasopharynx to the meninges. Patients without a spleen or with an immunoglobulin deficiency are also more prone to infections from encapsulated organisms such as pneumococci and meningococci. The opsonizing immunoglobulins coat the capsule, helping phagocytes in the spleen to remove them from the bloodstream. A patient presenting with multiple episodes of bacterial meningitis merits evaluation for these conditions.
In contrast, Listeria spp and, rarely, gramnegative bacteria enter the bloodstream through the gastrointestinal tract and then spread to the meninges.
Once in the subarachnoid space, bacteria elicit a profuse inflammatory response, which can be damaging.5 The inflammation in the subarachnoid space can extend along the Virchow-Robin spaces surrounding the blood vessels deep into the brain parenchyma. This perivascular inflammation can cause thrombosis in both the arterial and venous circulation.
Thus, the inflammation can lead to intracranial complications such as cerebral edema, hydrocephalus, and stroke. The complications of bacterial meningitis can be remembered by the acronym HACTIVE: hydrocephalus, abscess, cerebritis and cranial nerve lesions, thrombosis, infarct, ventriculitis and vasculopathy, and extra-axial collection.5,6
MICROBIOLOGY: WHEN TO SUSPECT DIFFERENT ORGANISMS
S pneumoniae: The most common cause in adults
Patients without a spleen and patients with either a primary or secondary immunoglobulin deficiency, including patients with multiple myeloma or human immunodeficiency virus infection, are at a higher risk of infection with this organism.
N meningitidis: More common in young adults
N meningitidis is easily transmitted and is associated with crowding, as in school dormitories and military barracks. People with congenital deficiencies of components of terminal complement are at greater risk for both meningococcal and gonococcal infections. Patients with recurrent episodes of Neisseria infection should be evaluated for complement deficiency.
Photos courtesy of Thomas Fraser, MD.
Figure 1. Petechial rash from Neisseria meningitides.
Meningococcal infection is more commonly associated with a rash. The most common rash of meningococcal meningitis is a very transient, maculopapular rash that appears early in the course of the disease. More pathognomonic is a petechial rash (Figure 1) with thrombocytopenia, which can very rapidly progress to purpura, ecchymosis, and disseminated intravascular coagulation. The petechial rash is evident in 60% of adults and up to 90% of children,7 and it is most likely to appear in dependent areas (such as the back of a patient lying down) and in areas of pressure, such as under the elastic band of underwear or stockings.
Listeria
Listeria infection is usually acquired through contaminated food such as raw vegetables, unpasteurized milk and cheese, and deli meats. From the gastrointestinal tract, it spreads to the bloodstream and then to the meninges.
Listeria is an intracellular pathogen; thus, people at greater risk are those with poor cell-mediated immunity due to immunosuppressant medications such as steroids or tumor necrosis factor inhibitors.
The rate of Listeria meningitis starts to increase with age, especially after age 50, probably due to immune senescence or decreased immunity with age.
Aerobic gram-negative bacilli
Gram-negative enteric bacilli usually cause meningitis after head trauma or neurosurgery and are very uncommon causes of community-acquired meningitis. Disseminated strongyloidiasis, also known as hyperinfection syndrome, should be suspected in any patient with community-acquired meningitis caused by enteric gram-negative bacilli.
Strongyloides stercoralis is a parasitic intestinal roundworm that is found in the tropics, in the subtropics, and in certain parts of the United States and Europe. The adult worm lives in the intestines and lays eggs, which hatch in the mucosa; the larvae are excreted in the stool. A small percentage of larvae penetrate the perianal skin and gut mucosa to cause an autoinfection. People may asymptomatically harbor the parasite for decades, then develop the hyperinfection syndrome when treated with immunosuppressive drugs such as steroids. In the hyperinfection syndrome a significant proportion of the larvae penetrate the gut mucosa to enter the bloodstream and travel throughout the body, including into the brain, carrying gram-negative bacteria with them.
The mortality rate of untreated hyperinfection syndrome can sometimes reach 100%.8 Thus, it is important to identify and treat the hyperinfection syndrome in the context of gram-negative bacillary meningitis.
SUSPECTED MENINGITIS: CLINICAL SCENARIO
A 36-year-old man presents to the emergency department with high fever, headache, and lethargy that developed over the past 24 hours. His temperature is 104°F (40°C), pulse 120 beats/min, respiratory rate 30/min, and blood pressure 130/70 mm Hg. He is oriented only to person and has nuchal rigidity. His white blood cell count is 30 × 109/L, with 20% bands.
The clinical questions that arise with such a patient are:
Does the patient have bacterial or viral meningitis?
Can we reliably rule out meningitis based on a history and physical examination?
Is a lumbar puncture for cerebrospinal fluid (CSF) analysis needed? How should these studies be interpreted?
Should computed tomography of the head be done before lumbar puncture?
Which antimicrobial drugs should be started empirically at the outset?
What is the role of steroids in treatment?
CLINICAL SIGNS AND SYMPTOMS
The classic triad of meningitis is fever, neck stiffness, and altered mental status. Other signs and symptoms that have been described are photophobia, headache, nausea, vomiting, focal neurologic symptoms, altered mental status, the Kernig sign (inability to allow full knee extension when the hip is flexed to a 90° angle), and the Brudzinski sign (spontaneous flexion of the hips during attempted passive flexion of the neck).
Can meningitis be ruled out if the patient does not have this classic presentation?
Unfortunately, only a few high-quality studies of the diagnostic accuracy of signs and symptoms of bacterial meningitis have been done. Fourteen retrospective studies examined this issue, but they were heterogeneous with respect to patient age, immunosuppression status, and clinical presentation, as well as to how meningitis was diagnosed (via culture or cerebrospinal fluid analysis), making the results difficult to interpret.9 Retrospective studies are more prone to bias, as they lack a control group, and examiner bias is more likely. Based on retrospective data, the combination of fever, neck stiffness, and altered mental status has a sensitivity of only 0.46.9
Two prospective studies examined symptoms and signs. Thomas et al10 evaluated 297 patients with “clinically suspected meningitis.” Unfortunately, in this study the physical examination was not standardized. In a study by Uchihara and Tsukagoshi,11 the measurement was more reliable, as they used a single examiner to evaluate patients presenting with fever and headache, but only 54 patients were studied.
Based on these prospective studies, the presence of nausea and vomiting, headache, or neck stiffness does not reliably rule in meningitis(Table 1).9 Similarly, the absence of these does not rule it out. The 95% confidence intervals (CIs) of the positive and negative likelihood ratios include the value 1. (A simple interpretation of that would be that the likelihood of finding these features is the same in patients with meningitis when compared with those without meningitis.9)
For the physical examination, the presence or absence of fever, the Kernig sign, or the Brudzinski sign were also inconclusive. The CIs of the positive and negative likelihood ratios, like those of the symptoms, included the value 1. Only one test done on physical examination looked promising in having diagnostic utility to rule out meningitis: the jolt accentuation test (performed by asking a patient with a headache to quickly move his or her head twice horizontally; the result is positive if the headache worsens). If the result is negative, meningitis is unlikely (negative likelihood ratio 0.05, 95% CI 0.01–0.35).9 However, a positive test is not useful in making the diagnosis. A caveat is that this is based on a single study.
In summary, the history and physical examination are not sufficient to determine whether a patient has meningitis. If a patient is suspected of having meningitis, a lumbar puncture is needed.
WORKUP AND DIAGNOSTIC TESTS
Which tests are needed?
Blood cultures should be drawn before antimicrobial treatment is started.12–14 Although positive only 19% to 70% of the time, they can help identify the pathogen.15–17
Lumbar puncture with CSF study is essential to make the diagnosis and to identify the organism and its susceptibility to various antibiotics. If lumbar puncture can be performed immediately, it should be done before starting antibiotics, to maximize the yield of cultures. Pediatric studies show that after starting antibiotics, complete sterilization of the cerebrospinal fluid can occur within 2 hours for N meningitides and within 4 hours for S pneumoniae.14 However, starting antimicrobials should not be delayed if a lumbar puncture cannot be done expeditiously.
Is computed tomography of the brain necessary before a lumbar puncture?
The rationale behind performing CT before lumbar puncture is to determine if the patient has elevated intracranial pressure, which would increase the risk of brain herniation due to lowering of the lumbar CSF pressure during lumbar puncture. For ethical and practical reasons, it would be difficult to evaluate this in a randomized clinical trial.
Hasbun et al18 performed a study to evaluate if any features on clinical presentation can predict abnormal findings on CT of the head suggestive of elevated intracranial pressure and thus the risk of herniation. The study included 301 adults with suspected meningitis. It found that abnormal findings on CT were unlikely if all of the following features were absent at baseline:
Immunocompromised state
History of central nervous system disease (mass lesion, stroke, or a focal infection)
New onset of seizure (≤ 1 week from presentation)
Specific abnormal neurologic findings (eg, an abnormal level of consciousness, inability to answer two consecutive questions correctly or to follow two consecutive commands, gaze palsy, abnormal visual fields, facial palsy, arm drift, leg drift, abnormal language).
Absence of these baseline features made it unlikely that CT would be abnormal (negative likelihood ratio 0.1, 95% CI 0.03–0.31).
Adapted from Tunkel AR, et al. Practice guidelines for the management of bacterial meningitis. Clin Infec Dis 2004; 39:1267–1284, with permission from the Infectious Diseases Society of America.
Figure 2.
According to the guidelines from the Infectious Diseases Society of America (IDSA),19 if none of those features is present, blood cultures and a lumbar puncture should be done immediately, followed by empiric antimicrobial therapy. If any of the features is present, blood cultures should be obtained first, then empiric antimicrobial therapy started, followed by CT of the brain to look for contraindications to a lumbar puncture (Figure 2).
What can lumbar puncture tell us?
Results of lumbar puncture studies can help determine whether meningitis is present and, if so, whether the cause is likely bacterial or viral.20
The opening pressure is elevated (usually > 180 mm H2O) in acute bacterial meningitis. The CSF white blood cell count is usually more than 1.0 × 109/L, consisting predominantly of neutrophils, in acute bacterial meningitis. In viral meningitis, it is usually less than 0.1 × 109/L, mostly lymphocytes.
Protein shows a mild to marked elevation in bacterial meningitis but is normal to elevated in viral meningitis.
The CSF glucose level is lower in bacterial meningitis than in viral meningitis.
The ratio of CSF glucose to blood glucose. Because the glucose levels in the CSF and the blood equilibrate, the ratio of CSF glucose to serum glucose has better diagnostic accuracy than the CSF glucose level alone. The equilibration takes place within a few hours, so the serum glucose level should be ordered at the same time lumbar puncture is done. The CSF glucose-blood glucose ratio is a better predictor of bacterial meningitis than the CSF white blood cell count. Bacterial meningitis is likely if the ratio is lower than 0.4.
Lactate levels are not usually measured, but a lactate level greater than 31.5 mg/dL (3.5 mmol/L) is predictive of meningitis, and a lower level makes the diagnosis unlikely.
The diagnostic accuracies (likelihood ratios) of the CSF tests were analyzed by Straus et al.21 The positive likelihood ratios for the CSF white blood cell count and for the CSF glucose-blood glucose ratio are greater than 10, but these tests have negative likelihood ratios of more than 0.1. (It is generally thought that a test with a positive likelihood ratio of more than 10 is considered good for ruling in a diagnosis, whereas one with a negative likelihood ratio of less than 0.1 is good for ruling out a diagnosis.) Thus, these tests are good to rule in bacterial meningitis, but not as good to rule it out. There are some data to show that CSF lactate and procalcitonin might be more sensitive in ruling out bacterial meningitis, but more studies are needed.22
Gram stain of the cerebrospinal fluid can be done quickly. If no bacteria are seen, the information is not helpful in ruling out bacterial meningitis (negative likelihood ratio 0.14, 95% CI 0.08–0.27). If it is positive, it is almost 100% specific for meningitis due to the organism seen (positive likelihood ratio 735, 95% CI 230–2,295).21
MANAGEMENT
Empiric antimicrobial therapy must be started as soon as feasible
Most studies of the timing of antimicrobial drugs were retrospective and included a very heterogeneous population. They were thus more prone to bias and confounding.23,24 Proulx et al,23 in a retrospective study, found that if antibiotics were given within 6 hours of the time the patient presented to the emergency department, the case fatality rate was only 5% to 6%. If treatment started 6 to 8 hours after presentation, the death rate was 45%, and if it started from 8 to 10 hours after presentation, the death rate was 75%. Most physicians would agree that starting antimicrobials early would be beneficial.
CSF concentrations of most antimicrobial drugs are considerably less than in the serum due to poor penetration of the blood-CSF barrier. Thus, the dose for treating meningitis is usually higher than the regular dose. For example, for the treatment of pneumococcal pneumonia, ceftriaxone (Rocephin) is used at a dose of 1 g every 24 hours, but for pneumococcal meningitis the dose is 2 g every 12 hours.
Empiric treatment of community-acquired bacterial meningitis in immunocompetent adults up to 50 years of age consists of a third-generation cephalosporin such as cefotaxime (Claforan) 2 g intravenously every 4 hours or ceftriaxone 2 g intravenously every 12 hours, which covers most S pneumoniae and N meningitides strains.19 The IDSA guidelines recommend adding vancomycin (Vancocin) empirically in suspected S pneumoniae meningitis due to concerns about drug-resistant pneumococcal strains.19 For vancomycin, 45 to 60 mg/kg intravenously per day divided into every-6-hour or every-8-hour doses would achieve better CSF concentrations.25
In patients over age 50 or those with a cell-mediated immunodeficiency, empiric therapy should also include ampicillin 2 g intravenously every 4 hours to cover Listeria.
It is important to tailor therapy to the results of Gram stain, culture, and susceptibility as they become available.
Role of corticosteroids
Glucocorticoids, especially dexamethasone (Decadron), have been well studied as adjunctive therapies in bacterial meningitis. The rationale behind their use is that the profuse inflammatory response to the bacterial components in the CSF by itself has deleterious effects, and steroids can reduce that.
In 2004, a Cochrane meta-analysis26 of five randomized clinical trials, including 623 adults with bacterial meningitis (234 with pneumococcal meningitis and 232 with meningococcal meningitis), found a significant reduction in the death rate for patients who received steroids: the death rate was 12% in patients who received steroids vs 22% in those who did not (odds ratio 0.6; 95% CI 0.40–0.81). This led to an IDSA practice guideline recommendation that in adults with suspected or proven pneumococcal meningitis, dexamethasone would be beneficial.19
But since then, many more studies have emerged from Europe, South America, Malawi, and Vietnam, and another Cochrane metaanalysis27 incorporated the new studies. Twenty-four studies involving 4,041 participants were included. Similar numbers of participants died in the corticosteroid and placebo groups (18% vs 20%; risk ratio [RR] 0.92, 95% CI 0.82–1.04, P = .18). A trend towards a lower mortality rate was noticed in adults receiving corticosteroids (RR 0.74, 95% CI 0.53–1.05, P = .09). In adults, corticosteroids were associated with lower rates of hearing loss (RR 0.74, 95% CI 0.56–0.98), and there was a trend towards fewer neurologic sequelae (RR 0.72, 95% CI 0.51–1.01). The benefits were shown in studies in adults in high-income countries, but the studies from low-income countries showed neither harm nor benefit. Based on these findings, the authors recommended the use of steroids in high-income countries, though the strength of the evidence was not optimal. The recommended steroid was dexamethasone 0.15 mg/kg intravenously every 6 hours for 4 days.
References
Swartz MN. Bacterial meningitis—a view of the past 90 years. N Engl J Med2004; 351:1826–1828.
Thigpen MC, Whitney CG, Messonnier NE, et al; Emerging Infections Programs Network. Bacterial meningitis in the United States, 1998–2007. N Engl J Med2011; 364:2010–2025.
Holmquist L, Russo CA, Elixhauser A. Meningitis-related hospitalizations in the United States, 2006. Statistical Brief #57. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville, MD, 2008. www.hcup-us.ahrq.gov/reports/statbriefs/sb57.jsp. Accessed May 4, 2012.
Schuchat A, Robinson K, Wenger JD, et al. Bacterial meningitis in the United States in 1995. N Engl J Med1997; 337:970–976.
Koedel U, Scheld WM, Pfister H-W. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis2002; 2:721–736.
Hughes DC, Raghavan A, Mordekar SR, Griffiths PD, Connolly DJ. Role of imaging in the diagnosis of acute bacterial meningitis and its complications. Postgrad Med J2010; 86:478–485.
Brouwer MC, Tunkel AR, van de Beek D. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin Microbiol Rev2010; 23:467–492.
Maguire JH. Intestinal nematodes (roundworms). In:Mandell G, Bennett J, Dolin R, editors. Principles and Practice of Infectious Diseases. Philadelphia: Elsevier, 2009:3577–3586.
Attia J, Hatala R, Cook DJ, Wong JG. Original article: does this adult patient have acute meningitis?In:Simel DL, Rennie D, editors. The Rational Clinical Examinatino: Evidence-Based Clinical Diagnosis. New York, NY: McGraw-Hill; 2009.
Thomas KE, Hasbun R, Jekel J, Quagliarello VJ. The diagnostic accuracy of Kernig’s sign, Brudzinski’s sign, and nuchal rigidity in adults with suspected meningitis. Clin Infect Dis2002; 35:46–52.
Uchihara T, Tsukagoshi H. Jolt accenulation of headache: the most sensitive sign of CSF pleocytosis. Headache1991; 31:167–171.
Geiseler PJ, Nelson KE, Levin S, Reddi KT, Moses VK. Community-acquired purulent meningitis: a review of 1,316 cases during the antibiotic era, 1954–1976. Rev Infect Dis1980; 2:725–745.
Talan DA, Hoffman JR, Yoshikawa TT, Overturf GD. Role of empiric parenteral antibiotics prior to lumbar puncture in suspected bacterial meningitis: state of the art. Rev Infect Dis1988; 10:365–376.
Kanegaye JT, Soliemanzadeh P, Bradley JS. Lumbar puncture in pediatric bacterial meningitis: defining the time interval for recovery of cerebrospinal fluid pathogens after parenteral antibiotic pretreatment. Pediatrics2001; 108:1169–1174.
Sigurdardóttir B, Björnsson OM, Jónsdóttir KE, Erlendsdóttir H, Gudmundsson S. Acute bacterial meningitis in adults. A 20-year overview. Arch Intern Med1997; 157:425–430.
Aronin SI, Peduzzi P, Quagliarello VJ. Community-acquired bacterial meningitis: risk stratification for adverse clinical outcome and effect of antibiotic timing. Ann Intern Med1998; 129:862–869.
Andersen J, Backer V, Voldsgaard P, Skinhój P, Wandall JH. Acute meningococcal meningitis: analysis of features of the disease according to the age of 255 patients. Copenhagen Meningitis Study Group. J Infect1997; 34:227–235.
Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med2001; 345:1727–1733.
Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis2004; 39:1267–1284.
Seehusen DA, Reeves MM, Fomin DA. Cerebrospinal fluid analysis. Am Fam Physician2003; 68:1103–1108.
Straus SE, Thorpe KE, Holroyd-Leduc J. How do I perform a lumbar puncture and analyze the results to diagnose bacterial meningitis?JAMA2006; 296:2010–2022.
Viallon A, Desseigne N, Marjollet O, et al. Meningitis in adult patients with a negative direct cerebrospinal fluid examination: value of cytochemical markers for differential diagnosis. Crit Care2011; 15:R136.
Proulx N, Fréchette D, Toye B, Chan J, Kravcik S. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM2005; 98:291–298.
Radetsky M. Duration of symptoms and outcome in bacterial meningitis: an analysis of causation and the implications of a delay in diagnosis. Pediatr Infect Dis J1992; 11:694–698.
Ricard JD, Wolff M, Lacherade JC, et al. Levels of vancomycin in cerebrospinal fluid of adult patients receiving adjunctive corticosteroids to treat pneumococcal meningitis: a prospective multicenter observational study. Clin Infect Dis2007; 44:250–255.
van de Beek D, de Gans J, McIntyre P, Prasad K. Steroids in adults with acute bacterial meningitis: a systematic review. Lancet Infect Dis2004; 4:139–143.
van de Beek D, Farrar JJ, de Gans J, et al. Adjunctive dexamethasone in bacterial meningitis: a meta-analysis of individual patient data. Lancet Neurol2010; 9:254–263.
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the author but are not peer-reviewed.
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the author but are not peer-reviewed.
Author and Disclosure Information
Adarsh Bhimraj, MD Head, Section of Neurologic Infectious Diseases, Department of Infectious Disease, Cleveland Clinic
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the author but are not peer-reviewed.
Although the incidence and rates of morbidity and death from acute community-acquired bacterial meningitis have dramatically declined, probably as a result of vaccination and better antimicrobial and adjuvant therapy, the disease still has a high toll. From 10% to 20% of people who contract it in the United States still die of it.1,2
In the United States, meningitis from all causes accounts for about 72,000 hospitalizations and up to $1.2 billion in hospital costs annually.3 However, the incidence of bacterial meningitis has declined from 3 to 5 per 100,000 per year a few decades ago to 1.3 to 2 per 100,000 per year currently.2 In less-developed countries, rates are much higher.
In the early 1900s in the United States, the death rate from bacterial meningitis was 80% to 100%. The use of intrathecal equine meningococcal antiserum during the first decades of the 1900s dramatically reduced the rate of death from meningococcal meningitis. With the advent of antimicrobial drugs in the 1930s and 1940s, the death rate from bacterial meningitis further declined.
The organisms that cause community-acquired bacterial meningitis differ somewhat by geographic region and by age. In a recent paper based on surveillance data, in the United States, from 1998 to 2007, the most common cause of bacterial meningitis among adults was Streptococcus pneumoniae. Among young adults, Neisseria meningitidis is nearly as common as S pneumoniae. The incidence of Listeria infections increases with age in adults.2
The epidemiologic features of bacterial meningitis have changed dramatically over the past decades with the advent of the Haemophilus influenzae vaccine. In 1986, about half the cases of acute bacterial meningitis were caused by H influenzae, but a decade later the incidence of H influenzae meningitis had been reduced by 94%.4
Meningitis is inflammation of the pia and arachnoid (the inner two layers of the meninges). Acute community-acquired meningitis can develop within hours to days and can be viral or bacterial. Viral meningitis usually has a good prognosis, whereas bacterial meningitis is associated with significant rates of morbidity and death, so it is critical to recognize and differentiate them promptly.
PATHOGENESIS
Most cases of community-acquired bacterial meningitis begin with colonization of the nasopharyngeal mucosa. In certain individuals this leads to mucosal invasion and bacteremia. Not all organisms that cause bacteremia are capable of breaching the blood-cerebrospinal fluid barrier to enter the subarachnoid space to cause meningitis. Very few organisms have this capacity, but N meningitidis and S pneumoniae do.5
Some patients are at higher risk of meningitis because of an abnormal communication between the nasopharynx and the subarachnoid space due either to trauma or a congenital anatomic abnormality. The organisms in these instances can directly spread from the nasopharynx to the meninges. Patients without a spleen or with an immunoglobulin deficiency are also more prone to infections from encapsulated organisms such as pneumococci and meningococci. The opsonizing immunoglobulins coat the capsule, helping phagocytes in the spleen to remove them from the bloodstream. A patient presenting with multiple episodes of bacterial meningitis merits evaluation for these conditions.
In contrast, Listeria spp and, rarely, gramnegative bacteria enter the bloodstream through the gastrointestinal tract and then spread to the meninges.
Once in the subarachnoid space, bacteria elicit a profuse inflammatory response, which can be damaging.5 The inflammation in the subarachnoid space can extend along the Virchow-Robin spaces surrounding the blood vessels deep into the brain parenchyma. This perivascular inflammation can cause thrombosis in both the arterial and venous circulation.
Thus, the inflammation can lead to intracranial complications such as cerebral edema, hydrocephalus, and stroke. The complications of bacterial meningitis can be remembered by the acronym HACTIVE: hydrocephalus, abscess, cerebritis and cranial nerve lesions, thrombosis, infarct, ventriculitis and vasculopathy, and extra-axial collection.5,6
MICROBIOLOGY: WHEN TO SUSPECT DIFFERENT ORGANISMS
S pneumoniae: The most common cause in adults
Patients without a spleen and patients with either a primary or secondary immunoglobulin deficiency, including patients with multiple myeloma or human immunodeficiency virus infection, are at a higher risk of infection with this organism.
N meningitidis: More common in young adults
N meningitidis is easily transmitted and is associated with crowding, as in school dormitories and military barracks. People with congenital deficiencies of components of terminal complement are at greater risk for both meningococcal and gonococcal infections. Patients with recurrent episodes of Neisseria infection should be evaluated for complement deficiency.
Photos courtesy of Thomas Fraser, MD.
Figure 1. Petechial rash from Neisseria meningitides.
Meningococcal infection is more commonly associated with a rash. The most common rash of meningococcal meningitis is a very transient, maculopapular rash that appears early in the course of the disease. More pathognomonic is a petechial rash (Figure 1) with thrombocytopenia, which can very rapidly progress to purpura, ecchymosis, and disseminated intravascular coagulation. The petechial rash is evident in 60% of adults and up to 90% of children,7 and it is most likely to appear in dependent areas (such as the back of a patient lying down) and in areas of pressure, such as under the elastic band of underwear or stockings.
Listeria
Listeria infection is usually acquired through contaminated food such as raw vegetables, unpasteurized milk and cheese, and deli meats. From the gastrointestinal tract, it spreads to the bloodstream and then to the meninges.
Listeria is an intracellular pathogen; thus, people at greater risk are those with poor cell-mediated immunity due to immunosuppressant medications such as steroids or tumor necrosis factor inhibitors.
The rate of Listeria meningitis starts to increase with age, especially after age 50, probably due to immune senescence or decreased immunity with age.
Aerobic gram-negative bacilli
Gram-negative enteric bacilli usually cause meningitis after head trauma or neurosurgery and are very uncommon causes of community-acquired meningitis. Disseminated strongyloidiasis, also known as hyperinfection syndrome, should be suspected in any patient with community-acquired meningitis caused by enteric gram-negative bacilli.
Strongyloides stercoralis is a parasitic intestinal roundworm that is found in the tropics, in the subtropics, and in certain parts of the United States and Europe. The adult worm lives in the intestines and lays eggs, which hatch in the mucosa; the larvae are excreted in the stool. A small percentage of larvae penetrate the perianal skin and gut mucosa to cause an autoinfection. People may asymptomatically harbor the parasite for decades, then develop the hyperinfection syndrome when treated with immunosuppressive drugs such as steroids. In the hyperinfection syndrome a significant proportion of the larvae penetrate the gut mucosa to enter the bloodstream and travel throughout the body, including into the brain, carrying gram-negative bacteria with them.
The mortality rate of untreated hyperinfection syndrome can sometimes reach 100%.8 Thus, it is important to identify and treat the hyperinfection syndrome in the context of gram-negative bacillary meningitis.
SUSPECTED MENINGITIS: CLINICAL SCENARIO
A 36-year-old man presents to the emergency department with high fever, headache, and lethargy that developed over the past 24 hours. His temperature is 104°F (40°C), pulse 120 beats/min, respiratory rate 30/min, and blood pressure 130/70 mm Hg. He is oriented only to person and has nuchal rigidity. His white blood cell count is 30 × 109/L, with 20% bands.
The clinical questions that arise with such a patient are:
Does the patient have bacterial or viral meningitis?
Can we reliably rule out meningitis based on a history and physical examination?
Is a lumbar puncture for cerebrospinal fluid (CSF) analysis needed? How should these studies be interpreted?
Should computed tomography of the head be done before lumbar puncture?
Which antimicrobial drugs should be started empirically at the outset?
What is the role of steroids in treatment?
CLINICAL SIGNS AND SYMPTOMS
The classic triad of meningitis is fever, neck stiffness, and altered mental status. Other signs and symptoms that have been described are photophobia, headache, nausea, vomiting, focal neurologic symptoms, altered mental status, the Kernig sign (inability to allow full knee extension when the hip is flexed to a 90° angle), and the Brudzinski sign (spontaneous flexion of the hips during attempted passive flexion of the neck).
Can meningitis be ruled out if the patient does not have this classic presentation?
Unfortunately, only a few high-quality studies of the diagnostic accuracy of signs and symptoms of bacterial meningitis have been done. Fourteen retrospective studies examined this issue, but they were heterogeneous with respect to patient age, immunosuppression status, and clinical presentation, as well as to how meningitis was diagnosed (via culture or cerebrospinal fluid analysis), making the results difficult to interpret.9 Retrospective studies are more prone to bias, as they lack a control group, and examiner bias is more likely. Based on retrospective data, the combination of fever, neck stiffness, and altered mental status has a sensitivity of only 0.46.9
Two prospective studies examined symptoms and signs. Thomas et al10 evaluated 297 patients with “clinically suspected meningitis.” Unfortunately, in this study the physical examination was not standardized. In a study by Uchihara and Tsukagoshi,11 the measurement was more reliable, as they used a single examiner to evaluate patients presenting with fever and headache, but only 54 patients were studied.
Based on these prospective studies, the presence of nausea and vomiting, headache, or neck stiffness does not reliably rule in meningitis(Table 1).9 Similarly, the absence of these does not rule it out. The 95% confidence intervals (CIs) of the positive and negative likelihood ratios include the value 1. (A simple interpretation of that would be that the likelihood of finding these features is the same in patients with meningitis when compared with those without meningitis.9)
For the physical examination, the presence or absence of fever, the Kernig sign, or the Brudzinski sign were also inconclusive. The CIs of the positive and negative likelihood ratios, like those of the symptoms, included the value 1. Only one test done on physical examination looked promising in having diagnostic utility to rule out meningitis: the jolt accentuation test (performed by asking a patient with a headache to quickly move his or her head twice horizontally; the result is positive if the headache worsens). If the result is negative, meningitis is unlikely (negative likelihood ratio 0.05, 95% CI 0.01–0.35).9 However, a positive test is not useful in making the diagnosis. A caveat is that this is based on a single study.
In summary, the history and physical examination are not sufficient to determine whether a patient has meningitis. If a patient is suspected of having meningitis, a lumbar puncture is needed.
WORKUP AND DIAGNOSTIC TESTS
Which tests are needed?
Blood cultures should be drawn before antimicrobial treatment is started.12–14 Although positive only 19% to 70% of the time, they can help identify the pathogen.15–17
Lumbar puncture with CSF study is essential to make the diagnosis and to identify the organism and its susceptibility to various antibiotics. If lumbar puncture can be performed immediately, it should be done before starting antibiotics, to maximize the yield of cultures. Pediatric studies show that after starting antibiotics, complete sterilization of the cerebrospinal fluid can occur within 2 hours for N meningitides and within 4 hours for S pneumoniae.14 However, starting antimicrobials should not be delayed if a lumbar puncture cannot be done expeditiously.
Is computed tomography of the brain necessary before a lumbar puncture?
The rationale behind performing CT before lumbar puncture is to determine if the patient has elevated intracranial pressure, which would increase the risk of brain herniation due to lowering of the lumbar CSF pressure during lumbar puncture. For ethical and practical reasons, it would be difficult to evaluate this in a randomized clinical trial.
Hasbun et al18 performed a study to evaluate if any features on clinical presentation can predict abnormal findings on CT of the head suggestive of elevated intracranial pressure and thus the risk of herniation. The study included 301 adults with suspected meningitis. It found that abnormal findings on CT were unlikely if all of the following features were absent at baseline:
Immunocompromised state
History of central nervous system disease (mass lesion, stroke, or a focal infection)
New onset of seizure (≤ 1 week from presentation)
Specific abnormal neurologic findings (eg, an abnormal level of consciousness, inability to answer two consecutive questions correctly or to follow two consecutive commands, gaze palsy, abnormal visual fields, facial palsy, arm drift, leg drift, abnormal language).
Absence of these baseline features made it unlikely that CT would be abnormal (negative likelihood ratio 0.1, 95% CI 0.03–0.31).
Adapted from Tunkel AR, et al. Practice guidelines for the management of bacterial meningitis. Clin Infec Dis 2004; 39:1267–1284, with permission from the Infectious Diseases Society of America.
Figure 2.
According to the guidelines from the Infectious Diseases Society of America (IDSA),19 if none of those features is present, blood cultures and a lumbar puncture should be done immediately, followed by empiric antimicrobial therapy. If any of the features is present, blood cultures should be obtained first, then empiric antimicrobial therapy started, followed by CT of the brain to look for contraindications to a lumbar puncture (Figure 2).
What can lumbar puncture tell us?
Results of lumbar puncture studies can help determine whether meningitis is present and, if so, whether the cause is likely bacterial or viral.20
The opening pressure is elevated (usually > 180 mm H2O) in acute bacterial meningitis. The CSF white blood cell count is usually more than 1.0 × 109/L, consisting predominantly of neutrophils, in acute bacterial meningitis. In viral meningitis, it is usually less than 0.1 × 109/L, mostly lymphocytes.
Protein shows a mild to marked elevation in bacterial meningitis but is normal to elevated in viral meningitis.
The CSF glucose level is lower in bacterial meningitis than in viral meningitis.
The ratio of CSF glucose to blood glucose. Because the glucose levels in the CSF and the blood equilibrate, the ratio of CSF glucose to serum glucose has better diagnostic accuracy than the CSF glucose level alone. The equilibration takes place within a few hours, so the serum glucose level should be ordered at the same time lumbar puncture is done. The CSF glucose-blood glucose ratio is a better predictor of bacterial meningitis than the CSF white blood cell count. Bacterial meningitis is likely if the ratio is lower than 0.4.
Lactate levels are not usually measured, but a lactate level greater than 31.5 mg/dL (3.5 mmol/L) is predictive of meningitis, and a lower level makes the diagnosis unlikely.
The diagnostic accuracies (likelihood ratios) of the CSF tests were analyzed by Straus et al.21 The positive likelihood ratios for the CSF white blood cell count and for the CSF glucose-blood glucose ratio are greater than 10, but these tests have negative likelihood ratios of more than 0.1. (It is generally thought that a test with a positive likelihood ratio of more than 10 is considered good for ruling in a diagnosis, whereas one with a negative likelihood ratio of less than 0.1 is good for ruling out a diagnosis.) Thus, these tests are good to rule in bacterial meningitis, but not as good to rule it out. There are some data to show that CSF lactate and procalcitonin might be more sensitive in ruling out bacterial meningitis, but more studies are needed.22
Gram stain of the cerebrospinal fluid can be done quickly. If no bacteria are seen, the information is not helpful in ruling out bacterial meningitis (negative likelihood ratio 0.14, 95% CI 0.08–0.27). If it is positive, it is almost 100% specific for meningitis due to the organism seen (positive likelihood ratio 735, 95% CI 230–2,295).21
MANAGEMENT
Empiric antimicrobial therapy must be started as soon as feasible
Most studies of the timing of antimicrobial drugs were retrospective and included a very heterogeneous population. They were thus more prone to bias and confounding.23,24 Proulx et al,23 in a retrospective study, found that if antibiotics were given within 6 hours of the time the patient presented to the emergency department, the case fatality rate was only 5% to 6%. If treatment started 6 to 8 hours after presentation, the death rate was 45%, and if it started from 8 to 10 hours after presentation, the death rate was 75%. Most physicians would agree that starting antimicrobials early would be beneficial.
CSF concentrations of most antimicrobial drugs are considerably less than in the serum due to poor penetration of the blood-CSF barrier. Thus, the dose for treating meningitis is usually higher than the regular dose. For example, for the treatment of pneumococcal pneumonia, ceftriaxone (Rocephin) is used at a dose of 1 g every 24 hours, but for pneumococcal meningitis the dose is 2 g every 12 hours.
Empiric treatment of community-acquired bacterial meningitis in immunocompetent adults up to 50 years of age consists of a third-generation cephalosporin such as cefotaxime (Claforan) 2 g intravenously every 4 hours or ceftriaxone 2 g intravenously every 12 hours, which covers most S pneumoniae and N meningitides strains.19 The IDSA guidelines recommend adding vancomycin (Vancocin) empirically in suspected S pneumoniae meningitis due to concerns about drug-resistant pneumococcal strains.19 For vancomycin, 45 to 60 mg/kg intravenously per day divided into every-6-hour or every-8-hour doses would achieve better CSF concentrations.25
In patients over age 50 or those with a cell-mediated immunodeficiency, empiric therapy should also include ampicillin 2 g intravenously every 4 hours to cover Listeria.
It is important to tailor therapy to the results of Gram stain, culture, and susceptibility as they become available.
Role of corticosteroids
Glucocorticoids, especially dexamethasone (Decadron), have been well studied as adjunctive therapies in bacterial meningitis. The rationale behind their use is that the profuse inflammatory response to the bacterial components in the CSF by itself has deleterious effects, and steroids can reduce that.
In 2004, a Cochrane meta-analysis26 of five randomized clinical trials, including 623 adults with bacterial meningitis (234 with pneumococcal meningitis and 232 with meningococcal meningitis), found a significant reduction in the death rate for patients who received steroids: the death rate was 12% in patients who received steroids vs 22% in those who did not (odds ratio 0.6; 95% CI 0.40–0.81). This led to an IDSA practice guideline recommendation that in adults with suspected or proven pneumococcal meningitis, dexamethasone would be beneficial.19
But since then, many more studies have emerged from Europe, South America, Malawi, and Vietnam, and another Cochrane metaanalysis27 incorporated the new studies. Twenty-four studies involving 4,041 participants were included. Similar numbers of participants died in the corticosteroid and placebo groups (18% vs 20%; risk ratio [RR] 0.92, 95% CI 0.82–1.04, P = .18). A trend towards a lower mortality rate was noticed in adults receiving corticosteroids (RR 0.74, 95% CI 0.53–1.05, P = .09). In adults, corticosteroids were associated with lower rates of hearing loss (RR 0.74, 95% CI 0.56–0.98), and there was a trend towards fewer neurologic sequelae (RR 0.72, 95% CI 0.51–1.01). The benefits were shown in studies in adults in high-income countries, but the studies from low-income countries showed neither harm nor benefit. Based on these findings, the authors recommended the use of steroids in high-income countries, though the strength of the evidence was not optimal. The recommended steroid was dexamethasone 0.15 mg/kg intravenously every 6 hours for 4 days.
Although the incidence and rates of morbidity and death from acute community-acquired bacterial meningitis have dramatically declined, probably as a result of vaccination and better antimicrobial and adjuvant therapy, the disease still has a high toll. From 10% to 20% of people who contract it in the United States still die of it.1,2
In the United States, meningitis from all causes accounts for about 72,000 hospitalizations and up to $1.2 billion in hospital costs annually.3 However, the incidence of bacterial meningitis has declined from 3 to 5 per 100,000 per year a few decades ago to 1.3 to 2 per 100,000 per year currently.2 In less-developed countries, rates are much higher.
In the early 1900s in the United States, the death rate from bacterial meningitis was 80% to 100%. The use of intrathecal equine meningococcal antiserum during the first decades of the 1900s dramatically reduced the rate of death from meningococcal meningitis. With the advent of antimicrobial drugs in the 1930s and 1940s, the death rate from bacterial meningitis further declined.
The organisms that cause community-acquired bacterial meningitis differ somewhat by geographic region and by age. In a recent paper based on surveillance data, in the United States, from 1998 to 2007, the most common cause of bacterial meningitis among adults was Streptococcus pneumoniae. Among young adults, Neisseria meningitidis is nearly as common as S pneumoniae. The incidence of Listeria infections increases with age in adults.2
The epidemiologic features of bacterial meningitis have changed dramatically over the past decades with the advent of the Haemophilus influenzae vaccine. In 1986, about half the cases of acute bacterial meningitis were caused by H influenzae, but a decade later the incidence of H influenzae meningitis had been reduced by 94%.4
Meningitis is inflammation of the pia and arachnoid (the inner two layers of the meninges). Acute community-acquired meningitis can develop within hours to days and can be viral or bacterial. Viral meningitis usually has a good prognosis, whereas bacterial meningitis is associated with significant rates of morbidity and death, so it is critical to recognize and differentiate them promptly.
PATHOGENESIS
Most cases of community-acquired bacterial meningitis begin with colonization of the nasopharyngeal mucosa. In certain individuals this leads to mucosal invasion and bacteremia. Not all organisms that cause bacteremia are capable of breaching the blood-cerebrospinal fluid barrier to enter the subarachnoid space to cause meningitis. Very few organisms have this capacity, but N meningitidis and S pneumoniae do.5
Some patients are at higher risk of meningitis because of an abnormal communication between the nasopharynx and the subarachnoid space due either to trauma or a congenital anatomic abnormality. The organisms in these instances can directly spread from the nasopharynx to the meninges. Patients without a spleen or with an immunoglobulin deficiency are also more prone to infections from encapsulated organisms such as pneumococci and meningococci. The opsonizing immunoglobulins coat the capsule, helping phagocytes in the spleen to remove them from the bloodstream. A patient presenting with multiple episodes of bacterial meningitis merits evaluation for these conditions.
In contrast, Listeria spp and, rarely, gramnegative bacteria enter the bloodstream through the gastrointestinal tract and then spread to the meninges.
Once in the subarachnoid space, bacteria elicit a profuse inflammatory response, which can be damaging.5 The inflammation in the subarachnoid space can extend along the Virchow-Robin spaces surrounding the blood vessels deep into the brain parenchyma. This perivascular inflammation can cause thrombosis in both the arterial and venous circulation.
Thus, the inflammation can lead to intracranial complications such as cerebral edema, hydrocephalus, and stroke. The complications of bacterial meningitis can be remembered by the acronym HACTIVE: hydrocephalus, abscess, cerebritis and cranial nerve lesions, thrombosis, infarct, ventriculitis and vasculopathy, and extra-axial collection.5,6
MICROBIOLOGY: WHEN TO SUSPECT DIFFERENT ORGANISMS
S pneumoniae: The most common cause in adults
Patients without a spleen and patients with either a primary or secondary immunoglobulin deficiency, including patients with multiple myeloma or human immunodeficiency virus infection, are at a higher risk of infection with this organism.
N meningitidis: More common in young adults
N meningitidis is easily transmitted and is associated with crowding, as in school dormitories and military barracks. People with congenital deficiencies of components of terminal complement are at greater risk for both meningococcal and gonococcal infections. Patients with recurrent episodes of Neisseria infection should be evaluated for complement deficiency.
Photos courtesy of Thomas Fraser, MD.
Figure 1. Petechial rash from Neisseria meningitides.
Meningococcal infection is more commonly associated with a rash. The most common rash of meningococcal meningitis is a very transient, maculopapular rash that appears early in the course of the disease. More pathognomonic is a petechial rash (Figure 1) with thrombocytopenia, which can very rapidly progress to purpura, ecchymosis, and disseminated intravascular coagulation. The petechial rash is evident in 60% of adults and up to 90% of children,7 and it is most likely to appear in dependent areas (such as the back of a patient lying down) and in areas of pressure, such as under the elastic band of underwear or stockings.
Listeria
Listeria infection is usually acquired through contaminated food such as raw vegetables, unpasteurized milk and cheese, and deli meats. From the gastrointestinal tract, it spreads to the bloodstream and then to the meninges.
Listeria is an intracellular pathogen; thus, people at greater risk are those with poor cell-mediated immunity due to immunosuppressant medications such as steroids or tumor necrosis factor inhibitors.
The rate of Listeria meningitis starts to increase with age, especially after age 50, probably due to immune senescence or decreased immunity with age.
Aerobic gram-negative bacilli
Gram-negative enteric bacilli usually cause meningitis after head trauma or neurosurgery and are very uncommon causes of community-acquired meningitis. Disseminated strongyloidiasis, also known as hyperinfection syndrome, should be suspected in any patient with community-acquired meningitis caused by enteric gram-negative bacilli.
Strongyloides stercoralis is a parasitic intestinal roundworm that is found in the tropics, in the subtropics, and in certain parts of the United States and Europe. The adult worm lives in the intestines and lays eggs, which hatch in the mucosa; the larvae are excreted in the stool. A small percentage of larvae penetrate the perianal skin and gut mucosa to cause an autoinfection. People may asymptomatically harbor the parasite for decades, then develop the hyperinfection syndrome when treated with immunosuppressive drugs such as steroids. In the hyperinfection syndrome a significant proportion of the larvae penetrate the gut mucosa to enter the bloodstream and travel throughout the body, including into the brain, carrying gram-negative bacteria with them.
The mortality rate of untreated hyperinfection syndrome can sometimes reach 100%.8 Thus, it is important to identify and treat the hyperinfection syndrome in the context of gram-negative bacillary meningitis.
SUSPECTED MENINGITIS: CLINICAL SCENARIO
A 36-year-old man presents to the emergency department with high fever, headache, and lethargy that developed over the past 24 hours. His temperature is 104°F (40°C), pulse 120 beats/min, respiratory rate 30/min, and blood pressure 130/70 mm Hg. He is oriented only to person and has nuchal rigidity. His white blood cell count is 30 × 109/L, with 20% bands.
The clinical questions that arise with such a patient are:
Does the patient have bacterial or viral meningitis?
Can we reliably rule out meningitis based on a history and physical examination?
Is a lumbar puncture for cerebrospinal fluid (CSF) analysis needed? How should these studies be interpreted?
Should computed tomography of the head be done before lumbar puncture?
Which antimicrobial drugs should be started empirically at the outset?
What is the role of steroids in treatment?
CLINICAL SIGNS AND SYMPTOMS
The classic triad of meningitis is fever, neck stiffness, and altered mental status. Other signs and symptoms that have been described are photophobia, headache, nausea, vomiting, focal neurologic symptoms, altered mental status, the Kernig sign (inability to allow full knee extension when the hip is flexed to a 90° angle), and the Brudzinski sign (spontaneous flexion of the hips during attempted passive flexion of the neck).
Can meningitis be ruled out if the patient does not have this classic presentation?
Unfortunately, only a few high-quality studies of the diagnostic accuracy of signs and symptoms of bacterial meningitis have been done. Fourteen retrospective studies examined this issue, but they were heterogeneous with respect to patient age, immunosuppression status, and clinical presentation, as well as to how meningitis was diagnosed (via culture or cerebrospinal fluid analysis), making the results difficult to interpret.9 Retrospective studies are more prone to bias, as they lack a control group, and examiner bias is more likely. Based on retrospective data, the combination of fever, neck stiffness, and altered mental status has a sensitivity of only 0.46.9
Two prospective studies examined symptoms and signs. Thomas et al10 evaluated 297 patients with “clinically suspected meningitis.” Unfortunately, in this study the physical examination was not standardized. In a study by Uchihara and Tsukagoshi,11 the measurement was more reliable, as they used a single examiner to evaluate patients presenting with fever and headache, but only 54 patients were studied.
Based on these prospective studies, the presence of nausea and vomiting, headache, or neck stiffness does not reliably rule in meningitis(Table 1).9 Similarly, the absence of these does not rule it out. The 95% confidence intervals (CIs) of the positive and negative likelihood ratios include the value 1. (A simple interpretation of that would be that the likelihood of finding these features is the same in patients with meningitis when compared with those without meningitis.9)
For the physical examination, the presence or absence of fever, the Kernig sign, or the Brudzinski sign were also inconclusive. The CIs of the positive and negative likelihood ratios, like those of the symptoms, included the value 1. Only one test done on physical examination looked promising in having diagnostic utility to rule out meningitis: the jolt accentuation test (performed by asking a patient with a headache to quickly move his or her head twice horizontally; the result is positive if the headache worsens). If the result is negative, meningitis is unlikely (negative likelihood ratio 0.05, 95% CI 0.01–0.35).9 However, a positive test is not useful in making the diagnosis. A caveat is that this is based on a single study.
In summary, the history and physical examination are not sufficient to determine whether a patient has meningitis. If a patient is suspected of having meningitis, a lumbar puncture is needed.
WORKUP AND DIAGNOSTIC TESTS
Which tests are needed?
Blood cultures should be drawn before antimicrobial treatment is started.12–14 Although positive only 19% to 70% of the time, they can help identify the pathogen.15–17
Lumbar puncture with CSF study is essential to make the diagnosis and to identify the organism and its susceptibility to various antibiotics. If lumbar puncture can be performed immediately, it should be done before starting antibiotics, to maximize the yield of cultures. Pediatric studies show that after starting antibiotics, complete sterilization of the cerebrospinal fluid can occur within 2 hours for N meningitides and within 4 hours for S pneumoniae.14 However, starting antimicrobials should not be delayed if a lumbar puncture cannot be done expeditiously.
Is computed tomography of the brain necessary before a lumbar puncture?
The rationale behind performing CT before lumbar puncture is to determine if the patient has elevated intracranial pressure, which would increase the risk of brain herniation due to lowering of the lumbar CSF pressure during lumbar puncture. For ethical and practical reasons, it would be difficult to evaluate this in a randomized clinical trial.
Hasbun et al18 performed a study to evaluate if any features on clinical presentation can predict abnormal findings on CT of the head suggestive of elevated intracranial pressure and thus the risk of herniation. The study included 301 adults with suspected meningitis. It found that abnormal findings on CT were unlikely if all of the following features were absent at baseline:
Immunocompromised state
History of central nervous system disease (mass lesion, stroke, or a focal infection)
New onset of seizure (≤ 1 week from presentation)
Specific abnormal neurologic findings (eg, an abnormal level of consciousness, inability to answer two consecutive questions correctly or to follow two consecutive commands, gaze palsy, abnormal visual fields, facial palsy, arm drift, leg drift, abnormal language).
Absence of these baseline features made it unlikely that CT would be abnormal (negative likelihood ratio 0.1, 95% CI 0.03–0.31).
Adapted from Tunkel AR, et al. Practice guidelines for the management of bacterial meningitis. Clin Infec Dis 2004; 39:1267–1284, with permission from the Infectious Diseases Society of America.
Figure 2.
According to the guidelines from the Infectious Diseases Society of America (IDSA),19 if none of those features is present, blood cultures and a lumbar puncture should be done immediately, followed by empiric antimicrobial therapy. If any of the features is present, blood cultures should be obtained first, then empiric antimicrobial therapy started, followed by CT of the brain to look for contraindications to a lumbar puncture (Figure 2).
What can lumbar puncture tell us?
Results of lumbar puncture studies can help determine whether meningitis is present and, if so, whether the cause is likely bacterial or viral.20
The opening pressure is elevated (usually > 180 mm H2O) in acute bacterial meningitis. The CSF white blood cell count is usually more than 1.0 × 109/L, consisting predominantly of neutrophils, in acute bacterial meningitis. In viral meningitis, it is usually less than 0.1 × 109/L, mostly lymphocytes.
Protein shows a mild to marked elevation in bacterial meningitis but is normal to elevated in viral meningitis.
The CSF glucose level is lower in bacterial meningitis than in viral meningitis.
The ratio of CSF glucose to blood glucose. Because the glucose levels in the CSF and the blood equilibrate, the ratio of CSF glucose to serum glucose has better diagnostic accuracy than the CSF glucose level alone. The equilibration takes place within a few hours, so the serum glucose level should be ordered at the same time lumbar puncture is done. The CSF glucose-blood glucose ratio is a better predictor of bacterial meningitis than the CSF white blood cell count. Bacterial meningitis is likely if the ratio is lower than 0.4.
Lactate levels are not usually measured, but a lactate level greater than 31.5 mg/dL (3.5 mmol/L) is predictive of meningitis, and a lower level makes the diagnosis unlikely.
The diagnostic accuracies (likelihood ratios) of the CSF tests were analyzed by Straus et al.21 The positive likelihood ratios for the CSF white blood cell count and for the CSF glucose-blood glucose ratio are greater than 10, but these tests have negative likelihood ratios of more than 0.1. (It is generally thought that a test with a positive likelihood ratio of more than 10 is considered good for ruling in a diagnosis, whereas one with a negative likelihood ratio of less than 0.1 is good for ruling out a diagnosis.) Thus, these tests are good to rule in bacterial meningitis, but not as good to rule it out. There are some data to show that CSF lactate and procalcitonin might be more sensitive in ruling out bacterial meningitis, but more studies are needed.22
Gram stain of the cerebrospinal fluid can be done quickly. If no bacteria are seen, the information is not helpful in ruling out bacterial meningitis (negative likelihood ratio 0.14, 95% CI 0.08–0.27). If it is positive, it is almost 100% specific for meningitis due to the organism seen (positive likelihood ratio 735, 95% CI 230–2,295).21
MANAGEMENT
Empiric antimicrobial therapy must be started as soon as feasible
Most studies of the timing of antimicrobial drugs were retrospective and included a very heterogeneous population. They were thus more prone to bias and confounding.23,24 Proulx et al,23 in a retrospective study, found that if antibiotics were given within 6 hours of the time the patient presented to the emergency department, the case fatality rate was only 5% to 6%. If treatment started 6 to 8 hours after presentation, the death rate was 45%, and if it started from 8 to 10 hours after presentation, the death rate was 75%. Most physicians would agree that starting antimicrobials early would be beneficial.
CSF concentrations of most antimicrobial drugs are considerably less than in the serum due to poor penetration of the blood-CSF barrier. Thus, the dose for treating meningitis is usually higher than the regular dose. For example, for the treatment of pneumococcal pneumonia, ceftriaxone (Rocephin) is used at a dose of 1 g every 24 hours, but for pneumococcal meningitis the dose is 2 g every 12 hours.
Empiric treatment of community-acquired bacterial meningitis in immunocompetent adults up to 50 years of age consists of a third-generation cephalosporin such as cefotaxime (Claforan) 2 g intravenously every 4 hours or ceftriaxone 2 g intravenously every 12 hours, which covers most S pneumoniae and N meningitides strains.19 The IDSA guidelines recommend adding vancomycin (Vancocin) empirically in suspected S pneumoniae meningitis due to concerns about drug-resistant pneumococcal strains.19 For vancomycin, 45 to 60 mg/kg intravenously per day divided into every-6-hour or every-8-hour doses would achieve better CSF concentrations.25
In patients over age 50 or those with a cell-mediated immunodeficiency, empiric therapy should also include ampicillin 2 g intravenously every 4 hours to cover Listeria.
It is important to tailor therapy to the results of Gram stain, culture, and susceptibility as they become available.
Role of corticosteroids
Glucocorticoids, especially dexamethasone (Decadron), have been well studied as adjunctive therapies in bacterial meningitis. The rationale behind their use is that the profuse inflammatory response to the bacterial components in the CSF by itself has deleterious effects, and steroids can reduce that.
In 2004, a Cochrane meta-analysis26 of five randomized clinical trials, including 623 adults with bacterial meningitis (234 with pneumococcal meningitis and 232 with meningococcal meningitis), found a significant reduction in the death rate for patients who received steroids: the death rate was 12% in patients who received steroids vs 22% in those who did not (odds ratio 0.6; 95% CI 0.40–0.81). This led to an IDSA practice guideline recommendation that in adults with suspected or proven pneumococcal meningitis, dexamethasone would be beneficial.19
But since then, many more studies have emerged from Europe, South America, Malawi, and Vietnam, and another Cochrane metaanalysis27 incorporated the new studies. Twenty-four studies involving 4,041 participants were included. Similar numbers of participants died in the corticosteroid and placebo groups (18% vs 20%; risk ratio [RR] 0.92, 95% CI 0.82–1.04, P = .18). A trend towards a lower mortality rate was noticed in adults receiving corticosteroids (RR 0.74, 95% CI 0.53–1.05, P = .09). In adults, corticosteroids were associated with lower rates of hearing loss (RR 0.74, 95% CI 0.56–0.98), and there was a trend towards fewer neurologic sequelae (RR 0.72, 95% CI 0.51–1.01). The benefits were shown in studies in adults in high-income countries, but the studies from low-income countries showed neither harm nor benefit. Based on these findings, the authors recommended the use of steroids in high-income countries, though the strength of the evidence was not optimal. The recommended steroid was dexamethasone 0.15 mg/kg intravenously every 6 hours for 4 days.
References
Swartz MN. Bacterial meningitis—a view of the past 90 years. N Engl J Med2004; 351:1826–1828.
Thigpen MC, Whitney CG, Messonnier NE, et al; Emerging Infections Programs Network. Bacterial meningitis in the United States, 1998–2007. N Engl J Med2011; 364:2010–2025.
Holmquist L, Russo CA, Elixhauser A. Meningitis-related hospitalizations in the United States, 2006. Statistical Brief #57. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville, MD, 2008. www.hcup-us.ahrq.gov/reports/statbriefs/sb57.jsp. Accessed May 4, 2012.
Schuchat A, Robinson K, Wenger JD, et al. Bacterial meningitis in the United States in 1995. N Engl J Med1997; 337:970–976.
Koedel U, Scheld WM, Pfister H-W. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis2002; 2:721–736.
Hughes DC, Raghavan A, Mordekar SR, Griffiths PD, Connolly DJ. Role of imaging in the diagnosis of acute bacterial meningitis and its complications. Postgrad Med J2010; 86:478–485.
Brouwer MC, Tunkel AR, van de Beek D. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin Microbiol Rev2010; 23:467–492.
Maguire JH. Intestinal nematodes (roundworms). In:Mandell G, Bennett J, Dolin R, editors. Principles and Practice of Infectious Diseases. Philadelphia: Elsevier, 2009:3577–3586.
Attia J, Hatala R, Cook DJ, Wong JG. Original article: does this adult patient have acute meningitis?In:Simel DL, Rennie D, editors. The Rational Clinical Examinatino: Evidence-Based Clinical Diagnosis. New York, NY: McGraw-Hill; 2009.
Thomas KE, Hasbun R, Jekel J, Quagliarello VJ. The diagnostic accuracy of Kernig’s sign, Brudzinski’s sign, and nuchal rigidity in adults with suspected meningitis. Clin Infect Dis2002; 35:46–52.
Uchihara T, Tsukagoshi H. Jolt accenulation of headache: the most sensitive sign of CSF pleocytosis. Headache1991; 31:167–171.
Geiseler PJ, Nelson KE, Levin S, Reddi KT, Moses VK. Community-acquired purulent meningitis: a review of 1,316 cases during the antibiotic era, 1954–1976. Rev Infect Dis1980; 2:725–745.
Talan DA, Hoffman JR, Yoshikawa TT, Overturf GD. Role of empiric parenteral antibiotics prior to lumbar puncture in suspected bacterial meningitis: state of the art. Rev Infect Dis1988; 10:365–376.
Kanegaye JT, Soliemanzadeh P, Bradley JS. Lumbar puncture in pediatric bacterial meningitis: defining the time interval for recovery of cerebrospinal fluid pathogens after parenteral antibiotic pretreatment. Pediatrics2001; 108:1169–1174.
Sigurdardóttir B, Björnsson OM, Jónsdóttir KE, Erlendsdóttir H, Gudmundsson S. Acute bacterial meningitis in adults. A 20-year overview. Arch Intern Med1997; 157:425–430.
Aronin SI, Peduzzi P, Quagliarello VJ. Community-acquired bacterial meningitis: risk stratification for adverse clinical outcome and effect of antibiotic timing. Ann Intern Med1998; 129:862–869.
Andersen J, Backer V, Voldsgaard P, Skinhój P, Wandall JH. Acute meningococcal meningitis: analysis of features of the disease according to the age of 255 patients. Copenhagen Meningitis Study Group. J Infect1997; 34:227–235.
Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med2001; 345:1727–1733.
Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis2004; 39:1267–1284.
Seehusen DA, Reeves MM, Fomin DA. Cerebrospinal fluid analysis. Am Fam Physician2003; 68:1103–1108.
Straus SE, Thorpe KE, Holroyd-Leduc J. How do I perform a lumbar puncture and analyze the results to diagnose bacterial meningitis?JAMA2006; 296:2010–2022.
Viallon A, Desseigne N, Marjollet O, et al. Meningitis in adult patients with a negative direct cerebrospinal fluid examination: value of cytochemical markers for differential diagnosis. Crit Care2011; 15:R136.
Proulx N, Fréchette D, Toye B, Chan J, Kravcik S. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM2005; 98:291–298.
Radetsky M. Duration of symptoms and outcome in bacterial meningitis: an analysis of causation and the implications of a delay in diagnosis. Pediatr Infect Dis J1992; 11:694–698.
Ricard JD, Wolff M, Lacherade JC, et al. Levels of vancomycin in cerebrospinal fluid of adult patients receiving adjunctive corticosteroids to treat pneumococcal meningitis: a prospective multicenter observational study. Clin Infect Dis2007; 44:250–255.
van de Beek D, de Gans J, McIntyre P, Prasad K. Steroids in adults with acute bacterial meningitis: a systematic review. Lancet Infect Dis2004; 4:139–143.
van de Beek D, Farrar JJ, de Gans J, et al. Adjunctive dexamethasone in bacterial meningitis: a meta-analysis of individual patient data. Lancet Neurol2010; 9:254–263.
References
Swartz MN. Bacterial meningitis—a view of the past 90 years. N Engl J Med2004; 351:1826–1828.
Thigpen MC, Whitney CG, Messonnier NE, et al; Emerging Infections Programs Network. Bacterial meningitis in the United States, 1998–2007. N Engl J Med2011; 364:2010–2025.
Holmquist L, Russo CA, Elixhauser A. Meningitis-related hospitalizations in the United States, 2006. Statistical Brief #57. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville, MD, 2008. www.hcup-us.ahrq.gov/reports/statbriefs/sb57.jsp. Accessed May 4, 2012.
Schuchat A, Robinson K, Wenger JD, et al. Bacterial meningitis in the United States in 1995. N Engl J Med1997; 337:970–976.
Koedel U, Scheld WM, Pfister H-W. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis2002; 2:721–736.
Hughes DC, Raghavan A, Mordekar SR, Griffiths PD, Connolly DJ. Role of imaging in the diagnosis of acute bacterial meningitis and its complications. Postgrad Med J2010; 86:478–485.
Brouwer MC, Tunkel AR, van de Beek D. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin Microbiol Rev2010; 23:467–492.
Maguire JH. Intestinal nematodes (roundworms). In:Mandell G, Bennett J, Dolin R, editors. Principles and Practice of Infectious Diseases. Philadelphia: Elsevier, 2009:3577–3586.
Attia J, Hatala R, Cook DJ, Wong JG. Original article: does this adult patient have acute meningitis?In:Simel DL, Rennie D, editors. The Rational Clinical Examinatino: Evidence-Based Clinical Diagnosis. New York, NY: McGraw-Hill; 2009.
Thomas KE, Hasbun R, Jekel J, Quagliarello VJ. The diagnostic accuracy of Kernig’s sign, Brudzinski’s sign, and nuchal rigidity in adults with suspected meningitis. Clin Infect Dis2002; 35:46–52.
Uchihara T, Tsukagoshi H. Jolt accenulation of headache: the most sensitive sign of CSF pleocytosis. Headache1991; 31:167–171.
Geiseler PJ, Nelson KE, Levin S, Reddi KT, Moses VK. Community-acquired purulent meningitis: a review of 1,316 cases during the antibiotic era, 1954–1976. Rev Infect Dis1980; 2:725–745.
Talan DA, Hoffman JR, Yoshikawa TT, Overturf GD. Role of empiric parenteral antibiotics prior to lumbar puncture in suspected bacterial meningitis: state of the art. Rev Infect Dis1988; 10:365–376.
Kanegaye JT, Soliemanzadeh P, Bradley JS. Lumbar puncture in pediatric bacterial meningitis: defining the time interval for recovery of cerebrospinal fluid pathogens after parenteral antibiotic pretreatment. Pediatrics2001; 108:1169–1174.
Sigurdardóttir B, Björnsson OM, Jónsdóttir KE, Erlendsdóttir H, Gudmundsson S. Acute bacterial meningitis in adults. A 20-year overview. Arch Intern Med1997; 157:425–430.
Aronin SI, Peduzzi P, Quagliarello VJ. Community-acquired bacterial meningitis: risk stratification for adverse clinical outcome and effect of antibiotic timing. Ann Intern Med1998; 129:862–869.
Andersen J, Backer V, Voldsgaard P, Skinhój P, Wandall JH. Acute meningococcal meningitis: analysis of features of the disease according to the age of 255 patients. Copenhagen Meningitis Study Group. J Infect1997; 34:227–235.
Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med2001; 345:1727–1733.
Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis2004; 39:1267–1284.
Seehusen DA, Reeves MM, Fomin DA. Cerebrospinal fluid analysis. Am Fam Physician2003; 68:1103–1108.
Straus SE, Thorpe KE, Holroyd-Leduc J. How do I perform a lumbar puncture and analyze the results to diagnose bacterial meningitis?JAMA2006; 296:2010–2022.
Viallon A, Desseigne N, Marjollet O, et al. Meningitis in adult patients with a negative direct cerebrospinal fluid examination: value of cytochemical markers for differential diagnosis. Crit Care2011; 15:R136.
Proulx N, Fréchette D, Toye B, Chan J, Kravcik S. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM2005; 98:291–298.
Radetsky M. Duration of symptoms and outcome in bacterial meningitis: an analysis of causation and the implications of a delay in diagnosis. Pediatr Infect Dis J1992; 11:694–698.
Ricard JD, Wolff M, Lacherade JC, et al. Levels of vancomycin in cerebrospinal fluid of adult patients receiving adjunctive corticosteroids to treat pneumococcal meningitis: a prospective multicenter observational study. Clin Infect Dis2007; 44:250–255.
van de Beek D, de Gans J, McIntyre P, Prasad K. Steroids in adults with acute bacterial meningitis: a systematic review. Lancet Infect Dis2004; 4:139–143.
van de Beek D, Farrar JJ, de Gans J, et al. Adjunctive dexamethasone in bacterial meningitis: a meta-analysis of individual patient data. Lancet Neurol2010; 9:254–263.
The most common organisms that cause community-acquired bacterial meningitis are Streptococcus pneumoniae and Neisseria meningitidis. The incidence of Listeria infection increases in patients over age 50 and in those with compromised cell-mediated immunity.
Symptoms and signs are not sensitive or specific enough to diagnose community-acquired bacterial meningitis. A lumbar puncture for cerebrospinal fluid studies is needed to reach the diagnosis, to identify the organism, and to determine antimicrobial susceptibilities.
Gram stain of cerebrospinal fluid may quickly identify the causative organism. It is not very sensitive, but it is specific.
Lumbar puncture should be performed as soon as possible. Computed tomography of the head is not necessary in all patients, only in immunocompromised patients and those who have features suggestive of or who are at risk of increased intracranial pressure.
Try to obtain blood and cerebrospinal fluid cultures before staring antimicrobial therapy, but do not delay therapy if obtaining them is not feasible.
Disallow All Ads
Alternative CME
Consolidated Pubs: Do Not Show Source Publication Logo
A 72-year-old man presented with abdominal cramping, diarrhea, intermittent flushing, asthenia, and a weight loss of 10 kg (22 lb) in the past 6 months. Physical examination revealed hepatosplenomegaly and an erythematous, maculopapular, confluent rash on the trunk (Figure 1) that displayed the Darier sign (redness, swelling, and itching in response to stroking in the involved area).
Laboratory analyses
Hemoglobin 9.8 g/dL (normal 13–17 g/dL)
White blood cell count 22.9 × 109/L (3.8–10)
Vitamin B12 1,730 pg/mL (220–900)
Serum tryptase 516 μg/L (5.5–13.5)
Beta-2 microglobulin 4.14 mg/L (1.39–2.11).
Radiologic evaluation
Figure 2. Radiologic evaluation showed diffuse osteosclerosis together with lytic and blastic areas (arrows).
Radiologic evaluation showed diffuse osteosclerosis with lytic and blastic areas (Figure 2).
Q: Which is the most likely diagnosis?
Carcinoid syndrome
Histiocytosis
Acute myeloblastic leukemia
Systemic mastocytosis
Chronic myeloblastic leukemia
A: The correct answer is systemic mastocytosis. The diagnosis was made according to the World Health Organization (WHO) diagnostic criteria for mastocytosis on the basis of the following findings in the bone marrow:
Figure 3. Bone marrow smear demonstrating increased numbers of abnormal mast cells (May-Grünwald-Giemsa stain, x 600).Morphologically abnormal mast cells characterized by large size, spindle shape and poorly granulated cytoplasm (Figure 3) together with criteria for refractory cytopenia and multilineage dysplasia
Diffuse infiltration by tryptase-positive mast cells as assessed by immunohistochemical study (Figure 4)
Figure 4. Bone marrow study demonstrating a massive infiltrate of abnormal mast cells (tryptase stain, x 200).One percent of mast cells that are immunophenotypically aberrant (CD25bright+), all of them showing an immature profile,1 associated with features of multilineage dysplasia2 as assessed by flow cytometry
The activating D816V KIT mutation, detected by peptide nucleic acid-mediated polymerase chain reaction clamping technique.3
MASTOCYTOSIS HAS SEVEN VARIANTS
Mastocytosis is a rare heterogeneous group of disorders characterized by proliferation and accumulation of abnormal mast cells in diverse organs and tissues, such as the skin, bone marrow, gastrointestinal tract, liver, spleen, or lymph nodes.4–6 The release of mast cell mediators causes a wide variety of symptoms, ranging from pruritus, flushing, abdominal cramping, and diarrhea to severe anaphylaxis with vascular collapse.7,8
The WHO defines seven variants6:
Cutaneous mastocytosis
Indolent systemic mastocytosis
Systemic mastocytosis with an associated (clonal) hematologic non-mast-cell disease (SM-AHNMD)
Aggressive systemic mastocytosis
Mast cell leukemia
Mast cell sarcoma
Extracutaneous mastocytoma.6
KIT mutation as a diagnostic criterion and prognostic factor
In most cases of systemic involvement, the clonal nature of the disease can be established by finding activating mutations of KIT, usually D816V, in lesions in the skin, bone marrow cells, or both.9 Apart from its value as a diagnostic criterion for systemic mastocytosis, KIT mutation has been reported to be strongly associated with progression of indolent systemic mastocytosis, including the development of myeloid malignancies, when the mutation is detected not only in mast cells but in all hematopoietic lineages.10
In cases of SM-AHNMD, a possible pathophysiologic relationship between the disorder in the mast cells and the disorder in other cells could be explained by a KIT mutation in early hematopoietic progenitor cells, which further evolve into phenotypically different subclones.
A rational management plan for mastocytosis must include carefully counselling the patient and care providers, avoiding factors that trigger acute release of mast cell mediators, and giving antimediator therapy such as oral cromolyn sodium (Gastrocrom), antihistamines, and leukotriene antagonists to relieve the symptoms caused by mast-cell-mediator release.11 In cases of SM-AHNMD, the clinical course and long-term prognosis are usually dominated by the concomitant hematologic malignancy, which should be treated as a separate entity.
CASE CONTINUED
Our patient’s bone marrow was analyzed for the KIT mutation in highly purified bone marrow cell subpopulations sorted by fluorescence-activated cell sorting. The mutation was detected in his mast cells, CD34+ cells, eosinophils, monocytes, neutrophils, lymphocytes, and nucleated erythroid precursors. According to the WHO recommendations, he had SMAHNMD, the associated hematologic disease being a myelodysplastic syndrome.
In view of his advanced age and concomitant myelodysplastic syndrome presenting with leukocytosis, we gave him hydroxyurea (Droxia; available in Spain as Hydrea) rather than other cytoreductive drugs as the first-line therapy. Additionally, we gave him corticosteroids in low doses, sodium cromolyn, and antihistamines to treat mastocytosis-related gastrointestinal symptoms. The patient was alive with stable disease 14 months after starting therapy.
References
Teodosio C, García-Montero AC, Jara-Acevedo M, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol2010; 125:719–726.
van de Loosdrecht AA, Alhan C, Béné MC, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica2009; 94:1124–1134.
Sotlar K, Escribano L, Landt O, et al. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol2003; 162:737–746.
Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res2001; 25:603–625.
Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest2007; 37:435–453.
Horny HP, Metcalfe DD, Bennet JM, et al. Mastocytosis. In:Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:54–63.
Castells M. Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin North Am2006; 26:465–485.
González de Olano D, de la Hoz Caballer B, Núñez López R, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy2007; 37:1547–1555.
Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood2006; 108:2366–2372.
Escribano L, Alvarez-Twose I, Sánchez-Muñoz L, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol2009; 124:514–521.
Escribano L, Akin C, Castells M, Schwartz LB. Current options in the treatment of mast cell mediator-related symptoms in mastocytosis. Inflamm Allergy Drug Targets2006; 5:61–77.
A 72-year-old man presented with abdominal cramping, diarrhea, intermittent flushing, asthenia, and a weight loss of 10 kg (22 lb) in the past 6 months. Physical examination revealed hepatosplenomegaly and an erythematous, maculopapular, confluent rash on the trunk (Figure 1) that displayed the Darier sign (redness, swelling, and itching in response to stroking in the involved area).
Laboratory analyses
Hemoglobin 9.8 g/dL (normal 13–17 g/dL)
White blood cell count 22.9 × 109/L (3.8–10)
Vitamin B12 1,730 pg/mL (220–900)
Serum tryptase 516 μg/L (5.5–13.5)
Beta-2 microglobulin 4.14 mg/L (1.39–2.11).
Radiologic evaluation
Figure 2. Radiologic evaluation showed diffuse osteosclerosis together with lytic and blastic areas (arrows).
Radiologic evaluation showed diffuse osteosclerosis with lytic and blastic areas (Figure 2).
Q: Which is the most likely diagnosis?
Carcinoid syndrome
Histiocytosis
Acute myeloblastic leukemia
Systemic mastocytosis
Chronic myeloblastic leukemia
A: The correct answer is systemic mastocytosis. The diagnosis was made according to the World Health Organization (WHO) diagnostic criteria for mastocytosis on the basis of the following findings in the bone marrow:
Figure 3. Bone marrow smear demonstrating increased numbers of abnormal mast cells (May-Grünwald-Giemsa stain, x 600).Morphologically abnormal mast cells characterized by large size, spindle shape and poorly granulated cytoplasm (Figure 3) together with criteria for refractory cytopenia and multilineage dysplasia
Diffuse infiltration by tryptase-positive mast cells as assessed by immunohistochemical study (Figure 4)
Figure 4. Bone marrow study demonstrating a massive infiltrate of abnormal mast cells (tryptase stain, x 200).One percent of mast cells that are immunophenotypically aberrant (CD25bright+), all of them showing an immature profile,1 associated with features of multilineage dysplasia2 as assessed by flow cytometry
The activating D816V KIT mutation, detected by peptide nucleic acid-mediated polymerase chain reaction clamping technique.3
MASTOCYTOSIS HAS SEVEN VARIANTS
Mastocytosis is a rare heterogeneous group of disorders characterized by proliferation and accumulation of abnormal mast cells in diverse organs and tissues, such as the skin, bone marrow, gastrointestinal tract, liver, spleen, or lymph nodes.4–6 The release of mast cell mediators causes a wide variety of symptoms, ranging from pruritus, flushing, abdominal cramping, and diarrhea to severe anaphylaxis with vascular collapse.7,8
The WHO defines seven variants6:
Cutaneous mastocytosis
Indolent systemic mastocytosis
Systemic mastocytosis with an associated (clonal) hematologic non-mast-cell disease (SM-AHNMD)
Aggressive systemic mastocytosis
Mast cell leukemia
Mast cell sarcoma
Extracutaneous mastocytoma.6
KIT mutation as a diagnostic criterion and prognostic factor
In most cases of systemic involvement, the clonal nature of the disease can be established by finding activating mutations of KIT, usually D816V, in lesions in the skin, bone marrow cells, or both.9 Apart from its value as a diagnostic criterion for systemic mastocytosis, KIT mutation has been reported to be strongly associated with progression of indolent systemic mastocytosis, including the development of myeloid malignancies, when the mutation is detected not only in mast cells but in all hematopoietic lineages.10
In cases of SM-AHNMD, a possible pathophysiologic relationship between the disorder in the mast cells and the disorder in other cells could be explained by a KIT mutation in early hematopoietic progenitor cells, which further evolve into phenotypically different subclones.
A rational management plan for mastocytosis must include carefully counselling the patient and care providers, avoiding factors that trigger acute release of mast cell mediators, and giving antimediator therapy such as oral cromolyn sodium (Gastrocrom), antihistamines, and leukotriene antagonists to relieve the symptoms caused by mast-cell-mediator release.11 In cases of SM-AHNMD, the clinical course and long-term prognosis are usually dominated by the concomitant hematologic malignancy, which should be treated as a separate entity.
CASE CONTINUED
Our patient’s bone marrow was analyzed for the KIT mutation in highly purified bone marrow cell subpopulations sorted by fluorescence-activated cell sorting. The mutation was detected in his mast cells, CD34+ cells, eosinophils, monocytes, neutrophils, lymphocytes, and nucleated erythroid precursors. According to the WHO recommendations, he had SMAHNMD, the associated hematologic disease being a myelodysplastic syndrome.
In view of his advanced age and concomitant myelodysplastic syndrome presenting with leukocytosis, we gave him hydroxyurea (Droxia; available in Spain as Hydrea) rather than other cytoreductive drugs as the first-line therapy. Additionally, we gave him corticosteroids in low doses, sodium cromolyn, and antihistamines to treat mastocytosis-related gastrointestinal symptoms. The patient was alive with stable disease 14 months after starting therapy.
A 72-year-old man presented with abdominal cramping, diarrhea, intermittent flushing, asthenia, and a weight loss of 10 kg (22 lb) in the past 6 months. Physical examination revealed hepatosplenomegaly and an erythematous, maculopapular, confluent rash on the trunk (Figure 1) that displayed the Darier sign (redness, swelling, and itching in response to stroking in the involved area).
Laboratory analyses
Hemoglobin 9.8 g/dL (normal 13–17 g/dL)
White blood cell count 22.9 × 109/L (3.8–10)
Vitamin B12 1,730 pg/mL (220–900)
Serum tryptase 516 μg/L (5.5–13.5)
Beta-2 microglobulin 4.14 mg/L (1.39–2.11).
Radiologic evaluation
Figure 2. Radiologic evaluation showed diffuse osteosclerosis together with lytic and blastic areas (arrows).
Radiologic evaluation showed diffuse osteosclerosis with lytic and blastic areas (Figure 2).
Q: Which is the most likely diagnosis?
Carcinoid syndrome
Histiocytosis
Acute myeloblastic leukemia
Systemic mastocytosis
Chronic myeloblastic leukemia
A: The correct answer is systemic mastocytosis. The diagnosis was made according to the World Health Organization (WHO) diagnostic criteria for mastocytosis on the basis of the following findings in the bone marrow:
Figure 3. Bone marrow smear demonstrating increased numbers of abnormal mast cells (May-Grünwald-Giemsa stain, x 600).Morphologically abnormal mast cells characterized by large size, spindle shape and poorly granulated cytoplasm (Figure 3) together with criteria for refractory cytopenia and multilineage dysplasia
Diffuse infiltration by tryptase-positive mast cells as assessed by immunohistochemical study (Figure 4)
Figure 4. Bone marrow study demonstrating a massive infiltrate of abnormal mast cells (tryptase stain, x 200).One percent of mast cells that are immunophenotypically aberrant (CD25bright+), all of them showing an immature profile,1 associated with features of multilineage dysplasia2 as assessed by flow cytometry
The activating D816V KIT mutation, detected by peptide nucleic acid-mediated polymerase chain reaction clamping technique.3
MASTOCYTOSIS HAS SEVEN VARIANTS
Mastocytosis is a rare heterogeneous group of disorders characterized by proliferation and accumulation of abnormal mast cells in diverse organs and tissues, such as the skin, bone marrow, gastrointestinal tract, liver, spleen, or lymph nodes.4–6 The release of mast cell mediators causes a wide variety of symptoms, ranging from pruritus, flushing, abdominal cramping, and diarrhea to severe anaphylaxis with vascular collapse.7,8
The WHO defines seven variants6:
Cutaneous mastocytosis
Indolent systemic mastocytosis
Systemic mastocytosis with an associated (clonal) hematologic non-mast-cell disease (SM-AHNMD)
Aggressive systemic mastocytosis
Mast cell leukemia
Mast cell sarcoma
Extracutaneous mastocytoma.6
KIT mutation as a diagnostic criterion and prognostic factor
In most cases of systemic involvement, the clonal nature of the disease can be established by finding activating mutations of KIT, usually D816V, in lesions in the skin, bone marrow cells, or both.9 Apart from its value as a diagnostic criterion for systemic mastocytosis, KIT mutation has been reported to be strongly associated with progression of indolent systemic mastocytosis, including the development of myeloid malignancies, when the mutation is detected not only in mast cells but in all hematopoietic lineages.10
In cases of SM-AHNMD, a possible pathophysiologic relationship between the disorder in the mast cells and the disorder in other cells could be explained by a KIT mutation in early hematopoietic progenitor cells, which further evolve into phenotypically different subclones.
A rational management plan for mastocytosis must include carefully counselling the patient and care providers, avoiding factors that trigger acute release of mast cell mediators, and giving antimediator therapy such as oral cromolyn sodium (Gastrocrom), antihistamines, and leukotriene antagonists to relieve the symptoms caused by mast-cell-mediator release.11 In cases of SM-AHNMD, the clinical course and long-term prognosis are usually dominated by the concomitant hematologic malignancy, which should be treated as a separate entity.
CASE CONTINUED
Our patient’s bone marrow was analyzed for the KIT mutation in highly purified bone marrow cell subpopulations sorted by fluorescence-activated cell sorting. The mutation was detected in his mast cells, CD34+ cells, eosinophils, monocytes, neutrophils, lymphocytes, and nucleated erythroid precursors. According to the WHO recommendations, he had SMAHNMD, the associated hematologic disease being a myelodysplastic syndrome.
In view of his advanced age and concomitant myelodysplastic syndrome presenting with leukocytosis, we gave him hydroxyurea (Droxia; available in Spain as Hydrea) rather than other cytoreductive drugs as the first-line therapy. Additionally, we gave him corticosteroids in low doses, sodium cromolyn, and antihistamines to treat mastocytosis-related gastrointestinal symptoms. The patient was alive with stable disease 14 months after starting therapy.
References
Teodosio C, García-Montero AC, Jara-Acevedo M, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol2010; 125:719–726.
van de Loosdrecht AA, Alhan C, Béné MC, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica2009; 94:1124–1134.
Sotlar K, Escribano L, Landt O, et al. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol2003; 162:737–746.
Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res2001; 25:603–625.
Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest2007; 37:435–453.
Horny HP, Metcalfe DD, Bennet JM, et al. Mastocytosis. In:Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:54–63.
Castells M. Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin North Am2006; 26:465–485.
González de Olano D, de la Hoz Caballer B, Núñez López R, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy2007; 37:1547–1555.
Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood2006; 108:2366–2372.
Escribano L, Alvarez-Twose I, Sánchez-Muñoz L, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol2009; 124:514–521.
Escribano L, Akin C, Castells M, Schwartz LB. Current options in the treatment of mast cell mediator-related symptoms in mastocytosis. Inflamm Allergy Drug Targets2006; 5:61–77.
References
Teodosio C, García-Montero AC, Jara-Acevedo M, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol2010; 125:719–726.
van de Loosdrecht AA, Alhan C, Béné MC, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica2009; 94:1124–1134.
Sotlar K, Escribano L, Landt O, et al. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol2003; 162:737–746.
Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res2001; 25:603–625.
Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest2007; 37:435–453.
Horny HP, Metcalfe DD, Bennet JM, et al. Mastocytosis. In:Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:54–63.
Castells M. Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin North Am2006; 26:465–485.
González de Olano D, de la Hoz Caballer B, Núñez López R, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy2007; 37:1547–1555.
Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood2006; 108:2366–2372.
Escribano L, Alvarez-Twose I, Sánchez-Muñoz L, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol2009; 124:514–521.
Escribano L, Akin C, Castells M, Schwartz LB. Current options in the treatment of mast cell mediator-related symptoms in mastocytosis. Inflamm Allergy Drug Targets2006; 5:61–77.
Measures of cost-effectiveness are used to compare the merits of diverse medical interventions. A novel drug for metastatic melanoma, for instance, can be compared with statin therapy for primary prevention of cardiovascular events, which in turn can be compared against a surgical procedure for pain, as all are described by a single number: dollars per life-year (or quality-adjusted life-year) gained. Presumably, this number tells practitioners and payers which interventions provide the most benefit for every dollar spent.
However, too often, studies of cost-effectiveness differ from one another. They can be based on data from different types of studies, such as randomized controlled trials, surveys of large payer databases, or single-center chart reviews. The comparison treatments may differ. And the treatments may be of unproven efficacy. In these cases, although the results are all expressed in dollars per life-year, we are comparing apples and oranges.
In the following discussion, I use three key contemporary examples to demonstrate problems central to cost-effectiveness analysis. Together, these examples show that cost-effectiveness, arguably our best tool for comparing apples and oranges, is a lot like apples and oranges itself. I conclude by proposing some solutions.
PROBLEMS WITH COST-EFFECTIVENESS: THREE EXAMPLES
Studies of three therapies highlight the dilemma of cost-effectiveness.
Example 1: Vertebroplasty
Studies of vertebroplasty, a treatment for osteoporotic vertebral fractures that involves injecting polymethylmethacrylate cement into the fractured bone, show the perils of calculating the cost-effectiveness of unproven therapies.
Vertebroplasty gained prominence during the first decade of the 2000s, but in 2009 it was found to be no better than a sham procedure.1,2
In 2008, one study reported that vertebroplasty was cheaper than medical management at 12 months and, thus, cost-effective.3 While this finding was certainly true for the regimen of medical management the authors examined, and while it may very well be true for other protocols for medical management, the finding obscures the fact that a sham procedure would be more cost-effective than either vertebroplasty or medical therapy—an unsettling conclusion.
Example 2: Exemestane
Another dilemma occurs when we can calculate cost-effectiveness for a particular outcome only.
Studies of exemestane (Aromasin), an aromatase inhibitor given to prevent breast cancer, show the difficulty. Recently, exemestane was shown to decrease the rate of breast cancer when used as primary prevention in postmenopausal women.4 What is the costeffectiveness of this therapy?
While we can calculate the dollars per invasive breast cancer averted, we cannot accurately calculate the dollars per life-year gained, as the trial’s end point was not the mortality rate. We can assume that the breast cancer deaths avoided are not negated by deaths incurred through other causes, but this may or may not prove true. Fibrates, for instance, may reduce the rate of cardiovascular death but increase deaths from noncardiac causes, providing no net benefit.5 Such long-term effects remain unknown in the breast cancer study.
Example 3: COX-2 inhibitors
Estimates of cost-effectiveness derived from randomized trials can differ from those derived from real-world studies. Studies of cyclooxygenase 2 (COX-2) inhibitors, which were touted as causing less gastrointestinal bleeding than other nonsteroidal anti-inflammatory drugs, show that cost-effectiveness analyses performed from randomized trials may not mirror dollars spent in real-world practice.
Estimates from randomized controlled trials indicate that a COX-2 inhibitor such as celecoxib (Celebrex) costs $20,000 to prevent one gastrointestinal hemorrhage. However, when calculated using real-world data, that number rises to over $100,000.6
TWO PROPOSED RULES FOR COST-EFFECTIVENESS ANALYSES
How do we reconcile these and related puzzles of cost-effectiveness? First, we should agree on what type of “cost-effectiveness” we are interested in. Most often, we want to know whether the real-world use of a therapy is financially rational. Thus, we are concerned with the effectiveness of therapies and not merely their efficacy in idealized clinical trials.
Furthermore, while real-world cost-effectiveness may change over time, particularly as pricing and delivery vary, we want some assurance that the therapy is truly better than placebo. Therefore, we should only calculate the cost-effectiveness of therapies that have previously demonstrated efficacy in properly controlled, randomized studies.7
To correct the deficiencies noted here, I propose two rules:
Cost-effectiveness should be calculated only for therapies that have been proven to work, and
These calculations should be done from the best available real-world data.
When both these conditions are met—ie, a therapy has proven efficacy, and we have data from its real-world use—cost-effectiveness analysis provides useful information for payers and practitioners. Then, indeed, a novel anticancer agent costing $30,000 per life-year gained can be compared against primary prevention with statin therapy in patients at elevated cardiovascular risk costing $20,000 per life-year gained.
CAN PREVENTION BE COMPARED WITH TREATMENT?
This leaves us with the final and most difficult question. Is it right to compare such things?
Having terminal cancer is a different experience than having high cholesterol, and this is the last apple and orange of cost-effectiveness. While a strict utilitarian view of medicine might find these cases indistinguishable, most practitioners and payers are not strict utilitarians. As a society, we tend to favor paying more to treat someone who is ill than paying an equivalent amount to prevent illness. Often, such a stance is criticized as a failure to invest in prevention and primary care, but another explanation is that the bias is a fundamental one of human risk-taking.
Cost-effectiveness is, to a certain degree, a slippery concept, and it is more likely to be “off” when a therapy is given broadly (to hundreds of thousands of people as opposed to hundreds) and taken in a decentralized fashion by individual patients (as opposed to directly observed therapy in an infusion suite). Accordingly, we may favor more expensive therapies, the cost-effectiveness of which can be estimated more precisely.
A recent meta-analysis of statins for primary prevention in high-risk patients found that they were not associated with improvement in the overall rate of death.8 Such a finding dramatically alters our impression of their cost-effectiveness and may explain the bias against investing in such therapies in the first place.
IMPROVING COST-EFFECTIVENESS RESEARCH
Studies of cost-effectiveness are not equivalent. Currently, such studies are apples and oranges, making difficult the very comparison that cost-effectiveness should facilitate. Knowing that a therapy is efficacious should be prerequisite to cost-effectiveness calculations, as should performing calculations under real-world conditions.
Regarding efficacy, it is inappropriate to calculate cost-effectiveness from trials that use only surrogate end points, or those that are improperly controlled.
For example, adding extended-release niacin to statin therapy may raise high-density lipoprotein cholesterol levels by 25%. Such an increase is, in turn, expected to confer a certain reduction in cardiovascular events and death. Thus, the cost-effectiveness of niacin might be calculated as $20,000 per life-year saved. However, adding extended-release niacin to statin therapy does not improve hard outcomes when directly measured,9 and the therapy is not efficacious at all. Its true “dollars per life-year saved” approaches infinity.
Studies that use historical controls, are observational, and are performed at single centers may also mislead us regarding a therapy’s efficacy. Tight glycemic control in intensive care patients initially seemed promising10,11 and cost-effective.12 However, several years later it was found to increase the mortality rate.13
“Real world” means that the best measures of cost-effectiveness will calculate the cost per life saved that the therapy achieves in clinical practice. Adherence to COX-2 inhibitors may not be as strict in the real world as it is in the carefully selected participants in randomized controlled trials, and, thus, the true costs may be higher. A drug that prevents breast cancer may have countervailing effects that may as yet be unknown, or compliance with it may wane over years. Thus, the most accurate measures of cost-effectiveness will examine therapies as best as they can function in typical practice and likely be derived from data sets of large payers or providers.
Finally, it remains an open and contentious issue whether the cost-effectiveness of primary prevention and the cost-effectiveness of treatment are comparable at all. We must continue to ponder and debate this philosophical question.
Certainly, these are the challenges of cost-effectiveness. Equally certain is that—with renewed consideration of the goals of such research, with stricter standards for future studies, and in an economic and political climate unable to sustain the status quo—the challenges must be surmounted.
References
Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med2009; 361:569–579.
Buchbinder R, Osborne RH, Ebeling PR, et al. A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med2009; 361:557–568.
Masala S, Ciarrapico AM, Konda D, Vinicola V, Mammucari M, Simonetti G. Cost-effectiveness of percutaneous vertebroplasty in osteoporotic vertebral fractures. Eur Spine J2008; 17:1242–1250.
Goss PE, Ingle JN, Alés-Martínez JE, et al; NCIC CTG MAP3 Study Investigators. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med2011; 364:2381–2391.
Studer M, Briel M, Leimenstoll B, Glass TR, Bucher HC. Effect of different antilipidemic agents and diets on mortality: a systematic review. Arch Intern Med2005; 165:725–730.
van Staa TP, Leufkens HG, Zhang B, Smeeth L. A comparison of cost effectiveness using data from randomized trials or actual clinical practice: selective cox-2 inhibitors as an example. PLoS Med2009; 6:e1000194.
Prasad V, Cifu A. A medical burden of proof: towards a new ethic. BioSocieties2012; 7:72–87.
Ray KK, Seshasai SR, Erqou S, et al. Statins and all-cause mortality in high-risk primary prevention: a meta-analysis of 11 randomized controlled trials involving 65,229 participants. Arch Intern Med2010; 170:1024–1031.
National Heart, Lung, and Blood Institute National Institutes of Health. AIM-HIGH: Blinded treatment phase of study stopped. http://www.aimhigh-heart.com/. Accessed January 31, 2012.
van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med2001; 345:1359–1367.
Van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med2006; 354:449–461.
Mesotten D, Van den Berghe G. Clinical potential of insulin therapy in critically ill patients. Drugs2003; 63:625–636.
NICE-SUGAR Study Investigators; Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med2009; 360:1283–1297.
Vinay Prasad, MD Department of Medicine, Northwestern University, Chicago, IL
Address: Vinay Prasad, MD, Deparment of Medicine, Northwestern University, 333 E. Ontario, Suite 901B, Chicago, IL 60611; e-mail v-prasad@md.northwestern.edu
Vinay Prasad, MD Department of Medicine, Northwestern University, Chicago, IL
Address: Vinay Prasad, MD, Deparment of Medicine, Northwestern University, 333 E. Ontario, Suite 901B, Chicago, IL 60611; e-mail v-prasad@md.northwestern.edu
Author and Disclosure Information
Vinay Prasad, MD Department of Medicine, Northwestern University, Chicago, IL
Address: Vinay Prasad, MD, Deparment of Medicine, Northwestern University, 333 E. Ontario, Suite 901B, Chicago, IL 60611; e-mail v-prasad@md.northwestern.edu
Measures of cost-effectiveness are used to compare the merits of diverse medical interventions. A novel drug for metastatic melanoma, for instance, can be compared with statin therapy for primary prevention of cardiovascular events, which in turn can be compared against a surgical procedure for pain, as all are described by a single number: dollars per life-year (or quality-adjusted life-year) gained. Presumably, this number tells practitioners and payers which interventions provide the most benefit for every dollar spent.
However, too often, studies of cost-effectiveness differ from one another. They can be based on data from different types of studies, such as randomized controlled trials, surveys of large payer databases, or single-center chart reviews. The comparison treatments may differ. And the treatments may be of unproven efficacy. In these cases, although the results are all expressed in dollars per life-year, we are comparing apples and oranges.
In the following discussion, I use three key contemporary examples to demonstrate problems central to cost-effectiveness analysis. Together, these examples show that cost-effectiveness, arguably our best tool for comparing apples and oranges, is a lot like apples and oranges itself. I conclude by proposing some solutions.
PROBLEMS WITH COST-EFFECTIVENESS: THREE EXAMPLES
Studies of three therapies highlight the dilemma of cost-effectiveness.
Example 1: Vertebroplasty
Studies of vertebroplasty, a treatment for osteoporotic vertebral fractures that involves injecting polymethylmethacrylate cement into the fractured bone, show the perils of calculating the cost-effectiveness of unproven therapies.
Vertebroplasty gained prominence during the first decade of the 2000s, but in 2009 it was found to be no better than a sham procedure.1,2
In 2008, one study reported that vertebroplasty was cheaper than medical management at 12 months and, thus, cost-effective.3 While this finding was certainly true for the regimen of medical management the authors examined, and while it may very well be true for other protocols for medical management, the finding obscures the fact that a sham procedure would be more cost-effective than either vertebroplasty or medical therapy—an unsettling conclusion.
Example 2: Exemestane
Another dilemma occurs when we can calculate cost-effectiveness for a particular outcome only.
Studies of exemestane (Aromasin), an aromatase inhibitor given to prevent breast cancer, show the difficulty. Recently, exemestane was shown to decrease the rate of breast cancer when used as primary prevention in postmenopausal women.4 What is the costeffectiveness of this therapy?
While we can calculate the dollars per invasive breast cancer averted, we cannot accurately calculate the dollars per life-year gained, as the trial’s end point was not the mortality rate. We can assume that the breast cancer deaths avoided are not negated by deaths incurred through other causes, but this may or may not prove true. Fibrates, for instance, may reduce the rate of cardiovascular death but increase deaths from noncardiac causes, providing no net benefit.5 Such long-term effects remain unknown in the breast cancer study.
Example 3: COX-2 inhibitors
Estimates of cost-effectiveness derived from randomized trials can differ from those derived from real-world studies. Studies of cyclooxygenase 2 (COX-2) inhibitors, which were touted as causing less gastrointestinal bleeding than other nonsteroidal anti-inflammatory drugs, show that cost-effectiveness analyses performed from randomized trials may not mirror dollars spent in real-world practice.
Estimates from randomized controlled trials indicate that a COX-2 inhibitor such as celecoxib (Celebrex) costs $20,000 to prevent one gastrointestinal hemorrhage. However, when calculated using real-world data, that number rises to over $100,000.6
TWO PROPOSED RULES FOR COST-EFFECTIVENESS ANALYSES
How do we reconcile these and related puzzles of cost-effectiveness? First, we should agree on what type of “cost-effectiveness” we are interested in. Most often, we want to know whether the real-world use of a therapy is financially rational. Thus, we are concerned with the effectiveness of therapies and not merely their efficacy in idealized clinical trials.
Furthermore, while real-world cost-effectiveness may change over time, particularly as pricing and delivery vary, we want some assurance that the therapy is truly better than placebo. Therefore, we should only calculate the cost-effectiveness of therapies that have previously demonstrated efficacy in properly controlled, randomized studies.7
To correct the deficiencies noted here, I propose two rules:
Cost-effectiveness should be calculated only for therapies that have been proven to work, and
These calculations should be done from the best available real-world data.
When both these conditions are met—ie, a therapy has proven efficacy, and we have data from its real-world use—cost-effectiveness analysis provides useful information for payers and practitioners. Then, indeed, a novel anticancer agent costing $30,000 per life-year gained can be compared against primary prevention with statin therapy in patients at elevated cardiovascular risk costing $20,000 per life-year gained.
CAN PREVENTION BE COMPARED WITH TREATMENT?
This leaves us with the final and most difficult question. Is it right to compare such things?
Having terminal cancer is a different experience than having high cholesterol, and this is the last apple and orange of cost-effectiveness. While a strict utilitarian view of medicine might find these cases indistinguishable, most practitioners and payers are not strict utilitarians. As a society, we tend to favor paying more to treat someone who is ill than paying an equivalent amount to prevent illness. Often, such a stance is criticized as a failure to invest in prevention and primary care, but another explanation is that the bias is a fundamental one of human risk-taking.
Cost-effectiveness is, to a certain degree, a slippery concept, and it is more likely to be “off” when a therapy is given broadly (to hundreds of thousands of people as opposed to hundreds) and taken in a decentralized fashion by individual patients (as opposed to directly observed therapy in an infusion suite). Accordingly, we may favor more expensive therapies, the cost-effectiveness of which can be estimated more precisely.
A recent meta-analysis of statins for primary prevention in high-risk patients found that they were not associated with improvement in the overall rate of death.8 Such a finding dramatically alters our impression of their cost-effectiveness and may explain the bias against investing in such therapies in the first place.
IMPROVING COST-EFFECTIVENESS RESEARCH
Studies of cost-effectiveness are not equivalent. Currently, such studies are apples and oranges, making difficult the very comparison that cost-effectiveness should facilitate. Knowing that a therapy is efficacious should be prerequisite to cost-effectiveness calculations, as should performing calculations under real-world conditions.
Regarding efficacy, it is inappropriate to calculate cost-effectiveness from trials that use only surrogate end points, or those that are improperly controlled.
For example, adding extended-release niacin to statin therapy may raise high-density lipoprotein cholesterol levels by 25%. Such an increase is, in turn, expected to confer a certain reduction in cardiovascular events and death. Thus, the cost-effectiveness of niacin might be calculated as $20,000 per life-year saved. However, adding extended-release niacin to statin therapy does not improve hard outcomes when directly measured,9 and the therapy is not efficacious at all. Its true “dollars per life-year saved” approaches infinity.
Studies that use historical controls, are observational, and are performed at single centers may also mislead us regarding a therapy’s efficacy. Tight glycemic control in intensive care patients initially seemed promising10,11 and cost-effective.12 However, several years later it was found to increase the mortality rate.13
“Real world” means that the best measures of cost-effectiveness will calculate the cost per life saved that the therapy achieves in clinical practice. Adherence to COX-2 inhibitors may not be as strict in the real world as it is in the carefully selected participants in randomized controlled trials, and, thus, the true costs may be higher. A drug that prevents breast cancer may have countervailing effects that may as yet be unknown, or compliance with it may wane over years. Thus, the most accurate measures of cost-effectiveness will examine therapies as best as they can function in typical practice and likely be derived from data sets of large payers or providers.
Finally, it remains an open and contentious issue whether the cost-effectiveness of primary prevention and the cost-effectiveness of treatment are comparable at all. We must continue to ponder and debate this philosophical question.
Certainly, these are the challenges of cost-effectiveness. Equally certain is that—with renewed consideration of the goals of such research, with stricter standards for future studies, and in an economic and political climate unable to sustain the status quo—the challenges must be surmounted.
Measures of cost-effectiveness are used to compare the merits of diverse medical interventions. A novel drug for metastatic melanoma, for instance, can be compared with statin therapy for primary prevention of cardiovascular events, which in turn can be compared against a surgical procedure for pain, as all are described by a single number: dollars per life-year (or quality-adjusted life-year) gained. Presumably, this number tells practitioners and payers which interventions provide the most benefit for every dollar spent.
However, too often, studies of cost-effectiveness differ from one another. They can be based on data from different types of studies, such as randomized controlled trials, surveys of large payer databases, or single-center chart reviews. The comparison treatments may differ. And the treatments may be of unproven efficacy. In these cases, although the results are all expressed in dollars per life-year, we are comparing apples and oranges.
In the following discussion, I use three key contemporary examples to demonstrate problems central to cost-effectiveness analysis. Together, these examples show that cost-effectiveness, arguably our best tool for comparing apples and oranges, is a lot like apples and oranges itself. I conclude by proposing some solutions.
PROBLEMS WITH COST-EFFECTIVENESS: THREE EXAMPLES
Studies of three therapies highlight the dilemma of cost-effectiveness.
Example 1: Vertebroplasty
Studies of vertebroplasty, a treatment for osteoporotic vertebral fractures that involves injecting polymethylmethacrylate cement into the fractured bone, show the perils of calculating the cost-effectiveness of unproven therapies.
Vertebroplasty gained prominence during the first decade of the 2000s, but in 2009 it was found to be no better than a sham procedure.1,2
In 2008, one study reported that vertebroplasty was cheaper than medical management at 12 months and, thus, cost-effective.3 While this finding was certainly true for the regimen of medical management the authors examined, and while it may very well be true for other protocols for medical management, the finding obscures the fact that a sham procedure would be more cost-effective than either vertebroplasty or medical therapy—an unsettling conclusion.
Example 2: Exemestane
Another dilemma occurs when we can calculate cost-effectiveness for a particular outcome only.
Studies of exemestane (Aromasin), an aromatase inhibitor given to prevent breast cancer, show the difficulty. Recently, exemestane was shown to decrease the rate of breast cancer when used as primary prevention in postmenopausal women.4 What is the costeffectiveness of this therapy?
While we can calculate the dollars per invasive breast cancer averted, we cannot accurately calculate the dollars per life-year gained, as the trial’s end point was not the mortality rate. We can assume that the breast cancer deaths avoided are not negated by deaths incurred through other causes, but this may or may not prove true. Fibrates, for instance, may reduce the rate of cardiovascular death but increase deaths from noncardiac causes, providing no net benefit.5 Such long-term effects remain unknown in the breast cancer study.
Example 3: COX-2 inhibitors
Estimates of cost-effectiveness derived from randomized trials can differ from those derived from real-world studies. Studies of cyclooxygenase 2 (COX-2) inhibitors, which were touted as causing less gastrointestinal bleeding than other nonsteroidal anti-inflammatory drugs, show that cost-effectiveness analyses performed from randomized trials may not mirror dollars spent in real-world practice.
Estimates from randomized controlled trials indicate that a COX-2 inhibitor such as celecoxib (Celebrex) costs $20,000 to prevent one gastrointestinal hemorrhage. However, when calculated using real-world data, that number rises to over $100,000.6
TWO PROPOSED RULES FOR COST-EFFECTIVENESS ANALYSES
How do we reconcile these and related puzzles of cost-effectiveness? First, we should agree on what type of “cost-effectiveness” we are interested in. Most often, we want to know whether the real-world use of a therapy is financially rational. Thus, we are concerned with the effectiveness of therapies and not merely their efficacy in idealized clinical trials.
Furthermore, while real-world cost-effectiveness may change over time, particularly as pricing and delivery vary, we want some assurance that the therapy is truly better than placebo. Therefore, we should only calculate the cost-effectiveness of therapies that have previously demonstrated efficacy in properly controlled, randomized studies.7
To correct the deficiencies noted here, I propose two rules:
Cost-effectiveness should be calculated only for therapies that have been proven to work, and
These calculations should be done from the best available real-world data.
When both these conditions are met—ie, a therapy has proven efficacy, and we have data from its real-world use—cost-effectiveness analysis provides useful information for payers and practitioners. Then, indeed, a novel anticancer agent costing $30,000 per life-year gained can be compared against primary prevention with statin therapy in patients at elevated cardiovascular risk costing $20,000 per life-year gained.
CAN PREVENTION BE COMPARED WITH TREATMENT?
This leaves us with the final and most difficult question. Is it right to compare such things?
Having terminal cancer is a different experience than having high cholesterol, and this is the last apple and orange of cost-effectiveness. While a strict utilitarian view of medicine might find these cases indistinguishable, most practitioners and payers are not strict utilitarians. As a society, we tend to favor paying more to treat someone who is ill than paying an equivalent amount to prevent illness. Often, such a stance is criticized as a failure to invest in prevention and primary care, but another explanation is that the bias is a fundamental one of human risk-taking.
Cost-effectiveness is, to a certain degree, a slippery concept, and it is more likely to be “off” when a therapy is given broadly (to hundreds of thousands of people as opposed to hundreds) and taken in a decentralized fashion by individual patients (as opposed to directly observed therapy in an infusion suite). Accordingly, we may favor more expensive therapies, the cost-effectiveness of which can be estimated more precisely.
A recent meta-analysis of statins for primary prevention in high-risk patients found that they were not associated with improvement in the overall rate of death.8 Such a finding dramatically alters our impression of their cost-effectiveness and may explain the bias against investing in such therapies in the first place.
IMPROVING COST-EFFECTIVENESS RESEARCH
Studies of cost-effectiveness are not equivalent. Currently, such studies are apples and oranges, making difficult the very comparison that cost-effectiveness should facilitate. Knowing that a therapy is efficacious should be prerequisite to cost-effectiveness calculations, as should performing calculations under real-world conditions.
Regarding efficacy, it is inappropriate to calculate cost-effectiveness from trials that use only surrogate end points, or those that are improperly controlled.
For example, adding extended-release niacin to statin therapy may raise high-density lipoprotein cholesterol levels by 25%. Such an increase is, in turn, expected to confer a certain reduction in cardiovascular events and death. Thus, the cost-effectiveness of niacin might be calculated as $20,000 per life-year saved. However, adding extended-release niacin to statin therapy does not improve hard outcomes when directly measured,9 and the therapy is not efficacious at all. Its true “dollars per life-year saved” approaches infinity.
Studies that use historical controls, are observational, and are performed at single centers may also mislead us regarding a therapy’s efficacy. Tight glycemic control in intensive care patients initially seemed promising10,11 and cost-effective.12 However, several years later it was found to increase the mortality rate.13
“Real world” means that the best measures of cost-effectiveness will calculate the cost per life saved that the therapy achieves in clinical practice. Adherence to COX-2 inhibitors may not be as strict in the real world as it is in the carefully selected participants in randomized controlled trials, and, thus, the true costs may be higher. A drug that prevents breast cancer may have countervailing effects that may as yet be unknown, or compliance with it may wane over years. Thus, the most accurate measures of cost-effectiveness will examine therapies as best as they can function in typical practice and likely be derived from data sets of large payers or providers.
Finally, it remains an open and contentious issue whether the cost-effectiveness of primary prevention and the cost-effectiveness of treatment are comparable at all. We must continue to ponder and debate this philosophical question.
Certainly, these are the challenges of cost-effectiveness. Equally certain is that—with renewed consideration of the goals of such research, with stricter standards for future studies, and in an economic and political climate unable to sustain the status quo—the challenges must be surmounted.
References
Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med2009; 361:569–579.
Buchbinder R, Osborne RH, Ebeling PR, et al. A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med2009; 361:557–568.
Masala S, Ciarrapico AM, Konda D, Vinicola V, Mammucari M, Simonetti G. Cost-effectiveness of percutaneous vertebroplasty in osteoporotic vertebral fractures. Eur Spine J2008; 17:1242–1250.
Goss PE, Ingle JN, Alés-Martínez JE, et al; NCIC CTG MAP3 Study Investigators. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med2011; 364:2381–2391.
Studer M, Briel M, Leimenstoll B, Glass TR, Bucher HC. Effect of different antilipidemic agents and diets on mortality: a systematic review. Arch Intern Med2005; 165:725–730.
van Staa TP, Leufkens HG, Zhang B, Smeeth L. A comparison of cost effectiveness using data from randomized trials or actual clinical practice: selective cox-2 inhibitors as an example. PLoS Med2009; 6:e1000194.
Prasad V, Cifu A. A medical burden of proof: towards a new ethic. BioSocieties2012; 7:72–87.
Ray KK, Seshasai SR, Erqou S, et al. Statins and all-cause mortality in high-risk primary prevention: a meta-analysis of 11 randomized controlled trials involving 65,229 participants. Arch Intern Med2010; 170:1024–1031.
National Heart, Lung, and Blood Institute National Institutes of Health. AIM-HIGH: Blinded treatment phase of study stopped. http://www.aimhigh-heart.com/. Accessed January 31, 2012.
van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med2001; 345:1359–1367.
Van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med2006; 354:449–461.
Mesotten D, Van den Berghe G. Clinical potential of insulin therapy in critically ill patients. Drugs2003; 63:625–636.
NICE-SUGAR Study Investigators; Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med2009; 360:1283–1297.
References
Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med2009; 361:569–579.
Buchbinder R, Osborne RH, Ebeling PR, et al. A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med2009; 361:557–568.
Masala S, Ciarrapico AM, Konda D, Vinicola V, Mammucari M, Simonetti G. Cost-effectiveness of percutaneous vertebroplasty in osteoporotic vertebral fractures. Eur Spine J2008; 17:1242–1250.
Goss PE, Ingle JN, Alés-Martínez JE, et al; NCIC CTG MAP3 Study Investigators. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med2011; 364:2381–2391.
Studer M, Briel M, Leimenstoll B, Glass TR, Bucher HC. Effect of different antilipidemic agents and diets on mortality: a systematic review. Arch Intern Med2005; 165:725–730.
van Staa TP, Leufkens HG, Zhang B, Smeeth L. A comparison of cost effectiveness using data from randomized trials or actual clinical practice: selective cox-2 inhibitors as an example. PLoS Med2009; 6:e1000194.
Prasad V, Cifu A. A medical burden of proof: towards a new ethic. BioSocieties2012; 7:72–87.
Ray KK, Seshasai SR, Erqou S, et al. Statins and all-cause mortality in high-risk primary prevention: a meta-analysis of 11 randomized controlled trials involving 65,229 participants. Arch Intern Med2010; 170:1024–1031.
National Heart, Lung, and Blood Institute National Institutes of Health. AIM-HIGH: Blinded treatment phase of study stopped. http://www.aimhigh-heart.com/. Accessed January 31, 2012.
van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med2001; 345:1359–1367.
Van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med2006; 354:449–461.
Mesotten D, Van den Berghe G. Clinical potential of insulin therapy in critically ill patients. Drugs2003; 63:625–636.
NICE-SUGAR Study Investigators; Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med2009; 360:1283–1297.
Perhaps 3% of the population has psoriasis. Thus, it is impossible to practice any aspect of internal medicine without encountering patients with this disease.
In this issue of the Journal, Dr. Jennifer Villaseñor-Park and her colleagues discuss the clinical patterns and management of psoriasis and the links between psoriasis and cardiovascular disease—links that should bind the internist and dermatologist in a shared mission of comanagement.
The connection between inflammation and atherosclerosis is now well known. Many of the same cellular and biochemical players have active roles in the inflammation of rheumatoid arthritis, systemic lupus erythematosus, psoriasis, and atherosclerosis. The observation that patients with inflammatory diseases have a higher prevalence of cardiovascular disease seems to strengthen this apparent link and supports the concept that drugs used to treat inflammation in the joints and skin might also reduce the burden of cardiovascular disease.
But addressing this risk is not so straightforward. Since the increased cardiovascular risk in rheumatoid arthritis and systemic lupus erythematosus is not completely explained by traditional risk factors, research is ongoing to identify the potential mechanisms of this risk, such as high-density lipoprotein particles modified by inflammation and high circulating levels of interferon, both of which may be atherogenic. It remains to be seen whether these and other potential nonclassic mediators of atherosclerosis can be targeted and cardiovascular events reduced.
But psoriasis is a little different. Compared with patients with rheumatoid arthritis and lupus (if they have not been affected by corticosteroid treatment), patients with psoriasis tend to be heavier and to have a higher prevalence of fatty liver disease and the metabolic syndrome. A debate continues as to whether psoriasis per se is a unique risk factor for cardiovascular disease or whether in fact these comorbidities constitute the major risk for cardiovascular events in patients with psoriasis.
The epidemiologists can continue to crunch the data in attempts to attribute the relative risks of poor outcome. But in the office, we should be vigilant and, in patients with psoriasis, should not ignore the traditional cardiovascular risk factors included in the metabolic syndrome, which is more prevalent in these patients.
Perhaps 3% of the population has psoriasis. Thus, it is impossible to practice any aspect of internal medicine without encountering patients with this disease.
In this issue of the Journal, Dr. Jennifer Villaseñor-Park and her colleagues discuss the clinical patterns and management of psoriasis and the links between psoriasis and cardiovascular disease—links that should bind the internist and dermatologist in a shared mission of comanagement.
The connection between inflammation and atherosclerosis is now well known. Many of the same cellular and biochemical players have active roles in the inflammation of rheumatoid arthritis, systemic lupus erythematosus, psoriasis, and atherosclerosis. The observation that patients with inflammatory diseases have a higher prevalence of cardiovascular disease seems to strengthen this apparent link and supports the concept that drugs used to treat inflammation in the joints and skin might also reduce the burden of cardiovascular disease.
But addressing this risk is not so straightforward. Since the increased cardiovascular risk in rheumatoid arthritis and systemic lupus erythematosus is not completely explained by traditional risk factors, research is ongoing to identify the potential mechanisms of this risk, such as high-density lipoprotein particles modified by inflammation and high circulating levels of interferon, both of which may be atherogenic. It remains to be seen whether these and other potential nonclassic mediators of atherosclerosis can be targeted and cardiovascular events reduced.
But psoriasis is a little different. Compared with patients with rheumatoid arthritis and lupus (if they have not been affected by corticosteroid treatment), patients with psoriasis tend to be heavier and to have a higher prevalence of fatty liver disease and the metabolic syndrome. A debate continues as to whether psoriasis per se is a unique risk factor for cardiovascular disease or whether in fact these comorbidities constitute the major risk for cardiovascular events in patients with psoriasis.
The epidemiologists can continue to crunch the data in attempts to attribute the relative risks of poor outcome. But in the office, we should be vigilant and, in patients with psoriasis, should not ignore the traditional cardiovascular risk factors included in the metabolic syndrome, which is more prevalent in these patients.
Perhaps 3% of the population has psoriasis. Thus, it is impossible to practice any aspect of internal medicine without encountering patients with this disease.
In this issue of the Journal, Dr. Jennifer Villaseñor-Park and her colleagues discuss the clinical patterns and management of psoriasis and the links between psoriasis and cardiovascular disease—links that should bind the internist and dermatologist in a shared mission of comanagement.
The connection between inflammation and atherosclerosis is now well known. Many of the same cellular and biochemical players have active roles in the inflammation of rheumatoid arthritis, systemic lupus erythematosus, psoriasis, and atherosclerosis. The observation that patients with inflammatory diseases have a higher prevalence of cardiovascular disease seems to strengthen this apparent link and supports the concept that drugs used to treat inflammation in the joints and skin might also reduce the burden of cardiovascular disease.
But addressing this risk is not so straightforward. Since the increased cardiovascular risk in rheumatoid arthritis and systemic lupus erythematosus is not completely explained by traditional risk factors, research is ongoing to identify the potential mechanisms of this risk, such as high-density lipoprotein particles modified by inflammation and high circulating levels of interferon, both of which may be atherogenic. It remains to be seen whether these and other potential nonclassic mediators of atherosclerosis can be targeted and cardiovascular events reduced.
But psoriasis is a little different. Compared with patients with rheumatoid arthritis and lupus (if they have not been affected by corticosteroid treatment), patients with psoriasis tend to be heavier and to have a higher prevalence of fatty liver disease and the metabolic syndrome. A debate continues as to whether psoriasis per se is a unique risk factor for cardiovascular disease or whether in fact these comorbidities constitute the major risk for cardiovascular events in patients with psoriasis.
The epidemiologists can continue to crunch the data in attempts to attribute the relative risks of poor outcome. But in the office, we should be vigilant and, in patients with psoriasis, should not ignore the traditional cardiovascular risk factors included in the metabolic syndrome, which is more prevalent in these patients.