User login
FDA grants drug accelerated approval for CLL

Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has granted accelerated approval for venetoclax (Venclexta) to treat patients with chronic lymphocytic leukemia (CLL) who have 17p deletion and have received at least one prior therapy.
Venetoclax will be available in the US within about a week, according to the companies developing the drug, AbbVie and Genentech (a member of the
Roche Group).
The companies said they plan to offer patient assistance programs for qualifying patients who wish to receive venetoclax.
Venetoclax (formerly ABT-199) is the first FDA-approved treatment that targets the BCL-2 protein, which is overexpressed in many patients with CLL.
The drug is indicated for daily use after 17p deletion is confirmed via the FDA-approved companion diagnostic Vysis CLL FISH probe kit, which is manufactured by Abbott Molecular.
The FDA granted venetoclax accelerated approval rather than traditional approval because the drug has not yet shown a clinical benefit. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of venetoclax for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted venetoclax breakthrough therapy designation, priority review status, and orphan drug designation.
Phase 2 trial
Results from the pivotal phase 2 trial of venetoclax (M13-982, NCT01889186) were presented at the 2015 ASH Annual Meeting. According to those data, the trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
Eighty-five patients responded to treatment, for an overall response rate of 79.4%. Eight patients (7.5%) achieved a complete response or complete response with incomplete count recovery, 3 (2.8%) had a near partial response, and 74 (69.2%) had a partial response. Twenty-two patients (20.6%) did not respond.
As of the ASH presentation, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72.0%, and the overall survival estimate was 86.7%.
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%). (About 22% of patients had neutropenia at baseline.)
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%). Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher infections.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Laboratory tumor lysis syndrome (TLS) occurred in 5 patients during the ramp-up period only. Two patients required a dose interruption of 1 day each. There were no clinical TLS events.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS.
Venetoclax is currently being evaluated in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL. ![]()
Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has granted accelerated approval for venetoclax (Venclexta) to treat patients with chronic lymphocytic leukemia (CLL) who have 17p deletion and have received at least one prior therapy.
Venetoclax will be available in the US within about a week, according to the companies developing the drug, AbbVie and Genentech (a member of the
Roche Group).
The companies said they plan to offer patient assistance programs for qualifying patients who wish to receive venetoclax.
Venetoclax (formerly ABT-199) is the first FDA-approved treatment that targets the BCL-2 protein, which is overexpressed in many patients with CLL.
The drug is indicated for daily use after 17p deletion is confirmed via the FDA-approved companion diagnostic Vysis CLL FISH probe kit, which is manufactured by Abbott Molecular.
The FDA granted venetoclax accelerated approval rather than traditional approval because the drug has not yet shown a clinical benefit. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of venetoclax for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted venetoclax breakthrough therapy designation, priority review status, and orphan drug designation.
Phase 2 trial
Results from the pivotal phase 2 trial of venetoclax (M13-982, NCT01889186) were presented at the 2015 ASH Annual Meeting. According to those data, the trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
Eighty-five patients responded to treatment, for an overall response rate of 79.4%. Eight patients (7.5%) achieved a complete response or complete response with incomplete count recovery, 3 (2.8%) had a near partial response, and 74 (69.2%) had a partial response. Twenty-two patients (20.6%) did not respond.
As of the ASH presentation, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72.0%, and the overall survival estimate was 86.7%.
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%). (About 22% of patients had neutropenia at baseline.)
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%). Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher infections.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Laboratory tumor lysis syndrome (TLS) occurred in 5 patients during the ramp-up period only. Two patients required a dose interruption of 1 day each. There were no clinical TLS events.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS.
Venetoclax is currently being evaluated in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL. ![]()
Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has granted accelerated approval for venetoclax (Venclexta) to treat patients with chronic lymphocytic leukemia (CLL) who have 17p deletion and have received at least one prior therapy.
Venetoclax will be available in the US within about a week, according to the companies developing the drug, AbbVie and Genentech (a member of the
Roche Group).
The companies said they plan to offer patient assistance programs for qualifying patients who wish to receive venetoclax.
Venetoclax (formerly ABT-199) is the first FDA-approved treatment that targets the BCL-2 protein, which is overexpressed in many patients with CLL.
The drug is indicated for daily use after 17p deletion is confirmed via the FDA-approved companion diagnostic Vysis CLL FISH probe kit, which is manufactured by Abbott Molecular.
The FDA granted venetoclax accelerated approval rather than traditional approval because the drug has not yet shown a clinical benefit. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of venetoclax for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted venetoclax breakthrough therapy designation, priority review status, and orphan drug designation.
Phase 2 trial
Results from the pivotal phase 2 trial of venetoclax (M13-982, NCT01889186) were presented at the 2015 ASH Annual Meeting. According to those data, the trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
Eighty-five patients responded to treatment, for an overall response rate of 79.4%. Eight patients (7.5%) achieved a complete response or complete response with incomplete count recovery, 3 (2.8%) had a near partial response, and 74 (69.2%) had a partial response. Twenty-two patients (20.6%) did not respond.
As of the ASH presentation, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72.0%, and the overall survival estimate was 86.7%.
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%). (About 22% of patients had neutropenia at baseline.)
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%). Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher infections.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Laboratory tumor lysis syndrome (TLS) occurred in 5 patients during the ramp-up period only. Two patients required a dose interruption of 1 day each. There were no clinical TLS events.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS.
Venetoclax is currently being evaluated in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL. ![]()
Team creates public repository of xenografts

Photo by Rhoda Baer
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project. ![]()
Photo by Rhoda Baer
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project. ![]()
Photo by Rhoda Baer
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project. ![]()
FDA approves venetoclax for CLL with 17p deletion
Venetoclax has been approved for the treatment of patients with chronic lymphocytic leukemia (CLL) who have a 17p deletion and who have been treated with a least one prior therapy, the Food and Drug Administration has announced.
The drug will be marketed as Venclexta, and is indicated for daily use after detection of a 17p deletion is confirmed through the use of the FDA-approved companion diagnostic test, the Vysis CLL FISH probe kit. A 17p deletion occurs in about 10% of patients with untreated CLL and in about 20% of patients with relapsed CLL. Venetoclax targets the B-cell lymphoma 2 (BCL-2) protein, according to the FDA press release.
“Up to half of people whose CLL progressed have 17p deletion,” Dr. Sandra Horning, chief medical officer and head of Global Product Development for Genentech, said in a press release issued by the company. Venclexta will be marketed by AbbVie and Genentech USA. The Vysis CLL FISH probe kit is manufactured by Abbott Molecular.
“For certain patients with CLL who have not had favorable outcomes with other therapies, Venclexta may provide a new option,” Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a press release issued by the FDA.
The approval was based on a clinical trial of 106 patients who had CLL and 17p deletions and who had received at least one prior therapy. Trial participants took oral venetoclax daily, beginning with a 20 mg dose that was increased over a 5-week period to 400 mg. A complete or partial remission of CLL occurred in 80% of trial participants. Data on venetoclax also was presented at the annual meeting of the American Society of Hematology.
The most common side effects of venetoclax include neutropenia, diarrhea, nausea, anemia, upper respiratory tract infection, thrombocytopenia, and fatigue.
The FDA granted the Venclexta application breakthrough therapy designation, priority review status, and accelerated approval for this indication. Venclexta also received orphan drug designation.
On Twitter @maryjodales
Venetoclax has been approved for the treatment of patients with chronic lymphocytic leukemia (CLL) who have a 17p deletion and who have been treated with a least one prior therapy, the Food and Drug Administration has announced.
The drug will be marketed as Venclexta, and is indicated for daily use after detection of a 17p deletion is confirmed through the use of the FDA-approved companion diagnostic test, the Vysis CLL FISH probe kit. A 17p deletion occurs in about 10% of patients with untreated CLL and in about 20% of patients with relapsed CLL. Venetoclax targets the B-cell lymphoma 2 (BCL-2) protein, according to the FDA press release.
“Up to half of people whose CLL progressed have 17p deletion,” Dr. Sandra Horning, chief medical officer and head of Global Product Development for Genentech, said in a press release issued by the company. Venclexta will be marketed by AbbVie and Genentech USA. The Vysis CLL FISH probe kit is manufactured by Abbott Molecular.
“For certain patients with CLL who have not had favorable outcomes with other therapies, Venclexta may provide a new option,” Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a press release issued by the FDA.
The approval was based on a clinical trial of 106 patients who had CLL and 17p deletions and who had received at least one prior therapy. Trial participants took oral venetoclax daily, beginning with a 20 mg dose that was increased over a 5-week period to 400 mg. A complete or partial remission of CLL occurred in 80% of trial participants. Data on venetoclax also was presented at the annual meeting of the American Society of Hematology.
The most common side effects of venetoclax include neutropenia, diarrhea, nausea, anemia, upper respiratory tract infection, thrombocytopenia, and fatigue.
The FDA granted the Venclexta application breakthrough therapy designation, priority review status, and accelerated approval for this indication. Venclexta also received orphan drug designation.
On Twitter @maryjodales
Venetoclax has been approved for the treatment of patients with chronic lymphocytic leukemia (CLL) who have a 17p deletion and who have been treated with a least one prior therapy, the Food and Drug Administration has announced.
The drug will be marketed as Venclexta, and is indicated for daily use after detection of a 17p deletion is confirmed through the use of the FDA-approved companion diagnostic test, the Vysis CLL FISH probe kit. A 17p deletion occurs in about 10% of patients with untreated CLL and in about 20% of patients with relapsed CLL. Venetoclax targets the B-cell lymphoma 2 (BCL-2) protein, according to the FDA press release.
“Up to half of people whose CLL progressed have 17p deletion,” Dr. Sandra Horning, chief medical officer and head of Global Product Development for Genentech, said in a press release issued by the company. Venclexta will be marketed by AbbVie and Genentech USA. The Vysis CLL FISH probe kit is manufactured by Abbott Molecular.
“For certain patients with CLL who have not had favorable outcomes with other therapies, Venclexta may provide a new option,” Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a press release issued by the FDA.
The approval was based on a clinical trial of 106 patients who had CLL and 17p deletions and who had received at least one prior therapy. Trial participants took oral venetoclax daily, beginning with a 20 mg dose that was increased over a 5-week period to 400 mg. A complete or partial remission of CLL occurred in 80% of trial participants. Data on venetoclax also was presented at the annual meeting of the American Society of Hematology.
The most common side effects of venetoclax include neutropenia, diarrhea, nausea, anemia, upper respiratory tract infection, thrombocytopenia, and fatigue.
The FDA granted the Venclexta application breakthrough therapy designation, priority review status, and accelerated approval for this indication. Venclexta also received orphan drug designation.
On Twitter @maryjodales
In myelodysplastic syndrome, improved tool for predicting death after HCT
A new risk-stratification tool goes one better than the standard tools used to predict survival in those undergoing allogeneic hematopoietic cell transplantation (allo HCT) for myelodysplastic syndrome, based on a study published online April 4 in the Journal of Clinical Oncology.
The concordance index for the new risk-stratification tool was modestly better at 0.575, compared with 0.538 for the standard International Prognostic Scoring System (IPSS) and 0.554 for the revised IPSS (IPSS-R), according to Dr. Brian C. Shaffer of Memorial Sloan Kettering Cancer Center, New York, and his colleagues who participate in the Center for International Blood and Marrow Transplant Research (CIBMTR) network.

“The proposed system generally agrees with the IPSS-R in the very high–risk subcategory; however, a significant portion of patients in high- and very high–risk IPSS-R groups were represented in the low- and intermediate-risk proposed scoring subcategories. The 3-year survival in patients classified as high risk with the IPSS-R was 75%; it was 57% in those classified as low or intermediate risk with the proposed system,” the researchers wrote.
Further, the “scoring system uses readily available clinical data and can be calculated quickly, facilitating patient consultation with respect to allo HCT, and may also be used to identify high-risk populations where interventions such as post–allo HCT maintenance therapies may be of benefit,” they wrote (J Clin Oncol. 2016 April 4. doi: 10.1200/JCO.2015.65.0515).
The data were obtained from the CIBMTR, a combined research program of the Medical College of Wisconsin and the National Marrow Donor Program. The CIBMTR comprises a voluntary network of more than 450 transplantation centers worldwide that contribute data on consecutive allo and autologous HCTs to a centralized statistical center.
The researchers applied the prognostic tool to 2,133 patients with MDS undergoing HLA-matched (n = 1,728) or -mismatched (n = 405) allo HCT. Factors prognostic of mortality were identified in a training subset (n = 1,151) of the HLA-matched cohort. A weighted score using these factors was then assigned to the validation cohort of 577 remaining patients undergoing HLA-matched allo HCT as well as to patients undergoing HLA-mismatched allo HCT. The training data set was used to develop a prognostic scoring system, and the validation data set was used to assess the prognostic ability of the scoring system, the researchers noted.
In the scoring system, 1 point was assigned for the following factors: Blood blasts greater than 3%, platelet levels of 50 × 109/L or less at transplantation, Karnofsky performance status less than 90%, comprehensive cytogenetic risk score of poor or very poor, and age 30-49 years. Two points were assigned for monosomal karyotype and age 50 years or older.
Based on the scoring system, 3-year overall survival after transplantation was 71% in patients with scores of 1 point, 49% with scores of 2-3, 41% with scores of 4-5, and 25% with scores of 6 or more. Increasing score was predictive of increased relapse and treatment-related mortality in the HLA-matched set and of relapse in the HLA-mismatched cohort.
To develop the scoring system, the researchers used a model that weighed patient age; sex; and Karnofsky performance status; disease stage at transplantation; comprehensive cytogenetic risk status; bone marrow and peripheral blood blast percentages; hemoglobin, neutrophil, and platelet counts at diagnosis and pretransplantation; lactate dehydrogenase at transplantation; pretransplantation therapy (hypomethylating agents, chemotherapy, neither, or both); time from diagnosis to transplantation; year of transplantation; conditioning regimen and regimen intensity (myeloablative v reduced intensity); donor–recipient sex match or mismatch; graft-versus-host disease prophylaxis; graft type (bone marrow vs. peripheral blood); presence of secondary myelodysplastic syndrome; and unrelated donor vs. related donor.
There were no significant differences in overall survival at 1, 3, and 5 years or in the 3-year incidences of relapse and treatment-related mortality in the training subset and the validation cohort.
Data on somatic mutations have become relevant in myelodysplastic syndrome prognostication and were missing from this analysis, the researchers wrote. “The next generation of prognostic tools will need to account for this information.”
Dr. Shaffer had no relevant financial disclosures.
On Twitter @maryjodales
A new risk-stratification tool goes one better than the standard tools used to predict survival in those undergoing allogeneic hematopoietic cell transplantation (allo HCT) for myelodysplastic syndrome, based on a study published online April 4 in the Journal of Clinical Oncology.
The concordance index for the new risk-stratification tool was modestly better at 0.575, compared with 0.538 for the standard International Prognostic Scoring System (IPSS) and 0.554 for the revised IPSS (IPSS-R), according to Dr. Brian C. Shaffer of Memorial Sloan Kettering Cancer Center, New York, and his colleagues who participate in the Center for International Blood and Marrow Transplant Research (CIBMTR) network.
“The proposed system generally agrees with the IPSS-R in the very high–risk subcategory; however, a significant portion of patients in high- and very high–risk IPSS-R groups were represented in the low- and intermediate-risk proposed scoring subcategories. The 3-year survival in patients classified as high risk with the IPSS-R was 75%; it was 57% in those classified as low or intermediate risk with the proposed system,” the researchers wrote.
Further, the “scoring system uses readily available clinical data and can be calculated quickly, facilitating patient consultation with respect to allo HCT, and may also be used to identify high-risk populations where interventions such as post–allo HCT maintenance therapies may be of benefit,” they wrote (J Clin Oncol. 2016 April 4. doi: 10.1200/JCO.2015.65.0515).
The data were obtained from the CIBMTR, a combined research program of the Medical College of Wisconsin and the National Marrow Donor Program. The CIBMTR comprises a voluntary network of more than 450 transplantation centers worldwide that contribute data on consecutive allo and autologous HCTs to a centralized statistical center.
The researchers applied the prognostic tool to 2,133 patients with MDS undergoing HLA-matched (n = 1,728) or -mismatched (n = 405) allo HCT. Factors prognostic of mortality were identified in a training subset (n = 1,151) of the HLA-matched cohort. A weighted score using these factors was then assigned to the validation cohort of 577 remaining patients undergoing HLA-matched allo HCT as well as to patients undergoing HLA-mismatched allo HCT. The training data set was used to develop a prognostic scoring system, and the validation data set was used to assess the prognostic ability of the scoring system, the researchers noted.
In the scoring system, 1 point was assigned for the following factors: Blood blasts greater than 3%, platelet levels of 50 × 109/L or less at transplantation, Karnofsky performance status less than 90%, comprehensive cytogenetic risk score of poor or very poor, and age 30-49 years. Two points were assigned for monosomal karyotype and age 50 years or older.
Based on the scoring system, 3-year overall survival after transplantation was 71% in patients with scores of 1 point, 49% with scores of 2-3, 41% with scores of 4-5, and 25% with scores of 6 or more. Increasing score was predictive of increased relapse and treatment-related mortality in the HLA-matched set and of relapse in the HLA-mismatched cohort.
To develop the scoring system, the researchers used a model that weighed patient age; sex; and Karnofsky performance status; disease stage at transplantation; comprehensive cytogenetic risk status; bone marrow and peripheral blood blast percentages; hemoglobin, neutrophil, and platelet counts at diagnosis and pretransplantation; lactate dehydrogenase at transplantation; pretransplantation therapy (hypomethylating agents, chemotherapy, neither, or both); time from diagnosis to transplantation; year of transplantation; conditioning regimen and regimen intensity (myeloablative v reduced intensity); donor–recipient sex match or mismatch; graft-versus-host disease prophylaxis; graft type (bone marrow vs. peripheral blood); presence of secondary myelodysplastic syndrome; and unrelated donor vs. related donor.
There were no significant differences in overall survival at 1, 3, and 5 years or in the 3-year incidences of relapse and treatment-related mortality in the training subset and the validation cohort.
Data on somatic mutations have become relevant in myelodysplastic syndrome prognostication and were missing from this analysis, the researchers wrote. “The next generation of prognostic tools will need to account for this information.”
Dr. Shaffer had no relevant financial disclosures.
On Twitter @maryjodales
A new risk-stratification tool goes one better than the standard tools used to predict survival in those undergoing allogeneic hematopoietic cell transplantation (allo HCT) for myelodysplastic syndrome, based on a study published online April 4 in the Journal of Clinical Oncology.
The concordance index for the new risk-stratification tool was modestly better at 0.575, compared with 0.538 for the standard International Prognostic Scoring System (IPSS) and 0.554 for the revised IPSS (IPSS-R), according to Dr. Brian C. Shaffer of Memorial Sloan Kettering Cancer Center, New York, and his colleagues who participate in the Center for International Blood and Marrow Transplant Research (CIBMTR) network.
“The proposed system generally agrees with the IPSS-R in the very high–risk subcategory; however, a significant portion of patients in high- and very high–risk IPSS-R groups were represented in the low- and intermediate-risk proposed scoring subcategories. The 3-year survival in patients classified as high risk with the IPSS-R was 75%; it was 57% in those classified as low or intermediate risk with the proposed system,” the researchers wrote.
Further, the “scoring system uses readily available clinical data and can be calculated quickly, facilitating patient consultation with respect to allo HCT, and may also be used to identify high-risk populations where interventions such as post–allo HCT maintenance therapies may be of benefit,” they wrote (J Clin Oncol. 2016 April 4. doi: 10.1200/JCO.2015.65.0515).
The data were obtained from the CIBMTR, a combined research program of the Medical College of Wisconsin and the National Marrow Donor Program. The CIBMTR comprises a voluntary network of more than 450 transplantation centers worldwide that contribute data on consecutive allo and autologous HCTs to a centralized statistical center.
The researchers applied the prognostic tool to 2,133 patients with MDS undergoing HLA-matched (n = 1,728) or -mismatched (n = 405) allo HCT. Factors prognostic of mortality were identified in a training subset (n = 1,151) of the HLA-matched cohort. A weighted score using these factors was then assigned to the validation cohort of 577 remaining patients undergoing HLA-matched allo HCT as well as to patients undergoing HLA-mismatched allo HCT. The training data set was used to develop a prognostic scoring system, and the validation data set was used to assess the prognostic ability of the scoring system, the researchers noted.
In the scoring system, 1 point was assigned for the following factors: Blood blasts greater than 3%, platelet levels of 50 × 109/L or less at transplantation, Karnofsky performance status less than 90%, comprehensive cytogenetic risk score of poor or very poor, and age 30-49 years. Two points were assigned for monosomal karyotype and age 50 years or older.
Based on the scoring system, 3-year overall survival after transplantation was 71% in patients with scores of 1 point, 49% with scores of 2-3, 41% with scores of 4-5, and 25% with scores of 6 or more. Increasing score was predictive of increased relapse and treatment-related mortality in the HLA-matched set and of relapse in the HLA-mismatched cohort.
To develop the scoring system, the researchers used a model that weighed patient age; sex; and Karnofsky performance status; disease stage at transplantation; comprehensive cytogenetic risk status; bone marrow and peripheral blood blast percentages; hemoglobin, neutrophil, and platelet counts at diagnosis and pretransplantation; lactate dehydrogenase at transplantation; pretransplantation therapy (hypomethylating agents, chemotherapy, neither, or both); time from diagnosis to transplantation; year of transplantation; conditioning regimen and regimen intensity (myeloablative v reduced intensity); donor–recipient sex match or mismatch; graft-versus-host disease prophylaxis; graft type (bone marrow vs. peripheral blood); presence of secondary myelodysplastic syndrome; and unrelated donor vs. related donor.
There were no significant differences in overall survival at 1, 3, and 5 years or in the 3-year incidences of relapse and treatment-related mortality in the training subset and the validation cohort.
Data on somatic mutations have become relevant in myelodysplastic syndrome prognostication and were missing from this analysis, the researchers wrote. “The next generation of prognostic tools will need to account for this information.”
Dr. Shaffer had no relevant financial disclosures.
On Twitter @maryjodales
FROM JCO
Key clinical point: A portion of patients with myelodysplastic syndrome in high- and very high–risk groups of the revised International Prognostic Scoring System (IPSS-R) were represented in the low- and intermediate-risk groups of the proposed scoring subcategories.
Major finding: The 3-year survival in patients classified as high risk with the IPSS-R was 75%; it was 57% in those classified as low or intermediate risk with the proposed system.
Data source: The Center for International Blood and Marrow Transplant Research (CIBMTR), a combined research program of the Medical College of Wisconsin and the National Marrow Donor Program. The CIBMTR comprises a voluntary network of more than 450 transplantation centers worldwide that contribute data on consecutive allo and autologous HCTs to a centralized statistical center.
Disclosures: Dr. Shaffer had no relevant financial disclosures.
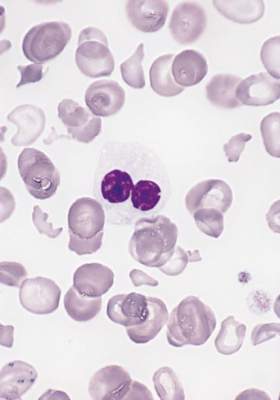
Combo appears effective against B-ALL

Combining a MEK inhibitor and a BCL-2/BCL-XL inhibitor may be a feasible treatment option for B-cell acute lymphoblastic leukemia (B-ALL), according to preclinical research published in Cell Death and Disease.
Researchers found that, when given alone, the MEK inhibitor trametinib did not block B-ALL cell growth.
And the BCL-2/BCL-XL inhibitors navitoclax (ABT-263) and venetoclax (ABT-199) did not prove particularly effective either.
However, combining trametinib with navitoclax or venetoclax successfully induced apoptosis in B-ALL cells.
“Cancer cells often outwit us by rewiring themselves, but this early research offers a promising idea to get ahead of them,” said study author Richard Marais, PhD, of the Cancer Research UK Manchester Institute.
“We’ll still need to do further research to prove that this is the case beyond cancer cells in the laboratory, and it may take many years before we see it in the clinic, but it’s the first step to finding a new, effective drug combination for B-cell acute lymphoblastic leukemia.”
Dr Marais and his colleagues found that, although the MEK/ERK pathway is activated in B-ALL cells driven by different oncogenes, MEK inhibition alone did not suppress B-ALL cell growth.
And although B-ALL cells were sensitive to treatment with navitoclax or venetoclax alone, the researchers did not see complete loss of cell viability at clinically achievable doses.
However, trametinib did synergize with either navitoclax or venetoclax to suppress proliferation and induce apoptosis in B-ALL cells.
Further investigation revealed that the resistance of B-ALL cells to BCL-2/BCL-XL inhibition is mediated by MCL-1. And the synergism between trametinib and navitoclax/venetoclax is mediated by the pro-apoptotic factor BIM.
BIM is dephosphorylated as a result of MEK inhibition, which allows it to bind to and neutralize MCL-1, thereby enhancing BCL-2/BCL-XL inhibitor-induced cell death.
The researchers said they observed this effect in B-ALL cells driven by a range of genetic abnormalities, so the combination of a MEK inhibitor and a BCL-2/BCL-XL inhibitor could have therapeutic potential in a range of B-ALL subtypes. ![]()
Combining a MEK inhibitor and a BCL-2/BCL-XL inhibitor may be a feasible treatment option for B-cell acute lymphoblastic leukemia (B-ALL), according to preclinical research published in Cell Death and Disease.
Researchers found that, when given alone, the MEK inhibitor trametinib did not block B-ALL cell growth.
And the BCL-2/BCL-XL inhibitors navitoclax (ABT-263) and venetoclax (ABT-199) did not prove particularly effective either.
However, combining trametinib with navitoclax or venetoclax successfully induced apoptosis in B-ALL cells.
“Cancer cells often outwit us by rewiring themselves, but this early research offers a promising idea to get ahead of them,” said study author Richard Marais, PhD, of the Cancer Research UK Manchester Institute.
“We’ll still need to do further research to prove that this is the case beyond cancer cells in the laboratory, and it may take many years before we see it in the clinic, but it’s the first step to finding a new, effective drug combination for B-cell acute lymphoblastic leukemia.”
Dr Marais and his colleagues found that, although the MEK/ERK pathway is activated in B-ALL cells driven by different oncogenes, MEK inhibition alone did not suppress B-ALL cell growth.
And although B-ALL cells were sensitive to treatment with navitoclax or venetoclax alone, the researchers did not see complete loss of cell viability at clinically achievable doses.
However, trametinib did synergize with either navitoclax or venetoclax to suppress proliferation and induce apoptosis in B-ALL cells.
Further investigation revealed that the resistance of B-ALL cells to BCL-2/BCL-XL inhibition is mediated by MCL-1. And the synergism between trametinib and navitoclax/venetoclax is mediated by the pro-apoptotic factor BIM.
BIM is dephosphorylated as a result of MEK inhibition, which allows it to bind to and neutralize MCL-1, thereby enhancing BCL-2/BCL-XL inhibitor-induced cell death.
The researchers said they observed this effect in B-ALL cells driven by a range of genetic abnormalities, so the combination of a MEK inhibitor and a BCL-2/BCL-XL inhibitor could have therapeutic potential in a range of B-ALL subtypes. ![]()
Combining a MEK inhibitor and a BCL-2/BCL-XL inhibitor may be a feasible treatment option for B-cell acute lymphoblastic leukemia (B-ALL), according to preclinical research published in Cell Death and Disease.
Researchers found that, when given alone, the MEK inhibitor trametinib did not block B-ALL cell growth.
And the BCL-2/BCL-XL inhibitors navitoclax (ABT-263) and venetoclax (ABT-199) did not prove particularly effective either.
However, combining trametinib with navitoclax or venetoclax successfully induced apoptosis in B-ALL cells.
“Cancer cells often outwit us by rewiring themselves, but this early research offers a promising idea to get ahead of them,” said study author Richard Marais, PhD, of the Cancer Research UK Manchester Institute.
“We’ll still need to do further research to prove that this is the case beyond cancer cells in the laboratory, and it may take many years before we see it in the clinic, but it’s the first step to finding a new, effective drug combination for B-cell acute lymphoblastic leukemia.”
Dr Marais and his colleagues found that, although the MEK/ERK pathway is activated in B-ALL cells driven by different oncogenes, MEK inhibition alone did not suppress B-ALL cell growth.
And although B-ALL cells were sensitive to treatment with navitoclax or venetoclax alone, the researchers did not see complete loss of cell viability at clinically achievable doses.
However, trametinib did synergize with either navitoclax or venetoclax to suppress proliferation and induce apoptosis in B-ALL cells.
Further investigation revealed that the resistance of B-ALL cells to BCL-2/BCL-XL inhibition is mediated by MCL-1. And the synergism between trametinib and navitoclax/venetoclax is mediated by the pro-apoptotic factor BIM.
BIM is dephosphorylated as a result of MEK inhibition, which allows it to bind to and neutralize MCL-1, thereby enhancing BCL-2/BCL-XL inhibitor-induced cell death.
The researchers said they observed this effect in B-ALL cells driven by a range of genetic abnormalities, so the combination of a MEK inhibitor and a BCL-2/BCL-XL inhibitor could have therapeutic potential in a range of B-ALL subtypes. ![]()
Why married cancer patients fare better

Photo by Alena Kratochvilova
Results from two new studies provide a possible explanation for the link between marital status and survival in cancer patients.
Previous studies have shown that married cancer patients are more likely to survive and tend to have longer survival times than unmarried cancer patients.
Now, a pair of studies published in Cancer suggest it is the social support a patient receives from a spouse that may improve the patient’s outcome.
In the first study, Scarlett Lin Gomez, PhD, of the Cancer Prevention Institute of California, and her colleagues assessed the impact of socioeconomic factors and marital status on survival in cancer patients.
The team found evidence to suggest that economic resources play a minimal role in explaining the inferior survival observed in unmarried cancer patients.
In the second study, María Elena Martínez, PhD, of UC San Diego Moores Cancer Center, and her colleagues assessed the roles that race/ethnicity, sex, and nativity play in the survival of married and unmarried cancer patients.
The group found that not being married was associated with higher mortality, but the association varied by race/ethnicity and sex. The researchers believe these differences can be explained by the differences in social support networks between racial/ethnic groups and between men and women.
Patient cohort
Both studies were conducted on the same cohort of patients from the California Cancer Registry.
The researchers studied 783,167 cancer patients—393,470 males and 389,697 females. They were diagnosed from 2000 through 2009 with a first primary, invasive cancer of the 10 most common sites of cancer-related death for each sex, which included leukemias and lymphomas.
The patients were followed through December 31, 2012. A total of 386,607 patients died from any cause—204,007 males and 182,600 females.
Economic factors
Dr Gomez and her colleagues evaluated health insurance status and neighborhood socioeconomic status for the nearly 800,000 patients.
The researchers found that unmarried cancer patients had a greater risk of death than married patients, and this risk was higher among males than females. The hazard ratio (HR) for males was 1.27 (95% CI, 1.26-1.29), and the HR for females was 1.19 (95% CI, 1.18-1.20, P-interaction <0.001).
When the researchers adjusted for insurance status and neighborhood socioeconomic status, the marital status HRs decreased to 1.22 (95% CI, 1.21–1.24) for males and 1.15 (95% CI, 1.14–1.16) for females.
Based on these results, the researchers concluded that the survival benefit of marriage operates independently of the economic resources evaluated in this study.
“While other studies have found similar protective effects associated with being married, ours is the first in a large, population-based setting to assess the extent to which economic resources explain these protective effects,” Dr Gomez said. “Our study provides evidence for social support as a key driver.”
Race/ethnicity, nativity, and sex
Dr Martínez and her colleagues found that all-cause mortality was higher in the unmarried patients than in the married patients, but this varied significantly according to race/ethnicity and sex.
Marriage conferred less of a survival benefit for women than for men. However, for both sexes, non-Hispanic whites benefitted the most from being married, and Hispanics and Asian Pacific Islanders benefitted less.
Among males, the adjusted HRs were 1.24 (95% CI, 1.23-1.26) in non-Hispanic whites, 1.20 (95% CI, 1.16-1.24) in blacks, 1.20 (95% CI, 1.17-1.23) in Hispanics, and 1.11 (95% CI, 1.07-1.15) in Asian Pacific Islanders.
In females, the adjusted HRs were 1.17 (95% CI, 1.15-1.18) in non-Hispanic whites, 1.09 (95% CI, 1.05-1.13) in blacks, 1.11 (95% CI, 1.08-1.14) in Hispanics, and 1.07 (95% CI, 1.04-1.11) in Asian Pacific Islanders.
The researchers also found that all-cause mortality associated with unmarried status was higher in US-born Asian Pacific Islander and Hispanic men and women relative to their foreign-born counterparts.
“The results suggest that the more acculturated you become to US culture, the more it impacts cancer survivorship,” Dr Martínez said. “Our hypothesis is that non-Hispanic whites don’t have the same social network as other cultures that have stronger bonds with family and friends outside of marriage.”
“As individuals acculturate, they tend to lose those bonds. It’s also been shown that women seek out help for health concerns more frequently than men, and women tend to remind spouses to see their physicians and live a healthy lifestyle.” ![]()
Photo by Alena Kratochvilova
Results from two new studies provide a possible explanation for the link between marital status and survival in cancer patients.
Previous studies have shown that married cancer patients are more likely to survive and tend to have longer survival times than unmarried cancer patients.
Now, a pair of studies published in Cancer suggest it is the social support a patient receives from a spouse that may improve the patient’s outcome.
In the first study, Scarlett Lin Gomez, PhD, of the Cancer Prevention Institute of California, and her colleagues assessed the impact of socioeconomic factors and marital status on survival in cancer patients.
The team found evidence to suggest that economic resources play a minimal role in explaining the inferior survival observed in unmarried cancer patients.
In the second study, María Elena Martínez, PhD, of UC San Diego Moores Cancer Center, and her colleagues assessed the roles that race/ethnicity, sex, and nativity play in the survival of married and unmarried cancer patients.
The group found that not being married was associated with higher mortality, but the association varied by race/ethnicity and sex. The researchers believe these differences can be explained by the differences in social support networks between racial/ethnic groups and between men and women.
Patient cohort
Both studies were conducted on the same cohort of patients from the California Cancer Registry.
The researchers studied 783,167 cancer patients—393,470 males and 389,697 females. They were diagnosed from 2000 through 2009 with a first primary, invasive cancer of the 10 most common sites of cancer-related death for each sex, which included leukemias and lymphomas.
The patients were followed through December 31, 2012. A total of 386,607 patients died from any cause—204,007 males and 182,600 females.
Economic factors
Dr Gomez and her colleagues evaluated health insurance status and neighborhood socioeconomic status for the nearly 800,000 patients.
The researchers found that unmarried cancer patients had a greater risk of death than married patients, and this risk was higher among males than females. The hazard ratio (HR) for males was 1.27 (95% CI, 1.26-1.29), and the HR for females was 1.19 (95% CI, 1.18-1.20, P-interaction <0.001).
When the researchers adjusted for insurance status and neighborhood socioeconomic status, the marital status HRs decreased to 1.22 (95% CI, 1.21–1.24) for males and 1.15 (95% CI, 1.14–1.16) for females.
Based on these results, the researchers concluded that the survival benefit of marriage operates independently of the economic resources evaluated in this study.
“While other studies have found similar protective effects associated with being married, ours is the first in a large, population-based setting to assess the extent to which economic resources explain these protective effects,” Dr Gomez said. “Our study provides evidence for social support as a key driver.”
Race/ethnicity, nativity, and sex
Dr Martínez and her colleagues found that all-cause mortality was higher in the unmarried patients than in the married patients, but this varied significantly according to race/ethnicity and sex.
Marriage conferred less of a survival benefit for women than for men. However, for both sexes, non-Hispanic whites benefitted the most from being married, and Hispanics and Asian Pacific Islanders benefitted less.
Among males, the adjusted HRs were 1.24 (95% CI, 1.23-1.26) in non-Hispanic whites, 1.20 (95% CI, 1.16-1.24) in blacks, 1.20 (95% CI, 1.17-1.23) in Hispanics, and 1.11 (95% CI, 1.07-1.15) in Asian Pacific Islanders.
In females, the adjusted HRs were 1.17 (95% CI, 1.15-1.18) in non-Hispanic whites, 1.09 (95% CI, 1.05-1.13) in blacks, 1.11 (95% CI, 1.08-1.14) in Hispanics, and 1.07 (95% CI, 1.04-1.11) in Asian Pacific Islanders.
The researchers also found that all-cause mortality associated with unmarried status was higher in US-born Asian Pacific Islander and Hispanic men and women relative to their foreign-born counterparts.
“The results suggest that the more acculturated you become to US culture, the more it impacts cancer survivorship,” Dr Martínez said. “Our hypothesis is that non-Hispanic whites don’t have the same social network as other cultures that have stronger bonds with family and friends outside of marriage.”
“As individuals acculturate, they tend to lose those bonds. It’s also been shown that women seek out help for health concerns more frequently than men, and women tend to remind spouses to see their physicians and live a healthy lifestyle.” ![]()
Photo by Alena Kratochvilova
Results from two new studies provide a possible explanation for the link between marital status and survival in cancer patients.
Previous studies have shown that married cancer patients are more likely to survive and tend to have longer survival times than unmarried cancer patients.
Now, a pair of studies published in Cancer suggest it is the social support a patient receives from a spouse that may improve the patient’s outcome.
In the first study, Scarlett Lin Gomez, PhD, of the Cancer Prevention Institute of California, and her colleagues assessed the impact of socioeconomic factors and marital status on survival in cancer patients.
The team found evidence to suggest that economic resources play a minimal role in explaining the inferior survival observed in unmarried cancer patients.
In the second study, María Elena Martínez, PhD, of UC San Diego Moores Cancer Center, and her colleagues assessed the roles that race/ethnicity, sex, and nativity play in the survival of married and unmarried cancer patients.
The group found that not being married was associated with higher mortality, but the association varied by race/ethnicity and sex. The researchers believe these differences can be explained by the differences in social support networks between racial/ethnic groups and between men and women.
Patient cohort
Both studies were conducted on the same cohort of patients from the California Cancer Registry.
The researchers studied 783,167 cancer patients—393,470 males and 389,697 females. They were diagnosed from 2000 through 2009 with a first primary, invasive cancer of the 10 most common sites of cancer-related death for each sex, which included leukemias and lymphomas.
The patients were followed through December 31, 2012. A total of 386,607 patients died from any cause—204,007 males and 182,600 females.
Economic factors
Dr Gomez and her colleagues evaluated health insurance status and neighborhood socioeconomic status for the nearly 800,000 patients.
The researchers found that unmarried cancer patients had a greater risk of death than married patients, and this risk was higher among males than females. The hazard ratio (HR) for males was 1.27 (95% CI, 1.26-1.29), and the HR for females was 1.19 (95% CI, 1.18-1.20, P-interaction <0.001).
When the researchers adjusted for insurance status and neighborhood socioeconomic status, the marital status HRs decreased to 1.22 (95% CI, 1.21–1.24) for males and 1.15 (95% CI, 1.14–1.16) for females.
Based on these results, the researchers concluded that the survival benefit of marriage operates independently of the economic resources evaluated in this study.
“While other studies have found similar protective effects associated with being married, ours is the first in a large, population-based setting to assess the extent to which economic resources explain these protective effects,” Dr Gomez said. “Our study provides evidence for social support as a key driver.”
Race/ethnicity, nativity, and sex
Dr Martínez and her colleagues found that all-cause mortality was higher in the unmarried patients than in the married patients, but this varied significantly according to race/ethnicity and sex.
Marriage conferred less of a survival benefit for women than for men. However, for both sexes, non-Hispanic whites benefitted the most from being married, and Hispanics and Asian Pacific Islanders benefitted less.
Among males, the adjusted HRs were 1.24 (95% CI, 1.23-1.26) in non-Hispanic whites, 1.20 (95% CI, 1.16-1.24) in blacks, 1.20 (95% CI, 1.17-1.23) in Hispanics, and 1.11 (95% CI, 1.07-1.15) in Asian Pacific Islanders.
In females, the adjusted HRs were 1.17 (95% CI, 1.15-1.18) in non-Hispanic whites, 1.09 (95% CI, 1.05-1.13) in blacks, 1.11 (95% CI, 1.08-1.14) in Hispanics, and 1.07 (95% CI, 1.04-1.11) in Asian Pacific Islanders.
The researchers also found that all-cause mortality associated with unmarried status was higher in US-born Asian Pacific Islander and Hispanic men and women relative to their foreign-born counterparts.
“The results suggest that the more acculturated you become to US culture, the more it impacts cancer survivorship,” Dr Martínez said. “Our hypothesis is that non-Hispanic whites don’t have the same social network as other cultures that have stronger bonds with family and friends outside of marriage.”
“As individuals acculturate, they tend to lose those bonds. It’s also been shown that women seek out help for health concerns more frequently than men, and women tend to remind spouses to see their physicians and live a healthy lifestyle.” ![]()
Inhibitor could overcome TKI resistance in Ph+ B-ALL

Results of preclinical research indicate that combining 2 kinase inhibitors may be a promising treatment strategy for Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia (Ph+ B-ALL).
Researchers found that combining a tyrosine kinase inhibitor (TKI) and an inhibitor of focal adhesion kinase (FAK) was “remarkably effective” against Ph+ B-ALL in vitro and in vivo.
The TKI dasatinib and the FAK inhibitor VS-4718 decreased leukemic cell survival and adhesion, inhibited tumor growth, and prolonged survival in mouse models of Ph+ B-ALL.
Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported their results in JCI Insight.
The researchers noted that patients with Ph+ ALL have shown resistance to TKI therapy, and this resistance has been tied to alterations in IKZF1.
As FAK expression is elevated in IKZF1-mutated leukemias, the team speculated that adding a FAK inhibitor to TKI therapy might lead to better results.
First, the researchers set out to confirm that FAK is overexpressed in Ph+ B-ALL. Their experiments revealed upregulation of the FAK pathway in Ph+ B-ALL cells, with further overexpression of FAK in IKZF1-mutated Ph+ B-ALL cells.
When they inhibited FAK with VS-4718, the team observed decreases in the survival, clonogenicity, and adhesion of IKZF1-mutated Ph+ B-ALL cells from both mice and humans.
Next, the researchers found that VS-4718 synergizes with the TKI dasatinib in vitro and in vivo.
In in vitro experiments with both mouse and human Ph+ B-ALL cells, the combination decreased cell survival and adhesion and inhibited downstream targets of FAK.
In mouse models of Ph+ B-ALL, VS-4718 proved ineffective when given alone.
However, the researchers said the combination of VS-4718 and dasatinib “dramatically” decreased leukemic burden and extended the lives of mice.
In fact, 1 long-term survivor achieved a complete remission that endured after treatment was stopped.
The researchers said these results suggest that targeting FAK with VS-4718 can overcome the deleterious effects of FAK overexpression in Ph+ B-ALL, potentiating responsiveness to TKIs. ![]()
Results of preclinical research indicate that combining 2 kinase inhibitors may be a promising treatment strategy for Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia (Ph+ B-ALL).
Researchers found that combining a tyrosine kinase inhibitor (TKI) and an inhibitor of focal adhesion kinase (FAK) was “remarkably effective” against Ph+ B-ALL in vitro and in vivo.
The TKI dasatinib and the FAK inhibitor VS-4718 decreased leukemic cell survival and adhesion, inhibited tumor growth, and prolonged survival in mouse models of Ph+ B-ALL.
Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported their results in JCI Insight.
The researchers noted that patients with Ph+ ALL have shown resistance to TKI therapy, and this resistance has been tied to alterations in IKZF1.
As FAK expression is elevated in IKZF1-mutated leukemias, the team speculated that adding a FAK inhibitor to TKI therapy might lead to better results.
First, the researchers set out to confirm that FAK is overexpressed in Ph+ B-ALL. Their experiments revealed upregulation of the FAK pathway in Ph+ B-ALL cells, with further overexpression of FAK in IKZF1-mutated Ph+ B-ALL cells.
When they inhibited FAK with VS-4718, the team observed decreases in the survival, clonogenicity, and adhesion of IKZF1-mutated Ph+ B-ALL cells from both mice and humans.
Next, the researchers found that VS-4718 synergizes with the TKI dasatinib in vitro and in vivo.
In in vitro experiments with both mouse and human Ph+ B-ALL cells, the combination decreased cell survival and adhesion and inhibited downstream targets of FAK.
In mouse models of Ph+ B-ALL, VS-4718 proved ineffective when given alone.
However, the researchers said the combination of VS-4718 and dasatinib “dramatically” decreased leukemic burden and extended the lives of mice.
In fact, 1 long-term survivor achieved a complete remission that endured after treatment was stopped.
The researchers said these results suggest that targeting FAK with VS-4718 can overcome the deleterious effects of FAK overexpression in Ph+ B-ALL, potentiating responsiveness to TKIs. ![]()
Results of preclinical research indicate that combining 2 kinase inhibitors may be a promising treatment strategy for Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia (Ph+ B-ALL).
Researchers found that combining a tyrosine kinase inhibitor (TKI) and an inhibitor of focal adhesion kinase (FAK) was “remarkably effective” against Ph+ B-ALL in vitro and in vivo.
The TKI dasatinib and the FAK inhibitor VS-4718 decreased leukemic cell survival and adhesion, inhibited tumor growth, and prolonged survival in mouse models of Ph+ B-ALL.
Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported their results in JCI Insight.
The researchers noted that patients with Ph+ ALL have shown resistance to TKI therapy, and this resistance has been tied to alterations in IKZF1.
As FAK expression is elevated in IKZF1-mutated leukemias, the team speculated that adding a FAK inhibitor to TKI therapy might lead to better results.
First, the researchers set out to confirm that FAK is overexpressed in Ph+ B-ALL. Their experiments revealed upregulation of the FAK pathway in Ph+ B-ALL cells, with further overexpression of FAK in IKZF1-mutated Ph+ B-ALL cells.
When they inhibited FAK with VS-4718, the team observed decreases in the survival, clonogenicity, and adhesion of IKZF1-mutated Ph+ B-ALL cells from both mice and humans.
Next, the researchers found that VS-4718 synergizes with the TKI dasatinib in vitro and in vivo.
In in vitro experiments with both mouse and human Ph+ B-ALL cells, the combination decreased cell survival and adhesion and inhibited downstream targets of FAK.
In mouse models of Ph+ B-ALL, VS-4718 proved ineffective when given alone.
However, the researchers said the combination of VS-4718 and dasatinib “dramatically” decreased leukemic burden and extended the lives of mice.
In fact, 1 long-term survivor achieved a complete remission that endured after treatment was stopped.
The researchers said these results suggest that targeting FAK with VS-4718 can overcome the deleterious effects of FAK overexpression in Ph+ B-ALL, potentiating responsiveness to TKIs. ![]()
EMA recommends orphan designation for cancer vaccine

The European Medicines Agency (EMA) has recommended orphan designation for the WT1 cancer vaccine galinpepimut-S as a treatment for patients
with acute myeloid leukemia (AML) and patients with malignant pleural mesothelioma (MPM).
The EMA’s opinion has been forwarded to the European Commission (EC), which makes the final decision.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
About the vaccine
The WT1 vaccine consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the WT1 antigen. The vaccine is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
Based on the vaccine’s mechanism and the accumulating evidence of activity in mid-stage trials, researchers believe the WT1 vaccine may have the potential to complement currently available therapies by destroying residual tumor cells of cancers in remission and providing ongoing immune surveillance for recurrent tumors.
The WT1 vaccine could potentially target more than 20 cancers that overexpress WT1, many of which are associated with relapse rates of up to 80% or more, as seen in patients with AML and MPM.
The vaccine is being developed by SELLAS Life Sciences Group. The company said that, in a phase 1 study, AML patients treated with the vaccine had a median overall survival of more than 3 years.
In a phase 2 trial of the vaccine, adult AML patients had a median overall survival of around 4 years. Data from the phase 2 trial are scheduled to be presented at the 2016 ASCO Annual Meeting.
SELLAS said it expects to begin a phase 3 trial of the vaccine in AML patients later this year. ![]()
The European Medicines Agency (EMA) has recommended orphan designation for the WT1 cancer vaccine galinpepimut-S as a treatment for patients
with acute myeloid leukemia (AML) and patients with malignant pleural mesothelioma (MPM).
The EMA’s opinion has been forwarded to the European Commission (EC), which makes the final decision.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
About the vaccine
The WT1 vaccine consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the WT1 antigen. The vaccine is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
Based on the vaccine’s mechanism and the accumulating evidence of activity in mid-stage trials, researchers believe the WT1 vaccine may have the potential to complement currently available therapies by destroying residual tumor cells of cancers in remission and providing ongoing immune surveillance for recurrent tumors.
The WT1 vaccine could potentially target more than 20 cancers that overexpress WT1, many of which are associated with relapse rates of up to 80% or more, as seen in patients with AML and MPM.
The vaccine is being developed by SELLAS Life Sciences Group. The company said that, in a phase 1 study, AML patients treated with the vaccine had a median overall survival of more than 3 years.
In a phase 2 trial of the vaccine, adult AML patients had a median overall survival of around 4 years. Data from the phase 2 trial are scheduled to be presented at the 2016 ASCO Annual Meeting.
SELLAS said it expects to begin a phase 3 trial of the vaccine in AML patients later this year. ![]()
The European Medicines Agency (EMA) has recommended orphan designation for the WT1 cancer vaccine galinpepimut-S as a treatment for patients
with acute myeloid leukemia (AML) and patients with malignant pleural mesothelioma (MPM).
The EMA’s opinion has been forwarded to the European Commission (EC), which makes the final decision.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
About the vaccine
The WT1 vaccine consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the WT1 antigen. The vaccine is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
Based on the vaccine’s mechanism and the accumulating evidence of activity in mid-stage trials, researchers believe the WT1 vaccine may have the potential to complement currently available therapies by destroying residual tumor cells of cancers in remission and providing ongoing immune surveillance for recurrent tumors.
The WT1 vaccine could potentially target more than 20 cancers that overexpress WT1, many of which are associated with relapse rates of up to 80% or more, as seen in patients with AML and MPM.
The vaccine is being developed by SELLAS Life Sciences Group. The company said that, in a phase 1 study, AML patients treated with the vaccine had a median overall survival of more than 3 years.
In a phase 2 trial of the vaccine, adult AML patients had a median overall survival of around 4 years. Data from the phase 2 trial are scheduled to be presented at the 2016 ASCO Annual Meeting.
SELLAS said it expects to begin a phase 3 trial of the vaccine in AML patients later this year. ![]()
Therapy may improve haplo-HSCT in leukemia patients

Photo by Bill Branson
VALENCIA, SPAIN—The adjunct T-cell therapy BPX-501 can make haploidentical hematopoietic stem cell transplant (haplo-HSCT) an “attractive option” for pediatric patients with acute leukemia, according to a presentation at the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT).
Acute leukemia patients who received BPX-501 after haplo-HSCT in a phase 1/2 trial tended to have favorable outcomes.
At a median follow-up of 7 months, 16 of the 17 patients were alive and disease-free.
There were several cases of graft-versus-host disease (GVHD), but nearly all of these resolved.
Franco Locatelli, MD, PhD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results at the EBMT meeting as abstract WP16.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, researchers reported results in 41 patients treated on this trial.
Dr Locatelli presented data on 17 patients with acute leukemias—13 with acute lymphoblastic leukemia and 4 with acute myeloid leukemia. Their median age at HSCT was 6.5 years (range, 0.9-16.1)
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis. All were in complete remission at the time of transplant.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only used in the event of uncontrollable GVHD.
Results
The median follow-up was 7 months (range, 1-15.6). The median time to platelet recovery was 11 days (range, 9-13), and the median time to neutrophil recovery was 17 days (range, 10-22).
Three patients developed skin-only acute GVHD, were treated with topical steroids, and the GVHD resolved. Two patients developed acute grade 3 GVHD, were treated with systemic steroids, and the GVHD resolved.
Two patients developed mild chronic GVHD, received systemic steroids, and the GVHD resolved. And 1 patient developed severe chronic GVHD, received systemic steroids and rimiducid, and the GVHD improved.
One patient relapsed. The estimated 1-year disease-free survival was 92.9%. Dr Locatelli noted that, although the follow-up is still limited, these results compare favorably to results in historical controls.
“These interim results continue to be very encouraging and indicate that a haploidentical transplant, with the addition of BPX-501-modified donor T cells, can be an attractive option for children in need of a transplant,” he said.
“Future studies will address the role of repeated infusions or higher numbers of BPX-501 cells in malignant patients with resistant disease.”
The BP-004 trial also included 24 patients with nonmalignant disorders. Results in these patients were presented at the EBMT meeting as abstract O007. ![]()
*Information in the abstract differs from that presented at the meeting.
Photo by Bill Branson
VALENCIA, SPAIN—The adjunct T-cell therapy BPX-501 can make haploidentical hematopoietic stem cell transplant (haplo-HSCT) an “attractive option” for pediatric patients with acute leukemia, according to a presentation at the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT).
Acute leukemia patients who received BPX-501 after haplo-HSCT in a phase 1/2 trial tended to have favorable outcomes.
At a median follow-up of 7 months, 16 of the 17 patients were alive and disease-free.
There were several cases of graft-versus-host disease (GVHD), but nearly all of these resolved.
Franco Locatelli, MD, PhD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results at the EBMT meeting as abstract WP16.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, researchers reported results in 41 patients treated on this trial.
Dr Locatelli presented data on 17 patients with acute leukemias—13 with acute lymphoblastic leukemia and 4 with acute myeloid leukemia. Their median age at HSCT was 6.5 years (range, 0.9-16.1)
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis. All were in complete remission at the time of transplant.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only used in the event of uncontrollable GVHD.
Results
The median follow-up was 7 months (range, 1-15.6). The median time to platelet recovery was 11 days (range, 9-13), and the median time to neutrophil recovery was 17 days (range, 10-22).
Three patients developed skin-only acute GVHD, were treated with topical steroids, and the GVHD resolved. Two patients developed acute grade 3 GVHD, were treated with systemic steroids, and the GVHD resolved.
Two patients developed mild chronic GVHD, received systemic steroids, and the GVHD resolved. And 1 patient developed severe chronic GVHD, received systemic steroids and rimiducid, and the GVHD improved.
One patient relapsed. The estimated 1-year disease-free survival was 92.9%. Dr Locatelli noted that, although the follow-up is still limited, these results compare favorably to results in historical controls.
“These interim results continue to be very encouraging and indicate that a haploidentical transplant, with the addition of BPX-501-modified donor T cells, can be an attractive option for children in need of a transplant,” he said.
“Future studies will address the role of repeated infusions or higher numbers of BPX-501 cells in malignant patients with resistant disease.”
The BP-004 trial also included 24 patients with nonmalignant disorders. Results in these patients were presented at the EBMT meeting as abstract O007. ![]()
*Information in the abstract differs from that presented at the meeting.
Photo by Bill Branson
VALENCIA, SPAIN—The adjunct T-cell therapy BPX-501 can make haploidentical hematopoietic stem cell transplant (haplo-HSCT) an “attractive option” for pediatric patients with acute leukemia, according to a presentation at the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT).
Acute leukemia patients who received BPX-501 after haplo-HSCT in a phase 1/2 trial tended to have favorable outcomes.
At a median follow-up of 7 months, 16 of the 17 patients were alive and disease-free.
There were several cases of graft-versus-host disease (GVHD), but nearly all of these resolved.
Franco Locatelli, MD, PhD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results at the EBMT meeting as abstract WP16.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, researchers reported results in 41 patients treated on this trial.
Dr Locatelli presented data on 17 patients with acute leukemias—13 with acute lymphoblastic leukemia and 4 with acute myeloid leukemia. Their median age at HSCT was 6.5 years (range, 0.9-16.1)
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis. All were in complete remission at the time of transplant.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only used in the event of uncontrollable GVHD.
Results
The median follow-up was 7 months (range, 1-15.6). The median time to platelet recovery was 11 days (range, 9-13), and the median time to neutrophil recovery was 17 days (range, 10-22).
Three patients developed skin-only acute GVHD, were treated with topical steroids, and the GVHD resolved. Two patients developed acute grade 3 GVHD, were treated with systemic steroids, and the GVHD resolved.
Two patients developed mild chronic GVHD, received systemic steroids, and the GVHD resolved. And 1 patient developed severe chronic GVHD, received systemic steroids and rimiducid, and the GVHD improved.
One patient relapsed. The estimated 1-year disease-free survival was 92.9%. Dr Locatelli noted that, although the follow-up is still limited, these results compare favorably to results in historical controls.
“These interim results continue to be very encouraging and indicate that a haploidentical transplant, with the addition of BPX-501-modified donor T cells, can be an attractive option for children in need of a transplant,” he said.
“Future studies will address the role of repeated infusions or higher numbers of BPX-501 cells in malignant patients with resistant disease.”
The BP-004 trial also included 24 patients with nonmalignant disorders. Results in these patients were presented at the EBMT meeting as abstract O007.
*Information in the abstract differs from that presented at the meeting.
Cyclophosphamide nets low rate of chronic GVHD after mobilized blood cell transplantation
High-dose cyclophosphamide is safe and effective when given as prophylaxis for chronic graft-versus-host disease (GVHD) to patients who have undergone transplantation of mobilized blood cells, finds a phase 2 trial reported in Blood.
Investigators led by Dr. Marco Mielcarek, medical director of the Adult Blood and Marrow Transplant Program and an oncologist at the Fred Hutchinson Cancer Research Center in Seattle, Washington, enrolled in the trial 43 patients with high-risk hematologic malignancies.

The patients underwent myeloablative conditioning followed by transplantation with growth factor–mobilized blood cells from related or unrelated donors, and were given high-dose cyclophosphamide on two early posttransplantation days.
Main results showed that the cumulative 1-year incidence of chronic GVHD was 16%, less than half of the roughly 35% seen historically with conventional immunosuppression.
Moreover, cyclophosphamide did not appear to compromise engraftment or control of the underlying malignancy. Only a single patient, one with an HLA-mismatched donor, had failure of primary engraftment; after amendment of the protocol to require HLA matching, there were no additional cases. Just 17% of patients experienced a recurrence of their malignancy by 2 years.
Taken together, the findings suggest that high-dose cyclophosphamide—as combined with two myeloablative conditioning options (to accommodate different malignancies) and with posttransplantation cyclosporine (to reduce the risk of acute GVHD)—may eliminate most of the drawbacks to using mobilized blood cells for transplantation, according to the investigators.
“If these findings are confirmed in future studies, HLA-matched mobilized blood cell transplantation may gain even greater acceptance and further replace marrow as a source of stem cells for most indications,” they maintain.
The patients studied had a median age of 43 years, and slightly more than half were in remission without minimal residual disease.
Blood cells were mobilized with granulocyte colony-stimulating factor (G-CSF). Overall, 28% of patients received grafts from related donors, while 72% received grafts from unrelated donors.
For pretransplant conditioning, patients received fludarabine and targeted busulfan, or total body irradiation with use of a minimum dose of 12 Gy.
The patients were given cyclophosphamide at 50 mg/kg per day on days 3 and 4 after transplantation. This was followed by cyclosporine starting on day 5.
The cumulative 1-year incidence of chronic GVHD as defined by National Institutes of Health criteria (i.e., that requiring systemic immunosuppressive therapy)—the trial’s primary endpoint—was 16%, which fell just short of the goal of 15% the investigators were aiming for (Blood. 2016;127:1502-8). Analyses failed to identify any predictors of this outcome.
Although the estimated cumulative incidence of grade 2 acute GVHD was high, at 77%, none of the patients developed grade 3 or 4 acute GVHD, according to the investigators, who disclosed that they had no competing financial interests.
The single patient who experienced failure of primary engraftment had familial myelodysplastic syndrome and had received a graft from an HLA A-antigen–mismatched unrelated donor.
The 2-year cumulative incidence of nonrelapse mortality was 14%, and the 2-year cumulative incidence of recurrent malignancy was 17%. Projected overall survival was 70%.
Among the 42 patients having at least a year of follow-up, 50% were alive and free of relapse without any systemic immunosuppression at 1 year after transplantation.
High-dose cyclophosphamide is safe and effective when given as prophylaxis for chronic graft-versus-host disease (GVHD) to patients who have undergone transplantation of mobilized blood cells, finds a phase 2 trial reported in Blood.
Investigators led by Dr. Marco Mielcarek, medical director of the Adult Blood and Marrow Transplant Program and an oncologist at the Fred Hutchinson Cancer Research Center in Seattle, Washington, enrolled in the trial 43 patients with high-risk hematologic malignancies.
The patients underwent myeloablative conditioning followed by transplantation with growth factor–mobilized blood cells from related or unrelated donors, and were given high-dose cyclophosphamide on two early posttransplantation days.
Main results showed that the cumulative 1-year incidence of chronic GVHD was 16%, less than half of the roughly 35% seen historically with conventional immunosuppression.
Moreover, cyclophosphamide did not appear to compromise engraftment or control of the underlying malignancy. Only a single patient, one with an HLA-mismatched donor, had failure of primary engraftment; after amendment of the protocol to require HLA matching, there were no additional cases. Just 17% of patients experienced a recurrence of their malignancy by 2 years.
Taken together, the findings suggest that high-dose cyclophosphamide—as combined with two myeloablative conditioning options (to accommodate different malignancies) and with posttransplantation cyclosporine (to reduce the risk of acute GVHD)—may eliminate most of the drawbacks to using mobilized blood cells for transplantation, according to the investigators.
“If these findings are confirmed in future studies, HLA-matched mobilized blood cell transplantation may gain even greater acceptance and further replace marrow as a source of stem cells for most indications,” they maintain.
The patients studied had a median age of 43 years, and slightly more than half were in remission without minimal residual disease.
Blood cells were mobilized with granulocyte colony-stimulating factor (G-CSF). Overall, 28% of patients received grafts from related donors, while 72% received grafts from unrelated donors.
For pretransplant conditioning, patients received fludarabine and targeted busulfan, or total body irradiation with use of a minimum dose of 12 Gy.
The patients were given cyclophosphamide at 50 mg/kg per day on days 3 and 4 after transplantation. This was followed by cyclosporine starting on day 5.
The cumulative 1-year incidence of chronic GVHD as defined by National Institutes of Health criteria (i.e., that requiring systemic immunosuppressive therapy)—the trial’s primary endpoint—was 16%, which fell just short of the goal of 15% the investigators were aiming for (Blood. 2016;127:1502-8). Analyses failed to identify any predictors of this outcome.
Although the estimated cumulative incidence of grade 2 acute GVHD was high, at 77%, none of the patients developed grade 3 or 4 acute GVHD, according to the investigators, who disclosed that they had no competing financial interests.
The single patient who experienced failure of primary engraftment had familial myelodysplastic syndrome and had received a graft from an HLA A-antigen–mismatched unrelated donor.
The 2-year cumulative incidence of nonrelapse mortality was 14%, and the 2-year cumulative incidence of recurrent malignancy was 17%. Projected overall survival was 70%.
Among the 42 patients having at least a year of follow-up, 50% were alive and free of relapse without any systemic immunosuppression at 1 year after transplantation.
High-dose cyclophosphamide is safe and effective when given as prophylaxis for chronic graft-versus-host disease (GVHD) to patients who have undergone transplantation of mobilized blood cells, finds a phase 2 trial reported in Blood.
Investigators led by Dr. Marco Mielcarek, medical director of the Adult Blood and Marrow Transplant Program and an oncologist at the Fred Hutchinson Cancer Research Center in Seattle, Washington, enrolled in the trial 43 patients with high-risk hematologic malignancies.
The patients underwent myeloablative conditioning followed by transplantation with growth factor–mobilized blood cells from related or unrelated donors, and were given high-dose cyclophosphamide on two early posttransplantation days.
Main results showed that the cumulative 1-year incidence of chronic GVHD was 16%, less than half of the roughly 35% seen historically with conventional immunosuppression.
Moreover, cyclophosphamide did not appear to compromise engraftment or control of the underlying malignancy. Only a single patient, one with an HLA-mismatched donor, had failure of primary engraftment; after amendment of the protocol to require HLA matching, there were no additional cases. Just 17% of patients experienced a recurrence of their malignancy by 2 years.
Taken together, the findings suggest that high-dose cyclophosphamide—as combined with two myeloablative conditioning options (to accommodate different malignancies) and with posttransplantation cyclosporine (to reduce the risk of acute GVHD)—may eliminate most of the drawbacks to using mobilized blood cells for transplantation, according to the investigators.
“If these findings are confirmed in future studies, HLA-matched mobilized blood cell transplantation may gain even greater acceptance and further replace marrow as a source of stem cells for most indications,” they maintain.
The patients studied had a median age of 43 years, and slightly more than half were in remission without minimal residual disease.
Blood cells were mobilized with granulocyte colony-stimulating factor (G-CSF). Overall, 28% of patients received grafts from related donors, while 72% received grafts from unrelated donors.
For pretransplant conditioning, patients received fludarabine and targeted busulfan, or total body irradiation with use of a minimum dose of 12 Gy.
The patients were given cyclophosphamide at 50 mg/kg per day on days 3 and 4 after transplantation. This was followed by cyclosporine starting on day 5.
The cumulative 1-year incidence of chronic GVHD as defined by National Institutes of Health criteria (i.e., that requiring systemic immunosuppressive therapy)—the trial’s primary endpoint—was 16%, which fell just short of the goal of 15% the investigators were aiming for (Blood. 2016;127:1502-8). Analyses failed to identify any predictors of this outcome.
Although the estimated cumulative incidence of grade 2 acute GVHD was high, at 77%, none of the patients developed grade 3 or 4 acute GVHD, according to the investigators, who disclosed that they had no competing financial interests.
The single patient who experienced failure of primary engraftment had familial myelodysplastic syndrome and had received a graft from an HLA A-antigen–mismatched unrelated donor.
The 2-year cumulative incidence of nonrelapse mortality was 14%, and the 2-year cumulative incidence of recurrent malignancy was 17%. Projected overall survival was 70%.
Among the 42 patients having at least a year of follow-up, 50% were alive and free of relapse without any systemic immunosuppression at 1 year after transplantation.
FROM BLOOD
Key clinical point: High-dose posttransplant cyclophosphamide is safe and effective for reducing the risk of chronic GVHD after mobilized blood cell transplantation.
Major finding: The cumulative 1-year incidence of chronic GVHD requiring immunosuppressive therapy was 16%.
Data source: A single-arm trial among 43 patients with high-risk hematologic malignancies undergoing growth factor–mobilized blood cell transplantation.
Disclosures: The authors disclosed that they have no competing financial interests.
