User login
Methylation patterns can predict survival in AML, team says

Credit: Lance Liotta
Researchers have found evidence to suggest that methylation patterns in hematopoietic stem cells (HSCs) can be used to determine prognosis in patients with acute myeloid leukemia (AML).
The team discovered that patients with methylation patterns resembling those of healthy individuals lived longer than patients with substantially different patterns.
If validated in clinical trials, this finding could be used to help physicians tailor treatment according to a patient’s needs.
Ulrich Steidl, MD, PhD, of the Albert Einstein College of Medicine in New York, and his colleagues described this research in The Journal of Clinical Investigation.
The investigators knew that aberrations in HSC methylation can prevent the cells from differentiating into mature blood cells, which leads to AML.
So they speculated that comparing how closely the methylation patterns in cells from AML patients resemble the patterns found in healthy individuals’ HSCs might foretell the patients’ response to treatment.
To find out, the researchers first looked at methylation patterns in HSCs from healthy individuals. The team found that most cytosines are methylated in healthy HSCs.
And where demethylation occurs, it’s mainly limited to one particular stage of HSC differentiation—the commitment step from short-term HSC to common myeloid progenitor.
The investigators then set out to identify loci with the most significant methylation changes across differentiation stages. Their analysis revealed a set of 561 loci that distinguished between the 4 stages of HSC development they investigated.
The team next wanted to determine whether the methylation status of these loci was affected in AML. So they developed an epigenetic signature score based on loci methylation. A patient’s score increased the more his methylation pattern differed from that of a healthy individual.
The researchers tested their scoring method using data from 3 cohorts of AML patients. In each of these groups, patients with low scores had approximately twice the median survival time of patients with high scores.
Specifically, the investigators evaluated AML patients in a trial testing 2 different doses of daunorubicin (Fernandez et al, NEJM 2009).
Among patients receiving lower-dose daunorubicin, those with lower epigenetic signature scores had a median overall survival (OS) of 19 months, compared with 10.8 months for patients with higher scores (P=0.0165).
The researchers observed similar results in the patients receiving a higher dose of daunorubicin. The median OS in the group with low epigenetic signature scores was 25.4 months, compared with 13.2 months in the group with high scores (P=0.0062).
Likewise, in a third cohort of AML patients, those with a low epigenetic signature score had significantly better OS than those with a high score—a median of 28.1 months and 14.9 months, respectively (P=0.0150).
The investigators performed the same analyses using a commitment-associated gene-expression signature. And they found their epigenetic signature was more effective at predicting patient survival.
Dr Steidl and his colleagues are now studying the genes found in the aberrant epigenetic signatures to determine if they play a role in causing AML. ![]()
Credit: Lance Liotta
Researchers have found evidence to suggest that methylation patterns in hematopoietic stem cells (HSCs) can be used to determine prognosis in patients with acute myeloid leukemia (AML).
The team discovered that patients with methylation patterns resembling those of healthy individuals lived longer than patients with substantially different patterns.
If validated in clinical trials, this finding could be used to help physicians tailor treatment according to a patient’s needs.
Ulrich Steidl, MD, PhD, of the Albert Einstein College of Medicine in New York, and his colleagues described this research in The Journal of Clinical Investigation.
The investigators knew that aberrations in HSC methylation can prevent the cells from differentiating into mature blood cells, which leads to AML.
So they speculated that comparing how closely the methylation patterns in cells from AML patients resemble the patterns found in healthy individuals’ HSCs might foretell the patients’ response to treatment.
To find out, the researchers first looked at methylation patterns in HSCs from healthy individuals. The team found that most cytosines are methylated in healthy HSCs.
And where demethylation occurs, it’s mainly limited to one particular stage of HSC differentiation—the commitment step from short-term HSC to common myeloid progenitor.
The investigators then set out to identify loci with the most significant methylation changes across differentiation stages. Their analysis revealed a set of 561 loci that distinguished between the 4 stages of HSC development they investigated.
The team next wanted to determine whether the methylation status of these loci was affected in AML. So they developed an epigenetic signature score based on loci methylation. A patient’s score increased the more his methylation pattern differed from that of a healthy individual.
The researchers tested their scoring method using data from 3 cohorts of AML patients. In each of these groups, patients with low scores had approximately twice the median survival time of patients with high scores.
Specifically, the investigators evaluated AML patients in a trial testing 2 different doses of daunorubicin (Fernandez et al, NEJM 2009).
Among patients receiving lower-dose daunorubicin, those with lower epigenetic signature scores had a median overall survival (OS) of 19 months, compared with 10.8 months for patients with higher scores (P=0.0165).
The researchers observed similar results in the patients receiving a higher dose of daunorubicin. The median OS in the group with low epigenetic signature scores was 25.4 months, compared with 13.2 months in the group with high scores (P=0.0062).
Likewise, in a third cohort of AML patients, those with a low epigenetic signature score had significantly better OS than those with a high score—a median of 28.1 months and 14.9 months, respectively (P=0.0150).
The investigators performed the same analyses using a commitment-associated gene-expression signature. And they found their epigenetic signature was more effective at predicting patient survival.
Dr Steidl and his colleagues are now studying the genes found in the aberrant epigenetic signatures to determine if they play a role in causing AML. ![]()
Credit: Lance Liotta
Researchers have found evidence to suggest that methylation patterns in hematopoietic stem cells (HSCs) can be used to determine prognosis in patients with acute myeloid leukemia (AML).
The team discovered that patients with methylation patterns resembling those of healthy individuals lived longer than patients with substantially different patterns.
If validated in clinical trials, this finding could be used to help physicians tailor treatment according to a patient’s needs.
Ulrich Steidl, MD, PhD, of the Albert Einstein College of Medicine in New York, and his colleagues described this research in The Journal of Clinical Investigation.
The investigators knew that aberrations in HSC methylation can prevent the cells from differentiating into mature blood cells, which leads to AML.
So they speculated that comparing how closely the methylation patterns in cells from AML patients resemble the patterns found in healthy individuals’ HSCs might foretell the patients’ response to treatment.
To find out, the researchers first looked at methylation patterns in HSCs from healthy individuals. The team found that most cytosines are methylated in healthy HSCs.
And where demethylation occurs, it’s mainly limited to one particular stage of HSC differentiation—the commitment step from short-term HSC to common myeloid progenitor.
The investigators then set out to identify loci with the most significant methylation changes across differentiation stages. Their analysis revealed a set of 561 loci that distinguished between the 4 stages of HSC development they investigated.
The team next wanted to determine whether the methylation status of these loci was affected in AML. So they developed an epigenetic signature score based on loci methylation. A patient’s score increased the more his methylation pattern differed from that of a healthy individual.
The researchers tested their scoring method using data from 3 cohorts of AML patients. In each of these groups, patients with low scores had approximately twice the median survival time of patients with high scores.
Specifically, the investigators evaluated AML patients in a trial testing 2 different doses of daunorubicin (Fernandez et al, NEJM 2009).
Among patients receiving lower-dose daunorubicin, those with lower epigenetic signature scores had a median overall survival (OS) of 19 months, compared with 10.8 months for patients with higher scores (P=0.0165).
The researchers observed similar results in the patients receiving a higher dose of daunorubicin. The median OS in the group with low epigenetic signature scores was 25.4 months, compared with 13.2 months in the group with high scores (P=0.0062).
Likewise, in a third cohort of AML patients, those with a low epigenetic signature score had significantly better OS than those with a high score—a median of 28.1 months and 14.9 months, respectively (P=0.0150).
The investigators performed the same analyses using a commitment-associated gene-expression signature. And they found their epigenetic signature was more effective at predicting patient survival.
Dr Steidl and his colleagues are now studying the genes found in the aberrant epigenetic signatures to determine if they play a role in causing AML. ![]()
Mouse model provides new insight into AML

Studies have suggested that mutations in isocitrate dehydrogenase-1 and 2 (IDH1 and IDH2) are present in approximately 20% of all acute myeloid leukemias (AMLs), and this implies that mutant IDH proteins are attractive drug targets.
With this in mind, a group of scientists generated a transgenic mouse model of the most common IDH2 mutation in human AML.
Experiments conducted with this model revealed that mutant IDH2 contributes to leukemia initiation and is required for the maintenance of leukemic cells in a living organism.
The researchers said these findings, published in Cell Stem Cell, confirm a potent oncogenic role for IDH2 and support its relevance as a therapeutic target for AML.
Furthermore, the model can be used to evaluate the pharmacological efficacy of IDH2 inhibitors, either alone or in combination with other compounds.
“The real hope is that we would one day be able to treat IDH2-mutant leukemia patients with a drug that targets this genetic abnormality,” said senior study author Pier Paolo Pandolfi, MD, PhD, of Beth Israel Deaconess Medical Center (BIDMC) in Boston.
He and his colleagues knew that IDH1 and IDH2 proteins are critical enzymes in the TCA cycle, which is centrally important to many biochemical pathways. Mutated forms of these proteins gain a novel ability to produce 2-hydroxyglutarate (2HG), a metabolite that has been shown to accumulate at high levels in cancer patients.
“Our goal was to generate an animal model of mutant IDH that was both inducible and reversible,” said Markus Reschke, PhD, also of BIDMC.
“This enabled us to address an important unanswered question: Does inhibition of mutant IDH proteins in active disease have an effect on tumor maintenance or progression in a living organism?”
The researchers studied 2 different models: a retroviral transduction model and a genetically engineered model in which IDH mice were crossed with mice harboring other leukemia-relevant mutations.
In the first model, the IDH mutation was combined with the oncogenes HoxA9 and Meis1a, 2 downstream targets of numerous pathways that are deregulated in AML.
The results showed evidence of differentiation within 2 weeks of genetic deinduction of mutant IDH. And 2 weeks later, 6 of 8 animals showed complete remission with elimination of any detectable leukemic cells.
The researchers said these results were both surprising and encouraging, demonstrating a situation in which IDH mutation occurs as an early event, and leukemic transformation occurs as a result of subsequent genetic hits.
“The retroviral model enabled us to observe that mutant IDH2 is essential for the maintenance of HoxA9/Meis1a-induced AML,” said Lev Kats, PhD, of BIDMC. “But this was still a surrogate model. This isn’t what happens in human patients, per se.”
The researchers therefore went on to develop a transgenic model that more closely recapitulates the genetics of human AML.
“By crossing the mutant IDH2 animals with other leukemia-relevant mutations, including mutations in the FMS-like tyrosine kinase 3 [FLT3], we observed that compound-mutant animals developed acute leukemias,” Dr Reschke said. “This exciting finding told us that mutant IDH2 contributes to leukemia initiation in vivo.”
As with the retroviral transduction model, genetic deinduction of mutant IDH2 in the context of a cooperating FLT3 mutation resulted in reduced proliferation and/or differentiation of leukemic cells, further demonstrating that mutant IDH2 expression is required for leukemia maintenance.
“This model has validated mutant IDH proteins as very strong candidates for continued development of targeted anticancer therapeutics,” Dr Pandolfi said. “The model will also be of paramount importance to study mechanisms of resistance to treatment that may occur.” ![]()
Studies have suggested that mutations in isocitrate dehydrogenase-1 and 2 (IDH1 and IDH2) are present in approximately 20% of all acute myeloid leukemias (AMLs), and this implies that mutant IDH proteins are attractive drug targets.
With this in mind, a group of scientists generated a transgenic mouse model of the most common IDH2 mutation in human AML.
Experiments conducted with this model revealed that mutant IDH2 contributes to leukemia initiation and is required for the maintenance of leukemic cells in a living organism.
The researchers said these findings, published in Cell Stem Cell, confirm a potent oncogenic role for IDH2 and support its relevance as a therapeutic target for AML.
Furthermore, the model can be used to evaluate the pharmacological efficacy of IDH2 inhibitors, either alone or in combination with other compounds.
“The real hope is that we would one day be able to treat IDH2-mutant leukemia patients with a drug that targets this genetic abnormality,” said senior study author Pier Paolo Pandolfi, MD, PhD, of Beth Israel Deaconess Medical Center (BIDMC) in Boston.
He and his colleagues knew that IDH1 and IDH2 proteins are critical enzymes in the TCA cycle, which is centrally important to many biochemical pathways. Mutated forms of these proteins gain a novel ability to produce 2-hydroxyglutarate (2HG), a metabolite that has been shown to accumulate at high levels in cancer patients.
“Our goal was to generate an animal model of mutant IDH that was both inducible and reversible,” said Markus Reschke, PhD, also of BIDMC.
“This enabled us to address an important unanswered question: Does inhibition of mutant IDH proteins in active disease have an effect on tumor maintenance or progression in a living organism?”
The researchers studied 2 different models: a retroviral transduction model and a genetically engineered model in which IDH mice were crossed with mice harboring other leukemia-relevant mutations.
In the first model, the IDH mutation was combined with the oncogenes HoxA9 and Meis1a, 2 downstream targets of numerous pathways that are deregulated in AML.
The results showed evidence of differentiation within 2 weeks of genetic deinduction of mutant IDH. And 2 weeks later, 6 of 8 animals showed complete remission with elimination of any detectable leukemic cells.
The researchers said these results were both surprising and encouraging, demonstrating a situation in which IDH mutation occurs as an early event, and leukemic transformation occurs as a result of subsequent genetic hits.
“The retroviral model enabled us to observe that mutant IDH2 is essential for the maintenance of HoxA9/Meis1a-induced AML,” said Lev Kats, PhD, of BIDMC. “But this was still a surrogate model. This isn’t what happens in human patients, per se.”
The researchers therefore went on to develop a transgenic model that more closely recapitulates the genetics of human AML.
“By crossing the mutant IDH2 animals with other leukemia-relevant mutations, including mutations in the FMS-like tyrosine kinase 3 [FLT3], we observed that compound-mutant animals developed acute leukemias,” Dr Reschke said. “This exciting finding told us that mutant IDH2 contributes to leukemia initiation in vivo.”
As with the retroviral transduction model, genetic deinduction of mutant IDH2 in the context of a cooperating FLT3 mutation resulted in reduced proliferation and/or differentiation of leukemic cells, further demonstrating that mutant IDH2 expression is required for leukemia maintenance.
“This model has validated mutant IDH proteins as very strong candidates for continued development of targeted anticancer therapeutics,” Dr Pandolfi said. “The model will also be of paramount importance to study mechanisms of resistance to treatment that may occur.” ![]()
Studies have suggested that mutations in isocitrate dehydrogenase-1 and 2 (IDH1 and IDH2) are present in approximately 20% of all acute myeloid leukemias (AMLs), and this implies that mutant IDH proteins are attractive drug targets.
With this in mind, a group of scientists generated a transgenic mouse model of the most common IDH2 mutation in human AML.
Experiments conducted with this model revealed that mutant IDH2 contributes to leukemia initiation and is required for the maintenance of leukemic cells in a living organism.
The researchers said these findings, published in Cell Stem Cell, confirm a potent oncogenic role for IDH2 and support its relevance as a therapeutic target for AML.
Furthermore, the model can be used to evaluate the pharmacological efficacy of IDH2 inhibitors, either alone or in combination with other compounds.
“The real hope is that we would one day be able to treat IDH2-mutant leukemia patients with a drug that targets this genetic abnormality,” said senior study author Pier Paolo Pandolfi, MD, PhD, of Beth Israel Deaconess Medical Center (BIDMC) in Boston.
He and his colleagues knew that IDH1 and IDH2 proteins are critical enzymes in the TCA cycle, which is centrally important to many biochemical pathways. Mutated forms of these proteins gain a novel ability to produce 2-hydroxyglutarate (2HG), a metabolite that has been shown to accumulate at high levels in cancer patients.
“Our goal was to generate an animal model of mutant IDH that was both inducible and reversible,” said Markus Reschke, PhD, also of BIDMC.
“This enabled us to address an important unanswered question: Does inhibition of mutant IDH proteins in active disease have an effect on tumor maintenance or progression in a living organism?”
The researchers studied 2 different models: a retroviral transduction model and a genetically engineered model in which IDH mice were crossed with mice harboring other leukemia-relevant mutations.
In the first model, the IDH mutation was combined with the oncogenes HoxA9 and Meis1a, 2 downstream targets of numerous pathways that are deregulated in AML.
The results showed evidence of differentiation within 2 weeks of genetic deinduction of mutant IDH. And 2 weeks later, 6 of 8 animals showed complete remission with elimination of any detectable leukemic cells.
The researchers said these results were both surprising and encouraging, demonstrating a situation in which IDH mutation occurs as an early event, and leukemic transformation occurs as a result of subsequent genetic hits.
“The retroviral model enabled us to observe that mutant IDH2 is essential for the maintenance of HoxA9/Meis1a-induced AML,” said Lev Kats, PhD, of BIDMC. “But this was still a surrogate model. This isn’t what happens in human patients, per se.”
The researchers therefore went on to develop a transgenic model that more closely recapitulates the genetics of human AML.
“By crossing the mutant IDH2 animals with other leukemia-relevant mutations, including mutations in the FMS-like tyrosine kinase 3 [FLT3], we observed that compound-mutant animals developed acute leukemias,” Dr Reschke said. “This exciting finding told us that mutant IDH2 contributes to leukemia initiation in vivo.”
As with the retroviral transduction model, genetic deinduction of mutant IDH2 in the context of a cooperating FLT3 mutation resulted in reduced proliferation and/or differentiation of leukemic cells, further demonstrating that mutant IDH2 expression is required for leukemia maintenance.
“This model has validated mutant IDH proteins as very strong candidates for continued development of targeted anticancer therapeutics,” Dr Pandolfi said. “The model will also be of paramount importance to study mechanisms of resistance to treatment that may occur.” ![]()
Mutation could be target for MDS/AML treatment
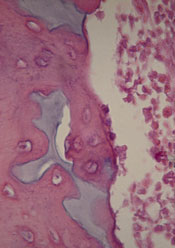
Scientists have found evidence to suggest that a genetic alteration in osteoblasts can induce acute myeloid leukemia (AML).
And this provides a potential therapeutic target for AML and myelodysplastic syndromes (MDS).
Stavroula Kousteni, PhD, of Columbia University Medical Center in New York, and her colleagues described these findings in Nature.
The researchers discovered that an activating mutation of beta-catenin in mouse osteoblasts induces AML.
This mutation leads to cancer in adjacent hematopoietic stem cells (HSCs) through a series of events. First, the mutated beta-catenin protein moves from its normal location on the exterior of the osteoblast to the cell’s nucleus, where it activates production of the protein jagged1.
Jagged1 proteins are then shipped to the osteoblast’s exterior membrane, where they can bind to Notch proteins—which activate signaling pathways—on neighboring HSCs. When this happens, Notch transmits signals inside the HSCs that enable leukemic transformation.
To confirm the role of jagged1 in AML development, the investigators removed 1 allele of jagged1 in osteoblasts. This decreased Notch signaling in Lin-Sca+c-Kit+ cells, rescued anemia and deregulation of HSC lineage differentiation, and prevented AML development.
The researchers then evaluated the effects of blocking Notch signaling using a gamma-secretase inhibitor. The treatment reversed hematopoietic deregulation and myeloid expansion in the blood, marrow, and spleens of the mice and reversed their AML.
“If the [process] works the same way in humans, our study suggests practical ways that we may be able to intervene with a drug or an antibody,” Dr Kousteni said.
With this in mind, she and her colleagues analyzed cells from 107 patients with AML or MDS. About 38% of the patients had changes in beta-catenin, jagged1, and Notch signaling that mirrored the changes in the mice. But none of the 56 healthy control subjects studied had these changes.
The investigators therefore concluded that these findings provide new insight into AML/MDS pathogenesis and may have implications for treatment. ![]()
Scientists have found evidence to suggest that a genetic alteration in osteoblasts can induce acute myeloid leukemia (AML).
And this provides a potential therapeutic target for AML and myelodysplastic syndromes (MDS).
Stavroula Kousteni, PhD, of Columbia University Medical Center in New York, and her colleagues described these findings in Nature.
The researchers discovered that an activating mutation of beta-catenin in mouse osteoblasts induces AML.
This mutation leads to cancer in adjacent hematopoietic stem cells (HSCs) through a series of events. First, the mutated beta-catenin protein moves from its normal location on the exterior of the osteoblast to the cell’s nucleus, where it activates production of the protein jagged1.
Jagged1 proteins are then shipped to the osteoblast’s exterior membrane, where they can bind to Notch proteins—which activate signaling pathways—on neighboring HSCs. When this happens, Notch transmits signals inside the HSCs that enable leukemic transformation.
To confirm the role of jagged1 in AML development, the investigators removed 1 allele of jagged1 in osteoblasts. This decreased Notch signaling in Lin-Sca+c-Kit+ cells, rescued anemia and deregulation of HSC lineage differentiation, and prevented AML development.
The researchers then evaluated the effects of blocking Notch signaling using a gamma-secretase inhibitor. The treatment reversed hematopoietic deregulation and myeloid expansion in the blood, marrow, and spleens of the mice and reversed their AML.
“If the [process] works the same way in humans, our study suggests practical ways that we may be able to intervene with a drug or an antibody,” Dr Kousteni said.
With this in mind, she and her colleagues analyzed cells from 107 patients with AML or MDS. About 38% of the patients had changes in beta-catenin, jagged1, and Notch signaling that mirrored the changes in the mice. But none of the 56 healthy control subjects studied had these changes.
The investigators therefore concluded that these findings provide new insight into AML/MDS pathogenesis and may have implications for treatment. ![]()
Scientists have found evidence to suggest that a genetic alteration in osteoblasts can induce acute myeloid leukemia (AML).
And this provides a potential therapeutic target for AML and myelodysplastic syndromes (MDS).
Stavroula Kousteni, PhD, of Columbia University Medical Center in New York, and her colleagues described these findings in Nature.
The researchers discovered that an activating mutation of beta-catenin in mouse osteoblasts induces AML.
This mutation leads to cancer in adjacent hematopoietic stem cells (HSCs) through a series of events. First, the mutated beta-catenin protein moves from its normal location on the exterior of the osteoblast to the cell’s nucleus, where it activates production of the protein jagged1.
Jagged1 proteins are then shipped to the osteoblast’s exterior membrane, where they can bind to Notch proteins—which activate signaling pathways—on neighboring HSCs. When this happens, Notch transmits signals inside the HSCs that enable leukemic transformation.
To confirm the role of jagged1 in AML development, the investigators removed 1 allele of jagged1 in osteoblasts. This decreased Notch signaling in Lin-Sca+c-Kit+ cells, rescued anemia and deregulation of HSC lineage differentiation, and prevented AML development.
The researchers then evaluated the effects of blocking Notch signaling using a gamma-secretase inhibitor. The treatment reversed hematopoietic deregulation and myeloid expansion in the blood, marrow, and spleens of the mice and reversed their AML.
“If the [process] works the same way in humans, our study suggests practical ways that we may be able to intervene with a drug or an antibody,” Dr Kousteni said.
With this in mind, she and her colleagues analyzed cells from 107 patients with AML or MDS. About 38% of the patients had changes in beta-catenin, jagged1, and Notch signaling that mirrored the changes in the mice. But none of the 56 healthy control subjects studied had these changes.
The investigators therefore concluded that these findings provide new insight into AML/MDS pathogenesis and may have implications for treatment. ![]()
AML scoring system could optimize treatment

Credit: NIGMS
A scoring system that combines genetic and epigenetic changes could help guide therapy for acute myeloid leukemia (AML), according to a study published in the Journal of Clinical Oncology.
The score is based on the presence of 7 mutated genes and DNA methylation.
For each of these genes, lower expression and higher DNA methylation were associated with better patient outcomes.
The investigators therefore believe this scoring system could guide treatment by identifying novel subsets of patients.
“To date, disease classification and prognostication for AML patients have been based largely on chromosomal and genetic markers,” said principal investigator Clara D. Bloomfield, MD, of The Ohio State University in Columbus.
“Epigenetic changes that affect gene expression have not been considered. Here, we show that epigenetic changes in previously recognized and prognostically important mutated genes can identify novel patient subgroups, which might better help guide therapy.”
Creating the score
Dr Bloomfield and her colleagues identified the 7-gene panel in 134 patients who were 60 and older, had cytogenetically normal AML (CN-AML), and had been treated on Cancer and Leukemia Group B/Alliance clinical trials.
The investigators used next-generation sequencing to analyze regions of methylated DNA associated with prognostically important genetic mutations. The 7 genes they identified are CD34, RHOC, SCRN1, F2RL1, FAM92A1, MIR155HG, and VWA8.
The team then developed a summary score based on the number of genes in the panel showing high expression.
And they applied the unweighted score to 126 of the aforementioned patients. Individuals with 1 or no highly expressed genes had a 96% complete remission (CR) rate, a 3-year disease-free survival (DFS) rate of 32%, and a 3-year overall survival (OS) rate of 39%.
Patients with 6 to 7 highly expressed genes, on the other hand, had a 25% CR rate, a 3-year DFS of 0%, and a 3-year OS of 4%.
Validating the system
The investigators also tested the score in 4 validation cohorts: older patients (age 60 and up) with primary AML (n=72), younger patients (59 and under) with primary AML (n=134), older patients with CN-AML (n=65), and younger patients with CN-AML (n=84).
“In both younger and older patients, those who had no highly expressed genes, or had one highly expressed gene, had the best outcomes,” said study author Guido Marcucci, MD, of The Ohio State University Comprehensive Cancer Center.
For the younger patients (with primary or CN-AML), individuals with 1 or no highly expressed genes had a 91% to 100% CR rate, a 3-year DFS of 60% to 65%, and a 3-year OS of 76% to 82%.
But younger patients with 6 to 7 highly expressed genes had a 53% to 71% CR rate, a 3-year DFS of 13% to 17%, and a 3-year OS of 7% to 24%.
For the older patients, individuals with 1 or no highly expressed genes had a 69% to 89% CR rate, a 3-year DFS of 42% (CN-AML only), and a 3-year OS of 44% to 46%.
Older patients with 6 to 7 highly expressed genes had a 50% CR rate (both types of AML), a 3-year DFS of 0% (CN-AML only), and a 3-year OS of 10% to 12%. DFS data were not evaluable for the older patients with primary AML due to the small sample size.
“Overall, our findings suggest that the unweighted summary score is a better model compared with all other prognostic markers and previously reported gene-expression profiles,” Dr Bloomfield concluded. ![]()
Credit: NIGMS
A scoring system that combines genetic and epigenetic changes could help guide therapy for acute myeloid leukemia (AML), according to a study published in the Journal of Clinical Oncology.
The score is based on the presence of 7 mutated genes and DNA methylation.
For each of these genes, lower expression and higher DNA methylation were associated with better patient outcomes.
The investigators therefore believe this scoring system could guide treatment by identifying novel subsets of patients.
“To date, disease classification and prognostication for AML patients have been based largely on chromosomal and genetic markers,” said principal investigator Clara D. Bloomfield, MD, of The Ohio State University in Columbus.
“Epigenetic changes that affect gene expression have not been considered. Here, we show that epigenetic changes in previously recognized and prognostically important mutated genes can identify novel patient subgroups, which might better help guide therapy.”
Creating the score
Dr Bloomfield and her colleagues identified the 7-gene panel in 134 patients who were 60 and older, had cytogenetically normal AML (CN-AML), and had been treated on Cancer and Leukemia Group B/Alliance clinical trials.
The investigators used next-generation sequencing to analyze regions of methylated DNA associated with prognostically important genetic mutations. The 7 genes they identified are CD34, RHOC, SCRN1, F2RL1, FAM92A1, MIR155HG, and VWA8.
The team then developed a summary score based on the number of genes in the panel showing high expression.
And they applied the unweighted score to 126 of the aforementioned patients. Individuals with 1 or no highly expressed genes had a 96% complete remission (CR) rate, a 3-year disease-free survival (DFS) rate of 32%, and a 3-year overall survival (OS) rate of 39%.
Patients with 6 to 7 highly expressed genes, on the other hand, had a 25% CR rate, a 3-year DFS of 0%, and a 3-year OS of 4%.
Validating the system
The investigators also tested the score in 4 validation cohorts: older patients (age 60 and up) with primary AML (n=72), younger patients (59 and under) with primary AML (n=134), older patients with CN-AML (n=65), and younger patients with CN-AML (n=84).
“In both younger and older patients, those who had no highly expressed genes, or had one highly expressed gene, had the best outcomes,” said study author Guido Marcucci, MD, of The Ohio State University Comprehensive Cancer Center.
For the younger patients (with primary or CN-AML), individuals with 1 or no highly expressed genes had a 91% to 100% CR rate, a 3-year DFS of 60% to 65%, and a 3-year OS of 76% to 82%.
But younger patients with 6 to 7 highly expressed genes had a 53% to 71% CR rate, a 3-year DFS of 13% to 17%, and a 3-year OS of 7% to 24%.
For the older patients, individuals with 1 or no highly expressed genes had a 69% to 89% CR rate, a 3-year DFS of 42% (CN-AML only), and a 3-year OS of 44% to 46%.
Older patients with 6 to 7 highly expressed genes had a 50% CR rate (both types of AML), a 3-year DFS of 0% (CN-AML only), and a 3-year OS of 10% to 12%. DFS data were not evaluable for the older patients with primary AML due to the small sample size.
“Overall, our findings suggest that the unweighted summary score is a better model compared with all other prognostic markers and previously reported gene-expression profiles,” Dr Bloomfield concluded. ![]()
Credit: NIGMS
A scoring system that combines genetic and epigenetic changes could help guide therapy for acute myeloid leukemia (AML), according to a study published in the Journal of Clinical Oncology.
The score is based on the presence of 7 mutated genes and DNA methylation.
For each of these genes, lower expression and higher DNA methylation were associated with better patient outcomes.
The investigators therefore believe this scoring system could guide treatment by identifying novel subsets of patients.
“To date, disease classification and prognostication for AML patients have been based largely on chromosomal and genetic markers,” said principal investigator Clara D. Bloomfield, MD, of The Ohio State University in Columbus.
“Epigenetic changes that affect gene expression have not been considered. Here, we show that epigenetic changes in previously recognized and prognostically important mutated genes can identify novel patient subgroups, which might better help guide therapy.”
Creating the score
Dr Bloomfield and her colleagues identified the 7-gene panel in 134 patients who were 60 and older, had cytogenetically normal AML (CN-AML), and had been treated on Cancer and Leukemia Group B/Alliance clinical trials.
The investigators used next-generation sequencing to analyze regions of methylated DNA associated with prognostically important genetic mutations. The 7 genes they identified are CD34, RHOC, SCRN1, F2RL1, FAM92A1, MIR155HG, and VWA8.
The team then developed a summary score based on the number of genes in the panel showing high expression.
And they applied the unweighted score to 126 of the aforementioned patients. Individuals with 1 or no highly expressed genes had a 96% complete remission (CR) rate, a 3-year disease-free survival (DFS) rate of 32%, and a 3-year overall survival (OS) rate of 39%.
Patients with 6 to 7 highly expressed genes, on the other hand, had a 25% CR rate, a 3-year DFS of 0%, and a 3-year OS of 4%.
Validating the system
The investigators also tested the score in 4 validation cohorts: older patients (age 60 and up) with primary AML (n=72), younger patients (59 and under) with primary AML (n=134), older patients with CN-AML (n=65), and younger patients with CN-AML (n=84).
“In both younger and older patients, those who had no highly expressed genes, or had one highly expressed gene, had the best outcomes,” said study author Guido Marcucci, MD, of The Ohio State University Comprehensive Cancer Center.
For the younger patients (with primary or CN-AML), individuals with 1 or no highly expressed genes had a 91% to 100% CR rate, a 3-year DFS of 60% to 65%, and a 3-year OS of 76% to 82%.
But younger patients with 6 to 7 highly expressed genes had a 53% to 71% CR rate, a 3-year DFS of 13% to 17%, and a 3-year OS of 7% to 24%.
For the older patients, individuals with 1 or no highly expressed genes had a 69% to 89% CR rate, a 3-year DFS of 42% (CN-AML only), and a 3-year OS of 44% to 46%.
Older patients with 6 to 7 highly expressed genes had a 50% CR rate (both types of AML), a 3-year DFS of 0% (CN-AML only), and a 3-year OS of 10% to 12%. DFS data were not evaluable for the older patients with primary AML due to the small sample size.
“Overall, our findings suggest that the unweighted summary score is a better model compared with all other prognostic markers and previously reported gene-expression profiles,” Dr Bloomfield concluded. ![]()
Lower-dose quizartinib diminishes QT events
NEW ORLEANS – Lower doses of quizartinib reduced worrisome QT-interval prolongation events without a loss of efficacy in patients with FLT3-ITD–positive relapsed or refractory acute myeloid leukemia, a phase II study shows.
In a 76-patient study, grade 2 QT-interval prolongation (QTcF) of more than 480-500 msec occurred in two patients (5%) on oral quizartinib 30 mg/day and in five patients (14%) on 60 mg/day, with no differences between groups in QTcF events of more than 500 msec (5% vs. 3%).
In addition, an increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on the 60-mg dose and in 3% of those on the 30-mg dose, Dr. Jorge Cortes reported at the annual meeting of the American Society of Hematology.
About a third of patients with acute myeloid leukemia (AML) will have FLT3 internal tandem duplications (FLT3-ITD), which are associated with early relapse and poor survival in AML. Quizartinib has shown the highest single-agent activity among FMS-like tyrosine kinase 3 (FLT3)-targeted agents in this population, according to Dr. Cortes.
At last year’s ASH meeting, investigators presented results from a phase II study in which the investigational agent elicited responses in both FLT3-positive and -negative relapsed/refractory AML. The unprecedented results at doses of 90, 135, and 200 mg were partially eclipsed, however, by respective 46%, 39%, and 92% increases in QTcF from baseline of more than 60 msec, noted Dr. Cortes, chair of the AML section, department of leukemia, University of Texas M.D. Anderson Cancer Center, Houston.
The current study randomized 76 patients to quizartinib 30 mg or 60 mg continuous daily dosing for primary AML or AML secondary to myelodysplastic syndrome that relapsed or was refractory to first-line salvage therapy or prior hematopoietic stem cell transplantation. The coprimary endpoints were rate of grade 2 QTc prolongation and the composite complete remission rate, which included complete remission (CR), CR with incomplete platelet recovery, and CR with incomplete hematologic recovery.
In all, 92% of patients had FLT3 internal tandem duplications, and 58 of 60 evaluable patients had intermediate or poor cytogenetic risk. Their mean age was 55 years. Two patients were randomized but not treated.
Treatment with the 30-mg and 60-mg doses resulted in a composite CR rate of 47%, Dr. Cortes said. The median duration of response was 4.1 weeks in the 30-mg group and 20 weeks in the 60-mg group.
Two patients (5%) on the 30-mg dose and 1 patient (3%) on the 60-mg dose achieved CR; 1 patient (3%) in the 60-mg group had a CR with incomplete platelet recovery; and 16 patients (42%) in each arm had a CR with incomplete hematologic recovery.
Partial responses were also seen in 5 patients (13%) in the 30-mg group and 9 (24%) in the 60-mg group.
These results compare favorably with composite CR rates of 47%, 45%, and 42% with the 90-, 135-, and 200-mg doses used in the earlier study, Dr. Cortes observed.
Median overall survival in the current study was 20.7 weeks in the lower-dose group and 25.4 weeks with the 60-mg dose.
Importantly, 34% of patients were successfully bridged to transplant, extending median survival to 31 weeks for those on 30 mg of quizartinib and to 28.1 weeks for those given 60 mg.
"This study demonstrates there is certainly sustained efficacy with these lower doses of quizartinib and a decreased QT signal at doses of 30 and 60 mg compared with the higher doses we’ve tested in the past," Dr. Cortes concluded.
Grade 3/4 adverse events were mainly anemia (39%) in the 30-mg group and febrile neutropenia (36%) in the 60-mg group. Three patients required dose reductions due to QTc prolongation.
A global phase III randomized study of quizartinib in FLT3-ITD–positive patients in first relapse is planned to start in early 2014, he said.
Development of quizartinib has been somewhat rocky, with Astellas Pharma announcing in March 2013 it was ending its collaboration with Ambit Biosciences to develop FLT3 inhibitors including quizartinib.
Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
NEW ORLEANS – Lower doses of quizartinib reduced worrisome QT-interval prolongation events without a loss of efficacy in patients with FLT3-ITD–positive relapsed or refractory acute myeloid leukemia, a phase II study shows.
In a 76-patient study, grade 2 QT-interval prolongation (QTcF) of more than 480-500 msec occurred in two patients (5%) on oral quizartinib 30 mg/day and in five patients (14%) on 60 mg/day, with no differences between groups in QTcF events of more than 500 msec (5% vs. 3%).
In addition, an increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on the 60-mg dose and in 3% of those on the 30-mg dose, Dr. Jorge Cortes reported at the annual meeting of the American Society of Hematology.
About a third of patients with acute myeloid leukemia (AML) will have FLT3 internal tandem duplications (FLT3-ITD), which are associated with early relapse and poor survival in AML. Quizartinib has shown the highest single-agent activity among FMS-like tyrosine kinase 3 (FLT3)-targeted agents in this population, according to Dr. Cortes.
At last year’s ASH meeting, investigators presented results from a phase II study in which the investigational agent elicited responses in both FLT3-positive and -negative relapsed/refractory AML. The unprecedented results at doses of 90, 135, and 200 mg were partially eclipsed, however, by respective 46%, 39%, and 92% increases in QTcF from baseline of more than 60 msec, noted Dr. Cortes, chair of the AML section, department of leukemia, University of Texas M.D. Anderson Cancer Center, Houston.
The current study randomized 76 patients to quizartinib 30 mg or 60 mg continuous daily dosing for primary AML or AML secondary to myelodysplastic syndrome that relapsed or was refractory to first-line salvage therapy or prior hematopoietic stem cell transplantation. The coprimary endpoints were rate of grade 2 QTc prolongation and the composite complete remission rate, which included complete remission (CR), CR with incomplete platelet recovery, and CR with incomplete hematologic recovery.
In all, 92% of patients had FLT3 internal tandem duplications, and 58 of 60 evaluable patients had intermediate or poor cytogenetic risk. Their mean age was 55 years. Two patients were randomized but not treated.
Treatment with the 30-mg and 60-mg doses resulted in a composite CR rate of 47%, Dr. Cortes said. The median duration of response was 4.1 weeks in the 30-mg group and 20 weeks in the 60-mg group.
Two patients (5%) on the 30-mg dose and 1 patient (3%) on the 60-mg dose achieved CR; 1 patient (3%) in the 60-mg group had a CR with incomplete platelet recovery; and 16 patients (42%) in each arm had a CR with incomplete hematologic recovery.
Partial responses were also seen in 5 patients (13%) in the 30-mg group and 9 (24%) in the 60-mg group.
These results compare favorably with composite CR rates of 47%, 45%, and 42% with the 90-, 135-, and 200-mg doses used in the earlier study, Dr. Cortes observed.
Median overall survival in the current study was 20.7 weeks in the lower-dose group and 25.4 weeks with the 60-mg dose.
Importantly, 34% of patients were successfully bridged to transplant, extending median survival to 31 weeks for those on 30 mg of quizartinib and to 28.1 weeks for those given 60 mg.
"This study demonstrates there is certainly sustained efficacy with these lower doses of quizartinib and a decreased QT signal at doses of 30 and 60 mg compared with the higher doses we’ve tested in the past," Dr. Cortes concluded.
Grade 3/4 adverse events were mainly anemia (39%) in the 30-mg group and febrile neutropenia (36%) in the 60-mg group. Three patients required dose reductions due to QTc prolongation.
A global phase III randomized study of quizartinib in FLT3-ITD–positive patients in first relapse is planned to start in early 2014, he said.
Development of quizartinib has been somewhat rocky, with Astellas Pharma announcing in March 2013 it was ending its collaboration with Ambit Biosciences to develop FLT3 inhibitors including quizartinib.
Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
NEW ORLEANS – Lower doses of quizartinib reduced worrisome QT-interval prolongation events without a loss of efficacy in patients with FLT3-ITD–positive relapsed or refractory acute myeloid leukemia, a phase II study shows.
In a 76-patient study, grade 2 QT-interval prolongation (QTcF) of more than 480-500 msec occurred in two patients (5%) on oral quizartinib 30 mg/day and in five patients (14%) on 60 mg/day, with no differences between groups in QTcF events of more than 500 msec (5% vs. 3%).
In addition, an increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on the 60-mg dose and in 3% of those on the 30-mg dose, Dr. Jorge Cortes reported at the annual meeting of the American Society of Hematology.
About a third of patients with acute myeloid leukemia (AML) will have FLT3 internal tandem duplications (FLT3-ITD), which are associated with early relapse and poor survival in AML. Quizartinib has shown the highest single-agent activity among FMS-like tyrosine kinase 3 (FLT3)-targeted agents in this population, according to Dr. Cortes.
At last year’s ASH meeting, investigators presented results from a phase II study in which the investigational agent elicited responses in both FLT3-positive and -negative relapsed/refractory AML. The unprecedented results at doses of 90, 135, and 200 mg were partially eclipsed, however, by respective 46%, 39%, and 92% increases in QTcF from baseline of more than 60 msec, noted Dr. Cortes, chair of the AML section, department of leukemia, University of Texas M.D. Anderson Cancer Center, Houston.
The current study randomized 76 patients to quizartinib 30 mg or 60 mg continuous daily dosing for primary AML or AML secondary to myelodysplastic syndrome that relapsed or was refractory to first-line salvage therapy or prior hematopoietic stem cell transplantation. The coprimary endpoints were rate of grade 2 QTc prolongation and the composite complete remission rate, which included complete remission (CR), CR with incomplete platelet recovery, and CR with incomplete hematologic recovery.
In all, 92% of patients had FLT3 internal tandem duplications, and 58 of 60 evaluable patients had intermediate or poor cytogenetic risk. Their mean age was 55 years. Two patients were randomized but not treated.
Treatment with the 30-mg and 60-mg doses resulted in a composite CR rate of 47%, Dr. Cortes said. The median duration of response was 4.1 weeks in the 30-mg group and 20 weeks in the 60-mg group.
Two patients (5%) on the 30-mg dose and 1 patient (3%) on the 60-mg dose achieved CR; 1 patient (3%) in the 60-mg group had a CR with incomplete platelet recovery; and 16 patients (42%) in each arm had a CR with incomplete hematologic recovery.
Partial responses were also seen in 5 patients (13%) in the 30-mg group and 9 (24%) in the 60-mg group.
These results compare favorably with composite CR rates of 47%, 45%, and 42% with the 90-, 135-, and 200-mg doses used in the earlier study, Dr. Cortes observed.
Median overall survival in the current study was 20.7 weeks in the lower-dose group and 25.4 weeks with the 60-mg dose.
Importantly, 34% of patients were successfully bridged to transplant, extending median survival to 31 weeks for those on 30 mg of quizartinib and to 28.1 weeks for those given 60 mg.
"This study demonstrates there is certainly sustained efficacy with these lower doses of quizartinib and a decreased QT signal at doses of 30 and 60 mg compared with the higher doses we’ve tested in the past," Dr. Cortes concluded.
Grade 3/4 adverse events were mainly anemia (39%) in the 30-mg group and febrile neutropenia (36%) in the 60-mg group. Three patients required dose reductions due to QTc prolongation.
A global phase III randomized study of quizartinib in FLT3-ITD–positive patients in first relapse is planned to start in early 2014, he said.
Development of quizartinib has been somewhat rocky, with Astellas Pharma announcing in March 2013 it was ending its collaboration with Ambit Biosciences to develop FLT3 inhibitors including quizartinib.
Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
AT ASH 2013
Major finding: An increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on quizartinib 60 mg and in 3% of those on 30 mg.
Data source: A prospective phase II study of 76 patients with relapsed/refractory AML.
Disclosures: Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
FDA provides steps to obtain ponatinib following suspension
The Food and Drug Administration has provided information on how health care professionals can ensure that patients who have benefitted from the leukemia drug ponatinib continue to have access to the treatment.
Less than a week after the agency announced that marketing and sales of the drug had been suspended because of the risk of life-threatening blood clots and severe narrowing of blood vessels associated with treatment, the FDA posted instructions on how to obtain emergency access to ponatinib through an Investigational New Drug (IND) application. This process entails contacting the FDA to obtain an emergency IND number for each patient who would benefit from continuing treatment and providing the manufacturer with that number to get ponatinib (Iclusig).
Health care professionals can obtain INDs for multiple patients during one phone call and should contact the agency at least 48-72 hours before the drug is needed, according to an FDA announcement.
The statement noted that health care professionals "may continue to use Iclusig for patients who they determine are responding to the drug and for whom the potential benefits outweigh the risks."
Ponatinib is a kinase inhibitor marketed by ARIAD Pharmaceuticals. It was approved in December 2012 for treating chronic myeloid leukemia and Philadelphia chromosome–positive acute lymphoblastic leukemia in adults.
Serious adverse events associated with ponatinib should be reported online to the FDA or by phone at 800-332-0178. Information about the IND program is available online. The FDA’s emergency IND telephone number is 301-796-7550 (between 8 a.m. and 4:30 p.m. EST, weekdays and 866-300-4374 after 4:30 p.m. EST. More information on how to obtain an IND for patients on ponatinib is available at http://www.fda.gov/Drugs/DrugSafety/ucm373040.htm.
The Food and Drug Administration has provided information on how health care professionals can ensure that patients who have benefitted from the leukemia drug ponatinib continue to have access to the treatment.
Less than a week after the agency announced that marketing and sales of the drug had been suspended because of the risk of life-threatening blood clots and severe narrowing of blood vessels associated with treatment, the FDA posted instructions on how to obtain emergency access to ponatinib through an Investigational New Drug (IND) application. This process entails contacting the FDA to obtain an emergency IND number for each patient who would benefit from continuing treatment and providing the manufacturer with that number to get ponatinib (Iclusig).
Health care professionals can obtain INDs for multiple patients during one phone call and should contact the agency at least 48-72 hours before the drug is needed, according to an FDA announcement.
The statement noted that health care professionals "may continue to use Iclusig for patients who they determine are responding to the drug and for whom the potential benefits outweigh the risks."
Ponatinib is a kinase inhibitor marketed by ARIAD Pharmaceuticals. It was approved in December 2012 for treating chronic myeloid leukemia and Philadelphia chromosome–positive acute lymphoblastic leukemia in adults.
Serious adverse events associated with ponatinib should be reported online to the FDA or by phone at 800-332-0178. Information about the IND program is available online. The FDA’s emergency IND telephone number is 301-796-7550 (between 8 a.m. and 4:30 p.m. EST, weekdays and 866-300-4374 after 4:30 p.m. EST. More information on how to obtain an IND for patients on ponatinib is available at http://www.fda.gov/Drugs/DrugSafety/ucm373040.htm.
The Food and Drug Administration has provided information on how health care professionals can ensure that patients who have benefitted from the leukemia drug ponatinib continue to have access to the treatment.
Less than a week after the agency announced that marketing and sales of the drug had been suspended because of the risk of life-threatening blood clots and severe narrowing of blood vessels associated with treatment, the FDA posted instructions on how to obtain emergency access to ponatinib through an Investigational New Drug (IND) application. This process entails contacting the FDA to obtain an emergency IND number for each patient who would benefit from continuing treatment and providing the manufacturer with that number to get ponatinib (Iclusig).
Health care professionals can obtain INDs for multiple patients during one phone call and should contact the agency at least 48-72 hours before the drug is needed, according to an FDA announcement.
The statement noted that health care professionals "may continue to use Iclusig for patients who they determine are responding to the drug and for whom the potential benefits outweigh the risks."
Ponatinib is a kinase inhibitor marketed by ARIAD Pharmaceuticals. It was approved in December 2012 for treating chronic myeloid leukemia and Philadelphia chromosome–positive acute lymphoblastic leukemia in adults.
Serious adverse events associated with ponatinib should be reported online to the FDA or by phone at 800-332-0178. Information about the IND program is available online. The FDA’s emergency IND telephone number is 301-796-7550 (between 8 a.m. and 4:30 p.m. EST, weekdays and 866-300-4374 after 4:30 p.m. EST. More information on how to obtain an IND for patients on ponatinib is available at http://www.fda.gov/Drugs/DrugSafety/ucm373040.htm.
Cord blood transplants improve juvenile myelomonocytic leukemia survival
Umbilical cord blood transplantation from unrelated donors offers a reasonable chance of cure for children with juvenile myelomonocytic leukemia, particularly in those diagnosed before age 2 years and where there is greater immunologic compatibility between donor and recipient.
A retrospective analysis of data from 110 children with juvenile myelomonocytic leukemia given single-unit, unrelated donor umbilical cord blood transplantation (UCBT) showed a 5-year disease-free survival rate of 44%, according to data published in Blood (2013;122:2135-41 [doi:10.1182/blood-2013-03-491589]).
"Our data document that a significant proportion of children with this disease, especially when receiving transplants from donors with limited HLA [human leukocyte antigens] disparity, can be cured with UCBT, thus indicating that this allograft can represent a suitable option for children with juvenile myelomonocytic leukemia lacking either a related donor or [an] unrelated donor of hematopoietic stem cells," wrote Dr. Franco Locatelli from the University of Pavia, Rome, and his colleagues.
"Compared with bone marrow transplantation, advantages of UCBT are represented by lower incidence and severity of graft-versus-host disease, easier procurement, and prompter availability of cord blood, and the possibility of using donors showing HLA disparities with the recipient," they said.
Patients’ median age at diagnosis was 1.4 years, and at transplantation was 2.2 years. While 16% of units were HLA matched with the recipient, 43% of units had one HLA disparity and 35% had two or three HLA disparities. Data on HLA compatibility were missing in 6 patients (6%).
The study found that children aged younger than 1.4 years at diagnosis had significantly better disease-free survival rates (hazard ratio, 0.42), while those who had one or no HLA disparities with the donor also had better survival rates (HR, 0.43).
Platelet recovery was achieved in 76 children, with the median time of 44 days required to reach a platelet count greater than 20 x 109/L, and by day 180, the cumulative incidence of platelet recovery was 71% plus or minus 6%.
A total of 28 children underwent splenectomy before transplantation, 88 received chemotherapy, 8 were given a reduced-intensity conditioning regimen, and the remainder were treated with myeloablative regimens. Nineteen children also received total body irradiation.
Disease recurrence was the major cause of treatment failure, with a 5-year cumulative incidence of relapse of 33% in the median follow-up period of 64 months.
Twenty-four children died of transplant-attributable causes during the follow-up period, while 45 patients were diagnosed with grade II to IV acute graft-versus-host disease.
The researchers noted an increased risk of transplantation-related mortality among those with monosomy 7 karyotype, and those who did not receive cytotoxic treatment before transplantation.
The authors declared they had no financial conflicts of interest.
Umbilical cord blood transplantation from unrelated donors offers a reasonable chance of cure for children with juvenile myelomonocytic leukemia, particularly in those diagnosed before age 2 years and where there is greater immunologic compatibility between donor and recipient.
A retrospective analysis of data from 110 children with juvenile myelomonocytic leukemia given single-unit, unrelated donor umbilical cord blood transplantation (UCBT) showed a 5-year disease-free survival rate of 44%, according to data published in Blood (2013;122:2135-41 [doi:10.1182/blood-2013-03-491589]).
"Our data document that a significant proportion of children with this disease, especially when receiving transplants from donors with limited HLA [human leukocyte antigens] disparity, can be cured with UCBT, thus indicating that this allograft can represent a suitable option for children with juvenile myelomonocytic leukemia lacking either a related donor or [an] unrelated donor of hematopoietic stem cells," wrote Dr. Franco Locatelli from the University of Pavia, Rome, and his colleagues.
"Compared with bone marrow transplantation, advantages of UCBT are represented by lower incidence and severity of graft-versus-host disease, easier procurement, and prompter availability of cord blood, and the possibility of using donors showing HLA disparities with the recipient," they said.
Patients’ median age at diagnosis was 1.4 years, and at transplantation was 2.2 years. While 16% of units were HLA matched with the recipient, 43% of units had one HLA disparity and 35% had two or three HLA disparities. Data on HLA compatibility were missing in 6 patients (6%).
The study found that children aged younger than 1.4 years at diagnosis had significantly better disease-free survival rates (hazard ratio, 0.42), while those who had one or no HLA disparities with the donor also had better survival rates (HR, 0.43).
Platelet recovery was achieved in 76 children, with the median time of 44 days required to reach a platelet count greater than 20 x 109/L, and by day 180, the cumulative incidence of platelet recovery was 71% plus or minus 6%.
A total of 28 children underwent splenectomy before transplantation, 88 received chemotherapy, 8 were given a reduced-intensity conditioning regimen, and the remainder were treated with myeloablative regimens. Nineteen children also received total body irradiation.
Disease recurrence was the major cause of treatment failure, with a 5-year cumulative incidence of relapse of 33% in the median follow-up period of 64 months.
Twenty-four children died of transplant-attributable causes during the follow-up period, while 45 patients were diagnosed with grade II to IV acute graft-versus-host disease.
The researchers noted an increased risk of transplantation-related mortality among those with monosomy 7 karyotype, and those who did not receive cytotoxic treatment before transplantation.
The authors declared they had no financial conflicts of interest.
Umbilical cord blood transplantation from unrelated donors offers a reasonable chance of cure for children with juvenile myelomonocytic leukemia, particularly in those diagnosed before age 2 years and where there is greater immunologic compatibility between donor and recipient.
A retrospective analysis of data from 110 children with juvenile myelomonocytic leukemia given single-unit, unrelated donor umbilical cord blood transplantation (UCBT) showed a 5-year disease-free survival rate of 44%, according to data published in Blood (2013;122:2135-41 [doi:10.1182/blood-2013-03-491589]).
"Our data document that a significant proportion of children with this disease, especially when receiving transplants from donors with limited HLA [human leukocyte antigens] disparity, can be cured with UCBT, thus indicating that this allograft can represent a suitable option for children with juvenile myelomonocytic leukemia lacking either a related donor or [an] unrelated donor of hematopoietic stem cells," wrote Dr. Franco Locatelli from the University of Pavia, Rome, and his colleagues.
"Compared with bone marrow transplantation, advantages of UCBT are represented by lower incidence and severity of graft-versus-host disease, easier procurement, and prompter availability of cord blood, and the possibility of using donors showing HLA disparities with the recipient," they said.
Patients’ median age at diagnosis was 1.4 years, and at transplantation was 2.2 years. While 16% of units were HLA matched with the recipient, 43% of units had one HLA disparity and 35% had two or three HLA disparities. Data on HLA compatibility were missing in 6 patients (6%).
The study found that children aged younger than 1.4 years at diagnosis had significantly better disease-free survival rates (hazard ratio, 0.42), while those who had one or no HLA disparities with the donor also had better survival rates (HR, 0.43).
Platelet recovery was achieved in 76 children, with the median time of 44 days required to reach a platelet count greater than 20 x 109/L, and by day 180, the cumulative incidence of platelet recovery was 71% plus or minus 6%.
A total of 28 children underwent splenectomy before transplantation, 88 received chemotherapy, 8 were given a reduced-intensity conditioning regimen, and the remainder were treated with myeloablative regimens. Nineteen children also received total body irradiation.
Disease recurrence was the major cause of treatment failure, with a 5-year cumulative incidence of relapse of 33% in the median follow-up period of 64 months.
Twenty-four children died of transplant-attributable causes during the follow-up period, while 45 patients were diagnosed with grade II to IV acute graft-versus-host disease.
The researchers noted an increased risk of transplantation-related mortality among those with monosomy 7 karyotype, and those who did not receive cytotoxic treatment before transplantation.
The authors declared they had no financial conflicts of interest.
FROM BLOOD
Major finding: Children aged younger than 1.4 years at diagnosis had significantly better disease-free survival rates (HR, 0.42), while those who had one or no HLA disparities with the donor also had better survival rates (HR, 0.43).
Data source: Retrospective cohort data analysis of 110 children with juvenile myelomonocytic leukemia.
Disclosures: No financial conflicts of interest disclosed.
More Extensive Gene Profiling Urged in AML, MDS
More detailed genetic profiling of patients with acute myeloid leukemia and of those with precursor myelodysplastic syndromes is likely to improve prognostic and therapeutic decision making, according to two separate studies published online March 14 in the New England Journal of Medicine.
In one study, investigators found that the presence of DNMT3A and NPM1 mutations and MLL translocations predicted an improved outcome when patients received high-dose daunorubicin instead of the standard dose in induction chemotherapy for acute myeloid leukemia (AML).
The results suggest that "mutational profiling can be used to determine which patients will benefit from dose-intensive induction therapy," wrote Jay P. Patel of the human oncology and pathogenesis program at Memorial Sloan-Kettering Cancer Center, New York, and his associates.
In the other study, researchers reported that "nearly all" of the bone marrow cells were clonally derived in paired samples of skin and bone marrow from seven patients with myelodysplastic syndromes (MDS) and secondary AML. Founding clones and daughter subclones in all seven paired samples had recurrent gene mutations, including at least one mutation in a coding gene.
"Although clonality is not sufficient to define malignant transformation, it is a cardinal manifestation of most human cancers, and our findings suggest that the myelodysplastic syndromes and secondary AML are both highly clonal hematologic cancers," said Dr. Matthew J. Walter of the departments of internal medicine and genetics at the Siteman Cancer Center, Washington University, St. Louis, and his associates.
Mutational Analysis of Trial Results
In the first study, researchers performed a more-extensive mutational analysis than is typically done to better discriminate among patients with different prognoses.
"Previous studies have suggested that mutational analysis of [the genes] CEBPA, NPM1, and FLT3-ITD can be used to stratify risk among patients with intermediate-risk AML," wrote Mr. Patel and his colleagues.
"We hypothesized that integrated mutational analysis of all known molecular alterations occurring in more than 5% of patients with AML would allow us to identify novel molecular markers of outcome ... and to identify molecularly defined subgroups of patients who would benefit from dose-intensified induction therapy."
For DNA extraction and profiling, the investigators used diagnostic samples of bone marrow and peripheral blood from 398 patients who were participating in the phase III ECOG (Eastern Cooperative Oncology Group) E1900 clinical trial in which two doses of induction therapy were tested. They found that 97.3% of the study subjects had mutations in 18 genes, and performed extensive mutational analysis of these 18 candidate genes.
The results led them to identify three distinct risk groups. Patients with favorable genetic profiles had a 3-year overall survival of 64% and had not yet reached a median survival; those with intermediate-risk genetic profiles had a 3-year survival of 42% and a median survival of 25 months; and those with unfavorable genetic profiles had a 3-year overall survival of 12% and a median survival of 10 months.
These findings were then validated in a separate group of 104 patients from the same clinical trial. The value of the genetic risk profiles was confirmed, with the favorable, intermediate, and unfavorable profiles accurately predicting patient outcomes independently of patient age, white cell count, induction dose, transplantation status, and type of postremission therapy, Mr. Patel and his colleagues said (N. Engl. J. Med. 2012 March 14 [doi:10.1056/NEJMoa1112304]).
Moreover, the 3-year overall survival rate in patients with a mutation in the DNMT3A or NPM1 genes or a MLL translocation was 44% with high-dose chemotherapy vs. 25% with the standard dose. In patients with other genotypes, it was 35% with the high-dose regimen and 39% with the standard dose.
"These data indicate that more detailed genetic analysis may lead to improved risk stratification and identification of patients who can benefit from more intensive induction chemotherapy. The challenge is to provide genetic information in a timely and affordable way and show that this information could prospectively influence treatment decisions," they noted.
Founding MDS Clones Persist in AML
In the second study, Dr. Walter and his associates used bone marrow biopsy specimens from seven patients who progressed from MDS to AML to define changes in the proportion of clonal cells and the genetic architecture that took place during that progression.
Several genes have already been identified that show recurrent mutations during this process, "but our understanding of the total number and clonal distribution of mutations in this disease is limited," they noted.
For each subject, DNA sequences were obtained from samples of normal skin, bone marrow obtained during the MDS stage, and bone marrow obtained during the secondary AML stage, to analyze mutations. In all seven samples, the founding clones (containing 182-660 mutations) persisted in the secondary samples, while acquiring at least one new mutation predicting translational consequences.
"We have found that the proportion of neoplastic bone marrow cells is indistinguishable [between] myelodysplastic-syndrome and secondary-AML samples, suggesting that the myelodysplastic syndromes are as clonal as secondary AML," Dr. Walter and his colleagues said (N. Engl. J. Med. 2012 March 14 [doi:10.1056/NEJMoa1106968]).
There are three major clinical implications, according to the authors.
First, MDS is currently distinguished from secondary AML based on hand counting of bone marrow myeloblasts – a method prone to inaccuracy but nonetheless relied upon to drive major treatment decisions. "Ultimately, identifying the patterns of pathogenic mutations and their clonality in bone marrow samples ... should lead to greater diagnostic certainty and improved prognostic algorithms," the investigators said.
Second, the dominant AML clone was derived from a founding MDS clone in every case, suggesting that "therapies targeted to these early mutations might be the most effective strategy for eliminating disease-propagating cells and improving the rate of response to traditional chemotherapy."
Third, it is possible that progression from MDS to AML "is driven not only by the presence of recurrent mutations ... but also by the clone ([that is], founding vs. daughter) in which they arise." Combining genotyping of samples with analysis of the clonal architecture "may yield more informative biomarkers and a better understanding of the pathogenesis of the myelodysplastic syndrome," Dr. Walter and his associates said.
Dr. Patel’s study was supported by the National Cancer Institute Physical Sciences Oncology Center, Gabrielle’s Angel Fund, the Starr Cancer Consortium, the Peter Solomon Fund, the American Society of Hematology, the Leukemia and Lymphoma Society, the Fund for Scientific Research Flanders, Burroughs Wellcome, the Sackler Center for Biomedical and Research Sciences, and the Howard Hughes Medical Institute. One of Dr. Patel’s associates reported ties to Agios, Incyte, and Novartis. Dr. Walter’s study was supported by the National Institutes of Health, the Howard Hughes Medical Institute, and the National Center for Research Resources. He and his associates reported no financial conflicts of interest.
The findings of Dr. Patel and colleagues "are sufficient to justify the expansion of the number of genetic mutations being examined in patients with AML at presentation, beyond the current analysis of [the] FLT3, NPM1, and CEBPA [genes]." These results also "challenge the field to address at what point data are compelling enough to change routine practice," said Dr. Lucy A. Godley.
The findings of Dr. Walter and colleagues also are challenging, since "it may be overwhelming for clinicians to receive a report with hundreds of gene mutations and expect them to make rational clinical decisions. An approach in which a fixed panel of genes was examined for mutations of particular clinical significance might be more affordable and the results easier to understand," she said.
Dr. Godley is in the section of hematology-oncology in the department of medicine at the University of Chicago. She reported ties to Celgene. These remarks were adapted from her editorial accompanying the two reports (N. Engl. J. Med. 2012 March 14 [doi:10.1056/NEJMe1200409]).
The findings of Dr. Patel and colleagues "are sufficient to justify the expansion of the number of genetic mutations being examined in patients with AML at presentation, beyond the current analysis of [the] FLT3, NPM1, and CEBPA [genes]." These results also "challenge the field to address at what point data are compelling enough to change routine practice," said Dr. Lucy A. Godley.
The findings of Dr. Walter and colleagues also are challenging, since "it may be overwhelming for clinicians to receive a report with hundreds of gene mutations and expect them to make rational clinical decisions. An approach in which a fixed panel of genes was examined for mutations of particular clinical significance might be more affordable and the results easier to understand," she said.
Dr. Godley is in the section of hematology-oncology in the department of medicine at the University of Chicago. She reported ties to Celgene. These remarks were adapted from her editorial accompanying the two reports (N. Engl. J. Med. 2012 March 14 [doi:10.1056/NEJMe1200409]).
The findings of Dr. Patel and colleagues "are sufficient to justify the expansion of the number of genetic mutations being examined in patients with AML at presentation, beyond the current analysis of [the] FLT3, NPM1, and CEBPA [genes]." These results also "challenge the field to address at what point data are compelling enough to change routine practice," said Dr. Lucy A. Godley.
The findings of Dr. Walter and colleagues also are challenging, since "it may be overwhelming for clinicians to receive a report with hundreds of gene mutations and expect them to make rational clinical decisions. An approach in which a fixed panel of genes was examined for mutations of particular clinical significance might be more affordable and the results easier to understand," she said.
Dr. Godley is in the section of hematology-oncology in the department of medicine at the University of Chicago. She reported ties to Celgene. These remarks were adapted from her editorial accompanying the two reports (N. Engl. J. Med. 2012 March 14 [doi:10.1056/NEJMe1200409]).
More detailed genetic profiling of patients with acute myeloid leukemia and of those with precursor myelodysplastic syndromes is likely to improve prognostic and therapeutic decision making, according to two separate studies published online March 14 in the New England Journal of Medicine.
In one study, investigators found that the presence of DNMT3A and NPM1 mutations and MLL translocations predicted an improved outcome when patients received high-dose daunorubicin instead of the standard dose in induction chemotherapy for acute myeloid leukemia (AML).
The results suggest that "mutational profiling can be used to determine which patients will benefit from dose-intensive induction therapy," wrote Jay P. Patel of the human oncology and pathogenesis program at Memorial Sloan-Kettering Cancer Center, New York, and his associates.
In the other study, researchers reported that "nearly all" of the bone marrow cells were clonally derived in paired samples of skin and bone marrow from seven patients with myelodysplastic syndromes (MDS) and secondary AML. Founding clones and daughter subclones in all seven paired samples had recurrent gene mutations, including at least one mutation in a coding gene.
"Although clonality is not sufficient to define malignant transformation, it is a cardinal manifestation of most human cancers, and our findings suggest that the myelodysplastic syndromes and secondary AML are both highly clonal hematologic cancers," said Dr. Matthew J. Walter of the departments of internal medicine and genetics at the Siteman Cancer Center, Washington University, St. Louis, and his associates.
Mutational Analysis of Trial Results
In the first study, researchers performed a more-extensive mutational analysis than is typically done to better discriminate among patients with different prognoses.
"Previous studies have suggested that mutational analysis of [the genes] CEBPA, NPM1, and FLT3-ITD can be used to stratify risk among patients with intermediate-risk AML," wrote Mr. Patel and his colleagues.
"We hypothesized that integrated mutational analysis of all known molecular alterations occurring in more than 5% of patients with AML would allow us to identify novel molecular markers of outcome ... and to identify molecularly defined subgroups of patients who would benefit from dose-intensified induction therapy."
For DNA extraction and profiling, the investigators used diagnostic samples of bone marrow and peripheral blood from 398 patients who were participating in the phase III ECOG (Eastern Cooperative Oncology Group) E1900 clinical trial in which two doses of induction therapy were tested. They found that 97.3% of the study subjects had mutations in 18 genes, and performed extensive mutational analysis of these 18 candidate genes.
The results led them to identify three distinct risk groups. Patients with favorable genetic profiles had a 3-year overall survival of 64% and had not yet reached a median survival; those with intermediate-risk genetic profiles had a 3-year survival of 42% and a median survival of 25 months; and those with unfavorable genetic profiles had a 3-year overall survival of 12% and a median survival of 10 months.
These findings were then validated in a separate group of 104 patients from the same clinical trial. The value of the genetic risk profiles was confirmed, with the favorable, intermediate, and unfavorable profiles accurately predicting patient outcomes independently of patient age, white cell count, induction dose, transplantation status, and type of postremission therapy, Mr. Patel and his colleagues said (N. Engl. J. Med. 2012 March 14 [doi:10.1056/NEJMoa1112304]).
Moreover, the 3-year overall survival rate in patients with a mutation in the DNMT3A or NPM1 genes or a MLL translocation was 44% with high-dose chemotherapy vs. 25% with the standard dose. In patients with other genotypes, it was 35% with the high-dose regimen and 39% with the standard dose.
"These data indicate that more detailed genetic analysis may lead to improved risk stratification and identification of patients who can benefit from more intensive induction chemotherapy. The challenge is to provide genetic information in a timely and affordable way and show that this information could prospectively influence treatment decisions," they noted.
Founding MDS Clones Persist in AML
In the second study, Dr. Walter and his associates used bone marrow biopsy specimens from seven patients who progressed from MDS to AML to define changes in the proportion of clonal cells and the genetic architecture that took place during that progression.
Several genes have already been identified that show recurrent mutations during this process, "but our understanding of the total number and clonal distribution of mutations in this disease is limited," they noted.
For each subject, DNA sequences were obtained from samples of normal skin, bone marrow obtained during the MDS stage, and bone marrow obtained during the secondary AML stage, to analyze mutations. In all seven samples, the founding clones (containing 182-660 mutations) persisted in the secondary samples, while acquiring at least one new mutation predicting translational consequences.
"We have found that the proportion of neoplastic bone marrow cells is indistinguishable [between] myelodysplastic-syndrome and secondary-AML samples, suggesting that the myelodysplastic syndromes are as clonal as secondary AML," Dr. Walter and his colleagues said (N. Engl. J. Med. 2012 March 14 [doi:10.1056/NEJMoa1106968]).
There are three major clinical implications, according to the authors.
First, MDS is currently distinguished from secondary AML based on hand counting of bone marrow myeloblasts – a method prone to inaccuracy but nonetheless relied upon to drive major treatment decisions. "Ultimately, identifying the patterns of pathogenic mutations and their clonality in bone marrow samples ... should lead to greater diagnostic certainty and improved prognostic algorithms," the investigators said.
Second, the dominant AML clone was derived from a founding MDS clone in every case, suggesting that "therapies targeted to these early mutations might be the most effective strategy for eliminating disease-propagating cells and improving the rate of response to traditional chemotherapy."
Third, it is possible that progression from MDS to AML "is driven not only by the presence of recurrent mutations ... but also by the clone ([that is], founding vs. daughter) in which they arise." Combining genotyping of samples with analysis of the clonal architecture "may yield more informative biomarkers and a better understanding of the pathogenesis of the myelodysplastic syndrome," Dr. Walter and his associates said.
Dr. Patel’s study was supported by the National Cancer Institute Physical Sciences Oncology Center, Gabrielle’s Angel Fund, the Starr Cancer Consortium, the Peter Solomon Fund, the American Society of Hematology, the Leukemia and Lymphoma Society, the Fund for Scientific Research Flanders, Burroughs Wellcome, the Sackler Center for Biomedical and Research Sciences, and the Howard Hughes Medical Institute. One of Dr. Patel’s associates reported ties to Agios, Incyte, and Novartis. Dr. Walter’s study was supported by the National Institutes of Health, the Howard Hughes Medical Institute, and the National Center for Research Resources. He and his associates reported no financial conflicts of interest.
More detailed genetic profiling of patients with acute myeloid leukemia and of those with precursor myelodysplastic syndromes is likely to improve prognostic and therapeutic decision making, according to two separate studies published online March 14 in the New England Journal of Medicine.
In one study, investigators found that the presence of DNMT3A and NPM1 mutations and MLL translocations predicted an improved outcome when patients received high-dose daunorubicin instead of the standard dose in induction chemotherapy for acute myeloid leukemia (AML).
The results suggest that "mutational profiling can be used to determine which patients will benefit from dose-intensive induction therapy," wrote Jay P. Patel of the human oncology and pathogenesis program at Memorial Sloan-Kettering Cancer Center, New York, and his associates.
In the other study, researchers reported that "nearly all" of the bone marrow cells were clonally derived in paired samples of skin and bone marrow from seven patients with myelodysplastic syndromes (MDS) and secondary AML. Founding clones and daughter subclones in all seven paired samples had recurrent gene mutations, including at least one mutation in a coding gene.
"Although clonality is not sufficient to define malignant transformation, it is a cardinal manifestation of most human cancers, and our findings suggest that the myelodysplastic syndromes and secondary AML are both highly clonal hematologic cancers," said Dr. Matthew J. Walter of the departments of internal medicine and genetics at the Siteman Cancer Center, Washington University, St. Louis, and his associates.
Mutational Analysis of Trial Results
In the first study, researchers performed a more-extensive mutational analysis than is typically done to better discriminate among patients with different prognoses.
"Previous studies have suggested that mutational analysis of [the genes] CEBPA, NPM1, and FLT3-ITD can be used to stratify risk among patients with intermediate-risk AML," wrote Mr. Patel and his colleagues.
"We hypothesized that integrated mutational analysis of all known molecular alterations occurring in more than 5% of patients with AML would allow us to identify novel molecular markers of outcome ... and to identify molecularly defined subgroups of patients who would benefit from dose-intensified induction therapy."
For DNA extraction and profiling, the investigators used diagnostic samples of bone marrow and peripheral blood from 398 patients who were participating in the phase III ECOG (Eastern Cooperative Oncology Group) E1900 clinical trial in which two doses of induction therapy were tested. They found that 97.3% of the study subjects had mutations in 18 genes, and performed extensive mutational analysis of these 18 candidate genes.
The results led them to identify three distinct risk groups. Patients with favorable genetic profiles had a 3-year overall survival of 64% and had not yet reached a median survival; those with intermediate-risk genetic profiles had a 3-year survival of 42% and a median survival of 25 months; and those with unfavorable genetic profiles had a 3-year overall survival of 12% and a median survival of 10 months.
These findings were then validated in a separate group of 104 patients from the same clinical trial. The value of the genetic risk profiles was confirmed, with the favorable, intermediate, and unfavorable profiles accurately predicting patient outcomes independently of patient age, white cell count, induction dose, transplantation status, and type of postremission therapy, Mr. Patel and his colleagues said (N. Engl. J. Med. 2012 March 14 [doi:10.1056/NEJMoa1112304]).
Moreover, the 3-year overall survival rate in patients with a mutation in the DNMT3A or NPM1 genes or a MLL translocation was 44% with high-dose chemotherapy vs. 25% with the standard dose. In patients with other genotypes, it was 35% with the high-dose regimen and 39% with the standard dose.
"These data indicate that more detailed genetic analysis may lead to improved risk stratification and identification of patients who can benefit from more intensive induction chemotherapy. The challenge is to provide genetic information in a timely and affordable way and show that this information could prospectively influence treatment decisions," they noted.
Founding MDS Clones Persist in AML
In the second study, Dr. Walter and his associates used bone marrow biopsy specimens from seven patients who progressed from MDS to AML to define changes in the proportion of clonal cells and the genetic architecture that took place during that progression.
Several genes have already been identified that show recurrent mutations during this process, "but our understanding of the total number and clonal distribution of mutations in this disease is limited," they noted.
For each subject, DNA sequences were obtained from samples of normal skin, bone marrow obtained during the MDS stage, and bone marrow obtained during the secondary AML stage, to analyze mutations. In all seven samples, the founding clones (containing 182-660 mutations) persisted in the secondary samples, while acquiring at least one new mutation predicting translational consequences.
"We have found that the proportion of neoplastic bone marrow cells is indistinguishable [between] myelodysplastic-syndrome and secondary-AML samples, suggesting that the myelodysplastic syndromes are as clonal as secondary AML," Dr. Walter and his colleagues said (N. Engl. J. Med. 2012 March 14 [doi:10.1056/NEJMoa1106968]).
There are three major clinical implications, according to the authors.
First, MDS is currently distinguished from secondary AML based on hand counting of bone marrow myeloblasts – a method prone to inaccuracy but nonetheless relied upon to drive major treatment decisions. "Ultimately, identifying the patterns of pathogenic mutations and their clonality in bone marrow samples ... should lead to greater diagnostic certainty and improved prognostic algorithms," the investigators said.
Second, the dominant AML clone was derived from a founding MDS clone in every case, suggesting that "therapies targeted to these early mutations might be the most effective strategy for eliminating disease-propagating cells and improving the rate of response to traditional chemotherapy."
Third, it is possible that progression from MDS to AML "is driven not only by the presence of recurrent mutations ... but also by the clone ([that is], founding vs. daughter) in which they arise." Combining genotyping of samples with analysis of the clonal architecture "may yield more informative biomarkers and a better understanding of the pathogenesis of the myelodysplastic syndrome," Dr. Walter and his associates said.
Dr. Patel’s study was supported by the National Cancer Institute Physical Sciences Oncology Center, Gabrielle’s Angel Fund, the Starr Cancer Consortium, the Peter Solomon Fund, the American Society of Hematology, the Leukemia and Lymphoma Society, the Fund for Scientific Research Flanders, Burroughs Wellcome, the Sackler Center for Biomedical and Research Sciences, and the Howard Hughes Medical Institute. One of Dr. Patel’s associates reported ties to Agios, Incyte, and Novartis. Dr. Walter’s study was supported by the National Institutes of Health, the Howard Hughes Medical Institute, and the National Center for Research Resources. He and his associates reported no financial conflicts of interest.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Major Finding: In one study 97.3% of samples from patients with AML had mutations in 18 genes. In the other, the founding clone in seven MDS samples persisted in all seven samples of secondary AML from the same patients.
Data Source: The first study was an extensive mutational analysis of 398 patients with AML, with findings confirmed in a validation cohort of 104 patients. The second was a whole-genome sequencing of paired samples of skin and bone marrow from seven patients who had MDS that progressed to secondary AML.
Disclosures: Dr. Patel’s study was supported by the National Cancer Institute Physical Sciences Oncology Center, Gabrielle’s Angel Fund, the Starr Cancer Consortium, the Peter Solomon Fund, the American Society of Hematology, the Leukemia and Lymphoma Society, the Fund for Scientific Research Flanders, Burroughs Wellcome, the Sackler Center for Biomedical and Research Sciences, and the Howard Hughes Medical Institute. One of Dr. Patel’s associates reported ties to Agios, Incyte, and Novartis. Dr. Walter’s study was supported by the National Institutes of Health, the Howard Hughes Medical Institute, and the National Center for Research Resources. He and his associates reported no financial conflicts of interest.
Brd4: Potential new target in AML
Researchers at Cold Spring Harbor Laboratory (CSHL) and 5 other laboratories may have identified a new drug target for the treatment of acute myeloid leukemia (AML).
They pinpointed a protein called Brd4, which contains a distinct region known as bromodomain and is a member of the BET protein family, known for regulating gene expression.
A new drug candidate that inhibits Brd4 was able to suppress AML in experimental models.
“The drug candidate not only displays remarkable anti-leukemia activity in aggressive disease models and against cells derived from patients with diverse, genetic subtypes of AML, but is also minimally toxic to non-cancerous cells,” said Chris Vakoc, MD, PhD, leader of the study at CSHL.
“The drug is currently being developed for therapeutic use for cancer patients by Tensha Therapeutics and is expected to enter clinical trials within 2 years.”
The team used RNAi screening in an AML murine model. The RNAi screen introduced small hairpin-shaped pieces of RNA (shRNA) that encode epigenetic proteins into mice that harbor leukemia-causing mutations.
The mice in this study carried the oncogene Nras and rearranged forms of the MLL gene, both of which are mutations often found in patients whose leukemia is resistant to standard chemotherapy.
The drinking water for the mice was also supplemented with doxycyline.
“Inducing shRNA that shuts down a gene required for the survival of leukemic cells can lead to complete disease remission,” said Johannes Zuber, MD, former postdoctoral researcher at CSHL. “This ability to use shRNA to simulate the effect of an anti-cancer drug illustrates the power of this approach.”
The team also identified another potential target in AML called Myb. The team found that suppressing the activity of Myb also eliminated AML in mice. The team screened more than 1000 shRNAs targeting 243 known epigenetic regulators of chromatin.
They homed in on Brd4 as a target and found that suppressing the protein led to the most dramatic changes in AML. The cell cycle was arrested, the leukemic cells died, leukemia progression was delayed, and the mice survived longer.
Previously, James Bradner, MD, at Dana-Farber Cancer Institute in Boston, and his research team had developed a small molecule inhibitor of Brd4 called JQ1. The two groups collaborated, reproducing the antileukemic effects in the Brd4 shRNA experiments. They consider JQ1 to be an ideal drug candidate.
Their findings were published online in Nature.![]()
Researchers at Cold Spring Harbor Laboratory (CSHL) and 5 other laboratories may have identified a new drug target for the treatment of acute myeloid leukemia (AML).
They pinpointed a protein called Brd4, which contains a distinct region known as bromodomain and is a member of the BET protein family, known for regulating gene expression.
A new drug candidate that inhibits Brd4 was able to suppress AML in experimental models.
“The drug candidate not only displays remarkable anti-leukemia activity in aggressive disease models and against cells derived from patients with diverse, genetic subtypes of AML, but is also minimally toxic to non-cancerous cells,” said Chris Vakoc, MD, PhD, leader of the study at CSHL.
“The drug is currently being developed for therapeutic use for cancer patients by Tensha Therapeutics and is expected to enter clinical trials within 2 years.”
The team used RNAi screening in an AML murine model. The RNAi screen introduced small hairpin-shaped pieces of RNA (shRNA) that encode epigenetic proteins into mice that harbor leukemia-causing mutations.
The mice in this study carried the oncogene Nras and rearranged forms of the MLL gene, both of which are mutations often found in patients whose leukemia is resistant to standard chemotherapy.
The drinking water for the mice was also supplemented with doxycyline.
“Inducing shRNA that shuts down a gene required for the survival of leukemic cells can lead to complete disease remission,” said Johannes Zuber, MD, former postdoctoral researcher at CSHL. “This ability to use shRNA to simulate the effect of an anti-cancer drug illustrates the power of this approach.”
The team also identified another potential target in AML called Myb. The team found that suppressing the activity of Myb also eliminated AML in mice. The team screened more than 1000 shRNAs targeting 243 known epigenetic regulators of chromatin.
They homed in on Brd4 as a target and found that suppressing the protein led to the most dramatic changes in AML. The cell cycle was arrested, the leukemic cells died, leukemia progression was delayed, and the mice survived longer.
Previously, James Bradner, MD, at Dana-Farber Cancer Institute in Boston, and his research team had developed a small molecule inhibitor of Brd4 called JQ1. The two groups collaborated, reproducing the antileukemic effects in the Brd4 shRNA experiments. They consider JQ1 to be an ideal drug candidate.
Their findings were published online in Nature.![]()
Researchers at Cold Spring Harbor Laboratory (CSHL) and 5 other laboratories may have identified a new drug target for the treatment of acute myeloid leukemia (AML).
They pinpointed a protein called Brd4, which contains a distinct region known as bromodomain and is a member of the BET protein family, known for regulating gene expression.
A new drug candidate that inhibits Brd4 was able to suppress AML in experimental models.
“The drug candidate not only displays remarkable anti-leukemia activity in aggressive disease models and against cells derived from patients with diverse, genetic subtypes of AML, but is also minimally toxic to non-cancerous cells,” said Chris Vakoc, MD, PhD, leader of the study at CSHL.
“The drug is currently being developed for therapeutic use for cancer patients by Tensha Therapeutics and is expected to enter clinical trials within 2 years.”
The team used RNAi screening in an AML murine model. The RNAi screen introduced small hairpin-shaped pieces of RNA (shRNA) that encode epigenetic proteins into mice that harbor leukemia-causing mutations.
The mice in this study carried the oncogene Nras and rearranged forms of the MLL gene, both of which are mutations often found in patients whose leukemia is resistant to standard chemotherapy.
The drinking water for the mice was also supplemented with doxycyline.
“Inducing shRNA that shuts down a gene required for the survival of leukemic cells can lead to complete disease remission,” said Johannes Zuber, MD, former postdoctoral researcher at CSHL. “This ability to use shRNA to simulate the effect of an anti-cancer drug illustrates the power of this approach.”
The team also identified another potential target in AML called Myb. The team found that suppressing the activity of Myb also eliminated AML in mice. The team screened more than 1000 shRNAs targeting 243 known epigenetic regulators of chromatin.
They homed in on Brd4 as a target and found that suppressing the protein led to the most dramatic changes in AML. The cell cycle was arrested, the leukemic cells died, leukemia progression was delayed, and the mice survived longer.
Previously, James Bradner, MD, at Dana-Farber Cancer Institute in Boston, and his research team had developed a small molecule inhibitor of Brd4 called JQ1. The two groups collaborated, reproducing the antileukemic effects in the Brd4 shRNA experiments. They consider JQ1 to be an ideal drug candidate.
Their findings were published online in Nature.![]()