User login
Acute monocular vision loss: Don’t lose sight of the differential
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.

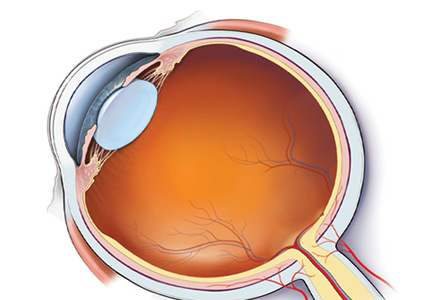
Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history

A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.

An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations

A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).

For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.
Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history
A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.
An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations
A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).
For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.
Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history
A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.
An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations
A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).
For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
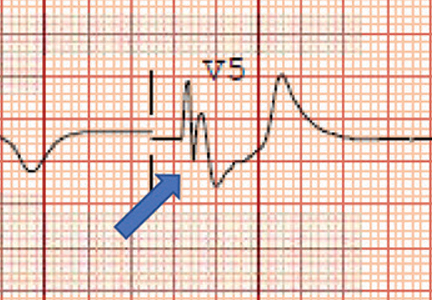
Osborn waves of hypothermia
A 40-year-old man was brought to the emergency department with altered mental status. His roommate had found him lying unconscious in snow on the lawn outside his residence. When the emergency medical services team arrived, they recorded a core body temperature of 28.3°C (82.9°F) and instituted advanced cardiac life support.
During transit to the hospital, the patient’s heart rhythm changed from asystole to ventricular fibrillation, and defibrillation was performed twice.
Upon his arrival at the emergency room, advanced life support was continued, resulting in return of spontaneous circulation, with slow, wide-complex QRS rhythm noted on electrocardiography (ECG).
On examination, the patient’s pupils were fixed and dilated. The extremities were cold to palpation. The core body temperature dropped to 27.7°C (81.9°F).
Laboratory test results showed severe acidemia (arterial pH 6.8), elevated aspartate aminotransferase and alanine aminotransferase levels, and elevated creatinine and troponin. The troponin was measured 3 times and rose from 0.8 ng/mL to 0.9 ng/mL. A urine toxicology screen was positive for cannabinoids and cocaine.
 is typically seen in precordial leads V3 to V6 and is a marker for hypothermia.")
ECG revealed J-point elevation (Osborn waves) in the precordial leads (Figure 1). A baseline electrocardiogram in the medical record from a previous admission had been normal.
An aggressive hypothermia protocol was initiated, but the patient died despite resuscitation efforts.
HYPOTHERMIA AND HEART RHYTHMS
Hypothermia—a core body temperature below 35°C (95°F)—causes generalized slowing of impulse conduction through cardiac tissues, shown on ECG as a prolongation of the PR, RR, QRS, and QT intervals.1
A characteristic feature is elevation of the J point, also called the J wave or Osborn wave, most prominent in precordial leads V2 to V5 and caused by abnormal membrane repolarization in the early phase. The degree of hypothermia correlates linearly with the amplitude of the Osborn wave.2,3
Laboratory tests can identify complications such as rhabdomyolysis, spontaneous bleeding, and lactic acidosis. Moderate to severe hypothermia may cause prolongation of all ECG intervals. Management requires resuscitation and rewarming.
Conditions to consider in the differential diagnosis are Brugada syndrome, hypercalcemia, and early repolarization syndrome.
- Doshi HH, Giudici MC. The EKG in hypothermia and hyperthermia. J Electrocardiol 2015; 48:203–208.
- Alsafwah S. Electrocardiographic changes in hypothermia. Heart Lung 2001; 30:161–163.
- Graham CA, McNaughton GW, Wyatt JP. The electrocardiogram in hypothermia. Wilderness Environ Med 2001; 12:232–235.
A 40-year-old man was brought to the emergency department with altered mental status. His roommate had found him lying unconscious in snow on the lawn outside his residence. When the emergency medical services team arrived, they recorded a core body temperature of 28.3°C (82.9°F) and instituted advanced cardiac life support.
During transit to the hospital, the patient’s heart rhythm changed from asystole to ventricular fibrillation, and defibrillation was performed twice.
Upon his arrival at the emergency room, advanced life support was continued, resulting in return of spontaneous circulation, with slow, wide-complex QRS rhythm noted on electrocardiography (ECG).
On examination, the patient’s pupils were fixed and dilated. The extremities were cold to palpation. The core body temperature dropped to 27.7°C (81.9°F).
Laboratory test results showed severe acidemia (arterial pH 6.8), elevated aspartate aminotransferase and alanine aminotransferase levels, and elevated creatinine and troponin. The troponin was measured 3 times and rose from 0.8 ng/mL to 0.9 ng/mL. A urine toxicology screen was positive for cannabinoids and cocaine.
ECG revealed J-point elevation (Osborn waves) in the precordial leads (Figure 1). A baseline electrocardiogram in the medical record from a previous admission had been normal.
An aggressive hypothermia protocol was initiated, but the patient died despite resuscitation efforts.
HYPOTHERMIA AND HEART RHYTHMS
Hypothermia—a core body temperature below 35°C (95°F)—causes generalized slowing of impulse conduction through cardiac tissues, shown on ECG as a prolongation of the PR, RR, QRS, and QT intervals.1
A characteristic feature is elevation of the J point, also called the J wave or Osborn wave, most prominent in precordial leads V2 to V5 and caused by abnormal membrane repolarization in the early phase. The degree of hypothermia correlates linearly with the amplitude of the Osborn wave.2,3
Laboratory tests can identify complications such as rhabdomyolysis, spontaneous bleeding, and lactic acidosis. Moderate to severe hypothermia may cause prolongation of all ECG intervals. Management requires resuscitation and rewarming.
Conditions to consider in the differential diagnosis are Brugada syndrome, hypercalcemia, and early repolarization syndrome.
A 40-year-old man was brought to the emergency department with altered mental status. His roommate had found him lying unconscious in snow on the lawn outside his residence. When the emergency medical services team arrived, they recorded a core body temperature of 28.3°C (82.9°F) and instituted advanced cardiac life support.
During transit to the hospital, the patient’s heart rhythm changed from asystole to ventricular fibrillation, and defibrillation was performed twice.
Upon his arrival at the emergency room, advanced life support was continued, resulting in return of spontaneous circulation, with slow, wide-complex QRS rhythm noted on electrocardiography (ECG).
On examination, the patient’s pupils were fixed and dilated. The extremities were cold to palpation. The core body temperature dropped to 27.7°C (81.9°F).
Laboratory test results showed severe acidemia (arterial pH 6.8), elevated aspartate aminotransferase and alanine aminotransferase levels, and elevated creatinine and troponin. The troponin was measured 3 times and rose from 0.8 ng/mL to 0.9 ng/mL. A urine toxicology screen was positive for cannabinoids and cocaine.
ECG revealed J-point elevation (Osborn waves) in the precordial leads (Figure 1). A baseline electrocardiogram in the medical record from a previous admission had been normal.
An aggressive hypothermia protocol was initiated, but the patient died despite resuscitation efforts.
HYPOTHERMIA AND HEART RHYTHMS
Hypothermia—a core body temperature below 35°C (95°F)—causes generalized slowing of impulse conduction through cardiac tissues, shown on ECG as a prolongation of the PR, RR, QRS, and QT intervals.1
A characteristic feature is elevation of the J point, also called the J wave or Osborn wave, most prominent in precordial leads V2 to V5 and caused by abnormal membrane repolarization in the early phase. The degree of hypothermia correlates linearly with the amplitude of the Osborn wave.2,3
Laboratory tests can identify complications such as rhabdomyolysis, spontaneous bleeding, and lactic acidosis. Moderate to severe hypothermia may cause prolongation of all ECG intervals. Management requires resuscitation and rewarming.
Conditions to consider in the differential diagnosis are Brugada syndrome, hypercalcemia, and early repolarization syndrome.
- Doshi HH, Giudici MC. The EKG in hypothermia and hyperthermia. J Electrocardiol 2015; 48:203–208.
- Alsafwah S. Electrocardiographic changes in hypothermia. Heart Lung 2001; 30:161–163.
- Graham CA, McNaughton GW, Wyatt JP. The electrocardiogram in hypothermia. Wilderness Environ Med 2001; 12:232–235.
- Doshi HH, Giudici MC. The EKG in hypothermia and hyperthermia. J Electrocardiol 2015; 48:203–208.
- Alsafwah S. Electrocardiographic changes in hypothermia. Heart Lung 2001; 30:161–163.
- Graham CA, McNaughton GW, Wyatt JP. The electrocardiogram in hypothermia. Wilderness Environ Med 2001; 12:232–235.
Haste Makes Waste
ANSWER
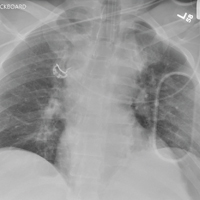
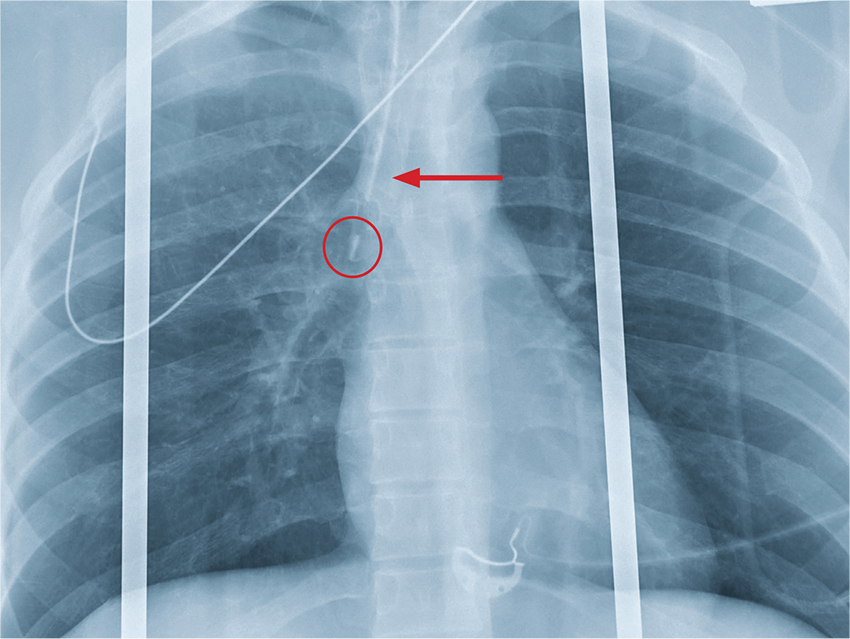
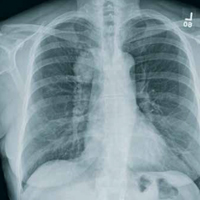
The chest radiograph shows an endotracheal tube within the right main stem bronchus. There is no evidence of any other acute pathology (eg, fracture, contusion, pneumothorax).
The tube needs to be withdrawn so that it sits just above the carina (see arrow). If not promptly addressed, incorrect placement of an endotracheal tube can lead to complications, including hypoxemia, pneumothorax, atelectasis, or complete collapse of the left lung.
ANSWER
The chest radiograph shows an endotracheal tube within the right main stem bronchus. There is no evidence of any other acute pathology (eg, fracture, contusion, pneumothorax).
The tube needs to be withdrawn so that it sits just above the carina (see arrow). If not promptly addressed, incorrect placement of an endotracheal tube can lead to complications, including hypoxemia, pneumothorax, atelectasis, or complete collapse of the left lung.
ANSWER
The chest radiograph shows an endotracheal tube within the right main stem bronchus. There is no evidence of any other acute pathology (eg, fracture, contusion, pneumothorax).
The tube needs to be withdrawn so that it sits just above the carina (see arrow). If not promptly addressed, incorrect placement of an endotracheal tube can lead to complications, including hypoxemia, pneumothorax, atelectasis, or complete collapse of the left lung.
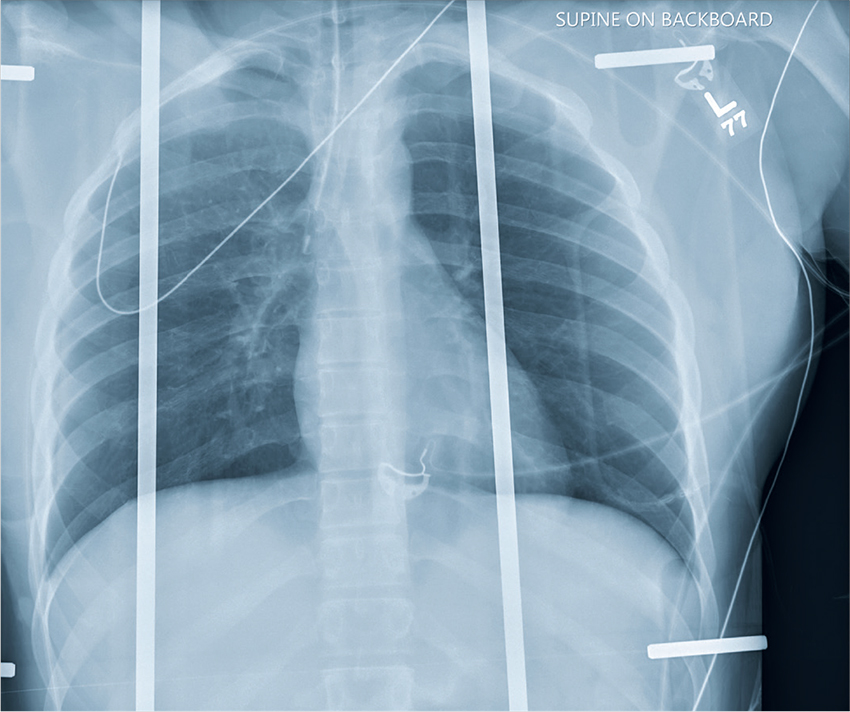
A woman who looks to be 30 years old is brought to your facility as a trauma code following a car accident. She was a restrained driver, traveling at a high speed when she lost control of her vehicle and hit a retaining wall.
When first responders arrived, the patient had extricated herself but demonstrated a decreased level of consciousness, severe respiratory distress, and a Glasgow Coma Scale score of 7. She was intubated at the scene by emergency medical personnel.
On evaluation, you note a young, intubated, unresponsive female. Her blood pressure is 90/50 mm Hg; heart rate, 90 beats/min; and O2 saturation, 100%. Rapid primary survey shows
A portable chest radiograph is obtained (shown). What is your impression?
The Not-So-Routine Physical
ANSWER
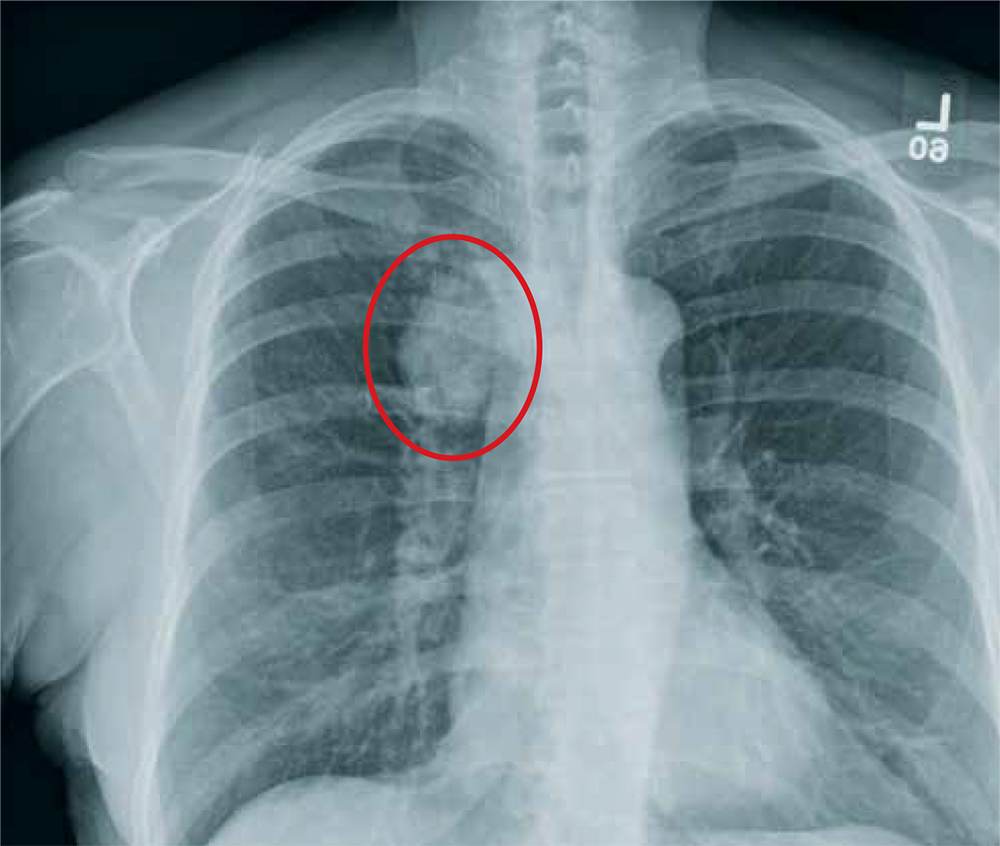
The radiograph shows a moderate-size mass, measuring about 5 × 3 cm, at the medial portion of the right upper lobe, within the paratracheal region. This lesion should be treated as a neoplasm until proven otherwise. Contrast-enhanced CT is warranted, as well as prompt referral to a cardiothoracic surgeon.
ANSWER
The radiograph shows a moderate-size mass, measuring about 5 × 3 cm, at the medial portion of the right upper lobe, within the paratracheal region. This lesion should be treated as a neoplasm until proven otherwise. Contrast-enhanced CT is warranted, as well as prompt referral to a cardiothoracic surgeon.
ANSWER
The radiograph shows a moderate-size mass, measuring about 5 × 3 cm, at the medial portion of the right upper lobe, within the paratracheal region. This lesion should be treated as a neoplasm until proven otherwise. Contrast-enhanced CT is warranted, as well as prompt referral to a cardiothoracic surgeon.
A 60-year-old woman wants to establish care as a new patient at your clinic. She presents for an annual physical and has no current complaints.
Her medical history is significant for hypertension and remote uterine cancer, which was treated with a hysterectomy. She does report smoking a half-pack to one pack of cigarettes per day for “about 30 to 40” years.
Vital signs are normal. Overall, the complete physical examination yields no abnormal findings. Routine bloodwork, 12-lead ECG, and a chest radiograph are ordered. The last is shown. What is your impression?
Don’t Always Rush to Rally Renal
Case
An otherwise healthy 20-month-old boy presented to the ED for evaluation after his father witnessed the child ingest a model race car fuel additive. According to the patient’s father, the boy was playing with several closed bottles that were stored in the garage, when he witnessed the boy open up and take a sip of a pink-colored fuel additive, which the father believed to contain 100% methanol. The patient’s father further noted that immediately after drinking the fluid, the patient spat and drooled, and had one episode of nonbloody emesis prior to arrival at the ED.
Initial vital signs at presentation were: blood pressure, 84/54 mm Hg; heart rate, 97 beats/min; respiratory rate, 24 breaths/min; and temperature 98°F. Oxygen saturation was 99% on room air. Physical examination was notable for mild erythema in the posterior oropharynx. Otherwise, the patient was acting appropriately for his age and in no acute distress. Laboratory studies were within normal limits, except for the following: serum anion gap, 18 mEq/L (reference range for children < 3 years old, 10-14 mEq/L); serum bicarbonate, 19 mmol/L (reference range for children 12-24 months, 17-25 mmol/L); and serum creatinine, 2.8 mg/dL (reference range for children 12 to 24 months, 0.2-0.5 mg/dL). A repeat creatinine test taken after bolus of fluid administration was 2.4 mg/dL. A renal ultrasound, performed to investigate the cause of the renal failure, was unremarkable.
What toxic exposures are of concern based on the clinical history?
The history of exposure to a liquid stored in a garage raises the likelihood of exposure to an automobile-related item such as diethylene glycol, ethylene glycol (EG), and methanol.
Diethylene Glycol. Diethylene glycol is an ingredient in brake and power steering fluids, and has toxic properties qualitatively similar to EG.
Ethylene Glycol. A clear, colorless, odorless fluid with a sweet taste, EG is an ingredient in radiator antifreeze, refrigerant fluid, coolants, and pesticides. Like methylene, EG reaches peak plasma concentration within 1 to 4 hours, but toxic clinical findings do not occur for 3 to 6 hours.1
Methanol. Methanol is a clear, colorless, alcohol found in antifreeze, windshield washer fluid, and race car fuel.2 Although methanol reaches peak plasma concentration in about 30 to 60 minutes, signs of systemic toxicity (ie, metabolic acidosis) typically take 6 to 12 hours to manifest.1
In both EG and methanol, there is a delay in toxic clinical findings because the parent compounds are not toxic in their initial form; rather, major toxicity is derived from their metabolites: formic acid and oxalic acid, respectively.
Other Toxins. Many other potentially toxic liquids are associated with a homeowner’s occupation or avocational interests. These include painting supplies (eg, industrial paints containing lead), gardening materials (eg, pesticides containing organophosphates), fuels (eg, gasoline, polychlorinated biphenyls in coolant, and lubricants), and cleaning supplies (eg, caustics, detergents, and air freshener).
Case Continuation
Since the patient’s elevated anion gap raised concerns for methanol or EG exposure, he was given fomepizole and transferred to a tertiary care children’s hospital for further management and possible hemodialysis. Upon arrival at the receiving hospital, the patient’s vital signs and physical examination remained unchanged. Repeat laboratory studies were notable for a creatinine level of 0.3 mg/dL. The patient’s father was instructed to retrieve the implicated bottle from home. An inspection of the bottle’s ingredients was notable for nitromethane, castor oil, and methanol.
What is nitromethane and what are its uses?
Nitromethane, the simplest nitro compound, is a colorless, viscous, lipid-soluble fluid.3 The polarity of nitromethane permits its use as a stabilizer in a number of chemical solvents, such as dry cleaning fluid, degreasers, and "super glue."4,5 Nitromethane is also commonly added to model-engine and drag-race fuels, which also contain methanol and castor oil.3 In this capacity, nitromethane functions as an oxygen carrier, allowing more efficient fuel use in combustion cylinders (compared to gasoline), thereby increasing the horsepower of the vehicle.6 It is therefore commonly added to fuel for drag racers, radio-controlled cars, and model aircrafts.4 In the small concentrations typically inadvertently ingested, the clinical effects of nitromethane itself are inconsequential.
What is the differential for creatinine elevation?
Creatinine itself is a normal breakdown product of muscle metabolism produced by spontaneous conversion from creatine and is found at a fairly constant serum level in proportion to muscle mass.7 Thus, as people age and muscle mass decreases, their baseline creatinine levels decrease proportionally.
Elimination. The majority of creatinine (85%-90%) is filtered and excreted by the kidneys, with the remaining 10% to 15% secreted by the tubules, allowing creatinine to be a surrogate measure of the glomerular filtration rate.7 Exogenous sources of creatine or creatinine include meat and creatine supplements, the latter of which are used as an "energy source" to enhance athletic performance.
Etiology. The etiology for an elevated serum creatinine concentration includes renal failure, both acute and chronic; volume depletion; hemorrhage (low blood volume); and medications, including diuretics, angiotensin converting enzyme inhibitors, angiotensin-receptor blockers, nonsteroidal anti-inflammatory drugs, and certain antibiotics. These etiologies can also be categorized as processes that increase creatinine production, decrease elimination (H2 antagonist and trimethoprim both inhibit the cation secretory pump in the tubules), or interfere with the creatinine assay (ketones, keto acids, lipemia, hemolysis, cephalosporins).7
Because creatinine is filtered so efficiently by the kidney, neither exogenous nor endogenous creatinine sources are expected to increase serum creatinine in the absence of renal dysfunction. However, transient elevation may occur in body builders who use extreme doses of creatine. Patients with rhabdomyolysis often develop elevated creatinine concentrations, but nearly always in the setting of myoglobinuric renal failure.
Jaffe Reaction and Enzymatic Methods. Serum creatinine can be measured using either the Jaffe reaction or the enzymatic method. In the Jaffe reaction, creatinine reacts with alkaline sodium picrate to form a red-orange chromophore, which absorbs light in the range of 470 to 550 nanometers on spectroscopy.6,8,9 The active methylene group on nitromethane also reacts with alkaline sodium picrate to form a chromophore which absorbs light in the same wavelength range.10 Thus, serum creatinine measurements via the Jaffe reaction are falsely elevated due to the cross-reactivity between nitromethane and alkaline sodium picrate. In some reported cases, there is a 20-fold increase in the measured serum creatinine in the presence of nitromethane; renal function, however, remains normal.5
This false reading seen in the Jaffe reaction can be avoided by utilizing the enzymatic method of creatinine measurement, a three-step process that ultimately produces hydrogen peroxide, which is measured and accurately correlates with serum creatinine—even in the presence of nitromethane.8 This distinction explains the dramatically different creatinine concentrations measured at the two institutions in this case.
Case Conclusion
The patient was monitored overnight at the children’s hospital. Repeat laboratory studies in the morning showed a normal creatinine level of 0.3 mg/dL and a negative methanol level. The patient was discharged home in the care of his father, who was instructed to follow-up with his son’s pediatrician. The father also received counseling on safe storage practices for dangerous chemicals.
1. Kruse JA. Methanol and ethylene glycol intoxication. Crit Care Clin. 2012;28(4):661-711. doi:10.1016/j.ccc.2012.07.002.
2. McMahon DM, Winstead S, Weant KA. Toxic alcohol ingestions: focus on ethylene glycol and methanol. Adv Emerg Nurs J. 2009;31(3):206-213. doi:10.1097/TME.0b013e3181ad8be8.
3. Cook MD, Clark RF. Creatinine elevation associated with nitromethane exposure: a marker of potential methanol toxicity. J Emerg Med. 2007;33(3):249-253. doi:10.1016/j.jemermed.2007.02.015.
4. Markofsky SB. Nitro compounds, aliphatic. In: Elvers B, ed. Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA; 2000. doi:10.1002/14356007.a17_401. [digital]
5. Mullins ME, Hammett-Stabler CA. Intoxication with nitromethane-containing fuels: don’t be "fueled" by the creatinine. J Toxicol Clin Toxicol. 1998;36(4):
315-320.
6. Ngo AS, Rowley F, Olson KR. Case files of the California poison control system, San Francisco division: blue thunder ingestion: methanol, nitromethane, and elevated creatinine. J Med Toxicol. 2010;6(1):67-71. doi:10.1007/s13181-010-0042-5.
7. Samra M, Abcar AC. False estimates of elevated creatinine. Perm J. 2012;16(2):51-52.
8. Booth C, Naidoo D, Rosenberg A, Kainer G. Elevated creatinine after ingestion of model aviation fuel: interference with the Jaffe reaction by nitromethane. J Paediatr Child Health. 1999;35(5):503-504.
9. de Lelis Medeiros de Morais C, Gomes de Lima KM. Determination and analytical validation of creatinine content in serum using image analysis by multivariate transfer calibration procedures. Anal Meth. 2015;7:6904-6910. doi:10.1039/C5AY01369K.
10. Killorn E, Lim RK, Rieder M. Apparent elevated creatinine after ingestion of nitromethane: interference with the Jaffe reaction. Ther Drug Monit. 2011;33(1):1-2. doi:10.1097/FTD.0b013e3181fe7e52.
Case
An otherwise healthy 20-month-old boy presented to the ED for evaluation after his father witnessed the child ingest a model race car fuel additive. According to the patient’s father, the boy was playing with several closed bottles that were stored in the garage, when he witnessed the boy open up and take a sip of a pink-colored fuel additive, which the father believed to contain 100% methanol. The patient’s father further noted that immediately after drinking the fluid, the patient spat and drooled, and had one episode of nonbloody emesis prior to arrival at the ED.
Initial vital signs at presentation were: blood pressure, 84/54 mm Hg; heart rate, 97 beats/min; respiratory rate, 24 breaths/min; and temperature 98°F. Oxygen saturation was 99% on room air. Physical examination was notable for mild erythema in the posterior oropharynx. Otherwise, the patient was acting appropriately for his age and in no acute distress. Laboratory studies were within normal limits, except for the following: serum anion gap, 18 mEq/L (reference range for children < 3 years old, 10-14 mEq/L); serum bicarbonate, 19 mmol/L (reference range for children 12-24 months, 17-25 mmol/L); and serum creatinine, 2.8 mg/dL (reference range for children 12 to 24 months, 0.2-0.5 mg/dL). A repeat creatinine test taken after bolus of fluid administration was 2.4 mg/dL. A renal ultrasound, performed to investigate the cause of the renal failure, was unremarkable.
What toxic exposures are of concern based on the clinical history?
The history of exposure to a liquid stored in a garage raises the likelihood of exposure to an automobile-related item such as diethylene glycol, ethylene glycol (EG), and methanol.
Diethylene Glycol. Diethylene glycol is an ingredient in brake and power steering fluids, and has toxic properties qualitatively similar to EG.
Ethylene Glycol. A clear, colorless, odorless fluid with a sweet taste, EG is an ingredient in radiator antifreeze, refrigerant fluid, coolants, and pesticides. Like methylene, EG reaches peak plasma concentration within 1 to 4 hours, but toxic clinical findings do not occur for 3 to 6 hours.1
Methanol. Methanol is a clear, colorless, alcohol found in antifreeze, windshield washer fluid, and race car fuel.2 Although methanol reaches peak plasma concentration in about 30 to 60 minutes, signs of systemic toxicity (ie, metabolic acidosis) typically take 6 to 12 hours to manifest.1
In both EG and methanol, there is a delay in toxic clinical findings because the parent compounds are not toxic in their initial form; rather, major toxicity is derived from their metabolites: formic acid and oxalic acid, respectively.
Other Toxins. Many other potentially toxic liquids are associated with a homeowner’s occupation or avocational interests. These include painting supplies (eg, industrial paints containing lead), gardening materials (eg, pesticides containing organophosphates), fuels (eg, gasoline, polychlorinated biphenyls in coolant, and lubricants), and cleaning supplies (eg, caustics, detergents, and air freshener).
Case Continuation
Since the patient’s elevated anion gap raised concerns for methanol or EG exposure, he was given fomepizole and transferred to a tertiary care children’s hospital for further management and possible hemodialysis. Upon arrival at the receiving hospital, the patient’s vital signs and physical examination remained unchanged. Repeat laboratory studies were notable for a creatinine level of 0.3 mg/dL. The patient’s father was instructed to retrieve the implicated bottle from home. An inspection of the bottle’s ingredients was notable for nitromethane, castor oil, and methanol.
What is nitromethane and what are its uses?
Nitromethane, the simplest nitro compound, is a colorless, viscous, lipid-soluble fluid.3 The polarity of nitromethane permits its use as a stabilizer in a number of chemical solvents, such as dry cleaning fluid, degreasers, and "super glue."4,5 Nitromethane is also commonly added to model-engine and drag-race fuels, which also contain methanol and castor oil.3 In this capacity, nitromethane functions as an oxygen carrier, allowing more efficient fuel use in combustion cylinders (compared to gasoline), thereby increasing the horsepower of the vehicle.6 It is therefore commonly added to fuel for drag racers, radio-controlled cars, and model aircrafts.4 In the small concentrations typically inadvertently ingested, the clinical effects of nitromethane itself are inconsequential.
What is the differential for creatinine elevation?
Creatinine itself is a normal breakdown product of muscle metabolism produced by spontaneous conversion from creatine and is found at a fairly constant serum level in proportion to muscle mass.7 Thus, as people age and muscle mass decreases, their baseline creatinine levels decrease proportionally.
Elimination. The majority of creatinine (85%-90%) is filtered and excreted by the kidneys, with the remaining 10% to 15% secreted by the tubules, allowing creatinine to be a surrogate measure of the glomerular filtration rate.7 Exogenous sources of creatine or creatinine include meat and creatine supplements, the latter of which are used as an "energy source" to enhance athletic performance.
Etiology. The etiology for an elevated serum creatinine concentration includes renal failure, both acute and chronic; volume depletion; hemorrhage (low blood volume); and medications, including diuretics, angiotensin converting enzyme inhibitors, angiotensin-receptor blockers, nonsteroidal anti-inflammatory drugs, and certain antibiotics. These etiologies can also be categorized as processes that increase creatinine production, decrease elimination (H2 antagonist and trimethoprim both inhibit the cation secretory pump in the tubules), or interfere with the creatinine assay (ketones, keto acids, lipemia, hemolysis, cephalosporins).7
Because creatinine is filtered so efficiently by the kidney, neither exogenous nor endogenous creatinine sources are expected to increase serum creatinine in the absence of renal dysfunction. However, transient elevation may occur in body builders who use extreme doses of creatine. Patients with rhabdomyolysis often develop elevated creatinine concentrations, but nearly always in the setting of myoglobinuric renal failure.
Jaffe Reaction and Enzymatic Methods. Serum creatinine can be measured using either the Jaffe reaction or the enzymatic method. In the Jaffe reaction, creatinine reacts with alkaline sodium picrate to form a red-orange chromophore, which absorbs light in the range of 470 to 550 nanometers on spectroscopy.6,8,9 The active methylene group on nitromethane also reacts with alkaline sodium picrate to form a chromophore which absorbs light in the same wavelength range.10 Thus, serum creatinine measurements via the Jaffe reaction are falsely elevated due to the cross-reactivity between nitromethane and alkaline sodium picrate. In some reported cases, there is a 20-fold increase in the measured serum creatinine in the presence of nitromethane; renal function, however, remains normal.5
This false reading seen in the Jaffe reaction can be avoided by utilizing the enzymatic method of creatinine measurement, a three-step process that ultimately produces hydrogen peroxide, which is measured and accurately correlates with serum creatinine—even in the presence of nitromethane.8 This distinction explains the dramatically different creatinine concentrations measured at the two institutions in this case.
Case Conclusion
The patient was monitored overnight at the children’s hospital. Repeat laboratory studies in the morning showed a normal creatinine level of 0.3 mg/dL and a negative methanol level. The patient was discharged home in the care of his father, who was instructed to follow-up with his son’s pediatrician. The father also received counseling on safe storage practices for dangerous chemicals.
Case
An otherwise healthy 20-month-old boy presented to the ED for evaluation after his father witnessed the child ingest a model race car fuel additive. According to the patient’s father, the boy was playing with several closed bottles that were stored in the garage, when he witnessed the boy open up and take a sip of a pink-colored fuel additive, which the father believed to contain 100% methanol. The patient’s father further noted that immediately after drinking the fluid, the patient spat and drooled, and had one episode of nonbloody emesis prior to arrival at the ED.
Initial vital signs at presentation were: blood pressure, 84/54 mm Hg; heart rate, 97 beats/min; respiratory rate, 24 breaths/min; and temperature 98°F. Oxygen saturation was 99% on room air. Physical examination was notable for mild erythema in the posterior oropharynx. Otherwise, the patient was acting appropriately for his age and in no acute distress. Laboratory studies were within normal limits, except for the following: serum anion gap, 18 mEq/L (reference range for children < 3 years old, 10-14 mEq/L); serum bicarbonate, 19 mmol/L (reference range for children 12-24 months, 17-25 mmol/L); and serum creatinine, 2.8 mg/dL (reference range for children 12 to 24 months, 0.2-0.5 mg/dL). A repeat creatinine test taken after bolus of fluid administration was 2.4 mg/dL. A renal ultrasound, performed to investigate the cause of the renal failure, was unremarkable.
What toxic exposures are of concern based on the clinical history?
The history of exposure to a liquid stored in a garage raises the likelihood of exposure to an automobile-related item such as diethylene glycol, ethylene glycol (EG), and methanol.
Diethylene Glycol. Diethylene glycol is an ingredient in brake and power steering fluids, and has toxic properties qualitatively similar to EG.
Ethylene Glycol. A clear, colorless, odorless fluid with a sweet taste, EG is an ingredient in radiator antifreeze, refrigerant fluid, coolants, and pesticides. Like methylene, EG reaches peak plasma concentration within 1 to 4 hours, but toxic clinical findings do not occur for 3 to 6 hours.1
Methanol. Methanol is a clear, colorless, alcohol found in antifreeze, windshield washer fluid, and race car fuel.2 Although methanol reaches peak plasma concentration in about 30 to 60 minutes, signs of systemic toxicity (ie, metabolic acidosis) typically take 6 to 12 hours to manifest.1
In both EG and methanol, there is a delay in toxic clinical findings because the parent compounds are not toxic in their initial form; rather, major toxicity is derived from their metabolites: formic acid and oxalic acid, respectively.
Other Toxins. Many other potentially toxic liquids are associated with a homeowner’s occupation or avocational interests. These include painting supplies (eg, industrial paints containing lead), gardening materials (eg, pesticides containing organophosphates), fuels (eg, gasoline, polychlorinated biphenyls in coolant, and lubricants), and cleaning supplies (eg, caustics, detergents, and air freshener).
Case Continuation
Since the patient’s elevated anion gap raised concerns for methanol or EG exposure, he was given fomepizole and transferred to a tertiary care children’s hospital for further management and possible hemodialysis. Upon arrival at the receiving hospital, the patient’s vital signs and physical examination remained unchanged. Repeat laboratory studies were notable for a creatinine level of 0.3 mg/dL. The patient’s father was instructed to retrieve the implicated bottle from home. An inspection of the bottle’s ingredients was notable for nitromethane, castor oil, and methanol.
What is nitromethane and what are its uses?
Nitromethane, the simplest nitro compound, is a colorless, viscous, lipid-soluble fluid.3 The polarity of nitromethane permits its use as a stabilizer in a number of chemical solvents, such as dry cleaning fluid, degreasers, and "super glue."4,5 Nitromethane is also commonly added to model-engine and drag-race fuels, which also contain methanol and castor oil.3 In this capacity, nitromethane functions as an oxygen carrier, allowing more efficient fuel use in combustion cylinders (compared to gasoline), thereby increasing the horsepower of the vehicle.6 It is therefore commonly added to fuel for drag racers, radio-controlled cars, and model aircrafts.4 In the small concentrations typically inadvertently ingested, the clinical effects of nitromethane itself are inconsequential.
What is the differential for creatinine elevation?
Creatinine itself is a normal breakdown product of muscle metabolism produced by spontaneous conversion from creatine and is found at a fairly constant serum level in proportion to muscle mass.7 Thus, as people age and muscle mass decreases, their baseline creatinine levels decrease proportionally.
Elimination. The majority of creatinine (85%-90%) is filtered and excreted by the kidneys, with the remaining 10% to 15% secreted by the tubules, allowing creatinine to be a surrogate measure of the glomerular filtration rate.7 Exogenous sources of creatine or creatinine include meat and creatine supplements, the latter of which are used as an "energy source" to enhance athletic performance.
Etiology. The etiology for an elevated serum creatinine concentration includes renal failure, both acute and chronic; volume depletion; hemorrhage (low blood volume); and medications, including diuretics, angiotensin converting enzyme inhibitors, angiotensin-receptor blockers, nonsteroidal anti-inflammatory drugs, and certain antibiotics. These etiologies can also be categorized as processes that increase creatinine production, decrease elimination (H2 antagonist and trimethoprim both inhibit the cation secretory pump in the tubules), or interfere with the creatinine assay (ketones, keto acids, lipemia, hemolysis, cephalosporins).7
Because creatinine is filtered so efficiently by the kidney, neither exogenous nor endogenous creatinine sources are expected to increase serum creatinine in the absence of renal dysfunction. However, transient elevation may occur in body builders who use extreme doses of creatine. Patients with rhabdomyolysis often develop elevated creatinine concentrations, but nearly always in the setting of myoglobinuric renal failure.
Jaffe Reaction and Enzymatic Methods. Serum creatinine can be measured using either the Jaffe reaction or the enzymatic method. In the Jaffe reaction, creatinine reacts with alkaline sodium picrate to form a red-orange chromophore, which absorbs light in the range of 470 to 550 nanometers on spectroscopy.6,8,9 The active methylene group on nitromethane also reacts with alkaline sodium picrate to form a chromophore which absorbs light in the same wavelength range.10 Thus, serum creatinine measurements via the Jaffe reaction are falsely elevated due to the cross-reactivity between nitromethane and alkaline sodium picrate. In some reported cases, there is a 20-fold increase in the measured serum creatinine in the presence of nitromethane; renal function, however, remains normal.5
This false reading seen in the Jaffe reaction can be avoided by utilizing the enzymatic method of creatinine measurement, a three-step process that ultimately produces hydrogen peroxide, which is measured and accurately correlates with serum creatinine—even in the presence of nitromethane.8 This distinction explains the dramatically different creatinine concentrations measured at the two institutions in this case.
Case Conclusion
The patient was monitored overnight at the children’s hospital. Repeat laboratory studies in the morning showed a normal creatinine level of 0.3 mg/dL and a negative methanol level. The patient was discharged home in the care of his father, who was instructed to follow-up with his son’s pediatrician. The father also received counseling on safe storage practices for dangerous chemicals.
1. Kruse JA. Methanol and ethylene glycol intoxication. Crit Care Clin. 2012;28(4):661-711. doi:10.1016/j.ccc.2012.07.002.
2. McMahon DM, Winstead S, Weant KA. Toxic alcohol ingestions: focus on ethylene glycol and methanol. Adv Emerg Nurs J. 2009;31(3):206-213. doi:10.1097/TME.0b013e3181ad8be8.
3. Cook MD, Clark RF. Creatinine elevation associated with nitromethane exposure: a marker of potential methanol toxicity. J Emerg Med. 2007;33(3):249-253. doi:10.1016/j.jemermed.2007.02.015.
4. Markofsky SB. Nitro compounds, aliphatic. In: Elvers B, ed. Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA; 2000. doi:10.1002/14356007.a17_401. [digital]
5. Mullins ME, Hammett-Stabler CA. Intoxication with nitromethane-containing fuels: don’t be "fueled" by the creatinine. J Toxicol Clin Toxicol. 1998;36(4):
315-320.
6. Ngo AS, Rowley F, Olson KR. Case files of the California poison control system, San Francisco division: blue thunder ingestion: methanol, nitromethane, and elevated creatinine. J Med Toxicol. 2010;6(1):67-71. doi:10.1007/s13181-010-0042-5.
7. Samra M, Abcar AC. False estimates of elevated creatinine. Perm J. 2012;16(2):51-52.
8. Booth C, Naidoo D, Rosenberg A, Kainer G. Elevated creatinine after ingestion of model aviation fuel: interference with the Jaffe reaction by nitromethane. J Paediatr Child Health. 1999;35(5):503-504.
9. de Lelis Medeiros de Morais C, Gomes de Lima KM. Determination and analytical validation of creatinine content in serum using image analysis by multivariate transfer calibration procedures. Anal Meth. 2015;7:6904-6910. doi:10.1039/C5AY01369K.
10. Killorn E, Lim RK, Rieder M. Apparent elevated creatinine after ingestion of nitromethane: interference with the Jaffe reaction. Ther Drug Monit. 2011;33(1):1-2. doi:10.1097/FTD.0b013e3181fe7e52.
1. Kruse JA. Methanol and ethylene glycol intoxication. Crit Care Clin. 2012;28(4):661-711. doi:10.1016/j.ccc.2012.07.002.
2. McMahon DM, Winstead S, Weant KA. Toxic alcohol ingestions: focus on ethylene glycol and methanol. Adv Emerg Nurs J. 2009;31(3):206-213. doi:10.1097/TME.0b013e3181ad8be8.
3. Cook MD, Clark RF. Creatinine elevation associated with nitromethane exposure: a marker of potential methanol toxicity. J Emerg Med. 2007;33(3):249-253. doi:10.1016/j.jemermed.2007.02.015.
4. Markofsky SB. Nitro compounds, aliphatic. In: Elvers B, ed. Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA; 2000. doi:10.1002/14356007.a17_401. [digital]
5. Mullins ME, Hammett-Stabler CA. Intoxication with nitromethane-containing fuels: don’t be "fueled" by the creatinine. J Toxicol Clin Toxicol. 1998;36(4):
315-320.
6. Ngo AS, Rowley F, Olson KR. Case files of the California poison control system, San Francisco division: blue thunder ingestion: methanol, nitromethane, and elevated creatinine. J Med Toxicol. 2010;6(1):67-71. doi:10.1007/s13181-010-0042-5.
7. Samra M, Abcar AC. False estimates of elevated creatinine. Perm J. 2012;16(2):51-52.
8. Booth C, Naidoo D, Rosenberg A, Kainer G. Elevated creatinine after ingestion of model aviation fuel: interference with the Jaffe reaction by nitromethane. J Paediatr Child Health. 1999;35(5):503-504.
9. de Lelis Medeiros de Morais C, Gomes de Lima KM. Determination and analytical validation of creatinine content in serum using image analysis by multivariate transfer calibration procedures. Anal Meth. 2015;7:6904-6910. doi:10.1039/C5AY01369K.
10. Killorn E, Lim RK, Rieder M. Apparent elevated creatinine after ingestion of nitromethane: interference with the Jaffe reaction. Ther Drug Monit. 2011;33(1):1-2. doi:10.1097/FTD.0b013e3181fe7e52.

Man, 32, With Severe Scrotal Pain and Swelling
IN THIS ARTICLE
- Lab values for case patient
- Differential diagnoses
- Case outcome
A 32-year-old man presents to the urgent care center at a community hospital with severe scrotal pain and swelling of five days’ duration. What began as mild left scrotal discomfort is now causing increasing pain, swelling, hematuria, dysuria, low-grade fever, and nausea, prompting him to seek medical attention.
The patient, who is a pipefitter in a hospital, was at work when his symptoms began. He denies any history of scrotal trauma, and his review of systems is otherwise unremarkable. His medical history is significant for mild hypertension and morbid obesity, but he is not immunocompromised. Two months ago, he had an excision and repair of a left ureterocele, for which he was treated prophylactically with ciprofloxacin for one week. He has a 3–pack-year history of smoking and consumes three alcoholic beverages per week. He denies illicit drug use and has no report of sexually transmitted infection.
Upon arrival to urgent care, the patient appears to be in moderate distress, with a blood pressure (BP) of 111/79 mm Hg; pulse, 104 beats/min; respiratory rate, 18 breaths/min-1; temperature, 100.1°F; and SpO2, 94%. Physical exam reveals left scrotal erythema, severe tenderness upon palpation, marked scrotal edema, and a slight amount of foul-smelling discharge seeping from a pinpoint opening in the left perineum (see Figure 1a). Given his scrotal presentation, he is quickly transferred to a regional emergency department (ED) for a urology consult.

In the ED, lab testing yields significant findings (see Table 1). His ECG demonstrates sinus tachycardia at 126 beats/min without rhythm or ST changes. His urinalysis reveals a cloudy appearance, a protein level of 100 mg/dL, and trace leukocyte esterase.
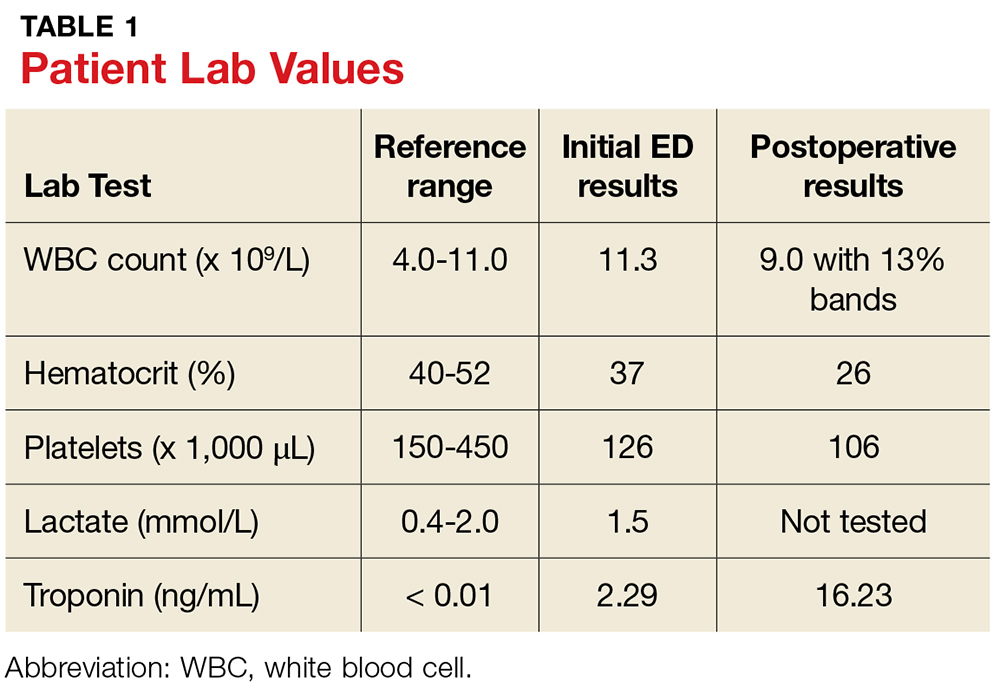
Urgent CT with contrast is obtained; it shows significant soft-tissue inflammatory changes in the left groin and scrotum that extend into the left thigh. In addition, a collection of fluid is seen in the inferior aspect of the left scrotal wall, indicating a probable abscess. There is no free air or lymphadenopathy.
Given the patient’s worsening condition and his apparent advancement to a systemic inflammatory response syndrome, surgical consult is obtained. He is diagnosed with a scrotal abscess and cellulitis; two blood and two scrotal cultures are obtained, and the patient is empirically started on IV ampicillin and gentamicin.
Two hours later, he has a BP of 122/74 mm Hg; pulse, 112 beats/min; respiratory rate, 20 breaths/min-1; and temperature, 103.1°F. His genital inflammation has advanced to the perineum and the left lower abdomen. The purulent, bloody, foul-smelling drainage from the opening in the left perineum is increasingly apparent. The patient is taken emergently to surgery for an incision and drainage, along with exploration of the scrotal abscess. During surgery, the patient is discovered to have Fournier’s gangrene.
DISCUSSION
Fournier’s gangrene (FG) is a necrotizing fasciitis of the perineal, perianal, and/or genital areas involving the superficial and deep fascial planes while sparing the deep muscular structures and overlying skin.1 A rare but potentially fatal disease, FG spreads at a rate of up to 3 cm/h.2,3
Mortality rates range from 7.5% to 88%, with the highest mortality occurring within the first 96 hours of hospitalization.1,4-7 Mortality is often related to the onset of sepsis.4,5 Survival requires early recognition; immediate, aggressive surgical debridement of all necrotic tissue; and concomitant, early administration of appropriate antibiotics.1,4,5,8 Mortality risk and prognosis are improved in patients younger than 60 with localized disease and no toxicity, along with sterile blood cultures.1
Risk Factors
FG is most commonly seen in males between the ages of 50 and 70, with a 10:1 male-to-female ratio.3,9 Impaired immunity typically increases a patient’s susceptibility to FG, with type 2 diabetes having the highest incidence (85% of patients).1,4,6,8,10 Other conditions that can increase the risk for FG include obesity, alcoholism, cirrhosis, cardiac disease, tobacco use, peripheral vascular disease, malignancy, chronic steroid use, renal insufficiency, IV drug abuse, and HIV.1,4,6,8,9,11
Trauma frequently initiates the infectious process,with urogenital trauma (eg, placement of urethral instrumentation, surgery, and urinary tract infection) being the main cause of bacterial introduction.1,3 Localized infection causes the development of an obliterative endarteritis, resulting in subcutaneous vascular ischemia, necrosis, and bacterial proliferation.3,7,9
Presentation and Diagnosis
Presenting symptoms of FG include intense, abrupt genital pain that is disproportionate to the physical exam findings.9 This rapidly escalates to include extreme swelling, erythema, bullae, discolored skin, and tissue crepitus with eventual necrosis.2,10 Lab results typically show leukocytosis > 18.0 × 109/L.4 The testicle and spermatic cord are generally unaffected (as in this patient), due to the anatomic relationship between the various layers of fascia within the scrotum and the anterior abdominal wall, as well as the independent blood supply of the compartmentalized testicular tissue.1-3
During an exam of the acute scrotum, the differential diagnosis includes cellulitis, scrotal abscess, acute epididymitis, and testicular torsion, with scrotal abscess being most frequently diagnosed (57% of patients).9,11,12 The distinguishing features of these diagnoses can be found in Table 2. Necrotizing fasciitis in the form of FG tends to be an unexpected, rare finding usually only diagnosed during the surgical draining of an abscess.12

CT is the test of choice to detect FG and determine the extent of its spread by identifying subcutaneous air/gas within the involved fascial planes.10,13 However, an incisional biopsy with culture is needed to confirm the diagnosis.3,9 Most patients with FG require an average of four surgeries (eg, reconstruction, skin grafting, and possibly colostomy if the infection has entered the peritoneal cavity) in order to eradicate the disease and achieve the best functional and cosmetic outcome.4
Etiology
About 83% of FG cases are polymicrobial infections comprised of enterobacter, enterococci, Escherichia coli, group A streptococci, pseudomonas, and clostridium, with symptoms evolving two to four days following the initial insult.4,7,11,14,15 Monomicrobial infections are much less common, but the symptoms progress even more rapidly.15 Methicillin-resistant Staphylococcus aureus (MRSA) necrotizing fasciitis infections occur in about 3% of monomicrobial cases.12 MRSA emerged in the early 2000s as an additional causative pathogen for polymicrobial necrotizing fasciitis infections.12,14,15 Prior to that time, S aureus strains were almost uniformly susceptible to penicillinase-resistant ß lactams.12
A distinction should be made between health care-associated (HA) MRSA and community-acquired (CA) MRSA due to treatment considerations. HA-MRSA infections are contracted through previous health care exposure (within the past year) and are less resistant to treatment.16,17 In contrast, CA-MRSA, which comprises 29% of MRSA cases, causes infections in previously healthy young patients without prior health care contact within the past year.16 CA-MRSA strains are more robust than HA-MRSA strains and can cause sepsis and other invasive, rapidly progressive, and possibly life-threatening infections due to the amount of tissue destruction and necrosis.16,18 Transmission of CA-MRSA is often associated with crowded environments, frequent skin-to-skin contact, compromised skin integrity, contaminated items or surfaces, and lack of cleanliness.16 Over the years, CA-MRSA has developed resistance to multiple antimicrobials; providers should therefore consider CA-MRSA on initial evaluation of necrotizing infections, to ensure appropriate initiation of treatment.12,16
CASE CONTINUED
Extensive debridement was completed down to healthy tissue in all affected areas (see Figure 1b). The necrotizing fasciitis had spared the left testicle and spermatic cord, and a colostomy was not required.
The patient’s initial postoperative vital signs were unremarkable, except for his BP (86/54 mm Hg). The patient was taken postoperatively to the surgical intensive care unit (SICU) with the diagnosis of FG. Aggressive IV fluids were administered for resuscitation, and he was closely monitored for increasing sepsis. Metronidazole was added for anaerobic and gram-positive coverage. His postoperative lab results can also be found in Table 1.
His ECG showed a normal sinus rhythm without ST changes, and he denied any cardiac symptoms. His physical exam was significant for mild pallor, dry mucus membranes, and a left scrotal and pelvic packed dressing. He was given two units of packed red blood cells for acute postoperative blood-loss anemia. The preliminary tissue culture results showed gram-positive cocci consistent with a staphylococcal infection; his antibiotics were then changed to IV ampicillin/sulbactam and clindamycin.
Approximately five hours postoperatively, an ECG suddenly showed acute ST elevation in leads II, II, and aVF, with reciprocal changes. The patient was diagnosed with an acute myocardial infarction (AMI). He denied any chest pain, shortness of breath, or diaphoresis. The SICU team initiated aspirin therapy and immediately contacted cardiology for an emergent coronary angiogram.
The angiogram and cardiac catheterization revealed an elevated left ventricular end diastolic (LVED) volume, inferior wall hypokinesis, a low-normal ejection fraction, and a 30% lesion in the first diagonal of his left anterior descending artery. A postprocedure echocardiogram demonstrated left ventricular (LV) ejection fraction of 50%, with LV hypokinesis in the inferior base and mild left atrial enlargement. The patient was started on metoprolol for myocardial protection and recovery.
Complications
Perioperative complications of FG, including AMI, must be considered due to the physiologic stress on the body.19 Most patients with perioperative AMI after noncardiac surgery do not experience ischemic symptoms.20
Growing evidence suggests the pivotal role of acute inflammation (postoperatively or from infection) as a precipitating event in AMI.20,21 Chemical mediators, such as inflammatory cytokines, endotoxins, and nitric oxide, may play a role in the development of an AMI.22
If cardiovascular disease and/or significant cardiovascular risk factors (ie, older age, male, cigarette smoking, cardiac family history, acute kidney injury) are present, the risk for AMI increases in the first two days following surgery.21,23 Acute infections and sepsis also initiate or increase systemic inflammatory activity via these same chemical mediators.21
Most suspected infectious agents also produce coronary artery sheer stress and destabilization of vulnerable plaques, leading to plaque rupture and thrombosis.19,24 Proinflammatory cytokines promote enhanced platelet activation and contribute to this thrombotic environment.21,23 Thrombus leads to obstructed coronary blood flow, myocardial ischemia, and finally, infarction.21
A reversible myocardial depression, cardiomyopathy, or myocardial ischemia may occur in patients with acute systemic infection or sepsis when the myocardium is functionally and structurally injured by these inflammatory chemical mediators.19,22-24 Characteristics of such a cardiomyopathy include left ventricle dilation with a low filling pressure, an abnormal increase in LVED volume, and a depressed ejection fraction.22
An acute infectious or septic process can raise troponin levels in 43% to 85% of patients.22,24 Troponin biomarkers can assist in predicting myocardial injury and events after surgery with nearly absolute myocardial tissue specificity.20 Cardiovascular involvement caused by myocardial injury–related sepsis is observed in up to 70% of patients in the ICU for these reasons.23 Therefore, providers should consider measuring troponin biomarkers during such infectious and septic processes, as this team did for the case patient. The providers were able to diagnose his AMI early and institute appropriate treatment measures to avoid extensive myocardial tissue damage.
Several studies have already demonstrated a correlation between pneumococcal pneumonia and an increased risk for AMI, and the same mechanisms are presumed responsible for any severe acute infectious state.21 More research is needed to understand the pathophysiology of AMI in sepsis and acute systemic infections.23
OUTCOME FOR THE CASE PATIENT
On postoperative day 2, the patient’s vital signs and lab results were normal. Additional lab results included an A1C of 5.2%. His ECG showed a resolving ST-elevation myocardial infarction (STEMI). The surgical wound had initiation of early granulation tissue without any further signs of necrosis.
A postoperative acute STEMI was unexpected in this patient, as his only risk factors included being male, mild hypertension, obesity, and tobacco use. At the time of his initial elevated troponin level, he had no cardiac symptoms or ECG changes. This initial high troponin level may have been stress-induced from the acute infectious process, and his acute inferior wall STEMI may have been secondary to a transient thrombotic event. The STEMI may then have resolved on its own during the cardiac catheterization with the administration of heparin, IV fluids, blood products, aspirin, or dye infiltration, thus enhancing reperfusion of the coronary artery system.
The final tissue culture showed MRSA. Given his job and his history of a genitourinary procedure, as well as the less fulminant form of disease and relatively quick recovery, it was likely HA-MRSA (rather than CA-MRSA). Only clindamycin was used for treatment.
The wound continued to have decreasing erythema, a reduction in tenderness, and evidence of viable, pink granulation tissue. HIV testing was not completed during his admission. The remainder of the patient’s hospital course was unremarkable, and he was discharged home with wound care, urology, and cardiology follow-up services.
CONCLUSION
Multiple factors contribute to a delayed or mistaken diagnosis of FG; it may be overlooked in the initial working diagnoses because of its low incidence and manifestations similar to those of other soft-tissue infections (eg, cellulitis, scrotal abscess). The cutaneous signs of FG often lag behind the disease manifestation, with minimal or no external presence while extensive internal tissue destruction is occurring. Constant review of symptoms is required when treating patients with soft-tissue infections, and early signs—such as pain out of proportion to physical findings—should prompt a clinician to include FG in the differential.
Early diagnosis with prompt debridement and antibiotic therapy are crucial to patient survival. Detecting FG within the first 24 hours is critical. Further differentiation between CA-MRSA and HA-MRSA can assist in patient recovery and survival by guiding appropriate antibiotic therapy. Perioperative risk assessment and serial troponin biomarkers may identify patients in need of intensive monitoring and management postoperatively to avoid an AMI, since patients may not experience ischemic symptoms.
1. Norton KS, Johnson LW, Perry T, et al. Management of Fournier’s gangrene: an eleven-year retrospective analysis of early recognition, diagnosis, and treatment. Am Surg. 2002;68(8):709-713.
2. Agostini T, Mori F, Perello R, et al. Successful combined approach to a severe Fournier’s gangrene. Indian J Plast Surg. 2014;47(1):132-136.
3. Cabrera G, March P. Fournier’s gangrene. Glendale, CA: Cinahl Information Systems; 2016.
4. Czymek R, Kujath P, Bruch HP, et al. Treatment, outcome and quality of life after Fournier’s gangrene: a multicentre study. Colorectal Dis. 2013;15(12):1529-1536.
5. Sugihara T, Yasunaga H, Horiguchi H, et al. Impact of surgical intervention timing on the case fatality rate for Fournier’s gangrene: an analysis of 379 cases. BJU Int. 2012;110(11c):E1096-1100.
6. Tuncel A, Keten T, Aslan Y, et al. Comparison of different scoring systems for outcome prediction in patients with Fournier’s gangrene: experience with 50 patients. Scand J Urol. 2014;48(4):393-399.
7. Taken K, Oncu MR, Ergun M, et al. Fournier’s gangrene: causes, presentation and survival of sixty-five patients. Pak J Med Sci. 2016;32(3):746-750.
8. Palvolgyi R, Kaji AH, Valeriano J, et al. Fournier’s gangrene: a model for early prediction. Am Surg. 2014;80(10):926-931.
9. Pais V, Santora T. Fournier gangrene. http://emedicine.medscape.com/article/2028899-overview. Accessed August 16, 2017.
10. Cottrill RR. A demonstration of clinical reasoning through a case of scrotal infection. Urol Nurs. 2013;33(1):33-37.
11. Summers A. Fournier’s gangrene. J Nurse Pract. 2014;10(8):582-587.
12. Miller LG, Perdreau-Remington F, Rieg G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med. 2005;352(14):1445-1453.
13. Gupta N, Zinn K, Bansal I, Weinstein R. Fournier’s gangrene: ultrasound or computed tomography? A letter to the editor. Med Ultrason. 2014;16(4):389-390.
14. Bjurlin MA, O’Grady T, Kim DY, et al. Causative pathogens, antibiotic sensitivity, resistance patterns, and severity in a contemporary series of Fournier’s gangrene. Urol. 2013;81(4):752-758.
15. Goh T, Goh LG. Pitfalls in diagnosing necrotizing fasciitis. https://psnet.ahrq.gov/webmm/case/329/pitfalls-in-diagnos ing-necrotizing-fasciitis. Accessed August 16, 2017.
16. Kale P, Dhawan B. The changing face of community-acquired methicillin-resistant Staphylococcus aureus. Indian J Med Microbiol. 2016;34(3):275-285.
17. CDC. Necrotizing fasciitis. www.cdc.gov/Features/NecrotizingFasciitis/index.html. Accessed August 16, 2017.
18. Barnes BE, Sampson DA. A literature review on community-acquired methicillin-resistant Staphylococcus aureus in the United States: clinical information for primary care nurse practitioners. J Am Acad Nurse Pract. 2011;23(1):23-32.
19. Madjid M, Vela D, Khalili-Tabrizi H, et al. Systemic infections cause exaggerated local inflammation in atherosclerotic coronary arteries. Clues to the triggering effect of acute infections on acute coronary syndromes. Tex Heart Inst J. 2007;34(1):11-18.
20. Devereaux PJ, Chan MTV, Alonso-Coello PA, et al; VISION Study Investigators. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA. 2012;307(21):2295-2304.
21. Corrales-Medina VF, Fatemi O, Serpa J, et al. The association between Staphylococcus aureus bacteremia and acute myocardial infarction. Scand J Infect Dis. 2009;41(6-7):511-514.
22. Romero-Bermejo FJ, Ruiz-Bailen M, Gil-Cebrian J, Huertos-Ranchal MJ. Sepsis-induced cardiomyopathy. Curr Cardiol Rev. 2011;7(3):163-183.
23. Smilowitz NR, Gupta N, Guo Y, Bangalore S. Comparison of outcomes of patients with sepsis with versus without acute myocardial infarction and comparison of invasive versus noninvasive management of the patients with infarction. Am J Cardiol. 2016;117(7):1065-1071.
24. Mattson M. Sepsis and cardiac disease: improving outcomes through recognition and management. Prog Cardiovasc Nurs. 2009;24(4):199-201.
25. Papadakis MA, McPhee SJ. Current Medical Diagnosis & Treatment. 54th ed. New York, NY: McGraw Hill Education; 2015:137-138, 151-152, 937.
26. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed August 16, 2017.
IN THIS ARTICLE
- Lab values for case patient
- Differential diagnoses
- Case outcome
A 32-year-old man presents to the urgent care center at a community hospital with severe scrotal pain and swelling of five days’ duration. What began as mild left scrotal discomfort is now causing increasing pain, swelling, hematuria, dysuria, low-grade fever, and nausea, prompting him to seek medical attention.
The patient, who is a pipefitter in a hospital, was at work when his symptoms began. He denies any history of scrotal trauma, and his review of systems is otherwise unremarkable. His medical history is significant for mild hypertension and morbid obesity, but he is not immunocompromised. Two months ago, he had an excision and repair of a left ureterocele, for which he was treated prophylactically with ciprofloxacin for one week. He has a 3–pack-year history of smoking and consumes three alcoholic beverages per week. He denies illicit drug use and has no report of sexually transmitted infection.
Upon arrival to urgent care, the patient appears to be in moderate distress, with a blood pressure (BP) of 111/79 mm Hg; pulse, 104 beats/min; respiratory rate, 18 breaths/min-1; temperature, 100.1°F; and SpO2, 94%. Physical exam reveals left scrotal erythema, severe tenderness upon palpation, marked scrotal edema, and a slight amount of foul-smelling discharge seeping from a pinpoint opening in the left perineum (see Figure 1a). Given his scrotal presentation, he is quickly transferred to a regional emergency department (ED) for a urology consult.
In the ED, lab testing yields significant findings (see Table 1). His ECG demonstrates sinus tachycardia at 126 beats/min without rhythm or ST changes. His urinalysis reveals a cloudy appearance, a protein level of 100 mg/dL, and trace leukocyte esterase.
Urgent CT with contrast is obtained; it shows significant soft-tissue inflammatory changes in the left groin and scrotum that extend into the left thigh. In addition, a collection of fluid is seen in the inferior aspect of the left scrotal wall, indicating a probable abscess. There is no free air or lymphadenopathy.
Given the patient’s worsening condition and his apparent advancement to a systemic inflammatory response syndrome, surgical consult is obtained. He is diagnosed with a scrotal abscess and cellulitis; two blood and two scrotal cultures are obtained, and the patient is empirically started on IV ampicillin and gentamicin.
Two hours later, he has a BP of 122/74 mm Hg; pulse, 112 beats/min; respiratory rate, 20 breaths/min-1; and temperature, 103.1°F. His genital inflammation has advanced to the perineum and the left lower abdomen. The purulent, bloody, foul-smelling drainage from the opening in the left perineum is increasingly apparent. The patient is taken emergently to surgery for an incision and drainage, along with exploration of the scrotal abscess. During surgery, the patient is discovered to have Fournier’s gangrene.
DISCUSSION
Fournier’s gangrene (FG) is a necrotizing fasciitis of the perineal, perianal, and/or genital areas involving the superficial and deep fascial planes while sparing the deep muscular structures and overlying skin.1 A rare but potentially fatal disease, FG spreads at a rate of up to 3 cm/h.2,3
Mortality rates range from 7.5% to 88%, with the highest mortality occurring within the first 96 hours of hospitalization.1,4-7 Mortality is often related to the onset of sepsis.4,5 Survival requires early recognition; immediate, aggressive surgical debridement of all necrotic tissue; and concomitant, early administration of appropriate antibiotics.1,4,5,8 Mortality risk and prognosis are improved in patients younger than 60 with localized disease and no toxicity, along with sterile blood cultures.1
Risk Factors
FG is most commonly seen in males between the ages of 50 and 70, with a 10:1 male-to-female ratio.3,9 Impaired immunity typically increases a patient’s susceptibility to FG, with type 2 diabetes having the highest incidence (85% of patients).1,4,6,8,10 Other conditions that can increase the risk for FG include obesity, alcoholism, cirrhosis, cardiac disease, tobacco use, peripheral vascular disease, malignancy, chronic steroid use, renal insufficiency, IV drug abuse, and HIV.1,4,6,8,9,11
Trauma frequently initiates the infectious process,with urogenital trauma (eg, placement of urethral instrumentation, surgery, and urinary tract infection) being the main cause of bacterial introduction.1,3 Localized infection causes the development of an obliterative endarteritis, resulting in subcutaneous vascular ischemia, necrosis, and bacterial proliferation.3,7,9
Presentation and Diagnosis
Presenting symptoms of FG include intense, abrupt genital pain that is disproportionate to the physical exam findings.9 This rapidly escalates to include extreme swelling, erythema, bullae, discolored skin, and tissue crepitus with eventual necrosis.2,10 Lab results typically show leukocytosis > 18.0 × 109/L.4 The testicle and spermatic cord are generally unaffected (as in this patient), due to the anatomic relationship between the various layers of fascia within the scrotum and the anterior abdominal wall, as well as the independent blood supply of the compartmentalized testicular tissue.1-3
During an exam of the acute scrotum, the differential diagnosis includes cellulitis, scrotal abscess, acute epididymitis, and testicular torsion, with scrotal abscess being most frequently diagnosed (57% of patients).9,11,12 The distinguishing features of these diagnoses can be found in Table 2. Necrotizing fasciitis in the form of FG tends to be an unexpected, rare finding usually only diagnosed during the surgical draining of an abscess.12
CT is the test of choice to detect FG and determine the extent of its spread by identifying subcutaneous air/gas within the involved fascial planes.10,13 However, an incisional biopsy with culture is needed to confirm the diagnosis.3,9 Most patients with FG require an average of four surgeries (eg, reconstruction, skin grafting, and possibly colostomy if the infection has entered the peritoneal cavity) in order to eradicate the disease and achieve the best functional and cosmetic outcome.4
Etiology
About 83% of FG cases are polymicrobial infections comprised of enterobacter, enterococci, Escherichia coli, group A streptococci, pseudomonas, and clostridium, with symptoms evolving two to four days following the initial insult.4,7,11,14,15 Monomicrobial infections are much less common, but the symptoms progress even more rapidly.15 Methicillin-resistant Staphylococcus aureus (MRSA) necrotizing fasciitis infections occur in about 3% of monomicrobial cases.12 MRSA emerged in the early 2000s as an additional causative pathogen for polymicrobial necrotizing fasciitis infections.12,14,15 Prior to that time, S aureus strains were almost uniformly susceptible to penicillinase-resistant ß lactams.12
A distinction should be made between health care-associated (HA) MRSA and community-acquired (CA) MRSA due to treatment considerations. HA-MRSA infections are contracted through previous health care exposure (within the past year) and are less resistant to treatment.16,17 In contrast, CA-MRSA, which comprises 29% of MRSA cases, causes infections in previously healthy young patients without prior health care contact within the past year.16 CA-MRSA strains are more robust than HA-MRSA strains and can cause sepsis and other invasive, rapidly progressive, and possibly life-threatening infections due to the amount of tissue destruction and necrosis.16,18 Transmission of CA-MRSA is often associated with crowded environments, frequent skin-to-skin contact, compromised skin integrity, contaminated items or surfaces, and lack of cleanliness.16 Over the years, CA-MRSA has developed resistance to multiple antimicrobials; providers should therefore consider CA-MRSA on initial evaluation of necrotizing infections, to ensure appropriate initiation of treatment.12,16
CASE CONTINUED
Extensive debridement was completed down to healthy tissue in all affected areas (see Figure 1b). The necrotizing fasciitis had spared the left testicle and spermatic cord, and a colostomy was not required.
The patient’s initial postoperative vital signs were unremarkable, except for his BP (86/54 mm Hg). The patient was taken postoperatively to the surgical intensive care unit (SICU) with the diagnosis of FG. Aggressive IV fluids were administered for resuscitation, and he was closely monitored for increasing sepsis. Metronidazole was added for anaerobic and gram-positive coverage. His postoperative lab results can also be found in Table 1.
His ECG showed a normal sinus rhythm without ST changes, and he denied any cardiac symptoms. His physical exam was significant for mild pallor, dry mucus membranes, and a left scrotal and pelvic packed dressing. He was given two units of packed red blood cells for acute postoperative blood-loss anemia. The preliminary tissue culture results showed gram-positive cocci consistent with a staphylococcal infection; his antibiotics were then changed to IV ampicillin/sulbactam and clindamycin.
Approximately five hours postoperatively, an ECG suddenly showed acute ST elevation in leads II, II, and aVF, with reciprocal changes. The patient was diagnosed with an acute myocardial infarction (AMI). He denied any chest pain, shortness of breath, or diaphoresis. The SICU team initiated aspirin therapy and immediately contacted cardiology for an emergent coronary angiogram.
The angiogram and cardiac catheterization revealed an elevated left ventricular end diastolic (LVED) volume, inferior wall hypokinesis, a low-normal ejection fraction, and a 30% lesion in the first diagonal of his left anterior descending artery. A postprocedure echocardiogram demonstrated left ventricular (LV) ejection fraction of 50%, with LV hypokinesis in the inferior base and mild left atrial enlargement. The patient was started on metoprolol for myocardial protection and recovery.
Complications
Perioperative complications of FG, including AMI, must be considered due to the physiologic stress on the body.19 Most patients with perioperative AMI after noncardiac surgery do not experience ischemic symptoms.20
Growing evidence suggests the pivotal role of acute inflammation (postoperatively or from infection) as a precipitating event in AMI.20,21 Chemical mediators, such as inflammatory cytokines, endotoxins, and nitric oxide, may play a role in the development of an AMI.22
If cardiovascular disease and/or significant cardiovascular risk factors (ie, older age, male, cigarette smoking, cardiac family history, acute kidney injury) are present, the risk for AMI increases in the first two days following surgery.21,23 Acute infections and sepsis also initiate or increase systemic inflammatory activity via these same chemical mediators.21
Most suspected infectious agents also produce coronary artery sheer stress and destabilization of vulnerable plaques, leading to plaque rupture and thrombosis.19,24 Proinflammatory cytokines promote enhanced platelet activation and contribute to this thrombotic environment.21,23 Thrombus leads to obstructed coronary blood flow, myocardial ischemia, and finally, infarction.21
A reversible myocardial depression, cardiomyopathy, or myocardial ischemia may occur in patients with acute systemic infection or sepsis when the myocardium is functionally and structurally injured by these inflammatory chemical mediators.19,22-24 Characteristics of such a cardiomyopathy include left ventricle dilation with a low filling pressure, an abnormal increase in LVED volume, and a depressed ejection fraction.22
An acute infectious or septic process can raise troponin levels in 43% to 85% of patients.22,24 Troponin biomarkers can assist in predicting myocardial injury and events after surgery with nearly absolute myocardial tissue specificity.20 Cardiovascular involvement caused by myocardial injury–related sepsis is observed in up to 70% of patients in the ICU for these reasons.23 Therefore, providers should consider measuring troponin biomarkers during such infectious and septic processes, as this team did for the case patient. The providers were able to diagnose his AMI early and institute appropriate treatment measures to avoid extensive myocardial tissue damage.
Several studies have already demonstrated a correlation between pneumococcal pneumonia and an increased risk for AMI, and the same mechanisms are presumed responsible for any severe acute infectious state.21 More research is needed to understand the pathophysiology of AMI in sepsis and acute systemic infections.23
OUTCOME FOR THE CASE PATIENT
On postoperative day 2, the patient’s vital signs and lab results were normal. Additional lab results included an A1C of 5.2%. His ECG showed a resolving ST-elevation myocardial infarction (STEMI). The surgical wound had initiation of early granulation tissue without any further signs of necrosis.
A postoperative acute STEMI was unexpected in this patient, as his only risk factors included being male, mild hypertension, obesity, and tobacco use. At the time of his initial elevated troponin level, he had no cardiac symptoms or ECG changes. This initial high troponin level may have been stress-induced from the acute infectious process, and his acute inferior wall STEMI may have been secondary to a transient thrombotic event. The STEMI may then have resolved on its own during the cardiac catheterization with the administration of heparin, IV fluids, blood products, aspirin, or dye infiltration, thus enhancing reperfusion of the coronary artery system.
The final tissue culture showed MRSA. Given his job and his history of a genitourinary procedure, as well as the less fulminant form of disease and relatively quick recovery, it was likely HA-MRSA (rather than CA-MRSA). Only clindamycin was used for treatment.
The wound continued to have decreasing erythema, a reduction in tenderness, and evidence of viable, pink granulation tissue. HIV testing was not completed during his admission. The remainder of the patient’s hospital course was unremarkable, and he was discharged home with wound care, urology, and cardiology follow-up services.
CONCLUSION
Multiple factors contribute to a delayed or mistaken diagnosis of FG; it may be overlooked in the initial working diagnoses because of its low incidence and manifestations similar to those of other soft-tissue infections (eg, cellulitis, scrotal abscess). The cutaneous signs of FG often lag behind the disease manifestation, with minimal or no external presence while extensive internal tissue destruction is occurring. Constant review of symptoms is required when treating patients with soft-tissue infections, and early signs—such as pain out of proportion to physical findings—should prompt a clinician to include FG in the differential.
Early diagnosis with prompt debridement and antibiotic therapy are crucial to patient survival. Detecting FG within the first 24 hours is critical. Further differentiation between CA-MRSA and HA-MRSA can assist in patient recovery and survival by guiding appropriate antibiotic therapy. Perioperative risk assessment and serial troponin biomarkers may identify patients in need of intensive monitoring and management postoperatively to avoid an AMI, since patients may not experience ischemic symptoms.
IN THIS ARTICLE
- Lab values for case patient
- Differential diagnoses
- Case outcome
A 32-year-old man presents to the urgent care center at a community hospital with severe scrotal pain and swelling of five days’ duration. What began as mild left scrotal discomfort is now causing increasing pain, swelling, hematuria, dysuria, low-grade fever, and nausea, prompting him to seek medical attention.
The patient, who is a pipefitter in a hospital, was at work when his symptoms began. He denies any history of scrotal trauma, and his review of systems is otherwise unremarkable. His medical history is significant for mild hypertension and morbid obesity, but he is not immunocompromised. Two months ago, he had an excision and repair of a left ureterocele, for which he was treated prophylactically with ciprofloxacin for one week. He has a 3–pack-year history of smoking and consumes three alcoholic beverages per week. He denies illicit drug use and has no report of sexually transmitted infection.
Upon arrival to urgent care, the patient appears to be in moderate distress, with a blood pressure (BP) of 111/79 mm Hg; pulse, 104 beats/min; respiratory rate, 18 breaths/min-1; temperature, 100.1°F; and SpO2, 94%. Physical exam reveals left scrotal erythema, severe tenderness upon palpation, marked scrotal edema, and a slight amount of foul-smelling discharge seeping from a pinpoint opening in the left perineum (see Figure 1a). Given his scrotal presentation, he is quickly transferred to a regional emergency department (ED) for a urology consult.
In the ED, lab testing yields significant findings (see Table 1). His ECG demonstrates sinus tachycardia at 126 beats/min without rhythm or ST changes. His urinalysis reveals a cloudy appearance, a protein level of 100 mg/dL, and trace leukocyte esterase.
Urgent CT with contrast is obtained; it shows significant soft-tissue inflammatory changes in the left groin and scrotum that extend into the left thigh. In addition, a collection of fluid is seen in the inferior aspect of the left scrotal wall, indicating a probable abscess. There is no free air or lymphadenopathy.
Given the patient’s worsening condition and his apparent advancement to a systemic inflammatory response syndrome, surgical consult is obtained. He is diagnosed with a scrotal abscess and cellulitis; two blood and two scrotal cultures are obtained, and the patient is empirically started on IV ampicillin and gentamicin.
Two hours later, he has a BP of 122/74 mm Hg; pulse, 112 beats/min; respiratory rate, 20 breaths/min-1; and temperature, 103.1°F. His genital inflammation has advanced to the perineum and the left lower abdomen. The purulent, bloody, foul-smelling drainage from the opening in the left perineum is increasingly apparent. The patient is taken emergently to surgery for an incision and drainage, along with exploration of the scrotal abscess. During surgery, the patient is discovered to have Fournier’s gangrene.
DISCUSSION
Fournier’s gangrene (FG) is a necrotizing fasciitis of the perineal, perianal, and/or genital areas involving the superficial and deep fascial planes while sparing the deep muscular structures and overlying skin.1 A rare but potentially fatal disease, FG spreads at a rate of up to 3 cm/h.2,3
Mortality rates range from 7.5% to 88%, with the highest mortality occurring within the first 96 hours of hospitalization.1,4-7 Mortality is often related to the onset of sepsis.4,5 Survival requires early recognition; immediate, aggressive surgical debridement of all necrotic tissue; and concomitant, early administration of appropriate antibiotics.1,4,5,8 Mortality risk and prognosis are improved in patients younger than 60 with localized disease and no toxicity, along with sterile blood cultures.1
Risk Factors
FG is most commonly seen in males between the ages of 50 and 70, with a 10:1 male-to-female ratio.3,9 Impaired immunity typically increases a patient’s susceptibility to FG, with type 2 diabetes having the highest incidence (85% of patients).1,4,6,8,10 Other conditions that can increase the risk for FG include obesity, alcoholism, cirrhosis, cardiac disease, tobacco use, peripheral vascular disease, malignancy, chronic steroid use, renal insufficiency, IV drug abuse, and HIV.1,4,6,8,9,11
Trauma frequently initiates the infectious process,with urogenital trauma (eg, placement of urethral instrumentation, surgery, and urinary tract infection) being the main cause of bacterial introduction.1,3 Localized infection causes the development of an obliterative endarteritis, resulting in subcutaneous vascular ischemia, necrosis, and bacterial proliferation.3,7,9
Presentation and Diagnosis
Presenting symptoms of FG include intense, abrupt genital pain that is disproportionate to the physical exam findings.9 This rapidly escalates to include extreme swelling, erythema, bullae, discolored skin, and tissue crepitus with eventual necrosis.2,10 Lab results typically show leukocytosis > 18.0 × 109/L.4 The testicle and spermatic cord are generally unaffected (as in this patient), due to the anatomic relationship between the various layers of fascia within the scrotum and the anterior abdominal wall, as well as the independent blood supply of the compartmentalized testicular tissue.1-3
During an exam of the acute scrotum, the differential diagnosis includes cellulitis, scrotal abscess, acute epididymitis, and testicular torsion, with scrotal abscess being most frequently diagnosed (57% of patients).9,11,12 The distinguishing features of these diagnoses can be found in Table 2. Necrotizing fasciitis in the form of FG tends to be an unexpected, rare finding usually only diagnosed during the surgical draining of an abscess.12
CT is the test of choice to detect FG and determine the extent of its spread by identifying subcutaneous air/gas within the involved fascial planes.10,13 However, an incisional biopsy with culture is needed to confirm the diagnosis.3,9 Most patients with FG require an average of four surgeries (eg, reconstruction, skin grafting, and possibly colostomy if the infection has entered the peritoneal cavity) in order to eradicate the disease and achieve the best functional and cosmetic outcome.4
Etiology
About 83% of FG cases are polymicrobial infections comprised of enterobacter, enterococci, Escherichia coli, group A streptococci, pseudomonas, and clostridium, with symptoms evolving two to four days following the initial insult.4,7,11,14,15 Monomicrobial infections are much less common, but the symptoms progress even more rapidly.15 Methicillin-resistant Staphylococcus aureus (MRSA) necrotizing fasciitis infections occur in about 3% of monomicrobial cases.12 MRSA emerged in the early 2000s as an additional causative pathogen for polymicrobial necrotizing fasciitis infections.12,14,15 Prior to that time, S aureus strains were almost uniformly susceptible to penicillinase-resistant ß lactams.12
A distinction should be made between health care-associated (HA) MRSA and community-acquired (CA) MRSA due to treatment considerations. HA-MRSA infections are contracted through previous health care exposure (within the past year) and are less resistant to treatment.16,17 In contrast, CA-MRSA, which comprises 29% of MRSA cases, causes infections in previously healthy young patients without prior health care contact within the past year.16 CA-MRSA strains are more robust than HA-MRSA strains and can cause sepsis and other invasive, rapidly progressive, and possibly life-threatening infections due to the amount of tissue destruction and necrosis.16,18 Transmission of CA-MRSA is often associated with crowded environments, frequent skin-to-skin contact, compromised skin integrity, contaminated items or surfaces, and lack of cleanliness.16 Over the years, CA-MRSA has developed resistance to multiple antimicrobials; providers should therefore consider CA-MRSA on initial evaluation of necrotizing infections, to ensure appropriate initiation of treatment.12,16
CASE CONTINUED
Extensive debridement was completed down to healthy tissue in all affected areas (see Figure 1b). The necrotizing fasciitis had spared the left testicle and spermatic cord, and a colostomy was not required.
The patient’s initial postoperative vital signs were unremarkable, except for his BP (86/54 mm Hg). The patient was taken postoperatively to the surgical intensive care unit (SICU) with the diagnosis of FG. Aggressive IV fluids were administered for resuscitation, and he was closely monitored for increasing sepsis. Metronidazole was added for anaerobic and gram-positive coverage. His postoperative lab results can also be found in Table 1.
His ECG showed a normal sinus rhythm without ST changes, and he denied any cardiac symptoms. His physical exam was significant for mild pallor, dry mucus membranes, and a left scrotal and pelvic packed dressing. He was given two units of packed red blood cells for acute postoperative blood-loss anemia. The preliminary tissue culture results showed gram-positive cocci consistent with a staphylococcal infection; his antibiotics were then changed to IV ampicillin/sulbactam and clindamycin.
Approximately five hours postoperatively, an ECG suddenly showed acute ST elevation in leads II, II, and aVF, with reciprocal changes. The patient was diagnosed with an acute myocardial infarction (AMI). He denied any chest pain, shortness of breath, or diaphoresis. The SICU team initiated aspirin therapy and immediately contacted cardiology for an emergent coronary angiogram.
The angiogram and cardiac catheterization revealed an elevated left ventricular end diastolic (LVED) volume, inferior wall hypokinesis, a low-normal ejection fraction, and a 30% lesion in the first diagonal of his left anterior descending artery. A postprocedure echocardiogram demonstrated left ventricular (LV) ejection fraction of 50%, with LV hypokinesis in the inferior base and mild left atrial enlargement. The patient was started on metoprolol for myocardial protection and recovery.
Complications
Perioperative complications of FG, including AMI, must be considered due to the physiologic stress on the body.19 Most patients with perioperative AMI after noncardiac surgery do not experience ischemic symptoms.20
Growing evidence suggests the pivotal role of acute inflammation (postoperatively or from infection) as a precipitating event in AMI.20,21 Chemical mediators, such as inflammatory cytokines, endotoxins, and nitric oxide, may play a role in the development of an AMI.22
If cardiovascular disease and/or significant cardiovascular risk factors (ie, older age, male, cigarette smoking, cardiac family history, acute kidney injury) are present, the risk for AMI increases in the first two days following surgery.21,23 Acute infections and sepsis also initiate or increase systemic inflammatory activity via these same chemical mediators.21
Most suspected infectious agents also produce coronary artery sheer stress and destabilization of vulnerable plaques, leading to plaque rupture and thrombosis.19,24 Proinflammatory cytokines promote enhanced platelet activation and contribute to this thrombotic environment.21,23 Thrombus leads to obstructed coronary blood flow, myocardial ischemia, and finally, infarction.21
A reversible myocardial depression, cardiomyopathy, or myocardial ischemia may occur in patients with acute systemic infection or sepsis when the myocardium is functionally and structurally injured by these inflammatory chemical mediators.19,22-24 Characteristics of such a cardiomyopathy include left ventricle dilation with a low filling pressure, an abnormal increase in LVED volume, and a depressed ejection fraction.22
An acute infectious or septic process can raise troponin levels in 43% to 85% of patients.22,24 Troponin biomarkers can assist in predicting myocardial injury and events after surgery with nearly absolute myocardial tissue specificity.20 Cardiovascular involvement caused by myocardial injury–related sepsis is observed in up to 70% of patients in the ICU for these reasons.23 Therefore, providers should consider measuring troponin biomarkers during such infectious and septic processes, as this team did for the case patient. The providers were able to diagnose his AMI early and institute appropriate treatment measures to avoid extensive myocardial tissue damage.
Several studies have already demonstrated a correlation between pneumococcal pneumonia and an increased risk for AMI, and the same mechanisms are presumed responsible for any severe acute infectious state.21 More research is needed to understand the pathophysiology of AMI in sepsis and acute systemic infections.23
OUTCOME FOR THE CASE PATIENT
On postoperative day 2, the patient’s vital signs and lab results were normal. Additional lab results included an A1C of 5.2%. His ECG showed a resolving ST-elevation myocardial infarction (STEMI). The surgical wound had initiation of early granulation tissue without any further signs of necrosis.
A postoperative acute STEMI was unexpected in this patient, as his only risk factors included being male, mild hypertension, obesity, and tobacco use. At the time of his initial elevated troponin level, he had no cardiac symptoms or ECG changes. This initial high troponin level may have been stress-induced from the acute infectious process, and his acute inferior wall STEMI may have been secondary to a transient thrombotic event. The STEMI may then have resolved on its own during the cardiac catheterization with the administration of heparin, IV fluids, blood products, aspirin, or dye infiltration, thus enhancing reperfusion of the coronary artery system.
The final tissue culture showed MRSA. Given his job and his history of a genitourinary procedure, as well as the less fulminant form of disease and relatively quick recovery, it was likely HA-MRSA (rather than CA-MRSA). Only clindamycin was used for treatment.
The wound continued to have decreasing erythema, a reduction in tenderness, and evidence of viable, pink granulation tissue. HIV testing was not completed during his admission. The remainder of the patient’s hospital course was unremarkable, and he was discharged home with wound care, urology, and cardiology follow-up services.
CONCLUSION
Multiple factors contribute to a delayed or mistaken diagnosis of FG; it may be overlooked in the initial working diagnoses because of its low incidence and manifestations similar to those of other soft-tissue infections (eg, cellulitis, scrotal abscess). The cutaneous signs of FG often lag behind the disease manifestation, with minimal or no external presence while extensive internal tissue destruction is occurring. Constant review of symptoms is required when treating patients with soft-tissue infections, and early signs—such as pain out of proportion to physical findings—should prompt a clinician to include FG in the differential.
Early diagnosis with prompt debridement and antibiotic therapy are crucial to patient survival. Detecting FG within the first 24 hours is critical. Further differentiation between CA-MRSA and HA-MRSA can assist in patient recovery and survival by guiding appropriate antibiotic therapy. Perioperative risk assessment and serial troponin biomarkers may identify patients in need of intensive monitoring and management postoperatively to avoid an AMI, since patients may not experience ischemic symptoms.
1. Norton KS, Johnson LW, Perry T, et al. Management of Fournier’s gangrene: an eleven-year retrospective analysis of early recognition, diagnosis, and treatment. Am Surg. 2002;68(8):709-713.
2. Agostini T, Mori F, Perello R, et al. Successful combined approach to a severe Fournier’s gangrene. Indian J Plast Surg. 2014;47(1):132-136.
3. Cabrera G, March P. Fournier’s gangrene. Glendale, CA: Cinahl Information Systems; 2016.
4. Czymek R, Kujath P, Bruch HP, et al. Treatment, outcome and quality of life after Fournier’s gangrene: a multicentre study. Colorectal Dis. 2013;15(12):1529-1536.
5. Sugihara T, Yasunaga H, Horiguchi H, et al. Impact of surgical intervention timing on the case fatality rate for Fournier’s gangrene: an analysis of 379 cases. BJU Int. 2012;110(11c):E1096-1100.
6. Tuncel A, Keten T, Aslan Y, et al. Comparison of different scoring systems for outcome prediction in patients with Fournier’s gangrene: experience with 50 patients. Scand J Urol. 2014;48(4):393-399.
7. Taken K, Oncu MR, Ergun M, et al. Fournier’s gangrene: causes, presentation and survival of sixty-five patients. Pak J Med Sci. 2016;32(3):746-750.
8. Palvolgyi R, Kaji AH, Valeriano J, et al. Fournier’s gangrene: a model for early prediction. Am Surg. 2014;80(10):926-931.
9. Pais V, Santora T. Fournier gangrene. http://emedicine.medscape.com/article/2028899-overview. Accessed August 16, 2017.
10. Cottrill RR. A demonstration of clinical reasoning through a case of scrotal infection. Urol Nurs. 2013;33(1):33-37.
11. Summers A. Fournier’s gangrene. J Nurse Pract. 2014;10(8):582-587.
12. Miller LG, Perdreau-Remington F, Rieg G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med. 2005;352(14):1445-1453.
13. Gupta N, Zinn K, Bansal I, Weinstein R. Fournier’s gangrene: ultrasound or computed tomography? A letter to the editor. Med Ultrason. 2014;16(4):389-390.
14. Bjurlin MA, O’Grady T, Kim DY, et al. Causative pathogens, antibiotic sensitivity, resistance patterns, and severity in a contemporary series of Fournier’s gangrene. Urol. 2013;81(4):752-758.
15. Goh T, Goh LG. Pitfalls in diagnosing necrotizing fasciitis. https://psnet.ahrq.gov/webmm/case/329/pitfalls-in-diagnos ing-necrotizing-fasciitis. Accessed August 16, 2017.
16. Kale P, Dhawan B. The changing face of community-acquired methicillin-resistant Staphylococcus aureus. Indian J Med Microbiol. 2016;34(3):275-285.
17. CDC. Necrotizing fasciitis. www.cdc.gov/Features/NecrotizingFasciitis/index.html. Accessed August 16, 2017.
18. Barnes BE, Sampson DA. A literature review on community-acquired methicillin-resistant Staphylococcus aureus in the United States: clinical information for primary care nurse practitioners. J Am Acad Nurse Pract. 2011;23(1):23-32.
19. Madjid M, Vela D, Khalili-Tabrizi H, et al. Systemic infections cause exaggerated local inflammation in atherosclerotic coronary arteries. Clues to the triggering effect of acute infections on acute coronary syndromes. Tex Heart Inst J. 2007;34(1):11-18.
20. Devereaux PJ, Chan MTV, Alonso-Coello PA, et al; VISION Study Investigators. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA. 2012;307(21):2295-2304.
21. Corrales-Medina VF, Fatemi O, Serpa J, et al. The association between Staphylococcus aureus bacteremia and acute myocardial infarction. Scand J Infect Dis. 2009;41(6-7):511-514.
22. Romero-Bermejo FJ, Ruiz-Bailen M, Gil-Cebrian J, Huertos-Ranchal MJ. Sepsis-induced cardiomyopathy. Curr Cardiol Rev. 2011;7(3):163-183.
23. Smilowitz NR, Gupta N, Guo Y, Bangalore S. Comparison of outcomes of patients with sepsis with versus without acute myocardial infarction and comparison of invasive versus noninvasive management of the patients with infarction. Am J Cardiol. 2016;117(7):1065-1071.
24. Mattson M. Sepsis and cardiac disease: improving outcomes through recognition and management. Prog Cardiovasc Nurs. 2009;24(4):199-201.
25. Papadakis MA, McPhee SJ. Current Medical Diagnosis & Treatment. 54th ed. New York, NY: McGraw Hill Education; 2015:137-138, 151-152, 937.
26. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed August 16, 2017.
1. Norton KS, Johnson LW, Perry T, et al. Management of Fournier’s gangrene: an eleven-year retrospective analysis of early recognition, diagnosis, and treatment. Am Surg. 2002;68(8):709-713.
2. Agostini T, Mori F, Perello R, et al. Successful combined approach to a severe Fournier’s gangrene. Indian J Plast Surg. 2014;47(1):132-136.
3. Cabrera G, March P. Fournier’s gangrene. Glendale, CA: Cinahl Information Systems; 2016.
4. Czymek R, Kujath P, Bruch HP, et al. Treatment, outcome and quality of life after Fournier’s gangrene: a multicentre study. Colorectal Dis. 2013;15(12):1529-1536.
5. Sugihara T, Yasunaga H, Horiguchi H, et al. Impact of surgical intervention timing on the case fatality rate for Fournier’s gangrene: an analysis of 379 cases. BJU Int. 2012;110(11c):E1096-1100.
6. Tuncel A, Keten T, Aslan Y, et al. Comparison of different scoring systems for outcome prediction in patients with Fournier’s gangrene: experience with 50 patients. Scand J Urol. 2014;48(4):393-399.
7. Taken K, Oncu MR, Ergun M, et al. Fournier’s gangrene: causes, presentation and survival of sixty-five patients. Pak J Med Sci. 2016;32(3):746-750.
8. Palvolgyi R, Kaji AH, Valeriano J, et al. Fournier’s gangrene: a model for early prediction. Am Surg. 2014;80(10):926-931.
9. Pais V, Santora T. Fournier gangrene. http://emedicine.medscape.com/article/2028899-overview. Accessed August 16, 2017.
10. Cottrill RR. A demonstration of clinical reasoning through a case of scrotal infection. Urol Nurs. 2013;33(1):33-37.
11. Summers A. Fournier’s gangrene. J Nurse Pract. 2014;10(8):582-587.
12. Miller LG, Perdreau-Remington F, Rieg G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med. 2005;352(14):1445-1453.
13. Gupta N, Zinn K, Bansal I, Weinstein R. Fournier’s gangrene: ultrasound or computed tomography? A letter to the editor. Med Ultrason. 2014;16(4):389-390.
14. Bjurlin MA, O’Grady T, Kim DY, et al. Causative pathogens, antibiotic sensitivity, resistance patterns, and severity in a contemporary series of Fournier’s gangrene. Urol. 2013;81(4):752-758.
15. Goh T, Goh LG. Pitfalls in diagnosing necrotizing fasciitis. https://psnet.ahrq.gov/webmm/case/329/pitfalls-in-diagnos ing-necrotizing-fasciitis. Accessed August 16, 2017.
16. Kale P, Dhawan B. The changing face of community-acquired methicillin-resistant Staphylococcus aureus. Indian J Med Microbiol. 2016;34(3):275-285.
17. CDC. Necrotizing fasciitis. www.cdc.gov/Features/NecrotizingFasciitis/index.html. Accessed August 16, 2017.
18. Barnes BE, Sampson DA. A literature review on community-acquired methicillin-resistant Staphylococcus aureus in the United States: clinical information for primary care nurse practitioners. J Am Acad Nurse Pract. 2011;23(1):23-32.
19. Madjid M, Vela D, Khalili-Tabrizi H, et al. Systemic infections cause exaggerated local inflammation in atherosclerotic coronary arteries. Clues to the triggering effect of acute infections on acute coronary syndromes. Tex Heart Inst J. 2007;34(1):11-18.
20. Devereaux PJ, Chan MTV, Alonso-Coello PA, et al; VISION Study Investigators. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA. 2012;307(21):2295-2304.
21. Corrales-Medina VF, Fatemi O, Serpa J, et al. The association between Staphylococcus aureus bacteremia and acute myocardial infarction. Scand J Infect Dis. 2009;41(6-7):511-514.
22. Romero-Bermejo FJ, Ruiz-Bailen M, Gil-Cebrian J, Huertos-Ranchal MJ. Sepsis-induced cardiomyopathy. Curr Cardiol Rev. 2011;7(3):163-183.
23. Smilowitz NR, Gupta N, Guo Y, Bangalore S. Comparison of outcomes of patients with sepsis with versus without acute myocardial infarction and comparison of invasive versus noninvasive management of the patients with infarction. Am J Cardiol. 2016;117(7):1065-1071.
24. Mattson M. Sepsis and cardiac disease: improving outcomes through recognition and management. Prog Cardiovasc Nurs. 2009;24(4):199-201.
25. Papadakis MA, McPhee SJ. Current Medical Diagnosis & Treatment. 54th ed. New York, NY: McGraw Hill Education; 2015:137-138, 151-152, 937.
26. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed August 16, 2017.
Heartburn or heart attack? A mimic of MI
A 71-year-old man with a history of hypertension, 4 prior myocardial infarctions (MIs), and well-compensated ischemic cardiomyopathy presented to the emergency department after 2 episodes of sharp pain in the left upper abdomen and chest. The episodes lasted 1 to 2 minutes and were not relieved by rest. Their location was similar to that of the pain he experienced with his MIs. He could not identify any exacerbating or ameliorating factors. The pain had resolved without specific therapy before he arrived.
He reported polydipsia and constipation over the past 2 weeks and generalized muscle weakness and acute exacerbations of chronic back pain in the past 2 days. Neither he nor a friend who accompanied him noticed any confusion. He had been taking as many as 15 calcium carbonate tablets a day for 6 weeks to self-treat dyspepsia refractory to once-daily ranitidine, and hydrochlorothiazide for his hypertension for 3 weeks.
FURTHER EVALUATION, CARDIOLOGY CONSULT
On physical examination, he had diffuse weakness, dry mucous membranes, and an irregular heart rhythm.

Laboratory testing showed the following:
- Troponin I 0.11 ng/mL (reference range ≤ 0.04); repeated, it was 0.12 ng/mL
- Serum creatinine 3.4 mg/dL (0.44–1.27) (9 months earlier it had been 0.99 mg/dL)
- Serum calcium 17.3 mg/dL (8.6–10.5)
- Parathyroid hormone 9 pg/mL (12–88)
- Serum bicarbonate 33 mmol/L (24–32); 2 weeks earlier, it had been 27 mmol/L.
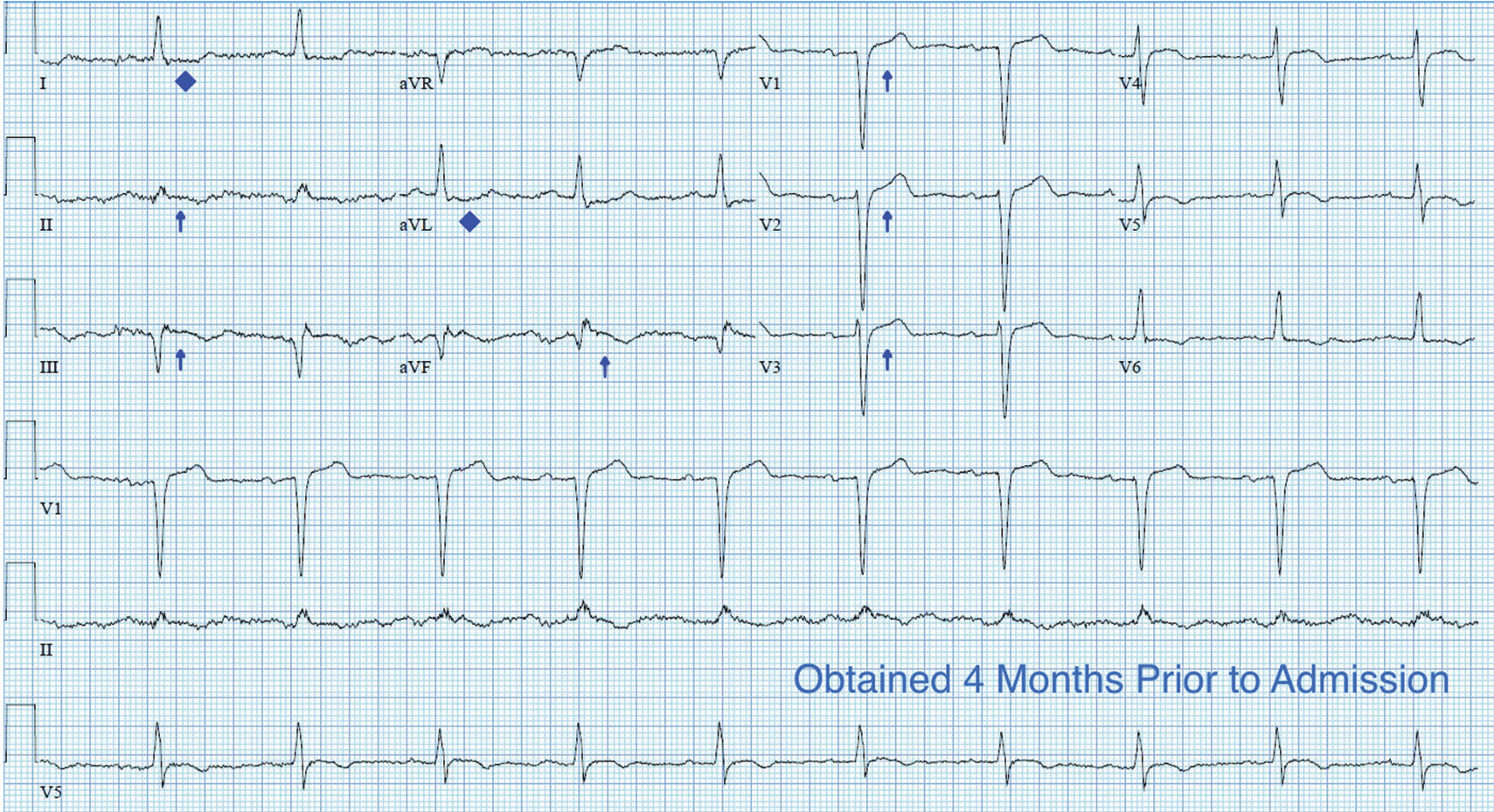
DIAGNOSIS: MILK-ALKALI SYNDROME
The diagnosis, based on the presentation and the results of the workup, was milk-alkali syndrome complicated by recent hydrochlorothiazide use. This syndrome consists of the triad of hypercalcemia, metabolic alkalosis, and acute kidney injury, all due to excessive ingestion of calcium and alkali, usually calcium carbonate.
His hydrochlorothiazide and calcium carbonate were discontinued. He was given intravenous normal saline and subcutaneous calcitonin, and his serum calcium level came down to 11.5 mg/dL within the next 24 hours. His dyspepsia was treated with pantoprazole.
The patient had no further episodes of chest pain, and the cardiology consult team again recommended against coronary angiography. Repeat ECG after the hypercalcemia resolved showed results identical to those 4 months before his admission. Two months later, his serum calcium level was 9.4 mg/dL and his creatinine level was 1.24 mg/dL.
A MIMIC OF STEMI
In numerous reported cases, these electrocardiographic findings coupled with chest pain led to misdiagnosis of STEMI.1–3 While STEMI and occasionally hypercalcemia can cause ST elevation, hypercalcemia causes a significant shortening of the corrected QT interval that is not associated with STEMI.4,5
Ultimately, the diagnosis of MI involves clinical, laboratory, and ECG findings, and if a strong clinical suspicion for myocardial ischemia exists, STEMI cannot reliably be distinguished from hypercalcemia by ECG alone. It is nonetheless important to be aware of this complication of hypercalcemia to avoid unnecessary cardiac interventions.
- Ashizawa N, Arakawa S, Koide Y, Toda G, Seto S, Yano K. Hypercalcemia due to vitamin D intoxication with clinical features mimicking acute myocardial infarction. Intern Med 2003; 42:340–344.
- Nishi SP, Barbagelata NA, Atar S, Birnbaum Y, Tuero E. Hypercalcemia-induced ST-segment elevation mimicking acute myocardial infarction. J Electrocardiol 2006; 39:298–300.
- Turnham S, Kilickap M, Kilinc S. ST segment elevation mimicking acute myocardial infarction in hypercalcemia. Heart 2005; 91:999.
- Nierenberg DW, Ransil BJ. Q-aTc interval as a clinical indicator of hypercalcemia. Am J Cardiol 1979; 44:243–248.
- Ahmed R, Hashiba K. Reliability of QT intervals as indicators of clinical hypercalcemia. Clin Cardiol 1988; 11:395–400.
A 71-year-old man with a history of hypertension, 4 prior myocardial infarctions (MIs), and well-compensated ischemic cardiomyopathy presented to the emergency department after 2 episodes of sharp pain in the left upper abdomen and chest. The episodes lasted 1 to 2 minutes and were not relieved by rest. Their location was similar to that of the pain he experienced with his MIs. He could not identify any exacerbating or ameliorating factors. The pain had resolved without specific therapy before he arrived.
He reported polydipsia and constipation over the past 2 weeks and generalized muscle weakness and acute exacerbations of chronic back pain in the past 2 days. Neither he nor a friend who accompanied him noticed any confusion. He had been taking as many as 15 calcium carbonate tablets a day for 6 weeks to self-treat dyspepsia refractory to once-daily ranitidine, and hydrochlorothiazide for his hypertension for 3 weeks.
FURTHER EVALUATION, CARDIOLOGY CONSULT
On physical examination, he had diffuse weakness, dry mucous membranes, and an irregular heart rhythm.
Laboratory testing showed the following:
- Troponin I 0.11 ng/mL (reference range ≤ 0.04); repeated, it was 0.12 ng/mL
- Serum creatinine 3.4 mg/dL (0.44–1.27) (9 months earlier it had been 0.99 mg/dL)
- Serum calcium 17.3 mg/dL (8.6–10.5)
- Parathyroid hormone 9 pg/mL (12–88)
- Serum bicarbonate 33 mmol/L (24–32); 2 weeks earlier, it had been 27 mmol/L.
DIAGNOSIS: MILK-ALKALI SYNDROME
The diagnosis, based on the presentation and the results of the workup, was milk-alkali syndrome complicated by recent hydrochlorothiazide use. This syndrome consists of the triad of hypercalcemia, metabolic alkalosis, and acute kidney injury, all due to excessive ingestion of calcium and alkali, usually calcium carbonate.
His hydrochlorothiazide and calcium carbonate were discontinued. He was given intravenous normal saline and subcutaneous calcitonin, and his serum calcium level came down to 11.5 mg/dL within the next 24 hours. His dyspepsia was treated with pantoprazole.
The patient had no further episodes of chest pain, and the cardiology consult team again recommended against coronary angiography. Repeat ECG after the hypercalcemia resolved showed results identical to those 4 months before his admission. Two months later, his serum calcium level was 9.4 mg/dL and his creatinine level was 1.24 mg/dL.
A MIMIC OF STEMI
In numerous reported cases, these electrocardiographic findings coupled with chest pain led to misdiagnosis of STEMI.1–3 While STEMI and occasionally hypercalcemia can cause ST elevation, hypercalcemia causes a significant shortening of the corrected QT interval that is not associated with STEMI.4,5
Ultimately, the diagnosis of MI involves clinical, laboratory, and ECG findings, and if a strong clinical suspicion for myocardial ischemia exists, STEMI cannot reliably be distinguished from hypercalcemia by ECG alone. It is nonetheless important to be aware of this complication of hypercalcemia to avoid unnecessary cardiac interventions.
A 71-year-old man with a history of hypertension, 4 prior myocardial infarctions (MIs), and well-compensated ischemic cardiomyopathy presented to the emergency department after 2 episodes of sharp pain in the left upper abdomen and chest. The episodes lasted 1 to 2 minutes and were not relieved by rest. Their location was similar to that of the pain he experienced with his MIs. He could not identify any exacerbating or ameliorating factors. The pain had resolved without specific therapy before he arrived.
He reported polydipsia and constipation over the past 2 weeks and generalized muscle weakness and acute exacerbations of chronic back pain in the past 2 days. Neither he nor a friend who accompanied him noticed any confusion. He had been taking as many as 15 calcium carbonate tablets a day for 6 weeks to self-treat dyspepsia refractory to once-daily ranitidine, and hydrochlorothiazide for his hypertension for 3 weeks.
FURTHER EVALUATION, CARDIOLOGY CONSULT
On physical examination, he had diffuse weakness, dry mucous membranes, and an irregular heart rhythm.
Laboratory testing showed the following:
- Troponin I 0.11 ng/mL (reference range ≤ 0.04); repeated, it was 0.12 ng/mL
- Serum creatinine 3.4 mg/dL (0.44–1.27) (9 months earlier it had been 0.99 mg/dL)
- Serum calcium 17.3 mg/dL (8.6–10.5)
- Parathyroid hormone 9 pg/mL (12–88)
- Serum bicarbonate 33 mmol/L (24–32); 2 weeks earlier, it had been 27 mmol/L.
DIAGNOSIS: MILK-ALKALI SYNDROME
The diagnosis, based on the presentation and the results of the workup, was milk-alkali syndrome complicated by recent hydrochlorothiazide use. This syndrome consists of the triad of hypercalcemia, metabolic alkalosis, and acute kidney injury, all due to excessive ingestion of calcium and alkali, usually calcium carbonate.
His hydrochlorothiazide and calcium carbonate were discontinued. He was given intravenous normal saline and subcutaneous calcitonin, and his serum calcium level came down to 11.5 mg/dL within the next 24 hours. His dyspepsia was treated with pantoprazole.
The patient had no further episodes of chest pain, and the cardiology consult team again recommended against coronary angiography. Repeat ECG after the hypercalcemia resolved showed results identical to those 4 months before his admission. Two months later, his serum calcium level was 9.4 mg/dL and his creatinine level was 1.24 mg/dL.
A MIMIC OF STEMI
In numerous reported cases, these electrocardiographic findings coupled with chest pain led to misdiagnosis of STEMI.1–3 While STEMI and occasionally hypercalcemia can cause ST elevation, hypercalcemia causes a significant shortening of the corrected QT interval that is not associated with STEMI.4,5
Ultimately, the diagnosis of MI involves clinical, laboratory, and ECG findings, and if a strong clinical suspicion for myocardial ischemia exists, STEMI cannot reliably be distinguished from hypercalcemia by ECG alone. It is nonetheless important to be aware of this complication of hypercalcemia to avoid unnecessary cardiac interventions.
- Ashizawa N, Arakawa S, Koide Y, Toda G, Seto S, Yano K. Hypercalcemia due to vitamin D intoxication with clinical features mimicking acute myocardial infarction. Intern Med 2003; 42:340–344.
- Nishi SP, Barbagelata NA, Atar S, Birnbaum Y, Tuero E. Hypercalcemia-induced ST-segment elevation mimicking acute myocardial infarction. J Electrocardiol 2006; 39:298–300.
- Turnham S, Kilickap M, Kilinc S. ST segment elevation mimicking acute myocardial infarction in hypercalcemia. Heart 2005; 91:999.
- Nierenberg DW, Ransil BJ. Q-aTc interval as a clinical indicator of hypercalcemia. Am J Cardiol 1979; 44:243–248.
- Ahmed R, Hashiba K. Reliability of QT intervals as indicators of clinical hypercalcemia. Clin Cardiol 1988; 11:395–400.
- Ashizawa N, Arakawa S, Koide Y, Toda G, Seto S, Yano K. Hypercalcemia due to vitamin D intoxication with clinical features mimicking acute myocardial infarction. Intern Med 2003; 42:340–344.
- Nishi SP, Barbagelata NA, Atar S, Birnbaum Y, Tuero E. Hypercalcemia-induced ST-segment elevation mimicking acute myocardial infarction. J Electrocardiol 2006; 39:298–300.
- Turnham S, Kilickap M, Kilinc S. ST segment elevation mimicking acute myocardial infarction in hypercalcemia. Heart 2005; 91:999.
- Nierenberg DW, Ransil BJ. Q-aTc interval as a clinical indicator of hypercalcemia. Am J Cardiol 1979; 44:243–248.
- Ahmed R, Hashiba K. Reliability of QT intervals as indicators of clinical hypercalcemia. Clin Cardiol 1988; 11:395–400.
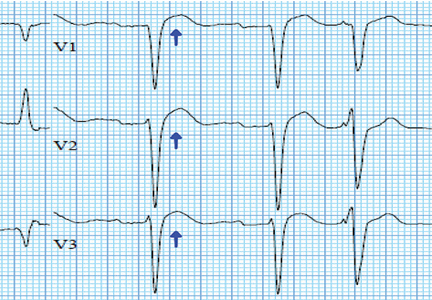
Watson, the game is a foot…or a palm
What common message do a 64-year-old woman with postoperative cognitive changes and an 83-year-old man with red palms have for us as physicians? As I read their clinical scenarios and the editorial by Westendorp, I was struck by the value and significance of informed clinical observation, an activity that I fear is going the way of the music CD and handwritten letters.
As I read the descriptions of these patients I was reminded of the internal satisfaction that I feel when I pick up a clinical or historical finding that directs me to a specific diagnosis and therapeutic recommendation. Sherlock Holmes I am not. Those satisfying pickups are infrequent, and I have no idea how many clues I have missed. I do know that most come from taking the time to perform a methodical physical examination, directed and informed by the patient’s recounted history. Some, like red palms or anisocoria, may be readily apparent and diagnostically useful—if the observer recognizes their potential significance. The 2 patients described in this issue of the Journal highlight the value of both observation and the knowledge and experience to place what we observe into a clinical context. Watson (the computer) can provide data regarding the potential significance of a physical finding, but only if someone first detects its existence.
Once it is recognized (or pointed out), we can all pull out our smartphones and Google “palmar erythema and disease,” and on our screen up pops liver disease, pregnancy, and assorted other conditions, including malignancies. But how many of us in our clinic, as opposed to the artificial scenario of reading it in the Journal or attending a clinicopathologic conference, will spontaneously recognize palmar erythema as a potentially relevant clinical finding?
For many physicians, the sense of professional satisfaction in making these observations is diminished. The professional joy gleaned from these moments has been diluted. We are in jeopardy of losing the passion for the professional work that we do as well as the intellectual and emotional satisfaction that accompanies a nuanced professional job well done, while focusing instead on our contracted jobs, frequently evaluated by our ability to meet commercial needs. The absence of emotional and intellectual satisfaction that should come from these collected moments of patient interaction and reflection undoubtedly contributes to the rising rate of physician burnout.
There are so many pressures on us in the office. Did I record that my new patient with known rheumatoid arthritis (who has had a recent MI and pneumonia and who has tried several biologic therapies without success and is in need of a creative change in her medication) has a cousin with hypothyroidism so I could include family history in my electronic medical record note and thus bill at a “desired” level of complexity? Did I use the appropriate catchphrase stating that over 50% of my time was spent in education of the patient (after collecting and reading for 30 minutes the stack of prior records, preparing to do battle with her insurance company to get the next therapy approved for coverage)?
There is little wonder that an observation of red palms gets missed or, if it is noted, that the Google search is never actually done. And when we do recognize the finding and its clinical significance, we often don’t take a moment to reflect and bask in the glow of a job well done, the satisfaction of successfully applying both our knowledge and experience to help resolve a clinical problem.
As Westendorp points out, bedside observation is still relevant. And I will add that there still should be joy in the intellectual pursuit of the job well done as well as the patient well managed. It takes more than a smartphone to know when and how to look at the palms and the eyes before typing in a Google search or consulting the digital (not the doctor) Watson. Those are skills to be proud of.
What common message do a 64-year-old woman with postoperative cognitive changes and an 83-year-old man with red palms have for us as physicians? As I read their clinical scenarios and the editorial by Westendorp, I was struck by the value and significance of informed clinical observation, an activity that I fear is going the way of the music CD and handwritten letters.
As I read the descriptions of these patients I was reminded of the internal satisfaction that I feel when I pick up a clinical or historical finding that directs me to a specific diagnosis and therapeutic recommendation. Sherlock Holmes I am not. Those satisfying pickups are infrequent, and I have no idea how many clues I have missed. I do know that most come from taking the time to perform a methodical physical examination, directed and informed by the patient’s recounted history. Some, like red palms or anisocoria, may be readily apparent and diagnostically useful—if the observer recognizes their potential significance. The 2 patients described in this issue of the Journal highlight the value of both observation and the knowledge and experience to place what we observe into a clinical context. Watson (the computer) can provide data regarding the potential significance of a physical finding, but only if someone first detects its existence.
Once it is recognized (or pointed out), we can all pull out our smartphones and Google “palmar erythema and disease,” and on our screen up pops liver disease, pregnancy, and assorted other conditions, including malignancies. But how many of us in our clinic, as opposed to the artificial scenario of reading it in the Journal or attending a clinicopathologic conference, will spontaneously recognize palmar erythema as a potentially relevant clinical finding?
For many physicians, the sense of professional satisfaction in making these observations is diminished. The professional joy gleaned from these moments has been diluted. We are in jeopardy of losing the passion for the professional work that we do as well as the intellectual and emotional satisfaction that accompanies a nuanced professional job well done, while focusing instead on our contracted jobs, frequently evaluated by our ability to meet commercial needs. The absence of emotional and intellectual satisfaction that should come from these collected moments of patient interaction and reflection undoubtedly contributes to the rising rate of physician burnout.
There are so many pressures on us in the office. Did I record that my new patient with known rheumatoid arthritis (who has had a recent MI and pneumonia and who has tried several biologic therapies without success and is in need of a creative change in her medication) has a cousin with hypothyroidism so I could include family history in my electronic medical record note and thus bill at a “desired” level of complexity? Did I use the appropriate catchphrase stating that over 50% of my time was spent in education of the patient (after collecting and reading for 30 minutes the stack of prior records, preparing to do battle with her insurance company to get the next therapy approved for coverage)?
There is little wonder that an observation of red palms gets missed or, if it is noted, that the Google search is never actually done. And when we do recognize the finding and its clinical significance, we often don’t take a moment to reflect and bask in the glow of a job well done, the satisfaction of successfully applying both our knowledge and experience to help resolve a clinical problem.
As Westendorp points out, bedside observation is still relevant. And I will add that there still should be joy in the intellectual pursuit of the job well done as well as the patient well managed. It takes more than a smartphone to know when and how to look at the palms and the eyes before typing in a Google search or consulting the digital (not the doctor) Watson. Those are skills to be proud of.
What common message do a 64-year-old woman with postoperative cognitive changes and an 83-year-old man with red palms have for us as physicians? As I read their clinical scenarios and the editorial by Westendorp, I was struck by the value and significance of informed clinical observation, an activity that I fear is going the way of the music CD and handwritten letters.
As I read the descriptions of these patients I was reminded of the internal satisfaction that I feel when I pick up a clinical or historical finding that directs me to a specific diagnosis and therapeutic recommendation. Sherlock Holmes I am not. Those satisfying pickups are infrequent, and I have no idea how many clues I have missed. I do know that most come from taking the time to perform a methodical physical examination, directed and informed by the patient’s recounted history. Some, like red palms or anisocoria, may be readily apparent and diagnostically useful—if the observer recognizes their potential significance. The 2 patients described in this issue of the Journal highlight the value of both observation and the knowledge and experience to place what we observe into a clinical context. Watson (the computer) can provide data regarding the potential significance of a physical finding, but only if someone first detects its existence.
Once it is recognized (or pointed out), we can all pull out our smartphones and Google “palmar erythema and disease,” and on our screen up pops liver disease, pregnancy, and assorted other conditions, including malignancies. But how many of us in our clinic, as opposed to the artificial scenario of reading it in the Journal or attending a clinicopathologic conference, will spontaneously recognize palmar erythema as a potentially relevant clinical finding?
For many physicians, the sense of professional satisfaction in making these observations is diminished. The professional joy gleaned from these moments has been diluted. We are in jeopardy of losing the passion for the professional work that we do as well as the intellectual and emotional satisfaction that accompanies a nuanced professional job well done, while focusing instead on our contracted jobs, frequently evaluated by our ability to meet commercial needs. The absence of emotional and intellectual satisfaction that should come from these collected moments of patient interaction and reflection undoubtedly contributes to the rising rate of physician burnout.
There are so many pressures on us in the office. Did I record that my new patient with known rheumatoid arthritis (who has had a recent MI and pneumonia and who has tried several biologic therapies without success and is in need of a creative change in her medication) has a cousin with hypothyroidism so I could include family history in my electronic medical record note and thus bill at a “desired” level of complexity? Did I use the appropriate catchphrase stating that over 50% of my time was spent in education of the patient (after collecting and reading for 30 minutes the stack of prior records, preparing to do battle with her insurance company to get the next therapy approved for coverage)?
There is little wonder that an observation of red palms gets missed or, if it is noted, that the Google search is never actually done. And when we do recognize the finding and its clinical significance, we often don’t take a moment to reflect and bask in the glow of a job well done, the satisfaction of successfully applying both our knowledge and experience to help resolve a clinical problem.
As Westendorp points out, bedside observation is still relevant. And I will add that there still should be joy in the intellectual pursuit of the job well done as well as the patient well managed. It takes more than a smartphone to know when and how to look at the palms and the eyes before typing in a Google search or consulting the digital (not the doctor) Watson. Those are skills to be proud of.

Another complication of cirrhosis
A 53-year-old Native American woman with a history of liver cirrhosis secondary to alcohol abuse presents to the emergency department after 2 days of diffuse abdominal pain and weakness. The pain was sudden in onset and has progressed relentlessly over the last day, reaching 9 on a scale of 10 in severity. Family members say that her oral intake has been decreased for the last 2 days, but she has had no fever, vomiting, change in bowel habit, blood in stool, or black stool. She has never undergone surgery, and has had one uncomplicated pregnancy.
Physical examination
Vital signs:
- Blood pressure 82/57 mm Hg
- Heart rate 96 beats per minute
- Temperature 37.3°C (99.1°F)
- Respiratory rate 16 per minute
- Oxygen saturation 92% while receiving oxygen at 2 L/minute.
The patient is somnolent and has scleral icterus. Her cardiopulmonary examination is normal. Her abdomen is tense, distended, and diffusely tender. She has bilateral +2 pitting edema in her lower extremities. She is oriented to person only and is noted to have asterixis. Her baseline Model for End-stage Liver Disease score is 18 points on a scale of 6 (less ill) to 40 (gravely ill).
Laboratory studies:
- Hemoglobin 9.8 g/dL (reference range 11.5–15.5)
- Platelet count 100 × 109/L (150–400)
- White blood cell count 9.9 × 109/L (3.7–11.0)
- Serum creatinine 1.06 mg/dL (0.58–0.96)
- Bilirubin 6.3 mg/dL (0.2–1.3)
- International normalized ratio of the prothrombin time 2.15 (0.8–1.2)
- Blood urea nitrogen 13 mg/dL (7–21)
- Serum albumin 2.7 g/dL (3.9–4.9).
Intravenous fluid resuscitation is initiated but the patient remains hypotensive, and on repeat laboratory testing 4 hours later her hemoglobin level has dropped to 7.3 mg/dL.
DIFFERENTIAL DIAGNOSIS
1. Which of the following are likely causes of this patient’s presentation?
- Splenic arterial aneurysm rupture
- Spontaneous bacterial peritonitis
- Variceal hemorrhage
- Portal vein thrombosis
- Abdominal aortic aneurysm rupture
Ruptured splenic artery aneurysm
Splenic artery aneurysms are the third most common intra-abdominal aneurysm, after those of the abdominal aorta and iliac artery.1 They are often asymptomatic and are being detected more frequently because of increased use of computed tomography (CT).2 Symptomatic splenic artery aneurysms may present with abdominal pain and have the potential to rupture, which can be life-threatening.3,4
This patient may have a ruptured splenic artery aneurysm, given her hemodynamic shock.
Spontaneous bacterial peritonitis
Ten percent to 20% of hospitalized patients with cirrhosis and ascites develop spontaneous bacterial peritonitis. Patients may present with ascites and abdominal pain, tenderness to palpation, fever, encephalopathy, or worsening liver and renal function.
Diagnostic paracentesis is paramount to delineate the cause of ascites; one should calculate the serum-ascites albumin gradient and obtain a cell count and culture of the ascitic fluid. The diagnosis of spontaneous bacterial peritonitis can be made if the ascitic fluid polymorphonuclear cell count is 0.25 × 109/L or higher, even if the ascitic fluid culture is negative.5,6 Simultaneous blood cultures should also be collected, as 50% of cases are associated with bacteremia.
The in-hospital mortality rate of an episode of spontaneous bacterial peritonitis has been reduced to 10% to 20% thanks to prompt diagnosis and empiric treatment with third-generation cephalosporins.7
Five percent of cases of infected ascites fluid are due to secondary bacterial peritonitis from a perforated viscus or a loculated abscess, which cannot be differentiated clinically from spontaneous bacterial peritonitis but can be diagnosed with CT.8
This patient may be presenting with septic shock secondary to either of these causes.
Variceal hemorrhage
Half of patients with cirrhosis have gastroesophageal varices due to portal hypertension. Endoscopic surveillance is warranted, as the risk of hemorrhage is 12% to 15% per year, and the mortality rate approaches 15% to 20% with each episode. Prompt resuscitation, diagnosis, and control of bleeding is paramount.
Esophagogastroduodenoscopy is used for both diagnosis and intervention. Short-term prophylactic use of antibiotics improves survival by preventing infections in the event bleeding recurs.9–11
Our patient may be presenting with hemodynamic shock from bleeding esophageal varices.
Portal vein thrombosis
Portal vein thrombosis is a common complication of cirrhosis, occurring in 5% to 28% of patients. The risk increases with the severity of liver disease and in association with hepatocellular carcinoma.12 Forty-three percent of cases are discovered incidentally in asymptomatic patients during ultrasonography, 39% present with upper gastrointestinal bleeding, and 18% present with abdominal pain.13,14
Portal vein thrombosis is the complete or partial obstruction of blood flow due to a thrombus in the lumen of the portal vein. Contrast ultrasonography and CT can be used to establish the diagnosis.15
Anticoagulation is recommended in cases of complete thrombosis in candidates for living-donor liver transplant and for those at risk of mesenteric ischemia because of the thrombus extending into the mesenteric veins. In symptomatic patients, the decision to initiate anticoagulation should be made on a case-by-case basis after appropriate screening and management of varices.16–18
Our patient’s thrombocytopenia reflects the severity of portal hypertension and increases her risk of portal vein thrombosis, but this is unlikely to be the sole cause of the hemodynamic compromise in this patient.
Ruptured abdominal aortic aneurysm
Rupture of an abdominal aortic aneurysm is a medical emergency, with a mortality rate approaching 90%. Risk factors for abdominal aortic aneurysms are smoking, male sex, age over 65, history of cardiovascular disease, hypertension, and a family history of abdominal aortic aneurysm, especially if a first-degree relative is affected.19 Endovascular repair is associated with lower rates of death and complications compared with open repair.20
The patient does not have any of those risk factors, making this diagnosis less likely.
CASE CONTINUED: RUPTURED SPLENIC ARTERY ANEURYSM

Emergency CT of the abdomen and pelvis with contrast enhancement shows a large left intraperitoneal hematoma with active extravasation from a ruptured splenic artery aneurysm (Figure 1). The patient receives packed red blood cells and fresh-frozen plasma before being transferred to our hospital.
2. Which of the following is false regarding splenic artery aneurysms?
- They are the most common type of splanchnic arterial aneurysm
- True aneurysms are more common than pseudoaneurysms
- Asymptomatic aneurysms are discovered incidentally during assessment for other radiographic indications
- Splenic artery aneurysm in portal hypertension is the result of athero-sclerotic changes to the vascular intima
Splenic artery aneurysm in portal hypertension is not the result of atherosclerotic change to the vascular intima.
Splenic artery aneurysms are the most common type of splanchnic artery aneurysm.1 True aneurysms involve all 3 layers of the arterial wall, ie, intima, media, and adventitia. Cirrhosis and portal hypertension are associated with true aneurysm formation. The proposed mechanism of aneurysm formation is increased splenic blood flow in response to portal congestion with resultant hemodynamic stress that disrupts arterial wall structure, leading to aneurysmal dilation.21
In earlier reports, the incidence of true splenic artery aneurysm in portal hypertension varied from 2.9% to 50%, the latter representing autopsy findings of small aneurysms that were found in the splenic hilum of patients with cirrhosis.22–25 The incidence of clinically significant aneurysms in cirrhosis is unknown but incidental asymptomatic aneurysm is being detected more frequently on imaging studies pursued for screening purposes.26
The risk of rupture is low, only 2% to 10% in older studies and likely even lower now due to increased incidental detection in asymptomatic patients.27 However, emergent management of rupture at a tertiary care facility is paramount, as the mortality rate of ruptured splenic artery aneurysm is 29% to 36%.1,26,28
Splenic artery pseudoaneurysm is rarer and has a different pathophysiologic process than true aneurysm. It usually arises in the setting of trauma, pancreatitis, or postsurgery.29,30 Pseudoaneurysm is more likely to rupture, owing to compromise in the vascular wall integrity.4,21,28 As a result, treatment is indicated for every pseudoaneurysm regardless of size.
RISK FACTORS FOR SPLENIC ARTERY ANEURYSM
3. Which of the following is true regarding our patient’s risk of splenic artery aneurysm?
- Liver cirrhosis and portal hypertension are her greatest risk factors for it
- Female sex and prior pregnancy are her greatest risk factors for it
- Being Native American makes it more likely that the patient has splenic artery aneurysm secondary to collagen vascular disease
- Her risk of rupture would diminish after receiving a liver transplant
Liver cirrhosis and portal hypertension are her greatest risk factors for splenic artery aneurysm.
Risk factors for true aneurysm include hypertension, atherosclerosis, portal hypertension with or without liver cirrhosis, liver transplant, third trimester of pregnancy, and multiparity.1,4,26,28,31 Splenic artery aneurysm is usually diagnosed in the sixth decade. It may be 4 times as common in women, given a hormonal influence.32 Cirrhosis is also associated with massive splenic artery aneurysm (≥ 5 cm). Although rare, massive splenic artery aneurysm is more frequent in men (the male-to-female ratio is 1.78:1) and has a heightened risk of rupture.28 The incidence of rupture increases to around 3% to 4% after liver transplant.33 Rare causes of true aneurysm include fibrodysplasia, collagen vascular disease (eg, Loeys-Dietz and type IV Ehler-Danlos syndromes), vasculitis (eg, polyarteritis nodosa due to amphetamine abuse), and mycotic aneurysms.24,25,28,29
This patient’s age, sex, and history of cirrhosis puts her at increased risk of splenic artery aneurysm. The risk of rupture is highest in the peripartum period and in patients with cirrhosis who become pregnant. Although being Native American portends an increased risk for collagen vascular disease, the latter is unlikely to be a contributing factor.
TREATMENT OF SPLENIC ARTERY ANEURYSM
4. Which of the following is false regarding treatment of splenic artery aneurysms?
- Aneurysms larger than 2 cm and those that are expanding require repair
- Treatment should be offered if the patient has symptoms attributable to the aneurysm
- Asymptomatic aneurysms in pregnant women can be followed with watchful waiting
- Minimally invasive therapies such as percutaneous embolization may be a good option in poor operative candidates
Asymptomatic aneurysms in pregnant women should not be followed with watchful waiting—they should be repaired, as rupture carries a maternal mortality rate of 75% and a fetal mortality rate of 95%.34
Complications of splenic artery aneurysm depend on the type of aneurysm and its predisposing factors. Indications for treatment of true aneurysms include:
- Symptoms attributable to the aneurysm (hence, the second answer choice above is true)
- Diameter 2 cm or greater or enlarging diameter (hence, the first answer choice is true)
- Women of childbearing age in anticipation of pregnancy
- Need for surgical intervention such as portocaval shunt and liver transplant.
Conservative management is associated with a late mortality risk of 4.9%.2 Interventional options include percutaneous embolization or stenting; or laparotomy with splenic artery ligation or excision with or without splenectomy.1,28,35–37
Endovascular and open surgical repair have both been used to treat splenic artery aneurysms. The method used depends on the patient’s surgical history and aneurysm anatomy such as splenic artery tortuosity hindering passage of a catheter. Open surgery is associated with longer intraoperative time and length of hospital stay and higher rates of 30-day mortality and perioperative morbidity.38–41 With endovascular repair, the complication of persistent or recurrent flow occurs in 3% to 5% of cases by 30 days; hence, postprocedural surveillance is recommended.42–44 Endovascular repair has a higher reintervention rate but may still be more cost-effective than open surgical repair.
Because patients with cirrhosis have a higher risk of surgical complications,45 elective endovascular treatment may be an option for patients with aneurysms at high risk of rupturing. Endovascular treatment of visceral aneurysms is associated with complications such as postembolization syndrome (fever, abdominal pain, pleural effusion, and pancreatitis), access site hematoma, splenic infarction, and persistent abdominal pain.42
Patients with cirrhosis as the cause of splenic artery aneurysm tend to need longer hospitalization after endovascular treatment, but there is insufficient evidence to suggest that they are at higher risk of other complications.37
CASE CONTINUED: SPLENIC ARTERY EMBOLIZATION

The patient undergoes emergency splenic artery embolization, performed by an interventional radiology team (Figure 2 and Figure 3). Over the next few days, her mental status improves and her abdominal pain resolves. Her hemoglobin level remains stable after the procedure.

The surgical and interventional radiology teams discuss the risk of repeat intervention with the patient and her family, who prefer a nonoperative approach. She is managed supportively in the intensive care unit and is finally discharged home in stable condition and is scheduled for outpatient follow-up.
SUSPECT THIS FATAL CONDITION
The low prevalence of ruptured splenic artery aneurysm may lead physicians to attribute septic shock to spontaneous bacterial peritonitis or hemorrhagic shock from gastroesophageal varices in patients with cirrhosis, but a high index of suspicion and early recognition of this rare disease can lead to timely diagnosis and treatment of this highly fatal complication.
KEY POINTS
- Splenic artery aneurysm is a common complication of cirrhosis, often diagnosed incidentally.
- Elective embolization should be considered for asymptomatic splenic artery aneurysms larger than 2 cm in diameter, clinically symptomatic aneurysms, women of childbearing age, and patients who are candidates for liver transplant.
- Although splenic artery aneurysm rupture is rare, it has a high mortality rate and warrants a high index of suspicion to institute prompt specialized intervention.
- We recommend that physicians consider splenic artery aneurysm rupture in their differential diagnoses in patients with liver cirrhosis presenting with abdominal pain, altered mental status, and hemodynamic shock.
- Bakhos CT, McIntosh BC, Nukta FA, et al. Staged arterial embolization and surgical resection of a giant splenic artery aneurysm. Ann Vasc Surg 2007; 21:208–210.
- Hogendoorn W, Lavida A, Hunink MG, et al. Open repair, endovascular repair, and conservative management of true splenic artery aneurysms. J Vasc Surg 2014; 60:1667–1676.e1.
- Algudkar A. Unruptured splenic artery aneurysm presenting as epigastric pain. JRSM Short Rep 2010; 1:24.
- Abbas MA, Stone WM, Fowl RJ, et al. Splenic artery aneurysms: two decades experience at Mayo Clinic. Ann Vasc Surg 2002; 16:442–449.
- Hoefs JC, Canawati HN, Sapico FL, Hopkins RR, Weiner J, Montgomerie JZ. Spontaneous bacterial peritonitis. Hepatology 1982; 2:399–407.
- Runyon BA, Hoefs JC. Culture-negative neutrocytic ascites: a variant of spontaneous bacterial peritonitis. Hepatology 1984; 4:1209–1211.
- Garcia-Tsao G. Spontaneous bacterial peritonitis: a historical perspective. J Hepatol 2004; 41:522–527.
- Soriano G, Castellote J, Alvarez C, et al. Secondary bacterial peritonitis in cirrhosis: a retrospective study of clinical and analytical characteristics, diagnosis and management. J Hepatol 2010; 52:39–44.
- D’Amico G, De Franchis R; Cooperative Study Group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003; 38:599–612.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W; Practice Guidelines Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007; 46:922–938.
- Tsochatzis EA, Senzolo M, Germani G, Gatt A, Burroughs AK. Systematic review: portal vein thrombosis in cirrhosis. Aliment Pharmacol Ther 2010; 31:366–374.
- Kobori L, van der Kolk MJ, de Jong KP, et al. Splenic artery aneurysms in liver transplant patients. Liver Transplant Group. J Hepatol 1997; 27:890–893.
- Manzano-Robleda Mdel C, Barranco-Fragoso B, Uribe M, Mendez-Sanchez N. Portal vein thrombosis: what is new? Ann Hepatol 2015; 14:20–27.
- Sarin SK, Philips CA, Kamath PS, et al. Toward a comprehensive new classification of portal vein thrombosis in patients with cirrhosis. Gastroenterology 2016; 151:574–577.e3.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Manzanet G, Sanjuan F, Orbis P, et al. Liver transplantation in patients with portal vein thrombosis. Liver Transpl 2001; 7:125–131.
- John BV, Konjeti R, Aggarwal A, et al. Impact of untreated portal vein thrombosis on pre and post liver transplant outcomes in cirrhosis. Ann Hepatol 2013; 12:952–958.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery/Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines. ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Associations for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (writing committee to develop guidelines for the management of patients with peripheral arterial disease)—summary of recommendations. J Vasc Interv Radiol 2006; 17:1383–1397.
- Schermerhorn ML, O’Malley AJ, Jhaveri A, Cotterill P, Pomposelli F, Landon BE. Endovascular vs open repair of abdominal aortic aneurysms in the Medicare population. N Engl J Med 2008; 358:464–474.
- Ohta M, Hashizume M, Ueno K, Tanoue K, Sugimachi K, Hasuo K. Hemodynamic study of splenic artery aneurysm in portal hypertension. Hepatogastroenterology 1994; 41:181–184.
- Sunagozaka H, Tsuji H, Mizukoshi E, et al. The development and clinical features of splenic aneurysm associated with liver cirrhosis. Liver Int 2006; 26:291–297.
- Manenti F, Williams R. Injection studies of the splenic vasculature in portal hypertension. Gut 1966; 7:175–180.
- Stanley JC, Fry WJ. Pathogenesis and clinical significance of splenic artery aneurysms. Surgery 1974; 76:898–909.
- Lee PC, Rhee RY, Gordon RY, Fung JJ, Webster MW. Management of splenic artery aneurysms: the significance of portal and essential hypertension. J Am Coll Surg 1999; 189:483–490.
- Al-Habbal Y, Christophi C, Muralidharan V. Aneurysms of the splenic artery—a review. Surgeon 2010; 8:223–231.
- Mattar SG, Lumsden AB. The management of splenic artery aneurysms: experience with 23 cases. Am J Surg 1995; 169:580–584.
- Akbulut S, Otan E. Management of giant splenic artery aneurysm: comprehensive literature review. Medicine (Baltimore) 2015; 94:e1016.
- Agrawal GA, Johnson PT, Fishman EK. Splenic artery aneurysms and pseudoaneurysms: clinical distinctions and CT appearances. AJR Am J Roentgenol 2007; 188:992–999.
- Tessier DJ, Stone WM, Fowl RJ, et al. Clinical features and management of splenic artery pseudoaneurysm: case series and cumulative review of literature. J Vasc Surg 2003; 38:969–974.
- Dave SP, Reis ED, Hossain A, Taub PJ, Kerstein MD, Hollier LH. Splenic artery aneurysm in the 1990s. Ann Vasc Surg 2000; 14:223–229.
- Parrish J, Maxwell C, Beecroft JR. Splenic artery aneurysm in pregnancy. J Obstet Gynaecol Can 2015; 37:816–818.
- Moon DB, Lee SG, Hwang S, et al. Characteristics and management of splenic artery aneurysms in adult living donor liver transplant recipients. Liver Transpl 2009; 15:1535–1541.
- Sadat U, Dar O, Walsh S, Varty K. Splenic artery aneurysms in pregnancy—a systematic review. Int J Surg 2008; 6:261–265.
- Geoghegan T, McAuley G, Snow A, Torreggiani WC. Emergency embolization of multiple splenic artery pseudoaneurysms associated with portal hypertension complicating cystic fibrosis. Australas Radiol 2007; 51(suppl):B337–B339.
- Jiang R, Ding X, Jian W, Jiang J, Hu S, Zhang Z. Combined endovascular embolization and open surgery for splenic artery aneurysm with arteriovenous fistula. Ann Vasc Surg 2016; 30:311.e1–311.e4.
- Naganuma M, Matsui H, Koizumi J, Fushimi K, Yasunaga H. Short-term outcomes following elective transcatheter arterial embolization for splenic artery aneurysms: data from a nationwide administrative database. Acta Radiol Open 2015; 4:2047981615574354.
- Batagini NC, El-Arousy H, Clair DG, Kirksey L. Open versus endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Ann Vasc Surg 2016; 35:1–8.
- Marone EM, Mascia D, Kahlberg A, Brioschi C, Tshomba Y, Chiesa R. Is open repair still the gold standard in visceral artery aneurysm management? Ann Vasc Surg 2011; 25:936–946.
- Sticco A, Aggarwal A, Shapiro M, Pratt A, Rissuci D, D'Ayala M. A comparison of open and endovascular treatment strategies for the management of splenic artery aneurysms. Vascular 2016; 24:487–491.
- Hogendoorn W, Lavida A, Hunink MG, et al. Cost-effectiveness of endovascular repair, open repair, and conservative management of splenic artery aneurysms. J Vasc Surg 2015; 61:1432–1440.
- Fankhauser GT, Stone WM, Naidu SG, et al; Mayo Vascular Research Center Consortium. The minimally invasive management of visceral artery aneurysms and pseudoaneurysms. J Vasc Surg 2011; 53:966–970.
- Lagana D, Carrafiello G, Mangini M, et al. Multimodal approach to endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Eur J Radiol 2006; 59:104–111.
- Guillon R, Garcier JM, Abergel A, et al. Management of splenic artery aneurysms and false aneurysms with endovascular treatment in 12 patients. Cardiovasc Intervent Radiol 2003; 26:256–260.
- Northup PG, Wanamaker RC, Lee VD, Adams RB, Berg CL. Model for end-stage liver disease (MELD) predicts nontransplant surgical mortality in patients with cirrhosis. Ann Surg 2005; 242:244–251.
A 53-year-old Native American woman with a history of liver cirrhosis secondary to alcohol abuse presents to the emergency department after 2 days of diffuse abdominal pain and weakness. The pain was sudden in onset and has progressed relentlessly over the last day, reaching 9 on a scale of 10 in severity. Family members say that her oral intake has been decreased for the last 2 days, but she has had no fever, vomiting, change in bowel habit, blood in stool, or black stool. She has never undergone surgery, and has had one uncomplicated pregnancy.
Physical examination
Vital signs:
- Blood pressure 82/57 mm Hg
- Heart rate 96 beats per minute
- Temperature 37.3°C (99.1°F)
- Respiratory rate 16 per minute
- Oxygen saturation 92% while receiving oxygen at 2 L/minute.
The patient is somnolent and has scleral icterus. Her cardiopulmonary examination is normal. Her abdomen is tense, distended, and diffusely tender. She has bilateral +2 pitting edema in her lower extremities. She is oriented to person only and is noted to have asterixis. Her baseline Model for End-stage Liver Disease score is 18 points on a scale of 6 (less ill) to 40 (gravely ill).
Laboratory studies:
- Hemoglobin 9.8 g/dL (reference range 11.5–15.5)
- Platelet count 100 × 109/L (150–400)
- White blood cell count 9.9 × 109/L (3.7–11.0)
- Serum creatinine 1.06 mg/dL (0.58–0.96)
- Bilirubin 6.3 mg/dL (0.2–1.3)
- International normalized ratio of the prothrombin time 2.15 (0.8–1.2)
- Blood urea nitrogen 13 mg/dL (7–21)
- Serum albumin 2.7 g/dL (3.9–4.9).
Intravenous fluid resuscitation is initiated but the patient remains hypotensive, and on repeat laboratory testing 4 hours later her hemoglobin level has dropped to 7.3 mg/dL.
DIFFERENTIAL DIAGNOSIS
1. Which of the following are likely causes of this patient’s presentation?
- Splenic arterial aneurysm rupture
- Spontaneous bacterial peritonitis
- Variceal hemorrhage
- Portal vein thrombosis
- Abdominal aortic aneurysm rupture
Ruptured splenic artery aneurysm
Splenic artery aneurysms are the third most common intra-abdominal aneurysm, after those of the abdominal aorta and iliac artery.1 They are often asymptomatic and are being detected more frequently because of increased use of computed tomography (CT).2 Symptomatic splenic artery aneurysms may present with abdominal pain and have the potential to rupture, which can be life-threatening.3,4
This patient may have a ruptured splenic artery aneurysm, given her hemodynamic shock.
Spontaneous bacterial peritonitis
Ten percent to 20% of hospitalized patients with cirrhosis and ascites develop spontaneous bacterial peritonitis. Patients may present with ascites and abdominal pain, tenderness to palpation, fever, encephalopathy, or worsening liver and renal function.
Diagnostic paracentesis is paramount to delineate the cause of ascites; one should calculate the serum-ascites albumin gradient and obtain a cell count and culture of the ascitic fluid. The diagnosis of spontaneous bacterial peritonitis can be made if the ascitic fluid polymorphonuclear cell count is 0.25 × 109/L or higher, even if the ascitic fluid culture is negative.5,6 Simultaneous blood cultures should also be collected, as 50% of cases are associated with bacteremia.
The in-hospital mortality rate of an episode of spontaneous bacterial peritonitis has been reduced to 10% to 20% thanks to prompt diagnosis and empiric treatment with third-generation cephalosporins.7
Five percent of cases of infected ascites fluid are due to secondary bacterial peritonitis from a perforated viscus or a loculated abscess, which cannot be differentiated clinically from spontaneous bacterial peritonitis but can be diagnosed with CT.8
This patient may be presenting with septic shock secondary to either of these causes.
Variceal hemorrhage
Half of patients with cirrhosis have gastroesophageal varices due to portal hypertension. Endoscopic surveillance is warranted, as the risk of hemorrhage is 12% to 15% per year, and the mortality rate approaches 15% to 20% with each episode. Prompt resuscitation, diagnosis, and control of bleeding is paramount.
Esophagogastroduodenoscopy is used for both diagnosis and intervention. Short-term prophylactic use of antibiotics improves survival by preventing infections in the event bleeding recurs.9–11
Our patient may be presenting with hemodynamic shock from bleeding esophageal varices.
Portal vein thrombosis
Portal vein thrombosis is a common complication of cirrhosis, occurring in 5% to 28% of patients. The risk increases with the severity of liver disease and in association with hepatocellular carcinoma.12 Forty-three percent of cases are discovered incidentally in asymptomatic patients during ultrasonography, 39% present with upper gastrointestinal bleeding, and 18% present with abdominal pain.13,14
Portal vein thrombosis is the complete or partial obstruction of blood flow due to a thrombus in the lumen of the portal vein. Contrast ultrasonography and CT can be used to establish the diagnosis.15
Anticoagulation is recommended in cases of complete thrombosis in candidates for living-donor liver transplant and for those at risk of mesenteric ischemia because of the thrombus extending into the mesenteric veins. In symptomatic patients, the decision to initiate anticoagulation should be made on a case-by-case basis after appropriate screening and management of varices.16–18
Our patient’s thrombocytopenia reflects the severity of portal hypertension and increases her risk of portal vein thrombosis, but this is unlikely to be the sole cause of the hemodynamic compromise in this patient.
Ruptured abdominal aortic aneurysm
Rupture of an abdominal aortic aneurysm is a medical emergency, with a mortality rate approaching 90%. Risk factors for abdominal aortic aneurysms are smoking, male sex, age over 65, history of cardiovascular disease, hypertension, and a family history of abdominal aortic aneurysm, especially if a first-degree relative is affected.19 Endovascular repair is associated with lower rates of death and complications compared with open repair.20
The patient does not have any of those risk factors, making this diagnosis less likely.
CASE CONTINUED: RUPTURED SPLENIC ARTERY ANEURYSM
Emergency CT of the abdomen and pelvis with contrast enhancement shows a large left intraperitoneal hematoma with active extravasation from a ruptured splenic artery aneurysm (Figure 1). The patient receives packed red blood cells and fresh-frozen plasma before being transferred to our hospital.
2. Which of the following is false regarding splenic artery aneurysms?
- They are the most common type of splanchnic arterial aneurysm
- True aneurysms are more common than pseudoaneurysms
- Asymptomatic aneurysms are discovered incidentally during assessment for other radiographic indications
- Splenic artery aneurysm in portal hypertension is the result of athero-sclerotic changes to the vascular intima
Splenic artery aneurysm in portal hypertension is not the result of atherosclerotic change to the vascular intima.
Splenic artery aneurysms are the most common type of splanchnic artery aneurysm.1 True aneurysms involve all 3 layers of the arterial wall, ie, intima, media, and adventitia. Cirrhosis and portal hypertension are associated with true aneurysm formation. The proposed mechanism of aneurysm formation is increased splenic blood flow in response to portal congestion with resultant hemodynamic stress that disrupts arterial wall structure, leading to aneurysmal dilation.21
In earlier reports, the incidence of true splenic artery aneurysm in portal hypertension varied from 2.9% to 50%, the latter representing autopsy findings of small aneurysms that were found in the splenic hilum of patients with cirrhosis.22–25 The incidence of clinically significant aneurysms in cirrhosis is unknown but incidental asymptomatic aneurysm is being detected more frequently on imaging studies pursued for screening purposes.26
The risk of rupture is low, only 2% to 10% in older studies and likely even lower now due to increased incidental detection in asymptomatic patients.27 However, emergent management of rupture at a tertiary care facility is paramount, as the mortality rate of ruptured splenic artery aneurysm is 29% to 36%.1,26,28
Splenic artery pseudoaneurysm is rarer and has a different pathophysiologic process than true aneurysm. It usually arises in the setting of trauma, pancreatitis, or postsurgery.29,30 Pseudoaneurysm is more likely to rupture, owing to compromise in the vascular wall integrity.4,21,28 As a result, treatment is indicated for every pseudoaneurysm regardless of size.
RISK FACTORS FOR SPLENIC ARTERY ANEURYSM
3. Which of the following is true regarding our patient’s risk of splenic artery aneurysm?
- Liver cirrhosis and portal hypertension are her greatest risk factors for it
- Female sex and prior pregnancy are her greatest risk factors for it
- Being Native American makes it more likely that the patient has splenic artery aneurysm secondary to collagen vascular disease
- Her risk of rupture would diminish after receiving a liver transplant
Liver cirrhosis and portal hypertension are her greatest risk factors for splenic artery aneurysm.
Risk factors for true aneurysm include hypertension, atherosclerosis, portal hypertension with or without liver cirrhosis, liver transplant, third trimester of pregnancy, and multiparity.1,4,26,28,31 Splenic artery aneurysm is usually diagnosed in the sixth decade. It may be 4 times as common in women, given a hormonal influence.32 Cirrhosis is also associated with massive splenic artery aneurysm (≥ 5 cm). Although rare, massive splenic artery aneurysm is more frequent in men (the male-to-female ratio is 1.78:1) and has a heightened risk of rupture.28 The incidence of rupture increases to around 3% to 4% after liver transplant.33 Rare causes of true aneurysm include fibrodysplasia, collagen vascular disease (eg, Loeys-Dietz and type IV Ehler-Danlos syndromes), vasculitis (eg, polyarteritis nodosa due to amphetamine abuse), and mycotic aneurysms.24,25,28,29
This patient’s age, sex, and history of cirrhosis puts her at increased risk of splenic artery aneurysm. The risk of rupture is highest in the peripartum period and in patients with cirrhosis who become pregnant. Although being Native American portends an increased risk for collagen vascular disease, the latter is unlikely to be a contributing factor.
TREATMENT OF SPLENIC ARTERY ANEURYSM
4. Which of the following is false regarding treatment of splenic artery aneurysms?
- Aneurysms larger than 2 cm and those that are expanding require repair
- Treatment should be offered if the patient has symptoms attributable to the aneurysm
- Asymptomatic aneurysms in pregnant women can be followed with watchful waiting
- Minimally invasive therapies such as percutaneous embolization may be a good option in poor operative candidates
Asymptomatic aneurysms in pregnant women should not be followed with watchful waiting—they should be repaired, as rupture carries a maternal mortality rate of 75% and a fetal mortality rate of 95%.34
Complications of splenic artery aneurysm depend on the type of aneurysm and its predisposing factors. Indications for treatment of true aneurysms include:
- Symptoms attributable to the aneurysm (hence, the second answer choice above is true)
- Diameter 2 cm or greater or enlarging diameter (hence, the first answer choice is true)
- Women of childbearing age in anticipation of pregnancy
- Need for surgical intervention such as portocaval shunt and liver transplant.
Conservative management is associated with a late mortality risk of 4.9%.2 Interventional options include percutaneous embolization or stenting; or laparotomy with splenic artery ligation or excision with or without splenectomy.1,28,35–37
Endovascular and open surgical repair have both been used to treat splenic artery aneurysms. The method used depends on the patient’s surgical history and aneurysm anatomy such as splenic artery tortuosity hindering passage of a catheter. Open surgery is associated with longer intraoperative time and length of hospital stay and higher rates of 30-day mortality and perioperative morbidity.38–41 With endovascular repair, the complication of persistent or recurrent flow occurs in 3% to 5% of cases by 30 days; hence, postprocedural surveillance is recommended.42–44 Endovascular repair has a higher reintervention rate but may still be more cost-effective than open surgical repair.
Because patients with cirrhosis have a higher risk of surgical complications,45 elective endovascular treatment may be an option for patients with aneurysms at high risk of rupturing. Endovascular treatment of visceral aneurysms is associated with complications such as postembolization syndrome (fever, abdominal pain, pleural effusion, and pancreatitis), access site hematoma, splenic infarction, and persistent abdominal pain.42
Patients with cirrhosis as the cause of splenic artery aneurysm tend to need longer hospitalization after endovascular treatment, but there is insufficient evidence to suggest that they are at higher risk of other complications.37
CASE CONTINUED: SPLENIC ARTERY EMBOLIZATION
The patient undergoes emergency splenic artery embolization, performed by an interventional radiology team (Figure 2 and Figure 3). Over the next few days, her mental status improves and her abdominal pain resolves. Her hemoglobin level remains stable after the procedure.
The surgical and interventional radiology teams discuss the risk of repeat intervention with the patient and her family, who prefer a nonoperative approach. She is managed supportively in the intensive care unit and is finally discharged home in stable condition and is scheduled for outpatient follow-up.
SUSPECT THIS FATAL CONDITION
The low prevalence of ruptured splenic artery aneurysm may lead physicians to attribute septic shock to spontaneous bacterial peritonitis or hemorrhagic shock from gastroesophageal varices in patients with cirrhosis, but a high index of suspicion and early recognition of this rare disease can lead to timely diagnosis and treatment of this highly fatal complication.
KEY POINTS
- Splenic artery aneurysm is a common complication of cirrhosis, often diagnosed incidentally.
- Elective embolization should be considered for asymptomatic splenic artery aneurysms larger than 2 cm in diameter, clinically symptomatic aneurysms, women of childbearing age, and patients who are candidates for liver transplant.
- Although splenic artery aneurysm rupture is rare, it has a high mortality rate and warrants a high index of suspicion to institute prompt specialized intervention.
- We recommend that physicians consider splenic artery aneurysm rupture in their differential diagnoses in patients with liver cirrhosis presenting with abdominal pain, altered mental status, and hemodynamic shock.
A 53-year-old Native American woman with a history of liver cirrhosis secondary to alcohol abuse presents to the emergency department after 2 days of diffuse abdominal pain and weakness. The pain was sudden in onset and has progressed relentlessly over the last day, reaching 9 on a scale of 10 in severity. Family members say that her oral intake has been decreased for the last 2 days, but she has had no fever, vomiting, change in bowel habit, blood in stool, or black stool. She has never undergone surgery, and has had one uncomplicated pregnancy.
Physical examination
Vital signs:
- Blood pressure 82/57 mm Hg
- Heart rate 96 beats per minute
- Temperature 37.3°C (99.1°F)
- Respiratory rate 16 per minute
- Oxygen saturation 92% while receiving oxygen at 2 L/minute.
The patient is somnolent and has scleral icterus. Her cardiopulmonary examination is normal. Her abdomen is tense, distended, and diffusely tender. She has bilateral +2 pitting edema in her lower extremities. She is oriented to person only and is noted to have asterixis. Her baseline Model for End-stage Liver Disease score is 18 points on a scale of 6 (less ill) to 40 (gravely ill).
Laboratory studies:
- Hemoglobin 9.8 g/dL (reference range 11.5–15.5)
- Platelet count 100 × 109/L (150–400)
- White blood cell count 9.9 × 109/L (3.7–11.0)
- Serum creatinine 1.06 mg/dL (0.58–0.96)
- Bilirubin 6.3 mg/dL (0.2–1.3)
- International normalized ratio of the prothrombin time 2.15 (0.8–1.2)
- Blood urea nitrogen 13 mg/dL (7–21)
- Serum albumin 2.7 g/dL (3.9–4.9).
Intravenous fluid resuscitation is initiated but the patient remains hypotensive, and on repeat laboratory testing 4 hours later her hemoglobin level has dropped to 7.3 mg/dL.
DIFFERENTIAL DIAGNOSIS
1. Which of the following are likely causes of this patient’s presentation?
- Splenic arterial aneurysm rupture
- Spontaneous bacterial peritonitis
- Variceal hemorrhage
- Portal vein thrombosis
- Abdominal aortic aneurysm rupture
Ruptured splenic artery aneurysm
Splenic artery aneurysms are the third most common intra-abdominal aneurysm, after those of the abdominal aorta and iliac artery.1 They are often asymptomatic and are being detected more frequently because of increased use of computed tomography (CT).2 Symptomatic splenic artery aneurysms may present with abdominal pain and have the potential to rupture, which can be life-threatening.3,4
This patient may have a ruptured splenic artery aneurysm, given her hemodynamic shock.
Spontaneous bacterial peritonitis
Ten percent to 20% of hospitalized patients with cirrhosis and ascites develop spontaneous bacterial peritonitis. Patients may present with ascites and abdominal pain, tenderness to palpation, fever, encephalopathy, or worsening liver and renal function.
Diagnostic paracentesis is paramount to delineate the cause of ascites; one should calculate the serum-ascites albumin gradient and obtain a cell count and culture of the ascitic fluid. The diagnosis of spontaneous bacterial peritonitis can be made if the ascitic fluid polymorphonuclear cell count is 0.25 × 109/L or higher, even if the ascitic fluid culture is negative.5,6 Simultaneous blood cultures should also be collected, as 50% of cases are associated with bacteremia.
The in-hospital mortality rate of an episode of spontaneous bacterial peritonitis has been reduced to 10% to 20% thanks to prompt diagnosis and empiric treatment with third-generation cephalosporins.7
Five percent of cases of infected ascites fluid are due to secondary bacterial peritonitis from a perforated viscus or a loculated abscess, which cannot be differentiated clinically from spontaneous bacterial peritonitis but can be diagnosed with CT.8
This patient may be presenting with septic shock secondary to either of these causes.
Variceal hemorrhage
Half of patients with cirrhosis have gastroesophageal varices due to portal hypertension. Endoscopic surveillance is warranted, as the risk of hemorrhage is 12% to 15% per year, and the mortality rate approaches 15% to 20% with each episode. Prompt resuscitation, diagnosis, and control of bleeding is paramount.
Esophagogastroduodenoscopy is used for both diagnosis and intervention. Short-term prophylactic use of antibiotics improves survival by preventing infections in the event bleeding recurs.9–11
Our patient may be presenting with hemodynamic shock from bleeding esophageal varices.
Portal vein thrombosis
Portal vein thrombosis is a common complication of cirrhosis, occurring in 5% to 28% of patients. The risk increases with the severity of liver disease and in association with hepatocellular carcinoma.12 Forty-three percent of cases are discovered incidentally in asymptomatic patients during ultrasonography, 39% present with upper gastrointestinal bleeding, and 18% present with abdominal pain.13,14
Portal vein thrombosis is the complete or partial obstruction of blood flow due to a thrombus in the lumen of the portal vein. Contrast ultrasonography and CT can be used to establish the diagnosis.15
Anticoagulation is recommended in cases of complete thrombosis in candidates for living-donor liver transplant and for those at risk of mesenteric ischemia because of the thrombus extending into the mesenteric veins. In symptomatic patients, the decision to initiate anticoagulation should be made on a case-by-case basis after appropriate screening and management of varices.16–18
Our patient’s thrombocytopenia reflects the severity of portal hypertension and increases her risk of portal vein thrombosis, but this is unlikely to be the sole cause of the hemodynamic compromise in this patient.
Ruptured abdominal aortic aneurysm
Rupture of an abdominal aortic aneurysm is a medical emergency, with a mortality rate approaching 90%. Risk factors for abdominal aortic aneurysms are smoking, male sex, age over 65, history of cardiovascular disease, hypertension, and a family history of abdominal aortic aneurysm, especially if a first-degree relative is affected.19 Endovascular repair is associated with lower rates of death and complications compared with open repair.20
The patient does not have any of those risk factors, making this diagnosis less likely.
CASE CONTINUED: RUPTURED SPLENIC ARTERY ANEURYSM
Emergency CT of the abdomen and pelvis with contrast enhancement shows a large left intraperitoneal hematoma with active extravasation from a ruptured splenic artery aneurysm (Figure 1). The patient receives packed red blood cells and fresh-frozen plasma before being transferred to our hospital.
2. Which of the following is false regarding splenic artery aneurysms?
- They are the most common type of splanchnic arterial aneurysm
- True aneurysms are more common than pseudoaneurysms
- Asymptomatic aneurysms are discovered incidentally during assessment for other radiographic indications
- Splenic artery aneurysm in portal hypertension is the result of athero-sclerotic changes to the vascular intima
Splenic artery aneurysm in portal hypertension is not the result of atherosclerotic change to the vascular intima.
Splenic artery aneurysms are the most common type of splanchnic artery aneurysm.1 True aneurysms involve all 3 layers of the arterial wall, ie, intima, media, and adventitia. Cirrhosis and portal hypertension are associated with true aneurysm formation. The proposed mechanism of aneurysm formation is increased splenic blood flow in response to portal congestion with resultant hemodynamic stress that disrupts arterial wall structure, leading to aneurysmal dilation.21
In earlier reports, the incidence of true splenic artery aneurysm in portal hypertension varied from 2.9% to 50%, the latter representing autopsy findings of small aneurysms that were found in the splenic hilum of patients with cirrhosis.22–25 The incidence of clinically significant aneurysms in cirrhosis is unknown but incidental asymptomatic aneurysm is being detected more frequently on imaging studies pursued for screening purposes.26
The risk of rupture is low, only 2% to 10% in older studies and likely even lower now due to increased incidental detection in asymptomatic patients.27 However, emergent management of rupture at a tertiary care facility is paramount, as the mortality rate of ruptured splenic artery aneurysm is 29% to 36%.1,26,28
Splenic artery pseudoaneurysm is rarer and has a different pathophysiologic process than true aneurysm. It usually arises in the setting of trauma, pancreatitis, or postsurgery.29,30 Pseudoaneurysm is more likely to rupture, owing to compromise in the vascular wall integrity.4,21,28 As a result, treatment is indicated for every pseudoaneurysm regardless of size.
RISK FACTORS FOR SPLENIC ARTERY ANEURYSM
3. Which of the following is true regarding our patient’s risk of splenic artery aneurysm?
- Liver cirrhosis and portal hypertension are her greatest risk factors for it
- Female sex and prior pregnancy are her greatest risk factors for it
- Being Native American makes it more likely that the patient has splenic artery aneurysm secondary to collagen vascular disease
- Her risk of rupture would diminish after receiving a liver transplant
Liver cirrhosis and portal hypertension are her greatest risk factors for splenic artery aneurysm.
Risk factors for true aneurysm include hypertension, atherosclerosis, portal hypertension with or without liver cirrhosis, liver transplant, third trimester of pregnancy, and multiparity.1,4,26,28,31 Splenic artery aneurysm is usually diagnosed in the sixth decade. It may be 4 times as common in women, given a hormonal influence.32 Cirrhosis is also associated with massive splenic artery aneurysm (≥ 5 cm). Although rare, massive splenic artery aneurysm is more frequent in men (the male-to-female ratio is 1.78:1) and has a heightened risk of rupture.28 The incidence of rupture increases to around 3% to 4% after liver transplant.33 Rare causes of true aneurysm include fibrodysplasia, collagen vascular disease (eg, Loeys-Dietz and type IV Ehler-Danlos syndromes), vasculitis (eg, polyarteritis nodosa due to amphetamine abuse), and mycotic aneurysms.24,25,28,29
This patient’s age, sex, and history of cirrhosis puts her at increased risk of splenic artery aneurysm. The risk of rupture is highest in the peripartum period and in patients with cirrhosis who become pregnant. Although being Native American portends an increased risk for collagen vascular disease, the latter is unlikely to be a contributing factor.
TREATMENT OF SPLENIC ARTERY ANEURYSM
4. Which of the following is false regarding treatment of splenic artery aneurysms?
- Aneurysms larger than 2 cm and those that are expanding require repair
- Treatment should be offered if the patient has symptoms attributable to the aneurysm
- Asymptomatic aneurysms in pregnant women can be followed with watchful waiting
- Minimally invasive therapies such as percutaneous embolization may be a good option in poor operative candidates
Asymptomatic aneurysms in pregnant women should not be followed with watchful waiting—they should be repaired, as rupture carries a maternal mortality rate of 75% and a fetal mortality rate of 95%.34
Complications of splenic artery aneurysm depend on the type of aneurysm and its predisposing factors. Indications for treatment of true aneurysms include:
- Symptoms attributable to the aneurysm (hence, the second answer choice above is true)
- Diameter 2 cm or greater or enlarging diameter (hence, the first answer choice is true)
- Women of childbearing age in anticipation of pregnancy
- Need for surgical intervention such as portocaval shunt and liver transplant.
Conservative management is associated with a late mortality risk of 4.9%.2 Interventional options include percutaneous embolization or stenting; or laparotomy with splenic artery ligation or excision with or without splenectomy.1,28,35–37
Endovascular and open surgical repair have both been used to treat splenic artery aneurysms. The method used depends on the patient’s surgical history and aneurysm anatomy such as splenic artery tortuosity hindering passage of a catheter. Open surgery is associated with longer intraoperative time and length of hospital stay and higher rates of 30-day mortality and perioperative morbidity.38–41 With endovascular repair, the complication of persistent or recurrent flow occurs in 3% to 5% of cases by 30 days; hence, postprocedural surveillance is recommended.42–44 Endovascular repair has a higher reintervention rate but may still be more cost-effective than open surgical repair.
Because patients with cirrhosis have a higher risk of surgical complications,45 elective endovascular treatment may be an option for patients with aneurysms at high risk of rupturing. Endovascular treatment of visceral aneurysms is associated with complications such as postembolization syndrome (fever, abdominal pain, pleural effusion, and pancreatitis), access site hematoma, splenic infarction, and persistent abdominal pain.42
Patients with cirrhosis as the cause of splenic artery aneurysm tend to need longer hospitalization after endovascular treatment, but there is insufficient evidence to suggest that they are at higher risk of other complications.37
CASE CONTINUED: SPLENIC ARTERY EMBOLIZATION
The patient undergoes emergency splenic artery embolization, performed by an interventional radiology team (Figure 2 and Figure 3). Over the next few days, her mental status improves and her abdominal pain resolves. Her hemoglobin level remains stable after the procedure.
The surgical and interventional radiology teams discuss the risk of repeat intervention with the patient and her family, who prefer a nonoperative approach. She is managed supportively in the intensive care unit and is finally discharged home in stable condition and is scheduled for outpatient follow-up.
SUSPECT THIS FATAL CONDITION
The low prevalence of ruptured splenic artery aneurysm may lead physicians to attribute septic shock to spontaneous bacterial peritonitis or hemorrhagic shock from gastroesophageal varices in patients with cirrhosis, but a high index of suspicion and early recognition of this rare disease can lead to timely diagnosis and treatment of this highly fatal complication.
KEY POINTS
- Splenic artery aneurysm is a common complication of cirrhosis, often diagnosed incidentally.
- Elective embolization should be considered for asymptomatic splenic artery aneurysms larger than 2 cm in diameter, clinically symptomatic aneurysms, women of childbearing age, and patients who are candidates for liver transplant.
- Although splenic artery aneurysm rupture is rare, it has a high mortality rate and warrants a high index of suspicion to institute prompt specialized intervention.
- We recommend that physicians consider splenic artery aneurysm rupture in their differential diagnoses in patients with liver cirrhosis presenting with abdominal pain, altered mental status, and hemodynamic shock.
- Bakhos CT, McIntosh BC, Nukta FA, et al. Staged arterial embolization and surgical resection of a giant splenic artery aneurysm. Ann Vasc Surg 2007; 21:208–210.
- Hogendoorn W, Lavida A, Hunink MG, et al. Open repair, endovascular repair, and conservative management of true splenic artery aneurysms. J Vasc Surg 2014; 60:1667–1676.e1.
- Algudkar A. Unruptured splenic artery aneurysm presenting as epigastric pain. JRSM Short Rep 2010; 1:24.
- Abbas MA, Stone WM, Fowl RJ, et al. Splenic artery aneurysms: two decades experience at Mayo Clinic. Ann Vasc Surg 2002; 16:442–449.
- Hoefs JC, Canawati HN, Sapico FL, Hopkins RR, Weiner J, Montgomerie JZ. Spontaneous bacterial peritonitis. Hepatology 1982; 2:399–407.
- Runyon BA, Hoefs JC. Culture-negative neutrocytic ascites: a variant of spontaneous bacterial peritonitis. Hepatology 1984; 4:1209–1211.
- Garcia-Tsao G. Spontaneous bacterial peritonitis: a historical perspective. J Hepatol 2004; 41:522–527.
- Soriano G, Castellote J, Alvarez C, et al. Secondary bacterial peritonitis in cirrhosis: a retrospective study of clinical and analytical characteristics, diagnosis and management. J Hepatol 2010; 52:39–44.
- D’Amico G, De Franchis R; Cooperative Study Group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003; 38:599–612.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W; Practice Guidelines Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007; 46:922–938.
- Tsochatzis EA, Senzolo M, Germani G, Gatt A, Burroughs AK. Systematic review: portal vein thrombosis in cirrhosis. Aliment Pharmacol Ther 2010; 31:366–374.
- Kobori L, van der Kolk MJ, de Jong KP, et al. Splenic artery aneurysms in liver transplant patients. Liver Transplant Group. J Hepatol 1997; 27:890–893.
- Manzano-Robleda Mdel C, Barranco-Fragoso B, Uribe M, Mendez-Sanchez N. Portal vein thrombosis: what is new? Ann Hepatol 2015; 14:20–27.
- Sarin SK, Philips CA, Kamath PS, et al. Toward a comprehensive new classification of portal vein thrombosis in patients with cirrhosis. Gastroenterology 2016; 151:574–577.e3.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Manzanet G, Sanjuan F, Orbis P, et al. Liver transplantation in patients with portal vein thrombosis. Liver Transpl 2001; 7:125–131.
- John BV, Konjeti R, Aggarwal A, et al. Impact of untreated portal vein thrombosis on pre and post liver transplant outcomes in cirrhosis. Ann Hepatol 2013; 12:952–958.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery/Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines. ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Associations for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (writing committee to develop guidelines for the management of patients with peripheral arterial disease)—summary of recommendations. J Vasc Interv Radiol 2006; 17:1383–1397.
- Schermerhorn ML, O’Malley AJ, Jhaveri A, Cotterill P, Pomposelli F, Landon BE. Endovascular vs open repair of abdominal aortic aneurysms in the Medicare population. N Engl J Med 2008; 358:464–474.
- Ohta M, Hashizume M, Ueno K, Tanoue K, Sugimachi K, Hasuo K. Hemodynamic study of splenic artery aneurysm in portal hypertension. Hepatogastroenterology 1994; 41:181–184.
- Sunagozaka H, Tsuji H, Mizukoshi E, et al. The development and clinical features of splenic aneurysm associated with liver cirrhosis. Liver Int 2006; 26:291–297.
- Manenti F, Williams R. Injection studies of the splenic vasculature in portal hypertension. Gut 1966; 7:175–180.
- Stanley JC, Fry WJ. Pathogenesis and clinical significance of splenic artery aneurysms. Surgery 1974; 76:898–909.
- Lee PC, Rhee RY, Gordon RY, Fung JJ, Webster MW. Management of splenic artery aneurysms: the significance of portal and essential hypertension. J Am Coll Surg 1999; 189:483–490.
- Al-Habbal Y, Christophi C, Muralidharan V. Aneurysms of the splenic artery—a review. Surgeon 2010; 8:223–231.
- Mattar SG, Lumsden AB. The management of splenic artery aneurysms: experience with 23 cases. Am J Surg 1995; 169:580–584.
- Akbulut S, Otan E. Management of giant splenic artery aneurysm: comprehensive literature review. Medicine (Baltimore) 2015; 94:e1016.
- Agrawal GA, Johnson PT, Fishman EK. Splenic artery aneurysms and pseudoaneurysms: clinical distinctions and CT appearances. AJR Am J Roentgenol 2007; 188:992–999.
- Tessier DJ, Stone WM, Fowl RJ, et al. Clinical features and management of splenic artery pseudoaneurysm: case series and cumulative review of literature. J Vasc Surg 2003; 38:969–974.
- Dave SP, Reis ED, Hossain A, Taub PJ, Kerstein MD, Hollier LH. Splenic artery aneurysm in the 1990s. Ann Vasc Surg 2000; 14:223–229.
- Parrish J, Maxwell C, Beecroft JR. Splenic artery aneurysm in pregnancy. J Obstet Gynaecol Can 2015; 37:816–818.
- Moon DB, Lee SG, Hwang S, et al. Characteristics and management of splenic artery aneurysms in adult living donor liver transplant recipients. Liver Transpl 2009; 15:1535–1541.
- Sadat U, Dar O, Walsh S, Varty K. Splenic artery aneurysms in pregnancy—a systematic review. Int J Surg 2008; 6:261–265.
- Geoghegan T, McAuley G, Snow A, Torreggiani WC. Emergency embolization of multiple splenic artery pseudoaneurysms associated with portal hypertension complicating cystic fibrosis. Australas Radiol 2007; 51(suppl):B337–B339.
- Jiang R, Ding X, Jian W, Jiang J, Hu S, Zhang Z. Combined endovascular embolization and open surgery for splenic artery aneurysm with arteriovenous fistula. Ann Vasc Surg 2016; 30:311.e1–311.e4.
- Naganuma M, Matsui H, Koizumi J, Fushimi K, Yasunaga H. Short-term outcomes following elective transcatheter arterial embolization for splenic artery aneurysms: data from a nationwide administrative database. Acta Radiol Open 2015; 4:2047981615574354.
- Batagini NC, El-Arousy H, Clair DG, Kirksey L. Open versus endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Ann Vasc Surg 2016; 35:1–8.
- Marone EM, Mascia D, Kahlberg A, Brioschi C, Tshomba Y, Chiesa R. Is open repair still the gold standard in visceral artery aneurysm management? Ann Vasc Surg 2011; 25:936–946.
- Sticco A, Aggarwal A, Shapiro M, Pratt A, Rissuci D, D'Ayala M. A comparison of open and endovascular treatment strategies for the management of splenic artery aneurysms. Vascular 2016; 24:487–491.
- Hogendoorn W, Lavida A, Hunink MG, et al. Cost-effectiveness of endovascular repair, open repair, and conservative management of splenic artery aneurysms. J Vasc Surg 2015; 61:1432–1440.
- Fankhauser GT, Stone WM, Naidu SG, et al; Mayo Vascular Research Center Consortium. The minimally invasive management of visceral artery aneurysms and pseudoaneurysms. J Vasc Surg 2011; 53:966–970.
- Lagana D, Carrafiello G, Mangini M, et al. Multimodal approach to endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Eur J Radiol 2006; 59:104–111.
- Guillon R, Garcier JM, Abergel A, et al. Management of splenic artery aneurysms and false aneurysms with endovascular treatment in 12 patients. Cardiovasc Intervent Radiol 2003; 26:256–260.
- Northup PG, Wanamaker RC, Lee VD, Adams RB, Berg CL. Model for end-stage liver disease (MELD) predicts nontransplant surgical mortality in patients with cirrhosis. Ann Surg 2005; 242:244–251.
- Bakhos CT, McIntosh BC, Nukta FA, et al. Staged arterial embolization and surgical resection of a giant splenic artery aneurysm. Ann Vasc Surg 2007; 21:208–210.
- Hogendoorn W, Lavida A, Hunink MG, et al. Open repair, endovascular repair, and conservative management of true splenic artery aneurysms. J Vasc Surg 2014; 60:1667–1676.e1.
- Algudkar A. Unruptured splenic artery aneurysm presenting as epigastric pain. JRSM Short Rep 2010; 1:24.
- Abbas MA, Stone WM, Fowl RJ, et al. Splenic artery aneurysms: two decades experience at Mayo Clinic. Ann Vasc Surg 2002; 16:442–449.
- Hoefs JC, Canawati HN, Sapico FL, Hopkins RR, Weiner J, Montgomerie JZ. Spontaneous bacterial peritonitis. Hepatology 1982; 2:399–407.
- Runyon BA, Hoefs JC. Culture-negative neutrocytic ascites: a variant of spontaneous bacterial peritonitis. Hepatology 1984; 4:1209–1211.
- Garcia-Tsao G. Spontaneous bacterial peritonitis: a historical perspective. J Hepatol 2004; 41:522–527.
- Soriano G, Castellote J, Alvarez C, et al. Secondary bacterial peritonitis in cirrhosis: a retrospective study of clinical and analytical characteristics, diagnosis and management. J Hepatol 2010; 52:39–44.
- D’Amico G, De Franchis R; Cooperative Study Group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003; 38:599–612.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W; Practice Guidelines Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007; 46:922–938.
- Tsochatzis EA, Senzolo M, Germani G, Gatt A, Burroughs AK. Systematic review: portal vein thrombosis in cirrhosis. Aliment Pharmacol Ther 2010; 31:366–374.
- Kobori L, van der Kolk MJ, de Jong KP, et al. Splenic artery aneurysms in liver transplant patients. Liver Transplant Group. J Hepatol 1997; 27:890–893.
- Manzano-Robleda Mdel C, Barranco-Fragoso B, Uribe M, Mendez-Sanchez N. Portal vein thrombosis: what is new? Ann Hepatol 2015; 14:20–27.
- Sarin SK, Philips CA, Kamath PS, et al. Toward a comprehensive new classification of portal vein thrombosis in patients with cirrhosis. Gastroenterology 2016; 151:574–577.e3.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Manzanet G, Sanjuan F, Orbis P, et al. Liver transplantation in patients with portal vein thrombosis. Liver Transpl 2001; 7:125–131.
- John BV, Konjeti R, Aggarwal A, et al. Impact of untreated portal vein thrombosis on pre and post liver transplant outcomes in cirrhosis. Ann Hepatol 2013; 12:952–958.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery/Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines. ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Associations for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (writing committee to develop guidelines for the management of patients with peripheral arterial disease)—summary of recommendations. J Vasc Interv Radiol 2006; 17:1383–1397.
- Schermerhorn ML, O’Malley AJ, Jhaveri A, Cotterill P, Pomposelli F, Landon BE. Endovascular vs open repair of abdominal aortic aneurysms in the Medicare population. N Engl J Med 2008; 358:464–474.
- Ohta M, Hashizume M, Ueno K, Tanoue K, Sugimachi K, Hasuo K. Hemodynamic study of splenic artery aneurysm in portal hypertension. Hepatogastroenterology 1994; 41:181–184.
- Sunagozaka H, Tsuji H, Mizukoshi E, et al. The development and clinical features of splenic aneurysm associated with liver cirrhosis. Liver Int 2006; 26:291–297.
- Manenti F, Williams R. Injection studies of the splenic vasculature in portal hypertension. Gut 1966; 7:175–180.
- Stanley JC, Fry WJ. Pathogenesis and clinical significance of splenic artery aneurysms. Surgery 1974; 76:898–909.
- Lee PC, Rhee RY, Gordon RY, Fung JJ, Webster MW. Management of splenic artery aneurysms: the significance of portal and essential hypertension. J Am Coll Surg 1999; 189:483–490.
- Al-Habbal Y, Christophi C, Muralidharan V. Aneurysms of the splenic artery—a review. Surgeon 2010; 8:223–231.
- Mattar SG, Lumsden AB. The management of splenic artery aneurysms: experience with 23 cases. Am J Surg 1995; 169:580–584.
- Akbulut S, Otan E. Management of giant splenic artery aneurysm: comprehensive literature review. Medicine (Baltimore) 2015; 94:e1016.
- Agrawal GA, Johnson PT, Fishman EK. Splenic artery aneurysms and pseudoaneurysms: clinical distinctions and CT appearances. AJR Am J Roentgenol 2007; 188:992–999.
- Tessier DJ, Stone WM, Fowl RJ, et al. Clinical features and management of splenic artery pseudoaneurysm: case series and cumulative review of literature. J Vasc Surg 2003; 38:969–974.
- Dave SP, Reis ED, Hossain A, Taub PJ, Kerstein MD, Hollier LH. Splenic artery aneurysm in the 1990s. Ann Vasc Surg 2000; 14:223–229.
- Parrish J, Maxwell C, Beecroft JR. Splenic artery aneurysm in pregnancy. J Obstet Gynaecol Can 2015; 37:816–818.
- Moon DB, Lee SG, Hwang S, et al. Characteristics and management of splenic artery aneurysms in adult living donor liver transplant recipients. Liver Transpl 2009; 15:1535–1541.
- Sadat U, Dar O, Walsh S, Varty K. Splenic artery aneurysms in pregnancy—a systematic review. Int J Surg 2008; 6:261–265.
- Geoghegan T, McAuley G, Snow A, Torreggiani WC. Emergency embolization of multiple splenic artery pseudoaneurysms associated with portal hypertension complicating cystic fibrosis. Australas Radiol 2007; 51(suppl):B337–B339.
- Jiang R, Ding X, Jian W, Jiang J, Hu S, Zhang Z. Combined endovascular embolization and open surgery for splenic artery aneurysm with arteriovenous fistula. Ann Vasc Surg 2016; 30:311.e1–311.e4.
- Naganuma M, Matsui H, Koizumi J, Fushimi K, Yasunaga H. Short-term outcomes following elective transcatheter arterial embolization for splenic artery aneurysms: data from a nationwide administrative database. Acta Radiol Open 2015; 4:2047981615574354.
- Batagini NC, El-Arousy H, Clair DG, Kirksey L. Open versus endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Ann Vasc Surg 2016; 35:1–8.
- Marone EM, Mascia D, Kahlberg A, Brioschi C, Tshomba Y, Chiesa R. Is open repair still the gold standard in visceral artery aneurysm management? Ann Vasc Surg 2011; 25:936–946.
- Sticco A, Aggarwal A, Shapiro M, Pratt A, Rissuci D, D'Ayala M. A comparison of open and endovascular treatment strategies for the management of splenic artery aneurysms. Vascular 2016; 24:487–491.
- Hogendoorn W, Lavida A, Hunink MG, et al. Cost-effectiveness of endovascular repair, open repair, and conservative management of splenic artery aneurysms. J Vasc Surg 2015; 61:1432–1440.
- Fankhauser GT, Stone WM, Naidu SG, et al; Mayo Vascular Research Center Consortium. The minimally invasive management of visceral artery aneurysms and pseudoaneurysms. J Vasc Surg 2011; 53:966–970.
- Lagana D, Carrafiello G, Mangini M, et al. Multimodal approach to endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Eur J Radiol 2006; 59:104–111.
- Guillon R, Garcier JM, Abergel A, et al. Management of splenic artery aneurysms and false aneurysms with endovascular treatment in 12 patients. Cardiovasc Intervent Radiol 2003; 26:256–260.
- Northup PG, Wanamaker RC, Lee VD, Adams RB, Berg CL. Model for end-stage liver disease (MELD) predicts nontransplant surgical mortality in patients with cirrhosis. Ann Surg 2005; 242:244–251.
When It All Comes Crashing Down
ANSWER
The radiograph shows that the patient is intubated. The lungs are clear overall. There is a fractured, slightly displaced left clavicle. Of concern, though, is the widened appearance of the mediastinum. In patients with blunt chest trauma, there should be a high index of suspicion for a great vessel injury, warranting a chest CT with contrast for further evaluation. Fortunately, in this patient's case, CT was negative.
ANSWER
The radiograph shows that the patient is intubated. The lungs are clear overall. There is a fractured, slightly displaced left clavicle. Of concern, though, is the widened appearance of the mediastinum. In patients with blunt chest trauma, there should be a high index of suspicion for a great vessel injury, warranting a chest CT with contrast for further evaluation. Fortunately, in this patient's case, CT was negative.
ANSWER
The radiograph shows that the patient is intubated. The lungs are clear overall. There is a fractured, slightly displaced left clavicle. Of concern, though, is the widened appearance of the mediastinum. In patients with blunt chest trauma, there should be a high index of suspicion for a great vessel injury, warranting a chest CT with contrast for further evaluation. Fortunately, in this patient's case, CT was negative.

A 40-year-old construction worker was remodeling a home when the roof collapsed. The patient’s head, face, and chest were reportedly struck by a large metal support beam. He was taken to a local facility, where he was found to have decreased level of consciousness and was combative. He was intubated for airway protection and sent to your facility for tertiary level of care.
History is limited. On arrival, you note a male patient who is intubated and sedated. His blood pressure is 90/60 mm Hg and his heart rate, 130 beats/min. A large laceration on his forehead and scalp has been primarily closed. His pupils are unequal, but both react. Neurologic exam is limited secondary to sedation.
As you complete your primary and secondary surveys, a portable chest radiograph is obtained (shown). What is your impression?