User login
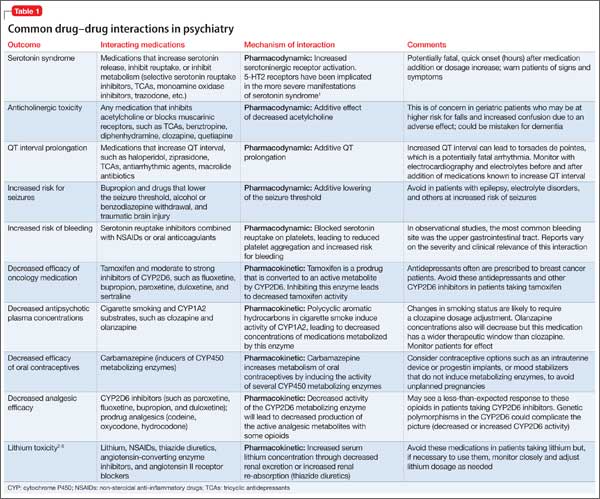
Avoiding common drug−drug interactions
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight

hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
Reducing medical comorbidity and mortality in severe mental illness
People with serious mental illness (SMI) have a life expectancy that is 25 years less than the general population, according to the Centers for Disease Control and Prevention.1 This disparity is partially a consequence of the lack of primary and preventive medical care for those with psychiatric illness. Decades of research have shown that people with SMI experience higher medical morbidity and mortality in addition to facing the stigma of mental illness.
This article aims to advance the idea that longitudinal “cross education” between primary care providers (PCPs) and behavioral health providers (BHPs) is essential in addressing this problem. BHPs include psychiatry clinics, which often are part of a university or large health systems; county-based community mental health programs; and independent mental health clinics that contract with public and private health plans to provide mental health services.
Although suicide and injury account for 40% of the excess mortality in schizophrenia, 60% can be attributed to cardiovascular disease, diabetes, respiratory diseases, and infection.2 Patients with SMI have 2 to 3 times the risk of diabetes, dyslipidemia, hypertension, and obesity.3,4 Furthermore, those with SMI consume more than one-third of tobacco products,5 and 50% to 80% of people with SMI smoke tobacco, an important reversible risk factor for cardiovascular disease.
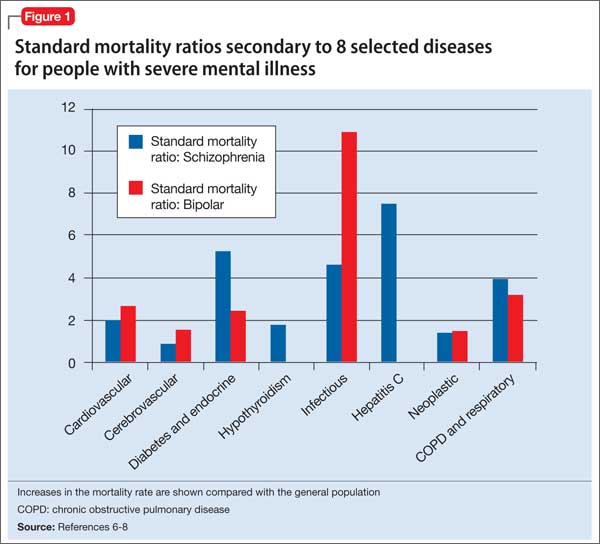
Figure 1 shows that people with SMI are at higher risk of dying from a chronic medical condition, such as cardiovascular disease, diabetes, chronic obstructive pulmonary disease, and hepatitis C6-8—many of which can be managed by primary and preventive medical interventions. These and other conditions often are not diagnosed or effectively managed in patients with SMI.
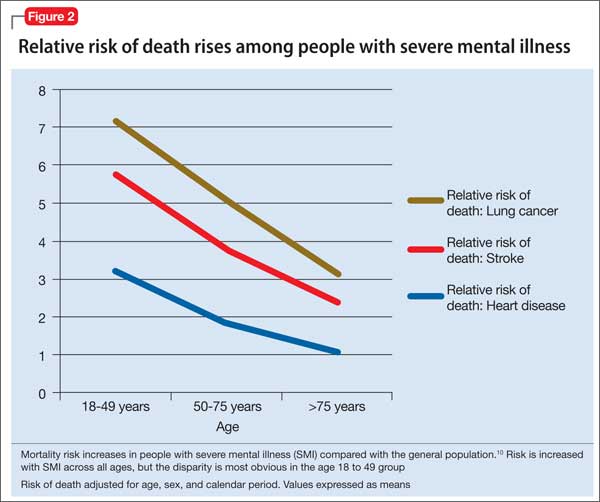
The high prevalence of metabolic syndrome and tobacco dependence among people with SMI accelerates development of cardiovascular disease, as shown by several studies. Bobes et al9 found that the prevalence of metabolic syndrome and cardiovascular risk among patients with SMI is similar to what is found in the general population at 10 to 15 years of greater age. Osborn et al10 demonstrated that people with SMI age 18 to 49 had a higher relative risk of death from coronary heart disease, stroke, and lung cancer than age-matched controls (Figure 2).
It can be said, therefore, that patients with SMI seem to “age” and die prematurely. To reduce this disparity, primary and preventive medical care—especially for cardiovascular disease—must be delivered earlier in life for those with SMI.
Iatrogenic causes of morbidity
Many psychiatric medications, especially second-generation antipsychotics (SGAs), could exacerbate cardiovascular and metabolic conditions by increasing the risk of weight gain, insulin resistance, and dyslipidemia. Antipsychotics that generally are considered to be more effective for refractory psychotic illness (eg, clozapine and olanzapine) are associated with the highest risk of metabolic syndrome. Simon et al11 found a dose-response relationship between olanzapine and clozapine serum concentrations and worsening metabolic outcomes. Valproic acid also can cause significant weight gain and could require monitoring similar to what is done with to SGAs, although there has been less clinical and research attention to this mood stabilizer.
The American Diabetes Association et al12 have published guidelines on monitoring antipsychotic-induced obesity and diabetes, but adoption of these guidelines has been slow. Mackin et al13 found that providers are slow to recognize the elevated rate of obesity and dyslipidemia among psychiatric patients, possibly because of “an alarmingly poor rate of monitoring of metabolic parameters.”
Treating adverse metabolic outcomes also seems to lag behind. The same study13 found that physical health parameters among psychiatric patients continue to become worse even when appropriate health care professionals were notified. Rates of nontreatment for diabetes, dyslipidemia, and hypertension were 30%, 60%, and 88% respectively, according to Nasrallah et al.14
Randomized controlled studies have shown that obesity and metabolic syndrome can be effectively managed using lifestyle and pharmacotherapeutic approaches,15,16 but more research is needed to test long-term outcomes and how to best incorporate these interventions. Newcomer et al17 found that gradually switching an antipsychotic with high risk of metabolic adverse effects to one with lower risk could reduce adverse metabolic outcomes; however, some patients returned to their prior antipsychotic because other medications did not effectively treat their schizophrenia symptoms. Therefore, physicians must pay careful attention to the trade-off between benefits and risks of antipsychotics and make treatment decisions on an individual basis.
Barriers to medical care
Research has demonstrated that patients with SMI receive less screening and fewer preventive medical services, especially blood pressure monitoring, vaccinations, mammography, lipid monitoring, and osteoporosis screening, compared with the general population (Table).18 Some barriers to preventive services could exist because of demographic factors and medical insurance coverage19 or medical providers’ discomfort with symptoms of SMI,20 although Mitchell et al21 found that disparities in mammography screening could not be explained by the presence of emotional distress in women with SMI.

DiMatteo et al22 reported that patients with SMI are 3 times more likely to be noncompliant with medical treatment. These patients also are less likely to receive sec ondary preventive medical care and invasive medical procedures. Those with SMI who experience acute myocardial infarction are less likely to receive drug therapy, such as a thrombolytic, aspirin, beta blocker, or angiotensin-converting enzyme inhibitor.23 They also are less likely to receive invasive cardiovascular procedures, including cardiac catheterization, angioplasty, and coronary artery bypass grafting.24
Therefore, not only are patients with SMI less likely to receive preventive care, they are also less likely to receive potentially lifesaving treatments for SMI. Because those with SMI might not be able to advocate for themselves in these matters, psychiatric clinicians can improve their patients’ lives by advocating for appropriate medical care despite multiple barriers.
Bridging the gap: Managing mental health in primary care
Research from the 1970s and 1980s demonstrated that most persons who sought help for depression or anxiety received treatment from their PCP, many of whom felt limited by their lack of behavioral health training. Moreover, many patients failed to receive a psychiatric diagnosis or adequate treatment, despite efforts to educate primary care physicians on appropriate diagnosis and treatment of mental illness.
Katon et al25 at the University of Washington developed the collaborative care model in the early 1990s to help improve treatment of depression in primary care settings. This model involved:
• case load review by psychiatrists
• use of nurses and other support staff to help monitor patients’ adherence and treatment response
• use of standardized tools such as the Patient Health Questionnaire to monitor symptoms
• enhancement of patient education with pamphlets or classes.
Studies evaluating the success of collaborative care models found overall improved outcomes, making it the only evidence-based model for integration of behavioral health and primary care.26 As a result, the collaborative care model has been implemented across the United States in primary care clinics and specialty care settings, such as obstetrics and gynecology.27
Regrettably, access to primary care has been hampered by:
• population growth
• a shortage of PCPs
• enrollment of a flood of new patients into the health care marketplace as a result of mandates of the Affordable Care Act (ACA).
In many settings, a psychiatrist might be the patient’s only consistent care provider, and could be thought of as a “primary care psychiatrist.”
To resolve this predicament, mental health professionals need to recognize the unique medical conditions faced by people with SMI, and also might need to provide treatment of common medical conditions, either directly or through collaborative arrangements. Psychiatrists who are capable of managing core medical issues likely will witness improved psychiatric and overall health outcomes in their patients. Consequently, psychiatrists and mental health professionals are increasingly called on to be advocates to improve access to medical services in patients with SMI and to participate in health systems reform.
Managing medical conditions in mental health settings
Although traditional collaborative care involves mental health providers working at primary care sites, other models have emerged that manage chronic disease in behavioral health settings. Federally funded grants for primary behavioral health care integration have allowed community mental health centers to partner with federally qualified health centers to provide on-site primary care services.28
In these models, care managers in mental health clinics:
• link patients to primary care services
• encourage lifestyle changes to improve their overall health
• identify and overcome barriers to receiving care
• track clinical outcomes in a registry format.
Currently, 126 mental health sites in the United States have received these grants and are working toward greater integration of primary care.
In addition, the ACA provided funding for “health homes” in non-primary care settings, which includes SMI. These health homes cannot provide direct primary care, but can deliver comprehensive care management, care coordination, health promotion, comprehensive transitional care services between facilities, individual and family support, and referral to community social support services. In these health homes, a PCP can act as a consultant to help establish priorities for disease management and improving health status.29 The PCP consultant also can support psychiatric staff and collaborate with providers who want to provide some direct care of medical conditions.30
Last, some behavioral health sites are choosing to apply for Federally Qualified Health Clinic status or add primary care services to their clinics, with the hope that sustainable funding will become available. Without additional funding to cover the limited reimbursement provided by public payers, such as Medicaid and Medicare, these models might be unsustainable. Current innovations in health care funding reform hopefully will offer solutions for sites to provide medical care in the natural “medical home” of the SMI population.
Bottom Line
Psychiatric providers are in a favorable position to develop and oversee a partnership with primary care physicians with the goal of addressing significant and often lethal health disparities among those with mental illness. Psychiatric providers must use evidence-based practices that include assessment and prevention of cardiopulmonary, metabolic, infectious, and oncologic disorders. True primary care–behavioral health integration must include longitudinal “cross education” and changes in health care policy, with an emphasis on decreasing morbidity and mortality in psychiatric patients.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.
2. Parks J, Svendsen D, Singer P, et al, eds. Morbidity and mortality in people with serious mental illness. Alexandria, VA: National Association of State Mental Health Program Directors (NASMHPD) Medical Directors Council; 2006.
3. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
4. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trails of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res. 2005;80(1):19-32.
5. Compton MT, Daumit GL, Druss BG. Cigarette smoking and overweight/obesity among individuals with serious mental illnesses: a preventive perspective. Harv Rev Psychiatry. 2006;14(2):212-222.
6. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131.
7. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156.
8. Carney CP, Jones L, Woolson RF. Medical comorbidity in women and men with schizophrenia: a population-based study. J Gen Intern Med. 2006;21(11):1133-1137.
9. Bobes J, Arango C, Aranda P, et al; CLAMORS Study Collaborative Group. Cardiovascular and metabolic risk in outpatients with schizoaffective disorder treated with antipsychotics; results from the CLAMORS study. Eur Psychiatry. 2012;27(4):267-274.
10. Osborn DP, Levy G, Nazareth I, et al. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s General Practice Research Database [Erratum in: Arch Gen Psychiatry. 2007;64(6):736]. Arch Gen Psychiatry. 2007;64(2):242-249.
11. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050.
12. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. Mackin P, Bishop DR, Watkinson HM. A prospective study of monitoring practices for metabolic disease in antipsychotic-treated community psychiatric patients. BMC Psychiatry. 2007;7:28.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Alvarez-Jiménez M, Hetrick SE, González-Blanch C, et al. Non-pharmacological management of antipsychotic-induced weight gain: systematic review and meta-analysis of randomized controlled trials. Br J Psychiatry. 2008; 193(2):101-107.
16. Maayan L, Vakhrusheva J, Correll CU. Effectiveness of medication used to attenuate antipsychotic-related weight gain and metabolic abnormalities: a systematic review and meta-analysis. Neuropsychopharmacology. 2010;35(7):1520-1530.
17. Newcomer JW, Weiden PJ, Buchanan RW. Switching antipsychotic medications to reduce adverse event burden in schizophrenia: establishing evidence-based practice. J Clin Psychiatry. 2013;74(11):1108-1120.
18. Lord O, Malone D, Mitchell AJ. Receipt of preventive medical care and medical screening for patients with mental illness: a comparative analysis. Gen Hosp Psychiatry. 2010;32(5):519-543.
19. Xiong GL, Iosif AM, Bermudes RA, et al. Preventive medical services use among community mental health patients with severe mental illness: the influence of gender and insurance coverage. Prim Care Companion J Clin Psychiatry. 2010;12(5). doi: 10.4088/PCC.09m00927gre.
20. Daub S. Turning toward treating the seriously mentally ill in primary care. Fam Syst Health. 2014;32(1):12-13.
21. Mitchell A, Pereira IE, Yadegarfar M, et al. Breast cancer screening in women with mental illness: comparative meta-analysis of mammography uptake. Br J Psychiatry. 2014;205(6):428-435.
22. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
23. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572.
24. Druss BG, Bradford DW, Rosenheck RA, et al. Mental disorders and use of cardiovascular procedures after myocardial infarction. JAMA. 2000;283(4):506-511.
25. Katon W, Unützer J, Wells K, et al. Collaborative depression care: history, evolution and ways to enhance dissemination and sustainability. Gen Hosp Psychiatry. 2010;32(5):456-464.
26. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525.
27. Katon W, Russo J, Reed SD, et al. A randomized trial of collaborative depression care in obstetrics and gynecology clinics: socioeconomic disadvantage and treatment response. Am J Psychiatry. 2015;172(1):32-40.
28. Substance Abuse and Mental Health Services Administration. Request for Applications (RFA) No. SM- 09-011. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2009.
29. Parks J. Behavioral health homes. In: Integrated care: working at the interface of primary care and behavioral health. Raney LE, ed. Arlington, VA: American Psychiatric Publishing; 2015:195.
30. Raney L. Integrated care: the evolving role of psychiatry in the era of health care reform. Psychiatr Serv. 2013;64(11):1076-1078.
People with serious mental illness (SMI) have a life expectancy that is 25 years less than the general population, according to the Centers for Disease Control and Prevention.1 This disparity is partially a consequence of the lack of primary and preventive medical care for those with psychiatric illness. Decades of research have shown that people with SMI experience higher medical morbidity and mortality in addition to facing the stigma of mental illness.
This article aims to advance the idea that longitudinal “cross education” between primary care providers (PCPs) and behavioral health providers (BHPs) is essential in addressing this problem. BHPs include psychiatry clinics, which often are part of a university or large health systems; county-based community mental health programs; and independent mental health clinics that contract with public and private health plans to provide mental health services.
Although suicide and injury account for 40% of the excess mortality in schizophrenia, 60% can be attributed to cardiovascular disease, diabetes, respiratory diseases, and infection.2 Patients with SMI have 2 to 3 times the risk of diabetes, dyslipidemia, hypertension, and obesity.3,4 Furthermore, those with SMI consume more than one-third of tobacco products,5 and 50% to 80% of people with SMI smoke tobacco, an important reversible risk factor for cardiovascular disease.
Figure 1 shows that people with SMI are at higher risk of dying from a chronic medical condition, such as cardiovascular disease, diabetes, chronic obstructive pulmonary disease, and hepatitis C6-8—many of which can be managed by primary and preventive medical interventions. These and other conditions often are not diagnosed or effectively managed in patients with SMI.
The high prevalence of metabolic syndrome and tobacco dependence among people with SMI accelerates development of cardiovascular disease, as shown by several studies. Bobes et al9 found that the prevalence of metabolic syndrome and cardiovascular risk among patients with SMI is similar to what is found in the general population at 10 to 15 years of greater age. Osborn et al10 demonstrated that people with SMI age 18 to 49 had a higher relative risk of death from coronary heart disease, stroke, and lung cancer than age-matched controls (Figure 2).
It can be said, therefore, that patients with SMI seem to “age” and die prematurely. To reduce this disparity, primary and preventive medical care—especially for cardiovascular disease—must be delivered earlier in life for those with SMI.
Iatrogenic causes of morbidity
Many psychiatric medications, especially second-generation antipsychotics (SGAs), could exacerbate cardiovascular and metabolic conditions by increasing the risk of weight gain, insulin resistance, and dyslipidemia. Antipsychotics that generally are considered to be more effective for refractory psychotic illness (eg, clozapine and olanzapine) are associated with the highest risk of metabolic syndrome. Simon et al11 found a dose-response relationship between olanzapine and clozapine serum concentrations and worsening metabolic outcomes. Valproic acid also can cause significant weight gain and could require monitoring similar to what is done with to SGAs, although there has been less clinical and research attention to this mood stabilizer.
The American Diabetes Association et al12 have published guidelines on monitoring antipsychotic-induced obesity and diabetes, but adoption of these guidelines has been slow. Mackin et al13 found that providers are slow to recognize the elevated rate of obesity and dyslipidemia among psychiatric patients, possibly because of “an alarmingly poor rate of monitoring of metabolic parameters.”
Treating adverse metabolic outcomes also seems to lag behind. The same study13 found that physical health parameters among psychiatric patients continue to become worse even when appropriate health care professionals were notified. Rates of nontreatment for diabetes, dyslipidemia, and hypertension were 30%, 60%, and 88% respectively, according to Nasrallah et al.14
Randomized controlled studies have shown that obesity and metabolic syndrome can be effectively managed using lifestyle and pharmacotherapeutic approaches,15,16 but more research is needed to test long-term outcomes and how to best incorporate these interventions. Newcomer et al17 found that gradually switching an antipsychotic with high risk of metabolic adverse effects to one with lower risk could reduce adverse metabolic outcomes; however, some patients returned to their prior antipsychotic because other medications did not effectively treat their schizophrenia symptoms. Therefore, physicians must pay careful attention to the trade-off between benefits and risks of antipsychotics and make treatment decisions on an individual basis.
Barriers to medical care
Research has demonstrated that patients with SMI receive less screening and fewer preventive medical services, especially blood pressure monitoring, vaccinations, mammography, lipid monitoring, and osteoporosis screening, compared with the general population (Table).18 Some barriers to preventive services could exist because of demographic factors and medical insurance coverage19 or medical providers’ discomfort with symptoms of SMI,20 although Mitchell et al21 found that disparities in mammography screening could not be explained by the presence of emotional distress in women with SMI.
DiMatteo et al22 reported that patients with SMI are 3 times more likely to be noncompliant with medical treatment. These patients also are less likely to receive sec ondary preventive medical care and invasive medical procedures. Those with SMI who experience acute myocardial infarction are less likely to receive drug therapy, such as a thrombolytic, aspirin, beta blocker, or angiotensin-converting enzyme inhibitor.23 They also are less likely to receive invasive cardiovascular procedures, including cardiac catheterization, angioplasty, and coronary artery bypass grafting.24
Therefore, not only are patients with SMI less likely to receive preventive care, they are also less likely to receive potentially lifesaving treatments for SMI. Because those with SMI might not be able to advocate for themselves in these matters, psychiatric clinicians can improve their patients’ lives by advocating for appropriate medical care despite multiple barriers.
Bridging the gap: Managing mental health in primary care
Research from the 1970s and 1980s demonstrated that most persons who sought help for depression or anxiety received treatment from their PCP, many of whom felt limited by their lack of behavioral health training. Moreover, many patients failed to receive a psychiatric diagnosis or adequate treatment, despite efforts to educate primary care physicians on appropriate diagnosis and treatment of mental illness.
Katon et al25 at the University of Washington developed the collaborative care model in the early 1990s to help improve treatment of depression in primary care settings. This model involved:
• case load review by psychiatrists
• use of nurses and other support staff to help monitor patients’ adherence and treatment response
• use of standardized tools such as the Patient Health Questionnaire to monitor symptoms
• enhancement of patient education with pamphlets or classes.
Studies evaluating the success of collaborative care models found overall improved outcomes, making it the only evidence-based model for integration of behavioral health and primary care.26 As a result, the collaborative care model has been implemented across the United States in primary care clinics and specialty care settings, such as obstetrics and gynecology.27
Regrettably, access to primary care has been hampered by:
• population growth
• a shortage of PCPs
• enrollment of a flood of new patients into the health care marketplace as a result of mandates of the Affordable Care Act (ACA).
In many settings, a psychiatrist might be the patient’s only consistent care provider, and could be thought of as a “primary care psychiatrist.”
To resolve this predicament, mental health professionals need to recognize the unique medical conditions faced by people with SMI, and also might need to provide treatment of common medical conditions, either directly or through collaborative arrangements. Psychiatrists who are capable of managing core medical issues likely will witness improved psychiatric and overall health outcomes in their patients. Consequently, psychiatrists and mental health professionals are increasingly called on to be advocates to improve access to medical services in patients with SMI and to participate in health systems reform.
Managing medical conditions in mental health settings
Although traditional collaborative care involves mental health providers working at primary care sites, other models have emerged that manage chronic disease in behavioral health settings. Federally funded grants for primary behavioral health care integration have allowed community mental health centers to partner with federally qualified health centers to provide on-site primary care services.28
In these models, care managers in mental health clinics:
• link patients to primary care services
• encourage lifestyle changes to improve their overall health
• identify and overcome barriers to receiving care
• track clinical outcomes in a registry format.
Currently, 126 mental health sites in the United States have received these grants and are working toward greater integration of primary care.
In addition, the ACA provided funding for “health homes” in non-primary care settings, which includes SMI. These health homes cannot provide direct primary care, but can deliver comprehensive care management, care coordination, health promotion, comprehensive transitional care services between facilities, individual and family support, and referral to community social support services. In these health homes, a PCP can act as a consultant to help establish priorities for disease management and improving health status.29 The PCP consultant also can support psychiatric staff and collaborate with providers who want to provide some direct care of medical conditions.30
Last, some behavioral health sites are choosing to apply for Federally Qualified Health Clinic status or add primary care services to their clinics, with the hope that sustainable funding will become available. Without additional funding to cover the limited reimbursement provided by public payers, such as Medicaid and Medicare, these models might be unsustainable. Current innovations in health care funding reform hopefully will offer solutions for sites to provide medical care in the natural “medical home” of the SMI population.
Bottom Line
Psychiatric providers are in a favorable position to develop and oversee a partnership with primary care physicians with the goal of addressing significant and often lethal health disparities among those with mental illness. Psychiatric providers must use evidence-based practices that include assessment and prevention of cardiopulmonary, metabolic, infectious, and oncologic disorders. True primary care–behavioral health integration must include longitudinal “cross education” and changes in health care policy, with an emphasis on decreasing morbidity and mortality in psychiatric patients.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
People with serious mental illness (SMI) have a life expectancy that is 25 years less than the general population, according to the Centers for Disease Control and Prevention.1 This disparity is partially a consequence of the lack of primary and preventive medical care for those with psychiatric illness. Decades of research have shown that people with SMI experience higher medical morbidity and mortality in addition to facing the stigma of mental illness.
This article aims to advance the idea that longitudinal “cross education” between primary care providers (PCPs) and behavioral health providers (BHPs) is essential in addressing this problem. BHPs include psychiatry clinics, which often are part of a university or large health systems; county-based community mental health programs; and independent mental health clinics that contract with public and private health plans to provide mental health services.
Although suicide and injury account for 40% of the excess mortality in schizophrenia, 60% can be attributed to cardiovascular disease, diabetes, respiratory diseases, and infection.2 Patients with SMI have 2 to 3 times the risk of diabetes, dyslipidemia, hypertension, and obesity.3,4 Furthermore, those with SMI consume more than one-third of tobacco products,5 and 50% to 80% of people with SMI smoke tobacco, an important reversible risk factor for cardiovascular disease.
Figure 1 shows that people with SMI are at higher risk of dying from a chronic medical condition, such as cardiovascular disease, diabetes, chronic obstructive pulmonary disease, and hepatitis C6-8—many of which can be managed by primary and preventive medical interventions. These and other conditions often are not diagnosed or effectively managed in patients with SMI.
The high prevalence of metabolic syndrome and tobacco dependence among people with SMI accelerates development of cardiovascular disease, as shown by several studies. Bobes et al9 found that the prevalence of metabolic syndrome and cardiovascular risk among patients with SMI is similar to what is found in the general population at 10 to 15 years of greater age. Osborn et al10 demonstrated that people with SMI age 18 to 49 had a higher relative risk of death from coronary heart disease, stroke, and lung cancer than age-matched controls (Figure 2).
It can be said, therefore, that patients with SMI seem to “age” and die prematurely. To reduce this disparity, primary and preventive medical care—especially for cardiovascular disease—must be delivered earlier in life for those with SMI.
Iatrogenic causes of morbidity
Many psychiatric medications, especially second-generation antipsychotics (SGAs), could exacerbate cardiovascular and metabolic conditions by increasing the risk of weight gain, insulin resistance, and dyslipidemia. Antipsychotics that generally are considered to be more effective for refractory psychotic illness (eg, clozapine and olanzapine) are associated with the highest risk of metabolic syndrome. Simon et al11 found a dose-response relationship between olanzapine and clozapine serum concentrations and worsening metabolic outcomes. Valproic acid also can cause significant weight gain and could require monitoring similar to what is done with to SGAs, although there has been less clinical and research attention to this mood stabilizer.
The American Diabetes Association et al12 have published guidelines on monitoring antipsychotic-induced obesity and diabetes, but adoption of these guidelines has been slow. Mackin et al13 found that providers are slow to recognize the elevated rate of obesity and dyslipidemia among psychiatric patients, possibly because of “an alarmingly poor rate of monitoring of metabolic parameters.”
Treating adverse metabolic outcomes also seems to lag behind. The same study13 found that physical health parameters among psychiatric patients continue to become worse even when appropriate health care professionals were notified. Rates of nontreatment for diabetes, dyslipidemia, and hypertension were 30%, 60%, and 88% respectively, according to Nasrallah et al.14
Randomized controlled studies have shown that obesity and metabolic syndrome can be effectively managed using lifestyle and pharmacotherapeutic approaches,15,16 but more research is needed to test long-term outcomes and how to best incorporate these interventions. Newcomer et al17 found that gradually switching an antipsychotic with high risk of metabolic adverse effects to one with lower risk could reduce adverse metabolic outcomes; however, some patients returned to their prior antipsychotic because other medications did not effectively treat their schizophrenia symptoms. Therefore, physicians must pay careful attention to the trade-off between benefits and risks of antipsychotics and make treatment decisions on an individual basis.
Barriers to medical care
Research has demonstrated that patients with SMI receive less screening and fewer preventive medical services, especially blood pressure monitoring, vaccinations, mammography, lipid monitoring, and osteoporosis screening, compared with the general population (Table).18 Some barriers to preventive services could exist because of demographic factors and medical insurance coverage19 or medical providers’ discomfort with symptoms of SMI,20 although Mitchell et al21 found that disparities in mammography screening could not be explained by the presence of emotional distress in women with SMI.
DiMatteo et al22 reported that patients with SMI are 3 times more likely to be noncompliant with medical treatment. These patients also are less likely to receive sec ondary preventive medical care and invasive medical procedures. Those with SMI who experience acute myocardial infarction are less likely to receive drug therapy, such as a thrombolytic, aspirin, beta blocker, or angiotensin-converting enzyme inhibitor.23 They also are less likely to receive invasive cardiovascular procedures, including cardiac catheterization, angioplasty, and coronary artery bypass grafting.24
Therefore, not only are patients with SMI less likely to receive preventive care, they are also less likely to receive potentially lifesaving treatments for SMI. Because those with SMI might not be able to advocate for themselves in these matters, psychiatric clinicians can improve their patients’ lives by advocating for appropriate medical care despite multiple barriers.
Bridging the gap: Managing mental health in primary care
Research from the 1970s and 1980s demonstrated that most persons who sought help for depression or anxiety received treatment from their PCP, many of whom felt limited by their lack of behavioral health training. Moreover, many patients failed to receive a psychiatric diagnosis or adequate treatment, despite efforts to educate primary care physicians on appropriate diagnosis and treatment of mental illness.
Katon et al25 at the University of Washington developed the collaborative care model in the early 1990s to help improve treatment of depression in primary care settings. This model involved:
• case load review by psychiatrists
• use of nurses and other support staff to help monitor patients’ adherence and treatment response
• use of standardized tools such as the Patient Health Questionnaire to monitor symptoms
• enhancement of patient education with pamphlets or classes.
Studies evaluating the success of collaborative care models found overall improved outcomes, making it the only evidence-based model for integration of behavioral health and primary care.26 As a result, the collaborative care model has been implemented across the United States in primary care clinics and specialty care settings, such as obstetrics and gynecology.27
Regrettably, access to primary care has been hampered by:
• population growth
• a shortage of PCPs
• enrollment of a flood of new patients into the health care marketplace as a result of mandates of the Affordable Care Act (ACA).
In many settings, a psychiatrist might be the patient’s only consistent care provider, and could be thought of as a “primary care psychiatrist.”
To resolve this predicament, mental health professionals need to recognize the unique medical conditions faced by people with SMI, and also might need to provide treatment of common medical conditions, either directly or through collaborative arrangements. Psychiatrists who are capable of managing core medical issues likely will witness improved psychiatric and overall health outcomes in their patients. Consequently, psychiatrists and mental health professionals are increasingly called on to be advocates to improve access to medical services in patients with SMI and to participate in health systems reform.
Managing medical conditions in mental health settings
Although traditional collaborative care involves mental health providers working at primary care sites, other models have emerged that manage chronic disease in behavioral health settings. Federally funded grants for primary behavioral health care integration have allowed community mental health centers to partner with federally qualified health centers to provide on-site primary care services.28
In these models, care managers in mental health clinics:
• link patients to primary care services
• encourage lifestyle changes to improve their overall health
• identify and overcome barriers to receiving care
• track clinical outcomes in a registry format.
Currently, 126 mental health sites in the United States have received these grants and are working toward greater integration of primary care.
In addition, the ACA provided funding for “health homes” in non-primary care settings, which includes SMI. These health homes cannot provide direct primary care, but can deliver comprehensive care management, care coordination, health promotion, comprehensive transitional care services between facilities, individual and family support, and referral to community social support services. In these health homes, a PCP can act as a consultant to help establish priorities for disease management and improving health status.29 The PCP consultant also can support psychiatric staff and collaborate with providers who want to provide some direct care of medical conditions.30
Last, some behavioral health sites are choosing to apply for Federally Qualified Health Clinic status or add primary care services to their clinics, with the hope that sustainable funding will become available. Without additional funding to cover the limited reimbursement provided by public payers, such as Medicaid and Medicare, these models might be unsustainable. Current innovations in health care funding reform hopefully will offer solutions for sites to provide medical care in the natural “medical home” of the SMI population.
Bottom Line
Psychiatric providers are in a favorable position to develop and oversee a partnership with primary care physicians with the goal of addressing significant and often lethal health disparities among those with mental illness. Psychiatric providers must use evidence-based practices that include assessment and prevention of cardiopulmonary, metabolic, infectious, and oncologic disorders. True primary care–behavioral health integration must include longitudinal “cross education” and changes in health care policy, with an emphasis on decreasing morbidity and mortality in psychiatric patients.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.
2. Parks J, Svendsen D, Singer P, et al, eds. Morbidity and mortality in people with serious mental illness. Alexandria, VA: National Association of State Mental Health Program Directors (NASMHPD) Medical Directors Council; 2006.
3. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
4. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trails of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res. 2005;80(1):19-32.
5. Compton MT, Daumit GL, Druss BG. Cigarette smoking and overweight/obesity among individuals with serious mental illnesses: a preventive perspective. Harv Rev Psychiatry. 2006;14(2):212-222.
6. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131.
7. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156.
8. Carney CP, Jones L, Woolson RF. Medical comorbidity in women and men with schizophrenia: a population-based study. J Gen Intern Med. 2006;21(11):1133-1137.
9. Bobes J, Arango C, Aranda P, et al; CLAMORS Study Collaborative Group. Cardiovascular and metabolic risk in outpatients with schizoaffective disorder treated with antipsychotics; results from the CLAMORS study. Eur Psychiatry. 2012;27(4):267-274.
10. Osborn DP, Levy G, Nazareth I, et al. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s General Practice Research Database [Erratum in: Arch Gen Psychiatry. 2007;64(6):736]. Arch Gen Psychiatry. 2007;64(2):242-249.
11. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050.
12. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. Mackin P, Bishop DR, Watkinson HM. A prospective study of monitoring practices for metabolic disease in antipsychotic-treated community psychiatric patients. BMC Psychiatry. 2007;7:28.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Alvarez-Jiménez M, Hetrick SE, González-Blanch C, et al. Non-pharmacological management of antipsychotic-induced weight gain: systematic review and meta-analysis of randomized controlled trials. Br J Psychiatry. 2008; 193(2):101-107.
16. Maayan L, Vakhrusheva J, Correll CU. Effectiveness of medication used to attenuate antipsychotic-related weight gain and metabolic abnormalities: a systematic review and meta-analysis. Neuropsychopharmacology. 2010;35(7):1520-1530.
17. Newcomer JW, Weiden PJ, Buchanan RW. Switching antipsychotic medications to reduce adverse event burden in schizophrenia: establishing evidence-based practice. J Clin Psychiatry. 2013;74(11):1108-1120.
18. Lord O, Malone D, Mitchell AJ. Receipt of preventive medical care and medical screening for patients with mental illness: a comparative analysis. Gen Hosp Psychiatry. 2010;32(5):519-543.
19. Xiong GL, Iosif AM, Bermudes RA, et al. Preventive medical services use among community mental health patients with severe mental illness: the influence of gender and insurance coverage. Prim Care Companion J Clin Psychiatry. 2010;12(5). doi: 10.4088/PCC.09m00927gre.
20. Daub S. Turning toward treating the seriously mentally ill in primary care. Fam Syst Health. 2014;32(1):12-13.
21. Mitchell A, Pereira IE, Yadegarfar M, et al. Breast cancer screening in women with mental illness: comparative meta-analysis of mammography uptake. Br J Psychiatry. 2014;205(6):428-435.
22. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
23. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572.
24. Druss BG, Bradford DW, Rosenheck RA, et al. Mental disorders and use of cardiovascular procedures after myocardial infarction. JAMA. 2000;283(4):506-511.
25. Katon W, Unützer J, Wells K, et al. Collaborative depression care: history, evolution and ways to enhance dissemination and sustainability. Gen Hosp Psychiatry. 2010;32(5):456-464.
26. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525.
27. Katon W, Russo J, Reed SD, et al. A randomized trial of collaborative depression care in obstetrics and gynecology clinics: socioeconomic disadvantage and treatment response. Am J Psychiatry. 2015;172(1):32-40.
28. Substance Abuse and Mental Health Services Administration. Request for Applications (RFA) No. SM- 09-011. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2009.
29. Parks J. Behavioral health homes. In: Integrated care: working at the interface of primary care and behavioral health. Raney LE, ed. Arlington, VA: American Psychiatric Publishing; 2015:195.
30. Raney L. Integrated care: the evolving role of psychiatry in the era of health care reform. Psychiatr Serv. 2013;64(11):1076-1078.
1. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.
2. Parks J, Svendsen D, Singer P, et al, eds. Morbidity and mortality in people with serious mental illness. Alexandria, VA: National Association of State Mental Health Program Directors (NASMHPD) Medical Directors Council; 2006.
3. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
4. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trails of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res. 2005;80(1):19-32.
5. Compton MT, Daumit GL, Druss BG. Cigarette smoking and overweight/obesity among individuals with serious mental illnesses: a preventive perspective. Harv Rev Psychiatry. 2006;14(2):212-222.
6. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131.
7. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156.
8. Carney CP, Jones L, Woolson RF. Medical comorbidity in women and men with schizophrenia: a population-based study. J Gen Intern Med. 2006;21(11):1133-1137.
9. Bobes J, Arango C, Aranda P, et al; CLAMORS Study Collaborative Group. Cardiovascular and metabolic risk in outpatients with schizoaffective disorder treated with antipsychotics; results from the CLAMORS study. Eur Psychiatry. 2012;27(4):267-274.
10. Osborn DP, Levy G, Nazareth I, et al. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s General Practice Research Database [Erratum in: Arch Gen Psychiatry. 2007;64(6):736]. Arch Gen Psychiatry. 2007;64(2):242-249.
11. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050.
12. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. Mackin P, Bishop DR, Watkinson HM. A prospective study of monitoring practices for metabolic disease in antipsychotic-treated community psychiatric patients. BMC Psychiatry. 2007;7:28.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Alvarez-Jiménez M, Hetrick SE, González-Blanch C, et al. Non-pharmacological management of antipsychotic-induced weight gain: systematic review and meta-analysis of randomized controlled trials. Br J Psychiatry. 2008; 193(2):101-107.
16. Maayan L, Vakhrusheva J, Correll CU. Effectiveness of medication used to attenuate antipsychotic-related weight gain and metabolic abnormalities: a systematic review and meta-analysis. Neuropsychopharmacology. 2010;35(7):1520-1530.
17. Newcomer JW, Weiden PJ, Buchanan RW. Switching antipsychotic medications to reduce adverse event burden in schizophrenia: establishing evidence-based practice. J Clin Psychiatry. 2013;74(11):1108-1120.
18. Lord O, Malone D, Mitchell AJ. Receipt of preventive medical care and medical screening for patients with mental illness: a comparative analysis. Gen Hosp Psychiatry. 2010;32(5):519-543.
19. Xiong GL, Iosif AM, Bermudes RA, et al. Preventive medical services use among community mental health patients with severe mental illness: the influence of gender and insurance coverage. Prim Care Companion J Clin Psychiatry. 2010;12(5). doi: 10.4088/PCC.09m00927gre.
20. Daub S. Turning toward treating the seriously mentally ill in primary care. Fam Syst Health. 2014;32(1):12-13.
21. Mitchell A, Pereira IE, Yadegarfar M, et al. Breast cancer screening in women with mental illness: comparative meta-analysis of mammography uptake. Br J Psychiatry. 2014;205(6):428-435.
22. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
23. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572.
24. Druss BG, Bradford DW, Rosenheck RA, et al. Mental disorders and use of cardiovascular procedures after myocardial infarction. JAMA. 2000;283(4):506-511.
25. Katon W, Unützer J, Wells K, et al. Collaborative depression care: history, evolution and ways to enhance dissemination and sustainability. Gen Hosp Psychiatry. 2010;32(5):456-464.
26. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525.
27. Katon W, Russo J, Reed SD, et al. A randomized trial of collaborative depression care in obstetrics and gynecology clinics: socioeconomic disadvantage and treatment response. Am J Psychiatry. 2015;172(1):32-40.
28. Substance Abuse and Mental Health Services Administration. Request for Applications (RFA) No. SM- 09-011. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2009.
29. Parks J. Behavioral health homes. In: Integrated care: working at the interface of primary care and behavioral health. Raney LE, ed. Arlington, VA: American Psychiatric Publishing; 2015:195.
30. Raney L. Integrated care: the evolving role of psychiatry in the era of health care reform. Psychiatr Serv. 2013;64(11):1076-1078.

Long-term data support use of FVIII treatment
TORONTO—The antihemophilic factor turoctocog alfa is safe and effective long-term, according to interim data from the phase 3 guardian 2 trial.
With more than 4 years of safety data, researchers have found turoctocog alfa to be well-tolerated in patients with hemophilia A.
The median annualized bleeding rate for patients on prophylactic treatment was 1.56 bleeds per patient per year. For patients who received turoctocog alfa on demand, a single injection stopped all bleeds.
And none of the patients developed factor VIII (FVIII) inhibitors.
Margareth Ozelo, MD, PhD, of the University of Campinas in Sao Paulo, Brazil and her colleagues presented these data at the ISTH 2015 Congress (abstract PO251-WED). The research is sponsored by Novo Nordisk, the company developing turoctocog alfa (as NovoEight).
Turoctocog alfa is a B-domain truncated recombinant human coagulation FVIII product indicated for the treatment and prevention of bleeding in patients with hemophilia A. The ongoing guardian 2 trial is a prospective safety and efficacy extension trial of the guardian 1 and guardian 3 studies.
Two hundred patients received turoctocog alfa in guardian 2. They had severe hemophilia A (FVIII activity ≤ 1%), no history of inhibitors, and had completed guardian 1, guardian 3, or a third pharmacokinetics trial. The patients’ mean age at first turoctocog alfa injection was 22.3 ± 14.4 years.
In guardian 1 and guardian 3, all patients switched from other FVIII products to turoctocog alfa prophylaxis every second day (adults/adolescents, 20–40 IU/kg; children, 25–50 IU/kg), or 3 times weekly (adults/adolescents, 20–50 IU/kg; children 25–60 IU/kg). Patients also received turoctocog alfa when bleeds arose.
Of the 200 patients enrolled on guardian 2, 133 were still participating in the trial at the interim cutoff date, December 31, 2013.
The interim analysis includes data for 451.6 patient-years and 72,320 days of exposure to turoctocog alfa. The total number of exposure days was 364.5
(range, 1-762) per patient for prophylaxis and 23.8 (range, 1-90) per patient for on-demand treatment.
The mean number of turoctocog alfa doses was 368.3 (range, 1-766) per patient for prophylaxis and 24.2 (range, 1-90) per patient for on-demand treatment.
Safety results
At the interim cutoff, none of the patients had developed FVIII inhibitors. Adverse events occurred in 84% of patients (n=168). The most common were
headache, nasopharyngitis, upper respiratory tract infection, and arthralgia.
Eight adverse events were considered possibly or probably related to turoctocog alfa in 5 patients (2.5%). These events were mild or moderate and included local swelling (n=1), increased aspartate aminotransferase (n=1), increased alanine aminotransferase (n=1), pain in extremity (n=1), musculoskeletal pain (n=1), lichenoid keratosis (n=1), and arthropathy (n=2).
There were 29 serious adverse events that were considered unlikely to be treatment-related. This included a death from subdural hemorrhage.
Efficacy results
For patients on prophylactic treatment (n=197), the median annualized bleeding rate was 1.56 bleeds per patient per year for all bleeds. It was 0.50 for
spontaneous bleeds, 0.49 for traumatic bleeds, 0.93 for joint bleeds, and 0.35 for nonjoint bleeds.
The success rate for treating bleeds during prophylaxis was 89.4%, and 90% of all bleeding episodes were successfully treated with 1 or 2 infusions of
turoctocog alfa. As for on-demand treatment, a single injection stopped all 73 bleeds.
“These interim results provide an extension to the body of evidence supporting the long-term use of NovoEight,” Dr Ozelo said. “For people with hemophilia A, finding treatments that are effective at preventing bleeding episodes long-term is essential.” ![]()
TORONTO—The antihemophilic factor turoctocog alfa is safe and effective long-term, according to interim data from the phase 3 guardian 2 trial.
With more than 4 years of safety data, researchers have found turoctocog alfa to be well-tolerated in patients with hemophilia A.
The median annualized bleeding rate for patients on prophylactic treatment was 1.56 bleeds per patient per year. For patients who received turoctocog alfa on demand, a single injection stopped all bleeds.
And none of the patients developed factor VIII (FVIII) inhibitors.
Margareth Ozelo, MD, PhD, of the University of Campinas in Sao Paulo, Brazil and her colleagues presented these data at the ISTH 2015 Congress (abstract PO251-WED). The research is sponsored by Novo Nordisk, the company developing turoctocog alfa (as NovoEight).
Turoctocog alfa is a B-domain truncated recombinant human coagulation FVIII product indicated for the treatment and prevention of bleeding in patients with hemophilia A. The ongoing guardian 2 trial is a prospective safety and efficacy extension trial of the guardian 1 and guardian 3 studies.
Two hundred patients received turoctocog alfa in guardian 2. They had severe hemophilia A (FVIII activity ≤ 1%), no history of inhibitors, and had completed guardian 1, guardian 3, or a third pharmacokinetics trial. The patients’ mean age at first turoctocog alfa injection was 22.3 ± 14.4 years.
In guardian 1 and guardian 3, all patients switched from other FVIII products to turoctocog alfa prophylaxis every second day (adults/adolescents, 20–40 IU/kg; children, 25–50 IU/kg), or 3 times weekly (adults/adolescents, 20–50 IU/kg; children 25–60 IU/kg). Patients also received turoctocog alfa when bleeds arose.
Of the 200 patients enrolled on guardian 2, 133 were still participating in the trial at the interim cutoff date, December 31, 2013.
The interim analysis includes data for 451.6 patient-years and 72,320 days of exposure to turoctocog alfa. The total number of exposure days was 364.5
(range, 1-762) per patient for prophylaxis and 23.8 (range, 1-90) per patient for on-demand treatment.
The mean number of turoctocog alfa doses was 368.3 (range, 1-766) per patient for prophylaxis and 24.2 (range, 1-90) per patient for on-demand treatment.
Safety results
At the interim cutoff, none of the patients had developed FVIII inhibitors. Adverse events occurred in 84% of patients (n=168). The most common were
headache, nasopharyngitis, upper respiratory tract infection, and arthralgia.
Eight adverse events were considered possibly or probably related to turoctocog alfa in 5 patients (2.5%). These events were mild or moderate and included local swelling (n=1), increased aspartate aminotransferase (n=1), increased alanine aminotransferase (n=1), pain in extremity (n=1), musculoskeletal pain (n=1), lichenoid keratosis (n=1), and arthropathy (n=2).
There were 29 serious adverse events that were considered unlikely to be treatment-related. This included a death from subdural hemorrhage.
Efficacy results
For patients on prophylactic treatment (n=197), the median annualized bleeding rate was 1.56 bleeds per patient per year for all bleeds. It was 0.50 for
spontaneous bleeds, 0.49 for traumatic bleeds, 0.93 for joint bleeds, and 0.35 for nonjoint bleeds.
The success rate for treating bleeds during prophylaxis was 89.4%, and 90% of all bleeding episodes were successfully treated with 1 or 2 infusions of
turoctocog alfa. As for on-demand treatment, a single injection stopped all 73 bleeds.
“These interim results provide an extension to the body of evidence supporting the long-term use of NovoEight,” Dr Ozelo said. “For people with hemophilia A, finding treatments that are effective at preventing bleeding episodes long-term is essential.” ![]()
TORONTO—The antihemophilic factor turoctocog alfa is safe and effective long-term, according to interim data from the phase 3 guardian 2 trial.
With more than 4 years of safety data, researchers have found turoctocog alfa to be well-tolerated in patients with hemophilia A.
The median annualized bleeding rate for patients on prophylactic treatment was 1.56 bleeds per patient per year. For patients who received turoctocog alfa on demand, a single injection stopped all bleeds.
And none of the patients developed factor VIII (FVIII) inhibitors.
Margareth Ozelo, MD, PhD, of the University of Campinas in Sao Paulo, Brazil and her colleagues presented these data at the ISTH 2015 Congress (abstract PO251-WED). The research is sponsored by Novo Nordisk, the company developing turoctocog alfa (as NovoEight).
Turoctocog alfa is a B-domain truncated recombinant human coagulation FVIII product indicated for the treatment and prevention of bleeding in patients with hemophilia A. The ongoing guardian 2 trial is a prospective safety and efficacy extension trial of the guardian 1 and guardian 3 studies.
Two hundred patients received turoctocog alfa in guardian 2. They had severe hemophilia A (FVIII activity ≤ 1%), no history of inhibitors, and had completed guardian 1, guardian 3, or a third pharmacokinetics trial. The patients’ mean age at first turoctocog alfa injection was 22.3 ± 14.4 years.
In guardian 1 and guardian 3, all patients switched from other FVIII products to turoctocog alfa prophylaxis every second day (adults/adolescents, 20–40 IU/kg; children, 25–50 IU/kg), or 3 times weekly (adults/adolescents, 20–50 IU/kg; children 25–60 IU/kg). Patients also received turoctocog alfa when bleeds arose.
Of the 200 patients enrolled on guardian 2, 133 were still participating in the trial at the interim cutoff date, December 31, 2013.
The interim analysis includes data for 451.6 patient-years and 72,320 days of exposure to turoctocog alfa. The total number of exposure days was 364.5
(range, 1-762) per patient for prophylaxis and 23.8 (range, 1-90) per patient for on-demand treatment.
The mean number of turoctocog alfa doses was 368.3 (range, 1-766) per patient for prophylaxis and 24.2 (range, 1-90) per patient for on-demand treatment.
Safety results
At the interim cutoff, none of the patients had developed FVIII inhibitors. Adverse events occurred in 84% of patients (n=168). The most common were
headache, nasopharyngitis, upper respiratory tract infection, and arthralgia.
Eight adverse events were considered possibly or probably related to turoctocog alfa in 5 patients (2.5%). These events were mild or moderate and included local swelling (n=1), increased aspartate aminotransferase (n=1), increased alanine aminotransferase (n=1), pain in extremity (n=1), musculoskeletal pain (n=1), lichenoid keratosis (n=1), and arthropathy (n=2).
There were 29 serious adverse events that were considered unlikely to be treatment-related. This included a death from subdural hemorrhage.
Efficacy results
For patients on prophylactic treatment (n=197), the median annualized bleeding rate was 1.56 bleeds per patient per year for all bleeds. It was 0.50 for
spontaneous bleeds, 0.49 for traumatic bleeds, 0.93 for joint bleeds, and 0.35 for nonjoint bleeds.
The success rate for treating bleeds during prophylaxis was 89.4%, and 90% of all bleeding episodes were successfully treated with 1 or 2 infusions of
turoctocog alfa. As for on-demand treatment, a single injection stopped all 73 bleeds.
“These interim results provide an extension to the body of evidence supporting the long-term use of NovoEight,” Dr Ozelo said. “For people with hemophilia A, finding treatments that are effective at preventing bleeding episodes long-term is essential.” ![]()
Insecticide can cause NHL, experts say

Photo by John Messina
The insecticide gamma-hexachlorocyclohexane (lindane) is carcinogenic, according to experts from the International Agency for Research on Cancer (IARC), the specialized cancer agency of the World Health Organization.
The experts said they found sufficient evidence that lindane, which is banned or restricted in most countries, can cause non-Hodgkin lymphoma (NHL).
The group also discovered that 2 other chemicals might cause NHL.
The evidence suggests the insecticide dichlorodiphenyltrichloroethane (DDT) is probably carcinogenic, and the herbicide 2,4-dichlorophenoxyacetic acid (2,4-D) is possibly carcinogenic.
A summary of these findings is available in The Lancet Oncology, and the experts’ detailed assessments will be published as Volume 113 of the IARC Monographs.
The group, which consisted of 26 experts convened by the IARC Monographs Programme, reviewed the latest scientific literature on lindane, DDT, and 2,4-D and used their findings to classify these 3 chemicals according to carcinogenicity.
The classification (Group 1, Group 2A, etc.) indicates the strength of the evidence that a substance causes cancer, not the level of risk associated with exposure. The Monographs Programme identifies cancer hazards even when risks are very low at current exposure levels, because new uses or unforeseen exposures could engender risks that are significantly higher.
Lindane
The experts classified lindane as carcinogenic to humans (Group 1), saying they found sufficient evidence that it can cause NHL. Large epidemiological studies of agricultural exposures in the US and Canada showed a 60% increased risk of NHL in people exposed to lindane.
Lindane has been used extensively for insect control, including in agriculture and for the treatment of human lice and scabies. High exposures have occurred among agricultural workers and pesticide applicators. However, the use of lindane is now banned or restricted in most countries.
DDT
The experts classified DDT as probably carcinogenic to humans (Group 2A), saying they found sufficient evidence that DDT causes cancer in experimental animals and limited evidence of DDT’s carcinogenicity in humans.
Epidemiological studies have shown positive associations between exposure to DDT and NHL, testicular cancer, and liver cancer.
There was also strong experimental evidence that DDT can suppress the immune system and disrupt sex hormones. However, overall, there was no association between breast cancer and DDT levels measured in samples of blood or fat.
DDT was introduced for the control of insect-borne diseases during World War II and was later applied widely to eradicate malaria and in agriculture. Most uses of DDT were banned in the 1970s. However, DDT and its breakdown products are highly persistent and can be found in the environment and in animal and human tissues throughout the world.
Exposure to DDT still occurs, mainly through diet. The remaining and essential use of DDT is for disease vector control, mainly for malaria. This use is strictly restricted under the Stockholm Convention.
2,4-D
The experts classified 2,4-D as possibly carcinogenic to humans (Group 2B), saying they had inadequate evidence in humans and limited evidence in experimental animals.
There is strong evidence that 2,4-D induces oxidative stress and moderate evidence that 2,4-D causes immunosuppression, based on in vivo and in vitro studies. However, epidemiological studies did not show strong or consistent increases in the risk of NHL or other cancers in relation to 2,4-D exposure.
Since its introduction in 1945, 2,4-D has been widely used to control weeds in agriculture, forestry, and urban and residential settings. Occupational exposures to 2,4-D can occur during manufacturing and application, and the general population can be exposed through food, water, dust, or residential application, and during spraying. ![]()
Photo by John Messina
The insecticide gamma-hexachlorocyclohexane (lindane) is carcinogenic, according to experts from the International Agency for Research on Cancer (IARC), the specialized cancer agency of the World Health Organization.
The experts said they found sufficient evidence that lindane, which is banned or restricted in most countries, can cause non-Hodgkin lymphoma (NHL).
The group also discovered that 2 other chemicals might cause NHL.
The evidence suggests the insecticide dichlorodiphenyltrichloroethane (DDT) is probably carcinogenic, and the herbicide 2,4-dichlorophenoxyacetic acid (2,4-D) is possibly carcinogenic.
A summary of these findings is available in The Lancet Oncology, and the experts’ detailed assessments will be published as Volume 113 of the IARC Monographs.
The group, which consisted of 26 experts convened by the IARC Monographs Programme, reviewed the latest scientific literature on lindane, DDT, and 2,4-D and used their findings to classify these 3 chemicals according to carcinogenicity.
The classification (Group 1, Group 2A, etc.) indicates the strength of the evidence that a substance causes cancer, not the level of risk associated with exposure. The Monographs Programme identifies cancer hazards even when risks are very low at current exposure levels, because new uses or unforeseen exposures could engender risks that are significantly higher.
Lindane
The experts classified lindane as carcinogenic to humans (Group 1), saying they found sufficient evidence that it can cause NHL. Large epidemiological studies of agricultural exposures in the US and Canada showed a 60% increased risk of NHL in people exposed to lindane.
Lindane has been used extensively for insect control, including in agriculture and for the treatment of human lice and scabies. High exposures have occurred among agricultural workers and pesticide applicators. However, the use of lindane is now banned or restricted in most countries.
DDT
The experts classified DDT as probably carcinogenic to humans (Group 2A), saying they found sufficient evidence that DDT causes cancer in experimental animals and limited evidence of DDT’s carcinogenicity in humans.
Epidemiological studies have shown positive associations between exposure to DDT and NHL, testicular cancer, and liver cancer.
There was also strong experimental evidence that DDT can suppress the immune system and disrupt sex hormones. However, overall, there was no association between breast cancer and DDT levels measured in samples of blood or fat.
DDT was introduced for the control of insect-borne diseases during World War II and was later applied widely to eradicate malaria and in agriculture. Most uses of DDT were banned in the 1970s. However, DDT and its breakdown products are highly persistent and can be found in the environment and in animal and human tissues throughout the world.
Exposure to DDT still occurs, mainly through diet. The remaining and essential use of DDT is for disease vector control, mainly for malaria. This use is strictly restricted under the Stockholm Convention.
2,4-D
The experts classified 2,4-D as possibly carcinogenic to humans (Group 2B), saying they had inadequate evidence in humans and limited evidence in experimental animals.
There is strong evidence that 2,4-D induces oxidative stress and moderate evidence that 2,4-D causes immunosuppression, based on in vivo and in vitro studies. However, epidemiological studies did not show strong or consistent increases in the risk of NHL or other cancers in relation to 2,4-D exposure.
Since its introduction in 1945, 2,4-D has been widely used to control weeds in agriculture, forestry, and urban and residential settings. Occupational exposures to 2,4-D can occur during manufacturing and application, and the general population can be exposed through food, water, dust, or residential application, and during spraying. ![]()
Photo by John Messina
The insecticide gamma-hexachlorocyclohexane (lindane) is carcinogenic, according to experts from the International Agency for Research on Cancer (IARC), the specialized cancer agency of the World Health Organization.
The experts said they found sufficient evidence that lindane, which is banned or restricted in most countries, can cause non-Hodgkin lymphoma (NHL).
The group also discovered that 2 other chemicals might cause NHL.
The evidence suggests the insecticide dichlorodiphenyltrichloroethane (DDT) is probably carcinogenic, and the herbicide 2,4-dichlorophenoxyacetic acid (2,4-D) is possibly carcinogenic.
A summary of these findings is available in The Lancet Oncology, and the experts’ detailed assessments will be published as Volume 113 of the IARC Monographs.
The group, which consisted of 26 experts convened by the IARC Monographs Programme, reviewed the latest scientific literature on lindane, DDT, and 2,4-D and used their findings to classify these 3 chemicals according to carcinogenicity.
The classification (Group 1, Group 2A, etc.) indicates the strength of the evidence that a substance causes cancer, not the level of risk associated with exposure. The Monographs Programme identifies cancer hazards even when risks are very low at current exposure levels, because new uses or unforeseen exposures could engender risks that are significantly higher.
Lindane
The experts classified lindane as carcinogenic to humans (Group 1), saying they found sufficient evidence that it can cause NHL. Large epidemiological studies of agricultural exposures in the US and Canada showed a 60% increased risk of NHL in people exposed to lindane.
Lindane has been used extensively for insect control, including in agriculture and for the treatment of human lice and scabies. High exposures have occurred among agricultural workers and pesticide applicators. However, the use of lindane is now banned or restricted in most countries.
DDT
The experts classified DDT as probably carcinogenic to humans (Group 2A), saying they found sufficient evidence that DDT causes cancer in experimental animals and limited evidence of DDT’s carcinogenicity in humans.
Epidemiological studies have shown positive associations between exposure to DDT and NHL, testicular cancer, and liver cancer.
There was also strong experimental evidence that DDT can suppress the immune system and disrupt sex hormones. However, overall, there was no association between breast cancer and DDT levels measured in samples of blood or fat.
DDT was introduced for the control of insect-borne diseases during World War II and was later applied widely to eradicate malaria and in agriculture. Most uses of DDT were banned in the 1970s. However, DDT and its breakdown products are highly persistent and can be found in the environment and in animal and human tissues throughout the world.
Exposure to DDT still occurs, mainly through diet. The remaining and essential use of DDT is for disease vector control, mainly for malaria. This use is strictly restricted under the Stockholm Convention.
2,4-D
The experts classified 2,4-D as possibly carcinogenic to humans (Group 2B), saying they had inadequate evidence in humans and limited evidence in experimental animals.
There is strong evidence that 2,4-D induces oxidative stress and moderate evidence that 2,4-D causes immunosuppression, based on in vivo and in vitro studies. However, epidemiological studies did not show strong or consistent increases in the risk of NHL or other cancers in relation to 2,4-D exposure.
Since its introduction in 1945, 2,4-D has been widely used to control weeds in agriculture, forestry, and urban and residential settings. Occupational exposures to 2,4-D can occur during manufacturing and application, and the general population can be exposed through food, water, dust, or residential application, and during spraying. ![]()
WCD: Secukinumab shows effectiveness for nail, palmoplantar psoriasis
VANCOUVER, B.C. – The interleukin-17A inhibitor secukinumab demonstrated the greatest improvement in nail psoriasis ever reported from a randomized, placebo-controlled trial in the phase IIIb TRANSFIGURE study, Dr. Kristian Reich reported at the World Congress of Dermatology.
At 198 patients, TRANSFIGURE is the largest-ever prospective study in patients with moderate to severe chronic plaque psoriasis and significant nail involvement. And while only the 16-week results are available thus far, when TRANSFIGURE is completed after a planned 132 weeks of treatment, it will also be the longest-ever study in the treatment of nail psoriasis, noted Dr. Reich, a dermatologist in group practice in Hamburg, Germany.

Elsewhere at WCD 2015, Dr. Alice B. Gottlieb presented the week 16 results of the phase IIIb GESTURE study, in which 205 psoriasis patients with moderate to severe psoriasis of the palms and soles were randomized to subcutaneous secukinumab (Cosentyx) at 150 or 300 mg or placebo. Dosing was weekly for the first 5 weeks and monthly thereafter.
The primary endpoint, a palmoplantar Investigator’s Global Assessment scale score of 0 or 1 – clear or almost clear – at week 16 was 33.3% with secukinumab at 300 mg, 22.1% at 150 mg, and 1.5% with placebo. The average reduction in palmoplantar PASI (Psoriasis Area Severity Index) score from baseline was 54.6% with high-dose and 35.3% with low-dose secukinumab, compared with 4.1% in placebo-treated controls, reported Dr. Gottlieb, professor and chair of dermatology at Tufts University, Boston.
Like the TRANSFIGURE trial, GESTURE will continue for 132 weeks, with the initial placebo-treated controls being randomized to secukinumab at 150 or 300 mg after week 16.
Dr. Reich reported that by 16 weeks in TRANSFIGURE, mean scores on the Nail Psoriasis Severity Index had improved by 45.3%, compared with baseline, in patients on secukinumab 300 mg, 37.9% in those on secukinumab 150 mg, and 10.8% with placebo.
The results on the skin were dramatic: a PASI 75 rate of 87.1% with secukinumab 300 mg, 77% with secukinumab 150 mg, and 5.1% with placebo. The PASI 100 response rate – meaning totally clear skin – was 31.9% with high-dose and 25.2% with lower-dose secukinumab, while there was a zero PASI 100 rate in controls.
The only adverse events more common than with placebo were nasopharyngitis and upper respiratory infections.
Dr. Reich predicted that as the ongoing TRANSFIGURE study continues well beyond 16 weeks, the nail psoriasis response rates will climb, since nails are so slow growing.
TRANSFIGURE and GESTURE are sponsored by Novartis, which markets secukinumab. Dr. Reich and Dr. Gottlieb reported having financial relationships with Novartis and numerous other pharmaceutical companies.
VANCOUVER, B.C. – The interleukin-17A inhibitor secukinumab demonstrated the greatest improvement in nail psoriasis ever reported from a randomized, placebo-controlled trial in the phase IIIb TRANSFIGURE study, Dr. Kristian Reich reported at the World Congress of Dermatology.
At 198 patients, TRANSFIGURE is the largest-ever prospective study in patients with moderate to severe chronic plaque psoriasis and significant nail involvement. And while only the 16-week results are available thus far, when TRANSFIGURE is completed after a planned 132 weeks of treatment, it will also be the longest-ever study in the treatment of nail psoriasis, noted Dr. Reich, a dermatologist in group practice in Hamburg, Germany.
Elsewhere at WCD 2015, Dr. Alice B. Gottlieb presented the week 16 results of the phase IIIb GESTURE study, in which 205 psoriasis patients with moderate to severe psoriasis of the palms and soles were randomized to subcutaneous secukinumab (Cosentyx) at 150 or 300 mg or placebo. Dosing was weekly for the first 5 weeks and monthly thereafter.
The primary endpoint, a palmoplantar Investigator’s Global Assessment scale score of 0 or 1 – clear or almost clear – at week 16 was 33.3% with secukinumab at 300 mg, 22.1% at 150 mg, and 1.5% with placebo. The average reduction in palmoplantar PASI (Psoriasis Area Severity Index) score from baseline was 54.6% with high-dose and 35.3% with low-dose secukinumab, compared with 4.1% in placebo-treated controls, reported Dr. Gottlieb, professor and chair of dermatology at Tufts University, Boston.
Like the TRANSFIGURE trial, GESTURE will continue for 132 weeks, with the initial placebo-treated controls being randomized to secukinumab at 150 or 300 mg after week 16.
Dr. Reich reported that by 16 weeks in TRANSFIGURE, mean scores on the Nail Psoriasis Severity Index had improved by 45.3%, compared with baseline, in patients on secukinumab 300 mg, 37.9% in those on secukinumab 150 mg, and 10.8% with placebo.
The results on the skin were dramatic: a PASI 75 rate of 87.1% with secukinumab 300 mg, 77% with secukinumab 150 mg, and 5.1% with placebo. The PASI 100 response rate – meaning totally clear skin – was 31.9% with high-dose and 25.2% with lower-dose secukinumab, while there was a zero PASI 100 rate in controls.
The only adverse events more common than with placebo were nasopharyngitis and upper respiratory infections.
Dr. Reich predicted that as the ongoing TRANSFIGURE study continues well beyond 16 weeks, the nail psoriasis response rates will climb, since nails are so slow growing.
TRANSFIGURE and GESTURE are sponsored by Novartis, which markets secukinumab. Dr. Reich and Dr. Gottlieb reported having financial relationships with Novartis and numerous other pharmaceutical companies.
VANCOUVER, B.C. – The interleukin-17A inhibitor secukinumab demonstrated the greatest improvement in nail psoriasis ever reported from a randomized, placebo-controlled trial in the phase IIIb TRANSFIGURE study, Dr. Kristian Reich reported at the World Congress of Dermatology.
At 198 patients, TRANSFIGURE is the largest-ever prospective study in patients with moderate to severe chronic plaque psoriasis and significant nail involvement. And while only the 16-week results are available thus far, when TRANSFIGURE is completed after a planned 132 weeks of treatment, it will also be the longest-ever study in the treatment of nail psoriasis, noted Dr. Reich, a dermatologist in group practice in Hamburg, Germany.
Elsewhere at WCD 2015, Dr. Alice B. Gottlieb presented the week 16 results of the phase IIIb GESTURE study, in which 205 psoriasis patients with moderate to severe psoriasis of the palms and soles were randomized to subcutaneous secukinumab (Cosentyx) at 150 or 300 mg or placebo. Dosing was weekly for the first 5 weeks and monthly thereafter.
The primary endpoint, a palmoplantar Investigator’s Global Assessment scale score of 0 or 1 – clear or almost clear – at week 16 was 33.3% with secukinumab at 300 mg, 22.1% at 150 mg, and 1.5% with placebo. The average reduction in palmoplantar PASI (Psoriasis Area Severity Index) score from baseline was 54.6% with high-dose and 35.3% with low-dose secukinumab, compared with 4.1% in placebo-treated controls, reported Dr. Gottlieb, professor and chair of dermatology at Tufts University, Boston.
Like the TRANSFIGURE trial, GESTURE will continue for 132 weeks, with the initial placebo-treated controls being randomized to secukinumab at 150 or 300 mg after week 16.
Dr. Reich reported that by 16 weeks in TRANSFIGURE, mean scores on the Nail Psoriasis Severity Index had improved by 45.3%, compared with baseline, in patients on secukinumab 300 mg, 37.9% in those on secukinumab 150 mg, and 10.8% with placebo.
The results on the skin were dramatic: a PASI 75 rate of 87.1% with secukinumab 300 mg, 77% with secukinumab 150 mg, and 5.1% with placebo. The PASI 100 response rate – meaning totally clear skin – was 31.9% with high-dose and 25.2% with lower-dose secukinumab, while there was a zero PASI 100 rate in controls.
The only adverse events more common than with placebo were nasopharyngitis and upper respiratory infections.
Dr. Reich predicted that as the ongoing TRANSFIGURE study continues well beyond 16 weeks, the nail psoriasis response rates will climb, since nails are so slow growing.
TRANSFIGURE and GESTURE are sponsored by Novartis, which markets secukinumab. Dr. Reich and Dr. Gottlieb reported having financial relationships with Novartis and numerous other pharmaceutical companies.
AT WCD 2015
Key clinical point: Two phase IIIb trials show secukinumab at 300 mg is the most effective drug ever formally studied for nail or palmoplantar psoriasis.
Major finding: At 16 weeks, secukinumab at 300 mg improved nail psoriasis by 45.3% and palmoplantar psoriasis by 33.3%.
Data source: The phase IIIb TRANSFIGURE and GESTURE studies, ongoing randomized, prospective, initially double-blind studies in which 198 patients with significant nail psoriasis and 205 with palmoplantar psoriasis received secukinumab at 150 or 300 mg or placebo. Both studies will continue out to 132 weeks.
Disclosures: TRANSFIGURE and GESTURE are sponsored by Novartis, which markets secukinumab. Dr. Reich and Dr. Gottlieb reported having financial relationships with Novartis and numerous other pharmaceutical companies.

EC approves edoxaban for patients with VTE, NVAF

Image by Andre E.X. Brown
The European Commission (EC) has approved edoxaban (Lixiana), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF who have one or more risk factors for stroke or systemic embolism, such as congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke, or transient ischemic attack.
The EC’s decision affects all 28 European Union member states, plus Iceland, Norway, and Liechtenstein. Edoxaban is already approved for use in the US, Japan, and Switzerland.
The EC based its approval of edoxaban on results of 2 phase 3 clinical trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
Image by Andre E.X. Brown
The European Commission (EC) has approved edoxaban (Lixiana), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF who have one or more risk factors for stroke or systemic embolism, such as congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke, or transient ischemic attack.
The EC’s decision affects all 28 European Union member states, plus Iceland, Norway, and Liechtenstein. Edoxaban is already approved for use in the US, Japan, and Switzerland.
The EC based its approval of edoxaban on results of 2 phase 3 clinical trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
Image by Andre E.X. Brown
The European Commission (EC) has approved edoxaban (Lixiana), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF who have one or more risk factors for stroke or systemic embolism, such as congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke, or transient ischemic attack.
The EC’s decision affects all 28 European Union member states, plus Iceland, Norway, and Liechtenstein. Edoxaban is already approved for use in the US, Japan, and Switzerland.
The EC based its approval of edoxaban on results of 2 phase 3 clinical trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
WCD: Smoking tied to worse occupational hand eczema
VANCOUVER, B.C. – Occupational hand eczema is worse and more persistent in smokers than nonsmokers, a large prospective cohort study found.
“Tobacco smoking is associated with work absenteeism and with not staying in the workforce due to occupational hand eczema. Smoking confers a worse prognosis and interferes with the outcome of prevention programs,” Dr. Richard Brans said at the World Congress of Dermatology.
Hand eczema is the most common occupational skin disease. Smoking might worsen signs and symptoms by inducing proinflammatory effects in the skin, said Dr. Brans, a dermatologist at the University of Osnabrück, Germany.
To better assess the link between smoking and hand eczema, he and his associates carried out a prospective 3-year study of 1,095 patients from throughout Germany. The patients initially had attended a 6-week residential treatment program for hand eczema that was followed by a 3-week outpatient program. Smokers comprised about half of the patients and resembled nonsmokers in terms of gender, general atopy, and degree of professional or occupational exposures, such as wetting or soiling the hands at work, Dr. Brans said. However, smokers were significantly younger than nonsmokers and were more likely to have allergic contact dermatitis, he noted.
The inpatient phase of the program markedly benefited both smokers and nonsmokers, but notably, smokers had significantly worse symptoms and signs of hand eczema at all time points assessed, Dr. Brans said. Furthermore, smokers missed an average of 37 days of work because of occupational hand eczema in the year before the program, compared with only 25 days for nonsmokers (P = .001), and smokers continued to miss more days of work because of hand eczema in the year after completing the program (P = .023), he reported. Significantly more smokers also left their professions because of their hand eczema, even after completing the prevention program (P = .021), he added.
The study found no link between number of cigarettes smoked per day and severity of hand eczema, Dr. Brans said. Smoking history was self-reported, and the study design excluded patients who changed their smoking behavior during follow-up, he noted. In addition, the researchers did not assess whether other factors associated with smoking might have confounded the association between smoking and severity of hand eczema, he said.
Dr. Brans reported no relevant disclosures.
VANCOUVER, B.C. – Occupational hand eczema is worse and more persistent in smokers than nonsmokers, a large prospective cohort study found.
“Tobacco smoking is associated with work absenteeism and with not staying in the workforce due to occupational hand eczema. Smoking confers a worse prognosis and interferes with the outcome of prevention programs,” Dr. Richard Brans said at the World Congress of Dermatology.
Hand eczema is the most common occupational skin disease. Smoking might worsen signs and symptoms by inducing proinflammatory effects in the skin, said Dr. Brans, a dermatologist at the University of Osnabrück, Germany.
To better assess the link between smoking and hand eczema, he and his associates carried out a prospective 3-year study of 1,095 patients from throughout Germany. The patients initially had attended a 6-week residential treatment program for hand eczema that was followed by a 3-week outpatient program. Smokers comprised about half of the patients and resembled nonsmokers in terms of gender, general atopy, and degree of professional or occupational exposures, such as wetting or soiling the hands at work, Dr. Brans said. However, smokers were significantly younger than nonsmokers and were more likely to have allergic contact dermatitis, he noted.
The inpatient phase of the program markedly benefited both smokers and nonsmokers, but notably, smokers had significantly worse symptoms and signs of hand eczema at all time points assessed, Dr. Brans said. Furthermore, smokers missed an average of 37 days of work because of occupational hand eczema in the year before the program, compared with only 25 days for nonsmokers (P = .001), and smokers continued to miss more days of work because of hand eczema in the year after completing the program (P = .023), he reported. Significantly more smokers also left their professions because of their hand eczema, even after completing the prevention program (P = .021), he added.
The study found no link between number of cigarettes smoked per day and severity of hand eczema, Dr. Brans said. Smoking history was self-reported, and the study design excluded patients who changed their smoking behavior during follow-up, he noted. In addition, the researchers did not assess whether other factors associated with smoking might have confounded the association between smoking and severity of hand eczema, he said.
Dr. Brans reported no relevant disclosures.
VANCOUVER, B.C. – Occupational hand eczema is worse and more persistent in smokers than nonsmokers, a large prospective cohort study found.
“Tobacco smoking is associated with work absenteeism and with not staying in the workforce due to occupational hand eczema. Smoking confers a worse prognosis and interferes with the outcome of prevention programs,” Dr. Richard Brans said at the World Congress of Dermatology.
Hand eczema is the most common occupational skin disease. Smoking might worsen signs and symptoms by inducing proinflammatory effects in the skin, said Dr. Brans, a dermatologist at the University of Osnabrück, Germany.
To better assess the link between smoking and hand eczema, he and his associates carried out a prospective 3-year study of 1,095 patients from throughout Germany. The patients initially had attended a 6-week residential treatment program for hand eczema that was followed by a 3-week outpatient program. Smokers comprised about half of the patients and resembled nonsmokers in terms of gender, general atopy, and degree of professional or occupational exposures, such as wetting or soiling the hands at work, Dr. Brans said. However, smokers were significantly younger than nonsmokers and were more likely to have allergic contact dermatitis, he noted.
The inpatient phase of the program markedly benefited both smokers and nonsmokers, but notably, smokers had significantly worse symptoms and signs of hand eczema at all time points assessed, Dr. Brans said. Furthermore, smokers missed an average of 37 days of work because of occupational hand eczema in the year before the program, compared with only 25 days for nonsmokers (P = .001), and smokers continued to miss more days of work because of hand eczema in the year after completing the program (P = .023), he reported. Significantly more smokers also left their professions because of their hand eczema, even after completing the prevention program (P = .021), he added.
The study found no link between number of cigarettes smoked per day and severity of hand eczema, Dr. Brans said. Smoking history was self-reported, and the study design excluded patients who changed their smoking behavior during follow-up, he noted. In addition, the researchers did not assess whether other factors associated with smoking might have confounded the association between smoking and severity of hand eczema, he said.
Dr. Brans reported no relevant disclosures.
AT WDC 2015
Key clinical point: Smoking might worsen the signs and symptoms of occupational hand eczema.
Major finding: Smokers had significantly worse symptoms and signs of hand eczema at all time points assessed.
Data source: Three-year prospective study of 1,095 smokers and nonsmokers with occupational hand eczema.
Disclosures: Dr. Brans reported no relevant conflicts of interest.
CVD becomes second-largest cause of death in U.K.
For the first time since the middle of the 20th century, cardiovascular disease is not the main cause of death overall in the United Kingdom, according to 2012 data published in Heart.
Cancer narrowly took the lead, with 29% of mortalities in 2012 having resulted from this disease, compared to the 28% of deaths that resulted from cardiovascular disease (CVD). But CVD remains the largest killer of women in the U.K.
In 2012, 28% of all female deaths and 32% of all male deaths were caused by CVD. The highest cause of mortality for men was cancer, with 32% of male deaths having resulted from that disease. A slightly smaller percentage of female deaths – 27% – was caused by cancer than by CVD. The Office for National Statistics (ONS), the National Records of Scotland, and the Northern Ireland Statistics and Research Agency provided the data.
Of the CVD deaths, 46%, or just under 73,500, were from coronary heart disease (CHD) and 26%, or about 41,000, were from stroke.
CVD caused more than a quarter of premature deaths – defined as deaths occurring in people younger than 75 – in men and 18% of premature deaths in women. CHD was the most common cause of premature death in U.K. men.
CVD death rates also varied per region of the United Kingdom, with higher percentages of the populations of Scotland and the north of England having died of CVD than the percentage of people living in the south of England who died from the disease, according to age-standardized death rates by local authorities. Glasgow City, Scotland, had the highest CVD mortality, with 144/100,0000 people having died prematurely and 400/100,000 people having died of the disease.
“The improvements in survival [of people with CVD] mean that there is now a high prevalence of people living with CVD,” according to Prachi Bhatnagar, Ph.D., and her colleagues.
The numbers of people suffering from CHD, stroke, atrial fibrillation and heart failure in the U.K. in 2012 and 2013 were approximately 2.3 million, 1.2 million, 1 million and 480,000, respectively, Quality of Outcomes Framework data suggest. The number of operations carried out to treat CHD is increasing in the United Kingdom, with greater than 90,000 percutaneous coronary interventions (PCIs) having been carried out in 2012 – more than twice as many as had been performed a decade earlier.
“CVD remains a substantial burden to the U.K., both in terms of health and economic costs,” according to the researchers.
Read the full study in Heart (doi:10.1136/heartjnl-2015-307516).
For the first time since the middle of the 20th century, cardiovascular disease is not the main cause of death overall in the United Kingdom, according to 2012 data published in Heart.
Cancer narrowly took the lead, with 29% of mortalities in 2012 having resulted from this disease, compared to the 28% of deaths that resulted from cardiovascular disease (CVD). But CVD remains the largest killer of women in the U.K.
In 2012, 28% of all female deaths and 32% of all male deaths were caused by CVD. The highest cause of mortality for men was cancer, with 32% of male deaths having resulted from that disease. A slightly smaller percentage of female deaths – 27% – was caused by cancer than by CVD. The Office for National Statistics (ONS), the National Records of Scotland, and the Northern Ireland Statistics and Research Agency provided the data.
Of the CVD deaths, 46%, or just under 73,500, were from coronary heart disease (CHD) and 26%, or about 41,000, were from stroke.
CVD caused more than a quarter of premature deaths – defined as deaths occurring in people younger than 75 – in men and 18% of premature deaths in women. CHD was the most common cause of premature death in U.K. men.
CVD death rates also varied per region of the United Kingdom, with higher percentages of the populations of Scotland and the north of England having died of CVD than the percentage of people living in the south of England who died from the disease, according to age-standardized death rates by local authorities. Glasgow City, Scotland, had the highest CVD mortality, with 144/100,0000 people having died prematurely and 400/100,000 people having died of the disease.
“The improvements in survival [of people with CVD] mean that there is now a high prevalence of people living with CVD,” according to Prachi Bhatnagar, Ph.D., and her colleagues.
The numbers of people suffering from CHD, stroke, atrial fibrillation and heart failure in the U.K. in 2012 and 2013 were approximately 2.3 million, 1.2 million, 1 million and 480,000, respectively, Quality of Outcomes Framework data suggest. The number of operations carried out to treat CHD is increasing in the United Kingdom, with greater than 90,000 percutaneous coronary interventions (PCIs) having been carried out in 2012 – more than twice as many as had been performed a decade earlier.
“CVD remains a substantial burden to the U.K., both in terms of health and economic costs,” according to the researchers.
Read the full study in Heart (doi:10.1136/heartjnl-2015-307516).
For the first time since the middle of the 20th century, cardiovascular disease is not the main cause of death overall in the United Kingdom, according to 2012 data published in Heart.
Cancer narrowly took the lead, with 29% of mortalities in 2012 having resulted from this disease, compared to the 28% of deaths that resulted from cardiovascular disease (CVD). But CVD remains the largest killer of women in the U.K.
In 2012, 28% of all female deaths and 32% of all male deaths were caused by CVD. The highest cause of mortality for men was cancer, with 32% of male deaths having resulted from that disease. A slightly smaller percentage of female deaths – 27% – was caused by cancer than by CVD. The Office for National Statistics (ONS), the National Records of Scotland, and the Northern Ireland Statistics and Research Agency provided the data.
Of the CVD deaths, 46%, or just under 73,500, were from coronary heart disease (CHD) and 26%, or about 41,000, were from stroke.
CVD caused more than a quarter of premature deaths – defined as deaths occurring in people younger than 75 – in men and 18% of premature deaths in women. CHD was the most common cause of premature death in U.K. men.
CVD death rates also varied per region of the United Kingdom, with higher percentages of the populations of Scotland and the north of England having died of CVD than the percentage of people living in the south of England who died from the disease, according to age-standardized death rates by local authorities. Glasgow City, Scotland, had the highest CVD mortality, with 144/100,0000 people having died prematurely and 400/100,000 people having died of the disease.
“The improvements in survival [of people with CVD] mean that there is now a high prevalence of people living with CVD,” according to Prachi Bhatnagar, Ph.D., and her colleagues.
The numbers of people suffering from CHD, stroke, atrial fibrillation and heart failure in the U.K. in 2012 and 2013 were approximately 2.3 million, 1.2 million, 1 million and 480,000, respectively, Quality of Outcomes Framework data suggest. The number of operations carried out to treat CHD is increasing in the United Kingdom, with greater than 90,000 percutaneous coronary interventions (PCIs) having been carried out in 2012 – more than twice as many as had been performed a decade earlier.
“CVD remains a substantial burden to the U.K., both in terms of health and economic costs,” according to the researchers.
Read the full study in Heart (doi:10.1136/heartjnl-2015-307516).
FROM HEART
CHMP recommends panobinostat for MM

Photo courtesy of the CDC
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending regulatory approval for panobinostat (Farydak) to treat relapsed and/or refractory multiple myeloma (MM).
The drug is indicated for use in combination with bortezomib and dexamethasone to treat adults with MM who have received at least 2 prior treatment regimens, including bortezomib and an immunomodulatory agent (IMiD).
Panobinostat is already approved for this indication in the US and Chile.
If the European Commission follows the CHMP’s recommendation, panobinostat will be the first histone deacetylase (HDAC) inhibitor approved to treat MM in the European Union.
The European Commission generally follows CHMP recommendations and delivers its final decision within 3 months of the recommendation. The decision will be applicable to all 28 European Union member states, plus Iceland, Norway, and Liechtenstein.
“Panobinostat is the first and only HDAC inhibitor recommended by the CHMP for the treatment of patients living with multiple myeloma who have progressed after standard-of-care therapy with bortezomib and an IMiD,” said Alessandro Riva, MD, Global Head of Oncology Development and Medical Affairs at Novartis Oncology, the company developing panobinostat.
“We are pleased with the positive CHMP opinion on panobinostat for previously treated patients because it brings us one step closer to providing a new treatment option for patients in need in Europe.”
The CHMP’s recommendation is based on efficacy and safety data in a subgroup analysis of 147 patients on the phase 3 PANORAMA-1 trial. Results from this analysis were recently presented at the 2015 ASCO Annual Meeting.
PANORAMA-1 was designed to compare panobinostat in combination with bortezomib and dexamethasone to bortezomib and dexamethasone alone in patients who had relapsed or relapsed and refractory MM.
At ASCO, researchers presented results in 147 patients who had received 2 or more prior treatment regimens, including bortezomib and an IMiD.
The patients who received panobinostat, bortezomib, and dexamethasone had a longer median progression-free survival than patients who received bortezomib and dexamethasone alone—12.5 months and 4.7 months, respectively (hazard ratio=0.47).
Common grade 3/4 adverse events in both treatment arms were thrombocytopenia, lymphopenia, neutropenia, diarrhea, asthenia/fatigue, and peripheral neuropathy. About 7% of patients in both arms died while on treatment. ![]()
Photo courtesy of the CDC
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending regulatory approval for panobinostat (Farydak) to treat relapsed and/or refractory multiple myeloma (MM).
The drug is indicated for use in combination with bortezomib and dexamethasone to treat adults with MM who have received at least 2 prior treatment regimens, including bortezomib and an immunomodulatory agent (IMiD).
Panobinostat is already approved for this indication in the US and Chile.
If the European Commission follows the CHMP’s recommendation, panobinostat will be the first histone deacetylase (HDAC) inhibitor approved to treat MM in the European Union.
The European Commission generally follows CHMP recommendations and delivers its final decision within 3 months of the recommendation. The decision will be applicable to all 28 European Union member states, plus Iceland, Norway, and Liechtenstein.
“Panobinostat is the first and only HDAC inhibitor recommended by the CHMP for the treatment of patients living with multiple myeloma who have progressed after standard-of-care therapy with bortezomib and an IMiD,” said Alessandro Riva, MD, Global Head of Oncology Development and Medical Affairs at Novartis Oncology, the company developing panobinostat.
“We are pleased with the positive CHMP opinion on panobinostat for previously treated patients because it brings us one step closer to providing a new treatment option for patients in need in Europe.”
The CHMP’s recommendation is based on efficacy and safety data in a subgroup analysis of 147 patients on the phase 3 PANORAMA-1 trial. Results from this analysis were recently presented at the 2015 ASCO Annual Meeting.
PANORAMA-1 was designed to compare panobinostat in combination with bortezomib and dexamethasone to bortezomib and dexamethasone alone in patients who had relapsed or relapsed and refractory MM.
At ASCO, researchers presented results in 147 patients who had received 2 or more prior treatment regimens, including bortezomib and an IMiD.
The patients who received panobinostat, bortezomib, and dexamethasone had a longer median progression-free survival than patients who received bortezomib and dexamethasone alone—12.5 months and 4.7 months, respectively (hazard ratio=0.47).
Common grade 3/4 adverse events in both treatment arms were thrombocytopenia, lymphopenia, neutropenia, diarrhea, asthenia/fatigue, and peripheral neuropathy. About 7% of patients in both arms died while on treatment. ![]()
Photo courtesy of the CDC
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending regulatory approval for panobinostat (Farydak) to treat relapsed and/or refractory multiple myeloma (MM).
The drug is indicated for use in combination with bortezomib and dexamethasone to treat adults with MM who have received at least 2 prior treatment regimens, including bortezomib and an immunomodulatory agent (IMiD).
Panobinostat is already approved for this indication in the US and Chile.
If the European Commission follows the CHMP’s recommendation, panobinostat will be the first histone deacetylase (HDAC) inhibitor approved to treat MM in the European Union.
The European Commission generally follows CHMP recommendations and delivers its final decision within 3 months of the recommendation. The decision will be applicable to all 28 European Union member states, plus Iceland, Norway, and Liechtenstein.
“Panobinostat is the first and only HDAC inhibitor recommended by the CHMP for the treatment of patients living with multiple myeloma who have progressed after standard-of-care therapy with bortezomib and an IMiD,” said Alessandro Riva, MD, Global Head of Oncology Development and Medical Affairs at Novartis Oncology, the company developing panobinostat.
“We are pleased with the positive CHMP opinion on panobinostat for previously treated patients because it brings us one step closer to providing a new treatment option for patients in need in Europe.”
The CHMP’s recommendation is based on efficacy and safety data in a subgroup analysis of 147 patients on the phase 3 PANORAMA-1 trial. Results from this analysis were recently presented at the 2015 ASCO Annual Meeting.
PANORAMA-1 was designed to compare panobinostat in combination with bortezomib and dexamethasone to bortezomib and dexamethasone alone in patients who had relapsed or relapsed and refractory MM.
At ASCO, researchers presented results in 147 patients who had received 2 or more prior treatment regimens, including bortezomib and an IMiD.
The patients who received panobinostat, bortezomib, and dexamethasone had a longer median progression-free survival than patients who received bortezomib and dexamethasone alone—12.5 months and 4.7 months, respectively (hazard ratio=0.47).
Common grade 3/4 adverse events in both treatment arms were thrombocytopenia, lymphopenia, neutropenia, diarrhea, asthenia/fatigue, and peripheral neuropathy. About 7% of patients in both arms died while on treatment. ![]()
Helping patients with insomnia to help themselves
A patient handout from the health services department of the University of California, Berkley titled “Insomnia Self-Care Guide” teaches patients how their behaviors might be contributing to their sleep difficulties. In addition to providing general information about sleep, the handout, which is available at http://www.uhs.berkeley.edu/home/healthtopics/PDF%20Handouts/Insomnia.pdf, also includes a checklist of questions about behaviors tied to insomnia, such as “Do you drink alcohol in the evenings?” and suggestions for inducing sleep, such as exercising daily.
A patient handout from the health services department of the University of California, Berkley titled “Insomnia Self-Care Guide” teaches patients how their behaviors might be contributing to their sleep difficulties. In addition to providing general information about sleep, the handout, which is available at http://www.uhs.berkeley.edu/home/healthtopics/PDF%20Handouts/Insomnia.pdf, also includes a checklist of questions about behaviors tied to insomnia, such as “Do you drink alcohol in the evenings?” and suggestions for inducing sleep, such as exercising daily.
A patient handout from the health services department of the University of California, Berkley titled “Insomnia Self-Care Guide” teaches patients how their behaviors might be contributing to their sleep difficulties. In addition to providing general information about sleep, the handout, which is available at http://www.uhs.berkeley.edu/home/healthtopics/PDF%20Handouts/Insomnia.pdf, also includes a checklist of questions about behaviors tied to insomnia, such as “Do you drink alcohol in the evenings?” and suggestions for inducing sleep, such as exercising daily.