User login
Drug reverses anticoagulant effect of dabigatran
TORONTO—Interim results of a phase 3 study suggest idarucizumab, a humanized antibody fragment, can reverse the anticoagulant effect of
dabigatran in real-world situations.
In the RE-VERSE AD trial, idarucizumab normalized diluted thrombin time (dTT) and ecarin clotting time (ECT) in a majority of patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
In addition, researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events.
These results have been published in NEJM and presented at the 2015 ISTH Congress (abstract LB005). The study was sponsored by Boehringer Ingelheim, the company developing idarucizumab and dabigatran.
“The interim analysis from RE-VERSE AD is important for healthcare professionals as it provides the first insights into the effect of a specific reversal agent to a non-vitamin K antagonist oral anticoagulant during real-world emergency situations,” said study investigator Charles Pollack, MD, of the University of Pennsylvania in Philadelphia.
Because RE-VERSE AD was designed to evaluate how idarucizumab would perform in real-world situations, severely ill or injured patients were eligible for enrollment. The interim analysis included data from 90 patients in emergency settings who were taking dabigatran and required reversal.
The patients were divided into 2 groups: those with uncontrolled or life-threatening bleeding complications, such as intracranial hemorrhage or severe trauma (group A, n=51), and patients requiring emergency surgery or an invasive procedure (group B, n=39).
The primary endpoint of the study is the degree to which 5 g of idarucizumab reversed the anticoagulant effect of dabigatran within 4 hours, measured by dTT and ECT.
The researchers were able to evaluate the percentage of reversal by dTT in 68 patients (40 in group A and 28 in group B) and the percentage of reversal by ECT in 81 patients (47 in group A and 34 in group B).
The dTT was normalized in 98% of evaluable patients in group A and 93% in group B. The ECT was normalized in 89% of evaluable patients in group A and 88% in group B.
At 12 hours and 24 hours, the dTT was below the upper limit of the normal range in 90% of evaluable patients in group A and 81% in group B. The ECT was below the upper limit of the normal range in 72% of evaluable group A patients and 54% of evaluable group B patients.
Among the 35 evaluable patients in group A, hemostasis was restored at a median of 11.4 hours. Among the 36 patients in group B who underwent a procedure, 33 had normal intraoperative hemostasis. Two patients had mildly abnormal hemostasis, and 1 patient had moderately abnormal hemostasis.
“As observed in earlier research in volunteers, idarucizumab reversed the anticoagulant effect of dabigatran in patients completely within minutes, even in those rare critical care situations studied in RE-VERSE AD,” Dr Pollack said.
“These data demonstrate that use of idarucizumab can help physicians focus on other vital aspects of emergency management beyond anticoagulant reversal in dabigatran-treated patients.”
Twenty-one patients (13 in group A and 8 in group B) experienced serious adverse events during the study.
This included 18 events that led to death, 5 thrombotic events, 2 cases of gastrointestinal hemorrhage, a postoperative wound infection, a case of delirium, a case of right ventricular failure, and a case of pulmonary edema. (Some patients had more than one serious adverse event.) ![]()
TORONTO—Interim results of a phase 3 study suggest idarucizumab, a humanized antibody fragment, can reverse the anticoagulant effect of
dabigatran in real-world situations.
In the RE-VERSE AD trial, idarucizumab normalized diluted thrombin time (dTT) and ecarin clotting time (ECT) in a majority of patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
In addition, researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events.
These results have been published in NEJM and presented at the 2015 ISTH Congress (abstract LB005). The study was sponsored by Boehringer Ingelheim, the company developing idarucizumab and dabigatran.
“The interim analysis from RE-VERSE AD is important for healthcare professionals as it provides the first insights into the effect of a specific reversal agent to a non-vitamin K antagonist oral anticoagulant during real-world emergency situations,” said study investigator Charles Pollack, MD, of the University of Pennsylvania in Philadelphia.
Because RE-VERSE AD was designed to evaluate how idarucizumab would perform in real-world situations, severely ill or injured patients were eligible for enrollment. The interim analysis included data from 90 patients in emergency settings who were taking dabigatran and required reversal.
The patients were divided into 2 groups: those with uncontrolled or life-threatening bleeding complications, such as intracranial hemorrhage or severe trauma (group A, n=51), and patients requiring emergency surgery or an invasive procedure (group B, n=39).
The primary endpoint of the study is the degree to which 5 g of idarucizumab reversed the anticoagulant effect of dabigatran within 4 hours, measured by dTT and ECT.
The researchers were able to evaluate the percentage of reversal by dTT in 68 patients (40 in group A and 28 in group B) and the percentage of reversal by ECT in 81 patients (47 in group A and 34 in group B).
The dTT was normalized in 98% of evaluable patients in group A and 93% in group B. The ECT was normalized in 89% of evaluable patients in group A and 88% in group B.
At 12 hours and 24 hours, the dTT was below the upper limit of the normal range in 90% of evaluable patients in group A and 81% in group B. The ECT was below the upper limit of the normal range in 72% of evaluable group A patients and 54% of evaluable group B patients.
Among the 35 evaluable patients in group A, hemostasis was restored at a median of 11.4 hours. Among the 36 patients in group B who underwent a procedure, 33 had normal intraoperative hemostasis. Two patients had mildly abnormal hemostasis, and 1 patient had moderately abnormal hemostasis.
“As observed in earlier research in volunteers, idarucizumab reversed the anticoagulant effect of dabigatran in patients completely within minutes, even in those rare critical care situations studied in RE-VERSE AD,” Dr Pollack said.
“These data demonstrate that use of idarucizumab can help physicians focus on other vital aspects of emergency management beyond anticoagulant reversal in dabigatran-treated patients.”
Twenty-one patients (13 in group A and 8 in group B) experienced serious adverse events during the study.
This included 18 events that led to death, 5 thrombotic events, 2 cases of gastrointestinal hemorrhage, a postoperative wound infection, a case of delirium, a case of right ventricular failure, and a case of pulmonary edema. (Some patients had more than one serious adverse event.) ![]()
TORONTO—Interim results of a phase 3 study suggest idarucizumab, a humanized antibody fragment, can reverse the anticoagulant effect of
dabigatran in real-world situations.
In the RE-VERSE AD trial, idarucizumab normalized diluted thrombin time (dTT) and ecarin clotting time (ECT) in a majority of patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
In addition, researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events.
These results have been published in NEJM and presented at the 2015 ISTH Congress (abstract LB005). The study was sponsored by Boehringer Ingelheim, the company developing idarucizumab and dabigatran.
“The interim analysis from RE-VERSE AD is important for healthcare professionals as it provides the first insights into the effect of a specific reversal agent to a non-vitamin K antagonist oral anticoagulant during real-world emergency situations,” said study investigator Charles Pollack, MD, of the University of Pennsylvania in Philadelphia.
Because RE-VERSE AD was designed to evaluate how idarucizumab would perform in real-world situations, severely ill or injured patients were eligible for enrollment. The interim analysis included data from 90 patients in emergency settings who were taking dabigatran and required reversal.
The patients were divided into 2 groups: those with uncontrolled or life-threatening bleeding complications, such as intracranial hemorrhage or severe trauma (group A, n=51), and patients requiring emergency surgery or an invasive procedure (group B, n=39).
The primary endpoint of the study is the degree to which 5 g of idarucizumab reversed the anticoagulant effect of dabigatran within 4 hours, measured by dTT and ECT.
The researchers were able to evaluate the percentage of reversal by dTT in 68 patients (40 in group A and 28 in group B) and the percentage of reversal by ECT in 81 patients (47 in group A and 34 in group B).
The dTT was normalized in 98% of evaluable patients in group A and 93% in group B. The ECT was normalized in 89% of evaluable patients in group A and 88% in group B.
At 12 hours and 24 hours, the dTT was below the upper limit of the normal range in 90% of evaluable patients in group A and 81% in group B. The ECT was below the upper limit of the normal range in 72% of evaluable group A patients and 54% of evaluable group B patients.
Among the 35 evaluable patients in group A, hemostasis was restored at a median of 11.4 hours. Among the 36 patients in group B who underwent a procedure, 33 had normal intraoperative hemostasis. Two patients had mildly abnormal hemostasis, and 1 patient had moderately abnormal hemostasis.
“As observed in earlier research in volunteers, idarucizumab reversed the anticoagulant effect of dabigatran in patients completely within minutes, even in those rare critical care situations studied in RE-VERSE AD,” Dr Pollack said.
“These data demonstrate that use of idarucizumab can help physicians focus on other vital aspects of emergency management beyond anticoagulant reversal in dabigatran-treated patients.”
Twenty-one patients (13 in group A and 8 in group B) experienced serious adverse events during the study.
This included 18 events that led to death, 5 thrombotic events, 2 cases of gastrointestinal hemorrhage, a postoperative wound infection, a case of delirium, a case of right ventricular failure, and a case of pulmonary edema. (Some patients had more than one serious adverse event.) ![]()
Thiotepa, rituximab improve response in CNS lymphoma

site of 13-ICML
LUGANO—Adding thiotepa and rituximab to the treatment of primary central nervous system (CNS) lymphoma is feasible from a safety perspective and has yielded promising results, according to new research.
An analysis of the IELSG 32 trial—in which patients received methotrexate and cytarabine alone, with rituximab, or with rituximab and thiotepa—has shown the 4-drug regimen improves responses and progression-free survival.
IELSG 32 builds on the foundation of the IELSG 20 trial to determine the best induction therapy for patients with primary CNS lymphoma. In IELSG 20, the complete response (CR) rate was higher among patients receiving high-dose methotrexate and cytarabine than in patients receiving methotrexate alone.
So the researchers decided to investigate whether rituximab and/or thiotepa added to the regimen would impact patient outcome. Rituximab has been associated with improved CR rates in primary CNS lymphoma, and thiotepa is active against aggressive lymphomas. Thiotepa is also able to cross the blood–brain barrier.
Andrés J.M. Ferreri, MD, of IRCCS Ospedale San Raffaele in Milan, Italy, reported the results of this research on behalf of the International Extranodal Lymphoma Study Group (IELSG) at the 13th International Conference on Malignant Lymphoma (abstract 009).
Treatment and toxicity
The investigators randomized 227 patients from 53 centers in 5 countries to 4 cycles of treatment in 3 cohorts:
- Arm A consisted of 4 cycles of methotrexate (3.5 g/m2) on day 1 and cytarabine (2 g/m2) twice a day on days 2 and 3 every 3 weeks.
- Arm B added rituximab (375 mg/m2) on days 5 and 0 to the regimen.
- Arm C added rituximab and thiotepa (30 mg/m2) on day 4 to the regimen.
Eight patients were excluded for various reasons, including incorrect diagnosis. So 219 patients received therapy, 75 in arm A, 69 in arm B, and 75 in arm C.
Patients were a median age of 58 years in arm A and 57 in arms B and C, and the majority were male. More than 80% of patients in all arms were intermediate- or high-risk according to IELSG. And all patients had the diffuse large B-cell lymphoma histotype.
Patients in arm A received 74% of the planned courses, those in arm B received 86%, and those in arm C received 91%. The relative dose intensity across all arms of each drug administered was not significantly different among the arms.
“As expected, hematologic toxicity was common, with grade 4 neutropenia (P=0.01) and thrombocytopenia (P=0.0001) being significantly more common for arm C,” Dr Ferreri said. “However, this was not associated with an increasing array of severe infections, interruptions, toxicity, or toxic deaths.”
Peripheral blood stem cell collection was successful in 94% of patients in arms A and C and 96% of patients in arm B.
Response rates
The overall response rate (ORR) was 72% in IELSG 32, compared to 54% in IELSG 20. But the CR rate was not significantly higher in IELSG 32 than in IELSG 20, at 34% and 31%, respectively.
“There are many explanations for this,” Dr Ferreri said. “But I think it was because there were a significantly greater number of patients with higher IELSG risk [in IELSG 32], a higher proportion of poor-prognosis patients.”
Patients in arm C had significantly higher response rates than patients in the other 2 arms, with a CR rate of 49%, a partial response (PR) rate of 37%, and an ORR of 87%.
This compared to a CR rate of 23% in arm A and 30% in arm B, a PR rate of 31% in arm A and 30% in arm B, and an ORR of 53% in arm A and 74% in arm B.
The investigators also analyzed activity according to IELSG risk.
“Arm C was significantly more active in all the 3 subgroups,” Dr Ferreri observed, “and importantly, the overall response rate and complete remission rate were similar for each arm in the 3 different risk groups.”
Second randomization and survival
One hundred and eighteen patients went on to a second randomization, 35 from arm A, 35 from arm B, and 48 from arm C.
Fifty-nine patients were randomized to the whole-brain radiotherapy cohort, and 59 to the carmustine-thiotepa-autologous stem cell transplant cohort.
At a median follow-up of 21 months, 110 patients remain failure free, 32% from arm A, 54% from arm B, and 65% from arm C. Failure in 97% of the cases was due to primary site involvement, usually the brain.
Fifty-six percent of the patients are still alive, 39% in arm A, 59% in arm B, and 69% in arm C.
Ninety-seven patients died, 73 from lymphoma, 15 from toxicity of the first-line treatment, 2 from toxicity of the salvage regimen, 2 from neurotoxicity, and 5 from other causes.
Dr Ferreri concluded that the MATRIX regimen—the addition of rituximab and thiotepa to methotrexate and cytarabine—was associated with significant improvements in CR rate, ORR, and progression-free and overall survival.
The addition of rituximab and thiotepa did not increase toxicity, with the exception of greater hematologic adverse events, nor did the drugs increase the rates of severe complications. The agents also allowed for high rates of successful stem cell collection. ![]()
site of 13-ICML
LUGANO—Adding thiotepa and rituximab to the treatment of primary central nervous system (CNS) lymphoma is feasible from a safety perspective and has yielded promising results, according to new research.
An analysis of the IELSG 32 trial—in which patients received methotrexate and cytarabine alone, with rituximab, or with rituximab and thiotepa—has shown the 4-drug regimen improves responses and progression-free survival.
IELSG 32 builds on the foundation of the IELSG 20 trial to determine the best induction therapy for patients with primary CNS lymphoma. In IELSG 20, the complete response (CR) rate was higher among patients receiving high-dose methotrexate and cytarabine than in patients receiving methotrexate alone.
So the researchers decided to investigate whether rituximab and/or thiotepa added to the regimen would impact patient outcome. Rituximab has been associated with improved CR rates in primary CNS lymphoma, and thiotepa is active against aggressive lymphomas. Thiotepa is also able to cross the blood–brain barrier.
Andrés J.M. Ferreri, MD, of IRCCS Ospedale San Raffaele in Milan, Italy, reported the results of this research on behalf of the International Extranodal Lymphoma Study Group (IELSG) at the 13th International Conference on Malignant Lymphoma (abstract 009).
Treatment and toxicity
The investigators randomized 227 patients from 53 centers in 5 countries to 4 cycles of treatment in 3 cohorts:
- Arm A consisted of 4 cycles of methotrexate (3.5 g/m2) on day 1 and cytarabine (2 g/m2) twice a day on days 2 and 3 every 3 weeks.
- Arm B added rituximab (375 mg/m2) on days 5 and 0 to the regimen.
- Arm C added rituximab and thiotepa (30 mg/m2) on day 4 to the regimen.
Eight patients were excluded for various reasons, including incorrect diagnosis. So 219 patients received therapy, 75 in arm A, 69 in arm B, and 75 in arm C.
Patients were a median age of 58 years in arm A and 57 in arms B and C, and the majority were male. More than 80% of patients in all arms were intermediate- or high-risk according to IELSG. And all patients had the diffuse large B-cell lymphoma histotype.
Patients in arm A received 74% of the planned courses, those in arm B received 86%, and those in arm C received 91%. The relative dose intensity across all arms of each drug administered was not significantly different among the arms.
“As expected, hematologic toxicity was common, with grade 4 neutropenia (P=0.01) and thrombocytopenia (P=0.0001) being significantly more common for arm C,” Dr Ferreri said. “However, this was not associated with an increasing array of severe infections, interruptions, toxicity, or toxic deaths.”
Peripheral blood stem cell collection was successful in 94% of patients in arms A and C and 96% of patients in arm B.
Response rates
The overall response rate (ORR) was 72% in IELSG 32, compared to 54% in IELSG 20. But the CR rate was not significantly higher in IELSG 32 than in IELSG 20, at 34% and 31%, respectively.
“There are many explanations for this,” Dr Ferreri said. “But I think it was because there were a significantly greater number of patients with higher IELSG risk [in IELSG 32], a higher proportion of poor-prognosis patients.”
Patients in arm C had significantly higher response rates than patients in the other 2 arms, with a CR rate of 49%, a partial response (PR) rate of 37%, and an ORR of 87%.
This compared to a CR rate of 23% in arm A and 30% in arm B, a PR rate of 31% in arm A and 30% in arm B, and an ORR of 53% in arm A and 74% in arm B.
The investigators also analyzed activity according to IELSG risk.
“Arm C was significantly more active in all the 3 subgroups,” Dr Ferreri observed, “and importantly, the overall response rate and complete remission rate were similar for each arm in the 3 different risk groups.”
Second randomization and survival
One hundred and eighteen patients went on to a second randomization, 35 from arm A, 35 from arm B, and 48 from arm C.
Fifty-nine patients were randomized to the whole-brain radiotherapy cohort, and 59 to the carmustine-thiotepa-autologous stem cell transplant cohort.
At a median follow-up of 21 months, 110 patients remain failure free, 32% from arm A, 54% from arm B, and 65% from arm C. Failure in 97% of the cases was due to primary site involvement, usually the brain.
Fifty-six percent of the patients are still alive, 39% in arm A, 59% in arm B, and 69% in arm C.
Ninety-seven patients died, 73 from lymphoma, 15 from toxicity of the first-line treatment, 2 from toxicity of the salvage regimen, 2 from neurotoxicity, and 5 from other causes.
Dr Ferreri concluded that the MATRIX regimen—the addition of rituximab and thiotepa to methotrexate and cytarabine—was associated with significant improvements in CR rate, ORR, and progression-free and overall survival.
The addition of rituximab and thiotepa did not increase toxicity, with the exception of greater hematologic adverse events, nor did the drugs increase the rates of severe complications. The agents also allowed for high rates of successful stem cell collection. ![]()
site of 13-ICML
LUGANO—Adding thiotepa and rituximab to the treatment of primary central nervous system (CNS) lymphoma is feasible from a safety perspective and has yielded promising results, according to new research.
An analysis of the IELSG 32 trial—in which patients received methotrexate and cytarabine alone, with rituximab, or with rituximab and thiotepa—has shown the 4-drug regimen improves responses and progression-free survival.
IELSG 32 builds on the foundation of the IELSG 20 trial to determine the best induction therapy for patients with primary CNS lymphoma. In IELSG 20, the complete response (CR) rate was higher among patients receiving high-dose methotrexate and cytarabine than in patients receiving methotrexate alone.
So the researchers decided to investigate whether rituximab and/or thiotepa added to the regimen would impact patient outcome. Rituximab has been associated with improved CR rates in primary CNS lymphoma, and thiotepa is active against aggressive lymphomas. Thiotepa is also able to cross the blood–brain barrier.
Andrés J.M. Ferreri, MD, of IRCCS Ospedale San Raffaele in Milan, Italy, reported the results of this research on behalf of the International Extranodal Lymphoma Study Group (IELSG) at the 13th International Conference on Malignant Lymphoma (abstract 009).
Treatment and toxicity
The investigators randomized 227 patients from 53 centers in 5 countries to 4 cycles of treatment in 3 cohorts:
- Arm A consisted of 4 cycles of methotrexate (3.5 g/m2) on day 1 and cytarabine (2 g/m2) twice a day on days 2 and 3 every 3 weeks.
- Arm B added rituximab (375 mg/m2) on days 5 and 0 to the regimen.
- Arm C added rituximab and thiotepa (30 mg/m2) on day 4 to the regimen.
Eight patients were excluded for various reasons, including incorrect diagnosis. So 219 patients received therapy, 75 in arm A, 69 in arm B, and 75 in arm C.
Patients were a median age of 58 years in arm A and 57 in arms B and C, and the majority were male. More than 80% of patients in all arms were intermediate- or high-risk according to IELSG. And all patients had the diffuse large B-cell lymphoma histotype.
Patients in arm A received 74% of the planned courses, those in arm B received 86%, and those in arm C received 91%. The relative dose intensity across all arms of each drug administered was not significantly different among the arms.
“As expected, hematologic toxicity was common, with grade 4 neutropenia (P=0.01) and thrombocytopenia (P=0.0001) being significantly more common for arm C,” Dr Ferreri said. “However, this was not associated with an increasing array of severe infections, interruptions, toxicity, or toxic deaths.”
Peripheral blood stem cell collection was successful in 94% of patients in arms A and C and 96% of patients in arm B.
Response rates
The overall response rate (ORR) was 72% in IELSG 32, compared to 54% in IELSG 20. But the CR rate was not significantly higher in IELSG 32 than in IELSG 20, at 34% and 31%, respectively.
“There are many explanations for this,” Dr Ferreri said. “But I think it was because there were a significantly greater number of patients with higher IELSG risk [in IELSG 32], a higher proportion of poor-prognosis patients.”
Patients in arm C had significantly higher response rates than patients in the other 2 arms, with a CR rate of 49%, a partial response (PR) rate of 37%, and an ORR of 87%.
This compared to a CR rate of 23% in arm A and 30% in arm B, a PR rate of 31% in arm A and 30% in arm B, and an ORR of 53% in arm A and 74% in arm B.
The investigators also analyzed activity according to IELSG risk.
“Arm C was significantly more active in all the 3 subgroups,” Dr Ferreri observed, “and importantly, the overall response rate and complete remission rate were similar for each arm in the 3 different risk groups.”
Second randomization and survival
One hundred and eighteen patients went on to a second randomization, 35 from arm A, 35 from arm B, and 48 from arm C.
Fifty-nine patients were randomized to the whole-brain radiotherapy cohort, and 59 to the carmustine-thiotepa-autologous stem cell transplant cohort.
At a median follow-up of 21 months, 110 patients remain failure free, 32% from arm A, 54% from arm B, and 65% from arm C. Failure in 97% of the cases was due to primary site involvement, usually the brain.
Fifty-six percent of the patients are still alive, 39% in arm A, 59% in arm B, and 69% in arm C.
Ninety-seven patients died, 73 from lymphoma, 15 from toxicity of the first-line treatment, 2 from toxicity of the salvage regimen, 2 from neurotoxicity, and 5 from other causes.
Dr Ferreri concluded that the MATRIX regimen—the addition of rituximab and thiotepa to methotrexate and cytarabine—was associated with significant improvements in CR rate, ORR, and progression-free and overall survival.
The addition of rituximab and thiotepa did not increase toxicity, with the exception of greater hematologic adverse events, nor did the drugs increase the rates of severe complications. The agents also allowed for high rates of successful stem cell collection. ![]()
Bridge therapy not necessary in AFib, team says

TORONTO—Bridge anticoagulant therapy appears to be unnecessary in patients with atrial fibrillation (AFib) undergoing elective surgery, according to researchers.
In the BRIDGE study, AFib patients who stopped all anticoagulant therapy before elective surgery had no higher risk of thrombosis and a lower risk of major bleeding than patients who were given bridge therapy with low-molecular weight heparin after stopping warfarin.
This research was presented at the 2015 ISTH Congress (abstract LB002) and published in NEJM.
“Bridging has been controversial because there has been a lack of data demonstrating that it’s necessary, so people don’t know what to do,” said study author Thomas L. Ortel, MD, PhD, of Duke University Medical Center in Durham, North Carolina.
“You can go to 5 different doctors, and some will bridge, and others won’t. It just depends on what they feel they can safely do. This trial gives a firm answer to that question.”
The trial enrolled 1884 patients with AFib and atrial flutter. Roughly half received bridge therapy with dalteparin, and the other half received a placebo while halting their warfarin for up to 13 days around their elective surgeries. Patients were followed for up to 37 days after their procedures.
Among patients who stopped all anticoagulant therapy, the incidence of arterial thrombosis was 0.4%, compared to 0.3% for patients who received bridge therapy (P=0.01 for noninferiority).
Major bleeding events were significantly less common among the non-bridging group. They occurred in 1.3% of patients who were not on anticoagulant therapy and 3.2% of patients who received bridge therapy (P=0.005 for superiority).
“Bridging does not improve the outcome for stroke prevention but increases the risk of major bleeding complications,” Dr Ortel said. “That’s the counter balance. We’re not doing patients any good, and we are potentially hurting them.”
Dr Ortel noted that these findings are specific to AFib patients who take warfarin and should not be generalized to other types of patients or other anticoagulants. But the results will be taken into consideration by organizations that develop guidelines.
“This is the first study to provide high-quality clinical trial data demonstrating that, for patients with atrial fibrillation who need a procedure and who need to come off warfarin, they can simply stop and restart,” Dr Ortel said. “They do not need to be bridged.” ![]()
TORONTO—Bridge anticoagulant therapy appears to be unnecessary in patients with atrial fibrillation (AFib) undergoing elective surgery, according to researchers.
In the BRIDGE study, AFib patients who stopped all anticoagulant therapy before elective surgery had no higher risk of thrombosis and a lower risk of major bleeding than patients who were given bridge therapy with low-molecular weight heparin after stopping warfarin.
This research was presented at the 2015 ISTH Congress (abstract LB002) and published in NEJM.
“Bridging has been controversial because there has been a lack of data demonstrating that it’s necessary, so people don’t know what to do,” said study author Thomas L. Ortel, MD, PhD, of Duke University Medical Center in Durham, North Carolina.
“You can go to 5 different doctors, and some will bridge, and others won’t. It just depends on what they feel they can safely do. This trial gives a firm answer to that question.”
The trial enrolled 1884 patients with AFib and atrial flutter. Roughly half received bridge therapy with dalteparin, and the other half received a placebo while halting their warfarin for up to 13 days around their elective surgeries. Patients were followed for up to 37 days after their procedures.
Among patients who stopped all anticoagulant therapy, the incidence of arterial thrombosis was 0.4%, compared to 0.3% for patients who received bridge therapy (P=0.01 for noninferiority).
Major bleeding events were significantly less common among the non-bridging group. They occurred in 1.3% of patients who were not on anticoagulant therapy and 3.2% of patients who received bridge therapy (P=0.005 for superiority).
“Bridging does not improve the outcome for stroke prevention but increases the risk of major bleeding complications,” Dr Ortel said. “That’s the counter balance. We’re not doing patients any good, and we are potentially hurting them.”
Dr Ortel noted that these findings are specific to AFib patients who take warfarin and should not be generalized to other types of patients or other anticoagulants. But the results will be taken into consideration by organizations that develop guidelines.
“This is the first study to provide high-quality clinical trial data demonstrating that, for patients with atrial fibrillation who need a procedure and who need to come off warfarin, they can simply stop and restart,” Dr Ortel said. “They do not need to be bridged.” ![]()
TORONTO—Bridge anticoagulant therapy appears to be unnecessary in patients with atrial fibrillation (AFib) undergoing elective surgery, according to researchers.
In the BRIDGE study, AFib patients who stopped all anticoagulant therapy before elective surgery had no higher risk of thrombosis and a lower risk of major bleeding than patients who were given bridge therapy with low-molecular weight heparin after stopping warfarin.
This research was presented at the 2015 ISTH Congress (abstract LB002) and published in NEJM.
“Bridging has been controversial because there has been a lack of data demonstrating that it’s necessary, so people don’t know what to do,” said study author Thomas L. Ortel, MD, PhD, of Duke University Medical Center in Durham, North Carolina.
“You can go to 5 different doctors, and some will bridge, and others won’t. It just depends on what they feel they can safely do. This trial gives a firm answer to that question.”
The trial enrolled 1884 patients with AFib and atrial flutter. Roughly half received bridge therapy with dalteparin, and the other half received a placebo while halting their warfarin for up to 13 days around their elective surgeries. Patients were followed for up to 37 days after their procedures.
Among patients who stopped all anticoagulant therapy, the incidence of arterial thrombosis was 0.4%, compared to 0.3% for patients who received bridge therapy (P=0.01 for noninferiority).
Major bleeding events were significantly less common among the non-bridging group. They occurred in 1.3% of patients who were not on anticoagulant therapy and 3.2% of patients who received bridge therapy (P=0.005 for superiority).
“Bridging does not improve the outcome for stroke prevention but increases the risk of major bleeding complications,” Dr Ortel said. “That’s the counter balance. We’re not doing patients any good, and we are potentially hurting them.”
Dr Ortel noted that these findings are specific to AFib patients who take warfarin and should not be generalized to other types of patients or other anticoagulants. But the results will be taken into consideration by organizations that develop guidelines.
“This is the first study to provide high-quality clinical trial data demonstrating that, for patients with atrial fibrillation who need a procedure and who need to come off warfarin, they can simply stop and restart,” Dr Ortel said. “They do not need to be bridged.” ![]()
FDA grants vaccine orphan designation for MM
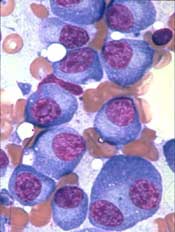
The US Food and Drug Administration (FDA) has granted a novel vaccine orphan designation as a treatment for multiple myeloma (MM).
The vaccine, known as ImMucin, targets the signal peptide domain of the MUC1 tumor antigen.
ImMucin works by “teaching” the immune system to identify and destroy cells that display a short, specific, 21-mer portion from MUC1, which appears on 90% of all cancer cells but not in patients’ blood.
Results of a phase 1/2 trial suggested that ImMucin was safe and active in MM patients. The trial included 15 MUC1-positive patients who had residual or biochemically progressive disease after autologous stem cell transplant.
The patients received 6 or 12 bi-weekly intradermal doses of ImMucin co-administered with human granulocyte-macrophage colony-stimulating factor.
The researchers said the vaccine was well-tolerated, as all adverse events were temporary, grade 1-2 in nature, and resolved spontaneously.
There was a significant decrease in soluble MUC1 levels in 9 patients, and 11 patients had stable disease or clinical improvement that persisted for 17.5 months to more than 41.3 months.
A follow-on study (which is ongoing) in patients who responded to ImMucin has shown that some patients can go more than 4 years without requiring any further treatment for their disease.
ImMucin is under development by Vaxil Biotherapeutics Ltd. The vaccine also has orphan designation as a treatment for MM in the European Union.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If the FDA approves ImMucin to treat patients with MM, orphan designation will provide Vaxil Biotherapeutics with 7 years of marketing exclusivity in the US. ![]()
The US Food and Drug Administration (FDA) has granted a novel vaccine orphan designation as a treatment for multiple myeloma (MM).
The vaccine, known as ImMucin, targets the signal peptide domain of the MUC1 tumor antigen.
ImMucin works by “teaching” the immune system to identify and destroy cells that display a short, specific, 21-mer portion from MUC1, which appears on 90% of all cancer cells but not in patients’ blood.
Results of a phase 1/2 trial suggested that ImMucin was safe and active in MM patients. The trial included 15 MUC1-positive patients who had residual or biochemically progressive disease after autologous stem cell transplant.
The patients received 6 or 12 bi-weekly intradermal doses of ImMucin co-administered with human granulocyte-macrophage colony-stimulating factor.
The researchers said the vaccine was well-tolerated, as all adverse events were temporary, grade 1-2 in nature, and resolved spontaneously.
There was a significant decrease in soluble MUC1 levels in 9 patients, and 11 patients had stable disease or clinical improvement that persisted for 17.5 months to more than 41.3 months.
A follow-on study (which is ongoing) in patients who responded to ImMucin has shown that some patients can go more than 4 years without requiring any further treatment for their disease.
ImMucin is under development by Vaxil Biotherapeutics Ltd. The vaccine also has orphan designation as a treatment for MM in the European Union.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If the FDA approves ImMucin to treat patients with MM, orphan designation will provide Vaxil Biotherapeutics with 7 years of marketing exclusivity in the US. ![]()
The US Food and Drug Administration (FDA) has granted a novel vaccine orphan designation as a treatment for multiple myeloma (MM).
The vaccine, known as ImMucin, targets the signal peptide domain of the MUC1 tumor antigen.
ImMucin works by “teaching” the immune system to identify and destroy cells that display a short, specific, 21-mer portion from MUC1, which appears on 90% of all cancer cells but not in patients’ blood.
Results of a phase 1/2 trial suggested that ImMucin was safe and active in MM patients. The trial included 15 MUC1-positive patients who had residual or biochemically progressive disease after autologous stem cell transplant.
The patients received 6 or 12 bi-weekly intradermal doses of ImMucin co-administered with human granulocyte-macrophage colony-stimulating factor.
The researchers said the vaccine was well-tolerated, as all adverse events were temporary, grade 1-2 in nature, and resolved spontaneously.
There was a significant decrease in soluble MUC1 levels in 9 patients, and 11 patients had stable disease or clinical improvement that persisted for 17.5 months to more than 41.3 months.
A follow-on study (which is ongoing) in patients who responded to ImMucin has shown that some patients can go more than 4 years without requiring any further treatment for their disease.
ImMucin is under development by Vaxil Biotherapeutics Ltd. The vaccine also has orphan designation as a treatment for MM in the European Union.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If the FDA approves ImMucin to treat patients with MM, orphan designation will provide Vaxil Biotherapeutics with 7 years of marketing exclusivity in the US. ![]()
Are clinical part-timers less well liked?
Despite all its glamour and opportunities to write columns like this one, primary care does not attract as many clinicians as it needs to provide for the aging population. Some have proposed that this is because when learners rotate with us, they witness frustration with preauthorizations and physician-patient relationships poisoned by opioid addiction – not the intangible spiritual fulfillment of long-term relationships with people who share their lives with us.
In addition, many primary care providers have other competing interests that take them away from practice. This trend will likely increase as practitioners work beyond the age of 65 years but at reduced hours. These demands naturally decrease patient access and can theoretically lead to dissatisfaction, which is potentially devastating if we are reimbursed based upon satisfaction scores.
So, do reduced hours frustrate patients?
Laura Panattoni, Ph.D., and her colleagues at the Palo Alto Medical Foundation Research Institute, Mountain View, Calif., evaluated the relationship between physicians’ clinical time, continuity of care, access to care, and patient satisfaction with the physician (J. Gen. Intern. Med. 2015;30:327-33). The study was a cross-section survey of physicians in family and internal medicine and their patients.
The investigators found that greater office time was directly associated with increased continuity and access but with lower patient satisfaction scores. Restated, reduced clinical hours were associated with improved patient satisfaction.
These findings are interesting and important at many levels. First, they suggest that clinicians who choose less than a full-time clinical obligation can keep their patients happy. Second, we can hypothesize that what is lost in continuity and access is made up for in effective communication delivered by clinicians who are happy themselves. Third, practice redesign should not require full-time commitment to deliver on the satisfaction side of the equation. The world is clamoring for alternative care models where electronic “touches” alleviate the pressure for “patients in rooms.” Studies have shown that up to 93% of patients would select a physician who allows them to communicate with them electronically. About 450,000 patients will see a doctor through the Internet this year. UnitedHealth Group started covering telemedicine and plans to expand this to 20 million customers next year.
I personally spend one-third of my time seeing patients in rooms, but I am electronically and telephonically accessible to them every day at all times. Maybe this helps keep my patients happy, despite me not being in the office every day.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified, practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow Dr. Ebbert on Twitter @jonebbert.
Despite all its glamour and opportunities to write columns like this one, primary care does not attract as many clinicians as it needs to provide for the aging population. Some have proposed that this is because when learners rotate with us, they witness frustration with preauthorizations and physician-patient relationships poisoned by opioid addiction – not the intangible spiritual fulfillment of long-term relationships with people who share their lives with us.
In addition, many primary care providers have other competing interests that take them away from practice. This trend will likely increase as practitioners work beyond the age of 65 years but at reduced hours. These demands naturally decrease patient access and can theoretically lead to dissatisfaction, which is potentially devastating if we are reimbursed based upon satisfaction scores.
So, do reduced hours frustrate patients?
Laura Panattoni, Ph.D., and her colleagues at the Palo Alto Medical Foundation Research Institute, Mountain View, Calif., evaluated the relationship between physicians’ clinical time, continuity of care, access to care, and patient satisfaction with the physician (J. Gen. Intern. Med. 2015;30:327-33). The study was a cross-section survey of physicians in family and internal medicine and their patients.
The investigators found that greater office time was directly associated with increased continuity and access but with lower patient satisfaction scores. Restated, reduced clinical hours were associated with improved patient satisfaction.
These findings are interesting and important at many levels. First, they suggest that clinicians who choose less than a full-time clinical obligation can keep their patients happy. Second, we can hypothesize that what is lost in continuity and access is made up for in effective communication delivered by clinicians who are happy themselves. Third, practice redesign should not require full-time commitment to deliver on the satisfaction side of the equation. The world is clamoring for alternative care models where electronic “touches” alleviate the pressure for “patients in rooms.” Studies have shown that up to 93% of patients would select a physician who allows them to communicate with them electronically. About 450,000 patients will see a doctor through the Internet this year. UnitedHealth Group started covering telemedicine and plans to expand this to 20 million customers next year.
I personally spend one-third of my time seeing patients in rooms, but I am electronically and telephonically accessible to them every day at all times. Maybe this helps keep my patients happy, despite me not being in the office every day.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified, practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow Dr. Ebbert on Twitter @jonebbert.
Despite all its glamour and opportunities to write columns like this one, primary care does not attract as many clinicians as it needs to provide for the aging population. Some have proposed that this is because when learners rotate with us, they witness frustration with preauthorizations and physician-patient relationships poisoned by opioid addiction – not the intangible spiritual fulfillment of long-term relationships with people who share their lives with us.
In addition, many primary care providers have other competing interests that take them away from practice. This trend will likely increase as practitioners work beyond the age of 65 years but at reduced hours. These demands naturally decrease patient access and can theoretically lead to dissatisfaction, which is potentially devastating if we are reimbursed based upon satisfaction scores.
So, do reduced hours frustrate patients?
Laura Panattoni, Ph.D., and her colleagues at the Palo Alto Medical Foundation Research Institute, Mountain View, Calif., evaluated the relationship between physicians’ clinical time, continuity of care, access to care, and patient satisfaction with the physician (J. Gen. Intern. Med. 2015;30:327-33). The study was a cross-section survey of physicians in family and internal medicine and their patients.
The investigators found that greater office time was directly associated with increased continuity and access but with lower patient satisfaction scores. Restated, reduced clinical hours were associated with improved patient satisfaction.
These findings are interesting and important at many levels. First, they suggest that clinicians who choose less than a full-time clinical obligation can keep their patients happy. Second, we can hypothesize that what is lost in continuity and access is made up for in effective communication delivered by clinicians who are happy themselves. Third, practice redesign should not require full-time commitment to deliver on the satisfaction side of the equation. The world is clamoring for alternative care models where electronic “touches” alleviate the pressure for “patients in rooms.” Studies have shown that up to 93% of patients would select a physician who allows them to communicate with them electronically. About 450,000 patients will see a doctor through the Internet this year. UnitedHealth Group started covering telemedicine and plans to expand this to 20 million customers next year.
I personally spend one-third of my time seeing patients in rooms, but I am electronically and telephonically accessible to them every day at all times. Maybe this helps keep my patients happy, despite me not being in the office every day.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified, practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow Dr. Ebbert on Twitter @jonebbert.

Anticoagulation Therapy Probably Not Needed While A-fib Patients Undergo Surgery
NEW YORK (Reuters Health) - When patients with atrialfibrillation need surgery or other invasive therapy, doctors can safely interrupt their warfarin therapy without offering a bridging anticoagulation regimen, according to a new U.S.-Canadian study.
The test of 1,884 patients treated at 108 centers found virtually identical rates of arterial thromboembolism in people who were switched to low-molecular-weight heparin and volunteers who got matching placebo twice daily immediately before and after the procedure.
In contrast, the incidence of major bleeding nearly tripled with bridging; it was 1.3% without heparin and 3.2% with the anticoagulant (p=0.005).
The study is "the first to give us really high-quality data. It allows us to make a recommendation that, at least with atrial fibrillation, you don't need to use bridging therapy," coauthor Dr. Thomas Ortel, chief of hematology at Duke University in Durham, North Carolina, told Reuters Health by phone.
"There may be certain high-risk patients where you may still wish to use it, but the vast majority of these patients really don't need anything done in the perioperative setting," he said.
The findings were presented Monday at the International Society on Thrombosis and Haemostasis 2015 Congress in Toronto and released online by the New England Journal of Medicine.
This is an issue doctors have wrestled with for years.
"Each year, this common clinical scenario affects approximately one in six warfarin-treated patients with atrial fibrillation," the researchers wrote in the Journal.
"Recommendations and guidelines have been conflicting," said Dr. Ortel. "We feel this is something more definitive."
In all patients, warfarin was halted five days before the procedure and restarted within 24 hours after the procedure.
In the bridging group, patients subcutaneously received 100 IU of dalteparin per kilogram of body weight or matching placebo twice daily, beginning three days before the procedure. Treatment was suspended 24 hours before the procedure.
Dalteparin/placebo therapy resumed within 24 hours after the procedure if that procedure carried a low risk of bleeding, and within 48 to 72 hours if the bleeding risk was high. Treatment continued until the international normalized ratio hit 2.0, usually for five to 10 days. Patients were followed for 30 to 37 days after the procedure.
In the placebo group, 0.4% developed an arterial thromboembolism versus 0.3% of the patients who received the heparin (p=0.01 for noninferiority). The rates were based on 1,813 patients who remained in the study until the end.
"For many of the patients it actually is not surprising because the risk of thromboembolism is pretty low," said Dr. Ortel. "But the issue has been that nobody has shown that you really can skip low-molecular-weight heparin as a bridge. There's quite a bit of conflicting thought out there among providers, as well as among patients, about whether something like this needs to be done."
"Major bleeding occurred in 1.3% of the patients (12 of 918) in the no-bridging group and 3.2% (29 of 895) in the bridging group, which indicated that no bridging was superior to bridging with regard to major bleeding," the researchers reported. None of the major bleeding episodes was fatal.
The two treatments produced no difference in the likelihood of death, acute myocardial infarction, pulmonary embolism ordeep vein thrombosis.
Dr. Ortel said the findings, if widely adopted, could save money by reducing the use of low-molecular-weight heparin and decreasing the expense of treating bleeding complications. "There are several different ways this could affect costs.
He cautioned that the study, known as BRIDGE, "does not apply to patients with prosthetic heart valves or patients with venous thromboembolism, like a deep vein thrombosis or pulmonary embolism. But for patients with atrial fibrillation, it's high-quality data that should indicate that people do not need to use anything to bridge these folks."
The National Heart, Lung, and Blood Institute funded this research. Ten coauthors reported relationships with pharmaceutical companies.
—Reuters Health
NEW YORK (Reuters Health) - When patients with atrialfibrillation need surgery or other invasive therapy, doctors can safely interrupt their warfarin therapy without offering a bridging anticoagulation regimen, according to a new U.S.-Canadian study.
The test of 1,884 patients treated at 108 centers found virtually identical rates of arterial thromboembolism in people who were switched to low-molecular-weight heparin and volunteers who got matching placebo twice daily immediately before and after the procedure.
In contrast, the incidence of major bleeding nearly tripled with bridging; it was 1.3% without heparin and 3.2% with the anticoagulant (p=0.005).
The study is "the first to give us really high-quality data. It allows us to make a recommendation that, at least with atrial fibrillation, you don't need to use bridging therapy," coauthor Dr. Thomas Ortel, chief of hematology at Duke University in Durham, North Carolina, told Reuters Health by phone.
"There may be certain high-risk patients where you may still wish to use it, but the vast majority of these patients really don't need anything done in the perioperative setting," he said.
The findings were presented Monday at the International Society on Thrombosis and Haemostasis 2015 Congress in Toronto and released online by the New England Journal of Medicine.
This is an issue doctors have wrestled with for years.
"Each year, this common clinical scenario affects approximately one in six warfarin-treated patients with atrial fibrillation," the researchers wrote in the Journal.
"Recommendations and guidelines have been conflicting," said Dr. Ortel. "We feel this is something more definitive."
In all patients, warfarin was halted five days before the procedure and restarted within 24 hours after the procedure.
In the bridging group, patients subcutaneously received 100 IU of dalteparin per kilogram of body weight or matching placebo twice daily, beginning three days before the procedure. Treatment was suspended 24 hours before the procedure.
Dalteparin/placebo therapy resumed within 24 hours after the procedure if that procedure carried a low risk of bleeding, and within 48 to 72 hours if the bleeding risk was high. Treatment continued until the international normalized ratio hit 2.0, usually for five to 10 days. Patients were followed for 30 to 37 days after the procedure.
In the placebo group, 0.4% developed an arterial thromboembolism versus 0.3% of the patients who received the heparin (p=0.01 for noninferiority). The rates were based on 1,813 patients who remained in the study until the end.
"For many of the patients it actually is not surprising because the risk of thromboembolism is pretty low," said Dr. Ortel. "But the issue has been that nobody has shown that you really can skip low-molecular-weight heparin as a bridge. There's quite a bit of conflicting thought out there among providers, as well as among patients, about whether something like this needs to be done."
"Major bleeding occurred in 1.3% of the patients (12 of 918) in the no-bridging group and 3.2% (29 of 895) in the bridging group, which indicated that no bridging was superior to bridging with regard to major bleeding," the researchers reported. None of the major bleeding episodes was fatal.
The two treatments produced no difference in the likelihood of death, acute myocardial infarction, pulmonary embolism ordeep vein thrombosis.
Dr. Ortel said the findings, if widely adopted, could save money by reducing the use of low-molecular-weight heparin and decreasing the expense of treating bleeding complications. "There are several different ways this could affect costs.
He cautioned that the study, known as BRIDGE, "does not apply to patients with prosthetic heart valves or patients with venous thromboembolism, like a deep vein thrombosis or pulmonary embolism. But for patients with atrial fibrillation, it's high-quality data that should indicate that people do not need to use anything to bridge these folks."
The National Heart, Lung, and Blood Institute funded this research. Ten coauthors reported relationships with pharmaceutical companies.
—Reuters Health
NEW YORK (Reuters Health) - When patients with atrialfibrillation need surgery or other invasive therapy, doctors can safely interrupt their warfarin therapy without offering a bridging anticoagulation regimen, according to a new U.S.-Canadian study.
The test of 1,884 patients treated at 108 centers found virtually identical rates of arterial thromboembolism in people who were switched to low-molecular-weight heparin and volunteers who got matching placebo twice daily immediately before and after the procedure.
In contrast, the incidence of major bleeding nearly tripled with bridging; it was 1.3% without heparin and 3.2% with the anticoagulant (p=0.005).
The study is "the first to give us really high-quality data. It allows us to make a recommendation that, at least with atrial fibrillation, you don't need to use bridging therapy," coauthor Dr. Thomas Ortel, chief of hematology at Duke University in Durham, North Carolina, told Reuters Health by phone.
"There may be certain high-risk patients where you may still wish to use it, but the vast majority of these patients really don't need anything done in the perioperative setting," he said.
The findings were presented Monday at the International Society on Thrombosis and Haemostasis 2015 Congress in Toronto and released online by the New England Journal of Medicine.
This is an issue doctors have wrestled with for years.
"Each year, this common clinical scenario affects approximately one in six warfarin-treated patients with atrial fibrillation," the researchers wrote in the Journal.
"Recommendations and guidelines have been conflicting," said Dr. Ortel. "We feel this is something more definitive."
In all patients, warfarin was halted five days before the procedure and restarted within 24 hours after the procedure.
In the bridging group, patients subcutaneously received 100 IU of dalteparin per kilogram of body weight or matching placebo twice daily, beginning three days before the procedure. Treatment was suspended 24 hours before the procedure.
Dalteparin/placebo therapy resumed within 24 hours after the procedure if that procedure carried a low risk of bleeding, and within 48 to 72 hours if the bleeding risk was high. Treatment continued until the international normalized ratio hit 2.0, usually for five to 10 days. Patients were followed for 30 to 37 days after the procedure.
In the placebo group, 0.4% developed an arterial thromboembolism versus 0.3% of the patients who received the heparin (p=0.01 for noninferiority). The rates were based on 1,813 patients who remained in the study until the end.
"For many of the patients it actually is not surprising because the risk of thromboembolism is pretty low," said Dr. Ortel. "But the issue has been that nobody has shown that you really can skip low-molecular-weight heparin as a bridge. There's quite a bit of conflicting thought out there among providers, as well as among patients, about whether something like this needs to be done."
"Major bleeding occurred in 1.3% of the patients (12 of 918) in the no-bridging group and 3.2% (29 of 895) in the bridging group, which indicated that no bridging was superior to bridging with regard to major bleeding," the researchers reported. None of the major bleeding episodes was fatal.
The two treatments produced no difference in the likelihood of death, acute myocardial infarction, pulmonary embolism ordeep vein thrombosis.
Dr. Ortel said the findings, if widely adopted, could save money by reducing the use of low-molecular-weight heparin and decreasing the expense of treating bleeding complications. "There are several different ways this could affect costs.
He cautioned that the study, known as BRIDGE, "does not apply to patients with prosthetic heart valves or patients with venous thromboembolism, like a deep vein thrombosis or pulmonary embolism. But for patients with atrial fibrillation, it's high-quality data that should indicate that people do not need to use anything to bridge these folks."
The National Heart, Lung, and Blood Institute funded this research. Ten coauthors reported relationships with pharmaceutical companies.
—Reuters Health
FDA advisors urge physician certification for flibanserin
Without the option of recommending physician certification as a condition for flibanserin approval, the Food and Drug Administration advisory panel vote might have shifted against approval of the drug for treating hypoactive sexual desire disorder in premenopausal women.
At a joint meeting of two FDA advisory panels in June, members of the Bone, Reproductive and Urologic Drugs Advisory Committee and the Drug Safety and Risk Management Advisory Committee voted 18-6 that the overall benefit-risk profile of flibanserin supported approval for treating hypoactive sexual desire disorder (HSDD) in premenopausal women, provided that certain risk management options beyond labeling were implemented. If approved by the FDA, flibanserin would be the first drug approved for treating HSDD.
Assurance that prescribers would be fully apprised of the serious risks of hypotension and syncope associated with the drug, exacerbation of those side effects when combined with alcohol or a CYP3A4 inhibitor – and the modest effects over placebo – was cited by several of the panelists who voted in favor of approval.
All of those voting in favor of approval chose the option of supporting approval “only if certain risk management options beyond labeling are implemented.” None of the panelists voted for the option of supporting approval with “labeling alone to manage the risks.”
The conditions include a risk management plan to address serious adverse effects associated with the drug, a requirement for physician certification, and postmarketing studies to further evaluate and monitor the drug’s safety and efficacy.
The risks of hypotension and syncope, and central nervous system depression are also exacerbated by moderate or strong CYP3A4 inhibitors, but the interaction with alcohol was raised as a particularly serious issue because of the high rate of alcohol use and binge drinking among women who would likely be treated with flibanserin, according to FDA reviewers.

The risk of drug interactions can be mitigated with drug interaction screening programs used in health care systems, such as in electronic medical records and pharmacies, while alcohol use is a patient-dependent behavior, is common among women, and therefore more difficult to control, Kimberly Lehrfeld, Pharm.D., a team leader in the division of risk management in the FDA’s office of medication error prevention and analysis, said at the meeting. Several panelists recommended that alcohol use be a contraindication.
Physician certification is among the Elements to Assure Safe Use or ETASU, which along with a medication guide and a communication plan for health care providers, are components of a Risk Evaluation and Mitigation Strategy (REMS), a way to help manage the risks of a drug or biologic while still making it available to patients who need it.
Physician certification is part of the REMS for drugs such as mifepristone (Mifeprex), thalidomide (Thalomid), and natalizumab (Tysabri).
“A risk strategy that gets physicians the information they need to use it properly is going to be key,” said Dr. Robert Silbergleit, who voted for approval.”A REMS strategy is going to be very important because I think that the most likely risks … are going to come from physicians who don’t use the drug properly because they’re not properly educated.”
Dr. Silbergleit, a professor in the department of emergency medicine at the University of Michigan, Ann Arbor, said he was also concerned about the marketing of the drug. “Clinicians may be in the situation where they have to counter direct-to-consumer marketing that could lead to misuse of the drug,” he said at the meeting.
Also voting for approval, Marjorie Shaw Phillips, R.Ph., pharmacy coordinator at Georgia Regents Medical Center in Augusta, said that everyone needs to be aware of the potential safety concerns. But while she does not think pharmacy registration would be beneficial, there is a role for the pharmacist to confirm that it’s an educated provider prescribing the drug and that they’ve discussed the risks with the patient.
She added that it will be important for physicians to set realistic expectations for patients.
“It’s not a magical little pink pill, and there are going to be a whole lot of women with sexual dysfunction for whom there’s no evidence that it’s going to benefit them,” she said at the meeting.
The panel did not specifically recommend pharmacy certification, but pharmacists would have to verify that the prescribing physicians are certified, if the drug is approved, an FDA official said at the meeting.
A decision from the FDA is expected in August. The FDA panelists reported having no relevant financial disclosures.
Sprout Pharmaceuticals, flibanserin’s manufacturer, said in a statement that the company looks forward “to continuing our work with the FDA as it completes its review of our new drug application, including the discussion of a Risks Evaluation and Mitigation Strategy.” If approved, the company plans to market flibanserin as “Addyi.”
Without the option of recommending physician certification as a condition for flibanserin approval, the Food and Drug Administration advisory panel vote might have shifted against approval of the drug for treating hypoactive sexual desire disorder in premenopausal women.
At a joint meeting of two FDA advisory panels in June, members of the Bone, Reproductive and Urologic Drugs Advisory Committee and the Drug Safety and Risk Management Advisory Committee voted 18-6 that the overall benefit-risk profile of flibanserin supported approval for treating hypoactive sexual desire disorder (HSDD) in premenopausal women, provided that certain risk management options beyond labeling were implemented. If approved by the FDA, flibanserin would be the first drug approved for treating HSDD.
Assurance that prescribers would be fully apprised of the serious risks of hypotension and syncope associated with the drug, exacerbation of those side effects when combined with alcohol or a CYP3A4 inhibitor – and the modest effects over placebo – was cited by several of the panelists who voted in favor of approval.
All of those voting in favor of approval chose the option of supporting approval “only if certain risk management options beyond labeling are implemented.” None of the panelists voted for the option of supporting approval with “labeling alone to manage the risks.”
The conditions include a risk management plan to address serious adverse effects associated with the drug, a requirement for physician certification, and postmarketing studies to further evaluate and monitor the drug’s safety and efficacy.
The risks of hypotension and syncope, and central nervous system depression are also exacerbated by moderate or strong CYP3A4 inhibitors, but the interaction with alcohol was raised as a particularly serious issue because of the high rate of alcohol use and binge drinking among women who would likely be treated with flibanserin, according to FDA reviewers.
The risk of drug interactions can be mitigated with drug interaction screening programs used in health care systems, such as in electronic medical records and pharmacies, while alcohol use is a patient-dependent behavior, is common among women, and therefore more difficult to control, Kimberly Lehrfeld, Pharm.D., a team leader in the division of risk management in the FDA’s office of medication error prevention and analysis, said at the meeting. Several panelists recommended that alcohol use be a contraindication.
Physician certification is among the Elements to Assure Safe Use or ETASU, which along with a medication guide and a communication plan for health care providers, are components of a Risk Evaluation and Mitigation Strategy (REMS), a way to help manage the risks of a drug or biologic while still making it available to patients who need it.
Physician certification is part of the REMS for drugs such as mifepristone (Mifeprex), thalidomide (Thalomid), and natalizumab (Tysabri).
“A risk strategy that gets physicians the information they need to use it properly is going to be key,” said Dr. Robert Silbergleit, who voted for approval.”A REMS strategy is going to be very important because I think that the most likely risks … are going to come from physicians who don’t use the drug properly because they’re not properly educated.”
Dr. Silbergleit, a professor in the department of emergency medicine at the University of Michigan, Ann Arbor, said he was also concerned about the marketing of the drug. “Clinicians may be in the situation where they have to counter direct-to-consumer marketing that could lead to misuse of the drug,” he said at the meeting.
Also voting for approval, Marjorie Shaw Phillips, R.Ph., pharmacy coordinator at Georgia Regents Medical Center in Augusta, said that everyone needs to be aware of the potential safety concerns. But while she does not think pharmacy registration would be beneficial, there is a role for the pharmacist to confirm that it’s an educated provider prescribing the drug and that they’ve discussed the risks with the patient.
She added that it will be important for physicians to set realistic expectations for patients.
“It’s not a magical little pink pill, and there are going to be a whole lot of women with sexual dysfunction for whom there’s no evidence that it’s going to benefit them,” she said at the meeting.
The panel did not specifically recommend pharmacy certification, but pharmacists would have to verify that the prescribing physicians are certified, if the drug is approved, an FDA official said at the meeting.
A decision from the FDA is expected in August. The FDA panelists reported having no relevant financial disclosures.
Sprout Pharmaceuticals, flibanserin’s manufacturer, said in a statement that the company looks forward “to continuing our work with the FDA as it completes its review of our new drug application, including the discussion of a Risks Evaluation and Mitigation Strategy.” If approved, the company plans to market flibanserin as “Addyi.”
Without the option of recommending physician certification as a condition for flibanserin approval, the Food and Drug Administration advisory panel vote might have shifted against approval of the drug for treating hypoactive sexual desire disorder in premenopausal women.
At a joint meeting of two FDA advisory panels in June, members of the Bone, Reproductive and Urologic Drugs Advisory Committee and the Drug Safety and Risk Management Advisory Committee voted 18-6 that the overall benefit-risk profile of flibanserin supported approval for treating hypoactive sexual desire disorder (HSDD) in premenopausal women, provided that certain risk management options beyond labeling were implemented. If approved by the FDA, flibanserin would be the first drug approved for treating HSDD.
Assurance that prescribers would be fully apprised of the serious risks of hypotension and syncope associated with the drug, exacerbation of those side effects when combined with alcohol or a CYP3A4 inhibitor – and the modest effects over placebo – was cited by several of the panelists who voted in favor of approval.
All of those voting in favor of approval chose the option of supporting approval “only if certain risk management options beyond labeling are implemented.” None of the panelists voted for the option of supporting approval with “labeling alone to manage the risks.”
The conditions include a risk management plan to address serious adverse effects associated with the drug, a requirement for physician certification, and postmarketing studies to further evaluate and monitor the drug’s safety and efficacy.
The risks of hypotension and syncope, and central nervous system depression are also exacerbated by moderate or strong CYP3A4 inhibitors, but the interaction with alcohol was raised as a particularly serious issue because of the high rate of alcohol use and binge drinking among women who would likely be treated with flibanserin, according to FDA reviewers.
The risk of drug interactions can be mitigated with drug interaction screening programs used in health care systems, such as in electronic medical records and pharmacies, while alcohol use is a patient-dependent behavior, is common among women, and therefore more difficult to control, Kimberly Lehrfeld, Pharm.D., a team leader in the division of risk management in the FDA’s office of medication error prevention and analysis, said at the meeting. Several panelists recommended that alcohol use be a contraindication.
Physician certification is among the Elements to Assure Safe Use or ETASU, which along with a medication guide and a communication plan for health care providers, are components of a Risk Evaluation and Mitigation Strategy (REMS), a way to help manage the risks of a drug or biologic while still making it available to patients who need it.
Physician certification is part of the REMS for drugs such as mifepristone (Mifeprex), thalidomide (Thalomid), and natalizumab (Tysabri).
“A risk strategy that gets physicians the information they need to use it properly is going to be key,” said Dr. Robert Silbergleit, who voted for approval.”A REMS strategy is going to be very important because I think that the most likely risks … are going to come from physicians who don’t use the drug properly because they’re not properly educated.”
Dr. Silbergleit, a professor in the department of emergency medicine at the University of Michigan, Ann Arbor, said he was also concerned about the marketing of the drug. “Clinicians may be in the situation where they have to counter direct-to-consumer marketing that could lead to misuse of the drug,” he said at the meeting.
Also voting for approval, Marjorie Shaw Phillips, R.Ph., pharmacy coordinator at Georgia Regents Medical Center in Augusta, said that everyone needs to be aware of the potential safety concerns. But while she does not think pharmacy registration would be beneficial, there is a role for the pharmacist to confirm that it’s an educated provider prescribing the drug and that they’ve discussed the risks with the patient.
She added that it will be important for physicians to set realistic expectations for patients.
“It’s not a magical little pink pill, and there are going to be a whole lot of women with sexual dysfunction for whom there’s no evidence that it’s going to benefit them,” she said at the meeting.
The panel did not specifically recommend pharmacy certification, but pharmacists would have to verify that the prescribing physicians are certified, if the drug is approved, an FDA official said at the meeting.
A decision from the FDA is expected in August. The FDA panelists reported having no relevant financial disclosures.
Sprout Pharmaceuticals, flibanserin’s manufacturer, said in a statement that the company looks forward “to continuing our work with the FDA as it completes its review of our new drug application, including the discussion of a Risks Evaluation and Mitigation Strategy.” If approved, the company plans to market flibanserin as “Addyi.”
Cardiovascular disease in oncology
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Measuring end-of-life care in oncology practices: learning from the care of the dying
Background There is increased interest among oncology and palliative professionals in providing appropriately timed hospice services for cancer patients. End of life (EoL) metrics have been included in oncology quality programs, but accurate EoL data and benchmarks are hard to obtain.
Objective To improve EoL care by measuring patterns of care among recently deceased patients.
Methods Care utilization among deceased patients was analyzed by using software integrated with patient electronic health records. The data was verified by chart review.
Results Of 179 cancer deaths, tumor registry data differed from chart review in 7% of cases with regard to dates and/or location of death. Institutional EoL metrics were significantly affected by a large number of patients (37%) with advanced illnesses who had clinical diagnoses of cancer made at the end of life, but who had not been managed by oncologists. This population of patients who had not been managed by oncologists was older, less likely to use hospice, and more likely to use the intensive care unit than were oncologist-managed cancer patients. Among the patients of individual oncologists, the median stay in hospice ranged from 6-28 days. Data collection and chart review took an average of 27 minutes per case with combined efforts by a data analyst and oncology practitioner.
Limitations Single institution with comprehensive electronic medical record; some patients were treated outside of the system.
Conclusion Acquiring accurate data on EoL metrics is time consuming. Compared with chart review, other data sources have inaccuracies and include some patients who have not been managed by oncologists. Accurate attribution to individual physicians requires chart review by an experienced clinician.
Click on the PDF icon at the top of this introduction to read the full article.
Background There is increased interest among oncology and palliative professionals in providing appropriately timed hospice services for cancer patients. End of life (EoL) metrics have been included in oncology quality programs, but accurate EoL data and benchmarks are hard to obtain.
Objective To improve EoL care by measuring patterns of care among recently deceased patients.
Methods Care utilization among deceased patients was analyzed by using software integrated with patient electronic health records. The data was verified by chart review.
Results Of 179 cancer deaths, tumor registry data differed from chart review in 7% of cases with regard to dates and/or location of death. Institutional EoL metrics were significantly affected by a large number of patients (37%) with advanced illnesses who had clinical diagnoses of cancer made at the end of life, but who had not been managed by oncologists. This population of patients who had not been managed by oncologists was older, less likely to use hospice, and more likely to use the intensive care unit than were oncologist-managed cancer patients. Among the patients of individual oncologists, the median stay in hospice ranged from 6-28 days. Data collection and chart review took an average of 27 minutes per case with combined efforts by a data analyst and oncology practitioner.
Limitations Single institution with comprehensive electronic medical record; some patients were treated outside of the system.
Conclusion Acquiring accurate data on EoL metrics is time consuming. Compared with chart review, other data sources have inaccuracies and include some patients who have not been managed by oncologists. Accurate attribution to individual physicians requires chart review by an experienced clinician.
Click on the PDF icon at the top of this introduction to read the full article.
Background There is increased interest among oncology and palliative professionals in providing appropriately timed hospice services for cancer patients. End of life (EoL) metrics have been included in oncology quality programs, but accurate EoL data and benchmarks are hard to obtain.
Objective To improve EoL care by measuring patterns of care among recently deceased patients.
Methods Care utilization among deceased patients was analyzed by using software integrated with patient electronic health records. The data was verified by chart review.
Results Of 179 cancer deaths, tumor registry data differed from chart review in 7% of cases with regard to dates and/or location of death. Institutional EoL metrics were significantly affected by a large number of patients (37%) with advanced illnesses who had clinical diagnoses of cancer made at the end of life, but who had not been managed by oncologists. This population of patients who had not been managed by oncologists was older, less likely to use hospice, and more likely to use the intensive care unit than were oncologist-managed cancer patients. Among the patients of individual oncologists, the median stay in hospice ranged from 6-28 days. Data collection and chart review took an average of 27 minutes per case with combined efforts by a data analyst and oncology practitioner.
Limitations Single institution with comprehensive electronic medical record; some patients were treated outside of the system.
Conclusion Acquiring accurate data on EoL metrics is time consuming. Compared with chart review, other data sources have inaccuracies and include some patients who have not been managed by oncologists. Accurate attribution to individual physicians requires chart review by an experienced clinician.
Click on the PDF icon at the top of this introduction to read the full article.
Impact of nurse navigation on timeliness of diagnostic medical services in patients with newly diagnosed lung cancer
Background The Summa Cancer Institute in Akron, Ohio, sought to improve access to and the timeliness of lung cancer care by hiring an oncology-certified nurse navigator. The nurse navigator was charged with coordinating diagnostic procedures and specialty oncology consultations, and with facilitating a multidisciplinary thoracic oncology tumor board.
Objective To test the hypothesis that nurse navigation would improve the timeliness of and access to diagnostic medical services among men and women with newly diagnosed lung cancer.
Methods A conducted a retrospective review of 460 patients with lung cancer to evaluate access to care and the timeliness of the care received in the non-navigated and nurse-navigated cohorts.
Results During December 2009-September 2013, the time between the suspicion of cancer on chest X-ray to treatment was 64 days. During October 2013-March 2014, the nurse navigator helped reduce that timespan to 45 days (P < .001).
Limitations Long-term follow-up on clinical outcomes remains premature.
Conclusion This finding attests to the successful implementation of nurse navigation to improve access and timeliness of lung cancer care in a community oncology practice.
Click on the PDF icon at the top of this introduction to read the full article.
Background The Summa Cancer Institute in Akron, Ohio, sought to improve access to and the timeliness of lung cancer care by hiring an oncology-certified nurse navigator. The nurse navigator was charged with coordinating diagnostic procedures and specialty oncology consultations, and with facilitating a multidisciplinary thoracic oncology tumor board.
Objective To test the hypothesis that nurse navigation would improve the timeliness of and access to diagnostic medical services among men and women with newly diagnosed lung cancer.
Methods A conducted a retrospective review of 460 patients with lung cancer to evaluate access to care and the timeliness of the care received in the non-navigated and nurse-navigated cohorts.
Results During December 2009-September 2013, the time between the suspicion of cancer on chest X-ray to treatment was 64 days. During October 2013-March 2014, the nurse navigator helped reduce that timespan to 45 days (P < .001).
Limitations Long-term follow-up on clinical outcomes remains premature.
Conclusion This finding attests to the successful implementation of nurse navigation to improve access and timeliness of lung cancer care in a community oncology practice.
Click on the PDF icon at the top of this introduction to read the full article.
Background The Summa Cancer Institute in Akron, Ohio, sought to improve access to and the timeliness of lung cancer care by hiring an oncology-certified nurse navigator. The nurse navigator was charged with coordinating diagnostic procedures and specialty oncology consultations, and with facilitating a multidisciplinary thoracic oncology tumor board.
Objective To test the hypothesis that nurse navigation would improve the timeliness of and access to diagnostic medical services among men and women with newly diagnosed lung cancer.
Methods A conducted a retrospective review of 460 patients with lung cancer to evaluate access to care and the timeliness of the care received in the non-navigated and nurse-navigated cohorts.
Results During December 2009-September 2013, the time between the suspicion of cancer on chest X-ray to treatment was 64 days. During October 2013-March 2014, the nurse navigator helped reduce that timespan to 45 days (P < .001).
Limitations Long-term follow-up on clinical outcomes remains premature.
Conclusion This finding attests to the successful implementation of nurse navigation to improve access and timeliness of lung cancer care in a community oncology practice.
Click on the PDF icon at the top of this introduction to read the full article.