User login
Targeted conjugate therapy kills ALL cells

Photo courtesy of the
University of California Davis
Researchers say they have developed a targeted conjugate therapy that harnesses a monoclonal antibody to deliver antisense DNA to acute lymphoblastic leukemia (ALL) cells.
Once delivered, the therapeutic DNA reduces levels of MXD3, a protein that helps cancer cells survive.
This conjugate therapy proved cytotoxic in ALL cell lines and showed promise in animal models, destroying ALL cells while limiting other damage.
“We’ve shown, for the first time, that anti-CD22 antibody-antisense conjugates are a potential therapeutic agent for ALL,” said Noriko Satake, MD, of the University of California Davis in Sacramento.
“This could be a new type of treatment that kills leukemia cells with few side effects.”
Dr Satake and her colleagues described the treatment in Molecular Medicine.
To create the therapy, the researchers attached antisense DNA that inhibits the MXD3 protein to an antibody that binds to CD22, a protein receptor expressed almost exclusively on ALL cells and normal B cells.
Once the antibody binds to CD22, the conjugate is drawn inside the cell, allowing the antisense molecule to prevent MXD3 production. Without this anti-apoptotic protein, cells are more prone to death.
The conjugate therapy was effective against ALL cell lines and primary ALL cells in a xenograft mouse model. Animals that received the therapy survived significantly longer than those in the control group.
While the conjugate therapy does target healthy B cells along with ALL cells, it is expected to leave hematopoietic stem cells and other tissues unharmed.
“Our novel conjugate is designed so that it does not harm hair, eyes, heart, kidneys, or other types of cells,” Dr Satake said.
She and her colleagues noted that, although this study shows the conjugate therapy can knock down MXD3, it is not clear exactly how this is accomplished. So the researchers plan to investigate the mechanism.
The team also plans to look into combining the conjugate therapy with other treatments. Because it hastens cell death, the conjugate could make traditional chemotherapy drugs more effective, and it might work against other cancers.
“You can see this as proof of principle,” Dr Satake said. “You could switch the target and substitute the antibody, which could be used to treat other cancers or even other diseases.”
This study was not industry-funded, but 4 study authors are employees and stockholders of Isis Pharmaceuticals. ![]()
Photo courtesy of the
University of California Davis
Researchers say they have developed a targeted conjugate therapy that harnesses a monoclonal antibody to deliver antisense DNA to acute lymphoblastic leukemia (ALL) cells.
Once delivered, the therapeutic DNA reduces levels of MXD3, a protein that helps cancer cells survive.
This conjugate therapy proved cytotoxic in ALL cell lines and showed promise in animal models, destroying ALL cells while limiting other damage.
“We’ve shown, for the first time, that anti-CD22 antibody-antisense conjugates are a potential therapeutic agent for ALL,” said Noriko Satake, MD, of the University of California Davis in Sacramento.
“This could be a new type of treatment that kills leukemia cells with few side effects.”
Dr Satake and her colleagues described the treatment in Molecular Medicine.
To create the therapy, the researchers attached antisense DNA that inhibits the MXD3 protein to an antibody that binds to CD22, a protein receptor expressed almost exclusively on ALL cells and normal B cells.
Once the antibody binds to CD22, the conjugate is drawn inside the cell, allowing the antisense molecule to prevent MXD3 production. Without this anti-apoptotic protein, cells are more prone to death.
The conjugate therapy was effective against ALL cell lines and primary ALL cells in a xenograft mouse model. Animals that received the therapy survived significantly longer than those in the control group.
While the conjugate therapy does target healthy B cells along with ALL cells, it is expected to leave hematopoietic stem cells and other tissues unharmed.
“Our novel conjugate is designed so that it does not harm hair, eyes, heart, kidneys, or other types of cells,” Dr Satake said.
She and her colleagues noted that, although this study shows the conjugate therapy can knock down MXD3, it is not clear exactly how this is accomplished. So the researchers plan to investigate the mechanism.
The team also plans to look into combining the conjugate therapy with other treatments. Because it hastens cell death, the conjugate could make traditional chemotherapy drugs more effective, and it might work against other cancers.
“You can see this as proof of principle,” Dr Satake said. “You could switch the target and substitute the antibody, which could be used to treat other cancers or even other diseases.”
This study was not industry-funded, but 4 study authors are employees and stockholders of Isis Pharmaceuticals. ![]()
Photo courtesy of the
University of California Davis
Researchers say they have developed a targeted conjugate therapy that harnesses a monoclonal antibody to deliver antisense DNA to acute lymphoblastic leukemia (ALL) cells.
Once delivered, the therapeutic DNA reduces levels of MXD3, a protein that helps cancer cells survive.
This conjugate therapy proved cytotoxic in ALL cell lines and showed promise in animal models, destroying ALL cells while limiting other damage.
“We’ve shown, for the first time, that anti-CD22 antibody-antisense conjugates are a potential therapeutic agent for ALL,” said Noriko Satake, MD, of the University of California Davis in Sacramento.
“This could be a new type of treatment that kills leukemia cells with few side effects.”
Dr Satake and her colleagues described the treatment in Molecular Medicine.
To create the therapy, the researchers attached antisense DNA that inhibits the MXD3 protein to an antibody that binds to CD22, a protein receptor expressed almost exclusively on ALL cells and normal B cells.
Once the antibody binds to CD22, the conjugate is drawn inside the cell, allowing the antisense molecule to prevent MXD3 production. Without this anti-apoptotic protein, cells are more prone to death.
The conjugate therapy was effective against ALL cell lines and primary ALL cells in a xenograft mouse model. Animals that received the therapy survived significantly longer than those in the control group.
While the conjugate therapy does target healthy B cells along with ALL cells, it is expected to leave hematopoietic stem cells and other tissues unharmed.
“Our novel conjugate is designed so that it does not harm hair, eyes, heart, kidneys, or other types of cells,” Dr Satake said.
She and her colleagues noted that, although this study shows the conjugate therapy can knock down MXD3, it is not clear exactly how this is accomplished. So the researchers plan to investigate the mechanism.
The team also plans to look into combining the conjugate therapy with other treatments. Because it hastens cell death, the conjugate could make traditional chemotherapy drugs more effective, and it might work against other cancers.
“You can see this as proof of principle,” Dr Satake said. “You could switch the target and substitute the antibody, which could be used to treat other cancers or even other diseases.”
This study was not industry-funded, but 4 study authors are employees and stockholders of Isis Pharmaceuticals. ![]()
CLL: Genetic aberrations predict poor treatment response in elderly
In elderly patients with chronic lymphocytic leukemia, complex karyotype abnormalities, certain KRAS and POT1 mutations, and newly discovered mutations in genes involved in the DNA damage response were found to predict a poor response to chlorambucil-based chemotherapy or chemoimmunotherapy and poor survival, according to a report in Blood.
These findings are from what the investigators described as the first comprehensive prospective analysis of chromosomal aberrations (including complex karyotype abnormalities), gene mutations, and clinical and biological features in elderly CLL patients who had multiple comorbidities. This patient population is generally considered ineligible for aggressive first-line agents such as fludarabine and cyclophosphamide, said Carmen Diana Herling, MD, of the Laboratory of Functional Genomics in Lymphoid Malignancies, University of Cologne (Germany), and her associates.
For their analysis, investigators studied 161 such patients enrolled in a clinical trial in which all were treated with chlorambucil alone, chlorambucil plus obinutuzumab, or chlorambucil plus rituximab. The median patient age was 75 years. Comprehensive genetic analyses were performed using peripheral blood drawn before the patients underwent treatment.
Karyotyping detected chromosomal aberrations in 68.68% of patients, while 31.2% carried translocations and 19.5% showed complex karyotypes. Gene sequencing detected 198 missense/nonsense mutations and other abnormalities in 76.4% of patients.
Dr. Herling and her associates found that complex karyotype abnormalities independently predicted poor response to chlorambucil and poor survival. “Thus, global karyotyping (i.e., by chromosome banding analysis) seems to substantially contribute to the identification of CLL patients with most adverse prognoses and should be considered a standard assessment in future CLL trials,” they said (Blood. 2016;128:395-404).
In addition, KRAS mutations correlated with a poor treatment response, particularly to rituximab. Targeting such patients for MEK, BRAF, or ERK inhibitors “might offer personalized treatment strategies to be investigated in such cases.”
Mutations in the POT1 gene also correlated with shorter survival after chlorambucil treatment. And finally, poor treatment response also correlated with previously unknown mutations in genes involved with the response to DNA damage. This “might contribute to the accumulation of genomic alterations and clonal evolution of CLL,” Dr. Herling and her associates said.
In elderly patients with chronic lymphocytic leukemia, complex karyotype abnormalities, certain KRAS and POT1 mutations, and newly discovered mutations in genes involved in the DNA damage response were found to predict a poor response to chlorambucil-based chemotherapy or chemoimmunotherapy and poor survival, according to a report in Blood.
These findings are from what the investigators described as the first comprehensive prospective analysis of chromosomal aberrations (including complex karyotype abnormalities), gene mutations, and clinical and biological features in elderly CLL patients who had multiple comorbidities. This patient population is generally considered ineligible for aggressive first-line agents such as fludarabine and cyclophosphamide, said Carmen Diana Herling, MD, of the Laboratory of Functional Genomics in Lymphoid Malignancies, University of Cologne (Germany), and her associates.
For their analysis, investigators studied 161 such patients enrolled in a clinical trial in which all were treated with chlorambucil alone, chlorambucil plus obinutuzumab, or chlorambucil plus rituximab. The median patient age was 75 years. Comprehensive genetic analyses were performed using peripheral blood drawn before the patients underwent treatment.
Karyotyping detected chromosomal aberrations in 68.68% of patients, while 31.2% carried translocations and 19.5% showed complex karyotypes. Gene sequencing detected 198 missense/nonsense mutations and other abnormalities in 76.4% of patients.
Dr. Herling and her associates found that complex karyotype abnormalities independently predicted poor response to chlorambucil and poor survival. “Thus, global karyotyping (i.e., by chromosome banding analysis) seems to substantially contribute to the identification of CLL patients with most adverse prognoses and should be considered a standard assessment in future CLL trials,” they said (Blood. 2016;128:395-404).
In addition, KRAS mutations correlated with a poor treatment response, particularly to rituximab. Targeting such patients for MEK, BRAF, or ERK inhibitors “might offer personalized treatment strategies to be investigated in such cases.”
Mutations in the POT1 gene also correlated with shorter survival after chlorambucil treatment. And finally, poor treatment response also correlated with previously unknown mutations in genes involved with the response to DNA damage. This “might contribute to the accumulation of genomic alterations and clonal evolution of CLL,” Dr. Herling and her associates said.
In elderly patients with chronic lymphocytic leukemia, complex karyotype abnormalities, certain KRAS and POT1 mutations, and newly discovered mutations in genes involved in the DNA damage response were found to predict a poor response to chlorambucil-based chemotherapy or chemoimmunotherapy and poor survival, according to a report in Blood.
These findings are from what the investigators described as the first comprehensive prospective analysis of chromosomal aberrations (including complex karyotype abnormalities), gene mutations, and clinical and biological features in elderly CLL patients who had multiple comorbidities. This patient population is generally considered ineligible for aggressive first-line agents such as fludarabine and cyclophosphamide, said Carmen Diana Herling, MD, of the Laboratory of Functional Genomics in Lymphoid Malignancies, University of Cologne (Germany), and her associates.
For their analysis, investigators studied 161 such patients enrolled in a clinical trial in which all were treated with chlorambucil alone, chlorambucil plus obinutuzumab, or chlorambucil plus rituximab. The median patient age was 75 years. Comprehensive genetic analyses were performed using peripheral blood drawn before the patients underwent treatment.
Karyotyping detected chromosomal aberrations in 68.68% of patients, while 31.2% carried translocations and 19.5% showed complex karyotypes. Gene sequencing detected 198 missense/nonsense mutations and other abnormalities in 76.4% of patients.
Dr. Herling and her associates found that complex karyotype abnormalities independently predicted poor response to chlorambucil and poor survival. “Thus, global karyotyping (i.e., by chromosome banding analysis) seems to substantially contribute to the identification of CLL patients with most adverse prognoses and should be considered a standard assessment in future CLL trials,” they said (Blood. 2016;128:395-404).
In addition, KRAS mutations correlated with a poor treatment response, particularly to rituximab. Targeting such patients for MEK, BRAF, or ERK inhibitors “might offer personalized treatment strategies to be investigated in such cases.”
Mutations in the POT1 gene also correlated with shorter survival after chlorambucil treatment. And finally, poor treatment response also correlated with previously unknown mutations in genes involved with the response to DNA damage. This “might contribute to the accumulation of genomic alterations and clonal evolution of CLL,” Dr. Herling and her associates said.
FROM BLOOD
Key clinical point: In elderly patients who have chronic lymphocytic leukemia and comorbidities, several genetic abnormalities predict a poor response to chlorambucil-based chemotherapy or chemoimmunotherapy.
Major finding: Complex karyotype abnormalities independently predicted poor response to chlorambucil and poor survival.
Data source: A series of karyotyping and other genetic studies involving 161 elderly patients with CLL and multiple comorbidities.
Disclosures: The participants in this study were drawn from a clinical trial funded by Hoffmann–La Roche; this analysis was supported by Volkswagenstiftung and grants from Deutsche Forschungsgemeinschaft, Deutsche Jose Carreras Leukamie Foundation, Helmholtz-Gemeinschaft, Else Kroner–Fresenius Foundation, and Deutsche Krebshilfe. Dr. Herling reported having no relevant financial disclosures; her associates reported ties to Hoffmann–La Roche.
CAR T-cell therapy eyed for CLL patients with residual disease
Four of eight patients with residual chronic lymphocytic leukemia (CLL) following initial chemotherapy had complete or partial responses to an outpatient therapy that used autologous T cells genetically targeted to the B cell–specific antigen CD19, Mark Blaine Geyer, MD, of Memorial Sloan Kettering Cancer Center, New York, reported at the annual meeting of the American Society of Clinical Oncology.
The therapy employing T cells genetically modified to express CD19-targeted 19-28z chimeric antigen receptors (CARs) was well tolerated but had limited observed efficacy, especially in patients with enlarged lymph nodes. The study goal was to find a safe dose of modified T cells for patients who have disease remaining after initial chemotherapy.
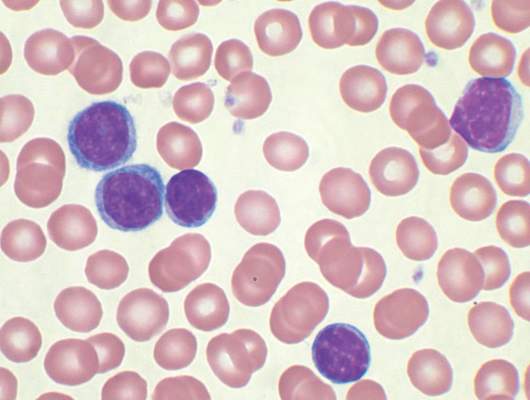
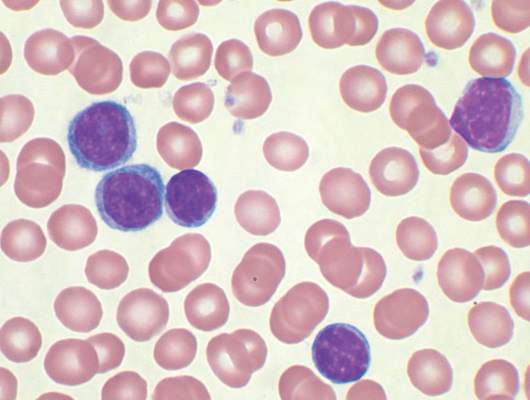
For the phase I dose escalation study (NCT01416974), Dr. Geyer and his associates enrolled eight CLL patients who had residual disease after upfront therapy consisting of six cycles of pentostatin, cyclophosphamide, and rituximab.
Five patients had clearly enlarged lymph nodes prior to T cell infusion.
Patients received cyclophosphamide 600 mg/m2 followed 2 days later by escalating doses of 19-28z T cells. Four of the five patients who received at least a 1 × 107 dose of 19-28z T cells/kg were admitted with fevers and mild cytokine release syndrome.
Maximal levels of CAR T cell persistence were detected at 8 weeks. With a median patient follow-up of 32 months, clinical complete response has been seen in two patients, partial response in two patients, and stable disease in one patient. Disease has progressed in three patients: one had a rising absolute lymphocyte count by the time of infusion and two had marrow response with progressive disease in lymph nodes. The median time to disease progression was 13.6 months, Dr. Geyer said.
Five of seven evaluable patients have received further CLL-directed therapy.
The researchers speculated that low-dose cyclophosphamide monotherapy, used before the CAR T-cell therapy, may be insufficient for lymphodepletion. Additionally, CAR T cell expansion and antitumor efficacy may be limited by a hostile CLL microenvironment. Strategies to enhance CAR T cell expansion and efficacy in patients with CLL are in preparation, Dr. Geyer reported.
Dr. Geyer had no financial disclosures. His colleagues reported various financial relationships with Juno Therapeutics, a developer of CAR technology.
On Twitter @maryjodales
Four of eight patients with residual chronic lymphocytic leukemia (CLL) following initial chemotherapy had complete or partial responses to an outpatient therapy that used autologous T cells genetically targeted to the B cell–specific antigen CD19, Mark Blaine Geyer, MD, of Memorial Sloan Kettering Cancer Center, New York, reported at the annual meeting of the American Society of Clinical Oncology.
The therapy employing T cells genetically modified to express CD19-targeted 19-28z chimeric antigen receptors (CARs) was well tolerated but had limited observed efficacy, especially in patients with enlarged lymph nodes. The study goal was to find a safe dose of modified T cells for patients who have disease remaining after initial chemotherapy.
For the phase I dose escalation study (NCT01416974), Dr. Geyer and his associates enrolled eight CLL patients who had residual disease after upfront therapy consisting of six cycles of pentostatin, cyclophosphamide, and rituximab.
Five patients had clearly enlarged lymph nodes prior to T cell infusion.
Patients received cyclophosphamide 600 mg/m2 followed 2 days later by escalating doses of 19-28z T cells. Four of the five patients who received at least a 1 × 107 dose of 19-28z T cells/kg were admitted with fevers and mild cytokine release syndrome.
Maximal levels of CAR T cell persistence were detected at 8 weeks. With a median patient follow-up of 32 months, clinical complete response has been seen in two patients, partial response in two patients, and stable disease in one patient. Disease has progressed in three patients: one had a rising absolute lymphocyte count by the time of infusion and two had marrow response with progressive disease in lymph nodes. The median time to disease progression was 13.6 months, Dr. Geyer said.
Five of seven evaluable patients have received further CLL-directed therapy.
The researchers speculated that low-dose cyclophosphamide monotherapy, used before the CAR T-cell therapy, may be insufficient for lymphodepletion. Additionally, CAR T cell expansion and antitumor efficacy may be limited by a hostile CLL microenvironment. Strategies to enhance CAR T cell expansion and efficacy in patients with CLL are in preparation, Dr. Geyer reported.
Dr. Geyer had no financial disclosures. His colleagues reported various financial relationships with Juno Therapeutics, a developer of CAR technology.
On Twitter @maryjodales
Four of eight patients with residual chronic lymphocytic leukemia (CLL) following initial chemotherapy had complete or partial responses to an outpatient therapy that used autologous T cells genetically targeted to the B cell–specific antigen CD19, Mark Blaine Geyer, MD, of Memorial Sloan Kettering Cancer Center, New York, reported at the annual meeting of the American Society of Clinical Oncology.
The therapy employing T cells genetically modified to express CD19-targeted 19-28z chimeric antigen receptors (CARs) was well tolerated but had limited observed efficacy, especially in patients with enlarged lymph nodes. The study goal was to find a safe dose of modified T cells for patients who have disease remaining after initial chemotherapy.
For the phase I dose escalation study (NCT01416974), Dr. Geyer and his associates enrolled eight CLL patients who had residual disease after upfront therapy consisting of six cycles of pentostatin, cyclophosphamide, and rituximab.
Five patients had clearly enlarged lymph nodes prior to T cell infusion.
Patients received cyclophosphamide 600 mg/m2 followed 2 days later by escalating doses of 19-28z T cells. Four of the five patients who received at least a 1 × 107 dose of 19-28z T cells/kg were admitted with fevers and mild cytokine release syndrome.
Maximal levels of CAR T cell persistence were detected at 8 weeks. With a median patient follow-up of 32 months, clinical complete response has been seen in two patients, partial response in two patients, and stable disease in one patient. Disease has progressed in three patients: one had a rising absolute lymphocyte count by the time of infusion and two had marrow response with progressive disease in lymph nodes. The median time to disease progression was 13.6 months, Dr. Geyer said.
Five of seven evaluable patients have received further CLL-directed therapy.
The researchers speculated that low-dose cyclophosphamide monotherapy, used before the CAR T-cell therapy, may be insufficient for lymphodepletion. Additionally, CAR T cell expansion and antitumor efficacy may be limited by a hostile CLL microenvironment. Strategies to enhance CAR T cell expansion and efficacy in patients with CLL are in preparation, Dr. Geyer reported.
Dr. Geyer had no financial disclosures. His colleagues reported various financial relationships with Juno Therapeutics, a developer of CAR technology.
On Twitter @maryjodales
FROM THE 2016 ASCO ANNUAL MEETING
Key clinical point: CAR T-cell therapy may be an option for chronic lymphocytic leukemia patients with residual disease after upfront therapy.
Major finding: Four of eight patients with residual CLL following initial chemotherapy had complete or partial responses to an outpatient therapy that used autologous T cells genetically targeted to the B cell–specific antigen CD19.
Data source: A phase I dose-finding and efficacy study in 8 patients with CLL.
Disclosures: Dr. Geyer had no financial disclosures. His colleagues reported various financial relationships with Juno Therapeutics, a developer of CAR technology.
Team identifies mutations contributing to APL

Image from the Armed Forces
Institute of Pathology
Researchers have identified genetic mutations that contribute to the onset of acute promyelocytic leukemia (APL), according to a paper published in Leukemia.
The team analyzed patient samples to identify somatic mutations that cooperate with the PML-RARA fusion gene in the pathogenesis of APL.
They performed whole-exome and targeted sequencing on 242 samples from APL patients, 165 who were newly diagnosed and 77 who had relapsed.
Samples from patients with newly diagnosed APL had recurrent mutations in FLT3, WT1, NRAS, and KRAS but rarely had mutations in other genes commonly mutated in myeloid leukemia.
The newly diagnosed samples also had loss-of-function mutations in ARID1A and ARID1B (members of a chromatin remodeling complex), which had not previously been identified in APL.
The researchers said the ARID1A and ARID1B mutations indicate dysregulation of epigenetic machinery in APL, and the mutations provide a subset of previously uncharacterized genes in leukemogenesis.
The team also found that knocking down ARID1B in the APL cell line NB4 resulted in large-scale activation of gene expression and reduced in vitro differentiation potential.
In the relapsed APL samples, the researchers discovered a set of mutations that were not observed in the newly diagnosed samples. Most prominently, mutations in RARA and PML were found to be exclusive to relapsed samples.
The team also found these mutations were largely acquired in 2 distinct patient groups—those treated at initial diagnosis with all-trans retinoic acid and those treated with arsenic trioxide.
“Our comprehensive study on the mutational landscape in a large cohort of primary and relapsed APL cases has enabled us to establish the molecular roadmap for APL, which is distinct from other subtypes of [acute myeloid leukemia],” said study author H. Phillip Koeffler, MD, of the Cancer Science Institute of Singapore.
“With an enhanced knowledge of the disease biology, we will be conducting further research to uncover the consequences of the novel mutations discovered, with an eventual goal of developing improved and targeted therapeutics.” ![]()
Image from the Armed Forces
Institute of Pathology
Researchers have identified genetic mutations that contribute to the onset of acute promyelocytic leukemia (APL), according to a paper published in Leukemia.
The team analyzed patient samples to identify somatic mutations that cooperate with the PML-RARA fusion gene in the pathogenesis of APL.
They performed whole-exome and targeted sequencing on 242 samples from APL patients, 165 who were newly diagnosed and 77 who had relapsed.
Samples from patients with newly diagnosed APL had recurrent mutations in FLT3, WT1, NRAS, and KRAS but rarely had mutations in other genes commonly mutated in myeloid leukemia.
The newly diagnosed samples also had loss-of-function mutations in ARID1A and ARID1B (members of a chromatin remodeling complex), which had not previously been identified in APL.
The researchers said the ARID1A and ARID1B mutations indicate dysregulation of epigenetic machinery in APL, and the mutations provide a subset of previously uncharacterized genes in leukemogenesis.
The team also found that knocking down ARID1B in the APL cell line NB4 resulted in large-scale activation of gene expression and reduced in vitro differentiation potential.
In the relapsed APL samples, the researchers discovered a set of mutations that were not observed in the newly diagnosed samples. Most prominently, mutations in RARA and PML were found to be exclusive to relapsed samples.
The team also found these mutations were largely acquired in 2 distinct patient groups—those treated at initial diagnosis with all-trans retinoic acid and those treated with arsenic trioxide.
“Our comprehensive study on the mutational landscape in a large cohort of primary and relapsed APL cases has enabled us to establish the molecular roadmap for APL, which is distinct from other subtypes of [acute myeloid leukemia],” said study author H. Phillip Koeffler, MD, of the Cancer Science Institute of Singapore.
“With an enhanced knowledge of the disease biology, we will be conducting further research to uncover the consequences of the novel mutations discovered, with an eventual goal of developing improved and targeted therapeutics.” ![]()
Image from the Armed Forces
Institute of Pathology
Researchers have identified genetic mutations that contribute to the onset of acute promyelocytic leukemia (APL), according to a paper published in Leukemia.
The team analyzed patient samples to identify somatic mutations that cooperate with the PML-RARA fusion gene in the pathogenesis of APL.
They performed whole-exome and targeted sequencing on 242 samples from APL patients, 165 who were newly diagnosed and 77 who had relapsed.
Samples from patients with newly diagnosed APL had recurrent mutations in FLT3, WT1, NRAS, and KRAS but rarely had mutations in other genes commonly mutated in myeloid leukemia.
The newly diagnosed samples also had loss-of-function mutations in ARID1A and ARID1B (members of a chromatin remodeling complex), which had not previously been identified in APL.
The researchers said the ARID1A and ARID1B mutations indicate dysregulation of epigenetic machinery in APL, and the mutations provide a subset of previously uncharacterized genes in leukemogenesis.
The team also found that knocking down ARID1B in the APL cell line NB4 resulted in large-scale activation of gene expression and reduced in vitro differentiation potential.
In the relapsed APL samples, the researchers discovered a set of mutations that were not observed in the newly diagnosed samples. Most prominently, mutations in RARA and PML were found to be exclusive to relapsed samples.
The team also found these mutations were largely acquired in 2 distinct patient groups—those treated at initial diagnosis with all-trans retinoic acid and those treated with arsenic trioxide.
“Our comprehensive study on the mutational landscape in a large cohort of primary and relapsed APL cases has enabled us to establish the molecular roadmap for APL, which is distinct from other subtypes of [acute myeloid leukemia],” said study author H. Phillip Koeffler, MD, of the Cancer Science Institute of Singapore.
“With an enhanced knowledge of the disease biology, we will be conducting further research to uncover the consequences of the novel mutations discovered, with an eventual goal of developing improved and targeted therapeutics.” ![]()
FDA clears kit for monitoring molecular response in CML

Photo by Juan D. Alfonso
The US Food and Drug Administration (FDA) has granted premarket clearance for the QuantideX® qPCR BCR-ABL IS Kit, a tool used to monitor molecular response (MR) in patients with chronic myeloid leukemia (CML).
The product is a quantitative polymerase chain reaction (qPCR)-based in vitro diagnostic test that quantifies BCR-ABL1 and ABL1 transcripts in total RNA from the whole blood of t(9;22)-positive CML patients expressing e13a2 and/or e14a2 fusion transcripts.
The QuantideX® qPCR BCR-ABL IS Kit is not designed to diagnose CML or monitor rare transcripts resulting from t(9;22).
The kit was cleared to run on the Applied Biosystems® 7500 Fast DX Real-Time PCR Instrument. Results are reported in International Scale (IS) values.
The QuantideX® qPCR BCR-ABL IS Kit was subjected to analytic and clinical review through the FDA’s de novo 510(k) premarket review pathway and secured clearance with a limit of detection of MR 4.7/0.002% IS (4.7 log molecular reduction from 100% IS).
The limit of detection was determined using real human RNA, not human-derived cell lines, ensuring that the assay reproducibly detects BCR-ABL1 RNA in at least 95% of patients at MR 4.7.
“In evaluating the QuantideX® qPCR BCR-ABL IS Kit, we confirmed the high level of sensitivity achieved for human clinical samples measured in our laboratory at MR 4.7 (0.002% IS),” said Y. Lynn. Wang, MD, PhD, of the University of Chicago Comprehensive Cancer Center.
“The configuration of the assay—multiplexed, single-lot reagents, efficient workflow, and direct IS reporting—provided the robustness, sensitivity, and data quality we believe to be unprecedented in the market today. The high level of sensitivity will contribute to the assessment of the depth and duration of clinical response to [tyrosine kinase inhibitors] and experimental therapies.”
The QuantideX® qPCR BCR-ABL IS Kit is now available for order in the US and Europe. The kit is a product of Asuragen, Inc. ![]()
Photo by Juan D. Alfonso
The US Food and Drug Administration (FDA) has granted premarket clearance for the QuantideX® qPCR BCR-ABL IS Kit, a tool used to monitor molecular response (MR) in patients with chronic myeloid leukemia (CML).
The product is a quantitative polymerase chain reaction (qPCR)-based in vitro diagnostic test that quantifies BCR-ABL1 and ABL1 transcripts in total RNA from the whole blood of t(9;22)-positive CML patients expressing e13a2 and/or e14a2 fusion transcripts.
The QuantideX® qPCR BCR-ABL IS Kit is not designed to diagnose CML or monitor rare transcripts resulting from t(9;22).
The kit was cleared to run on the Applied Biosystems® 7500 Fast DX Real-Time PCR Instrument. Results are reported in International Scale (IS) values.
The QuantideX® qPCR BCR-ABL IS Kit was subjected to analytic and clinical review through the FDA’s de novo 510(k) premarket review pathway and secured clearance with a limit of detection of MR 4.7/0.002% IS (4.7 log molecular reduction from 100% IS).
The limit of detection was determined using real human RNA, not human-derived cell lines, ensuring that the assay reproducibly detects BCR-ABL1 RNA in at least 95% of patients at MR 4.7.
“In evaluating the QuantideX® qPCR BCR-ABL IS Kit, we confirmed the high level of sensitivity achieved for human clinical samples measured in our laboratory at MR 4.7 (0.002% IS),” said Y. Lynn. Wang, MD, PhD, of the University of Chicago Comprehensive Cancer Center.
“The configuration of the assay—multiplexed, single-lot reagents, efficient workflow, and direct IS reporting—provided the robustness, sensitivity, and data quality we believe to be unprecedented in the market today. The high level of sensitivity will contribute to the assessment of the depth and duration of clinical response to [tyrosine kinase inhibitors] and experimental therapies.”
The QuantideX® qPCR BCR-ABL IS Kit is now available for order in the US and Europe. The kit is a product of Asuragen, Inc. ![]()
Photo by Juan D. Alfonso
The US Food and Drug Administration (FDA) has granted premarket clearance for the QuantideX® qPCR BCR-ABL IS Kit, a tool used to monitor molecular response (MR) in patients with chronic myeloid leukemia (CML).
The product is a quantitative polymerase chain reaction (qPCR)-based in vitro diagnostic test that quantifies BCR-ABL1 and ABL1 transcripts in total RNA from the whole blood of t(9;22)-positive CML patients expressing e13a2 and/or e14a2 fusion transcripts.
The QuantideX® qPCR BCR-ABL IS Kit is not designed to diagnose CML or monitor rare transcripts resulting from t(9;22).
The kit was cleared to run on the Applied Biosystems® 7500 Fast DX Real-Time PCR Instrument. Results are reported in International Scale (IS) values.
The QuantideX® qPCR BCR-ABL IS Kit was subjected to analytic and clinical review through the FDA’s de novo 510(k) premarket review pathway and secured clearance with a limit of detection of MR 4.7/0.002% IS (4.7 log molecular reduction from 100% IS).
The limit of detection was determined using real human RNA, not human-derived cell lines, ensuring that the assay reproducibly detects BCR-ABL1 RNA in at least 95% of patients at MR 4.7.
“In evaluating the QuantideX® qPCR BCR-ABL IS Kit, we confirmed the high level of sensitivity achieved for human clinical samples measured in our laboratory at MR 4.7 (0.002% IS),” said Y. Lynn. Wang, MD, PhD, of the University of Chicago Comprehensive Cancer Center.
“The configuration of the assay—multiplexed, single-lot reagents, efficient workflow, and direct IS reporting—provided the robustness, sensitivity, and data quality we believe to be unprecedented in the market today. The high level of sensitivity will contribute to the assessment of the depth and duration of clinical response to [tyrosine kinase inhibitors] and experimental therapies.”
The QuantideX® qPCR BCR-ABL IS Kit is now available for order in the US and Europe. The kit is a product of Asuragen, Inc. ![]()
Aleukemic acute lymphoblastic leukemia with unusual clinical features
Acute lymphoblastic leukemia is a neoplastic proliferation of lymphoblasts in the bone marrow. Normal hematopoiesis is affected, and symptoms from anemia (fatigue, breathlessness), leukopenia (recurrent infections) or thrombocytopenia (easy bruising, mucosal bleeding) are typically described in ALL. Hepatosplenomegaly and B-symptoms (fever, weight loss, and night sweats) are frequently seen. Presence of lymphoblasts in the peripheral smear is indicative of ALL, and a bone marrow biopsy finding of >25% lymphoblasts is confirmatory. Absence of peripheral lymphoblasts in a patient with acute leukemia is known as aleukemic leukemia. Aleukemic leukemia is uncommon, and most cases have described skin lesions from lymphoblast infiltration (leukemia cutis) in addition to bone marrow involvement.1 We report a case of aleukemic ALL in an adult presenting with unusual clinical features including bone pain, osteolytic lesions, hypercalcemia, and normal blood counts. To our knowledge, this is fifth such case ever reported in an adult patient.
Click on the PDF icon at the top of this introduction to read the full article.
Acute lymphoblastic leukemia is a neoplastic proliferation of lymphoblasts in the bone marrow. Normal hematopoiesis is affected, and symptoms from anemia (fatigue, breathlessness), leukopenia (recurrent infections) or thrombocytopenia (easy bruising, mucosal bleeding) are typically described in ALL. Hepatosplenomegaly and B-symptoms (fever, weight loss, and night sweats) are frequently seen. Presence of lymphoblasts in the peripheral smear is indicative of ALL, and a bone marrow biopsy finding of >25% lymphoblasts is confirmatory. Absence of peripheral lymphoblasts in a patient with acute leukemia is known as aleukemic leukemia. Aleukemic leukemia is uncommon, and most cases have described skin lesions from lymphoblast infiltration (leukemia cutis) in addition to bone marrow involvement.1 We report a case of aleukemic ALL in an adult presenting with unusual clinical features including bone pain, osteolytic lesions, hypercalcemia, and normal blood counts. To our knowledge, this is fifth such case ever reported in an adult patient.
Click on the PDF icon at the top of this introduction to read the full article.
Acute lymphoblastic leukemia is a neoplastic proliferation of lymphoblasts in the bone marrow. Normal hematopoiesis is affected, and symptoms from anemia (fatigue, breathlessness), leukopenia (recurrent infections) or thrombocytopenia (easy bruising, mucosal bleeding) are typically described in ALL. Hepatosplenomegaly and B-symptoms (fever, weight loss, and night sweats) are frequently seen. Presence of lymphoblasts in the peripheral smear is indicative of ALL, and a bone marrow biopsy finding of >25% lymphoblasts is confirmatory. Absence of peripheral lymphoblasts in a patient with acute leukemia is known as aleukemic leukemia. Aleukemic leukemia is uncommon, and most cases have described skin lesions from lymphoblast infiltration (leukemia cutis) in addition to bone marrow involvement.1 We report a case of aleukemic ALL in an adult presenting with unusual clinical features including bone pain, osteolytic lesions, hypercalcemia, and normal blood counts. To our knowledge, this is fifth such case ever reported in an adult patient.
Click on the PDF icon at the top of this introduction to read the full article.
Adolescent and young adult perceptions of cancer survivor care and supportive programming
Background Improvements in cancer therapy have led to an increasing number of adolescent and young adult (AYA) survivors of childhood cancers. Many survivors have ongoing needs for support and information that are not being met.
Objective To conduct a program evaluation to identify AYAs’ perceptions of survivor care services.
Methods Using a community-based approach, 157 AYA childhood cancer survivors (aged 15-30 years) completed a program evaluation survey to assess perceptions of the importance of survivor patient care services and supportive programming using a Likert scale (1, Not At All Important; 2, Of Little Importance; 3, Somewhat Important; 4, Important; 5, Very Important).
Results Receipt of a medical summary was ranked as the most important survivor patient care service (mean, 4.5; SD, 0.91). 70% of respondents reported interest in late-effects education. Informational mailings were the most valued form of supportive programming and were endorsed by 62% of AYAs. Older survivors were more likely to value workshops (P = .01-0.05), whereas those aged 19-22 years valued weekend retreats (P < .01) and social activities (P < .01). Survivors of brain/CNS tumors were more likely to value social activities (P = .03) and support groups (P = .03), compared with leukemia survivors.
Limitations Contact information from the hospital tumor registry was used, which limited the number of correct addresses.
Conclusion The greatest care needs reported by AYA survivors of childhood cancer are services such as generation of a medical summary, late-effects education, and survivor-focused follow-up care, which are provided through cancer survivor programs. Development of additional programming to engage and further educate and encourage AYA survivors will be important to reinforce their adherence with survivor care throughout adulthood.
Funding/Sponsorship LiveStrong Community Based Participatory Research Planning Grant
Click on the PDF icon at the top of this introduction to read the full article.
Background Improvements in cancer therapy have led to an increasing number of adolescent and young adult (AYA) survivors of childhood cancers. Many survivors have ongoing needs for support and information that are not being met.
Objective To conduct a program evaluation to identify AYAs’ perceptions of survivor care services.
Methods Using a community-based approach, 157 AYA childhood cancer survivors (aged 15-30 years) completed a program evaluation survey to assess perceptions of the importance of survivor patient care services and supportive programming using a Likert scale (1, Not At All Important; 2, Of Little Importance; 3, Somewhat Important; 4, Important; 5, Very Important).
Results Receipt of a medical summary was ranked as the most important survivor patient care service (mean, 4.5; SD, 0.91). 70% of respondents reported interest in late-effects education. Informational mailings were the most valued form of supportive programming and were endorsed by 62% of AYAs. Older survivors were more likely to value workshops (P = .01-0.05), whereas those aged 19-22 years valued weekend retreats (P < .01) and social activities (P < .01). Survivors of brain/CNS tumors were more likely to value social activities (P = .03) and support groups (P = .03), compared with leukemia survivors.
Limitations Contact information from the hospital tumor registry was used, which limited the number of correct addresses.
Conclusion The greatest care needs reported by AYA survivors of childhood cancer are services such as generation of a medical summary, late-effects education, and survivor-focused follow-up care, which are provided through cancer survivor programs. Development of additional programming to engage and further educate and encourage AYA survivors will be important to reinforce their adherence with survivor care throughout adulthood.
Funding/Sponsorship LiveStrong Community Based Participatory Research Planning Grant
Click on the PDF icon at the top of this introduction to read the full article.
Background Improvements in cancer therapy have led to an increasing number of adolescent and young adult (AYA) survivors of childhood cancers. Many survivors have ongoing needs for support and information that are not being met.
Objective To conduct a program evaluation to identify AYAs’ perceptions of survivor care services.
Methods Using a community-based approach, 157 AYA childhood cancer survivors (aged 15-30 years) completed a program evaluation survey to assess perceptions of the importance of survivor patient care services and supportive programming using a Likert scale (1, Not At All Important; 2, Of Little Importance; 3, Somewhat Important; 4, Important; 5, Very Important).
Results Receipt of a medical summary was ranked as the most important survivor patient care service (mean, 4.5; SD, 0.91). 70% of respondents reported interest in late-effects education. Informational mailings were the most valued form of supportive programming and were endorsed by 62% of AYAs. Older survivors were more likely to value workshops (P = .01-0.05), whereas those aged 19-22 years valued weekend retreats (P < .01) and social activities (P < .01). Survivors of brain/CNS tumors were more likely to value social activities (P = .03) and support groups (P = .03), compared with leukemia survivors.
Limitations Contact information from the hospital tumor registry was used, which limited the number of correct addresses.
Conclusion The greatest care needs reported by AYA survivors of childhood cancer are services such as generation of a medical summary, late-effects education, and survivor-focused follow-up care, which are provided through cancer survivor programs. Development of additional programming to engage and further educate and encourage AYA survivors will be important to reinforce their adherence with survivor care throughout adulthood.
Funding/Sponsorship LiveStrong Community Based Participatory Research Planning Grant
Click on the PDF icon at the top of this introduction to read the full article.
Delirium in advanced cancer may go undetected

patient and her father
Photo by Rhoda Baer
A new study indicates that delirium is relatively frequent and underdiagnosed in patients with advanced cancer visiting the emergency department.
The research showed that delirium was similarly common among older and younger patients.
According to researchers, this suggests that, in the setting of advanced cancer, all patients should be considered at higher risk for delirium.
The researchers reported their findings in Cancer.
For this study, the team assessed a random sample of 243 advanced cancer patients who presented to the emergency department. They were 19 to 89 years old.
All patients were assessed with 2 methods: the Confusion Assessment Method (CAM) to screen for delirium and the Memorial Delirium Assessment Scale (MDAS) to measure delirium severity (mild ≤15, moderate 16-22, and severe ≥23).
In all, 22 patients (9%) had CAM-positive delirium and a median MDAS score of 14. Among CAM-positive patients, delirium was mild in 18 (82%) and moderate in 4 (18%) according to the MDAS.
Of the 99 patients age 65 and older, 10 (10%) had CAM-positive delirium, compared with 12 (8%) of 144 patients younger than 65.
Emergency department physicians failed to detect delirium in 9 (41%) CAM-positive delirious patients.
“We found evidence of delirium in 1 of every 10 patients with advanced cancer who are treated in the emergency department,” said study author Knox Todd, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Given that we could only study patients who were able to give consent to enter our study, even 10% is likely to be a low estimate. We also identified many psychoactive medications that could have contributed to delirium, and sharing this information with treating oncologists may help them avoid such complications in the next patient they treat.” ![]()
patient and her father
Photo by Rhoda Baer
A new study indicates that delirium is relatively frequent and underdiagnosed in patients with advanced cancer visiting the emergency department.
The research showed that delirium was similarly common among older and younger patients.
According to researchers, this suggests that, in the setting of advanced cancer, all patients should be considered at higher risk for delirium.
The researchers reported their findings in Cancer.
For this study, the team assessed a random sample of 243 advanced cancer patients who presented to the emergency department. They were 19 to 89 years old.
All patients were assessed with 2 methods: the Confusion Assessment Method (CAM) to screen for delirium and the Memorial Delirium Assessment Scale (MDAS) to measure delirium severity (mild ≤15, moderate 16-22, and severe ≥23).
In all, 22 patients (9%) had CAM-positive delirium and a median MDAS score of 14. Among CAM-positive patients, delirium was mild in 18 (82%) and moderate in 4 (18%) according to the MDAS.
Of the 99 patients age 65 and older, 10 (10%) had CAM-positive delirium, compared with 12 (8%) of 144 patients younger than 65.
Emergency department physicians failed to detect delirium in 9 (41%) CAM-positive delirious patients.
“We found evidence of delirium in 1 of every 10 patients with advanced cancer who are treated in the emergency department,” said study author Knox Todd, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Given that we could only study patients who were able to give consent to enter our study, even 10% is likely to be a low estimate. We also identified many psychoactive medications that could have contributed to delirium, and sharing this information with treating oncologists may help them avoid such complications in the next patient they treat.” ![]()
patient and her father
Photo by Rhoda Baer
A new study indicates that delirium is relatively frequent and underdiagnosed in patients with advanced cancer visiting the emergency department.
The research showed that delirium was similarly common among older and younger patients.
According to researchers, this suggests that, in the setting of advanced cancer, all patients should be considered at higher risk for delirium.
The researchers reported their findings in Cancer.
For this study, the team assessed a random sample of 243 advanced cancer patients who presented to the emergency department. They were 19 to 89 years old.
All patients were assessed with 2 methods: the Confusion Assessment Method (CAM) to screen for delirium and the Memorial Delirium Assessment Scale (MDAS) to measure delirium severity (mild ≤15, moderate 16-22, and severe ≥23).
In all, 22 patients (9%) had CAM-positive delirium and a median MDAS score of 14. Among CAM-positive patients, delirium was mild in 18 (82%) and moderate in 4 (18%) according to the MDAS.
Of the 99 patients age 65 and older, 10 (10%) had CAM-positive delirium, compared with 12 (8%) of 144 patients younger than 65.
Emergency department physicians failed to detect delirium in 9 (41%) CAM-positive delirious patients.
“We found evidence of delirium in 1 of every 10 patients with advanced cancer who are treated in the emergency department,” said study author Knox Todd, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Given that we could only study patients who were able to give consent to enter our study, even 10% is likely to be a low estimate. We also identified many psychoactive medications that could have contributed to delirium, and sharing this information with treating oncologists may help them avoid such complications in the next patient they treat.” ![]()
Blood disorders prove costly for European economy

chemotherapy
Photo by Rhoda Baer
Malignant and non-malignant blood disorders cost 31 European countries a total of €23 billion in 2012, according to a pair of papers published in The Lancet Haematology.
Healthcare costs accounted for €16 billion of the total costs, with €7 billion for hospital inpatient care and €4 billion for medications.
Informal care (from friends and relatives) cost €1.6 billion, productivity losses due to mortality cost €2.5 billion, and morbidity cost €3 billion.
Researchers determined these figures by analyzing data from international health organizations (WHO and EUROSTAT), as well as national ministries of health and statistical institutes.
The team estimated the economic burden of malignant and non-malignant blood disorders in 2012 for all 28 countries in the European Union (EU), as well as Iceland, Norway, and Switzerland.
The costs considered were healthcare costs (primary care, accident and emergency care, hospital inpatient and outpatient care, and drugs), informal care costs (from friends and relatives), and productivity losses (due to premature death and people being unable to work due to illness).
Malignant blood disorders
In one paper, the researchers noted that the total economic cost of blood cancers to the 31 countries studied was €12 billion in 2012. Healthcare costs measured €7.3 billion (62% of total costs), productivity losses cost €3.6 billion (30%), and informal care cost €1 billion (8%).
In the 28 EU countries, blood cancers represented 8% of the total cancer costs (€143 billion), meaning that blood cancers are the fourth most expensive type of cancer after lung (15%), breast (12%), and colorectal (10%) cancers.
When considering healthcare costs alone, blood cancers were second only to breast cancers (12% vs 13% of healthcare costs for all cancers).
In 2012, blood cancers cost, on average, €14,674 per patient in the EU (€15,126 in all 31 countries), which is almost 2 times higher than the average cost per patient across all cancers (€7929 in the EU).
The researchers said this difference may be due to the longer length of hospital stay observed for patients with blood cancers (14 days, on average, compared to 8 days across all cancers).
Another potential reason is that blood cancers are increasingly treated with complex, long-term treatments (including stem cell transplants, multi-agent chemotherapy, and radiotherapy) and diagnosed via extensive procedures.
The costs of blood cancers varied widely between the countries studied, but the reasons for this were unclear. For instance, the average healthcare costs in Finland were nearly twice as high as in Belgium (€18,014 vs €9596), despite both countries having similar national income per capita.
Non-malignant blood disorders
In the other paper, the researchers said the total economic cost of non-malignant blood disorders to the 31 countries studied was €11 billion in 2012. Healthcare costs accounted for €8 billion (75% of total costs), productivity losses for €2 billion (19%), and informal care for €618 million (6%).
Averaged across the population studied, non-malignant blood disorders represented an annual healthcare cost of €159 per 10 citizens.
“Non-malignant blood disorders cost the European economy nearly as much as all blood cancers combined,” said Jose Leal, DPhil, of the University of Oxford in the UK.
“We found wide differences in the cost of treating blood disorders in different countries, likely linked to the significant differences in the access and delivery of care for patients with blood disorders. Our findings suggest there is a need to harmonize care of blood disorders across Europe in a cost-effective way.” ![]()
chemotherapy
Photo by Rhoda Baer
Malignant and non-malignant blood disorders cost 31 European countries a total of €23 billion in 2012, according to a pair of papers published in The Lancet Haematology.
Healthcare costs accounted for €16 billion of the total costs, with €7 billion for hospital inpatient care and €4 billion for medications.
Informal care (from friends and relatives) cost €1.6 billion, productivity losses due to mortality cost €2.5 billion, and morbidity cost €3 billion.
Researchers determined these figures by analyzing data from international health organizations (WHO and EUROSTAT), as well as national ministries of health and statistical institutes.
The team estimated the economic burden of malignant and non-malignant blood disorders in 2012 for all 28 countries in the European Union (EU), as well as Iceland, Norway, and Switzerland.
The costs considered were healthcare costs (primary care, accident and emergency care, hospital inpatient and outpatient care, and drugs), informal care costs (from friends and relatives), and productivity losses (due to premature death and people being unable to work due to illness).
Malignant blood disorders
In one paper, the researchers noted that the total economic cost of blood cancers to the 31 countries studied was €12 billion in 2012. Healthcare costs measured €7.3 billion (62% of total costs), productivity losses cost €3.6 billion (30%), and informal care cost €1 billion (8%).
In the 28 EU countries, blood cancers represented 8% of the total cancer costs (€143 billion), meaning that blood cancers are the fourth most expensive type of cancer after lung (15%), breast (12%), and colorectal (10%) cancers.
When considering healthcare costs alone, blood cancers were second only to breast cancers (12% vs 13% of healthcare costs for all cancers).
In 2012, blood cancers cost, on average, €14,674 per patient in the EU (€15,126 in all 31 countries), which is almost 2 times higher than the average cost per patient across all cancers (€7929 in the EU).
The researchers said this difference may be due to the longer length of hospital stay observed for patients with blood cancers (14 days, on average, compared to 8 days across all cancers).
Another potential reason is that blood cancers are increasingly treated with complex, long-term treatments (including stem cell transplants, multi-agent chemotherapy, and radiotherapy) and diagnosed via extensive procedures.
The costs of blood cancers varied widely between the countries studied, but the reasons for this were unclear. For instance, the average healthcare costs in Finland were nearly twice as high as in Belgium (€18,014 vs €9596), despite both countries having similar national income per capita.
Non-malignant blood disorders
In the other paper, the researchers said the total economic cost of non-malignant blood disorders to the 31 countries studied was €11 billion in 2012. Healthcare costs accounted for €8 billion (75% of total costs), productivity losses for €2 billion (19%), and informal care for €618 million (6%).
Averaged across the population studied, non-malignant blood disorders represented an annual healthcare cost of €159 per 10 citizens.
“Non-malignant blood disorders cost the European economy nearly as much as all blood cancers combined,” said Jose Leal, DPhil, of the University of Oxford in the UK.
“We found wide differences in the cost of treating blood disorders in different countries, likely linked to the significant differences in the access and delivery of care for patients with blood disorders. Our findings suggest there is a need to harmonize care of blood disorders across Europe in a cost-effective way.” ![]()
chemotherapy
Photo by Rhoda Baer
Malignant and non-malignant blood disorders cost 31 European countries a total of €23 billion in 2012, according to a pair of papers published in The Lancet Haematology.
Healthcare costs accounted for €16 billion of the total costs, with €7 billion for hospital inpatient care and €4 billion for medications.
Informal care (from friends and relatives) cost €1.6 billion, productivity losses due to mortality cost €2.5 billion, and morbidity cost €3 billion.
Researchers determined these figures by analyzing data from international health organizations (WHO and EUROSTAT), as well as national ministries of health and statistical institutes.
The team estimated the economic burden of malignant and non-malignant blood disorders in 2012 for all 28 countries in the European Union (EU), as well as Iceland, Norway, and Switzerland.
The costs considered were healthcare costs (primary care, accident and emergency care, hospital inpatient and outpatient care, and drugs), informal care costs (from friends and relatives), and productivity losses (due to premature death and people being unable to work due to illness).
Malignant blood disorders
In one paper, the researchers noted that the total economic cost of blood cancers to the 31 countries studied was €12 billion in 2012. Healthcare costs measured €7.3 billion (62% of total costs), productivity losses cost €3.6 billion (30%), and informal care cost €1 billion (8%).
In the 28 EU countries, blood cancers represented 8% of the total cancer costs (€143 billion), meaning that blood cancers are the fourth most expensive type of cancer after lung (15%), breast (12%), and colorectal (10%) cancers.
When considering healthcare costs alone, blood cancers were second only to breast cancers (12% vs 13% of healthcare costs for all cancers).
In 2012, blood cancers cost, on average, €14,674 per patient in the EU (€15,126 in all 31 countries), which is almost 2 times higher than the average cost per patient across all cancers (€7929 in the EU).
The researchers said this difference may be due to the longer length of hospital stay observed for patients with blood cancers (14 days, on average, compared to 8 days across all cancers).
Another potential reason is that blood cancers are increasingly treated with complex, long-term treatments (including stem cell transplants, multi-agent chemotherapy, and radiotherapy) and diagnosed via extensive procedures.
The costs of blood cancers varied widely between the countries studied, but the reasons for this were unclear. For instance, the average healthcare costs in Finland were nearly twice as high as in Belgium (€18,014 vs €9596), despite both countries having similar national income per capita.
Non-malignant blood disorders
In the other paper, the researchers said the total economic cost of non-malignant blood disorders to the 31 countries studied was €11 billion in 2012. Healthcare costs accounted for €8 billion (75% of total costs), productivity losses for €2 billion (19%), and informal care for €618 million (6%).
Averaged across the population studied, non-malignant blood disorders represented an annual healthcare cost of €159 per 10 citizens.
“Non-malignant blood disorders cost the European economy nearly as much as all blood cancers combined,” said Jose Leal, DPhil, of the University of Oxford in the UK.
“We found wide differences in the cost of treating blood disorders in different countries, likely linked to the significant differences in the access and delivery of care for patients with blood disorders. Our findings suggest there is a need to harmonize care of blood disorders across Europe in a cost-effective way.” ![]()
Study may explain how LSCs evade treatment
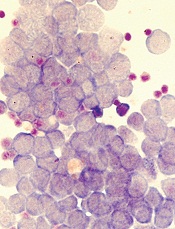
Image by Robert Paulson
New research suggests leukemia stem cells (LSCs) can “hide” in gonadal adipose tissue (GAT) and transform the tissue so they can survive treatment.
Experiments in a mouse model of chronic myeloid leukemia (CML) showed that LSCs are enriched in GAT.
While there, the LSCs create a microenvironment that supports leukemic growth and resistance to treatment, and expression of the fatty acid transporter CD36 makes LSCs particularly resistant.
Craig Jordan, PhD, of University of Colorado in Aurora, and his colleagues conducted this research and detailed their findings in Cell Stem Cell.
The researchers began by examining cancer cells found in GAT from mice with blast crisis CML. Rather than containing the expected mix of regular leukemia cells and LSCs, the tissue was enriched for LSCs.
And these GAT-resident LSCs used a different energy source than LSCs in the bone marrow microenvironment. The GAT-resident LSCs powered their survival and growth with fatty acids, manufacturing energy by the process of fatty acid oxidization.
In fact, the GAT-resident LSCs actively signaled fat to undergo lipolysis, which released fatty acids into the microenvironment.
“The basic biology was fascinating,” Dr Jordan said. “The tumor adapted the local environment to suit itself.”
Dr Jordan and his colleagues also found that CD36 played a role. CD36+ LSCs were enriched in GAT, were more likely to migrate to GAT than to bone marrow, and were protected from treatment by GAT.
The researchers tested the effects of several drugs (cytarabine, doxorubicin, etoposide, SN-38, irinotecan, and dasatinib) on CD36+ LSCs, CD36- LSCs, and bulk leukemia cells ex vivo.
Both CD36+ and CD36- LSCs were more resistant to treatment than bulk leukemia cells, but CD36+ LSCs were preferentially drug-resistant.
The researchers observed similar results in leukemic mice and found evidence to suggest that CD36 plays a similar role in patients with blast crisis CML and those with acute myeloid leukemia. ![]()
Image by Robert Paulson
New research suggests leukemia stem cells (LSCs) can “hide” in gonadal adipose tissue (GAT) and transform the tissue so they can survive treatment.
Experiments in a mouse model of chronic myeloid leukemia (CML) showed that LSCs are enriched in GAT.
While there, the LSCs create a microenvironment that supports leukemic growth and resistance to treatment, and expression of the fatty acid transporter CD36 makes LSCs particularly resistant.
Craig Jordan, PhD, of University of Colorado in Aurora, and his colleagues conducted this research and detailed their findings in Cell Stem Cell.
The researchers began by examining cancer cells found in GAT from mice with blast crisis CML. Rather than containing the expected mix of regular leukemia cells and LSCs, the tissue was enriched for LSCs.
And these GAT-resident LSCs used a different energy source than LSCs in the bone marrow microenvironment. The GAT-resident LSCs powered their survival and growth with fatty acids, manufacturing energy by the process of fatty acid oxidization.
In fact, the GAT-resident LSCs actively signaled fat to undergo lipolysis, which released fatty acids into the microenvironment.
“The basic biology was fascinating,” Dr Jordan said. “The tumor adapted the local environment to suit itself.”
Dr Jordan and his colleagues also found that CD36 played a role. CD36+ LSCs were enriched in GAT, were more likely to migrate to GAT than to bone marrow, and were protected from treatment by GAT.
The researchers tested the effects of several drugs (cytarabine, doxorubicin, etoposide, SN-38, irinotecan, and dasatinib) on CD36+ LSCs, CD36- LSCs, and bulk leukemia cells ex vivo.
Both CD36+ and CD36- LSCs were more resistant to treatment than bulk leukemia cells, but CD36+ LSCs were preferentially drug-resistant.
The researchers observed similar results in leukemic mice and found evidence to suggest that CD36 plays a similar role in patients with blast crisis CML and those with acute myeloid leukemia. ![]()
Image by Robert Paulson
New research suggests leukemia stem cells (LSCs) can “hide” in gonadal adipose tissue (GAT) and transform the tissue so they can survive treatment.
Experiments in a mouse model of chronic myeloid leukemia (CML) showed that LSCs are enriched in GAT.
While there, the LSCs create a microenvironment that supports leukemic growth and resistance to treatment, and expression of the fatty acid transporter CD36 makes LSCs particularly resistant.
Craig Jordan, PhD, of University of Colorado in Aurora, and his colleagues conducted this research and detailed their findings in Cell Stem Cell.
The researchers began by examining cancer cells found in GAT from mice with blast crisis CML. Rather than containing the expected mix of regular leukemia cells and LSCs, the tissue was enriched for LSCs.
And these GAT-resident LSCs used a different energy source than LSCs in the bone marrow microenvironment. The GAT-resident LSCs powered their survival and growth with fatty acids, manufacturing energy by the process of fatty acid oxidization.
In fact, the GAT-resident LSCs actively signaled fat to undergo lipolysis, which released fatty acids into the microenvironment.
“The basic biology was fascinating,” Dr Jordan said. “The tumor adapted the local environment to suit itself.”
Dr Jordan and his colleagues also found that CD36 played a role. CD36+ LSCs were enriched in GAT, were more likely to migrate to GAT than to bone marrow, and were protected from treatment by GAT.
The researchers tested the effects of several drugs (cytarabine, doxorubicin, etoposide, SN-38, irinotecan, and dasatinib) on CD36+ LSCs, CD36- LSCs, and bulk leukemia cells ex vivo.
Both CD36+ and CD36- LSCs were more resistant to treatment than bulk leukemia cells, but CD36+ LSCs were preferentially drug-resistant.
The researchers observed similar results in leukemic mice and found evidence to suggest that CD36 plays a similar role in patients with blast crisis CML and those with acute myeloid leukemia. ![]()