User login
Approach to asymptomatic creatine kinase elevation
Measuring serum creatine kinase (CK) is an important part of the evaluation of patients with muscle weakness or myalgia, and of assessing patients with myopathies or rhabdomyolysis. But elevated CK sometimes is an incidental finding in a patient without muscle-related symptoms or with only minimal nonspecific muscle symptoms (eg, cramps, spasms, fatigue) that do not significantly interfere with activities of daily living. This condition is sometimes referred to as “asymptomatic hyper-CK-emia.” Four other muscle enzymes that may also be elevated are aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, and aldolase.
This review focuses on the evaluation of patients with elevated CK without significant muscle-related symptoms and proposes an algorithm for this purpose (Figure 1).
CURRENT THRESHOLDS MAY BE LOW
What appears to be an elevated CK level may in fact be normal, and it is important to determine in the initial assessment whether a CK value is truly abnormal.

Most laboratories use the central 95% of observations in white people as a reference range for serum CK, assuming that levels have a gaussian (bell-shaped) distribution, which is usually about 0 to 200 IU/L. Using these parameters, an abnormal CK level was observed in 19% of men and 5% of women in a study of nearly 1,000 healthy young people,1 leading to overdiagnosis.
The actual distribution of serum CK levels in a healthy population is markedly skewed toward higher values and is nongaussian.1–3 A 97.5% normal threshold is associated with a much lower false-positive rate and is recommended by the European Federation of Neurological Societies (now the European Academy of Neurology).4 This group also recommends pursuing further investigation only for patients whose level is at least 1.5 times the upper limit of normal; this threshold results in only a small reduction in sensitivity.
CK levels vary significantly by sex and race.5 Possible reasons include differences in muscle mass or total body mass and inherited differences in the permeability of the sarcolemma to CK.6 There is also a small reduction in CK levels as people age.2
The European Federation of Neurological Societies suggests redefining elevated CK as values 1.5 times beyond the upper limit of normal. Based on a 97.5% threshold and normal values determined by Brewster et al3 for black and white men and women, the following thresholds can be used to help decide whether to pursue further evaluation4:
- White women—325 IU/L
- White men—504 IU/L
- Black women—621 IU/L
- Black men—1,200 IU/L
PHYSICAL ACTIVITY RAISES CK
CK levels transiently rise after exercise or heavy manual labor. Serum CK levels may increase to as much as 30 times the upper limit of normal within 24 hours of strenuous physical activity, then slowly decline over the next 7 days. The degree of CK elevation depends on the type and duration of exercise, with greater elevation in those who are untrained.2,4
In assessing asymptomatic or minimally symptomatic CK elevation, the test should be repeated after 7 days without exercise. A large community study in Norway found that repeat CK levels in people with incidentally discovered elevated CK were normal after 3 days of rest in 70% of cases.2
NONNEUROMUSCULAR CAUSES
NEED TO BE INVESTIGATED
Asymptomatic or minimally symptomatic elevated CK can be due to a primary neuromuscular disease or a variety of nonneuromuscular causes.

Patients who still have elevated CK after taking into account the 97.5% threshold, repeat testing after a week of rest, and a level more than 1.5 times the upper limit of normal for sex and race should first be evaluated for the many nonneuromuscular conditions that can cause elevated CK (Table 1).7–9
Cardiac causes should be evaluated by history and physical examination, electrocardiography, and possibly testing for cardiac troponins.
Drugs commonly elevate CK
Prescription drugs and supplements are an important and common cause of CK elevation, so it is important to carefully review medications the patient is taking.
Statins can cause myalgia, muscle weakness, and rhabdomyolysis. Up to 5% of users develop CK elevation, typically 2 to 10 times the upper limit of normal.10 CK usually drops after stopping statins but may require weeks to months to normalize. Rarely, statin users develop a serious immune-mediated necrotizing myopathy.11–13
The diversity of response to statin therapy appears to have a genetic basis. The SEARCH Collaborative Group14 conducted a genome-wide association study of 300,000 markers in 85 patients with definite or incipient myopathy and in 90 controls, all of whom were taking simvastatin 80 mg daily. They identified a single-nucleotide polymorphism in the SLCO1B1 gene on chromosome 12 that was strongly associated with a higher risk of statin-induced myopathy.
Patients with statin-related myopathy seem to have a higher frequency of occult metabolic muscle disease than the general population, also suggesting genetic susceptibility, although ascertainment bias could be a factor.14
Mechanisms of CK elevation in response to statins include increased muscle membrane fragility due to decreased cholesterol content, inhibition of isoprenoid production (a necessary step in the synthesis of membrane proteins), and depletion of ubiquinone, leading to mitochondrial dysfunction.
Macro CK: An abnormal enzyme complex
About 4% of patients with asymptomatic or minimally symptomatic elevated CK have “macro CK,” an enzyme complex with an atypically high molecular mass and reduced clearance, resulting in abnormally high blood levels of CK. Macro CK type 1 is more common and is found in up to 1.2% of the general population: complexes are composed of CK and immunoglobulin and are associated with autoimmune diseases.9,15 Macro CK type 2 complexes consist of CK and an undetermined protein and are associated with malignancies.
CK electrophoresis is required to detect macro CK. Types 1 and 2 can be distinguished by protein G affinity chromatography.9,15
Endocrine disorders
Muscle involvement in endocrine disorders often presents with muscle weakness in addition to muscle enzyme abnormalities.
Hypothyroidism often causes weakness, cramps, myalgia, and a mild to moderate serum CK elevation.16 Severe CK elevation has been reported to occur after vigorous exercise.17 Thyroid replacement usually results in normalization of serum CK levels in 1 to 2 months.18
Hyperthyroidism is typically associated with normal serum CK concentrations, but in rare cases it can cause rhabdomyolysis.19
NEUROMUSCULAR CAUSES ARE NOT ALWAYS WORTH PURSUING

Only after the nonneuromuscular causes of elevated CK have been ruled out should neuromuscular disorders be considered (Table 2). Evaluation with electromyography, nerve conduction studies, and muscle biopsy may lead to the diagnosis of a specific neuromuscular disorder: patients may be in the presymptomatic stage of disease and may or may not eventually develop muscle weakness or other symptoms.20,21
Is testing needed?
Most adult dystrophies and metabolic myopathies have no available treatment and their course is often benign, particularly if they present only with asymptomatic elevated CK. The value of a potentially extensive, expensive, and invasive evaluation for a specific neuromuscular cause should be weighed against the limited yield and treatment options. Moreover, specialized testing such as biochemical muscle enzyme analysis, sarcolemmal protein staining, and genetic testing are not available at all centers.
The European Federation of Neurological Societies guidelines recommend biopsy for patients with asymptomatic elevated CK who also have any of the following:
- Abnormal (myopathic) findings on electromyography
- CK more than three times the upper limit of normal
- Age less than 25
- Exercise intolerance.4
Idiopathic inflammatory myopathies rarely present with asymptomatic elevated CK.22–26 In one study,27 they were found in just 5% of patients with asymptomatic elevated CK.
Hypomyopathic dermatomyositis and inclusion body myositis can present with mild CK elevations with normal muscle strength, especially early in the disease course. A myositis subset of antisynthetase syndrome can present with mildly elevated CK and interstitial lung disease.27 Many of the inflammatory myopathies respond to treatment so are worth investigating.
In view of complexities in diagnosis of these conditions, one should proceed with testing only after discussing it with patients. Referral to a rheumatology specialist is preferred.
MUSCLE BIOPSY, ELECTROMYOGRAPHY, AND NERVE CONDUCTION STUDIES
Electromyography, nerve conduction studies, or muscle biopsy, or a combination of these tests, is usually needed to investigate neuromuscular causes of elevated CK.
Muscle biopsy abnormalities are found in about two-thirds of cases of asymptomatic elevated CK, but most abnormalities include nonspecific myopathic changes that are not diagnostic. A muscle biopsy that may include special stains for sarcolemmal proteins for muscular dystrophy and biochemical muscle enzyme analysis for metabolic myopathies is diagnostic in only 20% to 25% cases of asymptomatic elevated CK on average, with a variation between different series of 0% to 79%.7,21,27–33
Electromyography and nerve conduction studies alone add little to the workup of asymptomatic elevated CK apart from a modest negative predictive value and as a guide for muscle biopsy. For a very few neuromuscular disorders causing an elevated CK (eg, motor neuron disease, Charcot-Marie-Tooth disease, myotonic dystrophy), electromyography and nerve conduction studies could suffice to make the diagnosis.
Electromyography and nerve conduction studies detect abnormalities in nearly half of cases of asymptomatic CK elevation,7,21,27,28,30,31,33 but, as with biopsy, most changes are nonspecific. Although electromyography and nerve conduction studies can help distinguish primary neuropathic from myopathic disorders, the sensitivity and specificity are low for diagnosis. Normal studies do not rule out a condition, and abnormal studies are not diagnostic of a particular condition, although completely normal studies provide strong evidence against a severe neuromuscular disorder.
Combined testing
Using combined muscle biopsy, electromyography, and nerve conduction studies, the likelihood of making a diagnosis in patients with asymptomatic elevated CK is 28% on average (range of studies 4%–79%),2,7,21,26–28,30–32 and findings are nonspecific in 30% to 40% of cases. Findings are normal in about 30% to 40% of cases, which are thus diagnosed as idiopathic asymptomatic elevated CK.28–31,34
Prelle et al31 retrospectively reviewed the cases of 114 patients, ages 3 to 70, with incidentally discovered elevated CK and few or no symptoms, who underwent muscle biopsy, electromyography, and nerve conduction studies after nonneuromuscular causes were ruled out. Although muscle biopsy findings were abnormal in 39% of cases, a diagnosis was established in only 18% of cases after an extensive workup: the diagnosis was definitive in only 10% and included dystrophinopathies, metabolic myopathies, and rare noninflammatory myopathies. For the remaining 8%, the diagnosis was probable and included four cases of partial carnitine palmitoyl transferase deficiency, three cases of malignant hyperthermia, and two rare inherited disorders.
DNA testing
In women with a serum CK less than three times the upper limit of normal who have a family history of Duchenne or Becker muscular dystrophy, DNA analysis of blood lymphocytes identifies 70% of carriers.4
IDIOPATHIC ELEVATED SERUM CK
Rowland et al35 first coined the term “idiopathic hyper-CK-emia” and defined it as persistent elevation of serum CK despite a normal neurologic examination and testing, including electromyography, nerve conduction studies, and muscle biopsy.35,36 To receive this diagnosis, patients must also have no family history or clinical evidence of neuromuscular disease.
Idiopathic elevated serum CK is sometimes familial. In one study,37 elevated CK was found in family members of 13 of 28 unrelated probands. In the 13 families, 41 individuals had elevated CK. Genetic studies revealed that the condition is genetically heterogeneous and autosomal dominant in at least 60% of cases, with higher penetrance in men.
D’Adda et al26 followed 55 people with idiopathic elevated CK for 7 years. Ten percent were eventually diagnosed with a neuromuscular disorder, 10% developed malignancy, and the remaining 80% developed no new condition. The CK level normalized or decreased in many patients, but most continued to have persistent CK elevations with minimal or no symptoms.
- Lev EI, Tur-Kaspa I, Ashkenazy I, et al. Distribution of serum creatine kinase activity in young healthy persons. Clin Chim Acta 1999; 279:107–115.
- Lilleng H, Abeler K, Johnsen SH, et al. Variation of serum creatine kinase (CK) levels and prevalence of persistent hyperCKemia in a Norwegian normal population. The Tromsø Study. Neuromuscul Disord 2011; 21:494–500.
- Brewster LM, Mairuhu G, Sturk A, van Montfrans GA. Distribution of creatine kinase in the general population: implications for statin therapy. Am Heart J 2007; 154:655–661.
- Kyriakides T, Angelini C, Schaefer J, et al; European Federation of Neurological Societies. EFNS guidelines on the diagnostic approach to pauci- or asymptomatic hyperCKemia. Eur J Neurol 2010; 17:767–773.
- Prisant LM, Downton M, Watkins LO, et al. Efficacy and tolerability of lovastatin in 459 African-Americans with hypercholesterolemia. Am J Cardiol 1996; 78:420–444.
- Wong ET, Cobb C, Umehara MK, et al. Heterogeneity of serum creatine kinase activity among racial and gender groups of the population. Am J Clin Pathol 1983; 79:582–586.
- Brewster LM, de Visser M. Persistent hyperCKemia: fourteen patients studied in retrospect. Acta Neurol Scand 1988; 77:60–63.
- Weglinski MR, Wedel DJ, Engel AG. Malignant hyperthermia testing in patients with persistently increased serum creatine kinase levels. Anesth Analg 1997; 84:1038–1041.
- Galarraga B, Sinclair D, Fahie-Wilson MN, McCrae FC, Hull RG, Ledingham JM. A rare but important cause for a raised serum creatine kinase concentration: two case reports and a literature review. Rheumatology (Oxford) 2003; 42:186–188.
- Mancini GB, Tashakkor AY, Baker S, et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian Working Group Consensus update. Can J Cardiol 2013; 29:1553–1568.
- Arora R, Liebo M, Maldonado F. Statin-induced myopathy: the two faces of Janus. J Cardiovasc Pharmacol Ther 2006; 11:105–112.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Talbert RL. Safety issues with statin therapy. J Am Pharm Assoc (2003) 2006; 46:479–490.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Wyness SP, Hunsaker JJ, La’ulu SL, Rao LV, Roberts WL. Detection of macro-creatine kinase and macroamylase by polyethylene glycol precipitation and ultrafiltration methods. Clin Chim Acta 2011; 412:2052–2057.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Riggs JE. Acute exertional rhabdomyolysis in hypothyroidism: the result of a reversible defect in glycogenolysis? Mil Med 1990; 155:171–172.
- Mastaglia FL, Ojeda VJ, Sarnat HB, Kakulas BA. Myopathies associated with hypothyroidism: a review based upon 13 cases. Aust N Z J Med 1988; 18:799–806.
- Alshanti M, Eledrisi MS, Jones E. Rhabdomyolysis associated with hyperthyroidism. Am J Emerg Med 2001; 19:317.
- Rosalki SB. Serum enzymes in disease of skeletal muscle. Clin Lab Med 1989; 9:767–781.
- Joy JL, Oh SJ. Asymptomatic hyper-CK-emia: an electrophysiologic and histopathologic study. Muscle Nerve 1989; 12:206–209.
- Merlini L, Sabatelli P, Columbaro M, et al. Hyper-CK-emia as the sole manifestation of myotonic dystrophy type 2. Muscle Nerve 2005; 31:764–767.
- Eeg-Olofsson O, Kalimo H, Eeg-Olofsson KE, et al. Duchenne muscular dystrophy and idiopathic hyperCKemia in the same family. Eur J Paediatr Neurol 2008; 12:404–407.
- Dwianingsih EK, Takeshima Y, Itoh K, et al. A Japanese child with asymptomatic elevation of serum creatine kinase shows PTRF-CAVIN mutation matching with congenital generalized lipodystrophy type 4. Mol Genet Metab 2010; 101:233–237.
- Carbone I, Bruno C, Sotgia F, et al. Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology 2000; 54:1373–1376.
- D’Adda E, Sciacco M, Fruguglietti ME, et al. Follow-up of a large population of asymptomatic/oligosymptomatic hyperckemic subjects. J Neurol 2006; 253:1399–1403.
- Fernandez C, de Paula AM, Figarella-Branger D, et al. Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 2006; 66:1585–1587.
- Simmons Z, Peterlin BL, Boyer PJ, Towfighi J. Muscle biopsy in the evaluation of patients with modestly elevated creatine kinase levels. Muscle Nerve 2003; 27:242–244.
- Filosto M, Tonin P, Vattemi G, et al. The role of muscle biopsy in investigating isolated muscle pain. Neurology 2007; 68:181–186.
- Malandrini A, Orrico A, Gaudiano C, et al. Muscle biopsy and in vitro contracture test in subjects with idiopathic hyperCKemia. Anesthesiology 2008; 109:625–628.
- Prelle A, Tancredi L, Sciacco M, et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatine kinase levels. J Neurol 2002; 249:305–311.
- Dabby R, Sadeh M, Herman O, et al. Asymptomatic or minimally symptomatic hyperCKemia: histopathologic correlates. Isr Med Assoc J 2006; 8:110–113.
- Reijneveld JC, Notermans NC, Linssen WH, Wokke JH. Benign prognosis in idiopathic hyper-CK-emia. Muscle Nerve 2000; 23:575–579.
- Restivo DA, Pavone V, Nicotra A. Single-fiber electromyography in hyperCKemia: the value of fiber density. Neurol Sci 2012; 33:819–824.
- Rowland LP, Willner J, Cerri C, DiMauro S, Miranda A. Approaches to the membrane theory of Duchenne muscular dystrophy. In: Angelini C, Danielli GA, Fontanari D, editors. Muscular Dystrophy Research: Advances and New Trends, Amsterdam: Excerpta Medica; 1980:3–13.
- Reijneveld JC, Notermans NC, Linssen WH, Bär PR, Wokke JH. Hyper-CK-aemia revisited. Neuromuscul Disord 2001; 11:163–164.
- Capasso M, De Angelis MV, Di Muzio A, et al. Familial idiopathic hyper-CK-emia: an underrecognized condition. Muscle Nerve 2006; 33:760–765.
Measuring serum creatine kinase (CK) is an important part of the evaluation of patients with muscle weakness or myalgia, and of assessing patients with myopathies or rhabdomyolysis. But elevated CK sometimes is an incidental finding in a patient without muscle-related symptoms or with only minimal nonspecific muscle symptoms (eg, cramps, spasms, fatigue) that do not significantly interfere with activities of daily living. This condition is sometimes referred to as “asymptomatic hyper-CK-emia.” Four other muscle enzymes that may also be elevated are aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, and aldolase.
This review focuses on the evaluation of patients with elevated CK without significant muscle-related symptoms and proposes an algorithm for this purpose (Figure 1).
CURRENT THRESHOLDS MAY BE LOW
What appears to be an elevated CK level may in fact be normal, and it is important to determine in the initial assessment whether a CK value is truly abnormal.
Most laboratories use the central 95% of observations in white people as a reference range for serum CK, assuming that levels have a gaussian (bell-shaped) distribution, which is usually about 0 to 200 IU/L. Using these parameters, an abnormal CK level was observed in 19% of men and 5% of women in a study of nearly 1,000 healthy young people,1 leading to overdiagnosis.
The actual distribution of serum CK levels in a healthy population is markedly skewed toward higher values and is nongaussian.1–3 A 97.5% normal threshold is associated with a much lower false-positive rate and is recommended by the European Federation of Neurological Societies (now the European Academy of Neurology).4 This group also recommends pursuing further investigation only for patients whose level is at least 1.5 times the upper limit of normal; this threshold results in only a small reduction in sensitivity.
CK levels vary significantly by sex and race.5 Possible reasons include differences in muscle mass or total body mass and inherited differences in the permeability of the sarcolemma to CK.6 There is also a small reduction in CK levels as people age.2
The European Federation of Neurological Societies suggests redefining elevated CK as values 1.5 times beyond the upper limit of normal. Based on a 97.5% threshold and normal values determined by Brewster et al3 for black and white men and women, the following thresholds can be used to help decide whether to pursue further evaluation4:
- White women—325 IU/L
- White men—504 IU/L
- Black women—621 IU/L
- Black men—1,200 IU/L
PHYSICAL ACTIVITY RAISES CK
CK levels transiently rise after exercise or heavy manual labor. Serum CK levels may increase to as much as 30 times the upper limit of normal within 24 hours of strenuous physical activity, then slowly decline over the next 7 days. The degree of CK elevation depends on the type and duration of exercise, with greater elevation in those who are untrained.2,4
In assessing asymptomatic or minimally symptomatic CK elevation, the test should be repeated after 7 days without exercise. A large community study in Norway found that repeat CK levels in people with incidentally discovered elevated CK were normal after 3 days of rest in 70% of cases.2
NONNEUROMUSCULAR CAUSES
NEED TO BE INVESTIGATED
Asymptomatic or minimally symptomatic elevated CK can be due to a primary neuromuscular disease or a variety of nonneuromuscular causes.
Patients who still have elevated CK after taking into account the 97.5% threshold, repeat testing after a week of rest, and a level more than 1.5 times the upper limit of normal for sex and race should first be evaluated for the many nonneuromuscular conditions that can cause elevated CK (Table 1).7–9
Cardiac causes should be evaluated by history and physical examination, electrocardiography, and possibly testing for cardiac troponins.
Drugs commonly elevate CK
Prescription drugs and supplements are an important and common cause of CK elevation, so it is important to carefully review medications the patient is taking.
Statins can cause myalgia, muscle weakness, and rhabdomyolysis. Up to 5% of users develop CK elevation, typically 2 to 10 times the upper limit of normal.10 CK usually drops after stopping statins but may require weeks to months to normalize. Rarely, statin users develop a serious immune-mediated necrotizing myopathy.11–13
The diversity of response to statin therapy appears to have a genetic basis. The SEARCH Collaborative Group14 conducted a genome-wide association study of 300,000 markers in 85 patients with definite or incipient myopathy and in 90 controls, all of whom were taking simvastatin 80 mg daily. They identified a single-nucleotide polymorphism in the SLCO1B1 gene on chromosome 12 that was strongly associated with a higher risk of statin-induced myopathy.
Patients with statin-related myopathy seem to have a higher frequency of occult metabolic muscle disease than the general population, also suggesting genetic susceptibility, although ascertainment bias could be a factor.14
Mechanisms of CK elevation in response to statins include increased muscle membrane fragility due to decreased cholesterol content, inhibition of isoprenoid production (a necessary step in the synthesis of membrane proteins), and depletion of ubiquinone, leading to mitochondrial dysfunction.
Macro CK: An abnormal enzyme complex
About 4% of patients with asymptomatic or minimally symptomatic elevated CK have “macro CK,” an enzyme complex with an atypically high molecular mass and reduced clearance, resulting in abnormally high blood levels of CK. Macro CK type 1 is more common and is found in up to 1.2% of the general population: complexes are composed of CK and immunoglobulin and are associated with autoimmune diseases.9,15 Macro CK type 2 complexes consist of CK and an undetermined protein and are associated with malignancies.
CK electrophoresis is required to detect macro CK. Types 1 and 2 can be distinguished by protein G affinity chromatography.9,15
Endocrine disorders
Muscle involvement in endocrine disorders often presents with muscle weakness in addition to muscle enzyme abnormalities.
Hypothyroidism often causes weakness, cramps, myalgia, and a mild to moderate serum CK elevation.16 Severe CK elevation has been reported to occur after vigorous exercise.17 Thyroid replacement usually results in normalization of serum CK levels in 1 to 2 months.18
Hyperthyroidism is typically associated with normal serum CK concentrations, but in rare cases it can cause rhabdomyolysis.19
NEUROMUSCULAR CAUSES ARE NOT ALWAYS WORTH PURSUING
Only after the nonneuromuscular causes of elevated CK have been ruled out should neuromuscular disorders be considered (Table 2). Evaluation with electromyography, nerve conduction studies, and muscle biopsy may lead to the diagnosis of a specific neuromuscular disorder: patients may be in the presymptomatic stage of disease and may or may not eventually develop muscle weakness or other symptoms.20,21
Is testing needed?
Most adult dystrophies and metabolic myopathies have no available treatment and their course is often benign, particularly if they present only with asymptomatic elevated CK. The value of a potentially extensive, expensive, and invasive evaluation for a specific neuromuscular cause should be weighed against the limited yield and treatment options. Moreover, specialized testing such as biochemical muscle enzyme analysis, sarcolemmal protein staining, and genetic testing are not available at all centers.
The European Federation of Neurological Societies guidelines recommend biopsy for patients with asymptomatic elevated CK who also have any of the following:
- Abnormal (myopathic) findings on electromyography
- CK more than three times the upper limit of normal
- Age less than 25
- Exercise intolerance.4
Idiopathic inflammatory myopathies rarely present with asymptomatic elevated CK.22–26 In one study,27 they were found in just 5% of patients with asymptomatic elevated CK.
Hypomyopathic dermatomyositis and inclusion body myositis can present with mild CK elevations with normal muscle strength, especially early in the disease course. A myositis subset of antisynthetase syndrome can present with mildly elevated CK and interstitial lung disease.27 Many of the inflammatory myopathies respond to treatment so are worth investigating.
In view of complexities in diagnosis of these conditions, one should proceed with testing only after discussing it with patients. Referral to a rheumatology specialist is preferred.
MUSCLE BIOPSY, ELECTROMYOGRAPHY, AND NERVE CONDUCTION STUDIES
Electromyography, nerve conduction studies, or muscle biopsy, or a combination of these tests, is usually needed to investigate neuromuscular causes of elevated CK.
Muscle biopsy abnormalities are found in about two-thirds of cases of asymptomatic elevated CK, but most abnormalities include nonspecific myopathic changes that are not diagnostic. A muscle biopsy that may include special stains for sarcolemmal proteins for muscular dystrophy and biochemical muscle enzyme analysis for metabolic myopathies is diagnostic in only 20% to 25% cases of asymptomatic elevated CK on average, with a variation between different series of 0% to 79%.7,21,27–33
Electromyography and nerve conduction studies alone add little to the workup of asymptomatic elevated CK apart from a modest negative predictive value and as a guide for muscle biopsy. For a very few neuromuscular disorders causing an elevated CK (eg, motor neuron disease, Charcot-Marie-Tooth disease, myotonic dystrophy), electromyography and nerve conduction studies could suffice to make the diagnosis.
Electromyography and nerve conduction studies detect abnormalities in nearly half of cases of asymptomatic CK elevation,7,21,27,28,30,31,33 but, as with biopsy, most changes are nonspecific. Although electromyography and nerve conduction studies can help distinguish primary neuropathic from myopathic disorders, the sensitivity and specificity are low for diagnosis. Normal studies do not rule out a condition, and abnormal studies are not diagnostic of a particular condition, although completely normal studies provide strong evidence against a severe neuromuscular disorder.
Combined testing
Using combined muscle biopsy, electromyography, and nerve conduction studies, the likelihood of making a diagnosis in patients with asymptomatic elevated CK is 28% on average (range of studies 4%–79%),2,7,21,26–28,30–32 and findings are nonspecific in 30% to 40% of cases. Findings are normal in about 30% to 40% of cases, which are thus diagnosed as idiopathic asymptomatic elevated CK.28–31,34
Prelle et al31 retrospectively reviewed the cases of 114 patients, ages 3 to 70, with incidentally discovered elevated CK and few or no symptoms, who underwent muscle biopsy, electromyography, and nerve conduction studies after nonneuromuscular causes were ruled out. Although muscle biopsy findings were abnormal in 39% of cases, a diagnosis was established in only 18% of cases after an extensive workup: the diagnosis was definitive in only 10% and included dystrophinopathies, metabolic myopathies, and rare noninflammatory myopathies. For the remaining 8%, the diagnosis was probable and included four cases of partial carnitine palmitoyl transferase deficiency, three cases of malignant hyperthermia, and two rare inherited disorders.
DNA testing
In women with a serum CK less than three times the upper limit of normal who have a family history of Duchenne or Becker muscular dystrophy, DNA analysis of blood lymphocytes identifies 70% of carriers.4
IDIOPATHIC ELEVATED SERUM CK
Rowland et al35 first coined the term “idiopathic hyper-CK-emia” and defined it as persistent elevation of serum CK despite a normal neurologic examination and testing, including electromyography, nerve conduction studies, and muscle biopsy.35,36 To receive this diagnosis, patients must also have no family history or clinical evidence of neuromuscular disease.
Idiopathic elevated serum CK is sometimes familial. In one study,37 elevated CK was found in family members of 13 of 28 unrelated probands. In the 13 families, 41 individuals had elevated CK. Genetic studies revealed that the condition is genetically heterogeneous and autosomal dominant in at least 60% of cases, with higher penetrance in men.
D’Adda et al26 followed 55 people with idiopathic elevated CK for 7 years. Ten percent were eventually diagnosed with a neuromuscular disorder, 10% developed malignancy, and the remaining 80% developed no new condition. The CK level normalized or decreased in many patients, but most continued to have persistent CK elevations with minimal or no symptoms.
Measuring serum creatine kinase (CK) is an important part of the evaluation of patients with muscle weakness or myalgia, and of assessing patients with myopathies or rhabdomyolysis. But elevated CK sometimes is an incidental finding in a patient without muscle-related symptoms or with only minimal nonspecific muscle symptoms (eg, cramps, spasms, fatigue) that do not significantly interfere with activities of daily living. This condition is sometimes referred to as “asymptomatic hyper-CK-emia.” Four other muscle enzymes that may also be elevated are aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, and aldolase.
This review focuses on the evaluation of patients with elevated CK without significant muscle-related symptoms and proposes an algorithm for this purpose (Figure 1).
CURRENT THRESHOLDS MAY BE LOW
What appears to be an elevated CK level may in fact be normal, and it is important to determine in the initial assessment whether a CK value is truly abnormal.
Most laboratories use the central 95% of observations in white people as a reference range for serum CK, assuming that levels have a gaussian (bell-shaped) distribution, which is usually about 0 to 200 IU/L. Using these parameters, an abnormal CK level was observed in 19% of men and 5% of women in a study of nearly 1,000 healthy young people,1 leading to overdiagnosis.
The actual distribution of serum CK levels in a healthy population is markedly skewed toward higher values and is nongaussian.1–3 A 97.5% normal threshold is associated with a much lower false-positive rate and is recommended by the European Federation of Neurological Societies (now the European Academy of Neurology).4 This group also recommends pursuing further investigation only for patients whose level is at least 1.5 times the upper limit of normal; this threshold results in only a small reduction in sensitivity.
CK levels vary significantly by sex and race.5 Possible reasons include differences in muscle mass or total body mass and inherited differences in the permeability of the sarcolemma to CK.6 There is also a small reduction in CK levels as people age.2
The European Federation of Neurological Societies suggests redefining elevated CK as values 1.5 times beyond the upper limit of normal. Based on a 97.5% threshold and normal values determined by Brewster et al3 for black and white men and women, the following thresholds can be used to help decide whether to pursue further evaluation4:
- White women—325 IU/L
- White men—504 IU/L
- Black women—621 IU/L
- Black men—1,200 IU/L
PHYSICAL ACTIVITY RAISES CK
CK levels transiently rise after exercise or heavy manual labor. Serum CK levels may increase to as much as 30 times the upper limit of normal within 24 hours of strenuous physical activity, then slowly decline over the next 7 days. The degree of CK elevation depends on the type and duration of exercise, with greater elevation in those who are untrained.2,4
In assessing asymptomatic or minimally symptomatic CK elevation, the test should be repeated after 7 days without exercise. A large community study in Norway found that repeat CK levels in people with incidentally discovered elevated CK were normal after 3 days of rest in 70% of cases.2
NONNEUROMUSCULAR CAUSES
NEED TO BE INVESTIGATED
Asymptomatic or minimally symptomatic elevated CK can be due to a primary neuromuscular disease or a variety of nonneuromuscular causes.
Patients who still have elevated CK after taking into account the 97.5% threshold, repeat testing after a week of rest, and a level more than 1.5 times the upper limit of normal for sex and race should first be evaluated for the many nonneuromuscular conditions that can cause elevated CK (Table 1).7–9
Cardiac causes should be evaluated by history and physical examination, electrocardiography, and possibly testing for cardiac troponins.
Drugs commonly elevate CK
Prescription drugs and supplements are an important and common cause of CK elevation, so it is important to carefully review medications the patient is taking.
Statins can cause myalgia, muscle weakness, and rhabdomyolysis. Up to 5% of users develop CK elevation, typically 2 to 10 times the upper limit of normal.10 CK usually drops after stopping statins but may require weeks to months to normalize. Rarely, statin users develop a serious immune-mediated necrotizing myopathy.11–13
The diversity of response to statin therapy appears to have a genetic basis. The SEARCH Collaborative Group14 conducted a genome-wide association study of 300,000 markers in 85 patients with definite or incipient myopathy and in 90 controls, all of whom were taking simvastatin 80 mg daily. They identified a single-nucleotide polymorphism in the SLCO1B1 gene on chromosome 12 that was strongly associated with a higher risk of statin-induced myopathy.
Patients with statin-related myopathy seem to have a higher frequency of occult metabolic muscle disease than the general population, also suggesting genetic susceptibility, although ascertainment bias could be a factor.14
Mechanisms of CK elevation in response to statins include increased muscle membrane fragility due to decreased cholesterol content, inhibition of isoprenoid production (a necessary step in the synthesis of membrane proteins), and depletion of ubiquinone, leading to mitochondrial dysfunction.
Macro CK: An abnormal enzyme complex
About 4% of patients with asymptomatic or minimally symptomatic elevated CK have “macro CK,” an enzyme complex with an atypically high molecular mass and reduced clearance, resulting in abnormally high blood levels of CK. Macro CK type 1 is more common and is found in up to 1.2% of the general population: complexes are composed of CK and immunoglobulin and are associated with autoimmune diseases.9,15 Macro CK type 2 complexes consist of CK and an undetermined protein and are associated with malignancies.
CK electrophoresis is required to detect macro CK. Types 1 and 2 can be distinguished by protein G affinity chromatography.9,15
Endocrine disorders
Muscle involvement in endocrine disorders often presents with muscle weakness in addition to muscle enzyme abnormalities.
Hypothyroidism often causes weakness, cramps, myalgia, and a mild to moderate serum CK elevation.16 Severe CK elevation has been reported to occur after vigorous exercise.17 Thyroid replacement usually results in normalization of serum CK levels in 1 to 2 months.18
Hyperthyroidism is typically associated with normal serum CK concentrations, but in rare cases it can cause rhabdomyolysis.19
NEUROMUSCULAR CAUSES ARE NOT ALWAYS WORTH PURSUING
Only after the nonneuromuscular causes of elevated CK have been ruled out should neuromuscular disorders be considered (Table 2). Evaluation with electromyography, nerve conduction studies, and muscle biopsy may lead to the diagnosis of a specific neuromuscular disorder: patients may be in the presymptomatic stage of disease and may or may not eventually develop muscle weakness or other symptoms.20,21
Is testing needed?
Most adult dystrophies and metabolic myopathies have no available treatment and their course is often benign, particularly if they present only with asymptomatic elevated CK. The value of a potentially extensive, expensive, and invasive evaluation for a specific neuromuscular cause should be weighed against the limited yield and treatment options. Moreover, specialized testing such as biochemical muscle enzyme analysis, sarcolemmal protein staining, and genetic testing are not available at all centers.
The European Federation of Neurological Societies guidelines recommend biopsy for patients with asymptomatic elevated CK who also have any of the following:
- Abnormal (myopathic) findings on electromyography
- CK more than three times the upper limit of normal
- Age less than 25
- Exercise intolerance.4
Idiopathic inflammatory myopathies rarely present with asymptomatic elevated CK.22–26 In one study,27 they were found in just 5% of patients with asymptomatic elevated CK.
Hypomyopathic dermatomyositis and inclusion body myositis can present with mild CK elevations with normal muscle strength, especially early in the disease course. A myositis subset of antisynthetase syndrome can present with mildly elevated CK and interstitial lung disease.27 Many of the inflammatory myopathies respond to treatment so are worth investigating.
In view of complexities in diagnosis of these conditions, one should proceed with testing only after discussing it with patients. Referral to a rheumatology specialist is preferred.
MUSCLE BIOPSY, ELECTROMYOGRAPHY, AND NERVE CONDUCTION STUDIES
Electromyography, nerve conduction studies, or muscle biopsy, or a combination of these tests, is usually needed to investigate neuromuscular causes of elevated CK.
Muscle biopsy abnormalities are found in about two-thirds of cases of asymptomatic elevated CK, but most abnormalities include nonspecific myopathic changes that are not diagnostic. A muscle biopsy that may include special stains for sarcolemmal proteins for muscular dystrophy and biochemical muscle enzyme analysis for metabolic myopathies is diagnostic in only 20% to 25% cases of asymptomatic elevated CK on average, with a variation between different series of 0% to 79%.7,21,27–33
Electromyography and nerve conduction studies alone add little to the workup of asymptomatic elevated CK apart from a modest negative predictive value and as a guide for muscle biopsy. For a very few neuromuscular disorders causing an elevated CK (eg, motor neuron disease, Charcot-Marie-Tooth disease, myotonic dystrophy), electromyography and nerve conduction studies could suffice to make the diagnosis.
Electromyography and nerve conduction studies detect abnormalities in nearly half of cases of asymptomatic CK elevation,7,21,27,28,30,31,33 but, as with biopsy, most changes are nonspecific. Although electromyography and nerve conduction studies can help distinguish primary neuropathic from myopathic disorders, the sensitivity and specificity are low for diagnosis. Normal studies do not rule out a condition, and abnormal studies are not diagnostic of a particular condition, although completely normal studies provide strong evidence against a severe neuromuscular disorder.
Combined testing
Using combined muscle biopsy, electromyography, and nerve conduction studies, the likelihood of making a diagnosis in patients with asymptomatic elevated CK is 28% on average (range of studies 4%–79%),2,7,21,26–28,30–32 and findings are nonspecific in 30% to 40% of cases. Findings are normal in about 30% to 40% of cases, which are thus diagnosed as idiopathic asymptomatic elevated CK.28–31,34
Prelle et al31 retrospectively reviewed the cases of 114 patients, ages 3 to 70, with incidentally discovered elevated CK and few or no symptoms, who underwent muscle biopsy, electromyography, and nerve conduction studies after nonneuromuscular causes were ruled out. Although muscle biopsy findings were abnormal in 39% of cases, a diagnosis was established in only 18% of cases after an extensive workup: the diagnosis was definitive in only 10% and included dystrophinopathies, metabolic myopathies, and rare noninflammatory myopathies. For the remaining 8%, the diagnosis was probable and included four cases of partial carnitine palmitoyl transferase deficiency, three cases of malignant hyperthermia, and two rare inherited disorders.
DNA testing
In women with a serum CK less than three times the upper limit of normal who have a family history of Duchenne or Becker muscular dystrophy, DNA analysis of blood lymphocytes identifies 70% of carriers.4
IDIOPATHIC ELEVATED SERUM CK
Rowland et al35 first coined the term “idiopathic hyper-CK-emia” and defined it as persistent elevation of serum CK despite a normal neurologic examination and testing, including electromyography, nerve conduction studies, and muscle biopsy.35,36 To receive this diagnosis, patients must also have no family history or clinical evidence of neuromuscular disease.
Idiopathic elevated serum CK is sometimes familial. In one study,37 elevated CK was found in family members of 13 of 28 unrelated probands. In the 13 families, 41 individuals had elevated CK. Genetic studies revealed that the condition is genetically heterogeneous and autosomal dominant in at least 60% of cases, with higher penetrance in men.
D’Adda et al26 followed 55 people with idiopathic elevated CK for 7 years. Ten percent were eventually diagnosed with a neuromuscular disorder, 10% developed malignancy, and the remaining 80% developed no new condition. The CK level normalized or decreased in many patients, but most continued to have persistent CK elevations with minimal or no symptoms.
- Lev EI, Tur-Kaspa I, Ashkenazy I, et al. Distribution of serum creatine kinase activity in young healthy persons. Clin Chim Acta 1999; 279:107–115.
- Lilleng H, Abeler K, Johnsen SH, et al. Variation of serum creatine kinase (CK) levels and prevalence of persistent hyperCKemia in a Norwegian normal population. The Tromsø Study. Neuromuscul Disord 2011; 21:494–500.
- Brewster LM, Mairuhu G, Sturk A, van Montfrans GA. Distribution of creatine kinase in the general population: implications for statin therapy. Am Heart J 2007; 154:655–661.
- Kyriakides T, Angelini C, Schaefer J, et al; European Federation of Neurological Societies. EFNS guidelines on the diagnostic approach to pauci- or asymptomatic hyperCKemia. Eur J Neurol 2010; 17:767–773.
- Prisant LM, Downton M, Watkins LO, et al. Efficacy and tolerability of lovastatin in 459 African-Americans with hypercholesterolemia. Am J Cardiol 1996; 78:420–444.
- Wong ET, Cobb C, Umehara MK, et al. Heterogeneity of serum creatine kinase activity among racial and gender groups of the population. Am J Clin Pathol 1983; 79:582–586.
- Brewster LM, de Visser M. Persistent hyperCKemia: fourteen patients studied in retrospect. Acta Neurol Scand 1988; 77:60–63.
- Weglinski MR, Wedel DJ, Engel AG. Malignant hyperthermia testing in patients with persistently increased serum creatine kinase levels. Anesth Analg 1997; 84:1038–1041.
- Galarraga B, Sinclair D, Fahie-Wilson MN, McCrae FC, Hull RG, Ledingham JM. A rare but important cause for a raised serum creatine kinase concentration: two case reports and a literature review. Rheumatology (Oxford) 2003; 42:186–188.
- Mancini GB, Tashakkor AY, Baker S, et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian Working Group Consensus update. Can J Cardiol 2013; 29:1553–1568.
- Arora R, Liebo M, Maldonado F. Statin-induced myopathy: the two faces of Janus. J Cardiovasc Pharmacol Ther 2006; 11:105–112.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Talbert RL. Safety issues with statin therapy. J Am Pharm Assoc (2003) 2006; 46:479–490.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Wyness SP, Hunsaker JJ, La’ulu SL, Rao LV, Roberts WL. Detection of macro-creatine kinase and macroamylase by polyethylene glycol precipitation and ultrafiltration methods. Clin Chim Acta 2011; 412:2052–2057.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Riggs JE. Acute exertional rhabdomyolysis in hypothyroidism: the result of a reversible defect in glycogenolysis? Mil Med 1990; 155:171–172.
- Mastaglia FL, Ojeda VJ, Sarnat HB, Kakulas BA. Myopathies associated with hypothyroidism: a review based upon 13 cases. Aust N Z J Med 1988; 18:799–806.
- Alshanti M, Eledrisi MS, Jones E. Rhabdomyolysis associated with hyperthyroidism. Am J Emerg Med 2001; 19:317.
- Rosalki SB. Serum enzymes in disease of skeletal muscle. Clin Lab Med 1989; 9:767–781.
- Joy JL, Oh SJ. Asymptomatic hyper-CK-emia: an electrophysiologic and histopathologic study. Muscle Nerve 1989; 12:206–209.
- Merlini L, Sabatelli P, Columbaro M, et al. Hyper-CK-emia as the sole manifestation of myotonic dystrophy type 2. Muscle Nerve 2005; 31:764–767.
- Eeg-Olofsson O, Kalimo H, Eeg-Olofsson KE, et al. Duchenne muscular dystrophy and idiopathic hyperCKemia in the same family. Eur J Paediatr Neurol 2008; 12:404–407.
- Dwianingsih EK, Takeshima Y, Itoh K, et al. A Japanese child with asymptomatic elevation of serum creatine kinase shows PTRF-CAVIN mutation matching with congenital generalized lipodystrophy type 4. Mol Genet Metab 2010; 101:233–237.
- Carbone I, Bruno C, Sotgia F, et al. Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology 2000; 54:1373–1376.
- D’Adda E, Sciacco M, Fruguglietti ME, et al. Follow-up of a large population of asymptomatic/oligosymptomatic hyperckemic subjects. J Neurol 2006; 253:1399–1403.
- Fernandez C, de Paula AM, Figarella-Branger D, et al. Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 2006; 66:1585–1587.
- Simmons Z, Peterlin BL, Boyer PJ, Towfighi J. Muscle biopsy in the evaluation of patients with modestly elevated creatine kinase levels. Muscle Nerve 2003; 27:242–244.
- Filosto M, Tonin P, Vattemi G, et al. The role of muscle biopsy in investigating isolated muscle pain. Neurology 2007; 68:181–186.
- Malandrini A, Orrico A, Gaudiano C, et al. Muscle biopsy and in vitro contracture test in subjects with idiopathic hyperCKemia. Anesthesiology 2008; 109:625–628.
- Prelle A, Tancredi L, Sciacco M, et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatine kinase levels. J Neurol 2002; 249:305–311.
- Dabby R, Sadeh M, Herman O, et al. Asymptomatic or minimally symptomatic hyperCKemia: histopathologic correlates. Isr Med Assoc J 2006; 8:110–113.
- Reijneveld JC, Notermans NC, Linssen WH, Wokke JH. Benign prognosis in idiopathic hyper-CK-emia. Muscle Nerve 2000; 23:575–579.
- Restivo DA, Pavone V, Nicotra A. Single-fiber electromyography in hyperCKemia: the value of fiber density. Neurol Sci 2012; 33:819–824.
- Rowland LP, Willner J, Cerri C, DiMauro S, Miranda A. Approaches to the membrane theory of Duchenne muscular dystrophy. In: Angelini C, Danielli GA, Fontanari D, editors. Muscular Dystrophy Research: Advances and New Trends, Amsterdam: Excerpta Medica; 1980:3–13.
- Reijneveld JC, Notermans NC, Linssen WH, Bär PR, Wokke JH. Hyper-CK-aemia revisited. Neuromuscul Disord 2001; 11:163–164.
- Capasso M, De Angelis MV, Di Muzio A, et al. Familial idiopathic hyper-CK-emia: an underrecognized condition. Muscle Nerve 2006; 33:760–765.
- Lev EI, Tur-Kaspa I, Ashkenazy I, et al. Distribution of serum creatine kinase activity in young healthy persons. Clin Chim Acta 1999; 279:107–115.
- Lilleng H, Abeler K, Johnsen SH, et al. Variation of serum creatine kinase (CK) levels and prevalence of persistent hyperCKemia in a Norwegian normal population. The Tromsø Study. Neuromuscul Disord 2011; 21:494–500.
- Brewster LM, Mairuhu G, Sturk A, van Montfrans GA. Distribution of creatine kinase in the general population: implications for statin therapy. Am Heart J 2007; 154:655–661.
- Kyriakides T, Angelini C, Schaefer J, et al; European Federation of Neurological Societies. EFNS guidelines on the diagnostic approach to pauci- or asymptomatic hyperCKemia. Eur J Neurol 2010; 17:767–773.
- Prisant LM, Downton M, Watkins LO, et al. Efficacy and tolerability of lovastatin in 459 African-Americans with hypercholesterolemia. Am J Cardiol 1996; 78:420–444.
- Wong ET, Cobb C, Umehara MK, et al. Heterogeneity of serum creatine kinase activity among racial and gender groups of the population. Am J Clin Pathol 1983; 79:582–586.
- Brewster LM, de Visser M. Persistent hyperCKemia: fourteen patients studied in retrospect. Acta Neurol Scand 1988; 77:60–63.
- Weglinski MR, Wedel DJ, Engel AG. Malignant hyperthermia testing in patients with persistently increased serum creatine kinase levels. Anesth Analg 1997; 84:1038–1041.
- Galarraga B, Sinclair D, Fahie-Wilson MN, McCrae FC, Hull RG, Ledingham JM. A rare but important cause for a raised serum creatine kinase concentration: two case reports and a literature review. Rheumatology (Oxford) 2003; 42:186–188.
- Mancini GB, Tashakkor AY, Baker S, et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian Working Group Consensus update. Can J Cardiol 2013; 29:1553–1568.
- Arora R, Liebo M, Maldonado F. Statin-induced myopathy: the two faces of Janus. J Cardiovasc Pharmacol Ther 2006; 11:105–112.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Talbert RL. Safety issues with statin therapy. J Am Pharm Assoc (2003) 2006; 46:479–490.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Wyness SP, Hunsaker JJ, La’ulu SL, Rao LV, Roberts WL. Detection of macro-creatine kinase and macroamylase by polyethylene glycol precipitation and ultrafiltration methods. Clin Chim Acta 2011; 412:2052–2057.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Riggs JE. Acute exertional rhabdomyolysis in hypothyroidism: the result of a reversible defect in glycogenolysis? Mil Med 1990; 155:171–172.
- Mastaglia FL, Ojeda VJ, Sarnat HB, Kakulas BA. Myopathies associated with hypothyroidism: a review based upon 13 cases. Aust N Z J Med 1988; 18:799–806.
- Alshanti M, Eledrisi MS, Jones E. Rhabdomyolysis associated with hyperthyroidism. Am J Emerg Med 2001; 19:317.
- Rosalki SB. Serum enzymes in disease of skeletal muscle. Clin Lab Med 1989; 9:767–781.
- Joy JL, Oh SJ. Asymptomatic hyper-CK-emia: an electrophysiologic and histopathologic study. Muscle Nerve 1989; 12:206–209.
- Merlini L, Sabatelli P, Columbaro M, et al. Hyper-CK-emia as the sole manifestation of myotonic dystrophy type 2. Muscle Nerve 2005; 31:764–767.
- Eeg-Olofsson O, Kalimo H, Eeg-Olofsson KE, et al. Duchenne muscular dystrophy and idiopathic hyperCKemia in the same family. Eur J Paediatr Neurol 2008; 12:404–407.
- Dwianingsih EK, Takeshima Y, Itoh K, et al. A Japanese child with asymptomatic elevation of serum creatine kinase shows PTRF-CAVIN mutation matching with congenital generalized lipodystrophy type 4. Mol Genet Metab 2010; 101:233–237.
- Carbone I, Bruno C, Sotgia F, et al. Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology 2000; 54:1373–1376.
- D’Adda E, Sciacco M, Fruguglietti ME, et al. Follow-up of a large population of asymptomatic/oligosymptomatic hyperckemic subjects. J Neurol 2006; 253:1399–1403.
- Fernandez C, de Paula AM, Figarella-Branger D, et al. Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 2006; 66:1585–1587.
- Simmons Z, Peterlin BL, Boyer PJ, Towfighi J. Muscle biopsy in the evaluation of patients with modestly elevated creatine kinase levels. Muscle Nerve 2003; 27:242–244.
- Filosto M, Tonin P, Vattemi G, et al. The role of muscle biopsy in investigating isolated muscle pain. Neurology 2007; 68:181–186.
- Malandrini A, Orrico A, Gaudiano C, et al. Muscle biopsy and in vitro contracture test in subjects with idiopathic hyperCKemia. Anesthesiology 2008; 109:625–628.
- Prelle A, Tancredi L, Sciacco M, et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatine kinase levels. J Neurol 2002; 249:305–311.
- Dabby R, Sadeh M, Herman O, et al. Asymptomatic or minimally symptomatic hyperCKemia: histopathologic correlates. Isr Med Assoc J 2006; 8:110–113.
- Reijneveld JC, Notermans NC, Linssen WH, Wokke JH. Benign prognosis in idiopathic hyper-CK-emia. Muscle Nerve 2000; 23:575–579.
- Restivo DA, Pavone V, Nicotra A. Single-fiber electromyography in hyperCKemia: the value of fiber density. Neurol Sci 2012; 33:819–824.
- Rowland LP, Willner J, Cerri C, DiMauro S, Miranda A. Approaches to the membrane theory of Duchenne muscular dystrophy. In: Angelini C, Danielli GA, Fontanari D, editors. Muscular Dystrophy Research: Advances and New Trends, Amsterdam: Excerpta Medica; 1980:3–13.
- Reijneveld JC, Notermans NC, Linssen WH, Bär PR, Wokke JH. Hyper-CK-aemia revisited. Neuromuscul Disord 2001; 11:163–164.
- Capasso M, De Angelis MV, Di Muzio A, et al. Familial idiopathic hyper-CK-emia: an underrecognized condition. Muscle Nerve 2006; 33:760–765.
KEY POINTS
- Standard reference ranges for serum CK levels used by most laboratories are too low and lead to overdiagnosis of abnormal values.
- Serum CK levels are strongly affected by race, sex, and physical activity.
- A patient with truly elevated levels should be evaluated for a variety of nonneuromuscular causes, including endocrine disorders, metabolic disturbances, drug effects, and malignancy.
- Neuromuscular causes should be investigated only after ruling out nonneuromuscular causes and after considering whether potential benefits of a diagnosis outweigh the risks and expense of extensive testing.
Autoantibody-mediated encephalitis: Not just paraneoplastic, not just limbic, and not untreatable
A 79-year-old woman with a history of breast cancer in remission and hypertension presented to a local emergency department because of subacute memory loss and compulsive shopping. Her serum sodium concentration was 127 mmol/L (reference range 132–148). Computed tomography (CT) and magnetic resonance imaging (MRI) of the brain were normal, and she was sent home.
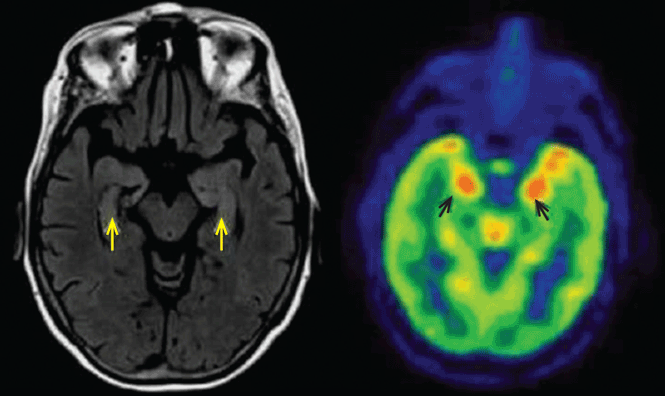
Three days later, she experienced a generalized tonic-clonic seizure that evolved into status epilepticus. She was intubated and admitted to the intensive care unit. Cerebrospinal fluid analysis was normal, and infectious causes of encephalitis were ruled out. MRI showed increased signal in both hippocampi (Figure 1). Her seizures were refractory to treatment, and she was given pentobarbital to induce a coma.
Serum evaluation of neuronal antibodies revealed elevated titers of the voltage-gated potassium channel (VGKC) complex antibody, with subsequent subtyping confirming the leucine-rich glioma-inactivated protein 1 (LGI1) protein as the antigenic target.

She received a 5-day course of intravenous immunoglobulin and methylprednisolone, pentobarbital was withdrawn, and the seizures did not recur, but weeks later she remained comatose. Positron emission tomography (PET) of the brain revealed hypermetabolism in the medial and anterior aspects of both temporal lobes. She underwent five sessions of plasma exchange, after which she began to improve and follow commands. She was ultimately discharged to an acute rehabilitation facility after a 4-week hospital stay.
She received infusions of intravenous immunoglobulin twice a month for 6 months. At her last follow-up visit, she was seizure-free and neurologically intact except for mild inattention.
NEWLY RECOGNIZED DISEASES
Although autoantibody-mediated encephalitic syndromes were first described more than 50 years ago,1,2 their autoimmune basis was not recognized until the early 1980s.3 In the past 10 years, a flood of novel clinical syndromes associated with neuronal autoantibodies has been described that may be markedly improved or even completely resolved with immunotherapy. In cases of unexplained seizure, encephalitis, or acute-onset psychiatric syndromes, suspecting these syndromes can lead to diagnosis, treatment, and a good outcome.
This review describes the key clinical autoantibody-mediated encephalitic syndromes, explains the better-characterized antibody associations, and discusses their diagnosis and treatment.
CLASSIFIED ANATOMICALLY, IMMUNOLOGICALLY, OR EPONYMOUSLY
Autoantibody-mediated encephalitis is also known as autoimmune-mediated encephalitis, autoimmune-mediated limbic encephalitis, and autoimmune synaptic encephalitis.
How to categorize these syndromes is still in flux: they can be listed by the area of the brain affected, the antibody involved, or the name of the discoverer (eg, Morvan syndrome).
Autoantibodies identified in autoimmune encephalitis fall under two broad categories:
- Those targeting intracellular (intranuclear or intracytoplasmic) antigens; the syndromes they cause are more likely to be paraneoplastic and less responsive to immunotherapy
- Those targeting antigens on the neuronal surface: the syndromes they cause are less likely to be paraneoplastic and are more responsive to immunotherapy.4
SYNDROMES DEFINED BY BRAIN AREA AFFECTED
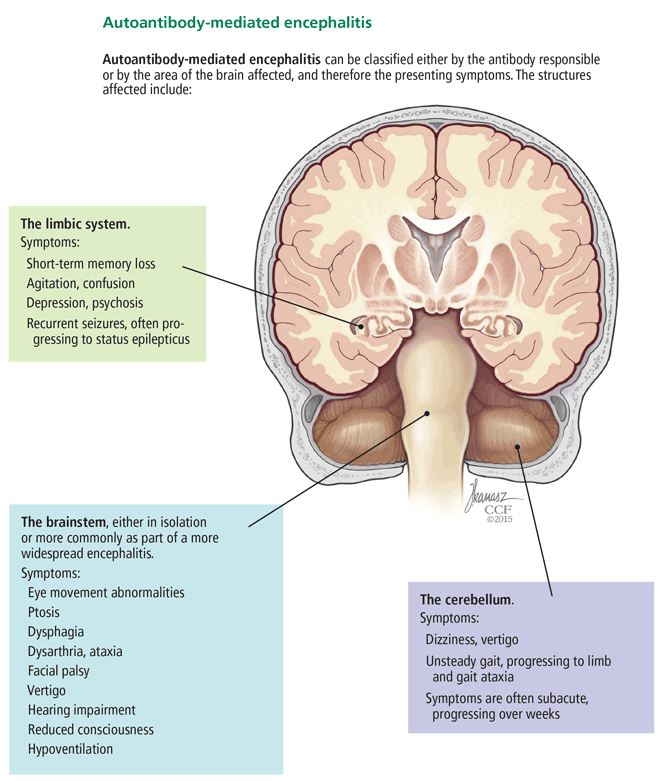
Below, we provide examples of neurologic syndromes of autoantibody-mediated encephalitis according to the region of the brain most affected, ie, the limbic system, the brainstem, or the cerebellum (Figure 2).
LIMBIC ENCEPHALITIS
Memory loss, behavioral changes, seizures
Patients with limbic encephalitis (such as the patient described in the vignette above) present with symptoms attributed to dysfunction of mesial temporal lobe structures, most notably the hippocampus. Prominent symptoms include short-term memory loss, behavioral disturbances such as agitation and confusion, and psychiatric problems such as depression and psychosis. Recurrent seizures are a salient feature and, not uncommonly, progress to status epilepticus.
Antibodies are not all cancer-associated
Cerebrospinal fluid analysis can be normal or show abnormalities suggesting immune activation, eg, slight pleocytosis, elevated protein, increased immunoglobulin G synthesis, and oligoclonal banding.5
In many cases, an autoantibody is found in the blood or in the cerebrospinal fluid. Some patients may express more than one autoantibody, so the traditional view of “one antibody, one syndrome” is incorrect.
Although initially identified as a rare paraneoplastic disorder, limbic encephalitis sometimes occurs in the absence of malignancy.
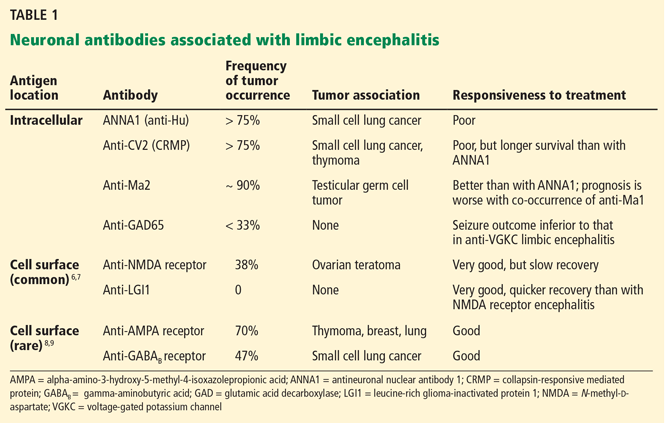
Multiple antibodies have been linked to the syndrome (Table 1).6–9 The “classic” antibodies initially found in paraneoplastic forms are now generally viewed as nonpathogenic, in part because they are directed against intracellular antigens. Neuronal injury in paraneoplastic limbic encephalitis is believed to be mediated by cytotoxic T lymphocytes, with neuronal autoantibodies being produced after the injury.4 Recently defined antibodies, such as those targeting the N-methyl-d-aspartate (NMDA) receptor6 and the LGI1 protein,7 are now understood to be common causes of limbic encephalitis.
Imaging usually shows limbic focal changes
Structural MRI or functional fluorodeoxyglucose (FDG)-PET imaging may show focal changes in limbic system structures, such as the mesial temporal lobes. It is now recognized that other cortical areas may be involved, and the term “limbic encephalitis” may give way to “cortical” or “focal encephalitis.”
In about 60% of patients, MRI shows hyperintense fluid-attenuated inversion recovery (FLAIR) or T2 signal changes in the mesial temporal lobes, likely reflecting inflammatory changes.4,10,11 On FDG-PET, hypermetabolism may be observed in the mesial temporal lobes early in the disease despite normal findings on MRI.12 Hypometabolism, either diffuse or localized to the mesial temporal lobes, eventually sets in, likely reflecting cytotoxic injury in the aftermath of prolonged inflammation or seizures.
Consider other causes
Before diagnosing limbic encephalitis, it is essential to evaluate for infectious meningoencephalitis, especially herpes simplex viral encephalitis. Thiamine deficiency (Wernicke encephalopathy), drug intoxication, prion disease, Hashimoto encephalopathy, tumor, and subclinical status epilepticus should also be considered. Some of these conditions are associated with the same neuronal autoantibodies detected in limbic encephalitis. Further complicating the picture, case reports have shown the presence of serum neuronal autoantibodies—VGKC complex13–15 and NMDA-receptor antibodies16,17—in confirmed cases of prion disease. In addition, adequately treated herpes simplex viral encephalitis can precipitate the production of NMDA-receptor antibodies and their characteristic syndrome.18–20
BRAINSTEM ENCEPHALITIS
The brainstem—the midbrain, pons, and medulla—can be affected, either in isolation or more commonly as part of a more widespread autoantibody-mediated encephalitis. Symptoms and signs include eye movement abnormalities, ptosis, dysphagia, dysarthria, ataxia, facial palsy, vertigo, hearing impairment, reduced consciousness, and hypoventilation.21
Anti-Hu, anti-Ri, and anti-Ma2 antibodies are most commonly associated with brainstem encephalitis (Table 2). Anti-Ma2-associated encephalitis may improve after a combination of immunotherapy and tumor removal21; the others have a poor prognosis.
Neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders most commonly involve demyelination affecting the optic nerves and spinal cord, leading to unilateral or bilateral optic neuritis and transverse myelitis spanning three or more vertebral segments.22 The initial clinical manifestation may be an encephalitic pattern, affecting predominantly the brainstem in a restricted fashion,22 or the central nervous system in a more diffuse pattern, mimicking either acute disseminated encephalomyelitis or, in less severe cases, posterior reversible encephalopathy syndrome.23
Testing for antiaquaporin-4 antibody, also known as neuromyelitis optica immunoglobulin G, is the single most decisive laboratory test for diagnosing neuromyelitis optica spectrum disorders, so serum and cerebrospinal fluid evaluation for this autoantibody should be considered when caring for a patient whose clinical picture suggests brainstem encephalitis.22
Bickerstaff brainstem encephalitis
Bickerstaff brainstem encephalitis was first described more than half a century ago in patients with postinfectious ataxia, ophthalmoparesis, and altered consciousness. This rare disease was later found to be associated with antiganglioside GQ1b (anti-GQ1b) autoantibody. MRI is normal in about 90% of cases, so recognizing the clinical presentation and analyzing anti-GQ1b serum titers are critical to diagnosis.
Recovery is usually spontaneous and complete and can be hastened by immunotherapy, especially intravenous immunoglobulin.24
Other causes of brainstem encephalitis
The differential diagnosis of a presentation of brainstem encephalitis includes:
- Infectious causes, the most common being Listeria species followed by enterovirus 71 and herpes simplex virus.25 Tuberculosis, brucellosis, and Whipple disease should also be considered.
- Primary central nervous system inflammatory and demyelinating conditions, eg, multiple sclerosis and acute disseminated encephalomyelitis.
- Systemic inflammatory conditions, eg, Behçet disease, systemic lupus erythematosus, and sarcoidosis.
- Direct brainstem neoplastic involvement, as might occur in primary central nervous system lymphoma or leptomeningeal carcinomatosis.
CEREBELLAR SYNDROME
Patients with autoantibody-mediated encephalitis localized predominantly to the cerebellum typically present with dizziness, vertigo, and unsteady gait, progressing eventually to limb and gait ataxia.4 Symptoms are often subacute, progressing over weeks.
Multiple neuronal autoantibodies have been found to occur with cerebellar encephalitis (Table 2). In most cases, they are paraneoplastic and considered not to be pathogenic, given the intracellular location of their target antigen.4 In such cases, the syndrome is more accurately described as autoantibody-associated rather than autoantibody-mediated. Only in a minority of cases have neuronal autoantibodies been demonstrated to be directly pathogenic, ie, antimetabotropic glutamate receptor type 1 (anti-mGluR1) antibody-associated cerebellitis26 and antiglutamic acid decarboxylase (anti-GAD)-associated cerebellar ataxia.27
Differential diagnosis of cerebellar syndromes
The differential diagnosis of autoantibody-associated cerebellar syndromes is broad and includes:
- Alcohol-induced atrophy
- Drug-induced cerebellar atrophy (eg, from lithium, phenytoin, gabapentin, metronidazole, amiodarone, carbamazepine)
- Vitamin B1 and E deficiency
- Hypothyroidism, hypoparathyroidism
- Neurodegenerative disease (eg, prion disease, multiple system atrophy)
- Parainfectious causes (eg, after infection with Epstein-Barr virus)
- Immune-mediated diseases (Miller-Fisher syndrome, associated with anti-GQ1b antibodies, and antigliadin-associated ataxia, which can occur in isolation or as part of celiac disease).4
SYNDROMES ASSOCIATED WITH SPECIFIC ANTIBODIES
A few of the autoantibody-mediated encephalitic syndromes have specific antibody associations and characteristic clinical presentations. The most prominent of these syndromes are VGKC complex antibody encephalitis (as in the patient described at the beginning of this article) and anti-NMDA receptor encephalitis.
VGKC COMPLEX ANTIBODY-MEDIATED LIMBIC ENCEPHALITIS
VGKC complex antibodies, initially reported to be associated with the peripheral nerve hyperexcitability disorder neuromyotonia, were subsequently found in Morvan syndrome.28,29 Patients with this syndrome often present with autonomic dysfunction and peripheral nerve hyperexcitability but also develop insomnia, confusion, hallucinations, and memory loss. Drawing on the clinical overlap between Morvan syndrome and limbic encephalitis, Buckley et al30 were the first to report VGKC complex antibodies in two cases of limbic encephalitis.
VGKC complex antibodies are now understood to be associated with a wide variety of neurologic conditions, including chronic idiopathic pain, epilepsy,31 movement disorders, cranial nerve abnormalities, autonomic dysfunction,32 and gut dysmotility.33 In contrast, these antibodies are rare in healthy people.34 Limbic encephalitis associated with VGKC complex antibody usually lacks cerebellar and brainstem dysfunction, which may help distinguish it from other types of autoantibody-mediated limbic encephalitis.12
VGKC complex antibody does not bind to the potassium channel itself. Instead it recognizes other constituents of the channel complex, most notably LGI1 and contactin-associated protein 2 (CASPR2). LGI1 antibody is more commonly associated with limbic encephalitis—as illustrated in our case study—in addition to a distinctive type of seizure affecting the arm and face (faciobrachial dystonic seizure).34 The CASPR2 antibody, on the other hand, more often correlates with peripheral nerve manifestations and Morvan syndrome.29 Hyponatremia is commonly seen on serum chemical analysis and provides a clue that these syndromes are present.12
Good response to immunotherapy
A critical change in therapy came as clinicians realized that seizures were often refractory to standard antiepileptic drugs but responded well to immunotherapies. On the basis of these observations, sera of patients with long-standing epilepsy have been reanalyzed to look for neuronal autoantibodies.31 These antibodies should be checked in cases of new-onset refractory status epilepticus of unknown origin that does not respond to antiepileptic medications.
About half of patients with VGKC complex antibody-mediated limbic encephalitis have normal findings on brain MRI.5 Seven of 10 patients who were prospectively followed for VGKC complex antibody-mediated faciobrachial dystonic seizures had normal brain MRIs.35
VGKC complex antibody-mediated limbic encephalitis does not usually recur.36 Most cases are nonparaneoplastic, as evidenced by failure to detect a single active tumor in 64 patients after a median follow-up of 3 years. The prognosis is generally favorable except in cases with coexisting tumors.12
ANTI-NMDA RECEPTOR ENCEPHALITIS
Often associated with ovarian teratoma
Anti-NMDA receptor encephalitis typically affects women in their 20s and 30s, and about half of patients have an ovarian teratoma. It can also occur in younger patients and in men, in whom it is less likely to be associated with a neoplasm.37
Typical initial symptoms include striking and often stereotyped neuropsychiatric disturbances manifesting as psychosis, confusion, seizures, and amnesia. After 1 to 2 weeks, new symptoms set in, including reduced consciousness, movement disorders (ranging from orolingualfacial dyskinesia to rigidity and choreoathetosis), autonomic dysfunction, and hypoventilation, often prompting admission to the intensive care unit.38
Although the outcome is favorable in most cases, recovery, in contrast to VGKC complex antibody-mediated limbic encephalitis, is slow and may take longer than 1 year. Up to a quarter of patients have a relapse, underscoring the importance of maintenance immunotherapy.
It is important to undertake an intensive search for possible ovarian and extraovarian teratomas in young women with this syndrome—including CT of the pelvis, vaginal ultrasonography, and PET imaging—as removal of the teratoma may be curative.37
DIAGNOSIS OF AUTOANTIBODY-MEDIATED ENCEPHALITIS
Critical to diagnosing autoantibody-mediated encephalitis is awareness of these disorders. Since antibody testing may be very specific and is not usually part of the standard batteries of tests, a high level of suspicion is needed. Patients may present to different specialists in different settings; therefore, clinicians in pediatrics, rheumatology, psychiatry, and intensive care medicine need to be aware of these syndromes to avoid delay and misdiagnosis.
Clinical features suggesting autoantibody-mediated encephalitis include:
- Acute or subacute onset of a neurologic syndrome
- New-onset refractory status epilepticus of unknown etiology
- Acute or subacute psychiatric illness with unexpected progression to neurologic symptoms or delirium
- Unusual movement disorders not conforming to standard syndromes
- Cognitive impairment, psychosis, or behavioral or language disorders with atypical findings on imaging or cerebrospinal fluid analysis.
Imaging. Diagnosis of autoantibody-mediated encephalitis focuses on evidence suggesting an inflammatory central nervous system syndrome. MRI may show hyperintense signals on T2, FLAIR, or diffusion-weighted imaging changes in various brain regions. In many cases, however, MRI is negative despite severe clinical symptoms. In a study of 72 patients suspected of having autoimmune dementia of various etiologies, including but not restricted to antineuronal surface antibody-mediated causes, Flanagan et al39 identified atypical neuroimaging findings in only 29%. PET imaging may show hypermetabolism in certain brain areas correlating to clinical syndromes but is often difficult to obtain in a timely fashion.
Cerebrospinal fluid is often abnormal, showing elevated protein, increased immunoglobulin G synthesis, or oligoclonal banding. As with imaging studies, the cerebrospinal fluid may be normal despite severe clinical manifestations.
Electroencephalography may show focal slowing or seizure activity. Neuropsychologic testing may show different patterns of abnormalities.
Antibody testing. None of these tests can be used in isolation, and the diagnosis of autoantibody-mediated encephalitis hinges on recognizing a clinical syndrome and ordering supportive testing. Specific antibodies are more likely in different clinical syndromes and should be sought (Table 3).
Patients who have autoantibody-mediated encephalitis may test negative for autoantibodies for many possible reasons:
- Blood testing for antibodies may be less sensitive than cerebrospinal fluid testing
- Antibody titers may vary in the course of the disease
- The patient may be expressing an antibody that is less often tested for (eg, anti-AMPA receptor or antigamma-aminobutyric acid B) or one that has not yet been isolated.
Evaluating for malignancy is recommended in all cases of autoantibody-mediated encephalitis. The initial workup may involve CT of the chest, abdomen, and pelvis, as well as mammography in women and serum prostate-specific antigen testing and testicular ultrasonography in men. Ordering FDG-PET in cases in which CT is negative or inconclusive increases cancer detection.40 If no cancer is found, close tumor surveillance—every 3 to 6 months—is recommended for at least 2 years.41
TREATMENT
Owing in large part to the rarity of autoantibody-mediated encephalitides, no randomized trials of therapy have been performed. Treatment at present is guided mostly by case series and expert consensus, which suggest first-line therapy with intravenous immunoglobulin, high-dose corticosteroids, plasmapheresis, or a combination.
Different syndromes and antibody-related disorders respond differently to therapy. Syndromes associated with antibodies against intracellular antigens tend to be more resistant to immune therapy than cell surface antigen-related syndromes.4
Tiered approach
Combined treatment with intravenous immunoglobulin and high-dose corticosteroids may be superior to treatment with steroids alone for LGI1-antibody mediated limbic encephalitis.42
In cases refractory to first-line (“tier 1”) therapy, second-line immunotherapy with drugs affecting B-cell populations (eg, rituximab, cyclophosphamide, and mycophenolate mofetil) has been used.
A tiered approach has been most extensively studied for anti-NMDA-receptor encephalitis, with better outcomes found using second-line therapy.43
Treatment strategies for these disorders will likely evolve over time with additional experience.
Outpatient management
Once the patient is discharged from the hospital, a multidisciplinary approach to care is recommended, including physical rehabilitation, speech therapy, neuropsychiatric and neuroimmunologic follow-up, and annual surveillance for malignancies.
- Brierley JB, Corsellis JAN, Hierons R, Nevin S. Subacute encephalitis of later adult life mainly affecting the limbic areas. Brain 1960; 83:357–368.
- Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain 1968; 91:481–496.
- Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol 1983; 14:609–613.
- Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn) 2012; 18:366–383.
- Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 2014; 76:168–184.
- Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61:25–36.
- Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010; 133:2734–2748.
- Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9:67–76.
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65:424–434.
- Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012; 83:638–645.
- Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 2005; 128:1764–1777.
- Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127:701–712.
- Jammoul A, Lederman RJ, Tavee J, Li Y. Presence of voltage-gated potassium channel complex antibody in a case of genetic prion disease. BMJ Case Rep 2014; pii:bcr2013201622.
- Angus-Leppan H, Rudge P, Mead S, Collinge J, Vincent A. Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 2013; 70:919–922.
- Fujita K, Yuasa T, Watanabe O, et al. Voltage-gated potassium channel complex antibodies in Creutzfeldt-Jakob disease. J Neurol 2012; 259:2249–2250.
- Fujita K, Yuasa T, Takahashi Y, et al. Antibodies to N-methyl-D-aspartate glutamate receptors in Creutzfeldt–Jakob disease patients. J Neuroimmunol 2012; 251:90–93.
- Mackay G, Ahmad K, Stone J, et al. NMDA receptor autoantibodies in sporadic Creutzfeldt-Jakob disease. J Neurol 2012; 259:1979–1981.
- Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus–1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology 2013; 81:1637–1639.
- Desena A, Graves D, Warnack W, Greenberg BM. Herpes simplex encephalitis as a potential cause of anti-N-methyl-D-aspartate receptor antibody encephalitis: report of 2 cases. JAMA Neurol 2014; 71:344–346.
- Armangue T, Leypoldt F, Málaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014; 75:317–323.
- Blaes F. Paraneoplastic brain stem encephalitis. Curr Treat Options Neurol 2013; 15:201–209.
- Wildemann B, Jarius S. The expanding range of autoimmune disorders of the nervous system. Lancet Neurol 2013; 12:22–24.
- Kim W, Kim SH, Lee SH, Li XF, Kim HJ. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler 2011; 17:1107–1112.
- Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013; 84:576–583.
- Jubelt B, Mihai C, Li MT, Veerapaneni P. Rhombencephalitis/brainstem encephalitis. Curr Neurol Neurosci Rep 2011; 11:543–552.
- Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000; 342:21–27.
- Ishida K, Mitoma H, Son SY, et al. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann Neurol 1999; 46:263–267.
- Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997; 41:238–246.
- Barber P, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage-gated K+ channel antibodies. Neurology 2000; 54:771–772.
- Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50:73–78.
- Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res 2006; 71:135–141.
- Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008; 70:1883–1890.
- Knowles CH, Lang B, Clover L, et al. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol 2002; 37:166–170.
- Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011; 69:892–900.
- Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013: 136:3151–3162.
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 2011; 10:759–772.
- Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011; 10:63–74.
- Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133:1655–1667.
- Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010; 85:881–897.
- Younes-Mhenni S, Janier MF, Cinotti L, et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain 2004; 127:2331–2338.
- Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011; 77:179–189.
- Shin YW, Lee ST, Shin JW, et al. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 2013; 265:75–81.
- Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013; 12:157–165.
A 79-year-old woman with a history of breast cancer in remission and hypertension presented to a local emergency department because of subacute memory loss and compulsive shopping. Her serum sodium concentration was 127 mmol/L (reference range 132–148). Computed tomography (CT) and magnetic resonance imaging (MRI) of the brain were normal, and she was sent home.
Three days later, she experienced a generalized tonic-clonic seizure that evolved into status epilepticus. She was intubated and admitted to the intensive care unit. Cerebrospinal fluid analysis was normal, and infectious causes of encephalitis were ruled out. MRI showed increased signal in both hippocampi (Figure 1). Her seizures were refractory to treatment, and she was given pentobarbital to induce a coma.
Serum evaluation of neuronal antibodies revealed elevated titers of the voltage-gated potassium channel (VGKC) complex antibody, with subsequent subtyping confirming the leucine-rich glioma-inactivated protein 1 (LGI1) protein as the antigenic target.
She received a 5-day course of intravenous immunoglobulin and methylprednisolone, pentobarbital was withdrawn, and the seizures did not recur, but weeks later she remained comatose. Positron emission tomography (PET) of the brain revealed hypermetabolism in the medial and anterior aspects of both temporal lobes. She underwent five sessions of plasma exchange, after which she began to improve and follow commands. She was ultimately discharged to an acute rehabilitation facility after a 4-week hospital stay.
She received infusions of intravenous immunoglobulin twice a month for 6 months. At her last follow-up visit, she was seizure-free and neurologically intact except for mild inattention.
NEWLY RECOGNIZED DISEASES
Although autoantibody-mediated encephalitic syndromes were first described more than 50 years ago,1,2 their autoimmune basis was not recognized until the early 1980s.3 In the past 10 years, a flood of novel clinical syndromes associated with neuronal autoantibodies has been described that may be markedly improved or even completely resolved with immunotherapy. In cases of unexplained seizure, encephalitis, or acute-onset psychiatric syndromes, suspecting these syndromes can lead to diagnosis, treatment, and a good outcome.
This review describes the key clinical autoantibody-mediated encephalitic syndromes, explains the better-characterized antibody associations, and discusses their diagnosis and treatment.
CLASSIFIED ANATOMICALLY, IMMUNOLOGICALLY, OR EPONYMOUSLY
Autoantibody-mediated encephalitis is also known as autoimmune-mediated encephalitis, autoimmune-mediated limbic encephalitis, and autoimmune synaptic encephalitis.
How to categorize these syndromes is still in flux: they can be listed by the area of the brain affected, the antibody involved, or the name of the discoverer (eg, Morvan syndrome).
Autoantibodies identified in autoimmune encephalitis fall under two broad categories:
- Those targeting intracellular (intranuclear or intracytoplasmic) antigens; the syndromes they cause are more likely to be paraneoplastic and less responsive to immunotherapy
- Those targeting antigens on the neuronal surface: the syndromes they cause are less likely to be paraneoplastic and are more responsive to immunotherapy.4
SYNDROMES DEFINED BY BRAIN AREA AFFECTED
Below, we provide examples of neurologic syndromes of autoantibody-mediated encephalitis according to the region of the brain most affected, ie, the limbic system, the brainstem, or the cerebellum (Figure 2).
LIMBIC ENCEPHALITIS
Memory loss, behavioral changes, seizures
Patients with limbic encephalitis (such as the patient described in the vignette above) present with symptoms attributed to dysfunction of mesial temporal lobe structures, most notably the hippocampus. Prominent symptoms include short-term memory loss, behavioral disturbances such as agitation and confusion, and psychiatric problems such as depression and psychosis. Recurrent seizures are a salient feature and, not uncommonly, progress to status epilepticus.
Antibodies are not all cancer-associated
Cerebrospinal fluid analysis can be normal or show abnormalities suggesting immune activation, eg, slight pleocytosis, elevated protein, increased immunoglobulin G synthesis, and oligoclonal banding.5
In many cases, an autoantibody is found in the blood or in the cerebrospinal fluid. Some patients may express more than one autoantibody, so the traditional view of “one antibody, one syndrome” is incorrect.
Although initially identified as a rare paraneoplastic disorder, limbic encephalitis sometimes occurs in the absence of malignancy.
Multiple antibodies have been linked to the syndrome (Table 1).6–9 The “classic” antibodies initially found in paraneoplastic forms are now generally viewed as nonpathogenic, in part because they are directed against intracellular antigens. Neuronal injury in paraneoplastic limbic encephalitis is believed to be mediated by cytotoxic T lymphocytes, with neuronal autoantibodies being produced after the injury.4 Recently defined antibodies, such as those targeting the N-methyl-d-aspartate (NMDA) receptor6 and the LGI1 protein,7 are now understood to be common causes of limbic encephalitis.
Imaging usually shows limbic focal changes
Structural MRI or functional fluorodeoxyglucose (FDG)-PET imaging may show focal changes in limbic system structures, such as the mesial temporal lobes. It is now recognized that other cortical areas may be involved, and the term “limbic encephalitis” may give way to “cortical” or “focal encephalitis.”
In about 60% of patients, MRI shows hyperintense fluid-attenuated inversion recovery (FLAIR) or T2 signal changes in the mesial temporal lobes, likely reflecting inflammatory changes.4,10,11 On FDG-PET, hypermetabolism may be observed in the mesial temporal lobes early in the disease despite normal findings on MRI.12 Hypometabolism, either diffuse or localized to the mesial temporal lobes, eventually sets in, likely reflecting cytotoxic injury in the aftermath of prolonged inflammation or seizures.
Consider other causes
Before diagnosing limbic encephalitis, it is essential to evaluate for infectious meningoencephalitis, especially herpes simplex viral encephalitis. Thiamine deficiency (Wernicke encephalopathy), drug intoxication, prion disease, Hashimoto encephalopathy, tumor, and subclinical status epilepticus should also be considered. Some of these conditions are associated with the same neuronal autoantibodies detected in limbic encephalitis. Further complicating the picture, case reports have shown the presence of serum neuronal autoantibodies—VGKC complex13–15 and NMDA-receptor antibodies16,17—in confirmed cases of prion disease. In addition, adequately treated herpes simplex viral encephalitis can precipitate the production of NMDA-receptor antibodies and their characteristic syndrome.18–20
BRAINSTEM ENCEPHALITIS
The brainstem—the midbrain, pons, and medulla—can be affected, either in isolation or more commonly as part of a more widespread autoantibody-mediated encephalitis. Symptoms and signs include eye movement abnormalities, ptosis, dysphagia, dysarthria, ataxia, facial palsy, vertigo, hearing impairment, reduced consciousness, and hypoventilation.21
Anti-Hu, anti-Ri, and anti-Ma2 antibodies are most commonly associated with brainstem encephalitis (Table 2). Anti-Ma2-associated encephalitis may improve after a combination of immunotherapy and tumor removal21; the others have a poor prognosis.
Neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders most commonly involve demyelination affecting the optic nerves and spinal cord, leading to unilateral or bilateral optic neuritis and transverse myelitis spanning three or more vertebral segments.22 The initial clinical manifestation may be an encephalitic pattern, affecting predominantly the brainstem in a restricted fashion,22 or the central nervous system in a more diffuse pattern, mimicking either acute disseminated encephalomyelitis or, in less severe cases, posterior reversible encephalopathy syndrome.23
Testing for antiaquaporin-4 antibody, also known as neuromyelitis optica immunoglobulin G, is the single most decisive laboratory test for diagnosing neuromyelitis optica spectrum disorders, so serum and cerebrospinal fluid evaluation for this autoantibody should be considered when caring for a patient whose clinical picture suggests brainstem encephalitis.22
Bickerstaff brainstem encephalitis
Bickerstaff brainstem encephalitis was first described more than half a century ago in patients with postinfectious ataxia, ophthalmoparesis, and altered consciousness. This rare disease was later found to be associated with antiganglioside GQ1b (anti-GQ1b) autoantibody. MRI is normal in about 90% of cases, so recognizing the clinical presentation and analyzing anti-GQ1b serum titers are critical to diagnosis.
Recovery is usually spontaneous and complete and can be hastened by immunotherapy, especially intravenous immunoglobulin.24
Other causes of brainstem encephalitis
The differential diagnosis of a presentation of brainstem encephalitis includes:
- Infectious causes, the most common being Listeria species followed by enterovirus 71 and herpes simplex virus.25 Tuberculosis, brucellosis, and Whipple disease should also be considered.
- Primary central nervous system inflammatory and demyelinating conditions, eg, multiple sclerosis and acute disseminated encephalomyelitis.
- Systemic inflammatory conditions, eg, Behçet disease, systemic lupus erythematosus, and sarcoidosis.
- Direct brainstem neoplastic involvement, as might occur in primary central nervous system lymphoma or leptomeningeal carcinomatosis.
CEREBELLAR SYNDROME
Patients with autoantibody-mediated encephalitis localized predominantly to the cerebellum typically present with dizziness, vertigo, and unsteady gait, progressing eventually to limb and gait ataxia.4 Symptoms are often subacute, progressing over weeks.
Multiple neuronal autoantibodies have been found to occur with cerebellar encephalitis (Table 2). In most cases, they are paraneoplastic and considered not to be pathogenic, given the intracellular location of their target antigen.4 In such cases, the syndrome is more accurately described as autoantibody-associated rather than autoantibody-mediated. Only in a minority of cases have neuronal autoantibodies been demonstrated to be directly pathogenic, ie, antimetabotropic glutamate receptor type 1 (anti-mGluR1) antibody-associated cerebellitis26 and antiglutamic acid decarboxylase (anti-GAD)-associated cerebellar ataxia.27
Differential diagnosis of cerebellar syndromes
The differential diagnosis of autoantibody-associated cerebellar syndromes is broad and includes:
- Alcohol-induced atrophy
- Drug-induced cerebellar atrophy (eg, from lithium, phenytoin, gabapentin, metronidazole, amiodarone, carbamazepine)
- Vitamin B1 and E deficiency
- Hypothyroidism, hypoparathyroidism
- Neurodegenerative disease (eg, prion disease, multiple system atrophy)
- Parainfectious causes (eg, after infection with Epstein-Barr virus)
- Immune-mediated diseases (Miller-Fisher syndrome, associated with anti-GQ1b antibodies, and antigliadin-associated ataxia, which can occur in isolation or as part of celiac disease).4
SYNDROMES ASSOCIATED WITH SPECIFIC ANTIBODIES
A few of the autoantibody-mediated encephalitic syndromes have specific antibody associations and characteristic clinical presentations. The most prominent of these syndromes are VGKC complex antibody encephalitis (as in the patient described at the beginning of this article) and anti-NMDA receptor encephalitis.
VGKC COMPLEX ANTIBODY-MEDIATED LIMBIC ENCEPHALITIS
VGKC complex antibodies, initially reported to be associated with the peripheral nerve hyperexcitability disorder neuromyotonia, were subsequently found in Morvan syndrome.28,29 Patients with this syndrome often present with autonomic dysfunction and peripheral nerve hyperexcitability but also develop insomnia, confusion, hallucinations, and memory loss. Drawing on the clinical overlap between Morvan syndrome and limbic encephalitis, Buckley et al30 were the first to report VGKC complex antibodies in two cases of limbic encephalitis.
VGKC complex antibodies are now understood to be associated with a wide variety of neurologic conditions, including chronic idiopathic pain, epilepsy,31 movement disorders, cranial nerve abnormalities, autonomic dysfunction,32 and gut dysmotility.33 In contrast, these antibodies are rare in healthy people.34 Limbic encephalitis associated with VGKC complex antibody usually lacks cerebellar and brainstem dysfunction, which may help distinguish it from other types of autoantibody-mediated limbic encephalitis.12
VGKC complex antibody does not bind to the potassium channel itself. Instead it recognizes other constituents of the channel complex, most notably LGI1 and contactin-associated protein 2 (CASPR2). LGI1 antibody is more commonly associated with limbic encephalitis—as illustrated in our case study—in addition to a distinctive type of seizure affecting the arm and face (faciobrachial dystonic seizure).34 The CASPR2 antibody, on the other hand, more often correlates with peripheral nerve manifestations and Morvan syndrome.29 Hyponatremia is commonly seen on serum chemical analysis and provides a clue that these syndromes are present.12
Good response to immunotherapy
A critical change in therapy came as clinicians realized that seizures were often refractory to standard antiepileptic drugs but responded well to immunotherapies. On the basis of these observations, sera of patients with long-standing epilepsy have been reanalyzed to look for neuronal autoantibodies.31 These antibodies should be checked in cases of new-onset refractory status epilepticus of unknown origin that does not respond to antiepileptic medications.
About half of patients with VGKC complex antibody-mediated limbic encephalitis have normal findings on brain MRI.5 Seven of 10 patients who were prospectively followed for VGKC complex antibody-mediated faciobrachial dystonic seizures had normal brain MRIs.35
VGKC complex antibody-mediated limbic encephalitis does not usually recur.36 Most cases are nonparaneoplastic, as evidenced by failure to detect a single active tumor in 64 patients after a median follow-up of 3 years. The prognosis is generally favorable except in cases with coexisting tumors.12
ANTI-NMDA RECEPTOR ENCEPHALITIS
Often associated with ovarian teratoma
Anti-NMDA receptor encephalitis typically affects women in their 20s and 30s, and about half of patients have an ovarian teratoma. It can also occur in younger patients and in men, in whom it is less likely to be associated with a neoplasm.37
Typical initial symptoms include striking and often stereotyped neuropsychiatric disturbances manifesting as psychosis, confusion, seizures, and amnesia. After 1 to 2 weeks, new symptoms set in, including reduced consciousness, movement disorders (ranging from orolingualfacial dyskinesia to rigidity and choreoathetosis), autonomic dysfunction, and hypoventilation, often prompting admission to the intensive care unit.38
Although the outcome is favorable in most cases, recovery, in contrast to VGKC complex antibody-mediated limbic encephalitis, is slow and may take longer than 1 year. Up to a quarter of patients have a relapse, underscoring the importance of maintenance immunotherapy.
It is important to undertake an intensive search for possible ovarian and extraovarian teratomas in young women with this syndrome—including CT of the pelvis, vaginal ultrasonography, and PET imaging—as removal of the teratoma may be curative.37
DIAGNOSIS OF AUTOANTIBODY-MEDIATED ENCEPHALITIS
Critical to diagnosing autoantibody-mediated encephalitis is awareness of these disorders. Since antibody testing may be very specific and is not usually part of the standard batteries of tests, a high level of suspicion is needed. Patients may present to different specialists in different settings; therefore, clinicians in pediatrics, rheumatology, psychiatry, and intensive care medicine need to be aware of these syndromes to avoid delay and misdiagnosis.
Clinical features suggesting autoantibody-mediated encephalitis include:
- Acute or subacute onset of a neurologic syndrome
- New-onset refractory status epilepticus of unknown etiology
- Acute or subacute psychiatric illness with unexpected progression to neurologic symptoms or delirium
- Unusual movement disorders not conforming to standard syndromes
- Cognitive impairment, psychosis, or behavioral or language disorders with atypical findings on imaging or cerebrospinal fluid analysis.
Imaging. Diagnosis of autoantibody-mediated encephalitis focuses on evidence suggesting an inflammatory central nervous system syndrome. MRI may show hyperintense signals on T2, FLAIR, or diffusion-weighted imaging changes in various brain regions. In many cases, however, MRI is negative despite severe clinical symptoms. In a study of 72 patients suspected of having autoimmune dementia of various etiologies, including but not restricted to antineuronal surface antibody-mediated causes, Flanagan et al39 identified atypical neuroimaging findings in only 29%. PET imaging may show hypermetabolism in certain brain areas correlating to clinical syndromes but is often difficult to obtain in a timely fashion.
Cerebrospinal fluid is often abnormal, showing elevated protein, increased immunoglobulin G synthesis, or oligoclonal banding. As with imaging studies, the cerebrospinal fluid may be normal despite severe clinical manifestations.
Electroencephalography may show focal slowing or seizure activity. Neuropsychologic testing may show different patterns of abnormalities.
Antibody testing. None of these tests can be used in isolation, and the diagnosis of autoantibody-mediated encephalitis hinges on recognizing a clinical syndrome and ordering supportive testing. Specific antibodies are more likely in different clinical syndromes and should be sought (Table 3).
Patients who have autoantibody-mediated encephalitis may test negative for autoantibodies for many possible reasons:
- Blood testing for antibodies may be less sensitive than cerebrospinal fluid testing
- Antibody titers may vary in the course of the disease
- The patient may be expressing an antibody that is less often tested for (eg, anti-AMPA receptor or antigamma-aminobutyric acid B) or one that has not yet been isolated.
Evaluating for malignancy is recommended in all cases of autoantibody-mediated encephalitis. The initial workup may involve CT of the chest, abdomen, and pelvis, as well as mammography in women and serum prostate-specific antigen testing and testicular ultrasonography in men. Ordering FDG-PET in cases in which CT is negative or inconclusive increases cancer detection.40 If no cancer is found, close tumor surveillance—every 3 to 6 months—is recommended for at least 2 years.41
TREATMENT
Owing in large part to the rarity of autoantibody-mediated encephalitides, no randomized trials of therapy have been performed. Treatment at present is guided mostly by case series and expert consensus, which suggest first-line therapy with intravenous immunoglobulin, high-dose corticosteroids, plasmapheresis, or a combination.
Different syndromes and antibody-related disorders respond differently to therapy. Syndromes associated with antibodies against intracellular antigens tend to be more resistant to immune therapy than cell surface antigen-related syndromes.4
Tiered approach
Combined treatment with intravenous immunoglobulin and high-dose corticosteroids may be superior to treatment with steroids alone for LGI1-antibody mediated limbic encephalitis.42
In cases refractory to first-line (“tier 1”) therapy, second-line immunotherapy with drugs affecting B-cell populations (eg, rituximab, cyclophosphamide, and mycophenolate mofetil) has been used.
A tiered approach has been most extensively studied for anti-NMDA-receptor encephalitis, with better outcomes found using second-line therapy.43
Treatment strategies for these disorders will likely evolve over time with additional experience.
Outpatient management
Once the patient is discharged from the hospital, a multidisciplinary approach to care is recommended, including physical rehabilitation, speech therapy, neuropsychiatric and neuroimmunologic follow-up, and annual surveillance for malignancies.
A 79-year-old woman with a history of breast cancer in remission and hypertension presented to a local emergency department because of subacute memory loss and compulsive shopping. Her serum sodium concentration was 127 mmol/L (reference range 132–148). Computed tomography (CT) and magnetic resonance imaging (MRI) of the brain were normal, and she was sent home.
Three days later, she experienced a generalized tonic-clonic seizure that evolved into status epilepticus. She was intubated and admitted to the intensive care unit. Cerebrospinal fluid analysis was normal, and infectious causes of encephalitis were ruled out. MRI showed increased signal in both hippocampi (Figure 1). Her seizures were refractory to treatment, and she was given pentobarbital to induce a coma.
Serum evaluation of neuronal antibodies revealed elevated titers of the voltage-gated potassium channel (VGKC) complex antibody, with subsequent subtyping confirming the leucine-rich glioma-inactivated protein 1 (LGI1) protein as the antigenic target.
She received a 5-day course of intravenous immunoglobulin and methylprednisolone, pentobarbital was withdrawn, and the seizures did not recur, but weeks later she remained comatose. Positron emission tomography (PET) of the brain revealed hypermetabolism in the medial and anterior aspects of both temporal lobes. She underwent five sessions of plasma exchange, after which she began to improve and follow commands. She was ultimately discharged to an acute rehabilitation facility after a 4-week hospital stay.
She received infusions of intravenous immunoglobulin twice a month for 6 months. At her last follow-up visit, she was seizure-free and neurologically intact except for mild inattention.
NEWLY RECOGNIZED DISEASES
Although autoantibody-mediated encephalitic syndromes were first described more than 50 years ago,1,2 their autoimmune basis was not recognized until the early 1980s.3 In the past 10 years, a flood of novel clinical syndromes associated with neuronal autoantibodies has been described that may be markedly improved or even completely resolved with immunotherapy. In cases of unexplained seizure, encephalitis, or acute-onset psychiatric syndromes, suspecting these syndromes can lead to diagnosis, treatment, and a good outcome.
This review describes the key clinical autoantibody-mediated encephalitic syndromes, explains the better-characterized antibody associations, and discusses their diagnosis and treatment.
CLASSIFIED ANATOMICALLY, IMMUNOLOGICALLY, OR EPONYMOUSLY
Autoantibody-mediated encephalitis is also known as autoimmune-mediated encephalitis, autoimmune-mediated limbic encephalitis, and autoimmune synaptic encephalitis.
How to categorize these syndromes is still in flux: they can be listed by the area of the brain affected, the antibody involved, or the name of the discoverer (eg, Morvan syndrome).
Autoantibodies identified in autoimmune encephalitis fall under two broad categories:
- Those targeting intracellular (intranuclear or intracytoplasmic) antigens; the syndromes they cause are more likely to be paraneoplastic and less responsive to immunotherapy
- Those targeting antigens on the neuronal surface: the syndromes they cause are less likely to be paraneoplastic and are more responsive to immunotherapy.4
SYNDROMES DEFINED BY BRAIN AREA AFFECTED
Below, we provide examples of neurologic syndromes of autoantibody-mediated encephalitis according to the region of the brain most affected, ie, the limbic system, the brainstem, or the cerebellum (Figure 2).
LIMBIC ENCEPHALITIS
Memory loss, behavioral changes, seizures
Patients with limbic encephalitis (such as the patient described in the vignette above) present with symptoms attributed to dysfunction of mesial temporal lobe structures, most notably the hippocampus. Prominent symptoms include short-term memory loss, behavioral disturbances such as agitation and confusion, and psychiatric problems such as depression and psychosis. Recurrent seizures are a salient feature and, not uncommonly, progress to status epilepticus.
Antibodies are not all cancer-associated
Cerebrospinal fluid analysis can be normal or show abnormalities suggesting immune activation, eg, slight pleocytosis, elevated protein, increased immunoglobulin G synthesis, and oligoclonal banding.5
In many cases, an autoantibody is found in the blood or in the cerebrospinal fluid. Some patients may express more than one autoantibody, so the traditional view of “one antibody, one syndrome” is incorrect.
Although initially identified as a rare paraneoplastic disorder, limbic encephalitis sometimes occurs in the absence of malignancy.
Multiple antibodies have been linked to the syndrome (Table 1).6–9 The “classic” antibodies initially found in paraneoplastic forms are now generally viewed as nonpathogenic, in part because they are directed against intracellular antigens. Neuronal injury in paraneoplastic limbic encephalitis is believed to be mediated by cytotoxic T lymphocytes, with neuronal autoantibodies being produced after the injury.4 Recently defined antibodies, such as those targeting the N-methyl-d-aspartate (NMDA) receptor6 and the LGI1 protein,7 are now understood to be common causes of limbic encephalitis.
Imaging usually shows limbic focal changes
Structural MRI or functional fluorodeoxyglucose (FDG)-PET imaging may show focal changes in limbic system structures, such as the mesial temporal lobes. It is now recognized that other cortical areas may be involved, and the term “limbic encephalitis” may give way to “cortical” or “focal encephalitis.”
In about 60% of patients, MRI shows hyperintense fluid-attenuated inversion recovery (FLAIR) or T2 signal changes in the mesial temporal lobes, likely reflecting inflammatory changes.4,10,11 On FDG-PET, hypermetabolism may be observed in the mesial temporal lobes early in the disease despite normal findings on MRI.12 Hypometabolism, either diffuse or localized to the mesial temporal lobes, eventually sets in, likely reflecting cytotoxic injury in the aftermath of prolonged inflammation or seizures.
Consider other causes
Before diagnosing limbic encephalitis, it is essential to evaluate for infectious meningoencephalitis, especially herpes simplex viral encephalitis. Thiamine deficiency (Wernicke encephalopathy), drug intoxication, prion disease, Hashimoto encephalopathy, tumor, and subclinical status epilepticus should also be considered. Some of these conditions are associated with the same neuronal autoantibodies detected in limbic encephalitis. Further complicating the picture, case reports have shown the presence of serum neuronal autoantibodies—VGKC complex13–15 and NMDA-receptor antibodies16,17—in confirmed cases of prion disease. In addition, adequately treated herpes simplex viral encephalitis can precipitate the production of NMDA-receptor antibodies and their characteristic syndrome.18–20
BRAINSTEM ENCEPHALITIS
The brainstem—the midbrain, pons, and medulla—can be affected, either in isolation or more commonly as part of a more widespread autoantibody-mediated encephalitis. Symptoms and signs include eye movement abnormalities, ptosis, dysphagia, dysarthria, ataxia, facial palsy, vertigo, hearing impairment, reduced consciousness, and hypoventilation.21
Anti-Hu, anti-Ri, and anti-Ma2 antibodies are most commonly associated with brainstem encephalitis (Table 2). Anti-Ma2-associated encephalitis may improve after a combination of immunotherapy and tumor removal21; the others have a poor prognosis.
Neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders most commonly involve demyelination affecting the optic nerves and spinal cord, leading to unilateral or bilateral optic neuritis and transverse myelitis spanning three or more vertebral segments.22 The initial clinical manifestation may be an encephalitic pattern, affecting predominantly the brainstem in a restricted fashion,22 or the central nervous system in a more diffuse pattern, mimicking either acute disseminated encephalomyelitis or, in less severe cases, posterior reversible encephalopathy syndrome.23
Testing for antiaquaporin-4 antibody, also known as neuromyelitis optica immunoglobulin G, is the single most decisive laboratory test for diagnosing neuromyelitis optica spectrum disorders, so serum and cerebrospinal fluid evaluation for this autoantibody should be considered when caring for a patient whose clinical picture suggests brainstem encephalitis.22
Bickerstaff brainstem encephalitis
Bickerstaff brainstem encephalitis was first described more than half a century ago in patients with postinfectious ataxia, ophthalmoparesis, and altered consciousness. This rare disease was later found to be associated with antiganglioside GQ1b (anti-GQ1b) autoantibody. MRI is normal in about 90% of cases, so recognizing the clinical presentation and analyzing anti-GQ1b serum titers are critical to diagnosis.
Recovery is usually spontaneous and complete and can be hastened by immunotherapy, especially intravenous immunoglobulin.24
Other causes of brainstem encephalitis
The differential diagnosis of a presentation of brainstem encephalitis includes:
- Infectious causes, the most common being Listeria species followed by enterovirus 71 and herpes simplex virus.25 Tuberculosis, brucellosis, and Whipple disease should also be considered.
- Primary central nervous system inflammatory and demyelinating conditions, eg, multiple sclerosis and acute disseminated encephalomyelitis.
- Systemic inflammatory conditions, eg, Behçet disease, systemic lupus erythematosus, and sarcoidosis.
- Direct brainstem neoplastic involvement, as might occur in primary central nervous system lymphoma or leptomeningeal carcinomatosis.
CEREBELLAR SYNDROME
Patients with autoantibody-mediated encephalitis localized predominantly to the cerebellum typically present with dizziness, vertigo, and unsteady gait, progressing eventually to limb and gait ataxia.4 Symptoms are often subacute, progressing over weeks.
Multiple neuronal autoantibodies have been found to occur with cerebellar encephalitis (Table 2). In most cases, they are paraneoplastic and considered not to be pathogenic, given the intracellular location of their target antigen.4 In such cases, the syndrome is more accurately described as autoantibody-associated rather than autoantibody-mediated. Only in a minority of cases have neuronal autoantibodies been demonstrated to be directly pathogenic, ie, antimetabotropic glutamate receptor type 1 (anti-mGluR1) antibody-associated cerebellitis26 and antiglutamic acid decarboxylase (anti-GAD)-associated cerebellar ataxia.27
Differential diagnosis of cerebellar syndromes
The differential diagnosis of autoantibody-associated cerebellar syndromes is broad and includes:
- Alcohol-induced atrophy
- Drug-induced cerebellar atrophy (eg, from lithium, phenytoin, gabapentin, metronidazole, amiodarone, carbamazepine)
- Vitamin B1 and E deficiency
- Hypothyroidism, hypoparathyroidism
- Neurodegenerative disease (eg, prion disease, multiple system atrophy)
- Parainfectious causes (eg, after infection with Epstein-Barr virus)
- Immune-mediated diseases (Miller-Fisher syndrome, associated with anti-GQ1b antibodies, and antigliadin-associated ataxia, which can occur in isolation or as part of celiac disease).4
SYNDROMES ASSOCIATED WITH SPECIFIC ANTIBODIES
A few of the autoantibody-mediated encephalitic syndromes have specific antibody associations and characteristic clinical presentations. The most prominent of these syndromes are VGKC complex antibody encephalitis (as in the patient described at the beginning of this article) and anti-NMDA receptor encephalitis.
VGKC COMPLEX ANTIBODY-MEDIATED LIMBIC ENCEPHALITIS
VGKC complex antibodies, initially reported to be associated with the peripheral nerve hyperexcitability disorder neuromyotonia, were subsequently found in Morvan syndrome.28,29 Patients with this syndrome often present with autonomic dysfunction and peripheral nerve hyperexcitability but also develop insomnia, confusion, hallucinations, and memory loss. Drawing on the clinical overlap between Morvan syndrome and limbic encephalitis, Buckley et al30 were the first to report VGKC complex antibodies in two cases of limbic encephalitis.
VGKC complex antibodies are now understood to be associated with a wide variety of neurologic conditions, including chronic idiopathic pain, epilepsy,31 movement disorders, cranial nerve abnormalities, autonomic dysfunction,32 and gut dysmotility.33 In contrast, these antibodies are rare in healthy people.34 Limbic encephalitis associated with VGKC complex antibody usually lacks cerebellar and brainstem dysfunction, which may help distinguish it from other types of autoantibody-mediated limbic encephalitis.12
VGKC complex antibody does not bind to the potassium channel itself. Instead it recognizes other constituents of the channel complex, most notably LGI1 and contactin-associated protein 2 (CASPR2). LGI1 antibody is more commonly associated with limbic encephalitis—as illustrated in our case study—in addition to a distinctive type of seizure affecting the arm and face (faciobrachial dystonic seizure).34 The CASPR2 antibody, on the other hand, more often correlates with peripheral nerve manifestations and Morvan syndrome.29 Hyponatremia is commonly seen on serum chemical analysis and provides a clue that these syndromes are present.12
Good response to immunotherapy
A critical change in therapy came as clinicians realized that seizures were often refractory to standard antiepileptic drugs but responded well to immunotherapies. On the basis of these observations, sera of patients with long-standing epilepsy have been reanalyzed to look for neuronal autoantibodies.31 These antibodies should be checked in cases of new-onset refractory status epilepticus of unknown origin that does not respond to antiepileptic medications.
About half of patients with VGKC complex antibody-mediated limbic encephalitis have normal findings on brain MRI.5 Seven of 10 patients who were prospectively followed for VGKC complex antibody-mediated faciobrachial dystonic seizures had normal brain MRIs.35
VGKC complex antibody-mediated limbic encephalitis does not usually recur.36 Most cases are nonparaneoplastic, as evidenced by failure to detect a single active tumor in 64 patients after a median follow-up of 3 years. The prognosis is generally favorable except in cases with coexisting tumors.12
ANTI-NMDA RECEPTOR ENCEPHALITIS
Often associated with ovarian teratoma
Anti-NMDA receptor encephalitis typically affects women in their 20s and 30s, and about half of patients have an ovarian teratoma. It can also occur in younger patients and in men, in whom it is less likely to be associated with a neoplasm.37
Typical initial symptoms include striking and often stereotyped neuropsychiatric disturbances manifesting as psychosis, confusion, seizures, and amnesia. After 1 to 2 weeks, new symptoms set in, including reduced consciousness, movement disorders (ranging from orolingualfacial dyskinesia to rigidity and choreoathetosis), autonomic dysfunction, and hypoventilation, often prompting admission to the intensive care unit.38
Although the outcome is favorable in most cases, recovery, in contrast to VGKC complex antibody-mediated limbic encephalitis, is slow and may take longer than 1 year. Up to a quarter of patients have a relapse, underscoring the importance of maintenance immunotherapy.
It is important to undertake an intensive search for possible ovarian and extraovarian teratomas in young women with this syndrome—including CT of the pelvis, vaginal ultrasonography, and PET imaging—as removal of the teratoma may be curative.37
DIAGNOSIS OF AUTOANTIBODY-MEDIATED ENCEPHALITIS
Critical to diagnosing autoantibody-mediated encephalitis is awareness of these disorders. Since antibody testing may be very specific and is not usually part of the standard batteries of tests, a high level of suspicion is needed. Patients may present to different specialists in different settings; therefore, clinicians in pediatrics, rheumatology, psychiatry, and intensive care medicine need to be aware of these syndromes to avoid delay and misdiagnosis.
Clinical features suggesting autoantibody-mediated encephalitis include:
- Acute or subacute onset of a neurologic syndrome
- New-onset refractory status epilepticus of unknown etiology
- Acute or subacute psychiatric illness with unexpected progression to neurologic symptoms or delirium
- Unusual movement disorders not conforming to standard syndromes
- Cognitive impairment, psychosis, or behavioral or language disorders with atypical findings on imaging or cerebrospinal fluid analysis.
Imaging. Diagnosis of autoantibody-mediated encephalitis focuses on evidence suggesting an inflammatory central nervous system syndrome. MRI may show hyperintense signals on T2, FLAIR, or diffusion-weighted imaging changes in various brain regions. In many cases, however, MRI is negative despite severe clinical symptoms. In a study of 72 patients suspected of having autoimmune dementia of various etiologies, including but not restricted to antineuronal surface antibody-mediated causes, Flanagan et al39 identified atypical neuroimaging findings in only 29%. PET imaging may show hypermetabolism in certain brain areas correlating to clinical syndromes but is often difficult to obtain in a timely fashion.
Cerebrospinal fluid is often abnormal, showing elevated protein, increased immunoglobulin G synthesis, or oligoclonal banding. As with imaging studies, the cerebrospinal fluid may be normal despite severe clinical manifestations.
Electroencephalography may show focal slowing or seizure activity. Neuropsychologic testing may show different patterns of abnormalities.
Antibody testing. None of these tests can be used in isolation, and the diagnosis of autoantibody-mediated encephalitis hinges on recognizing a clinical syndrome and ordering supportive testing. Specific antibodies are more likely in different clinical syndromes and should be sought (Table 3).
Patients who have autoantibody-mediated encephalitis may test negative for autoantibodies for many possible reasons:
- Blood testing for antibodies may be less sensitive than cerebrospinal fluid testing
- Antibody titers may vary in the course of the disease
- The patient may be expressing an antibody that is less often tested for (eg, anti-AMPA receptor or antigamma-aminobutyric acid B) or one that has not yet been isolated.
Evaluating for malignancy is recommended in all cases of autoantibody-mediated encephalitis. The initial workup may involve CT of the chest, abdomen, and pelvis, as well as mammography in women and serum prostate-specific antigen testing and testicular ultrasonography in men. Ordering FDG-PET in cases in which CT is negative or inconclusive increases cancer detection.40 If no cancer is found, close tumor surveillance—every 3 to 6 months—is recommended for at least 2 years.41
TREATMENT
Owing in large part to the rarity of autoantibody-mediated encephalitides, no randomized trials of therapy have been performed. Treatment at present is guided mostly by case series and expert consensus, which suggest first-line therapy with intravenous immunoglobulin, high-dose corticosteroids, plasmapheresis, or a combination.
Different syndromes and antibody-related disorders respond differently to therapy. Syndromes associated with antibodies against intracellular antigens tend to be more resistant to immune therapy than cell surface antigen-related syndromes.4
Tiered approach
Combined treatment with intravenous immunoglobulin and high-dose corticosteroids may be superior to treatment with steroids alone for LGI1-antibody mediated limbic encephalitis.42
In cases refractory to first-line (“tier 1”) therapy, second-line immunotherapy with drugs affecting B-cell populations (eg, rituximab, cyclophosphamide, and mycophenolate mofetil) has been used.
A tiered approach has been most extensively studied for anti-NMDA-receptor encephalitis, with better outcomes found using second-line therapy.43
Treatment strategies for these disorders will likely evolve over time with additional experience.
Outpatient management
Once the patient is discharged from the hospital, a multidisciplinary approach to care is recommended, including physical rehabilitation, speech therapy, neuropsychiatric and neuroimmunologic follow-up, and annual surveillance for malignancies.
- Brierley JB, Corsellis JAN, Hierons R, Nevin S. Subacute encephalitis of later adult life mainly affecting the limbic areas. Brain 1960; 83:357–368.
- Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain 1968; 91:481–496.
- Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol 1983; 14:609–613.
- Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn) 2012; 18:366–383.
- Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 2014; 76:168–184.
- Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61:25–36.
- Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010; 133:2734–2748.
- Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9:67–76.
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65:424–434.
- Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012; 83:638–645.
- Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 2005; 128:1764–1777.
- Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127:701–712.
- Jammoul A, Lederman RJ, Tavee J, Li Y. Presence of voltage-gated potassium channel complex antibody in a case of genetic prion disease. BMJ Case Rep 2014; pii:bcr2013201622.
- Angus-Leppan H, Rudge P, Mead S, Collinge J, Vincent A. Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 2013; 70:919–922.
- Fujita K, Yuasa T, Watanabe O, et al. Voltage-gated potassium channel complex antibodies in Creutzfeldt-Jakob disease. J Neurol 2012; 259:2249–2250.
- Fujita K, Yuasa T, Takahashi Y, et al. Antibodies to N-methyl-D-aspartate glutamate receptors in Creutzfeldt–Jakob disease patients. J Neuroimmunol 2012; 251:90–93.
- Mackay G, Ahmad K, Stone J, et al. NMDA receptor autoantibodies in sporadic Creutzfeldt-Jakob disease. J Neurol 2012; 259:1979–1981.
- Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus–1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology 2013; 81:1637–1639.
- Desena A, Graves D, Warnack W, Greenberg BM. Herpes simplex encephalitis as a potential cause of anti-N-methyl-D-aspartate receptor antibody encephalitis: report of 2 cases. JAMA Neurol 2014; 71:344–346.
- Armangue T, Leypoldt F, Málaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014; 75:317–323.
- Blaes F. Paraneoplastic brain stem encephalitis. Curr Treat Options Neurol 2013; 15:201–209.
- Wildemann B, Jarius S. The expanding range of autoimmune disorders of the nervous system. Lancet Neurol 2013; 12:22–24.
- Kim W, Kim SH, Lee SH, Li XF, Kim HJ. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler 2011; 17:1107–1112.
- Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013; 84:576–583.
- Jubelt B, Mihai C, Li MT, Veerapaneni P. Rhombencephalitis/brainstem encephalitis. Curr Neurol Neurosci Rep 2011; 11:543–552.
- Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000; 342:21–27.
- Ishida K, Mitoma H, Son SY, et al. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann Neurol 1999; 46:263–267.
- Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997; 41:238–246.
- Barber P, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage-gated K+ channel antibodies. Neurology 2000; 54:771–772.
- Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50:73–78.
- Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res 2006; 71:135–141.
- Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008; 70:1883–1890.
- Knowles CH, Lang B, Clover L, et al. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol 2002; 37:166–170.
- Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011; 69:892–900.
- Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013: 136:3151–3162.
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 2011; 10:759–772.
- Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011; 10:63–74.
- Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133:1655–1667.
- Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010; 85:881–897.
- Younes-Mhenni S, Janier MF, Cinotti L, et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain 2004; 127:2331–2338.
- Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011; 77:179–189.
- Shin YW, Lee ST, Shin JW, et al. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 2013; 265:75–81.
- Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013; 12:157–165.
- Brierley JB, Corsellis JAN, Hierons R, Nevin S. Subacute encephalitis of later adult life mainly affecting the limbic areas. Brain 1960; 83:357–368.
- Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain 1968; 91:481–496.
- Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol 1983; 14:609–613.
- Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn) 2012; 18:366–383.
- Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 2014; 76:168–184.
- Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61:25–36.
- Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010; 133:2734–2748.
- Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9:67–76.
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65:424–434.
- Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012; 83:638–645.
- Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 2005; 128:1764–1777.
- Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127:701–712.
- Jammoul A, Lederman RJ, Tavee J, Li Y. Presence of voltage-gated potassium channel complex antibody in a case of genetic prion disease. BMJ Case Rep 2014; pii:bcr2013201622.
- Angus-Leppan H, Rudge P, Mead S, Collinge J, Vincent A. Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 2013; 70:919–922.
- Fujita K, Yuasa T, Watanabe O, et al. Voltage-gated potassium channel complex antibodies in Creutzfeldt-Jakob disease. J Neurol 2012; 259:2249–2250.
- Fujita K, Yuasa T, Takahashi Y, et al. Antibodies to N-methyl-D-aspartate glutamate receptors in Creutzfeldt–Jakob disease patients. J Neuroimmunol 2012; 251:90–93.
- Mackay G, Ahmad K, Stone J, et al. NMDA receptor autoantibodies in sporadic Creutzfeldt-Jakob disease. J Neurol 2012; 259:1979–1981.
- Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus–1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology 2013; 81:1637–1639.
- Desena A, Graves D, Warnack W, Greenberg BM. Herpes simplex encephalitis as a potential cause of anti-N-methyl-D-aspartate receptor antibody encephalitis: report of 2 cases. JAMA Neurol 2014; 71:344–346.
- Armangue T, Leypoldt F, Málaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014; 75:317–323.
- Blaes F. Paraneoplastic brain stem encephalitis. Curr Treat Options Neurol 2013; 15:201–209.
- Wildemann B, Jarius S. The expanding range of autoimmune disorders of the nervous system. Lancet Neurol 2013; 12:22–24.
- Kim W, Kim SH, Lee SH, Li XF, Kim HJ. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler 2011; 17:1107–1112.
- Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013; 84:576–583.
- Jubelt B, Mihai C, Li MT, Veerapaneni P. Rhombencephalitis/brainstem encephalitis. Curr Neurol Neurosci Rep 2011; 11:543–552.
- Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000; 342:21–27.
- Ishida K, Mitoma H, Son SY, et al. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann Neurol 1999; 46:263–267.
- Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997; 41:238–246.
- Barber P, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage-gated K+ channel antibodies. Neurology 2000; 54:771–772.
- Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50:73–78.
- Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res 2006; 71:135–141.
- Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008; 70:1883–1890.
- Knowles CH, Lang B, Clover L, et al. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol 2002; 37:166–170.
- Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011; 69:892–900.
- Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013: 136:3151–3162.
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 2011; 10:759–772.
- Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011; 10:63–74.
- Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133:1655–1667.
- Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010; 85:881–897.
- Younes-Mhenni S, Janier MF, Cinotti L, et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain 2004; 127:2331–2338.
- Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011; 77:179–189.
- Shin YW, Lee ST, Shin JW, et al. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 2013; 265:75–81.
- Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013; 12:157–165.
KEY POINTS
- Autoantibody-mediated encephalitis accounts for a portion of cases of unexplained status epilepticus, encephalitis, and acute-onset psychiatric symptoms.
- Magnetic resonance imaging and cerebrospinal fluid analysis may be normal early in the disease course.
- Patients can express more than one autoantibody and present with more than one neuronal syndrome.
- Syndromes in which antibodies attack antigens on the surface of neurons are more likely to respond to immunotherapy than those involving intracellular antigens.
- Anti-N-methyl-d-aspartate receptor encephalitis typically presents with psychosis, seizures, and movement disorders in young women and is often associated with an ovarian teratoma.
- Limbic encephalitis, mediated by antibody to the voltage-gated potassium channel complex, is typically nonneoplastic and responds well to immunotherapy.
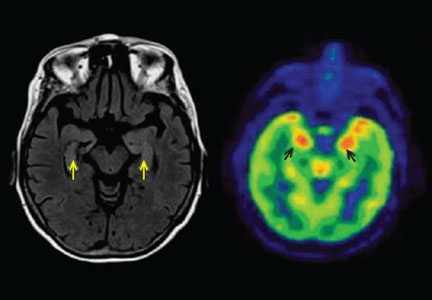
Not all abdominal pain is gastrointestinal
A 31-year-old woman presents to the office with a chief complaint of right mid-abdominal pain that began 1 day ago. She says she did not seek medical attention earlier because she had to be at work that morning and she thought the pain would resolve on its own.
She reports no fever, headache, anorexia, nausea, vomiting, malaise, loss of weight, melena, or changes in bowel habits. She describes the pain as sharp, localized to the right side, and radiating to the vulva upon sitting up. She denies any association of pain with current dietary habits or bowel function. She has no recollection of precipitating or alleviating factors, including the use of analgesics to reduce the pain.
On further discussion, she mentions that 1 year ago she began experiencing chronic abdominal pain, which she says is sometimes exacerbated by coughing, by standing for extended periods of time, and during menses, and is alleviated upon lying down.
She has regular menstrual periods, and her last one ended 7 days ago.
Her surgical history includes two uncomplicated cesarean deliveries. She does not use tobacco, alcohol, or illicit substances. She is not aware of any allergies to drugs or foods.
She appears to be in no acute distress and has been sitting quietly thus far. She seems to have positioned her hand on her abdomen over the corresponding area of pain.
On physical examination, vital signs are within normal limits, and she is alert and oriented to person, place, and time. Her sclerae are anicteric, and the pupils are equal, round, and reactive to light.
Cardiovascular and pulmonary examinations are also within normal limits. Examination of the abdomen elicits tenderness and guarding along the lateral border of the rectus abdominis muscle on the right side at the level of umbilicus, with no rebound tenderness or rigidity. The liver and spleen are not enlarged, and no abdominal mass is detected. No skin rash, joint swelling, or peripheral edema is noted. A neurologic examination is normal.
1. With the information provided, which of the following is least likely to be causing her symptoms?
- Chronic mesenteric ischemia
- Peptic ulcer
- Acute cholecystitis
- Slipping rib syndrome
CHRONIC MESENTERIC ISCHEMIA
Chronic mesenteric ischemia is the least likely diagnosis because the patient lacks risk factors for atherosclerosis and because she does not have postprandial pain, which is pathognomonic for chronic mesenteric ischemia. It is thought to be caused by a decrease in blood flow through the splanchnic vessels.1 Symptoms tend to arise after eating because of a postprandial increase in metabolic demands.1 These patients also often have atherosclerotic risk factors such as hypertension, hyperlipidemia, and smoking causing coronary artery disease, or a history of stroke.
The primary symptom is abdominal pain, most often described as achy, crampy, or spastic episodes of pain, usually occurring within 2 hours of eating.2 Weight loss is common, as patients can develop a fear of eating. Postprandial pain may also be associated with nausea, vomiting, and bloating.
Findings on clinical examination are usually less severe than the actual symptoms. Visceral duplex or multidetector computed tomography (CT) is an excellent tool to detect blood flow in potential stenotic vessels.2
PEPTIC ULCER DISEASE
Peptic ulcer disease is not a likely diagnosis in this patient because she has no history of taking nonsteroidal anti-inflammatory drugs (NSAIDs).
A study of US patients between 1997 and 2007 reported an annual incidence of peptic ulcer disease of 0.05% to 0.19% depending on the method of diagnosis.3 Peptic ulcer is thought to result from increased gastric acid secretion with a resultant inflammatory response, leading to erosion and ulceration.
The most common possible catalysts include Helicobacter pylori infection, NSAIDs, smoking, alcohol use, and hypersecretory states such as Zollinger-Ellison syndrome.4–6 Complications include internal bleeding, perforation causing peritonitis, and penetration to adjacent organs.
Pathophysiology
Peptic ulcer is the result of an increase in the normal level of gastric acid and a decrease in the protective ability of the gastric mucosa.7 Cytoprotection may be lost through a decrease in the products of arachidonic acid metabolism (eg, prostaglandins, which have a protective effect) or an increase in leukotriene B4 (LTB4), which has a damaging effect. Prostaglandins are thought not only to protect the normal gastric mucosa, but also to provide an antisecretory effect.
On the other hand, leukotrienes—specifically LTB4 and LTC4—are proinflammatory agents and can damage the gastric mucosa. NSAIDs enhance the production of leukotrienes through the 5-lipoxygenase pathway. The ability of LTB4 to cause degranulation and release of lysosomal enzymes may play a vital role in the inflammatory response to NSAIDs.8–10 LTC4 may promote gastric mucosal damage through a reduction of tissue perfusion resulting from the promotion of vascular stasis.8,11,12
Symptoms help differentiate ulcer type
The classic symptom is burning epigastric pain after meals. Pain that occurs immediately after meals is a classic symptom of gastric ulcer. Pain that occurs 2 to 3 hours after meals and that is relieved by food or antacids is a strong indicator of duodenal ulcer.13 Other symptoms include dyspepsia, bloating, distention, heartburn, and chest discomfort.13
Accurate diagnosis is vital in selecting the proper treatment. Diagnostic tests may include H pylori testing, upper-gastrointestinal endoscopy, and radiography with barium swallow.
CHOLECYSTITIS
In cholecystitis, the primary complaint is pain, usually in the right upper quadrant of the abdomen. Patients describe sudden, sharp, and intense pain that radiates to the back or shoulder. Patients may report pain after heavy meals, and some report nausea and vomiting. Cholecystitis is in the differential diagnosis of this patient because of the anatomic location of her pain.
The diagnosis is confirmed by imaging. Abdominal ultrasonography, technetium-99m hepatic iminodiacetic acid scanning, and CT are the most commonly used studies.14
Cholecystitis can be acute or chronic. Acute cholecystitis is categorized as calculous or acalculous. Calculous cholecystitis is multifactorial, but the primary cause is blockage of the cystic duct by gallstones.15 Other factors include irritants such as lysolecithin (released during bile stasis), which can trigger gallbladder inflammation,15–17 and infection.18
When the cystic duct is blocked, bile builds up inside the gallbladder, causing irritation and inflammation of the walls of the gallbladder.14
Acalculous cholecystitis, which resembles calculous cholecystitis but without the gallstones,19 accounts for 2% to 15% of all cases of acute cholecystitis.19,20 It has been observed in hospitalized critically ill patients, but it can also present in an outpatient setting, most often in elderly men with vascular disease.21 Causes include infection, trauma, and tumor obstruction, resulting in endothelial injury, gallbladder stasis, ischemia, and eventually necrosis.14,20,22,23
SLIPPING RIB SYNDROME
Slipping rib syndrome, also known as Tietze syndrome, is believed to be caused by hypermobile costal cartilage. The affected rib slips behind the rib above on contraction of the abdominal wall. This displacement increases the probability of costal nerve impingement and tissue inflammation producing unilateral, sharp, subcostal and upper-abdominal pain.
In this patient, slipping rib syndrome is a possible diagnosis because of the location of the pain and because the pain described by the patient is highly suggestive of neuropathic pain.
Slipping rib syndrome is diagnosed clinically by a “hooking” maneuver: the clinician hooks his or her fingers at the patient’s subcostal area, reproducing the pain by movement of the ribs anteriorly.24 When this test is performed in our patient the result is negative, ruling out slipping rib syndrome.
THE WORKUP CONTINUES
A complete blood cell count and comprehensive metabolic panel are within normal limits. Abdominal duplex ultrasonography reveals no celiac or mesenteric occlusions, thus ruling out chronic mesenteric ischemia.
Noncontrast CT shows no renal or ureteric stones and no evidence of bleeding in the urinary tract. CT with contrast shows no bowel distention, no evidence of hernia, and a normal appendix and ovaries.
2. After exclusion of the previous choices, which of the following is the most likely cause of her symptoms?
- Anterior cutaneous nerve entrapment syndrome (ACNES)
- Ovarian cyst
- Renal stones
- Appendicitis
- Ventral hernia
- Median arcuate ligament syndrome
ANTERIOR CUTANEOUS NERVE ENTRAPMENT SYNDROME
ACNES is the most likely diagnosis. A study published in 2013 indicated that many cases of functional abdominal pain may actually be undiagnosed cases of chronic abdominal wall pain such as ACNES.25 The condition, first described in 1972,26 is thought to be caused by thoracic cutaneous intercostal nerve entrapment between the abdominal muscles, causing pain at the point of entrapment.
The patient may present with pain that is either acute or chronic. Acute pain is localized more on the right side close to an old scar, or at the outer edge of the rectus abdominis muscle. The pain may vary from dull to burning to sharp; it can radiate horizontally in the upper half of the abdomen or obliquely in the lower half of the abdomen with movements such as twisting and sitting up.27
Despite the acute pain, patients are able to carry on daily functions. The pain may be alleviated by lying down.
The pain may be misdiagnosed as gynecologic or renal. In younger men, the pain may raise concern about hernia, and in older patients, cancer.27 Patients may complain of chronic intermittent pain, usually unilateral, and to a lesser extent bilateral.27
The anatomic location of the pain usually reflects the intercostal nerve involved. The pain is not related to eating or to bowel movements.25 Some patients report exacerbation upon coughing or standing, during menses, and with use of oral contraceptives.28,29 When inquiring about surgical history, it is common to find that the patient has had multiple abdominal surgical procedures.
On examination, the patient has nondistressing pain, with a hand often placed over the painful area.27 On firm palpation, a tender spot of less than 2 cm can be detected.
The diagnosis can be confirmed with a positive Carnett test. The patient lies supine on the examination table with the arms crossed over the chest, then elevates the head or the feet to tense the abdominal muscles.26,27 If doing so reproduces the pain (ie, a positive test), this increases the suspicion of ACNES; if the pain decreases or is not reproducible, an intra-abdominal cause is more likely.
If the pain is difficult to localize, the “pinch test” can be done by using the thumb and index finger to pinch and lift the skin of the abdomen, including the subcutaneous layer of fat, first on one side and then on the other. This helps determine the side with greater pain.27
OVARIAN CYSTS
Ovarian cysts are fluid-filled sacs on the surface of or within the ovary. They are often benign and require no intervention. However, 5% to 10% of US women with a suspicious ovarian mass undergo a surgical procedure, and 13% to 21% of these are found to have a malignancy.30,31
Ovarian cysts are usually painless unless complicated by rupture or bleeding. Patients who present with pain describe it as dull and aching and in the abdomen or pelvis. In rare cases, ovarian cysts can be large enough to cause pain from torsion. Other symptoms may include delayed menses and bleeding outside of the menstrual period.32–34
Ovarian cysts are thought to be caused by hormonal changes during the menstrual cycle. They can be detected during pelvic examination or during pelvic ultrasonography. Cysts that are primarily fluid-filled are generally benign and require no intervention. On the other hand, cysts composed of solid material require intervention.
Treatment depends on several factors, including size and type of cyst, the patient’s age, and whether torsion is present. Treatment can range from observation to medical or surgical management. Laparoscopic surgery is commonly used when surgical treatment is warranted.
RENAL STONES
From 10% to 15% of US adults develop a kidney stone at some time during their life.35 There is no single cause, but one factor that promotes stone formation is a greater amount of crystal-forming substances in the urine, such as calcium, oxalate, and uric acid.36 Most renal stones are calcium oxalate, uric acid, struvite, or cysteine.
Symptoms arise when the stone moves within the urinary tract. Patients present to the emergency room in severe distress, usually with flank pain that radiates to the lower abdomen or groin. The pain is episodic, fluctuates in intensity, and may present with dysuria, frequency, or urgency. It is also associated with nausea and vomiting.37
Renal stones are diagnosed through a series of laboratory and imaging studies. Imaging studies include plain radiography (which can miss small stones), renal sonography, and computed tomography without contrast.
APPENDICITIS
In the United States, the lifetime risk of developing appendicitis is 8.6% in men and 6.7% in women.38 Appendicitis is one of the most common reasons for emergency surgery.
Appendicitis is thought to result from obstruction by fecal matter blocking the opening of the appendix or from a viral infection (eg, with an adenovirus).39,40 The resulting bacterial growth can cause the appendix to become inflamed and purulent.
Patients typically present with umbilical or epigastric pain radiating to the right lower quadrant of the abdomen. Over time, the pain becomes sharper. Certain movements can exacerbate the pain, and lying down may alleviate it. Other symptoms are nausea, vomiting, loss of appetite, and low-grade fever.
Findings on the abdominal examination that help to confirm the diagnosis include rigidity and tenderness, classically localized to a point two-thirds of the way from the umbilicus to the anterior superior iliac spine. Rebound tenderness is usually present. Up to 25% of cases in some series presented atypically, with variable location and findings on physical examination (eg, bowel irregularities, indigestion, flatulence, generalized malaise). In addition to the physical examination, laboratory testing and imaging (ultrasonography, CT) may aid in confirming the diagnosis of appendicitis or any other cause of the pain.38
VENTRAL HERNIA
Ventral hernia is a bulging of abdominal organs or other tissues through a defect of the musculature of the abdominal wall. Ventral hernia is categorized by its location as epigastric, abdominal, or incisional. An open abdominal procedure is the cause in nearly 10% of cases41; the herniation occurs with weakening of the surgical scar.
Ventral hernia is usually detected on physical examination, and patients may present after noting a bulge in the abdominal wall. Symptoms vary. Some patients have no symptoms, while others have mild abdominal discomfort or severe abdominal pain as well as nausea and vomiting. Imaging with CT, ultrasonography, or magnetic resonance imaging helps confirm the diagnosis. Complications of ventral hernia include incarceration and bowel strangulation.
MEDIAN ARCUATE LIGAMENT SYNDROME
Median arcuate ligament syndrome is a challenging diagnosis and a very rare cause of abdominal pain. It is thought to be caused by celiac artery compression by fibroligamentous bands. Pain fluctuates with respiration and is greater during expiration.
Patients may present with recurrent episodes of crampy postprandial pain that cause them to avoid eating, resulting in weight loss. The pain may be associated with nausea, vomiting, and bloating.
The diagnosis is confirmed by duplex ultrasonography, angiography, or magnetic resonance angiography. Treatment is surgical division of the fibroligamentous band and crus, and this is often done laparascopically. In patients with severe persistent celiac artery stenosis, angioplasty and stenting may be considered.2
CASE CONTINUED
Before the physical examination, our patient identifies the location of her pain. A Carnett test is performed, as for ACNES: the patient is placed in the supine position and is instructed to cross both arms over her chest. In an effort to promote muscle tension, she is asked to elevate her head off the examination table, as if performing a mini sit-up, and as she does this, pressure is applied to the identified tender area. The pain is easily reproduced, further confirming involvement of the abdominal wall rather than the viscera. After this, electromyography shows abnormal findings. The patient is then referred to the pain management clinic for a diagnostic nerve block.
3. Which of the following is the first-line treatment of ACNES?
- Local injection of anesthetic
- Surgical neurectomy
LOCAL INJECTION OF ANESTHETIC
Local injection of anesthetic is the first-line treatment of ACNES.
Since ACNES is underdiagnosed, the patient may be less likely to be familiar with it. He or she should receive a detailed explanation of the condition and its management; this will help achieve a successful outcome.
Local anesthetic injection is used for both diagnosis and treatment; 2% lidocaine (or an equivalent) or dehydrated (absolute) alcohol or both can eliminate the pain caused by ACNES. The injection is commonly done under ultrasonographic guidance (Figure 1).42
Complete pain relief may be achieved with a single injection, but some patients require up to five injections.
The adjuvant use of corticosteroids in ACNES to reduce inflammation is controversial.
If anesthetic injections bring only minimal pain relief or if the patient has nerve entrapment in a scar, then surgical neurectomy is an option.43 The procedure is performed under local anesthesia, as the patient’s response aids in identifying the specific nerve or nerves involved.
RETURNING TO THE PATIENT
After a long discussion with our patient about ACNES and the treatment options, she agrees to undergo nerve block in the hope of relieving her pain. She receives a 0.5-mL injection of 2% lidocaine subcutaneously, and within minutes she reports relief of pain. She cannot believe that with a simple injection her pain was relieved. We advise her to return if her pain recurs or if new symptoms arise.
KEEP ACNES IN MIND
ACNES is one of the most commonly misdiagnosed conditions of patients presenting to the outpatient clinic with acute or chronic abdominal pain. This is because the focus is directed to intra-abdominal causes. But if ACNES is kept in consideration from the beginning of the patient encounter, extensive testing, time, and patient anxiety may be reduced significantly. A simple physical examination and the Carnett test aid in raising suspicion of ACNES. If ACNES is confirmed, ultrasonographically guided local anesthetic injection is both diagnostic and therapeutic.
- American Gastroenterological Association Medical Position Statement: Guidelines On Intestinal Ischemia. Gastroenterology 2000; 118:951–953.
- Bobadilla JL. Mesenteric ischemia. Surg Clin North Am 2013; 93:925–940.
- Sung JJ, Kuipers EJ, El-Serag HB. Systematic review: the global incidence and prevalence of peptic ulcer disease. Aliment Pharmacol Ther 2009; 29:938–946.
- Najm WI. Peptic ulcer disease. Prim Care 2011; 38:383–394.
- Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet 2009; 374:1449–1461.
- Chan FK, Leung WK. Peptic-ulcer disease. Lancet 2002; 360:933–941.
- Bright-Asare P, Habte T, Yirgou B, Benjamin J. Prostaglandins, H2-receptor antagonists and peptic ulcer disease. Drugs 1988; 35(suppl 3):1–9.
- Hudson N, Balsitis M, Everitt S, Hawkey CJ. Enhanced gastric mucosal leukotriene B4 synthesis in patients taking non-steroidal anti-inflammatory drugs. Gut 1993; 34:742–747.
- Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 1980; 286:264–265.
- Bokoch GM, Reed PW. Effect of various lipoxygenase metabolites of arachidonic acid on degranulation of polymorphonuclear leukocytes. J Biol Chem 1981; 256:5317–5320.
- Whittle BJ, Oren-Wolman N, Guth PH. Gastric vasoconstrictor actions of leukotriene C4, PGF2 alpha, and thromboxane mimetic U-46619 on rat submucosal microcirculation in vivo. Am J Physiol 1985; 248:G580–G586.
- Pihan G, Rogers C, Szabo S. Vascular injury in acute gastric mucosal damage. Mediatory role of leukotrienes. Dig Dis Sci 1988; 33:625–632.
- Ramakrishnan K, Salinas RC. Peptic ulcer disease. Am Fam Physician 2007; 76:1005–1012.
- Parmet S, Lynm C, Glass RM. JAMA patient page. Acute cholecystitis. JAMA 2003; 289:124.
- Roslyn JJ, DenBesten L, Thompson JE Jr, Silverman BF. Roles of lithogenic bile and cystic duct occlusion in the pathogenesis of acute cholecystitis. Am J Surg 1980; 140:126–130.
- Kaminski DL. Arachidonic acid metabolites in hepatobiliary physiology and disease. Gastroenterology 1989; 97:781–792.
- Jivegård L, Thornell E, Svanvik J. Pathophysiology of acute obstructive cholecystitis: implications for non-operative management. Br J Surg 1987; 74:1084–1086.
- Csendes A, Burdiles P, Maluenda F, Diaz JC, Csendes P, Mitru N. Simultaneous bacteriologic assessment of bile from gallbladder and common bile duct in control subjects and patients with gallstones and common duct stones. Arch Surg 1996; 131:389–394.
- Barie PS, Fischer E. Acute acalculous cholecystitis. J Am Coll Surg 1995; 180:232–244.
- Shapiro MJ, Luchtefeld WB, Kurzweil S, Kaminski DL, Durham RM, Mazuski JE. Acute acalculous cholecystitis in the critically ill. Am Surg 1994; 60:335–339.
- Savoca PE, Longo WE, Zucker KA, McMillen MM, Modlin IM. The increasing prevalence of acalculous cholecystitis in outpatients. Results of a 7-year study. Ann Surg 1990; 211:433–437.
- Gofrit O, Eid A, Pikarsky A, Lebensart PD, Pizov G, Rivkind A. Cholesterol embolisation causing chronic acalculous cholecystitis. Eur J Surg 1996; 162:243–245.
- McChesney JA, Northup PG, Bickston SJ. Acute acalculous cholecystitis associated with systemic sepsis and visceral arterial hypoperfusion: a case series and review of pathophysiology. Dig Dis Sci 2003; 48:1960–1967.
- Aeschlimann A, Kahn MF. Tietze’s syndrome: a critical review. Clin Exp Rheumatol 1990; 8:407–412.
- van Assen T, de Jager-Kievit JW, Scheltinga MR, Roumen RM. Chronic abdominal wall pain misdiagnosed as functional abdominal pain. J Am Board Fam Med 2013; 26:738–744.
- Akhnikh S, de Korte N, de Winter P. Anterior cutaneous nerve entrapment syndrome (ACNES): the forgotten diagnosis. Eur J Pediatr 2014; 173:445–449.
- Applegate WV. Abdominal cutaneous nerve entrapment syndrome (ACNES): a commonly overlooked cause of abdominal pain. Perm J 2002; 6:20–27.
- Grover M. UNC Center for Functional GI & Motility Disorders. Chronic abdominal wall pain: a missed diagnosis. www.med.unc.edu/ibs/files/educational-gi-handouts/Chronic%20Abdominal%20Pain.pdf. Accessed September 9, 2015.
- Greenbaum D, Dawson F, Watson R. Chronic abdominal wall pain (CAWP): a common but frequently overlooked disorder. Poster presented at the World Congress of Gastroenterology, Sydney, Australia, August 26–31, 1990.
- National Institutes of Health Consensus Development Conference Statement. Ovarian cancer: screening, treatment, and follow-up. Gynecol Oncol 1994; 55:S4–S14.
- Koonings PP, Campbell K, Mishell DR Jr, Grimes DA. Relative frequency of primary ovarian neoplasms: a 10-year review. Obstet Gynecol 1989; 74:921–926.
- Givens V, Mitchell GE, Harraway-Smith C, Reddy A, Maness DL. Diagnosis and management of adnexal masses. Am Fam Physician 2009; 80:815–820.
- Goff BA, Mandel L, Muntz HG, Melancon CH. Ovarian carcinoma diagnosis. Cancer 2000; 89:2068–2075.
- Friedman GD, Skilling JS, Udaltsova NV, Smith LH. Early symptoms of ovarian cancer: a case-control study without recall bias. Fam Pract 2005; 22:548–553.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976-1994. Kidney Int 2003; 63:1817–1823.
- Worcester EM, Coe FL. Clinical practice. Calcium kidney stones. N Engl J Med 2010; 363:954–963.
- Miller NL, Lingeman JE. Management of kidney stones. BMJ 2007; 334:468–472.
- Lewis SR, Mahony PJ, Simpson J. Appendicitis. BMJ 2011; 343:d5976.
- Lamps LW. Infectious causes of appendicitis. Infect Dis Clin North Am 2010; 24:995–1018.
- Reif RM. Viral appendicitis. Hum Pathol 1981; 12:193–196.
- Akkary E, Panait L, Roberts K, Duffy A, Bell R. Sutureless laparoscopic ventral hernia repair in obese patients. JSLS 2011; 15:154–159.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Randomized clinical trial of trigger point infiltration with lidocaine to diagnose anterior cutaneous nerve entrapment syndrome. Br J Surg 2013; 100:217–221.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Management of anterior cutaneous nerve entrapment syndrome in a cohort of 139 patients. Ann Surg 2011; 254:1054–1058.
A 31-year-old woman presents to the office with a chief complaint of right mid-abdominal pain that began 1 day ago. She says she did not seek medical attention earlier because she had to be at work that morning and she thought the pain would resolve on its own.
She reports no fever, headache, anorexia, nausea, vomiting, malaise, loss of weight, melena, or changes in bowel habits. She describes the pain as sharp, localized to the right side, and radiating to the vulva upon sitting up. She denies any association of pain with current dietary habits or bowel function. She has no recollection of precipitating or alleviating factors, including the use of analgesics to reduce the pain.
On further discussion, she mentions that 1 year ago she began experiencing chronic abdominal pain, which she says is sometimes exacerbated by coughing, by standing for extended periods of time, and during menses, and is alleviated upon lying down.
She has regular menstrual periods, and her last one ended 7 days ago.
Her surgical history includes two uncomplicated cesarean deliveries. She does not use tobacco, alcohol, or illicit substances. She is not aware of any allergies to drugs or foods.
She appears to be in no acute distress and has been sitting quietly thus far. She seems to have positioned her hand on her abdomen over the corresponding area of pain.
On physical examination, vital signs are within normal limits, and she is alert and oriented to person, place, and time. Her sclerae are anicteric, and the pupils are equal, round, and reactive to light.
Cardiovascular and pulmonary examinations are also within normal limits. Examination of the abdomen elicits tenderness and guarding along the lateral border of the rectus abdominis muscle on the right side at the level of umbilicus, with no rebound tenderness or rigidity. The liver and spleen are not enlarged, and no abdominal mass is detected. No skin rash, joint swelling, or peripheral edema is noted. A neurologic examination is normal.
1. With the information provided, which of the following is least likely to be causing her symptoms?
- Chronic mesenteric ischemia
- Peptic ulcer
- Acute cholecystitis
- Slipping rib syndrome
CHRONIC MESENTERIC ISCHEMIA
Chronic mesenteric ischemia is the least likely diagnosis because the patient lacks risk factors for atherosclerosis and because she does not have postprandial pain, which is pathognomonic for chronic mesenteric ischemia. It is thought to be caused by a decrease in blood flow through the splanchnic vessels.1 Symptoms tend to arise after eating because of a postprandial increase in metabolic demands.1 These patients also often have atherosclerotic risk factors such as hypertension, hyperlipidemia, and smoking causing coronary artery disease, or a history of stroke.
The primary symptom is abdominal pain, most often described as achy, crampy, or spastic episodes of pain, usually occurring within 2 hours of eating.2 Weight loss is common, as patients can develop a fear of eating. Postprandial pain may also be associated with nausea, vomiting, and bloating.
Findings on clinical examination are usually less severe than the actual symptoms. Visceral duplex or multidetector computed tomography (CT) is an excellent tool to detect blood flow in potential stenotic vessels.2
PEPTIC ULCER DISEASE
Peptic ulcer disease is not a likely diagnosis in this patient because she has no history of taking nonsteroidal anti-inflammatory drugs (NSAIDs).
A study of US patients between 1997 and 2007 reported an annual incidence of peptic ulcer disease of 0.05% to 0.19% depending on the method of diagnosis.3 Peptic ulcer is thought to result from increased gastric acid secretion with a resultant inflammatory response, leading to erosion and ulceration.
The most common possible catalysts include Helicobacter pylori infection, NSAIDs, smoking, alcohol use, and hypersecretory states such as Zollinger-Ellison syndrome.4–6 Complications include internal bleeding, perforation causing peritonitis, and penetration to adjacent organs.
Pathophysiology
Peptic ulcer is the result of an increase in the normal level of gastric acid and a decrease in the protective ability of the gastric mucosa.7 Cytoprotection may be lost through a decrease in the products of arachidonic acid metabolism (eg, prostaglandins, which have a protective effect) or an increase in leukotriene B4 (LTB4), which has a damaging effect. Prostaglandins are thought not only to protect the normal gastric mucosa, but also to provide an antisecretory effect.
On the other hand, leukotrienes—specifically LTB4 and LTC4—are proinflammatory agents and can damage the gastric mucosa. NSAIDs enhance the production of leukotrienes through the 5-lipoxygenase pathway. The ability of LTB4 to cause degranulation and release of lysosomal enzymes may play a vital role in the inflammatory response to NSAIDs.8–10 LTC4 may promote gastric mucosal damage through a reduction of tissue perfusion resulting from the promotion of vascular stasis.8,11,12
Symptoms help differentiate ulcer type
The classic symptom is burning epigastric pain after meals. Pain that occurs immediately after meals is a classic symptom of gastric ulcer. Pain that occurs 2 to 3 hours after meals and that is relieved by food or antacids is a strong indicator of duodenal ulcer.13 Other symptoms include dyspepsia, bloating, distention, heartburn, and chest discomfort.13
Accurate diagnosis is vital in selecting the proper treatment. Diagnostic tests may include H pylori testing, upper-gastrointestinal endoscopy, and radiography with barium swallow.
CHOLECYSTITIS
In cholecystitis, the primary complaint is pain, usually in the right upper quadrant of the abdomen. Patients describe sudden, sharp, and intense pain that radiates to the back or shoulder. Patients may report pain after heavy meals, and some report nausea and vomiting. Cholecystitis is in the differential diagnosis of this patient because of the anatomic location of her pain.
The diagnosis is confirmed by imaging. Abdominal ultrasonography, technetium-99m hepatic iminodiacetic acid scanning, and CT are the most commonly used studies.14
Cholecystitis can be acute or chronic. Acute cholecystitis is categorized as calculous or acalculous. Calculous cholecystitis is multifactorial, but the primary cause is blockage of the cystic duct by gallstones.15 Other factors include irritants such as lysolecithin (released during bile stasis), which can trigger gallbladder inflammation,15–17 and infection.18
When the cystic duct is blocked, bile builds up inside the gallbladder, causing irritation and inflammation of the walls of the gallbladder.14
Acalculous cholecystitis, which resembles calculous cholecystitis but without the gallstones,19 accounts for 2% to 15% of all cases of acute cholecystitis.19,20 It has been observed in hospitalized critically ill patients, but it can also present in an outpatient setting, most often in elderly men with vascular disease.21 Causes include infection, trauma, and tumor obstruction, resulting in endothelial injury, gallbladder stasis, ischemia, and eventually necrosis.14,20,22,23
SLIPPING RIB SYNDROME
Slipping rib syndrome, also known as Tietze syndrome, is believed to be caused by hypermobile costal cartilage. The affected rib slips behind the rib above on contraction of the abdominal wall. This displacement increases the probability of costal nerve impingement and tissue inflammation producing unilateral, sharp, subcostal and upper-abdominal pain.
In this patient, slipping rib syndrome is a possible diagnosis because of the location of the pain and because the pain described by the patient is highly suggestive of neuropathic pain.
Slipping rib syndrome is diagnosed clinically by a “hooking” maneuver: the clinician hooks his or her fingers at the patient’s subcostal area, reproducing the pain by movement of the ribs anteriorly.24 When this test is performed in our patient the result is negative, ruling out slipping rib syndrome.
THE WORKUP CONTINUES
A complete blood cell count and comprehensive metabolic panel are within normal limits. Abdominal duplex ultrasonography reveals no celiac or mesenteric occlusions, thus ruling out chronic mesenteric ischemia.
Noncontrast CT shows no renal or ureteric stones and no evidence of bleeding in the urinary tract. CT with contrast shows no bowel distention, no evidence of hernia, and a normal appendix and ovaries.
2. After exclusion of the previous choices, which of the following is the most likely cause of her symptoms?
- Anterior cutaneous nerve entrapment syndrome (ACNES)
- Ovarian cyst
- Renal stones
- Appendicitis
- Ventral hernia
- Median arcuate ligament syndrome
ANTERIOR CUTANEOUS NERVE ENTRAPMENT SYNDROME
ACNES is the most likely diagnosis. A study published in 2013 indicated that many cases of functional abdominal pain may actually be undiagnosed cases of chronic abdominal wall pain such as ACNES.25 The condition, first described in 1972,26 is thought to be caused by thoracic cutaneous intercostal nerve entrapment between the abdominal muscles, causing pain at the point of entrapment.
The patient may present with pain that is either acute or chronic. Acute pain is localized more on the right side close to an old scar, or at the outer edge of the rectus abdominis muscle. The pain may vary from dull to burning to sharp; it can radiate horizontally in the upper half of the abdomen or obliquely in the lower half of the abdomen with movements such as twisting and sitting up.27
Despite the acute pain, patients are able to carry on daily functions. The pain may be alleviated by lying down.
The pain may be misdiagnosed as gynecologic or renal. In younger men, the pain may raise concern about hernia, and in older patients, cancer.27 Patients may complain of chronic intermittent pain, usually unilateral, and to a lesser extent bilateral.27
The anatomic location of the pain usually reflects the intercostal nerve involved. The pain is not related to eating or to bowel movements.25 Some patients report exacerbation upon coughing or standing, during menses, and with use of oral contraceptives.28,29 When inquiring about surgical history, it is common to find that the patient has had multiple abdominal surgical procedures.
On examination, the patient has nondistressing pain, with a hand often placed over the painful area.27 On firm palpation, a tender spot of less than 2 cm can be detected.
The diagnosis can be confirmed with a positive Carnett test. The patient lies supine on the examination table with the arms crossed over the chest, then elevates the head or the feet to tense the abdominal muscles.26,27 If doing so reproduces the pain (ie, a positive test), this increases the suspicion of ACNES; if the pain decreases or is not reproducible, an intra-abdominal cause is more likely.
If the pain is difficult to localize, the “pinch test” can be done by using the thumb and index finger to pinch and lift the skin of the abdomen, including the subcutaneous layer of fat, first on one side and then on the other. This helps determine the side with greater pain.27
OVARIAN CYSTS
Ovarian cysts are fluid-filled sacs on the surface of or within the ovary. They are often benign and require no intervention. However, 5% to 10% of US women with a suspicious ovarian mass undergo a surgical procedure, and 13% to 21% of these are found to have a malignancy.30,31
Ovarian cysts are usually painless unless complicated by rupture or bleeding. Patients who present with pain describe it as dull and aching and in the abdomen or pelvis. In rare cases, ovarian cysts can be large enough to cause pain from torsion. Other symptoms may include delayed menses and bleeding outside of the menstrual period.32–34
Ovarian cysts are thought to be caused by hormonal changes during the menstrual cycle. They can be detected during pelvic examination or during pelvic ultrasonography. Cysts that are primarily fluid-filled are generally benign and require no intervention. On the other hand, cysts composed of solid material require intervention.
Treatment depends on several factors, including size and type of cyst, the patient’s age, and whether torsion is present. Treatment can range from observation to medical or surgical management. Laparoscopic surgery is commonly used when surgical treatment is warranted.
RENAL STONES
From 10% to 15% of US adults develop a kidney stone at some time during their life.35 There is no single cause, but one factor that promotes stone formation is a greater amount of crystal-forming substances in the urine, such as calcium, oxalate, and uric acid.36 Most renal stones are calcium oxalate, uric acid, struvite, or cysteine.
Symptoms arise when the stone moves within the urinary tract. Patients present to the emergency room in severe distress, usually with flank pain that radiates to the lower abdomen or groin. The pain is episodic, fluctuates in intensity, and may present with dysuria, frequency, or urgency. It is also associated with nausea and vomiting.37
Renal stones are diagnosed through a series of laboratory and imaging studies. Imaging studies include plain radiography (which can miss small stones), renal sonography, and computed tomography without contrast.
APPENDICITIS
In the United States, the lifetime risk of developing appendicitis is 8.6% in men and 6.7% in women.38 Appendicitis is one of the most common reasons for emergency surgery.
Appendicitis is thought to result from obstruction by fecal matter blocking the opening of the appendix or from a viral infection (eg, with an adenovirus).39,40 The resulting bacterial growth can cause the appendix to become inflamed and purulent.
Patients typically present with umbilical or epigastric pain radiating to the right lower quadrant of the abdomen. Over time, the pain becomes sharper. Certain movements can exacerbate the pain, and lying down may alleviate it. Other symptoms are nausea, vomiting, loss of appetite, and low-grade fever.
Findings on the abdominal examination that help to confirm the diagnosis include rigidity and tenderness, classically localized to a point two-thirds of the way from the umbilicus to the anterior superior iliac spine. Rebound tenderness is usually present. Up to 25% of cases in some series presented atypically, with variable location and findings on physical examination (eg, bowel irregularities, indigestion, flatulence, generalized malaise). In addition to the physical examination, laboratory testing and imaging (ultrasonography, CT) may aid in confirming the diagnosis of appendicitis or any other cause of the pain.38
VENTRAL HERNIA
Ventral hernia is a bulging of abdominal organs or other tissues through a defect of the musculature of the abdominal wall. Ventral hernia is categorized by its location as epigastric, abdominal, or incisional. An open abdominal procedure is the cause in nearly 10% of cases41; the herniation occurs with weakening of the surgical scar.
Ventral hernia is usually detected on physical examination, and patients may present after noting a bulge in the abdominal wall. Symptoms vary. Some patients have no symptoms, while others have mild abdominal discomfort or severe abdominal pain as well as nausea and vomiting. Imaging with CT, ultrasonography, or magnetic resonance imaging helps confirm the diagnosis. Complications of ventral hernia include incarceration and bowel strangulation.
MEDIAN ARCUATE LIGAMENT SYNDROME
Median arcuate ligament syndrome is a challenging diagnosis and a very rare cause of abdominal pain. It is thought to be caused by celiac artery compression by fibroligamentous bands. Pain fluctuates with respiration and is greater during expiration.
Patients may present with recurrent episodes of crampy postprandial pain that cause them to avoid eating, resulting in weight loss. The pain may be associated with nausea, vomiting, and bloating.
The diagnosis is confirmed by duplex ultrasonography, angiography, or magnetic resonance angiography. Treatment is surgical division of the fibroligamentous band and crus, and this is often done laparascopically. In patients with severe persistent celiac artery stenosis, angioplasty and stenting may be considered.2
CASE CONTINUED
Before the physical examination, our patient identifies the location of her pain. A Carnett test is performed, as for ACNES: the patient is placed in the supine position and is instructed to cross both arms over her chest. In an effort to promote muscle tension, she is asked to elevate her head off the examination table, as if performing a mini sit-up, and as she does this, pressure is applied to the identified tender area. The pain is easily reproduced, further confirming involvement of the abdominal wall rather than the viscera. After this, electromyography shows abnormal findings. The patient is then referred to the pain management clinic for a diagnostic nerve block.
3. Which of the following is the first-line treatment of ACNES?
- Local injection of anesthetic
- Surgical neurectomy
LOCAL INJECTION OF ANESTHETIC
Local injection of anesthetic is the first-line treatment of ACNES.
Since ACNES is underdiagnosed, the patient may be less likely to be familiar with it. He or she should receive a detailed explanation of the condition and its management; this will help achieve a successful outcome.
Local anesthetic injection is used for both diagnosis and treatment; 2% lidocaine (or an equivalent) or dehydrated (absolute) alcohol or both can eliminate the pain caused by ACNES. The injection is commonly done under ultrasonographic guidance (Figure 1).42
Complete pain relief may be achieved with a single injection, but some patients require up to five injections.
The adjuvant use of corticosteroids in ACNES to reduce inflammation is controversial.
If anesthetic injections bring only minimal pain relief or if the patient has nerve entrapment in a scar, then surgical neurectomy is an option.43 The procedure is performed under local anesthesia, as the patient’s response aids in identifying the specific nerve or nerves involved.
RETURNING TO THE PATIENT
After a long discussion with our patient about ACNES and the treatment options, she agrees to undergo nerve block in the hope of relieving her pain. She receives a 0.5-mL injection of 2% lidocaine subcutaneously, and within minutes she reports relief of pain. She cannot believe that with a simple injection her pain was relieved. We advise her to return if her pain recurs or if new symptoms arise.
KEEP ACNES IN MIND
ACNES is one of the most commonly misdiagnosed conditions of patients presenting to the outpatient clinic with acute or chronic abdominal pain. This is because the focus is directed to intra-abdominal causes. But if ACNES is kept in consideration from the beginning of the patient encounter, extensive testing, time, and patient anxiety may be reduced significantly. A simple physical examination and the Carnett test aid in raising suspicion of ACNES. If ACNES is confirmed, ultrasonographically guided local anesthetic injection is both diagnostic and therapeutic.
A 31-year-old woman presents to the office with a chief complaint of right mid-abdominal pain that began 1 day ago. She says she did not seek medical attention earlier because she had to be at work that morning and she thought the pain would resolve on its own.
She reports no fever, headache, anorexia, nausea, vomiting, malaise, loss of weight, melena, or changes in bowel habits. She describes the pain as sharp, localized to the right side, and radiating to the vulva upon sitting up. She denies any association of pain with current dietary habits or bowel function. She has no recollection of precipitating or alleviating factors, including the use of analgesics to reduce the pain.
On further discussion, she mentions that 1 year ago she began experiencing chronic abdominal pain, which she says is sometimes exacerbated by coughing, by standing for extended periods of time, and during menses, and is alleviated upon lying down.
She has regular menstrual periods, and her last one ended 7 days ago.
Her surgical history includes two uncomplicated cesarean deliveries. She does not use tobacco, alcohol, or illicit substances. She is not aware of any allergies to drugs or foods.
She appears to be in no acute distress and has been sitting quietly thus far. She seems to have positioned her hand on her abdomen over the corresponding area of pain.
On physical examination, vital signs are within normal limits, and she is alert and oriented to person, place, and time. Her sclerae are anicteric, and the pupils are equal, round, and reactive to light.
Cardiovascular and pulmonary examinations are also within normal limits. Examination of the abdomen elicits tenderness and guarding along the lateral border of the rectus abdominis muscle on the right side at the level of umbilicus, with no rebound tenderness or rigidity. The liver and spleen are not enlarged, and no abdominal mass is detected. No skin rash, joint swelling, or peripheral edema is noted. A neurologic examination is normal.
1. With the information provided, which of the following is least likely to be causing her symptoms?
- Chronic mesenteric ischemia
- Peptic ulcer
- Acute cholecystitis
- Slipping rib syndrome
CHRONIC MESENTERIC ISCHEMIA
Chronic mesenteric ischemia is the least likely diagnosis because the patient lacks risk factors for atherosclerosis and because she does not have postprandial pain, which is pathognomonic for chronic mesenteric ischemia. It is thought to be caused by a decrease in blood flow through the splanchnic vessels.1 Symptoms tend to arise after eating because of a postprandial increase in metabolic demands.1 These patients also often have atherosclerotic risk factors such as hypertension, hyperlipidemia, and smoking causing coronary artery disease, or a history of stroke.
The primary symptom is abdominal pain, most often described as achy, crampy, or spastic episodes of pain, usually occurring within 2 hours of eating.2 Weight loss is common, as patients can develop a fear of eating. Postprandial pain may also be associated with nausea, vomiting, and bloating.
Findings on clinical examination are usually less severe than the actual symptoms. Visceral duplex or multidetector computed tomography (CT) is an excellent tool to detect blood flow in potential stenotic vessels.2
PEPTIC ULCER DISEASE
Peptic ulcer disease is not a likely diagnosis in this patient because she has no history of taking nonsteroidal anti-inflammatory drugs (NSAIDs).
A study of US patients between 1997 and 2007 reported an annual incidence of peptic ulcer disease of 0.05% to 0.19% depending on the method of diagnosis.3 Peptic ulcer is thought to result from increased gastric acid secretion with a resultant inflammatory response, leading to erosion and ulceration.
The most common possible catalysts include Helicobacter pylori infection, NSAIDs, smoking, alcohol use, and hypersecretory states such as Zollinger-Ellison syndrome.4–6 Complications include internal bleeding, perforation causing peritonitis, and penetration to adjacent organs.
Pathophysiology
Peptic ulcer is the result of an increase in the normal level of gastric acid and a decrease in the protective ability of the gastric mucosa.7 Cytoprotection may be lost through a decrease in the products of arachidonic acid metabolism (eg, prostaglandins, which have a protective effect) or an increase in leukotriene B4 (LTB4), which has a damaging effect. Prostaglandins are thought not only to protect the normal gastric mucosa, but also to provide an antisecretory effect.
On the other hand, leukotrienes—specifically LTB4 and LTC4—are proinflammatory agents and can damage the gastric mucosa. NSAIDs enhance the production of leukotrienes through the 5-lipoxygenase pathway. The ability of LTB4 to cause degranulation and release of lysosomal enzymes may play a vital role in the inflammatory response to NSAIDs.8–10 LTC4 may promote gastric mucosal damage through a reduction of tissue perfusion resulting from the promotion of vascular stasis.8,11,12
Symptoms help differentiate ulcer type
The classic symptom is burning epigastric pain after meals. Pain that occurs immediately after meals is a classic symptom of gastric ulcer. Pain that occurs 2 to 3 hours after meals and that is relieved by food or antacids is a strong indicator of duodenal ulcer.13 Other symptoms include dyspepsia, bloating, distention, heartburn, and chest discomfort.13
Accurate diagnosis is vital in selecting the proper treatment. Diagnostic tests may include H pylori testing, upper-gastrointestinal endoscopy, and radiography with barium swallow.
CHOLECYSTITIS
In cholecystitis, the primary complaint is pain, usually in the right upper quadrant of the abdomen. Patients describe sudden, sharp, and intense pain that radiates to the back or shoulder. Patients may report pain after heavy meals, and some report nausea and vomiting. Cholecystitis is in the differential diagnosis of this patient because of the anatomic location of her pain.
The diagnosis is confirmed by imaging. Abdominal ultrasonography, technetium-99m hepatic iminodiacetic acid scanning, and CT are the most commonly used studies.14
Cholecystitis can be acute or chronic. Acute cholecystitis is categorized as calculous or acalculous. Calculous cholecystitis is multifactorial, but the primary cause is blockage of the cystic duct by gallstones.15 Other factors include irritants such as lysolecithin (released during bile stasis), which can trigger gallbladder inflammation,15–17 and infection.18
When the cystic duct is blocked, bile builds up inside the gallbladder, causing irritation and inflammation of the walls of the gallbladder.14
Acalculous cholecystitis, which resembles calculous cholecystitis but without the gallstones,19 accounts for 2% to 15% of all cases of acute cholecystitis.19,20 It has been observed in hospitalized critically ill patients, but it can also present in an outpatient setting, most often in elderly men with vascular disease.21 Causes include infection, trauma, and tumor obstruction, resulting in endothelial injury, gallbladder stasis, ischemia, and eventually necrosis.14,20,22,23
SLIPPING RIB SYNDROME
Slipping rib syndrome, also known as Tietze syndrome, is believed to be caused by hypermobile costal cartilage. The affected rib slips behind the rib above on contraction of the abdominal wall. This displacement increases the probability of costal nerve impingement and tissue inflammation producing unilateral, sharp, subcostal and upper-abdominal pain.
In this patient, slipping rib syndrome is a possible diagnosis because of the location of the pain and because the pain described by the patient is highly suggestive of neuropathic pain.
Slipping rib syndrome is diagnosed clinically by a “hooking” maneuver: the clinician hooks his or her fingers at the patient’s subcostal area, reproducing the pain by movement of the ribs anteriorly.24 When this test is performed in our patient the result is negative, ruling out slipping rib syndrome.
THE WORKUP CONTINUES
A complete blood cell count and comprehensive metabolic panel are within normal limits. Abdominal duplex ultrasonography reveals no celiac or mesenteric occlusions, thus ruling out chronic mesenteric ischemia.
Noncontrast CT shows no renal or ureteric stones and no evidence of bleeding in the urinary tract. CT with contrast shows no bowel distention, no evidence of hernia, and a normal appendix and ovaries.
2. After exclusion of the previous choices, which of the following is the most likely cause of her symptoms?
- Anterior cutaneous nerve entrapment syndrome (ACNES)
- Ovarian cyst
- Renal stones
- Appendicitis
- Ventral hernia
- Median arcuate ligament syndrome
ANTERIOR CUTANEOUS NERVE ENTRAPMENT SYNDROME
ACNES is the most likely diagnosis. A study published in 2013 indicated that many cases of functional abdominal pain may actually be undiagnosed cases of chronic abdominal wall pain such as ACNES.25 The condition, first described in 1972,26 is thought to be caused by thoracic cutaneous intercostal nerve entrapment between the abdominal muscles, causing pain at the point of entrapment.
The patient may present with pain that is either acute or chronic. Acute pain is localized more on the right side close to an old scar, or at the outer edge of the rectus abdominis muscle. The pain may vary from dull to burning to sharp; it can radiate horizontally in the upper half of the abdomen or obliquely in the lower half of the abdomen with movements such as twisting and sitting up.27
Despite the acute pain, patients are able to carry on daily functions. The pain may be alleviated by lying down.
The pain may be misdiagnosed as gynecologic or renal. In younger men, the pain may raise concern about hernia, and in older patients, cancer.27 Patients may complain of chronic intermittent pain, usually unilateral, and to a lesser extent bilateral.27
The anatomic location of the pain usually reflects the intercostal nerve involved. The pain is not related to eating or to bowel movements.25 Some patients report exacerbation upon coughing or standing, during menses, and with use of oral contraceptives.28,29 When inquiring about surgical history, it is common to find that the patient has had multiple abdominal surgical procedures.
On examination, the patient has nondistressing pain, with a hand often placed over the painful area.27 On firm palpation, a tender spot of less than 2 cm can be detected.
The diagnosis can be confirmed with a positive Carnett test. The patient lies supine on the examination table with the arms crossed over the chest, then elevates the head or the feet to tense the abdominal muscles.26,27 If doing so reproduces the pain (ie, a positive test), this increases the suspicion of ACNES; if the pain decreases or is not reproducible, an intra-abdominal cause is more likely.
If the pain is difficult to localize, the “pinch test” can be done by using the thumb and index finger to pinch and lift the skin of the abdomen, including the subcutaneous layer of fat, first on one side and then on the other. This helps determine the side with greater pain.27
OVARIAN CYSTS
Ovarian cysts are fluid-filled sacs on the surface of or within the ovary. They are often benign and require no intervention. However, 5% to 10% of US women with a suspicious ovarian mass undergo a surgical procedure, and 13% to 21% of these are found to have a malignancy.30,31
Ovarian cysts are usually painless unless complicated by rupture or bleeding. Patients who present with pain describe it as dull and aching and in the abdomen or pelvis. In rare cases, ovarian cysts can be large enough to cause pain from torsion. Other symptoms may include delayed menses and bleeding outside of the menstrual period.32–34
Ovarian cysts are thought to be caused by hormonal changes during the menstrual cycle. They can be detected during pelvic examination or during pelvic ultrasonography. Cysts that are primarily fluid-filled are generally benign and require no intervention. On the other hand, cysts composed of solid material require intervention.
Treatment depends on several factors, including size and type of cyst, the patient’s age, and whether torsion is present. Treatment can range from observation to medical or surgical management. Laparoscopic surgery is commonly used when surgical treatment is warranted.
RENAL STONES
From 10% to 15% of US adults develop a kidney stone at some time during their life.35 There is no single cause, but one factor that promotes stone formation is a greater amount of crystal-forming substances in the urine, such as calcium, oxalate, and uric acid.36 Most renal stones are calcium oxalate, uric acid, struvite, or cysteine.
Symptoms arise when the stone moves within the urinary tract. Patients present to the emergency room in severe distress, usually with flank pain that radiates to the lower abdomen or groin. The pain is episodic, fluctuates in intensity, and may present with dysuria, frequency, or urgency. It is also associated with nausea and vomiting.37
Renal stones are diagnosed through a series of laboratory and imaging studies. Imaging studies include plain radiography (which can miss small stones), renal sonography, and computed tomography without contrast.
APPENDICITIS
In the United States, the lifetime risk of developing appendicitis is 8.6% in men and 6.7% in women.38 Appendicitis is one of the most common reasons for emergency surgery.
Appendicitis is thought to result from obstruction by fecal matter blocking the opening of the appendix or from a viral infection (eg, with an adenovirus).39,40 The resulting bacterial growth can cause the appendix to become inflamed and purulent.
Patients typically present with umbilical or epigastric pain radiating to the right lower quadrant of the abdomen. Over time, the pain becomes sharper. Certain movements can exacerbate the pain, and lying down may alleviate it. Other symptoms are nausea, vomiting, loss of appetite, and low-grade fever.
Findings on the abdominal examination that help to confirm the diagnosis include rigidity and tenderness, classically localized to a point two-thirds of the way from the umbilicus to the anterior superior iliac spine. Rebound tenderness is usually present. Up to 25% of cases in some series presented atypically, with variable location and findings on physical examination (eg, bowel irregularities, indigestion, flatulence, generalized malaise). In addition to the physical examination, laboratory testing and imaging (ultrasonography, CT) may aid in confirming the diagnosis of appendicitis or any other cause of the pain.38
VENTRAL HERNIA
Ventral hernia is a bulging of abdominal organs or other tissues through a defect of the musculature of the abdominal wall. Ventral hernia is categorized by its location as epigastric, abdominal, or incisional. An open abdominal procedure is the cause in nearly 10% of cases41; the herniation occurs with weakening of the surgical scar.
Ventral hernia is usually detected on physical examination, and patients may present after noting a bulge in the abdominal wall. Symptoms vary. Some patients have no symptoms, while others have mild abdominal discomfort or severe abdominal pain as well as nausea and vomiting. Imaging with CT, ultrasonography, or magnetic resonance imaging helps confirm the diagnosis. Complications of ventral hernia include incarceration and bowel strangulation.
MEDIAN ARCUATE LIGAMENT SYNDROME
Median arcuate ligament syndrome is a challenging diagnosis and a very rare cause of abdominal pain. It is thought to be caused by celiac artery compression by fibroligamentous bands. Pain fluctuates with respiration and is greater during expiration.
Patients may present with recurrent episodes of crampy postprandial pain that cause them to avoid eating, resulting in weight loss. The pain may be associated with nausea, vomiting, and bloating.
The diagnosis is confirmed by duplex ultrasonography, angiography, or magnetic resonance angiography. Treatment is surgical division of the fibroligamentous band and crus, and this is often done laparascopically. In patients with severe persistent celiac artery stenosis, angioplasty and stenting may be considered.2
CASE CONTINUED
Before the physical examination, our patient identifies the location of her pain. A Carnett test is performed, as for ACNES: the patient is placed in the supine position and is instructed to cross both arms over her chest. In an effort to promote muscle tension, she is asked to elevate her head off the examination table, as if performing a mini sit-up, and as she does this, pressure is applied to the identified tender area. The pain is easily reproduced, further confirming involvement of the abdominal wall rather than the viscera. After this, electromyography shows abnormal findings. The patient is then referred to the pain management clinic for a diagnostic nerve block.
3. Which of the following is the first-line treatment of ACNES?
- Local injection of anesthetic
- Surgical neurectomy
LOCAL INJECTION OF ANESTHETIC
Local injection of anesthetic is the first-line treatment of ACNES.
Since ACNES is underdiagnosed, the patient may be less likely to be familiar with it. He or she should receive a detailed explanation of the condition and its management; this will help achieve a successful outcome.
Local anesthetic injection is used for both diagnosis and treatment; 2% lidocaine (or an equivalent) or dehydrated (absolute) alcohol or both can eliminate the pain caused by ACNES. The injection is commonly done under ultrasonographic guidance (Figure 1).42
Complete pain relief may be achieved with a single injection, but some patients require up to five injections.
The adjuvant use of corticosteroids in ACNES to reduce inflammation is controversial.
If anesthetic injections bring only minimal pain relief or if the patient has nerve entrapment in a scar, then surgical neurectomy is an option.43 The procedure is performed under local anesthesia, as the patient’s response aids in identifying the specific nerve or nerves involved.
RETURNING TO THE PATIENT
After a long discussion with our patient about ACNES and the treatment options, she agrees to undergo nerve block in the hope of relieving her pain. She receives a 0.5-mL injection of 2% lidocaine subcutaneously, and within minutes she reports relief of pain. She cannot believe that with a simple injection her pain was relieved. We advise her to return if her pain recurs or if new symptoms arise.
KEEP ACNES IN MIND
ACNES is one of the most commonly misdiagnosed conditions of patients presenting to the outpatient clinic with acute or chronic abdominal pain. This is because the focus is directed to intra-abdominal causes. But if ACNES is kept in consideration from the beginning of the patient encounter, extensive testing, time, and patient anxiety may be reduced significantly. A simple physical examination and the Carnett test aid in raising suspicion of ACNES. If ACNES is confirmed, ultrasonographically guided local anesthetic injection is both diagnostic and therapeutic.
- American Gastroenterological Association Medical Position Statement: Guidelines On Intestinal Ischemia. Gastroenterology 2000; 118:951–953.
- Bobadilla JL. Mesenteric ischemia. Surg Clin North Am 2013; 93:925–940.
- Sung JJ, Kuipers EJ, El-Serag HB. Systematic review: the global incidence and prevalence of peptic ulcer disease. Aliment Pharmacol Ther 2009; 29:938–946.
- Najm WI. Peptic ulcer disease. Prim Care 2011; 38:383–394.
- Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet 2009; 374:1449–1461.
- Chan FK, Leung WK. Peptic-ulcer disease. Lancet 2002; 360:933–941.
- Bright-Asare P, Habte T, Yirgou B, Benjamin J. Prostaglandins, H2-receptor antagonists and peptic ulcer disease. Drugs 1988; 35(suppl 3):1–9.
- Hudson N, Balsitis M, Everitt S, Hawkey CJ. Enhanced gastric mucosal leukotriene B4 synthesis in patients taking non-steroidal anti-inflammatory drugs. Gut 1993; 34:742–747.
- Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 1980; 286:264–265.
- Bokoch GM, Reed PW. Effect of various lipoxygenase metabolites of arachidonic acid on degranulation of polymorphonuclear leukocytes. J Biol Chem 1981; 256:5317–5320.
- Whittle BJ, Oren-Wolman N, Guth PH. Gastric vasoconstrictor actions of leukotriene C4, PGF2 alpha, and thromboxane mimetic U-46619 on rat submucosal microcirculation in vivo. Am J Physiol 1985; 248:G580–G586.
- Pihan G, Rogers C, Szabo S. Vascular injury in acute gastric mucosal damage. Mediatory role of leukotrienes. Dig Dis Sci 1988; 33:625–632.
- Ramakrishnan K, Salinas RC. Peptic ulcer disease. Am Fam Physician 2007; 76:1005–1012.
- Parmet S, Lynm C, Glass RM. JAMA patient page. Acute cholecystitis. JAMA 2003; 289:124.
- Roslyn JJ, DenBesten L, Thompson JE Jr, Silverman BF. Roles of lithogenic bile and cystic duct occlusion in the pathogenesis of acute cholecystitis. Am J Surg 1980; 140:126–130.
- Kaminski DL. Arachidonic acid metabolites in hepatobiliary physiology and disease. Gastroenterology 1989; 97:781–792.
- Jivegård L, Thornell E, Svanvik J. Pathophysiology of acute obstructive cholecystitis: implications for non-operative management. Br J Surg 1987; 74:1084–1086.
- Csendes A, Burdiles P, Maluenda F, Diaz JC, Csendes P, Mitru N. Simultaneous bacteriologic assessment of bile from gallbladder and common bile duct in control subjects and patients with gallstones and common duct stones. Arch Surg 1996; 131:389–394.
- Barie PS, Fischer E. Acute acalculous cholecystitis. J Am Coll Surg 1995; 180:232–244.
- Shapiro MJ, Luchtefeld WB, Kurzweil S, Kaminski DL, Durham RM, Mazuski JE. Acute acalculous cholecystitis in the critically ill. Am Surg 1994; 60:335–339.
- Savoca PE, Longo WE, Zucker KA, McMillen MM, Modlin IM. The increasing prevalence of acalculous cholecystitis in outpatients. Results of a 7-year study. Ann Surg 1990; 211:433–437.
- Gofrit O, Eid A, Pikarsky A, Lebensart PD, Pizov G, Rivkind A. Cholesterol embolisation causing chronic acalculous cholecystitis. Eur J Surg 1996; 162:243–245.
- McChesney JA, Northup PG, Bickston SJ. Acute acalculous cholecystitis associated with systemic sepsis and visceral arterial hypoperfusion: a case series and review of pathophysiology. Dig Dis Sci 2003; 48:1960–1967.
- Aeschlimann A, Kahn MF. Tietze’s syndrome: a critical review. Clin Exp Rheumatol 1990; 8:407–412.
- van Assen T, de Jager-Kievit JW, Scheltinga MR, Roumen RM. Chronic abdominal wall pain misdiagnosed as functional abdominal pain. J Am Board Fam Med 2013; 26:738–744.
- Akhnikh S, de Korte N, de Winter P. Anterior cutaneous nerve entrapment syndrome (ACNES): the forgotten diagnosis. Eur J Pediatr 2014; 173:445–449.
- Applegate WV. Abdominal cutaneous nerve entrapment syndrome (ACNES): a commonly overlooked cause of abdominal pain. Perm J 2002; 6:20–27.
- Grover M. UNC Center for Functional GI & Motility Disorders. Chronic abdominal wall pain: a missed diagnosis. www.med.unc.edu/ibs/files/educational-gi-handouts/Chronic%20Abdominal%20Pain.pdf. Accessed September 9, 2015.
- Greenbaum D, Dawson F, Watson R. Chronic abdominal wall pain (CAWP): a common but frequently overlooked disorder. Poster presented at the World Congress of Gastroenterology, Sydney, Australia, August 26–31, 1990.
- National Institutes of Health Consensus Development Conference Statement. Ovarian cancer: screening, treatment, and follow-up. Gynecol Oncol 1994; 55:S4–S14.
- Koonings PP, Campbell K, Mishell DR Jr, Grimes DA. Relative frequency of primary ovarian neoplasms: a 10-year review. Obstet Gynecol 1989; 74:921–926.
- Givens V, Mitchell GE, Harraway-Smith C, Reddy A, Maness DL. Diagnosis and management of adnexal masses. Am Fam Physician 2009; 80:815–820.
- Goff BA, Mandel L, Muntz HG, Melancon CH. Ovarian carcinoma diagnosis. Cancer 2000; 89:2068–2075.
- Friedman GD, Skilling JS, Udaltsova NV, Smith LH. Early symptoms of ovarian cancer: a case-control study without recall bias. Fam Pract 2005; 22:548–553.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976-1994. Kidney Int 2003; 63:1817–1823.
- Worcester EM, Coe FL. Clinical practice. Calcium kidney stones. N Engl J Med 2010; 363:954–963.
- Miller NL, Lingeman JE. Management of kidney stones. BMJ 2007; 334:468–472.
- Lewis SR, Mahony PJ, Simpson J. Appendicitis. BMJ 2011; 343:d5976.
- Lamps LW. Infectious causes of appendicitis. Infect Dis Clin North Am 2010; 24:995–1018.
- Reif RM. Viral appendicitis. Hum Pathol 1981; 12:193–196.
- Akkary E, Panait L, Roberts K, Duffy A, Bell R. Sutureless laparoscopic ventral hernia repair in obese patients. JSLS 2011; 15:154–159.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Randomized clinical trial of trigger point infiltration with lidocaine to diagnose anterior cutaneous nerve entrapment syndrome. Br J Surg 2013; 100:217–221.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Management of anterior cutaneous nerve entrapment syndrome in a cohort of 139 patients. Ann Surg 2011; 254:1054–1058.
- American Gastroenterological Association Medical Position Statement: Guidelines On Intestinal Ischemia. Gastroenterology 2000; 118:951–953.
- Bobadilla JL. Mesenteric ischemia. Surg Clin North Am 2013; 93:925–940.
- Sung JJ, Kuipers EJ, El-Serag HB. Systematic review: the global incidence and prevalence of peptic ulcer disease. Aliment Pharmacol Ther 2009; 29:938–946.
- Najm WI. Peptic ulcer disease. Prim Care 2011; 38:383–394.
- Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet 2009; 374:1449–1461.
- Chan FK, Leung WK. Peptic-ulcer disease. Lancet 2002; 360:933–941.
- Bright-Asare P, Habte T, Yirgou B, Benjamin J. Prostaglandins, H2-receptor antagonists and peptic ulcer disease. Drugs 1988; 35(suppl 3):1–9.
- Hudson N, Balsitis M, Everitt S, Hawkey CJ. Enhanced gastric mucosal leukotriene B4 synthesis in patients taking non-steroidal anti-inflammatory drugs. Gut 1993; 34:742–747.
- Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 1980; 286:264–265.
- Bokoch GM, Reed PW. Effect of various lipoxygenase metabolites of arachidonic acid on degranulation of polymorphonuclear leukocytes. J Biol Chem 1981; 256:5317–5320.
- Whittle BJ, Oren-Wolman N, Guth PH. Gastric vasoconstrictor actions of leukotriene C4, PGF2 alpha, and thromboxane mimetic U-46619 on rat submucosal microcirculation in vivo. Am J Physiol 1985; 248:G580–G586.
- Pihan G, Rogers C, Szabo S. Vascular injury in acute gastric mucosal damage. Mediatory role of leukotrienes. Dig Dis Sci 1988; 33:625–632.
- Ramakrishnan K, Salinas RC. Peptic ulcer disease. Am Fam Physician 2007; 76:1005–1012.
- Parmet S, Lynm C, Glass RM. JAMA patient page. Acute cholecystitis. JAMA 2003; 289:124.
- Roslyn JJ, DenBesten L, Thompson JE Jr, Silverman BF. Roles of lithogenic bile and cystic duct occlusion in the pathogenesis of acute cholecystitis. Am J Surg 1980; 140:126–130.
- Kaminski DL. Arachidonic acid metabolites in hepatobiliary physiology and disease. Gastroenterology 1989; 97:781–792.
- Jivegård L, Thornell E, Svanvik J. Pathophysiology of acute obstructive cholecystitis: implications for non-operative management. Br J Surg 1987; 74:1084–1086.
- Csendes A, Burdiles P, Maluenda F, Diaz JC, Csendes P, Mitru N. Simultaneous bacteriologic assessment of bile from gallbladder and common bile duct in control subjects and patients with gallstones and common duct stones. Arch Surg 1996; 131:389–394.
- Barie PS, Fischer E. Acute acalculous cholecystitis. J Am Coll Surg 1995; 180:232–244.
- Shapiro MJ, Luchtefeld WB, Kurzweil S, Kaminski DL, Durham RM, Mazuski JE. Acute acalculous cholecystitis in the critically ill. Am Surg 1994; 60:335–339.
- Savoca PE, Longo WE, Zucker KA, McMillen MM, Modlin IM. The increasing prevalence of acalculous cholecystitis in outpatients. Results of a 7-year study. Ann Surg 1990; 211:433–437.
- Gofrit O, Eid A, Pikarsky A, Lebensart PD, Pizov G, Rivkind A. Cholesterol embolisation causing chronic acalculous cholecystitis. Eur J Surg 1996; 162:243–245.
- McChesney JA, Northup PG, Bickston SJ. Acute acalculous cholecystitis associated with systemic sepsis and visceral arterial hypoperfusion: a case series and review of pathophysiology. Dig Dis Sci 2003; 48:1960–1967.
- Aeschlimann A, Kahn MF. Tietze’s syndrome: a critical review. Clin Exp Rheumatol 1990; 8:407–412.
- van Assen T, de Jager-Kievit JW, Scheltinga MR, Roumen RM. Chronic abdominal wall pain misdiagnosed as functional abdominal pain. J Am Board Fam Med 2013; 26:738–744.
- Akhnikh S, de Korte N, de Winter P. Anterior cutaneous nerve entrapment syndrome (ACNES): the forgotten diagnosis. Eur J Pediatr 2014; 173:445–449.
- Applegate WV. Abdominal cutaneous nerve entrapment syndrome (ACNES): a commonly overlooked cause of abdominal pain. Perm J 2002; 6:20–27.
- Grover M. UNC Center for Functional GI & Motility Disorders. Chronic abdominal wall pain: a missed diagnosis. www.med.unc.edu/ibs/files/educational-gi-handouts/Chronic%20Abdominal%20Pain.pdf. Accessed September 9, 2015.
- Greenbaum D, Dawson F, Watson R. Chronic abdominal wall pain (CAWP): a common but frequently overlooked disorder. Poster presented at the World Congress of Gastroenterology, Sydney, Australia, August 26–31, 1990.
- National Institutes of Health Consensus Development Conference Statement. Ovarian cancer: screening, treatment, and follow-up. Gynecol Oncol 1994; 55:S4–S14.
- Koonings PP, Campbell K, Mishell DR Jr, Grimes DA. Relative frequency of primary ovarian neoplasms: a 10-year review. Obstet Gynecol 1989; 74:921–926.
- Givens V, Mitchell GE, Harraway-Smith C, Reddy A, Maness DL. Diagnosis and management of adnexal masses. Am Fam Physician 2009; 80:815–820.
- Goff BA, Mandel L, Muntz HG, Melancon CH. Ovarian carcinoma diagnosis. Cancer 2000; 89:2068–2075.
- Friedman GD, Skilling JS, Udaltsova NV, Smith LH. Early symptoms of ovarian cancer: a case-control study without recall bias. Fam Pract 2005; 22:548–553.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976-1994. Kidney Int 2003; 63:1817–1823.
- Worcester EM, Coe FL. Clinical practice. Calcium kidney stones. N Engl J Med 2010; 363:954–963.
- Miller NL, Lingeman JE. Management of kidney stones. BMJ 2007; 334:468–472.
- Lewis SR, Mahony PJ, Simpson J. Appendicitis. BMJ 2011; 343:d5976.
- Lamps LW. Infectious causes of appendicitis. Infect Dis Clin North Am 2010; 24:995–1018.
- Reif RM. Viral appendicitis. Hum Pathol 1981; 12:193–196.
- Akkary E, Panait L, Roberts K, Duffy A, Bell R. Sutureless laparoscopic ventral hernia repair in obese patients. JSLS 2011; 15:154–159.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Randomized clinical trial of trigger point infiltration with lidocaine to diagnose anterior cutaneous nerve entrapment syndrome. Br J Surg 2013; 100:217–221.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Management of anterior cutaneous nerve entrapment syndrome in a cohort of 139 patients. Ann Surg 2011; 254:1054–1058.
Eventration of the diaphragm presenting as spontaneous pneumothorax
A 25-year-old man with a 2-day history of upper respiratory tract infection presents to the emergency department with the sudden onset of right-sided back and chest pain and shortness of breath after a severe coughing fit.
He is morbidly obese, is a long-time smoker, and has had recurrent exacerbations of asthma with frequent upper respiratory tract infections. He has no history of recent trauma.
A review of systems reveals no significant impairment in exercise tolerance. He has been able to continue doing manual labor at his job as a railroad worker.
Radiography shows a large right pneumothorax and an elevated right diaphragm (Figure 1). Computed tomography (CT) (Figure 2) reveals a right anterior apical pneumothorax with hypoplastic lung and significant elevation of the right diaphragm with fat, bowel, and kidney within the right thorax. He is hemodynamically stable and shows no signs of bowel obstruction.
The physical examination is normal except for diminished breath sounds on the right side. He is diagnosed with congenital diaphragmatic hernia and spontaneous pneumothorax. A 10-F locking pigtail catheter is inserted under CT guidance, leading to complete resolution of the pneumothorax. He is discharged home the next day with a plan for elective repair of the hernia.
Two months later, he returns for scheduled right thoracotomy to repair the hernia. However, while preparing the chest cavity, the surgeon finds no diaphragmatic hernia and no intra-abdominal content—but rather, a severely elevated and thinned-out diaphragm with uninterrupted continuity. The diagnosis is changed to congenital diaphragmatic eventration, and plication of the diaphragm is performed with a series of interrupted, pledgeted polypropylene sutures.
CONGENITAL EVENTRATION OF THE DIAPHRAGM
Congenital diaphragmatic eventration is a rare developmental defect of the central, muscular portion of the diaphragm. The true prevalence is not known, but early reports identified this condition in less than 0.1% of adult.1
Symptomatic patients usually experience dyspnea secondary to ventilation-perfusion mismatch resulting from chronic atelectasis and lung hypoplasia, as well as impaired ventilation resulting from the limited caudal migration of the diaphragm.2,3 Increased susceptibility to recurrent upper respiratory tract infections and pneumonia is also a common feature.
Although rare, spontaneous pneumothorax can develop in patients such as ours, whose lengthy history of smoking and asthma predisposed him to the development of emphysema-like blebs and bullae and to subsequent rupture of blebs brought on by vigorous coughing that caused an involuntary Valsalva maneuver.4
As in our patient, distinguishing congenital diaphragmatic eventration from hernia preoperatively can be difficult with plain chest radiography. Spiral CT with multiplanar reconstruction or with magnetic resonance imaging can help establish the diagnosis.3 However, a severely attenuated diaphragm can be difficult to visualize on CT, as in our patient, leading to a presumptive diagnosis of diaphragmatic hernia. In such situations, the diagnosis of eventration can only be made intraoperatively.
Surgical repair is indicated only for patients with symptoms. Other potential causes of the symptoms should first be ruled out, however, including primary pulmonary disease, cardiac dysfunction, and morbid obesity.
- Chin EF, Lynn RB. Surgery of eventration of the diaphragm. J Thorac Surg 1956; 32:6–14.
- Ridyard JB, Stewart RM. Regional lung function in unilateral diaphragmatic paralysis. Thorax 1976; 31:438–442.
- Shen C, Che G. Congenital eventration of hemidiaphragm in an adult. Ann Thorac Surg 2012; 94:e137–e139.
- Porpodis K, Zarogoulidis P, Spyratos D, et al. Pneumothorax and asthma. J Thorac Dis 2014; 6(suppl 1):S152–S161.
A 25-year-old man with a 2-day history of upper respiratory tract infection presents to the emergency department with the sudden onset of right-sided back and chest pain and shortness of breath after a severe coughing fit.
He is morbidly obese, is a long-time smoker, and has had recurrent exacerbations of asthma with frequent upper respiratory tract infections. He has no history of recent trauma.
A review of systems reveals no significant impairment in exercise tolerance. He has been able to continue doing manual labor at his job as a railroad worker.
Radiography shows a large right pneumothorax and an elevated right diaphragm (Figure 1). Computed tomography (CT) (Figure 2) reveals a right anterior apical pneumothorax with hypoplastic lung and significant elevation of the right diaphragm with fat, bowel, and kidney within the right thorax. He is hemodynamically stable and shows no signs of bowel obstruction.
The physical examination is normal except for diminished breath sounds on the right side. He is diagnosed with congenital diaphragmatic hernia and spontaneous pneumothorax. A 10-F locking pigtail catheter is inserted under CT guidance, leading to complete resolution of the pneumothorax. He is discharged home the next day with a plan for elective repair of the hernia.
Two months later, he returns for scheduled right thoracotomy to repair the hernia. However, while preparing the chest cavity, the surgeon finds no diaphragmatic hernia and no intra-abdominal content—but rather, a severely elevated and thinned-out diaphragm with uninterrupted continuity. The diagnosis is changed to congenital diaphragmatic eventration, and plication of the diaphragm is performed with a series of interrupted, pledgeted polypropylene sutures.
CONGENITAL EVENTRATION OF THE DIAPHRAGM
Congenital diaphragmatic eventration is a rare developmental defect of the central, muscular portion of the diaphragm. The true prevalence is not known, but early reports identified this condition in less than 0.1% of adult.1
Symptomatic patients usually experience dyspnea secondary to ventilation-perfusion mismatch resulting from chronic atelectasis and lung hypoplasia, as well as impaired ventilation resulting from the limited caudal migration of the diaphragm.2,3 Increased susceptibility to recurrent upper respiratory tract infections and pneumonia is also a common feature.
Although rare, spontaneous pneumothorax can develop in patients such as ours, whose lengthy history of smoking and asthma predisposed him to the development of emphysema-like blebs and bullae and to subsequent rupture of blebs brought on by vigorous coughing that caused an involuntary Valsalva maneuver.4
As in our patient, distinguishing congenital diaphragmatic eventration from hernia preoperatively can be difficult with plain chest radiography. Spiral CT with multiplanar reconstruction or with magnetic resonance imaging can help establish the diagnosis.3 However, a severely attenuated diaphragm can be difficult to visualize on CT, as in our patient, leading to a presumptive diagnosis of diaphragmatic hernia. In such situations, the diagnosis of eventration can only be made intraoperatively.
Surgical repair is indicated only for patients with symptoms. Other potential causes of the symptoms should first be ruled out, however, including primary pulmonary disease, cardiac dysfunction, and morbid obesity.
A 25-year-old man with a 2-day history of upper respiratory tract infection presents to the emergency department with the sudden onset of right-sided back and chest pain and shortness of breath after a severe coughing fit.
He is morbidly obese, is a long-time smoker, and has had recurrent exacerbations of asthma with frequent upper respiratory tract infections. He has no history of recent trauma.
A review of systems reveals no significant impairment in exercise tolerance. He has been able to continue doing manual labor at his job as a railroad worker.
Radiography shows a large right pneumothorax and an elevated right diaphragm (Figure 1). Computed tomography (CT) (Figure 2) reveals a right anterior apical pneumothorax with hypoplastic lung and significant elevation of the right diaphragm with fat, bowel, and kidney within the right thorax. He is hemodynamically stable and shows no signs of bowel obstruction.
The physical examination is normal except for diminished breath sounds on the right side. He is diagnosed with congenital diaphragmatic hernia and spontaneous pneumothorax. A 10-F locking pigtail catheter is inserted under CT guidance, leading to complete resolution of the pneumothorax. He is discharged home the next day with a plan for elective repair of the hernia.
Two months later, he returns for scheduled right thoracotomy to repair the hernia. However, while preparing the chest cavity, the surgeon finds no diaphragmatic hernia and no intra-abdominal content—but rather, a severely elevated and thinned-out diaphragm with uninterrupted continuity. The diagnosis is changed to congenital diaphragmatic eventration, and plication of the diaphragm is performed with a series of interrupted, pledgeted polypropylene sutures.
CONGENITAL EVENTRATION OF THE DIAPHRAGM
Congenital diaphragmatic eventration is a rare developmental defect of the central, muscular portion of the diaphragm. The true prevalence is not known, but early reports identified this condition in less than 0.1% of adult.1
Symptomatic patients usually experience dyspnea secondary to ventilation-perfusion mismatch resulting from chronic atelectasis and lung hypoplasia, as well as impaired ventilation resulting from the limited caudal migration of the diaphragm.2,3 Increased susceptibility to recurrent upper respiratory tract infections and pneumonia is also a common feature.
Although rare, spontaneous pneumothorax can develop in patients such as ours, whose lengthy history of smoking and asthma predisposed him to the development of emphysema-like blebs and bullae and to subsequent rupture of blebs brought on by vigorous coughing that caused an involuntary Valsalva maneuver.4
As in our patient, distinguishing congenital diaphragmatic eventration from hernia preoperatively can be difficult with plain chest radiography. Spiral CT with multiplanar reconstruction or with magnetic resonance imaging can help establish the diagnosis.3 However, a severely attenuated diaphragm can be difficult to visualize on CT, as in our patient, leading to a presumptive diagnosis of diaphragmatic hernia. In such situations, the diagnosis of eventration can only be made intraoperatively.
Surgical repair is indicated only for patients with symptoms. Other potential causes of the symptoms should first be ruled out, however, including primary pulmonary disease, cardiac dysfunction, and morbid obesity.
- Chin EF, Lynn RB. Surgery of eventration of the diaphragm. J Thorac Surg 1956; 32:6–14.
- Ridyard JB, Stewart RM. Regional lung function in unilateral diaphragmatic paralysis. Thorax 1976; 31:438–442.
- Shen C, Che G. Congenital eventration of hemidiaphragm in an adult. Ann Thorac Surg 2012; 94:e137–e139.
- Porpodis K, Zarogoulidis P, Spyratos D, et al. Pneumothorax and asthma. J Thorac Dis 2014; 6(suppl 1):S152–S161.
- Chin EF, Lynn RB. Surgery of eventration of the diaphragm. J Thorac Surg 1956; 32:6–14.
- Ridyard JB, Stewart RM. Regional lung function in unilateral diaphragmatic paralysis. Thorax 1976; 31:438–442.
- Shen C, Che G. Congenital eventration of hemidiaphragm in an adult. Ann Thorac Surg 2012; 94:e137–e139.
- Porpodis K, Zarogoulidis P, Spyratos D, et al. Pneumothorax and asthma. J Thorac Dis 2014; 6(suppl 1):S152–S161.
Bony bumps in the mouth
A 79-year-old woman with a long history of limited scleroderma was being evaluated in the rheumatology clinic. During routine examination of the oral cavity, masses were noted on her hard palate and on the lingual surface of both sides of the mandible (Figure 1). The masses had a bony consistency. The patient said that she had had these lumps for as long as she could remember, and that they were painless and had never caused any discomfort.
The masses were diagnosed as torus palatinus and torus mandibularis, localized benign overgrowths of cortical bone. The patient was reassured about the benign nature of these masses, and as they were asymptomatic, no further action was considered necessary.
TORUS PALATINUS AND TORUS MANDIBULARIS
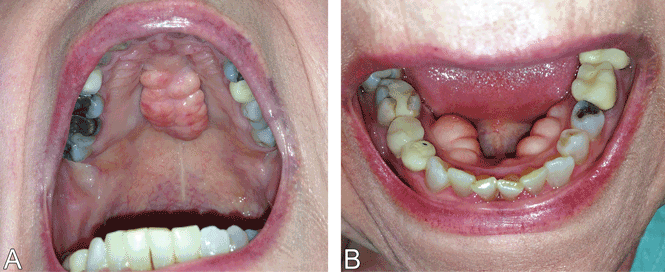
Torus palatinus and torus mandibularis are common exostoses of the mouth, ie, localized benign bony overgrowths arising from cortical bone.1 They are occasionally found incidentally during routine examination of the oral cavity. Patients should be reassured about the nonpathologic nature of this condition.
The condition is thought to be multifactorial, with causal factors including autosomal dominant inheritance, trauma, and lifestyle factors2 such as vitamin deficiency,3 a calcium-rich diet,3 fish consumption,4,5 and chewing on dry, raw, or frozen meat (as in Eskimo cultures).3 Masticatory hyperfunction and bruxism are thought to be risk factors.2,3
Epidemiologic studies indicate that oral tori are more common in women, and the prevalence varies considerably between geographic areas and ethnic groups.3 It is more common in Native Americans, Eskimos, Norwegians, and Thais.4
Torus palatinus is the most prevalent oral torus, occurring in 20% of the US population.6 It arises from the median raphe of the palatine bone and can vary in shape and size. Torus mandibularis is a protuberance arising in the premolar area of the lingual surface of the mandible.3 This form is much less common than torus palatinus, with a prevalence of 6%, and is bilateral in about 80% of cases.
Microscopic examination of tori reveal a mass of dense, lamellar, cortical bone with a small amount of fibrofatty marrow.1 An inner zone of trabecular bone may also be present.1
DIFFERENTIAL DIAGNOSIS
Oral tori must be differentiated from other growths in the mouth including fibromas, mucoceles, osteomas, osteochondromas, and osteoid osteomas.4 However, oral tori can usually be distinguished from other conditions on the basis of clinical findings alone. Biopsy may be warranted if there is doubt.4
Tori tend to grow gradually throughout life and do not have potential for malignant transformation.4 Although they are typically asymptomatic, removal is sometimes warranted for proper fitting of prostheses or for use in autogenous cortical bone grafting.5
- Neville BW, Douglas DD, Carl MA, Bouquot J. Developmental defects of the oral and maxillofacial region. In: Neville BW, Douglas DD, Carl MA, Bouquot J, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: WB Saunders; 2009:1–53.
- Eggen S. Torus mandibularis: an estimation of the degree of genetic determination. Acta Odontol Scand 1989; 47:409–415.
- Loukas M, Hulsberg P, Tubbs RS, et al. The tori of the mouth and ear: a review. Clin Anat 2013; 26:953–960.
- Ladizinski B, Lee KC. A nodular protuberance on the hard palate. JAMA 2014; 311:1558–1559.
- García-García AS, Martínez-González JM, Gómez-Font R, Soto-Rivadeneira A, Oviedo-Roldán L. Current status of the torus palatinus and torus mandibularis. Med Oral Patol Oral Cir Bucal 2010; 15:e353–e360.
- Larheim TA, Westesson PL. Facial growth disturbances. In: Maxillofacial Imaging. Berlin/Heidelberg: Springer-Verlag, 2008:231.
A 79-year-old woman with a long history of limited scleroderma was being evaluated in the rheumatology clinic. During routine examination of the oral cavity, masses were noted on her hard palate and on the lingual surface of both sides of the mandible (Figure 1). The masses had a bony consistency. The patient said that she had had these lumps for as long as she could remember, and that they were painless and had never caused any discomfort.
The masses were diagnosed as torus palatinus and torus mandibularis, localized benign overgrowths of cortical bone. The patient was reassured about the benign nature of these masses, and as they were asymptomatic, no further action was considered necessary.
TORUS PALATINUS AND TORUS MANDIBULARIS
Torus palatinus and torus mandibularis are common exostoses of the mouth, ie, localized benign bony overgrowths arising from cortical bone.1 They are occasionally found incidentally during routine examination of the oral cavity. Patients should be reassured about the nonpathologic nature of this condition.
The condition is thought to be multifactorial, with causal factors including autosomal dominant inheritance, trauma, and lifestyle factors2 such as vitamin deficiency,3 a calcium-rich diet,3 fish consumption,4,5 and chewing on dry, raw, or frozen meat (as in Eskimo cultures).3 Masticatory hyperfunction and bruxism are thought to be risk factors.2,3
Epidemiologic studies indicate that oral tori are more common in women, and the prevalence varies considerably between geographic areas and ethnic groups.3 It is more common in Native Americans, Eskimos, Norwegians, and Thais.4
Torus palatinus is the most prevalent oral torus, occurring in 20% of the US population.6 It arises from the median raphe of the palatine bone and can vary in shape and size. Torus mandibularis is a protuberance arising in the premolar area of the lingual surface of the mandible.3 This form is much less common than torus palatinus, with a prevalence of 6%, and is bilateral in about 80% of cases.
Microscopic examination of tori reveal a mass of dense, lamellar, cortical bone with a small amount of fibrofatty marrow.1 An inner zone of trabecular bone may also be present.1
DIFFERENTIAL DIAGNOSIS
Oral tori must be differentiated from other growths in the mouth including fibromas, mucoceles, osteomas, osteochondromas, and osteoid osteomas.4 However, oral tori can usually be distinguished from other conditions on the basis of clinical findings alone. Biopsy may be warranted if there is doubt.4
Tori tend to grow gradually throughout life and do not have potential for malignant transformation.4 Although they are typically asymptomatic, removal is sometimes warranted for proper fitting of prostheses or for use in autogenous cortical bone grafting.5
A 79-year-old woman with a long history of limited scleroderma was being evaluated in the rheumatology clinic. During routine examination of the oral cavity, masses were noted on her hard palate and on the lingual surface of both sides of the mandible (Figure 1). The masses had a bony consistency. The patient said that she had had these lumps for as long as she could remember, and that they were painless and had never caused any discomfort.
The masses were diagnosed as torus palatinus and torus mandibularis, localized benign overgrowths of cortical bone. The patient was reassured about the benign nature of these masses, and as they were asymptomatic, no further action was considered necessary.
TORUS PALATINUS AND TORUS MANDIBULARIS
Torus palatinus and torus mandibularis are common exostoses of the mouth, ie, localized benign bony overgrowths arising from cortical bone.1 They are occasionally found incidentally during routine examination of the oral cavity. Patients should be reassured about the nonpathologic nature of this condition.
The condition is thought to be multifactorial, with causal factors including autosomal dominant inheritance, trauma, and lifestyle factors2 such as vitamin deficiency,3 a calcium-rich diet,3 fish consumption,4,5 and chewing on dry, raw, or frozen meat (as in Eskimo cultures).3 Masticatory hyperfunction and bruxism are thought to be risk factors.2,3
Epidemiologic studies indicate that oral tori are more common in women, and the prevalence varies considerably between geographic areas and ethnic groups.3 It is more common in Native Americans, Eskimos, Norwegians, and Thais.4
Torus palatinus is the most prevalent oral torus, occurring in 20% of the US population.6 It arises from the median raphe of the palatine bone and can vary in shape and size. Torus mandibularis is a protuberance arising in the premolar area of the lingual surface of the mandible.3 This form is much less common than torus palatinus, with a prevalence of 6%, and is bilateral in about 80% of cases.
Microscopic examination of tori reveal a mass of dense, lamellar, cortical bone with a small amount of fibrofatty marrow.1 An inner zone of trabecular bone may also be present.1
DIFFERENTIAL DIAGNOSIS
Oral tori must be differentiated from other growths in the mouth including fibromas, mucoceles, osteomas, osteochondromas, and osteoid osteomas.4 However, oral tori can usually be distinguished from other conditions on the basis of clinical findings alone. Biopsy may be warranted if there is doubt.4
Tori tend to grow gradually throughout life and do not have potential for malignant transformation.4 Although they are typically asymptomatic, removal is sometimes warranted for proper fitting of prostheses or for use in autogenous cortical bone grafting.5
- Neville BW, Douglas DD, Carl MA, Bouquot J. Developmental defects of the oral and maxillofacial region. In: Neville BW, Douglas DD, Carl MA, Bouquot J, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: WB Saunders; 2009:1–53.
- Eggen S. Torus mandibularis: an estimation of the degree of genetic determination. Acta Odontol Scand 1989; 47:409–415.
- Loukas M, Hulsberg P, Tubbs RS, et al. The tori of the mouth and ear: a review. Clin Anat 2013; 26:953–960.
- Ladizinski B, Lee KC. A nodular protuberance on the hard palate. JAMA 2014; 311:1558–1559.
- García-García AS, Martínez-González JM, Gómez-Font R, Soto-Rivadeneira A, Oviedo-Roldán L. Current status of the torus palatinus and torus mandibularis. Med Oral Patol Oral Cir Bucal 2010; 15:e353–e360.
- Larheim TA, Westesson PL. Facial growth disturbances. In: Maxillofacial Imaging. Berlin/Heidelberg: Springer-Verlag, 2008:231.
- Neville BW, Douglas DD, Carl MA, Bouquot J. Developmental defects of the oral and maxillofacial region. In: Neville BW, Douglas DD, Carl MA, Bouquot J, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: WB Saunders; 2009:1–53.
- Eggen S. Torus mandibularis: an estimation of the degree of genetic determination. Acta Odontol Scand 1989; 47:409–415.
- Loukas M, Hulsberg P, Tubbs RS, et al. The tori of the mouth and ear: a review. Clin Anat 2013; 26:953–960.
- Ladizinski B, Lee KC. A nodular protuberance on the hard palate. JAMA 2014; 311:1558–1559.
- García-García AS, Martínez-González JM, Gómez-Font R, Soto-Rivadeneira A, Oviedo-Roldán L. Current status of the torus palatinus and torus mandibularis. Med Oral Patol Oral Cir Bucal 2010; 15:e353–e360.
- Larheim TA, Westesson PL. Facial growth disturbances. In: Maxillofacial Imaging. Berlin/Heidelberg: Springer-Verlag, 2008:231.
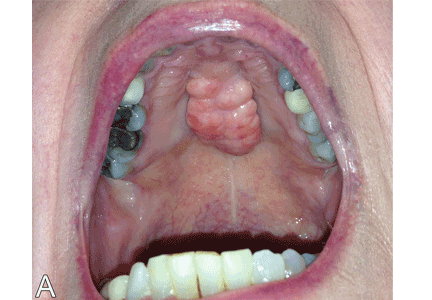
Vitamin B12 deficiency
To the Editor: In the article “An unusual cause of vitamin B12 and iron deficiency,”1 the diagnosis of vitamin B12 deficiency was made only by a vitamin B12 level of 108 pg/mL.
According to Harrison’s Principles of Internal Medicine, 18th edition, page 870, the diagnosis of vitamin B12 deficiency requires measurement of methylmalonic acid. Either this test was not performed on the 76-year-old woman described in the article, or the result was not entered. Without a methylmalonic acid level, the title of this article seems incorrect, or the article itself is incomplete by not including this level. The correct diagnosis of anemia due to an intestinal tapeworm was made by capsule endoscopy. She received appropriate therapy and her anemia cleared quickly.
If there is an updated concept for diagnosing vitamin B12 deficiency, I’m open to learning about it.
- Maithel S, Duong AK, Zhang J, Nguyen DL. An unusual cause of vitamin B12 and iron deficiency. Cleve Clin J Med 2015; 82:406–408.
To the Editor: In the article “An unusual cause of vitamin B12 and iron deficiency,”1 the diagnosis of vitamin B12 deficiency was made only by a vitamin B12 level of 108 pg/mL.
According to Harrison’s Principles of Internal Medicine, 18th edition, page 870, the diagnosis of vitamin B12 deficiency requires measurement of methylmalonic acid. Either this test was not performed on the 76-year-old woman described in the article, or the result was not entered. Without a methylmalonic acid level, the title of this article seems incorrect, or the article itself is incomplete by not including this level. The correct diagnosis of anemia due to an intestinal tapeworm was made by capsule endoscopy. She received appropriate therapy and her anemia cleared quickly.
If there is an updated concept for diagnosing vitamin B12 deficiency, I’m open to learning about it.
To the Editor: In the article “An unusual cause of vitamin B12 and iron deficiency,”1 the diagnosis of vitamin B12 deficiency was made only by a vitamin B12 level of 108 pg/mL.
According to Harrison’s Principles of Internal Medicine, 18th edition, page 870, the diagnosis of vitamin B12 deficiency requires measurement of methylmalonic acid. Either this test was not performed on the 76-year-old woman described in the article, or the result was not entered. Without a methylmalonic acid level, the title of this article seems incorrect, or the article itself is incomplete by not including this level. The correct diagnosis of anemia due to an intestinal tapeworm was made by capsule endoscopy. She received appropriate therapy and her anemia cleared quickly.
If there is an updated concept for diagnosing vitamin B12 deficiency, I’m open to learning about it.
- Maithel S, Duong AK, Zhang J, Nguyen DL. An unusual cause of vitamin B12 and iron deficiency. Cleve Clin J Med 2015; 82:406–408.
- Maithel S, Duong AK, Zhang J, Nguyen DL. An unusual cause of vitamin B12 and iron deficiency. Cleve Clin J Med 2015; 82:406–408.
In reply: Vitamin B12 deficiency
In Reply: We thank Dr. Phillips for his inquiry.
In general, serum vitamin B12 concentrations vary greatly, and we acknowledge that serum vitamin B12 may be normal in up to 5% of patients with documented B12 deficiency.1 In a prospective study of 1,599 patients, Matchar et al2 demonstrated that a single vitamin B12 level less than 200 pg/mL had a specificity greater than 95% at predicting vitamin B12 deficiency.2 We acknowledge that additional metabolite testing is necessary in equivocal cases in which the vitamin B12 level is between 200 and 300 pg/mL, which is often considered to be the normal range, but the patient has symptoms of vitamin B12 deficiency such as dementia and unexplained macrocytosis, and neurologic symptoms.3
Based on the patient’s symptoms of neuropathy and fatigue in conjunction with a vitamin B12 level well below 200 pg/mL, we believe that the diagnosis can be made.2,3 Nonetheless, although we did not mention it in our article, we did indeed send for a methylmalonic acid measurement at the time of the initial evaluation, and the level was elevated at 396 nmol/L (normal 87–318 nmol/L), further confirming her vitamin B12 deficiency.
- Naurath HJ, Joosten E, Riezler R, Stabler SP, Allen RH, Lindenbaum J. Effects of vitamin B12, folate, and vitamin B6 supplements in elderly people with normal serum vitamin concentrations. Lancet 1995; 346:85–89.
- Matchar DB, McCrory DC, Millington DS, Feussner JR. Performance of the serum cobalamin assay for diagnosis of cobalamin deficiency. Am J Med Sci 1994; 308:276–283.
- Stabler SP. Clinical practice. Vitamin B12 deficiency. N Eng J Med 2013; 368:149–160.
In Reply: We thank Dr. Phillips for his inquiry.
In general, serum vitamin B12 concentrations vary greatly, and we acknowledge that serum vitamin B12 may be normal in up to 5% of patients with documented B12 deficiency.1 In a prospective study of 1,599 patients, Matchar et al2 demonstrated that a single vitamin B12 level less than 200 pg/mL had a specificity greater than 95% at predicting vitamin B12 deficiency.2 We acknowledge that additional metabolite testing is necessary in equivocal cases in which the vitamin B12 level is between 200 and 300 pg/mL, which is often considered to be the normal range, but the patient has symptoms of vitamin B12 deficiency such as dementia and unexplained macrocytosis, and neurologic symptoms.3
Based on the patient’s symptoms of neuropathy and fatigue in conjunction with a vitamin B12 level well below 200 pg/mL, we believe that the diagnosis can be made.2,3 Nonetheless, although we did not mention it in our article, we did indeed send for a methylmalonic acid measurement at the time of the initial evaluation, and the level was elevated at 396 nmol/L (normal 87–318 nmol/L), further confirming her vitamin B12 deficiency.
In Reply: We thank Dr. Phillips for his inquiry.
In general, serum vitamin B12 concentrations vary greatly, and we acknowledge that serum vitamin B12 may be normal in up to 5% of patients with documented B12 deficiency.1 In a prospective study of 1,599 patients, Matchar et al2 demonstrated that a single vitamin B12 level less than 200 pg/mL had a specificity greater than 95% at predicting vitamin B12 deficiency.2 We acknowledge that additional metabolite testing is necessary in equivocal cases in which the vitamin B12 level is between 200 and 300 pg/mL, which is often considered to be the normal range, but the patient has symptoms of vitamin B12 deficiency such as dementia and unexplained macrocytosis, and neurologic symptoms.3
Based on the patient’s symptoms of neuropathy and fatigue in conjunction with a vitamin B12 level well below 200 pg/mL, we believe that the diagnosis can be made.2,3 Nonetheless, although we did not mention it in our article, we did indeed send for a methylmalonic acid measurement at the time of the initial evaluation, and the level was elevated at 396 nmol/L (normal 87–318 nmol/L), further confirming her vitamin B12 deficiency.
- Naurath HJ, Joosten E, Riezler R, Stabler SP, Allen RH, Lindenbaum J. Effects of vitamin B12, folate, and vitamin B6 supplements in elderly people with normal serum vitamin concentrations. Lancet 1995; 346:85–89.
- Matchar DB, McCrory DC, Millington DS, Feussner JR. Performance of the serum cobalamin assay for diagnosis of cobalamin deficiency. Am J Med Sci 1994; 308:276–283.
- Stabler SP. Clinical practice. Vitamin B12 deficiency. N Eng J Med 2013; 368:149–160.
- Naurath HJ, Joosten E, Riezler R, Stabler SP, Allen RH, Lindenbaum J. Effects of vitamin B12, folate, and vitamin B6 supplements in elderly people with normal serum vitamin concentrations. Lancet 1995; 346:85–89.
- Matchar DB, McCrory DC, Millington DS, Feussner JR. Performance of the serum cobalamin assay for diagnosis of cobalamin deficiency. Am J Med Sci 1994; 308:276–283.
- Stabler SP. Clinical practice. Vitamin B12 deficiency. N Eng J Med 2013; 368:149–160.
Preoperative testing
To the Editor: I read with great interest your 1-Minute Consult and the accompanying editorial on preoperative testing. I have long requested from my local hospitals the rationale for the long list of tests that used to be mandated for any surgery. I could not even get the courtesy of a reply from the department of anesthesia. For a while, in addition to the complete blood cell count and chemistry panel, one hospital demanded a urinalysis for cataract surgery.
Finally, without any explanation, the testing is now no longer mandated for cataract surgery but is still required for surgery such as the meniscus repair that was referenced.
These are not tests I want to order, but I am forced to order them or the surgery won’t be done. Certainly, in a diabetic patient or a patient treated with a complex regimen for hypertension, tests may be needed.
Thank you for the opportunity to comment.
To the Editor: I read with great interest your 1-Minute Consult and the accompanying editorial on preoperative testing. I have long requested from my local hospitals the rationale for the long list of tests that used to be mandated for any surgery. I could not even get the courtesy of a reply from the department of anesthesia. For a while, in addition to the complete blood cell count and chemistry panel, one hospital demanded a urinalysis for cataract surgery.
Finally, without any explanation, the testing is now no longer mandated for cataract surgery but is still required for surgery such as the meniscus repair that was referenced.
These are not tests I want to order, but I am forced to order them or the surgery won’t be done. Certainly, in a diabetic patient or a patient treated with a complex regimen for hypertension, tests may be needed.
Thank you for the opportunity to comment.
To the Editor: I read with great interest your 1-Minute Consult and the accompanying editorial on preoperative testing. I have long requested from my local hospitals the rationale for the long list of tests that used to be mandated for any surgery. I could not even get the courtesy of a reply from the department of anesthesia. For a while, in addition to the complete blood cell count and chemistry panel, one hospital demanded a urinalysis for cataract surgery.
Finally, without any explanation, the testing is now no longer mandated for cataract surgery but is still required for surgery such as the meniscus repair that was referenced.
These are not tests I want to order, but I am forced to order them or the surgery won’t be done. Certainly, in a diabetic patient or a patient treated with a complex regimen for hypertension, tests may be needed.
Thank you for the opportunity to comment.
Targeting gut flora to treat and prevent disease
› Encourage patients to eat a healthy diet that includes an adequate amount of soluble fiber to maintain a healthy, diverse microbiome. B
› Recommend combination probiotics to treat symptoms of irritable bowel syndrome. A
› Encourage patients to take probiotics containing Lactobacillus species to prevent antibiotic-associated diarrhea and Saccharomyces to prevent Clostridium difficile infection. A
› Recommend probiotics containing Lactobacillus species and/or Saccharomyces to treat acute infectious diarrhea. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 › Sheila S, age 27, has irritable bowel syndrome (IBS) and comes to your office for a follow-up visit. Over the past 6 months she has started taking a fiber supplement, drinking more water, and looking for links between stress and her symptoms. She has read about probiotics and wonders if you would consider recommending them in her situation.
CASE 2 › Mark M, age 45, has type 2 diabetes and is overweight. He is motivated to change his diet and has started to exercise more. He is taking metformin 2000 mg/d but his hemoglobin A1c remains slightly elevated at 7.2%. He heard on television that probiotics might help to keep him from needing to add another medication.
Most of the living organisms that comprise the human microbiome—all of the microbes that live on or in humans—are found in the gastrointestinal (GI) tract. The gut flora contribute 99% of the genetic material in the human body. The composition of the gut flora is remarkably diverse across the population; each individual has a unique microbial footprint. Within this microbial diversity, there appears to be a stable number of genes that are responsible for the major functions of the gut flora.1 These microbes:
- supply essential nutrients by breaking down complex carbohydrates;
- generate secondary bile acids that assist in digesting fats;2
- synthesize vitamins such as K, B12, folate, and biotin;3
- contribute to the defensive barrier in the colon by keeping pathogenic bacteria from crossing the colonic mucosa; and
- interact with our systemic immune system in a way that maintains a level of homeostasis, allowing for appropriate activation in the face of pathogens without developing autoimmunity.4
The gut flora also play a role in the communication between the central nervous system and the enteric nervous system by modulating the hormonal and neural pathways that have been labeled the “gut-brain axis.” The gut-brain axis has been associated with numerous disease states, including irritable bowel syndrome and certain psychiatric disorders.5
Researchers are investigating interventions that target the microbiome to increase microbial diversity and the presence of certain species to prevent or treat various diseases. The use of probiotics and dietary changes to increase intake of soluble fiber have been the most studied of these interventions. The thought is that these interventions can correct an imbalance, or dysbiosis, of the gut flora.6 Studies have shown that decreased microbial diversity is associated with elevations of certain disease markers (eg, adiposity, insulin, triglycerides, C–reactive protein)7 and that increases in soluble fiber lead to the greatest long-term improvement in microbial diversity.8 Fecal transplant—the transfer of a processed mixture of stool that contains “healthy” bacteria from a donor into the intestines of a patient—is being explored as a method of replacing colonic gut flora, but evidence is limited.
The following review takes a closer look at these options and identifies those that are most likely to benefit patients in the treatment—and prevention—of several diseases (TABLE 1).9-16
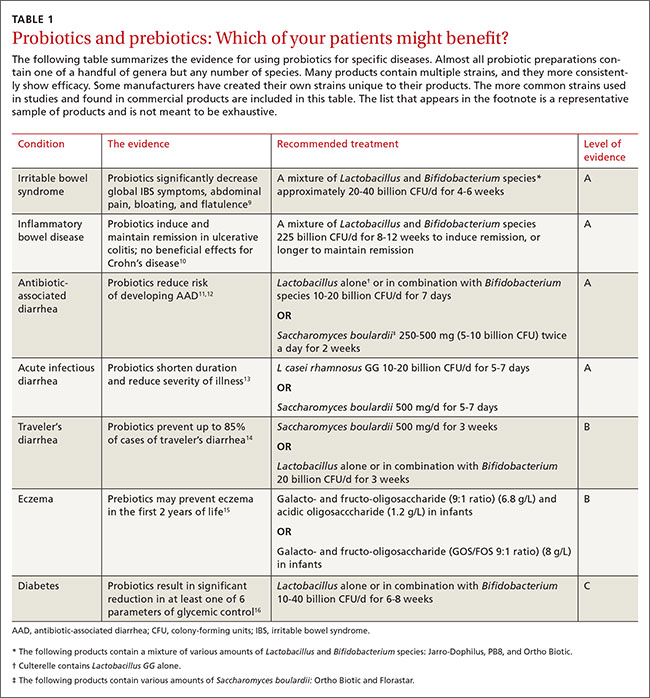
Evidence is best for using probiotics for digestive diseases
Dietary interventions for digestive diseases have long been studied, but are getting renewed attention for their potential impact on the microbiome.17 Beyond dietary modification, other similar treatment options include probiotics (live microorganisms thought to confer a beneficial effect on the host), prebiotics (non-digestible food ingredients, including oligosaccharides and inulin, thought to promote the growth of “helpful” gut flora), and synbiotics (combinations of the 2).18
Irritable bowel syndrome (IBS) is a heterogeneous disorder characterized by altered intestinal transit, low-grade colonic inflammation, and/or alterations in the gutbrain axis. Research has increasingly focused on recently discovered increases in intestinal immune activation, intestinal permeability, and alterations in the colonic microbiome (decreased diversity and increased pathogenic bacteria) associated with IBS.19
A meta-analysis of 43 randomized control trials (RCTs) found probiotics ranging from Lactobacillus to Saccharomyces can significantly decrease global IBS symptoms, abdominal pain, bloating, and flatulence.9 For a patient such as Ms. S, the evidence suggests a probiotic that contains a mixture of Lactobacillus and Bifidobacterium might help relieve her symptoms.9 In terms of dietary modifications, soluble fiber, which is already known to help treat IBS,20 has profound effects on improving microbiota diversity and in shifting the composition toward less pathogenic strains.21 The Institute of Medicine's daily recommended intake of soluble fiber is about 15 g/d.22
Inflammatory bowel disease (IBD) is caused by inflammation of the GI lining due to an overactive immune response. Evidence shows that patients with IBD have an altered microbial composition—specifically, an increase in bacteria that produce pro-inflammatory molecules and a decrease in bacteria that have a dampening effect on immune activation.23
Most studies evaluating probiotics as a treatment for IBD have been small and have used a wide variety of bacterial mixtures, which makes comparisons difficult. Recent meta-analyses found combination probiotics can both induce and maintain remission in patients with ulcerative colitis, but have no beneficial effects in Crohn’s disease.10 In a review of 9 case series of patients with IBD, fecal transplant reduced IBD symptoms, and patients were able to decrease medication use.24
Diarrheal illness. The human intestine is protected from diarrheal illness by healthy bacteria that block the actions of pathogenic bacteria. This mechanism is called colonization resistance. Moderate levels of evidence support the use of probiotics to prevent or treat several types of diarrheal illness.14
Antibiotic-associated diarrhea (AAD) is caused when antibiotic use alters the microbial balance. Recent meta-analyses have shown probiotics can prevent AAD and Clostridium difficile-associated diarrhea.11,12 Several case series and one RCT have found that fecal transplants are safe and efficacious for treating recurrent Clostridium difficile infection.25 Using probiotics to treat symptoms of AAD has been less studied.
Acute infectious diarrhea and traveler’s diarrhea (TD). A Cochrane review found that probiotics decreased the duration of diarrheal episodes by 25 hours, decreased the risk of an episode lasting more than 4 days by 59%, and led to one less diarrheal stool per day by the second day of the intervention.13 In a separate meta-analysis of 12 studies, probiotics significantly prevented 85% of cases of TD.14
Encouraging early evidence for several other illnesses
Metabolic disorders. Both animal and human studies support the theory that gut flora contribute to energy homeostasis, and in some genetically predisposed people dysbiosis may lead to obesity and diabetes. The traditional western diet4 and possibly decreased physical activity26 are major contributors to gut flora dysbiosis. Healthy bacteria in the gut break down soluble fiber into short chain fatty acids (SCFAs). SCFAs are associated with increased satiety, decreased food intake, lower levels of inflammation, and improvement in insulin signaling in adipose tissue. In addition to decreased SFCA production, dysbiosis also leads to increased lipid deposition through higher levels of lipoprotein lipase.27
Obesity. The bacteria in our gut affect energy metabolism. In patients with obesity, increased amounts of bacteria in the taxa Firmicutes and a corresponding decrease in Bacteroidetes is associated with an increased energy harvest and decreased SCFA production, which leads to a pro-inflammatory state.28 Probiotics that contain Bifidobacterium and Lactobacillus are thought to help correct this dysbiosis by increasing production of SCFAs.28
A recent meta-analysis of 4 RCTs found no significant difference between supplementation with probiotics and placebo on weight reduction.29 However, lower-quality studies with more subjects and longer duration have shown a statistically significant improvement in weight reduction with probiotic use compared to placebo.29
Diabetes. Although dietary interventions to improve glycemic control have long been an important cornerstone of treatment, probiotic supplementation to further alter gut flora composition is also being evaluated. Studies have found probiotics have largely beneficial effects on glycemic control, especially in animals. The largest systematic review to date looked at 33 studies, including 5 human trials. The human studies each found a significant reduction in at least one of 6 parameters of glycemic control (levels of fasting plasma glucose, postprandial blood glucose, glycated hemoglobin, insulin, insulin resistance, and onset of diabetes).16 It is unclear which probiotic strains confer benefit, and if those benefits are sustainable without dietary modification and increased physical activity.
Psychiatric illnesses. The gut-brain axis is thought to impact mental health by several mechanisms, including modulating the hypothalamic-pituitary-adrenal axis, activating the immune system, producing active metabolites, and affecting the vagus nerve. It is unclear which of these pathways may be clinically relevant.5,30 The few human studies that have looked for a potential link between gut flora and psychiatric illness have focused on depression and autism spectrum disorders (ASD).
Depression. Small studies comparing the microbiome composition of depressed patients vs healthy controls have found differences in patterns of both over- and underrepresented microbiota species in depressed patients, although the patterns across studies have been inconsistent.31,32 One small functional magnetic resonance imaging study of healthy women showed that a fermented milk product that contained probiotics affected activity in areas of the brain that control emotion and sensation.33 A few small studies have shown that patients who used probiotics had improved depression scores.34 Further studies are needed.
ASD. Children with ASD have GI disturbances—most commonly diarrhea, constipation, and/or bloating—more often than healthy controls.35,36 This association has led to speculation of a connection between the gut and brain. The microbial composition and diversity appears to be different in individuals with ASD; several studies have found an increase in Clostridia species.37
Research on probiotics for treating ASD has been primarily in preclinical models. Human studies of probiotics for ASD are lacking.38 Small studies on dietary modifications such as gluten-free and casein-free diets have had varying results; to what extent these dietary changes exert their influence via the intestinal microbiome is unknown.38
Eczema. Several studies have looked at the role of prebiotics and probiotics in reducing the risk for allergic disease. A 2013 Cochrane review found strong evidence that certain prebiotics can prevent eczema in children under age 2.15 There is limited evidence that probiotics may also play a role in preventing eczema.39,40 However, probiotics do not appear to be effective for treating eczema.41
Rheumatoid arthritis (RA). Patients with RA have a change in the balance of function of different T helper cells subsets, and several studies have shown that changes in the gut microbiome can affect this balance.42 A recent small study of patients with RA found that 75% of those with new onset RA had Prevotella copri bacteria as the predominant species, and patients with chronic RA had a decrease in Bacteroides species compared to healthy counterparts.42-44 The exact influence of gut flora dysbiosis on RA is unknown.45 Small studies suggest dietary changes may improve RA symptoms, while data on the use of probiotics to alleviate symptoms is mixed.46
What to tell patients about gut flora and health
There is increasing evidence that the gut microbiome and the genes contained therein have an impact on an individual’s health. (See TABLE 2 for additional resources.) The best preventive advice for patients and their families is to eat a diet rich in fruits and vegetables. This measure has well proven benefits beyond its potential effects on gut flora.

Correcting dysbiosis with diet or probiotics may play a role in treating chronic conditions; however, in many cases, further research is required to elucidate specific recommendations. In the meantime, given the safety profile of probiotics and dietary fiber, it is reasonable to consider using these interventions, particularly probiotics for treating IBS, ulcerative colitis, and acute infectious diarrhea; probiotics for preventing antibiotic-associated diarrhea and traveler’s diarrhea; and prebiotics for preventing eczema in high-risk infants.
CORRESPONDENCE
Jill Schneiderhan, MD, Family Medicine at Domino’s Farms, 24 Frank Lloyd Wright Dr., Lobby H, Suite 2300, Ann Arbor, MI 48105; jillsch@umich.edu.
1. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207-214.
2. Conlon MA, Bird AR. The impact of diet and lifestyle on gut microbiota and human health. Nutrients. 2015;7:17-44.
3. Nicholson JK, Holmes E, Kinross J, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262-1267.
4. Zhang YJ, Li S, Gan RY, et al. Impacts of gut bacteria on human health and diseases. Int J Mol Sci. 2015;16:7493-7519.
5. Tillisch K. The effects of gut microbiota on CNS function in humans. Gut Microbes. 2014;5:404-410.
6. Belizario JE, Napolitano M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front Microbiol. 2015;6:1050.
7. Le Chatelier E, Nielsen T, Qin J, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541-546.
8. Cotillard A, Kennedy SP, Kong LC, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500:585-588.
9. Ford AC, Quigley EM, Lacy BE, et al. Efficacy of prebiotics, probiotics, and synbiotics in irritable bowel syndrome and chronic idiopathic constipation: systematic review and meta-analysis. Am J Gastroenterol. 2014;109:1547-1561; quiz 1546,1562.
10. Fujiya M, Ueno N, Kohgo Y. Probiotic treatments for induction and maintenance of remission in inflammatory bowel diseases: a meta-analysis of randomized controlled trials. Clin J Gastroenterol. 2014;7(1):1-13.
11. Hempel S, Newberry SJ, Maher AR, et al. Probiotics for the prevention and treatment of antibiotic-associated diarrhea: a systematic review and meta-analysis. JAMA. 2012;307:1959-1969.
12. Szajewska H, Kolodziej M. Systematic review with meta-analysis: Saccharomyces boulardii in the prevention of antibiotic-associated diarrhoea. Aliment Pharmacol Ther. 2015;42:793–801.
13. Allen SJ, Martinez EG, Gregorio GV, et al. Probiotics for treating acute infectious diarrhoea. Cochrane Database Syst Rev. 2010(11):CD003048.
14. McFarland LV. Meta-analysis of probiotics for the prevention of traveler’s diarrhea. Travel Med Infect Dis. 2007;5:97-105.
15. Osborn DA, Sinn JK. Prebiotics in infants for prevention of allergy. The Cochrane Library. 2013. Cochrane Database Syst Rev. 2013;3:CD006474.
16. Razmpoosh E, Javadi M, Ejtahed HS, et al. Probiotics as beneficial agents in the management of diabetes mellitus: a systematic review. Diabetes Metab Res Rev. 2015. [Epub ahead of print].
17. Aguirre M, Eck A, Savelkoul PH, et al. Diet drives quick changes in the metabolic activity and composition of human gut microbiota in a validated in vitro gut model. Res Microbiol. 2015. [Epub ahead of print].
18. Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology. 2009;136:65-80.
19. Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: a clinical review. JAMA. 2015;313:949-958.
20. Moayyedi P, Quigley EM, Lacy BE, et al. The effect of fiber supplementation on irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol. 2014;109:1367-1374.
21. Simpson HL, Campbell BJ. Review article: dietary fibre-microbiota interactions. Aliment Pharmacol Ther. 2015;42:158-179.
22. Otten JJ, Hellwig JP, Meyers LD; Institute of Medicine of the National Academies. Dietary Reference Intakes: The essential guide to nutrient requirements. 2006. US Department of Agriculture Web site. Available at: http://www.nal.usda.gov/fnic/DRI/Essential_Guide/DRIEssentialGuideNutReq.pdf. Accessed December 8, 2015.
23. Hansen JJ, Sartor RB. Therapeutic manipulation of the microbiome in IBD: current results and future approaches. Curr Treat Options Gastroenterol. 2015;13:105-120.
24. Anderson JL, Edney RJ, Whelan K. Systematic review: faecal microbiota transplantation in the management of inflammatory bowel disease. Aliment Pharmacol Ther. 2012;36:503-516.
25. Cammarota G, Ianiro G, Gasbarrini A. Fecal microbiota transplantation for the treatment of Clostridium difficile infection: a systematic review. J Clin Gastroenterol. 2014;48:693-702.
26. Bermon S, Petriz B, Kajeniene A, et al. The microbiota: an exercise immunology perspective. Exerc Immunol Rev. 2015;21:70-79.
27. Hur KY, Lee MS. Gut microbiota and metabolic disorders. Diabetes Metab J. 2015;39:198-203.
28. Devaraj S, Hemarajata P, Versalovic J. The human gut microbiome and body metabolism: implications for obesity and diabetes. Clin Chem. 2013;59:617-628.
29. Park S, Bae JH. Probiotics for weight loss: a systematic review and meta-analysis. Nutr Res. 2015;35:566-575.
30. Petra AI, Panagiotidou S, Hatziagelaki E, et al. Gut-microbiotabrain axis and its effect on neuropsychiatric disorders with suspected immune dysregulation. Clin Ther. 2015;37:984-995.
31. Jiang H, Ling Z, Zhang Y, et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun. 2015;48:186-194.
32. Naseribafrouei A, Hestad K, Avershina E, et al. Correlation between the human fecal microbiota and depression. Neurogastroenterol Motil. 2014;26:1155-1162.
33. Tillisch K, Labus J, Kilpatrick L, et al. Consumption of fermented milk product with probiotic modulates brain activity. Gastroenterology. 2013;144:1394-1401.
34. Bested AC, Logan AC, Selhub EM. Intestinal microbiota, probiotics and mental health: from Metchnikoff to modern advances: part III - convergence toward clinical trials. Gut Pathog. 2013;5:4.
35. Krajmalnik-Brown R, Lozupone C, Kang DW, et al. Gut bacteria in children with autism spectrum disorders: challenges and promise of studying how a complex community influences a complex disease. Microb Ecol Health Dis. 2015;26:26914.
36. Buie T. Potential etiologic factors of microbiome disruption in autism. Clin Ther. 2015;37:976-983.
37. Cao X, Lin P, Jiang P, et al. Characteristics of the gastrointestinal microbiome in children with autism spectrum disorder: a systematic review. Shanghai Arch Psychiatry. 2013;25:342-353.
38. Frye RE, Slattery J, MacFabe DF, et al. Approaches to studying and manipulating the enteric microbiome to improve autism symptoms. Microb Ecol Health Dis. 2015;26:26878.
39. Osborn DA, Sinn JK. Probiotics in infants for prevention of allergic disease and food hypersensitivity. Cochrane Database Syst Rev. 2007;(4):CD006475.
40. Tang ML, Lahtinen SJ, Boyle RJ. Probiotics and prebiotics: clinical effects in allergic disease. Curr Opin Pediatr. 2010;22:626-634.
41. Boyle RJ, Bath-Hextall FJ, Leonardi-Bee J, et al. Probiotics for treating eczema. Cochrane Database Syst Rev. 2008;(4):CD006135.
42. Rogier R, Koenders MI, Abdollahi-Roodsaz S. Toll-like receptor mediated modulation of T cell response by commensal intestinal microbiota as a trigger for autoimmune arthritis. J Immunol Res. 2015;2015:527696.
43. Perez-Santiago Ja, Gianella Sa, Massanella Ma, et al. Gut Lactobacillales are associated with higher CD4 and less microbial translocation during HIV infection. AIDS. 2013;27:1921-1931.
44. Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202.
45. Scofield RH. Rheumatic diseases and the microbiome. Int J Rheum Dis. 2014;17:489-492.
46. Sandhya P, Danda D, Sharma D, et al. Does the buck stop with the bugs?: an overview of microbial dysbiosis in rheumatoid arthritis. Int J Rheum Dis. 2015. [Epub ahead of print].
› Encourage patients to eat a healthy diet that includes an adequate amount of soluble fiber to maintain a healthy, diverse microbiome. B
› Recommend combination probiotics to treat symptoms of irritable bowel syndrome. A
› Encourage patients to take probiotics containing Lactobacillus species to prevent antibiotic-associated diarrhea and Saccharomyces to prevent Clostridium difficile infection. A
› Recommend probiotics containing Lactobacillus species and/or Saccharomyces to treat acute infectious diarrhea. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 › Sheila S, age 27, has irritable bowel syndrome (IBS) and comes to your office for a follow-up visit. Over the past 6 months she has started taking a fiber supplement, drinking more water, and looking for links between stress and her symptoms. She has read about probiotics and wonders if you would consider recommending them in her situation.
CASE 2 › Mark M, age 45, has type 2 diabetes and is overweight. He is motivated to change his diet and has started to exercise more. He is taking metformin 2000 mg/d but his hemoglobin A1c remains slightly elevated at 7.2%. He heard on television that probiotics might help to keep him from needing to add another medication.
Most of the living organisms that comprise the human microbiome—all of the microbes that live on or in humans—are found in the gastrointestinal (GI) tract. The gut flora contribute 99% of the genetic material in the human body. The composition of the gut flora is remarkably diverse across the population; each individual has a unique microbial footprint. Within this microbial diversity, there appears to be a stable number of genes that are responsible for the major functions of the gut flora.1 These microbes:
- supply essential nutrients by breaking down complex carbohydrates;
- generate secondary bile acids that assist in digesting fats;2
- synthesize vitamins such as K, B12, folate, and biotin;3
- contribute to the defensive barrier in the colon by keeping pathogenic bacteria from crossing the colonic mucosa; and
- interact with our systemic immune system in a way that maintains a level of homeostasis, allowing for appropriate activation in the face of pathogens without developing autoimmunity.4
The gut flora also play a role in the communication between the central nervous system and the enteric nervous system by modulating the hormonal and neural pathways that have been labeled the “gut-brain axis.” The gut-brain axis has been associated with numerous disease states, including irritable bowel syndrome and certain psychiatric disorders.5
Researchers are investigating interventions that target the microbiome to increase microbial diversity and the presence of certain species to prevent or treat various diseases. The use of probiotics and dietary changes to increase intake of soluble fiber have been the most studied of these interventions. The thought is that these interventions can correct an imbalance, or dysbiosis, of the gut flora.6 Studies have shown that decreased microbial diversity is associated with elevations of certain disease markers (eg, adiposity, insulin, triglycerides, C–reactive protein)7 and that increases in soluble fiber lead to the greatest long-term improvement in microbial diversity.8 Fecal transplant—the transfer of a processed mixture of stool that contains “healthy” bacteria from a donor into the intestines of a patient—is being explored as a method of replacing colonic gut flora, but evidence is limited.
The following review takes a closer look at these options and identifies those that are most likely to benefit patients in the treatment—and prevention—of several diseases (TABLE 1).9-16
Evidence is best for using probiotics for digestive diseases
Dietary interventions for digestive diseases have long been studied, but are getting renewed attention for their potential impact on the microbiome.17 Beyond dietary modification, other similar treatment options include probiotics (live microorganisms thought to confer a beneficial effect on the host), prebiotics (non-digestible food ingredients, including oligosaccharides and inulin, thought to promote the growth of “helpful” gut flora), and synbiotics (combinations of the 2).18
Irritable bowel syndrome (IBS) is a heterogeneous disorder characterized by altered intestinal transit, low-grade colonic inflammation, and/or alterations in the gutbrain axis. Research has increasingly focused on recently discovered increases in intestinal immune activation, intestinal permeability, and alterations in the colonic microbiome (decreased diversity and increased pathogenic bacteria) associated with IBS.19
A meta-analysis of 43 randomized control trials (RCTs) found probiotics ranging from Lactobacillus to Saccharomyces can significantly decrease global IBS symptoms, abdominal pain, bloating, and flatulence.9 For a patient such as Ms. S, the evidence suggests a probiotic that contains a mixture of Lactobacillus and Bifidobacterium might help relieve her symptoms.9 In terms of dietary modifications, soluble fiber, which is already known to help treat IBS,20 has profound effects on improving microbiota diversity and in shifting the composition toward less pathogenic strains.21 The Institute of Medicine's daily recommended intake of soluble fiber is about 15 g/d.22
Inflammatory bowel disease (IBD) is caused by inflammation of the GI lining due to an overactive immune response. Evidence shows that patients with IBD have an altered microbial composition—specifically, an increase in bacteria that produce pro-inflammatory molecules and a decrease in bacteria that have a dampening effect on immune activation.23
Most studies evaluating probiotics as a treatment for IBD have been small and have used a wide variety of bacterial mixtures, which makes comparisons difficult. Recent meta-analyses found combination probiotics can both induce and maintain remission in patients with ulcerative colitis, but have no beneficial effects in Crohn’s disease.10 In a review of 9 case series of patients with IBD, fecal transplant reduced IBD symptoms, and patients were able to decrease medication use.24
Diarrheal illness. The human intestine is protected from diarrheal illness by healthy bacteria that block the actions of pathogenic bacteria. This mechanism is called colonization resistance. Moderate levels of evidence support the use of probiotics to prevent or treat several types of diarrheal illness.14
Antibiotic-associated diarrhea (AAD) is caused when antibiotic use alters the microbial balance. Recent meta-analyses have shown probiotics can prevent AAD and Clostridium difficile-associated diarrhea.11,12 Several case series and one RCT have found that fecal transplants are safe and efficacious for treating recurrent Clostridium difficile infection.25 Using probiotics to treat symptoms of AAD has been less studied.
Acute infectious diarrhea and traveler’s diarrhea (TD). A Cochrane review found that probiotics decreased the duration of diarrheal episodes by 25 hours, decreased the risk of an episode lasting more than 4 days by 59%, and led to one less diarrheal stool per day by the second day of the intervention.13 In a separate meta-analysis of 12 studies, probiotics significantly prevented 85% of cases of TD.14
Encouraging early evidence for several other illnesses
Metabolic disorders. Both animal and human studies support the theory that gut flora contribute to energy homeostasis, and in some genetically predisposed people dysbiosis may lead to obesity and diabetes. The traditional western diet4 and possibly decreased physical activity26 are major contributors to gut flora dysbiosis. Healthy bacteria in the gut break down soluble fiber into short chain fatty acids (SCFAs). SCFAs are associated with increased satiety, decreased food intake, lower levels of inflammation, and improvement in insulin signaling in adipose tissue. In addition to decreased SFCA production, dysbiosis also leads to increased lipid deposition through higher levels of lipoprotein lipase.27
Obesity. The bacteria in our gut affect energy metabolism. In patients with obesity, increased amounts of bacteria in the taxa Firmicutes and a corresponding decrease in Bacteroidetes is associated with an increased energy harvest and decreased SCFA production, which leads to a pro-inflammatory state.28 Probiotics that contain Bifidobacterium and Lactobacillus are thought to help correct this dysbiosis by increasing production of SCFAs.28
A recent meta-analysis of 4 RCTs found no significant difference between supplementation with probiotics and placebo on weight reduction.29 However, lower-quality studies with more subjects and longer duration have shown a statistically significant improvement in weight reduction with probiotic use compared to placebo.29
Diabetes. Although dietary interventions to improve glycemic control have long been an important cornerstone of treatment, probiotic supplementation to further alter gut flora composition is also being evaluated. Studies have found probiotics have largely beneficial effects on glycemic control, especially in animals. The largest systematic review to date looked at 33 studies, including 5 human trials. The human studies each found a significant reduction in at least one of 6 parameters of glycemic control (levels of fasting plasma glucose, postprandial blood glucose, glycated hemoglobin, insulin, insulin resistance, and onset of diabetes).16 It is unclear which probiotic strains confer benefit, and if those benefits are sustainable without dietary modification and increased physical activity.
Psychiatric illnesses. The gut-brain axis is thought to impact mental health by several mechanisms, including modulating the hypothalamic-pituitary-adrenal axis, activating the immune system, producing active metabolites, and affecting the vagus nerve. It is unclear which of these pathways may be clinically relevant.5,30 The few human studies that have looked for a potential link between gut flora and psychiatric illness have focused on depression and autism spectrum disorders (ASD).
Depression. Small studies comparing the microbiome composition of depressed patients vs healthy controls have found differences in patterns of both over- and underrepresented microbiota species in depressed patients, although the patterns across studies have been inconsistent.31,32 One small functional magnetic resonance imaging study of healthy women showed that a fermented milk product that contained probiotics affected activity in areas of the brain that control emotion and sensation.33 A few small studies have shown that patients who used probiotics had improved depression scores.34 Further studies are needed.
ASD. Children with ASD have GI disturbances—most commonly diarrhea, constipation, and/or bloating—more often than healthy controls.35,36 This association has led to speculation of a connection between the gut and brain. The microbial composition and diversity appears to be different in individuals with ASD; several studies have found an increase in Clostridia species.37
Research on probiotics for treating ASD has been primarily in preclinical models. Human studies of probiotics for ASD are lacking.38 Small studies on dietary modifications such as gluten-free and casein-free diets have had varying results; to what extent these dietary changes exert their influence via the intestinal microbiome is unknown.38
Eczema. Several studies have looked at the role of prebiotics and probiotics in reducing the risk for allergic disease. A 2013 Cochrane review found strong evidence that certain prebiotics can prevent eczema in children under age 2.15 There is limited evidence that probiotics may also play a role in preventing eczema.39,40 However, probiotics do not appear to be effective for treating eczema.41
Rheumatoid arthritis (RA). Patients with RA have a change in the balance of function of different T helper cells subsets, and several studies have shown that changes in the gut microbiome can affect this balance.42 A recent small study of patients with RA found that 75% of those with new onset RA had Prevotella copri bacteria as the predominant species, and patients with chronic RA had a decrease in Bacteroides species compared to healthy counterparts.42-44 The exact influence of gut flora dysbiosis on RA is unknown.45 Small studies suggest dietary changes may improve RA symptoms, while data on the use of probiotics to alleviate symptoms is mixed.46
What to tell patients about gut flora and health
There is increasing evidence that the gut microbiome and the genes contained therein have an impact on an individual’s health. (See TABLE 2 for additional resources.) The best preventive advice for patients and their families is to eat a diet rich in fruits and vegetables. This measure has well proven benefits beyond its potential effects on gut flora.
Correcting dysbiosis with diet or probiotics may play a role in treating chronic conditions; however, in many cases, further research is required to elucidate specific recommendations. In the meantime, given the safety profile of probiotics and dietary fiber, it is reasonable to consider using these interventions, particularly probiotics for treating IBS, ulcerative colitis, and acute infectious diarrhea; probiotics for preventing antibiotic-associated diarrhea and traveler’s diarrhea; and prebiotics for preventing eczema in high-risk infants.
CORRESPONDENCE
Jill Schneiderhan, MD, Family Medicine at Domino’s Farms, 24 Frank Lloyd Wright Dr., Lobby H, Suite 2300, Ann Arbor, MI 48105; jillsch@umich.edu.
› Encourage patients to eat a healthy diet that includes an adequate amount of soluble fiber to maintain a healthy, diverse microbiome. B
› Recommend combination probiotics to treat symptoms of irritable bowel syndrome. A
› Encourage patients to take probiotics containing Lactobacillus species to prevent antibiotic-associated diarrhea and Saccharomyces to prevent Clostridium difficile infection. A
› Recommend probiotics containing Lactobacillus species and/or Saccharomyces to treat acute infectious diarrhea. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 › Sheila S, age 27, has irritable bowel syndrome (IBS) and comes to your office for a follow-up visit. Over the past 6 months she has started taking a fiber supplement, drinking more water, and looking for links between stress and her symptoms. She has read about probiotics and wonders if you would consider recommending them in her situation.
CASE 2 › Mark M, age 45, has type 2 diabetes and is overweight. He is motivated to change his diet and has started to exercise more. He is taking metformin 2000 mg/d but his hemoglobin A1c remains slightly elevated at 7.2%. He heard on television that probiotics might help to keep him from needing to add another medication.
Most of the living organisms that comprise the human microbiome—all of the microbes that live on or in humans—are found in the gastrointestinal (GI) tract. The gut flora contribute 99% of the genetic material in the human body. The composition of the gut flora is remarkably diverse across the population; each individual has a unique microbial footprint. Within this microbial diversity, there appears to be a stable number of genes that are responsible for the major functions of the gut flora.1 These microbes:
- supply essential nutrients by breaking down complex carbohydrates;
- generate secondary bile acids that assist in digesting fats;2
- synthesize vitamins such as K, B12, folate, and biotin;3
- contribute to the defensive barrier in the colon by keeping pathogenic bacteria from crossing the colonic mucosa; and
- interact with our systemic immune system in a way that maintains a level of homeostasis, allowing for appropriate activation in the face of pathogens without developing autoimmunity.4
The gut flora also play a role in the communication between the central nervous system and the enteric nervous system by modulating the hormonal and neural pathways that have been labeled the “gut-brain axis.” The gut-brain axis has been associated with numerous disease states, including irritable bowel syndrome and certain psychiatric disorders.5
Researchers are investigating interventions that target the microbiome to increase microbial diversity and the presence of certain species to prevent or treat various diseases. The use of probiotics and dietary changes to increase intake of soluble fiber have been the most studied of these interventions. The thought is that these interventions can correct an imbalance, or dysbiosis, of the gut flora.6 Studies have shown that decreased microbial diversity is associated with elevations of certain disease markers (eg, adiposity, insulin, triglycerides, C–reactive protein)7 and that increases in soluble fiber lead to the greatest long-term improvement in microbial diversity.8 Fecal transplant—the transfer of a processed mixture of stool that contains “healthy” bacteria from a donor into the intestines of a patient—is being explored as a method of replacing colonic gut flora, but evidence is limited.
The following review takes a closer look at these options and identifies those that are most likely to benefit patients in the treatment—and prevention—of several diseases (TABLE 1).9-16
Evidence is best for using probiotics for digestive diseases
Dietary interventions for digestive diseases have long been studied, but are getting renewed attention for their potential impact on the microbiome.17 Beyond dietary modification, other similar treatment options include probiotics (live microorganisms thought to confer a beneficial effect on the host), prebiotics (non-digestible food ingredients, including oligosaccharides and inulin, thought to promote the growth of “helpful” gut flora), and synbiotics (combinations of the 2).18
Irritable bowel syndrome (IBS) is a heterogeneous disorder characterized by altered intestinal transit, low-grade colonic inflammation, and/or alterations in the gutbrain axis. Research has increasingly focused on recently discovered increases in intestinal immune activation, intestinal permeability, and alterations in the colonic microbiome (decreased diversity and increased pathogenic bacteria) associated with IBS.19
A meta-analysis of 43 randomized control trials (RCTs) found probiotics ranging from Lactobacillus to Saccharomyces can significantly decrease global IBS symptoms, abdominal pain, bloating, and flatulence.9 For a patient such as Ms. S, the evidence suggests a probiotic that contains a mixture of Lactobacillus and Bifidobacterium might help relieve her symptoms.9 In terms of dietary modifications, soluble fiber, which is already known to help treat IBS,20 has profound effects on improving microbiota diversity and in shifting the composition toward less pathogenic strains.21 The Institute of Medicine's daily recommended intake of soluble fiber is about 15 g/d.22
Inflammatory bowel disease (IBD) is caused by inflammation of the GI lining due to an overactive immune response. Evidence shows that patients with IBD have an altered microbial composition—specifically, an increase in bacteria that produce pro-inflammatory molecules and a decrease in bacteria that have a dampening effect on immune activation.23
Most studies evaluating probiotics as a treatment for IBD have been small and have used a wide variety of bacterial mixtures, which makes comparisons difficult. Recent meta-analyses found combination probiotics can both induce and maintain remission in patients with ulcerative colitis, but have no beneficial effects in Crohn’s disease.10 In a review of 9 case series of patients with IBD, fecal transplant reduced IBD symptoms, and patients were able to decrease medication use.24
Diarrheal illness. The human intestine is protected from diarrheal illness by healthy bacteria that block the actions of pathogenic bacteria. This mechanism is called colonization resistance. Moderate levels of evidence support the use of probiotics to prevent or treat several types of diarrheal illness.14
Antibiotic-associated diarrhea (AAD) is caused when antibiotic use alters the microbial balance. Recent meta-analyses have shown probiotics can prevent AAD and Clostridium difficile-associated diarrhea.11,12 Several case series and one RCT have found that fecal transplants are safe and efficacious for treating recurrent Clostridium difficile infection.25 Using probiotics to treat symptoms of AAD has been less studied.
Acute infectious diarrhea and traveler’s diarrhea (TD). A Cochrane review found that probiotics decreased the duration of diarrheal episodes by 25 hours, decreased the risk of an episode lasting more than 4 days by 59%, and led to one less diarrheal stool per day by the second day of the intervention.13 In a separate meta-analysis of 12 studies, probiotics significantly prevented 85% of cases of TD.14
Encouraging early evidence for several other illnesses
Metabolic disorders. Both animal and human studies support the theory that gut flora contribute to energy homeostasis, and in some genetically predisposed people dysbiosis may lead to obesity and diabetes. The traditional western diet4 and possibly decreased physical activity26 are major contributors to gut flora dysbiosis. Healthy bacteria in the gut break down soluble fiber into short chain fatty acids (SCFAs). SCFAs are associated with increased satiety, decreased food intake, lower levels of inflammation, and improvement in insulin signaling in adipose tissue. In addition to decreased SFCA production, dysbiosis also leads to increased lipid deposition through higher levels of lipoprotein lipase.27
Obesity. The bacteria in our gut affect energy metabolism. In patients with obesity, increased amounts of bacteria in the taxa Firmicutes and a corresponding decrease in Bacteroidetes is associated with an increased energy harvest and decreased SCFA production, which leads to a pro-inflammatory state.28 Probiotics that contain Bifidobacterium and Lactobacillus are thought to help correct this dysbiosis by increasing production of SCFAs.28
A recent meta-analysis of 4 RCTs found no significant difference between supplementation with probiotics and placebo on weight reduction.29 However, lower-quality studies with more subjects and longer duration have shown a statistically significant improvement in weight reduction with probiotic use compared to placebo.29
Diabetes. Although dietary interventions to improve glycemic control have long been an important cornerstone of treatment, probiotic supplementation to further alter gut flora composition is also being evaluated. Studies have found probiotics have largely beneficial effects on glycemic control, especially in animals. The largest systematic review to date looked at 33 studies, including 5 human trials. The human studies each found a significant reduction in at least one of 6 parameters of glycemic control (levels of fasting plasma glucose, postprandial blood glucose, glycated hemoglobin, insulin, insulin resistance, and onset of diabetes).16 It is unclear which probiotic strains confer benefit, and if those benefits are sustainable without dietary modification and increased physical activity.
Psychiatric illnesses. The gut-brain axis is thought to impact mental health by several mechanisms, including modulating the hypothalamic-pituitary-adrenal axis, activating the immune system, producing active metabolites, and affecting the vagus nerve. It is unclear which of these pathways may be clinically relevant.5,30 The few human studies that have looked for a potential link between gut flora and psychiatric illness have focused on depression and autism spectrum disorders (ASD).
Depression. Small studies comparing the microbiome composition of depressed patients vs healthy controls have found differences in patterns of both over- and underrepresented microbiota species in depressed patients, although the patterns across studies have been inconsistent.31,32 One small functional magnetic resonance imaging study of healthy women showed that a fermented milk product that contained probiotics affected activity in areas of the brain that control emotion and sensation.33 A few small studies have shown that patients who used probiotics had improved depression scores.34 Further studies are needed.
ASD. Children with ASD have GI disturbances—most commonly diarrhea, constipation, and/or bloating—more often than healthy controls.35,36 This association has led to speculation of a connection between the gut and brain. The microbial composition and diversity appears to be different in individuals with ASD; several studies have found an increase in Clostridia species.37
Research on probiotics for treating ASD has been primarily in preclinical models. Human studies of probiotics for ASD are lacking.38 Small studies on dietary modifications such as gluten-free and casein-free diets have had varying results; to what extent these dietary changes exert their influence via the intestinal microbiome is unknown.38
Eczema. Several studies have looked at the role of prebiotics and probiotics in reducing the risk for allergic disease. A 2013 Cochrane review found strong evidence that certain prebiotics can prevent eczema in children under age 2.15 There is limited evidence that probiotics may also play a role in preventing eczema.39,40 However, probiotics do not appear to be effective for treating eczema.41
Rheumatoid arthritis (RA). Patients with RA have a change in the balance of function of different T helper cells subsets, and several studies have shown that changes in the gut microbiome can affect this balance.42 A recent small study of patients with RA found that 75% of those with new onset RA had Prevotella copri bacteria as the predominant species, and patients with chronic RA had a decrease in Bacteroides species compared to healthy counterparts.42-44 The exact influence of gut flora dysbiosis on RA is unknown.45 Small studies suggest dietary changes may improve RA symptoms, while data on the use of probiotics to alleviate symptoms is mixed.46
What to tell patients about gut flora and health
There is increasing evidence that the gut microbiome and the genes contained therein have an impact on an individual’s health. (See TABLE 2 for additional resources.) The best preventive advice for patients and their families is to eat a diet rich in fruits and vegetables. This measure has well proven benefits beyond its potential effects on gut flora.
Correcting dysbiosis with diet or probiotics may play a role in treating chronic conditions; however, in many cases, further research is required to elucidate specific recommendations. In the meantime, given the safety profile of probiotics and dietary fiber, it is reasonable to consider using these interventions, particularly probiotics for treating IBS, ulcerative colitis, and acute infectious diarrhea; probiotics for preventing antibiotic-associated diarrhea and traveler’s diarrhea; and prebiotics for preventing eczema in high-risk infants.
CORRESPONDENCE
Jill Schneiderhan, MD, Family Medicine at Domino’s Farms, 24 Frank Lloyd Wright Dr., Lobby H, Suite 2300, Ann Arbor, MI 48105; jillsch@umich.edu.
1. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207-214.
2. Conlon MA, Bird AR. The impact of diet and lifestyle on gut microbiota and human health. Nutrients. 2015;7:17-44.
3. Nicholson JK, Holmes E, Kinross J, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262-1267.
4. Zhang YJ, Li S, Gan RY, et al. Impacts of gut bacteria on human health and diseases. Int J Mol Sci. 2015;16:7493-7519.
5. Tillisch K. The effects of gut microbiota on CNS function in humans. Gut Microbes. 2014;5:404-410.
6. Belizario JE, Napolitano M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front Microbiol. 2015;6:1050.
7. Le Chatelier E, Nielsen T, Qin J, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541-546.
8. Cotillard A, Kennedy SP, Kong LC, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500:585-588.
9. Ford AC, Quigley EM, Lacy BE, et al. Efficacy of prebiotics, probiotics, and synbiotics in irritable bowel syndrome and chronic idiopathic constipation: systematic review and meta-analysis. Am J Gastroenterol. 2014;109:1547-1561; quiz 1546,1562.
10. Fujiya M, Ueno N, Kohgo Y. Probiotic treatments for induction and maintenance of remission in inflammatory bowel diseases: a meta-analysis of randomized controlled trials. Clin J Gastroenterol. 2014;7(1):1-13.
11. Hempel S, Newberry SJ, Maher AR, et al. Probiotics for the prevention and treatment of antibiotic-associated diarrhea: a systematic review and meta-analysis. JAMA. 2012;307:1959-1969.
12. Szajewska H, Kolodziej M. Systematic review with meta-analysis: Saccharomyces boulardii in the prevention of antibiotic-associated diarrhoea. Aliment Pharmacol Ther. 2015;42:793–801.
13. Allen SJ, Martinez EG, Gregorio GV, et al. Probiotics for treating acute infectious diarrhoea. Cochrane Database Syst Rev. 2010(11):CD003048.
14. McFarland LV. Meta-analysis of probiotics for the prevention of traveler’s diarrhea. Travel Med Infect Dis. 2007;5:97-105.
15. Osborn DA, Sinn JK. Prebiotics in infants for prevention of allergy. The Cochrane Library. 2013. Cochrane Database Syst Rev. 2013;3:CD006474.
16. Razmpoosh E, Javadi M, Ejtahed HS, et al. Probiotics as beneficial agents in the management of diabetes mellitus: a systematic review. Diabetes Metab Res Rev. 2015. [Epub ahead of print].
17. Aguirre M, Eck A, Savelkoul PH, et al. Diet drives quick changes in the metabolic activity and composition of human gut microbiota in a validated in vitro gut model. Res Microbiol. 2015. [Epub ahead of print].
18. Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology. 2009;136:65-80.
19. Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: a clinical review. JAMA. 2015;313:949-958.
20. Moayyedi P, Quigley EM, Lacy BE, et al. The effect of fiber supplementation on irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol. 2014;109:1367-1374.
21. Simpson HL, Campbell BJ. Review article: dietary fibre-microbiota interactions. Aliment Pharmacol Ther. 2015;42:158-179.
22. Otten JJ, Hellwig JP, Meyers LD; Institute of Medicine of the National Academies. Dietary Reference Intakes: The essential guide to nutrient requirements. 2006. US Department of Agriculture Web site. Available at: http://www.nal.usda.gov/fnic/DRI/Essential_Guide/DRIEssentialGuideNutReq.pdf. Accessed December 8, 2015.
23. Hansen JJ, Sartor RB. Therapeutic manipulation of the microbiome in IBD: current results and future approaches. Curr Treat Options Gastroenterol. 2015;13:105-120.
24. Anderson JL, Edney RJ, Whelan K. Systematic review: faecal microbiota transplantation in the management of inflammatory bowel disease. Aliment Pharmacol Ther. 2012;36:503-516.
25. Cammarota G, Ianiro G, Gasbarrini A. Fecal microbiota transplantation for the treatment of Clostridium difficile infection: a systematic review. J Clin Gastroenterol. 2014;48:693-702.
26. Bermon S, Petriz B, Kajeniene A, et al. The microbiota: an exercise immunology perspective. Exerc Immunol Rev. 2015;21:70-79.
27. Hur KY, Lee MS. Gut microbiota and metabolic disorders. Diabetes Metab J. 2015;39:198-203.
28. Devaraj S, Hemarajata P, Versalovic J. The human gut microbiome and body metabolism: implications for obesity and diabetes. Clin Chem. 2013;59:617-628.
29. Park S, Bae JH. Probiotics for weight loss: a systematic review and meta-analysis. Nutr Res. 2015;35:566-575.
30. Petra AI, Panagiotidou S, Hatziagelaki E, et al. Gut-microbiotabrain axis and its effect on neuropsychiatric disorders with suspected immune dysregulation. Clin Ther. 2015;37:984-995.
31. Jiang H, Ling Z, Zhang Y, et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun. 2015;48:186-194.
32. Naseribafrouei A, Hestad K, Avershina E, et al. Correlation between the human fecal microbiota and depression. Neurogastroenterol Motil. 2014;26:1155-1162.
33. Tillisch K, Labus J, Kilpatrick L, et al. Consumption of fermented milk product with probiotic modulates brain activity. Gastroenterology. 2013;144:1394-1401.
34. Bested AC, Logan AC, Selhub EM. Intestinal microbiota, probiotics and mental health: from Metchnikoff to modern advances: part III - convergence toward clinical trials. Gut Pathog. 2013;5:4.
35. Krajmalnik-Brown R, Lozupone C, Kang DW, et al. Gut bacteria in children with autism spectrum disorders: challenges and promise of studying how a complex community influences a complex disease. Microb Ecol Health Dis. 2015;26:26914.
36. Buie T. Potential etiologic factors of microbiome disruption in autism. Clin Ther. 2015;37:976-983.
37. Cao X, Lin P, Jiang P, et al. Characteristics of the gastrointestinal microbiome in children with autism spectrum disorder: a systematic review. Shanghai Arch Psychiatry. 2013;25:342-353.
38. Frye RE, Slattery J, MacFabe DF, et al. Approaches to studying and manipulating the enteric microbiome to improve autism symptoms. Microb Ecol Health Dis. 2015;26:26878.
39. Osborn DA, Sinn JK. Probiotics in infants for prevention of allergic disease and food hypersensitivity. Cochrane Database Syst Rev. 2007;(4):CD006475.
40. Tang ML, Lahtinen SJ, Boyle RJ. Probiotics and prebiotics: clinical effects in allergic disease. Curr Opin Pediatr. 2010;22:626-634.
41. Boyle RJ, Bath-Hextall FJ, Leonardi-Bee J, et al. Probiotics for treating eczema. Cochrane Database Syst Rev. 2008;(4):CD006135.
42. Rogier R, Koenders MI, Abdollahi-Roodsaz S. Toll-like receptor mediated modulation of T cell response by commensal intestinal microbiota as a trigger for autoimmune arthritis. J Immunol Res. 2015;2015:527696.
43. Perez-Santiago Ja, Gianella Sa, Massanella Ma, et al. Gut Lactobacillales are associated with higher CD4 and less microbial translocation during HIV infection. AIDS. 2013;27:1921-1931.
44. Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202.
45. Scofield RH. Rheumatic diseases and the microbiome. Int J Rheum Dis. 2014;17:489-492.
46. Sandhya P, Danda D, Sharma D, et al. Does the buck stop with the bugs?: an overview of microbial dysbiosis in rheumatoid arthritis. Int J Rheum Dis. 2015. [Epub ahead of print].
1. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207-214.
2. Conlon MA, Bird AR. The impact of diet and lifestyle on gut microbiota and human health. Nutrients. 2015;7:17-44.
3. Nicholson JK, Holmes E, Kinross J, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262-1267.
4. Zhang YJ, Li S, Gan RY, et al. Impacts of gut bacteria on human health and diseases. Int J Mol Sci. 2015;16:7493-7519.
5. Tillisch K. The effects of gut microbiota on CNS function in humans. Gut Microbes. 2014;5:404-410.
6. Belizario JE, Napolitano M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front Microbiol. 2015;6:1050.
7. Le Chatelier E, Nielsen T, Qin J, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541-546.
8. Cotillard A, Kennedy SP, Kong LC, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500:585-588.
9. Ford AC, Quigley EM, Lacy BE, et al. Efficacy of prebiotics, probiotics, and synbiotics in irritable bowel syndrome and chronic idiopathic constipation: systematic review and meta-analysis. Am J Gastroenterol. 2014;109:1547-1561; quiz 1546,1562.
10. Fujiya M, Ueno N, Kohgo Y. Probiotic treatments for induction and maintenance of remission in inflammatory bowel diseases: a meta-analysis of randomized controlled trials. Clin J Gastroenterol. 2014;7(1):1-13.
11. Hempel S, Newberry SJ, Maher AR, et al. Probiotics for the prevention and treatment of antibiotic-associated diarrhea: a systematic review and meta-analysis. JAMA. 2012;307:1959-1969.
12. Szajewska H, Kolodziej M. Systematic review with meta-analysis: Saccharomyces boulardii in the prevention of antibiotic-associated diarrhoea. Aliment Pharmacol Ther. 2015;42:793–801.
13. Allen SJ, Martinez EG, Gregorio GV, et al. Probiotics for treating acute infectious diarrhoea. Cochrane Database Syst Rev. 2010(11):CD003048.
14. McFarland LV. Meta-analysis of probiotics for the prevention of traveler’s diarrhea. Travel Med Infect Dis. 2007;5:97-105.
15. Osborn DA, Sinn JK. Prebiotics in infants for prevention of allergy. The Cochrane Library. 2013. Cochrane Database Syst Rev. 2013;3:CD006474.
16. Razmpoosh E, Javadi M, Ejtahed HS, et al. Probiotics as beneficial agents in the management of diabetes mellitus: a systematic review. Diabetes Metab Res Rev. 2015. [Epub ahead of print].
17. Aguirre M, Eck A, Savelkoul PH, et al. Diet drives quick changes in the metabolic activity and composition of human gut microbiota in a validated in vitro gut model. Res Microbiol. 2015. [Epub ahead of print].
18. Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology. 2009;136:65-80.
19. Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: a clinical review. JAMA. 2015;313:949-958.
20. Moayyedi P, Quigley EM, Lacy BE, et al. The effect of fiber supplementation on irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol. 2014;109:1367-1374.
21. Simpson HL, Campbell BJ. Review article: dietary fibre-microbiota interactions. Aliment Pharmacol Ther. 2015;42:158-179.
22. Otten JJ, Hellwig JP, Meyers LD; Institute of Medicine of the National Academies. Dietary Reference Intakes: The essential guide to nutrient requirements. 2006. US Department of Agriculture Web site. Available at: http://www.nal.usda.gov/fnic/DRI/Essential_Guide/DRIEssentialGuideNutReq.pdf. Accessed December 8, 2015.
23. Hansen JJ, Sartor RB. Therapeutic manipulation of the microbiome in IBD: current results and future approaches. Curr Treat Options Gastroenterol. 2015;13:105-120.
24. Anderson JL, Edney RJ, Whelan K. Systematic review: faecal microbiota transplantation in the management of inflammatory bowel disease. Aliment Pharmacol Ther. 2012;36:503-516.
25. Cammarota G, Ianiro G, Gasbarrini A. Fecal microbiota transplantation for the treatment of Clostridium difficile infection: a systematic review. J Clin Gastroenterol. 2014;48:693-702.
26. Bermon S, Petriz B, Kajeniene A, et al. The microbiota: an exercise immunology perspective. Exerc Immunol Rev. 2015;21:70-79.
27. Hur KY, Lee MS. Gut microbiota and metabolic disorders. Diabetes Metab J. 2015;39:198-203.
28. Devaraj S, Hemarajata P, Versalovic J. The human gut microbiome and body metabolism: implications for obesity and diabetes. Clin Chem. 2013;59:617-628.
29. Park S, Bae JH. Probiotics for weight loss: a systematic review and meta-analysis. Nutr Res. 2015;35:566-575.
30. Petra AI, Panagiotidou S, Hatziagelaki E, et al. Gut-microbiotabrain axis and its effect on neuropsychiatric disorders with suspected immune dysregulation. Clin Ther. 2015;37:984-995.
31. Jiang H, Ling Z, Zhang Y, et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun. 2015;48:186-194.
32. Naseribafrouei A, Hestad K, Avershina E, et al. Correlation between the human fecal microbiota and depression. Neurogastroenterol Motil. 2014;26:1155-1162.
33. Tillisch K, Labus J, Kilpatrick L, et al. Consumption of fermented milk product with probiotic modulates brain activity. Gastroenterology. 2013;144:1394-1401.
34. Bested AC, Logan AC, Selhub EM. Intestinal microbiota, probiotics and mental health: from Metchnikoff to modern advances: part III - convergence toward clinical trials. Gut Pathog. 2013;5:4.
35. Krajmalnik-Brown R, Lozupone C, Kang DW, et al. Gut bacteria in children with autism spectrum disorders: challenges and promise of studying how a complex community influences a complex disease. Microb Ecol Health Dis. 2015;26:26914.
36. Buie T. Potential etiologic factors of microbiome disruption in autism. Clin Ther. 2015;37:976-983.
37. Cao X, Lin P, Jiang P, et al. Characteristics of the gastrointestinal microbiome in children with autism spectrum disorder: a systematic review. Shanghai Arch Psychiatry. 2013;25:342-353.
38. Frye RE, Slattery J, MacFabe DF, et al. Approaches to studying and manipulating the enteric microbiome to improve autism symptoms. Microb Ecol Health Dis. 2015;26:26878.
39. Osborn DA, Sinn JK. Probiotics in infants for prevention of allergic disease and food hypersensitivity. Cochrane Database Syst Rev. 2007;(4):CD006475.
40. Tang ML, Lahtinen SJ, Boyle RJ. Probiotics and prebiotics: clinical effects in allergic disease. Curr Opin Pediatr. 2010;22:626-634.
41. Boyle RJ, Bath-Hextall FJ, Leonardi-Bee J, et al. Probiotics for treating eczema. Cochrane Database Syst Rev. 2008;(4):CD006135.
42. Rogier R, Koenders MI, Abdollahi-Roodsaz S. Toll-like receptor mediated modulation of T cell response by commensal intestinal microbiota as a trigger for autoimmune arthritis. J Immunol Res. 2015;2015:527696.
43. Perez-Santiago Ja, Gianella Sa, Massanella Ma, et al. Gut Lactobacillales are associated with higher CD4 and less microbial translocation during HIV infection. AIDS. 2013;27:1921-1931.
44. Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202.
45. Scofield RH. Rheumatic diseases and the microbiome. Int J Rheum Dis. 2014;17:489-492.
46. Sandhya P, Danda D, Sharma D, et al. Does the buck stop with the bugs?: an overview of microbial dysbiosis in rheumatoid arthritis. Int J Rheum Dis. 2015. [Epub ahead of print].
Pediatric Dermatology Consult - January 2016
By Ellen S. Haddock and Lawrence F. Eichenfield, M.D.
Urticaria multiforme
Although not the most classic presentation, this patient’s migrating rash is most consistent with urticaria (hives). Urticaria is dermal edema which causes transient edematous and usually pruritic wheals.1,2 Each individual lesion lasts less than 24 hours and disappears without leaving a mark. Urticaria is caused by mast cell activation, which leads to release of antihistamines and other substances that increase capillary and venule permeability, allowing fluid to leak into the extravascular space.1 In children, mast cell activation is usually triggered by infections, drugs, or foods.1
Classic urticaria consists of large, pruritic plaques and may be associated with airway edema. However, urticaria also can present with annular and polycyclic lesions, which may be less pruritic and are not associated with airway edema.3 This “multiple redness” is distinct from erythema multiforme (EM), although often urticaria is confused with EM. Lesions of EM are annular and typically have purpuric or dusky centers, with each lesion lasting a minimum of 1 week.4 Annular lesions in urticaria usually do not have central duskiness or blisters. Often the centers of annular urticaria lesions are relatively normal and edges are raised. Some urticaria, especially in younger children as in this case, is sometimes called “urticaria multiforme” because the ecchymotic centers are reminiscent of, but distinct from, classic target lesions of EM. Urticaria multiforme is commonly misdiagnosed as EM, with 29% of patients originally misdiagnosed in one study.3
Urticaria multiforme occurs most commonly in infants and preschool-aged children,5 although it has been diagnosed in patients as old as 18 years.6 Patients often have had an antecedent bacterial or viral illness, recent treatment with antibiotics, or recent vaccination (67%, 44%, and 11% of patients, respectively, in one series).4 In contrast with classic urticaria, urticaria multiforme has not been associated with food allergy.4
In this case, urticaria multiforme was likely caused by a hypersensitivity reaction to amoxicillin. A reaction to nitrofurantoin was less likely because the patient had been taking it continuously for months without any complications.
Differential diagnosis
The differential diagnosis for urticaria multiforme includes EM and a serum sickness–like reaction. The main clue that this patient’s rash was a subtype of urticaria rather than EM was its transience, with individual lesions appearing and disappearing in less than a day.4 In contrast, the lesions of EM are fixed, persisting for a week or longer. While urticaria multiforme may have central ecchymosis (termed “hemorrhagic urticaria”) that looks similar to the dusky centers of EM lesions and persists longer than the transient edematous plaques,1 it resolves quickly with appropriate treatment.4 In contrast, the dusky centers of EM, which are caused by epidermal necrosis, take longer to resolve.4 Dermatographism, if present, would support a diagnosis of urticaria rather than EM. Similarly, facial or acral edema, if present, would support a diagnosis of urticaria multiforme; they are uncommon in EM. In contrast, any necrosis, blistering, or erosions in the centers of the annular lesions or on mucosal membranes would suggest EM, as necrosis, blistering, erosions, and mucosal involvement do not occur in urticaria multiforme.4 We stress that in EM, “the center of the lesion is the center of the action,” while in urticaria, wheals often have relatively normal centers.
Although both urticaria and EM lesions may be pruritic, any burning sensation is more suggestive of EM.4 Urticaria multiforme is often associated with antibiotics, vaccinations, and upper respiratory infections, while EM is most commonly associated with herpes simplex infection.4,7
Urticaria multiforme also may appear similar to a serum sickness–like reaction, which is another kind of hypersensitivity reaction triggered by the administration of antibiotics. It is most commonly associated with cefaclor, but also is associated with other antibiotics including amoxicillin.8 As with urticaria, hypersensitivity drug eruptions and serum sickness-like reactions may present with purpuric, polycyclic wheals with central clearing. However, as with EM, the lesions of serum sickness–like reactions are fixed, lasting for days to weeks.4 Facial or acral angioedema may occur in both urticaria multiforme and serum sickness–like reactions, but serum sickness–like reactions are not associated with dermatographism.4 Furthermore, serum sickness–like reactions are typically associated with high-grade fever, myalgia, arthralgia, and lymphadenopathy, which are not seen in urticaria multiforme.4,5
Diagnosis of urticaria multiforme usually can be made by history and physical exam, so lab testing and skin biopsy typically are not necessary.5 If performed, lab work may show modest elevation in erythrocyte sedimentation rate and C-reactive protein, but often these acute-phase reactants are within normal limits, and complete blood count and complete metabolic panel are unremarkable.3,5 Although urticaria multiforme often is associated with antecedent viral or bacterial infections, work-up for infectious etiology typically is not fruitful or helpful.4 If lesions are biopsied, the histology of urticaria multiforme is indistinguishable from other types of acute urticaria, showing dermal edema with perivascular lymphocytic infiltrate.9 In contrast, EM shows exocytosis, spongiosis, and epidermal necrosis.9
Treatment
The first step in managing urticaria multiforme is discontinuing any unnecessary antibiotic that could be triggering the hypersensitivity reaction. Urticaria multiforme typically resolves within 2 weeks without any treatment and responds to treatment with antihistamines within 24-28 hours.5 Treatment with a histamine1 (H1) blocker such as hydroxyzine, cetirizine, or diphenhydramine may be sufficient to resolve the eruption, but combination therapy with both an H1 blocker and an H2 blocker such as ranitidine can be helpful.4 Treatment with systemic corticosteroids usually is not necessary and should be reserved for severely symptomatic or refractory cases.4,9
One of the reasons that it is important to distinguish urticaria multiforme from EM is to avoid overtreatment with systemic steroids,3 which are rarely required for urticaria multiforme but are sometimes useful, although controversial, for EM.1 Additionally, the correct diagnosis is important for providing anticipatory guidance.6 Patients diagnosed with serum sickness–like reactions should be counseled to avoid unnecessary exposure to the culprit antibiotic in the future. Patients with urticaria multiforme who were taking an antibiotic at the onset of the eruption may consider avoiding the potential culprit antibiotic in the future, but it is important to keep in mind that urticaria multiforme is more strongly associated with antecedent infection than with antibiotic use, and so antibiotic avoidance may not be necessary unless justified by formal allergy testing. EM minor is more commonly associated with a herpes simplex virus infection than a drug reaction, so antibiotic use is less concerning, but patients should be counseled that recurrence is common and prophylactic treatment with acyclovir may be advised for recurrent disease.1
References
- “Neonatal and Infant Dermatology” (Elsevier Health Sciences: New York, 2014, pp. 456-70).
- CRIAI. 2006;30(1):003-012.
- Pediatr Dermatol. 1997;14(3):231-4.
- Pediatrics. 2007;119(5):e1177-83.
- Pediatr Dermatol. 2011;28(4):436-8.
- The Journal of Allergy and Clinical Immunology in Practice. 2013;1(5):520-1.
- Arch Dermatol. 1993;129(1):92-6.
- “The Hypersensitivity Syndromes” in Hurwitz Clinical Pediatric Dermatology. 4 ed. Elsevier: New York, 2011, pp. 455-84.
- J Clin Aesthet Dermatol. 2013;6(3):34-9.
Ms. Haddock is a medical student at University of California, San Diego School of Medicine and a research associate at Rady Children’s Hospital, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego and professor of medicine and pediatrics at UC San Diego School of Medicine. Dr. Eichenfield and Ms. Haddock said they have no relevant financial disclosures. Email pdnews@frontlinemedcom.com.
By Ellen S. Haddock and Lawrence F. Eichenfield, M.D.
Urticaria multiforme
Although not the most classic presentation, this patient’s migrating rash is most consistent with urticaria (hives). Urticaria is dermal edema which causes transient edematous and usually pruritic wheals.1,2 Each individual lesion lasts less than 24 hours and disappears without leaving a mark. Urticaria is caused by mast cell activation, which leads to release of antihistamines and other substances that increase capillary and venule permeability, allowing fluid to leak into the extravascular space.1 In children, mast cell activation is usually triggered by infections, drugs, or foods.1
Classic urticaria consists of large, pruritic plaques and may be associated with airway edema. However, urticaria also can present with annular and polycyclic lesions, which may be less pruritic and are not associated with airway edema.3 This “multiple redness” is distinct from erythema multiforme (EM), although often urticaria is confused with EM. Lesions of EM are annular and typically have purpuric or dusky centers, with each lesion lasting a minimum of 1 week.4 Annular lesions in urticaria usually do not have central duskiness or blisters. Often the centers of annular urticaria lesions are relatively normal and edges are raised. Some urticaria, especially in younger children as in this case, is sometimes called “urticaria multiforme” because the ecchymotic centers are reminiscent of, but distinct from, classic target lesions of EM. Urticaria multiforme is commonly misdiagnosed as EM, with 29% of patients originally misdiagnosed in one study.3
Urticaria multiforme occurs most commonly in infants and preschool-aged children,5 although it has been diagnosed in patients as old as 18 years.6 Patients often have had an antecedent bacterial or viral illness, recent treatment with antibiotics, or recent vaccination (67%, 44%, and 11% of patients, respectively, in one series).4 In contrast with classic urticaria, urticaria multiforme has not been associated with food allergy.4
In this case, urticaria multiforme was likely caused by a hypersensitivity reaction to amoxicillin. A reaction to nitrofurantoin was less likely because the patient had been taking it continuously for months without any complications.
Differential diagnosis
The differential diagnosis for urticaria multiforme includes EM and a serum sickness–like reaction. The main clue that this patient’s rash was a subtype of urticaria rather than EM was its transience, with individual lesions appearing and disappearing in less than a day.4 In contrast, the lesions of EM are fixed, persisting for a week or longer. While urticaria multiforme may have central ecchymosis (termed “hemorrhagic urticaria”) that looks similar to the dusky centers of EM lesions and persists longer than the transient edematous plaques,1 it resolves quickly with appropriate treatment.4 In contrast, the dusky centers of EM, which are caused by epidermal necrosis, take longer to resolve.4 Dermatographism, if present, would support a diagnosis of urticaria rather than EM. Similarly, facial or acral edema, if present, would support a diagnosis of urticaria multiforme; they are uncommon in EM. In contrast, any necrosis, blistering, or erosions in the centers of the annular lesions or on mucosal membranes would suggest EM, as necrosis, blistering, erosions, and mucosal involvement do not occur in urticaria multiforme.4 We stress that in EM, “the center of the lesion is the center of the action,” while in urticaria, wheals often have relatively normal centers.
Although both urticaria and EM lesions may be pruritic, any burning sensation is more suggestive of EM.4 Urticaria multiforme is often associated with antibiotics, vaccinations, and upper respiratory infections, while EM is most commonly associated with herpes simplex infection.4,7
Urticaria multiforme also may appear similar to a serum sickness–like reaction, which is another kind of hypersensitivity reaction triggered by the administration of antibiotics. It is most commonly associated with cefaclor, but also is associated with other antibiotics including amoxicillin.8 As with urticaria, hypersensitivity drug eruptions and serum sickness-like reactions may present with purpuric, polycyclic wheals with central clearing. However, as with EM, the lesions of serum sickness–like reactions are fixed, lasting for days to weeks.4 Facial or acral angioedema may occur in both urticaria multiforme and serum sickness–like reactions, but serum sickness–like reactions are not associated with dermatographism.4 Furthermore, serum sickness–like reactions are typically associated with high-grade fever, myalgia, arthralgia, and lymphadenopathy, which are not seen in urticaria multiforme.4,5
Diagnosis of urticaria multiforme usually can be made by history and physical exam, so lab testing and skin biopsy typically are not necessary.5 If performed, lab work may show modest elevation in erythrocyte sedimentation rate and C-reactive protein, but often these acute-phase reactants are within normal limits, and complete blood count and complete metabolic panel are unremarkable.3,5 Although urticaria multiforme often is associated with antecedent viral or bacterial infections, work-up for infectious etiology typically is not fruitful or helpful.4 If lesions are biopsied, the histology of urticaria multiforme is indistinguishable from other types of acute urticaria, showing dermal edema with perivascular lymphocytic infiltrate.9 In contrast, EM shows exocytosis, spongiosis, and epidermal necrosis.9
Treatment
The first step in managing urticaria multiforme is discontinuing any unnecessary antibiotic that could be triggering the hypersensitivity reaction. Urticaria multiforme typically resolves within 2 weeks without any treatment and responds to treatment with antihistamines within 24-28 hours.5 Treatment with a histamine1 (H1) blocker such as hydroxyzine, cetirizine, or diphenhydramine may be sufficient to resolve the eruption, but combination therapy with both an H1 blocker and an H2 blocker such as ranitidine can be helpful.4 Treatment with systemic corticosteroids usually is not necessary and should be reserved for severely symptomatic or refractory cases.4,9
One of the reasons that it is important to distinguish urticaria multiforme from EM is to avoid overtreatment with systemic steroids,3 which are rarely required for urticaria multiforme but are sometimes useful, although controversial, for EM.1 Additionally, the correct diagnosis is important for providing anticipatory guidance.6 Patients diagnosed with serum sickness–like reactions should be counseled to avoid unnecessary exposure to the culprit antibiotic in the future. Patients with urticaria multiforme who were taking an antibiotic at the onset of the eruption may consider avoiding the potential culprit antibiotic in the future, but it is important to keep in mind that urticaria multiforme is more strongly associated with antecedent infection than with antibiotic use, and so antibiotic avoidance may not be necessary unless justified by formal allergy testing. EM minor is more commonly associated with a herpes simplex virus infection than a drug reaction, so antibiotic use is less concerning, but patients should be counseled that recurrence is common and prophylactic treatment with acyclovir may be advised for recurrent disease.1
References
- “Neonatal and Infant Dermatology” (Elsevier Health Sciences: New York, 2014, pp. 456-70).
- CRIAI. 2006;30(1):003-012.
- Pediatr Dermatol. 1997;14(3):231-4.
- Pediatrics. 2007;119(5):e1177-83.
- Pediatr Dermatol. 2011;28(4):436-8.
- The Journal of Allergy and Clinical Immunology in Practice. 2013;1(5):520-1.
- Arch Dermatol. 1993;129(1):92-6.
- “The Hypersensitivity Syndromes” in Hurwitz Clinical Pediatric Dermatology. 4 ed. Elsevier: New York, 2011, pp. 455-84.
- J Clin Aesthet Dermatol. 2013;6(3):34-9.
Ms. Haddock is a medical student at University of California, San Diego School of Medicine and a research associate at Rady Children’s Hospital, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego and professor of medicine and pediatrics at UC San Diego School of Medicine. Dr. Eichenfield and Ms. Haddock said they have no relevant financial disclosures. Email pdnews@frontlinemedcom.com.
By Ellen S. Haddock and Lawrence F. Eichenfield, M.D.
Urticaria multiforme
Although not the most classic presentation, this patient’s migrating rash is most consistent with urticaria (hives). Urticaria is dermal edema which causes transient edematous and usually pruritic wheals.1,2 Each individual lesion lasts less than 24 hours and disappears without leaving a mark. Urticaria is caused by mast cell activation, which leads to release of antihistamines and other substances that increase capillary and venule permeability, allowing fluid to leak into the extravascular space.1 In children, mast cell activation is usually triggered by infections, drugs, or foods.1
Classic urticaria consists of large, pruritic plaques and may be associated with airway edema. However, urticaria also can present with annular and polycyclic lesions, which may be less pruritic and are not associated with airway edema.3 This “multiple redness” is distinct from erythema multiforme (EM), although often urticaria is confused with EM. Lesions of EM are annular and typically have purpuric or dusky centers, with each lesion lasting a minimum of 1 week.4 Annular lesions in urticaria usually do not have central duskiness or blisters. Often the centers of annular urticaria lesions are relatively normal and edges are raised. Some urticaria, especially in younger children as in this case, is sometimes called “urticaria multiforme” because the ecchymotic centers are reminiscent of, but distinct from, classic target lesions of EM. Urticaria multiforme is commonly misdiagnosed as EM, with 29% of patients originally misdiagnosed in one study.3
Urticaria multiforme occurs most commonly in infants and preschool-aged children,5 although it has been diagnosed in patients as old as 18 years.6 Patients often have had an antecedent bacterial or viral illness, recent treatment with antibiotics, or recent vaccination (67%, 44%, and 11% of patients, respectively, in one series).4 In contrast with classic urticaria, urticaria multiforme has not been associated with food allergy.4
In this case, urticaria multiforme was likely caused by a hypersensitivity reaction to amoxicillin. A reaction to nitrofurantoin was less likely because the patient had been taking it continuously for months without any complications.
Differential diagnosis
The differential diagnosis for urticaria multiforme includes EM and a serum sickness–like reaction. The main clue that this patient’s rash was a subtype of urticaria rather than EM was its transience, with individual lesions appearing and disappearing in less than a day.4 In contrast, the lesions of EM are fixed, persisting for a week or longer. While urticaria multiforme may have central ecchymosis (termed “hemorrhagic urticaria”) that looks similar to the dusky centers of EM lesions and persists longer than the transient edematous plaques,1 it resolves quickly with appropriate treatment.4 In contrast, the dusky centers of EM, which are caused by epidermal necrosis, take longer to resolve.4 Dermatographism, if present, would support a diagnosis of urticaria rather than EM. Similarly, facial or acral edema, if present, would support a diagnosis of urticaria multiforme; they are uncommon in EM. In contrast, any necrosis, blistering, or erosions in the centers of the annular lesions or on mucosal membranes would suggest EM, as necrosis, blistering, erosions, and mucosal involvement do not occur in urticaria multiforme.4 We stress that in EM, “the center of the lesion is the center of the action,” while in urticaria, wheals often have relatively normal centers.
Although both urticaria and EM lesions may be pruritic, any burning sensation is more suggestive of EM.4 Urticaria multiforme is often associated with antibiotics, vaccinations, and upper respiratory infections, while EM is most commonly associated with herpes simplex infection.4,7
Urticaria multiforme also may appear similar to a serum sickness–like reaction, which is another kind of hypersensitivity reaction triggered by the administration of antibiotics. It is most commonly associated with cefaclor, but also is associated with other antibiotics including amoxicillin.8 As with urticaria, hypersensitivity drug eruptions and serum sickness-like reactions may present with purpuric, polycyclic wheals with central clearing. However, as with EM, the lesions of serum sickness–like reactions are fixed, lasting for days to weeks.4 Facial or acral angioedema may occur in both urticaria multiforme and serum sickness–like reactions, but serum sickness–like reactions are not associated with dermatographism.4 Furthermore, serum sickness–like reactions are typically associated with high-grade fever, myalgia, arthralgia, and lymphadenopathy, which are not seen in urticaria multiforme.4,5
Diagnosis of urticaria multiforme usually can be made by history and physical exam, so lab testing and skin biopsy typically are not necessary.5 If performed, lab work may show modest elevation in erythrocyte sedimentation rate and C-reactive protein, but often these acute-phase reactants are within normal limits, and complete blood count and complete metabolic panel are unremarkable.3,5 Although urticaria multiforme often is associated with antecedent viral or bacterial infections, work-up for infectious etiology typically is not fruitful or helpful.4 If lesions are biopsied, the histology of urticaria multiforme is indistinguishable from other types of acute urticaria, showing dermal edema with perivascular lymphocytic infiltrate.9 In contrast, EM shows exocytosis, spongiosis, and epidermal necrosis.9
Treatment
The first step in managing urticaria multiforme is discontinuing any unnecessary antibiotic that could be triggering the hypersensitivity reaction. Urticaria multiforme typically resolves within 2 weeks without any treatment and responds to treatment with antihistamines within 24-28 hours.5 Treatment with a histamine1 (H1) blocker such as hydroxyzine, cetirizine, or diphenhydramine may be sufficient to resolve the eruption, but combination therapy with both an H1 blocker and an H2 blocker such as ranitidine can be helpful.4 Treatment with systemic corticosteroids usually is not necessary and should be reserved for severely symptomatic or refractory cases.4,9
One of the reasons that it is important to distinguish urticaria multiforme from EM is to avoid overtreatment with systemic steroids,3 which are rarely required for urticaria multiforme but are sometimes useful, although controversial, for EM.1 Additionally, the correct diagnosis is important for providing anticipatory guidance.6 Patients diagnosed with serum sickness–like reactions should be counseled to avoid unnecessary exposure to the culprit antibiotic in the future. Patients with urticaria multiforme who were taking an antibiotic at the onset of the eruption may consider avoiding the potential culprit antibiotic in the future, but it is important to keep in mind that urticaria multiforme is more strongly associated with antecedent infection than with antibiotic use, and so antibiotic avoidance may not be necessary unless justified by formal allergy testing. EM minor is more commonly associated with a herpes simplex virus infection than a drug reaction, so antibiotic use is less concerning, but patients should be counseled that recurrence is common and prophylactic treatment with acyclovir may be advised for recurrent disease.1
References
- “Neonatal and Infant Dermatology” (Elsevier Health Sciences: New York, 2014, pp. 456-70).
- CRIAI. 2006;30(1):003-012.
- Pediatr Dermatol. 1997;14(3):231-4.
- Pediatrics. 2007;119(5):e1177-83.
- Pediatr Dermatol. 2011;28(4):436-8.
- The Journal of Allergy and Clinical Immunology in Practice. 2013;1(5):520-1.
- Arch Dermatol. 1993;129(1):92-6.
- “The Hypersensitivity Syndromes” in Hurwitz Clinical Pediatric Dermatology. 4 ed. Elsevier: New York, 2011, pp. 455-84.
- J Clin Aesthet Dermatol. 2013;6(3):34-9.
Ms. Haddock is a medical student at University of California, San Diego School of Medicine and a research associate at Rady Children’s Hospital, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego and professor of medicine and pediatrics at UC San Diego School of Medicine. Dr. Eichenfield and Ms. Haddock said they have no relevant financial disclosures. Email pdnews@frontlinemedcom.com.
A 15-month-old male presents with a rash that began on his trunk 2 days ago and has spread to his arms, legs, and face. His parents say that the rash seems to migrate, with individual spots seeming to disappear and then reappear in new locations. The patient is playful and seems unbothered by the rash. He has a history of vesicoureteral reflux, for which he takes prophylactic nitrofurantoin daily. He was diagnosed with otitis media 7 days ago, for which he has been taking amoxicillin. On physical exam, the patient is afebrile. He has pink, edematous annular (ring-shaped) and polycyclic (composed of overlapping circles) plaques on his face, chest, abdomen, back, and upper and lower extremities. Some lesions appear vaguely targetoid with central clearing and raised borders. There is no mucosal involvement, joint involvement, or lymphadenopathy.
