User login
A Tough Egg to Crack
A 68-year-old woman presented to the emergency department with altered mental status. On the morning prior to admission, she was fully alert and oriented. Over the course of the day, she became more confused and somnolent, and by the evening, she was unarousable to voice. She had not fallen and had no head trauma.
Altered mental status may arise from metabolic (eg, hyponatremia), infectious (eg, urinary tract infection), structural (eg, subdural hematoma), or toxin-related (eg, adverse medication effect) processes. Any of these categories of encephalopathy can develop gradually over the course of a day.
One year prior, the patient was admitted for a similar episode of altered mental status. Asterixis and elevated transaminases prompted an abdominal ultrasound, which revealed a nodular liver and ascites. Paracentesis revealed a high serum-ascites albumin gradient. The diagnosis of cirrhosis was made based on these findings. Testing for viral hepatitis, autoimmune hepatitis, hemochromatosis, and Wilson’s disease were negative. Although steatosis was not detected on ultrasound, nonalcoholic fatty liver disease (NAFLD) was suspected based on the patient’s risk factors of hypertension and type 2 diabetes mellitus. She had four additional presentations of altered mental status with asterixis; each episode resolved with lactulose.
Other medical history included end-stage renal disease (ESRD) requiring hemodialysis. Her medications were labetalol, amlodipine, insulin, propranolol, lactulose, and rifaximin. She was originally from China and moved to the United States 10 years earlier. Given concerns about her ability to consistently take medications, she had moved to a long-term facility. She did not use alcohol, tobacco, or illicit substances.
The normalization of the patient’s mental status after lactulose treatment, especially in the context of recurrent episodes, is characteristic of hepatic encephalopathy, in which ammonia and other substances bypass hepatic metabolism and impair cerebral function. Hepatic encephalopathy is the most common cause of lactulose-responsive encephalopathy, and may recur in the setting of infection or nonadherence with lactulose and rifaximin. Other causes of lactulose-responsive encephalopathy include hyperammonemia caused by urease-producing bacterial infection (eg, Proteus), valproic acid toxicity, and urea cycle abnormalities.
Other causes of confusion with a self-limited course should be considered for the current episode. A postictal state is possible, but convulsions were not reported. The patient is at risk of hypoglycemia from insulin use and impaired gluconeogenesis due to cirrhosis and ESRD, but low blood sugar would have likely been detected at the time of hospitalization. Finally, she might have experienced episodic encephalopathy from ingestion of unreported medications or toxins, whose effects may have resolved with abstinence during hospitalization.
The patient’s temperature was 37.8°C, pulse 73 beats/minute, blood pressure 133/69 mmHg, respiratory rate 12 breaths/minute, and oxygen saturation 98% on ambient air. Her body mass index (BMI) was 19 kg/m2. She was somnolent but was moving all four extremities spontaneously. Her pupils were symmetric and reactive. There was no facial asymmetry. Biceps and patellar reflexes were 2+ bilaterally. Babinski sign was absent bilaterally. The patient could not cooperate with the assessment for asterixis. Her sclerae were anicteric. The jugular venous pressure was estimated at 13 cm of water. Her heart was regular with no murmurs. Her lungs were clear. She had a distended, nontender abdomen with caput medusae. She had symmetric pitting edema in her lower extremities up to the shins.
The elevated jugular venous pressure, lower extremity edema, and distended abdomen suggest volume overload. Jugular venous distention with clear lungs is characteristic of right ventricular failure from pulmonary hypertension, right ventricular myocardial infarction, tricuspid regurgitation, or constrictive pericarditis. However, chronic biventricular heart failure often presents in this manner and is more common than the aforementioned conditions. ESRD and cirrhosis may be contributing to the hypervolemia.
Although Asian patients may exhibit metabolic syndrome and NAFLD at a lower BMI than non-Asians, her BMI is uncharacteristically low for NAFLD, especially given the increased weight expected from volume overload. There are no signs of infection to account for worsening of hepatic encephalopathy.
Laboratory tests demonstrated a white blood cell count of 4400/µL with a normal differential, hemoglobin of 10.3 g/dL, and platelet count of 108,000 per cubic millimeter. Mean corpuscular volume was 103 fL. Basic metabolic panel was normal with the exception of blood urea nitrogen of 46 mg/dL and a creatinine of 6.4 mg/dL. Aspartate aminotransferase was 34 units/L, alanine aminotransferase 34 units/L, alkaline phosphatase 289 units/L (normal, 31-95), gamma-glutamyl transferase 104 units (GGT, normal, 12-43), total bilirubin 0.8 mg/dL, and albumin 2.5 g/dL (normal, 3.5-4.5). Pro-brain natriuretic peptide was 1429 pg/mL (normal, <100). The international normalized ratio (INR) was 1.0. Urinalysis showed trace proteinuria. The chest x-ray was normal. A noncontrast computed tomography (CT) of the head demonstrated no intracranial pathology. An abdominal ultrasound revealed a normal-sized nodular liver, a nonocclusive portal vein thrombus (PVT), splenomegaly (15 cm in length), and trace ascites. There was no biliary dilation, hepatic steatosis, or hepatic mass.
The evolving data set presents a mixed picture about the state of the liver. The distended abdominal wall veins, thrombocytopenia, and splenomegaly are commonly observed in advanced cirrhosis, but these findings reflect the associated portal hypertension and not the liver disease itself. The normal bilirubin and INR suggest preserved liver function and decrease the likelihood of cirrhosis being responsible for the portal hypertension. However, the elevated alkaline phosphatase and GGT levels suggest an infiltrative liver disease, such as lymphoma, sarcoidosis, or amyloidosis.
Furthermore, while a nodular liver on imaging is consistent with cirrhosis, no steatosis was noted to support the presumed diagnosis of NAFLD. One explanation for this discrepancy is that fatty infiltration may be absent when NAFLD-associated cirrhosis develops. In summary, there is evidence of liver disease, and there is evidence of portal hypertension, but there is no evidence of liver parenchymal failure. The key features of the latter – spider angiomata, palmar erythema, hyperbilirubinemia, and coagulopathy – are absent.
Noncirrhotic portal hypertension (NCPH) is an alternative explanation for the patient’s findings. NCPH is an elevation in the portal venous system pressure that arises from intrahepatic (but noncirrhotic) disease or from extrahepatic disease. Hepatic schistosomiasis is an example of intrahepatic but noncirrhotic portal hypertension. PVT that arises on account of a hypercoagulable condition (eg, abdominal malignancy, pancreatitis, or myeloproliferative disorders) is a prototype of extrahepatic NCPH. At this point, it is impossible to know if the PVT is a complication of NCPH or a cause of NCPH. PVT as a complication of cirrhosis is less likely.
An abdominal CT scan would better assess the hepatic parenchyma and exclude abdominal malignancies such as pancreatic adenocarcinoma. An echocardiogram is indicated to evaluate the cause of the elevated jugular venous pressure. A liver biopsy and measurement of portal venous pressure would help distinguish between cirrhotic and noncirrhotic portal hypertension.
Hepatitis A, B, and C serologies were negative as were antinuclear and antimitochondrial antibodies. Ferritin and ceruloplasmin levels were normal. A CT scan of the abdomen with contrast demonstrated a nodular liver contour, splenomegaly, and a nonocclusive PVT (Figure 1). A transthoracic echocardiogram showed normal biventricular systolic function and size, normal diastolic function, a pulmonary artery systolic pressure of 57 mmHg (normal, < 25), moderate tricuspid regurgitation, and no pericardial effusion or thickening. The patient’s confusion and somnolence resolved after two days of lactulose therapy. She denied the use of other medications, supplements, or herbs.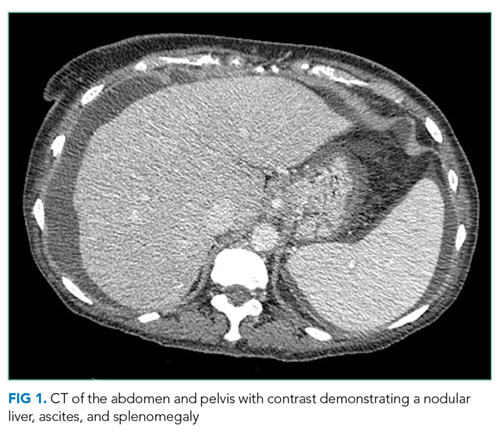
Pulmonary hypertension is usually a consequence of cardiopulmonary disease, but there is no exam or imaging evidence for left ventricular failure, mitral stenosis, obstructive lung disease, or interstitial lung disease. Portopulmonary hypertension (a form of pulmonary hypertension) can develop as a consequence of end-stage liver disease. The most common cause of hepatic encephalopathy due to portosystemic shunting is cirrhosis, but such shunting also arises in NCPH.
Schistosomiasis is the most common cause of NCPH worldwide. Parasite eggs trapped within the terminal portal venules cause inflammation, leading to fibrosis and intrahepatic portal hypertension. The liver becomes nodular on account of these changes, but the overall hepatic function is typically preserved. Portal hypertension, variceal bleeding, and pulmonary hypertension are common complications. The latter can arise from portosystemic shunting, which leads to embolization of schistosome eggs into the pulmonary circulation, where a granulomatous reaction ensues.
A percutaneous liver biopsy showed granulomatous inflammation and dilated portal venules consistent with increased resistance to venous inflow (Figure 2). There was no sinusoidal congestion to indicate impaired hepatic venous outflow. Mild sinusoidal and portal fibrosis and increased iron in Kupffer cells were noted. There was no evidence of cirrhosis or steatohepatitis. Stains for acid-fast bacilli and fungi were negative. 16S rDNA (a test assessing for bacterial DNA) and Mycobacterium tuberculosis polymerase chain reactions were negative. The biopsy confirmed the diagnosis of noncirrhotic portal hypertension.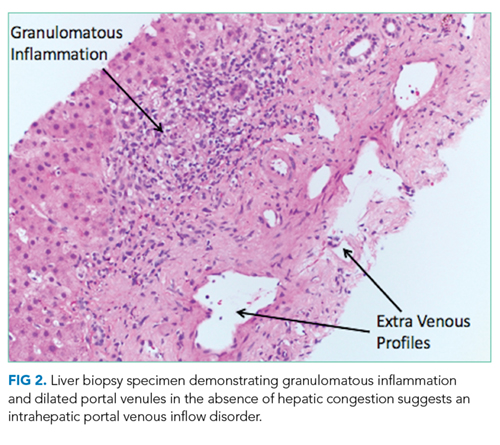
Hepatic granulomas can arise from infectious, immunologic, toxic, and malignant diseases. In the United States, immunologic disorders, such as sarcoidosis and primary biliary cholangitis, are the most common causes of granulomatous hepatitis. The patient lacks extrahepatic features of the former. The absence of bile duct injury and negative antimitochondrial antibody exclude the latter. None of the listed medications are commonly associated with hepatic granulomas. The ultrasound, CT scan, and biopsy did not reveal a granulomatous malignancy such as lymphoma.
Infections, such as brucellosis, Q fever, and tuberculosis, are common causes of granulomatous hepatitis in the developing world. Tuberculosis is prevalent in China, but the test results do not support tuberculosis as a unifying diagnosis.
Schistosomiasis accounts for the major clinical features (portal and pulmonary hypertension and preserved liver function) and hepatic pathology (ie, portal venous fibrosis with granulomatous inflammation) in this case and is prevalent in China, where the patient emigrated from. The biopsy specimen should be re-examined for schistosome eggs and serologic tests for schistosomiasis pursued.
Antibodies to human immunodeficiency virus, Brucella, Bartonella quintana, Bartonella henselae, Coxiella burnetii, Francisella tularensis, and Histoplasma were negative. Cryptococcal antigen and rapid plasma reagin were negative. IgG antibodies to Schistosoma were 0.21 units (normal, < 0.19 units). Based on the patient’s epidemiology, biopsy findings, and serology results, hepatic schistosomiasis was diagnosed. Praziquantel was prescribed. She continues to receive daily lactulose and rifaximin and has not had any episodes of encephalopathy in the year after discharge.
COMMENTARY
Portal hypertension arises when there is resistance to flow in the portal venous system. It is defined as a pressure gradient greater than 5 mmHg between the portal vein and the intra-abdominal portion of the inferior vena cava.1 Clinicians are familiar with the manifestations of portal hypertension – portosystemic shunting leading to encephalopathy and variceal hemorrhage, ascites, and splenomegaly with thrombocytopenia – because of their close association with cirrhosis. In developed countries, cirrhosis accounts for over 90% of cases of portal hypertension.1 In the remaining 10%, conditions such as portal vein thrombosis primarily affect the portal vasculature and increase resistance to portal blood flow while leaving hepatic synthetic function relatively spared (Figure 3). Therefore, cirrhosis cannot be inferred with certainty from signs of portal hypertension alone.
Liver biopsy is the gold standard for the diagnosis of cirrhosis, but this method is increasingly being replaced by noninvasive assessments of liver fibrosis, including imaging and scoring systems.2 Clinicians often infer cirrhosis from the combination of a known cause of liver injury, abnormal liver biochemical tests, evidence of liver dysfunction, and signs of portal hypertension.3 However, when signs of portal hypertension are present, but liver dysfunction cannot be established on physical exam (eg, palmar erythema, spider nevi, gynecomastia, and testicular atrophy) or laboratory testing (eg, low albumin, elevated INR, and elevated bilirubin), noncirrhotic causes of portal hypertension should be considered. In this case, the biopsy showed vascular changes that suggested impaired venous inflow without bridging fibrosis, which pointed to NCPH.
NCPH is categorized based on the location of resistance to blood flow: prehepatic (eg, portal vein thrombosis), intrahepatic (eg, schistosomiasis), and posthepatic (eg, right-sided heart failure).1 In our patient, the dilated portal venules (inflow) in the presence of normal hepatic vein outflow suggested an increased intrahepatic resistance to blood flow. This finding excluded a causal role of the portal vein thrombosis and prompted testing for schistosomiasis.
Schistosomiasis affects more than 200 million people worldwide and is prevalent in Sub-Saharan Africa, South America, Egypt, China, and Southeast Asia.4,5 Transmission occurs in fresh water, where the infectious form of the parasite is released from snails.4,6 Schistosome worms are not found in the United States, but as a result of immigration and travel, more than 400,000 people in the United States are estimated to be infected.5
Chronic schistosomiasis develops from the host’s granulomatous reaction to schistosome eggs whose location (depending on the species) leads to genitourinary, intestinal, hepatic, or rarely, neurologic disease.6 Hepatic schistosomiasis arises when eggs released in the portal venous system lodge in small portal venules and cause granulomatous inflammation, periportal fibrosis, and microvascular obstruction.6 The resultant portal hypertension develops insidiously, but the architecture and synthetic function of the liver is maintained until the very late stages of disease.6,7 Pulmonary hypertension can arise from the embolization of eggs to the pulmonary arterioles via portosystemic collaterals.
The demonstration of eggs in stool is the gold standard for the diagnosis of hepatic schistosomiasis, which is most commonly caused by Schistosoma mansoni and S. japonicum.7 Serologic assays provide evidence of infection or exposure but may cross-react with other helminths. Liver biopsy may reveal characteristic histopathologic findings, including granulomatous inflammation, distorted vasculature, and the deposition of collagen deposits in the periportal space, leading to “pipestem fibrosis.”8,9 If eggs cannot be detected on stool or histology, then serology, secondary histologic changes, and sometimes PCR are used to diagnose hepatic schistosomiasis. In our patient, the epidemiology, Schistosoma antibody titer, pulmonary hypertension, and liver biopsy with granulomatous inflammation, periportal fibrosis, and intrahepatic portal venule dilation were diagnostic of hepatic schistosomiasis.
The recurrent episodes of confusion which resolved with lactulose therapy were suggestive of hepatic encephalopathy, which results from shunting and accumulation of neurotoxic substances that would otherwise undergo hepatic metabolism.10 Clinicians are most familiar with hepatic encephalopathy in cirrhosis, where multiple liver functions – synthesis, excretion, metabolism, and circulation – simultaneously fail. NCPH represents a scenario where only the circulation is impaired, but this is sufficient to cause the portosystemic shunting that leads to encephalopathy. Our patient’s recurrent hepatic encephalopathy, despite adherence to lactulose and rifaximin and its resolution after praziquantel treatment, underscores the importance of addressing the underlying cause of portosystemic shunting.Associating portal hypertension with cirrhosis is efficient and accurate in many cases. However, when specific manifestations of cirrhosis are lacking, clinicians must decouple this association and pursue an alternative explanation for portal hypertension. The presence of some intrahepatic pathology (from schistosomiasis) but no cirrhosis made this case a particularly tough egg to crack.
Teaching Points
- In the developed world, 90% of portal hypertension is due to cirrhosis. Hepatic schistosomiasis is the most common cause of NCPH worldwide.
- Chronic schistosomiasis affects the gastrointestinal, hepatic, and genitourinary systems and causes significant global morbidity and mortality.
- Visualization of schistosome eggs is the diagnostic gold standard. Indirect testing such as schistosoma antibodies and secondary histologic changes may be required for the diagnosis in patients with a low burden of eggs.
Disclosures
Dr. Geha has no disclosures. Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. Dr. Peters’ spouse is employed by Hoffman-La Roche. Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME).
1. Sarin SK, Khanna R. Non-cirrhotic portal hypertension. Clin Liver Dis. 2014;18(2):451-76. doi: 10.1016/j.cld.2014.01.009. PubMed
2. Tapper EB, Lok AS. Use of liver imaging and biopsy in clinical practice. N Engl J Med. 2017;377(8):756-768. doi: 10.1056/NEJMra1610570. PubMed
3. Udell JA, Wang CS, Tinmouth J, et al. Does this patient with liver disease have cirrhosis? JAMA. 2012;307(8):832-42. doi: 10.1001/jama.2012.186. PubMed
4. Centers for Disease Control and Prevention. Parasites–Schistosomiasis. https://www.cdc.gov/parasites/schistosomiasis/. Accessed December 2, 2017.
5. Bica I, Hamer DH, Stadecker MJ. Hepatic schistosomiasis. Infect Dis Clin N Am. 2000;14(3):583-604. PubMed
6. Ross AG, Bartley PB, Sleigh AC, et al. Schistosomiasis. N Engl J Med. 2002;346(16):1212-20. doi: 10.1056/NEJMra012396. PubMed
7. Gray DJ, Ross AG, Li YS, McManus DP. Diagnosis and management of schistosomiasis. BMJ. 2011;342: 2561-2561. doi: doi.org/10.1136/bmj.d2651. PubMed
8. Manzella A, Ohtomo K, Monzawa S, Lim JH. Schistosomiasis of the liver. Abdom Imaging. 2008;33(2):144-50. doi: 10.1007/s00261-007-9329-7. PubMed
9. Gryseels B, Polman K, Clerinx J, Kestens L. Human schistosomiasis. Lancet. 2006;368(9541):1106-18. doi: 10.1016/S0140-6736(06)69440-3. PubMed
10. Blei AT, Córdoba J. Practice Parameters Committee of the American College of Gastroenterology. Hepatic encephalopathy. Am J Gastroenterol. 2001;96(7):1968. doi: 10.1111/j.1572-0241.2001.03964.x. PubMed
A 68-year-old woman presented to the emergency department with altered mental status. On the morning prior to admission, she was fully alert and oriented. Over the course of the day, she became more confused and somnolent, and by the evening, she was unarousable to voice. She had not fallen and had no head trauma.
Altered mental status may arise from metabolic (eg, hyponatremia), infectious (eg, urinary tract infection), structural (eg, subdural hematoma), or toxin-related (eg, adverse medication effect) processes. Any of these categories of encephalopathy can develop gradually over the course of a day.
One year prior, the patient was admitted for a similar episode of altered mental status. Asterixis and elevated transaminases prompted an abdominal ultrasound, which revealed a nodular liver and ascites. Paracentesis revealed a high serum-ascites albumin gradient. The diagnosis of cirrhosis was made based on these findings. Testing for viral hepatitis, autoimmune hepatitis, hemochromatosis, and Wilson’s disease were negative. Although steatosis was not detected on ultrasound, nonalcoholic fatty liver disease (NAFLD) was suspected based on the patient’s risk factors of hypertension and type 2 diabetes mellitus. She had four additional presentations of altered mental status with asterixis; each episode resolved with lactulose.
Other medical history included end-stage renal disease (ESRD) requiring hemodialysis. Her medications were labetalol, amlodipine, insulin, propranolol, lactulose, and rifaximin. She was originally from China and moved to the United States 10 years earlier. Given concerns about her ability to consistently take medications, she had moved to a long-term facility. She did not use alcohol, tobacco, or illicit substances.
The normalization of the patient’s mental status after lactulose treatment, especially in the context of recurrent episodes, is characteristic of hepatic encephalopathy, in which ammonia and other substances bypass hepatic metabolism and impair cerebral function. Hepatic encephalopathy is the most common cause of lactulose-responsive encephalopathy, and may recur in the setting of infection or nonadherence with lactulose and rifaximin. Other causes of lactulose-responsive encephalopathy include hyperammonemia caused by urease-producing bacterial infection (eg, Proteus), valproic acid toxicity, and urea cycle abnormalities.
Other causes of confusion with a self-limited course should be considered for the current episode. A postictal state is possible, but convulsions were not reported. The patient is at risk of hypoglycemia from insulin use and impaired gluconeogenesis due to cirrhosis and ESRD, but low blood sugar would have likely been detected at the time of hospitalization. Finally, she might have experienced episodic encephalopathy from ingestion of unreported medications or toxins, whose effects may have resolved with abstinence during hospitalization.
The patient’s temperature was 37.8°C, pulse 73 beats/minute, blood pressure 133/69 mmHg, respiratory rate 12 breaths/minute, and oxygen saturation 98% on ambient air. Her body mass index (BMI) was 19 kg/m2. She was somnolent but was moving all four extremities spontaneously. Her pupils were symmetric and reactive. There was no facial asymmetry. Biceps and patellar reflexes were 2+ bilaterally. Babinski sign was absent bilaterally. The patient could not cooperate with the assessment for asterixis. Her sclerae were anicteric. The jugular venous pressure was estimated at 13 cm of water. Her heart was regular with no murmurs. Her lungs were clear. She had a distended, nontender abdomen with caput medusae. She had symmetric pitting edema in her lower extremities up to the shins.
The elevated jugular venous pressure, lower extremity edema, and distended abdomen suggest volume overload. Jugular venous distention with clear lungs is characteristic of right ventricular failure from pulmonary hypertension, right ventricular myocardial infarction, tricuspid regurgitation, or constrictive pericarditis. However, chronic biventricular heart failure often presents in this manner and is more common than the aforementioned conditions. ESRD and cirrhosis may be contributing to the hypervolemia.
Although Asian patients may exhibit metabolic syndrome and NAFLD at a lower BMI than non-Asians, her BMI is uncharacteristically low for NAFLD, especially given the increased weight expected from volume overload. There are no signs of infection to account for worsening of hepatic encephalopathy.
Laboratory tests demonstrated a white blood cell count of 4400/µL with a normal differential, hemoglobin of 10.3 g/dL, and platelet count of 108,000 per cubic millimeter. Mean corpuscular volume was 103 fL. Basic metabolic panel was normal with the exception of blood urea nitrogen of 46 mg/dL and a creatinine of 6.4 mg/dL. Aspartate aminotransferase was 34 units/L, alanine aminotransferase 34 units/L, alkaline phosphatase 289 units/L (normal, 31-95), gamma-glutamyl transferase 104 units (GGT, normal, 12-43), total bilirubin 0.8 mg/dL, and albumin 2.5 g/dL (normal, 3.5-4.5). Pro-brain natriuretic peptide was 1429 pg/mL (normal, <100). The international normalized ratio (INR) was 1.0. Urinalysis showed trace proteinuria. The chest x-ray was normal. A noncontrast computed tomography (CT) of the head demonstrated no intracranial pathology. An abdominal ultrasound revealed a normal-sized nodular liver, a nonocclusive portal vein thrombus (PVT), splenomegaly (15 cm in length), and trace ascites. There was no biliary dilation, hepatic steatosis, or hepatic mass.
The evolving data set presents a mixed picture about the state of the liver. The distended abdominal wall veins, thrombocytopenia, and splenomegaly are commonly observed in advanced cirrhosis, but these findings reflect the associated portal hypertension and not the liver disease itself. The normal bilirubin and INR suggest preserved liver function and decrease the likelihood of cirrhosis being responsible for the portal hypertension. However, the elevated alkaline phosphatase and GGT levels suggest an infiltrative liver disease, such as lymphoma, sarcoidosis, or amyloidosis.
Furthermore, while a nodular liver on imaging is consistent with cirrhosis, no steatosis was noted to support the presumed diagnosis of NAFLD. One explanation for this discrepancy is that fatty infiltration may be absent when NAFLD-associated cirrhosis develops. In summary, there is evidence of liver disease, and there is evidence of portal hypertension, but there is no evidence of liver parenchymal failure. The key features of the latter – spider angiomata, palmar erythema, hyperbilirubinemia, and coagulopathy – are absent.
Noncirrhotic portal hypertension (NCPH) is an alternative explanation for the patient’s findings. NCPH is an elevation in the portal venous system pressure that arises from intrahepatic (but noncirrhotic) disease or from extrahepatic disease. Hepatic schistosomiasis is an example of intrahepatic but noncirrhotic portal hypertension. PVT that arises on account of a hypercoagulable condition (eg, abdominal malignancy, pancreatitis, or myeloproliferative disorders) is a prototype of extrahepatic NCPH. At this point, it is impossible to know if the PVT is a complication of NCPH or a cause of NCPH. PVT as a complication of cirrhosis is less likely.
An abdominal CT scan would better assess the hepatic parenchyma and exclude abdominal malignancies such as pancreatic adenocarcinoma. An echocardiogram is indicated to evaluate the cause of the elevated jugular venous pressure. A liver biopsy and measurement of portal venous pressure would help distinguish between cirrhotic and noncirrhotic portal hypertension.
Hepatitis A, B, and C serologies were negative as were antinuclear and antimitochondrial antibodies. Ferritin and ceruloplasmin levels were normal. A CT scan of the abdomen with contrast demonstrated a nodular liver contour, splenomegaly, and a nonocclusive PVT (Figure 1). A transthoracic echocardiogram showed normal biventricular systolic function and size, normal diastolic function, a pulmonary artery systolic pressure of 57 mmHg (normal, < 25), moderate tricuspid regurgitation, and no pericardial effusion or thickening. The patient’s confusion and somnolence resolved after two days of lactulose therapy. She denied the use of other medications, supplements, or herbs.
Pulmonary hypertension is usually a consequence of cardiopulmonary disease, but there is no exam or imaging evidence for left ventricular failure, mitral stenosis, obstructive lung disease, or interstitial lung disease. Portopulmonary hypertension (a form of pulmonary hypertension) can develop as a consequence of end-stage liver disease. The most common cause of hepatic encephalopathy due to portosystemic shunting is cirrhosis, but such shunting also arises in NCPH.
Schistosomiasis is the most common cause of NCPH worldwide. Parasite eggs trapped within the terminal portal venules cause inflammation, leading to fibrosis and intrahepatic portal hypertension. The liver becomes nodular on account of these changes, but the overall hepatic function is typically preserved. Portal hypertension, variceal bleeding, and pulmonary hypertension are common complications. The latter can arise from portosystemic shunting, which leads to embolization of schistosome eggs into the pulmonary circulation, where a granulomatous reaction ensues.
A percutaneous liver biopsy showed granulomatous inflammation and dilated portal venules consistent with increased resistance to venous inflow (Figure 2). There was no sinusoidal congestion to indicate impaired hepatic venous outflow. Mild sinusoidal and portal fibrosis and increased iron in Kupffer cells were noted. There was no evidence of cirrhosis or steatohepatitis. Stains for acid-fast bacilli and fungi were negative. 16S rDNA (a test assessing for bacterial DNA) and Mycobacterium tuberculosis polymerase chain reactions were negative. The biopsy confirmed the diagnosis of noncirrhotic portal hypertension.
Hepatic granulomas can arise from infectious, immunologic, toxic, and malignant diseases. In the United States, immunologic disorders, such as sarcoidosis and primary biliary cholangitis, are the most common causes of granulomatous hepatitis. The patient lacks extrahepatic features of the former. The absence of bile duct injury and negative antimitochondrial antibody exclude the latter. None of the listed medications are commonly associated with hepatic granulomas. The ultrasound, CT scan, and biopsy did not reveal a granulomatous malignancy such as lymphoma.
Infections, such as brucellosis, Q fever, and tuberculosis, are common causes of granulomatous hepatitis in the developing world. Tuberculosis is prevalent in China, but the test results do not support tuberculosis as a unifying diagnosis.
Schistosomiasis accounts for the major clinical features (portal and pulmonary hypertension and preserved liver function) and hepatic pathology (ie, portal venous fibrosis with granulomatous inflammation) in this case and is prevalent in China, where the patient emigrated from. The biopsy specimen should be re-examined for schistosome eggs and serologic tests for schistosomiasis pursued.
Antibodies to human immunodeficiency virus, Brucella, Bartonella quintana, Bartonella henselae, Coxiella burnetii, Francisella tularensis, and Histoplasma were negative. Cryptococcal antigen and rapid plasma reagin were negative. IgG antibodies to Schistosoma were 0.21 units (normal, < 0.19 units). Based on the patient’s epidemiology, biopsy findings, and serology results, hepatic schistosomiasis was diagnosed. Praziquantel was prescribed. She continues to receive daily lactulose and rifaximin and has not had any episodes of encephalopathy in the year after discharge.
COMMENTARY
Portal hypertension arises when there is resistance to flow in the portal venous system. It is defined as a pressure gradient greater than 5 mmHg between the portal vein and the intra-abdominal portion of the inferior vena cava.1 Clinicians are familiar with the manifestations of portal hypertension – portosystemic shunting leading to encephalopathy and variceal hemorrhage, ascites, and splenomegaly with thrombocytopenia – because of their close association with cirrhosis. In developed countries, cirrhosis accounts for over 90% of cases of portal hypertension.1 In the remaining 10%, conditions such as portal vein thrombosis primarily affect the portal vasculature and increase resistance to portal blood flow while leaving hepatic synthetic function relatively spared (Figure 3). Therefore, cirrhosis cannot be inferred with certainty from signs of portal hypertension alone.
Liver biopsy is the gold standard for the diagnosis of cirrhosis, but this method is increasingly being replaced by noninvasive assessments of liver fibrosis, including imaging and scoring systems.2 Clinicians often infer cirrhosis from the combination of a known cause of liver injury, abnormal liver biochemical tests, evidence of liver dysfunction, and signs of portal hypertension.3 However, when signs of portal hypertension are present, but liver dysfunction cannot be established on physical exam (eg, palmar erythema, spider nevi, gynecomastia, and testicular atrophy) or laboratory testing (eg, low albumin, elevated INR, and elevated bilirubin), noncirrhotic causes of portal hypertension should be considered. In this case, the biopsy showed vascular changes that suggested impaired venous inflow without bridging fibrosis, which pointed to NCPH.
NCPH is categorized based on the location of resistance to blood flow: prehepatic (eg, portal vein thrombosis), intrahepatic (eg, schistosomiasis), and posthepatic (eg, right-sided heart failure).1 In our patient, the dilated portal venules (inflow) in the presence of normal hepatic vein outflow suggested an increased intrahepatic resistance to blood flow. This finding excluded a causal role of the portal vein thrombosis and prompted testing for schistosomiasis.
Schistosomiasis affects more than 200 million people worldwide and is prevalent in Sub-Saharan Africa, South America, Egypt, China, and Southeast Asia.4,5 Transmission occurs in fresh water, where the infectious form of the parasite is released from snails.4,6 Schistosome worms are not found in the United States, but as a result of immigration and travel, more than 400,000 people in the United States are estimated to be infected.5
Chronic schistosomiasis develops from the host’s granulomatous reaction to schistosome eggs whose location (depending on the species) leads to genitourinary, intestinal, hepatic, or rarely, neurologic disease.6 Hepatic schistosomiasis arises when eggs released in the portal venous system lodge in small portal venules and cause granulomatous inflammation, periportal fibrosis, and microvascular obstruction.6 The resultant portal hypertension develops insidiously, but the architecture and synthetic function of the liver is maintained until the very late stages of disease.6,7 Pulmonary hypertension can arise from the embolization of eggs to the pulmonary arterioles via portosystemic collaterals.
The demonstration of eggs in stool is the gold standard for the diagnosis of hepatic schistosomiasis, which is most commonly caused by Schistosoma mansoni and S. japonicum.7 Serologic assays provide evidence of infection or exposure but may cross-react with other helminths. Liver biopsy may reveal characteristic histopathologic findings, including granulomatous inflammation, distorted vasculature, and the deposition of collagen deposits in the periportal space, leading to “pipestem fibrosis.”8,9 If eggs cannot be detected on stool or histology, then serology, secondary histologic changes, and sometimes PCR are used to diagnose hepatic schistosomiasis. In our patient, the epidemiology, Schistosoma antibody titer, pulmonary hypertension, and liver biopsy with granulomatous inflammation, periportal fibrosis, and intrahepatic portal venule dilation were diagnostic of hepatic schistosomiasis.
The recurrent episodes of confusion which resolved with lactulose therapy were suggestive of hepatic encephalopathy, which results from shunting and accumulation of neurotoxic substances that would otherwise undergo hepatic metabolism.10 Clinicians are most familiar with hepatic encephalopathy in cirrhosis, where multiple liver functions – synthesis, excretion, metabolism, and circulation – simultaneously fail. NCPH represents a scenario where only the circulation is impaired, but this is sufficient to cause the portosystemic shunting that leads to encephalopathy. Our patient’s recurrent hepatic encephalopathy, despite adherence to lactulose and rifaximin and its resolution after praziquantel treatment, underscores the importance of addressing the underlying cause of portosystemic shunting.Associating portal hypertension with cirrhosis is efficient and accurate in many cases. However, when specific manifestations of cirrhosis are lacking, clinicians must decouple this association and pursue an alternative explanation for portal hypertension. The presence of some intrahepatic pathology (from schistosomiasis) but no cirrhosis made this case a particularly tough egg to crack.
Teaching Points
- In the developed world, 90% of portal hypertension is due to cirrhosis. Hepatic schistosomiasis is the most common cause of NCPH worldwide.
- Chronic schistosomiasis affects the gastrointestinal, hepatic, and genitourinary systems and causes significant global morbidity and mortality.
- Visualization of schistosome eggs is the diagnostic gold standard. Indirect testing such as schistosoma antibodies and secondary histologic changes may be required for the diagnosis in patients with a low burden of eggs.
Disclosures
Dr. Geha has no disclosures. Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. Dr. Peters’ spouse is employed by Hoffman-La Roche. Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME).
A 68-year-old woman presented to the emergency department with altered mental status. On the morning prior to admission, she was fully alert and oriented. Over the course of the day, she became more confused and somnolent, and by the evening, she was unarousable to voice. She had not fallen and had no head trauma.
Altered mental status may arise from metabolic (eg, hyponatremia), infectious (eg, urinary tract infection), structural (eg, subdural hematoma), or toxin-related (eg, adverse medication effect) processes. Any of these categories of encephalopathy can develop gradually over the course of a day.
One year prior, the patient was admitted for a similar episode of altered mental status. Asterixis and elevated transaminases prompted an abdominal ultrasound, which revealed a nodular liver and ascites. Paracentesis revealed a high serum-ascites albumin gradient. The diagnosis of cirrhosis was made based on these findings. Testing for viral hepatitis, autoimmune hepatitis, hemochromatosis, and Wilson’s disease were negative. Although steatosis was not detected on ultrasound, nonalcoholic fatty liver disease (NAFLD) was suspected based on the patient’s risk factors of hypertension and type 2 diabetes mellitus. She had four additional presentations of altered mental status with asterixis; each episode resolved with lactulose.
Other medical history included end-stage renal disease (ESRD) requiring hemodialysis. Her medications were labetalol, amlodipine, insulin, propranolol, lactulose, and rifaximin. She was originally from China and moved to the United States 10 years earlier. Given concerns about her ability to consistently take medications, she had moved to a long-term facility. She did not use alcohol, tobacco, or illicit substances.
The normalization of the patient’s mental status after lactulose treatment, especially in the context of recurrent episodes, is characteristic of hepatic encephalopathy, in which ammonia and other substances bypass hepatic metabolism and impair cerebral function. Hepatic encephalopathy is the most common cause of lactulose-responsive encephalopathy, and may recur in the setting of infection or nonadherence with lactulose and rifaximin. Other causes of lactulose-responsive encephalopathy include hyperammonemia caused by urease-producing bacterial infection (eg, Proteus), valproic acid toxicity, and urea cycle abnormalities.
Other causes of confusion with a self-limited course should be considered for the current episode. A postictal state is possible, but convulsions were not reported. The patient is at risk of hypoglycemia from insulin use and impaired gluconeogenesis due to cirrhosis and ESRD, but low blood sugar would have likely been detected at the time of hospitalization. Finally, she might have experienced episodic encephalopathy from ingestion of unreported medications or toxins, whose effects may have resolved with abstinence during hospitalization.
The patient’s temperature was 37.8°C, pulse 73 beats/minute, blood pressure 133/69 mmHg, respiratory rate 12 breaths/minute, and oxygen saturation 98% on ambient air. Her body mass index (BMI) was 19 kg/m2. She was somnolent but was moving all four extremities spontaneously. Her pupils were symmetric and reactive. There was no facial asymmetry. Biceps and patellar reflexes were 2+ bilaterally. Babinski sign was absent bilaterally. The patient could not cooperate with the assessment for asterixis. Her sclerae were anicteric. The jugular venous pressure was estimated at 13 cm of water. Her heart was regular with no murmurs. Her lungs were clear. She had a distended, nontender abdomen with caput medusae. She had symmetric pitting edema in her lower extremities up to the shins.
The elevated jugular venous pressure, lower extremity edema, and distended abdomen suggest volume overload. Jugular venous distention with clear lungs is characteristic of right ventricular failure from pulmonary hypertension, right ventricular myocardial infarction, tricuspid regurgitation, or constrictive pericarditis. However, chronic biventricular heart failure often presents in this manner and is more common than the aforementioned conditions. ESRD and cirrhosis may be contributing to the hypervolemia.
Although Asian patients may exhibit metabolic syndrome and NAFLD at a lower BMI than non-Asians, her BMI is uncharacteristically low for NAFLD, especially given the increased weight expected from volume overload. There are no signs of infection to account for worsening of hepatic encephalopathy.
Laboratory tests demonstrated a white blood cell count of 4400/µL with a normal differential, hemoglobin of 10.3 g/dL, and platelet count of 108,000 per cubic millimeter. Mean corpuscular volume was 103 fL. Basic metabolic panel was normal with the exception of blood urea nitrogen of 46 mg/dL and a creatinine of 6.4 mg/dL. Aspartate aminotransferase was 34 units/L, alanine aminotransferase 34 units/L, alkaline phosphatase 289 units/L (normal, 31-95), gamma-glutamyl transferase 104 units (GGT, normal, 12-43), total bilirubin 0.8 mg/dL, and albumin 2.5 g/dL (normal, 3.5-4.5). Pro-brain natriuretic peptide was 1429 pg/mL (normal, <100). The international normalized ratio (INR) was 1.0. Urinalysis showed trace proteinuria. The chest x-ray was normal. A noncontrast computed tomography (CT) of the head demonstrated no intracranial pathology. An abdominal ultrasound revealed a normal-sized nodular liver, a nonocclusive portal vein thrombus (PVT), splenomegaly (15 cm in length), and trace ascites. There was no biliary dilation, hepatic steatosis, or hepatic mass.
The evolving data set presents a mixed picture about the state of the liver. The distended abdominal wall veins, thrombocytopenia, and splenomegaly are commonly observed in advanced cirrhosis, but these findings reflect the associated portal hypertension and not the liver disease itself. The normal bilirubin and INR suggest preserved liver function and decrease the likelihood of cirrhosis being responsible for the portal hypertension. However, the elevated alkaline phosphatase and GGT levels suggest an infiltrative liver disease, such as lymphoma, sarcoidosis, or amyloidosis.
Furthermore, while a nodular liver on imaging is consistent with cirrhosis, no steatosis was noted to support the presumed diagnosis of NAFLD. One explanation for this discrepancy is that fatty infiltration may be absent when NAFLD-associated cirrhosis develops. In summary, there is evidence of liver disease, and there is evidence of portal hypertension, but there is no evidence of liver parenchymal failure. The key features of the latter – spider angiomata, palmar erythema, hyperbilirubinemia, and coagulopathy – are absent.
Noncirrhotic portal hypertension (NCPH) is an alternative explanation for the patient’s findings. NCPH is an elevation in the portal venous system pressure that arises from intrahepatic (but noncirrhotic) disease or from extrahepatic disease. Hepatic schistosomiasis is an example of intrahepatic but noncirrhotic portal hypertension. PVT that arises on account of a hypercoagulable condition (eg, abdominal malignancy, pancreatitis, or myeloproliferative disorders) is a prototype of extrahepatic NCPH. At this point, it is impossible to know if the PVT is a complication of NCPH or a cause of NCPH. PVT as a complication of cirrhosis is less likely.
An abdominal CT scan would better assess the hepatic parenchyma and exclude abdominal malignancies such as pancreatic adenocarcinoma. An echocardiogram is indicated to evaluate the cause of the elevated jugular venous pressure. A liver biopsy and measurement of portal venous pressure would help distinguish between cirrhotic and noncirrhotic portal hypertension.
Hepatitis A, B, and C serologies were negative as were antinuclear and antimitochondrial antibodies. Ferritin and ceruloplasmin levels were normal. A CT scan of the abdomen with contrast demonstrated a nodular liver contour, splenomegaly, and a nonocclusive PVT (Figure 1). A transthoracic echocardiogram showed normal biventricular systolic function and size, normal diastolic function, a pulmonary artery systolic pressure of 57 mmHg (normal, < 25), moderate tricuspid regurgitation, and no pericardial effusion or thickening. The patient’s confusion and somnolence resolved after two days of lactulose therapy. She denied the use of other medications, supplements, or herbs.
Pulmonary hypertension is usually a consequence of cardiopulmonary disease, but there is no exam or imaging evidence for left ventricular failure, mitral stenosis, obstructive lung disease, or interstitial lung disease. Portopulmonary hypertension (a form of pulmonary hypertension) can develop as a consequence of end-stage liver disease. The most common cause of hepatic encephalopathy due to portosystemic shunting is cirrhosis, but such shunting also arises in NCPH.
Schistosomiasis is the most common cause of NCPH worldwide. Parasite eggs trapped within the terminal portal venules cause inflammation, leading to fibrosis and intrahepatic portal hypertension. The liver becomes nodular on account of these changes, but the overall hepatic function is typically preserved. Portal hypertension, variceal bleeding, and pulmonary hypertension are common complications. The latter can arise from portosystemic shunting, which leads to embolization of schistosome eggs into the pulmonary circulation, where a granulomatous reaction ensues.
A percutaneous liver biopsy showed granulomatous inflammation and dilated portal venules consistent with increased resistance to venous inflow (Figure 2). There was no sinusoidal congestion to indicate impaired hepatic venous outflow. Mild sinusoidal and portal fibrosis and increased iron in Kupffer cells were noted. There was no evidence of cirrhosis or steatohepatitis. Stains for acid-fast bacilli and fungi were negative. 16S rDNA (a test assessing for bacterial DNA) and Mycobacterium tuberculosis polymerase chain reactions were negative. The biopsy confirmed the diagnosis of noncirrhotic portal hypertension.
Hepatic granulomas can arise from infectious, immunologic, toxic, and malignant diseases. In the United States, immunologic disorders, such as sarcoidosis and primary biliary cholangitis, are the most common causes of granulomatous hepatitis. The patient lacks extrahepatic features of the former. The absence of bile duct injury and negative antimitochondrial antibody exclude the latter. None of the listed medications are commonly associated with hepatic granulomas. The ultrasound, CT scan, and biopsy did not reveal a granulomatous malignancy such as lymphoma.
Infections, such as brucellosis, Q fever, and tuberculosis, are common causes of granulomatous hepatitis in the developing world. Tuberculosis is prevalent in China, but the test results do not support tuberculosis as a unifying diagnosis.
Schistosomiasis accounts for the major clinical features (portal and pulmonary hypertension and preserved liver function) and hepatic pathology (ie, portal venous fibrosis with granulomatous inflammation) in this case and is prevalent in China, where the patient emigrated from. The biopsy specimen should be re-examined for schistosome eggs and serologic tests for schistosomiasis pursued.
Antibodies to human immunodeficiency virus, Brucella, Bartonella quintana, Bartonella henselae, Coxiella burnetii, Francisella tularensis, and Histoplasma were negative. Cryptococcal antigen and rapid plasma reagin were negative. IgG antibodies to Schistosoma were 0.21 units (normal, < 0.19 units). Based on the patient’s epidemiology, biopsy findings, and serology results, hepatic schistosomiasis was diagnosed. Praziquantel was prescribed. She continues to receive daily lactulose and rifaximin and has not had any episodes of encephalopathy in the year after discharge.
COMMENTARY
Portal hypertension arises when there is resistance to flow in the portal venous system. It is defined as a pressure gradient greater than 5 mmHg between the portal vein and the intra-abdominal portion of the inferior vena cava.1 Clinicians are familiar with the manifestations of portal hypertension – portosystemic shunting leading to encephalopathy and variceal hemorrhage, ascites, and splenomegaly with thrombocytopenia – because of their close association with cirrhosis. In developed countries, cirrhosis accounts for over 90% of cases of portal hypertension.1 In the remaining 10%, conditions such as portal vein thrombosis primarily affect the portal vasculature and increase resistance to portal blood flow while leaving hepatic synthetic function relatively spared (Figure 3). Therefore, cirrhosis cannot be inferred with certainty from signs of portal hypertension alone.
Liver biopsy is the gold standard for the diagnosis of cirrhosis, but this method is increasingly being replaced by noninvasive assessments of liver fibrosis, including imaging and scoring systems.2 Clinicians often infer cirrhosis from the combination of a known cause of liver injury, abnormal liver biochemical tests, evidence of liver dysfunction, and signs of portal hypertension.3 However, when signs of portal hypertension are present, but liver dysfunction cannot be established on physical exam (eg, palmar erythema, spider nevi, gynecomastia, and testicular atrophy) or laboratory testing (eg, low albumin, elevated INR, and elevated bilirubin), noncirrhotic causes of portal hypertension should be considered. In this case, the biopsy showed vascular changes that suggested impaired venous inflow without bridging fibrosis, which pointed to NCPH.
NCPH is categorized based on the location of resistance to blood flow: prehepatic (eg, portal vein thrombosis), intrahepatic (eg, schistosomiasis), and posthepatic (eg, right-sided heart failure).1 In our patient, the dilated portal venules (inflow) in the presence of normal hepatic vein outflow suggested an increased intrahepatic resistance to blood flow. This finding excluded a causal role of the portal vein thrombosis and prompted testing for schistosomiasis.
Schistosomiasis affects more than 200 million people worldwide and is prevalent in Sub-Saharan Africa, South America, Egypt, China, and Southeast Asia.4,5 Transmission occurs in fresh water, where the infectious form of the parasite is released from snails.4,6 Schistosome worms are not found in the United States, but as a result of immigration and travel, more than 400,000 people in the United States are estimated to be infected.5
Chronic schistosomiasis develops from the host’s granulomatous reaction to schistosome eggs whose location (depending on the species) leads to genitourinary, intestinal, hepatic, or rarely, neurologic disease.6 Hepatic schistosomiasis arises when eggs released in the portal venous system lodge in small portal venules and cause granulomatous inflammation, periportal fibrosis, and microvascular obstruction.6 The resultant portal hypertension develops insidiously, but the architecture and synthetic function of the liver is maintained until the very late stages of disease.6,7 Pulmonary hypertension can arise from the embolization of eggs to the pulmonary arterioles via portosystemic collaterals.
The demonstration of eggs in stool is the gold standard for the diagnosis of hepatic schistosomiasis, which is most commonly caused by Schistosoma mansoni and S. japonicum.7 Serologic assays provide evidence of infection or exposure but may cross-react with other helminths. Liver biopsy may reveal characteristic histopathologic findings, including granulomatous inflammation, distorted vasculature, and the deposition of collagen deposits in the periportal space, leading to “pipestem fibrosis.”8,9 If eggs cannot be detected on stool or histology, then serology, secondary histologic changes, and sometimes PCR are used to diagnose hepatic schistosomiasis. In our patient, the epidemiology, Schistosoma antibody titer, pulmonary hypertension, and liver biopsy with granulomatous inflammation, periportal fibrosis, and intrahepatic portal venule dilation were diagnostic of hepatic schistosomiasis.
The recurrent episodes of confusion which resolved with lactulose therapy were suggestive of hepatic encephalopathy, which results from shunting and accumulation of neurotoxic substances that would otherwise undergo hepatic metabolism.10 Clinicians are most familiar with hepatic encephalopathy in cirrhosis, where multiple liver functions – synthesis, excretion, metabolism, and circulation – simultaneously fail. NCPH represents a scenario where only the circulation is impaired, but this is sufficient to cause the portosystemic shunting that leads to encephalopathy. Our patient’s recurrent hepatic encephalopathy, despite adherence to lactulose and rifaximin and its resolution after praziquantel treatment, underscores the importance of addressing the underlying cause of portosystemic shunting.Associating portal hypertension with cirrhosis is efficient and accurate in many cases. However, when specific manifestations of cirrhosis are lacking, clinicians must decouple this association and pursue an alternative explanation for portal hypertension. The presence of some intrahepatic pathology (from schistosomiasis) but no cirrhosis made this case a particularly tough egg to crack.
Teaching Points
- In the developed world, 90% of portal hypertension is due to cirrhosis. Hepatic schistosomiasis is the most common cause of NCPH worldwide.
- Chronic schistosomiasis affects the gastrointestinal, hepatic, and genitourinary systems and causes significant global morbidity and mortality.
- Visualization of schistosome eggs is the diagnostic gold standard. Indirect testing such as schistosoma antibodies and secondary histologic changes may be required for the diagnosis in patients with a low burden of eggs.
Disclosures
Dr. Geha has no disclosures. Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. Dr. Peters’ spouse is employed by Hoffman-La Roche. Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME).
1. Sarin SK, Khanna R. Non-cirrhotic portal hypertension. Clin Liver Dis. 2014;18(2):451-76. doi: 10.1016/j.cld.2014.01.009. PubMed
2. Tapper EB, Lok AS. Use of liver imaging and biopsy in clinical practice. N Engl J Med. 2017;377(8):756-768. doi: 10.1056/NEJMra1610570. PubMed
3. Udell JA, Wang CS, Tinmouth J, et al. Does this patient with liver disease have cirrhosis? JAMA. 2012;307(8):832-42. doi: 10.1001/jama.2012.186. PubMed
4. Centers for Disease Control and Prevention. Parasites–Schistosomiasis. https://www.cdc.gov/parasites/schistosomiasis/. Accessed December 2, 2017.
5. Bica I, Hamer DH, Stadecker MJ. Hepatic schistosomiasis. Infect Dis Clin N Am. 2000;14(3):583-604. PubMed
6. Ross AG, Bartley PB, Sleigh AC, et al. Schistosomiasis. N Engl J Med. 2002;346(16):1212-20. doi: 10.1056/NEJMra012396. PubMed
7. Gray DJ, Ross AG, Li YS, McManus DP. Diagnosis and management of schistosomiasis. BMJ. 2011;342: 2561-2561. doi: doi.org/10.1136/bmj.d2651. PubMed
8. Manzella A, Ohtomo K, Monzawa S, Lim JH. Schistosomiasis of the liver. Abdom Imaging. 2008;33(2):144-50. doi: 10.1007/s00261-007-9329-7. PubMed
9. Gryseels B, Polman K, Clerinx J, Kestens L. Human schistosomiasis. Lancet. 2006;368(9541):1106-18. doi: 10.1016/S0140-6736(06)69440-3. PubMed
10. Blei AT, Córdoba J. Practice Parameters Committee of the American College of Gastroenterology. Hepatic encephalopathy. Am J Gastroenterol. 2001;96(7):1968. doi: 10.1111/j.1572-0241.2001.03964.x. PubMed
1. Sarin SK, Khanna R. Non-cirrhotic portal hypertension. Clin Liver Dis. 2014;18(2):451-76. doi: 10.1016/j.cld.2014.01.009. PubMed
2. Tapper EB, Lok AS. Use of liver imaging and biopsy in clinical practice. N Engl J Med. 2017;377(8):756-768. doi: 10.1056/NEJMra1610570. PubMed
3. Udell JA, Wang CS, Tinmouth J, et al. Does this patient with liver disease have cirrhosis? JAMA. 2012;307(8):832-42. doi: 10.1001/jama.2012.186. PubMed
4. Centers for Disease Control and Prevention. Parasites–Schistosomiasis. https://www.cdc.gov/parasites/schistosomiasis/. Accessed December 2, 2017.
5. Bica I, Hamer DH, Stadecker MJ. Hepatic schistosomiasis. Infect Dis Clin N Am. 2000;14(3):583-604. PubMed
6. Ross AG, Bartley PB, Sleigh AC, et al. Schistosomiasis. N Engl J Med. 2002;346(16):1212-20. doi: 10.1056/NEJMra012396. PubMed
7. Gray DJ, Ross AG, Li YS, McManus DP. Diagnosis and management of schistosomiasis. BMJ. 2011;342: 2561-2561. doi: doi.org/10.1136/bmj.d2651. PubMed
8. Manzella A, Ohtomo K, Monzawa S, Lim JH. Schistosomiasis of the liver. Abdom Imaging. 2008;33(2):144-50. doi: 10.1007/s00261-007-9329-7. PubMed
9. Gryseels B, Polman K, Clerinx J, Kestens L. Human schistosomiasis. Lancet. 2006;368(9541):1106-18. doi: 10.1016/S0140-6736(06)69440-3. PubMed
10. Blei AT, Córdoba J. Practice Parameters Committee of the American College of Gastroenterology. Hepatic encephalopathy. Am J Gastroenterol. 2001;96(7):1968. doi: 10.1111/j.1572-0241.2001.03964.x. PubMed
© 2018 Society of Hospital Medicine
Diagnosing the Treatment
A 70-year-old man presented to the emergency department with 5 days of decreased appetite, frequent urination, tremors, and memory difficulties. He also reported 9 months of malaise, generalized weakness, and weight loss. There was no history of fever, chills, nausea, diarrhea, constipation, pain, or focal neurologic complaints.
This patient exemplifies a common clinical challenge: an older adult with several possibly unrelated concerns. In many patients, a new presentation is usually either a different manifestation of a known condition (eg, a complication of an established malignancy) or the emergence of something they are at risk for based on health behavior or other characteristics (eg, lung cancer in a smoker). The diagnostic process in older adults can be complicated because many have, or are at risk for, multiple chronic conditions.
After reviewing the timeline of symptoms, the presence of 9 months of symptoms suggests a chronic and progressive underlying process, perhaps with subsequent superimposition of an acute problem. Although it is not certain whether chronic and acute symptoms are caused by the same process, this assumption is reasonable. The superimposition of acute symptoms on a chronic process may represent progression of the underlying condition or an acute complication of the underlying disease. However, the patient’s chronic symptoms of malaise, weakness, and weight loss are nonspecific.
Although malignancy is a consideration given the age of the patient and time course of symptoms, attributing the symptoms to a specific pattern of disease or building a cogent differential diagnosis is difficult until additional information is obtained. One strategy is to try to localize the findings to 1 or more organ systems; for example, given that tremors and memory difficulties localize to the central nervous system, neurodegenerative disorders, such as “Parkinson plus” syndromes, and cerebellar disease are possible. However, this tactic still leaves a relatively broad set of symptoms without an immediate and clear unifying cause.
The patient’s medical history included hyperlipidemia, peripheral neuropathy, prostate cancer, and papillary bladder cancer. The patient was admitted to the hospital 4 months earlier for severe sepsis presumed secondary to a urinary tract infection, although bacterial cultures were sterile. His social history was notable for a 50 pack-year smoking history. Outpatient medications included alfuzosin, gabapentin, simvastatin, hydrocodone, and cholecalciferol. He used a Bright Light Therapy lamp for 1 hour per week and occasionally used calcium carbonate for indigestion. The patient’s sister had a history of throat cancer.
On examination, the patient was detected with blood pressure of 104/56 mm Hg, pulse of 85 beats per minute, temperature of 98.2 °F, oxygen saturation of 97% on ambient air, and body mass index of 18 kg/m2. The patient appeared frail with mildly decreased strength in the upper and lower extremities bilaterally. The remainder of the physical examination was normal. Reflexes were symmetric, no tremors or rigidity was noted, sensation was intact to light touch, and the response to the Romberg maneuver was normal.
Past medical history is the cornerstone of the diagnostic process. The history of 2 different malignancies is the most striking element in this case. Papillary bladder cancer is usually a local process, but additional information is needed regarding its stage and previous treatment, including whether or not the patient received Bacille Calmette Guerin (BCG) vaccine, which can rarely be associated with infectious and inflammatory complications. Metastatic prostate cancer could certainly account for his symptomatology, and bladder outlet obstruction could explain the history of urinary frequency and probable urosepsis. His medication list suggested no obvious causes to explain his presentation, except that cholecalciferol and calcium carbonate, which when taken in excess, can cause hypercalcemia. This finding is of particular importance given that many of the patient’s symptoms, including polyuria, malaise, weakness, tremor, memory difficulties, anorexia, acute kidney injury and (indirectly) hypotension and weight loss, are also seen in patients with hypercalcemia. The relatively normal result of the neurologic examination decreases the probability of a primary neurologic disorder and increases the likelihood that his neurologic symptoms are due to a global systemic process. The relative hypotension and weight loss similarly support the possibility that the patient is experiencing a chronic and progressive process.
The differential diagnosis remains broad. An underlying malignancy would explain the chronic progressive course, and superimposed hypercalcemia would explain the acute symptoms of polyuria, tremor, and memory changes. Endocrinopathies including hyperthyroidism or adrenal insufficiency are other possibilities. A chronic progressive infection, such as tuberculosis, is possible, although no epidemiologic factors that increase his risk for this disease are present.
The patient had serum calcium of 14.5 mg/dL, ionized calcium of 3.46 mEq/L, albumin of 3.6 g/dL, BUN of 62 mg/dL, and creatinine of 3.9 mg/dL (all values were normal 3 months prior). His electrolytes and liver function were otherwise normal. Moreover, he had hemoglobin level of 10.5 mg/dL, white blood cell count of 4.8 × 109cells/L, and platelet count of 203 × 109 cells/L.
Until this point, only nonspecific findings were identified, leading to a broad differential diagnosis with little specificity. However, laboratory examinations confirm the suspected diagnosis of hypercalcemia, provide an opportunity to explain the patient’s symptoms, and offer a “lens” to narrow the differential diagnosis and guide the diagnostic evaluation. Hypercalcemia is most commonly secondary to primary hyperparathyroidism or malignancy. Primary hyperparathyroidism is unlikely in this patient given the relatively acute onset of symptoms. The degree of hypercalcemia is also atypical for primary hyperparathyroidism because it rarely exceeds 13 mg/dL, although the use of concurrent vitamin D and calcium supplementation could explain the high calcium level. Malignancy seems more likely given the degree of hypercalcemia in the setting of weight loss, tobacco use, and history of malignancy. Malignancy may cause hypercalcemia through multiple disparate mechanisms, including development of osteolytic bone metastases, elaboration of parathyroid hormone-related Peptide (PTHrP), increased production of 1,25-dihydroxyvitamin D, or, very rarely, ectopic production of parathyroid hormone (PTH). However, none of these mechanisms are particularly common in bladder or prostate cancer, which are the known malignancies in the patient. Other less likely and less common causes of hypercalcemia are also possible given the clinical clues, including vitamin D toxicity and milk alkali syndrome (vitamin D and calcium carbonate supplementation), multiple endocrine neoplasia (a sister with “throat cancer”), and granulomatous disease (weight loss). At this point, further laboratory evaluations would be helpful, specifically determination of PTH and PTHrP levels and serum and urine protein electrophoresis.
With respect to the patient’s past medical history, his Gleason 3 + 3 prostate cancer was diagnosed 12 years prior to admission and treated with external beam radiation therapy and brachytherapy. His bladder cancer was diagnosed 3 years before admission and treated with tumor resection followed by 2 rounds of intravesical BCG (iBCG), 1 round of mitomycin C, and 2 additional rounds of iBCG over the course of treatment spanning 2 years and 6 months. The treatment was complicated by urethral strictures requiring dilation, ureteral outlet obstruction requiring left ureteral stent placement, and multiple urinary tract infections.
The patient’s last round of iBCG was delivered 6 months prior to his current presentation. The patient’s hospital admission 4 months earlier for severe sepsis was presumed secondary to a urologic source considering that significant pyuria was noted on urinalysis and he was treated with meropenem, although bacterial cultures of blood and urine were sterile. From the time of discharge until his current presentation, he experienced progressive weakness and an approximately 50 lb weight loss.
The prior cancers and associated treatments of the patient may be involved in his current presentation. The simplest explanation would be metastatic disease with resultant hypercalcemia, which is atypical of either prostate or bladder cancer. The history of genitourinary surgery could predispose the patient to a chronic infection of the urinary tract with indolent organisms, such as a fungus, especially given the prior sepsis without clear etiology. However, the history would not explain the presence of hypercalcemia. Tuberculosis must thus be considered given the weight loss, hypercalcemia, and “sterile pyuria” of the patient. A more intriguing possibility is whether or not the patient’s constellation of signs and symptoms might be a late effect of iBCG. Intravesical BCG for treatment of localized bladder cancer is occasionally associated with complications. BCG is a modified live form of Mycobacterium bovis which invokes an intense inflammatory reaction when instilled into the bladder. These complications include disseminated infection and local complications, such as genitourinary infections. BCG infection might also explain the severe sepsis of unclear etiology that the patient had experienced 4 months earlier. Most interestingly, hypercalcemia has been described in the setting of BCG infection.
In the hospital, the patient received intravenous normal saline, furosemide, and pamidronate. Evaluation for hypercalcemia revealed appropriately suppressed PTH (8 mg/dL), and normal levels of PTHrP (<.74 pmol/L), prostate specific antigen (<.01 ng/mL), and morning cortisol (16.7 mcg/dL). Serum and urine electrophoresis did not show evidence for monoclonal gammopathy, and the 25-hydroxy vitamin D level (39.5 ng/mL) was within the normal limits
The suppressed PTH level makes primary hyperparathyroidism unlikely, the low PTHrP level decreases the probability of a paraneoplastic process, and the normal protein electrophoresis makes multiple myeloma unlikely. The presence of a significantly elevated 1,25-dihydroxy vitamin D level with a normal 25-hydroxy vitamin D level indicates extrarenal conversion of 25-hydroxy vitamin D by 1-hydroxylase as the etiology of hypercalcemia.
Lymphoma would appear to be the most likely diagnosis as it accounts for most of the clinical findings observed in the patient and is a fairly common disorder. Sarcoidosis is also reasonably common and would explain the laboratory abnormalities but is not usually associated with weight loss and frailty. Disseminated infections, such as tuberculosis, histoplasmosis, and coccidioidomycosis, are all possible, but the patient lacks key risk factors for these infections. A complication of iBCG is the most intriguing possibility and could account for many of the patient’s clinical findings, including the septic episode,
The bone survey was normal, the renal ultrasound examination showed nodular wall thickening of the bladder with areas of calcification, and the CT scan of the chest, abdomen, and pelvis showed an area of calcification in the superior portion of the bladder but no evidence of lymphadenopathy or masses to suggest lymphoma. Aerobic and anaerobic blood and urine cultures were sterile. The patient was discharged 12 days after admission with plans for further outpatient diagnostic evaluation. At this time, his serum calcium had stabilized at 10.5 mg/dL

DISCUSSION
Hypercalcemia is a common finding in both hospital and ambulatory settings. The classic symptoms associated with hypercalcemia are aptly summarized with the mnemonic “bones, stones, abdominal groans, and psychiatric overtones” (to represent the associated skeletal involvement, renal disease, gastrointestinal symptoms, and effects on the nervous system). However, the severity and type of symptoms vary depending on the degree of hypercalcemia, acuity of onset, and underlying etiology. The vast majority (90%) of hypercalcemia cases are due to primary hyperparathyroidism and malignancy.3 Measuring the PTH level is a key step in the diagnostic evaluation process. An isolated elevation of PTH confirms the presence of primary or possibly tertiary hyperparathyroidism. Low PTH concentrations (<20 pg/mL) occur in the settings of PTHrP or vitamin-D-mediated hypercalcemia such as hypervitaminosis D, malignancy, or granulomatous disease.
Elevated PTHrP occurs most commonly in squamous cell, renal, bladder, and ovarian carcinomas.3,4 Elevated levels of 25-hydroxy vitamin D can occur with excessive consumption of vitamin D-containing products and some herbal supplements. In this case, neither PTHrP nor 25-hydroxy vitamin D level was elevated, leading to an exhaustive search for other causes. Although iBCG treatment is a rare cause of hypercalcemia, 2 previous reports indicated the presence of hypercalcemia secondary to granuloma formation in treated patients.5,6
The finding of an elevated 1,25-dihydroxy vitamin D level was unexpected. As the discussant mentioned, this finding is associated with lymphoma and with granulomatous disorders that were not initially strong diagnostic considerations in the patient. A variety of granulomatous diseases can cause hypercalcemia. Sarcoidosis and tuberculosis are the most common, but berylliosis, fungal infections, Crohn’s disease, silicone exposure, and granulomatosis with polyangiitis may also be associated with hypercalcemia.7 The mechanism for hypercalcemia in these situations is increased intestinal calcium absorption mediated by inappropriately increased, PTH-independent, extrarenal calcitriol (1,25-dihydroxy vitamin D) production. Activated monocytes upregulate 25(OH)D-alpha-hydroxylase, converting 25-hydroxy vitamin D to 1,25-dihydroxy vitamin D. Concurrently, the elevated levels of gamma-interferon render macrophages resistant to the normal regulatory feedback mechanisms, thereby promoting the production and inhibiting the degradation of 1,25-dihydroxy vitamin D.8
The tuberculosis vaccine BCG is an attenuated form of M. bovis and was originally developed by Albert Calmette and Camille Guérin at the Pasteur Institute in Paris in the early 20th century. In addition to its use as a vaccine against tuberculosis, BCG can protect against other mycobacterial infections, help treat atopic conditions via stimulation of the Th1 cellular immune response, and has been used as an antineoplastic agent. To date, BCG remains the most effective agent available for intravesical treatment of superficial bladder cancer.9,10 Although iBCG therapy is considered relatively safe and well-tolerated, rare complications do occur. Localized symptoms (bladder irritation, hematuria) and/or flu-like symptoms are common immediately after instillation and thought to be related to the cellular immune response and inflammatory cascade triggered by mycobacterial antigens.11 Other adverse effects, such as infectious and noninfectious complications, may occur months to years after treatment with BCG, and the associated symptoms can be quite nonspecific. Infectious complications include mycobacterial prostatitis, orchiepididymitis, balantitis, pneumonia, hepatitis, nephritis, septic arthritis, osteomyelitis, infected orthopedic and vascular prostheses, endocarditis, and bacteremia. Traumatic catheterization is the most common risk factor for infection with BCG.11-13 Noninfectious complications include reactive arthritis, hypersensitivity pneumonitis, hemophagocytic lymphohistiocytosis (HLH), and sterile granulomatous infiltration of solid organs.
The protean and nonspecific nature of the adverse effects of iBCG treatment and the fact that complications can present weeks to years after instillation can make diagnosis quite challenging.14 Even if clinical suspicion is high, it may be difficult to definitively identify BCG as the underlying etiology because acid fast staining, culture, and even PCR can lead to falsely negative results.14,15 For this reason, biopsy and tissue culture are recommended to demonstrate granuloma formation and identify the presence of M. bovis.
Although no prospective studies have been conducted to assess the optimal therapy for BCG infection, opinion-based recommendations include cessation of BCG treatment, initiation of at least 3 tuberculostatic agents, and treatment for 3-12 months depending on the severity of the complications.11,14 M. bovis is susceptible to isoniazid, rifampin, and ethambutol as well as to fluoroquinolones, clarithromycin, aminoglycosides, and doxycycline; however, this organism is highly resistant to pyrazinamide due to single-point mutation.11,16
Although treatment with steroids is a standard approach for management of hypercalcemia in other granulomatous disorders and leads to rapid reduction in circulating levels of 1,25-dihydroxy vitamin D and serum calcium., specific evidence has not been established to support its efficacy and effectiveness in treating hypercalcemia and other complications due to M. bovis.17 Nevertheless, some experts recommend the use of steroids in conjunction with a multidrug tuberculostatic regimen in cases of septicemia and multiorgan failure due to M. bovis.12,14,18-20
In summary, this case illustrates the importance of making room in differential diagnosis to include iatrogenic complications. That is,
Teaching Points
- Complications of intravesical BCG treatment include manifestations of granulomatous diseases, such as hypercalcemia.
- When generating a differential diagnosis, medical providers should not only consider the possibility of a new disease process or the progression of a known comorbidity but also the potential of an adverse effect related to prior treatments.
- Medical providers should be wary of accepting previously made diagnoses, particularly when key pieces of objective data are lacking.
Disclosures
The authors have no financial or other conflicts of interest that might bias this work.
1. Geisel RE, Sakamoto K, Russell DG, Rhoades ER. In vivo activity of released cell wall lipids of Mycobacterium bovis bacillus Calmette-Guérin is due principally to trehalose mycolates. J Immunol. 2005;174(8):5007-5015. https://doi.org/10.4049/jimmunol.174.8.5007. PubMed
2. Ryll R, Kumazawa Y, Yano I. Immunological properties of trehalose dimycolate (cord factor) and other mycolic acid-containing glycolipids--a review. Microbiol Immunol. 2001;45(12):801-811. https://doi.org/10.1111/j.1348-0421.2001.tb01319.x. PubMed
3. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966. PubMed
4. Goldner W. Cancer-related hypercalcemia. J Oncol Pract. 2016;12(5):426-432. https://doi.org/10.1200/JOP.2016.011155. PubMed
5. Nayar N, Briscoe K. Systemic Bacillus Calmette-Guerin sepsis manifesting as hypercalcaemia and thrombocytopenia as a complication of intravesical Bacillus Calmette-Guerin therapy. Intern Med J. 2015;45(10):1091-1092. https://doi.org/10.1111/imj.12876. PubMed
6. Schattner A, Gilad A, Cohen J. Systemic granulomatosis and hypercalcaemia following intravesical bacillus Calmette–Guerin immunotherapy. J Intern Med. 2002;251(3):272-277. https://doi.org/10.1046/j.1365-2796.2002.00957.x. PubMed
7. Tebben PJ, Singh RJ, Kumar R. Vitamin D-mediated hypercalcemia: mechanisms, diagnosis, and treatment. Endocr Rev. 2016;37(5):521-547. https://doi.org/10.1210/er.2016-1070. PubMed
8. Nielsen CT, Andersen ÅB. Hypercalcemia and renal failure in a case of disseminated Mycobacterium marinum infection. Eur J Intern Med. 2016;20(2):e29-e31. https://doi.org/10.1016/j.ejim.2008.08.015. PubMed
9. Sylvester RJ. Bacillus Calmette-Guérin treatment of non-muscle invasive bladder cancer. Int J Urol. 2011;18(2):113-120. https://doi.org/10.1111/j.1442-2042.2010.02678.x.
10. Clark PE, Spiess P, Agarwal N, Al. E. NCCN Guidelines ® Insights Bladder Cancer, Version 2.2016 Featured Updates to the NCCN Guidelines. J Natl Compr Canc Netw. 2016;14(10):1213-1224. https://doi.org/10.6004/jnccn.2016.0131. PubMed
11. Decaestecker K, Oosterlinck W. Managing the adverse events of intravesical bacillus Calmette–Guérin therapy. Res Reports Urol. 2015;7:157-163. https://doi.org/10.2147/RRU.S63448. PubMed
12. Gandhi NM, Morales A, Lamm DL. Bacillus Calmette-Guerin immunotherapy for genitourinary cancer. BJU Int. 2013;112(3):288-297. https://doi.org/10.1111/j.1464-410X.2012.11754.x. PubMed
13. Brausi M, Oddens J, Sylvester R, et al. Side effects of bacillus calmette-guerin (BCG) in the treatment of intermediate- and high-risk Ta, T1 papillary carcinoma of the bladder: Results of the EORTC genito-urinary cancers group randomised phase 3 study comparing one-third dose with full dose and 1 year with 3 years of maintenance BCG. Eur Urol. 2014;65(1):69-76. https://doi.org/10.1016/j.eururo.2013.07.021. PubMed
14. Gonzalez OY, Musher DM, Brar I, et al. Spectrum of bacille Calmette-Guérin (BCG) infection after intravesical BCG immunotherapy. Clin Infect Dis. 2003;36(2):140-148. https://doi.org/10.1086/344908. PubMed
15. Pérez-Jacoiste Asín MA, Fernández-Ruiz M, López-Medrano F, et al. Bacillus Calmette-Guérin (BCG) infection following intravesical BCG administration as adjunctive therapy for bladder cancer. Medicine (Baltimore). 2014;93(17):236-254. https://doi.org/10.1097/MD.0000000000000119. PubMed
16. Durek C, Rüsch-Gerdes S, Jocham D, Böhle A. Sensitivity of BCG to modern antibiotics. Eur Urol. 2000;37(Suppl 1):21-25. https://doi.org/10.1159/000052378. PubMed
17. Sharma OP. Hypercalcemia in granulomatous disorders: a clinical review. Curr Opin Pulm Med. 2000;6(5):442-447. https://doi.org/10.1097/00063198-200009000-00010. PubMed
18. LeMense GP, Strange C. Granulomatous pneumonitis following intravesical BCG: what therapy is needed? Chest. 1994;106(5):1624-1626. https://doi.org/10.1378/chest.106.5.1624. PubMed
19. Nadasy KA, Patel RS, Emmett M, et al. Four cases of disseminated Mycobacterium bovis infection following intravesical BCG instillation for treatment of bladder carcinoma. South Med J. 2008;101(1):91-95. https://doi.org/10.1097/SMJ.0b013e31815d4047. PubMed
20. Macleod LC, Ngo TC, Gonzalgo ML. Complications of intravesical bacillus calmette-guérin. Can Urol Assoc J. 2014;8(7-8):E540-E544. https://doi.org/10.5489/cuaj.1411. PubMed
A 70-year-old man presented to the emergency department with 5 days of decreased appetite, frequent urination, tremors, and memory difficulties. He also reported 9 months of malaise, generalized weakness, and weight loss. There was no history of fever, chills, nausea, diarrhea, constipation, pain, or focal neurologic complaints.
This patient exemplifies a common clinical challenge: an older adult with several possibly unrelated concerns. In many patients, a new presentation is usually either a different manifestation of a known condition (eg, a complication of an established malignancy) or the emergence of something they are at risk for based on health behavior or other characteristics (eg, lung cancer in a smoker). The diagnostic process in older adults can be complicated because many have, or are at risk for, multiple chronic conditions.
After reviewing the timeline of symptoms, the presence of 9 months of symptoms suggests a chronic and progressive underlying process, perhaps with subsequent superimposition of an acute problem. Although it is not certain whether chronic and acute symptoms are caused by the same process, this assumption is reasonable. The superimposition of acute symptoms on a chronic process may represent progression of the underlying condition or an acute complication of the underlying disease. However, the patient’s chronic symptoms of malaise, weakness, and weight loss are nonspecific.
Although malignancy is a consideration given the age of the patient and time course of symptoms, attributing the symptoms to a specific pattern of disease or building a cogent differential diagnosis is difficult until additional information is obtained. One strategy is to try to localize the findings to 1 or more organ systems; for example, given that tremors and memory difficulties localize to the central nervous system, neurodegenerative disorders, such as “Parkinson plus” syndromes, and cerebellar disease are possible. However, this tactic still leaves a relatively broad set of symptoms without an immediate and clear unifying cause.
The patient’s medical history included hyperlipidemia, peripheral neuropathy, prostate cancer, and papillary bladder cancer. The patient was admitted to the hospital 4 months earlier for severe sepsis presumed secondary to a urinary tract infection, although bacterial cultures were sterile. His social history was notable for a 50 pack-year smoking history. Outpatient medications included alfuzosin, gabapentin, simvastatin, hydrocodone, and cholecalciferol. He used a Bright Light Therapy lamp for 1 hour per week and occasionally used calcium carbonate for indigestion. The patient’s sister had a history of throat cancer.
On examination, the patient was detected with blood pressure of 104/56 mm Hg, pulse of 85 beats per minute, temperature of 98.2 °F, oxygen saturation of 97% on ambient air, and body mass index of 18 kg/m2. The patient appeared frail with mildly decreased strength in the upper and lower extremities bilaterally. The remainder of the physical examination was normal. Reflexes were symmetric, no tremors or rigidity was noted, sensation was intact to light touch, and the response to the Romberg maneuver was normal.
Past medical history is the cornerstone of the diagnostic process. The history of 2 different malignancies is the most striking element in this case. Papillary bladder cancer is usually a local process, but additional information is needed regarding its stage and previous treatment, including whether or not the patient received Bacille Calmette Guerin (BCG) vaccine, which can rarely be associated with infectious and inflammatory complications. Metastatic prostate cancer could certainly account for his symptomatology, and bladder outlet obstruction could explain the history of urinary frequency and probable urosepsis. His medication list suggested no obvious causes to explain his presentation, except that cholecalciferol and calcium carbonate, which when taken in excess, can cause hypercalcemia. This finding is of particular importance given that many of the patient’s symptoms, including polyuria, malaise, weakness, tremor, memory difficulties, anorexia, acute kidney injury and (indirectly) hypotension and weight loss, are also seen in patients with hypercalcemia. The relatively normal result of the neurologic examination decreases the probability of a primary neurologic disorder and increases the likelihood that his neurologic symptoms are due to a global systemic process. The relative hypotension and weight loss similarly support the possibility that the patient is experiencing a chronic and progressive process.
The differential diagnosis remains broad. An underlying malignancy would explain the chronic progressive course, and superimposed hypercalcemia would explain the acute symptoms of polyuria, tremor, and memory changes. Endocrinopathies including hyperthyroidism or adrenal insufficiency are other possibilities. A chronic progressive infection, such as tuberculosis, is possible, although no epidemiologic factors that increase his risk for this disease are present.
The patient had serum calcium of 14.5 mg/dL, ionized calcium of 3.46 mEq/L, albumin of 3.6 g/dL, BUN of 62 mg/dL, and creatinine of 3.9 mg/dL (all values were normal 3 months prior). His electrolytes and liver function were otherwise normal. Moreover, he had hemoglobin level of 10.5 mg/dL, white blood cell count of 4.8 × 109cells/L, and platelet count of 203 × 109 cells/L.
Until this point, only nonspecific findings were identified, leading to a broad differential diagnosis with little specificity. However, laboratory examinations confirm the suspected diagnosis of hypercalcemia, provide an opportunity to explain the patient’s symptoms, and offer a “lens” to narrow the differential diagnosis and guide the diagnostic evaluation. Hypercalcemia is most commonly secondary to primary hyperparathyroidism or malignancy. Primary hyperparathyroidism is unlikely in this patient given the relatively acute onset of symptoms. The degree of hypercalcemia is also atypical for primary hyperparathyroidism because it rarely exceeds 13 mg/dL, although the use of concurrent vitamin D and calcium supplementation could explain the high calcium level. Malignancy seems more likely given the degree of hypercalcemia in the setting of weight loss, tobacco use, and history of malignancy. Malignancy may cause hypercalcemia through multiple disparate mechanisms, including development of osteolytic bone metastases, elaboration of parathyroid hormone-related Peptide (PTHrP), increased production of 1,25-dihydroxyvitamin D, or, very rarely, ectopic production of parathyroid hormone (PTH). However, none of these mechanisms are particularly common in bladder or prostate cancer, which are the known malignancies in the patient. Other less likely and less common causes of hypercalcemia are also possible given the clinical clues, including vitamin D toxicity and milk alkali syndrome (vitamin D and calcium carbonate supplementation), multiple endocrine neoplasia (a sister with “throat cancer”), and granulomatous disease (weight loss). At this point, further laboratory evaluations would be helpful, specifically determination of PTH and PTHrP levels and serum and urine protein electrophoresis.
With respect to the patient’s past medical history, his Gleason 3 + 3 prostate cancer was diagnosed 12 years prior to admission and treated with external beam radiation therapy and brachytherapy. His bladder cancer was diagnosed 3 years before admission and treated with tumor resection followed by 2 rounds of intravesical BCG (iBCG), 1 round of mitomycin C, and 2 additional rounds of iBCG over the course of treatment spanning 2 years and 6 months. The treatment was complicated by urethral strictures requiring dilation, ureteral outlet obstruction requiring left ureteral stent placement, and multiple urinary tract infections.
The patient’s last round of iBCG was delivered 6 months prior to his current presentation. The patient’s hospital admission 4 months earlier for severe sepsis was presumed secondary to a urologic source considering that significant pyuria was noted on urinalysis and he was treated with meropenem, although bacterial cultures of blood and urine were sterile. From the time of discharge until his current presentation, he experienced progressive weakness and an approximately 50 lb weight loss.
The prior cancers and associated treatments of the patient may be involved in his current presentation. The simplest explanation would be metastatic disease with resultant hypercalcemia, which is atypical of either prostate or bladder cancer. The history of genitourinary surgery could predispose the patient to a chronic infection of the urinary tract with indolent organisms, such as a fungus, especially given the prior sepsis without clear etiology. However, the history would not explain the presence of hypercalcemia. Tuberculosis must thus be considered given the weight loss, hypercalcemia, and “sterile pyuria” of the patient. A more intriguing possibility is whether or not the patient’s constellation of signs and symptoms might be a late effect of iBCG. Intravesical BCG for treatment of localized bladder cancer is occasionally associated with complications. BCG is a modified live form of Mycobacterium bovis which invokes an intense inflammatory reaction when instilled into the bladder. These complications include disseminated infection and local complications, such as genitourinary infections. BCG infection might also explain the severe sepsis of unclear etiology that the patient had experienced 4 months earlier. Most interestingly, hypercalcemia has been described in the setting of BCG infection.
In the hospital, the patient received intravenous normal saline, furosemide, and pamidronate. Evaluation for hypercalcemia revealed appropriately suppressed PTH (8 mg/dL), and normal levels of PTHrP (<.74 pmol/L), prostate specific antigen (<.01 ng/mL), and morning cortisol (16.7 mcg/dL). Serum and urine electrophoresis did not show evidence for monoclonal gammopathy, and the 25-hydroxy vitamin D level (39.5 ng/mL) was within the normal limits
The suppressed PTH level makes primary hyperparathyroidism unlikely, the low PTHrP level decreases the probability of a paraneoplastic process, and the normal protein electrophoresis makes multiple myeloma unlikely. The presence of a significantly elevated 1,25-dihydroxy vitamin D level with a normal 25-hydroxy vitamin D level indicates extrarenal conversion of 25-hydroxy vitamin D by 1-hydroxylase as the etiology of hypercalcemia.
Lymphoma would appear to be the most likely diagnosis as it accounts for most of the clinical findings observed in the patient and is a fairly common disorder. Sarcoidosis is also reasonably common and would explain the laboratory abnormalities but is not usually associated with weight loss and frailty. Disseminated infections, such as tuberculosis, histoplasmosis, and coccidioidomycosis, are all possible, but the patient lacks key risk factors for these infections. A complication of iBCG is the most intriguing possibility and could account for many of the patient’s clinical findings, including the septic episode,
The bone survey was normal, the renal ultrasound examination showed nodular wall thickening of the bladder with areas of calcification, and the CT scan of the chest, abdomen, and pelvis showed an area of calcification in the superior portion of the bladder but no evidence of lymphadenopathy or masses to suggest lymphoma. Aerobic and anaerobic blood and urine cultures were sterile. The patient was discharged 12 days after admission with plans for further outpatient diagnostic evaluation. At this time, his serum calcium had stabilized at 10.5 mg/dL
DISCUSSION
Hypercalcemia is a common finding in both hospital and ambulatory settings. The classic symptoms associated with hypercalcemia are aptly summarized with the mnemonic “bones, stones, abdominal groans, and psychiatric overtones” (to represent the associated skeletal involvement, renal disease, gastrointestinal symptoms, and effects on the nervous system). However, the severity and type of symptoms vary depending on the degree of hypercalcemia, acuity of onset, and underlying etiology. The vast majority (90%) of hypercalcemia cases are due to primary hyperparathyroidism and malignancy.3 Measuring the PTH level is a key step in the diagnostic evaluation process. An isolated elevation of PTH confirms the presence of primary or possibly tertiary hyperparathyroidism. Low PTH concentrations (<20 pg/mL) occur in the settings of PTHrP or vitamin-D-mediated hypercalcemia such as hypervitaminosis D, malignancy, or granulomatous disease.
Elevated PTHrP occurs most commonly in squamous cell, renal, bladder, and ovarian carcinomas.3,4 Elevated levels of 25-hydroxy vitamin D can occur with excessive consumption of vitamin D-containing products and some herbal supplements. In this case, neither PTHrP nor 25-hydroxy vitamin D level was elevated, leading to an exhaustive search for other causes. Although iBCG treatment is a rare cause of hypercalcemia, 2 previous reports indicated the presence of hypercalcemia secondary to granuloma formation in treated patients.5,6
The finding of an elevated 1,25-dihydroxy vitamin D level was unexpected. As the discussant mentioned, this finding is associated with lymphoma and with granulomatous disorders that were not initially strong diagnostic considerations in the patient. A variety of granulomatous diseases can cause hypercalcemia. Sarcoidosis and tuberculosis are the most common, but berylliosis, fungal infections, Crohn’s disease, silicone exposure, and granulomatosis with polyangiitis may also be associated with hypercalcemia.7 The mechanism for hypercalcemia in these situations is increased intestinal calcium absorption mediated by inappropriately increased, PTH-independent, extrarenal calcitriol (1,25-dihydroxy vitamin D) production. Activated monocytes upregulate 25(OH)D-alpha-hydroxylase, converting 25-hydroxy vitamin D to 1,25-dihydroxy vitamin D. Concurrently, the elevated levels of gamma-interferon render macrophages resistant to the normal regulatory feedback mechanisms, thereby promoting the production and inhibiting the degradation of 1,25-dihydroxy vitamin D.8
The tuberculosis vaccine BCG is an attenuated form of M. bovis and was originally developed by Albert Calmette and Camille Guérin at the Pasteur Institute in Paris in the early 20th century. In addition to its use as a vaccine against tuberculosis, BCG can protect against other mycobacterial infections, help treat atopic conditions via stimulation of the Th1 cellular immune response, and has been used as an antineoplastic agent. To date, BCG remains the most effective agent available for intravesical treatment of superficial bladder cancer.9,10 Although iBCG therapy is considered relatively safe and well-tolerated, rare complications do occur. Localized symptoms (bladder irritation, hematuria) and/or flu-like symptoms are common immediately after instillation and thought to be related to the cellular immune response and inflammatory cascade triggered by mycobacterial antigens.11 Other adverse effects, such as infectious and noninfectious complications, may occur months to years after treatment with BCG, and the associated symptoms can be quite nonspecific. Infectious complications include mycobacterial prostatitis, orchiepididymitis, balantitis, pneumonia, hepatitis, nephritis, septic arthritis, osteomyelitis, infected orthopedic and vascular prostheses, endocarditis, and bacteremia. Traumatic catheterization is the most common risk factor for infection with BCG.11-13 Noninfectious complications include reactive arthritis, hypersensitivity pneumonitis, hemophagocytic lymphohistiocytosis (HLH), and sterile granulomatous infiltration of solid organs.
The protean and nonspecific nature of the adverse effects of iBCG treatment and the fact that complications can present weeks to years after instillation can make diagnosis quite challenging.14 Even if clinical suspicion is high, it may be difficult to definitively identify BCG as the underlying etiology because acid fast staining, culture, and even PCR can lead to falsely negative results.14,15 For this reason, biopsy and tissue culture are recommended to demonstrate granuloma formation and identify the presence of M. bovis.
Although no prospective studies have been conducted to assess the optimal therapy for BCG infection, opinion-based recommendations include cessation of BCG treatment, initiation of at least 3 tuberculostatic agents, and treatment for 3-12 months depending on the severity of the complications.11,14 M. bovis is susceptible to isoniazid, rifampin, and ethambutol as well as to fluoroquinolones, clarithromycin, aminoglycosides, and doxycycline; however, this organism is highly resistant to pyrazinamide due to single-point mutation.11,16
Although treatment with steroids is a standard approach for management of hypercalcemia in other granulomatous disorders and leads to rapid reduction in circulating levels of 1,25-dihydroxy vitamin D and serum calcium., specific evidence has not been established to support its efficacy and effectiveness in treating hypercalcemia and other complications due to M. bovis.17 Nevertheless, some experts recommend the use of steroids in conjunction with a multidrug tuberculostatic regimen in cases of septicemia and multiorgan failure due to M. bovis.12,14,18-20
In summary, this case illustrates the importance of making room in differential diagnosis to include iatrogenic complications. That is,
Teaching Points
- Complications of intravesical BCG treatment include manifestations of granulomatous diseases, such as hypercalcemia.
- When generating a differential diagnosis, medical providers should not only consider the possibility of a new disease process or the progression of a known comorbidity but also the potential of an adverse effect related to prior treatments.
- Medical providers should be wary of accepting previously made diagnoses, particularly when key pieces of objective data are lacking.
Disclosures
The authors have no financial or other conflicts of interest that might bias this work.
A 70-year-old man presented to the emergency department with 5 days of decreased appetite, frequent urination, tremors, and memory difficulties. He also reported 9 months of malaise, generalized weakness, and weight loss. There was no history of fever, chills, nausea, diarrhea, constipation, pain, or focal neurologic complaints.
This patient exemplifies a common clinical challenge: an older adult with several possibly unrelated concerns. In many patients, a new presentation is usually either a different manifestation of a known condition (eg, a complication of an established malignancy) or the emergence of something they are at risk for based on health behavior or other characteristics (eg, lung cancer in a smoker). The diagnostic process in older adults can be complicated because many have, or are at risk for, multiple chronic conditions.
After reviewing the timeline of symptoms, the presence of 9 months of symptoms suggests a chronic and progressive underlying process, perhaps with subsequent superimposition of an acute problem. Although it is not certain whether chronic and acute symptoms are caused by the same process, this assumption is reasonable. The superimposition of acute symptoms on a chronic process may represent progression of the underlying condition or an acute complication of the underlying disease. However, the patient’s chronic symptoms of malaise, weakness, and weight loss are nonspecific.
Although malignancy is a consideration given the age of the patient and time course of symptoms, attributing the symptoms to a specific pattern of disease or building a cogent differential diagnosis is difficult until additional information is obtained. One strategy is to try to localize the findings to 1 or more organ systems; for example, given that tremors and memory difficulties localize to the central nervous system, neurodegenerative disorders, such as “Parkinson plus” syndromes, and cerebellar disease are possible. However, this tactic still leaves a relatively broad set of symptoms without an immediate and clear unifying cause.
The patient’s medical history included hyperlipidemia, peripheral neuropathy, prostate cancer, and papillary bladder cancer. The patient was admitted to the hospital 4 months earlier for severe sepsis presumed secondary to a urinary tract infection, although bacterial cultures were sterile. His social history was notable for a 50 pack-year smoking history. Outpatient medications included alfuzosin, gabapentin, simvastatin, hydrocodone, and cholecalciferol. He used a Bright Light Therapy lamp for 1 hour per week and occasionally used calcium carbonate for indigestion. The patient’s sister had a history of throat cancer.
On examination, the patient was detected with blood pressure of 104/56 mm Hg, pulse of 85 beats per minute, temperature of 98.2 °F, oxygen saturation of 97% on ambient air, and body mass index of 18 kg/m2. The patient appeared frail with mildly decreased strength in the upper and lower extremities bilaterally. The remainder of the physical examination was normal. Reflexes were symmetric, no tremors or rigidity was noted, sensation was intact to light touch, and the response to the Romberg maneuver was normal.
Past medical history is the cornerstone of the diagnostic process. The history of 2 different malignancies is the most striking element in this case. Papillary bladder cancer is usually a local process, but additional information is needed regarding its stage and previous treatment, including whether or not the patient received Bacille Calmette Guerin (BCG) vaccine, which can rarely be associated with infectious and inflammatory complications. Metastatic prostate cancer could certainly account for his symptomatology, and bladder outlet obstruction could explain the history of urinary frequency and probable urosepsis. His medication list suggested no obvious causes to explain his presentation, except that cholecalciferol and calcium carbonate, which when taken in excess, can cause hypercalcemia. This finding is of particular importance given that many of the patient’s symptoms, including polyuria, malaise, weakness, tremor, memory difficulties, anorexia, acute kidney injury and (indirectly) hypotension and weight loss, are also seen in patients with hypercalcemia. The relatively normal result of the neurologic examination decreases the probability of a primary neurologic disorder and increases the likelihood that his neurologic symptoms are due to a global systemic process. The relative hypotension and weight loss similarly support the possibility that the patient is experiencing a chronic and progressive process.
The differential diagnosis remains broad. An underlying malignancy would explain the chronic progressive course, and superimposed hypercalcemia would explain the acute symptoms of polyuria, tremor, and memory changes. Endocrinopathies including hyperthyroidism or adrenal insufficiency are other possibilities. A chronic progressive infection, such as tuberculosis, is possible, although no epidemiologic factors that increase his risk for this disease are present.
The patient had serum calcium of 14.5 mg/dL, ionized calcium of 3.46 mEq/L, albumin of 3.6 g/dL, BUN of 62 mg/dL, and creatinine of 3.9 mg/dL (all values were normal 3 months prior). His electrolytes and liver function were otherwise normal. Moreover, he had hemoglobin level of 10.5 mg/dL, white blood cell count of 4.8 × 109cells/L, and platelet count of 203 × 109 cells/L.
Until this point, only nonspecific findings were identified, leading to a broad differential diagnosis with little specificity. However, laboratory examinations confirm the suspected diagnosis of hypercalcemia, provide an opportunity to explain the patient’s symptoms, and offer a “lens” to narrow the differential diagnosis and guide the diagnostic evaluation. Hypercalcemia is most commonly secondary to primary hyperparathyroidism or malignancy. Primary hyperparathyroidism is unlikely in this patient given the relatively acute onset of symptoms. The degree of hypercalcemia is also atypical for primary hyperparathyroidism because it rarely exceeds 13 mg/dL, although the use of concurrent vitamin D and calcium supplementation could explain the high calcium level. Malignancy seems more likely given the degree of hypercalcemia in the setting of weight loss, tobacco use, and history of malignancy. Malignancy may cause hypercalcemia through multiple disparate mechanisms, including development of osteolytic bone metastases, elaboration of parathyroid hormone-related Peptide (PTHrP), increased production of 1,25-dihydroxyvitamin D, or, very rarely, ectopic production of parathyroid hormone (PTH). However, none of these mechanisms are particularly common in bladder or prostate cancer, which are the known malignancies in the patient. Other less likely and less common causes of hypercalcemia are also possible given the clinical clues, including vitamin D toxicity and milk alkali syndrome (vitamin D and calcium carbonate supplementation), multiple endocrine neoplasia (a sister with “throat cancer”), and granulomatous disease (weight loss). At this point, further laboratory evaluations would be helpful, specifically determination of PTH and PTHrP levels and serum and urine protein electrophoresis.
With respect to the patient’s past medical history, his Gleason 3 + 3 prostate cancer was diagnosed 12 years prior to admission and treated with external beam radiation therapy and brachytherapy. His bladder cancer was diagnosed 3 years before admission and treated with tumor resection followed by 2 rounds of intravesical BCG (iBCG), 1 round of mitomycin C, and 2 additional rounds of iBCG over the course of treatment spanning 2 years and 6 months. The treatment was complicated by urethral strictures requiring dilation, ureteral outlet obstruction requiring left ureteral stent placement, and multiple urinary tract infections.
The patient’s last round of iBCG was delivered 6 months prior to his current presentation. The patient’s hospital admission 4 months earlier for severe sepsis was presumed secondary to a urologic source considering that significant pyuria was noted on urinalysis and he was treated with meropenem, although bacterial cultures of blood and urine were sterile. From the time of discharge until his current presentation, he experienced progressive weakness and an approximately 50 lb weight loss.
The prior cancers and associated treatments of the patient may be involved in his current presentation. The simplest explanation would be metastatic disease with resultant hypercalcemia, which is atypical of either prostate or bladder cancer. The history of genitourinary surgery could predispose the patient to a chronic infection of the urinary tract with indolent organisms, such as a fungus, especially given the prior sepsis without clear etiology. However, the history would not explain the presence of hypercalcemia. Tuberculosis must thus be considered given the weight loss, hypercalcemia, and “sterile pyuria” of the patient. A more intriguing possibility is whether or not the patient’s constellation of signs and symptoms might be a late effect of iBCG. Intravesical BCG for treatment of localized bladder cancer is occasionally associated with complications. BCG is a modified live form of Mycobacterium bovis which invokes an intense inflammatory reaction when instilled into the bladder. These complications include disseminated infection and local complications, such as genitourinary infections. BCG infection might also explain the severe sepsis of unclear etiology that the patient had experienced 4 months earlier. Most interestingly, hypercalcemia has been described in the setting of BCG infection.
In the hospital, the patient received intravenous normal saline, furosemide, and pamidronate. Evaluation for hypercalcemia revealed appropriately suppressed PTH (8 mg/dL), and normal levels of PTHrP (<.74 pmol/L), prostate specific antigen (<.01 ng/mL), and morning cortisol (16.7 mcg/dL). Serum and urine electrophoresis did not show evidence for monoclonal gammopathy, and the 25-hydroxy vitamin D level (39.5 ng/mL) was within the normal limits
The suppressed PTH level makes primary hyperparathyroidism unlikely, the low PTHrP level decreases the probability of a paraneoplastic process, and the normal protein electrophoresis makes multiple myeloma unlikely. The presence of a significantly elevated 1,25-dihydroxy vitamin D level with a normal 25-hydroxy vitamin D level indicates extrarenal conversion of 25-hydroxy vitamin D by 1-hydroxylase as the etiology of hypercalcemia.
Lymphoma would appear to be the most likely diagnosis as it accounts for most of the clinical findings observed in the patient and is a fairly common disorder. Sarcoidosis is also reasonably common and would explain the laboratory abnormalities but is not usually associated with weight loss and frailty. Disseminated infections, such as tuberculosis, histoplasmosis, and coccidioidomycosis, are all possible, but the patient lacks key risk factors for these infections. A complication of iBCG is the most intriguing possibility and could account for many of the patient’s clinical findings, including the septic episode,
The bone survey was normal, the renal ultrasound examination showed nodular wall thickening of the bladder with areas of calcification, and the CT scan of the chest, abdomen, and pelvis showed an area of calcification in the superior portion of the bladder but no evidence of lymphadenopathy or masses to suggest lymphoma. Aerobic and anaerobic blood and urine cultures were sterile. The patient was discharged 12 days after admission with plans for further outpatient diagnostic evaluation. At this time, his serum calcium had stabilized at 10.5 mg/dL
DISCUSSION
Hypercalcemia is a common finding in both hospital and ambulatory settings. The classic symptoms associated with hypercalcemia are aptly summarized with the mnemonic “bones, stones, abdominal groans, and psychiatric overtones” (to represent the associated skeletal involvement, renal disease, gastrointestinal symptoms, and effects on the nervous system). However, the severity and type of symptoms vary depending on the degree of hypercalcemia, acuity of onset, and underlying etiology. The vast majority (90%) of hypercalcemia cases are due to primary hyperparathyroidism and malignancy.3 Measuring the PTH level is a key step in the diagnostic evaluation process. An isolated elevation of PTH confirms the presence of primary or possibly tertiary hyperparathyroidism. Low PTH concentrations (<20 pg/mL) occur in the settings of PTHrP or vitamin-D-mediated hypercalcemia such as hypervitaminosis D, malignancy, or granulomatous disease.
Elevated PTHrP occurs most commonly in squamous cell, renal, bladder, and ovarian carcinomas.3,4 Elevated levels of 25-hydroxy vitamin D can occur with excessive consumption of vitamin D-containing products and some herbal supplements. In this case, neither PTHrP nor 25-hydroxy vitamin D level was elevated, leading to an exhaustive search for other causes. Although iBCG treatment is a rare cause of hypercalcemia, 2 previous reports indicated the presence of hypercalcemia secondary to granuloma formation in treated patients.5,6
The finding of an elevated 1,25-dihydroxy vitamin D level was unexpected. As the discussant mentioned, this finding is associated with lymphoma and with granulomatous disorders that were not initially strong diagnostic considerations in the patient. A variety of granulomatous diseases can cause hypercalcemia. Sarcoidosis and tuberculosis are the most common, but berylliosis, fungal infections, Crohn’s disease, silicone exposure, and granulomatosis with polyangiitis may also be associated with hypercalcemia.7 The mechanism for hypercalcemia in these situations is increased intestinal calcium absorption mediated by inappropriately increased, PTH-independent, extrarenal calcitriol (1,25-dihydroxy vitamin D) production. Activated monocytes upregulate 25(OH)D-alpha-hydroxylase, converting 25-hydroxy vitamin D to 1,25-dihydroxy vitamin D. Concurrently, the elevated levels of gamma-interferon render macrophages resistant to the normal regulatory feedback mechanisms, thereby promoting the production and inhibiting the degradation of 1,25-dihydroxy vitamin D.8
The tuberculosis vaccine BCG is an attenuated form of M. bovis and was originally developed by Albert Calmette and Camille Guérin at the Pasteur Institute in Paris in the early 20th century. In addition to its use as a vaccine against tuberculosis, BCG can protect against other mycobacterial infections, help treat atopic conditions via stimulation of the Th1 cellular immune response, and has been used as an antineoplastic agent. To date, BCG remains the most effective agent available for intravesical treatment of superficial bladder cancer.9,10 Although iBCG therapy is considered relatively safe and well-tolerated, rare complications do occur. Localized symptoms (bladder irritation, hematuria) and/or flu-like symptoms are common immediately after instillation and thought to be related to the cellular immune response and inflammatory cascade triggered by mycobacterial antigens.11 Other adverse effects, such as infectious and noninfectious complications, may occur months to years after treatment with BCG, and the associated symptoms can be quite nonspecific. Infectious complications include mycobacterial prostatitis, orchiepididymitis, balantitis, pneumonia, hepatitis, nephritis, septic arthritis, osteomyelitis, infected orthopedic and vascular prostheses, endocarditis, and bacteremia. Traumatic catheterization is the most common risk factor for infection with BCG.11-13 Noninfectious complications include reactive arthritis, hypersensitivity pneumonitis, hemophagocytic lymphohistiocytosis (HLH), and sterile granulomatous infiltration of solid organs.
The protean and nonspecific nature of the adverse effects of iBCG treatment and the fact that complications can present weeks to years after instillation can make diagnosis quite challenging.14 Even if clinical suspicion is high, it may be difficult to definitively identify BCG as the underlying etiology because acid fast staining, culture, and even PCR can lead to falsely negative results.14,15 For this reason, biopsy and tissue culture are recommended to demonstrate granuloma formation and identify the presence of M. bovis.
Although no prospective studies have been conducted to assess the optimal therapy for BCG infection, opinion-based recommendations include cessation of BCG treatment, initiation of at least 3 tuberculostatic agents, and treatment for 3-12 months depending on the severity of the complications.11,14 M. bovis is susceptible to isoniazid, rifampin, and ethambutol as well as to fluoroquinolones, clarithromycin, aminoglycosides, and doxycycline; however, this organism is highly resistant to pyrazinamide due to single-point mutation.11,16
Although treatment with steroids is a standard approach for management of hypercalcemia in other granulomatous disorders and leads to rapid reduction in circulating levels of 1,25-dihydroxy vitamin D and serum calcium., specific evidence has not been established to support its efficacy and effectiveness in treating hypercalcemia and other complications due to M. bovis.17 Nevertheless, some experts recommend the use of steroids in conjunction with a multidrug tuberculostatic regimen in cases of septicemia and multiorgan failure due to M. bovis.12,14,18-20
In summary, this case illustrates the importance of making room in differential diagnosis to include iatrogenic complications. That is,
Teaching Points
- Complications of intravesical BCG treatment include manifestations of granulomatous diseases, such as hypercalcemia.
- When generating a differential diagnosis, medical providers should not only consider the possibility of a new disease process or the progression of a known comorbidity but also the potential of an adverse effect related to prior treatments.
- Medical providers should be wary of accepting previously made diagnoses, particularly when key pieces of objective data are lacking.
Disclosures
The authors have no financial or other conflicts of interest that might bias this work.
1. Geisel RE, Sakamoto K, Russell DG, Rhoades ER. In vivo activity of released cell wall lipids of Mycobacterium bovis bacillus Calmette-Guérin is due principally to trehalose mycolates. J Immunol. 2005;174(8):5007-5015. https://doi.org/10.4049/jimmunol.174.8.5007. PubMed
2. Ryll R, Kumazawa Y, Yano I. Immunological properties of trehalose dimycolate (cord factor) and other mycolic acid-containing glycolipids--a review. Microbiol Immunol. 2001;45(12):801-811. https://doi.org/10.1111/j.1348-0421.2001.tb01319.x. PubMed
3. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966. PubMed
4. Goldner W. Cancer-related hypercalcemia. J Oncol Pract. 2016;12(5):426-432. https://doi.org/10.1200/JOP.2016.011155. PubMed
5. Nayar N, Briscoe K. Systemic Bacillus Calmette-Guerin sepsis manifesting as hypercalcaemia and thrombocytopenia as a complication of intravesical Bacillus Calmette-Guerin therapy. Intern Med J. 2015;45(10):1091-1092. https://doi.org/10.1111/imj.12876. PubMed
6. Schattner A, Gilad A, Cohen J. Systemic granulomatosis and hypercalcaemia following intravesical bacillus Calmette–Guerin immunotherapy. J Intern Med. 2002;251(3):272-277. https://doi.org/10.1046/j.1365-2796.2002.00957.x. PubMed
7. Tebben PJ, Singh RJ, Kumar R. Vitamin D-mediated hypercalcemia: mechanisms, diagnosis, and treatment. Endocr Rev. 2016;37(5):521-547. https://doi.org/10.1210/er.2016-1070. PubMed
8. Nielsen CT, Andersen ÅB. Hypercalcemia and renal failure in a case of disseminated Mycobacterium marinum infection. Eur J Intern Med. 2016;20(2):e29-e31. https://doi.org/10.1016/j.ejim.2008.08.015. PubMed
9. Sylvester RJ. Bacillus Calmette-Guérin treatment of non-muscle invasive bladder cancer. Int J Urol. 2011;18(2):113-120. https://doi.org/10.1111/j.1442-2042.2010.02678.x.
10. Clark PE, Spiess P, Agarwal N, Al. E. NCCN Guidelines ® Insights Bladder Cancer, Version 2.2016 Featured Updates to the NCCN Guidelines. J Natl Compr Canc Netw. 2016;14(10):1213-1224. https://doi.org/10.6004/jnccn.2016.0131. PubMed
11. Decaestecker K, Oosterlinck W. Managing the adverse events of intravesical bacillus Calmette–Guérin therapy. Res Reports Urol. 2015;7:157-163. https://doi.org/10.2147/RRU.S63448. PubMed
12. Gandhi NM, Morales A, Lamm DL. Bacillus Calmette-Guerin immunotherapy for genitourinary cancer. BJU Int. 2013;112(3):288-297. https://doi.org/10.1111/j.1464-410X.2012.11754.x. PubMed
13. Brausi M, Oddens J, Sylvester R, et al. Side effects of bacillus calmette-guerin (BCG) in the treatment of intermediate- and high-risk Ta, T1 papillary carcinoma of the bladder: Results of the EORTC genito-urinary cancers group randomised phase 3 study comparing one-third dose with full dose and 1 year with 3 years of maintenance BCG. Eur Urol. 2014;65(1):69-76. https://doi.org/10.1016/j.eururo.2013.07.021. PubMed
14. Gonzalez OY, Musher DM, Brar I, et al. Spectrum of bacille Calmette-Guérin (BCG) infection after intravesical BCG immunotherapy. Clin Infect Dis. 2003;36(2):140-148. https://doi.org/10.1086/344908. PubMed
15. Pérez-Jacoiste Asín MA, Fernández-Ruiz M, López-Medrano F, et al. Bacillus Calmette-Guérin (BCG) infection following intravesical BCG administration as adjunctive therapy for bladder cancer. Medicine (Baltimore). 2014;93(17):236-254. https://doi.org/10.1097/MD.0000000000000119. PubMed
16. Durek C, Rüsch-Gerdes S, Jocham D, Böhle A. Sensitivity of BCG to modern antibiotics. Eur Urol. 2000;37(Suppl 1):21-25. https://doi.org/10.1159/000052378. PubMed
17. Sharma OP. Hypercalcemia in granulomatous disorders: a clinical review. Curr Opin Pulm Med. 2000;6(5):442-447. https://doi.org/10.1097/00063198-200009000-00010. PubMed
18. LeMense GP, Strange C. Granulomatous pneumonitis following intravesical BCG: what therapy is needed? Chest. 1994;106(5):1624-1626. https://doi.org/10.1378/chest.106.5.1624. PubMed
19. Nadasy KA, Patel RS, Emmett M, et al. Four cases of disseminated Mycobacterium bovis infection following intravesical BCG instillation for treatment of bladder carcinoma. South Med J. 2008;101(1):91-95. https://doi.org/10.1097/SMJ.0b013e31815d4047. PubMed
20. Macleod LC, Ngo TC, Gonzalgo ML. Complications of intravesical bacillus calmette-guérin. Can Urol Assoc J. 2014;8(7-8):E540-E544. https://doi.org/10.5489/cuaj.1411. PubMed
1. Geisel RE, Sakamoto K, Russell DG, Rhoades ER. In vivo activity of released cell wall lipids of Mycobacterium bovis bacillus Calmette-Guérin is due principally to trehalose mycolates. J Immunol. 2005;174(8):5007-5015. https://doi.org/10.4049/jimmunol.174.8.5007. PubMed
2. Ryll R, Kumazawa Y, Yano I. Immunological properties of trehalose dimycolate (cord factor) and other mycolic acid-containing glycolipids--a review. Microbiol Immunol. 2001;45(12):801-811. https://doi.org/10.1111/j.1348-0421.2001.tb01319.x. PubMed
3. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966. PubMed
4. Goldner W. Cancer-related hypercalcemia. J Oncol Pract. 2016;12(5):426-432. https://doi.org/10.1200/JOP.2016.011155. PubMed
5. Nayar N, Briscoe K. Systemic Bacillus Calmette-Guerin sepsis manifesting as hypercalcaemia and thrombocytopenia as a complication of intravesical Bacillus Calmette-Guerin therapy. Intern Med J. 2015;45(10):1091-1092. https://doi.org/10.1111/imj.12876. PubMed
6. Schattner A, Gilad A, Cohen J. Systemic granulomatosis and hypercalcaemia following intravesical bacillus Calmette–Guerin immunotherapy. J Intern Med. 2002;251(3):272-277. https://doi.org/10.1046/j.1365-2796.2002.00957.x. PubMed
7. Tebben PJ, Singh RJ, Kumar R. Vitamin D-mediated hypercalcemia: mechanisms, diagnosis, and treatment. Endocr Rev. 2016;37(5):521-547. https://doi.org/10.1210/er.2016-1070. PubMed
8. Nielsen CT, Andersen ÅB. Hypercalcemia and renal failure in a case of disseminated Mycobacterium marinum infection. Eur J Intern Med. 2016;20(2):e29-e31. https://doi.org/10.1016/j.ejim.2008.08.015. PubMed
9. Sylvester RJ. Bacillus Calmette-Guérin treatment of non-muscle invasive bladder cancer. Int J Urol. 2011;18(2):113-120. https://doi.org/10.1111/j.1442-2042.2010.02678.x.
10. Clark PE, Spiess P, Agarwal N, Al. E. NCCN Guidelines ® Insights Bladder Cancer, Version 2.2016 Featured Updates to the NCCN Guidelines. J Natl Compr Canc Netw. 2016;14(10):1213-1224. https://doi.org/10.6004/jnccn.2016.0131. PubMed
11. Decaestecker K, Oosterlinck W. Managing the adverse events of intravesical bacillus Calmette–Guérin therapy. Res Reports Urol. 2015;7:157-163. https://doi.org/10.2147/RRU.S63448. PubMed
12. Gandhi NM, Morales A, Lamm DL. Bacillus Calmette-Guerin immunotherapy for genitourinary cancer. BJU Int. 2013;112(3):288-297. https://doi.org/10.1111/j.1464-410X.2012.11754.x. PubMed
13. Brausi M, Oddens J, Sylvester R, et al. Side effects of bacillus calmette-guerin (BCG) in the treatment of intermediate- and high-risk Ta, T1 papillary carcinoma of the bladder: Results of the EORTC genito-urinary cancers group randomised phase 3 study comparing one-third dose with full dose and 1 year with 3 years of maintenance BCG. Eur Urol. 2014;65(1):69-76. https://doi.org/10.1016/j.eururo.2013.07.021. PubMed
14. Gonzalez OY, Musher DM, Brar I, et al. Spectrum of bacille Calmette-Guérin (BCG) infection after intravesical BCG immunotherapy. Clin Infect Dis. 2003;36(2):140-148. https://doi.org/10.1086/344908. PubMed
15. Pérez-Jacoiste Asín MA, Fernández-Ruiz M, López-Medrano F, et al. Bacillus Calmette-Guérin (BCG) infection following intravesical BCG administration as adjunctive therapy for bladder cancer. Medicine (Baltimore). 2014;93(17):236-254. https://doi.org/10.1097/MD.0000000000000119. PubMed
16. Durek C, Rüsch-Gerdes S, Jocham D, Böhle A. Sensitivity of BCG to modern antibiotics. Eur Urol. 2000;37(Suppl 1):21-25. https://doi.org/10.1159/000052378. PubMed
17. Sharma OP. Hypercalcemia in granulomatous disorders: a clinical review. Curr Opin Pulm Med. 2000;6(5):442-447. https://doi.org/10.1097/00063198-200009000-00010. PubMed
18. LeMense GP, Strange C. Granulomatous pneumonitis following intravesical BCG: what therapy is needed? Chest. 1994;106(5):1624-1626. https://doi.org/10.1378/chest.106.5.1624. PubMed
19. Nadasy KA, Patel RS, Emmett M, et al. Four cases of disseminated Mycobacterium bovis infection following intravesical BCG instillation for treatment of bladder carcinoma. South Med J. 2008;101(1):91-95. https://doi.org/10.1097/SMJ.0b013e31815d4047. PubMed
20. Macleod LC, Ngo TC, Gonzalgo ML. Complications of intravesical bacillus calmette-guérin. Can Urol Assoc J. 2014;8(7-8):E540-E544. https://doi.org/10.5489/cuaj.1411. PubMed
©2018 Society of Hospital Medicine
Tissue Isn’t the Issue
A 43-year-old man with a history of asplenia, hepatitis C, and nephrolithiasis reported right-flank pain. He described severe, sharp pain that came in waves and radiated to the right groin, associated with nausea and nonbloody emesis. He noted “pink urine” but no dysuria. He had 4prior similar episodes during which he had passed kidney stones, although stone analysis had never been performed. He denied having fevers or chills.
The patient had been involved in a remote motor vehicle accident complicated by splenic laceration, for which he underwent splenectomy. He was appropriately immunized. The patient also suffered from bipolar affective disorder and untreated chronic hepatitis C infection with no evidence of cirrhosis. He smoked one pack of tobacco per day for the last 10 years and reported distant alcohol and methamphetamine use.
Right-flank pain can arise from conditions affecting the lower thorax (effusion, pneumonia, pulmonary embolism), abdomen (hepatobiliary or intestinal disease), retroperitoneum (hemorrhage or infection), musculoskeletal system, peripheral nerves (herpes zoster), or the genitourinary system (pyelonephritis). Pain radiating to the groin, discolored urine (suggesting hematuria), and history of kidney stones increase the likelihood of renal colic from nephrolithiasis.
Less commonly, flank pain and hematuria may present as initial symptoms of renal cell carcinoma, renal infarction, or aortic dissection. The patient’s immunosuppression from asplenia and active injection drug use could predispose him to septic emboli to his kidneys. Prior trauma causing aortic injury could predispose himto subsequent dissection.
The patient appeared well with a heart rate of 100 beats per minute, blood pressure 122/76 mmHg, temperature 36.8°C, respiratory rate 16 breaths per minute, and oxygen saturation 96% on room air. His cardiopulmonary and abdominal examinations were normal, and he had no costovertebral angle tenderness. His skin was warm and dry without rashes. His white blood cell (WBC) count was 26,000/μL; absolute neutrophil count was 22,000/μL. Serum chemistries were normal, including creatinine 0.63 mg/dL, calcium 8.8 mg/dL, and phosphorus 3.1 mg/dL. Lactate was 0.8 mmol/L (reference range: 0-2.0 mmol/L). Urinalysis revealed large ketones, >50 red blood cells (RBC) per high power field (HPF), <5 WBC per HPF, 1+ calcium oxalate crystals and pH 6.0. A bedside ultrasound showed mild right hydronephrosis. Computed tomography (CT) with intravenous contrast of his abdomen and pelvis demonstrated diffuse, mildly prominent subcentimeter mesenteric lymphadenopathy and no kidney stones. He was treated with intravenous fluids and pain control, and was discharged with a presumptive diagnosis of a passed kidney stone.
A passed stone would not explain this degree of leukocytosis. The CT results reduce the likelihood of a renal neoplasm, renal infarction, or pyelonephritis. Mesenteric lymphadenopathy is nonspecific, but it may signal underlying infection or malignancy with spread to lymph nodes, or it may be part of a systemic disorder causing generalized lymphadenopathy. Malignant causes of mesenteric lymphadenopathy (with no apparent primary tumor) include testicular cancer, lymphoma, and primary urogenital neoplasms.
The lower extremity nodules are consistent with erythema nodosum, which may be observed in numerous infectious and noninfectious illnesses. The rapid tempo of this febrile illness mandates early consideration of infection. Splenectomized patients are at risk for overwhelming post-splenectomy infection from encapsulated organisms, although this risk is significantly mitigated with appropriate immunization. The patient is at risk of bacterial endocarditis, which could explain his fevers and polyarthritis, although plaques, pustules, and oral ulcers would be unusual. Disseminated gonococcal infection causes fevers, oral lesions, polyarthritis and pustular skin lesions, but plaques are uncommon. Disseminated mycobacterial and fungal infections may cause oral ulcers, but affected patients tend to be severely ill and have profound immunosuppression. Secondary syphilis may account for many of the findings; however, oral ulcers would be unusual, and the rash tends to be more widespread, with a predilection for the palms and soles. Human immunodeficiency virus (HIV) can cause oral ulcers and is the chief viral etiology to consider.
Noninfectious illnesses to consider include neoplasms and connective tissue diseases. Malignancy would be unlikely to manifest this abruptly or produce a paraneoplastic disorder with these features.
The patient described severe fatigue and drenching night sweats for two months prior to admission. He denied dyspnea or cough. He was born in the southwestern United States and had lived in California for almost a decade. He had been incarcerated for a few years and released three years prior. He had intermittently lived in homeless shelters, but currently lived alone in downtown San Francisco. He had traveled remotely to the Caribbean, and more recently traveled frequently to the Central Valley in California. The patient formerly worked as a pipe-fitter and welder. He denied animal exposure or recent sick contacts. He was sexually active with women, and intermittently used barrier protection.
His years in the southwestern United States may have exposed the patient to blastomycosis or histoplasmosis; both can mimic mycobacterial disease. Blastomycosis demonstrates a slightly stronger predilection for spreading to the bones, genitourinary tract, and central nervous system, whereas histoplasmosis is a more frequent cause of polyarthrtitis and mesenteric adenopathy. The patient’s travel to the Central Valley, California raises the possibility of coccidioidomycosis, which typically starts with pulmonary disease prior to dissemination to bones, skin, and other less common sites. Pipe-fitters are predisposed to asbestos-related illnesses, including lung cancer and mesothelioma, which would not explain this patient’s presentation. Incarceration and high-risk sexual practices increase his risk for tuberculosis, HIV, and syphilis. Widespread skin involvement is more characteristic of syphilis or primary HIV infection than of disseminated fungal or mycobacterial infection.
WBC measured 29,000/uL with a neutrophilic predominance. His peripheral blood smear was unremarkable. A comprehensive metabolic panel was normal. Lactate dehydrogenase (LDH) was 317 U/L (reference range 140-280 U/L). Erythrocyte sedimentation rate (ESR) was 39 mm/hr (reference range < 20 mm/hr) and C-reactive protein (CRP) was 66 mg/L (reference range <6.3 mg/L). Blood, urine, and throat cultures were sent. Chest radiograph showed clear lungs without adenopathy. Ankle and knee radiographs identified small effusions bilaterally without bony abnormalities. CT of his brain showed a small, hypodense lesion in the right lacrimal gland. A lumbar puncture with cerebrospinal fluid (CSF) analysis showed absence of RBCs; WBC, 2/µL; protein, 35 mg/dL; glucose, 62 mg/dL; negative gram stain. CSF bacterial and fungal cultures, venereal disease research laboratory (VDRL), herpes simplex virus polymerase chain reaction (HSV PCR), and cryptococcal antigen were sent for laboratory analysis. The patient was started on vancomycin and aztreonam.
Lesions of the lacrimal gland feature multiple causes, including autoimmune diseases (Sjögren’s, Behçet’s disease), granulomatous diseases (sarcoidosis, granulomatosis with polyangiitis), neoplasms (salivary gland tumors, lymphoma), and infections. Initiating broad-spectrum antibiotics is reasonable while awaiting additional information from blood and urine cultures, serologies for HIV and syphilis, and purified protein derivative or interferon-gamma release assay (IGRA).
If these tests fail to reveal a diagnosis, the search for atypical infections and noninfectious possibilities should expand.
The patient continued to have intermittent fevers, sweats, and malaise over the next 3 days. All bacterial and fungal cultures remained negative, and antibiotics were discontinued. Rheumatoid factor, anticyclic citrullinated peptide, antinuclear antibody, and cryoglobulins were negative. Serum C3, C4, and angiotensin-converting enzyme (ACE) levels were normal. A rapid plasma reagin (RPR), HIV antibody, IGRA, and serum antibodies for Coccidioides, histoplasmosis, and West Nile virus were negative. Urine nucleic acid amplification testing for gonorrhea and chlamydia was negative. CSF VDRL, HSV PCR and cryptococcal antigen were negative. HSV culture from an oral ulcer showed no growth. The patient had a reactive hepatitis C antibody with a viral load of 3 million virus equivalents/mL.
The additional test results lower

The most likely diagnosis is Löfgren’s syndrome, a variant of sarcoidosis characterized by erythema nodosum, bilateral hilar lymphadenopathy, and polyarthralgias or polyarthritis. Löfgren’s syndrome may include fevers, uveitis, widespread skin lesions and other systemic manifestations. Sarcoidosis could explain the lacrimal gland lesion, and could manifest with recurrent kidney stones. Oral lesions may occur in sarcoidosis. A normal serum ACE level may be observed in up to half of patients. The lack of visualized granulomas on the submental node FNA may reflect sampling error, lower likelihood of visualizing granulomas on FNA (compared with excisional biopsy), or biopsy location (hilar nodes are more likely to demonstrate sarcoid granulomas).
Although Löfgren’s syndrome is often self-limited, treatment can ameliorate symptoms. Nonsteroidal anti-inflammatory medication can be tried first, with prednisone reserved for refractory cases.
The constellation of bilateral hilar adenopathy, arthritis, and erythema nodosum was consistent with Löfgren’s syndrome, further supported by granulomatous infiltrates on biopsy. The patient’s symptoms resolved with naproxen. He was scheduled for follow-up in dermatology and rheumatology clinics and was referred to hepatology for management of hepatitis C.
COMMENTARY
Sarcoidosis is a multisystem granulomatous disease of unclear etiology. The disease derives its name from Boeck’s 1899 report describing benign cutaneous lesions that resembled sarcomas.1 Sarcoidosis most commonly manifests as bilateral hilar adenopathy and pulmonary infiltrates, but may impact any tissue or organ, including the eyes, nonhilar lymph nodes, liver, spleen, joints, mucous membranes, and skin. Nephrolithiasis may result from hypercalcemia and/or hypercalciuria (related to granulomatous production of 1,25 vitamin D) and can be the presenting feature of sarcoidosis.2 Less common presentations include neurologic sarcoidosis (which can present with seizures, aseptic meningitis, encephalopathy, neuroendocrine dysfunction, myelopathy and peripheral neuropathies), cardiac sarcoidosis (which may present with arrhythmias, valvular dysfunction, heart failure, ischemia, or pericardial disease), and Heerfordt syndrome (the constellation of parotid gland enlargement, facial palsy, anterior uveitis, and fever). Sarcoidosis may mimic other diseases, including malignancy, idiopathic pulmonary fibrosis, and infiltrative tuberculosis.3 Sarcoidosis-like reactions have occurred in response to malignancy and medications.4
The patient’s rash demonstrated a predilection for areas of prior scarring, which has a limited differential diagnosis. Keloids and hypertrophic scars occur at sites of former surgical wounds, lacerations, or areas of inflammation. Pruritic urticarial papules and plaques of pregnancy (PUPPP) is a benign inflammatory condition where papules cluster in areas of prior striae. Cutaneous lesions of Behçet’s syndrome display pathergy, where pustular response is observed at sites of injury. Granulomatous infiltration in sarcoidosis may demonstrate a predilection for scars and tattoos (ie, scar or tattoo sarcoidosis).5 Sarcoidosis can have other cutaneous manifestations, including psoriaform, ulcerative, or erythrodermic lesions; subcutaneous nodules; scarring or nonscarring alopecia; and lupus pernio – violaceous, nodular and plaque-like lesions on the nose, earlobes, cheeks, and digits.5
Löfgren’s syndrome is a distinct variant of sarcoidosis.In 1952, Dr. Löfgren described a case series of patients with bilateral hilar lymphadenopathy and coexisting erythema nodosum and polyarthralgia.6 The epidemiology favors young women.7 Patients with Löfgren’s syndrome present acutely (as in this case), which differs from the typical subacute course observed with sarcoidosis. In addition to the classic presentation described above, patients with Löfgren’s syndrome may demonstrate additional manifestations of sarcoidosis, including fevers, peripheral adenopathy, arthritis, and granulomatous skin lesions. Painful symptoms may require short-term anti-inflammatory treatments. Most patients do not require systemic immunosuppression. Symptoms usually decrease over several months, and the majority of patients experience complete remission within years. Rare recurrences have been described up to several years.8
In confirming the diagnosis of sarcoidosis, current guidelines recommend exclusion of other diseases that present similarly, a work-up that generally includes compatible laboratory tests and imaging, and histologic demonstration of noncaseating granulomas.9 However, Löfgren’s syndrome is a notable exception. The constellation of fever, bilateral hilar adenopathy, polyarthralgia, and erythema nodosum suffices to diagnose Löfgren’s syndrome as long as the disease remits rapidly and spontaneously.9 Thus, in this case, although granulomatous infiltrates were confirmed on biopsy, the diagnosis of Löfgren’s syndrome could have been based on clinical and radiologic features alone.
KEY LEARNING POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly presents with bilateral hilar adenopathy and pulmonary infiltrates but can also present atypically, including with nephrolithiasis from hypercalcemia, neurologic syndromes, and cardiac involvement.
- Löfgren’s syndrome, a variant of sarcoidosis, is characterized by relatively acute onset of fevers, erythema nodosum, bilateral hilar adenopathy, and polyarthralgia or polyarthritis. Most patients recover and manifest complete remission.
- A limited differential exists for rashes with a predilection for areas of tattoos and prior scarring, including keloids, PUPPP, Behçet’s disease, and granulomatous infiltration.
Disclosure
There are no conflicts of interest or financial disclosures to report.
1. Multiple Benign Sarcoids of the Skin. JAMA. 1899;XXXIII(26):1620-1621.
2. Rizzato G, Fraioli P, Montemurro L. Nephrolithiasis as a presenting feature of chronic sarcoidosis. Thorax. 1995;50(5):555-559. PubMed
3. Romanov V. Atypical variants of clinical course of sarcoidosis. Eur Respir J. 2014;44(58):3782. PubMed
4. Arish N, Kuint R, Sapir E, et al. Characteristics of Sarcoidosis in Patients with Previous Malignancy: Causality or Coincidence? Respiration. 2017;93(4):247-252. PubMed
5. Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25(3):295-302. PubMed
6. Löfgren S. The Bilateral Hilar Lymphoma Syndrome. Acta Med Scand. 1952;142(4):265-273. PubMed
7. Mañá J, Gómez-Vaquero C, Montero A et al. Löfgren’s syndrome revisited: a study of 186 patients. Am J Med. 1999;107(3):240-245. PubMed
8. Gran J, Bohmer E. Acute Sarcoid Arthritis: A Favourable Outcome? Scand J Rheumatol. 1996;25(2):70-73. PubMed
9. American Thoracic Society. Statement on Sarcoidosis. Am J Respir Crit Care Med. 1999;160:736-755.Otate voluptiatia qui aut iur, utendi quiae incipis m PubMed
A 43-year-old man with a history of asplenia, hepatitis C, and nephrolithiasis reported right-flank pain. He described severe, sharp pain that came in waves and radiated to the right groin, associated with nausea and nonbloody emesis. He noted “pink urine” but no dysuria. He had 4prior similar episodes during which he had passed kidney stones, although stone analysis had never been performed. He denied having fevers or chills.
The patient had been involved in a remote motor vehicle accident complicated by splenic laceration, for which he underwent splenectomy. He was appropriately immunized. The patient also suffered from bipolar affective disorder and untreated chronic hepatitis C infection with no evidence of cirrhosis. He smoked one pack of tobacco per day for the last 10 years and reported distant alcohol and methamphetamine use.
Right-flank pain can arise from conditions affecting the lower thorax (effusion, pneumonia, pulmonary embolism), abdomen (hepatobiliary or intestinal disease), retroperitoneum (hemorrhage or infection), musculoskeletal system, peripheral nerves (herpes zoster), or the genitourinary system (pyelonephritis). Pain radiating to the groin, discolored urine (suggesting hematuria), and history of kidney stones increase the likelihood of renal colic from nephrolithiasis.
Less commonly, flank pain and hematuria may present as initial symptoms of renal cell carcinoma, renal infarction, or aortic dissection. The patient’s immunosuppression from asplenia and active injection drug use could predispose him to septic emboli to his kidneys. Prior trauma causing aortic injury could predispose himto subsequent dissection.
The patient appeared well with a heart rate of 100 beats per minute, blood pressure 122/76 mmHg, temperature 36.8°C, respiratory rate 16 breaths per minute, and oxygen saturation 96% on room air. His cardiopulmonary and abdominal examinations were normal, and he had no costovertebral angle tenderness. His skin was warm and dry without rashes. His white blood cell (WBC) count was 26,000/μL; absolute neutrophil count was 22,000/μL. Serum chemistries were normal, including creatinine 0.63 mg/dL, calcium 8.8 mg/dL, and phosphorus 3.1 mg/dL. Lactate was 0.8 mmol/L (reference range: 0-2.0 mmol/L). Urinalysis revealed large ketones, >50 red blood cells (RBC) per high power field (HPF), <5 WBC per HPF, 1+ calcium oxalate crystals and pH 6.0. A bedside ultrasound showed mild right hydronephrosis. Computed tomography (CT) with intravenous contrast of his abdomen and pelvis demonstrated diffuse, mildly prominent subcentimeter mesenteric lymphadenopathy and no kidney stones. He was treated with intravenous fluids and pain control, and was discharged with a presumptive diagnosis of a passed kidney stone.
A passed stone would not explain this degree of leukocytosis. The CT results reduce the likelihood of a renal neoplasm, renal infarction, or pyelonephritis. Mesenteric lymphadenopathy is nonspecific, but it may signal underlying infection or malignancy with spread to lymph nodes, or it may be part of a systemic disorder causing generalized lymphadenopathy. Malignant causes of mesenteric lymphadenopathy (with no apparent primary tumor) include testicular cancer, lymphoma, and primary urogenital neoplasms.
The lower extremity nodules are consistent with erythema nodosum, which may be observed in numerous infectious and noninfectious illnesses. The rapid tempo of this febrile illness mandates early consideration of infection. Splenectomized patients are at risk for overwhelming post-splenectomy infection from encapsulated organisms, although this risk is significantly mitigated with appropriate immunization. The patient is at risk of bacterial endocarditis, which could explain his fevers and polyarthritis, although plaques, pustules, and oral ulcers would be unusual. Disseminated gonococcal infection causes fevers, oral lesions, polyarthritis and pustular skin lesions, but plaques are uncommon. Disseminated mycobacterial and fungal infections may cause oral ulcers, but affected patients tend to be severely ill and have profound immunosuppression. Secondary syphilis may account for many of the findings; however, oral ulcers would be unusual, and the rash tends to be more widespread, with a predilection for the palms and soles. Human immunodeficiency virus (HIV) can cause oral ulcers and is the chief viral etiology to consider.
Noninfectious illnesses to consider include neoplasms and connective tissue diseases. Malignancy would be unlikely to manifest this abruptly or produce a paraneoplastic disorder with these features.
The patient described severe fatigue and drenching night sweats for two months prior to admission. He denied dyspnea or cough. He was born in the southwestern United States and had lived in California for almost a decade. He had been incarcerated for a few years and released three years prior. He had intermittently lived in homeless shelters, but currently lived alone in downtown San Francisco. He had traveled remotely to the Caribbean, and more recently traveled frequently to the Central Valley in California. The patient formerly worked as a pipe-fitter and welder. He denied animal exposure or recent sick contacts. He was sexually active with women, and intermittently used barrier protection.
His years in the southwestern United States may have exposed the patient to blastomycosis or histoplasmosis; both can mimic mycobacterial disease. Blastomycosis demonstrates a slightly stronger predilection for spreading to the bones, genitourinary tract, and central nervous system, whereas histoplasmosis is a more frequent cause of polyarthrtitis and mesenteric adenopathy. The patient’s travel to the Central Valley, California raises the possibility of coccidioidomycosis, which typically starts with pulmonary disease prior to dissemination to bones, skin, and other less common sites. Pipe-fitters are predisposed to asbestos-related illnesses, including lung cancer and mesothelioma, which would not explain this patient’s presentation. Incarceration and high-risk sexual practices increase his risk for tuberculosis, HIV, and syphilis. Widespread skin involvement is more characteristic of syphilis or primary HIV infection than of disseminated fungal or mycobacterial infection.
WBC measured 29,000/uL with a neutrophilic predominance. His peripheral blood smear was unremarkable. A comprehensive metabolic panel was normal. Lactate dehydrogenase (LDH) was 317 U/L (reference range 140-280 U/L). Erythrocyte sedimentation rate (ESR) was 39 mm/hr (reference range < 20 mm/hr) and C-reactive protein (CRP) was 66 mg/L (reference range <6.3 mg/L). Blood, urine, and throat cultures were sent. Chest radiograph showed clear lungs without adenopathy. Ankle and knee radiographs identified small effusions bilaterally without bony abnormalities. CT of his brain showed a small, hypodense lesion in the right lacrimal gland. A lumbar puncture with cerebrospinal fluid (CSF) analysis showed absence of RBCs; WBC, 2/µL; protein, 35 mg/dL; glucose, 62 mg/dL; negative gram stain. CSF bacterial and fungal cultures, venereal disease research laboratory (VDRL), herpes simplex virus polymerase chain reaction (HSV PCR), and cryptococcal antigen were sent for laboratory analysis. The patient was started on vancomycin and aztreonam.
Lesions of the lacrimal gland feature multiple causes, including autoimmune diseases (Sjögren’s, Behçet’s disease), granulomatous diseases (sarcoidosis, granulomatosis with polyangiitis), neoplasms (salivary gland tumors, lymphoma), and infections. Initiating broad-spectrum antibiotics is reasonable while awaiting additional information from blood and urine cultures, serologies for HIV and syphilis, and purified protein derivative or interferon-gamma release assay (IGRA).
If these tests fail to reveal a diagnosis, the search for atypical infections and noninfectious possibilities should expand.
The patient continued to have intermittent fevers, sweats, and malaise over the next 3 days. All bacterial and fungal cultures remained negative, and antibiotics were discontinued. Rheumatoid factor, anticyclic citrullinated peptide, antinuclear antibody, and cryoglobulins were negative. Serum C3, C4, and angiotensin-converting enzyme (ACE) levels were normal. A rapid plasma reagin (RPR), HIV antibody, IGRA, and serum antibodies for Coccidioides, histoplasmosis, and West Nile virus were negative. Urine nucleic acid amplification testing for gonorrhea and chlamydia was negative. CSF VDRL, HSV PCR and cryptococcal antigen were negative. HSV culture from an oral ulcer showed no growth. The patient had a reactive hepatitis C antibody with a viral load of 3 million virus equivalents/mL.
The additional test results lower
The most likely diagnosis is Löfgren’s syndrome, a variant of sarcoidosis characterized by erythema nodosum, bilateral hilar lymphadenopathy, and polyarthralgias or polyarthritis. Löfgren’s syndrome may include fevers, uveitis, widespread skin lesions and other systemic manifestations. Sarcoidosis could explain the lacrimal gland lesion, and could manifest with recurrent kidney stones. Oral lesions may occur in sarcoidosis. A normal serum ACE level may be observed in up to half of patients. The lack of visualized granulomas on the submental node FNA may reflect sampling error, lower likelihood of visualizing granulomas on FNA (compared with excisional biopsy), or biopsy location (hilar nodes are more likely to demonstrate sarcoid granulomas).
Although Löfgren’s syndrome is often self-limited, treatment can ameliorate symptoms. Nonsteroidal anti-inflammatory medication can be tried first, with prednisone reserved for refractory cases.
The constellation of bilateral hilar adenopathy, arthritis, and erythema nodosum was consistent with Löfgren’s syndrome, further supported by granulomatous infiltrates on biopsy. The patient’s symptoms resolved with naproxen. He was scheduled for follow-up in dermatology and rheumatology clinics and was referred to hepatology for management of hepatitis C.
COMMENTARY
Sarcoidosis is a multisystem granulomatous disease of unclear etiology. The disease derives its name from Boeck’s 1899 report describing benign cutaneous lesions that resembled sarcomas.1 Sarcoidosis most commonly manifests as bilateral hilar adenopathy and pulmonary infiltrates, but may impact any tissue or organ, including the eyes, nonhilar lymph nodes, liver, spleen, joints, mucous membranes, and skin. Nephrolithiasis may result from hypercalcemia and/or hypercalciuria (related to granulomatous production of 1,25 vitamin D) and can be the presenting feature of sarcoidosis.2 Less common presentations include neurologic sarcoidosis (which can present with seizures, aseptic meningitis, encephalopathy, neuroendocrine dysfunction, myelopathy and peripheral neuropathies), cardiac sarcoidosis (which may present with arrhythmias, valvular dysfunction, heart failure, ischemia, or pericardial disease), and Heerfordt syndrome (the constellation of parotid gland enlargement, facial palsy, anterior uveitis, and fever). Sarcoidosis may mimic other diseases, including malignancy, idiopathic pulmonary fibrosis, and infiltrative tuberculosis.3 Sarcoidosis-like reactions have occurred in response to malignancy and medications.4
The patient’s rash demonstrated a predilection for areas of prior scarring, which has a limited differential diagnosis. Keloids and hypertrophic scars occur at sites of former surgical wounds, lacerations, or areas of inflammation. Pruritic urticarial papules and plaques of pregnancy (PUPPP) is a benign inflammatory condition where papules cluster in areas of prior striae. Cutaneous lesions of Behçet’s syndrome display pathergy, where pustular response is observed at sites of injury. Granulomatous infiltration in sarcoidosis may demonstrate a predilection for scars and tattoos (ie, scar or tattoo sarcoidosis).5 Sarcoidosis can have other cutaneous manifestations, including psoriaform, ulcerative, or erythrodermic lesions; subcutaneous nodules; scarring or nonscarring alopecia; and lupus pernio – violaceous, nodular and plaque-like lesions on the nose, earlobes, cheeks, and digits.5
Löfgren’s syndrome is a distinct variant of sarcoidosis.In 1952, Dr. Löfgren described a case series of patients with bilateral hilar lymphadenopathy and coexisting erythema nodosum and polyarthralgia.6 The epidemiology favors young women.7 Patients with Löfgren’s syndrome present acutely (as in this case), which differs from the typical subacute course observed with sarcoidosis. In addition to the classic presentation described above, patients with Löfgren’s syndrome may demonstrate additional manifestations of sarcoidosis, including fevers, peripheral adenopathy, arthritis, and granulomatous skin lesions. Painful symptoms may require short-term anti-inflammatory treatments. Most patients do not require systemic immunosuppression. Symptoms usually decrease over several months, and the majority of patients experience complete remission within years. Rare recurrences have been described up to several years.8
In confirming the diagnosis of sarcoidosis, current guidelines recommend exclusion of other diseases that present similarly, a work-up that generally includes compatible laboratory tests and imaging, and histologic demonstration of noncaseating granulomas.9 However, Löfgren’s syndrome is a notable exception. The constellation of fever, bilateral hilar adenopathy, polyarthralgia, and erythema nodosum suffices to diagnose Löfgren’s syndrome as long as the disease remits rapidly and spontaneously.9 Thus, in this case, although granulomatous infiltrates were confirmed on biopsy, the diagnosis of Löfgren’s syndrome could have been based on clinical and radiologic features alone.
KEY LEARNING POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly presents with bilateral hilar adenopathy and pulmonary infiltrates but can also present atypically, including with nephrolithiasis from hypercalcemia, neurologic syndromes, and cardiac involvement.
- Löfgren’s syndrome, a variant of sarcoidosis, is characterized by relatively acute onset of fevers, erythema nodosum, bilateral hilar adenopathy, and polyarthralgia or polyarthritis. Most patients recover and manifest complete remission.
- A limited differential exists for rashes with a predilection for areas of tattoos and prior scarring, including keloids, PUPPP, Behçet’s disease, and granulomatous infiltration.
Disclosure
There are no conflicts of interest or financial disclosures to report.
A 43-year-old man with a history of asplenia, hepatitis C, and nephrolithiasis reported right-flank pain. He described severe, sharp pain that came in waves and radiated to the right groin, associated with nausea and nonbloody emesis. He noted “pink urine” but no dysuria. He had 4prior similar episodes during which he had passed kidney stones, although stone analysis had never been performed. He denied having fevers or chills.
The patient had been involved in a remote motor vehicle accident complicated by splenic laceration, for which he underwent splenectomy. He was appropriately immunized. The patient also suffered from bipolar affective disorder and untreated chronic hepatitis C infection with no evidence of cirrhosis. He smoked one pack of tobacco per day for the last 10 years and reported distant alcohol and methamphetamine use.
Right-flank pain can arise from conditions affecting the lower thorax (effusion, pneumonia, pulmonary embolism), abdomen (hepatobiliary or intestinal disease), retroperitoneum (hemorrhage or infection), musculoskeletal system, peripheral nerves (herpes zoster), or the genitourinary system (pyelonephritis). Pain radiating to the groin, discolored urine (suggesting hematuria), and history of kidney stones increase the likelihood of renal colic from nephrolithiasis.
Less commonly, flank pain and hematuria may present as initial symptoms of renal cell carcinoma, renal infarction, or aortic dissection. The patient’s immunosuppression from asplenia and active injection drug use could predispose him to septic emboli to his kidneys. Prior trauma causing aortic injury could predispose himto subsequent dissection.
The patient appeared well with a heart rate of 100 beats per minute, blood pressure 122/76 mmHg, temperature 36.8°C, respiratory rate 16 breaths per minute, and oxygen saturation 96% on room air. His cardiopulmonary and abdominal examinations were normal, and he had no costovertebral angle tenderness. His skin was warm and dry without rashes. His white blood cell (WBC) count was 26,000/μL; absolute neutrophil count was 22,000/μL. Serum chemistries were normal, including creatinine 0.63 mg/dL, calcium 8.8 mg/dL, and phosphorus 3.1 mg/dL. Lactate was 0.8 mmol/L (reference range: 0-2.0 mmol/L). Urinalysis revealed large ketones, >50 red blood cells (RBC) per high power field (HPF), <5 WBC per HPF, 1+ calcium oxalate crystals and pH 6.0. A bedside ultrasound showed mild right hydronephrosis. Computed tomography (CT) with intravenous contrast of his abdomen and pelvis demonstrated diffuse, mildly prominent subcentimeter mesenteric lymphadenopathy and no kidney stones. He was treated with intravenous fluids and pain control, and was discharged with a presumptive diagnosis of a passed kidney stone.
A passed stone would not explain this degree of leukocytosis. The CT results reduce the likelihood of a renal neoplasm, renal infarction, or pyelonephritis. Mesenteric lymphadenopathy is nonspecific, but it may signal underlying infection or malignancy with spread to lymph nodes, or it may be part of a systemic disorder causing generalized lymphadenopathy. Malignant causes of mesenteric lymphadenopathy (with no apparent primary tumor) include testicular cancer, lymphoma, and primary urogenital neoplasms.
The lower extremity nodules are consistent with erythema nodosum, which may be observed in numerous infectious and noninfectious illnesses. The rapid tempo of this febrile illness mandates early consideration of infection. Splenectomized patients are at risk for overwhelming post-splenectomy infection from encapsulated organisms, although this risk is significantly mitigated with appropriate immunization. The patient is at risk of bacterial endocarditis, which could explain his fevers and polyarthritis, although plaques, pustules, and oral ulcers would be unusual. Disseminated gonococcal infection causes fevers, oral lesions, polyarthritis and pustular skin lesions, but plaques are uncommon. Disseminated mycobacterial and fungal infections may cause oral ulcers, but affected patients tend to be severely ill and have profound immunosuppression. Secondary syphilis may account for many of the findings; however, oral ulcers would be unusual, and the rash tends to be more widespread, with a predilection for the palms and soles. Human immunodeficiency virus (HIV) can cause oral ulcers and is the chief viral etiology to consider.
Noninfectious illnesses to consider include neoplasms and connective tissue diseases. Malignancy would be unlikely to manifest this abruptly or produce a paraneoplastic disorder with these features.
The patient described severe fatigue and drenching night sweats for two months prior to admission. He denied dyspnea or cough. He was born in the southwestern United States and had lived in California for almost a decade. He had been incarcerated for a few years and released three years prior. He had intermittently lived in homeless shelters, but currently lived alone in downtown San Francisco. He had traveled remotely to the Caribbean, and more recently traveled frequently to the Central Valley in California. The patient formerly worked as a pipe-fitter and welder. He denied animal exposure or recent sick contacts. He was sexually active with women, and intermittently used barrier protection.
His years in the southwestern United States may have exposed the patient to blastomycosis or histoplasmosis; both can mimic mycobacterial disease. Blastomycosis demonstrates a slightly stronger predilection for spreading to the bones, genitourinary tract, and central nervous system, whereas histoplasmosis is a more frequent cause of polyarthrtitis and mesenteric adenopathy. The patient’s travel to the Central Valley, California raises the possibility of coccidioidomycosis, which typically starts with pulmonary disease prior to dissemination to bones, skin, and other less common sites. Pipe-fitters are predisposed to asbestos-related illnesses, including lung cancer and mesothelioma, which would not explain this patient’s presentation. Incarceration and high-risk sexual practices increase his risk for tuberculosis, HIV, and syphilis. Widespread skin involvement is more characteristic of syphilis or primary HIV infection than of disseminated fungal or mycobacterial infection.
WBC measured 29,000/uL with a neutrophilic predominance. His peripheral blood smear was unremarkable. A comprehensive metabolic panel was normal. Lactate dehydrogenase (LDH) was 317 U/L (reference range 140-280 U/L). Erythrocyte sedimentation rate (ESR) was 39 mm/hr (reference range < 20 mm/hr) and C-reactive protein (CRP) was 66 mg/L (reference range <6.3 mg/L). Blood, urine, and throat cultures were sent. Chest radiograph showed clear lungs without adenopathy. Ankle and knee radiographs identified small effusions bilaterally without bony abnormalities. CT of his brain showed a small, hypodense lesion in the right lacrimal gland. A lumbar puncture with cerebrospinal fluid (CSF) analysis showed absence of RBCs; WBC, 2/µL; protein, 35 mg/dL; glucose, 62 mg/dL; negative gram stain. CSF bacterial and fungal cultures, venereal disease research laboratory (VDRL), herpes simplex virus polymerase chain reaction (HSV PCR), and cryptococcal antigen were sent for laboratory analysis. The patient was started on vancomycin and aztreonam.
Lesions of the lacrimal gland feature multiple causes, including autoimmune diseases (Sjögren’s, Behçet’s disease), granulomatous diseases (sarcoidosis, granulomatosis with polyangiitis), neoplasms (salivary gland tumors, lymphoma), and infections. Initiating broad-spectrum antibiotics is reasonable while awaiting additional information from blood and urine cultures, serologies for HIV and syphilis, and purified protein derivative or interferon-gamma release assay (IGRA).
If these tests fail to reveal a diagnosis, the search for atypical infections and noninfectious possibilities should expand.
The patient continued to have intermittent fevers, sweats, and malaise over the next 3 days. All bacterial and fungal cultures remained negative, and antibiotics were discontinued. Rheumatoid factor, anticyclic citrullinated peptide, antinuclear antibody, and cryoglobulins were negative. Serum C3, C4, and angiotensin-converting enzyme (ACE) levels were normal. A rapid plasma reagin (RPR), HIV antibody, IGRA, and serum antibodies for Coccidioides, histoplasmosis, and West Nile virus were negative. Urine nucleic acid amplification testing for gonorrhea and chlamydia was negative. CSF VDRL, HSV PCR and cryptococcal antigen were negative. HSV culture from an oral ulcer showed no growth. The patient had a reactive hepatitis C antibody with a viral load of 3 million virus equivalents/mL.
The additional test results lower
The most likely diagnosis is Löfgren’s syndrome, a variant of sarcoidosis characterized by erythema nodosum, bilateral hilar lymphadenopathy, and polyarthralgias or polyarthritis. Löfgren’s syndrome may include fevers, uveitis, widespread skin lesions and other systemic manifestations. Sarcoidosis could explain the lacrimal gland lesion, and could manifest with recurrent kidney stones. Oral lesions may occur in sarcoidosis. A normal serum ACE level may be observed in up to half of patients. The lack of visualized granulomas on the submental node FNA may reflect sampling error, lower likelihood of visualizing granulomas on FNA (compared with excisional biopsy), or biopsy location (hilar nodes are more likely to demonstrate sarcoid granulomas).
Although Löfgren’s syndrome is often self-limited, treatment can ameliorate symptoms. Nonsteroidal anti-inflammatory medication can be tried first, with prednisone reserved for refractory cases.
The constellation of bilateral hilar adenopathy, arthritis, and erythema nodosum was consistent with Löfgren’s syndrome, further supported by granulomatous infiltrates on biopsy. The patient’s symptoms resolved with naproxen. He was scheduled for follow-up in dermatology and rheumatology clinics and was referred to hepatology for management of hepatitis C.
COMMENTARY
Sarcoidosis is a multisystem granulomatous disease of unclear etiology. The disease derives its name from Boeck’s 1899 report describing benign cutaneous lesions that resembled sarcomas.1 Sarcoidosis most commonly manifests as bilateral hilar adenopathy and pulmonary infiltrates, but may impact any tissue or organ, including the eyes, nonhilar lymph nodes, liver, spleen, joints, mucous membranes, and skin. Nephrolithiasis may result from hypercalcemia and/or hypercalciuria (related to granulomatous production of 1,25 vitamin D) and can be the presenting feature of sarcoidosis.2 Less common presentations include neurologic sarcoidosis (which can present with seizures, aseptic meningitis, encephalopathy, neuroendocrine dysfunction, myelopathy and peripheral neuropathies), cardiac sarcoidosis (which may present with arrhythmias, valvular dysfunction, heart failure, ischemia, or pericardial disease), and Heerfordt syndrome (the constellation of parotid gland enlargement, facial palsy, anterior uveitis, and fever). Sarcoidosis may mimic other diseases, including malignancy, idiopathic pulmonary fibrosis, and infiltrative tuberculosis.3 Sarcoidosis-like reactions have occurred in response to malignancy and medications.4
The patient’s rash demonstrated a predilection for areas of prior scarring, which has a limited differential diagnosis. Keloids and hypertrophic scars occur at sites of former surgical wounds, lacerations, or areas of inflammation. Pruritic urticarial papules and plaques of pregnancy (PUPPP) is a benign inflammatory condition where papules cluster in areas of prior striae. Cutaneous lesions of Behçet’s syndrome display pathergy, where pustular response is observed at sites of injury. Granulomatous infiltration in sarcoidosis may demonstrate a predilection for scars and tattoos (ie, scar or tattoo sarcoidosis).5 Sarcoidosis can have other cutaneous manifestations, including psoriaform, ulcerative, or erythrodermic lesions; subcutaneous nodules; scarring or nonscarring alopecia; and lupus pernio – violaceous, nodular and plaque-like lesions on the nose, earlobes, cheeks, and digits.5
Löfgren’s syndrome is a distinct variant of sarcoidosis.In 1952, Dr. Löfgren described a case series of patients with bilateral hilar lymphadenopathy and coexisting erythema nodosum and polyarthralgia.6 The epidemiology favors young women.7 Patients with Löfgren’s syndrome present acutely (as in this case), which differs from the typical subacute course observed with sarcoidosis. In addition to the classic presentation described above, patients with Löfgren’s syndrome may demonstrate additional manifestations of sarcoidosis, including fevers, peripheral adenopathy, arthritis, and granulomatous skin lesions. Painful symptoms may require short-term anti-inflammatory treatments. Most patients do not require systemic immunosuppression. Symptoms usually decrease over several months, and the majority of patients experience complete remission within years. Rare recurrences have been described up to several years.8
In confirming the diagnosis of sarcoidosis, current guidelines recommend exclusion of other diseases that present similarly, a work-up that generally includes compatible laboratory tests and imaging, and histologic demonstration of noncaseating granulomas.9 However, Löfgren’s syndrome is a notable exception. The constellation of fever, bilateral hilar adenopathy, polyarthralgia, and erythema nodosum suffices to diagnose Löfgren’s syndrome as long as the disease remits rapidly and spontaneously.9 Thus, in this case, although granulomatous infiltrates were confirmed on biopsy, the diagnosis of Löfgren’s syndrome could have been based on clinical and radiologic features alone.
KEY LEARNING POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly presents with bilateral hilar adenopathy and pulmonary infiltrates but can also present atypically, including with nephrolithiasis from hypercalcemia, neurologic syndromes, and cardiac involvement.
- Löfgren’s syndrome, a variant of sarcoidosis, is characterized by relatively acute onset of fevers, erythema nodosum, bilateral hilar adenopathy, and polyarthralgia or polyarthritis. Most patients recover and manifest complete remission.
- A limited differential exists for rashes with a predilection for areas of tattoos and prior scarring, including keloids, PUPPP, Behçet’s disease, and granulomatous infiltration.
Disclosure
There are no conflicts of interest or financial disclosures to report.
1. Multiple Benign Sarcoids of the Skin. JAMA. 1899;XXXIII(26):1620-1621.
2. Rizzato G, Fraioli P, Montemurro L. Nephrolithiasis as a presenting feature of chronic sarcoidosis. Thorax. 1995;50(5):555-559. PubMed
3. Romanov V. Atypical variants of clinical course of sarcoidosis. Eur Respir J. 2014;44(58):3782. PubMed
4. Arish N, Kuint R, Sapir E, et al. Characteristics of Sarcoidosis in Patients with Previous Malignancy: Causality or Coincidence? Respiration. 2017;93(4):247-252. PubMed
5. Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25(3):295-302. PubMed
6. Löfgren S. The Bilateral Hilar Lymphoma Syndrome. Acta Med Scand. 1952;142(4):265-273. PubMed
7. Mañá J, Gómez-Vaquero C, Montero A et al. Löfgren’s syndrome revisited: a study of 186 patients. Am J Med. 1999;107(3):240-245. PubMed
8. Gran J, Bohmer E. Acute Sarcoid Arthritis: A Favourable Outcome? Scand J Rheumatol. 1996;25(2):70-73. PubMed
9. American Thoracic Society. Statement on Sarcoidosis. Am J Respir Crit Care Med. 1999;160:736-755.Otate voluptiatia qui aut iur, utendi quiae incipis m PubMed
1. Multiple Benign Sarcoids of the Skin. JAMA. 1899;XXXIII(26):1620-1621.
2. Rizzato G, Fraioli P, Montemurro L. Nephrolithiasis as a presenting feature of chronic sarcoidosis. Thorax. 1995;50(5):555-559. PubMed
3. Romanov V. Atypical variants of clinical course of sarcoidosis. Eur Respir J. 2014;44(58):3782. PubMed
4. Arish N, Kuint R, Sapir E, et al. Characteristics of Sarcoidosis in Patients with Previous Malignancy: Causality or Coincidence? Respiration. 2017;93(4):247-252. PubMed
5. Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25(3):295-302. PubMed
6. Löfgren S. The Bilateral Hilar Lymphoma Syndrome. Acta Med Scand. 1952;142(4):265-273. PubMed
7. Mañá J, Gómez-Vaquero C, Montero A et al. Löfgren’s syndrome revisited: a study of 186 patients. Am J Med. 1999;107(3):240-245. PubMed
8. Gran J, Bohmer E. Acute Sarcoid Arthritis: A Favourable Outcome? Scand J Rheumatol. 1996;25(2):70-73. PubMed
9. American Thoracic Society. Statement on Sarcoidosis. Am J Respir Crit Care Med. 1999;160:736-755.Otate voluptiatia qui aut iur, utendi quiae incipis m PubMed
© 2018 Society of Hospital Medicine
Scratching Beneath the Surface
A 62-year-old man with severe chronic obstructive pulmonary disease (COPD; forced expiratory volume during the first second [FEV1] 40% predicted) and type 2 diabetes mellitus presented to a Veterans Affairs emergency department (ED) with a steadily worsening cough of 4-months’ duration. He also reported subjective fevers, sputum production, shortness of breath, and unintentional 20-pound weight loss. He denied chills, chest pain, nausea, or vomiting.
Cough is classified as acute, subacute, or chronic based on duration of less than 3 weeks, between 3-8 weeks, and greater than 8 weeks, respectively. Common causes of chronic cough include bronchitis, acid reflux, cough-variant asthma, and a side effect of angiotensin converting enzyme inhibitors. Unintentional weight loss suggests a serious disorder, including indolent infection, end-stage COPD, malignancy, and autoimmune causes. Among patients with chronic bronchitis, the microbiology of sputum is often mixed with commensal respiratory flora, including Streptococcus pneumoniae and Haemophilus species. When these organisms are not recovered in sputa, or when patients fail to respond to empiric treatment, the differential diagnosis should be broadened to include pulmonary tuberculosis, nontuberculous mycobacterial infection, lung abscess, pulmonary nocardiosis, or pertussis.
An exposure and social history can focus the differential. For example, coccidioidomycosis or histoplasmosis may present indolently, but have distinct geographic distributions. Bird fanciers may acquire hypersensitivity pneumonitis, psittacosis, or cryptococcosis. Risk factors including smoking history, corticosteroid use, uncontrolled diabetes, and ill contacts should be assessed.
He was discharged from the ED twice in the last 2 weeks after presenting with similar symptoms. On each occasion, he was treated for presumed COPD exacerbations with nebulized albuterol and ipratropium, methylprednisolone followed by oral prednisone, and azithromycin, which did not lead to improvement. Over the last 3 days, he developed lower extremity edema, orthopnea, and dyspnea at rest. He reported worsening fatigue, night sweats, and anorexia. He denied any sick contacts.
Two diagnostic issues have emerged. His edema, orthopnea, and dyspnea at rest suggest a new cause of hypervolemia, perhaps caused by sodium retention from corticosteroids, pulmonary edema from valvular or myocardial disease, or renal failure. More concerning is that he has been treated with azithromycin twice recently but still has night sweats, fatigue, and anorexia. The presence of weight loss despite extracellular volume accumulation suggests an indolent systemic illness. Infection with macrolide-resistant organisms, such as nocardia, mycobacteria, or endemic mycoses, remains high on the differential diagnosis.
His past medical history included hypertension, untreated chronic hepatitis C, tobacco dependence, alcohol use disorder, and extraction of 8 decayed teeth 2 months earlier. He served in a noncombat role during the Vietnam War. He consumed 12 beers weekly with a remote history of alcoholism which required rehabilitation, reported a 50 pack-year smoking history, and denied intravenous (IV) drug use. He lived with an appropriately vaccinated dog and denied recent insect or animal exposures. He had a cat that passed away from an unknown illness 3 years prior. He was in a monogamous relationship with his girlfriend of 35 years. His father had coronary disease. His medications included glyburide, hydrochlorothiazide, lisinopril, theophylline, and meloxicam. Chronic cough, weight loss, diabetes, alcoholism, and history of dental disease raise concern for lung abscess. Oral microbiota such as Streptococcus viridans and Actinomycetes are usually harmless, but when aspirated repeatedly, such as during alcohol intoxication, may evolve into a lung abscess via bronchogenic spread. The combination of unintentional weight loss and smoking history raises concern for lung malignancy. Small cell lung cancer can present with paraneoplastic Cushing’s syndrome and could explain the patient’s volume overload. Finally, human immunodeficiency virus (HIV) serostatus should be determined in all adult patients.
His temperature was 37 °C, blood pressure 161/69 mm Hg, pulse 104 beats per minute, respiratory rate 20 breaths per minute, and oxygen saturation was 95% on room air. On examination, he was an unkempt, ill-appearing man. He had poor dentition, but no oral ulcers or petechiae. Pulmonary exam revealed diffuse rhonchi and scattered wheezes. He developed dyspnea after speaking 2 sentences. Cardiovascular exam showed regular tachycardia, normal S1 and S2 heart sounds, and both an S3 and S4 gallop. A grade III/VI holosystolic murmur at the left lower sternal border with apical radiation, and an early, grade III/IV diastolic murmur at the right upper sternal border were present. Neck exam showed jugular venous distention (JVD) 8 cm above the right clavicle. Lower extremities showed symmetric 3+ pitting edema to the knees. His abdomen was soft, nondistended, and without hepatosplenomegaly. There was no lymphadenopathy. Skin exam showed small, healed excoriations on his anterior shins, forearms, and knuckles. There were no petechiae, Janeway lesions, or Osler’s nodes.
These exam findings change the differential substantially. New regurgitant murmurs strongly suggest infective endocarditis (IE). A diastolic murmur is never normal and suggests aortic regurgitation. The holosystolic murmur with apical radiation suggests mitral regurgitation. Cutaneous stigmata should always be sought, but are found in fewer than half of cases of subacute IE, and their absence does not rule out this diagnosis. Disheveled hygiene and excoriations suggest a skin source of infection, and poor dentition is concerning for an oral source. For the moment, the source does not matter. His clinical condition is serious: tachycardia, JVD, edema, and two-sentence dyspnea indicate congestive heart failure. Even before labs and imaging return, inpatient admission is warranted.
Serum sodium concentration was 140 mEq/L, potassium 3.7 mEq/L, chloride 103 mEq/L, bicarbonate 30 mEq/L, blood urea nitrogen (BUN) 26 mg/dL, creatinine 0.8 mg/dL, glucose 120 mg/dL, and calcium 9.0 mg/dL. The white blood cell count was 7100/µL, hemoglobin 11.8 g/dL, and platelet count 101 K/µL. Brain natriuretic peptide (BNP) was 785 pg/mL (reference range 0-100 pg/mL), aspartate aminotransferase 77 U/L, alanine aminotransferase 57 U/L, alkaline phosphatase 125 U/L, total bilirubin 0.8 mg/dL, total protein 7.7 g/dL, and albumin 3.7 g/dL. Erythrocyte sedimentation (ESR) rate was 38 mm/hour (reference range 0-25 mm/hour) and C-reactive protein (CRP) 0.62 mg/dL (reference range <1.0 mg/dL). Cardiac troponins were 0.03 ng/mL (reference range <0.04 ng/mL). Screening for HIV was negative. Urinalysis showed trace blood by dipstick, but no glucose, protein, dysmorphic red blood cells, or casts. Two sets of peripheral blood cultures were drawn. Two sets of blood cultures from his previous ED visits were negative (drawn 6 and 14 days prior).
These laboratory values are nonspecific, and the differential remains unchanged, with top concern for IE, then lung abscess. Ideally, 3 sets of cultures drawn greater than 12 hours apart should be obtained because the likelihood of pathogen detection rises with the volume of blood tested. Thrombocytopenia and microscopic hematuria suggest microangiopathic hemolytic anemia, and a peripheral blood smear should be examined for schistocytes. Glomerulonephritis from immune complex deposition can occur in IE, but is unlikely with a normal serum creatinine and lack of proteinuria, dysmorphic red blood cells, or casts. The elevated BNP suggests cardiac strain due to a regurgitant valve. ESR and CRP are rarely helpful in this situation, and perhaps previous treatment with azithromycin and steroids prevented significant elevation.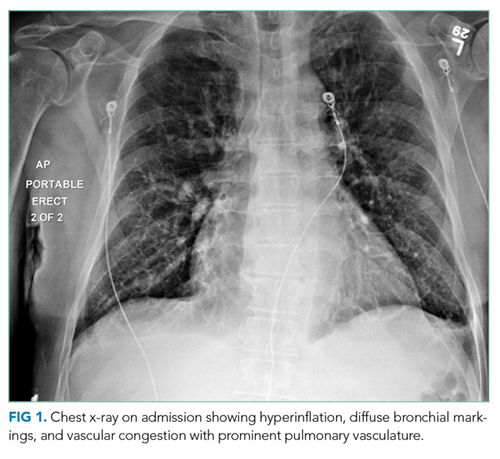
His chest x-ray is not consistent with acute or chronic pulmonary infection. His symptoms, EKG, edema, and improvement with diuresis support the diagnosis of congestive heart failure. The leading diagnosis is left-sided IE, and antimicrobial therapy should not be delayed for the sake of awaiting positive blood cultures. He should immediately receive empiric antibiotics to cover gram-positive bacteria (Methicillin-resistant Staphylococcus aureus, Methicillin-sensitive S. aureus, coagulase-negative staphylococci, and enterococci) and Haemophilus species, Actinobacillus actinomycetemcomitans, Cardiobacterium hominis, Eikenella species, and Kingella kingae (the HACEK group). In accordance with Infectious Diseases Society of America (IDSA) practice guidelines, he should empirically receive IV vancomycin plus ceftriaxone and urgently undergo echocardiography.
Transthoracic echocardiogram (TTE) showed severe aortic insufficiency, aortic valve vegetations, and raised suspicion for a moderate-sized vegetation on the anterior leaflet of the mitral valve. There was moderate mitral insufficiency, moderate tricuspid insufficiency, and an elevated right ventricular systolic pressure of 50 mm Hg. The left ventricle showed concentric hypertrophy with an ejection fraction of 55%. A previous echocardiogram 2 years prior showed mild mitral insufficiency, but no aneurysm or aortic insufficiency. Blood cultures from admission yielded no growth.
Due to concern for IE, blood cultures were repeated, and IV vancomycin, IV ceftriaxone, and IV gentamicin were initiated. Azithromycin and prednisone were discontinued. His respiratory status continued to improve with IV furosemide, albuterol, ipratropium, and supportive care.
TTE inadequately visualizes the mitral valve, but is useful for tricuspid valve assessment because the right ventricle is closer to the chest wall. Transesophageal echocardiography (TEE) is indicated for a more detailed assessment of the left heart valves for vegetations and perivalvar abscesses. The new regurgitant murmurs satisfy a major criterion of the modified Duke criteria, and valvar vegetations suggests IE. He does not yet fulfill the other major modified Duke criterion for IE, nor does he satisfy enough minor criteria because there are no diagnostic vascular, microbiologic, or immunologic phenomena. However, no diagnostic rubric is perfect, and these results should not supersede clinical judgment. Despite the absence of positive cultures, the concern for bacterial IE remains high. The absence of embolic phenomena fits best with subacute rather than acute IE. Three negative blood cultures to date suggest a fastidious organism is responsible, although oral flora remain on the differential.
There is rarely a need to “hold” blood cultures for prolonged periods because modern instruments typically yield positive results within 7 days for most bacteria, including the HACEK group. Blood culture-negative endocarditis (BCNE) is considered when 3 sets of cultures are negative for at least 5 days. In this situation, one should consider other microorganisms based on the patient’s exposure history. Only certain species with complex growth requirements, such as Brucella and Bartonella, require prolonged holds. Revisiting his exposure history would be helpful in deciding whether serologic testing warranted. If he recalls exposure to parturient animals, then Coxiella is worth pursuing; if he has been bitten by lice, then B. quintana rises as a possibility; if the scratches on his limbs are from recent cat scratches, then B. henselae becomes more likely. Both C. burnetti and Bartonella endocarditis might be partially treated by his courses of azithromycin, confounding the picture.
If the infectious work-up is ultimately negative, one could then consider other etiologies of endocarditis, such as nonbacterial thrombotic endocarditis, which is seen in the context of malignancy and systemic lupus erythematosus (Libman-Sacks endocarditis). Other mimickers of IE include myxomatous valve degeneration, ruptured mitral chordae, and eosinophilic heart disease (Löffler’s endocarditis).
A transesophageal echocardiogram confirmed the presence of small echodensities on the aortic valve’s right and left coronary cusps, consistent with vegetations. The vegetation on the anterior leaflet of the mitral valve from the TTE also showed an aneurysm with a small perforation (Figure 2).
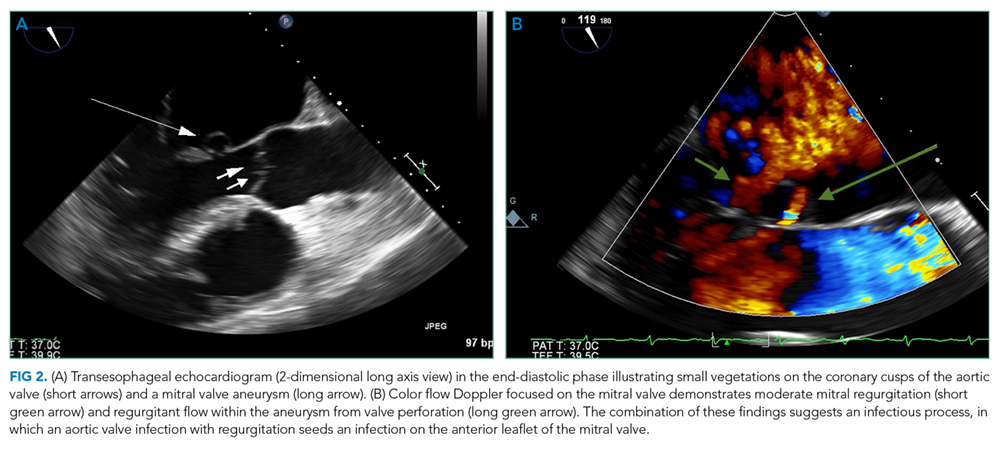
The combination of aortic regurgitation and the mitral valve aneurysm supports IE, because the aortic regurgitant jet directly strikes the anterior mitral valve leaflet, seeding the valve with infection from the aortic cusps. A positive serum PCR is diagnostic, but if it had been negative or unavailable, the serology would remain very helpful. In this context, the elevated IgG titer implicates B. henselae, the agent responsible for cat scratch disease (CSD). Out of context, these titers would not be diagnostic, because anti-Bartonella IgG may be increased due to a prior subclinical episode of CSD. Anti-Bartonella IgM is an unreliable indicator of recent infection because it may wane within weeks, and this IgG titer is higher than what is observed with most remote infections.
Revisiting previous cat exposure is warranted. He lost his cat to an illness 3 years prior, however it would be appropriate to inquire about other animals, such as a stray kitten with fleas, which his skin scratches suggest. Up to 50% of all cats in flea endemic regions harbor Bartonella and are asymptomatic. Rarely, dogs can serve as reservoirs of this organism, with a presumed transmission route via flea, louse, or tick. Regardless of the route of infection, treatment should be focused on B. henselae IE.
Azithromycin can treat CSD, and its use for his presumed COPD exacerbation may have temporized his infection. However, azithromycin monotherapy is not recommended for B. henselae IE. Treatment is usually with 2 antibiotics, including an aminoglycoside (gentamicin) for the first 2 weeks, combined with either a tetracycline, a macrolide, or a beta-lactam for a minimum of 4-6 weeks. Oral rifampin can be considered if gentamicin is not tolerated. After completing IV treatment, an additional 6 months of oral doxycycline or azithromycin should be considered, especially for those who have not undergone valve surgery.
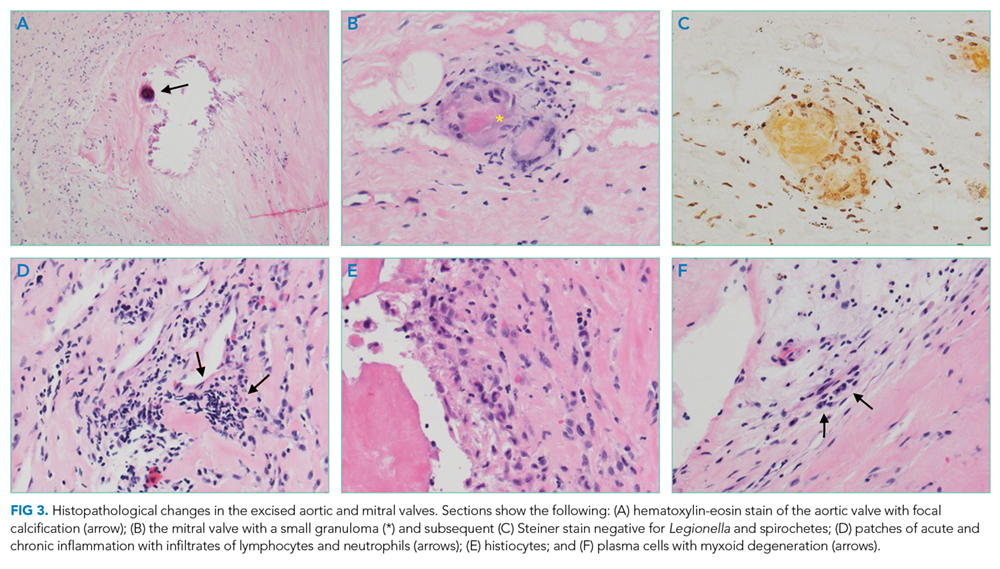
The mitral valve aneurysm, abscesses, and heart failure warranted valve replacement. Surgery should be considered for all patients with Bartonella IE, primarily because delayed diagnosis often leads to irreversible valve damage. Ideally, surgically explanted tissue should be divided into 2 portions: half should be sent to pathology and stained with H&E, Warthin-Starry, and Steiner staining procedures, while the other half should be sent for culture, and then PCR if stains are negative.
His symptoms are compatible with subacute IE, which is typically more difficult to diagnose than acute IE due to its insidious onset. He meets criteria for blood culture negative IE based on 3 sets of negative blood cultures for greater than 5 days and major criteria for IE. The pathologic changes are consistent with B. henselae infection.
DISCUSSION
The incidence of IE in the United States is 40,000 cases per year1 with an in-hospital mortality of 15%-20% and a 1-year mortality of up to 40%.2,3 Five to 20% of patients with IE never develop positive blood cultures4 due to receipt of antibiotics prior to culture, inadequate microbiologic testing, or infection caused by noncultivable bacteria (eg, Tropheryma whipplei), fastidious extracellular bacteria (eg, HACEK group and nutritionally variant streptococci), or by intracellular pathogens with complex nutrient requirements (eg, Bartonella, Chlamydia, Brucella, or Coxiella). Previous administration of antibiotics reduces the likelihood of isolating an organism by 35%-40%.5 Patients meeting criteria for BCNE should prompt consideration of serologic testing. The most prevalent pathogens vary globally, and incidence data in the US is scarce. Worldwide, the majority of BCNE cases are caused by Coxiella, Bartonella, and Brucella species.6,7
When clinical suspicion for IE remains high despite negative cultures, detailed history can uncover clues and guide additional testing. For example, contact with contaminated milk products or farm animals are associated with Brucella, Coxiella, and Erysipelothrix species IE.7,8 Bartonella species are zoonotic gram-negative bacilli with a tropism for endothelial cells and are transmitted by arthropod vectors (ie, fleas, lice, ticks, and sandflies), cat scratches, or cat bites. Bartonella may account for 3%-4% of all cases of IE, most of which are due to B. henselae and B. quintana.7, 9 Underlying heart valve disease, alcoholism, cirrhosis, and homelessness are associated with B. henselae endocarditis.10
Diagnostic criteria are lacking for B. henselae IE, and the modified Duke criteria is of limited utility for diagnosing Bartonella IE because blood cultures are often negative and echocardiographic evidence of vegetation is not always apparent. Serology plays a critical role in the diagnosis of Bartonella infections. The addition of positive serology, Western blot or PCR for B. henselae and B. quintana as a major criterion in the modified Duke criteria for IE has been proposed but has not yet been formally accepted.9 For B. henselae IE, an IgG titer of ≥1:800 has been recommended as a cutoff for subacute IE because it combines a high specificity and positive predictive value along with reasonable sensitivity and negative predictive value in this situation.9 The humoral immune response rises over time, and thus acute IE due to Bartonella may not generate a substantial IgG titer. Interestingly, because of the indolent nature of this pathogen, most cases of IE present once IgG titers have begun to rise. Serum PCR testing has shown a sensitivity and specificity of 58% and 100%, respectively.11 Isolation by blood culture requires specific growth media and prolonged incubation, with a sensitivity as low as 20% and 30% for blood and tissue, respectively.10 The microbiology laboratory should be notified of suspected Bartonella to intensify efforts to cultivate this organism. If infection with Coxiella or Brucella is suspected, the lab should also be informed, both to increase diagnostic yield and to trigger enhanced biosafety precautions when handling the specimens. Despite attempts to optimize the yield, up to 75% of Bartonella IE may remain culture negative,12,13 making it difficult to meet the current major modified Duke criterion of positive blood cultures. H&E staining of valve tissue infected with Bartonella commonly reveals increased inflammation, fibrosis, and calcified granulomas relative to endocarditis from other causes.14 The Warthin-Starry silver stain can identify small, darkly staining bacteria in more than 75% of Bartonella endocarditis; however, this stain is not specific for Bartonella species.9
This case highlights the challenge of diagnosing subacute IE because this patient received antibiotics and steroids prior to presentation, clouding the clinical picture. Although he did not exhibit textbook signs of endocarditis, his symptoms (new onset heart failure and new regurgitant murmurs) prioritized the diagnosis. The combination of elevated serum titers, positive PCR, valve granulomas and abscesses on TEE, and pathology findings led the discussant to the correct diagnosis. Scratching beneath the surface revealed his penchant for cats, but this was only considered a key epidemiological feature later in his clinical course.
TEACHING POINTS
- Subacute IE typically presents with indolent constitutional symptoms over a course of weeks to months, whereas acute IE causes a rapid onset of fevers, rigors, and is more likely to exhibit embolic phenomena.
- Epidemiologic features specific to Bartonella species include alcoholism, cirrhosis, dog or cat exposure, homelessness, and body lice, and should be considered in suspected cases of BCNE.
- If suspicion for endocarditis remains high and animal exposure is elicited, then serologic and PCR testing for fastidious organisms should be strongly considered. The most common causes of BCNE include Coxiella, Bartonella, and Brucella species.
- The modified Duke criteria do not incorporate Bartonella within the diagnostic schema. Presentation is usually late and often requires valve replacement.
Acknowledgments
The authors thank Dr. Michael Pfeiffer from the Pennsylvania State Hershey Heart and Vascular Institute for providing his expertise in diagnostic echocardiography.
Disclosure
There are no conflicts of interest or financial disclosures to report.
1. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893. PubMed
2. Breitschwerdt EB, Kordick DL. Bartonella infection in animals: carriership, reservoir potential, pathogenicity, and zoonotic potential for human infection. Clin Microbiol Rev. 2000;13(3):428-438. PubMed
3. Heller R, Artois M, Xemar V, et al. Prevalence of Bartonella henselae and Bartonella clarridgeiae in stray cats. J Clin Microbiol. 1997;35(6):1327-1331. PubMed
4. Bor DH, Woolhandler S, Nardin R, Brusch J, Himmelsein DU. Infective endocarditis in the U.S., 1998-2009: a nationwide study. PLoS One. 2013;8(3):e60033. PubMed
5. Bashore TM, Cabell C, Fowler, V Jr., Update on infective endocarditis. Curr Probl Cardiol. 2006;31(4):274-352. PubMed
6. Werner M, Andersson R, Olaison L, Hogevik H. A clinical study of culture-negative endocarditis. Medicine (Baltimore). 2003;82(4):263-273. PubMed
7. Baddour LM, Wilson WR, Bayer AS, et al. American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective Endocarditis in Adults: Diagnosis, Antimicrobial Therapy, and Management of Complications: A Scientific Statement for Healthcare Professionals From the American Heart Association. Circulation. 2015; 132(15):1435-1486. PubMed
8. Tunkel AR, Kaye D. Endocarditis with negative blood cultures. N Engl J Med. 1992;326(18):1215-1217. PubMed
9. Okaro U, Addisu A, Casanas B, Anderson B. Bartonella Species, an Emerging Cause of Blood-Culture-Negative Endocarditis. Clin Microbiol Rev. 2017;30(3):709-746. PubMed
10. Houpikian P, Raoult D. Blood culture-negative endocarditis in a reference center: etiologic diagnosis of 348 cases. Medicine (Baltimore). 2005;84(3):162-173. PubMed
11. Sanogo YO, Zeaiter Z, Caruso G, et al. Bartonella henselae in Ixodes ricinus ticks (Acari: Ixodida) removed from humans, Belluno province, Italy. Emerg Infect Dis. 2003;9(3):329-332. PubMed
12. Raoult D, Fournier PE, DrancourtM, et al. Diagnosis of 22 new cases of Bartonella endocarditis. Ann Intern Med. 1996;125(8):646-652. PubMed
13. La Scola B, Raoult D. Culture of Bartonella quintana and Bartonella henselae from human samples: a 5-year experience (1993 to 1998). J Clin Microbiol. 1999;37(6):1899-1905. PubMed
14. Lepidi H, Fournier PE, Raoult D. Quantitative analysis of valvular lesions during Bartonella endocarditis. Am J Clin Pathol. 2000;114(6):880-889. PubMed
A 62-year-old man with severe chronic obstructive pulmonary disease (COPD; forced expiratory volume during the first second [FEV1] 40% predicted) and type 2 diabetes mellitus presented to a Veterans Affairs emergency department (ED) with a steadily worsening cough of 4-months’ duration. He also reported subjective fevers, sputum production, shortness of breath, and unintentional 20-pound weight loss. He denied chills, chest pain, nausea, or vomiting.
Cough is classified as acute, subacute, or chronic based on duration of less than 3 weeks, between 3-8 weeks, and greater than 8 weeks, respectively. Common causes of chronic cough include bronchitis, acid reflux, cough-variant asthma, and a side effect of angiotensin converting enzyme inhibitors. Unintentional weight loss suggests a serious disorder, including indolent infection, end-stage COPD, malignancy, and autoimmune causes. Among patients with chronic bronchitis, the microbiology of sputum is often mixed with commensal respiratory flora, including Streptococcus pneumoniae and Haemophilus species. When these organisms are not recovered in sputa, or when patients fail to respond to empiric treatment, the differential diagnosis should be broadened to include pulmonary tuberculosis, nontuberculous mycobacterial infection, lung abscess, pulmonary nocardiosis, or pertussis.
An exposure and social history can focus the differential. For example, coccidioidomycosis or histoplasmosis may present indolently, but have distinct geographic distributions. Bird fanciers may acquire hypersensitivity pneumonitis, psittacosis, or cryptococcosis. Risk factors including smoking history, corticosteroid use, uncontrolled diabetes, and ill contacts should be assessed.
He was discharged from the ED twice in the last 2 weeks after presenting with similar symptoms. On each occasion, he was treated for presumed COPD exacerbations with nebulized albuterol and ipratropium, methylprednisolone followed by oral prednisone, and azithromycin, which did not lead to improvement. Over the last 3 days, he developed lower extremity edema, orthopnea, and dyspnea at rest. He reported worsening fatigue, night sweats, and anorexia. He denied any sick contacts.
Two diagnostic issues have emerged. His edema, orthopnea, and dyspnea at rest suggest a new cause of hypervolemia, perhaps caused by sodium retention from corticosteroids, pulmonary edema from valvular or myocardial disease, or renal failure. More concerning is that he has been treated with azithromycin twice recently but still has night sweats, fatigue, and anorexia. The presence of weight loss despite extracellular volume accumulation suggests an indolent systemic illness. Infection with macrolide-resistant organisms, such as nocardia, mycobacteria, or endemic mycoses, remains high on the differential diagnosis.
His past medical history included hypertension, untreated chronic hepatitis C, tobacco dependence, alcohol use disorder, and extraction of 8 decayed teeth 2 months earlier. He served in a noncombat role during the Vietnam War. He consumed 12 beers weekly with a remote history of alcoholism which required rehabilitation, reported a 50 pack-year smoking history, and denied intravenous (IV) drug use. He lived with an appropriately vaccinated dog and denied recent insect or animal exposures. He had a cat that passed away from an unknown illness 3 years prior. He was in a monogamous relationship with his girlfriend of 35 years. His father had coronary disease. His medications included glyburide, hydrochlorothiazide, lisinopril, theophylline, and meloxicam. Chronic cough, weight loss, diabetes, alcoholism, and history of dental disease raise concern for lung abscess. Oral microbiota such as Streptococcus viridans and Actinomycetes are usually harmless, but when aspirated repeatedly, such as during alcohol intoxication, may evolve into a lung abscess via bronchogenic spread. The combination of unintentional weight loss and smoking history raises concern for lung malignancy. Small cell lung cancer can present with paraneoplastic Cushing’s syndrome and could explain the patient’s volume overload. Finally, human immunodeficiency virus (HIV) serostatus should be determined in all adult patients.
His temperature was 37 °C, blood pressure 161/69 mm Hg, pulse 104 beats per minute, respiratory rate 20 breaths per minute, and oxygen saturation was 95% on room air. On examination, he was an unkempt, ill-appearing man. He had poor dentition, but no oral ulcers or petechiae. Pulmonary exam revealed diffuse rhonchi and scattered wheezes. He developed dyspnea after speaking 2 sentences. Cardiovascular exam showed regular tachycardia, normal S1 and S2 heart sounds, and both an S3 and S4 gallop. A grade III/VI holosystolic murmur at the left lower sternal border with apical radiation, and an early, grade III/IV diastolic murmur at the right upper sternal border were present. Neck exam showed jugular venous distention (JVD) 8 cm above the right clavicle. Lower extremities showed symmetric 3+ pitting edema to the knees. His abdomen was soft, nondistended, and without hepatosplenomegaly. There was no lymphadenopathy. Skin exam showed small, healed excoriations on his anterior shins, forearms, and knuckles. There were no petechiae, Janeway lesions, or Osler’s nodes.
These exam findings change the differential substantially. New regurgitant murmurs strongly suggest infective endocarditis (IE). A diastolic murmur is never normal and suggests aortic regurgitation. The holosystolic murmur with apical radiation suggests mitral regurgitation. Cutaneous stigmata should always be sought, but are found in fewer than half of cases of subacute IE, and their absence does not rule out this diagnosis. Disheveled hygiene and excoriations suggest a skin source of infection, and poor dentition is concerning for an oral source. For the moment, the source does not matter. His clinical condition is serious: tachycardia, JVD, edema, and two-sentence dyspnea indicate congestive heart failure. Even before labs and imaging return, inpatient admission is warranted.
Serum sodium concentration was 140 mEq/L, potassium 3.7 mEq/L, chloride 103 mEq/L, bicarbonate 30 mEq/L, blood urea nitrogen (BUN) 26 mg/dL, creatinine 0.8 mg/dL, glucose 120 mg/dL, and calcium 9.0 mg/dL. The white blood cell count was 7100/µL, hemoglobin 11.8 g/dL, and platelet count 101 K/µL. Brain natriuretic peptide (BNP) was 785 pg/mL (reference range 0-100 pg/mL), aspartate aminotransferase 77 U/L, alanine aminotransferase 57 U/L, alkaline phosphatase 125 U/L, total bilirubin 0.8 mg/dL, total protein 7.7 g/dL, and albumin 3.7 g/dL. Erythrocyte sedimentation (ESR) rate was 38 mm/hour (reference range 0-25 mm/hour) and C-reactive protein (CRP) 0.62 mg/dL (reference range <1.0 mg/dL). Cardiac troponins were 0.03 ng/mL (reference range <0.04 ng/mL). Screening for HIV was negative. Urinalysis showed trace blood by dipstick, but no glucose, protein, dysmorphic red blood cells, or casts. Two sets of peripheral blood cultures were drawn. Two sets of blood cultures from his previous ED visits were negative (drawn 6 and 14 days prior).
These laboratory values are nonspecific, and the differential remains unchanged, with top concern for IE, then lung abscess. Ideally, 3 sets of cultures drawn greater than 12 hours apart should be obtained because the likelihood of pathogen detection rises with the volume of blood tested. Thrombocytopenia and microscopic hematuria suggest microangiopathic hemolytic anemia, and a peripheral blood smear should be examined for schistocytes. Glomerulonephritis from immune complex deposition can occur in IE, but is unlikely with a normal serum creatinine and lack of proteinuria, dysmorphic red blood cells, or casts. The elevated BNP suggests cardiac strain due to a regurgitant valve. ESR and CRP are rarely helpful in this situation, and perhaps previous treatment with azithromycin and steroids prevented significant elevation.
His chest x-ray is not consistent with acute or chronic pulmonary infection. His symptoms, EKG, edema, and improvement with diuresis support the diagnosis of congestive heart failure. The leading diagnosis is left-sided IE, and antimicrobial therapy should not be delayed for the sake of awaiting positive blood cultures. He should immediately receive empiric antibiotics to cover gram-positive bacteria (Methicillin-resistant Staphylococcus aureus, Methicillin-sensitive S. aureus, coagulase-negative staphylococci, and enterococci) and Haemophilus species, Actinobacillus actinomycetemcomitans, Cardiobacterium hominis, Eikenella species, and Kingella kingae (the HACEK group). In accordance with Infectious Diseases Society of America (IDSA) practice guidelines, he should empirically receive IV vancomycin plus ceftriaxone and urgently undergo echocardiography.
Transthoracic echocardiogram (TTE) showed severe aortic insufficiency, aortic valve vegetations, and raised suspicion for a moderate-sized vegetation on the anterior leaflet of the mitral valve. There was moderate mitral insufficiency, moderate tricuspid insufficiency, and an elevated right ventricular systolic pressure of 50 mm Hg. The left ventricle showed concentric hypertrophy with an ejection fraction of 55%. A previous echocardiogram 2 years prior showed mild mitral insufficiency, but no aneurysm or aortic insufficiency. Blood cultures from admission yielded no growth.
Due to concern for IE, blood cultures were repeated, and IV vancomycin, IV ceftriaxone, and IV gentamicin were initiated. Azithromycin and prednisone were discontinued. His respiratory status continued to improve with IV furosemide, albuterol, ipratropium, and supportive care.
TTE inadequately visualizes the mitral valve, but is useful for tricuspid valve assessment because the right ventricle is closer to the chest wall. Transesophageal echocardiography (TEE) is indicated for a more detailed assessment of the left heart valves for vegetations and perivalvar abscesses. The new regurgitant murmurs satisfy a major criterion of the modified Duke criteria, and valvar vegetations suggests IE. He does not yet fulfill the other major modified Duke criterion for IE, nor does he satisfy enough minor criteria because there are no diagnostic vascular, microbiologic, or immunologic phenomena. However, no diagnostic rubric is perfect, and these results should not supersede clinical judgment. Despite the absence of positive cultures, the concern for bacterial IE remains high. The absence of embolic phenomena fits best with subacute rather than acute IE. Three negative blood cultures to date suggest a fastidious organism is responsible, although oral flora remain on the differential.
There is rarely a need to “hold” blood cultures for prolonged periods because modern instruments typically yield positive results within 7 days for most bacteria, including the HACEK group. Blood culture-negative endocarditis (BCNE) is considered when 3 sets of cultures are negative for at least 5 days. In this situation, one should consider other microorganisms based on the patient’s exposure history. Only certain species with complex growth requirements, such as Brucella and Bartonella, require prolonged holds. Revisiting his exposure history would be helpful in deciding whether serologic testing warranted. If he recalls exposure to parturient animals, then Coxiella is worth pursuing; if he has been bitten by lice, then B. quintana rises as a possibility; if the scratches on his limbs are from recent cat scratches, then B. henselae becomes more likely. Both C. burnetti and Bartonella endocarditis might be partially treated by his courses of azithromycin, confounding the picture.
If the infectious work-up is ultimately negative, one could then consider other etiologies of endocarditis, such as nonbacterial thrombotic endocarditis, which is seen in the context of malignancy and systemic lupus erythematosus (Libman-Sacks endocarditis). Other mimickers of IE include myxomatous valve degeneration, ruptured mitral chordae, and eosinophilic heart disease (Löffler’s endocarditis).
A transesophageal echocardiogram confirmed the presence of small echodensities on the aortic valve’s right and left coronary cusps, consistent with vegetations. The vegetation on the anterior leaflet of the mitral valve from the TTE also showed an aneurysm with a small perforation (Figure 2).
The combination of aortic regurgitation and the mitral valve aneurysm supports IE, because the aortic regurgitant jet directly strikes the anterior mitral valve leaflet, seeding the valve with infection from the aortic cusps. A positive serum PCR is diagnostic, but if it had been negative or unavailable, the serology would remain very helpful. In this context, the elevated IgG titer implicates B. henselae, the agent responsible for cat scratch disease (CSD). Out of context, these titers would not be diagnostic, because anti-Bartonella IgG may be increased due to a prior subclinical episode of CSD. Anti-Bartonella IgM is an unreliable indicator of recent infection because it may wane within weeks, and this IgG titer is higher than what is observed with most remote infections.
Revisiting previous cat exposure is warranted. He lost his cat to an illness 3 years prior, however it would be appropriate to inquire about other animals, such as a stray kitten with fleas, which his skin scratches suggest. Up to 50% of all cats in flea endemic regions harbor Bartonella and are asymptomatic. Rarely, dogs can serve as reservoirs of this organism, with a presumed transmission route via flea, louse, or tick. Regardless of the route of infection, treatment should be focused on B. henselae IE.
Azithromycin can treat CSD, and its use for his presumed COPD exacerbation may have temporized his infection. However, azithromycin monotherapy is not recommended for B. henselae IE. Treatment is usually with 2 antibiotics, including an aminoglycoside (gentamicin) for the first 2 weeks, combined with either a tetracycline, a macrolide, or a beta-lactam for a minimum of 4-6 weeks. Oral rifampin can be considered if gentamicin is not tolerated. After completing IV treatment, an additional 6 months of oral doxycycline or azithromycin should be considered, especially for those who have not undergone valve surgery.
The mitral valve aneurysm, abscesses, and heart failure warranted valve replacement. Surgery should be considered for all patients with Bartonella IE, primarily because delayed diagnosis often leads to irreversible valve damage. Ideally, surgically explanted tissue should be divided into 2 portions: half should be sent to pathology and stained with H&E, Warthin-Starry, and Steiner staining procedures, while the other half should be sent for culture, and then PCR if stains are negative.
His symptoms are compatible with subacute IE, which is typically more difficult to diagnose than acute IE due to its insidious onset. He meets criteria for blood culture negative IE based on 3 sets of negative blood cultures for greater than 5 days and major criteria for IE. The pathologic changes are consistent with B. henselae infection.
DISCUSSION
The incidence of IE in the United States is 40,000 cases per year1 with an in-hospital mortality of 15%-20% and a 1-year mortality of up to 40%.2,3 Five to 20% of patients with IE never develop positive blood cultures4 due to receipt of antibiotics prior to culture, inadequate microbiologic testing, or infection caused by noncultivable bacteria (eg, Tropheryma whipplei), fastidious extracellular bacteria (eg, HACEK group and nutritionally variant streptococci), or by intracellular pathogens with complex nutrient requirements (eg, Bartonella, Chlamydia, Brucella, or Coxiella). Previous administration of antibiotics reduces the likelihood of isolating an organism by 35%-40%.5 Patients meeting criteria for BCNE should prompt consideration of serologic testing. The most prevalent pathogens vary globally, and incidence data in the US is scarce. Worldwide, the majority of BCNE cases are caused by Coxiella, Bartonella, and Brucella species.6,7
When clinical suspicion for IE remains high despite negative cultures, detailed history can uncover clues and guide additional testing. For example, contact with contaminated milk products or farm animals are associated with Brucella, Coxiella, and Erysipelothrix species IE.7,8 Bartonella species are zoonotic gram-negative bacilli with a tropism for endothelial cells and are transmitted by arthropod vectors (ie, fleas, lice, ticks, and sandflies), cat scratches, or cat bites. Bartonella may account for 3%-4% of all cases of IE, most of which are due to B. henselae and B. quintana.7, 9 Underlying heart valve disease, alcoholism, cirrhosis, and homelessness are associated with B. henselae endocarditis.10
Diagnostic criteria are lacking for B. henselae IE, and the modified Duke criteria is of limited utility for diagnosing Bartonella IE because blood cultures are often negative and echocardiographic evidence of vegetation is not always apparent. Serology plays a critical role in the diagnosis of Bartonella infections. The addition of positive serology, Western blot or PCR for B. henselae and B. quintana as a major criterion in the modified Duke criteria for IE has been proposed but has not yet been formally accepted.9 For B. henselae IE, an IgG titer of ≥1:800 has been recommended as a cutoff for subacute IE because it combines a high specificity and positive predictive value along with reasonable sensitivity and negative predictive value in this situation.9 The humoral immune response rises over time, and thus acute IE due to Bartonella may not generate a substantial IgG titer. Interestingly, because of the indolent nature of this pathogen, most cases of IE present once IgG titers have begun to rise. Serum PCR testing has shown a sensitivity and specificity of 58% and 100%, respectively.11 Isolation by blood culture requires specific growth media and prolonged incubation, with a sensitivity as low as 20% and 30% for blood and tissue, respectively.10 The microbiology laboratory should be notified of suspected Bartonella to intensify efforts to cultivate this organism. If infection with Coxiella or Brucella is suspected, the lab should also be informed, both to increase diagnostic yield and to trigger enhanced biosafety precautions when handling the specimens. Despite attempts to optimize the yield, up to 75% of Bartonella IE may remain culture negative,12,13 making it difficult to meet the current major modified Duke criterion of positive blood cultures. H&E staining of valve tissue infected with Bartonella commonly reveals increased inflammation, fibrosis, and calcified granulomas relative to endocarditis from other causes.14 The Warthin-Starry silver stain can identify small, darkly staining bacteria in more than 75% of Bartonella endocarditis; however, this stain is not specific for Bartonella species.9
This case highlights the challenge of diagnosing subacute IE because this patient received antibiotics and steroids prior to presentation, clouding the clinical picture. Although he did not exhibit textbook signs of endocarditis, his symptoms (new onset heart failure and new regurgitant murmurs) prioritized the diagnosis. The combination of elevated serum titers, positive PCR, valve granulomas and abscesses on TEE, and pathology findings led the discussant to the correct diagnosis. Scratching beneath the surface revealed his penchant for cats, but this was only considered a key epidemiological feature later in his clinical course.
TEACHING POINTS
- Subacute IE typically presents with indolent constitutional symptoms over a course of weeks to months, whereas acute IE causes a rapid onset of fevers, rigors, and is more likely to exhibit embolic phenomena.
- Epidemiologic features specific to Bartonella species include alcoholism, cirrhosis, dog or cat exposure, homelessness, and body lice, and should be considered in suspected cases of BCNE.
- If suspicion for endocarditis remains high and animal exposure is elicited, then serologic and PCR testing for fastidious organisms should be strongly considered. The most common causes of BCNE include Coxiella, Bartonella, and Brucella species.
- The modified Duke criteria do not incorporate Bartonella within the diagnostic schema. Presentation is usually late and often requires valve replacement.
Acknowledgments
The authors thank Dr. Michael Pfeiffer from the Pennsylvania State Hershey Heart and Vascular Institute for providing his expertise in diagnostic echocardiography.
Disclosure
There are no conflicts of interest or financial disclosures to report.
A 62-year-old man with severe chronic obstructive pulmonary disease (COPD; forced expiratory volume during the first second [FEV1] 40% predicted) and type 2 diabetes mellitus presented to a Veterans Affairs emergency department (ED) with a steadily worsening cough of 4-months’ duration. He also reported subjective fevers, sputum production, shortness of breath, and unintentional 20-pound weight loss. He denied chills, chest pain, nausea, or vomiting.
Cough is classified as acute, subacute, or chronic based on duration of less than 3 weeks, between 3-8 weeks, and greater than 8 weeks, respectively. Common causes of chronic cough include bronchitis, acid reflux, cough-variant asthma, and a side effect of angiotensin converting enzyme inhibitors. Unintentional weight loss suggests a serious disorder, including indolent infection, end-stage COPD, malignancy, and autoimmune causes. Among patients with chronic bronchitis, the microbiology of sputum is often mixed with commensal respiratory flora, including Streptococcus pneumoniae and Haemophilus species. When these organisms are not recovered in sputa, or when patients fail to respond to empiric treatment, the differential diagnosis should be broadened to include pulmonary tuberculosis, nontuberculous mycobacterial infection, lung abscess, pulmonary nocardiosis, or pertussis.
An exposure and social history can focus the differential. For example, coccidioidomycosis or histoplasmosis may present indolently, but have distinct geographic distributions. Bird fanciers may acquire hypersensitivity pneumonitis, psittacosis, or cryptococcosis. Risk factors including smoking history, corticosteroid use, uncontrolled diabetes, and ill contacts should be assessed.
He was discharged from the ED twice in the last 2 weeks after presenting with similar symptoms. On each occasion, he was treated for presumed COPD exacerbations with nebulized albuterol and ipratropium, methylprednisolone followed by oral prednisone, and azithromycin, which did not lead to improvement. Over the last 3 days, he developed lower extremity edema, orthopnea, and dyspnea at rest. He reported worsening fatigue, night sweats, and anorexia. He denied any sick contacts.
Two diagnostic issues have emerged. His edema, orthopnea, and dyspnea at rest suggest a new cause of hypervolemia, perhaps caused by sodium retention from corticosteroids, pulmonary edema from valvular or myocardial disease, or renal failure. More concerning is that he has been treated with azithromycin twice recently but still has night sweats, fatigue, and anorexia. The presence of weight loss despite extracellular volume accumulation suggests an indolent systemic illness. Infection with macrolide-resistant organisms, such as nocardia, mycobacteria, or endemic mycoses, remains high on the differential diagnosis.
His past medical history included hypertension, untreated chronic hepatitis C, tobacco dependence, alcohol use disorder, and extraction of 8 decayed teeth 2 months earlier. He served in a noncombat role during the Vietnam War. He consumed 12 beers weekly with a remote history of alcoholism which required rehabilitation, reported a 50 pack-year smoking history, and denied intravenous (IV) drug use. He lived with an appropriately vaccinated dog and denied recent insect or animal exposures. He had a cat that passed away from an unknown illness 3 years prior. He was in a monogamous relationship with his girlfriend of 35 years. His father had coronary disease. His medications included glyburide, hydrochlorothiazide, lisinopril, theophylline, and meloxicam. Chronic cough, weight loss, diabetes, alcoholism, and history of dental disease raise concern for lung abscess. Oral microbiota such as Streptococcus viridans and Actinomycetes are usually harmless, but when aspirated repeatedly, such as during alcohol intoxication, may evolve into a lung abscess via bronchogenic spread. The combination of unintentional weight loss and smoking history raises concern for lung malignancy. Small cell lung cancer can present with paraneoplastic Cushing’s syndrome and could explain the patient’s volume overload. Finally, human immunodeficiency virus (HIV) serostatus should be determined in all adult patients.
His temperature was 37 °C, blood pressure 161/69 mm Hg, pulse 104 beats per minute, respiratory rate 20 breaths per minute, and oxygen saturation was 95% on room air. On examination, he was an unkempt, ill-appearing man. He had poor dentition, but no oral ulcers or petechiae. Pulmonary exam revealed diffuse rhonchi and scattered wheezes. He developed dyspnea after speaking 2 sentences. Cardiovascular exam showed regular tachycardia, normal S1 and S2 heart sounds, and both an S3 and S4 gallop. A grade III/VI holosystolic murmur at the left lower sternal border with apical radiation, and an early, grade III/IV diastolic murmur at the right upper sternal border were present. Neck exam showed jugular venous distention (JVD) 8 cm above the right clavicle. Lower extremities showed symmetric 3+ pitting edema to the knees. His abdomen was soft, nondistended, and without hepatosplenomegaly. There was no lymphadenopathy. Skin exam showed small, healed excoriations on his anterior shins, forearms, and knuckles. There were no petechiae, Janeway lesions, or Osler’s nodes.
These exam findings change the differential substantially. New regurgitant murmurs strongly suggest infective endocarditis (IE). A diastolic murmur is never normal and suggests aortic regurgitation. The holosystolic murmur with apical radiation suggests mitral regurgitation. Cutaneous stigmata should always be sought, but are found in fewer than half of cases of subacute IE, and their absence does not rule out this diagnosis. Disheveled hygiene and excoriations suggest a skin source of infection, and poor dentition is concerning for an oral source. For the moment, the source does not matter. His clinical condition is serious: tachycardia, JVD, edema, and two-sentence dyspnea indicate congestive heart failure. Even before labs and imaging return, inpatient admission is warranted.
Serum sodium concentration was 140 mEq/L, potassium 3.7 mEq/L, chloride 103 mEq/L, bicarbonate 30 mEq/L, blood urea nitrogen (BUN) 26 mg/dL, creatinine 0.8 mg/dL, glucose 120 mg/dL, and calcium 9.0 mg/dL. The white blood cell count was 7100/µL, hemoglobin 11.8 g/dL, and platelet count 101 K/µL. Brain natriuretic peptide (BNP) was 785 pg/mL (reference range 0-100 pg/mL), aspartate aminotransferase 77 U/L, alanine aminotransferase 57 U/L, alkaline phosphatase 125 U/L, total bilirubin 0.8 mg/dL, total protein 7.7 g/dL, and albumin 3.7 g/dL. Erythrocyte sedimentation (ESR) rate was 38 mm/hour (reference range 0-25 mm/hour) and C-reactive protein (CRP) 0.62 mg/dL (reference range <1.0 mg/dL). Cardiac troponins were 0.03 ng/mL (reference range <0.04 ng/mL). Screening for HIV was negative. Urinalysis showed trace blood by dipstick, but no glucose, protein, dysmorphic red blood cells, or casts. Two sets of peripheral blood cultures were drawn. Two sets of blood cultures from his previous ED visits were negative (drawn 6 and 14 days prior).
These laboratory values are nonspecific, and the differential remains unchanged, with top concern for IE, then lung abscess. Ideally, 3 sets of cultures drawn greater than 12 hours apart should be obtained because the likelihood of pathogen detection rises with the volume of blood tested. Thrombocytopenia and microscopic hematuria suggest microangiopathic hemolytic anemia, and a peripheral blood smear should be examined for schistocytes. Glomerulonephritis from immune complex deposition can occur in IE, but is unlikely with a normal serum creatinine and lack of proteinuria, dysmorphic red blood cells, or casts. The elevated BNP suggests cardiac strain due to a regurgitant valve. ESR and CRP are rarely helpful in this situation, and perhaps previous treatment with azithromycin and steroids prevented significant elevation.
His chest x-ray is not consistent with acute or chronic pulmonary infection. His symptoms, EKG, edema, and improvement with diuresis support the diagnosis of congestive heart failure. The leading diagnosis is left-sided IE, and antimicrobial therapy should not be delayed for the sake of awaiting positive blood cultures. He should immediately receive empiric antibiotics to cover gram-positive bacteria (Methicillin-resistant Staphylococcus aureus, Methicillin-sensitive S. aureus, coagulase-negative staphylococci, and enterococci) and Haemophilus species, Actinobacillus actinomycetemcomitans, Cardiobacterium hominis, Eikenella species, and Kingella kingae (the HACEK group). In accordance with Infectious Diseases Society of America (IDSA) practice guidelines, he should empirically receive IV vancomycin plus ceftriaxone and urgently undergo echocardiography.
Transthoracic echocardiogram (TTE) showed severe aortic insufficiency, aortic valve vegetations, and raised suspicion for a moderate-sized vegetation on the anterior leaflet of the mitral valve. There was moderate mitral insufficiency, moderate tricuspid insufficiency, and an elevated right ventricular systolic pressure of 50 mm Hg. The left ventricle showed concentric hypertrophy with an ejection fraction of 55%. A previous echocardiogram 2 years prior showed mild mitral insufficiency, but no aneurysm or aortic insufficiency. Blood cultures from admission yielded no growth.
Due to concern for IE, blood cultures were repeated, and IV vancomycin, IV ceftriaxone, and IV gentamicin were initiated. Azithromycin and prednisone were discontinued. His respiratory status continued to improve with IV furosemide, albuterol, ipratropium, and supportive care.
TTE inadequately visualizes the mitral valve, but is useful for tricuspid valve assessment because the right ventricle is closer to the chest wall. Transesophageal echocardiography (TEE) is indicated for a more detailed assessment of the left heart valves for vegetations and perivalvar abscesses. The new regurgitant murmurs satisfy a major criterion of the modified Duke criteria, and valvar vegetations suggests IE. He does not yet fulfill the other major modified Duke criterion for IE, nor does he satisfy enough minor criteria because there are no diagnostic vascular, microbiologic, or immunologic phenomena. However, no diagnostic rubric is perfect, and these results should not supersede clinical judgment. Despite the absence of positive cultures, the concern for bacterial IE remains high. The absence of embolic phenomena fits best with subacute rather than acute IE. Three negative blood cultures to date suggest a fastidious organism is responsible, although oral flora remain on the differential.
There is rarely a need to “hold” blood cultures for prolonged periods because modern instruments typically yield positive results within 7 days for most bacteria, including the HACEK group. Blood culture-negative endocarditis (BCNE) is considered when 3 sets of cultures are negative for at least 5 days. In this situation, one should consider other microorganisms based on the patient’s exposure history. Only certain species with complex growth requirements, such as Brucella and Bartonella, require prolonged holds. Revisiting his exposure history would be helpful in deciding whether serologic testing warranted. If he recalls exposure to parturient animals, then Coxiella is worth pursuing; if he has been bitten by lice, then B. quintana rises as a possibility; if the scratches on his limbs are from recent cat scratches, then B. henselae becomes more likely. Both C. burnetti and Bartonella endocarditis might be partially treated by his courses of azithromycin, confounding the picture.
If the infectious work-up is ultimately negative, one could then consider other etiologies of endocarditis, such as nonbacterial thrombotic endocarditis, which is seen in the context of malignancy and systemic lupus erythematosus (Libman-Sacks endocarditis). Other mimickers of IE include myxomatous valve degeneration, ruptured mitral chordae, and eosinophilic heart disease (Löffler’s endocarditis).
A transesophageal echocardiogram confirmed the presence of small echodensities on the aortic valve’s right and left coronary cusps, consistent with vegetations. The vegetation on the anterior leaflet of the mitral valve from the TTE also showed an aneurysm with a small perforation (Figure 2).
The combination of aortic regurgitation and the mitral valve aneurysm supports IE, because the aortic regurgitant jet directly strikes the anterior mitral valve leaflet, seeding the valve with infection from the aortic cusps. A positive serum PCR is diagnostic, but if it had been negative or unavailable, the serology would remain very helpful. In this context, the elevated IgG titer implicates B. henselae, the agent responsible for cat scratch disease (CSD). Out of context, these titers would not be diagnostic, because anti-Bartonella IgG may be increased due to a prior subclinical episode of CSD. Anti-Bartonella IgM is an unreliable indicator of recent infection because it may wane within weeks, and this IgG titer is higher than what is observed with most remote infections.
Revisiting previous cat exposure is warranted. He lost his cat to an illness 3 years prior, however it would be appropriate to inquire about other animals, such as a stray kitten with fleas, which his skin scratches suggest. Up to 50% of all cats in flea endemic regions harbor Bartonella and are asymptomatic. Rarely, dogs can serve as reservoirs of this organism, with a presumed transmission route via flea, louse, or tick. Regardless of the route of infection, treatment should be focused on B. henselae IE.
Azithromycin can treat CSD, and its use for his presumed COPD exacerbation may have temporized his infection. However, azithromycin monotherapy is not recommended for B. henselae IE. Treatment is usually with 2 antibiotics, including an aminoglycoside (gentamicin) for the first 2 weeks, combined with either a tetracycline, a macrolide, or a beta-lactam for a minimum of 4-6 weeks. Oral rifampin can be considered if gentamicin is not tolerated. After completing IV treatment, an additional 6 months of oral doxycycline or azithromycin should be considered, especially for those who have not undergone valve surgery.
The mitral valve aneurysm, abscesses, and heart failure warranted valve replacement. Surgery should be considered for all patients with Bartonella IE, primarily because delayed diagnosis often leads to irreversible valve damage. Ideally, surgically explanted tissue should be divided into 2 portions: half should be sent to pathology and stained with H&E, Warthin-Starry, and Steiner staining procedures, while the other half should be sent for culture, and then PCR if stains are negative.
His symptoms are compatible with subacute IE, which is typically more difficult to diagnose than acute IE due to its insidious onset. He meets criteria for blood culture negative IE based on 3 sets of negative blood cultures for greater than 5 days and major criteria for IE. The pathologic changes are consistent with B. henselae infection.
DISCUSSION
The incidence of IE in the United States is 40,000 cases per year1 with an in-hospital mortality of 15%-20% and a 1-year mortality of up to 40%.2,3 Five to 20% of patients with IE never develop positive blood cultures4 due to receipt of antibiotics prior to culture, inadequate microbiologic testing, or infection caused by noncultivable bacteria (eg, Tropheryma whipplei), fastidious extracellular bacteria (eg, HACEK group and nutritionally variant streptococci), or by intracellular pathogens with complex nutrient requirements (eg, Bartonella, Chlamydia, Brucella, or Coxiella). Previous administration of antibiotics reduces the likelihood of isolating an organism by 35%-40%.5 Patients meeting criteria for BCNE should prompt consideration of serologic testing. The most prevalent pathogens vary globally, and incidence data in the US is scarce. Worldwide, the majority of BCNE cases are caused by Coxiella, Bartonella, and Brucella species.6,7
When clinical suspicion for IE remains high despite negative cultures, detailed history can uncover clues and guide additional testing. For example, contact with contaminated milk products or farm animals are associated with Brucella, Coxiella, and Erysipelothrix species IE.7,8 Bartonella species are zoonotic gram-negative bacilli with a tropism for endothelial cells and are transmitted by arthropod vectors (ie, fleas, lice, ticks, and sandflies), cat scratches, or cat bites. Bartonella may account for 3%-4% of all cases of IE, most of which are due to B. henselae and B. quintana.7, 9 Underlying heart valve disease, alcoholism, cirrhosis, and homelessness are associated with B. henselae endocarditis.10
Diagnostic criteria are lacking for B. henselae IE, and the modified Duke criteria is of limited utility for diagnosing Bartonella IE because blood cultures are often negative and echocardiographic evidence of vegetation is not always apparent. Serology plays a critical role in the diagnosis of Bartonella infections. The addition of positive serology, Western blot or PCR for B. henselae and B. quintana as a major criterion in the modified Duke criteria for IE has been proposed but has not yet been formally accepted.9 For B. henselae IE, an IgG titer of ≥1:800 has been recommended as a cutoff for subacute IE because it combines a high specificity and positive predictive value along with reasonable sensitivity and negative predictive value in this situation.9 The humoral immune response rises over time, and thus acute IE due to Bartonella may not generate a substantial IgG titer. Interestingly, because of the indolent nature of this pathogen, most cases of IE present once IgG titers have begun to rise. Serum PCR testing has shown a sensitivity and specificity of 58% and 100%, respectively.11 Isolation by blood culture requires specific growth media and prolonged incubation, with a sensitivity as low as 20% and 30% for blood and tissue, respectively.10 The microbiology laboratory should be notified of suspected Bartonella to intensify efforts to cultivate this organism. If infection with Coxiella or Brucella is suspected, the lab should also be informed, both to increase diagnostic yield and to trigger enhanced biosafety precautions when handling the specimens. Despite attempts to optimize the yield, up to 75% of Bartonella IE may remain culture negative,12,13 making it difficult to meet the current major modified Duke criterion of positive blood cultures. H&E staining of valve tissue infected with Bartonella commonly reveals increased inflammation, fibrosis, and calcified granulomas relative to endocarditis from other causes.14 The Warthin-Starry silver stain can identify small, darkly staining bacteria in more than 75% of Bartonella endocarditis; however, this stain is not specific for Bartonella species.9
This case highlights the challenge of diagnosing subacute IE because this patient received antibiotics and steroids prior to presentation, clouding the clinical picture. Although he did not exhibit textbook signs of endocarditis, his symptoms (new onset heart failure and new regurgitant murmurs) prioritized the diagnosis. The combination of elevated serum titers, positive PCR, valve granulomas and abscesses on TEE, and pathology findings led the discussant to the correct diagnosis. Scratching beneath the surface revealed his penchant for cats, but this was only considered a key epidemiological feature later in his clinical course.
TEACHING POINTS
- Subacute IE typically presents with indolent constitutional symptoms over a course of weeks to months, whereas acute IE causes a rapid onset of fevers, rigors, and is more likely to exhibit embolic phenomena.
- Epidemiologic features specific to Bartonella species include alcoholism, cirrhosis, dog or cat exposure, homelessness, and body lice, and should be considered in suspected cases of BCNE.
- If suspicion for endocarditis remains high and animal exposure is elicited, then serologic and PCR testing for fastidious organisms should be strongly considered. The most common causes of BCNE include Coxiella, Bartonella, and Brucella species.
- The modified Duke criteria do not incorporate Bartonella within the diagnostic schema. Presentation is usually late and often requires valve replacement.
Acknowledgments
The authors thank Dr. Michael Pfeiffer from the Pennsylvania State Hershey Heart and Vascular Institute for providing his expertise in diagnostic echocardiography.
Disclosure
There are no conflicts of interest or financial disclosures to report.
1. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893. PubMed
2. Breitschwerdt EB, Kordick DL. Bartonella infection in animals: carriership, reservoir potential, pathogenicity, and zoonotic potential for human infection. Clin Microbiol Rev. 2000;13(3):428-438. PubMed
3. Heller R, Artois M, Xemar V, et al. Prevalence of Bartonella henselae and Bartonella clarridgeiae in stray cats. J Clin Microbiol. 1997;35(6):1327-1331. PubMed
4. Bor DH, Woolhandler S, Nardin R, Brusch J, Himmelsein DU. Infective endocarditis in the U.S., 1998-2009: a nationwide study. PLoS One. 2013;8(3):e60033. PubMed
5. Bashore TM, Cabell C, Fowler, V Jr., Update on infective endocarditis. Curr Probl Cardiol. 2006;31(4):274-352. PubMed
6. Werner M, Andersson R, Olaison L, Hogevik H. A clinical study of culture-negative endocarditis. Medicine (Baltimore). 2003;82(4):263-273. PubMed
7. Baddour LM, Wilson WR, Bayer AS, et al. American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective Endocarditis in Adults: Diagnosis, Antimicrobial Therapy, and Management of Complications: A Scientific Statement for Healthcare Professionals From the American Heart Association. Circulation. 2015; 132(15):1435-1486. PubMed
8. Tunkel AR, Kaye D. Endocarditis with negative blood cultures. N Engl J Med. 1992;326(18):1215-1217. PubMed
9. Okaro U, Addisu A, Casanas B, Anderson B. Bartonella Species, an Emerging Cause of Blood-Culture-Negative Endocarditis. Clin Microbiol Rev. 2017;30(3):709-746. PubMed
10. Houpikian P, Raoult D. Blood culture-negative endocarditis in a reference center: etiologic diagnosis of 348 cases. Medicine (Baltimore). 2005;84(3):162-173. PubMed
11. Sanogo YO, Zeaiter Z, Caruso G, et al. Bartonella henselae in Ixodes ricinus ticks (Acari: Ixodida) removed from humans, Belluno province, Italy. Emerg Infect Dis. 2003;9(3):329-332. PubMed
12. Raoult D, Fournier PE, DrancourtM, et al. Diagnosis of 22 new cases of Bartonella endocarditis. Ann Intern Med. 1996;125(8):646-652. PubMed
13. La Scola B, Raoult D. Culture of Bartonella quintana and Bartonella henselae from human samples: a 5-year experience (1993 to 1998). J Clin Microbiol. 1999;37(6):1899-1905. PubMed
14. Lepidi H, Fournier PE, Raoult D. Quantitative analysis of valvular lesions during Bartonella endocarditis. Am J Clin Pathol. 2000;114(6):880-889. PubMed
1. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893. PubMed
2. Breitschwerdt EB, Kordick DL. Bartonella infection in animals: carriership, reservoir potential, pathogenicity, and zoonotic potential for human infection. Clin Microbiol Rev. 2000;13(3):428-438. PubMed
3. Heller R, Artois M, Xemar V, et al. Prevalence of Bartonella henselae and Bartonella clarridgeiae in stray cats. J Clin Microbiol. 1997;35(6):1327-1331. PubMed
4. Bor DH, Woolhandler S, Nardin R, Brusch J, Himmelsein DU. Infective endocarditis in the U.S., 1998-2009: a nationwide study. PLoS One. 2013;8(3):e60033. PubMed
5. Bashore TM, Cabell C, Fowler, V Jr., Update on infective endocarditis. Curr Probl Cardiol. 2006;31(4):274-352. PubMed
6. Werner M, Andersson R, Olaison L, Hogevik H. A clinical study of culture-negative endocarditis. Medicine (Baltimore). 2003;82(4):263-273. PubMed
7. Baddour LM, Wilson WR, Bayer AS, et al. American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective Endocarditis in Adults: Diagnosis, Antimicrobial Therapy, and Management of Complications: A Scientific Statement for Healthcare Professionals From the American Heart Association. Circulation. 2015; 132(15):1435-1486. PubMed
8. Tunkel AR, Kaye D. Endocarditis with negative blood cultures. N Engl J Med. 1992;326(18):1215-1217. PubMed
9. Okaro U, Addisu A, Casanas B, Anderson B. Bartonella Species, an Emerging Cause of Blood-Culture-Negative Endocarditis. Clin Microbiol Rev. 2017;30(3):709-746. PubMed
10. Houpikian P, Raoult D. Blood culture-negative endocarditis in a reference center: etiologic diagnosis of 348 cases. Medicine (Baltimore). 2005;84(3):162-173. PubMed
11. Sanogo YO, Zeaiter Z, Caruso G, et al. Bartonella henselae in Ixodes ricinus ticks (Acari: Ixodida) removed from humans, Belluno province, Italy. Emerg Infect Dis. 2003;9(3):329-332. PubMed
12. Raoult D, Fournier PE, DrancourtM, et al. Diagnosis of 22 new cases of Bartonella endocarditis. Ann Intern Med. 1996;125(8):646-652. PubMed
13. La Scola B, Raoult D. Culture of Bartonella quintana and Bartonella henselae from human samples: a 5-year experience (1993 to 1998). J Clin Microbiol. 1999;37(6):1899-1905. PubMed
14. Lepidi H, Fournier PE, Raoult D. Quantitative analysis of valvular lesions during Bartonella endocarditis. Am J Clin Pathol. 2000;114(6):880-889. PubMed
© 2018 Society of Hospital Medicine
Near and Far
A previously healthy 30-year-old woman presented to the emergency department with 2 weeks of weakness.
True muscle weakness must be distinguished from the more common causes of asthenia. Many systemic disorders produce fatigue, with resulting functional limitation that is often interpreted by patients as weakness. Initial history should focus on conditions producing fatigue, such as cardiopulmonary disease, anemia, connective tissue disease, depression or cachexia related to malignancy, infection, or other inflammatory states. Careful questioning may reveal evidence of dyspnea, poor exercise tolerance, or joint pain as an alternative to actual loss of muscle power. If true weakness is still suspected, attention should be focused on the pattern, onset, anatomic site, and progression of weakness. Muscle weakness is often characterized by difficulty with specific tasks, such as climbing stairs, rising from a chair, raising a hand, or using cutlery. The physical examination is critical in determining whether weakness is due to true loss of motor power. The differential diagnosis of weakness is broad and includes neurologic, infectious, endocrine, inflammatory, genetic, metabolic, and drug-induced etiologies.
She initially experienced 3 days of mild cramps and soreness in her thighs. She then developed weakness that began in her thighs and progressed to involve her lower legs and upper and lower arms. She had difficulty combing her hair. She required the use of her arms to get up from a chair. She grasped onto objects to aid in ambulation around the house. In addition, she described 1 year of moderate fatigue but no fever, weight loss, dyspnea, dysphagia, visual changes, paresthesias, bowel or bladder incontinence, back pain, or preceding gastrointestinal or respiratory illness. She had experienced diffuse intermittent hives, most prominent in her chest and upper arms, for the past several weeks.
History certainly supports true weakness but will need to be confirmed on examination. The distribution began as proximal but now appears diffuse. The presence of myalgia and cramping raises the possibility of noninflammatory myopathies, which are usually more insidious in onset. A severe electrolyte disturbance would be possible, based on the diffuse nature of weakness that was preceded by cramping. The distribution of weakness and lack of bowel or bladder incontinence is reassuring and suggests against a spinal cord disorder; however, a high index of suspicion must be maintained for myelopathy because delayed treatment might result in irreversible paralysis.
The patient’s course also includes hives. Common causes of hives include infections and allergic reactions to medications, foods, and insect stings. Urticaria may also result from systemic disorders, such as vasculitis, lupus, lymphoma, mastocytosis, and paraproteinemias, which can be associated with weakness and fatigue. Although severe weakness in combination with hives makes an infectious and allergic reaction less likely, we still seek to ascertain if the evolving chief complaints of weakness and hives are the result of a single unifying and evolving multisystem disorder or are distinct and unrelated processes.
Her past medical history included fibromyalgia, kidney stones, and gastroesophageal reflux disease. One week prior to presentation, she was prescribed prednisone 60 mg daily for the treatment of hives; the dose had been tapered to 40 mg at presentation, with mild improvement of hives. She recently started doxepin for fibromyalgia and insomnia. She lived at home with her husband and 8-year-old child. She worked as a clerk in a pest control office and denied any pesticide exposure. She denied tobacco, alcohol, or illicit drug use. Her family history included systemic lupus erythematosus (SLE) in her mother and maternal aunt.
Glucocorticoids are associated with myopathy; however, the weakness preceded steroid therapy. Thus, unless there was unknown exposure to high-dose steroid medication to treat recurrent episodes of urticaria earlier in her course, glucocorticoid-related myopathy is unlikely. Fibromyalgia might cause the perception of weakness from pain. However, the history of difficulty combing her hair and rising from a chair suggests actual loss of motor power. The side effects of her medications, such as newly started doxepin, must be reviewed. A family history of SLE raises concern for rheumatologic conditions; however, one might expect improvement with steroid therapy.
On physical examination, her temperature was 36.9 °C, blood pressure 126/93 mmHg, pulse 81 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 100% on ambient air. Her cardiopulmonary examination was normal. Her abdomen was nontender and without hepatosplenomegaly. Her strength was 2 out of 5 in proximal and distal legs, bilaterally, and 4 out of 5 in proximal and distal upper extremities. She had normal muscle tone without fasciculations or atrophy. Her joints were without edema, erythema, or impaired range of motion. She had normal sensation to light touch in arms and legs. Her reflexes were 2+ in the patellar, Achilles, and brachioradialis tendons. She had no lymphadenopathy, mucosal ulcerations, or alopecia. A skin examination revealed smooth, slightly elevated, and faded pink wheals that were diffuse but most prominent in upper arms and chest.
Physical examination confirms the presence of true muscle weakness. The differential diagnosis is narrowed by several findings, both positive and negative, elicited in the examination. The diffuse nature of the weakness eliminates focal central nervous system lesions, such as stroke, intracranial mass lesions, or demyelinating white matter foci. Combining this finding with normal reflexes and history of preceding myalgias makes electrolyte-induced and inflammatory (eg, polymyositis) myopathies more likely. The normal deep tendon reflexes and the absence of a delayed relaxation phase lower the likelihood of hypothyroidism.
Diseases originating from the neuromuscular junction, such as myasthenia gravis, may also present with weakness and normal reflexes, although this pattern of weakness would be atypical; myasthenia gravis classically presents with fatigable weakness and ocular findings of diplopia and/or ptosis. First-tier testing should include a complete blood count to evaluate for eosinophilia, comprehensive metabolic panel, and urinalysis for myoglobinuria, thyroid stimulating hormone, and muscle enzymes.
Results of a complete blood count demonstrated a leukocyte count of 16.1 k/uL with 82% neutrophils, 13% lymphocytes, 5% monocytes, and 0% eosinophils. Hemoglobin was 13.2 g/dL, and platelet count 226 k/uL. Sodium was 136 mmol/L, potassium 1.5 mmol/L, chloride 115 mmol/L, bicarbonate 12 mmol/L, blood urea nitrogen 26 mg/dL, creatinine 1.0 mg/dL (baseline creatinine: 0.6), and glucose 102 mg/dL. Calcium was 9.4 mg/dL, magnesium 2.6 mg/dL, phosphorus 1.8 mg/dL, CK 501 U/L (normal: 40-230), and TSH 5.48 uIU/mL (normal: 0.5-4). Aspartate aminotransferase was 64 U/L, alanine aminotransferase 23 U/L, alkaline phosphatase 66 U/L, bilirubin 0.9 mg/dL, albumin 3.8 g/dL, and total protein 8.7 g/dL (normal: 6.2-7.8). Human immunodeficiency virus antibody screen was negative. An electrocardiogram revealed normal sinus rhythm, flattened T waves, and prominent U waves.
Potassium losses are classically categorized into 1 of 3 groups: renal losses, gastrointestinal losses, or transcellular shifts. Without a clear history of diuretic use, renal losses may not be apparent on history and examination. In contrast, gastrointestinal losses are almost always evidenced by a history of vomiting and/or diarrhea, with rare exceptions, including unreported laxative abuse or surreptitious vomiting. Transcellular potassium shifts can be seen in states of increased insulin or beta-adrenergic activity and alkalosis and result from both primary and secondary causes of hypokalemic periodic paralysis.
The presence of a reduced serum bicarbonate and elevated chloride concentration suggests a normal anion gap metabolic acidosis. Many conditions associated with normal anion gap metabolic acidosis are evident by history, such as diarrhea. In enigmatic cases such as this, it will be important to take a stepwise approach that includes an evaluation for urinary potassium losses and assessment of acid-base status. An unexplained normal anion gap metabolic acidosis combined with hypokalemia raises suspicion for a distal renal tubular acidosis (RTA). Additional testing to evaluate for a possible RTA should include the assessment of urinary electrolytes and urinary pH. The hypokalemia explains her weakness, but the etiology of such profound hypokalemia is not evident, nor is it clear how it relates to her hives.
The severity of the hypokalemia, combined with electrocardiogram changes, necessitates rapid intravenous potassium repletion, telemetry monitoring, and frequent serum potassium measurement. Treatment of her metabolic acidosis is more nuanced and depends upon both the severity of disturbance and the suspicion of whether the etiology is transcellular shift, potassium depletion, or both.
Urine studies demonstrated a urine specific gravity of 1.006 (normal: 1.001-1.030), urine pH was 6.5 (normal: 5-6.5), trace leukocyte esterase, negative nitrite, 30 mg/dL of protein (normal: <15), sodium 64 mmol/L (normal: 40-220), potassium 17 mmol/L (normal: 25-125), and chloride 71 mmol/L (normal: 110-250). Urine microscopy demonstrated 3 red blood cells per high power field (normal: 0-1), 4 white blood cells per high power field (normal: 0-4), 4+ bacteria per high power field, and no red blood cell casts. Urine protein-to-creatinine ratio was 1.6. C3 and C4 complement levels were 53 mg/dL (normal: 80-165) and 12 mg/dL (normal: 15-49), respectively. C-reactive protein was <0.5 (normal: 0-0.9), and erythrocyte sedimentation rate was 16 mm/hour (normal: 0-20).
A calculation of the urine anion gap (UAG; [urine sodium + urine potassium] – urine chloride) yields a UAG of 10 mq/L. A positive UAG, together with a nongap metabolic acidosis, should prompt the consideration of RTA. The normal renal response to acidosis is to reduce the urine pH to less than 5.3 through an increase in hydrogen ion excretion in the form of ammonium. A urine pH of 6.5 is highly suggestive of type 1 (distal) RTA and its associated impairment of distal acidification. Treatment with sodium bicarbonate to correct the acidosis and associated complications is warranted.
A distal RTA would account for her past medical history of renal stones. Acidemia promotes both increased calcium phosphate release from bone (with subsequent hypercalciuria) and enhanced citrate reabsorption in the proximal renal tubules, leading to decreased urinary citrate. Citrate inhibits calcium stone formation. The increased calcium load to renal tubules in addition to decreased urinary citrate both lead to increased precipitation of calcium stones in the genitourinary tract.
A diagnosis of distal RTA should prompt evaluation for specific etiologies, such as
Her antinuclear antibody titer was >1:1280 (normal: <80). Anti-SSA and -SSB antibodies were both positive, with a titer >100 (normal: <20). Rheumatoid factor was positive at 22 IU/mL (normal: 0-14). Anti-smith, anti-double stranded DNA, and anti-ribonucleoprotein antibodies were negative.
Sjögren’s syndrome appears to be the ultimate etiology of this patient’s distal RTA. The diagnosis of Sjögren’s is more classically made in the presence of lacrimal and/or salivary dysfunction and confirmed with compatible autoantibodies. In the absence of dry eyes or dry mouth, attention should be focused on her skin findings. Cutaneous vasculitis does occur in a small percentage of Sjögren’s syndrome cases. Urticarial lesions have been reported in this subset, and skin biopsy would further support the diagnosis.
Treatment of Sjögren’s syndrome with immunosuppressive therapy may ameliorate renal parenchymal pathology and improve her profound metabolic disturbances.
On further questioning, she described several months of mild xerostomia, which resulted in increased consumption of fluids. She did not have keratoconjunctivitis sicca. Biopsy of her urticarial rash demonstrated a leukocytoclastic vasculitis with eosinophilic infiltration (Figure 1). Renal biopsy with hematoxylin and eosin staining, immunofluorescence, and electron microscopy demonstrated an immune complex-mediated glomerulonephritis and moderate tubulointerstitial nephritis (Figure 2). A diagnosis of Sjögren’s syndrome was made based on the patient’s xerostomia, high titers of antinuclear antibodies, SSA and SSB antibodies, positive rheumatoid factor, hypocomplementemia, and systemic manifestations associated with Sjögren’s syndrome, including distal RTA, nephrolithiasis, and hives, with histologic evidence of leukocytoclastic vasculitis.
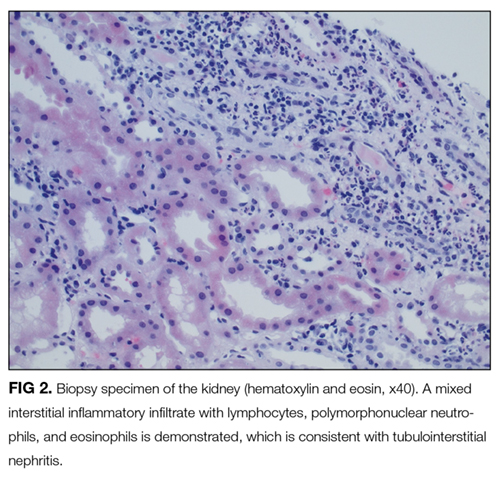
She received aggressive potassium and bicarbonate repletion and, several days later, had normalization of both. Her weakness and myalgia rapidly improved concomitantly with the correction of her hypokalemia. Five days later she was ambulating independently and discharged with potassium citrate and prednisone therapy. She had improved fatigue and rash at a 1-month follow-up with rheumatology. As an outpatient, she was started on azathioprine and slowly tapered off her steroids. Over the next several months, she had normal potassium, bicarbonate, and renal function, although she did require lithotripsy for an obstructive renal stone.
COMMENTARY
RTA should be considered in the differential diagnosis of an unexplained normal anion gap metabolic acidosis. There are 3 major types of RTAs, with different characteristics. In type 1 (distal) RTA, the primary defect is impaired distal acidification of the urine. Distal RTA commonly presents with hypokalemia, calciuria (often presenting as renal stones), and a positive UAG.1 In type 2 (proximal) RTA, the primary defect is impaired bicarbonate reabsorption, leading to bicarbonate wasting in the urine. Proximal RTAs can be secondary to an isolated defect in bicarbonate reabsorption or generalized proximal renal tubule dysfunction (Fanconi syndrome).1 A type 4 RTA is characterized by hypoaldosteronism, presenting usually with a mild nonanion gap metabolic acidosis and hyperkalemia. This patient’s history of renal stones, hypokalemia, and positive UAG supported a type 1 (distal) RTA. Distal RTA is often idiopathic, but initial evaluation should include a review of medications and investigation into an underlying systemic disorder (eg, plasma cell dyscrasia or autoimmune disease). This would include eliciting a possible history of xerostomia and xerophthalmia, together with testing of SSA (Ro) and SSB (La) antibodies, to assess for Sjögren’s syndrome. In addition, checking serum calcium to assess for hyperparathyroidism or familial idiopathic hypercalciuria and a review of medications, such as lithium and amphotericin,1 may uncover other secondary causes of distal RTA.
While Sjögren’s syndrome primarily affects salivary and lacrimal glands, leading to dry mouth and dry eyes, respectively, extraglandular manifestations are common, with fatigue and arthralgia occurring in half of patients. Extra-glandular involvement also often includes the skin and kidneys but can affect several other organ systems, including the central nervous system, heart, lungs, bone marrow, and lymph nodes.2
There are many cutaneous manifestations of Sjögren’s syndrome.3 Xerosis, or xeroderma, is the most common and is characterized by dry, scaly skin. Cutaneous vasculitis can occur in 10% of patients with Sjögren’s syndrome and often presents with palpable purpura or diffuse urticarial lesions, as in our patient.4 Erythematous maculopapules and cryoglobulinemic vasculitis may also occur.4 A less common skin manifestation is annular erythema, presenting as an indurated, ring-like lesion.5
Chronic tubulointerstitial nephritis is the most common renal manifestation of Sjögren’s syndrome.6 This often pre-sents with a mild elevated serum creatinine and a distal RTA, leading to hypokalemia, as in the case discussed. Distal RTA is well described, occurring in one-quarter of patients with Sjögren’s syndrome.7 The pathophysiology leading to distal RTA in Sjögren’s syndrome is thought to arise from autoimmune injury to the H(+)-ATPase pump in the renal collecting tubules, leading to decreased distal proton secretion.8,9 Younger adults with Sjögren’s syndrome, in the third and fourth decades of life, have a predilection to develop tubulointerstitial inflammation, distal RTA, and nephrolithiasis, as in the present case.6 Sjögren’s syndrome less commonly presents with membranoproliferative glomerulonephritis or membranous nephropathy.10,11 Cryoglobulinemia-associated hypocomplementemia and glomerulonephritis may also occur with Sjögren’s syndrome, yet glomerular lesions are less common than is tubulointerstitial inflammation. The patient discussed had proteinuria and evidence of immune complex-mediated glomerulonephritis.
Treatment of sicca symptoms is generally supportive. It includes artificial tears, encouragement of good hydration, salivary stimulants, and maintaining good oral hygiene. Pilocarpine, a cholinergic parasympathomimetic agent, is approved by the Food and Drug Administration to treat dry mouth associated with Sjögren’s syndrome. The treatment of extraglandular manifestations depends on the organ(s) involved. More severe presentations, such as vasculitis and glomerulonephritis, often require immunosuppressive therapy with systemic glucocorticoids, cyclophosphamide, azathioprine, or other immunosuppressive agents,12 including rituximab. RTA often necessitates treatment with oral bicarbonate and supplemental potassium repletion.
The base rate of disease (ie, prevalence of disease) influences a diagnostician’s pretest probability of a given diagnosis. The discussant briefly considered rare causes of hives (eg, vasculitis) but appropriately fine-tuned their differential for the patient’s hypokalemia and RTA. Once the diagnosis of Sjögren’s syndrome was made with certainty, the clinician was able to revisit the patient’s rash with a new lens. Urticarial vasculitis suddenly became a plausible consideration, despite its rarity (compared to allergic causes of hives) because of the direct link to the underlying autoimmune condition, which affected both the proximal muscles and distal nephrons.
TEACHING POINTS
- Evaluation of patients with weakness starts with determining true muscle weakness (ie, pathology involving the brain, spinal cord, peripheral nerve, neuromuscular junction, and/or muscle) from asthenia.
- Distal RTA should be considered in patients with a nonanion gap metabolic acidosis and hypokalemia.
- Sjögren’s syndrome has many extraglandular clinical manifestations, including vasculitis, urticaria, tubulointerstitial renal inflammation, glomerulonephritis, and lymphoma.
Acknowledgment
The authors thank Virgilius Cornea, MD, for his interpretation of the pathologic images.
Disclosure
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
1. Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13(8):2160-2170. PubMed
2. Asmussen K, Andersen V, Bendixen G, Schiødt M, Oxholm P. A new model for classification of disease manifestations in primary Sjögren’s syndrome: evaluation in a retrospective long-term study. J Intern Med. 1996;239(6):475-482. PubMed
3. Kittridge A, Routhouska SB, Korman NJ. Dermatologic manifestations of Sjögren syndrome. J Cutan Med Surg. 2011;15(1):8-14. PubMed
4. Ramos-Casals M, Anaya JM, García-Carrasco M, et al. Cutaneous vasculitis in primary Sjögren syndrome: classification and clinical significance of 52 patients. Medicine (Baltimore). 2004; 83(2):96-106. PubMed
5. Katayama I, Kotobuki Y, Kiyohara E, Murota H. Annular erythema associated with Sjögren’s syndrome: review of the literature on the management and clinical analysis of skin lesions. Mod Rheumatol. 2010;20(2):123-129. PubMed
6. Maripuri S, Grande JP, Osborn TG, et al. Renal involvement in primary Sjögren’s syndrome: a clinicopathologic study. Clin J Am Soc Nephrol. 2009;4(9):1423-1431. PubMed
7. Pun KK, Wong CK, Tsui EY, Tam SC, Kung AW, Wang CC. Hypokalemic periodic paralysis due to the Sjögren syndrome in Chinese patients. Ann Intern Med. 1989;110(5):405-406. PubMed
8. Cohen EP, Bastani B, Cohen MR, Kolner S, Hemken P, Gluck SL. Absence of H(+)-ATPase in cortical collecting tubules of a patient with Sjogren’s syndrome and distal renal tubular acidosis. J Am Soc Nephrol. 1992;3(2):264-271. PubMed
9. Bastani B, Haragsim L, Gluck S, Siamopoulos KC. Lack of H-ATPase in distal nephron causing hypokalaemic distal RTA in a patient with Sjögren’s syndrome. Nephrol Dial Transplant. 1995;10(6):908-909. PubMed
10. Cortez MS, Sturgill BC, Bolton WK. Membranoproliferative glomerulonephritis with primary Sjögren’s syndrome. Am J Kidney Dis. 1995;25(4):632-636. PubMed
11. Baba A, Hara S, Sato Y, Yamada K, Fujimoto S, Eto T. [Three patients with nephrotic syndrome due to membranous nephropathy complicated by Sjögren’s syndrome]. Nihon Jinzo Gakkai Shi. 2005;47(8):882-886. PubMed
12. Thanou-Stavraki A, James JA. Primary Sjogren’s syndrome: current and prospective therapies. Semin Arthritis Rheum. 2008;37(5):273-292. PubMed
A previously healthy 30-year-old woman presented to the emergency department with 2 weeks of weakness.
True muscle weakness must be distinguished from the more common causes of asthenia. Many systemic disorders produce fatigue, with resulting functional limitation that is often interpreted by patients as weakness. Initial history should focus on conditions producing fatigue, such as cardiopulmonary disease, anemia, connective tissue disease, depression or cachexia related to malignancy, infection, or other inflammatory states. Careful questioning may reveal evidence of dyspnea, poor exercise tolerance, or joint pain as an alternative to actual loss of muscle power. If true weakness is still suspected, attention should be focused on the pattern, onset, anatomic site, and progression of weakness. Muscle weakness is often characterized by difficulty with specific tasks, such as climbing stairs, rising from a chair, raising a hand, or using cutlery. The physical examination is critical in determining whether weakness is due to true loss of motor power. The differential diagnosis of weakness is broad and includes neurologic, infectious, endocrine, inflammatory, genetic, metabolic, and drug-induced etiologies.
She initially experienced 3 days of mild cramps and soreness in her thighs. She then developed weakness that began in her thighs and progressed to involve her lower legs and upper and lower arms. She had difficulty combing her hair. She required the use of her arms to get up from a chair. She grasped onto objects to aid in ambulation around the house. In addition, she described 1 year of moderate fatigue but no fever, weight loss, dyspnea, dysphagia, visual changes, paresthesias, bowel or bladder incontinence, back pain, or preceding gastrointestinal or respiratory illness. She had experienced diffuse intermittent hives, most prominent in her chest and upper arms, for the past several weeks.
History certainly supports true weakness but will need to be confirmed on examination. The distribution began as proximal but now appears diffuse. The presence of myalgia and cramping raises the possibility of noninflammatory myopathies, which are usually more insidious in onset. A severe electrolyte disturbance would be possible, based on the diffuse nature of weakness that was preceded by cramping. The distribution of weakness and lack of bowel or bladder incontinence is reassuring and suggests against a spinal cord disorder; however, a high index of suspicion must be maintained for myelopathy because delayed treatment might result in irreversible paralysis.
The patient’s course also includes hives. Common causes of hives include infections and allergic reactions to medications, foods, and insect stings. Urticaria may also result from systemic disorders, such as vasculitis, lupus, lymphoma, mastocytosis, and paraproteinemias, which can be associated with weakness and fatigue. Although severe weakness in combination with hives makes an infectious and allergic reaction less likely, we still seek to ascertain if the evolving chief complaints of weakness and hives are the result of a single unifying and evolving multisystem disorder or are distinct and unrelated processes.
Her past medical history included fibromyalgia, kidney stones, and gastroesophageal reflux disease. One week prior to presentation, she was prescribed prednisone 60 mg daily for the treatment of hives; the dose had been tapered to 40 mg at presentation, with mild improvement of hives. She recently started doxepin for fibromyalgia and insomnia. She lived at home with her husband and 8-year-old child. She worked as a clerk in a pest control office and denied any pesticide exposure. She denied tobacco, alcohol, or illicit drug use. Her family history included systemic lupus erythematosus (SLE) in her mother and maternal aunt.
Glucocorticoids are associated with myopathy; however, the weakness preceded steroid therapy. Thus, unless there was unknown exposure to high-dose steroid medication to treat recurrent episodes of urticaria earlier in her course, glucocorticoid-related myopathy is unlikely. Fibromyalgia might cause the perception of weakness from pain. However, the history of difficulty combing her hair and rising from a chair suggests actual loss of motor power. The side effects of her medications, such as newly started doxepin, must be reviewed. A family history of SLE raises concern for rheumatologic conditions; however, one might expect improvement with steroid therapy.
On physical examination, her temperature was 36.9 °C, blood pressure 126/93 mmHg, pulse 81 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 100% on ambient air. Her cardiopulmonary examination was normal. Her abdomen was nontender and without hepatosplenomegaly. Her strength was 2 out of 5 in proximal and distal legs, bilaterally, and 4 out of 5 in proximal and distal upper extremities. She had normal muscle tone without fasciculations or atrophy. Her joints were without edema, erythema, or impaired range of motion. She had normal sensation to light touch in arms and legs. Her reflexes were 2+ in the patellar, Achilles, and brachioradialis tendons. She had no lymphadenopathy, mucosal ulcerations, or alopecia. A skin examination revealed smooth, slightly elevated, and faded pink wheals that were diffuse but most prominent in upper arms and chest.
Physical examination confirms the presence of true muscle weakness. The differential diagnosis is narrowed by several findings, both positive and negative, elicited in the examination. The diffuse nature of the weakness eliminates focal central nervous system lesions, such as stroke, intracranial mass lesions, or demyelinating white matter foci. Combining this finding with normal reflexes and history of preceding myalgias makes electrolyte-induced and inflammatory (eg, polymyositis) myopathies more likely. The normal deep tendon reflexes and the absence of a delayed relaxation phase lower the likelihood of hypothyroidism.
Diseases originating from the neuromuscular junction, such as myasthenia gravis, may also present with weakness and normal reflexes, although this pattern of weakness would be atypical; myasthenia gravis classically presents with fatigable weakness and ocular findings of diplopia and/or ptosis. First-tier testing should include a complete blood count to evaluate for eosinophilia, comprehensive metabolic panel, and urinalysis for myoglobinuria, thyroid stimulating hormone, and muscle enzymes.
Results of a complete blood count demonstrated a leukocyte count of 16.1 k/uL with 82% neutrophils, 13% lymphocytes, 5% monocytes, and 0% eosinophils. Hemoglobin was 13.2 g/dL, and platelet count 226 k/uL. Sodium was 136 mmol/L, potassium 1.5 mmol/L, chloride 115 mmol/L, bicarbonate 12 mmol/L, blood urea nitrogen 26 mg/dL, creatinine 1.0 mg/dL (baseline creatinine: 0.6), and glucose 102 mg/dL. Calcium was 9.4 mg/dL, magnesium 2.6 mg/dL, phosphorus 1.8 mg/dL, CK 501 U/L (normal: 40-230), and TSH 5.48 uIU/mL (normal: 0.5-4). Aspartate aminotransferase was 64 U/L, alanine aminotransferase 23 U/L, alkaline phosphatase 66 U/L, bilirubin 0.9 mg/dL, albumin 3.8 g/dL, and total protein 8.7 g/dL (normal: 6.2-7.8). Human immunodeficiency virus antibody screen was negative. An electrocardiogram revealed normal sinus rhythm, flattened T waves, and prominent U waves.
Potassium losses are classically categorized into 1 of 3 groups: renal losses, gastrointestinal losses, or transcellular shifts. Without a clear history of diuretic use, renal losses may not be apparent on history and examination. In contrast, gastrointestinal losses are almost always evidenced by a history of vomiting and/or diarrhea, with rare exceptions, including unreported laxative abuse or surreptitious vomiting. Transcellular potassium shifts can be seen in states of increased insulin or beta-adrenergic activity and alkalosis and result from both primary and secondary causes of hypokalemic periodic paralysis.
The presence of a reduced serum bicarbonate and elevated chloride concentration suggests a normal anion gap metabolic acidosis. Many conditions associated with normal anion gap metabolic acidosis are evident by history, such as diarrhea. In enigmatic cases such as this, it will be important to take a stepwise approach that includes an evaluation for urinary potassium losses and assessment of acid-base status. An unexplained normal anion gap metabolic acidosis combined with hypokalemia raises suspicion for a distal renal tubular acidosis (RTA). Additional testing to evaluate for a possible RTA should include the assessment of urinary electrolytes and urinary pH. The hypokalemia explains her weakness, but the etiology of such profound hypokalemia is not evident, nor is it clear how it relates to her hives.
The severity of the hypokalemia, combined with electrocardiogram changes, necessitates rapid intravenous potassium repletion, telemetry monitoring, and frequent serum potassium measurement. Treatment of her metabolic acidosis is more nuanced and depends upon both the severity of disturbance and the suspicion of whether the etiology is transcellular shift, potassium depletion, or both.
Urine studies demonstrated a urine specific gravity of 1.006 (normal: 1.001-1.030), urine pH was 6.5 (normal: 5-6.5), trace leukocyte esterase, negative nitrite, 30 mg/dL of protein (normal: <15), sodium 64 mmol/L (normal: 40-220), potassium 17 mmol/L (normal: 25-125), and chloride 71 mmol/L (normal: 110-250). Urine microscopy demonstrated 3 red blood cells per high power field (normal: 0-1), 4 white blood cells per high power field (normal: 0-4), 4+ bacteria per high power field, and no red blood cell casts. Urine protein-to-creatinine ratio was 1.6. C3 and C4 complement levels were 53 mg/dL (normal: 80-165) and 12 mg/dL (normal: 15-49), respectively. C-reactive protein was <0.5 (normal: 0-0.9), and erythrocyte sedimentation rate was 16 mm/hour (normal: 0-20).
A calculation of the urine anion gap (UAG; [urine sodium + urine potassium] – urine chloride) yields a UAG of 10 mq/L. A positive UAG, together with a nongap metabolic acidosis, should prompt the consideration of RTA. The normal renal response to acidosis is to reduce the urine pH to less than 5.3 through an increase in hydrogen ion excretion in the form of ammonium. A urine pH of 6.5 is highly suggestive of type 1 (distal) RTA and its associated impairment of distal acidification. Treatment with sodium bicarbonate to correct the acidosis and associated complications is warranted.
A distal RTA would account for her past medical history of renal stones. Acidemia promotes both increased calcium phosphate release from bone (with subsequent hypercalciuria) and enhanced citrate reabsorption in the proximal renal tubules, leading to decreased urinary citrate. Citrate inhibits calcium stone formation. The increased calcium load to renal tubules in addition to decreased urinary citrate both lead to increased precipitation of calcium stones in the genitourinary tract.
A diagnosis of distal RTA should prompt evaluation for specific etiologies, such as
Her antinuclear antibody titer was >1:1280 (normal: <80). Anti-SSA and -SSB antibodies were both positive, with a titer >100 (normal: <20). Rheumatoid factor was positive at 22 IU/mL (normal: 0-14). Anti-smith, anti-double stranded DNA, and anti-ribonucleoprotein antibodies were negative.
Sjögren’s syndrome appears to be the ultimate etiology of this patient’s distal RTA. The diagnosis of Sjögren’s is more classically made in the presence of lacrimal and/or salivary dysfunction and confirmed with compatible autoantibodies. In the absence of dry eyes or dry mouth, attention should be focused on her skin findings. Cutaneous vasculitis does occur in a small percentage of Sjögren’s syndrome cases. Urticarial lesions have been reported in this subset, and skin biopsy would further support the diagnosis.
Treatment of Sjögren’s syndrome with immunosuppressive therapy may ameliorate renal parenchymal pathology and improve her profound metabolic disturbances.
On further questioning, she described several months of mild xerostomia, which resulted in increased consumption of fluids. She did not have keratoconjunctivitis sicca. Biopsy of her urticarial rash demonstrated a leukocytoclastic vasculitis with eosinophilic infiltration (Figure 1). Renal biopsy with hematoxylin and eosin staining, immunofluorescence, and electron microscopy demonstrated an immune complex-mediated glomerulonephritis and moderate tubulointerstitial nephritis (Figure 2). A diagnosis of Sjögren’s syndrome was made based on the patient’s xerostomia, high titers of antinuclear antibodies, SSA and SSB antibodies, positive rheumatoid factor, hypocomplementemia, and systemic manifestations associated with Sjögren’s syndrome, including distal RTA, nephrolithiasis, and hives, with histologic evidence of leukocytoclastic vasculitis.
She received aggressive potassium and bicarbonate repletion and, several days later, had normalization of both. Her weakness and myalgia rapidly improved concomitantly with the correction of her hypokalemia. Five days later she was ambulating independently and discharged with potassium citrate and prednisone therapy. She had improved fatigue and rash at a 1-month follow-up with rheumatology. As an outpatient, she was started on azathioprine and slowly tapered off her steroids. Over the next several months, she had normal potassium, bicarbonate, and renal function, although she did require lithotripsy for an obstructive renal stone.
COMMENTARY
RTA should be considered in the differential diagnosis of an unexplained normal anion gap metabolic acidosis. There are 3 major types of RTAs, with different characteristics. In type 1 (distal) RTA, the primary defect is impaired distal acidification of the urine. Distal RTA commonly presents with hypokalemia, calciuria (often presenting as renal stones), and a positive UAG.1 In type 2 (proximal) RTA, the primary defect is impaired bicarbonate reabsorption, leading to bicarbonate wasting in the urine. Proximal RTAs can be secondary to an isolated defect in bicarbonate reabsorption or generalized proximal renal tubule dysfunction (Fanconi syndrome).1 A type 4 RTA is characterized by hypoaldosteronism, presenting usually with a mild nonanion gap metabolic acidosis and hyperkalemia. This patient’s history of renal stones, hypokalemia, and positive UAG supported a type 1 (distal) RTA. Distal RTA is often idiopathic, but initial evaluation should include a review of medications and investigation into an underlying systemic disorder (eg, plasma cell dyscrasia or autoimmune disease). This would include eliciting a possible history of xerostomia and xerophthalmia, together with testing of SSA (Ro) and SSB (La) antibodies, to assess for Sjögren’s syndrome. In addition, checking serum calcium to assess for hyperparathyroidism or familial idiopathic hypercalciuria and a review of medications, such as lithium and amphotericin,1 may uncover other secondary causes of distal RTA.
While Sjögren’s syndrome primarily affects salivary and lacrimal glands, leading to dry mouth and dry eyes, respectively, extraglandular manifestations are common, with fatigue and arthralgia occurring in half of patients. Extra-glandular involvement also often includes the skin and kidneys but can affect several other organ systems, including the central nervous system, heart, lungs, bone marrow, and lymph nodes.2
There are many cutaneous manifestations of Sjögren’s syndrome.3 Xerosis, or xeroderma, is the most common and is characterized by dry, scaly skin. Cutaneous vasculitis can occur in 10% of patients with Sjögren’s syndrome and often presents with palpable purpura or diffuse urticarial lesions, as in our patient.4 Erythematous maculopapules and cryoglobulinemic vasculitis may also occur.4 A less common skin manifestation is annular erythema, presenting as an indurated, ring-like lesion.5
Chronic tubulointerstitial nephritis is the most common renal manifestation of Sjögren’s syndrome.6 This often pre-sents with a mild elevated serum creatinine and a distal RTA, leading to hypokalemia, as in the case discussed. Distal RTA is well described, occurring in one-quarter of patients with Sjögren’s syndrome.7 The pathophysiology leading to distal RTA in Sjögren’s syndrome is thought to arise from autoimmune injury to the H(+)-ATPase pump in the renal collecting tubules, leading to decreased distal proton secretion.8,9 Younger adults with Sjögren’s syndrome, in the third and fourth decades of life, have a predilection to develop tubulointerstitial inflammation, distal RTA, and nephrolithiasis, as in the present case.6 Sjögren’s syndrome less commonly presents with membranoproliferative glomerulonephritis or membranous nephropathy.10,11 Cryoglobulinemia-associated hypocomplementemia and glomerulonephritis may also occur with Sjögren’s syndrome, yet glomerular lesions are less common than is tubulointerstitial inflammation. The patient discussed had proteinuria and evidence of immune complex-mediated glomerulonephritis.
Treatment of sicca symptoms is generally supportive. It includes artificial tears, encouragement of good hydration, salivary stimulants, and maintaining good oral hygiene. Pilocarpine, a cholinergic parasympathomimetic agent, is approved by the Food and Drug Administration to treat dry mouth associated with Sjögren’s syndrome. The treatment of extraglandular manifestations depends on the organ(s) involved. More severe presentations, such as vasculitis and glomerulonephritis, often require immunosuppressive therapy with systemic glucocorticoids, cyclophosphamide, azathioprine, or other immunosuppressive agents,12 including rituximab. RTA often necessitates treatment with oral bicarbonate and supplemental potassium repletion.
The base rate of disease (ie, prevalence of disease) influences a diagnostician’s pretest probability of a given diagnosis. The discussant briefly considered rare causes of hives (eg, vasculitis) but appropriately fine-tuned their differential for the patient’s hypokalemia and RTA. Once the diagnosis of Sjögren’s syndrome was made with certainty, the clinician was able to revisit the patient’s rash with a new lens. Urticarial vasculitis suddenly became a plausible consideration, despite its rarity (compared to allergic causes of hives) because of the direct link to the underlying autoimmune condition, which affected both the proximal muscles and distal nephrons.
TEACHING POINTS
- Evaluation of patients with weakness starts with determining true muscle weakness (ie, pathology involving the brain, spinal cord, peripheral nerve, neuromuscular junction, and/or muscle) from asthenia.
- Distal RTA should be considered in patients with a nonanion gap metabolic acidosis and hypokalemia.
- Sjögren’s syndrome has many extraglandular clinical manifestations, including vasculitis, urticaria, tubulointerstitial renal inflammation, glomerulonephritis, and lymphoma.
Acknowledgment
The authors thank Virgilius Cornea, MD, for his interpretation of the pathologic images.
Disclosure
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
A previously healthy 30-year-old woman presented to the emergency department with 2 weeks of weakness.
True muscle weakness must be distinguished from the more common causes of asthenia. Many systemic disorders produce fatigue, with resulting functional limitation that is often interpreted by patients as weakness. Initial history should focus on conditions producing fatigue, such as cardiopulmonary disease, anemia, connective tissue disease, depression or cachexia related to malignancy, infection, or other inflammatory states. Careful questioning may reveal evidence of dyspnea, poor exercise tolerance, or joint pain as an alternative to actual loss of muscle power. If true weakness is still suspected, attention should be focused on the pattern, onset, anatomic site, and progression of weakness. Muscle weakness is often characterized by difficulty with specific tasks, such as climbing stairs, rising from a chair, raising a hand, or using cutlery. The physical examination is critical in determining whether weakness is due to true loss of motor power. The differential diagnosis of weakness is broad and includes neurologic, infectious, endocrine, inflammatory, genetic, metabolic, and drug-induced etiologies.
She initially experienced 3 days of mild cramps and soreness in her thighs. She then developed weakness that began in her thighs and progressed to involve her lower legs and upper and lower arms. She had difficulty combing her hair. She required the use of her arms to get up from a chair. She grasped onto objects to aid in ambulation around the house. In addition, she described 1 year of moderate fatigue but no fever, weight loss, dyspnea, dysphagia, visual changes, paresthesias, bowel or bladder incontinence, back pain, or preceding gastrointestinal or respiratory illness. She had experienced diffuse intermittent hives, most prominent in her chest and upper arms, for the past several weeks.
History certainly supports true weakness but will need to be confirmed on examination. The distribution began as proximal but now appears diffuse. The presence of myalgia and cramping raises the possibility of noninflammatory myopathies, which are usually more insidious in onset. A severe electrolyte disturbance would be possible, based on the diffuse nature of weakness that was preceded by cramping. The distribution of weakness and lack of bowel or bladder incontinence is reassuring and suggests against a spinal cord disorder; however, a high index of suspicion must be maintained for myelopathy because delayed treatment might result in irreversible paralysis.
The patient’s course also includes hives. Common causes of hives include infections and allergic reactions to medications, foods, and insect stings. Urticaria may also result from systemic disorders, such as vasculitis, lupus, lymphoma, mastocytosis, and paraproteinemias, which can be associated with weakness and fatigue. Although severe weakness in combination with hives makes an infectious and allergic reaction less likely, we still seek to ascertain if the evolving chief complaints of weakness and hives are the result of a single unifying and evolving multisystem disorder or are distinct and unrelated processes.
Her past medical history included fibromyalgia, kidney stones, and gastroesophageal reflux disease. One week prior to presentation, she was prescribed prednisone 60 mg daily for the treatment of hives; the dose had been tapered to 40 mg at presentation, with mild improvement of hives. She recently started doxepin for fibromyalgia and insomnia. She lived at home with her husband and 8-year-old child. She worked as a clerk in a pest control office and denied any pesticide exposure. She denied tobacco, alcohol, or illicit drug use. Her family history included systemic lupus erythematosus (SLE) in her mother and maternal aunt.
Glucocorticoids are associated with myopathy; however, the weakness preceded steroid therapy. Thus, unless there was unknown exposure to high-dose steroid medication to treat recurrent episodes of urticaria earlier in her course, glucocorticoid-related myopathy is unlikely. Fibromyalgia might cause the perception of weakness from pain. However, the history of difficulty combing her hair and rising from a chair suggests actual loss of motor power. The side effects of her medications, such as newly started doxepin, must be reviewed. A family history of SLE raises concern for rheumatologic conditions; however, one might expect improvement with steroid therapy.
On physical examination, her temperature was 36.9 °C, blood pressure 126/93 mmHg, pulse 81 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 100% on ambient air. Her cardiopulmonary examination was normal. Her abdomen was nontender and without hepatosplenomegaly. Her strength was 2 out of 5 in proximal and distal legs, bilaterally, and 4 out of 5 in proximal and distal upper extremities. She had normal muscle tone without fasciculations or atrophy. Her joints were without edema, erythema, or impaired range of motion. She had normal sensation to light touch in arms and legs. Her reflexes were 2+ in the patellar, Achilles, and brachioradialis tendons. She had no lymphadenopathy, mucosal ulcerations, or alopecia. A skin examination revealed smooth, slightly elevated, and faded pink wheals that were diffuse but most prominent in upper arms and chest.
Physical examination confirms the presence of true muscle weakness. The differential diagnosis is narrowed by several findings, both positive and negative, elicited in the examination. The diffuse nature of the weakness eliminates focal central nervous system lesions, such as stroke, intracranial mass lesions, or demyelinating white matter foci. Combining this finding with normal reflexes and history of preceding myalgias makes electrolyte-induced and inflammatory (eg, polymyositis) myopathies more likely. The normal deep tendon reflexes and the absence of a delayed relaxation phase lower the likelihood of hypothyroidism.
Diseases originating from the neuromuscular junction, such as myasthenia gravis, may also present with weakness and normal reflexes, although this pattern of weakness would be atypical; myasthenia gravis classically presents with fatigable weakness and ocular findings of diplopia and/or ptosis. First-tier testing should include a complete blood count to evaluate for eosinophilia, comprehensive metabolic panel, and urinalysis for myoglobinuria, thyroid stimulating hormone, and muscle enzymes.
Results of a complete blood count demonstrated a leukocyte count of 16.1 k/uL with 82% neutrophils, 13% lymphocytes, 5% monocytes, and 0% eosinophils. Hemoglobin was 13.2 g/dL, and platelet count 226 k/uL. Sodium was 136 mmol/L, potassium 1.5 mmol/L, chloride 115 mmol/L, bicarbonate 12 mmol/L, blood urea nitrogen 26 mg/dL, creatinine 1.0 mg/dL (baseline creatinine: 0.6), and glucose 102 mg/dL. Calcium was 9.4 mg/dL, magnesium 2.6 mg/dL, phosphorus 1.8 mg/dL, CK 501 U/L (normal: 40-230), and TSH 5.48 uIU/mL (normal: 0.5-4). Aspartate aminotransferase was 64 U/L, alanine aminotransferase 23 U/L, alkaline phosphatase 66 U/L, bilirubin 0.9 mg/dL, albumin 3.8 g/dL, and total protein 8.7 g/dL (normal: 6.2-7.8). Human immunodeficiency virus antibody screen was negative. An electrocardiogram revealed normal sinus rhythm, flattened T waves, and prominent U waves.
Potassium losses are classically categorized into 1 of 3 groups: renal losses, gastrointestinal losses, or transcellular shifts. Without a clear history of diuretic use, renal losses may not be apparent on history and examination. In contrast, gastrointestinal losses are almost always evidenced by a history of vomiting and/or diarrhea, with rare exceptions, including unreported laxative abuse or surreptitious vomiting. Transcellular potassium shifts can be seen in states of increased insulin or beta-adrenergic activity and alkalosis and result from both primary and secondary causes of hypokalemic periodic paralysis.
The presence of a reduced serum bicarbonate and elevated chloride concentration suggests a normal anion gap metabolic acidosis. Many conditions associated with normal anion gap metabolic acidosis are evident by history, such as diarrhea. In enigmatic cases such as this, it will be important to take a stepwise approach that includes an evaluation for urinary potassium losses and assessment of acid-base status. An unexplained normal anion gap metabolic acidosis combined with hypokalemia raises suspicion for a distal renal tubular acidosis (RTA). Additional testing to evaluate for a possible RTA should include the assessment of urinary electrolytes and urinary pH. The hypokalemia explains her weakness, but the etiology of such profound hypokalemia is not evident, nor is it clear how it relates to her hives.
The severity of the hypokalemia, combined with electrocardiogram changes, necessitates rapid intravenous potassium repletion, telemetry monitoring, and frequent serum potassium measurement. Treatment of her metabolic acidosis is more nuanced and depends upon both the severity of disturbance and the suspicion of whether the etiology is transcellular shift, potassium depletion, or both.
Urine studies demonstrated a urine specific gravity of 1.006 (normal: 1.001-1.030), urine pH was 6.5 (normal: 5-6.5), trace leukocyte esterase, negative nitrite, 30 mg/dL of protein (normal: <15), sodium 64 mmol/L (normal: 40-220), potassium 17 mmol/L (normal: 25-125), and chloride 71 mmol/L (normal: 110-250). Urine microscopy demonstrated 3 red blood cells per high power field (normal: 0-1), 4 white blood cells per high power field (normal: 0-4), 4+ bacteria per high power field, and no red blood cell casts. Urine protein-to-creatinine ratio was 1.6. C3 and C4 complement levels were 53 mg/dL (normal: 80-165) and 12 mg/dL (normal: 15-49), respectively. C-reactive protein was <0.5 (normal: 0-0.9), and erythrocyte sedimentation rate was 16 mm/hour (normal: 0-20).
A calculation of the urine anion gap (UAG; [urine sodium + urine potassium] – urine chloride) yields a UAG of 10 mq/L. A positive UAG, together with a nongap metabolic acidosis, should prompt the consideration of RTA. The normal renal response to acidosis is to reduce the urine pH to less than 5.3 through an increase in hydrogen ion excretion in the form of ammonium. A urine pH of 6.5 is highly suggestive of type 1 (distal) RTA and its associated impairment of distal acidification. Treatment with sodium bicarbonate to correct the acidosis and associated complications is warranted.
A distal RTA would account for her past medical history of renal stones. Acidemia promotes both increased calcium phosphate release from bone (with subsequent hypercalciuria) and enhanced citrate reabsorption in the proximal renal tubules, leading to decreased urinary citrate. Citrate inhibits calcium stone formation. The increased calcium load to renal tubules in addition to decreased urinary citrate both lead to increased precipitation of calcium stones in the genitourinary tract.
A diagnosis of distal RTA should prompt evaluation for specific etiologies, such as
Her antinuclear antibody titer was >1:1280 (normal: <80). Anti-SSA and -SSB antibodies were both positive, with a titer >100 (normal: <20). Rheumatoid factor was positive at 22 IU/mL (normal: 0-14). Anti-smith, anti-double stranded DNA, and anti-ribonucleoprotein antibodies were negative.
Sjögren’s syndrome appears to be the ultimate etiology of this patient’s distal RTA. The diagnosis of Sjögren’s is more classically made in the presence of lacrimal and/or salivary dysfunction and confirmed with compatible autoantibodies. In the absence of dry eyes or dry mouth, attention should be focused on her skin findings. Cutaneous vasculitis does occur in a small percentage of Sjögren’s syndrome cases. Urticarial lesions have been reported in this subset, and skin biopsy would further support the diagnosis.
Treatment of Sjögren’s syndrome with immunosuppressive therapy may ameliorate renal parenchymal pathology and improve her profound metabolic disturbances.
On further questioning, she described several months of mild xerostomia, which resulted in increased consumption of fluids. She did not have keratoconjunctivitis sicca. Biopsy of her urticarial rash demonstrated a leukocytoclastic vasculitis with eosinophilic infiltration (Figure 1). Renal biopsy with hematoxylin and eosin staining, immunofluorescence, and electron microscopy demonstrated an immune complex-mediated glomerulonephritis and moderate tubulointerstitial nephritis (Figure 2). A diagnosis of Sjögren’s syndrome was made based on the patient’s xerostomia, high titers of antinuclear antibodies, SSA and SSB antibodies, positive rheumatoid factor, hypocomplementemia, and systemic manifestations associated with Sjögren’s syndrome, including distal RTA, nephrolithiasis, and hives, with histologic evidence of leukocytoclastic vasculitis.
She received aggressive potassium and bicarbonate repletion and, several days later, had normalization of both. Her weakness and myalgia rapidly improved concomitantly with the correction of her hypokalemia. Five days later she was ambulating independently and discharged with potassium citrate and prednisone therapy. She had improved fatigue and rash at a 1-month follow-up with rheumatology. As an outpatient, she was started on azathioprine and slowly tapered off her steroids. Over the next several months, she had normal potassium, bicarbonate, and renal function, although she did require lithotripsy for an obstructive renal stone.
COMMENTARY
RTA should be considered in the differential diagnosis of an unexplained normal anion gap metabolic acidosis. There are 3 major types of RTAs, with different characteristics. In type 1 (distal) RTA, the primary defect is impaired distal acidification of the urine. Distal RTA commonly presents with hypokalemia, calciuria (often presenting as renal stones), and a positive UAG.1 In type 2 (proximal) RTA, the primary defect is impaired bicarbonate reabsorption, leading to bicarbonate wasting in the urine. Proximal RTAs can be secondary to an isolated defect in bicarbonate reabsorption or generalized proximal renal tubule dysfunction (Fanconi syndrome).1 A type 4 RTA is characterized by hypoaldosteronism, presenting usually with a mild nonanion gap metabolic acidosis and hyperkalemia. This patient’s history of renal stones, hypokalemia, and positive UAG supported a type 1 (distal) RTA. Distal RTA is often idiopathic, but initial evaluation should include a review of medications and investigation into an underlying systemic disorder (eg, plasma cell dyscrasia or autoimmune disease). This would include eliciting a possible history of xerostomia and xerophthalmia, together with testing of SSA (Ro) and SSB (La) antibodies, to assess for Sjögren’s syndrome. In addition, checking serum calcium to assess for hyperparathyroidism or familial idiopathic hypercalciuria and a review of medications, such as lithium and amphotericin,1 may uncover other secondary causes of distal RTA.
While Sjögren’s syndrome primarily affects salivary and lacrimal glands, leading to dry mouth and dry eyes, respectively, extraglandular manifestations are common, with fatigue and arthralgia occurring in half of patients. Extra-glandular involvement also often includes the skin and kidneys but can affect several other organ systems, including the central nervous system, heart, lungs, bone marrow, and lymph nodes.2
There are many cutaneous manifestations of Sjögren’s syndrome.3 Xerosis, or xeroderma, is the most common and is characterized by dry, scaly skin. Cutaneous vasculitis can occur in 10% of patients with Sjögren’s syndrome and often presents with palpable purpura or diffuse urticarial lesions, as in our patient.4 Erythematous maculopapules and cryoglobulinemic vasculitis may also occur.4 A less common skin manifestation is annular erythema, presenting as an indurated, ring-like lesion.5
Chronic tubulointerstitial nephritis is the most common renal manifestation of Sjögren’s syndrome.6 This often pre-sents with a mild elevated serum creatinine and a distal RTA, leading to hypokalemia, as in the case discussed. Distal RTA is well described, occurring in one-quarter of patients with Sjögren’s syndrome.7 The pathophysiology leading to distal RTA in Sjögren’s syndrome is thought to arise from autoimmune injury to the H(+)-ATPase pump in the renal collecting tubules, leading to decreased distal proton secretion.8,9 Younger adults with Sjögren’s syndrome, in the third and fourth decades of life, have a predilection to develop tubulointerstitial inflammation, distal RTA, and nephrolithiasis, as in the present case.6 Sjögren’s syndrome less commonly presents with membranoproliferative glomerulonephritis or membranous nephropathy.10,11 Cryoglobulinemia-associated hypocomplementemia and glomerulonephritis may also occur with Sjögren’s syndrome, yet glomerular lesions are less common than is tubulointerstitial inflammation. The patient discussed had proteinuria and evidence of immune complex-mediated glomerulonephritis.
Treatment of sicca symptoms is generally supportive. It includes artificial tears, encouragement of good hydration, salivary stimulants, and maintaining good oral hygiene. Pilocarpine, a cholinergic parasympathomimetic agent, is approved by the Food and Drug Administration to treat dry mouth associated with Sjögren’s syndrome. The treatment of extraglandular manifestations depends on the organ(s) involved. More severe presentations, such as vasculitis and glomerulonephritis, often require immunosuppressive therapy with systemic glucocorticoids, cyclophosphamide, azathioprine, or other immunosuppressive agents,12 including rituximab. RTA often necessitates treatment with oral bicarbonate and supplemental potassium repletion.
The base rate of disease (ie, prevalence of disease) influences a diagnostician’s pretest probability of a given diagnosis. The discussant briefly considered rare causes of hives (eg, vasculitis) but appropriately fine-tuned their differential for the patient’s hypokalemia and RTA. Once the diagnosis of Sjögren’s syndrome was made with certainty, the clinician was able to revisit the patient’s rash with a new lens. Urticarial vasculitis suddenly became a plausible consideration, despite its rarity (compared to allergic causes of hives) because of the direct link to the underlying autoimmune condition, which affected both the proximal muscles and distal nephrons.
TEACHING POINTS
- Evaluation of patients with weakness starts with determining true muscle weakness (ie, pathology involving the brain, spinal cord, peripheral nerve, neuromuscular junction, and/or muscle) from asthenia.
- Distal RTA should be considered in patients with a nonanion gap metabolic acidosis and hypokalemia.
- Sjögren’s syndrome has many extraglandular clinical manifestations, including vasculitis, urticaria, tubulointerstitial renal inflammation, glomerulonephritis, and lymphoma.
Acknowledgment
The authors thank Virgilius Cornea, MD, for his interpretation of the pathologic images.
Disclosure
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
1. Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13(8):2160-2170. PubMed
2. Asmussen K, Andersen V, Bendixen G, Schiødt M, Oxholm P. A new model for classification of disease manifestations in primary Sjögren’s syndrome: evaluation in a retrospective long-term study. J Intern Med. 1996;239(6):475-482. PubMed
3. Kittridge A, Routhouska SB, Korman NJ. Dermatologic manifestations of Sjögren syndrome. J Cutan Med Surg. 2011;15(1):8-14. PubMed
4. Ramos-Casals M, Anaya JM, García-Carrasco M, et al. Cutaneous vasculitis in primary Sjögren syndrome: classification and clinical significance of 52 patients. Medicine (Baltimore). 2004; 83(2):96-106. PubMed
5. Katayama I, Kotobuki Y, Kiyohara E, Murota H. Annular erythema associated with Sjögren’s syndrome: review of the literature on the management and clinical analysis of skin lesions. Mod Rheumatol. 2010;20(2):123-129. PubMed
6. Maripuri S, Grande JP, Osborn TG, et al. Renal involvement in primary Sjögren’s syndrome: a clinicopathologic study. Clin J Am Soc Nephrol. 2009;4(9):1423-1431. PubMed
7. Pun KK, Wong CK, Tsui EY, Tam SC, Kung AW, Wang CC. Hypokalemic periodic paralysis due to the Sjögren syndrome in Chinese patients. Ann Intern Med. 1989;110(5):405-406. PubMed
8. Cohen EP, Bastani B, Cohen MR, Kolner S, Hemken P, Gluck SL. Absence of H(+)-ATPase in cortical collecting tubules of a patient with Sjogren’s syndrome and distal renal tubular acidosis. J Am Soc Nephrol. 1992;3(2):264-271. PubMed
9. Bastani B, Haragsim L, Gluck S, Siamopoulos KC. Lack of H-ATPase in distal nephron causing hypokalaemic distal RTA in a patient with Sjögren’s syndrome. Nephrol Dial Transplant. 1995;10(6):908-909. PubMed
10. Cortez MS, Sturgill BC, Bolton WK. Membranoproliferative glomerulonephritis with primary Sjögren’s syndrome. Am J Kidney Dis. 1995;25(4):632-636. PubMed
11. Baba A, Hara S, Sato Y, Yamada K, Fujimoto S, Eto T. [Three patients with nephrotic syndrome due to membranous nephropathy complicated by Sjögren’s syndrome]. Nihon Jinzo Gakkai Shi. 2005;47(8):882-886. PubMed
12. Thanou-Stavraki A, James JA. Primary Sjogren’s syndrome: current and prospective therapies. Semin Arthritis Rheum. 2008;37(5):273-292. PubMed
1. Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13(8):2160-2170. PubMed
2. Asmussen K, Andersen V, Bendixen G, Schiødt M, Oxholm P. A new model for classification of disease manifestations in primary Sjögren’s syndrome: evaluation in a retrospective long-term study. J Intern Med. 1996;239(6):475-482. PubMed
3. Kittridge A, Routhouska SB, Korman NJ. Dermatologic manifestations of Sjögren syndrome. J Cutan Med Surg. 2011;15(1):8-14. PubMed
4. Ramos-Casals M, Anaya JM, García-Carrasco M, et al. Cutaneous vasculitis in primary Sjögren syndrome: classification and clinical significance of 52 patients. Medicine (Baltimore). 2004; 83(2):96-106. PubMed
5. Katayama I, Kotobuki Y, Kiyohara E, Murota H. Annular erythema associated with Sjögren’s syndrome: review of the literature on the management and clinical analysis of skin lesions. Mod Rheumatol. 2010;20(2):123-129. PubMed
6. Maripuri S, Grande JP, Osborn TG, et al. Renal involvement in primary Sjögren’s syndrome: a clinicopathologic study. Clin J Am Soc Nephrol. 2009;4(9):1423-1431. PubMed
7. Pun KK, Wong CK, Tsui EY, Tam SC, Kung AW, Wang CC. Hypokalemic periodic paralysis due to the Sjögren syndrome in Chinese patients. Ann Intern Med. 1989;110(5):405-406. PubMed
8. Cohen EP, Bastani B, Cohen MR, Kolner S, Hemken P, Gluck SL. Absence of H(+)-ATPase in cortical collecting tubules of a patient with Sjogren’s syndrome and distal renal tubular acidosis. J Am Soc Nephrol. 1992;3(2):264-271. PubMed
9. Bastani B, Haragsim L, Gluck S, Siamopoulos KC. Lack of H-ATPase in distal nephron causing hypokalaemic distal RTA in a patient with Sjögren’s syndrome. Nephrol Dial Transplant. 1995;10(6):908-909. PubMed
10. Cortez MS, Sturgill BC, Bolton WK. Membranoproliferative glomerulonephritis with primary Sjögren’s syndrome. Am J Kidney Dis. 1995;25(4):632-636. PubMed
11. Baba A, Hara S, Sato Y, Yamada K, Fujimoto S, Eto T. [Three patients with nephrotic syndrome due to membranous nephropathy complicated by Sjögren’s syndrome]. Nihon Jinzo Gakkai Shi. 2005;47(8):882-886. PubMed
12. Thanou-Stavraki A, James JA. Primary Sjogren’s syndrome: current and prospective therapies. Semin Arthritis Rheum. 2008;37(5):273-292. PubMed
© 2017 Society of Hospital Medicine
Off Target But Hitting the Mark
A 32-year-old woman presented to the emergency department (ED) with 3 months of abdominal pain and 1 week of vomiting.
The differential diagnosis of abdominal pain is broad. This presentation could be caused by disorders of the gastrointestinal (GI), gynecologic, urinary, or, less likely, the neuromuscular systems. The presence of vomiting supports a GI cause. Pregnancy should be excluded in any woman of childbearing age presenting with abdominal pain.
Characteristics of the pain, including location, temporal characteristics, severity, and aggravating and alleviating factors, can narrow the differential diagnosis. The past medical history, including prior surgeries, menstrual, and obstetric history, is also critical.
Approximately 3 months prior to presentation, she reported a tick bite that had evolved into a circumferential targetoid rash. Her primary care provider performed serologic testing for Lyme disease, which was negative, and prescribed doxycycline, which she stopped after a week because of nausea and diffuse, achy, and constant abdominal pain. After initial improvement, symptoms recurred a week prior to presentation. The nausea was now associated with intractable vomiting and anorexia. She denied hematemesis or coffee ground emesis. Her abdominal pain intensified and radiated to her back. She lost 10 pounds over the past week. She denied headache, constipation, diarrhea, blood per rectum, melena, dysuria, vaginal discharge, or rash. She reported chills and temperatures up to 37.8 ° C at home.
She had a history of migraine headaches for which she took ibuprofen occasionally but took no other prescription or over-the-counter medications. She had never smoked, consumed 2 alcoholic beverages a month, and denied illicit drug use. She lived with her boyfriend on a farm in Indiana where she raised chickens, rabbits, and ducks.
The patient dates the onset of nausea and abdominal pain to a course of doxycycline, presumably prescribed for early Lyme disease, which was stopped after only 1 week. GI side effects, including nausea, vomiting, and upper abdominal pain, are common with doxycycline and may account for the early symptoms. However, these symptoms typically resolve promptly with drug discontinuation. Doxycycline may rarely cause esophageal and gastric ulcers, which could explain her symptoms.
Fewer than half of patients with erythema migrans caused by Lyme disease are seropositive at presentation, as there has been insufficient time for antibodies to develop. Lyme disease typically affects the skin, joints, heart, and nervous system and only rarely affects the GI tract. Acute Lyme disease can cause intestinal pseudoobstruction, splenomegaly, and mild hepatitis. Although Lyme disease is unlikely to be the cause of the current symptoms, serologic testing should be repeated and should be positive if the patient now has early disseminated disease.
Patients with Lyme disease are occasionally coinfected with a second organism. Ixodes scapularis, the tick that transmits Lyme disease in the Northeast and Midwest, can be coinfected with Babesia microti, a red cell parasite. Babesiosis can persist for months and presents with fever, malaise, and many other nonspecific symptoms, including some that this patient has: anorexia, weight loss, abdominal pain, and vomiting.
The history of migraine and intractable vomiting suggests the possibility of cyclic vomiting syndrome. This syndrome is characterized by episodic bouts of vomiting lasting from hours to as long as a week. The vomiting is often accompanied by abdominal pain and occasionally headaches. Episodes are separated by asymptomatic periods that may last months. Cyclic vomiting syndrome can occur at any age but is more common in children, those with a personal or family history of migraines, and heavy users of cannabis. At least 3 stereotypical episodes are required to make the diagnosis, so a history of prior similar symptoms should be explored.
The differential diagnosis of abdominal pain and vomiting should stay broad until a comprehensive physical exam and initial laboratory tests are performed. Volume status should be assessed by estimating jugular venous pressure and by obtaining supine and standing blood pressure measurements. The abdomen should be examined carefully, and the presence or absence of hepatomegaly, splenomegaly, masses, and ascites should be specifically noted. The presence of bradycardia, oligoarticular arthritis, or neuropathy could provide supporting evidence for Lyme disease. Pregnancy is less likely given the diffuse and persistent nature of the pain but should still be excluded.
On physical examination, she was distressed, writhing on the bed, and appearing comfortable only on her side with her knees flexed. Her temperature was 36.5 ° C, heart rate 83 beats per minute, respiratory rate 18 breaths per minute, blood pressure 143/77 mmHg, and oxygen saturation 94% while breathing ambient air. Her abdomen was diffusely tender, most markedly in the epigastrium. Abdominal rigidity, rebound tenderness, and costovertebral tenderness were absent. There was no rash; the previously reported targetoid skin lesion was no longer present. The remainder of the exam was normal.
Laboratory evaluation showed a white count of 7900/mm3, hemoglobin 14.3 gm/dL with normocytic indices, and a platelet count of 175,000/mm3. Sodium was 130 mmol/L, potassium was 3.1 mmol/L, bicarbonate 26 mmol/L, blood urea nitrogen 15 mg/dL, creatinine 0.6 mg/dL, and glucose 92 mg/dL. Serum calcium, aspartate aminotransferase, alanine aminotransferase, bilirubin, and lipase were normal. A urine pregnancy test was negative. Urine analysis was negative for nitrites and leukocyte esterase. Abdominal and pelvic computed tomography (CT) scan with intravenous (IV) contrast performed 3 days prior at an outside ED revealed a 3.4 centimeter left ovarian cyst. A subsequent transvaginal ultrasound was negative for cyst torsion and confirmed appropriate placement of an intrauterine device.
The absence of abdominal rigidity and rebound tenderness does not exclude peritonitis. A normal white blood cell count also does not reliably exclude serious intraabdominal pathology. However, the CT scan argues strongly against many common causes of abdominal pain, including appendicitis, diverticulitis, perforated ulcer, intestinal obstruction, and malignancy, assuming the symptoms have not changed since it was performed.
The patient’s laboratory studies argue against biliary obstruction, pancreatitis, pregnancy, hypercalcemia, and ongoing urinary tract infection. Patients with functional gallbladder disorders may have normal laboratory and CT findings but typically have recurrent, biliary-colic-type pain. The low serum potassium, a high blood urea nitrogen to creatinine ratio, and a low serum sodium reflect her significant vomiting. The hyponatremia is consistent with the appropriate release of antidiuretic hormone (ADH) in the setting of volume depletion. She should receive isotonic fluids plus potassium in addition to symptomatic treatment of pain and nausea. Given the severity and duration of symptoms, an esophagogastroduodenoscopy (EGD) should be performed to exclude GI mucosal disease, including peptic ulcer disease and gastritis, which may not be evident on the CT scan.
Additional diagnoses should be considered at this point. This patient has exposure to chickens, ducks, rabbits, and ticks as well as reported chills and mild temperature elevation at home. Tularemia, which can be transmitted by tick bites or exposure to infected rabbits, can cause a prolonged illness. Some patients have abdominal pain, anorexia, nausea, and weight loss, although fever is usually more prominent. Tularemia is uncommon and most frequently seen in the south-central part of the United States but has been reported throughout the country. She should be queried regarding additional exposures, including well water to assess her risk for Campylobacter infection.
Opiate withdrawal can present with pain and vomiting, but she reports no opiate use and lacks other findings such pupillary dilation or piloerection. Given the prevalence of opiate abuse, however, a toxicology screen should be performed. Hypercalcemia and diabetic ketoacidosis as metabolic causes of abdominal pain have been ruled out by her laboratory values. If no other cause is identified, other metabolic etiologies like Addison disease, familial Mediterranean fever, or porphyria should be considered.
Cyclic vomiting syndrome should still be on the differential. It is a diagnosis of exclusion requiring a history of recurrent, stereotypical episodes, which should be explicitly explored.
The patient was admitted to a medical unit by the hospitalist service and received IV normal saline, parenteral potassium, and IV pantoprazole. She underwent an EGD that revealed minor erosions in the antrum of the stomach. Biopsies were obtained.
Seven hours after the endoscopy, the patient had a brief period of confusion followed by a generalized tonic-clonic seizure lasting 1 minute. A head CT without contrast was negative for any focal abnormality. Repeat laboratory evaluation revealed that serum sodium was 125 mmol/L, and serum glucose was 113 mg/dL. She was transferred to the progressive care unit and received IV levetiracetam.
The endoscopy excluded structural abnormalities of the stomach and duodenum. The patient now has an additional problem, seizure, which needs to be incorporated in the diagnostic reasoning.
Seizures can be caused by the rapid development of severe hyponatremia, with serum sodium levels usually less than 120 mmol/L. Seizures caused by hyponatremia are typically preceded by headache and lethargy, as the intracellular movement of excess water causes cerebral edema. Hyponatremia is unlikely to be the cause of her seizure but should nevertheless be evaluated with a urine sodium concentration and serum and urine osmolality. If she is euvolemic, the IV fluids should be stopped and her free water intake should be restricted to avoid worsening the hyponatremia, as it is potentially caused by the syndrome of inappropriate ADH (SIADH).
There are many other possible causes for new onset seizures in adults, including brain tumor, head trauma, alcohol withdrawal, medications, and central nervous system infection, including Lyme disease. Lyme serologies should be repeated.
In this patient, it is likely that the seizure is a manifestation of the same illness that is causing her vomiting and abdominal pain. Seizure is not a feature of cyclic vomiting syndrome in adults. It is also not a feature of tularemia, adrenal insufficiency, or opioid withdrawal.
Acute intermittent porphyria (AIP) can cause both abdominal and neurologic problems. Hyponatremia is common during acute attacks, caused by either the inappropriate release of ADH or the appropriate release of the hormone if there is fluid loss. AIP is a rare diagnosis but could explain the uncommon combination of abdominal pain, vomiting, seizure, and hyponatremia. A spot urine porphobilinogen test should be sent to assess for AIP.
Additional laboratory studies were sent. Serum osmolality was 269 mosm/kg with a corresponding urine osmolality of 699 mosm/kg. A random urine sodium was 145 mEq/L. Thyroid stimulating hormone and cosyntropin stimulating testing were normal. IgM and IgG antibodies to Borrelia burgdorferi were negative. Urine porphobilinogen was sent. An electroencephalogram did not reveal epileptiform discharges. Magnetic resonance imaging (MRI) of the brain was significant for T2/FLAIR hyperintensity in the cortex and subcortical white matter of the occipital lobes bilaterally. Hypertonic saline and fluid restriction were initiated.
The patient’s labs are consistent with SIADH. Excessive ADH release because of volume depletion and consequent hyponatremia should have improved rapidly with the administration of saline. The high urine sodium suggests that she is now volume replete, while the high urine osmolality is consistent with the presence of excessive ADH in the absence of appropriate stimuli. In the context of normal thyroid and adrenal function, the hyponatremia is likely due to the SIADH.
Negative serologic testing for Lyme disease, 3 months after the onset of rash, excludes this diagnosis.
The MRI findings are consistent with posterior reversible encephalopathy syndrome (PRES), a clinicoradiographic syndrome of headache, altered mental status, seizure, and/or vision loss with associated white matter abnormalities of the posterior cerebral hemispheres. PRES has been reported with AIP as well as other disorders, most commonly hypertensive encephalopathy, eclampsia, and immunosuppressive drug use.
The patient’s sodium improved with fluid restriction and the administration of hypertonic saline. There was no recurrence of seizure activity. Amlodipine was initiated for blood pressure readings as high as 156/106 mmHg. A hepatobiliary scan revealed a gallbladder ejection fraction of 13%. Biopsies from her endoscopy revealed nonspecific inflammation without the presence of Helicobacter pylori. The patient was discharged home 7 days after admission after stabilization of serum sodium, improvement in her abdominal pain, and tolerance of oral intake. A plan was made for outpatient cholecystectomy.
Many causes of abdominal pain have been excluded and the remaining diagnostic possibility, porphyria, is rare. The clinicians have revisited their differential and considered other causes of abdominal pain, including functional gallbladder disorders. However, chronic cholecystitis (or functional gallbladder disorder) is not this patient’s primary problem. The diffuse, severe, and constant abdominal pain prior to admission is not typical of biliary pain, and many medical conditions and drugs, including amlodipine, can lead to a positive hepatobiliary scan. Chronic cholecystitis would not explain her seizure.
AIP remains at the top of the differential for this young woman. A urine porphobilinogen has been sent and must be followed up prior to any further workup or surgery.
One week after discharge, the patient’s urine porphobilinogen resulted at 172.8 mCmol/ (upper limits of normal 8.8). Sequencing analysis for genes coding the enzymes involved in the synthetic pathway for heme were sent. Hydroxymethylbilane synthase, coproporphyrinogen oxidase, and protoporphyrinogen oxidase mutation assays were all normal. Despite the normal genetic assays, the diagnosis of AIP was made on the basis of the clinical presentation and elevated urine porphobilinogen. The patient was referred to a hematologist and initiated on oral glucose supplements and hematin infusions.
DISCUSSION
Although abdominal pain has a broad differential, the combination of abdominal pain and neurologic or psychiatric symptoms should suggest the possibility of porphyria, especially if symptoms are recurrent or unexplained. The porphyrias are a group of disorders caused by defects in the synthetic pathway of heme, leading to an overproduction and accumulation of precursors. Heme is a component of multiple proteins, including hemoglobin, myoglobin, and the cytochrome P450 enzymes. Although it is synthesized in all tissues, the bone marrow and liver are the organs most actively involved. The porphyrias can be classified according to the primary site of the overproduction and accumulation of heme precursors (liver vs bone marrow). Although there is overlap between the 2 groups, hepatic porphyrias often present with acute neurovisceral symptoms, while the erythropoietic porphyrias often cause cutaneous photosensitivity.1
AIP is the most common hepatic porphyria with a prevalence of 1 in 20,000 in Caucasians of Western European descent.1 AIP is caused by a defect in the gene that encodes porphobilinogen deaminase, leading to the accumulation of porphobilinogen.1 The cardinal manifestation is an acute porphyric attack. While the precise mechanisms underlying the symptoms are unknown, the accumulating metabolites may be directly neurotoxic.2 Attacks are precipitated by factors that induce heme synthesis, including caloric restriction, alcohol, and certain medications, particularly those that upregulate cyP450. The most commonly implicated drugs are anesthetics, antiepileptics, sulfonamides, rifampin, and estrogen and progesterone. Attacks can also be precipitated by changes in endogenous sex hormone levels, like the increase in progesterone seen in the luteal phase of the menstrual cycle, which may account for the higher incidence of symptomatic attacks in women.3
Acute attacks of AIP may have a wide variety of presentations; the disease was referred to as the “little imitator” in the early 20th century.4 The most common symptom is acute, severe abdominal pain, which may mimic an acute abdomen. Because the pain is neuropathic rather than inflammatory, abdominal tenderness, rebound, fever, and leukocytosis are usually absent, as they were in this patient. Abdominal pain is often accompanied by neuropsychiatric symptoms, including sensory and motor neuropathy, anxiety, hallucinations, delirium, and altered level of consciousness. Seizure occurs in 20% of cases. Involvement of the autonomic nervous system causes tachycardia and new onset hypertension in the majority of patients as well as restlessness and tremor. Hyponatremia, mediated by the syndrome of inappropriate ADH secretion, occurs in nearly a third of patients.5,6 MRI findings consistent with PRES have also been described in AIP.7
The diagnosis of AIP is often delayed; diagnosis later in the disease course is associated with a poorer prognosis.8 Reported intervals between presentation and diagnosis range from several months to as long as 20 years.9 Associating the use of medications, caloric restriction, or the menstrual cycle with the exacerbation of symptoms or darkening of urine can help prompt an earlier diagnosis.6
AIP can be diagnosed by detecting a greater than 5-fold elevation of urinary porphobilinogen excretion in conjunction with the typical symptoms of an acute attack.5 Renal dysfunction causes urinary excretion of PBG to fall and serum levels to rise.10 Serum PBG levels should therefore be sent when AIP is suspected in the setting of renal dysfunction. The primary role of genetic testing in a patient who has AIP confirmed clinically and biochemically is to assist in genetic counseling and to identify asymptomatic family members.11 Genetic testing is not required to confirm the diagnosis and does not help prognosticate. It is unusual that a mutation was not detected in this case, as the current sensitivity of genetic testing is 97% to 100%.11
There are 4 principles of management of an acute porphyric attack. First, any precipitating factors such as medications should be stopped. Second, abdominal pain should be treated appropriately with opioids, if necessary. Third, if autonomic dysfunction is present, beta-blockers or clonidine should be given to treat hypertension.5 Finally, glucose and/or hemin should be administered to downregulate aminolevulinic acid (ALA) synthase by negative feedback. Downregulation of ALA synthase decreases the accumulation of the neurotoxic porphyrin precursors ALA and PBG.5 For patients with mild symptoms, glucose alone (300-500 g/d) may be enough to abort the attack.12 This can be achieved via a high-carbohydrate diet in those able to tolerate oral intake or via continuous infusions of dextrose containing fluids.5 For more severe attacks with associated polyneuropathy, respiratory muscle weakness, or seizures, or for attacks that are not resolving, heme preparations dosed at 3 to 4 mg/kg/d for 3 to 4 days are indicated.5
The recent diagnosis of acute Lyme disease was a distractor in this presentation. In Lyme endemic areas, patients with erythema migrans are treated based on the clinical presentation rather than serologic testing.13 Although this patient took only 1 week of doxycycline, testing during this hospitalization showed that she had either been cured early or had not had Lyme disease in the first place. There is no known association between Lyme disease and the porphyrias, and doxycycline is not a common precipitant of AIP attacks.14 However, the GI side effects of doxycycline may have decreased caloric intake and ultimately provoked the patient’s first attack of AIP. The clinicians in this case appropriately avoided the “target” but hit the mark by correctly diagnosing AIP.
KEY POINTS
- Consider AIP in patients with unexplained abdominal pain, especially when accompanied by neuropsychiatric symptoms and autonomic lability.
- Diagnose AIP by sending a urine PBG during a suspected acute attack.
- Treat AIP acutely by removing precipitants, treating abdominal pain, and initiating dextrose-containing fluids and hemin infusions to downregulate ALA synthase.
Acknowledgments
The authors thank the patient who enthusiastically supported the writing of this report.
Disclosure
Warren Gavin, MD has disclosed participation in expert testimony. The authors have no financial or other conflicts of interest to disclose.
1. Desnick RJ, Balwani M. The Porphyrias. In: Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J, eds. Harrison’s Principles of Internal Medicine, 19th Edition. New York: McGraw-Hill; 2015. http://accessmedicine.mhmedical.com.proxy.medlib.uits.iu.edu/content.aspx?bookid=1130&Sectionid=79754263. Accessed June 14, 2016.
2. Bissell DM, Lai JC, Meister RK, Blanc PD. Role of Delta-aminolevulinic Acid in the Symptoms of Acute Porphyria. Am J Med. 2015;128(3):313-317. PubMed
3. Bonkovsky HL, Guo JT, Hou W, Li T, Narang T, Thapar M. Porphyrin and Heme Metabolism and the Porphyrias. Compr Physiol. 2013;3(1):365-401. PubMed
4. Crimlisk HL. The little imitator--porphyria: a neuropsychiatric disorder. J Neurol Neurosurg Psychiatry. 1997;62(4):319-328. PubMed
5. Pischik E, Kauppinen R. An update of clinical management of acute intermittent porphyria. Appl Clin Genet. 2015;8:201-214. PubMed
6. Ventura P, Cappellini MD, Biolcati G, Guida CC, Rocchi E; Gruppo Italiano Porfiria (GrIP). A challenging diagnosis for potential fatal diseases: recommendations for diagnosing acute porphyrias. Eur J Intern Med. 2014;25(6):497-505. PubMed
7. Dagens A, Gilhooley MJ. Acute intermittent porphyria leading to posterior reversible encephalopathy syndrome (PRES): a rare cause of abdominal pain and seizures. BMJ Case Rep. 2016:bcr2016215350. PubMed
8. Pischik E, Bulyanitsa A, Kazakov V, Kauppinen R. Clinical features predictive of a poor prognosis in acute porphyria. J Neurol. 2004;251(12):1538-1541. PubMed
9. Sack GH. Acute intermittent porphyria. JAMA. 1990;264(10):1290-1293. PubMed
10. Sardh E, Andersson DEH, Henrichson A, Harper P. Porphyrin precursors and porphyrins in three patients with acute intermittent porphyria and end-stage renal disease under different therapy regimes. Cell Mol Biol (Noisy-le-grand). 2009;55(1):66-71. PubMed
11. Whatley SD, Badminton MN. Role of genetic testing in the management of patients with inherited porphyria and their families. Ann Clin Biochem. 2013;50(3):204-216. PubMed
12. Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142(6):439-450. PubMed
13. Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43(9):1089-1134. PubMed
14. American Porphyria Foundation. Drug database. http://www.porphyriafoundation.com/drug-database. Accessed July 21, 2017.
A 32-year-old woman presented to the emergency department (ED) with 3 months of abdominal pain and 1 week of vomiting.
The differential diagnosis of abdominal pain is broad. This presentation could be caused by disorders of the gastrointestinal (GI), gynecologic, urinary, or, less likely, the neuromuscular systems. The presence of vomiting supports a GI cause. Pregnancy should be excluded in any woman of childbearing age presenting with abdominal pain.
Characteristics of the pain, including location, temporal characteristics, severity, and aggravating and alleviating factors, can narrow the differential diagnosis. The past medical history, including prior surgeries, menstrual, and obstetric history, is also critical.
Approximately 3 months prior to presentation, she reported a tick bite that had evolved into a circumferential targetoid rash. Her primary care provider performed serologic testing for Lyme disease, which was negative, and prescribed doxycycline, which she stopped after a week because of nausea and diffuse, achy, and constant abdominal pain. After initial improvement, symptoms recurred a week prior to presentation. The nausea was now associated with intractable vomiting and anorexia. She denied hematemesis or coffee ground emesis. Her abdominal pain intensified and radiated to her back. She lost 10 pounds over the past week. She denied headache, constipation, diarrhea, blood per rectum, melena, dysuria, vaginal discharge, or rash. She reported chills and temperatures up to 37.8 ° C at home.
She had a history of migraine headaches for which she took ibuprofen occasionally but took no other prescription or over-the-counter medications. She had never smoked, consumed 2 alcoholic beverages a month, and denied illicit drug use. She lived with her boyfriend on a farm in Indiana where she raised chickens, rabbits, and ducks.
The patient dates the onset of nausea and abdominal pain to a course of doxycycline, presumably prescribed for early Lyme disease, which was stopped after only 1 week. GI side effects, including nausea, vomiting, and upper abdominal pain, are common with doxycycline and may account for the early symptoms. However, these symptoms typically resolve promptly with drug discontinuation. Doxycycline may rarely cause esophageal and gastric ulcers, which could explain her symptoms.
Fewer than half of patients with erythema migrans caused by Lyme disease are seropositive at presentation, as there has been insufficient time for antibodies to develop. Lyme disease typically affects the skin, joints, heart, and nervous system and only rarely affects the GI tract. Acute Lyme disease can cause intestinal pseudoobstruction, splenomegaly, and mild hepatitis. Although Lyme disease is unlikely to be the cause of the current symptoms, serologic testing should be repeated and should be positive if the patient now has early disseminated disease.
Patients with Lyme disease are occasionally coinfected with a second organism. Ixodes scapularis, the tick that transmits Lyme disease in the Northeast and Midwest, can be coinfected with Babesia microti, a red cell parasite. Babesiosis can persist for months and presents with fever, malaise, and many other nonspecific symptoms, including some that this patient has: anorexia, weight loss, abdominal pain, and vomiting.
The history of migraine and intractable vomiting suggests the possibility of cyclic vomiting syndrome. This syndrome is characterized by episodic bouts of vomiting lasting from hours to as long as a week. The vomiting is often accompanied by abdominal pain and occasionally headaches. Episodes are separated by asymptomatic periods that may last months. Cyclic vomiting syndrome can occur at any age but is more common in children, those with a personal or family history of migraines, and heavy users of cannabis. At least 3 stereotypical episodes are required to make the diagnosis, so a history of prior similar symptoms should be explored.
The differential diagnosis of abdominal pain and vomiting should stay broad until a comprehensive physical exam and initial laboratory tests are performed. Volume status should be assessed by estimating jugular venous pressure and by obtaining supine and standing blood pressure measurements. The abdomen should be examined carefully, and the presence or absence of hepatomegaly, splenomegaly, masses, and ascites should be specifically noted. The presence of bradycardia, oligoarticular arthritis, or neuropathy could provide supporting evidence for Lyme disease. Pregnancy is less likely given the diffuse and persistent nature of the pain but should still be excluded.
On physical examination, she was distressed, writhing on the bed, and appearing comfortable only on her side with her knees flexed. Her temperature was 36.5 ° C, heart rate 83 beats per minute, respiratory rate 18 breaths per minute, blood pressure 143/77 mmHg, and oxygen saturation 94% while breathing ambient air. Her abdomen was diffusely tender, most markedly in the epigastrium. Abdominal rigidity, rebound tenderness, and costovertebral tenderness were absent. There was no rash; the previously reported targetoid skin lesion was no longer present. The remainder of the exam was normal.
Laboratory evaluation showed a white count of 7900/mm3, hemoglobin 14.3 gm/dL with normocytic indices, and a platelet count of 175,000/mm3. Sodium was 130 mmol/L, potassium was 3.1 mmol/L, bicarbonate 26 mmol/L, blood urea nitrogen 15 mg/dL, creatinine 0.6 mg/dL, and glucose 92 mg/dL. Serum calcium, aspartate aminotransferase, alanine aminotransferase, bilirubin, and lipase were normal. A urine pregnancy test was negative. Urine analysis was negative for nitrites and leukocyte esterase. Abdominal and pelvic computed tomography (CT) scan with intravenous (IV) contrast performed 3 days prior at an outside ED revealed a 3.4 centimeter left ovarian cyst. A subsequent transvaginal ultrasound was negative for cyst torsion and confirmed appropriate placement of an intrauterine device.
The absence of abdominal rigidity and rebound tenderness does not exclude peritonitis. A normal white blood cell count also does not reliably exclude serious intraabdominal pathology. However, the CT scan argues strongly against many common causes of abdominal pain, including appendicitis, diverticulitis, perforated ulcer, intestinal obstruction, and malignancy, assuming the symptoms have not changed since it was performed.
The patient’s laboratory studies argue against biliary obstruction, pancreatitis, pregnancy, hypercalcemia, and ongoing urinary tract infection. Patients with functional gallbladder disorders may have normal laboratory and CT findings but typically have recurrent, biliary-colic-type pain. The low serum potassium, a high blood urea nitrogen to creatinine ratio, and a low serum sodium reflect her significant vomiting. The hyponatremia is consistent with the appropriate release of antidiuretic hormone (ADH) in the setting of volume depletion. She should receive isotonic fluids plus potassium in addition to symptomatic treatment of pain and nausea. Given the severity and duration of symptoms, an esophagogastroduodenoscopy (EGD) should be performed to exclude GI mucosal disease, including peptic ulcer disease and gastritis, which may not be evident on the CT scan.
Additional diagnoses should be considered at this point. This patient has exposure to chickens, ducks, rabbits, and ticks as well as reported chills and mild temperature elevation at home. Tularemia, which can be transmitted by tick bites or exposure to infected rabbits, can cause a prolonged illness. Some patients have abdominal pain, anorexia, nausea, and weight loss, although fever is usually more prominent. Tularemia is uncommon and most frequently seen in the south-central part of the United States but has been reported throughout the country. She should be queried regarding additional exposures, including well water to assess her risk for Campylobacter infection.
Opiate withdrawal can present with pain and vomiting, but she reports no opiate use and lacks other findings such pupillary dilation or piloerection. Given the prevalence of opiate abuse, however, a toxicology screen should be performed. Hypercalcemia and diabetic ketoacidosis as metabolic causes of abdominal pain have been ruled out by her laboratory values. If no other cause is identified, other metabolic etiologies like Addison disease, familial Mediterranean fever, or porphyria should be considered.
Cyclic vomiting syndrome should still be on the differential. It is a diagnosis of exclusion requiring a history of recurrent, stereotypical episodes, which should be explicitly explored.
The patient was admitted to a medical unit by the hospitalist service and received IV normal saline, parenteral potassium, and IV pantoprazole. She underwent an EGD that revealed minor erosions in the antrum of the stomach. Biopsies were obtained.
Seven hours after the endoscopy, the patient had a brief period of confusion followed by a generalized tonic-clonic seizure lasting 1 minute. A head CT without contrast was negative for any focal abnormality. Repeat laboratory evaluation revealed that serum sodium was 125 mmol/L, and serum glucose was 113 mg/dL. She was transferred to the progressive care unit and received IV levetiracetam.
The endoscopy excluded structural abnormalities of the stomach and duodenum. The patient now has an additional problem, seizure, which needs to be incorporated in the diagnostic reasoning.
Seizures can be caused by the rapid development of severe hyponatremia, with serum sodium levels usually less than 120 mmol/L. Seizures caused by hyponatremia are typically preceded by headache and lethargy, as the intracellular movement of excess water causes cerebral edema. Hyponatremia is unlikely to be the cause of her seizure but should nevertheless be evaluated with a urine sodium concentration and serum and urine osmolality. If she is euvolemic, the IV fluids should be stopped and her free water intake should be restricted to avoid worsening the hyponatremia, as it is potentially caused by the syndrome of inappropriate ADH (SIADH).
There are many other possible causes for new onset seizures in adults, including brain tumor, head trauma, alcohol withdrawal, medications, and central nervous system infection, including Lyme disease. Lyme serologies should be repeated.
In this patient, it is likely that the seizure is a manifestation of the same illness that is causing her vomiting and abdominal pain. Seizure is not a feature of cyclic vomiting syndrome in adults. It is also not a feature of tularemia, adrenal insufficiency, or opioid withdrawal.
Acute intermittent porphyria (AIP) can cause both abdominal and neurologic problems. Hyponatremia is common during acute attacks, caused by either the inappropriate release of ADH or the appropriate release of the hormone if there is fluid loss. AIP is a rare diagnosis but could explain the uncommon combination of abdominal pain, vomiting, seizure, and hyponatremia. A spot urine porphobilinogen test should be sent to assess for AIP.
Additional laboratory studies were sent. Serum osmolality was 269 mosm/kg with a corresponding urine osmolality of 699 mosm/kg. A random urine sodium was 145 mEq/L. Thyroid stimulating hormone and cosyntropin stimulating testing were normal. IgM and IgG antibodies to Borrelia burgdorferi were negative. Urine porphobilinogen was sent. An electroencephalogram did not reveal epileptiform discharges. Magnetic resonance imaging (MRI) of the brain was significant for T2/FLAIR hyperintensity in the cortex and subcortical white matter of the occipital lobes bilaterally. Hypertonic saline and fluid restriction were initiated.
The patient’s labs are consistent with SIADH. Excessive ADH release because of volume depletion and consequent hyponatremia should have improved rapidly with the administration of saline. The high urine sodium suggests that she is now volume replete, while the high urine osmolality is consistent with the presence of excessive ADH in the absence of appropriate stimuli. In the context of normal thyroid and adrenal function, the hyponatremia is likely due to the SIADH.
Negative serologic testing for Lyme disease, 3 months after the onset of rash, excludes this diagnosis.
The MRI findings are consistent with posterior reversible encephalopathy syndrome (PRES), a clinicoradiographic syndrome of headache, altered mental status, seizure, and/or vision loss with associated white matter abnormalities of the posterior cerebral hemispheres. PRES has been reported with AIP as well as other disorders, most commonly hypertensive encephalopathy, eclampsia, and immunosuppressive drug use.
The patient’s sodium improved with fluid restriction and the administration of hypertonic saline. There was no recurrence of seizure activity. Amlodipine was initiated for blood pressure readings as high as 156/106 mmHg. A hepatobiliary scan revealed a gallbladder ejection fraction of 13%. Biopsies from her endoscopy revealed nonspecific inflammation without the presence of Helicobacter pylori. The patient was discharged home 7 days after admission after stabilization of serum sodium, improvement in her abdominal pain, and tolerance of oral intake. A plan was made for outpatient cholecystectomy.
Many causes of abdominal pain have been excluded and the remaining diagnostic possibility, porphyria, is rare. The clinicians have revisited their differential and considered other causes of abdominal pain, including functional gallbladder disorders. However, chronic cholecystitis (or functional gallbladder disorder) is not this patient’s primary problem. The diffuse, severe, and constant abdominal pain prior to admission is not typical of biliary pain, and many medical conditions and drugs, including amlodipine, can lead to a positive hepatobiliary scan. Chronic cholecystitis would not explain her seizure.
AIP remains at the top of the differential for this young woman. A urine porphobilinogen has been sent and must be followed up prior to any further workup or surgery.
One week after discharge, the patient’s urine porphobilinogen resulted at 172.8 mCmol/ (upper limits of normal 8.8). Sequencing analysis for genes coding the enzymes involved in the synthetic pathway for heme were sent. Hydroxymethylbilane synthase, coproporphyrinogen oxidase, and protoporphyrinogen oxidase mutation assays were all normal. Despite the normal genetic assays, the diagnosis of AIP was made on the basis of the clinical presentation and elevated urine porphobilinogen. The patient was referred to a hematologist and initiated on oral glucose supplements and hematin infusions.
DISCUSSION
Although abdominal pain has a broad differential, the combination of abdominal pain and neurologic or psychiatric symptoms should suggest the possibility of porphyria, especially if symptoms are recurrent or unexplained. The porphyrias are a group of disorders caused by defects in the synthetic pathway of heme, leading to an overproduction and accumulation of precursors. Heme is a component of multiple proteins, including hemoglobin, myoglobin, and the cytochrome P450 enzymes. Although it is synthesized in all tissues, the bone marrow and liver are the organs most actively involved. The porphyrias can be classified according to the primary site of the overproduction and accumulation of heme precursors (liver vs bone marrow). Although there is overlap between the 2 groups, hepatic porphyrias often present with acute neurovisceral symptoms, while the erythropoietic porphyrias often cause cutaneous photosensitivity.1
AIP is the most common hepatic porphyria with a prevalence of 1 in 20,000 in Caucasians of Western European descent.1 AIP is caused by a defect in the gene that encodes porphobilinogen deaminase, leading to the accumulation of porphobilinogen.1 The cardinal manifestation is an acute porphyric attack. While the precise mechanisms underlying the symptoms are unknown, the accumulating metabolites may be directly neurotoxic.2 Attacks are precipitated by factors that induce heme synthesis, including caloric restriction, alcohol, and certain medications, particularly those that upregulate cyP450. The most commonly implicated drugs are anesthetics, antiepileptics, sulfonamides, rifampin, and estrogen and progesterone. Attacks can also be precipitated by changes in endogenous sex hormone levels, like the increase in progesterone seen in the luteal phase of the menstrual cycle, which may account for the higher incidence of symptomatic attacks in women.3
Acute attacks of AIP may have a wide variety of presentations; the disease was referred to as the “little imitator” in the early 20th century.4 The most common symptom is acute, severe abdominal pain, which may mimic an acute abdomen. Because the pain is neuropathic rather than inflammatory, abdominal tenderness, rebound, fever, and leukocytosis are usually absent, as they were in this patient. Abdominal pain is often accompanied by neuropsychiatric symptoms, including sensory and motor neuropathy, anxiety, hallucinations, delirium, and altered level of consciousness. Seizure occurs in 20% of cases. Involvement of the autonomic nervous system causes tachycardia and new onset hypertension in the majority of patients as well as restlessness and tremor. Hyponatremia, mediated by the syndrome of inappropriate ADH secretion, occurs in nearly a third of patients.5,6 MRI findings consistent with PRES have also been described in AIP.7
The diagnosis of AIP is often delayed; diagnosis later in the disease course is associated with a poorer prognosis.8 Reported intervals between presentation and diagnosis range from several months to as long as 20 years.9 Associating the use of medications, caloric restriction, or the menstrual cycle with the exacerbation of symptoms or darkening of urine can help prompt an earlier diagnosis.6
AIP can be diagnosed by detecting a greater than 5-fold elevation of urinary porphobilinogen excretion in conjunction with the typical symptoms of an acute attack.5 Renal dysfunction causes urinary excretion of PBG to fall and serum levels to rise.10 Serum PBG levels should therefore be sent when AIP is suspected in the setting of renal dysfunction. The primary role of genetic testing in a patient who has AIP confirmed clinically and biochemically is to assist in genetic counseling and to identify asymptomatic family members.11 Genetic testing is not required to confirm the diagnosis and does not help prognosticate. It is unusual that a mutation was not detected in this case, as the current sensitivity of genetic testing is 97% to 100%.11
There are 4 principles of management of an acute porphyric attack. First, any precipitating factors such as medications should be stopped. Second, abdominal pain should be treated appropriately with opioids, if necessary. Third, if autonomic dysfunction is present, beta-blockers or clonidine should be given to treat hypertension.5 Finally, glucose and/or hemin should be administered to downregulate aminolevulinic acid (ALA) synthase by negative feedback. Downregulation of ALA synthase decreases the accumulation of the neurotoxic porphyrin precursors ALA and PBG.5 For patients with mild symptoms, glucose alone (300-500 g/d) may be enough to abort the attack.12 This can be achieved via a high-carbohydrate diet in those able to tolerate oral intake or via continuous infusions of dextrose containing fluids.5 For more severe attacks with associated polyneuropathy, respiratory muscle weakness, or seizures, or for attacks that are not resolving, heme preparations dosed at 3 to 4 mg/kg/d for 3 to 4 days are indicated.5
The recent diagnosis of acute Lyme disease was a distractor in this presentation. In Lyme endemic areas, patients with erythema migrans are treated based on the clinical presentation rather than serologic testing.13 Although this patient took only 1 week of doxycycline, testing during this hospitalization showed that she had either been cured early or had not had Lyme disease in the first place. There is no known association between Lyme disease and the porphyrias, and doxycycline is not a common precipitant of AIP attacks.14 However, the GI side effects of doxycycline may have decreased caloric intake and ultimately provoked the patient’s first attack of AIP. The clinicians in this case appropriately avoided the “target” but hit the mark by correctly diagnosing AIP.
KEY POINTS
- Consider AIP in patients with unexplained abdominal pain, especially when accompanied by neuropsychiatric symptoms and autonomic lability.
- Diagnose AIP by sending a urine PBG during a suspected acute attack.
- Treat AIP acutely by removing precipitants, treating abdominal pain, and initiating dextrose-containing fluids and hemin infusions to downregulate ALA synthase.
Acknowledgments
The authors thank the patient who enthusiastically supported the writing of this report.
Disclosure
Warren Gavin, MD has disclosed participation in expert testimony. The authors have no financial or other conflicts of interest to disclose.
A 32-year-old woman presented to the emergency department (ED) with 3 months of abdominal pain and 1 week of vomiting.
The differential diagnosis of abdominal pain is broad. This presentation could be caused by disorders of the gastrointestinal (GI), gynecologic, urinary, or, less likely, the neuromuscular systems. The presence of vomiting supports a GI cause. Pregnancy should be excluded in any woman of childbearing age presenting with abdominal pain.
Characteristics of the pain, including location, temporal characteristics, severity, and aggravating and alleviating factors, can narrow the differential diagnosis. The past medical history, including prior surgeries, menstrual, and obstetric history, is also critical.
Approximately 3 months prior to presentation, she reported a tick bite that had evolved into a circumferential targetoid rash. Her primary care provider performed serologic testing for Lyme disease, which was negative, and prescribed doxycycline, which she stopped after a week because of nausea and diffuse, achy, and constant abdominal pain. After initial improvement, symptoms recurred a week prior to presentation. The nausea was now associated with intractable vomiting and anorexia. She denied hematemesis or coffee ground emesis. Her abdominal pain intensified and radiated to her back. She lost 10 pounds over the past week. She denied headache, constipation, diarrhea, blood per rectum, melena, dysuria, vaginal discharge, or rash. She reported chills and temperatures up to 37.8 ° C at home.
She had a history of migraine headaches for which she took ibuprofen occasionally but took no other prescription or over-the-counter medications. She had never smoked, consumed 2 alcoholic beverages a month, and denied illicit drug use. She lived with her boyfriend on a farm in Indiana where she raised chickens, rabbits, and ducks.
The patient dates the onset of nausea and abdominal pain to a course of doxycycline, presumably prescribed for early Lyme disease, which was stopped after only 1 week. GI side effects, including nausea, vomiting, and upper abdominal pain, are common with doxycycline and may account for the early symptoms. However, these symptoms typically resolve promptly with drug discontinuation. Doxycycline may rarely cause esophageal and gastric ulcers, which could explain her symptoms.
Fewer than half of patients with erythema migrans caused by Lyme disease are seropositive at presentation, as there has been insufficient time for antibodies to develop. Lyme disease typically affects the skin, joints, heart, and nervous system and only rarely affects the GI tract. Acute Lyme disease can cause intestinal pseudoobstruction, splenomegaly, and mild hepatitis. Although Lyme disease is unlikely to be the cause of the current symptoms, serologic testing should be repeated and should be positive if the patient now has early disseminated disease.
Patients with Lyme disease are occasionally coinfected with a second organism. Ixodes scapularis, the tick that transmits Lyme disease in the Northeast and Midwest, can be coinfected with Babesia microti, a red cell parasite. Babesiosis can persist for months and presents with fever, malaise, and many other nonspecific symptoms, including some that this patient has: anorexia, weight loss, abdominal pain, and vomiting.
The history of migraine and intractable vomiting suggests the possibility of cyclic vomiting syndrome. This syndrome is characterized by episodic bouts of vomiting lasting from hours to as long as a week. The vomiting is often accompanied by abdominal pain and occasionally headaches. Episodes are separated by asymptomatic periods that may last months. Cyclic vomiting syndrome can occur at any age but is more common in children, those with a personal or family history of migraines, and heavy users of cannabis. At least 3 stereotypical episodes are required to make the diagnosis, so a history of prior similar symptoms should be explored.
The differential diagnosis of abdominal pain and vomiting should stay broad until a comprehensive physical exam and initial laboratory tests are performed. Volume status should be assessed by estimating jugular venous pressure and by obtaining supine and standing blood pressure measurements. The abdomen should be examined carefully, and the presence or absence of hepatomegaly, splenomegaly, masses, and ascites should be specifically noted. The presence of bradycardia, oligoarticular arthritis, or neuropathy could provide supporting evidence for Lyme disease. Pregnancy is less likely given the diffuse and persistent nature of the pain but should still be excluded.
On physical examination, she was distressed, writhing on the bed, and appearing comfortable only on her side with her knees flexed. Her temperature was 36.5 ° C, heart rate 83 beats per minute, respiratory rate 18 breaths per minute, blood pressure 143/77 mmHg, and oxygen saturation 94% while breathing ambient air. Her abdomen was diffusely tender, most markedly in the epigastrium. Abdominal rigidity, rebound tenderness, and costovertebral tenderness were absent. There was no rash; the previously reported targetoid skin lesion was no longer present. The remainder of the exam was normal.
Laboratory evaluation showed a white count of 7900/mm3, hemoglobin 14.3 gm/dL with normocytic indices, and a platelet count of 175,000/mm3. Sodium was 130 mmol/L, potassium was 3.1 mmol/L, bicarbonate 26 mmol/L, blood urea nitrogen 15 mg/dL, creatinine 0.6 mg/dL, and glucose 92 mg/dL. Serum calcium, aspartate aminotransferase, alanine aminotransferase, bilirubin, and lipase were normal. A urine pregnancy test was negative. Urine analysis was negative for nitrites and leukocyte esterase. Abdominal and pelvic computed tomography (CT) scan with intravenous (IV) contrast performed 3 days prior at an outside ED revealed a 3.4 centimeter left ovarian cyst. A subsequent transvaginal ultrasound was negative for cyst torsion and confirmed appropriate placement of an intrauterine device.
The absence of abdominal rigidity and rebound tenderness does not exclude peritonitis. A normal white blood cell count also does not reliably exclude serious intraabdominal pathology. However, the CT scan argues strongly against many common causes of abdominal pain, including appendicitis, diverticulitis, perforated ulcer, intestinal obstruction, and malignancy, assuming the symptoms have not changed since it was performed.
The patient’s laboratory studies argue against biliary obstruction, pancreatitis, pregnancy, hypercalcemia, and ongoing urinary tract infection. Patients with functional gallbladder disorders may have normal laboratory and CT findings but typically have recurrent, biliary-colic-type pain. The low serum potassium, a high blood urea nitrogen to creatinine ratio, and a low serum sodium reflect her significant vomiting. The hyponatremia is consistent with the appropriate release of antidiuretic hormone (ADH) in the setting of volume depletion. She should receive isotonic fluids plus potassium in addition to symptomatic treatment of pain and nausea. Given the severity and duration of symptoms, an esophagogastroduodenoscopy (EGD) should be performed to exclude GI mucosal disease, including peptic ulcer disease and gastritis, which may not be evident on the CT scan.
Additional diagnoses should be considered at this point. This patient has exposure to chickens, ducks, rabbits, and ticks as well as reported chills and mild temperature elevation at home. Tularemia, which can be transmitted by tick bites or exposure to infected rabbits, can cause a prolonged illness. Some patients have abdominal pain, anorexia, nausea, and weight loss, although fever is usually more prominent. Tularemia is uncommon and most frequently seen in the south-central part of the United States but has been reported throughout the country. She should be queried regarding additional exposures, including well water to assess her risk for Campylobacter infection.
Opiate withdrawal can present with pain and vomiting, but she reports no opiate use and lacks other findings such pupillary dilation or piloerection. Given the prevalence of opiate abuse, however, a toxicology screen should be performed. Hypercalcemia and diabetic ketoacidosis as metabolic causes of abdominal pain have been ruled out by her laboratory values. If no other cause is identified, other metabolic etiologies like Addison disease, familial Mediterranean fever, or porphyria should be considered.
Cyclic vomiting syndrome should still be on the differential. It is a diagnosis of exclusion requiring a history of recurrent, stereotypical episodes, which should be explicitly explored.
The patient was admitted to a medical unit by the hospitalist service and received IV normal saline, parenteral potassium, and IV pantoprazole. She underwent an EGD that revealed minor erosions in the antrum of the stomach. Biopsies were obtained.
Seven hours after the endoscopy, the patient had a brief period of confusion followed by a generalized tonic-clonic seizure lasting 1 minute. A head CT without contrast was negative for any focal abnormality. Repeat laboratory evaluation revealed that serum sodium was 125 mmol/L, and serum glucose was 113 mg/dL. She was transferred to the progressive care unit and received IV levetiracetam.
The endoscopy excluded structural abnormalities of the stomach and duodenum. The patient now has an additional problem, seizure, which needs to be incorporated in the diagnostic reasoning.
Seizures can be caused by the rapid development of severe hyponatremia, with serum sodium levels usually less than 120 mmol/L. Seizures caused by hyponatremia are typically preceded by headache and lethargy, as the intracellular movement of excess water causes cerebral edema. Hyponatremia is unlikely to be the cause of her seizure but should nevertheless be evaluated with a urine sodium concentration and serum and urine osmolality. If she is euvolemic, the IV fluids should be stopped and her free water intake should be restricted to avoid worsening the hyponatremia, as it is potentially caused by the syndrome of inappropriate ADH (SIADH).
There are many other possible causes for new onset seizures in adults, including brain tumor, head trauma, alcohol withdrawal, medications, and central nervous system infection, including Lyme disease. Lyme serologies should be repeated.
In this patient, it is likely that the seizure is a manifestation of the same illness that is causing her vomiting and abdominal pain. Seizure is not a feature of cyclic vomiting syndrome in adults. It is also not a feature of tularemia, adrenal insufficiency, or opioid withdrawal.
Acute intermittent porphyria (AIP) can cause both abdominal and neurologic problems. Hyponatremia is common during acute attacks, caused by either the inappropriate release of ADH or the appropriate release of the hormone if there is fluid loss. AIP is a rare diagnosis but could explain the uncommon combination of abdominal pain, vomiting, seizure, and hyponatremia. A spot urine porphobilinogen test should be sent to assess for AIP.
Additional laboratory studies were sent. Serum osmolality was 269 mosm/kg with a corresponding urine osmolality of 699 mosm/kg. A random urine sodium was 145 mEq/L. Thyroid stimulating hormone and cosyntropin stimulating testing were normal. IgM and IgG antibodies to Borrelia burgdorferi were negative. Urine porphobilinogen was sent. An electroencephalogram did not reveal epileptiform discharges. Magnetic resonance imaging (MRI) of the brain was significant for T2/FLAIR hyperintensity in the cortex and subcortical white matter of the occipital lobes bilaterally. Hypertonic saline and fluid restriction were initiated.
The patient’s labs are consistent with SIADH. Excessive ADH release because of volume depletion and consequent hyponatremia should have improved rapidly with the administration of saline. The high urine sodium suggests that she is now volume replete, while the high urine osmolality is consistent with the presence of excessive ADH in the absence of appropriate stimuli. In the context of normal thyroid and adrenal function, the hyponatremia is likely due to the SIADH.
Negative serologic testing for Lyme disease, 3 months after the onset of rash, excludes this diagnosis.
The MRI findings are consistent with posterior reversible encephalopathy syndrome (PRES), a clinicoradiographic syndrome of headache, altered mental status, seizure, and/or vision loss with associated white matter abnormalities of the posterior cerebral hemispheres. PRES has been reported with AIP as well as other disorders, most commonly hypertensive encephalopathy, eclampsia, and immunosuppressive drug use.
The patient’s sodium improved with fluid restriction and the administration of hypertonic saline. There was no recurrence of seizure activity. Amlodipine was initiated for blood pressure readings as high as 156/106 mmHg. A hepatobiliary scan revealed a gallbladder ejection fraction of 13%. Biopsies from her endoscopy revealed nonspecific inflammation without the presence of Helicobacter pylori. The patient was discharged home 7 days after admission after stabilization of serum sodium, improvement in her abdominal pain, and tolerance of oral intake. A plan was made for outpatient cholecystectomy.
Many causes of abdominal pain have been excluded and the remaining diagnostic possibility, porphyria, is rare. The clinicians have revisited their differential and considered other causes of abdominal pain, including functional gallbladder disorders. However, chronic cholecystitis (or functional gallbladder disorder) is not this patient’s primary problem. The diffuse, severe, and constant abdominal pain prior to admission is not typical of biliary pain, and many medical conditions and drugs, including amlodipine, can lead to a positive hepatobiliary scan. Chronic cholecystitis would not explain her seizure.
AIP remains at the top of the differential for this young woman. A urine porphobilinogen has been sent and must be followed up prior to any further workup or surgery.
One week after discharge, the patient’s urine porphobilinogen resulted at 172.8 mCmol/ (upper limits of normal 8.8). Sequencing analysis for genes coding the enzymes involved in the synthetic pathway for heme were sent. Hydroxymethylbilane synthase, coproporphyrinogen oxidase, and protoporphyrinogen oxidase mutation assays were all normal. Despite the normal genetic assays, the diagnosis of AIP was made on the basis of the clinical presentation and elevated urine porphobilinogen. The patient was referred to a hematologist and initiated on oral glucose supplements and hematin infusions.
DISCUSSION
Although abdominal pain has a broad differential, the combination of abdominal pain and neurologic or psychiatric symptoms should suggest the possibility of porphyria, especially if symptoms are recurrent or unexplained. The porphyrias are a group of disorders caused by defects in the synthetic pathway of heme, leading to an overproduction and accumulation of precursors. Heme is a component of multiple proteins, including hemoglobin, myoglobin, and the cytochrome P450 enzymes. Although it is synthesized in all tissues, the bone marrow and liver are the organs most actively involved. The porphyrias can be classified according to the primary site of the overproduction and accumulation of heme precursors (liver vs bone marrow). Although there is overlap between the 2 groups, hepatic porphyrias often present with acute neurovisceral symptoms, while the erythropoietic porphyrias often cause cutaneous photosensitivity.1
AIP is the most common hepatic porphyria with a prevalence of 1 in 20,000 in Caucasians of Western European descent.1 AIP is caused by a defect in the gene that encodes porphobilinogen deaminase, leading to the accumulation of porphobilinogen.1 The cardinal manifestation is an acute porphyric attack. While the precise mechanisms underlying the symptoms are unknown, the accumulating metabolites may be directly neurotoxic.2 Attacks are precipitated by factors that induce heme synthesis, including caloric restriction, alcohol, and certain medications, particularly those that upregulate cyP450. The most commonly implicated drugs are anesthetics, antiepileptics, sulfonamides, rifampin, and estrogen and progesterone. Attacks can also be precipitated by changes in endogenous sex hormone levels, like the increase in progesterone seen in the luteal phase of the menstrual cycle, which may account for the higher incidence of symptomatic attacks in women.3
Acute attacks of AIP may have a wide variety of presentations; the disease was referred to as the “little imitator” in the early 20th century.4 The most common symptom is acute, severe abdominal pain, which may mimic an acute abdomen. Because the pain is neuropathic rather than inflammatory, abdominal tenderness, rebound, fever, and leukocytosis are usually absent, as they were in this patient. Abdominal pain is often accompanied by neuropsychiatric symptoms, including sensory and motor neuropathy, anxiety, hallucinations, delirium, and altered level of consciousness. Seizure occurs in 20% of cases. Involvement of the autonomic nervous system causes tachycardia and new onset hypertension in the majority of patients as well as restlessness and tremor. Hyponatremia, mediated by the syndrome of inappropriate ADH secretion, occurs in nearly a third of patients.5,6 MRI findings consistent with PRES have also been described in AIP.7
The diagnosis of AIP is often delayed; diagnosis later in the disease course is associated with a poorer prognosis.8 Reported intervals between presentation and diagnosis range from several months to as long as 20 years.9 Associating the use of medications, caloric restriction, or the menstrual cycle with the exacerbation of symptoms or darkening of urine can help prompt an earlier diagnosis.6
AIP can be diagnosed by detecting a greater than 5-fold elevation of urinary porphobilinogen excretion in conjunction with the typical symptoms of an acute attack.5 Renal dysfunction causes urinary excretion of PBG to fall and serum levels to rise.10 Serum PBG levels should therefore be sent when AIP is suspected in the setting of renal dysfunction. The primary role of genetic testing in a patient who has AIP confirmed clinically and biochemically is to assist in genetic counseling and to identify asymptomatic family members.11 Genetic testing is not required to confirm the diagnosis and does not help prognosticate. It is unusual that a mutation was not detected in this case, as the current sensitivity of genetic testing is 97% to 100%.11
There are 4 principles of management of an acute porphyric attack. First, any precipitating factors such as medications should be stopped. Second, abdominal pain should be treated appropriately with opioids, if necessary. Third, if autonomic dysfunction is present, beta-blockers or clonidine should be given to treat hypertension.5 Finally, glucose and/or hemin should be administered to downregulate aminolevulinic acid (ALA) synthase by negative feedback. Downregulation of ALA synthase decreases the accumulation of the neurotoxic porphyrin precursors ALA and PBG.5 For patients with mild symptoms, glucose alone (300-500 g/d) may be enough to abort the attack.12 This can be achieved via a high-carbohydrate diet in those able to tolerate oral intake or via continuous infusions of dextrose containing fluids.5 For more severe attacks with associated polyneuropathy, respiratory muscle weakness, or seizures, or for attacks that are not resolving, heme preparations dosed at 3 to 4 mg/kg/d for 3 to 4 days are indicated.5
The recent diagnosis of acute Lyme disease was a distractor in this presentation. In Lyme endemic areas, patients with erythema migrans are treated based on the clinical presentation rather than serologic testing.13 Although this patient took only 1 week of doxycycline, testing during this hospitalization showed that she had either been cured early or had not had Lyme disease in the first place. There is no known association between Lyme disease and the porphyrias, and doxycycline is not a common precipitant of AIP attacks.14 However, the GI side effects of doxycycline may have decreased caloric intake and ultimately provoked the patient’s first attack of AIP. The clinicians in this case appropriately avoided the “target” but hit the mark by correctly diagnosing AIP.
KEY POINTS
- Consider AIP in patients with unexplained abdominal pain, especially when accompanied by neuropsychiatric symptoms and autonomic lability.
- Diagnose AIP by sending a urine PBG during a suspected acute attack.
- Treat AIP acutely by removing precipitants, treating abdominal pain, and initiating dextrose-containing fluids and hemin infusions to downregulate ALA synthase.
Acknowledgments
The authors thank the patient who enthusiastically supported the writing of this report.
Disclosure
Warren Gavin, MD has disclosed participation in expert testimony. The authors have no financial or other conflicts of interest to disclose.
1. Desnick RJ, Balwani M. The Porphyrias. In: Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J, eds. Harrison’s Principles of Internal Medicine, 19th Edition. New York: McGraw-Hill; 2015. http://accessmedicine.mhmedical.com.proxy.medlib.uits.iu.edu/content.aspx?bookid=1130&Sectionid=79754263. Accessed June 14, 2016.
2. Bissell DM, Lai JC, Meister RK, Blanc PD. Role of Delta-aminolevulinic Acid in the Symptoms of Acute Porphyria. Am J Med. 2015;128(3):313-317. PubMed
3. Bonkovsky HL, Guo JT, Hou W, Li T, Narang T, Thapar M. Porphyrin and Heme Metabolism and the Porphyrias. Compr Physiol. 2013;3(1):365-401. PubMed
4. Crimlisk HL. The little imitator--porphyria: a neuropsychiatric disorder. J Neurol Neurosurg Psychiatry. 1997;62(4):319-328. PubMed
5. Pischik E, Kauppinen R. An update of clinical management of acute intermittent porphyria. Appl Clin Genet. 2015;8:201-214. PubMed
6. Ventura P, Cappellini MD, Biolcati G, Guida CC, Rocchi E; Gruppo Italiano Porfiria (GrIP). A challenging diagnosis for potential fatal diseases: recommendations for diagnosing acute porphyrias. Eur J Intern Med. 2014;25(6):497-505. PubMed
7. Dagens A, Gilhooley MJ. Acute intermittent porphyria leading to posterior reversible encephalopathy syndrome (PRES): a rare cause of abdominal pain and seizures. BMJ Case Rep. 2016:bcr2016215350. PubMed
8. Pischik E, Bulyanitsa A, Kazakov V, Kauppinen R. Clinical features predictive of a poor prognosis in acute porphyria. J Neurol. 2004;251(12):1538-1541. PubMed
9. Sack GH. Acute intermittent porphyria. JAMA. 1990;264(10):1290-1293. PubMed
10. Sardh E, Andersson DEH, Henrichson A, Harper P. Porphyrin precursors and porphyrins in three patients with acute intermittent porphyria and end-stage renal disease under different therapy regimes. Cell Mol Biol (Noisy-le-grand). 2009;55(1):66-71. PubMed
11. Whatley SD, Badminton MN. Role of genetic testing in the management of patients with inherited porphyria and their families. Ann Clin Biochem. 2013;50(3):204-216. PubMed
12. Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142(6):439-450. PubMed
13. Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43(9):1089-1134. PubMed
14. American Porphyria Foundation. Drug database. http://www.porphyriafoundation.com/drug-database. Accessed July 21, 2017.
1. Desnick RJ, Balwani M. The Porphyrias. In: Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J, eds. Harrison’s Principles of Internal Medicine, 19th Edition. New York: McGraw-Hill; 2015. http://accessmedicine.mhmedical.com.proxy.medlib.uits.iu.edu/content.aspx?bookid=1130&Sectionid=79754263. Accessed June 14, 2016.
2. Bissell DM, Lai JC, Meister RK, Blanc PD. Role of Delta-aminolevulinic Acid in the Symptoms of Acute Porphyria. Am J Med. 2015;128(3):313-317. PubMed
3. Bonkovsky HL, Guo JT, Hou W, Li T, Narang T, Thapar M. Porphyrin and Heme Metabolism and the Porphyrias. Compr Physiol. 2013;3(1):365-401. PubMed
4. Crimlisk HL. The little imitator--porphyria: a neuropsychiatric disorder. J Neurol Neurosurg Psychiatry. 1997;62(4):319-328. PubMed
5. Pischik E, Kauppinen R. An update of clinical management of acute intermittent porphyria. Appl Clin Genet. 2015;8:201-214. PubMed
6. Ventura P, Cappellini MD, Biolcati G, Guida CC, Rocchi E; Gruppo Italiano Porfiria (GrIP). A challenging diagnosis for potential fatal diseases: recommendations for diagnosing acute porphyrias. Eur J Intern Med. 2014;25(6):497-505. PubMed
7. Dagens A, Gilhooley MJ. Acute intermittent porphyria leading to posterior reversible encephalopathy syndrome (PRES): a rare cause of abdominal pain and seizures. BMJ Case Rep. 2016:bcr2016215350. PubMed
8. Pischik E, Bulyanitsa A, Kazakov V, Kauppinen R. Clinical features predictive of a poor prognosis in acute porphyria. J Neurol. 2004;251(12):1538-1541. PubMed
9. Sack GH. Acute intermittent porphyria. JAMA. 1990;264(10):1290-1293. PubMed
10. Sardh E, Andersson DEH, Henrichson A, Harper P. Porphyrin precursors and porphyrins in three patients with acute intermittent porphyria and end-stage renal disease under different therapy regimes. Cell Mol Biol (Noisy-le-grand). 2009;55(1):66-71. PubMed
11. Whatley SD, Badminton MN. Role of genetic testing in the management of patients with inherited porphyria and their families. Ann Clin Biochem. 2013;50(3):204-216. PubMed
12. Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142(6):439-450. PubMed
13. Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43(9):1089-1134. PubMed
14. American Porphyria Foundation. Drug database. http://www.porphyriafoundation.com/drug-database. Accessed July 21, 2017.
© 2018 Society of Hospital Medicine
A Strong Diagnosis of Weakness
A 52-year-old man presented with bilateral weakness in all extremities. He noted the gradual onset of progressive muscle weakness 6 months prior to presentation. He reported generalized fatigue and difficulty with climbing stairs and carrying heavy objects.
Initial considerations of chronic weakness and fatigue are myopathy, polyneuropathy, medications, malignancy, endocrinopathies, human immunodeficiency virus (HIV), neuromuscular junction dysfunction, and central nervous system (CNS) disorders, such as amyotrophic lateral sclerosis (ALS) or multiple sclerosis (MS). Symmetrical muscle involvement and proximal weakness make myopathy most likely. Polyneuropathy, such as chronic inflammatory demyelinating polyneuropathy (CIDP), is less likely but still possible given the slowly progressive course. The use of medications that can cause myopathy should be explored, including colchicine, steroids, and statins. Gathering further history should focus on risk factors for HIV, as well as alcohol and illicit drug use. Malignancy can cause paraneoplastic myopathy. The review of systems should include symptoms of endocrinopathies, such as thyrotoxicosis and hypothyroidism. Fluctuations in weakness and dysphagia or ocular symptoms would suggest myasthenia gravis (MG). The time course and symmetrical weakness make a central disorder, such as ALS or MS, unlikely.
His past medical history was notable for pulmonary tuberculosis diagnosed at the age of 6 years, which was treated with hospitalization and an unknown medication regimen. He was not taking medications prior to this admission. His family history was significant for diabetes mellitus in both parents. He denied sick contacts. He was sexually active with his wife. He denied the use of tobacco and illicit drugs but endorsed alcohol consumption on a daily basis over the last 32 years. He reported no fluctuation in his symptoms, muscle or joint pains, rash, fevers, chills, diaphoresis, chest pain, dyspnea, abdominal pain, diarrhea, paresthesias, weight loss, or night sweats. He had never had a colonoscopy.
Painless progressive weakness of the limbs without sensory deficit is typical of a myopathy. Though CIDP can present with only motor weakness, the majority of patients have sensory symptoms, making this less likely. Although chronic alcohol abuse can cause myopathy, it seems less likely because other neurologic complications, such as sensory polyneuropathy or ataxia, would be expected. A review of systems does not suggest a thyroid disorder or malignancy, although this does not preclude an evaluation for both. The absence of fluctuations in weakness argues against MG. Though ALS, MG, MS, and CIDP are less likely, a neurologic exam is crucial in excluding them. The hallmark of ALS is upper motor neuron (UMN) and lower motor neuron signs in the absence of sensory symptoms and signs, while global hyporeflexia would be expected in CIDP, and fatigability on repeated power testing would be expected in MG. Neurologic findings disseminated in space (neuro-anatomically) would be expected in MS.
On physical examination, the patient had a temperature of 36.9°C, heart rate of 70 beats per minute, and regular respiratory rate of 10 breaths per minute, blood pressure 130/80 mmHg, and oxygen saturation 98% while breathing ambient air. Auscultation of the heart and lungs revealed normal findings. The abdomen was soft, nontender, and without masses or organomegaly. Neurologic examination disclosed bilateral symmetric upper and lower extremity weakness with positive Gower sign. Muscle strength scores of the bilateral biceps brachii, iliopsoas, and digitis extensor were between 4 and 5 without fatigability. Grasping power was impaired. Deep tendon reflexes were preserved, and there were no UMN signs. There was no tenderness to palpation in any muscle groups. Sensory testing was normal. Skin and lymph examinations were without abnormality. The rest of the physical examination was unremarkable.
Gower sign, characteristic of but not specific to muscular dystrophy, indicates proximal muscle weakness of lower extremities, wherein hands and arms are used to walk up the body into an upright position. The exam also reveals distal weakness as shown by reduced hand grasp. Symmetrical proximal weakness of all extremities without sensory deficits suggests a myopathic process, albeit one with some distal involvement. The absence of UMN signs argues against ALS, lack of fatigability argues against MG, and the absence of CNS or sensory deficits argues against MS.
Because myopathy is most likely, the next step would be to determine if this is an idiopathic inflammatory myopathy, such as polymyositis (PM) or dermatomyositis (DM), secondary inflammatory myopathy, or noninflammatory myopathy due to endocrinopathies. The time course is consistent with an inflammatory myopathy, such as PM or DM. Inclusion body myositis (IBM), another inflammatory myopathy, presents much more insidiously over years and tends to be asymmetric compared to PM. The absence of myalgia, arthralgia, rash, and gastrointestinal symptoms makes myopathy as a component of a connective tissue disease, such as systemic lupus erythematosus, or a mixed connective tissue disease unlikely. The next steps would be laboratory testing of muscle enzymes, complete blood count, biochemical profile, and antinuclear antibody (ANA).
Laboratory studies revealed a white blood cell count of 4460/mm3 with normal differential, hemoglobin 12.5 g/dL, and platelet count 345,000/mm3. Creatinine was 0.87 mg/dL, aspartate aminotransferase 61 IU/mL, alanine aminotransferase 45 IU/mL, and creatine kinase (CK) 529 U/L (normal range, 38-174 U/L). Other liver function enzymes were normal. Biochemistry studies disclosed normal sodium, potassium, glucose, calcium, and magnesium levels. Dipstick urinalysis revealed blood and protein, and the microscopic examination of urinary sediment was unremarkable without the presence of erythrocytes. Twenty-four-hour creatinine clearance was 106 mL/min (normal range, 97-137 mL/min). Chest radiography was unrevealing.
The modest increase in CK, evidence of myoglobinuria, and proteinuria can all occur with an inflammatory or metabolic myopathy. The combination of proximal and distal weakness, coupled with only a modestly elevated CK, makes IBM more likely than PM, as PM usually presents with proximal weakness and much higher CK values. Normal skin examination makes DM less likely, as skin manifestations are generally found at time of presentation. The onset of symptoms after age 50 and the patient being male also favor IBM, though a longer time course would be expected. Definitively distinguishing IBM from PM is important because treatment and prognosis differ.
Thyroid function and HIV testing should be obtained. ANA, more common in PM than in IBM, should be checked because these myopathies can be associated with other autoimmune diseases. Imaging is generally not essential, although magnetic resonance imaging (MRI) of the thighs may help to differentiate IBM from PM. Electromyography (EMG) should be done to determine the pattern of myopathy and select muscle biopsy sites.
Additional testing revealed a normal thyroid stimulating hormone level. HIV and ANA were negative. Serum aldolase level was 19 IU/L (normal range, 2.7-5.9 IU/L), myoglobin 277 ng/mL (normal range, 28-72 ng/mL), lactate dehydrogenase 416 IU/mL (normal range, 119-229 IU/mL), and C-reactive protein 0.32 mg/dL. An EMG revealed mild myogenic changes in all extremities. An MRI of the left brachial muscle revealed multiple scattered high-signal lesions.
The EMG and MRI findings are consistent with an inflammatory myopathy. The modest elevation in muscle enzymes and negative ANA are more consistent with IBM since most patients with PM or DM are ANA positive. Muscle biopsy can be very helpful in establishing the etiology of myopathy.
Malignancy is associated with DM and PM in about 9% and 4% of patients, respectively. The common cancers associated with these conditions are adenocarcinomas of the ovary, cervix, lung, pancreas, and stomach. Most cancers are diagnosed around the time of myositis diagnosis, although they can precede or follow by years. Idiopathic IBM is not associated with cancer.
Open surgical muscle biopsy of the left biceps brachii was performed. Light microscopic examination disclosed interstitial edema and noncaseating granulomas. Immunostaining revealed an increase in the number of cluster of differentiation (CD) 4+ T cells. Caseating granulomas and Langhans giant cells were not present (Figure 3).

Tuberculin reaction and interferon-γ-release assay were negative. Staining for AFB and fungi was negative. ANCA, rheumatoid factor (RF), anti-Ro/SSA, anti-La/SSB, anti-Sm, anti-RNP, and anti-Jo-1 were all negative or unremarkable. Serum angiotensin converting enzyme (ACE) level was 155.6 U/L (normal range, 7-25 U/L). Twenty-four-hour urine analysis revealed calcium excretion of 517.7 mg/day (normal range, 58-450 mg/day), β2-microglobulin 69,627 ug/day (normal range, <254 ug/day), and N-acetyl-D-glucosamine 95.3 U/day (normal range, <5.1 U/day) with a normal creatinine clearance. Serum intact parathyroid hormone level (PTH) was 5 pg/mL (normal range, 10-65 pg/mL), and 25-hydroxyvitamin D level was 51.1 ng/mL (normal range, 30-80 ng/mL). A CT of the thorax revealed a small ground-glass density lesion in the left lower lobe but no hilar or mediastinal lymphadenopathy.
Negative ANCA, RF, and autoantibodies exclude systemic vasculitis and connective tissue disease as causes of GM. Hypercalciuria is suggestive of granulomatous production of calcitriol, which, in turn, suppresses PTH. Hypercalcemia is not common in patients with sarcoidosis, but hypercalciuria occurs frequently. Serum ACE is a marker associated with sarcoidosis, but its diagnostic and prognostic utility is unclear.
Though there is a concern for sarcoidosis, this diagnosis can only be confidently made by finding noncaseating granulomas on a background of compatible clinical and radiologic findings after alternate possible etiologies are excluded. The chest CT reveals a small ground-glass density lesion without hilar adenopathy. These findings, though not incompatible, are not typical for pulmonary sarcoidosis. Therefore, finding noncaeseating granulomas in a second organ system would point toward systemic sarcoidosis as a unifying diagnosis. Bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial biopsy has a reasonable yield even in the absence of hilar adenopathy or typical parenchymal findings. A CD4/CD8 T-cell ratio of 2 or more on BAL provides supportive evidence for sarcoidosis.
It is reasonable to start empiric glucocorticoids for GM given that the AFB and fungal stains on histopathology are negative and that there is no evidence of lymphoma.
The CD4/CD8 T-cell ratio greater than 2, combined with the absence of neutrophils and eosinophils on BAL, is helpful in distinguishing sarcoidosis from other pulmonary diseases. This patient’s inflammatory myopathy was revealed to be a rare initial manifestation of systemic sarcoidosis.
DISCUSSION
Weakness is a common symptom of muscle disorders such as myopathies and muscular dystrophy. Idiopathic inflammatory myopathies include PM, DM, and others.1,2 These usually present with proximal-dominant muscle weakness, decreased endurance, and muscle inflammation. A diagnosis is made according to symptoms in combination with diagnostic examinations, including elevated serum CK levels, abnormal EMG findings, and histopathology of skeletal muscle biopsy specimens.
Sarcoidosis, a multisystem disorder of unknown etiology, is characterized histopathologically by noncaseating granulomas in affected organs.3 It typically affects young adults, with incidence peaking at 20 to 39 years of age. Although any organ may be involved, the disorder usually presents with 1 or more common abnormalities, including bilateral hilar lymphadenopathy, lung lesions, and skin and eye involvement. Musculoskeletal involvement is less common. It is estimated that skeletal muscle is involved in 50% to 80% of patients with sarcoidosis but is rarely symptomatic (0.5% to 2.5%).4-6
In this patient, weakness was distributed in both proximal and distal muscles, yet proximal weakness is the most characteristic feature in PM and DM. Therefore, sarcoidosis should be considered in the differential diagnosis of idiopathic inflammatory myopathies, especially when weakness accompanies abnormalities in other organs typically affected by sarcoidosis.
Myoglobinuria often is observed in rhabdomyolysis and inflammatory myopathies, conditions that produce high levels of serum CK and myoglobin. Myoglobinuria, often accompanied by the elevation of urinary β2-microglobulin and N-acetyl-D-glucosamine levels, can induce tubulointerstitial damage, which leads to acute kidney injury. In this case, however, these abnormal kidney findings were observed without high levels of serum CK or myoglobin. This suggests the potential for other causes of tubulointerstitial damage, such as granulomatous interstitial nephritis in renal sarcoidosis.3
Another characteristic abnormality was the elevation of urinary calcium excretion, which indicated an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis. In sarcoidosis, hypercalciuria occurs in 40% of patients, hypercalcemia in 11%, and renal calculi in 10%.3,7 Hypercalciuria, for this patient, was important in arriving at the correct diagnosis after the gallium scan was obtained given the dearth of other typical features of sarcoidosis.
Although muscle biopsy is essential, imaging studies for idiopathic inflammatory myopathy are considered useful tools to narrow the differential diagnosis. The use of MRI of the skeletal muscle is helpful to both identify an adequate muscle for biopsy and demonstrate the pattern of affected muscles beyond clinical appearance, which aids in excluding, for example, muscular dystrophies.8,9
FDG PET/CT is a very sensitive imaging modality used to detect neoplastic lesions and has been widely used to screen for occult neoplasms and detect metastases.10-12 It is also useful for detecting inflammation in patients with osteomyelitis, metastatic infectious diseases, rheumatoid arthritis, vasculitis, inflammatory bowel diseases, fever of unknown origin, and sarcoidosis.11,12 In PM and DM, however, the sensitivity of FDG PET/CT for detection of myositis is reportedly lower than that of EMG and MRI.13 Similarly, gallium scintigraphy is usually performed to examine the disease activity of interstitial pneumonia or to detect malignancy. Previous literature and this case show that the striking images of gallium scintigraphy and FDG PET/CT have utility, not only for detection of sarcoid myopathy but also for the evaluation of treatment efficacy.14-17 Characteristic imaging findings on FDG PET/CT have been described as a “tiger man” appearance.17
For the treatment of sarcoid myopathy, systemic glucocorticoids are used for patients with symptomatic acute or chronic forms. The standard doses of prednisolone used for other forms of idiopathic inflammatory myopathies are usually administered.3-6 In general, the response of acute sarcoid myopathy to glucocorticoid therapy is favorable, and the clinical course is usually benign. However, the course in chronic sarcoid myopathy can be unpredictable with exacerbations. Given the lack of randomized trials of this therapy and because glucocorticoids themselves can cause steroid-induced myopathy, they are not used for asymptomatic patients.
In the end, astute clinical thinking, deductive reasoning, and pattern recognition were all instrumental in making this strong diagnosis of weakness.
KEY TEACHING POINTS
- Proximal muscle–dominant weakness is the characteristic feature in inflammatory myopathies like PM and DM. Myopathy causing proximal and distal weakness is more characteristic of sarcoidosis, IBM, alcohol, and statins.
- Elevations of urinary Times New Romanβ2-microglobulin and N-acetyl-D-glucosamine are often observed in inflammatory muscle diseases because of myoglobin-induced tubulointerstitial damage. These findings may also be caused by other conditions that affect the tubules, such as lupus nephritis, Sjogren’s syndrome, or renal sarcoidosis.
- Hypercalciuria in a patient with myopathy could suggest an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis.
- The striking uptake within systemic skeletal striated muscles on gallium scintigraphy and “tiger man” appearance on FDG PET/CT are characteristic features of acute sarcoid myopathy; these are not common in other inflammatory myopathies.
Disclosure
Drs. Sudo, Wada, Narita, Mba, and Houchens have no conflicts of interest to disclose.
1. Vincze M, Danko K. Idiopathic inflammatory myopathies. Best Pract Res Clin Rheumatol. 2012;26:25-45. PubMed
2. Carstens PO, Schmidt J. Diagnosis, pathogenesis, and treatment of myositis: recent advances. Clin Exp Immunol. 2014;175:425-438. PubMed
3. Lannuzzi MC, Rhbicki BA, Teirstein AS. Sarcoidosis. N Eng J Med. 2007;357:2153-2165. PubMed
4. Baydur A, Pandya K, Sharma OP, et al. Control of ventilation, respiratory muscle strength, and granulomatous involvement of skeletal muscle in patients with sarcoidosis. Chest. 1993;103:396-402. PubMed
5. Zisman DA, Biermann JS, Martinez FJ, et al. Sarcoidosis presenting as a tumorlike muscular lesion. Case report and review of the literature. Medicine (Baltimore). 1999;78:112-122. PubMed
6. Fayad F, Liote F, Berenbaum F, et al. Muscle involvement in sarcoidosis: a retrospective and followup studies. J Rheumatol. 2006;33:98-103. PubMed
7. Berliner AR, Haas M, Choi MJ. Sarcoidosis: the nephrologist’s perspective. Am J Kidney Dis. 2006;48:856-870. PubMed
8. Otake S, Ishigaki T. Musular sarcoidosis. Semin Musculoskelet Radiol. 2001;5:167-170. PubMed
9. Otake S, Imagumbai N, Suzuki M, et al. MR imaging of muscular sarcoidosis after steroid therapy. Eur Radiol. 1998;8:1651-1653. PubMed
10. Hoffman JM, Gambhir SS. Molecular imaging: The vision and opportunity for radiology in the future. Radiology. 2007;244:39-47. PubMed
11. Basu S, Zhuang H, Torigian DA, et al. Functional imaging of inflammatory diseases using nuclear medicine techniques. Semin Nucl Med. 2009;39:124-145. PubMed
12. Gotthardt M, Cleeker-Rovers CP, Boerman OC, et al. Imaging of inflammation by PET, conventional scintigraphy, and other imaging techniques. J Nucl Med. 2010;51:1937-1949. PubMed
13. Owada T, Maezawa R, Kurasawa K, et al. Detection of inflammatory lesions by F-18 fluorodeoxyglucose positron emission tomography in patients with polymyositis and dermatomyositis. J Rheumatol. 2012;39:1659-1665. PubMed
14. Liem IH, Drent M, Antevska E, et al. Intense muscle uptake of gallium-67 in a patient with sarcoidosis. J Nucl Med. 1998;39:1605-1607. PubMed
15. Suehiro S, Shiokawa S, Taniguchi S, et al. Gallium-67 scintigraphy in the diagnosis and management of chronic sarcoid myopathy. Clin Rheumatol. 2003;22:146-148. PubMed
16. Marie I, Josse S, Lahaxe L, et al. Clinical images: muscle sarcoidosis demonstrated on positron emission tomography. Arthritis Rheum. 2009;60:2847. PubMed
17. Wieers G, Lhommel R, Lecouvet F, et al. A tiger man. Lancet. 2012;380:1859. PubMed
A 52-year-old man presented with bilateral weakness in all extremities. He noted the gradual onset of progressive muscle weakness 6 months prior to presentation. He reported generalized fatigue and difficulty with climbing stairs and carrying heavy objects.
Initial considerations of chronic weakness and fatigue are myopathy, polyneuropathy, medications, malignancy, endocrinopathies, human immunodeficiency virus (HIV), neuromuscular junction dysfunction, and central nervous system (CNS) disorders, such as amyotrophic lateral sclerosis (ALS) or multiple sclerosis (MS). Symmetrical muscle involvement and proximal weakness make myopathy most likely. Polyneuropathy, such as chronic inflammatory demyelinating polyneuropathy (CIDP), is less likely but still possible given the slowly progressive course. The use of medications that can cause myopathy should be explored, including colchicine, steroids, and statins. Gathering further history should focus on risk factors for HIV, as well as alcohol and illicit drug use. Malignancy can cause paraneoplastic myopathy. The review of systems should include symptoms of endocrinopathies, such as thyrotoxicosis and hypothyroidism. Fluctuations in weakness and dysphagia or ocular symptoms would suggest myasthenia gravis (MG). The time course and symmetrical weakness make a central disorder, such as ALS or MS, unlikely.
His past medical history was notable for pulmonary tuberculosis diagnosed at the age of 6 years, which was treated with hospitalization and an unknown medication regimen. He was not taking medications prior to this admission. His family history was significant for diabetes mellitus in both parents. He denied sick contacts. He was sexually active with his wife. He denied the use of tobacco and illicit drugs but endorsed alcohol consumption on a daily basis over the last 32 years. He reported no fluctuation in his symptoms, muscle or joint pains, rash, fevers, chills, diaphoresis, chest pain, dyspnea, abdominal pain, diarrhea, paresthesias, weight loss, or night sweats. He had never had a colonoscopy.
Painless progressive weakness of the limbs without sensory deficit is typical of a myopathy. Though CIDP can present with only motor weakness, the majority of patients have sensory symptoms, making this less likely. Although chronic alcohol abuse can cause myopathy, it seems less likely because other neurologic complications, such as sensory polyneuropathy or ataxia, would be expected. A review of systems does not suggest a thyroid disorder or malignancy, although this does not preclude an evaluation for both. The absence of fluctuations in weakness argues against MG. Though ALS, MG, MS, and CIDP are less likely, a neurologic exam is crucial in excluding them. The hallmark of ALS is upper motor neuron (UMN) and lower motor neuron signs in the absence of sensory symptoms and signs, while global hyporeflexia would be expected in CIDP, and fatigability on repeated power testing would be expected in MG. Neurologic findings disseminated in space (neuro-anatomically) would be expected in MS.
On physical examination, the patient had a temperature of 36.9°C, heart rate of 70 beats per minute, and regular respiratory rate of 10 breaths per minute, blood pressure 130/80 mmHg, and oxygen saturation 98% while breathing ambient air. Auscultation of the heart and lungs revealed normal findings. The abdomen was soft, nontender, and without masses or organomegaly. Neurologic examination disclosed bilateral symmetric upper and lower extremity weakness with positive Gower sign. Muscle strength scores of the bilateral biceps brachii, iliopsoas, and digitis extensor were between 4 and 5 without fatigability. Grasping power was impaired. Deep tendon reflexes were preserved, and there were no UMN signs. There was no tenderness to palpation in any muscle groups. Sensory testing was normal. Skin and lymph examinations were without abnormality. The rest of the physical examination was unremarkable.
Gower sign, characteristic of but not specific to muscular dystrophy, indicates proximal muscle weakness of lower extremities, wherein hands and arms are used to walk up the body into an upright position. The exam also reveals distal weakness as shown by reduced hand grasp. Symmetrical proximal weakness of all extremities without sensory deficits suggests a myopathic process, albeit one with some distal involvement. The absence of UMN signs argues against ALS, lack of fatigability argues against MG, and the absence of CNS or sensory deficits argues against MS.
Because myopathy is most likely, the next step would be to determine if this is an idiopathic inflammatory myopathy, such as polymyositis (PM) or dermatomyositis (DM), secondary inflammatory myopathy, or noninflammatory myopathy due to endocrinopathies. The time course is consistent with an inflammatory myopathy, such as PM or DM. Inclusion body myositis (IBM), another inflammatory myopathy, presents much more insidiously over years and tends to be asymmetric compared to PM. The absence of myalgia, arthralgia, rash, and gastrointestinal symptoms makes myopathy as a component of a connective tissue disease, such as systemic lupus erythematosus, or a mixed connective tissue disease unlikely. The next steps would be laboratory testing of muscle enzymes, complete blood count, biochemical profile, and antinuclear antibody (ANA).
Laboratory studies revealed a white blood cell count of 4460/mm3 with normal differential, hemoglobin 12.5 g/dL, and platelet count 345,000/mm3. Creatinine was 0.87 mg/dL, aspartate aminotransferase 61 IU/mL, alanine aminotransferase 45 IU/mL, and creatine kinase (CK) 529 U/L (normal range, 38-174 U/L). Other liver function enzymes were normal. Biochemistry studies disclosed normal sodium, potassium, glucose, calcium, and magnesium levels. Dipstick urinalysis revealed blood and protein, and the microscopic examination of urinary sediment was unremarkable without the presence of erythrocytes. Twenty-four-hour creatinine clearance was 106 mL/min (normal range, 97-137 mL/min). Chest radiography was unrevealing.
The modest increase in CK, evidence of myoglobinuria, and proteinuria can all occur with an inflammatory or metabolic myopathy. The combination of proximal and distal weakness, coupled with only a modestly elevated CK, makes IBM more likely than PM, as PM usually presents with proximal weakness and much higher CK values. Normal skin examination makes DM less likely, as skin manifestations are generally found at time of presentation. The onset of symptoms after age 50 and the patient being male also favor IBM, though a longer time course would be expected. Definitively distinguishing IBM from PM is important because treatment and prognosis differ.
Thyroid function and HIV testing should be obtained. ANA, more common in PM than in IBM, should be checked because these myopathies can be associated with other autoimmune diseases. Imaging is generally not essential, although magnetic resonance imaging (MRI) of the thighs may help to differentiate IBM from PM. Electromyography (EMG) should be done to determine the pattern of myopathy and select muscle biopsy sites.
Additional testing revealed a normal thyroid stimulating hormone level. HIV and ANA were negative. Serum aldolase level was 19 IU/L (normal range, 2.7-5.9 IU/L), myoglobin 277 ng/mL (normal range, 28-72 ng/mL), lactate dehydrogenase 416 IU/mL (normal range, 119-229 IU/mL), and C-reactive protein 0.32 mg/dL. An EMG revealed mild myogenic changes in all extremities. An MRI of the left brachial muscle revealed multiple scattered high-signal lesions.
The EMG and MRI findings are consistent with an inflammatory myopathy. The modest elevation in muscle enzymes and negative ANA are more consistent with IBM since most patients with PM or DM are ANA positive. Muscle biopsy can be very helpful in establishing the etiology of myopathy.
Malignancy is associated with DM and PM in about 9% and 4% of patients, respectively. The common cancers associated with these conditions are adenocarcinomas of the ovary, cervix, lung, pancreas, and stomach. Most cancers are diagnosed around the time of myositis diagnosis, although they can precede or follow by years. Idiopathic IBM is not associated with cancer.
Open surgical muscle biopsy of the left biceps brachii was performed. Light microscopic examination disclosed interstitial edema and noncaseating granulomas. Immunostaining revealed an increase in the number of cluster of differentiation (CD) 4+ T cells. Caseating granulomas and Langhans giant cells were not present (Figure 3).
Tuberculin reaction and interferon-γ-release assay were negative. Staining for AFB and fungi was negative. ANCA, rheumatoid factor (RF), anti-Ro/SSA, anti-La/SSB, anti-Sm, anti-RNP, and anti-Jo-1 were all negative or unremarkable. Serum angiotensin converting enzyme (ACE) level was 155.6 U/L (normal range, 7-25 U/L). Twenty-four-hour urine analysis revealed calcium excretion of 517.7 mg/day (normal range, 58-450 mg/day), β2-microglobulin 69,627 ug/day (normal range, <254 ug/day), and N-acetyl-D-glucosamine 95.3 U/day (normal range, <5.1 U/day) with a normal creatinine clearance. Serum intact parathyroid hormone level (PTH) was 5 pg/mL (normal range, 10-65 pg/mL), and 25-hydroxyvitamin D level was 51.1 ng/mL (normal range, 30-80 ng/mL). A CT of the thorax revealed a small ground-glass density lesion in the left lower lobe but no hilar or mediastinal lymphadenopathy.
Negative ANCA, RF, and autoantibodies exclude systemic vasculitis and connective tissue disease as causes of GM. Hypercalciuria is suggestive of granulomatous production of calcitriol, which, in turn, suppresses PTH. Hypercalcemia is not common in patients with sarcoidosis, but hypercalciuria occurs frequently. Serum ACE is a marker associated with sarcoidosis, but its diagnostic and prognostic utility is unclear.
Though there is a concern for sarcoidosis, this diagnosis can only be confidently made by finding noncaseating granulomas on a background of compatible clinical and radiologic findings after alternate possible etiologies are excluded. The chest CT reveals a small ground-glass density lesion without hilar adenopathy. These findings, though not incompatible, are not typical for pulmonary sarcoidosis. Therefore, finding noncaeseating granulomas in a second organ system would point toward systemic sarcoidosis as a unifying diagnosis. Bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial biopsy has a reasonable yield even in the absence of hilar adenopathy or typical parenchymal findings. A CD4/CD8 T-cell ratio of 2 or more on BAL provides supportive evidence for sarcoidosis.
It is reasonable to start empiric glucocorticoids for GM given that the AFB and fungal stains on histopathology are negative and that there is no evidence of lymphoma.
The CD4/CD8 T-cell ratio greater than 2, combined with the absence of neutrophils and eosinophils on BAL, is helpful in distinguishing sarcoidosis from other pulmonary diseases. This patient’s inflammatory myopathy was revealed to be a rare initial manifestation of systemic sarcoidosis.
DISCUSSION
Weakness is a common symptom of muscle disorders such as myopathies and muscular dystrophy. Idiopathic inflammatory myopathies include PM, DM, and others.1,2 These usually present with proximal-dominant muscle weakness, decreased endurance, and muscle inflammation. A diagnosis is made according to symptoms in combination with diagnostic examinations, including elevated serum CK levels, abnormal EMG findings, and histopathology of skeletal muscle biopsy specimens.
Sarcoidosis, a multisystem disorder of unknown etiology, is characterized histopathologically by noncaseating granulomas in affected organs.3 It typically affects young adults, with incidence peaking at 20 to 39 years of age. Although any organ may be involved, the disorder usually presents with 1 or more common abnormalities, including bilateral hilar lymphadenopathy, lung lesions, and skin and eye involvement. Musculoskeletal involvement is less common. It is estimated that skeletal muscle is involved in 50% to 80% of patients with sarcoidosis but is rarely symptomatic (0.5% to 2.5%).4-6
In this patient, weakness was distributed in both proximal and distal muscles, yet proximal weakness is the most characteristic feature in PM and DM. Therefore, sarcoidosis should be considered in the differential diagnosis of idiopathic inflammatory myopathies, especially when weakness accompanies abnormalities in other organs typically affected by sarcoidosis.
Myoglobinuria often is observed in rhabdomyolysis and inflammatory myopathies, conditions that produce high levels of serum CK and myoglobin. Myoglobinuria, often accompanied by the elevation of urinary β2-microglobulin and N-acetyl-D-glucosamine levels, can induce tubulointerstitial damage, which leads to acute kidney injury. In this case, however, these abnormal kidney findings were observed without high levels of serum CK or myoglobin. This suggests the potential for other causes of tubulointerstitial damage, such as granulomatous interstitial nephritis in renal sarcoidosis.3
Another characteristic abnormality was the elevation of urinary calcium excretion, which indicated an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis. In sarcoidosis, hypercalciuria occurs in 40% of patients, hypercalcemia in 11%, and renal calculi in 10%.3,7 Hypercalciuria, for this patient, was important in arriving at the correct diagnosis after the gallium scan was obtained given the dearth of other typical features of sarcoidosis.
Although muscle biopsy is essential, imaging studies for idiopathic inflammatory myopathy are considered useful tools to narrow the differential diagnosis. The use of MRI of the skeletal muscle is helpful to both identify an adequate muscle for biopsy and demonstrate the pattern of affected muscles beyond clinical appearance, which aids in excluding, for example, muscular dystrophies.8,9
FDG PET/CT is a very sensitive imaging modality used to detect neoplastic lesions and has been widely used to screen for occult neoplasms and detect metastases.10-12 It is also useful for detecting inflammation in patients with osteomyelitis, metastatic infectious diseases, rheumatoid arthritis, vasculitis, inflammatory bowel diseases, fever of unknown origin, and sarcoidosis.11,12 In PM and DM, however, the sensitivity of FDG PET/CT for detection of myositis is reportedly lower than that of EMG and MRI.13 Similarly, gallium scintigraphy is usually performed to examine the disease activity of interstitial pneumonia or to detect malignancy. Previous literature and this case show that the striking images of gallium scintigraphy and FDG PET/CT have utility, not only for detection of sarcoid myopathy but also for the evaluation of treatment efficacy.14-17 Characteristic imaging findings on FDG PET/CT have been described as a “tiger man” appearance.17
For the treatment of sarcoid myopathy, systemic glucocorticoids are used for patients with symptomatic acute or chronic forms. The standard doses of prednisolone used for other forms of idiopathic inflammatory myopathies are usually administered.3-6 In general, the response of acute sarcoid myopathy to glucocorticoid therapy is favorable, and the clinical course is usually benign. However, the course in chronic sarcoid myopathy can be unpredictable with exacerbations. Given the lack of randomized trials of this therapy and because glucocorticoids themselves can cause steroid-induced myopathy, they are not used for asymptomatic patients.
In the end, astute clinical thinking, deductive reasoning, and pattern recognition were all instrumental in making this strong diagnosis of weakness.
KEY TEACHING POINTS
- Proximal muscle–dominant weakness is the characteristic feature in inflammatory myopathies like PM and DM. Myopathy causing proximal and distal weakness is more characteristic of sarcoidosis, IBM, alcohol, and statins.
- Elevations of urinary Times New Romanβ2-microglobulin and N-acetyl-D-glucosamine are often observed in inflammatory muscle diseases because of myoglobin-induced tubulointerstitial damage. These findings may also be caused by other conditions that affect the tubules, such as lupus nephritis, Sjogren’s syndrome, or renal sarcoidosis.
- Hypercalciuria in a patient with myopathy could suggest an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis.
- The striking uptake within systemic skeletal striated muscles on gallium scintigraphy and “tiger man” appearance on FDG PET/CT are characteristic features of acute sarcoid myopathy; these are not common in other inflammatory myopathies.
Disclosure
Drs. Sudo, Wada, Narita, Mba, and Houchens have no conflicts of interest to disclose.
A 52-year-old man presented with bilateral weakness in all extremities. He noted the gradual onset of progressive muscle weakness 6 months prior to presentation. He reported generalized fatigue and difficulty with climbing stairs and carrying heavy objects.
Initial considerations of chronic weakness and fatigue are myopathy, polyneuropathy, medications, malignancy, endocrinopathies, human immunodeficiency virus (HIV), neuromuscular junction dysfunction, and central nervous system (CNS) disorders, such as amyotrophic lateral sclerosis (ALS) or multiple sclerosis (MS). Symmetrical muscle involvement and proximal weakness make myopathy most likely. Polyneuropathy, such as chronic inflammatory demyelinating polyneuropathy (CIDP), is less likely but still possible given the slowly progressive course. The use of medications that can cause myopathy should be explored, including colchicine, steroids, and statins. Gathering further history should focus on risk factors for HIV, as well as alcohol and illicit drug use. Malignancy can cause paraneoplastic myopathy. The review of systems should include symptoms of endocrinopathies, such as thyrotoxicosis and hypothyroidism. Fluctuations in weakness and dysphagia or ocular symptoms would suggest myasthenia gravis (MG). The time course and symmetrical weakness make a central disorder, such as ALS or MS, unlikely.
His past medical history was notable for pulmonary tuberculosis diagnosed at the age of 6 years, which was treated with hospitalization and an unknown medication regimen. He was not taking medications prior to this admission. His family history was significant for diabetes mellitus in both parents. He denied sick contacts. He was sexually active with his wife. He denied the use of tobacco and illicit drugs but endorsed alcohol consumption on a daily basis over the last 32 years. He reported no fluctuation in his symptoms, muscle or joint pains, rash, fevers, chills, diaphoresis, chest pain, dyspnea, abdominal pain, diarrhea, paresthesias, weight loss, or night sweats. He had never had a colonoscopy.
Painless progressive weakness of the limbs without sensory deficit is typical of a myopathy. Though CIDP can present with only motor weakness, the majority of patients have sensory symptoms, making this less likely. Although chronic alcohol abuse can cause myopathy, it seems less likely because other neurologic complications, such as sensory polyneuropathy or ataxia, would be expected. A review of systems does not suggest a thyroid disorder or malignancy, although this does not preclude an evaluation for both. The absence of fluctuations in weakness argues against MG. Though ALS, MG, MS, and CIDP are less likely, a neurologic exam is crucial in excluding them. The hallmark of ALS is upper motor neuron (UMN) and lower motor neuron signs in the absence of sensory symptoms and signs, while global hyporeflexia would be expected in CIDP, and fatigability on repeated power testing would be expected in MG. Neurologic findings disseminated in space (neuro-anatomically) would be expected in MS.
On physical examination, the patient had a temperature of 36.9°C, heart rate of 70 beats per minute, and regular respiratory rate of 10 breaths per minute, blood pressure 130/80 mmHg, and oxygen saturation 98% while breathing ambient air. Auscultation of the heart and lungs revealed normal findings. The abdomen was soft, nontender, and without masses or organomegaly. Neurologic examination disclosed bilateral symmetric upper and lower extremity weakness with positive Gower sign. Muscle strength scores of the bilateral biceps brachii, iliopsoas, and digitis extensor were between 4 and 5 without fatigability. Grasping power was impaired. Deep tendon reflexes were preserved, and there were no UMN signs. There was no tenderness to palpation in any muscle groups. Sensory testing was normal. Skin and lymph examinations were without abnormality. The rest of the physical examination was unremarkable.
Gower sign, characteristic of but not specific to muscular dystrophy, indicates proximal muscle weakness of lower extremities, wherein hands and arms are used to walk up the body into an upright position. The exam also reveals distal weakness as shown by reduced hand grasp. Symmetrical proximal weakness of all extremities without sensory deficits suggests a myopathic process, albeit one with some distal involvement. The absence of UMN signs argues against ALS, lack of fatigability argues against MG, and the absence of CNS or sensory deficits argues against MS.
Because myopathy is most likely, the next step would be to determine if this is an idiopathic inflammatory myopathy, such as polymyositis (PM) or dermatomyositis (DM), secondary inflammatory myopathy, or noninflammatory myopathy due to endocrinopathies. The time course is consistent with an inflammatory myopathy, such as PM or DM. Inclusion body myositis (IBM), another inflammatory myopathy, presents much more insidiously over years and tends to be asymmetric compared to PM. The absence of myalgia, arthralgia, rash, and gastrointestinal symptoms makes myopathy as a component of a connective tissue disease, such as systemic lupus erythematosus, or a mixed connective tissue disease unlikely. The next steps would be laboratory testing of muscle enzymes, complete blood count, biochemical profile, and antinuclear antibody (ANA).
Laboratory studies revealed a white blood cell count of 4460/mm3 with normal differential, hemoglobin 12.5 g/dL, and platelet count 345,000/mm3. Creatinine was 0.87 mg/dL, aspartate aminotransferase 61 IU/mL, alanine aminotransferase 45 IU/mL, and creatine kinase (CK) 529 U/L (normal range, 38-174 U/L). Other liver function enzymes were normal. Biochemistry studies disclosed normal sodium, potassium, glucose, calcium, and magnesium levels. Dipstick urinalysis revealed blood and protein, and the microscopic examination of urinary sediment was unremarkable without the presence of erythrocytes. Twenty-four-hour creatinine clearance was 106 mL/min (normal range, 97-137 mL/min). Chest radiography was unrevealing.
The modest increase in CK, evidence of myoglobinuria, and proteinuria can all occur with an inflammatory or metabolic myopathy. The combination of proximal and distal weakness, coupled with only a modestly elevated CK, makes IBM more likely than PM, as PM usually presents with proximal weakness and much higher CK values. Normal skin examination makes DM less likely, as skin manifestations are generally found at time of presentation. The onset of symptoms after age 50 and the patient being male also favor IBM, though a longer time course would be expected. Definitively distinguishing IBM from PM is important because treatment and prognosis differ.
Thyroid function and HIV testing should be obtained. ANA, more common in PM than in IBM, should be checked because these myopathies can be associated with other autoimmune diseases. Imaging is generally not essential, although magnetic resonance imaging (MRI) of the thighs may help to differentiate IBM from PM. Electromyography (EMG) should be done to determine the pattern of myopathy and select muscle biopsy sites.
Additional testing revealed a normal thyroid stimulating hormone level. HIV and ANA were negative. Serum aldolase level was 19 IU/L (normal range, 2.7-5.9 IU/L), myoglobin 277 ng/mL (normal range, 28-72 ng/mL), lactate dehydrogenase 416 IU/mL (normal range, 119-229 IU/mL), and C-reactive protein 0.32 mg/dL. An EMG revealed mild myogenic changes in all extremities. An MRI of the left brachial muscle revealed multiple scattered high-signal lesions.
The EMG and MRI findings are consistent with an inflammatory myopathy. The modest elevation in muscle enzymes and negative ANA are more consistent with IBM since most patients with PM or DM are ANA positive. Muscle biopsy can be very helpful in establishing the etiology of myopathy.
Malignancy is associated with DM and PM in about 9% and 4% of patients, respectively. The common cancers associated with these conditions are adenocarcinomas of the ovary, cervix, lung, pancreas, and stomach. Most cancers are diagnosed around the time of myositis diagnosis, although they can precede or follow by years. Idiopathic IBM is not associated with cancer.
Open surgical muscle biopsy of the left biceps brachii was performed. Light microscopic examination disclosed interstitial edema and noncaseating granulomas. Immunostaining revealed an increase in the number of cluster of differentiation (CD) 4+ T cells. Caseating granulomas and Langhans giant cells were not present (Figure 3).
Tuberculin reaction and interferon-γ-release assay were negative. Staining for AFB and fungi was negative. ANCA, rheumatoid factor (RF), anti-Ro/SSA, anti-La/SSB, anti-Sm, anti-RNP, and anti-Jo-1 were all negative or unremarkable. Serum angiotensin converting enzyme (ACE) level was 155.6 U/L (normal range, 7-25 U/L). Twenty-four-hour urine analysis revealed calcium excretion of 517.7 mg/day (normal range, 58-450 mg/day), β2-microglobulin 69,627 ug/day (normal range, <254 ug/day), and N-acetyl-D-glucosamine 95.3 U/day (normal range, <5.1 U/day) with a normal creatinine clearance. Serum intact parathyroid hormone level (PTH) was 5 pg/mL (normal range, 10-65 pg/mL), and 25-hydroxyvitamin D level was 51.1 ng/mL (normal range, 30-80 ng/mL). A CT of the thorax revealed a small ground-glass density lesion in the left lower lobe but no hilar or mediastinal lymphadenopathy.
Negative ANCA, RF, and autoantibodies exclude systemic vasculitis and connective tissue disease as causes of GM. Hypercalciuria is suggestive of granulomatous production of calcitriol, which, in turn, suppresses PTH. Hypercalcemia is not common in patients with sarcoidosis, but hypercalciuria occurs frequently. Serum ACE is a marker associated with sarcoidosis, but its diagnostic and prognostic utility is unclear.
Though there is a concern for sarcoidosis, this diagnosis can only be confidently made by finding noncaseating granulomas on a background of compatible clinical and radiologic findings after alternate possible etiologies are excluded. The chest CT reveals a small ground-glass density lesion without hilar adenopathy. These findings, though not incompatible, are not typical for pulmonary sarcoidosis. Therefore, finding noncaeseating granulomas in a second organ system would point toward systemic sarcoidosis as a unifying diagnosis. Bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial biopsy has a reasonable yield even in the absence of hilar adenopathy or typical parenchymal findings. A CD4/CD8 T-cell ratio of 2 or more on BAL provides supportive evidence for sarcoidosis.
It is reasonable to start empiric glucocorticoids for GM given that the AFB and fungal stains on histopathology are negative and that there is no evidence of lymphoma.
The CD4/CD8 T-cell ratio greater than 2, combined with the absence of neutrophils and eosinophils on BAL, is helpful in distinguishing sarcoidosis from other pulmonary diseases. This patient’s inflammatory myopathy was revealed to be a rare initial manifestation of systemic sarcoidosis.
DISCUSSION
Weakness is a common symptom of muscle disorders such as myopathies and muscular dystrophy. Idiopathic inflammatory myopathies include PM, DM, and others.1,2 These usually present with proximal-dominant muscle weakness, decreased endurance, and muscle inflammation. A diagnosis is made according to symptoms in combination with diagnostic examinations, including elevated serum CK levels, abnormal EMG findings, and histopathology of skeletal muscle biopsy specimens.
Sarcoidosis, a multisystem disorder of unknown etiology, is characterized histopathologically by noncaseating granulomas in affected organs.3 It typically affects young adults, with incidence peaking at 20 to 39 years of age. Although any organ may be involved, the disorder usually presents with 1 or more common abnormalities, including bilateral hilar lymphadenopathy, lung lesions, and skin and eye involvement. Musculoskeletal involvement is less common. It is estimated that skeletal muscle is involved in 50% to 80% of patients with sarcoidosis but is rarely symptomatic (0.5% to 2.5%).4-6
In this patient, weakness was distributed in both proximal and distal muscles, yet proximal weakness is the most characteristic feature in PM and DM. Therefore, sarcoidosis should be considered in the differential diagnosis of idiopathic inflammatory myopathies, especially when weakness accompanies abnormalities in other organs typically affected by sarcoidosis.
Myoglobinuria often is observed in rhabdomyolysis and inflammatory myopathies, conditions that produce high levels of serum CK and myoglobin. Myoglobinuria, often accompanied by the elevation of urinary β2-microglobulin and N-acetyl-D-glucosamine levels, can induce tubulointerstitial damage, which leads to acute kidney injury. In this case, however, these abnormal kidney findings were observed without high levels of serum CK or myoglobin. This suggests the potential for other causes of tubulointerstitial damage, such as granulomatous interstitial nephritis in renal sarcoidosis.3
Another characteristic abnormality was the elevation of urinary calcium excretion, which indicated an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis. In sarcoidosis, hypercalciuria occurs in 40% of patients, hypercalcemia in 11%, and renal calculi in 10%.3,7 Hypercalciuria, for this patient, was important in arriving at the correct diagnosis after the gallium scan was obtained given the dearth of other typical features of sarcoidosis.
Although muscle biopsy is essential, imaging studies for idiopathic inflammatory myopathy are considered useful tools to narrow the differential diagnosis. The use of MRI of the skeletal muscle is helpful to both identify an adequate muscle for biopsy and demonstrate the pattern of affected muscles beyond clinical appearance, which aids in excluding, for example, muscular dystrophies.8,9
FDG PET/CT is a very sensitive imaging modality used to detect neoplastic lesions and has been widely used to screen for occult neoplasms and detect metastases.10-12 It is also useful for detecting inflammation in patients with osteomyelitis, metastatic infectious diseases, rheumatoid arthritis, vasculitis, inflammatory bowel diseases, fever of unknown origin, and sarcoidosis.11,12 In PM and DM, however, the sensitivity of FDG PET/CT for detection of myositis is reportedly lower than that of EMG and MRI.13 Similarly, gallium scintigraphy is usually performed to examine the disease activity of interstitial pneumonia or to detect malignancy. Previous literature and this case show that the striking images of gallium scintigraphy and FDG PET/CT have utility, not only for detection of sarcoid myopathy but also for the evaluation of treatment efficacy.14-17 Characteristic imaging findings on FDG PET/CT have been described as a “tiger man” appearance.17
For the treatment of sarcoid myopathy, systemic glucocorticoids are used for patients with symptomatic acute or chronic forms. The standard doses of prednisolone used for other forms of idiopathic inflammatory myopathies are usually administered.3-6 In general, the response of acute sarcoid myopathy to glucocorticoid therapy is favorable, and the clinical course is usually benign. However, the course in chronic sarcoid myopathy can be unpredictable with exacerbations. Given the lack of randomized trials of this therapy and because glucocorticoids themselves can cause steroid-induced myopathy, they are not used for asymptomatic patients.
In the end, astute clinical thinking, deductive reasoning, and pattern recognition were all instrumental in making this strong diagnosis of weakness.
KEY TEACHING POINTS
- Proximal muscle–dominant weakness is the characteristic feature in inflammatory myopathies like PM and DM. Myopathy causing proximal and distal weakness is more characteristic of sarcoidosis, IBM, alcohol, and statins.
- Elevations of urinary Times New Romanβ2-microglobulin and N-acetyl-D-glucosamine are often observed in inflammatory muscle diseases because of myoglobin-induced tubulointerstitial damage. These findings may also be caused by other conditions that affect the tubules, such as lupus nephritis, Sjogren’s syndrome, or renal sarcoidosis.
- Hypercalciuria in a patient with myopathy could suggest an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis.
- The striking uptake within systemic skeletal striated muscles on gallium scintigraphy and “tiger man” appearance on FDG PET/CT are characteristic features of acute sarcoid myopathy; these are not common in other inflammatory myopathies.
Disclosure
Drs. Sudo, Wada, Narita, Mba, and Houchens have no conflicts of interest to disclose.
1. Vincze M, Danko K. Idiopathic inflammatory myopathies. Best Pract Res Clin Rheumatol. 2012;26:25-45. PubMed
2. Carstens PO, Schmidt J. Diagnosis, pathogenesis, and treatment of myositis: recent advances. Clin Exp Immunol. 2014;175:425-438. PubMed
3. Lannuzzi MC, Rhbicki BA, Teirstein AS. Sarcoidosis. N Eng J Med. 2007;357:2153-2165. PubMed
4. Baydur A, Pandya K, Sharma OP, et al. Control of ventilation, respiratory muscle strength, and granulomatous involvement of skeletal muscle in patients with sarcoidosis. Chest. 1993;103:396-402. PubMed
5. Zisman DA, Biermann JS, Martinez FJ, et al. Sarcoidosis presenting as a tumorlike muscular lesion. Case report and review of the literature. Medicine (Baltimore). 1999;78:112-122. PubMed
6. Fayad F, Liote F, Berenbaum F, et al. Muscle involvement in sarcoidosis: a retrospective and followup studies. J Rheumatol. 2006;33:98-103. PubMed
7. Berliner AR, Haas M, Choi MJ. Sarcoidosis: the nephrologist’s perspective. Am J Kidney Dis. 2006;48:856-870. PubMed
8. Otake S, Ishigaki T. Musular sarcoidosis. Semin Musculoskelet Radiol. 2001;5:167-170. PubMed
9. Otake S, Imagumbai N, Suzuki M, et al. MR imaging of muscular sarcoidosis after steroid therapy. Eur Radiol. 1998;8:1651-1653. PubMed
10. Hoffman JM, Gambhir SS. Molecular imaging: The vision and opportunity for radiology in the future. Radiology. 2007;244:39-47. PubMed
11. Basu S, Zhuang H, Torigian DA, et al. Functional imaging of inflammatory diseases using nuclear medicine techniques. Semin Nucl Med. 2009;39:124-145. PubMed
12. Gotthardt M, Cleeker-Rovers CP, Boerman OC, et al. Imaging of inflammation by PET, conventional scintigraphy, and other imaging techniques. J Nucl Med. 2010;51:1937-1949. PubMed
13. Owada T, Maezawa R, Kurasawa K, et al. Detection of inflammatory lesions by F-18 fluorodeoxyglucose positron emission tomography in patients with polymyositis and dermatomyositis. J Rheumatol. 2012;39:1659-1665. PubMed
14. Liem IH, Drent M, Antevska E, et al. Intense muscle uptake of gallium-67 in a patient with sarcoidosis. J Nucl Med. 1998;39:1605-1607. PubMed
15. Suehiro S, Shiokawa S, Taniguchi S, et al. Gallium-67 scintigraphy in the diagnosis and management of chronic sarcoid myopathy. Clin Rheumatol. 2003;22:146-148. PubMed
16. Marie I, Josse S, Lahaxe L, et al. Clinical images: muscle sarcoidosis demonstrated on positron emission tomography. Arthritis Rheum. 2009;60:2847. PubMed
17. Wieers G, Lhommel R, Lecouvet F, et al. A tiger man. Lancet. 2012;380:1859. PubMed
1. Vincze M, Danko K. Idiopathic inflammatory myopathies. Best Pract Res Clin Rheumatol. 2012;26:25-45. PubMed
2. Carstens PO, Schmidt J. Diagnosis, pathogenesis, and treatment of myositis: recent advances. Clin Exp Immunol. 2014;175:425-438. PubMed
3. Lannuzzi MC, Rhbicki BA, Teirstein AS. Sarcoidosis. N Eng J Med. 2007;357:2153-2165. PubMed
4. Baydur A, Pandya K, Sharma OP, et al. Control of ventilation, respiratory muscle strength, and granulomatous involvement of skeletal muscle in patients with sarcoidosis. Chest. 1993;103:396-402. PubMed
5. Zisman DA, Biermann JS, Martinez FJ, et al. Sarcoidosis presenting as a tumorlike muscular lesion. Case report and review of the literature. Medicine (Baltimore). 1999;78:112-122. PubMed
6. Fayad F, Liote F, Berenbaum F, et al. Muscle involvement in sarcoidosis: a retrospective and followup studies. J Rheumatol. 2006;33:98-103. PubMed
7. Berliner AR, Haas M, Choi MJ. Sarcoidosis: the nephrologist’s perspective. Am J Kidney Dis. 2006;48:856-870. PubMed
8. Otake S, Ishigaki T. Musular sarcoidosis. Semin Musculoskelet Radiol. 2001;5:167-170. PubMed
9. Otake S, Imagumbai N, Suzuki M, et al. MR imaging of muscular sarcoidosis after steroid therapy. Eur Radiol. 1998;8:1651-1653. PubMed
10. Hoffman JM, Gambhir SS. Molecular imaging: The vision and opportunity for radiology in the future. Radiology. 2007;244:39-47. PubMed
11. Basu S, Zhuang H, Torigian DA, et al. Functional imaging of inflammatory diseases using nuclear medicine techniques. Semin Nucl Med. 2009;39:124-145. PubMed
12. Gotthardt M, Cleeker-Rovers CP, Boerman OC, et al. Imaging of inflammation by PET, conventional scintigraphy, and other imaging techniques. J Nucl Med. 2010;51:1937-1949. PubMed
13. Owada T, Maezawa R, Kurasawa K, et al. Detection of inflammatory lesions by F-18 fluorodeoxyglucose positron emission tomography in patients with polymyositis and dermatomyositis. J Rheumatol. 2012;39:1659-1665. PubMed
14. Liem IH, Drent M, Antevska E, et al. Intense muscle uptake of gallium-67 in a patient with sarcoidosis. J Nucl Med. 1998;39:1605-1607. PubMed
15. Suehiro S, Shiokawa S, Taniguchi S, et al. Gallium-67 scintigraphy in the diagnosis and management of chronic sarcoid myopathy. Clin Rheumatol. 2003;22:146-148. PubMed
16. Marie I, Josse S, Lahaxe L, et al. Clinical images: muscle sarcoidosis demonstrated on positron emission tomography. Arthritis Rheum. 2009;60:2847. PubMed
17. Wieers G, Lhommel R, Lecouvet F, et al. A tiger man. Lancet. 2012;380:1859. PubMed
© 2017 Society of Hospital Medicine
Thinking Outside the Checkbox
A 34-year-old, previously healthy Japanese man developed a dry cough. He did not have dyspnea, nasal discharge, sore throat, facial pain, nasal congestion, or postnasal drip. His symptoms persisted despite several courses of antibiotics (from different physicians), including clarithromycin, minocycline, and levofloxacin. A chest x-ray after 2 months of symptoms and a noncontrast chest computed tomography (CT) after 4 months of symptoms were normal, and bacterial and mycobacterial sputum cultures were sterile. Treatment with salmeterol and fluticasone was ineffective.
The persistence of a cough for longer than 8 weeks constitutes chronic cough. The initial negative review of systems argues against several of the usual etiologies. The lack of nasal discharge, sore throat, facial pain, nasal congestion, and postnasal drip lessens the probability of upper airway cough syndrome. The absence of dyspnea decreases the likelihood of congestive heart failure, asthma, or chronic obstructive pulmonary disease. Additional history should include whether the patient has orthopnea, paroxysmal nocturnal dyspnea, or a reduced exercise tolerance.
The persistence of symptoms despite multiple courses of antibiotics suggests that the process is inflammatory but not infectious, that the infection is not susceptible to the selected antibiotics, that the antibiotics cannot penetrate the site of infection, or that the ongoing symptoms are related to the antibiotics themselves. Pathogens that may cause chronic cough for months include mycobacteria, fungi (eg, Aspergillus , endemic mycoses), and parasites (eg, Strongyloides , Paragonimus ). Even when appropriately treated, many infections may result in a prolonged cough (eg, pertussis). The fluoroquinolone and macrolide exposure may have suppressed the mycobacterial cultures. The lack of response to salmeterol and fluticasone lessens the probability of asthma.
After 4 months of symptoms, his cough worsened, and he developed dysphagia and odynophagia, particularly when he initiated swallowing. He experienced daily fevers with temperatures between 38.0°C and 38.5°C. A repeat chest x-ray was normal. His white blood cell count was 14,200 per μL, and the C-reactive protein (CRP) was 12.91 mg/dL (normal <0.24 mg/dL). His symptoms did not improve with additional courses of clarithromycin, levofloxacin, or moxifloxacin. After 5 months of symptoms, he was referred to the internal medicine clinic of a teaching hospital in Japan.
The patient’s fevers, leukocytosis, and elevated CRP signal an inflammatory process, but whether it is infectious or not remains uncertain. The normal repeat chest x-ray lessens the likelihood of a pulmonary infection. Difficulty with initiating a swallow characterizes oropharyngeal dysphagia which features coughing or choking with oral intake and is typically caused by neuromuscular conditions like stroke, amyotrophic lateral sclerosis, or myasthenia gravis. The coexistence of oropharyngeal dysphagia and odynophagia may indicate pharyngitis, a retropharyngeal or parapharyngeal abscess, or oropharyngeal cancer.
Esophageal dysphagia occurs several seconds following swallow initiation and may arise with mucosal, smooth muscle, or neuromuscular diseases of the esophagus. Concomitant dysphagia and odynophagia may indicate esophageal spasm or esophagitis. Causes of esophagitis include infection (eg, candidiasis, herpes simplex virus [HSV], cytomegalovirus [CMV], or human immunodeficiency virus [HIV]), infiltration (eg, eosinophilic esophagitis), or irritation (eg, from medication, caustic ingestion, or gastroesophageal reflux). He is at risk for esophageal candidiasis following multiple courses of antibiotics. Esophageal dysphagia occurring with liquids and solids may indicate disordered motility, as opposed to dysphagia with solids alone, which may signal endoluminal obstruction.
At his outpatient evaluation, he denied headache, vision changes, chest pain, hemoptysis, palpitations, abdominal pain, dysuria, musculoskeletal symptoms, anorexia, or symptoms of gastroesophageal reflux. He did not have chills, rigors, or night sweats, but he had lost 3.4 kg in 5 months. He had not traveled within or outside of Japan in many years and was not involved in outdoor activities. He was engaged to and monogamous with his female partner of 5 years. He smoked 10 cigarettes per day for 14 years but stopped smoking during the last 2 months on account of his symptoms. He drank 6 beers per month and worked as a researcher at a chemical company but did not have any inhalational exposures.
His weight loss could be from reduced caloric intake due to dysphagia and odynophagia or may reflect an energy deficit related to chronic illness and inflammatory state. His smoking history increases his risk of bronchopulmonary infection and malignancy. Bronchogenic carcinoma may present with chronic cough, fevers, weight loss, or dysphagia from external compression by lymphadenopathy or mediastinal disease; however, his young age and recent chest CT results make lung cancer unlikely.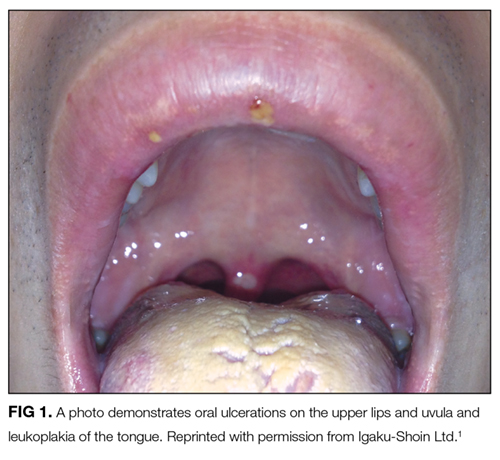
The white coating on his tongue could reflect oral leukoplakia, a reactive and potentially precancerous process that typically manifests as patches or plaques on oral mucosa. It can be distinguished from candidiasis, which scrapes off using a tongue blade. The extensive tongue coating is consistent with oral candidiasis. Potential predispositions include inhaled corticosteroids, antibiotic exposure, and/or an undiagnosed immunodeficiency syndrome (eg, HIV).
The initial diagnostic branch point for nontraumatic oral ulcers is infectious versus noninfectious. Infections that cause oral ulcers include HSV, CMV, and syphilis. The appearance and occurrence of the ulcers on freely moveable mucosa are consistent with aphthous stomatitis. Recurrent aphthous ulcers may occur in autoimmune diseases, including Behçet disease, Crohn disease, celiac sprue, and reactive arthritis. An endoscopy should be considered to detect esophageal ulcerations or esophageal candidiasis.
The rash may indicate folliculitis, usually attributable to Staphylococcus aureus or to Pseudomonas in the setting of recreational water exposure. Broad-spectrum antibiotics or immunodeficiency predisposes to candida folliculitis, while systemic candidiasis may cause metastatic skin lesions. The most common cutaneous manifestation of Behçet disease is erythema nodosum, but follicular and papulopustular lesions are also characteristic.

Pulmonary nodules are caused by infections, noninfectious inflammation, and malignancy. Infectious causes of pulmonary nodules include septic emboli, bacterial abscesses, and mycobacterial and fungal infection; noninfectious inflammatory causes include vasculitis (eg, granulomatosis with polyangiitis), rheumatoid arthritis, sarcoidosis, and lymphomatoid granulomatosis. Although additional culture data, serologic testing, and tuberculin skin testing or an interferon-gamma release assay may help to exclude these infections, the chronicity of symptoms, and lack of response to multiple antibiotic courses favor a noninfectious etiology.
Thickening of the aorta and left pulmonary artery may arise from an infectious, infiltrative, or inflammatory process. Arterial infections arise from direct inoculation, such as catheterization, trauma, or a contiguous site of infection, or from embolic seeding of atherosclerotic plaques or aneurysms. Malignant and nonmalignant processes, including sarcomas, lymphomas, histiocytoses (eg, Erdheim–Chester disease), and IgG4-related disease, may infiltrate the vascular walls. He has no evidence of visceral organ involvement to suggest these multisystem diagnoses.
The combined involvement of the aorta and pulmonary artery suggest a large-vessel vasculitis. Giant cell arteritis is exceedingly rare in patients younger than 50. Takayasu arteritis is a large-vessel vasculitis that predominantly affects women and may present with hypertension, arterial bruits, or discrepant blood pressure between arms, none of which were reported in this case. Behçet disease affects blood vessels of all sizes, including the aorta and pulmonary vasculature. His fevers, oral ulcers, perifollicular rash, and lymphadenopathy are consistent with this diagnosis, although he lacks the genital ulcers that occur in the majority of patients. Pulmonary nodules in Behçet disease arise from pulmonary or pleural vasculitis, resulting in focal inflammation, hemorrhage, or infarction. An ophthalmologic examination for uveitis and a pathergy test would support this diagnosis.
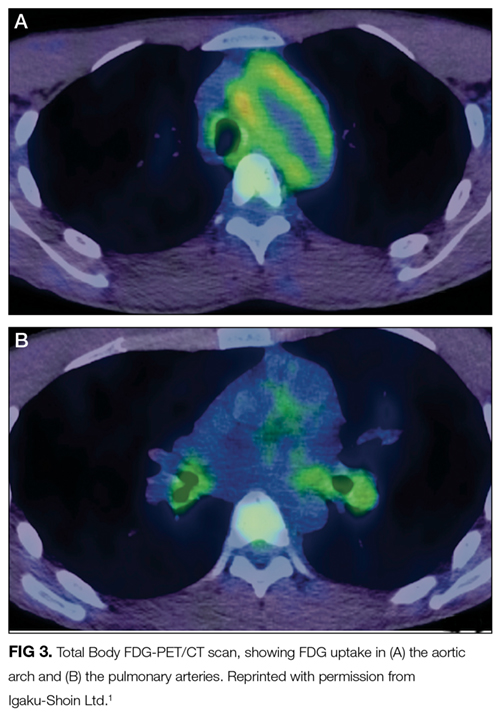
FDG accumulation in the aorta and pulmonary arteries signals large-vessel inflammation. The lack of FDG-avidity of the ground-glass opacities and nodular lesion suggests that these are not metabolically active tumors or infections but may be sequelae of the underlying disease, such as a hemorrhage or infarction from vasculitis. Sarcoidosis could account for the lung findings, but large-vessel vasculopathy would be exceedingly uncommon. Microscopic polyangiitis and granulomatosis with polyangiitis also cause pulmonary and vascular inflammation, but the nonreactive ANCA, absence of sinus disease, and normal urinalysis and kidney function make pauci-immune vasculitis unlikely. While the large-vessel involvement is consistent with Takayasu arteritis, the oral ulcers and rash are not.
Despite the absence of uveitis and the negative pathergy test, his oral aphthosis, papulopustular rash, and large-vessel vasculitis make Behçet disease the likely diagnosis. Behçet disease is most strongly associated with HLA B51, although other HLA haplotypes (including HLA A26 and HLA B52) are frequent in Behçet disease as well. As aortitis and pulmonary vasculitis can be associated with substantial morbidity and mortality, an urgent consultation with a rheumatologist regarding the initiation of immunosuppression is warranted.
Based on the mucocutaneous lesions, radiologic findings consistent with large-vessel vasculitis, and positive HLA A26 and HLA B52, he was diagnosed with Behçet disease. After 1 week of treatment with prednisolone 60 mg daily, his cough resolved and the oral aphthous ulcers and papulopustular rash improved. One month later, a chest CT showed significant reduction of the wall thickening of the aorta, its branches, and of the left pulmonary artery. The nodular lesion in the left lower lobe was unchanged, but the ground-glass opacities in the left upper lobe had disappeared.
When prednisolone was tapered down to 17.5 mg, his dry cough and low-grade fevers recurred, along with a slight elevation of inflammatory markers, and a ground-glass opacity appeared on the periphery of the left upper lobe. A sputum culture and fungal antigens were negative. His cough improved with the resumption of the previous dose of prednisolone. He remained symptom-free after 2 years of treatment with azathioprine 150 mg daily and prednisolone 2 mg daily and is now only treated with azathioprine.
DISCUSSION
Behçet disease is a multisystem vasculitis involving blood vessels of all sizes in the arterial and venous circulation that presents with oral and genital ulcers, ocular abnormalities (uveitis, retinitis), skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”), pathergy, and vascular lesions (thrombophlebitis, thrombosis, and aneurysm).
This patient presented with a chronic cough from pulmonary involvement by Behçet disease. The most common presenting symptom in a study of 47 patients with Behçet disease with pulmonary arteriopathy was hemoptysis followed by a nonbloody cough.2 Among these patients with pulmonary artery aneurysm, thrombosis, or both, 40 (85%) had nodules caused by infarction or inflammation and 21 (45%) had ground-glass opacities attributed to intraparenchymal hemorrhage. There are several case reports of chronic cough attributed to large-vessel vasculitis.3-5 Although the pathology of vasculitis-related cough is not fully understood, the inflammation of large vessels (aorta and pulmonary arteries) adjacent to the tracheobronchial tree may irritate regional cough receptors.3
Disease classification criteria are common in rheumatologic diseases; these criteria are developed to categorize patients for research studies and are not intended to diagnose individual patients.6 The classification criteria favor increased specificity at the expense of sensitivity to avoid misclassifying patients as having a disease, which would compromise the results of research studies. For instance, a study assessing a treatment for Behçet disease must exclude patients with inflammatory bowel disease, as these distinct patient populations may demonstrate discrepant responses to the investigative therapy. The specificity and homogeneity favored by classification criteria make those criteria inappropriate to rely on exclusively for the diagnosis of individual patients.7 The symptoms of many autoimmune diseases develop sequentially over time. Waiting for a patient with active, multisystem vasculitis to fulfill all of the Behçet disease classification criteria can lead to the harmful withholding of disease-modifying treatment.
The diagnosis of Behçet disease is made on clinical grounds; there is no gold standard test or histopathologic finding, and classification criteria remain imperfect. Although classification criteria help clinicians understand cardinal disease features, they cannot substitute for the more complex clinical reasoning required to establish a working diagnosis. The clinician must understand the pretest probability of disease, consider the presence or absence of characteristic features, exclude competing diagnoses, and decipher the risk-to-benefit ratio of therapeutic options and the urgency of treatment when assigning a diagnostic label. This patient’s pneumonitis, mucocutaneous changes, aortopathy, and compatible HLA typing (coupled with the exclusion of infectious diseases) were sufficient to diagnose Behçet disease. This case reminds us that classification criteria serve as a starting point, not as an end point, and that clinicians must ultimately make diagnoses and initiate treatment by thinking outside the checkbox.
TEACHING POINTS
- Large-vessel vasculitis is a rare cause of chronic cough.
- Although the most well-recognized signs of Behçet disease include genital and oral ulcers and uveitis, patients may also present with less common manifestations such as skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”) and vascular lesions of the artery (arteritis and aneurysm) and veins (thrombophlebitis and thrombosis).
- Classification criteria capture cardinal features of a disease but favor specificity over sensitivity and should not serve as a checklist for diagnosing a patient.
Acknowledgment
A brief version of this case was published as a case report in the Journal of Integrated Medicine 2013;23(12):1014-1017. Images from that publication were republished here with the permission of the publisher (Igaku-Shoin Ltd).
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. All other authors have nothing to disclose.
1. Kanamori M, Kubo T, Sakemi H. What’s your diagnosis? [in Japanese] J Integrated Med. 2013; 23 (12):1014-1017.
2. Seyahi E, Melikoglu M, Akman C, et al. Pulmonary artery involvement and associated lung disease in Behçet disease: a series of 47 patients. Medicine (Baltimore). 2012;91(1):35-48. PubMed
3. Olopade CO, Sekosan M, Schraufnagel DE. Giant cell arteritis manifesting as chronic cough and fever of unknown origin. Mayo Clin Proc. 1997;72(11):1048-1050. PubMed
4. Hellmann DB. Temporal arteritis: a cough, toothache, and tongue infarction. JAMA. 2002;287(22):2996-3000. PubMed
5. Karagiannis A, Mathiopoulou L, Tziomalos K, et al. Dry cough as first manifestation of giant-cell arteritis. J Am Geriatr Soc. 2006;54(12):1957-1958. PubMed
6. Aggarwal R, Ringold S, Khanna D, et al. Distinctions between diagnostic and classification criteria? Arthritis Care Res (Hoboken). 2015;67(7):891-897. PubMed
7. Rao JK, Allen NB, Pincus T. Limitations of the 1990 American College of Rheumatology classification criteria in the diagnosis of vasculitis. Ann Intern Med. 1998;129(5):345-352. PubMed
8. Davatchi F, Sadeghi Abdollahi B, Shahram F, Chams-Davatchi C, Shams H, Nadji A. Classification and Diagnosis Criteria for Behçet’s Disease. In: Emmi L, ed. Behçet’s Syndrome. From Pathogenesis to Treatment. Milan, Italy: Springer; 2014:189-198.
9. Criteria for diagnosis of Behcet’s disease. International Study Group for Behçet’s Disease. Lancet. 1990;335(8697):1078-1080. PubMed
10. Davatchi F, Assaad-Khalil S, Calamia KT, et al. The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338–347. PubMed
11. Suzuki Kurokawa M, Suzuki N. Behçet’s disease. Clin Exp Med. 2004;4(1):10-20. PubMed
A 34-year-old, previously healthy Japanese man developed a dry cough. He did not have dyspnea, nasal discharge, sore throat, facial pain, nasal congestion, or postnasal drip. His symptoms persisted despite several courses of antibiotics (from different physicians), including clarithromycin, minocycline, and levofloxacin. A chest x-ray after 2 months of symptoms and a noncontrast chest computed tomography (CT) after 4 months of symptoms were normal, and bacterial and mycobacterial sputum cultures were sterile. Treatment with salmeterol and fluticasone was ineffective.
The persistence of a cough for longer than 8 weeks constitutes chronic cough. The initial negative review of systems argues against several of the usual etiologies. The lack of nasal discharge, sore throat, facial pain, nasal congestion, and postnasal drip lessens the probability of upper airway cough syndrome. The absence of dyspnea decreases the likelihood of congestive heart failure, asthma, or chronic obstructive pulmonary disease. Additional history should include whether the patient has orthopnea, paroxysmal nocturnal dyspnea, or a reduced exercise tolerance.
The persistence of symptoms despite multiple courses of antibiotics suggests that the process is inflammatory but not infectious, that the infection is not susceptible to the selected antibiotics, that the antibiotics cannot penetrate the site of infection, or that the ongoing symptoms are related to the antibiotics themselves. Pathogens that may cause chronic cough for months include mycobacteria, fungi (eg, Aspergillus , endemic mycoses), and parasites (eg, Strongyloides , Paragonimus ). Even when appropriately treated, many infections may result in a prolonged cough (eg, pertussis). The fluoroquinolone and macrolide exposure may have suppressed the mycobacterial cultures. The lack of response to salmeterol and fluticasone lessens the probability of asthma.
After 4 months of symptoms, his cough worsened, and he developed dysphagia and odynophagia, particularly when he initiated swallowing. He experienced daily fevers with temperatures between 38.0°C and 38.5°C. A repeat chest x-ray was normal. His white blood cell count was 14,200 per μL, and the C-reactive protein (CRP) was 12.91 mg/dL (normal <0.24 mg/dL). His symptoms did not improve with additional courses of clarithromycin, levofloxacin, or moxifloxacin. After 5 months of symptoms, he was referred to the internal medicine clinic of a teaching hospital in Japan.
The patient’s fevers, leukocytosis, and elevated CRP signal an inflammatory process, but whether it is infectious or not remains uncertain. The normal repeat chest x-ray lessens the likelihood of a pulmonary infection. Difficulty with initiating a swallow characterizes oropharyngeal dysphagia which features coughing or choking with oral intake and is typically caused by neuromuscular conditions like stroke, amyotrophic lateral sclerosis, or myasthenia gravis. The coexistence of oropharyngeal dysphagia and odynophagia may indicate pharyngitis, a retropharyngeal or parapharyngeal abscess, or oropharyngeal cancer.
Esophageal dysphagia occurs several seconds following swallow initiation and may arise with mucosal, smooth muscle, or neuromuscular diseases of the esophagus. Concomitant dysphagia and odynophagia may indicate esophageal spasm or esophagitis. Causes of esophagitis include infection (eg, candidiasis, herpes simplex virus [HSV], cytomegalovirus [CMV], or human immunodeficiency virus [HIV]), infiltration (eg, eosinophilic esophagitis), or irritation (eg, from medication, caustic ingestion, or gastroesophageal reflux). He is at risk for esophageal candidiasis following multiple courses of antibiotics. Esophageal dysphagia occurring with liquids and solids may indicate disordered motility, as opposed to dysphagia with solids alone, which may signal endoluminal obstruction.
At his outpatient evaluation, he denied headache, vision changes, chest pain, hemoptysis, palpitations, abdominal pain, dysuria, musculoskeletal symptoms, anorexia, or symptoms of gastroesophageal reflux. He did not have chills, rigors, or night sweats, but he had lost 3.4 kg in 5 months. He had not traveled within or outside of Japan in many years and was not involved in outdoor activities. He was engaged to and monogamous with his female partner of 5 years. He smoked 10 cigarettes per day for 14 years but stopped smoking during the last 2 months on account of his symptoms. He drank 6 beers per month and worked as a researcher at a chemical company but did not have any inhalational exposures.
His weight loss could be from reduced caloric intake due to dysphagia and odynophagia or may reflect an energy deficit related to chronic illness and inflammatory state. His smoking history increases his risk of bronchopulmonary infection and malignancy. Bronchogenic carcinoma may present with chronic cough, fevers, weight loss, or dysphagia from external compression by lymphadenopathy or mediastinal disease; however, his young age and recent chest CT results make lung cancer unlikely.
The white coating on his tongue could reflect oral leukoplakia, a reactive and potentially precancerous process that typically manifests as patches or plaques on oral mucosa. It can be distinguished from candidiasis, which scrapes off using a tongue blade. The extensive tongue coating is consistent with oral candidiasis. Potential predispositions include inhaled corticosteroids, antibiotic exposure, and/or an undiagnosed immunodeficiency syndrome (eg, HIV).
The initial diagnostic branch point for nontraumatic oral ulcers is infectious versus noninfectious. Infections that cause oral ulcers include HSV, CMV, and syphilis. The appearance and occurrence of the ulcers on freely moveable mucosa are consistent with aphthous stomatitis. Recurrent aphthous ulcers may occur in autoimmune diseases, including Behçet disease, Crohn disease, celiac sprue, and reactive arthritis. An endoscopy should be considered to detect esophageal ulcerations or esophageal candidiasis.
The rash may indicate folliculitis, usually attributable to Staphylococcus aureus or to Pseudomonas in the setting of recreational water exposure. Broad-spectrum antibiotics or immunodeficiency predisposes to candida folliculitis, while systemic candidiasis may cause metastatic skin lesions. The most common cutaneous manifestation of Behçet disease is erythema nodosum, but follicular and papulopustular lesions are also characteristic.
Pulmonary nodules are caused by infections, noninfectious inflammation, and malignancy. Infectious causes of pulmonary nodules include septic emboli, bacterial abscesses, and mycobacterial and fungal infection; noninfectious inflammatory causes include vasculitis (eg, granulomatosis with polyangiitis), rheumatoid arthritis, sarcoidosis, and lymphomatoid granulomatosis. Although additional culture data, serologic testing, and tuberculin skin testing or an interferon-gamma release assay may help to exclude these infections, the chronicity of symptoms, and lack of response to multiple antibiotic courses favor a noninfectious etiology.
Thickening of the aorta and left pulmonary artery may arise from an infectious, infiltrative, or inflammatory process. Arterial infections arise from direct inoculation, such as catheterization, trauma, or a contiguous site of infection, or from embolic seeding of atherosclerotic plaques or aneurysms. Malignant and nonmalignant processes, including sarcomas, lymphomas, histiocytoses (eg, Erdheim–Chester disease), and IgG4-related disease, may infiltrate the vascular walls. He has no evidence of visceral organ involvement to suggest these multisystem diagnoses.
The combined involvement of the aorta and pulmonary artery suggest a large-vessel vasculitis. Giant cell arteritis is exceedingly rare in patients younger than 50. Takayasu arteritis is a large-vessel vasculitis that predominantly affects women and may present with hypertension, arterial bruits, or discrepant blood pressure between arms, none of which were reported in this case. Behçet disease affects blood vessels of all sizes, including the aorta and pulmonary vasculature. His fevers, oral ulcers, perifollicular rash, and lymphadenopathy are consistent with this diagnosis, although he lacks the genital ulcers that occur in the majority of patients. Pulmonary nodules in Behçet disease arise from pulmonary or pleural vasculitis, resulting in focal inflammation, hemorrhage, or infarction. An ophthalmologic examination for uveitis and a pathergy test would support this diagnosis.
FDG accumulation in the aorta and pulmonary arteries signals large-vessel inflammation. The lack of FDG-avidity of the ground-glass opacities and nodular lesion suggests that these are not metabolically active tumors or infections but may be sequelae of the underlying disease, such as a hemorrhage or infarction from vasculitis. Sarcoidosis could account for the lung findings, but large-vessel vasculopathy would be exceedingly uncommon. Microscopic polyangiitis and granulomatosis with polyangiitis also cause pulmonary and vascular inflammation, but the nonreactive ANCA, absence of sinus disease, and normal urinalysis and kidney function make pauci-immune vasculitis unlikely. While the large-vessel involvement is consistent with Takayasu arteritis, the oral ulcers and rash are not.
Despite the absence of uveitis and the negative pathergy test, his oral aphthosis, papulopustular rash, and large-vessel vasculitis make Behçet disease the likely diagnosis. Behçet disease is most strongly associated with HLA B51, although other HLA haplotypes (including HLA A26 and HLA B52) are frequent in Behçet disease as well. As aortitis and pulmonary vasculitis can be associated with substantial morbidity and mortality, an urgent consultation with a rheumatologist regarding the initiation of immunosuppression is warranted.
Based on the mucocutaneous lesions, radiologic findings consistent with large-vessel vasculitis, and positive HLA A26 and HLA B52, he was diagnosed with Behçet disease. After 1 week of treatment with prednisolone 60 mg daily, his cough resolved and the oral aphthous ulcers and papulopustular rash improved. One month later, a chest CT showed significant reduction of the wall thickening of the aorta, its branches, and of the left pulmonary artery. The nodular lesion in the left lower lobe was unchanged, but the ground-glass opacities in the left upper lobe had disappeared.
When prednisolone was tapered down to 17.5 mg, his dry cough and low-grade fevers recurred, along with a slight elevation of inflammatory markers, and a ground-glass opacity appeared on the periphery of the left upper lobe. A sputum culture and fungal antigens were negative. His cough improved with the resumption of the previous dose of prednisolone. He remained symptom-free after 2 years of treatment with azathioprine 150 mg daily and prednisolone 2 mg daily and is now only treated with azathioprine.
DISCUSSION
Behçet disease is a multisystem vasculitis involving blood vessels of all sizes in the arterial and venous circulation that presents with oral and genital ulcers, ocular abnormalities (uveitis, retinitis), skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”), pathergy, and vascular lesions (thrombophlebitis, thrombosis, and aneurysm).
This patient presented with a chronic cough from pulmonary involvement by Behçet disease. The most common presenting symptom in a study of 47 patients with Behçet disease with pulmonary arteriopathy was hemoptysis followed by a nonbloody cough.2 Among these patients with pulmonary artery aneurysm, thrombosis, or both, 40 (85%) had nodules caused by infarction or inflammation and 21 (45%) had ground-glass opacities attributed to intraparenchymal hemorrhage. There are several case reports of chronic cough attributed to large-vessel vasculitis.3-5 Although the pathology of vasculitis-related cough is not fully understood, the inflammation of large vessels (aorta and pulmonary arteries) adjacent to the tracheobronchial tree may irritate regional cough receptors.3
Disease classification criteria are common in rheumatologic diseases; these criteria are developed to categorize patients for research studies and are not intended to diagnose individual patients.6 The classification criteria favor increased specificity at the expense of sensitivity to avoid misclassifying patients as having a disease, which would compromise the results of research studies. For instance, a study assessing a treatment for Behçet disease must exclude patients with inflammatory bowel disease, as these distinct patient populations may demonstrate discrepant responses to the investigative therapy. The specificity and homogeneity favored by classification criteria make those criteria inappropriate to rely on exclusively for the diagnosis of individual patients.7 The symptoms of many autoimmune diseases develop sequentially over time. Waiting for a patient with active, multisystem vasculitis to fulfill all of the Behçet disease classification criteria can lead to the harmful withholding of disease-modifying treatment.
The diagnosis of Behçet disease is made on clinical grounds; there is no gold standard test or histopathologic finding, and classification criteria remain imperfect. Although classification criteria help clinicians understand cardinal disease features, they cannot substitute for the more complex clinical reasoning required to establish a working diagnosis. The clinician must understand the pretest probability of disease, consider the presence or absence of characteristic features, exclude competing diagnoses, and decipher the risk-to-benefit ratio of therapeutic options and the urgency of treatment when assigning a diagnostic label. This patient’s pneumonitis, mucocutaneous changes, aortopathy, and compatible HLA typing (coupled with the exclusion of infectious diseases) were sufficient to diagnose Behçet disease. This case reminds us that classification criteria serve as a starting point, not as an end point, and that clinicians must ultimately make diagnoses and initiate treatment by thinking outside the checkbox.
TEACHING POINTS
- Large-vessel vasculitis is a rare cause of chronic cough.
- Although the most well-recognized signs of Behçet disease include genital and oral ulcers and uveitis, patients may also present with less common manifestations such as skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”) and vascular lesions of the artery (arteritis and aneurysm) and veins (thrombophlebitis and thrombosis).
- Classification criteria capture cardinal features of a disease but favor specificity over sensitivity and should not serve as a checklist for diagnosing a patient.
Acknowledgment
A brief version of this case was published as a case report in the Journal of Integrated Medicine 2013;23(12):1014-1017. Images from that publication were republished here with the permission of the publisher (Igaku-Shoin Ltd).
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. All other authors have nothing to disclose.
A 34-year-old, previously healthy Japanese man developed a dry cough. He did not have dyspnea, nasal discharge, sore throat, facial pain, nasal congestion, or postnasal drip. His symptoms persisted despite several courses of antibiotics (from different physicians), including clarithromycin, minocycline, and levofloxacin. A chest x-ray after 2 months of symptoms and a noncontrast chest computed tomography (CT) after 4 months of symptoms were normal, and bacterial and mycobacterial sputum cultures were sterile. Treatment with salmeterol and fluticasone was ineffective.
The persistence of a cough for longer than 8 weeks constitutes chronic cough. The initial negative review of systems argues against several of the usual etiologies. The lack of nasal discharge, sore throat, facial pain, nasal congestion, and postnasal drip lessens the probability of upper airway cough syndrome. The absence of dyspnea decreases the likelihood of congestive heart failure, asthma, or chronic obstructive pulmonary disease. Additional history should include whether the patient has orthopnea, paroxysmal nocturnal dyspnea, or a reduced exercise tolerance.
The persistence of symptoms despite multiple courses of antibiotics suggests that the process is inflammatory but not infectious, that the infection is not susceptible to the selected antibiotics, that the antibiotics cannot penetrate the site of infection, or that the ongoing symptoms are related to the antibiotics themselves. Pathogens that may cause chronic cough for months include mycobacteria, fungi (eg, Aspergillus , endemic mycoses), and parasites (eg, Strongyloides , Paragonimus ). Even when appropriately treated, many infections may result in a prolonged cough (eg, pertussis). The fluoroquinolone and macrolide exposure may have suppressed the mycobacterial cultures. The lack of response to salmeterol and fluticasone lessens the probability of asthma.
After 4 months of symptoms, his cough worsened, and he developed dysphagia and odynophagia, particularly when he initiated swallowing. He experienced daily fevers with temperatures between 38.0°C and 38.5°C. A repeat chest x-ray was normal. His white blood cell count was 14,200 per μL, and the C-reactive protein (CRP) was 12.91 mg/dL (normal <0.24 mg/dL). His symptoms did not improve with additional courses of clarithromycin, levofloxacin, or moxifloxacin. After 5 months of symptoms, he was referred to the internal medicine clinic of a teaching hospital in Japan.
The patient’s fevers, leukocytosis, and elevated CRP signal an inflammatory process, but whether it is infectious or not remains uncertain. The normal repeat chest x-ray lessens the likelihood of a pulmonary infection. Difficulty with initiating a swallow characterizes oropharyngeal dysphagia which features coughing or choking with oral intake and is typically caused by neuromuscular conditions like stroke, amyotrophic lateral sclerosis, or myasthenia gravis. The coexistence of oropharyngeal dysphagia and odynophagia may indicate pharyngitis, a retropharyngeal or parapharyngeal abscess, or oropharyngeal cancer.
Esophageal dysphagia occurs several seconds following swallow initiation and may arise with mucosal, smooth muscle, or neuromuscular diseases of the esophagus. Concomitant dysphagia and odynophagia may indicate esophageal spasm or esophagitis. Causes of esophagitis include infection (eg, candidiasis, herpes simplex virus [HSV], cytomegalovirus [CMV], or human immunodeficiency virus [HIV]), infiltration (eg, eosinophilic esophagitis), or irritation (eg, from medication, caustic ingestion, or gastroesophageal reflux). He is at risk for esophageal candidiasis following multiple courses of antibiotics. Esophageal dysphagia occurring with liquids and solids may indicate disordered motility, as opposed to dysphagia with solids alone, which may signal endoluminal obstruction.
At his outpatient evaluation, he denied headache, vision changes, chest pain, hemoptysis, palpitations, abdominal pain, dysuria, musculoskeletal symptoms, anorexia, or symptoms of gastroesophageal reflux. He did not have chills, rigors, or night sweats, but he had lost 3.4 kg in 5 months. He had not traveled within or outside of Japan in many years and was not involved in outdoor activities. He was engaged to and monogamous with his female partner of 5 years. He smoked 10 cigarettes per day for 14 years but stopped smoking during the last 2 months on account of his symptoms. He drank 6 beers per month and worked as a researcher at a chemical company but did not have any inhalational exposures.
His weight loss could be from reduced caloric intake due to dysphagia and odynophagia or may reflect an energy deficit related to chronic illness and inflammatory state. His smoking history increases his risk of bronchopulmonary infection and malignancy. Bronchogenic carcinoma may present with chronic cough, fevers, weight loss, or dysphagia from external compression by lymphadenopathy or mediastinal disease; however, his young age and recent chest CT results make lung cancer unlikely.
The white coating on his tongue could reflect oral leukoplakia, a reactive and potentially precancerous process that typically manifests as patches or plaques on oral mucosa. It can be distinguished from candidiasis, which scrapes off using a tongue blade. The extensive tongue coating is consistent with oral candidiasis. Potential predispositions include inhaled corticosteroids, antibiotic exposure, and/or an undiagnosed immunodeficiency syndrome (eg, HIV).
The initial diagnostic branch point for nontraumatic oral ulcers is infectious versus noninfectious. Infections that cause oral ulcers include HSV, CMV, and syphilis. The appearance and occurrence of the ulcers on freely moveable mucosa are consistent with aphthous stomatitis. Recurrent aphthous ulcers may occur in autoimmune diseases, including Behçet disease, Crohn disease, celiac sprue, and reactive arthritis. An endoscopy should be considered to detect esophageal ulcerations or esophageal candidiasis.
The rash may indicate folliculitis, usually attributable to Staphylococcus aureus or to Pseudomonas in the setting of recreational water exposure. Broad-spectrum antibiotics or immunodeficiency predisposes to candida folliculitis, while systemic candidiasis may cause metastatic skin lesions. The most common cutaneous manifestation of Behçet disease is erythema nodosum, but follicular and papulopustular lesions are also characteristic.
Pulmonary nodules are caused by infections, noninfectious inflammation, and malignancy. Infectious causes of pulmonary nodules include septic emboli, bacterial abscesses, and mycobacterial and fungal infection; noninfectious inflammatory causes include vasculitis (eg, granulomatosis with polyangiitis), rheumatoid arthritis, sarcoidosis, and lymphomatoid granulomatosis. Although additional culture data, serologic testing, and tuberculin skin testing or an interferon-gamma release assay may help to exclude these infections, the chronicity of symptoms, and lack of response to multiple antibiotic courses favor a noninfectious etiology.
Thickening of the aorta and left pulmonary artery may arise from an infectious, infiltrative, or inflammatory process. Arterial infections arise from direct inoculation, such as catheterization, trauma, or a contiguous site of infection, or from embolic seeding of atherosclerotic plaques or aneurysms. Malignant and nonmalignant processes, including sarcomas, lymphomas, histiocytoses (eg, Erdheim–Chester disease), and IgG4-related disease, may infiltrate the vascular walls. He has no evidence of visceral organ involvement to suggest these multisystem diagnoses.
The combined involvement of the aorta and pulmonary artery suggest a large-vessel vasculitis. Giant cell arteritis is exceedingly rare in patients younger than 50. Takayasu arteritis is a large-vessel vasculitis that predominantly affects women and may present with hypertension, arterial bruits, or discrepant blood pressure between arms, none of which were reported in this case. Behçet disease affects blood vessels of all sizes, including the aorta and pulmonary vasculature. His fevers, oral ulcers, perifollicular rash, and lymphadenopathy are consistent with this diagnosis, although he lacks the genital ulcers that occur in the majority of patients. Pulmonary nodules in Behçet disease arise from pulmonary or pleural vasculitis, resulting in focal inflammation, hemorrhage, or infarction. An ophthalmologic examination for uveitis and a pathergy test would support this diagnosis.
FDG accumulation in the aorta and pulmonary arteries signals large-vessel inflammation. The lack of FDG-avidity of the ground-glass opacities and nodular lesion suggests that these are not metabolically active tumors or infections but may be sequelae of the underlying disease, such as a hemorrhage or infarction from vasculitis. Sarcoidosis could account for the lung findings, but large-vessel vasculopathy would be exceedingly uncommon. Microscopic polyangiitis and granulomatosis with polyangiitis also cause pulmonary and vascular inflammation, but the nonreactive ANCA, absence of sinus disease, and normal urinalysis and kidney function make pauci-immune vasculitis unlikely. While the large-vessel involvement is consistent with Takayasu arteritis, the oral ulcers and rash are not.
Despite the absence of uveitis and the negative pathergy test, his oral aphthosis, papulopustular rash, and large-vessel vasculitis make Behçet disease the likely diagnosis. Behçet disease is most strongly associated with HLA B51, although other HLA haplotypes (including HLA A26 and HLA B52) are frequent in Behçet disease as well. As aortitis and pulmonary vasculitis can be associated with substantial morbidity and mortality, an urgent consultation with a rheumatologist regarding the initiation of immunosuppression is warranted.
Based on the mucocutaneous lesions, radiologic findings consistent with large-vessel vasculitis, and positive HLA A26 and HLA B52, he was diagnosed with Behçet disease. After 1 week of treatment with prednisolone 60 mg daily, his cough resolved and the oral aphthous ulcers and papulopustular rash improved. One month later, a chest CT showed significant reduction of the wall thickening of the aorta, its branches, and of the left pulmonary artery. The nodular lesion in the left lower lobe was unchanged, but the ground-glass opacities in the left upper lobe had disappeared.
When prednisolone was tapered down to 17.5 mg, his dry cough and low-grade fevers recurred, along with a slight elevation of inflammatory markers, and a ground-glass opacity appeared on the periphery of the left upper lobe. A sputum culture and fungal antigens were negative. His cough improved with the resumption of the previous dose of prednisolone. He remained symptom-free after 2 years of treatment with azathioprine 150 mg daily and prednisolone 2 mg daily and is now only treated with azathioprine.
DISCUSSION
Behçet disease is a multisystem vasculitis involving blood vessels of all sizes in the arterial and venous circulation that presents with oral and genital ulcers, ocular abnormalities (uveitis, retinitis), skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”), pathergy, and vascular lesions (thrombophlebitis, thrombosis, and aneurysm).
This patient presented with a chronic cough from pulmonary involvement by Behçet disease. The most common presenting symptom in a study of 47 patients with Behçet disease with pulmonary arteriopathy was hemoptysis followed by a nonbloody cough.2 Among these patients with pulmonary artery aneurysm, thrombosis, or both, 40 (85%) had nodules caused by infarction or inflammation and 21 (45%) had ground-glass opacities attributed to intraparenchymal hemorrhage. There are several case reports of chronic cough attributed to large-vessel vasculitis.3-5 Although the pathology of vasculitis-related cough is not fully understood, the inflammation of large vessels (aorta and pulmonary arteries) adjacent to the tracheobronchial tree may irritate regional cough receptors.3
Disease classification criteria are common in rheumatologic diseases; these criteria are developed to categorize patients for research studies and are not intended to diagnose individual patients.6 The classification criteria favor increased specificity at the expense of sensitivity to avoid misclassifying patients as having a disease, which would compromise the results of research studies. For instance, a study assessing a treatment for Behçet disease must exclude patients with inflammatory bowel disease, as these distinct patient populations may demonstrate discrepant responses to the investigative therapy. The specificity and homogeneity favored by classification criteria make those criteria inappropriate to rely on exclusively for the diagnosis of individual patients.7 The symptoms of many autoimmune diseases develop sequentially over time. Waiting for a patient with active, multisystem vasculitis to fulfill all of the Behçet disease classification criteria can lead to the harmful withholding of disease-modifying treatment.
The diagnosis of Behçet disease is made on clinical grounds; there is no gold standard test or histopathologic finding, and classification criteria remain imperfect. Although classification criteria help clinicians understand cardinal disease features, they cannot substitute for the more complex clinical reasoning required to establish a working diagnosis. The clinician must understand the pretest probability of disease, consider the presence or absence of characteristic features, exclude competing diagnoses, and decipher the risk-to-benefit ratio of therapeutic options and the urgency of treatment when assigning a diagnostic label. This patient’s pneumonitis, mucocutaneous changes, aortopathy, and compatible HLA typing (coupled with the exclusion of infectious diseases) were sufficient to diagnose Behçet disease. This case reminds us that classification criteria serve as a starting point, not as an end point, and that clinicians must ultimately make diagnoses and initiate treatment by thinking outside the checkbox.
TEACHING POINTS
- Large-vessel vasculitis is a rare cause of chronic cough.
- Although the most well-recognized signs of Behçet disease include genital and oral ulcers and uveitis, patients may also present with less common manifestations such as skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”) and vascular lesions of the artery (arteritis and aneurysm) and veins (thrombophlebitis and thrombosis).
- Classification criteria capture cardinal features of a disease but favor specificity over sensitivity and should not serve as a checklist for diagnosing a patient.
Acknowledgment
A brief version of this case was published as a case report in the Journal of Integrated Medicine 2013;23(12):1014-1017. Images from that publication were republished here with the permission of the publisher (Igaku-Shoin Ltd).
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. All other authors have nothing to disclose.
1. Kanamori M, Kubo T, Sakemi H. What’s your diagnosis? [in Japanese] J Integrated Med. 2013; 23 (12):1014-1017.
2. Seyahi E, Melikoglu M, Akman C, et al. Pulmonary artery involvement and associated lung disease in Behçet disease: a series of 47 patients. Medicine (Baltimore). 2012;91(1):35-48. PubMed
3. Olopade CO, Sekosan M, Schraufnagel DE. Giant cell arteritis manifesting as chronic cough and fever of unknown origin. Mayo Clin Proc. 1997;72(11):1048-1050. PubMed
4. Hellmann DB. Temporal arteritis: a cough, toothache, and tongue infarction. JAMA. 2002;287(22):2996-3000. PubMed
5. Karagiannis A, Mathiopoulou L, Tziomalos K, et al. Dry cough as first manifestation of giant-cell arteritis. J Am Geriatr Soc. 2006;54(12):1957-1958. PubMed
6. Aggarwal R, Ringold S, Khanna D, et al. Distinctions between diagnostic and classification criteria? Arthritis Care Res (Hoboken). 2015;67(7):891-897. PubMed
7. Rao JK, Allen NB, Pincus T. Limitations of the 1990 American College of Rheumatology classification criteria in the diagnosis of vasculitis. Ann Intern Med. 1998;129(5):345-352. PubMed
8. Davatchi F, Sadeghi Abdollahi B, Shahram F, Chams-Davatchi C, Shams H, Nadji A. Classification and Diagnosis Criteria for Behçet’s Disease. In: Emmi L, ed. Behçet’s Syndrome. From Pathogenesis to Treatment. Milan, Italy: Springer; 2014:189-198.
9. Criteria for diagnosis of Behcet’s disease. International Study Group for Behçet’s Disease. Lancet. 1990;335(8697):1078-1080. PubMed
10. Davatchi F, Assaad-Khalil S, Calamia KT, et al. The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338–347. PubMed
11. Suzuki Kurokawa M, Suzuki N. Behçet’s disease. Clin Exp Med. 2004;4(1):10-20. PubMed
1. Kanamori M, Kubo T, Sakemi H. What’s your diagnosis? [in Japanese] J Integrated Med. 2013; 23 (12):1014-1017.
2. Seyahi E, Melikoglu M, Akman C, et al. Pulmonary artery involvement and associated lung disease in Behçet disease: a series of 47 patients. Medicine (Baltimore). 2012;91(1):35-48. PubMed
3. Olopade CO, Sekosan M, Schraufnagel DE. Giant cell arteritis manifesting as chronic cough and fever of unknown origin. Mayo Clin Proc. 1997;72(11):1048-1050. PubMed
4. Hellmann DB. Temporal arteritis: a cough, toothache, and tongue infarction. JAMA. 2002;287(22):2996-3000. PubMed
5. Karagiannis A, Mathiopoulou L, Tziomalos K, et al. Dry cough as first manifestation of giant-cell arteritis. J Am Geriatr Soc. 2006;54(12):1957-1958. PubMed
6. Aggarwal R, Ringold S, Khanna D, et al. Distinctions between diagnostic and classification criteria? Arthritis Care Res (Hoboken). 2015;67(7):891-897. PubMed
7. Rao JK, Allen NB, Pincus T. Limitations of the 1990 American College of Rheumatology classification criteria in the diagnosis of vasculitis. Ann Intern Med. 1998;129(5):345-352. PubMed
8. Davatchi F, Sadeghi Abdollahi B, Shahram F, Chams-Davatchi C, Shams H, Nadji A. Classification and Diagnosis Criteria for Behçet’s Disease. In: Emmi L, ed. Behçet’s Syndrome. From Pathogenesis to Treatment. Milan, Italy: Springer; 2014:189-198.
9. Criteria for diagnosis of Behcet’s disease. International Study Group for Behçet’s Disease. Lancet. 1990;335(8697):1078-1080. PubMed
10. Davatchi F, Assaad-Khalil S, Calamia KT, et al. The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338–347. PubMed
11. Suzuki Kurokawa M, Suzuki N. Behçet’s disease. Clin Exp Med. 2004;4(1):10-20. PubMed
© 2018 Society of Hospital Medicine
A Howling Cause of Pancytopenia
A 15-year-old African American girl presented to the emergency department with 3 days of fever, sore throat, nausea, vomiting, and poor appetite. She reported a 4-week history of fatigue, right hand pain and swelling, and a 6-kilogram weight loss for which she had seen her primary care provider several times. She reported no recent travel, sick contacts, or new medications.
It appears that there are potentially at least 2 separate problems: an acute one (past 3 days) and a more chronic one (past 4 weeks). These 2 problems may be directly related (ie, acute worsening of the more chronic problem), indirectly related (ie, the more chronic problem is leading to increased susceptibility to the acute problem, for instance, an evolving immunodeficiency predisposing to an opportunistic infection), or “true, true, but unrelated.” The clinical challenge is to keep one’s mind open to each of these potential scenarios and to avoid the tendency to focus on one of the problems and not pay enough attention to the other. Occam’s razor likely does not apply here.
Numerous common and typically transient diseases could cause the symptoms of the past 3 days, particularly infectious etiologies such as streptococcal pharyngitis or a viral infection. One cannot forget about these possibilities while contemplating the more worrisome symptoms of the past 4 weeks, especially weight loss in a growing adolescent. Patients may unintentionally lose weight for a variety of reasons, which can be broadly categorized by decreased caloric supply, gastrointestinal losses or malabsorption, and increased caloric demand; these categories are not mutually exclusive.
Lastly, 1 symptom may provide a more specific direction: the right hand pain and swelling of the past 4 weeks. More specifics, including the extent of the hand swelling, other areas of involvement, and the nature of her pain, will be helpful.
Her temperature was 99.5°F, heart rate 100 beats per minute, respiratory rate 18 breaths per minute, oxygen saturation 95% while breathing ambient air, blood pressure 99/56 mmHg, weight 44 kilograms, height 161 centimeters, and body mass index 17. She appeared generally ill and underweight. She had edematous and violaceous eyelids, dry cracked lips, and pharyngeal erythema with ulcerations of the hard palate. She had nontender cervical and inguinal lymphadenopathy. Her abdomen was tender to palpation in the lower quadrants without guarding or rebound; there was no organomegaly. A right knee effusion with overlying warmth was present without redness or decreased range of motion. She also had an enlarged third proximal interphalangeal joint and loss of palpable metacarpal phalangeal joint landmarks on her right hand. She was noted to be using her arms to move her legs when repositioning in bed.
These exam findings clearly point toward a systemic process but not 1 specific diagnosis. The presence of at least 2 inflamed joints points toward rheumatologic/inflammatory or infectious diseases. Localized edema (eyelids and right metacarpal phalangeal joints), oral ulcers, possible myositis, and arthritis point toward a systemic vasculitis (eg, granulomatosis with polyangiitis, Behçet disease). While Kawasaki disease is also a systemic vasculitis, the presence of oral ulcers and generalized lymphadenopathy argues against it. Inflammatory myopathies like polymyositis, and especially juvenile dermatomyositis, fit many aspects of this presentation with the violaceous eyelids and possible myositis, though no other cutaneous stigmata of this disease are evident (eg, no Gottron’s papules). Polyarthritis, violaceous eyelids, and possible myositis could be consistent with systemic lupus erythematosus (SLE).
The presence of oral ulcers and arthritis make other systemic inflammatory conditions, such as inflammatory bowel disease with arthritis and autoimmune- or infection-related hepatitis, possible. Infectious etiologies alone or in combination with a rheumatologic process are also possible given fevers and lymphadenopathy. In particular, herpesvirus infections (Epstein-Barr virus [EBV], cytomegalovirus [CMV], herpes simplex virus, or human herpes virus 6), human immunodeficiency virus (HIV), hepatitis C virus (HCV), and syphilis can cause oral ulcers and lymphadenopathy. Other potential infectious etiologies include subacute bacterial endocarditis and disseminated gonococcal infection given the presence of polyarthritis, but these infections are less likely as they do not explain all of the symptoms.
In summary, the differential diagnosis is broad and should be prioritized to consider systemic inflammatory conditions, including autoimmune and infectious (especially viral) syndromes, and initial work-up should focus on these etiologies.

The pancytopenia is obviously notable. It raises the possibility that the oral ulcerations are due to the neutropenia rather than a primary disease manifestation. Other possible causes of pancytopenia include SLE, antiphospholipid antibody syndrome, and related rheumatologic diagnoses, including hemophagocytic lymphohistiocytosis (HLH). Given her age and subacute presentation, secondary forms of HLH seem more likely than primary (genetic) forms, which typically present within the first few years of life. Secondary forms of HLH can occur in association with rheumatic diseases and are then referred to as Macrophage Activation Syndrome (MAS). The most common rheumatologic diseases associated with MAS are systemic juvenile idiopathic arthritis, SLE, and Kawasaki disease. Secondary HLH can also occur with infectious diseases, particularly viral infections such as EBV. It is also important to consider thrombotic thrombocytopenic purpura and other forms of thrombotic microangiopathy, especially if her violaceous eyelids actually represent purpura. The presence of pancytopenia also expands the differential diagnosis to include leukemia, lymphoma, and other oncologic diseases. After obtaining results from pending infectious disease studies, additional diagnostic work-up should include examination of the bone marrow and a peripheral blood smear to evaluate for hemophagocytosis and/or malignancy. Testing for double-stranded DNA antibodies and antinuclear antibodies (ANA) should be sent to evaluate for SLE, and antiphospholipid antibodies should also be checked. Renal function must also be evaluated.
Additional laboratory work-up revealed a reticulocyte count of 0.2%, a positive Coombs immunoglobulin G (IgG) test, haptoglobin less than 80 mg/L, and lactate dehydrogenase (LDH) 25.2 µkat/L (1509 units/L); coagulation studies were normal. Her chemistries showed electrolytes, blood urea nitrogen, and creatinine were within normal limits; her aspartate aminotransferase was 216 units/L, and alanine aminotransferase was 56 units/L. Her spot urine protein-to-creatinine ratio was 1.28. Complement and inflammatory studies showed C3 0.14 g/L (14 mg/dL, normal 83-151 mg/dL), C4 0.05 g/L (5 mg/dL, normal 13-37 mg/dL), erythrocyte sedimentation rate (ESR) 103 mm/hr (normal 0-20 mm/hr), and C-reactive protein (CRP) 3.2 mg/L (normal 0.7-1.7 mg/L). Additional studies showed elevated triglycerides (376 mg/dL), elevated creatine kinase (2437 units/L), and elevated ferritin (22,295.5 ng/mL). An ANA screen and specific autoantibody studies were sent, including antidouble stranded DNA antibody, antiribonucleoprotein antibody, anti-Smith antibody, anti-Ro antibody, and anti-La antibody. A bone marrow biopsy was performed.
The hematologic studies provide a mixed picture. There is evidence of an autoimmune hemolytic anemia (AIHA). Typically, AIHA is associated with reticulocytosis rather than reticulocytopenia. Reticulocytopenia can occur in AIHA, however, because of antibodies directed against erythroid precursors or if 2 processes are occurring simultaneously—ie, AIHA plus bone marrow destructive/failure process. The latter scenario is more likely here. Specifically, the pancytopenia, elevated triglycerides, and extreme hyperferritinemia strongly support the diagnosis of HLH. The very low C3 and C4 suggest a complement-consumptive process, and SLE is the most likely etiology. Proteinuria and Coombs-positive anemia are also features of SLE. The discordance between the ESR (markedly elevated) and CRP (mild elevation) is surprising in the setting of systemic inflammation. However, her other clinical features are consistent with marked systemic inflammation, and it is important not to dismiss a likely diagnosis simply on the basis of a few incongruous features. At this point, the diagnosis of SLE complicated by secondary HLH is favored, remembering that both these entities can be triggered by a viral infection. Therefore, diligent follow-up of the aforementioned specific autoantibody studies and the bone marrow biopsy is the next logical step, along with the still-pending infectious disease studies.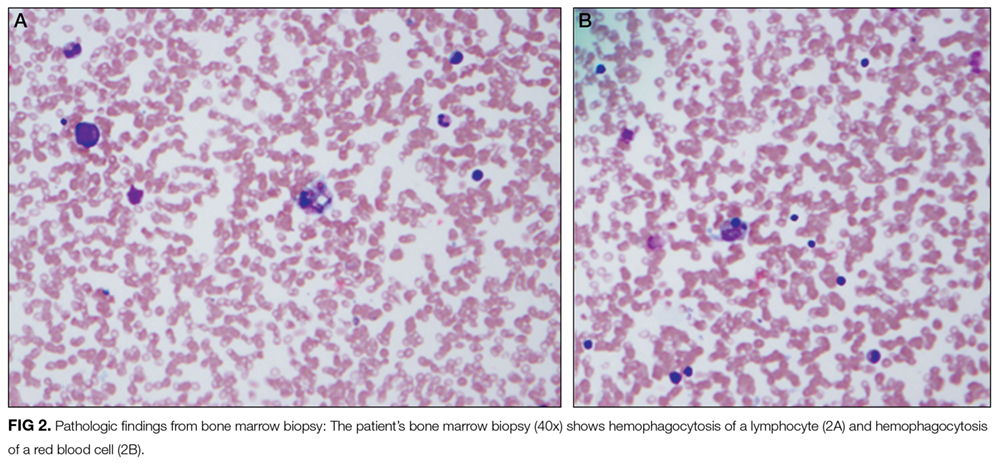
These findings are consistent with the diagnosis of SLE complicated by secondary HLH (ie, MAS). It remains possible, but unlikely, that the patient has genetic or familial HLH (fHLH), as this entity is exceedingly rare with distinct underlying genetic aberrations separate from SLE. Ideally, the NK cell function studies would be repeated after the current episode of HLH is controlled and the patient is off of immunosuppressive therapies, but this will likely not be possible given the underlying SLE. Patients with fHLH have reduced or absent NK cell function at baseline (ie, not only during an acute episode of HLH and not because of immunosuppressive medications). Alternatively, one could consider genetic testing for fHLH. The clinical importance of doing this is that patients with fHLH are candidates for bone marrow or stem cell transplantation. There currently is not a published standard of care for the work-up and management of MAS in children with rheumatic disease, so the decision to repeat NK cell function testing and/or genetic testing would be left to the discretion of the treating physician and would depend on the patient’s ongoing clinical course.
The patient required red blood cell and platelet transfusions. She received pulse dose intravenous methylprednisolone for treatment of SLE and MAS; she clinically improved within 48 hours of starting steroids. Cyclosporine was added for management of MAS. The patient was transitioned to oral corticosteroids and discharged home. All cell counts normalized within 1 month of discharge. She was weaned off corticosteroids and cyclosporine was discontinued. Her maintenance SLE therapy includes hydroxychloroquine and mycophenolate mofetil.
COMMENTARY
Because the differential diagnosis for new-onset pancytopenia encompasses many diseases across several medical subspecialties, a thorough history and physical exam are necessary to form a tailored clinical approach.1 The primary causes of pediatric pancytopenia vary depending on geographic location because of the local prevalence of infectious agents and nutritional deficiency patterns. A retrospective study investigating the primary cause of pancytopenia in children without existing malignancy presenting to a US tertiary care hospital found that 64% of cases were due to infection, 28% were due to hematologic disease (most frequently aplastic anemia), and 8% were due to miscellaneous etiologies, including adverse drug reactions and autoimmune diseases.2 In contrast, the most common cause of pancytopenia in pediatric patients presenting to a tertiary care hospital in India was megaloblastic anemia (28%), followed by infections (21%), acute leukemia (21%), and aplastic anemia (20%).3 While clinicians do (and should) consider malignancy as a cause of pancytopenia, there is sparse literature regarding the frequency of pancytopenia associated with the presentations of childhood malignancies.4 A study of pediatric patients with acute lymphoblastic anemia found that only 11% of newly diagnosed patients had pancytopenia at initial presentation.4
There are no official guidelines for the work-up of pediatric pancytopenia from any of the academic societies. Depending on the clinical history, initial laboratory investigation for pediatric pancytopenia may include complete blood cell count with differential, reticulocyte count, peripheral blood smear, complete metabolic panel, hemolysis labs (haptoglobin, LDH, Coombs test) and inflammatory markers (ESR, CRP, fibrinogen). Further investigation to clarify the specific etiology of pancytopenia can be guided by the results of these initial tests.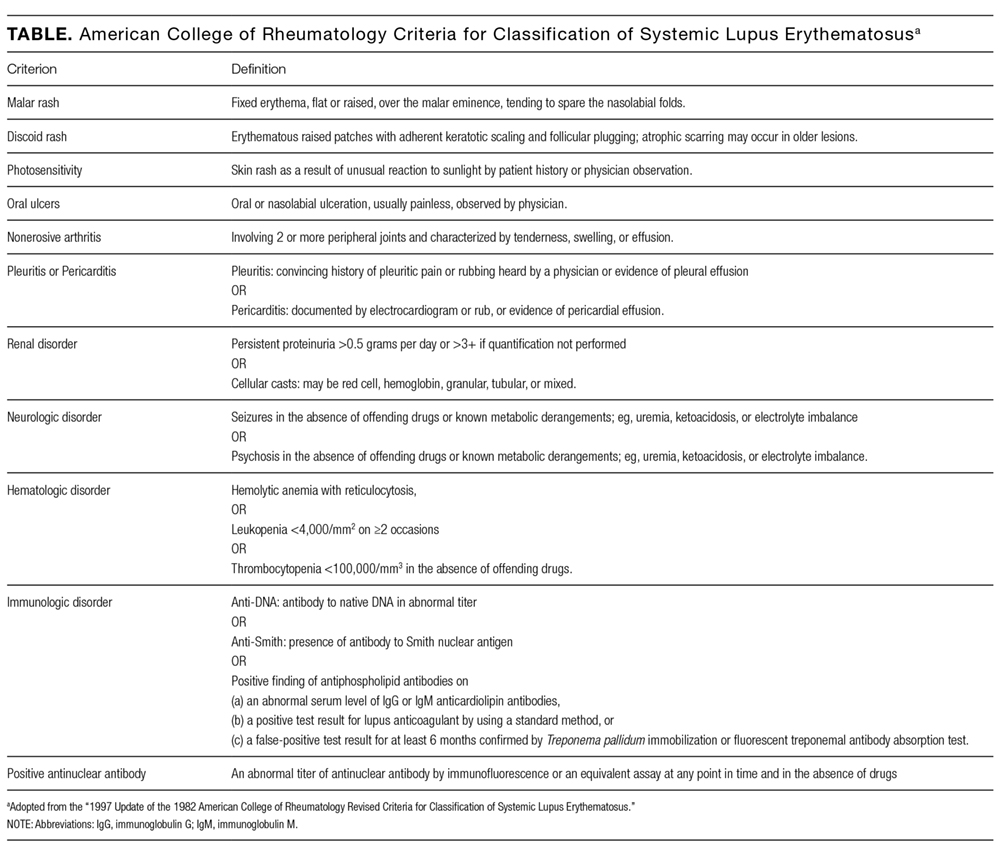
Hematologic abnormalities are present in greater than 70% of pediatric SLE cases.10,11 The pathogenesis of hematologic abnormalities in SLE is heterogeneous, involving actions of autoreactive lymphocytes, autoantibodies, and proinflammatory cytokines that can disrupt bone marrow production and cause peripheral blood cell destruction.12,13 While pancytopenia is common in children with SLE, other coexisting diagnoses should be considered in patients with SLE and pancytopenia. Concurrent diagnoses that can lead to pancytopenia in patients with SLE include infection, pharmacologic side effects, and secondary HLH,14,15 each of which has differing implications for prognosis and treatment.
Secondary HLH is a severe and often acute complication of systemic inflammatory disorders caused by the proliferation and activation of T cells and macrophages, leading to an enhanced inflammatory state. When HLH occurs in the setting of an underlying autoimmune or autoinflammatory process, it is typically termed MAS. MAS affects an estimated 0.9% to 4.6% of patients with SLE.16 Early diagnosis and treatment of MAS is important because MAS can be rapidly fatal, with a mortality rate of 8% to 20% in pediatric patients.17,18 Clinical features of MAS include physical exam findings of fever and splenomegaly as well as laboratory abnormalities, including pancytopenia, elevated ferritin, elevated triglycerides, and low fibrinogen.18 A bone marrow biopsy showing hemophagocytosis in the absence of malignancy is diagnostic of MAS. Although a bone marrow biopsy is not required to diagnose MAS, it is often obtained to exclude other etiologies of pancytopenia such as malignancy.19 Specialized diagnostic testing for MAS includes NK cell counts and functional studies, including expression of perforin and granzyme B (NK cell proteins triggering apoptosis in target cells), soluble IL-2R (marker of activated lymphocytes), and CD163 (transmembrane protein of hemophagocytic macrophages). There is no standardized protocol for treating MAS.20 It is most commonly treated with highdose corticosteroids; additional agents, including cyclosporine and biologic therapies, are also utilized.16,20
KEY POINTS
- Children with SLE tend to have greater involvement of major organ systems and more rapid accrual of organ damage than adults with SLE. Therefore, it is sometimes necessary to initiate immunosuppressive therapies before full diagnostic criteria are met, provided that malignancy and infection have been ruled out.
- While pancytopenia is common in pediatric patients with SLE, providers should make sure to consider coexisting diagnoses such as infection and MAS, both of which require different treatment strategies.
- It is important to consider HLH/MAS early in the work-up of pancytopenia, because early diagnosis and treatment improves clinical outcomes. Obtaining a ferritin level can aid in the work-up of pancytopenia because it is both a sensitive and specific marker of HLH/MAS when dramatically elevated.
Disclosure
The authors report no conflicts of interest.
1. Weinzierl EP, Arber DA. The Differential Diagnosis and Bone Marrow Evaluation of New-Onset Pancytopenia. Am J Clin Pathol. 2012;139(1):9-29. doi:10.1309/AJCP50AEEYGREWUZ. PubMed
2. Pine M, Walter AW. Pancytopenia in hospitalized children: a five-year review. J Pediatr Hematol Oncol. 2010;32(5):e192-e194. doi:10.1097/MPH.0b013e3181e03082. PubMed
3. Bhatnagar SK. Pancytopenia in Children: Etiological Profile. J Trop Pediatr. 2005;51(4):236-239. doi:10.1093/tropej/fmi010. PubMed
4. Kulkarni KP, Marwaha RK. Acute lymphoblastic leukemia with pancytopenia at presentation: clinical correlates, prognostic impact, and association with survival. J Pediatr Hematol Oncol. 2013;35(7):573-576. doi:10.1097/MPH.0b013e31829d46f3. PubMed
5. Holubar, K. Terminology and iconography of lupus erythematosus: A historical vignette. Am J Dermatopathol. 1980;2(3):239-242. PubMed
6. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. PubMed
7. Petri M, Orbai, A, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686. doi:10.1002/art.34473. PubMed
8. Tucker L. Review: Making the diagnosis of systemic lupus erythematosus in children and adolescents. Lupus. 2007;16(8):546-549. doi:10.1177/0961203307078068. PubMed
9. Brunner HI, Gladman DD, Ibañez D, Urowitz MD, Silverman ED. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58(2):556-562. doi:10.1002/art.23204. PubMed
10. Benseler SM, Silverman ED. Systemic Lupus Erythematosus. Rheum Dis Clin North Am. 2007;33(3):471-498. doi:10.1016/j.rdc.2007.07.008. PubMed
11. Gokce M, Bilginer Y, Besbas N, et al. Hematological features of pediatric systemic lupus erythematosus: suggesting management strategies in children. Lupus. 2012;21(8):878-884. doi:10.1177/0961203312443721. PubMed
12. Voulgarelis M, Giannouli S, Tasidou A, Anagnostou D, Ziakas PD, Tzioufas AG. Bone marrow histological findings in systemic lupus erythematosus with hematologic abnormalities: A clinicopathological study. Am J Hematol. 2006;81(8):590-597. doi:10.1002/ajh.20593. PubMed
13. Pereira RM, Velloso ER, Menezes Y, Gualandro S, Vassalo J, Yoshinari NH. Bone marrow findings in systemic lupus erythematosus patients with peripheral cytopenias. Clin Rheumatol. 1998;17(3):219-222. PubMed
14. Avčin T, Tse SML, Schneider R, Ngan B, Silverman ED. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006;148(5):683-686. doi:10.1016/j.jpeds.2005.12.070. PubMed
15. Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and Long-Term Outcome of 15 Episodes of Systemic Lupus Erythematosus-Associated Hemophagocytic Syndrome. Medicine. 2006;85(3):169-182. doi:10.1097/01.md.0000224708.62510.d1. PubMed
16. Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology. 2008;47(11):1686-1691. doi:10.1093/rheumatology/ken342. PubMed
17. Stephan JL. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285-1292. doi:10.1093/rheumatology/40.11.1285. PubMed
18. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421-426. PubMed
19. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039. PubMed
20. Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol. 2012;31(8):1223-1230. doi:10.1007/s10067-012-1998-0. PubMed
A 15-year-old African American girl presented to the emergency department with 3 days of fever, sore throat, nausea, vomiting, and poor appetite. She reported a 4-week history of fatigue, right hand pain and swelling, and a 6-kilogram weight loss for which she had seen her primary care provider several times. She reported no recent travel, sick contacts, or new medications.
It appears that there are potentially at least 2 separate problems: an acute one (past 3 days) and a more chronic one (past 4 weeks). These 2 problems may be directly related (ie, acute worsening of the more chronic problem), indirectly related (ie, the more chronic problem is leading to increased susceptibility to the acute problem, for instance, an evolving immunodeficiency predisposing to an opportunistic infection), or “true, true, but unrelated.” The clinical challenge is to keep one’s mind open to each of these potential scenarios and to avoid the tendency to focus on one of the problems and not pay enough attention to the other. Occam’s razor likely does not apply here.
Numerous common and typically transient diseases could cause the symptoms of the past 3 days, particularly infectious etiologies such as streptococcal pharyngitis or a viral infection. One cannot forget about these possibilities while contemplating the more worrisome symptoms of the past 4 weeks, especially weight loss in a growing adolescent. Patients may unintentionally lose weight for a variety of reasons, which can be broadly categorized by decreased caloric supply, gastrointestinal losses or malabsorption, and increased caloric demand; these categories are not mutually exclusive.
Lastly, 1 symptom may provide a more specific direction: the right hand pain and swelling of the past 4 weeks. More specifics, including the extent of the hand swelling, other areas of involvement, and the nature of her pain, will be helpful.
Her temperature was 99.5°F, heart rate 100 beats per minute, respiratory rate 18 breaths per minute, oxygen saturation 95% while breathing ambient air, blood pressure 99/56 mmHg, weight 44 kilograms, height 161 centimeters, and body mass index 17. She appeared generally ill and underweight. She had edematous and violaceous eyelids, dry cracked lips, and pharyngeal erythema with ulcerations of the hard palate. She had nontender cervical and inguinal lymphadenopathy. Her abdomen was tender to palpation in the lower quadrants without guarding or rebound; there was no organomegaly. A right knee effusion with overlying warmth was present without redness or decreased range of motion. She also had an enlarged third proximal interphalangeal joint and loss of palpable metacarpal phalangeal joint landmarks on her right hand. She was noted to be using her arms to move her legs when repositioning in bed.
These exam findings clearly point toward a systemic process but not 1 specific diagnosis. The presence of at least 2 inflamed joints points toward rheumatologic/inflammatory or infectious diseases. Localized edema (eyelids and right metacarpal phalangeal joints), oral ulcers, possible myositis, and arthritis point toward a systemic vasculitis (eg, granulomatosis with polyangiitis, Behçet disease). While Kawasaki disease is also a systemic vasculitis, the presence of oral ulcers and generalized lymphadenopathy argues against it. Inflammatory myopathies like polymyositis, and especially juvenile dermatomyositis, fit many aspects of this presentation with the violaceous eyelids and possible myositis, though no other cutaneous stigmata of this disease are evident (eg, no Gottron’s papules). Polyarthritis, violaceous eyelids, and possible myositis could be consistent with systemic lupus erythematosus (SLE).
The presence of oral ulcers and arthritis make other systemic inflammatory conditions, such as inflammatory bowel disease with arthritis and autoimmune- or infection-related hepatitis, possible. Infectious etiologies alone or in combination with a rheumatologic process are also possible given fevers and lymphadenopathy. In particular, herpesvirus infections (Epstein-Barr virus [EBV], cytomegalovirus [CMV], herpes simplex virus, or human herpes virus 6), human immunodeficiency virus (HIV), hepatitis C virus (HCV), and syphilis can cause oral ulcers and lymphadenopathy. Other potential infectious etiologies include subacute bacterial endocarditis and disseminated gonococcal infection given the presence of polyarthritis, but these infections are less likely as they do not explain all of the symptoms.
In summary, the differential diagnosis is broad and should be prioritized to consider systemic inflammatory conditions, including autoimmune and infectious (especially viral) syndromes, and initial work-up should focus on these etiologies.
The pancytopenia is obviously notable. It raises the possibility that the oral ulcerations are due to the neutropenia rather than a primary disease manifestation. Other possible causes of pancytopenia include SLE, antiphospholipid antibody syndrome, and related rheumatologic diagnoses, including hemophagocytic lymphohistiocytosis (HLH). Given her age and subacute presentation, secondary forms of HLH seem more likely than primary (genetic) forms, which typically present within the first few years of life. Secondary forms of HLH can occur in association with rheumatic diseases and are then referred to as Macrophage Activation Syndrome (MAS). The most common rheumatologic diseases associated with MAS are systemic juvenile idiopathic arthritis, SLE, and Kawasaki disease. Secondary HLH can also occur with infectious diseases, particularly viral infections such as EBV. It is also important to consider thrombotic thrombocytopenic purpura and other forms of thrombotic microangiopathy, especially if her violaceous eyelids actually represent purpura. The presence of pancytopenia also expands the differential diagnosis to include leukemia, lymphoma, and other oncologic diseases. After obtaining results from pending infectious disease studies, additional diagnostic work-up should include examination of the bone marrow and a peripheral blood smear to evaluate for hemophagocytosis and/or malignancy. Testing for double-stranded DNA antibodies and antinuclear antibodies (ANA) should be sent to evaluate for SLE, and antiphospholipid antibodies should also be checked. Renal function must also be evaluated.
Additional laboratory work-up revealed a reticulocyte count of 0.2%, a positive Coombs immunoglobulin G (IgG) test, haptoglobin less than 80 mg/L, and lactate dehydrogenase (LDH) 25.2 µkat/L (1509 units/L); coagulation studies were normal. Her chemistries showed electrolytes, blood urea nitrogen, and creatinine were within normal limits; her aspartate aminotransferase was 216 units/L, and alanine aminotransferase was 56 units/L. Her spot urine protein-to-creatinine ratio was 1.28. Complement and inflammatory studies showed C3 0.14 g/L (14 mg/dL, normal 83-151 mg/dL), C4 0.05 g/L (5 mg/dL, normal 13-37 mg/dL), erythrocyte sedimentation rate (ESR) 103 mm/hr (normal 0-20 mm/hr), and C-reactive protein (CRP) 3.2 mg/L (normal 0.7-1.7 mg/L). Additional studies showed elevated triglycerides (376 mg/dL), elevated creatine kinase (2437 units/L), and elevated ferritin (22,295.5 ng/mL). An ANA screen and specific autoantibody studies were sent, including antidouble stranded DNA antibody, antiribonucleoprotein antibody, anti-Smith antibody, anti-Ro antibody, and anti-La antibody. A bone marrow biopsy was performed.
The hematologic studies provide a mixed picture. There is evidence of an autoimmune hemolytic anemia (AIHA). Typically, AIHA is associated with reticulocytosis rather than reticulocytopenia. Reticulocytopenia can occur in AIHA, however, because of antibodies directed against erythroid precursors or if 2 processes are occurring simultaneously—ie, AIHA plus bone marrow destructive/failure process. The latter scenario is more likely here. Specifically, the pancytopenia, elevated triglycerides, and extreme hyperferritinemia strongly support the diagnosis of HLH. The very low C3 and C4 suggest a complement-consumptive process, and SLE is the most likely etiology. Proteinuria and Coombs-positive anemia are also features of SLE. The discordance between the ESR (markedly elevated) and CRP (mild elevation) is surprising in the setting of systemic inflammation. However, her other clinical features are consistent with marked systemic inflammation, and it is important not to dismiss a likely diagnosis simply on the basis of a few incongruous features. At this point, the diagnosis of SLE complicated by secondary HLH is favored, remembering that both these entities can be triggered by a viral infection. Therefore, diligent follow-up of the aforementioned specific autoantibody studies and the bone marrow biopsy is the next logical step, along with the still-pending infectious disease studies.
These findings are consistent with the diagnosis of SLE complicated by secondary HLH (ie, MAS). It remains possible, but unlikely, that the patient has genetic or familial HLH (fHLH), as this entity is exceedingly rare with distinct underlying genetic aberrations separate from SLE. Ideally, the NK cell function studies would be repeated after the current episode of HLH is controlled and the patient is off of immunosuppressive therapies, but this will likely not be possible given the underlying SLE. Patients with fHLH have reduced or absent NK cell function at baseline (ie, not only during an acute episode of HLH and not because of immunosuppressive medications). Alternatively, one could consider genetic testing for fHLH. The clinical importance of doing this is that patients with fHLH are candidates for bone marrow or stem cell transplantation. There currently is not a published standard of care for the work-up and management of MAS in children with rheumatic disease, so the decision to repeat NK cell function testing and/or genetic testing would be left to the discretion of the treating physician and would depend on the patient’s ongoing clinical course.
The patient required red blood cell and platelet transfusions. She received pulse dose intravenous methylprednisolone for treatment of SLE and MAS; she clinically improved within 48 hours of starting steroids. Cyclosporine was added for management of MAS. The patient was transitioned to oral corticosteroids and discharged home. All cell counts normalized within 1 month of discharge. She was weaned off corticosteroids and cyclosporine was discontinued. Her maintenance SLE therapy includes hydroxychloroquine and mycophenolate mofetil.
COMMENTARY
Because the differential diagnosis for new-onset pancytopenia encompasses many diseases across several medical subspecialties, a thorough history and physical exam are necessary to form a tailored clinical approach.1 The primary causes of pediatric pancytopenia vary depending on geographic location because of the local prevalence of infectious agents and nutritional deficiency patterns. A retrospective study investigating the primary cause of pancytopenia in children without existing malignancy presenting to a US tertiary care hospital found that 64% of cases were due to infection, 28% were due to hematologic disease (most frequently aplastic anemia), and 8% were due to miscellaneous etiologies, including adverse drug reactions and autoimmune diseases.2 In contrast, the most common cause of pancytopenia in pediatric patients presenting to a tertiary care hospital in India was megaloblastic anemia (28%), followed by infections (21%), acute leukemia (21%), and aplastic anemia (20%).3 While clinicians do (and should) consider malignancy as a cause of pancytopenia, there is sparse literature regarding the frequency of pancytopenia associated with the presentations of childhood malignancies.4 A study of pediatric patients with acute lymphoblastic anemia found that only 11% of newly diagnosed patients had pancytopenia at initial presentation.4
There are no official guidelines for the work-up of pediatric pancytopenia from any of the academic societies. Depending on the clinical history, initial laboratory investigation for pediatric pancytopenia may include complete blood cell count with differential, reticulocyte count, peripheral blood smear, complete metabolic panel, hemolysis labs (haptoglobin, LDH, Coombs test) and inflammatory markers (ESR, CRP, fibrinogen). Further investigation to clarify the specific etiology of pancytopenia can be guided by the results of these initial tests.
Hematologic abnormalities are present in greater than 70% of pediatric SLE cases.10,11 The pathogenesis of hematologic abnormalities in SLE is heterogeneous, involving actions of autoreactive lymphocytes, autoantibodies, and proinflammatory cytokines that can disrupt bone marrow production and cause peripheral blood cell destruction.12,13 While pancytopenia is common in children with SLE, other coexisting diagnoses should be considered in patients with SLE and pancytopenia. Concurrent diagnoses that can lead to pancytopenia in patients with SLE include infection, pharmacologic side effects, and secondary HLH,14,15 each of which has differing implications for prognosis and treatment.
Secondary HLH is a severe and often acute complication of systemic inflammatory disorders caused by the proliferation and activation of T cells and macrophages, leading to an enhanced inflammatory state. When HLH occurs in the setting of an underlying autoimmune or autoinflammatory process, it is typically termed MAS. MAS affects an estimated 0.9% to 4.6% of patients with SLE.16 Early diagnosis and treatment of MAS is important because MAS can be rapidly fatal, with a mortality rate of 8% to 20% in pediatric patients.17,18 Clinical features of MAS include physical exam findings of fever and splenomegaly as well as laboratory abnormalities, including pancytopenia, elevated ferritin, elevated triglycerides, and low fibrinogen.18 A bone marrow biopsy showing hemophagocytosis in the absence of malignancy is diagnostic of MAS. Although a bone marrow biopsy is not required to diagnose MAS, it is often obtained to exclude other etiologies of pancytopenia such as malignancy.19 Specialized diagnostic testing for MAS includes NK cell counts and functional studies, including expression of perforin and granzyme B (NK cell proteins triggering apoptosis in target cells), soluble IL-2R (marker of activated lymphocytes), and CD163 (transmembrane protein of hemophagocytic macrophages). There is no standardized protocol for treating MAS.20 It is most commonly treated with highdose corticosteroids; additional agents, including cyclosporine and biologic therapies, are also utilized.16,20
KEY POINTS
- Children with SLE tend to have greater involvement of major organ systems and more rapid accrual of organ damage than adults with SLE. Therefore, it is sometimes necessary to initiate immunosuppressive therapies before full diagnostic criteria are met, provided that malignancy and infection have been ruled out.
- While pancytopenia is common in pediatric patients with SLE, providers should make sure to consider coexisting diagnoses such as infection and MAS, both of which require different treatment strategies.
- It is important to consider HLH/MAS early in the work-up of pancytopenia, because early diagnosis and treatment improves clinical outcomes. Obtaining a ferritin level can aid in the work-up of pancytopenia because it is both a sensitive and specific marker of HLH/MAS when dramatically elevated.
Disclosure
The authors report no conflicts of interest.
A 15-year-old African American girl presented to the emergency department with 3 days of fever, sore throat, nausea, vomiting, and poor appetite. She reported a 4-week history of fatigue, right hand pain and swelling, and a 6-kilogram weight loss for which she had seen her primary care provider several times. She reported no recent travel, sick contacts, or new medications.
It appears that there are potentially at least 2 separate problems: an acute one (past 3 days) and a more chronic one (past 4 weeks). These 2 problems may be directly related (ie, acute worsening of the more chronic problem), indirectly related (ie, the more chronic problem is leading to increased susceptibility to the acute problem, for instance, an evolving immunodeficiency predisposing to an opportunistic infection), or “true, true, but unrelated.” The clinical challenge is to keep one’s mind open to each of these potential scenarios and to avoid the tendency to focus on one of the problems and not pay enough attention to the other. Occam’s razor likely does not apply here.
Numerous common and typically transient diseases could cause the symptoms of the past 3 days, particularly infectious etiologies such as streptococcal pharyngitis or a viral infection. One cannot forget about these possibilities while contemplating the more worrisome symptoms of the past 4 weeks, especially weight loss in a growing adolescent. Patients may unintentionally lose weight for a variety of reasons, which can be broadly categorized by decreased caloric supply, gastrointestinal losses or malabsorption, and increased caloric demand; these categories are not mutually exclusive.
Lastly, 1 symptom may provide a more specific direction: the right hand pain and swelling of the past 4 weeks. More specifics, including the extent of the hand swelling, other areas of involvement, and the nature of her pain, will be helpful.
Her temperature was 99.5°F, heart rate 100 beats per minute, respiratory rate 18 breaths per minute, oxygen saturation 95% while breathing ambient air, blood pressure 99/56 mmHg, weight 44 kilograms, height 161 centimeters, and body mass index 17. She appeared generally ill and underweight. She had edematous and violaceous eyelids, dry cracked lips, and pharyngeal erythema with ulcerations of the hard palate. She had nontender cervical and inguinal lymphadenopathy. Her abdomen was tender to palpation in the lower quadrants without guarding or rebound; there was no organomegaly. A right knee effusion with overlying warmth was present without redness or decreased range of motion. She also had an enlarged third proximal interphalangeal joint and loss of palpable metacarpal phalangeal joint landmarks on her right hand. She was noted to be using her arms to move her legs when repositioning in bed.
These exam findings clearly point toward a systemic process but not 1 specific diagnosis. The presence of at least 2 inflamed joints points toward rheumatologic/inflammatory or infectious diseases. Localized edema (eyelids and right metacarpal phalangeal joints), oral ulcers, possible myositis, and arthritis point toward a systemic vasculitis (eg, granulomatosis with polyangiitis, Behçet disease). While Kawasaki disease is also a systemic vasculitis, the presence of oral ulcers and generalized lymphadenopathy argues against it. Inflammatory myopathies like polymyositis, and especially juvenile dermatomyositis, fit many aspects of this presentation with the violaceous eyelids and possible myositis, though no other cutaneous stigmata of this disease are evident (eg, no Gottron’s papules). Polyarthritis, violaceous eyelids, and possible myositis could be consistent with systemic lupus erythematosus (SLE).
The presence of oral ulcers and arthritis make other systemic inflammatory conditions, such as inflammatory bowel disease with arthritis and autoimmune- or infection-related hepatitis, possible. Infectious etiologies alone or in combination with a rheumatologic process are also possible given fevers and lymphadenopathy. In particular, herpesvirus infections (Epstein-Barr virus [EBV], cytomegalovirus [CMV], herpes simplex virus, or human herpes virus 6), human immunodeficiency virus (HIV), hepatitis C virus (HCV), and syphilis can cause oral ulcers and lymphadenopathy. Other potential infectious etiologies include subacute bacterial endocarditis and disseminated gonococcal infection given the presence of polyarthritis, but these infections are less likely as they do not explain all of the symptoms.
In summary, the differential diagnosis is broad and should be prioritized to consider systemic inflammatory conditions, including autoimmune and infectious (especially viral) syndromes, and initial work-up should focus on these etiologies.
The pancytopenia is obviously notable. It raises the possibility that the oral ulcerations are due to the neutropenia rather than a primary disease manifestation. Other possible causes of pancytopenia include SLE, antiphospholipid antibody syndrome, and related rheumatologic diagnoses, including hemophagocytic lymphohistiocytosis (HLH). Given her age and subacute presentation, secondary forms of HLH seem more likely than primary (genetic) forms, which typically present within the first few years of life. Secondary forms of HLH can occur in association with rheumatic diseases and are then referred to as Macrophage Activation Syndrome (MAS). The most common rheumatologic diseases associated with MAS are systemic juvenile idiopathic arthritis, SLE, and Kawasaki disease. Secondary HLH can also occur with infectious diseases, particularly viral infections such as EBV. It is also important to consider thrombotic thrombocytopenic purpura and other forms of thrombotic microangiopathy, especially if her violaceous eyelids actually represent purpura. The presence of pancytopenia also expands the differential diagnosis to include leukemia, lymphoma, and other oncologic diseases. After obtaining results from pending infectious disease studies, additional diagnostic work-up should include examination of the bone marrow and a peripheral blood smear to evaluate for hemophagocytosis and/or malignancy. Testing for double-stranded DNA antibodies and antinuclear antibodies (ANA) should be sent to evaluate for SLE, and antiphospholipid antibodies should also be checked. Renal function must also be evaluated.
Additional laboratory work-up revealed a reticulocyte count of 0.2%, a positive Coombs immunoglobulin G (IgG) test, haptoglobin less than 80 mg/L, and lactate dehydrogenase (LDH) 25.2 µkat/L (1509 units/L); coagulation studies were normal. Her chemistries showed electrolytes, blood urea nitrogen, and creatinine were within normal limits; her aspartate aminotransferase was 216 units/L, and alanine aminotransferase was 56 units/L. Her spot urine protein-to-creatinine ratio was 1.28. Complement and inflammatory studies showed C3 0.14 g/L (14 mg/dL, normal 83-151 mg/dL), C4 0.05 g/L (5 mg/dL, normal 13-37 mg/dL), erythrocyte sedimentation rate (ESR) 103 mm/hr (normal 0-20 mm/hr), and C-reactive protein (CRP) 3.2 mg/L (normal 0.7-1.7 mg/L). Additional studies showed elevated triglycerides (376 mg/dL), elevated creatine kinase (2437 units/L), and elevated ferritin (22,295.5 ng/mL). An ANA screen and specific autoantibody studies were sent, including antidouble stranded DNA antibody, antiribonucleoprotein antibody, anti-Smith antibody, anti-Ro antibody, and anti-La antibody. A bone marrow biopsy was performed.
The hematologic studies provide a mixed picture. There is evidence of an autoimmune hemolytic anemia (AIHA). Typically, AIHA is associated with reticulocytosis rather than reticulocytopenia. Reticulocytopenia can occur in AIHA, however, because of antibodies directed against erythroid precursors or if 2 processes are occurring simultaneously—ie, AIHA plus bone marrow destructive/failure process. The latter scenario is more likely here. Specifically, the pancytopenia, elevated triglycerides, and extreme hyperferritinemia strongly support the diagnosis of HLH. The very low C3 and C4 suggest a complement-consumptive process, and SLE is the most likely etiology. Proteinuria and Coombs-positive anemia are also features of SLE. The discordance between the ESR (markedly elevated) and CRP (mild elevation) is surprising in the setting of systemic inflammation. However, her other clinical features are consistent with marked systemic inflammation, and it is important not to dismiss a likely diagnosis simply on the basis of a few incongruous features. At this point, the diagnosis of SLE complicated by secondary HLH is favored, remembering that both these entities can be triggered by a viral infection. Therefore, diligent follow-up of the aforementioned specific autoantibody studies and the bone marrow biopsy is the next logical step, along with the still-pending infectious disease studies.
These findings are consistent with the diagnosis of SLE complicated by secondary HLH (ie, MAS). It remains possible, but unlikely, that the patient has genetic or familial HLH (fHLH), as this entity is exceedingly rare with distinct underlying genetic aberrations separate from SLE. Ideally, the NK cell function studies would be repeated after the current episode of HLH is controlled and the patient is off of immunosuppressive therapies, but this will likely not be possible given the underlying SLE. Patients with fHLH have reduced or absent NK cell function at baseline (ie, not only during an acute episode of HLH and not because of immunosuppressive medications). Alternatively, one could consider genetic testing for fHLH. The clinical importance of doing this is that patients with fHLH are candidates for bone marrow or stem cell transplantation. There currently is not a published standard of care for the work-up and management of MAS in children with rheumatic disease, so the decision to repeat NK cell function testing and/or genetic testing would be left to the discretion of the treating physician and would depend on the patient’s ongoing clinical course.
The patient required red blood cell and platelet transfusions. She received pulse dose intravenous methylprednisolone for treatment of SLE and MAS; she clinically improved within 48 hours of starting steroids. Cyclosporine was added for management of MAS. The patient was transitioned to oral corticosteroids and discharged home. All cell counts normalized within 1 month of discharge. She was weaned off corticosteroids and cyclosporine was discontinued. Her maintenance SLE therapy includes hydroxychloroquine and mycophenolate mofetil.
COMMENTARY
Because the differential diagnosis for new-onset pancytopenia encompasses many diseases across several medical subspecialties, a thorough history and physical exam are necessary to form a tailored clinical approach.1 The primary causes of pediatric pancytopenia vary depending on geographic location because of the local prevalence of infectious agents and nutritional deficiency patterns. A retrospective study investigating the primary cause of pancytopenia in children without existing malignancy presenting to a US tertiary care hospital found that 64% of cases were due to infection, 28% were due to hematologic disease (most frequently aplastic anemia), and 8% were due to miscellaneous etiologies, including adverse drug reactions and autoimmune diseases.2 In contrast, the most common cause of pancytopenia in pediatric patients presenting to a tertiary care hospital in India was megaloblastic anemia (28%), followed by infections (21%), acute leukemia (21%), and aplastic anemia (20%).3 While clinicians do (and should) consider malignancy as a cause of pancytopenia, there is sparse literature regarding the frequency of pancytopenia associated with the presentations of childhood malignancies.4 A study of pediatric patients with acute lymphoblastic anemia found that only 11% of newly diagnosed patients had pancytopenia at initial presentation.4
There are no official guidelines for the work-up of pediatric pancytopenia from any of the academic societies. Depending on the clinical history, initial laboratory investigation for pediatric pancytopenia may include complete blood cell count with differential, reticulocyte count, peripheral blood smear, complete metabolic panel, hemolysis labs (haptoglobin, LDH, Coombs test) and inflammatory markers (ESR, CRP, fibrinogen). Further investigation to clarify the specific etiology of pancytopenia can be guided by the results of these initial tests.
Hematologic abnormalities are present in greater than 70% of pediatric SLE cases.10,11 The pathogenesis of hematologic abnormalities in SLE is heterogeneous, involving actions of autoreactive lymphocytes, autoantibodies, and proinflammatory cytokines that can disrupt bone marrow production and cause peripheral blood cell destruction.12,13 While pancytopenia is common in children with SLE, other coexisting diagnoses should be considered in patients with SLE and pancytopenia. Concurrent diagnoses that can lead to pancytopenia in patients with SLE include infection, pharmacologic side effects, and secondary HLH,14,15 each of which has differing implications for prognosis and treatment.
Secondary HLH is a severe and often acute complication of systemic inflammatory disorders caused by the proliferation and activation of T cells and macrophages, leading to an enhanced inflammatory state. When HLH occurs in the setting of an underlying autoimmune or autoinflammatory process, it is typically termed MAS. MAS affects an estimated 0.9% to 4.6% of patients with SLE.16 Early diagnosis and treatment of MAS is important because MAS can be rapidly fatal, with a mortality rate of 8% to 20% in pediatric patients.17,18 Clinical features of MAS include physical exam findings of fever and splenomegaly as well as laboratory abnormalities, including pancytopenia, elevated ferritin, elevated triglycerides, and low fibrinogen.18 A bone marrow biopsy showing hemophagocytosis in the absence of malignancy is diagnostic of MAS. Although a bone marrow biopsy is not required to diagnose MAS, it is often obtained to exclude other etiologies of pancytopenia such as malignancy.19 Specialized diagnostic testing for MAS includes NK cell counts and functional studies, including expression of perforin and granzyme B (NK cell proteins triggering apoptosis in target cells), soluble IL-2R (marker of activated lymphocytes), and CD163 (transmembrane protein of hemophagocytic macrophages). There is no standardized protocol for treating MAS.20 It is most commonly treated with highdose corticosteroids; additional agents, including cyclosporine and biologic therapies, are also utilized.16,20
KEY POINTS
- Children with SLE tend to have greater involvement of major organ systems and more rapid accrual of organ damage than adults with SLE. Therefore, it is sometimes necessary to initiate immunosuppressive therapies before full diagnostic criteria are met, provided that malignancy and infection have been ruled out.
- While pancytopenia is common in pediatric patients with SLE, providers should make sure to consider coexisting diagnoses such as infection and MAS, both of which require different treatment strategies.
- It is important to consider HLH/MAS early in the work-up of pancytopenia, because early diagnosis and treatment improves clinical outcomes. Obtaining a ferritin level can aid in the work-up of pancytopenia because it is both a sensitive and specific marker of HLH/MAS when dramatically elevated.
Disclosure
The authors report no conflicts of interest.
1. Weinzierl EP, Arber DA. The Differential Diagnosis and Bone Marrow Evaluation of New-Onset Pancytopenia. Am J Clin Pathol. 2012;139(1):9-29. doi:10.1309/AJCP50AEEYGREWUZ. PubMed
2. Pine M, Walter AW. Pancytopenia in hospitalized children: a five-year review. J Pediatr Hematol Oncol. 2010;32(5):e192-e194. doi:10.1097/MPH.0b013e3181e03082. PubMed
3. Bhatnagar SK. Pancytopenia in Children: Etiological Profile. J Trop Pediatr. 2005;51(4):236-239. doi:10.1093/tropej/fmi010. PubMed
4. Kulkarni KP, Marwaha RK. Acute lymphoblastic leukemia with pancytopenia at presentation: clinical correlates, prognostic impact, and association with survival. J Pediatr Hematol Oncol. 2013;35(7):573-576. doi:10.1097/MPH.0b013e31829d46f3. PubMed
5. Holubar, K. Terminology and iconography of lupus erythematosus: A historical vignette. Am J Dermatopathol. 1980;2(3):239-242. PubMed
6. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. PubMed
7. Petri M, Orbai, A, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686. doi:10.1002/art.34473. PubMed
8. Tucker L. Review: Making the diagnosis of systemic lupus erythematosus in children and adolescents. Lupus. 2007;16(8):546-549. doi:10.1177/0961203307078068. PubMed
9. Brunner HI, Gladman DD, Ibañez D, Urowitz MD, Silverman ED. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58(2):556-562. doi:10.1002/art.23204. PubMed
10. Benseler SM, Silverman ED. Systemic Lupus Erythematosus. Rheum Dis Clin North Am. 2007;33(3):471-498. doi:10.1016/j.rdc.2007.07.008. PubMed
11. Gokce M, Bilginer Y, Besbas N, et al. Hematological features of pediatric systemic lupus erythematosus: suggesting management strategies in children. Lupus. 2012;21(8):878-884. doi:10.1177/0961203312443721. PubMed
12. Voulgarelis M, Giannouli S, Tasidou A, Anagnostou D, Ziakas PD, Tzioufas AG. Bone marrow histological findings in systemic lupus erythematosus with hematologic abnormalities: A clinicopathological study. Am J Hematol. 2006;81(8):590-597. doi:10.1002/ajh.20593. PubMed
13. Pereira RM, Velloso ER, Menezes Y, Gualandro S, Vassalo J, Yoshinari NH. Bone marrow findings in systemic lupus erythematosus patients with peripheral cytopenias. Clin Rheumatol. 1998;17(3):219-222. PubMed
14. Avčin T, Tse SML, Schneider R, Ngan B, Silverman ED. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006;148(5):683-686. doi:10.1016/j.jpeds.2005.12.070. PubMed
15. Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and Long-Term Outcome of 15 Episodes of Systemic Lupus Erythematosus-Associated Hemophagocytic Syndrome. Medicine. 2006;85(3):169-182. doi:10.1097/01.md.0000224708.62510.d1. PubMed
16. Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology. 2008;47(11):1686-1691. doi:10.1093/rheumatology/ken342. PubMed
17. Stephan JL. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285-1292. doi:10.1093/rheumatology/40.11.1285. PubMed
18. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421-426. PubMed
19. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039. PubMed
20. Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol. 2012;31(8):1223-1230. doi:10.1007/s10067-012-1998-0. PubMed
1. Weinzierl EP, Arber DA. The Differential Diagnosis and Bone Marrow Evaluation of New-Onset Pancytopenia. Am J Clin Pathol. 2012;139(1):9-29. doi:10.1309/AJCP50AEEYGREWUZ. PubMed
2. Pine M, Walter AW. Pancytopenia in hospitalized children: a five-year review. J Pediatr Hematol Oncol. 2010;32(5):e192-e194. doi:10.1097/MPH.0b013e3181e03082. PubMed
3. Bhatnagar SK. Pancytopenia in Children: Etiological Profile. J Trop Pediatr. 2005;51(4):236-239. doi:10.1093/tropej/fmi010. PubMed
4. Kulkarni KP, Marwaha RK. Acute lymphoblastic leukemia with pancytopenia at presentation: clinical correlates, prognostic impact, and association with survival. J Pediatr Hematol Oncol. 2013;35(7):573-576. doi:10.1097/MPH.0b013e31829d46f3. PubMed
5. Holubar, K. Terminology and iconography of lupus erythematosus: A historical vignette. Am J Dermatopathol. 1980;2(3):239-242. PubMed
6. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. PubMed
7. Petri M, Orbai, A, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686. doi:10.1002/art.34473. PubMed
8. Tucker L. Review: Making the diagnosis of systemic lupus erythematosus in children and adolescents. Lupus. 2007;16(8):546-549. doi:10.1177/0961203307078068. PubMed
9. Brunner HI, Gladman DD, Ibañez D, Urowitz MD, Silverman ED. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58(2):556-562. doi:10.1002/art.23204. PubMed
10. Benseler SM, Silverman ED. Systemic Lupus Erythematosus. Rheum Dis Clin North Am. 2007;33(3):471-498. doi:10.1016/j.rdc.2007.07.008. PubMed
11. Gokce M, Bilginer Y, Besbas N, et al. Hematological features of pediatric systemic lupus erythematosus: suggesting management strategies in children. Lupus. 2012;21(8):878-884. doi:10.1177/0961203312443721. PubMed
12. Voulgarelis M, Giannouli S, Tasidou A, Anagnostou D, Ziakas PD, Tzioufas AG. Bone marrow histological findings in systemic lupus erythematosus with hematologic abnormalities: A clinicopathological study. Am J Hematol. 2006;81(8):590-597. doi:10.1002/ajh.20593. PubMed
13. Pereira RM, Velloso ER, Menezes Y, Gualandro S, Vassalo J, Yoshinari NH. Bone marrow findings in systemic lupus erythematosus patients with peripheral cytopenias. Clin Rheumatol. 1998;17(3):219-222. PubMed
14. Avčin T, Tse SML, Schneider R, Ngan B, Silverman ED. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006;148(5):683-686. doi:10.1016/j.jpeds.2005.12.070. PubMed
15. Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and Long-Term Outcome of 15 Episodes of Systemic Lupus Erythematosus-Associated Hemophagocytic Syndrome. Medicine. 2006;85(3):169-182. doi:10.1097/01.md.0000224708.62510.d1. PubMed
16. Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology. 2008;47(11):1686-1691. doi:10.1093/rheumatology/ken342. PubMed
17. Stephan JL. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285-1292. doi:10.1093/rheumatology/40.11.1285. PubMed
18. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421-426. PubMed
19. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039. PubMed
20. Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol. 2012;31(8):1223-1230. doi:10.1007/s10067-012-1998-0. PubMed
© 2018 Society of Hospital Medicine