User login
Chemo-free induction regimen shines in MCL
SAN DIEGO – A chemotherapy-free induction regimen of ibrutinib and rituximab was well tolerated and achieved an overall response rate of 100% among patients with newly diagnosed mantle cell lymphoma (MCL), according to results of a small single-center phase II trial.
A total of 72% of patients had complete responses to induction, while 28% had partial responses, and 100% had complete responses to consolidation, reported Michael Wang, MD, at the annual meeting of the American Society of Hematology. The findings highlight a “window of opportunity” to effectively treat de novo MCL in young, fit patients while potentially sparing them from repeated cycles of intensive chemoimmunotherapy, the investigators noted.
Established treatments for MCL include eight cycles of rituximab–hyper-CVAD (hyper-fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) alternating with rituximab–methotrexate–Ara-C (cytarabine). Median overall survival with this regimen exceeds 10 years, but during that decade, more than 6% of patients will develop myeloid neoplasms related to treatment, noted Dr. Wang of the University of Texas MD Anderson Cancer Center in Houston.
For the study, patients up to 65 years old with newly diagnosed, untreated MCL underwent induction with continuous daily ibrutinib (560 mg), plus rituximab (375 mg/m2) administered weekly for 4 weeks during cycle 1 and on day 1 of cycles 3-12. Consolidation consisted of rituximab plus hyper-CVAD, alternating every 28 days with rituximab plus high-dose methotrexate–Ara-C. Complete responders to induction received four cycles of chemoimmunotherapy, while progressors and partial responders received chemoimmunotherapy for two cycles beyond the point of complete remission.
Among 36 evaluable patients, 28% had a partial response and the rest had complete responses to induction. Moreover, all 19 patients who completed consolidation had a complete response. During induction, the most common toxicities were fatigue, diarrhea, rash, and myalgia, nearly all of which were mild or moderate in severity. During consolidation, two patients developed severe neutropenia, one patient developed severe febrile neutropenia, and one developed a severe increase in liver enzymes. There were no treatment-related deaths.
“The toxicity after intensive immune-chemotherapy in shortened cycles [is] much improved compared to historical controls, but longer follow-up is needed,” the researchers wrote. “This unprecedented efficacy and safety may provide a window of opportunity for less chemoimmunotherapy needed for consolidation.”
Dr. Wang disclosed ties to Janssen, Onyx, BeiGene, Kite Pharma, Asana Biosciences, Juno Therapeutics, Acerta Pharma, and Celgene.
SAN DIEGO – A chemotherapy-free induction regimen of ibrutinib and rituximab was well tolerated and achieved an overall response rate of 100% among patients with newly diagnosed mantle cell lymphoma (MCL), according to results of a small single-center phase II trial.
A total of 72% of patients had complete responses to induction, while 28% had partial responses, and 100% had complete responses to consolidation, reported Michael Wang, MD, at the annual meeting of the American Society of Hematology. The findings highlight a “window of opportunity” to effectively treat de novo MCL in young, fit patients while potentially sparing them from repeated cycles of intensive chemoimmunotherapy, the investigators noted.
Established treatments for MCL include eight cycles of rituximab–hyper-CVAD (hyper-fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) alternating with rituximab–methotrexate–Ara-C (cytarabine). Median overall survival with this regimen exceeds 10 years, but during that decade, more than 6% of patients will develop myeloid neoplasms related to treatment, noted Dr. Wang of the University of Texas MD Anderson Cancer Center in Houston.
For the study, patients up to 65 years old with newly diagnosed, untreated MCL underwent induction with continuous daily ibrutinib (560 mg), plus rituximab (375 mg/m2) administered weekly for 4 weeks during cycle 1 and on day 1 of cycles 3-12. Consolidation consisted of rituximab plus hyper-CVAD, alternating every 28 days with rituximab plus high-dose methotrexate–Ara-C. Complete responders to induction received four cycles of chemoimmunotherapy, while progressors and partial responders received chemoimmunotherapy for two cycles beyond the point of complete remission.
Among 36 evaluable patients, 28% had a partial response and the rest had complete responses to induction. Moreover, all 19 patients who completed consolidation had a complete response. During induction, the most common toxicities were fatigue, diarrhea, rash, and myalgia, nearly all of which were mild or moderate in severity. During consolidation, two patients developed severe neutropenia, one patient developed severe febrile neutropenia, and one developed a severe increase in liver enzymes. There were no treatment-related deaths.
“The toxicity after intensive immune-chemotherapy in shortened cycles [is] much improved compared to historical controls, but longer follow-up is needed,” the researchers wrote. “This unprecedented efficacy and safety may provide a window of opportunity for less chemoimmunotherapy needed for consolidation.”
Dr. Wang disclosed ties to Janssen, Onyx, BeiGene, Kite Pharma, Asana Biosciences, Juno Therapeutics, Acerta Pharma, and Celgene.
SAN DIEGO – A chemotherapy-free induction regimen of ibrutinib and rituximab was well tolerated and achieved an overall response rate of 100% among patients with newly diagnosed mantle cell lymphoma (MCL), according to results of a small single-center phase II trial.
A total of 72% of patients had complete responses to induction, while 28% had partial responses, and 100% had complete responses to consolidation, reported Michael Wang, MD, at the annual meeting of the American Society of Hematology. The findings highlight a “window of opportunity” to effectively treat de novo MCL in young, fit patients while potentially sparing them from repeated cycles of intensive chemoimmunotherapy, the investigators noted.
Established treatments for MCL include eight cycles of rituximab–hyper-CVAD (hyper-fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) alternating with rituximab–methotrexate–Ara-C (cytarabine). Median overall survival with this regimen exceeds 10 years, but during that decade, more than 6% of patients will develop myeloid neoplasms related to treatment, noted Dr. Wang of the University of Texas MD Anderson Cancer Center in Houston.
For the study, patients up to 65 years old with newly diagnosed, untreated MCL underwent induction with continuous daily ibrutinib (560 mg), plus rituximab (375 mg/m2) administered weekly for 4 weeks during cycle 1 and on day 1 of cycles 3-12. Consolidation consisted of rituximab plus hyper-CVAD, alternating every 28 days with rituximab plus high-dose methotrexate–Ara-C. Complete responders to induction received four cycles of chemoimmunotherapy, while progressors and partial responders received chemoimmunotherapy for two cycles beyond the point of complete remission.
Among 36 evaluable patients, 28% had a partial response and the rest had complete responses to induction. Moreover, all 19 patients who completed consolidation had a complete response. During induction, the most common toxicities were fatigue, diarrhea, rash, and myalgia, nearly all of which were mild or moderate in severity. During consolidation, two patients developed severe neutropenia, one patient developed severe febrile neutropenia, and one developed a severe increase in liver enzymes. There were no treatment-related deaths.
“The toxicity after intensive immune-chemotherapy in shortened cycles [is] much improved compared to historical controls, but longer follow-up is needed,” the researchers wrote. “This unprecedented efficacy and safety may provide a window of opportunity for less chemoimmunotherapy needed for consolidation.”
Dr. Wang disclosed ties to Janssen, Onyx, BeiGene, Kite Pharma, Asana Biosciences, Juno Therapeutics, Acerta Pharma, and Celgene.
AT ASH 2016
Key clinical point: Induction with ibrutinib and rituximab achieved a 100% response rate in newly diagnosed mantle cell lymphoma, enabling patients to receive less intensive consolidation.
Major finding: Rates of complete response were 72% for induction and 100% for induction plus consolidation.
Data source: A single-center phase II trial of 36 patients with newly diagnosed, untreated mantle cell lymphoma.
Disclosures: Dr. Wang disclosed ties to Janssen, Onyx, BeiGene, Kite Pharma, Asana Biosciences, Juno Therapeutics, Acerta Pharma, and Celgene.
Rituximab vanquished MRD in mantle cell lymphoma
SAN DIEGO – Rituximab can at least temporarily vanquish minimal residual disease (MRD) in mantle cell lymphoma (MCL) patients who relapse after induction therapy and autologous stem cell transplantation (ASCT), researchers reported at the annual meeting of the American Society of Hematology.
Of 58 patients whose MCL relapsed after induction therapy and ASCT, 82% converted back to an MRD-negative state after receiving 4 weekly doses of rituximab (375 mg/m2), Arne Kolstad, MD, PhD, and his associates. The data “strongly suggest that preemptive rituximab treatment delayed clinical relapse in MCL,” they wrote in their abstract. They recommended molecular and clinical monitoring after ASCT, not only “as an alternative to maintenance therapy for all MCL patients” but to identify MRD-positive candidates for clinical trials.
The study was an analysis of the Nordic Lymphoma Group phase II MCL2 and MCL3 trials (NTC 00514475), in which patients received six alternating cycles of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab) and R-Ara-C (rituximab-cytarabine). followed by high-dose ASCT. In MCL3, responders who fell short of complete remission also received intensification with yttium-90 ibritumomab tiuxetan (0.4 mCi/kg) 1 week before treatment with BEAM/C (carmustine, etoposide, cytarabine, and melphalan or cyclophosphamide). Patients were evaluated 2-3 months after completing ASCT, and then every 6 months for 5 years or until relapse. Survivors were followed for a median of 8.5 years, noted Dr. Kolstad, who is with Oslo University Hospital in Norway.
Among 183 patients who underwent polymerase chain reaction–based testing for markers of MRD, median time to molecular relapse was 55 months. However, the relapse-free survival curve did not plateau – patients in all risk groups continued to relapse 5-10 years after undergoing ASCT, the researchers said. “Hence, it is fair to consider MCL as a chronic incurable lymphoma entity, and novel approaches will be necessary to change the natural course of this disease,” they wrote.
After controlling for potential confounders, significant predictors of molecular relapse included high MCL international prognostic index at diagnosis (hazard ratio, 1.9; 95% confidence interval 1.4-2.7; P = .0001) and detection of MRD before patients underwent ASCT (HR, 2.5; 95% CI, 1.5-4.1; P = .0005). Minimal residual disease predicted clinical relapse and shorter survival (P less than .001 for both associations). In contrast, the 86 patients who remained in continuous molecular remission had a 76% chance of having at least a 10-year clinical remission, the investigators said.
Minimal residual disease was assessed by testing bone marrow and blood samples with combined standard nested and quantitative real-time polymerase chain reaction (PCR) for Bcl-1 or IgH rearrangement. They defined molecular relapse as conversion from a negative to a positive result on standard nested PCR, or, for patients who were MRD positive after ASCT, as a more than fivefold rise in real-time quantitative PCR levels in two consecutive bone marrow samples.
Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
SAN DIEGO – Rituximab can at least temporarily vanquish minimal residual disease (MRD) in mantle cell lymphoma (MCL) patients who relapse after induction therapy and autologous stem cell transplantation (ASCT), researchers reported at the annual meeting of the American Society of Hematology.
Of 58 patients whose MCL relapsed after induction therapy and ASCT, 82% converted back to an MRD-negative state after receiving 4 weekly doses of rituximab (375 mg/m2), Arne Kolstad, MD, PhD, and his associates. The data “strongly suggest that preemptive rituximab treatment delayed clinical relapse in MCL,” they wrote in their abstract. They recommended molecular and clinical monitoring after ASCT, not only “as an alternative to maintenance therapy for all MCL patients” but to identify MRD-positive candidates for clinical trials.
The study was an analysis of the Nordic Lymphoma Group phase II MCL2 and MCL3 trials (NTC 00514475), in which patients received six alternating cycles of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab) and R-Ara-C (rituximab-cytarabine). followed by high-dose ASCT. In MCL3, responders who fell short of complete remission also received intensification with yttium-90 ibritumomab tiuxetan (0.4 mCi/kg) 1 week before treatment with BEAM/C (carmustine, etoposide, cytarabine, and melphalan or cyclophosphamide). Patients were evaluated 2-3 months after completing ASCT, and then every 6 months for 5 years or until relapse. Survivors were followed for a median of 8.5 years, noted Dr. Kolstad, who is with Oslo University Hospital in Norway.
Among 183 patients who underwent polymerase chain reaction–based testing for markers of MRD, median time to molecular relapse was 55 months. However, the relapse-free survival curve did not plateau – patients in all risk groups continued to relapse 5-10 years after undergoing ASCT, the researchers said. “Hence, it is fair to consider MCL as a chronic incurable lymphoma entity, and novel approaches will be necessary to change the natural course of this disease,” they wrote.
After controlling for potential confounders, significant predictors of molecular relapse included high MCL international prognostic index at diagnosis (hazard ratio, 1.9; 95% confidence interval 1.4-2.7; P = .0001) and detection of MRD before patients underwent ASCT (HR, 2.5; 95% CI, 1.5-4.1; P = .0005). Minimal residual disease predicted clinical relapse and shorter survival (P less than .001 for both associations). In contrast, the 86 patients who remained in continuous molecular remission had a 76% chance of having at least a 10-year clinical remission, the investigators said.
Minimal residual disease was assessed by testing bone marrow and blood samples with combined standard nested and quantitative real-time polymerase chain reaction (PCR) for Bcl-1 or IgH rearrangement. They defined molecular relapse as conversion from a negative to a positive result on standard nested PCR, or, for patients who were MRD positive after ASCT, as a more than fivefold rise in real-time quantitative PCR levels in two consecutive bone marrow samples.
Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
SAN DIEGO – Rituximab can at least temporarily vanquish minimal residual disease (MRD) in mantle cell lymphoma (MCL) patients who relapse after induction therapy and autologous stem cell transplantation (ASCT), researchers reported at the annual meeting of the American Society of Hematology.
Of 58 patients whose MCL relapsed after induction therapy and ASCT, 82% converted back to an MRD-negative state after receiving 4 weekly doses of rituximab (375 mg/m2), Arne Kolstad, MD, PhD, and his associates. The data “strongly suggest that preemptive rituximab treatment delayed clinical relapse in MCL,” they wrote in their abstract. They recommended molecular and clinical monitoring after ASCT, not only “as an alternative to maintenance therapy for all MCL patients” but to identify MRD-positive candidates for clinical trials.
The study was an analysis of the Nordic Lymphoma Group phase II MCL2 and MCL3 trials (NTC 00514475), in which patients received six alternating cycles of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab) and R-Ara-C (rituximab-cytarabine). followed by high-dose ASCT. In MCL3, responders who fell short of complete remission also received intensification with yttium-90 ibritumomab tiuxetan (0.4 mCi/kg) 1 week before treatment with BEAM/C (carmustine, etoposide, cytarabine, and melphalan or cyclophosphamide). Patients were evaluated 2-3 months after completing ASCT, and then every 6 months for 5 years or until relapse. Survivors were followed for a median of 8.5 years, noted Dr. Kolstad, who is with Oslo University Hospital in Norway.
Among 183 patients who underwent polymerase chain reaction–based testing for markers of MRD, median time to molecular relapse was 55 months. However, the relapse-free survival curve did not plateau – patients in all risk groups continued to relapse 5-10 years after undergoing ASCT, the researchers said. “Hence, it is fair to consider MCL as a chronic incurable lymphoma entity, and novel approaches will be necessary to change the natural course of this disease,” they wrote.
After controlling for potential confounders, significant predictors of molecular relapse included high MCL international prognostic index at diagnosis (hazard ratio, 1.9; 95% confidence interval 1.4-2.7; P = .0001) and detection of MRD before patients underwent ASCT (HR, 2.5; 95% CI, 1.5-4.1; P = .0005). Minimal residual disease predicted clinical relapse and shorter survival (P less than .001 for both associations). In contrast, the 86 patients who remained in continuous molecular remission had a 76% chance of having at least a 10-year clinical remission, the investigators said.
Minimal residual disease was assessed by testing bone marrow and blood samples with combined standard nested and quantitative real-time polymerase chain reaction (PCR) for Bcl-1 or IgH rearrangement. They defined molecular relapse as conversion from a negative to a positive result on standard nested PCR, or, for patients who were MRD positive after ASCT, as a more than fivefold rise in real-time quantitative PCR levels in two consecutive bone marrow samples.
Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
AT ASH 2016
Key clinical point:
Major finding: Among 58 patients who relapsed after induction therapy and autologous stem cell transplantation, 82% converted back to an MRD-negative state with 4 weekly doses of rituximab (375 mg/m2).
Data source: A study of 183 patients with mantle cell lymphoma from the Nordic MCL2 and MCL3 trials.
Disclosures: Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
VIDEO: Obinutuzumab bests rituximab for PFS in follicular lymphoma
SAN DIEGO – For patients with indolent non-Hodgkin lymphoma, adding the anti-CD20 antibody rituximab to a standard-combination chemotherapy regimen resulted in significant improvements in survival, compared with chemotherapy alone. Obinutuzumab (Gazyva), a second-generation anti-CD20 antibody touted as the heir apparent to rituximab, is being explored in various combinations for the treatment of indolent lymphomas, including follicular lymphoma and marginal zone lymphoma.
In this video interview from the annual meeting of the American Society of Hematology, Robert Marcus, FRCP, of King’s College Hospital, London, discussed results of the phase III GALLIUM study, in which patients with untreated follicular lymphoma were randomly assigned to one of three chemotherapy regimens with either obinutuzumab or rituximab. The primary endpoint of investigator-assessed 3-year progression-free survival (PFS) at a median follow-up of 34.5 months was 80% for patients with follicular lymphoma treated with obinutuzumab and one of three standard chemotherapy regimens, compared with 73.3% for patients treated with rituximab and chemotherapy. This difference translated into a hazard ratio (HR) favoring obinutuzumab of 0.68 (P = .0012).
Respective 3-year overall survival rates at 3 years were similar, however, at 94% and 92.1% (HR, 0.75; P = .21).
The GALLIUM trial is sponsored by F. Hoffmann-La Roche. Dr. Marcus disclosed consulting with and receiving honoraria from the company, and relationships with other companies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – For patients with indolent non-Hodgkin lymphoma, adding the anti-CD20 antibody rituximab to a standard-combination chemotherapy regimen resulted in significant improvements in survival, compared with chemotherapy alone. Obinutuzumab (Gazyva), a second-generation anti-CD20 antibody touted as the heir apparent to rituximab, is being explored in various combinations for the treatment of indolent lymphomas, including follicular lymphoma and marginal zone lymphoma.
In this video interview from the annual meeting of the American Society of Hematology, Robert Marcus, FRCP, of King’s College Hospital, London, discussed results of the phase III GALLIUM study, in which patients with untreated follicular lymphoma were randomly assigned to one of three chemotherapy regimens with either obinutuzumab or rituximab. The primary endpoint of investigator-assessed 3-year progression-free survival (PFS) at a median follow-up of 34.5 months was 80% for patients with follicular lymphoma treated with obinutuzumab and one of three standard chemotherapy regimens, compared with 73.3% for patients treated with rituximab and chemotherapy. This difference translated into a hazard ratio (HR) favoring obinutuzumab of 0.68 (P = .0012).
Respective 3-year overall survival rates at 3 years were similar, however, at 94% and 92.1% (HR, 0.75; P = .21).
The GALLIUM trial is sponsored by F. Hoffmann-La Roche. Dr. Marcus disclosed consulting with and receiving honoraria from the company, and relationships with other companies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – For patients with indolent non-Hodgkin lymphoma, adding the anti-CD20 antibody rituximab to a standard-combination chemotherapy regimen resulted in significant improvements in survival, compared with chemotherapy alone. Obinutuzumab (Gazyva), a second-generation anti-CD20 antibody touted as the heir apparent to rituximab, is being explored in various combinations for the treatment of indolent lymphomas, including follicular lymphoma and marginal zone lymphoma.
In this video interview from the annual meeting of the American Society of Hematology, Robert Marcus, FRCP, of King’s College Hospital, London, discussed results of the phase III GALLIUM study, in which patients with untreated follicular lymphoma were randomly assigned to one of three chemotherapy regimens with either obinutuzumab or rituximab. The primary endpoint of investigator-assessed 3-year progression-free survival (PFS) at a median follow-up of 34.5 months was 80% for patients with follicular lymphoma treated with obinutuzumab and one of three standard chemotherapy regimens, compared with 73.3% for patients treated with rituximab and chemotherapy. This difference translated into a hazard ratio (HR) favoring obinutuzumab of 0.68 (P = .0012).
Respective 3-year overall survival rates at 3 years were similar, however, at 94% and 92.1% (HR, 0.75; P = .21).
The GALLIUM trial is sponsored by F. Hoffmann-La Roche. Dr. Marcus disclosed consulting with and receiving honoraria from the company, and relationships with other companies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ASH 2016

Antibody face-off in follicular lymphoma gives PFS, but not OS, edge to obinutuzumab
SAN DIEGO – Obinutuzumab, a second-generation anti-CD20 antibody touted as the heir apparent to rituximab, offered a progression-free survival (PFS) edge over rituximab when combined with standard chemotherapy in patients with previously untreated advanced follicular lymphoma.

But other clinicians and investigators who
attended the presentation of the GALLIUM data at a plenary session during the American Society of Hematology annual meeting indicated that despite the data, they weren’t ready to make a switch to the newer, costlier antibody.
“I feel that it is not convincing for practice-changing,” said Kanti R. Rai, MD, professor of medicine and molecular medicine at Hofstra University, Hempstead, N.Y.
“Unless we have evidence of a survival advantage in indolent disease, progression-free survivorship is not an adequate reason to jump to another antibody,” he said in an interview.
In GALLIUM, the primary endpoint of investigator-assessed 3-year PFS at a median follow-up of 34.5 months was 80% for patients with follicular lymphoma treated with obinutuzumab and one of three standard chemotherapy regimens, compared with 73.3% for patients treated with rituximab and chemotherapy. This difference translated into a hazard ratio of 0.68 favoring obinutuzumab (P = .0012).
Respective 3-year overall survival rates were similar, however, at 94% and 92.1% (HR, 0.75; P = .21).
Indolent lymphoma trial
The GALLIUM trial is a phase III study comparing obinutuzumab with rituximab when paired with one of three standard chemotherapy regimens for indolent non-Hodgkin lymphomas, including follicular lymphoma and splenic, nodal, or extranodal marginal zone lymphoma. Dr. Marcus presented data on patients with follicular lymphoma only.
The antibodies were delivered in combination with either CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone; 33.1% of patients), CVP (cyclophosphamide, vincristine, prednisone; 9.8%) or bendamustine alone (B; 57.1%) as the chemotherapy backbone. The choice of regimen was at the discretion of the treating center.
A total of 1,202 patients with follicular lymphoma were enrolled and randomized to treatment and were included in an intention-to-treat analysis.
The treatment arms were well balanced with regard to distribution of patients characteristics, with approximately 21% in each arm having Follicular Lymphoma International Prognostic Index low-risk disease; 37% having intermediate-risk disease; and 34% having high-risk disease.
Roughly half of patients in each arm had bone marrow involvement, and two-thirds had extranodal involvement.
Obinutuzumab was dosed 1,000 mg IV on days 1, 8, and 15 of cycle one, and either on day 1 of cycles two through eight every 3 weeks, or every 4 weeks during cycles two through six.
Overall response rates at the end of induction were 86.9% with rituximab and 88.5% with obinutuzumab, with complete responses of 23.8% and 19.5%, respectively.
As noted before, investigator-assessed PFS favored obinutuzumab, as did PFS assessed by independent reviewer, at 81.9% vs. 77.9% for rituximab (HR, 0.71; P = .0138).
The newer antibody also had a slight edge in time to new treatment, with 87.1% of patients on obinutuzumab not starting on new therapy, compared with 81.2% of patients on rituximab.
More bendamustine deaths
Nearly all patients in each arm had an adverse event, with grade 3 or greater events occurring in 74.6% of patients on obinutuzumab vs. 67.8% on rituximab. Rates of neutropenia, leukopenia, febrile neutropenia, infusion reactions, and thrombocytopenia were all slightly higher with obinutuzumab. Grade 3 or greater infections occurred in 20% with obinutuzumab, compared with 15.6% with rituximab.
“What we did note, however, was a high level of mortality in patients receiving either obinutuzumab-based therapy or rituximab-based therapy, which were no different between the two arms and were somewhat higher than one might expect from patients receiving induction treatment in follicular lymphoma. Hence, we did a more detailed analysis of safety by treatment regimen,” Dr. Marcus said.
There were more deaths among patients treated with bendamustine (5.6% for patients in the B-obinutuzumab cohort, and 4.4% of patients in the B-rituximab cohort) vs. 1.6% and 2.0%, respectively, for patients on CHOP, and 1.6 and 1.8% for patients on CVP.
Dose effect?
John P. Leonard, MD, from Cornell University, New York , who introduced Dr. Marcus, commented that PFS may not be the ideal endpoint for patients with follicular lymphoma.

He pointed out that in trials comparing rituximab with obinutuzumab for other diseases, results have been mixed, with obinutuzumab showing superiority in chronic lymphocytic leukemia, but in data presented elsewhere at ASH 2016, obinutuzumab was not superior to rituximab for treatment of diffuse large B-cell lymphoma.
“One question is whether obinutuzumab, which is generally administered at a higher mg dose to patients, is in fact a better antibody or if it is in fact a dose effect,” he said.
In response to a similar question following his presentation, Dr. Marcus replied that, despite sharing a target, the two antibodies are different, with different mechanisms of action. He also noted that there is no evidence to suggest that rituximab potency would be greater in follicular lymphoma if it were given at higher doses.
The GALLIUM trial is sponsored by Hoffmann-La Roche, Dr, Marcus disclosed consulting with and receiving honoraria from the company, and relationships with other companies.
SAN DIEGO – Obinutuzumab, a second-generation anti-CD20 antibody touted as the heir apparent to rituximab, offered a progression-free survival (PFS) edge over rituximab when combined with standard chemotherapy in patients with previously untreated advanced follicular lymphoma.
But other clinicians and investigators who
attended the presentation of the GALLIUM data at a plenary session during the American Society of Hematology annual meeting indicated that despite the data, they weren’t ready to make a switch to the newer, costlier antibody.
“I feel that it is not convincing for practice-changing,” said Kanti R. Rai, MD, professor of medicine and molecular medicine at Hofstra University, Hempstead, N.Y.
“Unless we have evidence of a survival advantage in indolent disease, progression-free survivorship is not an adequate reason to jump to another antibody,” he said in an interview.
In GALLIUM, the primary endpoint of investigator-assessed 3-year PFS at a median follow-up of 34.5 months was 80% for patients with follicular lymphoma treated with obinutuzumab and one of three standard chemotherapy regimens, compared with 73.3% for patients treated with rituximab and chemotherapy. This difference translated into a hazard ratio of 0.68 favoring obinutuzumab (P = .0012).
Respective 3-year overall survival rates were similar, however, at 94% and 92.1% (HR, 0.75; P = .21).
Indolent lymphoma trial
The GALLIUM trial is a phase III study comparing obinutuzumab with rituximab when paired with one of three standard chemotherapy regimens for indolent non-Hodgkin lymphomas, including follicular lymphoma and splenic, nodal, or extranodal marginal zone lymphoma. Dr. Marcus presented data on patients with follicular lymphoma only.
The antibodies were delivered in combination with either CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone; 33.1% of patients), CVP (cyclophosphamide, vincristine, prednisone; 9.8%) or bendamustine alone (B; 57.1%) as the chemotherapy backbone. The choice of regimen was at the discretion of the treating center.
A total of 1,202 patients with follicular lymphoma were enrolled and randomized to treatment and were included in an intention-to-treat analysis.
The treatment arms were well balanced with regard to distribution of patients characteristics, with approximately 21% in each arm having Follicular Lymphoma International Prognostic Index low-risk disease; 37% having intermediate-risk disease; and 34% having high-risk disease.
Roughly half of patients in each arm had bone marrow involvement, and two-thirds had extranodal involvement.
Obinutuzumab was dosed 1,000 mg IV on days 1, 8, and 15 of cycle one, and either on day 1 of cycles two through eight every 3 weeks, or every 4 weeks during cycles two through six.
Overall response rates at the end of induction were 86.9% with rituximab and 88.5% with obinutuzumab, with complete responses of 23.8% and 19.5%, respectively.
As noted before, investigator-assessed PFS favored obinutuzumab, as did PFS assessed by independent reviewer, at 81.9% vs. 77.9% for rituximab (HR, 0.71; P = .0138).
The newer antibody also had a slight edge in time to new treatment, with 87.1% of patients on obinutuzumab not starting on new therapy, compared with 81.2% of patients on rituximab.
More bendamustine deaths
Nearly all patients in each arm had an adverse event, with grade 3 or greater events occurring in 74.6% of patients on obinutuzumab vs. 67.8% on rituximab. Rates of neutropenia, leukopenia, febrile neutropenia, infusion reactions, and thrombocytopenia were all slightly higher with obinutuzumab. Grade 3 or greater infections occurred in 20% with obinutuzumab, compared with 15.6% with rituximab.
“What we did note, however, was a high level of mortality in patients receiving either obinutuzumab-based therapy or rituximab-based therapy, which were no different between the two arms and were somewhat higher than one might expect from patients receiving induction treatment in follicular lymphoma. Hence, we did a more detailed analysis of safety by treatment regimen,” Dr. Marcus said.
There were more deaths among patients treated with bendamustine (5.6% for patients in the B-obinutuzumab cohort, and 4.4% of patients in the B-rituximab cohort) vs. 1.6% and 2.0%, respectively, for patients on CHOP, and 1.6 and 1.8% for patients on CVP.
Dose effect?
John P. Leonard, MD, from Cornell University, New York , who introduced Dr. Marcus, commented that PFS may not be the ideal endpoint for patients with follicular lymphoma.
He pointed out that in trials comparing rituximab with obinutuzumab for other diseases, results have been mixed, with obinutuzumab showing superiority in chronic lymphocytic leukemia, but in data presented elsewhere at ASH 2016, obinutuzumab was not superior to rituximab for treatment of diffuse large B-cell lymphoma.
“One question is whether obinutuzumab, which is generally administered at a higher mg dose to patients, is in fact a better antibody or if it is in fact a dose effect,” he said.
In response to a similar question following his presentation, Dr. Marcus replied that, despite sharing a target, the two antibodies are different, with different mechanisms of action. He also noted that there is no evidence to suggest that rituximab potency would be greater in follicular lymphoma if it were given at higher doses.
The GALLIUM trial is sponsored by Hoffmann-La Roche, Dr, Marcus disclosed consulting with and receiving honoraria from the company, and relationships with other companies.
SAN DIEGO – Obinutuzumab, a second-generation anti-CD20 antibody touted as the heir apparent to rituximab, offered a progression-free survival (PFS) edge over rituximab when combined with standard chemotherapy in patients with previously untreated advanced follicular lymphoma.
But other clinicians and investigators who
attended the presentation of the GALLIUM data at a plenary session during the American Society of Hematology annual meeting indicated that despite the data, they weren’t ready to make a switch to the newer, costlier antibody.
“I feel that it is not convincing for practice-changing,” said Kanti R. Rai, MD, professor of medicine and molecular medicine at Hofstra University, Hempstead, N.Y.
“Unless we have evidence of a survival advantage in indolent disease, progression-free survivorship is not an adequate reason to jump to another antibody,” he said in an interview.
In GALLIUM, the primary endpoint of investigator-assessed 3-year PFS at a median follow-up of 34.5 months was 80% for patients with follicular lymphoma treated with obinutuzumab and one of three standard chemotherapy regimens, compared with 73.3% for patients treated with rituximab and chemotherapy. This difference translated into a hazard ratio of 0.68 favoring obinutuzumab (P = .0012).
Respective 3-year overall survival rates were similar, however, at 94% and 92.1% (HR, 0.75; P = .21).
Indolent lymphoma trial
The GALLIUM trial is a phase III study comparing obinutuzumab with rituximab when paired with one of three standard chemotherapy regimens for indolent non-Hodgkin lymphomas, including follicular lymphoma and splenic, nodal, or extranodal marginal zone lymphoma. Dr. Marcus presented data on patients with follicular lymphoma only.
The antibodies were delivered in combination with either CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone; 33.1% of patients), CVP (cyclophosphamide, vincristine, prednisone; 9.8%) or bendamustine alone (B; 57.1%) as the chemotherapy backbone. The choice of regimen was at the discretion of the treating center.
A total of 1,202 patients with follicular lymphoma were enrolled and randomized to treatment and were included in an intention-to-treat analysis.
The treatment arms were well balanced with regard to distribution of patients characteristics, with approximately 21% in each arm having Follicular Lymphoma International Prognostic Index low-risk disease; 37% having intermediate-risk disease; and 34% having high-risk disease.
Roughly half of patients in each arm had bone marrow involvement, and two-thirds had extranodal involvement.
Obinutuzumab was dosed 1,000 mg IV on days 1, 8, and 15 of cycle one, and either on day 1 of cycles two through eight every 3 weeks, or every 4 weeks during cycles two through six.
Overall response rates at the end of induction were 86.9% with rituximab and 88.5% with obinutuzumab, with complete responses of 23.8% and 19.5%, respectively.
As noted before, investigator-assessed PFS favored obinutuzumab, as did PFS assessed by independent reviewer, at 81.9% vs. 77.9% for rituximab (HR, 0.71; P = .0138).
The newer antibody also had a slight edge in time to new treatment, with 87.1% of patients on obinutuzumab not starting on new therapy, compared with 81.2% of patients on rituximab.
More bendamustine deaths
Nearly all patients in each arm had an adverse event, with grade 3 or greater events occurring in 74.6% of patients on obinutuzumab vs. 67.8% on rituximab. Rates of neutropenia, leukopenia, febrile neutropenia, infusion reactions, and thrombocytopenia were all slightly higher with obinutuzumab. Grade 3 or greater infections occurred in 20% with obinutuzumab, compared with 15.6% with rituximab.
“What we did note, however, was a high level of mortality in patients receiving either obinutuzumab-based therapy or rituximab-based therapy, which were no different between the two arms and were somewhat higher than one might expect from patients receiving induction treatment in follicular lymphoma. Hence, we did a more detailed analysis of safety by treatment regimen,” Dr. Marcus said.
There were more deaths among patients treated with bendamustine (5.6% for patients in the B-obinutuzumab cohort, and 4.4% of patients in the B-rituximab cohort) vs. 1.6% and 2.0%, respectively, for patients on CHOP, and 1.6 and 1.8% for patients on CVP.
Dose effect?
John P. Leonard, MD, from Cornell University, New York , who introduced Dr. Marcus, commented that PFS may not be the ideal endpoint for patients with follicular lymphoma.
He pointed out that in trials comparing rituximab with obinutuzumab for other diseases, results have been mixed, with obinutuzumab showing superiority in chronic lymphocytic leukemia, but in data presented elsewhere at ASH 2016, obinutuzumab was not superior to rituximab for treatment of diffuse large B-cell lymphoma.
“One question is whether obinutuzumab, which is generally administered at a higher mg dose to patients, is in fact a better antibody or if it is in fact a dose effect,” he said.
In response to a similar question following his presentation, Dr. Marcus replied that, despite sharing a target, the two antibodies are different, with different mechanisms of action. He also noted that there is no evidence to suggest that rituximab potency would be greater in follicular lymphoma if it were given at higher doses.
The GALLIUM trial is sponsored by Hoffmann-La Roche, Dr, Marcus disclosed consulting with and receiving honoraria from the company, and relationships with other companies.
AT ASH 2016
Key clinical point: Obinutuzumab plus chemotherapy was associated with better 3-year progression-free survival in patients with untreated follicular lymphoma.
Major finding: Obinutuzumab/chemo was associated with a hazard ratio for investigator-assessed PFS of 0.68 (P = .0012)
Data source: Randomized phase III trial in 1202 patients with previously untreated follicular lymphoma.
Disclosures: The GALLIUM trial was sponsored by Hoffmann-La Roche. Dr. Marcus disclosed consulting with and receiving honoraria from the company, and relationships with other companies.
Ibrutinib bodes well for relapsed mantle-cell lymphoma
Progression-free survival was significantly better when patients with relapsed or refractory mantle-cell lymphoma were treated with oral ibrutinib than with intravenous temsirolimus, based on results from 280 patients in an international, randomized, open-label phase III trial.
Study subjects had undergone one or more previous rituximab-containing chemotherapy regimens to receive intravenous temsirolimus or oral ibrutinib at a daily dose of 560 mg.
Compared with temsirolimus, ibrutinib resulted in a 57% reduction in the risk of disease progression or death at a median follow-up of 20 months. Median progression-free survival – the trial’s primary endpoint – was 14.6 months for the ibrutinib group and 6.2 months for the temsirolimus group.
Ibrutinib was also better tolerated, with 68% of patients having grade 3 or higher treatment-emergent adverse events as compared to 87% of patients in the temsirolimus group, despite a median 4-fold longer treatment duration for the ibrutinib group than the temsirolimus group. Additionally, 6% of patients discontinued ibrutinib because of adverse events versus 26% in the temsirolimus group, reported Dr. Martin Dreyling of Klinikum der Universität in Munich, Germany, and his associates.
Based on results of the Functional Assessment of Cancer Therapy-Lymphoma (FACT-Lym) questionnaire, ibrutinib was associated with greater and more rapid improvements, and also with less worsening in lymphoma symptoms, as measured by the lymphoma subscale of the FACT-Lym (Lancet. 2016;387:770-78).
Ibrutinib, a first-in-class oral inhibitor of Bruton’s tyrosine kinase, is approved in the United States and the European Union at a dose of 560 mg per day for patients with mantle cell lymphoma who have received at least one previous line of therapy.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is approved in the European Union for relapsed or refractory mantle-cell lymphoma, but does not have FDA approval for this indication.
The study, funded by Janssen, is ongoing. Future research, the investigators say, should examine ibrutinib-based combination approaches for patients with relapsed or refractory mantle-cell lymphoma and in front-line therapy.
Dr. Dreyling reported grants and personal fees from Janssen and Pfizer outside of the study. Several other authors reported grants from Janssen during the study and financial ties to the company.
The findings from this phase III trial clearly establish ibrutinib as a new standard for treatment of relapsed mantle-cell lymphoma. Within the next 2 years, many expect the agent will find its way into the frontline setting for treatment of mantle cell lymphoma in combination with standard chemotherapy, based on results of another already completed phase III trial (the SHINE trial).
Despite this remarkable progress, however, mantle-cell lymphoma remains incurable. Roughly 30%-40% of people with the disease will not respond to ibrutinib, and even among responders relapse seems inevitable.
Mantle-cell lymphoma has been a model for accelerated development of novel drugs. Ibrutinib was developed with tremendous speed, and the FDA’s approval of the agent in 2013 based on findings from a non-pivotal phase II trial was surprising to everyone other than the participating patients and physicians. Hopefully the resources mobilized to bring ibrutinib so far, so fast, will continue to be available to help us learn how best to use the drug.
Dr. Peter Martin is with the department of medicine at Weill Cornell Medical College in New York. His comments are excerpted from an editorial that accompanied the study in The Lancet. Dr. Martin reported that he is a consultant for Janssen and has received honoraria from the company for speaking.
The findings from this phase III trial clearly establish ibrutinib as a new standard for treatment of relapsed mantle-cell lymphoma. Within the next 2 years, many expect the agent will find its way into the frontline setting for treatment of mantle cell lymphoma in combination with standard chemotherapy, based on results of another already completed phase III trial (the SHINE trial).
Despite this remarkable progress, however, mantle-cell lymphoma remains incurable. Roughly 30%-40% of people with the disease will not respond to ibrutinib, and even among responders relapse seems inevitable.
Mantle-cell lymphoma has been a model for accelerated development of novel drugs. Ibrutinib was developed with tremendous speed, and the FDA’s approval of the agent in 2013 based on findings from a non-pivotal phase II trial was surprising to everyone other than the participating patients and physicians. Hopefully the resources mobilized to bring ibrutinib so far, so fast, will continue to be available to help us learn how best to use the drug.
Dr. Peter Martin is with the department of medicine at Weill Cornell Medical College in New York. His comments are excerpted from an editorial that accompanied the study in The Lancet. Dr. Martin reported that he is a consultant for Janssen and has received honoraria from the company for speaking.
The findings from this phase III trial clearly establish ibrutinib as a new standard for treatment of relapsed mantle-cell lymphoma. Within the next 2 years, many expect the agent will find its way into the frontline setting for treatment of mantle cell lymphoma in combination with standard chemotherapy, based on results of another already completed phase III trial (the SHINE trial).
Despite this remarkable progress, however, mantle-cell lymphoma remains incurable. Roughly 30%-40% of people with the disease will not respond to ibrutinib, and even among responders relapse seems inevitable.
Mantle-cell lymphoma has been a model for accelerated development of novel drugs. Ibrutinib was developed with tremendous speed, and the FDA’s approval of the agent in 2013 based on findings from a non-pivotal phase II trial was surprising to everyone other than the participating patients and physicians. Hopefully the resources mobilized to bring ibrutinib so far, so fast, will continue to be available to help us learn how best to use the drug.
Dr. Peter Martin is with the department of medicine at Weill Cornell Medical College in New York. His comments are excerpted from an editorial that accompanied the study in The Lancet. Dr. Martin reported that he is a consultant for Janssen and has received honoraria from the company for speaking.
Progression-free survival was significantly better when patients with relapsed or refractory mantle-cell lymphoma were treated with oral ibrutinib than with intravenous temsirolimus, based on results from 280 patients in an international, randomized, open-label phase III trial.
Study subjects had undergone one or more previous rituximab-containing chemotherapy regimens to receive intravenous temsirolimus or oral ibrutinib at a daily dose of 560 mg.
Compared with temsirolimus, ibrutinib resulted in a 57% reduction in the risk of disease progression or death at a median follow-up of 20 months. Median progression-free survival – the trial’s primary endpoint – was 14.6 months for the ibrutinib group and 6.2 months for the temsirolimus group.
Ibrutinib was also better tolerated, with 68% of patients having grade 3 or higher treatment-emergent adverse events as compared to 87% of patients in the temsirolimus group, despite a median 4-fold longer treatment duration for the ibrutinib group than the temsirolimus group. Additionally, 6% of patients discontinued ibrutinib because of adverse events versus 26% in the temsirolimus group, reported Dr. Martin Dreyling of Klinikum der Universität in Munich, Germany, and his associates.
Based on results of the Functional Assessment of Cancer Therapy-Lymphoma (FACT-Lym) questionnaire, ibrutinib was associated with greater and more rapid improvements, and also with less worsening in lymphoma symptoms, as measured by the lymphoma subscale of the FACT-Lym (Lancet. 2016;387:770-78).
Ibrutinib, a first-in-class oral inhibitor of Bruton’s tyrosine kinase, is approved in the United States and the European Union at a dose of 560 mg per day for patients with mantle cell lymphoma who have received at least one previous line of therapy.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is approved in the European Union for relapsed or refractory mantle-cell lymphoma, but does not have FDA approval for this indication.
The study, funded by Janssen, is ongoing. Future research, the investigators say, should examine ibrutinib-based combination approaches for patients with relapsed or refractory mantle-cell lymphoma and in front-line therapy.
Dr. Dreyling reported grants and personal fees from Janssen and Pfizer outside of the study. Several other authors reported grants from Janssen during the study and financial ties to the company.
Progression-free survival was significantly better when patients with relapsed or refractory mantle-cell lymphoma were treated with oral ibrutinib than with intravenous temsirolimus, based on results from 280 patients in an international, randomized, open-label phase III trial.
Study subjects had undergone one or more previous rituximab-containing chemotherapy regimens to receive intravenous temsirolimus or oral ibrutinib at a daily dose of 560 mg.
Compared with temsirolimus, ibrutinib resulted in a 57% reduction in the risk of disease progression or death at a median follow-up of 20 months. Median progression-free survival – the trial’s primary endpoint – was 14.6 months for the ibrutinib group and 6.2 months for the temsirolimus group.
Ibrutinib was also better tolerated, with 68% of patients having grade 3 or higher treatment-emergent adverse events as compared to 87% of patients in the temsirolimus group, despite a median 4-fold longer treatment duration for the ibrutinib group than the temsirolimus group. Additionally, 6% of patients discontinued ibrutinib because of adverse events versus 26% in the temsirolimus group, reported Dr. Martin Dreyling of Klinikum der Universität in Munich, Germany, and his associates.
Based on results of the Functional Assessment of Cancer Therapy-Lymphoma (FACT-Lym) questionnaire, ibrutinib was associated with greater and more rapid improvements, and also with less worsening in lymphoma symptoms, as measured by the lymphoma subscale of the FACT-Lym (Lancet. 2016;387:770-78).
Ibrutinib, a first-in-class oral inhibitor of Bruton’s tyrosine kinase, is approved in the United States and the European Union at a dose of 560 mg per day for patients with mantle cell lymphoma who have received at least one previous line of therapy.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is approved in the European Union for relapsed or refractory mantle-cell lymphoma, but does not have FDA approval for this indication.
The study, funded by Janssen, is ongoing. Future research, the investigators say, should examine ibrutinib-based combination approaches for patients with relapsed or refractory mantle-cell lymphoma and in front-line therapy.
Dr. Dreyling reported grants and personal fees from Janssen and Pfizer outside of the study. Several other authors reported grants from Janssen during the study and financial ties to the company.
FROM THE LANCET
Key clinical point: Ibrutinib significantly improved progression-free survival, compared with temsirolimus in patients with relapsed or refractory mantle-cell lymphoma.
Major finding: Median progression-free survival was 14.6 months with ibrutinib and 6.2 months with temsirolimus.
Data source: A randomized open-label phase III trial (ongoing) that randomized 280 patients to each treatment group.
Disclosures: The study was funded by Janssen. Dr. Dreyling reported grants and personal fees from Janssen and Pfizer outside of the study, and other authors reported grants from Janssen during the study and financial ties to the company.
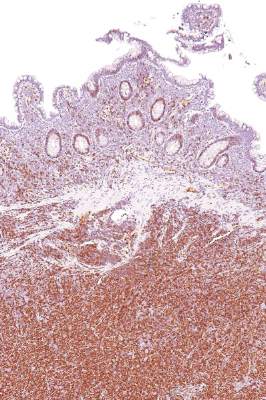
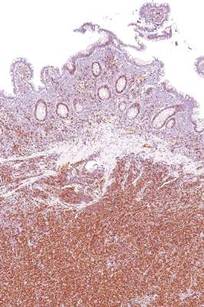
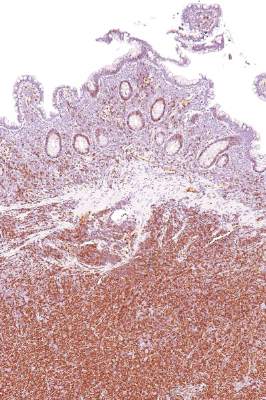
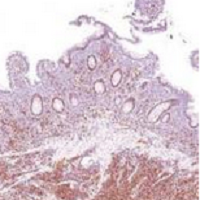
Ki-67 bests cytology, growth pattern as prognostic factor for MCL
Evaluating routinely available histopathological prognostic features from more than 500 MCL patients in prospective trials, researchers found that the Ki-67 index is a better prognostic factor than are cytology and growth pattern in mantle-cell lymphoma (MCL). In addition, the combination of the Ki-67 index with the Mantle Cell Lymphoma International Prognostic Index [MIPI] defined four prognostic groups with better discrimination than did MIPI or the two-category biologic MIPI (MIPI-b) alone.
Higher Ki-67 index was associated with poorer overall survival (OS) (hazard ratio [HR], 1.24 per 10% increase; P less than .001) and progression-free survival (PFS) (HR, 1.17; P less than .001). Consistent with an earlier, population-based study, results showed prognostic value for a 30% cutoff of the Ki-67 index. Quantitative levels below 30% provided no additional prognostic information.
“The Ki-67 index remains the only routinely available independent prognostic factor in addition to MIPI. In contrast to cytology and growth pattern, the Ki-67 evaluation has been standardized for routine application,” wrote Dr. Eva Hoster of University Hospital Munich, and colleagues. “The modified combination of Ki-67 index and MIPI integrates the most important clinical and biologic markers currently available in clinical routine and was shown to allow a simple and powerful risk stratification superior to MIPI and MIPI-b in our evaluation,” they added (J Clin Oncol. 2016 Feb. 29. doi: 10.1200/jco.63.8387).
Blastoid cytology was associated with inferior 5-year OS compared with nonblastoid cytology (35% vs. 68%; HR, 2.35; P less than .001) and PFS (29% vs. 44%; HR, 1.58; P = .007), but the effect was largely accounted for by a generally higher Ki-67 index in blastoid MCL. Diffuse growth pattern was associated slightly worse 5-year OS (61% vs. 72%; HR, 1.38; P = .048) and PFS (38% vs. 49%; HR, 1.25; P = .087), but the effect was largely explained by MIPI score.
Combining dichotomized Ki-67 (above or below 30%) with MIPI risk groups defined four prognostic groups by the sum of weights (total 0 to 3): Ki-67 of 30% or more (weight 1), intermediate-risk MIPI (weight 1), and high-risk MIPI (weight 2). The 5-year OS rates for the four groups ranged from 17% to 85%, with OS hazard ratios greater than 2 between adjacent risk groups.
The study analyzed pooled data from two randomized trials initiated in 2004 by the European Mantle Cell Lymphoma Network, MCL Younger and MCL Elderly. In total, 508 patients of median age 62 years were included. The proportion of low-risk, intermediate-risk, and high-risk MIPI were 41%, 35%, and 24%, respectively.
Research was supported in part by Roche. Dr. Hoster reported receiving funding from Roche Pharma AG and Celgene. Several of her coauthors reported ties to industry.
Evaluating routinely available histopathological prognostic features from more than 500 MCL patients in prospective trials, researchers found that the Ki-67 index is a better prognostic factor than are cytology and growth pattern in mantle-cell lymphoma (MCL). In addition, the combination of the Ki-67 index with the Mantle Cell Lymphoma International Prognostic Index [MIPI] defined four prognostic groups with better discrimination than did MIPI or the two-category biologic MIPI (MIPI-b) alone.
Higher Ki-67 index was associated with poorer overall survival (OS) (hazard ratio [HR], 1.24 per 10% increase; P less than .001) and progression-free survival (PFS) (HR, 1.17; P less than .001). Consistent with an earlier, population-based study, results showed prognostic value for a 30% cutoff of the Ki-67 index. Quantitative levels below 30% provided no additional prognostic information.
“The Ki-67 index remains the only routinely available independent prognostic factor in addition to MIPI. In contrast to cytology and growth pattern, the Ki-67 evaluation has been standardized for routine application,” wrote Dr. Eva Hoster of University Hospital Munich, and colleagues. “The modified combination of Ki-67 index and MIPI integrates the most important clinical and biologic markers currently available in clinical routine and was shown to allow a simple and powerful risk stratification superior to MIPI and MIPI-b in our evaluation,” they added (J Clin Oncol. 2016 Feb. 29. doi: 10.1200/jco.63.8387).
Blastoid cytology was associated with inferior 5-year OS compared with nonblastoid cytology (35% vs. 68%; HR, 2.35; P less than .001) and PFS (29% vs. 44%; HR, 1.58; P = .007), but the effect was largely accounted for by a generally higher Ki-67 index in blastoid MCL. Diffuse growth pattern was associated slightly worse 5-year OS (61% vs. 72%; HR, 1.38; P = .048) and PFS (38% vs. 49%; HR, 1.25; P = .087), but the effect was largely explained by MIPI score.
Combining dichotomized Ki-67 (above or below 30%) with MIPI risk groups defined four prognostic groups by the sum of weights (total 0 to 3): Ki-67 of 30% or more (weight 1), intermediate-risk MIPI (weight 1), and high-risk MIPI (weight 2). The 5-year OS rates for the four groups ranged from 17% to 85%, with OS hazard ratios greater than 2 between adjacent risk groups.
The study analyzed pooled data from two randomized trials initiated in 2004 by the European Mantle Cell Lymphoma Network, MCL Younger and MCL Elderly. In total, 508 patients of median age 62 years were included. The proportion of low-risk, intermediate-risk, and high-risk MIPI were 41%, 35%, and 24%, respectively.
Research was supported in part by Roche. Dr. Hoster reported receiving funding from Roche Pharma AG and Celgene. Several of her coauthors reported ties to industry.
Evaluating routinely available histopathological prognostic features from more than 500 MCL patients in prospective trials, researchers found that the Ki-67 index is a better prognostic factor than are cytology and growth pattern in mantle-cell lymphoma (MCL). In addition, the combination of the Ki-67 index with the Mantle Cell Lymphoma International Prognostic Index [MIPI] defined four prognostic groups with better discrimination than did MIPI or the two-category biologic MIPI (MIPI-b) alone.
Higher Ki-67 index was associated with poorer overall survival (OS) (hazard ratio [HR], 1.24 per 10% increase; P less than .001) and progression-free survival (PFS) (HR, 1.17; P less than .001). Consistent with an earlier, population-based study, results showed prognostic value for a 30% cutoff of the Ki-67 index. Quantitative levels below 30% provided no additional prognostic information.
“The Ki-67 index remains the only routinely available independent prognostic factor in addition to MIPI. In contrast to cytology and growth pattern, the Ki-67 evaluation has been standardized for routine application,” wrote Dr. Eva Hoster of University Hospital Munich, and colleagues. “The modified combination of Ki-67 index and MIPI integrates the most important clinical and biologic markers currently available in clinical routine and was shown to allow a simple and powerful risk stratification superior to MIPI and MIPI-b in our evaluation,” they added (J Clin Oncol. 2016 Feb. 29. doi: 10.1200/jco.63.8387).
Blastoid cytology was associated with inferior 5-year OS compared with nonblastoid cytology (35% vs. 68%; HR, 2.35; P less than .001) and PFS (29% vs. 44%; HR, 1.58; P = .007), but the effect was largely accounted for by a generally higher Ki-67 index in blastoid MCL. Diffuse growth pattern was associated slightly worse 5-year OS (61% vs. 72%; HR, 1.38; P = .048) and PFS (38% vs. 49%; HR, 1.25; P = .087), but the effect was largely explained by MIPI score.
Combining dichotomized Ki-67 (above or below 30%) with MIPI risk groups defined four prognostic groups by the sum of weights (total 0 to 3): Ki-67 of 30% or more (weight 1), intermediate-risk MIPI (weight 1), and high-risk MIPI (weight 2). The 5-year OS rates for the four groups ranged from 17% to 85%, with OS hazard ratios greater than 2 between adjacent risk groups.
The study analyzed pooled data from two randomized trials initiated in 2004 by the European Mantle Cell Lymphoma Network, MCL Younger and MCL Elderly. In total, 508 patients of median age 62 years were included. The proportion of low-risk, intermediate-risk, and high-risk MIPI were 41%, 35%, and 24%, respectively.
Research was supported in part by Roche. Dr. Hoster reported receiving funding from Roche Pharma AG and Celgene. Several of her coauthors reported ties to industry.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: The Ki-67 index was superior to cytology and growth pattern as a prognostic factor in mantle-cell lymphoma (MCL).
Major finding: Higher Ki-67 index was associated with poorer overall survival (hazard ratio [HR], 1.24 per 10% increase; P less than .001) and progression-free survival (HR, 1.17; P less than .001).
Data source: Pooled data from two randomized trials initiated in 2004 by the European Mantle Cell Lymphoma Network, MCL Younger and MCL Elderly, included 508 patients.
Disclosures: Research was supported in part by Roche. Dr. Hoster reported receiving funding from Roche Pharma AG and Celgene. Several of her coauthors reported ties to industry.
Lenalidomide plus rituximab achieves 87% response rate
First-line combination biologic therapy with lenalidomide plus rituximab produced an 87% overall response rate in stage 3-4 mantle cell lymphoma, in an industry-sponsored, phase II clinical trial reported online Nov. 5 in the New England Journal of Medicine.
Mantle cell lymphoma is generally incurable, and patients have a median survival of 4-5 years. Initial therapy is usually very intensive, involving high-dose chemotherapy and hematopoietic cell transplantation. Since the malignancy primarily affects older adults who aren’t suitable candidates for intensive regimens, treatment “remains a clinical challenge,” said Dr. Jia Ruan of the Meyer Cancer Center and the division of biostatistics and epidemiology, Weill Cornell Medical College and New York-Presbyterian Hospital, New York, and her associates.
Reasoning that biologic therapy might offer effective disease control with fewer and less intense adverse effects, the investigators performed the open-label, single-group trial over a 3-year period. They treated 38 patients whose mean age was 65 years (range, 42-86 years), most of whom were at intermediate or high risk for imminent progression. These participants received a 12-cycle induction phase of lenalidomide plus rituximab, followed by a maintenance phase until disease progressed, unacceptable adverse effects developed, or patients withdrew from the study. The median follow-up was 30 months (range, 10-42 months).
The primary endpoint – overall response rate – was 87% in the intention-to-treat population, and the complete response rate was 61%. The number of complete responses increased over time with continuing treatment: the median time to a partial response was 3 months, and the median time to a complete response was 11 months. Two-year progression-free survival was 85%, and 2-year overall survival was 97%, the investigators said (New Engl. J. Med. 2015 Nov 5. doi: 10.1056/NEJMoa1505237).
Only eight patients showed progression of mantle cell lymphoma while taking lenalidomide plus rituximab, two of whom died from their disease. The other six patients responded to second-line therapy and remain alive, indicating that this first-line combination biologic therapy doesn’t compromise outcomes after subsequent treatments, Dr. Ruan and her associates said.
Almost as important as these favorable survival results were the findings concerning adverse effects. Scores on several quality-of-life measures either remained stable or improved throughout the induction and maintenance phases of treatment. As expected, grade 3 or 4 hematologic adverse effects included neutropenia (50% of patients), thrombocytopenia (13%), and anemia (11%), all of which resolved; grade 3 or 4 nonhematologic adverse effects included rash (29%), tumor flare (11%), serum sickness (8%), and fatigue (8%), all of which also resolved. All the serious infections that developed during the maintenance phase of treatment, which included pneumonia, cholangitis, and West Nile viral encephalitis, also resolved with antibiotics and supportive care.
Secondary cancers that developed during follow-up included two squamous cell skin cancers, one basal cell skin cancer, two cases of melanoma in situ, one Merkel cell carcinoma, and one pancreatic cancer.
“Our data show that a lower-intensity approach for initial therapy than that usually used in the case of patients with this cancer can be highly active, with durable responses observed in most patients,” Dr. Ruan and her associates said.
This study was supported by Celgene, maker of lenalidomide, and a Weill Cornell Medical College Clinical Translational Science Center grant. Dr. Ruan reported ties to Celgene, and her associates reported ties to numerous industry sources.
First-line combination biologic therapy with lenalidomide plus rituximab produced an 87% overall response rate in stage 3-4 mantle cell lymphoma, in an industry-sponsored, phase II clinical trial reported online Nov. 5 in the New England Journal of Medicine.
Mantle cell lymphoma is generally incurable, and patients have a median survival of 4-5 years. Initial therapy is usually very intensive, involving high-dose chemotherapy and hematopoietic cell transplantation. Since the malignancy primarily affects older adults who aren’t suitable candidates for intensive regimens, treatment “remains a clinical challenge,” said Dr. Jia Ruan of the Meyer Cancer Center and the division of biostatistics and epidemiology, Weill Cornell Medical College and New York-Presbyterian Hospital, New York, and her associates.
Reasoning that biologic therapy might offer effective disease control with fewer and less intense adverse effects, the investigators performed the open-label, single-group trial over a 3-year period. They treated 38 patients whose mean age was 65 years (range, 42-86 years), most of whom were at intermediate or high risk for imminent progression. These participants received a 12-cycle induction phase of lenalidomide plus rituximab, followed by a maintenance phase until disease progressed, unacceptable adverse effects developed, or patients withdrew from the study. The median follow-up was 30 months (range, 10-42 months).
The primary endpoint – overall response rate – was 87% in the intention-to-treat population, and the complete response rate was 61%. The number of complete responses increased over time with continuing treatment: the median time to a partial response was 3 months, and the median time to a complete response was 11 months. Two-year progression-free survival was 85%, and 2-year overall survival was 97%, the investigators said (New Engl. J. Med. 2015 Nov 5. doi: 10.1056/NEJMoa1505237).
Only eight patients showed progression of mantle cell lymphoma while taking lenalidomide plus rituximab, two of whom died from their disease. The other six patients responded to second-line therapy and remain alive, indicating that this first-line combination biologic therapy doesn’t compromise outcomes after subsequent treatments, Dr. Ruan and her associates said.
Almost as important as these favorable survival results were the findings concerning adverse effects. Scores on several quality-of-life measures either remained stable or improved throughout the induction and maintenance phases of treatment. As expected, grade 3 or 4 hematologic adverse effects included neutropenia (50% of patients), thrombocytopenia (13%), and anemia (11%), all of which resolved; grade 3 or 4 nonhematologic adverse effects included rash (29%), tumor flare (11%), serum sickness (8%), and fatigue (8%), all of which also resolved. All the serious infections that developed during the maintenance phase of treatment, which included pneumonia, cholangitis, and West Nile viral encephalitis, also resolved with antibiotics and supportive care.
Secondary cancers that developed during follow-up included two squamous cell skin cancers, one basal cell skin cancer, two cases of melanoma in situ, one Merkel cell carcinoma, and one pancreatic cancer.
“Our data show that a lower-intensity approach for initial therapy than that usually used in the case of patients with this cancer can be highly active, with durable responses observed in most patients,” Dr. Ruan and her associates said.
This study was supported by Celgene, maker of lenalidomide, and a Weill Cornell Medical College Clinical Translational Science Center grant. Dr. Ruan reported ties to Celgene, and her associates reported ties to numerous industry sources.
First-line combination biologic therapy with lenalidomide plus rituximab produced an 87% overall response rate in stage 3-4 mantle cell lymphoma, in an industry-sponsored, phase II clinical trial reported online Nov. 5 in the New England Journal of Medicine.
Mantle cell lymphoma is generally incurable, and patients have a median survival of 4-5 years. Initial therapy is usually very intensive, involving high-dose chemotherapy and hematopoietic cell transplantation. Since the malignancy primarily affects older adults who aren’t suitable candidates for intensive regimens, treatment “remains a clinical challenge,” said Dr. Jia Ruan of the Meyer Cancer Center and the division of biostatistics and epidemiology, Weill Cornell Medical College and New York-Presbyterian Hospital, New York, and her associates.
Reasoning that biologic therapy might offer effective disease control with fewer and less intense adverse effects, the investigators performed the open-label, single-group trial over a 3-year period. They treated 38 patients whose mean age was 65 years (range, 42-86 years), most of whom were at intermediate or high risk for imminent progression. These participants received a 12-cycle induction phase of lenalidomide plus rituximab, followed by a maintenance phase until disease progressed, unacceptable adverse effects developed, or patients withdrew from the study. The median follow-up was 30 months (range, 10-42 months).
The primary endpoint – overall response rate – was 87% in the intention-to-treat population, and the complete response rate was 61%. The number of complete responses increased over time with continuing treatment: the median time to a partial response was 3 months, and the median time to a complete response was 11 months. Two-year progression-free survival was 85%, and 2-year overall survival was 97%, the investigators said (New Engl. J. Med. 2015 Nov 5. doi: 10.1056/NEJMoa1505237).
Only eight patients showed progression of mantle cell lymphoma while taking lenalidomide plus rituximab, two of whom died from their disease. The other six patients responded to second-line therapy and remain alive, indicating that this first-line combination biologic therapy doesn’t compromise outcomes after subsequent treatments, Dr. Ruan and her associates said.
Almost as important as these favorable survival results were the findings concerning adverse effects. Scores on several quality-of-life measures either remained stable or improved throughout the induction and maintenance phases of treatment. As expected, grade 3 or 4 hematologic adverse effects included neutropenia (50% of patients), thrombocytopenia (13%), and anemia (11%), all of which resolved; grade 3 or 4 nonhematologic adverse effects included rash (29%), tumor flare (11%), serum sickness (8%), and fatigue (8%), all of which also resolved. All the serious infections that developed during the maintenance phase of treatment, which included pneumonia, cholangitis, and West Nile viral encephalitis, also resolved with antibiotics and supportive care.
Secondary cancers that developed during follow-up included two squamous cell skin cancers, one basal cell skin cancer, two cases of melanoma in situ, one Merkel cell carcinoma, and one pancreatic cancer.
“Our data show that a lower-intensity approach for initial therapy than that usually used in the case of patients with this cancer can be highly active, with durable responses observed in most patients,” Dr. Ruan and her associates said.
This study was supported by Celgene, maker of lenalidomide, and a Weill Cornell Medical College Clinical Translational Science Center grant. Dr. Ruan reported ties to Celgene, and her associates reported ties to numerous industry sources.
FROM NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: First-line combination biologic therapy with lenalidomide plus rituximab produced an 87% overall response rate in stage 3 or 4 mantle cell lymphoma.
Major finding: The primary endpoint – overall response rate – was 87%, and the complete response rate was 61%.
Data source: A multicenter, industry-sponsored, open-label, phase II study involving 38 patients with mantle cell lymphoma followed for a median of 30 months.
Disclosures: This study was supported by Celgene, maker of lenalidomide, and a Weill Cornell Medical College Clinical Translational Science Center grant. Dr. Ruan reported ties to Celgene, and her associates reported ties to numerous industry sources.
RBAC500 safe, effective for elderly patients with mantle cell lymphoma
Reducing the dose of cytarabine from 800 mg/m2 to 500 mg/m2 allowed a regimen of rituximab, bendamustine, and cytarabine to be safely administered as first-line therapy to elderly patients who had mantle cell lymphoma and were not candidates for autologous stem cell transplant, according to Dr. Carlo Visco of the San Bortolo Hospital in Vicenza, Italy.
“Hematologic toxicity was substantially reduced, compared to the earlier study, Dr. Visco said, calling the R-BAC500 regimen “a highly effective treatment” for patients with mantle cell lymphoma.
Speaking at the at the International Congress on Malignant Lymphoma in Lugano, Switzerland, Dr. Visco noted the “encouraging results, but high hematologic toxicity” seen in a previous study that employed the higher cytarabine dose. In that previous study, transient grades 3-4 thrombocytopenia occurred in 76% of cycles.
In an attempt to reduce hematologic toxicity, the Fondazione Italiana Linfomi designed a phase II trial in which the cytarabine dose was lowered to 500 mg/m2 (R-BAC500). The administration schedule of cytarabine (on days 2-4) and the other components of the original regimen (rituximab, 375 mg/m2, on day 1 and bendamustine, 70 mg/m2, on days 2 and 3) remained unchanged.
The 57 study subjects, median age 71, had newly diagnosed mantle cell lymphoma, and were not eligible for autologous transplant as determined by the comprehensive geriatric assessment; 75% of the patients were males and 91% had Ann Arbor stage III/IV disease.
The Mantle Cell International Prognostic Index (MIPI) was low in 15%, intermediate in 40%, and high in 45%; 9% had the blastoid variant of the disease.
The primary endpoints were complete remission rate, as measured by 18-fluorodeoxyglucose–PET, according to Cheson criteria 2007, and safety. Secondary endpoints included molecular response rate, progression-free survival, and overall survival.
The overall response rate was 96%, and the complete remission rate was 93%. The molecular response rate at the end of treatment was 76% on peripheral blood and 55% on bone marrow samples. With a median follow-up of 18 months, the projected 2-year progression-free survival was 83%, and the overall survival was 91% without maintenance therapy.
Nearly all patients, 53 of 57, received at least four cycles of therapy, and 36 had six cycles. Treatment was discontinued because of toxicity (primarily hematologic) in 15 patients. Only one patient discontinued because of progressive disease.
Grade 3 or 4 neutropenia and thrombocytopenia were observed in about half of administered cycles. Febrile neutropenia occurred in 6%. Extrahematologic toxicity was mainly cardiac (5%).
BR is a commonly used regimen for older, less fit patients with MCL. Inclusion of high dose cytarabine appears to be beneficial n younger patients with MCL, particularly in induction pre-SCT. The FIL has been investigating intermediate doses of cytarabine combined, rather than alternating, with BR. This phase 2 study utilized cytarabine 500 mg/m2 daily x 3 with BR (slightly lower than standard dose bendamustine). The patient population was older with predominantly intermediate-high MIPI, yet results were impressive, particularly the PET negative rate of 93% and marrow MRD negative rate of 55%. Follow-up is short, but remissions do appear durable. Concerns are the high number of patients unable to complete planned therapy, the high rate of grade 3 and 4 cytopenias, and the frequency of visits required for close blood count monitoring and blood product support.
BR is a commonly used regimen for older, less fit patients with MCL. Inclusion of high dose cytarabine appears to be beneficial n younger patients with MCL, particularly in induction pre-SCT. The FIL has been investigating intermediate doses of cytarabine combined, rather than alternating, with BR. This phase 2 study utilized cytarabine 500 mg/m2 daily x 3 with BR (slightly lower than standard dose bendamustine). The patient population was older with predominantly intermediate-high MIPI, yet results were impressive, particularly the PET negative rate of 93% and marrow MRD negative rate of 55%. Follow-up is short, but remissions do appear durable. Concerns are the high number of patients unable to complete planned therapy, the high rate of grade 3 and 4 cytopenias, and the frequency of visits required for close blood count monitoring and blood product support.
BR is a commonly used regimen for older, less fit patients with MCL. Inclusion of high dose cytarabine appears to be beneficial n younger patients with MCL, particularly in induction pre-SCT. The FIL has been investigating intermediate doses of cytarabine combined, rather than alternating, with BR. This phase 2 study utilized cytarabine 500 mg/m2 daily x 3 with BR (slightly lower than standard dose bendamustine). The patient population was older with predominantly intermediate-high MIPI, yet results were impressive, particularly the PET negative rate of 93% and marrow MRD negative rate of 55%. Follow-up is short, but remissions do appear durable. Concerns are the high number of patients unable to complete planned therapy, the high rate of grade 3 and 4 cytopenias, and the frequency of visits required for close blood count monitoring and blood product support.
Reducing the dose of cytarabine from 800 mg/m2 to 500 mg/m2 allowed a regimen of rituximab, bendamustine, and cytarabine to be safely administered as first-line therapy to elderly patients who had mantle cell lymphoma and were not candidates for autologous stem cell transplant, according to Dr. Carlo Visco of the San Bortolo Hospital in Vicenza, Italy.
“Hematologic toxicity was substantially reduced, compared to the earlier study, Dr. Visco said, calling the R-BAC500 regimen “a highly effective treatment” for patients with mantle cell lymphoma.
Speaking at the at the International Congress on Malignant Lymphoma in Lugano, Switzerland, Dr. Visco noted the “encouraging results, but high hematologic toxicity” seen in a previous study that employed the higher cytarabine dose. In that previous study, transient grades 3-4 thrombocytopenia occurred in 76% of cycles.
In an attempt to reduce hematologic toxicity, the Fondazione Italiana Linfomi designed a phase II trial in which the cytarabine dose was lowered to 500 mg/m2 (R-BAC500). The administration schedule of cytarabine (on days 2-4) and the other components of the original regimen (rituximab, 375 mg/m2, on day 1 and bendamustine, 70 mg/m2, on days 2 and 3) remained unchanged.
The 57 study subjects, median age 71, had newly diagnosed mantle cell lymphoma, and were not eligible for autologous transplant as determined by the comprehensive geriatric assessment; 75% of the patients were males and 91% had Ann Arbor stage III/IV disease.
The Mantle Cell International Prognostic Index (MIPI) was low in 15%, intermediate in 40%, and high in 45%; 9% had the blastoid variant of the disease.
The primary endpoints were complete remission rate, as measured by 18-fluorodeoxyglucose–PET, according to Cheson criteria 2007, and safety. Secondary endpoints included molecular response rate, progression-free survival, and overall survival.
The overall response rate was 96%, and the complete remission rate was 93%. The molecular response rate at the end of treatment was 76% on peripheral blood and 55% on bone marrow samples. With a median follow-up of 18 months, the projected 2-year progression-free survival was 83%, and the overall survival was 91% without maintenance therapy.
Nearly all patients, 53 of 57, received at least four cycles of therapy, and 36 had six cycles. Treatment was discontinued because of toxicity (primarily hematologic) in 15 patients. Only one patient discontinued because of progressive disease.
Grade 3 or 4 neutropenia and thrombocytopenia were observed in about half of administered cycles. Febrile neutropenia occurred in 6%. Extrahematologic toxicity was mainly cardiac (5%).
Reducing the dose of cytarabine from 800 mg/m2 to 500 mg/m2 allowed a regimen of rituximab, bendamustine, and cytarabine to be safely administered as first-line therapy to elderly patients who had mantle cell lymphoma and were not candidates for autologous stem cell transplant, according to Dr. Carlo Visco of the San Bortolo Hospital in Vicenza, Italy.
“Hematologic toxicity was substantially reduced, compared to the earlier study, Dr. Visco said, calling the R-BAC500 regimen “a highly effective treatment” for patients with mantle cell lymphoma.
Speaking at the at the International Congress on Malignant Lymphoma in Lugano, Switzerland, Dr. Visco noted the “encouraging results, but high hematologic toxicity” seen in a previous study that employed the higher cytarabine dose. In that previous study, transient grades 3-4 thrombocytopenia occurred in 76% of cycles.
In an attempt to reduce hematologic toxicity, the Fondazione Italiana Linfomi designed a phase II trial in which the cytarabine dose was lowered to 500 mg/m2 (R-BAC500). The administration schedule of cytarabine (on days 2-4) and the other components of the original regimen (rituximab, 375 mg/m2, on day 1 and bendamustine, 70 mg/m2, on days 2 and 3) remained unchanged.
The 57 study subjects, median age 71, had newly diagnosed mantle cell lymphoma, and were not eligible for autologous transplant as determined by the comprehensive geriatric assessment; 75% of the patients were males and 91% had Ann Arbor stage III/IV disease.
The Mantle Cell International Prognostic Index (MIPI) was low in 15%, intermediate in 40%, and high in 45%; 9% had the blastoid variant of the disease.
The primary endpoints were complete remission rate, as measured by 18-fluorodeoxyglucose–PET, according to Cheson criteria 2007, and safety. Secondary endpoints included molecular response rate, progression-free survival, and overall survival.
The overall response rate was 96%, and the complete remission rate was 93%. The molecular response rate at the end of treatment was 76% on peripheral blood and 55% on bone marrow samples. With a median follow-up of 18 months, the projected 2-year progression-free survival was 83%, and the overall survival was 91% without maintenance therapy.
Nearly all patients, 53 of 57, received at least four cycles of therapy, and 36 had six cycles. Treatment was discontinued because of toxicity (primarily hematologic) in 15 patients. Only one patient discontinued because of progressive disease.
Grade 3 or 4 neutropenia and thrombocytopenia were observed in about half of administered cycles. Febrile neutropenia occurred in 6%. Extrahematologic toxicity was mainly cardiac (5%).
FROM 13-ICML
Key clinical point: Reducing the dose of cytarabine from 800 mg/m2 to 500 mg/m2 allowed a regimen of rituximab, bendamustine, and cytarabine to be safely administered as first-line therapy to elderly patients with mantle cell lymphoma.
Major finding: Nearly all patients, 53 of 57, received at least four cycles of therapy, and 36 had six cycles. Treatment was discontinued because of toxicity (primarily hematologic) in 15 patients.
Data source: 57 study subjects, median age 71, who had newly diagnosed mantle cell lymphoma and were not eligible for autologous transplant as determined by the comprehensive geriatric assessment.
Disclosures: The trial was conducted by the Fondazione Italiana Linfomi. There were no relevant financial disclosures.
Bendamustine regimen may be induction-therapy option in mantle cell lymphoma
Rituximab plus bendamustine may prove to be an induction-therapy option for younger patients with mantle cell lymphoma, Dr. Richard Chen and his colleagues in a SWOG (Southwest Oncology Group) trial reported at the International Congress on Malignant Lymphoma in Lugano, Switzerland.
Compared with a more aggressive combination regimen, a rituximab plus bendamustine (Treanda) option is a simple regimen that can be given in an outpatient setting and was associated with fewer adverse events and similar 2-year outcomes, the researchers found. The more aggressive regimen, however, was associated with lower-than-expected stem cell mobilization rates and the trial was prematurely closed, allowing no significant results.
For this study, two induction-therapy regimens were compared in 53 patients with untreated stage III or IV (or bulky stage II) mantle cell lymphoma. All patients were less than age 65 years and received rituximab (R) in combination with one of two regimens: 18 patients received four cycles of R-HyperCVAD + methotrexate + cytarabine (R-HyperCVAD/MTX/ARA-C) and 35 patients received six cycles of R-bendamustine.
The overall response rate was 94% with R-HyperCVAD/MTX/ARA-C and 86% with R-bendamustine; the complete response rates were 31% and 43%, respectively; the partial response rates were 62% and 43%, respectively, Dr. Chen and his associates reported.
The median follow-up for surviving patients is nearly 24 months. The estimated 2-year progression-free survival was 87% for patients in both treatment groups.
Significantly higher rates of bone marrow toxicity occurred in the group receiving the R-HyperCVAD/MTX/ARA-C regimen, compared with the bendamustine regimen. Grade 3 and 4 thrombocytopenia occurred in 69% given R-HyperCVAD/MTX/ARA-C and 17% given R-bendamustine. Anemia affected 56% of those given R-HyperCVAD/MTX/ARA-C and 8.6% given R-bendamustine. Neutropenia was seen in 63% given R-HyperCVAD/MTX/ARA-C and 34% of patients given R-bendamustine. Febrile neutropenia occurred in 31% given R-HyperCVAD/MTX/ARA-C and 14% given R-bendamustine.
The study was discontinued prematurely because of the low mobilization of stem cells at the transplant phase of the study in patients given R-HyperCVAD/MTX/ARA-C. Just 4 of 16 patients on R-HyperCVAD/MTX/ARA-C and 21 of 35 patients given R-bendamustine underwent autologous stem cell transplants.
The R-bendamustine regimen seems less myelosuppressive. Because of the premature closure of the trial, the study did not reach statistical significance for 2-year progression-free survival, the researchers reported. Since bendamustine in combination with rituximab was associated with lower rates of hematologic toxicity, however, it warrants further study as an induction regimen, they concluded.
Young, fit patients with mantle cell lymphoma (MCL) are often treated with intensive, though non-curative, therapy. While some centers still use R-HyperCVAD/MA alone, most use alternating R-CHOP-based and high dose cytarabine-based regimens, followed by SCT. The U.S. Intergroup trial, led by SWOG, was designed to gather information about a strategy using a limited number of cycles of R-HyperCVAD/MA followed by SCT, and an alternative strategy using an effective but less-intense induction, bendamustine-rituximab (BR), also followed by SCT. The R-HyperCVAD/MA arm was closed early due to difficulties with stem cell collection. While there are technical reasons for this that likely could be overcome, results with other pre-SCT regimens are good enough that this is not likely to be further studied. The BR followed by SCT arm was closed after accrual of 35 patients, enough to get a sense that this was feasible, although it will be important to see further updates regarding how many of these patients did go on to SCT, and their ultimate outcomes. A key question is whether a study comparing BR induction with a different, commonly used intense regimen pre-SCT is worth the commitment of resources, given the range of novel agents now available for MCL.
Dr. Mitchell Smith is a medical oncologist affiliated with the Cleveland Clinic.
Young, fit patients with mantle cell lymphoma (MCL) are often treated with intensive, though non-curative, therapy. While some centers still use R-HyperCVAD/MA alone, most use alternating R-CHOP-based and high dose cytarabine-based regimens, followed by SCT. The U.S. Intergroup trial, led by SWOG, was designed to gather information about a strategy using a limited number of cycles of R-HyperCVAD/MA followed by SCT, and an alternative strategy using an effective but less-intense induction, bendamustine-rituximab (BR), also followed by SCT. The R-HyperCVAD/MA arm was closed early due to difficulties with stem cell collection. While there are technical reasons for this that likely could be overcome, results with other pre-SCT regimens are good enough that this is not likely to be further studied. The BR followed by SCT arm was closed after accrual of 35 patients, enough to get a sense that this was feasible, although it will be important to see further updates regarding how many of these patients did go on to SCT, and their ultimate outcomes. A key question is whether a study comparing BR induction with a different, commonly used intense regimen pre-SCT is worth the commitment of resources, given the range of novel agents now available for MCL.
Dr. Mitchell Smith is a medical oncologist affiliated with the Cleveland Clinic.
Young, fit patients with mantle cell lymphoma (MCL) are often treated with intensive, though non-curative, therapy. While some centers still use R-HyperCVAD/MA alone, most use alternating R-CHOP-based and high dose cytarabine-based regimens, followed by SCT. The U.S. Intergroup trial, led by SWOG, was designed to gather information about a strategy using a limited number of cycles of R-HyperCVAD/MA followed by SCT, and an alternative strategy using an effective but less-intense induction, bendamustine-rituximab (BR), also followed by SCT. The R-HyperCVAD/MA arm was closed early due to difficulties with stem cell collection. While there are technical reasons for this that likely could be overcome, results with other pre-SCT regimens are good enough that this is not likely to be further studied. The BR followed by SCT arm was closed after accrual of 35 patients, enough to get a sense that this was feasible, although it will be important to see further updates regarding how many of these patients did go on to SCT, and their ultimate outcomes. A key question is whether a study comparing BR induction with a different, commonly used intense regimen pre-SCT is worth the commitment of resources, given the range of novel agents now available for MCL.
Dr. Mitchell Smith is a medical oncologist affiliated with the Cleveland Clinic.
Rituximab plus bendamustine may prove to be an induction-therapy option for younger patients with mantle cell lymphoma, Dr. Richard Chen and his colleagues in a SWOG (Southwest Oncology Group) trial reported at the International Congress on Malignant Lymphoma in Lugano, Switzerland.
Compared with a more aggressive combination regimen, a rituximab plus bendamustine (Treanda) option is a simple regimen that can be given in an outpatient setting and was associated with fewer adverse events and similar 2-year outcomes, the researchers found. The more aggressive regimen, however, was associated with lower-than-expected stem cell mobilization rates and the trial was prematurely closed, allowing no significant results.
For this study, two induction-therapy regimens were compared in 53 patients with untreated stage III or IV (or bulky stage II) mantle cell lymphoma. All patients were less than age 65 years and received rituximab (R) in combination with one of two regimens: 18 patients received four cycles of R-HyperCVAD + methotrexate + cytarabine (R-HyperCVAD/MTX/ARA-C) and 35 patients received six cycles of R-bendamustine.
The overall response rate was 94% with R-HyperCVAD/MTX/ARA-C and 86% with R-bendamustine; the complete response rates were 31% and 43%, respectively; the partial response rates were 62% and 43%, respectively, Dr. Chen and his associates reported.
The median follow-up for surviving patients is nearly 24 months. The estimated 2-year progression-free survival was 87% for patients in both treatment groups.
Significantly higher rates of bone marrow toxicity occurred in the group receiving the R-HyperCVAD/MTX/ARA-C regimen, compared with the bendamustine regimen. Grade 3 and 4 thrombocytopenia occurred in 69% given R-HyperCVAD/MTX/ARA-C and 17% given R-bendamustine. Anemia affected 56% of those given R-HyperCVAD/MTX/ARA-C and 8.6% given R-bendamustine. Neutropenia was seen in 63% given R-HyperCVAD/MTX/ARA-C and 34% of patients given R-bendamustine. Febrile neutropenia occurred in 31% given R-HyperCVAD/MTX/ARA-C and 14% given R-bendamustine.
The study was discontinued prematurely because of the low mobilization of stem cells at the transplant phase of the study in patients given R-HyperCVAD/MTX/ARA-C. Just 4 of 16 patients on R-HyperCVAD/MTX/ARA-C and 21 of 35 patients given R-bendamustine underwent autologous stem cell transplants.
The R-bendamustine regimen seems less myelosuppressive. Because of the premature closure of the trial, the study did not reach statistical significance for 2-year progression-free survival, the researchers reported. Since bendamustine in combination with rituximab was associated with lower rates of hematologic toxicity, however, it warrants further study as an induction regimen, they concluded.
Rituximab plus bendamustine may prove to be an induction-therapy option for younger patients with mantle cell lymphoma, Dr. Richard Chen and his colleagues in a SWOG (Southwest Oncology Group) trial reported at the International Congress on Malignant Lymphoma in Lugano, Switzerland.
Compared with a more aggressive combination regimen, a rituximab plus bendamustine (Treanda) option is a simple regimen that can be given in an outpatient setting and was associated with fewer adverse events and similar 2-year outcomes, the researchers found. The more aggressive regimen, however, was associated with lower-than-expected stem cell mobilization rates and the trial was prematurely closed, allowing no significant results.
For this study, two induction-therapy regimens were compared in 53 patients with untreated stage III or IV (or bulky stage II) mantle cell lymphoma. All patients were less than age 65 years and received rituximab (R) in combination with one of two regimens: 18 patients received four cycles of R-HyperCVAD + methotrexate + cytarabine (R-HyperCVAD/MTX/ARA-C) and 35 patients received six cycles of R-bendamustine.
The overall response rate was 94% with R-HyperCVAD/MTX/ARA-C and 86% with R-bendamustine; the complete response rates were 31% and 43%, respectively; the partial response rates were 62% and 43%, respectively, Dr. Chen and his associates reported.
The median follow-up for surviving patients is nearly 24 months. The estimated 2-year progression-free survival was 87% for patients in both treatment groups.
Significantly higher rates of bone marrow toxicity occurred in the group receiving the R-HyperCVAD/MTX/ARA-C regimen, compared with the bendamustine regimen. Grade 3 and 4 thrombocytopenia occurred in 69% given R-HyperCVAD/MTX/ARA-C and 17% given R-bendamustine. Anemia affected 56% of those given R-HyperCVAD/MTX/ARA-C and 8.6% given R-bendamustine. Neutropenia was seen in 63% given R-HyperCVAD/MTX/ARA-C and 34% of patients given R-bendamustine. Febrile neutropenia occurred in 31% given R-HyperCVAD/MTX/ARA-C and 14% given R-bendamustine.
The study was discontinued prematurely because of the low mobilization of stem cells at the transplant phase of the study in patients given R-HyperCVAD/MTX/ARA-C. Just 4 of 16 patients on R-HyperCVAD/MTX/ARA-C and 21 of 35 patients given R-bendamustine underwent autologous stem cell transplants.
The R-bendamustine regimen seems less myelosuppressive. Because of the premature closure of the trial, the study did not reach statistical significance for 2-year progression-free survival, the researchers reported. Since bendamustine in combination with rituximab was associated with lower rates of hematologic toxicity, however, it warrants further study as an induction regimen, they concluded.
FROM 13-ICML
Key clinical point: Rituximab plus bendamustine may prove to be an option for induction therapy prior to autologous stem cell transplant in patients with mantle cell lymphoma.
Major finding: The overall response rate was 94% with R-HyperCVAD/MTX/ARA-C and 86% with R-bendamustine; the complete response rates were 31% and 43%, respectively; the partial response rates were 62% and 43%, respectively.
Data source: 53 patients with untreated stage III or IV (or bulky stage II) mantle cell lymphoma.
Disclosures: The investigators did not report any conflicts.
Swapping bortezomib for vincristine improves PFS in mantle cell lymphoma
CHICAGO – Tweaking the R-CHOP recipe to substitute bortezomib for vincristine may bring clinical benefit to patients with newly diagnosed mantle cell lymphoma who are ineligible for bone marrow transplant.
In a randomized phase III trial in patients with MCL who could not undergo bone marrow transplant due to age or comorbidities, those patients who received a combination of rituximab, doxorubicin, bortezomib (Velcade), cyclophosphamide, and prednisone (the VR-CAP regimen) had significantly better progression-free survival than did patients treated with R-CHOP, the same regimen but with vincristine instead of bortezomib.
The VR-CAP regimen "could be considered a new standard of care for newly diagnosed mantle cell lymphoma patients not considered for intensive treatment and bone marrow transplant," Dr. Franco Cavalli reported at the annual meeting of the American Society of Clinical Oncology.
The bortezomib-containing regimen was associated with more grade 3 or 4 toxicities than standard R-CHOP, but adverse events were manageable, and most patients in each study arm were able to stay on chemotherapy for all prescribed cycles, said Dr. Cavalli of the Oncology Institute of Southern Switzerland, Bellinzona.
R-CHOP is a standard frontline therapy for patients with MCL who are deemed to be ineligible for intensive therapy and/or bone marrow transplant. But the regimen offers only limited progression-free survival (PFS) in this population, Dr. Cavalli said.
Because bortezomib is approved for the treatment of relapsed MCL in the United States and 53 other nations, the authors investigated whether it could improve outcomes when given to patients with newly diagnosed disease.
The LYM-3002 trial was a phase III study conducted at 128 centers in 28 countries in Europe, Asia, the Americas, and Africa. Patients with newly diagnosed MCL stage II-IV, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2, and who were ineligible or not considered for bone marrow transplant, were randomized to receive either R-CHOP or VR-CAP. In R-CHOP, vincristine 1.4 mg/m2 was delivered to a maximum of 2 mg intravenously on day 1 of each cycle. In VR-CAP, bortezomib 1.3 mg/m2 was delivered via intravenous infusion on days 1, 4, 8, and 11 of each cycle. Patients were assigned to receive at least six cycles of therapy, with an additional two cycles possible if investigator-assessed responses were first documented at the end of cycle 6.
A total of 487 patients (244 assigned to R-CHOP and 243 to VR-CAP) were included in the intention-to-treat analysis.
At a median follow-up of 40 months, median PFS, the primary endpoint, was 24.7 months in the VR-CAP arm, compared with 14.4 months for R-CHOP (hazard ratio, 0.63; P less than .001), as judged by an independent review committee. Investigator-rated PFS was 30.7 months vs. 16.1 months, respectively (HR, 0.51; P less than .001).
Clinical responses according to International Working Group revised response criteria for malignant lymphoma included overall response rates (complete response, complete unconfirmed response, and partial response) of 90% in the R-CHOP–treated patients and 92% in those who received VR-CAP.
However, there was a higher proportion of combined complete response and complete unconfirmed response in the VR-CAP group: 42% for R-CHOP vs. 53% for VR-CAP (odds ratio, 1.69; P = .007).
Median time to response was also shorter with VR-CAP (1.6 vs. 1.4 months; HR, 1.54; P less than .001).
Independent reviewer-rated median time-to-progression was 16.1 months for R-CHOP vs. 30.5 months for VR-CAP (HR, 0.58; P less than .001). Median time to next therapy was 24.8 vs. 44.5 months, respectively (HR, 0.50; P less than .001), and median treatment-free interval was 20.5 vs. 40.6 months (HR, 0.50; P less than .001).
Median overall survival was 56.3 months among R-CHOP–treated patients, vs. not reached among VR-CAP–treated patients.
Grade 3 or higher drug-related adverse events occurred in 85% and 93% of patients, respectively. The events were considered serious in 21% of R-CHOP–treated patients and in 33% of VR-CAP–treated patients. In all, 7% of patients on R-CHOP and 9% of those on VR-CAP discontinued therapy because of adverse events.
Grade 3 adverse events were more frequent with VR-CAP and included neutropenia, leukopenia, lymphopenia, and thrombocytopenia, the last of which occurred in 6% of patients on R-CHOP, compared with 57% for VR-CAP. Despite this difference, however, rates of grade 3 or higher bleeding were similar between the groups, occurring in 1.2% and 1.7%, respectively.
The invited discussant, Dr. Michael E. Williams, chief of hematology/oncology at the University of Virginia Cancer Center, Charlottesville, commented that the study provides proof of principle "that if you add an active single agent and substitute bortezomib for vincristine, which would appear to be a less active agent, that you can certainly improve PFS significantly."
Dr. Williams said that it remains to be seen, however, whether, as Dr. Cavalli suggested, certain treatment strategies could be used to lower the incidence of drug-related adverse events and improve PFS rates further, such as the use of subcutaneous rather than intravenous bortezomib, different dosing schedules, or rituximab in the maintenance phase.
The study was supported by Janssen Global Services and Millennium. Dr. Cavalli disclosed receiving travel support for attending the ASCO annual meeting, but reported having no other conflicts of interest. Dr. Williams disclosed consulting/advising for Millennium and receiving research funding from Janssen and Millennium.
CHICAGO – Tweaking the R-CHOP recipe to substitute bortezomib for vincristine may bring clinical benefit to patients with newly diagnosed mantle cell lymphoma who are ineligible for bone marrow transplant.
In a randomized phase III trial in patients with MCL who could not undergo bone marrow transplant due to age or comorbidities, those patients who received a combination of rituximab, doxorubicin, bortezomib (Velcade), cyclophosphamide, and prednisone (the VR-CAP regimen) had significantly better progression-free survival than did patients treated with R-CHOP, the same regimen but with vincristine instead of bortezomib.
The VR-CAP regimen "could be considered a new standard of care for newly diagnosed mantle cell lymphoma patients not considered for intensive treatment and bone marrow transplant," Dr. Franco Cavalli reported at the annual meeting of the American Society of Clinical Oncology.
The bortezomib-containing regimen was associated with more grade 3 or 4 toxicities than standard R-CHOP, but adverse events were manageable, and most patients in each study arm were able to stay on chemotherapy for all prescribed cycles, said Dr. Cavalli of the Oncology Institute of Southern Switzerland, Bellinzona.
R-CHOP is a standard frontline therapy for patients with MCL who are deemed to be ineligible for intensive therapy and/or bone marrow transplant. But the regimen offers only limited progression-free survival (PFS) in this population, Dr. Cavalli said.
Because bortezomib is approved for the treatment of relapsed MCL in the United States and 53 other nations, the authors investigated whether it could improve outcomes when given to patients with newly diagnosed disease.
The LYM-3002 trial was a phase III study conducted at 128 centers in 28 countries in Europe, Asia, the Americas, and Africa. Patients with newly diagnosed MCL stage II-IV, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2, and who were ineligible or not considered for bone marrow transplant, were randomized to receive either R-CHOP or VR-CAP. In R-CHOP, vincristine 1.4 mg/m2 was delivered to a maximum of 2 mg intravenously on day 1 of each cycle. In VR-CAP, bortezomib 1.3 mg/m2 was delivered via intravenous infusion on days 1, 4, 8, and 11 of each cycle. Patients were assigned to receive at least six cycles of therapy, with an additional two cycles possible if investigator-assessed responses were first documented at the end of cycle 6.
A total of 487 patients (244 assigned to R-CHOP and 243 to VR-CAP) were included in the intention-to-treat analysis.
At a median follow-up of 40 months, median PFS, the primary endpoint, was 24.7 months in the VR-CAP arm, compared with 14.4 months for R-CHOP (hazard ratio, 0.63; P less than .001), as judged by an independent review committee. Investigator-rated PFS was 30.7 months vs. 16.1 months, respectively (HR, 0.51; P less than .001).
Clinical responses according to International Working Group revised response criteria for malignant lymphoma included overall response rates (complete response, complete unconfirmed response, and partial response) of 90% in the R-CHOP–treated patients and 92% in those who received VR-CAP.
However, there was a higher proportion of combined complete response and complete unconfirmed response in the VR-CAP group: 42% for R-CHOP vs. 53% for VR-CAP (odds ratio, 1.69; P = .007).
Median time to response was also shorter with VR-CAP (1.6 vs. 1.4 months; HR, 1.54; P less than .001).
Independent reviewer-rated median time-to-progression was 16.1 months for R-CHOP vs. 30.5 months for VR-CAP (HR, 0.58; P less than .001). Median time to next therapy was 24.8 vs. 44.5 months, respectively (HR, 0.50; P less than .001), and median treatment-free interval was 20.5 vs. 40.6 months (HR, 0.50; P less than .001).
Median overall survival was 56.3 months among R-CHOP–treated patients, vs. not reached among VR-CAP–treated patients.
Grade 3 or higher drug-related adverse events occurred in 85% and 93% of patients, respectively. The events were considered serious in 21% of R-CHOP–treated patients and in 33% of VR-CAP–treated patients. In all, 7% of patients on R-CHOP and 9% of those on VR-CAP discontinued therapy because of adverse events.
Grade 3 adverse events were more frequent with VR-CAP and included neutropenia, leukopenia, lymphopenia, and thrombocytopenia, the last of which occurred in 6% of patients on R-CHOP, compared with 57% for VR-CAP. Despite this difference, however, rates of grade 3 or higher bleeding were similar between the groups, occurring in 1.2% and 1.7%, respectively.
The invited discussant, Dr. Michael E. Williams, chief of hematology/oncology at the University of Virginia Cancer Center, Charlottesville, commented that the study provides proof of principle "that if you add an active single agent and substitute bortezomib for vincristine, which would appear to be a less active agent, that you can certainly improve PFS significantly."
Dr. Williams said that it remains to be seen, however, whether, as Dr. Cavalli suggested, certain treatment strategies could be used to lower the incidence of drug-related adverse events and improve PFS rates further, such as the use of subcutaneous rather than intravenous bortezomib, different dosing schedules, or rituximab in the maintenance phase.
The study was supported by Janssen Global Services and Millennium. Dr. Cavalli disclosed receiving travel support for attending the ASCO annual meeting, but reported having no other conflicts of interest. Dr. Williams disclosed consulting/advising for Millennium and receiving research funding from Janssen and Millennium.
CHICAGO – Tweaking the R-CHOP recipe to substitute bortezomib for vincristine may bring clinical benefit to patients with newly diagnosed mantle cell lymphoma who are ineligible for bone marrow transplant.
In a randomized phase III trial in patients with MCL who could not undergo bone marrow transplant due to age or comorbidities, those patients who received a combination of rituximab, doxorubicin, bortezomib (Velcade), cyclophosphamide, and prednisone (the VR-CAP regimen) had significantly better progression-free survival than did patients treated with R-CHOP, the same regimen but with vincristine instead of bortezomib.
The VR-CAP regimen "could be considered a new standard of care for newly diagnosed mantle cell lymphoma patients not considered for intensive treatment and bone marrow transplant," Dr. Franco Cavalli reported at the annual meeting of the American Society of Clinical Oncology.
The bortezomib-containing regimen was associated with more grade 3 or 4 toxicities than standard R-CHOP, but adverse events were manageable, and most patients in each study arm were able to stay on chemotherapy for all prescribed cycles, said Dr. Cavalli of the Oncology Institute of Southern Switzerland, Bellinzona.
R-CHOP is a standard frontline therapy for patients with MCL who are deemed to be ineligible for intensive therapy and/or bone marrow transplant. But the regimen offers only limited progression-free survival (PFS) in this population, Dr. Cavalli said.
Because bortezomib is approved for the treatment of relapsed MCL in the United States and 53 other nations, the authors investigated whether it could improve outcomes when given to patients with newly diagnosed disease.
The LYM-3002 trial was a phase III study conducted at 128 centers in 28 countries in Europe, Asia, the Americas, and Africa. Patients with newly diagnosed MCL stage II-IV, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2, and who were ineligible or not considered for bone marrow transplant, were randomized to receive either R-CHOP or VR-CAP. In R-CHOP, vincristine 1.4 mg/m2 was delivered to a maximum of 2 mg intravenously on day 1 of each cycle. In VR-CAP, bortezomib 1.3 mg/m2 was delivered via intravenous infusion on days 1, 4, 8, and 11 of each cycle. Patients were assigned to receive at least six cycles of therapy, with an additional two cycles possible if investigator-assessed responses were first documented at the end of cycle 6.
A total of 487 patients (244 assigned to R-CHOP and 243 to VR-CAP) were included in the intention-to-treat analysis.
At a median follow-up of 40 months, median PFS, the primary endpoint, was 24.7 months in the VR-CAP arm, compared with 14.4 months for R-CHOP (hazard ratio, 0.63; P less than .001), as judged by an independent review committee. Investigator-rated PFS was 30.7 months vs. 16.1 months, respectively (HR, 0.51; P less than .001).
Clinical responses according to International Working Group revised response criteria for malignant lymphoma included overall response rates (complete response, complete unconfirmed response, and partial response) of 90% in the R-CHOP–treated patients and 92% in those who received VR-CAP.
However, there was a higher proportion of combined complete response and complete unconfirmed response in the VR-CAP group: 42% for R-CHOP vs. 53% for VR-CAP (odds ratio, 1.69; P = .007).
Median time to response was also shorter with VR-CAP (1.6 vs. 1.4 months; HR, 1.54; P less than .001).
Independent reviewer-rated median time-to-progression was 16.1 months for R-CHOP vs. 30.5 months for VR-CAP (HR, 0.58; P less than .001). Median time to next therapy was 24.8 vs. 44.5 months, respectively (HR, 0.50; P less than .001), and median treatment-free interval was 20.5 vs. 40.6 months (HR, 0.50; P less than .001).
Median overall survival was 56.3 months among R-CHOP–treated patients, vs. not reached among VR-CAP–treated patients.
Grade 3 or higher drug-related adverse events occurred in 85% and 93% of patients, respectively. The events were considered serious in 21% of R-CHOP–treated patients and in 33% of VR-CAP–treated patients. In all, 7% of patients on R-CHOP and 9% of those on VR-CAP discontinued therapy because of adverse events.
Grade 3 adverse events were more frequent with VR-CAP and included neutropenia, leukopenia, lymphopenia, and thrombocytopenia, the last of which occurred in 6% of patients on R-CHOP, compared with 57% for VR-CAP. Despite this difference, however, rates of grade 3 or higher bleeding were similar between the groups, occurring in 1.2% and 1.7%, respectively.
The invited discussant, Dr. Michael E. Williams, chief of hematology/oncology at the University of Virginia Cancer Center, Charlottesville, commented that the study provides proof of principle "that if you add an active single agent and substitute bortezomib for vincristine, which would appear to be a less active agent, that you can certainly improve PFS significantly."
Dr. Williams said that it remains to be seen, however, whether, as Dr. Cavalli suggested, certain treatment strategies could be used to lower the incidence of drug-related adverse events and improve PFS rates further, such as the use of subcutaneous rather than intravenous bortezomib, different dosing schedules, or rituximab in the maintenance phase.
The study was supported by Janssen Global Services and Millennium. Dr. Cavalli disclosed receiving travel support for attending the ASCO annual meeting, but reported having no other conflicts of interest. Dr. Williams disclosed consulting/advising for Millennium and receiving research funding from Janssen and Millennium.
AT THE ASCO ANNUAL MEETING 2014
Key clinical point: Substituting bortezomib for vincristine may bring clinical benefit to patients with newly diagnosed mantle cell lymphoma who are ineligible for bone marrow transplant.
Major finding: At a median follow-up of 40 months, median progression-free survival was 24.7 months in the bortezomib-containing VR-CAP arm, compared with 14.4 months for R-CHOP, a significant difference.
Data source: Randomized, open-label phase III study in 487 patients with newly diagnosed mantle cell lymphoma.
Disclosures: The study was supported by Janssen Global Services and Millennium. Dr. Cavalli disclosed receiving travel support for attending the ASCO annual meeting, but reported having no other conflicts of interest. Dr. Williams disclosed consulting/advising for Millennium and receiving research funding from Janssen and Millennium.