User login
CASE REPORT
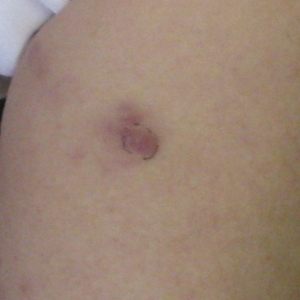
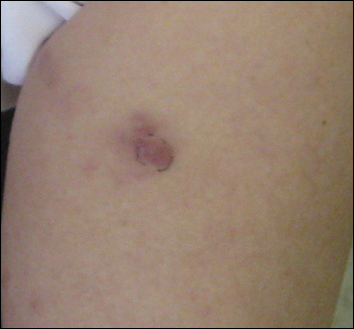
A 74-year-old woman presented with a painful lesion on the left lower leg that was getting larger and more edematous and erythematous over the last 5 months. She experienced numbness and burning of the left lower leg 1 year prior to the development of the lesion. A review of her medical history revealed an otherwise healthy woman with no constitutional symptoms of fever, chills, nausea, vomiting, diarrhea, or chest pain. The patient did not exhibit mucosal, genital, or nail involvement. Physical examination revealed a group of four 1-cm, ill-defined, irregularly bordered, violaceous plaques on the left anterior tibial leg with faint surrounding erythematous to violaceous patches (Figure 1). The plaques were tender to palpation with no bleeding or drainage.
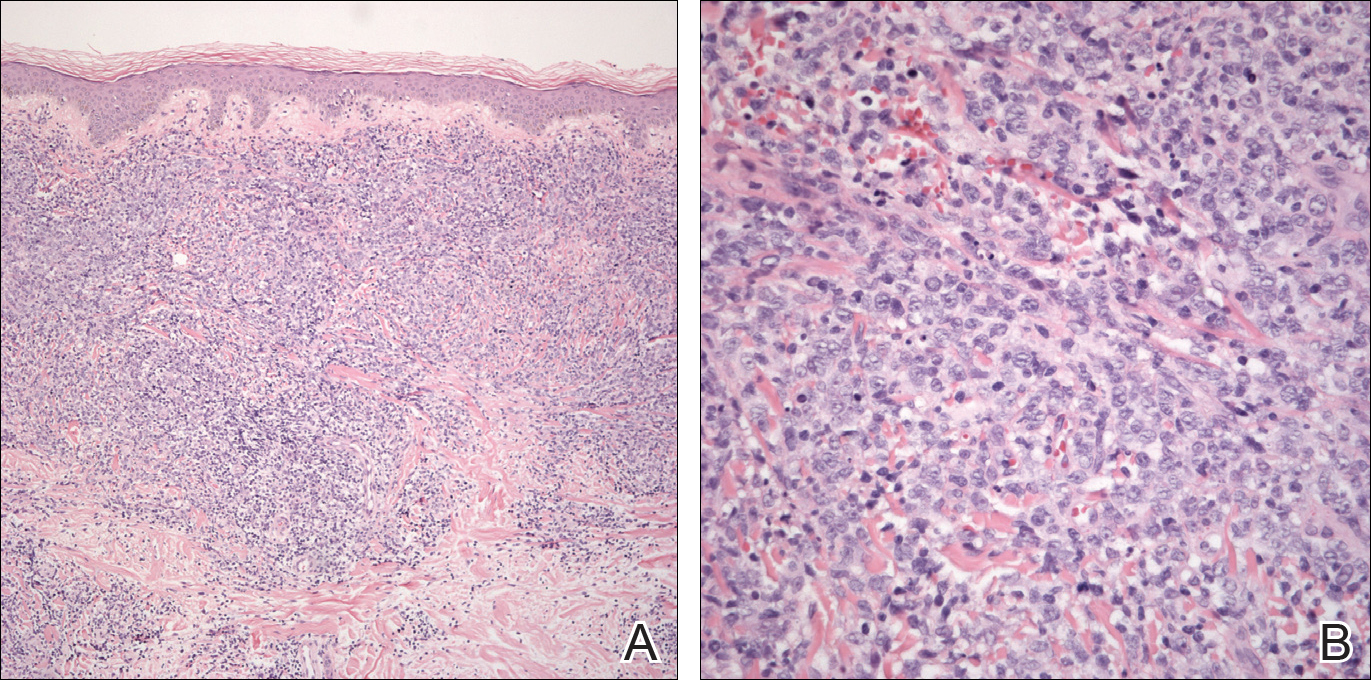
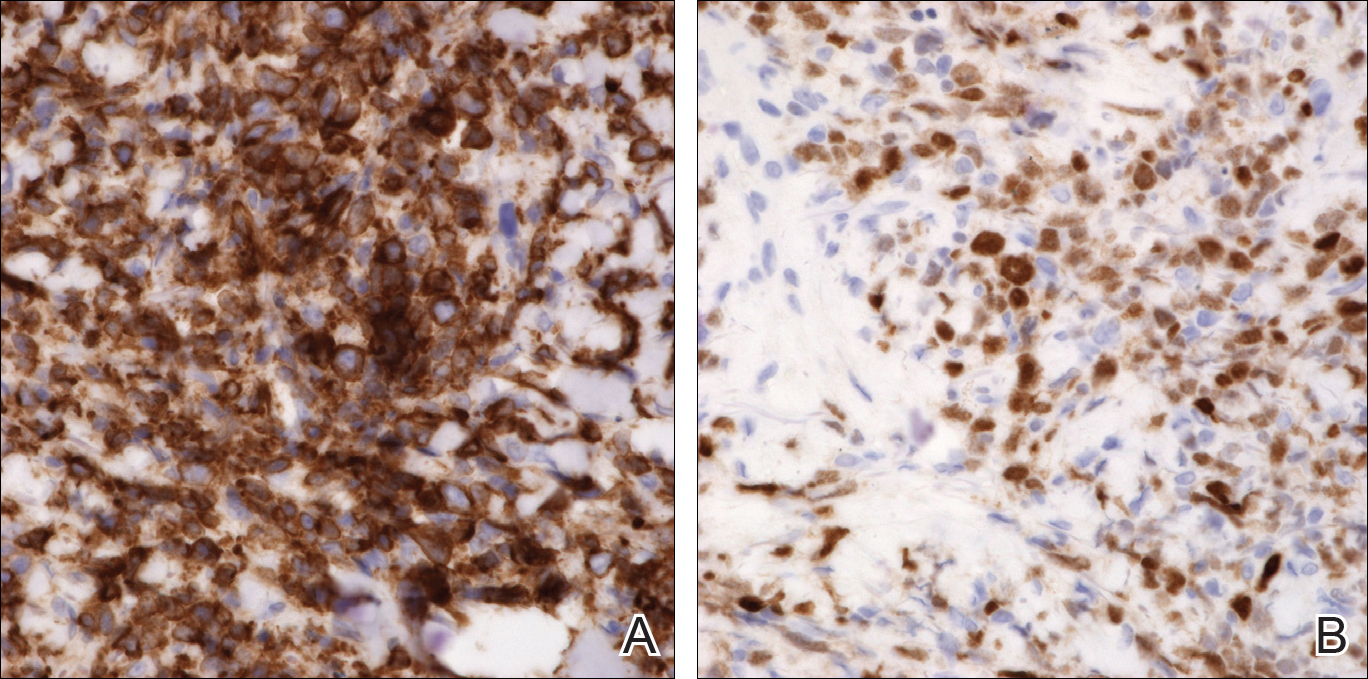
An 8.0-mm punch biopsy of the lesion was obtained. Hematoxylin and eosin staining on low-power magnification demonstrated a diffuse lymphocytic inflammatory infiltrate in the dermis and subcutis. Notable sparing of the subepidermal area (free grenz zone) was present (Figure 2A). On higher power, centroblasts and immunoblasts were visualized alongside extravasated red blood cells (Figure 2B). A diagnosis of primary cutaneous diffuse large B-cell lymphoma, leg type (DLBCLLT) was made. Various immunohistochemical stains confirmed the diagnosis, including B-cell lymphoma 2 (BCL-2)(Figure 3A) and multiple myeloma oncogene 1 (MUM-1)(Figure 3B), which were highly positive in our patient. The patient had a negative bone marrow biopsy and positron emission tomography scan. She was started on rituximab infusions and multiple radiation treatments. At 2-year follow-up the lymphoma continued to recur despite radiation therapy.
COMMENT
Incidence and Clinical Characteristics
Primary cutaneous DLBCLLT is an intermediately aggressive form of primary cutaneous B-cell lymphoma (CBCL) that accounts for approximately 10% to 20% of all primary CBCLs and 1% to 3% of all cutaneous lymphomas.1 Diffuse large B-cell lymphoma, leg type primarily affects elderly patients (median age, 70 years). Women are more commonly affected. Clinically, primary cutaneous DLBCLLT presents as red-brown to bluish nodules or tumors on one or both distal legs.
Histopathology
The diagnosis of DLBCLLT is best made histologically. There is a dense inflammatory infiltrate present in the dermis and subcutis that may extend upward into the dermoepidermal junction. Often a subepidermal free grenz zone may be seen, and adnexal structures may be destroyed. This infiltrate is composed of confluent sheets of large round cells including centroblasts and immunoblasts.2 Centroblasts are large cells that have nuclei with several small nucleoli adhering to the membrane, while immunoblasts are large round cells containing nuclei with large central nucleoli. Both centroblasts and immunoblasts stain positively for BCL-2. Centrocytes typically are absent. Staining for BCL-2 can be important in distinguishing DLBCLLT from other forms of CBCL. Diffuse large B-cell lymphoma, leg type also can demonstrate clusters of large atypical cells in the epidermis simulating epidermotropism and Pautrier microabscesses. Neoplastic cells in this condition may express monoclonal surface and cytoplasmic immunoglobulins. Primary cutaneous DLBCLLT typically is positive for B-cell markers CD20 and CD79a. Additionally, MUM-1/IRF4 (interferon regulatory factor 4) and forkhead box protein 1 (FOXP1) are strongly expressed by most patients, which helps distinguish it from other forms of CBCL.
Treatment
Diffuse large B-cell lymphoma, leg type is a relatively aggressive form of CBCL that requires more aggressive treatment than the conservative watchful waiting of some of the more indolent forms of primary CBCL. One regimen involves using cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab. Local chemotherapy or radiation with rituximab is another treatment option.1,2 In patients with severe comorbidities, rituximab alone may be administered. The prognosis for DLBCLLT is not as favorable as other types of primary CBCL, with an estimated 5-year survival rate of approximately 50%.2
Differential Diagnosis
Lymphomas are malignancies of the lymphocytes that may be subdivided depending on the organ of origin. Both primary nodal lymphomas and primary cutaneous lymphomas exist. Primary nodal lymphomas arise from the lymph nodes and are divided into Hodgkin and non-Hodgkin lymphomas. There are 2 major types of primary cutaneous lymphomas: cutaneous T-cell lymphoma (CTCL) and CBCL. Most primary cutaneous lymphomas are CTCLs, accounting for 75% to 80%.3
Pseudolymphoma
Pseudolymphoma is an inflammatory condition that may histologically mimic cutaneous lymphoma but has a benign clinical course. Pseudolymphoma is not a specific disease but rather is a reactive lymphoproliferative response to a known or unknown stimulus.4 Pseudolymphoma can be broken down into 2 or 3 major categories: cutaneous B-cell pseudolymphoma; cutaneous T-cell pseudolymphoma; and debatably lymphomatoid papulosis, a chronic, self-remitting, papulonecrotic condition that resembles lymphoma histologically but clinically appears benign. It is unknown if lymphomatoid papulosis represents a pseudolymphoma or a true lymphoma. Lymphomatoid papulosis may represent an early indolent form of CTCL.4
Pseudolymphomas can be triggered by a variety of causes. Most cases are idiopathic, and a causative stimulus is never identified. Drugs are known to cause many cases of pseudolymphoma, either by a causing a hypersensitivity reaction or by depressing immunosurveillance.5 Pseudolymphomas may result from exogenous stimuli such as jewelry, tattoo dyes, injectable fillers (eg, silicone), insect bites, vaccines, and trauma.6,7 Lastly, infections in the form of Borrelia, varicella, and molluscum contagiosum can potentially cause pseudolymphomas.4
Clinically, pseudolymphomas may demonstrate a B-cell or T-cell pattern. In cutaneous B-cell pseudolymphomas, asymptomatic solitary erythematous, violaceous, or flesh-colored nodules appear on the face, followed by the chest and arms. Cutaneous T-cell pseudolymphomas present with erythematous patches that are more likely to be symptomatic.4
Histologically, pseudolymphomas also are classified as demonstrating B-cell or T-cell patterns. The nodular inflammatory infiltrate of cutaneous B-cell pseudolymphoma corresponds with its clinically apparent nodules. It can be distinguished from lymphoma in that it is not solely a lymphocytic infiltrate but rather a mixed infiltrate including histiocytes, lymphocytes, eosinophils, and plasma cells. Additionally, cutaneous B-cell pseudolymphoma does not penetrate the dermis as deeply as CBCL.8 Cutaneous T-cell pseudolymphoma is more difficult to distinguish from CTCL because it also demonstrates a bandlike lymphocytic infiltrate in the papillary dermis with epidermotropism.9
Treatment must address the underlying cause of pseudolymphoma for resolution. Other treatment options include surgery, cryotherapy, local radiotherapy, topical steroids, and topical immunomodulators. Spontaneous resolution also can occur. The prognosis is better when a known trigger is eliminated, though idiopathic pseudolymphomas may be chronic in nature. It is important to rule out concurrent cutaneous lymphoma or rare transformation into cutaneous lymphoma.
Cutaneous T-Cell Lymphoma
Cutaneous T-cell lymphomas are a diverse group of neoplasms that account for most cutaneous lymphomas seen by dermatologists. In 1806, the first case of CTCL in the form of mycosis fungoides (MF) was described by Jean Louis Alibert. Mycosis fungoides represents the most common form of CTCL, accounting for approximately 50% of all primary cutaneous lymphomas.10 Mycosis fungoides was named after its morphological resemblance to mushrooms. Although not all cases exhibit a classic progression, MF is known for its stepwise progression from patch stage to tumor stage.
Clinically, lesions typically begin as patches that progress to plaques and finally tumors. This progression may not always occur and often can take years to decades to progress. Patches are characterized by erythematous, finely scaling lesions that may be easily confused with eczema or psoriasis. Lesions occur primarily in a swimming trunk distribution.
Mycosis fungoides histologically demonstrates a bandlike lymphocytic infiltrate with epidermotropism, which occurs when lymphocytes infiltrate the epidermis without spongiosis. These lymphocytes are larger, darker, and more angulated than normal lymphocytes. Intraepidermal nests of these atypical lymphocytes creating Pautrier microabscesses may be present. Tumor-stage lesions demonstrate diminished epidermotropism with dense sheets of lymphocytes in the dermis, and fat cells with cerebriform nuclei are present.
Therapies for MF may control the disease but may not prolong patients’ lives. Topical corticosteroids, phototherapy, and radiotherapy are options for skin-targeting therapies. Systemic chemotherapy and biological response modifiers also are viable treatment options. Prognosis for MF is poor.
There are a few notable variants of MF that are important to consider. Sézary syndrome is an erythrodermic variant of MF characterized by atypical Sézary cells. Clinically, it presents with generalized erythroderma with leonine facies, facial edema, and alopecia with associated symptoms of burning and pruritus. Histologically, Sézary syndrome is similar to MF with an increased CD4:CD8 ratio.10 Sézary syndrome may be treated with methotrexate or photopheresis, but the prognosis remains poor with an average survival of 5 years.
Cutaneous B-Cell Lymphoma
There are 5 types of primary CBCL: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone B-cell lymphoma; primary cutaneous diffuse large B-cell lymphoma, other; precursor B-cell lymphoblastic lymphoma; and primary cutaneous DLBCLLT, which was seen in our patient.11
Primary cutaneous follicle center lymphoma is an indolent neoplastic proliferation in the skin. Clinically, it presents with solitary or grouped pinkish purple papules, plaques, or nodules on the trunk with surrounding patches of erythema.3 Lesions located on the back are referred to as Crosti lymphoma. Histopathology reveals a lymphocytic infiltrate with a diffuse follicular pattern and large round centroblasts, centrocytes, and immunoblasts with epidermal sparing. Tumor cells stain positively for κ or λ light chains, as well as CD20, CD79a, and B-cell lymphoma 6 (BCL-6); however, staining for the protein product of BCL-2 may be negative, which differentiates this form of CBCL from primary nodal B-cell lymphoma. Staining for MUM-1 may be negative, which contrasts with the strong expression seen in DLBCLLT. The follicular pattern of follicle center lymphoma stains positive for CD10, but the diffuse pattern may be CD10 negative. The prognosis for primary cutaneous follicle center lymphoma is favorable, but the recurrence rate is up to 50%.3 Treatment includes local radiotherapy or surgical excision.
Primary cutaneous marginal zone B-cell lymphoma is another indolent primary CBCL subtype that is closely related to mucosa-associated lymphoid tissue lymphomas and arises in areas of acrodermatitis chronica atrophicans and Borrelia infection. Clinically, it presents with recurrent, asymptomatic, red-brown papules, plaques, and nodules of the arms and legs. Histologically, there is a patchy infiltrate in the dermis and subcutis with sparing of the epidermis with pale-staining cells with indented nuclei, along with plasma cells and eosinophils. Primary cutaneous marginal zone B-cell lymphoma typically does not demonstrate epidermotropism. Centrocyte cells stain positively for CD20, CD79a, and BCL-2. The prognosis of primary cutaneous marginal zone B-cell lymphoma is favorable. Treatment is similar to primary cutaneous follicle center lymphoma with surgical excision, radiotherapy, and surveillance being the main modalities.
Primary cutaneous diffuse large B-cell lymphoma, other is an intermediately aggressive form of primary CBCL that is thought to be related to primary cutaneous DLBCLLT. Clinically, it presents with indurated erythematous to violaceous plaques on the trunk and thighs that may resemble a vascular tumor or panniculitis.2,12 Histopathologically, this form of lymphoma presents with a round cell morphology without BCL-2 expression, which distinguishes it from DLBCLLT. If limited to skin, the prognosis is better than the systemic form but is still less favorable than other forms of CBCL.
Precursor B-cell lymphoblastic lymphoma is an extremely rare type of CBCL that potentially can occur in the skin. It primarily affects children and young adults. Clinically, it presents as a solitary large erythematous tumor of the head. Histol
CONCLUSION
We present a rare case of primary cutaneous DLBCLLT. Our case demonstrates the classic presentation of primary cutaneous DLBCLLT in a 74-year-old woman with a tumor on the lower left leg. Histologically, a dense dermal and subcutis infiltrate of centroblasts and immunoblasts with a grenz zone was present. Immunostaining in our patient was consistent with characteristic findings in the literature, staining highly positive for BCL-2 and MUM-1. Primary cutaneous DLBCLLT is an extremely rare and unique form of cutaneous lymphoma that can have potentially fatal consequences if undiagnosed; therefore, clinicians must take great care to make the correct diagnosis based on a knowledge of the clinical and immunohistochemical findings of DLBCLLT.
- Sokol L, Naghashpour M, Glass LF. Primary cutaneous B-cell lymphomas: recent advances in diagnosis and management. Cancer Control. 2012;19:236-244.
- Grange F, Beylot-Barry M, Courville P, et al. Primary cutaneous diffuse large B-cell lymphoma, leg type: clinicopathologic features and prognostic analysis in 60 cases. Arch Dermatol. 2007;143:1144-1150.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:2768-3785.
- Brodell RT, Santa Cruz DJ. Cutaneous pseudolymphomas. Dermatol Clin. 1985;3:719-734.
- Albrecht J, Fine LA, Piette W. Drug-associated lymphoma and pseudolymphoma: recognition and management. Dermatol Clin. 2007;25:233-244; vii.
- Maubec E, Pinquier L, Viguier M, et al. Vaccination-induced cutaneous pseudolymphoma. J Am Acad Dermatol. 2005;52:623-629.
- Kluger N, Vermeulen C, Moguelet P, et al. Cutaneous lymphoid hyperplasia (pseudolymphoma) in tattoos: a case series of seven patients. J Eur Acad Dermatol Venereol. 2010;24:208-213.
- Burg G, Kerl H, Schmoeckel C. Differentiation between malignant B-cell lymphomas and pseudolymphomas of the skin. J Dermatol Surg Oncol. 1984;10:271-275.
- Ploysangam T, Breneman DL, Mutasim DF. Cutaneous pseudolymphomas. J Am Acad Dermatol. 1998;38(6, pt 1):877-895; quiz 896-897.
- Diamandidou E, Cohen PR, Kurzrock R. Mycosis fungoides and Sézary syndrome. Blood. 1996;88:2385-2409.
- Kempf W, Ralfkiaer E, Duncan LM, et al. Cutaneous marginal zone B-cell lymphoma. In: LeBoit P, Burg G, Weedon D, et al, eds. Pathology and Genetics of Skin Tumors. Lyon, France: IARC Press; 2006:194-195.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in cutaneous large B-cell lymphomas: a European multicentric study. J Clin Oncol. 2001;19:3602-3610.
- Chimenti S, Fink-Puches R, Peris K, et al. Cutaneous involvement in lymphoblastic lymphoma. J Cutan Pathol. 1999;26:379-385.
CASE REPORT
A 74-year-old woman presented with a painful lesion on the left lower leg that was getting larger and more edematous and erythematous over the last 5 months. She experienced numbness and burning of the left lower leg 1 year prior to the development of the lesion. A review of her medical history revealed an otherwise healthy woman with no constitutional symptoms of fever, chills, nausea, vomiting, diarrhea, or chest pain. The patient did not exhibit mucosal, genital, or nail involvement. Physical examination revealed a group of four 1-cm, ill-defined, irregularly bordered, violaceous plaques on the left anterior tibial leg with faint surrounding erythematous to violaceous patches (Figure 1). The plaques were tender to palpation with no bleeding or drainage.
An 8.0-mm punch biopsy of the lesion was obtained. Hematoxylin and eosin staining on low-power magnification demonstrated a diffuse lymphocytic inflammatory infiltrate in the dermis and subcutis. Notable sparing of the subepidermal area (free grenz zone) was present (Figure 2A). On higher power, centroblasts and immunoblasts were visualized alongside extravasated red blood cells (Figure 2B). A diagnosis of primary cutaneous diffuse large B-cell lymphoma, leg type (DLBCLLT) was made. Various immunohistochemical stains confirmed the diagnosis, including B-cell lymphoma 2 (BCL-2)(Figure 3A) and multiple myeloma oncogene 1 (MUM-1)(Figure 3B), which were highly positive in our patient. The patient had a negative bone marrow biopsy and positron emission tomography scan. She was started on rituximab infusions and multiple radiation treatments. At 2-year follow-up the lymphoma continued to recur despite radiation therapy.
COMMENT
Incidence and Clinical Characteristics
Primary cutaneous DLBCLLT is an intermediately aggressive form of primary cutaneous B-cell lymphoma (CBCL) that accounts for approximately 10% to 20% of all primary CBCLs and 1% to 3% of all cutaneous lymphomas.1 Diffuse large B-cell lymphoma, leg type primarily affects elderly patients (median age, 70 years). Women are more commonly affected. Clinically, primary cutaneous DLBCLLT presents as red-brown to bluish nodules or tumors on one or both distal legs.
Histopathology
The diagnosis of DLBCLLT is best made histologically. There is a dense inflammatory infiltrate present in the dermis and subcutis that may extend upward into the dermoepidermal junction. Often a subepidermal free grenz zone may be seen, and adnexal structures may be destroyed. This infiltrate is composed of confluent sheets of large round cells including centroblasts and immunoblasts.2 Centroblasts are large cells that have nuclei with several small nucleoli adhering to the membrane, while immunoblasts are large round cells containing nuclei with large central nucleoli. Both centroblasts and immunoblasts stain positively for BCL-2. Centrocytes typically are absent. Staining for BCL-2 can be important in distinguishing DLBCLLT from other forms of CBCL. Diffuse large B-cell lymphoma, leg type also can demonstrate clusters of large atypical cells in the epidermis simulating epidermotropism and Pautrier microabscesses. Neoplastic cells in this condition may express monoclonal surface and cytoplasmic immunoglobulins. Primary cutaneous DLBCLLT typically is positive for B-cell markers CD20 and CD79a. Additionally, MUM-1/IRF4 (interferon regulatory factor 4) and forkhead box protein 1 (FOXP1) are strongly expressed by most patients, which helps distinguish it from other forms of CBCL.
Treatment
Diffuse large B-cell lymphoma, leg type is a relatively aggressive form of CBCL that requires more aggressive treatment than the conservative watchful waiting of some of the more indolent forms of primary CBCL. One regimen involves using cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab. Local chemotherapy or radiation with rituximab is another treatment option.1,2 In patients with severe comorbidities, rituximab alone may be administered. The prognosis for DLBCLLT is not as favorable as other types of primary CBCL, with an estimated 5-year survival rate of approximately 50%.2
Differential Diagnosis
Lymphomas are malignancies of the lymphocytes that may be subdivided depending on the organ of origin. Both primary nodal lymphomas and primary cutaneous lymphomas exist. Primary nodal lymphomas arise from the lymph nodes and are divided into Hodgkin and non-Hodgkin lymphomas. There are 2 major types of primary cutaneous lymphomas: cutaneous T-cell lymphoma (CTCL) and CBCL. Most primary cutaneous lymphomas are CTCLs, accounting for 75% to 80%.3
Pseudolymphoma
Pseudolymphoma is an inflammatory condition that may histologically mimic cutaneous lymphoma but has a benign clinical course. Pseudolymphoma is not a specific disease but rather is a reactive lymphoproliferative response to a known or unknown stimulus.4 Pseudolymphoma can be broken down into 2 or 3 major categories: cutaneous B-cell pseudolymphoma; cutaneous T-cell pseudolymphoma; and debatably lymphomatoid papulosis, a chronic, self-remitting, papulonecrotic condition that resembles lymphoma histologically but clinically appears benign. It is unknown if lymphomatoid papulosis represents a pseudolymphoma or a true lymphoma. Lymphomatoid papulosis may represent an early indolent form of CTCL.4
Pseudolymphomas can be triggered by a variety of causes. Most cases are idiopathic, and a causative stimulus is never identified. Drugs are known to cause many cases of pseudolymphoma, either by a causing a hypersensitivity reaction or by depressing immunosurveillance.5 Pseudolymphomas may result from exogenous stimuli such as jewelry, tattoo dyes, injectable fillers (eg, silicone), insect bites, vaccines, and trauma.6,7 Lastly, infections in the form of Borrelia, varicella, and molluscum contagiosum can potentially cause pseudolymphomas.4
Clinically, pseudolymphomas may demonstrate a B-cell or T-cell pattern. In cutaneous B-cell pseudolymphomas, asymptomatic solitary erythematous, violaceous, or flesh-colored nodules appear on the face, followed by the chest and arms. Cutaneous T-cell pseudolymphomas present with erythematous patches that are more likely to be symptomatic.4
Histologically, pseudolymphomas also are classified as demonstrating B-cell or T-cell patterns. The nodular inflammatory infiltrate of cutaneous B-cell pseudolymphoma corresponds with its clinically apparent nodules. It can be distinguished from lymphoma in that it is not solely a lymphocytic infiltrate but rather a mixed infiltrate including histiocytes, lymphocytes, eosinophils, and plasma cells. Additionally, cutaneous B-cell pseudolymphoma does not penetrate the dermis as deeply as CBCL.8 Cutaneous T-cell pseudolymphoma is more difficult to distinguish from CTCL because it also demonstrates a bandlike lymphocytic infiltrate in the papillary dermis with epidermotropism.9
Treatment must address the underlying cause of pseudolymphoma for resolution. Other treatment options include surgery, cryotherapy, local radiotherapy, topical steroids, and topical immunomodulators. Spontaneous resolution also can occur. The prognosis is better when a known trigger is eliminated, though idiopathic pseudolymphomas may be chronic in nature. It is important to rule out concurrent cutaneous lymphoma or rare transformation into cutaneous lymphoma.
Cutaneous T-Cell Lymphoma
Cutaneous T-cell lymphomas are a diverse group of neoplasms that account for most cutaneous lymphomas seen by dermatologists. In 1806, the first case of CTCL in the form of mycosis fungoides (MF) was described by Jean Louis Alibert. Mycosis fungoides represents the most common form of CTCL, accounting for approximately 50% of all primary cutaneous lymphomas.10 Mycosis fungoides was named after its morphological resemblance to mushrooms. Although not all cases exhibit a classic progression, MF is known for its stepwise progression from patch stage to tumor stage.
Clinically, lesions typically begin as patches that progress to plaques and finally tumors. This progression may not always occur and often can take years to decades to progress. Patches are characterized by erythematous, finely scaling lesions that may be easily confused with eczema or psoriasis. Lesions occur primarily in a swimming trunk distribution.
Mycosis fungoides histologically demonstrates a bandlike lymphocytic infiltrate with epidermotropism, which occurs when lymphocytes infiltrate the epidermis without spongiosis. These lymphocytes are larger, darker, and more angulated than normal lymphocytes. Intraepidermal nests of these atypical lymphocytes creating Pautrier microabscesses may be present. Tumor-stage lesions demonstrate diminished epidermotropism with dense sheets of lymphocytes in the dermis, and fat cells with cerebriform nuclei are present.
Therapies for MF may control the disease but may not prolong patients’ lives. Topical corticosteroids, phototherapy, and radiotherapy are options for skin-targeting therapies. Systemic chemotherapy and biological response modifiers also are viable treatment options. Prognosis for MF is poor.
There are a few notable variants of MF that are important to consider. Sézary syndrome is an erythrodermic variant of MF characterized by atypical Sézary cells. Clinically, it presents with generalized erythroderma with leonine facies, facial edema, and alopecia with associated symptoms of burning and pruritus. Histologically, Sézary syndrome is similar to MF with an increased CD4:CD8 ratio.10 Sézary syndrome may be treated with methotrexate or photopheresis, but the prognosis remains poor with an average survival of 5 years.
Cutaneous B-Cell Lymphoma
There are 5 types of primary CBCL: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone B-cell lymphoma; primary cutaneous diffuse large B-cell lymphoma, other; precursor B-cell lymphoblastic lymphoma; and primary cutaneous DLBCLLT, which was seen in our patient.11
Primary cutaneous follicle center lymphoma is an indolent neoplastic proliferation in the skin. Clinically, it presents with solitary or grouped pinkish purple papules, plaques, or nodules on the trunk with surrounding patches of erythema.3 Lesions located on the back are referred to as Crosti lymphoma. Histopathology reveals a lymphocytic infiltrate with a diffuse follicular pattern and large round centroblasts, centrocytes, and immunoblasts with epidermal sparing. Tumor cells stain positively for κ or λ light chains, as well as CD20, CD79a, and B-cell lymphoma 6 (BCL-6); however, staining for the protein product of BCL-2 may be negative, which differentiates this form of CBCL from primary nodal B-cell lymphoma. Staining for MUM-1 may be negative, which contrasts with the strong expression seen in DLBCLLT. The follicular pattern of follicle center lymphoma stains positive for CD10, but the diffuse pattern may be CD10 negative. The prognosis for primary cutaneous follicle center lymphoma is favorable, but the recurrence rate is up to 50%.3 Treatment includes local radiotherapy or surgical excision.
Primary cutaneous marginal zone B-cell lymphoma is another indolent primary CBCL subtype that is closely related to mucosa-associated lymphoid tissue lymphomas and arises in areas of acrodermatitis chronica atrophicans and Borrelia infection. Clinically, it presents with recurrent, asymptomatic, red-brown papules, plaques, and nodules of the arms and legs. Histologically, there is a patchy infiltrate in the dermis and subcutis with sparing of the epidermis with pale-staining cells with indented nuclei, along with plasma cells and eosinophils. Primary cutaneous marginal zone B-cell lymphoma typically does not demonstrate epidermotropism. Centrocyte cells stain positively for CD20, CD79a, and BCL-2. The prognosis of primary cutaneous marginal zone B-cell lymphoma is favorable. Treatment is similar to primary cutaneous follicle center lymphoma with surgical excision, radiotherapy, and surveillance being the main modalities.
Primary cutaneous diffuse large B-cell lymphoma, other is an intermediately aggressive form of primary CBCL that is thought to be related to primary cutaneous DLBCLLT. Clinically, it presents with indurated erythematous to violaceous plaques on the trunk and thighs that may resemble a vascular tumor or panniculitis.2,12 Histopathologically, this form of lymphoma presents with a round cell morphology without BCL-2 expression, which distinguishes it from DLBCLLT. If limited to skin, the prognosis is better than the systemic form but is still less favorable than other forms of CBCL.
Precursor B-cell lymphoblastic lymphoma is an extremely rare type of CBCL that potentially can occur in the skin. It primarily affects children and young adults. Clinically, it presents as a solitary large erythematous tumor of the head. Histol
CONCLUSION
We present a rare case of primary cutaneous DLBCLLT. Our case demonstrates the classic presentation of primary cutaneous DLBCLLT in a 74-year-old woman with a tumor on the lower left leg. Histologically, a dense dermal and subcutis infiltrate of centroblasts and immunoblasts with a grenz zone was present. Immunostaining in our patient was consistent with characteristic findings in the literature, staining highly positive for BCL-2 and MUM-1. Primary cutaneous DLBCLLT is an extremely rare and unique form of cutaneous lymphoma that can have potentially fatal consequences if undiagnosed; therefore, clinicians must take great care to make the correct diagnosis based on a knowledge of the clinical and immunohistochemical findings of DLBCLLT.
CASE REPORT
A 74-year-old woman presented with a painful lesion on the left lower leg that was getting larger and more edematous and erythematous over the last 5 months. She experienced numbness and burning of the left lower leg 1 year prior to the development of the lesion. A review of her medical history revealed an otherwise healthy woman with no constitutional symptoms of fever, chills, nausea, vomiting, diarrhea, or chest pain. The patient did not exhibit mucosal, genital, or nail involvement. Physical examination revealed a group of four 1-cm, ill-defined, irregularly bordered, violaceous plaques on the left anterior tibial leg with faint surrounding erythematous to violaceous patches (Figure 1). The plaques were tender to palpation with no bleeding or drainage.
An 8.0-mm punch biopsy of the lesion was obtained. Hematoxylin and eosin staining on low-power magnification demonstrated a diffuse lymphocytic inflammatory infiltrate in the dermis and subcutis. Notable sparing of the subepidermal area (free grenz zone) was present (Figure 2A). On higher power, centroblasts and immunoblasts were visualized alongside extravasated red blood cells (Figure 2B). A diagnosis of primary cutaneous diffuse large B-cell lymphoma, leg type (DLBCLLT) was made. Various immunohistochemical stains confirmed the diagnosis, including B-cell lymphoma 2 (BCL-2)(Figure 3A) and multiple myeloma oncogene 1 (MUM-1)(Figure 3B), which were highly positive in our patient. The patient had a negative bone marrow biopsy and positron emission tomography scan. She was started on rituximab infusions and multiple radiation treatments. At 2-year follow-up the lymphoma continued to recur despite radiation therapy.
COMMENT
Incidence and Clinical Characteristics
Primary cutaneous DLBCLLT is an intermediately aggressive form of primary cutaneous B-cell lymphoma (CBCL) that accounts for approximately 10% to 20% of all primary CBCLs and 1% to 3% of all cutaneous lymphomas.1 Diffuse large B-cell lymphoma, leg type primarily affects elderly patients (median age, 70 years). Women are more commonly affected. Clinically, primary cutaneous DLBCLLT presents as red-brown to bluish nodules or tumors on one or both distal legs.
Histopathology
The diagnosis of DLBCLLT is best made histologically. There is a dense inflammatory infiltrate present in the dermis and subcutis that may extend upward into the dermoepidermal junction. Often a subepidermal free grenz zone may be seen, and adnexal structures may be destroyed. This infiltrate is composed of confluent sheets of large round cells including centroblasts and immunoblasts.2 Centroblasts are large cells that have nuclei with several small nucleoli adhering to the membrane, while immunoblasts are large round cells containing nuclei with large central nucleoli. Both centroblasts and immunoblasts stain positively for BCL-2. Centrocytes typically are absent. Staining for BCL-2 can be important in distinguishing DLBCLLT from other forms of CBCL. Diffuse large B-cell lymphoma, leg type also can demonstrate clusters of large atypical cells in the epidermis simulating epidermotropism and Pautrier microabscesses. Neoplastic cells in this condition may express monoclonal surface and cytoplasmic immunoglobulins. Primary cutaneous DLBCLLT typically is positive for B-cell markers CD20 and CD79a. Additionally, MUM-1/IRF4 (interferon regulatory factor 4) and forkhead box protein 1 (FOXP1) are strongly expressed by most patients, which helps distinguish it from other forms of CBCL.
Treatment
Diffuse large B-cell lymphoma, leg type is a relatively aggressive form of CBCL that requires more aggressive treatment than the conservative watchful waiting of some of the more indolent forms of primary CBCL. One regimen involves using cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab. Local chemotherapy or radiation with rituximab is another treatment option.1,2 In patients with severe comorbidities, rituximab alone may be administered. The prognosis for DLBCLLT is not as favorable as other types of primary CBCL, with an estimated 5-year survival rate of approximately 50%.2
Differential Diagnosis
Lymphomas are malignancies of the lymphocytes that may be subdivided depending on the organ of origin. Both primary nodal lymphomas and primary cutaneous lymphomas exist. Primary nodal lymphomas arise from the lymph nodes and are divided into Hodgkin and non-Hodgkin lymphomas. There are 2 major types of primary cutaneous lymphomas: cutaneous T-cell lymphoma (CTCL) and CBCL. Most primary cutaneous lymphomas are CTCLs, accounting for 75% to 80%.3
Pseudolymphoma
Pseudolymphoma is an inflammatory condition that may histologically mimic cutaneous lymphoma but has a benign clinical course. Pseudolymphoma is not a specific disease but rather is a reactive lymphoproliferative response to a known or unknown stimulus.4 Pseudolymphoma can be broken down into 2 or 3 major categories: cutaneous B-cell pseudolymphoma; cutaneous T-cell pseudolymphoma; and debatably lymphomatoid papulosis, a chronic, self-remitting, papulonecrotic condition that resembles lymphoma histologically but clinically appears benign. It is unknown if lymphomatoid papulosis represents a pseudolymphoma or a true lymphoma. Lymphomatoid papulosis may represent an early indolent form of CTCL.4
Pseudolymphomas can be triggered by a variety of causes. Most cases are idiopathic, and a causative stimulus is never identified. Drugs are known to cause many cases of pseudolymphoma, either by a causing a hypersensitivity reaction or by depressing immunosurveillance.5 Pseudolymphomas may result from exogenous stimuli such as jewelry, tattoo dyes, injectable fillers (eg, silicone), insect bites, vaccines, and trauma.6,7 Lastly, infections in the form of Borrelia, varicella, and molluscum contagiosum can potentially cause pseudolymphomas.4
Clinically, pseudolymphomas may demonstrate a B-cell or T-cell pattern. In cutaneous B-cell pseudolymphomas, asymptomatic solitary erythematous, violaceous, or flesh-colored nodules appear on the face, followed by the chest and arms. Cutaneous T-cell pseudolymphomas present with erythematous patches that are more likely to be symptomatic.4
Histologically, pseudolymphomas also are classified as demonstrating B-cell or T-cell patterns. The nodular inflammatory infiltrate of cutaneous B-cell pseudolymphoma corresponds with its clinically apparent nodules. It can be distinguished from lymphoma in that it is not solely a lymphocytic infiltrate but rather a mixed infiltrate including histiocytes, lymphocytes, eosinophils, and plasma cells. Additionally, cutaneous B-cell pseudolymphoma does not penetrate the dermis as deeply as CBCL.8 Cutaneous T-cell pseudolymphoma is more difficult to distinguish from CTCL because it also demonstrates a bandlike lymphocytic infiltrate in the papillary dermis with epidermotropism.9
Treatment must address the underlying cause of pseudolymphoma for resolution. Other treatment options include surgery, cryotherapy, local radiotherapy, topical steroids, and topical immunomodulators. Spontaneous resolution also can occur. The prognosis is better when a known trigger is eliminated, though idiopathic pseudolymphomas may be chronic in nature. It is important to rule out concurrent cutaneous lymphoma or rare transformation into cutaneous lymphoma.
Cutaneous T-Cell Lymphoma
Cutaneous T-cell lymphomas are a diverse group of neoplasms that account for most cutaneous lymphomas seen by dermatologists. In 1806, the first case of CTCL in the form of mycosis fungoides (MF) was described by Jean Louis Alibert. Mycosis fungoides represents the most common form of CTCL, accounting for approximately 50% of all primary cutaneous lymphomas.10 Mycosis fungoides was named after its morphological resemblance to mushrooms. Although not all cases exhibit a classic progression, MF is known for its stepwise progression from patch stage to tumor stage.
Clinically, lesions typically begin as patches that progress to plaques and finally tumors. This progression may not always occur and often can take years to decades to progress. Patches are characterized by erythematous, finely scaling lesions that may be easily confused with eczema or psoriasis. Lesions occur primarily in a swimming trunk distribution.
Mycosis fungoides histologically demonstrates a bandlike lymphocytic infiltrate with epidermotropism, which occurs when lymphocytes infiltrate the epidermis without spongiosis. These lymphocytes are larger, darker, and more angulated than normal lymphocytes. Intraepidermal nests of these atypical lymphocytes creating Pautrier microabscesses may be present. Tumor-stage lesions demonstrate diminished epidermotropism with dense sheets of lymphocytes in the dermis, and fat cells with cerebriform nuclei are present.
Therapies for MF may control the disease but may not prolong patients’ lives. Topical corticosteroids, phototherapy, and radiotherapy are options for skin-targeting therapies. Systemic chemotherapy and biological response modifiers also are viable treatment options. Prognosis for MF is poor.
There are a few notable variants of MF that are important to consider. Sézary syndrome is an erythrodermic variant of MF characterized by atypical Sézary cells. Clinically, it presents with generalized erythroderma with leonine facies, facial edema, and alopecia with associated symptoms of burning and pruritus. Histologically, Sézary syndrome is similar to MF with an increased CD4:CD8 ratio.10 Sézary syndrome may be treated with methotrexate or photopheresis, but the prognosis remains poor with an average survival of 5 years.
Cutaneous B-Cell Lymphoma
There are 5 types of primary CBCL: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone B-cell lymphoma; primary cutaneous diffuse large B-cell lymphoma, other; precursor B-cell lymphoblastic lymphoma; and primary cutaneous DLBCLLT, which was seen in our patient.11
Primary cutaneous follicle center lymphoma is an indolent neoplastic proliferation in the skin. Clinically, it presents with solitary or grouped pinkish purple papules, plaques, or nodules on the trunk with surrounding patches of erythema.3 Lesions located on the back are referred to as Crosti lymphoma. Histopathology reveals a lymphocytic infiltrate with a diffuse follicular pattern and large round centroblasts, centrocytes, and immunoblasts with epidermal sparing. Tumor cells stain positively for κ or λ light chains, as well as CD20, CD79a, and B-cell lymphoma 6 (BCL-6); however, staining for the protein product of BCL-2 may be negative, which differentiates this form of CBCL from primary nodal B-cell lymphoma. Staining for MUM-1 may be negative, which contrasts with the strong expression seen in DLBCLLT. The follicular pattern of follicle center lymphoma stains positive for CD10, but the diffuse pattern may be CD10 negative. The prognosis for primary cutaneous follicle center lymphoma is favorable, but the recurrence rate is up to 50%.3 Treatment includes local radiotherapy or surgical excision.
Primary cutaneous marginal zone B-cell lymphoma is another indolent primary CBCL subtype that is closely related to mucosa-associated lymphoid tissue lymphomas and arises in areas of acrodermatitis chronica atrophicans and Borrelia infection. Clinically, it presents with recurrent, asymptomatic, red-brown papules, plaques, and nodules of the arms and legs. Histologically, there is a patchy infiltrate in the dermis and subcutis with sparing of the epidermis with pale-staining cells with indented nuclei, along with plasma cells and eosinophils. Primary cutaneous marginal zone B-cell lymphoma typically does not demonstrate epidermotropism. Centrocyte cells stain positively for CD20, CD79a, and BCL-2. The prognosis of primary cutaneous marginal zone B-cell lymphoma is favorable. Treatment is similar to primary cutaneous follicle center lymphoma with surgical excision, radiotherapy, and surveillance being the main modalities.
Primary cutaneous diffuse large B-cell lymphoma, other is an intermediately aggressive form of primary CBCL that is thought to be related to primary cutaneous DLBCLLT. Clinically, it presents with indurated erythematous to violaceous plaques on the trunk and thighs that may resemble a vascular tumor or panniculitis.2,12 Histopathologically, this form of lymphoma presents with a round cell morphology without BCL-2 expression, which distinguishes it from DLBCLLT. If limited to skin, the prognosis is better than the systemic form but is still less favorable than other forms of CBCL.
Precursor B-cell lymphoblastic lymphoma is an extremely rare type of CBCL that potentially can occur in the skin. It primarily affects children and young adults. Clinically, it presents as a solitary large erythematous tumor of the head. Histol
CONCLUSION
We present a rare case of primary cutaneous DLBCLLT. Our case demonstrates the classic presentation of primary cutaneous DLBCLLT in a 74-year-old woman with a tumor on the lower left leg. Histologically, a dense dermal and subcutis infiltrate of centroblasts and immunoblasts with a grenz zone was present. Immunostaining in our patient was consistent with characteristic findings in the literature, staining highly positive for BCL-2 and MUM-1. Primary cutaneous DLBCLLT is an extremely rare and unique form of cutaneous lymphoma that can have potentially fatal consequences if undiagnosed; therefore, clinicians must take great care to make the correct diagnosis based on a knowledge of the clinical and immunohistochemical findings of DLBCLLT.
- Sokol L, Naghashpour M, Glass LF. Primary cutaneous B-cell lymphomas: recent advances in diagnosis and management. Cancer Control. 2012;19:236-244.
- Grange F, Beylot-Barry M, Courville P, et al. Primary cutaneous diffuse large B-cell lymphoma, leg type: clinicopathologic features and prognostic analysis in 60 cases. Arch Dermatol. 2007;143:1144-1150.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:2768-3785.
- Brodell RT, Santa Cruz DJ. Cutaneous pseudolymphomas. Dermatol Clin. 1985;3:719-734.
- Albrecht J, Fine LA, Piette W. Drug-associated lymphoma and pseudolymphoma: recognition and management. Dermatol Clin. 2007;25:233-244; vii.
- Maubec E, Pinquier L, Viguier M, et al. Vaccination-induced cutaneous pseudolymphoma. J Am Acad Dermatol. 2005;52:623-629.
- Kluger N, Vermeulen C, Moguelet P, et al. Cutaneous lymphoid hyperplasia (pseudolymphoma) in tattoos: a case series of seven patients. J Eur Acad Dermatol Venereol. 2010;24:208-213.
- Burg G, Kerl H, Schmoeckel C. Differentiation between malignant B-cell lymphomas and pseudolymphomas of the skin. J Dermatol Surg Oncol. 1984;10:271-275.
- Ploysangam T, Breneman DL, Mutasim DF. Cutaneous pseudolymphomas. J Am Acad Dermatol. 1998;38(6, pt 1):877-895; quiz 896-897.
- Diamandidou E, Cohen PR, Kurzrock R. Mycosis fungoides and Sézary syndrome. Blood. 1996;88:2385-2409.
- Kempf W, Ralfkiaer E, Duncan LM, et al. Cutaneous marginal zone B-cell lymphoma. In: LeBoit P, Burg G, Weedon D, et al, eds. Pathology and Genetics of Skin Tumors. Lyon, France: IARC Press; 2006:194-195.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in cutaneous large B-cell lymphomas: a European multicentric study. J Clin Oncol. 2001;19:3602-3610.
- Chimenti S, Fink-Puches R, Peris K, et al. Cutaneous involvement in lymphoblastic lymphoma. J Cutan Pathol. 1999;26:379-385.
- Sokol L, Naghashpour M, Glass LF. Primary cutaneous B-cell lymphomas: recent advances in diagnosis and management. Cancer Control. 2012;19:236-244.
- Grange F, Beylot-Barry M, Courville P, et al. Primary cutaneous diffuse large B-cell lymphoma, leg type: clinicopathologic features and prognostic analysis in 60 cases. Arch Dermatol. 2007;143:1144-1150.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:2768-3785.
- Brodell RT, Santa Cruz DJ. Cutaneous pseudolymphomas. Dermatol Clin. 1985;3:719-734.
- Albrecht J, Fine LA, Piette W. Drug-associated lymphoma and pseudolymphoma: recognition and management. Dermatol Clin. 2007;25:233-244; vii.
- Maubec E, Pinquier L, Viguier M, et al. Vaccination-induced cutaneous pseudolymphoma. J Am Acad Dermatol. 2005;52:623-629.
- Kluger N, Vermeulen C, Moguelet P, et al. Cutaneous lymphoid hyperplasia (pseudolymphoma) in tattoos: a case series of seven patients. J Eur Acad Dermatol Venereol. 2010;24:208-213.
- Burg G, Kerl H, Schmoeckel C. Differentiation between malignant B-cell lymphomas and pseudolymphomas of the skin. J Dermatol Surg Oncol. 1984;10:271-275.
- Ploysangam T, Breneman DL, Mutasim DF. Cutaneous pseudolymphomas. J Am Acad Dermatol. 1998;38(6, pt 1):877-895; quiz 896-897.
- Diamandidou E, Cohen PR, Kurzrock R. Mycosis fungoides and Sézary syndrome. Blood. 1996;88:2385-2409.
- Kempf W, Ralfkiaer E, Duncan LM, et al. Cutaneous marginal zone B-cell lymphoma. In: LeBoit P, Burg G, Weedon D, et al, eds. Pathology and Genetics of Skin Tumors. Lyon, France: IARC Press; 2006:194-195.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in cutaneous large B-cell lymphomas: a European multicentric study. J Clin Oncol. 2001;19:3602-3610.
- Chimenti S, Fink-Puches R, Peris K, et al. Cutaneous involvement in lymphoblastic lymphoma. J Cutan Pathol. 1999;26:379-385.
Practice Points
- Primary cutaneous diffuse large B-cell lymphoma, leg type (DLBCLLT) is characterized by the presence of large round cells on histopathology.
- There are potentially fatal consequences if undiagnosed; therefore, clinicians must take great care to make the correct diagnosis based on a knowledge of the clinical and immunohistochemical findings of DLBCLLT.