User login
Grand Rounds: Man, 65, With Heart Failure Symptoms
A black man, age 65, with no known history of cardiopulmonary disease presented with acute-onset exertional dyspnea and lower extremity edema. He also reported an episode of syncope, as well as occasional dizziness and abdominal bloating. He said he experienced exertional dyspnea while doing a routine step aerobic exercise. His exercise regimen included distance walking, yoga, and aerobics four to five days per week.
The patient’s medical history was remarkable for a single episode of a bleeding ulcer in previous years, low back pain, shoulder pain, and a septic arthritic hip. His social history was negative for use of tobacco, alcohol, or illegal drugs. He was married and had two biological daughters with fairly unremarkable medical histories. The patient had earned a master’s degree, worked full-time in the insurance business, and was an avid worldwide traveler. He reported diminished quality of life as a result of his acute-onset heart failure symptoms, which reduced his ability to exercise routinely, work full-time, or travel.
The patient’s sudden experience of exertional dyspnea prompted him to visit his primary care provider, who ordered an ECG that demonstrated low voltage patterns and a first-degree atrioventricular (AV) block. Subsequent stress echocardiography showed generalized thickening of the left ventricular myocardium. Posterior wall thickness measured 1.7 cm (normal range, 0.6 to 1.1 cm), septal thickness measured 1.9 cm (normal, 0.6 to 1.1 cm), and ejection fraction was 65%. The stress echocardiogram also showed a speckling pattern (brightly scattered spots) on the myocardium.
Although stress echocardiography results were negative for ischemic disease, the patient did experience dyspnea during the exam. He underwent cardiac catheterization, which indicated normal coronary arteries.
Additional diagnostic studies included cardiac MRI with and without contrast, which showed nulling of the heart muscle and delayed patchy hyperenhancement; this suggested myocardial tissue abnormality as result of amyloid fibril deposition.1 Both pulmonary and tricuspid aortic valves were normal, with no evidence of stenosis. No regional wall motion abnormalities were noted.
Laboratory findings during the work-up were lipid panel, unremarkable; complete blood count (CBC), mild anemia and leukopenia; and urinalysis, positive for proteinuria. Brain natriuretic peptide (BNP) was measured at 686 pg/mL (normal, 0.0 to 100 pg/mL), indicating moderate heart failure. A peripheral blood smear was negative for monoclonal plasma cells.
The patient’s physical exam was unremarkable except for 2+ pedal edema bilaterally. In consideration of normal coronary arteries on cardiac catheterization, the patient’s heart failure symptoms, and stress echocardiography abnormalities, a heart biopsy was ordered. An endomyocardial biopsy with Congo Red stain demonstrated an apple-green birefringent pattern viewed under high-definition polarized light microscope, which was consistent with amyloid deposition.2
The patient was given a diagnosis of primary amyloidosis by his local cardiologist despite negative findings on the peripheral blood smear for monoclonal plasma cells (which are typically found in primary amyloidosis).3 He presented to an institution well-known for its expertise in amyloidosis, for a second opinion. There, the diagnosis was negated, based on reevaluation of the patient’s previous heart specimen through immunohistochemical studies. These studies were positive for serum amyloid P, which is suggestive of transthyretin (TTR) or familial amyloidosis.4 Genetic testing revealed a familial amyloidosis DNA sequence analysis with the Val122Ile variant (ie, isoleucine for valine at position 1225). With the correct diagnosis confirmed, the patient was referred to another highly regarded institution to begin a work-up for cardiac transplantation. Meanwhile, he was cautiously treated with the loop diuretic furosemide to manage his shortness of breath and peripheral edema.
Fifteen months later (13 weeks after being listed for transplant), the patient underwent successful cardiac transplantation.
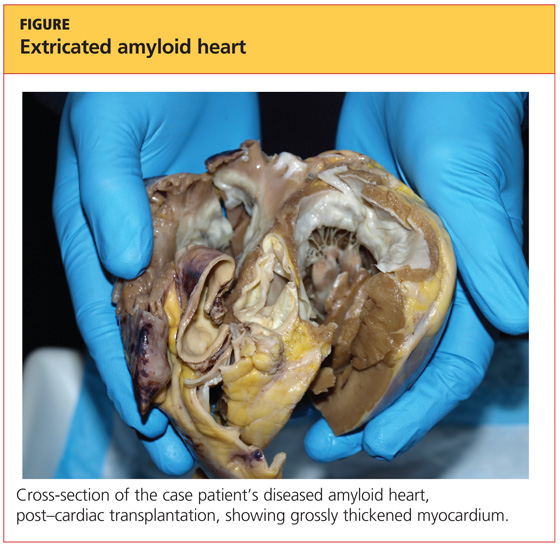
On pathologic review of the patient’s extricated heart, the myocardium was found to be grossly thickened (see figure, above) and weighed 540 g; the average adult heart weighs 300 to 350 g, depending on the patient’s size.6 Congo Red staining showed extensive amyloid deposits with infiltration throughout the myocardium.
Ninety percent of the amyloid deposits were interstitial, 5% were in the vessels, and 5% were noted in a nodular pattern. The left ventricular cavity showed dilated and thickened walls. Intramural and extramural blood vessels were infiltrated with amyloid as well.
Six months after transplantation, the patient underwent diagnostic testing to assess the function and structure of his new heart. Cardiac catheterization was negative for coronary artery disease. Thirteen months posttransplantation, endomyocardial biopsy with Congo Red stain was negative for amyloid deposition or organ rejection.
About 24 months posttransplantation, the patient was taking tacrolimus, pravastatin, pantoprazole, dapsone, propanolol, colchicine, and donepezil. Stress echocardiography demonstrated normal right and left ventricular systolic function; no wall-motion abnormalities or left ventricular hypertrophy were detected, and the right atrium was of normal size. There was abnormal structural enlargement of the left atrium at the site of anastamosis—a common finding in cardiac transplant patients. The aortic, tricuspid, and mitral valves were all normal.
At that time, it was decided not to repeat endomyocardial biopsy because of normal results on molecular expression testing (a noninvasive technique called AlloMap®7-9), which is performed to assess for heart transplant rejection. The patient’s lipid panel remained within normal limits. CBC indicated persistent anemia and leukopenia. Urine protein and BNP test results were not available.
Since undergoing cardiac transplantation, the patient has resumed his normal routine activities, including some type of exercise five days per week. He said his diet is maintained in moderation. He denied shortness of breath, chest pain, dizziness, or edema. He has returned to full-time employment and has vacationed in Croatia, Italy, and Central America.
DISCUSSION
Familial amyloidosis is an autosomal dominant disease characterized by the production of mutated proteins, most commonly ATTR. Presence of the ATTR Val 122Ile allele has been reported in 3.9% of all black Americans, and in one study, 23% of black Americans diagnosed with cardiac amyloidosis at autopsy were heterozygous for this variant allele.10-12 ATTR Val122Ile usually manifests in the fifth
or sixth decade of life with its characteristic presentation of infiltrative/restrictive cardiomyopathy,13 resulting in heart failure and sometimes peripheral neuropathy.10,11,14
Pathophysiology
In patients with ATTR Val122Ile, cardiomyopathy results from the deposition of mutant protein fibrils in the cardiac muscle, leading to restricted heart wall motion10,15 and stiffening of the cardiac ventricles, with subsequent disruption of the diastolic filling properties of the cardiac muscle.16 Fluid overload and heart failure follow.3,10,17 The atrium of the heart dilates, and the walls of the ventricles become thickened and fibrous.18 Liepnieks and Benson6 reported that the cadaver heart of one Val122Ile patient infiltrated with amyloid protein fibrils weighed 725 g—more than double the weight of an average adult heart.
In patients with cardiac amyloidosis, ECG can detect arrhythmia, and echocardiography shows cardiac enlargement; however, as in the case patient, cardiac catheterization shows normal coronary arteries.15,16,19,20 Thus, previously healthy patients who present with heart failure and negative results on cardiac catheterization should undergo further work-up for cardiac amyloidosis.19
Amyloidosis affects all populations globally.10 In systemic amyloidosis, amyloid releases into the plasma, infiltrating and impairing multiple organs. Poor survival has been reported in patients with heart failure symptoms resulting from amyloid deposition.20,21
Types of Amyloidosis
Primary amyloidosis, the most common of the three amyloidosis types, can be systemic or localized.22 It occurs when protein fibrils, developed from immunoglobin light chains or monoclonal plasma cells and measuring 7 to 10 nm in diameter, adhere to the heart, kidneys, peripheral nerves, eyes, and other organs.5,11,20,23,24 Known for its relation to multiple myeloma,19 primary amyloidosis is associated with a poor prognosis.3,10
Secondary amyloidosis results from a chronic inflammatory disorder, such as rheumatoid arthritis or ankylosing spondylitis—conditions that trigger the production of amyloid proteins.3,10 This type has also been associated with substance abuse and AIDS.23
Familial or hereditary amyloidosis, according to Benson,10 is a group of diseases, each resulting from mutation in a specific protein. In the United States, the most common type of familial amyloidosis is ATTR.11 More than 100 mutant types of ATTR proteins have been identified, each involving a specific nationality or group of nationalities.10,11,23
Mutant ATTR amyloid, when deposited in specific organs, leads to their dysfunction and ultimate failure.6 ATTR may affect the cardiac, gastric, renal, ophthalmic, or nervous system. Depending on the ATTR variant, the resulting clinical features are age- and time-dependent, with onset most common between the third and fifth decade of life.10
Prevalence
The prevalence of ATTR Val 122Ile amyloidosis is reportedly high in West Africa, and in the US African-American population (3.9%).4,11,14,25,26 In a study conducted at a county hospital in Indianapolis, 3% of black newborns were found positive for ATTR Val122Ile through DNA sampling of umbilical cord blood.25 These statistics are of concern, as ATTR amyloidosis could be a significant health concern in a patient population that is already medically underserved.
Yamashita et al25 estimate that 1.35 million Americans of African-American descent may be affected by ATTR Val122Ile and vulnerable to restrictive cardiomyopathy–related heart failure and death. At the very least, this disorder can impair quality of life, especially in the presence of other comorbid conditions.
Clinical Presentation
The presence of exertional syncope at presentation is ominous, as it may be a marker of severe restrictive cardiomyopathy, postural hypotension due to excessive diuresis or autonomic neuropathy, ventricular arrhythmias from localized hypoperfusion, and rarely from cardiac tamponade due to pericardial involvement.27 Despite widespread involvement of the conduction system in specimens at autopsy, high-grade IV block is unusual.3
Diagnostic Studies
ECG. Both ECG and Holter monitoring can detect the arrhythmias and conduction disturbances (eg, first-degree AV block, low voltage patterns) associated with cardiac amyloidosis. Patients often experience syncopal and near-syncopal episodes as a result of conduction disturbances.28 Patients with conditions such as cardiac amyloidosis who present with severe heart failure are at high risk for sudden death secondary to conduction disturbances. Many have benefited from implanted defibrillators.29
Echocardiography. In patients with ATTR Val122Ile cardiac amyloidosis, echocardiography reveals thickened ventricular walls (ie, measuring ≥ 15 mm; normal, ≤ 11 mm).19 Amyloid-restrictive cardiomyopathy is associated with a marked dissociation between short- and long-axis systolic function, in cases in which left ventricular ejection fraction is normal.30
Echocardiography may demonstrate the characteristic specular or granular sparkling appearance that signifies advanced disease.15,21 Only a minority have this pattern in the myocardium, however, and changes in echocardiographic technology have made this finding less noticeable.30
MRI. Among more recently used diagnostic studies, cardiac magnetic resonance (CMR) has been reported to demonstrate late gadolinium enhancement (LGE) in perhaps 80% of patients with familial amyloidosis and cardiac involvement (as determined through biopsy and Congo Red stain). LGE-CMR shows darkening of the cardiac tissue, a common occurrence in amyloidosis.31
LGE is associated with increased thickness of both left and right ventricles, lower ECG voltage patterns, elevated BNP, and elevated troponin T.31,32 Globally, LGE is associated with the worst prognosis in patients with cardiac amyloidosis. Use of LGE-CMR testing can help facilitate early detection of cardiac amyloidosis in patients who may be vulnerable to cardiac damage.31
Cardiac catheterization. In patients with cardiac amyloidosis, cardiac catheterization usually shows normal coronary arteries.3
Diagnosis
Early diagnosis of ATTR cardiac amyloidosis is crucial to the patient’s survival; it should be ruled out in any African-American patient with unexplained heart failure and echocardiography showing increased wall thickness with a nondilated left ventricular cavity. Additional clues include significant proteinuria, hepatomegaly disproportionate to the degree of heart failure, or corresponding neuropathy. Known family history of the disease, along with variant type, allows for a prompt and correct diagnosis.10
It has been reported that most clinicians who encounter heart failure, particularly in black patients, do not consider amyloidosis in the differential diagnosis, because of the high prevalence of hypertension and congestive heart failure in this population.10,15,19 As a result, amyloidosis often goes undiagnosed.19
Findings of enlarged and thickened cardiac walls on echocardiography but normal coronary arteries on cardiac catheterization should alert the treating clinician to further work-up for cardiac amyloidosis.19 In such a patient, according to Kristen et al,21 endomyocardial biopsy with Congo Red staining is the gold standard for diagnosis of amyloidosis.
In ATTR Val122Ile familial amyloidosis, it is unclear whether patients who are homozygous for the disease present with symptoms at earlier onset with more progressive illness or die sooner than those who are heterozygous.33 Nevertheless, once the diagnosis is confirmed, it is important to determine the patient’s specific variant type by DNA testing so that appropriate treatment can be initiated and the patient’s prognosis evaluated.5,10,13
Treatment
For familial amyloidosis in general, some researchers advocate liver transplantation to remove the source of mutant amyloid protein and stop all deposition of amyloid fibrils; this procedure can be followed later by transplantation of other affected organs (including the heart).5,23 Maurer et al34 have reported improved one-year survival rates among patients with ATTR amyloidosis who underwent both cardiac and liver transplantation: 75%, versus 23% in patients who did not receive transplanted organs.
Management of cardiac amyloidosis usually requires a twofold approach: treating associated congestive heart failure, and preventing further deposition of amyloid.24 In the case patient (as in most patients with ATTR amyloidosis), heart transplantation was deemed the only life-sustaining treatment option.11,19,33
Pharmacotherapeutic options are limited for patients with ATTR Val122Ile familial amyloidosis. Conventional heart failure agents (eg, ACE inhibitors, angiotensin receptor blockers, digoxin, β-blockers, calcium channel blockers) can exacerbate heart failure symptoms, leading to a potentially life-threatening arrhythmia.3,11,19,24,35 Amyloid fibrils bind to digitalis, increasing susceptibility to digitalis toxicity; and to nifedipine, causing hemodynamic deterioration. Verapamil should be avoided, as it may induce severe left ventricular dysfunction. ACE inhibitors often provoke profound hypotension in primary amyloidosis.24,35
Diuretics, too (eg, furosemide, as was prescribed for the case patient), must be used with caution.3 These agents have been used to treat fluid overload and the resulting peripheral edema and shortness of breath found in ATTR Val122Ile patients who experience heart failure.36 According to Dubrey et al,5 cautious use of diuretics is necessary for management of heart failure symptoms in these patients.
Because the risk for intracardiac thrombus is high, anticoagulation (using agents other than β-blockers or calcium channel blockers) should be implemented unless compelling risks are involved.11,24 Amiodarone is relatively well tolerated for ventricular tachydysrhythmias and in atrial fibrillation if the goal is maintaining sinus rhythm.37
Regarding heart transplantation in patients with familial amyloidosis, Jacob et al33 hypothesize that since mutant amyloid protein is synthesized by the liver, it would take approximately 50 years for a transplanted heart to become affected by amyloid deposition. In a 59-year-old Afro-Caribbean man with familial amyloidosis who underwent cardiac transplantation, Hamour et al11 reported that the donor heart remained amyloid-free three years posttransplantation, as demonstrated by serial cardiac biopsy.
On the Horizon
Clinical trials are now under way to examine pharmacotherapeutic options for patients with ATTR amyloidosis. Now being examined in clinical trials, for example, is Fx-1006A, a drug that stabilizes ATTR and prevents the misfolding of the amyloid protein fibril, in turn preventing it from binding to the target organ.38 Similarly, ALN-TTR, a drug believed to prevent disease manifestation and possibly facilitate disease regression, is being investigated in early human trials.39
Additionally, the use of genetic testing is recommended in at-risk individuals to identify the TTR gene. Affected patients may benefit from prophylactic medical management, which would halt amyloidogenesis of TTR—and possibly treat the condition as well.35 Pharmacotherapeutic agents like diflunisal, an NSAID, antagonize the aggregation of TTR protein and hinder formation of the amyloid fibrils.40
CONCLUSION
ATTR Val122Ile familial amyloidosis is a rare disorder that causes abnormal synthesis of amyloid protein in the liver, which then infiltrates the cardiac structure, leading to restrictive cardiomyopathy and progressive heart failure. Patients who present with symptoms of heart failure, cardiac enlargement on echocardiography, and a finding of granular speckling patterns, though not specific on echocardiography, should prompt the health care provider to refer the patient to a cardiologist familiar with cardiac amyloidosis for further work-up.
Diagnosed patients must undergo genetic testing to determine the specific variant type so that prompt treatment can be initiated. In patients with ATTR Val122Ile familial amyloidosis, the treatment of choice is cardiac transplantation. Although the mutant amyloid protein continues to be synthesized in the liver, the donor heart is unlikely to become affected by this substance for many years. Appropriately treated patients can maintain good quality of life, free of heart failure.
REFERENCES
1. Lim RP, Srichai MB, Lee VS. Non-ischemic causes of delayed myocardial hyperenhancement on MRI. AJR Am J Roentgenol. 2007;188 (6):1675-1681.
2. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17(3-4):101-104.
3. Kendall H. Cardiac amyloidosis. Crit Care Nurse. 2010;30(2):16-23.
4. Eriksson M, Büttener J, Todorov T, et al. Prevalence of germline mutations in the TTR gene in a consecutive series of surgical pathology specimens with AATR amyloid. Am J Surg Pathol. 2009;33(1):58-65.
5. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
6. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277-282.
7. XDx Expression Diagnostics. Allomap®: molecular expression testing (2004). www.allomap.com. Accessed May 14, 2012.
8. Mandras SA, Crespo J, Patel HM. Innovative application of immunologic principles in heart transplantation. Ochsner J. 2010;10(4):231-235.
9. Yamani MH, Taylor DO, Rodriguez R, et al. Transplant vasculopathy is associated with increased AlloMap gene expression score. J Heart Lung Transplant. 2007;26(4):403-406.
10. Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol. 2003;17(6):909-927.
11. Hamour IM, Lachmann HJ, Goodman HJ, et al. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant. 2008;8(5):1056-1059.
12. Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336(7):466-473.
13. Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52(4):347-361.
14. Askanas V, Engel WK, McFerrin J, Vattemi G. Transthyretin Val122Ile, accumulated Abeta, and inclusion-body myositis aspects in cultured muscle. Neurology. 2003;61(2):257-260.
15. Hamidi Asl K, Nakamura M, Yamashita T, Benson MD. Cardiac amyloidoses associated with the transthyretin lle122 mutation in a Caucasian family. Amyloid. 2001;8(4):263-269.
16. Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24(3): 214-220.
17. Nihoyannopoulos P, Dawson D. Restrictive cardiomyopathies. Eur J Echocardiogr. 2009;10 (8):iii23-iii33.
18. Bruce J. Getting to the heart of cardiomyopathies. Nursing. 2005;35(8):44-47.
19. Falk RH. The neglected entity of familial cardiac amyloidosis in African Americans. Ethn Dis. 2002;12(1):141-143.
20. Piper C, Butz T, Farr M, et al. How to diagnose cardiac amyloidosis early: impact of ECG, tissue Doppler echocardiography, and myocardial biopsy. Amyloid. 2010;17(1):1-9.
21. Kristen AV, Meyer FJ, Perz JB, et al. Risk stratification in cardiac amyloidosis: novel approaches. Transplantation. 2005;80(1 suppl):S151-S155.
22. Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid. 2007;14(3):179-183.
23. Picken MM. New insights into systemic amyloidosis: the importance of diagnosis of specific type. Curr Opin Nephrol Hypertens. 2007; 16(3):196-203.
24. Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124(9):1079-1085.
25. Yamashita T, Asl KH, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile 122 allele frequency in an African-American population. Amyloid. 2005;12(2):127-130.
26. Benson MD, Yazaki M, Magy N. Laboratory assessment of transthyretin amyloidosis. Clin Chem Lab Med. 2002;40(12):1262-1265.
27. Chamarthi B, Dubrey SW, Cha K, et al. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol. 1997;80(9):1242-1245.
28. Correia MJ, Coutinho CA, Conceiçao I, et al. Role of heart rate variability in the assessment of autonomic dysfunction in type I familial amyloidotic polyneuropathy. Folia Cardiol. 2005;12(suppl C):459-462.
29. Kadish A, Mehra M. Heart failure devices: Implantable cardioverter-defibrillators and biventricular pacing therapy. Circulation. 2005; 111(24):3327-3335.
30. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43(3):410-415.
31. Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3(2):155-164.
32. Fealey ME, Edwards WD, Buadi FK, et al. Echocardiographic features of cardiac amyloidosis presenting as endomyocardial disease in a 54-year-old male. J Cardiol. 2009;54(1):162-166.
33. Jacob EK, Edwards WD, Zucker M, et al. Homozygous transthyretin mutation in an African American male. J Mol Diagn. 2007; 9(1):127-131.
34. Maurer MS, Raina A, Hesdorffer C, et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83(5):539-545.
35. Buxbaum J, Alexander A, Koziol J, et al. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African-Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159(5):864-870.
36. Rose BD, Colucci WS. Use of diuretics in heart failure (2010). www.uptodate.com/con tents/use-of-diuretics-in-patients-with-heart-failure. Accessed May 14, 2012.
37. Trappe H-J. Treating critical supraventricular and ventricular arrhythmias. J Emerg Trauma Shock. 2010;3(2):143-152.
38. Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14(30):3219-3230.
39. Alnylam Pharmaceuticals. TTR Amyloidosis: ALN-TTR (2011). www.alnylam.com/Programs-and-Pipeline/Programs/index.php. Accessed May 14, 2012.
40. Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin: potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355-374.
A black man, age 65, with no known history of cardiopulmonary disease presented with acute-onset exertional dyspnea and lower extremity edema. He also reported an episode of syncope, as well as occasional dizziness and abdominal bloating. He said he experienced exertional dyspnea while doing a routine step aerobic exercise. His exercise regimen included distance walking, yoga, and aerobics four to five days per week.
The patient’s medical history was remarkable for a single episode of a bleeding ulcer in previous years, low back pain, shoulder pain, and a septic arthritic hip. His social history was negative for use of tobacco, alcohol, or illegal drugs. He was married and had two biological daughters with fairly unremarkable medical histories. The patient had earned a master’s degree, worked full-time in the insurance business, and was an avid worldwide traveler. He reported diminished quality of life as a result of his acute-onset heart failure symptoms, which reduced his ability to exercise routinely, work full-time, or travel.
The patient’s sudden experience of exertional dyspnea prompted him to visit his primary care provider, who ordered an ECG that demonstrated low voltage patterns and a first-degree atrioventricular (AV) block. Subsequent stress echocardiography showed generalized thickening of the left ventricular myocardium. Posterior wall thickness measured 1.7 cm (normal range, 0.6 to 1.1 cm), septal thickness measured 1.9 cm (normal, 0.6 to 1.1 cm), and ejection fraction was 65%. The stress echocardiogram also showed a speckling pattern (brightly scattered spots) on the myocardium.
Although stress echocardiography results were negative for ischemic disease, the patient did experience dyspnea during the exam. He underwent cardiac catheterization, which indicated normal coronary arteries.
Additional diagnostic studies included cardiac MRI with and without contrast, which showed nulling of the heart muscle and delayed patchy hyperenhancement; this suggested myocardial tissue abnormality as result of amyloid fibril deposition.1 Both pulmonary and tricuspid aortic valves were normal, with no evidence of stenosis. No regional wall motion abnormalities were noted.
Laboratory findings during the work-up were lipid panel, unremarkable; complete blood count (CBC), mild anemia and leukopenia; and urinalysis, positive for proteinuria. Brain natriuretic peptide (BNP) was measured at 686 pg/mL (normal, 0.0 to 100 pg/mL), indicating moderate heart failure. A peripheral blood smear was negative for monoclonal plasma cells.
The patient’s physical exam was unremarkable except for 2+ pedal edema bilaterally. In consideration of normal coronary arteries on cardiac catheterization, the patient’s heart failure symptoms, and stress echocardiography abnormalities, a heart biopsy was ordered. An endomyocardial biopsy with Congo Red stain demonstrated an apple-green birefringent pattern viewed under high-definition polarized light microscope, which was consistent with amyloid deposition.2
The patient was given a diagnosis of primary amyloidosis by his local cardiologist despite negative findings on the peripheral blood smear for monoclonal plasma cells (which are typically found in primary amyloidosis).3 He presented to an institution well-known for its expertise in amyloidosis, for a second opinion. There, the diagnosis was negated, based on reevaluation of the patient’s previous heart specimen through immunohistochemical studies. These studies were positive for serum amyloid P, which is suggestive of transthyretin (TTR) or familial amyloidosis.4 Genetic testing revealed a familial amyloidosis DNA sequence analysis with the Val122Ile variant (ie, isoleucine for valine at position 1225). With the correct diagnosis confirmed, the patient was referred to another highly regarded institution to begin a work-up for cardiac transplantation. Meanwhile, he was cautiously treated with the loop diuretic furosemide to manage his shortness of breath and peripheral edema.
Fifteen months later (13 weeks after being listed for transplant), the patient underwent successful cardiac transplantation.
On pathologic review of the patient’s extricated heart, the myocardium was found to be grossly thickened (see figure, above) and weighed 540 g; the average adult heart weighs 300 to 350 g, depending on the patient’s size.6 Congo Red staining showed extensive amyloid deposits with infiltration throughout the myocardium.
Ninety percent of the amyloid deposits were interstitial, 5% were in the vessels, and 5% were noted in a nodular pattern. The left ventricular cavity showed dilated and thickened walls. Intramural and extramural blood vessels were infiltrated with amyloid as well.
Six months after transplantation, the patient underwent diagnostic testing to assess the function and structure of his new heart. Cardiac catheterization was negative for coronary artery disease. Thirteen months posttransplantation, endomyocardial biopsy with Congo Red stain was negative for amyloid deposition or organ rejection.
About 24 months posttransplantation, the patient was taking tacrolimus, pravastatin, pantoprazole, dapsone, propanolol, colchicine, and donepezil. Stress echocardiography demonstrated normal right and left ventricular systolic function; no wall-motion abnormalities or left ventricular hypertrophy were detected, and the right atrium was of normal size. There was abnormal structural enlargement of the left atrium at the site of anastamosis—a common finding in cardiac transplant patients. The aortic, tricuspid, and mitral valves were all normal.
At that time, it was decided not to repeat endomyocardial biopsy because of normal results on molecular expression testing (a noninvasive technique called AlloMap®7-9), which is performed to assess for heart transplant rejection. The patient’s lipid panel remained within normal limits. CBC indicated persistent anemia and leukopenia. Urine protein and BNP test results were not available.
Since undergoing cardiac transplantation, the patient has resumed his normal routine activities, including some type of exercise five days per week. He said his diet is maintained in moderation. He denied shortness of breath, chest pain, dizziness, or edema. He has returned to full-time employment and has vacationed in Croatia, Italy, and Central America.
DISCUSSION
Familial amyloidosis is an autosomal dominant disease characterized by the production of mutated proteins, most commonly ATTR. Presence of the ATTR Val 122Ile allele has been reported in 3.9% of all black Americans, and in one study, 23% of black Americans diagnosed with cardiac amyloidosis at autopsy were heterozygous for this variant allele.10-12 ATTR Val122Ile usually manifests in the fifth
or sixth decade of life with its characteristic presentation of infiltrative/restrictive cardiomyopathy,13 resulting in heart failure and sometimes peripheral neuropathy.10,11,14
Pathophysiology
In patients with ATTR Val122Ile, cardiomyopathy results from the deposition of mutant protein fibrils in the cardiac muscle, leading to restricted heart wall motion10,15 and stiffening of the cardiac ventricles, with subsequent disruption of the diastolic filling properties of the cardiac muscle.16 Fluid overload and heart failure follow.3,10,17 The atrium of the heart dilates, and the walls of the ventricles become thickened and fibrous.18 Liepnieks and Benson6 reported that the cadaver heart of one Val122Ile patient infiltrated with amyloid protein fibrils weighed 725 g—more than double the weight of an average adult heart.
In patients with cardiac amyloidosis, ECG can detect arrhythmia, and echocardiography shows cardiac enlargement; however, as in the case patient, cardiac catheterization shows normal coronary arteries.15,16,19,20 Thus, previously healthy patients who present with heart failure and negative results on cardiac catheterization should undergo further work-up for cardiac amyloidosis.19
Amyloidosis affects all populations globally.10 In systemic amyloidosis, amyloid releases into the plasma, infiltrating and impairing multiple organs. Poor survival has been reported in patients with heart failure symptoms resulting from amyloid deposition.20,21
Types of Amyloidosis
Primary amyloidosis, the most common of the three amyloidosis types, can be systemic or localized.22 It occurs when protein fibrils, developed from immunoglobin light chains or monoclonal plasma cells and measuring 7 to 10 nm in diameter, adhere to the heart, kidneys, peripheral nerves, eyes, and other organs.5,11,20,23,24 Known for its relation to multiple myeloma,19 primary amyloidosis is associated with a poor prognosis.3,10
Secondary amyloidosis results from a chronic inflammatory disorder, such as rheumatoid arthritis or ankylosing spondylitis—conditions that trigger the production of amyloid proteins.3,10 This type has also been associated with substance abuse and AIDS.23
Familial or hereditary amyloidosis, according to Benson,10 is a group of diseases, each resulting from mutation in a specific protein. In the United States, the most common type of familial amyloidosis is ATTR.11 More than 100 mutant types of ATTR proteins have been identified, each involving a specific nationality or group of nationalities.10,11,23
Mutant ATTR amyloid, when deposited in specific organs, leads to their dysfunction and ultimate failure.6 ATTR may affect the cardiac, gastric, renal, ophthalmic, or nervous system. Depending on the ATTR variant, the resulting clinical features are age- and time-dependent, with onset most common between the third and fifth decade of life.10
Prevalence
The prevalence of ATTR Val 122Ile amyloidosis is reportedly high in West Africa, and in the US African-American population (3.9%).4,11,14,25,26 In a study conducted at a county hospital in Indianapolis, 3% of black newborns were found positive for ATTR Val122Ile through DNA sampling of umbilical cord blood.25 These statistics are of concern, as ATTR amyloidosis could be a significant health concern in a patient population that is already medically underserved.
Yamashita et al25 estimate that 1.35 million Americans of African-American descent may be affected by ATTR Val122Ile and vulnerable to restrictive cardiomyopathy–related heart failure and death. At the very least, this disorder can impair quality of life, especially in the presence of other comorbid conditions.
Clinical Presentation
The presence of exertional syncope at presentation is ominous, as it may be a marker of severe restrictive cardiomyopathy, postural hypotension due to excessive diuresis or autonomic neuropathy, ventricular arrhythmias from localized hypoperfusion, and rarely from cardiac tamponade due to pericardial involvement.27 Despite widespread involvement of the conduction system in specimens at autopsy, high-grade IV block is unusual.3
Diagnostic Studies
ECG. Both ECG and Holter monitoring can detect the arrhythmias and conduction disturbances (eg, first-degree AV block, low voltage patterns) associated with cardiac amyloidosis. Patients often experience syncopal and near-syncopal episodes as a result of conduction disturbances.28 Patients with conditions such as cardiac amyloidosis who present with severe heart failure are at high risk for sudden death secondary to conduction disturbances. Many have benefited from implanted defibrillators.29
Echocardiography. In patients with ATTR Val122Ile cardiac amyloidosis, echocardiography reveals thickened ventricular walls (ie, measuring ≥ 15 mm; normal, ≤ 11 mm).19 Amyloid-restrictive cardiomyopathy is associated with a marked dissociation between short- and long-axis systolic function, in cases in which left ventricular ejection fraction is normal.30
Echocardiography may demonstrate the characteristic specular or granular sparkling appearance that signifies advanced disease.15,21 Only a minority have this pattern in the myocardium, however, and changes in echocardiographic technology have made this finding less noticeable.30
MRI. Among more recently used diagnostic studies, cardiac magnetic resonance (CMR) has been reported to demonstrate late gadolinium enhancement (LGE) in perhaps 80% of patients with familial amyloidosis and cardiac involvement (as determined through biopsy and Congo Red stain). LGE-CMR shows darkening of the cardiac tissue, a common occurrence in amyloidosis.31
LGE is associated with increased thickness of both left and right ventricles, lower ECG voltage patterns, elevated BNP, and elevated troponin T.31,32 Globally, LGE is associated with the worst prognosis in patients with cardiac amyloidosis. Use of LGE-CMR testing can help facilitate early detection of cardiac amyloidosis in patients who may be vulnerable to cardiac damage.31
Cardiac catheterization. In patients with cardiac amyloidosis, cardiac catheterization usually shows normal coronary arteries.3
Diagnosis
Early diagnosis of ATTR cardiac amyloidosis is crucial to the patient’s survival; it should be ruled out in any African-American patient with unexplained heart failure and echocardiography showing increased wall thickness with a nondilated left ventricular cavity. Additional clues include significant proteinuria, hepatomegaly disproportionate to the degree of heart failure, or corresponding neuropathy. Known family history of the disease, along with variant type, allows for a prompt and correct diagnosis.10
It has been reported that most clinicians who encounter heart failure, particularly in black patients, do not consider amyloidosis in the differential diagnosis, because of the high prevalence of hypertension and congestive heart failure in this population.10,15,19 As a result, amyloidosis often goes undiagnosed.19
Findings of enlarged and thickened cardiac walls on echocardiography but normal coronary arteries on cardiac catheterization should alert the treating clinician to further work-up for cardiac amyloidosis.19 In such a patient, according to Kristen et al,21 endomyocardial biopsy with Congo Red staining is the gold standard for diagnosis of amyloidosis.
In ATTR Val122Ile familial amyloidosis, it is unclear whether patients who are homozygous for the disease present with symptoms at earlier onset with more progressive illness or die sooner than those who are heterozygous.33 Nevertheless, once the diagnosis is confirmed, it is important to determine the patient’s specific variant type by DNA testing so that appropriate treatment can be initiated and the patient’s prognosis evaluated.5,10,13
Treatment
For familial amyloidosis in general, some researchers advocate liver transplantation to remove the source of mutant amyloid protein and stop all deposition of amyloid fibrils; this procedure can be followed later by transplantation of other affected organs (including the heart).5,23 Maurer et al34 have reported improved one-year survival rates among patients with ATTR amyloidosis who underwent both cardiac and liver transplantation: 75%, versus 23% in patients who did not receive transplanted organs.
Management of cardiac amyloidosis usually requires a twofold approach: treating associated congestive heart failure, and preventing further deposition of amyloid.24 In the case patient (as in most patients with ATTR amyloidosis), heart transplantation was deemed the only life-sustaining treatment option.11,19,33
Pharmacotherapeutic options are limited for patients with ATTR Val122Ile familial amyloidosis. Conventional heart failure agents (eg, ACE inhibitors, angiotensin receptor blockers, digoxin, β-blockers, calcium channel blockers) can exacerbate heart failure symptoms, leading to a potentially life-threatening arrhythmia.3,11,19,24,35 Amyloid fibrils bind to digitalis, increasing susceptibility to digitalis toxicity; and to nifedipine, causing hemodynamic deterioration. Verapamil should be avoided, as it may induce severe left ventricular dysfunction. ACE inhibitors often provoke profound hypotension in primary amyloidosis.24,35
Diuretics, too (eg, furosemide, as was prescribed for the case patient), must be used with caution.3 These agents have been used to treat fluid overload and the resulting peripheral edema and shortness of breath found in ATTR Val122Ile patients who experience heart failure.36 According to Dubrey et al,5 cautious use of diuretics is necessary for management of heart failure symptoms in these patients.
Because the risk for intracardiac thrombus is high, anticoagulation (using agents other than β-blockers or calcium channel blockers) should be implemented unless compelling risks are involved.11,24 Amiodarone is relatively well tolerated for ventricular tachydysrhythmias and in atrial fibrillation if the goal is maintaining sinus rhythm.37
Regarding heart transplantation in patients with familial amyloidosis, Jacob et al33 hypothesize that since mutant amyloid protein is synthesized by the liver, it would take approximately 50 years for a transplanted heart to become affected by amyloid deposition. In a 59-year-old Afro-Caribbean man with familial amyloidosis who underwent cardiac transplantation, Hamour et al11 reported that the donor heart remained amyloid-free three years posttransplantation, as demonstrated by serial cardiac biopsy.
On the Horizon
Clinical trials are now under way to examine pharmacotherapeutic options for patients with ATTR amyloidosis. Now being examined in clinical trials, for example, is Fx-1006A, a drug that stabilizes ATTR and prevents the misfolding of the amyloid protein fibril, in turn preventing it from binding to the target organ.38 Similarly, ALN-TTR, a drug believed to prevent disease manifestation and possibly facilitate disease regression, is being investigated in early human trials.39
Additionally, the use of genetic testing is recommended in at-risk individuals to identify the TTR gene. Affected patients may benefit from prophylactic medical management, which would halt amyloidogenesis of TTR—and possibly treat the condition as well.35 Pharmacotherapeutic agents like diflunisal, an NSAID, antagonize the aggregation of TTR protein and hinder formation of the amyloid fibrils.40
CONCLUSION
ATTR Val122Ile familial amyloidosis is a rare disorder that causes abnormal synthesis of amyloid protein in the liver, which then infiltrates the cardiac structure, leading to restrictive cardiomyopathy and progressive heart failure. Patients who present with symptoms of heart failure, cardiac enlargement on echocardiography, and a finding of granular speckling patterns, though not specific on echocardiography, should prompt the health care provider to refer the patient to a cardiologist familiar with cardiac amyloidosis for further work-up.
Diagnosed patients must undergo genetic testing to determine the specific variant type so that prompt treatment can be initiated. In patients with ATTR Val122Ile familial amyloidosis, the treatment of choice is cardiac transplantation. Although the mutant amyloid protein continues to be synthesized in the liver, the donor heart is unlikely to become affected by this substance for many years. Appropriately treated patients can maintain good quality of life, free of heart failure.
REFERENCES
1. Lim RP, Srichai MB, Lee VS. Non-ischemic causes of delayed myocardial hyperenhancement on MRI. AJR Am J Roentgenol. 2007;188 (6):1675-1681.
2. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17(3-4):101-104.
3. Kendall H. Cardiac amyloidosis. Crit Care Nurse. 2010;30(2):16-23.
4. Eriksson M, Büttener J, Todorov T, et al. Prevalence of germline mutations in the TTR gene in a consecutive series of surgical pathology specimens with AATR amyloid. Am J Surg Pathol. 2009;33(1):58-65.
5. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
6. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277-282.
7. XDx Expression Diagnostics. Allomap®: molecular expression testing (2004). www.allomap.com. Accessed May 14, 2012.
8. Mandras SA, Crespo J, Patel HM. Innovative application of immunologic principles in heart transplantation. Ochsner J. 2010;10(4):231-235.
9. Yamani MH, Taylor DO, Rodriguez R, et al. Transplant vasculopathy is associated with increased AlloMap gene expression score. J Heart Lung Transplant. 2007;26(4):403-406.
10. Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol. 2003;17(6):909-927.
11. Hamour IM, Lachmann HJ, Goodman HJ, et al. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant. 2008;8(5):1056-1059.
12. Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336(7):466-473.
13. Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52(4):347-361.
14. Askanas V, Engel WK, McFerrin J, Vattemi G. Transthyretin Val122Ile, accumulated Abeta, and inclusion-body myositis aspects in cultured muscle. Neurology. 2003;61(2):257-260.
15. Hamidi Asl K, Nakamura M, Yamashita T, Benson MD. Cardiac amyloidoses associated with the transthyretin lle122 mutation in a Caucasian family. Amyloid. 2001;8(4):263-269.
16. Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24(3): 214-220.
17. Nihoyannopoulos P, Dawson D. Restrictive cardiomyopathies. Eur J Echocardiogr. 2009;10 (8):iii23-iii33.
18. Bruce J. Getting to the heart of cardiomyopathies. Nursing. 2005;35(8):44-47.
19. Falk RH. The neglected entity of familial cardiac amyloidosis in African Americans. Ethn Dis. 2002;12(1):141-143.
20. Piper C, Butz T, Farr M, et al. How to diagnose cardiac amyloidosis early: impact of ECG, tissue Doppler echocardiography, and myocardial biopsy. Amyloid. 2010;17(1):1-9.
21. Kristen AV, Meyer FJ, Perz JB, et al. Risk stratification in cardiac amyloidosis: novel approaches. Transplantation. 2005;80(1 suppl):S151-S155.
22. Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid. 2007;14(3):179-183.
23. Picken MM. New insights into systemic amyloidosis: the importance of diagnosis of specific type. Curr Opin Nephrol Hypertens. 2007; 16(3):196-203.
24. Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124(9):1079-1085.
25. Yamashita T, Asl KH, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile 122 allele frequency in an African-American population. Amyloid. 2005;12(2):127-130.
26. Benson MD, Yazaki M, Magy N. Laboratory assessment of transthyretin amyloidosis. Clin Chem Lab Med. 2002;40(12):1262-1265.
27. Chamarthi B, Dubrey SW, Cha K, et al. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol. 1997;80(9):1242-1245.
28. Correia MJ, Coutinho CA, Conceiçao I, et al. Role of heart rate variability in the assessment of autonomic dysfunction in type I familial amyloidotic polyneuropathy. Folia Cardiol. 2005;12(suppl C):459-462.
29. Kadish A, Mehra M. Heart failure devices: Implantable cardioverter-defibrillators and biventricular pacing therapy. Circulation. 2005; 111(24):3327-3335.
30. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43(3):410-415.
31. Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3(2):155-164.
32. Fealey ME, Edwards WD, Buadi FK, et al. Echocardiographic features of cardiac amyloidosis presenting as endomyocardial disease in a 54-year-old male. J Cardiol. 2009;54(1):162-166.
33. Jacob EK, Edwards WD, Zucker M, et al. Homozygous transthyretin mutation in an African American male. J Mol Diagn. 2007; 9(1):127-131.
34. Maurer MS, Raina A, Hesdorffer C, et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83(5):539-545.
35. Buxbaum J, Alexander A, Koziol J, et al. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African-Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159(5):864-870.
36. Rose BD, Colucci WS. Use of diuretics in heart failure (2010). www.uptodate.com/con tents/use-of-diuretics-in-patients-with-heart-failure. Accessed May 14, 2012.
37. Trappe H-J. Treating critical supraventricular and ventricular arrhythmias. J Emerg Trauma Shock. 2010;3(2):143-152.
38. Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14(30):3219-3230.
39. Alnylam Pharmaceuticals. TTR Amyloidosis: ALN-TTR (2011). www.alnylam.com/Programs-and-Pipeline/Programs/index.php. Accessed May 14, 2012.
40. Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin: potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355-374.
A black man, age 65, with no known history of cardiopulmonary disease presented with acute-onset exertional dyspnea and lower extremity edema. He also reported an episode of syncope, as well as occasional dizziness and abdominal bloating. He said he experienced exertional dyspnea while doing a routine step aerobic exercise. His exercise regimen included distance walking, yoga, and aerobics four to five days per week.
The patient’s medical history was remarkable for a single episode of a bleeding ulcer in previous years, low back pain, shoulder pain, and a septic arthritic hip. His social history was negative for use of tobacco, alcohol, or illegal drugs. He was married and had two biological daughters with fairly unremarkable medical histories. The patient had earned a master’s degree, worked full-time in the insurance business, and was an avid worldwide traveler. He reported diminished quality of life as a result of his acute-onset heart failure symptoms, which reduced his ability to exercise routinely, work full-time, or travel.
The patient’s sudden experience of exertional dyspnea prompted him to visit his primary care provider, who ordered an ECG that demonstrated low voltage patterns and a first-degree atrioventricular (AV) block. Subsequent stress echocardiography showed generalized thickening of the left ventricular myocardium. Posterior wall thickness measured 1.7 cm (normal range, 0.6 to 1.1 cm), septal thickness measured 1.9 cm (normal, 0.6 to 1.1 cm), and ejection fraction was 65%. The stress echocardiogram also showed a speckling pattern (brightly scattered spots) on the myocardium.
Although stress echocardiography results were negative for ischemic disease, the patient did experience dyspnea during the exam. He underwent cardiac catheterization, which indicated normal coronary arteries.
Additional diagnostic studies included cardiac MRI with and without contrast, which showed nulling of the heart muscle and delayed patchy hyperenhancement; this suggested myocardial tissue abnormality as result of amyloid fibril deposition.1 Both pulmonary and tricuspid aortic valves were normal, with no evidence of stenosis. No regional wall motion abnormalities were noted.
Laboratory findings during the work-up were lipid panel, unremarkable; complete blood count (CBC), mild anemia and leukopenia; and urinalysis, positive for proteinuria. Brain natriuretic peptide (BNP) was measured at 686 pg/mL (normal, 0.0 to 100 pg/mL), indicating moderate heart failure. A peripheral blood smear was negative for monoclonal plasma cells.
The patient’s physical exam was unremarkable except for 2+ pedal edema bilaterally. In consideration of normal coronary arteries on cardiac catheterization, the patient’s heart failure symptoms, and stress echocardiography abnormalities, a heart biopsy was ordered. An endomyocardial biopsy with Congo Red stain demonstrated an apple-green birefringent pattern viewed under high-definition polarized light microscope, which was consistent with amyloid deposition.2
The patient was given a diagnosis of primary amyloidosis by his local cardiologist despite negative findings on the peripheral blood smear for monoclonal plasma cells (which are typically found in primary amyloidosis).3 He presented to an institution well-known for its expertise in amyloidosis, for a second opinion. There, the diagnosis was negated, based on reevaluation of the patient’s previous heart specimen through immunohistochemical studies. These studies were positive for serum amyloid P, which is suggestive of transthyretin (TTR) or familial amyloidosis.4 Genetic testing revealed a familial amyloidosis DNA sequence analysis with the Val122Ile variant (ie, isoleucine for valine at position 1225). With the correct diagnosis confirmed, the patient was referred to another highly regarded institution to begin a work-up for cardiac transplantation. Meanwhile, he was cautiously treated with the loop diuretic furosemide to manage his shortness of breath and peripheral edema.
Fifteen months later (13 weeks after being listed for transplant), the patient underwent successful cardiac transplantation.
On pathologic review of the patient’s extricated heart, the myocardium was found to be grossly thickened (see figure, above) and weighed 540 g; the average adult heart weighs 300 to 350 g, depending on the patient’s size.6 Congo Red staining showed extensive amyloid deposits with infiltration throughout the myocardium.
Ninety percent of the amyloid deposits were interstitial, 5% were in the vessels, and 5% were noted in a nodular pattern. The left ventricular cavity showed dilated and thickened walls. Intramural and extramural blood vessels were infiltrated with amyloid as well.
Six months after transplantation, the patient underwent diagnostic testing to assess the function and structure of his new heart. Cardiac catheterization was negative for coronary artery disease. Thirteen months posttransplantation, endomyocardial biopsy with Congo Red stain was negative for amyloid deposition or organ rejection.
About 24 months posttransplantation, the patient was taking tacrolimus, pravastatin, pantoprazole, dapsone, propanolol, colchicine, and donepezil. Stress echocardiography demonstrated normal right and left ventricular systolic function; no wall-motion abnormalities or left ventricular hypertrophy were detected, and the right atrium was of normal size. There was abnormal structural enlargement of the left atrium at the site of anastamosis—a common finding in cardiac transplant patients. The aortic, tricuspid, and mitral valves were all normal.
At that time, it was decided not to repeat endomyocardial biopsy because of normal results on molecular expression testing (a noninvasive technique called AlloMap®7-9), which is performed to assess for heart transplant rejection. The patient’s lipid panel remained within normal limits. CBC indicated persistent anemia and leukopenia. Urine protein and BNP test results were not available.
Since undergoing cardiac transplantation, the patient has resumed his normal routine activities, including some type of exercise five days per week. He said his diet is maintained in moderation. He denied shortness of breath, chest pain, dizziness, or edema. He has returned to full-time employment and has vacationed in Croatia, Italy, and Central America.
DISCUSSION
Familial amyloidosis is an autosomal dominant disease characterized by the production of mutated proteins, most commonly ATTR. Presence of the ATTR Val 122Ile allele has been reported in 3.9% of all black Americans, and in one study, 23% of black Americans diagnosed with cardiac amyloidosis at autopsy were heterozygous for this variant allele.10-12 ATTR Val122Ile usually manifests in the fifth
or sixth decade of life with its characteristic presentation of infiltrative/restrictive cardiomyopathy,13 resulting in heart failure and sometimes peripheral neuropathy.10,11,14
Pathophysiology
In patients with ATTR Val122Ile, cardiomyopathy results from the deposition of mutant protein fibrils in the cardiac muscle, leading to restricted heart wall motion10,15 and stiffening of the cardiac ventricles, with subsequent disruption of the diastolic filling properties of the cardiac muscle.16 Fluid overload and heart failure follow.3,10,17 The atrium of the heart dilates, and the walls of the ventricles become thickened and fibrous.18 Liepnieks and Benson6 reported that the cadaver heart of one Val122Ile patient infiltrated with amyloid protein fibrils weighed 725 g—more than double the weight of an average adult heart.
In patients with cardiac amyloidosis, ECG can detect arrhythmia, and echocardiography shows cardiac enlargement; however, as in the case patient, cardiac catheterization shows normal coronary arteries.15,16,19,20 Thus, previously healthy patients who present with heart failure and negative results on cardiac catheterization should undergo further work-up for cardiac amyloidosis.19
Amyloidosis affects all populations globally.10 In systemic amyloidosis, amyloid releases into the plasma, infiltrating and impairing multiple organs. Poor survival has been reported in patients with heart failure symptoms resulting from amyloid deposition.20,21
Types of Amyloidosis
Primary amyloidosis, the most common of the three amyloidosis types, can be systemic or localized.22 It occurs when protein fibrils, developed from immunoglobin light chains or monoclonal plasma cells and measuring 7 to 10 nm in diameter, adhere to the heart, kidneys, peripheral nerves, eyes, and other organs.5,11,20,23,24 Known for its relation to multiple myeloma,19 primary amyloidosis is associated with a poor prognosis.3,10
Secondary amyloidosis results from a chronic inflammatory disorder, such as rheumatoid arthritis or ankylosing spondylitis—conditions that trigger the production of amyloid proteins.3,10 This type has also been associated with substance abuse and AIDS.23
Familial or hereditary amyloidosis, according to Benson,10 is a group of diseases, each resulting from mutation in a specific protein. In the United States, the most common type of familial amyloidosis is ATTR.11 More than 100 mutant types of ATTR proteins have been identified, each involving a specific nationality or group of nationalities.10,11,23
Mutant ATTR amyloid, when deposited in specific organs, leads to their dysfunction and ultimate failure.6 ATTR may affect the cardiac, gastric, renal, ophthalmic, or nervous system. Depending on the ATTR variant, the resulting clinical features are age- and time-dependent, with onset most common between the third and fifth decade of life.10
Prevalence
The prevalence of ATTR Val 122Ile amyloidosis is reportedly high in West Africa, and in the US African-American population (3.9%).4,11,14,25,26 In a study conducted at a county hospital in Indianapolis, 3% of black newborns were found positive for ATTR Val122Ile through DNA sampling of umbilical cord blood.25 These statistics are of concern, as ATTR amyloidosis could be a significant health concern in a patient population that is already medically underserved.
Yamashita et al25 estimate that 1.35 million Americans of African-American descent may be affected by ATTR Val122Ile and vulnerable to restrictive cardiomyopathy–related heart failure and death. At the very least, this disorder can impair quality of life, especially in the presence of other comorbid conditions.
Clinical Presentation
The presence of exertional syncope at presentation is ominous, as it may be a marker of severe restrictive cardiomyopathy, postural hypotension due to excessive diuresis or autonomic neuropathy, ventricular arrhythmias from localized hypoperfusion, and rarely from cardiac tamponade due to pericardial involvement.27 Despite widespread involvement of the conduction system in specimens at autopsy, high-grade IV block is unusual.3
Diagnostic Studies
ECG. Both ECG and Holter monitoring can detect the arrhythmias and conduction disturbances (eg, first-degree AV block, low voltage patterns) associated with cardiac amyloidosis. Patients often experience syncopal and near-syncopal episodes as a result of conduction disturbances.28 Patients with conditions such as cardiac amyloidosis who present with severe heart failure are at high risk for sudden death secondary to conduction disturbances. Many have benefited from implanted defibrillators.29
Echocardiography. In patients with ATTR Val122Ile cardiac amyloidosis, echocardiography reveals thickened ventricular walls (ie, measuring ≥ 15 mm; normal, ≤ 11 mm).19 Amyloid-restrictive cardiomyopathy is associated with a marked dissociation between short- and long-axis systolic function, in cases in which left ventricular ejection fraction is normal.30
Echocardiography may demonstrate the characteristic specular or granular sparkling appearance that signifies advanced disease.15,21 Only a minority have this pattern in the myocardium, however, and changes in echocardiographic technology have made this finding less noticeable.30
MRI. Among more recently used diagnostic studies, cardiac magnetic resonance (CMR) has been reported to demonstrate late gadolinium enhancement (LGE) in perhaps 80% of patients with familial amyloidosis and cardiac involvement (as determined through biopsy and Congo Red stain). LGE-CMR shows darkening of the cardiac tissue, a common occurrence in amyloidosis.31
LGE is associated with increased thickness of both left and right ventricles, lower ECG voltage patterns, elevated BNP, and elevated troponin T.31,32 Globally, LGE is associated with the worst prognosis in patients with cardiac amyloidosis. Use of LGE-CMR testing can help facilitate early detection of cardiac amyloidosis in patients who may be vulnerable to cardiac damage.31
Cardiac catheterization. In patients with cardiac amyloidosis, cardiac catheterization usually shows normal coronary arteries.3
Diagnosis
Early diagnosis of ATTR cardiac amyloidosis is crucial to the patient’s survival; it should be ruled out in any African-American patient with unexplained heart failure and echocardiography showing increased wall thickness with a nondilated left ventricular cavity. Additional clues include significant proteinuria, hepatomegaly disproportionate to the degree of heart failure, or corresponding neuropathy. Known family history of the disease, along with variant type, allows for a prompt and correct diagnosis.10
It has been reported that most clinicians who encounter heart failure, particularly in black patients, do not consider amyloidosis in the differential diagnosis, because of the high prevalence of hypertension and congestive heart failure in this population.10,15,19 As a result, amyloidosis often goes undiagnosed.19
Findings of enlarged and thickened cardiac walls on echocardiography but normal coronary arteries on cardiac catheterization should alert the treating clinician to further work-up for cardiac amyloidosis.19 In such a patient, according to Kristen et al,21 endomyocardial biopsy with Congo Red staining is the gold standard for diagnosis of amyloidosis.
In ATTR Val122Ile familial amyloidosis, it is unclear whether patients who are homozygous for the disease present with symptoms at earlier onset with more progressive illness or die sooner than those who are heterozygous.33 Nevertheless, once the diagnosis is confirmed, it is important to determine the patient’s specific variant type by DNA testing so that appropriate treatment can be initiated and the patient’s prognosis evaluated.5,10,13
Treatment
For familial amyloidosis in general, some researchers advocate liver transplantation to remove the source of mutant amyloid protein and stop all deposition of amyloid fibrils; this procedure can be followed later by transplantation of other affected organs (including the heart).5,23 Maurer et al34 have reported improved one-year survival rates among patients with ATTR amyloidosis who underwent both cardiac and liver transplantation: 75%, versus 23% in patients who did not receive transplanted organs.
Management of cardiac amyloidosis usually requires a twofold approach: treating associated congestive heart failure, and preventing further deposition of amyloid.24 In the case patient (as in most patients with ATTR amyloidosis), heart transplantation was deemed the only life-sustaining treatment option.11,19,33
Pharmacotherapeutic options are limited for patients with ATTR Val122Ile familial amyloidosis. Conventional heart failure agents (eg, ACE inhibitors, angiotensin receptor blockers, digoxin, β-blockers, calcium channel blockers) can exacerbate heart failure symptoms, leading to a potentially life-threatening arrhythmia.3,11,19,24,35 Amyloid fibrils bind to digitalis, increasing susceptibility to digitalis toxicity; and to nifedipine, causing hemodynamic deterioration. Verapamil should be avoided, as it may induce severe left ventricular dysfunction. ACE inhibitors often provoke profound hypotension in primary amyloidosis.24,35
Diuretics, too (eg, furosemide, as was prescribed for the case patient), must be used with caution.3 These agents have been used to treat fluid overload and the resulting peripheral edema and shortness of breath found in ATTR Val122Ile patients who experience heart failure.36 According to Dubrey et al,5 cautious use of diuretics is necessary for management of heart failure symptoms in these patients.
Because the risk for intracardiac thrombus is high, anticoagulation (using agents other than β-blockers or calcium channel blockers) should be implemented unless compelling risks are involved.11,24 Amiodarone is relatively well tolerated for ventricular tachydysrhythmias and in atrial fibrillation if the goal is maintaining sinus rhythm.37
Regarding heart transplantation in patients with familial amyloidosis, Jacob et al33 hypothesize that since mutant amyloid protein is synthesized by the liver, it would take approximately 50 years for a transplanted heart to become affected by amyloid deposition. In a 59-year-old Afro-Caribbean man with familial amyloidosis who underwent cardiac transplantation, Hamour et al11 reported that the donor heart remained amyloid-free three years posttransplantation, as demonstrated by serial cardiac biopsy.
On the Horizon
Clinical trials are now under way to examine pharmacotherapeutic options for patients with ATTR amyloidosis. Now being examined in clinical trials, for example, is Fx-1006A, a drug that stabilizes ATTR and prevents the misfolding of the amyloid protein fibril, in turn preventing it from binding to the target organ.38 Similarly, ALN-TTR, a drug believed to prevent disease manifestation and possibly facilitate disease regression, is being investigated in early human trials.39
Additionally, the use of genetic testing is recommended in at-risk individuals to identify the TTR gene. Affected patients may benefit from prophylactic medical management, which would halt amyloidogenesis of TTR—and possibly treat the condition as well.35 Pharmacotherapeutic agents like diflunisal, an NSAID, antagonize the aggregation of TTR protein and hinder formation of the amyloid fibrils.40
CONCLUSION
ATTR Val122Ile familial amyloidosis is a rare disorder that causes abnormal synthesis of amyloid protein in the liver, which then infiltrates the cardiac structure, leading to restrictive cardiomyopathy and progressive heart failure. Patients who present with symptoms of heart failure, cardiac enlargement on echocardiography, and a finding of granular speckling patterns, though not specific on echocardiography, should prompt the health care provider to refer the patient to a cardiologist familiar with cardiac amyloidosis for further work-up.
Diagnosed patients must undergo genetic testing to determine the specific variant type so that prompt treatment can be initiated. In patients with ATTR Val122Ile familial amyloidosis, the treatment of choice is cardiac transplantation. Although the mutant amyloid protein continues to be synthesized in the liver, the donor heart is unlikely to become affected by this substance for many years. Appropriately treated patients can maintain good quality of life, free of heart failure.
REFERENCES
1. Lim RP, Srichai MB, Lee VS. Non-ischemic causes of delayed myocardial hyperenhancement on MRI. AJR Am J Roentgenol. 2007;188 (6):1675-1681.
2. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17(3-4):101-104.
3. Kendall H. Cardiac amyloidosis. Crit Care Nurse. 2010;30(2):16-23.
4. Eriksson M, Büttener J, Todorov T, et al. Prevalence of germline mutations in the TTR gene in a consecutive series of surgical pathology specimens with AATR amyloid. Am J Surg Pathol. 2009;33(1):58-65.
5. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
6. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277-282.
7. XDx Expression Diagnostics. Allomap®: molecular expression testing (2004). www.allomap.com. Accessed May 14, 2012.
8. Mandras SA, Crespo J, Patel HM. Innovative application of immunologic principles in heart transplantation. Ochsner J. 2010;10(4):231-235.
9. Yamani MH, Taylor DO, Rodriguez R, et al. Transplant vasculopathy is associated with increased AlloMap gene expression score. J Heart Lung Transplant. 2007;26(4):403-406.
10. Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol. 2003;17(6):909-927.
11. Hamour IM, Lachmann HJ, Goodman HJ, et al. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant. 2008;8(5):1056-1059.
12. Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336(7):466-473.
13. Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52(4):347-361.
14. Askanas V, Engel WK, McFerrin J, Vattemi G. Transthyretin Val122Ile, accumulated Abeta, and inclusion-body myositis aspects in cultured muscle. Neurology. 2003;61(2):257-260.
15. Hamidi Asl K, Nakamura M, Yamashita T, Benson MD. Cardiac amyloidoses associated with the transthyretin lle122 mutation in a Caucasian family. Amyloid. 2001;8(4):263-269.
16. Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24(3): 214-220.
17. Nihoyannopoulos P, Dawson D. Restrictive cardiomyopathies. Eur J Echocardiogr. 2009;10 (8):iii23-iii33.
18. Bruce J. Getting to the heart of cardiomyopathies. Nursing. 2005;35(8):44-47.
19. Falk RH. The neglected entity of familial cardiac amyloidosis in African Americans. Ethn Dis. 2002;12(1):141-143.
20. Piper C, Butz T, Farr M, et al. How to diagnose cardiac amyloidosis early: impact of ECG, tissue Doppler echocardiography, and myocardial biopsy. Amyloid. 2010;17(1):1-9.
21. Kristen AV, Meyer FJ, Perz JB, et al. Risk stratification in cardiac amyloidosis: novel approaches. Transplantation. 2005;80(1 suppl):S151-S155.
22. Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid. 2007;14(3):179-183.
23. Picken MM. New insights into systemic amyloidosis: the importance of diagnosis of specific type. Curr Opin Nephrol Hypertens. 2007; 16(3):196-203.
24. Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124(9):1079-1085.
25. Yamashita T, Asl KH, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile 122 allele frequency in an African-American population. Amyloid. 2005;12(2):127-130.
26. Benson MD, Yazaki M, Magy N. Laboratory assessment of transthyretin amyloidosis. Clin Chem Lab Med. 2002;40(12):1262-1265.
27. Chamarthi B, Dubrey SW, Cha K, et al. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol. 1997;80(9):1242-1245.
28. Correia MJ, Coutinho CA, Conceiçao I, et al. Role of heart rate variability in the assessment of autonomic dysfunction in type I familial amyloidotic polyneuropathy. Folia Cardiol. 2005;12(suppl C):459-462.
29. Kadish A, Mehra M. Heart failure devices: Implantable cardioverter-defibrillators and biventricular pacing therapy. Circulation. 2005; 111(24):3327-3335.
30. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43(3):410-415.
31. Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3(2):155-164.
32. Fealey ME, Edwards WD, Buadi FK, et al. Echocardiographic features of cardiac amyloidosis presenting as endomyocardial disease in a 54-year-old male. J Cardiol. 2009;54(1):162-166.
33. Jacob EK, Edwards WD, Zucker M, et al. Homozygous transthyretin mutation in an African American male. J Mol Diagn. 2007; 9(1):127-131.
34. Maurer MS, Raina A, Hesdorffer C, et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83(5):539-545.
35. Buxbaum J, Alexander A, Koziol J, et al. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African-Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159(5):864-870.
36. Rose BD, Colucci WS. Use of diuretics in heart failure (2010). www.uptodate.com/con tents/use-of-diuretics-in-patients-with-heart-failure. Accessed May 14, 2012.
37. Trappe H-J. Treating critical supraventricular and ventricular arrhythmias. J Emerg Trauma Shock. 2010;3(2):143-152.
38. Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14(30):3219-3230.
39. Alnylam Pharmaceuticals. TTR Amyloidosis: ALN-TTR (2011). www.alnylam.com/Programs-and-Pipeline/Programs/index.php. Accessed May 14, 2012.
40. Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin: potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355-374.