User login
A medication change, then involuntary lip smacking and tongue rolling
CASE Insurer denies drug coverage
Ms. X, age 65, has a 35-year history of bipolar I disorder (BD I) characterized by psychotic mania and severe suicidal depression. For the past year, her symptoms have been well controlled with aripiprazole, 5 mg/d; trazodone, 50 mg at bedtime; and citalopram, 20 mg/d. Because her health insurance has changed, Ms. X asks to be switched to an alternative antipsychotic because the new provider denied coverage of aripiprazole.
While taking aripiprazole, Ms. X did not report any extrapyramidal side effects, including tardive dyskinesia. Her Abnormal Involuntary Movement Scale (AIMS) score is 4. No significant abnormal movements were noted on examination during previous medication management sessions.
We decide to replace aripiprazole with quetiapine, 50 mg/d. At a 2-week follow-up visit, Ms. X is noted to have euphoric mood and reduced need to sleep, flight of ideas, increased talkativeness, and paranoia. We also notice that she has significant tongue rolling and lip smacking, which she says started 10 days after changing from aripiprazole to quetiapine. Her AIMS score is 17.
What could be causing Ms. X’s tongue rolling and lip smacking?
a) an irreversible syndrome usually starting after 1 or 2 years of continuous exposure to antipsychotics
b) a self-limited condition expected to resolve completely within 12 weeks
c) an acute manifestation of an antipsychotic that can respond to an anticholinergic agent
d) none of the above
The authors’ observations
Tardive dyskinesia (TD) refers to at least moderate abnormal involuntary movements in ≥1 areas of the body or at least mild movements in ≥2 areas of the body, developing after ≥3 months of cumulative exposure (continuous or discontinuous) to dopamine D2 receptor-blocking agents.1 AIMS is a 14-item, clinician-administered questionnaire designed to evaluate such movements and track their severity over time. The first 10 items are rated on 5-point scale (0 = none; 1 = minimal; 2 = mild; 3 = moderate; 4 = severe), with items 1 to 4 assessing orofacial movements, 5 to 7 assessing extremity and truncal movements, and 8 to 10 assessing overall severity, impairment, and subjective distress. Items 11 to 13 assess dental status because lack of teeth can result in oral movements mimicking TDs. The last item assesses whether these movements disappear during sleep.
HISTORY Poor response
Ms. X was given a diagnosis of BD I at age 30; she first started taking antipsychotics 10 years later. Previous psychotropic trials included lamotrigine, divalproex sodium, risperidone, and ziprasidone, which were ineffective or poorly tolerated. Her medical history includes obstructive sleep apnea, narcolepsy, type 2 diabetes mellitus, hypertension, dyslipidemia, fibromyalgia, gastroesophageal reflux disease, and hypothyroidism. She takes metformin, omeprazole, pravastatin, carvedilol, insulin, levothyroxine, methylphenidate (for hypersomnia), and enalapril.
What is the next best step in management?
a) discontinue quetiapine
b) replace quetiapine with clozapine
c) increase quetiapine to target manic symptoms and reassess in a few weeks
d) continue quetiapine and treat abnormal movements with benztropine
TREATMENT Increase dosage
We increase quetiapine to 150 mg/d to target Ms. X’s manic symptoms. She is scheduled for a follow-up visit in 4 weeks but is instructed to return to the clinic earlier if her manic symptoms do not improve. At the 4-week follow-up visit, Ms. X does not have any abnormal movements and her manic symptoms have resolved. Her AIMS score is 4. Her husband reports that her abnormal movements resolved 4 days after increasing quetiapine to 150 mg/d.
The authors’ observations
Second-generation antipsychotics are known to have a lower risk of extrapyramidal adverse reactions compared with older first-generation antipsychotics.2,3 TD differs from other extrapyramidal symptoms (EPS) because of its delayed onset. Risk factors for TD include:
• female sex
• age >50
• history of brain damage
• long-term antipsychotic use
• diagnosis of a mood disorder.
Gardos et al4 described 2 other forms of delayed dyskinesias related to antipsychotic use but resulting from antipsychotic discontinuation: withdrawal dyskinesia and covert dyskinesia. Evidence for these types of antipsychotic discontinuation syndromes mostly is anecdotal.5,6Table 1 highlights 3 different types of dyskinesias and their management.
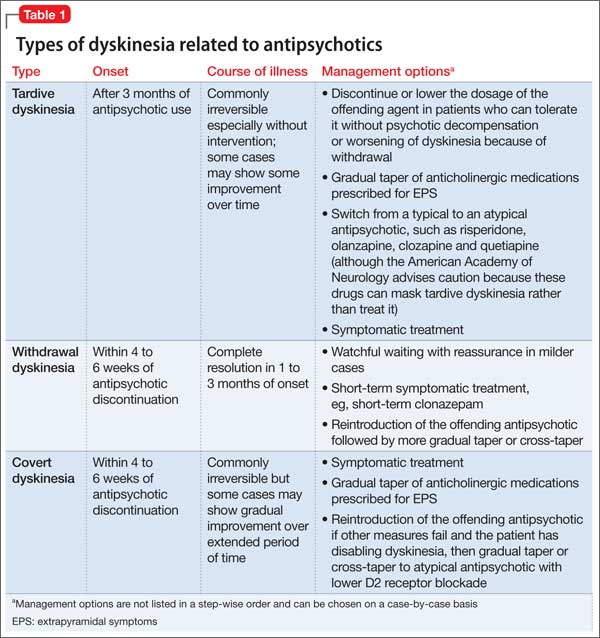
Withdrawal dyskinesia has been described as a syndrome resembling TD that appears after discontinuation or dosage reduction of an antipsychotic in a patient who does not have an earlier TD diagnosis.7 The prevalence of withdrawal dyskinesia among patients undergoing antipsychotic discontinuation is approximately 30%.8 Cases of withdrawal dyskinesia are self-limited and resolve in 1 to 3 months.9,10 We believe that Ms. X’s movement disorder was withdrawal dyskinesia from aripiprazole because her symptoms started 10 days after the drug was discontinued, and was self-limited and reversible.
Similar to TD, withdrawal dyskinesia can present in different forms:
• tongue protrusion movements
• facial grimacing
• ticks
• chorea
• tremors
• athetosis
• involuntary vocalizations
• abnormal movements of hands and legs
• “dyspnea” due to involvement of respiratory musculature.5,11
There may be a sex difference in duration of withdrawal dyskinesias, because symptoms persist longer in females.9
Although covert dyskinesia also develops after discontinuation or dosage reduction of a dopamine-blocking agent, the symptoms usually are permanent, and could require reintroducing the antipsychotic or management with evidence-based treatments for TD, such as tetrabenazine or amantadine.6,12
What is the cause of Ms. X’s abnormal involuntary movements?
a) quetiapine-induced D2 receptor hypersensitivity
b) aripiprazole-induced cholinergic overactivity
c) quetiapine-induced cholinergic overactivity
d) aripiprazole-induced D2 receptor hypersensitivity
The authors’ observations
Pathophysiology of this condition is unknown but different theories have been proposed. D2 receptor up-regulation and hypersensitivity to compensate for chronic D2 receptor blockade by antipsychotics is a commonly cited theory.7,13 Discontinuation of an antipsychotic can make this D2 receptor up-regulation and hypersensitivity manifest as withdrawal dyskinesia by creating a temporary hyperdopaminergic state in basal ganglia. Other theories implicate decrease of γ-aminobutyric acid (GABA) in the globus pallidus (GP) and substantia nigra (SN) regions of the brain, and oxidative damage to GABAergic interneurons in GP and SN from excess production of catecholamines in response to chronic dopamine blockade.14
It has been proposed that patients with withdrawal dyskinesia might be in an early phase of D2 receptor modulation that, if continued because of use of the antipsychotic implicated in withdrawal dyskinesia, can lead to development of TD.4,7,8 A feature of withdrawal dyskinesia that differentiates it from TD is that it usually remits spontaneously within several weeks to a few months.4,7 Because of this characteristic, Schultz et al8 propose that, if withdrawal dyskinesia is identified early in treatment, it may be possible to prevent development of persistent TD.
Look carefully for dyskinetic movements in patients who have recently discontinued or decreased the dosage of their antipsychotic. Non-compliance and partial compliance are common problems among patients taking an antipsychotic.15 Therefore, careful watchfulness for withdrawal dyskinesias at all times can be beneficial. Inquiring about recent history of these dyskinesias in such patients is probably more useful than an exam because the dyskinesias may not be evident on exam when these patients show up for their follow-up visit, because of their self-limited nature.8
Treatment options
If a patient is noted to have a withdrawal-emergent dyskinesia, a clinician has options to prevent TD, including:
• decreasing the dosage of the antipsychotic
• switching from a typical antipsychotic to an atypical antipsychotic
• switching from one atypical to another with lesser affinity for striatal D2 receptor, such as clozapine or quetiapine.16,17
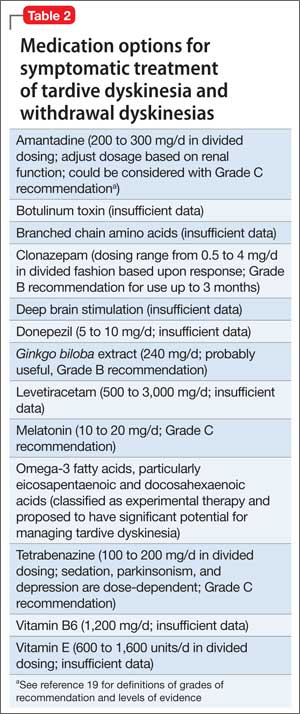
In addition, researchers are investigating the use of vitamin B6, Ginkgo biloba, amantadine, levetiracetam, melatonin, tetrabenazine, zonisamide, branched chain amino acids, clonazepam, and vitamin E as treatment alternatives for TD.
Tetrabenazine acts by blocking vesicular monoamine transporter type 2, thereby inhibiting release of monoamines, including dopamine into synaptic cleft area in basal ganglia.18 Clonazepam’s benefit for TD relates to its facilitation of GABAergic neurotransmission, because reduced GABAergic transmission in GP and SN has been associ ated with hyperkinetic movements, including TD.14Ginkgo biloba and melatonin exert their beneficial effects in TD through their antioxidant function.14
The agents listed in Table 219 could be used on a short-term basis for symptomatic treatment of withdrawal dyskinesias.1,18,20
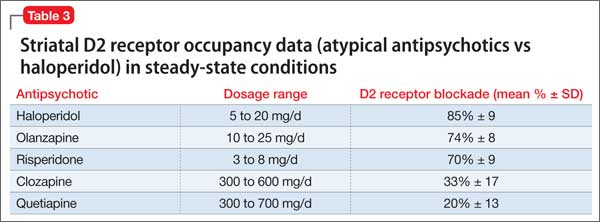
Withdrawal dyskinesia has been reported with aripiprazole discontinuation and is thought to be related to aripiprazole’s strong affinity for D2 receptors.21 Aripiprazole at dosages of 15 to 30 mg/d can occupy more than 80% of the striatal D2 dopamine receptors. The dosage of ≥30 mg/d can lead to receptor occupancy of >90%.22 Studies have shown that EPS correlate with D2 receptor occupancy in steady-state conditions, and occupancy exceeding 80% results in these symptoms.22
Compared with aripiprazole, quetiapine has weak affinity for D2 receptors (Table 3), making it an unlikely culprit if dyskinesia emerges within 2 weeks of initation.22 We believe that, in Ms. X’s case, quetiapine might have masked the severity of aripiprazole withdrawal dyskinesia by causing some degree of D2 receptor blockade. It may have decreased the duration of withdrawal dyskinesia by the same effect on D2 receptors. It may have lasted longer if aripiprazole was not replaced by another antipsychotic. This is particularly evident because dyskinesia improved quickly when quetiapine was titrated to 150 mg/d. The higher quetiapine dosage of 150 mg/d is closer to 5 mg/d of aripiprazole in terms of D2 receptor occupancy and affinity. However, quetiapine is weaker than aripiprazole in terms of D2 receptor occupancy at all dosages, and therefore less likely to cause EPS.16
Summing up
Withdrawal dyskinesia in the absence of a history of TD is a common symptom of antipsychotic discontinuation or dosage reduction after long-term use of an antipsychotic. It is more commonly seen with antipsychotics with high D2 receptor occupancy, and has been hypothesized to be related to D2 receptor supersensitivity to ambient dopamine, resulting as a compensatory response to chronic D2 blockade by this class of medication.
Evidence suggests that reversible withdrawal dyskinesia could represent a prodrome to irreversible TD. Therefore, keeping a watchful eye for these movements during the exam, along with specific inquiry about withdrawal dyskinesias while taking a history at every follow-up visit, is important because doing so can:
• inform the clinician about partial compliance or noncompliance to these medications, which could lead to treatment failure
• help prevent development of irreversible TD syndrome.
Ms. X’s case reminds clinicians (1) to be aware of this unexpected side effect occurring even with second-generation antipsychotics and (2) that they should consider EPS in patients while they are discontinuing their drugs. Furthermore, it is important for clinical and medicolegal reasons to inform our patients that different forms of dyskinesias can be potential side effects of antipsychotics.
Bottom Line
Dyskinesias can result from withdrawal of both typical and atypical antipsychotics, and usually are self-limited. Withdrawal dyskinesia may represent a prodrome to tardive dyskinesia; early recognition may aid in preventing development of persistent tardive dyskinesia.
Related Resources
• Abnormal Involuntary Movement Scale. http://www.cqaimh.org/pdf/toolaims.pdf.
• Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing, Inc; 2012.
• Tarsay D. Tardive dyskinesia: prevention and treatment. http:// www.uptodate.com/contents/tardive-dyskinesia-prevention-and-treatment?topicKey=NEURO%2F4908&elapsedTimeMs=3 &view=print&displayedView=full#.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Benztropine • Cogentin
Carvedilol • Coreg
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Divalproex sodium • Depakote
Donepezil • Aricept
Enalapril • Vasotec
Haloperidol • Haldol
Lamotrigine • Lamictal
Levetiracetam • Keppra
Levothyroxine • Levoxyl, Synthroid
Metformin • Glucophage
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Omeprazole • Prilosec
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trazodone • Desyrel, Oleptro
Ziprasidone • Geodon
Zonisamide • Zonegran
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bhidayasiri R1, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
2. Dolder CR, Jeste DV. Incidence of tardive dyskinesia with typical versus atypical antipsychotics in very high risk patients. Biol Psychiatry. 2003;53(12):1142-1145.
3. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
4. Gardos G, Cole JO, Tarsy D. Withdrawal syndromes associated with antipsychotic drugs. Am J Psychiatry. 1978;135(11):1321-1324.
5. Salomon C, Hamilton B. Antipsychotic discontinuation syndromes: a narrative review of the evidence and its integration into Australian mental health nursing textbooks. Int J Ment Health Nurs. 2014;23(1):69-78.
6. Moseley CN, Simpson-Khanna HA, Catalano G, et al. Covert dyskinesia associated with aripiprazole: a case report and review of the literature. Clin Neuropharmacol. 2013;36(4):128-130.
7. Anand VS, Dewan MJ. Withdrawal-emergent dyskinesia in a patient on risperidone undergoing dosage reduction. Ann Clin Psychiatry. 1996;8(3):179-182.
8. Schultz SK, Miller DD, Arndt S, et al. Withdrawal-emergent dyskinesia in patients with schizophrenia during antipsychotic discontinuation. Biol Psychiatry. 1995;38(11):713-719.
9. Degkwitz R, Bauer MP, Gruber M, et al. Time relationship between the appearance of persisting extrapyramidal hyperkineses and psychotic recurrences following sudden interruption of prolonged neuroleptic therapy of chronic schizophrenic patients [in German]. Arzneimittelforschung. 1970;20(7):890-893.
10. Sethi KD. Tardive dyskinesias. In: Adler CH, Ahlskog JE, eds. Parkinson’s disease and movement disorders: diagnosis and treatment guidelines for the practicing physician. New York, NY: Humana Press; 2000:331-338.
11. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
12. Horváth K, Aschermann Z, Komoly S, et al. Treatment of tardive syndromes [in Hungarian]. Psychiatr Hung. 2014;29(2):214-224.
13. Samaha AN, Seeman P, Stewart J, et al. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci. 2007;27(11):2979-2986.
14. Thelma B, Srivastava V, Tiwari AK. Genetic underpinnings of tardive dyskinesia: passing the baton to pharmacogenetics. Pharmacogenomics. 2008;9(9):1285-1306.
15. Keith SJ, Kane JM. Partial compliance and patient consequences in schizophrenia: our patients can do better. J Clin Psychiatry. 2003;64(11):1308-1315.
16. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
17. Farah A. Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry. 2005;7(6):268-274.
18. Rana AQ, Chaudry ZM, Blanchet PJ. New and emerging treatments for symptomatic tardive dyskinesia. Drug Des Devel Ther. 2013;7:1329-1340.
19. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
20. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics. 2014;11(1):166-176.
21. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
22. Pani L, Pira L, Marchese G. Antipsychotic efficacy: relationship to optimal D2-receptor occupancy. Eur Psychiatry. 2007;22(5):267-275.
CASE Insurer denies drug coverage
Ms. X, age 65, has a 35-year history of bipolar I disorder (BD I) characterized by psychotic mania and severe suicidal depression. For the past year, her symptoms have been well controlled with aripiprazole, 5 mg/d; trazodone, 50 mg at bedtime; and citalopram, 20 mg/d. Because her health insurance has changed, Ms. X asks to be switched to an alternative antipsychotic because the new provider denied coverage of aripiprazole.
While taking aripiprazole, Ms. X did not report any extrapyramidal side effects, including tardive dyskinesia. Her Abnormal Involuntary Movement Scale (AIMS) score is 4. No significant abnormal movements were noted on examination during previous medication management sessions.
We decide to replace aripiprazole with quetiapine, 50 mg/d. At a 2-week follow-up visit, Ms. X is noted to have euphoric mood and reduced need to sleep, flight of ideas, increased talkativeness, and paranoia. We also notice that she has significant tongue rolling and lip smacking, which she says started 10 days after changing from aripiprazole to quetiapine. Her AIMS score is 17.
What could be causing Ms. X’s tongue rolling and lip smacking?
a) an irreversible syndrome usually starting after 1 or 2 years of continuous exposure to antipsychotics
b) a self-limited condition expected to resolve completely within 12 weeks
c) an acute manifestation of an antipsychotic that can respond to an anticholinergic agent
d) none of the above
The authors’ observations
Tardive dyskinesia (TD) refers to at least moderate abnormal involuntary movements in ≥1 areas of the body or at least mild movements in ≥2 areas of the body, developing after ≥3 months of cumulative exposure (continuous or discontinuous) to dopamine D2 receptor-blocking agents.1 AIMS is a 14-item, clinician-administered questionnaire designed to evaluate such movements and track their severity over time. The first 10 items are rated on 5-point scale (0 = none; 1 = minimal; 2 = mild; 3 = moderate; 4 = severe), with items 1 to 4 assessing orofacial movements, 5 to 7 assessing extremity and truncal movements, and 8 to 10 assessing overall severity, impairment, and subjective distress. Items 11 to 13 assess dental status because lack of teeth can result in oral movements mimicking TDs. The last item assesses whether these movements disappear during sleep.
HISTORY Poor response
Ms. X was given a diagnosis of BD I at age 30; she first started taking antipsychotics 10 years later. Previous psychotropic trials included lamotrigine, divalproex sodium, risperidone, and ziprasidone, which were ineffective or poorly tolerated. Her medical history includes obstructive sleep apnea, narcolepsy, type 2 diabetes mellitus, hypertension, dyslipidemia, fibromyalgia, gastroesophageal reflux disease, and hypothyroidism. She takes metformin, omeprazole, pravastatin, carvedilol, insulin, levothyroxine, methylphenidate (for hypersomnia), and enalapril.
What is the next best step in management?
a) discontinue quetiapine
b) replace quetiapine with clozapine
c) increase quetiapine to target manic symptoms and reassess in a few weeks
d) continue quetiapine and treat abnormal movements with benztropine
TREATMENT Increase dosage
We increase quetiapine to 150 mg/d to target Ms. X’s manic symptoms. She is scheduled for a follow-up visit in 4 weeks but is instructed to return to the clinic earlier if her manic symptoms do not improve. At the 4-week follow-up visit, Ms. X does not have any abnormal movements and her manic symptoms have resolved. Her AIMS score is 4. Her husband reports that her abnormal movements resolved 4 days after increasing quetiapine to 150 mg/d.
The authors’ observations
Second-generation antipsychotics are known to have a lower risk of extrapyramidal adverse reactions compared with older first-generation antipsychotics.2,3 TD differs from other extrapyramidal symptoms (EPS) because of its delayed onset. Risk factors for TD include:
• female sex
• age >50
• history of brain damage
• long-term antipsychotic use
• diagnosis of a mood disorder.
Gardos et al4 described 2 other forms of delayed dyskinesias related to antipsychotic use but resulting from antipsychotic discontinuation: withdrawal dyskinesia and covert dyskinesia. Evidence for these types of antipsychotic discontinuation syndromes mostly is anecdotal.5,6Table 1 highlights 3 different types of dyskinesias and their management.
Withdrawal dyskinesia has been described as a syndrome resembling TD that appears after discontinuation or dosage reduction of an antipsychotic in a patient who does not have an earlier TD diagnosis.7 The prevalence of withdrawal dyskinesia among patients undergoing antipsychotic discontinuation is approximately 30%.8 Cases of withdrawal dyskinesia are self-limited and resolve in 1 to 3 months.9,10 We believe that Ms. X’s movement disorder was withdrawal dyskinesia from aripiprazole because her symptoms started 10 days after the drug was discontinued, and was self-limited and reversible.
Similar to TD, withdrawal dyskinesia can present in different forms:
• tongue protrusion movements
• facial grimacing
• ticks
• chorea
• tremors
• athetosis
• involuntary vocalizations
• abnormal movements of hands and legs
• “dyspnea” due to involvement of respiratory musculature.5,11
There may be a sex difference in duration of withdrawal dyskinesias, because symptoms persist longer in females.9
Although covert dyskinesia also develops after discontinuation or dosage reduction of a dopamine-blocking agent, the symptoms usually are permanent, and could require reintroducing the antipsychotic or management with evidence-based treatments for TD, such as tetrabenazine or amantadine.6,12
What is the cause of Ms. X’s abnormal involuntary movements?
a) quetiapine-induced D2 receptor hypersensitivity
b) aripiprazole-induced cholinergic overactivity
c) quetiapine-induced cholinergic overactivity
d) aripiprazole-induced D2 receptor hypersensitivity
The authors’ observations
Pathophysiology of this condition is unknown but different theories have been proposed. D2 receptor up-regulation and hypersensitivity to compensate for chronic D2 receptor blockade by antipsychotics is a commonly cited theory.7,13 Discontinuation of an antipsychotic can make this D2 receptor up-regulation and hypersensitivity manifest as withdrawal dyskinesia by creating a temporary hyperdopaminergic state in basal ganglia. Other theories implicate decrease of γ-aminobutyric acid (GABA) in the globus pallidus (GP) and substantia nigra (SN) regions of the brain, and oxidative damage to GABAergic interneurons in GP and SN from excess production of catecholamines in response to chronic dopamine blockade.14
It has been proposed that patients with withdrawal dyskinesia might be in an early phase of D2 receptor modulation that, if continued because of use of the antipsychotic implicated in withdrawal dyskinesia, can lead to development of TD.4,7,8 A feature of withdrawal dyskinesia that differentiates it from TD is that it usually remits spontaneously within several weeks to a few months.4,7 Because of this characteristic, Schultz et al8 propose that, if withdrawal dyskinesia is identified early in treatment, it may be possible to prevent development of persistent TD.
Look carefully for dyskinetic movements in patients who have recently discontinued or decreased the dosage of their antipsychotic. Non-compliance and partial compliance are common problems among patients taking an antipsychotic.15 Therefore, careful watchfulness for withdrawal dyskinesias at all times can be beneficial. Inquiring about recent history of these dyskinesias in such patients is probably more useful than an exam because the dyskinesias may not be evident on exam when these patients show up for their follow-up visit, because of their self-limited nature.8
Treatment options
If a patient is noted to have a withdrawal-emergent dyskinesia, a clinician has options to prevent TD, including:
• decreasing the dosage of the antipsychotic
• switching from a typical antipsychotic to an atypical antipsychotic
• switching from one atypical to another with lesser affinity for striatal D2 receptor, such as clozapine or quetiapine.16,17
In addition, researchers are investigating the use of vitamin B6, Ginkgo biloba, amantadine, levetiracetam, melatonin, tetrabenazine, zonisamide, branched chain amino acids, clonazepam, and vitamin E as treatment alternatives for TD.
Tetrabenazine acts by blocking vesicular monoamine transporter type 2, thereby inhibiting release of monoamines, including dopamine into synaptic cleft area in basal ganglia.18 Clonazepam’s benefit for TD relates to its facilitation of GABAergic neurotransmission, because reduced GABAergic transmission in GP and SN has been associ ated with hyperkinetic movements, including TD.14Ginkgo biloba and melatonin exert their beneficial effects in TD through their antioxidant function.14
The agents listed in Table 219 could be used on a short-term basis for symptomatic treatment of withdrawal dyskinesias.1,18,20
Withdrawal dyskinesia has been reported with aripiprazole discontinuation and is thought to be related to aripiprazole’s strong affinity for D2 receptors.21 Aripiprazole at dosages of 15 to 30 mg/d can occupy more than 80% of the striatal D2 dopamine receptors. The dosage of ≥30 mg/d can lead to receptor occupancy of >90%.22 Studies have shown that EPS correlate with D2 receptor occupancy in steady-state conditions, and occupancy exceeding 80% results in these symptoms.22
Compared with aripiprazole, quetiapine has weak affinity for D2 receptors (Table 3), making it an unlikely culprit if dyskinesia emerges within 2 weeks of initation.22 We believe that, in Ms. X’s case, quetiapine might have masked the severity of aripiprazole withdrawal dyskinesia by causing some degree of D2 receptor blockade. It may have decreased the duration of withdrawal dyskinesia by the same effect on D2 receptors. It may have lasted longer if aripiprazole was not replaced by another antipsychotic. This is particularly evident because dyskinesia improved quickly when quetiapine was titrated to 150 mg/d. The higher quetiapine dosage of 150 mg/d is closer to 5 mg/d of aripiprazole in terms of D2 receptor occupancy and affinity. However, quetiapine is weaker than aripiprazole in terms of D2 receptor occupancy at all dosages, and therefore less likely to cause EPS.16
Summing up
Withdrawal dyskinesia in the absence of a history of TD is a common symptom of antipsychotic discontinuation or dosage reduction after long-term use of an antipsychotic. It is more commonly seen with antipsychotics with high D2 receptor occupancy, and has been hypothesized to be related to D2 receptor supersensitivity to ambient dopamine, resulting as a compensatory response to chronic D2 blockade by this class of medication.
Evidence suggests that reversible withdrawal dyskinesia could represent a prodrome to irreversible TD. Therefore, keeping a watchful eye for these movements during the exam, along with specific inquiry about withdrawal dyskinesias while taking a history at every follow-up visit, is important because doing so can:
• inform the clinician about partial compliance or noncompliance to these medications, which could lead to treatment failure
• help prevent development of irreversible TD syndrome.
Ms. X’s case reminds clinicians (1) to be aware of this unexpected side effect occurring even with second-generation antipsychotics and (2) that they should consider EPS in patients while they are discontinuing their drugs. Furthermore, it is important for clinical and medicolegal reasons to inform our patients that different forms of dyskinesias can be potential side effects of antipsychotics.
Bottom Line
Dyskinesias can result from withdrawal of both typical and atypical antipsychotics, and usually are self-limited. Withdrawal dyskinesia may represent a prodrome to tardive dyskinesia; early recognition may aid in preventing development of persistent tardive dyskinesia.
Related Resources
• Abnormal Involuntary Movement Scale. http://www.cqaimh.org/pdf/toolaims.pdf.
• Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing, Inc; 2012.
• Tarsay D. Tardive dyskinesia: prevention and treatment. http:// www.uptodate.com/contents/tardive-dyskinesia-prevention-and-treatment?topicKey=NEURO%2F4908&elapsedTimeMs=3 &view=print&displayedView=full#.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Benztropine • Cogentin
Carvedilol • Coreg
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Divalproex sodium • Depakote
Donepezil • Aricept
Enalapril • Vasotec
Haloperidol • Haldol
Lamotrigine • Lamictal
Levetiracetam • Keppra
Levothyroxine • Levoxyl, Synthroid
Metformin • Glucophage
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Omeprazole • Prilosec
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trazodone • Desyrel, Oleptro
Ziprasidone • Geodon
Zonisamide • Zonegran
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Insurer denies drug coverage
Ms. X, age 65, has a 35-year history of bipolar I disorder (BD I) characterized by psychotic mania and severe suicidal depression. For the past year, her symptoms have been well controlled with aripiprazole, 5 mg/d; trazodone, 50 mg at bedtime; and citalopram, 20 mg/d. Because her health insurance has changed, Ms. X asks to be switched to an alternative antipsychotic because the new provider denied coverage of aripiprazole.
While taking aripiprazole, Ms. X did not report any extrapyramidal side effects, including tardive dyskinesia. Her Abnormal Involuntary Movement Scale (AIMS) score is 4. No significant abnormal movements were noted on examination during previous medication management sessions.
We decide to replace aripiprazole with quetiapine, 50 mg/d. At a 2-week follow-up visit, Ms. X is noted to have euphoric mood and reduced need to sleep, flight of ideas, increased talkativeness, and paranoia. We also notice that she has significant tongue rolling and lip smacking, which she says started 10 days after changing from aripiprazole to quetiapine. Her AIMS score is 17.
What could be causing Ms. X’s tongue rolling and lip smacking?
a) an irreversible syndrome usually starting after 1 or 2 years of continuous exposure to antipsychotics
b) a self-limited condition expected to resolve completely within 12 weeks
c) an acute manifestation of an antipsychotic that can respond to an anticholinergic agent
d) none of the above
The authors’ observations
Tardive dyskinesia (TD) refers to at least moderate abnormal involuntary movements in ≥1 areas of the body or at least mild movements in ≥2 areas of the body, developing after ≥3 months of cumulative exposure (continuous or discontinuous) to dopamine D2 receptor-blocking agents.1 AIMS is a 14-item, clinician-administered questionnaire designed to evaluate such movements and track their severity over time. The first 10 items are rated on 5-point scale (0 = none; 1 = minimal; 2 = mild; 3 = moderate; 4 = severe), with items 1 to 4 assessing orofacial movements, 5 to 7 assessing extremity and truncal movements, and 8 to 10 assessing overall severity, impairment, and subjective distress. Items 11 to 13 assess dental status because lack of teeth can result in oral movements mimicking TDs. The last item assesses whether these movements disappear during sleep.
HISTORY Poor response
Ms. X was given a diagnosis of BD I at age 30; she first started taking antipsychotics 10 years later. Previous psychotropic trials included lamotrigine, divalproex sodium, risperidone, and ziprasidone, which were ineffective or poorly tolerated. Her medical history includes obstructive sleep apnea, narcolepsy, type 2 diabetes mellitus, hypertension, dyslipidemia, fibromyalgia, gastroesophageal reflux disease, and hypothyroidism. She takes metformin, omeprazole, pravastatin, carvedilol, insulin, levothyroxine, methylphenidate (for hypersomnia), and enalapril.
What is the next best step in management?
a) discontinue quetiapine
b) replace quetiapine with clozapine
c) increase quetiapine to target manic symptoms and reassess in a few weeks
d) continue quetiapine and treat abnormal movements with benztropine
TREATMENT Increase dosage
We increase quetiapine to 150 mg/d to target Ms. X’s manic symptoms. She is scheduled for a follow-up visit in 4 weeks but is instructed to return to the clinic earlier if her manic symptoms do not improve. At the 4-week follow-up visit, Ms. X does not have any abnormal movements and her manic symptoms have resolved. Her AIMS score is 4. Her husband reports that her abnormal movements resolved 4 days after increasing quetiapine to 150 mg/d.
The authors’ observations
Second-generation antipsychotics are known to have a lower risk of extrapyramidal adverse reactions compared with older first-generation antipsychotics.2,3 TD differs from other extrapyramidal symptoms (EPS) because of its delayed onset. Risk factors for TD include:
• female sex
• age >50
• history of brain damage
• long-term antipsychotic use
• diagnosis of a mood disorder.
Gardos et al4 described 2 other forms of delayed dyskinesias related to antipsychotic use but resulting from antipsychotic discontinuation: withdrawal dyskinesia and covert dyskinesia. Evidence for these types of antipsychotic discontinuation syndromes mostly is anecdotal.5,6Table 1 highlights 3 different types of dyskinesias and their management.
Withdrawal dyskinesia has been described as a syndrome resembling TD that appears after discontinuation or dosage reduction of an antipsychotic in a patient who does not have an earlier TD diagnosis.7 The prevalence of withdrawal dyskinesia among patients undergoing antipsychotic discontinuation is approximately 30%.8 Cases of withdrawal dyskinesia are self-limited and resolve in 1 to 3 months.9,10 We believe that Ms. X’s movement disorder was withdrawal dyskinesia from aripiprazole because her symptoms started 10 days after the drug was discontinued, and was self-limited and reversible.
Similar to TD, withdrawal dyskinesia can present in different forms:
• tongue protrusion movements
• facial grimacing
• ticks
• chorea
• tremors
• athetosis
• involuntary vocalizations
• abnormal movements of hands and legs
• “dyspnea” due to involvement of respiratory musculature.5,11
There may be a sex difference in duration of withdrawal dyskinesias, because symptoms persist longer in females.9
Although covert dyskinesia also develops after discontinuation or dosage reduction of a dopamine-blocking agent, the symptoms usually are permanent, and could require reintroducing the antipsychotic or management with evidence-based treatments for TD, such as tetrabenazine or amantadine.6,12
What is the cause of Ms. X’s abnormal involuntary movements?
a) quetiapine-induced D2 receptor hypersensitivity
b) aripiprazole-induced cholinergic overactivity
c) quetiapine-induced cholinergic overactivity
d) aripiprazole-induced D2 receptor hypersensitivity
The authors’ observations
Pathophysiology of this condition is unknown but different theories have been proposed. D2 receptor up-regulation and hypersensitivity to compensate for chronic D2 receptor blockade by antipsychotics is a commonly cited theory.7,13 Discontinuation of an antipsychotic can make this D2 receptor up-regulation and hypersensitivity manifest as withdrawal dyskinesia by creating a temporary hyperdopaminergic state in basal ganglia. Other theories implicate decrease of γ-aminobutyric acid (GABA) in the globus pallidus (GP) and substantia nigra (SN) regions of the brain, and oxidative damage to GABAergic interneurons in GP and SN from excess production of catecholamines in response to chronic dopamine blockade.14
It has been proposed that patients with withdrawal dyskinesia might be in an early phase of D2 receptor modulation that, if continued because of use of the antipsychotic implicated in withdrawal dyskinesia, can lead to development of TD.4,7,8 A feature of withdrawal dyskinesia that differentiates it from TD is that it usually remits spontaneously within several weeks to a few months.4,7 Because of this characteristic, Schultz et al8 propose that, if withdrawal dyskinesia is identified early in treatment, it may be possible to prevent development of persistent TD.
Look carefully for dyskinetic movements in patients who have recently discontinued or decreased the dosage of their antipsychotic. Non-compliance and partial compliance are common problems among patients taking an antipsychotic.15 Therefore, careful watchfulness for withdrawal dyskinesias at all times can be beneficial. Inquiring about recent history of these dyskinesias in such patients is probably more useful than an exam because the dyskinesias may not be evident on exam when these patients show up for their follow-up visit, because of their self-limited nature.8
Treatment options
If a patient is noted to have a withdrawal-emergent dyskinesia, a clinician has options to prevent TD, including:
• decreasing the dosage of the antipsychotic
• switching from a typical antipsychotic to an atypical antipsychotic
• switching from one atypical to another with lesser affinity for striatal D2 receptor, such as clozapine or quetiapine.16,17
In addition, researchers are investigating the use of vitamin B6, Ginkgo biloba, amantadine, levetiracetam, melatonin, tetrabenazine, zonisamide, branched chain amino acids, clonazepam, and vitamin E as treatment alternatives for TD.
Tetrabenazine acts by blocking vesicular monoamine transporter type 2, thereby inhibiting release of monoamines, including dopamine into synaptic cleft area in basal ganglia.18 Clonazepam’s benefit for TD relates to its facilitation of GABAergic neurotransmission, because reduced GABAergic transmission in GP and SN has been associ ated with hyperkinetic movements, including TD.14Ginkgo biloba and melatonin exert their beneficial effects in TD through their antioxidant function.14
The agents listed in Table 219 could be used on a short-term basis for symptomatic treatment of withdrawal dyskinesias.1,18,20
Withdrawal dyskinesia has been reported with aripiprazole discontinuation and is thought to be related to aripiprazole’s strong affinity for D2 receptors.21 Aripiprazole at dosages of 15 to 30 mg/d can occupy more than 80% of the striatal D2 dopamine receptors. The dosage of ≥30 mg/d can lead to receptor occupancy of >90%.22 Studies have shown that EPS correlate with D2 receptor occupancy in steady-state conditions, and occupancy exceeding 80% results in these symptoms.22
Compared with aripiprazole, quetiapine has weak affinity for D2 receptors (Table 3), making it an unlikely culprit if dyskinesia emerges within 2 weeks of initation.22 We believe that, in Ms. X’s case, quetiapine might have masked the severity of aripiprazole withdrawal dyskinesia by causing some degree of D2 receptor blockade. It may have decreased the duration of withdrawal dyskinesia by the same effect on D2 receptors. It may have lasted longer if aripiprazole was not replaced by another antipsychotic. This is particularly evident because dyskinesia improved quickly when quetiapine was titrated to 150 mg/d. The higher quetiapine dosage of 150 mg/d is closer to 5 mg/d of aripiprazole in terms of D2 receptor occupancy and affinity. However, quetiapine is weaker than aripiprazole in terms of D2 receptor occupancy at all dosages, and therefore less likely to cause EPS.16
Summing up
Withdrawal dyskinesia in the absence of a history of TD is a common symptom of antipsychotic discontinuation or dosage reduction after long-term use of an antipsychotic. It is more commonly seen with antipsychotics with high D2 receptor occupancy, and has been hypothesized to be related to D2 receptor supersensitivity to ambient dopamine, resulting as a compensatory response to chronic D2 blockade by this class of medication.
Evidence suggests that reversible withdrawal dyskinesia could represent a prodrome to irreversible TD. Therefore, keeping a watchful eye for these movements during the exam, along with specific inquiry about withdrawal dyskinesias while taking a history at every follow-up visit, is important because doing so can:
• inform the clinician about partial compliance or noncompliance to these medications, which could lead to treatment failure
• help prevent development of irreversible TD syndrome.
Ms. X’s case reminds clinicians (1) to be aware of this unexpected side effect occurring even with second-generation antipsychotics and (2) that they should consider EPS in patients while they are discontinuing their drugs. Furthermore, it is important for clinical and medicolegal reasons to inform our patients that different forms of dyskinesias can be potential side effects of antipsychotics.
Bottom Line
Dyskinesias can result from withdrawal of both typical and atypical antipsychotics, and usually are self-limited. Withdrawal dyskinesia may represent a prodrome to tardive dyskinesia; early recognition may aid in preventing development of persistent tardive dyskinesia.
Related Resources
• Abnormal Involuntary Movement Scale. http://www.cqaimh.org/pdf/toolaims.pdf.
• Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing, Inc; 2012.
• Tarsay D. Tardive dyskinesia: prevention and treatment. http:// www.uptodate.com/contents/tardive-dyskinesia-prevention-and-treatment?topicKey=NEURO%2F4908&elapsedTimeMs=3 &view=print&displayedView=full#.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Benztropine • Cogentin
Carvedilol • Coreg
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Divalproex sodium • Depakote
Donepezil • Aricept
Enalapril • Vasotec
Haloperidol • Haldol
Lamotrigine • Lamictal
Levetiracetam • Keppra
Levothyroxine • Levoxyl, Synthroid
Metformin • Glucophage
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Omeprazole • Prilosec
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trazodone • Desyrel, Oleptro
Ziprasidone • Geodon
Zonisamide • Zonegran
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bhidayasiri R1, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
2. Dolder CR, Jeste DV. Incidence of tardive dyskinesia with typical versus atypical antipsychotics in very high risk patients. Biol Psychiatry. 2003;53(12):1142-1145.
3. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
4. Gardos G, Cole JO, Tarsy D. Withdrawal syndromes associated with antipsychotic drugs. Am J Psychiatry. 1978;135(11):1321-1324.
5. Salomon C, Hamilton B. Antipsychotic discontinuation syndromes: a narrative review of the evidence and its integration into Australian mental health nursing textbooks. Int J Ment Health Nurs. 2014;23(1):69-78.
6. Moseley CN, Simpson-Khanna HA, Catalano G, et al. Covert dyskinesia associated with aripiprazole: a case report and review of the literature. Clin Neuropharmacol. 2013;36(4):128-130.
7. Anand VS, Dewan MJ. Withdrawal-emergent dyskinesia in a patient on risperidone undergoing dosage reduction. Ann Clin Psychiatry. 1996;8(3):179-182.
8. Schultz SK, Miller DD, Arndt S, et al. Withdrawal-emergent dyskinesia in patients with schizophrenia during antipsychotic discontinuation. Biol Psychiatry. 1995;38(11):713-719.
9. Degkwitz R, Bauer MP, Gruber M, et al. Time relationship between the appearance of persisting extrapyramidal hyperkineses and psychotic recurrences following sudden interruption of prolonged neuroleptic therapy of chronic schizophrenic patients [in German]. Arzneimittelforschung. 1970;20(7):890-893.
10. Sethi KD. Tardive dyskinesias. In: Adler CH, Ahlskog JE, eds. Parkinson’s disease and movement disorders: diagnosis and treatment guidelines for the practicing physician. New York, NY: Humana Press; 2000:331-338.
11. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
12. Horváth K, Aschermann Z, Komoly S, et al. Treatment of tardive syndromes [in Hungarian]. Psychiatr Hung. 2014;29(2):214-224.
13. Samaha AN, Seeman P, Stewart J, et al. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci. 2007;27(11):2979-2986.
14. Thelma B, Srivastava V, Tiwari AK. Genetic underpinnings of tardive dyskinesia: passing the baton to pharmacogenetics. Pharmacogenomics. 2008;9(9):1285-1306.
15. Keith SJ, Kane JM. Partial compliance and patient consequences in schizophrenia: our patients can do better. J Clin Psychiatry. 2003;64(11):1308-1315.
16. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
17. Farah A. Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry. 2005;7(6):268-274.
18. Rana AQ, Chaudry ZM, Blanchet PJ. New and emerging treatments for symptomatic tardive dyskinesia. Drug Des Devel Ther. 2013;7:1329-1340.
19. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
20. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics. 2014;11(1):166-176.
21. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
22. Pani L, Pira L, Marchese G. Antipsychotic efficacy: relationship to optimal D2-receptor occupancy. Eur Psychiatry. 2007;22(5):267-275.
1. Bhidayasiri R1, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
2. Dolder CR, Jeste DV. Incidence of tardive dyskinesia with typical versus atypical antipsychotics in very high risk patients. Biol Psychiatry. 2003;53(12):1142-1145.
3. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
4. Gardos G, Cole JO, Tarsy D. Withdrawal syndromes associated with antipsychotic drugs. Am J Psychiatry. 1978;135(11):1321-1324.
5. Salomon C, Hamilton B. Antipsychotic discontinuation syndromes: a narrative review of the evidence and its integration into Australian mental health nursing textbooks. Int J Ment Health Nurs. 2014;23(1):69-78.
6. Moseley CN, Simpson-Khanna HA, Catalano G, et al. Covert dyskinesia associated with aripiprazole: a case report and review of the literature. Clin Neuropharmacol. 2013;36(4):128-130.
7. Anand VS, Dewan MJ. Withdrawal-emergent dyskinesia in a patient on risperidone undergoing dosage reduction. Ann Clin Psychiatry. 1996;8(3):179-182.
8. Schultz SK, Miller DD, Arndt S, et al. Withdrawal-emergent dyskinesia in patients with schizophrenia during antipsychotic discontinuation. Biol Psychiatry. 1995;38(11):713-719.
9. Degkwitz R, Bauer MP, Gruber M, et al. Time relationship between the appearance of persisting extrapyramidal hyperkineses and psychotic recurrences following sudden interruption of prolonged neuroleptic therapy of chronic schizophrenic patients [in German]. Arzneimittelforschung. 1970;20(7):890-893.
10. Sethi KD. Tardive dyskinesias. In: Adler CH, Ahlskog JE, eds. Parkinson’s disease and movement disorders: diagnosis and treatment guidelines for the practicing physician. New York, NY: Humana Press; 2000:331-338.
11. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
12. Horváth K, Aschermann Z, Komoly S, et al. Treatment of tardive syndromes [in Hungarian]. Psychiatr Hung. 2014;29(2):214-224.
13. Samaha AN, Seeman P, Stewart J, et al. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci. 2007;27(11):2979-2986.
14. Thelma B, Srivastava V, Tiwari AK. Genetic underpinnings of tardive dyskinesia: passing the baton to pharmacogenetics. Pharmacogenomics. 2008;9(9):1285-1306.
15. Keith SJ, Kane JM. Partial compliance and patient consequences in schizophrenia: our patients can do better. J Clin Psychiatry. 2003;64(11):1308-1315.
16. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
17. Farah A. Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry. 2005;7(6):268-274.
18. Rana AQ, Chaudry ZM, Blanchet PJ. New and emerging treatments for symptomatic tardive dyskinesia. Drug Des Devel Ther. 2013;7:1329-1340.
19. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
20. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics. 2014;11(1):166-176.
21. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
22. Pani L, Pira L, Marchese G. Antipsychotic efficacy: relationship to optimal D2-receptor occupancy. Eur Psychiatry. 2007;22(5):267-275.
Ingenol mebutate helped clear actinic keratoses
Cryosurgery followed by topical ingenol mebutate cleared extensive regions of actinic keratosis, which helped reveal residual squamous cell carcinomas, according to a report in the August issue of the Journal of Drugs in Dermatology.
The findings show that ingenol mebutate can clear multiple AKs and reduce the number of scarring biopsies required to identify SCCs, said Dr. Miriam S. Bettencourt, a dermatologist in group practice in Henderson, Nev. “In our dermatology clinic, many of the patients with a long history of AK who were treated with ingenol mebutate used sequentially after cryosurgery have achieved complete or partial clearance of AKs.”
Ingenol mebutate gel after cryosurgery cleared AKs more effectively than cryosurgery alone in a recent phase III trial (J Drugs Dermatol. 2014 Jun;13[6]741-7), Dr. Bettencourt noted. She described six men and one woman who each had at least 10 recurrent or hyperkeratotic AKs and previously had undergone cryosurgery. She treated all patients with cryosurgery, followed 2 weeks later by two or three once-daily applications of ingenol mebutate gel at strengths of 0.05% or 0.015%, respectively (J Drugs Dermatol. 2015 Aug;14[8];813-8). One course of ingenol mebutate gel cleared 50%-100% of AKs, Dr. Bettencourt said. She treated residual AKs with cryosurgery, and five patients also received at least one more course of ingenol mebutate to re-treat a partially cleared area or to treat a separate area. Shave biopsies of 10 residual suspicious lesions taken 3-8 months later all revealed invasive SCCs, which were treated with Mohs micrographic surgery (MMS). “These lesions may have been preexisting at the time of topical treatment but not readily recognized as suspicious in the heavily actinically damaged skin, in which suspected or small SCCs may be adjacent to or obscured by AKs,” she said. “Alternatively, these tumors may have been spontaneous new SCCs. In either case, we suggest that effective clearance of AKs from the palette of sun-damaged skin with ingenol mebutate permitted prompt recognition of these lesions as suspicious, and led to further diagnosis and treatment with MMS.”
All patients developed mild to moderate localized redness, flaking, and crusting starting on the second day of ingenol mebutate treatment and resolving within a week of finishing the course, Dr. Bettencourt said.
She reported that she had no relevant financial conflicts.
Cryosurgery followed by topical ingenol mebutate cleared extensive regions of actinic keratosis, which helped reveal residual squamous cell carcinomas, according to a report in the August issue of the Journal of Drugs in Dermatology.
The findings show that ingenol mebutate can clear multiple AKs and reduce the number of scarring biopsies required to identify SCCs, said Dr. Miriam S. Bettencourt, a dermatologist in group practice in Henderson, Nev. “In our dermatology clinic, many of the patients with a long history of AK who were treated with ingenol mebutate used sequentially after cryosurgery have achieved complete or partial clearance of AKs.”
Ingenol mebutate gel after cryosurgery cleared AKs more effectively than cryosurgery alone in a recent phase III trial (J Drugs Dermatol. 2014 Jun;13[6]741-7), Dr. Bettencourt noted. She described six men and one woman who each had at least 10 recurrent or hyperkeratotic AKs and previously had undergone cryosurgery. She treated all patients with cryosurgery, followed 2 weeks later by two or three once-daily applications of ingenol mebutate gel at strengths of 0.05% or 0.015%, respectively (J Drugs Dermatol. 2015 Aug;14[8];813-8). One course of ingenol mebutate gel cleared 50%-100% of AKs, Dr. Bettencourt said. She treated residual AKs with cryosurgery, and five patients also received at least one more course of ingenol mebutate to re-treat a partially cleared area or to treat a separate area. Shave biopsies of 10 residual suspicious lesions taken 3-8 months later all revealed invasive SCCs, which were treated with Mohs micrographic surgery (MMS). “These lesions may have been preexisting at the time of topical treatment but not readily recognized as suspicious in the heavily actinically damaged skin, in which suspected or small SCCs may be adjacent to or obscured by AKs,” she said. “Alternatively, these tumors may have been spontaneous new SCCs. In either case, we suggest that effective clearance of AKs from the palette of sun-damaged skin with ingenol mebutate permitted prompt recognition of these lesions as suspicious, and led to further diagnosis and treatment with MMS.”
All patients developed mild to moderate localized redness, flaking, and crusting starting on the second day of ingenol mebutate treatment and resolving within a week of finishing the course, Dr. Bettencourt said.
She reported that she had no relevant financial conflicts.
Cryosurgery followed by topical ingenol mebutate cleared extensive regions of actinic keratosis, which helped reveal residual squamous cell carcinomas, according to a report in the August issue of the Journal of Drugs in Dermatology.
The findings show that ingenol mebutate can clear multiple AKs and reduce the number of scarring biopsies required to identify SCCs, said Dr. Miriam S. Bettencourt, a dermatologist in group practice in Henderson, Nev. “In our dermatology clinic, many of the patients with a long history of AK who were treated with ingenol mebutate used sequentially after cryosurgery have achieved complete or partial clearance of AKs.”
Ingenol mebutate gel after cryosurgery cleared AKs more effectively than cryosurgery alone in a recent phase III trial (J Drugs Dermatol. 2014 Jun;13[6]741-7), Dr. Bettencourt noted. She described six men and one woman who each had at least 10 recurrent or hyperkeratotic AKs and previously had undergone cryosurgery. She treated all patients with cryosurgery, followed 2 weeks later by two or three once-daily applications of ingenol mebutate gel at strengths of 0.05% or 0.015%, respectively (J Drugs Dermatol. 2015 Aug;14[8];813-8). One course of ingenol mebutate gel cleared 50%-100% of AKs, Dr. Bettencourt said. She treated residual AKs with cryosurgery, and five patients also received at least one more course of ingenol mebutate to re-treat a partially cleared area or to treat a separate area. Shave biopsies of 10 residual suspicious lesions taken 3-8 months later all revealed invasive SCCs, which were treated with Mohs micrographic surgery (MMS). “These lesions may have been preexisting at the time of topical treatment but not readily recognized as suspicious in the heavily actinically damaged skin, in which suspected or small SCCs may be adjacent to or obscured by AKs,” she said. “Alternatively, these tumors may have been spontaneous new SCCs. In either case, we suggest that effective clearance of AKs from the palette of sun-damaged skin with ingenol mebutate permitted prompt recognition of these lesions as suspicious, and led to further diagnosis and treatment with MMS.”
All patients developed mild to moderate localized redness, flaking, and crusting starting on the second day of ingenol mebutate treatment and resolving within a week of finishing the course, Dr. Bettencourt said.
She reported that she had no relevant financial conflicts.
FROM THE JOURNAL OF DRUGS IN DERMATOLOGY
Key clinical point: Several courses of cryosurgery and ingenol mebutate helped clear actinic keratoses, helping a clinician identify residual squamous cell carcinomas.
Major finding: Lesion counts dropped by 50%-100% after cryosurgery followed by one to three courses of ingenol mebutate gel.
Data source: A case series of seven patients who had multiple AKs and 10 SCCs.
Disclosures: Dr. Bettencourt reported that she had no relevant financial conflicts.
August 2015 Quiz 2
ANSWER: D
Critique
This patient has vitamin B12 deficiency, which is common in the elderly. In addition, gastrectomy can produce cobalamin deficiency due to lack of gastrin and pepsin resulting in impaired release of dietary B12 from ingested proteins. Also, the lack of intrinsic factor will result in impaired absorption of B12. B12 and folate are required to metabolize homocysteine to methionine. Therefore, with deficiency of either folate or B12, there is an increase in serum homocysteine levels. B12 is also a cofactor in the synthesis of succinyl-CoA from methylmalonyl-CoA and therefore, with B12 deficiency, methylmalonic acid levels are also elevated. Hypoglycemia would not explain this constellation of symptoms. Microscopic colitis causes diarrhea but does not cause dementia or cognitive impairment, glossitis, or taste disturbances. The dominant micronutrient deficiencies with celiac disease are iron and calcium malabsorption, and while B12 deficiency is possible with extensive disease, it is not seen as commonly, and celiac would not be the most likely etiology for her B12 deficiency.
Reference
1. Sumner, A.E., Chin, M.M., Abrahm, J.L., et al. Elevated methylmalonic acid and total homocysteine levels show high prevalence of B12 deficiency after gastric surgery. Ann. Intern. Med. 1996;124:469.
ANSWER: D
Critique
This patient has vitamin B12 deficiency, which is common in the elderly. In addition, gastrectomy can produce cobalamin deficiency due to lack of gastrin and pepsin resulting in impaired release of dietary B12 from ingested proteins. Also, the lack of intrinsic factor will result in impaired absorption of B12. B12 and folate are required to metabolize homocysteine to methionine. Therefore, with deficiency of either folate or B12, there is an increase in serum homocysteine levels. B12 is also a cofactor in the synthesis of succinyl-CoA from methylmalonyl-CoA and therefore, with B12 deficiency, methylmalonic acid levels are also elevated. Hypoglycemia would not explain this constellation of symptoms. Microscopic colitis causes diarrhea but does not cause dementia or cognitive impairment, glossitis, or taste disturbances. The dominant micronutrient deficiencies with celiac disease are iron and calcium malabsorption, and while B12 deficiency is possible with extensive disease, it is not seen as commonly, and celiac would not be the most likely etiology for her B12 deficiency.
Reference
1. Sumner, A.E., Chin, M.M., Abrahm, J.L., et al. Elevated methylmalonic acid and total homocysteine levels show high prevalence of B12 deficiency after gastric surgery. Ann. Intern. Med. 1996;124:469.
ANSWER: D
Critique
This patient has vitamin B12 deficiency, which is common in the elderly. In addition, gastrectomy can produce cobalamin deficiency due to lack of gastrin and pepsin resulting in impaired release of dietary B12 from ingested proteins. Also, the lack of intrinsic factor will result in impaired absorption of B12. B12 and folate are required to metabolize homocysteine to methionine. Therefore, with deficiency of either folate or B12, there is an increase in serum homocysteine levels. B12 is also a cofactor in the synthesis of succinyl-CoA from methylmalonyl-CoA and therefore, with B12 deficiency, methylmalonic acid levels are also elevated. Hypoglycemia would not explain this constellation of symptoms. Microscopic colitis causes diarrhea but does not cause dementia or cognitive impairment, glossitis, or taste disturbances. The dominant micronutrient deficiencies with celiac disease are iron and calcium malabsorption, and while B12 deficiency is possible with extensive disease, it is not seen as commonly, and celiac would not be the most likely etiology for her B12 deficiency.
Reference
1. Sumner, A.E., Chin, M.M., Abrahm, J.L., et al. Elevated methylmalonic acid and total homocysteine levels show high prevalence of B12 deficiency after gastric surgery. Ann. Intern. Med. 1996;124:469.
August 2015 Quiz 1
ANSWER: D
Critique
Cystic fibrosis (CF) is the correct diagnosis here even in the absence of respiratory symptoms; failure to thrive with malabsorption, elevated liver chemistries, and protein malnutrition (low serum albumin) are all suggestive of CF. Additionally, profound hypoalbuminemia and anemia have been reported with the use of soy protein-based formulas in infants with CF. Although celiac disease can have a very early onset, this may obviously only follow ingestion of gluten, so it is not a diagnostic possibility in the case of this formula-fed child. Poor feeding technique is a cause of failure to thrive in early infancy, but here we have good oral intake also suggesting an absorption issue. Giardiasis may have caused this child's symptoms, as this parasitic infection may result in malabsorption, but at this early age this is a highly unlikely explanation, especially in developed countries. Milk protein allergy-induced enteropathy is also possible in this case but is less likely with heme-negative stool and elevated liver chemistries.
Reference
1. Messick, J. A 21st century approach to cystic fibrosis: optimizing outcomes across the disease spectrum. J. Pediatr. Gastroenterol. Nutr. 2010;51(Suppl 7):S1-7.
ANSWER: D
Critique
Cystic fibrosis (CF) is the correct diagnosis here even in the absence of respiratory symptoms; failure to thrive with malabsorption, elevated liver chemistries, and protein malnutrition (low serum albumin) are all suggestive of CF. Additionally, profound hypoalbuminemia and anemia have been reported with the use of soy protein-based formulas in infants with CF. Although celiac disease can have a very early onset, this may obviously only follow ingestion of gluten, so it is not a diagnostic possibility in the case of this formula-fed child. Poor feeding technique is a cause of failure to thrive in early infancy, but here we have good oral intake also suggesting an absorption issue. Giardiasis may have caused this child's symptoms, as this parasitic infection may result in malabsorption, but at this early age this is a highly unlikely explanation, especially in developed countries. Milk protein allergy-induced enteropathy is also possible in this case but is less likely with heme-negative stool and elevated liver chemistries.
Reference
1. Messick, J. A 21st century approach to cystic fibrosis: optimizing outcomes across the disease spectrum. J. Pediatr. Gastroenterol. Nutr. 2010;51(Suppl 7):S1-7.
ANSWER: D
Critique
Cystic fibrosis (CF) is the correct diagnosis here even in the absence of respiratory symptoms; failure to thrive with malabsorption, elevated liver chemistries, and protein malnutrition (low serum albumin) are all suggestive of CF. Additionally, profound hypoalbuminemia and anemia have been reported with the use of soy protein-based formulas in infants with CF. Although celiac disease can have a very early onset, this may obviously only follow ingestion of gluten, so it is not a diagnostic possibility in the case of this formula-fed child. Poor feeding technique is a cause of failure to thrive in early infancy, but here we have good oral intake also suggesting an absorption issue. Giardiasis may have caused this child's symptoms, as this parasitic infection may result in malabsorption, but at this early age this is a highly unlikely explanation, especially in developed countries. Milk protein allergy-induced enteropathy is also possible in this case but is less likely with heme-negative stool and elevated liver chemistries.
Reference
1. Messick, J. A 21st century approach to cystic fibrosis: optimizing outcomes across the disease spectrum. J. Pediatr. Gastroenterol. Nutr. 2010;51(Suppl 7):S1-7.
What does molecular imaging reveal about the causes of ADHD and the potential for better management?
Attention-deficit/hyperactivity disorder (ADHD) is one of the most common pediatric psychiatric disorders, occurring in approximately 5% of children.1 The disorder persists into adulthood in about one-half of those who are affected in childhood.2 In adults and children, diagnosis continues to be based on the examiner’s subjective assessment. (Box 13-9 describes how ADHD presents a complicated, moving target for the diagnostician.)

Patients who have ADHD are rarely studied with imaging; there are no established imaging findings associated with an ADHD diagnosis. Over the past 20 years, however, significant research has shown that molecular alterations along the dopaminergic−frontostriatal pathways occur in association with the behavioral constellation of ADHD symptoms—suggesting a pathophysiologic mechanism for this disorder.
In this article, we describe molecular findings from nuclear medicine imaging in ADHD. We also summarize imaging evidence for dysfunction of the dopaminergic-frontostriatal neural circuits as central in the pathophysiology of ADHD, with special focus on the dopamine reuptake transporter (DaT). Box 210,11 reviews our key observations and looks at the future of imaging in the management of ADHD.

Dopaminergic theory of ADHD
The executive functions that are disordered in ADHD (impulse control, judgment, maintaining attention) are thought to be centered in the infraorbital, dorsolateral, and medial frontal lobes. Neurotransmitters that have been implicated in the pathophysiology of ADHD include norepinephrine12 and dopamine13; medications that selectively block reuptake of these neurotransmitters are used to treat ADHD.14,15 Only the dopamine system has been extensively evaluated with molecular imaging techniques.
Because methylphenidate, a potent selective dopamine reuptake inhibitor, has been shown to reduce disordered executive functional behaviors in ADHD, considerable imaging research has focused on the dopaminergic neural circuits in the frontostriatal regions of the brain. The dopaminergic theory of ADHD is based on the hypothesis that alterations in the density or function of these circuits are responsible for behaviors that constitute ADHD.
Despite decades of efforts to delineate the underlying pathophysiology and neurochemistry of ADHD, no single unifying theory accounts for all imaging findings in all patients. This might be in part because of imprecision inherent in psychiatric diagnoses that are based on subjective observations. The behavioral criteria for ADHD can manifest in several disorders. For example, anxiety-related symptoms seen in posttraumatic stress disorder, social anxiety disorder, and panic disorder also present as behaviors similar to those in ADHD diagnostic criteria.
Molecular imaging might provide a window into the underlying pathophysiology of ADHD and, by identifying objective findings, (1) allow for patient stratification based on underlying physiologic subtypes, (2) refine diagnostic criteria, and (3) predict treatment response.
Nuclear medicine findings
In general, nuclear medicine investigations of ADHD can be divided into studies of changes in regional cerebral blood flow (rCBF) or glucose metabolism (rCGM) and those that have assessed the concentration of synaptic structures, using highly specific radiolabeled ligands. Both kinds of studies provide limited anatomic resolution, unless co-registered with MRI or CT scans and either single photon emission computed tomography (SPECT) or positron emission tomography (PET).
Synaptic imaging using radiolabeled ligands with high biologic specificity for synaptic structures has high molecular resolution—that is, radiolabeled ligands used for selective imaging of the dopamine transporter or receptor do not identify serotonin transporters or receptors, and vice versa. (Details of SPECT and PET techniques are beyond the scope of this article but can be found in standard nuclear medicine textbooks.)
SPECT and PET of rCBF
Early investigations of rCBF in ADHD were performed using inhaled radioactive xenon-133 gas.16 Later, rCBF was assessed using fat-soluble radiolabeled ligands that rapidly distribute in the brain in proportion to blood flow by crossing the blood−brain barrier. Labeled with radioactive 99m-technetium, these ligands cross rapidly into brain cells after IV injection. Once intracellular, covalent bonds within the ligands cleave into 2 charged particles that do not easily recross the cell membrane. There is little redistribution of tracer after initial uptake.
The imaging data set that results can be reconstructed as (1) surface images, on which defects indicate areas of reduced rCBF, or (2) tomographic slices on which color scales indicate relative rCBF values (Figure 1). Because of the minimal redistribution of the tracer, SPECT images obtained 1 or 2 hours after injection provide a snapshot of rCBF at the time tracer is injected. Patients can be injected under various conditions, such as at rest with eyes and ears open in a dimly lit, quiet room, and then under cognitive stress (Figure 2), such as performing a computer-based attention and impulse control task, or during stimulant treatment.


Numerous investigators have found reduced frontal or striatal rCBF, or both, in patients with ADHD, unilaterally on the right17 or left,18,19 or bilaterally.20 Additionally, with stimulant therapy, normalization of striatal and frontal rCBF has been demonstrated14,19—changes that correlate with resolution of behavioral symptoms of ADHD with stimulant treatment.21
SPECT of 32 boys with previously untreated ADHD. Kim et al21 found that the presence of reduced right or left, or both, frontal rCBF, which normalized with 8 weeks of stimulant therapy, predicted symptom improvement in 85% of patients. Absence of improvement of reduced frontal rCBF had a 75% negative predictive value for treatment response. (Additionally, hyperperfusion of the somatosensory cortex has been demonstrated in children with ADHD,16,22 suggesting increased responsiveness to extraneous environmental input.)
SPECT of 40 untreated pediatric patients compared with 17 age-matched controls. Using SPECT, Lee et al23 reported rCBF reductions in the orbitofrontal cortex and the medial temporal gyrus of participants; reductions corresponded to areas of motor and impulsivity control. The researchers also demonstrated increased rCBF in the somatosensory area.
After methylphenidate treatment, blood flow to these areas normalized, and rCBF to higher visual and superior prefrontal areas decreased. Substantial clinical improvement occurred in 64% of patients—suggesting methylphenidate treatment of ADHD works by (1) increasing function of areas of the brain that control impulses, motor activity, and attention, and (2) reducing function to sensory areas that lead to distraction by extraneous environmental sensory input.
O-15-labeled water PET of 10 adults with ADHD. Schweitzer et al24 found that participants who demonstrated improvement in behavioral symptoms with chronic stimulant therapy had reduced rCBF in the striata at baseline—again, suggesting that baseline hypometabolism in the striata is associated with ADHD.
PET of regional cerebral glucose metabolism
Cerebral metabolism requires a constant supply of glucose; regional differences in cerebral glucose metabolism can be assessed directly with positron-emitting F-18-fluoro-2-deoxyglucose. Although metabolically inert, this agent is transported intracellularly similar to glucose; once phosphorylated within brain cells, however, it can no longer undergo further metabolism or redistribution.
Studies using PET to assess rCGM were some of the earliest molecular imaging applications in ADHD. Zametkin et al25 reported low global cerebral glucose utilization in adults, but not adolescents,26 with ADHD. However, further study, with normalization of the PET data, confirmed reduced rCGM in the left prefrontal cortex in both adolescents26 and adults,27 indicating hypometabolism of cortical areas associated with impulse control and attention in ADHD. In adolescents, symptom severity was inversely related to rCBF in the left anterior frontal cortex.
Synaptic imaging
Nuclear imaging has been used to study several components of the striatal dopaminergic synapse, including:
• dopamine substrates, using fluorine- 18-labeled dopa or carbon-11-labeled dopa
• dopamine receptors, using carbon- 11-labeled raclopride or iodine-123 iodobenzamide
• the tDaT, using iodine-123 ioflupane, 99m-technetium TRODAT, or carbon-11 cocaine (Figure 3).
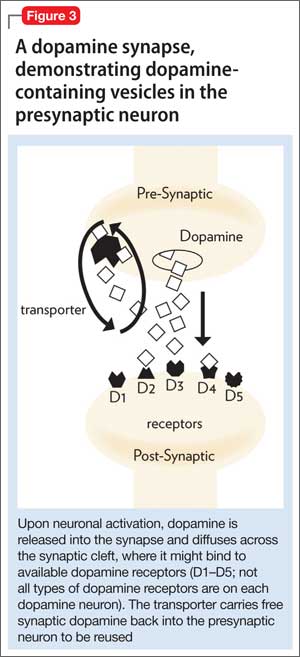
All of these synaptic imaging agents were used mainly as research tools until 2011, when the FDA approved the SPECT imaging agent iodine-123 ioflupane (DaTscan) for clinical use in assessment of Parkinson’s disease.28 This commercially available agent has high specificity for the DaT, with little background activity noted on SPECT imaging (Figure 4).
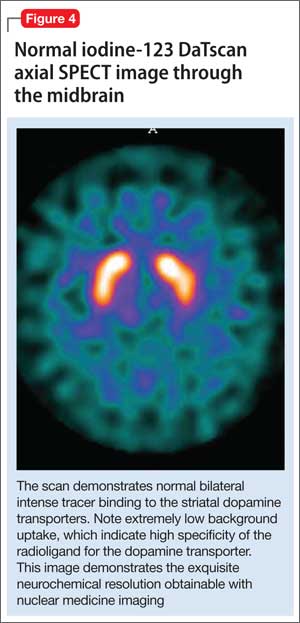
Dopamine transporter imaging
Because the site of action of methylphenidate is the DaT, imaging this component of the striatal dopaminergic synapse has been an area of intense investigation in ADHD. Located almost exclusively in the striata, DaT reduces synaptic concentrations of dopamine by means of reuptake channels in the cell membrane.29 By reversibly binding to, and occupying sites on, the DaT, methylphenidate impedes dopamine reuptake, which results in increased availability of dopamine at the synapse.30
By demonstrating an increase in striatal DaT density in patients with ADHD— first reported by Dougherty et al31 using iodine-123 altropane (a dopaminergic uptake inhibitor) in 6 adults with ADHD—investigators have hypothesized that excessive expression of the DaT protein in the striata, which may result from genetic or environmental factors, is a central causative agent of ADHD.32 Subsequent studies, however, have yielded contradictory findings: Hesse et al,33 using SPECT imaging, and Volkow et al,34 using carbon-11 cocaine PET imaging, found reduced DaT density in, respectively, 9 and 26 patients with ADHD.
To clarify the role of DaT levels in the etiology of ADHD and to explain discrepant results, Fusar-Poli et al35 performed a meta-analysis of 9 published papers that reported the results of DaT imaging in a total of 169 ADHD patients and 129 controls. They noted that these studies included 6 different imaging agents and protocols. Patients were stimulant therapy-naïve (n = 137) or drug-free (refrained from stimulant therapy for a time [n = 32]). The team found that the degree of elevation of the striatal DaT concentration correlated with a history of stimulant exposure, and that the drug-naïve group had a reduced DaT level.
Fusar-Poli’s hypothesis? Elevated DaT levels result from up-regulation in the presence of chronic methylphenidate therapy, which accounts for early reports that demonstrated increased striatal DaT density. Clinically, up-regulation might explain the lack of sustained relief of behavioral symptoms with stimulant therapy in 20% of patients with ADHD who showed clinical improvement initially.36
Only limited conclusions can be drawn about the role of DaT levels in ADHD, given the small number of patients studied in published reports. In addition, the Fusar-Poli meta-analysis has come under strong criticism because of methodological errors with improper patient inclusion and characterization of treatment status,37 calling into question the investigators’ conclusions.
Does the DaT level hold promise for practice? Despite a lack of clarity about the significance of DaT level in the etiology of ADHD, knowledge of a patient’s level might prove useful in predicting which patients will respond to methylphenidate. Namely, several researchers have found that:
• an elevated baseline level of DaT (before stimulant therapy) correlates with robust clinical response
• absence of an elevated baseline DaT level suggests that symptomatic improvement with stimulant therapy in unlikely.38-40
Dresel et al38 evaluated 17 drug-naïve adults, newly diagnosed with ADHD, using 99m-technetium TRODAT SPECT before and after methylphenidate therapy. They found a 15% increase in specific DaT binding in patients with ADHD, compared with controls, at baseline. After treatment, the researchers observed a 28% reduction in specific DaT binding—a significant change from baseline that correlated with behavioral response.
Study: SPECT in 18 adults with ADHD given methylphenidate. Krause39 used the same SPECT agent to study 18 adults before they received methylphenidate and 10 weeks after treatment. Participants were categorized as responders or nonresponders based on clinical assessment of ADHD symptoms after those 10 weeks. All 12 responders had an elevated striatal DaT concentration at baseline. Of the 6 nonresponders, 5 had a normal level of striatal DaT compared with age-matched controls.
Study: 22 Adult ADHD patients evaluated with 99m-technetium TRODAT SPECT. The same group of investigators40 presented imaging findings in 22 additional adult patients. Seventeen had an elevated striatal DaT level, 16 of whom responded to stimulant therapy. The remaining 5 patients had reduced striatal DaT at baseline; none had a good clinical response to methylphenidate.
The positive clinical response to methylphenidate in 67%37 and 77%40 of patients is in good agreement with results from larger studies, which reported that approximately 75% of patients with ADHD show prompt clinical improvement with stimulants.41 Improvement might be related to an increase in functioning of the frontostriatal dopaminergic circuit that is seen with stimulant therapy. Increased availability of dopamine at the synapse, resulting from stimulant blockade of the dopamine reuptake transporter, produces increased dopamine neurotransmission and increased activation of frontostriatal circuits.
In another study, rCBF in frontostriatal circuits was determined to be inversely proportional to DaT density; rCBF normalized with stimulant therapy.42
Will imaging pave the way for therapeutic stratification? Baseline determinations of striatal DaT concentration with SPECT imaging might make it possible to stratify patients with ADHD symptoms into those likely to show significant behavioral symptom response to methylphenidate and those who are not likely to respond. There might be an objective imaging finding—striatal DaT density—that allows clinicians to distinguish stimulant-responsive ADHD from stimulant-unresponsive ADHD.
Dopamine substrate imaging
Radiolabeled dopa (carbon-11 or fluorine-18) is transported into presynaptic dopaminergic neurons in the striatum, where it is decarboxylated, converted to radio-dopamine, and stored within vesicles until released in response to neuronal excitation. Semi-quantitative assessment is achieved with calculation of specific (striatal) to nonspecific (background) uptake ratios. Increased values are thought to indicate increased density of dopaminergic neurons.43
Ernst et al44 reported a 50% decrease in specific fluorine-18 dopa uptake in the left prefrontal cortex in 17 drug-naïve adults with ADHD, compared with 23 controls. The same team reported increased midbrain fluorine-18 dopa levels in 10 adolescents with ADHD—48% higher, overall, than what was seen in 10 controls.43 They hypothesized that these opposite results were the results of a reduction in the dopaminergic neuronal density in adults, which might be part of the natural history of ADHD, or a normal age-related reduction in neuronal density, or both. Increased dopa levels in the team’s adolescent group were hypothesized to reflect up-regulation in dopamine synthesis due to low synaptic dopamine concentrations that might result from increased dopamine reuptake.
Dopamine-receptor imaging
The 5 distinct dopamine receptors (D1, D2, D3, D4, and D5) can be grouped into 2 subtypes, based on their coupling with G proteins. D1 and D5 constitute a group; D2, D3, and D4, a second group.
The D1 receptor is the most common dopamine receptor in the brain and is widely distributed in the striatum and prefrontal cerebral cortex. D1 receptor knockout mice demonstrate hyperactivity and poorer performance on learning tasks and are used as an animal model for ADHD.45 D1 has been imaged using C-11 SCH 23390 PET46 in rats, but its role in ADHD has yet to be evaluated. D5 is the most recently cloned and most widely distributed of the known dopamine receptors; however, there are no imaging studies of the D5 receptor.13
D2 receptors are present in presynaptic and postsynaptic neurons47 in the neocortex, substantia nigra, nucleus accumbens, and olfactory tubercle, as well as in other structures.48 Presynaptic D2 receptors act as autoregulators, inhibiting dopaminergic synthesis, firing rate, and release.49
Using C-11 raclopride PET imaging, Lou et al50 reported high D2/3 receptor availability in adolescents who had a history of perinatal cerebral ischemia. They found that this availability is associated with an increase in the severity of ADHD symptoms. They proposed that the increase in “empty” receptor density might have been caused by perinatal ischemia-induced presynaptic dopaminergic neuronal loss or an increase in presynaptic dopamine reuptake (Figure 550). Either mechanism could result in up-regulation in postsynaptic D2/3 receptors.
Volkow et al51 reported that D2 receptor density correlated with methylphenidate-induced changes in rCBF in frontal and temporal lobes in humans. They postulated that the variable therapeutic effects of methylphenidate seen in ADHD patients might be related to variations in baseline D2 receptor availability.
Lou et al50 reported elevated D2 receptor density, demonstrated using carbon-11 raclopride, in children with ADHD, compared with normal adults.
Further support for a relationship between D2-receptor density and symptomatic improvement with methylphenidate in ADHD was presented by Ilgin et al52 using iodine-123 iodobenzamide SPECT. They found elevated D2 receptor levels in 9 drug-naïve children with ADHD, which is 20% to 60% above what is seen in unaffected children. They noted that these patients showed improvement in hyperactivity when treated with methylphenidate.
In a similar study of 20 drug-naïve adults, Volkow et al53 found that durable symptomatic improvement with methylphenidate therapy was associated with increased D2 receptor availability.
Summing up
Striatal DaT is the most likely synaptic target for stratifying patients with ADHD, now that a dopamine transporter imaging agent is available commercially. Stratification might allow for refinement in the diagnostic categorization of ADHD, with introduction of stimulant-responsive and stimulant-unresponsive subtypes that are based on DaT imaging findings.
Bottom Line
Given recent advances showing molecular alterations in the dopaminergic-frontostriatal pathway as central to attention-deficit/hyperactivity disorder, molecular imaging might be useful as an objective study for diagnosis.
Related Resources
• Schweitzer JB, Lee DO, Hanford RB, et al. A positron emission tomography study of methylphenidate in adults with ADHD: alterations in resting blood flow and predicting treatment response. Neuropsychopharmacology. 2003;28(5):967-973.
• Raz A. Brain imaging data of ADHD. Psychiatric Times. http://www.psychiatrictimes.com/adhd/brain-imaging-data-adhd.
Drug Brand Names
Iodine-123 ioflupane • Methylphenidate • Ritalin DaTscan
Acknowledgment
Kylee M. L. Unsdorfer, a medical student at Northeast Ohio Medical University, helped prepare the manuscript of this article.
Disclosures
Dr. Thacker reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Binkovitz received 4 doses of ioflupane I123I (DaTscan) from General Electric for investigator-initiated research, used for animal imaging in 2012.
1. Polanczyk G, de Lima MS, Horta BL, et al. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry. 2007;164(6):942-948.
2. Simon V, Czobor P, Bálint S, et al. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry. 2009;194(3):204-211.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Berger I. Diagnosis of attention deficit hyperactivity disorder: much ado about something. Isr Med Assoc J. 2011;13(9):571-574.
5. Schonwald A, Lechner E. Attention deficit/hyperactivity disorder: complexities and controversies. Curr Opin Pediatr. 2006;18(2):189-195.
6. Rousseau C, Measham T, Bathiche-Suidan M. DSM IV, culture and child psychiatry. J Can Acad Child Adolesc Psychiatry. 2008;17(2):69-75.
7. Taylor-Klaus E. Bringing the ADHD debate into sharper focus: part 1. The Huffington Post. http:// www.huffingtonpost.com/elaine-taylorklaus/adhd-debate_b_4571097.html. Updated March 17, 2014. Accessed August 18, 2015.
8. Sweeney CT, Sembower MA, Ertischek MD, et al. Nonmedical use of prescription ADHD stimulants and preexisting patterns of drug abuse. J Addict Dis. 2013;32(1):1-10.
9. Hitt E. Multiple reports of ADHD drug shortages. Medscape. http://www.medscape.com/viewarticle/742686. Published May 13, 2011. Accessed June 4, 2015.
10. Rubia K, Alegria AA, Cubillo AI, et al. Effects of stimulants on brain function in attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. Biol Psychiatry. 2014;76(8):616-628.
11. Cortese S, Kelly C, Chabernaud C, et al. Toward systems neuroscience of ADHD: a meta-analysis of 55 fMRI studies. Am J Psychiatry. 2012;169(10):1038-1055.
12. Garnock-Jones KP, Keating GM. Atomoxetine: a review of its use in attention-deficit hyperactivity disorder in children and adolescents. Paediatr Drugs. 2009;11(3):203-226.
13. Wu J, Xiao H, Sun H, et al. Role of dopamine receptors in ADHD: a systematic meta-analysis. Mol Neurobiol. 2012; 45(3):605-620.
14. Del Campo N, Chamberlain SR, Sahakian BJ, et al. The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2011;69(12):e145-e157.
15. Berridge CW, Devilbiss DM. Psychostimulants as cognitive enhancers: the prefrontal cortex, catecholamines, and attention-deficit/hyperactivity disorder. Biol Psychiatry. 2011;69(12):e101-e111.
16. Lou HC, Henriksen L, Bruhn P. Focal cerebral hypoperfusion in children with dysphasia and/or attention deficit disorder. Arch Neurol. 1984;41(8):825-829.
17. Gustafsson P, Thernlund G, Ryding E, et al. Associations between cerebral blood-flow measured by single photon emission computed tomography (SPECT), electro-encephalogram (EEG), behaviour symptoms, cognition and neurological soft signs in children with attention-deficit hyperactivity disorder (ADHD). Acta Paediatr. 2000;89(7):830-835.
18. Sieg KG, Gaffney GR, Preston DF, et al. SPECT brain imaging abnormalities in attention deficit hyperactivity disorder. Clin Nucl Med. 1995;20(1):55-60.
19. Spalletta G, Pasini A, Pau F, et al. Prefrontal blood flow dysregulation in drug naive ADHD children without structural abnormalities. J Neural Transm. 2001;108(10):1203-1216.
20. Amen DG, Carmichael BD. High-resolution brain SPECT imaging in ADHD. Ann Clin Psychiatry. 1997;9(2):81-86.
21. Kim BN, Lee JS, Cho SC, et al. Methylphenidate increased regional cerebral blood flow in subjects with attention deficit/hyperactivity disorder. Yonsei Med J. 2001;42(1):19-29.
22. Lou HC, Henriksen L, Bruhn P, et al. Striatal dysfunction in attention deficit and hyperkinetic disorder. Arch Neurol. 1989;46(1):48-52.
23. Lee JS, Kim BN, Kang E, et al. Regional cerebral blood flow in children with attention deficit hyperactivity disorder: comparison before and after methylphenidate treatment. Hum Brain Mapp. 2005;24(3):157-164.
24. Schweitzer JB, Lee DO, Hanford RB, et al. A positron emission tomography study of methylphenidate in adults with ADHD: alterations in resting blood flow and predicting treatment response. Neuropsychopharmacology. 2003;28(5):967-973.
25. Zametkin AJ, Nordahl TE, Gross M, et al. Cerebral glucose metabolism in adults with hyperactivity of childhood onset. N Engl J Med. 1990;323(20):1361-1366.
26. Zametkin AJ, Liebenauer LL, Fitzgerald GA, et al. Brain metabolism in teenagers with attention-deficit hyperactivity disorder. Arch Gen Psychiatry. 1993;50(5):333-340.
27. Ernst M, Zametkin AJ, Matochik JA, et al. Effects of intravenous dextroamphetamine on brain metabolism in adults with attention-deficit hyperactivity disorder (ADHD). Preliminary findings. Psychopharmacol Bull. 1994;30(2):219-225.
28. Janssen M. Dopamine transporter (DaT) SPECT imaging. MI Gateway. 2012;6(1):1-3. http://interactive.snm.org/ docs/MI_Gateway_Newsletter_2012-1%20Dopamine%20 Transporter%20SPECT%20Imaging.pdf. Accessed August 18, 2015.
29. Volkow ND, Wang GJ, Fowler JS, et al. Dopamine transporter occupancies in the human brain induced by therapeutic doses of oral methylphenidate. Am J Psychiatry. 1998;155(10):1325-1331.
30. Volkow ND, Wang G, Fowler JS, et al. Therapeutic doses of oral methylphenidate significantly increase extracellular dopamine in the human brain. J Neurosci. 2001;21(2):RC121.
31. Dougherty DD, Bonab AA, Spencer TJ, et al. Dopamine transporter density in patients with attention deficit hyperactivity disorder. Lancet. 1999;354(9196):2132-2133.
32. Li JJ, Lee SS. Interaction of dopamine transporter gene and observed parenting behaviors on attention-deficit/ hyperactivity disorder: a structural equation modeling approach. J Clin Child Adolesc Psychol. 2013;42(2):174-186.
33. Hesse S, Ballaschke O, Barthel H, et al. Dopamine transporter imaging in adult patients with attention-deficit/ hyperactivity disorder. Psychiatry Res. 2009;171(2):120-128.
34. Volkow ND, Wang GJ, Kollins SH, et al. Evaluating dopamine reward pathway in ADHD: clinical implications. JAMA. 2009;302(10):1084-1091.
35. Fusar-Poli P, Rubia K, Rossi G, et al. Striatal dopamine transporter alterations in ADHD: pathophysiology or adaptation to psychostimulants? A meta-analysis. Am J Psychiatry. 2012;169(3):264-272.
36. Wang GJ, Volkow ND, Wigal T, et al. Long-term stimulant treatment affects brain dopamine transporter level in patients with attention deficit hyperactive disorder. PLoS One. 2013;8(5):e63023.
37. Spencer TJ, Madras BK, Fischman AJ, et al. Striatal dopamine transporter binding in adults with ADHD. Am J Psychiatry. 2012;169(6):665; author reply 666.
38. Dresel S, Krause J, Krause KH, et al. Attention deficit hyperactivity disorder: binding of [99mTc]TRODAT-1 to the dopamine transporter before and after methylphenidate treatment. Eur J Nucl Med. 2000;27(10):1518-1524.
39. Krause J, la Fougere C, Krause KH, et al. Influence of striatal dopamine transporter availability on the response to methylphenidate in adult patients with ADHD. Eur Arch Psychiatry Clin Neurosci. 2005;255(6):428-431.
40. la Fougère C, Krause J, Krause KH, et al. Value of 99mTc-TRODAT-1 SPECT to predict clinical response to methylphenidate treatment in adults with attention deficit hyperactivity disorder. Nucl Med Commun. 2006;27(9):733-737.
41. MTA Cooperative Group. National Institute of Mental Health Multimodal Treatment Study of ADHD follow-up: 24-month outcomes of treatment strategies for attention-deficit/hyperactivity disorder. Pediatrics. 2004;113(4):754-761.
42. da Silva N Jr, Szobot CM, Anselmi CE, et al. Attention deficit/hyperactivity disorder: is there a correlation between dopamine transporter density and cerebral blood flow? Clin Nucl Med. 2011;36(8):656-660.
43. Ernst M, Zametkin AJ, Matochik JA, et al. High midbrain [18F]DOPA accumulation in children with attention deficit hyperactivity disorder. Am J Psychiatry. 1999;156(8):1209-1215.
44. Ernst M, Zametkin AJ, Matochik JA, et al. DOPA decarboxylase activity in attention deficit hyperactivity disorder adults. A [fluorine-18]fluorodopa positron emission tomographic study. J Neurosci. 1998;18(15):5901-5907.
45. Xu M, Moratalla R, Gold LH, et al. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell. 1994;79(4):729-742.
46. Goodwin RJ, Mackay CL, Nilsson A, et al. Qualitative and quantitative MALDI imaging of the positron emission tomography ligands raclopride (a D2 dopamine antagonist) and SCH 23390 (a D1 dopamine antagonist) in rat brain tissue sections using a solvent-free dry matrix application method. Anal Chem. 2011;83(24):9694-9701.
47. Negyessy L, Goldman-Rakic PS. Subcellular localization of the dopamine D2 receptor and coexistence with the calcium-binding protein neuronal calcium sensor-1 in the primate prefrontal cortex. J Comp Neurol. 2005;488(4):464-475.
48. Boyson SJ, McGonigle P, Molinoff PB. Quantitative autoradiographic localization of the D1 and D2 subtypes of dopamine receptors in rat brain. J Neurosci. 1986;6(11):3177-3188.
49. Doi M, Yujnovsky I, Hirayama J, et al. Impaired light masking in dopamine D2 receptor-null mice. Nat Neurosci. 2006;9(6):732-734.
50. Lou HC, Rosa P, Pryds O, et al. ADHD: increased dopamine receptor availability linked to attention deficit and low neonatal cerebral blood flow. Dev Med Child Neurol. 2004;46(3):179-183.
51. Volkow ND, Wang GJ, Fowler JS, et al. Effects of methylphenidate on regional brain glucose metabolism in humans: relationship to dopamine D2 receptors. Am J Psychiatry. 1997;154(1):50-55.
52. Ilgin N, Senol S, Gucuyener K, et al. Is increased D2 receptor availability associated with response to stimulant medication in ADHD. Dev Med Child Neurol. 2001;43(11):755-760.
53. Volkow ND, Wang GJ, Tomasi D, et al. Methylphenidate-elicited dopamine increases in ventral striatum are associated with long-term symptom improvement in adults with attention deficit hyperactivity disorder. J Neurosci. 2012;32(3):841-849.
Attention-deficit/hyperactivity disorder (ADHD) is one of the most common pediatric psychiatric disorders, occurring in approximately 5% of children.1 The disorder persists into adulthood in about one-half of those who are affected in childhood.2 In adults and children, diagnosis continues to be based on the examiner’s subjective assessment. (Box 13-9 describes how ADHD presents a complicated, moving target for the diagnostician.)
Patients who have ADHD are rarely studied with imaging; there are no established imaging findings associated with an ADHD diagnosis. Over the past 20 years, however, significant research has shown that molecular alterations along the dopaminergic−frontostriatal pathways occur in association with the behavioral constellation of ADHD symptoms—suggesting a pathophysiologic mechanism for this disorder.
In this article, we describe molecular findings from nuclear medicine imaging in ADHD. We also summarize imaging evidence for dysfunction of the dopaminergic-frontostriatal neural circuits as central in the pathophysiology of ADHD, with special focus on the dopamine reuptake transporter (DaT). Box 210,11 reviews our key observations and looks at the future of imaging in the management of ADHD.
Dopaminergic theory of ADHD
The executive functions that are disordered in ADHD (impulse control, judgment, maintaining attention) are thought to be centered in the infraorbital, dorsolateral, and medial frontal lobes. Neurotransmitters that have been implicated in the pathophysiology of ADHD include norepinephrine12 and dopamine13; medications that selectively block reuptake of these neurotransmitters are used to treat ADHD.14,15 Only the dopamine system has been extensively evaluated with molecular imaging techniques.
Because methylphenidate, a potent selective dopamine reuptake inhibitor, has been shown to reduce disordered executive functional behaviors in ADHD, considerable imaging research has focused on the dopaminergic neural circuits in the frontostriatal regions of the brain. The dopaminergic theory of ADHD is based on the hypothesis that alterations in the density or function of these circuits are responsible for behaviors that constitute ADHD.
Despite decades of efforts to delineate the underlying pathophysiology and neurochemistry of ADHD, no single unifying theory accounts for all imaging findings in all patients. This might be in part because of imprecision inherent in psychiatric diagnoses that are based on subjective observations. The behavioral criteria for ADHD can manifest in several disorders. For example, anxiety-related symptoms seen in posttraumatic stress disorder, social anxiety disorder, and panic disorder also present as behaviors similar to those in ADHD diagnostic criteria.
Molecular imaging might provide a window into the underlying pathophysiology of ADHD and, by identifying objective findings, (1) allow for patient stratification based on underlying physiologic subtypes, (2) refine diagnostic criteria, and (3) predict treatment response.
Nuclear medicine findings
In general, nuclear medicine investigations of ADHD can be divided into studies of changes in regional cerebral blood flow (rCBF) or glucose metabolism (rCGM) and those that have assessed the concentration of synaptic structures, using highly specific radiolabeled ligands. Both kinds of studies provide limited anatomic resolution, unless co-registered with MRI or CT scans and either single photon emission computed tomography (SPECT) or positron emission tomography (PET).
Synaptic imaging using radiolabeled ligands with high biologic specificity for synaptic structures has high molecular resolution—that is, radiolabeled ligands used for selective imaging of the dopamine transporter or receptor do not identify serotonin transporters or receptors, and vice versa. (Details of SPECT and PET techniques are beyond the scope of this article but can be found in standard nuclear medicine textbooks.)
SPECT and PET of rCBF
Early investigations of rCBF in ADHD were performed using inhaled radioactive xenon-133 gas.16 Later, rCBF was assessed using fat-soluble radiolabeled ligands that rapidly distribute in the brain in proportion to blood flow by crossing the blood−brain barrier. Labeled with radioactive 99m-technetium, these ligands cross rapidly into brain cells after IV injection. Once intracellular, covalent bonds within the ligands cleave into 2 charged particles that do not easily recross the cell membrane. There is little redistribution of tracer after initial uptake.
The imaging data set that results can be reconstructed as (1) surface images, on which defects indicate areas of reduced rCBF, or (2) tomographic slices on which color scales indicate relative rCBF values (Figure 1). Because of the minimal redistribution of the tracer, SPECT images obtained 1 or 2 hours after injection provide a snapshot of rCBF at the time tracer is injected. Patients can be injected under various conditions, such as at rest with eyes and ears open in a dimly lit, quiet room, and then under cognitive stress (Figure 2), such as performing a computer-based attention and impulse control task, or during stimulant treatment.
Numerous investigators have found reduced frontal or striatal rCBF, or both, in patients with ADHD, unilaterally on the right17 or left,18,19 or bilaterally.20 Additionally, with stimulant therapy, normalization of striatal and frontal rCBF has been demonstrated14,19—changes that correlate with resolution of behavioral symptoms of ADHD with stimulant treatment.21
SPECT of 32 boys with previously untreated ADHD. Kim et al21 found that the presence of reduced right or left, or both, frontal rCBF, which normalized with 8 weeks of stimulant therapy, predicted symptom improvement in 85% of patients. Absence of improvement of reduced frontal rCBF had a 75% negative predictive value for treatment response. (Additionally, hyperperfusion of the somatosensory cortex has been demonstrated in children with ADHD,16,22 suggesting increased responsiveness to extraneous environmental input.)
SPECT of 40 untreated pediatric patients compared with 17 age-matched controls. Using SPECT, Lee et al23 reported rCBF reductions in the orbitofrontal cortex and the medial temporal gyrus of participants; reductions corresponded to areas of motor and impulsivity control. The researchers also demonstrated increased rCBF in the somatosensory area.
After methylphenidate treatment, blood flow to these areas normalized, and rCBF to higher visual and superior prefrontal areas decreased. Substantial clinical improvement occurred in 64% of patients—suggesting methylphenidate treatment of ADHD works by (1) increasing function of areas of the brain that control impulses, motor activity, and attention, and (2) reducing function to sensory areas that lead to distraction by extraneous environmental sensory input.
O-15-labeled water PET of 10 adults with ADHD. Schweitzer et al24 found that participants who demonstrated improvement in behavioral symptoms with chronic stimulant therapy had reduced rCBF in the striata at baseline—again, suggesting that baseline hypometabolism in the striata is associated with ADHD.
PET of regional cerebral glucose metabolism
Cerebral metabolism requires a constant supply of glucose; regional differences in cerebral glucose metabolism can be assessed directly with positron-emitting F-18-fluoro-2-deoxyglucose. Although metabolically inert, this agent is transported intracellularly similar to glucose; once phosphorylated within brain cells, however, it can no longer undergo further metabolism or redistribution.
Studies using PET to assess rCGM were some of the earliest molecular imaging applications in ADHD. Zametkin et al25 reported low global cerebral glucose utilization in adults, but not adolescents,26 with ADHD. However, further study, with normalization of the PET data, confirmed reduced rCGM in the left prefrontal cortex in both adolescents26 and adults,27 indicating hypometabolism of cortical areas associated with impulse control and attention in ADHD. In adolescents, symptom severity was inversely related to rCBF in the left anterior frontal cortex.
Synaptic imaging
Nuclear imaging has been used to study several components of the striatal dopaminergic synapse, including:
• dopamine substrates, using fluorine- 18-labeled dopa or carbon-11-labeled dopa
• dopamine receptors, using carbon- 11-labeled raclopride or iodine-123 iodobenzamide
• the tDaT, using iodine-123 ioflupane, 99m-technetium TRODAT, or carbon-11 cocaine (Figure 3).
All of these synaptic imaging agents were used mainly as research tools until 2011, when the FDA approved the SPECT imaging agent iodine-123 ioflupane (DaTscan) for clinical use in assessment of Parkinson’s disease.28 This commercially available agent has high specificity for the DaT, with little background activity noted on SPECT imaging (Figure 4).
Dopamine transporter imaging
Because the site of action of methylphenidate is the DaT, imaging this component of the striatal dopaminergic synapse has been an area of intense investigation in ADHD. Located almost exclusively in the striata, DaT reduces synaptic concentrations of dopamine by means of reuptake channels in the cell membrane.29 By reversibly binding to, and occupying sites on, the DaT, methylphenidate impedes dopamine reuptake, which results in increased availability of dopamine at the synapse.30
By demonstrating an increase in striatal DaT density in patients with ADHD— first reported by Dougherty et al31 using iodine-123 altropane (a dopaminergic uptake inhibitor) in 6 adults with ADHD—investigators have hypothesized that excessive expression of the DaT protein in the striata, which may result from genetic or environmental factors, is a central causative agent of ADHD.32 Subsequent studies, however, have yielded contradictory findings: Hesse et al,33 using SPECT imaging, and Volkow et al,34 using carbon-11 cocaine PET imaging, found reduced DaT density in, respectively, 9 and 26 patients with ADHD.
To clarify the role of DaT levels in the etiology of ADHD and to explain discrepant results, Fusar-Poli et al35 performed a meta-analysis of 9 published papers that reported the results of DaT imaging in a total of 169 ADHD patients and 129 controls. They noted that these studies included 6 different imaging agents and protocols. Patients were stimulant therapy-naïve (n = 137) or drug-free (refrained from stimulant therapy for a time [n = 32]). The team found that the degree of elevation of the striatal DaT concentration correlated with a history of stimulant exposure, and that the drug-naïve group had a reduced DaT level.
Fusar-Poli’s hypothesis? Elevated DaT levels result from up-regulation in the presence of chronic methylphenidate therapy, which accounts for early reports that demonstrated increased striatal DaT density. Clinically, up-regulation might explain the lack of sustained relief of behavioral symptoms with stimulant therapy in 20% of patients with ADHD who showed clinical improvement initially.36
Only limited conclusions can be drawn about the role of DaT levels in ADHD, given the small number of patients studied in published reports. In addition, the Fusar-Poli meta-analysis has come under strong criticism because of methodological errors with improper patient inclusion and characterization of treatment status,37 calling into question the investigators’ conclusions.
Does the DaT level hold promise for practice? Despite a lack of clarity about the significance of DaT level in the etiology of ADHD, knowledge of a patient’s level might prove useful in predicting which patients will respond to methylphenidate. Namely, several researchers have found that:
• an elevated baseline level of DaT (before stimulant therapy) correlates with robust clinical response
• absence of an elevated baseline DaT level suggests that symptomatic improvement with stimulant therapy in unlikely.38-40
Dresel et al38 evaluated 17 drug-naïve adults, newly diagnosed with ADHD, using 99m-technetium TRODAT SPECT before and after methylphenidate therapy. They found a 15% increase in specific DaT binding in patients with ADHD, compared with controls, at baseline. After treatment, the researchers observed a 28% reduction in specific DaT binding—a significant change from baseline that correlated with behavioral response.
Study: SPECT in 18 adults with ADHD given methylphenidate. Krause39 used the same SPECT agent to study 18 adults before they received methylphenidate and 10 weeks after treatment. Participants were categorized as responders or nonresponders based on clinical assessment of ADHD symptoms after those 10 weeks. All 12 responders had an elevated striatal DaT concentration at baseline. Of the 6 nonresponders, 5 had a normal level of striatal DaT compared with age-matched controls.
Study: 22 Adult ADHD patients evaluated with 99m-technetium TRODAT SPECT. The same group of investigators40 presented imaging findings in 22 additional adult patients. Seventeen had an elevated striatal DaT level, 16 of whom responded to stimulant therapy. The remaining 5 patients had reduced striatal DaT at baseline; none had a good clinical response to methylphenidate.
The positive clinical response to methylphenidate in 67%37 and 77%40 of patients is in good agreement with results from larger studies, which reported that approximately 75% of patients with ADHD show prompt clinical improvement with stimulants.41 Improvement might be related to an increase in functioning of the frontostriatal dopaminergic circuit that is seen with stimulant therapy. Increased availability of dopamine at the synapse, resulting from stimulant blockade of the dopamine reuptake transporter, produces increased dopamine neurotransmission and increased activation of frontostriatal circuits.
In another study, rCBF in frontostriatal circuits was determined to be inversely proportional to DaT density; rCBF normalized with stimulant therapy.42
Will imaging pave the way for therapeutic stratification? Baseline determinations of striatal DaT concentration with SPECT imaging might make it possible to stratify patients with ADHD symptoms into those likely to show significant behavioral symptom response to methylphenidate and those who are not likely to respond. There might be an objective imaging finding—striatal DaT density—that allows clinicians to distinguish stimulant-responsive ADHD from stimulant-unresponsive ADHD.
Dopamine substrate imaging
Radiolabeled dopa (carbon-11 or fluorine-18) is transported into presynaptic dopaminergic neurons in the striatum, where it is decarboxylated, converted to radio-dopamine, and stored within vesicles until released in response to neuronal excitation. Semi-quantitative assessment is achieved with calculation of specific (striatal) to nonspecific (background) uptake ratios. Increased values are thought to indicate increased density of dopaminergic neurons.43
Ernst et al44 reported a 50% decrease in specific fluorine-18 dopa uptake in the left prefrontal cortex in 17 drug-naïve adults with ADHD, compared with 23 controls. The same team reported increased midbrain fluorine-18 dopa levels in 10 adolescents with ADHD—48% higher, overall, than what was seen in 10 controls.43 They hypothesized that these opposite results were the results of a reduction in the dopaminergic neuronal density in adults, which might be part of the natural history of ADHD, or a normal age-related reduction in neuronal density, or both. Increased dopa levels in the team’s adolescent group were hypothesized to reflect up-regulation in dopamine synthesis due to low synaptic dopamine concentrations that might result from increased dopamine reuptake.
Dopamine-receptor imaging
The 5 distinct dopamine receptors (D1, D2, D3, D4, and D5) can be grouped into 2 subtypes, based on their coupling with G proteins. D1 and D5 constitute a group; D2, D3, and D4, a second group.
The D1 receptor is the most common dopamine receptor in the brain and is widely distributed in the striatum and prefrontal cerebral cortex. D1 receptor knockout mice demonstrate hyperactivity and poorer performance on learning tasks and are used as an animal model for ADHD.45 D1 has been imaged using C-11 SCH 23390 PET46 in rats, but its role in ADHD has yet to be evaluated. D5 is the most recently cloned and most widely distributed of the known dopamine receptors; however, there are no imaging studies of the D5 receptor.13
D2 receptors are present in presynaptic and postsynaptic neurons47 in the neocortex, substantia nigra, nucleus accumbens, and olfactory tubercle, as well as in other structures.48 Presynaptic D2 receptors act as autoregulators, inhibiting dopaminergic synthesis, firing rate, and release.49
Using C-11 raclopride PET imaging, Lou et al50 reported high D2/3 receptor availability in adolescents who had a history of perinatal cerebral ischemia. They found that this availability is associated with an increase in the severity of ADHD symptoms. They proposed that the increase in “empty” receptor density might have been caused by perinatal ischemia-induced presynaptic dopaminergic neuronal loss or an increase in presynaptic dopamine reuptake (Figure 550). Either mechanism could result in up-regulation in postsynaptic D2/3 receptors.
Volkow et al51 reported that D2 receptor density correlated with methylphenidate-induced changes in rCBF in frontal and temporal lobes in humans. They postulated that the variable therapeutic effects of methylphenidate seen in ADHD patients might be related to variations in baseline D2 receptor availability.
Lou et al50 reported elevated D2 receptor density, demonstrated using carbon-11 raclopride, in children with ADHD, compared with normal adults.
Further support for a relationship between D2-receptor density and symptomatic improvement with methylphenidate in ADHD was presented by Ilgin et al52 using iodine-123 iodobenzamide SPECT. They found elevated D2 receptor levels in 9 drug-naïve children with ADHD, which is 20% to 60% above what is seen in unaffected children. They noted that these patients showed improvement in hyperactivity when treated with methylphenidate.
In a similar study of 20 drug-naïve adults, Volkow et al53 found that durable symptomatic improvement with methylphenidate therapy was associated with increased D2 receptor availability.
Summing up
Striatal DaT is the most likely synaptic target for stratifying patients with ADHD, now that a dopamine transporter imaging agent is available commercially. Stratification might allow for refinement in the diagnostic categorization of ADHD, with introduction of stimulant-responsive and stimulant-unresponsive subtypes that are based on DaT imaging findings.
Bottom Line
Given recent advances showing molecular alterations in the dopaminergic-frontostriatal pathway as central to attention-deficit/hyperactivity disorder, molecular imaging might be useful as an objective study for diagnosis.
Related Resources
• Schweitzer JB, Lee DO, Hanford RB, et al. A positron emission tomography study of methylphenidate in adults with ADHD: alterations in resting blood flow and predicting treatment response. Neuropsychopharmacology. 2003;28(5):967-973.
• Raz A. Brain imaging data of ADHD. Psychiatric Times. http://www.psychiatrictimes.com/adhd/brain-imaging-data-adhd.
Drug Brand Names
Iodine-123 ioflupane • Methylphenidate • Ritalin DaTscan
Acknowledgment
Kylee M. L. Unsdorfer, a medical student at Northeast Ohio Medical University, helped prepare the manuscript of this article.
Disclosures
Dr. Thacker reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Binkovitz received 4 doses of ioflupane I123I (DaTscan) from General Electric for investigator-initiated research, used for animal imaging in 2012.
Attention-deficit/hyperactivity disorder (ADHD) is one of the most common pediatric psychiatric disorders, occurring in approximately 5% of children.1 The disorder persists into adulthood in about one-half of those who are affected in childhood.2 In adults and children, diagnosis continues to be based on the examiner’s subjective assessment. (Box 13-9 describes how ADHD presents a complicated, moving target for the diagnostician.)
Patients who have ADHD are rarely studied with imaging; there are no established imaging findings associated with an ADHD diagnosis. Over the past 20 years, however, significant research has shown that molecular alterations along the dopaminergic−frontostriatal pathways occur in association with the behavioral constellation of ADHD symptoms—suggesting a pathophysiologic mechanism for this disorder.
In this article, we describe molecular findings from nuclear medicine imaging in ADHD. We also summarize imaging evidence for dysfunction of the dopaminergic-frontostriatal neural circuits as central in the pathophysiology of ADHD, with special focus on the dopamine reuptake transporter (DaT). Box 210,11 reviews our key observations and looks at the future of imaging in the management of ADHD.
Dopaminergic theory of ADHD
The executive functions that are disordered in ADHD (impulse control, judgment, maintaining attention) are thought to be centered in the infraorbital, dorsolateral, and medial frontal lobes. Neurotransmitters that have been implicated in the pathophysiology of ADHD include norepinephrine12 and dopamine13; medications that selectively block reuptake of these neurotransmitters are used to treat ADHD.14,15 Only the dopamine system has been extensively evaluated with molecular imaging techniques.
Because methylphenidate, a potent selective dopamine reuptake inhibitor, has been shown to reduce disordered executive functional behaviors in ADHD, considerable imaging research has focused on the dopaminergic neural circuits in the frontostriatal regions of the brain. The dopaminergic theory of ADHD is based on the hypothesis that alterations in the density or function of these circuits are responsible for behaviors that constitute ADHD.
Despite decades of efforts to delineate the underlying pathophysiology and neurochemistry of ADHD, no single unifying theory accounts for all imaging findings in all patients. This might be in part because of imprecision inherent in psychiatric diagnoses that are based on subjective observations. The behavioral criteria for ADHD can manifest in several disorders. For example, anxiety-related symptoms seen in posttraumatic stress disorder, social anxiety disorder, and panic disorder also present as behaviors similar to those in ADHD diagnostic criteria.
Molecular imaging might provide a window into the underlying pathophysiology of ADHD and, by identifying objective findings, (1) allow for patient stratification based on underlying physiologic subtypes, (2) refine diagnostic criteria, and (3) predict treatment response.
Nuclear medicine findings
In general, nuclear medicine investigations of ADHD can be divided into studies of changes in regional cerebral blood flow (rCBF) or glucose metabolism (rCGM) and those that have assessed the concentration of synaptic structures, using highly specific radiolabeled ligands. Both kinds of studies provide limited anatomic resolution, unless co-registered with MRI or CT scans and either single photon emission computed tomography (SPECT) or positron emission tomography (PET).
Synaptic imaging using radiolabeled ligands with high biologic specificity for synaptic structures has high molecular resolution—that is, radiolabeled ligands used for selective imaging of the dopamine transporter or receptor do not identify serotonin transporters or receptors, and vice versa. (Details of SPECT and PET techniques are beyond the scope of this article but can be found in standard nuclear medicine textbooks.)
SPECT and PET of rCBF
Early investigations of rCBF in ADHD were performed using inhaled radioactive xenon-133 gas.16 Later, rCBF was assessed using fat-soluble radiolabeled ligands that rapidly distribute in the brain in proportion to blood flow by crossing the blood−brain barrier. Labeled with radioactive 99m-technetium, these ligands cross rapidly into brain cells after IV injection. Once intracellular, covalent bonds within the ligands cleave into 2 charged particles that do not easily recross the cell membrane. There is little redistribution of tracer after initial uptake.
The imaging data set that results can be reconstructed as (1) surface images, on which defects indicate areas of reduced rCBF, or (2) tomographic slices on which color scales indicate relative rCBF values (Figure 1). Because of the minimal redistribution of the tracer, SPECT images obtained 1 or 2 hours after injection provide a snapshot of rCBF at the time tracer is injected. Patients can be injected under various conditions, such as at rest with eyes and ears open in a dimly lit, quiet room, and then under cognitive stress (Figure 2), such as performing a computer-based attention and impulse control task, or during stimulant treatment.
Numerous investigators have found reduced frontal or striatal rCBF, or both, in patients with ADHD, unilaterally on the right17 or left,18,19 or bilaterally.20 Additionally, with stimulant therapy, normalization of striatal and frontal rCBF has been demonstrated14,19—changes that correlate with resolution of behavioral symptoms of ADHD with stimulant treatment.21
SPECT of 32 boys with previously untreated ADHD. Kim et al21 found that the presence of reduced right or left, or both, frontal rCBF, which normalized with 8 weeks of stimulant therapy, predicted symptom improvement in 85% of patients. Absence of improvement of reduced frontal rCBF had a 75% negative predictive value for treatment response. (Additionally, hyperperfusion of the somatosensory cortex has been demonstrated in children with ADHD,16,22 suggesting increased responsiveness to extraneous environmental input.)
SPECT of 40 untreated pediatric patients compared with 17 age-matched controls. Using SPECT, Lee et al23 reported rCBF reductions in the orbitofrontal cortex and the medial temporal gyrus of participants; reductions corresponded to areas of motor and impulsivity control. The researchers also demonstrated increased rCBF in the somatosensory area.
After methylphenidate treatment, blood flow to these areas normalized, and rCBF to higher visual and superior prefrontal areas decreased. Substantial clinical improvement occurred in 64% of patients—suggesting methylphenidate treatment of ADHD works by (1) increasing function of areas of the brain that control impulses, motor activity, and attention, and (2) reducing function to sensory areas that lead to distraction by extraneous environmental sensory input.
O-15-labeled water PET of 10 adults with ADHD. Schweitzer et al24 found that participants who demonstrated improvement in behavioral symptoms with chronic stimulant therapy had reduced rCBF in the striata at baseline—again, suggesting that baseline hypometabolism in the striata is associated with ADHD.
PET of regional cerebral glucose metabolism
Cerebral metabolism requires a constant supply of glucose; regional differences in cerebral glucose metabolism can be assessed directly with positron-emitting F-18-fluoro-2-deoxyglucose. Although metabolically inert, this agent is transported intracellularly similar to glucose; once phosphorylated within brain cells, however, it can no longer undergo further metabolism or redistribution.
Studies using PET to assess rCGM were some of the earliest molecular imaging applications in ADHD. Zametkin et al25 reported low global cerebral glucose utilization in adults, but not adolescents,26 with ADHD. However, further study, with normalization of the PET data, confirmed reduced rCGM in the left prefrontal cortex in both adolescents26 and adults,27 indicating hypometabolism of cortical areas associated with impulse control and attention in ADHD. In adolescents, symptom severity was inversely related to rCBF in the left anterior frontal cortex.
Synaptic imaging
Nuclear imaging has been used to study several components of the striatal dopaminergic synapse, including:
• dopamine substrates, using fluorine- 18-labeled dopa or carbon-11-labeled dopa
• dopamine receptors, using carbon- 11-labeled raclopride or iodine-123 iodobenzamide
• the tDaT, using iodine-123 ioflupane, 99m-technetium TRODAT, or carbon-11 cocaine (Figure 3).
All of these synaptic imaging agents were used mainly as research tools until 2011, when the FDA approved the SPECT imaging agent iodine-123 ioflupane (DaTscan) for clinical use in assessment of Parkinson’s disease.28 This commercially available agent has high specificity for the DaT, with little background activity noted on SPECT imaging (Figure 4).
Dopamine transporter imaging
Because the site of action of methylphenidate is the DaT, imaging this component of the striatal dopaminergic synapse has been an area of intense investigation in ADHD. Located almost exclusively in the striata, DaT reduces synaptic concentrations of dopamine by means of reuptake channels in the cell membrane.29 By reversibly binding to, and occupying sites on, the DaT, methylphenidate impedes dopamine reuptake, which results in increased availability of dopamine at the synapse.30
By demonstrating an increase in striatal DaT density in patients with ADHD— first reported by Dougherty et al31 using iodine-123 altropane (a dopaminergic uptake inhibitor) in 6 adults with ADHD—investigators have hypothesized that excessive expression of the DaT protein in the striata, which may result from genetic or environmental factors, is a central causative agent of ADHD.32 Subsequent studies, however, have yielded contradictory findings: Hesse et al,33 using SPECT imaging, and Volkow et al,34 using carbon-11 cocaine PET imaging, found reduced DaT density in, respectively, 9 and 26 patients with ADHD.
To clarify the role of DaT levels in the etiology of ADHD and to explain discrepant results, Fusar-Poli et al35 performed a meta-analysis of 9 published papers that reported the results of DaT imaging in a total of 169 ADHD patients and 129 controls. They noted that these studies included 6 different imaging agents and protocols. Patients were stimulant therapy-naïve (n = 137) or drug-free (refrained from stimulant therapy for a time [n = 32]). The team found that the degree of elevation of the striatal DaT concentration correlated with a history of stimulant exposure, and that the drug-naïve group had a reduced DaT level.
Fusar-Poli’s hypothesis? Elevated DaT levels result from up-regulation in the presence of chronic methylphenidate therapy, which accounts for early reports that demonstrated increased striatal DaT density. Clinically, up-regulation might explain the lack of sustained relief of behavioral symptoms with stimulant therapy in 20% of patients with ADHD who showed clinical improvement initially.36
Only limited conclusions can be drawn about the role of DaT levels in ADHD, given the small number of patients studied in published reports. In addition, the Fusar-Poli meta-analysis has come under strong criticism because of methodological errors with improper patient inclusion and characterization of treatment status,37 calling into question the investigators’ conclusions.
Does the DaT level hold promise for practice? Despite a lack of clarity about the significance of DaT level in the etiology of ADHD, knowledge of a patient’s level might prove useful in predicting which patients will respond to methylphenidate. Namely, several researchers have found that:
• an elevated baseline level of DaT (before stimulant therapy) correlates with robust clinical response
• absence of an elevated baseline DaT level suggests that symptomatic improvement with stimulant therapy in unlikely.38-40
Dresel et al38 evaluated 17 drug-naïve adults, newly diagnosed with ADHD, using 99m-technetium TRODAT SPECT before and after methylphenidate therapy. They found a 15% increase in specific DaT binding in patients with ADHD, compared with controls, at baseline. After treatment, the researchers observed a 28% reduction in specific DaT binding—a significant change from baseline that correlated with behavioral response.
Study: SPECT in 18 adults with ADHD given methylphenidate. Krause39 used the same SPECT agent to study 18 adults before they received methylphenidate and 10 weeks after treatment. Participants were categorized as responders or nonresponders based on clinical assessment of ADHD symptoms after those 10 weeks. All 12 responders had an elevated striatal DaT concentration at baseline. Of the 6 nonresponders, 5 had a normal level of striatal DaT compared with age-matched controls.
Study: 22 Adult ADHD patients evaluated with 99m-technetium TRODAT SPECT. The same group of investigators40 presented imaging findings in 22 additional adult patients. Seventeen had an elevated striatal DaT level, 16 of whom responded to stimulant therapy. The remaining 5 patients had reduced striatal DaT at baseline; none had a good clinical response to methylphenidate.
The positive clinical response to methylphenidate in 67%37 and 77%40 of patients is in good agreement with results from larger studies, which reported that approximately 75% of patients with ADHD show prompt clinical improvement with stimulants.41 Improvement might be related to an increase in functioning of the frontostriatal dopaminergic circuit that is seen with stimulant therapy. Increased availability of dopamine at the synapse, resulting from stimulant blockade of the dopamine reuptake transporter, produces increased dopamine neurotransmission and increased activation of frontostriatal circuits.
In another study, rCBF in frontostriatal circuits was determined to be inversely proportional to DaT density; rCBF normalized with stimulant therapy.42
Will imaging pave the way for therapeutic stratification? Baseline determinations of striatal DaT concentration with SPECT imaging might make it possible to stratify patients with ADHD symptoms into those likely to show significant behavioral symptom response to methylphenidate and those who are not likely to respond. There might be an objective imaging finding—striatal DaT density—that allows clinicians to distinguish stimulant-responsive ADHD from stimulant-unresponsive ADHD.
Dopamine substrate imaging
Radiolabeled dopa (carbon-11 or fluorine-18) is transported into presynaptic dopaminergic neurons in the striatum, where it is decarboxylated, converted to radio-dopamine, and stored within vesicles until released in response to neuronal excitation. Semi-quantitative assessment is achieved with calculation of specific (striatal) to nonspecific (background) uptake ratios. Increased values are thought to indicate increased density of dopaminergic neurons.43
Ernst et al44 reported a 50% decrease in specific fluorine-18 dopa uptake in the left prefrontal cortex in 17 drug-naïve adults with ADHD, compared with 23 controls. The same team reported increased midbrain fluorine-18 dopa levels in 10 adolescents with ADHD—48% higher, overall, than what was seen in 10 controls.43 They hypothesized that these opposite results were the results of a reduction in the dopaminergic neuronal density in adults, which might be part of the natural history of ADHD, or a normal age-related reduction in neuronal density, or both. Increased dopa levels in the team’s adolescent group were hypothesized to reflect up-regulation in dopamine synthesis due to low synaptic dopamine concentrations that might result from increased dopamine reuptake.
Dopamine-receptor imaging
The 5 distinct dopamine receptors (D1, D2, D3, D4, and D5) can be grouped into 2 subtypes, based on their coupling with G proteins. D1 and D5 constitute a group; D2, D3, and D4, a second group.
The D1 receptor is the most common dopamine receptor in the brain and is widely distributed in the striatum and prefrontal cerebral cortex. D1 receptor knockout mice demonstrate hyperactivity and poorer performance on learning tasks and are used as an animal model for ADHD.45 D1 has been imaged using C-11 SCH 23390 PET46 in rats, but its role in ADHD has yet to be evaluated. D5 is the most recently cloned and most widely distributed of the known dopamine receptors; however, there are no imaging studies of the D5 receptor.13
D2 receptors are present in presynaptic and postsynaptic neurons47 in the neocortex, substantia nigra, nucleus accumbens, and olfactory tubercle, as well as in other structures.48 Presynaptic D2 receptors act as autoregulators, inhibiting dopaminergic synthesis, firing rate, and release.49
Using C-11 raclopride PET imaging, Lou et al50 reported high D2/3 receptor availability in adolescents who had a history of perinatal cerebral ischemia. They found that this availability is associated with an increase in the severity of ADHD symptoms. They proposed that the increase in “empty” receptor density might have been caused by perinatal ischemia-induced presynaptic dopaminergic neuronal loss or an increase in presynaptic dopamine reuptake (Figure 550). Either mechanism could result in up-regulation in postsynaptic D2/3 receptors.
Volkow et al51 reported that D2 receptor density correlated with methylphenidate-induced changes in rCBF in frontal and temporal lobes in humans. They postulated that the variable therapeutic effects of methylphenidate seen in ADHD patients might be related to variations in baseline D2 receptor availability.
Lou et al50 reported elevated D2 receptor density, demonstrated using carbon-11 raclopride, in children with ADHD, compared with normal adults.
Further support for a relationship between D2-receptor density and symptomatic improvement with methylphenidate in ADHD was presented by Ilgin et al52 using iodine-123 iodobenzamide SPECT. They found elevated D2 receptor levels in 9 drug-naïve children with ADHD, which is 20% to 60% above what is seen in unaffected children. They noted that these patients showed improvement in hyperactivity when treated with methylphenidate.
In a similar study of 20 drug-naïve adults, Volkow et al53 found that durable symptomatic improvement with methylphenidate therapy was associated with increased D2 receptor availability.
Summing up
Striatal DaT is the most likely synaptic target for stratifying patients with ADHD, now that a dopamine transporter imaging agent is available commercially. Stratification might allow for refinement in the diagnostic categorization of ADHD, with introduction of stimulant-responsive and stimulant-unresponsive subtypes that are based on DaT imaging findings.
Bottom Line
Given recent advances showing molecular alterations in the dopaminergic-frontostriatal pathway as central to attention-deficit/hyperactivity disorder, molecular imaging might be useful as an objective study for diagnosis.
Related Resources
• Schweitzer JB, Lee DO, Hanford RB, et al. A positron emission tomography study of methylphenidate in adults with ADHD: alterations in resting blood flow and predicting treatment response. Neuropsychopharmacology. 2003;28(5):967-973.
• Raz A. Brain imaging data of ADHD. Psychiatric Times. http://www.psychiatrictimes.com/adhd/brain-imaging-data-adhd.
Drug Brand Names
Iodine-123 ioflupane • Methylphenidate • Ritalin DaTscan
Acknowledgment
Kylee M. L. Unsdorfer, a medical student at Northeast Ohio Medical University, helped prepare the manuscript of this article.
Disclosures
Dr. Thacker reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Binkovitz received 4 doses of ioflupane I123I (DaTscan) from General Electric for investigator-initiated research, used for animal imaging in 2012.
1. Polanczyk G, de Lima MS, Horta BL, et al. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry. 2007;164(6):942-948.
2. Simon V, Czobor P, Bálint S, et al. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry. 2009;194(3):204-211.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Berger I. Diagnosis of attention deficit hyperactivity disorder: much ado about something. Isr Med Assoc J. 2011;13(9):571-574.
5. Schonwald A, Lechner E. Attention deficit/hyperactivity disorder: complexities and controversies. Curr Opin Pediatr. 2006;18(2):189-195.
6. Rousseau C, Measham T, Bathiche-Suidan M. DSM IV, culture and child psychiatry. J Can Acad Child Adolesc Psychiatry. 2008;17(2):69-75.
7. Taylor-Klaus E. Bringing the ADHD debate into sharper focus: part 1. The Huffington Post. http:// www.huffingtonpost.com/elaine-taylorklaus/adhd-debate_b_4571097.html. Updated March 17, 2014. Accessed August 18, 2015.
8. Sweeney CT, Sembower MA, Ertischek MD, et al. Nonmedical use of prescription ADHD stimulants and preexisting patterns of drug abuse. J Addict Dis. 2013;32(1):1-10.
9. Hitt E. Multiple reports of ADHD drug shortages. Medscape. http://www.medscape.com/viewarticle/742686. Published May 13, 2011. Accessed June 4, 2015.
10. Rubia K, Alegria AA, Cubillo AI, et al. Effects of stimulants on brain function in attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. Biol Psychiatry. 2014;76(8):616-628.
11. Cortese S, Kelly C, Chabernaud C, et al. Toward systems neuroscience of ADHD: a meta-analysis of 55 fMRI studies. Am J Psychiatry. 2012;169(10):1038-1055.
12. Garnock-Jones KP, Keating GM. Atomoxetine: a review of its use in attention-deficit hyperactivity disorder in children and adolescents. Paediatr Drugs. 2009;11(3):203-226.
13. Wu J, Xiao H, Sun H, et al. Role of dopamine receptors in ADHD: a systematic meta-analysis. Mol Neurobiol. 2012; 45(3):605-620.
14. Del Campo N, Chamberlain SR, Sahakian BJ, et al. The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2011;69(12):e145-e157.
15. Berridge CW, Devilbiss DM. Psychostimulants as cognitive enhancers: the prefrontal cortex, catecholamines, and attention-deficit/hyperactivity disorder. Biol Psychiatry. 2011;69(12):e101-e111.
16. Lou HC, Henriksen L, Bruhn P. Focal cerebral hypoperfusion in children with dysphasia and/or attention deficit disorder. Arch Neurol. 1984;41(8):825-829.
17. Gustafsson P, Thernlund G, Ryding E, et al. Associations between cerebral blood-flow measured by single photon emission computed tomography (SPECT), electro-encephalogram (EEG), behaviour symptoms, cognition and neurological soft signs in children with attention-deficit hyperactivity disorder (ADHD). Acta Paediatr. 2000;89(7):830-835.
18. Sieg KG, Gaffney GR, Preston DF, et al. SPECT brain imaging abnormalities in attention deficit hyperactivity disorder. Clin Nucl Med. 1995;20(1):55-60.
19. Spalletta G, Pasini A, Pau F, et al. Prefrontal blood flow dysregulation in drug naive ADHD children without structural abnormalities. J Neural Transm. 2001;108(10):1203-1216.
20. Amen DG, Carmichael BD. High-resolution brain SPECT imaging in ADHD. Ann Clin Psychiatry. 1997;9(2):81-86.
21. Kim BN, Lee JS, Cho SC, et al. Methylphenidate increased regional cerebral blood flow in subjects with attention deficit/hyperactivity disorder. Yonsei Med J. 2001;42(1):19-29.
22. Lou HC, Henriksen L, Bruhn P, et al. Striatal dysfunction in attention deficit and hyperkinetic disorder. Arch Neurol. 1989;46(1):48-52.
23. Lee JS, Kim BN, Kang E, et al. Regional cerebral blood flow in children with attention deficit hyperactivity disorder: comparison before and after methylphenidate treatment. Hum Brain Mapp. 2005;24(3):157-164.
24. Schweitzer JB, Lee DO, Hanford RB, et al. A positron emission tomography study of methylphenidate in adults with ADHD: alterations in resting blood flow and predicting treatment response. Neuropsychopharmacology. 2003;28(5):967-973.
25. Zametkin AJ, Nordahl TE, Gross M, et al. Cerebral glucose metabolism in adults with hyperactivity of childhood onset. N Engl J Med. 1990;323(20):1361-1366.
26. Zametkin AJ, Liebenauer LL, Fitzgerald GA, et al. Brain metabolism in teenagers with attention-deficit hyperactivity disorder. Arch Gen Psychiatry. 1993;50(5):333-340.
27. Ernst M, Zametkin AJ, Matochik JA, et al. Effects of intravenous dextroamphetamine on brain metabolism in adults with attention-deficit hyperactivity disorder (ADHD). Preliminary findings. Psychopharmacol Bull. 1994;30(2):219-225.
28. Janssen M. Dopamine transporter (DaT) SPECT imaging. MI Gateway. 2012;6(1):1-3. http://interactive.snm.org/ docs/MI_Gateway_Newsletter_2012-1%20Dopamine%20 Transporter%20SPECT%20Imaging.pdf. Accessed August 18, 2015.
29. Volkow ND, Wang GJ, Fowler JS, et al. Dopamine transporter occupancies in the human brain induced by therapeutic doses of oral methylphenidate. Am J Psychiatry. 1998;155(10):1325-1331.
30. Volkow ND, Wang G, Fowler JS, et al. Therapeutic doses of oral methylphenidate significantly increase extracellular dopamine in the human brain. J Neurosci. 2001;21(2):RC121.
31. Dougherty DD, Bonab AA, Spencer TJ, et al. Dopamine transporter density in patients with attention deficit hyperactivity disorder. Lancet. 1999;354(9196):2132-2133.
32. Li JJ, Lee SS. Interaction of dopamine transporter gene and observed parenting behaviors on attention-deficit/ hyperactivity disorder: a structural equation modeling approach. J Clin Child Adolesc Psychol. 2013;42(2):174-186.
33. Hesse S, Ballaschke O, Barthel H, et al. Dopamine transporter imaging in adult patients with attention-deficit/ hyperactivity disorder. Psychiatry Res. 2009;171(2):120-128.
34. Volkow ND, Wang GJ, Kollins SH, et al. Evaluating dopamine reward pathway in ADHD: clinical implications. JAMA. 2009;302(10):1084-1091.
35. Fusar-Poli P, Rubia K, Rossi G, et al. Striatal dopamine transporter alterations in ADHD: pathophysiology or adaptation to psychostimulants? A meta-analysis. Am J Psychiatry. 2012;169(3):264-272.
36. Wang GJ, Volkow ND, Wigal T, et al. Long-term stimulant treatment affects brain dopamine transporter level in patients with attention deficit hyperactive disorder. PLoS One. 2013;8(5):e63023.
37. Spencer TJ, Madras BK, Fischman AJ, et al. Striatal dopamine transporter binding in adults with ADHD. Am J Psychiatry. 2012;169(6):665; author reply 666.
38. Dresel S, Krause J, Krause KH, et al. Attention deficit hyperactivity disorder: binding of [99mTc]TRODAT-1 to the dopamine transporter before and after methylphenidate treatment. Eur J Nucl Med. 2000;27(10):1518-1524.
39. Krause J, la Fougere C, Krause KH, et al. Influence of striatal dopamine transporter availability on the response to methylphenidate in adult patients with ADHD. Eur Arch Psychiatry Clin Neurosci. 2005;255(6):428-431.
40. la Fougère C, Krause J, Krause KH, et al. Value of 99mTc-TRODAT-1 SPECT to predict clinical response to methylphenidate treatment in adults with attention deficit hyperactivity disorder. Nucl Med Commun. 2006;27(9):733-737.
41. MTA Cooperative Group. National Institute of Mental Health Multimodal Treatment Study of ADHD follow-up: 24-month outcomes of treatment strategies for attention-deficit/hyperactivity disorder. Pediatrics. 2004;113(4):754-761.
42. da Silva N Jr, Szobot CM, Anselmi CE, et al. Attention deficit/hyperactivity disorder: is there a correlation between dopamine transporter density and cerebral blood flow? Clin Nucl Med. 2011;36(8):656-660.
43. Ernst M, Zametkin AJ, Matochik JA, et al. High midbrain [18F]DOPA accumulation in children with attention deficit hyperactivity disorder. Am J Psychiatry. 1999;156(8):1209-1215.
44. Ernst M, Zametkin AJ, Matochik JA, et al. DOPA decarboxylase activity in attention deficit hyperactivity disorder adults. A [fluorine-18]fluorodopa positron emission tomographic study. J Neurosci. 1998;18(15):5901-5907.
45. Xu M, Moratalla R, Gold LH, et al. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell. 1994;79(4):729-742.
46. Goodwin RJ, Mackay CL, Nilsson A, et al. Qualitative and quantitative MALDI imaging of the positron emission tomography ligands raclopride (a D2 dopamine antagonist) and SCH 23390 (a D1 dopamine antagonist) in rat brain tissue sections using a solvent-free dry matrix application method. Anal Chem. 2011;83(24):9694-9701.
47. Negyessy L, Goldman-Rakic PS. Subcellular localization of the dopamine D2 receptor and coexistence with the calcium-binding protein neuronal calcium sensor-1 in the primate prefrontal cortex. J Comp Neurol. 2005;488(4):464-475.
48. Boyson SJ, McGonigle P, Molinoff PB. Quantitative autoradiographic localization of the D1 and D2 subtypes of dopamine receptors in rat brain. J Neurosci. 1986;6(11):3177-3188.
49. Doi M, Yujnovsky I, Hirayama J, et al. Impaired light masking in dopamine D2 receptor-null mice. Nat Neurosci. 2006;9(6):732-734.
50. Lou HC, Rosa P, Pryds O, et al. ADHD: increased dopamine receptor availability linked to attention deficit and low neonatal cerebral blood flow. Dev Med Child Neurol. 2004;46(3):179-183.
51. Volkow ND, Wang GJ, Fowler JS, et al. Effects of methylphenidate on regional brain glucose metabolism in humans: relationship to dopamine D2 receptors. Am J Psychiatry. 1997;154(1):50-55.
52. Ilgin N, Senol S, Gucuyener K, et al. Is increased D2 receptor availability associated with response to stimulant medication in ADHD. Dev Med Child Neurol. 2001;43(11):755-760.
53. Volkow ND, Wang GJ, Tomasi D, et al. Methylphenidate-elicited dopamine increases in ventral striatum are associated with long-term symptom improvement in adults with attention deficit hyperactivity disorder. J Neurosci. 2012;32(3):841-849.
1. Polanczyk G, de Lima MS, Horta BL, et al. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry. 2007;164(6):942-948.
2. Simon V, Czobor P, Bálint S, et al. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry. 2009;194(3):204-211.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Berger I. Diagnosis of attention deficit hyperactivity disorder: much ado about something. Isr Med Assoc J. 2011;13(9):571-574.
5. Schonwald A, Lechner E. Attention deficit/hyperactivity disorder: complexities and controversies. Curr Opin Pediatr. 2006;18(2):189-195.
6. Rousseau C, Measham T, Bathiche-Suidan M. DSM IV, culture and child psychiatry. J Can Acad Child Adolesc Psychiatry. 2008;17(2):69-75.
7. Taylor-Klaus E. Bringing the ADHD debate into sharper focus: part 1. The Huffington Post. http:// www.huffingtonpost.com/elaine-taylorklaus/adhd-debate_b_4571097.html. Updated March 17, 2014. Accessed August 18, 2015.
8. Sweeney CT, Sembower MA, Ertischek MD, et al. Nonmedical use of prescription ADHD stimulants and preexisting patterns of drug abuse. J Addict Dis. 2013;32(1):1-10.
9. Hitt E. Multiple reports of ADHD drug shortages. Medscape. http://www.medscape.com/viewarticle/742686. Published May 13, 2011. Accessed June 4, 2015.
10. Rubia K, Alegria AA, Cubillo AI, et al. Effects of stimulants on brain function in attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. Biol Psychiatry. 2014;76(8):616-628.
11. Cortese S, Kelly C, Chabernaud C, et al. Toward systems neuroscience of ADHD: a meta-analysis of 55 fMRI studies. Am J Psychiatry. 2012;169(10):1038-1055.
12. Garnock-Jones KP, Keating GM. Atomoxetine: a review of its use in attention-deficit hyperactivity disorder in children and adolescents. Paediatr Drugs. 2009;11(3):203-226.
13. Wu J, Xiao H, Sun H, et al. Role of dopamine receptors in ADHD: a systematic meta-analysis. Mol Neurobiol. 2012; 45(3):605-620.
14. Del Campo N, Chamberlain SR, Sahakian BJ, et al. The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2011;69(12):e145-e157.
15. Berridge CW, Devilbiss DM. Psychostimulants as cognitive enhancers: the prefrontal cortex, catecholamines, and attention-deficit/hyperactivity disorder. Biol Psychiatry. 2011;69(12):e101-e111.
16. Lou HC, Henriksen L, Bruhn P. Focal cerebral hypoperfusion in children with dysphasia and/or attention deficit disorder. Arch Neurol. 1984;41(8):825-829.
17. Gustafsson P, Thernlund G, Ryding E, et al. Associations between cerebral blood-flow measured by single photon emission computed tomography (SPECT), electro-encephalogram (EEG), behaviour symptoms, cognition and neurological soft signs in children with attention-deficit hyperactivity disorder (ADHD). Acta Paediatr. 2000;89(7):830-835.
18. Sieg KG, Gaffney GR, Preston DF, et al. SPECT brain imaging abnormalities in attention deficit hyperactivity disorder. Clin Nucl Med. 1995;20(1):55-60.
19. Spalletta G, Pasini A, Pau F, et al. Prefrontal blood flow dysregulation in drug naive ADHD children without structural abnormalities. J Neural Transm. 2001;108(10):1203-1216.
20. Amen DG, Carmichael BD. High-resolution brain SPECT imaging in ADHD. Ann Clin Psychiatry. 1997;9(2):81-86.
21. Kim BN, Lee JS, Cho SC, et al. Methylphenidate increased regional cerebral blood flow in subjects with attention deficit/hyperactivity disorder. Yonsei Med J. 2001;42(1):19-29.
22. Lou HC, Henriksen L, Bruhn P, et al. Striatal dysfunction in attention deficit and hyperkinetic disorder. Arch Neurol. 1989;46(1):48-52.
23. Lee JS, Kim BN, Kang E, et al. Regional cerebral blood flow in children with attention deficit hyperactivity disorder: comparison before and after methylphenidate treatment. Hum Brain Mapp. 2005;24(3):157-164.
24. Schweitzer JB, Lee DO, Hanford RB, et al. A positron emission tomography study of methylphenidate in adults with ADHD: alterations in resting blood flow and predicting treatment response. Neuropsychopharmacology. 2003;28(5):967-973.
25. Zametkin AJ, Nordahl TE, Gross M, et al. Cerebral glucose metabolism in adults with hyperactivity of childhood onset. N Engl J Med. 1990;323(20):1361-1366.
26. Zametkin AJ, Liebenauer LL, Fitzgerald GA, et al. Brain metabolism in teenagers with attention-deficit hyperactivity disorder. Arch Gen Psychiatry. 1993;50(5):333-340.
27. Ernst M, Zametkin AJ, Matochik JA, et al. Effects of intravenous dextroamphetamine on brain metabolism in adults with attention-deficit hyperactivity disorder (ADHD). Preliminary findings. Psychopharmacol Bull. 1994;30(2):219-225.
28. Janssen M. Dopamine transporter (DaT) SPECT imaging. MI Gateway. 2012;6(1):1-3. http://interactive.snm.org/ docs/MI_Gateway_Newsletter_2012-1%20Dopamine%20 Transporter%20SPECT%20Imaging.pdf. Accessed August 18, 2015.
29. Volkow ND, Wang GJ, Fowler JS, et al. Dopamine transporter occupancies in the human brain induced by therapeutic doses of oral methylphenidate. Am J Psychiatry. 1998;155(10):1325-1331.
30. Volkow ND, Wang G, Fowler JS, et al. Therapeutic doses of oral methylphenidate significantly increase extracellular dopamine in the human brain. J Neurosci. 2001;21(2):RC121.
31. Dougherty DD, Bonab AA, Spencer TJ, et al. Dopamine transporter density in patients with attention deficit hyperactivity disorder. Lancet. 1999;354(9196):2132-2133.
32. Li JJ, Lee SS. Interaction of dopamine transporter gene and observed parenting behaviors on attention-deficit/ hyperactivity disorder: a structural equation modeling approach. J Clin Child Adolesc Psychol. 2013;42(2):174-186.
33. Hesse S, Ballaschke O, Barthel H, et al. Dopamine transporter imaging in adult patients with attention-deficit/ hyperactivity disorder. Psychiatry Res. 2009;171(2):120-128.
34. Volkow ND, Wang GJ, Kollins SH, et al. Evaluating dopamine reward pathway in ADHD: clinical implications. JAMA. 2009;302(10):1084-1091.
35. Fusar-Poli P, Rubia K, Rossi G, et al. Striatal dopamine transporter alterations in ADHD: pathophysiology or adaptation to psychostimulants? A meta-analysis. Am J Psychiatry. 2012;169(3):264-272.
36. Wang GJ, Volkow ND, Wigal T, et al. Long-term stimulant treatment affects brain dopamine transporter level in patients with attention deficit hyperactive disorder. PLoS One. 2013;8(5):e63023.
37. Spencer TJ, Madras BK, Fischman AJ, et al. Striatal dopamine transporter binding in adults with ADHD. Am J Psychiatry. 2012;169(6):665; author reply 666.
38. Dresel S, Krause J, Krause KH, et al. Attention deficit hyperactivity disorder: binding of [99mTc]TRODAT-1 to the dopamine transporter before and after methylphenidate treatment. Eur J Nucl Med. 2000;27(10):1518-1524.
39. Krause J, la Fougere C, Krause KH, et al. Influence of striatal dopamine transporter availability on the response to methylphenidate in adult patients with ADHD. Eur Arch Psychiatry Clin Neurosci. 2005;255(6):428-431.
40. la Fougère C, Krause J, Krause KH, et al. Value of 99mTc-TRODAT-1 SPECT to predict clinical response to methylphenidate treatment in adults with attention deficit hyperactivity disorder. Nucl Med Commun. 2006;27(9):733-737.
41. MTA Cooperative Group. National Institute of Mental Health Multimodal Treatment Study of ADHD follow-up: 24-month outcomes of treatment strategies for attention-deficit/hyperactivity disorder. Pediatrics. 2004;113(4):754-761.
42. da Silva N Jr, Szobot CM, Anselmi CE, et al. Attention deficit/hyperactivity disorder: is there a correlation between dopamine transporter density and cerebral blood flow? Clin Nucl Med. 2011;36(8):656-660.
43. Ernst M, Zametkin AJ, Matochik JA, et al. High midbrain [18F]DOPA accumulation in children with attention deficit hyperactivity disorder. Am J Psychiatry. 1999;156(8):1209-1215.
44. Ernst M, Zametkin AJ, Matochik JA, et al. DOPA decarboxylase activity in attention deficit hyperactivity disorder adults. A [fluorine-18]fluorodopa positron emission tomographic study. J Neurosci. 1998;18(15):5901-5907.
45. Xu M, Moratalla R, Gold LH, et al. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell. 1994;79(4):729-742.
46. Goodwin RJ, Mackay CL, Nilsson A, et al. Qualitative and quantitative MALDI imaging of the positron emission tomography ligands raclopride (a D2 dopamine antagonist) and SCH 23390 (a D1 dopamine antagonist) in rat brain tissue sections using a solvent-free dry matrix application method. Anal Chem. 2011;83(24):9694-9701.
47. Negyessy L, Goldman-Rakic PS. Subcellular localization of the dopamine D2 receptor and coexistence with the calcium-binding protein neuronal calcium sensor-1 in the primate prefrontal cortex. J Comp Neurol. 2005;488(4):464-475.
48. Boyson SJ, McGonigle P, Molinoff PB. Quantitative autoradiographic localization of the D1 and D2 subtypes of dopamine receptors in rat brain. J Neurosci. 1986;6(11):3177-3188.
49. Doi M, Yujnovsky I, Hirayama J, et al. Impaired light masking in dopamine D2 receptor-null mice. Nat Neurosci. 2006;9(6):732-734.
50. Lou HC, Rosa P, Pryds O, et al. ADHD: increased dopamine receptor availability linked to attention deficit and low neonatal cerebral blood flow. Dev Med Child Neurol. 2004;46(3):179-183.
51. Volkow ND, Wang GJ, Fowler JS, et al. Effects of methylphenidate on regional brain glucose metabolism in humans: relationship to dopamine D2 receptors. Am J Psychiatry. 1997;154(1):50-55.
52. Ilgin N, Senol S, Gucuyener K, et al. Is increased D2 receptor availability associated with response to stimulant medication in ADHD. Dev Med Child Neurol. 2001;43(11):755-760.
53. Volkow ND, Wang GJ, Tomasi D, et al. Methylphenidate-elicited dopamine increases in ventral striatum are associated with long-term symptom improvement in adults with attention deficit hyperactivity disorder. J Neurosci. 2012;32(3):841-849.
Guidelines ineffective for reprocessing of endoscopes
Reprocessing guidelines currently in place for clinical gastrointestinal endoscopes need to be reevaluated and updated as soon as possible, according to a study published in the American Journal of Infection Control (doi: 10.1016/j.ajic.2015.03.003).
Procedures that follow current guidelines do not fully decontaminate the colonoscopes and gastroscopes after they’re used for procedures, leaving behind microbe levels that are well above the accepted benchmark standards.
“Previous studies show that cleaning endoscope channels is laborious and time consuming, and technicians often skip steps,” wrote the study’s authors – led by Cori L. Ofstead, MSPH, of Ofstead & Associates, St. Paul, Minn. “This is concerning, given that the procedure-related risk of endoscopy may be higher than previously thought [and] without objective verification, clinicians can unknowingly use contaminated endoscopes for procedures.”
The study, conducted at the Mayo Clinic in Rochester, Minn., examined the use of 15 endoscopes involved in 60 separate procedures over the period of Nov. 4-8, 2013, to determine how each endoscope was treated after an operation and whether these cleaning procedures followed guidelines and effectively decontaminated the devices.
“Endoscope testing was performed in a dedicated room adjacent to the procedure room, which allowed for rapid sampling and testing [and] barrier separation [minimized] potential for environmental cross-contamination.” the authors explained.
They maintained aseptic environmental conditions while gathering data, using disinfectant wipes on surfaces, using disposable absorbent pads, and restricting room access.
The researchers wore gloves, impervious gowns, face masks with splash protection, hair nets, and shoe covers; the gloves were changed between each sample collection, and gowns were changed between endoscope collections.
Microbial cultures and rapid indicator tests were conducted for ATP, protein, hemoglobin, and carbohydrate residue. Viable microbe samples were collected from 92% of bedside-cleaned endoscopes, 46% of manually cleaned endoscopes, 64% of “high-level disinfected” endoscopes, and 9% of stored endoscopes. Results from rapid indicator tests showed that some form of contamination was present on 100% of bedside-cleaned endoscopes, 92% of manually cleaned endoscopes, 73% of high-level disinfected endoscopes, and 82% of stored endoscopes which had viable microbes.
“Current guidelines rely on visual inspection to verify cleaning [but] visible contamination was never present, potentially because blood and feces may be difficult to discern from endoscopes’ black exteriors, and microscopic organisms cannot be seen by the naked eye,” the researchers wrote.
Consequently, Ms. Ofstead and colleagues called for a reexamination of current guidelines to move away from the eyeball test, saying that the results found here should pave the way for future studies to devise methods of more effectively cleaning and disinfecting endoscopes in a shorter amount of time.
“In the meantime, methods to ensure effectiveness of reprocessing practices are needed, including the potential use of routine monitoring with rapid indicators and microbiologic cultures,” they suggested.
Ms. Ofstead is president and CEO of Ofstead & Associates, which has received research funding and speaking honoraria related to endoscopic procedures from 3M, Advanced Sterilization Products, Medivators, Invendo Medical, Boston Scientific, and Steris. Three coauthors are employed by Ofstead & Associates, but have no other disclosures to report. The remaining coauthors had no disclosures to report. Funding was provided by 3M, Mayo Clinic, and Ofstead & Associates.
Reprocessing guidelines currently in place for clinical gastrointestinal endoscopes need to be reevaluated and updated as soon as possible, according to a study published in the American Journal of Infection Control (doi: 10.1016/j.ajic.2015.03.003).
Procedures that follow current guidelines do not fully decontaminate the colonoscopes and gastroscopes after they’re used for procedures, leaving behind microbe levels that are well above the accepted benchmark standards.
“Previous studies show that cleaning endoscope channels is laborious and time consuming, and technicians often skip steps,” wrote the study’s authors – led by Cori L. Ofstead, MSPH, of Ofstead & Associates, St. Paul, Minn. “This is concerning, given that the procedure-related risk of endoscopy may be higher than previously thought [and] without objective verification, clinicians can unknowingly use contaminated endoscopes for procedures.”
The study, conducted at the Mayo Clinic in Rochester, Minn., examined the use of 15 endoscopes involved in 60 separate procedures over the period of Nov. 4-8, 2013, to determine how each endoscope was treated after an operation and whether these cleaning procedures followed guidelines and effectively decontaminated the devices.
“Endoscope testing was performed in a dedicated room adjacent to the procedure room, which allowed for rapid sampling and testing [and] barrier separation [minimized] potential for environmental cross-contamination.” the authors explained.
They maintained aseptic environmental conditions while gathering data, using disinfectant wipes on surfaces, using disposable absorbent pads, and restricting room access.
The researchers wore gloves, impervious gowns, face masks with splash protection, hair nets, and shoe covers; the gloves were changed between each sample collection, and gowns were changed between endoscope collections.
Microbial cultures and rapid indicator tests were conducted for ATP, protein, hemoglobin, and carbohydrate residue. Viable microbe samples were collected from 92% of bedside-cleaned endoscopes, 46% of manually cleaned endoscopes, 64% of “high-level disinfected” endoscopes, and 9% of stored endoscopes. Results from rapid indicator tests showed that some form of contamination was present on 100% of bedside-cleaned endoscopes, 92% of manually cleaned endoscopes, 73% of high-level disinfected endoscopes, and 82% of stored endoscopes which had viable microbes.
“Current guidelines rely on visual inspection to verify cleaning [but] visible contamination was never present, potentially because blood and feces may be difficult to discern from endoscopes’ black exteriors, and microscopic organisms cannot be seen by the naked eye,” the researchers wrote.
Consequently, Ms. Ofstead and colleagues called for a reexamination of current guidelines to move away from the eyeball test, saying that the results found here should pave the way for future studies to devise methods of more effectively cleaning and disinfecting endoscopes in a shorter amount of time.
“In the meantime, methods to ensure effectiveness of reprocessing practices are needed, including the potential use of routine monitoring with rapid indicators and microbiologic cultures,” they suggested.
Ms. Ofstead is president and CEO of Ofstead & Associates, which has received research funding and speaking honoraria related to endoscopic procedures from 3M, Advanced Sterilization Products, Medivators, Invendo Medical, Boston Scientific, and Steris. Three coauthors are employed by Ofstead & Associates, but have no other disclosures to report. The remaining coauthors had no disclosures to report. Funding was provided by 3M, Mayo Clinic, and Ofstead & Associates.
Reprocessing guidelines currently in place for clinical gastrointestinal endoscopes need to be reevaluated and updated as soon as possible, according to a study published in the American Journal of Infection Control (doi: 10.1016/j.ajic.2015.03.003).
Procedures that follow current guidelines do not fully decontaminate the colonoscopes and gastroscopes after they’re used for procedures, leaving behind microbe levels that are well above the accepted benchmark standards.
“Previous studies show that cleaning endoscope channels is laborious and time consuming, and technicians often skip steps,” wrote the study’s authors – led by Cori L. Ofstead, MSPH, of Ofstead & Associates, St. Paul, Minn. “This is concerning, given that the procedure-related risk of endoscopy may be higher than previously thought [and] without objective verification, clinicians can unknowingly use contaminated endoscopes for procedures.”
The study, conducted at the Mayo Clinic in Rochester, Minn., examined the use of 15 endoscopes involved in 60 separate procedures over the period of Nov. 4-8, 2013, to determine how each endoscope was treated after an operation and whether these cleaning procedures followed guidelines and effectively decontaminated the devices.
“Endoscope testing was performed in a dedicated room adjacent to the procedure room, which allowed for rapid sampling and testing [and] barrier separation [minimized] potential for environmental cross-contamination.” the authors explained.
They maintained aseptic environmental conditions while gathering data, using disinfectant wipes on surfaces, using disposable absorbent pads, and restricting room access.
The researchers wore gloves, impervious gowns, face masks with splash protection, hair nets, and shoe covers; the gloves were changed between each sample collection, and gowns were changed between endoscope collections.
Microbial cultures and rapid indicator tests were conducted for ATP, protein, hemoglobin, and carbohydrate residue. Viable microbe samples were collected from 92% of bedside-cleaned endoscopes, 46% of manually cleaned endoscopes, 64% of “high-level disinfected” endoscopes, and 9% of stored endoscopes. Results from rapid indicator tests showed that some form of contamination was present on 100% of bedside-cleaned endoscopes, 92% of manually cleaned endoscopes, 73% of high-level disinfected endoscopes, and 82% of stored endoscopes which had viable microbes.
“Current guidelines rely on visual inspection to verify cleaning [but] visible contamination was never present, potentially because blood and feces may be difficult to discern from endoscopes’ black exteriors, and microscopic organisms cannot be seen by the naked eye,” the researchers wrote.
Consequently, Ms. Ofstead and colleagues called for a reexamination of current guidelines to move away from the eyeball test, saying that the results found here should pave the way for future studies to devise methods of more effectively cleaning and disinfecting endoscopes in a shorter amount of time.
“In the meantime, methods to ensure effectiveness of reprocessing practices are needed, including the potential use of routine monitoring with rapid indicators and microbiologic cultures,” they suggested.
Ms. Ofstead is president and CEO of Ofstead & Associates, which has received research funding and speaking honoraria related to endoscopic procedures from 3M, Advanced Sterilization Products, Medivators, Invendo Medical, Boston Scientific, and Steris. Three coauthors are employed by Ofstead & Associates, but have no other disclosures to report. The remaining coauthors had no disclosures to report. Funding was provided by 3M, Mayo Clinic, and Ofstead & Associates.
FDA approves ReShape intragastric balloon device
The first intragastric balloon–based device designed to help obese people lose weight has been approved by the Food and Drug Administration, providing a treatment option that is less invasive than bariatric surgery and gastric banding.
The FDA approved the ReShape Integrated Dual Balloon System on July 28, for “weight reduction when used in conjunction with diet and exercise, in obese patients with a body mass index (BMI) of 30-40 kg/m2 and one or more obesity-related comorbid conditions,” in adults who have not been able to lose weight with diet and exercise alone, according to the agency’s approval letter. Laparoscopic gastric banding is indicated for patients with a BMI of at least 40 kg/m2 (or at least 30 kg/m2 in people with one or more obesity-related comorbidities) and bariatric surgery is usually recommended for patients with a BMI of at least 40 kg/m2 (or at least 35 kg/m2 in people with at least one obesity-related comorbidity).

The ReShape device is made up of two attached balloons that are placed in the stomach through a minimally invasive endoscopic procedure, where they are filled with about 2 cups of saline and methylene blue dye, under mild sedation; the balloons are sealed with mineral oil and left in place for up to 6 months. If a balloon ruptures, the dye appears in the urine. When it is time to remove the balloons, they are deflated then removed using another endoscopic procedure.
The device was evaluated in a pivotal study at eight U.S. sites of over 300 mostly female obese patients whose mean age was about 44 years; their mean weight was about 209-213 pounds, and their mean BMI was about 35 kg/m2; 187 received the device and 139 had the endoscopy only. All participants were on a medically managed diet and exercise program. At 6 months, those in the device group had lost a mean of about 24% of their weight, vs. a mean of about 11% among controls, a statistically significant difference (P = .0041). Those who had lost weight at 6 months “maintained 60% of this weight loss through 48 weeks of follow-up,” according to the FDA.
After placement of the device, common adverse events were vomiting, nausea, and abdominal pain, but most symptoms resolved within 30 days, according to the FDA. The development of gastric ulcerations is described as the “most worrisome” device-related risk, but “there were no unanticipated adverse device effects, no deaths, no intestinal obstructions, and no gastric perforations” in the study.
Among the 265 patients who received the device (those initially enrolled in the pivotal trial plus 78 who were in the control group and opted to receive the device after the first 6 months), 20 (7.5%) experienced severe adverse events; vomiting was the most common, in 4.5%. Serious events included gastric ulcers in two patients (0.8%) at 19 and at 97 days after the device was placed; in both cases, the device was removed. Almost 15% of those who received the device had to have it removed because of an adverse event. The rate of gastric ulcers after a minor change was made to the device was 10%; and the rate of balloon deflations without migration was 6%.
The FDA summary of the approval refers to the “marginal benefit of weight loss” among those in the treatment group, compared with controls, but adds that the decision to approve the device “is based in part on the limited options available to patients with mild to moderate obesity who have failed other means for conservative weight loss.”
While the effectiveness of the device is better than what would be expected with diet and exercise or pharmacologic therapy,” it is “substantially less than what would be expected with gastric banding or other surgical interventions.” The list of contraindications includes previous gastrointestinal surgery “with sequelae,” such as an obstruction or adhesions; previous bariatric surgery; any GI inflammatory disease, severe coagulopathy; and women who are pregnant or breastfeeding.
“The company plans to make the ReShape procedure available to patients first in select markets, as physicians and allied health professionals are trained in the procedure and support program to optimize patient outcome,” according to the company’s statement announcing approval.
The ReShape device has been available in Europe since 2007.
Information posted by the FDA, including labeling for professionals, is available at www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/pma/pma.cfm?num=P140012.
The AGA Center for GI Innovation and Technology is committed to supporting the development of new devices and their introduction to the market in a safe and efficient manner. The center has been working with the FDA for the past several years to help the agency determine how to assess obesity devices.
“It is gratifying to see that the FDA continues to facilitate technologies to address significant public health concerns,” said chair of the center, Dr. Michael L. Kochman, FACP, AGAF. “The AGA Center for GI Innovation and Technology’s obesity-focused meeting, held in conjunction with the FDA, and the annual AGA Tech Summits have helped inform the discussion surrounding the new and novel devices in the obesity and metabolism space.”
The first intragastric balloon–based device designed to help obese people lose weight has been approved by the Food and Drug Administration, providing a treatment option that is less invasive than bariatric surgery and gastric banding.
The FDA approved the ReShape Integrated Dual Balloon System on July 28, for “weight reduction when used in conjunction with diet and exercise, in obese patients with a body mass index (BMI) of 30-40 kg/m2 and one or more obesity-related comorbid conditions,” in adults who have not been able to lose weight with diet and exercise alone, according to the agency’s approval letter. Laparoscopic gastric banding is indicated for patients with a BMI of at least 40 kg/m2 (or at least 30 kg/m2 in people with one or more obesity-related comorbidities) and bariatric surgery is usually recommended for patients with a BMI of at least 40 kg/m2 (or at least 35 kg/m2 in people with at least one obesity-related comorbidity).
The ReShape device is made up of two attached balloons that are placed in the stomach through a minimally invasive endoscopic procedure, where they are filled with about 2 cups of saline and methylene blue dye, under mild sedation; the balloons are sealed with mineral oil and left in place for up to 6 months. If a balloon ruptures, the dye appears in the urine. When it is time to remove the balloons, they are deflated then removed using another endoscopic procedure.
The device was evaluated in a pivotal study at eight U.S. sites of over 300 mostly female obese patients whose mean age was about 44 years; their mean weight was about 209-213 pounds, and their mean BMI was about 35 kg/m2; 187 received the device and 139 had the endoscopy only. All participants were on a medically managed diet and exercise program. At 6 months, those in the device group had lost a mean of about 24% of their weight, vs. a mean of about 11% among controls, a statistically significant difference (P = .0041). Those who had lost weight at 6 months “maintained 60% of this weight loss through 48 weeks of follow-up,” according to the FDA.
After placement of the device, common adverse events were vomiting, nausea, and abdominal pain, but most symptoms resolved within 30 days, according to the FDA. The development of gastric ulcerations is described as the “most worrisome” device-related risk, but “there were no unanticipated adverse device effects, no deaths, no intestinal obstructions, and no gastric perforations” in the study.
Among the 265 patients who received the device (those initially enrolled in the pivotal trial plus 78 who were in the control group and opted to receive the device after the first 6 months), 20 (7.5%) experienced severe adverse events; vomiting was the most common, in 4.5%. Serious events included gastric ulcers in two patients (0.8%) at 19 and at 97 days after the device was placed; in both cases, the device was removed. Almost 15% of those who received the device had to have it removed because of an adverse event. The rate of gastric ulcers after a minor change was made to the device was 10%; and the rate of balloon deflations without migration was 6%.
The FDA summary of the approval refers to the “marginal benefit of weight loss” among those in the treatment group, compared with controls, but adds that the decision to approve the device “is based in part on the limited options available to patients with mild to moderate obesity who have failed other means for conservative weight loss.”
While the effectiveness of the device is better than what would be expected with diet and exercise or pharmacologic therapy,” it is “substantially less than what would be expected with gastric banding or other surgical interventions.” The list of contraindications includes previous gastrointestinal surgery “with sequelae,” such as an obstruction or adhesions; previous bariatric surgery; any GI inflammatory disease, severe coagulopathy; and women who are pregnant or breastfeeding.
“The company plans to make the ReShape procedure available to patients first in select markets, as physicians and allied health professionals are trained in the procedure and support program to optimize patient outcome,” according to the company’s statement announcing approval.
The ReShape device has been available in Europe since 2007.
Information posted by the FDA, including labeling for professionals, is available at www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/pma/pma.cfm?num=P140012.
The AGA Center for GI Innovation and Technology is committed to supporting the development of new devices and their introduction to the market in a safe and efficient manner. The center has been working with the FDA for the past several years to help the agency determine how to assess obesity devices.
“It is gratifying to see that the FDA continues to facilitate technologies to address significant public health concerns,” said chair of the center, Dr. Michael L. Kochman, FACP, AGAF. “The AGA Center for GI Innovation and Technology’s obesity-focused meeting, held in conjunction with the FDA, and the annual AGA Tech Summits have helped inform the discussion surrounding the new and novel devices in the obesity and metabolism space.”
The first intragastric balloon–based device designed to help obese people lose weight has been approved by the Food and Drug Administration, providing a treatment option that is less invasive than bariatric surgery and gastric banding.
The FDA approved the ReShape Integrated Dual Balloon System on July 28, for “weight reduction when used in conjunction with diet and exercise, in obese patients with a body mass index (BMI) of 30-40 kg/m2 and one or more obesity-related comorbid conditions,” in adults who have not been able to lose weight with diet and exercise alone, according to the agency’s approval letter. Laparoscopic gastric banding is indicated for patients with a BMI of at least 40 kg/m2 (or at least 30 kg/m2 in people with one or more obesity-related comorbidities) and bariatric surgery is usually recommended for patients with a BMI of at least 40 kg/m2 (or at least 35 kg/m2 in people with at least one obesity-related comorbidity).
The ReShape device is made up of two attached balloons that are placed in the stomach through a minimally invasive endoscopic procedure, where they are filled with about 2 cups of saline and methylene blue dye, under mild sedation; the balloons are sealed with mineral oil and left in place for up to 6 months. If a balloon ruptures, the dye appears in the urine. When it is time to remove the balloons, they are deflated then removed using another endoscopic procedure.
The device was evaluated in a pivotal study at eight U.S. sites of over 300 mostly female obese patients whose mean age was about 44 years; their mean weight was about 209-213 pounds, and their mean BMI was about 35 kg/m2; 187 received the device and 139 had the endoscopy only. All participants were on a medically managed diet and exercise program. At 6 months, those in the device group had lost a mean of about 24% of their weight, vs. a mean of about 11% among controls, a statistically significant difference (P = .0041). Those who had lost weight at 6 months “maintained 60% of this weight loss through 48 weeks of follow-up,” according to the FDA.
After placement of the device, common adverse events were vomiting, nausea, and abdominal pain, but most symptoms resolved within 30 days, according to the FDA. The development of gastric ulcerations is described as the “most worrisome” device-related risk, but “there were no unanticipated adverse device effects, no deaths, no intestinal obstructions, and no gastric perforations” in the study.
Among the 265 patients who received the device (those initially enrolled in the pivotal trial plus 78 who were in the control group and opted to receive the device after the first 6 months), 20 (7.5%) experienced severe adverse events; vomiting was the most common, in 4.5%. Serious events included gastric ulcers in two patients (0.8%) at 19 and at 97 days after the device was placed; in both cases, the device was removed. Almost 15% of those who received the device had to have it removed because of an adverse event. The rate of gastric ulcers after a minor change was made to the device was 10%; and the rate of balloon deflations without migration was 6%.
The FDA summary of the approval refers to the “marginal benefit of weight loss” among those in the treatment group, compared with controls, but adds that the decision to approve the device “is based in part on the limited options available to patients with mild to moderate obesity who have failed other means for conservative weight loss.”
While the effectiveness of the device is better than what would be expected with diet and exercise or pharmacologic therapy,” it is “substantially less than what would be expected with gastric banding or other surgical interventions.” The list of contraindications includes previous gastrointestinal surgery “with sequelae,” such as an obstruction or adhesions; previous bariatric surgery; any GI inflammatory disease, severe coagulopathy; and women who are pregnant or breastfeeding.
“The company plans to make the ReShape procedure available to patients first in select markets, as physicians and allied health professionals are trained in the procedure and support program to optimize patient outcome,” according to the company’s statement announcing approval.
The ReShape device has been available in Europe since 2007.
Information posted by the FDA, including labeling for professionals, is available at www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/pma/pma.cfm?num=P140012.
The AGA Center for GI Innovation and Technology is committed to supporting the development of new devices and their introduction to the market in a safe and efficient manner. The center has been working with the FDA for the past several years to help the agency determine how to assess obesity devices.
“It is gratifying to see that the FDA continues to facilitate technologies to address significant public health concerns,” said chair of the center, Dr. Michael L. Kochman, FACP, AGAF. “The AGA Center for GI Innovation and Technology’s obesity-focused meeting, held in conjunction with the FDA, and the annual AGA Tech Summits have helped inform the discussion surrounding the new and novel devices in the obesity and metabolism space.”

3-Month paliperidone palmitate for preventing relapse in schizophrenia
A 3-month paliperidone palmitate (PPM-3) extended-release injectable suspension was approved by the FDA in May 2015 for preventing relapse among patients with schizophrenia, under the brand name Invega Trinza (Table 1). Administered 4 times a year, PPM-3 provides the longest interval of any approved long-acting injectable antipsychotic (LAIA). PPM-3 can be administered to patients with schizophrenia who have been taking 1-month paliperidone palmitate (PPM-1) extended-release injectable suspension (brand name, Invega Sustenna), once a month, for at least 4 months.

How it works
PPM-3 is a LAIA injection. Because of its low solubility in water, paliperidone palmitate dissolves slowly once injected before being hydrolyzed as paliperidone and absorbed into the bloodstream. From time of release on Day 1, PPM-3 remains active for as long 18 months.
PPM-3 reaches a maximum plasma concentration between Day 30 and Day 33. In clinical trials, PPM-3 had a median half-life of 84 to 95 days when injected into the deltoid muscle and a median half-life of 118 to 139 days when injected into the gluteal muscle.
Paliperidone is not extensively metabolized in the liver. Although results of a study suggest that cytochrome P450 (CYP) 2D6 and CYP3A4 might play a role in metabolizing paliperidone, there is no evidence that it has a significant role.
Dosing and administration
PPM-3 is administered intramuscularly by a licensed health care professional, once every 3 months. The recommended dosage is based on the patient’s previous dosage of PPM-1 (Table 2).
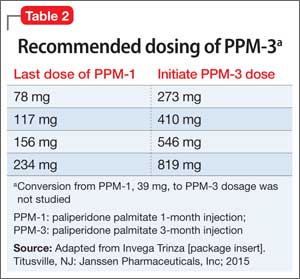
See the prescribing information for administration instructions.
Efficacy
The efficacy of PPM-3 was assessed in a long-term double-blind, placebo-controlled, randomized-withdrawal trial in adult patients with acute symptoms (previously treated with an oral antipsychotic) or adequately treated with a LAIA, either PPM-1 or another agent; patients receiving PPM-1, 39 mg, injections were ineligible. All patients entering the study received PPM-1 in place of the next scheduled injection.
The study comprised 3 treatment periods:
• 17-Week flexible-dose open-label period with PPM-1 (ie, first part of a 29-week open-label stabilization phase): Patients (N = 506) received PPM-1 with a flexible dose based on symptom response, tolerability, and medication history. Patients had to achieve a Positive and Negative Syndrome Scale (PANSS) total score of <70 at Week 17 to enter the second phase.
• 12-Week open-label with PPM-3 (ie, second part of the 29-week open-label stabilization phase): Patients (N = 379) received a single injection of PPM-3 that was 3.5 times the last dose of PPM-1. Patients had to achieve a PANSS total score of <70 and ≤4 for 7 specific PANSS items.
• A variable length double-blind treatment period: Patients (N = 305) were randomized 1:1 to continue treatment with PPM-3 (273 mg, 410 mg, 546 mg, or 819 mg) or placebo (administered once every 12 weeks) until relapse, early withdrawal, or end of the study. The primary efficacy measure was time to first relapse, defined as psychiatric hospitalization, ≥25% increase or a 10-point increase in total PANSS score on 2 consecutive assessments, deliberate self-injury, violent behavior, suicidal or homicidal ideation, or a score of ≥5 (if the maximum baseline score was ≤3) or ≥6 (if the maximum baseline score was 4) on 2 consecutive assessments of the specific PANSS items.
Among the patients in the third treatment period, 23% of those who received placebo and 7.4% of those who received PPM-3 experienced a relapse event. The time to relapse was significantly longer for patients who received PPM-3 than for those who received placebo.
See Table 3 for adverse reactions reported in patients who received PPM-3 and those taking placebo in the study.
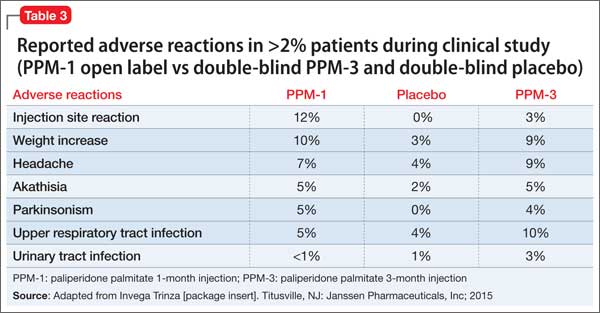
Contraindications
Allergic reactions. Patients who have a hypersensitivity to paliperidone, risperidone, or their components should not receive PPM-3. Anaphylactic reactions have been reported in patients who previously tolerated risperidone or oral paliperidone, which could be significant because the drug is slowly released over 3 months. Other adverse reactions, including angioedema, ileus, swollen tongue, thrombotic thrombocytopenic purpura, urinary incontinence, and urinary retention, were reported post-approval of paliperidone; however, these adverse effects were reported voluntarily from an unknown population size and, therefore, it is unknown whether there is a causal relationship to the drug or its frequency.
Drug-drug interactions. Although paliperidone is not expected to cause drug– drug interactions with medications that are metabolized by CYP isoenzymes, it is recommended to avoid using a strong inducer of CYP3A4 and/or P-glycoprotein.
Overdose. When assessing treatment options and recovery, consider the half-life of PPM-3 and its long-lasting effects.
Because PPM-3 is administered by a licensed health care provider, the potential for overdose is low. However, if overdose occurs, general treatment and management measures should be employed as with overdose of any drug and the possibility of multiple drug overdose should be considered. There is no specific antidote to paliperidone. Contact a certified poison control center for guidance on managing paliperidone and PPM-3 overdose. Generally, management consists of supportive care.
Black-box warning in dementia. As with all atypical antipsychotics, the black-box warning for PPM-3 states that it is not approved for, and should not be used in, patients with dementia-related psychosis. An analysis of placebo-controlled studies revealed that patients taking an antipsychotic had (1) 1.6 to 1.7 times the risk of death than those who received placebo and (2) a higher incidence of cerebrovascular adverse reactions.
Adverse reactions
The safety profile of PPM-3 is similar to that of PPM-1. The most common adverse reactions are:
• reaction at the injection site
• weight gain
• headache
• upper respiratory tract infection
• akathisia
• parkinsonism.
See the full prescribing information for a complete list of adverse effects.
Related Resources
• Sedky K, Nazir R, Lindenmayer JP, et al. Paliperidone palmitate: once monthly treatment option for schizophrenia. Current Psychiatry. 2010;9(3):48-49.
• Berwaerts J, Liu Y, Gopal S, et al. Efficacy and safety of the 3-month formulation of paliperidone palmitate vs placebo for relapse prevention of schizophrenia: a randomized clinical trial [published online March 29, 2015]. JAMA Psychiatry. doi: 10.1001/jamapsychiatry.2015.0241.
Drug Brand Names
Paliperidone palmitate • Invega Sustenna, Invega Trinza
Risperidone • Risperdal
Source: Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2015.
A 3-month paliperidone palmitate (PPM-3) extended-release injectable suspension was approved by the FDA in May 2015 for preventing relapse among patients with schizophrenia, under the brand name Invega Trinza (Table 1). Administered 4 times a year, PPM-3 provides the longest interval of any approved long-acting injectable antipsychotic (LAIA). PPM-3 can be administered to patients with schizophrenia who have been taking 1-month paliperidone palmitate (PPM-1) extended-release injectable suspension (brand name, Invega Sustenna), once a month, for at least 4 months.
How it works
PPM-3 is a LAIA injection. Because of its low solubility in water, paliperidone palmitate dissolves slowly once injected before being hydrolyzed as paliperidone and absorbed into the bloodstream. From time of release on Day 1, PPM-3 remains active for as long 18 months.
PPM-3 reaches a maximum plasma concentration between Day 30 and Day 33. In clinical trials, PPM-3 had a median half-life of 84 to 95 days when injected into the deltoid muscle and a median half-life of 118 to 139 days when injected into the gluteal muscle.
Paliperidone is not extensively metabolized in the liver. Although results of a study suggest that cytochrome P450 (CYP) 2D6 and CYP3A4 might play a role in metabolizing paliperidone, there is no evidence that it has a significant role.
Dosing and administration
PPM-3 is administered intramuscularly by a licensed health care professional, once every 3 months. The recommended dosage is based on the patient’s previous dosage of PPM-1 (Table 2).
See the prescribing information for administration instructions.
Efficacy
The efficacy of PPM-3 was assessed in a long-term double-blind, placebo-controlled, randomized-withdrawal trial in adult patients with acute symptoms (previously treated with an oral antipsychotic) or adequately treated with a LAIA, either PPM-1 or another agent; patients receiving PPM-1, 39 mg, injections were ineligible. All patients entering the study received PPM-1 in place of the next scheduled injection.
The study comprised 3 treatment periods:
• 17-Week flexible-dose open-label period with PPM-1 (ie, first part of a 29-week open-label stabilization phase): Patients (N = 506) received PPM-1 with a flexible dose based on symptom response, tolerability, and medication history. Patients had to achieve a Positive and Negative Syndrome Scale (PANSS) total score of <70 at Week 17 to enter the second phase.
• 12-Week open-label with PPM-3 (ie, second part of the 29-week open-label stabilization phase): Patients (N = 379) received a single injection of PPM-3 that was 3.5 times the last dose of PPM-1. Patients had to achieve a PANSS total score of <70 and ≤4 for 7 specific PANSS items.
• A variable length double-blind treatment period: Patients (N = 305) were randomized 1:1 to continue treatment with PPM-3 (273 mg, 410 mg, 546 mg, or 819 mg) or placebo (administered once every 12 weeks) until relapse, early withdrawal, or end of the study. The primary efficacy measure was time to first relapse, defined as psychiatric hospitalization, ≥25% increase or a 10-point increase in total PANSS score on 2 consecutive assessments, deliberate self-injury, violent behavior, suicidal or homicidal ideation, or a score of ≥5 (if the maximum baseline score was ≤3) or ≥6 (if the maximum baseline score was 4) on 2 consecutive assessments of the specific PANSS items.
Among the patients in the third treatment period, 23% of those who received placebo and 7.4% of those who received PPM-3 experienced a relapse event. The time to relapse was significantly longer for patients who received PPM-3 than for those who received placebo.
See Table 3 for adverse reactions reported in patients who received PPM-3 and those taking placebo in the study.
Contraindications
Allergic reactions. Patients who have a hypersensitivity to paliperidone, risperidone, or their components should not receive PPM-3. Anaphylactic reactions have been reported in patients who previously tolerated risperidone or oral paliperidone, which could be significant because the drug is slowly released over 3 months. Other adverse reactions, including angioedema, ileus, swollen tongue, thrombotic thrombocytopenic purpura, urinary incontinence, and urinary retention, were reported post-approval of paliperidone; however, these adverse effects were reported voluntarily from an unknown population size and, therefore, it is unknown whether there is a causal relationship to the drug or its frequency.
Drug-drug interactions. Although paliperidone is not expected to cause drug– drug interactions with medications that are metabolized by CYP isoenzymes, it is recommended to avoid using a strong inducer of CYP3A4 and/or P-glycoprotein.
Overdose. When assessing treatment options and recovery, consider the half-life of PPM-3 and its long-lasting effects.
Because PPM-3 is administered by a licensed health care provider, the potential for overdose is low. However, if overdose occurs, general treatment and management measures should be employed as with overdose of any drug and the possibility of multiple drug overdose should be considered. There is no specific antidote to paliperidone. Contact a certified poison control center for guidance on managing paliperidone and PPM-3 overdose. Generally, management consists of supportive care.
Black-box warning in dementia. As with all atypical antipsychotics, the black-box warning for PPM-3 states that it is not approved for, and should not be used in, patients with dementia-related psychosis. An analysis of placebo-controlled studies revealed that patients taking an antipsychotic had (1) 1.6 to 1.7 times the risk of death than those who received placebo and (2) a higher incidence of cerebrovascular adverse reactions.
Adverse reactions
The safety profile of PPM-3 is similar to that of PPM-1. The most common adverse reactions are:
• reaction at the injection site
• weight gain
• headache
• upper respiratory tract infection
• akathisia
• parkinsonism.
See the full prescribing information for a complete list of adverse effects.
Related Resources
• Sedky K, Nazir R, Lindenmayer JP, et al. Paliperidone palmitate: once monthly treatment option for schizophrenia. Current Psychiatry. 2010;9(3):48-49.
• Berwaerts J, Liu Y, Gopal S, et al. Efficacy and safety of the 3-month formulation of paliperidone palmitate vs placebo for relapse prevention of schizophrenia: a randomized clinical trial [published online March 29, 2015]. JAMA Psychiatry. doi: 10.1001/jamapsychiatry.2015.0241.
Drug Brand Names
Paliperidone palmitate • Invega Sustenna, Invega Trinza
Risperidone • Risperdal
A 3-month paliperidone palmitate (PPM-3) extended-release injectable suspension was approved by the FDA in May 2015 for preventing relapse among patients with schizophrenia, under the brand name Invega Trinza (Table 1). Administered 4 times a year, PPM-3 provides the longest interval of any approved long-acting injectable antipsychotic (LAIA). PPM-3 can be administered to patients with schizophrenia who have been taking 1-month paliperidone palmitate (PPM-1) extended-release injectable suspension (brand name, Invega Sustenna), once a month, for at least 4 months.
How it works
PPM-3 is a LAIA injection. Because of its low solubility in water, paliperidone palmitate dissolves slowly once injected before being hydrolyzed as paliperidone and absorbed into the bloodstream. From time of release on Day 1, PPM-3 remains active for as long 18 months.
PPM-3 reaches a maximum plasma concentration between Day 30 and Day 33. In clinical trials, PPM-3 had a median half-life of 84 to 95 days when injected into the deltoid muscle and a median half-life of 118 to 139 days when injected into the gluteal muscle.
Paliperidone is not extensively metabolized in the liver. Although results of a study suggest that cytochrome P450 (CYP) 2D6 and CYP3A4 might play a role in metabolizing paliperidone, there is no evidence that it has a significant role.
Dosing and administration
PPM-3 is administered intramuscularly by a licensed health care professional, once every 3 months. The recommended dosage is based on the patient’s previous dosage of PPM-1 (Table 2).
See the prescribing information for administration instructions.
Efficacy
The efficacy of PPM-3 was assessed in a long-term double-blind, placebo-controlled, randomized-withdrawal trial in adult patients with acute symptoms (previously treated with an oral antipsychotic) or adequately treated with a LAIA, either PPM-1 or another agent; patients receiving PPM-1, 39 mg, injections were ineligible. All patients entering the study received PPM-1 in place of the next scheduled injection.
The study comprised 3 treatment periods:
• 17-Week flexible-dose open-label period with PPM-1 (ie, first part of a 29-week open-label stabilization phase): Patients (N = 506) received PPM-1 with a flexible dose based on symptom response, tolerability, and medication history. Patients had to achieve a Positive and Negative Syndrome Scale (PANSS) total score of <70 at Week 17 to enter the second phase.
• 12-Week open-label with PPM-3 (ie, second part of the 29-week open-label stabilization phase): Patients (N = 379) received a single injection of PPM-3 that was 3.5 times the last dose of PPM-1. Patients had to achieve a PANSS total score of <70 and ≤4 for 7 specific PANSS items.
• A variable length double-blind treatment period: Patients (N = 305) were randomized 1:1 to continue treatment with PPM-3 (273 mg, 410 mg, 546 mg, or 819 mg) or placebo (administered once every 12 weeks) until relapse, early withdrawal, or end of the study. The primary efficacy measure was time to first relapse, defined as psychiatric hospitalization, ≥25% increase or a 10-point increase in total PANSS score on 2 consecutive assessments, deliberate self-injury, violent behavior, suicidal or homicidal ideation, or a score of ≥5 (if the maximum baseline score was ≤3) or ≥6 (if the maximum baseline score was 4) on 2 consecutive assessments of the specific PANSS items.
Among the patients in the third treatment period, 23% of those who received placebo and 7.4% of those who received PPM-3 experienced a relapse event. The time to relapse was significantly longer for patients who received PPM-3 than for those who received placebo.
See Table 3 for adverse reactions reported in patients who received PPM-3 and those taking placebo in the study.
Contraindications
Allergic reactions. Patients who have a hypersensitivity to paliperidone, risperidone, or their components should not receive PPM-3. Anaphylactic reactions have been reported in patients who previously tolerated risperidone or oral paliperidone, which could be significant because the drug is slowly released over 3 months. Other adverse reactions, including angioedema, ileus, swollen tongue, thrombotic thrombocytopenic purpura, urinary incontinence, and urinary retention, were reported post-approval of paliperidone; however, these adverse effects were reported voluntarily from an unknown population size and, therefore, it is unknown whether there is a causal relationship to the drug or its frequency.
Drug-drug interactions. Although paliperidone is not expected to cause drug– drug interactions with medications that are metabolized by CYP isoenzymes, it is recommended to avoid using a strong inducer of CYP3A4 and/or P-glycoprotein.
Overdose. When assessing treatment options and recovery, consider the half-life of PPM-3 and its long-lasting effects.
Because PPM-3 is administered by a licensed health care provider, the potential for overdose is low. However, if overdose occurs, general treatment and management measures should be employed as with overdose of any drug and the possibility of multiple drug overdose should be considered. There is no specific antidote to paliperidone. Contact a certified poison control center for guidance on managing paliperidone and PPM-3 overdose. Generally, management consists of supportive care.
Black-box warning in dementia. As with all atypical antipsychotics, the black-box warning for PPM-3 states that it is not approved for, and should not be used in, patients with dementia-related psychosis. An analysis of placebo-controlled studies revealed that patients taking an antipsychotic had (1) 1.6 to 1.7 times the risk of death than those who received placebo and (2) a higher incidence of cerebrovascular adverse reactions.
Adverse reactions
The safety profile of PPM-3 is similar to that of PPM-1. The most common adverse reactions are:
• reaction at the injection site
• weight gain
• headache
• upper respiratory tract infection
• akathisia
• parkinsonism.
See the full prescribing information for a complete list of adverse effects.
Related Resources
• Sedky K, Nazir R, Lindenmayer JP, et al. Paliperidone palmitate: once monthly treatment option for schizophrenia. Current Psychiatry. 2010;9(3):48-49.
• Berwaerts J, Liu Y, Gopal S, et al. Efficacy and safety of the 3-month formulation of paliperidone palmitate vs placebo for relapse prevention of schizophrenia: a randomized clinical trial [published online March 29, 2015]. JAMA Psychiatry. doi: 10.1001/jamapsychiatry.2015.0241.
Drug Brand Names
Paliperidone palmitate • Invega Sustenna, Invega Trinza
Risperidone • Risperdal
Source: Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2015.
Source: Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2015.
Head pain and psychiatric illness: Applying the biopsychosocial model to care
More than 45% of people worldwide suffer from headache at some point in their life.1 Head pain can lead to disability and functional decline, yet headache disorders often are underdiagnosed and poorly assessed. For example, 60% of migraine and tension-type headaches go undiagnosed and 50% of persons suffering from migraine have severe functional disability or require bed rest.2-4
Because head pain can be associated with secondary medical and psychiatric conditions, diagnosis can be challenging. This article reviews the medical and psychological aspects of major headaches and assists with clinical assessment. We present clinical interviewing tools and a diagram to enhance focused, efficient assessment and inform treatment plans.
Classification of headache
Headache is a common complaint, yet it is often underdiagnosed and ineffectively treated. The World Health Organization estimates that, globally, 50% of people with headache self-treat their pain.5 The International Headache Society classifies headache as primary or secondary; approximately 90% of complaints are from primary headache.6
Assessment and diagnosis of headache can be complex because of overlapping, subjective symptoms. It is important to have a general understanding of primary and secondary causes of headache so that interrelated symptoms do not obscure the most accurate diagnosis and effective treatment course. Although most headache complaints are benign, ruling out secondary causes helps gauge the likelihood of developing severe sequelae from underlying pathology.
By definition, primary headaches are idiopathic and commonly include migraine, tension-type, cluster, and hemicrania continua headache. Secondary headaches have an underlying pathology, which could improve by targeting the disorder. Common secondary causes of headache include:
• trauma
• vascular abnormalities
• structural abnormalities
• chemical (including medications)
• inflammation or infection
• metabolic conditions
• diseases of the neck and pericranial and intracranial structures
• psychiatric conditions.
Table 1 illustrates common causes of head pain. More definitive criteria for symptoms and diagnosis can be found in the International Classification of Headache Disorders.7

Primary headache
Tension-type is the most common primary headache, accounting for more than one-half of all headaches.7 Patients usually describe a tight pain in a bilateral band-like distribution, which could be caused by sustained neck muscle contraction. Pain usually builds in intensity and can last 30 minutes to several days. There is a well-established association between emotional stress or depression and the development of tension-type headaches.8
Migraine typically causes pulsating pain in a localized area of the head that lasts as long as 72 hours and can be associated with nausea, vomiting, photophobia, phono-phobia, and aura. Patients report varying precipitating factors but commonly cite certain foods, menstruation, and sleep deprivation. Although rare, migraine with aura has been linked to ischemic stroke; most cases have been reported in female smokers and oral contraceptive users age <45.9
Because migraines can be debilitating, some patients—typically those with ≥4 attacks a month—opt for prophylactic medication. Effective prophylactics include amitriptyline, propranolol, divalproex sodium, and topiramate, which should be monitored closely and given a trial for several months before switching to another drug. Commonly used abortive treatments include triptans and anti-emetics such as metoclopramide.
Meperidine and ketorolac are popular second-line agents for migraine. Botulinum toxin A also has been used in severe cases to reduce the number of headache days in chronic migraine patients.6
Cluster headache is rare, but typically exhibits repeated burning and intense unilateral periorbital or retro-orbital pain that lasts 15 minutes to 3 hours over several weeks. Men are predominantly affected. Cluster headaches typically improve with oxygen treatment.
Biopsychosocial model of head pain
The biomedical model has helped iden tify pathophysiological pain mechanisms and pharmacotherapeutic agents for headache. However, during assessment, limiting one’s attention to the linear relationship between pathology, mechanism of action, and pain oversimplifies common questions clinicians face when assessing chronic head pain.
Advancements in the last 3 decades have expanded the conceptualization of head pain to integrate sociocultural, environmental, behavioral, affective, cognitive, and biological variables—otherwise known as the biopsychosocial model.10,11 The biopsychosocial model is a multidimensional theory that helps answer difficult clinical assessment questions and complex patient presentations (Table 2).10-13 Many unusual responses to pain treatment, questionable validity of pain behavior, and disproportionate pain perception and functional decline are explained by non-pathophysiological and non-biomechanical models.

Psychiatric comorbidity and head pain
Psychiatric conditions are highly prevalent among persons with primary headache. Verri et al14 found that 90% of chronic daily headache patients had ≥1 psychiatric condition; depression and anxiety were most common. Of concern, 1 study found that headache is associated with increased frequency of suicidal ideation among patients with chronic pain.15 It is critical for clinicians to screen for psychiatric comorbidities in patients with chronic headache. Conversely, clinicians might want to screen for headache in their patients with psychiatric illness.
Migraine. Mood disorders are common among patients who suffer from migraine. The rate of depression is 2 to 4 times higher in those with migraine compared with healthy controls.16,17 In a large-scale study, patients with migraine had a 1.9-fold higher risk (compared with controls) of having a comorbid depressive episode; a 2-fold higher risk of manic episodes; and a 3-fold higher risk of both mania and depression.18 In a study of 62 inpatients, Fasmer19 reported that 46% of patients with unipolar depression and 44% of patients with bipolar disorder experienced migraine (77% of the bipolar disorder patients with migraine had bipolar II disorder). Patients with migraine are at increased risk of suicide attempts (odds ratio 4.3; 95% CI, 1.2-15.7).20
Tension-type headache. The relationship between psychiatric comorbidity in tension-type headache is well established. In contrast to what is seen with migraines, Puca et al21 found a higher prevalence of anxiety disorders (52.5%) than depressive disorders (36.4%) in patients with tension-type headache. Generalized anxiety disorder was one of the most prevalent anxiety conditions (83.3%), and dysthymia was the most prevalent mood disorder (45.6%). In the same study, 21.7% of patients were found to have a comorbid somatoform disorder.21
Emotional and cognitive factors can co-occur in patients with tension-type headache and a comorbid psychiatric condition. For example, difficulty identifying or recognizing emotions—commonly referred to as alexithymia—has been linked to tension-type headache.22 Additionally, maladaptive cognitive appraisal of stress is more common among patients with tension-type headache when compared with those without headaches.23 Being mindful of and recognizing these co-occurring emotional and cognitive factors will help clinicians construct a more accurate assessment and effective behavioral treatment plan.
Clinical assessment with a useful mnemonic
Clinical assessment of psychiatric illness is essential when evaluating chronic pain patients. Using the acronym AMPS (Anxiety, Mood, Psychosis, and Substance use disorders) (Table 3) is an efficient way for the clinician to ask pertinent questions regarding common psychiatric conditions that could have a direct effect on chronic pain.24 Head pain can be more intense when combined with untreated anxiety, depression, psychosis, or a substance use disorder. Untreated anxiety, for example, can amplify sympathetic response to pain and complicate treatment.

Investigating head pain patients for an underlying mood disorder is essential to providing successful treatment. Consider:
• starting psychotherapy modalities that address both pain and psychiatric illness, such as cognitive-behavioral therapy (CBT)
• reframing unhelpful pain beliefs
• managing activity-rest levels
• biofeedback
• supportive group therapy
• reducing family members’ reinforcement of the patient’s pain behavior or sick role.25
Assessing for somatic symptom disorders
In addition to using the AMPS approach for psychiatric assessment, clinicians should evaluate for somatization, which can present as head pain. Somatic symptom disorders (SSD) are a class of conditions that are impacted by affective, cognitive, and reinforcing factors that might or might not be consciously or intentionally produced. Patients with an SSD have somatic symptoms that are distressing or cause significant disruption of daily life because of excessive thoughts, feelings, or behaviors related to the somatic symptoms, for ≥6 months. The Figure outlines SSD, related conditions, and their respective prominent symptoms to assist in the differential diagnosis.26
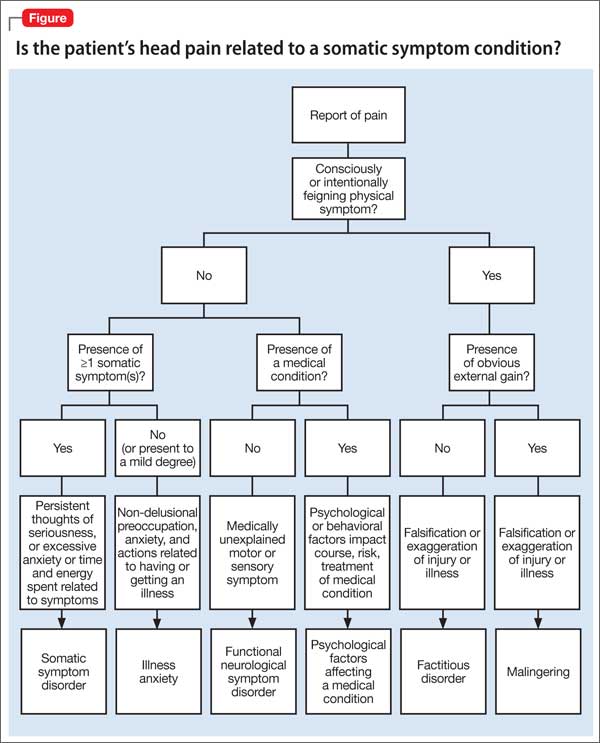
Note that some headache conditions present with severe distress because of their abrupt onset and severity of symptoms (eg, cluster headaches). Therefore, the expectation and likelihood of psychological disturbance should be factored into a diagnosis of SSD and related conditions as seen in the Figure.
Secondary factors of unusual pain behavior or treatment response. The role of thoughts, affect, and behaviors is clinically meaningful in understanding SSD and similar conditions. Specific questions about cultural beliefs and rituals as they relate to exacerbations of head pain are of value. Table 413,27 lists behavioral, cognitive, and affective dimensions of head pain using the biopsychosocial model, and further clarifies common questions that arise with unusual pain response and complex patient presentations, which were outlined in the beginning of the article.
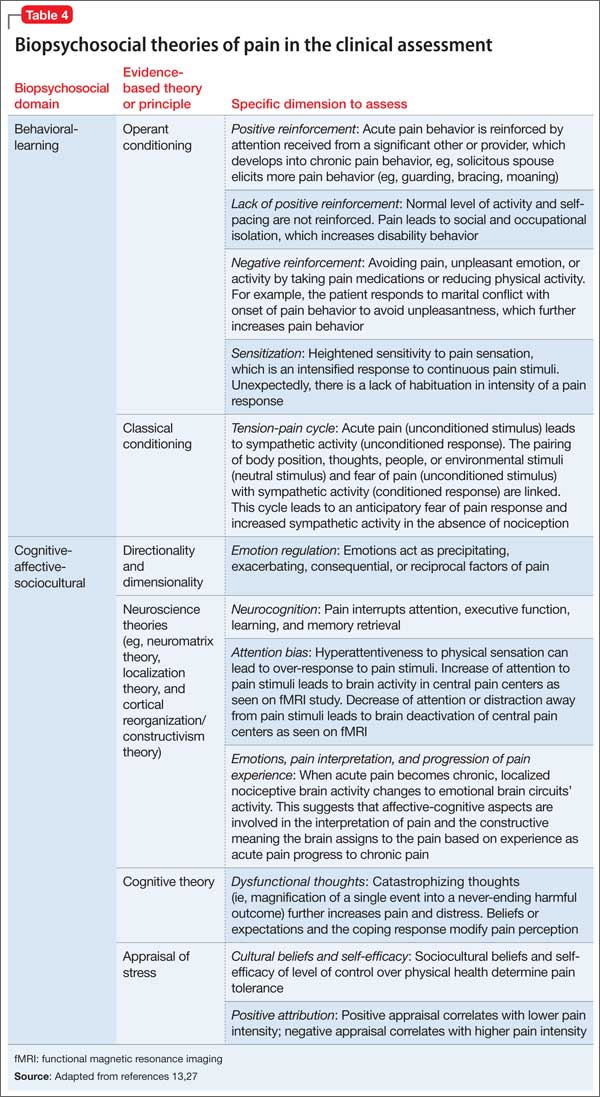
Because depression and anxiety can be comorbid with head pain, it is important to recognize psychological factors that contribute to pain perception. Indifference or denial of emotional stress as a result of severe pain and disability can imply a somatization process, which could suggest emotional disconnection or dissociation from somatic functioning.28 This finding can be a component of alexithymia, in which a person is disconnected from emotions and how emotions impact the body. Therefore, recognizing alexithymia assists in identifying psychological factors when patients deny mood symptoms, particularly in tension-type headache.
Functional assessment to rule out the disproportional impact of pain on daily activities is helpful in understanding the somatization process. Neurocognitive functioning should be assessed, particularly because frontal and subcortical dysregulation has been observed in head pain sufferers.29,30 Patients with cognitive changes as a result of a medical illness (eg, stroke, head concussion, brain tumor, or seizures) are especially at risk for neurocognitive dysfunction.
Neuropsychological assessment can be useful, not only to assess neurocognitive functioning (eg, Repeated Battery for the Assessment of Neuropsychological Status) but to identify objective test profiles associated with altered motivation (eg, Rey 15-Item Test, Minnesota Multiphasic Personality Inventory-2-Restructured Form F Scale, Personality Assessment Inventory [PAI] Negative Impression Management) and somatization processes (eg, PAI Somatization Scale). These instruments help to identify the severity of psychiatric and neurocognitive symptoms by comparing scores to normative (eg, healthy control group), clinical (eg, somatization, traumatic brain injury, mild cognitive impairment), and altered motivation (eg, persons instructed to exaggerate symptoms) databases.
If the clinician pursues neurocognitive assessment, direct referral to a neuropsychologist, referral to neurologist, or administration of a cognitive screening tool such as the Montreal Cognitive Assessment, Saint Louis University Mental Status, or Cognitive Log is recommended. If the cognitive screening is positive, next steps include: referring for full neuropsychological assessment, which includes complete cognitive and motor testing, personality testing, and integration of neuroimaging data (eg, MRI, CT scans, and/or EEG).
Assessing the patients’ self-talk or thought patterns as they describe their head pain will help clinicians understand belief systems that may be distorting the reality of the medical condition. For example, a patient might report that “my pain feels like someone is hitting me with an axe”; this is a catastrophic thought that can distort the clarity and perceptibility of pain. Encouraging patients to monitor and analyze their anxiety and associated negative thoughts is an important strategy for improving mood and decreasing somatization. Recording daily thoughts and CBT can help the patient identify and appropriately address his (her) cognitive distortions and futile thinking.
When implementing a treatment plan for somatization disorder, we propose the mnemonic device CARE MD:
• CBT
• Assess (by ruling out a medical cause for somatic complaints)
• Regular visits
• Empathy
• Med-psych interface (help the patient connect physical complaints and emotional stressors)
• Do no harm.
Clinical recommendations
Chronic head pain can be debilitating; psychodiagnostic assessment should therefore be considered an important part of the diagnosis and treatment plan. After ruling out common and emergent primary or secondary causes of head pain, consider psychiatric comorbidities. Depression and anxiety have a strong bidirectional relationship with chronic headache; therefore, we suggest evaluating patients with the intention of alleviating both psychiatric symptoms and head pain.
It is important to diligently assess for common psychiatric comorbidities; using the AMPS and CARE MD mnemonics, along with screening for somatization disorders, is an easy and effective way to evaluate for relevant psychiatric conditions associated with chronic head pain. Because many patients have unusual and complicated responses to head pain that can be explained by non-pathophysiological and non-biomechanical models, using the biopsychosocial model is essential for effective diagnosis, assessment, and treatment. Abortive and prophylactic medical interventions, as well as behavioral, sociocultural, and cognitive assessment, are vital to a comprehensive treatment approach.
Bottom Line
The psychodiagnostic assessment can help the astute clinician identify comorbid psychiatric conditions, psychological factors, and somatic symptoms to develop a comprehensive biopsychosocial treatment plan for patients with chronic head pain. Rule out primary and secondary causes of pain and screen for somatization disorders. Consider medication and psychotherapeutic treatment options.
Related Resources
• Pompili M, Di Cosimo D, Innamorati M, et al. Psychiatric comorbidity in patients with chronic daily headache and migraine: a selective overview including personality traits and suicide risk. J Headache Pain. 2009;10(4):283-290.
• Sinclair AJ, Sturrock A, Davies B, et al. Headache management: pharmacological approaches [published online July 3, 2015]. Pract Neurol. doi: 10.1136/practneurol-2015-001167.
Drug Brand Names
Amitriptyline • Elavil Meperidine • Demerol
Botulinum toxin A • Botox Metoclopramide • Reglan
Divalproex sodium • Depakote Propranolol • Inderide
Ketorolac • Toradol Topiramate • Topamax
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stovner LJ, Hagen K, Jensen R, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27(3):193-210.
2. Lipton RB, Stewart WF, Diamond S, et al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41(7):646-657.
3. The World Health Report 2001: Mental health: new understanding new hope. Geneva, Switzerland: World Health Organization; 2001.
4. World Health Organization. The global burden of disease: 2004 update. http://www.who.int/healthinfo/global_burden_ disease/GBD_report_2004update_full.pdf. Published 2004. Accessed July 31, 2015.
5. World Health Organization. Headache disorders. http:// www.who.int/mediacentre/factsheets/fs277/en/. Published October 2012. Accessed July 31, 2015.
6. Clinch C. Evaluation & management of headache. In: South- Paul JE, Matheny SC, Lewis EL. eds. Current diagnosis & treatment in family medicine, 4th ed. New York, NY: McGraw-Hill; 2015:293-297.
7. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629-808.
8. Mawet J, Kurth T, Ayata C. Migraine and stroke: in search of shared mechanisms. Cephalalgia. 2015;35(2):165-181.
9. Janke EA, Holroyd KA, Romanek K. Depression increases onset of tension-type headache following laboratory stress. Pain. 2004;111(3):230-238.
10. Engel GL. The need for a new medical model: a challenge for biomedicine. Science. 1977;196(4286):129-136.
11. Engel GL. The clinical application of the biopsychosocial model. Am J Psychiatry. 1980;137(5):535-544.
12. Turk DC, Flor H. Chronic pain: a biobehavioral perspective. In: Gatchel RJ, Turk DC, eds. Psychosocial factors in pain: critical perspectives. New York, NY: Guilford Press; 1999:18-34.
13. Andrasik F, Flor H, Turk DC. An expanded view of psychological aspects in head pain: the biopsychosocial model. Neurol Sci. 2005;26(suppl 2):s87-s91.
14. Verri AP, Proietti Cecchini A, Galli C, et al. Psychiatric comorbidity in chronic daily headache. Cephalalgia. 1998; 18(suppl 21):45-49.
15. Ilgen MA, Zivin K, McCammon RJ, et al. Pain and suicidal thoughts, plans and attempts in the United States. Gen Hosp Psychiatry. 2008;30(6):521-527.
16. Kowacs F, Socal MP, Ziomkowski SC, et al. Symptoms of depression and anxiety, and screening for mental disorders in migrainous patients. Cephalalgia. 2003;23(2):79-89.
17. Hamelsky SW, Lipton RB. Psychiatric comorbidity of migraine. Headache. 2006;46(9):1327-1333.
18. Nguyen TV, Low NC. Comorbidity of migraine and mood episodes in a nationally representative population-based sample. Headache. 2013;53(3):498-506.
19. Fasmer OB. The prevalence of migraine in patients with bipolar and unipolar affective disorders. Cephalalgia. 2001; 21(9):894-899.
20. Breslau N. Migraine, suicidal ideation, and suicide attempts. Neurology. 1992;42(2):392-395.
21. Puca F, Genco S, Prudenzano MP, et al. Psychiatric comorbidity and psychosocial stress in patients with tension-type headache from headache centers in Italy. Cephalalgia. 1999;19(3):159-164.
22. Yücel B, Kora K, Ozyalçín S, et al. Depression, automatic thoughts, alexithymia, and assertiveness in patients with tension-type headache. Headache. 2002;42(3):194-199.
23. Wittrock DA, Myers TC. The comparison of individuals with recurrent tension-type headache and headache-free controls in physiological response, appraisal, and coping with stressors: a review of the literature. Ann Behav Med. 1998;20(2):118-134.
24. Onate J, Xiong G, McCarron R. The primary care psychiatric interview. In: McCarron R, Xiong G, Bourgeois J. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott, Williams and Wilkins; 2009:3-4.
25. Songer D. Psychotherapeutic approaches in the treatment of pain. Psychiatry (Edgmont). 2005;2(5):19-24.
26. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
27. Hashmi JA, Baliki MN, Huang L, et al. Shape shifting pain: chronification of back pain shifts brain representation from nociceptive to emotional circuits. Brain. 2013;136(pt 9):2751-2768.
28. Packard RC. Conversion headache. Headache. 1980;20(5):266-268.
29. Mongini F, Keller R, Deregibus A, et al. Frontal lobe dysfunction in patients with chronic migraine: a clinical-neuropsychological study. Psychiatry Res. 2005;133(1):101-106.
30. Martelli MF, Grayson RL, Zasler ND. Posttraumatic headache: neuropsychological and psychological effects and treatment implications. J Head Trauma Rehabil. 1999;14(1):49-69.
More than 45% of people worldwide suffer from headache at some point in their life.1 Head pain can lead to disability and functional decline, yet headache disorders often are underdiagnosed and poorly assessed. For example, 60% of migraine and tension-type headaches go undiagnosed and 50% of persons suffering from migraine have severe functional disability or require bed rest.2-4
Because head pain can be associated with secondary medical and psychiatric conditions, diagnosis can be challenging. This article reviews the medical and psychological aspects of major headaches and assists with clinical assessment. We present clinical interviewing tools and a diagram to enhance focused, efficient assessment and inform treatment plans.
Classification of headache
Headache is a common complaint, yet it is often underdiagnosed and ineffectively treated. The World Health Organization estimates that, globally, 50% of people with headache self-treat their pain.5 The International Headache Society classifies headache as primary or secondary; approximately 90% of complaints are from primary headache.6
Assessment and diagnosis of headache can be complex because of overlapping, subjective symptoms. It is important to have a general understanding of primary and secondary causes of headache so that interrelated symptoms do not obscure the most accurate diagnosis and effective treatment course. Although most headache complaints are benign, ruling out secondary causes helps gauge the likelihood of developing severe sequelae from underlying pathology.
By definition, primary headaches are idiopathic and commonly include migraine, tension-type, cluster, and hemicrania continua headache. Secondary headaches have an underlying pathology, which could improve by targeting the disorder. Common secondary causes of headache include:
• trauma
• vascular abnormalities
• structural abnormalities
• chemical (including medications)
• inflammation or infection
• metabolic conditions
• diseases of the neck and pericranial and intracranial structures
• psychiatric conditions.
Table 1 illustrates common causes of head pain. More definitive criteria for symptoms and diagnosis can be found in the International Classification of Headache Disorders.7
Primary headache
Tension-type is the most common primary headache, accounting for more than one-half of all headaches.7 Patients usually describe a tight pain in a bilateral band-like distribution, which could be caused by sustained neck muscle contraction. Pain usually builds in intensity and can last 30 minutes to several days. There is a well-established association between emotional stress or depression and the development of tension-type headaches.8
Migraine typically causes pulsating pain in a localized area of the head that lasts as long as 72 hours and can be associated with nausea, vomiting, photophobia, phono-phobia, and aura. Patients report varying precipitating factors but commonly cite certain foods, menstruation, and sleep deprivation. Although rare, migraine with aura has been linked to ischemic stroke; most cases have been reported in female smokers and oral contraceptive users age <45.9
Because migraines can be debilitating, some patients—typically those with ≥4 attacks a month—opt for prophylactic medication. Effective prophylactics include amitriptyline, propranolol, divalproex sodium, and topiramate, which should be monitored closely and given a trial for several months before switching to another drug. Commonly used abortive treatments include triptans and anti-emetics such as metoclopramide.
Meperidine and ketorolac are popular second-line agents for migraine. Botulinum toxin A also has been used in severe cases to reduce the number of headache days in chronic migraine patients.6
Cluster headache is rare, but typically exhibits repeated burning and intense unilateral periorbital or retro-orbital pain that lasts 15 minutes to 3 hours over several weeks. Men are predominantly affected. Cluster headaches typically improve with oxygen treatment.
Biopsychosocial model of head pain
The biomedical model has helped iden tify pathophysiological pain mechanisms and pharmacotherapeutic agents for headache. However, during assessment, limiting one’s attention to the linear relationship between pathology, mechanism of action, and pain oversimplifies common questions clinicians face when assessing chronic head pain.
Advancements in the last 3 decades have expanded the conceptualization of head pain to integrate sociocultural, environmental, behavioral, affective, cognitive, and biological variables—otherwise known as the biopsychosocial model.10,11 The biopsychosocial model is a multidimensional theory that helps answer difficult clinical assessment questions and complex patient presentations (Table 2).10-13 Many unusual responses to pain treatment, questionable validity of pain behavior, and disproportionate pain perception and functional decline are explained by non-pathophysiological and non-biomechanical models.
Psychiatric comorbidity and head pain
Psychiatric conditions are highly prevalent among persons with primary headache. Verri et al14 found that 90% of chronic daily headache patients had ≥1 psychiatric condition; depression and anxiety were most common. Of concern, 1 study found that headache is associated with increased frequency of suicidal ideation among patients with chronic pain.15 It is critical for clinicians to screen for psychiatric comorbidities in patients with chronic headache. Conversely, clinicians might want to screen for headache in their patients with psychiatric illness.
Migraine. Mood disorders are common among patients who suffer from migraine. The rate of depression is 2 to 4 times higher in those with migraine compared with healthy controls.16,17 In a large-scale study, patients with migraine had a 1.9-fold higher risk (compared with controls) of having a comorbid depressive episode; a 2-fold higher risk of manic episodes; and a 3-fold higher risk of both mania and depression.18 In a study of 62 inpatients, Fasmer19 reported that 46% of patients with unipolar depression and 44% of patients with bipolar disorder experienced migraine (77% of the bipolar disorder patients with migraine had bipolar II disorder). Patients with migraine are at increased risk of suicide attempts (odds ratio 4.3; 95% CI, 1.2-15.7).20
Tension-type headache. The relationship between psychiatric comorbidity in tension-type headache is well established. In contrast to what is seen with migraines, Puca et al21 found a higher prevalence of anxiety disorders (52.5%) than depressive disorders (36.4%) in patients with tension-type headache. Generalized anxiety disorder was one of the most prevalent anxiety conditions (83.3%), and dysthymia was the most prevalent mood disorder (45.6%). In the same study, 21.7% of patients were found to have a comorbid somatoform disorder.21
Emotional and cognitive factors can co-occur in patients with tension-type headache and a comorbid psychiatric condition. For example, difficulty identifying or recognizing emotions—commonly referred to as alexithymia—has been linked to tension-type headache.22 Additionally, maladaptive cognitive appraisal of stress is more common among patients with tension-type headache when compared with those without headaches.23 Being mindful of and recognizing these co-occurring emotional and cognitive factors will help clinicians construct a more accurate assessment and effective behavioral treatment plan.
Clinical assessment with a useful mnemonic
Clinical assessment of psychiatric illness is essential when evaluating chronic pain patients. Using the acronym AMPS (Anxiety, Mood, Psychosis, and Substance use disorders) (Table 3) is an efficient way for the clinician to ask pertinent questions regarding common psychiatric conditions that could have a direct effect on chronic pain.24 Head pain can be more intense when combined with untreated anxiety, depression, psychosis, or a substance use disorder. Untreated anxiety, for example, can amplify sympathetic response to pain and complicate treatment.
Investigating head pain patients for an underlying mood disorder is essential to providing successful treatment. Consider:
• starting psychotherapy modalities that address both pain and psychiatric illness, such as cognitive-behavioral therapy (CBT)
• reframing unhelpful pain beliefs
• managing activity-rest levels
• biofeedback
• supportive group therapy
• reducing family members’ reinforcement of the patient’s pain behavior or sick role.25
Assessing for somatic symptom disorders
In addition to using the AMPS approach for psychiatric assessment, clinicians should evaluate for somatization, which can present as head pain. Somatic symptom disorders (SSD) are a class of conditions that are impacted by affective, cognitive, and reinforcing factors that might or might not be consciously or intentionally produced. Patients with an SSD have somatic symptoms that are distressing or cause significant disruption of daily life because of excessive thoughts, feelings, or behaviors related to the somatic symptoms, for ≥6 months. The Figure outlines SSD, related conditions, and their respective prominent symptoms to assist in the differential diagnosis.26
Note that some headache conditions present with severe distress because of their abrupt onset and severity of symptoms (eg, cluster headaches). Therefore, the expectation and likelihood of psychological disturbance should be factored into a diagnosis of SSD and related conditions as seen in the Figure.
Secondary factors of unusual pain behavior or treatment response. The role of thoughts, affect, and behaviors is clinically meaningful in understanding SSD and similar conditions. Specific questions about cultural beliefs and rituals as they relate to exacerbations of head pain are of value. Table 413,27 lists behavioral, cognitive, and affective dimensions of head pain using the biopsychosocial model, and further clarifies common questions that arise with unusual pain response and complex patient presentations, which were outlined in the beginning of the article.
Because depression and anxiety can be comorbid with head pain, it is important to recognize psychological factors that contribute to pain perception. Indifference or denial of emotional stress as a result of severe pain and disability can imply a somatization process, which could suggest emotional disconnection or dissociation from somatic functioning.28 This finding can be a component of alexithymia, in which a person is disconnected from emotions and how emotions impact the body. Therefore, recognizing alexithymia assists in identifying psychological factors when patients deny mood symptoms, particularly in tension-type headache.
Functional assessment to rule out the disproportional impact of pain on daily activities is helpful in understanding the somatization process. Neurocognitive functioning should be assessed, particularly because frontal and subcortical dysregulation has been observed in head pain sufferers.29,30 Patients with cognitive changes as a result of a medical illness (eg, stroke, head concussion, brain tumor, or seizures) are especially at risk for neurocognitive dysfunction.
Neuropsychological assessment can be useful, not only to assess neurocognitive functioning (eg, Repeated Battery for the Assessment of Neuropsychological Status) but to identify objective test profiles associated with altered motivation (eg, Rey 15-Item Test, Minnesota Multiphasic Personality Inventory-2-Restructured Form F Scale, Personality Assessment Inventory [PAI] Negative Impression Management) and somatization processes (eg, PAI Somatization Scale). These instruments help to identify the severity of psychiatric and neurocognitive symptoms by comparing scores to normative (eg, healthy control group), clinical (eg, somatization, traumatic brain injury, mild cognitive impairment), and altered motivation (eg, persons instructed to exaggerate symptoms) databases.
If the clinician pursues neurocognitive assessment, direct referral to a neuropsychologist, referral to neurologist, or administration of a cognitive screening tool such as the Montreal Cognitive Assessment, Saint Louis University Mental Status, or Cognitive Log is recommended. If the cognitive screening is positive, next steps include: referring for full neuropsychological assessment, which includes complete cognitive and motor testing, personality testing, and integration of neuroimaging data (eg, MRI, CT scans, and/or EEG).
Assessing the patients’ self-talk or thought patterns as they describe their head pain will help clinicians understand belief systems that may be distorting the reality of the medical condition. For example, a patient might report that “my pain feels like someone is hitting me with an axe”; this is a catastrophic thought that can distort the clarity and perceptibility of pain. Encouraging patients to monitor and analyze their anxiety and associated negative thoughts is an important strategy for improving mood and decreasing somatization. Recording daily thoughts and CBT can help the patient identify and appropriately address his (her) cognitive distortions and futile thinking.
When implementing a treatment plan for somatization disorder, we propose the mnemonic device CARE MD:
• CBT
• Assess (by ruling out a medical cause for somatic complaints)
• Regular visits
• Empathy
• Med-psych interface (help the patient connect physical complaints and emotional stressors)
• Do no harm.
Clinical recommendations
Chronic head pain can be debilitating; psychodiagnostic assessment should therefore be considered an important part of the diagnosis and treatment plan. After ruling out common and emergent primary or secondary causes of head pain, consider psychiatric comorbidities. Depression and anxiety have a strong bidirectional relationship with chronic headache; therefore, we suggest evaluating patients with the intention of alleviating both psychiatric symptoms and head pain.
It is important to diligently assess for common psychiatric comorbidities; using the AMPS and CARE MD mnemonics, along with screening for somatization disorders, is an easy and effective way to evaluate for relevant psychiatric conditions associated with chronic head pain. Because many patients have unusual and complicated responses to head pain that can be explained by non-pathophysiological and non-biomechanical models, using the biopsychosocial model is essential for effective diagnosis, assessment, and treatment. Abortive and prophylactic medical interventions, as well as behavioral, sociocultural, and cognitive assessment, are vital to a comprehensive treatment approach.
Bottom Line
The psychodiagnostic assessment can help the astute clinician identify comorbid psychiatric conditions, psychological factors, and somatic symptoms to develop a comprehensive biopsychosocial treatment plan for patients with chronic head pain. Rule out primary and secondary causes of pain and screen for somatization disorders. Consider medication and psychotherapeutic treatment options.
Related Resources
• Pompili M, Di Cosimo D, Innamorati M, et al. Psychiatric comorbidity in patients with chronic daily headache and migraine: a selective overview including personality traits and suicide risk. J Headache Pain. 2009;10(4):283-290.
• Sinclair AJ, Sturrock A, Davies B, et al. Headache management: pharmacological approaches [published online July 3, 2015]. Pract Neurol. doi: 10.1136/practneurol-2015-001167.
Drug Brand Names
Amitriptyline • Elavil Meperidine • Demerol
Botulinum toxin A • Botox Metoclopramide • Reglan
Divalproex sodium • Depakote Propranolol • Inderide
Ketorolac • Toradol Topiramate • Topamax
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
More than 45% of people worldwide suffer from headache at some point in their life.1 Head pain can lead to disability and functional decline, yet headache disorders often are underdiagnosed and poorly assessed. For example, 60% of migraine and tension-type headaches go undiagnosed and 50% of persons suffering from migraine have severe functional disability or require bed rest.2-4
Because head pain can be associated with secondary medical and psychiatric conditions, diagnosis can be challenging. This article reviews the medical and psychological aspects of major headaches and assists with clinical assessment. We present clinical interviewing tools and a diagram to enhance focused, efficient assessment and inform treatment plans.
Classification of headache
Headache is a common complaint, yet it is often underdiagnosed and ineffectively treated. The World Health Organization estimates that, globally, 50% of people with headache self-treat their pain.5 The International Headache Society classifies headache as primary or secondary; approximately 90% of complaints are from primary headache.6
Assessment and diagnosis of headache can be complex because of overlapping, subjective symptoms. It is important to have a general understanding of primary and secondary causes of headache so that interrelated symptoms do not obscure the most accurate diagnosis and effective treatment course. Although most headache complaints are benign, ruling out secondary causes helps gauge the likelihood of developing severe sequelae from underlying pathology.
By definition, primary headaches are idiopathic and commonly include migraine, tension-type, cluster, and hemicrania continua headache. Secondary headaches have an underlying pathology, which could improve by targeting the disorder. Common secondary causes of headache include:
• trauma
• vascular abnormalities
• structural abnormalities
• chemical (including medications)
• inflammation or infection
• metabolic conditions
• diseases of the neck and pericranial and intracranial structures
• psychiatric conditions.
Table 1 illustrates common causes of head pain. More definitive criteria for symptoms and diagnosis can be found in the International Classification of Headache Disorders.7
Primary headache
Tension-type is the most common primary headache, accounting for more than one-half of all headaches.7 Patients usually describe a tight pain in a bilateral band-like distribution, which could be caused by sustained neck muscle contraction. Pain usually builds in intensity and can last 30 minutes to several days. There is a well-established association between emotional stress or depression and the development of tension-type headaches.8
Migraine typically causes pulsating pain in a localized area of the head that lasts as long as 72 hours and can be associated with nausea, vomiting, photophobia, phono-phobia, and aura. Patients report varying precipitating factors but commonly cite certain foods, menstruation, and sleep deprivation. Although rare, migraine with aura has been linked to ischemic stroke; most cases have been reported in female smokers and oral contraceptive users age <45.9
Because migraines can be debilitating, some patients—typically those with ≥4 attacks a month—opt for prophylactic medication. Effective prophylactics include amitriptyline, propranolol, divalproex sodium, and topiramate, which should be monitored closely and given a trial for several months before switching to another drug. Commonly used abortive treatments include triptans and anti-emetics such as metoclopramide.
Meperidine and ketorolac are popular second-line agents for migraine. Botulinum toxin A also has been used in severe cases to reduce the number of headache days in chronic migraine patients.6
Cluster headache is rare, but typically exhibits repeated burning and intense unilateral periorbital or retro-orbital pain that lasts 15 minutes to 3 hours over several weeks. Men are predominantly affected. Cluster headaches typically improve with oxygen treatment.
Biopsychosocial model of head pain
The biomedical model has helped iden tify pathophysiological pain mechanisms and pharmacotherapeutic agents for headache. However, during assessment, limiting one’s attention to the linear relationship between pathology, mechanism of action, and pain oversimplifies common questions clinicians face when assessing chronic head pain.
Advancements in the last 3 decades have expanded the conceptualization of head pain to integrate sociocultural, environmental, behavioral, affective, cognitive, and biological variables—otherwise known as the biopsychosocial model.10,11 The biopsychosocial model is a multidimensional theory that helps answer difficult clinical assessment questions and complex patient presentations (Table 2).10-13 Many unusual responses to pain treatment, questionable validity of pain behavior, and disproportionate pain perception and functional decline are explained by non-pathophysiological and non-biomechanical models.
Psychiatric comorbidity and head pain
Psychiatric conditions are highly prevalent among persons with primary headache. Verri et al14 found that 90% of chronic daily headache patients had ≥1 psychiatric condition; depression and anxiety were most common. Of concern, 1 study found that headache is associated with increased frequency of suicidal ideation among patients with chronic pain.15 It is critical for clinicians to screen for psychiatric comorbidities in patients with chronic headache. Conversely, clinicians might want to screen for headache in their patients with psychiatric illness.
Migraine. Mood disorders are common among patients who suffer from migraine. The rate of depression is 2 to 4 times higher in those with migraine compared with healthy controls.16,17 In a large-scale study, patients with migraine had a 1.9-fold higher risk (compared with controls) of having a comorbid depressive episode; a 2-fold higher risk of manic episodes; and a 3-fold higher risk of both mania and depression.18 In a study of 62 inpatients, Fasmer19 reported that 46% of patients with unipolar depression and 44% of patients with bipolar disorder experienced migraine (77% of the bipolar disorder patients with migraine had bipolar II disorder). Patients with migraine are at increased risk of suicide attempts (odds ratio 4.3; 95% CI, 1.2-15.7).20
Tension-type headache. The relationship between psychiatric comorbidity in tension-type headache is well established. In contrast to what is seen with migraines, Puca et al21 found a higher prevalence of anxiety disorders (52.5%) than depressive disorders (36.4%) in patients with tension-type headache. Generalized anxiety disorder was one of the most prevalent anxiety conditions (83.3%), and dysthymia was the most prevalent mood disorder (45.6%). In the same study, 21.7% of patients were found to have a comorbid somatoform disorder.21
Emotional and cognitive factors can co-occur in patients with tension-type headache and a comorbid psychiatric condition. For example, difficulty identifying or recognizing emotions—commonly referred to as alexithymia—has been linked to tension-type headache.22 Additionally, maladaptive cognitive appraisal of stress is more common among patients with tension-type headache when compared with those without headaches.23 Being mindful of and recognizing these co-occurring emotional and cognitive factors will help clinicians construct a more accurate assessment and effective behavioral treatment plan.
Clinical assessment with a useful mnemonic
Clinical assessment of psychiatric illness is essential when evaluating chronic pain patients. Using the acronym AMPS (Anxiety, Mood, Psychosis, and Substance use disorders) (Table 3) is an efficient way for the clinician to ask pertinent questions regarding common psychiatric conditions that could have a direct effect on chronic pain.24 Head pain can be more intense when combined with untreated anxiety, depression, psychosis, or a substance use disorder. Untreated anxiety, for example, can amplify sympathetic response to pain and complicate treatment.
Investigating head pain patients for an underlying mood disorder is essential to providing successful treatment. Consider:
• starting psychotherapy modalities that address both pain and psychiatric illness, such as cognitive-behavioral therapy (CBT)
• reframing unhelpful pain beliefs
• managing activity-rest levels
• biofeedback
• supportive group therapy
• reducing family members’ reinforcement of the patient’s pain behavior or sick role.25
Assessing for somatic symptom disorders
In addition to using the AMPS approach for psychiatric assessment, clinicians should evaluate for somatization, which can present as head pain. Somatic symptom disorders (SSD) are a class of conditions that are impacted by affective, cognitive, and reinforcing factors that might or might not be consciously or intentionally produced. Patients with an SSD have somatic symptoms that are distressing or cause significant disruption of daily life because of excessive thoughts, feelings, or behaviors related to the somatic symptoms, for ≥6 months. The Figure outlines SSD, related conditions, and their respective prominent symptoms to assist in the differential diagnosis.26
Note that some headache conditions present with severe distress because of their abrupt onset and severity of symptoms (eg, cluster headaches). Therefore, the expectation and likelihood of psychological disturbance should be factored into a diagnosis of SSD and related conditions as seen in the Figure.
Secondary factors of unusual pain behavior or treatment response. The role of thoughts, affect, and behaviors is clinically meaningful in understanding SSD and similar conditions. Specific questions about cultural beliefs and rituals as they relate to exacerbations of head pain are of value. Table 413,27 lists behavioral, cognitive, and affective dimensions of head pain using the biopsychosocial model, and further clarifies common questions that arise with unusual pain response and complex patient presentations, which were outlined in the beginning of the article.
Because depression and anxiety can be comorbid with head pain, it is important to recognize psychological factors that contribute to pain perception. Indifference or denial of emotional stress as a result of severe pain and disability can imply a somatization process, which could suggest emotional disconnection or dissociation from somatic functioning.28 This finding can be a component of alexithymia, in which a person is disconnected from emotions and how emotions impact the body. Therefore, recognizing alexithymia assists in identifying psychological factors when patients deny mood symptoms, particularly in tension-type headache.
Functional assessment to rule out the disproportional impact of pain on daily activities is helpful in understanding the somatization process. Neurocognitive functioning should be assessed, particularly because frontal and subcortical dysregulation has been observed in head pain sufferers.29,30 Patients with cognitive changes as a result of a medical illness (eg, stroke, head concussion, brain tumor, or seizures) are especially at risk for neurocognitive dysfunction.
Neuropsychological assessment can be useful, not only to assess neurocognitive functioning (eg, Repeated Battery for the Assessment of Neuropsychological Status) but to identify objective test profiles associated with altered motivation (eg, Rey 15-Item Test, Minnesota Multiphasic Personality Inventory-2-Restructured Form F Scale, Personality Assessment Inventory [PAI] Negative Impression Management) and somatization processes (eg, PAI Somatization Scale). These instruments help to identify the severity of psychiatric and neurocognitive symptoms by comparing scores to normative (eg, healthy control group), clinical (eg, somatization, traumatic brain injury, mild cognitive impairment), and altered motivation (eg, persons instructed to exaggerate symptoms) databases.
If the clinician pursues neurocognitive assessment, direct referral to a neuropsychologist, referral to neurologist, or administration of a cognitive screening tool such as the Montreal Cognitive Assessment, Saint Louis University Mental Status, or Cognitive Log is recommended. If the cognitive screening is positive, next steps include: referring for full neuropsychological assessment, which includes complete cognitive and motor testing, personality testing, and integration of neuroimaging data (eg, MRI, CT scans, and/or EEG).
Assessing the patients’ self-talk or thought patterns as they describe their head pain will help clinicians understand belief systems that may be distorting the reality of the medical condition. For example, a patient might report that “my pain feels like someone is hitting me with an axe”; this is a catastrophic thought that can distort the clarity and perceptibility of pain. Encouraging patients to monitor and analyze their anxiety and associated negative thoughts is an important strategy for improving mood and decreasing somatization. Recording daily thoughts and CBT can help the patient identify and appropriately address his (her) cognitive distortions and futile thinking.
When implementing a treatment plan for somatization disorder, we propose the mnemonic device CARE MD:
• CBT
• Assess (by ruling out a medical cause for somatic complaints)
• Regular visits
• Empathy
• Med-psych interface (help the patient connect physical complaints and emotional stressors)
• Do no harm.
Clinical recommendations
Chronic head pain can be debilitating; psychodiagnostic assessment should therefore be considered an important part of the diagnosis and treatment plan. After ruling out common and emergent primary or secondary causes of head pain, consider psychiatric comorbidities. Depression and anxiety have a strong bidirectional relationship with chronic headache; therefore, we suggest evaluating patients with the intention of alleviating both psychiatric symptoms and head pain.
It is important to diligently assess for common psychiatric comorbidities; using the AMPS and CARE MD mnemonics, along with screening for somatization disorders, is an easy and effective way to evaluate for relevant psychiatric conditions associated with chronic head pain. Because many patients have unusual and complicated responses to head pain that can be explained by non-pathophysiological and non-biomechanical models, using the biopsychosocial model is essential for effective diagnosis, assessment, and treatment. Abortive and prophylactic medical interventions, as well as behavioral, sociocultural, and cognitive assessment, are vital to a comprehensive treatment approach.
Bottom Line
The psychodiagnostic assessment can help the astute clinician identify comorbid psychiatric conditions, psychological factors, and somatic symptoms to develop a comprehensive biopsychosocial treatment plan for patients with chronic head pain. Rule out primary and secondary causes of pain and screen for somatization disorders. Consider medication and psychotherapeutic treatment options.
Related Resources
• Pompili M, Di Cosimo D, Innamorati M, et al. Psychiatric comorbidity in patients with chronic daily headache and migraine: a selective overview including personality traits and suicide risk. J Headache Pain. 2009;10(4):283-290.
• Sinclair AJ, Sturrock A, Davies B, et al. Headache management: pharmacological approaches [published online July 3, 2015]. Pract Neurol. doi: 10.1136/practneurol-2015-001167.
Drug Brand Names
Amitriptyline • Elavil Meperidine • Demerol
Botulinum toxin A • Botox Metoclopramide • Reglan
Divalproex sodium • Depakote Propranolol • Inderide
Ketorolac • Toradol Topiramate • Topamax
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stovner LJ, Hagen K, Jensen R, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27(3):193-210.
2. Lipton RB, Stewart WF, Diamond S, et al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41(7):646-657.
3. The World Health Report 2001: Mental health: new understanding new hope. Geneva, Switzerland: World Health Organization; 2001.
4. World Health Organization. The global burden of disease: 2004 update. http://www.who.int/healthinfo/global_burden_ disease/GBD_report_2004update_full.pdf. Published 2004. Accessed July 31, 2015.
5. World Health Organization. Headache disorders. http:// www.who.int/mediacentre/factsheets/fs277/en/. Published October 2012. Accessed July 31, 2015.
6. Clinch C. Evaluation & management of headache. In: South- Paul JE, Matheny SC, Lewis EL. eds. Current diagnosis & treatment in family medicine, 4th ed. New York, NY: McGraw-Hill; 2015:293-297.
7. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629-808.
8. Mawet J, Kurth T, Ayata C. Migraine and stroke: in search of shared mechanisms. Cephalalgia. 2015;35(2):165-181.
9. Janke EA, Holroyd KA, Romanek K. Depression increases onset of tension-type headache following laboratory stress. Pain. 2004;111(3):230-238.
10. Engel GL. The need for a new medical model: a challenge for biomedicine. Science. 1977;196(4286):129-136.
11. Engel GL. The clinical application of the biopsychosocial model. Am J Psychiatry. 1980;137(5):535-544.
12. Turk DC, Flor H. Chronic pain: a biobehavioral perspective. In: Gatchel RJ, Turk DC, eds. Psychosocial factors in pain: critical perspectives. New York, NY: Guilford Press; 1999:18-34.
13. Andrasik F, Flor H, Turk DC. An expanded view of psychological aspects in head pain: the biopsychosocial model. Neurol Sci. 2005;26(suppl 2):s87-s91.
14. Verri AP, Proietti Cecchini A, Galli C, et al. Psychiatric comorbidity in chronic daily headache. Cephalalgia. 1998; 18(suppl 21):45-49.
15. Ilgen MA, Zivin K, McCammon RJ, et al. Pain and suicidal thoughts, plans and attempts in the United States. Gen Hosp Psychiatry. 2008;30(6):521-527.
16. Kowacs F, Socal MP, Ziomkowski SC, et al. Symptoms of depression and anxiety, and screening for mental disorders in migrainous patients. Cephalalgia. 2003;23(2):79-89.
17. Hamelsky SW, Lipton RB. Psychiatric comorbidity of migraine. Headache. 2006;46(9):1327-1333.
18. Nguyen TV, Low NC. Comorbidity of migraine and mood episodes in a nationally representative population-based sample. Headache. 2013;53(3):498-506.
19. Fasmer OB. The prevalence of migraine in patients with bipolar and unipolar affective disorders. Cephalalgia. 2001; 21(9):894-899.
20. Breslau N. Migraine, suicidal ideation, and suicide attempts. Neurology. 1992;42(2):392-395.
21. Puca F, Genco S, Prudenzano MP, et al. Psychiatric comorbidity and psychosocial stress in patients with tension-type headache from headache centers in Italy. Cephalalgia. 1999;19(3):159-164.
22. Yücel B, Kora K, Ozyalçín S, et al. Depression, automatic thoughts, alexithymia, and assertiveness in patients with tension-type headache. Headache. 2002;42(3):194-199.
23. Wittrock DA, Myers TC. The comparison of individuals with recurrent tension-type headache and headache-free controls in physiological response, appraisal, and coping with stressors: a review of the literature. Ann Behav Med. 1998;20(2):118-134.
24. Onate J, Xiong G, McCarron R. The primary care psychiatric interview. In: McCarron R, Xiong G, Bourgeois J. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott, Williams and Wilkins; 2009:3-4.
25. Songer D. Psychotherapeutic approaches in the treatment of pain. Psychiatry (Edgmont). 2005;2(5):19-24.
26. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
27. Hashmi JA, Baliki MN, Huang L, et al. Shape shifting pain: chronification of back pain shifts brain representation from nociceptive to emotional circuits. Brain. 2013;136(pt 9):2751-2768.
28. Packard RC. Conversion headache. Headache. 1980;20(5):266-268.
29. Mongini F, Keller R, Deregibus A, et al. Frontal lobe dysfunction in patients with chronic migraine: a clinical-neuropsychological study. Psychiatry Res. 2005;133(1):101-106.
30. Martelli MF, Grayson RL, Zasler ND. Posttraumatic headache: neuropsychological and psychological effects and treatment implications. J Head Trauma Rehabil. 1999;14(1):49-69.
1. Stovner LJ, Hagen K, Jensen R, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27(3):193-210.
2. Lipton RB, Stewart WF, Diamond S, et al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41(7):646-657.
3. The World Health Report 2001: Mental health: new understanding new hope. Geneva, Switzerland: World Health Organization; 2001.
4. World Health Organization. The global burden of disease: 2004 update. http://www.who.int/healthinfo/global_burden_ disease/GBD_report_2004update_full.pdf. Published 2004. Accessed July 31, 2015.
5. World Health Organization. Headache disorders. http:// www.who.int/mediacentre/factsheets/fs277/en/. Published October 2012. Accessed July 31, 2015.
6. Clinch C. Evaluation & management of headache. In: South- Paul JE, Matheny SC, Lewis EL. eds. Current diagnosis & treatment in family medicine, 4th ed. New York, NY: McGraw-Hill; 2015:293-297.
7. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629-808.
8. Mawet J, Kurth T, Ayata C. Migraine and stroke: in search of shared mechanisms. Cephalalgia. 2015;35(2):165-181.
9. Janke EA, Holroyd KA, Romanek K. Depression increases onset of tension-type headache following laboratory stress. Pain. 2004;111(3):230-238.
10. Engel GL. The need for a new medical model: a challenge for biomedicine. Science. 1977;196(4286):129-136.
11. Engel GL. The clinical application of the biopsychosocial model. Am J Psychiatry. 1980;137(5):535-544.
12. Turk DC, Flor H. Chronic pain: a biobehavioral perspective. In: Gatchel RJ, Turk DC, eds. Psychosocial factors in pain: critical perspectives. New York, NY: Guilford Press; 1999:18-34.
13. Andrasik F, Flor H, Turk DC. An expanded view of psychological aspects in head pain: the biopsychosocial model. Neurol Sci. 2005;26(suppl 2):s87-s91.
14. Verri AP, Proietti Cecchini A, Galli C, et al. Psychiatric comorbidity in chronic daily headache. Cephalalgia. 1998; 18(suppl 21):45-49.
15. Ilgen MA, Zivin K, McCammon RJ, et al. Pain and suicidal thoughts, plans and attempts in the United States. Gen Hosp Psychiatry. 2008;30(6):521-527.
16. Kowacs F, Socal MP, Ziomkowski SC, et al. Symptoms of depression and anxiety, and screening for mental disorders in migrainous patients. Cephalalgia. 2003;23(2):79-89.
17. Hamelsky SW, Lipton RB. Psychiatric comorbidity of migraine. Headache. 2006;46(9):1327-1333.
18. Nguyen TV, Low NC. Comorbidity of migraine and mood episodes in a nationally representative population-based sample. Headache. 2013;53(3):498-506.
19. Fasmer OB. The prevalence of migraine in patients with bipolar and unipolar affective disorders. Cephalalgia. 2001; 21(9):894-899.
20. Breslau N. Migraine, suicidal ideation, and suicide attempts. Neurology. 1992;42(2):392-395.
21. Puca F, Genco S, Prudenzano MP, et al. Psychiatric comorbidity and psychosocial stress in patients with tension-type headache from headache centers in Italy. Cephalalgia. 1999;19(3):159-164.
22. Yücel B, Kora K, Ozyalçín S, et al. Depression, automatic thoughts, alexithymia, and assertiveness in patients with tension-type headache. Headache. 2002;42(3):194-199.
23. Wittrock DA, Myers TC. The comparison of individuals with recurrent tension-type headache and headache-free controls in physiological response, appraisal, and coping with stressors: a review of the literature. Ann Behav Med. 1998;20(2):118-134.
24. Onate J, Xiong G, McCarron R. The primary care psychiatric interview. In: McCarron R, Xiong G, Bourgeois J. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott, Williams and Wilkins; 2009:3-4.
25. Songer D. Psychotherapeutic approaches in the treatment of pain. Psychiatry (Edgmont). 2005;2(5):19-24.
26. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
27. Hashmi JA, Baliki MN, Huang L, et al. Shape shifting pain: chronification of back pain shifts brain representation from nociceptive to emotional circuits. Brain. 2013;136(pt 9):2751-2768.
28. Packard RC. Conversion headache. Headache. 1980;20(5):266-268.
29. Mongini F, Keller R, Deregibus A, et al. Frontal lobe dysfunction in patients with chronic migraine: a clinical-neuropsychological study. Psychiatry Res. 2005;133(1):101-106.
30. Martelli MF, Grayson RL, Zasler ND. Posttraumatic headache: neuropsychological and psychological effects and treatment implications. J Head Trauma Rehabil. 1999;14(1):49-69.

Rx: Treating chronic medical vulnerability in the mentally ill
With few exceptions, I have found that patients who have chronic moderate or severe mental illness tend to be relatively more vulnerable in terms of (1) receiving suboptimal primary medical care and (2) suffering a resulting increase in morbidity, mortality, and disability.
Across the board, I’ve found, psychiatrists are more likely to treat patients who are chronically vulnerable.
Why are they so vulnerable?
The unique vulnerability of patients with severe mental illness stems from several causative factors:
• the stigma attached to mental illness
• poor implementation of parity in reimbursement for mental health services
• a suboptimal-sized mental health workforce
• related poor patient-centered support
• most important, these patients’ lack of primary and preventive medical care.
Here are a few examples that demonstrate how dire the situation is:
Smoking cigarettes is one of the most dangerous modifiable risk factors for vascular disease and early death. People with mental illness smoke almost half (44%) of the cigarettes sold in the United States and are twice as likely to smoke than those who do not have a mental illness.1,2
HIV infection is at least 2 or 3 times more prevalent among people with severe mental illness as it is in the general population.3
Hepatitis C infection is at least twice as prevalent in people with a diagnosis of schizophrenia as it is in the general population.4
Schizophrenia. As many as 60% of premature deaths among people with schizophrenia are attributable to a medical illness.5 For example, those with schizophrenia have an increased 10-year cardiac mortality; comparatively higher rates of hypertension, diabetes, and smoking; and, on average, a lower level of high-density lipoprotein cholesterol. Nasrallah et al reported that the rate of untreated hypertension among patients with schizophrenia is 62.4%.6
Premature death. People who have a diagnosis of severe mental illness are at risk of dying prematurely by as much as 25 years.5,7-10
Who should take the lead?
How can psychiatrists address this ongoing vulnerability within the mentally ill patient population, and advocate for their patients? A comprehensive answer to this question is beyond the scope of this article, but I can offer this prescription for your consideration.
Be an advocate. You, as a psychiatrist, are well positioned to counter the mental health-related stigma and advocate for implementation of mental health parity nationwide. In addition to participating in community education and outreach, become a member of, and get involved in, established organizations, such as the American Psychiatric Association, that advocates for psychiatric patients at all levels.
Keep patients connected. Make sure your patients are connected with a primary care provider, and use your psychotherapeutic skills to help patients understand the importance of receiving primary and secondary preventive medical care.
Monitor health and disease. As a physician first and a psychiatrist second, closely monitor your patients for general medical conditions that are related to the presence and treatment of psychiatric disorders. Consider routinely reviewing pertinent lab work with patients—even results of tests ordered by a primary care provider (eg, the metabolic panel and a thyroid-stimulating hormone level in patients taking lithium).
Collaborate with your primary care colleagues; they need your help as much as you can use their help! Make sure your patients witness this collaboration, because it mirrors how you would like them to interact with their primary care provider.
Educate yourself. Education in the essentials of psychiatry-based preventive medical care is key, as we work to more effectively address the increased disability, morbidity, mortality, and overall vulnerability in our patients. Stay “current” on general medical topics by reading the “Med/Psych Update” section of Current Psychiatry and relevant articles in other clinical guides to both integrated and preventive medicine.11
1. Lasser K, Boyd JW, Woolhandler S, et al. Smoking and mental illness: a population-based prevalence study. JAMA. 2000;284(20):2606-2610.
2. Grant BF, Hasin DS, Chou SP, et al. Nicotine dependence and psychiatric disorders in the United States: results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry. 2004;61(11):1107-1115.
3. Meade CS, Sikkema KJ. HIV risk behavior among adults with severe mental illness: a systematic review. Clin Psychol Rev. 2005;25(4):433-457.
4. Dinwiddie SH, Shicker L, Newman T. Prevalence of hepatitis C among psychiatric patients in the public sector. Am J Psychiatry. 2003;160(1):172-174.
5. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131.
6. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
7. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.
8. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572.
9. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156.
10. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
11. McCarron RM, Xiong G, Keenan CR, et al. Preventive medical care in psychiatry: a practical guide for clinicians. Arlington, VA: American Psychiatric Publishing; 2014.
With few exceptions, I have found that patients who have chronic moderate or severe mental illness tend to be relatively more vulnerable in terms of (1) receiving suboptimal primary medical care and (2) suffering a resulting increase in morbidity, mortality, and disability.
Across the board, I’ve found, psychiatrists are more likely to treat patients who are chronically vulnerable.
Why are they so vulnerable?
The unique vulnerability of patients with severe mental illness stems from several causative factors:
• the stigma attached to mental illness
• poor implementation of parity in reimbursement for mental health services
• a suboptimal-sized mental health workforce
• related poor patient-centered support
• most important, these patients’ lack of primary and preventive medical care.
Here are a few examples that demonstrate how dire the situation is:
Smoking cigarettes is one of the most dangerous modifiable risk factors for vascular disease and early death. People with mental illness smoke almost half (44%) of the cigarettes sold in the United States and are twice as likely to smoke than those who do not have a mental illness.1,2
HIV infection is at least 2 or 3 times more prevalent among people with severe mental illness as it is in the general population.3
Hepatitis C infection is at least twice as prevalent in people with a diagnosis of schizophrenia as it is in the general population.4
Schizophrenia. As many as 60% of premature deaths among people with schizophrenia are attributable to a medical illness.5 For example, those with schizophrenia have an increased 10-year cardiac mortality; comparatively higher rates of hypertension, diabetes, and smoking; and, on average, a lower level of high-density lipoprotein cholesterol. Nasrallah et al reported that the rate of untreated hypertension among patients with schizophrenia is 62.4%.6
Premature death. People who have a diagnosis of severe mental illness are at risk of dying prematurely by as much as 25 years.5,7-10
Who should take the lead?
How can psychiatrists address this ongoing vulnerability within the mentally ill patient population, and advocate for their patients? A comprehensive answer to this question is beyond the scope of this article, but I can offer this prescription for your consideration.
Be an advocate. You, as a psychiatrist, are well positioned to counter the mental health-related stigma and advocate for implementation of mental health parity nationwide. In addition to participating in community education and outreach, become a member of, and get involved in, established organizations, such as the American Psychiatric Association, that advocates for psychiatric patients at all levels.
Keep patients connected. Make sure your patients are connected with a primary care provider, and use your psychotherapeutic skills to help patients understand the importance of receiving primary and secondary preventive medical care.
Monitor health and disease. As a physician first and a psychiatrist second, closely monitor your patients for general medical conditions that are related to the presence and treatment of psychiatric disorders. Consider routinely reviewing pertinent lab work with patients—even results of tests ordered by a primary care provider (eg, the metabolic panel and a thyroid-stimulating hormone level in patients taking lithium).
Collaborate with your primary care colleagues; they need your help as much as you can use their help! Make sure your patients witness this collaboration, because it mirrors how you would like them to interact with their primary care provider.
Educate yourself. Education in the essentials of psychiatry-based preventive medical care is key, as we work to more effectively address the increased disability, morbidity, mortality, and overall vulnerability in our patients. Stay “current” on general medical topics by reading the “Med/Psych Update” section of Current Psychiatry and relevant articles in other clinical guides to both integrated and preventive medicine.11
With few exceptions, I have found that patients who have chronic moderate or severe mental illness tend to be relatively more vulnerable in terms of (1) receiving suboptimal primary medical care and (2) suffering a resulting increase in morbidity, mortality, and disability.
Across the board, I’ve found, psychiatrists are more likely to treat patients who are chronically vulnerable.
Why are they so vulnerable?
The unique vulnerability of patients with severe mental illness stems from several causative factors:
• the stigma attached to mental illness
• poor implementation of parity in reimbursement for mental health services
• a suboptimal-sized mental health workforce
• related poor patient-centered support
• most important, these patients’ lack of primary and preventive medical care.
Here are a few examples that demonstrate how dire the situation is:
Smoking cigarettes is one of the most dangerous modifiable risk factors for vascular disease and early death. People with mental illness smoke almost half (44%) of the cigarettes sold in the United States and are twice as likely to smoke than those who do not have a mental illness.1,2
HIV infection is at least 2 or 3 times more prevalent among people with severe mental illness as it is in the general population.3
Hepatitis C infection is at least twice as prevalent in people with a diagnosis of schizophrenia as it is in the general population.4
Schizophrenia. As many as 60% of premature deaths among people with schizophrenia are attributable to a medical illness.5 For example, those with schizophrenia have an increased 10-year cardiac mortality; comparatively higher rates of hypertension, diabetes, and smoking; and, on average, a lower level of high-density lipoprotein cholesterol. Nasrallah et al reported that the rate of untreated hypertension among patients with schizophrenia is 62.4%.6
Premature death. People who have a diagnosis of severe mental illness are at risk of dying prematurely by as much as 25 years.5,7-10
Who should take the lead?
How can psychiatrists address this ongoing vulnerability within the mentally ill patient population, and advocate for their patients? A comprehensive answer to this question is beyond the scope of this article, but I can offer this prescription for your consideration.
Be an advocate. You, as a psychiatrist, are well positioned to counter the mental health-related stigma and advocate for implementation of mental health parity nationwide. In addition to participating in community education and outreach, become a member of, and get involved in, established organizations, such as the American Psychiatric Association, that advocates for psychiatric patients at all levels.
Keep patients connected. Make sure your patients are connected with a primary care provider, and use your psychotherapeutic skills to help patients understand the importance of receiving primary and secondary preventive medical care.
Monitor health and disease. As a physician first and a psychiatrist second, closely monitor your patients for general medical conditions that are related to the presence and treatment of psychiatric disorders. Consider routinely reviewing pertinent lab work with patients—even results of tests ordered by a primary care provider (eg, the metabolic panel and a thyroid-stimulating hormone level in patients taking lithium).
Collaborate with your primary care colleagues; they need your help as much as you can use their help! Make sure your patients witness this collaboration, because it mirrors how you would like them to interact with their primary care provider.
Educate yourself. Education in the essentials of psychiatry-based preventive medical care is key, as we work to more effectively address the increased disability, morbidity, mortality, and overall vulnerability in our patients. Stay “current” on general medical topics by reading the “Med/Psych Update” section of Current Psychiatry and relevant articles in other clinical guides to both integrated and preventive medicine.11
1. Lasser K, Boyd JW, Woolhandler S, et al. Smoking and mental illness: a population-based prevalence study. JAMA. 2000;284(20):2606-2610.
2. Grant BF, Hasin DS, Chou SP, et al. Nicotine dependence and psychiatric disorders in the United States: results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry. 2004;61(11):1107-1115.
3. Meade CS, Sikkema KJ. HIV risk behavior among adults with severe mental illness: a systematic review. Clin Psychol Rev. 2005;25(4):433-457.
4. Dinwiddie SH, Shicker L, Newman T. Prevalence of hepatitis C among psychiatric patients in the public sector. Am J Psychiatry. 2003;160(1):172-174.
5. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131.
6. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
7. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.
8. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572.
9. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156.
10. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
11. McCarron RM, Xiong G, Keenan CR, et al. Preventive medical care in psychiatry: a practical guide for clinicians. Arlington, VA: American Psychiatric Publishing; 2014.
1. Lasser K, Boyd JW, Woolhandler S, et al. Smoking and mental illness: a population-based prevalence study. JAMA. 2000;284(20):2606-2610.
2. Grant BF, Hasin DS, Chou SP, et al. Nicotine dependence and psychiatric disorders in the United States: results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry. 2004;61(11):1107-1115.
3. Meade CS, Sikkema KJ. HIV risk behavior among adults with severe mental illness: a systematic review. Clin Psychol Rev. 2005;25(4):433-457.
4. Dinwiddie SH, Shicker L, Newman T. Prevalence of hepatitis C among psychiatric patients in the public sector. Am J Psychiatry. 2003;160(1):172-174.
5. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131.
6. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
7. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.
8. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572.
9. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156.
10. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
11. McCarron RM, Xiong G, Keenan CR, et al. Preventive medical care in psychiatry: a practical guide for clinicians. Arlington, VA: American Psychiatric Publishing; 2014.
