User login
Targeting CD98 to treat AML
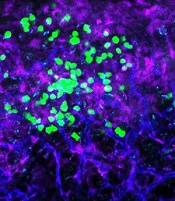
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
Team maps genomic landscape of CBF-AML

whole-genome sequencing
Photo courtesy of the US
Food and Drug Administration
Whole-genome and whole-exome sequencing has provided new insight into the pathogenesis and development of core-binding factor acute myeloid leukemia (CBF-AML), according to researchers.
The team said their work has revealed “dramatic” differences in the genomic landscape of CBF-AMLs that contribute to the diversity of this disease.
The researchers reported their findings in Nature Genetics.
“We set out to understand the genetic variations that contribute to the development of CBF-AML using whole-exome and whole-genome sequencing,” said study author Jeffery Klco, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our goal was to define a detailed mutational landscape to understand better the genetic changes that contribute to disease.”
Dr Klco and his colleagues sequenced samples from 87 children and 78 adults with CBF-AML. Eighty-five of the patients had the RUNX1-RUNX1T1 subtype, and 80 had the CBFB-MYH11 subtype.
Development and relapse
The researchers identified several genes with mutations that may contribute to CBF-AML development, including CCND2, DHX15, ASXL2, ZBTB7A, and MGA.
“Many of the mutations we identified interfered with molecular signaling or epigenetic factors,” said study author Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital.
“Some of the mutations, like ASXL2, are epigenetic regulators that modify the local state of chromatin,” Dr Klco noted. “Others, like ZBTB7A, appear to act like tumor suppressors.”
The researchers also compared mutations present at diagnosis and relapse in an attempt to understand how CBF-AML changes over time.
Their results suggested that KMT2C mutations are associated with relapse. Of the 4 patients in this study who had KMT2C mutations, 3 relapsed in less than 12 months, and the fourth had residual disease after a course of remission-induction therapy.
Similarities and differences
The researchers found a similar mutational landscape in adults and children with CBF-AML but differences between patients with the RUNX1-RUNX1T1 and CBFB-MYH11 subtypes.
NRAS was the most frequently mutated gene in CBF-AMLs, but NRAS mutations were more common in CBFB-MYH11 AML than RUNX1-RUNX1T1 AML. The same was true for mutations in NF1 and WT1.
Patients with both subtypes of CBF-AML had mutations in NRAS, KIT, NF1, WT1, FLT3, KRAS, MGA, TTN, CCND2, KDM6A, PHIP, TET2, HCN1, KMT2C, and SETD2.
But only patients with CBFB-MYH11 AML had mutations in PTPN11.
Only patients with RUNX1-RUNX1T1 AML had mutations in ASXL2, ZBTB7A, EZH2, SMC1A, DHX15, RAD21, CBL, DNM2, CSF3R, GIGYF2, SMC3, and ZNF687.
The researchers said their findings suggest a range of mutations may play roles in CBF-AML, but additional research is needed to confirm their precise function in the disease.
Further studies are already underway to fully evaluate the contributions of the different genes as well as the roles of the newly identified genetic alterations in CBF-AML. ![]()
whole-genome sequencing
Photo courtesy of the US
Food and Drug Administration
Whole-genome and whole-exome sequencing has provided new insight into the pathogenesis and development of core-binding factor acute myeloid leukemia (CBF-AML), according to researchers.
The team said their work has revealed “dramatic” differences in the genomic landscape of CBF-AMLs that contribute to the diversity of this disease.
The researchers reported their findings in Nature Genetics.
“We set out to understand the genetic variations that contribute to the development of CBF-AML using whole-exome and whole-genome sequencing,” said study author Jeffery Klco, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our goal was to define a detailed mutational landscape to understand better the genetic changes that contribute to disease.”
Dr Klco and his colleagues sequenced samples from 87 children and 78 adults with CBF-AML. Eighty-five of the patients had the RUNX1-RUNX1T1 subtype, and 80 had the CBFB-MYH11 subtype.
Development and relapse
The researchers identified several genes with mutations that may contribute to CBF-AML development, including CCND2, DHX15, ASXL2, ZBTB7A, and MGA.
“Many of the mutations we identified interfered with molecular signaling or epigenetic factors,” said study author Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital.
“Some of the mutations, like ASXL2, are epigenetic regulators that modify the local state of chromatin,” Dr Klco noted. “Others, like ZBTB7A, appear to act like tumor suppressors.”
The researchers also compared mutations present at diagnosis and relapse in an attempt to understand how CBF-AML changes over time.
Their results suggested that KMT2C mutations are associated with relapse. Of the 4 patients in this study who had KMT2C mutations, 3 relapsed in less than 12 months, and the fourth had residual disease after a course of remission-induction therapy.
Similarities and differences
The researchers found a similar mutational landscape in adults and children with CBF-AML but differences between patients with the RUNX1-RUNX1T1 and CBFB-MYH11 subtypes.
NRAS was the most frequently mutated gene in CBF-AMLs, but NRAS mutations were more common in CBFB-MYH11 AML than RUNX1-RUNX1T1 AML. The same was true for mutations in NF1 and WT1.
Patients with both subtypes of CBF-AML had mutations in NRAS, KIT, NF1, WT1, FLT3, KRAS, MGA, TTN, CCND2, KDM6A, PHIP, TET2, HCN1, KMT2C, and SETD2.
But only patients with CBFB-MYH11 AML had mutations in PTPN11.
Only patients with RUNX1-RUNX1T1 AML had mutations in ASXL2, ZBTB7A, EZH2, SMC1A, DHX15, RAD21, CBL, DNM2, CSF3R, GIGYF2, SMC3, and ZNF687.
The researchers said their findings suggest a range of mutations may play roles in CBF-AML, but additional research is needed to confirm their precise function in the disease.
Further studies are already underway to fully evaluate the contributions of the different genes as well as the roles of the newly identified genetic alterations in CBF-AML. ![]()
whole-genome sequencing
Photo courtesy of the US
Food and Drug Administration
Whole-genome and whole-exome sequencing has provided new insight into the pathogenesis and development of core-binding factor acute myeloid leukemia (CBF-AML), according to researchers.
The team said their work has revealed “dramatic” differences in the genomic landscape of CBF-AMLs that contribute to the diversity of this disease.
The researchers reported their findings in Nature Genetics.
“We set out to understand the genetic variations that contribute to the development of CBF-AML using whole-exome and whole-genome sequencing,” said study author Jeffery Klco, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our goal was to define a detailed mutational landscape to understand better the genetic changes that contribute to disease.”
Dr Klco and his colleagues sequenced samples from 87 children and 78 adults with CBF-AML. Eighty-five of the patients had the RUNX1-RUNX1T1 subtype, and 80 had the CBFB-MYH11 subtype.
Development and relapse
The researchers identified several genes with mutations that may contribute to CBF-AML development, including CCND2, DHX15, ASXL2, ZBTB7A, and MGA.
“Many of the mutations we identified interfered with molecular signaling or epigenetic factors,” said study author Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital.
“Some of the mutations, like ASXL2, are epigenetic regulators that modify the local state of chromatin,” Dr Klco noted. “Others, like ZBTB7A, appear to act like tumor suppressors.”
The researchers also compared mutations present at diagnosis and relapse in an attempt to understand how CBF-AML changes over time.
Their results suggested that KMT2C mutations are associated with relapse. Of the 4 patients in this study who had KMT2C mutations, 3 relapsed in less than 12 months, and the fourth had residual disease after a course of remission-induction therapy.
Similarities and differences
The researchers found a similar mutational landscape in adults and children with CBF-AML but differences between patients with the RUNX1-RUNX1T1 and CBFB-MYH11 subtypes.
NRAS was the most frequently mutated gene in CBF-AMLs, but NRAS mutations were more common in CBFB-MYH11 AML than RUNX1-RUNX1T1 AML. The same was true for mutations in NF1 and WT1.
Patients with both subtypes of CBF-AML had mutations in NRAS, KIT, NF1, WT1, FLT3, KRAS, MGA, TTN, CCND2, KDM6A, PHIP, TET2, HCN1, KMT2C, and SETD2.
But only patients with CBFB-MYH11 AML had mutations in PTPN11.
Only patients with RUNX1-RUNX1T1 AML had mutations in ASXL2, ZBTB7A, EZH2, SMC1A, DHX15, RAD21, CBL, DNM2, CSF3R, GIGYF2, SMC3, and ZNF687.
The researchers said their findings suggest a range of mutations may play roles in CBF-AML, but additional research is needed to confirm their precise function in the disease.
Further studies are already underway to fully evaluate the contributions of the different genes as well as the roles of the newly identified genetic alterations in CBF-AML. ![]()
Androgen improved survival in elderly AML patients
Elderly patients with acute myeloid leukemia (AML) generally have a poor prognosis, but maintenance therapy with an androgen, norethandrolone, significantly improved survival in this population, investigators report in the Journal of Clinical Oncology.
The 5-year disease-free survival was almost double in patients who received norethandrolone (31.2% vs 16.2%), and event-free survival (EFS) and overall survival (OS) were also markedly improved.
A number of hypotheses could explain why norethandrolone improves outcomes in this population. “Because androgen supplementation was initiated when the tumoral mass was decreased, it is possible that norethandrolone decreased the proliferation of remaining blast cells and/or their genetic instability in restoring proper telomere length,” wrote Arnaud Pigneux, MD, PhD, of the Centre Hospitalier Universitaire, Bordeaux (France), and his coauthors. “In addition, as in aplastic anemia, a beneficial effect could be exerted by androgens on normal hematopoietic cells recovering after the end of the postinduction aplastic phase,” they said (J Clin Oncol. 2016 Oct 17. doi: 10.1200/JCO.2016.67.6213)
The majority of patients with AML are older than 60 years of age at the time of their diagnosis, and the prognosis is particularly poor for a number of reasons, including a greater intolerance to intensive chemotherapies and the presence of comorbidities. In addition, older patients also frequently present with unfavorable prognostic features, including multidrug resistant phenotypes or poor-risk cytogenetics.
In this study, Dr. Pigneux and his team enrolled 330 patients with AML de novo or secondary to chemotherapy or radiotherapy, who were 60 years of age or older. Induction therapy of idarubicin 8 mg/m2 on days 1 to 5, cytarabine 100 mg/m2 on days 1 to 7, and lomustine 200 mg/m2 on day 1 was administered.
Those who achieved complete or partial remission received six reinduction courses, alternating idarubicin and cytarabine, and a regimen of methotrexate and mercaptopurine. The cohort was then randomized to receive norethandrolone 10 or 20 mg/day (arm A) according to body weight, or no norethandrolone (arm B) for a 2-year maintenance therapy regimen.
Disease-free survival (DFS) was significantly improved in patients who received androgen therapy, but there was no difference between groups in patients with a high WBC count.
The 5-year DFS was 39.2% for arm A versus 15.1% in arm B for patients with a low WBC count, and 14.3% in arm A versus 20.8% in arm B for patients with a high WBC count.
From the time of inclusion, the 5-year EFS and OS were 21.5% (95% confidence interval, 15.5%-28.1%) and 26.3% (95% CI, 19.7%-33.2%), respectively, in arm A versus 12.9% (95% CI, 8.3%-18.5%) and 17.2% (95% CI, 11.9%-23.4%), respectively, in arm B.
For patients with unfavorable cytogenetics (n = 78), outcomes were better if they received norethandrolone.
The 5-year DFS for this subset was 15.79% in arm A versus 7.69% in arm B. For patients with low- or intermediate-risk cytogenetics, the 5-year DFS was 39.73% in arm A versus 19.72% in arm B.
No funding source was disclosed. Dr. Pigneux has disclosed consulting or advisory roles with Amgen, Celgene, MSD, and Novartis, and disclosed receiving funding for travel and accommodations expenses from Amgen and Pfizer. Several of the authors have disclosed relationships with industry.
Elderly patients with acute myeloid leukemia (AML) generally have a poor prognosis, but maintenance therapy with an androgen, norethandrolone, significantly improved survival in this population, investigators report in the Journal of Clinical Oncology.
The 5-year disease-free survival was almost double in patients who received norethandrolone (31.2% vs 16.2%), and event-free survival (EFS) and overall survival (OS) were also markedly improved.
A number of hypotheses could explain why norethandrolone improves outcomes in this population. “Because androgen supplementation was initiated when the tumoral mass was decreased, it is possible that norethandrolone decreased the proliferation of remaining blast cells and/or their genetic instability in restoring proper telomere length,” wrote Arnaud Pigneux, MD, PhD, of the Centre Hospitalier Universitaire, Bordeaux (France), and his coauthors. “In addition, as in aplastic anemia, a beneficial effect could be exerted by androgens on normal hematopoietic cells recovering after the end of the postinduction aplastic phase,” they said (J Clin Oncol. 2016 Oct 17. doi: 10.1200/JCO.2016.67.6213)
The majority of patients with AML are older than 60 years of age at the time of their diagnosis, and the prognosis is particularly poor for a number of reasons, including a greater intolerance to intensive chemotherapies and the presence of comorbidities. In addition, older patients also frequently present with unfavorable prognostic features, including multidrug resistant phenotypes or poor-risk cytogenetics.
In this study, Dr. Pigneux and his team enrolled 330 patients with AML de novo or secondary to chemotherapy or radiotherapy, who were 60 years of age or older. Induction therapy of idarubicin 8 mg/m2 on days 1 to 5, cytarabine 100 mg/m2 on days 1 to 7, and lomustine 200 mg/m2 on day 1 was administered.
Those who achieved complete or partial remission received six reinduction courses, alternating idarubicin and cytarabine, and a regimen of methotrexate and mercaptopurine. The cohort was then randomized to receive norethandrolone 10 or 20 mg/day (arm A) according to body weight, or no norethandrolone (arm B) for a 2-year maintenance therapy regimen.
Disease-free survival (DFS) was significantly improved in patients who received androgen therapy, but there was no difference between groups in patients with a high WBC count.
The 5-year DFS was 39.2% for arm A versus 15.1% in arm B for patients with a low WBC count, and 14.3% in arm A versus 20.8% in arm B for patients with a high WBC count.
From the time of inclusion, the 5-year EFS and OS were 21.5% (95% confidence interval, 15.5%-28.1%) and 26.3% (95% CI, 19.7%-33.2%), respectively, in arm A versus 12.9% (95% CI, 8.3%-18.5%) and 17.2% (95% CI, 11.9%-23.4%), respectively, in arm B.
For patients with unfavorable cytogenetics (n = 78), outcomes were better if they received norethandrolone.
The 5-year DFS for this subset was 15.79% in arm A versus 7.69% in arm B. For patients with low- or intermediate-risk cytogenetics, the 5-year DFS was 39.73% in arm A versus 19.72% in arm B.
No funding source was disclosed. Dr. Pigneux has disclosed consulting or advisory roles with Amgen, Celgene, MSD, and Novartis, and disclosed receiving funding for travel and accommodations expenses from Amgen and Pfizer. Several of the authors have disclosed relationships with industry.
Elderly patients with acute myeloid leukemia (AML) generally have a poor prognosis, but maintenance therapy with an androgen, norethandrolone, significantly improved survival in this population, investigators report in the Journal of Clinical Oncology.
The 5-year disease-free survival was almost double in patients who received norethandrolone (31.2% vs 16.2%), and event-free survival (EFS) and overall survival (OS) were also markedly improved.
A number of hypotheses could explain why norethandrolone improves outcomes in this population. “Because androgen supplementation was initiated when the tumoral mass was decreased, it is possible that norethandrolone decreased the proliferation of remaining blast cells and/or their genetic instability in restoring proper telomere length,” wrote Arnaud Pigneux, MD, PhD, of the Centre Hospitalier Universitaire, Bordeaux (France), and his coauthors. “In addition, as in aplastic anemia, a beneficial effect could be exerted by androgens on normal hematopoietic cells recovering after the end of the postinduction aplastic phase,” they said (J Clin Oncol. 2016 Oct 17. doi: 10.1200/JCO.2016.67.6213)
The majority of patients with AML are older than 60 years of age at the time of their diagnosis, and the prognosis is particularly poor for a number of reasons, including a greater intolerance to intensive chemotherapies and the presence of comorbidities. In addition, older patients also frequently present with unfavorable prognostic features, including multidrug resistant phenotypes or poor-risk cytogenetics.
In this study, Dr. Pigneux and his team enrolled 330 patients with AML de novo or secondary to chemotherapy or radiotherapy, who were 60 years of age or older. Induction therapy of idarubicin 8 mg/m2 on days 1 to 5, cytarabine 100 mg/m2 on days 1 to 7, and lomustine 200 mg/m2 on day 1 was administered.
Those who achieved complete or partial remission received six reinduction courses, alternating idarubicin and cytarabine, and a regimen of methotrexate and mercaptopurine. The cohort was then randomized to receive norethandrolone 10 or 20 mg/day (arm A) according to body weight, or no norethandrolone (arm B) for a 2-year maintenance therapy regimen.
Disease-free survival (DFS) was significantly improved in patients who received androgen therapy, but there was no difference between groups in patients with a high WBC count.
The 5-year DFS was 39.2% for arm A versus 15.1% in arm B for patients with a low WBC count, and 14.3% in arm A versus 20.8% in arm B for patients with a high WBC count.
From the time of inclusion, the 5-year EFS and OS were 21.5% (95% confidence interval, 15.5%-28.1%) and 26.3% (95% CI, 19.7%-33.2%), respectively, in arm A versus 12.9% (95% CI, 8.3%-18.5%) and 17.2% (95% CI, 11.9%-23.4%), respectively, in arm B.
For patients with unfavorable cytogenetics (n = 78), outcomes were better if they received norethandrolone.
The 5-year DFS for this subset was 15.79% in arm A versus 7.69% in arm B. For patients with low- or intermediate-risk cytogenetics, the 5-year DFS was 39.73% in arm A versus 19.72% in arm B.
No funding source was disclosed. Dr. Pigneux has disclosed consulting or advisory roles with Amgen, Celgene, MSD, and Novartis, and disclosed receiving funding for travel and accommodations expenses from Amgen and Pfizer. Several of the authors have disclosed relationships with industry.
Key clinical point: Maintenance therapy with norethandrolone improved outcomes in elderly patients with acute myeloid leukemia.
Major finding: 5-year disease-free survival was 31.2% (for the norethandrolone group) vs. 16.2% and event-free survival was 21.5% and 12.9%, respectively.
Data source: Prospective, randomized, multicenter, open-label phase III study that included 330 patients with AML.
Disclosures: No funding source was disclosed. Dr. Pigneux has disclosed consulting or advisory roles with Amgen, Celgene, MSD, and Novartis, and disclosed receiving funding for travel and accommodations expenses from Amgen and Pfizer. Several of the authors have also disclosed relationships with industry.
Compound could treat a range of blood cancers

Image by Ed Uthman
A new compound has shown promise for treating hematologic malignancies and other cancers, according to preclinical research published in Nature.
The compound, known as S63845, targets the BCL2 family protein MCL1.
Investigators said their research on S63845 provides the first clear evidence that inhibiting MCL1 is effective in targeting several cancer types, including leukemia, lymphoma, and multiple myeloma (MM).
“MCL1 is important for many cancers because it is a pro-survival protein that allows the cancerous cells to evade the process of programmed cell death that normally removes cancer cells from the body,” said study author Guillaume Lessene, PhD, of the Walter and Eliza Hall Institute in Melbourne, Australia.
“Extensive studies performed in a variety of cancer models have shown that S63845 potently targets cancer cells dependent on MCL1 for their survival.”
About S63845
Dr Lessene and his colleagues said S63845 binds with high affinity to the BH3-binding groove of MCL1. And the compound kills MCL1-dependent cancer cells by activating the BAX/BAK-dependent mitochondrial apoptotic pathway.
In solid tumors, S63845 often wasn’t effective enough on its own. However, when the compound was combined with various kinase inhibitors, it induced a “potent cytotoxic response” in breast cancer, lung cancer, and melanoma cells.
In hematologic malignancies, S63845 proved effective when given alone.
Myeloma
The investigators said 17 of 25 MM cell lines tested were highly sensitive to S63845 (IC50 < 0.1 μ M), 6 cell lines were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 2 cell lines were insensitive (IC50 > 1 μ M).
The team also administered S63845 (at 25 mg/kg) to mice with MM. Seven of 8 mice had complete regression at 100 days after treatment.
Lymphoma
The investigators tested S63845 in 11 cell lines representative of human lymphomas. Five were highly sensitive to the compound (IC50 < 0.1 μ M), 3 were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 3 were insensitive (IC50 > 1 μ M).
The team also tested S63845 in 7 c-MYC-driven human Burkitt lymphoma cell lines and found the compound exhibited “potent cytotoxic activity” in all of them.
The investigators then tested S63845 in a c-MYC-driven mouse lymphoma model. They noted that both tumor cells and normal tissues express mouse MCL1 protein in this model.
Treatment with S63845 (25 mg/kg) for 5 consecutive days cured 70% of these mice, and the investigators said there were no evident side effects in normal tissues.
Leukemia
The investigators tested S63845 in 5 chronic myeloid leukemia cell lines, and none of them were sensitive to the compound.
However, the team also tested S63845 in 8 acute myeloid leukemia (AML) cell lines, and all of them were sensitive to the compound (IC50 4–233 nM).
When S63845 was given to mice with AML (25 mg/kg), 6 of the 8 mice achieved complete remission after 80 days.
The investigators also tested S63845 in 25 freshly derived samples from patients with AML. The team said these samples displayed a wide range of responses to S63845.
The most sensitive samples required 100- to 1000-fold less drug (to induce apoptosis) than the resistant samples or normal CD34+ progenitor cells.
Development/funding
S63845 was discovered through a collaboration between 2 pharmaceutical companies—Servier, which is headquartered in France, and Vernalis (R&D), which is based in the UK.
“[C]linical development of a MCL1 inhibitor should be launched in the near future,” said Olivier Geneste, director of oncology research at Servier.
The current research was supported through a collaboration with Servier and through funding from the National Health and Medical Research Council of Australia, the Leukemia and Lymphoma Society, Cancer Council Victoria, the Kay Kendall Leukemia Fund, Victorian Cancer Agency, Australian Cancer Research Foundation, the Victorian Government Operational Infrastructure Scheme, and the estate of Anthony Redstone. ![]()
Image by Ed Uthman
A new compound has shown promise for treating hematologic malignancies and other cancers, according to preclinical research published in Nature.
The compound, known as S63845, targets the BCL2 family protein MCL1.
Investigators said their research on S63845 provides the first clear evidence that inhibiting MCL1 is effective in targeting several cancer types, including leukemia, lymphoma, and multiple myeloma (MM).
“MCL1 is important for many cancers because it is a pro-survival protein that allows the cancerous cells to evade the process of programmed cell death that normally removes cancer cells from the body,” said study author Guillaume Lessene, PhD, of the Walter and Eliza Hall Institute in Melbourne, Australia.
“Extensive studies performed in a variety of cancer models have shown that S63845 potently targets cancer cells dependent on MCL1 for their survival.”
About S63845
Dr Lessene and his colleagues said S63845 binds with high affinity to the BH3-binding groove of MCL1. And the compound kills MCL1-dependent cancer cells by activating the BAX/BAK-dependent mitochondrial apoptotic pathway.
In solid tumors, S63845 often wasn’t effective enough on its own. However, when the compound was combined with various kinase inhibitors, it induced a “potent cytotoxic response” in breast cancer, lung cancer, and melanoma cells.
In hematologic malignancies, S63845 proved effective when given alone.
Myeloma
The investigators said 17 of 25 MM cell lines tested were highly sensitive to S63845 (IC50 < 0.1 μ M), 6 cell lines were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 2 cell lines were insensitive (IC50 > 1 μ M).
The team also administered S63845 (at 25 mg/kg) to mice with MM. Seven of 8 mice had complete regression at 100 days after treatment.
Lymphoma
The investigators tested S63845 in 11 cell lines representative of human lymphomas. Five were highly sensitive to the compound (IC50 < 0.1 μ M), 3 were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 3 were insensitive (IC50 > 1 μ M).
The team also tested S63845 in 7 c-MYC-driven human Burkitt lymphoma cell lines and found the compound exhibited “potent cytotoxic activity” in all of them.
The investigators then tested S63845 in a c-MYC-driven mouse lymphoma model. They noted that both tumor cells and normal tissues express mouse MCL1 protein in this model.
Treatment with S63845 (25 mg/kg) for 5 consecutive days cured 70% of these mice, and the investigators said there were no evident side effects in normal tissues.
Leukemia
The investigators tested S63845 in 5 chronic myeloid leukemia cell lines, and none of them were sensitive to the compound.
However, the team also tested S63845 in 8 acute myeloid leukemia (AML) cell lines, and all of them were sensitive to the compound (IC50 4–233 nM).
When S63845 was given to mice with AML (25 mg/kg), 6 of the 8 mice achieved complete remission after 80 days.
The investigators also tested S63845 in 25 freshly derived samples from patients with AML. The team said these samples displayed a wide range of responses to S63845.
The most sensitive samples required 100- to 1000-fold less drug (to induce apoptosis) than the resistant samples or normal CD34+ progenitor cells.
Development/funding
S63845 was discovered through a collaboration between 2 pharmaceutical companies—Servier, which is headquartered in France, and Vernalis (R&D), which is based in the UK.
“[C]linical development of a MCL1 inhibitor should be launched in the near future,” said Olivier Geneste, director of oncology research at Servier.
The current research was supported through a collaboration with Servier and through funding from the National Health and Medical Research Council of Australia, the Leukemia and Lymphoma Society, Cancer Council Victoria, the Kay Kendall Leukemia Fund, Victorian Cancer Agency, Australian Cancer Research Foundation, the Victorian Government Operational Infrastructure Scheme, and the estate of Anthony Redstone. ![]()
Image by Ed Uthman
A new compound has shown promise for treating hematologic malignancies and other cancers, according to preclinical research published in Nature.
The compound, known as S63845, targets the BCL2 family protein MCL1.
Investigators said their research on S63845 provides the first clear evidence that inhibiting MCL1 is effective in targeting several cancer types, including leukemia, lymphoma, and multiple myeloma (MM).
“MCL1 is important for many cancers because it is a pro-survival protein that allows the cancerous cells to evade the process of programmed cell death that normally removes cancer cells from the body,” said study author Guillaume Lessene, PhD, of the Walter and Eliza Hall Institute in Melbourne, Australia.
“Extensive studies performed in a variety of cancer models have shown that S63845 potently targets cancer cells dependent on MCL1 for their survival.”
About S63845
Dr Lessene and his colleagues said S63845 binds with high affinity to the BH3-binding groove of MCL1. And the compound kills MCL1-dependent cancer cells by activating the BAX/BAK-dependent mitochondrial apoptotic pathway.
In solid tumors, S63845 often wasn’t effective enough on its own. However, when the compound was combined with various kinase inhibitors, it induced a “potent cytotoxic response” in breast cancer, lung cancer, and melanoma cells.
In hematologic malignancies, S63845 proved effective when given alone.
Myeloma
The investigators said 17 of 25 MM cell lines tested were highly sensitive to S63845 (IC50 < 0.1 μ M), 6 cell lines were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 2 cell lines were insensitive (IC50 > 1 μ M).
The team also administered S63845 (at 25 mg/kg) to mice with MM. Seven of 8 mice had complete regression at 100 days after treatment.
Lymphoma
The investigators tested S63845 in 11 cell lines representative of human lymphomas. Five were highly sensitive to the compound (IC50 < 0.1 μ M), 3 were moderately sensitive (0.1 μ M < IC50 < 1 μ M), and 3 were insensitive (IC50 > 1 μ M).
The team also tested S63845 in 7 c-MYC-driven human Burkitt lymphoma cell lines and found the compound exhibited “potent cytotoxic activity” in all of them.
The investigators then tested S63845 in a c-MYC-driven mouse lymphoma model. They noted that both tumor cells and normal tissues express mouse MCL1 protein in this model.
Treatment with S63845 (25 mg/kg) for 5 consecutive days cured 70% of these mice, and the investigators said there were no evident side effects in normal tissues.
Leukemia
The investigators tested S63845 in 5 chronic myeloid leukemia cell lines, and none of them were sensitive to the compound.
However, the team also tested S63845 in 8 acute myeloid leukemia (AML) cell lines, and all of them were sensitive to the compound (IC50 4–233 nM).
When S63845 was given to mice with AML (25 mg/kg), 6 of the 8 mice achieved complete remission after 80 days.
The investigators also tested S63845 in 25 freshly derived samples from patients with AML. The team said these samples displayed a wide range of responses to S63845.
The most sensitive samples required 100- to 1000-fold less drug (to induce apoptosis) than the resistant samples or normal CD34+ progenitor cells.
Development/funding
S63845 was discovered through a collaboration between 2 pharmaceutical companies—Servier, which is headquartered in France, and Vernalis (R&D), which is based in the UK.
“[C]linical development of a MCL1 inhibitor should be launched in the near future,” said Olivier Geneste, director of oncology research at Servier.
The current research was supported through a collaboration with Servier and through funding from the National Health and Medical Research Council of Australia, the Leukemia and Lymphoma Society, Cancer Council Victoria, the Kay Kendall Leukemia Fund, Victorian Cancer Agency, Australian Cancer Research Foundation, the Victorian Government Operational Infrastructure Scheme, and the estate of Anthony Redstone. ![]()
Combo produces CR/CRis in FLT3-ITD AML

Photo by Bill Branson
A 2-drug combination has shown promise for treating patients with FLT3-ITD acute myeloid leukemia (AML), according to research published in Science Translational Medicine.
Researchers found that omacetaxine mepesuccinate (formerly known as homoharringtonine) exhibits preferential antileukemic activity against FLT3-ITD AML.
Subsequent preclinical experiments revealed that omacetaxine synergizes with sorafenib and other FLT3 inhibitors.
So researchers tested omacetaxine in combination with sorafenib in a phase 2 trial of patients with FLT3-ITD AML.
The combination produced complete responses (CRs) or CRs with incomplete hematologic recovery (CRis) in a majority of patients, and researchers said the treatment was well-tolerated.
Anskar Y. H. Leung, MD, PhD, of The University of Hong Kong, and his colleagues conducted this research.
The team first performed an in vitro screen on AML patient samples to determine their responses to various drugs.
One of the compounds tested, the protein translation inhibitor omacetaxine mepesuccinate, showed strong antileukemic effects against FLT3-ITD AML. In fact, omacetaxine preferentially inhibited the growth of FLT3-ITD cell lines.
The researchers then found that omacetaxine synergizes with sorafenib and other FLT3 inhibitors to suppress leukemia growth in FLT3-ITD AML cell lines.
Omacetaxine and sorafenib in combination also prolonged survival in mouse models of FLT3-ITD AML (mice transplanted with MV4-11 or MOLM-13 cells).
Phase 2 trial
The researchers went on to test omacetaxine and sorafenib in a phase 2 trial. The trial enrolled 24 patients with FLT3-ITD AML and a median age of 50 (range, 21-76).
Most of the patients had relapsed or refractory disease, but 2 were unsuitable for induction chemotherapy because of advanced age and comorbidities.
The patients received omacetaxine and sorafenib continuously until intolerance, disease progression, or allogeneic hematopoietic stem cell transplant (HSCT).
Twenty patients (83.3%) achieved a CR or CRi at a median of 22 days (range, 18-55). Three patients did not respond, and 1 patient experienced a near-CRi—a reduction of blasts without complete clearance.
Fifteen of the responders relapsed, but 3 of these patients received omacetaxine and sorafenib again and achieved a CRi. One patient received re-treatment and failed to achieve a response.
Seven patients proceeded to HSCT after receiving omacetaxine and sorafenib.
At a median follow-up of 7.1 months (range, 2.2 to 20.5), 4 patients were still in CR/CRi (3 patients after HSCT), and 1 patient who had relapsed was still alive.
The remaining 19 patients had died—14 due to relapse, 4 due to non-response (1 after re-treatment), and 1 due to HSCT.
The median leukemia-free survival was 88 days (range, 9-510), and the median overall survival was 228 days (range, 53 to 615).
Adverse events occurring after treatment with omacetaxine and sorafenib included fever (n=14), rash (n=8), hand-foot-skin reactions (n=6), pneumonia (n=2), neutropenic fever (n=1), and bacteremia (n=1).
The researchers said this study validated the principle and clinical relevance of in vitro drug testing and identified a drug combination that might improve the treatment of FLT3-ITD AML. ![]()
Photo by Bill Branson
A 2-drug combination has shown promise for treating patients with FLT3-ITD acute myeloid leukemia (AML), according to research published in Science Translational Medicine.
Researchers found that omacetaxine mepesuccinate (formerly known as homoharringtonine) exhibits preferential antileukemic activity against FLT3-ITD AML.
Subsequent preclinical experiments revealed that omacetaxine synergizes with sorafenib and other FLT3 inhibitors.
So researchers tested omacetaxine in combination with sorafenib in a phase 2 trial of patients with FLT3-ITD AML.
The combination produced complete responses (CRs) or CRs with incomplete hematologic recovery (CRis) in a majority of patients, and researchers said the treatment was well-tolerated.
Anskar Y. H. Leung, MD, PhD, of The University of Hong Kong, and his colleagues conducted this research.
The team first performed an in vitro screen on AML patient samples to determine their responses to various drugs.
One of the compounds tested, the protein translation inhibitor omacetaxine mepesuccinate, showed strong antileukemic effects against FLT3-ITD AML. In fact, omacetaxine preferentially inhibited the growth of FLT3-ITD cell lines.
The researchers then found that omacetaxine synergizes with sorafenib and other FLT3 inhibitors to suppress leukemia growth in FLT3-ITD AML cell lines.
Omacetaxine and sorafenib in combination also prolonged survival in mouse models of FLT3-ITD AML (mice transplanted with MV4-11 or MOLM-13 cells).
Phase 2 trial
The researchers went on to test omacetaxine and sorafenib in a phase 2 trial. The trial enrolled 24 patients with FLT3-ITD AML and a median age of 50 (range, 21-76).
Most of the patients had relapsed or refractory disease, but 2 were unsuitable for induction chemotherapy because of advanced age and comorbidities.
The patients received omacetaxine and sorafenib continuously until intolerance, disease progression, or allogeneic hematopoietic stem cell transplant (HSCT).
Twenty patients (83.3%) achieved a CR or CRi at a median of 22 days (range, 18-55). Three patients did not respond, and 1 patient experienced a near-CRi—a reduction of blasts without complete clearance.
Fifteen of the responders relapsed, but 3 of these patients received omacetaxine and sorafenib again and achieved a CRi. One patient received re-treatment and failed to achieve a response.
Seven patients proceeded to HSCT after receiving omacetaxine and sorafenib.
At a median follow-up of 7.1 months (range, 2.2 to 20.5), 4 patients were still in CR/CRi (3 patients after HSCT), and 1 patient who had relapsed was still alive.
The remaining 19 patients had died—14 due to relapse, 4 due to non-response (1 after re-treatment), and 1 due to HSCT.
The median leukemia-free survival was 88 days (range, 9-510), and the median overall survival was 228 days (range, 53 to 615).
Adverse events occurring after treatment with omacetaxine and sorafenib included fever (n=14), rash (n=8), hand-foot-skin reactions (n=6), pneumonia (n=2), neutropenic fever (n=1), and bacteremia (n=1).
The researchers said this study validated the principle and clinical relevance of in vitro drug testing and identified a drug combination that might improve the treatment of FLT3-ITD AML. ![]()
Photo by Bill Branson
A 2-drug combination has shown promise for treating patients with FLT3-ITD acute myeloid leukemia (AML), according to research published in Science Translational Medicine.
Researchers found that omacetaxine mepesuccinate (formerly known as homoharringtonine) exhibits preferential antileukemic activity against FLT3-ITD AML.
Subsequent preclinical experiments revealed that omacetaxine synergizes with sorafenib and other FLT3 inhibitors.
So researchers tested omacetaxine in combination with sorafenib in a phase 2 trial of patients with FLT3-ITD AML.
The combination produced complete responses (CRs) or CRs with incomplete hematologic recovery (CRis) in a majority of patients, and researchers said the treatment was well-tolerated.
Anskar Y. H. Leung, MD, PhD, of The University of Hong Kong, and his colleagues conducted this research.
The team first performed an in vitro screen on AML patient samples to determine their responses to various drugs.
One of the compounds tested, the protein translation inhibitor omacetaxine mepesuccinate, showed strong antileukemic effects against FLT3-ITD AML. In fact, omacetaxine preferentially inhibited the growth of FLT3-ITD cell lines.
The researchers then found that omacetaxine synergizes with sorafenib and other FLT3 inhibitors to suppress leukemia growth in FLT3-ITD AML cell lines.
Omacetaxine and sorafenib in combination also prolonged survival in mouse models of FLT3-ITD AML (mice transplanted with MV4-11 or MOLM-13 cells).
Phase 2 trial
The researchers went on to test omacetaxine and sorafenib in a phase 2 trial. The trial enrolled 24 patients with FLT3-ITD AML and a median age of 50 (range, 21-76).
Most of the patients had relapsed or refractory disease, but 2 were unsuitable for induction chemotherapy because of advanced age and comorbidities.
The patients received omacetaxine and sorafenib continuously until intolerance, disease progression, or allogeneic hematopoietic stem cell transplant (HSCT).
Twenty patients (83.3%) achieved a CR or CRi at a median of 22 days (range, 18-55). Three patients did not respond, and 1 patient experienced a near-CRi—a reduction of blasts without complete clearance.
Fifteen of the responders relapsed, but 3 of these patients received omacetaxine and sorafenib again and achieved a CRi. One patient received re-treatment and failed to achieve a response.
Seven patients proceeded to HSCT after receiving omacetaxine and sorafenib.
At a median follow-up of 7.1 months (range, 2.2 to 20.5), 4 patients were still in CR/CRi (3 patients after HSCT), and 1 patient who had relapsed was still alive.
The remaining 19 patients had died—14 due to relapse, 4 due to non-response (1 after re-treatment), and 1 due to HSCT.
The median leukemia-free survival was 88 days (range, 9-510), and the median overall survival was 228 days (range, 53 to 615).
Adverse events occurring after treatment with omacetaxine and sorafenib included fever (n=14), rash (n=8), hand-foot-skin reactions (n=6), pneumonia (n=2), neutropenic fever (n=1), and bacteremia (n=1).
The researchers said this study validated the principle and clinical relevance of in vitro drug testing and identified a drug combination that might improve the treatment of FLT3-ITD AML. ![]()
Work reveals potential therapeutic targets in AML

By adapting CRISPR-Cas9 technology and using it to screen the leukemia genome, researchers have identified hundreds of potential therapeutic targets for acute myeloid leukemia (AML).
The group’s work revealed nearly 500 genes, many of which had not been identified previously, that might serve as targets for AML treatment.
Subsequent experiments showed that targeting one of the genes, KAT2A, can destroy AML cells without harming normal blood cells.
This research was published in Cell Reports.
For this study, the researchers used CRISPR-Cas9 gene-editing technology to screen leukemia cells for vulnerable points. The team said they refined the technology so they could disrupt all genes in the leukemia cell genome individually.
This allowed the researchers to identify those genes whose disruption was detrimental to the growth and survival of AML cells, particularly the AML cell lines MOLM-13, HL-60, OCI-AML2, OCI-AML3, and MV4-11.
“Previous studies showed proof of principle, but this is one of the first systematic attempts to identify the genetic vulnerabilities of AML,” said study author Kosuke Yusa, PhD, of Wellcome Trust Sanger Institute in Hinxton, Cambridge, UK.
“We have improved and applied CRISPR-Cas9 technology to look at what actually kills cells.”
In this way, the researchers identified 492 genes that are essential for AML cell survival, including 227 genes that are druggable.
The team noted that a handful of the genes they identified—including DOT1L, BCL2, and MEN1—are already established therapeutic targets, but most of them are not.
The researchers chose to perform additional experiments with one of the genes they identified, KAT2A, to demonstrate the validity of their findings.
KAT2A was one of 66 genes that were essential to 3 or more of the AML cell lines studied. KAT2A was essential for survival in MOLM-13, OCI-AML2, and OCI-AML3.
The team inhibited KAT2A in vitro using genetic and drug-based techniques. Results showed that disrupting KAT2A inhibited the growth and survival of AML cells but did not affect normal blood cells.
“This is an exciting finding, as KAT2A inhibition worked on a number of primary AML cells with diverse genotypes,” said study author Konstantinos Tzelepis, a PhD student at Wellcome Trust Sanger Institute.
“Whilst the gene needs to be studied in greater depth to understand its potential for use in the clinic, we show that targeting KAT2A destroyed AML cells in the laboratory while sparing healthy blood cells.”
The researchers also targeted KAT2A in transgenic mice. The team observed a significant reduction in AML cell expansion and a significant improvement in survival when KAT2A was disrupted.
“This research has led to the identification of many potential gene targets for future AML therapy, which we are making available to other researchers to explore,” said study author George Vassiliou, PhD, of Wellcome Trust Sanger Institute.
“Whilst KAT2A inhibition now needs to be investigated as a treatment strategy for acute myeloid leukemia, there are many more candidates to pursue by the leukemia research community. Our hope is that this work will lead to more effective treatments against AML that will improve both the survival and the quality of life of patients.” ![]()
By adapting CRISPR-Cas9 technology and using it to screen the leukemia genome, researchers have identified hundreds of potential therapeutic targets for acute myeloid leukemia (AML).
The group’s work revealed nearly 500 genes, many of which had not been identified previously, that might serve as targets for AML treatment.
Subsequent experiments showed that targeting one of the genes, KAT2A, can destroy AML cells without harming normal blood cells.
This research was published in Cell Reports.
For this study, the researchers used CRISPR-Cas9 gene-editing technology to screen leukemia cells for vulnerable points. The team said they refined the technology so they could disrupt all genes in the leukemia cell genome individually.
This allowed the researchers to identify those genes whose disruption was detrimental to the growth and survival of AML cells, particularly the AML cell lines MOLM-13, HL-60, OCI-AML2, OCI-AML3, and MV4-11.
“Previous studies showed proof of principle, but this is one of the first systematic attempts to identify the genetic vulnerabilities of AML,” said study author Kosuke Yusa, PhD, of Wellcome Trust Sanger Institute in Hinxton, Cambridge, UK.
“We have improved and applied CRISPR-Cas9 technology to look at what actually kills cells.”
In this way, the researchers identified 492 genes that are essential for AML cell survival, including 227 genes that are druggable.
The team noted that a handful of the genes they identified—including DOT1L, BCL2, and MEN1—are already established therapeutic targets, but most of them are not.
The researchers chose to perform additional experiments with one of the genes they identified, KAT2A, to demonstrate the validity of their findings.
KAT2A was one of 66 genes that were essential to 3 or more of the AML cell lines studied. KAT2A was essential for survival in MOLM-13, OCI-AML2, and OCI-AML3.
The team inhibited KAT2A in vitro using genetic and drug-based techniques. Results showed that disrupting KAT2A inhibited the growth and survival of AML cells but did not affect normal blood cells.
“This is an exciting finding, as KAT2A inhibition worked on a number of primary AML cells with diverse genotypes,” said study author Konstantinos Tzelepis, a PhD student at Wellcome Trust Sanger Institute.
“Whilst the gene needs to be studied in greater depth to understand its potential for use in the clinic, we show that targeting KAT2A destroyed AML cells in the laboratory while sparing healthy blood cells.”
The researchers also targeted KAT2A in transgenic mice. The team observed a significant reduction in AML cell expansion and a significant improvement in survival when KAT2A was disrupted.
“This research has led to the identification of many potential gene targets for future AML therapy, which we are making available to other researchers to explore,” said study author George Vassiliou, PhD, of Wellcome Trust Sanger Institute.
“Whilst KAT2A inhibition now needs to be investigated as a treatment strategy for acute myeloid leukemia, there are many more candidates to pursue by the leukemia research community. Our hope is that this work will lead to more effective treatments against AML that will improve both the survival and the quality of life of patients.” ![]()
By adapting CRISPR-Cas9 technology and using it to screen the leukemia genome, researchers have identified hundreds of potential therapeutic targets for acute myeloid leukemia (AML).
The group’s work revealed nearly 500 genes, many of which had not been identified previously, that might serve as targets for AML treatment.
Subsequent experiments showed that targeting one of the genes, KAT2A, can destroy AML cells without harming normal blood cells.
This research was published in Cell Reports.
For this study, the researchers used CRISPR-Cas9 gene-editing technology to screen leukemia cells for vulnerable points. The team said they refined the technology so they could disrupt all genes in the leukemia cell genome individually.
This allowed the researchers to identify those genes whose disruption was detrimental to the growth and survival of AML cells, particularly the AML cell lines MOLM-13, HL-60, OCI-AML2, OCI-AML3, and MV4-11.
“Previous studies showed proof of principle, but this is one of the first systematic attempts to identify the genetic vulnerabilities of AML,” said study author Kosuke Yusa, PhD, of Wellcome Trust Sanger Institute in Hinxton, Cambridge, UK.
“We have improved and applied CRISPR-Cas9 technology to look at what actually kills cells.”
In this way, the researchers identified 492 genes that are essential for AML cell survival, including 227 genes that are druggable.
The team noted that a handful of the genes they identified—including DOT1L, BCL2, and MEN1—are already established therapeutic targets, but most of them are not.
The researchers chose to perform additional experiments with one of the genes they identified, KAT2A, to demonstrate the validity of their findings.
KAT2A was one of 66 genes that were essential to 3 or more of the AML cell lines studied. KAT2A was essential for survival in MOLM-13, OCI-AML2, and OCI-AML3.
The team inhibited KAT2A in vitro using genetic and drug-based techniques. Results showed that disrupting KAT2A inhibited the growth and survival of AML cells but did not affect normal blood cells.
“This is an exciting finding, as KAT2A inhibition worked on a number of primary AML cells with diverse genotypes,” said study author Konstantinos Tzelepis, a PhD student at Wellcome Trust Sanger Institute.
“Whilst the gene needs to be studied in greater depth to understand its potential for use in the clinic, we show that targeting KAT2A destroyed AML cells in the laboratory while sparing healthy blood cells.”
The researchers also targeted KAT2A in transgenic mice. The team observed a significant reduction in AML cell expansion and a significant improvement in survival when KAT2A was disrupted.
“This research has led to the identification of many potential gene targets for future AML therapy, which we are making available to other researchers to explore,” said study author George Vassiliou, PhD, of Wellcome Trust Sanger Institute.
“Whilst KAT2A inhibition now needs to be investigated as a treatment strategy for acute myeloid leukemia, there are many more candidates to pursue by the leukemia research community. Our hope is that this work will lead to more effective treatments against AML that will improve both the survival and the quality of life of patients.” ![]()
Proteins may be therapeutic targets for AML subtype
Image by Eric Smith
Preclinical research suggests a pair of histone-modifying proteins may be promising therapeutic targets for NPM1-mutated acute myeloid leukemia (AML).
Investigators found that these proteins—MLL and DOT1L—play key roles in NPM1-mutated AML.
Pharmacologic inhibition of either protein alone produced anti-leukemic activity in vitro and in vivo, but inhibiting both proteins together had a more profound effect.
Michael Kühn, MD, of the Mainz University Medical Center in Mainz, Germany, and his colleagues reported these findings in Cancer Discovery.
The investigators noted that nearly all NPM1-mutated AMLs are characterized by aberrant HOX expression, and FLT3 is concomitantly mutated in roughly 60% of these cases. However, it hasn’t been clear how mutant NPM1 cells maintain aberrant gene expression.
With this study, Dr Kühn and his colleagues showed that MLL1 and DOT1L control HOX and FLT3 expression and differentiation in NPM1-mutated AML.
The investigators were able to demonstrate that survival of NPM1-mutated AML cells depends on these 2 proteins. And NPM1-mutated AML is “exceptionally dependent” on the menin binding site in MLL1.
The team tested MI-503, a menin–MLL1 inhibitor, and the DOT1L inhibitor EPZ4777 in human and murine models of NPM1-mutated AML.
Each of the drugs reduced the activity of HOX genes in NPM1-mutated AML cells, but combining the drugs resulted in near-complete inactivation of HOX genes.
When given alone, EPZ4777 and MI-503 each reduced the proliferation and colony-forming potential of NPM1-mutated AML cells in vitro. And each of the drugs prolonged survival in mouse models of NPM1-mutated AML.
However, EPZ4777 and MI-503 given in combination significantly delayed the onset of leukemia and significantly prolonged survival when compared to either drug given alone.
The investigators said this suggests that inhibiting both DOT1L and menin–MLL1 affects leukemia-initiating cells, and this approach represents the first molecularly targeted treatment of NPM1-mutated AML that works by reversing a key mechanism of leukemogenesis.
They added that this research paves the way for trials assessing EPZ4777 and MI-503 in patients with NPM1-mutated AML. ![]()
Image by Eric Smith
Preclinical research suggests a pair of histone-modifying proteins may be promising therapeutic targets for NPM1-mutated acute myeloid leukemia (AML).
Investigators found that these proteins—MLL and DOT1L—play key roles in NPM1-mutated AML.
Pharmacologic inhibition of either protein alone produced anti-leukemic activity in vitro and in vivo, but inhibiting both proteins together had a more profound effect.
Michael Kühn, MD, of the Mainz University Medical Center in Mainz, Germany, and his colleagues reported these findings in Cancer Discovery.
The investigators noted that nearly all NPM1-mutated AMLs are characterized by aberrant HOX expression, and FLT3 is concomitantly mutated in roughly 60% of these cases. However, it hasn’t been clear how mutant NPM1 cells maintain aberrant gene expression.
With this study, Dr Kühn and his colleagues showed that MLL1 and DOT1L control HOX and FLT3 expression and differentiation in NPM1-mutated AML.
The investigators were able to demonstrate that survival of NPM1-mutated AML cells depends on these 2 proteins. And NPM1-mutated AML is “exceptionally dependent” on the menin binding site in MLL1.
The team tested MI-503, a menin–MLL1 inhibitor, and the DOT1L inhibitor EPZ4777 in human and murine models of NPM1-mutated AML.
Each of the drugs reduced the activity of HOX genes in NPM1-mutated AML cells, but combining the drugs resulted in near-complete inactivation of HOX genes.
When given alone, EPZ4777 and MI-503 each reduced the proliferation and colony-forming potential of NPM1-mutated AML cells in vitro. And each of the drugs prolonged survival in mouse models of NPM1-mutated AML.
However, EPZ4777 and MI-503 given in combination significantly delayed the onset of leukemia and significantly prolonged survival when compared to either drug given alone.
The investigators said this suggests that inhibiting both DOT1L and menin–MLL1 affects leukemia-initiating cells, and this approach represents the first molecularly targeted treatment of NPM1-mutated AML that works by reversing a key mechanism of leukemogenesis.
They added that this research paves the way for trials assessing EPZ4777 and MI-503 in patients with NPM1-mutated AML. ![]()
Image by Eric Smith
Preclinical research suggests a pair of histone-modifying proteins may be promising therapeutic targets for NPM1-mutated acute myeloid leukemia (AML).
Investigators found that these proteins—MLL and DOT1L—play key roles in NPM1-mutated AML.
Pharmacologic inhibition of either protein alone produced anti-leukemic activity in vitro and in vivo, but inhibiting both proteins together had a more profound effect.
Michael Kühn, MD, of the Mainz University Medical Center in Mainz, Germany, and his colleagues reported these findings in Cancer Discovery.
The investigators noted that nearly all NPM1-mutated AMLs are characterized by aberrant HOX expression, and FLT3 is concomitantly mutated in roughly 60% of these cases. However, it hasn’t been clear how mutant NPM1 cells maintain aberrant gene expression.
With this study, Dr Kühn and his colleagues showed that MLL1 and DOT1L control HOX and FLT3 expression and differentiation in NPM1-mutated AML.
The investigators were able to demonstrate that survival of NPM1-mutated AML cells depends on these 2 proteins. And NPM1-mutated AML is “exceptionally dependent” on the menin binding site in MLL1.
The team tested MI-503, a menin–MLL1 inhibitor, and the DOT1L inhibitor EPZ4777 in human and murine models of NPM1-mutated AML.
Each of the drugs reduced the activity of HOX genes in NPM1-mutated AML cells, but combining the drugs resulted in near-complete inactivation of HOX genes.
When given alone, EPZ4777 and MI-503 each reduced the proliferation and colony-forming potential of NPM1-mutated AML cells in vitro. And each of the drugs prolonged survival in mouse models of NPM1-mutated AML.
However, EPZ4777 and MI-503 given in combination significantly delayed the onset of leukemia and significantly prolonged survival when compared to either drug given alone.
The investigators said this suggests that inhibiting both DOT1L and menin–MLL1 affects leukemia-initiating cells, and this approach represents the first molecularly targeted treatment of NPM1-mutated AML that works by reversing a key mechanism of leukemogenesis.
They added that this research paves the way for trials assessing EPZ4777 and MI-503 in patients with NPM1-mutated AML. ![]()
Combo could treat AML, other cancers
A novel combination has shown promise for treating acute myeloid leukemia (AML) and other cancers, according to preclinical research published in Cancer Cell.
Researchers found that combining a DNMT inhibitor and a PARP inhibitor greatly increases the drugs’ anti-tumor activity, and the combination could be effective in malignancies that are not responsive to PARP inhibitors or DNMT inhibitors alone.
Experiments showed that, when combined, the 2 types of inhibitors cause interactions that significantly disrupt cancer cells’ ability to survive DNA damage.
“Our preclinical data suggest that combining low doses of these inhibitors will enhance the clinical effects of both drugs as a potential treatment for patients with AML,” said study author Feyruz V. Rassool, PhD, of the University of Maryland School of Medicine in Baltimore.
“Moreover, our initial data suggest that subtypes of AML with a poor prognosis are likely to be sensitive to this new therapeutic approach.”
Dr Rassool and her colleagues assessed the activity of a DNMT inhibitor—decitabine or 5-azacytidine—in combination with a PARP inhibitor—veliparib or talazoparib—against AML and breast cancer.
In both AML and breast cancer cells, combination treatment increased cytotoxicity and decreased clonogenicity, compared to treatment with either type of inhibitor alone.
The combination of 5-azacytidine and talazoparib produced “very robust responses” in 2 mouse models of AML (MV411 and MOLM14), according to the researchers.
“[It was] somewhat of a surprise that leukemia cells were this sensitive to the combination treatment,” said study author Stephen B. Baylin, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins Medical Institutions in Baltimore.
“And if further research confirms our findings, it looks like it also could be useful for breast cancer and ovarian cancers for which PARP inhibitors have not been useful as yet.”
How the inhibitors work together
Dr Baylin noted that PARP helps repair naturally occurring breaks in strands of DNA, and some cancers rely more frequently on PARP than others.
“[PARP inhibitors] work according to how intensely and durably the PARP enzyme is trapped at certain DNA damage sites,” he explained. “If you can ramp up the duration and intensity of this trapping, you could potentially increase the efficacy of the drug[s].”
“We figured that if we pair 5-azacytidine and a PARP inhibitor like talazoparib, we may be able to increase PARP trapping at DNA damage sites.”
That’s because 5-azacytidine blocks proteins that attach gene-regulating methyl groups to DNA and traps those proteins on DNA. The proteins blocked by 5-azacytidine also interact with PARP enzymes at DNA damage sites.
In fact, the researchers did find that combining 5-azacytidine and talazoparib increased the time that PARP was trapped at sites of DNA damage in cancer cells. The time was extended from 30 minutes to 3-6 hours after treatment.
Next steps
Based on the results of this research, a clinical trial is planned to test whether low doses of decitabine and talazoparib can be safely combined and whether this therapy will be effective in AML patients.
The researchers are especially interested in testing the combination in patients who cannot receive intensive chemotherapy, whose leukemia is resistant to treatment, or who have relapsed after treatment.
“This is really a new paradigm mechanism that is being translated into a clinical trial,” Dr Rassool said. “It’s not just putting 2 drugs together.”
“We have shown in the laboratory that the proteins that these inhibitors target actually interact, so the effects of these inhibitors are enhanced through this interaction. Therein lies the novelty of this new approach.” ![]()
A novel combination has shown promise for treating acute myeloid leukemia (AML) and other cancers, according to preclinical research published in Cancer Cell.
Researchers found that combining a DNMT inhibitor and a PARP inhibitor greatly increases the drugs’ anti-tumor activity, and the combination could be effective in malignancies that are not responsive to PARP inhibitors or DNMT inhibitors alone.
Experiments showed that, when combined, the 2 types of inhibitors cause interactions that significantly disrupt cancer cells’ ability to survive DNA damage.
“Our preclinical data suggest that combining low doses of these inhibitors will enhance the clinical effects of both drugs as a potential treatment for patients with AML,” said study author Feyruz V. Rassool, PhD, of the University of Maryland School of Medicine in Baltimore.
“Moreover, our initial data suggest that subtypes of AML with a poor prognosis are likely to be sensitive to this new therapeutic approach.”
Dr Rassool and her colleagues assessed the activity of a DNMT inhibitor—decitabine or 5-azacytidine—in combination with a PARP inhibitor—veliparib or talazoparib—against AML and breast cancer.
In both AML and breast cancer cells, combination treatment increased cytotoxicity and decreased clonogenicity, compared to treatment with either type of inhibitor alone.
The combination of 5-azacytidine and talazoparib produced “very robust responses” in 2 mouse models of AML (MV411 and MOLM14), according to the researchers.
“[It was] somewhat of a surprise that leukemia cells were this sensitive to the combination treatment,” said study author Stephen B. Baylin, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins Medical Institutions in Baltimore.
“And if further research confirms our findings, it looks like it also could be useful for breast cancer and ovarian cancers for which PARP inhibitors have not been useful as yet.”
How the inhibitors work together
Dr Baylin noted that PARP helps repair naturally occurring breaks in strands of DNA, and some cancers rely more frequently on PARP than others.
“[PARP inhibitors] work according to how intensely and durably the PARP enzyme is trapped at certain DNA damage sites,” he explained. “If you can ramp up the duration and intensity of this trapping, you could potentially increase the efficacy of the drug[s].”
“We figured that if we pair 5-azacytidine and a PARP inhibitor like talazoparib, we may be able to increase PARP trapping at DNA damage sites.”
That’s because 5-azacytidine blocks proteins that attach gene-regulating methyl groups to DNA and traps those proteins on DNA. The proteins blocked by 5-azacytidine also interact with PARP enzymes at DNA damage sites.
In fact, the researchers did find that combining 5-azacytidine and talazoparib increased the time that PARP was trapped at sites of DNA damage in cancer cells. The time was extended from 30 minutes to 3-6 hours after treatment.
Next steps
Based on the results of this research, a clinical trial is planned to test whether low doses of decitabine and talazoparib can be safely combined and whether this therapy will be effective in AML patients.
The researchers are especially interested in testing the combination in patients who cannot receive intensive chemotherapy, whose leukemia is resistant to treatment, or who have relapsed after treatment.
“This is really a new paradigm mechanism that is being translated into a clinical trial,” Dr Rassool said. “It’s not just putting 2 drugs together.”
“We have shown in the laboratory that the proteins that these inhibitors target actually interact, so the effects of these inhibitors are enhanced through this interaction. Therein lies the novelty of this new approach.” ![]()
A novel combination has shown promise for treating acute myeloid leukemia (AML) and other cancers, according to preclinical research published in Cancer Cell.
Researchers found that combining a DNMT inhibitor and a PARP inhibitor greatly increases the drugs’ anti-tumor activity, and the combination could be effective in malignancies that are not responsive to PARP inhibitors or DNMT inhibitors alone.
Experiments showed that, when combined, the 2 types of inhibitors cause interactions that significantly disrupt cancer cells’ ability to survive DNA damage.
“Our preclinical data suggest that combining low doses of these inhibitors will enhance the clinical effects of both drugs as a potential treatment for patients with AML,” said study author Feyruz V. Rassool, PhD, of the University of Maryland School of Medicine in Baltimore.
“Moreover, our initial data suggest that subtypes of AML with a poor prognosis are likely to be sensitive to this new therapeutic approach.”
Dr Rassool and her colleagues assessed the activity of a DNMT inhibitor—decitabine or 5-azacytidine—in combination with a PARP inhibitor—veliparib or talazoparib—against AML and breast cancer.
In both AML and breast cancer cells, combination treatment increased cytotoxicity and decreased clonogenicity, compared to treatment with either type of inhibitor alone.
The combination of 5-azacytidine and talazoparib produced “very robust responses” in 2 mouse models of AML (MV411 and MOLM14), according to the researchers.
“[It was] somewhat of a surprise that leukemia cells were this sensitive to the combination treatment,” said study author Stephen B. Baylin, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins Medical Institutions in Baltimore.
“And if further research confirms our findings, it looks like it also could be useful for breast cancer and ovarian cancers for which PARP inhibitors have not been useful as yet.”
How the inhibitors work together
Dr Baylin noted that PARP helps repair naturally occurring breaks in strands of DNA, and some cancers rely more frequently on PARP than others.
“[PARP inhibitors] work according to how intensely and durably the PARP enzyme is trapped at certain DNA damage sites,” he explained. “If you can ramp up the duration and intensity of this trapping, you could potentially increase the efficacy of the drug[s].”
“We figured that if we pair 5-azacytidine and a PARP inhibitor like talazoparib, we may be able to increase PARP trapping at DNA damage sites.”
That’s because 5-azacytidine blocks proteins that attach gene-regulating methyl groups to DNA and traps those proteins on DNA. The proteins blocked by 5-azacytidine also interact with PARP enzymes at DNA damage sites.
In fact, the researchers did find that combining 5-azacytidine and talazoparib increased the time that PARP was trapped at sites of DNA damage in cancer cells. The time was extended from 30 minutes to 3-6 hours after treatment.
Next steps
Based on the results of this research, a clinical trial is planned to test whether low doses of decitabine and talazoparib can be safely combined and whether this therapy will be effective in AML patients.
The researchers are especially interested in testing the combination in patients who cannot receive intensive chemotherapy, whose leukemia is resistant to treatment, or who have relapsed after treatment.
“This is really a new paradigm mechanism that is being translated into a clinical trial,” Dr Rassool said. “It’s not just putting 2 drugs together.”
“We have shown in the laboratory that the proteins that these inhibitors target actually interact, so the effects of these inhibitors are enhanced through this interaction. Therein lies the novelty of this new approach.”
‘Practice-changing’ treatments emerging in AML
NEW YORK—We are “finally” making progress in the treatment of acute myeloid leukemia (AML), according to a speaker at the NCCN 11th Annual Congress: Hematologic Malignancies.
Jessica K. Altman, MD, said a number of developments have resulted in improved AML treatment, including a better understanding of biology and prognostic assessment, continued advances in transplant, and updating standard treatments and incorporating novel agents in both relapsed/refractory and newly diagnosed patients.
“There are a couple of practice-changing treatments in acute myeloid leukemia, 2 of which happened over the last decade: daunorubicin intensification and the use of FLT3 inhibitors,” said Dr Altman, an associate professor at Northwestern University Feinberg School of Medicine in Chicago, Illinois.
Dr Altman went on to explain that novel therapies for AML can be divided into 2 basic categories. There are agents that don’t depend on mutation status (like daunorubicin) and those that are mutation-specific (like FLT3 inhibitors).
Therapies not dependent on mutational complexity
The therapies that are not dependent on mutational complexity include anti-CD33 antibodies, BCL‐2 inhibitors, a dose-intensified anthracycline regimen, and different formulations of 7+3, including CPX‐351.
Escalated daunorubicin
Randomized trials of escalated daunorubicin (90 vs 45 mg/m2) have demonstrated benefit in complete responses (CRs) and overall survival (OS) in intermediate-risk patients and patients with core-binding factor mutation. They have demonstrated benefit in OS in FLT3 ITD+ patients.
In patients up to 65 years of age, 60–90 mg/m2 of daunorubicin is now standard.
“It’s still not clear to me—and I don’t know if it ever will be—if 90 is equivalent to 60,” Dr Altman said.
CPX-351
CPX-351 is a liposomal formulation of cytarabine and daunorubicin. In a randomized, phase 3 study of older adults with secondary AML, the median OS was 9.56 months for patients treated with CPX-351 and 5.95 months for patients on the 7+3 regimen (P=0.005).
The median event-free survival was significantly better with CPX-351 (P=0.021), as was the rate of CR + CR with incomplete blood count recovery (CRi). The rate of CR + CRi was 47.7% with CPX-351 and 33.3% for 7+3 (P=0.016).
A similar number of patients went on to transplant in each arm. Grade 3-5 adverse events were similar in frequency and severity in both arms—92% with CPX-351 vs 91% with 7+3.
SGN-CD33A
CD33 is not a new target in myeloid leukemia, Dr Altman pointed out. Gemtuzumab ozogamicin has been studied, approved by the US Food and Drug Administration, and then withdrawn.
However, an increasing number of studies with gemtuzumab are underway, she said, and the agent may once again have a place in the AML armamentarium.
The newest CD33 construct is SGN-CD33A, a stable dipeptide linker that enables uniform drug loading of a pyrrolobenzodiazepine dimer that crosslinks DNA and leads to cell death.
“Single-agent data was quite promising,” Dr Altman noted, with a CR + CRi rate of 41% in previously treated patients and 58% in 12 treatment-naïve patients.
These results prompted a combination study of SGN-CD33A with hypomethylating agents.
“Results were higher than expected with a hypomethylating agent,” Dr Altman pointed out.
The CR + CRi + CR with incomplete platelet recovery was 58%. And the median relapse-free survival was 7.7 months.
A phase 3 randomized trial of SGN-CD33A is planned.
Venetoclax
BCL-2 inhibitors are the fourth type of agent not dependent on mutation complexity. Venetoclax (ABT‐199) is a small‐molecule BCL-2 inhibitor that leads to the initiation of apoptosis.
In a phase 1b trial of venetoclax in combination with a hypomethylating agent, the overall CR rate was 35%, and the CRi rate was 35%.
“Again, higher than what would be expected with a hypomethylating agent alone,” Dr Altman said.
In a phase 1b/2 trial of venetoclax in combination with low‐dose cytarabine, the CR + CRi rate was 54%. Patients responded even if they had prior exposure to hypomethylating agents.
Mutation-specific novel agents
The FLT3 inhibitor midostaurin and the IDH inhibitors AG-120 and AG-221 are among the most exciting mutation-specific agents and the ones most progressed, according to Dr Altman.
FLT3-ITD is mutated in about 30% of AML patients and carries an unfavorable prognosis, and the IDH mutation occurs in about 10% and confers a favorable prognosis.
Midostaurin
A phase 3, randomized, double-blind study of daunorubicin/cytarabine induction and high-dose cytarabine consolidation with midostaurin (PKC412) or placebo had a 59% CR rate by day 60 in the midostaurin arm, compared with 53% in the placebo arm.
“The CR rate was slightly higher in the midostaurin arm,” Dr Altman said, “but what’s the most remarkable about this study is the difference in overall survival.”
The median OS in the midostaurin arm was 74.7 months, compared with 25.6 months in the placebo arm (P=0.0074).
“The major take-home message from this clinical trial,” Dr Altman said, “is that midostaurin improved the overall survival when added to standard therapy and represents a new standard of care.”
AG-120 and AG-221
Two IDH inhibitors that have substantial data available are the IDH1 inhibitor AG-120 and the IDH2 inhibitor AG-221.
As of October 2015, 78 patients had been treated with AG-120 in a phase 1 trial, yielding an overall response rate of 35% and a CR rate of 15%.
As of September 2015, 209 patients had been treated with AG-221 in a phase 1/2 trial, and 66 are still on study. The overall response rate was 37% in 159 adults with relapsed/refractory AML, with a median duration of response of 6.9 months. The CR rate was 18%.
Investigators have initiated a phase 3 study of AG-221 compared to conventional care regimens.
NEW YORK—We are “finally” making progress in the treatment of acute myeloid leukemia (AML), according to a speaker at the NCCN 11th Annual Congress: Hematologic Malignancies.
Jessica K. Altman, MD, said a number of developments have resulted in improved AML treatment, including a better understanding of biology and prognostic assessment, continued advances in transplant, and updating standard treatments and incorporating novel agents in both relapsed/refractory and newly diagnosed patients.
“There are a couple of practice-changing treatments in acute myeloid leukemia, 2 of which happened over the last decade: daunorubicin intensification and the use of FLT3 inhibitors,” said Dr Altman, an associate professor at Northwestern University Feinberg School of Medicine in Chicago, Illinois.
Dr Altman went on to explain that novel therapies for AML can be divided into 2 basic categories. There are agents that don’t depend on mutation status (like daunorubicin) and those that are mutation-specific (like FLT3 inhibitors).
Therapies not dependent on mutational complexity
The therapies that are not dependent on mutational complexity include anti-CD33 antibodies, BCL‐2 inhibitors, a dose-intensified anthracycline regimen, and different formulations of 7+3, including CPX‐351.
Escalated daunorubicin
Randomized trials of escalated daunorubicin (90 vs 45 mg/m2) have demonstrated benefit in complete responses (CRs) and overall survival (OS) in intermediate-risk patients and patients with core-binding factor mutation. They have demonstrated benefit in OS in FLT3 ITD+ patients.
In patients up to 65 years of age, 60–90 mg/m2 of daunorubicin is now standard.
“It’s still not clear to me—and I don’t know if it ever will be—if 90 is equivalent to 60,” Dr Altman said.
CPX-351
CPX-351 is a liposomal formulation of cytarabine and daunorubicin. In a randomized, phase 3 study of older adults with secondary AML, the median OS was 9.56 months for patients treated with CPX-351 and 5.95 months for patients on the 7+3 regimen (P=0.005).
The median event-free survival was significantly better with CPX-351 (P=0.021), as was the rate of CR + CR with incomplete blood count recovery (CRi). The rate of CR + CRi was 47.7% with CPX-351 and 33.3% for 7+3 (P=0.016).
A similar number of patients went on to transplant in each arm. Grade 3-5 adverse events were similar in frequency and severity in both arms—92% with CPX-351 vs 91% with 7+3.
SGN-CD33A
CD33 is not a new target in myeloid leukemia, Dr Altman pointed out. Gemtuzumab ozogamicin has been studied, approved by the US Food and Drug Administration, and then withdrawn.
However, an increasing number of studies with gemtuzumab are underway, she said, and the agent may once again have a place in the AML armamentarium.
The newest CD33 construct is SGN-CD33A, a stable dipeptide linker that enables uniform drug loading of a pyrrolobenzodiazepine dimer that crosslinks DNA and leads to cell death.
“Single-agent data was quite promising,” Dr Altman noted, with a CR + CRi rate of 41% in previously treated patients and 58% in 12 treatment-naïve patients.
These results prompted a combination study of SGN-CD33A with hypomethylating agents.
“Results were higher than expected with a hypomethylating agent,” Dr Altman pointed out.
The CR + CRi + CR with incomplete platelet recovery was 58%. And the median relapse-free survival was 7.7 months.
A phase 3 randomized trial of SGN-CD33A is planned.
Venetoclax
BCL-2 inhibitors are the fourth type of agent not dependent on mutation complexity. Venetoclax (ABT‐199) is a small‐molecule BCL-2 inhibitor that leads to the initiation of apoptosis.
In a phase 1b trial of venetoclax in combination with a hypomethylating agent, the overall CR rate was 35%, and the CRi rate was 35%.
“Again, higher than what would be expected with a hypomethylating agent alone,” Dr Altman said.
In a phase 1b/2 trial of venetoclax in combination with low‐dose cytarabine, the CR + CRi rate was 54%. Patients responded even if they had prior exposure to hypomethylating agents.
Mutation-specific novel agents
The FLT3 inhibitor midostaurin and the IDH inhibitors AG-120 and AG-221 are among the most exciting mutation-specific agents and the ones most progressed, according to Dr Altman.
FLT3-ITD is mutated in about 30% of AML patients and carries an unfavorable prognosis, and the IDH mutation occurs in about 10% and confers a favorable prognosis.
Midostaurin
A phase 3, randomized, double-blind study of daunorubicin/cytarabine induction and high-dose cytarabine consolidation with midostaurin (PKC412) or placebo had a 59% CR rate by day 60 in the midostaurin arm, compared with 53% in the placebo arm.
“The CR rate was slightly higher in the midostaurin arm,” Dr Altman said, “but what’s the most remarkable about this study is the difference in overall survival.”
The median OS in the midostaurin arm was 74.7 months, compared with 25.6 months in the placebo arm (P=0.0074).
“The major take-home message from this clinical trial,” Dr Altman said, “is that midostaurin improved the overall survival when added to standard therapy and represents a new standard of care.”
AG-120 and AG-221
Two IDH inhibitors that have substantial data available are the IDH1 inhibitor AG-120 and the IDH2 inhibitor AG-221.
As of October 2015, 78 patients had been treated with AG-120 in a phase 1 trial, yielding an overall response rate of 35% and a CR rate of 15%.
As of September 2015, 209 patients had been treated with AG-221 in a phase 1/2 trial, and 66 are still on study. The overall response rate was 37% in 159 adults with relapsed/refractory AML, with a median duration of response of 6.9 months. The CR rate was 18%.
Investigators have initiated a phase 3 study of AG-221 compared to conventional care regimens.
NEW YORK—We are “finally” making progress in the treatment of acute myeloid leukemia (AML), according to a speaker at the NCCN 11th Annual Congress: Hematologic Malignancies.
Jessica K. Altman, MD, said a number of developments have resulted in improved AML treatment, including a better understanding of biology and prognostic assessment, continued advances in transplant, and updating standard treatments and incorporating novel agents in both relapsed/refractory and newly diagnosed patients.
“There are a couple of practice-changing treatments in acute myeloid leukemia, 2 of which happened over the last decade: daunorubicin intensification and the use of FLT3 inhibitors,” said Dr Altman, an associate professor at Northwestern University Feinberg School of Medicine in Chicago, Illinois.
Dr Altman went on to explain that novel therapies for AML can be divided into 2 basic categories. There are agents that don’t depend on mutation status (like daunorubicin) and those that are mutation-specific (like FLT3 inhibitors).
Therapies not dependent on mutational complexity
The therapies that are not dependent on mutational complexity include anti-CD33 antibodies, BCL‐2 inhibitors, a dose-intensified anthracycline regimen, and different formulations of 7+3, including CPX‐351.
Escalated daunorubicin
Randomized trials of escalated daunorubicin (90 vs 45 mg/m2) have demonstrated benefit in complete responses (CRs) and overall survival (OS) in intermediate-risk patients and patients with core-binding factor mutation. They have demonstrated benefit in OS in FLT3 ITD+ patients.
In patients up to 65 years of age, 60–90 mg/m2 of daunorubicin is now standard.
“It’s still not clear to me—and I don’t know if it ever will be—if 90 is equivalent to 60,” Dr Altman said.
CPX-351
CPX-351 is a liposomal formulation of cytarabine and daunorubicin. In a randomized, phase 3 study of older adults with secondary AML, the median OS was 9.56 months for patients treated with CPX-351 and 5.95 months for patients on the 7+3 regimen (P=0.005).
The median event-free survival was significantly better with CPX-351 (P=0.021), as was the rate of CR + CR with incomplete blood count recovery (CRi). The rate of CR + CRi was 47.7% with CPX-351 and 33.3% for 7+3 (P=0.016).
A similar number of patients went on to transplant in each arm. Grade 3-5 adverse events were similar in frequency and severity in both arms—92% with CPX-351 vs 91% with 7+3.
SGN-CD33A
CD33 is not a new target in myeloid leukemia, Dr Altman pointed out. Gemtuzumab ozogamicin has been studied, approved by the US Food and Drug Administration, and then withdrawn.
However, an increasing number of studies with gemtuzumab are underway, she said, and the agent may once again have a place in the AML armamentarium.
The newest CD33 construct is SGN-CD33A, a stable dipeptide linker that enables uniform drug loading of a pyrrolobenzodiazepine dimer that crosslinks DNA and leads to cell death.
“Single-agent data was quite promising,” Dr Altman noted, with a CR + CRi rate of 41% in previously treated patients and 58% in 12 treatment-naïve patients.
These results prompted a combination study of SGN-CD33A with hypomethylating agents.
“Results were higher than expected with a hypomethylating agent,” Dr Altman pointed out.
The CR + CRi + CR with incomplete platelet recovery was 58%. And the median relapse-free survival was 7.7 months.
A phase 3 randomized trial of SGN-CD33A is planned.
Venetoclax
BCL-2 inhibitors are the fourth type of agent not dependent on mutation complexity. Venetoclax (ABT‐199) is a small‐molecule BCL-2 inhibitor that leads to the initiation of apoptosis.
In a phase 1b trial of venetoclax in combination with a hypomethylating agent, the overall CR rate was 35%, and the CRi rate was 35%.
“Again, higher than what would be expected with a hypomethylating agent alone,” Dr Altman said.
In a phase 1b/2 trial of venetoclax in combination with low‐dose cytarabine, the CR + CRi rate was 54%. Patients responded even if they had prior exposure to hypomethylating agents.
Mutation-specific novel agents
The FLT3 inhibitor midostaurin and the IDH inhibitors AG-120 and AG-221 are among the most exciting mutation-specific agents and the ones most progressed, according to Dr Altman.
FLT3-ITD is mutated in about 30% of AML patients and carries an unfavorable prognosis, and the IDH mutation occurs in about 10% and confers a favorable prognosis.
Midostaurin
A phase 3, randomized, double-blind study of daunorubicin/cytarabine induction and high-dose cytarabine consolidation with midostaurin (PKC412) or placebo had a 59% CR rate by day 60 in the midostaurin arm, compared with 53% in the placebo arm.
“The CR rate was slightly higher in the midostaurin arm,” Dr Altman said, “but what’s the most remarkable about this study is the difference in overall survival.”
The median OS in the midostaurin arm was 74.7 months, compared with 25.6 months in the placebo arm (P=0.0074).
“The major take-home message from this clinical trial,” Dr Altman said, “is that midostaurin improved the overall survival when added to standard therapy and represents a new standard of care.”
AG-120 and AG-221
Two IDH inhibitors that have substantial data available are the IDH1 inhibitor AG-120 and the IDH2 inhibitor AG-221.
As of October 2015, 78 patients had been treated with AG-120 in a phase 1 trial, yielding an overall response rate of 35% and a CR rate of 15%.
As of September 2015, 209 patients had been treated with AG-221 in a phase 1/2 trial, and 66 are still on study. The overall response rate was 37% in 159 adults with relapsed/refractory AML, with a median duration of response of 6.9 months. The CR rate was 18%.
Investigators have initiated a phase 3 study of AG-221 compared to conventional care regimens.
Inflammation may predict transformation to AML

Inflammatory signaling in mesenchymal niche cells can be used to predict the transformation from pre-leukemic syndrome to acute myeloid leukemia (AML), according to preclinical research published in Cell Stem Cell.
“This discovery sheds new light on the long-standing association between inflammation and cancer,” said study author Marc Raaijmakers, MD, PhD, of the Erasmus MC Cancer Institute in Rotterdam, Netherlands.
“The elucidation of the molecular mechanism underlying this concept opens the prospect of improved diagnosis of patients at increased risk for the development of leukemia and the potential of future, niche-targeted therapy to delay or prevent the development of leukemia.”
In a previous study, Dr Raaijmakers and his colleagues discovered that mutations in mesenchymal progenitor cells can induce myelodysplasia in mice and promote the development of AML.
With the current study, the researchers wanted to build upon those findings by identifying the underlying mechanisms and determining their relevance to human disease.
So the team performed massive parallel RNA sequencing of mesenchymal cells in mice with Shwachman-Diamond syndrome and bone marrow samples from patients with Shwachman-Diamond syndrome, Diamond-Blackfan anemia, and myelodysplastic syndromes (MDS).
The researchers found that mesenchymal cells in these pre-leukemic disorders are under stress. The stress leads to the release of inflammatory molecules called S100A8 and S100A9, which cause mitochondrial and DNA damage in hematopoietic stem and progenitor cells.
The team also found that activation of this inflammatory pathway in mesenchymal cells predicted the development of AML and clinical outcomes in patients with MDS.
Leukemic evolution occurred in 29.4% (5/17) of MDS patients whose mesenchymal cells overexpressed S100A8/9 and 14.2% (4/28) of MDS patients without S100A8/9 overexpression.
The time to leukemic evolution and the length of progression-free survival were both significantly shorter in niche S100A8/9+ patients than niche S100A8/9- patients.
The average time to leukemic evolution was 3.4 months and 18.5 months, respectively (P=0.03). And the median progression-free survival was 11.5 months and 53 months, respectively (P=0.03)
The researchers believe these findings, if confirmed in subsequent studies, could lead to the development of tests to identify patients with pre-leukemic syndromes who have a high risk of developing AML.
“These high-risk patients could be treated more aggressively at an earlier stage, thereby preventing or slowing down disease progression,” Dr Raaijmakers said. “Moreover, the findings suggest that new drugs targeting the inflammatory pathway should be tested in future preclinical studies.”
Inflammatory signaling in mesenchymal niche cells can be used to predict the transformation from pre-leukemic syndrome to acute myeloid leukemia (AML), according to preclinical research published in Cell Stem Cell.
“This discovery sheds new light on the long-standing association between inflammation and cancer,” said study author Marc Raaijmakers, MD, PhD, of the Erasmus MC Cancer Institute in Rotterdam, Netherlands.
“The elucidation of the molecular mechanism underlying this concept opens the prospect of improved diagnosis of patients at increased risk for the development of leukemia and the potential of future, niche-targeted therapy to delay or prevent the development of leukemia.”
In a previous study, Dr Raaijmakers and his colleagues discovered that mutations in mesenchymal progenitor cells can induce myelodysplasia in mice and promote the development of AML.
With the current study, the researchers wanted to build upon those findings by identifying the underlying mechanisms and determining their relevance to human disease.
So the team performed massive parallel RNA sequencing of mesenchymal cells in mice with Shwachman-Diamond syndrome and bone marrow samples from patients with Shwachman-Diamond syndrome, Diamond-Blackfan anemia, and myelodysplastic syndromes (MDS).
The researchers found that mesenchymal cells in these pre-leukemic disorders are under stress. The stress leads to the release of inflammatory molecules called S100A8 and S100A9, which cause mitochondrial and DNA damage in hematopoietic stem and progenitor cells.
The team also found that activation of this inflammatory pathway in mesenchymal cells predicted the development of AML and clinical outcomes in patients with MDS.
Leukemic evolution occurred in 29.4% (5/17) of MDS patients whose mesenchymal cells overexpressed S100A8/9 and 14.2% (4/28) of MDS patients without S100A8/9 overexpression.
The time to leukemic evolution and the length of progression-free survival were both significantly shorter in niche S100A8/9+ patients than niche S100A8/9- patients.
The average time to leukemic evolution was 3.4 months and 18.5 months, respectively (P=0.03). And the median progression-free survival was 11.5 months and 53 months, respectively (P=0.03)
The researchers believe these findings, if confirmed in subsequent studies, could lead to the development of tests to identify patients with pre-leukemic syndromes who have a high risk of developing AML.
“These high-risk patients could be treated more aggressively at an earlier stage, thereby preventing or slowing down disease progression,” Dr Raaijmakers said. “Moreover, the findings suggest that new drugs targeting the inflammatory pathway should be tested in future preclinical studies.”
Inflammatory signaling in mesenchymal niche cells can be used to predict the transformation from pre-leukemic syndrome to acute myeloid leukemia (AML), according to preclinical research published in Cell Stem Cell.
“This discovery sheds new light on the long-standing association between inflammation and cancer,” said study author Marc Raaijmakers, MD, PhD, of the Erasmus MC Cancer Institute in Rotterdam, Netherlands.
“The elucidation of the molecular mechanism underlying this concept opens the prospect of improved diagnosis of patients at increased risk for the development of leukemia and the potential of future, niche-targeted therapy to delay or prevent the development of leukemia.”
In a previous study, Dr Raaijmakers and his colleagues discovered that mutations in mesenchymal progenitor cells can induce myelodysplasia in mice and promote the development of AML.
With the current study, the researchers wanted to build upon those findings by identifying the underlying mechanisms and determining their relevance to human disease.
So the team performed massive parallel RNA sequencing of mesenchymal cells in mice with Shwachman-Diamond syndrome and bone marrow samples from patients with Shwachman-Diamond syndrome, Diamond-Blackfan anemia, and myelodysplastic syndromes (MDS).
The researchers found that mesenchymal cells in these pre-leukemic disorders are under stress. The stress leads to the release of inflammatory molecules called S100A8 and S100A9, which cause mitochondrial and DNA damage in hematopoietic stem and progenitor cells.
The team also found that activation of this inflammatory pathway in mesenchymal cells predicted the development of AML and clinical outcomes in patients with MDS.
Leukemic evolution occurred in 29.4% (5/17) of MDS patients whose mesenchymal cells overexpressed S100A8/9 and 14.2% (4/28) of MDS patients without S100A8/9 overexpression.
The time to leukemic evolution and the length of progression-free survival were both significantly shorter in niche S100A8/9+ patients than niche S100A8/9- patients.
The average time to leukemic evolution was 3.4 months and 18.5 months, respectively (P=0.03). And the median progression-free survival was 11.5 months and 53 months, respectively (P=0.03)
The researchers believe these findings, if confirmed in subsequent studies, could lead to the development of tests to identify patients with pre-leukemic syndromes who have a high risk of developing AML.
“These high-risk patients could be treated more aggressively at an earlier stage, thereby preventing or slowing down disease progression,” Dr Raaijmakers said. “Moreover, the findings suggest that new drugs targeting the inflammatory pathway should be tested in future preclinical studies.”