User login
ACO Insider: Not ready for an ACO? Think CPC+
The Centers for Medicare & Medicaid Services in April announced its newest initiative, Comprehensive Primary Care Plus, to target primary care practices of varying capabilities to participate in an innovative payment model designed to support the delivery of comprehensive primary care that rewards value and quality.
“Strengthening primary care is critical to an effective health care system,” said Patrick Conway, MD, CMS deputy administrator and chief medical officer. “By supporting primary care doctors and clinicians to spend time with patients, serve patients’ needs outside of the office visit, and better coordinate care with specialists, we can continue to build a health care system that results in healthier people and smarter spending of our health care dollars.”

As readers of this column know, these are also the engines of accountable care organization success. So, if you and your patient-centered medical home are not in a Medicare ACO, this gets you going on high-value activities – and pays you monthly to do it.
The rub is that once you are in the Medicare Shared Savings Program, you can’t continue with this initiative. But, it’s a great “on ramp” to prep you for ACO success. You get monthly payments instead of waiting 18 months for shared savings that you may or may not get under the Medicare Shared Savings Program.
CPC+ is an advanced primary medical home model, created from lessons learned in the Comprehensive Primary Care Initiative and the Multi-Payer Advanced Primary Care Practice Demonstration. Similar to these programs, multi-payer engagement is an essential component of the model.
In the CPC+ model, the CMS intends to nationally solicit a variety of payers committed to strengthening primary care in up to 20 regions and accept up to 5,000 practices to participate in those regions. The CPC+ program is further evidence that primary care should not only be a fundamental component to moving our health care system to one that awards clinicians based on the quality, not quantity, of care they give patients, but that payment redesign must provide flexibility to accommodate the diverse needs of primary care practices.
What to know about payment
To provide this flexibility and to attract practices of varying capabilities and levels of experience, the CPC+ program offers two tracks with different payment options, which include a monthly care management fee, comprehensive primary care payments, and performance-based incentive payments.
In track 1, the CMS will pay practices a risk-adjusted prospective monthly care management fee ($15 per beneficiary per month [PBPM] average across four risk tiers), in addition to the fee-for-service payments under the Medicare Physician Fee Schedule for activities.
In track 2, the Medicare monthly care management fees will average $28 PBPM across five risk tiers, which includes a $100 care management fee to support care for patients with the most complex needs. Instead of full Medicare fee-for-service payments for evaluation and management services, track 2 practices will receive a hybrid of reduced Medicare fee-for-service payments and up-front comprehensive primary care payments for those services.
In addition, the CMS is providing incentive payments at $2.50 PBPM for track 1 and $4 PBPM for track 2, based on practice performance on utilization metrics and quality, measured at the practice level. While these payments are prepaid at the beginning of a performance year, they are subject to recoupment if the practice does not meet thresholds for quality and utilization performance.
What to know about participation
To participate, your practice must be located within 1 of the 20 regional geographic areas selected by the CMS and must serve not only Medicare beneficiaries, but patients covered by one or more additional participating payers.
You may apply for either track 1 or track 2, but participation for the entire 5-year period will be within a single track.
All practices will be expected to deliver a set of five comprehensive primary care functions and have certified electronic health record technology capabilities. Track 2 practices will be expected to focus on a core set of advance capabilities for health information technology and must submit a letter of support from their health IT vendors. The CMS may require a track 2 applicant to participate in track 1.
Participating in the CPC+ program limits your ability to fully participate in or utilize other CMS initiatives, models, or demonstrations, however – including the Medicare Shared Savings Program and Next Generation ACO, or bill for the chronic care management fee. This is a big trade-off for practices well down the value transformation path, but an opportunity for those getting started.
Although the shift to payment for improved population health can herald the golden age of primary care, you cannot default on this opportunity through inaction. It is urgent that you choose a path to value-care delivery. CPC+ provides the ability for greater cash flow and flexibility for primary care practices to deliver high-quality, whole-person patient-centered care.
Mr. Bobbitt is head of the health law group at the Smith Anderson law firm in Raleigh, N.C. He is president of Value Health Partners, LLC, a health care strategic consulting company. He has years of experience assisting physicians to form integrated delivery systems and prepare for the value-based compensation era. Mr. Parker is a member of the health law group at Smith Anderson and works with Mr. Bobbitt to guide physicians regarding preparing for value-based care. This article is meant to be educational and does not constitute legal advice. For additional information, readers may contact the author at bbobbitt@smithlaw.com or 919-821-6612.
The Centers for Medicare & Medicaid Services in April announced its newest initiative, Comprehensive Primary Care Plus, to target primary care practices of varying capabilities to participate in an innovative payment model designed to support the delivery of comprehensive primary care that rewards value and quality.
“Strengthening primary care is critical to an effective health care system,” said Patrick Conway, MD, CMS deputy administrator and chief medical officer. “By supporting primary care doctors and clinicians to spend time with patients, serve patients’ needs outside of the office visit, and better coordinate care with specialists, we can continue to build a health care system that results in healthier people and smarter spending of our health care dollars.”
As readers of this column know, these are also the engines of accountable care organization success. So, if you and your patient-centered medical home are not in a Medicare ACO, this gets you going on high-value activities – and pays you monthly to do it.
The rub is that once you are in the Medicare Shared Savings Program, you can’t continue with this initiative. But, it’s a great “on ramp” to prep you for ACO success. You get monthly payments instead of waiting 18 months for shared savings that you may or may not get under the Medicare Shared Savings Program.
CPC+ is an advanced primary medical home model, created from lessons learned in the Comprehensive Primary Care Initiative and the Multi-Payer Advanced Primary Care Practice Demonstration. Similar to these programs, multi-payer engagement is an essential component of the model.
In the CPC+ model, the CMS intends to nationally solicit a variety of payers committed to strengthening primary care in up to 20 regions and accept up to 5,000 practices to participate in those regions. The CPC+ program is further evidence that primary care should not only be a fundamental component to moving our health care system to one that awards clinicians based on the quality, not quantity, of care they give patients, but that payment redesign must provide flexibility to accommodate the diverse needs of primary care practices.
What to know about payment
To provide this flexibility and to attract practices of varying capabilities and levels of experience, the CPC+ program offers two tracks with different payment options, which include a monthly care management fee, comprehensive primary care payments, and performance-based incentive payments.
In track 1, the CMS will pay practices a risk-adjusted prospective monthly care management fee ($15 per beneficiary per month [PBPM] average across four risk tiers), in addition to the fee-for-service payments under the Medicare Physician Fee Schedule for activities.
In track 2, the Medicare monthly care management fees will average $28 PBPM across five risk tiers, which includes a $100 care management fee to support care for patients with the most complex needs. Instead of full Medicare fee-for-service payments for evaluation and management services, track 2 practices will receive a hybrid of reduced Medicare fee-for-service payments and up-front comprehensive primary care payments for those services.
In addition, the CMS is providing incentive payments at $2.50 PBPM for track 1 and $4 PBPM for track 2, based on practice performance on utilization metrics and quality, measured at the practice level. While these payments are prepaid at the beginning of a performance year, they are subject to recoupment if the practice does not meet thresholds for quality and utilization performance.
What to know about participation
To participate, your practice must be located within 1 of the 20 regional geographic areas selected by the CMS and must serve not only Medicare beneficiaries, but patients covered by one or more additional participating payers.
You may apply for either track 1 or track 2, but participation for the entire 5-year period will be within a single track.
All practices will be expected to deliver a set of five comprehensive primary care functions and have certified electronic health record technology capabilities. Track 2 practices will be expected to focus on a core set of advance capabilities for health information technology and must submit a letter of support from their health IT vendors. The CMS may require a track 2 applicant to participate in track 1.
Participating in the CPC+ program limits your ability to fully participate in or utilize other CMS initiatives, models, or demonstrations, however – including the Medicare Shared Savings Program and Next Generation ACO, or bill for the chronic care management fee. This is a big trade-off for practices well down the value transformation path, but an opportunity for those getting started.
Although the shift to payment for improved population health can herald the golden age of primary care, you cannot default on this opportunity through inaction. It is urgent that you choose a path to value-care delivery. CPC+ provides the ability for greater cash flow and flexibility for primary care practices to deliver high-quality, whole-person patient-centered care.
Mr. Bobbitt is head of the health law group at the Smith Anderson law firm in Raleigh, N.C. He is president of Value Health Partners, LLC, a health care strategic consulting company. He has years of experience assisting physicians to form integrated delivery systems and prepare for the value-based compensation era. Mr. Parker is a member of the health law group at Smith Anderson and works with Mr. Bobbitt to guide physicians regarding preparing for value-based care. This article is meant to be educational and does not constitute legal advice. For additional information, readers may contact the author at bbobbitt@smithlaw.com or 919-821-6612.
The Centers for Medicare & Medicaid Services in April announced its newest initiative, Comprehensive Primary Care Plus, to target primary care practices of varying capabilities to participate in an innovative payment model designed to support the delivery of comprehensive primary care that rewards value and quality.
“Strengthening primary care is critical to an effective health care system,” said Patrick Conway, MD, CMS deputy administrator and chief medical officer. “By supporting primary care doctors and clinicians to spend time with patients, serve patients’ needs outside of the office visit, and better coordinate care with specialists, we can continue to build a health care system that results in healthier people and smarter spending of our health care dollars.”
As readers of this column know, these are also the engines of accountable care organization success. So, if you and your patient-centered medical home are not in a Medicare ACO, this gets you going on high-value activities – and pays you monthly to do it.
The rub is that once you are in the Medicare Shared Savings Program, you can’t continue with this initiative. But, it’s a great “on ramp” to prep you for ACO success. You get monthly payments instead of waiting 18 months for shared savings that you may or may not get under the Medicare Shared Savings Program.
CPC+ is an advanced primary medical home model, created from lessons learned in the Comprehensive Primary Care Initiative and the Multi-Payer Advanced Primary Care Practice Demonstration. Similar to these programs, multi-payer engagement is an essential component of the model.
In the CPC+ model, the CMS intends to nationally solicit a variety of payers committed to strengthening primary care in up to 20 regions and accept up to 5,000 practices to participate in those regions. The CPC+ program is further evidence that primary care should not only be a fundamental component to moving our health care system to one that awards clinicians based on the quality, not quantity, of care they give patients, but that payment redesign must provide flexibility to accommodate the diverse needs of primary care practices.
What to know about payment
To provide this flexibility and to attract practices of varying capabilities and levels of experience, the CPC+ program offers two tracks with different payment options, which include a monthly care management fee, comprehensive primary care payments, and performance-based incentive payments.
In track 1, the CMS will pay practices a risk-adjusted prospective monthly care management fee ($15 per beneficiary per month [PBPM] average across four risk tiers), in addition to the fee-for-service payments under the Medicare Physician Fee Schedule for activities.
In track 2, the Medicare monthly care management fees will average $28 PBPM across five risk tiers, which includes a $100 care management fee to support care for patients with the most complex needs. Instead of full Medicare fee-for-service payments for evaluation and management services, track 2 practices will receive a hybrid of reduced Medicare fee-for-service payments and up-front comprehensive primary care payments for those services.
In addition, the CMS is providing incentive payments at $2.50 PBPM for track 1 and $4 PBPM for track 2, based on practice performance on utilization metrics and quality, measured at the practice level. While these payments are prepaid at the beginning of a performance year, they are subject to recoupment if the practice does not meet thresholds for quality and utilization performance.
What to know about participation
To participate, your practice must be located within 1 of the 20 regional geographic areas selected by the CMS and must serve not only Medicare beneficiaries, but patients covered by one or more additional participating payers.
You may apply for either track 1 or track 2, but participation for the entire 5-year period will be within a single track.
All practices will be expected to deliver a set of five comprehensive primary care functions and have certified electronic health record technology capabilities. Track 2 practices will be expected to focus on a core set of advance capabilities for health information technology and must submit a letter of support from their health IT vendors. The CMS may require a track 2 applicant to participate in track 1.
Participating in the CPC+ program limits your ability to fully participate in or utilize other CMS initiatives, models, or demonstrations, however – including the Medicare Shared Savings Program and Next Generation ACO, or bill for the chronic care management fee. This is a big trade-off for practices well down the value transformation path, but an opportunity for those getting started.
Although the shift to payment for improved population health can herald the golden age of primary care, you cannot default on this opportunity through inaction. It is urgent that you choose a path to value-care delivery. CPC+ provides the ability for greater cash flow and flexibility for primary care practices to deliver high-quality, whole-person patient-centered care.
Mr. Bobbitt is head of the health law group at the Smith Anderson law firm in Raleigh, N.C. He is president of Value Health Partners, LLC, a health care strategic consulting company. He has years of experience assisting physicians to form integrated delivery systems and prepare for the value-based compensation era. Mr. Parker is a member of the health law group at Smith Anderson and works with Mr. Bobbitt to guide physicians regarding preparing for value-based care. This article is meant to be educational and does not constitute legal advice. For additional information, readers may contact the author at bbobbitt@smithlaw.com or 919-821-6612.

Adding ipilimumab to chemotherapy did not boost overall survival in small-cell lung cancer
For patients with extensive-stage small-cell lung cancer, adding the anti–CTLA-4 antibody ipilimumab (Yervoy) to etoposide and platinum did not improve overall survival, compared with administering etoposide and platinum alone, investigators reported in the Journal of Clinical Oncology.
“Although exploratory in nature, chemotherapy plus ipilimumab did not demonstrate significant improvement in other endpoints, and no subgroups demonstrated greater benefit versus chemotherapy alone,” added Martin Reck, MD, of LungenClinic Grosshansdorf (Germany), and his associates.
The randomized phase III study enrolled 1,132 chemotherapy-naive patients who were randomly assigned to either chemotherapy plus ipilimumab (etoposide plus investigator’s choice of cisplatin or carboplatin during cycles one to four, and ipilimumab during cycles three to six) or to chemotherapy plus placebo (etoposide plus investigator’s choice of cisplatin or carboplatin during cycles one to four, and placebo during cycles three to six). Patients with a complete or partial response during induction could undergo prophylactic cranial irradiation before starting maintenance ipilimumab (10 mg/kg) or placebo administered every 12 weeks, the investigators reported (J Clin Oncol. 2016 July 26. doi:10.1200/JCO.2016.67.6601).
Among 954 patients who received at least one dose of study therapy, the primary endpoint, median overall survival, was 11.0 months for the ipilimumab-chemotherapy arm and 10.9 months for the placebo arm (hazard ratio, 0.9; 95% confidence interval, 0.8-1.1; P = .4). “Across most prespecified patient subgroups, hazard ratios for overall survival [also] did not seem to favor one treatment arm,” the researchers wrote.
Ipilimumab also failed to achieve a meaningful increase in median progression-free survival, compared with placebo (4.6 months and 4.4 months, respectively). There was only one complete response in the ipilimumab-chemotherapy group and none in the chemotherapy-placebo group. A total of 62% of patients achieved a partial response in each group, and the median duration of response was 4 months in the ipilimumab-chemotherapy group (95% CI, 3.3-4.2 months) versus 3.5 months (95% CI, 3.3-4.1 months) in the chemotherapy-placebo group.
There were no new or unexpected safety signals, but ipilimumab was associated with higher rates of overall and severe-grade diarrhea, colitis, and rash. The five treatment-related deaths in the ipilimumab-chemotherapy group included two from colitis, two from sepsis, and one from liver toxicity. “In the chemotherapy plus placebo arm, there were two treatment-related deaths, one resulting from sepsis and one from bone marrow suppression,” the researchers wrote. Treatment-related discontinuations also were more common with ipilimumab plus chemotherapy (18%) than with chemotherapy plus placebo (2%).
“To date, PD-1 inhibitors, alone or in combination with CTLA-4 inhibitors, show the most promise in small-cell lung cancer,” the investigators said. Multiple trials are exploring the use of these agents as maintenance therapy or in second-line settings, they added.
Bristol-Myers Squibb makes ipilimumab, funded the study, and helped collect and analyze the data. Dr. Reck disclosed financial ties to Bristol-Myers Squibb, Hoffmann-La Roche, Eli Lilly, Merck Sharp & Dohme, and several other pharmaceutical companies.
For patients with extensive-stage small-cell lung cancer, adding the anti–CTLA-4 antibody ipilimumab (Yervoy) to etoposide and platinum did not improve overall survival, compared with administering etoposide and platinum alone, investigators reported in the Journal of Clinical Oncology.
“Although exploratory in nature, chemotherapy plus ipilimumab did not demonstrate significant improvement in other endpoints, and no subgroups demonstrated greater benefit versus chemotherapy alone,” added Martin Reck, MD, of LungenClinic Grosshansdorf (Germany), and his associates.
The randomized phase III study enrolled 1,132 chemotherapy-naive patients who were randomly assigned to either chemotherapy plus ipilimumab (etoposide plus investigator’s choice of cisplatin or carboplatin during cycles one to four, and ipilimumab during cycles three to six) or to chemotherapy plus placebo (etoposide plus investigator’s choice of cisplatin or carboplatin during cycles one to four, and placebo during cycles three to six). Patients with a complete or partial response during induction could undergo prophylactic cranial irradiation before starting maintenance ipilimumab (10 mg/kg) or placebo administered every 12 weeks, the investigators reported (J Clin Oncol. 2016 July 26. doi:10.1200/JCO.2016.67.6601).
Among 954 patients who received at least one dose of study therapy, the primary endpoint, median overall survival, was 11.0 months for the ipilimumab-chemotherapy arm and 10.9 months for the placebo arm (hazard ratio, 0.9; 95% confidence interval, 0.8-1.1; P = .4). “Across most prespecified patient subgroups, hazard ratios for overall survival [also] did not seem to favor one treatment arm,” the researchers wrote.
Ipilimumab also failed to achieve a meaningful increase in median progression-free survival, compared with placebo (4.6 months and 4.4 months, respectively). There was only one complete response in the ipilimumab-chemotherapy group and none in the chemotherapy-placebo group. A total of 62% of patients achieved a partial response in each group, and the median duration of response was 4 months in the ipilimumab-chemotherapy group (95% CI, 3.3-4.2 months) versus 3.5 months (95% CI, 3.3-4.1 months) in the chemotherapy-placebo group.
There were no new or unexpected safety signals, but ipilimumab was associated with higher rates of overall and severe-grade diarrhea, colitis, and rash. The five treatment-related deaths in the ipilimumab-chemotherapy group included two from colitis, two from sepsis, and one from liver toxicity. “In the chemotherapy plus placebo arm, there were two treatment-related deaths, one resulting from sepsis and one from bone marrow suppression,” the researchers wrote. Treatment-related discontinuations also were more common with ipilimumab plus chemotherapy (18%) than with chemotherapy plus placebo (2%).
“To date, PD-1 inhibitors, alone or in combination with CTLA-4 inhibitors, show the most promise in small-cell lung cancer,” the investigators said. Multiple trials are exploring the use of these agents as maintenance therapy or in second-line settings, they added.
Bristol-Myers Squibb makes ipilimumab, funded the study, and helped collect and analyze the data. Dr. Reck disclosed financial ties to Bristol-Myers Squibb, Hoffmann-La Roche, Eli Lilly, Merck Sharp & Dohme, and several other pharmaceutical companies.
For patients with extensive-stage small-cell lung cancer, adding the anti–CTLA-4 antibody ipilimumab (Yervoy) to etoposide and platinum did not improve overall survival, compared with administering etoposide and platinum alone, investigators reported in the Journal of Clinical Oncology.
“Although exploratory in nature, chemotherapy plus ipilimumab did not demonstrate significant improvement in other endpoints, and no subgroups demonstrated greater benefit versus chemotherapy alone,” added Martin Reck, MD, of LungenClinic Grosshansdorf (Germany), and his associates.
The randomized phase III study enrolled 1,132 chemotherapy-naive patients who were randomly assigned to either chemotherapy plus ipilimumab (etoposide plus investigator’s choice of cisplatin or carboplatin during cycles one to four, and ipilimumab during cycles three to six) or to chemotherapy plus placebo (etoposide plus investigator’s choice of cisplatin or carboplatin during cycles one to four, and placebo during cycles three to six). Patients with a complete or partial response during induction could undergo prophylactic cranial irradiation before starting maintenance ipilimumab (10 mg/kg) or placebo administered every 12 weeks, the investigators reported (J Clin Oncol. 2016 July 26. doi:10.1200/JCO.2016.67.6601).
Among 954 patients who received at least one dose of study therapy, the primary endpoint, median overall survival, was 11.0 months for the ipilimumab-chemotherapy arm and 10.9 months for the placebo arm (hazard ratio, 0.9; 95% confidence interval, 0.8-1.1; P = .4). “Across most prespecified patient subgroups, hazard ratios for overall survival [also] did not seem to favor one treatment arm,” the researchers wrote.
Ipilimumab also failed to achieve a meaningful increase in median progression-free survival, compared with placebo (4.6 months and 4.4 months, respectively). There was only one complete response in the ipilimumab-chemotherapy group and none in the chemotherapy-placebo group. A total of 62% of patients achieved a partial response in each group, and the median duration of response was 4 months in the ipilimumab-chemotherapy group (95% CI, 3.3-4.2 months) versus 3.5 months (95% CI, 3.3-4.1 months) in the chemotherapy-placebo group.
There were no new or unexpected safety signals, but ipilimumab was associated with higher rates of overall and severe-grade diarrhea, colitis, and rash. The five treatment-related deaths in the ipilimumab-chemotherapy group included two from colitis, two from sepsis, and one from liver toxicity. “In the chemotherapy plus placebo arm, there were two treatment-related deaths, one resulting from sepsis and one from bone marrow suppression,” the researchers wrote. Treatment-related discontinuations also were more common with ipilimumab plus chemotherapy (18%) than with chemotherapy plus placebo (2%).
“To date, PD-1 inhibitors, alone or in combination with CTLA-4 inhibitors, show the most promise in small-cell lung cancer,” the investigators said. Multiple trials are exploring the use of these agents as maintenance therapy or in second-line settings, they added.
Bristol-Myers Squibb makes ipilimumab, funded the study, and helped collect and analyze the data. Dr. Reck disclosed financial ties to Bristol-Myers Squibb, Hoffmann-La Roche, Eli Lilly, Merck Sharp & Dohme, and several other pharmaceutical companies.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: The anti–CTLA-4 antibody ipilimumab (Yervoy) missed its primary endpoint in a trial of patients with extensive-stage small-cell lung cancer.
Major finding: The median overall survival, was 11.0 months for the ipilimumab-chemotherapy arm and 10.9 months for the placebo arm (hazard ratio, 0.9; 95% confidence interval, 0.8-1.1; P = .4).
Data source: A randomized phase III trial of 954 patients.
Disclosures: Bristol-Myers Squibb funded the study and helped collect and analyze the study results. Dr. Reck disclosed financial ties to Bristol-Myers Squibb, Hoffmann-La Roche, Eli Lilly, Merck Sharp & Dohme, and several other pharmaceutical companies.
VIDEO: Minimal disease activity criteria sought for axial spondyloarthritis
DENVER – U.S. rheumatologists plan to develop new criteria to define minimal disease activity in patients with axial spondyloarthritis to better gauge in routine clinical practice how well these patients respond to treatment.
A score on the Ankylosing Spondylitis Disease Activity Score (ASDAS) of less than 1.3 is the most commonly used measure today of minimal disease activity, but the ASDAS isn’t suitable for point-of-care assessment in routine practice because of the need for a C-reactive protein level or erythrocyte sedimentation rate.

“In the clinic, ASDAS is very difficult or next to impossible to do” because it needs CRP or ESR, which are “never available at the point of care,” Atul A. Deodhar, MD, said in an interview at the annual meeting of the Spondyloarthritis Research and Treatment Network (SPARTAN). Another limitation of ASDAS is that it focuses exclusively on musculoskeletal measures and does not take into account extra-articular manifestations of axial spondyloarthritis such as those in the eye, gastrointestinal tract, or skin.
“We would like a clinical measurement that might tell us that a patient is appropriately treated and at minimal disease activity,” said Dr. Deodhar, professor of medicine and medical director of the rheumatology clinics at Oregon Health & Science University in Portland.
As outgoing chair of SPARTAN, Dr. Deodhar introduced a proposal that SPARTAN develop new minimal disease activity criteria for patients with axial spondyloarthritis, presenting the rationale for this project at the meeting along with the incoming chair Lianne S. Gensler, MD.
“We want a way to measure disease activity at a stage that is not full remission but with enough of a reduction in disease activity to make a difference,” said Dr. Gensler, director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco. Another frequently used gauge of minimal disease activity in axial spondyloarthritis, the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), is flawed by being totally based on subjective measurements without input from the attending rheumatologist, Dr. Gensler said.
The SPARTAN leadership will try to partner in this effort with the OMERACT (Outcome Measures in Rheumatology) program, the Assessment of Spondyloarthritis International Society (ASAS), or both, but if necessary SPARTAN will develop new minimal disease activity criteria on its own, Dr. Deodhar said.
Dr. Deodhar has received research support from 10 drug companies. Dr. Gensler has been a consultant to or has received research support from AbbVie, Amgen, Janssen, Novartis, and UCB.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
DENVER – U.S. rheumatologists plan to develop new criteria to define minimal disease activity in patients with axial spondyloarthritis to better gauge in routine clinical practice how well these patients respond to treatment.
A score on the Ankylosing Spondylitis Disease Activity Score (ASDAS) of less than 1.3 is the most commonly used measure today of minimal disease activity, but the ASDAS isn’t suitable for point-of-care assessment in routine practice because of the need for a C-reactive protein level or erythrocyte sedimentation rate.
“In the clinic, ASDAS is very difficult or next to impossible to do” because it needs CRP or ESR, which are “never available at the point of care,” Atul A. Deodhar, MD, said in an interview at the annual meeting of the Spondyloarthritis Research and Treatment Network (SPARTAN). Another limitation of ASDAS is that it focuses exclusively on musculoskeletal measures and does not take into account extra-articular manifestations of axial spondyloarthritis such as those in the eye, gastrointestinal tract, or skin.
“We would like a clinical measurement that might tell us that a patient is appropriately treated and at minimal disease activity,” said Dr. Deodhar, professor of medicine and medical director of the rheumatology clinics at Oregon Health & Science University in Portland.
As outgoing chair of SPARTAN, Dr. Deodhar introduced a proposal that SPARTAN develop new minimal disease activity criteria for patients with axial spondyloarthritis, presenting the rationale for this project at the meeting along with the incoming chair Lianne S. Gensler, MD.
“We want a way to measure disease activity at a stage that is not full remission but with enough of a reduction in disease activity to make a difference,” said Dr. Gensler, director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco. Another frequently used gauge of minimal disease activity in axial spondyloarthritis, the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), is flawed by being totally based on subjective measurements without input from the attending rheumatologist, Dr. Gensler said.
The SPARTAN leadership will try to partner in this effort with the OMERACT (Outcome Measures in Rheumatology) program, the Assessment of Spondyloarthritis International Society (ASAS), or both, but if necessary SPARTAN will develop new minimal disease activity criteria on its own, Dr. Deodhar said.
Dr. Deodhar has received research support from 10 drug companies. Dr. Gensler has been a consultant to or has received research support from AbbVie, Amgen, Janssen, Novartis, and UCB.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
DENVER – U.S. rheumatologists plan to develop new criteria to define minimal disease activity in patients with axial spondyloarthritis to better gauge in routine clinical practice how well these patients respond to treatment.
A score on the Ankylosing Spondylitis Disease Activity Score (ASDAS) of less than 1.3 is the most commonly used measure today of minimal disease activity, but the ASDAS isn’t suitable for point-of-care assessment in routine practice because of the need for a C-reactive protein level or erythrocyte sedimentation rate.
“In the clinic, ASDAS is very difficult or next to impossible to do” because it needs CRP or ESR, which are “never available at the point of care,” Atul A. Deodhar, MD, said in an interview at the annual meeting of the Spondyloarthritis Research and Treatment Network (SPARTAN). Another limitation of ASDAS is that it focuses exclusively on musculoskeletal measures and does not take into account extra-articular manifestations of axial spondyloarthritis such as those in the eye, gastrointestinal tract, or skin.
“We would like a clinical measurement that might tell us that a patient is appropriately treated and at minimal disease activity,” said Dr. Deodhar, professor of medicine and medical director of the rheumatology clinics at Oregon Health & Science University in Portland.
As outgoing chair of SPARTAN, Dr. Deodhar introduced a proposal that SPARTAN develop new minimal disease activity criteria for patients with axial spondyloarthritis, presenting the rationale for this project at the meeting along with the incoming chair Lianne S. Gensler, MD.
“We want a way to measure disease activity at a stage that is not full remission but with enough of a reduction in disease activity to make a difference,” said Dr. Gensler, director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco. Another frequently used gauge of minimal disease activity in axial spondyloarthritis, the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), is flawed by being totally based on subjective measurements without input from the attending rheumatologist, Dr. Gensler said.
The SPARTAN leadership will try to partner in this effort with the OMERACT (Outcome Measures in Rheumatology) program, the Assessment of Spondyloarthritis International Society (ASAS), or both, but if necessary SPARTAN will develop new minimal disease activity criteria on its own, Dr. Deodhar said.
Dr. Deodhar has received research support from 10 drug companies. Dr. Gensler has been a consultant to or has received research support from AbbVie, Amgen, Janssen, Novartis, and UCB.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM THE 2016 SPARTAN ANNUAL MEETING

Migraine With Aura Increases Risk of Venous Thromboembolism
SAN DIEGO—Migraine with aura is associated with increased risk of venous thromboembolism, but migraine without aura is not, according to research presented at the 58th Annual Scientific Meeting of the American Headache Society. The investigators plan to identify the mechanism of the increased risk, which they hypothesize could involve hormones, genes, or prothrombotic states.
Many previous studies have focused on the associations between migraine and atherothrombosis, migraine and stroke, and migraine and myocardial infarction. Results generally have indicated that gender and migraine status (ie, migraine with or without aura) modify the risk of these outcomes. Fewer studies have examined the relationship between migraine and venous thromboembolism, however. Most of the published studies of this topic have focused on specific subgroups of patients and lacked data on the general population of migraineurs.
Kuan-Po Peng, MD, a neurologist at Taipei Veterans Hospital in Taiwan, and colleagues sought to evaluate the risk of venous thromboembolism in the general migraine population and to identify the subgroup of patients at greatest risk. They collected data from the Taiwan National Health Insurance Research Database, which includes information for nearly 750,000 patients with migraine. To increase diagnostic accuracy, the researchers identified patients with neurologist-diagnosed migraine. They also used a diagnosis code plus a composite of anticoagulation prescriptions to identify patients with venous thromboembolism.

Kuan-Po Peng, MD
Dr. Peng and colleagues identified a cohort of migraineurs and a cohort of patients without headache. Each cohort included 102,159 patients, and both were matched by sex and propensity score. The researchers followed both cohorts for approximately four years and observed cases of venous thromboembolism. They used Cox proportional hazards regression analyses to calculate adjusted hazard ratios (aHRs) for the two study arms.
In all, 226 migraineurs and 203 patients without headache had venous thromboembolism. “To our surprise, we found that, overall, the risk of venous thromboembolism is not increased in the migraine population,” said Dr. Peng. The aHR of venous thromboembolism was 1.12 among migraineurs. Subgroup analysis by migraine subtype indicated an elevated risk of venous thromboembolism in patients with migraine with aura (aHR, 2.42), but not in patients with migraine without aura (aHR, 0.81). When the investigators analyzed the data by gender, they found a higher risk of venous thromboembolism among female migraineurs with aura, compared with their male counterparts (aHR, 2.81 vs 1.81).
Dr. Peng’s group conducted sensitivity analyses, excluding anticoagulant use, contraceptive use, major surgery, pregnancy, and other risk factors for venous thromboembolism. In these analyses, the risk of venous thromboembolism was similar to that in the primary analysis.
“We are interested in the mechanism behind this specific association,” said Dr. Peng. “There might be a role for estrogen because … all the patients at higher risk are female, and estrogen is a known risk factor for venous thromboembolism.” The investigators, however, saw no difference in the association between migraine with aura and venous thromboembolism when comparing women younger than 50 (an age used to mark the beginning of menopause) and those older than 50. The low number of cases of venous thromboembolism, and consequent lack of power, may explain this result, said Dr. Peng.
—Erik Greb
SAN DIEGO—Migraine with aura is associated with increased risk of venous thromboembolism, but migraine without aura is not, according to research presented at the 58th Annual Scientific Meeting of the American Headache Society. The investigators plan to identify the mechanism of the increased risk, which they hypothesize could involve hormones, genes, or prothrombotic states.
Many previous studies have focused on the associations between migraine and atherothrombosis, migraine and stroke, and migraine and myocardial infarction. Results generally have indicated that gender and migraine status (ie, migraine with or without aura) modify the risk of these outcomes. Fewer studies have examined the relationship between migraine and venous thromboembolism, however. Most of the published studies of this topic have focused on specific subgroups of patients and lacked data on the general population of migraineurs.
Kuan-Po Peng, MD, a neurologist at Taipei Veterans Hospital in Taiwan, and colleagues sought to evaluate the risk of venous thromboembolism in the general migraine population and to identify the subgroup of patients at greatest risk. They collected data from the Taiwan National Health Insurance Research Database, which includes information for nearly 750,000 patients with migraine. To increase diagnostic accuracy, the researchers identified patients with neurologist-diagnosed migraine. They also used a diagnosis code plus a composite of anticoagulation prescriptions to identify patients with venous thromboembolism.
Kuan-Po Peng, MD
Dr. Peng and colleagues identified a cohort of migraineurs and a cohort of patients without headache. Each cohort included 102,159 patients, and both were matched by sex and propensity score. The researchers followed both cohorts for approximately four years and observed cases of venous thromboembolism. They used Cox proportional hazards regression analyses to calculate adjusted hazard ratios (aHRs) for the two study arms.
In all, 226 migraineurs and 203 patients without headache had venous thromboembolism. “To our surprise, we found that, overall, the risk of venous thromboembolism is not increased in the migraine population,” said Dr. Peng. The aHR of venous thromboembolism was 1.12 among migraineurs. Subgroup analysis by migraine subtype indicated an elevated risk of venous thromboembolism in patients with migraine with aura (aHR, 2.42), but not in patients with migraine without aura (aHR, 0.81). When the investigators analyzed the data by gender, they found a higher risk of venous thromboembolism among female migraineurs with aura, compared with their male counterparts (aHR, 2.81 vs 1.81).
Dr. Peng’s group conducted sensitivity analyses, excluding anticoagulant use, contraceptive use, major surgery, pregnancy, and other risk factors for venous thromboembolism. In these analyses, the risk of venous thromboembolism was similar to that in the primary analysis.
“We are interested in the mechanism behind this specific association,” said Dr. Peng. “There might be a role for estrogen because … all the patients at higher risk are female, and estrogen is a known risk factor for venous thromboembolism.” The investigators, however, saw no difference in the association between migraine with aura and venous thromboembolism when comparing women younger than 50 (an age used to mark the beginning of menopause) and those older than 50. The low number of cases of venous thromboembolism, and consequent lack of power, may explain this result, said Dr. Peng.
—Erik Greb
SAN DIEGO—Migraine with aura is associated with increased risk of venous thromboembolism, but migraine without aura is not, according to research presented at the 58th Annual Scientific Meeting of the American Headache Society. The investigators plan to identify the mechanism of the increased risk, which they hypothesize could involve hormones, genes, or prothrombotic states.
Many previous studies have focused on the associations between migraine and atherothrombosis, migraine and stroke, and migraine and myocardial infarction. Results generally have indicated that gender and migraine status (ie, migraine with or without aura) modify the risk of these outcomes. Fewer studies have examined the relationship between migraine and venous thromboembolism, however. Most of the published studies of this topic have focused on specific subgroups of patients and lacked data on the general population of migraineurs.
Kuan-Po Peng, MD, a neurologist at Taipei Veterans Hospital in Taiwan, and colleagues sought to evaluate the risk of venous thromboembolism in the general migraine population and to identify the subgroup of patients at greatest risk. They collected data from the Taiwan National Health Insurance Research Database, which includes information for nearly 750,000 patients with migraine. To increase diagnostic accuracy, the researchers identified patients with neurologist-diagnosed migraine. They also used a diagnosis code plus a composite of anticoagulation prescriptions to identify patients with venous thromboembolism.
Kuan-Po Peng, MD
Dr. Peng and colleagues identified a cohort of migraineurs and a cohort of patients without headache. Each cohort included 102,159 patients, and both were matched by sex and propensity score. The researchers followed both cohorts for approximately four years and observed cases of venous thromboembolism. They used Cox proportional hazards regression analyses to calculate adjusted hazard ratios (aHRs) for the two study arms.
In all, 226 migraineurs and 203 patients without headache had venous thromboembolism. “To our surprise, we found that, overall, the risk of venous thromboembolism is not increased in the migraine population,” said Dr. Peng. The aHR of venous thromboembolism was 1.12 among migraineurs. Subgroup analysis by migraine subtype indicated an elevated risk of venous thromboembolism in patients with migraine with aura (aHR, 2.42), but not in patients with migraine without aura (aHR, 0.81). When the investigators analyzed the data by gender, they found a higher risk of venous thromboembolism among female migraineurs with aura, compared with their male counterparts (aHR, 2.81 vs 1.81).
Dr. Peng’s group conducted sensitivity analyses, excluding anticoagulant use, contraceptive use, major surgery, pregnancy, and other risk factors for venous thromboembolism. In these analyses, the risk of venous thromboembolism was similar to that in the primary analysis.
“We are interested in the mechanism behind this specific association,” said Dr. Peng. “There might be a role for estrogen because … all the patients at higher risk are female, and estrogen is a known risk factor for venous thromboembolism.” The investigators, however, saw no difference in the association between migraine with aura and venous thromboembolism when comparing women younger than 50 (an age used to mark the beginning of menopause) and those older than 50. The low number of cases of venous thromboembolism, and consequent lack of power, may explain this result, said Dr. Peng.
—Erik Greb

Promoting your older patient’s healthy ‘brain aging’

Adjusting the dosing of antipsychotics and other psychotropics in renal disease

Triptorelin doesn’t prevent ovarian failure in young women treated for lymphoma
Protective treatment with the GnRH agonist triptorelin failed to prevent chemotherapy-induced premature ovarian failure in young women with lymphoma, according to a report published in the Journal of Clinical Oncology.
This type of protective therapy has been used for at least 20 years but is still controversial. Some clinical practice guidelines endorse the practice, but others do not. The evidence is both sparse and ambiguous, said Isabelle Demeestere, MD, of the research laboratory on human reproduction, Université Libre de Bruxelles (Belgium), and her associates.
In what they described as “the first randomized clinical trial providing accurate information on ovarian function and fertility after a median of 5 years of follow-up,” the investigators assessed 67 young women (median age, 26 years) treated for Hodgkin’s or non-Hodgkin’s lymphoma at 15 cancer centers in France, Belgium, and Italy. These patients had been randomly assigned to receive chemotherapy plus triptorelin (32 women) or chemotherapy plus placebo injections (35 women) at baseline.
Premature ovarian failure, as measured by elevated FSH levels, occurred in six of the GnRH group and eight of the control group, a nonsignificant difference. Moreover, the rate of recovery of ovarian function, as measured by at least one FSH level of 15 IU/L or less, was the same between the two study groups. And 17 women (53%) in the GnRH group eventually achieved pregnancy, which is not significantly different from the 43% pregnancy rate in the control group (15 of 35 women).
Five women – two in the GnRH group and three in the control group – achieved pregnancy during long-term follow-up, even though they had been classified as having premature ovarian failure. This “confirms the possibility of incidental ovarian cycle recovery, leading to fertility restoration several years after treatment in this young population,” Dr. Demeestere and her associates wrote (J Clin Oncol. 2016. [doi:10.1200/JCO.2015.65.884]).
The study findings “suggest caution” regarding guidelines that recommend using GnRH agonists to preserve fertility in young women undergoing chemotherapy, they added.
Protective treatment with the GnRH agonist triptorelin failed to prevent chemotherapy-induced premature ovarian failure in young women with lymphoma, according to a report published in the Journal of Clinical Oncology.
This type of protective therapy has been used for at least 20 years but is still controversial. Some clinical practice guidelines endorse the practice, but others do not. The evidence is both sparse and ambiguous, said Isabelle Demeestere, MD, of the research laboratory on human reproduction, Université Libre de Bruxelles (Belgium), and her associates.
In what they described as “the first randomized clinical trial providing accurate information on ovarian function and fertility after a median of 5 years of follow-up,” the investigators assessed 67 young women (median age, 26 years) treated for Hodgkin’s or non-Hodgkin’s lymphoma at 15 cancer centers in France, Belgium, and Italy. These patients had been randomly assigned to receive chemotherapy plus triptorelin (32 women) or chemotherapy plus placebo injections (35 women) at baseline.
Premature ovarian failure, as measured by elevated FSH levels, occurred in six of the GnRH group and eight of the control group, a nonsignificant difference. Moreover, the rate of recovery of ovarian function, as measured by at least one FSH level of 15 IU/L or less, was the same between the two study groups. And 17 women (53%) in the GnRH group eventually achieved pregnancy, which is not significantly different from the 43% pregnancy rate in the control group (15 of 35 women).
Five women – two in the GnRH group and three in the control group – achieved pregnancy during long-term follow-up, even though they had been classified as having premature ovarian failure. This “confirms the possibility of incidental ovarian cycle recovery, leading to fertility restoration several years after treatment in this young population,” Dr. Demeestere and her associates wrote (J Clin Oncol. 2016. [doi:10.1200/JCO.2015.65.884]).
The study findings “suggest caution” regarding guidelines that recommend using GnRH agonists to preserve fertility in young women undergoing chemotherapy, they added.
Protective treatment with the GnRH agonist triptorelin failed to prevent chemotherapy-induced premature ovarian failure in young women with lymphoma, according to a report published in the Journal of Clinical Oncology.
This type of protective therapy has been used for at least 20 years but is still controversial. Some clinical practice guidelines endorse the practice, but others do not. The evidence is both sparse and ambiguous, said Isabelle Demeestere, MD, of the research laboratory on human reproduction, Université Libre de Bruxelles (Belgium), and her associates.
In what they described as “the first randomized clinical trial providing accurate information on ovarian function and fertility after a median of 5 years of follow-up,” the investigators assessed 67 young women (median age, 26 years) treated for Hodgkin’s or non-Hodgkin’s lymphoma at 15 cancer centers in France, Belgium, and Italy. These patients had been randomly assigned to receive chemotherapy plus triptorelin (32 women) or chemotherapy plus placebo injections (35 women) at baseline.
Premature ovarian failure, as measured by elevated FSH levels, occurred in six of the GnRH group and eight of the control group, a nonsignificant difference. Moreover, the rate of recovery of ovarian function, as measured by at least one FSH level of 15 IU/L or less, was the same between the two study groups. And 17 women (53%) in the GnRH group eventually achieved pregnancy, which is not significantly different from the 43% pregnancy rate in the control group (15 of 35 women).
Five women – two in the GnRH group and three in the control group – achieved pregnancy during long-term follow-up, even though they had been classified as having premature ovarian failure. This “confirms the possibility of incidental ovarian cycle recovery, leading to fertility restoration several years after treatment in this young population,” Dr. Demeestere and her associates wrote (J Clin Oncol. 2016. [doi:10.1200/JCO.2015.65.884]).
The study findings “suggest caution” regarding guidelines that recommend using GnRH agonists to preserve fertility in young women undergoing chemotherapy, they added.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: The GnRH agonist triptorelin failed to prevent chemotherapy-induced premature ovarian failure in young women with lymphoma.
Major finding: Premature ovarian failure occurred in six of the GnRH group and eight of the control group, a nonsignificant difference.
Data source: A multicenter, randomized clinical trial involving 67 women followed for 5 years.
Disclosures: This study was supported by Fonds National de la Recherche Scientifique and Ipsen Pharmaceutical Group. Dr. Demeestere reported having no relevant financial disclosures; her associates reported ties to numerous industry sources.
Skin Examinations for Early Melanoma Detection


When the diagnosis is hard to swallow, take these management steps
CASE REPORTMr. C, age 72, reports a lack of desire to swallow food. He denies feeling a lump in his throat. Over the past 6 months, he lost >30 lb.
The patient had a similar episode 2 years ago, which resolved without intervention. The death of his wife recently has led to isolation and lack of desire to swallow food.
Testing with standard food samples to elicit eating behaviors is normal. Electromyography and video fluoroscopy test results show no abnormalities.
What is phagophobia?The case of Mr. C brings to light the condition known as phagophobia—a sensation of not being able to swallow. Phagophobia mimics oral apraxia; pharyngoesophageal and neurologic functions as well as the ability to speak remain intact, however.1
It is estimated that about 6% of the adult general population reports dysphagia.2 About 47% of patients with dysphagic complaints do not show motor-manometric or radiological abnormalities of the upper digestive tract. A number of psychiatric conditions, including panic disorder, obsessive-compulsive disorder, social phobia, anorexia nervosa, globus hystericus, hypersensitive gag reflex, and posttraumatic stress disorder can simulate this condition.3
When Barofsky and Fontaine4 compared phagophobia patients with other subjects—healthy controls, anorexia nervosa restrictors, dysphagic patients with esophageal obstruction, dysphagic patients with motility disturbance, and patients with non-motility non-obstructive dysphagia—they found that patients with psychogenic dysphagia did not appear to have an eating disorder. However, they did have a clinically significant level of psychological distress, particularly anxiety.
Diagnostic tools and management stepsThere are a number of approaches to assess your patient’s fear of swallowing (Table,5-7 page 68). Non-invasive assessment tools along with educational modalities usually are tried alone or together with psychopharmacological intervention. It is, however, imperative that you have an empathetic and understanding approach to such patients. When patients have confidence in the clinician they tend to respond more effectively with such approaches.

Investigations4 include questionnaires (swallow disorder history, Eating Disorder Inventory-2, and Symptom Checklist–90-R); weight assessment; testing with standardized food samples to elicit eating behaviors; self-reports; electromyography; and videofluoroscopy.
Education and reassurance includes individual demonstration of swallowing, combined with group therapy, exercises, and reassurance. Patients benefit from advice on how to maximize sensation within the oropharynx to increase taste, perception of temperature, and texture stimulation.8
Behavioral intervention involves practicing slow breathing and muscle relaxation techniques to gradually increase bite size and reduce the amount of time spent chewing each bite.
Introspection therapycomprises psychoeducation, cognitive restructuring, and in vivo and introspective exposure; helps patients replace anxiety-producing thoughts with probability estimation and decatastrophizing. Introspective exposure targets the fear of choking by having the patient create sensations of throat tightening by holding a swallow in mid-action and by rapid swallowing. In vivo exposure targets the fear of swallowing by having the patient practice feeding foods (such as semi-solid easy-to-swallow choices), in and outside of the session.6
Aversion therapy requires that you pinch the patient’s hand while he (she) chews, and release the hand when he swallows.
Psychopharmacotherapeutic intervention. A number of medications can be used to help, such as imipramine up to 150 mg; desipramine, up to 150 mg; or lorazepam, 0.25 mg, twice daily, to address anxiety or panic symptoms.
Acknowledgment
Duy Li, BS, and Yu Hsuan Liao, BS, contributed to the development of the manuscript of this article.
1. Evans IM, Pia P. Phagophobia: behavioral treatment of a complex case involving fear of fear. Clinical Case Studies. 2011;10(1):37-52.
2. Kim CH, Hsu JJ, Williams DE, et al. A prospective psychological evaluation of patients with dysphagia of various etiologies. Dysphagia. 1996;11(1):34-40.
3. McNally RJ. Choking phobia: a review of the literature. Compr Psychiatry. 1994;35(1):83-89.
4. Barofsky I, Fontaine KR. Do psychogenic dysphagia patients have an eating disorder? Dysphagia. 1998;13(1):24-27.
5. Bishop LC, Riley WT. The psychiatric management of the globus syndrome. Gen Hosp Psychiatry. 1988;10(3):214-219.
6. Ball SG, Otto MW. Cognitive-behavioral treatment of choking phobia: 3 case studies Psychother Psychosom. 1994;62(3-4):207-211.
7. Epstein SJ, Deyoub P. Hypnotherapy for fear of choking: treatment implications of a case report. Int J Clin Hypn. 1981;29(2):117-127.
8. Scemes S, Wielenska RC, Savoia MG, et al. Choking phobia: full remission following behavior therapy. Rev Bras Psiquiatr. 2009;31(3):257-260.
CASE REPORTMr. C, age 72, reports a lack of desire to swallow food. He denies feeling a lump in his throat. Over the past 6 months, he lost >30 lb.
The patient had a similar episode 2 years ago, which resolved without intervention. The death of his wife recently has led to isolation and lack of desire to swallow food.
Testing with standard food samples to elicit eating behaviors is normal. Electromyography and video fluoroscopy test results show no abnormalities.
What is phagophobia?The case of Mr. C brings to light the condition known as phagophobia—a sensation of not being able to swallow. Phagophobia mimics oral apraxia; pharyngoesophageal and neurologic functions as well as the ability to speak remain intact, however.1
It is estimated that about 6% of the adult general population reports dysphagia.2 About 47% of patients with dysphagic complaints do not show motor-manometric or radiological abnormalities of the upper digestive tract. A number of psychiatric conditions, including panic disorder, obsessive-compulsive disorder, social phobia, anorexia nervosa, globus hystericus, hypersensitive gag reflex, and posttraumatic stress disorder can simulate this condition.3
When Barofsky and Fontaine4 compared phagophobia patients with other subjects—healthy controls, anorexia nervosa restrictors, dysphagic patients with esophageal obstruction, dysphagic patients with motility disturbance, and patients with non-motility non-obstructive dysphagia—they found that patients with psychogenic dysphagia did not appear to have an eating disorder. However, they did have a clinically significant level of psychological distress, particularly anxiety.
Diagnostic tools and management stepsThere are a number of approaches to assess your patient’s fear of swallowing (Table,5-7 page 68). Non-invasive assessment tools along with educational modalities usually are tried alone or together with psychopharmacological intervention. It is, however, imperative that you have an empathetic and understanding approach to such patients. When patients have confidence in the clinician they tend to respond more effectively with such approaches.
Investigations4 include questionnaires (swallow disorder history, Eating Disorder Inventory-2, and Symptom Checklist–90-R); weight assessment; testing with standardized food samples to elicit eating behaviors; self-reports; electromyography; and videofluoroscopy.
Education and reassurance includes individual demonstration of swallowing, combined with group therapy, exercises, and reassurance. Patients benefit from advice on how to maximize sensation within the oropharynx to increase taste, perception of temperature, and texture stimulation.8
Behavioral intervention involves practicing slow breathing and muscle relaxation techniques to gradually increase bite size and reduce the amount of time spent chewing each bite.
Introspection therapycomprises psychoeducation, cognitive restructuring, and in vivo and introspective exposure; helps patients replace anxiety-producing thoughts with probability estimation and decatastrophizing. Introspective exposure targets the fear of choking by having the patient create sensations of throat tightening by holding a swallow in mid-action and by rapid swallowing. In vivo exposure targets the fear of swallowing by having the patient practice feeding foods (such as semi-solid easy-to-swallow choices), in and outside of the session.6
Aversion therapy requires that you pinch the patient’s hand while he (she) chews, and release the hand when he swallows.
Psychopharmacotherapeutic intervention. A number of medications can be used to help, such as imipramine up to 150 mg; desipramine, up to 150 mg; or lorazepam, 0.25 mg, twice daily, to address anxiety or panic symptoms.
Acknowledgment
Duy Li, BS, and Yu Hsuan Liao, BS, contributed to the development of the manuscript of this article.
CASE REPORTMr. C, age 72, reports a lack of desire to swallow food. He denies feeling a lump in his throat. Over the past 6 months, he lost >30 lb.
The patient had a similar episode 2 years ago, which resolved without intervention. The death of his wife recently has led to isolation and lack of desire to swallow food.
Testing with standard food samples to elicit eating behaviors is normal. Electromyography and video fluoroscopy test results show no abnormalities.
What is phagophobia?The case of Mr. C brings to light the condition known as phagophobia—a sensation of not being able to swallow. Phagophobia mimics oral apraxia; pharyngoesophageal and neurologic functions as well as the ability to speak remain intact, however.1
It is estimated that about 6% of the adult general population reports dysphagia.2 About 47% of patients with dysphagic complaints do not show motor-manometric or radiological abnormalities of the upper digestive tract. A number of psychiatric conditions, including panic disorder, obsessive-compulsive disorder, social phobia, anorexia nervosa, globus hystericus, hypersensitive gag reflex, and posttraumatic stress disorder can simulate this condition.3
When Barofsky and Fontaine4 compared phagophobia patients with other subjects—healthy controls, anorexia nervosa restrictors, dysphagic patients with esophageal obstruction, dysphagic patients with motility disturbance, and patients with non-motility non-obstructive dysphagia—they found that patients with psychogenic dysphagia did not appear to have an eating disorder. However, they did have a clinically significant level of psychological distress, particularly anxiety.
Diagnostic tools and management stepsThere are a number of approaches to assess your patient’s fear of swallowing (Table,5-7 page 68). Non-invasive assessment tools along with educational modalities usually are tried alone or together with psychopharmacological intervention. It is, however, imperative that you have an empathetic and understanding approach to such patients. When patients have confidence in the clinician they tend to respond more effectively with such approaches.
Investigations4 include questionnaires (swallow disorder history, Eating Disorder Inventory-2, and Symptom Checklist–90-R); weight assessment; testing with standardized food samples to elicit eating behaviors; self-reports; electromyography; and videofluoroscopy.
Education and reassurance includes individual demonstration of swallowing, combined with group therapy, exercises, and reassurance. Patients benefit from advice on how to maximize sensation within the oropharynx to increase taste, perception of temperature, and texture stimulation.8
Behavioral intervention involves practicing slow breathing and muscle relaxation techniques to gradually increase bite size and reduce the amount of time spent chewing each bite.
Introspection therapycomprises psychoeducation, cognitive restructuring, and in vivo and introspective exposure; helps patients replace anxiety-producing thoughts with probability estimation and decatastrophizing. Introspective exposure targets the fear of choking by having the patient create sensations of throat tightening by holding a swallow in mid-action and by rapid swallowing. In vivo exposure targets the fear of swallowing by having the patient practice feeding foods (such as semi-solid easy-to-swallow choices), in and outside of the session.6
Aversion therapy requires that you pinch the patient’s hand while he (she) chews, and release the hand when he swallows.
Psychopharmacotherapeutic intervention. A number of medications can be used to help, such as imipramine up to 150 mg; desipramine, up to 150 mg; or lorazepam, 0.25 mg, twice daily, to address anxiety or panic symptoms.
Acknowledgment
Duy Li, BS, and Yu Hsuan Liao, BS, contributed to the development of the manuscript of this article.
1. Evans IM, Pia P. Phagophobia: behavioral treatment of a complex case involving fear of fear. Clinical Case Studies. 2011;10(1):37-52.
2. Kim CH, Hsu JJ, Williams DE, et al. A prospective psychological evaluation of patients with dysphagia of various etiologies. Dysphagia. 1996;11(1):34-40.
3. McNally RJ. Choking phobia: a review of the literature. Compr Psychiatry. 1994;35(1):83-89.
4. Barofsky I, Fontaine KR. Do psychogenic dysphagia patients have an eating disorder? Dysphagia. 1998;13(1):24-27.
5. Bishop LC, Riley WT. The psychiatric management of the globus syndrome. Gen Hosp Psychiatry. 1988;10(3):214-219.
6. Ball SG, Otto MW. Cognitive-behavioral treatment of choking phobia: 3 case studies Psychother Psychosom. 1994;62(3-4):207-211.
7. Epstein SJ, Deyoub P. Hypnotherapy for fear of choking: treatment implications of a case report. Int J Clin Hypn. 1981;29(2):117-127.
8. Scemes S, Wielenska RC, Savoia MG, et al. Choking phobia: full remission following behavior therapy. Rev Bras Psiquiatr. 2009;31(3):257-260.
1. Evans IM, Pia P. Phagophobia: behavioral treatment of a complex case involving fear of fear. Clinical Case Studies. 2011;10(1):37-52.
2. Kim CH, Hsu JJ, Williams DE, et al. A prospective psychological evaluation of patients with dysphagia of various etiologies. Dysphagia. 1996;11(1):34-40.
3. McNally RJ. Choking phobia: a review of the literature. Compr Psychiatry. 1994;35(1):83-89.
4. Barofsky I, Fontaine KR. Do psychogenic dysphagia patients have an eating disorder? Dysphagia. 1998;13(1):24-27.
5. Bishop LC, Riley WT. The psychiatric management of the globus syndrome. Gen Hosp Psychiatry. 1988;10(3):214-219.
6. Ball SG, Otto MW. Cognitive-behavioral treatment of choking phobia: 3 case studies Psychother Psychosom. 1994;62(3-4):207-211.
7. Epstein SJ, Deyoub P. Hypnotherapy for fear of choking: treatment implications of a case report. Int J Clin Hypn. 1981;29(2):117-127.
8. Scemes S, Wielenska RC, Savoia MG, et al. Choking phobia: full remission following behavior therapy. Rev Bras Psiquiatr. 2009;31(3):257-260.
When to adjust the dosing of psychotropics in patients with renal impairment
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
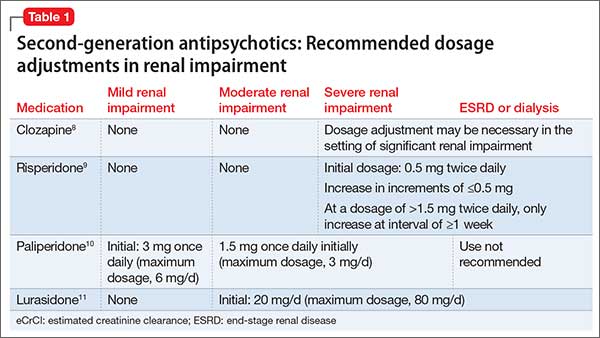
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
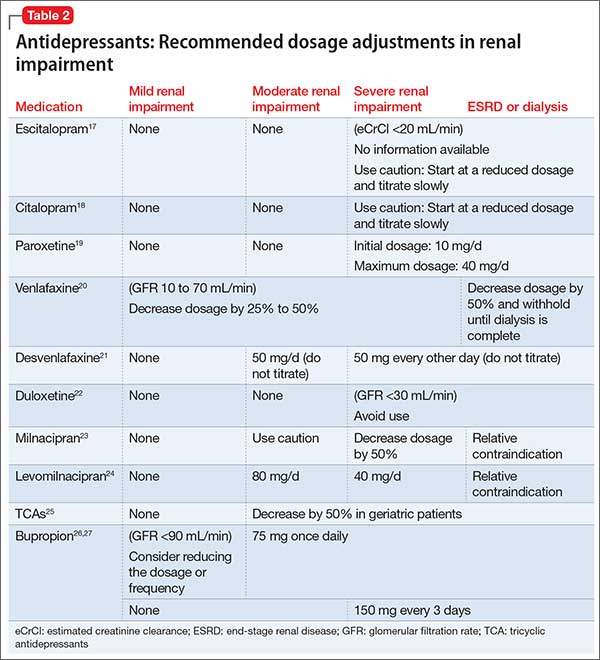
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
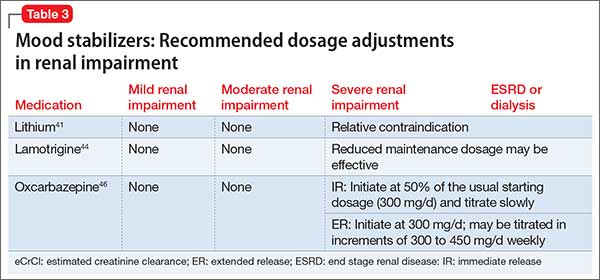
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.