User login
VIDEO: 33A + ‘7 + 3’ equals good remission numbers in untreated AML
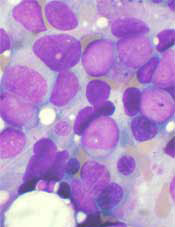
SAN DIEGO – Call it “7+3+1”: an experimental induction regimen combining standard chemotherapy with an antibody drug conjugate induced rapid and deep remissions in a majority of patients with newly diagnosed acute myeloid leukemia in a small study.
Among 42 evaluable patients with previously untreated AML, the combination of cytarabine and an anthracycline (7+3, also known as 3+7), and the investigational antibody drug conjugate vadastuximab talirine was associated with a 60% complete remission (CR) rate, and 17% complete remission with incomplete recovery of platelets (CRi), reported Harry P. Erba, MD, PhD, of the University of Alabama at Birmingham, who discussed the findings in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
“In 1973, 43 years ago, the first paper was published on what we still continue to use as the initial therapy for a very aggressive cancer, acute myeloid leukemia,” he said at a briefing at the American Society of Hematology annual meeting.
“Nothing has been shown yet to be superior to that, despite four decades of clinical research,” he added.
Recent studies have suggested that depth of postinduction remissions, specifically being minimal residual disease (MRD)-negative, is associated with improved survival, he noted.
Vadastuximab talirine (33A, for short) is an antibody-drug conjugate targeted to CD33, which is expressed in approximately 90% of AML cells. The drug is designed to deliver a cytotoxic agent to myeloid leukemia cells.
As reported previously, 33A, in combination with a hypomethylating agent (decitabine or azacitidine) in 49 evaluable patients, was associated with a composite CR/CRi rate of 71%; the rates of CR/CRi were similar regardless of the partner agent used.
The overall response rate in that study was 76%, with responses seen among higher-risk patients, including remissions in 16 of 22 patients with underlying myelodysplasia, and in 15 of 18 patients with adverse cytogenetics.
Rapid complete remissions
In the phase Ib trial reported at ASH 2016 by Dr. Erba, adults aged 18-65 years with untreated primary or secondary AML (except acute promyelocytic leukemia) were enrolled.
The patients received 33A in combination with 7+3 induction therapy (cytarabine 100 mg/m2 and daunorubicin 60 mg/m2) on days 1 and 4 of a 28-day treatment cycle. Patients were assessed for response on days 1 and 28 according to International Working Group Criteria.
Second induction regimens and postremission therapies were permitted at the investigators discretion, and did not include 33A.
The median patient age was 45.5 years. The patients had generally good performance status (Eastern Cooperative Oncology Group 0 or 1). In all, 17% of patients had secondary AML. In all, 12% had favorable cytogenetic risk disease, 50% had intermediate risk, and 36% had adverse risk. Ten percent of patients had NPM1 mutated disease, and 14% had FLT-3 mutations.
As noted, the composite CR/CRi rate was 76%, consisting of 60% CR and 17% CRI.
All five patients with favorable risk disease had a CR. The rate of CR/CRi was 86% among patients with intermediate-risk disease, and 60 for those with adverse-risk disease.
Of the 32 patients who achieved a CR or CRi, 94% did so after 1 cycle of therapy, and 25 were MRD negative, as evaluated by an independent laboratory using 10-color multi-parameter flow cytometry.
Treatment-related adverse hematologic events included febrile neutropenia (primarily grade 3) in 43% of patients, thrombocytopenia (mostly grade 4) in 38%, anemia (all grade 3) in 24%, and neutropenia (mostly grade 4) in 17%. Other treatment related events were similar to those seen with 7 + 3 alone, and included nausea, diarrhea, decreased appetite and fatigue, mostly grade 1 or 2. One patient had a grade 3 irreversible hepatic toxicity.
The death rate was 2%.
“What we felt we showed is that we were able to combine active doses of 33A with 7 + 3. The doses here were less than the doses used as a single agent, but all doses used in our phase 1b study, including lower doses that what we actually used here, showed complete remissions as a single agent.”
33A “added acceptable on-target myelosuppression. We saw platelet counts recovering to over 100,000, and neutrophils over 1,000 by about four-and-a-half to five weeks, which we felt was reasonable, and patients were able to go on to get post-remission therapy.
A randomized phase II trial comparing 33A and 7+3 to 7+3 alone is slated to launch in the first quarter of 2017.
Dr. Erba disclosed serving as a consultant to and receiving research funding from Seattle Genetics, which supported the study.
SAN DIEGO – Call it “7+3+1”: an experimental induction regimen combining standard chemotherapy with an antibody drug conjugate induced rapid and deep remissions in a majority of patients with newly diagnosed acute myeloid leukemia in a small study.
Among 42 evaluable patients with previously untreated AML, the combination of cytarabine and an anthracycline (7+3, also known as 3+7), and the investigational antibody drug conjugate vadastuximab talirine was associated with a 60% complete remission (CR) rate, and 17% complete remission with incomplete recovery of platelets (CRi), reported Harry P. Erba, MD, PhD, of the University of Alabama at Birmingham, who discussed the findings in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
“In 1973, 43 years ago, the first paper was published on what we still continue to use as the initial therapy for a very aggressive cancer, acute myeloid leukemia,” he said at a briefing at the American Society of Hematology annual meeting.
“Nothing has been shown yet to be superior to that, despite four decades of clinical research,” he added.
Recent studies have suggested that depth of postinduction remissions, specifically being minimal residual disease (MRD)-negative, is associated with improved survival, he noted.
Vadastuximab talirine (33A, for short) is an antibody-drug conjugate targeted to CD33, which is expressed in approximately 90% of AML cells. The drug is designed to deliver a cytotoxic agent to myeloid leukemia cells.
As reported previously, 33A, in combination with a hypomethylating agent (decitabine or azacitidine) in 49 evaluable patients, was associated with a composite CR/CRi rate of 71%; the rates of CR/CRi were similar regardless of the partner agent used.
The overall response rate in that study was 76%, with responses seen among higher-risk patients, including remissions in 16 of 22 patients with underlying myelodysplasia, and in 15 of 18 patients with adverse cytogenetics.
Rapid complete remissions
In the phase Ib trial reported at ASH 2016 by Dr. Erba, adults aged 18-65 years with untreated primary or secondary AML (except acute promyelocytic leukemia) were enrolled.
The patients received 33A in combination with 7+3 induction therapy (cytarabine 100 mg/m2 and daunorubicin 60 mg/m2) on days 1 and 4 of a 28-day treatment cycle. Patients were assessed for response on days 1 and 28 according to International Working Group Criteria.
Second induction regimens and postremission therapies were permitted at the investigators discretion, and did not include 33A.
The median patient age was 45.5 years. The patients had generally good performance status (Eastern Cooperative Oncology Group 0 or 1). In all, 17% of patients had secondary AML. In all, 12% had favorable cytogenetic risk disease, 50% had intermediate risk, and 36% had adverse risk. Ten percent of patients had NPM1 mutated disease, and 14% had FLT-3 mutations.
As noted, the composite CR/CRi rate was 76%, consisting of 60% CR and 17% CRI.
All five patients with favorable risk disease had a CR. The rate of CR/CRi was 86% among patients with intermediate-risk disease, and 60 for those with adverse-risk disease.
Of the 32 patients who achieved a CR or CRi, 94% did so after 1 cycle of therapy, and 25 were MRD negative, as evaluated by an independent laboratory using 10-color multi-parameter flow cytometry.
Treatment-related adverse hematologic events included febrile neutropenia (primarily grade 3) in 43% of patients, thrombocytopenia (mostly grade 4) in 38%, anemia (all grade 3) in 24%, and neutropenia (mostly grade 4) in 17%. Other treatment related events were similar to those seen with 7 + 3 alone, and included nausea, diarrhea, decreased appetite and fatigue, mostly grade 1 or 2. One patient had a grade 3 irreversible hepatic toxicity.
The death rate was 2%.
“What we felt we showed is that we were able to combine active doses of 33A with 7 + 3. The doses here were less than the doses used as a single agent, but all doses used in our phase 1b study, including lower doses that what we actually used here, showed complete remissions as a single agent.”
33A “added acceptable on-target myelosuppression. We saw platelet counts recovering to over 100,000, and neutrophils over 1,000 by about four-and-a-half to five weeks, which we felt was reasonable, and patients were able to go on to get post-remission therapy.
A randomized phase II trial comparing 33A and 7+3 to 7+3 alone is slated to launch in the first quarter of 2017.
Dr. Erba disclosed serving as a consultant to and receiving research funding from Seattle Genetics, which supported the study.
SAN DIEGO – Call it “7+3+1”: an experimental induction regimen combining standard chemotherapy with an antibody drug conjugate induced rapid and deep remissions in a majority of patients with newly diagnosed acute myeloid leukemia in a small study.
Among 42 evaluable patients with previously untreated AML, the combination of cytarabine and an anthracycline (7+3, also known as 3+7), and the investigational antibody drug conjugate vadastuximab talirine was associated with a 60% complete remission (CR) rate, and 17% complete remission with incomplete recovery of platelets (CRi), reported Harry P. Erba, MD, PhD, of the University of Alabama at Birmingham, who discussed the findings in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
“In 1973, 43 years ago, the first paper was published on what we still continue to use as the initial therapy for a very aggressive cancer, acute myeloid leukemia,” he said at a briefing at the American Society of Hematology annual meeting.
“Nothing has been shown yet to be superior to that, despite four decades of clinical research,” he added.
Recent studies have suggested that depth of postinduction remissions, specifically being minimal residual disease (MRD)-negative, is associated with improved survival, he noted.
Vadastuximab talirine (33A, for short) is an antibody-drug conjugate targeted to CD33, which is expressed in approximately 90% of AML cells. The drug is designed to deliver a cytotoxic agent to myeloid leukemia cells.
As reported previously, 33A, in combination with a hypomethylating agent (decitabine or azacitidine) in 49 evaluable patients, was associated with a composite CR/CRi rate of 71%; the rates of CR/CRi were similar regardless of the partner agent used.
The overall response rate in that study was 76%, with responses seen among higher-risk patients, including remissions in 16 of 22 patients with underlying myelodysplasia, and in 15 of 18 patients with adverse cytogenetics.
Rapid complete remissions
In the phase Ib trial reported at ASH 2016 by Dr. Erba, adults aged 18-65 years with untreated primary or secondary AML (except acute promyelocytic leukemia) were enrolled.
The patients received 33A in combination with 7+3 induction therapy (cytarabine 100 mg/m2 and daunorubicin 60 mg/m2) on days 1 and 4 of a 28-day treatment cycle. Patients were assessed for response on days 1 and 28 according to International Working Group Criteria.
Second induction regimens and postremission therapies were permitted at the investigators discretion, and did not include 33A.
The median patient age was 45.5 years. The patients had generally good performance status (Eastern Cooperative Oncology Group 0 or 1). In all, 17% of patients had secondary AML. In all, 12% had favorable cytogenetic risk disease, 50% had intermediate risk, and 36% had adverse risk. Ten percent of patients had NPM1 mutated disease, and 14% had FLT-3 mutations.
As noted, the composite CR/CRi rate was 76%, consisting of 60% CR and 17% CRI.
All five patients with favorable risk disease had a CR. The rate of CR/CRi was 86% among patients with intermediate-risk disease, and 60 for those with adverse-risk disease.
Of the 32 patients who achieved a CR or CRi, 94% did so after 1 cycle of therapy, and 25 were MRD negative, as evaluated by an independent laboratory using 10-color multi-parameter flow cytometry.
Treatment-related adverse hematologic events included febrile neutropenia (primarily grade 3) in 43% of patients, thrombocytopenia (mostly grade 4) in 38%, anemia (all grade 3) in 24%, and neutropenia (mostly grade 4) in 17%. Other treatment related events were similar to those seen with 7 + 3 alone, and included nausea, diarrhea, decreased appetite and fatigue, mostly grade 1 or 2. One patient had a grade 3 irreversible hepatic toxicity.
The death rate was 2%.
“What we felt we showed is that we were able to combine active doses of 33A with 7 + 3. The doses here were less than the doses used as a single agent, but all doses used in our phase 1b study, including lower doses that what we actually used here, showed complete remissions as a single agent.”
33A “added acceptable on-target myelosuppression. We saw platelet counts recovering to over 100,000, and neutrophils over 1,000 by about four-and-a-half to five weeks, which we felt was reasonable, and patients were able to go on to get post-remission therapy.
A randomized phase II trial comparing 33A and 7+3 to 7+3 alone is slated to launch in the first quarter of 2017.
Dr. Erba disclosed serving as a consultant to and receiving research funding from Seattle Genetics, which supported the study.
AT ASH 2016
Key clinical point: Deep remissions following induction therapy with AML are associated with better survival outcomes.
Major finding: Adding the antibody drug conjugate vadastuximab talirine (33A) to 7+3 induction therapy induced complete or near-complete remissions 76% of patients with newly diagnosed acute myeloid leukemia.
Data source: Phase Ib study in 42 patients with previously untreated primary or secondary AML.
Disclosures: Dr. Erba disclosed serving as a consultant to and receiving research funding from Seattle Genetics, which supported the study.

Decitabine produces responses in high-risk MDS, AML

receiving chemotherapy
Photo by Rhoda Baer
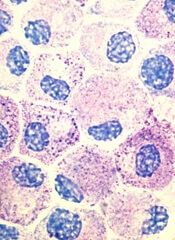
Patients with TP53-mutated myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML) may benefit from treatment with decitabine, according to a study published in NEJM.
All patients in this study who had TP53 mutations responded to decitabine.
Although these responses were not durable, the patients’ median overall survival was similar to that of patients with lower-risk disease who received decitabine.
“The findings need to be validated in a larger trial, but they do suggest that TP53 mutations can reliably predict responses to decitabine, potentially prolonging survival in this ultra-high-risk group of patients and providing a bridge to transplantation in some patients who might not otherwise be candidates,” said study author Timothy J. Ley, MD, of Washington University School of Medicine in St. Louis, Missouri.
For this study, Dr Ley and his colleagues analyzed 116 patients—54 with AML, 36 with relapsed AML, and 26 with MDS.
Eighty-four of the patients were enrolled in a prospective trial and received decitabine at a dose of 20 mg/m2/day for 10 consecutive days in monthly cycles. Thirty-two additional patients received decitabine on different protocols.
To determine whether genetic mutations could be used to predict responses to decitabine, the researchers performed enhanced exome or gene-panel sequencing in 67 of the patients. The team also performed sequencing at multiple time points to evaluate patterns of mutation clearance in 54 patients.
Response
Thirteen percent of patients (n=15) achieved a complete response (CR), 21% (n=24) had a CR with incomplete count recovery, 5% (n=6) had a morphologic CR with hematologic improvement, and 7% (n=8) had a morphologic CR without hematologic improvement.
Eight percent of patients (n=9) had a partial response, 20% (n=23) had stable disease, and 16% (n=19) had progressive disease.
There were 21 patients with TP53 mutations, and all of them achieved bone marrow blast clearance with less than 5% blasts.
Nineteen percent (n=4) had a CR, 43% (n=9) had a CR with incomplete count recovery, 24% (n=5) had morphologic CR with hematologic improvement, and 14% (n=3) had morphologic CR without hematologic improvement.
“What’s really unique here is that all the patients in the study with TP53 mutations had a response to decitabine and achieved an initial remission,” Dr Ley said.
“With standard aggressive chemotherapy, we only see about 20% to 30% of these patients achieving remission, which is the critical first step to have a chance to cure patients with additional therapies.”
Dr Ley and his colleagues also found that patients in this study were likely to respond to decitabine if they were considered “unfavorable risk” based on extensive chromosomal rearrangements. (Many of these patients also had TP53 mutations.)
Indeed, 67% (29/43) of patients with an unfavorable risk had less than 5% blasts after treatment with decitabine, compared with 34% (24/71) of patients with intermediate or favorable risk.
“The challenge with using decitabine has been knowing which patients are most likely to respond,” said study author Amanda Cashen, MD, of Washington University School of Medicine.
“The value of this study is the comprehensive mutational analysis that helps us figure out which patients are likely to benefit. This information opens the door to using decitabine in a more targeted fashion to treat not just older patients, but also younger patients who carry TP53 mutations.”
Survival and next steps
The researchers found that responses to decitabine were usually short-lived. The drug did not provide complete mutation clearance, which led to relapse.
“Remissions with decitabine typically don’t last long, and no one was cured with this drug,” Dr Ley noted. “But patients who responded to decitabine live longer than what you would expect with aggressive chemotherapy, and that can mean something. Some people live a year or 2 and with a good quality of life because the chemotherapy is not too toxic.”
The median overall survival was 11.6 months among patients with unfavorable risk and 10 months among patients with favorable or intermediate risk (P=0.29).
The median overall survival was 12.7 months among patients with TP53 mutations and 15.4 months among patients with wild-type TP53 (P=0.79).
“It’s important to note that patients with an extremely poor prognosis in this relatively small study had the same survival outcomes as patients facing a better prognosis, which is encouraging,” said study author John Welch, MD, PhD, of Washington University School of Medicine.
“We don’t yet understand why patients with TP53 mutations consistently respond to decitabine, and more work is needed to understand that phenomenon. We’re now planning a larger trial to evaluate decitabine in AML patients of all ages who carry TP53 mutations. It’s exciting to think we may have a therapy that has the potential to improve response rates in this group of high-risk patients.” ![]()
receiving chemotherapy
Photo by Rhoda Baer
Patients with TP53-mutated myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML) may benefit from treatment with decitabine, according to a study published in NEJM.
All patients in this study who had TP53 mutations responded to decitabine.
Although these responses were not durable, the patients’ median overall survival was similar to that of patients with lower-risk disease who received decitabine.
“The findings need to be validated in a larger trial, but they do suggest that TP53 mutations can reliably predict responses to decitabine, potentially prolonging survival in this ultra-high-risk group of patients and providing a bridge to transplantation in some patients who might not otherwise be candidates,” said study author Timothy J. Ley, MD, of Washington University School of Medicine in St. Louis, Missouri.
For this study, Dr Ley and his colleagues analyzed 116 patients—54 with AML, 36 with relapsed AML, and 26 with MDS.
Eighty-four of the patients were enrolled in a prospective trial and received decitabine at a dose of 20 mg/m2/day for 10 consecutive days in monthly cycles. Thirty-two additional patients received decitabine on different protocols.
To determine whether genetic mutations could be used to predict responses to decitabine, the researchers performed enhanced exome or gene-panel sequencing in 67 of the patients. The team also performed sequencing at multiple time points to evaluate patterns of mutation clearance in 54 patients.
Response
Thirteen percent of patients (n=15) achieved a complete response (CR), 21% (n=24) had a CR with incomplete count recovery, 5% (n=6) had a morphologic CR with hematologic improvement, and 7% (n=8) had a morphologic CR without hematologic improvement.
Eight percent of patients (n=9) had a partial response, 20% (n=23) had stable disease, and 16% (n=19) had progressive disease.
There were 21 patients with TP53 mutations, and all of them achieved bone marrow blast clearance with less than 5% blasts.
Nineteen percent (n=4) had a CR, 43% (n=9) had a CR with incomplete count recovery, 24% (n=5) had morphologic CR with hematologic improvement, and 14% (n=3) had morphologic CR without hematologic improvement.
“What’s really unique here is that all the patients in the study with TP53 mutations had a response to decitabine and achieved an initial remission,” Dr Ley said.
“With standard aggressive chemotherapy, we only see about 20% to 30% of these patients achieving remission, which is the critical first step to have a chance to cure patients with additional therapies.”
Dr Ley and his colleagues also found that patients in this study were likely to respond to decitabine if they were considered “unfavorable risk” based on extensive chromosomal rearrangements. (Many of these patients also had TP53 mutations.)
Indeed, 67% (29/43) of patients with an unfavorable risk had less than 5% blasts after treatment with decitabine, compared with 34% (24/71) of patients with intermediate or favorable risk.
“The challenge with using decitabine has been knowing which patients are most likely to respond,” said study author Amanda Cashen, MD, of Washington University School of Medicine.
“The value of this study is the comprehensive mutational analysis that helps us figure out which patients are likely to benefit. This information opens the door to using decitabine in a more targeted fashion to treat not just older patients, but also younger patients who carry TP53 mutations.”
Survival and next steps
The researchers found that responses to decitabine were usually short-lived. The drug did not provide complete mutation clearance, which led to relapse.
“Remissions with decitabine typically don’t last long, and no one was cured with this drug,” Dr Ley noted. “But patients who responded to decitabine live longer than what you would expect with aggressive chemotherapy, and that can mean something. Some people live a year or 2 and with a good quality of life because the chemotherapy is not too toxic.”
The median overall survival was 11.6 months among patients with unfavorable risk and 10 months among patients with favorable or intermediate risk (P=0.29).
The median overall survival was 12.7 months among patients with TP53 mutations and 15.4 months among patients with wild-type TP53 (P=0.79).
“It’s important to note that patients with an extremely poor prognosis in this relatively small study had the same survival outcomes as patients facing a better prognosis, which is encouraging,” said study author John Welch, MD, PhD, of Washington University School of Medicine.
“We don’t yet understand why patients with TP53 mutations consistently respond to decitabine, and more work is needed to understand that phenomenon. We’re now planning a larger trial to evaluate decitabine in AML patients of all ages who carry TP53 mutations. It’s exciting to think we may have a therapy that has the potential to improve response rates in this group of high-risk patients.” ![]()
receiving chemotherapy
Photo by Rhoda Baer
Patients with TP53-mutated myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML) may benefit from treatment with decitabine, according to a study published in NEJM.
All patients in this study who had TP53 mutations responded to decitabine.
Although these responses were not durable, the patients’ median overall survival was similar to that of patients with lower-risk disease who received decitabine.
“The findings need to be validated in a larger trial, but they do suggest that TP53 mutations can reliably predict responses to decitabine, potentially prolonging survival in this ultra-high-risk group of patients and providing a bridge to transplantation in some patients who might not otherwise be candidates,” said study author Timothy J. Ley, MD, of Washington University School of Medicine in St. Louis, Missouri.
For this study, Dr Ley and his colleagues analyzed 116 patients—54 with AML, 36 with relapsed AML, and 26 with MDS.
Eighty-four of the patients were enrolled in a prospective trial and received decitabine at a dose of 20 mg/m2/day for 10 consecutive days in monthly cycles. Thirty-two additional patients received decitabine on different protocols.
To determine whether genetic mutations could be used to predict responses to decitabine, the researchers performed enhanced exome or gene-panel sequencing in 67 of the patients. The team also performed sequencing at multiple time points to evaluate patterns of mutation clearance in 54 patients.
Response
Thirteen percent of patients (n=15) achieved a complete response (CR), 21% (n=24) had a CR with incomplete count recovery, 5% (n=6) had a morphologic CR with hematologic improvement, and 7% (n=8) had a morphologic CR without hematologic improvement.
Eight percent of patients (n=9) had a partial response, 20% (n=23) had stable disease, and 16% (n=19) had progressive disease.
There were 21 patients with TP53 mutations, and all of them achieved bone marrow blast clearance with less than 5% blasts.
Nineteen percent (n=4) had a CR, 43% (n=9) had a CR with incomplete count recovery, 24% (n=5) had morphologic CR with hematologic improvement, and 14% (n=3) had morphologic CR without hematologic improvement.
“What’s really unique here is that all the patients in the study with TP53 mutations had a response to decitabine and achieved an initial remission,” Dr Ley said.
“With standard aggressive chemotherapy, we only see about 20% to 30% of these patients achieving remission, which is the critical first step to have a chance to cure patients with additional therapies.”
Dr Ley and his colleagues also found that patients in this study were likely to respond to decitabine if they were considered “unfavorable risk” based on extensive chromosomal rearrangements. (Many of these patients also had TP53 mutations.)
Indeed, 67% (29/43) of patients with an unfavorable risk had less than 5% blasts after treatment with decitabine, compared with 34% (24/71) of patients with intermediate or favorable risk.
“The challenge with using decitabine has been knowing which patients are most likely to respond,” said study author Amanda Cashen, MD, of Washington University School of Medicine.
“The value of this study is the comprehensive mutational analysis that helps us figure out which patients are likely to benefit. This information opens the door to using decitabine in a more targeted fashion to treat not just older patients, but also younger patients who carry TP53 mutations.”
Survival and next steps
The researchers found that responses to decitabine were usually short-lived. The drug did not provide complete mutation clearance, which led to relapse.
“Remissions with decitabine typically don’t last long, and no one was cured with this drug,” Dr Ley noted. “But patients who responded to decitabine live longer than what you would expect with aggressive chemotherapy, and that can mean something. Some people live a year or 2 and with a good quality of life because the chemotherapy is not too toxic.”
The median overall survival was 11.6 months among patients with unfavorable risk and 10 months among patients with favorable or intermediate risk (P=0.29).
The median overall survival was 12.7 months among patients with TP53 mutations and 15.4 months among patients with wild-type TP53 (P=0.79).
“It’s important to note that patients with an extremely poor prognosis in this relatively small study had the same survival outcomes as patients facing a better prognosis, which is encouraging,” said study author John Welch, MD, PhD, of Washington University School of Medicine.
“We don’t yet understand why patients with TP53 mutations consistently respond to decitabine, and more work is needed to understand that phenomenon. We’re now planning a larger trial to evaluate decitabine in AML patients of all ages who carry TP53 mutations. It’s exciting to think we may have a therapy that has the potential to improve response rates in this group of high-risk patients.” ![]()
EC grants drug orphan status for AML, sarcoma
The European Commission (EC) has granted orphan drug designation to crenolanib for the treatment of acute myeloid leukemia (AML) and soft tissue sarcoma.
Crenolanib is a benzimidazole type I kinase inhibitor that selectively inhibits signaling of wild-type and mutant isoforms of FLT3 and PDGFRα/β.
The drug is under investigation as a treatment for multiple cancers. It is being developed by Arog Pharmaceuticals, Inc.
Results from a phase 2 trial of crenolanib in relapsed/refractory, FLT3+ AML were presented at the 2016 ASCO Annual Meeting.
About orphan designation
The EC grants orphan designation to therapies intended to treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides companies developing such drugs with regulatory and financial incentives, including protocol assistance, 10 years of market exclusivity once the drug is approved, and, in some cases, reductions in fees. ![]()
The European Commission (EC) has granted orphan drug designation to crenolanib for the treatment of acute myeloid leukemia (AML) and soft tissue sarcoma.
Crenolanib is a benzimidazole type I kinase inhibitor that selectively inhibits signaling of wild-type and mutant isoforms of FLT3 and PDGFRα/β.
The drug is under investigation as a treatment for multiple cancers. It is being developed by Arog Pharmaceuticals, Inc.
Results from a phase 2 trial of crenolanib in relapsed/refractory, FLT3+ AML were presented at the 2016 ASCO Annual Meeting.
About orphan designation
The EC grants orphan designation to therapies intended to treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides companies developing such drugs with regulatory and financial incentives, including protocol assistance, 10 years of market exclusivity once the drug is approved, and, in some cases, reductions in fees. ![]()
The European Commission (EC) has granted orphan drug designation to crenolanib for the treatment of acute myeloid leukemia (AML) and soft tissue sarcoma.
Crenolanib is a benzimidazole type I kinase inhibitor that selectively inhibits signaling of wild-type and mutant isoforms of FLT3 and PDGFRα/β.
The drug is under investigation as a treatment for multiple cancers. It is being developed by Arog Pharmaceuticals, Inc.
Results from a phase 2 trial of crenolanib in relapsed/refractory, FLT3+ AML were presented at the 2016 ASCO Annual Meeting.
About orphan designation
The EC grants orphan designation to therapies intended to treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides companies developing such drugs with regulatory and financial incentives, including protocol assistance, 10 years of market exclusivity once the drug is approved, and, in some cases, reductions in fees. ![]()
Decitabine elicits favorable response in high-risk AML/MDS
Single-agent therapy with decitabine elicited favorable responses in patients with acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) who had cytogenetic abnormalities associated with an unfavorable risk profile, a study showed.
Even though the responses were not long lasting, the overall survival rates were similar to those of AML patients who had an intermediate-risk cytogenetic profile and who had received the same treatment regimen.
Adult AML patients with karyotypes associated with unfavorable risk and older patients with AML (aged 60 or older) generally have poor outcomes, with a median survival in the range of 1 year. Those with AML and TP53 mutations tend to be older (median age, 61-67 years), and nearly all patients have karyotypes associated with unfavorable risk. These patients have particularly dismal outcomes, with median survival in the range of 4-6 months, if they receive cytotoxic chemotherapy.
In their study, John S Welch, MD, of Washington University, St Louis, and his colleagues evaluated somatic mutations and their relationships to clinical responses in 84 adult patients with AML or MDS who received treatment with decitabine as monotherapy.
Decitabine was administered at a dose of 20 mg/m2 of body surface area per day for 10 consecutive days in monthly cycles. An extension cohort that included 32 additional patients also were treated with decitabine but in different protocols.
Of the entire cohort of 116 patients, 15 patients (13%) achieved a complete remission, and 38 patients had bone marrow blast clearance with less than 5% blasts (complete remission with incomplete count recovery or morphologic complete remission). The overall response rate was 46%. In addition, 9 patients (8%), achieved a partial response, stable disease was observed in 23 patients (20%), and progressive disease was seen in 19 patients (16%).
Clinical responses were strongly correlated with karyotypes associated with unfavorable risk and the presence of TP53 mutations. Bone marrow blast clearance (complete remission, complete remission with incomplete count recovery, or morphologic complete remission) occurred in 29 of 43 patients with karyotypes associated with unfavorable risk (67%), compared with 24 of 71 patients with karyotypes associated with intermediate or favorable risk (34%). The same pattern was observed in all patients with TP53 mutations (100%) versus 32 of 78 patients with wildtype TP53 mutations (41%).
“Additional studies will be required to determine whether these differences in survival are truly due to improved responses associated with decitabine or whether conventional chemotherapy with an anthracycline and cytarabine actually decreases the rate of survival among patients with unfavorable-risk cytogenetic profiles,” wrote Dr. Welch and his colleagues.
The study was supported by the Specialized Program of Research Excellence in AML of the National Cancer Institute and the Genomics of AML Program Project. Dr. Welch had no disclosures, and several of his coauthors reported relationships with industry.
The current study raises the important question of the definition of a response. Complete remission conventionally entails bone marrow with less than 5% blasts and normalization of blood counts; absent these, remission is considered incomplete. After various confounding factors are taken into account, a complete response with cytotoxic therapy is associated with longer remissions and longer survival than is complete remission with incomplete count recovery.
The authors of this paper considered a response to be blast clearance to less than 5%, but complete remission was seen in only 4 of the 21 patients with TP53 mutations who fulfilled this criterion. The mutant allele burden was also similar in patients who had a response, regardless of blood count recovery.
In contrast, measurable residual disease is considerably more frequent in patients with complete remission with incomplete count recovery than in patients with complete remission who have received cytotoxic therapy, indicating that more data are needed on subsequent clinical outcomes according to whether clearance of blasts is accompanied by count recovery in patients with AML and TP53 mutations who have received decitabine.
AML “targeted-therapy” trials typically involve one drug, and this policy is called into question by the diverse molecular architecture (and brief remissions) observed in this trial. The trial by Welch et al. points to inevitable, rational replacement of large trials in which homogeneous therapy is administered for a heterogeneous disease by smaller, subgroup-specific trials.
The article also suggests questions that are likely to complicate this future.
Dr. Elihu Estey is with the division of hematology, University of Washington Medical Center, and the clinical research division, Fred Hutchinson Cancer Research Center, Seattle. He had no disclosures. These remarks were taken from an editorial accompanying Dr. Welch’s paper (N Engl J Med. 2016;375:2023-36).
The current study raises the important question of the definition of a response. Complete remission conventionally entails bone marrow with less than 5% blasts and normalization of blood counts; absent these, remission is considered incomplete. After various confounding factors are taken into account, a complete response with cytotoxic therapy is associated with longer remissions and longer survival than is complete remission with incomplete count recovery.
The authors of this paper considered a response to be blast clearance to less than 5%, but complete remission was seen in only 4 of the 21 patients with TP53 mutations who fulfilled this criterion. The mutant allele burden was also similar in patients who had a response, regardless of blood count recovery.
In contrast, measurable residual disease is considerably more frequent in patients with complete remission with incomplete count recovery than in patients with complete remission who have received cytotoxic therapy, indicating that more data are needed on subsequent clinical outcomes according to whether clearance of blasts is accompanied by count recovery in patients with AML and TP53 mutations who have received decitabine.
AML “targeted-therapy” trials typically involve one drug, and this policy is called into question by the diverse molecular architecture (and brief remissions) observed in this trial. The trial by Welch et al. points to inevitable, rational replacement of large trials in which homogeneous therapy is administered for a heterogeneous disease by smaller, subgroup-specific trials.
The article also suggests questions that are likely to complicate this future.
Dr. Elihu Estey is with the division of hematology, University of Washington Medical Center, and the clinical research division, Fred Hutchinson Cancer Research Center, Seattle. He had no disclosures. These remarks were taken from an editorial accompanying Dr. Welch’s paper (N Engl J Med. 2016;375:2023-36).
The current study raises the important question of the definition of a response. Complete remission conventionally entails bone marrow with less than 5% blasts and normalization of blood counts; absent these, remission is considered incomplete. After various confounding factors are taken into account, a complete response with cytotoxic therapy is associated with longer remissions and longer survival than is complete remission with incomplete count recovery.
The authors of this paper considered a response to be blast clearance to less than 5%, but complete remission was seen in only 4 of the 21 patients with TP53 mutations who fulfilled this criterion. The mutant allele burden was also similar in patients who had a response, regardless of blood count recovery.
In contrast, measurable residual disease is considerably more frequent in patients with complete remission with incomplete count recovery than in patients with complete remission who have received cytotoxic therapy, indicating that more data are needed on subsequent clinical outcomes according to whether clearance of blasts is accompanied by count recovery in patients with AML and TP53 mutations who have received decitabine.
AML “targeted-therapy” trials typically involve one drug, and this policy is called into question by the diverse molecular architecture (and brief remissions) observed in this trial. The trial by Welch et al. points to inevitable, rational replacement of large trials in which homogeneous therapy is administered for a heterogeneous disease by smaller, subgroup-specific trials.
The article also suggests questions that are likely to complicate this future.
Dr. Elihu Estey is with the division of hematology, University of Washington Medical Center, and the clinical research division, Fred Hutchinson Cancer Research Center, Seattle. He had no disclosures. These remarks were taken from an editorial accompanying Dr. Welch’s paper (N Engl J Med. 2016;375:2023-36).
Single-agent therapy with decitabine elicited favorable responses in patients with acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) who had cytogenetic abnormalities associated with an unfavorable risk profile, a study showed.
Even though the responses were not long lasting, the overall survival rates were similar to those of AML patients who had an intermediate-risk cytogenetic profile and who had received the same treatment regimen.
Adult AML patients with karyotypes associated with unfavorable risk and older patients with AML (aged 60 or older) generally have poor outcomes, with a median survival in the range of 1 year. Those with AML and TP53 mutations tend to be older (median age, 61-67 years), and nearly all patients have karyotypes associated with unfavorable risk. These patients have particularly dismal outcomes, with median survival in the range of 4-6 months, if they receive cytotoxic chemotherapy.
In their study, John S Welch, MD, of Washington University, St Louis, and his colleagues evaluated somatic mutations and their relationships to clinical responses in 84 adult patients with AML or MDS who received treatment with decitabine as monotherapy.
Decitabine was administered at a dose of 20 mg/m2 of body surface area per day for 10 consecutive days in monthly cycles. An extension cohort that included 32 additional patients also were treated with decitabine but in different protocols.
Of the entire cohort of 116 patients, 15 patients (13%) achieved a complete remission, and 38 patients had bone marrow blast clearance with less than 5% blasts (complete remission with incomplete count recovery or morphologic complete remission). The overall response rate was 46%. In addition, 9 patients (8%), achieved a partial response, stable disease was observed in 23 patients (20%), and progressive disease was seen in 19 patients (16%).
Clinical responses were strongly correlated with karyotypes associated with unfavorable risk and the presence of TP53 mutations. Bone marrow blast clearance (complete remission, complete remission with incomplete count recovery, or morphologic complete remission) occurred in 29 of 43 patients with karyotypes associated with unfavorable risk (67%), compared with 24 of 71 patients with karyotypes associated with intermediate or favorable risk (34%). The same pattern was observed in all patients with TP53 mutations (100%) versus 32 of 78 patients with wildtype TP53 mutations (41%).
“Additional studies will be required to determine whether these differences in survival are truly due to improved responses associated with decitabine or whether conventional chemotherapy with an anthracycline and cytarabine actually decreases the rate of survival among patients with unfavorable-risk cytogenetic profiles,” wrote Dr. Welch and his colleagues.
The study was supported by the Specialized Program of Research Excellence in AML of the National Cancer Institute and the Genomics of AML Program Project. Dr. Welch had no disclosures, and several of his coauthors reported relationships with industry.
Single-agent therapy with decitabine elicited favorable responses in patients with acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) who had cytogenetic abnormalities associated with an unfavorable risk profile, a study showed.
Even though the responses were not long lasting, the overall survival rates were similar to those of AML patients who had an intermediate-risk cytogenetic profile and who had received the same treatment regimen.
Adult AML patients with karyotypes associated with unfavorable risk and older patients with AML (aged 60 or older) generally have poor outcomes, with a median survival in the range of 1 year. Those with AML and TP53 mutations tend to be older (median age, 61-67 years), and nearly all patients have karyotypes associated with unfavorable risk. These patients have particularly dismal outcomes, with median survival in the range of 4-6 months, if they receive cytotoxic chemotherapy.
In their study, John S Welch, MD, of Washington University, St Louis, and his colleagues evaluated somatic mutations and their relationships to clinical responses in 84 adult patients with AML or MDS who received treatment with decitabine as monotherapy.
Decitabine was administered at a dose of 20 mg/m2 of body surface area per day for 10 consecutive days in monthly cycles. An extension cohort that included 32 additional patients also were treated with decitabine but in different protocols.
Of the entire cohort of 116 patients, 15 patients (13%) achieved a complete remission, and 38 patients had bone marrow blast clearance with less than 5% blasts (complete remission with incomplete count recovery or morphologic complete remission). The overall response rate was 46%. In addition, 9 patients (8%), achieved a partial response, stable disease was observed in 23 patients (20%), and progressive disease was seen in 19 patients (16%).
Clinical responses were strongly correlated with karyotypes associated with unfavorable risk and the presence of TP53 mutations. Bone marrow blast clearance (complete remission, complete remission with incomplete count recovery, or morphologic complete remission) occurred in 29 of 43 patients with karyotypes associated with unfavorable risk (67%), compared with 24 of 71 patients with karyotypes associated with intermediate or favorable risk (34%). The same pattern was observed in all patients with TP53 mutations (100%) versus 32 of 78 patients with wildtype TP53 mutations (41%).
“Additional studies will be required to determine whether these differences in survival are truly due to improved responses associated with decitabine or whether conventional chemotherapy with an anthracycline and cytarabine actually decreases the rate of survival among patients with unfavorable-risk cytogenetic profiles,” wrote Dr. Welch and his colleagues.
The study was supported by the Specialized Program of Research Excellence in AML of the National Cancer Institute and the Genomics of AML Program Project. Dr. Welch had no disclosures, and several of his coauthors reported relationships with industry.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: Of 116 patients, 53 (46%) experienced bone marrow blast clearance (less than 5% blasts), and response rates were higher among those with an unfavorable cytogenetic risk profile.
Data source: A prospective single-center clinical trial that evaluated single-agent decitabine in 84 adult patients with AML or MDS, with an extension cohort.
Disclosures: The study was supported by the Specialized Program of Research Excellence in AML of the National Cancer Institute and the Genomics of AML Program Project. Dr. Welch had no disclosures, and several of his coauthors reported relationships with industry.
The complex genetic landscape of AML
A unifying genetic basis has been sought to explain the complex and heterogeneous nature of myeloid neoplasms since before Janet Rowley’s quinacrine banding discovered the Philadelphia chromosome (Nature. 1973;243[5405]:290-3). In the decades following that discovery, groundbreaking work has uncovered new chromosomal abnormalities, new gene fusions, new recurrent mutations – often with prognostic implications, but rarely with therapeutic ones.
The recent work by Elli Papaemmanuil, PhD, of Memorial Sloan Kettering Cancer Center, New York, and her colleagues reaffirms the genetic heterogeneity of AML based on molecular profiling of patients from three large European trials. Yet the most insightful aspect of this reclassification is not just the detail of the genetic resolution but the realization that, even within a gene such as NRAS, the genetic background for acquisition of a codon 12/13 mutation is mutually exclusive with clones where NRAS codon 61 occurs.
When speaking with relapsed patients, I often say that, while we are very good at cutting down trees in AML, we still have not done very well with getting rid of the roots. Admittedly, this metaphor grossly oversimplifies cancer stem cell biology, but it gets at the real importance of the work by Dr. Papaemmanuil and her colleagues. The interactions of gene mutations such as NPM1 and DNMT3A are not uncommon and their co-mutation in isolation has an intermediate prognosis. The clonal acquisition of a codon 12/13 mutation in NRAS seems to result in a more favorable prognosis – lending to the likelihood that the tumor is simply more chemosensitive. In contrast, the acquisition of FLT3-ITD by the NPM1/DNMT3A co-mutant clone results in a very poor prognosis likely due to chemoresistance.
The real power of this study’s findings is the potential for building a toolbox of agents to push against the innate clonal selection and force the “tree” to grow in a direction that is detrimental to its survival. One could consider using FLT3 inhibitors in the wild-type setting of a genetic background primed towards FLT3-ITD evolution to prevent this resistant outgrowth. Of course, such an approach needs to be studied first in a laboratory setting, but similar therapeutic strategies have been applied to BRAF in melanoma. Peter Nowell urged “controlling the evolutionary process in tumors before it reaches the late stage,” and this new ordinal understanding of AML may help to do just that.
vinya@mskcc.org
On Twitter @thedoctorisvin
A unifying genetic basis has been sought to explain the complex and heterogeneous nature of myeloid neoplasms since before Janet Rowley’s quinacrine banding discovered the Philadelphia chromosome (Nature. 1973;243[5405]:290-3). In the decades following that discovery, groundbreaking work has uncovered new chromosomal abnormalities, new gene fusions, new recurrent mutations – often with prognostic implications, but rarely with therapeutic ones.
The recent work by Elli Papaemmanuil, PhD, of Memorial Sloan Kettering Cancer Center, New York, and her colleagues reaffirms the genetic heterogeneity of AML based on molecular profiling of patients from three large European trials. Yet the most insightful aspect of this reclassification is not just the detail of the genetic resolution but the realization that, even within a gene such as NRAS, the genetic background for acquisition of a codon 12/13 mutation is mutually exclusive with clones where NRAS codon 61 occurs.
When speaking with relapsed patients, I often say that, while we are very good at cutting down trees in AML, we still have not done very well with getting rid of the roots. Admittedly, this metaphor grossly oversimplifies cancer stem cell biology, but it gets at the real importance of the work by Dr. Papaemmanuil and her colleagues. The interactions of gene mutations such as NPM1 and DNMT3A are not uncommon and their co-mutation in isolation has an intermediate prognosis. The clonal acquisition of a codon 12/13 mutation in NRAS seems to result in a more favorable prognosis – lending to the likelihood that the tumor is simply more chemosensitive. In contrast, the acquisition of FLT3-ITD by the NPM1/DNMT3A co-mutant clone results in a very poor prognosis likely due to chemoresistance.
The real power of this study’s findings is the potential for building a toolbox of agents to push against the innate clonal selection and force the “tree” to grow in a direction that is detrimental to its survival. One could consider using FLT3 inhibitors in the wild-type setting of a genetic background primed towards FLT3-ITD evolution to prevent this resistant outgrowth. Of course, such an approach needs to be studied first in a laboratory setting, but similar therapeutic strategies have been applied to BRAF in melanoma. Peter Nowell urged “controlling the evolutionary process in tumors before it reaches the late stage,” and this new ordinal understanding of AML may help to do just that.
vinya@mskcc.org
On Twitter @thedoctorisvin
A unifying genetic basis has been sought to explain the complex and heterogeneous nature of myeloid neoplasms since before Janet Rowley’s quinacrine banding discovered the Philadelphia chromosome (Nature. 1973;243[5405]:290-3). In the decades following that discovery, groundbreaking work has uncovered new chromosomal abnormalities, new gene fusions, new recurrent mutations – often with prognostic implications, but rarely with therapeutic ones.
The recent work by Elli Papaemmanuil, PhD, of Memorial Sloan Kettering Cancer Center, New York, and her colleagues reaffirms the genetic heterogeneity of AML based on molecular profiling of patients from three large European trials. Yet the most insightful aspect of this reclassification is not just the detail of the genetic resolution but the realization that, even within a gene such as NRAS, the genetic background for acquisition of a codon 12/13 mutation is mutually exclusive with clones where NRAS codon 61 occurs.
When speaking with relapsed patients, I often say that, while we are very good at cutting down trees in AML, we still have not done very well with getting rid of the roots. Admittedly, this metaphor grossly oversimplifies cancer stem cell biology, but it gets at the real importance of the work by Dr. Papaemmanuil and her colleagues. The interactions of gene mutations such as NPM1 and DNMT3A are not uncommon and their co-mutation in isolation has an intermediate prognosis. The clonal acquisition of a codon 12/13 mutation in NRAS seems to result in a more favorable prognosis – lending to the likelihood that the tumor is simply more chemosensitive. In contrast, the acquisition of FLT3-ITD by the NPM1/DNMT3A co-mutant clone results in a very poor prognosis likely due to chemoresistance.
The real power of this study’s findings is the potential for building a toolbox of agents to push against the innate clonal selection and force the “tree” to grow in a direction that is detrimental to its survival. One could consider using FLT3 inhibitors in the wild-type setting of a genetic background primed towards FLT3-ITD evolution to prevent this resistant outgrowth. Of course, such an approach needs to be studied first in a laboratory setting, but similar therapeutic strategies have been applied to BRAF in melanoma. Peter Nowell urged “controlling the evolutionary process in tumors before it reaches the late stage,” and this new ordinal understanding of AML may help to do just that.
vinya@mskcc.org
On Twitter @thedoctorisvin
FDA grants priority review for midostaurin
The US Food and Drug Administration (FDA) has granted priority review for the new drug application for midostaurin (PKC412) as a treatment for advanced systemic mastocytosis (SM) and newly diagnosed, FLT3-mutated acute myeloid leukemia (AML).
The FDA has also accepted for review the premarket approval application for the midostaurin FLT3 companion diagnostic, which is designed to help identify patients who may have a FLT3 mutation and could potentially benefit from treatment with midostaurin.
Midostaurin is being developed by Novartis. The companion diagnostic is being developed by Novartis and Invivoscribe Technologies, Inc.
About priority review
The FDA grants priority review to applications for therapies that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it. The goal in the standard review process is to take action within 10 months.
About midostaurin
Midostaurin is an oral, multi-targeted kinase inhibitor. The drug was granted breakthrough therapy designation by the FDA earlier this year for newly diagnosed, FLT3-mutated AML.
According to Novartis, the new drug application submission for midostaurin includes data from the largest clinical trials conducted to date in advanced SM and newly diagnosed, FLT3-mutated AML.
Midostaurin in AML
In the phase 3 RATIFY trial, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in adults younger than 60 with FLT3-mutated AML. Results from this trial were presented at the 2015 ASH Annual Meeting.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.0074).
There was no significant difference in the overall rate of grade 3 or higher hematologic and non-hematologic adverse events in midostaurin arm and the placebo arm. Similarly, there was no significant difference in treatment-related deaths between the arms.
Midostaurin in SM
Data from the phase 2 study of midostaurin in patients with advanced SM were published in NEJM in June.
The drug produced a 60% overall response rate, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose of midostaurin.
Access to midostaurin
Since midostaurin remains investigational, both within the US and globally, Novartis opened a Global Individual Patient Program (compassionate use program) and, in the US, an Expanded Treatment Protocol, to provide access to midostaurin for eligible patients with newly diagnosed AML and advanced SM.
Physicians who want to request midostaurin for eligible patients can contact a Novartis medical representative in their respective countries. In the US, physicians can call 1-888-NOW-NOVA (1-888-669-6682) for more information. ![]()
The US Food and Drug Administration (FDA) has granted priority review for the new drug application for midostaurin (PKC412) as a treatment for advanced systemic mastocytosis (SM) and newly diagnosed, FLT3-mutated acute myeloid leukemia (AML).
The FDA has also accepted for review the premarket approval application for the midostaurin FLT3 companion diagnostic, which is designed to help identify patients who may have a FLT3 mutation and could potentially benefit from treatment with midostaurin.
Midostaurin is being developed by Novartis. The companion diagnostic is being developed by Novartis and Invivoscribe Technologies, Inc.
About priority review
The FDA grants priority review to applications for therapies that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it. The goal in the standard review process is to take action within 10 months.
About midostaurin
Midostaurin is an oral, multi-targeted kinase inhibitor. The drug was granted breakthrough therapy designation by the FDA earlier this year for newly diagnosed, FLT3-mutated AML.
According to Novartis, the new drug application submission for midostaurin includes data from the largest clinical trials conducted to date in advanced SM and newly diagnosed, FLT3-mutated AML.
Midostaurin in AML
In the phase 3 RATIFY trial, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in adults younger than 60 with FLT3-mutated AML. Results from this trial were presented at the 2015 ASH Annual Meeting.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.0074).
There was no significant difference in the overall rate of grade 3 or higher hematologic and non-hematologic adverse events in midostaurin arm and the placebo arm. Similarly, there was no significant difference in treatment-related deaths between the arms.
Midostaurin in SM
Data from the phase 2 study of midostaurin in patients with advanced SM were published in NEJM in June.
The drug produced a 60% overall response rate, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose of midostaurin.
Access to midostaurin
Since midostaurin remains investigational, both within the US and globally, Novartis opened a Global Individual Patient Program (compassionate use program) and, in the US, an Expanded Treatment Protocol, to provide access to midostaurin for eligible patients with newly diagnosed AML and advanced SM.
Physicians who want to request midostaurin for eligible patients can contact a Novartis medical representative in their respective countries. In the US, physicians can call 1-888-NOW-NOVA (1-888-669-6682) for more information. ![]()
The US Food and Drug Administration (FDA) has granted priority review for the new drug application for midostaurin (PKC412) as a treatment for advanced systemic mastocytosis (SM) and newly diagnosed, FLT3-mutated acute myeloid leukemia (AML).
The FDA has also accepted for review the premarket approval application for the midostaurin FLT3 companion diagnostic, which is designed to help identify patients who may have a FLT3 mutation and could potentially benefit from treatment with midostaurin.
Midostaurin is being developed by Novartis. The companion diagnostic is being developed by Novartis and Invivoscribe Technologies, Inc.
About priority review
The FDA grants priority review to applications for therapies that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it. The goal in the standard review process is to take action within 10 months.
About midostaurin
Midostaurin is an oral, multi-targeted kinase inhibitor. The drug was granted breakthrough therapy designation by the FDA earlier this year for newly diagnosed, FLT3-mutated AML.
According to Novartis, the new drug application submission for midostaurin includes data from the largest clinical trials conducted to date in advanced SM and newly diagnosed, FLT3-mutated AML.
Midostaurin in AML
In the phase 3 RATIFY trial, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in adults younger than 60 with FLT3-mutated AML. Results from this trial were presented at the 2015 ASH Annual Meeting.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.0074).
There was no significant difference in the overall rate of grade 3 or higher hematologic and non-hematologic adverse events in midostaurin arm and the placebo arm. Similarly, there was no significant difference in treatment-related deaths between the arms.
Midostaurin in SM
Data from the phase 2 study of midostaurin in patients with advanced SM were published in NEJM in June.
The drug produced a 60% overall response rate, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose of midostaurin.
Access to midostaurin
Since midostaurin remains investigational, both within the US and globally, Novartis opened a Global Individual Patient Program (compassionate use program) and, in the US, an Expanded Treatment Protocol, to provide access to midostaurin for eligible patients with newly diagnosed AML and advanced SM.
Physicians who want to request midostaurin for eligible patients can contact a Novartis medical representative in their respective countries. In the US, physicians can call 1-888-NOW-NOVA (1-888-669-6682) for more information. ![]()
EMA recommends orphan status for drug in AML

The European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products (COMP) has recommended that BP1001 receive orphan designation as a treatment for acute myeloid leukemia (AML).
BP1001 (liposomal Grb2 antisense) is a neutral-charge, liposome-incorporated, antisense drug designed to inhibit protein synthesis of growth factor receptor bound protein 2 (Grb2).
BP1001 is being developed by Bio-Path Holdings, Inc.
According to Bio-Path, inhibition of Grb2 by BP1001 represents a significant advance in treating cancers with activated tyrosine kinases using a target not druggable with small molecule inhibitors.
Research has suggested that Grb2 plays an essential role in cancer cell activation via the RAS pathway. Grb2 bridges signals between activated and mutated tyrosine kinases, such as Flt3, c-Kit, and Bcr-Abl, and the Ras pathway, leading to activation of the ERK and AKT proteins.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The European Commission typically makes a decision within 30 days.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
Trials of BP1001
Bio-Path has completed a phase 1 trial of BP1001 in patients with relapsed/refractory AML, chronic myeloid leukemia, and myelodysplastic syndromes.
The company has also completed the safety segment of a phase 2 trial in which BP1001 is being investigated in combination with low-dose ara-C to treat AML.
Bio-Path recently released data from these studies.
The phase 1 study included patients who had received an average of 6 prior therapies.
The patients received 8 doses of BP1001 over 4 weeks, escalating to a maximum dose of 90 mg/m2. There were no dose-limiting toxicities, and Bio-Path said the drug was well tolerated.
Of the 18 evaluable patients with circulating blasts, 83% responded to BP1001. The average reduction in circulating blasts was 67%.
The phase 2 trial included patients with relapsed/refractory AML. There were 3 evaluable patients in each of 2 dosing cohorts—60 mg/m2 and 90 mg/m2. Patients received BP1001 twice a week for 4 weeks.
Five of the patients responded—3 with a complete response and 2 with a partial response. There were no adverse events attributed to BP1001, and the maximum-tolerated dose was not reached. ![]()
The European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products (COMP) has recommended that BP1001 receive orphan designation as a treatment for acute myeloid leukemia (AML).
BP1001 (liposomal Grb2 antisense) is a neutral-charge, liposome-incorporated, antisense drug designed to inhibit protein synthesis of growth factor receptor bound protein 2 (Grb2).
BP1001 is being developed by Bio-Path Holdings, Inc.
According to Bio-Path, inhibition of Grb2 by BP1001 represents a significant advance in treating cancers with activated tyrosine kinases using a target not druggable with small molecule inhibitors.
Research has suggested that Grb2 plays an essential role in cancer cell activation via the RAS pathway. Grb2 bridges signals between activated and mutated tyrosine kinases, such as Flt3, c-Kit, and Bcr-Abl, and the Ras pathway, leading to activation of the ERK and AKT proteins.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The European Commission typically makes a decision within 30 days.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
Trials of BP1001
Bio-Path has completed a phase 1 trial of BP1001 in patients with relapsed/refractory AML, chronic myeloid leukemia, and myelodysplastic syndromes.
The company has also completed the safety segment of a phase 2 trial in which BP1001 is being investigated in combination with low-dose ara-C to treat AML.
Bio-Path recently released data from these studies.
The phase 1 study included patients who had received an average of 6 prior therapies.
The patients received 8 doses of BP1001 over 4 weeks, escalating to a maximum dose of 90 mg/m2. There were no dose-limiting toxicities, and Bio-Path said the drug was well tolerated.
Of the 18 evaluable patients with circulating blasts, 83% responded to BP1001. The average reduction in circulating blasts was 67%.
The phase 2 trial included patients with relapsed/refractory AML. There were 3 evaluable patients in each of 2 dosing cohorts—60 mg/m2 and 90 mg/m2. Patients received BP1001 twice a week for 4 weeks.
Five of the patients responded—3 with a complete response and 2 with a partial response. There were no adverse events attributed to BP1001, and the maximum-tolerated dose was not reached. ![]()
The European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products (COMP) has recommended that BP1001 receive orphan designation as a treatment for acute myeloid leukemia (AML).
BP1001 (liposomal Grb2 antisense) is a neutral-charge, liposome-incorporated, antisense drug designed to inhibit protein synthesis of growth factor receptor bound protein 2 (Grb2).
BP1001 is being developed by Bio-Path Holdings, Inc.
According to Bio-Path, inhibition of Grb2 by BP1001 represents a significant advance in treating cancers with activated tyrosine kinases using a target not druggable with small molecule inhibitors.
Research has suggested that Grb2 plays an essential role in cancer cell activation via the RAS pathway. Grb2 bridges signals between activated and mutated tyrosine kinases, such as Flt3, c-Kit, and Bcr-Abl, and the Ras pathway, leading to activation of the ERK and AKT proteins.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The European Commission typically makes a decision within 30 days.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
Trials of BP1001
Bio-Path has completed a phase 1 trial of BP1001 in patients with relapsed/refractory AML, chronic myeloid leukemia, and myelodysplastic syndromes.
The company has also completed the safety segment of a phase 2 trial in which BP1001 is being investigated in combination with low-dose ara-C to treat AML.
Bio-Path recently released data from these studies.
The phase 1 study included patients who had received an average of 6 prior therapies.
The patients received 8 doses of BP1001 over 4 weeks, escalating to a maximum dose of 90 mg/m2. There were no dose-limiting toxicities, and Bio-Path said the drug was well tolerated.
Of the 18 evaluable patients with circulating blasts, 83% responded to BP1001. The average reduction in circulating blasts was 67%.
The phase 2 trial included patients with relapsed/refractory AML. There were 3 evaluable patients in each of 2 dosing cohorts—60 mg/m2 and 90 mg/m2. Patients received BP1001 twice a week for 4 weeks.
Five of the patients responded—3 with a complete response and 2 with a partial response. There were no adverse events attributed to BP1001, and the maximum-tolerated dose was not reached. ![]()
Investigational AML drugs boosted remission rates
Two investigational drugs appear to be improving outcomes for patients with acute myeloid leukemia (AML), based on results reported in separate abstracts of studies that will be featured at press conferences to be held during the annual meeting of the American Society of Hematology.
In the first study, induction therapy with the investigational drug CPX-351 (Vyxeos), a liposomal formulation of cytarabine and daunorubicin, allowed a higher proportion of patients over age 60 with secondary AML to qualify for allogeneic hematopoietic cell transplants. Those patients went on to have improved survival, compared with patients who received standard 7+3 cytarabine and daunorubicin, Jeffrey E. Lancet, MD, of the H. Lee Moffitt Cancer Center and Research Institute, Tampa, and his colleagues reported in abstract 906.
The finding that CPX-351 may be an effective bridge to successful transplant for older patients with newly diagnosed secondary AML comes from an exploratory analysis of a phase III study comparing induction therapy with CPX-351 and standard cytarabine and daunorubicin. Initial data from the randomized open-label study, reported last June at the annual meeting of the American Society of Clinical Oncology, indicated CPX-351 significantly improved overall survival, event-free survival, and treatment response without an increase in 60-day mortality or in the frequency and severity of adverse events, compared with the standard 7+3 regimen of cytarabine and daunorubicin.
The data to be presented at ASH 2016 will examine the outcomes of 52 patients in the CPX-351 arm and 39 patients in the standard cytarabine and daunorubicin arm who underwent allogeneic hematopoietic cell transplantation (HCT) after induction. Data reported in the abstract indicate that 18 of the 52 patients in the CPX-351 arm and 26 of the 39 patients in the standard cytarabine and daunorubicin arm have died. The median survival time was 10.25 months with standard therapy; median survival has not yet been reached in the CPX-351 arm. The results indicate 53% fewer deaths occurred within 100 days of transplant in the CPX-351 group.
Newly diagnosed secondary AML was defined as having a history of prior cytotoxic treatment, antecedent myelodysplastic syndrome (MDS) with or without prior treatment with hypomethylating agents, or AML with World Health Organization–defined MDS-related cytogenetic abnormalities.
For the trial, conducted over 2 years at 39 U.S. and Canadian sites, 153 patients were randomized to the CPX-351 arm and 156 randomized to the standard therapy arm. Of 125 patients who had a complete response (CR) or a CR with incomplete (CRi) platelet or neutrophil recovery, 91 underwent allogeneic HCT: 52 (34%) from the CPX-351 arm and 39 (25%) from the standard therapy arm. Each arm had a similar percentage of patients who underwent transplant in CR/CRi status; however, the CPX-351 arm contained a higher percentage of patients age 70 and older (31% vs. 15%). Mortality at 100 days after transplant was 9.6% for patients in the CPX-351 arm and 20.5% for patients in the standard therapy arm. Deaths that occurred within 100 days after allogeneic HCT were due to refractory AML (CPX-351, 3.8%; standard therapy, 7.7%); graft vs. host disease (CPX-351, 3.8%; standard therapy, 2.6%); or renal, respiratory, or multiorgan failure, or septic shock (CPX-351, 0 for each; standard therapy, 2.6% for each), or the cause of death was unknown (CPX-351, 1.9%; standard therapy, 0).
For the 91 patients who had transplants, those in the CPX-351 arm had markedly better overall survival (hazard ratio, 0.46; P = .0046). The time-dependent Cox hazard ratio for overall survival in the CPX-351 arm vs. the 7+3 arm was 0.51 (95% confidence interval, 0.35–0.75; P = .0007).
In the phase Ib trial of vadastuximab talirine, 42 patients received the drug on days 1 and 4 of standard 7+3 cytarabine and daunorubicin induction therapy. Most patients had intermediate (40%) or adverse (43%) cytogenetic risk by Medical Research Council criteria, and 17% of patients had secondary AML. Response was assessed on days 15 and 28; MRD was assessed centrally by bone marrow examination using a multiparametric flow cytometric assay. The investigator chose whether to do a second induction regimen and any postremission therapies, which did not include additional administration of vadastuximab talirine.
Of the 40 patients who could be evaluated for efficacy, 24 (60%) had a CR and 7 (18%) had a CRi, and 4 (10%) reached a morphologic leukemia-free state. Nearly all (94%) of CR and CRi responses occurred after one cycle of induction therapy, and 23 of the 31 patients who reached CR or CRi achieved MRD-negative status.
Extramedullary adverse events, including hepatic toxicity, and induction mortality rates were similar to reported rates for 7+3 cytarabine and daunorubicin alone. All patients had grade 4 myelosuppression. In patients who achieved CR or CRi, the estimated median time to count recovery from day 1 of therapy was 33 days for neutrophils and 35 days for platelets. The 30- and 60-day mortality rates were 0% and 7%, respectively.
An alternative schedule of single-day dosing on day 1 is under investigation, and enrollment continues.
The CPX-351 (Vyxeos) study was supported by the drug’s maker, Celator Pharmaceuticals, which is a subsidiary of Jazz Pharmaceuticals. Dr. Lancet is a consultant to Celator as well as numerous other drug companies. Several of his colleagues disclosed a wide variety of relationships with drug companies, including Celator. Two of the study investigators disclosed employment by and equity ownership in Celator.
The vadastuximab talirine study was sponsored by the drug’s maker, Seattle Genetics. Dr. Erba disclosed a wide variety of relationships with drug companies, including research funding from Seattle Genetics. His colleagues had a similar wide variety of relationships, and two disclosed employment by and equity ownership in Seattle Genetics.
Abstract 906 Survival Following Allogeneic Hematopoietic Cell Transplantation in Older High-Risk Acute Myeloid Leukemia Patients Initially Treated With CPX-351 Liposome Injection Versus Standard Cytarabine and Daunorubicin: Subgroup Analysis of a Large Phase III Trial, will be presented in session 616 at 4:00 p.m. on Monday, Dec. 5.
Abstract 211 A Phase Ib Study of Vadastuximab Talirine in Combination With 7+3 Induction Therapy for Patients With Newly Diagnosed Acute Myeloid Leukemia (AML) will be presented in session 613 at 4:00 p.m. on Saturday, Dec. 3.
mdales@frontlinemedcom.com
On Twitter @maryjodales
Two investigational drugs appear to be improving outcomes for patients with acute myeloid leukemia (AML), based on results reported in separate abstracts of studies that will be featured at press conferences to be held during the annual meeting of the American Society of Hematology.
In the first study, induction therapy with the investigational drug CPX-351 (Vyxeos), a liposomal formulation of cytarabine and daunorubicin, allowed a higher proportion of patients over age 60 with secondary AML to qualify for allogeneic hematopoietic cell transplants. Those patients went on to have improved survival, compared with patients who received standard 7+3 cytarabine and daunorubicin, Jeffrey E. Lancet, MD, of the H. Lee Moffitt Cancer Center and Research Institute, Tampa, and his colleagues reported in abstract 906.
The finding that CPX-351 may be an effective bridge to successful transplant for older patients with newly diagnosed secondary AML comes from an exploratory analysis of a phase III study comparing induction therapy with CPX-351 and standard cytarabine and daunorubicin. Initial data from the randomized open-label study, reported last June at the annual meeting of the American Society of Clinical Oncology, indicated CPX-351 significantly improved overall survival, event-free survival, and treatment response without an increase in 60-day mortality or in the frequency and severity of adverse events, compared with the standard 7+3 regimen of cytarabine and daunorubicin.
The data to be presented at ASH 2016 will examine the outcomes of 52 patients in the CPX-351 arm and 39 patients in the standard cytarabine and daunorubicin arm who underwent allogeneic hematopoietic cell transplantation (HCT) after induction. Data reported in the abstract indicate that 18 of the 52 patients in the CPX-351 arm and 26 of the 39 patients in the standard cytarabine and daunorubicin arm have died. The median survival time was 10.25 months with standard therapy; median survival has not yet been reached in the CPX-351 arm. The results indicate 53% fewer deaths occurred within 100 days of transplant in the CPX-351 group.
Newly diagnosed secondary AML was defined as having a history of prior cytotoxic treatment, antecedent myelodysplastic syndrome (MDS) with or without prior treatment with hypomethylating agents, or AML with World Health Organization–defined MDS-related cytogenetic abnormalities.
For the trial, conducted over 2 years at 39 U.S. and Canadian sites, 153 patients were randomized to the CPX-351 arm and 156 randomized to the standard therapy arm. Of 125 patients who had a complete response (CR) or a CR with incomplete (CRi) platelet or neutrophil recovery, 91 underwent allogeneic HCT: 52 (34%) from the CPX-351 arm and 39 (25%) from the standard therapy arm. Each arm had a similar percentage of patients who underwent transplant in CR/CRi status; however, the CPX-351 arm contained a higher percentage of patients age 70 and older (31% vs. 15%). Mortality at 100 days after transplant was 9.6% for patients in the CPX-351 arm and 20.5% for patients in the standard therapy arm. Deaths that occurred within 100 days after allogeneic HCT were due to refractory AML (CPX-351, 3.8%; standard therapy, 7.7%); graft vs. host disease (CPX-351, 3.8%; standard therapy, 2.6%); or renal, respiratory, or multiorgan failure, or septic shock (CPX-351, 0 for each; standard therapy, 2.6% for each), or the cause of death was unknown (CPX-351, 1.9%; standard therapy, 0).
For the 91 patients who had transplants, those in the CPX-351 arm had markedly better overall survival (hazard ratio, 0.46; P = .0046). The time-dependent Cox hazard ratio for overall survival in the CPX-351 arm vs. the 7+3 arm was 0.51 (95% confidence interval, 0.35–0.75; P = .0007).
In the phase Ib trial of vadastuximab talirine, 42 patients received the drug on days 1 and 4 of standard 7+3 cytarabine and daunorubicin induction therapy. Most patients had intermediate (40%) or adverse (43%) cytogenetic risk by Medical Research Council criteria, and 17% of patients had secondary AML. Response was assessed on days 15 and 28; MRD was assessed centrally by bone marrow examination using a multiparametric flow cytometric assay. The investigator chose whether to do a second induction regimen and any postremission therapies, which did not include additional administration of vadastuximab talirine.
Of the 40 patients who could be evaluated for efficacy, 24 (60%) had a CR and 7 (18%) had a CRi, and 4 (10%) reached a morphologic leukemia-free state. Nearly all (94%) of CR and CRi responses occurred after one cycle of induction therapy, and 23 of the 31 patients who reached CR or CRi achieved MRD-negative status.
Extramedullary adverse events, including hepatic toxicity, and induction mortality rates were similar to reported rates for 7+3 cytarabine and daunorubicin alone. All patients had grade 4 myelosuppression. In patients who achieved CR or CRi, the estimated median time to count recovery from day 1 of therapy was 33 days for neutrophils and 35 days for platelets. The 30- and 60-day mortality rates were 0% and 7%, respectively.
An alternative schedule of single-day dosing on day 1 is under investigation, and enrollment continues.
The CPX-351 (Vyxeos) study was supported by the drug’s maker, Celator Pharmaceuticals, which is a subsidiary of Jazz Pharmaceuticals. Dr. Lancet is a consultant to Celator as well as numerous other drug companies. Several of his colleagues disclosed a wide variety of relationships with drug companies, including Celator. Two of the study investigators disclosed employment by and equity ownership in Celator.
The vadastuximab talirine study was sponsored by the drug’s maker, Seattle Genetics. Dr. Erba disclosed a wide variety of relationships with drug companies, including research funding from Seattle Genetics. His colleagues had a similar wide variety of relationships, and two disclosed employment by and equity ownership in Seattle Genetics.
Abstract 906 Survival Following Allogeneic Hematopoietic Cell Transplantation in Older High-Risk Acute Myeloid Leukemia Patients Initially Treated With CPX-351 Liposome Injection Versus Standard Cytarabine and Daunorubicin: Subgroup Analysis of a Large Phase III Trial, will be presented in session 616 at 4:00 p.m. on Monday, Dec. 5.
Abstract 211 A Phase Ib Study of Vadastuximab Talirine in Combination With 7+3 Induction Therapy for Patients With Newly Diagnosed Acute Myeloid Leukemia (AML) will be presented in session 613 at 4:00 p.m. on Saturday, Dec. 3.
mdales@frontlinemedcom.com
On Twitter @maryjodales
Two investigational drugs appear to be improving outcomes for patients with acute myeloid leukemia (AML), based on results reported in separate abstracts of studies that will be featured at press conferences to be held during the annual meeting of the American Society of Hematology.
In the first study, induction therapy with the investigational drug CPX-351 (Vyxeos), a liposomal formulation of cytarabine and daunorubicin, allowed a higher proportion of patients over age 60 with secondary AML to qualify for allogeneic hematopoietic cell transplants. Those patients went on to have improved survival, compared with patients who received standard 7+3 cytarabine and daunorubicin, Jeffrey E. Lancet, MD, of the H. Lee Moffitt Cancer Center and Research Institute, Tampa, and his colleagues reported in abstract 906.
The finding that CPX-351 may be an effective bridge to successful transplant for older patients with newly diagnosed secondary AML comes from an exploratory analysis of a phase III study comparing induction therapy with CPX-351 and standard cytarabine and daunorubicin. Initial data from the randomized open-label study, reported last June at the annual meeting of the American Society of Clinical Oncology, indicated CPX-351 significantly improved overall survival, event-free survival, and treatment response without an increase in 60-day mortality or in the frequency and severity of adverse events, compared with the standard 7+3 regimen of cytarabine and daunorubicin.
The data to be presented at ASH 2016 will examine the outcomes of 52 patients in the CPX-351 arm and 39 patients in the standard cytarabine and daunorubicin arm who underwent allogeneic hematopoietic cell transplantation (HCT) after induction. Data reported in the abstract indicate that 18 of the 52 patients in the CPX-351 arm and 26 of the 39 patients in the standard cytarabine and daunorubicin arm have died. The median survival time was 10.25 months with standard therapy; median survival has not yet been reached in the CPX-351 arm. The results indicate 53% fewer deaths occurred within 100 days of transplant in the CPX-351 group.
Newly diagnosed secondary AML was defined as having a history of prior cytotoxic treatment, antecedent myelodysplastic syndrome (MDS) with or without prior treatment with hypomethylating agents, or AML with World Health Organization–defined MDS-related cytogenetic abnormalities.
For the trial, conducted over 2 years at 39 U.S. and Canadian sites, 153 patients were randomized to the CPX-351 arm and 156 randomized to the standard therapy arm. Of 125 patients who had a complete response (CR) or a CR with incomplete (CRi) platelet or neutrophil recovery, 91 underwent allogeneic HCT: 52 (34%) from the CPX-351 arm and 39 (25%) from the standard therapy arm. Each arm had a similar percentage of patients who underwent transplant in CR/CRi status; however, the CPX-351 arm contained a higher percentage of patients age 70 and older (31% vs. 15%). Mortality at 100 days after transplant was 9.6% for patients in the CPX-351 arm and 20.5% for patients in the standard therapy arm. Deaths that occurred within 100 days after allogeneic HCT were due to refractory AML (CPX-351, 3.8%; standard therapy, 7.7%); graft vs. host disease (CPX-351, 3.8%; standard therapy, 2.6%); or renal, respiratory, or multiorgan failure, or septic shock (CPX-351, 0 for each; standard therapy, 2.6% for each), or the cause of death was unknown (CPX-351, 1.9%; standard therapy, 0).
For the 91 patients who had transplants, those in the CPX-351 arm had markedly better overall survival (hazard ratio, 0.46; P = .0046). The time-dependent Cox hazard ratio for overall survival in the CPX-351 arm vs. the 7+3 arm was 0.51 (95% confidence interval, 0.35–0.75; P = .0007).
In the phase Ib trial of vadastuximab talirine, 42 patients received the drug on days 1 and 4 of standard 7+3 cytarabine and daunorubicin induction therapy. Most patients had intermediate (40%) or adverse (43%) cytogenetic risk by Medical Research Council criteria, and 17% of patients had secondary AML. Response was assessed on days 15 and 28; MRD was assessed centrally by bone marrow examination using a multiparametric flow cytometric assay. The investigator chose whether to do a second induction regimen and any postremission therapies, which did not include additional administration of vadastuximab talirine.
Of the 40 patients who could be evaluated for efficacy, 24 (60%) had a CR and 7 (18%) had a CRi, and 4 (10%) reached a morphologic leukemia-free state. Nearly all (94%) of CR and CRi responses occurred after one cycle of induction therapy, and 23 of the 31 patients who reached CR or CRi achieved MRD-negative status.
Extramedullary adverse events, including hepatic toxicity, and induction mortality rates were similar to reported rates for 7+3 cytarabine and daunorubicin alone. All patients had grade 4 myelosuppression. In patients who achieved CR or CRi, the estimated median time to count recovery from day 1 of therapy was 33 days for neutrophils and 35 days for platelets. The 30- and 60-day mortality rates were 0% and 7%, respectively.
An alternative schedule of single-day dosing on day 1 is under investigation, and enrollment continues.
The CPX-351 (Vyxeos) study was supported by the drug’s maker, Celator Pharmaceuticals, which is a subsidiary of Jazz Pharmaceuticals. Dr. Lancet is a consultant to Celator as well as numerous other drug companies. Several of his colleagues disclosed a wide variety of relationships with drug companies, including Celator. Two of the study investigators disclosed employment by and equity ownership in Celator.
The vadastuximab talirine study was sponsored by the drug’s maker, Seattle Genetics. Dr. Erba disclosed a wide variety of relationships with drug companies, including research funding from Seattle Genetics. His colleagues had a similar wide variety of relationships, and two disclosed employment by and equity ownership in Seattle Genetics.
Abstract 906 Survival Following Allogeneic Hematopoietic Cell Transplantation in Older High-Risk Acute Myeloid Leukemia Patients Initially Treated With CPX-351 Liposome Injection Versus Standard Cytarabine and Daunorubicin: Subgroup Analysis of a Large Phase III Trial, will be presented in session 616 at 4:00 p.m. on Monday, Dec. 5.
Abstract 211 A Phase Ib Study of Vadastuximab Talirine in Combination With 7+3 Induction Therapy for Patients With Newly Diagnosed Acute Myeloid Leukemia (AML) will be presented in session 613 at 4:00 p.m. on Saturday, Dec. 3.
mdales@frontlinemedcom.com
On Twitter @maryjodales
FROM ASH 2016
Combating drug resistance in FLT3-mutated AML
with autophagosomes (green),
a process that happens during
mitophagy in cancer cells
treated with FLT3 inhibitor
Image from Besim Ogretmen
and Mohammed Dany/MUSC
Research published in Blood has revealed a mechanism that confers treatment resistance in FLT3-mutated acute myeloid leukemia (AML), as well as a drug that might overcome that resistance.
Researchers found that ceramide-dependent mitophagy plays a key role in drug-mediated AML cell death.
“Ceramide, a pro-cell-death lipid, kills cancer cells by causing them to eat their own mitochondria,” explained study author Besim Ogretmen, PhD, of the Medical University of South Carolina (MUSC) Hollings Cancer Center in Charleston, South Carolina.
AML cells with FLT3-ITD inhibit ceramide synthesis and thereby become resistant to cell death. FLT3 inhibitors have been developed to combat this resistance, but they’ve fallen short of expectations.
“Unfortunately, regardless of the inhibitor, the problem of resistance to FLT3-targeted therapy has persisted,” said study author Mohammed Dany, an MD/PhD student at MUSC.
However, Dany, Dr Ogretmen, and their colleagues were able to overcome this resistance with a synthetic ceramide analogue known as LCL-461.
In vitro, the drug reactivated mitophagy and killed AML cells that were resistant to treatment with the FLT3 inhibitor crenolanib.
In mice with crenolanib-resistant human AML xenografts, LCL-461 eliminated AML cells from the bone marrow.
A positively charged molecule, LCL-461 is attracted to the mitochondria of cancer cells, which become negatively charged through the Warburg effect. The researchers said this limits off-target effects that can occur with less specific inhibitors of FLT3 signaling.
Furthermore, Dr Ogretmen’s lab has tested the safety of LCL-461 in previous studies and reported that it had no major side effects at therapeutically active doses.
Dr Ogretmen and his colleagues’ next step is to perform large animal studies with LCL-461.
“We are very excited about this,” Dr Ogretmen said. “Head and neck cancers also respond to this drug very well. What we are trying to do is really cure cancer one disease at a time, and we are digging and digging to understand the mechanisms of how these cancer cells escape therapeutic interventions so that we can find mechanism-based therapeutics to have more tools for treatment.”
LCL-461 was developed at MUSC. The MUSC Foundation for Research Development has patented the drug and licensed it to Charleston-based startup SphingoGene, Inc. ![]()
with autophagosomes (green),
a process that happens during
mitophagy in cancer cells
treated with FLT3 inhibitor
Image from Besim Ogretmen
and Mohammed Dany/MUSC
Research published in Blood has revealed a mechanism that confers treatment resistance in FLT3-mutated acute myeloid leukemia (AML), as well as a drug that might overcome that resistance.
Researchers found that ceramide-dependent mitophagy plays a key role in drug-mediated AML cell death.
“Ceramide, a pro-cell-death lipid, kills cancer cells by causing them to eat their own mitochondria,” explained study author Besim Ogretmen, PhD, of the Medical University of South Carolina (MUSC) Hollings Cancer Center in Charleston, South Carolina.
AML cells with FLT3-ITD inhibit ceramide synthesis and thereby become resistant to cell death. FLT3 inhibitors have been developed to combat this resistance, but they’ve fallen short of expectations.
“Unfortunately, regardless of the inhibitor, the problem of resistance to FLT3-targeted therapy has persisted,” said study author Mohammed Dany, an MD/PhD student at MUSC.
However, Dany, Dr Ogretmen, and their colleagues were able to overcome this resistance with a synthetic ceramide analogue known as LCL-461.
In vitro, the drug reactivated mitophagy and killed AML cells that were resistant to treatment with the FLT3 inhibitor crenolanib.
In mice with crenolanib-resistant human AML xenografts, LCL-461 eliminated AML cells from the bone marrow.
A positively charged molecule, LCL-461 is attracted to the mitochondria of cancer cells, which become negatively charged through the Warburg effect. The researchers said this limits off-target effects that can occur with less specific inhibitors of FLT3 signaling.
Furthermore, Dr Ogretmen’s lab has tested the safety of LCL-461 in previous studies and reported that it had no major side effects at therapeutically active doses.
Dr Ogretmen and his colleagues’ next step is to perform large animal studies with LCL-461.
“We are very excited about this,” Dr Ogretmen said. “Head and neck cancers also respond to this drug very well. What we are trying to do is really cure cancer one disease at a time, and we are digging and digging to understand the mechanisms of how these cancer cells escape therapeutic interventions so that we can find mechanism-based therapeutics to have more tools for treatment.”
LCL-461 was developed at MUSC. The MUSC Foundation for Research Development has patented the drug and licensed it to Charleston-based startup SphingoGene, Inc. ![]()
with autophagosomes (green),
a process that happens during
mitophagy in cancer cells
treated with FLT3 inhibitor
Image from Besim Ogretmen
and Mohammed Dany/MUSC
Research published in Blood has revealed a mechanism that confers treatment resistance in FLT3-mutated acute myeloid leukemia (AML), as well as a drug that might overcome that resistance.
Researchers found that ceramide-dependent mitophagy plays a key role in drug-mediated AML cell death.
“Ceramide, a pro-cell-death lipid, kills cancer cells by causing them to eat their own mitochondria,” explained study author Besim Ogretmen, PhD, of the Medical University of South Carolina (MUSC) Hollings Cancer Center in Charleston, South Carolina.
AML cells with FLT3-ITD inhibit ceramide synthesis and thereby become resistant to cell death. FLT3 inhibitors have been developed to combat this resistance, but they’ve fallen short of expectations.
“Unfortunately, regardless of the inhibitor, the problem of resistance to FLT3-targeted therapy has persisted,” said study author Mohammed Dany, an MD/PhD student at MUSC.
However, Dany, Dr Ogretmen, and their colleagues were able to overcome this resistance with a synthetic ceramide analogue known as LCL-461.
In vitro, the drug reactivated mitophagy and killed AML cells that were resistant to treatment with the FLT3 inhibitor crenolanib.
In mice with crenolanib-resistant human AML xenografts, LCL-461 eliminated AML cells from the bone marrow.
A positively charged molecule, LCL-461 is attracted to the mitochondria of cancer cells, which become negatively charged through the Warburg effect. The researchers said this limits off-target effects that can occur with less specific inhibitors of FLT3 signaling.
Furthermore, Dr Ogretmen’s lab has tested the safety of LCL-461 in previous studies and reported that it had no major side effects at therapeutically active doses.
Dr Ogretmen and his colleagues’ next step is to perform large animal studies with LCL-461.
“We are very excited about this,” Dr Ogretmen said. “Head and neck cancers also respond to this drug very well. What we are trying to do is really cure cancer one disease at a time, and we are digging and digging to understand the mechanisms of how these cancer cells escape therapeutic interventions so that we can find mechanism-based therapeutics to have more tools for treatment.”
LCL-461 was developed at MUSC. The MUSC Foundation for Research Development has patented the drug and licensed it to Charleston-based startup SphingoGene, Inc. ![]()
Team targets fructose to treat AML

Photo from the University
of Hawaii Cancer Center
Researchers say they have discovered a novel treatment strategy for acute myeloid leukemia (AML)—inhibiting fructose utilization.
The team discovered that AML cells are prone to using fructose for energy, and increased fructose utilization predicts poor treatment outcomes in AML patients.
The researchers also found that 2,5-anhydro-D-mannitol (2,5-AM) could inhibit fructose use in AML cells and therefore hinder their growth.
Wei Jia, PhD, of the University of Hawaii Cancer Center in Honolulu, and his colleagues reported these findings in Cancer Cell.
The researchers said their findings highlight the unique ability of AML cells to switch their energy supply from glucose to fructose, when glucose is in short supply.
Fructose is the second most abundant blood sugar in the body and is used as a glucose alternative by AML cells to retain energy. After the switch, AML cells begin to multiply faster.
The study revealed a potential treatment by stopping the glucose transporter GLUT5. This restricts the AML energy supply and effectively slows AML growth.
To target GLUT5, the researchers used 2,5-AM, a fructose analog with high affinity for GLUT5.
The team tested 2,5-AM in AML cells with enhanced fructose utilization and found the drug significantly suppressed fructose-induced proliferation, colony growth, and migration in the absence of glucose or when glucose levels were low.
The researchers tested 2,5-AM in 4 different AML cell lines and found the drug suppressed fructose-induced cell proliferation in a dose-dependent manner in all of the cell lines under glucose-limiting conditions. However, 2,5-AM had little effect on glucose-induced cell proliferation.
The team tested 2,5-AM in normal monocytes as well. They said the drug had a negligible effect on glucose-induced cell growth.
“Our normal cells hardly rely on fructose for growth,” Dr Jia noted. “This makes the fructose transport in cancer cells an attractive drug target.”
Finally, the researchers tested 2,5-AM in combination with ara-C in the 4 AML cell lines. They observed a synergistic effect between the drugs in all cell lines in the absence of glucose or when glucose levels were low.
“We are in the process of developing a GLUT5 inhibitor, thus cutting the cancer cells’ energy source and eventually killing them,” Dr Jia said. “The new GLUT5 inhibitor can potentially be used alone or in addition to the current chemotherapy drugs to enhance anticancer effects.” ![]()
Photo from the University
of Hawaii Cancer Center
Researchers say they have discovered a novel treatment strategy for acute myeloid leukemia (AML)—inhibiting fructose utilization.
The team discovered that AML cells are prone to using fructose for energy, and increased fructose utilization predicts poor treatment outcomes in AML patients.
The researchers also found that 2,5-anhydro-D-mannitol (2,5-AM) could inhibit fructose use in AML cells and therefore hinder their growth.
Wei Jia, PhD, of the University of Hawaii Cancer Center in Honolulu, and his colleagues reported these findings in Cancer Cell.
The researchers said their findings highlight the unique ability of AML cells to switch their energy supply from glucose to fructose, when glucose is in short supply.
Fructose is the second most abundant blood sugar in the body and is used as a glucose alternative by AML cells to retain energy. After the switch, AML cells begin to multiply faster.
The study revealed a potential treatment by stopping the glucose transporter GLUT5. This restricts the AML energy supply and effectively slows AML growth.
To target GLUT5, the researchers used 2,5-AM, a fructose analog with high affinity for GLUT5.
The team tested 2,5-AM in AML cells with enhanced fructose utilization and found the drug significantly suppressed fructose-induced proliferation, colony growth, and migration in the absence of glucose or when glucose levels were low.
The researchers tested 2,5-AM in 4 different AML cell lines and found the drug suppressed fructose-induced cell proliferation in a dose-dependent manner in all of the cell lines under glucose-limiting conditions. However, 2,5-AM had little effect on glucose-induced cell proliferation.
The team tested 2,5-AM in normal monocytes as well. They said the drug had a negligible effect on glucose-induced cell growth.
“Our normal cells hardly rely on fructose for growth,” Dr Jia noted. “This makes the fructose transport in cancer cells an attractive drug target.”
Finally, the researchers tested 2,5-AM in combination with ara-C in the 4 AML cell lines. They observed a synergistic effect between the drugs in all cell lines in the absence of glucose or when glucose levels were low.
“We are in the process of developing a GLUT5 inhibitor, thus cutting the cancer cells’ energy source and eventually killing them,” Dr Jia said. “The new GLUT5 inhibitor can potentially be used alone or in addition to the current chemotherapy drugs to enhance anticancer effects.” ![]()
Photo from the University
of Hawaii Cancer Center
Researchers say they have discovered a novel treatment strategy for acute myeloid leukemia (AML)—inhibiting fructose utilization.
The team discovered that AML cells are prone to using fructose for energy, and increased fructose utilization predicts poor treatment outcomes in AML patients.
The researchers also found that 2,5-anhydro-D-mannitol (2,5-AM) could inhibit fructose use in AML cells and therefore hinder their growth.
Wei Jia, PhD, of the University of Hawaii Cancer Center in Honolulu, and his colleagues reported these findings in Cancer Cell.
The researchers said their findings highlight the unique ability of AML cells to switch their energy supply from glucose to fructose, when glucose is in short supply.
Fructose is the second most abundant blood sugar in the body and is used as a glucose alternative by AML cells to retain energy. After the switch, AML cells begin to multiply faster.
The study revealed a potential treatment by stopping the glucose transporter GLUT5. This restricts the AML energy supply and effectively slows AML growth.
To target GLUT5, the researchers used 2,5-AM, a fructose analog with high affinity for GLUT5.
The team tested 2,5-AM in AML cells with enhanced fructose utilization and found the drug significantly suppressed fructose-induced proliferation, colony growth, and migration in the absence of glucose or when glucose levels were low.
The researchers tested 2,5-AM in 4 different AML cell lines and found the drug suppressed fructose-induced cell proliferation in a dose-dependent manner in all of the cell lines under glucose-limiting conditions. However, 2,5-AM had little effect on glucose-induced cell proliferation.
The team tested 2,5-AM in normal monocytes as well. They said the drug had a negligible effect on glucose-induced cell growth.
“Our normal cells hardly rely on fructose for growth,” Dr Jia noted. “This makes the fructose transport in cancer cells an attractive drug target.”
Finally, the researchers tested 2,5-AM in combination with ara-C in the 4 AML cell lines. They observed a synergistic effect between the drugs in all cell lines in the absence of glucose or when glucose levels were low.
“We are in the process of developing a GLUT5 inhibitor, thus cutting the cancer cells’ energy source and eventually killing them,” Dr Jia said. “The new GLUT5 inhibitor can potentially be used alone or in addition to the current chemotherapy drugs to enhance anticancer effects.” ![]()