User login
American Heart Association (AHA): Scientific Sessions 2013
Advanced pacing slows AF progression in bradycardia
DALLAS – Enhanced pacemaker technology sharply reduced progression of nonpermanent atrial fibrillation to permanent AF in patients with bradycardia and sinus node disease in the large international, randomized MINERVA trial.
In the MINERVA (Minimize Right Ventricular Pacing to Prevent Atrial Fibrillation and Heart Failure) study, the use of dual-chamber pacemakers containing features for atrial preventive pacing and atrial antitachycardia pacing, called DDDRPs, plus managed ventricular pacing (MVP) resulted in a 61% reduction in the incidence of permanent AF during 2 years of prospective follow-up, compared with conventional dual-chamber pacing, Dr. Giuseppe Boriani reported at the American Heart Association scientific sessions.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
In addition, patients in the enhanced-pacing group experienced a 52% reduction in AF-related hospitalizations and emergency department visits, as well as a 49% decrease in atrial cardioversions relative to controls, added Dr. Boriani of the University of Bologna (Italy).
"This is the first study to show that using these enhanced pacing features in combination not only delays the AF disease progression, but also has an impact on health care utilization," he noted. "With this solid evidence of beneficial results, I think the practice guidelines probably should be updated to state this type of device should be implanted for sinus node disease in patients with a history of atrial fibrillation."

MINERVA included 1,166 patients at 63 European, Asian, and Middle Eastern centers; all had bradycardia, sinus node disease, and a history of nonpermanent AF. They received a commercially available Medtronic dual-chamber pacemaker that was randomized single-blind to operate using both the DDDRP and MVP features, the MVP alone, or conventional dual-chamber pacing with neither enhanced feature.
At 2 years, the primary composite endpoint of death, cardiovascular hospitalization, or permanent AF had occurred in 19.8% of the DDDRP plus MVP group. That amounted to a significant 26% reduction in the endpoint compared with the 26.5% rate in the controls on standard dual-chamber pacing. The 21.4% rate in patients whose device used only the MVP algorithm was not significantly different from the rate in controls.
Most strikingly, the 2-year incidence of progression to permanent AF was 3.8% in patients with the enhanced pacing features, compared with 9.2% in those assigned to conventional dual-chamber pacing, for a 61% relative risk reduction. This means 20 patients would need to be treated for 2 years with a pacemaker utilizing the advanced pacing features in order to prevent one additional case of permanent AF.
In addition, patients whose pacemakers utilized rate response and antitachycardia pacing plus MVP reported better quality of life and less fatigue than did those in the other two study arms.
Dr. Boriani estimated that of the 170,000 patients each year in the United States who undergo pacemaker implantation for bradycardia, roughly 25,000 have sinus node disease and a history of AF and would thus now be considered strong candidates for the enhanced pacing features.
Discussant Dr. Anthony Tang explained that it has been known for years that pacemaker algorithms designed to increase the atrial pacing rate above the intrinsic sinus rate can prevent events that trigger AF. The problem has been that this atrial pacing can have the side effect of also increasing ventricular pacing, which can initiate AF, thus canceling the advantage of the atrial preventive pacing. The MVP algorithm decreases this unnecessary pacing in the right ventricle, with the resultant benefits seen in MINERVA.

In his view, the enhanced pacing features used in MINERVA represent a very good option for patients with severe sinus node dysfunction but preserved atrioventricular node conduction, that is, patients who need atrial pacing for long sinus pauses but don’t need much ventricular pacing.
In contrast, for patients with an indication for a pacemaker but who only occasionally need pacing, such as those with a rare sinus pause or occasional AV block, a simple single-chamber ventricular pacemaker will generally do. For those with second- or third-degree AV block with left ventricular dysfunction and who need both atrial and ventricular pacing most of the time, a cardiac resynchronization therapy device plus either a pacemaker or a defibrillator is appropriate, depending upon the severity of the left ventricular dysfunction, according to Dr. Tang, professor of medicine and chair of cardiovascular population health at Western University, London, Ont.
Discussant Dr. Timothy J. Gardner wasn’t bowled over by the number-needed-to-treat (NNT) of 20 in MINERVA.
"That seems a very modest impact. I wonder from a health care/societal benefit standpoint whether an NNT of 20 for a more complex and expensive technology is appropriate," challenged Dr. Gardner, a heart surgeon and medical director of the Christiana Care Center for Heart and Vascular Health in Newark, Del.
Dr. Boriani replied that the benefits seen with advanced pacing in this 2-year study will only become more pronounced with longer follow-up.
"Permanent atrial fibrillation is the later stage of irreversible AF, when the arrhythmia becomes untreatable. And it is a marker for adverse events, including heart failure, stroke, and death," he said.
The MINERVA trial was sponsored by Medtronic, which markets the proprietary enhanced pacing technology. Dr. Boriani reported having no relevant financial interests.
DALLAS – Enhanced pacemaker technology sharply reduced progression of nonpermanent atrial fibrillation to permanent AF in patients with bradycardia and sinus node disease in the large international, randomized MINERVA trial.
In the MINERVA (Minimize Right Ventricular Pacing to Prevent Atrial Fibrillation and Heart Failure) study, the use of dual-chamber pacemakers containing features for atrial preventive pacing and atrial antitachycardia pacing, called DDDRPs, plus managed ventricular pacing (MVP) resulted in a 61% reduction in the incidence of permanent AF during 2 years of prospective follow-up, compared with conventional dual-chamber pacing, Dr. Giuseppe Boriani reported at the American Heart Association scientific sessions.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
In addition, patients in the enhanced-pacing group experienced a 52% reduction in AF-related hospitalizations and emergency department visits, as well as a 49% decrease in atrial cardioversions relative to controls, added Dr. Boriani of the University of Bologna (Italy).
"This is the first study to show that using these enhanced pacing features in combination not only delays the AF disease progression, but also has an impact on health care utilization," he noted. "With this solid evidence of beneficial results, I think the practice guidelines probably should be updated to state this type of device should be implanted for sinus node disease in patients with a history of atrial fibrillation."
MINERVA included 1,166 patients at 63 European, Asian, and Middle Eastern centers; all had bradycardia, sinus node disease, and a history of nonpermanent AF. They received a commercially available Medtronic dual-chamber pacemaker that was randomized single-blind to operate using both the DDDRP and MVP features, the MVP alone, or conventional dual-chamber pacing with neither enhanced feature.
At 2 years, the primary composite endpoint of death, cardiovascular hospitalization, or permanent AF had occurred in 19.8% of the DDDRP plus MVP group. That amounted to a significant 26% reduction in the endpoint compared with the 26.5% rate in the controls on standard dual-chamber pacing. The 21.4% rate in patients whose device used only the MVP algorithm was not significantly different from the rate in controls.
Most strikingly, the 2-year incidence of progression to permanent AF was 3.8% in patients with the enhanced pacing features, compared with 9.2% in those assigned to conventional dual-chamber pacing, for a 61% relative risk reduction. This means 20 patients would need to be treated for 2 years with a pacemaker utilizing the advanced pacing features in order to prevent one additional case of permanent AF.
In addition, patients whose pacemakers utilized rate response and antitachycardia pacing plus MVP reported better quality of life and less fatigue than did those in the other two study arms.
Dr. Boriani estimated that of the 170,000 patients each year in the United States who undergo pacemaker implantation for bradycardia, roughly 25,000 have sinus node disease and a history of AF and would thus now be considered strong candidates for the enhanced pacing features.
Discussant Dr. Anthony Tang explained that it has been known for years that pacemaker algorithms designed to increase the atrial pacing rate above the intrinsic sinus rate can prevent events that trigger AF. The problem has been that this atrial pacing can have the side effect of also increasing ventricular pacing, which can initiate AF, thus canceling the advantage of the atrial preventive pacing. The MVP algorithm decreases this unnecessary pacing in the right ventricle, with the resultant benefits seen in MINERVA.
In his view, the enhanced pacing features used in MINERVA represent a very good option for patients with severe sinus node dysfunction but preserved atrioventricular node conduction, that is, patients who need atrial pacing for long sinus pauses but don’t need much ventricular pacing.
In contrast, for patients with an indication for a pacemaker but who only occasionally need pacing, such as those with a rare sinus pause or occasional AV block, a simple single-chamber ventricular pacemaker will generally do. For those with second- or third-degree AV block with left ventricular dysfunction and who need both atrial and ventricular pacing most of the time, a cardiac resynchronization therapy device plus either a pacemaker or a defibrillator is appropriate, depending upon the severity of the left ventricular dysfunction, according to Dr. Tang, professor of medicine and chair of cardiovascular population health at Western University, London, Ont.
Discussant Dr. Timothy J. Gardner wasn’t bowled over by the number-needed-to-treat (NNT) of 20 in MINERVA.
"That seems a very modest impact. I wonder from a health care/societal benefit standpoint whether an NNT of 20 for a more complex and expensive technology is appropriate," challenged Dr. Gardner, a heart surgeon and medical director of the Christiana Care Center for Heart and Vascular Health in Newark, Del.
Dr. Boriani replied that the benefits seen with advanced pacing in this 2-year study will only become more pronounced with longer follow-up.
"Permanent atrial fibrillation is the later stage of irreversible AF, when the arrhythmia becomes untreatable. And it is a marker for adverse events, including heart failure, stroke, and death," he said.
The MINERVA trial was sponsored by Medtronic, which markets the proprietary enhanced pacing technology. Dr. Boriani reported having no relevant financial interests.
DALLAS – Enhanced pacemaker technology sharply reduced progression of nonpermanent atrial fibrillation to permanent AF in patients with bradycardia and sinus node disease in the large international, randomized MINERVA trial.
In the MINERVA (Minimize Right Ventricular Pacing to Prevent Atrial Fibrillation and Heart Failure) study, the use of dual-chamber pacemakers containing features for atrial preventive pacing and atrial antitachycardia pacing, called DDDRPs, plus managed ventricular pacing (MVP) resulted in a 61% reduction in the incidence of permanent AF during 2 years of prospective follow-up, compared with conventional dual-chamber pacing, Dr. Giuseppe Boriani reported at the American Heart Association scientific sessions.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
In addition, patients in the enhanced-pacing group experienced a 52% reduction in AF-related hospitalizations and emergency department visits, as well as a 49% decrease in atrial cardioversions relative to controls, added Dr. Boriani of the University of Bologna (Italy).
"This is the first study to show that using these enhanced pacing features in combination not only delays the AF disease progression, but also has an impact on health care utilization," he noted. "With this solid evidence of beneficial results, I think the practice guidelines probably should be updated to state this type of device should be implanted for sinus node disease in patients with a history of atrial fibrillation."
MINERVA included 1,166 patients at 63 European, Asian, and Middle Eastern centers; all had bradycardia, sinus node disease, and a history of nonpermanent AF. They received a commercially available Medtronic dual-chamber pacemaker that was randomized single-blind to operate using both the DDDRP and MVP features, the MVP alone, or conventional dual-chamber pacing with neither enhanced feature.
At 2 years, the primary composite endpoint of death, cardiovascular hospitalization, or permanent AF had occurred in 19.8% of the DDDRP plus MVP group. That amounted to a significant 26% reduction in the endpoint compared with the 26.5% rate in the controls on standard dual-chamber pacing. The 21.4% rate in patients whose device used only the MVP algorithm was not significantly different from the rate in controls.
Most strikingly, the 2-year incidence of progression to permanent AF was 3.8% in patients with the enhanced pacing features, compared with 9.2% in those assigned to conventional dual-chamber pacing, for a 61% relative risk reduction. This means 20 patients would need to be treated for 2 years with a pacemaker utilizing the advanced pacing features in order to prevent one additional case of permanent AF.
In addition, patients whose pacemakers utilized rate response and antitachycardia pacing plus MVP reported better quality of life and less fatigue than did those in the other two study arms.
Dr. Boriani estimated that of the 170,000 patients each year in the United States who undergo pacemaker implantation for bradycardia, roughly 25,000 have sinus node disease and a history of AF and would thus now be considered strong candidates for the enhanced pacing features.
Discussant Dr. Anthony Tang explained that it has been known for years that pacemaker algorithms designed to increase the atrial pacing rate above the intrinsic sinus rate can prevent events that trigger AF. The problem has been that this atrial pacing can have the side effect of also increasing ventricular pacing, which can initiate AF, thus canceling the advantage of the atrial preventive pacing. The MVP algorithm decreases this unnecessary pacing in the right ventricle, with the resultant benefits seen in MINERVA.
In his view, the enhanced pacing features used in MINERVA represent a very good option for patients with severe sinus node dysfunction but preserved atrioventricular node conduction, that is, patients who need atrial pacing for long sinus pauses but don’t need much ventricular pacing.
In contrast, for patients with an indication for a pacemaker but who only occasionally need pacing, such as those with a rare sinus pause or occasional AV block, a simple single-chamber ventricular pacemaker will generally do. For those with second- or third-degree AV block with left ventricular dysfunction and who need both atrial and ventricular pacing most of the time, a cardiac resynchronization therapy device plus either a pacemaker or a defibrillator is appropriate, depending upon the severity of the left ventricular dysfunction, according to Dr. Tang, professor of medicine and chair of cardiovascular population health at Western University, London, Ont.
Discussant Dr. Timothy J. Gardner wasn’t bowled over by the number-needed-to-treat (NNT) of 20 in MINERVA.
"That seems a very modest impact. I wonder from a health care/societal benefit standpoint whether an NNT of 20 for a more complex and expensive technology is appropriate," challenged Dr. Gardner, a heart surgeon and medical director of the Christiana Care Center for Heart and Vascular Health in Newark, Del.
Dr. Boriani replied that the benefits seen with advanced pacing in this 2-year study will only become more pronounced with longer follow-up.
"Permanent atrial fibrillation is the later stage of irreversible AF, when the arrhythmia becomes untreatable. And it is a marker for adverse events, including heart failure, stroke, and death," he said.
The MINERVA trial was sponsored by Medtronic, which markets the proprietary enhanced pacing technology. Dr. Boriani reported having no relevant financial interests.
AT THE AHA SCIENTIFIC SESSIONS
Major finding: Patients with bradycardia, sinus node disease, and a history of atrial fibrillation who were implanted with a dual-chamber pacemaker utilizing enhanced pacing features showed a 61% reduction in progression to permanent AF over 2 years, compared with those randomized to conventional dual-chamber pacing.
Data source: The MINERVA trial, a single-blind, randomized, international, prospective, 2-year trial conducted in 1,166 patients.
Disclosures: The MINERVA trial was sponsored by Medtronic, which markets the proprietary enhanced pacing technology. Dr. Boriani reported having no relevant financial interests.

Substrate ablation shows no advantage for A fib
DALLAS – A substrate-based ablation strategy failed to show an efficacy advantage over conventional circumferential pulmonary vein isolation for eliminating persistent or paroxysmal atrial fibrillation in a multicenter randomized trial with 232 patients.
High-frequency source ablation (HFSA), a strategy that targets rotors in the atrial substrate responsible for maintaining fibrillation episodes, did not reach statistical significance for noninferiority compared with circumferential pulmonary vein isolation (CPVI) after 6 months in patients with paroxysmal atrial fibrillation (AF), the study’s primary endpoint, Dr. Felipe Atienza said at the American Heart Association scientific sessions.

In enrolled patients with persistent atrial fibrillation, adding HFSA to CPVI "offered no incremental value with a trend for an increase in complications," after 6 months in a study designed to test superiority, said Dr. Atienza, an electrophysiologist at Gregorio Marañón University Hospital in Madrid.
But a ray of promise came from 1-year follow-up of the paroxysmal atrial fibrillation patients, half of the patients enrolled in the study. (The other patients had persistent atrial fibrillation.) At that time, HFSA treatment achieved statistically significant noninferiority compared with CPVI for freedom from atrial fibrillation and freedom from atrial tachyarrhythmia, with a significantly lower rate of serious adverse events.
Although the results mean that pulmonary vein isolation remains the standard of care for treating atrial fibrillation patients who fail to respond to drug treatment, substrate-based ablation showed enough potential in the study to remain a viable candidate for further testing, experts said.
"In this trial, HFSA did not improve outcomes in patients with paroxysmal or persistent AF, but the fact that HFSA treatment was equivalent to CPVI in patients with paroxysmal AF keeps the AF substrate and trigger argument alive," commented Dr. Mark S. Link, professor and codirector of the New England Cardiac Arrhythmia Center at Tufts Medical Center Boston, and designated discussant for the report.
"The way to go is to run further investigations like this to try to find and eliminate the substrates of atrial fibrillation instead of ablating everywhere," said Dr. Atienza.
The RADAR-AF (Radiofrequency Ablation of Drivers of Atrial Fibrillation) trial enrolled 115 patients with paroxysmal AF and 117 patients with persistent AF at 11 hospitals in Spain. The researchers randomized the patients with paroxysmal AF to treatment with either HFSA or CPVI; they randomized patients with persistent AF to treatment with both HFSA and CPVI or to CPVI alone. They used software developed by St. Jude Medical to identify the sites for HFSA.
The study’s primary endpoint was freedom from AF while off of antiarrhythmic drugs at 6 months. In the paroxysmal patients, this endpoint was reached by 73% of patients treated with HFSA and 83% treated with CPVI, which failed to meet the prespecified criteria for noninferiority. However, after 1 year the rate of freedom from AF was 69% in patients in both groups, and the rate of freedom from atrial tachyarrhythmia in both groups was 59%, rates that met the criteria for noninferiority. In addition, after 1 year the cumulative rate of serious adverse events was 9% in the HFSA group and 24% in the CPVI group, a statistically significant difference.
Among the patients with persistent atrial fibrillation, the rate of freedom from AF after 6 months was 61% in patients treated with both techniques and 60% in those treated with CPVI alone, which failed to show superiority. Efficacy rates were also similar in both arms after 1 year, and the rate of serious adverse events was 24% in patients treated with both HFSA and CPVI, compared with 10% among those treated by CPVI only.
The RADAR-AF trial was sponsored by St. Jude Medical, which developed the rotor-mapping software used for substrate ablation in the study. Dr. Atienza and Dr. Link said they had no relevant financial disclosures.
On Twitter @mitchelzoler
The hypothesis underlying the RADAR-AF trial is that while atrial fibrillation is initiated by focal triggers, it is maintained in the atrial substrate, specifically by rotors in the substrate. The concept is that if you can ablate the substrate correctly, it will prevent episodes of sustained atrial fibrillation and move us beyond the ceiling we currently have with pulmonary vein isolation, which has about a 70% success rate.

|
Mitchel L. Zoler/IMNG Medical Media
|
The results from RADAR-AF do not kill the notion that the substrate is important, especially because in patients with paroxysmal atrial fibrillation just ablating the substrate did as well after 1 year as standard pulmonary vein isolation.
The findings also indicate that the technique tested for identifying rotors in the substrate is inadequate, and we need a better way to do this. Several ongoing clinical trials are looking at this. We just do not have adequate tools right now to know where to ablate the substrate.
The most important thing is that the RADAR-AF results produced a signal that the substrate is important.
Dr. Mark S. Link is professor of medicine and codirector of the New England Cardiac Arrhythmia Center at Tufts Medical Center in Boston. He made these comments as the designated discussant for the report and during a press conference. He said he had no relevant financial disclosures.
RADAR-AF, Radiofrequency Ablation of Drivers of Atrial Fibrillation trial,
The hypothesis underlying the RADAR-AF trial is that while atrial fibrillation is initiated by focal triggers, it is maintained in the atrial substrate, specifically by rotors in the substrate. The concept is that if you can ablate the substrate correctly, it will prevent episodes of sustained atrial fibrillation and move us beyond the ceiling we currently have with pulmonary vein isolation, which has about a 70% success rate.
|
|
Mitchel L. Zoler/IMNG Medical Media
|
The results from RADAR-AF do not kill the notion that the substrate is important, especially because in patients with paroxysmal atrial fibrillation just ablating the substrate did as well after 1 year as standard pulmonary vein isolation.
The findings also indicate that the technique tested for identifying rotors in the substrate is inadequate, and we need a better way to do this. Several ongoing clinical trials are looking at this. We just do not have adequate tools right now to know where to ablate the substrate.
The most important thing is that the RADAR-AF results produced a signal that the substrate is important.
Dr. Mark S. Link is professor of medicine and codirector of the New England Cardiac Arrhythmia Center at Tufts Medical Center in Boston. He made these comments as the designated discussant for the report and during a press conference. He said he had no relevant financial disclosures.
The hypothesis underlying the RADAR-AF trial is that while atrial fibrillation is initiated by focal triggers, it is maintained in the atrial substrate, specifically by rotors in the substrate. The concept is that if you can ablate the substrate correctly, it will prevent episodes of sustained atrial fibrillation and move us beyond the ceiling we currently have with pulmonary vein isolation, which has about a 70% success rate.
|
|
Mitchel L. Zoler/IMNG Medical Media
|
The results from RADAR-AF do not kill the notion that the substrate is important, especially because in patients with paroxysmal atrial fibrillation just ablating the substrate did as well after 1 year as standard pulmonary vein isolation.
The findings also indicate that the technique tested for identifying rotors in the substrate is inadequate, and we need a better way to do this. Several ongoing clinical trials are looking at this. We just do not have adequate tools right now to know where to ablate the substrate.
The most important thing is that the RADAR-AF results produced a signal that the substrate is important.
Dr. Mark S. Link is professor of medicine and codirector of the New England Cardiac Arrhythmia Center at Tufts Medical Center in Boston. He made these comments as the designated discussant for the report and during a press conference. He said he had no relevant financial disclosures.
DALLAS – A substrate-based ablation strategy failed to show an efficacy advantage over conventional circumferential pulmonary vein isolation for eliminating persistent or paroxysmal atrial fibrillation in a multicenter randomized trial with 232 patients.
High-frequency source ablation (HFSA), a strategy that targets rotors in the atrial substrate responsible for maintaining fibrillation episodes, did not reach statistical significance for noninferiority compared with circumferential pulmonary vein isolation (CPVI) after 6 months in patients with paroxysmal atrial fibrillation (AF), the study’s primary endpoint, Dr. Felipe Atienza said at the American Heart Association scientific sessions.
In enrolled patients with persistent atrial fibrillation, adding HFSA to CPVI "offered no incremental value with a trend for an increase in complications," after 6 months in a study designed to test superiority, said Dr. Atienza, an electrophysiologist at Gregorio Marañón University Hospital in Madrid.
But a ray of promise came from 1-year follow-up of the paroxysmal atrial fibrillation patients, half of the patients enrolled in the study. (The other patients had persistent atrial fibrillation.) At that time, HFSA treatment achieved statistically significant noninferiority compared with CPVI for freedom from atrial fibrillation and freedom from atrial tachyarrhythmia, with a significantly lower rate of serious adverse events.
Although the results mean that pulmonary vein isolation remains the standard of care for treating atrial fibrillation patients who fail to respond to drug treatment, substrate-based ablation showed enough potential in the study to remain a viable candidate for further testing, experts said.
"In this trial, HFSA did not improve outcomes in patients with paroxysmal or persistent AF, but the fact that HFSA treatment was equivalent to CPVI in patients with paroxysmal AF keeps the AF substrate and trigger argument alive," commented Dr. Mark S. Link, professor and codirector of the New England Cardiac Arrhythmia Center at Tufts Medical Center Boston, and designated discussant for the report.
"The way to go is to run further investigations like this to try to find and eliminate the substrates of atrial fibrillation instead of ablating everywhere," said Dr. Atienza.
The RADAR-AF (Radiofrequency Ablation of Drivers of Atrial Fibrillation) trial enrolled 115 patients with paroxysmal AF and 117 patients with persistent AF at 11 hospitals in Spain. The researchers randomized the patients with paroxysmal AF to treatment with either HFSA or CPVI; they randomized patients with persistent AF to treatment with both HFSA and CPVI or to CPVI alone. They used software developed by St. Jude Medical to identify the sites for HFSA.
The study’s primary endpoint was freedom from AF while off of antiarrhythmic drugs at 6 months. In the paroxysmal patients, this endpoint was reached by 73% of patients treated with HFSA and 83% treated with CPVI, which failed to meet the prespecified criteria for noninferiority. However, after 1 year the rate of freedom from AF was 69% in patients in both groups, and the rate of freedom from atrial tachyarrhythmia in both groups was 59%, rates that met the criteria for noninferiority. In addition, after 1 year the cumulative rate of serious adverse events was 9% in the HFSA group and 24% in the CPVI group, a statistically significant difference.
Among the patients with persistent atrial fibrillation, the rate of freedom from AF after 6 months was 61% in patients treated with both techniques and 60% in those treated with CPVI alone, which failed to show superiority. Efficacy rates were also similar in both arms after 1 year, and the rate of serious adverse events was 24% in patients treated with both HFSA and CPVI, compared with 10% among those treated by CPVI only.
The RADAR-AF trial was sponsored by St. Jude Medical, which developed the rotor-mapping software used for substrate ablation in the study. Dr. Atienza and Dr. Link said they had no relevant financial disclosures.
On Twitter @mitchelzoler
DALLAS – A substrate-based ablation strategy failed to show an efficacy advantage over conventional circumferential pulmonary vein isolation for eliminating persistent or paroxysmal atrial fibrillation in a multicenter randomized trial with 232 patients.
High-frequency source ablation (HFSA), a strategy that targets rotors in the atrial substrate responsible for maintaining fibrillation episodes, did not reach statistical significance for noninferiority compared with circumferential pulmonary vein isolation (CPVI) after 6 months in patients with paroxysmal atrial fibrillation (AF), the study’s primary endpoint, Dr. Felipe Atienza said at the American Heart Association scientific sessions.
In enrolled patients with persistent atrial fibrillation, adding HFSA to CPVI "offered no incremental value with a trend for an increase in complications," after 6 months in a study designed to test superiority, said Dr. Atienza, an electrophysiologist at Gregorio Marañón University Hospital in Madrid.
But a ray of promise came from 1-year follow-up of the paroxysmal atrial fibrillation patients, half of the patients enrolled in the study. (The other patients had persistent atrial fibrillation.) At that time, HFSA treatment achieved statistically significant noninferiority compared with CPVI for freedom from atrial fibrillation and freedom from atrial tachyarrhythmia, with a significantly lower rate of serious adverse events.
Although the results mean that pulmonary vein isolation remains the standard of care for treating atrial fibrillation patients who fail to respond to drug treatment, substrate-based ablation showed enough potential in the study to remain a viable candidate for further testing, experts said.
"In this trial, HFSA did not improve outcomes in patients with paroxysmal or persistent AF, but the fact that HFSA treatment was equivalent to CPVI in patients with paroxysmal AF keeps the AF substrate and trigger argument alive," commented Dr. Mark S. Link, professor and codirector of the New England Cardiac Arrhythmia Center at Tufts Medical Center Boston, and designated discussant for the report.
"The way to go is to run further investigations like this to try to find and eliminate the substrates of atrial fibrillation instead of ablating everywhere," said Dr. Atienza.
The RADAR-AF (Radiofrequency Ablation of Drivers of Atrial Fibrillation) trial enrolled 115 patients with paroxysmal AF and 117 patients with persistent AF at 11 hospitals in Spain. The researchers randomized the patients with paroxysmal AF to treatment with either HFSA or CPVI; they randomized patients with persistent AF to treatment with both HFSA and CPVI or to CPVI alone. They used software developed by St. Jude Medical to identify the sites for HFSA.
The study’s primary endpoint was freedom from AF while off of antiarrhythmic drugs at 6 months. In the paroxysmal patients, this endpoint was reached by 73% of patients treated with HFSA and 83% treated with CPVI, which failed to meet the prespecified criteria for noninferiority. However, after 1 year the rate of freedom from AF was 69% in patients in both groups, and the rate of freedom from atrial tachyarrhythmia in both groups was 59%, rates that met the criteria for noninferiority. In addition, after 1 year the cumulative rate of serious adverse events was 9% in the HFSA group and 24% in the CPVI group, a statistically significant difference.
Among the patients with persistent atrial fibrillation, the rate of freedom from AF after 6 months was 61% in patients treated with both techniques and 60% in those treated with CPVI alone, which failed to show superiority. Efficacy rates were also similar in both arms after 1 year, and the rate of serious adverse events was 24% in patients treated with both HFSA and CPVI, compared with 10% among those treated by CPVI only.
The RADAR-AF trial was sponsored by St. Jude Medical, which developed the rotor-mapping software used for substrate ablation in the study. Dr. Atienza and Dr. Link said they had no relevant financial disclosures.
On Twitter @mitchelzoler
RADAR-AF, Radiofrequency Ablation of Drivers of Atrial Fibrillation trial,
RADAR-AF, Radiofrequency Ablation of Drivers of Atrial Fibrillation trial,
AT THE AHA SCIENTIFIC SESSIONS
Major finding: Substrate ablation failed to show efficacy compared with conventional pulmonary vein isolation for patients with atrial fibrillation.
Data source: The RADAR-AF trial, which enrolled 232 patients with paroxysmal or persistent atrial fibrillation at 11 Spanish Hospitals.
Disclosures: The study was sponsored by St. Jude Medical, which developed the rotor-mapping software used for substrate ablation in the study. Dr. Atienza and Dr. Link said they had no relevant financial disclosures.

TOPCAT: Spironolactone cuts hospitalizations for diastolic heart failure
DALLAS – Spironolactone did not hit a home run in the large international "treatment of preserved cardiac function heart failure with an aldosterone antagonist" (TOPCAT) trial, but it did knock out a solid single in the form of significantly reduced hospitalizations for this extremely common, chronic, high morbidity/mortality condition.
It was this positive result for an important prespecified secondary outcome that enabled TOPCAT to avoid becoming roadkill. Technically, TOPCAT was a negative clinical trial in that spironolactone did not significantly outperform placebo on the primary composite outcome of cardiovascular mortality, heart failure hospitalization, or aborted cardiac arrest.
Yet that negative primary outcome was controversial: The aldosterone antagonist actually showed a significant positive result for the composite endpoint in North and South American participants, yet the results were resoundingly negative – and also considerably out of whack with the characteristic arc of progressive heart failure – among the nearly one-half of TOPCAT participants in Russia and the Republic of Georgia.
"What happened in Russia and Georgia we just don’t understand," Dr. Bertram Pitt, TOPCAT steering committee chair, said in an interview, shaking his head. "The event rate with placebo in Eastern Europe was so low it’s not compatible with anything we know about heart failure. The signs and symptoms of HFpEF [heart failure with preserved ejection fraction] are nonspecific; they can be due to obesity, lung disease, and other things. Clearly there are some people getting into the major trials of HFpEF that probably don’t have it."
TOPCAT was a randomized, double-blind clinical trial comprising 3,445 participants with symptomatic HFpEF at 250 sites in the United States and five other countries. They were randomized to spironolactone or placebo and followed prospectively for a mean of 3.3 years. The starting dose of the aldosterone antagonist was 15 mg/day, with a target of 30 mg/day. The drug could be titrated within the range of 15-45 mg/day. Eight months into the trial, the mean daily dose was 25 mg.

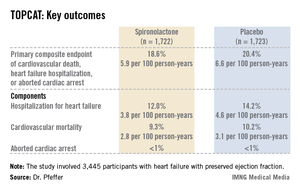
Presenting the TOPCAT results at the American Heart Association scientific sessions, Dr. Marc A. Pfeffer noted that the primary composite endpoint occurred in 20.4% of placebo-treated controls and 18.6% on spironolactone, a statistically nonsignificant difference. In contrast, the 17% reduction in the rate of hospitalization for HFpEF in the spironolactone group relative to controls was significant (P = .04). Moreover, the spironolactone-treated patients had a collective 394 HFpEF hospitalizations, markedly fewer than the 475 in controls. This translated to a hospitalization for heart failure with a preserved left ventricular ejection fraction occurring at a rate of 3.8 per 100 person-years in patients randomized to spironolactone, compared with the 4.6 per 100 person-years in placebo-treated controls.
Hyperkalemia in excess of 5.5 mmol/L occurred in 18.7% of the spironolactone group, twice the rate of controls (9.1%). And the incidence of a creatinine level more than double the upper limit of normal was 49% greater in the spironolactone group. That said, neither of these laboratory abnormalities resulted in any serious adverse consequences because investigators adjusted the dose in response, explained Dr. Pfeffer, professor of medicine at Harvard University, Boston.
He drew special attention to two points: The primary composite event rate in placebo-treated patients in the Americas was 31.8% consistent with what has been seen in other studies of HFpEF – compared to a mere 8.4% in Eastern Europe. And patients who qualified for TOPCAT on the basis of an elevated natriuretic peptide level had a primary endpoint rate of 15.9% with spironolactone, a highly significant 35% reduction compared with the 23.6% in controls, suggesting that an elevated baseline natriuretic peptide level may be a biomarker useful in identifying those HFpEF patients most likely to respond to an aldosterone antagonist.
Dr. Pfeffer said that because the pharmaceutical industry has zero interest in spironolactone and the National Heart, Lung, and Blood Institute (NHLBI), which sponsored TOPCAT, has finite resources, he doubts there will be any further large studies of the drug in HFpEF.
"One is going to have to make decisions based on this trial. I don’t see another trial behind us," the cardiologist said.
And while TOPCAT was flawed, he said it contains a compelling message for clinicians: "I think we have an important finding here. We’re very confident that with this generic medication, which costs pennies per day, we can reduce hospitalizations for heart failure, which are the major burden in patients with HFpEF."
Discussant Dr. Margaret M. Redfield was more cautious. While there was a sound rationale for studying spironolactone in HFpEF based upon its impressive benefits in systolic heart failure as shown in earlier landmark clinical trials, given that the drug didn’t result in a significant reduction in all-cause hospitalizations, she said she wants to see evidence of improved patient-centered outcomes, such as quality of life, before prescribing spironolactone for HFpEF.
"One has to worry that the problems with worsening renal function and hyperkalemia may be much more common in clinical practice than in the highly monitored environment of a clinical trial," added Dr. Redfield, professor of medicine at the Mayo Clinic, Rochester, Minn.

Other heart failure experts not involved in TOPCAT took a more positive view of the study.
Dr. Clyde W. Yancy, AHA spokesperson, said in an interview that HFpEF is probably even more common than systolic heart failure, and it is a disease for which physicians have had no proven-effective treatments.
"You can almost justifiably say, ‘Let’s just look at the Americas, where patients had event rates that are consistent with the disease we think we understand.’ And it looks like there’s a signal there. I think that there’s enough evidence in TOPCAT for the clinical pragmatist to be able to be comfortable that spironolactone is probably beneficial and can be given safely. If you are convinced you can give it safely in your own clinical scenario, I think you should use it," said Dr. Yancy, professor of medicine and chief of cardiology at Northwestern University, Chicago.
Dr. Marco Metra, a cardiologist at the University of Brescia (Italy), said he interprets the TOPCAT results as an indication for prescribing spironolactone, at least in HFpEF patients with high natriuretic peptide levels or multiple heart failure hospitalizations.
Dr. Pitt, emeritus professor of cardiovascular medicine at the University of Michigan, Ann Arbor, emphasized that in prescribing spironolactone, whether for HFpEF or systolic heart failure, it’s obligatory to measure potassium and creatinine levels at baseline, when changing the dose, and at each routine follow-up visit, titrating in response to the results. Failure to do so is akin to prescribing warfarin for a patient with atrial fibrillation and then never measuring the INR (international normalized ratio) he said.
TOPCAT was sponsored by the National, Heart, Lung, and Blood Institute. Dr. Pfeffer and Dr. Pitt serve as consultants to numerous pharmaceutical companies that had no involvement in the trial.
DALLAS – Spironolactone did not hit a home run in the large international "treatment of preserved cardiac function heart failure with an aldosterone antagonist" (TOPCAT) trial, but it did knock out a solid single in the form of significantly reduced hospitalizations for this extremely common, chronic, high morbidity/mortality condition.
It was this positive result for an important prespecified secondary outcome that enabled TOPCAT to avoid becoming roadkill. Technically, TOPCAT was a negative clinical trial in that spironolactone did not significantly outperform placebo on the primary composite outcome of cardiovascular mortality, heart failure hospitalization, or aborted cardiac arrest.
Yet that negative primary outcome was controversial: The aldosterone antagonist actually showed a significant positive result for the composite endpoint in North and South American participants, yet the results were resoundingly negative – and also considerably out of whack with the characteristic arc of progressive heart failure – among the nearly one-half of TOPCAT participants in Russia and the Republic of Georgia.
"What happened in Russia and Georgia we just don’t understand," Dr. Bertram Pitt, TOPCAT steering committee chair, said in an interview, shaking his head. "The event rate with placebo in Eastern Europe was so low it’s not compatible with anything we know about heart failure. The signs and symptoms of HFpEF [heart failure with preserved ejection fraction] are nonspecific; they can be due to obesity, lung disease, and other things. Clearly there are some people getting into the major trials of HFpEF that probably don’t have it."
TOPCAT was a randomized, double-blind clinical trial comprising 3,445 participants with symptomatic HFpEF at 250 sites in the United States and five other countries. They were randomized to spironolactone or placebo and followed prospectively for a mean of 3.3 years. The starting dose of the aldosterone antagonist was 15 mg/day, with a target of 30 mg/day. The drug could be titrated within the range of 15-45 mg/day. Eight months into the trial, the mean daily dose was 25 mg.
Presenting the TOPCAT results at the American Heart Association scientific sessions, Dr. Marc A. Pfeffer noted that the primary composite endpoint occurred in 20.4% of placebo-treated controls and 18.6% on spironolactone, a statistically nonsignificant difference. In contrast, the 17% reduction in the rate of hospitalization for HFpEF in the spironolactone group relative to controls was significant (P = .04). Moreover, the spironolactone-treated patients had a collective 394 HFpEF hospitalizations, markedly fewer than the 475 in controls. This translated to a hospitalization for heart failure with a preserved left ventricular ejection fraction occurring at a rate of 3.8 per 100 person-years in patients randomized to spironolactone, compared with the 4.6 per 100 person-years in placebo-treated controls.
Hyperkalemia in excess of 5.5 mmol/L occurred in 18.7% of the spironolactone group, twice the rate of controls (9.1%). And the incidence of a creatinine level more than double the upper limit of normal was 49% greater in the spironolactone group. That said, neither of these laboratory abnormalities resulted in any serious adverse consequences because investigators adjusted the dose in response, explained Dr. Pfeffer, professor of medicine at Harvard University, Boston.
He drew special attention to two points: The primary composite event rate in placebo-treated patients in the Americas was 31.8% consistent with what has been seen in other studies of HFpEF – compared to a mere 8.4% in Eastern Europe. And patients who qualified for TOPCAT on the basis of an elevated natriuretic peptide level had a primary endpoint rate of 15.9% with spironolactone, a highly significant 35% reduction compared with the 23.6% in controls, suggesting that an elevated baseline natriuretic peptide level may be a biomarker useful in identifying those HFpEF patients most likely to respond to an aldosterone antagonist.
Dr. Pfeffer said that because the pharmaceutical industry has zero interest in spironolactone and the National Heart, Lung, and Blood Institute (NHLBI), which sponsored TOPCAT, has finite resources, he doubts there will be any further large studies of the drug in HFpEF.
"One is going to have to make decisions based on this trial. I don’t see another trial behind us," the cardiologist said.
And while TOPCAT was flawed, he said it contains a compelling message for clinicians: "I think we have an important finding here. We’re very confident that with this generic medication, which costs pennies per day, we can reduce hospitalizations for heart failure, which are the major burden in patients with HFpEF."
Discussant Dr. Margaret M. Redfield was more cautious. While there was a sound rationale for studying spironolactone in HFpEF based upon its impressive benefits in systolic heart failure as shown in earlier landmark clinical trials, given that the drug didn’t result in a significant reduction in all-cause hospitalizations, she said she wants to see evidence of improved patient-centered outcomes, such as quality of life, before prescribing spironolactone for HFpEF.
"One has to worry that the problems with worsening renal function and hyperkalemia may be much more common in clinical practice than in the highly monitored environment of a clinical trial," added Dr. Redfield, professor of medicine at the Mayo Clinic, Rochester, Minn.
Other heart failure experts not involved in TOPCAT took a more positive view of the study.
Dr. Clyde W. Yancy, AHA spokesperson, said in an interview that HFpEF is probably even more common than systolic heart failure, and it is a disease for which physicians have had no proven-effective treatments.
"You can almost justifiably say, ‘Let’s just look at the Americas, where patients had event rates that are consistent with the disease we think we understand.’ And it looks like there’s a signal there. I think that there’s enough evidence in TOPCAT for the clinical pragmatist to be able to be comfortable that spironolactone is probably beneficial and can be given safely. If you are convinced you can give it safely in your own clinical scenario, I think you should use it," said Dr. Yancy, professor of medicine and chief of cardiology at Northwestern University, Chicago.
Dr. Marco Metra, a cardiologist at the University of Brescia (Italy), said he interprets the TOPCAT results as an indication for prescribing spironolactone, at least in HFpEF patients with high natriuretic peptide levels or multiple heart failure hospitalizations.
Dr. Pitt, emeritus professor of cardiovascular medicine at the University of Michigan, Ann Arbor, emphasized that in prescribing spironolactone, whether for HFpEF or systolic heart failure, it’s obligatory to measure potassium and creatinine levels at baseline, when changing the dose, and at each routine follow-up visit, titrating in response to the results. Failure to do so is akin to prescribing warfarin for a patient with atrial fibrillation and then never measuring the INR (international normalized ratio) he said.
TOPCAT was sponsored by the National, Heart, Lung, and Blood Institute. Dr. Pfeffer and Dr. Pitt serve as consultants to numerous pharmaceutical companies that had no involvement in the trial.
DALLAS – Spironolactone did not hit a home run in the large international "treatment of preserved cardiac function heart failure with an aldosterone antagonist" (TOPCAT) trial, but it did knock out a solid single in the form of significantly reduced hospitalizations for this extremely common, chronic, high morbidity/mortality condition.
It was this positive result for an important prespecified secondary outcome that enabled TOPCAT to avoid becoming roadkill. Technically, TOPCAT was a negative clinical trial in that spironolactone did not significantly outperform placebo on the primary composite outcome of cardiovascular mortality, heart failure hospitalization, or aborted cardiac arrest.
Yet that negative primary outcome was controversial: The aldosterone antagonist actually showed a significant positive result for the composite endpoint in North and South American participants, yet the results were resoundingly negative – and also considerably out of whack with the characteristic arc of progressive heart failure – among the nearly one-half of TOPCAT participants in Russia and the Republic of Georgia.
"What happened in Russia and Georgia we just don’t understand," Dr. Bertram Pitt, TOPCAT steering committee chair, said in an interview, shaking his head. "The event rate with placebo in Eastern Europe was so low it’s not compatible with anything we know about heart failure. The signs and symptoms of HFpEF [heart failure with preserved ejection fraction] are nonspecific; they can be due to obesity, lung disease, and other things. Clearly there are some people getting into the major trials of HFpEF that probably don’t have it."
TOPCAT was a randomized, double-blind clinical trial comprising 3,445 participants with symptomatic HFpEF at 250 sites in the United States and five other countries. They were randomized to spironolactone or placebo and followed prospectively for a mean of 3.3 years. The starting dose of the aldosterone antagonist was 15 mg/day, with a target of 30 mg/day. The drug could be titrated within the range of 15-45 mg/day. Eight months into the trial, the mean daily dose was 25 mg.
Presenting the TOPCAT results at the American Heart Association scientific sessions, Dr. Marc A. Pfeffer noted that the primary composite endpoint occurred in 20.4% of placebo-treated controls and 18.6% on spironolactone, a statistically nonsignificant difference. In contrast, the 17% reduction in the rate of hospitalization for HFpEF in the spironolactone group relative to controls was significant (P = .04). Moreover, the spironolactone-treated patients had a collective 394 HFpEF hospitalizations, markedly fewer than the 475 in controls. This translated to a hospitalization for heart failure with a preserved left ventricular ejection fraction occurring at a rate of 3.8 per 100 person-years in patients randomized to spironolactone, compared with the 4.6 per 100 person-years in placebo-treated controls.
Hyperkalemia in excess of 5.5 mmol/L occurred in 18.7% of the spironolactone group, twice the rate of controls (9.1%). And the incidence of a creatinine level more than double the upper limit of normal was 49% greater in the spironolactone group. That said, neither of these laboratory abnormalities resulted in any serious adverse consequences because investigators adjusted the dose in response, explained Dr. Pfeffer, professor of medicine at Harvard University, Boston.
He drew special attention to two points: The primary composite event rate in placebo-treated patients in the Americas was 31.8% consistent with what has been seen in other studies of HFpEF – compared to a mere 8.4% in Eastern Europe. And patients who qualified for TOPCAT on the basis of an elevated natriuretic peptide level had a primary endpoint rate of 15.9% with spironolactone, a highly significant 35% reduction compared with the 23.6% in controls, suggesting that an elevated baseline natriuretic peptide level may be a biomarker useful in identifying those HFpEF patients most likely to respond to an aldosterone antagonist.
Dr. Pfeffer said that because the pharmaceutical industry has zero interest in spironolactone and the National Heart, Lung, and Blood Institute (NHLBI), which sponsored TOPCAT, has finite resources, he doubts there will be any further large studies of the drug in HFpEF.
"One is going to have to make decisions based on this trial. I don’t see another trial behind us," the cardiologist said.
And while TOPCAT was flawed, he said it contains a compelling message for clinicians: "I think we have an important finding here. We’re very confident that with this generic medication, which costs pennies per day, we can reduce hospitalizations for heart failure, which are the major burden in patients with HFpEF."
Discussant Dr. Margaret M. Redfield was more cautious. While there was a sound rationale for studying spironolactone in HFpEF based upon its impressive benefits in systolic heart failure as shown in earlier landmark clinical trials, given that the drug didn’t result in a significant reduction in all-cause hospitalizations, she said she wants to see evidence of improved patient-centered outcomes, such as quality of life, before prescribing spironolactone for HFpEF.
"One has to worry that the problems with worsening renal function and hyperkalemia may be much more common in clinical practice than in the highly monitored environment of a clinical trial," added Dr. Redfield, professor of medicine at the Mayo Clinic, Rochester, Minn.
Other heart failure experts not involved in TOPCAT took a more positive view of the study.
Dr. Clyde W. Yancy, AHA spokesperson, said in an interview that HFpEF is probably even more common than systolic heart failure, and it is a disease for which physicians have had no proven-effective treatments.
"You can almost justifiably say, ‘Let’s just look at the Americas, where patients had event rates that are consistent with the disease we think we understand.’ And it looks like there’s a signal there. I think that there’s enough evidence in TOPCAT for the clinical pragmatist to be able to be comfortable that spironolactone is probably beneficial and can be given safely. If you are convinced you can give it safely in your own clinical scenario, I think you should use it," said Dr. Yancy, professor of medicine and chief of cardiology at Northwestern University, Chicago.
Dr. Marco Metra, a cardiologist at the University of Brescia (Italy), said he interprets the TOPCAT results as an indication for prescribing spironolactone, at least in HFpEF patients with high natriuretic peptide levels or multiple heart failure hospitalizations.
Dr. Pitt, emeritus professor of cardiovascular medicine at the University of Michigan, Ann Arbor, emphasized that in prescribing spironolactone, whether for HFpEF or systolic heart failure, it’s obligatory to measure potassium and creatinine levels at baseline, when changing the dose, and at each routine follow-up visit, titrating in response to the results. Failure to do so is akin to prescribing warfarin for a patient with atrial fibrillation and then never measuring the INR (international normalized ratio) he said.
TOPCAT was sponsored by the National, Heart, Lung, and Blood Institute. Dr. Pfeffer and Dr. Pitt serve as consultants to numerous pharmaceutical companies that had no involvement in the trial.
AT THE AHA SCIENTIFIC SESSIONS
Major finding: Hospitalization for heart failure with a preserved left ventricular ejection fraction occurred at a rate of 3.8 per 100 person-years in patients randomized to spironolactone, a significant 17% risk reduction compared with the 4.6 per 100 person-years in placebo-treated controls.
Data source: TOPCAT, a randomized, double-blind, six-country clinical trial involving 3,445 patients with symptomatic heart failure and an ejection fraction of 45% or more.
Disclosures:. TOPCAT was sponsored by the National, Heart, Lung, and Blood Institute. Dr. Pfeffer and Dr. Pitt serve as consultants to pharmaceutical companies that had no involvement in the trial.

Guideline authors, AHA-ACC leaders confident in risk calculator
DALLAS – In the face of high-profile criticism of the new U.S. cholesterol-management guidelines released by the American College of Cardiology and American Heart Association on Nov. 12, the physicians who crafted the guidelines, as well as the leadership of the two organizations, stood firmly behind the work, reiterating that it is a big step forward in the battle against atherosclerotic cardiovascular disease.
During a last-minute press conference called on Monday morning, Nov. 18, to address concerns that first bubbled up over the prior weekend, a series of representatives from the guideline-writing panel stressed that the new cardiovascular-risk-calculation formula introduced in the guidelines reflected the best and most comprehensive evidence available today. They also stressed that the guidelines do not advocate simply using the formula and a rigid risk-threshold to decide whether or not a patient should receive a statin, but instead tell physicians and their patients to use the risk calculator as the starting point for a discussion of the risks and benefits that statin treatment might pose for each person.

"I think these are these are the most carefully vetted guidelines ever published," said Dr. Mariell Jessup, AHA president. "We’re confident that they are based on the best evidence."
"We intend to move forward with implementation of these guidelines," said Dr. Sidney Smith, professor of medicine at the University of North Carolina in Chapel Hill and chair of the ACC/AHA subcommittee on prevention guidelines.
The controversy began with an analysis run by two Harvard researchers starting on the day the guidelines and risk calculator came out that applied the new risk calculator to three databases at their immediate disposal, the Women’s Health Study, the Physicians’ Health Study, and the Women’s Health Initiative Observational Study. They found that the risk calculator overestimated the 10-year rate of atherosclerotic cardiovascular disease (ASCVD) event by 75%-150%, doubling the actual, observed risk in these three cohorts. The authors of the analysis, Dr. Paul M. Ridker and Nancy R. Cook, Sc.D., of Harvard Medical School and Brigham and Women’s Hospital, both in Boston, published their results in a brief article published online in the Lancet on Nov. 19.

Based on their calculations, "it is possible that as many as 40% to 50% of the 33 million Americans targeted by the new guidelines for statin therapy do not actually have risk thresholds exceeding the 7.5% level suggested for treatment," the two researchers wrote. "Miscalibration to this extent should be reconciled and addressed ... before these new prediction models are widely implemented."
Their analysis was made available to the New York Times over the weekend, which led to a prominent story in the newspaper’s Nov. 18 edition that called this overestimate by the risk calculator a "major embarrassment," and quoted some prominent cardiologists who also called for a delay in implementation of the new guidelines and risk calculator.
The researchers who devised the calculator responded by acknowledging the flaws in the device, something they had already done in their manuscript, but stressed that it represented a major improvement over the risk calculator from the prior guidelines, the third edition of the Adult Treatment Panel (ATP III) used for the past 12 years, particularly because of its inclusion of strokes as a ASCVD endpoint and its reliance on databases that had substantial numbers of African Americans, two major features missing from ATP III.
A risk calculator "will never be perfect, but this is a huge step ahead from where we were 12 years ago," said Dr. Donald Lloyd-Jones, professor of preventive medicine at Northwestern University in Chicago and cochair of the panel that developed the risk calculator. "We made it better for women [by including stroke as an outcome], for African Americans, and for white men, too. We feel very confident that we are in a great place now, but as more data become available, we’re very happy to see if we can improve it further. You can certainly criticize the calculator, but I don’t know of anything that’s better," he said in an interview.
Other experts who led development of the new guidelines also stressed that they do not call for blindly prescribing a statin to every person whose risk calculates at or above 7.5%.
"No one said that patients automatically get a statin. We said that there needs to be a risk discussion between the patient and physician, because sometimes the numbers make sense and sometimes they don’t," said Dr. Neil Stone, Robert Bonow Professor in the division of medicine-cardiology at Northwestern University and chair of the ACC/AHA panel that wrote the new guidelines. "For the first time, we built into the guidelines the unique judgment of physicians, and patients’ personal preferences." Risk calculation "is the start of a discussion" between a physician and patient, not the endpoint. He also noted that the guidelines included a built-in "buffer" because "we had evidence that statins are effective even for patients with a 5% risk" for an ASCVD event over the subsequent 10 years.
The guidelines say that it is "reasonable to offer" statin treatment to a middle-age person with a 7.5% or greater 10-year risk for a ASCVD event and no other risks that warrant statin treatment and that before starting statin treatment, it is "reasonable" for a physician and patient to have a discussion that covers the patient’s individual risk level, the potential risks and benefits of statin treatment, and patient preference.
Aside from their critique of the risk calculator, Dr. Ridker and Dr. Cook applauded the overall guidelines in their commentary, calling them "a major step in the right direction."
On Twitter @mitchelzoler
DALLAS – In the face of high-profile criticism of the new U.S. cholesterol-management guidelines released by the American College of Cardiology and American Heart Association on Nov. 12, the physicians who crafted the guidelines, as well as the leadership of the two organizations, stood firmly behind the work, reiterating that it is a big step forward in the battle against atherosclerotic cardiovascular disease.
During a last-minute press conference called on Monday morning, Nov. 18, to address concerns that first bubbled up over the prior weekend, a series of representatives from the guideline-writing panel stressed that the new cardiovascular-risk-calculation formula introduced in the guidelines reflected the best and most comprehensive evidence available today. They also stressed that the guidelines do not advocate simply using the formula and a rigid risk-threshold to decide whether or not a patient should receive a statin, but instead tell physicians and their patients to use the risk calculator as the starting point for a discussion of the risks and benefits that statin treatment might pose for each person.
"I think these are these are the most carefully vetted guidelines ever published," said Dr. Mariell Jessup, AHA president. "We’re confident that they are based on the best evidence."
"We intend to move forward with implementation of these guidelines," said Dr. Sidney Smith, professor of medicine at the University of North Carolina in Chapel Hill and chair of the ACC/AHA subcommittee on prevention guidelines.
The controversy began with an analysis run by two Harvard researchers starting on the day the guidelines and risk calculator came out that applied the new risk calculator to three databases at their immediate disposal, the Women’s Health Study, the Physicians’ Health Study, and the Women’s Health Initiative Observational Study. They found that the risk calculator overestimated the 10-year rate of atherosclerotic cardiovascular disease (ASCVD) event by 75%-150%, doubling the actual, observed risk in these three cohorts. The authors of the analysis, Dr. Paul M. Ridker and Nancy R. Cook, Sc.D., of Harvard Medical School and Brigham and Women’s Hospital, both in Boston, published their results in a brief article published online in the Lancet on Nov. 19.
Based on their calculations, "it is possible that as many as 40% to 50% of the 33 million Americans targeted by the new guidelines for statin therapy do not actually have risk thresholds exceeding the 7.5% level suggested for treatment," the two researchers wrote. "Miscalibration to this extent should be reconciled and addressed ... before these new prediction models are widely implemented."
Their analysis was made available to the New York Times over the weekend, which led to a prominent story in the newspaper’s Nov. 18 edition that called this overestimate by the risk calculator a "major embarrassment," and quoted some prominent cardiologists who also called for a delay in implementation of the new guidelines and risk calculator.
The researchers who devised the calculator responded by acknowledging the flaws in the device, something they had already done in their manuscript, but stressed that it represented a major improvement over the risk calculator from the prior guidelines, the third edition of the Adult Treatment Panel (ATP III) used for the past 12 years, particularly because of its inclusion of strokes as a ASCVD endpoint and its reliance on databases that had substantial numbers of African Americans, two major features missing from ATP III.
A risk calculator "will never be perfect, but this is a huge step ahead from where we were 12 years ago," said Dr. Donald Lloyd-Jones, professor of preventive medicine at Northwestern University in Chicago and cochair of the panel that developed the risk calculator. "We made it better for women [by including stroke as an outcome], for African Americans, and for white men, too. We feel very confident that we are in a great place now, but as more data become available, we’re very happy to see if we can improve it further. You can certainly criticize the calculator, but I don’t know of anything that’s better," he said in an interview.
Other experts who led development of the new guidelines also stressed that they do not call for blindly prescribing a statin to every person whose risk calculates at or above 7.5%.
"No one said that patients automatically get a statin. We said that there needs to be a risk discussion between the patient and physician, because sometimes the numbers make sense and sometimes they don’t," said Dr. Neil Stone, Robert Bonow Professor in the division of medicine-cardiology at Northwestern University and chair of the ACC/AHA panel that wrote the new guidelines. "For the first time, we built into the guidelines the unique judgment of physicians, and patients’ personal preferences." Risk calculation "is the start of a discussion" between a physician and patient, not the endpoint. He also noted that the guidelines included a built-in "buffer" because "we had evidence that statins are effective even for patients with a 5% risk" for an ASCVD event over the subsequent 10 years.
The guidelines say that it is "reasonable to offer" statin treatment to a middle-age person with a 7.5% or greater 10-year risk for a ASCVD event and no other risks that warrant statin treatment and that before starting statin treatment, it is "reasonable" for a physician and patient to have a discussion that covers the patient’s individual risk level, the potential risks and benefits of statin treatment, and patient preference.
Aside from their critique of the risk calculator, Dr. Ridker and Dr. Cook applauded the overall guidelines in their commentary, calling them "a major step in the right direction."
On Twitter @mitchelzoler
DALLAS – In the face of high-profile criticism of the new U.S. cholesterol-management guidelines released by the American College of Cardiology and American Heart Association on Nov. 12, the physicians who crafted the guidelines, as well as the leadership of the two organizations, stood firmly behind the work, reiterating that it is a big step forward in the battle against atherosclerotic cardiovascular disease.
During a last-minute press conference called on Monday morning, Nov. 18, to address concerns that first bubbled up over the prior weekend, a series of representatives from the guideline-writing panel stressed that the new cardiovascular-risk-calculation formula introduced in the guidelines reflected the best and most comprehensive evidence available today. They also stressed that the guidelines do not advocate simply using the formula and a rigid risk-threshold to decide whether or not a patient should receive a statin, but instead tell physicians and their patients to use the risk calculator as the starting point for a discussion of the risks and benefits that statin treatment might pose for each person.
"I think these are these are the most carefully vetted guidelines ever published," said Dr. Mariell Jessup, AHA president. "We’re confident that they are based on the best evidence."
"We intend to move forward with implementation of these guidelines," said Dr. Sidney Smith, professor of medicine at the University of North Carolina in Chapel Hill and chair of the ACC/AHA subcommittee on prevention guidelines.
The controversy began with an analysis run by two Harvard researchers starting on the day the guidelines and risk calculator came out that applied the new risk calculator to three databases at their immediate disposal, the Women’s Health Study, the Physicians’ Health Study, and the Women’s Health Initiative Observational Study. They found that the risk calculator overestimated the 10-year rate of atherosclerotic cardiovascular disease (ASCVD) event by 75%-150%, doubling the actual, observed risk in these three cohorts. The authors of the analysis, Dr. Paul M. Ridker and Nancy R. Cook, Sc.D., of Harvard Medical School and Brigham and Women’s Hospital, both in Boston, published their results in a brief article published online in the Lancet on Nov. 19.
Based on their calculations, "it is possible that as many as 40% to 50% of the 33 million Americans targeted by the new guidelines for statin therapy do not actually have risk thresholds exceeding the 7.5% level suggested for treatment," the two researchers wrote. "Miscalibration to this extent should be reconciled and addressed ... before these new prediction models are widely implemented."
Their analysis was made available to the New York Times over the weekend, which led to a prominent story in the newspaper’s Nov. 18 edition that called this overestimate by the risk calculator a "major embarrassment," and quoted some prominent cardiologists who also called for a delay in implementation of the new guidelines and risk calculator.
The researchers who devised the calculator responded by acknowledging the flaws in the device, something they had already done in their manuscript, but stressed that it represented a major improvement over the risk calculator from the prior guidelines, the third edition of the Adult Treatment Panel (ATP III) used for the past 12 years, particularly because of its inclusion of strokes as a ASCVD endpoint and its reliance on databases that had substantial numbers of African Americans, two major features missing from ATP III.
A risk calculator "will never be perfect, but this is a huge step ahead from where we were 12 years ago," said Dr. Donald Lloyd-Jones, professor of preventive medicine at Northwestern University in Chicago and cochair of the panel that developed the risk calculator. "We made it better for women [by including stroke as an outcome], for African Americans, and for white men, too. We feel very confident that we are in a great place now, but as more data become available, we’re very happy to see if we can improve it further. You can certainly criticize the calculator, but I don’t know of anything that’s better," he said in an interview.
Other experts who led development of the new guidelines also stressed that they do not call for blindly prescribing a statin to every person whose risk calculates at or above 7.5%.
"No one said that patients automatically get a statin. We said that there needs to be a risk discussion between the patient and physician, because sometimes the numbers make sense and sometimes they don’t," said Dr. Neil Stone, Robert Bonow Professor in the division of medicine-cardiology at Northwestern University and chair of the ACC/AHA panel that wrote the new guidelines. "For the first time, we built into the guidelines the unique judgment of physicians, and patients’ personal preferences." Risk calculation "is the start of a discussion" between a physician and patient, not the endpoint. He also noted that the guidelines included a built-in "buffer" because "we had evidence that statins are effective even for patients with a 5% risk" for an ASCVD event over the subsequent 10 years.
The guidelines say that it is "reasonable to offer" statin treatment to a middle-age person with a 7.5% or greater 10-year risk for a ASCVD event and no other risks that warrant statin treatment and that before starting statin treatment, it is "reasonable" for a physician and patient to have a discussion that covers the patient’s individual risk level, the potential risks and benefits of statin treatment, and patient preference.
Aside from their critique of the risk calculator, Dr. Ridker and Dr. Cook applauded the overall guidelines in their commentary, calling them "a major step in the right direction."
On Twitter @mitchelzoler
EXPERT OPINION AT THE AHA SCIENTIFIC SESSIONS

Anticoagulant edoxaban comparable with warfarin in atrial fib
Two doses of the once-daily, oral factor Xa inhibitor edoxaban were noninferior to warfarin in preventing stroke or systemic embolism in patients with atrial fibrillation in the phase III ENGAGE AF-TIMI 48 trial.
The primary endpoint of stroke or embolic events was seen in 1.50% of patients per year on well-controlled warfarin vs. 1.18% with edoxaban 60 mg (hazard ratio, 0.79; P less than .001 for noninferiority) and 1.61% with edoxaban 30 mg (HR, 1.07; P = .005 for noninferiority), Dr. Robert P. Giugliano reported at the American Heart Association scientific sessions.
Edoxaban also bested oral warfarin on the study’s principal safety endpoint of major bleeding and significantly reduced bleeding and death from cardiovascular causes in the study, published simultaneously with the presentation (N. Engl. J. Med. 2013 Nov. 19 [doi:10.1056/NEJMoa1310907]).

Edoxaban is currently approved only in Japan (Lixiana) for venous thromboembolism (VTE) prevention after major orthopedic surgery, although regulatory filings are expected in the United States, Europe, and Japan in 2014 following recent positive results phase III results for the treatment and prevention of recurrent symptomatic VTE.
Though positive, the results of ENGAGE AF-TIMI 48 (Effective Anticoagulation With Factor Xa Next Generation in Atrial Fibrillation – Thrombolysis in Myocardial Infarction 48) do little to distinguish edoxaban from other novel oral anticoagulants entering the market, including the factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis), and the twice-daily direct thrombin inhibitor dabigatran (Pradaxa).
Approval of the lower dose of edoxaban is unlikely as it barely meets noninferiority for the primary endpoint in the intention-to-treat analysis (upper 95% confidence interval bound of 1.34; noninferiority margin of 1.38) – and ischemic stroke was significantly worse than warfarin (1.77% per year vs. 1.25% per year; HR, 1.41; P less than .001), observed cardiologist Dr. Sanjay Kaul of Cedars-Sinai Medical Center, Los Angeles.
High-dose edoxaban had the same ischemic stroke rate as warfarin, 1.25%.
Annualized rates of hemorrhagic stroke were 0.47% with warfarin, compared with 0.26% with high-dose edoxaban (HR, 0.54) and 0.16% with low-dose edoxaban (HR, 0.33; both P less than .001).
High-dose edoxaban also failed to meet superiority over warfarin for the primary endpoint in a prespecified intention-to-treat analysis (1.57% vs. 1.80%; HR, 0.87; P = .08), so a superiority claim won’t be allowed.
"Marketability might be an issue, as the treatment advantage is not overwhelming," Dr. Kaul said in an interview. "Being the fourth kid on the block with no unique advantage might challenge its acceptability in clinical practice and marketability."
ENGAGE AF-TIMI 48 enrolled 21,105 patients with moderate- to high-risk atrial fibrillation. One-quarter had paroxysmal atrial fibrillation, median follow-up was 2.8 years, and the warfarin group was in the therapeutic range for a median of 68.4% of the treatment period.
Major bleeding occurred in 3.43% of patients per year with warfarin vs. 2.75% with edoxaban 60 mg (HR, 0.80) and in 1.61% with edoxaban 30 mg (HR, 0.47; both P less than .001), reported Dr. Giugliano of Brigham and Women’s Hospital and Harvard Medical School, in Boston. The corresponding annualized rates of death from cardiovascular causes were, respectively, 3.17% vs. 2.74% and 2.71%, both nonsignificant differences.
Rates of the key composite secondary endpoint of stroke, systemic embolism, or death from cardiovascular causes were 4.43% with warfarin vs. 3.85% with high-dose edoxaban (HR, 0.87%; P = .005) and 4.23% for low-dose edoxaban, which failed to reach statistical significance (HR, 0.95; P = .32), he noted.
Dr. Giugliano and his coauthors reported grant support and other financial relationships with Daiichi Sankyo, the study sponsor. Dr. Kaul reported stock in Johnson & Johnson and consultancy for Boehringer Ingelheim.

|
| Dr. Jun Chiong |
Dr. Jun Chiong, FCCP, comments: The direct factor Xa (FXa) inhibitors have gained popularity for use in several indications, most notably for the prevention of stroke in patients with atrial fibrillation. There are several advantages of using these agents, particularly on the PT/INR monitoring which are challenging, especially to stroke patients with limited activities of daily living.
The Achilles' heel of these novel oral anticoagulants is the lack of an antidote, although research is ongoing currently. Widespread use of these agents will mainly depend onits their formulary status in health plans as we are moving toward affordable and value-based care.
|
|
| Dr. Jun Chiong |
Dr. Jun Chiong, FCCP, comments: The direct factor Xa (FXa) inhibitors have gained popularity for use in several indications, most notably for the prevention of stroke in patients with atrial fibrillation. There are several advantages of using these agents, particularly on the PT/INR monitoring which are challenging, especially to stroke patients with limited activities of daily living.
The Achilles' heel of these novel oral anticoagulants is the lack of an antidote, although research is ongoing currently. Widespread use of these agents will mainly depend onits their formulary status in health plans as we are moving toward affordable and value-based care.
|
|
| Dr. Jun Chiong |
Dr. Jun Chiong, FCCP, comments: The direct factor Xa (FXa) inhibitors have gained popularity for use in several indications, most notably for the prevention of stroke in patients with atrial fibrillation. There are several advantages of using these agents, particularly on the PT/INR monitoring which are challenging, especially to stroke patients with limited activities of daily living.
The Achilles' heel of these novel oral anticoagulants is the lack of an antidote, although research is ongoing currently. Widespread use of these agents will mainly depend onits their formulary status in health plans as we are moving toward affordable and value-based care.
Two doses of the once-daily, oral factor Xa inhibitor edoxaban were noninferior to warfarin in preventing stroke or systemic embolism in patients with atrial fibrillation in the phase III ENGAGE AF-TIMI 48 trial.
The primary endpoint of stroke or embolic events was seen in 1.50% of patients per year on well-controlled warfarin vs. 1.18% with edoxaban 60 mg (hazard ratio, 0.79; P less than .001 for noninferiority) and 1.61% with edoxaban 30 mg (HR, 1.07; P = .005 for noninferiority), Dr. Robert P. Giugliano reported at the American Heart Association scientific sessions.
Edoxaban also bested oral warfarin on the study’s principal safety endpoint of major bleeding and significantly reduced bleeding and death from cardiovascular causes in the study, published simultaneously with the presentation (N. Engl. J. Med. 2013 Nov. 19 [doi:10.1056/NEJMoa1310907]).
Edoxaban is currently approved only in Japan (Lixiana) for venous thromboembolism (VTE) prevention after major orthopedic surgery, although regulatory filings are expected in the United States, Europe, and Japan in 2014 following recent positive results phase III results for the treatment and prevention of recurrent symptomatic VTE.
Though positive, the results of ENGAGE AF-TIMI 48 (Effective Anticoagulation With Factor Xa Next Generation in Atrial Fibrillation – Thrombolysis in Myocardial Infarction 48) do little to distinguish edoxaban from other novel oral anticoagulants entering the market, including the factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis), and the twice-daily direct thrombin inhibitor dabigatran (Pradaxa).
Approval of the lower dose of edoxaban is unlikely as it barely meets noninferiority for the primary endpoint in the intention-to-treat analysis (upper 95% confidence interval bound of 1.34; noninferiority margin of 1.38) – and ischemic stroke was significantly worse than warfarin (1.77% per year vs. 1.25% per year; HR, 1.41; P less than .001), observed cardiologist Dr. Sanjay Kaul of Cedars-Sinai Medical Center, Los Angeles.
High-dose edoxaban had the same ischemic stroke rate as warfarin, 1.25%.
Annualized rates of hemorrhagic stroke were 0.47% with warfarin, compared with 0.26% with high-dose edoxaban (HR, 0.54) and 0.16% with low-dose edoxaban (HR, 0.33; both P less than .001).
High-dose edoxaban also failed to meet superiority over warfarin for the primary endpoint in a prespecified intention-to-treat analysis (1.57% vs. 1.80%; HR, 0.87; P = .08), so a superiority claim won’t be allowed.
"Marketability might be an issue, as the treatment advantage is not overwhelming," Dr. Kaul said in an interview. "Being the fourth kid on the block with no unique advantage might challenge its acceptability in clinical practice and marketability."
ENGAGE AF-TIMI 48 enrolled 21,105 patients with moderate- to high-risk atrial fibrillation. One-quarter had paroxysmal atrial fibrillation, median follow-up was 2.8 years, and the warfarin group was in the therapeutic range for a median of 68.4% of the treatment period.
Major bleeding occurred in 3.43% of patients per year with warfarin vs. 2.75% with edoxaban 60 mg (HR, 0.80) and in 1.61% with edoxaban 30 mg (HR, 0.47; both P less than .001), reported Dr. Giugliano of Brigham and Women’s Hospital and Harvard Medical School, in Boston. The corresponding annualized rates of death from cardiovascular causes were, respectively, 3.17% vs. 2.74% and 2.71%, both nonsignificant differences.
Rates of the key composite secondary endpoint of stroke, systemic embolism, or death from cardiovascular causes were 4.43% with warfarin vs. 3.85% with high-dose edoxaban (HR, 0.87%; P = .005) and 4.23% for low-dose edoxaban, which failed to reach statistical significance (HR, 0.95; P = .32), he noted.
Dr. Giugliano and his coauthors reported grant support and other financial relationships with Daiichi Sankyo, the study sponsor. Dr. Kaul reported stock in Johnson & Johnson and consultancy for Boehringer Ingelheim.
Two doses of the once-daily, oral factor Xa inhibitor edoxaban were noninferior to warfarin in preventing stroke or systemic embolism in patients with atrial fibrillation in the phase III ENGAGE AF-TIMI 48 trial.
The primary endpoint of stroke or embolic events was seen in 1.50% of patients per year on well-controlled warfarin vs. 1.18% with edoxaban 60 mg (hazard ratio, 0.79; P less than .001 for noninferiority) and 1.61% with edoxaban 30 mg (HR, 1.07; P = .005 for noninferiority), Dr. Robert P. Giugliano reported at the American Heart Association scientific sessions.
Edoxaban also bested oral warfarin on the study’s principal safety endpoint of major bleeding and significantly reduced bleeding and death from cardiovascular causes in the study, published simultaneously with the presentation (N. Engl. J. Med. 2013 Nov. 19 [doi:10.1056/NEJMoa1310907]).
Edoxaban is currently approved only in Japan (Lixiana) for venous thromboembolism (VTE) prevention after major orthopedic surgery, although regulatory filings are expected in the United States, Europe, and Japan in 2014 following recent positive results phase III results for the treatment and prevention of recurrent symptomatic VTE.
Though positive, the results of ENGAGE AF-TIMI 48 (Effective Anticoagulation With Factor Xa Next Generation in Atrial Fibrillation – Thrombolysis in Myocardial Infarction 48) do little to distinguish edoxaban from other novel oral anticoagulants entering the market, including the factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis), and the twice-daily direct thrombin inhibitor dabigatran (Pradaxa).
Approval of the lower dose of edoxaban is unlikely as it barely meets noninferiority for the primary endpoint in the intention-to-treat analysis (upper 95% confidence interval bound of 1.34; noninferiority margin of 1.38) – and ischemic stroke was significantly worse than warfarin (1.77% per year vs. 1.25% per year; HR, 1.41; P less than .001), observed cardiologist Dr. Sanjay Kaul of Cedars-Sinai Medical Center, Los Angeles.
High-dose edoxaban had the same ischemic stroke rate as warfarin, 1.25%.
Annualized rates of hemorrhagic stroke were 0.47% with warfarin, compared with 0.26% with high-dose edoxaban (HR, 0.54) and 0.16% with low-dose edoxaban (HR, 0.33; both P less than .001).
High-dose edoxaban also failed to meet superiority over warfarin for the primary endpoint in a prespecified intention-to-treat analysis (1.57% vs. 1.80%; HR, 0.87; P = .08), so a superiority claim won’t be allowed.
"Marketability might be an issue, as the treatment advantage is not overwhelming," Dr. Kaul said in an interview. "Being the fourth kid on the block with no unique advantage might challenge its acceptability in clinical practice and marketability."
ENGAGE AF-TIMI 48 enrolled 21,105 patients with moderate- to high-risk atrial fibrillation. One-quarter had paroxysmal atrial fibrillation, median follow-up was 2.8 years, and the warfarin group was in the therapeutic range for a median of 68.4% of the treatment period.
Major bleeding occurred in 3.43% of patients per year with warfarin vs. 2.75% with edoxaban 60 mg (HR, 0.80) and in 1.61% with edoxaban 30 mg (HR, 0.47; both P less than .001), reported Dr. Giugliano of Brigham and Women’s Hospital and Harvard Medical School, in Boston. The corresponding annualized rates of death from cardiovascular causes were, respectively, 3.17% vs. 2.74% and 2.71%, both nonsignificant differences.
Rates of the key composite secondary endpoint of stroke, systemic embolism, or death from cardiovascular causes were 4.43% with warfarin vs. 3.85% with high-dose edoxaban (HR, 0.87%; P = .005) and 4.23% for low-dose edoxaban, which failed to reach statistical significance (HR, 0.95; P = .32), he noted.
Dr. Giugliano and his coauthors reported grant support and other financial relationships with Daiichi Sankyo, the study sponsor. Dr. Kaul reported stock in Johnson & Johnson and consultancy for Boehringer Ingelheim.
FROM THE AHA SCIENTIFIC SESSIONS
Major finding: The annualized rate of stroke or embolic events was 1.50% with warfarin vs. 1.18% with edoxaban 60 mg (HR, 0.79; P less than .001) and 1.61% with edoxaban 30 mg (HR, 1.07, P .005).
Data source: A prospective, phase III study in 21,105 patients with moderate- to high-risk atrial fibrillation.
Disclosures: Dr. Giugliano and his coauthors reported grant support and other financial relationships with Daiichi Sankyo, the study sponsor. Dr. Kaul reported stock in Johnson & Johnson and consultancy for Boehringer Ingelheim.

Varespladib raises MI risk after recent acute coronary syndrome
Far from reducing the risk of cardiovascular events in patients with recent acute coronary syndrome, the sPLA2 inhibitor varespladib actually raised the risk of MI in an international randomized clinical trial.
The drug did exert beneficial effects on inflammatory and lipid biomarkers as expected, "which theoretically should have translated to a lower propensity to plaque rupture" in the 16 weeks following ACS. But the study was terminated early "for futility" after an interim analysis of the outcomes for only 212 of the 5,012 randomized patients, said Dr. Stephen J. Nicholls and his associates in the VISTA-16 clinical trial, which was reported simultaneously at the American Heart Association scientific sessions and published online in JAMA (2013 Nov. 18 [doi:10.1001/jama.2013.282836]).
Compared with placebo, varespladib caused an excess of MIs and of the composite outcome of cardiovascular mortality, MI, and stroke. "These findings suggest that short-term sPLA2 inhibition with varespladib is harmful following ACS," said Dr. Nicholls of the South Australian Health and Medical Research Institute, Adelaide, and his colleagues.

"Our findings ... reemphasize that identification of a circulating marker of cardiovascular risk does not necessarily imply that pharmacologic suppression or inhibition of the marker will reduce risk," they noted.
Varespladib is a nonspecific inhibitor of the family of secretory phospholipase A2 (sPLA2) enzymes that are involved in inflammation, some of which are present in atherosclerotic lesions and myocardial areas injured by ischemia. In initial studies, the drug reduced circulating levels of sPLA2 by more than 90%, lowered LDL cholesterol, and decreased C-reactive protein in ACS patients.
The VISTA-16 (Vascular Inflammation Suppression to Treat Acute coronary syndrome for 16 weeks) trial, sponsored by Anthera Pharmaceuticals, the maker of varespladib, was designed to assess the drug’s efficacy at preventing CV events during the high-risk period of the 4 months immediately following ACS.
A total of 5,145 patients were enrolled at 362 medical centers in 17 countries over the course of 3 years, within hours of presenting with ACS. They had a high prevalence of CV risk factors and established atherosclerotic disease, and most were already receiving appropriate therapy with antiplatelet agents, statins, beta-blockers, ACE inhibitors, or receptor blockers. Eighty percent underwent coronary revascularization after the index event.
The primary endpoint was a composite of CV mortality, nonfatal MI, nonfatal stroke, or unstable angina requiring hospitalization. When the trial was terminated, outcomes in 212 patients had been documented after a mean of 13.4 weeks of treatment. The primary endpoint occurred in 6.1% of patients who took varespladib, compared with 5.1% of those who took placebo (hazard ratio, 1.25), the investigators said.
A secondary outcome, a composite of CV mortality, MI, and stroke, occurred in 4.6% of patients who took varespladib and 3.8% of those who took placebo (HR, 1.36).
"This was due primarily to a greater incidence of MI in the varespladib group compared with the placebo group (3.4% vs 2.2%; HR, 1.66)," the researchers said.
Six months after patients had discontinued study treatment, all-cause mortality was 2.7% in the varespladib group and 2.0% in the placebo group.
Twice as many patients taking the active drug (2.8%) as taking placebo (1.4%) discontinued treatment because of adverse or serious adverse events.
The reasons for varespladib’s adverse effect on MI are not yet known. "Whether this represents an adverse effect of the varespladib molecule or a consequence of pan-sPLA2 inhibition remains to be determined," Dr. Nicholls and his associates said.
The VISTA-16 trial was funded by Anthera Pharmaceuticals. Dr. Nicholls and his associates reported numerous ties to industry sources.
Far from reducing the risk of cardiovascular events in patients with recent acute coronary syndrome, the sPLA2 inhibitor varespladib actually raised the risk of MI in an international randomized clinical trial.
The drug did exert beneficial effects on inflammatory and lipid biomarkers as expected, "which theoretically should have translated to a lower propensity to plaque rupture" in the 16 weeks following ACS. But the study was terminated early "for futility" after an interim analysis of the outcomes for only 212 of the 5,012 randomized patients, said Dr. Stephen J. Nicholls and his associates in the VISTA-16 clinical trial, which was reported simultaneously at the American Heart Association scientific sessions and published online in JAMA (2013 Nov. 18 [doi:10.1001/jama.2013.282836]).
Compared with placebo, varespladib caused an excess of MIs and of the composite outcome of cardiovascular mortality, MI, and stroke. "These findings suggest that short-term sPLA2 inhibition with varespladib is harmful following ACS," said Dr. Nicholls of the South Australian Health and Medical Research Institute, Adelaide, and his colleagues.
"Our findings ... reemphasize that identification of a circulating marker of cardiovascular risk does not necessarily imply that pharmacologic suppression or inhibition of the marker will reduce risk," they noted.
Varespladib is a nonspecific inhibitor of the family of secretory phospholipase A2 (sPLA2) enzymes that are involved in inflammation, some of which are present in atherosclerotic lesions and myocardial areas injured by ischemia. In initial studies, the drug reduced circulating levels of sPLA2 by more than 90%, lowered LDL cholesterol, and decreased C-reactive protein in ACS patients.
The VISTA-16 (Vascular Inflammation Suppression to Treat Acute coronary syndrome for 16 weeks) trial, sponsored by Anthera Pharmaceuticals, the maker of varespladib, was designed to assess the drug’s efficacy at preventing CV events during the high-risk period of the 4 months immediately following ACS.
A total of 5,145 patients were enrolled at 362 medical centers in 17 countries over the course of 3 years, within hours of presenting with ACS. They had a high prevalence of CV risk factors and established atherosclerotic disease, and most were already receiving appropriate therapy with antiplatelet agents, statins, beta-blockers, ACE inhibitors, or receptor blockers. Eighty percent underwent coronary revascularization after the index event.
The primary endpoint was a composite of CV mortality, nonfatal MI, nonfatal stroke, or unstable angina requiring hospitalization. When the trial was terminated, outcomes in 212 patients had been documented after a mean of 13.4 weeks of treatment. The primary endpoint occurred in 6.1% of patients who took varespladib, compared with 5.1% of those who took placebo (hazard ratio, 1.25), the investigators said.
A secondary outcome, a composite of CV mortality, MI, and stroke, occurred in 4.6% of patients who took varespladib and 3.8% of those who took placebo (HR, 1.36).
"This was due primarily to a greater incidence of MI in the varespladib group compared with the placebo group (3.4% vs 2.2%; HR, 1.66)," the researchers said.
Six months after patients had discontinued study treatment, all-cause mortality was 2.7% in the varespladib group and 2.0% in the placebo group.
Twice as many patients taking the active drug (2.8%) as taking placebo (1.4%) discontinued treatment because of adverse or serious adverse events.
The reasons for varespladib’s adverse effect on MI are not yet known. "Whether this represents an adverse effect of the varespladib molecule or a consequence of pan-sPLA2 inhibition remains to be determined," Dr. Nicholls and his associates said.
The VISTA-16 trial was funded by Anthera Pharmaceuticals. Dr. Nicholls and his associates reported numerous ties to industry sources.
Far from reducing the risk of cardiovascular events in patients with recent acute coronary syndrome, the sPLA2 inhibitor varespladib actually raised the risk of MI in an international randomized clinical trial.
The drug did exert beneficial effects on inflammatory and lipid biomarkers as expected, "which theoretically should have translated to a lower propensity to plaque rupture" in the 16 weeks following ACS. But the study was terminated early "for futility" after an interim analysis of the outcomes for only 212 of the 5,012 randomized patients, said Dr. Stephen J. Nicholls and his associates in the VISTA-16 clinical trial, which was reported simultaneously at the American Heart Association scientific sessions and published online in JAMA (2013 Nov. 18 [doi:10.1001/jama.2013.282836]).
Compared with placebo, varespladib caused an excess of MIs and of the composite outcome of cardiovascular mortality, MI, and stroke. "These findings suggest that short-term sPLA2 inhibition with varespladib is harmful following ACS," said Dr. Nicholls of the South Australian Health and Medical Research Institute, Adelaide, and his colleagues.
"Our findings ... reemphasize that identification of a circulating marker of cardiovascular risk does not necessarily imply that pharmacologic suppression or inhibition of the marker will reduce risk," they noted.
Varespladib is a nonspecific inhibitor of the family of secretory phospholipase A2 (sPLA2) enzymes that are involved in inflammation, some of which are present in atherosclerotic lesions and myocardial areas injured by ischemia. In initial studies, the drug reduced circulating levels of sPLA2 by more than 90%, lowered LDL cholesterol, and decreased C-reactive protein in ACS patients.
The VISTA-16 (Vascular Inflammation Suppression to Treat Acute coronary syndrome for 16 weeks) trial, sponsored by Anthera Pharmaceuticals, the maker of varespladib, was designed to assess the drug’s efficacy at preventing CV events during the high-risk period of the 4 months immediately following ACS.
A total of 5,145 patients were enrolled at 362 medical centers in 17 countries over the course of 3 years, within hours of presenting with ACS. They had a high prevalence of CV risk factors and established atherosclerotic disease, and most were already receiving appropriate therapy with antiplatelet agents, statins, beta-blockers, ACE inhibitors, or receptor blockers. Eighty percent underwent coronary revascularization after the index event.
The primary endpoint was a composite of CV mortality, nonfatal MI, nonfatal stroke, or unstable angina requiring hospitalization. When the trial was terminated, outcomes in 212 patients had been documented after a mean of 13.4 weeks of treatment. The primary endpoint occurred in 6.1% of patients who took varespladib, compared with 5.1% of those who took placebo (hazard ratio, 1.25), the investigators said.
A secondary outcome, a composite of CV mortality, MI, and stroke, occurred in 4.6% of patients who took varespladib and 3.8% of those who took placebo (HR, 1.36).
"This was due primarily to a greater incidence of MI in the varespladib group compared with the placebo group (3.4% vs 2.2%; HR, 1.66)," the researchers said.
Six months after patients had discontinued study treatment, all-cause mortality was 2.7% in the varespladib group and 2.0% in the placebo group.
Twice as many patients taking the active drug (2.8%) as taking placebo (1.4%) discontinued treatment because of adverse or serious adverse events.
The reasons for varespladib’s adverse effect on MI are not yet known. "Whether this represents an adverse effect of the varespladib molecule or a consequence of pan-sPLA2 inhibition remains to be determined," Dr. Nicholls and his associates said.
The VISTA-16 trial was funded by Anthera Pharmaceuticals. Dr. Nicholls and his associates reported numerous ties to industry sources.
FROM THE AHA SCIENTIFIC SESSIONS
Major Finding: Patients who took varespladib had a significantly higher rate of MI (3.4%) than those who took placebo (2.2%), with a hazard ratio of 1.66.
Data Source: A double-blind, randomized clinical trial comparing varespladib against placebo in 5,012 ACS patients, which was terminated early when final outcomes were available for just 212 patients.
Disclosures: The VISTA-16 trial was funded by Anthera Pharmaceuticals. Dr. Nicholls and his associates reported numerous ties to industry sources.

CORAL: No added benefit with renal stenting vs. medication alone
Adding renal-artery stenting to comprehensive medical therapy did not confer significant benefit with respect to the prevention of clinical events in patients with atherosclerotic renal-artery stenosis and hypertension or chronic kidney disease in the randomized controlled CORAL trial.
During a median follow-up of 43 months in the multicenter, open-label trial, the rate of a primary composite endpoint consisting of death from cardiovascular or renal causes, myocardial infarction, stroke, hospitalization for heart failure, progressive renal insufficiency, or the need for renal replacement therapy did not differ significantly between 459 patients randomized to receive medical therapy plus renal-artery stenting, and 472 patients randomized to receive medical therapy alone (35.1% and 35.8%, respectively; hazard ratio with stenting, 0.94), Dr. Christopher J. Cooper reported at the American Heart Association scientific sessions.

Furthermore, no significant differences were seen between the treatment groups in the rates of the individual components of the composite primary endpoint, or in all-cause mortality. There was, however, a consistent, modest improvement in systolic blood pressure in the stent group (–2.33 mm Hg vs. the medication only group), noted Dr. Cooper of the University of Toledo (Ohio) and his coinvestigators.
The findings of the Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial were published online simultaneously with the Nov. 18 presentation (N. Engl. J. Med. 2013 Nov. 18 [doi:10.1056/NE/NEJMoa1310753]).
CORAL comprised adults with a mean age of 69 years who had atherosclerotic renal-artery stenosis and either chronic kidney disease or systolic hypertension (despite taking at least two antihypertensive drugs). They were recruited during May 2005–January 2010.
Mandated medications as per the study protocol (unless contraindicated) included the angiotensin II type-1 receptor blocker candesartan with or without hydrochlorothiazide, and the combination agent amlodipine-atorvastatin dose-adjusted on the basis of blood pressure and lipid status.
The findings are important because renal artery stenosis is present in 1%-5% of people with hypertension, and in up to 7% of those over age 65 years, the investigators noted.
Although uncontrolled studies in the 1990s suggested that renal-artery angioplasty or stenting results in significant reductions in systolic blood pressure and stabilization of chronic kidney disease (findings which led to a 364% increase in the number of procedures among Medicare beneficiaries between 1996 and 2000), subsequent randomized trials failed to show a benefit of renal-artery angioplasty on blood pressure or of renal-artery stenting on kidney function, they noted.
The current study is among the first to specifically assess clinical outcomes, and based on the findings, "it is clear that medical therapy without stenting is the preferred management strategy for the majority of people with atherosclerotic renal-artery stenosis," they said.
"A key issue in the interpretation of our results is whether the medical therapy that was given to CORAL participants can be replicated in clinical practice. The medical therapy in our study included the use of an angiotensin-receptor blocker, with or without a thiazide-type diuretic, with the addition of amlodipine for blood-pressure control. In addition, participants received antiplatelet therapy and atorvastatin for management of lipid levels, and diabetes was managed according to clinical practice guidelines," they wrote, noting that with this regimen, "patients who received medical treatment alone had remarkably good cardiovascular and renal outcomes, despite their advanced age and the high rates of hypertension, diabetes, chronic kidney disease, and other coexisting cardiovascular conditions."
The National Heart, Lung, and Blood Institute and the National Institutes of Health funded the trial, with support from Cordis – which donated a short-tip Angioguard device – and Pfizer. AstraZeneca and Pfizer donated medications. Dr. Cooper reported having no conflicts of interest. Detailed disclosures for all investigators are available with the full text of the article at nejm.org.
Adding renal-artery stenting to comprehensive medical therapy did not confer significant benefit with respect to the prevention of clinical events in patients with atherosclerotic renal-artery stenosis and hypertension or chronic kidney disease in the randomized controlled CORAL trial.
During a median follow-up of 43 months in the multicenter, open-label trial, the rate of a primary composite endpoint consisting of death from cardiovascular or renal causes, myocardial infarction, stroke, hospitalization for heart failure, progressive renal insufficiency, or the need for renal replacement therapy did not differ significantly between 459 patients randomized to receive medical therapy plus renal-artery stenting, and 472 patients randomized to receive medical therapy alone (35.1% and 35.8%, respectively; hazard ratio with stenting, 0.94), Dr. Christopher J. Cooper reported at the American Heart Association scientific sessions.
Furthermore, no significant differences were seen between the treatment groups in the rates of the individual components of the composite primary endpoint, or in all-cause mortality. There was, however, a consistent, modest improvement in systolic blood pressure in the stent group (–2.33 mm Hg vs. the medication only group), noted Dr. Cooper of the University of Toledo (Ohio) and his coinvestigators.
The findings of the Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial were published online simultaneously with the Nov. 18 presentation (N. Engl. J. Med. 2013 Nov. 18 [doi:10.1056/NE/NEJMoa1310753]).
CORAL comprised adults with a mean age of 69 years who had atherosclerotic renal-artery stenosis and either chronic kidney disease or systolic hypertension (despite taking at least two antihypertensive drugs). They were recruited during May 2005–January 2010.
Mandated medications as per the study protocol (unless contraindicated) included the angiotensin II type-1 receptor blocker candesartan with or without hydrochlorothiazide, and the combination agent amlodipine-atorvastatin dose-adjusted on the basis of blood pressure and lipid status.
The findings are important because renal artery stenosis is present in 1%-5% of people with hypertension, and in up to 7% of those over age 65 years, the investigators noted.
Although uncontrolled studies in the 1990s suggested that renal-artery angioplasty or stenting results in significant reductions in systolic blood pressure and stabilization of chronic kidney disease (findings which led to a 364% increase in the number of procedures among Medicare beneficiaries between 1996 and 2000), subsequent randomized trials failed to show a benefit of renal-artery angioplasty on blood pressure or of renal-artery stenting on kidney function, they noted.
The current study is among the first to specifically assess clinical outcomes, and based on the findings, "it is clear that medical therapy without stenting is the preferred management strategy for the majority of people with atherosclerotic renal-artery stenosis," they said.
"A key issue in the interpretation of our results is whether the medical therapy that was given to CORAL participants can be replicated in clinical practice. The medical therapy in our study included the use of an angiotensin-receptor blocker, with or without a thiazide-type diuretic, with the addition of amlodipine for blood-pressure control. In addition, participants received antiplatelet therapy and atorvastatin for management of lipid levels, and diabetes was managed according to clinical practice guidelines," they wrote, noting that with this regimen, "patients who received medical treatment alone had remarkably good cardiovascular and renal outcomes, despite their advanced age and the high rates of hypertension, diabetes, chronic kidney disease, and other coexisting cardiovascular conditions."
The National Heart, Lung, and Blood Institute and the National Institutes of Health funded the trial, with support from Cordis – which donated a short-tip Angioguard device – and Pfizer. AstraZeneca and Pfizer donated medications. Dr. Cooper reported having no conflicts of interest. Detailed disclosures for all investigators are available with the full text of the article at nejm.org.
Adding renal-artery stenting to comprehensive medical therapy did not confer significant benefit with respect to the prevention of clinical events in patients with atherosclerotic renal-artery stenosis and hypertension or chronic kidney disease in the randomized controlled CORAL trial.
During a median follow-up of 43 months in the multicenter, open-label trial, the rate of a primary composite endpoint consisting of death from cardiovascular or renal causes, myocardial infarction, stroke, hospitalization for heart failure, progressive renal insufficiency, or the need for renal replacement therapy did not differ significantly between 459 patients randomized to receive medical therapy plus renal-artery stenting, and 472 patients randomized to receive medical therapy alone (35.1% and 35.8%, respectively; hazard ratio with stenting, 0.94), Dr. Christopher J. Cooper reported at the American Heart Association scientific sessions.
Furthermore, no significant differences were seen between the treatment groups in the rates of the individual components of the composite primary endpoint, or in all-cause mortality. There was, however, a consistent, modest improvement in systolic blood pressure in the stent group (–2.33 mm Hg vs. the medication only group), noted Dr. Cooper of the University of Toledo (Ohio) and his coinvestigators.
The findings of the Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial were published online simultaneously with the Nov. 18 presentation (N. Engl. J. Med. 2013 Nov. 18 [doi:10.1056/NE/NEJMoa1310753]).
CORAL comprised adults with a mean age of 69 years who had atherosclerotic renal-artery stenosis and either chronic kidney disease or systolic hypertension (despite taking at least two antihypertensive drugs). They were recruited during May 2005–January 2010.
Mandated medications as per the study protocol (unless contraindicated) included the angiotensin II type-1 receptor blocker candesartan with or without hydrochlorothiazide, and the combination agent amlodipine-atorvastatin dose-adjusted on the basis of blood pressure and lipid status.
The findings are important because renal artery stenosis is present in 1%-5% of people with hypertension, and in up to 7% of those over age 65 years, the investigators noted.
Although uncontrolled studies in the 1990s suggested that renal-artery angioplasty or stenting results in significant reductions in systolic blood pressure and stabilization of chronic kidney disease (findings which led to a 364% increase in the number of procedures among Medicare beneficiaries between 1996 and 2000), subsequent randomized trials failed to show a benefit of renal-artery angioplasty on blood pressure or of renal-artery stenting on kidney function, they noted.
The current study is among the first to specifically assess clinical outcomes, and based on the findings, "it is clear that medical therapy without stenting is the preferred management strategy for the majority of people with atherosclerotic renal-artery stenosis," they said.
"A key issue in the interpretation of our results is whether the medical therapy that was given to CORAL participants can be replicated in clinical practice. The medical therapy in our study included the use of an angiotensin-receptor blocker, with or without a thiazide-type diuretic, with the addition of amlodipine for blood-pressure control. In addition, participants received antiplatelet therapy and atorvastatin for management of lipid levels, and diabetes was managed according to clinical practice guidelines," they wrote, noting that with this regimen, "patients who received medical treatment alone had remarkably good cardiovascular and renal outcomes, despite their advanced age and the high rates of hypertension, diabetes, chronic kidney disease, and other coexisting cardiovascular conditions."
The National Heart, Lung, and Blood Institute and the National Institutes of Health funded the trial, with support from Cordis – which donated a short-tip Angioguard device – and Pfizer. AstraZeneca and Pfizer donated medications. Dr. Cooper reported having no conflicts of interest. Detailed disclosures for all investigators are available with the full text of the article at nejm.org.
FROM THE AHA SCIENTIFIC SESSIONS
Major finding: The rate of a primary composite endpoint did not differ significantly between the renal-artery stent and control groups (35.1% and 35.8%, respectively; hazard ratio with stenting, 0.94).
Data source: The randomized controlled CORAL trial involving 931 patients.
Disclosures: The National Heart, Lung, and Blood Institute and the National Institutes of Health funded the trial, with support from Cordis – which donated a short-tip Angioguard device – and Pfizer. AstraZeneca and Pfizer donated medications. Dr. Cooper reported having no conflicts of interest. Detailed disclosures for all investigators are available with the full text of the article at NEJM.org.

BP therapy didn’t boost survival in ischemic stroke
DALLAS – Immediate blood pressure reduction in hypertensive acute ischemic stroke patients did not reduce death and disability after 14 days, but the strategy was safe and did not worsen patient’s 14-day outcomes, a randomized controlled study has shown.
The findings suggest that, among patients with relatively mild acute ischemic strokes and a systolic blood pressure of 140-219 mm Hg, "the decision to lower blood pressure with antihypertensive treatment should be based on individual clinical judgment," Dr. Jiang He said at the American Heart Association scientific sessions.
Elevated blood pressure immediately following an ischemic stroke poses a risk of hemorrhagic conversion or cerebral edema, but an elevated blood pressure also might be protective by forcing more blood into the penumbra around the stroke site. Blood pressure reduction measures are not appropriate for patients treated by reperfusion, but they are considered necessary for patients with "markedly elevated" blood pressure, generally defined as a systolic pressure of 220 mm Hg or higher, he said.
U.S. guidelines on stroke management published earlier this year noted that the data to guide recommendations for treating less severe arterial hypertension, in the range studied in this trial, are "inconclusive or conflicting," and that "the benefit of treating arterial hypertension in the setting of acute ischemic stroke is not well established" (Stroke 2013;44:870-947). Some U.S. clinicians, however, take steps to reduce moderately elevated blood pressure in acute ischemic stroke patients, especially when systolic pressures are at or close to 200 mm Hg.
Based on the new findings, Dr. Sila proposed in her formal comments a strategy for managing patients with mild ischemic strokes who do not undergo reperfusion treatment and have a systolic pressure of 140-219 mm Hg more than 15 hours after their stroke onset and no major-vessel stenosis or occlusion. She suggested that a "reasonable" goal was to lower blood pressure by 10%-15% over the first 24 hours of treatment, with a goal blood pressure of less than 140/90 mm Hg within the next 7 days.
The China Antihypertensive Trial in Acute Ischemic Stroke (CATIS) randomized 4,071 patients aged 22 years or older with a confirmed ischemic stroke who did not undergo reperfusion treatment at 26 hospitals in China. Their average age was 62 years, they were seen an average of 15 hours after their stroke onset, and they had a median National Institutes of Health Stroke Scale score of 4.
The 2,038 patients randomized to blood pressure reduction received an intravenous angiotensin-converting enzyme inhibitor, enalapril, as their first-line treatment, followed by a calcium channel blocker as second-line treatment and a diuretic as a third-line agent. The objective was to reduce systolic pressure by 10%-25% within the first 24 hours, with a goal blood pressure of less than 140/90 mm Hg after 7 days.
The treatments were effective, resulting in an average 13% reduction in blood pressure in treated patients after 24 hours (an average drop of 22 mm Hg), and an average systolic pressure of 137 mm Hg after 7 days. The control patients had an average 7% reduction in their systolic pressure, an average reduction of 13 mm Hg after 24 hours, and an average systolic pressure of 147 mm Hg after 7 days. All of the differences were significant.
The study’s primary endpoint was the rate of death or major disability (a modified Rankin score of 3 or higher) at 14 days or at the time of hospital discharge. This endpoint occurred in 34% of patients in both the intervention and control arms, reported Dr. He, professor of epidemiology at Tulane University in New Orleans. After 14 days or at discharge, the average modified Rankin score was 2 for patients in both treatment arms.
The report by Dr. He and his associates was published online in JAMA (2013 Nov. 17 [doi:10.1001/jama.2013.282543]) concurrently with his presentation.
Dr. He and Dr. Sila said that they had no disclosures.
On Twitter @mitchelzoler
Elevated blood pressure occurs in most patients with acute ischemic stroke, and it may be harmful but might also be protective. The results of this study, the largest reported trial of blood pressure management in acute ischemic stroke patients, add something to our clinical judgment for the subset of patients who match those enrolled in the study: patients beyond the hyper-acute phase with a mild stroke (average NIH Stroke Scale score of 4), no major-vessel occlusions, moderately elevated blood pressure, and no reperfusion treatment.

|
Mitchel L. Zoler/IMNG Medical Media
|
In this trial, an average blood pressure reduction of 12.7% after 24 hours did not improve the primary endpoint but it did not worsen recovery. That is reassuring. It shows that we can push the envelope a little in our use of blood pressure reduction. In current U.S. practice, some clinicians will try to reduce blood pressure in patients with moderately elevated pressure. Many U.S. stroke units are comfortable with blood pressure monitoring and with the target of less than 180 mm Hg used for patients treated with tissue plasminogen activator. These protocols are often also applied to ischemic stroke patients not treated with fibrinolysis because it is part of their usual practice.
My take on the findings is that, for the types of patients enrolled in this study, this strategy of more active blood pressure management to get pressure down to a goal level could be a safe option.
Dr. Cathy Sila is professor of neurology and director of the stroke and cerebrovascular center at Case Medical Center in Cleveland. She made these comments as the designated discussant for the report and in an interview.
Elevated blood pressure occurs in most patients with acute ischemic stroke, and it may be harmful but might also be protective. The results of this study, the largest reported trial of blood pressure management in acute ischemic stroke patients, add something to our clinical judgment for the subset of patients who match those enrolled in the study: patients beyond the hyper-acute phase with a mild stroke (average NIH Stroke Scale score of 4), no major-vessel occlusions, moderately elevated blood pressure, and no reperfusion treatment.
|
|
Mitchel L. Zoler/IMNG Medical Media
|
In this trial, an average blood pressure reduction of 12.7% after 24 hours did not improve the primary endpoint but it did not worsen recovery. That is reassuring. It shows that we can push the envelope a little in our use of blood pressure reduction. In current U.S. practice, some clinicians will try to reduce blood pressure in patients with moderately elevated pressure. Many U.S. stroke units are comfortable with blood pressure monitoring and with the target of less than 180 mm Hg used for patients treated with tissue plasminogen activator. These protocols are often also applied to ischemic stroke patients not treated with fibrinolysis because it is part of their usual practice.
My take on the findings is that, for the types of patients enrolled in this study, this strategy of more active blood pressure management to get pressure down to a goal level could be a safe option.
Dr. Cathy Sila is professor of neurology and director of the stroke and cerebrovascular center at Case Medical Center in Cleveland. She made these comments as the designated discussant for the report and in an interview.
Elevated blood pressure occurs in most patients with acute ischemic stroke, and it may be harmful but might also be protective. The results of this study, the largest reported trial of blood pressure management in acute ischemic stroke patients, add something to our clinical judgment for the subset of patients who match those enrolled in the study: patients beyond the hyper-acute phase with a mild stroke (average NIH Stroke Scale score of 4), no major-vessel occlusions, moderately elevated blood pressure, and no reperfusion treatment.
|
|
Mitchel L. Zoler/IMNG Medical Media
|
In this trial, an average blood pressure reduction of 12.7% after 24 hours did not improve the primary endpoint but it did not worsen recovery. That is reassuring. It shows that we can push the envelope a little in our use of blood pressure reduction. In current U.S. practice, some clinicians will try to reduce blood pressure in patients with moderately elevated pressure. Many U.S. stroke units are comfortable with blood pressure monitoring and with the target of less than 180 mm Hg used for patients treated with tissue plasminogen activator. These protocols are often also applied to ischemic stroke patients not treated with fibrinolysis because it is part of their usual practice.
My take on the findings is that, for the types of patients enrolled in this study, this strategy of more active blood pressure management to get pressure down to a goal level could be a safe option.
Dr. Cathy Sila is professor of neurology and director of the stroke and cerebrovascular center at Case Medical Center in Cleveland. She made these comments as the designated discussant for the report and in an interview.
DALLAS – Immediate blood pressure reduction in hypertensive acute ischemic stroke patients did not reduce death and disability after 14 days, but the strategy was safe and did not worsen patient’s 14-day outcomes, a randomized controlled study has shown.
The findings suggest that, among patients with relatively mild acute ischemic strokes and a systolic blood pressure of 140-219 mm Hg, "the decision to lower blood pressure with antihypertensive treatment should be based on individual clinical judgment," Dr. Jiang He said at the American Heart Association scientific sessions.
Elevated blood pressure immediately following an ischemic stroke poses a risk of hemorrhagic conversion or cerebral edema, but an elevated blood pressure also might be protective by forcing more blood into the penumbra around the stroke site. Blood pressure reduction measures are not appropriate for patients treated by reperfusion, but they are considered necessary for patients with "markedly elevated" blood pressure, generally defined as a systolic pressure of 220 mm Hg or higher, he said.
U.S. guidelines on stroke management published earlier this year noted that the data to guide recommendations for treating less severe arterial hypertension, in the range studied in this trial, are "inconclusive or conflicting," and that "the benefit of treating arterial hypertension in the setting of acute ischemic stroke is not well established" (Stroke 2013;44:870-947). Some U.S. clinicians, however, take steps to reduce moderately elevated blood pressure in acute ischemic stroke patients, especially when systolic pressures are at or close to 200 mm Hg.
Based on the new findings, Dr. Sila proposed in her formal comments a strategy for managing patients with mild ischemic strokes who do not undergo reperfusion treatment and have a systolic pressure of 140-219 mm Hg more than 15 hours after their stroke onset and no major-vessel stenosis or occlusion. She suggested that a "reasonable" goal was to lower blood pressure by 10%-15% over the first 24 hours of treatment, with a goal blood pressure of less than 140/90 mm Hg within the next 7 days.
The China Antihypertensive Trial in Acute Ischemic Stroke (CATIS) randomized 4,071 patients aged 22 years or older with a confirmed ischemic stroke who did not undergo reperfusion treatment at 26 hospitals in China. Their average age was 62 years, they were seen an average of 15 hours after their stroke onset, and they had a median National Institutes of Health Stroke Scale score of 4.
The 2,038 patients randomized to blood pressure reduction received an intravenous angiotensin-converting enzyme inhibitor, enalapril, as their first-line treatment, followed by a calcium channel blocker as second-line treatment and a diuretic as a third-line agent. The objective was to reduce systolic pressure by 10%-25% within the first 24 hours, with a goal blood pressure of less than 140/90 mm Hg after 7 days.
The treatments were effective, resulting in an average 13% reduction in blood pressure in treated patients after 24 hours (an average drop of 22 mm Hg), and an average systolic pressure of 137 mm Hg after 7 days. The control patients had an average 7% reduction in their systolic pressure, an average reduction of 13 mm Hg after 24 hours, and an average systolic pressure of 147 mm Hg after 7 days. All of the differences were significant.
The study’s primary endpoint was the rate of death or major disability (a modified Rankin score of 3 or higher) at 14 days or at the time of hospital discharge. This endpoint occurred in 34% of patients in both the intervention and control arms, reported Dr. He, professor of epidemiology at Tulane University in New Orleans. After 14 days or at discharge, the average modified Rankin score was 2 for patients in both treatment arms.
The report by Dr. He and his associates was published online in JAMA (2013 Nov. 17 [doi:10.1001/jama.2013.282543]) concurrently with his presentation.
Dr. He and Dr. Sila said that they had no disclosures.
On Twitter @mitchelzoler
DALLAS – Immediate blood pressure reduction in hypertensive acute ischemic stroke patients did not reduce death and disability after 14 days, but the strategy was safe and did not worsen patient’s 14-day outcomes, a randomized controlled study has shown.
The findings suggest that, among patients with relatively mild acute ischemic strokes and a systolic blood pressure of 140-219 mm Hg, "the decision to lower blood pressure with antihypertensive treatment should be based on individual clinical judgment," Dr. Jiang He said at the American Heart Association scientific sessions.
Elevated blood pressure immediately following an ischemic stroke poses a risk of hemorrhagic conversion or cerebral edema, but an elevated blood pressure also might be protective by forcing more blood into the penumbra around the stroke site. Blood pressure reduction measures are not appropriate for patients treated by reperfusion, but they are considered necessary for patients with "markedly elevated" blood pressure, generally defined as a systolic pressure of 220 mm Hg or higher, he said.
U.S. guidelines on stroke management published earlier this year noted that the data to guide recommendations for treating less severe arterial hypertension, in the range studied in this trial, are "inconclusive or conflicting," and that "the benefit of treating arterial hypertension in the setting of acute ischemic stroke is not well established" (Stroke 2013;44:870-947). Some U.S. clinicians, however, take steps to reduce moderately elevated blood pressure in acute ischemic stroke patients, especially when systolic pressures are at or close to 200 mm Hg.
Based on the new findings, Dr. Sila proposed in her formal comments a strategy for managing patients with mild ischemic strokes who do not undergo reperfusion treatment and have a systolic pressure of 140-219 mm Hg more than 15 hours after their stroke onset and no major-vessel stenosis or occlusion. She suggested that a "reasonable" goal was to lower blood pressure by 10%-15% over the first 24 hours of treatment, with a goal blood pressure of less than 140/90 mm Hg within the next 7 days.
The China Antihypertensive Trial in Acute Ischemic Stroke (CATIS) randomized 4,071 patients aged 22 years or older with a confirmed ischemic stroke who did not undergo reperfusion treatment at 26 hospitals in China. Their average age was 62 years, they were seen an average of 15 hours after their stroke onset, and they had a median National Institutes of Health Stroke Scale score of 4.
The 2,038 patients randomized to blood pressure reduction received an intravenous angiotensin-converting enzyme inhibitor, enalapril, as their first-line treatment, followed by a calcium channel blocker as second-line treatment and a diuretic as a third-line agent. The objective was to reduce systolic pressure by 10%-25% within the first 24 hours, with a goal blood pressure of less than 140/90 mm Hg after 7 days.
The treatments were effective, resulting in an average 13% reduction in blood pressure in treated patients after 24 hours (an average drop of 22 mm Hg), and an average systolic pressure of 137 mm Hg after 7 days. The control patients had an average 7% reduction in their systolic pressure, an average reduction of 13 mm Hg after 24 hours, and an average systolic pressure of 147 mm Hg after 7 days. All of the differences were significant.
The study’s primary endpoint was the rate of death or major disability (a modified Rankin score of 3 or higher) at 14 days or at the time of hospital discharge. This endpoint occurred in 34% of patients in both the intervention and control arms, reported Dr. He, professor of epidemiology at Tulane University in New Orleans. After 14 days or at discharge, the average modified Rankin score was 2 for patients in both treatment arms.
The report by Dr. He and his associates was published online in JAMA (2013 Nov. 17 [doi:10.1001/jama.2013.282543]) concurrently with his presentation.
Dr. He and Dr. Sila said that they had no disclosures.
On Twitter @mitchelzoler
AT THE AHA SCIENTIFIC SESSIONS
Major finding: Blood pressure lowering resulted in a similar rate of death and major disability as placebo after 14 days.
Data source: The CATIS trial randomized 4,071 patients with mild acute ischemic strokes to a blood pressure-lowering strategy or placebo at 26 Chinese hospitals.
Disclosures: Dr. He and Dr. Sila said that they had no disclosures.

Mitral valve repair no better than replacement
Mitral valve repair was no better than chordal-sparing mitral valve replacement in the first randomized clinical trial attempting to settle the controversy over which procedure is superior for treating functional ischemic mitral regurgitation, which was simultaneously reported at the annual scientific sessions of the American Heart Association and online Nov. 18 in the New England Journal of Medicine.
In the past few years, the use of mitral valve repair has far exceeded that of mitral valve replacement for this indication, largely on the basis of reports that the repair procedure yields lower operative mortality, improved left ventricular function, and higher long-term survival rates. In particular, a 2011 meta-analysis found a 35% lower relative risk of death in the long term with mitral valve repair, compared with replacement, said Dr. Michael A. Acker and his associates in the Cardiothoracic Surgical Trials Network (CTSN).

But in their multicenter study directly comparing the two procedures in 251 patients with severe functional ischemic mitral regurgitation, there was no significant difference between the surgeries in left ventricular end-systolic volume index at 1 year, nor in mortality at either 1 month or 1 year.
Moreover, study participants who underwent mitral valve repair showed a disturbing excess in the rate of recurrence of mitral regurgitation at 1 year, with a rate that was 30 percentage points higher than that among patients who underwent mitral valve replacement. "This lack of durability in correction of mitral regurgitation is disconcerting, given its reported association with further progression and long-term negative outcomes," said Dr. Acker of the division of cardiovascular surgery, University of Pennsylvania, Philadelphia, and his associates.
Functional ischemic mitral regurgitation, a "high-prevalence" condition affecting an estimated 2-3 million Americans, differs from primary degenerative mitral regurgitation in that the valve leaflets themselves remain normal while the defect occurs in the myocardium. "Ischemic mitral regurgitation is a consequence of adverse left ventricular remodeling after myocardial injury, with enlargement of the left ventricular chamber and mitral annulus, apical and lateral migration of the papillary muscles, leaflet retethering, and reduced closing forces.
"These processes lead to malcoaptation of the leaflets and variable degrees of mitral regurgitation that can fluctuate dynamically as a function of volume status, afterload, heart rhythm, and residual ischemia," the researchers said.
Current practice guidelines recommend mitral valve repair or chordal-sparing mitral valve replacement for severe regurgitation unresponsive to medical therapy, but do not specify which procedure is preferred because there is no conclusive evidence demonstrating the superiority of one over the other. "Recently, the field has embraced mitral valve repair over replacement," even without such evidence, Dr. Acker and his colleagues said.
The CTSN performed this study at 22 medical centers to assess the relative benefits of the two surgeries, with 126 patients randomized to undergo mitral valve repair and 125 to undergo replacement that included complete preservation of the subvalvular apparatus.
The primary endpoint was the degree of LV reverse remodeling, as measured by the left ventricular end-systolic volume index (LVESVI) on transthoracic echocardiography, at 1 year. The mean LVESVI was not significantly different between the repair group (54.6 mL per square meter) and the replacement group (60.7 mL per square meter), reflecting decreases of 6.6 mL per square meter and 6.8 mL per square meter, respectively, the investigators said (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1312808]).
The median between group difference in the change in LVESVI score after surgery also was not clinically significant.
However, 32.6% of patients who underwent mitral valve repair had a recurrence of regurgitation within 1 year, compared with only 2.3% of those who had mitral valve replacement. Three patients in the repair group required reoperation, compared with none in the replacement group.
There were no significant differences in cumulative mortality, 30-day postoperative mortality, or 1-year mortality between the two study groups, and no significant difference in a composite endpoint of major adverse cardiac or cerebrovascular events.
Rates of serious adverse events were similar, and the durations of hospitalization were similar between the two study groups, as were rates of readmission. All measures of quality of life and functional status on two different assessment tools also were similar.
"Our findings contradict much of the published literature on this topic, which reports several advantages to mitral valve repair over replacement, including lower operative mortality, improved left ventricular function, and higher rates of long-term survival," Dr. Acker and his associates noted.
The evolution of the valve replacement procedure, which now includes chordal sparing, "may account for the improved results we observed, as compared with previous studies, since the retention of the internal architectural support of the left ventricle may preserve contractile efficiency and reduce left ventricular dilatation and dysfunction," they said.
This study was funded by the National Heart, Lung, and Blood Institute, the National Institute of Neurological Diseases and Stroke, and the Canadian Institutes of Health Research. Dr. Acker and his associates reported no financial conflicts of interest.
Mitral valve repair was no better than chordal-sparing mitral valve replacement in the first randomized clinical trial attempting to settle the controversy over which procedure is superior for treating functional ischemic mitral regurgitation, which was simultaneously reported at the annual scientific sessions of the American Heart Association and online Nov. 18 in the New England Journal of Medicine.
In the past few years, the use of mitral valve repair has far exceeded that of mitral valve replacement for this indication, largely on the basis of reports that the repair procedure yields lower operative mortality, improved left ventricular function, and higher long-term survival rates. In particular, a 2011 meta-analysis found a 35% lower relative risk of death in the long term with mitral valve repair, compared with replacement, said Dr. Michael A. Acker and his associates in the Cardiothoracic Surgical Trials Network (CTSN).
But in their multicenter study directly comparing the two procedures in 251 patients with severe functional ischemic mitral regurgitation, there was no significant difference between the surgeries in left ventricular end-systolic volume index at 1 year, nor in mortality at either 1 month or 1 year.
Moreover, study participants who underwent mitral valve repair showed a disturbing excess in the rate of recurrence of mitral regurgitation at 1 year, with a rate that was 30 percentage points higher than that among patients who underwent mitral valve replacement. "This lack of durability in correction of mitral regurgitation is disconcerting, given its reported association with further progression and long-term negative outcomes," said Dr. Acker of the division of cardiovascular surgery, University of Pennsylvania, Philadelphia, and his associates.
Functional ischemic mitral regurgitation, a "high-prevalence" condition affecting an estimated 2-3 million Americans, differs from primary degenerative mitral regurgitation in that the valve leaflets themselves remain normal while the defect occurs in the myocardium. "Ischemic mitral regurgitation is a consequence of adverse left ventricular remodeling after myocardial injury, with enlargement of the left ventricular chamber and mitral annulus, apical and lateral migration of the papillary muscles, leaflet retethering, and reduced closing forces.
"These processes lead to malcoaptation of the leaflets and variable degrees of mitral regurgitation that can fluctuate dynamically as a function of volume status, afterload, heart rhythm, and residual ischemia," the researchers said.
Current practice guidelines recommend mitral valve repair or chordal-sparing mitral valve replacement for severe regurgitation unresponsive to medical therapy, but do not specify which procedure is preferred because there is no conclusive evidence demonstrating the superiority of one over the other. "Recently, the field has embraced mitral valve repair over replacement," even without such evidence, Dr. Acker and his colleagues said.
The CTSN performed this study at 22 medical centers to assess the relative benefits of the two surgeries, with 126 patients randomized to undergo mitral valve repair and 125 to undergo replacement that included complete preservation of the subvalvular apparatus.
The primary endpoint was the degree of LV reverse remodeling, as measured by the left ventricular end-systolic volume index (LVESVI) on transthoracic echocardiography, at 1 year. The mean LVESVI was not significantly different between the repair group (54.6 mL per square meter) and the replacement group (60.7 mL per square meter), reflecting decreases of 6.6 mL per square meter and 6.8 mL per square meter, respectively, the investigators said (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1312808]).
The median between group difference in the change in LVESVI score after surgery also was not clinically significant.
However, 32.6% of patients who underwent mitral valve repair had a recurrence of regurgitation within 1 year, compared with only 2.3% of those who had mitral valve replacement. Three patients in the repair group required reoperation, compared with none in the replacement group.
There were no significant differences in cumulative mortality, 30-day postoperative mortality, or 1-year mortality between the two study groups, and no significant difference in a composite endpoint of major adverse cardiac or cerebrovascular events.
Rates of serious adverse events were similar, and the durations of hospitalization were similar between the two study groups, as were rates of readmission. All measures of quality of life and functional status on two different assessment tools also were similar.
"Our findings contradict much of the published literature on this topic, which reports several advantages to mitral valve repair over replacement, including lower operative mortality, improved left ventricular function, and higher rates of long-term survival," Dr. Acker and his associates noted.
The evolution of the valve replacement procedure, which now includes chordal sparing, "may account for the improved results we observed, as compared with previous studies, since the retention of the internal architectural support of the left ventricle may preserve contractile efficiency and reduce left ventricular dilatation and dysfunction," they said.
This study was funded by the National Heart, Lung, and Blood Institute, the National Institute of Neurological Diseases and Stroke, and the Canadian Institutes of Health Research. Dr. Acker and his associates reported no financial conflicts of interest.
Mitral valve repair was no better than chordal-sparing mitral valve replacement in the first randomized clinical trial attempting to settle the controversy over which procedure is superior for treating functional ischemic mitral regurgitation, which was simultaneously reported at the annual scientific sessions of the American Heart Association and online Nov. 18 in the New England Journal of Medicine.
In the past few years, the use of mitral valve repair has far exceeded that of mitral valve replacement for this indication, largely on the basis of reports that the repair procedure yields lower operative mortality, improved left ventricular function, and higher long-term survival rates. In particular, a 2011 meta-analysis found a 35% lower relative risk of death in the long term with mitral valve repair, compared with replacement, said Dr. Michael A. Acker and his associates in the Cardiothoracic Surgical Trials Network (CTSN).
But in their multicenter study directly comparing the two procedures in 251 patients with severe functional ischemic mitral regurgitation, there was no significant difference between the surgeries in left ventricular end-systolic volume index at 1 year, nor in mortality at either 1 month or 1 year.
Moreover, study participants who underwent mitral valve repair showed a disturbing excess in the rate of recurrence of mitral regurgitation at 1 year, with a rate that was 30 percentage points higher than that among patients who underwent mitral valve replacement. "This lack of durability in correction of mitral regurgitation is disconcerting, given its reported association with further progression and long-term negative outcomes," said Dr. Acker of the division of cardiovascular surgery, University of Pennsylvania, Philadelphia, and his associates.
Functional ischemic mitral regurgitation, a "high-prevalence" condition affecting an estimated 2-3 million Americans, differs from primary degenerative mitral regurgitation in that the valve leaflets themselves remain normal while the defect occurs in the myocardium. "Ischemic mitral regurgitation is a consequence of adverse left ventricular remodeling after myocardial injury, with enlargement of the left ventricular chamber and mitral annulus, apical and lateral migration of the papillary muscles, leaflet retethering, and reduced closing forces.
"These processes lead to malcoaptation of the leaflets and variable degrees of mitral regurgitation that can fluctuate dynamically as a function of volume status, afterload, heart rhythm, and residual ischemia," the researchers said.
Current practice guidelines recommend mitral valve repair or chordal-sparing mitral valve replacement for severe regurgitation unresponsive to medical therapy, but do not specify which procedure is preferred because there is no conclusive evidence demonstrating the superiority of one over the other. "Recently, the field has embraced mitral valve repair over replacement," even without such evidence, Dr. Acker and his colleagues said.
The CTSN performed this study at 22 medical centers to assess the relative benefits of the two surgeries, with 126 patients randomized to undergo mitral valve repair and 125 to undergo replacement that included complete preservation of the subvalvular apparatus.
The primary endpoint was the degree of LV reverse remodeling, as measured by the left ventricular end-systolic volume index (LVESVI) on transthoracic echocardiography, at 1 year. The mean LVESVI was not significantly different between the repair group (54.6 mL per square meter) and the replacement group (60.7 mL per square meter), reflecting decreases of 6.6 mL per square meter and 6.8 mL per square meter, respectively, the investigators said (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1312808]).
The median between group difference in the change in LVESVI score after surgery also was not clinically significant.
However, 32.6% of patients who underwent mitral valve repair had a recurrence of regurgitation within 1 year, compared with only 2.3% of those who had mitral valve replacement. Three patients in the repair group required reoperation, compared with none in the replacement group.
There were no significant differences in cumulative mortality, 30-day postoperative mortality, or 1-year mortality between the two study groups, and no significant difference in a composite endpoint of major adverse cardiac or cerebrovascular events.
Rates of serious adverse events were similar, and the durations of hospitalization were similar between the two study groups, as were rates of readmission. All measures of quality of life and functional status on two different assessment tools also were similar.
"Our findings contradict much of the published literature on this topic, which reports several advantages to mitral valve repair over replacement, including lower operative mortality, improved left ventricular function, and higher rates of long-term survival," Dr. Acker and his associates noted.
The evolution of the valve replacement procedure, which now includes chordal sparing, "may account for the improved results we observed, as compared with previous studies, since the retention of the internal architectural support of the left ventricle may preserve contractile efficiency and reduce left ventricular dilatation and dysfunction," they said.
This study was funded by the National Heart, Lung, and Blood Institute, the National Institute of Neurological Diseases and Stroke, and the Canadian Institutes of Health Research. Dr. Acker and his associates reported no financial conflicts of interest.
FROM THE AHA SCIENTIFIC SESSIONS
Major Finding: The primary end point – the degree of LV reverse remodeling, as measured by the left ventricular end-systolic volume index on transthoracic echocardiography at 1 year – was not significantly different between the repair group (54.6 mL per square meter) and the replacement group (60.7 mL per square meter).
Data Source: A randomized clinical trial involving 126 patients with severe functional ischemic mitral regurgitation who underwent mitral valve repair and 125 who underwent chordal-sparing mitral valve replacement, and who were followed for 1 year.
Disclosures: This study was funded by the National Heart, Lung, and Blood Institute, the National Institute of Neurological Diseases and Stroke, and the Canadian Institutes of Health Research. Dr. Acker and his associates reported no financial conflicts of interest.

Intervention boosts post-acute coronary syndrome medication adherence, but not outcomes
A multifaceted intervention was effective for improving postdischarge medication adherence among patients hospitalized for acute coronary syndrome but had no effect on achievement of blood pressure and cholesterol level goals, in a prospective randomized clinical trial.
The intervention, which involved pharmacist-led medication reconciliation and tailoring, patient education, collaboration between pharmacists and physicians, and voice messaging reminders, was associated with an 89.3% medication adherence rate among 122 patients randomized to the intervention groups, compared with a 73.9% adherence rate among 119 patients randomized to receive usual care, Dr. P. Michael Ho of the Denver Veterans Administration Medical Center, reported on Nov. 17 at the American Heart Association scientific sessions.
The mean proportion of days covered was significantly higher in the intervention group than in the usual care group (0.94 vs. 0.87 days), and a greater proportion of patients in the intervention group were adherent to clopidogrel (86.8% vs. 70.7%), statins (93.2% vs. 71.3%), and ACE inhibitors and angiotensin receptor blockers (93.1% vs. 81.7%). No significant difference was seen between the groups with respect to use of beta-blockers (88.1% vs. 84.8%), Dr. Ho reported. The findings were published simultaneously with the presentation (JAMA Intern Med Nov.18 [doi:10.1001/jamainternmed.2013.12944]).
Despite the improved adherence to treatment, no differences were seen in the percentage of patients achieving blood pressure goals or LDL cholesterol goals, although there was a trend toward greater blood pressure control (58.6% vs. 48.9%), decline in systolic blood pressure (–12 vs. –4 mm Hg), and decline in diastolic blood pressure (–5 vs. –3 mm Hg) for intervention vs. usual care patients, respectively, he said.
Also, adherent patient were more likely to get follow-up LDL cholesterol laboratory evaluations, and "accordingly we saw declines in LDL-C levels in both intervention and usual care patients, which may have attenuated the intervention effect on LDL-C levels," he wrote in the JAMA article, noting that additional studies in larger patient samples are needed to assess the association between adherence and these clinical outcomes.
The cost of the intervention, based on the pharmacist time spent, cardiologist time spent, and pill box cost, was about $360/patient. Medication cost differences did not differ significantly between the intervention and usual care groups.
Patients in the study had been hospitalized for acute coronary syndrome at Department of Veterans Affairs Medical Centers in Denver, Seattle, Durham, N.C., or Little Rock, Ark., and were discharged during 2010 or 2011. They were randomized to the intervention or usual care group prior to discharge.
The intervention, which lasted 1 year following discharge, involved:
• Medication reconciliation and tailoring by a pharmacist who met with patients via an in-person clinic visit or by telephone. The pharmacist addressed medication problems or adverse effects, reconciled differences in medications, provided a pill box and instructions for use if needed, and called a month later to follow up, synchronize refill dates, answer questions, and encourage adherence.
• Patient education at the point of discharge, and afterward by the pharmacist at 1-week and 1-month visits. Thereafter, educational messages were provided via automated voice messages and pharmacist phone calls.
• Collaborative care involving pharmacist and primary care clinician or cardiologist communications.
• Voice messaging at regularly scheduled intervals to remind patients about medications and refills.
Studies show that postdischarge adherence to cardioprotective drug regimens is often poor among patients hospitalized for acute coronary syndrome. For example, several studies have shown that a third of those who were taking their cardiac medications at discharge discontinued at least one medication by 1 month, and only about 60% were still taking statin medications at 1 year. Cohort and population-based studies show that lower adherence to cardioprotective drug regimens after acute MI are associated with a higher 1-year and long-term mortality, Dr. Ho noted.
In the current study, the intervention improved the proportion of adherent patients by about 15% and the mean adherence to the four medications combined by about 7%, Dr. Ho said, noting that the findings, along with those from prior intervention studies, "provide an increasing evidence base of interventions to improve adherence to cardiac medication regimens after ACS discharge."
"It will be important for us to continue to follow up patients longer to assess whether the higher adherence in the intervention group translates into improved clinical outcomes," he wrote, noting that he and his colleagues plan to follow the patients beyond the initial 12 months to assess for differences in outcomes and costs in the longer term.
This study is limited by a number of factors, including the largely male patient population at the VA medical centers where it was conducted, and the use of pharmacy refill data rather than pill counts to assess adherence.
This study was funded by Veterans Health Administration Health Service Research & Development Investigator-Initiated Awards. The authors reported having no disclosures.
In an editor’s note, Dr. Rita F. Redberg called the work "interesting and careful," and noted that the findings demonstrate an absolute increase of 11% in adherence to statins, ACE inhibitors, and angiotensin receptor blockers, and a 3% increase in adherence to beta-blockers.
The cost estimate of $360/patient is based on the "relatively lower costs of services in the VA system," but if applied to every patient with acute coronary syndrome in the United States (about 2.5 million annually, by recent estimates), the costs of such an intervention would add $1 billion annually to health care costs, she noted.
"For many reasons, the relatively modest increases in already high rates of medication regimen adherence in the patients studied may not translate into improved outcomes even if maintained for 3-5 years or longer. Of course, we hope that they do. But before recommending investment in this strategy, it would be prudent to know that patient outcomes will actually improve," she wrote.
Dr. Redberg of the department of medicine and women’s cardiovascular services at the University of California, San Francisco, is editor of JAMA Internal Medicine.
In an editor’s note, Dr. Rita F. Redberg called the work "interesting and careful," and noted that the findings demonstrate an absolute increase of 11% in adherence to statins, ACE inhibitors, and angiotensin receptor blockers, and a 3% increase in adherence to beta-blockers.
The cost estimate of $360/patient is based on the "relatively lower costs of services in the VA system," but if applied to every patient with acute coronary syndrome in the United States (about 2.5 million annually, by recent estimates), the costs of such an intervention would add $1 billion annually to health care costs, she noted.
"For many reasons, the relatively modest increases in already high rates of medication regimen adherence in the patients studied may not translate into improved outcomes even if maintained for 3-5 years or longer. Of course, we hope that they do. But before recommending investment in this strategy, it would be prudent to know that patient outcomes will actually improve," she wrote.
Dr. Redberg of the department of medicine and women’s cardiovascular services at the University of California, San Francisco, is editor of JAMA Internal Medicine.
In an editor’s note, Dr. Rita F. Redberg called the work "interesting and careful," and noted that the findings demonstrate an absolute increase of 11% in adherence to statins, ACE inhibitors, and angiotensin receptor blockers, and a 3% increase in adherence to beta-blockers.
The cost estimate of $360/patient is based on the "relatively lower costs of services in the VA system," but if applied to every patient with acute coronary syndrome in the United States (about 2.5 million annually, by recent estimates), the costs of such an intervention would add $1 billion annually to health care costs, she noted.
"For many reasons, the relatively modest increases in already high rates of medication regimen adherence in the patients studied may not translate into improved outcomes even if maintained for 3-5 years or longer. Of course, we hope that they do. But before recommending investment in this strategy, it would be prudent to know that patient outcomes will actually improve," she wrote.
Dr. Redberg of the department of medicine and women’s cardiovascular services at the University of California, San Francisco, is editor of JAMA Internal Medicine.
A multifaceted intervention was effective for improving postdischarge medication adherence among patients hospitalized for acute coronary syndrome but had no effect on achievement of blood pressure and cholesterol level goals, in a prospective randomized clinical trial.
The intervention, which involved pharmacist-led medication reconciliation and tailoring, patient education, collaboration between pharmacists and physicians, and voice messaging reminders, was associated with an 89.3% medication adherence rate among 122 patients randomized to the intervention groups, compared with a 73.9% adherence rate among 119 patients randomized to receive usual care, Dr. P. Michael Ho of the Denver Veterans Administration Medical Center, reported on Nov. 17 at the American Heart Association scientific sessions.
The mean proportion of days covered was significantly higher in the intervention group than in the usual care group (0.94 vs. 0.87 days), and a greater proportion of patients in the intervention group were adherent to clopidogrel (86.8% vs. 70.7%), statins (93.2% vs. 71.3%), and ACE inhibitors and angiotensin receptor blockers (93.1% vs. 81.7%). No significant difference was seen between the groups with respect to use of beta-blockers (88.1% vs. 84.8%), Dr. Ho reported. The findings were published simultaneously with the presentation (JAMA Intern Med Nov.18 [doi:10.1001/jamainternmed.2013.12944]).
Despite the improved adherence to treatment, no differences were seen in the percentage of patients achieving blood pressure goals or LDL cholesterol goals, although there was a trend toward greater blood pressure control (58.6% vs. 48.9%), decline in systolic blood pressure (–12 vs. –4 mm Hg), and decline in diastolic blood pressure (–5 vs. –3 mm Hg) for intervention vs. usual care patients, respectively, he said.
Also, adherent patient were more likely to get follow-up LDL cholesterol laboratory evaluations, and "accordingly we saw declines in LDL-C levels in both intervention and usual care patients, which may have attenuated the intervention effect on LDL-C levels," he wrote in the JAMA article, noting that additional studies in larger patient samples are needed to assess the association between adherence and these clinical outcomes.
The cost of the intervention, based on the pharmacist time spent, cardiologist time spent, and pill box cost, was about $360/patient. Medication cost differences did not differ significantly between the intervention and usual care groups.
Patients in the study had been hospitalized for acute coronary syndrome at Department of Veterans Affairs Medical Centers in Denver, Seattle, Durham, N.C., or Little Rock, Ark., and were discharged during 2010 or 2011. They were randomized to the intervention or usual care group prior to discharge.
The intervention, which lasted 1 year following discharge, involved:
• Medication reconciliation and tailoring by a pharmacist who met with patients via an in-person clinic visit or by telephone. The pharmacist addressed medication problems or adverse effects, reconciled differences in medications, provided a pill box and instructions for use if needed, and called a month later to follow up, synchronize refill dates, answer questions, and encourage adherence.
• Patient education at the point of discharge, and afterward by the pharmacist at 1-week and 1-month visits. Thereafter, educational messages were provided via automated voice messages and pharmacist phone calls.
• Collaborative care involving pharmacist and primary care clinician or cardiologist communications.
• Voice messaging at regularly scheduled intervals to remind patients about medications and refills.
Studies show that postdischarge adherence to cardioprotective drug regimens is often poor among patients hospitalized for acute coronary syndrome. For example, several studies have shown that a third of those who were taking their cardiac medications at discharge discontinued at least one medication by 1 month, and only about 60% were still taking statin medications at 1 year. Cohort and population-based studies show that lower adherence to cardioprotective drug regimens after acute MI are associated with a higher 1-year and long-term mortality, Dr. Ho noted.
In the current study, the intervention improved the proportion of adherent patients by about 15% and the mean adherence to the four medications combined by about 7%, Dr. Ho said, noting that the findings, along with those from prior intervention studies, "provide an increasing evidence base of interventions to improve adherence to cardiac medication regimens after ACS discharge."
"It will be important for us to continue to follow up patients longer to assess whether the higher adherence in the intervention group translates into improved clinical outcomes," he wrote, noting that he and his colleagues plan to follow the patients beyond the initial 12 months to assess for differences in outcomes and costs in the longer term.
This study is limited by a number of factors, including the largely male patient population at the VA medical centers where it was conducted, and the use of pharmacy refill data rather than pill counts to assess adherence.
This study was funded by Veterans Health Administration Health Service Research & Development Investigator-Initiated Awards. The authors reported having no disclosures.
A multifaceted intervention was effective for improving postdischarge medication adherence among patients hospitalized for acute coronary syndrome but had no effect on achievement of blood pressure and cholesterol level goals, in a prospective randomized clinical trial.
The intervention, which involved pharmacist-led medication reconciliation and tailoring, patient education, collaboration between pharmacists and physicians, and voice messaging reminders, was associated with an 89.3% medication adherence rate among 122 patients randomized to the intervention groups, compared with a 73.9% adherence rate among 119 patients randomized to receive usual care, Dr. P. Michael Ho of the Denver Veterans Administration Medical Center, reported on Nov. 17 at the American Heart Association scientific sessions.
The mean proportion of days covered was significantly higher in the intervention group than in the usual care group (0.94 vs. 0.87 days), and a greater proportion of patients in the intervention group were adherent to clopidogrel (86.8% vs. 70.7%), statins (93.2% vs. 71.3%), and ACE inhibitors and angiotensin receptor blockers (93.1% vs. 81.7%). No significant difference was seen between the groups with respect to use of beta-blockers (88.1% vs. 84.8%), Dr. Ho reported. The findings were published simultaneously with the presentation (JAMA Intern Med Nov.18 [doi:10.1001/jamainternmed.2013.12944]).
Despite the improved adherence to treatment, no differences were seen in the percentage of patients achieving blood pressure goals or LDL cholesterol goals, although there was a trend toward greater blood pressure control (58.6% vs. 48.9%), decline in systolic blood pressure (–12 vs. –4 mm Hg), and decline in diastolic blood pressure (–5 vs. –3 mm Hg) for intervention vs. usual care patients, respectively, he said.
Also, adherent patient were more likely to get follow-up LDL cholesterol laboratory evaluations, and "accordingly we saw declines in LDL-C levels in both intervention and usual care patients, which may have attenuated the intervention effect on LDL-C levels," he wrote in the JAMA article, noting that additional studies in larger patient samples are needed to assess the association between adherence and these clinical outcomes.
The cost of the intervention, based on the pharmacist time spent, cardiologist time spent, and pill box cost, was about $360/patient. Medication cost differences did not differ significantly between the intervention and usual care groups.
Patients in the study had been hospitalized for acute coronary syndrome at Department of Veterans Affairs Medical Centers in Denver, Seattle, Durham, N.C., or Little Rock, Ark., and were discharged during 2010 or 2011. They were randomized to the intervention or usual care group prior to discharge.
The intervention, which lasted 1 year following discharge, involved:
• Medication reconciliation and tailoring by a pharmacist who met with patients via an in-person clinic visit or by telephone. The pharmacist addressed medication problems or adverse effects, reconciled differences in medications, provided a pill box and instructions for use if needed, and called a month later to follow up, synchronize refill dates, answer questions, and encourage adherence.
• Patient education at the point of discharge, and afterward by the pharmacist at 1-week and 1-month visits. Thereafter, educational messages were provided via automated voice messages and pharmacist phone calls.
• Collaborative care involving pharmacist and primary care clinician or cardiologist communications.
• Voice messaging at regularly scheduled intervals to remind patients about medications and refills.
Studies show that postdischarge adherence to cardioprotective drug regimens is often poor among patients hospitalized for acute coronary syndrome. For example, several studies have shown that a third of those who were taking their cardiac medications at discharge discontinued at least one medication by 1 month, and only about 60% were still taking statin medications at 1 year. Cohort and population-based studies show that lower adherence to cardioprotective drug regimens after acute MI are associated with a higher 1-year and long-term mortality, Dr. Ho noted.
In the current study, the intervention improved the proportion of adherent patients by about 15% and the mean adherence to the four medications combined by about 7%, Dr. Ho said, noting that the findings, along with those from prior intervention studies, "provide an increasing evidence base of interventions to improve adherence to cardiac medication regimens after ACS discharge."
"It will be important for us to continue to follow up patients longer to assess whether the higher adherence in the intervention group translates into improved clinical outcomes," he wrote, noting that he and his colleagues plan to follow the patients beyond the initial 12 months to assess for differences in outcomes and costs in the longer term.
This study is limited by a number of factors, including the largely male patient population at the VA medical centers where it was conducted, and the use of pharmacy refill data rather than pill counts to assess adherence.
This study was funded by Veterans Health Administration Health Service Research & Development Investigator-Initiated Awards. The authors reported having no disclosures.
FROM THE AHA SCIENTIFIC SESSIONS
Major finding: The intervention improved the proportion of adherent patients by 15%, and improved the mean adherence to the four medications combined by about 7%.
Data source: A prospective, randomized, clinical trial involving 241 patients.
Disclosures: This study was funded by Veterans Health Administration Health Service Research & Development Investigator-Initiated Awards. The authors reported having no disclosures.