User login
Building and Maintaining a Successful Inflammatory Bowel Disease Practice
Anyone can build a successful inflammatory bowel disease (IBD) practice. To do so requires commitment and focus in the area of IBD including both Crohn’s disease and ulcerative colitis. It also requires a fundamental knowledge of medicine as well as a desire to excel and learn all that one can in these areas. Given the high number of stakeholders, good interpersonal skills are vital. Establishing an IBD practice provides an opportunity to make a big difference in peoples’ lives and the age range of impact is about the broadest in all of medical practice. The more resources you have, the greater the potential impact of your care. Table 1 lists resources that are useful to provide optimal IBD patient care.

You, the gastroenterologist, is the most important resource for the patient. Medical school, residency, fellowship, and “postgraduate” training serves as the foundation for your wealth of knowledge. Maximizing your training is of value, and this can be done by being part of an academic program, keeping abreast of current literature, and attending meetings and post-graduate courses. AGA offers a variety of publications (http://www.gastro.org/journals-and-publications) and continued training opportunities (http://www.gastro.org/education).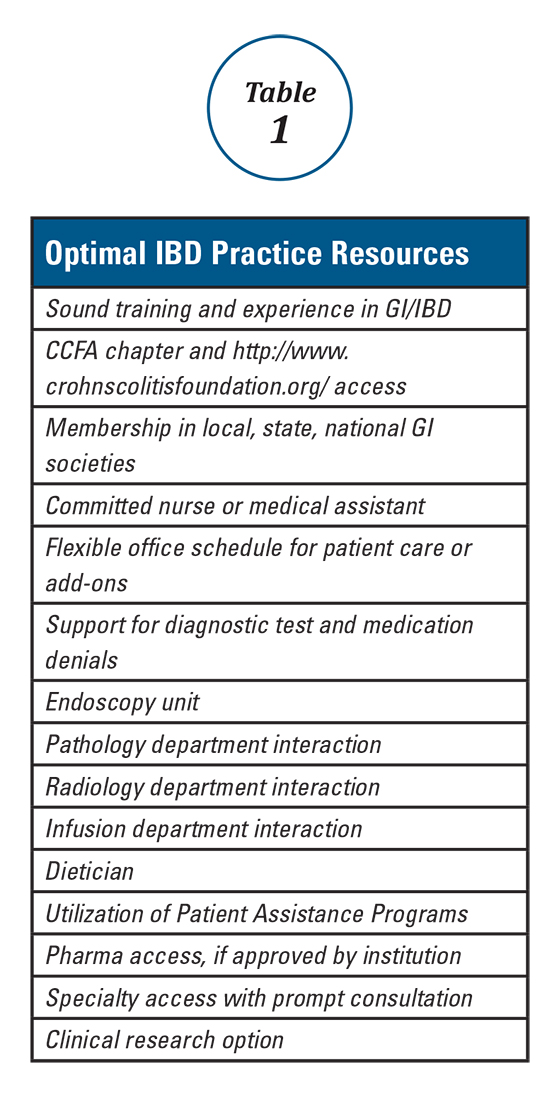
One further point regarding scheduling is that one must be willing and able to see patients urgently, rather than sending them to the emergency room. ERs are appropriate for true emergencies, but are not an ideal place for care when an IBD patient has a flare and requires prompt follow-up. I try to avoid ER visits for my patients unless they are vomiting, have severe abdominal pain, significant bleeding or have clear signs of toxicity. In an ER, abdominal pain equals a CT scan; one should consider seeing these patients in the office and triaging accordingly.
With the increasing requirements of managed care and restrictive medical plans, there has been a similar rise in the frequency of diagnostic test as well as procedure and medication denials. Re-approval and recertification of biologics and other medications have become common, which can add a great deal to your workload and that of your staff. Integration of endoscopy, pathology, and imaging (e.g., ultrasound, CT/CTE) improves response time, dialogue, and can have a positive impact on care. Office infusion allows for a better integration of this service into your practice. There is typically better communication with the infusion nurse(s) and better expedited care as well as fewer cancellations for minor infections. This all helps avoid infusion procedure delays. Infliximab, vedolizumab, ustekinumab, and lyophilized certolizumab pegol as well as intravenous iron administration can also expand services and enhance quality.
Having a medical assistant, nurse, and others in your practice to assist with patient services and care is a must. There will be many phone calls, emails, and other interactions regarding appointments, consults, routine lab testing, radiology testing, standard medications, biologics, and other treatments that necessitate an effective team-approach. For this role, either a nurse or an experienced medical assistant would be well-suited. Additional support staff and services can also aid our IBD patients. A dietitian knowledgeable in IBD and practical dietary options can, in many instances, prove invaluable. Understanding and utilizing pharma-sponsored “Patient Assistance Programs” provides drug access for the 10-20% (or more) of patients who do not have insurance or biologic coverage. Having specialty access and collegiality with colorectal surgeons, general surgeons, OB/GYNs, dermatologists, hematologists, oncologists, and others is important to expedite consults and provide collaborative care. Finally, offering clinical research options improves access for patients with limited and no coverage and also helps provide needed options for all IBD patients.
This brief overview has hopefully given you some insight into how to provide a higher level of evaluation and care for our IBD patients. These approaches have allowed me to build and maintain a successful IBD practice, and I hope that the integration of some or all of these strategies help you to build and sustain a successful IBD practice.
Dr. Wolf is director of IBD research, Atlanta Gastroenterology Associates.
Anyone can build a successful inflammatory bowel disease (IBD) practice. To do so requires commitment and focus in the area of IBD including both Crohn’s disease and ulcerative colitis. It also requires a fundamental knowledge of medicine as well as a desire to excel and learn all that one can in these areas. Given the high number of stakeholders, good interpersonal skills are vital. Establishing an IBD practice provides an opportunity to make a big difference in peoples’ lives and the age range of impact is about the broadest in all of medical practice. The more resources you have, the greater the potential impact of your care. Table 1 lists resources that are useful to provide optimal IBD patient care.
You, the gastroenterologist, is the most important resource for the patient. Medical school, residency, fellowship, and “postgraduate” training serves as the foundation for your wealth of knowledge. Maximizing your training is of value, and this can be done by being part of an academic program, keeping abreast of current literature, and attending meetings and post-graduate courses. AGA offers a variety of publications (http://www.gastro.org/journals-and-publications) and continued training opportunities (http://www.gastro.org/education).
One further point regarding scheduling is that one must be willing and able to see patients urgently, rather than sending them to the emergency room. ERs are appropriate for true emergencies, but are not an ideal place for care when an IBD patient has a flare and requires prompt follow-up. I try to avoid ER visits for my patients unless they are vomiting, have severe abdominal pain, significant bleeding or have clear signs of toxicity. In an ER, abdominal pain equals a CT scan; one should consider seeing these patients in the office and triaging accordingly.
With the increasing requirements of managed care and restrictive medical plans, there has been a similar rise in the frequency of diagnostic test as well as procedure and medication denials. Re-approval and recertification of biologics and other medications have become common, which can add a great deal to your workload and that of your staff. Integration of endoscopy, pathology, and imaging (e.g., ultrasound, CT/CTE) improves response time, dialogue, and can have a positive impact on care. Office infusion allows for a better integration of this service into your practice. There is typically better communication with the infusion nurse(s) and better expedited care as well as fewer cancellations for minor infections. This all helps avoid infusion procedure delays. Infliximab, vedolizumab, ustekinumab, and lyophilized certolizumab pegol as well as intravenous iron administration can also expand services and enhance quality.
Having a medical assistant, nurse, and others in your practice to assist with patient services and care is a must. There will be many phone calls, emails, and other interactions regarding appointments, consults, routine lab testing, radiology testing, standard medications, biologics, and other treatments that necessitate an effective team-approach. For this role, either a nurse or an experienced medical assistant would be well-suited. Additional support staff and services can also aid our IBD patients. A dietitian knowledgeable in IBD and practical dietary options can, in many instances, prove invaluable. Understanding and utilizing pharma-sponsored “Patient Assistance Programs” provides drug access for the 10-20% (or more) of patients who do not have insurance or biologic coverage. Having specialty access and collegiality with colorectal surgeons, general surgeons, OB/GYNs, dermatologists, hematologists, oncologists, and others is important to expedite consults and provide collaborative care. Finally, offering clinical research options improves access for patients with limited and no coverage and also helps provide needed options for all IBD patients.
This brief overview has hopefully given you some insight into how to provide a higher level of evaluation and care for our IBD patients. These approaches have allowed me to build and maintain a successful IBD practice, and I hope that the integration of some or all of these strategies help you to build and sustain a successful IBD practice.
Dr. Wolf is director of IBD research, Atlanta Gastroenterology Associates.
Anyone can build a successful inflammatory bowel disease (IBD) practice. To do so requires commitment and focus in the area of IBD including both Crohn’s disease and ulcerative colitis. It also requires a fundamental knowledge of medicine as well as a desire to excel and learn all that one can in these areas. Given the high number of stakeholders, good interpersonal skills are vital. Establishing an IBD practice provides an opportunity to make a big difference in peoples’ lives and the age range of impact is about the broadest in all of medical practice. The more resources you have, the greater the potential impact of your care. Table 1 lists resources that are useful to provide optimal IBD patient care.
You, the gastroenterologist, is the most important resource for the patient. Medical school, residency, fellowship, and “postgraduate” training serves as the foundation for your wealth of knowledge. Maximizing your training is of value, and this can be done by being part of an academic program, keeping abreast of current literature, and attending meetings and post-graduate courses. AGA offers a variety of publications (http://www.gastro.org/journals-and-publications) and continued training opportunities (http://www.gastro.org/education).
One further point regarding scheduling is that one must be willing and able to see patients urgently, rather than sending them to the emergency room. ERs are appropriate for true emergencies, but are not an ideal place for care when an IBD patient has a flare and requires prompt follow-up. I try to avoid ER visits for my patients unless they are vomiting, have severe abdominal pain, significant bleeding or have clear signs of toxicity. In an ER, abdominal pain equals a CT scan; one should consider seeing these patients in the office and triaging accordingly.
With the increasing requirements of managed care and restrictive medical plans, there has been a similar rise in the frequency of diagnostic test as well as procedure and medication denials. Re-approval and recertification of biologics and other medications have become common, which can add a great deal to your workload and that of your staff. Integration of endoscopy, pathology, and imaging (e.g., ultrasound, CT/CTE) improves response time, dialogue, and can have a positive impact on care. Office infusion allows for a better integration of this service into your practice. There is typically better communication with the infusion nurse(s) and better expedited care as well as fewer cancellations for minor infections. This all helps avoid infusion procedure delays. Infliximab, vedolizumab, ustekinumab, and lyophilized certolizumab pegol as well as intravenous iron administration can also expand services and enhance quality.
Having a medical assistant, nurse, and others in your practice to assist with patient services and care is a must. There will be many phone calls, emails, and other interactions regarding appointments, consults, routine lab testing, radiology testing, standard medications, biologics, and other treatments that necessitate an effective team-approach. For this role, either a nurse or an experienced medical assistant would be well-suited. Additional support staff and services can also aid our IBD patients. A dietitian knowledgeable in IBD and practical dietary options can, in many instances, prove invaluable. Understanding and utilizing pharma-sponsored “Patient Assistance Programs” provides drug access for the 10-20% (or more) of patients who do not have insurance or biologic coverage. Having specialty access and collegiality with colorectal surgeons, general surgeons, OB/GYNs, dermatologists, hematologists, oncologists, and others is important to expedite consults and provide collaborative care. Finally, offering clinical research options improves access for patients with limited and no coverage and also helps provide needed options for all IBD patients.
This brief overview has hopefully given you some insight into how to provide a higher level of evaluation and care for our IBD patients. These approaches have allowed me to build and maintain a successful IBD practice, and I hope that the integration of some or all of these strategies help you to build and sustain a successful IBD practice.
Dr. Wolf is director of IBD research, Atlanta Gastroenterology Associates.
Health Maintenance and Preventive Care in Patients with Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) consists of two chronic inflammatory diseases, Crohn’s disease (CD) and ulcerative colitis (UC), as well as a small category of patients (~10%) who have atypical features called IBD-unclassified (IBD-U) or indeterminate colitis. The prevalence of IBD ranges from 0.3% to 0.5% overall in North America and Europe.1 In North America, the incidences of CD and UC are estimated to be 3.1 to 14.6 per 100,000 person-years and 2.2 to 14.3 cases per 100,000 person-years, respectively; similar rates are seen in Europe.2 However, incidences up to 19.2 and 20.2 per 100,000 for UC and CD, respectively, have been reported in Canada.3,4 The incidences of both UC and CD are increasing over time in Western countries and in rapidly industrializing countries throughout Asia and South America.5-8

Influenza vaccine and pneumococcal vaccine
Influenza A and B outbreaks are commonly seen during the fall and early spring and risk factors for pneumonia and hospitalization include older age, chronic medical conditions, and immunosuppression. The CDC now recommend annual influenza vaccination for all individuals older than six months. For patients on immunosuppression, the vaccine administered should be the inactivated vaccine, as live attenuated vaccines should not be administered to these patients.

In IBD patients, the influenza and pneumococcal vaccines are both well tolerated without an increased rate of adverse effects over the general population and without an increased risk of IBD flares after vaccination.12 A common question for patients on biologic therapy is whether the vaccine should be timed at a specific point in the dose cycle. For infliximab, and likely other biologics, the timing does not change the vaccine immunogenicity and patients should be given these vaccines regardless of where they are in the cycle of administration of their biologic.13 In addition, there is significant response to influenza and pneumococcal vaccines in patients on combination therapy with immunomodulators and anti-TNFs and concerns about a lack of response to vaccines should not discourage vaccination since benefits are still acquired by patients even if immunogenicity is somewhat decreased.14,15
Other vaccinations
In addition to the influenza and pneumococcal vaccines, adult and pediatric patients with IBD should follow the ACIP recommendations for tetanus, diphtheria, pertussis (Tdap), Td boosters, hepatitis A, hepatitis B, human papilloma virus (HPV), and meningococcal vaccinations.16,17
Live vaccines including measles mumps rubella (MMR), varicella, and zoster vaccines are in general contraindicated in immunosuppressed patients on corticosteroids, azathioprine/6-mercaptopurine, methotrexate, anti-TNF, and anti-integrin biologics. An inactive varicella-zoster vaccine will likely be available in the near future and may obviate the need for the live vaccine, which is an important development given the increased risk of zoster in patients with IBD on immunosuppression.18
Osteoporosis screening

Skin cancer screening
Multiple studies have demonstrated that immunosuppression, especially with methotrexate and azathioprine/6-mercaptopurine (6MP) is a risk factor for the development of initial and recurrent non-melanoma skin cancer (NMSC) in IBD patients, the data for biologics are less definitive.23-25 In addition, biologics are associated with increased risk of melanoma in IBD.26 The elevated risk of skin cancer begins in the first year of treatment with thiopurines and may continue after discontinuation. On the basis of this data, screening for melanoma and NMSC is recommended in IBD patients on immunosuppression. Especially for patients on thiopurines it is reasonable for the initial dermatologist visit to occur in the first year of treatment and thereafter with at least annual visits for a full body skin examination. In addition, it is reasonable to recommend regular sunscreen use and protective clothing such as hats.
Cervical cancer screening
A recent meta-analysis shows that women with IBD on immunosuppression have an increased risk of cervical high grade dysplasia and cervical cancer.27 HPV is the major risk factor for cervical cancer and is necessary for its development. The current American College of Gynecology guidelines for women on immunosuppression are to start cervical cancer screening at 21 and annual screening thereafter with Pap and HPV testing.28
Smoking
Smoking has well known associations with poor outcomes in the general population such as increased risk of lung and pancreatic cancers, as well as high risk of cardiovascular disease. In addition, smoking has risks specific to IBD. In CD, smoking is associated with increased disease activity, increased risk of post-operative recurrence, and increased severity of disease.29 Smoking cessation is associated with improved long-term disease outcomes and less risk.30 Making it a point to regularly discuss smoking cessation and partnering with PCPs to offer evidence-based quitting aids may be one of our most significant and beneficial interventions.
Depression and anxiety
Several studies have shown high levels of depression and anxiety in IBD patients and higher levels of depression are associated with increased symptoms, clinical recurrence, poor quality of life and decreased social support.31-33 A recent systematic review of several studies suggested that antidepressants use in IBD patients benefits their mental health and may improve their clinical course as well.34 As such, screening for depression and anxiety regularly and either offering treatment or referral to psychiatrists and psychologists for further management is recommended.10
Conclusion
Patients with IBD frequently develop long-term relationships with their gastroenterologists due to their lifelong chronic disease. It is therefore incumbent on us to be attentive to issues related to IBD patients’ preventive care and collaborate with PCPs to coordinate care for our patients since many of these interventions have both short-term and long-term benefits.
Dr. Chachu is assistant professor and gastroenterologist at Duke University, Durham, N.C.
References
1. Kaplan GG, Ng SC. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology. 2017;152(2):313-21.e2.
2. Loftus EV, Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126(6):1504-17.
3. Bernstein CN, Wajda A, Svenson LW, et al. The Epidemiology of Inflammatory Bowel Disease in Canada: A Population-Based Study. The American journal of gastroenterology. 2006;101(7):1559-68.
4. Lowe AM, Roy PO, M BP, et al. Epidemiology of Crohn’s disease in Quebec, Canada. Inflammatory bowel diseases. 2009;15(3):429-35.
5. Kappelman MD, Rifas-Shiman SL, Kleinman K, et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2007;5(12):1424-9.
6. Kappelman MD, Moore KR, Allen JK, et al. Recent trends in the prevalence of Crohn’s disease and ulcerative colitis in a commercially insured US population. Digestive diseases and sciences. 2013;58(2):519-25.
7. Ng SC, Kaplan G, Banerjee R, et al. 78 Incidence and Phenotype of Inflammatory Bowel Disease From 13 Countries in Asia-Pacific: Results From the Asia-Pacific Crohn’s and Colitis Epidemiologic Study 2011-2013. Gastroenterology.150(4):S21.
8. Parente JML, Coy CSR, Campelo V, et al. Inflammatory bowel disease in an underdeveloped region of Northeastern Brazil. World Journal of Gastroenterology : WJG. 2015;21(4):1197-206.
9. Selby L, Kane S, Wilson J, et al. Receipt of preventive health services by IBD patients is significantly lower than by primary care patients. Inflammatory bowel diseases. 2008;14(2):253-8.
10. Farraye FA, Melmed GY, Lichtenstein GR, et al. ACG Clinical Guideline: Preventive Care in Inflammatory Bowel Disease. The American journal of gastroenterology. 2017;112(2):241-58.
11. Long MD, Martin C, Sandler RS, et al. Increased risk of pneumonia among patients with inflammatory bowel disease. The American journal of gastroenterology. 2013;108(2):240-8.
12. Rahier JF, Papay P, Salleron J, et al. H1N1 vaccines in a large observational cohort of patients with inflammatory bowel disease treated with immunomodulators and biological therapy. Gut. 2011;60(4):456-62.
13. deBruyn J, Fonseca K, Ghosh S, et al. Immunogenicity of Influenza Vaccine for Patients with Inflammatory Bowel Disease on Maintenance Infliximab Therapy: A Randomized Trial. Inflammatory bowel diseases. 2016;22(3):638-47.
14. Brezinschek HP, Hofstaetter T, Leeb BF, et al. Immunization of patients with rheumatoid arthritis with antitumor necrosis factor alpha therapy and methotrexate. Current opinion in rheumatology. 2008;20(3):295-9.
15. Kaine JL, Kivitz AJ, Birbara C, et al. Immune responses following administration of influenza and pneumococcal vaccines to patients with rheumatoid arthritis receiving adalimumab. J Rheumatol. 2007;34(2):272-9.
16. Kim DK, Riley LE, Harriman KH, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Adults Aged 19 Years or Older - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):136-8.
17. Robinson CL, Romero JR, Kempe A, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Children and Adolescents Aged 18 Years or Younger - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):134-5.
18. Cullen G, Baden RP, Cheifetz AS. Varicella zoster virus infection in inflammatory bowel disease. Inflammatory bowel diseases. 2012;18(12):2392-403.
19. Card T, West J, Hubbard R, et al. Hip fractures in patients with inflammatory bowel disease and their relationship to corticosteroid use: a population based cohort study. Gut. 2004;53(2):251-5.
20. Casals-Seoane F, Chaparro M, Mate J, et al. Clinical Course of Bone Metabolism Disorders in Patients with Inflammatory Bowel Disease: A 5-Year Prospective Study. Inflammatory bowel diseases. 2016;22(8):1929-36.
21. Melek J, Sakuraba A. Efficacy and safety of medical therapy for low bone mineral density in patients with inflammatory bowel disease: a meta-analysis and systematic review. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2014;12(1):32-44.e5.
22. Cosman F, de Beur SJ, LeBoff MS, et al. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporosis International. 2014;25(10):2359-81.
23. Peyrin-Biroulet L, Khosrotehrani K, Carrat F, et al. Increased risk for nonmelanoma skin cancers in patients who receive thiopurines for inflammatory bowel disease. Gastroenterology. 2011;141(5):1621-28.e1-5.
24. Long MD, Herfarth HH, Pipkin CA, et al. Increased risk for non-melanoma skin cancer in patients with inflammatory bowel disease. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2010;8(3):268-74.
25. Scott FI, Mamtani R, Brensinger CM, et al. Risk of Nonmelanoma Skin Cancer Associated With the Use of Immunosuppressant and Biologic Agents in Patients With a History of Autoimmune Disease and Nonmelanoma Skin Cancer. JAMA dermatology. 2016;152(2):164-72.
26. Long MD, Martin CF, Pipkin CA, et al. Risk of melanoma and nonmelanoma skin cancer among patients with inflammatory bowel disease. Gastroenterology. 2012;143(2):390-9.e1.
27. Allegretti JR, Barnes EL, Cameron A. Are patients with inflammatory bowel disease on chronic immunosuppressive therapy at increased risk of cervical high-grade dysplasia/cancer? A meta-analysis. Inflammatory bowel diseases. 2015;21(5):1089-97.
28. Practice Bulletin No. 168: Cervical Cancer Screening and Prevention. Obstetrics and gynecology. 2016;128(4):e111-30.
29. Ryan WR, Allan RN, Yamamoto T, et al. Crohn’s disease patients who quit smoking have a reduced risk of reoperation for recurrence. American journal of surgery. 2004;187(2):219-25.
30. Cosnes J, Beaugerie L, Carbonnel F, et al. Smoking cessation and the course of Crohn’s disease: an intervention study. Gastroenterology. 2001;120(5):1093-9.
31. Fuller-Thomson E, Sulman J. Depression and inflammatory bowel disease: findings from two nationally representative Canadian surveys. Inflammatory bowel diseases. 2006;12(8):697-707.
32. Walker EA, Gelfand MD, Gelfand AN, et al. The relationship of current psychiatric disorder to functional disability and distress in patients with inflammatory bowel disease. General hospital psychiatry. 1996;18(4):220-9.
33. Mikocka-Walus A, Pittet V, Rossel J-B, et al. Symptoms of Depression and Anxiety Are Independently Associated With Clinical Recurrence of Inflammatory Bowel Disease. Clinical Gastroenterology and Hepatology.14(6):829-35.e1.
34. Macer BJD, Prady SL, Mikocka-Walus A. Antidepressants in Inflammatory Bowel Disease: A Systematic Review. Inflammatory bowel diseases. 2017;23(4):534-50.
Inflammatory bowel disease (IBD) consists of two chronic inflammatory diseases, Crohn’s disease (CD) and ulcerative colitis (UC), as well as a small category of patients (~10%) who have atypical features called IBD-unclassified (IBD-U) or indeterminate colitis. The prevalence of IBD ranges from 0.3% to 0.5% overall in North America and Europe.1 In North America, the incidences of CD and UC are estimated to be 3.1 to 14.6 per 100,000 person-years and 2.2 to 14.3 cases per 100,000 person-years, respectively; similar rates are seen in Europe.2 However, incidences up to 19.2 and 20.2 per 100,000 for UC and CD, respectively, have been reported in Canada.3,4 The incidences of both UC and CD are increasing over time in Western countries and in rapidly industrializing countries throughout Asia and South America.5-8
Influenza vaccine and pneumococcal vaccine
Influenza A and B outbreaks are commonly seen during the fall and early spring and risk factors for pneumonia and hospitalization include older age, chronic medical conditions, and immunosuppression. The CDC now recommend annual influenza vaccination for all individuals older than six months. For patients on immunosuppression, the vaccine administered should be the inactivated vaccine, as live attenuated vaccines should not be administered to these patients.
In IBD patients, the influenza and pneumococcal vaccines are both well tolerated without an increased rate of adverse effects over the general population and without an increased risk of IBD flares after vaccination.12 A common question for patients on biologic therapy is whether the vaccine should be timed at a specific point in the dose cycle. For infliximab, and likely other biologics, the timing does not change the vaccine immunogenicity and patients should be given these vaccines regardless of where they are in the cycle of administration of their biologic.13 In addition, there is significant response to influenza and pneumococcal vaccines in patients on combination therapy with immunomodulators and anti-TNFs and concerns about a lack of response to vaccines should not discourage vaccination since benefits are still acquired by patients even if immunogenicity is somewhat decreased.14,15
Other vaccinations
In addition to the influenza and pneumococcal vaccines, adult and pediatric patients with IBD should follow the ACIP recommendations for tetanus, diphtheria, pertussis (Tdap), Td boosters, hepatitis A, hepatitis B, human papilloma virus (HPV), and meningococcal vaccinations.16,17
Live vaccines including measles mumps rubella (MMR), varicella, and zoster vaccines are in general contraindicated in immunosuppressed patients on corticosteroids, azathioprine/6-mercaptopurine, methotrexate, anti-TNF, and anti-integrin biologics. An inactive varicella-zoster vaccine will likely be available in the near future and may obviate the need for the live vaccine, which is an important development given the increased risk of zoster in patients with IBD on immunosuppression.18
Osteoporosis screening
Skin cancer screening
Multiple studies have demonstrated that immunosuppression, especially with methotrexate and azathioprine/6-mercaptopurine (6MP) is a risk factor for the development of initial and recurrent non-melanoma skin cancer (NMSC) in IBD patients, the data for biologics are less definitive.23-25 In addition, biologics are associated with increased risk of melanoma in IBD.26 The elevated risk of skin cancer begins in the first year of treatment with thiopurines and may continue after discontinuation. On the basis of this data, screening for melanoma and NMSC is recommended in IBD patients on immunosuppression. Especially for patients on thiopurines it is reasonable for the initial dermatologist visit to occur in the first year of treatment and thereafter with at least annual visits for a full body skin examination. In addition, it is reasonable to recommend regular sunscreen use and protective clothing such as hats.
Cervical cancer screening
A recent meta-analysis shows that women with IBD on immunosuppression have an increased risk of cervical high grade dysplasia and cervical cancer.27 HPV is the major risk factor for cervical cancer and is necessary for its development. The current American College of Gynecology guidelines for women on immunosuppression are to start cervical cancer screening at 21 and annual screening thereafter with Pap and HPV testing.28
Smoking
Smoking has well known associations with poor outcomes in the general population such as increased risk of lung and pancreatic cancers, as well as high risk of cardiovascular disease. In addition, smoking has risks specific to IBD. In CD, smoking is associated with increased disease activity, increased risk of post-operative recurrence, and increased severity of disease.29 Smoking cessation is associated with improved long-term disease outcomes and less risk.30 Making it a point to regularly discuss smoking cessation and partnering with PCPs to offer evidence-based quitting aids may be one of our most significant and beneficial interventions.
Depression and anxiety
Several studies have shown high levels of depression and anxiety in IBD patients and higher levels of depression are associated with increased symptoms, clinical recurrence, poor quality of life and decreased social support.31-33 A recent systematic review of several studies suggested that antidepressants use in IBD patients benefits their mental health and may improve their clinical course as well.34 As such, screening for depression and anxiety regularly and either offering treatment or referral to psychiatrists and psychologists for further management is recommended.10
Conclusion
Patients with IBD frequently develop long-term relationships with their gastroenterologists due to their lifelong chronic disease. It is therefore incumbent on us to be attentive to issues related to IBD patients’ preventive care and collaborate with PCPs to coordinate care for our patients since many of these interventions have both short-term and long-term benefits.
Dr. Chachu is assistant professor and gastroenterologist at Duke University, Durham, N.C.
References
1. Kaplan GG, Ng SC. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology. 2017;152(2):313-21.e2.
2. Loftus EV, Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126(6):1504-17.
3. Bernstein CN, Wajda A, Svenson LW, et al. The Epidemiology of Inflammatory Bowel Disease in Canada: A Population-Based Study. The American journal of gastroenterology. 2006;101(7):1559-68.
4. Lowe AM, Roy PO, M BP, et al. Epidemiology of Crohn’s disease in Quebec, Canada. Inflammatory bowel diseases. 2009;15(3):429-35.
5. Kappelman MD, Rifas-Shiman SL, Kleinman K, et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2007;5(12):1424-9.
6. Kappelman MD, Moore KR, Allen JK, et al. Recent trends in the prevalence of Crohn’s disease and ulcerative colitis in a commercially insured US population. Digestive diseases and sciences. 2013;58(2):519-25.
7. Ng SC, Kaplan G, Banerjee R, et al. 78 Incidence and Phenotype of Inflammatory Bowel Disease From 13 Countries in Asia-Pacific: Results From the Asia-Pacific Crohn’s and Colitis Epidemiologic Study 2011-2013. Gastroenterology.150(4):S21.
8. Parente JML, Coy CSR, Campelo V, et al. Inflammatory bowel disease in an underdeveloped region of Northeastern Brazil. World Journal of Gastroenterology : WJG. 2015;21(4):1197-206.
9. Selby L, Kane S, Wilson J, et al. Receipt of preventive health services by IBD patients is significantly lower than by primary care patients. Inflammatory bowel diseases. 2008;14(2):253-8.
10. Farraye FA, Melmed GY, Lichtenstein GR, et al. ACG Clinical Guideline: Preventive Care in Inflammatory Bowel Disease. The American journal of gastroenterology. 2017;112(2):241-58.
11. Long MD, Martin C, Sandler RS, et al. Increased risk of pneumonia among patients with inflammatory bowel disease. The American journal of gastroenterology. 2013;108(2):240-8.
12. Rahier JF, Papay P, Salleron J, et al. H1N1 vaccines in a large observational cohort of patients with inflammatory bowel disease treated with immunomodulators and biological therapy. Gut. 2011;60(4):456-62.
13. deBruyn J, Fonseca K, Ghosh S, et al. Immunogenicity of Influenza Vaccine for Patients with Inflammatory Bowel Disease on Maintenance Infliximab Therapy: A Randomized Trial. Inflammatory bowel diseases. 2016;22(3):638-47.
14. Brezinschek HP, Hofstaetter T, Leeb BF, et al. Immunization of patients with rheumatoid arthritis with antitumor necrosis factor alpha therapy and methotrexate. Current opinion in rheumatology. 2008;20(3):295-9.
15. Kaine JL, Kivitz AJ, Birbara C, et al. Immune responses following administration of influenza and pneumococcal vaccines to patients with rheumatoid arthritis receiving adalimumab. J Rheumatol. 2007;34(2):272-9.
16. Kim DK, Riley LE, Harriman KH, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Adults Aged 19 Years or Older - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):136-8.
17. Robinson CL, Romero JR, Kempe A, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Children and Adolescents Aged 18 Years or Younger - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):134-5.
18. Cullen G, Baden RP, Cheifetz AS. Varicella zoster virus infection in inflammatory bowel disease. Inflammatory bowel diseases. 2012;18(12):2392-403.
19. Card T, West J, Hubbard R, et al. Hip fractures in patients with inflammatory bowel disease and their relationship to corticosteroid use: a population based cohort study. Gut. 2004;53(2):251-5.
20. Casals-Seoane F, Chaparro M, Mate J, et al. Clinical Course of Bone Metabolism Disorders in Patients with Inflammatory Bowel Disease: A 5-Year Prospective Study. Inflammatory bowel diseases. 2016;22(8):1929-36.
21. Melek J, Sakuraba A. Efficacy and safety of medical therapy for low bone mineral density in patients with inflammatory bowel disease: a meta-analysis and systematic review. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2014;12(1):32-44.e5.
22. Cosman F, de Beur SJ, LeBoff MS, et al. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporosis International. 2014;25(10):2359-81.
23. Peyrin-Biroulet L, Khosrotehrani K, Carrat F, et al. Increased risk for nonmelanoma skin cancers in patients who receive thiopurines for inflammatory bowel disease. Gastroenterology. 2011;141(5):1621-28.e1-5.
24. Long MD, Herfarth HH, Pipkin CA, et al. Increased risk for non-melanoma skin cancer in patients with inflammatory bowel disease. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2010;8(3):268-74.
25. Scott FI, Mamtani R, Brensinger CM, et al. Risk of Nonmelanoma Skin Cancer Associated With the Use of Immunosuppressant and Biologic Agents in Patients With a History of Autoimmune Disease and Nonmelanoma Skin Cancer. JAMA dermatology. 2016;152(2):164-72.
26. Long MD, Martin CF, Pipkin CA, et al. Risk of melanoma and nonmelanoma skin cancer among patients with inflammatory bowel disease. Gastroenterology. 2012;143(2):390-9.e1.
27. Allegretti JR, Barnes EL, Cameron A. Are patients with inflammatory bowel disease on chronic immunosuppressive therapy at increased risk of cervical high-grade dysplasia/cancer? A meta-analysis. Inflammatory bowel diseases. 2015;21(5):1089-97.
28. Practice Bulletin No. 168: Cervical Cancer Screening and Prevention. Obstetrics and gynecology. 2016;128(4):e111-30.
29. Ryan WR, Allan RN, Yamamoto T, et al. Crohn’s disease patients who quit smoking have a reduced risk of reoperation for recurrence. American journal of surgery. 2004;187(2):219-25.
30. Cosnes J, Beaugerie L, Carbonnel F, et al. Smoking cessation and the course of Crohn’s disease: an intervention study. Gastroenterology. 2001;120(5):1093-9.
31. Fuller-Thomson E, Sulman J. Depression and inflammatory bowel disease: findings from two nationally representative Canadian surveys. Inflammatory bowel diseases. 2006;12(8):697-707.
32. Walker EA, Gelfand MD, Gelfand AN, et al. The relationship of current psychiatric disorder to functional disability and distress in patients with inflammatory bowel disease. General hospital psychiatry. 1996;18(4):220-9.
33. Mikocka-Walus A, Pittet V, Rossel J-B, et al. Symptoms of Depression and Anxiety Are Independently Associated With Clinical Recurrence of Inflammatory Bowel Disease. Clinical Gastroenterology and Hepatology.14(6):829-35.e1.
34. Macer BJD, Prady SL, Mikocka-Walus A. Antidepressants in Inflammatory Bowel Disease: A Systematic Review. Inflammatory bowel diseases. 2017;23(4):534-50.
Inflammatory bowel disease (IBD) consists of two chronic inflammatory diseases, Crohn’s disease (CD) and ulcerative colitis (UC), as well as a small category of patients (~10%) who have atypical features called IBD-unclassified (IBD-U) or indeterminate colitis. The prevalence of IBD ranges from 0.3% to 0.5% overall in North America and Europe.1 In North America, the incidences of CD and UC are estimated to be 3.1 to 14.6 per 100,000 person-years and 2.2 to 14.3 cases per 100,000 person-years, respectively; similar rates are seen in Europe.2 However, incidences up to 19.2 and 20.2 per 100,000 for UC and CD, respectively, have been reported in Canada.3,4 The incidences of both UC and CD are increasing over time in Western countries and in rapidly industrializing countries throughout Asia and South America.5-8
Influenza vaccine and pneumococcal vaccine
Influenza A and B outbreaks are commonly seen during the fall and early spring and risk factors for pneumonia and hospitalization include older age, chronic medical conditions, and immunosuppression. The CDC now recommend annual influenza vaccination for all individuals older than six months. For patients on immunosuppression, the vaccine administered should be the inactivated vaccine, as live attenuated vaccines should not be administered to these patients.
In IBD patients, the influenza and pneumococcal vaccines are both well tolerated without an increased rate of adverse effects over the general population and without an increased risk of IBD flares after vaccination.12 A common question for patients on biologic therapy is whether the vaccine should be timed at a specific point in the dose cycle. For infliximab, and likely other biologics, the timing does not change the vaccine immunogenicity and patients should be given these vaccines regardless of where they are in the cycle of administration of their biologic.13 In addition, there is significant response to influenza and pneumococcal vaccines in patients on combination therapy with immunomodulators and anti-TNFs and concerns about a lack of response to vaccines should not discourage vaccination since benefits are still acquired by patients even if immunogenicity is somewhat decreased.14,15
Other vaccinations
In addition to the influenza and pneumococcal vaccines, adult and pediatric patients with IBD should follow the ACIP recommendations for tetanus, diphtheria, pertussis (Tdap), Td boosters, hepatitis A, hepatitis B, human papilloma virus (HPV), and meningococcal vaccinations.16,17
Live vaccines including measles mumps rubella (MMR), varicella, and zoster vaccines are in general contraindicated in immunosuppressed patients on corticosteroids, azathioprine/6-mercaptopurine, methotrexate, anti-TNF, and anti-integrin biologics. An inactive varicella-zoster vaccine will likely be available in the near future and may obviate the need for the live vaccine, which is an important development given the increased risk of zoster in patients with IBD on immunosuppression.18
Osteoporosis screening
Skin cancer screening
Multiple studies have demonstrated that immunosuppression, especially with methotrexate and azathioprine/6-mercaptopurine (6MP) is a risk factor for the development of initial and recurrent non-melanoma skin cancer (NMSC) in IBD patients, the data for biologics are less definitive.23-25 In addition, biologics are associated with increased risk of melanoma in IBD.26 The elevated risk of skin cancer begins in the first year of treatment with thiopurines and may continue after discontinuation. On the basis of this data, screening for melanoma and NMSC is recommended in IBD patients on immunosuppression. Especially for patients on thiopurines it is reasonable for the initial dermatologist visit to occur in the first year of treatment and thereafter with at least annual visits for a full body skin examination. In addition, it is reasonable to recommend regular sunscreen use and protective clothing such as hats.
Cervical cancer screening
A recent meta-analysis shows that women with IBD on immunosuppression have an increased risk of cervical high grade dysplasia and cervical cancer.27 HPV is the major risk factor for cervical cancer and is necessary for its development. The current American College of Gynecology guidelines for women on immunosuppression are to start cervical cancer screening at 21 and annual screening thereafter with Pap and HPV testing.28
Smoking
Smoking has well known associations with poor outcomes in the general population such as increased risk of lung and pancreatic cancers, as well as high risk of cardiovascular disease. In addition, smoking has risks specific to IBD. In CD, smoking is associated with increased disease activity, increased risk of post-operative recurrence, and increased severity of disease.29 Smoking cessation is associated with improved long-term disease outcomes and less risk.30 Making it a point to regularly discuss smoking cessation and partnering with PCPs to offer evidence-based quitting aids may be one of our most significant and beneficial interventions.
Depression and anxiety
Several studies have shown high levels of depression and anxiety in IBD patients and higher levels of depression are associated with increased symptoms, clinical recurrence, poor quality of life and decreased social support.31-33 A recent systematic review of several studies suggested that antidepressants use in IBD patients benefits their mental health and may improve their clinical course as well.34 As such, screening for depression and anxiety regularly and either offering treatment or referral to psychiatrists and psychologists for further management is recommended.10
Conclusion
Patients with IBD frequently develop long-term relationships with their gastroenterologists due to their lifelong chronic disease. It is therefore incumbent on us to be attentive to issues related to IBD patients’ preventive care and collaborate with PCPs to coordinate care for our patients since many of these interventions have both short-term and long-term benefits.
Dr. Chachu is assistant professor and gastroenterologist at Duke University, Durham, N.C.
References
1. Kaplan GG, Ng SC. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology. 2017;152(2):313-21.e2.
2. Loftus EV, Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126(6):1504-17.
3. Bernstein CN, Wajda A, Svenson LW, et al. The Epidemiology of Inflammatory Bowel Disease in Canada: A Population-Based Study. The American journal of gastroenterology. 2006;101(7):1559-68.
4. Lowe AM, Roy PO, M BP, et al. Epidemiology of Crohn’s disease in Quebec, Canada. Inflammatory bowel diseases. 2009;15(3):429-35.
5. Kappelman MD, Rifas-Shiman SL, Kleinman K, et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2007;5(12):1424-9.
6. Kappelman MD, Moore KR, Allen JK, et al. Recent trends in the prevalence of Crohn’s disease and ulcerative colitis in a commercially insured US population. Digestive diseases and sciences. 2013;58(2):519-25.
7. Ng SC, Kaplan G, Banerjee R, et al. 78 Incidence and Phenotype of Inflammatory Bowel Disease From 13 Countries in Asia-Pacific: Results From the Asia-Pacific Crohn’s and Colitis Epidemiologic Study 2011-2013. Gastroenterology.150(4):S21.
8. Parente JML, Coy CSR, Campelo V, et al. Inflammatory bowel disease in an underdeveloped region of Northeastern Brazil. World Journal of Gastroenterology : WJG. 2015;21(4):1197-206.
9. Selby L, Kane S, Wilson J, et al. Receipt of preventive health services by IBD patients is significantly lower than by primary care patients. Inflammatory bowel diseases. 2008;14(2):253-8.
10. Farraye FA, Melmed GY, Lichtenstein GR, et al. ACG Clinical Guideline: Preventive Care in Inflammatory Bowel Disease. The American journal of gastroenterology. 2017;112(2):241-58.
11. Long MD, Martin C, Sandler RS, et al. Increased risk of pneumonia among patients with inflammatory bowel disease. The American journal of gastroenterology. 2013;108(2):240-8.
12. Rahier JF, Papay P, Salleron J, et al. H1N1 vaccines in a large observational cohort of patients with inflammatory bowel disease treated with immunomodulators and biological therapy. Gut. 2011;60(4):456-62.
13. deBruyn J, Fonseca K, Ghosh S, et al. Immunogenicity of Influenza Vaccine for Patients with Inflammatory Bowel Disease on Maintenance Infliximab Therapy: A Randomized Trial. Inflammatory bowel diseases. 2016;22(3):638-47.
14. Brezinschek HP, Hofstaetter T, Leeb BF, et al. Immunization of patients with rheumatoid arthritis with antitumor necrosis factor alpha therapy and methotrexate. Current opinion in rheumatology. 2008;20(3):295-9.
15. Kaine JL, Kivitz AJ, Birbara C, et al. Immune responses following administration of influenza and pneumococcal vaccines to patients with rheumatoid arthritis receiving adalimumab. J Rheumatol. 2007;34(2):272-9.
16. Kim DK, Riley LE, Harriman KH, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Adults Aged 19 Years or Older - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):136-8.
17. Robinson CL, Romero JR, Kempe A, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Children and Adolescents Aged 18 Years or Younger - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):134-5.
18. Cullen G, Baden RP, Cheifetz AS. Varicella zoster virus infection in inflammatory bowel disease. Inflammatory bowel diseases. 2012;18(12):2392-403.
19. Card T, West J, Hubbard R, et al. Hip fractures in patients with inflammatory bowel disease and their relationship to corticosteroid use: a population based cohort study. Gut. 2004;53(2):251-5.
20. Casals-Seoane F, Chaparro M, Mate J, et al. Clinical Course of Bone Metabolism Disorders in Patients with Inflammatory Bowel Disease: A 5-Year Prospective Study. Inflammatory bowel diseases. 2016;22(8):1929-36.
21. Melek J, Sakuraba A. Efficacy and safety of medical therapy for low bone mineral density in patients with inflammatory bowel disease: a meta-analysis and systematic review. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2014;12(1):32-44.e5.
22. Cosman F, de Beur SJ, LeBoff MS, et al. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporosis International. 2014;25(10):2359-81.
23. Peyrin-Biroulet L, Khosrotehrani K, Carrat F, et al. Increased risk for nonmelanoma skin cancers in patients who receive thiopurines for inflammatory bowel disease. Gastroenterology. 2011;141(5):1621-28.e1-5.
24. Long MD, Herfarth HH, Pipkin CA, et al. Increased risk for non-melanoma skin cancer in patients with inflammatory bowel disease. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2010;8(3):268-74.
25. Scott FI, Mamtani R, Brensinger CM, et al. Risk of Nonmelanoma Skin Cancer Associated With the Use of Immunosuppressant and Biologic Agents in Patients With a History of Autoimmune Disease and Nonmelanoma Skin Cancer. JAMA dermatology. 2016;152(2):164-72.
26. Long MD, Martin CF, Pipkin CA, et al. Risk of melanoma and nonmelanoma skin cancer among patients with inflammatory bowel disease. Gastroenterology. 2012;143(2):390-9.e1.
27. Allegretti JR, Barnes EL, Cameron A. Are patients with inflammatory bowel disease on chronic immunosuppressive therapy at increased risk of cervical high-grade dysplasia/cancer? A meta-analysis. Inflammatory bowel diseases. 2015;21(5):1089-97.
28. Practice Bulletin No. 168: Cervical Cancer Screening and Prevention. Obstetrics and gynecology. 2016;128(4):e111-30.
29. Ryan WR, Allan RN, Yamamoto T, et al. Crohn’s disease patients who quit smoking have a reduced risk of reoperation for recurrence. American journal of surgery. 2004;187(2):219-25.
30. Cosnes J, Beaugerie L, Carbonnel F, et al. Smoking cessation and the course of Crohn’s disease: an intervention study. Gastroenterology. 2001;120(5):1093-9.
31. Fuller-Thomson E, Sulman J. Depression and inflammatory bowel disease: findings from two nationally representative Canadian surveys. Inflammatory bowel diseases. 2006;12(8):697-707.
32. Walker EA, Gelfand MD, Gelfand AN, et al. The relationship of current psychiatric disorder to functional disability and distress in patients with inflammatory bowel disease. General hospital psychiatry. 1996;18(4):220-9.
33. Mikocka-Walus A, Pittet V, Rossel J-B, et al. Symptoms of Depression and Anxiety Are Independently Associated With Clinical Recurrence of Inflammatory Bowel Disease. Clinical Gastroenterology and Hepatology.14(6):829-35.e1.
34. Macer BJD, Prady SL, Mikocka-Walus A. Antidepressants in Inflammatory Bowel Disease: A Systematic Review. Inflammatory bowel diseases. 2017;23(4):534-50.
The 'Nuts and Bolts' of Drug Concentration Monitoring in IBD
Introduction
Anti–tumor necrosis factor (anti-TNF) therapy is the cornerstone of inflammatory bowel disease (IBD) treatment.1 Nevertheless, up to 30% of patients show no clinical benefit, considered as primary non-responders, while another 50% lose response over time and need to escalate or discontinue anti-TNF therapy due to either pharmacokinetic (PK) or pharmacodynamic issues.2 Therapeutic drug monitoring (TDM), defined as the assessment of drug concentration and anti-drug antibodies (ADA), is emerging as a new therapeutic strategy to better explain, manage, and hopefully prevent these undesired clinical outcomes.3 Moreover, numerous studies have shown that higher serum anti-TNF drug concentrations both during maintenance and induction therapy are associated with favorable objective therapeutic outcomes, suggesting of a ‘treat-to-trough’ in addition to a ‘treat-to-target’ therapeutic approach.4-6 This concept of TDM is not new in IBD. TDM has also been used for optimizing thiopurines.7 This brief review will discuss a practical approach to the use of TDM in IBD with a focus on its use with anti-TNF therapies. 
Reactive TDM of anti-TNF therapy
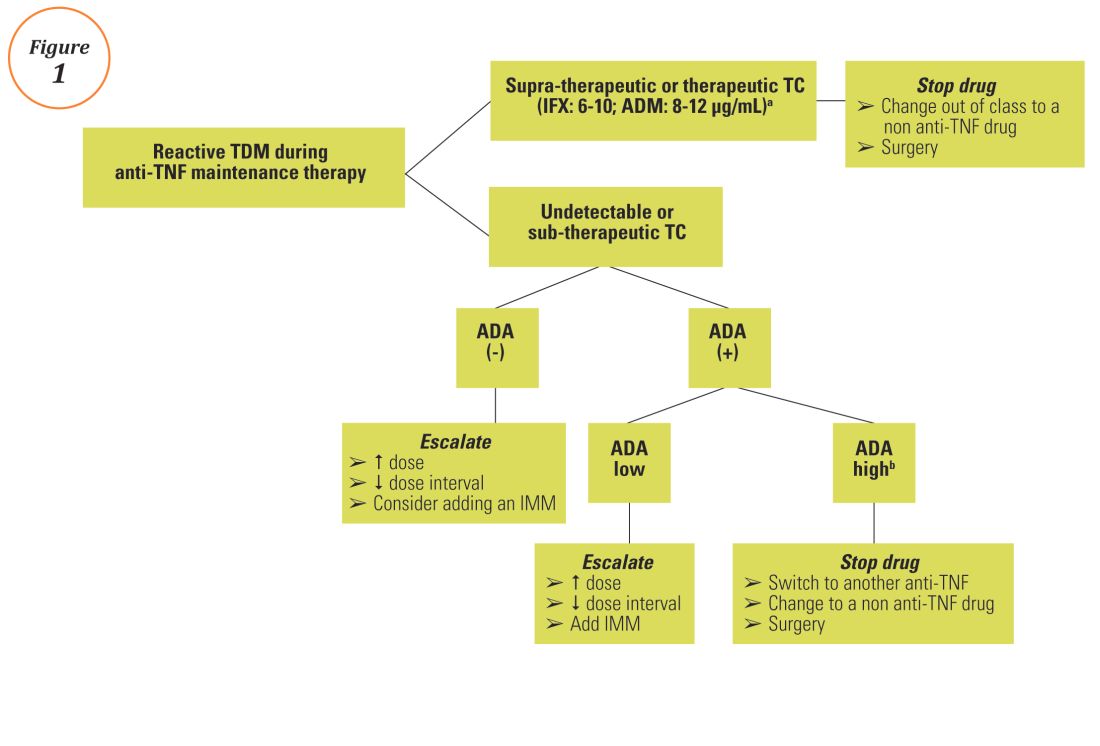
Reactive TDM more rationally guides therapeutic decisions for dealing with loss of response to anti-TNF therapy in IBD and is actually more cost-effective.8,9 Patients with sub-therapeutic or undetectable drug concentrations without ADA derive more benefit from dose escalation (increasing the dose or decreasing the interval) compared to those switched to another anti-TNF agent. On the other hand, patients with therapeutic or supra-therapeutic drug concentrations have better outcomes when changing to a medication with a different mechanism of action (as their disease is probably no longer TNF-driven).3 A recent study showed that trough concentration of adalimumab >4.5 mcg/mL or infliximab >3.8 mcg/mL at time of loss of response identifies patients who benefit more from alternative therapies rather than dose escalation or switching to another anti-TNF agent.10 In clinical practice, in order to fully optimize the original anti-TNF, we will typically dose optimize patients to drug concentrations of infliximab and adalimumab to >10 mcg/mL before giving up and changing medications. Moreover, patients with high ADA titer have better outcomes when switched to another anti-TNF rather than undergo further dose escalation.3 Vande Casteele et al, showed that antibodies to infliximab (ATI) >9.1 U/mL at time of loss of response resulted in a likelihood ratio of 3.6 for an unsuccessful intervention, defined as the need to initiate corticosteroids, immunomodulators (IMM), or other medications or infliximab discontinuation within two infusions after the intervention (shorten of infusion intervals, dose increase to 10 mg/kg, or a combination of both).11 A proposed treatment algorithm for using reactive TDM for anti-TNF therapy is shown in Figure 1.
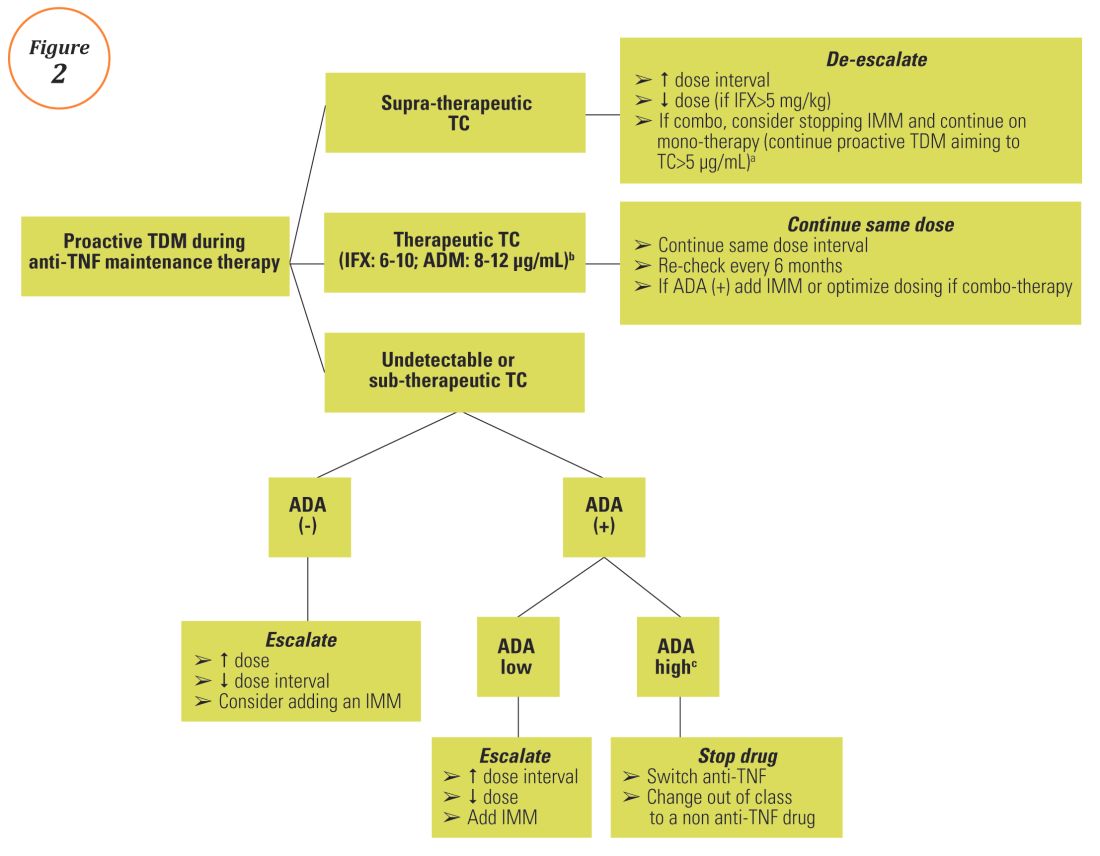
Proactive TDM of anti-TNF therapy

TDM of thiopurines
Anti-TNF TDM assays
Conclusions
A growing body of evidence demonstrates the clinical utility of TDM of anti-TNF therapy in IBD clinical practice and a move towards personalized medicine, as it is now clear that “one dose does not fit all patients.” Nevertheless, before a TDM-based approach can be widely implemented and emerge as the new standard-of-care for anti-TNF therapy in IBD, several barriers regarding cost issues (insurance coverage and out of pocket expenses), time lag from serum sampling to test results (typically 5 to 10 days), proper interpretation and application of the results, type of assay used, and the optimal timing of serum collection should be overcome. Initiatives are already underway including the development of accurate, easily accessible, and affordable rapid assays that will allow anti-TNF concentration measurement at the point-of-care site and software-decision support tools or ‘dashboards’ that will incorporate a predictive PK model based on patient and disease characteristics.29,30 Additionally, more data from well-designed prospective studies and randomized controlled trials regarding both induction and maintenance treatment and for all available biologics (originators and biosimilars) are urgently needed. A panel consisting of members of the Building Research in Inflammatory Bowel Disease Globally research alliance (www.BRIDGeIBD.com), and recognized leaders in the field of TDM in IBD has recently published recommendations that help clinicians on the appropriate timing and best way to interpret and respond to TDM results depending on the specific clinical scenario.31
Funding: KP received a fellowship grant from the Hellenic Group for the study of IBD.
Potential competing interests: K.P.: nothing to disclose; A.S.C: received consultancy fees from AbbVie, Janssen, UCB, Takeda, Prometheus, and Pfizer.
Dr. Papamichail is a research fellow and Dr. Cheifetz is the director of the Center for Inflammatory Bowel Diseases, division of gastroenterology, Beth-Israel Deaconess Medical Center, Harvard Medical School, Boston. Dr. Papamichail received a fellowship grant from the Hellenic Group for the study of IBD. Dr. Cheifetz received consultancy fees from AbbVie, Janssen, UCB, Takeda, Prometheus, and Pfizer.
References
1. Miligkos M, Papamichael K, Casteele NV, et al. Efficacy and safety profile of anti-tumor necrosis factor-alpha versus anti-integrin agents for the treatment of Crohn’s disease: a network meta-analysis of indirect comparisons. Clin Ther. 2016;38(6):1342-1358.e6
2. Papamichael K, Gils A, Rutgeerts P, et al. Role for therapeutic drug monitoring during induction therapy with TNF antagonists in IBD: evolution in the definition and management of primary nonresponse. Inflamm Bowel Dis. 2015;21(1):182-97
3. Papamichael K, Cheifetz AS. Use of anti-TNF drug levels to optimise patient management. Frontline Gastroenterol 2016;7;289-300.
4. Papamichael K, Baert F, Tops S, et al. Post-Induction Adalimumab concentration is associated with short-term mucosal healing in patients with ulcerative colitis. J Crohns Colitis 2017;11:53-59
5. Papamichael K, Van Stappen T, Vande Casteele N, et al. Infliximab concentration thresholds during induction therapy are associated with short-term mucosal healing in patients with ulcerative colitis. Clin Gastroenterol Hepatol 2016;14:543-9.
6. Ungar B, Levy I, Yavne Y, et al. Optimizing Anti-TNF-Alpha Therapy: Serum levels of infliximab and adalimumab are associated with mucosal healing in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol 2016;14:550-557.e2.
7. Singh N, Dubinsky MC. Therapeutic drug monitoring in children and young adults with inflammatory bowel disease: a practical approach. Gastroenterol Hepatol (NY). 2015;11:48-55.
8. Steenholdt C, Brynskov J, Thomsen OØ, et al. Individualised therapy is more cost-effective than dose intensification in patients with Crohn’s disease who lose response to anti-TNF treatment: a randomised, controlled trial. Gut 2014;63:919-27.
9. Velayos FS, Kahn JG, Sandborn WJ, et al. A test-based strategy is more cost effective than empiric dose escalation for patients with Crohn’s disease who lose responsiveness to infliximab. Clin Gastroenterol Hepatol 2013;11:654–66.
10. Yanai H, Lichtenstein L, Assa A, et al. Levels of drug and antidrug antibodies are associated with outcome of interventions after loss of response to infliximab or adalimumab. Clin Gastroenterol Hepatol 2015;13:522-30.
11. Casteele NV, Gils A, Singh S, et al. Antibody response to infliximab and its impact on pharmacokinetics can be transient. Am J Gastroenterol 2013;108:962-71.
12. Vaughn BP, Martinez-Vazquez M, Patwardhan VR, et al. Proactive therapeutic concentration monitoring of infliximab may improve outcomes for patients with inflammatory bowel disease: results from a pilot observational study. Inflamm Bowel Dis 2014;20:1996-2003.
13. Vande Casteele N, Ferrante M, Van Assche G, et al. Trough concentrations of infliximab guide dosing for patients with inflammatory bowel disease. Gastroenterology 2015;148:1320-9.e3.
14. Adedokun OJ, Sandborn WJ, Feagan BG, et al. Association between serum concentration of infliximab and efficacy in adult patients with ulcerative colitis. Gastroenterology 2014;147:1296–307.e5.
15. Cornillie F, Hanauer SB, Diamond RH, et al. Postinduction serum infliximab trough level and decrease of C-reactive protein level are associated with durable sustained response to infliximab: a retrospective analysis of the ACCENT I trial. Gut 2014;63:1721–7.
16. Arias MT, Vande Casteele N, Vermeire S, et al. A panel to predict long-term outcome of infliximab therapy for patients with ulcerative colitis. Clin Gastroenterol Hepatol 2015;13:531–8.
17. Singh N, Rosenthal CJ, Melmed GY, et al Early infliximab trough levels are associated with persistent remission in pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis 2014;20:1708-13.
18. Baert F, Vande Casteele N, Tops S, et al. Prior response to infliximab and early serum drug concentrations predict effects of adalimumab in ulcerative colitis. Aliment Pharmacol Ther 2014;40:1324–32.
19. Baert F, Kondragunta V, Lockton S, et al. Antibodies to adalimumab are associated with future inflammation in Crohn’s patients receiving maintenance adalimumab therapy: a post hoc analysis of the Karmiris trial. Gut 2016;65:1126–31.
20. Colombel JF, Sandborn WJ, Allez M, et al. Association between plasma concentrations of certolizumab pegol and endoscopic outcomes of patients with Crohn’s disease. Clin Gastroenterol Hepatol. 2014;12:423-31.e1
21. Pariente B, Laharie D. Review article: why, when and how to de-escalate therapy in inflammatory bowel diseases. Aliment Pharmacol Ther 2014;40:338–53.
22. Baert F, Drobne D, Gils A, et al. Early trough levels and antibodies to infliximab predict safety and success of reinitiation of infliximab therapy. Clin Gastroenterol Hepatol 2014;12:1474-81.e2
23. Osterman MT, Kundu R, Lichtenstein GR, Lewis JD. Association of 6-thioguanine nucleotide levels and inflammatory bowel disease activity: a meta-analysis. Gastroenterology 2006;130:1047-53
24. Dassopoulos T, Dubinsky MC, Bentsen JL, et al. Randomised clinical trial: individualised vs. weight-based dosing of azathioprine in Crohn’s disease. Aliment Pharmacol Ther 2014;39:163-175.
25. Waljee AK, Joyce JC, Wang S, et al. Algorithms outperform metabolite tests in predicting response of patients with inflammatory bowel disease to thiopurines. Clin Gastroenterol Hepatol 2010;8:143-150.
26. Yarur A, Kubiliun M, Czul F, et al. Concentrations of 6-thioguanine nucleotide correlate with trough levels of infliximab in patients with inflammatory bowel disease on combination therapy. Clin Gastroenterol Hepatol. 2015;13:1118-1124.
27. Marini JC, Sendecki J, Cornillie F, et al. Comparisons of serum infliximab and antibodies-to-infliximab tests used in inflammatory bowel disease clinical trials of Remicade®.AAPS J. 2016 Sep 6. [Epub ahead of print]. DOI: 10.1208/s12248-016-9981-3
28. Gils A, Vande Casteele N, Poppe R, et al. Development of a universal anti-adalimumab antibody standard for interlaboratory harmonization. Ther Drug Monit. 2014;36:669-673.
29. Van Stappen T, Bollen L, Vande Casteele N, et al. Rapid test for infliximab drug concentration allows immediate dose adaptation. Clin Transl Gastroenterol 2016;7:e206
30. Dubinsky MC, Phan BL, Singh N, et al. Pharmacokinetic dashboard-recommended dosing is different than standard of care dosing in infliximab-treated pediatric IBD patients. AAPS J. 2016 Oct 13. [Epub ahead of print]
31. Melmed GY, Irving PM, Jones J, et al. Appropriateness of testing for anti-tumor necrosis factor agent and antibody concentrations, and interpretation of results. Clin Gastroenterol Hepatol 2016;14:1302-9.
32. Baert F, Noman M, Vermeire S, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N Engl J Med 2003;348:601-8.
33. Drobne D, Bossuyt P, Breynaert C, et al. Withdrawal of immunomodulators after co-treatment does not reduce trough level of infliximab in patients with Crohn’s disease. Clin Gastroenterol Hepatol 2015;13:514-21.e4.
Introduction
Anti–tumor necrosis factor (anti-TNF) therapy is the cornerstone of inflammatory bowel disease (IBD) treatment.1 Nevertheless, up to 30% of patients show no clinical benefit, considered as primary non-responders, while another 50% lose response over time and need to escalate or discontinue anti-TNF therapy due to either pharmacokinetic (PK) or pharmacodynamic issues.2 Therapeutic drug monitoring (TDM), defined as the assessment of drug concentration and anti-drug antibodies (ADA), is emerging as a new therapeutic strategy to better explain, manage, and hopefully prevent these undesired clinical outcomes.3 Moreover, numerous studies have shown that higher serum anti-TNF drug concentrations both during maintenance and induction therapy are associated with favorable objective therapeutic outcomes, suggesting of a ‘treat-to-trough’ in addition to a ‘treat-to-target’ therapeutic approach.4-6 This concept of TDM is not new in IBD. TDM has also been used for optimizing thiopurines.7 This brief review will discuss a practical approach to the use of TDM in IBD with a focus on its use with anti-TNF therapies.
Reactive TDM of anti-TNF therapy
Reactive TDM more rationally guides therapeutic decisions for dealing with loss of response to anti-TNF therapy in IBD and is actually more cost-effective.8,9 Patients with sub-therapeutic or undetectable drug concentrations without ADA derive more benefit from dose escalation (increasing the dose or decreasing the interval) compared to those switched to another anti-TNF agent. On the other hand, patients with therapeutic or supra-therapeutic drug concentrations have better outcomes when changing to a medication with a different mechanism of action (as their disease is probably no longer TNF-driven).3 A recent study showed that trough concentration of adalimumab >4.5 mcg/mL or infliximab >3.8 mcg/mL at time of loss of response identifies patients who benefit more from alternative therapies rather than dose escalation or switching to another anti-TNF agent.10 In clinical practice, in order to fully optimize the original anti-TNF, we will typically dose optimize patients to drug concentrations of infliximab and adalimumab to >10 mcg/mL before giving up and changing medications. Moreover, patients with high ADA titer have better outcomes when switched to another anti-TNF rather than undergo further dose escalation.3 Vande Casteele et al, showed that antibodies to infliximab (ATI) >9.1 U/mL at time of loss of response resulted in a likelihood ratio of 3.6 for an unsuccessful intervention, defined as the need to initiate corticosteroids, immunomodulators (IMM), or other medications or infliximab discontinuation within two infusions after the intervention (shorten of infusion intervals, dose increase to 10 mg/kg, or a combination of both).11 A proposed treatment algorithm for using reactive TDM for anti-TNF therapy is shown in Figure 1.
Proactive TDM of anti-TNF therapy
TDM of thiopurines
Anti-TNF TDM assays
Conclusions
A growing body of evidence demonstrates the clinical utility of TDM of anti-TNF therapy in IBD clinical practice and a move towards personalized medicine, as it is now clear that “one dose does not fit all patients.” Nevertheless, before a TDM-based approach can be widely implemented and emerge as the new standard-of-care for anti-TNF therapy in IBD, several barriers regarding cost issues (insurance coverage and out of pocket expenses), time lag from serum sampling to test results (typically 5 to 10 days), proper interpretation and application of the results, type of assay used, and the optimal timing of serum collection should be overcome. Initiatives are already underway including the development of accurate, easily accessible, and affordable rapid assays that will allow anti-TNF concentration measurement at the point-of-care site and software-decision support tools or ‘dashboards’ that will incorporate a predictive PK model based on patient and disease characteristics.29,30 Additionally, more data from well-designed prospective studies and randomized controlled trials regarding both induction and maintenance treatment and for all available biologics (originators and biosimilars) are urgently needed. A panel consisting of members of the Building Research in Inflammatory Bowel Disease Globally research alliance (www.BRIDGeIBD.com), and recognized leaders in the field of TDM in IBD has recently published recommendations that help clinicians on the appropriate timing and best way to interpret and respond to TDM results depending on the specific clinical scenario.31
Funding: KP received a fellowship grant from the Hellenic Group for the study of IBD.
Potential competing interests: K.P.: nothing to disclose; A.S.C: received consultancy fees from AbbVie, Janssen, UCB, Takeda, Prometheus, and Pfizer.
Dr. Papamichail is a research fellow and Dr. Cheifetz is the director of the Center for Inflammatory Bowel Diseases, division of gastroenterology, Beth-Israel Deaconess Medical Center, Harvard Medical School, Boston. Dr. Papamichail received a fellowship grant from the Hellenic Group for the study of IBD. Dr. Cheifetz received consultancy fees from AbbVie, Janssen, UCB, Takeda, Prometheus, and Pfizer.
References
1. Miligkos M, Papamichael K, Casteele NV, et al. Efficacy and safety profile of anti-tumor necrosis factor-alpha versus anti-integrin agents for the treatment of Crohn’s disease: a network meta-analysis of indirect comparisons. Clin Ther. 2016;38(6):1342-1358.e6
2. Papamichael K, Gils A, Rutgeerts P, et al. Role for therapeutic drug monitoring during induction therapy with TNF antagonists in IBD: evolution in the definition and management of primary nonresponse. Inflamm Bowel Dis. 2015;21(1):182-97
3. Papamichael K, Cheifetz AS. Use of anti-TNF drug levels to optimise patient management. Frontline Gastroenterol 2016;7;289-300.
4. Papamichael K, Baert F, Tops S, et al. Post-Induction Adalimumab concentration is associated with short-term mucosal healing in patients with ulcerative colitis. J Crohns Colitis 2017;11:53-59
5. Papamichael K, Van Stappen T, Vande Casteele N, et al. Infliximab concentration thresholds during induction therapy are associated with short-term mucosal healing in patients with ulcerative colitis. Clin Gastroenterol Hepatol 2016;14:543-9.
6. Ungar B, Levy I, Yavne Y, et al. Optimizing Anti-TNF-Alpha Therapy: Serum levels of infliximab and adalimumab are associated with mucosal healing in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol 2016;14:550-557.e2.
7. Singh N, Dubinsky MC. Therapeutic drug monitoring in children and young adults with inflammatory bowel disease: a practical approach. Gastroenterol Hepatol (NY). 2015;11:48-55.
8. Steenholdt C, Brynskov J, Thomsen OØ, et al. Individualised therapy is more cost-effective than dose intensification in patients with Crohn’s disease who lose response to anti-TNF treatment: a randomised, controlled trial. Gut 2014;63:919-27.
9. Velayos FS, Kahn JG, Sandborn WJ, et al. A test-based strategy is more cost effective than empiric dose escalation for patients with Crohn’s disease who lose responsiveness to infliximab. Clin Gastroenterol Hepatol 2013;11:654–66.
10. Yanai H, Lichtenstein L, Assa A, et al. Levels of drug and antidrug antibodies are associated with outcome of interventions after loss of response to infliximab or adalimumab. Clin Gastroenterol Hepatol 2015;13:522-30.
11. Casteele NV, Gils A, Singh S, et al. Antibody response to infliximab and its impact on pharmacokinetics can be transient. Am J Gastroenterol 2013;108:962-71.
12. Vaughn BP, Martinez-Vazquez M, Patwardhan VR, et al. Proactive therapeutic concentration monitoring of infliximab may improve outcomes for patients with inflammatory bowel disease: results from a pilot observational study. Inflamm Bowel Dis 2014;20:1996-2003.
13. Vande Casteele N, Ferrante M, Van Assche G, et al. Trough concentrations of infliximab guide dosing for patients with inflammatory bowel disease. Gastroenterology 2015;148:1320-9.e3.
14. Adedokun OJ, Sandborn WJ, Feagan BG, et al. Association between serum concentration of infliximab and efficacy in adult patients with ulcerative colitis. Gastroenterology 2014;147:1296–307.e5.
15. Cornillie F, Hanauer SB, Diamond RH, et al. Postinduction serum infliximab trough level and decrease of C-reactive protein level are associated with durable sustained response to infliximab: a retrospective analysis of the ACCENT I trial. Gut 2014;63:1721–7.
16. Arias MT, Vande Casteele N, Vermeire S, et al. A panel to predict long-term outcome of infliximab therapy for patients with ulcerative colitis. Clin Gastroenterol Hepatol 2015;13:531–8.
17. Singh N, Rosenthal CJ, Melmed GY, et al Early infliximab trough levels are associated with persistent remission in pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis 2014;20:1708-13.
18. Baert F, Vande Casteele N, Tops S, et al. Prior response to infliximab and early serum drug concentrations predict effects of adalimumab in ulcerative colitis. Aliment Pharmacol Ther 2014;40:1324–32.
19. Baert F, Kondragunta V, Lockton S, et al. Antibodies to adalimumab are associated with future inflammation in Crohn’s patients receiving maintenance adalimumab therapy: a post hoc analysis of the Karmiris trial. Gut 2016;65:1126–31.
20. Colombel JF, Sandborn WJ, Allez M, et al. Association between plasma concentrations of certolizumab pegol and endoscopic outcomes of patients with Crohn’s disease. Clin Gastroenterol Hepatol. 2014;12:423-31.e1
21. Pariente B, Laharie D. Review article: why, when and how to de-escalate therapy in inflammatory bowel diseases. Aliment Pharmacol Ther 2014;40:338–53.
22. Baert F, Drobne D, Gils A, et al. Early trough levels and antibodies to infliximab predict safety and success of reinitiation of infliximab therapy. Clin Gastroenterol Hepatol 2014;12:1474-81.e2
23. Osterman MT, Kundu R, Lichtenstein GR, Lewis JD. Association of 6-thioguanine nucleotide levels and inflammatory bowel disease activity: a meta-analysis. Gastroenterology 2006;130:1047-53
24. Dassopoulos T, Dubinsky MC, Bentsen JL, et al. Randomised clinical trial: individualised vs. weight-based dosing of azathioprine in Crohn’s disease. Aliment Pharmacol Ther 2014;39:163-175.
25. Waljee AK, Joyce JC, Wang S, et al. Algorithms outperform metabolite tests in predicting response of patients with inflammatory bowel disease to thiopurines. Clin Gastroenterol Hepatol 2010;8:143-150.
26. Yarur A, Kubiliun M, Czul F, et al. Concentrations of 6-thioguanine nucleotide correlate with trough levels of infliximab in patients with inflammatory bowel disease on combination therapy. Clin Gastroenterol Hepatol. 2015;13:1118-1124.
27. Marini JC, Sendecki J, Cornillie F, et al. Comparisons of serum infliximab and antibodies-to-infliximab tests used in inflammatory bowel disease clinical trials of Remicade®.AAPS J. 2016 Sep 6. [Epub ahead of print]. DOI: 10.1208/s12248-016-9981-3
28. Gils A, Vande Casteele N, Poppe R, et al. Development of a universal anti-adalimumab antibody standard for interlaboratory harmonization. Ther Drug Monit. 2014;36:669-673.
29. Van Stappen T, Bollen L, Vande Casteele N, et al. Rapid test for infliximab drug concentration allows immediate dose adaptation. Clin Transl Gastroenterol 2016;7:e206
30. Dubinsky MC, Phan BL, Singh N, et al. Pharmacokinetic dashboard-recommended dosing is different than standard of care dosing in infliximab-treated pediatric IBD patients. AAPS J. 2016 Oct 13. [Epub ahead of print]
31. Melmed GY, Irving PM, Jones J, et al. Appropriateness of testing for anti-tumor necrosis factor agent and antibody concentrations, and interpretation of results. Clin Gastroenterol Hepatol 2016;14:1302-9.
32. Baert F, Noman M, Vermeire S, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N Engl J Med 2003;348:601-8.
33. Drobne D, Bossuyt P, Breynaert C, et al. Withdrawal of immunomodulators after co-treatment does not reduce trough level of infliximab in patients with Crohn’s disease. Clin Gastroenterol Hepatol 2015;13:514-21.e4.
Introduction
Anti–tumor necrosis factor (anti-TNF) therapy is the cornerstone of inflammatory bowel disease (IBD) treatment.1 Nevertheless, up to 30% of patients show no clinical benefit, considered as primary non-responders, while another 50% lose response over time and need to escalate or discontinue anti-TNF therapy due to either pharmacokinetic (PK) or pharmacodynamic issues.2 Therapeutic drug monitoring (TDM), defined as the assessment of drug concentration and anti-drug antibodies (ADA), is emerging as a new therapeutic strategy to better explain, manage, and hopefully prevent these undesired clinical outcomes.3 Moreover, numerous studies have shown that higher serum anti-TNF drug concentrations both during maintenance and induction therapy are associated with favorable objective therapeutic outcomes, suggesting of a ‘treat-to-trough’ in addition to a ‘treat-to-target’ therapeutic approach.4-6 This concept of TDM is not new in IBD. TDM has also been used for optimizing thiopurines.7 This brief review will discuss a practical approach to the use of TDM in IBD with a focus on its use with anti-TNF therapies.
Reactive TDM of anti-TNF therapy
Reactive TDM more rationally guides therapeutic decisions for dealing with loss of response to anti-TNF therapy in IBD and is actually more cost-effective.8,9 Patients with sub-therapeutic or undetectable drug concentrations without ADA derive more benefit from dose escalation (increasing the dose or decreasing the interval) compared to those switched to another anti-TNF agent. On the other hand, patients with therapeutic or supra-therapeutic drug concentrations have better outcomes when changing to a medication with a different mechanism of action (as their disease is probably no longer TNF-driven).3 A recent study showed that trough concentration of adalimumab >4.5 mcg/mL or infliximab >3.8 mcg/mL at time of loss of response identifies patients who benefit more from alternative therapies rather than dose escalation or switching to another anti-TNF agent.10 In clinical practice, in order to fully optimize the original anti-TNF, we will typically dose optimize patients to drug concentrations of infliximab and adalimumab to >10 mcg/mL before giving up and changing medications. Moreover, patients with high ADA titer have better outcomes when switched to another anti-TNF rather than undergo further dose escalation.3 Vande Casteele et al, showed that antibodies to infliximab (ATI) >9.1 U/mL at time of loss of response resulted in a likelihood ratio of 3.6 for an unsuccessful intervention, defined as the need to initiate corticosteroids, immunomodulators (IMM), or other medications or infliximab discontinuation within two infusions after the intervention (shorten of infusion intervals, dose increase to 10 mg/kg, or a combination of both).11 A proposed treatment algorithm for using reactive TDM for anti-TNF therapy is shown in Figure 1.
Proactive TDM of anti-TNF therapy
TDM of thiopurines
Anti-TNF TDM assays
Conclusions
A growing body of evidence demonstrates the clinical utility of TDM of anti-TNF therapy in IBD clinical practice and a move towards personalized medicine, as it is now clear that “one dose does not fit all patients.” Nevertheless, before a TDM-based approach can be widely implemented and emerge as the new standard-of-care for anti-TNF therapy in IBD, several barriers regarding cost issues (insurance coverage and out of pocket expenses), time lag from serum sampling to test results (typically 5 to 10 days), proper interpretation and application of the results, type of assay used, and the optimal timing of serum collection should be overcome. Initiatives are already underway including the development of accurate, easily accessible, and affordable rapid assays that will allow anti-TNF concentration measurement at the point-of-care site and software-decision support tools or ‘dashboards’ that will incorporate a predictive PK model based on patient and disease characteristics.29,30 Additionally, more data from well-designed prospective studies and randomized controlled trials regarding both induction and maintenance treatment and for all available biologics (originators and biosimilars) are urgently needed. A panel consisting of members of the Building Research in Inflammatory Bowel Disease Globally research alliance (www.BRIDGeIBD.com), and recognized leaders in the field of TDM in IBD has recently published recommendations that help clinicians on the appropriate timing and best way to interpret and respond to TDM results depending on the specific clinical scenario.31
Funding: KP received a fellowship grant from the Hellenic Group for the study of IBD.
Potential competing interests: K.P.: nothing to disclose; A.S.C: received consultancy fees from AbbVie, Janssen, UCB, Takeda, Prometheus, and Pfizer.
Dr. Papamichail is a research fellow and Dr. Cheifetz is the director of the Center for Inflammatory Bowel Diseases, division of gastroenterology, Beth-Israel Deaconess Medical Center, Harvard Medical School, Boston. Dr. Papamichail received a fellowship grant from the Hellenic Group for the study of IBD. Dr. Cheifetz received consultancy fees from AbbVie, Janssen, UCB, Takeda, Prometheus, and Pfizer.
References
1. Miligkos M, Papamichael K, Casteele NV, et al. Efficacy and safety profile of anti-tumor necrosis factor-alpha versus anti-integrin agents for the treatment of Crohn’s disease: a network meta-analysis of indirect comparisons. Clin Ther. 2016;38(6):1342-1358.e6
2. Papamichael K, Gils A, Rutgeerts P, et al. Role for therapeutic drug monitoring during induction therapy with TNF antagonists in IBD: evolution in the definition and management of primary nonresponse. Inflamm Bowel Dis. 2015;21(1):182-97
3. Papamichael K, Cheifetz AS. Use of anti-TNF drug levels to optimise patient management. Frontline Gastroenterol 2016;7;289-300.
4. Papamichael K, Baert F, Tops S, et al. Post-Induction Adalimumab concentration is associated with short-term mucosal healing in patients with ulcerative colitis. J Crohns Colitis 2017;11:53-59
5. Papamichael K, Van Stappen T, Vande Casteele N, et al. Infliximab concentration thresholds during induction therapy are associated with short-term mucosal healing in patients with ulcerative colitis. Clin Gastroenterol Hepatol 2016;14:543-9.
6. Ungar B, Levy I, Yavne Y, et al. Optimizing Anti-TNF-Alpha Therapy: Serum levels of infliximab and adalimumab are associated with mucosal healing in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol 2016;14:550-557.e2.
7. Singh N, Dubinsky MC. Therapeutic drug monitoring in children and young adults with inflammatory bowel disease: a practical approach. Gastroenterol Hepatol (NY). 2015;11:48-55.
8. Steenholdt C, Brynskov J, Thomsen OØ, et al. Individualised therapy is more cost-effective than dose intensification in patients with Crohn’s disease who lose response to anti-TNF treatment: a randomised, controlled trial. Gut 2014;63:919-27.
9. Velayos FS, Kahn JG, Sandborn WJ, et al. A test-based strategy is more cost effective than empiric dose escalation for patients with Crohn’s disease who lose responsiveness to infliximab. Clin Gastroenterol Hepatol 2013;11:654–66.
10. Yanai H, Lichtenstein L, Assa A, et al. Levels of drug and antidrug antibodies are associated with outcome of interventions after loss of response to infliximab or adalimumab. Clin Gastroenterol Hepatol 2015;13:522-30.
11. Casteele NV, Gils A, Singh S, et al. Antibody response to infliximab and its impact on pharmacokinetics can be transient. Am J Gastroenterol 2013;108:962-71.
12. Vaughn BP, Martinez-Vazquez M, Patwardhan VR, et al. Proactive therapeutic concentration monitoring of infliximab may improve outcomes for patients with inflammatory bowel disease: results from a pilot observational study. Inflamm Bowel Dis 2014;20:1996-2003.
13. Vande Casteele N, Ferrante M, Van Assche G, et al. Trough concentrations of infliximab guide dosing for patients with inflammatory bowel disease. Gastroenterology 2015;148:1320-9.e3.
14. Adedokun OJ, Sandborn WJ, Feagan BG, et al. Association between serum concentration of infliximab and efficacy in adult patients with ulcerative colitis. Gastroenterology 2014;147:1296–307.e5.
15. Cornillie F, Hanauer SB, Diamond RH, et al. Postinduction serum infliximab trough level and decrease of C-reactive protein level are associated with durable sustained response to infliximab: a retrospective analysis of the ACCENT I trial. Gut 2014;63:1721–7.
16. Arias MT, Vande Casteele N, Vermeire S, et al. A panel to predict long-term outcome of infliximab therapy for patients with ulcerative colitis. Clin Gastroenterol Hepatol 2015;13:531–8.
17. Singh N, Rosenthal CJ, Melmed GY, et al Early infliximab trough levels are associated with persistent remission in pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis 2014;20:1708-13.
18. Baert F, Vande Casteele N, Tops S, et al. Prior response to infliximab and early serum drug concentrations predict effects of adalimumab in ulcerative colitis. Aliment Pharmacol Ther 2014;40:1324–32.
19. Baert F, Kondragunta V, Lockton S, et al. Antibodies to adalimumab are associated with future inflammation in Crohn’s patients receiving maintenance adalimumab therapy: a post hoc analysis of the Karmiris trial. Gut 2016;65:1126–31.
20. Colombel JF, Sandborn WJ, Allez M, et al. Association between plasma concentrations of certolizumab pegol and endoscopic outcomes of patients with Crohn’s disease. Clin Gastroenterol Hepatol. 2014;12:423-31.e1
21. Pariente B, Laharie D. Review article: why, when and how to de-escalate therapy in inflammatory bowel diseases. Aliment Pharmacol Ther 2014;40:338–53.
22. Baert F, Drobne D, Gils A, et al. Early trough levels and antibodies to infliximab predict safety and success of reinitiation of infliximab therapy. Clin Gastroenterol Hepatol 2014;12:1474-81.e2
23. Osterman MT, Kundu R, Lichtenstein GR, Lewis JD. Association of 6-thioguanine nucleotide levels and inflammatory bowel disease activity: a meta-analysis. Gastroenterology 2006;130:1047-53
24. Dassopoulos T, Dubinsky MC, Bentsen JL, et al. Randomised clinical trial: individualised vs. weight-based dosing of azathioprine in Crohn’s disease. Aliment Pharmacol Ther 2014;39:163-175.
25. Waljee AK, Joyce JC, Wang S, et al. Algorithms outperform metabolite tests in predicting response of patients with inflammatory bowel disease to thiopurines. Clin Gastroenterol Hepatol 2010;8:143-150.
26. Yarur A, Kubiliun M, Czul F, et al. Concentrations of 6-thioguanine nucleotide correlate with trough levels of infliximab in patients with inflammatory bowel disease on combination therapy. Clin Gastroenterol Hepatol. 2015;13:1118-1124.
27. Marini JC, Sendecki J, Cornillie F, et al. Comparisons of serum infliximab and antibodies-to-infliximab tests used in inflammatory bowel disease clinical trials of Remicade®.AAPS J. 2016 Sep 6. [Epub ahead of print]. DOI: 10.1208/s12248-016-9981-3
28. Gils A, Vande Casteele N, Poppe R, et al. Development of a universal anti-adalimumab antibody standard for interlaboratory harmonization. Ther Drug Monit. 2014;36:669-673.
29. Van Stappen T, Bollen L, Vande Casteele N, et al. Rapid test for infliximab drug concentration allows immediate dose adaptation. Clin Transl Gastroenterol 2016;7:e206
30. Dubinsky MC, Phan BL, Singh N, et al. Pharmacokinetic dashboard-recommended dosing is different than standard of care dosing in infliximab-treated pediatric IBD patients. AAPS J. 2016 Oct 13. [Epub ahead of print]
31. Melmed GY, Irving PM, Jones J, et al. Appropriateness of testing for anti-tumor necrosis factor agent and antibody concentrations, and interpretation of results. Clin Gastroenterol Hepatol 2016;14:1302-9.
32. Baert F, Noman M, Vermeire S, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N Engl J Med 2003;348:601-8.
33. Drobne D, Bossuyt P, Breynaert C, et al. Withdrawal of immunomodulators after co-treatment does not reduce trough level of infliximab in patients with Crohn’s disease. Clin Gastroenterol Hepatol 2015;13:514-21.e4.
News from the AGA
Introducing a New, Private Community Just for AGA’s Trainee and Early Career Members
Networking is an important part of your career, between connecting with mentors, gaining valuable referrals and tackling that next rung on the career ladder. AGA created the Early Career Group in the AGA Community to help you connect and network through the forum and directory, but also to provide education tools you’re not going to find anywhere else.
In case you haven’t yet taken a tour, the group creates an open dialogue for trainees and early career members up to seven years out of training. Each month will host a new theme and corresponding presentation, webinars, journal articles or tip sheets, as well as other member-only online events, such as forums with leading experts in the field.
Also, the group’s event calendar will help you stay on top of important deadlines, conferences and possibly even local meet-ups.
Visit http://Community.Gastro.org/EarlyCareerGroup/ today to take advantage of this collaboration space created just for you.
18 GIs to Watch: The Newest Class of AGA Future Leaders
AGA has announced the second class of its Future Leaders Program, which was created in 2015 to identify early career gastroenterologists who have the potential to make a significant impact on the specialty. The 18 gastroenterologists selected to participate in the 2017-2018 program stood out for their current achievements, commitment to advancing the field, and potential for future success.
“AGA relies heavily on the engagement and expertise of volunteer leaders to develop programs that continue to advance our specialty and support our members through changes to the health-care delivery landscape,” said Suzanne Rose, MD, MSEd, AGAF, co-program chair for the AGA Future Leaders Program. “The newest class of AGA Future Leaders shows exceptional promise and dedication to the field, and we look forward to working with these rising stars to cultivate the future leaders of AGA and the field of gastroenterology.”
The AGA Future Leaders Program provides a pathway within AGA for selected participants who seek opportunities to support the gastroenterology profession, advance their careers, connect with potential mentors and develop the leadership skills necessary to serve the organization. During this year-long program, participants will receive leadership training and work closely with AGA mentors on projects linked to AGA’s Strategic Plan.
AGA is pleased to announce the second class of the Future Leaders program:
- Arthur Beyder, MD, PhD, Assistant Professor, Mayo Clinic-Rochester
- Brigid S. Boland, MD, Assistant Adjunct Professor of Medicine, University of California, San Diego
- Lea Ann Chen, MD, Assistant Professor of Medicine, New York University School of Medicine, NY
- Bruno P. Chumpitazi, MD, MPH, Director, Neurogastroenterology and Motility Program, Texas Children’s Hospital/Baylor College of Medicine, Houston, TX
- Matthew A. Ciorba, MD, Assistant Professor of Medicine, Washington University in St. Louis, MO
- Katherine S. Garman, MD, Assistant Professor of Medicine, Duke University Medical Center, Durham, NC
- Christina Y. Ha, MD, Assistant Professor of Medicine, University of Los Angeles, David Geffen School of Medicine, CA
- Bryson W. Katona, MD, MS, PhD, Instructor, University of Pennsylvania, Philadelphia
- Peter S. Liang, MD, MPH, Instructor, NYU/Manhattan VA, New York, NY
- Folasade P. May, MD, PhD, MPhil, Assistant Professor of Medicine, David Geffen School of Medicine at the University of California, Los Angeles; Department of Veterans Affairs, Los Angeles, CA
- Marty M. Meyer, MD, Gastroenterologist, The Ohio State University, Columbus, OH
- Susan N. Ramdhaney, MD, AGAF, Gastroenterologist, President Comprehensive Digestive Care, Manhasset, NY
- Jonathan A. Rosenberg, MD, Gastroenterologist, Illinois Gastroenterology Group, Highland Park
- N. Jewel Samadder, MD, Assistant Professor of Medicine, Huntsman Cancer Institute, Salt Lake City, UT
- Siddharth Singh, MD, Assistant Professor of Medicine, University of California, San Diego
- Maria I. Vazquez-Roque, MD, MSc, Gastroenterologist, Mayo Clinic, Jacksonville, FL
- Sachin B. Wani, MD, Associate Professor of Medicine, University of Colorado, Aurora
- Jennifer Weiss, MD, MS, Assistant Professor, University of Wisconsin School of Medicine and Public Health, Madison
Learn more about the AGA Future Leaders program on the AGA website: www.gastro.org.
New AGA Guidelines
AGA recently released new clinical guidelines that provide evidence-based recommendations to help guide your clinical practice decisions based on rigorous systematic reviews of the medical literature.
AGA Institute Guideline on the Management of Crohn’s Disease After Surgical Resection: AGA developed this guideline, technical review and Clinical Decision Support Tool to outline strategies to reduce disease recurrence in Crohn’s disease patients who have achieved remission following bowel resection. Prevention of endoscopic recurrence, a strong surrogate measure of surgical recurrence, was evaluated for the development of the guideline.
The guidelines are intended to reduce practice variation and promote high-value care. The current evidence supports the early prophylactic use of thiopurines and/or anti-TNF therapy in patients who are at higher risk for clinical recurrence. However, some patients at lower risk may opt for close endoscopic monitoring instead. Although all patients should undergo ileocolonoscopy at six to 12 months after surgical resection, surveillance for endoscopic recurrence is most important for patients not on any pharmacological prophylaxis. In general, those with endoscopic recurrence should undergo treatment with anti-TNF and/or thiopurine therapy.
This guideline is available in the January issue of Gastroenterology.
AGA Institute Guidelines for the Diagnosis and Management of Acute Liver Failure: AGA developed this guideline and technical review to provide recommendations about controversial diagnostic and treatment strategies and predictive models for outcome of acute liver failure (ALF), which have arisen since acute liver failure is difficult to study in randomized clinical trials.
Recommendations include a strong recommendation for the use of N-acetyl cysteine (NAC) in patients with ALF related to acetaminophen, but there remains a lack of data to allow recommendations for testing for Wilson’s disease and varicella zoster virus in patients with ALF. Although there are low-quality data, because there are therapies that may be beneficial in patients with ALF, recommendations to test for herpes simplex virus and autoimmune hepatitis are supported, as is hepatitis E virus testing in pregnant women with ALF.
This guideline is available in the February issue of Gastroenterology.
Announcing New Crohn’s & Colitis Congress
AGA and the Crohn’s & Colitis Foundation are partnering to co-sponsor a new annual conference for health-care professionals and researchers. By joining the nation’s leading IBD patient organization with the premier GI professional organization, this will be the must-attend IBD conference, bringing state-of-the-art comprehensive care together with the latest research to advance prevention, treatment and cures for IBD patients.
Save the date – Jan. 18-20, 2018, in Las Vegas. Get ready to expand your knowledge, network with other leaders, and be inspired! Stay tuned for our website launch and more details coming this spring.
Introducing a New, Private Community Just for AGA’s Trainee and Early Career Members
Networking is an important part of your career, between connecting with mentors, gaining valuable referrals and tackling that next rung on the career ladder. AGA created the Early Career Group in the AGA Community to help you connect and network through the forum and directory, but also to provide education tools you’re not going to find anywhere else.
In case you haven’t yet taken a tour, the group creates an open dialogue for trainees and early career members up to seven years out of training. Each month will host a new theme and corresponding presentation, webinars, journal articles or tip sheets, as well as other member-only online events, such as forums with leading experts in the field.
Also, the group’s event calendar will help you stay on top of important deadlines, conferences and possibly even local meet-ups.
Visit http://Community.Gastro.org/EarlyCareerGroup/ today to take advantage of this collaboration space created just for you.
18 GIs to Watch: The Newest Class of AGA Future Leaders
AGA has announced the second class of its Future Leaders Program, which was created in 2015 to identify early career gastroenterologists who have the potential to make a significant impact on the specialty. The 18 gastroenterologists selected to participate in the 2017-2018 program stood out for their current achievements, commitment to advancing the field, and potential for future success.
“AGA relies heavily on the engagement and expertise of volunteer leaders to develop programs that continue to advance our specialty and support our members through changes to the health-care delivery landscape,” said Suzanne Rose, MD, MSEd, AGAF, co-program chair for the AGA Future Leaders Program. “The newest class of AGA Future Leaders shows exceptional promise and dedication to the field, and we look forward to working with these rising stars to cultivate the future leaders of AGA and the field of gastroenterology.”
The AGA Future Leaders Program provides a pathway within AGA for selected participants who seek opportunities to support the gastroenterology profession, advance their careers, connect with potential mentors and develop the leadership skills necessary to serve the organization. During this year-long program, participants will receive leadership training and work closely with AGA mentors on projects linked to AGA’s Strategic Plan.
AGA is pleased to announce the second class of the Future Leaders program:
- Arthur Beyder, MD, PhD, Assistant Professor, Mayo Clinic-Rochester
- Brigid S. Boland, MD, Assistant Adjunct Professor of Medicine, University of California, San Diego
- Lea Ann Chen, MD, Assistant Professor of Medicine, New York University School of Medicine, NY
- Bruno P. Chumpitazi, MD, MPH, Director, Neurogastroenterology and Motility Program, Texas Children’s Hospital/Baylor College of Medicine, Houston, TX
- Matthew A. Ciorba, MD, Assistant Professor of Medicine, Washington University in St. Louis, MO
- Katherine S. Garman, MD, Assistant Professor of Medicine, Duke University Medical Center, Durham, NC
- Christina Y. Ha, MD, Assistant Professor of Medicine, University of Los Angeles, David Geffen School of Medicine, CA
- Bryson W. Katona, MD, MS, PhD, Instructor, University of Pennsylvania, Philadelphia
- Peter S. Liang, MD, MPH, Instructor, NYU/Manhattan VA, New York, NY
- Folasade P. May, MD, PhD, MPhil, Assistant Professor of Medicine, David Geffen School of Medicine at the University of California, Los Angeles; Department of Veterans Affairs, Los Angeles, CA
- Marty M. Meyer, MD, Gastroenterologist, The Ohio State University, Columbus, OH
- Susan N. Ramdhaney, MD, AGAF, Gastroenterologist, President Comprehensive Digestive Care, Manhasset, NY
- Jonathan A. Rosenberg, MD, Gastroenterologist, Illinois Gastroenterology Group, Highland Park
- N. Jewel Samadder, MD, Assistant Professor of Medicine, Huntsman Cancer Institute, Salt Lake City, UT
- Siddharth Singh, MD, Assistant Professor of Medicine, University of California, San Diego
- Maria I. Vazquez-Roque, MD, MSc, Gastroenterologist, Mayo Clinic, Jacksonville, FL
- Sachin B. Wani, MD, Associate Professor of Medicine, University of Colorado, Aurora
- Jennifer Weiss, MD, MS, Assistant Professor, University of Wisconsin School of Medicine and Public Health, Madison
Learn more about the AGA Future Leaders program on the AGA website: www.gastro.org.
New AGA Guidelines
AGA recently released new clinical guidelines that provide evidence-based recommendations to help guide your clinical practice decisions based on rigorous systematic reviews of the medical literature.
AGA Institute Guideline on the Management of Crohn’s Disease After Surgical Resection: AGA developed this guideline, technical review and Clinical Decision Support Tool to outline strategies to reduce disease recurrence in Crohn’s disease patients who have achieved remission following bowel resection. Prevention of endoscopic recurrence, a strong surrogate measure of surgical recurrence, was evaluated for the development of the guideline.
The guidelines are intended to reduce practice variation and promote high-value care. The current evidence supports the early prophylactic use of thiopurines and/or anti-TNF therapy in patients who are at higher risk for clinical recurrence. However, some patients at lower risk may opt for close endoscopic monitoring instead. Although all patients should undergo ileocolonoscopy at six to 12 months after surgical resection, surveillance for endoscopic recurrence is most important for patients not on any pharmacological prophylaxis. In general, those with endoscopic recurrence should undergo treatment with anti-TNF and/or thiopurine therapy.
This guideline is available in the January issue of Gastroenterology.
AGA Institute Guidelines for the Diagnosis and Management of Acute Liver Failure: AGA developed this guideline and technical review to provide recommendations about controversial diagnostic and treatment strategies and predictive models for outcome of acute liver failure (ALF), which have arisen since acute liver failure is difficult to study in randomized clinical trials.
Recommendations include a strong recommendation for the use of N-acetyl cysteine (NAC) in patients with ALF related to acetaminophen, but there remains a lack of data to allow recommendations for testing for Wilson’s disease and varicella zoster virus in patients with ALF. Although there are low-quality data, because there are therapies that may be beneficial in patients with ALF, recommendations to test for herpes simplex virus and autoimmune hepatitis are supported, as is hepatitis E virus testing in pregnant women with ALF.
This guideline is available in the February issue of Gastroenterology.
Announcing New Crohn’s & Colitis Congress
AGA and the Crohn’s & Colitis Foundation are partnering to co-sponsor a new annual conference for health-care professionals and researchers. By joining the nation’s leading IBD patient organization with the premier GI professional organization, this will be the must-attend IBD conference, bringing state-of-the-art comprehensive care together with the latest research to advance prevention, treatment and cures for IBD patients.
Save the date – Jan. 18-20, 2018, in Las Vegas. Get ready to expand your knowledge, network with other leaders, and be inspired! Stay tuned for our website launch and more details coming this spring.
Introducing a New, Private Community Just for AGA’s Trainee and Early Career Members
Networking is an important part of your career, between connecting with mentors, gaining valuable referrals and tackling that next rung on the career ladder. AGA created the Early Career Group in the AGA Community to help you connect and network through the forum and directory, but also to provide education tools you’re not going to find anywhere else.
In case you haven’t yet taken a tour, the group creates an open dialogue for trainees and early career members up to seven years out of training. Each month will host a new theme and corresponding presentation, webinars, journal articles or tip sheets, as well as other member-only online events, such as forums with leading experts in the field.
Also, the group’s event calendar will help you stay on top of important deadlines, conferences and possibly even local meet-ups.
Visit http://Community.Gastro.org/EarlyCareerGroup/ today to take advantage of this collaboration space created just for you.
18 GIs to Watch: The Newest Class of AGA Future Leaders
AGA has announced the second class of its Future Leaders Program, which was created in 2015 to identify early career gastroenterologists who have the potential to make a significant impact on the specialty. The 18 gastroenterologists selected to participate in the 2017-2018 program stood out for their current achievements, commitment to advancing the field, and potential for future success.
“AGA relies heavily on the engagement and expertise of volunteer leaders to develop programs that continue to advance our specialty and support our members through changes to the health-care delivery landscape,” said Suzanne Rose, MD, MSEd, AGAF, co-program chair for the AGA Future Leaders Program. “The newest class of AGA Future Leaders shows exceptional promise and dedication to the field, and we look forward to working with these rising stars to cultivate the future leaders of AGA and the field of gastroenterology.”
The AGA Future Leaders Program provides a pathway within AGA for selected participants who seek opportunities to support the gastroenterology profession, advance their careers, connect with potential mentors and develop the leadership skills necessary to serve the organization. During this year-long program, participants will receive leadership training and work closely with AGA mentors on projects linked to AGA’s Strategic Plan.
AGA is pleased to announce the second class of the Future Leaders program:
- Arthur Beyder, MD, PhD, Assistant Professor, Mayo Clinic-Rochester
- Brigid S. Boland, MD, Assistant Adjunct Professor of Medicine, University of California, San Diego
- Lea Ann Chen, MD, Assistant Professor of Medicine, New York University School of Medicine, NY
- Bruno P. Chumpitazi, MD, MPH, Director, Neurogastroenterology and Motility Program, Texas Children’s Hospital/Baylor College of Medicine, Houston, TX
- Matthew A. Ciorba, MD, Assistant Professor of Medicine, Washington University in St. Louis, MO
- Katherine S. Garman, MD, Assistant Professor of Medicine, Duke University Medical Center, Durham, NC
- Christina Y. Ha, MD, Assistant Professor of Medicine, University of Los Angeles, David Geffen School of Medicine, CA
- Bryson W. Katona, MD, MS, PhD, Instructor, University of Pennsylvania, Philadelphia
- Peter S. Liang, MD, MPH, Instructor, NYU/Manhattan VA, New York, NY
- Folasade P. May, MD, PhD, MPhil, Assistant Professor of Medicine, David Geffen School of Medicine at the University of California, Los Angeles; Department of Veterans Affairs, Los Angeles, CA
- Marty M. Meyer, MD, Gastroenterologist, The Ohio State University, Columbus, OH
- Susan N. Ramdhaney, MD, AGAF, Gastroenterologist, President Comprehensive Digestive Care, Manhasset, NY
- Jonathan A. Rosenberg, MD, Gastroenterologist, Illinois Gastroenterology Group, Highland Park
- N. Jewel Samadder, MD, Assistant Professor of Medicine, Huntsman Cancer Institute, Salt Lake City, UT
- Siddharth Singh, MD, Assistant Professor of Medicine, University of California, San Diego
- Maria I. Vazquez-Roque, MD, MSc, Gastroenterologist, Mayo Clinic, Jacksonville, FL
- Sachin B. Wani, MD, Associate Professor of Medicine, University of Colorado, Aurora
- Jennifer Weiss, MD, MS, Assistant Professor, University of Wisconsin School of Medicine and Public Health, Madison
Learn more about the AGA Future Leaders program on the AGA website: www.gastro.org.
New AGA Guidelines
AGA recently released new clinical guidelines that provide evidence-based recommendations to help guide your clinical practice decisions based on rigorous systematic reviews of the medical literature.
AGA Institute Guideline on the Management of Crohn’s Disease After Surgical Resection: AGA developed this guideline, technical review and Clinical Decision Support Tool to outline strategies to reduce disease recurrence in Crohn’s disease patients who have achieved remission following bowel resection. Prevention of endoscopic recurrence, a strong surrogate measure of surgical recurrence, was evaluated for the development of the guideline.
The guidelines are intended to reduce practice variation and promote high-value care. The current evidence supports the early prophylactic use of thiopurines and/or anti-TNF therapy in patients who are at higher risk for clinical recurrence. However, some patients at lower risk may opt for close endoscopic monitoring instead. Although all patients should undergo ileocolonoscopy at six to 12 months after surgical resection, surveillance for endoscopic recurrence is most important for patients not on any pharmacological prophylaxis. In general, those with endoscopic recurrence should undergo treatment with anti-TNF and/or thiopurine therapy.
This guideline is available in the January issue of Gastroenterology.
AGA Institute Guidelines for the Diagnosis and Management of Acute Liver Failure: AGA developed this guideline and technical review to provide recommendations about controversial diagnostic and treatment strategies and predictive models for outcome of acute liver failure (ALF), which have arisen since acute liver failure is difficult to study in randomized clinical trials.
Recommendations include a strong recommendation for the use of N-acetyl cysteine (NAC) in patients with ALF related to acetaminophen, but there remains a lack of data to allow recommendations for testing for Wilson’s disease and varicella zoster virus in patients with ALF. Although there are low-quality data, because there are therapies that may be beneficial in patients with ALF, recommendations to test for herpes simplex virus and autoimmune hepatitis are supported, as is hepatitis E virus testing in pregnant women with ALF.
This guideline is available in the February issue of Gastroenterology.
Announcing New Crohn’s & Colitis Congress
AGA and the Crohn’s & Colitis Foundation are partnering to co-sponsor a new annual conference for health-care professionals and researchers. By joining the nation’s leading IBD patient organization with the premier GI professional organization, this will be the must-attend IBD conference, bringing state-of-the-art comprehensive care together with the latest research to advance prevention, treatment and cures for IBD patients.
Save the date – Jan. 18-20, 2018, in Las Vegas. Get ready to expand your knowledge, network with other leaders, and be inspired! Stay tuned for our website launch and more details coming this spring.

An 87-Year-Old Woman With Recurrent Dysphagia
The correct answer is C: lymphocytic esophagitis.
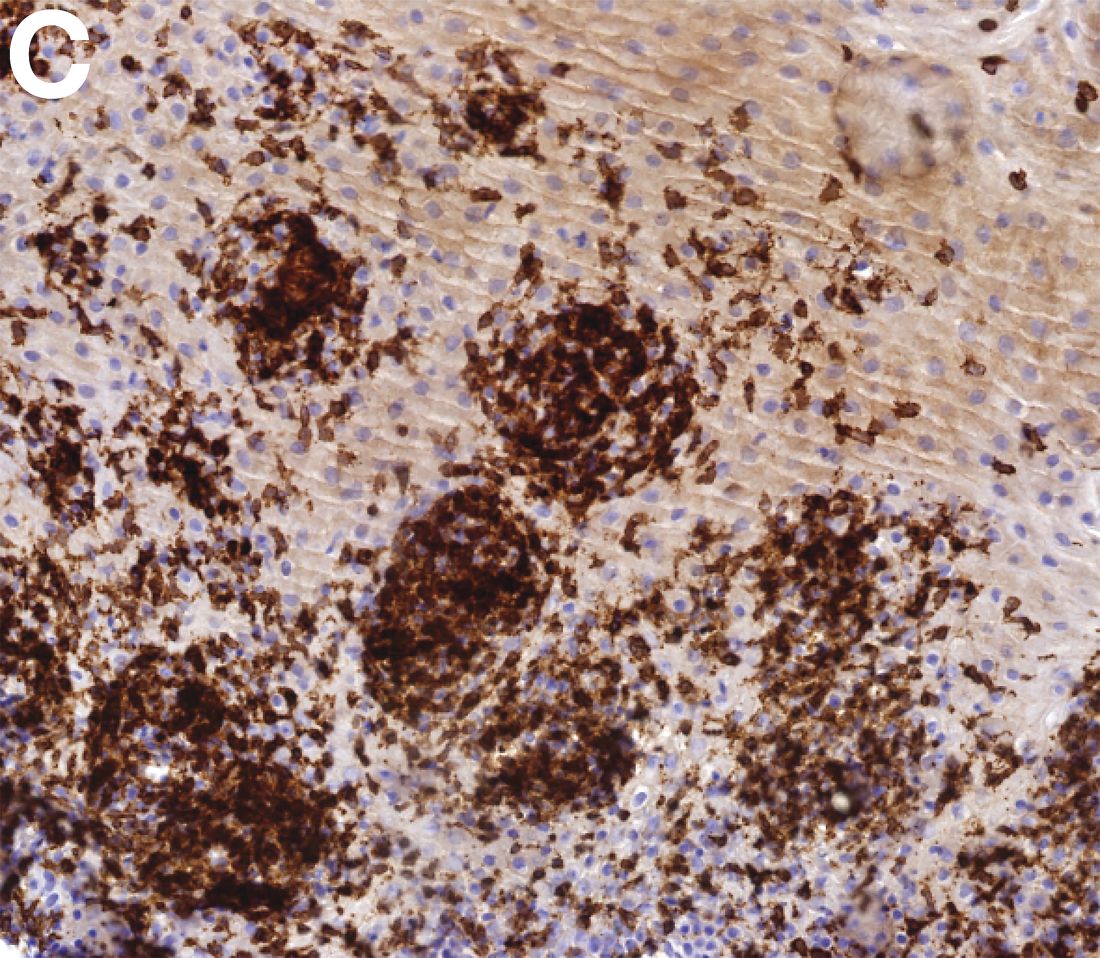
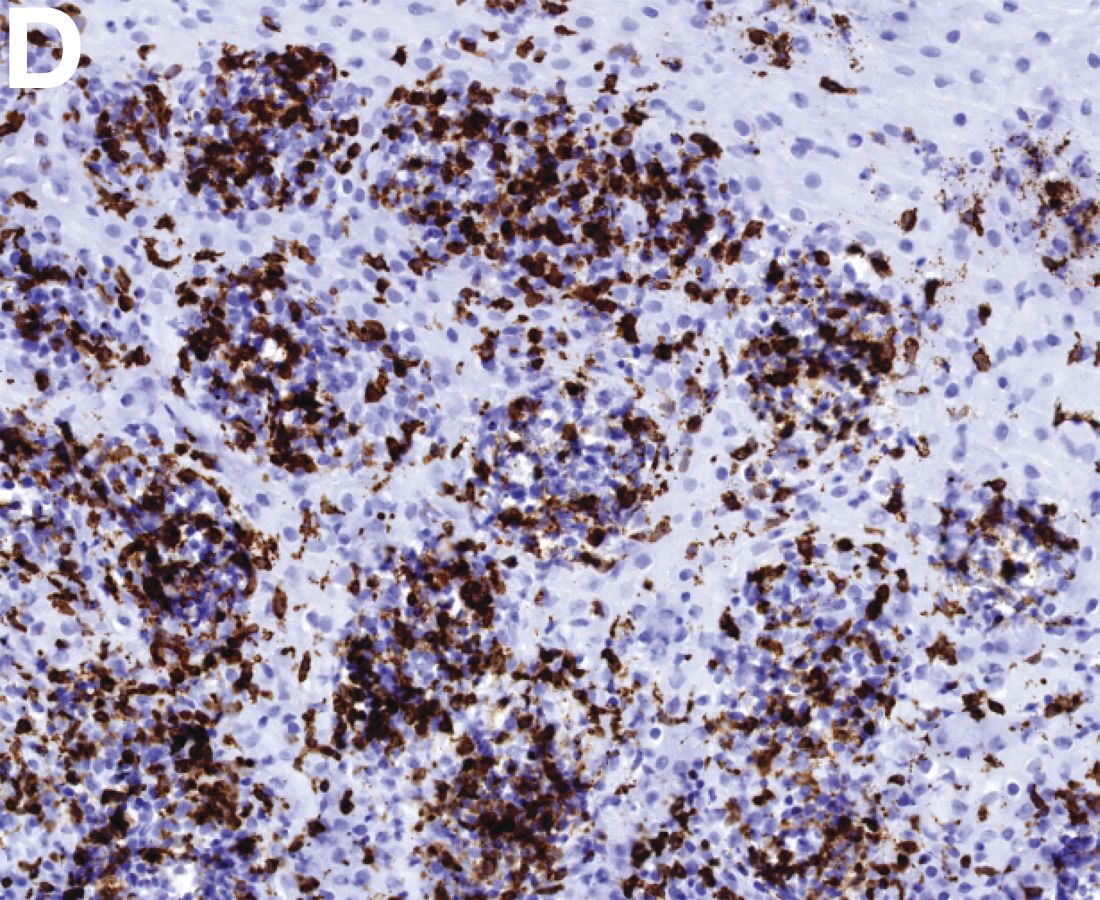
References
1. Rubio, C.A., Sjodahl, K., Lagergren, J. Lymphocytic esophagitis: A histologic subset of chronic esophagitis. Am J Clin Pathol. 2006;125:432-7.
2. Cohen, S., Saxena, A., Waljee, A.K., et al. Lymphocytic esophagitis: A diagnosis of increasing frequency. J Clin Gastroenterol. 2012;46:828-32.
3. Haque, S., Genta, R.M. Lymphocytic oesophagitis: Clinicopathological aspects of an emerging condition. Gut. 2012;61:1108-14.
This article has an accompanying continuing medical education activity, also eligible for MOC credit (see Gastroenterology website for details). Learning Objective: Upon completion of this teaching case and questions, the learners will be able to identify one typical clinical and endoscopic presentation of the entity lymphocytic esophagitis, distinguish its histological pattern from other esophageal disorders and recognize a variety of other clinical presentations of this condition.
The correct answer is C: lymphocytic esophagitis.
References
1. Rubio, C.A., Sjodahl, K., Lagergren, J. Lymphocytic esophagitis: A histologic subset of chronic esophagitis. Am J Clin Pathol. 2006;125:432-7.
2. Cohen, S., Saxena, A., Waljee, A.K., et al. Lymphocytic esophagitis: A diagnosis of increasing frequency. J Clin Gastroenterol. 2012;46:828-32.
3. Haque, S., Genta, R.M. Lymphocytic oesophagitis: Clinicopathological aspects of an emerging condition. Gut. 2012;61:1108-14.
This article has an accompanying continuing medical education activity, also eligible for MOC credit (see Gastroenterology website for details). Learning Objective: Upon completion of this teaching case and questions, the learners will be able to identify one typical clinical and endoscopic presentation of the entity lymphocytic esophagitis, distinguish its histological pattern from other esophageal disorders and recognize a variety of other clinical presentations of this condition.
The correct answer is C: lymphocytic esophagitis.
References
1. Rubio, C.A., Sjodahl, K., Lagergren, J. Lymphocytic esophagitis: A histologic subset of chronic esophagitis. Am J Clin Pathol. 2006;125:432-7.
2. Cohen, S., Saxena, A., Waljee, A.K., et al. Lymphocytic esophagitis: A diagnosis of increasing frequency. J Clin Gastroenterol. 2012;46:828-32.
3. Haque, S., Genta, R.M. Lymphocytic oesophagitis: Clinicopathological aspects of an emerging condition. Gut. 2012;61:1108-14.
This article has an accompanying continuing medical education activity, also eligible for MOC credit (see Gastroenterology website for details). Learning Objective: Upon completion of this teaching case and questions, the learners will be able to identify one typical clinical and endoscopic presentation of the entity lymphocytic esophagitis, distinguish its histological pattern from other esophageal disorders and recognize a variety of other clinical presentations of this condition.
Previously Published in Gastroenterology (2016;151:1085-6)
An 87-year-old woman was referred due to dysphagia that had been present for several years. Three years prior to this presentation she had undergone an esophagogastroduodenoscopy (EGD) on the same indication showing a proximal and a distal esophageal benign-appearing stricture but no signs of esophagitis. Both were dilated and biopsied. Histopathology showed infiltration with lymphocytes and neutrophilic granulocytes, and superficially fungal hyphae and spores. No predominance of eosinophilic granulocytes was noted. A proton-pump inhibitor was prescribed and she was scheduled for a control gastroscopy, but was lost to follow-up. She was otherwise healthy without any allergies.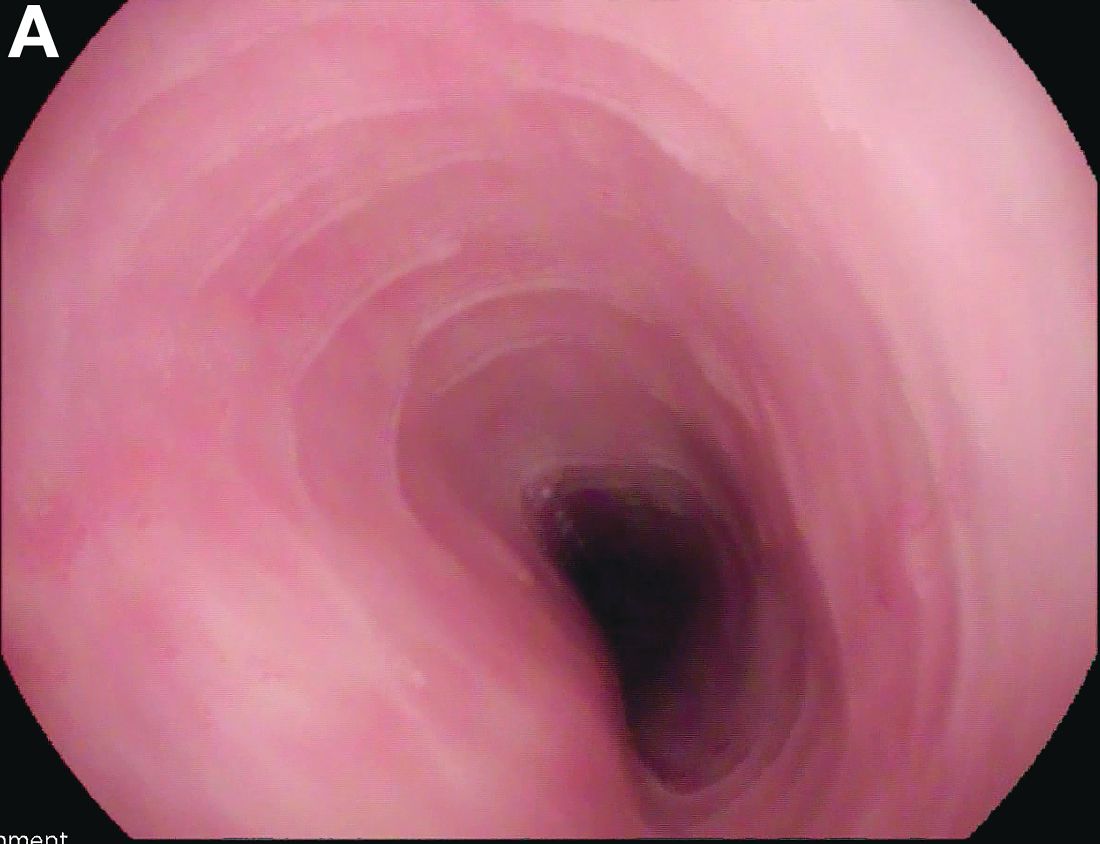
Upon re-presentation, she was under treatment with pantoprazole 40 mg OD. Upon EGD a spiral-shaped proximal esophageal stricture with normal-appearing mucosa only passable with a nasal endoscope was observed. The rest of the esophagus was seen with mucosal concentric rings (Figure A; video). The esophageal mucosa was otherwise endoscopically normal throughout. Biopsies were taken from the distal and proximal esophagus. Balloon dilation of the proximal stricture was performed (CRE, Boston Scientific) to 13.5 mm (video). Subsequently, a standard gastroscope could be passed to the duodenum revealing normal-appearing gastric and duodenal mucosa.
Dr. Havre and Dr. Kalaitzakis are in the Endoscopy Unit of Copenhagen University Hospital/Herlev, University of Copenhagen. Ms. Hallager is in the department of pathology, Copenhagen University Hospital/Herlev. The authors disclose no conflicts.
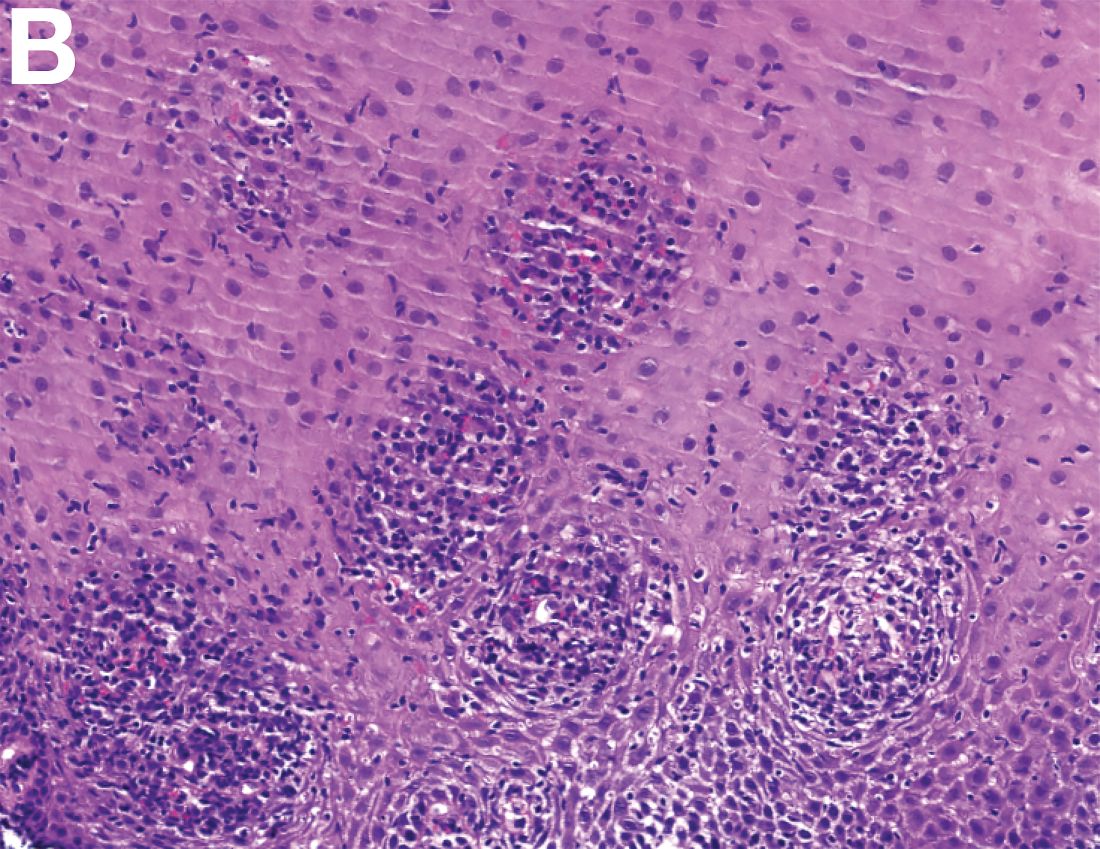
Special IBD-themed issue
Dear Colleagues,
Inflammatory bowel disease (IBD) is becoming an increasingly important part of GI practice and it is certainly an exciting time to be involved in the field. While new IBD therapeutics often get most of the attention, there are many other issues surrounding IBD care that are important for all of us. This special IBD-themed issue of The New Gastroenterologist provides expert opinions addressing some of these other, important issues that are critical to both the care of IBD patients and the development of an effective IBD practice.

In this issue of The New Gastroenterologist, we also have several articles that will be very helpful to those who either have or are developing a practice with a significant IBD focus. First, Douglas Wolf (Atlanta Gastroenterology Associates) discusses the steps necessary to build a successful IBD practice, and, additionally, Nitin Gupta (University of Mississippi Medical Center) provides some useful tips to help physicians start collaborations with industry.
As MACRA looms over us all, it is only a matter of time before we will all have to firmly understand its intricacies. The implementation of MACRA and MIPS will undoubtedly affect quality measures in IBD and to help all of us understand the complexities of this issue, Ryan McConnell and Fernando Velayos (University of California, San Francisco) provide an overview of quality measures in IBD. Finally, although treatment, monitoring, and quality are all important in the care of IBD patients, so also are the relationships that we develop with our IBD patients. To give us input on this topic from a patient perspective, a group of IBD patients from the Crohn’s and Colitis Foundation of America address what we as physicians can do to enhance our doctor-patient relationships.
If you want to read The New Gastroenterologist “on the go,” please download our free app, or read our electronic version on www.mdedge.com/gihepnews or www.gastro.org. Additionally, if you have other topics you would be interested in reading about, or if you are interested in contributing to future issues, please e-mail me at bryson.katona@uphs.upenn.edu or The New Gastroenterologist’s managing editor Ryan Farrell at rfarrell@gastro.org.
Sincerely,
Bryson W. Katona, MD, PhD
Editor-In-Chief
Bryson W. Katona is an instructor of medicine in the division of gastroenterology at the University of Pennsylvania
Dear Colleagues,
Inflammatory bowel disease (IBD) is becoming an increasingly important part of GI practice and it is certainly an exciting time to be involved in the field. While new IBD therapeutics often get most of the attention, there are many other issues surrounding IBD care that are important for all of us. This special IBD-themed issue of The New Gastroenterologist provides expert opinions addressing some of these other, important issues that are critical to both the care of IBD patients and the development of an effective IBD practice.
In this issue of The New Gastroenterologist, we also have several articles that will be very helpful to those who either have or are developing a practice with a significant IBD focus. First, Douglas Wolf (Atlanta Gastroenterology Associates) discusses the steps necessary to build a successful IBD practice, and, additionally, Nitin Gupta (University of Mississippi Medical Center) provides some useful tips to help physicians start collaborations with industry.
As MACRA looms over us all, it is only a matter of time before we will all have to firmly understand its intricacies. The implementation of MACRA and MIPS will undoubtedly affect quality measures in IBD and to help all of us understand the complexities of this issue, Ryan McConnell and Fernando Velayos (University of California, San Francisco) provide an overview of quality measures in IBD. Finally, although treatment, monitoring, and quality are all important in the care of IBD patients, so also are the relationships that we develop with our IBD patients. To give us input on this topic from a patient perspective, a group of IBD patients from the Crohn’s and Colitis Foundation of America address what we as physicians can do to enhance our doctor-patient relationships.
If you want to read The New Gastroenterologist “on the go,” please download our free app, or read our electronic version on www.mdedge.com/gihepnews or www.gastro.org. Additionally, if you have other topics you would be interested in reading about, or if you are interested in contributing to future issues, please e-mail me at bryson.katona@uphs.upenn.edu or The New Gastroenterologist’s managing editor Ryan Farrell at rfarrell@gastro.org.
Sincerely,
Bryson W. Katona, MD, PhD
Editor-In-Chief
Bryson W. Katona is an instructor of medicine in the division of gastroenterology at the University of Pennsylvania
Dear Colleagues,
Inflammatory bowel disease (IBD) is becoming an increasingly important part of GI practice and it is certainly an exciting time to be involved in the field. While new IBD therapeutics often get most of the attention, there are many other issues surrounding IBD care that are important for all of us. This special IBD-themed issue of The New Gastroenterologist provides expert opinions addressing some of these other, important issues that are critical to both the care of IBD patients and the development of an effective IBD practice.
In this issue of The New Gastroenterologist, we also have several articles that will be very helpful to those who either have or are developing a practice with a significant IBD focus. First, Douglas Wolf (Atlanta Gastroenterology Associates) discusses the steps necessary to build a successful IBD practice, and, additionally, Nitin Gupta (University of Mississippi Medical Center) provides some useful tips to help physicians start collaborations with industry.
As MACRA looms over us all, it is only a matter of time before we will all have to firmly understand its intricacies. The implementation of MACRA and MIPS will undoubtedly affect quality measures in IBD and to help all of us understand the complexities of this issue, Ryan McConnell and Fernando Velayos (University of California, San Francisco) provide an overview of quality measures in IBD. Finally, although treatment, monitoring, and quality are all important in the care of IBD patients, so also are the relationships that we develop with our IBD patients. To give us input on this topic from a patient perspective, a group of IBD patients from the Crohn’s and Colitis Foundation of America address what we as physicians can do to enhance our doctor-patient relationships.
If you want to read The New Gastroenterologist “on the go,” please download our free app, or read our electronic version on www.mdedge.com/gihepnews or www.gastro.org. Additionally, if you have other topics you would be interested in reading about, or if you are interested in contributing to future issues, please e-mail me at bryson.katona@uphs.upenn.edu or The New Gastroenterologist’s managing editor Ryan Farrell at rfarrell@gastro.org.
Sincerely,
Bryson W. Katona, MD, PhD
Editor-In-Chief
Bryson W. Katona is an instructor of medicine in the division of gastroenterology at the University of Pennsylvania
Neoadjuvant and Adjuvant Therapy for Gastric Cancer
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
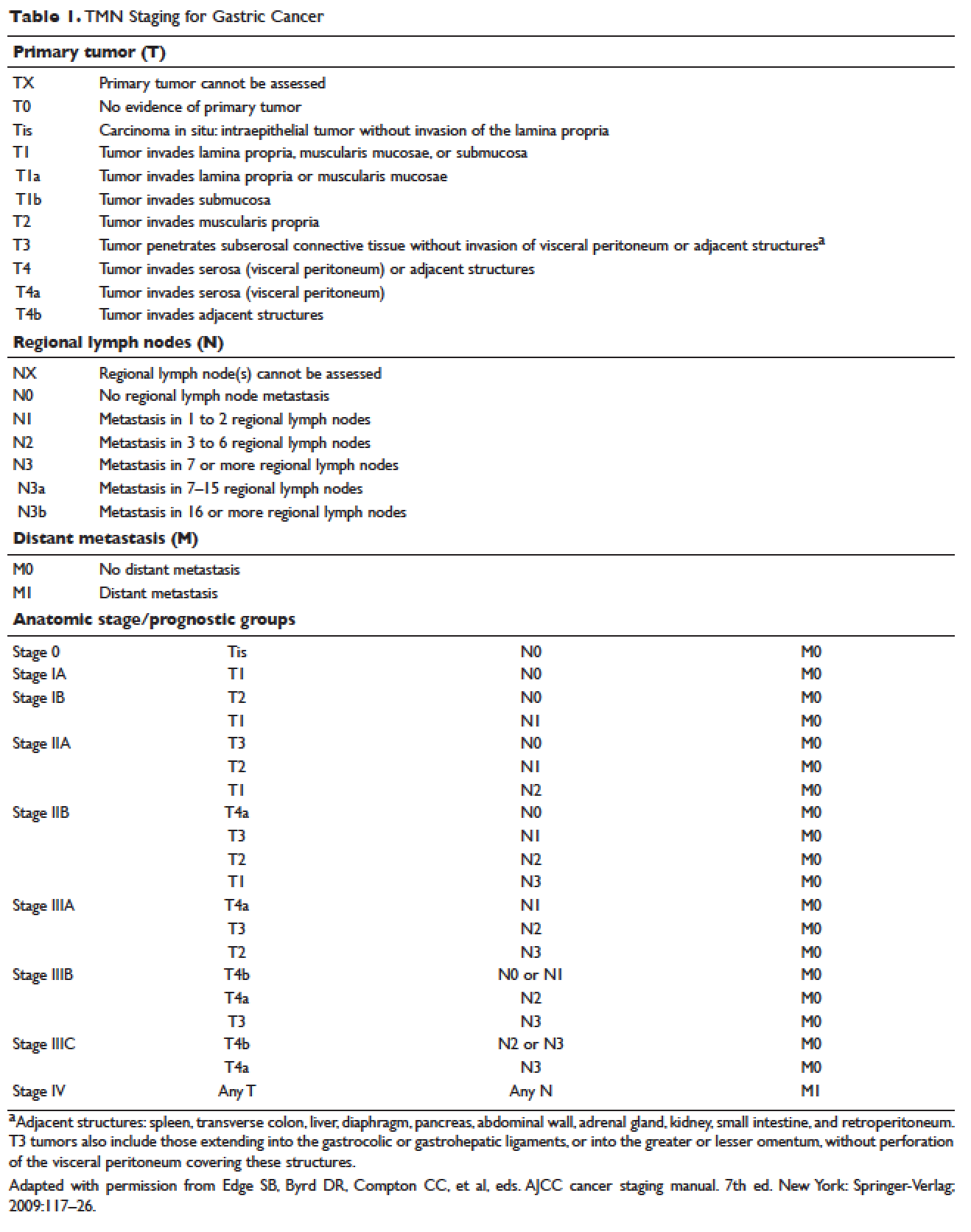
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
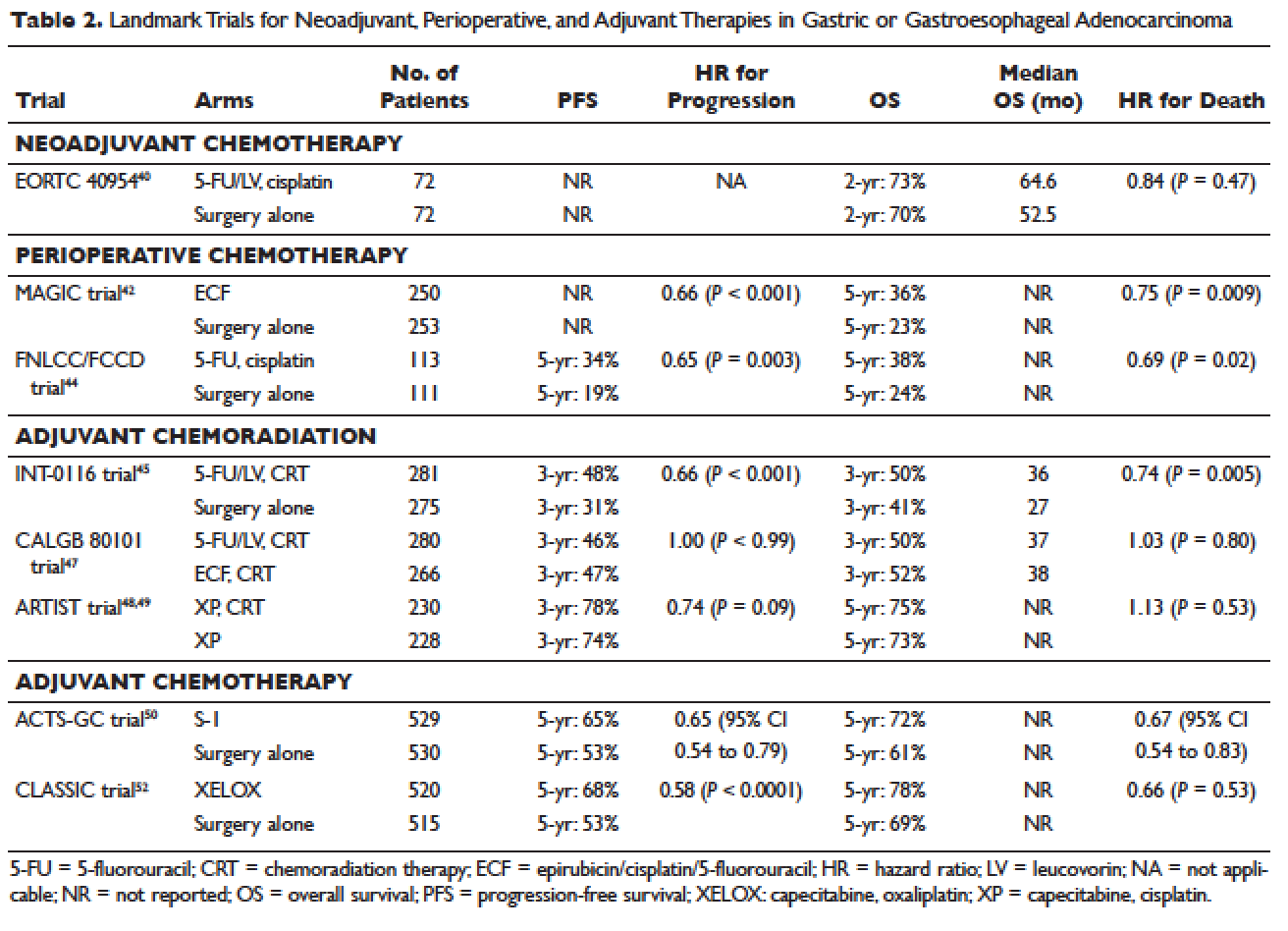
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
Hemophilia A and B: An Overview
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).

MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
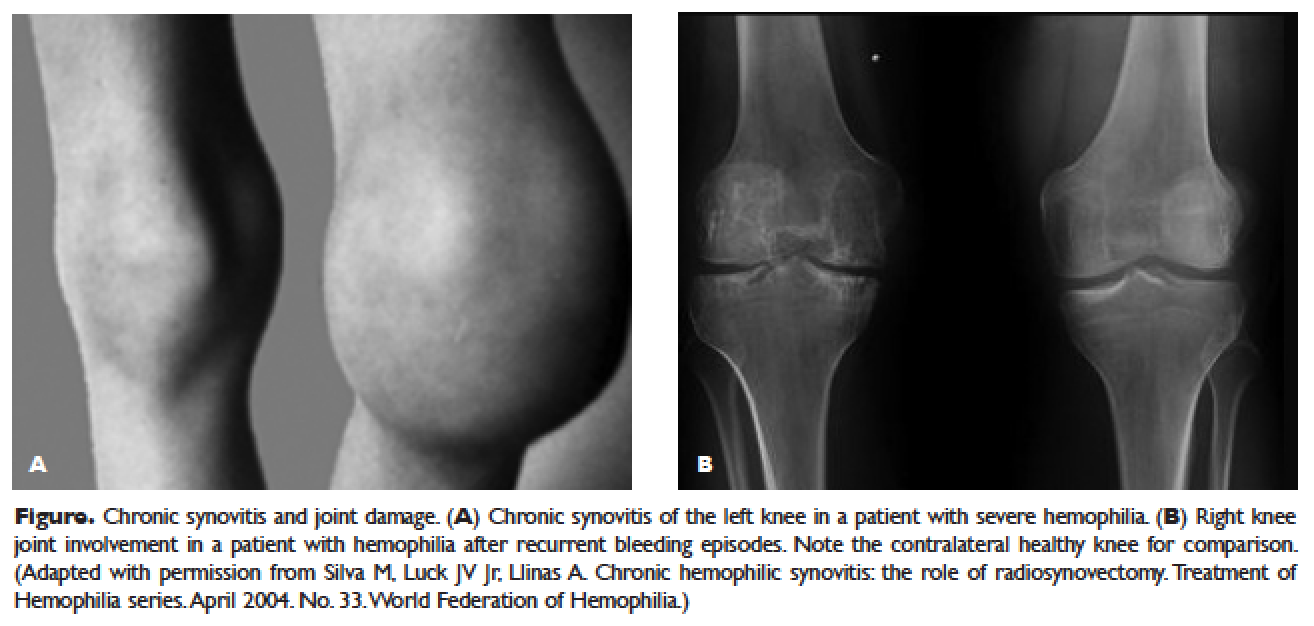
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
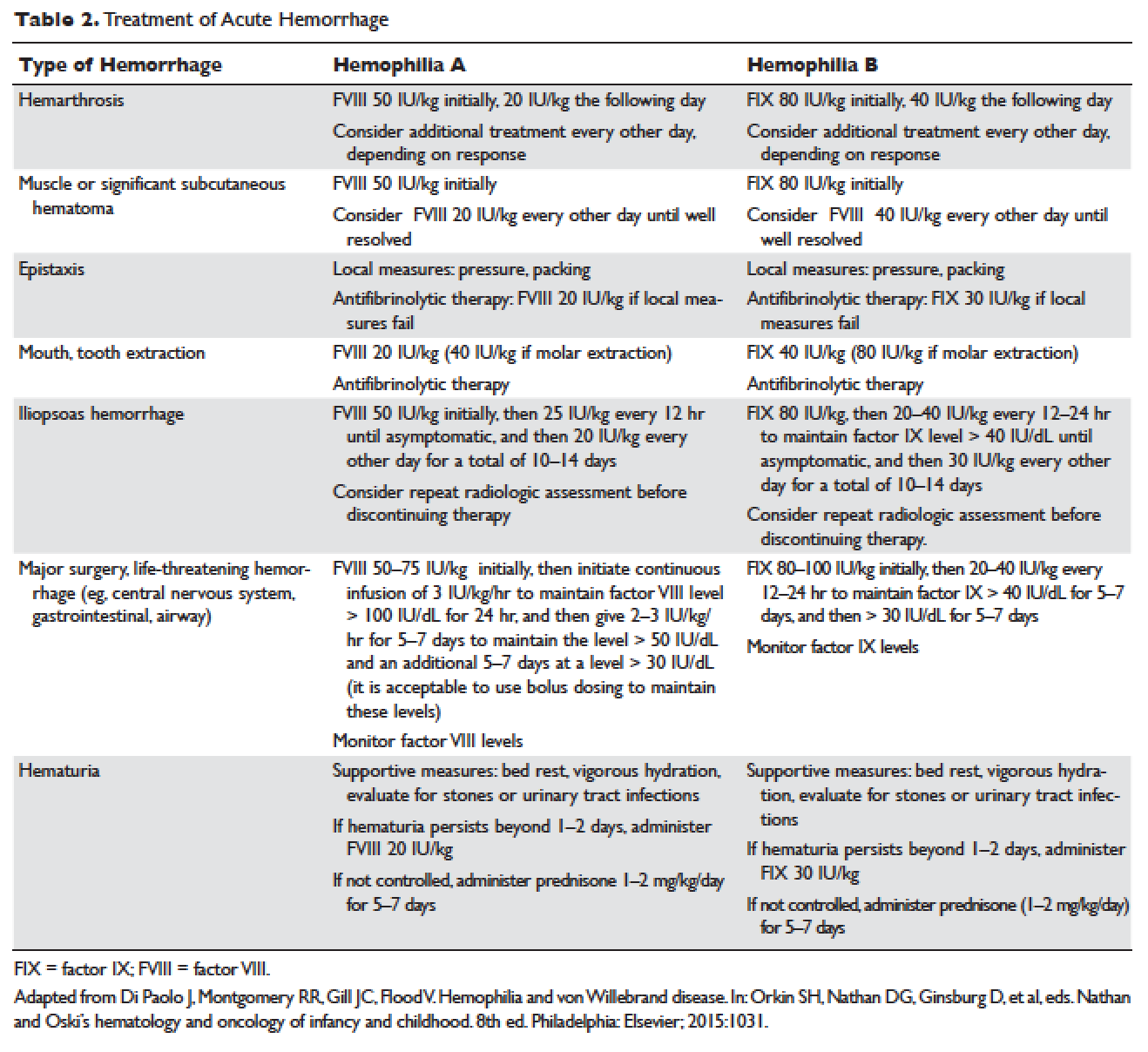
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
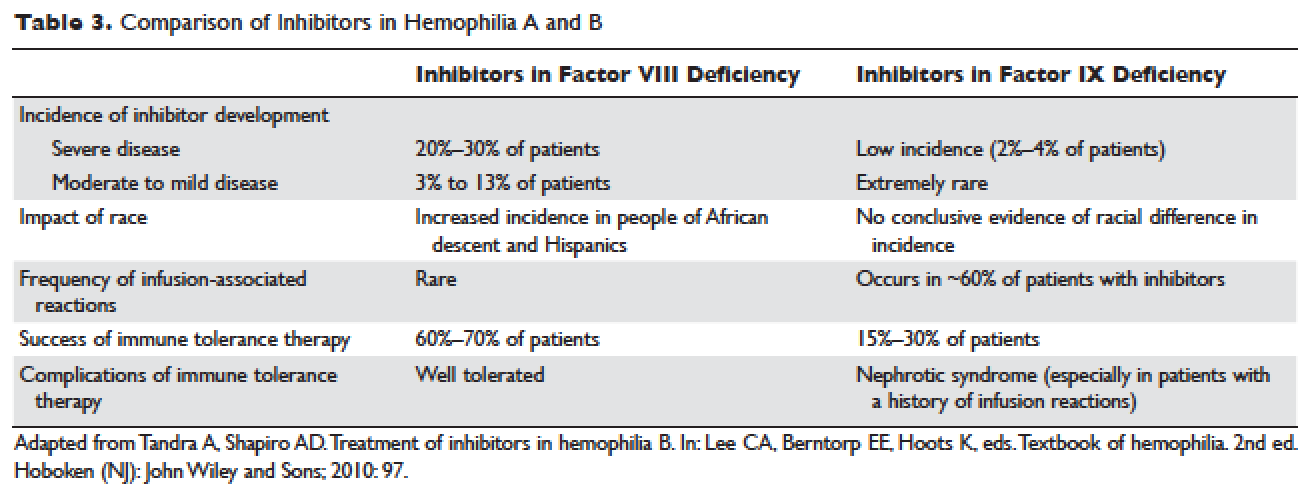
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.

Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82

ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).
MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.
Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82
ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).
MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.
Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82
ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
Mantle Cell Lymphoma
INTRODUCTION
Mantle cell lymphoma (MCL) is an uncommon, distinct clinical subtype of non-Hodgkin lymphoma (NHL) that comprises approximately 8% of all lymphoma diagnoses in the United States and Europe.1,2 Considered incurable, MCL often presents in advanced stages, particularly with involvement of the lymph nodes, spleen, bone marrow, and gastrointestinal tract in the form of lymphomatous polyps. MCL disproportionately affects males, and incidence rises with age, with a median age at diagnosis of 68 years.2 Historically, the prognosis of patients with MCL has been among the poorest among B-cell lymphoma patients, with a median overall survival (OS) of 3 to 5 years, and time to treatment failure (TTF) of 18 to 24 months, although this is improving in the modern era.3 Less frequently, patients with MCL display isolated bone marrow, peripheral blood, and splenic involvement. These cases tend to behave more indolently with longer survival.4,5 Recent advances in therapy have dramatically impacted treatment alternatives and outcomes for MCL. As such, the therapeutic and prognostic landscape of MCL is evolving rapidly.
PATHOGENESIS
The histologic diagnosis of MCL by morphology alone is often challenging. Accurate diagnosis relies on immunohistochemical staining for the purposes of immunophenotyping.6 MCL typically expresses B-cell markers CD5 and CD20, and lacks both CD10 and CD23. The genetic hallmark of MCL is the t(11;14) (q13;q32) chromosomal translocation leading to upregulation of the cyclin D1 protein, a critical regulator of the G1 phase of the cell cycle. Specifically, the t(11;14) translocation, present in virtually all cases of MCL, juxtaposes the proto-oncogene CCND1 to the immunoglobulin heavy chain gene.7 Consequently, cyclin D1, normally not expressed in B lymphocytes, becomes constitutively overexpressed. This alteration is thought to facilitate the deregulation of the cell cycle at the G1-S phase transition.8
Gene expression profiling studies have underscored the importance of cell cycle deregulation in MCL, and high proliferation is associated with a worse prognosis.9 More than 50% of the genes associated with poor outcomes were derived from the “proliferation signature” that was more highly expressed in dividing cells. In the seminal Rosenwald study, a gene expression–based outcome model was constructed in which the proliferation signature average represents a linear variable that assigns a discrete probability of survival to an individual patient.9 The proliferative index, or proliferative signature, of MCL can be estimated by the percentage of Ki-67–positive cells present in the tumor through immunohistochemistry. This is often used as a marker of poor outcomes, and as a surrogate for the proliferative signature in MCL that can be incorporated into clinical practice (as opposed to gene expression profiling). Statistically significant differences in OS have emerged between groups of MCL patients with Ki-67–positive cells comprising less than 30% of their tumor sample (favorable) and those with Ki-67–positive cells comprising 30% or greater (unfavorable).10
Recent data has also identified the importance of the transcription factor SOX 11 (SRY-related HMG-box), which regulates multiple cellular transcriptional events, including cell proliferation and differentiation, apoptosis, and angiogenesis.11 MCL expressing SOX 11 behaves more aggressively than MCL variants lacking SOX 11 expression, and tends to accumulate more genetic alterations.12 Moreover, lack of SOX 11 expression characterizes a subset of MCL that does not carry the t(11;14) translocation.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 62-year-old man with a history of diabetes mellitus and hypertension presents with cervical lymphadenopathy, fatigue, and early satiety over the past several months. He is otherwise in good health. His Eastern Cooperative Oncology Group (ECOG) performance status is 1. On physical examination, 3-cm lymphadenopathy in the bilateral cervical chain is noted. Bilateral axillary lymph nodes measure 2 to 4 cm. His spleen is enlarged and is palpable at approximately 5 cm below the costal margin. A complete blood count reveals a total white blood cell (WBC) count of 14,000 cells/μL, with 68% lymphocytes and a normal distribution of neutrophils. Hemoglobin is 11 g/dL, and platelet count is 112,000/μL. The lactate dehydrogenase (LDH) level is 322 U/L (upper limit of normal: 225 U/L).
• How is MCL diagnosed?
Diagnosis of MCL requires review by expert hematopathologists.13 Whenever possible, an excisional biopsy should be performed for the adequate characterization of lymph node architecture and evaluation by immunohistochemistry. Aside from the characteristic expression of CD5 and CD20 and absence of CD23, MCL should express cyclin D1, which reflects t(11;14). If cyclin D1 is inconclusive or unavailable, fluorescent in situ hybridization (FISH) for t(11;14) should be performed.8 Patients often have circulating malignant lymphocytes, or leukemic phase MCL. Flow cytometry of the peripheral blood can detect traditional surface markers, and FISH can also be performed on circulating abnormal lymphocytes.
For disease staging, bone marrow biopsy and aspiration are required. Radiographic staging using computed tomography (CT) scans and/or positron emission tomography (PET) scans had traditionally followed the Ann Arbor staging system, but recently the Lugano classification has emerged, which delineates only early or advanced stage.14 Gastrointestinal evaluation of MCL with endoscopy and colonoscopy with blind biopsies has been recommended to evaluate for the presence of lymphomatous polyps, but this is not an absolute requirement.15
RISK STRATIFICATION
At diagnosis, patients should undergo risk stratification in order to understand prognosis and possibly guide treatment. In MCL, the MCL international prognostic index (MIPI) is used. The MIPI is a prognostic tool developed exclusively for patients with MCL using data from 455 patients with advanced-stage MCL treated within 3 European clinical trials.16 The MIPI classified patients into risk groups based on age, ECOG performance status, LDH level, and WBC count. Patients were categorized into low-risk (44% of patients, median OS not reached), intermediate-risk (35%, median OS 51 months), and high-risk groups (21%, median OS 29 months). This is done through a logarithmic calculation, which can be accessed through online calculators (a prototype example can be found at www.qxmd.com/calculate-online/hematology/prognosis-mantle-cell-lymphoma-mipi). Cell proliferation using the Ki-67 index was evaluated in an exploratory analysis (the biologic [“B”] MIPI), and also demonstrated strong prognostic relevance.16 Currently, treatment of MCL patients is not stratified by MIPI outside of a clinical trial, but this useful tool assists in assessing patient prognosis and has been validated for use with both conventional chemoimmunotherapy and in the setting of autologous stem cell transplant (autoSCT).16,17 At this point in time, the MIPI score is not used to stratify treatment, although some clinical trials are incorporating the use of the MIPI score at diagnosis. Nonetheless, given its prognostic importance, the MIPI should be performed for all MCL patients undergoing staging and evaluation for treatment to establish disease risk.
As noted, the proliferative signature, represented by the Ki-67 protein, is also highly prognostic in MCL. Ki-67 is expressed in the late G1, S, G2, and M phases of the cell cycle. The Ki-67 index is defined by the hematopathologist as the percentage of lymphoma cells staining positive for Ki-67 protein, based on the number of cells per high-power field. There is significant interobserver variability in this process, which can be minimized by assessing Ki-67 quantitatively using computer software. The prognostic significance of Ki-67 at diagnosis was established in large studies of MCL patient cohorts, with survival differing by up to 3 years.18,19 Determann et al demonstrated the utility of the proliferative index in patients with MCL treated with standard chemoimmunotherapy.10 In this study, 249 patients with advanced-stage MCL treated within randomized trials conducted by the European MCL Network were analyzed. The Ki-67 index was found to be extremely prognostic of OS, independent of other clinical risk factors, including the MIPI score. As a continuous variable, Ki-67 indices of greater than 10% correlated with poor outcomes. The Ki-67 index has also been confirmed as prognostic in relapsed MCL.20 It is important to note that, as a unique feature, the Ki-67 index has remained an independent prognostic factor, even when incorporated into the “B” MIPI.
TREATMENT
CASE CONTINUED
The patient undergoes an excisional biopsy of a cervical lymph node, which demonstrates an abnormal proliferation of small-medium–sized lymphocytes with slightly irregular nuclear contours. Immunohistochemistry shows that the abnormal lymphocytes are positive for CD20 and CD5, negative for CD10 and CD23, and diffusely positive for cyclin D1, consistent with a diagnosis of MCL. The proliferative index, as measured by the Ki-67 immunostain, is 40%. A bone marrow aspirate and biopsy are then obtained, which show a clonal population of B lymphocytes expressing the same immunophenotype as the lymph node (positive for CD20 and CD5, negative for CD10 and CD23, cyclin D1 positive). A CT scan of the neck, chest, abdomen, and pelvis with contrast is obtained, along with a PET scan. These studies identify extensive hypermetabolic lymphadenopathy in the bilateral cervical chains, supraclavicular areas, mediastinum, and hilum. Mesenteric lymph nodes are also enlarged and hypermetabolic, as are retroperitoneal lymph nodes. The spleen is noted to be enlarged with multiple hypermetabolic lesions. Based on the presence of extensive lymphadenopathy as well as bone marrow involvement, the patient is diagnosed with stage IV MCL. He undergoes risk-stratification with the MIPI. His MIPI score is 6.3, high risk.
• What is the approach to upfront therapy for MCL?
FRONTLINE THERAPY
Role of Watchful Waiting
A small proportion of MCL patients have indolent disease that can be observed. This population is more likely to have leukemic-phase MCL with circulating lymphocytes, splenomegaly, and bone marrow involvement and absent or minimal lymphadenopathy.4,5 A retrospective study of 97 patients established that deferment of initial therapy in MCL is acceptable in some patients.5 In this study, approximately one third of patients with MCL were observed for more than 3 months before initiating systemic therapy, and the median time to treatment for the observation group was 12 months. Most patients undergoing observation had a low-risk MIPI. Patients were not harmed by observation, as no OS differences were observed among groups. This study underscores that deferred treatment can be an acceptable alternative in selected MCL patients for a short period of time. In practice, the type of patient who would be appropriate for this approach is someone who is frail, elderly, and with multiple comorbidities. Additionally, expectant observation could be considered for patients with limited-stage or low-volume MCL, low Ki-67 index, and low-risk MIPI scores.
Approach to Therapy
Treatment of MCL is generally approached by evaluating patient age and fitness for treatment. While there is no accepted standard, for younger patients healthy enough to tolerate aggressive approaches, treatment often involves an intensive cytarabine-containing regimen, which is consolidated with an autoSCT. This approach results in the longest remission duration, with some series suggesting a plateau in survival after 5 years, with no relapses.21 Nonintensive conventional chemotherapy alone is often reserved for the frailer or older patient. Given that remission durations with chemotherapy alone in MCL are short, goals of treatment focus on maximizing benefit and remission duration and minimizing risk of toxicity.
Standard Chemotherapy: Elderly and/or Frail Patients
Conventional chemotherapy alone for the treatment of MCL results in a 70% to 85% overall response rate (ORR) and 7% to 30% complete response (CR) rate.22 Rituximab, a mouse humanized monoclonal IgG1 anti-CD20 antibody, is used as standard of care in combination with chemotherapy, since its addition has been found to increase response rates and extend both progression-free survival (PFS) and OS compared to chemotherapy alone.23,24 However, chemoimmunotherapy approaches do not provide long-term control of MCL and are considered noncurative. Various regimens have been studied and include anthracycline-containing regimens such as R-CHOP (rituximab with cyclophosphamide, doxorubicin, vincristine, prednisone),22 combination chemotherapy with antimetabolites such as R-hyper-CVAD (hyper-fractionated rituximab with cyclophosphamide, vincristine, doxorubicin, dexamethasone, alternating with methotrexate and cytarabine),25 purine analogue–based regimens such as R-FC (rituximab with fludarabine and cyclophosphamide),26 bortezomib-containing regimens,27 and alkylator-based treatment with BR (bendamustine and rituximab) (Table 1).28,29 Among these, the most commonly used are R-CHOP and BR.
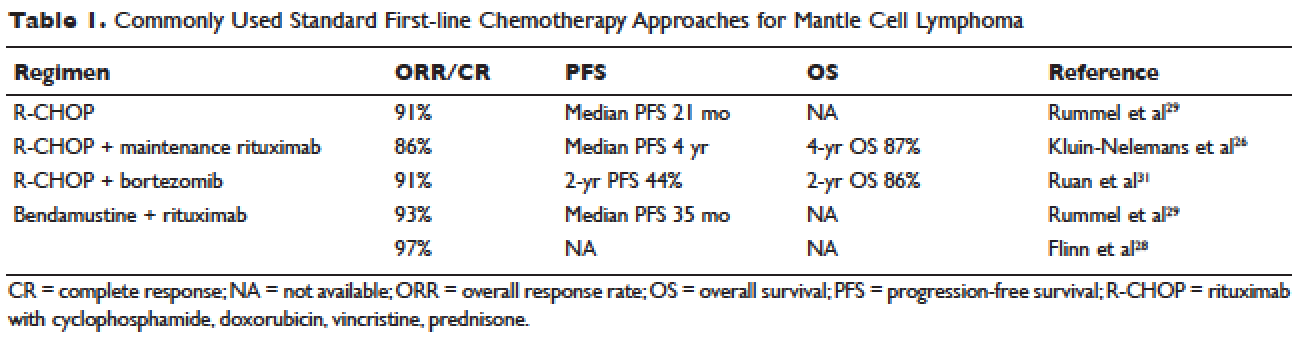
Two large randomized studies compared R-CHOP for 6 cycles to BR for 6 cycles in patients with indolent NHL and MCL. Among MCL patients, BR resulted in superior PFS compared to R-CHOP (69 months versus 26 months) but no benefit in OS.28,29 The ORR to R-CHOP was approximately 90%, with a PFS of 21 months in the Rummel et al study.29 This study included more than 80 centers in Germany and enrolled 549 patients with MCL, follicular lymphoma, small lymphocytic lymphoma, marginal zone lymphoma, and Waldenström macroglobulinemia. Patients were randomized in a 1:1 fashion. Among these, 46 patients received BR and 48 received R-CHOP (18% for both, respectively). It should be noted that patients in the BR group had significantly less toxicity and experienced fewer side effects than did those in the R-CHOP group. Similarly, BR-treated patients had a lower frequency of hematologic side effects and infections of any grade. However, drug-associated skin reactions and allergies were more common with BR compared to R-CHOP. The study by Flinn and colleagues was an international randomized, noninferiority phase 3 study designed to evaluate the efficacy and safety of BR compared with R-CHOP or R-CVP (rituximab plus cyclophosphamide, vincristine, and prednisone) for treatment-naive patients with MCL or other indolent NHL. The primary endpoint was CR. In this study, BR was found to be noninferior to R-CHOP and R-CVP based on CR rate (31% versus 25%, respectively; P = 0.0225). Response rates in general were high: 97% for BR and 91% for R-CHOP/R-CVP (P = 0.0102). Here, BR-treated patients experienced more nausea, emesis, and drug-induced hypersensitivity compared to the R-CHOP and R-CVP groups.
Another approach studied in older patients is the use of R-CHOP with rituximab maintenance. In a large European study, 560 patients 60 years of age or older with advanced-stage MCL were randomly assigned to either R-FC (rituximab, fludarabine, and cyclophosphamide) every 28 days for 6 cycles, or R-CHOP every 21 days for 8 cycles. Patients who had a response then underwent a second randomization, with one group receiving rituximab maintenance therapy. Maintenance was continued until progression of disease. Patients in this study were not eligible for high-dose chemotherapy and autoSCT. The study found that rates of CR were similar with both R-FC and R-CHOP (40% and 34%, respectively; P = 0.10). However, the R-FC arm underperformed in several arenas. Disease progression occurred more frequently with R-FC (14% versus 5% with R-CHOP), and OS was shorter (4-year OS, 47% versus 62%; P = 0.005, respectively). More patients also died in the R-FC group, and there was greater hematologic toxicity compared to R-CHOP. At 4 years, 58% of the patients receiving rituximab remained in remission. Among patients who responded to R-CHOP, rituximab maintenance led to a benefit in OS, reducing the risk of progression or death by 45%.26 At this time, studies are ongoing to establish the benefit of rituximab maintenance after BR.
Bendamustine in combination with other agents has also been studied in the frontline setting. Visco and colleagues evaluated the combination of bendamustine with rituximab and cytarabine (R-BAC) in older patients with MCL (age 65 or older).63 This phase 2, two-stage study enrolled 40 patients and had a dose-finding arm for cytarabine in combination with BR. It permitted relapsed/refractory patients, but 50% had newly diagnosed, previously untreated MCL. The regimen had an impressive ORR of 100%, with CR rates of 95% for previously untreated patients. PFS at 2 years was 95%. R-BAC was well tolerated, with the primary toxicity being reversible myelosuppression.
BR was combined with the proteasome inhibitor bortezomib and dexamethasone in a phase 2 study.64 This Lymphoma Study Association (LYSA) study evaluated 76 patients with newly diagnosed MCL older than age 65 years. BR was administered in standard doses (bendamustine 90 mg/m2 on days 1 and 2 and rituximab 375 mg/m² IV on day 1) and bortezomib was administered subcutaneously on days 1, 4, 8, and 11, with acyclovir for viral prophylaxis. Patients received 6 cycles. The ORR was 87% and the CR was 60%. Patients experienced toxicity, and not all bortezomib doses were administered due to neurotoxic or hematologic side effects.
A randomized phase 3 study compared R-CHOP to the VR-CAP regimen (R-CHOP regimen but bortezomib replaces vincristine on days 1, 4, 8, 11, at 1.3 mg/m2) in 487 newly diagnosed MCL patients.27 Median PFS was superior in the VR-CAP group compared with R-CHOP (14.4 months versus 24.7 months, respectively). Additionally, rates of CR were superior in the VR-CAP group (53% compared to 42% with R-CHOP). However, there was more hematologic toxicity with VR-CAP. On the basis of these findings, the U.S. Food and Drug Administration approved bortezomib for the frontline treatment of MCL.
Other chemoimmunotherapy combinations containing bortezomib have been studied in frontline MCL treatment, with promising results. These include bortezomib in combination with R-CHOP or modified R-hyper-CVAD, as well as bortezomib in combination with CHOP-like treatments and purine analogues.27,30–32 The ongoing ECOG 1411 study is currently evaluating bortezomib added to BR for induction therapy of newly diagnosed MCL in a 4-arm randomized trial. Patients receive BR with or without bortezomib during induction and are then randomly assigned to maintenance with either rituximab alone or rituximab with lenalidomide. Other novel combination agents are actively being studied in frontline MCL treatment, including lenalidomide and rituximab and BR with lenalidomide.
Intensification of Therapy and AutoSCT: Fitter and/or Younger Patients
Short response duration has created the need for post-remission therapy in MCL. One approach to improve remission duration in MCL is to intensify induction through the use of cytarabine-containing regimens and/or consolidation with high-dose chemotherapy, typically using BEAM (carmustine, etoposide, cytarabine, melphalan) and autoSCT (Table 2). The cytarabine-containing R-hyper-CVAD regimen, developed at the MD Anderson Cancer Center, resulted in a 97% ORR and an 87% CR rate, with TTF of nearly 5 years. However, nearly one third of patients were unable to complete treatment due to toxicity, and 5 patients developed secondary myelodysplastic syndrome or acute myeloid leukemia.33 The feasibility of this R-hyper-CVAD regimen was tested in a multicenter cooperative group setting, but similar results were not seen; in this study, nearly 40% of patients were unable to complete the full scheduled course of treatment due to toxicity.34
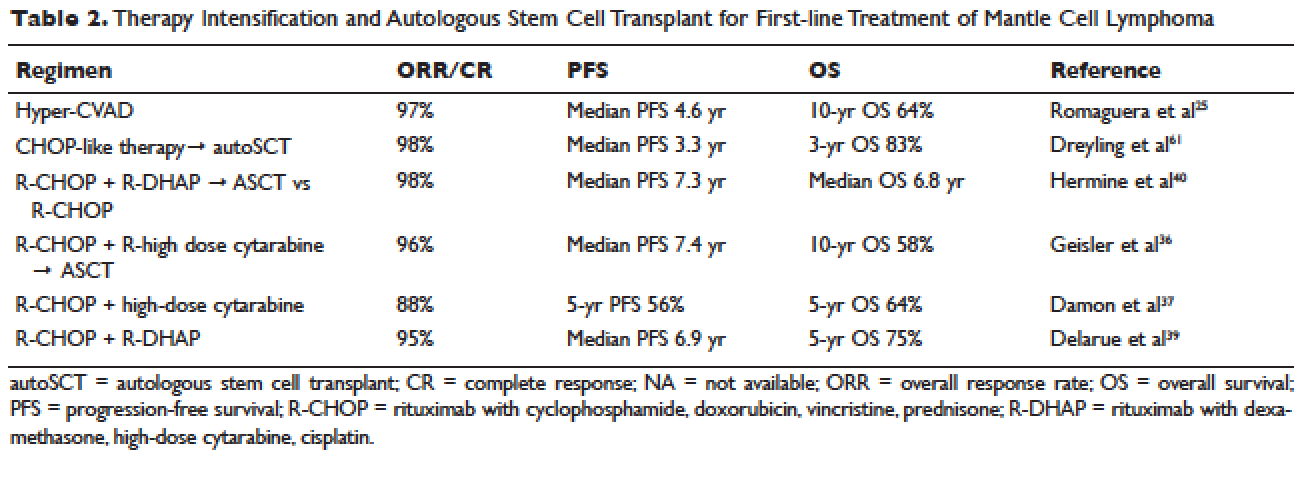
Other ways to intensify therapy in MCL involve adding a second non-cross-resistant cytarabine-containing regimen to R-CHOP after remission, such as DHAP (dexamethasone, high-dose cytarabine, cisplatin), followed by consolidation with an autoSCT. A retrospective registry from the National Comprehensive Cancer Network sought to compare the efficacy of different treatment approaches in the frontline setting. They studied 167 patients with MCL and compared 4 groups: treatment with R-hyper-CVAD, either with or without autoSCT, and treatment with R-CHOP, either with or without autoSCT. This study found that in patients younger than 65, R-CHOP followed by autoSCT or R-hyper-CVAD without autoSCT resulted in similar PF and OS, but was superior to R-CHOP alone for newly diagnosed MCL patients.35 These data support more intensive regimens in younger and fitter patients. Several other prospective and randomized studies have demonstrated clinical benefit for patients with MCL undergoing autoSCT in first remission. Of particular importance is the seminal phase 3 study of the European MCL Network, which established the role of autoSCT in this setting.61 In this prospective randomized trial involving 122 newly diagnosed MCL patients who responded to CHOP-like induction, patients in CR derived a greater benefit from autoSCT.
More recent studies have demonstrated similar benefits using cytarabine-based autoSCT. The Nordic MCL2 study evaluated 160 patients using R-CHOP, alternating with rituximab and high-dose cytarabine, followed by autoSCT. This study used “maxi-CHOP,” an augmented CHOP regimen (cyclophosphamide 1200 mg/m2, doxorubicin 75 mg/m2, but standard doses of vincristine [2 mg] and prednisone [100 mg days 1–5]), alternating with 4 infusions of cytarabine at 2 g/m2 and standard doses of rituximab (375 mg/m2). Patients then received conditioning with BEAM and autoSCT. Patients were evaluated for the presence of minimal residual disease (MRD) and for the t(11;14) or clonal immunoglobulin heavy chain gene rearrangement with polymerase chain reaction (PCR). Patients with MRD were offered therapy with rituximab at 375 mg/m2 weekly for 4 doses. This combination resulted in 10-year OS rates of 58%.36 In a multicenter study involving 78 patients from the Cancer and Leukemia Group B (CALGB), R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT resulted in a 5-year OS of 64%.37 A single-arm phase 2 study from the Netherlands also tested R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT. Nonhematologic toxicities were 22% after high-dose cytarabine, and 55% after BEAM. The ORR was 70%, with a 64% CR rate and 66% OS at 4 years.38 The French GELA group used 3 cycles of R-CHOP and 3 cycles of R-DHAP in a phase 2 study of young (under age 66) MCL patients. Following R-CHOP, the ORR was 93%, and following R-DHAP the ORR was 95%. Five-year OSA was 75%.39 A large randomized phase 3 study by Hermine and colleagues of the EMCLN confirmed the benefit of this approach in 497 patients with newly diagnosed MCL. R-CHOP for 6 cycles followed by autoSCT was compared to R-CHOP for 3 cycles alternating with R-DHAP for 3 cycles and autoSCT with a cytarabine-based conditioning regimen. The addition of cytarabine significantly increased rates of CR, TTF, and OS, without increasing toxicity.40
CASE CONTINUED
The patient is treated with R-CHOP chemotherapy for 3 cycles followed by R-DHAP. His course is complicated by mild tinnitus and acute kidney injury from cisplatin that promptly resolves. Three weeks following treatment, a restaging PET/CT scan shows resolution of all lymphadenopathy, with no hypermetabolic uptake, consistent with a complete remission. A repeat bone marrow biopsy shows no involvement with MCL. He subsequently undergoes an autoSCT, and restaging CT/PET 3 months following autoSCT shows continued remission. He is monitored every 3 to 6 months over the next several years.
He has a 4.5-year disease remission, after which he develops growing palpable lymphadenopathy on exam and progressive anemia and thrombocytopenia. A bone marrow biopsy is repeated, which shows recurrent MCL. Restaging diagnostic imaging with a CT scan reveals lymphadenopathy above and below the diaphragm. An axillary lymph node biopsy also demonstrates recurrent MCL. At this time the patient is otherwise in fairly good health, except for feeling fatigued. His ECOG performance status is 1. He begins therapy with bortezomib at a dose of 1.3 mg/m2 intravenously on days 1, 4, 8, and 11 for 6 cycles. His treatment course is complicated by painful sensory peripheral neuropathy of the bilateral lower extremities. Restaging studies at the completion of therapy demonstrate that he has achieved a partial response, with a 50% reduction in the size of involved lymphadenopathy and some residual areas of hypermetabolic uptake. His peripheral cytopenias improve moderately.
• What are the therapeutic options for relapsed MCL?
TREATMENT OF RELAPSED MCL
Single-Agent and Combination Chemotherapy
Whenever possible, and since there is no standard, patients with relapsed MCL should be offered a clinical trial. Outside of a clinical study, many of the treatment regimens used at diagnosis can also be applied in the relapsed setting. In relapsed MCL, Rummel et al showed that BR for 4 cycles resulted in an ORR of 90%, with a CR of 60%. The median PFS was 24 months.41 Bortezomib, an inhibitor of the proteasome-ubiquitin pathway, leads to apoptosis and cell cycle arrest in MCL.42 Multiple studies have evaluated bortezomib both as a single agent and in combination for patients with relapsed MCL. In 2006, bortezomib became the first agent approved by the FDA in relapsed or refractory MCL, based on the phase 2 PINNACLE study. This prospective multicenter study involving 155 patients demonstrated an ORR of 33%, CR rate of 8%, and median treatment duration of 9 months. The median time to progression was 6 months.43 Subsequently, bortezomib-containing combinations evolved. In a multicenter study of relapsed and refractory indolent NHL and MCL, Friedberg and colleagues evaluated bortezomib in combination with BR.44 In the MCL cohort, the ORR was 71%. These promising results led to the study of this combination in the frontline setting. The ongoing ECOG 1411 study is using BR for the frontline treatment of MCL with or without bortezomib as induction. This study also includes rituximab maintenance, and randomizes patients to undergo maintenance with or without the immunomodulator lenalidomide. Bortezomib has been associated with herpes simplex and herpes zoster reactivation. Neuropathy has also been observed with bortezomib, which can be attenuated by administering it subcutaneously.
Lenalidomide is an immunomodulatory agent derived from thalidomide. It has significant activity and is a mainstay of treatment in multiple myeloma. Lenalidomide acts by enhancing cellular immunity, has antiproliferative effects, and inhibits T-cell function leading to growth inhibitory effects in the tumor microenvironment.45 In MCL, lenalidomide has demonstrated clinical activity both as a single agent and in combination, as well as in preclinical studies establishing its pro-apoptotic effects.46 The pivotal EMERGE study evaluated monotherapy with lenalidomide in heavily pretreated relapsed and refractory MCL. This multicenter international study of 134 patents reported an ORR of 28% with a 7.5% CR rate and median PFS of 4 months. All patients had relapsed or progressed following bortezomib. This led to the approval of lenalidomide by the FDA in 2013 for the treatment of patients with MCL whose disease relapsed or progressed following 2 prior therapies, one of which included bortezomib.47 Lenalidomide has been associated with neutropenia, secondary cancers, and deep venous thrombosis.
In combination with other agents in the relapsed setting, lenalidomide shows broader activity. A phase 1/2 study by Wang and colleagues demonstrated an ORR of 57%; the median response duration was 19 months when lenalidomide was combined with rituximab for relapsed/refractory MCL.48
Novel Therapies
More recently, novel treatment approaches have been tested in MCL based on an increased understanding of aberrant signaling pathways in this disease (Table 3). Constitutive activation of B-cell receptor signaling is critical for the survival and proliferation of lymphomas, and has led to the development of targeted agents inhibiting B-cell receptor–associated protein kinases. Bruton’s tyrosine kinase (BTK) is one essential component of the B-cell receptor.49 In particular, proteins upstream of the BTK pathway have been implicated in growth and proliferation of MCL, suggesting that inhibition of BTK may impede lymphomagenesis.50 Ibrutinib is an oral inhibitor of BTK, and demonstrates activity in multiple lymphoma subtypes. In a phase 1 study of ibrutinib in relapsed and refractory hematologic malignancies, an ORR of 60% was observed in 50 evaluable patients, with 16% CR. Median PFS was 13 months. Among these, 7 of 9 patients with MCL responded, including 3 CRs.51 Given these promising results, a phase 2 multicenter study evaluating ibrutinib in relapsed and refractory MCL was completed.52 At a dose of 560 mg daily, the response rate was 68%, with CR of 21%. The most common observed treatment-related side effects included diarrhea, fatigue, and nausea. Neutropenia and thrombocytopenia were also observed. Of importance, 5% of patients had grade 3 or higher bleeding events, including subdural hematoma, gastrointestinal bleeding, and hematuria. The estimated OS rate was 58% at 18 months. On the basis of this study, the FDA approved ibrutinib for relapsed and refractory MCL in November 2013.
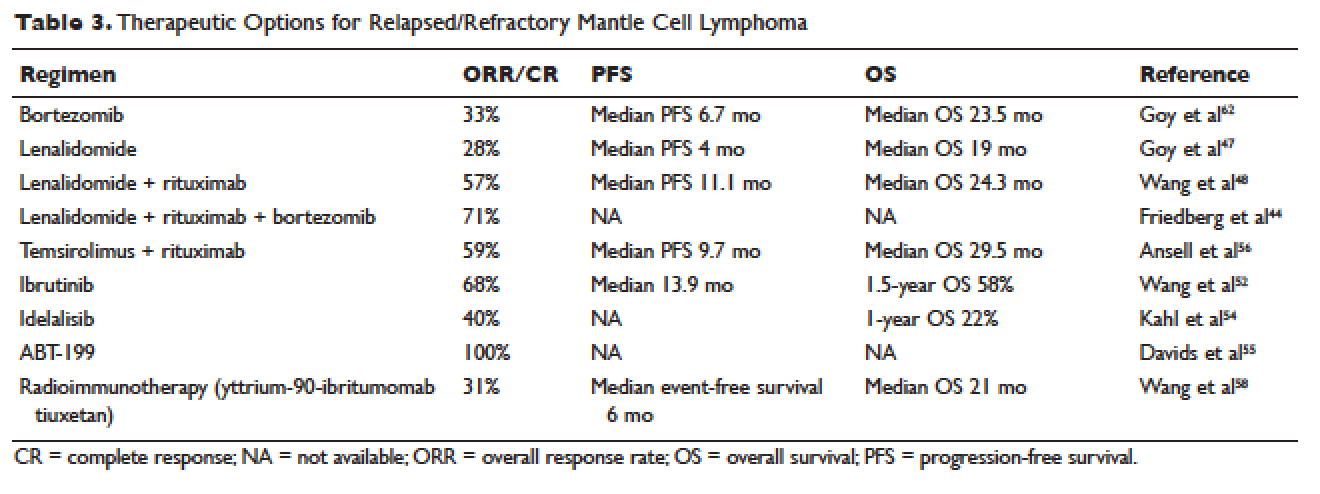
The PI3K pathway is another survival pathway that is dysfunctional in several hematologic disorders, including MCL. Overexpression of PI3K and its downstream targets contributes to MCL pathogenesis.53 Idelalisib is an oral small molecule inhibitor of the delta isoform of PI3K that is dosed daily; it was approved by the FDA for the treatment of relapsed and refractory follicular lymphoma, small lymphocytic lymphoma, and chronic lymphocytic leukemia. It is being further evaluated in MCL. A dose-escalation phase 1 study in heavily pre-treated MCL patients established safety and tolerability.54 Efficacy analysis showed an ORR of 40%, CR of 5%, and 1-year OS of 22%. Further phase 2 studies testing idelalisib as a single agent and in combination for MCL are ongoing. Side effects of idelalisib include elevated liver enzymes, pneumonitis, and diarrhea.
The BCL family of proteins is involved in both pro-and anti-apoptotic functions. BCL2 is an intracellular protein that blocks apoptosis. ABT-199 is an oral BCL2 inhibitor that in early clinical trials has shown very promising activity in MCL. In a phase 1 study of 31 relapsed and refractory NHL patients, all 8 MCL patients (100% ORR) responded to ABT-199 therapy.55 Given these promising initial results, multiple studies evaluating ABT-199 are ongoing in MCL as part of first-line treatment as well as for relapsed disease. ABT-199 has been implicated in tumor lysis syndrome, and in early studies of chronic lymphocytic leukemia, fatal tumor lysis was observed.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus has been evaluated in relapsed MCL. It is given weekly at 250 mg intravenously. Response rates to single-agent temsirolimus are approximately 20% to 35%, and are higher when combined with rituximab.56,57 The phase 2 study evaluating temsirolimus as a single agent enrolled 35 heavily pre-treated patients. ORR was 38% with only 1 CR. The duration of response was 7 months. Temsirolimus is approved for relapsed MCL in Europe but not in the United States. Similar to the other targeted agents, temsirolimus is actively being studied in combination with other active agents in MCL. Adverse effects noted with temsirolimus include diarrhea, stomatitis, and rash. Thrombocytopenia requiring dose reductions is another frequently observed complication.
Radioimmunotherapy
Radioimmunotherapy (RIT) has been studied extensively in MCL. RIT consists of anti-CD 20 antibodies coupled to radioactive particles that deliver radiation to targeted cells, minimizing toxicity to surrounding tissues. RIT is not used as frequently in the modern era as it had been in the past. At this time, only yttrium-90-ibritumomab tiuxetan is available.
RIT has been evaluated in MCL both at the time of relapse58 and more recently, as part of a conditioning regimen prior to autoSCT, with good tolerability.65–67 Averse events noted with RIT include hematologic toxicity (can be prolonged), hypothyroidism, and in rare cases, myelodysplastic syndrome and acute leukemia. The bone marrow must have less than 25% involvement with disease prior to administration. Wang and colleagues evaluated yttrium-90-ibritumomab tiuxetan in 34 heavily pretreated patients with MCL.58 They observed an ORR of 31%. The median event-free survival (EFS) was 6 months, but in patients achieving either CR or PR, EFS was 28 months. A 21-month OS was noted.
In the upfront setting, RIT has been added as a mechanism of intensification. A recent Nordic group study of RIT with autoSCT did not find benefit with the addition of RIT.59 An ECOG study recently added yttrium-90-ibritumomab tiuxetan after CHOP chemotherapy in the upfront treatment of MCL, with good tolerability.55 However, when added to R-hyper-CVAD, the combination had unexpected high rates of hematologic toxicity, including grade 3/4 cytopenias and an unacceptably high rate of secondary malignancies.68
AutoSCT or Allogeneic Transplant
While many studies noted above have established the beneficial role of autoSCT in MCL in first remission, the role of allogeneic transplant (alloSCT) in MCL remains controversial. A recent large retrospective study conducted by the Center for International Blood and Marrow Transplant Research (CIBMTR) evaluated 519 patients with MCL who underwent both autoSCT and alloSCT.60 Patients were grouped into an early cohort (transplant in first PR or CR, and 2 or fewer treatments) and late cohort (all other patients). The analysis had mature follow up. A multivariate analysis demonstrated that early autoSCT was associated with superior outcomes compared to autoSCT performed later. While it was not possible to demonstrate a survival benefit favoring autoSCT over reduced intensity (RIC) alloSCT, patients transplanted later in their disease course had shorter OS. For patients receiving autoSCT in CR 1 following only 1 prior line of therapy, OS at 5 years was 75% and PFS was 70%. Patients undergoing RIC followed by alloSCT had fewer relapses, but this was negated by higher nonrelapse mortality (25%), resulting in a PFS similar to autoSCT.
CASE CONCLUSION
After treatment with bortezomib the patient is well for 9 months. Subsequently, however, he develops increasing lymphadenopathy and progressive fatigue. He is then started on lenalidomide 25 mg orally daily for 21 out of 28 days. He experiences significant fatigue with lenalidomide and prolonged neutropenia requiring dose delays, despite dose modification to 10 mg orally daily. He requires discontinuation of lenalidomide. Given persistent disease, the patient then begins treatment with ibrutinib. Within a few days of starting ibrutinib therapy, he experiences a marked but transient leukocytosis. Two months later, the patient’s palpable lymphadenopathy has decreased, and his anemia and thrombocytopenia related to MCL are improving. He has tolerated treatment well. His course has been complicated only by a mild, pruritic maculopapular eruption on his chest, back, and arms, that was responsive to topical low-dose steroids. He remains on ibrutinib 1 year later.
CONCLUSION
Advances in our understanding of MCL treatment are revolutionizing the approach to this once deadly disease. Over the next several years, these gains will weave themselves into the current treatment paradigm and likely alter the treatment landscape for MCL as we know it.
- Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
- Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer 2008;113:791–8.
- Geisler CH. Front-line treatment of mantle cell lymphoma. Haematologica 2010;95:1241–3.
- Fernandez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res 2010;70:1408–18.
- Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
- Bertoni F, Ponzoni M. The cellular origin of mantle cell lymphoma. Int J Biochem Cell Biol 2007;39:1747–53.
- de Boer CJ, van Krieken JH, Kluin-Nelemans HC, et al. Cyclin D1 messenger RNA overexpression as a marker for mantle cell lymphoma. Oncogene 1995;10:1833–40.
- Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest 2012;122:3416–23.
- Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185–97.
- Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood 2008;111:2385–7.
- Vegliante MC, Palomero J, Perez-Galan P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood 2013;121:2175–85.
- Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 2013;110:18250–5.
- Zelenetz AD, Abramson JS, Advani RH, et al. Non- Hodgkin’s lymphomas. J Natl Compr Canc Netw 2011;9: 484–560.
- Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
- Zelenetz AD, Abramson JS, Advani RH, et al. NCCN Clinical Practice Guidelines in Oncology: non-Hodgkin’s lymphomas. J Natl Compr Canc Netw 2010;8:288–334.
- Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
- Geisler CH, Kolstad A, Laurell A, et al. The Mantle Cell Lymphoma International Prognostic Index (MIPI) is superior to the International Prognostic Index (IPI) in predicting survival following intensive first-line immunochemotherapy and autologous stem cell transplantation (ASCT). Blood 2010;115:1530–3.
- Tiemann M, Schrader C, Klapper W, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 2005;131:29–38.
- Raty R, Franssila K, Joensuu H, et al. Ki-67 expression level, histological subtype, and the International Prognostic Index as outcome predictors in mantle cell lymphoma. Eur J Haematol 2002;69:11–20.
- Vogt N, Klapper W. Variability in morphology and cell proliferation in sequential biopsies of mantle cell lymphoma at diagnosis and relapse: clinical correlation and insights into disease progression. Histopathology 2013;62:334–42.
- Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 2008;112:2687–93.
- Howard OM, Gribben JG, Neuberg DS, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol 2002;20:1288–94.
- Griffiths R, Mikhael J, Gleeson M, et al. Addition of rituximab to chemotherapy alone as first-line therapy improves overall survival in elderly patients with mantle cell lymphoma. Blood 2011;118:4808–16.
- Lenz G, Dreyling M, Hoster E, et al. immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
- Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol 2005;23:7013–23.
- Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
- Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 2015;372:944–53.
- Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
- Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
- Houot R, Le Gouill S, Ojeda Uribe M, et al. Combination of rituximab, bortezomib, doxorubicin, dexamethasone and chlorambucil (RiPAD+C) as first-line therapy for elderly mantle cell lymphoma patients: results of a phase II trial from the GOELAMS. Ann Oncol 2012;23:1555–61.
- Ruan J, Martin P, Furman RR, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol 2011;29:690–7.
- Chang JE, Li H, Smith MR, et al. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: an Eastern Cooperative Oncology Group study (E1405). Blood 2014; 123:1665–73.
- Romaguera JE, Fayad LE, Feng L, et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol 2010;150:200–8.
- Bernstein SH, Epner E, Unger JM, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol 2013;24:1587–93.
- LaCasce AS, Vandergrift JL, Rodriguez MA, et al. Comparative outcome of initial therapy for younger patients with mantle cell lymphoma: an analysis from the NCCN NHL Database. Blood 2012;119:2093–9.
- Geisler CH, Kolstad A, Laurell A, et al. Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol 2012;158:355–62.
- Damon LE, Johnson JL, Niedzwiecki D, et al. Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol 2009;27:6101–8.
- van ‘t Veer MB, de Jong D, MacKenzie M, et al. High-dose Ara-C and beam with autograft rescue in R-CHOP responsive mantle cell lymphoma patients. Br J Haematol 2009;144:524–30.
- Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
- Hermine O, Hoster E, Walewski J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) increases overall survival when compared to 6 courses of CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: final analysis of the MCL Younger Trial of the European Mantle Cell Lymphoma Network (MCL net). In: American Society of Hematology Proceedings. December 8–11, 2012; Atlanta, GA. Abstract 151.
- Rummel MJ, Al-Batran SE, Kim SZ, et al. Bendamustine plus rituximab is effective and has a favorable toxicity profile in the treatment of mantle cell and low-grade non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:3383–9.
- Pham LV, Tamayo AT, Yoshimura LC, et al. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol 2003;171:88–95.
- Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
- Friedberg JW, Vose JM, Kelly JL, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood 2011;117:2807–12.
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–22.
- Qian Z, Zhang L, Cai Z, et al. Lenalidomide synergizes with dexamethasone to induce growth arrest and apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leuk Res 2011;35:380–6.
- Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
- Wang M, Fayad L, Wagner-Bartak N, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 2012;13:716–23.
- Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 2012;31: 119–32.
- Rinaldi A, Kwee I, Taborelli M, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol 2006;132:303–16.
- Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31:88–94.
- Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
- Rudelius M, Pittaluga S, Nishizuka S, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006;108: 1668–76.
- Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014;123:3398–405.
- Davids MS, Seymour JF, Gerecitano JF, et al. Updated results of a phase I first in human study of the BCL-2inhibitor ABT-199 in patients with relapsed/refractory NHL. J Clin Oncol 31, 2013 (suppl; abstr 8520).
- Ansell SM, Tang H, Kurtin PJ, et al. Temsirolimus and rituximab in patients with relapsed or refractory mantle cell lymphoma: a phase 2 study. Lancet Oncol 2011;12:361–8.
- Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–56.
- Wang M, Oki Y, Pro B, et al. Phase II study of yttrium-90-ibritumomab tiuxetan in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27:5213–8.
- Kolstad A, Laurell A, Jerkeman M, et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood 2014;123:2953–9.
- Fenske TS, Zhang MJ, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
- Dreyling M, Lenz G, Hoster E, et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 2005;105:2677–84.
- Goy A, Younes A, McLaughlin P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:667–75.
- Visco C, Finotto S, Zambello R, et al. Combination of rituximab, bendamustine, and cytarabine for patients with mantle-cell non-Hodgkin lymphoma ineligible for intensive regimens or autologous transplantation. J Clin Oncol 2013;10;31:1442–9.
- Gressin R, Callanan M, Daguindau N, et al. The Ribvd regimen (Rituximab IV, Bendamustine IV, Velcade SC, Dexamethasone IV) offers a high complete response rate In elderly patients with untreated mantle cell lymphoma. Preliminary results of the Lysa trial “Lymphome Du Manteau 2010 SA.” Blood 2013;122:370.
- Krishnan A, Nademanee A, Fung HC, et al. Phase II trial of a transplantation regimen of yttrium-90 ibritumomab tiuxetan and high-dose chemotherapy in patients with non-Hodgkin’s lymphoma. J Clin Oncol 2008;26:90–5.
- Nademanee A, Forman S, Molina A, et al. A phase 1/2 trial of high-dose yttrium-90-ibritumomab tiuxetan in combination with high-dose etoposide and cyclophosphamide followed by autologous stem cell transplantation in patients with poor-risk or relapsed non-Hodgkinlymphoma. Blood 2005;106:2896–902.
- Shimoni A, Avivi I, Rowe JM, et al. A randomized study comparing yttrium-90 ibritumomab tiuxetan (Zevalin) and high-dose BEAM chemotherapy versus BEAM alone as the conditioning regimen before autologous stem cell transplantation in patients with aggressive lymphoma. Cancer 2012;118:4706–14.
- Arranz R, García-Noblejas A, Grande C, et al. First-line treatment with rituximab-hyperCVAD alternating with rituximab-methotrexate-cytarabine and followed by consolidation with 90Y-ibritumomab-tiuxetan in patients with mantle cell lymphoma. Results of a multicenter, phase 2 pilot trial from the GELTAMO group. Haematologica 2013;98:1563-70.
INTRODUCTION
Mantle cell lymphoma (MCL) is an uncommon, distinct clinical subtype of non-Hodgkin lymphoma (NHL) that comprises approximately 8% of all lymphoma diagnoses in the United States and Europe.1,2 Considered incurable, MCL often presents in advanced stages, particularly with involvement of the lymph nodes, spleen, bone marrow, and gastrointestinal tract in the form of lymphomatous polyps. MCL disproportionately affects males, and incidence rises with age, with a median age at diagnosis of 68 years.2 Historically, the prognosis of patients with MCL has been among the poorest among B-cell lymphoma patients, with a median overall survival (OS) of 3 to 5 years, and time to treatment failure (TTF) of 18 to 24 months, although this is improving in the modern era.3 Less frequently, patients with MCL display isolated bone marrow, peripheral blood, and splenic involvement. These cases tend to behave more indolently with longer survival.4,5 Recent advances in therapy have dramatically impacted treatment alternatives and outcomes for MCL. As such, the therapeutic and prognostic landscape of MCL is evolving rapidly.
PATHOGENESIS
The histologic diagnosis of MCL by morphology alone is often challenging. Accurate diagnosis relies on immunohistochemical staining for the purposes of immunophenotyping.6 MCL typically expresses B-cell markers CD5 and CD20, and lacks both CD10 and CD23. The genetic hallmark of MCL is the t(11;14) (q13;q32) chromosomal translocation leading to upregulation of the cyclin D1 protein, a critical regulator of the G1 phase of the cell cycle. Specifically, the t(11;14) translocation, present in virtually all cases of MCL, juxtaposes the proto-oncogene CCND1 to the immunoglobulin heavy chain gene.7 Consequently, cyclin D1, normally not expressed in B lymphocytes, becomes constitutively overexpressed. This alteration is thought to facilitate the deregulation of the cell cycle at the G1-S phase transition.8
Gene expression profiling studies have underscored the importance of cell cycle deregulation in MCL, and high proliferation is associated with a worse prognosis.9 More than 50% of the genes associated with poor outcomes were derived from the “proliferation signature” that was more highly expressed in dividing cells. In the seminal Rosenwald study, a gene expression–based outcome model was constructed in which the proliferation signature average represents a linear variable that assigns a discrete probability of survival to an individual patient.9 The proliferative index, or proliferative signature, of MCL can be estimated by the percentage of Ki-67–positive cells present in the tumor through immunohistochemistry. This is often used as a marker of poor outcomes, and as a surrogate for the proliferative signature in MCL that can be incorporated into clinical practice (as opposed to gene expression profiling). Statistically significant differences in OS have emerged between groups of MCL patients with Ki-67–positive cells comprising less than 30% of their tumor sample (favorable) and those with Ki-67–positive cells comprising 30% or greater (unfavorable).10
Recent data has also identified the importance of the transcription factor SOX 11 (SRY-related HMG-box), which regulates multiple cellular transcriptional events, including cell proliferation and differentiation, apoptosis, and angiogenesis.11 MCL expressing SOX 11 behaves more aggressively than MCL variants lacking SOX 11 expression, and tends to accumulate more genetic alterations.12 Moreover, lack of SOX 11 expression characterizes a subset of MCL that does not carry the t(11;14) translocation.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 62-year-old man with a history of diabetes mellitus and hypertension presents with cervical lymphadenopathy, fatigue, and early satiety over the past several months. He is otherwise in good health. His Eastern Cooperative Oncology Group (ECOG) performance status is 1. On physical examination, 3-cm lymphadenopathy in the bilateral cervical chain is noted. Bilateral axillary lymph nodes measure 2 to 4 cm. His spleen is enlarged and is palpable at approximately 5 cm below the costal margin. A complete blood count reveals a total white blood cell (WBC) count of 14,000 cells/μL, with 68% lymphocytes and a normal distribution of neutrophils. Hemoglobin is 11 g/dL, and platelet count is 112,000/μL. The lactate dehydrogenase (LDH) level is 322 U/L (upper limit of normal: 225 U/L).
• How is MCL diagnosed?
Diagnosis of MCL requires review by expert hematopathologists.13 Whenever possible, an excisional biopsy should be performed for the adequate characterization of lymph node architecture and evaluation by immunohistochemistry. Aside from the characteristic expression of CD5 and CD20 and absence of CD23, MCL should express cyclin D1, which reflects t(11;14). If cyclin D1 is inconclusive or unavailable, fluorescent in situ hybridization (FISH) for t(11;14) should be performed.8 Patients often have circulating malignant lymphocytes, or leukemic phase MCL. Flow cytometry of the peripheral blood can detect traditional surface markers, and FISH can also be performed on circulating abnormal lymphocytes.
For disease staging, bone marrow biopsy and aspiration are required. Radiographic staging using computed tomography (CT) scans and/or positron emission tomography (PET) scans had traditionally followed the Ann Arbor staging system, but recently the Lugano classification has emerged, which delineates only early or advanced stage.14 Gastrointestinal evaluation of MCL with endoscopy and colonoscopy with blind biopsies has been recommended to evaluate for the presence of lymphomatous polyps, but this is not an absolute requirement.15
RISK STRATIFICATION
At diagnosis, patients should undergo risk stratification in order to understand prognosis and possibly guide treatment. In MCL, the MCL international prognostic index (MIPI) is used. The MIPI is a prognostic tool developed exclusively for patients with MCL using data from 455 patients with advanced-stage MCL treated within 3 European clinical trials.16 The MIPI classified patients into risk groups based on age, ECOG performance status, LDH level, and WBC count. Patients were categorized into low-risk (44% of patients, median OS not reached), intermediate-risk (35%, median OS 51 months), and high-risk groups (21%, median OS 29 months). This is done through a logarithmic calculation, which can be accessed through online calculators (a prototype example can be found at www.qxmd.com/calculate-online/hematology/prognosis-mantle-cell-lymphoma-mipi). Cell proliferation using the Ki-67 index was evaluated in an exploratory analysis (the biologic [“B”] MIPI), and also demonstrated strong prognostic relevance.16 Currently, treatment of MCL patients is not stratified by MIPI outside of a clinical trial, but this useful tool assists in assessing patient prognosis and has been validated for use with both conventional chemoimmunotherapy and in the setting of autologous stem cell transplant (autoSCT).16,17 At this point in time, the MIPI score is not used to stratify treatment, although some clinical trials are incorporating the use of the MIPI score at diagnosis. Nonetheless, given its prognostic importance, the MIPI should be performed for all MCL patients undergoing staging and evaluation for treatment to establish disease risk.
As noted, the proliferative signature, represented by the Ki-67 protein, is also highly prognostic in MCL. Ki-67 is expressed in the late G1, S, G2, and M phases of the cell cycle. The Ki-67 index is defined by the hematopathologist as the percentage of lymphoma cells staining positive for Ki-67 protein, based on the number of cells per high-power field. There is significant interobserver variability in this process, which can be minimized by assessing Ki-67 quantitatively using computer software. The prognostic significance of Ki-67 at diagnosis was established in large studies of MCL patient cohorts, with survival differing by up to 3 years.18,19 Determann et al demonstrated the utility of the proliferative index in patients with MCL treated with standard chemoimmunotherapy.10 In this study, 249 patients with advanced-stage MCL treated within randomized trials conducted by the European MCL Network were analyzed. The Ki-67 index was found to be extremely prognostic of OS, independent of other clinical risk factors, including the MIPI score. As a continuous variable, Ki-67 indices of greater than 10% correlated with poor outcomes. The Ki-67 index has also been confirmed as prognostic in relapsed MCL.20 It is important to note that, as a unique feature, the Ki-67 index has remained an independent prognostic factor, even when incorporated into the “B” MIPI.
TREATMENT
CASE CONTINUED
The patient undergoes an excisional biopsy of a cervical lymph node, which demonstrates an abnormal proliferation of small-medium–sized lymphocytes with slightly irregular nuclear contours. Immunohistochemistry shows that the abnormal lymphocytes are positive for CD20 and CD5, negative for CD10 and CD23, and diffusely positive for cyclin D1, consistent with a diagnosis of MCL. The proliferative index, as measured by the Ki-67 immunostain, is 40%. A bone marrow aspirate and biopsy are then obtained, which show a clonal population of B lymphocytes expressing the same immunophenotype as the lymph node (positive for CD20 and CD5, negative for CD10 and CD23, cyclin D1 positive). A CT scan of the neck, chest, abdomen, and pelvis with contrast is obtained, along with a PET scan. These studies identify extensive hypermetabolic lymphadenopathy in the bilateral cervical chains, supraclavicular areas, mediastinum, and hilum. Mesenteric lymph nodes are also enlarged and hypermetabolic, as are retroperitoneal lymph nodes. The spleen is noted to be enlarged with multiple hypermetabolic lesions. Based on the presence of extensive lymphadenopathy as well as bone marrow involvement, the patient is diagnosed with stage IV MCL. He undergoes risk-stratification with the MIPI. His MIPI score is 6.3, high risk.
• What is the approach to upfront therapy for MCL?
FRONTLINE THERAPY
Role of Watchful Waiting
A small proportion of MCL patients have indolent disease that can be observed. This population is more likely to have leukemic-phase MCL with circulating lymphocytes, splenomegaly, and bone marrow involvement and absent or minimal lymphadenopathy.4,5 A retrospective study of 97 patients established that deferment of initial therapy in MCL is acceptable in some patients.5 In this study, approximately one third of patients with MCL were observed for more than 3 months before initiating systemic therapy, and the median time to treatment for the observation group was 12 months. Most patients undergoing observation had a low-risk MIPI. Patients were not harmed by observation, as no OS differences were observed among groups. This study underscores that deferred treatment can be an acceptable alternative in selected MCL patients for a short period of time. In practice, the type of patient who would be appropriate for this approach is someone who is frail, elderly, and with multiple comorbidities. Additionally, expectant observation could be considered for patients with limited-stage or low-volume MCL, low Ki-67 index, and low-risk MIPI scores.
Approach to Therapy
Treatment of MCL is generally approached by evaluating patient age and fitness for treatment. While there is no accepted standard, for younger patients healthy enough to tolerate aggressive approaches, treatment often involves an intensive cytarabine-containing regimen, which is consolidated with an autoSCT. This approach results in the longest remission duration, with some series suggesting a plateau in survival after 5 years, with no relapses.21 Nonintensive conventional chemotherapy alone is often reserved for the frailer or older patient. Given that remission durations with chemotherapy alone in MCL are short, goals of treatment focus on maximizing benefit and remission duration and minimizing risk of toxicity.
Standard Chemotherapy: Elderly and/or Frail Patients
Conventional chemotherapy alone for the treatment of MCL results in a 70% to 85% overall response rate (ORR) and 7% to 30% complete response (CR) rate.22 Rituximab, a mouse humanized monoclonal IgG1 anti-CD20 antibody, is used as standard of care in combination with chemotherapy, since its addition has been found to increase response rates and extend both progression-free survival (PFS) and OS compared to chemotherapy alone.23,24 However, chemoimmunotherapy approaches do not provide long-term control of MCL and are considered noncurative. Various regimens have been studied and include anthracycline-containing regimens such as R-CHOP (rituximab with cyclophosphamide, doxorubicin, vincristine, prednisone),22 combination chemotherapy with antimetabolites such as R-hyper-CVAD (hyper-fractionated rituximab with cyclophosphamide, vincristine, doxorubicin, dexamethasone, alternating with methotrexate and cytarabine),25 purine analogue–based regimens such as R-FC (rituximab with fludarabine and cyclophosphamide),26 bortezomib-containing regimens,27 and alkylator-based treatment with BR (bendamustine and rituximab) (Table 1).28,29 Among these, the most commonly used are R-CHOP and BR.
Two large randomized studies compared R-CHOP for 6 cycles to BR for 6 cycles in patients with indolent NHL and MCL. Among MCL patients, BR resulted in superior PFS compared to R-CHOP (69 months versus 26 months) but no benefit in OS.28,29 The ORR to R-CHOP was approximately 90%, with a PFS of 21 months in the Rummel et al study.29 This study included more than 80 centers in Germany and enrolled 549 patients with MCL, follicular lymphoma, small lymphocytic lymphoma, marginal zone lymphoma, and Waldenström macroglobulinemia. Patients were randomized in a 1:1 fashion. Among these, 46 patients received BR and 48 received R-CHOP (18% for both, respectively). It should be noted that patients in the BR group had significantly less toxicity and experienced fewer side effects than did those in the R-CHOP group. Similarly, BR-treated patients had a lower frequency of hematologic side effects and infections of any grade. However, drug-associated skin reactions and allergies were more common with BR compared to R-CHOP. The study by Flinn and colleagues was an international randomized, noninferiority phase 3 study designed to evaluate the efficacy and safety of BR compared with R-CHOP or R-CVP (rituximab plus cyclophosphamide, vincristine, and prednisone) for treatment-naive patients with MCL or other indolent NHL. The primary endpoint was CR. In this study, BR was found to be noninferior to R-CHOP and R-CVP based on CR rate (31% versus 25%, respectively; P = 0.0225). Response rates in general were high: 97% for BR and 91% for R-CHOP/R-CVP (P = 0.0102). Here, BR-treated patients experienced more nausea, emesis, and drug-induced hypersensitivity compared to the R-CHOP and R-CVP groups.
Another approach studied in older patients is the use of R-CHOP with rituximab maintenance. In a large European study, 560 patients 60 years of age or older with advanced-stage MCL were randomly assigned to either R-FC (rituximab, fludarabine, and cyclophosphamide) every 28 days for 6 cycles, or R-CHOP every 21 days for 8 cycles. Patients who had a response then underwent a second randomization, with one group receiving rituximab maintenance therapy. Maintenance was continued until progression of disease. Patients in this study were not eligible for high-dose chemotherapy and autoSCT. The study found that rates of CR were similar with both R-FC and R-CHOP (40% and 34%, respectively; P = 0.10). However, the R-FC arm underperformed in several arenas. Disease progression occurred more frequently with R-FC (14% versus 5% with R-CHOP), and OS was shorter (4-year OS, 47% versus 62%; P = 0.005, respectively). More patients also died in the R-FC group, and there was greater hematologic toxicity compared to R-CHOP. At 4 years, 58% of the patients receiving rituximab remained in remission. Among patients who responded to R-CHOP, rituximab maintenance led to a benefit in OS, reducing the risk of progression or death by 45%.26 At this time, studies are ongoing to establish the benefit of rituximab maintenance after BR.
Bendamustine in combination with other agents has also been studied in the frontline setting. Visco and colleagues evaluated the combination of bendamustine with rituximab and cytarabine (R-BAC) in older patients with MCL (age 65 or older).63 This phase 2, two-stage study enrolled 40 patients and had a dose-finding arm for cytarabine in combination with BR. It permitted relapsed/refractory patients, but 50% had newly diagnosed, previously untreated MCL. The regimen had an impressive ORR of 100%, with CR rates of 95% for previously untreated patients. PFS at 2 years was 95%. R-BAC was well tolerated, with the primary toxicity being reversible myelosuppression.
BR was combined with the proteasome inhibitor bortezomib and dexamethasone in a phase 2 study.64 This Lymphoma Study Association (LYSA) study evaluated 76 patients with newly diagnosed MCL older than age 65 years. BR was administered in standard doses (bendamustine 90 mg/m2 on days 1 and 2 and rituximab 375 mg/m² IV on day 1) and bortezomib was administered subcutaneously on days 1, 4, 8, and 11, with acyclovir for viral prophylaxis. Patients received 6 cycles. The ORR was 87% and the CR was 60%. Patients experienced toxicity, and not all bortezomib doses were administered due to neurotoxic or hematologic side effects.
A randomized phase 3 study compared R-CHOP to the VR-CAP regimen (R-CHOP regimen but bortezomib replaces vincristine on days 1, 4, 8, 11, at 1.3 mg/m2) in 487 newly diagnosed MCL patients.27 Median PFS was superior in the VR-CAP group compared with R-CHOP (14.4 months versus 24.7 months, respectively). Additionally, rates of CR were superior in the VR-CAP group (53% compared to 42% with R-CHOP). However, there was more hematologic toxicity with VR-CAP. On the basis of these findings, the U.S. Food and Drug Administration approved bortezomib for the frontline treatment of MCL.
Other chemoimmunotherapy combinations containing bortezomib have been studied in frontline MCL treatment, with promising results. These include bortezomib in combination with R-CHOP or modified R-hyper-CVAD, as well as bortezomib in combination with CHOP-like treatments and purine analogues.27,30–32 The ongoing ECOG 1411 study is currently evaluating bortezomib added to BR for induction therapy of newly diagnosed MCL in a 4-arm randomized trial. Patients receive BR with or without bortezomib during induction and are then randomly assigned to maintenance with either rituximab alone or rituximab with lenalidomide. Other novel combination agents are actively being studied in frontline MCL treatment, including lenalidomide and rituximab and BR with lenalidomide.
Intensification of Therapy and AutoSCT: Fitter and/or Younger Patients
Short response duration has created the need for post-remission therapy in MCL. One approach to improve remission duration in MCL is to intensify induction through the use of cytarabine-containing regimens and/or consolidation with high-dose chemotherapy, typically using BEAM (carmustine, etoposide, cytarabine, melphalan) and autoSCT (Table 2). The cytarabine-containing R-hyper-CVAD regimen, developed at the MD Anderson Cancer Center, resulted in a 97% ORR and an 87% CR rate, with TTF of nearly 5 years. However, nearly one third of patients were unable to complete treatment due to toxicity, and 5 patients developed secondary myelodysplastic syndrome or acute myeloid leukemia.33 The feasibility of this R-hyper-CVAD regimen was tested in a multicenter cooperative group setting, but similar results were not seen; in this study, nearly 40% of patients were unable to complete the full scheduled course of treatment due to toxicity.34
Other ways to intensify therapy in MCL involve adding a second non-cross-resistant cytarabine-containing regimen to R-CHOP after remission, such as DHAP (dexamethasone, high-dose cytarabine, cisplatin), followed by consolidation with an autoSCT. A retrospective registry from the National Comprehensive Cancer Network sought to compare the efficacy of different treatment approaches in the frontline setting. They studied 167 patients with MCL and compared 4 groups: treatment with R-hyper-CVAD, either with or without autoSCT, and treatment with R-CHOP, either with or without autoSCT. This study found that in patients younger than 65, R-CHOP followed by autoSCT or R-hyper-CVAD without autoSCT resulted in similar PF and OS, but was superior to R-CHOP alone for newly diagnosed MCL patients.35 These data support more intensive regimens in younger and fitter patients. Several other prospective and randomized studies have demonstrated clinical benefit for patients with MCL undergoing autoSCT in first remission. Of particular importance is the seminal phase 3 study of the European MCL Network, which established the role of autoSCT in this setting.61 In this prospective randomized trial involving 122 newly diagnosed MCL patients who responded to CHOP-like induction, patients in CR derived a greater benefit from autoSCT.
More recent studies have demonstrated similar benefits using cytarabine-based autoSCT. The Nordic MCL2 study evaluated 160 patients using R-CHOP, alternating with rituximab and high-dose cytarabine, followed by autoSCT. This study used “maxi-CHOP,” an augmented CHOP regimen (cyclophosphamide 1200 mg/m2, doxorubicin 75 mg/m2, but standard doses of vincristine [2 mg] and prednisone [100 mg days 1–5]), alternating with 4 infusions of cytarabine at 2 g/m2 and standard doses of rituximab (375 mg/m2). Patients then received conditioning with BEAM and autoSCT. Patients were evaluated for the presence of minimal residual disease (MRD) and for the t(11;14) or clonal immunoglobulin heavy chain gene rearrangement with polymerase chain reaction (PCR). Patients with MRD were offered therapy with rituximab at 375 mg/m2 weekly for 4 doses. This combination resulted in 10-year OS rates of 58%.36 In a multicenter study involving 78 patients from the Cancer and Leukemia Group B (CALGB), R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT resulted in a 5-year OS of 64%.37 A single-arm phase 2 study from the Netherlands also tested R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT. Nonhematologic toxicities were 22% after high-dose cytarabine, and 55% after BEAM. The ORR was 70%, with a 64% CR rate and 66% OS at 4 years.38 The French GELA group used 3 cycles of R-CHOP and 3 cycles of R-DHAP in a phase 2 study of young (under age 66) MCL patients. Following R-CHOP, the ORR was 93%, and following R-DHAP the ORR was 95%. Five-year OSA was 75%.39 A large randomized phase 3 study by Hermine and colleagues of the EMCLN confirmed the benefit of this approach in 497 patients with newly diagnosed MCL. R-CHOP for 6 cycles followed by autoSCT was compared to R-CHOP for 3 cycles alternating with R-DHAP for 3 cycles and autoSCT with a cytarabine-based conditioning regimen. The addition of cytarabine significantly increased rates of CR, TTF, and OS, without increasing toxicity.40
CASE CONTINUED
The patient is treated with R-CHOP chemotherapy for 3 cycles followed by R-DHAP. His course is complicated by mild tinnitus and acute kidney injury from cisplatin that promptly resolves. Three weeks following treatment, a restaging PET/CT scan shows resolution of all lymphadenopathy, with no hypermetabolic uptake, consistent with a complete remission. A repeat bone marrow biopsy shows no involvement with MCL. He subsequently undergoes an autoSCT, and restaging CT/PET 3 months following autoSCT shows continued remission. He is monitored every 3 to 6 months over the next several years.
He has a 4.5-year disease remission, after which he develops growing palpable lymphadenopathy on exam and progressive anemia and thrombocytopenia. A bone marrow biopsy is repeated, which shows recurrent MCL. Restaging diagnostic imaging with a CT scan reveals lymphadenopathy above and below the diaphragm. An axillary lymph node biopsy also demonstrates recurrent MCL. At this time the patient is otherwise in fairly good health, except for feeling fatigued. His ECOG performance status is 1. He begins therapy with bortezomib at a dose of 1.3 mg/m2 intravenously on days 1, 4, 8, and 11 for 6 cycles. His treatment course is complicated by painful sensory peripheral neuropathy of the bilateral lower extremities. Restaging studies at the completion of therapy demonstrate that he has achieved a partial response, with a 50% reduction in the size of involved lymphadenopathy and some residual areas of hypermetabolic uptake. His peripheral cytopenias improve moderately.
• What are the therapeutic options for relapsed MCL?
TREATMENT OF RELAPSED MCL
Single-Agent and Combination Chemotherapy
Whenever possible, and since there is no standard, patients with relapsed MCL should be offered a clinical trial. Outside of a clinical study, many of the treatment regimens used at diagnosis can also be applied in the relapsed setting. In relapsed MCL, Rummel et al showed that BR for 4 cycles resulted in an ORR of 90%, with a CR of 60%. The median PFS was 24 months.41 Bortezomib, an inhibitor of the proteasome-ubiquitin pathway, leads to apoptosis and cell cycle arrest in MCL.42 Multiple studies have evaluated bortezomib both as a single agent and in combination for patients with relapsed MCL. In 2006, bortezomib became the first agent approved by the FDA in relapsed or refractory MCL, based on the phase 2 PINNACLE study. This prospective multicenter study involving 155 patients demonstrated an ORR of 33%, CR rate of 8%, and median treatment duration of 9 months. The median time to progression was 6 months.43 Subsequently, bortezomib-containing combinations evolved. In a multicenter study of relapsed and refractory indolent NHL and MCL, Friedberg and colleagues evaluated bortezomib in combination with BR.44 In the MCL cohort, the ORR was 71%. These promising results led to the study of this combination in the frontline setting. The ongoing ECOG 1411 study is using BR for the frontline treatment of MCL with or without bortezomib as induction. This study also includes rituximab maintenance, and randomizes patients to undergo maintenance with or without the immunomodulator lenalidomide. Bortezomib has been associated with herpes simplex and herpes zoster reactivation. Neuropathy has also been observed with bortezomib, which can be attenuated by administering it subcutaneously.
Lenalidomide is an immunomodulatory agent derived from thalidomide. It has significant activity and is a mainstay of treatment in multiple myeloma. Lenalidomide acts by enhancing cellular immunity, has antiproliferative effects, and inhibits T-cell function leading to growth inhibitory effects in the tumor microenvironment.45 In MCL, lenalidomide has demonstrated clinical activity both as a single agent and in combination, as well as in preclinical studies establishing its pro-apoptotic effects.46 The pivotal EMERGE study evaluated monotherapy with lenalidomide in heavily pretreated relapsed and refractory MCL. This multicenter international study of 134 patents reported an ORR of 28% with a 7.5% CR rate and median PFS of 4 months. All patients had relapsed or progressed following bortezomib. This led to the approval of lenalidomide by the FDA in 2013 for the treatment of patients with MCL whose disease relapsed or progressed following 2 prior therapies, one of which included bortezomib.47 Lenalidomide has been associated with neutropenia, secondary cancers, and deep venous thrombosis.
In combination with other agents in the relapsed setting, lenalidomide shows broader activity. A phase 1/2 study by Wang and colleagues demonstrated an ORR of 57%; the median response duration was 19 months when lenalidomide was combined with rituximab for relapsed/refractory MCL.48
Novel Therapies
More recently, novel treatment approaches have been tested in MCL based on an increased understanding of aberrant signaling pathways in this disease (Table 3). Constitutive activation of B-cell receptor signaling is critical for the survival and proliferation of lymphomas, and has led to the development of targeted agents inhibiting B-cell receptor–associated protein kinases. Bruton’s tyrosine kinase (BTK) is one essential component of the B-cell receptor.49 In particular, proteins upstream of the BTK pathway have been implicated in growth and proliferation of MCL, suggesting that inhibition of BTK may impede lymphomagenesis.50 Ibrutinib is an oral inhibitor of BTK, and demonstrates activity in multiple lymphoma subtypes. In a phase 1 study of ibrutinib in relapsed and refractory hematologic malignancies, an ORR of 60% was observed in 50 evaluable patients, with 16% CR. Median PFS was 13 months. Among these, 7 of 9 patients with MCL responded, including 3 CRs.51 Given these promising results, a phase 2 multicenter study evaluating ibrutinib in relapsed and refractory MCL was completed.52 At a dose of 560 mg daily, the response rate was 68%, with CR of 21%. The most common observed treatment-related side effects included diarrhea, fatigue, and nausea. Neutropenia and thrombocytopenia were also observed. Of importance, 5% of patients had grade 3 or higher bleeding events, including subdural hematoma, gastrointestinal bleeding, and hematuria. The estimated OS rate was 58% at 18 months. On the basis of this study, the FDA approved ibrutinib for relapsed and refractory MCL in November 2013.
The PI3K pathway is another survival pathway that is dysfunctional in several hematologic disorders, including MCL. Overexpression of PI3K and its downstream targets contributes to MCL pathogenesis.53 Idelalisib is an oral small molecule inhibitor of the delta isoform of PI3K that is dosed daily; it was approved by the FDA for the treatment of relapsed and refractory follicular lymphoma, small lymphocytic lymphoma, and chronic lymphocytic leukemia. It is being further evaluated in MCL. A dose-escalation phase 1 study in heavily pre-treated MCL patients established safety and tolerability.54 Efficacy analysis showed an ORR of 40%, CR of 5%, and 1-year OS of 22%. Further phase 2 studies testing idelalisib as a single agent and in combination for MCL are ongoing. Side effects of idelalisib include elevated liver enzymes, pneumonitis, and diarrhea.
The BCL family of proteins is involved in both pro-and anti-apoptotic functions. BCL2 is an intracellular protein that blocks apoptosis. ABT-199 is an oral BCL2 inhibitor that in early clinical trials has shown very promising activity in MCL. In a phase 1 study of 31 relapsed and refractory NHL patients, all 8 MCL patients (100% ORR) responded to ABT-199 therapy.55 Given these promising initial results, multiple studies evaluating ABT-199 are ongoing in MCL as part of first-line treatment as well as for relapsed disease. ABT-199 has been implicated in tumor lysis syndrome, and in early studies of chronic lymphocytic leukemia, fatal tumor lysis was observed.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus has been evaluated in relapsed MCL. It is given weekly at 250 mg intravenously. Response rates to single-agent temsirolimus are approximately 20% to 35%, and are higher when combined with rituximab.56,57 The phase 2 study evaluating temsirolimus as a single agent enrolled 35 heavily pre-treated patients. ORR was 38% with only 1 CR. The duration of response was 7 months. Temsirolimus is approved for relapsed MCL in Europe but not in the United States. Similar to the other targeted agents, temsirolimus is actively being studied in combination with other active agents in MCL. Adverse effects noted with temsirolimus include diarrhea, stomatitis, and rash. Thrombocytopenia requiring dose reductions is another frequently observed complication.
Radioimmunotherapy
Radioimmunotherapy (RIT) has been studied extensively in MCL. RIT consists of anti-CD 20 antibodies coupled to radioactive particles that deliver radiation to targeted cells, minimizing toxicity to surrounding tissues. RIT is not used as frequently in the modern era as it had been in the past. At this time, only yttrium-90-ibritumomab tiuxetan is available.
RIT has been evaluated in MCL both at the time of relapse58 and more recently, as part of a conditioning regimen prior to autoSCT, with good tolerability.65–67 Averse events noted with RIT include hematologic toxicity (can be prolonged), hypothyroidism, and in rare cases, myelodysplastic syndrome and acute leukemia. The bone marrow must have less than 25% involvement with disease prior to administration. Wang and colleagues evaluated yttrium-90-ibritumomab tiuxetan in 34 heavily pretreated patients with MCL.58 They observed an ORR of 31%. The median event-free survival (EFS) was 6 months, but in patients achieving either CR or PR, EFS was 28 months. A 21-month OS was noted.
In the upfront setting, RIT has been added as a mechanism of intensification. A recent Nordic group study of RIT with autoSCT did not find benefit with the addition of RIT.59 An ECOG study recently added yttrium-90-ibritumomab tiuxetan after CHOP chemotherapy in the upfront treatment of MCL, with good tolerability.55 However, when added to R-hyper-CVAD, the combination had unexpected high rates of hematologic toxicity, including grade 3/4 cytopenias and an unacceptably high rate of secondary malignancies.68
AutoSCT or Allogeneic Transplant
While many studies noted above have established the beneficial role of autoSCT in MCL in first remission, the role of allogeneic transplant (alloSCT) in MCL remains controversial. A recent large retrospective study conducted by the Center for International Blood and Marrow Transplant Research (CIBMTR) evaluated 519 patients with MCL who underwent both autoSCT and alloSCT.60 Patients were grouped into an early cohort (transplant in first PR or CR, and 2 or fewer treatments) and late cohort (all other patients). The analysis had mature follow up. A multivariate analysis demonstrated that early autoSCT was associated with superior outcomes compared to autoSCT performed later. While it was not possible to demonstrate a survival benefit favoring autoSCT over reduced intensity (RIC) alloSCT, patients transplanted later in their disease course had shorter OS. For patients receiving autoSCT in CR 1 following only 1 prior line of therapy, OS at 5 years was 75% and PFS was 70%. Patients undergoing RIC followed by alloSCT had fewer relapses, but this was negated by higher nonrelapse mortality (25%), resulting in a PFS similar to autoSCT.
CASE CONCLUSION
After treatment with bortezomib the patient is well for 9 months. Subsequently, however, he develops increasing lymphadenopathy and progressive fatigue. He is then started on lenalidomide 25 mg orally daily for 21 out of 28 days. He experiences significant fatigue with lenalidomide and prolonged neutropenia requiring dose delays, despite dose modification to 10 mg orally daily. He requires discontinuation of lenalidomide. Given persistent disease, the patient then begins treatment with ibrutinib. Within a few days of starting ibrutinib therapy, he experiences a marked but transient leukocytosis. Two months later, the patient’s palpable lymphadenopathy has decreased, and his anemia and thrombocytopenia related to MCL are improving. He has tolerated treatment well. His course has been complicated only by a mild, pruritic maculopapular eruption on his chest, back, and arms, that was responsive to topical low-dose steroids. He remains on ibrutinib 1 year later.
CONCLUSION
Advances in our understanding of MCL treatment are revolutionizing the approach to this once deadly disease. Over the next several years, these gains will weave themselves into the current treatment paradigm and likely alter the treatment landscape for MCL as we know it.
INTRODUCTION
Mantle cell lymphoma (MCL) is an uncommon, distinct clinical subtype of non-Hodgkin lymphoma (NHL) that comprises approximately 8% of all lymphoma diagnoses in the United States and Europe.1,2 Considered incurable, MCL often presents in advanced stages, particularly with involvement of the lymph nodes, spleen, bone marrow, and gastrointestinal tract in the form of lymphomatous polyps. MCL disproportionately affects males, and incidence rises with age, with a median age at diagnosis of 68 years.2 Historically, the prognosis of patients with MCL has been among the poorest among B-cell lymphoma patients, with a median overall survival (OS) of 3 to 5 years, and time to treatment failure (TTF) of 18 to 24 months, although this is improving in the modern era.3 Less frequently, patients with MCL display isolated bone marrow, peripheral blood, and splenic involvement. These cases tend to behave more indolently with longer survival.4,5 Recent advances in therapy have dramatically impacted treatment alternatives and outcomes for MCL. As such, the therapeutic and prognostic landscape of MCL is evolving rapidly.
PATHOGENESIS
The histologic diagnosis of MCL by morphology alone is often challenging. Accurate diagnosis relies on immunohistochemical staining for the purposes of immunophenotyping.6 MCL typically expresses B-cell markers CD5 and CD20, and lacks both CD10 and CD23. The genetic hallmark of MCL is the t(11;14) (q13;q32) chromosomal translocation leading to upregulation of the cyclin D1 protein, a critical regulator of the G1 phase of the cell cycle. Specifically, the t(11;14) translocation, present in virtually all cases of MCL, juxtaposes the proto-oncogene CCND1 to the immunoglobulin heavy chain gene.7 Consequently, cyclin D1, normally not expressed in B lymphocytes, becomes constitutively overexpressed. This alteration is thought to facilitate the deregulation of the cell cycle at the G1-S phase transition.8
Gene expression profiling studies have underscored the importance of cell cycle deregulation in MCL, and high proliferation is associated with a worse prognosis.9 More than 50% of the genes associated with poor outcomes were derived from the “proliferation signature” that was more highly expressed in dividing cells. In the seminal Rosenwald study, a gene expression–based outcome model was constructed in which the proliferation signature average represents a linear variable that assigns a discrete probability of survival to an individual patient.9 The proliferative index, or proliferative signature, of MCL can be estimated by the percentage of Ki-67–positive cells present in the tumor through immunohistochemistry. This is often used as a marker of poor outcomes, and as a surrogate for the proliferative signature in MCL that can be incorporated into clinical practice (as opposed to gene expression profiling). Statistically significant differences in OS have emerged between groups of MCL patients with Ki-67–positive cells comprising less than 30% of their tumor sample (favorable) and those with Ki-67–positive cells comprising 30% or greater (unfavorable).10
Recent data has also identified the importance of the transcription factor SOX 11 (SRY-related HMG-box), which regulates multiple cellular transcriptional events, including cell proliferation and differentiation, apoptosis, and angiogenesis.11 MCL expressing SOX 11 behaves more aggressively than MCL variants lacking SOX 11 expression, and tends to accumulate more genetic alterations.12 Moreover, lack of SOX 11 expression characterizes a subset of MCL that does not carry the t(11;14) translocation.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 62-year-old man with a history of diabetes mellitus and hypertension presents with cervical lymphadenopathy, fatigue, and early satiety over the past several months. He is otherwise in good health. His Eastern Cooperative Oncology Group (ECOG) performance status is 1. On physical examination, 3-cm lymphadenopathy in the bilateral cervical chain is noted. Bilateral axillary lymph nodes measure 2 to 4 cm. His spleen is enlarged and is palpable at approximately 5 cm below the costal margin. A complete blood count reveals a total white blood cell (WBC) count of 14,000 cells/μL, with 68% lymphocytes and a normal distribution of neutrophils. Hemoglobin is 11 g/dL, and platelet count is 112,000/μL. The lactate dehydrogenase (LDH) level is 322 U/L (upper limit of normal: 225 U/L).
• How is MCL diagnosed?
Diagnosis of MCL requires review by expert hematopathologists.13 Whenever possible, an excisional biopsy should be performed for the adequate characterization of lymph node architecture and evaluation by immunohistochemistry. Aside from the characteristic expression of CD5 and CD20 and absence of CD23, MCL should express cyclin D1, which reflects t(11;14). If cyclin D1 is inconclusive or unavailable, fluorescent in situ hybridization (FISH) for t(11;14) should be performed.8 Patients often have circulating malignant lymphocytes, or leukemic phase MCL. Flow cytometry of the peripheral blood can detect traditional surface markers, and FISH can also be performed on circulating abnormal lymphocytes.
For disease staging, bone marrow biopsy and aspiration are required. Radiographic staging using computed tomography (CT) scans and/or positron emission tomography (PET) scans had traditionally followed the Ann Arbor staging system, but recently the Lugano classification has emerged, which delineates only early or advanced stage.14 Gastrointestinal evaluation of MCL with endoscopy and colonoscopy with blind biopsies has been recommended to evaluate for the presence of lymphomatous polyps, but this is not an absolute requirement.15
RISK STRATIFICATION
At diagnosis, patients should undergo risk stratification in order to understand prognosis and possibly guide treatment. In MCL, the MCL international prognostic index (MIPI) is used. The MIPI is a prognostic tool developed exclusively for patients with MCL using data from 455 patients with advanced-stage MCL treated within 3 European clinical trials.16 The MIPI classified patients into risk groups based on age, ECOG performance status, LDH level, and WBC count. Patients were categorized into low-risk (44% of patients, median OS not reached), intermediate-risk (35%, median OS 51 months), and high-risk groups (21%, median OS 29 months). This is done through a logarithmic calculation, which can be accessed through online calculators (a prototype example can be found at www.qxmd.com/calculate-online/hematology/prognosis-mantle-cell-lymphoma-mipi). Cell proliferation using the Ki-67 index was evaluated in an exploratory analysis (the biologic [“B”] MIPI), and also demonstrated strong prognostic relevance.16 Currently, treatment of MCL patients is not stratified by MIPI outside of a clinical trial, but this useful tool assists in assessing patient prognosis and has been validated for use with both conventional chemoimmunotherapy and in the setting of autologous stem cell transplant (autoSCT).16,17 At this point in time, the MIPI score is not used to stratify treatment, although some clinical trials are incorporating the use of the MIPI score at diagnosis. Nonetheless, given its prognostic importance, the MIPI should be performed for all MCL patients undergoing staging and evaluation for treatment to establish disease risk.
As noted, the proliferative signature, represented by the Ki-67 protein, is also highly prognostic in MCL. Ki-67 is expressed in the late G1, S, G2, and M phases of the cell cycle. The Ki-67 index is defined by the hematopathologist as the percentage of lymphoma cells staining positive for Ki-67 protein, based on the number of cells per high-power field. There is significant interobserver variability in this process, which can be minimized by assessing Ki-67 quantitatively using computer software. The prognostic significance of Ki-67 at diagnosis was established in large studies of MCL patient cohorts, with survival differing by up to 3 years.18,19 Determann et al demonstrated the utility of the proliferative index in patients with MCL treated with standard chemoimmunotherapy.10 In this study, 249 patients with advanced-stage MCL treated within randomized trials conducted by the European MCL Network were analyzed. The Ki-67 index was found to be extremely prognostic of OS, independent of other clinical risk factors, including the MIPI score. As a continuous variable, Ki-67 indices of greater than 10% correlated with poor outcomes. The Ki-67 index has also been confirmed as prognostic in relapsed MCL.20 It is important to note that, as a unique feature, the Ki-67 index has remained an independent prognostic factor, even when incorporated into the “B” MIPI.
TREATMENT
CASE CONTINUED
The patient undergoes an excisional biopsy of a cervical lymph node, which demonstrates an abnormal proliferation of small-medium–sized lymphocytes with slightly irregular nuclear contours. Immunohistochemistry shows that the abnormal lymphocytes are positive for CD20 and CD5, negative for CD10 and CD23, and diffusely positive for cyclin D1, consistent with a diagnosis of MCL. The proliferative index, as measured by the Ki-67 immunostain, is 40%. A bone marrow aspirate and biopsy are then obtained, which show a clonal population of B lymphocytes expressing the same immunophenotype as the lymph node (positive for CD20 and CD5, negative for CD10 and CD23, cyclin D1 positive). A CT scan of the neck, chest, abdomen, and pelvis with contrast is obtained, along with a PET scan. These studies identify extensive hypermetabolic lymphadenopathy in the bilateral cervical chains, supraclavicular areas, mediastinum, and hilum. Mesenteric lymph nodes are also enlarged and hypermetabolic, as are retroperitoneal lymph nodes. The spleen is noted to be enlarged with multiple hypermetabolic lesions. Based on the presence of extensive lymphadenopathy as well as bone marrow involvement, the patient is diagnosed with stage IV MCL. He undergoes risk-stratification with the MIPI. His MIPI score is 6.3, high risk.
• What is the approach to upfront therapy for MCL?
FRONTLINE THERAPY
Role of Watchful Waiting
A small proportion of MCL patients have indolent disease that can be observed. This population is more likely to have leukemic-phase MCL with circulating lymphocytes, splenomegaly, and bone marrow involvement and absent or minimal lymphadenopathy.4,5 A retrospective study of 97 patients established that deferment of initial therapy in MCL is acceptable in some patients.5 In this study, approximately one third of patients with MCL were observed for more than 3 months before initiating systemic therapy, and the median time to treatment for the observation group was 12 months. Most patients undergoing observation had a low-risk MIPI. Patients were not harmed by observation, as no OS differences were observed among groups. This study underscores that deferred treatment can be an acceptable alternative in selected MCL patients for a short period of time. In practice, the type of patient who would be appropriate for this approach is someone who is frail, elderly, and with multiple comorbidities. Additionally, expectant observation could be considered for patients with limited-stage or low-volume MCL, low Ki-67 index, and low-risk MIPI scores.
Approach to Therapy
Treatment of MCL is generally approached by evaluating patient age and fitness for treatment. While there is no accepted standard, for younger patients healthy enough to tolerate aggressive approaches, treatment often involves an intensive cytarabine-containing regimen, which is consolidated with an autoSCT. This approach results in the longest remission duration, with some series suggesting a plateau in survival after 5 years, with no relapses.21 Nonintensive conventional chemotherapy alone is often reserved for the frailer or older patient. Given that remission durations with chemotherapy alone in MCL are short, goals of treatment focus on maximizing benefit and remission duration and minimizing risk of toxicity.
Standard Chemotherapy: Elderly and/or Frail Patients
Conventional chemotherapy alone for the treatment of MCL results in a 70% to 85% overall response rate (ORR) and 7% to 30% complete response (CR) rate.22 Rituximab, a mouse humanized monoclonal IgG1 anti-CD20 antibody, is used as standard of care in combination with chemotherapy, since its addition has been found to increase response rates and extend both progression-free survival (PFS) and OS compared to chemotherapy alone.23,24 However, chemoimmunotherapy approaches do not provide long-term control of MCL and are considered noncurative. Various regimens have been studied and include anthracycline-containing regimens such as R-CHOP (rituximab with cyclophosphamide, doxorubicin, vincristine, prednisone),22 combination chemotherapy with antimetabolites such as R-hyper-CVAD (hyper-fractionated rituximab with cyclophosphamide, vincristine, doxorubicin, dexamethasone, alternating with methotrexate and cytarabine),25 purine analogue–based regimens such as R-FC (rituximab with fludarabine and cyclophosphamide),26 bortezomib-containing regimens,27 and alkylator-based treatment with BR (bendamustine and rituximab) (Table 1).28,29 Among these, the most commonly used are R-CHOP and BR.
Two large randomized studies compared R-CHOP for 6 cycles to BR for 6 cycles in patients with indolent NHL and MCL. Among MCL patients, BR resulted in superior PFS compared to R-CHOP (69 months versus 26 months) but no benefit in OS.28,29 The ORR to R-CHOP was approximately 90%, with a PFS of 21 months in the Rummel et al study.29 This study included more than 80 centers in Germany and enrolled 549 patients with MCL, follicular lymphoma, small lymphocytic lymphoma, marginal zone lymphoma, and Waldenström macroglobulinemia. Patients were randomized in a 1:1 fashion. Among these, 46 patients received BR and 48 received R-CHOP (18% for both, respectively). It should be noted that patients in the BR group had significantly less toxicity and experienced fewer side effects than did those in the R-CHOP group. Similarly, BR-treated patients had a lower frequency of hematologic side effects and infections of any grade. However, drug-associated skin reactions and allergies were more common with BR compared to R-CHOP. The study by Flinn and colleagues was an international randomized, noninferiority phase 3 study designed to evaluate the efficacy and safety of BR compared with R-CHOP or R-CVP (rituximab plus cyclophosphamide, vincristine, and prednisone) for treatment-naive patients with MCL or other indolent NHL. The primary endpoint was CR. In this study, BR was found to be noninferior to R-CHOP and R-CVP based on CR rate (31% versus 25%, respectively; P = 0.0225). Response rates in general were high: 97% for BR and 91% for R-CHOP/R-CVP (P = 0.0102). Here, BR-treated patients experienced more nausea, emesis, and drug-induced hypersensitivity compared to the R-CHOP and R-CVP groups.
Another approach studied in older patients is the use of R-CHOP with rituximab maintenance. In a large European study, 560 patients 60 years of age or older with advanced-stage MCL were randomly assigned to either R-FC (rituximab, fludarabine, and cyclophosphamide) every 28 days for 6 cycles, or R-CHOP every 21 days for 8 cycles. Patients who had a response then underwent a second randomization, with one group receiving rituximab maintenance therapy. Maintenance was continued until progression of disease. Patients in this study were not eligible for high-dose chemotherapy and autoSCT. The study found that rates of CR were similar with both R-FC and R-CHOP (40% and 34%, respectively; P = 0.10). However, the R-FC arm underperformed in several arenas. Disease progression occurred more frequently with R-FC (14% versus 5% with R-CHOP), and OS was shorter (4-year OS, 47% versus 62%; P = 0.005, respectively). More patients also died in the R-FC group, and there was greater hematologic toxicity compared to R-CHOP. At 4 years, 58% of the patients receiving rituximab remained in remission. Among patients who responded to R-CHOP, rituximab maintenance led to a benefit in OS, reducing the risk of progression or death by 45%.26 At this time, studies are ongoing to establish the benefit of rituximab maintenance after BR.
Bendamustine in combination with other agents has also been studied in the frontline setting. Visco and colleagues evaluated the combination of bendamustine with rituximab and cytarabine (R-BAC) in older patients with MCL (age 65 or older).63 This phase 2, two-stage study enrolled 40 patients and had a dose-finding arm for cytarabine in combination with BR. It permitted relapsed/refractory patients, but 50% had newly diagnosed, previously untreated MCL. The regimen had an impressive ORR of 100%, with CR rates of 95% for previously untreated patients. PFS at 2 years was 95%. R-BAC was well tolerated, with the primary toxicity being reversible myelosuppression.
BR was combined with the proteasome inhibitor bortezomib and dexamethasone in a phase 2 study.64 This Lymphoma Study Association (LYSA) study evaluated 76 patients with newly diagnosed MCL older than age 65 years. BR was administered in standard doses (bendamustine 90 mg/m2 on days 1 and 2 and rituximab 375 mg/m² IV on day 1) and bortezomib was administered subcutaneously on days 1, 4, 8, and 11, with acyclovir for viral prophylaxis. Patients received 6 cycles. The ORR was 87% and the CR was 60%. Patients experienced toxicity, and not all bortezomib doses were administered due to neurotoxic or hematologic side effects.
A randomized phase 3 study compared R-CHOP to the VR-CAP regimen (R-CHOP regimen but bortezomib replaces vincristine on days 1, 4, 8, 11, at 1.3 mg/m2) in 487 newly diagnosed MCL patients.27 Median PFS was superior in the VR-CAP group compared with R-CHOP (14.4 months versus 24.7 months, respectively). Additionally, rates of CR were superior in the VR-CAP group (53% compared to 42% with R-CHOP). However, there was more hematologic toxicity with VR-CAP. On the basis of these findings, the U.S. Food and Drug Administration approved bortezomib for the frontline treatment of MCL.
Other chemoimmunotherapy combinations containing bortezomib have been studied in frontline MCL treatment, with promising results. These include bortezomib in combination with R-CHOP or modified R-hyper-CVAD, as well as bortezomib in combination with CHOP-like treatments and purine analogues.27,30–32 The ongoing ECOG 1411 study is currently evaluating bortezomib added to BR for induction therapy of newly diagnosed MCL in a 4-arm randomized trial. Patients receive BR with or without bortezomib during induction and are then randomly assigned to maintenance with either rituximab alone or rituximab with lenalidomide. Other novel combination agents are actively being studied in frontline MCL treatment, including lenalidomide and rituximab and BR with lenalidomide.
Intensification of Therapy and AutoSCT: Fitter and/or Younger Patients
Short response duration has created the need for post-remission therapy in MCL. One approach to improve remission duration in MCL is to intensify induction through the use of cytarabine-containing regimens and/or consolidation with high-dose chemotherapy, typically using BEAM (carmustine, etoposide, cytarabine, melphalan) and autoSCT (Table 2). The cytarabine-containing R-hyper-CVAD regimen, developed at the MD Anderson Cancer Center, resulted in a 97% ORR and an 87% CR rate, with TTF of nearly 5 years. However, nearly one third of patients were unable to complete treatment due to toxicity, and 5 patients developed secondary myelodysplastic syndrome or acute myeloid leukemia.33 The feasibility of this R-hyper-CVAD regimen was tested in a multicenter cooperative group setting, but similar results were not seen; in this study, nearly 40% of patients were unable to complete the full scheduled course of treatment due to toxicity.34
Other ways to intensify therapy in MCL involve adding a second non-cross-resistant cytarabine-containing regimen to R-CHOP after remission, such as DHAP (dexamethasone, high-dose cytarabine, cisplatin), followed by consolidation with an autoSCT. A retrospective registry from the National Comprehensive Cancer Network sought to compare the efficacy of different treatment approaches in the frontline setting. They studied 167 patients with MCL and compared 4 groups: treatment with R-hyper-CVAD, either with or without autoSCT, and treatment with R-CHOP, either with or without autoSCT. This study found that in patients younger than 65, R-CHOP followed by autoSCT or R-hyper-CVAD without autoSCT resulted in similar PF and OS, but was superior to R-CHOP alone for newly diagnosed MCL patients.35 These data support more intensive regimens in younger and fitter patients. Several other prospective and randomized studies have demonstrated clinical benefit for patients with MCL undergoing autoSCT in first remission. Of particular importance is the seminal phase 3 study of the European MCL Network, which established the role of autoSCT in this setting.61 In this prospective randomized trial involving 122 newly diagnosed MCL patients who responded to CHOP-like induction, patients in CR derived a greater benefit from autoSCT.
More recent studies have demonstrated similar benefits using cytarabine-based autoSCT. The Nordic MCL2 study evaluated 160 patients using R-CHOP, alternating with rituximab and high-dose cytarabine, followed by autoSCT. This study used “maxi-CHOP,” an augmented CHOP regimen (cyclophosphamide 1200 mg/m2, doxorubicin 75 mg/m2, but standard doses of vincristine [2 mg] and prednisone [100 mg days 1–5]), alternating with 4 infusions of cytarabine at 2 g/m2 and standard doses of rituximab (375 mg/m2). Patients then received conditioning with BEAM and autoSCT. Patients were evaluated for the presence of minimal residual disease (MRD) and for the t(11;14) or clonal immunoglobulin heavy chain gene rearrangement with polymerase chain reaction (PCR). Patients with MRD were offered therapy with rituximab at 375 mg/m2 weekly for 4 doses. This combination resulted in 10-year OS rates of 58%.36 In a multicenter study involving 78 patients from the Cancer and Leukemia Group B (CALGB), R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT resulted in a 5-year OS of 64%.37 A single-arm phase 2 study from the Netherlands also tested R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT. Nonhematologic toxicities were 22% after high-dose cytarabine, and 55% after BEAM. The ORR was 70%, with a 64% CR rate and 66% OS at 4 years.38 The French GELA group used 3 cycles of R-CHOP and 3 cycles of R-DHAP in a phase 2 study of young (under age 66) MCL patients. Following R-CHOP, the ORR was 93%, and following R-DHAP the ORR was 95%. Five-year OSA was 75%.39 A large randomized phase 3 study by Hermine and colleagues of the EMCLN confirmed the benefit of this approach in 497 patients with newly diagnosed MCL. R-CHOP for 6 cycles followed by autoSCT was compared to R-CHOP for 3 cycles alternating with R-DHAP for 3 cycles and autoSCT with a cytarabine-based conditioning regimen. The addition of cytarabine significantly increased rates of CR, TTF, and OS, without increasing toxicity.40
CASE CONTINUED
The patient is treated with R-CHOP chemotherapy for 3 cycles followed by R-DHAP. His course is complicated by mild tinnitus and acute kidney injury from cisplatin that promptly resolves. Three weeks following treatment, a restaging PET/CT scan shows resolution of all lymphadenopathy, with no hypermetabolic uptake, consistent with a complete remission. A repeat bone marrow biopsy shows no involvement with MCL. He subsequently undergoes an autoSCT, and restaging CT/PET 3 months following autoSCT shows continued remission. He is monitored every 3 to 6 months over the next several years.
He has a 4.5-year disease remission, after which he develops growing palpable lymphadenopathy on exam and progressive anemia and thrombocytopenia. A bone marrow biopsy is repeated, which shows recurrent MCL. Restaging diagnostic imaging with a CT scan reveals lymphadenopathy above and below the diaphragm. An axillary lymph node biopsy also demonstrates recurrent MCL. At this time the patient is otherwise in fairly good health, except for feeling fatigued. His ECOG performance status is 1. He begins therapy with bortezomib at a dose of 1.3 mg/m2 intravenously on days 1, 4, 8, and 11 for 6 cycles. His treatment course is complicated by painful sensory peripheral neuropathy of the bilateral lower extremities. Restaging studies at the completion of therapy demonstrate that he has achieved a partial response, with a 50% reduction in the size of involved lymphadenopathy and some residual areas of hypermetabolic uptake. His peripheral cytopenias improve moderately.
• What are the therapeutic options for relapsed MCL?
TREATMENT OF RELAPSED MCL
Single-Agent and Combination Chemotherapy
Whenever possible, and since there is no standard, patients with relapsed MCL should be offered a clinical trial. Outside of a clinical study, many of the treatment regimens used at diagnosis can also be applied in the relapsed setting. In relapsed MCL, Rummel et al showed that BR for 4 cycles resulted in an ORR of 90%, with a CR of 60%. The median PFS was 24 months.41 Bortezomib, an inhibitor of the proteasome-ubiquitin pathway, leads to apoptosis and cell cycle arrest in MCL.42 Multiple studies have evaluated bortezomib both as a single agent and in combination for patients with relapsed MCL. In 2006, bortezomib became the first agent approved by the FDA in relapsed or refractory MCL, based on the phase 2 PINNACLE study. This prospective multicenter study involving 155 patients demonstrated an ORR of 33%, CR rate of 8%, and median treatment duration of 9 months. The median time to progression was 6 months.43 Subsequently, bortezomib-containing combinations evolved. In a multicenter study of relapsed and refractory indolent NHL and MCL, Friedberg and colleagues evaluated bortezomib in combination with BR.44 In the MCL cohort, the ORR was 71%. These promising results led to the study of this combination in the frontline setting. The ongoing ECOG 1411 study is using BR for the frontline treatment of MCL with or without bortezomib as induction. This study also includes rituximab maintenance, and randomizes patients to undergo maintenance with or without the immunomodulator lenalidomide. Bortezomib has been associated with herpes simplex and herpes zoster reactivation. Neuropathy has also been observed with bortezomib, which can be attenuated by administering it subcutaneously.
Lenalidomide is an immunomodulatory agent derived from thalidomide. It has significant activity and is a mainstay of treatment in multiple myeloma. Lenalidomide acts by enhancing cellular immunity, has antiproliferative effects, and inhibits T-cell function leading to growth inhibitory effects in the tumor microenvironment.45 In MCL, lenalidomide has demonstrated clinical activity both as a single agent and in combination, as well as in preclinical studies establishing its pro-apoptotic effects.46 The pivotal EMERGE study evaluated monotherapy with lenalidomide in heavily pretreated relapsed and refractory MCL. This multicenter international study of 134 patents reported an ORR of 28% with a 7.5% CR rate and median PFS of 4 months. All patients had relapsed or progressed following bortezomib. This led to the approval of lenalidomide by the FDA in 2013 for the treatment of patients with MCL whose disease relapsed or progressed following 2 prior therapies, one of which included bortezomib.47 Lenalidomide has been associated with neutropenia, secondary cancers, and deep venous thrombosis.
In combination with other agents in the relapsed setting, lenalidomide shows broader activity. A phase 1/2 study by Wang and colleagues demonstrated an ORR of 57%; the median response duration was 19 months when lenalidomide was combined with rituximab for relapsed/refractory MCL.48
Novel Therapies
More recently, novel treatment approaches have been tested in MCL based on an increased understanding of aberrant signaling pathways in this disease (Table 3). Constitutive activation of B-cell receptor signaling is critical for the survival and proliferation of lymphomas, and has led to the development of targeted agents inhibiting B-cell receptor–associated protein kinases. Bruton’s tyrosine kinase (BTK) is one essential component of the B-cell receptor.49 In particular, proteins upstream of the BTK pathway have been implicated in growth and proliferation of MCL, suggesting that inhibition of BTK may impede lymphomagenesis.50 Ibrutinib is an oral inhibitor of BTK, and demonstrates activity in multiple lymphoma subtypes. In a phase 1 study of ibrutinib in relapsed and refractory hematologic malignancies, an ORR of 60% was observed in 50 evaluable patients, with 16% CR. Median PFS was 13 months. Among these, 7 of 9 patients with MCL responded, including 3 CRs.51 Given these promising results, a phase 2 multicenter study evaluating ibrutinib in relapsed and refractory MCL was completed.52 At a dose of 560 mg daily, the response rate was 68%, with CR of 21%. The most common observed treatment-related side effects included diarrhea, fatigue, and nausea. Neutropenia and thrombocytopenia were also observed. Of importance, 5% of patients had grade 3 or higher bleeding events, including subdural hematoma, gastrointestinal bleeding, and hematuria. The estimated OS rate was 58% at 18 months. On the basis of this study, the FDA approved ibrutinib for relapsed and refractory MCL in November 2013.
The PI3K pathway is another survival pathway that is dysfunctional in several hematologic disorders, including MCL. Overexpression of PI3K and its downstream targets contributes to MCL pathogenesis.53 Idelalisib is an oral small molecule inhibitor of the delta isoform of PI3K that is dosed daily; it was approved by the FDA for the treatment of relapsed and refractory follicular lymphoma, small lymphocytic lymphoma, and chronic lymphocytic leukemia. It is being further evaluated in MCL. A dose-escalation phase 1 study in heavily pre-treated MCL patients established safety and tolerability.54 Efficacy analysis showed an ORR of 40%, CR of 5%, and 1-year OS of 22%. Further phase 2 studies testing idelalisib as a single agent and in combination for MCL are ongoing. Side effects of idelalisib include elevated liver enzymes, pneumonitis, and diarrhea.
The BCL family of proteins is involved in both pro-and anti-apoptotic functions. BCL2 is an intracellular protein that blocks apoptosis. ABT-199 is an oral BCL2 inhibitor that in early clinical trials has shown very promising activity in MCL. In a phase 1 study of 31 relapsed and refractory NHL patients, all 8 MCL patients (100% ORR) responded to ABT-199 therapy.55 Given these promising initial results, multiple studies evaluating ABT-199 are ongoing in MCL as part of first-line treatment as well as for relapsed disease. ABT-199 has been implicated in tumor lysis syndrome, and in early studies of chronic lymphocytic leukemia, fatal tumor lysis was observed.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus has been evaluated in relapsed MCL. It is given weekly at 250 mg intravenously. Response rates to single-agent temsirolimus are approximately 20% to 35%, and are higher when combined with rituximab.56,57 The phase 2 study evaluating temsirolimus as a single agent enrolled 35 heavily pre-treated patients. ORR was 38% with only 1 CR. The duration of response was 7 months. Temsirolimus is approved for relapsed MCL in Europe but not in the United States. Similar to the other targeted agents, temsirolimus is actively being studied in combination with other active agents in MCL. Adverse effects noted with temsirolimus include diarrhea, stomatitis, and rash. Thrombocytopenia requiring dose reductions is another frequently observed complication.
Radioimmunotherapy
Radioimmunotherapy (RIT) has been studied extensively in MCL. RIT consists of anti-CD 20 antibodies coupled to radioactive particles that deliver radiation to targeted cells, minimizing toxicity to surrounding tissues. RIT is not used as frequently in the modern era as it had been in the past. At this time, only yttrium-90-ibritumomab tiuxetan is available.
RIT has been evaluated in MCL both at the time of relapse58 and more recently, as part of a conditioning regimen prior to autoSCT, with good tolerability.65–67 Averse events noted with RIT include hematologic toxicity (can be prolonged), hypothyroidism, and in rare cases, myelodysplastic syndrome and acute leukemia. The bone marrow must have less than 25% involvement with disease prior to administration. Wang and colleagues evaluated yttrium-90-ibritumomab tiuxetan in 34 heavily pretreated patients with MCL.58 They observed an ORR of 31%. The median event-free survival (EFS) was 6 months, but in patients achieving either CR or PR, EFS was 28 months. A 21-month OS was noted.
In the upfront setting, RIT has been added as a mechanism of intensification. A recent Nordic group study of RIT with autoSCT did not find benefit with the addition of RIT.59 An ECOG study recently added yttrium-90-ibritumomab tiuxetan after CHOP chemotherapy in the upfront treatment of MCL, with good tolerability.55 However, when added to R-hyper-CVAD, the combination had unexpected high rates of hematologic toxicity, including grade 3/4 cytopenias and an unacceptably high rate of secondary malignancies.68
AutoSCT or Allogeneic Transplant
While many studies noted above have established the beneficial role of autoSCT in MCL in first remission, the role of allogeneic transplant (alloSCT) in MCL remains controversial. A recent large retrospective study conducted by the Center for International Blood and Marrow Transplant Research (CIBMTR) evaluated 519 patients with MCL who underwent both autoSCT and alloSCT.60 Patients were grouped into an early cohort (transplant in first PR or CR, and 2 or fewer treatments) and late cohort (all other patients). The analysis had mature follow up. A multivariate analysis demonstrated that early autoSCT was associated with superior outcomes compared to autoSCT performed later. While it was not possible to demonstrate a survival benefit favoring autoSCT over reduced intensity (RIC) alloSCT, patients transplanted later in their disease course had shorter OS. For patients receiving autoSCT in CR 1 following only 1 prior line of therapy, OS at 5 years was 75% and PFS was 70%. Patients undergoing RIC followed by alloSCT had fewer relapses, but this was negated by higher nonrelapse mortality (25%), resulting in a PFS similar to autoSCT.
CASE CONCLUSION
After treatment with bortezomib the patient is well for 9 months. Subsequently, however, he develops increasing lymphadenopathy and progressive fatigue. He is then started on lenalidomide 25 mg orally daily for 21 out of 28 days. He experiences significant fatigue with lenalidomide and prolonged neutropenia requiring dose delays, despite dose modification to 10 mg orally daily. He requires discontinuation of lenalidomide. Given persistent disease, the patient then begins treatment with ibrutinib. Within a few days of starting ibrutinib therapy, he experiences a marked but transient leukocytosis. Two months later, the patient’s palpable lymphadenopathy has decreased, and his anemia and thrombocytopenia related to MCL are improving. He has tolerated treatment well. His course has been complicated only by a mild, pruritic maculopapular eruption on his chest, back, and arms, that was responsive to topical low-dose steroids. He remains on ibrutinib 1 year later.
CONCLUSION
Advances in our understanding of MCL treatment are revolutionizing the approach to this once deadly disease. Over the next several years, these gains will weave themselves into the current treatment paradigm and likely alter the treatment landscape for MCL as we know it.
- Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
- Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer 2008;113:791–8.
- Geisler CH. Front-line treatment of mantle cell lymphoma. Haematologica 2010;95:1241–3.
- Fernandez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res 2010;70:1408–18.
- Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
- Bertoni F, Ponzoni M. The cellular origin of mantle cell lymphoma. Int J Biochem Cell Biol 2007;39:1747–53.
- de Boer CJ, van Krieken JH, Kluin-Nelemans HC, et al. Cyclin D1 messenger RNA overexpression as a marker for mantle cell lymphoma. Oncogene 1995;10:1833–40.
- Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest 2012;122:3416–23.
- Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185–97.
- Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood 2008;111:2385–7.
- Vegliante MC, Palomero J, Perez-Galan P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood 2013;121:2175–85.
- Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 2013;110:18250–5.
- Zelenetz AD, Abramson JS, Advani RH, et al. Non- Hodgkin’s lymphomas. J Natl Compr Canc Netw 2011;9: 484–560.
- Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
- Zelenetz AD, Abramson JS, Advani RH, et al. NCCN Clinical Practice Guidelines in Oncology: non-Hodgkin’s lymphomas. J Natl Compr Canc Netw 2010;8:288–334.
- Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
- Geisler CH, Kolstad A, Laurell A, et al. The Mantle Cell Lymphoma International Prognostic Index (MIPI) is superior to the International Prognostic Index (IPI) in predicting survival following intensive first-line immunochemotherapy and autologous stem cell transplantation (ASCT). Blood 2010;115:1530–3.
- Tiemann M, Schrader C, Klapper W, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 2005;131:29–38.
- Raty R, Franssila K, Joensuu H, et al. Ki-67 expression level, histological subtype, and the International Prognostic Index as outcome predictors in mantle cell lymphoma. Eur J Haematol 2002;69:11–20.
- Vogt N, Klapper W. Variability in morphology and cell proliferation in sequential biopsies of mantle cell lymphoma at diagnosis and relapse: clinical correlation and insights into disease progression. Histopathology 2013;62:334–42.
- Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 2008;112:2687–93.
- Howard OM, Gribben JG, Neuberg DS, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol 2002;20:1288–94.
- Griffiths R, Mikhael J, Gleeson M, et al. Addition of rituximab to chemotherapy alone as first-line therapy improves overall survival in elderly patients with mantle cell lymphoma. Blood 2011;118:4808–16.
- Lenz G, Dreyling M, Hoster E, et al. immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
- Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol 2005;23:7013–23.
- Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
- Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 2015;372:944–53.
- Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
- Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
- Houot R, Le Gouill S, Ojeda Uribe M, et al. Combination of rituximab, bortezomib, doxorubicin, dexamethasone and chlorambucil (RiPAD+C) as first-line therapy for elderly mantle cell lymphoma patients: results of a phase II trial from the GOELAMS. Ann Oncol 2012;23:1555–61.
- Ruan J, Martin P, Furman RR, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol 2011;29:690–7.
- Chang JE, Li H, Smith MR, et al. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: an Eastern Cooperative Oncology Group study (E1405). Blood 2014; 123:1665–73.
- Romaguera JE, Fayad LE, Feng L, et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol 2010;150:200–8.
- Bernstein SH, Epner E, Unger JM, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol 2013;24:1587–93.
- LaCasce AS, Vandergrift JL, Rodriguez MA, et al. Comparative outcome of initial therapy for younger patients with mantle cell lymphoma: an analysis from the NCCN NHL Database. Blood 2012;119:2093–9.
- Geisler CH, Kolstad A, Laurell A, et al. Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol 2012;158:355–62.
- Damon LE, Johnson JL, Niedzwiecki D, et al. Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol 2009;27:6101–8.
- van ‘t Veer MB, de Jong D, MacKenzie M, et al. High-dose Ara-C and beam with autograft rescue in R-CHOP responsive mantle cell lymphoma patients. Br J Haematol 2009;144:524–30.
- Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
- Hermine O, Hoster E, Walewski J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) increases overall survival when compared to 6 courses of CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: final analysis of the MCL Younger Trial of the European Mantle Cell Lymphoma Network (MCL net). In: American Society of Hematology Proceedings. December 8–11, 2012; Atlanta, GA. Abstract 151.
- Rummel MJ, Al-Batran SE, Kim SZ, et al. Bendamustine plus rituximab is effective and has a favorable toxicity profile in the treatment of mantle cell and low-grade non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:3383–9.
- Pham LV, Tamayo AT, Yoshimura LC, et al. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol 2003;171:88–95.
- Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
- Friedberg JW, Vose JM, Kelly JL, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood 2011;117:2807–12.
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–22.
- Qian Z, Zhang L, Cai Z, et al. Lenalidomide synergizes with dexamethasone to induce growth arrest and apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leuk Res 2011;35:380–6.
- Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
- Wang M, Fayad L, Wagner-Bartak N, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 2012;13:716–23.
- Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 2012;31: 119–32.
- Rinaldi A, Kwee I, Taborelli M, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol 2006;132:303–16.
- Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31:88–94.
- Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
- Rudelius M, Pittaluga S, Nishizuka S, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006;108: 1668–76.
- Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014;123:3398–405.
- Davids MS, Seymour JF, Gerecitano JF, et al. Updated results of a phase I first in human study of the BCL-2inhibitor ABT-199 in patients with relapsed/refractory NHL. J Clin Oncol 31, 2013 (suppl; abstr 8520).
- Ansell SM, Tang H, Kurtin PJ, et al. Temsirolimus and rituximab in patients with relapsed or refractory mantle cell lymphoma: a phase 2 study. Lancet Oncol 2011;12:361–8.
- Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–56.
- Wang M, Oki Y, Pro B, et al. Phase II study of yttrium-90-ibritumomab tiuxetan in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27:5213–8.
- Kolstad A, Laurell A, Jerkeman M, et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood 2014;123:2953–9.
- Fenske TS, Zhang MJ, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
- Dreyling M, Lenz G, Hoster E, et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 2005;105:2677–84.
- Goy A, Younes A, McLaughlin P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:667–75.
- Visco C, Finotto S, Zambello R, et al. Combination of rituximab, bendamustine, and cytarabine for patients with mantle-cell non-Hodgkin lymphoma ineligible for intensive regimens or autologous transplantation. J Clin Oncol 2013;10;31:1442–9.
- Gressin R, Callanan M, Daguindau N, et al. The Ribvd regimen (Rituximab IV, Bendamustine IV, Velcade SC, Dexamethasone IV) offers a high complete response rate In elderly patients with untreated mantle cell lymphoma. Preliminary results of the Lysa trial “Lymphome Du Manteau 2010 SA.” Blood 2013;122:370.
- Krishnan A, Nademanee A, Fung HC, et al. Phase II trial of a transplantation regimen of yttrium-90 ibritumomab tiuxetan and high-dose chemotherapy in patients with non-Hodgkin’s lymphoma. J Clin Oncol 2008;26:90–5.
- Nademanee A, Forman S, Molina A, et al. A phase 1/2 trial of high-dose yttrium-90-ibritumomab tiuxetan in combination with high-dose etoposide and cyclophosphamide followed by autologous stem cell transplantation in patients with poor-risk or relapsed non-Hodgkinlymphoma. Blood 2005;106:2896–902.
- Shimoni A, Avivi I, Rowe JM, et al. A randomized study comparing yttrium-90 ibritumomab tiuxetan (Zevalin) and high-dose BEAM chemotherapy versus BEAM alone as the conditioning regimen before autologous stem cell transplantation in patients with aggressive lymphoma. Cancer 2012;118:4706–14.
- Arranz R, García-Noblejas A, Grande C, et al. First-line treatment with rituximab-hyperCVAD alternating with rituximab-methotrexate-cytarabine and followed by consolidation with 90Y-ibritumomab-tiuxetan in patients with mantle cell lymphoma. Results of a multicenter, phase 2 pilot trial from the GELTAMO group. Haematologica 2013;98:1563-70.
- Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
- Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer 2008;113:791–8.
- Geisler CH. Front-line treatment of mantle cell lymphoma. Haematologica 2010;95:1241–3.
- Fernandez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res 2010;70:1408–18.
- Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
- Bertoni F, Ponzoni M. The cellular origin of mantle cell lymphoma. Int J Biochem Cell Biol 2007;39:1747–53.
- de Boer CJ, van Krieken JH, Kluin-Nelemans HC, et al. Cyclin D1 messenger RNA overexpression as a marker for mantle cell lymphoma. Oncogene 1995;10:1833–40.
- Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest 2012;122:3416–23.
- Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185–97.
- Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood 2008;111:2385–7.
- Vegliante MC, Palomero J, Perez-Galan P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood 2013;121:2175–85.
- Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 2013;110:18250–5.
- Zelenetz AD, Abramson JS, Advani RH, et al. Non- Hodgkin’s lymphomas. J Natl Compr Canc Netw 2011;9: 484–560.
- Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
- Zelenetz AD, Abramson JS, Advani RH, et al. NCCN Clinical Practice Guidelines in Oncology: non-Hodgkin’s lymphomas. J Natl Compr Canc Netw 2010;8:288–334.
- Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
- Geisler CH, Kolstad A, Laurell A, et al. The Mantle Cell Lymphoma International Prognostic Index (MIPI) is superior to the International Prognostic Index (IPI) in predicting survival following intensive first-line immunochemotherapy and autologous stem cell transplantation (ASCT). Blood 2010;115:1530–3.
- Tiemann M, Schrader C, Klapper W, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 2005;131:29–38.
- Raty R, Franssila K, Joensuu H, et al. Ki-67 expression level, histological subtype, and the International Prognostic Index as outcome predictors in mantle cell lymphoma. Eur J Haematol 2002;69:11–20.
- Vogt N, Klapper W. Variability in morphology and cell proliferation in sequential biopsies of mantle cell lymphoma at diagnosis and relapse: clinical correlation and insights into disease progression. Histopathology 2013;62:334–42.
- Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 2008;112:2687–93.
- Howard OM, Gribben JG, Neuberg DS, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol 2002;20:1288–94.
- Griffiths R, Mikhael J, Gleeson M, et al. Addition of rituximab to chemotherapy alone as first-line therapy improves overall survival in elderly patients with mantle cell lymphoma. Blood 2011;118:4808–16.
- Lenz G, Dreyling M, Hoster E, et al. immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
- Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol 2005;23:7013–23.
- Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
- Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 2015;372:944–53.
- Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
- Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
- Houot R, Le Gouill S, Ojeda Uribe M, et al. Combination of rituximab, bortezomib, doxorubicin, dexamethasone and chlorambucil (RiPAD+C) as first-line therapy for elderly mantle cell lymphoma patients: results of a phase II trial from the GOELAMS. Ann Oncol 2012;23:1555–61.
- Ruan J, Martin P, Furman RR, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol 2011;29:690–7.
- Chang JE, Li H, Smith MR, et al. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: an Eastern Cooperative Oncology Group study (E1405). Blood 2014; 123:1665–73.
- Romaguera JE, Fayad LE, Feng L, et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol 2010;150:200–8.
- Bernstein SH, Epner E, Unger JM, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol 2013;24:1587–93.
- LaCasce AS, Vandergrift JL, Rodriguez MA, et al. Comparative outcome of initial therapy for younger patients with mantle cell lymphoma: an analysis from the NCCN NHL Database. Blood 2012;119:2093–9.
- Geisler CH, Kolstad A, Laurell A, et al. Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol 2012;158:355–62.
- Damon LE, Johnson JL, Niedzwiecki D, et al. Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol 2009;27:6101–8.
- van ‘t Veer MB, de Jong D, MacKenzie M, et al. High-dose Ara-C and beam with autograft rescue in R-CHOP responsive mantle cell lymphoma patients. Br J Haematol 2009;144:524–30.
- Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
- Hermine O, Hoster E, Walewski J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) increases overall survival when compared to 6 courses of CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: final analysis of the MCL Younger Trial of the European Mantle Cell Lymphoma Network (MCL net). In: American Society of Hematology Proceedings. December 8–11, 2012; Atlanta, GA. Abstract 151.
- Rummel MJ, Al-Batran SE, Kim SZ, et al. Bendamustine plus rituximab is effective and has a favorable toxicity profile in the treatment of mantle cell and low-grade non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:3383–9.
- Pham LV, Tamayo AT, Yoshimura LC, et al. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol 2003;171:88–95.
- Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
- Friedberg JW, Vose JM, Kelly JL, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood 2011;117:2807–12.
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–22.
- Qian Z, Zhang L, Cai Z, et al. Lenalidomide synergizes with dexamethasone to induce growth arrest and apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leuk Res 2011;35:380–6.
- Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
- Wang M, Fayad L, Wagner-Bartak N, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 2012;13:716–23.
- Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 2012;31: 119–32.
- Rinaldi A, Kwee I, Taborelli M, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol 2006;132:303–16.
- Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31:88–94.
- Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
- Rudelius M, Pittaluga S, Nishizuka S, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006;108: 1668–76.
- Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014;123:3398–405.
- Davids MS, Seymour JF, Gerecitano JF, et al. Updated results of a phase I first in human study of the BCL-2inhibitor ABT-199 in patients with relapsed/refractory NHL. J Clin Oncol 31, 2013 (suppl; abstr 8520).
- Ansell SM, Tang H, Kurtin PJ, et al. Temsirolimus and rituximab in patients with relapsed or refractory mantle cell lymphoma: a phase 2 study. Lancet Oncol 2011;12:361–8.
- Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–56.
- Wang M, Oki Y, Pro B, et al. Phase II study of yttrium-90-ibritumomab tiuxetan in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27:5213–8.
- Kolstad A, Laurell A, Jerkeman M, et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood 2014;123:2953–9.
- Fenske TS, Zhang MJ, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
- Dreyling M, Lenz G, Hoster E, et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 2005;105:2677–84.
- Goy A, Younes A, McLaughlin P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:667–75.
- Visco C, Finotto S, Zambello R, et al. Combination of rituximab, bendamustine, and cytarabine for patients with mantle-cell non-Hodgkin lymphoma ineligible for intensive regimens or autologous transplantation. J Clin Oncol 2013;10;31:1442–9.
- Gressin R, Callanan M, Daguindau N, et al. The Ribvd regimen (Rituximab IV, Bendamustine IV, Velcade SC, Dexamethasone IV) offers a high complete response rate In elderly patients with untreated mantle cell lymphoma. Preliminary results of the Lysa trial “Lymphome Du Manteau 2010 SA.” Blood 2013;122:370.
- Krishnan A, Nademanee A, Fung HC, et al. Phase II trial of a transplantation regimen of yttrium-90 ibritumomab tiuxetan and high-dose chemotherapy in patients with non-Hodgkin’s lymphoma. J Clin Oncol 2008;26:90–5.
- Nademanee A, Forman S, Molina A, et al. A phase 1/2 trial of high-dose yttrium-90-ibritumomab tiuxetan in combination with high-dose etoposide and cyclophosphamide followed by autologous stem cell transplantation in patients with poor-risk or relapsed non-Hodgkinlymphoma. Blood 2005;106:2896–902.
- Shimoni A, Avivi I, Rowe JM, et al. A randomized study comparing yttrium-90 ibritumomab tiuxetan (Zevalin) and high-dose BEAM chemotherapy versus BEAM alone as the conditioning regimen before autologous stem cell transplantation in patients with aggressive lymphoma. Cancer 2012;118:4706–14.
- Arranz R, García-Noblejas A, Grande C, et al. First-line treatment with rituximab-hyperCVAD alternating with rituximab-methotrexate-cytarabine and followed by consolidation with 90Y-ibritumomab-tiuxetan in patients with mantle cell lymphoma. Results of a multicenter, phase 2 pilot trial from the GELTAMO group. Haematologica 2013;98:1563-70.
Unique, multi-omic profile found in children with autism and functional GI disorders
The gut microbiomes of children with autism spectrum disorder (ASD) and functional gastrointestinal disorders (FGID) had significantly higher levels of several Clostridium species and lower concentrations of other bacteria compared with neurotypical children with and without FGIDs, which correlated with increases in inflammatory cytokines, decreased tryptophan, and increased serotonin, according to a small, single-center, cross-sectional study.
This “unique multi-omic profile [was] specific to ASD-FGID and ASD-FGID with abdominal pain,” wrote Ruth Ann Luna, PhD, of Texas Children’s Microbiome Center at Texas Children’s Hospital, Houston, and her associates. The report was published online in Cellular and Molecular Gastroenterology and Hepatology (doi: 10.1016/j.jcmgh.2016.11.008).
Children with ASD are at increased risk for FGIDs such as functional constipation, nonretentive fecal incontinence, functional abdominal pain, abdominal migraines, and irritable bowel syndrome, compared with their neurotypical peers. Changes in the gut microbiome can affect immunologic pathways and the balance between tryptophan and serotonin. This altered “microbial-gut-brain axis” has been reported in both ASD and FGID, suggesting “that altered gut-brain communications not only may play a role in the increased occurrence of FGIDs in ASD individuals, but could advance our understanding of potential risk factors for FGID in the ASD community,” the researchers wrote.
Previous studies of stool specimens have found higher levels of several species of Clostridium in pediatric ASD compared with neurotypical children. To confirm and expand on that work, the investigators examined microbial and neuroimmune markers in rectal biopsies and blood specimens from 14 children with ASD-FGID, 15 neurotypical children with FGID, and 6 asymptomatic neurotypical children. Participants were recruited from Nationwide Children’s Hospital in Columbus, Ohio. The researchers quantified microbial 16S ribosomal DNA community signatures, cytokines, chemokines, and serotonergic metabolites, and correlated results with parental responses to the Questionnaire on Pediatric Gastrointestinal Symptoms–Rome III version.
The ASD-FGID group had significantly higher numbers for ribosomal DNA sequences for Clostridium lituseburense (P = .002), Lachnoclostridium bolteae (P = .02), Lachnoclostridium hathewayi (P = .03), Clostridium aldenense (P = .04), and Oscillospira plautii (P = .04), compared with neurotypical children with and without FGID. Children with ASD-FGID also had significantly lower levels of Dorea formicigenerans (P = .006), Blautia luti (P = .02), and Sutterella species (P = .03). “Overall, our identification of clostridial species aligns with previous autism studies that have identified microbiome alterations,” the researchers noted.
They also looked specifically at abdominal pain. Children with ASD-FGID and abdominal pain had significantly higher gut mucosal levels of Turicibacter sanguinis (P = .03), Clostridium aldenense (P = .004), Clostridium lituseburense (P = .003), Oscillospira plautii (P = .01), Clostridium disporicum (P = .049), and Clostridium tertium (P = .045) than did any other subgroup, the investigators found. Patients with both ASD-FGID and abdominal pain also had significantly higher levels of C. aldenense (P = .03), O. plautii (P = .04), Tyzzerella species (P = .045), and Parasutterella excrementihominis (P = .04) than did ASD-FGID patients without abdominal pain.
Both C. disporicum and C. tertium correlated with increases in the proinflammatory cytokines IL6 and interferon-gamma. Levels of these cytokines were highest in patients with ASD-FGID, and IL6 was highest of all among children with ASD-FGID with abdominal pain. Another proinflammatory cytokine, IL17A, also correlated with Clostridia species that were enriched in children with ASD-FGID. Both IL6 and IL17A have been implicated in autism-like phenotypes in rodents, the researchers noted. Several other cytokines also were linked to ASD-FGID, and abdominal pain correlated significantly with increases in MCP-1 (P = .03) and eotaxin (P = .03).
Gut mucosal levels of tryptophan were significantly lower among children with ASD-FGID compared with neurotypical children, either with (P = .006) or without (P = .009) FGID. In contrast, gut mucosal levels of 5-HIAA, the primary metabolite of serotonin, were significantly higher among children with ASD-FGID compared with neurotypical children (P = .01). Increased 5-HIAA also correlated significantly with abdominal pain (P = .04). Six species of bacteria correlated significantly with tryptophan or serotonin, implicating the gut microbiome in the serotonin pathway.
“Although these initial findings are correlative, these data form the framework for future studies targeting tryptophan-serotonin metabolism and inflammatory pathways in FGID in ASD,” the researchers concluded.
The U.S. Department of Health and Human Services funded the work. The investigators had no relevant disclosures.
Autism-spectrum disorder is a serious and increasingly prevalent developmental behavior disorder often accompanied and aggravated by a range of gastrointestinal and cognitive dysfunctions. Its etiology probably involves maternal diet and inflammatory events that alter central nervous system neurodevelopment critical to the cognition of social interaction. Candidate causal products of these events include the cytokines IL-6 and IL-17A, and certain bioactive amines, notably serotonin. Functional gastrointestinal disorders share these same molecules as biomarkers and disease modifiers, probably elicited in part by the intestinal microbiome. Hence, the comorbidity in ASD suggests these two disease processes are etiologically related.
The study by Luna and colleagues tightens the case for a microbial hub and serotonin and cytokine spokes in the gastrointestinal dysfunction of ASD: elevated mucosal tissue levels of select microbial taxa, mainly members of the genus Clostridium, and mucosal production of cytokines and serotonin-pathway bioamines associated with these and other select microbial species. Important and challenging questions loom ahead. What are the direct mucosal cell types and functions targeted of this network for the microbiota, and via what microbial products? Might they elicit epithelial or mucosal hematopoietic cell cytokine production that in turn causes mucosal bioamine secretion? And, what associated microbiota and products are just secondarily altered and not causally involved? The exciting study of Luna and colleagues raises confidence for this path ahead, and its promise for clarifying ASD pathogenesis and uncovering targetable elements for intervention.
Jonathan Braun, MD, PhD, is professor and chair of pathology and laboratory medicine, UCLA David Geffen School of Medicine, UCLA Health System, Los Angeles. He has no conflicts of interest.
Autism-spectrum disorder is a serious and increasingly prevalent developmental behavior disorder often accompanied and aggravated by a range of gastrointestinal and cognitive dysfunctions. Its etiology probably involves maternal diet and inflammatory events that alter central nervous system neurodevelopment critical to the cognition of social interaction. Candidate causal products of these events include the cytokines IL-6 and IL-17A, and certain bioactive amines, notably serotonin. Functional gastrointestinal disorders share these same molecules as biomarkers and disease modifiers, probably elicited in part by the intestinal microbiome. Hence, the comorbidity in ASD suggests these two disease processes are etiologically related.
The study by Luna and colleagues tightens the case for a microbial hub and serotonin and cytokine spokes in the gastrointestinal dysfunction of ASD: elevated mucosal tissue levels of select microbial taxa, mainly members of the genus Clostridium, and mucosal production of cytokines and serotonin-pathway bioamines associated with these and other select microbial species. Important and challenging questions loom ahead. What are the direct mucosal cell types and functions targeted of this network for the microbiota, and via what microbial products? Might they elicit epithelial or mucosal hematopoietic cell cytokine production that in turn causes mucosal bioamine secretion? And, what associated microbiota and products are just secondarily altered and not causally involved? The exciting study of Luna and colleagues raises confidence for this path ahead, and its promise for clarifying ASD pathogenesis and uncovering targetable elements for intervention.
Jonathan Braun, MD, PhD, is professor and chair of pathology and laboratory medicine, UCLA David Geffen School of Medicine, UCLA Health System, Los Angeles. He has no conflicts of interest.
Autism-spectrum disorder is a serious and increasingly prevalent developmental behavior disorder often accompanied and aggravated by a range of gastrointestinal and cognitive dysfunctions. Its etiology probably involves maternal diet and inflammatory events that alter central nervous system neurodevelopment critical to the cognition of social interaction. Candidate causal products of these events include the cytokines IL-6 and IL-17A, and certain bioactive amines, notably serotonin. Functional gastrointestinal disorders share these same molecules as biomarkers and disease modifiers, probably elicited in part by the intestinal microbiome. Hence, the comorbidity in ASD suggests these two disease processes are etiologically related.
The study by Luna and colleagues tightens the case for a microbial hub and serotonin and cytokine spokes in the gastrointestinal dysfunction of ASD: elevated mucosal tissue levels of select microbial taxa, mainly members of the genus Clostridium, and mucosal production of cytokines and serotonin-pathway bioamines associated with these and other select microbial species. Important and challenging questions loom ahead. What are the direct mucosal cell types and functions targeted of this network for the microbiota, and via what microbial products? Might they elicit epithelial or mucosal hematopoietic cell cytokine production that in turn causes mucosal bioamine secretion? And, what associated microbiota and products are just secondarily altered and not causally involved? The exciting study of Luna and colleagues raises confidence for this path ahead, and its promise for clarifying ASD pathogenesis and uncovering targetable elements for intervention.
Jonathan Braun, MD, PhD, is professor and chair of pathology and laboratory medicine, UCLA David Geffen School of Medicine, UCLA Health System, Los Angeles. He has no conflicts of interest.
The gut microbiomes of children with autism spectrum disorder (ASD) and functional gastrointestinal disorders (FGID) had significantly higher levels of several Clostridium species and lower concentrations of other bacteria compared with neurotypical children with and without FGIDs, which correlated with increases in inflammatory cytokines, decreased tryptophan, and increased serotonin, according to a small, single-center, cross-sectional study.
This “unique multi-omic profile [was] specific to ASD-FGID and ASD-FGID with abdominal pain,” wrote Ruth Ann Luna, PhD, of Texas Children’s Microbiome Center at Texas Children’s Hospital, Houston, and her associates. The report was published online in Cellular and Molecular Gastroenterology and Hepatology (doi: 10.1016/j.jcmgh.2016.11.008).
Children with ASD are at increased risk for FGIDs such as functional constipation, nonretentive fecal incontinence, functional abdominal pain, abdominal migraines, and irritable bowel syndrome, compared with their neurotypical peers. Changes in the gut microbiome can affect immunologic pathways and the balance between tryptophan and serotonin. This altered “microbial-gut-brain axis” has been reported in both ASD and FGID, suggesting “that altered gut-brain communications not only may play a role in the increased occurrence of FGIDs in ASD individuals, but could advance our understanding of potential risk factors for FGID in the ASD community,” the researchers wrote.
Previous studies of stool specimens have found higher levels of several species of Clostridium in pediatric ASD compared with neurotypical children. To confirm and expand on that work, the investigators examined microbial and neuroimmune markers in rectal biopsies and blood specimens from 14 children with ASD-FGID, 15 neurotypical children with FGID, and 6 asymptomatic neurotypical children. Participants were recruited from Nationwide Children’s Hospital in Columbus, Ohio. The researchers quantified microbial 16S ribosomal DNA community signatures, cytokines, chemokines, and serotonergic metabolites, and correlated results with parental responses to the Questionnaire on Pediatric Gastrointestinal Symptoms–Rome III version.
The ASD-FGID group had significantly higher numbers for ribosomal DNA sequences for Clostridium lituseburense (P = .002), Lachnoclostridium bolteae (P = .02), Lachnoclostridium hathewayi (P = .03), Clostridium aldenense (P = .04), and Oscillospira plautii (P = .04), compared with neurotypical children with and without FGID. Children with ASD-FGID also had significantly lower levels of Dorea formicigenerans (P = .006), Blautia luti (P = .02), and Sutterella species (P = .03). “Overall, our identification of clostridial species aligns with previous autism studies that have identified microbiome alterations,” the researchers noted.
They also looked specifically at abdominal pain. Children with ASD-FGID and abdominal pain had significantly higher gut mucosal levels of Turicibacter sanguinis (P = .03), Clostridium aldenense (P = .004), Clostridium lituseburense (P = .003), Oscillospira plautii (P = .01), Clostridium disporicum (P = .049), and Clostridium tertium (P = .045) than did any other subgroup, the investigators found. Patients with both ASD-FGID and abdominal pain also had significantly higher levels of C. aldenense (P = .03), O. plautii (P = .04), Tyzzerella species (P = .045), and Parasutterella excrementihominis (P = .04) than did ASD-FGID patients without abdominal pain.
Both C. disporicum and C. tertium correlated with increases in the proinflammatory cytokines IL6 and interferon-gamma. Levels of these cytokines were highest in patients with ASD-FGID, and IL6 was highest of all among children with ASD-FGID with abdominal pain. Another proinflammatory cytokine, IL17A, also correlated with Clostridia species that were enriched in children with ASD-FGID. Both IL6 and IL17A have been implicated in autism-like phenotypes in rodents, the researchers noted. Several other cytokines also were linked to ASD-FGID, and abdominal pain correlated significantly with increases in MCP-1 (P = .03) and eotaxin (P = .03).
Gut mucosal levels of tryptophan were significantly lower among children with ASD-FGID compared with neurotypical children, either with (P = .006) or without (P = .009) FGID. In contrast, gut mucosal levels of 5-HIAA, the primary metabolite of serotonin, were significantly higher among children with ASD-FGID compared with neurotypical children (P = .01). Increased 5-HIAA also correlated significantly with abdominal pain (P = .04). Six species of bacteria correlated significantly with tryptophan or serotonin, implicating the gut microbiome in the serotonin pathway.
“Although these initial findings are correlative, these data form the framework for future studies targeting tryptophan-serotonin metabolism and inflammatory pathways in FGID in ASD,” the researchers concluded.
The U.S. Department of Health and Human Services funded the work. The investigators had no relevant disclosures.
The gut microbiomes of children with autism spectrum disorder (ASD) and functional gastrointestinal disorders (FGID) had significantly higher levels of several Clostridium species and lower concentrations of other bacteria compared with neurotypical children with and without FGIDs, which correlated with increases in inflammatory cytokines, decreased tryptophan, and increased serotonin, according to a small, single-center, cross-sectional study.
This “unique multi-omic profile [was] specific to ASD-FGID and ASD-FGID with abdominal pain,” wrote Ruth Ann Luna, PhD, of Texas Children’s Microbiome Center at Texas Children’s Hospital, Houston, and her associates. The report was published online in Cellular and Molecular Gastroenterology and Hepatology (doi: 10.1016/j.jcmgh.2016.11.008).
Children with ASD are at increased risk for FGIDs such as functional constipation, nonretentive fecal incontinence, functional abdominal pain, abdominal migraines, and irritable bowel syndrome, compared with their neurotypical peers. Changes in the gut microbiome can affect immunologic pathways and the balance between tryptophan and serotonin. This altered “microbial-gut-brain axis” has been reported in both ASD and FGID, suggesting “that altered gut-brain communications not only may play a role in the increased occurrence of FGIDs in ASD individuals, but could advance our understanding of potential risk factors for FGID in the ASD community,” the researchers wrote.
Previous studies of stool specimens have found higher levels of several species of Clostridium in pediatric ASD compared with neurotypical children. To confirm and expand on that work, the investigators examined microbial and neuroimmune markers in rectal biopsies and blood specimens from 14 children with ASD-FGID, 15 neurotypical children with FGID, and 6 asymptomatic neurotypical children. Participants were recruited from Nationwide Children’s Hospital in Columbus, Ohio. The researchers quantified microbial 16S ribosomal DNA community signatures, cytokines, chemokines, and serotonergic metabolites, and correlated results with parental responses to the Questionnaire on Pediatric Gastrointestinal Symptoms–Rome III version.
The ASD-FGID group had significantly higher numbers for ribosomal DNA sequences for Clostridium lituseburense (P = .002), Lachnoclostridium bolteae (P = .02), Lachnoclostridium hathewayi (P = .03), Clostridium aldenense (P = .04), and Oscillospira plautii (P = .04), compared with neurotypical children with and without FGID. Children with ASD-FGID also had significantly lower levels of Dorea formicigenerans (P = .006), Blautia luti (P = .02), and Sutterella species (P = .03). “Overall, our identification of clostridial species aligns with previous autism studies that have identified microbiome alterations,” the researchers noted.
They also looked specifically at abdominal pain. Children with ASD-FGID and abdominal pain had significantly higher gut mucosal levels of Turicibacter sanguinis (P = .03), Clostridium aldenense (P = .004), Clostridium lituseburense (P = .003), Oscillospira plautii (P = .01), Clostridium disporicum (P = .049), and Clostridium tertium (P = .045) than did any other subgroup, the investigators found. Patients with both ASD-FGID and abdominal pain also had significantly higher levels of C. aldenense (P = .03), O. plautii (P = .04), Tyzzerella species (P = .045), and Parasutterella excrementihominis (P = .04) than did ASD-FGID patients without abdominal pain.
Both C. disporicum and C. tertium correlated with increases in the proinflammatory cytokines IL6 and interferon-gamma. Levels of these cytokines were highest in patients with ASD-FGID, and IL6 was highest of all among children with ASD-FGID with abdominal pain. Another proinflammatory cytokine, IL17A, also correlated with Clostridia species that were enriched in children with ASD-FGID. Both IL6 and IL17A have been implicated in autism-like phenotypes in rodents, the researchers noted. Several other cytokines also were linked to ASD-FGID, and abdominal pain correlated significantly with increases in MCP-1 (P = .03) and eotaxin (P = .03).
Gut mucosal levels of tryptophan were significantly lower among children with ASD-FGID compared with neurotypical children, either with (P = .006) or without (P = .009) FGID. In contrast, gut mucosal levels of 5-HIAA, the primary metabolite of serotonin, were significantly higher among children with ASD-FGID compared with neurotypical children (P = .01). Increased 5-HIAA also correlated significantly with abdominal pain (P = .04). Six species of bacteria correlated significantly with tryptophan or serotonin, implicating the gut microbiome in the serotonin pathway.
“Although these initial findings are correlative, these data form the framework for future studies targeting tryptophan-serotonin metabolism and inflammatory pathways in FGID in ASD,” the researchers concluded.
The U.S. Department of Health and Human Services funded the work. The investigators had no relevant disclosures.
FROM CELLULAR AND MOLECULAR GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: The mucosal microbiome of children with comorbid autism spectrum disorder and functional gastrointestinal disorders significantly differed from that of neurotypical children with and without FGIDs, and these differences correlated with altered levels of inflammatory cytokines, tryptophan, and serotonin.
Major finding: Children with ASD-FGID had significant increases in Clostridium lituseburense (P = .002), Lachnoclostridium bolteae (P = .02), Lachnoclostridium hathewayi (P = .03), Clostridium aldenense (P = .04), and Oscillospira plautii (P = .04), and significant decreases in Dorea formicigenerans (P = .006), Blautia luti (P = .020), and Sutterella species (P = .025). The ASD-FGID phenotype was characterized by significantly lower gut levels of tryptophan, with higher levels of the serotonin metabolite 5-HIAA, and with several proinflammatory cytokines. Several bacterial species correlated with tryptophan, serotonin, or proinflammatory cytokines.
Data source: A single-center cross-sectional study of 14 children with ASD-FGID and 21 neurotypical children, of whom 15 had FGIDs.
Disclosures: The U.S. Department of Health and Human Services funded the work. The investigators had no relevant disclosures.