User login
AML leads percent gains in 5-year survival among leukemias
Over the 60-year span from the early 1950s to 2013, the 5-year survival rate for all leukemias increased by 500%, according to data from the Surveillance, Epidemiology, and End Results Program.
For 2008-2013, the 5-year relative survival rate for all leukemias was 60.1%, compared with 10% during 1950-1954, said Ali H. Mokdad, PhD, and his associates at the Institute for Health Metrics and Evaluation at the University of Washington, Seattle (JAMA 2017;317[4]:388-406).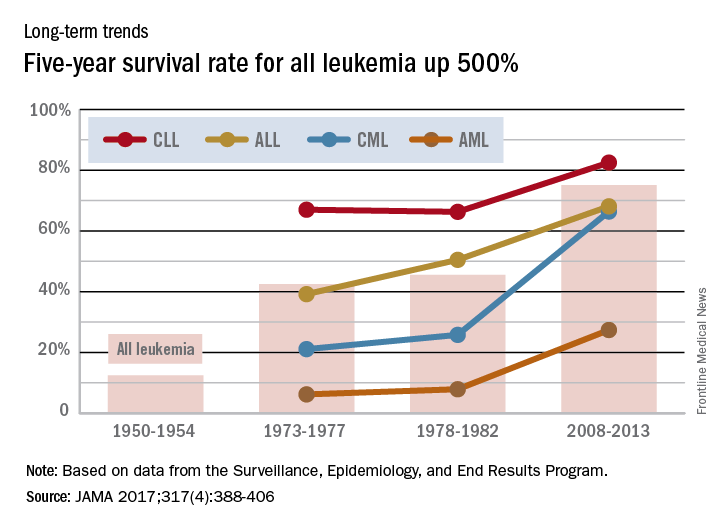
Over the 60-year span from the early 1950s to 2013, the 5-year survival rate for all leukemias increased by 500%, according to data from the Surveillance, Epidemiology, and End Results Program.
For 2008-2013, the 5-year relative survival rate for all leukemias was 60.1%, compared with 10% during 1950-1954, said Ali H. Mokdad, PhD, and his associates at the Institute for Health Metrics and Evaluation at the University of Washington, Seattle (JAMA 2017;317[4]:388-406).
Over the 60-year span from the early 1950s to 2013, the 5-year survival rate for all leukemias increased by 500%, according to data from the Surveillance, Epidemiology, and End Results Program.
For 2008-2013, the 5-year relative survival rate for all leukemias was 60.1%, compared with 10% during 1950-1954, said Ali H. Mokdad, PhD, and his associates at the Institute for Health Metrics and Evaluation at the University of Washington, Seattle (JAMA 2017;317[4]:388-406).
New recommendations for managing adult AML

Photo courtesy of CDC
The European LeukemiaNet (ELN) has released updated recommendations for the diagnosis and treatment of acute myeloid leukemia (AML) in adults.
The recommendations include revised ELN genetic categories, a proposed response category based on minimal residual disease status, and a proposed category for progressive disease for clinical trials.
They also include the updated World Health Organization classification of myeloid neoplasms and acute leukemia.
The recommendations are published in Blood.
“These guidelines are an important update of the current and widely used recommendations for managing AML, for constructing clinical trials, and for predicting outcomes of AML patients,” said Clara D. Bloomfield, MD, of The Ohio State University Comprehensive Cancer Center in Columbus.
“They will be the new standard of care and will replace the 2010 ELN recommendations for managing AML patients and designing clinical trials.”
Dr Bloomfield said updating the ELN recommendations was prompted by new insights into the molecular and genomic causes of AML, by the development of new genetic tests and tests for detecting minimal residual disease, and by the development of novel anti-leukemic agents.
Changes of note, according to Dr Bloomfield, are that there are now 3 genetic risk categories rather than 4, and the FLT3-ITD mutation has been added as a marker of risk.
In addition, “complete remission with no evidence of minimal residual disease” is a new proposed response category. This criterion requires that genetic markers present at diagnosis are no longer detectable.
“It is no longer good enough to examine bone marrow samples and say the leukemia is gone,” Dr Bloomfield said. “We must also see the loss of genetic markers.”
Another change is that “progressive disease” is a new provisional response category to be used in clinical trials only. The purpose of the category is to harmonize the various definitions of progressive disease that are used in different clinical trials. ![]()
Photo courtesy of CDC
The European LeukemiaNet (ELN) has released updated recommendations for the diagnosis and treatment of acute myeloid leukemia (AML) in adults.
The recommendations include revised ELN genetic categories, a proposed response category based on minimal residual disease status, and a proposed category for progressive disease for clinical trials.
They also include the updated World Health Organization classification of myeloid neoplasms and acute leukemia.
The recommendations are published in Blood.
“These guidelines are an important update of the current and widely used recommendations for managing AML, for constructing clinical trials, and for predicting outcomes of AML patients,” said Clara D. Bloomfield, MD, of The Ohio State University Comprehensive Cancer Center in Columbus.
“They will be the new standard of care and will replace the 2010 ELN recommendations for managing AML patients and designing clinical trials.”
Dr Bloomfield said updating the ELN recommendations was prompted by new insights into the molecular and genomic causes of AML, by the development of new genetic tests and tests for detecting minimal residual disease, and by the development of novel anti-leukemic agents.
Changes of note, according to Dr Bloomfield, are that there are now 3 genetic risk categories rather than 4, and the FLT3-ITD mutation has been added as a marker of risk.
In addition, “complete remission with no evidence of minimal residual disease” is a new proposed response category. This criterion requires that genetic markers present at diagnosis are no longer detectable.
“It is no longer good enough to examine bone marrow samples and say the leukemia is gone,” Dr Bloomfield said. “We must also see the loss of genetic markers.”
Another change is that “progressive disease” is a new provisional response category to be used in clinical trials only. The purpose of the category is to harmonize the various definitions of progressive disease that are used in different clinical trials. ![]()
Photo courtesy of CDC
The European LeukemiaNet (ELN) has released updated recommendations for the diagnosis and treatment of acute myeloid leukemia (AML) in adults.
The recommendations include revised ELN genetic categories, a proposed response category based on minimal residual disease status, and a proposed category for progressive disease for clinical trials.
They also include the updated World Health Organization classification of myeloid neoplasms and acute leukemia.
The recommendations are published in Blood.
“These guidelines are an important update of the current and widely used recommendations for managing AML, for constructing clinical trials, and for predicting outcomes of AML patients,” said Clara D. Bloomfield, MD, of The Ohio State University Comprehensive Cancer Center in Columbus.
“They will be the new standard of care and will replace the 2010 ELN recommendations for managing AML patients and designing clinical trials.”
Dr Bloomfield said updating the ELN recommendations was prompted by new insights into the molecular and genomic causes of AML, by the development of new genetic tests and tests for detecting minimal residual disease, and by the development of novel anti-leukemic agents.
Changes of note, according to Dr Bloomfield, are that there are now 3 genetic risk categories rather than 4, and the FLT3-ITD mutation has been added as a marker of risk.
In addition, “complete remission with no evidence of minimal residual disease” is a new proposed response category. This criterion requires that genetic markers present at diagnosis are no longer detectable.
“It is no longer good enough to examine bone marrow samples and say the leukemia is gone,” Dr Bloomfield said. “We must also see the loss of genetic markers.”
Another change is that “progressive disease” is a new provisional response category to be used in clinical trials only. The purpose of the category is to harmonize the various definitions of progressive disease that are used in different clinical trials. ![]()
Distress linked to higher risk of death from leukemia, other cancers

patient and her father
Photo by Rhoda Baer
A study published in The BMJ suggests that higher levels of psychological distress (anxiety and depression) may be associated with an increased risk of death from leukemia and other cancers.
The findings are observational, so no firm conclusions about cause and effect can be drawn.
However, the researchers said the findings add to the growing evidence that psychological distress could have some predictive capacity for certain physical conditions.
There is some evidence that psychological distress (anxiety and depression) is related to increased rates of cardiovascular disease, but links with different types of cancer are either unclear or untested.
So David Batty, PhD, of University College London in the UK, and his colleagues set out to examine if psychological distress is a potential predictor of site-specific cancer mortality.
The researchers analyzed data from 16 studies (13 from England and 3 from Scotland), which started between 1994 and 2008. The data included 163,363 men and women age 16 or over who were free from cancer at the start of the study.
Psychological distress scores were measured using the general health questionnaire (GHQ-12), and participants were monitored for an average of 9.5 years. During this time, there were 4353 deaths from cancer.
Several factors that could have influenced the results were taken into account, including age, sex, education, socioeconomic status, body mass index, smoking, and alcohol intake.
“After statistical control for these factors, the results show that, compared with people in the least distressed group, death rates in the most distressed group were consistently higher for cancer of the bowel, prostate, pancreas, and esophagus and for leukemia,” Dr Batty said.
He and his colleagues pointed out that this association may also be affected by reverse causality, where undiagnosed (early) cancer might have had an underlying impact on mood.
In a bid to correct for this, the team conducted a further analysis excluding study participants who died in the first 5 years of follow-up, but this made no difference to the findings. The links between distress and cancer remained.
Specifically, compared to people in the least distressed group (GHQ-12 score 0-6), death rates in the most distressed group (score 7-12) were consistently increased for cancer of all sites combined, with a multivariable adjusted hazard ratio (HR) of 1.32.
Death rates were also increased for colorectal (HR=1.84), prostate (HR=2.42), pancreatic (HR=2.76), and esophageal cancers (HR=2.59), as well as for leukemia (HR=3.86).
“Our findings contribute to the evidence that poor mental health might have some predictive capacity for certain physical diseases,” Dr Batty said, “but we are a long way off from knowing if these relationships are truly causal.” ![]()
patient and her father
Photo by Rhoda Baer
A study published in The BMJ suggests that higher levels of psychological distress (anxiety and depression) may be associated with an increased risk of death from leukemia and other cancers.
The findings are observational, so no firm conclusions about cause and effect can be drawn.
However, the researchers said the findings add to the growing evidence that psychological distress could have some predictive capacity for certain physical conditions.
There is some evidence that psychological distress (anxiety and depression) is related to increased rates of cardiovascular disease, but links with different types of cancer are either unclear or untested.
So David Batty, PhD, of University College London in the UK, and his colleagues set out to examine if psychological distress is a potential predictor of site-specific cancer mortality.
The researchers analyzed data from 16 studies (13 from England and 3 from Scotland), which started between 1994 and 2008. The data included 163,363 men and women age 16 or over who were free from cancer at the start of the study.
Psychological distress scores were measured using the general health questionnaire (GHQ-12), and participants were monitored for an average of 9.5 years. During this time, there were 4353 deaths from cancer.
Several factors that could have influenced the results were taken into account, including age, sex, education, socioeconomic status, body mass index, smoking, and alcohol intake.
“After statistical control for these factors, the results show that, compared with people in the least distressed group, death rates in the most distressed group were consistently higher for cancer of the bowel, prostate, pancreas, and esophagus and for leukemia,” Dr Batty said.
He and his colleagues pointed out that this association may also be affected by reverse causality, where undiagnosed (early) cancer might have had an underlying impact on mood.
In a bid to correct for this, the team conducted a further analysis excluding study participants who died in the first 5 years of follow-up, but this made no difference to the findings. The links between distress and cancer remained.
Specifically, compared to people in the least distressed group (GHQ-12 score 0-6), death rates in the most distressed group (score 7-12) were consistently increased for cancer of all sites combined, with a multivariable adjusted hazard ratio (HR) of 1.32.
Death rates were also increased for colorectal (HR=1.84), prostate (HR=2.42), pancreatic (HR=2.76), and esophageal cancers (HR=2.59), as well as for leukemia (HR=3.86).
“Our findings contribute to the evidence that poor mental health might have some predictive capacity for certain physical diseases,” Dr Batty said, “but we are a long way off from knowing if these relationships are truly causal.” ![]()
patient and her father
Photo by Rhoda Baer
A study published in The BMJ suggests that higher levels of psychological distress (anxiety and depression) may be associated with an increased risk of death from leukemia and other cancers.
The findings are observational, so no firm conclusions about cause and effect can be drawn.
However, the researchers said the findings add to the growing evidence that psychological distress could have some predictive capacity for certain physical conditions.
There is some evidence that psychological distress (anxiety and depression) is related to increased rates of cardiovascular disease, but links with different types of cancer are either unclear or untested.
So David Batty, PhD, of University College London in the UK, and his colleagues set out to examine if psychological distress is a potential predictor of site-specific cancer mortality.
The researchers analyzed data from 16 studies (13 from England and 3 from Scotland), which started between 1994 and 2008. The data included 163,363 men and women age 16 or over who were free from cancer at the start of the study.
Psychological distress scores were measured using the general health questionnaire (GHQ-12), and participants were monitored for an average of 9.5 years. During this time, there were 4353 deaths from cancer.
Several factors that could have influenced the results were taken into account, including age, sex, education, socioeconomic status, body mass index, smoking, and alcohol intake.
“After statistical control for these factors, the results show that, compared with people in the least distressed group, death rates in the most distressed group were consistently higher for cancer of the bowel, prostate, pancreas, and esophagus and for leukemia,” Dr Batty said.
He and his colleagues pointed out that this association may also be affected by reverse causality, where undiagnosed (early) cancer might have had an underlying impact on mood.
In a bid to correct for this, the team conducted a further analysis excluding study participants who died in the first 5 years of follow-up, but this made no difference to the findings. The links between distress and cancer remained.
Specifically, compared to people in the least distressed group (GHQ-12 score 0-6), death rates in the most distressed group (score 7-12) were consistently increased for cancer of all sites combined, with a multivariable adjusted hazard ratio (HR) of 1.32.
Death rates were also increased for colorectal (HR=1.84), prostate (HR=2.42), pancreatic (HR=2.76), and esophageal cancers (HR=2.59), as well as for leukemia (HR=3.86).
“Our findings contribute to the evidence that poor mental health might have some predictive capacity for certain physical diseases,” Dr Batty said, “but we are a long way off from knowing if these relationships are truly causal.” ![]()
Donor CAR T cells bridge to HSCT in infants with ALL
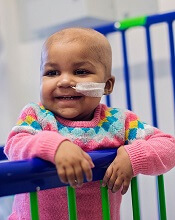
treated with UCART19
Photo courtesy of GOSH
New research suggests that “universal,” donor-derived, chimeric antigen receptor (CAR) T cells may be a viable treatment option for very young children who do not have sufficient healthy T cells for autologous CAR T-cell therapy.
The universal CAR T-cell therapy, known as UCART19, was given to 2 infants with relapsed/refractory B-cell acute lymphoblastic leukemia (ALL) who had previously
exhausted all other treatment options.
Both infants achieved remission after UCART19 and were able to proceed to transplant.
Both were still alive and leukemia-free at last follow-up—12 months and 18 months after UCART19, respectively.
Waseem Qasim, MBBS, PhD, of University College London’s Institute of Child Health and Great Ormond Street Hospital (GOSH) in London, UK, and his colleagues reported these results in Science Translational Medicine.
About the therapy
UCART19 consists of donor T cells modified using transcription activator-like effector nucleases. The cells are programmed to target CD19 and be insensitive to alemtuzumab. That way, a patient can receive alemtuzumab to prevent rejection of HLA-mismatched cells.
UCART19 was under development by Cellectis but is now being developed by Servier and Pfizer Inc. Pfizer has exclusive rights to develop and commercialize UCART19 in the US, while Servier retains exclusive rights for all other countries.
In Science Translational Medicine, Dr Qasim and his colleagues reported results in the first patients ever treated with UCART19. That research was funded, in part, by Cellectis.
Subject 1
The first patient was an 11-month-old, mixed-race infant with high-risk, t(11;19), CD19+ B-ALL. She had already failed chemotherapy, an allogeneic hematopoietic stem cell transplant (HSCT), and blinatumomab.
Prior to UCART19, she received a dose of vincristine and asparaginase and 7 days of dexamethasone, followed by cytoreduction with fludarabine, cyclophosphamide, and alemtuzumab. She then received a single dose of UCART19 (4.6 × 106/kg).
The patient had neutrophil recovery by day 30, although this was dependent on granulocyte colony-stimulating factor. After that, she developed multilineage cytopenia that persisted until she underwent a second allogeneic HSCT.
Prior to the second HSCT, the patient achieved cytogenetic and molecular remission but also developed grade 2 skin graft-vs-host disease. This was managed with systemic steroids.
The child received rituximab and conditioning with antithymocyte globulin, fludarabine, cyclophosphamide, thiotepa, and total body irradiation, followed by an HSCT from the original mismatched, unrelated donor.
The child achieved a complete remission and has remained minimal residual disease-negative, with full donor chimerism and normalized lymphocyte profiles, at 18 months after UCART19.
Results with this patient were previously reported by GOSH in November 2015 and at the 2015 ASH Annual Meeting.
Subject 2
The second patient was a 16-month-old Caucasian infant who had been diagnosed at 4 weeks of age with high-risk, congenital, mixed lineage leukemia–rearranged B-ALL.
She had already undergone HSCT from a matched, unrelated donor but relapsed. She did not respond to subsequent blinatumomab.
After these failed treatments, the patient received fludarabine, cyclophosphamide, and alemtuzumab, followed by a single infusion of UCART19 (4.0 × 106/kg).
She developed an erythematous rash after treatment, but this was immediately responsive to topical steroids.
The child achieved donor-derived neutrophil recovery and went on to receive a transplant from the same matched, unrelated donor as her previous HSCT.
The child received the transplant within 10 weeks of UCART19 therapy, after receiving rituximab and conditioning with antithymocyte globulin, fludarabine, cyclophosphamide, and total body irradiation.
At 12 months after UCART19, the child remains minimal residual disease-negative and “clinically well.”
“Both infants who have had this treatment have been at home for some time and are doing well,” Dr Qasim said. “We continue to monitor them closely, and, while we have reduced the frequency of their hospital visits and checks, we will still need to keep an eye on them for some time.”
Dr Qasim added that there are phase 1 trials of UCART19 underway for children and adults with chronic lymphocytic leukemia and acute lymphoblastic leukemia. ![]()
treated with UCART19
Photo courtesy of GOSH
New research suggests that “universal,” donor-derived, chimeric antigen receptor (CAR) T cells may be a viable treatment option for very young children who do not have sufficient healthy T cells for autologous CAR T-cell therapy.
The universal CAR T-cell therapy, known as UCART19, was given to 2 infants with relapsed/refractory B-cell acute lymphoblastic leukemia (ALL) who had previously
exhausted all other treatment options.
Both infants achieved remission after UCART19 and were able to proceed to transplant.
Both were still alive and leukemia-free at last follow-up—12 months and 18 months after UCART19, respectively.
Waseem Qasim, MBBS, PhD, of University College London’s Institute of Child Health and Great Ormond Street Hospital (GOSH) in London, UK, and his colleagues reported these results in Science Translational Medicine.
About the therapy
UCART19 consists of donor T cells modified using transcription activator-like effector nucleases. The cells are programmed to target CD19 and be insensitive to alemtuzumab. That way, a patient can receive alemtuzumab to prevent rejection of HLA-mismatched cells.
UCART19 was under development by Cellectis but is now being developed by Servier and Pfizer Inc. Pfizer has exclusive rights to develop and commercialize UCART19 in the US, while Servier retains exclusive rights for all other countries.
In Science Translational Medicine, Dr Qasim and his colleagues reported results in the first patients ever treated with UCART19. That research was funded, in part, by Cellectis.
Subject 1
The first patient was an 11-month-old, mixed-race infant with high-risk, t(11;19), CD19+ B-ALL. She had already failed chemotherapy, an allogeneic hematopoietic stem cell transplant (HSCT), and blinatumomab.
Prior to UCART19, she received a dose of vincristine and asparaginase and 7 days of dexamethasone, followed by cytoreduction with fludarabine, cyclophosphamide, and alemtuzumab. She then received a single dose of UCART19 (4.6 × 106/kg).
The patient had neutrophil recovery by day 30, although this was dependent on granulocyte colony-stimulating factor. After that, she developed multilineage cytopenia that persisted until she underwent a second allogeneic HSCT.
Prior to the second HSCT, the patient achieved cytogenetic and molecular remission but also developed grade 2 skin graft-vs-host disease. This was managed with systemic steroids.
The child received rituximab and conditioning with antithymocyte globulin, fludarabine, cyclophosphamide, thiotepa, and total body irradiation, followed by an HSCT from the original mismatched, unrelated donor.
The child achieved a complete remission and has remained minimal residual disease-negative, with full donor chimerism and normalized lymphocyte profiles, at 18 months after UCART19.
Results with this patient were previously reported by GOSH in November 2015 and at the 2015 ASH Annual Meeting.
Subject 2
The second patient was a 16-month-old Caucasian infant who had been diagnosed at 4 weeks of age with high-risk, congenital, mixed lineage leukemia–rearranged B-ALL.
She had already undergone HSCT from a matched, unrelated donor but relapsed. She did not respond to subsequent blinatumomab.
After these failed treatments, the patient received fludarabine, cyclophosphamide, and alemtuzumab, followed by a single infusion of UCART19 (4.0 × 106/kg).
She developed an erythematous rash after treatment, but this was immediately responsive to topical steroids.
The child achieved donor-derived neutrophil recovery and went on to receive a transplant from the same matched, unrelated donor as her previous HSCT.
The child received the transplant within 10 weeks of UCART19 therapy, after receiving rituximab and conditioning with antithymocyte globulin, fludarabine, cyclophosphamide, and total body irradiation.
At 12 months after UCART19, the child remains minimal residual disease-negative and “clinically well.”
“Both infants who have had this treatment have been at home for some time and are doing well,” Dr Qasim said. “We continue to monitor them closely, and, while we have reduced the frequency of their hospital visits and checks, we will still need to keep an eye on them for some time.”
Dr Qasim added that there are phase 1 trials of UCART19 underway for children and adults with chronic lymphocytic leukemia and acute lymphoblastic leukemia. ![]()
treated with UCART19
Photo courtesy of GOSH
New research suggests that “universal,” donor-derived, chimeric antigen receptor (CAR) T cells may be a viable treatment option for very young children who do not have sufficient healthy T cells for autologous CAR T-cell therapy.
The universal CAR T-cell therapy, known as UCART19, was given to 2 infants with relapsed/refractory B-cell acute lymphoblastic leukemia (ALL) who had previously
exhausted all other treatment options.
Both infants achieved remission after UCART19 and were able to proceed to transplant.
Both were still alive and leukemia-free at last follow-up—12 months and 18 months after UCART19, respectively.
Waseem Qasim, MBBS, PhD, of University College London’s Institute of Child Health and Great Ormond Street Hospital (GOSH) in London, UK, and his colleagues reported these results in Science Translational Medicine.
About the therapy
UCART19 consists of donor T cells modified using transcription activator-like effector nucleases. The cells are programmed to target CD19 and be insensitive to alemtuzumab. That way, a patient can receive alemtuzumab to prevent rejection of HLA-mismatched cells.
UCART19 was under development by Cellectis but is now being developed by Servier and Pfizer Inc. Pfizer has exclusive rights to develop and commercialize UCART19 in the US, while Servier retains exclusive rights for all other countries.
In Science Translational Medicine, Dr Qasim and his colleagues reported results in the first patients ever treated with UCART19. That research was funded, in part, by Cellectis.
Subject 1
The first patient was an 11-month-old, mixed-race infant with high-risk, t(11;19), CD19+ B-ALL. She had already failed chemotherapy, an allogeneic hematopoietic stem cell transplant (HSCT), and blinatumomab.
Prior to UCART19, she received a dose of vincristine and asparaginase and 7 days of dexamethasone, followed by cytoreduction with fludarabine, cyclophosphamide, and alemtuzumab. She then received a single dose of UCART19 (4.6 × 106/kg).
The patient had neutrophil recovery by day 30, although this was dependent on granulocyte colony-stimulating factor. After that, she developed multilineage cytopenia that persisted until she underwent a second allogeneic HSCT.
Prior to the second HSCT, the patient achieved cytogenetic and molecular remission but also developed grade 2 skin graft-vs-host disease. This was managed with systemic steroids.
The child received rituximab and conditioning with antithymocyte globulin, fludarabine, cyclophosphamide, thiotepa, and total body irradiation, followed by an HSCT from the original mismatched, unrelated donor.
The child achieved a complete remission and has remained minimal residual disease-negative, with full donor chimerism and normalized lymphocyte profiles, at 18 months after UCART19.
Results with this patient were previously reported by GOSH in November 2015 and at the 2015 ASH Annual Meeting.
Subject 2
The second patient was a 16-month-old Caucasian infant who had been diagnosed at 4 weeks of age with high-risk, congenital, mixed lineage leukemia–rearranged B-ALL.
She had already undergone HSCT from a matched, unrelated donor but relapsed. She did not respond to subsequent blinatumomab.
After these failed treatments, the patient received fludarabine, cyclophosphamide, and alemtuzumab, followed by a single infusion of UCART19 (4.0 × 106/kg).
She developed an erythematous rash after treatment, but this was immediately responsive to topical steroids.
The child achieved donor-derived neutrophil recovery and went on to receive a transplant from the same matched, unrelated donor as her previous HSCT.
The child received the transplant within 10 weeks of UCART19 therapy, after receiving rituximab and conditioning with antithymocyte globulin, fludarabine, cyclophosphamide, and total body irradiation.
At 12 months after UCART19, the child remains minimal residual disease-negative and “clinically well.”
“Both infants who have had this treatment have been at home for some time and are doing well,” Dr Qasim said. “We continue to monitor them closely, and, while we have reduced the frequency of their hospital visits and checks, we will still need to keep an eye on them for some time.”
Dr Qasim added that there are phase 1 trials of UCART19 underway for children and adults with chronic lymphocytic leukemia and acute lymphoblastic leukemia. ![]()
Study quantifies 5-year survival rates for blood cancers

chemotherapy
Photo by Rhoda Baer
A new study shows that 5-year survival rates for US patients with hematologic malignancies have increased greatly since the 1950s, but there is still room for improvement, particularly for patients with acute myeloid leukemia (AML).
Researchers found the absolute difference in improvement for 5-year survival from 1950-1954 to 2008-2013 ranged from 38.2% for non-Hodgkin lymphoma (NHL) to 56.6% for Hodgkin lymphoma.
And although the 5-year survival rate for Hodgkin lymphoma patients reached 86.6% for 2008-2013, the 5-year survival rate for patients with AML only reached 27.4%.
This study also revealed large disparities in overall cancer mortality rates between different counties across the country.
Ali H. Mokdad, PhD, of the Institute for Health Metrics and Evaluation in Seattle, Washington, and his colleagues reported these findings in JAMA.
Overall cancer deaths
The researchers found there were 19,511,910 cancer deaths recorded in the US between 1980 and 2014. Cancer mortality decreased by 20.1% between 1980 and 2014, from 240.2 deaths per 100,000 people to 192.0 deaths per 100,000 people.
In 1980, cancer mortality ranged from 130.6 per 100,000 in Summit County, Colorado, to 386.9 per 100,000 in North Slope Borough, Alaska.
In 2014, cancer mortality ranged from 70.7 per 100,000 in Summit County, Colorado, to 503.1 per 100,000 in Union County, Florida.
“Such significant disparities among US counties is unacceptable,” Dr Mokdad said. “Every person should have access to early screenings for cancer, as well as adequate treatment.”
Mortality rates for hematologic malignancies
In 2014, the mortality rates, per 100,000 people, for hematologic malignancies were:
- 0.4 for Hodgkin lymphoma (rank out of all cancers, 27)
- 8.3 for NHL (rank, 7)
- 3.9 for multiple myeloma (rank, 16)
- 9.0 for all leukemias (rank, 6)
- 0.7 for acute lymphoid leukemia (ALL)
- 2.6 for chronic lymphoid leukemia (CLL)
- 5.1 for AML
- 0.6 for chronic myeloid leukemia (CML).
The leukemia subtypes were not assigned a rank.
5-year survival rates for hematologic malignancies
Hodgkin lymphoma
- 30% for 1950-54
- 68.6% for 1973-77
- 72.1% for 1978-82
- 86.6% for 2008-2013
- Absolute difference (between the first and latest year of data), 56.6%.
NHL
- 33% for 1950-54
- 45.3% for 1973-77
- 48.7% for 1978-82
- 71.2% for 2008-2013
- Absolute difference, 38.2%.
Multiple myeloma
- 6% for 1950-54
- 23.4% for 1973-77
- 26.6% for 1978-82
- 49.8% for 2008-2013
- Absolute difference, 43.8%.
Leukemia
- 10% for 1950-54
- 34% for 1973-77
- 36.3% for 1978-82
- 60.1% for 2008-2013
- Absolute difference, 50.1%.
ALL
- 39.2% for 1973-77
- 50.5% for 1978-82
- 68.1% for 2008-2013
- Absolute difference, 28.9%.
CLL
- 67% for 1973-77
- 66.3% for 1978-82
- 82.5% for 2008-2013
- Absolute difference, 15.5%.
AML
- 6.2% for 1973-77
- 7.9% for 1978-82
- 27.4% for 2008-2013
- Absolute difference, 21.2%.
CML
- 21.1% for 1973-77
- 25.8% for 1978-82
- 66.4% for 2008-2013
- Absolute difference, 45.3%.
For the leukemia subtypes, there was no data for 1950 to 1954. ![]()
chemotherapy
Photo by Rhoda Baer
A new study shows that 5-year survival rates for US patients with hematologic malignancies have increased greatly since the 1950s, but there is still room for improvement, particularly for patients with acute myeloid leukemia (AML).
Researchers found the absolute difference in improvement for 5-year survival from 1950-1954 to 2008-2013 ranged from 38.2% for non-Hodgkin lymphoma (NHL) to 56.6% for Hodgkin lymphoma.
And although the 5-year survival rate for Hodgkin lymphoma patients reached 86.6% for 2008-2013, the 5-year survival rate for patients with AML only reached 27.4%.
This study also revealed large disparities in overall cancer mortality rates between different counties across the country.
Ali H. Mokdad, PhD, of the Institute for Health Metrics and Evaluation in Seattle, Washington, and his colleagues reported these findings in JAMA.
Overall cancer deaths
The researchers found there were 19,511,910 cancer deaths recorded in the US between 1980 and 2014. Cancer mortality decreased by 20.1% between 1980 and 2014, from 240.2 deaths per 100,000 people to 192.0 deaths per 100,000 people.
In 1980, cancer mortality ranged from 130.6 per 100,000 in Summit County, Colorado, to 386.9 per 100,000 in North Slope Borough, Alaska.
In 2014, cancer mortality ranged from 70.7 per 100,000 in Summit County, Colorado, to 503.1 per 100,000 in Union County, Florida.
“Such significant disparities among US counties is unacceptable,” Dr Mokdad said. “Every person should have access to early screenings for cancer, as well as adequate treatment.”
Mortality rates for hematologic malignancies
In 2014, the mortality rates, per 100,000 people, for hematologic malignancies were:
- 0.4 for Hodgkin lymphoma (rank out of all cancers, 27)
- 8.3 for NHL (rank, 7)
- 3.9 for multiple myeloma (rank, 16)
- 9.0 for all leukemias (rank, 6)
- 0.7 for acute lymphoid leukemia (ALL)
- 2.6 for chronic lymphoid leukemia (CLL)
- 5.1 for AML
- 0.6 for chronic myeloid leukemia (CML).
The leukemia subtypes were not assigned a rank.
5-year survival rates for hematologic malignancies
Hodgkin lymphoma
- 30% for 1950-54
- 68.6% for 1973-77
- 72.1% for 1978-82
- 86.6% for 2008-2013
- Absolute difference (between the first and latest year of data), 56.6%.
NHL
- 33% for 1950-54
- 45.3% for 1973-77
- 48.7% for 1978-82
- 71.2% for 2008-2013
- Absolute difference, 38.2%.
Multiple myeloma
- 6% for 1950-54
- 23.4% for 1973-77
- 26.6% for 1978-82
- 49.8% for 2008-2013
- Absolute difference, 43.8%.
Leukemia
- 10% for 1950-54
- 34% for 1973-77
- 36.3% for 1978-82
- 60.1% for 2008-2013
- Absolute difference, 50.1%.
ALL
- 39.2% for 1973-77
- 50.5% for 1978-82
- 68.1% for 2008-2013
- Absolute difference, 28.9%.
CLL
- 67% for 1973-77
- 66.3% for 1978-82
- 82.5% for 2008-2013
- Absolute difference, 15.5%.
AML
- 6.2% for 1973-77
- 7.9% for 1978-82
- 27.4% for 2008-2013
- Absolute difference, 21.2%.
CML
- 21.1% for 1973-77
- 25.8% for 1978-82
- 66.4% for 2008-2013
- Absolute difference, 45.3%.
For the leukemia subtypes, there was no data for 1950 to 1954. ![]()
chemotherapy
Photo by Rhoda Baer
A new study shows that 5-year survival rates for US patients with hematologic malignancies have increased greatly since the 1950s, but there is still room for improvement, particularly for patients with acute myeloid leukemia (AML).
Researchers found the absolute difference in improvement for 5-year survival from 1950-1954 to 2008-2013 ranged from 38.2% for non-Hodgkin lymphoma (NHL) to 56.6% for Hodgkin lymphoma.
And although the 5-year survival rate for Hodgkin lymphoma patients reached 86.6% for 2008-2013, the 5-year survival rate for patients with AML only reached 27.4%.
This study also revealed large disparities in overall cancer mortality rates between different counties across the country.
Ali H. Mokdad, PhD, of the Institute for Health Metrics and Evaluation in Seattle, Washington, and his colleagues reported these findings in JAMA.
Overall cancer deaths
The researchers found there were 19,511,910 cancer deaths recorded in the US between 1980 and 2014. Cancer mortality decreased by 20.1% between 1980 and 2014, from 240.2 deaths per 100,000 people to 192.0 deaths per 100,000 people.
In 1980, cancer mortality ranged from 130.6 per 100,000 in Summit County, Colorado, to 386.9 per 100,000 in North Slope Borough, Alaska.
In 2014, cancer mortality ranged from 70.7 per 100,000 in Summit County, Colorado, to 503.1 per 100,000 in Union County, Florida.
“Such significant disparities among US counties is unacceptable,” Dr Mokdad said. “Every person should have access to early screenings for cancer, as well as adequate treatment.”
Mortality rates for hematologic malignancies
In 2014, the mortality rates, per 100,000 people, for hematologic malignancies were:
- 0.4 for Hodgkin lymphoma (rank out of all cancers, 27)
- 8.3 for NHL (rank, 7)
- 3.9 for multiple myeloma (rank, 16)
- 9.0 for all leukemias (rank, 6)
- 0.7 for acute lymphoid leukemia (ALL)
- 2.6 for chronic lymphoid leukemia (CLL)
- 5.1 for AML
- 0.6 for chronic myeloid leukemia (CML).
The leukemia subtypes were not assigned a rank.
5-year survival rates for hematologic malignancies
Hodgkin lymphoma
- 30% for 1950-54
- 68.6% for 1973-77
- 72.1% for 1978-82
- 86.6% for 2008-2013
- Absolute difference (between the first and latest year of data), 56.6%.
NHL
- 33% for 1950-54
- 45.3% for 1973-77
- 48.7% for 1978-82
- 71.2% for 2008-2013
- Absolute difference, 38.2%.
Multiple myeloma
- 6% for 1950-54
- 23.4% for 1973-77
- 26.6% for 1978-82
- 49.8% for 2008-2013
- Absolute difference, 43.8%.
Leukemia
- 10% for 1950-54
- 34% for 1973-77
- 36.3% for 1978-82
- 60.1% for 2008-2013
- Absolute difference, 50.1%.
ALL
- 39.2% for 1973-77
- 50.5% for 1978-82
- 68.1% for 2008-2013
- Absolute difference, 28.9%.
CLL
- 67% for 1973-77
- 66.3% for 1978-82
- 82.5% for 2008-2013
- Absolute difference, 15.5%.
AML
- 6.2% for 1973-77
- 7.9% for 1978-82
- 27.4% for 2008-2013
- Absolute difference, 21.2%.
CML
- 21.1% for 1973-77
- 25.8% for 1978-82
- 66.4% for 2008-2013
- Absolute difference, 45.3%.
For the leukemia subtypes, there was no data for 1950 to 1954. ![]()
Ruxolitinib beats best available care for hematocrit control in polycythemia
For patients with polycythemia vera without splenomegaly who had inadequate responses to hydroxyurea, targeted therapy with the Janus kinase (JAK) inhibitor ruxolitinib (Jakafi) offered better control of hematocrit and better improvement of symptoms than did the best available therapy, according to results of a multinational randomized phase IIIb trial.
Among 74 patients randomly assigned to receive ruxolitinib, 46 (62%) achieved hematocrit control, compared with 14 of 75 patients (19%) assigned to receive one of several different options lumped into the best available therapy category, noted investigators led by Francesco Passamonti, MD, of the University of Insubria in Varese, Italy.
“Ruxolitinib also led to an improved symptom burden and quality of life. Patients treated with ruxolitinib experienced improvements in all polycythemia vera–associated symptoms, including pruritus, whereas patients treated with best available therapy experienced worsening of most symptoms,” they wrote.
Unlike cytoreductive therapies such as hydroxyurea or pegylated interferon, ruxolitinib works by inhibition of JAK1 and JAK2 signaling. A majority of patients with polycythemia vera have an activating JAK2 mutation that leads to overactivation of the JAK-STAT signaling pathway, resulting in erythrocytosis, the hallmark symptom of polycythemia vera, and associated vascular complications.
“In some patients, conventional therapies can lose effectiveness over time. Although hydroxyurea is well tolerated in most patients, about 15%-20% of patients become resistant or intolerant, with hydroxyurea resistance affecting survival and increasing the risk of progression to myelofibrosis. Additionally, patients who are intolerant of hydroxyurea can have adverse side effects, such as drug-induced fever, mouth ulcers, leg ulcers, and skin malignancies, which necessitate discontinuation of first-line therapy,” the investigators noted.
RESPONSE trials
Ruxolitinib had previously been shown in the RESPONSE study to be superior to the best available therapy for controlling hematocrit and for improving splenomegaly and other symptoms in patients with polycythemia vera and disease-associated splenomegaly who had an inadequate response or unacceptable toxicities from treatment with hydroxyurea.
In the currently reported study, dubbed RESPONSE-2, patients 18 and older with polycythemia vera with no palpable splenomegaly who were intolerant of hydroxyurea or had disease that was resistant to it were randomized to receive either oral ruxolitinib 10 mg twice daily, or best available therapy at the investigators’ discretion. Best available therapy consisted of either hydroxyurea at the maximum tolerated dose, interferon or pegylated interferon, pipobroman, anagrelide, approved immunomodulators, or no cytoreductive treatment.
As noted, hematocrit control at week 28, the primary endpoint, was significantly higher among patients on ruxolitinib, with an odds ratio (OR) of 7.28 (P less than .0001).
Hematocrit levels among patients on ruxolitinib group decreased from a mean of 42.8% at baseline to 40.2% at week 28. In contrast, hematocrit in the best available therapy group increased from a mean of 42.7% to 44.9% at week 28.
Fewer patients on ruxolitinib required phlebotomy procedures during the 28 weeks of the study compared with patients on best available therapy, and of those patients who did undergo phlebotomy, fewer of those in the ruxolitinib group underwent more than two procedures. There were a total of 98 phlebotomies among best available care patients, vs. 19 among ruxolitinib patients.
Complete hematologic remissions, a secondary endpoint, occurred in 23% of patients on ruxolitinib, compared with 5% of those on best available care (OR 5.58, P = .0019).
The most frequent hematologic adverse events of any grade were anemia, which occurred in 10 patients on ruxolitinib (none grade 3 or greater) vs. two on best available therapy (one grade 3), and thrombocytopenia occurring in two (both grade 1 or 2) and six patients (three grade 1 or 2, two grade 3, and one grade 4) respectively.
Grade 3 or 4 nonhematologic adverse events included hypertension in five patients on ruxolitinib vs. three on best available care, and pruritus in none vs. two, respectively.
Two patients died; both were in the best available therapy group.
“Although the short follow-up of this study precludes any conclusions about vascular complications, an important finding is that patients treated with ruxolitinib in both RESPONSE and RESPONSE-2 had fewer thromboembolic events compared with those given best available therapy; there were two thromboembolic events with ruxolitinib (one in each study) versus nine with best available therapy across both studies (six in RESPONSE and three in RESPONSE-2). This finding could have been attributable to better control of hematocrit or white blood cell count with ruxolitinib, given that baseline risk factors were similar in both treatment groups,” the investigators wrote.
The findings from the two studies support the use of ruxolitinib as a standard of care for second-line therapy of patients with polycythemia vera following treatment with hydroxyurea, they contended.
Several coauthors disclosed ties to Novartis, which supported the study. Two are Novartis employees; one was previously employed by the company.
Ruxolitinib has now been assessed in two clinical trials in polycythemia vera without myelofibrosis. Francesco Passamonti and colleagues report the results of RESPONSE-2, a randomized, open-label, phase IIIb trial of ruxolitinib in patients with polycythemia vera without splenomegaly, who were intolerant of or unresponsive to hydroxyurea, versus the best available therapy (usually hydroxyurea), making this trial – like the previous RESPONSE trial, in which the effect of ruxolitinib was examined in hydroxyurea-intolerant or unresponsive patients with polycythemia vera with splenomegaly – a referendum on hydroxyurea. On the basis of their age, most RESPONSE-2 patients were defined as so-called high-risk patients, and were phlebotomy dependent.
Unsurprisingly, for the primary endpoint of patients achieving hematocrit control, ruxolitinib was superior to best available therapy (46 [62%] of 74 patients in the ruxolitinib group vs. 14 [19%] of 75 in the best available therapy group; odds ratio, 7.28 [95% CI 3.43–15.45]; P less than .0001), and also for the key secondary endpoint for patients achieving complete hematologic remission (23% vs .5%).
Symptom control, including pruritus, was superior in the ruxolitinib group, and adverse events were more common in the best available therapy group. The authors concluded that ruxolitinib “could be considered a standard of care for second-line therapy in this post-hydroxyurea patient population”
However, I challenge this conclusion. First, the consensus that hydroxyurea is first-line therapy for polycythemia vera is not evidence based. Indeed, a trial using hydroxyurea in patients with polycythemia vera to achieve European LeukemiaNet criteria for complete hematologic remission did not result in better survival or less thrombosis compared with the expected survival of patients with similar disease characteristics.
Second, the primary endpoint of RESPONSE-2 was phlebotomy control but the appropriate control group (a phlebotomy-only group) was not included, nor was phlebotomy control or hematologic remission achieved with ruxolitinib in all patients. Ruxolitinib is an expensive drug, but phlebotomy is an inexpensive and immediately effective procedure, and it is unlikely that insurers would support the use of ruxolitinib in polycythemia vera for hematocrit control without proof of greater efficacy than phlebotomy therapy.
Third, no study of ruxolitinib in polycythemia vera has capitalized on the observation, based on both clinical and gene-expression data, that patients with polycythemia vera are not all alike; male and female patients differ clinically and in gene expression, and the disease is indolent in some patients and aggressive in others, who also differ in gene expression. Thus, it should not be presumed that all patients with polycythemia vera will require ruxolitinib therapy or that all patients who do receive this treatment will respond similarly. Finally, polycythemia vera is an hematopoietic stem-cell disorder and, so far, ruxolitinib does not seem to affect hematopoietic stem cell behavior. At present, pegylated interferon is the only drug that targets hematopoietic stem cells and produces hematologic and molecular remission, although not in all patients and, like ruxolitinib, we still do not know how best to use it. Thus, following the data recorded in the RESPONSE trials we now have access to two non-myelotoxic therapies (ruxolitinib and pegylated interferon) to treat a disease whose natural history is measured in decades but whose patients do not all have the same genetic background or require the same level of myelosuppression – a setting most appropriate for precision medicine.
Jerry L. Spivak, MD, is with Johns Hopkins University in Baltimore. His remarks were excerpted from an accompanying editorial.
Ruxolitinib has now been assessed in two clinical trials in polycythemia vera without myelofibrosis. Francesco Passamonti and colleagues report the results of RESPONSE-2, a randomized, open-label, phase IIIb trial of ruxolitinib in patients with polycythemia vera without splenomegaly, who were intolerant of or unresponsive to hydroxyurea, versus the best available therapy (usually hydroxyurea), making this trial – like the previous RESPONSE trial, in which the effect of ruxolitinib was examined in hydroxyurea-intolerant or unresponsive patients with polycythemia vera with splenomegaly – a referendum on hydroxyurea. On the basis of their age, most RESPONSE-2 patients were defined as so-called high-risk patients, and were phlebotomy dependent.
Unsurprisingly, for the primary endpoint of patients achieving hematocrit control, ruxolitinib was superior to best available therapy (46 [62%] of 74 patients in the ruxolitinib group vs. 14 [19%] of 75 in the best available therapy group; odds ratio, 7.28 [95% CI 3.43–15.45]; P less than .0001), and also for the key secondary endpoint for patients achieving complete hematologic remission (23% vs .5%).
Symptom control, including pruritus, was superior in the ruxolitinib group, and adverse events were more common in the best available therapy group. The authors concluded that ruxolitinib “could be considered a standard of care for second-line therapy in this post-hydroxyurea patient population”
However, I challenge this conclusion. First, the consensus that hydroxyurea is first-line therapy for polycythemia vera is not evidence based. Indeed, a trial using hydroxyurea in patients with polycythemia vera to achieve European LeukemiaNet criteria for complete hematologic remission did not result in better survival or less thrombosis compared with the expected survival of patients with similar disease characteristics.
Second, the primary endpoint of RESPONSE-2 was phlebotomy control but the appropriate control group (a phlebotomy-only group) was not included, nor was phlebotomy control or hematologic remission achieved with ruxolitinib in all patients. Ruxolitinib is an expensive drug, but phlebotomy is an inexpensive and immediately effective procedure, and it is unlikely that insurers would support the use of ruxolitinib in polycythemia vera for hematocrit control without proof of greater efficacy than phlebotomy therapy.
Third, no study of ruxolitinib in polycythemia vera has capitalized on the observation, based on both clinical and gene-expression data, that patients with polycythemia vera are not all alike; male and female patients differ clinically and in gene expression, and the disease is indolent in some patients and aggressive in others, who also differ in gene expression. Thus, it should not be presumed that all patients with polycythemia vera will require ruxolitinib therapy or that all patients who do receive this treatment will respond similarly. Finally, polycythemia vera is an hematopoietic stem-cell disorder and, so far, ruxolitinib does not seem to affect hematopoietic stem cell behavior. At present, pegylated interferon is the only drug that targets hematopoietic stem cells and produces hematologic and molecular remission, although not in all patients and, like ruxolitinib, we still do not know how best to use it. Thus, following the data recorded in the RESPONSE trials we now have access to two non-myelotoxic therapies (ruxolitinib and pegylated interferon) to treat a disease whose natural history is measured in decades but whose patients do not all have the same genetic background or require the same level of myelosuppression – a setting most appropriate for precision medicine.
Jerry L. Spivak, MD, is with Johns Hopkins University in Baltimore. His remarks were excerpted from an accompanying editorial.
Ruxolitinib has now been assessed in two clinical trials in polycythemia vera without myelofibrosis. Francesco Passamonti and colleagues report the results of RESPONSE-2, a randomized, open-label, phase IIIb trial of ruxolitinib in patients with polycythemia vera without splenomegaly, who were intolerant of or unresponsive to hydroxyurea, versus the best available therapy (usually hydroxyurea), making this trial – like the previous RESPONSE trial, in which the effect of ruxolitinib was examined in hydroxyurea-intolerant or unresponsive patients with polycythemia vera with splenomegaly – a referendum on hydroxyurea. On the basis of their age, most RESPONSE-2 patients were defined as so-called high-risk patients, and were phlebotomy dependent.
Unsurprisingly, for the primary endpoint of patients achieving hematocrit control, ruxolitinib was superior to best available therapy (46 [62%] of 74 patients in the ruxolitinib group vs. 14 [19%] of 75 in the best available therapy group; odds ratio, 7.28 [95% CI 3.43–15.45]; P less than .0001), and also for the key secondary endpoint for patients achieving complete hematologic remission (23% vs .5%).
Symptom control, including pruritus, was superior in the ruxolitinib group, and adverse events were more common in the best available therapy group. The authors concluded that ruxolitinib “could be considered a standard of care for second-line therapy in this post-hydroxyurea patient population”
However, I challenge this conclusion. First, the consensus that hydroxyurea is first-line therapy for polycythemia vera is not evidence based. Indeed, a trial using hydroxyurea in patients with polycythemia vera to achieve European LeukemiaNet criteria for complete hematologic remission did not result in better survival or less thrombosis compared with the expected survival of patients with similar disease characteristics.
Second, the primary endpoint of RESPONSE-2 was phlebotomy control but the appropriate control group (a phlebotomy-only group) was not included, nor was phlebotomy control or hematologic remission achieved with ruxolitinib in all patients. Ruxolitinib is an expensive drug, but phlebotomy is an inexpensive and immediately effective procedure, and it is unlikely that insurers would support the use of ruxolitinib in polycythemia vera for hematocrit control without proof of greater efficacy than phlebotomy therapy.
Third, no study of ruxolitinib in polycythemia vera has capitalized on the observation, based on both clinical and gene-expression data, that patients with polycythemia vera are not all alike; male and female patients differ clinically and in gene expression, and the disease is indolent in some patients and aggressive in others, who also differ in gene expression. Thus, it should not be presumed that all patients with polycythemia vera will require ruxolitinib therapy or that all patients who do receive this treatment will respond similarly. Finally, polycythemia vera is an hematopoietic stem-cell disorder and, so far, ruxolitinib does not seem to affect hematopoietic stem cell behavior. At present, pegylated interferon is the only drug that targets hematopoietic stem cells and produces hematologic and molecular remission, although not in all patients and, like ruxolitinib, we still do not know how best to use it. Thus, following the data recorded in the RESPONSE trials we now have access to two non-myelotoxic therapies (ruxolitinib and pegylated interferon) to treat a disease whose natural history is measured in decades but whose patients do not all have the same genetic background or require the same level of myelosuppression – a setting most appropriate for precision medicine.
Jerry L. Spivak, MD, is with Johns Hopkins University in Baltimore. His remarks were excerpted from an accompanying editorial.
For patients with polycythemia vera without splenomegaly who had inadequate responses to hydroxyurea, targeted therapy with the Janus kinase (JAK) inhibitor ruxolitinib (Jakafi) offered better control of hematocrit and better improvement of symptoms than did the best available therapy, according to results of a multinational randomized phase IIIb trial.
Among 74 patients randomly assigned to receive ruxolitinib, 46 (62%) achieved hematocrit control, compared with 14 of 75 patients (19%) assigned to receive one of several different options lumped into the best available therapy category, noted investigators led by Francesco Passamonti, MD, of the University of Insubria in Varese, Italy.
“Ruxolitinib also led to an improved symptom burden and quality of life. Patients treated with ruxolitinib experienced improvements in all polycythemia vera–associated symptoms, including pruritus, whereas patients treated with best available therapy experienced worsening of most symptoms,” they wrote.
Unlike cytoreductive therapies such as hydroxyurea or pegylated interferon, ruxolitinib works by inhibition of JAK1 and JAK2 signaling. A majority of patients with polycythemia vera have an activating JAK2 mutation that leads to overactivation of the JAK-STAT signaling pathway, resulting in erythrocytosis, the hallmark symptom of polycythemia vera, and associated vascular complications.
“In some patients, conventional therapies can lose effectiveness over time. Although hydroxyurea is well tolerated in most patients, about 15%-20% of patients become resistant or intolerant, with hydroxyurea resistance affecting survival and increasing the risk of progression to myelofibrosis. Additionally, patients who are intolerant of hydroxyurea can have adverse side effects, such as drug-induced fever, mouth ulcers, leg ulcers, and skin malignancies, which necessitate discontinuation of first-line therapy,” the investigators noted.
RESPONSE trials
Ruxolitinib had previously been shown in the RESPONSE study to be superior to the best available therapy for controlling hematocrit and for improving splenomegaly and other symptoms in patients with polycythemia vera and disease-associated splenomegaly who had an inadequate response or unacceptable toxicities from treatment with hydroxyurea.
In the currently reported study, dubbed RESPONSE-2, patients 18 and older with polycythemia vera with no palpable splenomegaly who were intolerant of hydroxyurea or had disease that was resistant to it were randomized to receive either oral ruxolitinib 10 mg twice daily, or best available therapy at the investigators’ discretion. Best available therapy consisted of either hydroxyurea at the maximum tolerated dose, interferon or pegylated interferon, pipobroman, anagrelide, approved immunomodulators, or no cytoreductive treatment.
As noted, hematocrit control at week 28, the primary endpoint, was significantly higher among patients on ruxolitinib, with an odds ratio (OR) of 7.28 (P less than .0001).
Hematocrit levels among patients on ruxolitinib group decreased from a mean of 42.8% at baseline to 40.2% at week 28. In contrast, hematocrit in the best available therapy group increased from a mean of 42.7% to 44.9% at week 28.
Fewer patients on ruxolitinib required phlebotomy procedures during the 28 weeks of the study compared with patients on best available therapy, and of those patients who did undergo phlebotomy, fewer of those in the ruxolitinib group underwent more than two procedures. There were a total of 98 phlebotomies among best available care patients, vs. 19 among ruxolitinib patients.
Complete hematologic remissions, a secondary endpoint, occurred in 23% of patients on ruxolitinib, compared with 5% of those on best available care (OR 5.58, P = .0019).
The most frequent hematologic adverse events of any grade were anemia, which occurred in 10 patients on ruxolitinib (none grade 3 or greater) vs. two on best available therapy (one grade 3), and thrombocytopenia occurring in two (both grade 1 or 2) and six patients (three grade 1 or 2, two grade 3, and one grade 4) respectively.
Grade 3 or 4 nonhematologic adverse events included hypertension in five patients on ruxolitinib vs. three on best available care, and pruritus in none vs. two, respectively.
Two patients died; both were in the best available therapy group.
“Although the short follow-up of this study precludes any conclusions about vascular complications, an important finding is that patients treated with ruxolitinib in both RESPONSE and RESPONSE-2 had fewer thromboembolic events compared with those given best available therapy; there were two thromboembolic events with ruxolitinib (one in each study) versus nine with best available therapy across both studies (six in RESPONSE and three in RESPONSE-2). This finding could have been attributable to better control of hematocrit or white blood cell count with ruxolitinib, given that baseline risk factors were similar in both treatment groups,” the investigators wrote.
The findings from the two studies support the use of ruxolitinib as a standard of care for second-line therapy of patients with polycythemia vera following treatment with hydroxyurea, they contended.
Several coauthors disclosed ties to Novartis, which supported the study. Two are Novartis employees; one was previously employed by the company.
For patients with polycythemia vera without splenomegaly who had inadequate responses to hydroxyurea, targeted therapy with the Janus kinase (JAK) inhibitor ruxolitinib (Jakafi) offered better control of hematocrit and better improvement of symptoms than did the best available therapy, according to results of a multinational randomized phase IIIb trial.
Among 74 patients randomly assigned to receive ruxolitinib, 46 (62%) achieved hematocrit control, compared with 14 of 75 patients (19%) assigned to receive one of several different options lumped into the best available therapy category, noted investigators led by Francesco Passamonti, MD, of the University of Insubria in Varese, Italy.
“Ruxolitinib also led to an improved symptom burden and quality of life. Patients treated with ruxolitinib experienced improvements in all polycythemia vera–associated symptoms, including pruritus, whereas patients treated with best available therapy experienced worsening of most symptoms,” they wrote.
Unlike cytoreductive therapies such as hydroxyurea or pegylated interferon, ruxolitinib works by inhibition of JAK1 and JAK2 signaling. A majority of patients with polycythemia vera have an activating JAK2 mutation that leads to overactivation of the JAK-STAT signaling pathway, resulting in erythrocytosis, the hallmark symptom of polycythemia vera, and associated vascular complications.
“In some patients, conventional therapies can lose effectiveness over time. Although hydroxyurea is well tolerated in most patients, about 15%-20% of patients become resistant or intolerant, with hydroxyurea resistance affecting survival and increasing the risk of progression to myelofibrosis. Additionally, patients who are intolerant of hydroxyurea can have adverse side effects, such as drug-induced fever, mouth ulcers, leg ulcers, and skin malignancies, which necessitate discontinuation of first-line therapy,” the investigators noted.
RESPONSE trials
Ruxolitinib had previously been shown in the RESPONSE study to be superior to the best available therapy for controlling hematocrit and for improving splenomegaly and other symptoms in patients with polycythemia vera and disease-associated splenomegaly who had an inadequate response or unacceptable toxicities from treatment with hydroxyurea.
In the currently reported study, dubbed RESPONSE-2, patients 18 and older with polycythemia vera with no palpable splenomegaly who were intolerant of hydroxyurea or had disease that was resistant to it were randomized to receive either oral ruxolitinib 10 mg twice daily, or best available therapy at the investigators’ discretion. Best available therapy consisted of either hydroxyurea at the maximum tolerated dose, interferon or pegylated interferon, pipobroman, anagrelide, approved immunomodulators, or no cytoreductive treatment.
As noted, hematocrit control at week 28, the primary endpoint, was significantly higher among patients on ruxolitinib, with an odds ratio (OR) of 7.28 (P less than .0001).
Hematocrit levels among patients on ruxolitinib group decreased from a mean of 42.8% at baseline to 40.2% at week 28. In contrast, hematocrit in the best available therapy group increased from a mean of 42.7% to 44.9% at week 28.
Fewer patients on ruxolitinib required phlebotomy procedures during the 28 weeks of the study compared with patients on best available therapy, and of those patients who did undergo phlebotomy, fewer of those in the ruxolitinib group underwent more than two procedures. There were a total of 98 phlebotomies among best available care patients, vs. 19 among ruxolitinib patients.
Complete hematologic remissions, a secondary endpoint, occurred in 23% of patients on ruxolitinib, compared with 5% of those on best available care (OR 5.58, P = .0019).
The most frequent hematologic adverse events of any grade were anemia, which occurred in 10 patients on ruxolitinib (none grade 3 or greater) vs. two on best available therapy (one grade 3), and thrombocytopenia occurring in two (both grade 1 or 2) and six patients (three grade 1 or 2, two grade 3, and one grade 4) respectively.
Grade 3 or 4 nonhematologic adverse events included hypertension in five patients on ruxolitinib vs. three on best available care, and pruritus in none vs. two, respectively.
Two patients died; both were in the best available therapy group.
“Although the short follow-up of this study precludes any conclusions about vascular complications, an important finding is that patients treated with ruxolitinib in both RESPONSE and RESPONSE-2 had fewer thromboembolic events compared with those given best available therapy; there were two thromboembolic events with ruxolitinib (one in each study) versus nine with best available therapy across both studies (six in RESPONSE and three in RESPONSE-2). This finding could have been attributable to better control of hematocrit or white blood cell count with ruxolitinib, given that baseline risk factors were similar in both treatment groups,” the investigators wrote.
The findings from the two studies support the use of ruxolitinib as a standard of care for second-line therapy of patients with polycythemia vera following treatment with hydroxyurea, they contended.
Several coauthors disclosed ties to Novartis, which supported the study. Two are Novartis employees; one was previously employed by the company.
FROM LANCET ONCOLOGY
Key clinical point: Ruxolitinib was superior to best available care for hematocrit control among patients with polycythemia vera without splenomegaly.
Major finding: In the ruxolitinib group, 62% had control of hematocrit at week 28, vs. 19% on best available care.
Data source: Randomized trial of 149 adults with polycythemia vera in the absence of palpable splenomegaly.
Disclosures: Several coauthors disclosed ties to Novartis, which supported the study. Two are Novartis employees; one was previously employed by the company.
Improving the efficacy of obinutuzumab

Preclinical research suggests that immune stimulation through Toll-like receptor 7 (TLR7) agonism can enhance the efficacy of obinutuzumab in lymphoma.
Researchers found that combining the anti-CD20 monoclonal antibody obinutuzumab with the TLR7 agonist R848 improved survival in lab mice with lymphoma.
The combination also demonstrated efficacy against chronic lymphocytic leukemia (CLL) cells in vitro.
Tim Illidge, PhD, MBBS, of the University of Manchester in the UK, and his colleagues reported these findings in the journal Leukemia.
The research was funded by the Kay Kendall Leukaemia Fund and Cancer Research UK in collaboration with Roche Pharmaceutical Research and Early Development.
The researchers said they initially found that R848 activates immune cells in vivo and enhances obinutuzumab-mediated antitumor effector mechanisms in vitro.
The team therefore went on to test R848 and obinutuzumab in C57Bl/6 mice bearing human CD20+ lymphoma (EL4). The mice received obinutuzumab modified to express the murine glycoengineered IgG2a Fc region (m2a) starting 1 day after tumor inoculation and systemic R848 once weekly for 4 weeks.
The researchers found that monotherapy with either obinutuzumab or R848 significantly improved survival compared to control (P<0.0001), but only 8% to 15% of mice that received monotherapy were long-term survivors (living more than 90 days).
Mice that received obinutuzumab in combination with R848 had significantly better survival than mice that received either monotherapy (P<0.0001). And about 70% of mice receiving the combination remained tumor-free out to 95 days.
Furthermore, long-term survivors that had received the combination treatment were protected from tumor re-challenge.
The researchers also tested the combination in a second model—human CD20 transgenic mice, which express the human CD20 antigen on normal B cells. The team said this model is more akin to the clinical situation.
The mice received treatment 7 days after the inoculation of EL4hCD20 cells. Mice that received obinutuzumab monotherapy had significantly better survival than control mice (P=0.02), but there were no long-term survivors. For mice that received R848 monotherapy, survival was not significantly different from that of controls.
Mice that received R848 in combination with obinutuzumab had significantly better survival than mice that received obinutuzumab alone (P=0.003).
In fact, 6 of the 12 mice that received the combination were long-term survivors. And 5 of these mice rejected tumor re-challenge.
“We were excited when we discovered that combining obinutuzumab with TLR7 activation significantly enhanced survival of animals with lymphoma by effectively eradicating tumors,” Dr Illidge said. “Clearly, more work needs to be done to assess the impact of this combination on humans, but this study is, nevertheless, very promising.”
The researchers said the primary antitumor activity of the combination is dependent on natural killer cells and CD4 helper T cells but not on CD8 killer T cells.
“While the combination therapy was highly effective, CD8 killer T cells did not play a major role in the therapy,” said Eleanor Cheadle, PhD, also of the University of Manchester.
“Given the important role that killer T cells can play in long-term protection from tumor regrowth, we are looking at ways to enhance activation of these cells after obinutuzumab therapy.”
The researchers also found that, in vitro, R848 significantly enhanced natural killer cell-mediated antibody-dependent cellular cytotoxicity against obinutuzumab-opsonized CLL cells and significantly increased non-specific, antibody-independent killing of CLL cells. ![]()
Preclinical research suggests that immune stimulation through Toll-like receptor 7 (TLR7) agonism can enhance the efficacy of obinutuzumab in lymphoma.
Researchers found that combining the anti-CD20 monoclonal antibody obinutuzumab with the TLR7 agonist R848 improved survival in lab mice with lymphoma.
The combination also demonstrated efficacy against chronic lymphocytic leukemia (CLL) cells in vitro.
Tim Illidge, PhD, MBBS, of the University of Manchester in the UK, and his colleagues reported these findings in the journal Leukemia.
The research was funded by the Kay Kendall Leukaemia Fund and Cancer Research UK in collaboration with Roche Pharmaceutical Research and Early Development.
The researchers said they initially found that R848 activates immune cells in vivo and enhances obinutuzumab-mediated antitumor effector mechanisms in vitro.
The team therefore went on to test R848 and obinutuzumab in C57Bl/6 mice bearing human CD20+ lymphoma (EL4). The mice received obinutuzumab modified to express the murine glycoengineered IgG2a Fc region (m2a) starting 1 day after tumor inoculation and systemic R848 once weekly for 4 weeks.
The researchers found that monotherapy with either obinutuzumab or R848 significantly improved survival compared to control (P<0.0001), but only 8% to 15% of mice that received monotherapy were long-term survivors (living more than 90 days).
Mice that received obinutuzumab in combination with R848 had significantly better survival than mice that received either monotherapy (P<0.0001). And about 70% of mice receiving the combination remained tumor-free out to 95 days.
Furthermore, long-term survivors that had received the combination treatment were protected from tumor re-challenge.
The researchers also tested the combination in a second model—human CD20 transgenic mice, which express the human CD20 antigen on normal B cells. The team said this model is more akin to the clinical situation.
The mice received treatment 7 days after the inoculation of EL4hCD20 cells. Mice that received obinutuzumab monotherapy had significantly better survival than control mice (P=0.02), but there were no long-term survivors. For mice that received R848 monotherapy, survival was not significantly different from that of controls.
Mice that received R848 in combination with obinutuzumab had significantly better survival than mice that received obinutuzumab alone (P=0.003).
In fact, 6 of the 12 mice that received the combination were long-term survivors. And 5 of these mice rejected tumor re-challenge.
“We were excited when we discovered that combining obinutuzumab with TLR7 activation significantly enhanced survival of animals with lymphoma by effectively eradicating tumors,” Dr Illidge said. “Clearly, more work needs to be done to assess the impact of this combination on humans, but this study is, nevertheless, very promising.”
The researchers said the primary antitumor activity of the combination is dependent on natural killer cells and CD4 helper T cells but not on CD8 killer T cells.
“While the combination therapy was highly effective, CD8 killer T cells did not play a major role in the therapy,” said Eleanor Cheadle, PhD, also of the University of Manchester.
“Given the important role that killer T cells can play in long-term protection from tumor regrowth, we are looking at ways to enhance activation of these cells after obinutuzumab therapy.”
The researchers also found that, in vitro, R848 significantly enhanced natural killer cell-mediated antibody-dependent cellular cytotoxicity against obinutuzumab-opsonized CLL cells and significantly increased non-specific, antibody-independent killing of CLL cells. ![]()
Preclinical research suggests that immune stimulation through Toll-like receptor 7 (TLR7) agonism can enhance the efficacy of obinutuzumab in lymphoma.
Researchers found that combining the anti-CD20 monoclonal antibody obinutuzumab with the TLR7 agonist R848 improved survival in lab mice with lymphoma.
The combination also demonstrated efficacy against chronic lymphocytic leukemia (CLL) cells in vitro.
Tim Illidge, PhD, MBBS, of the University of Manchester in the UK, and his colleagues reported these findings in the journal Leukemia.
The research was funded by the Kay Kendall Leukaemia Fund and Cancer Research UK in collaboration with Roche Pharmaceutical Research and Early Development.
The researchers said they initially found that R848 activates immune cells in vivo and enhances obinutuzumab-mediated antitumor effector mechanisms in vitro.
The team therefore went on to test R848 and obinutuzumab in C57Bl/6 mice bearing human CD20+ lymphoma (EL4). The mice received obinutuzumab modified to express the murine glycoengineered IgG2a Fc region (m2a) starting 1 day after tumor inoculation and systemic R848 once weekly for 4 weeks.
The researchers found that monotherapy with either obinutuzumab or R848 significantly improved survival compared to control (P<0.0001), but only 8% to 15% of mice that received monotherapy were long-term survivors (living more than 90 days).
Mice that received obinutuzumab in combination with R848 had significantly better survival than mice that received either monotherapy (P<0.0001). And about 70% of mice receiving the combination remained tumor-free out to 95 days.
Furthermore, long-term survivors that had received the combination treatment were protected from tumor re-challenge.
The researchers also tested the combination in a second model—human CD20 transgenic mice, which express the human CD20 antigen on normal B cells. The team said this model is more akin to the clinical situation.
The mice received treatment 7 days after the inoculation of EL4hCD20 cells. Mice that received obinutuzumab monotherapy had significantly better survival than control mice (P=0.02), but there were no long-term survivors. For mice that received R848 monotherapy, survival was not significantly different from that of controls.
Mice that received R848 in combination with obinutuzumab had significantly better survival than mice that received obinutuzumab alone (P=0.003).
In fact, 6 of the 12 mice that received the combination were long-term survivors. And 5 of these mice rejected tumor re-challenge.
“We were excited when we discovered that combining obinutuzumab with TLR7 activation significantly enhanced survival of animals with lymphoma by effectively eradicating tumors,” Dr Illidge said. “Clearly, more work needs to be done to assess the impact of this combination on humans, but this study is, nevertheless, very promising.”
The researchers said the primary antitumor activity of the combination is dependent on natural killer cells and CD4 helper T cells but not on CD8 killer T cells.
“While the combination therapy was highly effective, CD8 killer T cells did not play a major role in the therapy,” said Eleanor Cheadle, PhD, also of the University of Manchester.
“Given the important role that killer T cells can play in long-term protection from tumor regrowth, we are looking at ways to enhance activation of these cells after obinutuzumab therapy.”
The researchers also found that, in vitro, R848 significantly enhanced natural killer cell-mediated antibody-dependent cellular cytotoxicity against obinutuzumab-opsonized CLL cells and significantly increased non-specific, antibody-independent killing of CLL cells. ![]()
New recommendations for pediatric AMKL
Image courtesy of St. Jude
Children’s Research Hospital
and Tina Motroni
Research has revealed genetic alterations that may prove useful for predicting treatment outcomes in pediatric patients with acute megakaryoblastic leukemia (AMKL) who do not have Down syndrome.
The study suggests that 3
genetic alterations can be used to identify high-risk patients who may benefit
from allogeneic stem cell transplant, and 1 alteration may identify
low-risk patients who require less chemotherapy than their peers.
Researchers said these findings, published in Nature Genetics, support revised diagnostic screening and treatment recommendations for pediatric AMKL.
“Because long-term survival for pediatric AMKL patients without Down syndrome is poor, just 14% to 34%, the standard recommendation by many pediatric oncologists has been to treat all patients with allogeneic stem cell transplantation during their first remission,” said study author Tanja Gruber, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“In this study, we identified several genetic alterations that are important predictors of treatment success. All newly identified pediatric AMKL patients without Down syndrome should be screened for these prognostic indicators at diagnosis. The results will help identify which patients need allogeneic stem cell transplants during their first remission and which do not.”
This study involved next-generation sequencing of the whole exome or RNA of 99 AMKL patients without Down syndrome (75 pediatric and 24 adult patients). Along with the sequencing data, researchers analyzed patients’ gene expression and long-term survival.
Results
The results showed that non-Down syndrome pediatric AMKL can be divided into 7 subgroups based on the underlying genetic alteration, pattern of gene expression, and treatment outcome.
The subgroups include the newly identified HOX subgroup. About 15% of the pediatric patients in this study were in the HOX subgroup, which is characterized by several different HOX fusion genes.
The researchers also identified cooperating mutations that help fuel AMKL in different subgroups. The cooperating mutations include changes in the RB1 gene and recurring mutations in the RAS and JAK pathways.
In addition, the study showed that 3 genetic alterations—CBFA2T3-GLIS2, KMT2A rearrangements, and NUP98-KDM5A—are associated with reduced survival in pediatric AMKL subtypes.
The researchers said patients with these alterations may benefit from allogeneic stem cell transplants, so non-Down syndrome pediatric AMKL patients should be screened for these alterations at diagnosis.
The team also recommended testing AMKL patients for mutations in the GATA1 gene. GATA1 mutations are a hallmark of AMKL in children with Down syndrome, who almost always survive the leukemia. In this study, AMKL patients with GATA1 mutations and no fusion gene had the same favorable outcomes.
“The results raise the possibility that pediatric AMKL patients without Down syndrome who have mutations in GATA1 may benefit from the same reduced chemotherapy used to treat the leukemia in patients with Down syndrome,” Dr Gruber said.
These revised diagnostic screening and treatment recommendations are being implemented at St. Jude.
This study also showed that adults with AMKL lacked recurrent fusion genes. The most common mutations found in the adult patients were in TP53 (21%), cohesin genes (17%), splicing factor genes (17%), ASXL genes (17%), and DNMT3A (13%). About 4% had GATA1 mutations. ![]()
Image courtesy of St. Jude
Children’s Research Hospital
and Tina Motroni
Research has revealed genetic alterations that may prove useful for predicting treatment outcomes in pediatric patients with acute megakaryoblastic leukemia (AMKL) who do not have Down syndrome.
The study suggests that 3
genetic alterations can be used to identify high-risk patients who may benefit
from allogeneic stem cell transplant, and 1 alteration may identify
low-risk patients who require less chemotherapy than their peers.
Researchers said these findings, published in Nature Genetics, support revised diagnostic screening and treatment recommendations for pediatric AMKL.
“Because long-term survival for pediatric AMKL patients without Down syndrome is poor, just 14% to 34%, the standard recommendation by many pediatric oncologists has been to treat all patients with allogeneic stem cell transplantation during their first remission,” said study author Tanja Gruber, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“In this study, we identified several genetic alterations that are important predictors of treatment success. All newly identified pediatric AMKL patients without Down syndrome should be screened for these prognostic indicators at diagnosis. The results will help identify which patients need allogeneic stem cell transplants during their first remission and which do not.”
This study involved next-generation sequencing of the whole exome or RNA of 99 AMKL patients without Down syndrome (75 pediatric and 24 adult patients). Along with the sequencing data, researchers analyzed patients’ gene expression and long-term survival.
Results
The results showed that non-Down syndrome pediatric AMKL can be divided into 7 subgroups based on the underlying genetic alteration, pattern of gene expression, and treatment outcome.
The subgroups include the newly identified HOX subgroup. About 15% of the pediatric patients in this study were in the HOX subgroup, which is characterized by several different HOX fusion genes.
The researchers also identified cooperating mutations that help fuel AMKL in different subgroups. The cooperating mutations include changes in the RB1 gene and recurring mutations in the RAS and JAK pathways.
In addition, the study showed that 3 genetic alterations—CBFA2T3-GLIS2, KMT2A rearrangements, and NUP98-KDM5A—are associated with reduced survival in pediatric AMKL subtypes.
The researchers said patients with these alterations may benefit from allogeneic stem cell transplants, so non-Down syndrome pediatric AMKL patients should be screened for these alterations at diagnosis.
The team also recommended testing AMKL patients for mutations in the GATA1 gene. GATA1 mutations are a hallmark of AMKL in children with Down syndrome, who almost always survive the leukemia. In this study, AMKL patients with GATA1 mutations and no fusion gene had the same favorable outcomes.
“The results raise the possibility that pediatric AMKL patients without Down syndrome who have mutations in GATA1 may benefit from the same reduced chemotherapy used to treat the leukemia in patients with Down syndrome,” Dr Gruber said.
These revised diagnostic screening and treatment recommendations are being implemented at St. Jude.
This study also showed that adults with AMKL lacked recurrent fusion genes. The most common mutations found in the adult patients were in TP53 (21%), cohesin genes (17%), splicing factor genes (17%), ASXL genes (17%), and DNMT3A (13%). About 4% had GATA1 mutations. ![]()
Image courtesy of St. Jude
Children’s Research Hospital
and Tina Motroni
Research has revealed genetic alterations that may prove useful for predicting treatment outcomes in pediatric patients with acute megakaryoblastic leukemia (AMKL) who do not have Down syndrome.
The study suggests that 3
genetic alterations can be used to identify high-risk patients who may benefit
from allogeneic stem cell transplant, and 1 alteration may identify
low-risk patients who require less chemotherapy than their peers.
Researchers said these findings, published in Nature Genetics, support revised diagnostic screening and treatment recommendations for pediatric AMKL.
“Because long-term survival for pediatric AMKL patients without Down syndrome is poor, just 14% to 34%, the standard recommendation by many pediatric oncologists has been to treat all patients with allogeneic stem cell transplantation during their first remission,” said study author Tanja Gruber, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“In this study, we identified several genetic alterations that are important predictors of treatment success. All newly identified pediatric AMKL patients without Down syndrome should be screened for these prognostic indicators at diagnosis. The results will help identify which patients need allogeneic stem cell transplants during their first remission and which do not.”
This study involved next-generation sequencing of the whole exome or RNA of 99 AMKL patients without Down syndrome (75 pediatric and 24 adult patients). Along with the sequencing data, researchers analyzed patients’ gene expression and long-term survival.
Results
The results showed that non-Down syndrome pediatric AMKL can be divided into 7 subgroups based on the underlying genetic alteration, pattern of gene expression, and treatment outcome.
The subgroups include the newly identified HOX subgroup. About 15% of the pediatric patients in this study were in the HOX subgroup, which is characterized by several different HOX fusion genes.
The researchers also identified cooperating mutations that help fuel AMKL in different subgroups. The cooperating mutations include changes in the RB1 gene and recurring mutations in the RAS and JAK pathways.
In addition, the study showed that 3 genetic alterations—CBFA2T3-GLIS2, KMT2A rearrangements, and NUP98-KDM5A—are associated with reduced survival in pediatric AMKL subtypes.
The researchers said patients with these alterations may benefit from allogeneic stem cell transplants, so non-Down syndrome pediatric AMKL patients should be screened for these alterations at diagnosis.
The team also recommended testing AMKL patients for mutations in the GATA1 gene. GATA1 mutations are a hallmark of AMKL in children with Down syndrome, who almost always survive the leukemia. In this study, AMKL patients with GATA1 mutations and no fusion gene had the same favorable outcomes.
“The results raise the possibility that pediatric AMKL patients without Down syndrome who have mutations in GATA1 may benefit from the same reduced chemotherapy used to treat the leukemia in patients with Down syndrome,” Dr Gruber said.
These revised diagnostic screening and treatment recommendations are being implemented at St. Jude.
This study also showed that adults with AMKL lacked recurrent fusion genes. The most common mutations found in the adult patients were in TP53 (21%), cohesin genes (17%), splicing factor genes (17%), ASXL genes (17%), and DNMT3A (13%). About 4% had GATA1 mutations. ![]()
What Do Parasites Have to Do With Leukemia?
Parasites have been shown to have both pro- and antitumor effects. Malaria parasites (Plasmodium spp) are among those known to have this possible “bidirectional role” in carcinogenesis, say researchers from Aix-Marseille Université in France. They reviewed the current thinking on whether malaria—a worldwide killer—can be useful in cancer prevention and treatment.
Positive relationships between malaria and virus-associated cancers are relatively well documented, the researchers say. Evidence suggests that malaria can alter immune responses by modulating both humoral and cell-mediated immunity. Plasmodium-related cancers are primarily lymphoproliferative, vulnerable to virus reactivation. Epstein-Barr virus (EBV), for example, has been observed in lymphatic and hematologic tumors such as Hodgkin disease and T cell lymphoma, and malaria can reactivate EBV.
In animal studies, malarial infection with Plasmodium berghei (P berghei) increased the rate of spontaneous leukemia. In one study, concurrent infection with P berghei increased the incidence of malignant lymphoma in mice injected with Moloney leukemogenic virus.
On the other hand, Plasmodium spp also produces proteins that demonstrate certain anti-oncogenic effects, they note. The researchers suggest that using proteins in cancer treatment should be explored, adding that it’s a “safer approach than the inoculation of wild type Plasmodium.” Positive parasite-induced effects against cancers of the hematopoietic and lymphoid tissues are mentioned only for 2 species and those only in a decades-old study. Based on current knowledge, the researchers say, the antitumor effects observed are attributable to modifications to the host immune response. Thus, their characteristics and locations within the host can be highly diverse.
All in all, the researchers conclude, the growing evidence is opening intriguing pathways for using one ill to cure another.
Source:
Faure E. Parasitology. 2016;143(14):1811-1823.
Parasites have been shown to have both pro- and antitumor effects. Malaria parasites (Plasmodium spp) are among those known to have this possible “bidirectional role” in carcinogenesis, say researchers from Aix-Marseille Université in France. They reviewed the current thinking on whether malaria—a worldwide killer—can be useful in cancer prevention and treatment.
Positive relationships between malaria and virus-associated cancers are relatively well documented, the researchers say. Evidence suggests that malaria can alter immune responses by modulating both humoral and cell-mediated immunity. Plasmodium-related cancers are primarily lymphoproliferative, vulnerable to virus reactivation. Epstein-Barr virus (EBV), for example, has been observed in lymphatic and hematologic tumors such as Hodgkin disease and T cell lymphoma, and malaria can reactivate EBV.
In animal studies, malarial infection with Plasmodium berghei (P berghei) increased the rate of spontaneous leukemia. In one study, concurrent infection with P berghei increased the incidence of malignant lymphoma in mice injected with Moloney leukemogenic virus.
On the other hand, Plasmodium spp also produces proteins that demonstrate certain anti-oncogenic effects, they note. The researchers suggest that using proteins in cancer treatment should be explored, adding that it’s a “safer approach than the inoculation of wild type Plasmodium.” Positive parasite-induced effects against cancers of the hematopoietic and lymphoid tissues are mentioned only for 2 species and those only in a decades-old study. Based on current knowledge, the researchers say, the antitumor effects observed are attributable to modifications to the host immune response. Thus, their characteristics and locations within the host can be highly diverse.
All in all, the researchers conclude, the growing evidence is opening intriguing pathways for using one ill to cure another.
Source:
Faure E. Parasitology. 2016;143(14):1811-1823.
Parasites have been shown to have both pro- and antitumor effects. Malaria parasites (Plasmodium spp) are among those known to have this possible “bidirectional role” in carcinogenesis, say researchers from Aix-Marseille Université in France. They reviewed the current thinking on whether malaria—a worldwide killer—can be useful in cancer prevention and treatment.
Positive relationships between malaria and virus-associated cancers are relatively well documented, the researchers say. Evidence suggests that malaria can alter immune responses by modulating both humoral and cell-mediated immunity. Plasmodium-related cancers are primarily lymphoproliferative, vulnerable to virus reactivation. Epstein-Barr virus (EBV), for example, has been observed in lymphatic and hematologic tumors such as Hodgkin disease and T cell lymphoma, and malaria can reactivate EBV.
In animal studies, malarial infection with Plasmodium berghei (P berghei) increased the rate of spontaneous leukemia. In one study, concurrent infection with P berghei increased the incidence of malignant lymphoma in mice injected with Moloney leukemogenic virus.
On the other hand, Plasmodium spp also produces proteins that demonstrate certain anti-oncogenic effects, they note. The researchers suggest that using proteins in cancer treatment should be explored, adding that it’s a “safer approach than the inoculation of wild type Plasmodium.” Positive parasite-induced effects against cancers of the hematopoietic and lymphoid tissues are mentioned only for 2 species and those only in a decades-old study. Based on current knowledge, the researchers say, the antitumor effects observed are attributable to modifications to the host immune response. Thus, their characteristics and locations within the host can be highly diverse.
All in all, the researchers conclude, the growing evidence is opening intriguing pathways for using one ill to cure another.
Source:
Faure E. Parasitology. 2016;143(14):1811-1823.
Clonal hematopoiesis increases risk for therapy-related cancers
Small pre-leukemic clones left behind after treatment for non-myeloid malignancies appear to increase the risk for therapy-related myelodysplasia or leukemia, report investigators in two studies.
An analysis of peripheral blood samples taken from patients at the time of their primary cancer diagnosis and bone marrow samples taken at the time of a later therapy-related myeloid neoplasm diagnosis showed that 10 of 14 patients (71%) had clonal hematopoiesis before starting on cytotoxic chemotherapy. In contrast, clonal hematopoiesis was detected in pre-treatment samples of only 17 of 54 controls (31%), reported Koichi Takahashi, MD, and colleagues from the University of Texas MD Anderson Cancer Center in Houston.
“Preleukemic clonal hematopoiesis is common in patients with therapy-related myeloid neoplasms at the time of their primary cancer diagnosis and before they have been exposed to treatment. Our results suggest that clonal hematopoiesis could be used as a predictive marker to identify patients with cancer who are at risk of developing therapy-related myeloid neoplasms,” they wrote (Lancet Oncol 2017; 18: 100–11).
In a separate study, investigators from the Moffitt Cancer Center in Tampa, Florida, found in a nested case-control study that patients with therapy-related myeloid neoplasms were more likely than controls to have clonal hematopoiesis of indeterminate potential (CHIP), and that the CHIP was often present before exposure to chemotherapy.
“We recorded a significantly higher prevalence of CHIP in individuals who developed therapy-related myeloid neoplasms (cases) than in those who did not (controls); however, around 27% of individuals with CHIP did not develop therapy-related myeloid neoplasms, suggesting that this feature alone should not be used to determine a patient’s suitability for chemotherapy,” wrote Nancy K. Gillis, PharmD, and colleagues (Lancet Oncol 2017; 18:112-21).
Risk factors examined
Dr. Takahashi and colleagues noted that previous studies have identified several treatment-related risk factors as being associated with therapy-related myeloid dysplasia or leukemia, including the use of alkylating agents, topoisomerase II inhibitors, and high-dose chemotherapy with autologous stem-cell transplantation.
“By contrast, little is known about patient-specific risk factors. Older age was shown to increase the risk of therapy-related myeloid neoplasms. Several germline polymorphisms have also been associated with this risk, but none have been validated. As such, no predictive biomarkers exist for therapy-related myeloid neoplasms,” they wrote.
They performed a retrospective case-control study comparing patients treated for a primary cancer at their center from 1997 through 2015 who subsequently developed a myeloid neoplasm with controls treated during the same period. Controls were age-matched patients treated with combination chemotherapy for lymphoma who did not develop a therapy-related myeloid malignancy after at least 5 years of follow-up.
In addition, the investigators further explored the association between clonal hematopoiesis and therapy-related cancers in an external cohort of patients with lymphoma treated in a randomized trial at their center from 1999 through 2001. That trial compared the CHOP regimen (cyclophosphamide, doxorubicin, vincristine and prednisone) with and without melatonin.
To detect clonal hematopoiesis in pre-treatment peripheral blood, the investigators used molecular barcode sequencing of 32 genes. They also used targeted gene sequencing on bone marrow samples from cases to investigate clonal evolution from clonal hematopoiesis to the development of therapy-related myeloid neoplasms.
As noted before, 10 of 14 cases had evidence of pre-treatment clonal hematopoiesis, compared with 17 of 54 controls. For both cases and controls, the cumulative incidence of therapy-related myeloid cancers after 5 years was significantly higher among those with baseline clonal hematopoiesis, at 30% vs. 7% for patients without it (P = .016).
Five of 74 patients in the external cohort (7%) went on to develop therapy-related myeloid neoplasms, and of this group, four (80%) had clonal hematopoiesis at baseline. In contrast, of the 69 patients who did not develop therapy-related cancers, 11 (16%) had baseline clonal hematopoiesis.
In a multivariate model using data from the external cohort, clonal hematopoiesis was significantly associated with risk for therapy-related myeloid neoplasms, with a hazard ratio of 13.7 (P = .013).
Elderly patient study
Dr. Gillis and her colleagues conducted a nested, case-control, proof-of-concept study to compare the prevalence of CHIP between patients with cancer who later developed therapy-related myeloid neoplasms (cases) and patients who did not (controls).
The cases were identified from an internal biobank of 123,357 patients, and included all patients who were diagnosed with a primary cancer, treated with chemotherapy, and subsequently developed a therapy-related myeloid neoplasm. The patients had to be 70 or older at the time of either primary or therapy-related cancer diagnosis with peripheral blood or mononuclear samples collected before the diagnosis of the second cancer.
Controls were patients diagnosed with a primary malignancy at age 70 or older who had chemotherapy but did not develop therapy-related myeloid neoplasms. Every case was matched with at least four controls selected for sex, primary tumor type, age at diagnosis, smoking status, chemotherapy drug class, and duration of follow up.
They used sequential targeted and whole-exome sequencing to assess clonal evolution in cases for whom paired CHIP and therapy-related myeloid neoplasm samples were available.
They identified a total of 13 cases and 56 controls. Among all patients, CHIP was seen in 23 (33%). In contrast, previous studies have shown a prevalence of CHIP among older patients without cancer of about 10%, the authors note in their article.
The prevalence of CHIP was significantly higher among cases than among controls, occurring in 8 of 13 cases (62%) vs 15 of 56 controls (27%; P = .024). The odds ratio for therapy-related neoplasms with CHIP was 5.75 (P = .013).
The most commonly mutated genes were TET2 and TP53 among cases, and TET2 among controls.
“The distribution of CHIP-related gene mutations differs between individuals with therapy-related myeloid neoplasm and those without, suggesting that mutation-specific differences might exist in therapy-related myeloid neoplasm risk,” the investigators write.
Dr. Takahashi’s study was supported by the Cancer Prevention Research Institute of Texas, Red and Charline McCombs Institute for the Early Detection and Treatment of Cancer, The National Institutes of Health through MD Anderson Cancer Center Support Grant, and the MD Anderson MDS & AML Moon Shots Program. Dr. Gillis’ study was internally funded. Dr. Takahasi and colleagues reported no competing financial interests. Two of Dr. Gillis’ colleagues reported grants or fees from several drug companies.
The real importance of the work reported by Gillis and colleagues and Takahashi and colleagues will come when therapies exist that can effectively eradicate nascent clonal hematopoiesis, thereby preventing therapy-related myeloid neoplasm evolution in at-risk patients.
Although high-intensity treatments, such as anthracycline-based induction chemotherapy, can eradicate myeloid clones, their effectiveness in clearing TP53-mutant cells is limited, and it is difficult to imagine intense approaches having a favorable risk–benefit balance in patients whose clonal hematopoiesis might never become a problem. Existing lower-intensity therapies for myeloid neoplasms such as DNA hypomethylating agents are not curative and often do not result in the reduction of VAF [variant allele frequencies] even when hematopoietic improvement occurs during therapy, so such agents would not be expected to eliminate pre-therapy-related myeloid neoplasm clones (although this hypothesis might still be worth testing, given that the emergence of therapy-related myeloid neoplasm could at least be delayed – even if not entirely prevented – with azacitidine or decitabine).
More promising are strategies that change the bone marrow microenvironment or break the immune tolerance of abnormal clones, although the use of these approaches for myeloid neoplasia is still in the very early stages. Although no method yet exists to reliably eliminate the preleukemic clones that can give rise to therapy-related myeloid neoplasms, identification of higher risk patients could still affect monitoring practices, such as the frequency of clinical assessments. Molecular genetic panels are expensive at present but are becoming less so. Because VAF assessment by next-generation sequencing is quantitative and proportional to clone size, serial assessment could identify patients whose mutant clones are large and expanding and who therefore warrant closer monitoring or enrollment in so-called preventive hematology trials.
David P. Steensma, MD, is with the Dana-Farber Cancer Institute, Harvard Medical School, Boston. His remarks were excerpted from an accompanying editorial.
The real importance of the work reported by Gillis and colleagues and Takahashi and colleagues will come when therapies exist that can effectively eradicate nascent clonal hematopoiesis, thereby preventing therapy-related myeloid neoplasm evolution in at-risk patients.
Although high-intensity treatments, such as anthracycline-based induction chemotherapy, can eradicate myeloid clones, their effectiveness in clearing TP53-mutant cells is limited, and it is difficult to imagine intense approaches having a favorable risk–benefit balance in patients whose clonal hematopoiesis might never become a problem. Existing lower-intensity therapies for myeloid neoplasms such as DNA hypomethylating agents are not curative and often do not result in the reduction of VAF [variant allele frequencies] even when hematopoietic improvement occurs during therapy, so such agents would not be expected to eliminate pre-therapy-related myeloid neoplasm clones (although this hypothesis might still be worth testing, given that the emergence of therapy-related myeloid neoplasm could at least be delayed – even if not entirely prevented – with azacitidine or decitabine).
More promising are strategies that change the bone marrow microenvironment or break the immune tolerance of abnormal clones, although the use of these approaches for myeloid neoplasia is still in the very early stages. Although no method yet exists to reliably eliminate the preleukemic clones that can give rise to therapy-related myeloid neoplasms, identification of higher risk patients could still affect monitoring practices, such as the frequency of clinical assessments. Molecular genetic panels are expensive at present but are becoming less so. Because VAF assessment by next-generation sequencing is quantitative and proportional to clone size, serial assessment could identify patients whose mutant clones are large and expanding and who therefore warrant closer monitoring or enrollment in so-called preventive hematology trials.
David P. Steensma, MD, is with the Dana-Farber Cancer Institute, Harvard Medical School, Boston. His remarks were excerpted from an accompanying editorial.
The real importance of the work reported by Gillis and colleagues and Takahashi and colleagues will come when therapies exist that can effectively eradicate nascent clonal hematopoiesis, thereby preventing therapy-related myeloid neoplasm evolution in at-risk patients.
Although high-intensity treatments, such as anthracycline-based induction chemotherapy, can eradicate myeloid clones, their effectiveness in clearing TP53-mutant cells is limited, and it is difficult to imagine intense approaches having a favorable risk–benefit balance in patients whose clonal hematopoiesis might never become a problem. Existing lower-intensity therapies for myeloid neoplasms such as DNA hypomethylating agents are not curative and often do not result in the reduction of VAF [variant allele frequencies] even when hematopoietic improvement occurs during therapy, so such agents would not be expected to eliminate pre-therapy-related myeloid neoplasm clones (although this hypothesis might still be worth testing, given that the emergence of therapy-related myeloid neoplasm could at least be delayed – even if not entirely prevented – with azacitidine or decitabine).
More promising are strategies that change the bone marrow microenvironment or break the immune tolerance of abnormal clones, although the use of these approaches for myeloid neoplasia is still in the very early stages. Although no method yet exists to reliably eliminate the preleukemic clones that can give rise to therapy-related myeloid neoplasms, identification of higher risk patients could still affect monitoring practices, such as the frequency of clinical assessments. Molecular genetic panels are expensive at present but are becoming less so. Because VAF assessment by next-generation sequencing is quantitative and proportional to clone size, serial assessment could identify patients whose mutant clones are large and expanding and who therefore warrant closer monitoring or enrollment in so-called preventive hematology trials.
David P. Steensma, MD, is with the Dana-Farber Cancer Institute, Harvard Medical School, Boston. His remarks were excerpted from an accompanying editorial.
Small pre-leukemic clones left behind after treatment for non-myeloid malignancies appear to increase the risk for therapy-related myelodysplasia or leukemia, report investigators in two studies.
An analysis of peripheral blood samples taken from patients at the time of their primary cancer diagnosis and bone marrow samples taken at the time of a later therapy-related myeloid neoplasm diagnosis showed that 10 of 14 patients (71%) had clonal hematopoiesis before starting on cytotoxic chemotherapy. In contrast, clonal hematopoiesis was detected in pre-treatment samples of only 17 of 54 controls (31%), reported Koichi Takahashi, MD, and colleagues from the University of Texas MD Anderson Cancer Center in Houston.
“Preleukemic clonal hematopoiesis is common in patients with therapy-related myeloid neoplasms at the time of their primary cancer diagnosis and before they have been exposed to treatment. Our results suggest that clonal hematopoiesis could be used as a predictive marker to identify patients with cancer who are at risk of developing therapy-related myeloid neoplasms,” they wrote (Lancet Oncol 2017; 18: 100–11).
In a separate study, investigators from the Moffitt Cancer Center in Tampa, Florida, found in a nested case-control study that patients with therapy-related myeloid neoplasms were more likely than controls to have clonal hematopoiesis of indeterminate potential (CHIP), and that the CHIP was often present before exposure to chemotherapy.
“We recorded a significantly higher prevalence of CHIP in individuals who developed therapy-related myeloid neoplasms (cases) than in those who did not (controls); however, around 27% of individuals with CHIP did not develop therapy-related myeloid neoplasms, suggesting that this feature alone should not be used to determine a patient’s suitability for chemotherapy,” wrote Nancy K. Gillis, PharmD, and colleagues (Lancet Oncol 2017; 18:112-21).
Risk factors examined
Dr. Takahashi and colleagues noted that previous studies have identified several treatment-related risk factors as being associated with therapy-related myeloid dysplasia or leukemia, including the use of alkylating agents, topoisomerase II inhibitors, and high-dose chemotherapy with autologous stem-cell transplantation.
“By contrast, little is known about patient-specific risk factors. Older age was shown to increase the risk of therapy-related myeloid neoplasms. Several germline polymorphisms have also been associated with this risk, but none have been validated. As such, no predictive biomarkers exist for therapy-related myeloid neoplasms,” they wrote.
They performed a retrospective case-control study comparing patients treated for a primary cancer at their center from 1997 through 2015 who subsequently developed a myeloid neoplasm with controls treated during the same period. Controls were age-matched patients treated with combination chemotherapy for lymphoma who did not develop a therapy-related myeloid malignancy after at least 5 years of follow-up.
In addition, the investigators further explored the association between clonal hematopoiesis and therapy-related cancers in an external cohort of patients with lymphoma treated in a randomized trial at their center from 1999 through 2001. That trial compared the CHOP regimen (cyclophosphamide, doxorubicin, vincristine and prednisone) with and without melatonin.
To detect clonal hematopoiesis in pre-treatment peripheral blood, the investigators used molecular barcode sequencing of 32 genes. They also used targeted gene sequencing on bone marrow samples from cases to investigate clonal evolution from clonal hematopoiesis to the development of therapy-related myeloid neoplasms.
As noted before, 10 of 14 cases had evidence of pre-treatment clonal hematopoiesis, compared with 17 of 54 controls. For both cases and controls, the cumulative incidence of therapy-related myeloid cancers after 5 years was significantly higher among those with baseline clonal hematopoiesis, at 30% vs. 7% for patients without it (P = .016).
Five of 74 patients in the external cohort (7%) went on to develop therapy-related myeloid neoplasms, and of this group, four (80%) had clonal hematopoiesis at baseline. In contrast, of the 69 patients who did not develop therapy-related cancers, 11 (16%) had baseline clonal hematopoiesis.
In a multivariate model using data from the external cohort, clonal hematopoiesis was significantly associated with risk for therapy-related myeloid neoplasms, with a hazard ratio of 13.7 (P = .013).
Elderly patient study
Dr. Gillis and her colleagues conducted a nested, case-control, proof-of-concept study to compare the prevalence of CHIP between patients with cancer who later developed therapy-related myeloid neoplasms (cases) and patients who did not (controls).
The cases were identified from an internal biobank of 123,357 patients, and included all patients who were diagnosed with a primary cancer, treated with chemotherapy, and subsequently developed a therapy-related myeloid neoplasm. The patients had to be 70 or older at the time of either primary or therapy-related cancer diagnosis with peripheral blood or mononuclear samples collected before the diagnosis of the second cancer.
Controls were patients diagnosed with a primary malignancy at age 70 or older who had chemotherapy but did not develop therapy-related myeloid neoplasms. Every case was matched with at least four controls selected for sex, primary tumor type, age at diagnosis, smoking status, chemotherapy drug class, and duration of follow up.
They used sequential targeted and whole-exome sequencing to assess clonal evolution in cases for whom paired CHIP and therapy-related myeloid neoplasm samples were available.
They identified a total of 13 cases and 56 controls. Among all patients, CHIP was seen in 23 (33%). In contrast, previous studies have shown a prevalence of CHIP among older patients without cancer of about 10%, the authors note in their article.
The prevalence of CHIP was significantly higher among cases than among controls, occurring in 8 of 13 cases (62%) vs 15 of 56 controls (27%; P = .024). The odds ratio for therapy-related neoplasms with CHIP was 5.75 (P = .013).
The most commonly mutated genes were TET2 and TP53 among cases, and TET2 among controls.
“The distribution of CHIP-related gene mutations differs between individuals with therapy-related myeloid neoplasm and those without, suggesting that mutation-specific differences might exist in therapy-related myeloid neoplasm risk,” the investigators write.
Dr. Takahashi’s study was supported by the Cancer Prevention Research Institute of Texas, Red and Charline McCombs Institute for the Early Detection and Treatment of Cancer, The National Institutes of Health through MD Anderson Cancer Center Support Grant, and the MD Anderson MDS & AML Moon Shots Program. Dr. Gillis’ study was internally funded. Dr. Takahasi and colleagues reported no competing financial interests. Two of Dr. Gillis’ colleagues reported grants or fees from several drug companies.
Small pre-leukemic clones left behind after treatment for non-myeloid malignancies appear to increase the risk for therapy-related myelodysplasia or leukemia, report investigators in two studies.
An analysis of peripheral blood samples taken from patients at the time of their primary cancer diagnosis and bone marrow samples taken at the time of a later therapy-related myeloid neoplasm diagnosis showed that 10 of 14 patients (71%) had clonal hematopoiesis before starting on cytotoxic chemotherapy. In contrast, clonal hematopoiesis was detected in pre-treatment samples of only 17 of 54 controls (31%), reported Koichi Takahashi, MD, and colleagues from the University of Texas MD Anderson Cancer Center in Houston.
“Preleukemic clonal hematopoiesis is common in patients with therapy-related myeloid neoplasms at the time of their primary cancer diagnosis and before they have been exposed to treatment. Our results suggest that clonal hematopoiesis could be used as a predictive marker to identify patients with cancer who are at risk of developing therapy-related myeloid neoplasms,” they wrote (Lancet Oncol 2017; 18: 100–11).
In a separate study, investigators from the Moffitt Cancer Center in Tampa, Florida, found in a nested case-control study that patients with therapy-related myeloid neoplasms were more likely than controls to have clonal hematopoiesis of indeterminate potential (CHIP), and that the CHIP was often present before exposure to chemotherapy.
“We recorded a significantly higher prevalence of CHIP in individuals who developed therapy-related myeloid neoplasms (cases) than in those who did not (controls); however, around 27% of individuals with CHIP did not develop therapy-related myeloid neoplasms, suggesting that this feature alone should not be used to determine a patient’s suitability for chemotherapy,” wrote Nancy K. Gillis, PharmD, and colleagues (Lancet Oncol 2017; 18:112-21).
Risk factors examined
Dr. Takahashi and colleagues noted that previous studies have identified several treatment-related risk factors as being associated with therapy-related myeloid dysplasia or leukemia, including the use of alkylating agents, topoisomerase II inhibitors, and high-dose chemotherapy with autologous stem-cell transplantation.
“By contrast, little is known about patient-specific risk factors. Older age was shown to increase the risk of therapy-related myeloid neoplasms. Several germline polymorphisms have also been associated with this risk, but none have been validated. As such, no predictive biomarkers exist for therapy-related myeloid neoplasms,” they wrote.
They performed a retrospective case-control study comparing patients treated for a primary cancer at their center from 1997 through 2015 who subsequently developed a myeloid neoplasm with controls treated during the same period. Controls were age-matched patients treated with combination chemotherapy for lymphoma who did not develop a therapy-related myeloid malignancy after at least 5 years of follow-up.
In addition, the investigators further explored the association between clonal hematopoiesis and therapy-related cancers in an external cohort of patients with lymphoma treated in a randomized trial at their center from 1999 through 2001. That trial compared the CHOP regimen (cyclophosphamide, doxorubicin, vincristine and prednisone) with and without melatonin.
To detect clonal hematopoiesis in pre-treatment peripheral blood, the investigators used molecular barcode sequencing of 32 genes. They also used targeted gene sequencing on bone marrow samples from cases to investigate clonal evolution from clonal hematopoiesis to the development of therapy-related myeloid neoplasms.
As noted before, 10 of 14 cases had evidence of pre-treatment clonal hematopoiesis, compared with 17 of 54 controls. For both cases and controls, the cumulative incidence of therapy-related myeloid cancers after 5 years was significantly higher among those with baseline clonal hematopoiesis, at 30% vs. 7% for patients without it (P = .016).
Five of 74 patients in the external cohort (7%) went on to develop therapy-related myeloid neoplasms, and of this group, four (80%) had clonal hematopoiesis at baseline. In contrast, of the 69 patients who did not develop therapy-related cancers, 11 (16%) had baseline clonal hematopoiesis.
In a multivariate model using data from the external cohort, clonal hematopoiesis was significantly associated with risk for therapy-related myeloid neoplasms, with a hazard ratio of 13.7 (P = .013).
Elderly patient study
Dr. Gillis and her colleagues conducted a nested, case-control, proof-of-concept study to compare the prevalence of CHIP between patients with cancer who later developed therapy-related myeloid neoplasms (cases) and patients who did not (controls).
The cases were identified from an internal biobank of 123,357 patients, and included all patients who were diagnosed with a primary cancer, treated with chemotherapy, and subsequently developed a therapy-related myeloid neoplasm. The patients had to be 70 or older at the time of either primary or therapy-related cancer diagnosis with peripheral blood or mononuclear samples collected before the diagnosis of the second cancer.
Controls were patients diagnosed with a primary malignancy at age 70 or older who had chemotherapy but did not develop therapy-related myeloid neoplasms. Every case was matched with at least four controls selected for sex, primary tumor type, age at diagnosis, smoking status, chemotherapy drug class, and duration of follow up.
They used sequential targeted and whole-exome sequencing to assess clonal evolution in cases for whom paired CHIP and therapy-related myeloid neoplasm samples were available.
They identified a total of 13 cases and 56 controls. Among all patients, CHIP was seen in 23 (33%). In contrast, previous studies have shown a prevalence of CHIP among older patients without cancer of about 10%, the authors note in their article.
The prevalence of CHIP was significantly higher among cases than among controls, occurring in 8 of 13 cases (62%) vs 15 of 56 controls (27%; P = .024). The odds ratio for therapy-related neoplasms with CHIP was 5.75 (P = .013).
The most commonly mutated genes were TET2 and TP53 among cases, and TET2 among controls.
“The distribution of CHIP-related gene mutations differs between individuals with therapy-related myeloid neoplasm and those without, suggesting that mutation-specific differences might exist in therapy-related myeloid neoplasm risk,” the investigators write.
Dr. Takahashi’s study was supported by the Cancer Prevention Research Institute of Texas, Red and Charline McCombs Institute for the Early Detection and Treatment of Cancer, The National Institutes of Health through MD Anderson Cancer Center Support Grant, and the MD Anderson MDS & AML Moon Shots Program. Dr. Gillis’ study was internally funded. Dr. Takahasi and colleagues reported no competing financial interests. Two of Dr. Gillis’ colleagues reported grants or fees from several drug companies.
FROM LANCET ONCOLOGY
Key clinical point: Pre-therapy clonal hematopoiesis is associated with increased risk for therapy-related myeloid neoplasms.
Major finding: In two studies, the incidence of therapy-related myeloid neoplasms was higher among patients with clonal hematopoiesis at baseline.
Data source: Retrospective case-control studies.
Disclosures: Dr. Takahashi’s study was supported by the Cancer Prevention Research Institute of Texas, Red and Charline McCombs Institute for the Early Detection and Treatment of Cancer, The National Institutes of Health through MD Anderson Cancer Center Support Grant, and the MD Anderson MDS & AML Moon Shots Program. Dr. Gillis’ study was internally funded. Dr. Takahasi and colleagues reported no competing financial interests. Two of Dr. Gillis’ colleagues reported grants or fees from several drug companies.