User login
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
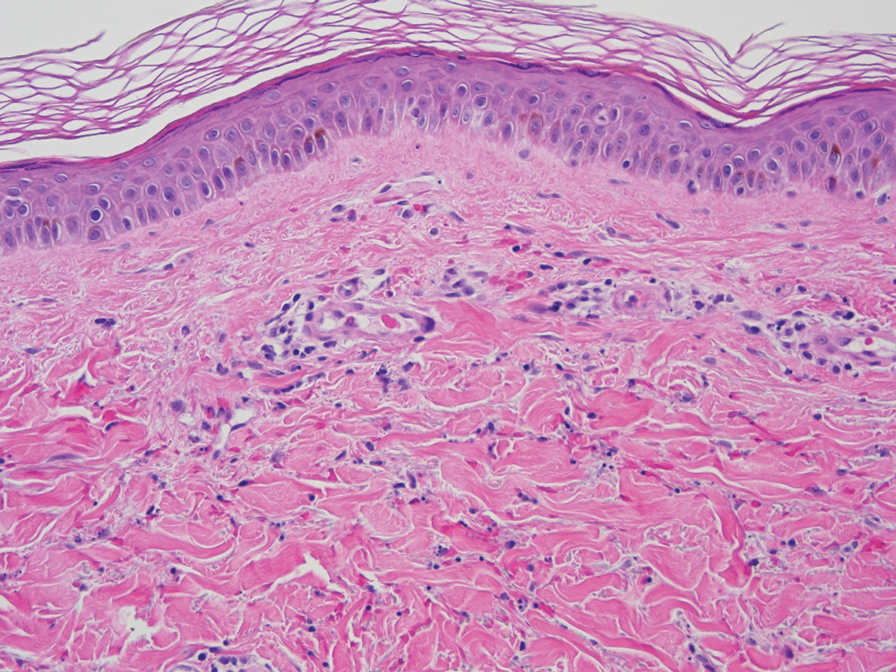
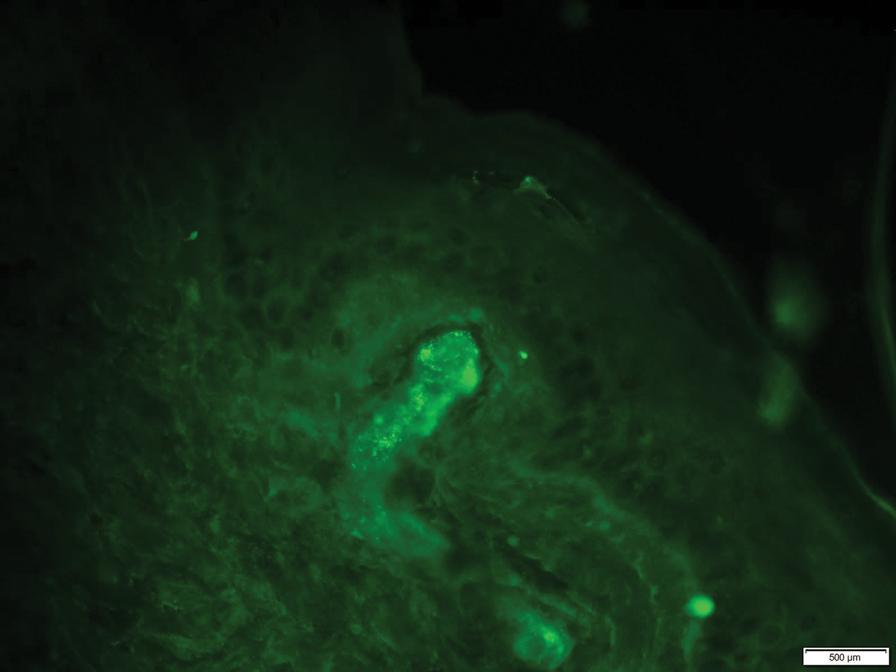
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
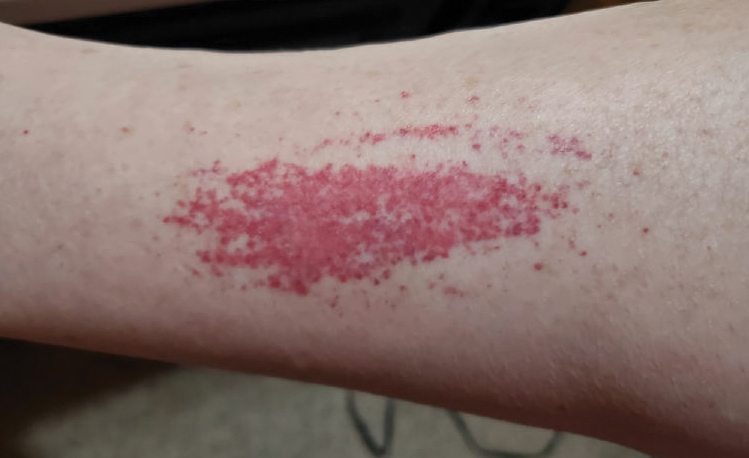
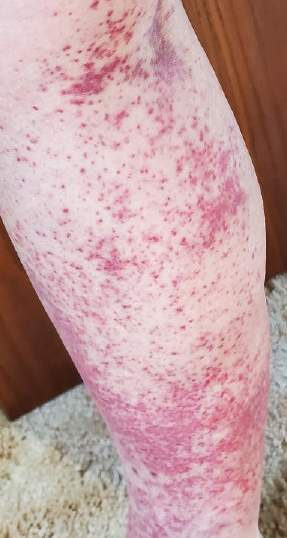
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Practice Points
- Elevation of the erythrocyte sedimentation rate (ESR) is nonspecific for inflammation and may be observed in the setting of increased immunoglobulin levels.
- Patients with elevated ESR and clinical evidence of recurrent petechiae and purpura should be screened for monoclonal and polyclonal gammopathies.
