User login
Health Canada approves new use for brentuximab vedotin

Health Canada has issued a non-conditional marketing authorization for brentuximab vedotin (Adcetris).
This means the drug is now approved for use as consolidation after autologous stem cell transplant (ASCT) in patients with Hodgkin lymphoma (HL) who have an increased risk of relapse or progression.
Brentuximab vedotin previously received approval with conditions in Canada to treat HL patients who relapse after ASCT or HL patients who are not ASCT candidates and relapse after at least 2 multi-agent chemotherapy regimens.
Brentuximab vedotin also has conditional approval in Canada to treat patients with systemic anaplastic large-cell lymphoma who relapse after at least 1 multi-agent chemotherapy regimen.
Brentuximab vedotin is an antibody-drug conjugate consisting of an anti-CD30 monoclonal antibody attached by a protease-cleavable linker to a microtubule disrupting agent, monomethyl auristatin E.
Brentuximab vedotin has marketing authorization in 67 countries for the treatment of relapsed or refractory HL and systemic anaplastic large-cell lymphoma.
Seattle Genetics and Takeda are jointly developing brentuximab vedotin. Seattle Genetics has US and Canadian commercialization rights, and Takeda has rights to commercialize the drug in the rest of the world.
AETHERA trial
Health Canada’s decision to extend the marketing authorization of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015.
The study enrolled 329 HL patients at risk of relapse or progression—165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
Health Canada has issued a non-conditional marketing authorization for brentuximab vedotin (Adcetris).
This means the drug is now approved for use as consolidation after autologous stem cell transplant (ASCT) in patients with Hodgkin lymphoma (HL) who have an increased risk of relapse or progression.
Brentuximab vedotin previously received approval with conditions in Canada to treat HL patients who relapse after ASCT or HL patients who are not ASCT candidates and relapse after at least 2 multi-agent chemotherapy regimens.
Brentuximab vedotin also has conditional approval in Canada to treat patients with systemic anaplastic large-cell lymphoma who relapse after at least 1 multi-agent chemotherapy regimen.
Brentuximab vedotin is an antibody-drug conjugate consisting of an anti-CD30 monoclonal antibody attached by a protease-cleavable linker to a microtubule disrupting agent, monomethyl auristatin E.
Brentuximab vedotin has marketing authorization in 67 countries for the treatment of relapsed or refractory HL and systemic anaplastic large-cell lymphoma.
Seattle Genetics and Takeda are jointly developing brentuximab vedotin. Seattle Genetics has US and Canadian commercialization rights, and Takeda has rights to commercialize the drug in the rest of the world.
AETHERA trial
Health Canada’s decision to extend the marketing authorization of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015.
The study enrolled 329 HL patients at risk of relapse or progression—165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
Health Canada has issued a non-conditional marketing authorization for brentuximab vedotin (Adcetris).
This means the drug is now approved for use as consolidation after autologous stem cell transplant (ASCT) in patients with Hodgkin lymphoma (HL) who have an increased risk of relapse or progression.
Brentuximab vedotin previously received approval with conditions in Canada to treat HL patients who relapse after ASCT or HL patients who are not ASCT candidates and relapse after at least 2 multi-agent chemotherapy regimens.
Brentuximab vedotin also has conditional approval in Canada to treat patients with systemic anaplastic large-cell lymphoma who relapse after at least 1 multi-agent chemotherapy regimen.
Brentuximab vedotin is an antibody-drug conjugate consisting of an anti-CD30 monoclonal antibody attached by a protease-cleavable linker to a microtubule disrupting agent, monomethyl auristatin E.
Brentuximab vedotin has marketing authorization in 67 countries for the treatment of relapsed or refractory HL and systemic anaplastic large-cell lymphoma.
Seattle Genetics and Takeda are jointly developing brentuximab vedotin. Seattle Genetics has US and Canadian commercialization rights, and Takeda has rights to commercialize the drug in the rest of the world.
AETHERA trial
Health Canada’s decision to extend the marketing authorization of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015.
The study enrolled 329 HL patients at risk of relapse or progression—165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
Survey reveals change in public perception of clinical trials

More Americans are viewing clinical trials in a positive light, according to a survey commissioned by Research!America and the Association of Clinical Research Organizations (ACRO).
The results of the survey, which included 1000 participants and was conducted this month, reveal a shift in public attitudes about clinical trials since a similar survey was commissioned in 2013.
Thirty-seven percent of respondents involved in the current survey said they would “very likely” enroll in a clinical trial if their doctor recommended it. This represents an 11% increase from 2013.
Eighty-four percent of current survey respondents said they are willing to share personal health information, assuming that appropriate privacy protections are in place, so researchers can better understand diseases and develop new ways to prevent, treat, and cure them. This is a 10% increase from 2013.
When asked how much they admire people who volunteer for clinical trials, 46% of current respondents said “a great deal,” which is a 9% increase from 2013.
“More and more Americans appear to recognize the value of clinical trials—a very positive sign—but stubborn barriers to participation remain in place,” said Mary Woolley, president and CEO of Research!America.
“Development of incentives to drive more discussions between patients and healthcare professionals about the importance of participating in trials could encourage both ill and healthy individuals to view this as a routine health behavior.”
“We are pleased to have joined with Research!America in commissioning this important survey and gratified that the public’s knowledge of and attitudes toward clinical trials have moved in positive directions since 2013,” said Doug Peddicord, executive director of ACRO.
“The option to participate in a clinical trial, when appropriate, should be a routine part of the healthcare encounter, and ACRO will continue to work with Research!America and others to spread that message to doctors and patients alike.”
Survey results
The survey of 1000 US adults was conducted by Zogby Analytics for Research!America and ACRO in July 2017. The margin of error is +/- 3.1 percentage points.
Ninety percent of survey respondents said clinical trials are important to advancing science, and 87% said trials are important to improving the nation’s health. Seventy-five percent said taking part in clinical trials is as valuable to the US healthcare system as giving blood.
Eighty-six percent of respondents said healthcare professionals should discuss clinical trials with patients diagnosed with a disease as part of their standard care.
And 44% of respondents said clinical trial participation should be a routine health behavior, whether a person is healthy or ill, similar to getting an annual checkup with a healthcare provider.
Men (48%) were significantly more likely than women (39%) to say trial participation should be routine. A larger percentage of 18- to 29-year-olds (53%) and 30- to 49-year olds (48%) were in favor of routine trial participation, compared to people ages 50 to 64 (38%) and those 65 and older (34%).
Eighty percent of respondents said they had heard of a clinical trial, and 18% said they or someone in their family had participated in one.
Seventy-four percent of respondents said they would participate in a clinical trial if asked by someone they trust.
However, respondents were split on whether it’s important for everyone to take part in a clinical trial if asked. Forty-four percent said it is important, 45% disagreed, and 12% were not sure.
More than half of respondents (55%) said people don’t participate in clinical trials because of lack of awareness and information, 43% said it’s because trials are viewed as “too risky,” 41% said it’s due to a lack of information about the process, 38% said it’s due to a lack of trust, and 34% said it’s due to the risk of adverse health effects.
Nearly two-thirds of respondents (64%) said a doctor or healthcare provider is a reliable source for clinical trial information.
Respondents said doctors and other healthcare providers (44%), followed by the government (23%), have the greatest responsibility in educating the public about clinical trials. However, 74% said no healthcare professional has ever talked to them about medical research.
Seventy-two percent of respondents said they would be likely to use technology such as apps and monitoring devices to share their personal health data for clinical research.
Eighty-eight percent said clinical trial participants should have access to trial results. And 47% said they would like having clinical trial information/data/results delivered through their phone. ![]()
More Americans are viewing clinical trials in a positive light, according to a survey commissioned by Research!America and the Association of Clinical Research Organizations (ACRO).
The results of the survey, which included 1000 participants and was conducted this month, reveal a shift in public attitudes about clinical trials since a similar survey was commissioned in 2013.
Thirty-seven percent of respondents involved in the current survey said they would “very likely” enroll in a clinical trial if their doctor recommended it. This represents an 11% increase from 2013.
Eighty-four percent of current survey respondents said they are willing to share personal health information, assuming that appropriate privacy protections are in place, so researchers can better understand diseases and develop new ways to prevent, treat, and cure them. This is a 10% increase from 2013.
When asked how much they admire people who volunteer for clinical trials, 46% of current respondents said “a great deal,” which is a 9% increase from 2013.
“More and more Americans appear to recognize the value of clinical trials—a very positive sign—but stubborn barriers to participation remain in place,” said Mary Woolley, president and CEO of Research!America.
“Development of incentives to drive more discussions between patients and healthcare professionals about the importance of participating in trials could encourage both ill and healthy individuals to view this as a routine health behavior.”
“We are pleased to have joined with Research!America in commissioning this important survey and gratified that the public’s knowledge of and attitudes toward clinical trials have moved in positive directions since 2013,” said Doug Peddicord, executive director of ACRO.
“The option to participate in a clinical trial, when appropriate, should be a routine part of the healthcare encounter, and ACRO will continue to work with Research!America and others to spread that message to doctors and patients alike.”
Survey results
The survey of 1000 US adults was conducted by Zogby Analytics for Research!America and ACRO in July 2017. The margin of error is +/- 3.1 percentage points.
Ninety percent of survey respondents said clinical trials are important to advancing science, and 87% said trials are important to improving the nation’s health. Seventy-five percent said taking part in clinical trials is as valuable to the US healthcare system as giving blood.
Eighty-six percent of respondents said healthcare professionals should discuss clinical trials with patients diagnosed with a disease as part of their standard care.
And 44% of respondents said clinical trial participation should be a routine health behavior, whether a person is healthy or ill, similar to getting an annual checkup with a healthcare provider.
Men (48%) were significantly more likely than women (39%) to say trial participation should be routine. A larger percentage of 18- to 29-year-olds (53%) and 30- to 49-year olds (48%) were in favor of routine trial participation, compared to people ages 50 to 64 (38%) and those 65 and older (34%).
Eighty percent of respondents said they had heard of a clinical trial, and 18% said they or someone in their family had participated in one.
Seventy-four percent of respondents said they would participate in a clinical trial if asked by someone they trust.
However, respondents were split on whether it’s important for everyone to take part in a clinical trial if asked. Forty-four percent said it is important, 45% disagreed, and 12% were not sure.
More than half of respondents (55%) said people don’t participate in clinical trials because of lack of awareness and information, 43% said it’s because trials are viewed as “too risky,” 41% said it’s due to a lack of information about the process, 38% said it’s due to a lack of trust, and 34% said it’s due to the risk of adverse health effects.
Nearly two-thirds of respondents (64%) said a doctor or healthcare provider is a reliable source for clinical trial information.
Respondents said doctors and other healthcare providers (44%), followed by the government (23%), have the greatest responsibility in educating the public about clinical trials. However, 74% said no healthcare professional has ever talked to them about medical research.
Seventy-two percent of respondents said they would be likely to use technology such as apps and monitoring devices to share their personal health data for clinical research.
Eighty-eight percent said clinical trial participants should have access to trial results. And 47% said they would like having clinical trial information/data/results delivered through their phone. ![]()
More Americans are viewing clinical trials in a positive light, according to a survey commissioned by Research!America and the Association of Clinical Research Organizations (ACRO).
The results of the survey, which included 1000 participants and was conducted this month, reveal a shift in public attitudes about clinical trials since a similar survey was commissioned in 2013.
Thirty-seven percent of respondents involved in the current survey said they would “very likely” enroll in a clinical trial if their doctor recommended it. This represents an 11% increase from 2013.
Eighty-four percent of current survey respondents said they are willing to share personal health information, assuming that appropriate privacy protections are in place, so researchers can better understand diseases and develop new ways to prevent, treat, and cure them. This is a 10% increase from 2013.
When asked how much they admire people who volunteer for clinical trials, 46% of current respondents said “a great deal,” which is a 9% increase from 2013.
“More and more Americans appear to recognize the value of clinical trials—a very positive sign—but stubborn barriers to participation remain in place,” said Mary Woolley, president and CEO of Research!America.
“Development of incentives to drive more discussions between patients and healthcare professionals about the importance of participating in trials could encourage both ill and healthy individuals to view this as a routine health behavior.”
“We are pleased to have joined with Research!America in commissioning this important survey and gratified that the public’s knowledge of and attitudes toward clinical trials have moved in positive directions since 2013,” said Doug Peddicord, executive director of ACRO.
“The option to participate in a clinical trial, when appropriate, should be a routine part of the healthcare encounter, and ACRO will continue to work with Research!America and others to spread that message to doctors and patients alike.”
Survey results
The survey of 1000 US adults was conducted by Zogby Analytics for Research!America and ACRO in July 2017. The margin of error is +/- 3.1 percentage points.
Ninety percent of survey respondents said clinical trials are important to advancing science, and 87% said trials are important to improving the nation’s health. Seventy-five percent said taking part in clinical trials is as valuable to the US healthcare system as giving blood.
Eighty-six percent of respondents said healthcare professionals should discuss clinical trials with patients diagnosed with a disease as part of their standard care.
And 44% of respondents said clinical trial participation should be a routine health behavior, whether a person is healthy or ill, similar to getting an annual checkup with a healthcare provider.
Men (48%) were significantly more likely than women (39%) to say trial participation should be routine. A larger percentage of 18- to 29-year-olds (53%) and 30- to 49-year olds (48%) were in favor of routine trial participation, compared to people ages 50 to 64 (38%) and those 65 and older (34%).
Eighty percent of respondents said they had heard of a clinical trial, and 18% said they or someone in their family had participated in one.
Seventy-four percent of respondents said they would participate in a clinical trial if asked by someone they trust.
However, respondents were split on whether it’s important for everyone to take part in a clinical trial if asked. Forty-four percent said it is important, 45% disagreed, and 12% were not sure.
More than half of respondents (55%) said people don’t participate in clinical trials because of lack of awareness and information, 43% said it’s because trials are viewed as “too risky,” 41% said it’s due to a lack of information about the process, 38% said it’s due to a lack of trust, and 34% said it’s due to the risk of adverse health effects.
Nearly two-thirds of respondents (64%) said a doctor or healthcare provider is a reliable source for clinical trial information.
Respondents said doctors and other healthcare providers (44%), followed by the government (23%), have the greatest responsibility in educating the public about clinical trials. However, 74% said no healthcare professional has ever talked to them about medical research.
Seventy-two percent of respondents said they would be likely to use technology such as apps and monitoring devices to share their personal health data for clinical research.
Eighty-eight percent said clinical trial participants should have access to trial results. And 47% said they would like having clinical trial information/data/results delivered through their phone. ![]()
Predicting response to treatment in AML, MDS

Researchers say they have determined which patients will respond to treatment with SY-1425, a retinoic acid receptor alpha (RARα) agonist.
The team discovered a subset of patients with acute myeloid leukemia (AML) who had a super-enhancer associated with the RARA gene, which is predictive of response to SY-1425.
The researchers also identified a subset of patients with myelodysplastic syndromes (MDS) who had high expression of the RARA gene.
And experiments showed that RARA-high MDS had a similar response to SY-1425 as that seen in AML driven by the RARA super-enhancer.
Ravindra Majeti MD, PhD, of Stanford University School of Medicine in California, and colleagues reported these findings in Cancer Discovery. Employees of Syros Pharmaceuticals, the company developing SY-1425, were also involved in this research.
In collaboration with the Majeti lab, Syros used its gene control platform to analyze 66 AML patients’ tumor samples. In this way, the researchers identified 6 distinct patient subsets based on super-enhancer profiles, including 1 enriched for a super-enhancer associated with the RARA gene.
The team found that super-enhancer profiles were strongly associated with survival outcomes, often independent of known genetic mutations in AML.
The RARA super-enhancer was associated with high expression of the RARA gene, which codes for a transcription factor targeted by SY-1425.
The RARA super-enhancer was predictive of response to SY-1425. In AML cells with high RARA expression, SY-1425 reduced proliferation and promoted differentiation.
Moreover, SY-1425 decreased tumor burden and prolonged survival in patient-derived xenograft models of AML with high RARA expression. However, there was no effect on AML cells or models with low RARA expression.
The researchers said SY-1425 induced profound transcriptional changes promoting cell differentiation in AML cells with high RARA expression, but the drug produced little to no transcriptional changes in AML cells with low RARA expression.
DHRS3 was the most strongly and rapidly induced gene in response to treatment with SY-1425. This led to the identification of DHRS3 induction as an early indicator of whether SY-1425 is affecting the targeted biology in defined subsets of AML and MDS patients. It is therefore used as a pharmacodynamic marker in the ongoing phase 2 trial of SY-1425.
In this trial, researchers are assessing the safety and efficacy of SY-1425 as a single agent in 4 AML and MDS patient populations, as well as testing SY-1425 in combination with azacitidine in newly diagnosed AML patients who are not suitable candidates for standard chemotherapy. ![]()
Researchers say they have determined which patients will respond to treatment with SY-1425, a retinoic acid receptor alpha (RARα) agonist.
The team discovered a subset of patients with acute myeloid leukemia (AML) who had a super-enhancer associated with the RARA gene, which is predictive of response to SY-1425.
The researchers also identified a subset of patients with myelodysplastic syndromes (MDS) who had high expression of the RARA gene.
And experiments showed that RARA-high MDS had a similar response to SY-1425 as that seen in AML driven by the RARA super-enhancer.
Ravindra Majeti MD, PhD, of Stanford University School of Medicine in California, and colleagues reported these findings in Cancer Discovery. Employees of Syros Pharmaceuticals, the company developing SY-1425, were also involved in this research.
In collaboration with the Majeti lab, Syros used its gene control platform to analyze 66 AML patients’ tumor samples. In this way, the researchers identified 6 distinct patient subsets based on super-enhancer profiles, including 1 enriched for a super-enhancer associated with the RARA gene.
The team found that super-enhancer profiles were strongly associated with survival outcomes, often independent of known genetic mutations in AML.
The RARA super-enhancer was associated with high expression of the RARA gene, which codes for a transcription factor targeted by SY-1425.
The RARA super-enhancer was predictive of response to SY-1425. In AML cells with high RARA expression, SY-1425 reduced proliferation and promoted differentiation.
Moreover, SY-1425 decreased tumor burden and prolonged survival in patient-derived xenograft models of AML with high RARA expression. However, there was no effect on AML cells or models with low RARA expression.
The researchers said SY-1425 induced profound transcriptional changes promoting cell differentiation in AML cells with high RARA expression, but the drug produced little to no transcriptional changes in AML cells with low RARA expression.
DHRS3 was the most strongly and rapidly induced gene in response to treatment with SY-1425. This led to the identification of DHRS3 induction as an early indicator of whether SY-1425 is affecting the targeted biology in defined subsets of AML and MDS patients. It is therefore used as a pharmacodynamic marker in the ongoing phase 2 trial of SY-1425.
In this trial, researchers are assessing the safety and efficacy of SY-1425 as a single agent in 4 AML and MDS patient populations, as well as testing SY-1425 in combination with azacitidine in newly diagnosed AML patients who are not suitable candidates for standard chemotherapy. ![]()
Researchers say they have determined which patients will respond to treatment with SY-1425, a retinoic acid receptor alpha (RARα) agonist.
The team discovered a subset of patients with acute myeloid leukemia (AML) who had a super-enhancer associated with the RARA gene, which is predictive of response to SY-1425.
The researchers also identified a subset of patients with myelodysplastic syndromes (MDS) who had high expression of the RARA gene.
And experiments showed that RARA-high MDS had a similar response to SY-1425 as that seen in AML driven by the RARA super-enhancer.
Ravindra Majeti MD, PhD, of Stanford University School of Medicine in California, and colleagues reported these findings in Cancer Discovery. Employees of Syros Pharmaceuticals, the company developing SY-1425, were also involved in this research.
In collaboration with the Majeti lab, Syros used its gene control platform to analyze 66 AML patients’ tumor samples. In this way, the researchers identified 6 distinct patient subsets based on super-enhancer profiles, including 1 enriched for a super-enhancer associated with the RARA gene.
The team found that super-enhancer profiles were strongly associated with survival outcomes, often independent of known genetic mutations in AML.
The RARA super-enhancer was associated with high expression of the RARA gene, which codes for a transcription factor targeted by SY-1425.
The RARA super-enhancer was predictive of response to SY-1425. In AML cells with high RARA expression, SY-1425 reduced proliferation and promoted differentiation.
Moreover, SY-1425 decreased tumor burden and prolonged survival in patient-derived xenograft models of AML with high RARA expression. However, there was no effect on AML cells or models with low RARA expression.
The researchers said SY-1425 induced profound transcriptional changes promoting cell differentiation in AML cells with high RARA expression, but the drug produced little to no transcriptional changes in AML cells with low RARA expression.
DHRS3 was the most strongly and rapidly induced gene in response to treatment with SY-1425. This led to the identification of DHRS3 induction as an early indicator of whether SY-1425 is affecting the targeted biology in defined subsets of AML and MDS patients. It is therefore used as a pharmacodynamic marker in the ongoing phase 2 trial of SY-1425.
In this trial, researchers are assessing the safety and efficacy of SY-1425 as a single agent in 4 AML and MDS patient populations, as well as testing SY-1425 in combination with azacitidine in newly diagnosed AML patients who are not suitable candidates for standard chemotherapy. ![]()
England, Scotland to shorten deferral for high-risk blood donors
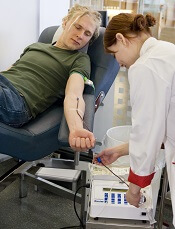
England and Scotland are changing some of their policies regarding blood donation, shortening the deferral periods for donors who engage in “high-risk” sexual behavior.
Wales is considering making the same changes to its blood donation policies but has not yet made a commitment to do so.
The policy changes will mean that men who have sex with men (MSM), commercial sex workers, and people with sexual partners who have a high risk of sexually transmitted infections (including those who have been sexually active in areas where HIV is common) will be able to donate blood after 3 months have passed since their last sexual activity.
At present, MSMs and individuals with high-risk sexual partners can only donate blood after 12 months have passed since their last sexual activity, and commercial sex workers are permanently banned from giving blood.
The change in deferral periods will begin to take effect in England in early 2018 and in Scotland in November 2017. Until then, the existing rules still apply.
The Advisory Committee on the Safety of Blood, Tissues and Organs (SaBTO), which advises UK ministers and health departments, recommended the aforementioned policy changes following a review of blood donor criteria and risk assessment of certain behaviors.
SaBTO also recommended shortening the deferral period for potential donors who have undergone acupuncture, piercing, tattooing, and endoscopy, as well as those with a history of non-prescribed injectable drug use.
However, this change requires changing UK legislation, in addition to a derogation from or amendments to current European Union legislation.
“We welcome the review by SaBTO and the recommendations,” said Moira Carter, Associate Director of Donor Services and Transport for the Scottish National Blood Transfusion Service.
“The updates for donor eligibility will allow more people the opportunity to give blood. The changes take into account the latest available medical and scientific evidence about the risk of acquiring infections that can be passed on in blood, along with evidence supporting the reliability of the blood screening tests we use.”
“We’re pleased that the lifetime ban on former and current sex workers has been lifted, and the deferral period is now in line with other deferrals based on sexual behavior,” said Alex Phillips, Blood Donations Policy Lead at Terrence Higgins Trust in London.
“We know from our research that the majority of sex workers take great care of their sexual health, with 98% of sex workers we asked rating their sexual health as very important, 76% having a sexual health check up every 3 months, and 98% knowing their HIV status.”
“Medical evidence is, of course, constantly and quickly being updated, so it’s important that the deferral periods are regularly reviewed in line with the latest evidence. We therefore hope that today’s changes will pave the way for more progress as further evidence becomes available.” ![]()
England and Scotland are changing some of their policies regarding blood donation, shortening the deferral periods for donors who engage in “high-risk” sexual behavior.
Wales is considering making the same changes to its blood donation policies but has not yet made a commitment to do so.
The policy changes will mean that men who have sex with men (MSM), commercial sex workers, and people with sexual partners who have a high risk of sexually transmitted infections (including those who have been sexually active in areas where HIV is common) will be able to donate blood after 3 months have passed since their last sexual activity.
At present, MSMs and individuals with high-risk sexual partners can only donate blood after 12 months have passed since their last sexual activity, and commercial sex workers are permanently banned from giving blood.
The change in deferral periods will begin to take effect in England in early 2018 and in Scotland in November 2017. Until then, the existing rules still apply.
The Advisory Committee on the Safety of Blood, Tissues and Organs (SaBTO), which advises UK ministers and health departments, recommended the aforementioned policy changes following a review of blood donor criteria and risk assessment of certain behaviors.
SaBTO also recommended shortening the deferral period for potential donors who have undergone acupuncture, piercing, tattooing, and endoscopy, as well as those with a history of non-prescribed injectable drug use.
However, this change requires changing UK legislation, in addition to a derogation from or amendments to current European Union legislation.
“We welcome the review by SaBTO and the recommendations,” said Moira Carter, Associate Director of Donor Services and Transport for the Scottish National Blood Transfusion Service.
“The updates for donor eligibility will allow more people the opportunity to give blood. The changes take into account the latest available medical and scientific evidence about the risk of acquiring infections that can be passed on in blood, along with evidence supporting the reliability of the blood screening tests we use.”
“We’re pleased that the lifetime ban on former and current sex workers has been lifted, and the deferral period is now in line with other deferrals based on sexual behavior,” said Alex Phillips, Blood Donations Policy Lead at Terrence Higgins Trust in London.
“We know from our research that the majority of sex workers take great care of their sexual health, with 98% of sex workers we asked rating their sexual health as very important, 76% having a sexual health check up every 3 months, and 98% knowing their HIV status.”
“Medical evidence is, of course, constantly and quickly being updated, so it’s important that the deferral periods are regularly reviewed in line with the latest evidence. We therefore hope that today’s changes will pave the way for more progress as further evidence becomes available.” ![]()
England and Scotland are changing some of their policies regarding blood donation, shortening the deferral periods for donors who engage in “high-risk” sexual behavior.
Wales is considering making the same changes to its blood donation policies but has not yet made a commitment to do so.
The policy changes will mean that men who have sex with men (MSM), commercial sex workers, and people with sexual partners who have a high risk of sexually transmitted infections (including those who have been sexually active in areas where HIV is common) will be able to donate blood after 3 months have passed since their last sexual activity.
At present, MSMs and individuals with high-risk sexual partners can only donate blood after 12 months have passed since their last sexual activity, and commercial sex workers are permanently banned from giving blood.
The change in deferral periods will begin to take effect in England in early 2018 and in Scotland in November 2017. Until then, the existing rules still apply.
The Advisory Committee on the Safety of Blood, Tissues and Organs (SaBTO), which advises UK ministers and health departments, recommended the aforementioned policy changes following a review of blood donor criteria and risk assessment of certain behaviors.
SaBTO also recommended shortening the deferral period for potential donors who have undergone acupuncture, piercing, tattooing, and endoscopy, as well as those with a history of non-prescribed injectable drug use.
However, this change requires changing UK legislation, in addition to a derogation from or amendments to current European Union legislation.
“We welcome the review by SaBTO and the recommendations,” said Moira Carter, Associate Director of Donor Services and Transport for the Scottish National Blood Transfusion Service.
“The updates for donor eligibility will allow more people the opportunity to give blood. The changes take into account the latest available medical and scientific evidence about the risk of acquiring infections that can be passed on in blood, along with evidence supporting the reliability of the blood screening tests we use.”
“We’re pleased that the lifetime ban on former and current sex workers has been lifted, and the deferral period is now in line with other deferrals based on sexual behavior,” said Alex Phillips, Blood Donations Policy Lead at Terrence Higgins Trust in London.
“We know from our research that the majority of sex workers take great care of their sexual health, with 98% of sex workers we asked rating their sexual health as very important, 76% having a sexual health check up every 3 months, and 98% knowing their HIV status.”
“Medical evidence is, of course, constantly and quickly being updated, so it’s important that the deferral periods are regularly reviewed in line with the latest evidence. We therefore hope that today’s changes will pave the way for more progress as further evidence becomes available.” ![]()
KRD appears more active, less safe than KCD in newly diagnosed MM

MADRID—New research suggests one triplet regimen may be more active but also more toxic than another in patients with newly diagnosed multiple myeloma (MM).
In the FORTE trial, the combination of carfilzomib, lenalidomide, and dexamethasone (KRD) produced higher response rates than carfilzomib, cyclophosphamide, and dexamethasone (KCD).
However, treatment with KRD also produced significantly more grade 3/4 non-hematologic adverse events (AEs) and serious AEs than the KCD regimen.
Francesca Gay, MD, PhD, of University of Torino in Italy, presented these results, from a planned interim analysis of FORTE, at the 22nd Congress of the European Hematology Association (EHA) as abstract S410.
The trial enrolled 477 MM patients younger than 65 years of age.
The patients were randomized to receive:
- Four 28-day KCD cycles (n=154)
Carfilzomib at 20 mg/m2 (for cycles 1 and 2) or 36 mg/m2 (for subsequent cycles) on days 1, 2, 8, 9, 15, and 16
Cyclophosphamide at 300 mg/m2 on days 1, 8, and 15
Dexamethasone at 20 mg on days 1, 2, 8, 9, 15, 16, 22, and 23
- Four 28-day KRD cycles (n=309)
Carfilzomib and dexamethasone as above
Lenalidomide at 25 mg on days 1-21
After the fourth induction cycle, all patients received cyclophosphamide at 2 g/m2, followed by peripheral blood stem cell (PBSC) collection for autologous stem cell transplant.
Patient characteristics
The median age was 57 (range, 52-62) in the KCD arm and the KRD arm (range, 51-62). In both arms, 55% of patients were male.
Roughly half of patients in both arms had an ISS stage of I (52% in the KCD arm and 51% in the KRD arm), 31% of patients in both arms were stage II, and 17% were stage III.
Fifty-eight percent of patients in the KCD arm and 59% in the KRD arm did not have t(4;14), t(14;16), or del17. Thirty-one percent and 26%, respectively, did have one of these cytogenetic abnormalities. (For the remaining patients, cytogenetic risk data were missing.)
PBSC mobilization and response
The median number of PBSCs collected was 8.6 x 106/kg in the KCD arm and 6.3 x 106/kg in the KRD arm. Plerixafor was required in 6% of patients in the KCD arm and 28% of patients in the KRD arm (P<0.001).
Ninety-two percent of patients in the KCD arm and 96% in the KRD arm had a partial response or better. Sixty-one percent and 74%, respectively, had a very good partial response (P=0.01). Six percent and 15%, respectively, had a complete response.
Safety
Grade 3/4 hematologic AEs and serious AEs (in the KRD and KCD arms, respectively) were thrombocytopenia (2% and 1%), neutropenia (6% and 5%), and anemia (2% and 3%). Seven percent of KRD patients and 6% of KCD patients had at least 1 grade 3/4 or serious hematologic AE.
Grade 3/4 non-hematologic AEs and serious AEs (in the KRD and KCD arms, respectively) included dermatologic events (8% and 1%, P<0.001), renal events (1% and 2%), fever (4% and 1%), infections (5% and 3%), gastrointestinal AEs (2% and 1%), hepatic events (8% and 1%, P<0.001), venous thromboembolism (1% and 0%), hypertension (3% and 2%), and cardiac events (1% and 2%).
Thirty-two percent of KRD patients and 16% of KCD patients had at least 1 grade 3/4 or serious non-hematologic AE (P<0.001).
Four percent of patients in the KRD arm and 2% in the KCD arm discontinued treatment due to AEs. Fifteen percent and 6%, respectively, reduced the dose of at least 1 drug (P=0.005).
One percent of patients in the KRD arm and 2% in the KCD arm died from AEs. In the KCD arm, 1 patient had a sudden death, and 1 died of pneumonia. In the KRD arm, there was 1 sudden death in a patient with sepsis, 1 patient died of infection, and 1 died of cardiac arrest (this patient previously discontinued treatment due to renal failure). ![]()
MADRID—New research suggests one triplet regimen may be more active but also more toxic than another in patients with newly diagnosed multiple myeloma (MM).
In the FORTE trial, the combination of carfilzomib, lenalidomide, and dexamethasone (KRD) produced higher response rates than carfilzomib, cyclophosphamide, and dexamethasone (KCD).
However, treatment with KRD also produced significantly more grade 3/4 non-hematologic adverse events (AEs) and serious AEs than the KCD regimen.
Francesca Gay, MD, PhD, of University of Torino in Italy, presented these results, from a planned interim analysis of FORTE, at the 22nd Congress of the European Hematology Association (EHA) as abstract S410.
The trial enrolled 477 MM patients younger than 65 years of age.
The patients were randomized to receive:
- Four 28-day KCD cycles (n=154)
Carfilzomib at 20 mg/m2 (for cycles 1 and 2) or 36 mg/m2 (for subsequent cycles) on days 1, 2, 8, 9, 15, and 16
Cyclophosphamide at 300 mg/m2 on days 1, 8, and 15
Dexamethasone at 20 mg on days 1, 2, 8, 9, 15, 16, 22, and 23
- Four 28-day KRD cycles (n=309)
Carfilzomib and dexamethasone as above
Lenalidomide at 25 mg on days 1-21
After the fourth induction cycle, all patients received cyclophosphamide at 2 g/m2, followed by peripheral blood stem cell (PBSC) collection for autologous stem cell transplant.
Patient characteristics
The median age was 57 (range, 52-62) in the KCD arm and the KRD arm (range, 51-62). In both arms, 55% of patients were male.
Roughly half of patients in both arms had an ISS stage of I (52% in the KCD arm and 51% in the KRD arm), 31% of patients in both arms were stage II, and 17% were stage III.
Fifty-eight percent of patients in the KCD arm and 59% in the KRD arm did not have t(4;14), t(14;16), or del17. Thirty-one percent and 26%, respectively, did have one of these cytogenetic abnormalities. (For the remaining patients, cytogenetic risk data were missing.)
PBSC mobilization and response
The median number of PBSCs collected was 8.6 x 106/kg in the KCD arm and 6.3 x 106/kg in the KRD arm. Plerixafor was required in 6% of patients in the KCD arm and 28% of patients in the KRD arm (P<0.001).
Ninety-two percent of patients in the KCD arm and 96% in the KRD arm had a partial response or better. Sixty-one percent and 74%, respectively, had a very good partial response (P=0.01). Six percent and 15%, respectively, had a complete response.
Safety
Grade 3/4 hematologic AEs and serious AEs (in the KRD and KCD arms, respectively) were thrombocytopenia (2% and 1%), neutropenia (6% and 5%), and anemia (2% and 3%). Seven percent of KRD patients and 6% of KCD patients had at least 1 grade 3/4 or serious hematologic AE.
Grade 3/4 non-hematologic AEs and serious AEs (in the KRD and KCD arms, respectively) included dermatologic events (8% and 1%, P<0.001), renal events (1% and 2%), fever (4% and 1%), infections (5% and 3%), gastrointestinal AEs (2% and 1%), hepatic events (8% and 1%, P<0.001), venous thromboembolism (1% and 0%), hypertension (3% and 2%), and cardiac events (1% and 2%).
Thirty-two percent of KRD patients and 16% of KCD patients had at least 1 grade 3/4 or serious non-hematologic AE (P<0.001).
Four percent of patients in the KRD arm and 2% in the KCD arm discontinued treatment due to AEs. Fifteen percent and 6%, respectively, reduced the dose of at least 1 drug (P=0.005).
One percent of patients in the KRD arm and 2% in the KCD arm died from AEs. In the KCD arm, 1 patient had a sudden death, and 1 died of pneumonia. In the KRD arm, there was 1 sudden death in a patient with sepsis, 1 patient died of infection, and 1 died of cardiac arrest (this patient previously discontinued treatment due to renal failure). ![]()
MADRID—New research suggests one triplet regimen may be more active but also more toxic than another in patients with newly diagnosed multiple myeloma (MM).
In the FORTE trial, the combination of carfilzomib, lenalidomide, and dexamethasone (KRD) produced higher response rates than carfilzomib, cyclophosphamide, and dexamethasone (KCD).
However, treatment with KRD also produced significantly more grade 3/4 non-hematologic adverse events (AEs) and serious AEs than the KCD regimen.
Francesca Gay, MD, PhD, of University of Torino in Italy, presented these results, from a planned interim analysis of FORTE, at the 22nd Congress of the European Hematology Association (EHA) as abstract S410.
The trial enrolled 477 MM patients younger than 65 years of age.
The patients were randomized to receive:
- Four 28-day KCD cycles (n=154)
Carfilzomib at 20 mg/m2 (for cycles 1 and 2) or 36 mg/m2 (for subsequent cycles) on days 1, 2, 8, 9, 15, and 16
Cyclophosphamide at 300 mg/m2 on days 1, 8, and 15
Dexamethasone at 20 mg on days 1, 2, 8, 9, 15, 16, 22, and 23
- Four 28-day KRD cycles (n=309)
Carfilzomib and dexamethasone as above
Lenalidomide at 25 mg on days 1-21
After the fourth induction cycle, all patients received cyclophosphamide at 2 g/m2, followed by peripheral blood stem cell (PBSC) collection for autologous stem cell transplant.
Patient characteristics
The median age was 57 (range, 52-62) in the KCD arm and the KRD arm (range, 51-62). In both arms, 55% of patients were male.
Roughly half of patients in both arms had an ISS stage of I (52% in the KCD arm and 51% in the KRD arm), 31% of patients in both arms were stage II, and 17% were stage III.
Fifty-eight percent of patients in the KCD arm and 59% in the KRD arm did not have t(4;14), t(14;16), or del17. Thirty-one percent and 26%, respectively, did have one of these cytogenetic abnormalities. (For the remaining patients, cytogenetic risk data were missing.)
PBSC mobilization and response
The median number of PBSCs collected was 8.6 x 106/kg in the KCD arm and 6.3 x 106/kg in the KRD arm. Plerixafor was required in 6% of patients in the KCD arm and 28% of patients in the KRD arm (P<0.001).
Ninety-two percent of patients in the KCD arm and 96% in the KRD arm had a partial response or better. Sixty-one percent and 74%, respectively, had a very good partial response (P=0.01). Six percent and 15%, respectively, had a complete response.
Safety
Grade 3/4 hematologic AEs and serious AEs (in the KRD and KCD arms, respectively) were thrombocytopenia (2% and 1%), neutropenia (6% and 5%), and anemia (2% and 3%). Seven percent of KRD patients and 6% of KCD patients had at least 1 grade 3/4 or serious hematologic AE.
Grade 3/4 non-hematologic AEs and serious AEs (in the KRD and KCD arms, respectively) included dermatologic events (8% and 1%, P<0.001), renal events (1% and 2%), fever (4% and 1%), infections (5% and 3%), gastrointestinal AEs (2% and 1%), hepatic events (8% and 1%, P<0.001), venous thromboembolism (1% and 0%), hypertension (3% and 2%), and cardiac events (1% and 2%).
Thirty-two percent of KRD patients and 16% of KCD patients had at least 1 grade 3/4 or serious non-hematologic AE (P<0.001).
Four percent of patients in the KRD arm and 2% in the KCD arm discontinued treatment due to AEs. Fifteen percent and 6%, respectively, reduced the dose of at least 1 drug (P=0.005).
One percent of patients in the KRD arm and 2% in the KCD arm died from AEs. In the KCD arm, 1 patient had a sudden death, and 1 died of pneumonia. In the KRD arm, there was 1 sudden death in a patient with sepsis, 1 patient died of infection, and 1 died of cardiac arrest (this patient previously discontinued treatment due to renal failure). ![]()
High ORR with sequential regimen in CLL

LUGANO, SWITZERLAND—A sequential treatment regimen can produce a high overall response rate (ORR) in patients with treatment-naïve (TN) or relapsed/refractory (RR) chronic lymphocytic leukemia (CLL), results of the CLL2-BAG study suggest.
Patients who received bendamustine followed by obinutuzumab and venetoclax achieved an ORR of 95%, and 87% of them were negative for minimal residual disease (MRD) in the peripheral blood.
In addition, this regimen did not lead to cumulative or unexpected toxicity, according to study investigator Paula Cramer, MD, of University Hospital of Cologne in Germany.
Dr Cramer presented results from CLL2-BAG at the 14th International Conference on Malignant Lymphoma (ICML). The trial was sponsored by the German CLL Study Group.
CLL2-BAG included 63 patients—34 with TN and 29 with RR CLL. The median age was 58 (range, 43-76) and 61 (range, 28-77), respectively.
Thirty-five percent of TN patients had bulky disease (>5 cm), as did 45% of RR patients. Twelve percent and 10%, respectively, had massive splenomegaly (>20 cm)
Twenty-one percent of TN patients and 35% of RR patients were Binet stage A. Thirty-two percent and 21%, respectively, were stage B, and 47% and 45%, respectively, were stage C.
Treatment
Patients first underwent debulking with bendamustine, given at 70 mg/m2 on days 1 and 2 for two 28-day cycles. They then received induction with obinutuzumab and venetoclax for six 28-day cycles.
In cycle 1, patients received obinutuzumab at 100 mg or 900 mg on day 1 or 2 and 1000 mg on days 8 and 15. For cycles 2-6, patients received 1000 mg of obinutuzumab on day 1.
In cycle 2, patients received venetoclax at 20 mg on days 1-7, 50 mg on days 8-14, 100 mg on days 15-21, and 200 mg on days 22-28. For cycles 3-6, they received venetoclax at 400 mg on days 1-28.
Patients also received maintenance with obinutuzumab and venetoclax for 2 to 8 cycles with a duration of 84 days. Obinutuzumab was given at 1000 mg on day 1 of each cycle, and venetoclax was given at 400 mg on days 1-84.
Maintenance was continued until patients completed 24 months of maintenance therapy, until 3 months after patients achieved a complete response (CR) or CR with incomplete count recovery (CRi) and MRD negativity, until progression of CLL or start of new CLL treatment, or until unacceptable toxicity.
ORR, MRD, and survival
At the end of induction, the ORR was 95% in the entire population—100% among TN patients and 90% among RR patients.
The rate of CR was 8%, 9%, and 7%, respectively. The rate of unconfirmed/clinical CR/CRi was 32%, 41%, and 21%, respectively.
Five percent of patients progressed, all of them in the RR group.
Eighty-seven percent of evaluable patients were MRD negative (<10-4) in the peripheral blood, including 91% of TN patients and 83% of RR patients. Two patients (3%) were missing data on MRD status in peripheral blood.
Thirteen percent of all evaluable patients were MRD negative in the bone marrow, including 12% of TN patients and 14% of RR patients. The remaining patients (87%, 88%, and 86%, respectively) were missing data on MRD status in the bone marrow.
The progression-free survival at 15 months was 100% in the TN patients and 83% in the RR patients.
Adverse events
After debulking, 28.1% of TN patients and 46.7% of RR patients had experienced adverse events (AEs).
These included (in TN and RR patients, respectively) neutropenia (9.4% and 13.3%), anemia (6.3% and 20.0%), thrombocytopenia (6.3% and 6.7%), infections (6.3% and 6.7%), coronary artery disorders (1 TN patient, 3.1%), rash (1 TN patient, 3.1%), tumor lysis syndrome (1 TN patient, 3.1%), vomiting (1 TN patient, 3.1%), and pyrexia (1 RR patient, 6.7%).
After induction, 54.3% of TN patients and 80.6% of RR patients had experienced AEs.
These included (in TN and RR patients, respectively) neutropenia (34.3% and 54.8%), infections (8.6% and 29.0%), thrombocytopenia (2.9% and 22.6%), infusion-related reactions (5 RR patients, 16.1%), neoplasms (2.9% and 9.7%), hypertension (2.9% and 6.5%), coronary artery disorders (2.9% and 6.5%), anemia (3 RR patients, 9.7%), and tumor lysis syndrome (2 RR patients, 6.5%). ![]()
LUGANO, SWITZERLAND—A sequential treatment regimen can produce a high overall response rate (ORR) in patients with treatment-naïve (TN) or relapsed/refractory (RR) chronic lymphocytic leukemia (CLL), results of the CLL2-BAG study suggest.
Patients who received bendamustine followed by obinutuzumab and venetoclax achieved an ORR of 95%, and 87% of them were negative for minimal residual disease (MRD) in the peripheral blood.
In addition, this regimen did not lead to cumulative or unexpected toxicity, according to study investigator Paula Cramer, MD, of University Hospital of Cologne in Germany.
Dr Cramer presented results from CLL2-BAG at the 14th International Conference on Malignant Lymphoma (ICML). The trial was sponsored by the German CLL Study Group.
CLL2-BAG included 63 patients—34 with TN and 29 with RR CLL. The median age was 58 (range, 43-76) and 61 (range, 28-77), respectively.
Thirty-five percent of TN patients had bulky disease (>5 cm), as did 45% of RR patients. Twelve percent and 10%, respectively, had massive splenomegaly (>20 cm)
Twenty-one percent of TN patients and 35% of RR patients were Binet stage A. Thirty-two percent and 21%, respectively, were stage B, and 47% and 45%, respectively, were stage C.
Treatment
Patients first underwent debulking with bendamustine, given at 70 mg/m2 on days 1 and 2 for two 28-day cycles. They then received induction with obinutuzumab and venetoclax for six 28-day cycles.
In cycle 1, patients received obinutuzumab at 100 mg or 900 mg on day 1 or 2 and 1000 mg on days 8 and 15. For cycles 2-6, patients received 1000 mg of obinutuzumab on day 1.
In cycle 2, patients received venetoclax at 20 mg on days 1-7, 50 mg on days 8-14, 100 mg on days 15-21, and 200 mg on days 22-28. For cycles 3-6, they received venetoclax at 400 mg on days 1-28.
Patients also received maintenance with obinutuzumab and venetoclax for 2 to 8 cycles with a duration of 84 days. Obinutuzumab was given at 1000 mg on day 1 of each cycle, and venetoclax was given at 400 mg on days 1-84.
Maintenance was continued until patients completed 24 months of maintenance therapy, until 3 months after patients achieved a complete response (CR) or CR with incomplete count recovery (CRi) and MRD negativity, until progression of CLL or start of new CLL treatment, or until unacceptable toxicity.
ORR, MRD, and survival
At the end of induction, the ORR was 95% in the entire population—100% among TN patients and 90% among RR patients.
The rate of CR was 8%, 9%, and 7%, respectively. The rate of unconfirmed/clinical CR/CRi was 32%, 41%, and 21%, respectively.
Five percent of patients progressed, all of them in the RR group.
Eighty-seven percent of evaluable patients were MRD negative (<10-4) in the peripheral blood, including 91% of TN patients and 83% of RR patients. Two patients (3%) were missing data on MRD status in peripheral blood.
Thirteen percent of all evaluable patients were MRD negative in the bone marrow, including 12% of TN patients and 14% of RR patients. The remaining patients (87%, 88%, and 86%, respectively) were missing data on MRD status in the bone marrow.
The progression-free survival at 15 months was 100% in the TN patients and 83% in the RR patients.
Adverse events
After debulking, 28.1% of TN patients and 46.7% of RR patients had experienced adverse events (AEs).
These included (in TN and RR patients, respectively) neutropenia (9.4% and 13.3%), anemia (6.3% and 20.0%), thrombocytopenia (6.3% and 6.7%), infections (6.3% and 6.7%), coronary artery disorders (1 TN patient, 3.1%), rash (1 TN patient, 3.1%), tumor lysis syndrome (1 TN patient, 3.1%), vomiting (1 TN patient, 3.1%), and pyrexia (1 RR patient, 6.7%).
After induction, 54.3% of TN patients and 80.6% of RR patients had experienced AEs.
These included (in TN and RR patients, respectively) neutropenia (34.3% and 54.8%), infections (8.6% and 29.0%), thrombocytopenia (2.9% and 22.6%), infusion-related reactions (5 RR patients, 16.1%), neoplasms (2.9% and 9.7%), hypertension (2.9% and 6.5%), coronary artery disorders (2.9% and 6.5%), anemia (3 RR patients, 9.7%), and tumor lysis syndrome (2 RR patients, 6.5%). ![]()
LUGANO, SWITZERLAND—A sequential treatment regimen can produce a high overall response rate (ORR) in patients with treatment-naïve (TN) or relapsed/refractory (RR) chronic lymphocytic leukemia (CLL), results of the CLL2-BAG study suggest.
Patients who received bendamustine followed by obinutuzumab and venetoclax achieved an ORR of 95%, and 87% of them were negative for minimal residual disease (MRD) in the peripheral blood.
In addition, this regimen did not lead to cumulative or unexpected toxicity, according to study investigator Paula Cramer, MD, of University Hospital of Cologne in Germany.
Dr Cramer presented results from CLL2-BAG at the 14th International Conference on Malignant Lymphoma (ICML). The trial was sponsored by the German CLL Study Group.
CLL2-BAG included 63 patients—34 with TN and 29 with RR CLL. The median age was 58 (range, 43-76) and 61 (range, 28-77), respectively.
Thirty-five percent of TN patients had bulky disease (>5 cm), as did 45% of RR patients. Twelve percent and 10%, respectively, had massive splenomegaly (>20 cm)
Twenty-one percent of TN patients and 35% of RR patients were Binet stage A. Thirty-two percent and 21%, respectively, were stage B, and 47% and 45%, respectively, were stage C.
Treatment
Patients first underwent debulking with bendamustine, given at 70 mg/m2 on days 1 and 2 for two 28-day cycles. They then received induction with obinutuzumab and venetoclax for six 28-day cycles.
In cycle 1, patients received obinutuzumab at 100 mg or 900 mg on day 1 or 2 and 1000 mg on days 8 and 15. For cycles 2-6, patients received 1000 mg of obinutuzumab on day 1.
In cycle 2, patients received venetoclax at 20 mg on days 1-7, 50 mg on days 8-14, 100 mg on days 15-21, and 200 mg on days 22-28. For cycles 3-6, they received venetoclax at 400 mg on days 1-28.
Patients also received maintenance with obinutuzumab and venetoclax for 2 to 8 cycles with a duration of 84 days. Obinutuzumab was given at 1000 mg on day 1 of each cycle, and venetoclax was given at 400 mg on days 1-84.
Maintenance was continued until patients completed 24 months of maintenance therapy, until 3 months after patients achieved a complete response (CR) or CR with incomplete count recovery (CRi) and MRD negativity, until progression of CLL or start of new CLL treatment, or until unacceptable toxicity.
ORR, MRD, and survival
At the end of induction, the ORR was 95% in the entire population—100% among TN patients and 90% among RR patients.
The rate of CR was 8%, 9%, and 7%, respectively. The rate of unconfirmed/clinical CR/CRi was 32%, 41%, and 21%, respectively.
Five percent of patients progressed, all of them in the RR group.
Eighty-seven percent of evaluable patients were MRD negative (<10-4) in the peripheral blood, including 91% of TN patients and 83% of RR patients. Two patients (3%) were missing data on MRD status in peripheral blood.
Thirteen percent of all evaluable patients were MRD negative in the bone marrow, including 12% of TN patients and 14% of RR patients. The remaining patients (87%, 88%, and 86%, respectively) were missing data on MRD status in the bone marrow.
The progression-free survival at 15 months was 100% in the TN patients and 83% in the RR patients.
Adverse events
After debulking, 28.1% of TN patients and 46.7% of RR patients had experienced adverse events (AEs).
These included (in TN and RR patients, respectively) neutropenia (9.4% and 13.3%), anemia (6.3% and 20.0%), thrombocytopenia (6.3% and 6.7%), infections (6.3% and 6.7%), coronary artery disorders (1 TN patient, 3.1%), rash (1 TN patient, 3.1%), tumor lysis syndrome (1 TN patient, 3.1%), vomiting (1 TN patient, 3.1%), and pyrexia (1 RR patient, 6.7%).
After induction, 54.3% of TN patients and 80.6% of RR patients had experienced AEs.
These included (in TN and RR patients, respectively) neutropenia (34.3% and 54.8%), infections (8.6% and 29.0%), thrombocytopenia (2.9% and 22.6%), infusion-related reactions (5 RR patients, 16.1%), neoplasms (2.9% and 9.7%), hypertension (2.9% and 6.5%), coronary artery disorders (2.9% and 6.5%), anemia (3 RR patients, 9.7%), and tumor lysis syndrome (2 RR patients, 6.5%). ![]()
Tests reveal risk of passing on SCD, other diseases

Quest Diagnostics has announced the US launch of QHerit™, a genetic screening service that helps people of multiple ethnicities identify their risk of passing on heritable disorders to their offspring.
The QHerit Pan-Ethnic Expanded Carrier Screen is a panel of tests for the 22 heritable diseases cited in new screening guidelines from the American College of Gynecology (ACOG).
Among the diseases are alpha-thalassemia, Fanconi anemia, and beta-hemoglobinopathies, including sickle cell disease (SCD).
Traditionally, genetic carrier screening has been used for at-risk populations based on specific ancestry assumptions and focused on only a few likely disorders with higher prevalence associated with that ethnicity.
In its new guidelines, ACOG recommends offering pan-ethnic, expanded carrier, and ethnic-specific screening for all women considering pregnancy. The guidelines also state that the partner of a woman who tests positive may be a candidate for screening.
QHerit screens women and men for clinically relevant variants of genes for disorders that could have potentially devastating consequences, result in early death, or create a need for significant early intervention.
The disorders covered by QHerit include:
| Disease | Race/ethnicity | |||
| Alpha-thalassemia | Mediterranean, Middle East, Southeast Asian, African, Chinese, Asian Indian | |||
| Beta-
hemoglobinopathies (including SCD) |
Mediterranean, Middle East, Southeast Asian, African, Chinese, Asian Indian | |||
| Bloom syndrome | Ashkenazi Jewish descent (AJ) | |||
| Canavan disease | AJ and non-AJ | |||
| Cystic fibrosis | African American, AJ, Asian American, Hispanic American, non-Hispanic Caucasian | |||
| Dihydrolipoamide
dehydrogenase deficiency |
AJ | |||
| Familial dysautonomia | AJ | |||
| Familial hyperinsulinism | AJ | |||
| Fanconi anemia Type C | AJ | |||
| Fragile X syndrome | Females | |||
| Gaucher disease | AJ | |||
| Glycogen storage
disease Type Ia |
AJ, Caucasian | |||
| Joubert syndrome 2 | AJ | |||
| Maple syrup urine
disease |
AJ | |||
| Mucolipidosis Type IV | AJ | |||
| Nemaline myopathy | AJ | |||
| Niemann-Pick
disease Types A & B |
AJ | |||
| Spinal muscular
atrophy |
African American, AJ, Asian, Caucasian, Hispanic | |||
| Tay-Sachs disease | AJ, French Canadian, general population | |||
| Usher syndrome,
Type IF |
AJ | |||
| Usher syndrome,
Type IIIA |
AJ | |||
| Walker-Warburg
syndrome |
AJ | |||
QHerit is now available for order by US physicians. For more information, visit www.QHerit.com. ![]()
Quest Diagnostics has announced the US launch of QHerit™, a genetic screening service that helps people of multiple ethnicities identify their risk of passing on heritable disorders to their offspring.
The QHerit Pan-Ethnic Expanded Carrier Screen is a panel of tests for the 22 heritable diseases cited in new screening guidelines from the American College of Gynecology (ACOG).
Among the diseases are alpha-thalassemia, Fanconi anemia, and beta-hemoglobinopathies, including sickle cell disease (SCD).
Traditionally, genetic carrier screening has been used for at-risk populations based on specific ancestry assumptions and focused on only a few likely disorders with higher prevalence associated with that ethnicity.
In its new guidelines, ACOG recommends offering pan-ethnic, expanded carrier, and ethnic-specific screening for all women considering pregnancy. The guidelines also state that the partner of a woman who tests positive may be a candidate for screening.
QHerit screens women and men for clinically relevant variants of genes for disorders that could have potentially devastating consequences, result in early death, or create a need for significant early intervention.
The disorders covered by QHerit include:
| Disease | Race/ethnicity | |||
| Alpha-thalassemia | Mediterranean, Middle East, Southeast Asian, African, Chinese, Asian Indian | |||
| Beta-
hemoglobinopathies (including SCD) |
Mediterranean, Middle East, Southeast Asian, African, Chinese, Asian Indian | |||
| Bloom syndrome | Ashkenazi Jewish descent (AJ) | |||
| Canavan disease | AJ and non-AJ | |||
| Cystic fibrosis | African American, AJ, Asian American, Hispanic American, non-Hispanic Caucasian | |||
| Dihydrolipoamide
dehydrogenase deficiency |
AJ | |||
| Familial dysautonomia | AJ | |||
| Familial hyperinsulinism | AJ | |||
| Fanconi anemia Type C | AJ | |||
| Fragile X syndrome | Females | |||
| Gaucher disease | AJ | |||
| Glycogen storage
disease Type Ia |
AJ, Caucasian | |||
| Joubert syndrome 2 | AJ | |||
| Maple syrup urine
disease |
AJ | |||
| Mucolipidosis Type IV | AJ | |||
| Nemaline myopathy | AJ | |||
| Niemann-Pick
disease Types A & B |
AJ | |||
| Spinal muscular
atrophy |
African American, AJ, Asian, Caucasian, Hispanic | |||
| Tay-Sachs disease | AJ, French Canadian, general population | |||
| Usher syndrome,
Type IF |
AJ | |||
| Usher syndrome,
Type IIIA |
AJ | |||
| Walker-Warburg
syndrome |
AJ | |||
QHerit is now available for order by US physicians. For more information, visit www.QHerit.com. ![]()
Quest Diagnostics has announced the US launch of QHerit™, a genetic screening service that helps people of multiple ethnicities identify their risk of passing on heritable disorders to their offspring.
The QHerit Pan-Ethnic Expanded Carrier Screen is a panel of tests for the 22 heritable diseases cited in new screening guidelines from the American College of Gynecology (ACOG).
Among the diseases are alpha-thalassemia, Fanconi anemia, and beta-hemoglobinopathies, including sickle cell disease (SCD).
Traditionally, genetic carrier screening has been used for at-risk populations based on specific ancestry assumptions and focused on only a few likely disorders with higher prevalence associated with that ethnicity.
In its new guidelines, ACOG recommends offering pan-ethnic, expanded carrier, and ethnic-specific screening for all women considering pregnancy. The guidelines also state that the partner of a woman who tests positive may be a candidate for screening.
QHerit screens women and men for clinically relevant variants of genes for disorders that could have potentially devastating consequences, result in early death, or create a need for significant early intervention.
The disorders covered by QHerit include:
| Disease | Race/ethnicity | |||
| Alpha-thalassemia | Mediterranean, Middle East, Southeast Asian, African, Chinese, Asian Indian | |||
| Beta-
hemoglobinopathies (including SCD) |
Mediterranean, Middle East, Southeast Asian, African, Chinese, Asian Indian | |||
| Bloom syndrome | Ashkenazi Jewish descent (AJ) | |||
| Canavan disease | AJ and non-AJ | |||
| Cystic fibrosis | African American, AJ, Asian American, Hispanic American, non-Hispanic Caucasian | |||
| Dihydrolipoamide
dehydrogenase deficiency |
AJ | |||
| Familial dysautonomia | AJ | |||
| Familial hyperinsulinism | AJ | |||
| Fanconi anemia Type C | AJ | |||
| Fragile X syndrome | Females | |||
| Gaucher disease | AJ | |||
| Glycogen storage
disease Type Ia |
AJ, Caucasian | |||
| Joubert syndrome 2 | AJ | |||
| Maple syrup urine
disease |
AJ | |||
| Mucolipidosis Type IV | AJ | |||
| Nemaline myopathy | AJ | |||
| Niemann-Pick
disease Types A & B |
AJ | |||
| Spinal muscular
atrophy |
African American, AJ, Asian, Caucasian, Hispanic | |||
| Tay-Sachs disease | AJ, French Canadian, general population | |||
| Usher syndrome,
Type IF |
AJ | |||
| Usher syndrome,
Type IIIA |
AJ | |||
| Walker-Warburg
syndrome |
AJ | |||
QHerit is now available for order by US physicians. For more information, visit www.QHerit.com.
CHMP recommends midostaurin for FLT3+ AML, SM

The European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP) is recommending approval for midostaurin (Rydapt®) as a treatment for acute myeloid leukemia (AML) and systemic mastocytosis (SM).
If approved by the European Commission, midostaurin would be used in combination with standard daunorubicin and cytarabine induction and high-dose cytarabine consolidation—followed by midostaurin maintenance for patients in complete response—in adults with newly diagnosed acute myeloid leukemia (AML) who are FLT3-mutation-positive.
Midostaurin would also be approved to treat adults with aggressive SM, SM with associated hematological neoplasm (SM-AHN), and mast cell leukemia (MCL).
If approved, midostaurin would be the first targeted treatment available in the European Union for newly diagnosed FLT3+ AML patients and advanced SM patients.
The European Commission typically adheres to the CHMP’s recommendations and delivers its final decision within 2 to 3 months’ of the CHMP’s recommendation. The decision will be applicable to all member states of the European Union, plus Iceland, Liechtenstein, and Norway.
Midostaurin in AML
The CHMP’s recommendation for midostaurin in AML is based on results from the phase 3 RATIFY trial, which were recently published in NEJM.
In RATIFY, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in 717 adults younger than age 60 who had FLT3-mutated AML.
The median overall survival was significantly longer in the midostaurin arm than the placebo arm—74.7 months and 25.6 months, respectively (hazard ratio=0.77, P=0.016).
And the median event-free survival was significantly longer in the midostaurin arm than the placebo arm—8.2 months and 3.0 months, respectively (hazard ratio=0.78, P=0.004).
The most frequent adverse events (AEs) in the midostaurin arm (occurring in at least 20% of patients) were febrile neutropenia, nausea, vomiting, mucositis, headache, musculoskeletal pain, petechiae, device-related infection, epistaxis, hyperglycemia, and upper respiratory tract infections.
The most frequent grade 3/4 AEs (occurring in at least 10% of patients) were febrile neutropenia, device-related infection, and mucositis. Nine percent of patients in the midostaurin arm stopped treatment due to AEs, as did 6% in the placebo arm.
Midostaurin in advanced SM
The CHMP’s recommendation for midostaurin in advanced SM was based on results from a pair of phase 2, single-arm studies, hereafter referred to as Study 2 and Study 3.
Data from Study 2 were published in NEJM in June 2016, and data from Study 3 were presented at the 2010 ASH Annual Meeting.
Study 2 included 116 patients, 115 of whom were evaluable for response.
The overall response rate (ORR) was 17% in the entire cohort, 31% among patients with ASM, 11% among patients with SM-AHN, and 19% among patients with MCL. The complete response rates were 2%, 6%, 0%, and 5%, respectively.
Study 3 included 26 patients with advanced SM. In 3 of the patients, the subtype of SM was unconfirmed.
Among the 17 patients with SM-AHN, there were 10 responses (ORR=59%), including 1 partial response and 9 major responses. In the 6 patients with MCL, there were 2 responses (ORR=33%), which included 1 partial response and 1 major response.
In both studies combined, there were 142 adults with ASM, SM-AHN, or MCL.
The most frequent AEs (excluding laboratory abnormalities) that occurred in at least 20% of these patients were nausea, vomiting, diarrhea, edema, musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, constipation, pyrexia, headache, and dyspnea.
The most frequent grade 3 or higher AEs (excluding laboratory abnormalities) that occurred in at least 5% of patients were fatigue, sepsis, gastrointestinal hemorrhage, pneumonia, diarrhea, febrile neutropenia, edema, dyspnea, nausea, vomiting, abdominal pain, and renal insufficiency.
Serious AEs occurred in 68% of patients, most commonly infections and gastrointestinal disorders. Twenty-one percent of patients discontinued treatment due to AEs, the most frequent of which were infection, nausea or vomiting, QT prolongation, and gastrointestinal hemorrhage.
The European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP) is recommending approval for midostaurin (Rydapt®) as a treatment for acute myeloid leukemia (AML) and systemic mastocytosis (SM).
If approved by the European Commission, midostaurin would be used in combination with standard daunorubicin and cytarabine induction and high-dose cytarabine consolidation—followed by midostaurin maintenance for patients in complete response—in adults with newly diagnosed acute myeloid leukemia (AML) who are FLT3-mutation-positive.
Midostaurin would also be approved to treat adults with aggressive SM, SM with associated hematological neoplasm (SM-AHN), and mast cell leukemia (MCL).
If approved, midostaurin would be the first targeted treatment available in the European Union for newly diagnosed FLT3+ AML patients and advanced SM patients.
The European Commission typically adheres to the CHMP’s recommendations and delivers its final decision within 2 to 3 months’ of the CHMP’s recommendation. The decision will be applicable to all member states of the European Union, plus Iceland, Liechtenstein, and Norway.
Midostaurin in AML
The CHMP’s recommendation for midostaurin in AML is based on results from the phase 3 RATIFY trial, which were recently published in NEJM.
In RATIFY, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in 717 adults younger than age 60 who had FLT3-mutated AML.
The median overall survival was significantly longer in the midostaurin arm than the placebo arm—74.7 months and 25.6 months, respectively (hazard ratio=0.77, P=0.016).
And the median event-free survival was significantly longer in the midostaurin arm than the placebo arm—8.2 months and 3.0 months, respectively (hazard ratio=0.78, P=0.004).
The most frequent adverse events (AEs) in the midostaurin arm (occurring in at least 20% of patients) were febrile neutropenia, nausea, vomiting, mucositis, headache, musculoskeletal pain, petechiae, device-related infection, epistaxis, hyperglycemia, and upper respiratory tract infections.
The most frequent grade 3/4 AEs (occurring in at least 10% of patients) were febrile neutropenia, device-related infection, and mucositis. Nine percent of patients in the midostaurin arm stopped treatment due to AEs, as did 6% in the placebo arm.
Midostaurin in advanced SM
The CHMP’s recommendation for midostaurin in advanced SM was based on results from a pair of phase 2, single-arm studies, hereafter referred to as Study 2 and Study 3.
Data from Study 2 were published in NEJM in June 2016, and data from Study 3 were presented at the 2010 ASH Annual Meeting.
Study 2 included 116 patients, 115 of whom were evaluable for response.
The overall response rate (ORR) was 17% in the entire cohort, 31% among patients with ASM, 11% among patients with SM-AHN, and 19% among patients with MCL. The complete response rates were 2%, 6%, 0%, and 5%, respectively.
Study 3 included 26 patients with advanced SM. In 3 of the patients, the subtype of SM was unconfirmed.
Among the 17 patients with SM-AHN, there were 10 responses (ORR=59%), including 1 partial response and 9 major responses. In the 6 patients with MCL, there were 2 responses (ORR=33%), which included 1 partial response and 1 major response.
In both studies combined, there were 142 adults with ASM, SM-AHN, or MCL.
The most frequent AEs (excluding laboratory abnormalities) that occurred in at least 20% of these patients were nausea, vomiting, diarrhea, edema, musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, constipation, pyrexia, headache, and dyspnea.
The most frequent grade 3 or higher AEs (excluding laboratory abnormalities) that occurred in at least 5% of patients were fatigue, sepsis, gastrointestinal hemorrhage, pneumonia, diarrhea, febrile neutropenia, edema, dyspnea, nausea, vomiting, abdominal pain, and renal insufficiency.
Serious AEs occurred in 68% of patients, most commonly infections and gastrointestinal disorders. Twenty-one percent of patients discontinued treatment due to AEs, the most frequent of which were infection, nausea or vomiting, QT prolongation, and gastrointestinal hemorrhage.
The European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP) is recommending approval for midostaurin (Rydapt®) as a treatment for acute myeloid leukemia (AML) and systemic mastocytosis (SM).
If approved by the European Commission, midostaurin would be used in combination with standard daunorubicin and cytarabine induction and high-dose cytarabine consolidation—followed by midostaurin maintenance for patients in complete response—in adults with newly diagnosed acute myeloid leukemia (AML) who are FLT3-mutation-positive.
Midostaurin would also be approved to treat adults with aggressive SM, SM with associated hematological neoplasm (SM-AHN), and mast cell leukemia (MCL).
If approved, midostaurin would be the first targeted treatment available in the European Union for newly diagnosed FLT3+ AML patients and advanced SM patients.
The European Commission typically adheres to the CHMP’s recommendations and delivers its final decision within 2 to 3 months’ of the CHMP’s recommendation. The decision will be applicable to all member states of the European Union, plus Iceland, Liechtenstein, and Norway.
Midostaurin in AML
The CHMP’s recommendation for midostaurin in AML is based on results from the phase 3 RATIFY trial, which were recently published in NEJM.
In RATIFY, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in 717 adults younger than age 60 who had FLT3-mutated AML.
The median overall survival was significantly longer in the midostaurin arm than the placebo arm—74.7 months and 25.6 months, respectively (hazard ratio=0.77, P=0.016).
And the median event-free survival was significantly longer in the midostaurin arm than the placebo arm—8.2 months and 3.0 months, respectively (hazard ratio=0.78, P=0.004).
The most frequent adverse events (AEs) in the midostaurin arm (occurring in at least 20% of patients) were febrile neutropenia, nausea, vomiting, mucositis, headache, musculoskeletal pain, petechiae, device-related infection, epistaxis, hyperglycemia, and upper respiratory tract infections.
The most frequent grade 3/4 AEs (occurring in at least 10% of patients) were febrile neutropenia, device-related infection, and mucositis. Nine percent of patients in the midostaurin arm stopped treatment due to AEs, as did 6% in the placebo arm.
Midostaurin in advanced SM
The CHMP’s recommendation for midostaurin in advanced SM was based on results from a pair of phase 2, single-arm studies, hereafter referred to as Study 2 and Study 3.
Data from Study 2 were published in NEJM in June 2016, and data from Study 3 were presented at the 2010 ASH Annual Meeting.
Study 2 included 116 patients, 115 of whom were evaluable for response.
The overall response rate (ORR) was 17% in the entire cohort, 31% among patients with ASM, 11% among patients with SM-AHN, and 19% among patients with MCL. The complete response rates were 2%, 6%, 0%, and 5%, respectively.
Study 3 included 26 patients with advanced SM. In 3 of the patients, the subtype of SM was unconfirmed.
Among the 17 patients with SM-AHN, there were 10 responses (ORR=59%), including 1 partial response and 9 major responses. In the 6 patients with MCL, there were 2 responses (ORR=33%), which included 1 partial response and 1 major response.
In both studies combined, there were 142 adults with ASM, SM-AHN, or MCL.
The most frequent AEs (excluding laboratory abnormalities) that occurred in at least 20% of these patients were nausea, vomiting, diarrhea, edema, musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, constipation, pyrexia, headache, and dyspnea.
The most frequent grade 3 or higher AEs (excluding laboratory abnormalities) that occurred in at least 5% of patients were fatigue, sepsis, gastrointestinal hemorrhage, pneumonia, diarrhea, febrile neutropenia, edema, dyspnea, nausea, vomiting, abdominal pain, and renal insufficiency.
Serious AEs occurred in 68% of patients, most commonly infections and gastrointestinal disorders. Twenty-one percent of patients discontinued treatment due to AEs, the most frequent of which were infection, nausea or vomiting, QT prolongation, and gastrointestinal hemorrhage.
CHMP recommends new indication for obinutuzumab

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the marketing authorization for obinutuzumab (Gazyvaro).
The new proposed indication is for obinutuzumab in combination with chemotherapy for patients with previously untreated, advanced follicular lymphoma (FL). This would be followed by obinutuzumab maintenance in patients who achieved a response.
The European Commission typically adheres to the CHMP’s recommendations and delivers its final decision within 2 to 3 months’ of the CHMP’s recommendation.
The decision will be applicable to all member states of the European Union, plus Iceland, Liechtenstein, and Norway.
If approved for this new indication, obinutuzumab will be authorized for use in the European Economic Area as follows:
- In combination with chlorambucil for the treatment of adults with previously untreated chronic lymphocytic leukemia and comorbidities making them unsuitable for full-dose fludarabine-based therapy.
- In combination with bendamustine, followed by obinutuzumab maintenance, for the treatment of patients with FL who did not respond to, or who progressed during or up to 6 months after, treatment with rituximab or a rituximab-containing regimen.
- In combination with chemotherapy, followed by obinutuzumab maintenance in responders, for the treatment of patients with previously untreated, advanced FL.
GALLIUM trial
The CHMP’s recommendation is based on results of the phase 3 GALLIUM trial, which were presented at the 2016 ASH Annual Meeting.
The study enrolled 1401 patients with previously untreated, indolent non-Hodgkin lymphoma, including 1202 with FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
Patients who received obinutuzumab had significantly better progression-free survival than patients who received rituximab. The 3-year progression-free survival rate was 73.3% in the rituximab arm and 80% in the obinutuzumab arm (hazard ratio [HR]=0.66, P=0.0012).
There was no significant difference between the treatment arms with regard to overall survival. The 3-year overall survival was 92.1% in the rituximab arm and 94% in the obinutuzumab arm (HR=0.75, P=0.21).
The overall incidence of adverse events (AEs) was 98.3% in the rituximab arm and 99.5% in the obinutuzumab arm. The incidence of serious AEs was 39.9% and 46.1%, respectively.
The incidence of grade 3 or higher AEs was higher among patients who received obinutuzumab.
Grade 3 or higher AEs occurring in at least 5% of patients in either arm (rituximab and obinutuzumab, respectively) included neutropenia (67.8% and 74.6%), leukopenia (37.9% and 43.9%), febrile neutropenia (4.9% and 6.9%), infections and infestations (3.7% and 6.7%), and thrombocytopenia (2.7% and 6.1%).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the marketing authorization for obinutuzumab (Gazyvaro).
The new proposed indication is for obinutuzumab in combination with chemotherapy for patients with previously untreated, advanced follicular lymphoma (FL). This would be followed by obinutuzumab maintenance in patients who achieved a response.
The European Commission typically adheres to the CHMP’s recommendations and delivers its final decision within 2 to 3 months’ of the CHMP’s recommendation.
The decision will be applicable to all member states of the European Union, plus Iceland, Liechtenstein, and Norway.
If approved for this new indication, obinutuzumab will be authorized for use in the European Economic Area as follows:
- In combination with chlorambucil for the treatment of adults with previously untreated chronic lymphocytic leukemia and comorbidities making them unsuitable for full-dose fludarabine-based therapy.
- In combination with bendamustine, followed by obinutuzumab maintenance, for the treatment of patients with FL who did not respond to, or who progressed during or up to 6 months after, treatment with rituximab or a rituximab-containing regimen.
- In combination with chemotherapy, followed by obinutuzumab maintenance in responders, for the treatment of patients with previously untreated, advanced FL.
GALLIUM trial
The CHMP’s recommendation is based on results of the phase 3 GALLIUM trial, which were presented at the 2016 ASH Annual Meeting.
The study enrolled 1401 patients with previously untreated, indolent non-Hodgkin lymphoma, including 1202 with FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
Patients who received obinutuzumab had significantly better progression-free survival than patients who received rituximab. The 3-year progression-free survival rate was 73.3% in the rituximab arm and 80% in the obinutuzumab arm (hazard ratio [HR]=0.66, P=0.0012).
There was no significant difference between the treatment arms with regard to overall survival. The 3-year overall survival was 92.1% in the rituximab arm and 94% in the obinutuzumab arm (HR=0.75, P=0.21).
The overall incidence of adverse events (AEs) was 98.3% in the rituximab arm and 99.5% in the obinutuzumab arm. The incidence of serious AEs was 39.9% and 46.1%, respectively.
The incidence of grade 3 or higher AEs was higher among patients who received obinutuzumab.
Grade 3 or higher AEs occurring in at least 5% of patients in either arm (rituximab and obinutuzumab, respectively) included neutropenia (67.8% and 74.6%), leukopenia (37.9% and 43.9%), febrile neutropenia (4.9% and 6.9%), infections and infestations (3.7% and 6.7%), and thrombocytopenia (2.7% and 6.1%).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the marketing authorization for obinutuzumab (Gazyvaro).
The new proposed indication is for obinutuzumab in combination with chemotherapy for patients with previously untreated, advanced follicular lymphoma (FL). This would be followed by obinutuzumab maintenance in patients who achieved a response.
The European Commission typically adheres to the CHMP’s recommendations and delivers its final decision within 2 to 3 months’ of the CHMP’s recommendation.
The decision will be applicable to all member states of the European Union, plus Iceland, Liechtenstein, and Norway.
If approved for this new indication, obinutuzumab will be authorized for use in the European Economic Area as follows:
- In combination with chlorambucil for the treatment of adults with previously untreated chronic lymphocytic leukemia and comorbidities making them unsuitable for full-dose fludarabine-based therapy.
- In combination with bendamustine, followed by obinutuzumab maintenance, for the treatment of patients with FL who did not respond to, or who progressed during or up to 6 months after, treatment with rituximab or a rituximab-containing regimen.
- In combination with chemotherapy, followed by obinutuzumab maintenance in responders, for the treatment of patients with previously untreated, advanced FL.
GALLIUM trial
The CHMP’s recommendation is based on results of the phase 3 GALLIUM trial, which were presented at the 2016 ASH Annual Meeting.
The study enrolled 1401 patients with previously untreated, indolent non-Hodgkin lymphoma, including 1202 with FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
Patients who received obinutuzumab had significantly better progression-free survival than patients who received rituximab. The 3-year progression-free survival rate was 73.3% in the rituximab arm and 80% in the obinutuzumab arm (hazard ratio [HR]=0.66, P=0.0012).
There was no significant difference between the treatment arms with regard to overall survival. The 3-year overall survival was 92.1% in the rituximab arm and 94% in the obinutuzumab arm (HR=0.75, P=0.21).
The overall incidence of adverse events (AEs) was 98.3% in the rituximab arm and 99.5% in the obinutuzumab arm. The incidence of serious AEs was 39.9% and 46.1%, respectively.
The incidence of grade 3 or higher AEs was higher among patients who received obinutuzumab.
Grade 3 or higher AEs occurring in at least 5% of patients in either arm (rituximab and obinutuzumab, respectively) included neutropenia (67.8% and 74.6%), leukopenia (37.9% and 43.9%), febrile neutropenia (4.9% and 6.9%), infections and infestations (3.7% and 6.7%), and thrombocytopenia (2.7% and 6.1%).
Modified, CB-derived NK cells target CLL and BL

Natural killer (NK) cells derived from cord blood (CB) can be modified to fight chronic lymphocytic leukemia (CLL) and Burkitt lymphoma (BL), according to research published in Leukemia.
Using a viral vector, researchers transduced CB-derived NK cells with a chimeric antigen receptor (CAR), interleukin-15 (IL-15), and an inducible caspase-9-based suicide gene.
The CAR directed the NK cells to kill CD19-expressing cells, IL-15 prolonged the NK cells’ survival and enhanced their antitumor activity, and the suicide gene allowed researchers to kill off the NK cells in the event of a severe inflammatory response.
“Natural killer cells are the immune system’s most potent killers, but they are short-lived, and cancers manage to evade a patient’s own NK cells to progress,” said study author Katy Rezvani, MD, PhD, of The University of Texas MD Anderson Cancer Center in Houston.
“Our cord-blood derived NK cells, genetically equipped with a receptor that focuses them on B-cell malignancies and with interleukin-15 to help them persist longer—potentially for months instead of 2 or 3 weeks—are designed to address these challenges.”
Dr Rezvani and her colleagues tested their CB-derived CAR NK cells in primary CLL cells and a mouse model of BL. Compared to unmodified NK cells, the CAR NK cells killed malignant cells more efficiently and extended the survival of mice.
Another experiment showed the CB-derived CAR NK cells killed CLL cells more efficiently than NK cells that were taken from CLL patients and modified in the same way. The researchers said this highlights the need to transplant CAR-engineered NK cells derived from healthy CB rather than using a patient’s own cells.
Additional experiments in the mouse model of BL showed that a single infusion of low-dose CAR NK cells resulted in prolonged survival.
When CAR NK cells were given at a higher dose, none of the mice died of BL. Half of them survived 100 days and beyond. However, all mice treated with other types of NK cells died by day 41.
Some mice treated with the higher dose of CAR NK cells died of cytokine release syndrome (CRS).
To counteract this toxicity, the researchers activated the suicide gene (iC9) via treatment with a small-molecule dimerizer, AP1903.
The team found that adding as little as 10 nM of AP1903 to cell cultures induced apoptosis/necrosis of the CAR NK cells within 4 hours but had no effect on non-CAR CB-derived NK cells. Similar results were observed in the mouse model.
Next steps
A phase 1/2 trial of these CB-derived CAR NK cells opened at MD Anderson in June for patients with relapsed or refractory CLL, acute lymphocytic leukemia, or non-Hodgkin lymphoma.
Dr Rezvani, the principal investigator of the trial, said the protocol calls for vigilance for signs of CRS, treatment with steroids and tocilizumab for low-grade CRS, and AP1903 added to activate the suicide gene for grade 3 or 4 CRS.
She and her colleagues noted that CB-derived NK cells do not cause graft-vs-host disease. Therefore, they can be an off-the-shelf product, prepared in advance with the necessary receptor and given to patients promptly.
The researchers are developing CB-derived CAR NK cells for other targets in hematologic and solid tumor malignancies.
MD Anderson and the researchers have intellectual property related to the engineered NK cells, which is being managed in accordance with the institution’s conflict-of-interest rules.
Funding for this research was provided by MD Anderson’s Moon Shots Program, the National Cancer Institute of the National Institutes of Health Cancer Center Support Grant (CA016672) to MD Anderson, and grants from the Leukemia and Lymphoma Society and the American Cancer Society.
Natural killer (NK) cells derived from cord blood (CB) can be modified to fight chronic lymphocytic leukemia (CLL) and Burkitt lymphoma (BL), according to research published in Leukemia.
Using a viral vector, researchers transduced CB-derived NK cells with a chimeric antigen receptor (CAR), interleukin-15 (IL-15), and an inducible caspase-9-based suicide gene.
The CAR directed the NK cells to kill CD19-expressing cells, IL-15 prolonged the NK cells’ survival and enhanced their antitumor activity, and the suicide gene allowed researchers to kill off the NK cells in the event of a severe inflammatory response.
“Natural killer cells are the immune system’s most potent killers, but they are short-lived, and cancers manage to evade a patient’s own NK cells to progress,” said study author Katy Rezvani, MD, PhD, of The University of Texas MD Anderson Cancer Center in Houston.
“Our cord-blood derived NK cells, genetically equipped with a receptor that focuses them on B-cell malignancies and with interleukin-15 to help them persist longer—potentially for months instead of 2 or 3 weeks—are designed to address these challenges.”
Dr Rezvani and her colleagues tested their CB-derived CAR NK cells in primary CLL cells and a mouse model of BL. Compared to unmodified NK cells, the CAR NK cells killed malignant cells more efficiently and extended the survival of mice.
Another experiment showed the CB-derived CAR NK cells killed CLL cells more efficiently than NK cells that were taken from CLL patients and modified in the same way. The researchers said this highlights the need to transplant CAR-engineered NK cells derived from healthy CB rather than using a patient’s own cells.
Additional experiments in the mouse model of BL showed that a single infusion of low-dose CAR NK cells resulted in prolonged survival.
When CAR NK cells were given at a higher dose, none of the mice died of BL. Half of them survived 100 days and beyond. However, all mice treated with other types of NK cells died by day 41.
Some mice treated with the higher dose of CAR NK cells died of cytokine release syndrome (CRS).
To counteract this toxicity, the researchers activated the suicide gene (iC9) via treatment with a small-molecule dimerizer, AP1903.
The team found that adding as little as 10 nM of AP1903 to cell cultures induced apoptosis/necrosis of the CAR NK cells within 4 hours but had no effect on non-CAR CB-derived NK cells. Similar results were observed in the mouse model.
Next steps
A phase 1/2 trial of these CB-derived CAR NK cells opened at MD Anderson in June for patients with relapsed or refractory CLL, acute lymphocytic leukemia, or non-Hodgkin lymphoma.
Dr Rezvani, the principal investigator of the trial, said the protocol calls for vigilance for signs of CRS, treatment with steroids and tocilizumab for low-grade CRS, and AP1903 added to activate the suicide gene for grade 3 or 4 CRS.
She and her colleagues noted that CB-derived NK cells do not cause graft-vs-host disease. Therefore, they can be an off-the-shelf product, prepared in advance with the necessary receptor and given to patients promptly.
The researchers are developing CB-derived CAR NK cells for other targets in hematologic and solid tumor malignancies.
MD Anderson and the researchers have intellectual property related to the engineered NK cells, which is being managed in accordance with the institution’s conflict-of-interest rules.
Funding for this research was provided by MD Anderson’s Moon Shots Program, the National Cancer Institute of the National Institutes of Health Cancer Center Support Grant (CA016672) to MD Anderson, and grants from the Leukemia and Lymphoma Society and the American Cancer Society.
Natural killer (NK) cells derived from cord blood (CB) can be modified to fight chronic lymphocytic leukemia (CLL) and Burkitt lymphoma (BL), according to research published in Leukemia.
Using a viral vector, researchers transduced CB-derived NK cells with a chimeric antigen receptor (CAR), interleukin-15 (IL-15), and an inducible caspase-9-based suicide gene.
The CAR directed the NK cells to kill CD19-expressing cells, IL-15 prolonged the NK cells’ survival and enhanced their antitumor activity, and the suicide gene allowed researchers to kill off the NK cells in the event of a severe inflammatory response.
“Natural killer cells are the immune system’s most potent killers, but they are short-lived, and cancers manage to evade a patient’s own NK cells to progress,” said study author Katy Rezvani, MD, PhD, of The University of Texas MD Anderson Cancer Center in Houston.
“Our cord-blood derived NK cells, genetically equipped with a receptor that focuses them on B-cell malignancies and with interleukin-15 to help them persist longer—potentially for months instead of 2 or 3 weeks—are designed to address these challenges.”
Dr Rezvani and her colleagues tested their CB-derived CAR NK cells in primary CLL cells and a mouse model of BL. Compared to unmodified NK cells, the CAR NK cells killed malignant cells more efficiently and extended the survival of mice.
Another experiment showed the CB-derived CAR NK cells killed CLL cells more efficiently than NK cells that were taken from CLL patients and modified in the same way. The researchers said this highlights the need to transplant CAR-engineered NK cells derived from healthy CB rather than using a patient’s own cells.
Additional experiments in the mouse model of BL showed that a single infusion of low-dose CAR NK cells resulted in prolonged survival.
When CAR NK cells were given at a higher dose, none of the mice died of BL. Half of them survived 100 days and beyond. However, all mice treated with other types of NK cells died by day 41.
Some mice treated with the higher dose of CAR NK cells died of cytokine release syndrome (CRS).
To counteract this toxicity, the researchers activated the suicide gene (iC9) via treatment with a small-molecule dimerizer, AP1903.
The team found that adding as little as 10 nM of AP1903 to cell cultures induced apoptosis/necrosis of the CAR NK cells within 4 hours but had no effect on non-CAR CB-derived NK cells. Similar results were observed in the mouse model.
Next steps
A phase 1/2 trial of these CB-derived CAR NK cells opened at MD Anderson in June for patients with relapsed or refractory CLL, acute lymphocytic leukemia, or non-Hodgkin lymphoma.
Dr Rezvani, the principal investigator of the trial, said the protocol calls for vigilance for signs of CRS, treatment with steroids and tocilizumab for low-grade CRS, and AP1903 added to activate the suicide gene for grade 3 or 4 CRS.
She and her colleagues noted that CB-derived NK cells do not cause graft-vs-host disease. Therefore, they can be an off-the-shelf product, prepared in advance with the necessary receptor and given to patients promptly.
The researchers are developing CB-derived CAR NK cells for other targets in hematologic and solid tumor malignancies.
MD Anderson and the researchers have intellectual property related to the engineered NK cells, which is being managed in accordance with the institution’s conflict-of-interest rules.
Funding for this research was provided by MD Anderson’s Moon Shots Program, the National Cancer Institute of the National Institutes of Health Cancer Center Support Grant (CA016672) to MD Anderson, and grants from the Leukemia and Lymphoma Society and the American Cancer Society.